/



























































































































































































































































































































































































































































































































Теги: медицина фармакопея
Год: 2001




Текст
МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ’Я УКРАЇНИ
Державний департамент з контролю за якістю,
безпекою та виробництвом лікарських засобів і виробів
медичного призначення
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
перше видання
Введено в дію з 1 жовтня 2001 року
наказом Міністра охорони здоров’я України
від 12 березня 2001 року № 95
Розроблено Державним підприємством
«Науково-експертний фармакопейний центр»
на підставі Європейської Фармакопеї
Харків 2001
ЗМІСТ
1. РЕДАКЦІЙНА КОЛЕГІЯ ДЕРЖАВНОЇ ФАРМАКОПЕЇ УКРАЇНИ і
II. СЕКРЕТАРІА Т РЕДАКЦІЙНОЇ КОЛЕГІЇ ДЕРЖАВНОЇ
ФАРМАКОПЕЇ УКРАЇНИ уп
III. ВІДПОВІДАЛЬНІ ОСОБИ уш
IV. ОРГАНІЗАЦІЇ ТА УСТАНОВИ УКРАЇНИ, ЩО БЕРУТЬ УЧАСТЬ
У РОЗРОБЦІ ДЕРЖАВНОЇ ФАРМАКОПЕЇ УКРА ЇНИ їх
V. ВСТУП хі
ЗАГАЛЬНІ СТАТТІ 1
1. ЗАГАЛЬНІ ЗАУВАЖЕННЯ З
1.1. ЗАГАЛЬНІ ПОЛОЖЕННЯ З
1.2. ІНШІ ПОЛОЖЕННЯ, ЩО ПОШИРЮЮТЬСЯ НА ЗАГАЛЬНІ
СТА ТТІЙ МОНОГРА ФІЇ З
1.3. ЗАГАЛЬНІ СТАТТІ 4
1.4. МОНОГРАФІЇ 5
1.5. СКОРОЧЕННЯ ТА ПОЗНАЧЕННЯ 8
1.6. ОДИНИЦІ МІЖНАРОДНОЇ СИСТЕМИ (СІ), ВИКОРИСТОВУВАНІ
У ФАРМАКОПЕЇ, І ЇХНЯ ВІДПОВІДНІСТЬ ІНШИМ ОДИНИЦЯМ 9
2. МЕТОДИ АНАЛІЗУ 13
2.1. ОБЛАДНАННЯ 13
2.1.2. Порівняльна таблиця пористості скляних фільтрів 13
2.1.4. Сита 13
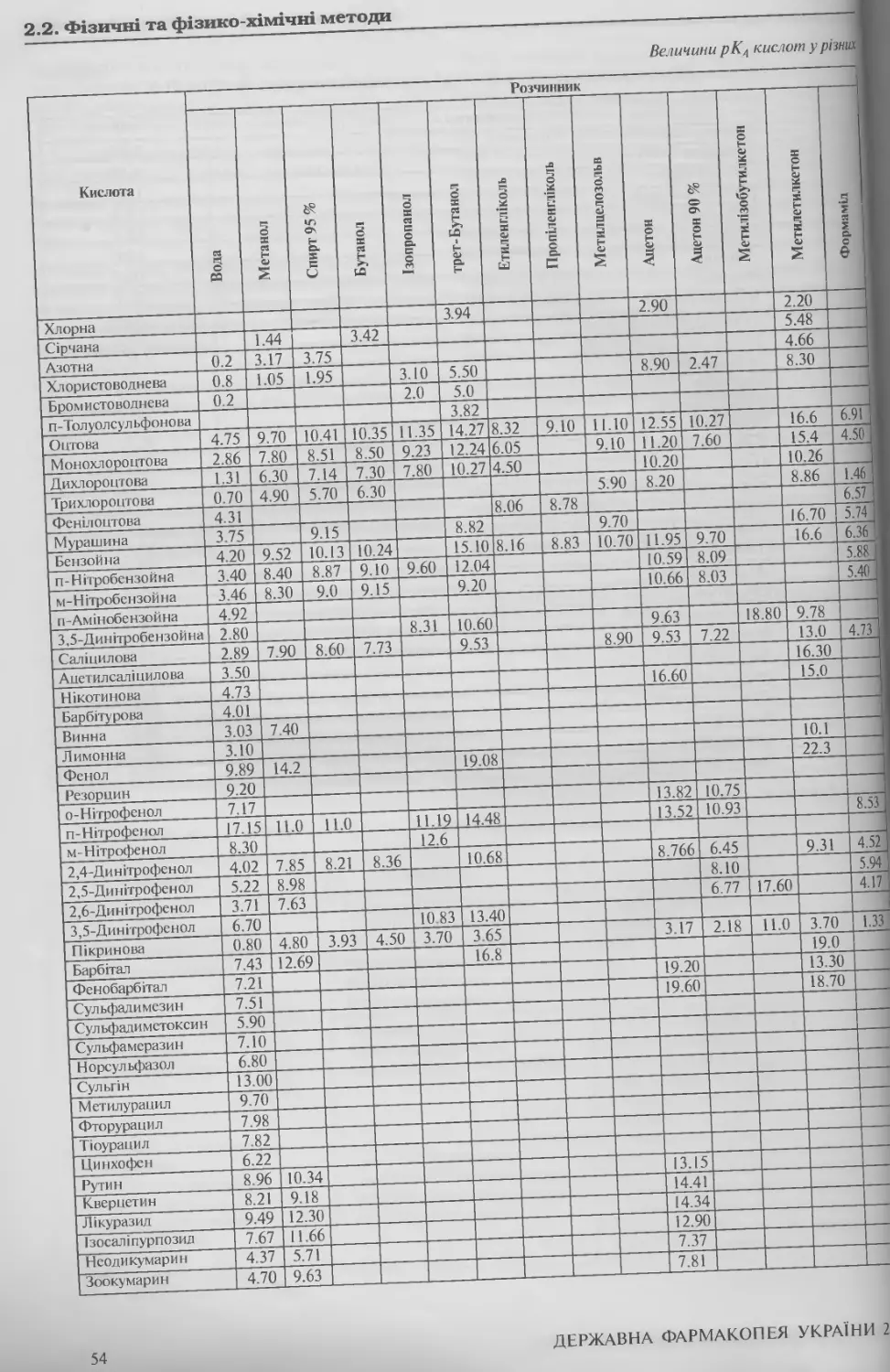
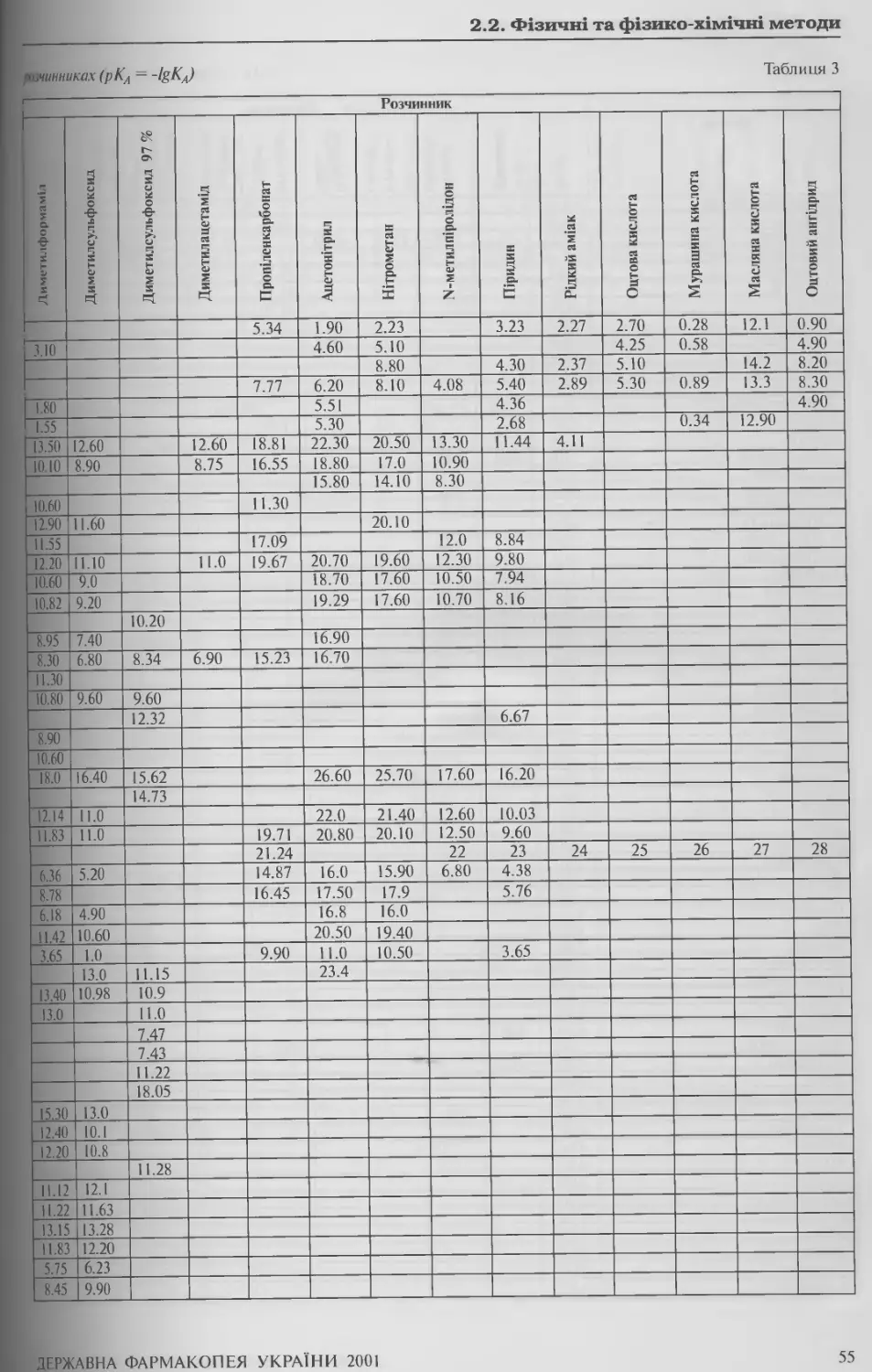
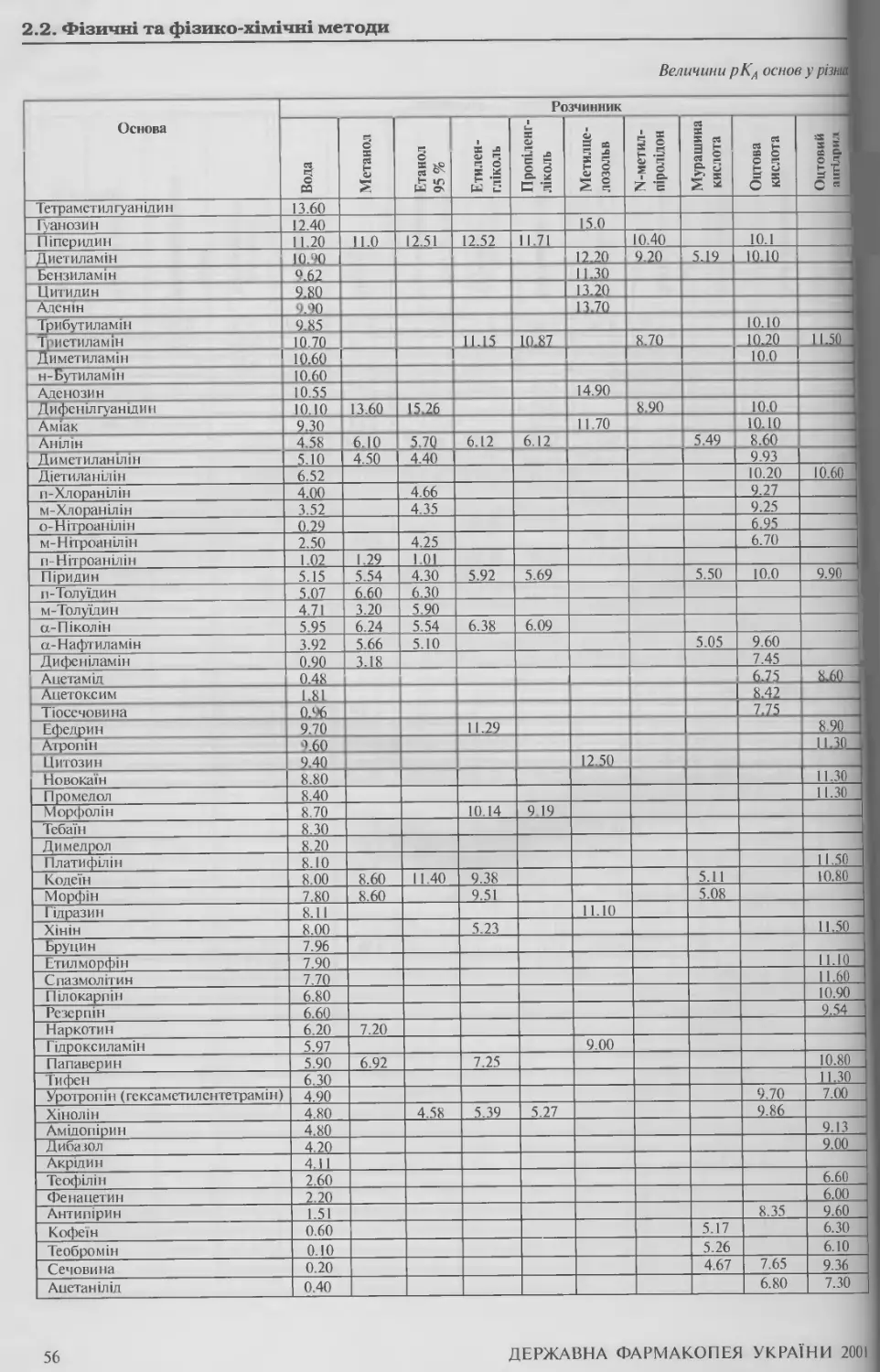
2.2. ФІЗИЧНІ ТА ФІЗИКО-ХІМІЧНІ МЕТОДИ 15
2.2.1 Визначення прозорості і ступеня каламутності рідин 15
2.2.2. Визначення ступеня забарвлення рідин 15
2.2.3. Потенціометричне визначення рН 17
2.2.4. Залежність між реакцією розчину, приблизним
значенням рН і кольором індикаторів 19
2.2.5 Відносна густина 19
2.2.6. Показник заломлення (індекс рефракції) 21
2.2.7. Оптичне обертання 22
2.2.8. В’язкість 23
2.2.9. Метод капілярної віскозиметрії 23
2.2.10. Метод ротаційної віскозиметрії 24
2.2.11. Температурні межі перегонки 25
2.2.13. Визначення води методом відгону 26
2.2.14. Температура плавлення — капілярний метод 27
2.2.15. Температура плавлення — відкритий капілярний метод 27
2.2.16. Температура плавлення — метод миттєвого плавлення 28
2.2.17. Температура краплепадіння 28
2.2.18. Температура тверднення 29
2.2.19. Амперометричне титрування ЗО
2.2.20. Потенціометричне титрування ЗО
2.2.22. Атомно-емісійна спектрометрія 32
2.2.23. Атомно-абсорбційна спектрометрія 32
2.2.24. Абсорбційна спектрофотометрія в інфрачервоній області 34
2.2.25. Абсорбційна спектрофотометрія в ультрафіолетовій
і видимій областях 36
2.2.27. Тонкошарова хроматографія 41
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2.28. Газова хроматографія 44
2.2.29. Рідинна хроматографія 47
2.2.32. Втрата в масі при висушуванні 49
2.2.35. Осмоляльність 50
N Титрування у неводних розчинниках1^ 51
N Валідапія аналітичних методик і випробувань1^ 58
2.3. ІДЕНТИФІКАЦІЯ 68
2.3.1. Реакції ідентифікації на іони і функціональні групи 68
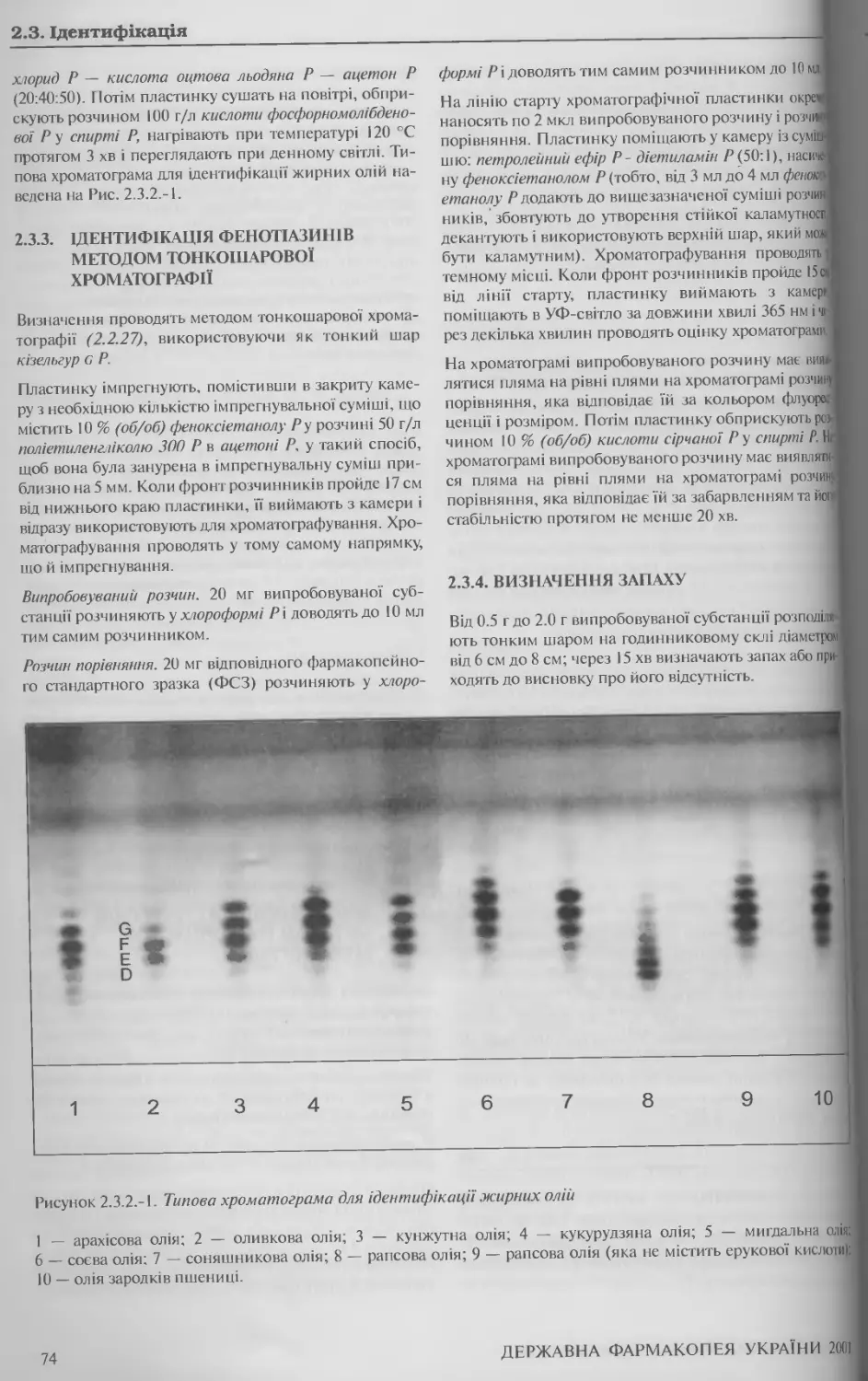
2.3.2. Ідентифікація жирних олій методом тонкошарової
хроматографії 73
2.3.3. Ідентифікація фенотіазинів методом тонкошарової
хроматографії 74
2.3.4. Визначення запаху 74
2.4. ВИПРОБУВАННЯ НА ГРАНИЧНИЙ ВМІСТ ДОМІШОК 75
2.4.1. Амонію солі 75
2.4.2. Арсен 75
2.4.3. Кальцій 76
2.4.4. Хлориди 76
2.4.5. Фториди 77
2.4.6. Магній 77
2.4.7. Магній і лужноземельні метали 77
2.4.8. Важкі метали 78
2.4.9. Залізо 80
2.4.10. Свинець у цукрах 80
2.4.11. Фосфати 80
2.4.12. Калій 80
2.4.13. Сульфати 81
2.4.14. Сульфатна зола 81
2.4.15. Нікель у поліолах 81
2.4.16. Загальна зола 81
2.4.17. Алюміній 82
2.4.18. Вільний формальдегід 82
2.4.19. Лужні домішки у жирних оліях 82
2.4.20. Антиоксиданти у жирних оліях 83
2.4.21. Сторонні олії у жирних оліях методом тонкошарової хроматографії 84
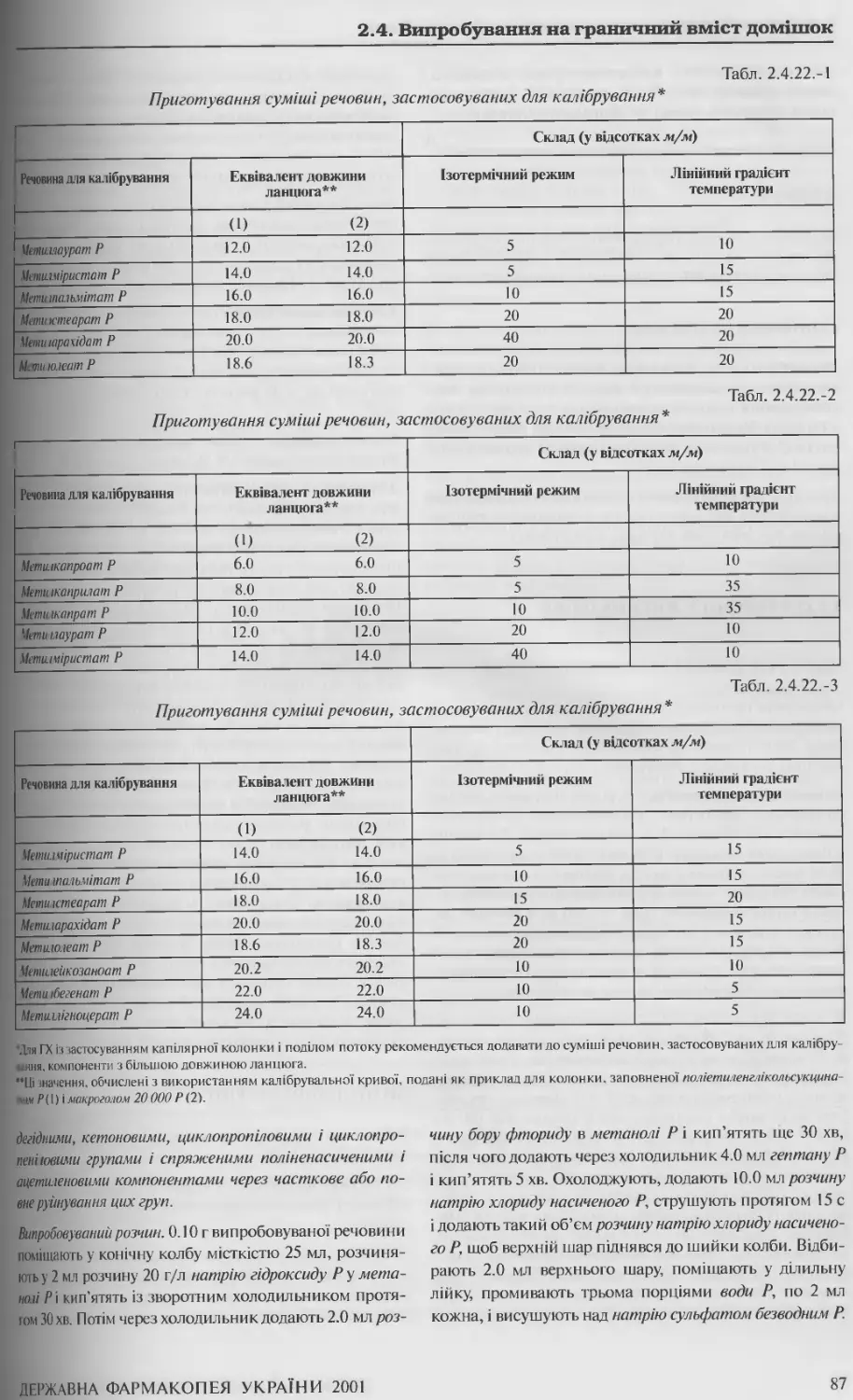
2.4.22. Сторонні олії у жирних оліях методом газової хроматографії 85
2.4.23. Стерини у жирних оліях 88
2.4.25. Залишкові кількості етиленоксиду і діоксану 90
2.4.26. 1Ч,Г<-диметиланілін 91
2.4.27. Нікель у гідрогенізованих рослинних оліях 92
2.4.28. 2-Етилгексанова кислота 92
2.4.29. Цинк 93
2.4.30. Речовини, що легко обвуглюються 93
2.5. МЕТОДИ КІЛЬКІСНОГО ВИЗНАЧЕННЯ 94
2.5.1. Кислотне число 94
2.5.2. Ефірне число 94
2.5.3. Гідроксильне число 94
2.5.4. Йодне число 95
2.5.5. Перекисне число 96
2.5.6. Число омилення 97
2.5.7. Неомилювані речовини 97
2.5.9. Визначення азоту після мінералізації сірчаною кислотою 98
2.5.11. Комплексометричне титрування 98
2.5.12. Визначення води напівмікрометодом (метод К. Фішера) 99
2.6. БІОЛОГІЧНІ ВИПРОБУВАННЯ 101
2.6.1. Стерильність 101
2.6.8. Пірогени 107
2.6.9. Аномальна токсичність 109
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6.11. Депресорні речовини 110
2.6.12. Випробування мікробіологічної чистоти нестерильних лікарських
засобів (визначення загального числа життєздатних аеробних
мікроорганізмів) 111
2.6.13. Випробування мікробіологічної чистоти нестерильних лікарських
засобів (випробування на окремі види мікроорганізмів) 115
2.6.14. Бактеріальні ендотоксини 127
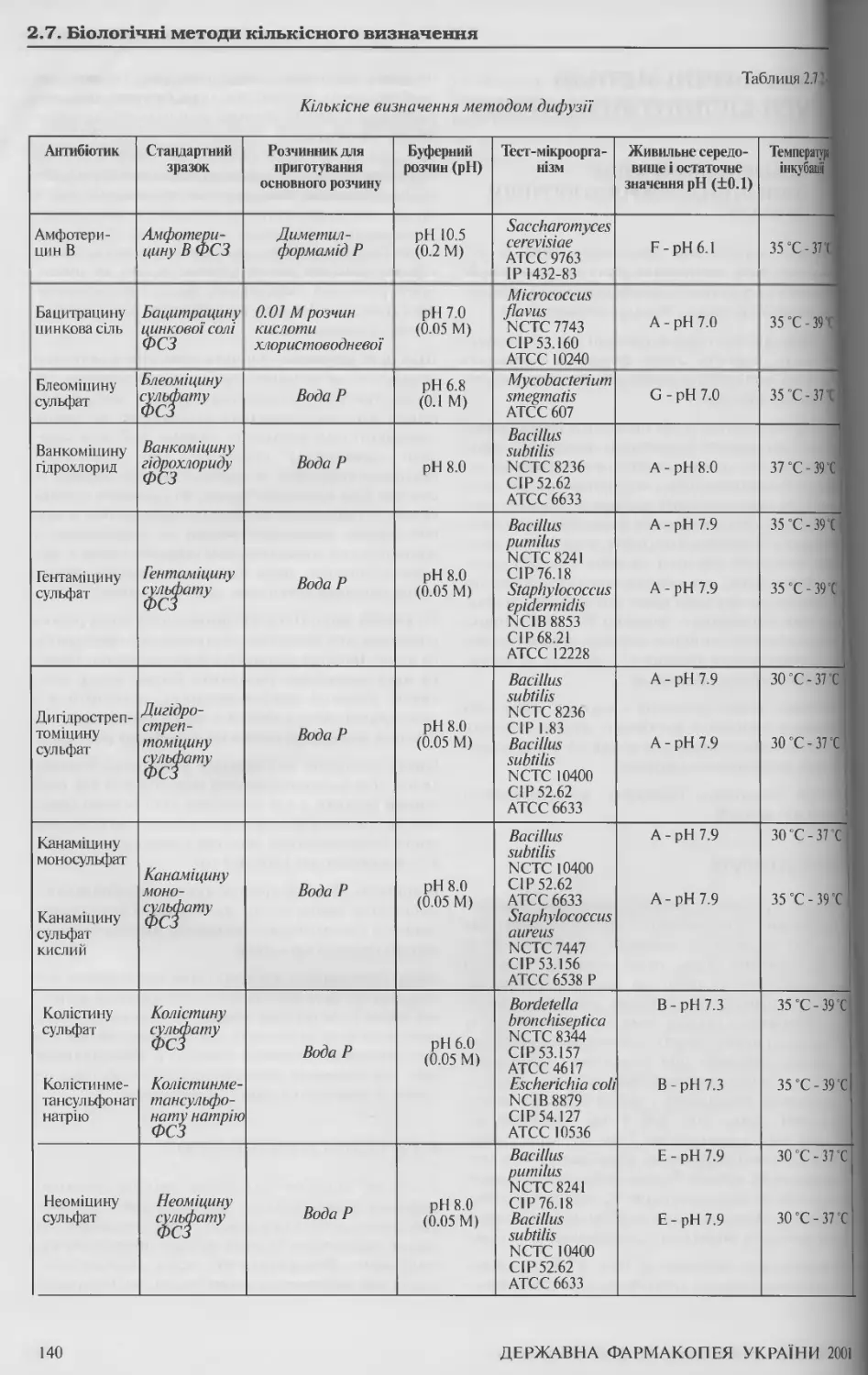
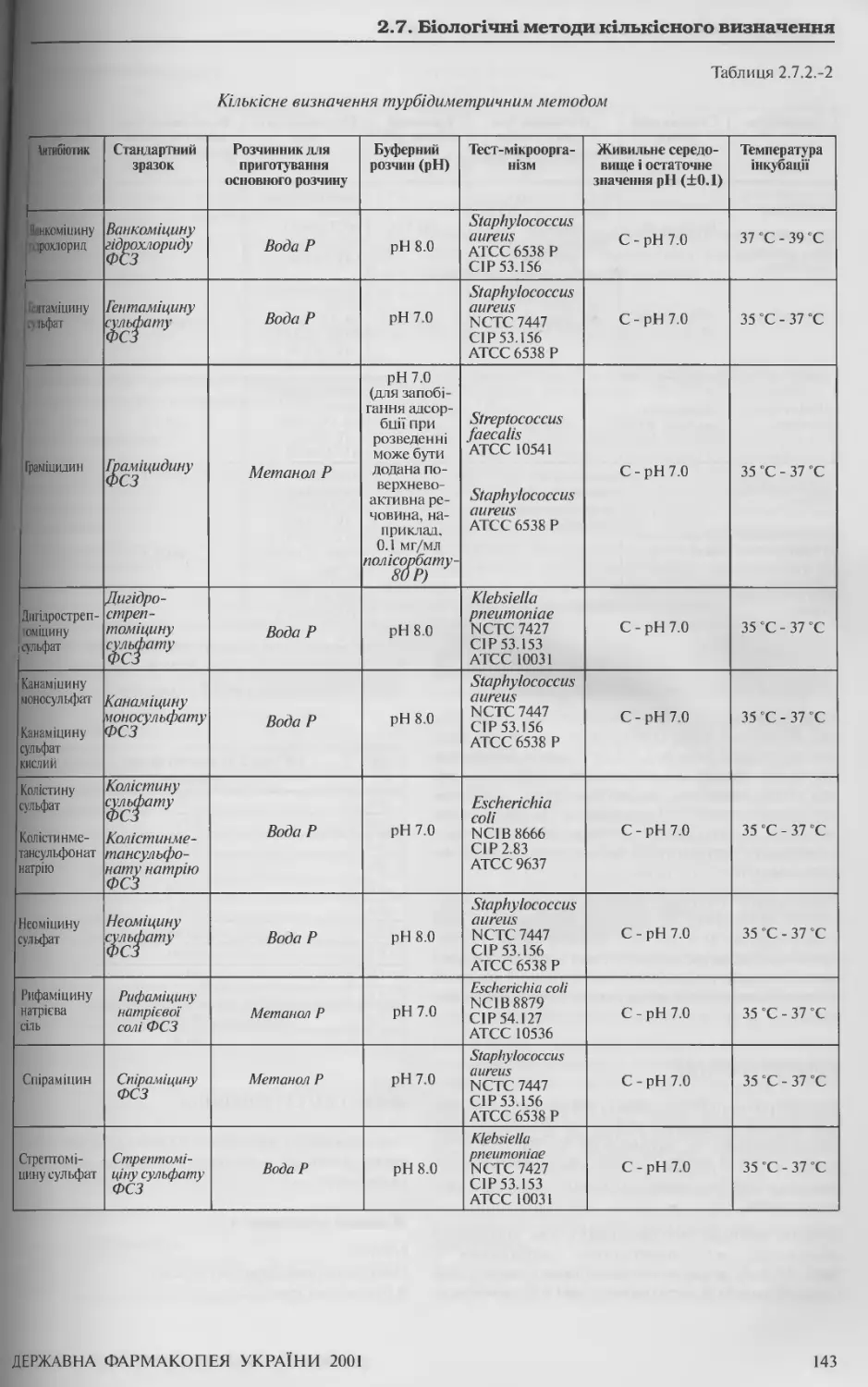
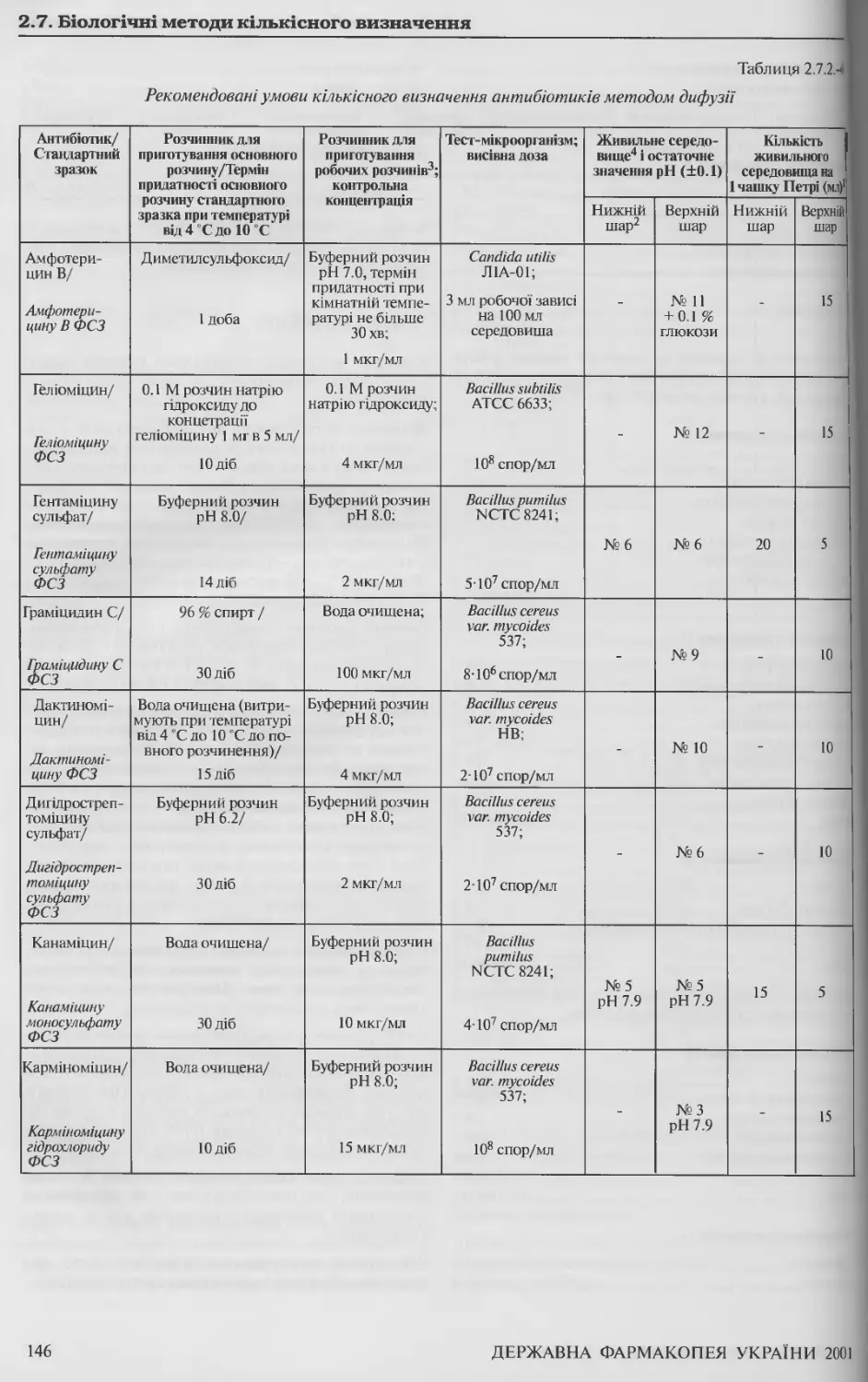
2.7. БІОЛОГІЧНІ МЕТОДИ КІЛЬКІСНОГО ВИЗНА ЧЕННЯ 139
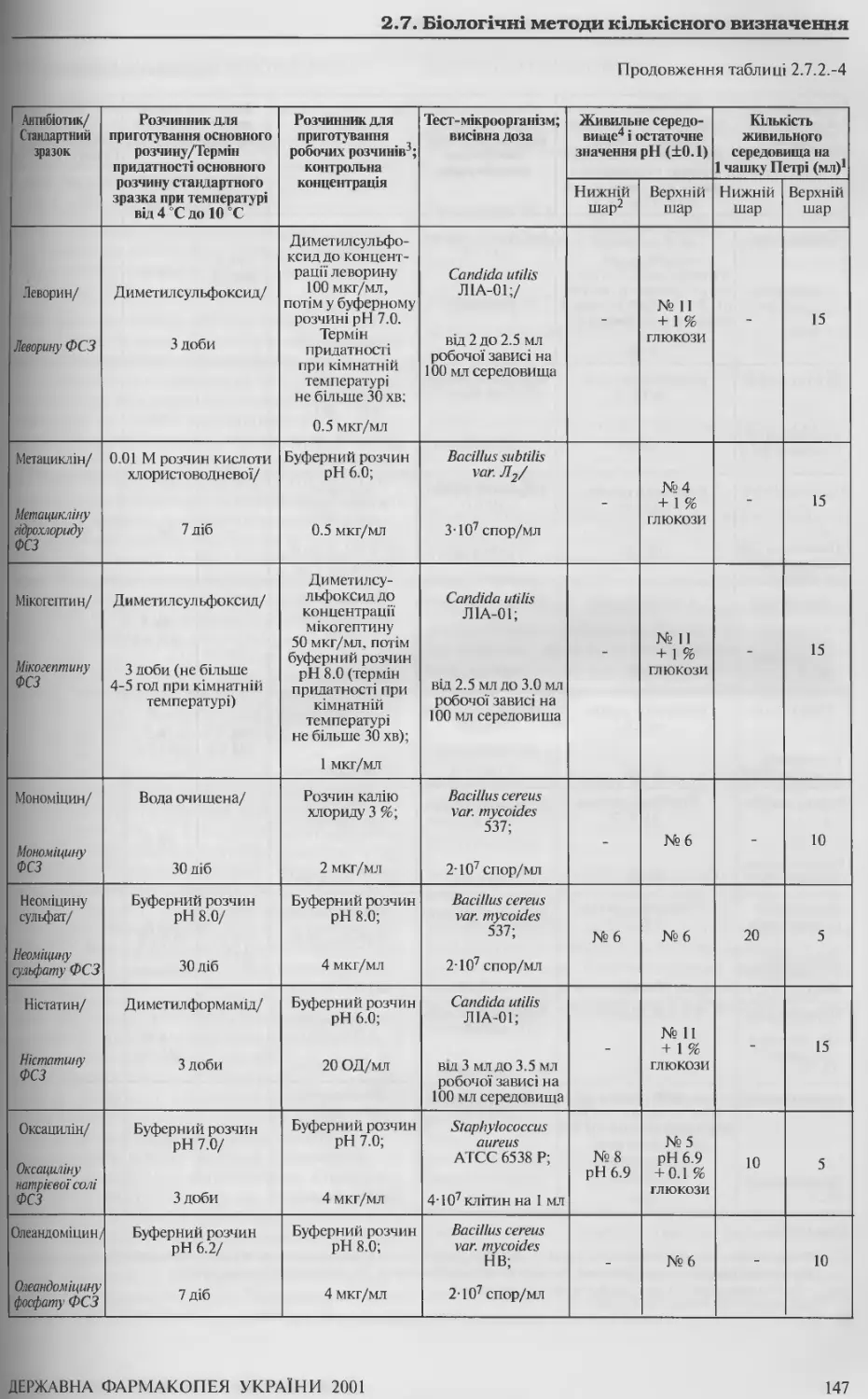
2.7.2 Кількісне визначення антибіотиків мікробіологічним методом 139
2.9. ФАРМАКО-ТЕХНОЛОГІЧНІ ВИПРОБУВАННЯ 151
2.9.1. Розпадання таблеток і капсул 151
2.9.2. Розпадання супозиторіїв і песаріїв 151
2.9.3. Тест "Розчинення" для твердих дозованих форм 153
2.9.5. Однорідність маси для одиниці дозованого лікарського засобу 157
2.9.6 Однорідність вмісту діючої речовини в одиниці дозованого
лікарського засобу 158
2.9.7. Стираність таблеток без оболонки 160
2.9.8. Стійкість таблеток до роздавлювання 161
2.9.12. Ситовий аналіз 162
2.9.13. Визначення розміру часток порошків методом мікроскопії 162
2.9.15. Насипний об’єм 162
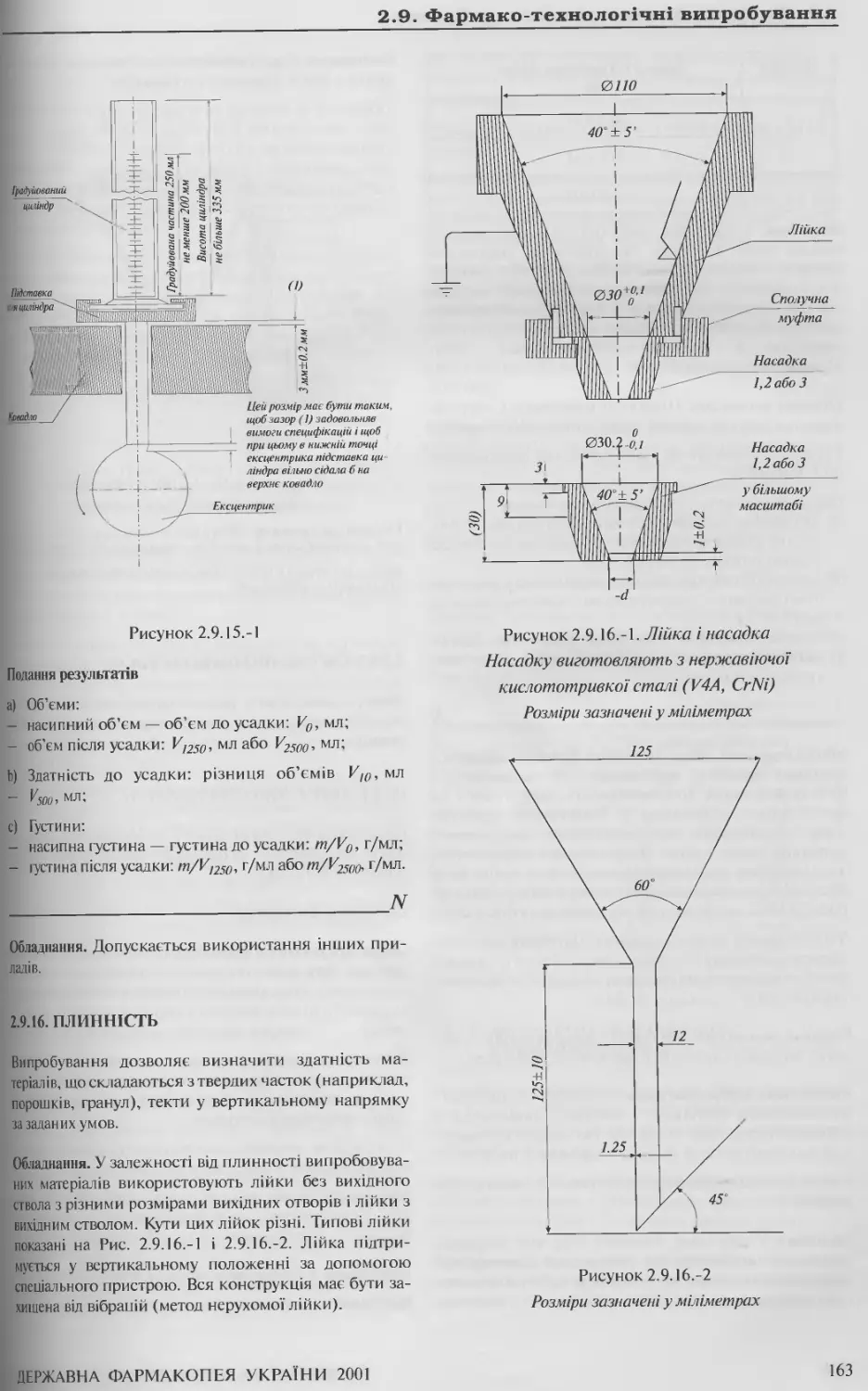
2.9.16 Плинність 163
2.9.17. Об’єм, що витягається 164
2.9.19. Механічні включення: невидимі частки 165
2.9.20. Механічні включення: видимі частки 166
2.9.21. Механічні включення: метод мікроскопії 166
4. РЕАКТИВИ 169
4.1. РЕАКТИВИ, ЕТАЛОННІ РОЗЧИНИ, БУФЕРНІ РОЗЧИНИ 169
4.1 1. Реактиви 169
4.1.2. Еталонні розчини для випробувань на граничний вміст домішок 279
4.1.3. Буферні розчини 284
4.2. РЕАКТИВИ, ТИТРОВАНІ РОЗЧИНИ ДЛЯ ОБ’ЄМНОГО АНАЛІЗУ 290
4.2.1. Вихідні стандартні речовини для титрованих розчинів 290
4.2.2. Титровані розчини 290
5. ЗАГАЛЬНІ ТЕКСТИ 297
5.1.1. Методи приготування стерильних продуктів 297
5.1.2. Біологічні індикатори стерилізації 299
5.1.3. Ефективність антимікробних консервантів 301
5.1.4. Мікробіологічна чистота лікарських засобів 303
5.4. ЗАЛИШКОВІ КІЛЬКОСТІ ОРГАНІЧНИХ РОЗЧИННИКІВ" 306
МОНОГРАФІЇ 311
Аланін 313
Аміаку розчин концентрований 314
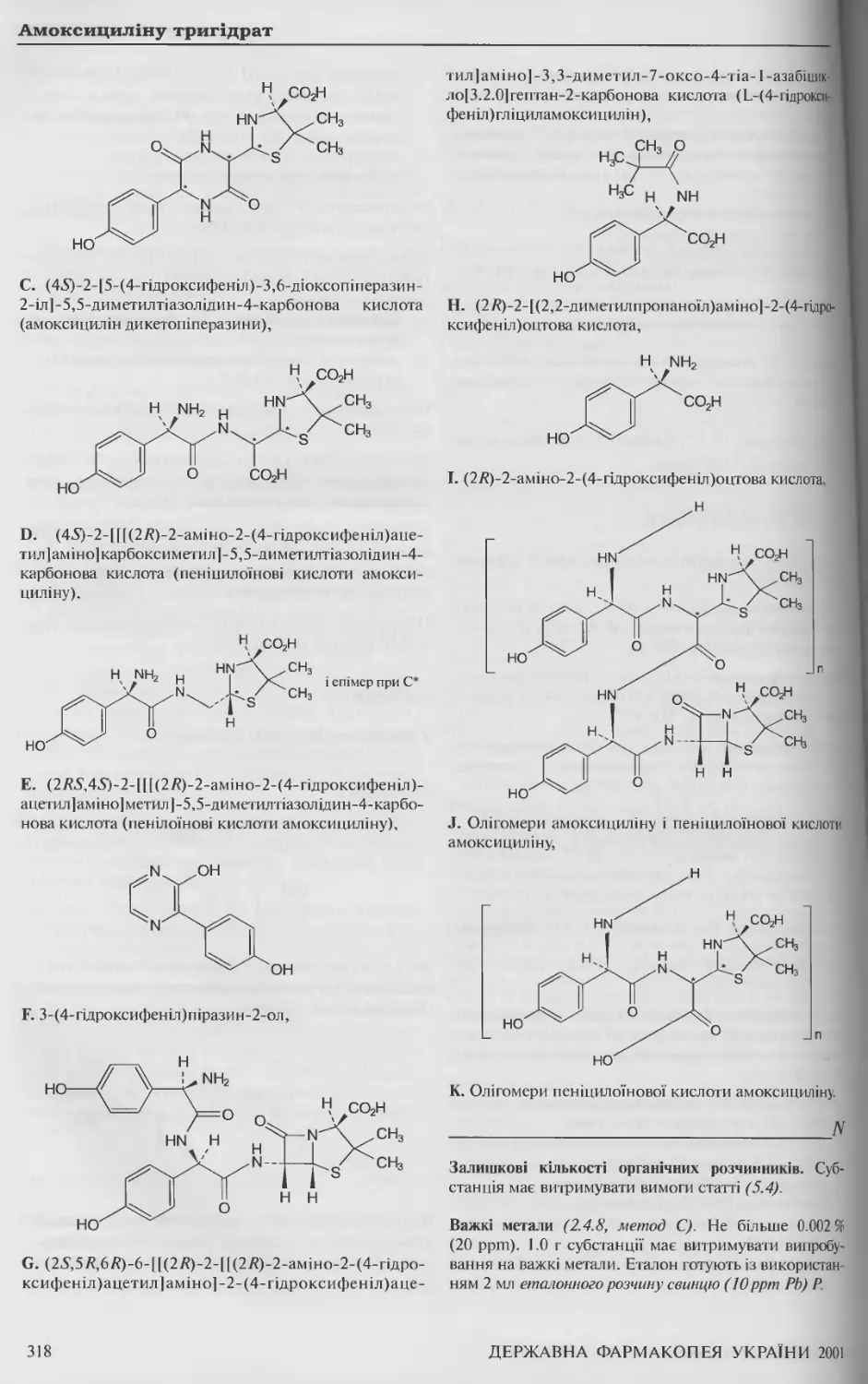
Амоксициліну тригідрат 315
Аргінін 319
Аргініну гідрохлорид 320
Атенолол 322
Атропіну сульфат 323
Ацетилцистеїн 325
Бензилпеніциліну калієва сіль 329
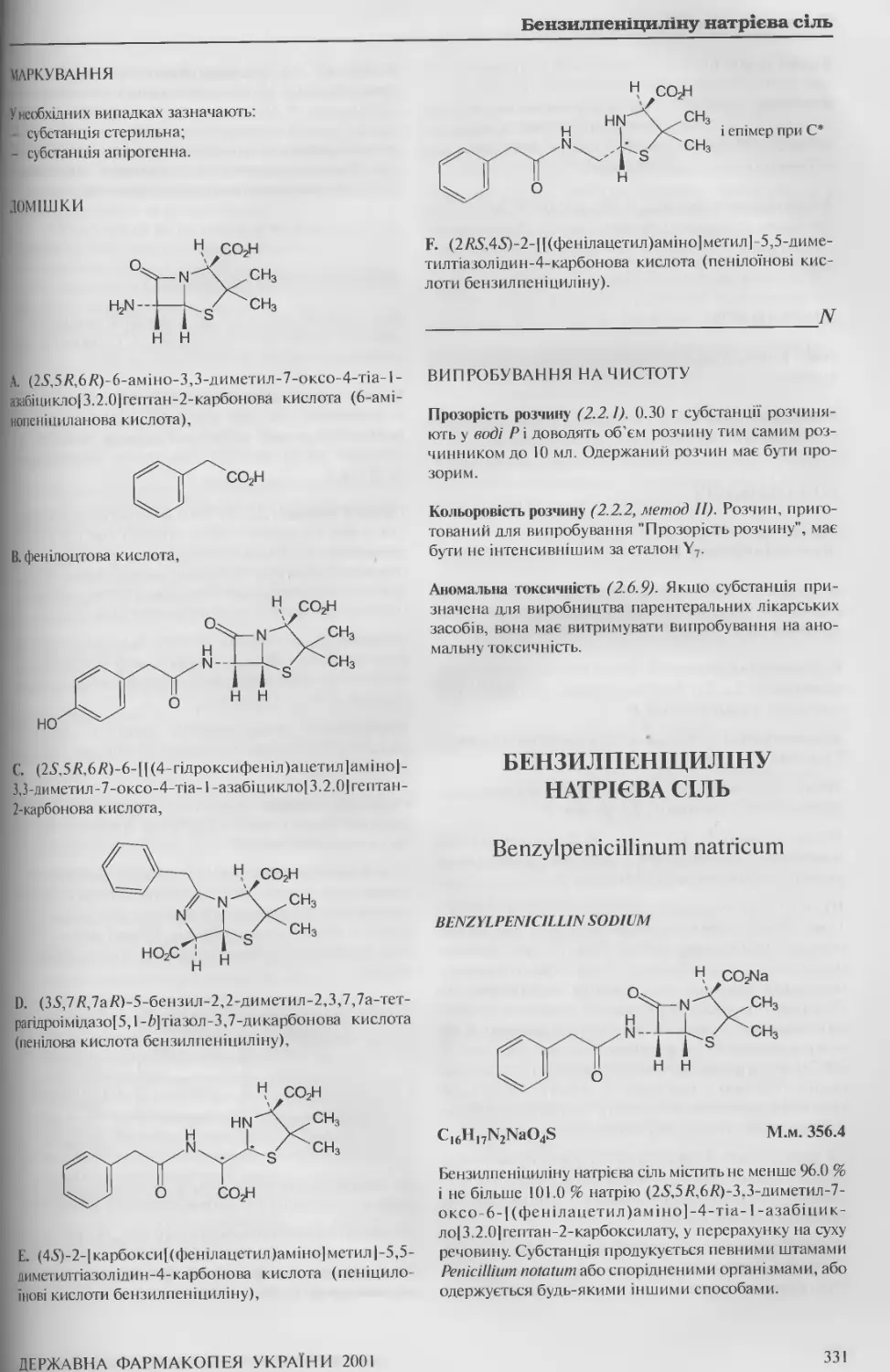
Бензилпеніциліну натрієва сіль 331
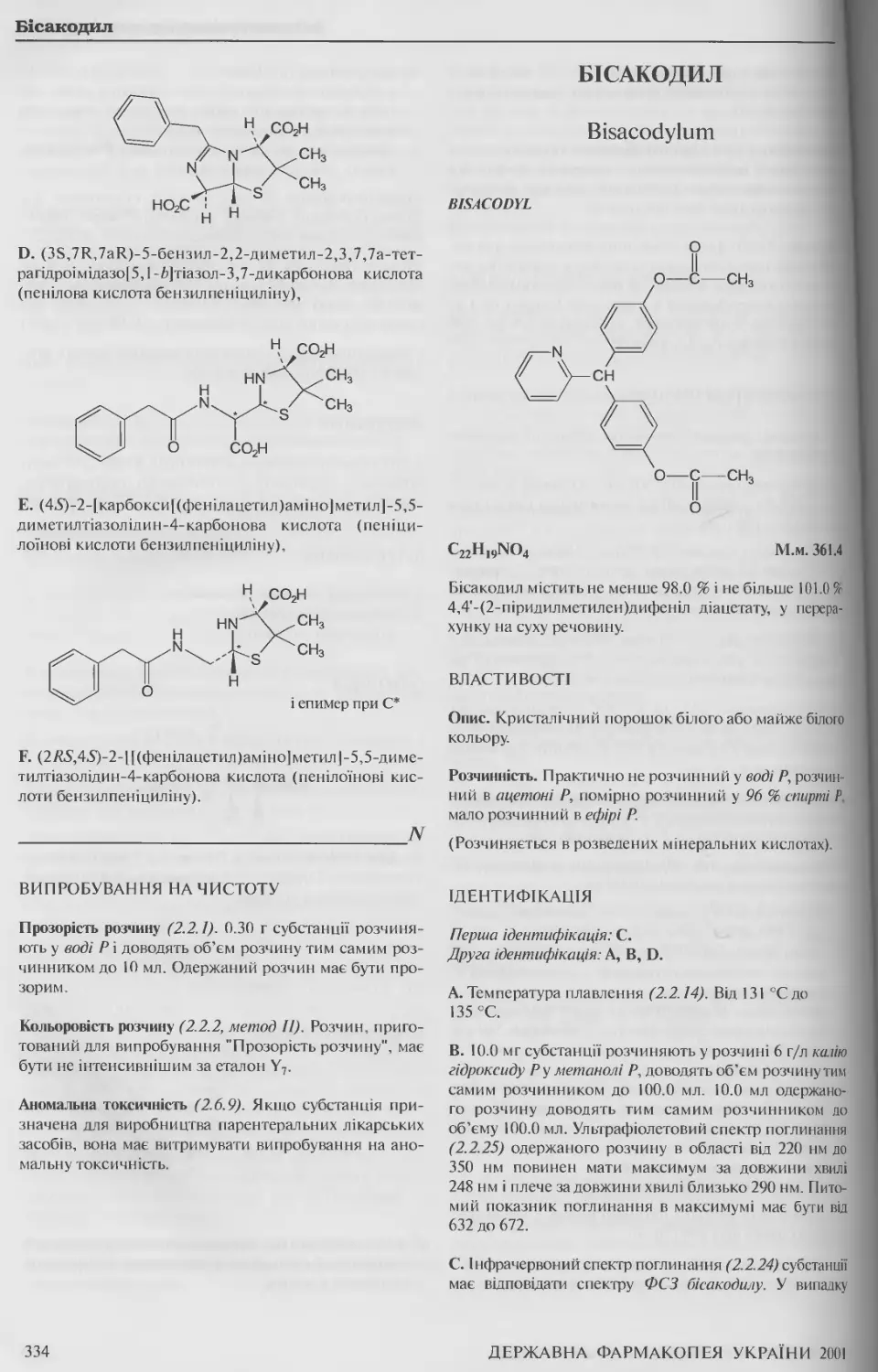
Бісакодил 334
Бромгексину гідрохлорид 335
Бупренорфіну гідрохлорид 336
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Валін 339
Ванілін 340
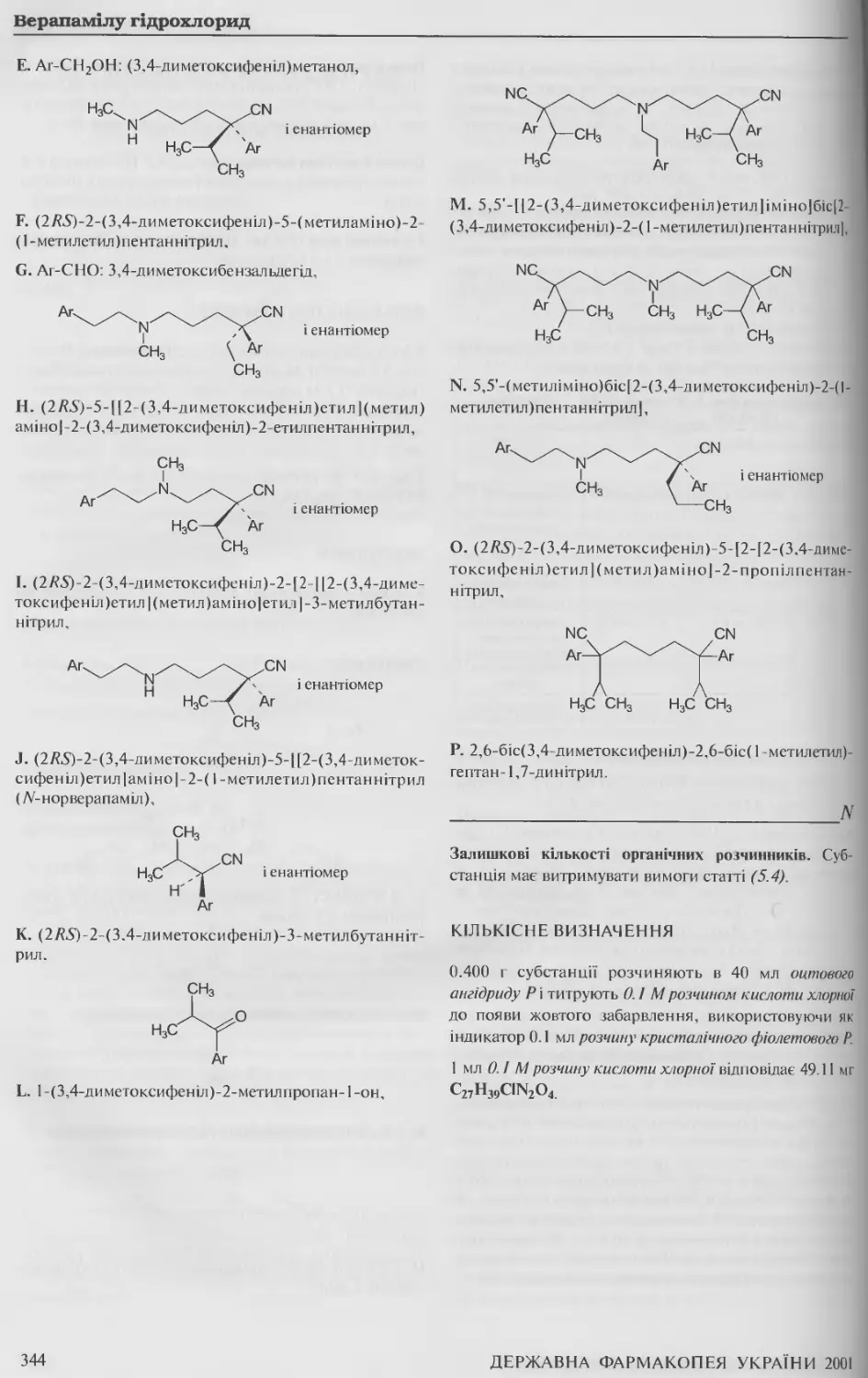
Верапамілу гідрохлорид 342
Галоперидол 345
Гентаміцину сульфат 347
Гістидин 349
Гістидину гідрохлорид моногідрат 351
Гліцерин 353
Гліцерин (85 %) 355
Гліцерину тринітрату розчин 357
Гліцин 359
Глюкоза безводна 360
Діазепам 363
Динатрію фосфат дигідрат 364
Динатрію фосфат додекагідрат 365
Дисульфірам 366
Допаміну гідрохлорид 367
Еконазолу нітрат 369
Етилморфіну гідрохлорид 370
Етилолеат 371
Ізолейцин 373
Ізосорбіду динітрат розведений 374
Кальцію глюконат 379
Кальцію глюконат для ін’єкцій 380
Камфора рацемічна 382
Канаміцину моносульфат 383
Каптоприл 385
Кетаміну гідрохлорид 386
Кислота аскорбінова 388
Кислота аспарагінова 389
Кислота ацетилсаліцилова 391
Кислота борна 392
Кислота глутамінова 393
Кислота лимонна безводна 395
Кислота нікотинова 396
Кислота сорбінова 397
Клонідину гідрохлорид 398
Кофеїн 399
Кофеїну моногідрат 400
Лейцин 403
Лізину гідрохлорид 404
Лінкоміцину гідрохлорид 406
Магнію оксид важкий 409
Магнію оксид легкий 410
Метилпарагідроксибензоат 411
Метіонін 412
Налоксону гідрохлорид дигідрат 415
Напроксен 417
Натрію дигідрофосфат дигідрат 418
Натрію диклофенак 419
Натрію метабісульфіт 420
Натрію тетраборат 421
Натрію фторид 422
Натрію цитрат 423
Нікотинамід 424
Нітразепам 425
Нітрофурал 426
Оксазепам 429
Орнітину гідрохлорид14 430
Пентоксифілін 433
Піперазину адипінат 434
Піридоксину гідрохлорид 435
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Прокаїнаміду гідрохлорид 437
Прокаїну гідрохлорид 438
Пролін 439
Прометазину гідрохлорид 441
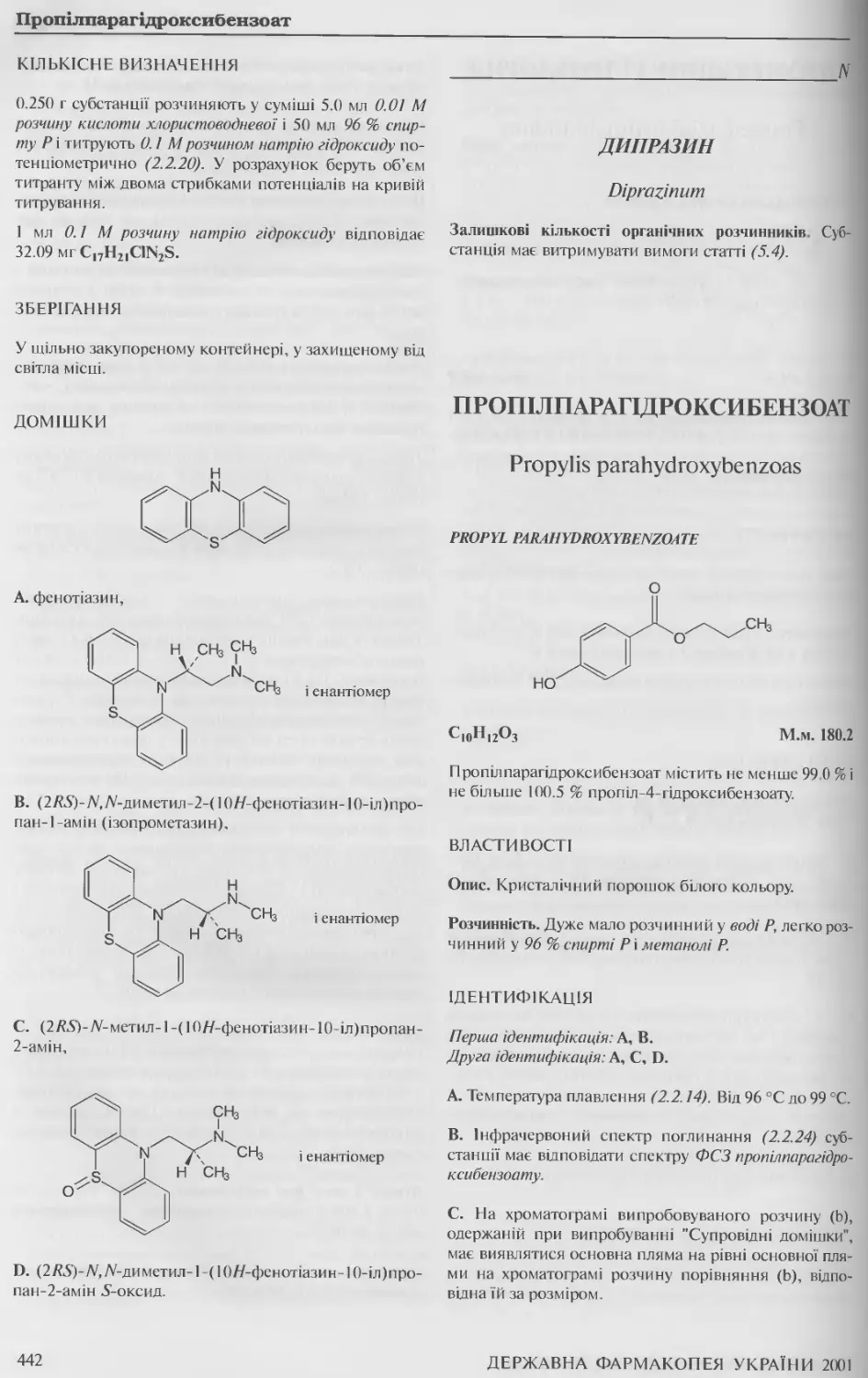
Пропілпарагідроксибензоат 442
Резорцин 445
Рибофлавін 446
Серин 449
Спирт ізопропіловий 450
Твердий жир 453
Тіаміну гідробромід14 454
Тіаміну гідрохлорид 456
Тирозин 458
Треонін 459
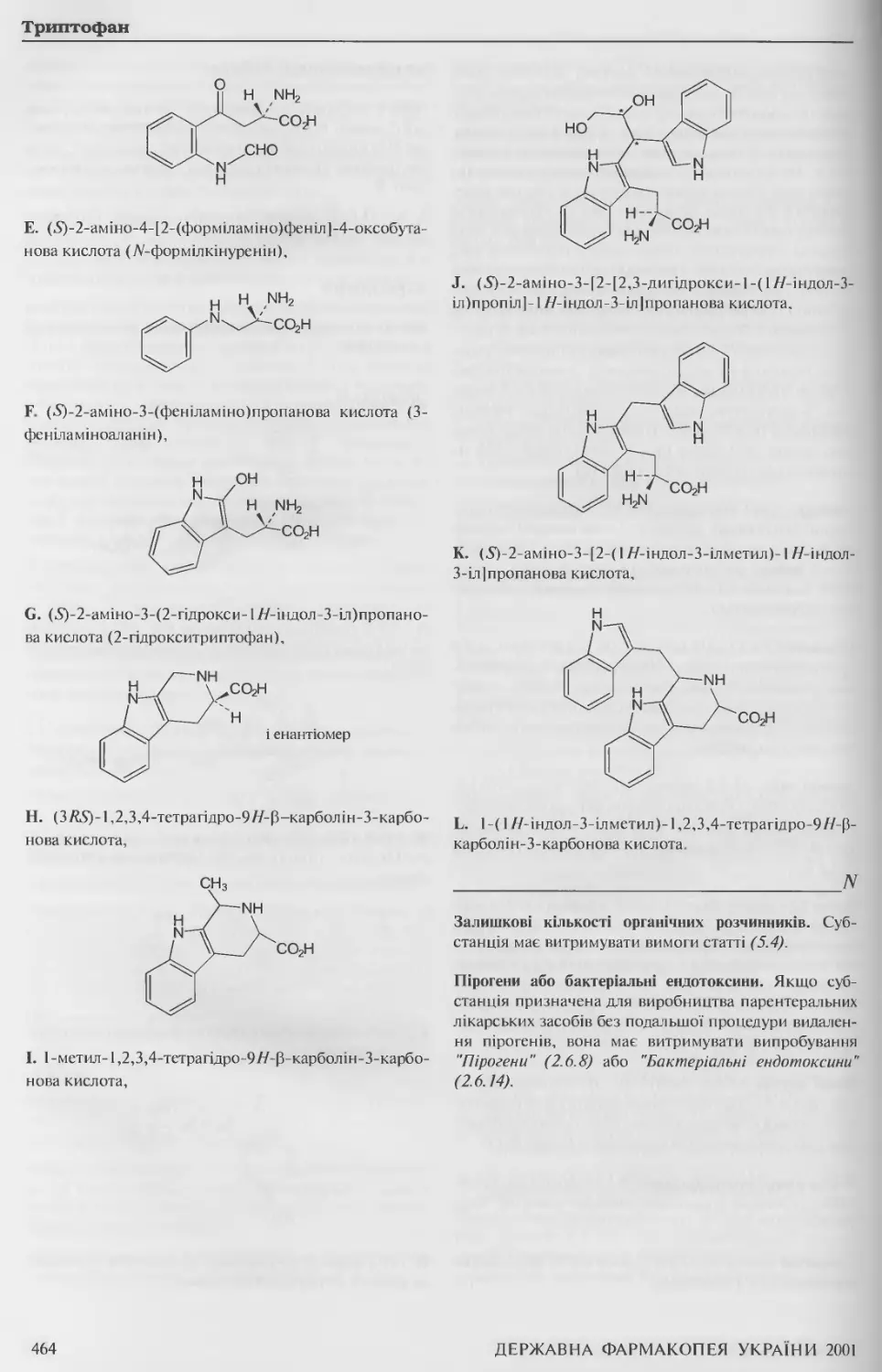
Триптофан 461
Фенілаланін 465
Фенол 466
Фторурацил 469
Фуросемід 470
Хлорамін 473
Хлорпромазину гідрохлорид 474
Цефалексин 477
Цефтриаксону натрієва сіль 479
Циклофосфамід 481
Цистеїн14 483
ЗАГАЛЬНІ СТАТТІ НА ЛІКАРСЬКІ ФОРМИ ТА СУБСТАНЦІЇ 485
Вушні лікарські засоби 487
Гранули 488
Екстракти 490
Капсули 493
Лікарські засоби для вагінального застосування 495
Лікарські засоби для парентерального застосування 497
Лікарські засоби для ректального застосування 502
Лікарські засоби, що знаходяться під тиском 506
М’які лікарські засоби для місцевого застосування14 507
Назальні лікарські засоби 511
Настойки 513
Очні лікарські засоби 515
Піни медичні 518
Порошки для зовнішнього застосування 519
Порошки для орального застосування 521
Рідкі лікарські засоби для орального застосування 522
Субстанції14 524
Таблетки 527
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Організації та установи
IV. ОРГАНІЗАЦІЇ ТА УСТАНОВИ УКРАЇНИ,
ЩО БЕРУТЬ УЧАСТЬ У РОЗРОБЦІ ДЕРЖАВНОЇ
ФАРМАКОПЕЇ УКРАЇНИ
1. “Біолік”, ЗАТ, Харків
1. “Біостимулятор", дочірнє підприємство Державної акціонерної компанії "Укрмедпром”, Одеса
3. “Вітаміни”, ВАТ, Умань
4. Вінницький державний медичний університет
5. “Галичфарм”, АТВТ, Львів
6. Державна інспекція з контролю якості лікарських засобів МОЗ України, Київ
7. Державне підприємство "Науково-експертний фармакопейний центр", Харків
8. Державне підприємство "Центр імунобіологічних препаратів", Київ
9. Державний Департамент з контролю за якістю, безпекою та виробництвом лікарських засобів і виробів
медичного призначення, Київ
10. Державний науковий центр лікарських засобів МОЗ і НАН України, Харків
11. Державний фармакологічний центр МОЗ України, Київ
12. “Дніпрофарм”, ВАТ, Дніпропетровськ
13. Дослідна станція лікарських рослин Української академії аграрних наук, Березоточа
14. Дослідний завод ДНЦЛЗ, дочірнє підприємство Державної акціонерної компанії "Укрмедпром", Харків
15. Запорізький державний медичний університет, Запоріжжя
16. Інститут біоорганічної хімії та нафтохімії НАН України, Київ
17. Інститут гігієни та медичної екології АМН України, Київ
18. Інститут дерматології та венерології АМН України, Харків
19. Інститут екогігієни та токсикології ім. Л.І. Медведя, Київ
20. Інститут експериментальної патології, онкології і радіобіології ім. Р.Є. Кавецького, Київ
21. Інститут епідеміології та інфекційних захворювань ім. Л.В. Громашевського АМН України, Київ
22. Інститут кардіології ім. академіка М.Д. Стражеско АМН України, Київ
23. Інститут медичної радіології ім. С.П. Григор'єва АМН України. Харків
24. Інститут мікробіології і вірусології ім. Д.К. Заболотного НАН України, Київ
25. Інститут мікробіології та імунології ім. 1.1. Мечнікова АМН України, Харків
26. Інститут органічної хімії НАН України, Київ
27. Інститут очних хвороб та тканинної терапії ім. В.П. Філатова АМН України, Одеса
28. Інститут проблем ендокринної патології ім. В.Я. Данилевського АМН України, Харків
29. Інститут сорбції та проблем ендоекології НАН України, Київ
ЗО. Інститут терапії АМН України, Харків
31. Інститут фармакології та токсикології АМН України, Київ
32. Інститут хімії поверхні НАН України. Київ
33. “ІнтерХім”, ВАТ СП, Одеса
34. “Київмедпрепарат”, АП, Київ
35. Київська медична академія післядипломної освіти ім. П.Л. Шупика
36. “Лубнифарм”, СП ВАТ, Лубни
37. Львівський державний медичний університет ім. Данила Галицького
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
їх
Організації та установи
38. Міністерство охорони здоров'я України, Київ
39. Науковий центр радіаційної медицини АМН України, Київ
40. Науково-виробничий центр "Борщагівський ХФЗ”, ЗАТ, Київ
41. Національна фармацевтична академія України, Харків
42. Національний ботанічний сад ім. М.М. Гришка НАН України, Київ
43. Національний медичний університет ім. 0.0 Богомольця, Київ
44. Нікітський ботанічний сад Національного наукового центру, Ялта
45. Підприємство “Фарма Старт”, Київ
46. ТОВ “НІР”, Київ
47. Українська військово-медична академія, Київ
48. Українська медична стоматологічна академія, Полтава
49. Український науково-дослідний інститут харчування, Київ
50. “Фармак”, ВАТ, Київ
51. Фармацевтична асоціація України, Київ
52. Фармацевтична фірма “Дарниця”, ЗАТ, Київ
53. Фармацевтична фірма “Здоров'я”, ВАТ, Харків
54. Фізико-хімічний інститут ім. О.В. Богатського НАН України, Одеса
55. Харківське державне фармацевтичне підприємство “Здоров'я народу” Державної акціонерної компані
"Укрмедпром”
56. Харківський медичний університет
57. Центральна лабораторія з аналізу якості лікарських засобів МОЗ України, Київ
х
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 200
Вступ
V. ВСТУП
Відповідно до Закону України "Про лікарські засоби", стаття 2, Державна Фармакопея України — це правовий акт,
який містить загальні вимоги до лікарських засобів, фармакопейні статті (монографії), а також методики контро-
лю якості лікарських засобів.
Державна Фармакопея має законодавчий характер. Її вимоги, що висуваються долікарських засобів, є обов'язковими
для всіх підприємств і установ України, незалежно від їх форми власності, що виробляють, зберігають, контролюють,
реалізують і застосовують лікарські засоби
До Вашої уваги пропонується перший випуск Державної Фармакопеї України (ДФУ). Із введенням у дію ДФУ
втрачає свою силу в Україні Державна Фармакопея СРСР XI видання (ГФ XI) як основний нормативний доку-
мент, що регламентує питання контролю і якості лікарських засобів. Втрачають свою силу також ті статті Держав-
ної Фармакопеї СРСР X видання, які ще мали чинність до теперішнього часу.
Головною відмінністю Державної Фармакопеї України від ГФ XI є те, шо вона повністю гармонізована з Євро-
пейською Фармакопеєю.
Згідно з Постановою Кабінету Міністрів № 244 від 19.03.97р. Україна взяла курс на інтеграцію до Європейсько-
го Співтовариства. З лютого 1998 року Україна є спостерігачем у Європейській Фармакопеї. Відповідно до цього
Державна Фармакопея України має бути гармонізована з Європейською Фармакопеєю.
Європейська Фармакопея передбачає обов'язкове виробництво лікарських засобів відповідно до вимог належної
виробничої практики (СМР). В даний час в Україні ще не створені умови для переходу на обов'язкове виконання
цих вимог. Це доводиться якоюсь мірою компенсувати жорсткістю вимог до якості кінцевого продукту. Така прак-
тика була прийнята в колишньому СРСР і зберігається ще вданий час в Україні. Відмова від таких додаткових ви-
мог при відсутності СМР означала б істотне зниження в деяких випадках вимог до якості лікарських засобів. Крім
того, це суперечило б вже укладеним міждержавним угодам у рамках Міждержавної комісії зі стандартизації,
реєстрації і контролю якості лікарських засобів, виробів медичного призначення і медичної техніки лержав-учас-
ниць СНД.
Тому при розробці ДФУ відповідні статті Європейської Фармакопеї були доповнені вимогами, що враховують спе-
цифіку сучасного стану фармацевтичного виробництва України. Цс призвело до побудови загальних і окремих
статей (монографій) ДФУ у вигляді двох взаємозалежних частин — європейської частини, ідентичної відповідній
статті Європейської Фармакопеї, і національної, що відбиває національну специфіку України. Така схема побудо-
ви Фармакопеї прийнята і в інших країнах Європейського Співтовариства, наприклад, у Великобританії.
Національна частина не суперечить європейській, а містить додаткові вимоги (які зараз вже є чинними в Україні)
для лікарських засобів, що не випускаються за вимогами СМР, встановленими у Європейському Співтоваристві.
Це, насамперед, вимоги ГФ XI і міждержавних документів, підписаних у рамках Міждержавної комісії зі стандар-
тизації, реєстрації і контролю якості лікарських засобів, виробів медичного призначення і медичної техніки дер-
жав-учасниць СНД — у тому випадку, якщо вони доповнюють європейські. Тому відповідність вимогам ДФУ ав-
томатично означає відповідність вимогам цих документів. У той же час, відповідність вимогам цих документів не
завжди може означати автоматичну відповідність вимогам ДФУ, оскільки ДФУ включає в себе також і вимоги
Європейської Фармакопеї. У національну частину включені також додаткові інформаційні матеріали й альтерна-
тивні методики. Вимоги національної частини не поширюються на лікарські засоби, що випускаються в умовах
СМР, визнаних у Європейському Співтоваристві.
Така концепція побудови ДФУ була узгоджена Фармакопейним центром із Європейською Фармакопеєю. При пе-
реході України зі спостерігачів у постійні члени Європейської Фармакопеї національна частина буде виключена з
наступних видань ДФУ і її доповнень.
В ДФУ максимально урахований стиль побудови Європейської Фармакопеї. Усі формули, літерні позначення, ци-
фровий матеріал, одиниці виміру, нумерація розділів і т.д. подані в редакції Європейської Фармакопеї. Хімічні на-
зви дані в редакції, максимально наближеній до європейської. Це пов’язано з тим, шо велика частина субстанцій
у даний час імпортується в Україну відповідно до вимог Європейської Фармакопеї. Тому інші хімічні назви мо-
жуть призвести до утруднень в ідентифікації продукту. Максимально наближені до Європейської Фармакопеї і на-
зви монографій і реактивів. При цьому наводяться також відповідні вітчизняні синоніми.
Порівняно з ГФ XI зміст ДФУ більш систематизований і чітко поділяється на такі розділи: "Загальні зауваження",
"Методи аналізу", "Матеріали для контейнерів і контейнери"(даний розділ знаходиться в стадії розробки), "Реак-
тиви", "Загальні тексти", "Субстанції та лікарські форми" (загальні статті), "Монографії" (на субстанції).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
хі
Вступ
У методи аналізу включені фармако-технологічні випробування, що у ГФ XI звичайно включалися в загальні
статті на лікарські форми. Слід зазначити включення в розділ "Методи аналізу" нової загальної статті "Валідація
аналітичних методик і випробувань". Характерною особливістю ДФУ є також поява нового розділу "Загальні тек-
сти", куди включені загальні статті, що мають у значній мірі інформаційний характер.
Оскільки всі статті ДФУ грунтуються на Європейській Фармакопеї, то ДФУ суттєво відрізняється від ГФ XI: вико-
ристовуються різні еталони ступеня забарвлення, системи назв титрованих розчинів і т.д. Тому в текстах усіх
аналітичних нормативних документів (АНД) необхідно наводити посилання тільки на ДФУ. Рівнобіжні посилан-
ня на ГФ XI при цьому неприпустимі. У тих випадках, коли усе ж необхідно використовувати якісь матеріали, ре-
активи, еталони і т.д. з ГФ XI, їхнє описання або приготування необхідно цілком наводити в тексті АНД Виняток
робиться для лікарської рослинної сировини, якість якої, до виходу відповідних статей ДФУ, може контролювати-
ся за монографіями ГФ XI з використанням відповідних загальних статей ГФ XI.
Європейська Фармакопея перевидається кожні п’ять років із щорічними доповненнями й змінами. Для
збереження гармонізації з Європейською Фармакопеєю передбачається в такі самі терміни проводити
перевидання й доповнення Державної Фармакопеї України.
хп
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
1. Загальні зауваження
1. ЗАГАЛЬНІ ЗАУВАЖЕННЯ
1.1. ЗАГАЛЬНІ ПОЛОЖЕННЯ
Положення статті "Загальні зауваження"поширюють-
ся на всі загальні статті, монографії та інші матеріали
Державної Фармакопеї України.
У матеріалах Державної Фармакопеї України слово
"Фармакопея" без уточнень означає Державну Фарма-
копею України. Поряд із цим може також використо-
вуватися офіційне скорочення ДФУ
Посилання в матеріалах Фармакопеї на якусь статтю
і/абоїї розділ означає, що продукт відповідає вимогам
цієї статті. Назва статті, на яку дається посилання,
і/або її номер звичайно виділені курсивом.
Готовий лікарський засіб має відповідати вимогам
Фармакопеї протягом терміну його придатності.
Термін придатності і дата, з якої він має відраховува-
тись, погоджується компетентним уповноваженим
органом на підставі експериментальних досліджень із
стабільності даного готового лікарського засобу. Будь-
який інший продукт (субстанція, допоміжна речовина
тощо) має відповідати вимогам Фармакопеї протягом
усього періоду його використання.
Вимоги монографії є обов’язковими, якщо немає
спеціальних застережень у статті "Загальні зауваження "
або в даній монографії. Загальні статті стають обов’яз-
ковими, коли на них наводиться посилання в тій або
іншій монографії або загальній статті, якщо не зробле-
но спеціального застереження, що посилання наво-
диться винятково як інформація або рекомендація.
Діючі речовини (субстанції), допоміжні речовини, го-
тові лікарські засоби та інші продукти, що описують-
ся в монографіях Фармакопеї, призначені для викори-
стання людиною та у ветеринарії (якщо немає інших
зазначень).
Якщо продукт не відповідає усім без винятку вимогам
монографії Фармакопеї, він не є виробом фармако-
пейної якості. Це не означає, що для підтвердження
відповідності виробу вимогам Фармакопеї виробник
має перед випуском продукту провести всі випробу-
вання, згадані в монографії. Деякі дані можуть бути
взяті виробником, наприклад, із валідаційних випро-
бувань у поєднанні з результатами контролю процесу
виробництва даного продукту. Такий підхід, якщо
компетентні уповноважені органи вважають його
обгрунтованим, не суперечить необхідності від-
повідності вимогам Фармакопеї.
Випробування та методики кількісного визначення,
наведені у Фармакопеї, є офіційними методиками,
проте за узгодженням із компетентним уповноваже-
ним органом можуть використовуватися й інші мето-
дики, за умови, що ці методики дають результати, які
відповідають фармакопейним методикам. У випадку
сумнівів або розбіжностей вирішальною є фармако-
пейна методика.
Субстанції, допоміжні речовини та інші продукти, на
які поширюються вимоги Фармакопеї, можуть вико-
ристовуватися в різних цілях (наприклад, для одер-
жання парентеральних лікарських засобів або табле-
тованих лікарських форм тощо). Коли щодо цього не-
має зазначень у відповідній монографії, її вимоги по-
ширюються на продукт незалежно від цілей його за-
стосування. У деяких випадках, зокрема, у випадку
допоміжних речовин, монографія може бути доповне-
на переліком характеристик, важливих для викорис-
тання даної речовини; цей перелік додається як
інформація та рекомендації. Для інформації можуть
також бути наведені методики контролю однієї або
декількох таких характеристик
Загальна стаття на ту чи іншу лікарську форму поши-
рюється на всі лікарські засоби, виготовлені у вигляді
цієї лікарської форми. Для конкретного лікарського
засобу вимоги відповідної загальної статті не обов’яз-
ково є вичерпними і можуть бути доповнені компе-
тентним уповноваженим органом.
Прийнята термінологія. Термін "компетентний уповно-
важений орган" означає національний, наднаціональ-
ний або міжнародний орган (організацію), уповнова-
жений приймати рішення з відповідних питань. Це
може бути, наприклад, національний фармакопейний
орган, інстанція, шо ліцензує, або офіційна контроль-
на лабораторія.
Словосполучення ”якшо немає інших зазначень в ок-
ремій статті”1 означає, шо вимоги загальної статті ма-
ють бути виконані, якщо тільки компетентний упов-
новажений орган не вніс у ці вимоги зміни, про що
зазначається в окремій статті.
У деяких загальних статтях і монографіях Фармакопеї
при описанні реактиву, мікроорганізму, методики то-
що використовується термін "підхожий". Якщо при
цьому критерії їхньої придатності не сформульовані,
придатність конкретних реактивів, методик тощо, ви-
користовуваних в АНД, має бути обгрунтована компе-
тентному уповноваженому органу.
1.2. ІНШІ ПОЛОЖЕННЯ,
ЩО ПОШИРЮЮТЬСЯ
НА ЗАГАЛЬНІ СТАТТІ
Й МОНОГРАФІЇ
Кількість речовини. При описанні кількісного визна-
чення або випробування з чисельно заданими межами
кількість речовини, необхідна для проведення випро-
' Під окремою статтею мають на увазі монографію на субстанцію
Фармакопеї або аналітичний нормативний документ (АНД), за-
тверджений компетентним уповноваженим органом.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
1. Загальні зауваження
бування, зазначена приблизно. Насправді вона може
відхилятися в межах ±10% від зазначеної кількості.
Необхідно взяти точну наважку аналізованої речови-
ни (або відміряти її будь-яким іншим способом) і всі
обчислення робити для цієї точної кількості речови-
ни. Якщо межі випробування задані не чисельно, а
визначаються шляхом порівняння зі стандартом за тих
самих умов, для випробування беруть точно зазначену
кількість речовини. Реактиви завжди беруть у точно
зазначених кількостях.
Якщо значення маси наважок або об’ємів не викорис-
товують для подальших розрахунків, то точність
їхнього взяття (відмірювання, відважування) має по-
годжуватися із зазначеною в статті точністю. Точність
зважування має бути +5 одиниць після останньої за
значеної цифри; наприклад, наважку 0.25 г слід ро-
зуміти як таку, що лежить в інтервалі від 0.245 г до 0.255 г.
Об’єми відміряють у такий спосіб. Якщо після десят-
кової точки стоїть 0 або число, шо закінчується 0 (на
приклад, 10.0 мл або 0.50 мл), необхідний об’єм
відміряють за допомогою піпетки, мірної колби або
бюретки. В інших випадках можна використовувати
градуйований мірний циліндр або градуйовану піпет-
ку. Мікролітри відміряють за допомогою мікропіпетки
або мікрошприца. Необхідно, проте, відзначити, що в
деяких випадках точність, із якою зазначають
кількість речовини, не відповідає числу значущих
цифр при зазначенні конкретної чисельної межі. Зва-
жування і вимірювання здійснюють у цьому випадку з
більш високою точністю.
Обладнання й аналітичні операції. Скляний мірний по-
суд має відповідати вимогам Класу А Міжнародних
стандартів, виданих Міжнародною організацією зі
стандартизації (180).
Аналітичні операції, якщо немає інших зазначень,
здійснюють при температурі від 15 °С до 25 °С.
Порівняльні випробування, якшо немає інших зазна-
чень, проводять з використанням пробірок із безбарв-
ного прозорого нейтрального скла із плоскою основою
і внутрішнім діаметром 16 мм. Порівнюють однакові
об’єми рідин на білому (або, якщо необхідно, на чорно-
му) фоні. Випробування проводять у розсіяному світлі
Якщо для проведення випробування або кількісного
визначення необхідно використовувати розчинник із
розчиненим у ньому індикатором і при цьому не пе-
редбачено контрольного досліду, то цей розчинник
попередньо нейтралізують за цим індикатором.
Водяна баня. Якщо немає інших зазначень, то мається
на увазі баня з киплячою водою. Можна використову-
вати й інші способи, якщо вони гарантовано забезпе-
чують температуру, близьку, але не переважаючу 100 °С
(або іншу зазначену температуру).
Висушування і прожарювання до постійної маси. Ре-
зультати двох послідовних зважувань мають відрізня-
тися не більш як на 0.5 мг; інтервал часу між двома
зважуваннями визначається властивостями й кіль-
кістю висушуваного/прожарюваного залишку. У тих
випадках, коли потрібне висушування "в ексикаторі"
або "у вакуумі", воно здійснюється відповідно до умов,
описаних у 2.2.32. "Втрата в масі при висушуванні".
РЕАКТИВИ
Надійність результатів, одержуваних за допомогою
описаних у Фармакопеї аналітичних операцій, зале-
жить, зокрема, від якості використовуваних реактивів.
Реактиви описані в загальній статті 4. "Реактиви". Пе-
редбачуваний ступінь чистоти — не нижче ч.д.а. (апа-
Іуїїсаі £гаде). Для деяких реактивів опис включає ви-
пробування для визначення придатності.
РОЗЧИННИКИ
Якщо для розчинів не зазначений розчинник, то ма-
ються на увазі водні розчини. Для проведення описа-
них у Фармакопеї аналітичних операцій і для приготу-
вання реактивів використовують воду, яка відповідає
вимогам окремої статті "Вода очищена". Термін "вода
дистильована" означає "вода очищена", отримана
шляхом дистиляції.
Термін "етанол" без уточнень означає абсолютний
спирз Термін ”96 % спирт" без уточнень означає ети-
ловий спирт, який містить приблизно 96 об’ємних
відсотків етанолу. Інші ступені розведення познача-
ються терміном "спирт" із вказівкою вмісту етанолу в
об’ємних відсотках.
СПОСОБИ ВИРАЖЕННЯ КОНЦЕНТРАЦІЇ
Залежно від контексту вираз ”%" може мати одне з
двох значень:
— масовий відсоток (м/м) - число грамів речовини у
100 грамах кінцевого продукту;
— об’ємний відсоток (об/об) - число мілілітрів речо-
вини у 100 мілілітрах кінцевого продукту.
Позначення “ррт” (частин на мільйон) передбачає
масове співвідношення
ТЕМПЕРАТУРА
Крім конкретного зазначення температури викорис-
товуються також такі терміни;
Глибоке охолодження нижче -15°С
У холодильнику від 2 °С до 8 °С
У холодному чи
прохолодному місці від 8 °С до 15 °С
При кімнатній температурі від 15 °С до 25 °С
1.3. ЗАГАЛЬНІ СТАТТІ
КОНТЕЙНЕРИ
Матеріали, використовувані для контейнерів, описані
в загальній статті 3. "Контейнери".
Для матеріалів, використовуваних для виробництва кон-
тейнерів, особливо для полімерних мазеріалів, наводять
загальні назви, кожна з яких охоплює ряд матеріалів, що
відрізняються як властивостями основного компонента,
4
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
1. Загальні зауваження
так і використовуваними добавками. Випробування та
межі нормування залежать від конкретного складу ма-
теріалу й таким чином застосовні тільки за умови, що
матеріал відповідає вступній частині до його спе-
цифікації. За узгодженням із компетентним уповнова-
женим органом можуть використовуватися матеріали
інших складів, а також випробування для них.
Специфікації на контейнери, включені до статті З,
розроблялися для всіх контейнерів зазначеної кате-
горії. Проте, з огляду на велику розмаїтість існуючих
контейнерів 1 можливість появи нових контейнерів,
публікація специфікації не виключає можливості ви
користання контейнерів, що відповідають іншій спе-
цифікації, якщо це обгрунтовано та узгоджено з ком-
петентним уповноваженим органом.
У статтях Фармакопеї можуть даватися посилання на
визначення і специфікації контейнерів. У розділах
"Визначення", "Виробництво" загальних статей на
лікарські форми може міститися вимога щодо викори-
стання певного типу контейнера. У розділі "Зберіган-
ня" деяких статей може вказуватися тип рекомендова-
ного контейнера
1.4. МОНОГРАФІЇ
НАЗВИ
Крім назв українською мовою, наводиться також ла-
тинська назва. Ця назва може використовуватися у
відповідних випадках замість української назви так са-
мо, як і будь-який інший синонім, що його визнав
еквівалентним компетентний уповноважений орган.
ВІДНОСНІ АТОМНІ Й МОЛЕКУЛЯРНІ МАСИ
Відносна атомна маса (А.м.) або відносна молекуляр-
на маса (М.м.) зазначаються, коли це необхідно, на
початку монографії. Відносну масу й графічну форму-
лу наводять як інформаційний матеріал.
ВСТУПНА ЧАСТИНА МОНОГРАФІЙ
У вступній частині, що йде після назви монографії,
наводиться офіційне визначення субстанції, готового
лікарського засобу або іншого продукту, що є предме-
том монографії.
Межі вмісту. Якщо зазначені межі вмісту, то це межі,
одержані з використанням методу, зазначеного в
розділі "Кількісне визначення".
Лікарські засоби рослинного походження. У моно-
графіях на лікарські засоби рослинного походження
вступна частина включає зазначення предмета моно-
графії. Це може бути, наприклад, рослинна сировина
у вихідному вигляді або рослинна сировина, подрібне-
на на порошок. Якщо монографія поширюється на
декілька варіантів, наприклад, на обидва із зазначе-
них, то про це попереджають у вступній частині.
ВИРОБНИЦТВО
Інформація в розділі "Виробництво" має привернути
увагу до деяких важливих аспектів процесу вироб-
ництва і не обов’язково є вичерпною. Інструкції, що в
ньому містяться, адресовані виробнику. Вони можуть
стосуватися, наприклад, джерела матеріалів, процесу
виробництва, його валідації й контролю, до по-
стадійного контролю, а також випробувань, які ви-
робник має проводити перед випуском для кожної
серії продукту або для обраних серій. Ці положення не
обов’язково мають бути підтверджені незалежним
аналітиком за допомогою аналізу кінцевого продукту.
Компетентним уповноваженим органом може бути
встановлено, що наведені в даному розділі інструкції
були виконані. Такий висновок може бути зроблений
на підставі перевірки одержаних від виробника даних,
або при інспектуванні виробництва чи при випробу-
ванні відповідних зразків.
Відсутність розділу "Виробництво" не означає, що ас-
пекти процесу виробництва, відзначені више, не по-
гребують уваги. Будь-який описаний у Фармакопеї
продукт має вироблятися відповідно до принципів на-
лежної виробничої практики (НВП, ОМР) і відпо-
відних міжнародних угод, а також національних й над-
національних законів, що поширюються на продукти,
призначені для людини або використовувані у ветери-
нарії.
У розділі "Виробництво" у монографії на вакцину мо-
жуть бути зазначені властивості штаму й тестові мето-
ди для підтвердження цих властивостей. Ці методи на-
водяться для інформації як приклад.
ВЛАСТИВОСТІ
Інформація, наведена в цьому розділі, має рекомен-
даційний характер.
Розчинність. Для зазначення розчинності в даному
підрозділі використовуються описові терміни, які в
температурному інтервалі від 15 °С до 25 °С мають
зміст, зазначенний у Табл. 1.4-1.
Таблиця 1.4.-І
Термін Приблизна кількість розчинника (мл), необхідна для розчинення 1 г речовини
Дуже легко розчинний до 1
Легко розчинний більше 1 до 10
Розчинний 10 до 30
Помірно розчинний ЗО до 100
Мало розчинний 100 до 1000
Дуже мало розчинний 1000 до 10 000
Практично не розчинний 10 000
Частково розчинний Термін використовується для характеристики сумішей, які містять розчинні та не розчинні компоненти
Змішується з... Термін використовується для характеристики рідин. що змішуються із зазначеним розчинником у будь-яких співвідношеннях
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
5
1. Загальні зауваження
ІДЕНТИФІКАЦІЯ
Наведені в цьому розділі випробування не розраховані
на повне підтвердження хімічної структури або складу
продукту. Вони призначені для підтвердження з прий-
нятним ступенем вірогідності того, шо продукт
відповідає інформації, наведеній на етикетці.
У деяких монографіях є підрозділи "Перша іден-
тифікація" та "Друга ідентифікація”. Звичайно вико-
ристовують першу ідентифікацію. Якщо є гарантія то-
го, що дана серія субстанції була раніше сертифікова-
на на відповідність усім вимогам монографії, випробу-
вання з другого підрозділу можуть використовуватися
замість випробувань із першого підрозділу.
ВИПРОБУВАННЯ ТА КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Область застосування. Ці вимоги не розраховані на
охоплення всіх можливих домішок. Зокрема, із того,
що домішка не визначається за допомогою описаних
випробувань, не слід робити висновок, що вона допу-
стима, якщо здоровий глузд і належна фармацевтична
практика не допускають її наявності. Див. також ниж-
че в розділі "Домішки”.
Розрахунки. Якщо при проведенні обчислень
потрібний перерахунок на суху або безводну речовину
чи домовлена якась інша умова, то втрату в масі при
висушуванні, вміст води або інший показник визнача-
ють за допомогою методу, описаного в монографії.
Межі. Зазначувані межі грунтуються на результатах,
одержаних у рамках звичайної аналітичної практики;
у них уже враховано звичайні аналітичні похибки, до-
пустимий розкид при виробництві й приготуванні, а
також погіршення якості в процесі зберігання у ме-
жах, які вважаються прийнятними. При визначенні
відповідності продукту вимогам монографії до зазна-
чених меж не мають додаватися будь-які додаткові до-
пуски.
Результат, одержаний у випробуванні, округляють до
зазначеної у межі кількості значущих цифр (якщо не-
має інших зазначень). При цьому останню цифру
збільшують на одиницю, якщо цифра, яку відкидають
при округленні, більша або дорівнює п’яти. Якщо ци-
фра, яку відкидають при округленні, менша п’яти, ос-
танню цифру лишають незмінною.
Зазначення і допустима межа домішок. Приблизний до-
пустимий вміст домішки або суми домішок може бути
зазначений у дужках тільки для інформації. Якщо для
даної домішки не передбачене використання стан-
дартного зразка, її вміст може бути виражений, вихо-
дячи з номінальної концентрації речовини, викорис-
товуваної для приготування зазначеного в монографії
розчину порівняння (якщо немає інших зазначень).
Лікарські засоби рослинного походження. Для лікарського
засобу рослинного походження сульфатна зола, загальна
зола, розчинні у воді сторонні речовини, розчинні в
спирті сторонні речовини, вміст води, вміст ефірних
олій та вміст діючих речовин обчислюють у розрахунку
на лікарський засіб, який не було спеціально висушено
(якщо немає інших зазначень у монографії).
Еквіваленти. У тих випадках, коли наводиться еквіва-
лент, він дається з такою кількістю значущих цифр,
яка потрібна в даній монографії.
ЗБЕРІГАННЯ
Інформація і рекомендації, наведені в розділі
"Зберігання", не є вичерпними фармакопейними ви-
могами, і компетентні уповноважені органи можуть
зазначати конкретні умови зберігання, обов’язкові
для виконання.
Описані у Фармакопеї продукти слід зберігати таким
чином, щоб запобігти їхньому забрудненню і, по мож-
ливості, розкладанню. Якщо рекомендуються особ-
ливі умови зберігання, включаючи тип контейнера
(див. вище розділ "Контейнери") і температурні межі,
ці рекомендації наводяться в монографії.
Нижче роз’яснюються вирази, використовувані в мо-
нографіях у розділі "Зберігання”.
"Захищати від вологи". Продукт має зберігатися в
повітронепроникному контейнері. При розкритті
контейнера у вологій атмосфері необхідно виявляти
обережність. Якщо необхідно, низький вміст вологи
можна підтримувати за допомогою осушувальних ре-
човин, за умови, що їхній прямий контакт із продук-
том буде виключений.
"Узахищеному від світла місці". Одне з трьох: або кон-
тейнер має бути виготовлений із матеріалу, який до-
статньою мірою поглинає світло, здатне спричинити
фотохімічні перетворення; або контейнер має бути
вміщений у зовнішній контейнер, що забезпечує та-
кий захист; або лікарська речовина має зберігатися в
місці, яке виключає можливість попадання такого
світла.
МАРКУВАННЯ
Маркування є предметом національних і наднаціональ-
них законодавств, а також міжнародних угод. Таким чи-
ном, інформація в розділі "Маркування" не претендує на
повноту. Вона орієнтована насамперед на фармакопейні
цілі, і обов’язковими є лише положення, необхідні для
підтвердження відповідності продукту статті. Вся інша
інформація має рекомендаційний характер. У тих випад-
ках, коли у Фармакопеї вживається термін "етикетка",
відповідна інформація може бути зазначена на контей-
нері, на упаковці або у вкладиші, залежно від рішення
компетентного уповноваженого органа.
ЗАСТЕРЕЖЕННЯ
Описувані в статтях Фармакопеї продукти і реактиви
можуть виявитися небезпечними для здоров’я, якщо
6
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
1. Загальні зауваження
не вживати необхідних заходів. У всіх випадках слід
дотримуватися принципів належної лабораторної
практики (НЛП, СЕР), а також відповідних положень
техніки безпеки. У деякі статті включені спеціальні за-
значення про необхідні запобіжні заходи. Але
відсутність таких зазначень не слід трактувати як
відсутність будь-якого ризику.
ДОМІШКИ
У монографії може бути наведений перелік усіх відо-
мих і потенційних домішок, для яких показано, що
вони контролюються випробуваннями. Цей перелік
може бути поділений на дві частини: "Конкретно за-
значувані домішки" та "Інші виявлювані домішки". У
першу частину включають домішки, які раніше вже
були кваліфіковані компетентним уповноваженим ор-
ганом. У цей перелік також включають домішки, для
яких відомо, що вони можуть утворюватися в резуль-
таті природного метаболізму. В другу частину переліку
включають потенційні домішки, які не були виявлені
в жодному зі зразків субстанції за час розробки моно-
графії або вміст яких не перевищував 0.1 %, але які, в
принципі, детектуються за допомогою наведених ви-
пробувань.
ФІЗИЧНІ ХАРАКТЕРИСТИКИ
У монографії може також наводитись як інформація
та рекомендації перелік фізичних характеристик, шо
не належатьдо офіційних вимог, але все ж важливі при
використанні продукту (див. вище /. 1. Загальні поло-
ження).
аналізу, ні будь-яка інша додаткова інформація не на-
дається. Не вказується також дата "Придатний до..
гарантується стабільність препарату в момент відправ-
лення і можливість його використання протягом шес-
ти місяців, якщо не розкупорений контейнер збері-
гається в умовах, зазначених у супровідній докумен-
тації. Після закінчення цього терміну треба прокон-
сультуватися в уповноваженому фармакопейному ор-
гані. Стабільність вмісту розкритого контейнера не га-
рантується.
Хімічні стандартні зразки. Абревіатура ФСЗ означає
хімічні стандартні зразки, установлені Фармакопеєю.
Деякі хімічні стандартні речовини використовуються
для мікробіологічного кількісного визначення ан-
тибіотиків. У цьому випадку їхня активність вира
жається в Міжнародних одиницях (МО) таким же чи-
ном, як для біологічних стандартних препаратів, і за-
значається на упаковці або в супровідному документі.
Біологічні стандартні препарати. Більшість згадуваних у
Фармакопеї біологічних стандартних препаратів — це
відповідні Міжнародні стандарти і стандартні препа-
рати, установлені Всесвітньою організацією охорони
здоров’я (ВООЗ). Оскільки вони, як правило, до-
ступні в обмежених кількостях, Фармакопея встано-
вила в тих випадках, коли це доцільно, свої біологічні
стандартні препарати (БСП). їхня активність вираже-
на, коли це можливо, у МО.
Еталонні спектри. Еталонний спектр супроводжується
інформацією про умови приготування випробовува-
ного зразка і запису спектра.
СТАНДАРТНІ ЗРАЗКИ, СТАНДАРТНІ ПРЕПАРА-
ТИ ТА ЕТАЛОННІ СПЕКТРИ
Деякі монографії передбачають використання стан-
дартних зразків, стандартних препаратів або еталон-
них спектрів. Вони розроблені з урахуванням їхнього
призначення і їх слід використовувати так, як припи-
сує Фармакопея. За інших обставин вони можуть вия-
витися непридатними.
Стандартні зразки, стандартні препарати та еталонні
спектри уводяться вдію уповноваженим фармакопей-
ним органом. Повний перелік може бути одержаний у
зазначеній організації. Ці стандартні матеріали є
офіційними у випадку арбітражу.
Робочі стандартні зразки можуть використовуватися
для проведення поточних аналізів за умови, що вони
відкалібровані за Фармакопейними стандартними
зразками (ФСЗ).
Уся інформація, необхідна для правильного викорис-
тання стандартного зразка або стандартного препара-
ту. наводиться на упаковці, або у вкладиші, або в су-
провідній документації. Якщо не зазначено жодних
умов висушування, стандарт треба використовувати в
такому вигляді, у якому він одержаний. Ні сертифікат
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
7
1. Загальні зауваження
1.5. СКОРОЧЕННЯ ТА ПОЗНАЧЕННЯ
АБРЕВІАТУРИ ТА СИМ ВОЛ И
КОЛЕКЦІЇ МІКРООРГАНІЗМІВ
А Оптична густина
л 1% , .
л і См Питомии показник поглинання
А.м. Відносна атомна маса
кіі, Питоме оптичне обертання
БСП Біологічний стандартний препарат
ФСЗ Фармакопейний стандартний зразок
(ПО V,
и20 Відносна густина
МО Міжнародні одиниці
X Довжина хвилі
М Молярність
М.м. Відносна молекулярна маса
Показник заломлення
ЄФО Європейська фармакопейна одиниця
ррт Одна частина на мільйон частин
Р Речовина або розчин, зазначені в статті
"Реактиви"
К] Використовується в ТШХ для позначен-
ня відношення шляху, пройденого речо-
виною, до шляху, пройденого фронтом
розчинника
РО Вихідні стандартні речовини для вста-
новлення титру титрованих розчинів в
об’ємному аналізі
АТСС Американська колекція типових культур
Атегісап Туре Сиііиге Соїіесііоп
12301 РагкІаіУП Огіує
КоскУІПе, МО 20852, Ц8А
С.І.Р. Колекція Пастерівського інституту
(штами бактерій)
Соїіесііоп сіє 1’1п8іііпі Райеиг (8Ігаіп8 оГ
Ьасіегіа)
І.Р. Пастерівський інститут (штами інших
мікроорганізмів)
ІП8ІІІШ РаМеп г (8ігаіп8 оГ оіЬег тісгоог-
§апІ8Ш8)
Бегуісе де 1а Соїіесііоп N3000316 де
Сиїїиге де Місгоог£апІ8те8 (С.ЬІ.С.М.)
25, гие ди Оосіеиг-Коих
Г-75015 РагІ8, Егапсе
N0^16 Національна колекція промислових і
морських бактерій
№ііопз! Соїіесііоп оГ Іпдшігіаі апд
Магіпе Взсіегіз Бід
23 81 Масйаг Огіує
АЬегдееп АВ2 1К¥, Сгеаі Вгіїаіп
№СРЕ Національна колекція патогенних грибів
Каїіопаї Соїіесііоп оГ Радю£епіс Еипф
Еопдоп 8с1юо1 оГ Нуріепе апд Тгорісаі
Медісіпе
Керреї 8ігееІ
Еопдоп\УС1Е 7НТ, Сгеаі Вгіїаіп
МСТС Національна колекція типових культур
N36003! Соїіесііоп оГТуре Си11пге8
Сепігаї РпЬІіс Неаіій БаЬогаіогу
Соїіпдаїе Ауепие
Еопдоп №\У9 5НТ, Огеаі Вгіїаіп
N€¥0 Національна колекція дріжджових куль-
тур
Кдііопаї Соїіесііоп оГ¥еа8І Си1іиге8
АЕКС Гоод Ке8еагсй ІП8ІіІШе
Соіпеу Бапе
ГмоілмісЕї N^4 7 БА, Сгеаі Вгіїаіп
8.8.1. Державний інститут сироваток
8іаіеп8 8егит Іп8ІіІпІ
80 Атарег Воиіеуагд, Сорепііа^еп,
Оептагк
8
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
1. Загальні зауваження
1.6. ОДИНИЦІ МІЖНАРОДНОЇ СИСТЕМИ (СІ), ВИКОРИСТОВУВАНІ
У ФАРМАКОПЕЇ, І ЇХНЯ ВІДПОВІДНІСТЬ ІНШИМ одиницям
МІЖНАРОДНА СИСТЕМА ОДИНИЦЬ (СІ)
Міжнародна система одиниць складається із трьох класів одиниць фізичних величин, а саме: основні одиниці,
похідні одиниці та допоміжні одиниці. Основні одиниці та їхні визначення наведені в Табл. 1.6.-1.
Фізичні величини, шо входять у систему, але визначаються через основні розміри цієї системи, називаються
похідними величинами системи. Деякі з таких похідних величин мають свої назви й символи. Одиниці таких ве-
личин, використовуваних Фармакопеєю, наведені в Табл. 1.6.-2.
Деякі важливі й широко використовувані одиниці, що не входять у СІ, наведені в Табл. 1.6.-3.
Множні префікси для утворення десяткових часткових і кратних одиниць наведені в Табл. 1.6.-4.
Основні одиниці СІ
Таблиця 1.6.-1
Величина Одиниця Визначення
Найменування Символ Найменування Символ
Довжина / метр м Один метр являє собою довжину шляху, який проходить світло в вакуумі за 1/299 792 458 частину секунди
Маса т кілограм кг Один кілограм дорівнює масі міжнародного еталона - кілограм
Час ( секунда с Одна секунда являє собою сумарну тривалість 9 192 631 770 періодів випромінювання, які відповідають переходу між двома надтонкими рівнями основного стану атома цезію-133
Сила електричного струму І ампер А Один ампер являє собою такий постійний струм, який, проходячи по двох точно паралельних провідниках нескінченої довжини та нехтовно малого кругового перерізу, розташованих на відстані одного метра у вакуумі, спричиняє між цими провідниками силу взаємодії, яка дорівнює 2-10"7 ньютона на один метр довжини
Абсолютна температура Т кельвін К Один кельвін являє собою 1/273.16 частину від абсолютної температури потрійної точки води
Кількість речовини п моль м Один моль являє собою кількість речовини, яка містить таку саму кількість найпростіших часток, яка міститься у 0.012 кілограма вуглецю-12(*)
Сила світла IV кандела кд Кандела являє собою інтенсивність світіння в даному напрямку від джерела, яке випромінює монохроматичне випромінювання з частотою 540-1012 герц і такого джерела, інтенсивність якого в цьому напрямку становить 1/683 вата на один стереорадіан
(*)Якщо використано молі, то треба вказувати, до чого вони відносяться, наприклад, атоми, молекули, іони, електрони чи
інші частки або певні групи таких об’єктів.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
9
1. Загальні зауваження
Таблиця 1.6.-2
Одиниці СІ Європейської Фармакопеї та їх відповідність іншим одиницям
Величина Одиниця Перетворення інших одиниць у одиниці СІ
Найменування Символ Найменування Символ Вираження в основних одиницях СІ Вираження в інших одиницях СІ
Хвильове число V одиниця на один метр 1/м М’1
Довжина хвилі Л мікрометр мкм 10 6 м
нанометр нм І0“9м
Площа А,8 квадратний метр м2 м2
Об’єм V кубічний метр м3 м3 1 мл = 1 СМ3= 10’6М3
Частота V герц Гц с'1
Густина р кілограм на кубічний метр кг/м3 кг-м"3 1 г/мл = 1г/см3 = 103-кгм'3
Швидкість V метр за секунду м/с мс 1
Сила р ньютон Н мкгс-2 1 дин= ІГ’СМ’С 2 = 10’5 Н 1 кр = 9.806 65 Н
Тиск р паскаль Па М'*-КГ.С 2 Нм-2 1 дин/см2 = 10_| Па = ІО^Н м-2 1 атм = 101 325 Па= 101.325 кПа 1 бар = 105 Па = 0.1 МПа 1 мм рт.ст. = 133.322 387 Па 1 Торр= 133.322 368 Па 1 р«і = 6 894 757 кПа
Динамічна в’язкість п паскаль- секунла Па-с м’кгс’1 Нс-м-2 1 П= КНПа ^ Ю-'Н-с м-2 1сП = 1 мПас
Кінетична в’язкість V квадратний метр на секунду м2/с м -с Па с м3-кг'* Нмскг1 1 Ст = 1 см2 -с'^КН-м2 -с1
Енергія ]¥ джоуль Дж м2кг-с’2 Нм 1 ерг=1 см2-г-с-2=1 дин-см=10-1 Дж Ікал = 4.1868 Дж
Потік електромагнітного випромінювання р ват Вт м2-кг-с-3 Нм-с1 Джс1 1 ерг/с = 1 дин-см-с-1 = 10-7 Вт = = 10~7 Н-м-с"1 = Ю-7 Дж-с-1
Поглинута доза іонізуючого випромінювання 0 грей Гр М2-С’2 Дж-кг1 1 рад = 10~2 Гр
Електричний потенціал, електрорушійна сила Є вольт В м2-кг-с‘3-А'1 ВтА1
Електричний опір Р ом Ом м2-кг-с’3А 2 БА'1
Кількість електрики 0 кулон Кл Ас
Радіоактивність речовини А бекерель Бк с_| 1 Ки=37 109 Бк=37 109с-‘
Молярна концентрація с моль на кубічний метр моль/м3 моль-м-3 1 моль/л= 1 М= 1 моль/дм3 = =103моль-м-3
Масова концентрація Р кілограм на кубічний метр кг/м3 кг-м’3 1 г/л=1 г/дм3 = 1 кгм-3
10
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
1. Загальні зауваження
Таблиця 1.6.-З
Одиниці, використовувані нарівні з Міжнародною системою одиниць
Величина Одиниця Значення в одиницях СІ
Час хвилина хв 1 хв = 60 с
година гол 1 год = 60 хв = 3600 с
доба доба 1 доба = 24 год = 86 400 с
Кут на площині градус 0 1" = (тг/180) рад
Об’єм літр л 1 л = 1 ДМ3 = 10'3 м3
Маса тонна т 1 т = 103 кг
Частота обертання _ обертів за хвилину об/хв 1 об/хв = (1/60) с'1 * * * У * * *
Таблиця 1.6.-4
Множники та префікси для утворення десяткових кратних і часткових одиниць
Множник Префікс Позначення Множник Префікс Позначення
1018 екза Е 10 1 деци Д
1015 пета Р 10 2 санти с
10і2 тера Т 10 3 мілі м
109 гіга Г 10-6 мікро мк
106 мега М 10 9 нано н
103 кіло к ю12 піко п
102 гекто г 10~15 фемто ф
10і дека Да 10 1Х атто а
ПРИМІТКИ
1. У Фармакопеї для позначення температури за
Цельсієм використовується символ і. Температура за
Цельсієм визначається згідно з рівнянням і = Т - То ,
де То = 273.15 К. Температура за Цельсієм виражається
в градусах Цельсія (символ °С). Один "градус Цельсія"
дорівнює одному кельвіну.
2. Практичні вирази для концентрацій, використову-
ваних у Фармакопеї, визначені у Загальних зауважен-
нях.
3. Радіан являє собою плоский кут, що вирізає на колі
дугу, довжина якої дорівнює радіусу кола.
4. У Фармакопеї умови центрифугування визначають-
ся відцентровим прискоренням по відношенню до
прискорення вільного падіння (§), яке береться
рівним § = 9.806 65 м-С’2.
5. У Фармакопеї деякі величини використовуються без
розмірності, як, наприклад, відносна густина (2.2.5),
оптична густина (2.2.25), питомий показник поглинан-
ня (2.2.25) та показник заломлення (2.2.6); так само як
і величини, виражені в інших одиницях, як, наприклад,
питомий показник оптичного обертання (2.2.7).
6. Мікрокатал визначається як ензиматична ак-
тивність, яка за зазначених умов призводить до пе-
ретворення (наприклад, до гідролізу) одного мікромо-
ля субстрату за одну секунду.
__________________________________________N
1.1. ЗАГАЛЬНІ ПОЛОЖЕННЯ
Усі загальні статті на методи аналізу, лікарські форми і
фармако-технологічні випробування (далі просто за-
гальні статті), а також окремі статті на лікарські суб-
станції, що входять до ДФУ, діляться на дві категорії:
гармонізовані з Європейською Фармакопеєю (ЄФ) і
національні, позначені літерою N.
Усі загальні й окремі статті ДФУ, гармонізовані з ЄФ,
побудовані в такому форматі.
НАЗВА
Адаптований переклад відповідного матеріалу
Європейської Фармакопеї
Національна частина: додаткові випробування,
інформаційні та інші матеріали
У деяких випадках національна частина може бути
відсутньою.
У тому випадку, коли виробництво лікарського засобу
не проводиться відповідно до вимог належної вироб-
ничої практики (НВП, СМР), встановлених в Євро-
пейському Співтоваристві, до даного лікарського за-
собу ставляться альтернативні вимоги, зазначені в
національній частині статті, на що подається зазна-
чення відразу після риси.
Нумерація загальних статей ДФУ, там де вона наявна,
збігається з нумерацією відповідних загальних статей
ЄФ, Загальні статті, не описані в ЄФ, винесено в
кінець відповідного розділу. Загальні статті на суб-
станції та лікарські форми розташовані за алфавітом.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
11
1. Загальні зауваження
1.3. ІНШІ ПОЛОЖЕННЯ, що
ПОШИРЮЮТЬСЯ
НА ЗАГАЛЬНІ ТА ОКРЕМІ
СТАТТІ
ТЕМПЕРАТУРА
Крім термінів, наведених також такі терміни: вище, ви користовують
Теплий від 40 ’С до 50 ’С
Гарячий від Температура 80 ’С до 90 ’С
"водяної бані" від Температура "льодяної бані" 98 ’С до 100 ’С 0 ’С
СПОСОБИ ВИРАЖЕННЯ КОНЦЕНТРАЦІЇ
Якщо зазначено, що при приготуванні суміші розчин-
ників їх беруть у співвідношенні (а:Ь), то мається на
увазі співвідношення об’ємів. Наприклад, співвідно-
шення гексан - бензол (1:3) означає, що змішують
один об’єм гексану з трьома об’ємами бензолу.
1.4. МОНОГРАФІЇ
Вимоги національної частини монографії не є обов’яз-
ковими для продуктів, що мають сертифікат відповід-
ності ЄФ.
ВИРОБНИЦТВО
Описані у Фармакопеї продукти можуть вироблятися
відповідно до вимог, прийнятих в Україні.
ВЛАСТИВОСТІ
Якщо на продукт, описаний у монографії, немає Сер-
тифіката відповідності ЄФ або ДФУ, то інформація,
наведена в цьому розділі, за відсутності інших зазна-
чень, являє собою вимоги, за винятком інформації,
наведеної в дужках.
Розчинність. Для визначення розчинності наважку
субстанції вносять у відміряну кількість розчинника і
безупинно струшують протягом 10 хв при (20±2) °С.
Попередньо зразок може бути розтертий. Для
повільно розчинних зразків, які потребують для свого
розчинення більше 10 хв, допускається також нагрі-
вання на водяній бані до ЗО °С; спостереження роблять
після охолодження розчину до (20+2) °С і енергійного
струшування протягом 1 -2 хв.
Якщо зазначено, що субстанція розчинна в жирних
оліях, то мається на увазі, що вона розчинна в будь- І
якій олії, яка належить до класу жирних олій.
ВИПРОБУВАННЯ ТА КІЛЬКІСНІ ВИЗНАЧЕННЯ
Якщо у випробуваннях із використанням хромато-
графічних методів після наведеного об’єму розчину,
який уводять або наносять, у мікролітрах, у дужках І
зазначено кількість речовини в мікрограмах, то
мається на увазі приблизна кількість.
Коли зазначено, шо випробування проводять "у захи-
щеному від світла місці", це означає, що слід вжити за-
ходів до запобігання прямому сонячному світлу, будь-
якому іншому яскравому світлу, а також виключити
попадання ультрафіолетового світла, наприклад, шля-
хом використання посуду зі спеціального скла, роботи
в затемненій кімнаті тощо.
СТАНДАРТНІ ЗРАЗКИ, СТАНДАРТНІ ПРЕПАРА-
ТИ ТА ЕТАЛОННІ СПЕКТРИ
Фармакопейні стандартні зразки (ФСЗ) — не стан-
дартні зразки, впроваджені Європейською Фармако-
пеєю (ЕР СК8) або Фармакопеєю України (ФСЗ ДФУ).
КОЛЕКЦІЇ МІКРООРГАНІЗМІВ
Українська колекція мікроорганізмів
Інститут мікробіології ім. Д. К. Заболотного
НАН України
01000, Київ, вул. Заболотного 154
Колекція штамів Російського музею патогенних
бактерій
Государственньїй институт стандартизации и
контроля медицинских биологических препаратов
им. Л. А. Тарасевича
121002, Москва, Сивцев-Вражек, 41
12 ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.1. Обладнання
2. МЕТОДИ АНАЛІЗУ
Сита з квадратними отворами виготовляють із відпо- відних матеріалів. Для неаналітичних процедур також 2.1. ОБЛАДНАННЯ. можуть бути використані сита з круглими отворами, діаметр яких у 1.25 рази перевищує розмір сторони квадратного отвору сита відповідного номера. Не має 2.1. 2. ПОРІВНЯЛЬНА ТАБЛИЦЯ ПОРИСТОСТІ бути взаємодії між матеріалом, з якого виготовлене СКЛЯНИХ ФІЛЬТРІВ!1). сито, і речовиною, яку просіюють. Здрібненість зазна- чають в окремій статті, використовуючи номер сита, Можливе таке застосування фільтрів: відповідний номінальному розміру сторони отвору в мікрометрах, який наводиться у дужках після назви < 2.5 Бактеріологічне фільтрування. речовини 4—10 Ультратонка фільтрація, відокремлен- ня мікроорганізмів великого діаметра. Максимальний допуск!3' для розміру отвору (+Х) об- 10 — 40 Аналітична фільтрація, дуже тонка числюють за формулою: фільтрація ртуті, дуже тонке диспер- гування газів. о.75) 40— 100 Тонка фільтрація, фільтрація ртуті, X = -—л—-— + 4((о0-25) , тонке диспергування газів. 100— 160 Груба фільтрація, диспергування і промивання газів, використання де: як підкладки для інших фільтруючих ~ номінальний розмір отвору, матеріалів. 160 — 500 Дуже груба фільтрація часток, дис- При цьому не має бути отворів, розмір яких псреви- пергування і промивання газів. щує номінальний розмір більш як на величину X. Таблиця 2.1.2.-1
Пористість (2) фільтра (Ф. Євр.) Максимальний діаметр пор у мікрометрах Німеччина Франція Велико- британія N
— менше 1.0 — — — ПОРІ.О
1.6 менше 1.6 51 — — ПОР1.6
— 1-2.5 5 — 5
— 1.6-3 — — — ПОРЗ.О
4 1.6-4 — — —
— 4-6 — 5 —
— 3-10 — — — ПОРЮ
10 4— 10 41 — 4
16 10-16 4 4 — ПОРІб
40 16-40 3 3 3 ПОР40
— 40 - 50 — — 2
100 40 - 100 2 2 — ПОР 100
— 100- 120 — — 1
160 100-160 1 1 — ПОР160
— 150-200 0 0 —
250 160 - 250 — — — ПОР250
— 200 - 500 — 00 —
500^ 250 - 500 — — — ПОР500
О).
(1) Дані межі є приблизними. (2) Європейська Фармакопея прийняла систему, запропоновану Міжнародною Організацією зі Стандартизації (IX (3) Див. Міжнародний стандарт ІХО 3310/1(1975).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
13
2.1. Обладнання
Допуск для середнього значення розміру отвору (±¥)
обчислюють за формулою:
При цьому середній розмір отвору не має відхилятися
від номінального розміру більш як на величину ±К
Діаметр <і дроту, застосовуваного для плетіння метале-
вої дротяної тканини, уставленої у раму, подано у
Табл. 2.1.4,-1. Діаметр дроту має знаходитися у межах
від с!тіг, до дтах Ці межі відповідають допускам (±15%)
від рекомендованого номінального діаметра. Діаметр
дроту в ситах має бути однаковим по всьому ситу.
Проміжний допуск (+2) обчислюють за формулою:
Х+ ¥
2
При ньому не більш як 6 % загального числа отворів
можуть мати розміри між "номінальний + X' і "номі-
нальний + 7".
Таблиця 2.1.4.-1
Номер сита (номінальний розмір отвору, мкм) Допуск для отвору, мкм Діаметр дроту, мкм
Максимальний допуск для отвору Допуск для сере- днього значення розміру отвору Проміжний допуск Рекомендований номінальний діаметр Допустима межа
+х ±У +2 а $тах ^тіп
11200 770 350 560 2500 2900 2100
8000 600 250 430 2000 2300 1700
5600 470 180 320 1600 1900 1300
4000 370 130 250 1400 1700 1200
2800 290 90 190 1120 1300 950
2000 230 70 150 900 1040 770
1400 180 50 ПО 710 820 600
1000 140 зо 90 560 640 480
710 112 25 69 450 520 380
500 89 18 54 315 360 270
355 72 13 43 224 260 190
250 58 9.9 34 160 190 130
180 47 7.6 27 125 150 106
125 38 5.8 22 90 104 77
90 32 4.6 18 63 72 54
63 26 3.7 15 45 52 38
45 22 3.1 13 32 37 27
38 — — — зо 35 24
14
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
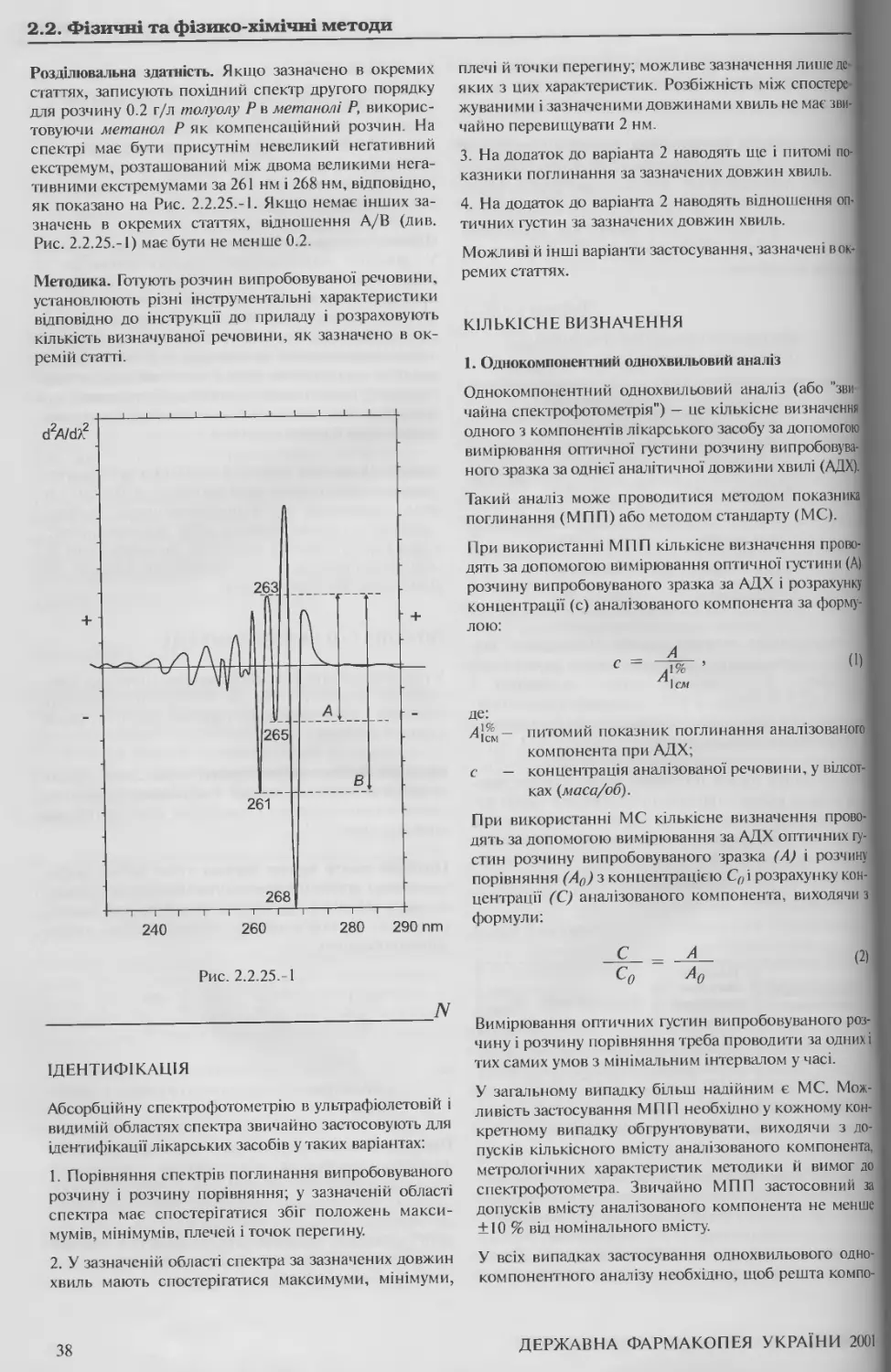
2.2. Фізичні та фізико-хімічні методи
2.2. ФІЗИЧНІ ТА ФІЗИКО-ХІМІЧНІ
МЕТОДИ
2.2.1. ВИЗНАЧЕННЯ ПРОЗОРОСТІ І СТУПЕНЯ
КАЛАМУТНОСТІ РІДИН
Для визначення прозорості і ступеня каламутності
рідин використовують однакові пробірки з безбарвно-
го прозорого нейтрального скла з плоским дном, що
мають внутрішній діаметр від 15 мм до 25 мм. 40-мм
шар випробовуваної рідини порівнюють із 40-мм ша-
ром свіжоприготованого, як описано нижче, еталона.
Порівняння рідин проводять у розсіяному денному
світлі через 5 хв після приготування еталона, перегля-
даючи зразки вздовж вертикальної осі пробірок на
чорному фоні. Розсіяння світла має бути таким, щоб
еталон І легко відрізнявся від води, а еталон II легко
відрізнявся від еталона І.
Випробовувану рідину вважають прозорою, якщо во-
на витримує порівняння з водою Р або розчинником,
використовуваним при приготуванні випробовуваної
рідини при перегляді за описаних вище умов, або її ка-
ламутність не перевищує каламутності еталона І.
РЕАКТИВИ
Розчин гідразину сульфату. 1.0 г гідразину сульфату Р
розчиняють у воді Р і доводять об’єм розчину водою Р
до 100 0 мл. Розчин витримують протягом 4-6 год.
Розчин гексаметилентетраміну. 2.5 г гексаметилентет-
раміну 73розчиняють у 25.0 мл води Ру колбі місткістю
100 мл зі скляною притертою пробкою.
Вихідна суспензія. 25.0 мл розчину гідразину сульфату
додають до приготованого розчину гексаметилентет-
раміну, перемішують і лишають на 24 год. Суспензія
стабільна протягом 2 міс при зберіганні у скляному
посуді, шо не має дефектів поверхні. Суспензія не має
прилипати до скла, і її необхідно ретельно збовтувати
перед використанням.
Основна суспензія. 15.0 мл вихідної суспензії поміща-
ють у колбу місткістю 1000.0 мл і доводять водою Р до
позначки. Термін придатності основної суспензії
24 год.
Еталони. Приготування еталонів проводять відпо-
відно до Табл.2.2.1-1. Основну суспензію і воду Р пе-
ремішують і струшують безпосередньо перед викорис-
танням.
Таблиця 2.2.1.-1
Еталон
І II III IV
Основна суспензія 5.0 мл 10.0 мл 30.0 мл 50.0 мл
Вода Р 95.0 мл 90.0 мл 70.0 мл 50.0 мл
2.2.2. ВИЗНАЧЕННЯ СТУПЕНЯ
ЗАБАРВЛЕННЯ РІДИН
Визначення ступеня забарвлення рідин в ряду корич-
невий - жовтий - червоний проводять візуально шля-
хом порівняння з відповідними еталонами одним з
двох нижче наведених методів, шо зазначають в ок-
ремій статті.
Розчин вважають безбарвним, якщо він витримує
порівняння з водою Р чи розчинником, або забарвле-
ний не більш інтенсивно, ніж еталон В9.
МЕТОД І
2.0 мл випробовуваної рідини порівнюють з 2.0 мл во-
ди Р або розчинника, або еталона (див. Таблиці ета-
лонів), зазначеного в окремій статті, використовуючи
однакові пробірки з безбарвного прозорого нейтраль-
ного скла з зовнішнім діаметром 12 мм. Порівняння
забарвлення проводять у розсіяному денному світлі,
переглядаючи зразки горизонтально (перпендикуляр-
но вісі пробірок) на білому фоні.
МЕТОД II
40-мм шар випробовуваної рідини порівнюють з
40-мм шаром води Рабо розчинника, або еталона (див.
Таблиці еталонів), зазначеного в окремій статті, вико-
ристовуючи однакові пробірки з безбарвного прозо-
рого нейтрального скла з плоским дном, які мають
внутрішній діаметр від 15 мм до 25 мм. Порівняння за-
барвлення проводять у розсіяному денному світлі, пе-
реглядаючи зразки вздовж вертикальної осі пробірок
на білому фоні.
РЕАКТИВИ
Вихідні розчини
Жовтий розчин. 46 г заліза(Ш) хлориду Р поміщають у
мірну колбу місткістю 1000 мл, розчиняють у 900 мл
суміші кислота хлористоводнева Р — вода Р (25:975),
доводять об’єм розчину цією самою сумішшю до по-
значки і перемішують. Визначають концентрацію
одержаного розчину і розбавляють розчин цією самою
сумішшю таким чином, щоб вміст ЕеС13,6Н2О в І мл
становив 45.0 мг.
Розчин зберігають у захищеному від світла місці.
Визначення концентрації. 10.0 мл одержаного розчину
поміщають у конічну колбу з притертою скляною
пробкою місткістю 250 мл, додають 15 мл води Р, 5 мл
кислоти хлористоводневої Р і 4 г калію йодиду Р, колбу
закривають і залишають на 15 хв у темному місці. До-
дають 100 мл води Р і йод, що виділився, титрують
0.1 М розчином натрію тіосульфату, додаючи на-
прикінці титрування як індикатор 0.5 мл розчину крох-
малю Р.
1 мл 0.1 М розчину натрію тіосульфату відповідає
27.03 мг ГеС13,6Н2О.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
15
2.2. Фізичні та фізико-хімічні методи
Червоний розчин. 60 г кобальту хлориду Р поміщають у
мірну колбу місткістю 1000 мл, розчиняють у 900 мл
суміші: кислота хлористоводнева Р— вода Р (25:975),
доводять об’єм розчину цією самою сумішшю до по-
значки і перемішують. Визначають концентрацію
одержаного розчину і розбавляють розчин цією самою
сумішшю таким чином, щоб вміст СоС12,6Н2О в 1 мл
становив 59.5 мг.
Визначення концентрації. 5.0 мл одержаного розчину
поміщають у конічну колбу місткістю 250 мл з притер-
тою скляною пробкою, додають 5 мл розчину перокси-
ду водню розведеного Р і 10 мл розчину 300 г/л натрію
гідроксиду Р, обережно кип’ятять 10 хв, охолоджують і
додають 60 мл кислоти сірчаної розведеної Р і 2 г калію
йодиду Р. Колбу закривають і обережно струшують до
повного розчинення осаду. Йод, що виділився, титру-
ють 0.1 М розчином натрію тіосульфату, додаючи на-
прикінці титрування як індикатор 0.5 мл розчину крох-
малю Р і титрують до блідо-рожевого забарвлення.
1 мл 0.1 М розчину натрію тіосульфату відповідає
23.79 мг СоС12,6Н2О.
Блакитний розчин. 63 г міді сульфату Р помішають
у мірну колбу місткістю 1000 мл, розчиняють у 900 мл
суміші: кислота хлористоводнева Р— вода Р (25:975),
доводять об’єм розчину цією самою сумішшю до по-
значки і перемішують. Визначають концентрацію
одержаного розчину і розбавляють розчин цією самою
сумішшю таким чином, щоб вміст Си8О4,5Н2О в
1 мл становив 62.4 мг.
Визначення концентрації. 10.0 мл одержаного розчину
поміщають у конічну колбу місткістю 250 мл з притер-
тою скляною пробкою, додають 50 мл води Р, 12 мл
кислоти оцтової розведеної Р і 3 г калію йодиду Р. Йод,
що виділився, титрують 0.1 М розчином натрію
тіосульфату, додаючи наприкінці титрування як
індикатор 0.5 мл розчину крохмалю Р і титрують до
блідо-коричневого забарвлення.
1 мл 0.1 М розчину натрію тіосульфату відповідає
24.97 мг Си$О4,5Н2О.
Таблиця 2.2.2,-1
Основні розчини
Основні розчини
П’ять основних розчинів готують з використанням
трьох вихідних розчинів як зазначено у Табл. 2.2.2,-1.
Еталони для методів І і II
Еталони готують з п’яти основних розчинів.
Таблиця 2.2.2.-2
Еталони шкали В
Об’єм у мілілітрах
Еталон Основний розчин В Розчин кислоти хлористоводневої (10 г/л НСІ)
в. 75.0 25.0
в2 50.0 50.0
Вз 37.5 62.5
в4 25.0 75.0
В5 12.5 87.5
Вб 5.0 95.0
в7 2.5 97.5
Ви 1.5 98.5
В» 1.0 99.0
Таблиця 2.2.2.-З
Еталони шкали В¥
Об’єм у мілілітрах
Еталон Основний розчин В¥ Розчин кислоти хлористоводневої (10 г/л НСІ)
В¥( 100.0 0.0
В¥2 75.0 25.0
В¥3 50.0 50.0
В% 25.0 75.0
В¥5 12.5 87.5
ВХ, 5.0 95.0
В¥7 2.5 97.5
Об’єм у мілілітрах
Основний розчин Жовтий розчин Червоний розчин Блакитний розчин Розчин кислоти хлористо- водневої (10 г/л неї)
В(корич- невий) 3.0 3.0 2.4 1.6
В¥(корич- нювато- жовтий) 2.4 1.0 0.4 6.2
V (жовтий) 2.4 0.6 0.0 7.0
О¥ (зелену- вато- жовтий) 9.6 0.2 0.2 0.0
К (чер- воний) 1.0 2.0 0.0 7.0
Таблиця 2.2.2.-4
Еталони шкали ¥
Об’єм у мілілітрах
Еталон Основний розчин ¥ Розчин кислоти хлористоводневої (10 г/л НСІ)
¥| 100.0 0.0
¥2 75.0 25.0
¥3 50.0 50.0
¥4 25.0 75.0
¥5 12.5 87.5
¥6 5.0 95.0
¥7 2.5 97.5
16
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
Таблиця 2.2.2.-5
Еталони шкали 6¥
Об’єм у мілілітрах
Еталон Основний розчин С¥ Розчин кислоти хлористоводневої (10 г/л НСІ)
С¥, 25.0 75.0
С¥2 15.0 85.0
С¥3 8.5 91.5
С% 5.0 95.0
С¥5 3.0 97.0
с\ 1.5 98.5
С¥7 0.75 99.25
Таблиця 2.2.2.-6
Еталони шкали К
Об’єм у мілілітрах
Еталон Основний розчин К Розчин кислоти хлористоводневої (10 г/л НСІ)
К| 100.0 0.0
К2 75.0 25.0
Кз 50.0 50.0
Щ 37.5 62.5
25.0 75.0
Кб 12.5 87.5
К7 5.0 95.0
ЗБЕРІГАННЯ
Еталони для визначення ступеня забарвлення рідин за
методом І зберігають в запаяних пробірках з безбарв-
ного прозорого нейтрального скла із зовнішнім діаме-
тром 12 мм, у захищеному від світла місці.
Еталони, які використовують для визначення ступеня
забарвлення рідин за методом II, готують з відповідних
основних розчинів безпоссредньо перед використанням.
____________________________________________N
Порівняння ступеня забарвлення рідини з еталонами
(В, В¥, ¥, О¥, К)|_3 звичайно проводять за методом І;
якщо використовують еталони (В, В¥, ¥, Сі¥. К.)4^9, за-
стосовують метод II.
Ступінь забарвлення випробовуваного зразка не
має перевищувати ступінь забарвлення відповідно-
го еталона. Колір випробовуваного зразка має бути
максимально наближений до кольору відповідного
еталона.
ЗБЕРІГАННЯ
Термін придатності вихідних і основних розчинів 1 рік.
2.2.3. ПОТЕНЦІОМЕТРИЧНЕ
ВИЗНАЧЕННЯ рН
рН — число, яке умовно характеризує концентрацію
іонів водню у водних розчинах. На практиці рН визна-
чають експериментально. рН випробовуваного розчи-
ну пов’язане з рН стандартного розчину (рН5) таким
рівнянням:
Рн = Рн5 - —.А,
к
де:
Е — потенціал електрода у випробовуваному
розчині, у вольтах;
Е5 — потенціал того самого електрода в розчині з
відомим рН (рН5), у вольтах.
Температурний коефіцієнт (к), виражений у вольтах,
при будь-якій температурі може бути розрахований за
формулою:
к = 0.05916 + 0.000198(1-25 °С).
Таблиця 2.2.3.-1
Значення к при різних температурах
Температура °С к
15 0.0572
20 0.0582
25 0.0592
зо 0.0601
35 0.0611
Потенціометричне визначення рН проводять шляхом
вимірювання різниці потенціалів між двома відпо-
відними електродами, зануреними у випробовуваний
розчин: один з електродів чутливий до іонів водню
(звичайно скляний електрод), другий — електрод
порівняння (наприклад, насичений каломельний
електрод).
Прилад. Вимірювальним приладом є вольтметр з вхід-
ним опором принаймні у 100 разів більшим за опір
використовуваних електродів. Прилад звичайно гра-
дуюється в одиницях рН і повинен мати таку чут-
ливість, щоб можна було виявити відмінність при-
наймні 0.05 одиниць рН або 0.003 В.
Методика. Усі виміри проводять при тій самій темпе-
ратурі в інтервалі від 20 °С до 25 °С, якщо немає інших
зазначень в окремій статті. Табл. 2.2.3.-2 показує за-
лежність значень рН від температури для різних стан-
дартних буферних розчинів, використовуваних для
калібрування. Якщо необхідно, враховують темпера-
турні поправки відповідно до інструкції підприємст-
ва-виробника. Прилад калібрують за допомогою бу-
ферного розчину калію гідрофталату (первинний стан-
дарт) і одного з буферних розчинів з іншим значенням
рН (краще одного з наведених у Табл. 2.2.3.-2). Пока-
зання приладу для третього буферного розчину з
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
17
2.2. Фізичні та фізико-хімічні методи
проміжним значенням рН не мають відрізнятись
більш як на 0.05 одиниць рН від табличного значення
рН цього розчину. Електроди занурюють у випробову-
ваний розчин і вимірюють рН у тих самих умовах, що
і для буферних розчинів.
Якщо прилад використовують часто, його калібруван-
ня проводять регулярно. У противному разі калібру-
вання приладу має проводитися перед кожним ви-
міром.
Усі випробовувані розчини і стандартні буферні роз-
чини мають бути приготовані на воді, вільній від діок-
сиду вуглецю, Р.
Приготування стандартних буферних розчинів
0.05 М розчин калію тетраоксалату. 12 61 г
КС4Н3О8,2Н2О розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 1000.0 мл.
Насичений при 25 °С розчин калію гідротартрату.
Надлишок КС4Н5О6 енергійно струшують з водою Р
при 25 °С. Фільтрують або декантують. Розчин вико-
ристовують свіжоприготованим.
0.05 М розчин калію дигідроцитрату. 11.41 г КС6Н7О7
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 1000.0 мл. Розчин використову-
ють свіжоприготованим.
0.05 М розчин калію гідрофталату. 10.13 г КС8Н5О4, по-
передньо висушеного при температурі від 110 °С до
135 °С до постійної маси, розчиняють у воді Лі доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
0.025 Мрозчин калію дигідрофосфату і 0025 Мрозчин
натрію гідрофосфату. 3.39 г КН2РО4 і 3.53 г №2НРО4,
попередньо висушених протягом 2 год при темпера-
турі від 110 °С до 130 °С до постійної маси, розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 1000.0 мл.
0.0087 М розчин калію дигідрофосфату і 0.0303 Мроз-
чин натрію гідрофосфату. 1.18 г КН2РО4 і 4.30 і
№2Н РО4, попередньо висушених при температурі вії
110 °С до 130 °С до постійної маси, розчиняють у во-
ді Р і доводять об’єм розчину тим самим розчиннико^
до 1000.0 мл.
0.01 М розчин натрію тетраборату. 3.80 г №2В4О7
ЮН2О розчиняють у воді Р \ доводять об’єм розчин;
тим самим розчинником до 1000.0 мл Зберігають, за
хищаючи від діоксиду вуглецю.
0.025 М розчин натрію карбонату і 0.025 М розчиї
натрію гідрокарбонату. 2.64 г N33003 і 2.09 г №НСО
розчиняють у воді Р ї доводять об’єм розчину тим са
мим розчинником до 1000.0 мл.
________________________________________________Д
Водневим показником (рН) називається від’ємний де
сятковий логарифм активності іонів водню.
рН = -1§ан+.
Прилад. Підготовку приладу, електродної системи, ;
також калібрування приладу проводять відповідно Д(
інструкції підприємства-виробника.
Допускається використання як електрода порівнянні
хлорсрібного електрода, а також застосування елект
ролітичного містка.
Методика. На додаток до стандартних буферних роз
чинів, описаних вище, для калібрування приладу до
пускається використання насиченого при 25 °С розчин
кальцію гідроксиду.
Приготування стандартних буферних розчинів
Для приготування стандартних буферних розчинії
можуть бути використані реактиви кваліфікації "Длі
Таблиця 2.2.3.-:
рН стандартних буферних розчинів при різних температурах
Тем- 0.05 М Насиче- 0.05 М 0.05 М 0.025 М 0.0087 М о.оїм 0.025 М
пера- розчин ний при розчин розчин розчин ка- розчин ка- розчин розчин
тура"С калію 25 °С калію калію лію дигід- лію дигід- натрію натрію
тетра- розчин дигідро- гідро- рофосфа- рофосфа- тетра- карбо-
оксала- калію гід- цит- фта- ту і 0.025 М туї борату нашу і 0.025 М
ту ротарт- риту лату 0.0303 м
риту розчин розчин розчин
натрію натрію натрію
гідро- фосфату гідро- фосфату гідро- карбо-
нату
КС4Н3О8, кс4н5о6 КСбНтО7 кс8н5о4 КН2РО4 + КН2РО4 +
2Н2О Ка2ІІРО4 Ка2Н РО4 ЮН2О КаІІСО3
15 1.67 3.80 4.00 6.90 7 45 9.28 10.12
20 1.68 3.79 4.00 6.88 7 43 9.23 10.06
25 1.68 3.56 3.78 4.01 6.87 7.41 9.18 10.01
ЗО 1.68 3.55 3.77 4.02 6.85 7.40 9.14 9.97
35 1.69 3.55 3.76 4.02 6.84 7.39 9.10 9.93
(і) ДрН +0.001 -0.0014 -0.0022 +0.0012 -0.0028 -0.0028 -0.0082 -0.0096
де
<0 - змінювання рН на градус за Цельсієм
18
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 200
2.2. Фізичні та фізико-хімічні методи
рН-метрії", х.ч., ч.д.а. або реактиви імпортного вироб-
ництва відповідної чистоти.
Насичений при 25 °С розчин кальцію гідроксиду.
Са(ОН)2 струшують протягом години з водою Р при
25 °С і після відстоювання декантують або фільтру-
ють Зберігають, захищаючи від діоксиду вуглецю.
Таблиця 2.2.3.-2.1
рН насиченого при 25 °С розчину кальцію гідроксиду
Температура "С РН
15 12.81
20 12.63
25 12.45
ЗО 12.29
35 12.13
Допускається вимірювання рН у змішаних водно-ор-
ганічних розчинниках. У цьому випадку, а також для
деяких колоїдних систем одержані значення рН є
умовними.
2.2.4. ЗАЛЕЖНІСТЬ МІЖ РЕАКЦІЄЮ
РОЗЧИНУ, ПРИБЛИЗНИМ ЗНАЧЕННЯМ
рН І КОЛЬОРОМ ІНДИКАТОРІВ
До 10 мл випробовуваного розчину додають 0.1 мл
розчину індикатора, крім винятків, зазначених у
Табл. 2.2.4,-1.
___________________________________________N
Додаткова інформація зазначена у Табл. 2.2.4.-2.
2.2.5. ВІДНОСНА ГУСТИНА
Відносна густина речовини являє собою відношен-
ня маси певного об’єму цієї речовини до маси рівного
йому об’єму води при температурі 20 °С.
Відносну густину визначають за допомогою пікно-
метра, густиноміра. гідростатичних вагів або ареомет-
ра з точністю до числа десяткових знаків, зазначених в
окремій статті. Атмосферним тиском при зважуванні
нехтують, бо зв’язана з цим помилка не перевищує
одиниці у третьому десятковому знаку.
Звичайно використовують два інші визначення.
Відносна густина сї^' речовини являє собою відно-
шення маси певного об’єму цієї речовини при темпе-
Таблиця 2.2.4.-1
Реакція розчину7 рН Індикатор Колір
Лужна >8 Лакмусовий папір Тимоловий синій Синій Сірий або фіолетово-синій
Слабколужна 8.0- 10.0 Фенолфталеїн1 Тимоловий синій Від безбарвного до рожевого Сірий
Сильнолужна > 10 Фенолфталеїновий папір Тимоловий синій Червоний Фіолетово-синій
Нейтральна 6.0-8.0 Метиловий червоний Феноловий червоний1 Жовтий Жовтий або рожевий
Нейтральна за тропеоліном ОО >3.0 Тропеолін ОО Жовтий
Нейтральна за диметиловим ЖОВТИМ >4.0 Диметиловий жовтий1 Жовтий, червоний після додавання 0.1 мл 0.1 М розчину кислоти
Нейтральна іа метиловим червоним 4.5 - 6.0 Метиловий червоний Оранжево -черво ний
Нейтральна за фенолфталеїном < 8.0 Фенолфталеїн1 Безбарвний; рожевий або червоний після додавання 0.05 мл 0.1 М розчину основи
Кисла <6 Метиловий червоний Бромтимоловий синій2 Оранжевий або червоний Жовтий
Слабкокисла 4.0 - 6.0 Метиловий червоний Бромкрезоловий зелений Оранжевий Зелений або синій
Сильнокисла <4 Конго червоного папір Диметиловий жовтий1 Зелений або синій Оранжевий або червоний
1 Використовують 0.05 мл.
2 Використовують розчин бромтимолоеого синього Р1.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
19
2.2. Фізичні та фізико-хімічні методи
Таблиця 2.2.4.-3
Інтервали рН і зміни кольору індикаторів
Назва індикатора Інтервал рН пере- ходу кольору Зміна кольору
Метиловий фіолетовий 0.1-1.5 Жовтий — зелений
Малахітовий зелений 0.1-2.0 Жовтий — зеленувато-блакитний
Крезоловий червоний 0.2-1.8 Червоний — жовтий
Крезоловий пурпуровий 1.8-2.8 Рожево-червоний — жовтий
Тимоловий синій 1.2-2.8 Червоний — жовтий
Метиловий фіолетовий 1.5-3.2 Зелений — фіолетовий
Диметиловий жовтий 3.0-4.0 Червоний — жовтий
Метиловий оранжевий 3.0-4.4 Червоний — жовтий
Бромфеноловий синій 3.0-4.6 Жовтий — синій
Конго червоний 3.0-5.2 Синьо-фіолетовий — червоний
Бромкрезоловий зелений (синій) 3.8-5.4 Жовтий — синій
Алізариновий червоний С 4.6-6.0 Жовтий — пурпурово-червоний
Метиловий червоний 4.2-6.2 Червоний — жовтий
Лакмоїд 4.4-Б.2 Червоний — синій
Бромкрезоловий пурпуровий 5.2-6.8 Жовтий — пурпуровий
Бромтимоловий синій 6.0-7.6 Жовтий — синій
Нейтральний червоний 6.8-8.0 Червоний — жовтий
Феноловий червоний 6.8-8.4 Жовтий — червоний
Крезоловий червоний 7.2-8.8 Жовтий — пурпурово-червоний
а- Нафтолфталеїн 7.4-8.6 Жовтувато-рожевий — зеленувато-синій
Крезоловий пурпуровий 7.4-9.0 Жовтий — фіолетовий
Тимоловий синій 8.0-9.6 Жовтий — синій
Тимолфталеїн 9.4-10.6 Безбарвний — синій
Алізариновий жовтий Р 10.0-12.0 Світло-жовтий — червоно-оранжевий
Малахітовий зелений 11.4-13.0 Зеленувато-блакитний — безбарвний
Індигокармін 11.6-14.0 Синій — жовтий
ратурі 20 °С до маси рівного йому об’єму води при тем-
пературі 4 °С.
Густина р20 речовини — це відношення маси речовини
до її об’єму при температурі 20 °С. Густину виражають
у кілограмах на кубічний метр (1 кг м"* 3 = 10 3 г см 3).
Числові співвідношення між відносною густиною і гус-
тиною, у кілограмах на кубічний метр, виражаються так:
р20 = 998.202
або
0$ = 1.001 80- Ю’3р2О
р20 = 999.972 $
або
4°= 1.000 03- ю-3р20
4°= 0.998 230
N
У тих випадках, коли для речовини регламентують
значення густини, її визначення проводять одним
з нижченаведених методів, якщо немає інших зазна-
чень в окремій статті.
Метод 1. Застосовують у випадку визначення густини
рідин з точністю до 0.001.
Чистий сухий пікнометр зважують з точністю до
0.0002 г, заповнюють за допомогою сухої лійки дисти-
льованою водою трохи вище позначки, закривають
пробкою і витримують протягом 20 хв у термостаті, в
якому підтримують постійну температуру води 20 °С з
точністю до 0.1 °С. При цій температурі рівень води у
пікнометрі доводять до позначки, швидко відбираючи
надлишок води за допомогою піпетки або згорнутої в
трубку смужки фільтрувального паперу. Пікнометр
знову закривають пробкою і витримують у термостаті
ще 10 хв, перевіряючи положення меніска по відно-
20
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
шенню до позначки. Потім пікнометр виймають з тер-
мостата, фільтрувальним папером витирають
внутрішню поверхню шийки пікнометра, а також
увесь пікнометр ззовні, лишають під склом аналітич-
них ватів протягом 10 хв і зважують з тією самою
точністю.
Пікнометр звільняють від води, висушують, споліску-
ючи послідовно спиртом і ефіром (сушити пікнометр
шляхом нагрівання не допускається), видаляють за-
лишки ефіру продуванням повітря, заповнюють
пікнометр випробовуваною рідиною і потім прово-
дять ті самі операції, що й з дистильованою водою.
Густину р20 (г/см3) обчислюють за формулою:
(т7 - лг)-0.99703
Р20 = —---------------- + 0.0012 ,
ш т1 - т
де:
т — маса порожнього пікнометра, у грамах;
т} — маса пікнометра з дистильованою водою, у
грамах;
т, — маса пікнометра з випробовуваною ріди-
ною, у грамах;
0.99703— шачення густини води при 20 °С
(у г/см3 з врахуванням густини повітря);
0.0012 — густина повітря при 20 °С і барометрично-
му тиску 1011 гПа (760 мм рт. ст.).
Метод 2. Застосовують у випадку визначення густини
рідин з точністю до 0.01.
Випробовувану рідину поміщають у циліндр і при тем-
пературі рідини 20 °С обережно опускають в неї чис-
тий сухий ареометр, на шкалі якого передбачена
очікувана величина густини. Ареометр не випускають
з рук, доки не стане очевидним, що він плаває; при
цьому необхідно стежити, щоб ареометр не торкався
стінок і дна циліндра. Відлік проводять через 3-4 хв
після занурення за поділкою на шкалі ареометра,
відповідною нижньому меніску рідини (при відліку
око мас бути на рівні меніска).
Примітки.
1. Визначення густини сильнолетких речовин ареоме-
тром не допускається.
2. У випадку визначення густини темнозабарвлених
рідин відлік проводять за верхнім меніском.
Метод 3. Застосовують для визначення густини твер-
дих жирів і воску. Точно зважують порожній пікно-
метр, потім зважують той самий пікнометр, наповне-
ний дистильованою водою, температура якої 20 °С.
Після цього воду видаляють і пікнометр висушують.
Усі операції проводять, дотримуючи умов, зазначених
у методі 1.
У пікнометр уливають задопомогою піпетки або неве-
ликої лійки з тонковідтягнутим кінцем розплавлений
жир або віск у такій кількості, щоб він займав І/3-1/2
об’єму пікнометра. Пікнометр ставлять на 1 год без
пробки в гарячу воду, потім охолоджують до 20 °С і зва-
жують; доводять до позначки дистильованою водою
при 20 °С, витирають насухо і знову зважують. У
обох фазах на поверхні їхнього поділу не має бути
бульбашок повітря.
Густину обчислюють за формулою:
(щ7 - т)-0.99703
Р20 = ,----------------- + 0.0012 ,
(/«! + т2) - (т + т3)
де:
т — маса порожнього пікнометра, у грамах;
/И] — маса пікнометра з дистильованою водою, у
грамах;
т2 — маса пікнометра з жиром, у грамах;
т3 — маса пікнометра з жиром і водою, у грамах.
2.2.6. ПОКАЗНИК ЗАЛОМЛЕННЯ
(ІНДЕКС РЕФРАКЦІЇ)
Показник заломлення ^середовища відносно повітря
дорівнює відношенню синуса кута падіння променя
світла в повітрі до синуса кута заломлення променя
світла в даному середовищі.
Якщо немає інших зазначень в окремій статті, визна-
чення показника заломлення проводять при темпера-
турі (20±0.5) °С за довжини хвилі лінії Б спектра
натрію (А = 589.3 нм); показник заломлення, визначе-
ний за таких умов, позначають індексом п2^.
Рефрактометри звичайно визначають критичний кут.
У таких приладах основною частиною є призма з відо-
мим показником заломлення, що перебуває в контакті
з аналізованою рідиною.
Для калібрування приладів використовують еталонні
рідини, зазначені у Табл. 2.2.6,-1. Значення показника
заломлення кожної еталонної рідини зазначається на
етикетці.
Таблиця 2.2.6,-1
Еталонна рідина Дл/Дґ (температурний коефіцієнт)
Триметилпентан ФСЗ - 0.00049
Толуол ФСЗ - 0.00056
Метилнафталін ФСЗ - 0.00048
При використанні білого світла рефрактометри мають
бути обладнані компенсаційною системою. Прилад
має давати показання з точністю як мінімум до
третього десяткового знака і забезпечувати можли-
вість проведення операцій при заданій температурі.
Ціна поділки термометра не має перевищувати 0.5 °С.
____________________________________________N
Показник заломлення залежить від ^мператури і дов-
жини хвилі світла, за якої здійснюють визначення. У
розчинах показник заломлення залежить також від
концентрації речовини і природи розчинника.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
21
2.2. Фізичні та фізико-хімічні методи
Визначення показника заломлення застосовується
для установлення справжності і чистоти речовини.
Метод застосовують також для визначення концент-
рації речовини в розчині, яку знаходять за графіком
залежності показника заломлення від концентрації. У
цьому випадку точність виміру показника заломлення
має бути не нижче ±2.10-4. На графіку вибирають
інтервал концентрацій, у якому дотримана лінійна за-
лежність між показником заломлення і концент-
рацією. У цьому інтервалі концентрацію можна об-
числити за формулою:
ратурі 20 °С у розчині випробовуваної речовини і роз
рахований для товщини шару 1 дециметр у перерахуй
ку на вміст 1 грама речовини в 1 мілілітрі розчину,
питомого оптичного обертання речовини в розчин
завжди зазначають використовуваний розчинник
концентрацію розчину.
У Фармакопеї питоме оптичне обертання виражають
градус-мілілітрах на дециметр-грамм [(°)- мл-дм-1- г*
Перерахунок питомого обертання в одиницях з
Міжнародною системою в одиниці, використовуваь
Фармакопеєю, проводять за формулою:
Р
[<*„,]{ = И-0.1745
де:
X — концентрація розчину;
п — показник заломлення розчину;
«о — показник заломлення розчинника при тій
самій температурі;
Р — фактор, що дорівнює величині приросту по-
казника заломлення при збільшенні концент-
рації на 1 % (встановлюється експериментально).
Для калібрування рефрактометрів використовують
еталонні рідини, зазначені у Табл. 2.2.6.-1. Допус-
кається калібрування за однією з еталонних рідин, що
додаються до приладів, або за дистильованою водою,
для якої Лр = 1.3330 (Дя/Л/ = - 0.000085).
У деяких випадках, зазначених в окремій статті, ку
обертання може бути виміряний при температура:
відмінних від 20 °С і за інших довжин хвиль.
Використовуваний поляриметр має забезпечува'
вимірювання з точністю до 0.01°. Шкалу звичайно п
ревіряють за допомогою сертифікованих кварцові
пластинок. Лінійність шкали може бути перевірена
допомогою розчинів сахарози.
2.2.7. ОПТИЧНЕ ОБЕРТАННЯ
Оптичне обертання — це властивість речовини обер-
тати площину поляризації поляризованого світла.
Питоме оптичне обертання , виражене в
радіанах (рад), являє собою обертання, викликане ша-
ром рідини або розчину завтовшки 1 метр, який
містить 1 кілограм оптично активної речовини в 1
метрі кубічному при проходженні крізь нього поляри-
зованого світла з довжиною хвилі Л при температурі і.
Для практичних цілей питоме оптичне обертання
[о!/л]^ звичайно виражають у мілірадіан-метрах квад-
ратних на кілограм (мрад-м1
У Фармакопеї використовують такі визначення.
Кут оптичного обертання рідких речовин являє собою
кут обертання а, виражений у градусах (°), площини
поляризації за довжини хвилі лінії Б спектра натрію
(А. = 589.3 нм), виміряний при температурі 20 °С у тов-
щині шару 1 дециметр. Для розчинів спосіб приготу-
вання зазначають в окремій статті.
Питоме оптичне обертання [о!]р рідини являє собою
кут обертання а, виражений у градусах (°), площини
поляризації за довжини хвилі лінії Б спектра натрію
бк = 589.3 нм), виміряний при температурі 20 °С, роз-
рахований для товщини шару випробовуваної речови-
ни 1 дециметр і поділений на густину, виражену в гра-
мах на кубічний сантиметр.
Питоме оптичне обертання [а]□ речовини в розчині
являє собою кут обертання а, виражений у граду-
сах (°), площини поляризації за довжини хвилі лінії Б
спектра натрію (А. = 589.3 нм), виміряний при темпе-
Методика. Визначають нуль поляриметра і кут обе|
тання площини поляризації за довжини хвилі лінії
спектра натрію бк = 589.3 нм) при температу
(20+0.5) °С. Вимірювання оптичного обертання м<
жуть проводитися при інших температурах лише в ті
випадках, якщо в окремій статті зазначений епос
урахування температури. Визначають нуль приладу
закритою трубкою; для рідин — з порожньою труї
кою; для розчинів твердих речовин — з трубкою, з:
повненою відповідним розчинником. Проводять 1
менше п’яти вимірів і розраховують середнє значенн
Питоме оптичне обертання обчислюють за форму
ми, позначаючи праве і ліве обертання відповідно
Для рідин:
г і20-
Мр -
а
/• Р2о
Для речовин у розчині:
г т20-
Мр -
1000 • а
де:
с — концентрація розчину, у г/л.
Вміст с або с'розчиненої речовини, у г/л або у ві.
ках
(м/м), відповідно, обчислюють за формулами:
с
1000 • а
/|аІО
100-а
/• [а]р • Р20
де:
а
І
Р20
кут обертання, виміряний при темпераї
(20±0.5) °С, у градусах;
довжина поляриметричної трубки, у де
метрах;
густина при температурі 20 °С, у грамах на
бічний сантиметр.
22
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ
2.2. Фізичні та фізико-хімічні методи
У фармакопейному аналізі густину заміняють віднос-
ною густиною (2.2.5).
2.2.8. В’ЯЗКІСТЬ
Динамічна в’язкість або коефіцієнт в’язкості г/ — це
тангенціальна сила, гцо припадає на одиницю по-
верхні і називається також напругою зсуву' т, виражена
\ паскалях (Па), яку необхідно докласти для того, щоб
перемістити шар рідини площею 1 м2 зі швид-
кістю (V) 1 метр за секунду (м с '), який знаходиться
на відстані (х) 1 метр відносно другого шару, паралель-
но площині ковзання.
Величина сК/сіх являє собою градієнт швидкості і обу-
мовлює швидкість зсуву £), виражену в обернених се-
кундах (с1). таким чином:
Л = т/Б
Одиницею динамічної в’язкості є паскаль-секунда
(Па с). Найчастіше використовується дольна одиниця
міліпаскаль-секунда (мПа-с).
Кінематичну в’язкість V, виражену в метрах квадрат-
них на секунду (м2-с ')’ одержують діленням величини
динамічної в’язкості г/ на густину рідини р, виражену
в кілограмах на метр кубічний (кг-м3), виміряну при
тій самій температурі, таким чином:
V = Л/Р
Кінематичну в’язкість звичайно виражають у мілімет-
рах квадратних на секунду (ммк-1).
Для визначення в’язкості ньютонівських рідин можна
використати капілярний віскозиметр; для визначення
в’язкості як ньютонівських, так і неньютонівських
рідин можна використати ротаційний віскозиметр.
Допускається використання й інших віскозиметрів за
умови, що точність і правильність вимірів будуть не
гіршими, ніж у випадку використання віскозиметрів,
описаних нижче.
_______________________________________________N
В'язкість (внутрішнє тертя) — властивість текучих тіл
чинити опір переміщенню однієї їхньої частини
відносно іншої.
Текучі тіла можуть мати ньютонівський тип течії.
Ньютонівськими рідинами називають системи, в’яз-
кість яких не залежить від напруги зсуву і є постійною
величиною відповідно до закону Ньютона.
Неньютонівські рідини не підпадають під дію закону
Ньютона, їхня в’язкість залежить від напруги зсуву.
Для ньютонівських рідин розрізняють динамічну,
кінематичну, відносну, питому, зведену і характерис-
тичну в’язкості. Для неньютонівських рідин характер-
на, головним чином, структурна в’язкість.
Структурна (ефективна або уявна) в’язкість —
в’язкість при даній напрузі зсуву. У ряді випадків тре-
ба визначити в’язкість однієї рідини відносно іншої —
відносну в’язкість г)ЬІДН.
Часто використовують питому в’язкість цпит, що по-
казує, який внесок у в’язкість розчину робить присут-
ність у ньому розчиненої речовини:
де:
Г) — в’язкість розчину,
г)0 — в’язкість розчинника.
Питому в’язкість, віднесену до одиниці концентрації
розчину, називають зведеною в’язкістю Т)ЗВЄД:
ДіВЄД ^ПИТ /с ’
де:
с — концентрація розчину.
Для розчинів полімерів в’язкість є функцією молеку-
лярних мас, форми, розмірів і гнучкості макромоле-
кул. Щоб визначити структурні характеристики полі-
мерів, зведену в’язкість екстраполюють до нульової
концентрації. У такому випадку уводиться поняття ха-
рактеристичної в ’язкості | :
[77] = 1ітт7звед = Іігп
2.2.9. МЕТОД КАПІЛЯРНОЇ ВІСКОЗИМЕТРІЇ
Визначення в’язкості проводять, використовуючи
підхожий капілярний віскозиметр, при температурі
(20±0.1) °С, якщо не зазначена інша температура в ок-
ремій статті. Час витікання рідини від однієї позначки
віскозиметра до другої вимірюється секундоміром з
точністю до однієї п’ятої секунди. Одержані дані є
прийнятними за умови, що результати двох послідо-
вних вимірювань відрізняються не більш як на 1 %.
Час витікання випробовуваної рідини визначають як
середнє не менш як трьох вимірів.
Динамічну в’язкість ц (2.2.8), виражену в міліпаскаль-
секундах (мПас), обчислюють за формулою:
П = крї,
де:
к — стала віскозиметра, у міліметрах квадратних
на секунду квадратну (мм2 -с-2);
р — густина випробовуваної рідини, у міліграмах
на міліметр кубічний (мг-мм-3), одержана
множенням відносної густини (г?2о) на 0.9982;
г — час витікання випробовуваної рідини, у секун-
дах (с).
Сталу к визначають з використанням відповідної
фармакопейної рідини для калібрування віскозиметрів.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
23
2.2. Фізичні та фізико-хімічні методи
Кінематичну в’язкість, виражену у міліметрах
квадратних на секунду (мм2-с -•), обчислюють за
формулою:
V = кі
Визначення в’язкості може проводитися за допомо-
гою приладу*1' (Рис. 2.2.9.-1), характеристики якого
зазначені у Табл. 2.2.9.-1.
Методика. Випробовувану рідину, що має температуру
20 °С, якщо в окремій статті не зазначена інша
температура, заливають у віскозиметр через трубку (Е)
у такій кількості, щоб заповнити розшир (А), але при
цьому рівень рідини у розширі (В) має залишатись
нижче виходу до вентиляційної трубки (М).
Віскозиметр у вертикальному положенні занурюють у
водяну баню при температурі (20±0.1) сС, якщо в
окремій статті не зазначена інша температура, утри-
муючи його в цьому положенні не менше ЗО хв для
встановлення температурної рівноваги. Трубку (М)
закривають і підвищують рівень рідини у трубці (ІУ)
таким чином, щоб він знаходився приблизно на 8 мм
вище від позначки (Е). Утримують рідину на цьому
рівні, закривши трубку (К) і відкривши трубку (М).
Потім відкривають трубку (М) і вимірюють час, за який
рівень рідини знизиться від позначки (Е) до позначки
(Е), секундоміром з точністю до однієї п’ятої секунди.
Рисунок 2.2 9.-1. Віскозиметр з висячим рівнем
Розміри зазначені у міліметрах
________________________________________________Л
Поряд з приладом, описаним више, допускається ви-
користання віскозиметрів капілярних скляних з вися-
чим рівнем*2', параметри якого аналогічні до наведе-1
ного на Рис. 2.2.9.-1. В’язкість вимірюють відповідноз І
інструкцією щодо застосування віскозиметра.
Для визначення відносної в’язкості рідини вимірюють і
час іОсе/, витікання між верхньою і нижньою познач-1
кою віскозиметра тієї рідини, відносно якої проводять
виміри Т)еІдн. Потім у тому ж чистому і сухому віскози-1
метрі за тих же умов визначають час витікання ЦІ
випробуваної рідини.
Одночасно вимірюють густину рідин, що вивчаються,
пікнометром (р0, р) при тій самій температурі, при
якій визначають в’язкість, і обчислюють відносну
в’язкість за формулою:
_ ї сер Р
~ *0сер- Р0
Для визначення характеристичної в’язкості готують
не менше п’яти досліджуваних розчинів різної кон-
центрації. При цьому має виконуватись умова можли-
вості застосування методу лінійної екстраполяції зве-
деної в’язкості до нульової концентрації, тобто треба
обирати мінімальні концентрації розчину у межах чут-
ливості й точності методу вимірювання. Для кожної
концентрації розчину визначають р і г)відн і розрахову-
ють зведену в’язкість. Потім будують залежність Т]зеед
від концентрації с і графічно або лінійним методом
найменших квадратів екстраполюють зведену в’яз-
кість до нульової концентрації, тобто знаходять харак-
теристичну в’язкість.
2.2.10. МЕТОД РОТАЦІЙНОЇ ВІСКОЗИМЕТРІЇ
Принцип дії найчастіше використовуваних ро-
таційних віскозиметрів полягає у вимірюванні сили
зсуву в рідкому середовищі, розташованому між двома
коаксіальними циліндрами, один з яких обертається
двигуном, а другий обертається за допомогою першо-
го. В’язкість (структурна, ефективна або уявна в’яз-
кість) характеризується кутом (М), на який повер-
тається другий циліндр; цей кут пропорційний мо-
менту сили, вираженому у ньютон-метрах (Н-м).
У випадку ламінарного потоку динамічну в’язкість г),
виражену в паскаль-секундах (Па с), обчислюють за
формулою:
1 1
де:
й — глибина занурення другого циліндра у рідк
середовище, у метрах;
<') У Державній Фармакопеї України (за аналогією з Європейською Фармакопеєю) описується система, запропонована Міжнародної
організацією із стандартизації.
<2' Наприклад, віскозиметр капілярний скляний типу ВПЖ-1.
24
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 20С
2.2. Фізичні та фізико-хімічні методи
Таблиця 2.2.9.-1
Розмір Номінальна стала віскозиметра Діапазон кінематичної в’язкості Внутрішній діаметр трубки К Об’єм розширення с Внутрішній діаметр трубки N
ММ2'С 2 ММ2СЧ ММ (±2%) мл (± 5 %) ММ
І 0.01 3.5-10 0.64 5.6 2.8-3.2
1А 0.03 6-30 0.84 5.6 2.8-3.2
2 0.1 20-100 1.15 5.6 2.8-3.2
2А 0.3 60-300 1.51 5.6 2.8-3.2
3 1.0 200-1000 2.06 5.6 3.7-4.3
ЗА 3.0 600-3000 2.74 5.6 4.6-5.4
4 10 2000-10 000 3.70 5.6 4.6-5.4
4А зо 6000-30 000 4.07 5.6 5.6-6.4
5 100 20 000-100 000 6.76 5.6 6.8-7.5
Мінімальний час витікання має становити 350 с у випадку розміру 1 і 200 с - у решті випадків.
ЛА — радіус меншого з циліндрів, у метрах;
кв — радіус більшого з циліндрів, у метрах;
а» — кутова швидкість, у радіанах на секунду.
Стала віскозиметра №> може бути визначена при
різних швидкостях обертання з використанням фар-
макопейних рідин для калібрування віскозиметрів.
При цьому в’язкість розраховується за формулою:
Методика. В’язкість вимірюють відповідно до
інструкції щодо застосування ротаційного віскозиме-
тра. Температуру, при якій вимірюють в’язкість, зазна-
чають в окремій статті. Для псевдопластичних та
інших неньютонівських систем в окремій статті зазна-
чають тип віскозиметра і кутову швидкість або
швидкість зсуву, при яких здійснюють виміри. Якщо
неможливо одержати точне значення зазначеної
швидкості, виміри проводять при дещо більшій або
дещо меншій швидкостях. Одержані результати інтер-
полюють.
___________________________________________N
Поряд з ротаційними віскозиметрами, принцип дії
яких описано у розділі 2.2.10, допускається викорис-
тання інших типів ротаційних віскозиметрів з коаксі-
альними циліндрами, в яких обертається лише один
циліндр.
2.2.11. ТЕМПЕРАТУРНІ МЕЖІ ПЕРЕГОНКИ
Температурні межі перегонки являють собою інтервал
температур, приведений для тиску 101.3 кПа
(760 мм рт. ст.), в межах якого переганяється рідина
або певна її фракція у передбачених умовах.
Прилад. Прилад (див. Рис. 2.2.11.-1) складається з пе-
регонної колби (А), прямого холодильника (В),
приєднаного до колби з бічної сторони, і вставної
трубки (алонжа) (С), приєднаної до кінця холодиль-
ника. У горловину колби поміщають термометр таким
чином, щоб кінець ртутного резервуара знаходився на
5 мм нижче від нижнього краю приєднання відвідної
трубки перегонної колби. Застосовують термометр з
діапазоном шкали близько 50 СС і ціною поділки
0.2 °С. Під час випробування колбу, включаючи і гор-
ловину, захищають від охолодження підхожим екра-
ном.
Методика. 50.0 мл випробовуваної рідини і декілька
шматочків пористого матеріалу поміщають у кол-
бу (А). Для рідин, що киплять при температурі нижче
150 °С, необхідне охолодження циркулюючою во-
дою. Колбу нагрівають таким чином, щоб швидко
досягти кипіння, і відмічають температуру, при якій у
циліндр потрапляють перші краплі відгону. Установ-
люють нагрівання, що забезпечує перегонку від
2 мл до 3 мл за хвилину і відмічають температуру, при
якій уся рідина або передбачена фракція, об’єм якої
вимірюють при температурі 20 °С, відігнані. Відгін
збирають у циліндр місткістю 50 мл з ціною поділки
1 мл.
Вносять поправку в спостережувані температури для
приведення до нормального тиску за формулою:
І} = 12 + Аг(101.3 - Ь),
де:
Г| — виправлена температура, у градусах Цельсія;
/2 — спостережувана температура при тиску Ь, у
градусах Цельсія;
к — поправочний коефіцієнт, у випадку необ-
хідності, зТабл. 2.2.11.-1;
Ь — барометричний тиск під час перегонки, у кіло-
паскалях.
О’ До промислових віскозиметрів додається таблиця із значеннями сталих віскозиметрів залежно від площі поверхні циліндрів і
швидкості їхнього обертання
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
25
2.2. Фізичн;
НЗИКО-Х1М1ЧН1 методи
Кінемату
квадрат Е*
форм І чГ
Таблиця 2.2.11.-1
правки для приведення
ального тиску
Коефіцієнт поправки к
0.30
е-яГио °С 0.34
вд140 ’Сдо 190 ’С 0 38
від 190 ’ С до 240 ’ С 041
вище 240 ’С 0.45
де:
Рисунок 2.2.11.-1. Прилад для визначення темпера-
турних меж перегонки
Розміри зазначені у міліметрах
2.2.13. ВИЗНАЧЕННЯ ВОДИ МЕТОДОМ
ВІДГОНУ
Прилад (Рис. 2.2.13.-1) складається зі скляної кол-
би (А), з’єднаної трубкою (і)) з циліндричною труб-
кою (В), спорядженою градуйованим приймачем (Е) і
зворотним холодильником (С). Ціна поділки прийма-
ча (Е) 0.1 мл. Як джерело нагрівання переважно вико-
ристовують електричний нагрівач з реостататом або
масляну баню. Верхня частина колби і сполучна труб-
ка можуть бути покриті теплоізоляцією.
Методика. Приймач і холодильник приладу очища-
ють, ретельно промивають водою і висушують.
200 мл толуолу Р і близько 2 мл води Р поміщають у су-
ху колбу і відганяють протягом 2 год. Колбу залишають
для охолодження протягом ЗО хв і записують об’єм во-
ди з точністю до 0.05 мл. У колбу поміщають кількість
речовини, зважену з точністю до 1 %, шо містить при-
близно від 2 мл до 3 мл води. Якщо речовина має пас-
топодібну консистенцію, її зважують у човнику з мета-
левої фольги. До колби вносять декілька шматочків
пористого матеріалу й обережно нагрівають протягом
15 хв. Коли толуол починає кипіти, відганяють зі
швидкістю близько двох крапель за секунду, доки
більша частина води не відгониться, а потім підвищу-
ють швидкість відгону до близько чотирьох крапель за
секунду. Коли вода відгониться повністю, внутрішню
трубку холодильника промивають толуолом Р. Нагрі-
—
вання продовжують ще 5 хв, потім нагрівач прибира-
ють, дають приймачу охолонути до кімнатної темпера-
тури і струшують усі краплі води, шо прилипли до
стінок приймача. Після повного розділення води і то-
луолу записують об’єм води і обчислюють її відсотко-
вий вміст у речовині за формулою:
1ОО(/72 /;і)
т
т — маса випробовуваної речовини, у грамах;
П\ — об’єм води, одержаної при першому відгоні, у
мілілітрах;
п2 — загальний об’єм відігнаної води, одержаної у
двох відгонах, у мілілітрах.
Рисунок 2.2.13.-1. Прилад для визначення води
методом відгону
Розміри зазначені у міліметрах
26
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
_____________________________________________N
Кип'ятіння припиняють, коли об’єм води у приймачі
перестане збільшуватися і верхній шар розчинника у
приймачі стане прозорим.
2.2.14. ТЕМПЕРАТУРА ПЛАВЛЕННЯ -
КАПІЛЯРНИЙ МЕТОД
Температура плавлення, визначена капілярним мето-
дом, являє собою температуру, за якої остання тверда
часточка згущеного стовпчика речовини в капілярній
трубці переходить у рідку фазу.
Якщо немає інших зазначень в окремій статті, той са-
мий прилад і методику застосовують для визначення
інших показників, таких як утворення меніска або
діапазону плавлення, що характеризують поведінку
речовини при плавленні.
Прилад. Складовими частинами приладу є:
— підхожа скляна посудина, що містить рідину (на-
приклад, воду, вазелінове або силіконове масло),
яка використовується як баня і оснащена підхо-
жим пристроєм для нагрівання;
— підхожий пристрій для перемішування, який за-
безпечує однорідність температури всередині бані;
- підхожий термометр з позначкою занурення і
ціною поділки не більше 0.5 °С. Різниця між верх-
ньою і нижньою поділками термометра у ділянці
вимірюваної температури — не більше 100 °С;
— запаяні з одного кінця капілярні трубки з безлуж-
ного міцного скла діаметром від 0.9 мм до 1.1 мм і
товщиною стінок від 0.10 мм до 0.15 мм.
Методика. Якщо немає інших зазначень в окремій
статті, тонко роздрібнену на порошок речовину су-
шать у вакуумі (2.2.32, спосіб Ь) над силікагелем безвод-
ним Р протягом 24 год. Достатню кількість речовини
поміщають у капілярну трубку до одержання згущено-
го стовпчика заввишки від 4 мм до 6 мм. Підвищують
температуру бані до температури приблизно на 10 °С
нижчої за передбачувану температуру плавлення і
потім продовжують нагрівання із швидкістю близько
1 °С за хвилину. Коли температура досягне значення
на 5 °С нижчого від передбачуваної температури плав-
лення, капілярну трубку поміщають у прилад. При ви-
користанні приладу, описаного вище, капілярну труб-
ку занурюють у баню так, щоб її запаяний кінець зна-
ходився на рівні центра кульки термометра, позначка
занурення якого знаходиться на рівні поверхні рідини.
Відмічають температуру, за якої остання тверда час-
точка перейде у рідку фазу.
Калібрування приладу. Для калібрування приладу ви-
користовують стандартні речовини Всесвітньої ор-
ганізації охорони здоров’я або інші відповідні речови-
ни, підхожі для таких цілей.
_____________________________________________N
Капілярний метод застосовують для визначення тем-
ператури плавлення твердих речовин, легко перетво-
рюваних на порошок.
Необхідне ущільнення речовини при заповненні
капілярної трубки можна одержати, якщо її кілька
разів кинути запаяним кінцем униз у скляну трубку за-
вдовжки не менше 1.0 м, поставлену вертикально на
тверду поверхню.
Проводять не менше двох визначень. За температуру
плавлення беруть середнє значення. Розбіжність між
визначеннями не має перевищувати 1 °С.
Допускається застосування інших приладів, які вико-
ристовують капілярний метод, якщо показано, що
точність і правильність вимірів будуть не гіршими, ніж
у випадку застосування приладу, описаного више.
Увесь процес плавлення проходить протягом певного
проміжку часу і певного інтервалу температур: почат-
ком плавлення — появою першої краплі рідини і
кінцем плавлення (температурою плавлення) — по-
вним переходом речовини у рідкий стан. Цей інтервал
температур, який називається діапазоном плавлення,
не має перевищувати 2 °С, якщо немає інших зазна-
чень в окремій статті.
Цілий ряд органічних сполук при плавленні розкла-
дається (відбувається різка зміна зовнішнього вигляду
речовини, наприклад, спінення). Таку температуру
називають температурою розкладання. Вона значною
мірою залежить від швидкості нагрівання, тому при
визначенні температури розкладання в окремих стат-
тях зазначають швидкість нагрівання.
Поряд із визначенням температури плавлення, якщо
немає інших зазначень в окремій статті, прилад і мето-
дику, описані в цьому розділі, застосовують для визна-
чення діапазону плавлення або температури розкла-
дання.
2.2.15. ТЕМПЕРАТУРА ПЛАВЛЕННЯ -
ВІДКРИТИЙ КАПІЛЯРНИЙ МЕТОД
Для деяких речовин визначають температуру розрід-
ження, яку звичайно називають температурою плав-
лення. Визначення проводять таким методом.
Використовують скляну капілярну трубку, відкриту з
обох кінців, завдовжки близько 80 мм, зовнішнім
діаметром від 1.4 мм до 1.5 мм і внутрішнім діаметром
від 1.0 мм до 1.2 мм.
Речовину, попередньо оброблену, як зазначено в ок-
ремій статті, поміщають у кожну з п’яти капілярних
трубок у кількості достатній для формування в кожній
трубці стовпчика заввишки близько 10 мм. Трубки за-
лишають на певний час при температурі, зазначеній в
окремій статті.
Прикріплюють одну з капілярних трубок до термоме-
тра з ціною поділки 0.2 °С таким чином, щоб речови-
на знаходилась у безпосередній близькості до кульки
термометра.
Термометр з прикріпленою капілярною трубкою
поміщають у склянку так, щоб відстань між дном
склянки і нижньою частиною кульки термометра
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
27
2.2. Фізичні та фізико-хімічні методи
становила 1 см. Склянку наповнюють водою так, щоб
висота шару становила 5 см. Підвищують температуру
із швидкістю 1 °С за хвилину.
о
За температуру плавлення беруть температуру, за якої
речовина починає підніматись капілярною трубкою.
Повторюють цю операцію з чотирма іншими капіляр-
ними трубками і розраховують результат як середнє з
п’яти показань.
Відкритий капілярний метод застосовують для речо-
вин, які мають аморфну структуру, не розтираються на
порошок і плавляться нижче температури кипіння во-
ди, таких як жири, віск, парафін, вазелін, смоли. У тих
випадках, коли стовпчик речовини не піднімається в
капілярі, за температуру плавлення беруть температуру,
за якої стовпчик речовини в капілярі стає прозорим.
2.2.16. ТЕМПЕРАТУРА ПЛАВЛЕННЯ -
МЕТОД МИТТЄВОГО ПЛАВЛЕННЯ
Температуру плавлення за цим методом обчислюють
за формулою:
6 + {2
температуру Г|, за якої речовина плавиться миттєво
при стиканні з металом. Зупиняють нагрівання. Під
час охолодження через рівні проміжки часу кидають
кілька часточок речовини на поверхню блока, очища-
ючи її після кожного випробування. Записують темпе-
ратуру Г2, за якої речовина миттєво припиняє плави-
тись при стиканні з металом.
Калібрування приладу. Для калібрування приладу ви-
користовують стандартні речовини Всесвітньої ор-
ганізації охорони здоров’я або інші відповідні речови-
ни, підхожі для таких цілей.
_____________________________________________л
Метод миттєвого плавлення застосовують для твердих
речовин, які легко перетворюються на порошок.
2.2.17. ТЕМПЕРАТУРА КРАПЛЕПАДІННЯ
Температура краплепадіння являє собою температуру, за
якої в умовах, наведених нижче, перша крапля розплав-
леної випробовуваної речовини падає з чашечки.
Прилад. Прилад (Рис. 2.2.17.-1) складається з двох ме-
талевих гільз (А) і (В), з’єднаних одна з одною за допо-
могою нарізки. Гільза (А) прикріплена до ртутного
термометра.
ґ, — перша температура;
Г2 — друга температура, які визначаються в умовах,
наведених нижче.
Прилад. Прилад складається з металевого блока, виго-
товленого з матеріалу, який має високу тепло-
провідність і не взаємодіє з випробовуваною речови-
ною, наприклад, з латуні. Верхня поверхня блока має
бути плоскою і ретельно відполірованою. Блок
рівномірно нагрівають по всій масі газовим пальни-
ком з мікрорегулюванням або електричним нагріва-
чем з тонким регулюванням. Блок має достатньо ши-
року циліндричну порожнину для розміщення термо-
метра, стовпчик ртуті якого має знаходитись в одному
і тому ж положенні як при калібруванні, так і при
визначенні температури плавлення випробовуваної
речовини. Циліндрична порожнина розміщена пара-
лельно до відполірованої верхньої поверхні блока і на
відстані близько 3 мм від неї. Прилад калібрують, ви-
користовуючи підхожі речовини з відомою температу-
рою плавлення.
Методика. Блок швидко нагрівають до температури на
10 °С нижчої за передбачувану температуру плавлення
і потім установлюють швидкість нагрівання близько 1 °С
за хвилину. Декілька часток тонко роздрібненої на по-
рошок речовини, висушеної у вакуумі (2.2.32, спосібЬ)
над силікагелем безводним Р протягом 24 год, кидають
через рівні проміжки часу на поверхню блока в безпо-
середній близькості до кульки термометра, очишаючи
поверхню після кожного випробування. Записують
Рисунок 2.2.17,-1. Прилад для визначення
температури краплепадіння
Розміри зазначені у міліметрах
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
У нижній частині гільзи (В) за допомогою двох
уші.іьнювачів (Е) вільно закріплена металева чашечка
(Е). Точне положення чашечки визначається фіксато-
рами (В) завдовжки у 2 мм. які використовуються та-
кож для центрування термометра. Отвір (С) у стінці
гільзи (В) призначений для вирівнювання тиску.
Відвідна поверхня чашечки має бути плоскою, а краї
вихідного отвору — під прямим кутом до поверхні.
Нижня частина ртутного термометра має форму і
розмір, як показано на рисунку; термометр градуйова-
ний від 0 °С до 110 °С і відстань на шкалі в 1 мм відпо-
відає різниці температур в 1 °С. Ртутна кулька термо-
метра має діаметр (3.5±0.2) мм і висоту (6.0±0.3) мм.
Прилад встановлюють по осі пробірки завдовжки
близько 200 мм із зовнішнім діаметром близько 40 мм.
Прилад прикріплюють до пробірки за допомогою
пробки, в яку вставлений термометр і яка має бічний
проріз Отвір чашечки має знаходитися на відстані
близько 15 мм від дна пробірки. Увесь пристрій зану-
рюють у склянку місткістю близько 1 л, заповнену во-
дою. Дно пробірки має знаходитись на відстані близь-
ко 25 мм від дна склянки. Рівень води має досягати
верхньої частини гільзи (А). Для рівномірного роз-
поділу температури у склянці використовують мішалку.
Методика. Заповнюють чашечку по вінця нерозплав-
леною випробовуваною речовиною, якщо немає
інших зазначень в окремій статті. Надлишок речовини
видаляють з обох боків шпателем. Після того як гільзи
(А) і (В) з’єднані, проштовхують чашечку всередину на
її місце в гільзі (В) до упору. Видаляють шпателем ре-
човину, видавлену термометром. Прилад помішають у
водяну баню, як описано вище. Водяну баню нагріва-
ють до температури приблизно на 10 СС нижчої перед-
бачуваної температури краплепадіння і установлюють
швидкість нагрівання близько 1 °С за хвилину.
Відмічають температуру падіння першої краплі. Про-
водять не менше трьох визначень, щоразу з новим
зразком речовини. Різниця між показаннями не має
перевищувати З °С. Середнє з одержаних значень яв-
ляє собою температуру краплепадіння.
___________________________________________N
Визначення температури краплепадіння проводять
для речовин, які не розтираються на порошок і плав-
ляться нижче температури кипіння води, таких як жи-
ри, віск, парафін, вазелін, смоли.
2.2.18. ТЕМПЕРАТУРА ТВЕРДНЕННЯ
Температура тверднення являє собою максимальну
температуру, при якій відбувається тверднення пере-
охолодженої рідини.
Прилад. Прилад (Рис. 2.2.18.-1) складається з пробірки
для проведення випробування діаметром близько 25 мм
і іавдовжки близько 150 мм, поміщеної всередині
іншої пробірки діаметром близько 40 мм і завдовжки
близько 160 мм. Внутрішня пробірка закрита проб-
кою, спорядженою термометром завдовжки близько
175 мм з ціною поділки 0.2 °С, який закріплений та-
ким чином, щоб ртутна кулька знаходилася на відстані
близько 15 мм від дна пробірки. У пробці є отвір, крізь
який проходить вал мішалки, виготовлений із скляно-
го стрижня або іншого підхожого матеріалу, зігнутий
на кінці під прямим кутом у вигляді петлі, зовнішній
діаметр якої близько 18 мм. Внутрішню пробірку ра-
зом з зовнішньою пробіркою розташовують у центрі
склянки місткістю 1 л, що містить підхожу охолодну
рідину, рівень якої знаходиться у межах 20 мм від верх-
нього краю склянки. Охолодна баня також має бути
споряджена термометром.
Рисунок 2.2.18.-1. Прилад для визначення
температури тверднення
Розміри зазначені у міліметрах
Методика. У внутрішню пробірку помішають достат-
ню кількість рідини або попередньо розплавленої ви-
пробовуваної речовини, щоб покрити ртутну кульку
термометра, і при швидкому охолодженні визначають
приблизну температуру тверднення. Внутрішню
пробірку помішають у водяну баню з температурою
на 5 °С вищю за приблизно визначену температуру
тверднення до повного розплавлення кристалів.
Потім заповнюють склянку водою або насиченим
розчином натрію хлориду з температурою на 5 °С
нижче очікуваної температури тверднення. Внутріш-
ню пробірку разом із зовнішньою поміщають у
склянку, ретельно перемішують вміст до початку по-
яви кристалів і витримують до повного тверднення.
Позначають найбільш високу температуру, спостере-
жувану під час тверднення.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
29
2.2. Фізичні та фізико-хімічні методи
_____________________________________________N
Методика. Термометр закріплюють таким чином, щоб
ртутна кулька знаходилася посередині шару випробо-
вуваної речовини.
Температуру позначають кожні ЗО с. Спочатку відбу-
вається поступове зниження температури, потім, при
появі твердої фази, вона залишається деякий час
постійною або підвищується перед тим, як стати
постійною, а потім знову знижується. Позначають
найбільш високу температуру, що залишається корот-
кий час постійною з початку тверднення речовини.
Якщо речовина залишається рідкою при очікуваній
температурі тверднення, то тверднення викликають
потиранням об стінки внутрішньої пробірки термоме-
тром або внесенням невеликої кількості раніше одер-
жаної застиглої речовини.
2.2.19. АМПЕРОМЕТРИЧНЕ ТИТРУВАННЯ
При амперометричному титруванні кінцеву точку ви-
значають за зміною струму між зануреними у
випробовуваний розчин електродами (один з них, що
поляризується, індикаторний, а другий, що не поля-
ризується. електрод порівняння або два індикаторних
електроди, що поляризуються) як функції від кіль-
кості доданого титранту при постійній і контрольо-
ваній різниці потенціалів. Потенціал індикаторного
електрода має забезпечити граничний дифузійний
струм для електрохімічно активної сполуки.
Прилад. Прилад складається з джерела постійного
струму з регульованою напругою і чутливого мікроам-
перметра. Система, щодетектує. звичайно складається
з індикаторного електрода (наприклад, платинового,
ртутного крапельного, дискового, що обертається, або
графітового електрода) і електрода порівняння (на-
приклад, каломельного або хлорсрібного електрода).
Іноді використовують триелектродну систему, що
складається з індикаторного електрода, електрода
порівняння і поляризованого допоміжного електрода.
Методика. Електроди поміщають у випробовуваний
розчин, установлюють постійний потенціал, зазначе-
ний в окремій статті, і додають титрант порціями. За
значеннями сили початкового струму і одержаними у
процесі титрування будують графік залежності сили
струму від кількості титранту, шо додається. Титрант
додають послідовно, не менше як трьома порціями,
що складають у сумі 80 % від теоретичного об’єму,
відповідного передбачуваній точні еквівалентності.
Три одержаних значення сили струму мають укладати-
ся на пряму. Продовжують послідовно додавати тит-
рант після передбачуваної точки еквівалентності не
менше трьох разів. Одержані значення мають уклада-
тися на пряму. Точка перетину цих двох прямих являє
кінцеву точку титрування.
При амперометричному титруванні з двома індика-
торними електродами реєструють усю криву титру-
вання і визначають кінцеву точку.
_____________________________________________Лі
При амперометричному титруванні з двома індикатор-
ними електродами (без електрода порівняння) обидва
електроди виконані з одного і того самого матеріалу і
мають однакову відносно невелику поверхню. У цьому
випадку реєструють усю криву титрування і визначають
кінцеву точку за мінімальним значенням сили струму.
Найбільша точність амперометричного титрування до-
сягається при потенціалі на індикаторному електроді,
відповідному граничному дифузійному струму.
При амперометричному титруванні, як правило, кон-
центрація титранту в 10-20 разів перевищує концент-
рацію визначуваної речовини.
У фармакопейному аналізі амперометричне титруван-
ня доцільно застосовувати у нітритометрії, при визна-
ченні води напівмікрометодом (за К.Фішером) та :
йодометрії.
У окремих статтях треба зазначати параметри, необхідні
для коректного відтворення методики, наприклад: типи
електродів; потенціал, що задається; масу наважки пре-
парату; концентрацію титранту; температуру і т.д.
2.2.20. ПОТЕНЦІОМЕТРИЧНЕ ТИТРУВАННЯ
При потенціометричному титруванні кінцеву точку
титрування знаходять, вимірюючи електрорушійну
силу (е.р.с.) електродної пари, що складається з інди-
каторного електрода і електрода порівняння або двох
індикаторних електродів, занурених у випробовува-
ний розчин як функцію кількості доданого титранту
Е.р.с. звичайно вимірюють при нульовому або прак-
тично нульовому струмі.
Прилад. Використовуваний прилад (простий потен-
ціометр або електронний пристрій) включає вольт
метр з розрізненням близько мілівольта.
Вибір індикаторного електрода залежить від природи
визначуваної речовини. Цей електрод може бути скля-
ним або металевим (наприклад, платиновим, золотим,
срібним або ртутним). Електродом порівняння зви-
чайно є каломельний або хлорсрібний електрод.
Для кислотно-основного титрування, якщо немає ін-
ших зазначень в окремій статті, використовують сис-
тему скляного і каломельного або скляного і хлорсріб-
ного електродів.
Методика. Будують графік залежності зміни е.р.с. від
кількості доданого титранту, продовжуючи додавати
титрант понад передбачувану точку еквівалентності.
Кінцева точка відповідає різкій зміні е.р.с.
____________________________________________ТУ
Потенціометричне титрування звичайно дає більш
точні результати за індикаторне, особливо при аналізі
каламутних і забарвлених розчинів, дозволяє автома-
тизувати процес титрування.
Як правило, електродну пару занурюють у випробову-
ваний розчин, крім випадків, коли іони з електрода
ЗО
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
порівняння заважають титруванню. У цьому випадку
електрод порівняння сполучають з випробовуваним
розчином електролітичним мостом.
Прилад. Вимірювання е р.с. проводять за допомогою
внсокоомних потенціометрів (рН-метрів, іонометрів).
Потенціометричне титрування застосовують для ана-
лізу. іаснованого на таких типах реакцій: нейт-
ралізації, осадження, комплексоутворення, окиснен-
ня-відновлення. Вибір електродів залежить від типу
аналітичної реакції. Індикаторний електрол вибира-
ють так, шоб його потенціал закономірно змінювався
при перебігу хімічної реакції між титрованими іонами
та іонами титранту (Табл.2.2.20.-1).
Методика. Об’єм титранту в точці еквівалентності Уекв
може бути визначений такими способами:
1) за графіком кривої титрування у координа тах [ У;Е\,
застосовуючи метод дотичних;
2) за графіком [ V ; д^ 1 ,
де:
ДЕ — зміна е.р.с.;
ЛІ — відповідний приріст об’єму титранту.
При цьому точці еквівалентності відповідає макси-
мальне значення ;
ДЕ
3) розрахунковим шляхом за максимальним значен-
ням Д£ і відповідно Л( ЕЕ \ як зазначено у
ДГ 1 ДГ '
Табл.2.2.20.-2 і формулі розрахунку.
Еквівалентний об’єм титранту Уекв обчислюють за фор-
мулою:
де
И/ — об’єм титранту, відповідний останньому пози-
тивному (негативному) значенню величини Ау,
У2 — об’єм титранту, відповідний першому негатив-
ному (позитивному) значенню величини Ау.
Ау = Д(-~р-) — прирости величин д^ .
При проходженні через точку еквівалентності Ау
змінює знак на протилежний.
Таблиця 2.2.20.-1
Електродні системи для потенціометричного титрування
Тип аналітичної реакції Індикаторні електроди Електроди порівняння Застосування
Кислотно-основний Скляний Хлорсрібний, кало- мельний Титрування кислот, основ і солей
Осадження Срібний, сульфідсрібний Хлорсрібний, кало- мельний, скляний Титрування галогенідів, родані- дів, ціанідів, сульфідів
Комплексономсіричний Ртутний, іонселек- тивний Хлорсрібний, кало- мельний, скляний Титрування катіонів, що утво- рюють міцні комплексонати
Окисно- відновний Платиновий Хлорсрібний, кало- мельний, скляний Титрування відновників різними окисниками, наприклад, бро- матом, біхроматом, пермангана- том, йодом і церієм (IV). Титрування окисників різними відновниками, наприклад, арсе- нітом, тіосульфатом і нітритом.
Таблиця 2.2.20.-2
ґь мл ДЕ Е. піВ ДЕ ДЕ ДЕ
5.00 250
0.1 13 130
5.10 263 + 150
0.1 28 280
5.20 291 +720
0.1 100 1000
5.30 391 -450
0.1 55 550
5.40 446 -330
0.1 22 220
5.50 468 -120
0.1 10 100
5.60 478
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
31
2.2. Фізичні та фізико-хімічні методи
Приклад:
Уекв = 5.20 + (5.30 - 5.20) 720 7_2°(_450) = 5.26 мл
Потенціометричне титрування може бути автоматизо-
ване шляхом застосування приладів двох типів: таких,
що використовують математичний аналіз кривої тит-
рування, або таких, що припиняють додавання тит-
ранту, коли досягається е.р.с. електродної пари,
відповідна точці еквівалентності.
2.2.22. АТОМНО-ЕМІСІЙНА СПЕКТРОМЕТРІЯ
Атомно-емісійна спектрометрія — це метод визначен-
ня вмісту хімічного елемента в зразку, що випробо-
вується, шляхом вимірювання інтенсивності однієї з
емісійних ліній атомної пари елемента. Визначення
проводять за довжини хвилі, яка відповідає вибраній
емісійній лінії.
Прилад. Головними складовими частинами приладу є:
генератор атомної пари елемента, який визначається
(полум’я, плазма, дуга і т.п.), монохроматор і детектор.
Якщо генератором атомної пари є полум’я, як розчин-
нику для приготування випробовуваного і розчинів
порівняння краще віддати перевагу воді Р. Як розчин-
ники можуть також використовуватись органічні роз-
чинники, якщо вони не впливають на стабільність по-
лум’я.
Методика. Атомно-емісійний спектрометр виводять
на режим відповідно до інструкції підприємства-виго-
товлювача і встановлюють потрібну довжину хвилі. До
генератора атомної пари вводять холостий розчин і
настроюють реєструючий пристрій на нульове зна-
чення. Вводять розчин порівняння елемента, що виз-
начається, з найбільшою концентрацією і настроюють
прилад так, щоб одержати сигнал, що реєструється, в
оптимальному діапазоні вимірювань.
Визначення проводять шляхом порівняння інтенсив-
ностей емісії випробовуваного розчину і розчинів
порівняння з відомими концентраціями елемента, що
визначається методом калібрувальної кривої (метод 1),
або методом стандартних добавок (метод 2).
МЕТОД 1 - МЕТОД КАЛІБРУВАЛЬНОЇ КРИВОЇ
Випробовуваний розчин готують, як зазначено в ок-
ремій статті. Не менше трьох розчинів порівняння
елемента, що визначається, готують так, щоб діапазон
концентрацій цих розчинів включав очікуване зна-
чення концентрації елемента, що визначається, у ви-
пробовуваному розчині. Будь-які реагенти, які вико-
ристовуються у приготуванні випробовуваного розчи-
ну, додають у холостий і розчини порівняння у таких
же кількостях, як і випробовуваний розчин.
Випробовуваний розчин і кожний розчин порівняння
вводять до генератора не менше трьох разів і щоразу
реєструють усталене показання. Після введення ви-
пробовуваного і розчинів порівняння щоразу проми- і
вають прилад холостим розчином і перевіряють, щоб І
показання реєструючого пристрою поверталися до
початкового значення.
Будують калібрувальну криву залежності середніх зна-
чень емісії розчинів порівняння від концентрації і І
визначають концентрацію елемента у випробовувано-
му розчині за побудованою калібрувальною кривою.
МЕТОД 2 - МЕТОД СТАНДАРТНИХ ДОБАВОК
Випробовуваний розчин готують, як зазначено в ок-
ремій статті. Рівні об’єми випробовуваного розчину
поміщають не менш як у три мірні колби однакового
об’єму. У всі колби, крім однієї, додають об’єми, що
пропорційно збільшуються, еталонного розчину еле-
мента, який визначається, і розчинником доводять
вміст кожної колби до позначки. При цьому значення
емісії розчинів із стандартними добавками (розчинів
порівняння) має знаходитися у лінійній ділянці
калібрувальної кривої.
Випробовуваний розчин і кожний розчин із стандарт-
ною добавкою вводять до генератора не менше трьох
разів і щоразу реєструють усталене показання. Після
введення випробовуваного розчину і розчинів з добав-
ками щоразу промивають прилад холостим розчином і
перевіряють, щоб показання реєструючого пристрою
поверталися до нульового значення.
Обчислюють параметри лінійного рівняння прямої
залежності середніх значень емісії розчинів від кон-
центрації методом найменших квадратів і обрахову-
ють концентрацію елемента, що визначається, у ви-
пробовуваному розчині.
Допускається визначення концентрації з використан-
ням графічного методу. Задля цього по осі ординат
відкладають середні значення емісії випробовуваного
розчину і розчинів із стандартними добавками, а по
осі абсцис — концентрації стандартних добавок еле-
мента, що визначається. Екстраполюють лінію, що
проходить через одержані точки, до перетину з віссю
абсцис. Концентрація елемента, що визначається, у
випробовуваному розчині дорівнює відстані між одер-
жаною точкою і початком координат.
У випадку використання техніки введення твердих
проб умови проведення визначень мают ь зазначатися
в окремій статті.
2.2.23. АТОМНО-АБСОРБЦІЙНА
СПЕКТРОМЕТРІЯ
Атомно-абсорбційна спектрометрія — це метод визна-
чення вмісту хімічного елемента у випробовуваному
зразку за допомогою вимірювання абсорбції ви-
промінювання атомною парою визначуваного еле-
мента. Визначення проводять за довжини хвилі,
відповідній вибраній абсорбційній лінії.
Прилад. Головними складовими частинами приладу є:
джерело випромінювання, генератор атомної пари
32
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
елемента, що визначається (полум’я, піч та ін.), моно-
хроматор і детектор.
Спосіб введення зразка залежить від типу використо-
вуваного генератора. Якщо генератором атомної пари
є полум’я, як розчинник для приготування випробо-
вуваного і розчинів порівняння краще використовува-
ти воду Р. Як розчинник можуть також використову-
ватися органічні розчинники, якщо вони не вплива-
ють на стабільність полум’я. При використовуванні
печі зразок може бути введений до генератора у ви-
гляді розчину в воді Рабо в органічному розчиннику,
також може бути застосована техніка введення твер-
дих проб.
Атомну пару можна одержати також поза спектромет-
ром, наприклад, методом холодної пари для визначен-
ня ртуті або гідридним методом. При визначенні ртуті
атомна пара, одержана хімічним відновленням, вно-
ситься потоком інертного газу в абсорбційну кювету,
розташовану на оптичному шляху приладу. У випадку
використання гідридного методу одержують гідрид
елемента, який визначається, він або змішується з га-
зом, що живить пальник, або вноситься інертним га-
зом у нагріту абсорбційну кювету, де відбувається ди-
соціація гідриду до атомів.
Методика. Атомно-абсорбційний спектрометр виво-
дять на режим відповідно до інструкції підприємства-
виготовлювача й установлюють потрібну довжину
хвилі. У генератор атомної пари вводять холостий роз-
чин і настроюють реєструючий пристрій на максималь-
не світлопропускання. Вводять розчин порівняння ви-
значуваного елемента з найбільшою концентрацією і
настроюють прилад так, щоб одержати реєструючий
сигнал в оптимальному діапазоні вимірювань.
Визначення здійснюють шляхом порівняння величи-
ни поглинання випробовуваного розчину і розчинів
порівняння з відомими концентраціями елемента, що
визначається, або методом калібрувальної кривої (ме-
тод 1), або методом стандартних добавок (метод 2).
МЕТОД 1 - МЕТОД КАЛІБРУВАЛЬНОЇ КРИВОЇ
Випробовуваний розчин готують, як зазначено в ок-
ремій статті. Не менше трьох розчинів порівняння
елемента, що визначається, готують так, щоб діапазон
концентрацій цих розчинів включав очікуване зна-
чення концентрації визначуваного елемента у випро-
бовуваному розчині. Будь-які реагенти, що викорис-
товуються у приготуванні випробовуваного розчину,
додають до холостого і розчинів порівняння у таких же
кількостях, як і у випробовуваний розчин.
Випробовуваний розчин і кожний розчин порівняння
вводять до генератора не менше трьох разів і щоразу
реєструють усталене показання. Після введення ви-
пробовуваного і розчинів порівняння щоразу проми-
вають прилад холостим розчином і перевіряють, щоб
показання реєструючого пристрою поверталися до
початкового значення.
У випадку використання печі як генератора атомної
пари між вимірами її відпалюють.
Будують калібрувальну криву залежності середніх зна-
чень поглинання розчинів порівняння від концент-
рації і визначають концентрацію елемента за значен-
ням поглинання випробовуваного розчину за побудо-
ваною калібрувальною кривою.
У випадку використання техніки введення твердих
проб умови проведення визначень мають бути зазна-
чені в окремій статті.
МЕТОД 2 - МЕТОД СТАНДАРТНИХ ДОБАВОК
Випробовуваний розчин готують, як зазначено в ок-
ремій статті. Рівні об’єми випробовуваного розчину
поміщають не менш як у три мірні колби однакового
об’єму. У всі колби, крім однієї, додають об’єми, що
пропорційно збільшуються, еталонного розчину еле-
мента, який визначається, і розчинником доводять
вміст кожної колби до позначки. При цьому значення
поглинання розчинів зі стандартними добавками
(розчинів порівняння) має знаходитися у лінійній
ділянці калібрувальної кривої.
Випробовуваний розчин і кожний розчин зі стандарт-
ною добавкою вводять до генератора не менше трьох
разів і щоразу реєструють усталене показання. Після
введення випробовуваного розчину і розчинів з добав-
ками щоразу промивають прилад холостим розчином і
перевіряють, щоб показання реєструючого пристрою
поверталися до початкового значення.
У випадку використання печі як генератора атомної
пари між вимірами її відпалюють.
Обчислюють параметри лінійного рівняння прямої
залежності середніх значень поглинання розчинів від
концентрації методом найменших квадратів і обрахо-
вують концентрацію елемента, що визначається, у ви-
пробовуваному розчині.
Допускається визначення концентрації з використан-
ням графічного методу. Задля цього по осі ординат
відкладають середні значення поглинання випробову-
ваного розчину і розчинів зі стандартними добавками,
а по осі абсцис — концентрації стандартних добавок
елемента, що визначається. Екстраполюють лінію, що
проходить через одержані точки, до перетину з віссю
абсцис. Концентрація елемента, шо визначається, у
випробовуваному розчині дорівнює відстані між одер-
жаною точкою і початком координат.
У випадку використання техніки введення твердих
проб умови проведення визначень мають бути зазна-
чені в окремій статті.
_____________________________________________N
Для визначення концентрації елемента в аналізовано-
му зразку поряд з методом 1 і методом 2 допускається
використання методу порівняння і методу обмежую-
чих розчинів або інших валідованих методів.
Загальні вимоги до проведення атомно-емісійного і атом-
но-абсорбційного аналізу. Для обчислення параметрів
лінійної робочої ділянки калібрувальних кривих реко-
мендується як у методі 1, так і в методі 2 використову-
вати метод найменших квадратів.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
33
2.2. Фізичні та фізико-хімічні методи
Послідовність введення випробовуваного і стандарт-
них розчинів до генератора має бути зазначена, при
необхідності, в окремій статті.
До методики атомно-емісійного (абсорбційного) ви-
значення рекомендується включити тест "Перевірка
придатності системи". Одним з параметрів даного тесту
є відносне стандартне відхилення значення емісійного
(абсорбційного) сигналу, який не перевищує за значен-
ням величини, зазначеної в окремій статті
У процесі проведення атомно-емісійних (абсорб-
ційних) визначень рекомендується підтверджувати
незмінність кута нахилу лінійної робочої області
калібрувальної кривої.
Реактиви й еталонні розчини. Вода має бути деіонізова-
ною на іонообмінних смолах і продистильованою без-
посередньо перед вживанням і відповідати за чисто-
тою воді Р.
Нижче наведені розчини солей, катіони яких позна-
чені назвами елементів, найбільш часто нормованих у
фармацевтичному аналізі.
Кальцій. 1.001 г кальцію карбонату Р, висушеного до
постійної маси при температурі 105 °С, розчиняють у
25 мл ЇМ розчину кислоти хлористоводневої і дово-
дять об’єм розчину водою Р до 1000 0 мл. Розчин
кальцію містить 400 мкг іонів Са в 1 мл.
Термін придатності розчину 1 міс, зберігання при
кімнатній температурі.
Калій. 1.1440 г калію хлориду Р, висушеного до постій-
ної маси при температурі 130 °С, розчиняють у неве-
ликій кількості води Р і доводять об’єм розчину во-
дою Р до 1000.0 мл. Розчин калію містить 600 мкг
іонів К в 1 мл.
Термін придатності розчину 2 міс, зберігання при
кімнатній температурі
Натрій. 0.5084 г натрію хлориду Р, висушеного до
постійної маси при температурі 130 °С, розчиняють у
невеликій кількості води Р і доводять об’єм розчину
водою Р до 1000.0 мл. Розчин натрію містить 200 мкг
іонів ІМа в 1 мл.
Термін придатності розчину 2 міс, зберігання при
кімнатній температурі.
Цинк. 2.5 г цинку Р розчиняють у 20 мл 6 Мрозчину кис-
лоти хлористоводневої і доводять об’єм розчину водою Р
до 500.0 мл. Розчин цинку містить 5 мг іонів 7п в 1 мл
Термін придатності розчину 2 міс, зберігання при
кімнатній температурі.
Свинець. 0.1600 г свинцю нітрату Р розчиняють у 5 мл
кислоти азотної Р і доводять об’єм розчину водою Рдо
1000.0 мл. Розчин свинцю містить 100 мкг іонів РЬ в
1 мл.
Термін придатності розчину 2 міс, зберігання при
кімнатній температурі.
Мідь. 1.000 г міді Р розчиняють у невеликому об’ємі
кислоти азотної Рі доводять розчином 10 г/л кислоти
азотної Р до 1000.0 мл Розчин міді містить 1 мг
іонів Си в 1 мл.
Термін придатності розчину 2 міс, зберігання при
кімнатній температурі.
Допускається використання інших реактивів і еталонних
розчинів дтя спектрального аналізу, атестованих компе-
тентним уповноваженим органом або визнаних ним.
Еталонні, а також приготовані на їхній основі розчини
порівняння зберігають у посуді, що дозволяє зберігати
концентрацію цих розчинів незмінною (наприклад, у
посуді з кварцу, тефлону і т.п.). Комірки і тиглі для
озолення проб мають бути виготовлені з кварцу.
2.2.24. АБСОРБЦІЙНА СПЕКТРОФОТОМЕТРІЯ
В ІНФРАЧЕРВОНІЙ ОБЛАСТІ
Інфрачервоні спектрофотометри застосовують для за-
пису спектрів в області від 4000 см-1 до 670 см’1 (від
2.5 мкм до 15 мкм), а у деяких випадках до 200 см-' (до
50 мкм).
У спектрофотометрах з Фур’є-перетворюванням ви-
користовується поліхроматичне випромінювання і
розраховується спектр у заданій області частот шля-
хом Фур’є-перетворювання вихідних даних. Також
можуть бути використані спектрофотометри, забезпе-
чені оптичною системою, здатною виділяти монохро-
матичне випромінювання у вимірюваній області. Зви-
чайно спектр подається як функція пропускання, тоб-
то відношення інтенсивності випромінювання, що
пройшло, до падаючого на зразок.
Оптичну густину (Л) визначають як десятковий лога-
рифм оберненої величини пропускання (Т):
А = Іо810(1/П = 1о8|0(/0//),
де:
Т=1/Іо,
10 — інтенсивність випромінювання, падаючого на
речовину;
1 — інтенсивність випромінювання, що пройшло
крізь речовину.
ПІДГОТУВАННЯ ЗРАЗКА
Для запису пропускання або поглинання субстанцію
готують за однією з таких методик.
Рідини. Рідини досліджують або у формі плівки між
двома пластинками, прозорими для інфрачервоного
випромінювання, або в кюветі з відповідною товщи-
ною шару, також прозорою для інфрачервоного ви-
промінювання.
Рідини або тверді речовини у розчині. Готують розчин
випробовуваної субстанції у підхожому розчиннику.
Вибирають концентрацію речовини і товщину шару
кювети, які дозволяють одержати задовільний спектр.
Звичайно добрі результати одержують при концент-
раціях від 10 г/л до 100 г/л за товщини шару від 0.5 мм
до 0.1 мм. Поглинання розчинника компенсують
34
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
шляхом поміщення у канал порівняння аналогічної
кювети, яка містить вибраний розчинник.
Тверді речовини. Тверді речовини досліджують диспер-
юваними у підхожій рідині у вигляді суспензії або у
твердому стані (диски з галогенідів лужних металів).
Якщо іазначено в окремій статті, формують плівку з
розплавленої маси між двома пластинами, прозорими
іля інфрачервоного випромінювання.
а) Суспензія. Невелику кількість субстанції, призначе-
ної для випробування, розтирають із мінімальною
кількістю вазелінового масла Р або іншої підхожої
рідини, звичайно від 5 мг до 10 мг субстанції достати ьо
для одержання придатної суспензії. Одержану сус-
пензію стискують між двома пластинками, прозорими
дія інфрачервоного випромінювання.
її} Лиски Від 1 мг до 2 мг субстанції, призначеної для
випробування, розтирають з 300-400 мг, якщо немає
інших зазначень, ретельно здрібненого калію бро-
міду Рабо калію хлориду Р. Звичайно цих кількостей
".остатньо для одержання диска діаметром 13 мм і спе-
ктра відповідної інтенсивності. Суміш ретельно пере-
тирають, домагаючись необхідної однорідності, і пре-
сують при тиску близько 800 М Па (8 т ем 2) у вакуумі.
Причиною утворення неякісних дисків можуть бути
такіфактори, як недостатнє або надмірне розтирання,
вологість або інші домішки у дисперсійному середо-
вищі іі недостатнє здрібнення часток.
Диск не придатний для випробування, якщо він при
візуальному огляді неоднорідний на прозорість або
якщо пропускання при 2000 см4 (5 мкм) становить
менше 75 % без компенсації при відсутності спе-
цифічної смуги поглинання речовини.
Гази. Гази досліджують у кюветі, прозорій для інфра-
червоного випромінювання з довжиною оптичного
шляху близько 100 мм. Кювету відкачують і заповню-
ють через кран або за допомогою голчатого клапана
через газову лінію між кюветою і контейнером з суб-
станцією, призначеною для випробування.
Якщо необхідно, доводять тиск у кюветі до атмосфер-
ного використовуючи газ, прозорий для інфрачерво-
ного випромінювання (наприклад, азот Р або ар-
гон Р). Заважаючий вплив поглинання води, вуглецю
діоксиду або інших атмосферних газів виключають
шляхом вміщення у канал порівняння ідентичної кю-
вети яка або вакуумована, або заповнена газом, про-
зорим для інфрачервоного випромінювання.
Запис багаторазового відбиття
Субстанцію готують за однією з таких методик.
а) Субстанцію розчиняють у підхожому розчиннику за
умов, описаних в окремій статті. Розчин випаровують
на пластинці талію бромід-йодиду або на іншій при-
датній пластинці.
в) Субстанцію поміщають на пластинку талію бромід-
йодиду або на іншу підхожу пластинку таким чином,
щоб одержати гомогенний контакт.
ІДЕНТИФІКАЦІЯ З ВИКОРИСТАННЯМ
СТАНДАРТНИХ ЗРАЗКІВ
Зразки випробовуваної субстанції і стандартної речо-
вини готують за однією і тією самою методикою і за-
писують спектри в області від 4000 см 1 до 670 см1 (від
2.5 мкм до 15 мкм) за одних і тих самих умов. Мініму-
ми пропускання (максимуми поглинання) у спектрах
випробовуваної субстанції мають відповідати за поло-
женням і відносною величиною таким у спектрі стан-
дартного зразка.
Якщо спектри, одержані у твердому стані, показують
відмінність у положенні мінімумів пропускання (мак-
симумів поглинання), то зразок випробовуваної суб-
станції і стандартний зразок обробляють одним і тим
самим засобом так, щоб вони кристалізувались або
виходили в одній і тій самій формі, або обробляють
способом, зазначеним в окремій статті, а потім зніма-
ють спектри.
ІДЕНТИФІКАЦІЯ З ВИКОРИСТАННЯМ
ЕТАЛОННИХ СПЕКТРІВ
Контроль розрізнювальної здатності. Записують спектр
плівки полістиролу завтовшки 0.04 мм. Різниця х (див.
Рис. 2.2.24,-1) між відсотком пропускання у максиму-
мі пропускання А при 2870 см1 (3.48 мкм) і мінімумі
пропускання В при 2851 см 1 (3.51 мкм) має бути
більше 18. Різниця у між відсотком пропускання у
максимумі пропускання С при 1589 см 1 (6.29 мкм) і
мінімумі пропускання О при 1583 см 1 (6.32 мкм) має
бути більше 12.
Рисунок 2.2.24.-1. Типовий спектр полістиролу,
використовуваного для перевірки
розрізнювальної здатності
Перевірка шкали хвильових чисел. Шкала хвильових
чисел може бути перевірена з використанням плівки
полістиролу, яка має мінімуми пропускання (макси-
муми поглинання) при хвильових числах (см1), наве-
дених у Табл. 2.2.24.-1.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
35
2.2. Фізичні та фізико-хімічні методи
Таблиця 2.2.24-1
Мінімуми пропускання (допустимі межі)
плівки полістироле
3060.0 (1 1.5) см-і
___________2849.5(1 1.5) см-і_________
___________1942.9 (1 1.5) см-і________
___________1601.2(11.0)см-і___________
___________1583.0(11.0) см-і__________
___________1154.5(11.0) см-і__________
1028.3(1 1.0) см-1
Методика. Субстанцію готують до випробування
згідно з інструкцією, доданою до еталонного спектра.
Використовуючи умови, за яких проводилася перевір-
ка розрізнювальної здатності, записують спектр ви-
пробовуваної субстанції і поверх нього смуги погли-
нання полістирольної плівки при 2849.5 см 1
(3.51 мкм), 1601.2 см-1 (6.25 мкм) і 1028.3 см 1
(9.72 мкм). Порівнюють два спектри (еталонний і
спектр випробовуваної субстанції) і смуги поглинання
плівки полістиролу, зазначені вище. Положення зна-
чущих смуг у спектрі випробовуваної субстанції і ета-
лонному спектрі мають відповідати в межах 0.5 % від
шкали хвильових чисел. Відносна величина смуг обох
спектрів має узгоджуватися між собою.
ДОМІШКИ У ГАЗАХ
Для аналізу домішок використовують кювету, прозору
для інфрачервоного випромінювання і таку, що має
відповідну довжину оптичного шляху (наприклад, від
1 м до 20 м). Кювету заповнюють так, як зазначено у
розділі "Гази". Для визначення і кількісної оцінки
домішок використовують методики, зазначені в окре-
мих статтях.
____________________________________________N
Спектрофотометрію в ІЧ-області спектру звичайно
використовують для ідентифікації, а також для кон-
тролю домішок у субстанціях.
Кожний інфрачервоний спектр характеризується
серією смуг поглинання, максимуми яких визнача-
ються хвильовим числом V або довжиною хвилі X і
інтенсивністю максимумів поглинання. Хвильове
число V, вимірюване в обернених сантиметрах (см1),
. . ІО4
визначається із співвідношення у =--- ,
А.
де:
А — довжина хвилі у мікрометрах (мкм).
ІДЕНТИФІКАЦІЯ З ВИКОРИСТАННЯМ
ЕТАЛОННИХ СПЕКТРІВ
Перевірка шкали хвильових чисел. Для перевірки шка-
ли хвильових чисел можуть використовуватися мето-
дики, наведені в інструкції до приладу.
2.2.25. АБСОРБЦІЙНА СПЕКТРОФОТОМЕТРІЯ
В УЛЬТРАФІОЛЕТОВІЙ І ВИДИМІЙ
ОБЛАСТЯХ
Визначення оптичної густини. Оптична гусі ина (А) роз-
чину являє собою десятковий логарифм оберненої ве-
личини пропускання (Т) для монохроматичного ви-
промінювання і виражається співвідношенням:
А = 1оЕ10(1/Г) = 1о§І0(/о//),
Т= І/Іо,
де:
Іо — інтенсивність падаючого монохроматичного
випромінювання;
/ — інтенсивність монохроматичного випроміню-
вання, яке пройшло.
За відсутністю інших фізико-хімічних факторів
виміряна оптична густина (А) пропорційна довжині
шляху (Ь), крізь який проходить випромінювання, і
концентрації (с) речовини у розчині відповідно з рів-
нянням:
А = е с Ь ,
де:
є — молярний показник поглинання;
Ь — довжина оптичного шляху, у сантиметрах;
с — концентрація речовини в розчині, у молях на
літр.
Величина Л{см являє собою питомий показник погли-
нання, тобто оптичну густину розчину речовини з
концентрацією 10 г/л у кюветі з товщиною шару 1 см,
тобто:
А1% = Ю £.
Ісм М.м
Якщо немає інших зазначень в окремій статті,
вимірювання оптичної густини проводять за зазначе-
ної довжини хвилі з використанням кювети зав-
довжки 1 см і при температурі (20± І) °С. Якщо немає
інших зазначень в окремій статті, вимірювання прово-
дять у порівнянні з тим самим розчинником або тією
самою сумішшю розчинників, у якій розчинено речо-
вину. Оптична густина розчинника, виміряна проти
повітря за зазначеної довжини хвилі, не має переви-
щувати 0.4 і бажано, щоб вона була менше за 0.2.
Спектр поглинання представляють у такий спосіб,
щоб оптична густина або її деяка функція були наве-
дені по осі ординат, а довжина хвилі або деяка функція
від довжини хвилі — по осі абсцис.
Якщо в окремій статті наводять лише одне значення
для положення максимуму поглинання, то це означає,
що одержане значення максимуму не має відрізнятися
від зазначеного більше як на ±2 нм.
Прилад. Спектрофотометр, призначений для вимірю-
вань в ультрафіолетовій і видимій областях спектра,
36
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
складається з оптичної системи, яка виділяє монохро-
матичне випромінювання в області від 200 нм до
№0 нм, і пристрою для вимірювання оптичної густини.
Перевірка шкали довжин хвиль. Для перевірки шкали
довжин хвиль використовують лінії водневої або дей-
терієвої розрядної лампи або лінії пари ртуті, а також
максимуми поглинання розчину гольмію перхлорату Р,
які подані у Табл. 2.2 25.-1. Допустиме відхилення
складає ±1 нм для ультрафіолетового і ±3 нм для ви-
димого діапазонів.
Таблиця 2.2.25,-1
Максимуми поглинання (або емісії)
для перевірки шкали довжин хвиль
241.15 нм (Но) 404.66 нм (Не)
253.7 нм (Не) 435.83 нм (Не)
287.15 нм (Но) 486.0 нм (ОР)
302.25 нм (Не) 486.1 нм(НР)
313.16 нм (Не) 536.3 нм (Но)
334.15 нм (Н§) 546.07 нм (Не)
361.5 нм(Но) 576.96 нм (Не)
365.48 нм (Не) 579.07 нм (Не)
Перевірка шкали оптичної густини. Перевіряють зна-
чення оптичних густин, використовуючи розчин калію
діхромату Р за довжин хвиль, зазначених у
Табл. 2.2.25.-2. У Табл. 2.2.25.-2 наведені точні значен-
ня питомого показника поглинання і його допустимі
межі для кожної довжини хвилі.
Для перевірки шкали оптичних густин використову-
ють розчин калію діхромату, приготований таким чи-
ном. Від 57.0 мг до 63.0 мг (точну наважку) калію
діхромату Р, попередньо висушеного до постійної ма-
си при температурі 130 °С, розчиняють у 0.005 Мроз-
чині кислоти сірчаної і доводять до 1000.0 мл цим са-
мим розчинником.
Таблиця 2.2.25.-2
Довжина хвилі, у манометрах Питомий показник поглинання АХ% А Іс.м 1 % Допустимі межі А ।
235 124.5 від 122.9 до 126.2
257 144.5 від 142.8 до 146.2
313 48.6 від 47.0 до 50.3
350 107.3 від 105.6 до 109.0
Граничний рівень розсіяного світла. Розсіяне світло мо-
же бути визначене за даної довжини хвилі з викорис-
танням відповідних фільтрів або розчинів: наприклад,
оптична густина розчину 12 г/л калію хлориду Ру кю-
веті з товщиною шару 1 см за 200 нм при використанні
води Ряк компенсаційного розчину має бути більше 2.
Розділювальна здатність (для якісного аналізу). Якщо
зазначено в окремих статтях, то визначають розділю-
вальну здатність спектрофотометра таким чином. За-
писують спектр 0.02 % (об/об) розчину толуолу Р у
гексані Р. Мінімально допустиме значення відношен-
ня оптичної густини у максимумі поглинання за
269 нм до оптичної густини в мінімумі поглинання за
266 нм зазначають в окремій статті.
Ширина спектральної щілини (для кількісного аналізу).
У випадку використання спектрофотометра із
змінною шириною спектральної щілини за вибраної
довжини хвилі можливі похибки, пов’язані з шири-
ною цієї щілини. Для їхнього виключення ширина
спектральної щілини має бути малою у порівнянні з
напівшириною смуги поглинання й у той самий час
має бути максимально велика для одержання високо-
го рівня /0. Отже ширина щілини має бути такою, щоб
подальше її зменшення не змінювало величину
вимірюваної оптичної густини.
Кювети. Допустимі варіації у товщині шару викорис-
товуваних кювет мають бути не більше ±0.005 см. Кю-
вети, призначені для випробовуваного і компен-
саційного розчинів, повинні мати однакове пропус-
кання (або оптичну густину) при заповненні тим са-
мим розчинником. У протилежному випадку цю
відмінність треба враховувати.
ПОХІДНА СПЕКТРОФОТОМЕТРІЯ
У похідній спектрофотометрії використовується пере-
творення вихідного спектра поглинання (нульовий
порядок) у похідні спектри першого, другого і більш
високих порядків.
Похідний спектр першого порядку являє собою графік
залежності градієнта кривої поглинання (швидкість
зміни оптичної густини З ДОВЖИНОЮ ХВИЛІ, СІЛ/СІА) від
довжини хвилі.
Похідний спектр другого порядку являє собою графік
залежності кривизни спектра поглинання від довжи-
ни хвилі (с12Л/с1А2). Друга похідна за будь-якої довжи-
ни хвилі А пов’язана з концентрацією таким
співвідношенням.
а А “ А\см сЬ ае ,,
---т~ = ----5-------- = —Т~ ’с
СІЇ5 сії? 10 ІЇК
де:
с' — концентрація поглинаючого розчину, у грамах
на літр.
Прилад. Використовують спектрофотометр, який
відповідає зазначеним вище вимогам і оснащений
аналоговим резистентно-ємнісним диференціюючим
модулем, або цифровим диференціатором, або інши-
ми засобами одержання похідних спектрів. Деякі ме-
тоди одержання похідних спектрів другого порядку
зрушують їх відносно спектра нульового порядку, що
треба враховувати там, де це необхідно.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
37
2.2. Фізичні та фізико-хімічні методи
Розділювальна здатність. Якщо зазначено в окремих
статтях, записують похідний спектр другого порядку
для розчину 0.2 г/л толуолу Р в метанолі Р, викорис-
товуючи метанол Р як компенсаційний розчин. На
спектрі має бути присутнім невеликий негативний
екстремум, розташований між двома великими нега-
тивними екстремумами за 261 нм і 268 нм, відповідно,
як показано на Рис. 2.2.25.-1. Якщо немає інших за-
значень в окремих статтях, відношення А/В (див.
Рис. 2.2.25.-1) має бути не менше 0.2.
Методика. Готують розчин випробовуваної речовини,
установлюють різні інструментальні характеристики
відповідно до інструкції до приладу і розраховують
кількість визначуваної речовини, як зазначено в ок-
ремій статті.
ІДЕНТИФІКАЦІЯ
Абсорбційну спектрофотометрію в ультрафіолетовій і
видимій областях спектра звичайно застосовують для
ідентифікації лікарських засобів у таких варіантах:
1. Порівняння спектрів поглинання випробовуваного
розчину і розчину порівняння; у зазначеній області
спектра має спостерігатися збіг положень макси-
мумів, мінімумів, плечей і точок перегину.
2. У зазначеній області спектра за зазначених довжин
хвиль мають спостерігатися максимуми, мінімуми,
плечі й точки перегину; можливе зазначення лише де-1
яких з цих характеристик. Розбіжність між спостере
жуваними і зазначеними довжинами хвиль не має зви-1
чайно перевищувати 2 нм.
3. На додаток до варіанта 2 наводять ще і питомі по-1
казники поглинання за зазначених довжин хвиль.
4. На додаток до варіанта 2 наводять відношення оп- і
тичних густин за зазначених довжин хвиль.
Можливі й інші варіанти застосування, зазначені вок-
ремих статтях.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
1. Одиокомпоненіний однохвильовий аналіз
Однокомпонентний однохвильовий аналіз (або "зви
чайна спектрофотометрія") — це кількісне визначення
одного з компонентів лікарського засобу за допомогою
вимірювання оптичної густини розчину випробовува-
ного зразка за однієї аналітичної довжини хвилі (АДХ).
Такий аналіз може проводитися методом показника
поглинання (МПП) або методом стандарту (МС).
При використанні МПП кількісне визначення прово-
дять за допомогою вимірювання оптичної густини (А)
розчину випробовуваного зразка за АДХ і розрахунку
концентрації (с) аналізованого компонента за форму-
лою:
де:
^їсм~ питомий показник поглинання аналізованого
компонента при АДХ;
с — концентрація аналізованої речовини, у відсот-
ках (маса/об).
При використанні МС кількісне визначення прово-
дять за допомогою вимірювання за АДХ оптичних гу-
стин розчину випробовуваного зразка (А) і розчину
порівняння (Ао) з концентрацією Сп і розрахунку кон-
центрації (С) аналізованого компонента, виходячи з
формули:
С = А (2)
Со ^0
Вимірювання оптичних густин випробовуваного роз-
чину і розчину порівняння треба проводити за одних і
тих самих умов з мінімальним інтервалом у часі.
У загальному випадку більш надійним є МС. Мож-
ливість застосування МПП необхідно у кожному кон-
кретному випадку обгрунтовувати, виходячи з до-
пусків кількісного вмісту аналізованого компонента,
метрологічних характеристик методики й вимог до
спектрофотометра. Звичайно МПП застосовний за
допусків вмісту аналізованого компонента не менше
±10 % від номінального вмісту.
У всіх випадках застосування однохвильового одно-
компонентного аналізу необхідно, щоб решта компо-
38
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
нентів препарату не чинили істотного впливу на ре-
зультати. Звичайно частка їхнього сумарного погли-
нання в оптичному поглинанні зразка за АДХ не має
перевищувати десятої частини допусків вмісту аналі-
званого компонента.
2. Багатокомпонентний спектрофотометричний аналіз
Багатокомпонентний спектрофотометричний аналіз
застосовують для одночасного кількісного визначен-
ня компонентів лікарських засобів.
У звичайній спектрофотометрії похибка власне спект-
рофотометричних вимірювань ("спектрофотометрич-
на похибка") мало залежить від типу аналізованої ре-
човини і вибору аналітичної довжини хвилі, а визна-
чається класом спектрофотометра і не перевищує зви-
чайно 0.5 %>. З урахуванням похибок приготування
розчинів це призводить до сумарної похибки аналізу,
яка не перевищує звичайно 1 %.
На відміну від звичайної спектрофотометрії, спектро-
фотометрична похибка багатокомпонентного аналізу
визначається не лише класом приладу, але й сильно
алежить від складу аналізованого лікарського засобу і
особливо вибору аналітичних довжин хвиль. Ця по-
хибка може бути охарактеризована коефіцієнтом
підсилення (К), який показує, у скільки разів спектро-
фотометрична похибка визначення даної речовини в
аналізованій суміші за допомогою багатокомпонент-
ної спектрофотометрії перевищує спектрофотомет-
ричну похибку визначення цієї самої речовини у чис-
тому розчині (без інших компонентів) методом зви-
чайної спектрофотометрії. Способи розрахунку ко-
ефіцієнтів підсилення для кожного компонента при
використанні різних методів наведені нижче.
Звичайними є величини К = 5-10, але можливі й зна-
чення К = 100 і більше, що може призводити до за-
гальної похибки аналізу, що складає десятки і навіть
сотні відсотків.
Для одержання надійних результатів при кількісному
визначенні лікарських засобів коефіцієнти підсилен-
ня К не мають звичайно перевищувати 5.
Тому прогноз похибки визначення і порівняння її з до-
пусками вмісту аналізованого компонента є обов’язко-
вою умовою при обгрунтуванні застосовності методик
багатокомпонентної спектрофотометрії. Якщо немає
відповідного обгрунтування, то має витримуватися та-
ке співвідношення між повною відносною похибкою
кількісного визначення Аого компонента аналізова-
ного зразка (Дк г %) і допусками (±В %) вмісту цього
компонента в зразку:
А,* (3)
А»,-2'5^' <4)
де:
5ҐІ(г- відносне генеральне стандартне відхилення
повної похибки кількісного визначення &-ОГО
компонента аналізованого зразка.
Кількісне визначення у багатокомпонентному спект-
рофотометричному аналізі грунтується звичайно на
використанні рівняння:
т
А. = X Е с і = 1...Н , (5)
' 7=1 '7 7
де:
А, — оптична густина випробовуваного розчину за
/-ої довжини хвилі;
Еу — показник поглинання (залежний від способу
вираження концентрації) у'-ого компонента
зразка за 1-ої аналітичної довжини хвилі;
Су — концентраціяу-ого компонента зразка.
Для розв’язання даного рівняння можуть застосовува-
тися різні підходи, серед яких можна виділити три ос-
новних: метод найменших квадратів (МНК), модифіко-
ваний метод найменших квадратів (ММНК) і метод
відношення розрахованих концентрацій (МВРК).
2.1. Метод найменших квадратів. Метод найменших
квадратів (МНК) є узагальненням методу показника
поглинання однохвильового однокомпонентного
аналізу. У рамках МНК розв’язання рівняння (3) має
вигляд:
с, = .-А. , к = \...т. (6)
к । кі і
Розрахункові коефіцієнти аМНК знаходять у відповід-
ності з матричним співвідношенням:
аМНК = (Ег Е)'1ЕТ, <7)
де:
аМНК— матриця розрахункових коефіцієнтів;
Е — матриця показників поглинання;
Т — символ транспонування.
Вибір аналітичних довжин хвиль (АДХ). Дивіться вибір
АДХ для модифікованого методу найменших квадратів.
Прогноз похибки визначення. Повна похибка кількіс-
ного визначення Е-ого компонента за допомогою
МНК визначається із співвідношення:
52, = (<жк)2 • (52 + 52 ) + 52 (8)
ск,г к А,г Е,г У,г ' '
Л &
„ а,.А.
к'' )2 . (9)
К /=і С,
к
де:
8скг~ відносне стандартне відхилення повної похиб-
ки кількісного визначення /с-ого компонента
зразка;
А}1 — оптична густина розчину модельної суміші
зразка, яка містить номінальні концентрації
усіх компонентів;
Ск — номінальна концентрація Л-ого компонента
зразка в модельній суміші;
г — відносне стандартне відхилення збіжності оп-
тичної густини на спектрофотометрі (включа-
ючи кюветну похибку);
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
39
2.2. Фізичні та фізико-хімічні методи
8Ег— відносне стандартне відхилення правильності
оптичної густини на спектрофотометрі;
5Иг— відносне стандартне відхилення похибки при-
готування розчинів.
Величини 8Аг і 5/г відомі з паспортних даних спект-
рофотометра, а 8Уг оцінюють, виходячи з похибок
взяття наважок і розведень.
При цьому мають виконуватися співвідношення (3-4).
Перевагою МНКє те, що його застосування не вима-
гає використання стандартних зразків. Однак через
значну похибку правильності оптичної густини (з
Табл. 2.2.25 -2 видно, що величини 8Ег можуть дося-
гати декількох відсотків) та її неконтрольованості по-
вна похибка аналізу за допомогою МНК може досяга-
ти десяти і більше відсотків, що робить МНК не-
надійним методом. Його застосування ставить дуже
високі вимоги до спектрофотометрів і до рівня роботи
аналітичного персоналу, тому він застосовний звичай-
но лише у наукових дослідженнях на стадії розробки
лікарських засобів за великих коливань у концент-
раціях аналізованих компонентів.
2.2. Модифікований метод найменших квадратів. Мо-
дифікований метод найменших квадратів є одним з
варіантів узагальнення методу стандарту на випадок
багатокомпонентної спектрофотометрії і грунтується
на рівняннях:
Я т Ф т
а = —'-г =Хг.. -Д- = Хг..х., (іо)
л5' Лї'7 с 7=17 7
і }
І = 1...П.
= Е..-с« і = (11)
де змінні мають той же зміст, що й у рівняннях (5) і (9),
величини Гц являють собою інформаційні коефіцієнти, а
Лу 100 являє собою концентрацію./-ого компонента пре-
парату у відсотках до його номінального вмісту.
Розв’язання рівняння (10) має вигляд’
X, = X а*,мнк Д, /=1...и, (12)
де розрахункові коефіцієнти аммнк знаходять за мат-
ричним рівнянням:
аммнк= (ГТ_ гу\гТ ' (13)
Вибір аналітичних довжин хвиль (АДХ). АДХ знахо-
дять, виходячи з критерію мінімуму коефіцієнта
підсилення КММИК, одержуваного зі співвідношення:
(№™)2 =^2=£
;=і 7 ;=і
£ (аММНКу2 , (|4)
де:
Л) — коефіцієнт підсилення для ./-ого компонента
зразка.
У випадку аналізу двох сполук за двома довжинами!
хвиль АДХ можна знаходити з умови максимумі
інформаційних коефіцієнтів кожного компонента зі
однієї з двох довжин хвиль.
Схема проведення аналізу. Готують модельну суміш, шо
містить усі компоненти препарату точно у номіналь-І
них концентраціях (розчин порівняння). Проводять
необхідні розведення, вимірюють поперемінно оп-
тичні густини випробовуваного розчину і розчину
порівняння при АДХ і проводять розрахунок за
рівняннями (10-11).
Прогноз похибки визначення. Повну похибку кількіс-
ного визначення 6-ого компонента за допомогою
ММНК визначають із співвідношення:
№.
ск,г
= 7.\(К ммнк)2 82
к 7 А,г
(15)
Має виконуватися співвідношення (3).
Недоліком ММНК є необхідність готування точно:
номінальної суміші зразка, однак він є най-
надійнішим і найточнішим з усіх багатохвильових ме-
тодів кількісного визначення лікарських засобів.
Окремий випадок — кількісне визначення одного компо-
нента суміші за однією довжиною хвилі. Якщо інфор-
маційний коефіцієнт (г,Д 6-ого компонента за ї-ої до-
вжини хвилі значно перевершує всі інші, то кількісне
визначення цього компонента можна проводити за
спрощеною формулою:
%А=Д (16)
Максимальна похибка такого наближення не
перевершує 2-В-(1 - гік). Дане наближення є обгрун-
тованим за гік > 0 95.
2.3. Метод відношення розрахованих концентрацій. Ме-
тод відношення розрахованих концентрацій (МВРК)
є одним з варіантів узагальнення методу стандарту на
випадок багатокомпонентної спектрофотометрії і
грунтується на допущенні, що відношення концент-
рацій, розрахованих для випробовуваного розчину і
розчину порівняння, є більш точним за самі розрахо-
вані концентрації, тобто:
де ак1 — коефіцієнти розрахункової матриці, одержані
за допомогою МНК (рівняння (7)) або іншими мето-
дами цифрової фільтрації, наприклад, методом
похідної спектрофотометрії. За відсутності фонового
поглинання найточнішим є застосування МНК.
Вибір аналітичних довжин хвиль (АДХ). Дивіться вибір
АДХ у ММНК.
Процедура проведення аналізу. Така сама як для
ММНК, але для виготовлення модельної суміші мож-
40
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
на використовувати концентрації компонентів,
близькі (а не точно рівні) до номінальних. Розрахунок
концентрацій проводять за рівнянням (17).
Прогноз похибки визначення (у випадку використання
МНК) проводять за співвідношенням:
А2 = 2-|(А'/нк)2 З’2 + А2 1. (18)
ск.г і к А,г У.г1
Має виконуватися співвідношення (3).
МВРК менш точний за ММНК, але він не вимагає
приготування точно номінальної суміші й тому
простіший у застосуванні.
ПОХІДНА СПЕКТРОФОТОМЕТРІЯ
Похідну спектрофотометрію використовують для
кількісного визначення звичайно у тих випадках, коли
є фонове поглинання, викликане присутністю речо-
вин, вміст яких не регламентується. Вона може
астосовуватися у двох варіантах.
У першому випадку одержання похідної проводить
сам аналітик за допомогою похідних поліномів, які
підставляються у рівняння (17) замість величин акі.
Прогноз похибки концентрації має враховувати у цьо-
му випадку похибку неповного перетворення в нуль
поглинання інших компонентів суміші.
У другому випадку безпосередньо використовують зна-
чення похідних, одержуваних на спектрофотометрі.
Процедура аналізу при цьому аналогічна застосуванню
методу стандарту у звичайній спектроє]ютометрїї, але
іамість оптичних густин використовують похідні.
2.2.27. ТОНКОШАРОВА ХРОМАТОГРАФІЯ
Тонкошарова хроматографія являє собою метод
розділення, в якому використовується нерухома фаза,
шо складається з підхожого матеріалу, нанесеного у ви-
гляді стандартизованого тонкого шару і зафіксованого
на основі (пластинці або пластині) із скла, металу або
пластмаси. Перед хроматографуванням розчини речо-
вин. що аналізуються, наносять на пластинку. Розді-
лення засноване на процесах адсорбції, розподілу, іон-
ного обміну або на їхній комбінації і здійснюєт ься за
допомогою переміщення в тонкому шарі (нерухомій
фазі) досліджуваних речовин, розчинених у розчинни-
ку або у відповідній суміші розчинників (рухомій фазі).
ОБЛАДНАННЯ
Пластинки. Хроматографування проводять з викорис-
танням пластинок, одержаних як описано у розділі
4.1.1. "Реактиви".
Попередня підготовка пластинок. У деяких випадках
може знадобитися промивання пластинок перед хро-
чатографуванням, яке може бути виконане за допомо-
гою попереднього елюювання чистих пластинок у
підхожому розчиннику Пластинки можуть бути також
імпрегновані (просякнуті) за допомогою таких проце-
дур, як елюювання, занурення або обприскування.
Перед використанням пластинки активують, якщо
необхідно, за допомогою нагрівання в термостаті при
температурі від 100 °С до 105 °С протягом і год.
Хроматографічна камера являє собою ємність з інерт-
ного прозорого матеріалу із щільно припасованою
кришкою і з плоским дном або дном з двома жолоба-
ми, відповідними за розміром пластинкам, що вико-
ристовуються. Для горизонтального елюювання хро-
матографічна камера має жолоб для рухомої фази і до-
датково містить пристрій для подачі рухомої фази до
нерухомої фази.
Мікропіпетки, мікрошприци, калібровані капіляри або
інші пристрої, придатні для нанесення розчинів
Пристрій для виявлення або гасіння флуоресценції.
Проявні реактиви — для виявлення розділених речовин
за допомогою обприскування, оброблення парою або
занурення.
МЕТОДИКА
Вертикальне елюювання. Стінки хроматографічної ка-
мери вистилають фільтрувальним папером. Рухому
фазу наливають у камеру в кількості, достатній для то-
го, щоб після змочування фільтрувального паперу по-
крити дно камери шаром рідини, необхідним для хро-
матографування. Для насичення хроматографічну ка-
меру з рухомою фазою закривають кришкою і витри-
мують протягом 1 год при температурі від 20 °С до
25 °С.
Об’єми розчинів аналізованих речовин, зазначені в
окремій статті, наносять невеликими порціями, одер-
жуючи смуги або круглі плями на підхожій відстані від
нижнього краю і від бічних країв пластинки. Розчини
наносять на лінію, паралельну нижньому краю плас-
тинки, з відстанню не менше 10 мм між пробами.
Після випарування розчинників з нанесених проб
пластинку поміщають у хроматографічну камеру яко-
мога більше вертикально, стежачи за тим, щоб плями
або смуги знаходилися вище поверхні рухомої фази.
Камеру закривають, залишають її при температурі від
20 °С до 25 °С у захищеному від прямих сонячних про-
менів місці. Після того, як рухома фаза пройде
відстань, зазначену в окремій статті, пластинку вий-
мають, сушать і виявляють плями способом, зазначе-
ним в окремій статті.
У випадку двомірної хроматографії після першого
хроматографування пластинку сушать і виконують
друге хроматографування у напрямку, перпендикуляр-
ному першому.
Горизонтальне елюювання. Об’єми розчинів досліджу-
ваних речовин, зазначені в окремій статті, наносять
невеликими порціями, одержуючи круглі плями (від
1 мм до 2 мм у діаметрі) або смуги (завдовжки від 5 мм
до 10 мм і завширшки від 1 мм до 2 мм) на підхожій
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
41
2.2. Фізичн'
Ьізико-хімічні методи
краю і від бічних країв пластин-
іа лінію, паралельну нижньому
овалом не менш як 5 мм між
ників з нанесених проб у жо-
__сри уводять за допомогою
_^г«^бстатню кількість рухомої фази,
^_^**Тїластинку горизонтально в хромато-
0^Ж|і!чну камеру і приєднують пристрій для подачі ру-
хомої фази у відповідності до інструкції виробника. Як-
що зазначено в окремій статті, пластинку елююють, по-
чинаючи одночасно з двох кінців. Камеру закривають і
проводять хроматографування при температурі від
20 °С до 25 °С. Після того, як рухома фаза пройде
відстань, зазначену в окремій статті, пластинку вийма-
ють, сушать і виявляють плями зазначеним способом.
У випадку двомірної хроматографії після першого
хроматографування пластинку сушать і виконують
друге хроматографування у напрямку, перпендикуляр-
ному першому.
ВІЗУАЛЬНА ОЦІНКА
Ідентифікація. Основну пляму на хроматограмі, одер-
жаній для випробовуваного розчину, порівнюють візу-
ально з відповідною плямою на хроматограмі, одер-
жаній для розчину стандартного зразка (розчину
порівняння), порівнюючи забарвлення (колір флуо-
ресценції), розмір і величину утримування (Д) обох
плям.
Величину утримування (Кг) визначають як відношен-
ня відстані віл точки нанесення плями до верхньої
кромки плями після хроматографування до відстані,
пройденої фронтом розчинника від точки нанесення.
Перевірка розділювальної здатності нерухомої фази для
ідентифікації. Звичайно для оцінки придатності досить
випробування на придатність нерухомої фази, описа-
ного у розділі 4.1.1. "Реактиви". В особливих випадках
додаткові вимоги зазначають в окремих статтях.
Випробування на супровідні домішки. Додаткову пляму
(плями) на хроматограмі, одержаній для випробовува-
ного розчину, порівнюють візуально з відповідною
плямою (плямами) на хроматограмі, одержаній для
розчину порівняння. Як стандартний зразок для виго-
товлення розчину порівняння використовують як са-
му домішку (домішки), так і різні розведення випро-
бовуваного розчину.
Перевірка розділювальної здатності. Вимоги для пе-
ревірки розділювальної здатності наводять у
відповідних окремих статтях.
Перевірка чутливості. Чутливість вважається за-
довільною, якщо пляма або смуга чітко виявляються
на хроматограмі, одержаній з найбільш розведеним
розчином порівняння.
КІЛЬКІСНІ ВИМІРЮВАННЯ
Вимоги розділення наводять в окремих статтях.
У тому випадку, коли речовини, розділювані методо»11
тонкошарової хроматографії, поглинають або флуоЯ
ресціюють в ультрафіолетовому або видимому світа, І
їх можна кількісно визначити безпосередньо на плас-1
тинці, використовуючи підхоже обладнання. Для цьо-1
го вимірюють відбиття або пропускання падаючого І
світла, пересуваючи пластинку або вимірюючий!
пристрій. Аналогічно, використовуючи підхоже оп-І
тичне обладнання, можна вимірювати флуорес-І
ценцію. Речовини, які містять радіонукліди, можуть 1
бути кількісно визначені трьома способами:
— безпосередньо на пластинці — пересуванням плас-1
тинки вздовж підхожого лічильника радіоактив-І
ності або лічильника радіоактивності уздовж плас-1
тини (див. Радіофармацевтичні препарати 125); І
— розрізанням пластинки на смуги і вимірюванням І
радіоактивності на кожній смузі, використовуючи!
підхожий лічильник радіоактивності;
— зіскрібанням нерухомої фази, розчиненням її її
підхожому сцинтиляційному коктейлі й вимірю-|
ванням радіоактивності з використанням рідинно-1
го сцинтиляційного лічильника.
Обладнання. Обладнання для вимірювань безпосеред-І
ньо на пластинці включає в себе:
— пристрій для прямого нанесення у певному місці
пластинки необхідної кількості речовини;
— механічний пристрій для пересування пластинки
або вимірюваного пристрою вздовж вісей X або У;
— самопис й інтегратор або комп’ютер;
— для речовин, поглинаючих або флуоресціюючих в уль-
трафіолетовому або видимому світлі: для вимірю-
вання відбиття або пропускання використовуються
фотометр з джерелом світла, оптичний пристрій,
генеруючий монохроматичне світло, і фотокомірка
відповідної чутливості; у тому випадку, коли
вимірюється флуоресценція, потрібний додатково
монохроматичний фільтр для вибору відповідної
спектральної області випромінюваного світла;
— для речовин, що містять радіонукліди: підхожий
лічильник радіоактивності; для нього необхідно
перевірити лінійний діапазон.
Методика. Готують способом, зазначеним в окремій
статті, розчин аналізованої речовини (випробовува-
ний розчин) і, якщо необхідно, розчини стандартних
зразків аналізованих речовин у тому самому розчин-
нику (розчини порівняння). Наносять однаковий
об’єм кожного розчину на пластинку і хроматографу-
ють.
Для речовин, поглинаючих або флуоресціюючих в ульт-
рафіолетовому або видимому світлі. Готують і нано-
сять не менше трьох розчинів порівняння, концент-
рації яких охоплюють очікуване значення концент-
рації у випробовуваному розчині (близько 80 %, 100 %
і 120 % від цієї концентрації). Обприскують, якщо не-
обхідно, зазначеним реактивом і реєструють відбиття,
пропускання або флуоресценцію на хроматограмах,
одержаних для випробовуваного розчину і розчинів
порівняння. За одержаними даними розраховують
кількість речовини у випробовуваному розчині.
Для речовин, що містять радіонукліди. Готують і нано-
сять випробовуваний розчин, що містить близько
42
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
Кл) с очікуваного значення концентрації. Вимірюють
ііоактивність як функцію довжини шляху і запису-
ють радіоактивність кожного одержаного піка у від-
сотках від сумарної радіоактивності.
Якшо немає інших зазначень в окремій статті, резуль-
іати визначення вважають недійсними, якщо ко-
ефіцієнт розділення (Кх) між вимірюваними на хрома-
тограмі піками менше 1.0.
Коефіцієнт розділення (К5) обчислюють за формулою:
1.18^-^)
^0.5а + ^0.5/>
ле
- — відстані вздовж базової лінії між точкою
нанесення зразка і перпендикулярами,
опущеними з вершин двох сусідніх піків,
у міліметрах;
~ ширина піків на половині висоти, у
міліметрах.
У випадку встановлення граничного вмісту домішок
з.! допомогою фотометричного детектора важливим
параметром для визначення межі виявлення є відно-
шення сигнал/шум (8/М)'.
8 = 2Н
N И
де:
Н - висота піка аналізованого компонента на хро-
матограмі розчину порівняння, виміряна від
вершини до базової лінії; базова лінія
вимірюється при цьому в інтервалі двадцяти
ширин піка, виміряних на половині його ви-
соти;
/і максимальна амплітуда фонового шуму на
хроматограмі холостого розчину, спостережу-
вана у тому самому інтервалі, що і для випро-
бовуваного розчину.
______________________________________________N
ОБЛАДНАННЯ
Пластинки. Допускається використання пластинок,
приготованих у промислових умовах, якщо вони
відповідають вимогам розділу 4.1.1. "Реактиви", а та-
кож витримують вимоги тесту "Перевірка придатності
хроматографічної системи", описані в окремій статті.
Хроматографічна камера. У необхідних випадках допу-
скається використання хроматографічних камер
інших типів з описом їх в окремих статтях.
Допускаються інші умови активації пластинок, опи-
сані в окремих статтях.
МЕТОДИКА
Якщо немає інших зазначень в окремій статті, хрома-
тографічне розділення виконують висхідним спосо-
бом у насиченій атмосфері.
Краще використовувати такі рухомі фази, які забезпе-
чують величини випробовуваних сполук у межах
від 0.3 до 0.7.
Якшо немає інших зазначень в окремій статті, плями або
смуги наносять на відстані не менше 15 мм від нижньо-
го краю і не менше 10 мм від бічних країв пластинки.
Шар рідини у хроматографічній камері має бути та-
ким, щоб після поміщення в нього пластинки плями
або смуги знаходилися над рівнем рідини.
Якщо умови насичення хроматографічної камери, на-
несення плям або хроматографування відрізняються
від зазначених вище, вони мають бути описані в ок-
ремій статті.
ПЕРЕВІРКА ПРИДАТНОСТІ
ХРОМАТОГРАФІЧНОЇ СИСТЕМИ
Результати аналізу методом тонкошарової хромато-
графії (ТШХ) вважаються вірогідними, якщо викону-
ються вимоги тесту "Перевірка придатності хромато-
графічної системи".
Хроматографічна система вважається придатною, як-
шо:
— на хроматограмі розчину порівняння, використо-
вуваного для перевірки придатності хромато-
графічної системи, чітко діляться плями зазначе-
них в окремій статті речовин;
— /^основної плями на хроматограмі випробовува-
ного розчину має бути близько величини, зазначе-
ної в окремій статті;
— на хроматограмі розчину порівняння, використо-
вуваного для перевірки чутливості хромато-
графічної системи, має бути чітко видно пляму.
СПОСОБИ ОЦІНКИ ВМІСТУ ДОМІШОК
МЕТОДОМ ТШХ
При контролі домішок небажане включення до окре-
мої статті вимоги відсутності плями контрольованої
домішки на хроматограмі випробовуваного розчину.
Методом ТШХ можуть контролюватися як конкретно
зазначувані (специфічні) домішки, так і загальний
вміст домішок.
КОНТРОЛЬ СПЕЦИФІЧНИХ ДОМІШОК
Контроль специфічних домішок застосовують у тих
випадках, коли вміст якихось конкретних домішок,
що виникають у процесі виробництва препарату або
при його зберіганні, має бути обмежений через їхню
токсичність або з інших міркувань.
При контролі специфічних домішок звичайно вико-
ристовують порівняння плям домішок, що регламен-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
43
2.2. Фізичні та фізико-хімічні методи
туються, на хроматограмах випробовуваного розчину і
розчинів порівняння.
Типова регламентація вмісту домішки виглядає в цьо-
му випадку таким чином:
На хроматограмі випробовуваного розчину, крім основ-
ної плями, допускається наявність додаткової плями,
розташованої на рівні плями на хроматограмі розчину
порівняння і не перевищуючої її за величиною и інтен-
сивністю поглинання або забарвлення (... %).
КОНТРОЛЬ ЗАГАЛЬНОГО ВМІСТУ ДОМІШОК
У тих випадках, коли немає підстав вважати якісь до-
мішки особливо токсичними, часто не так важливо
знати їхній справжній вміст. Важливо знати, що цей
вміст не перевершує певний рівень. У таких випадках
використовують метод внутрішньої нормалізації — як
розчини порівняння звичайно використовують розчи-
ни самої випробовуваної субстанції різної концентра-
ції, а вміст домішок знаходять у перерахунку на цю
субстанцію.
У залежності від кількості різних розчинів субстанції,
що наносять на хроматограму у вигляді розчинів
порівняння, контроль загального вмісту домішок мо-
же бути однорівневим, дворівневим і трирівневим.
Типова регламентація вмісту домішки в однорівнево-
му варіанті виглядає таким чином:
"На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної плями, не має перевищувати за
розміром и інтенсивністю поглинання або забарвлення
пляму на хроматограмі розчину порівняння (... %)".
Типова регламентація вмісту домішки у дворівневому
варіанті виглядає таким чином:
"На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної плями, не має перевищувати за
розміром и інтенсивністю поглинання або забарвлення
основну пляму на хроматограмі розчину порівнян-
ня 1 (... %), і тільки одна пляма може бути інтен-
сивнішою за пляму на хроматограмі розчину порівнян-
ня 2(...%)".
Типова регламентація вмісту домішки у трирівневому
варіанті виглядає таким чином:
"На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної плями, не має перевищувати за
розміром й інтенсивністю поглинання або забарвлення
пляму на хроматограмі розчину порівняння 1 (... %), і
тільки одна пляма може бути інтенсивнішою за пляму
на хроматограмі розчину порівняння 2 (... %), і не
більше ... плям можуть бути інтенсивнішими за пляму
на хроматограмірозчину порівняння 3 (... %)".
У дво- і трирівневому варіантах можлива регламен-
тація і загальної суми домішок.
2.2.28. ГАЗОВА ХРОМАТОГРАФІЯ
Газова хроматографія (ГХ) являє собою метод
розділення, у якому рухомою фазою є газ (газ-носій),
а нерухомою фазою, помішеною у колонку, є тверд;
речовина або рідина, що нанесені на твердий інертний
носій або рівномірно покривають внутрішні стінк»
колонки.
Газова хроматографія заснована на механізмах ад-
сорбції і/або розподілу.
Обладнання. Обладнання складається з системи по
дачі газу, пристрою вводу проби, хроматографічної ко-
лонки, детектора і реєструючого пристрою. Колонки
звичайно виготовляють із скла або нержавіючої сталі йІ
заповнюють нерухомою фазою. Газ-носій проходить із!
заданою швидкістю через блок вводу проби, колонку
а потім через детектор.
Визначення проводять при постійній температурі або
у відповідності із заданою температурною програмою
Використовуваний детектор має забезпечувати визна-
чення тих кількостей аналізованих речовин, які елюю-,
ються з колонки. Звичайно детектування засноване на
ефектах іонізації у полум’ї, теплопровідності, тер-
моіонному ефекті або на ефекті захвату електронів.
Методика. Колонку, пристрій вводу проби і детектор
термостатують при зазначеній температурі. Готують!
випробовуваний розчин і розчин(и) порівняння, як
зазначено в окремій статті. Використовуючи розчини
порівняння, настроюють прилад і добирають об'єми
проб, що вводяться і дозволяють одержати необхідний
сигнал. Виконують повторні введення для перевірки
збіжності сигналу і перевіряють, якщо необхідно, чис-
ло теоретичних тарілок.
Вводять розчини і реєструють результати хроматогра-
фування. Для перевірки збіжності сигналу виконують
повторні введення. Визначають площі піків аналізова-
них компонентів. У випадку, якщо коефіцієнт си-
метрії, обчислений, як описано нижче, має значення
від 0.8 до 1.20, допускається визначення за висоток
піків. При використанні програмування температурні
необхідно проводити визначення за площами піків.
При використанні внутрішнього стандарту треба
упевнитися, що жоден з піків, що ВІДНОСЯТЬСЯ Д(
аналізованої речовини або її домішки, не маскується а
піком внутрішнього стандарту.
З одержаних значень обчислюють вміст аналізованого
компонента або компонентів. Якщо зазначено в ок-
ремій статті, відсотковий вміст одного або декільком
компонентів аналізованої проби визначають за допо
могою обчислення відсоткової частки площі вілло
відного піка або піків у сумарній плоті усіх піків, ви-
ключаючи піки розчинників або доданих реактивів
(метод внутрішньої нормалізації). У цих випадках ре-
комендується використання широкодіапазонного
підсилювача і автоматичного інтегратора.
Коефіцієнт симетрії піка може бути обчислений за
формулою:
^0.05
2/1 ’ І
де:
^0.05 — ширина піка на одній двадцятій висоти піка;
44
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
к — відстань між перпендикуляром, опущеним з
максимуму піка, і передньою межею піка на
одній двадцятій висоти піка.
Коефіцієнт розділення (К,) може бути обчислений за
формулою:
^0.5а + ^0.5Ь
1КЬ > {Ка ,
де:
<№'^0 ~ відстані вздовж базової лінії від точки
вводу проби до перпендикулярів, опу-
щених з максимумів двох сусідніх піків, у
міліметрах;
/>0 5«І^0 5і — ширина піків на половині їхньої висоти,
у міліметрах.
Якщо немає інших зазначень в окремій статті,
результати аналізу вважаються вірогідними, якщо
коефіцієнт розділення для вимірюваних піків на
хроматограмі більше 1.0.
Число теоретичних тарілок (п) може бути обчислене з
даних, одержаних в ізотермічному режимі, за форму-
лою:
де:
відстань уздовж базової лінії від точки вводу
проби до перпендикуляра, опущеного з мак-
симуму піка аналізованої речовини, у мілімет-
рах;
/>п, - ширина піка на половині висоти, у міліметрах.
Коефіцієнт ємності к' (відомий також як коефіцієнт
розподілу мас /),„) визначають як:
І _ ш_ крнф 22
и,п КРРФ
де:
КРНФ - кількість розчиненої речовини у нерухомій
фазі;
КРРФ - кількість розчиненої речовини у рухомій
фазі;
К - рівноважний коефіцієнт розподілу;
К - об’єм нерухомої фази;
ИПІ - об’єм рухомої фази.
Коефіцієнт ємності компонента може бути визначе-
ний з даних хроматограми за формулою:
де:
Іц - відстань уздовж базової лінії від точки вводу про-
би до перпендикуляра, опушеного з максимуму
піка аналізованого компонента, у міліметрах;
— відстань уздовж базової лінії від точки вводу
проби до перпендикуляра, опущеного з макси-
муму піка неутримуваного компонента, у мілі-
метрах.
Відношення сигнал/шум (8/М) обчислюють за форму-
лою:
де:
Н — висота піка відповідного компонента на хро-
матограмі. одержаній для зазначеного розчину
порівняння;
Ап — абсолютне значення найбільшої флуктуації
шуму базової лінії на хроматограмі холостого
розчину, спостережуване на проміжку, рівному
двадцятикратній ширині на напіввисоті піка
на хроматограмі розчину порівняння,
розміщеному рівномірно навколо місця розта-
шування піка.
ПАРОФАЗНА ГАЗОВА ХРОМАТОГРАФІЯ
Парофазна газова хроматографія є методом, найбільш
придатним для розділення і визначення летких спо-
лук, присутніх у твердих або рідких зразках. Метод за-
снований на аналізі парової фази, що перебуває у
рівновазі з твердою або рідкою фазою.
Обладнання. Обладнання складається з газового хро-
матографа, оснащеного блоком вводу парової фази,
що знаходиться над випробовуваним зразком.
Пристрій вводу може бути приєднаний до блока, що
автоматично контролює і регулює тиск і температуру.
Якщо необхідно, використовують пристрій для вида-
лення розчинників.
Аналізовану пробу вводять у контейнер, оснащений
придатною пробкою і клапанною системою, яка регу-
лює проходження газу-носія. Контейнер поміщають у
термостатовану камеру з температурою, що установ-
люється відповідно до властивостей аналізованого
зразка.
Пробу витримують при заданій температурі протягом
часу, достатнього для установлення рівноваги між
твердою або рідкою фазою і паровою фазою.
У контейнер вводять газ-носій і після закінчення за-
значеного часу відкривають клапан, щоб газ надходив
у хроматографічну колонку, переносячи з собою ком-
поненти, що перейшли в парову фазу.
Замість використання хроматографа, спеціально ос-
нащеного блоком вводу парової фази, можливе також
використання герметичних шприців і хроматографа
без зазначеного пристрою. У ньому випадку рівновага
установлюється в окремій камері, і парова фаза пере-
носиться в колонку з дотриманням необхідних засте-
режних заходів для запобігання будь-яких змін рівно-
важного складу.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
45
2.2. Фізичні та фізико-хімічні методи
Методика. Настроюють прилад для одержання не-
обхідного сигналу, використовуючи підготовані зраз-
ки порівняння.
а) Метод прямого калібрування
В однакові контейнери нарізно поміщають аналізова-
ну пробу і кожний із зразків порівняння, приготовані,
як зазначено в окремій статті, уникаючи контакту між
пристроєм для вводу проб і зразками.
Контейнери герметично закривають і поміщають у
термостатовану камеру з температурою і тиском, за-
значеними в окремій статті. Після установлення
рівноваги парову фазу хроматографують у зазначених
умовах.
б) Метод стандартних добавок
Рівні об’єми аналізованої проби поміщають в однакові
зазначені в окремій статті контейнери. В усі контейне-
ри, крім одного, додають зазначені кількості розчину
порівняння, шо містить відому концентрацію аналізо-
ваної речовини, для одержання ряду зразків з концент-
раціями цієї речовини, шо рівномірно збільшуються.
Контейнери герметично закривають і поміщають у
термостатовану камеру з температурою і тиском, за-
значеними в окремій статті. Після установлення
рівноваги хроматографують парову фазу в зазначених
умовах.
Рівняння лінійної залежності розраховують методом
найменших квадратів. За одержаним рівнянням ви-
значають концентрацію аналізованої речовини у ви-
пробовуваній пробі.
Допускається визначення концентрації з використан-
ням графічного методу. Для цього по осі ординат
відкладають середні значення одержаних результатів,
а по осі абсцис — концентрації стандартних добавок
аналізованої речовини. Екстраполюють лінію, що
проходить через одержані точки, до перетину з віссю
абсцис. Відстань між цією точкою і початком коорди-
нат являє собою концентрацію аналізованої речовини
у випробовуваному розчині.
с) Метод послідовних доборів
Застосування даного методу описують в окремій
статті.
_____________________________________________N
Обладнання. Звичайно детектування засноване на
ефектах іонізації у полум’ї, теплопровідності, тер-
моіонному ефекті або на ефекті захвату електронів.
Використання детекторів, заснованих на інших прин-
ципах, вимагає відповідного обгрунтування. При цьо-
му належить детально зазначати режим їхньої роботи.
МЕТОДИКА
Перед використанням колонка має бути кондиціоно-
вана.
ІДЕНТИФІКАЦІЯ
Відносний час утримування — це відношення часу ут-
римування аналізованої речовини до часу утримував-’
ня речовини, прийнятої за стандарт.
Ідентифікацію звичайно проводять одним із таких
способів:
1) порівняння часів утримування аналізованої речо-
вини у випробовуваній пробі і розчині порівняння;
2) порівняння відносних часів утримування аналізо-
ваної речовини у випробовуваній пробі і розчині
порівняння;
3) порівняння хроматограми випробовуваної проби з
хроматограмою розчину порівняння або з хромато-
грамою, наведеною в окремій статті.
Звичайно використовують перший спосіб. Другим
спосіб доцільно використовувати, якщо можлива
невідтворюваність умов хроматографування. Третій
спосіб може застосовуватися для препаратів рослин-
ного і тваринного походження.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Абсолютне калібрування. Якщо немає інших зазначень
в окремій статті, випробовуваний розчин і розчин
порівняння поперемінно хроматографують на газово-
му хроматографі, одержуючи не менше п’яти хромате-1
грам, в умовах, зазначених в окремій статті. Для ви-
пробовуваного розчину і розчину порівняння розра-
ховують середні значення площ або висот піків аналі-1
зованої речовини. За одержаними середніми значен-
нями розраховують концентрацію аналізованої речо-
вини у випробовуваному розчині.
Метод внутрішнього стандарту. Для кожної хромато-
грами спочатку розраховують відношення площі або
висоти піка аналізованої речовини до площі або висо-
ти піка внутрішнього стандарту. Одержані відношення
усереднюють для випробовуваного розчину і розчину
порівняння і за знайденими середніми значеннями
визначають концентрацію аналізованої речовини у
випробовуваному розчині.
КОНТРОЛЬ ДОМІШОК
Для контролю домішок звичайно використовують такі
підходи.
7. Кількісне визначення домішки з використанням раз
чину порівняння з відомою концентрацією домішки
(звичайно у варіанті абсолютного калібрування). Такий
підхід передбачає однаковий відгук домішки у присут-
ності й відсутності основної речовини.
2. Метод внутрішньої нормалізації. Такий підхід пе-
редбачає виконання лінійності в широкому діапазоні
й може вимагати урахування відмінностей у відгуках
домішки і основної речовини. Його часто застосову-
ють для визначення суми домішок. При цьому суму
площ усіх піків на хроматограмі (без урахування піка
розчинника) беруть за 100 % і вміст кожної конкретної
46
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
домішки або суми домішок знаходять як частку площі
піка цієї домішки або суми площ піків домішок у за-
гальній сумі площ усіх піків на хроматограмі.
3. Порівняння з розведеним розчином основної речовини.
4. Метод стандартних добавок. До аналізованої проби
іоіають відому кількість домішки. За даними хрома-
тографування випробовуваної проби і випробовуваної
проби з добавкою визначають вміст домішки. Для
підвищення точності можливе використання методу
внутрішнього стандарту.
УМОВИ ХРОМАТОГРАФІЧНОГО АНАЛІЗУ
У методиці рекомендується зазначати такі умови
аналізу:
- розміри хроматографічної колонки і матеріал, із
якого вона виготовлена;
- тип нерухомої фази та її кількість;
- тип твердого носія і розмір його часток;
- температуру колонки, блока вводу проб і детектора;
газ-носій і його витрата;
- тип детектора;
необхідність використання автосамплера;
- коефіцієнт поділу потоку (для капілярних коло-
нок).
Якщо введення проби здійснюється не у випарник, а
безпосередньо в колонку, належить давати відповідне
зазначення у методиці, наведеній в окремій статті.
Придатність хроматографічної системи. Результати
аналізу вважаються вірогідними, якшо виконуються
вимоги теста "Перевірка придатності хроматографіч-
ної системи". Даний тест звичайно проводять з вико-
ристанням розчинів порівняння.
Якшо немає інших зазначень в окремій статті, хрома-
тографічна система вважається придатною, якшо ви-
конуються такі умови:
відносні часи утримування зазначених речовин
мають бути близько регламентованих значень;
- число теоретичних тарілок (ефективність хрома-
тографічної системи) має бути не менше регла-
ментованої величини;
коефіцієнт розділення зазначених піків, розрахо-
ваний з хроматограм розчину порівняння, має бу-
ти не менше зазначеної величини;
- відносне стандартне відхилення, розраховане для
висоти або площі зазначеного піка або їхніх відно-
шень до висоти або площі піка внутрішнього стан-
дарту з хроматограм розчину порівняння, має бути
не більше регламентованої величини; для розра-
хунку відносного стандартного відхилення вико-
ристовують дані п’яти паралельних хроматограм;
якщо потрібне відносне стандартне відхилення
перевищує 2.0 %, його розрахунок проводять, ви-
користовуючи дані шести або більше паралельних
хроматограм;
- коефіцієнт симетрії зазначеного піка, розрахова-
ний з хроматограм розчину порівняння, має бути у
межах, зазначених в окремій статті.
Для забезпечення виконання вимог тесту "Перевірка
придатності хроматографічної системи" допускається
модифікація хроматографічних умов, описаних в ок-
ремій статті (змін^ температури термостата колонок
і/або витрати газу-носія).
ПАРОФАЗНА ГАЗОВА ХРОМАТОГРАФІЯ
Обладнання. Можливе використання інших способів
вводу парової фази у хроматографічну колонку.
2.2.29. РІДИННА ХРОМАТОГРАФІЯ
Рідинна хроматографія (РХ) являє собою метод
розділення, у якому рухомою фазою є рідина, а неру-
хомою фазою, поміщеною у колонку, є тонкодисперс-
на тверда речовина або рідина, нанесена на твердий
тонкодисперсний носій, або твердий тонкодисперс-
ний носій, хімічно модифікований шляхом уведення
органічних груп.
Рідинна хроматографія заснована на механізмах ад-
сорбції. розподілу, іонного обміну або розділення за
розмірами молекул.
Обладнання. Обладнання звичайно складається з сис-
теми подавання рухомої фази, блока вводу проби (з
використанням шприца або петлевого дозатора), хро-
матографічної колонки, детектора і реєструючого
пристрою. Рухома фаза звичайно подається під тис-
ком з однієї або декількох посудин і протікає через
блок вводу проби, колонку, а потім через детектор із
заданою швидкістю.
Температуру хроматографічної колонки підтримують
постійною. Склад рухомої фази, залежно від зазначе-
ного в окремій статті, може або залишатися постійним
протягом всього аналізу (ізократичне елюювання),
або може змінюватися відповідно до заданої програми
(градієнтне елюювання).
Використовуваний детектор має забезпечувати визна-
чення тих кількостей аналізованих речовин, які елюю-
ються з колонки. Звичайно для детектування викори-
стовують абсорбційну спектрофотометрію; викорис-
товують також диференціальну рефрактометрію, флу-
ориметрію. спалювання і електрохімічні методи.
Методика. Колонку врівноважують при зазначеному
складі рухомої фази. Готують випробовуваний розчин і
розчин(и) порівняння, як зазначено в окремій статті.
Розчини не мають містити твердих часток. Використову-
ючи розчини порівняння, настроюють прилад і підбира-
ють об’єми проб, що вводяться, які дозволяють одержа-
ти необхідний (адекватний) сигнал. Виконують по-
вторні введення для перевірки збіжності сигналу і пе-
ревіряють, якщо необхідно, число теоретичних тарілок.
Вводять розчини і реєструють результати хроматогра-
фування. Для перевірки збіжності сигналу виконують
повторні введення. Визначають площі піків аналізова-
них компонентів. У випадку, якщо коефіцієнт си-
метрії, обчислений, як описано нижче, має значення
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
47
2.2. Фізичні та фізико-хімічні методи
від 0.8 до 1.20, допускається проводити визначення за
висотою піків. При використанні градієнтного елюю-
вання необхідно проводити визначення за площами
піків. При використанні внутрішнього стандарту тре-
ба переконатися, що жоден з піків аналізованої речо-
вини або його домішки не маскується піком внутріш-
нього стандарту.
З одержаних значень обчислюють вміст визначувано-
го компонента або компонентів. Якщо зазначено в ок-
ремій статті, відсотковий вміст одного або декількох
компонентів аналізованої проби визначають за допо-
могою обчислення відсоткової частки площі
відповідного піка або піків до сумарної площі всіх
піків, виключаючи піки розчинників або доданих ре-
активів (метод внутрішньої нормалізації). У цих ви-
падках рекомендується використання широкодіапа-
зонного підсилювача і автоматичного інтегратора.
Коефіцієнт симетрії піка може бути обчислений за
формулою:
^0.05
2А
де:
/>0(І5— ширина піка на одній двадцятій висоти піка;
А — відстань між перпендикуляром, опущеним з
максимуму піка, і передньою межею піка на
одній двадцятій висоти піка.
Коефіцієнт розділення (К8) може бути обчислений за
формулою:
п _ ~ */?о)
5 ^0.5о + ^0.5/>
?/?Л > 1Ка ,
де:
Ікь * {Ка ~ відстані уздовж базової лінії від точки
введення проби до перпендикулярів,
опущених з максимумів двох сусідніх
піків, у міліметрах;
/>С5а і />0 5Л - ширина піків на половині висоти, у
міліметрах.
Якщо немає інших зазначень в окремій статті, резуль-
тати аналізу вважаються вірогідними, якщо ко-
ефіцієнт розділення для вимірюваних піків на хрома-
тограмі більше 1.0
Коефіцієнт ємності к’ (відомий також як коефіцієнт
розподілу мас £>,„) визначають як:
крнф .22
КРРФ ' Ут
де:
КРНФ - кількість розчиненої речовини у нерухомій
фазі;
КРРФ — кількість розчиненої речовини у рухомій
фазі;
К — рівноважний коефіцієнт розподілу;
Ц — об’єм нерухомої фази;
Ит — об’єм рухомої фази.
Коефіцієнт ємності компонента може бути визначе-
ний з даних хроматограми за формулою:
£>,„ = к' =
" 7/?'
ґ/?
де:
відстань уздовж базової лінії від точки введен-
ня проби до перпендикуляра, опущеного
максимуму піка аналізованого компонента,
міліметрах;
відстань уздовж базової лінії від точки введем
проби до
максимуму
піка
у міліметрах.
перпендикуляра,
опущеного
неутримуваного
компонента
Відношення сигнал/шум (8/М) обчислюють за форму
лою:
5/7У =
п
де:
Н
висота піка відповідного компонента на хро
матограмі, одержаній для зазначеного розчину
порівняння;
абсолютне значення найбільшої флуктуації
шуму базової лінії на хроматограмі холостого
розчину, спостережуване на проміжку, шо
дорівнює двадцятикратній ширині на напівви
соті піка на хроматограмі розчину порівняння
розміщеному рівномірно навколо місця розта
шування піка.
Число теоретичних тарілок (п) може бути обчислене з
даних, одержаних в ізократичному режимі, за форму-
лою:
ІДЕНТИФІКАЦІЯ
Відносний час утримування
це
відношення
часу ут
римування
аналізованої речовини до
часу утримував
де:
ір — відстань уздовж базової лінії від точки введен-
ня проби до перпендикуляра, опущеного з
максимуму піка аналізованої речовини, у
міліметрах;
Ьс 5 — ширина піка на половині висоти, у міліметрах.
ня речовини, взятої за стандарт.
Ідентифікацію звичайно проводять одним
способів:
1)
порівняння часів утримування
вини у випробовуваній
пробі
із таких
аналізованої
й розчині
речо
порівняй
ня;
48
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
2001
2.2. Фізичні та фізико-хімічні методи
2) порівняння відносних часів утримування аналізо-
ваної речовини у випробовуваній пробі й розчині
порівняння;
Й порівняння хроматограми випробовуваної проби з
хроматограмою розчину порівняння або з хрома-
тограмою, наведеною в окремій статті.
Звичайно використовують перший спосіб. Другий
спосіб доцільно використовувати, якщо можлива
невідтворюваність умов хроматографування. Третій
спосіб може застосовуватися для препаратів рослин-
ного і тваринного походження.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Абсолютне калібрування. Якщо немає інших зазначень
в окремій статті, випробовуваний розчин і розчин
порівняння поперемінно хроматографують на рідин-
ному хроматографі, одержуючи не менше п’яти хро-
матограм, в умовах, зазначених в окремій статті. Для
випробовуваного розчину і розчину порівняння роз-
раховують середні значення площ або висот піків
аналізованої речовини. За одержаними середніми зна-
ченнями розраховують концентрацію аналізованої ре-
човини у випробовуваному розчині.
Метод внутрішнього стандарту. Для кожної хромато-
грами спочатку розраховують відношення площі або
висоти піка аналізованої речовини до площі або висо-
ти піка внутрішнього стандарту. Одержані відношення
усереднюють для випробовуваного розчину і розчину
порівняння і за знайденими середніми значеннями
визначають концентрацію аналізованої речовини у
випробовуваному розчині.
КОНТРОЛЬ ДОМІШОК
Дія контролю домішок звичайно використовують такі
підходи.
І. Кількісне визначення домішки з використанням роз-
чину порівняння з відомою концентрацією домішки
(звичайно у варіанті абсолютного калібрування). Та-
кий підхід передбачає однаковий відгук домішки у
присутності й у відсутності основної речовини.
2. Метод внутрішньої нормалізації. Такий підхід перед-
бачає виконання лінійності у широкому діапазоні й
може вимагати врахування відмінностей у відгуках
домішки й основної речовини. Його часто застосову-
ють для визначення суми домішок. При цьому суму
п.іош усіх піків на хроматограмі (без врахування піка
розчинника) беруть за 100 % і вміст кожної конкретної
домішки або суми домішок знаходять як частку площі
піка цієї домішки або суми площ піків домішок у за-
гальній сумі площ усіх піків на хроматограмі.
3. Порівняння з розведеним розчином основної речовини.
4. Метод стандартних добавок. До аналізованої проби
додають відому кількість домішки. За даними хрома-
тографування випробовуваної проби і випробовуваної
проби з добавкою визначають вміст домішки. Для
підвищення точності можливе використання методу
внутрішнього стандарту.
УМОВИ ХРОМАТОГРАФІЧНОГО АНАЛІЗУ
У методиці рекомендується зазначати такі умови
аналізу:
— розміри хроматографічної колонки і матеріал, з
якого вона виготовлена;
— тип нерухомої фази і, якщо необхідно, її ко-
мерційну марку;
— розмір часток нерухомої фази;
— температуру колонки;
— швидкість і склад рухомої фази;
— тип детектора.
Придатність хроматографічної системи. Результати
аналізу вважаються вірогідними, якщо виконуються
вимоги теста "Перевірка придатності хроматографіч-
ної системи". Даний тест звичайно проводять з вико-
ристанням розчинів порівняння.
Якщо немає інших зазначень в окремій статті, хрома-
тографічна система вважається придатною, якщо ви-
конуються такі умови:
— відносні часи утримування зазначених речовин ма-
ють бути близькими до зазначених величин;
— число теоретичних тарілок (ефективність хромато-
графічної системи), розраховане за зазначеним
піком, має бути не менше зазначеної величини;
— коефіцієнт розділення зазначених піків, розрахова-
ний з хроматограм розчину порівняння, має бути не
менше зазначеної величини;
— відносне стандартне відхилення, розраховане для
висоти або площі зазначеного піка або їхніх відно-
шень до висоти або площі піка внутрішнього стан-
дарту з хроматограм розчину порівняння, має бути
не більше зазначеної величини; для розрахунку
відносного стандартного відхилення використову-
ють дані звичайно п’яти паралельних хроматограм;
— коефіцієнт симетрії зазначеного піка, розрахований
з хроматограм розчину порівняння, має бути у ме-
жах, зазначених в окремій статті.
Для забезпечення виконання вимог тесту "Перевірка
придатності хроматографічної системи" допускається
модифікація хроматографічних умов, описаних в ок-
ремій статті.
2.2.32. ВТРАТА В МАСІ ПРИ ВИСУШУВАННІ
Визначення втрати в масі при висушуванні проводять
одним з наведених способів і виражають у відсотках
(маса/маса).
Методика. Зазначену в окремій статті кількість випро-
бовуваної речовини поміщають у зважений бюкс, по-
передньо висушений за умов, описаних для випробо-
вуваної речовини. Речовину сушать до постійної маси
або протягом часу, зазначеного в окремій статті, од-
ним з таких способів:
а) "в ексикаторі": висушування проводять над фос-
фору(У) оксидом Рза атмосферного тиску і кімнат-
ної температури;
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
49
2.2. Фізичні та фізико-хімічні методи
Ь) "у вакуумі": висушування проводять над фосфо-
ру(У) оксидом Р за тиску від 1.5 кПа до 2.5 кПа і
кімнатної температури;
с) "у вакуумі в межах зазначеного температурного
інтервалу": висушування над фосфору(У) окси-
дом Р за тиску від 1.5 кПа до 2.5 кПа і температу-
ри. зазначеної в окремій статті;
б) "в межах зазначеного температурного інтервалу":
висушування у сушильній шафі за температурного
інтервалу, зазначеного в окремій статті;
е) "під високим вакуумом": висушування над фосфо-
ру(У) оксидом Рза тиску не більше 0.1 кПа і темпе-
ратури, зазначеної в окремій статті.
Якщо зазначені інші умови, використовувана методи-
ка повністю описується в окремій статті.
2.2.35. ОСМОЛЯЛЬНІСТЬ
Осмоляльність — це показник, що дозволяє оцінити
сумарний вклад різних розчинених речовин в осмо-
тичний тиск розчину. Наближений розрахунок осмо-
ляльності водного розчину здійснюють за форму-
лою:
= ошФ ,
де:
о — сумарне число іонів, які утворюються з однієї
молекули розчиненої речовини в результаті
дисоціації. Якщо розчинена речовина не ди-
соціює на іони, о=1;
т — модальність розчину, тобто число молів розчи-
неної речовини на кілограм розчинника;
Ф — моляльний осмотичний коефіцієнт, який вра-
ховує взаємодію між іонами протилежного
знака у розчині й залежить від величини т. У
міру ускладнення складу розчину усклад-
нюється і визначення величини Ф.
Одиницею осмоляльності є осмоль на кілограм роз-
чинника (осмоль/кг), але на практиці звичайно вико-
ристовується одиниця міліосмоль на кілограм розчин-
ника (мосмоль/кг).
Осмоляльність визначають за зниженням темпераіу
ри замерзання розчину, якщо немає інших зазначень,
окремій статті. Залежність між осмоляльністю і зн
женням температури замерзання АГ виражаю;
співвідношенням:
• 1000 мосмоль/кг
Прилад. Складовими частинами приладу (осмометра)
— пристрій для охолодження посудини з виміри
вальною кюветою;
— система для вимірювання температури, що ста
дається з чутливого до температури опору (термі.
тора) з відповідним пристроєм для вимірювані»
струму або різниці потенціалів. Вимірювальні'
пристрій може бути градуйованим у градусах зни-І
ження температури або безпосередньо в одинице
осмоляльності;
— як правило, пристрій для перемішування зразка
Методика. Готують стандартні розчини відповідною
Табл. 2.2.35,-1. Установлюють нульове значення на
шкалі приладу, використовуючи воду Р. ПроводяЛ
калібрування приладу, використовуючи стандарт
розчини: поміщають від 50 мкл до 250 мкл стандарт-
ного розчину у вимірювальну кювету і починаютьохЛ
лодження системи. Щоб запобігти переохолодженн'
вимірювальний пристрій, як правило, програмуютьн
роботу при температурах більш низьких, ніж очікув
не кріоскопічне зниження температури.
Відповідний пристрій вказує надосягнсння рівноваг'
Перед кожним вимірюванням кювету обполіскуюЛ
відповідним стандартним розчином.
Ті самі операції здійснюють з випробовуваним розчі
ном. При цьому перед кожним вимірюванням кювет
обполіскують випробовуваним розчином.
Результати або безпосередньо визначають за інкалой І
приладу, або розраховують за виміряним зниження»
температури замерзання. Результати вважаюті
вірогідними, якшо одержане значення осмоляльносп
випробовуваного розчину не виходить за межі знамен,
осмоляльності двох стандартних розчинів, використа-
них для калібрування.
Стандартні розчини для калібрування осмометра
Таблиця 2.2.35.-І
Маса натрію хлориду Р, у грамах на кіло- грам води Р Фактична осмоляльність (мосмоль/кг) Теоретична осмоляльність ідеального розчину (мосмоль/кг) Моляльний осмотич- ний коефіцієнт Кріоскопічне зни- ження температури ("О
3.087 100 105.67 0.9463 0.186
6.260 200 214.20 0.9337 0.372
9.463 300 323.83 0.9264 0.558
12.684 400 434.07 0.9215 0.744
15.916 500 544.66 0.9180 0.930
19.147 600 655.24 0.9157 1.116 |
22.380 700 765.86 0.9140 1.302
50
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
______________________________________________N
Поряд з поняттям "осмоляльність" у практиці викори-
стовується поняття "осмолярність". Аналогічно до ос-
мо.іяльності осмолярність — показник, шо дозволяє
оцінити сумарний вклад різних розчинених речовин в
осмотичний тиск розчину.
Дані показники близькі й відрізняються один від од-
ного лише іншим способом вираження концентрації
розчинів — моляльної і молярної.
Осмоляльність — кількість осмолів на 1 кг розчинника.
Осмолярність — кількість осмолів на 1 л розчину.
Для ідеальних розчинів маса осмоля, у грамах, являє
собою відношення грам-молекулярної маси речовини
до числа часток або іонів, що утворюються при його
розчиненні.
Дія розведених розчинів, близьких до ідеальних, обидві
величини можуть бути розрахованими теоретично.
Осмолярність ідеальних розчинів може бути розахова-
на за формулою:
концентрація речовини кількість часток
Осмолярність =-------------------------------- ,
молекулярна маса
де:
концентрація речовини — кількість розчиненої ре-
човини на літр розчину, у грамах;
кількість часток — число часток або іонів, що ут-
ворюються при розчиненні однієї молекули речовини.
Одиницею осмолярності є осмоль на літр розчину (ос-
моль/л), але на практиці звичайно використовується
одиниця міліосмоль на літр розчину (мосмоль/л).
При підвищенні концентрації розчину взаємодія між
частками речовини зростає і фактична осмолярність
знижується у порівнянні з осмолярністю ідеального
розчину. Теоретичний розрахунок осмолярності роз-
чинів речовин з великою молекулярною масою (на-
приклад, білкових гідролізатів) і висококонцентрова-
них розчинів неможливий. У таких випадках визнача-
ють осмоляльність експериментальним шляхом за
зниженням температури замерзання розчину або за
зниженням тиску пари над розчином. Зниження тем-
ператури замерзання на 1.86 °С і зниження тиску пари
на 0.3 мм рт.ст. при температурі 25 °С відповідає 1 ос-
молю на 1 кг води.
Розчини, що дорівнюють за осмоляльністю (осмо-
лярністю) 0.9 % розчину натрію хлориду, називають
ізотонічними.
Поряд з приладом, описаним вище, для визначення
осмоляльності можна використовувати також осмо-
метри, засновані на вимірюванні тиску пари над роз-
чином. Вони вимагають малого об’єму проби, що уво-
диться (близько 5 мкл), при цьому точність і пра-
вильність визначення порівняна з величинами, одер-
жуваними на приладах, заснованих на вимірюванні
температури замерзання.
У осмометрах, заснованих на вимірюванні зниження
температури замерзання, об’єм проби зразка, який
уводиться, можна варіювати.
Указання значення осмоляльності (осмолярності) у
маркуванні препаратів. Значення осмоляльності (осмо-
лярності) необхідно зазначати на етикетках інфузій-
них розчинів.
ТИТРУВАННЯ У НЕВОДНИХ
РОЗЧИННИКАХ N
Метод кислотно-основного титрування у неводних
розчинниках застосовується для кількісного визна-
чення речовин, що являють собою кислоти, основи
або солі, титрування яких у воді утруднене або немож-
ливе через слабкі кислотно-основні властивості або
малу розчинність.
У неводних розчинниках різко змінюються кислотно-
основні властивості різних речовин. У залежності від
розчинника та сама речовина може бути кислотою, ос-
новою або взагалі не виявляти кислотно-основних влас-
тивостей. Застосування різних розчинників дозволяє уп-
равляти кислотно-основними властивостями речовин.
Вибір розчинника здійснюється на підставі величин
констант титрування КТ або їхніх від’ємних лога-
рифмів рКт. Величина рКт є критерієм можливості і
точності титрування. Чим більше ця величина, тим
кращі умови титрування. Константа титрування
визначається двома основними величинами: констан-
тою дисоціації розчиненої речовини (КА — для кислот,
Кв — для основ) і константою розчинника — іонним
добутком середовища (К{).
— Для титрування кислот:
Кі
Кт = — або рКт = рК= - рКА;
лл
— Для титрування основ:
«і
Кт = „ ‘ або рКт = рК{ - рКв або рКт = рКА;
в
— Для титрування суміші двох кислот:
Кл(і)
кт = абор/Гг = рКА(2} - рКА(Х}
— Для титрування суміші двох основ:
Аі(і)
= КА(2) аб° = ^^4(1) - РКд(2)
Індекси 1 і 2 позначають порядок нейтралізації.
Значення величин іонних добутків для ряду розчинни-
ків і константи дисоціації кислот та основ у воді й різних
неводних розчинниках наведені у Табл. 2, 3, 4. Ці таб-
лиці дозволяють швидко визначати рКт. Задачу вибо-
ру розчинника допомагає розв’язати також лінійна за-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
51
2.2. Фізичні та фізико-хімічні методи
лежність рКт кислот і основ, що належать до однієї
групи хімічних сполук, у воді й неводних розчинни-
ках. Це дозволяє визначати рКт кислот і основ у не-
водних розчинниках, якщо відомі їхні рКт у воді.
Як кислоти можна титрувати: карбонові кислоти, фе-
ноли, барбітурати, сульфаміди, амінокислоти та ін.
Як основи можна титрувати: аміни, азотвмісні гетеро-
циклічні сполуки, аміди, четвертинні амонієві основи
та ін.
Оптимальні умови титрування для слабких кислот до-
сягаються в основних неводних розчинниках, таких
як піридин, диметилформамід; для слабких основ у
кислих неводних розчинниках, таких як оцтова кис-
лота і оцтовий ангідрид.
Солі деяких органічних і мінеральних кислот можуть
бути відтитровані як основи в кислих розчинниках.
У випадку титрування солей галогенводневих кислот
перед титруванням додають звичайно розчин ртуті
окисної ацетату для зв’язування іонів галогенів у спо-
луки, що мало дисоціюють. При використанні оцто-
вого ангідриду як розчинника можливе титрування
солей галогенводневих кислот, переважно хлоридів,
без додавання ртуті окисної ацетату.
Для роздільного титрування сумішей кислот або
сумішей основ використовують диференціюючі роз-
чинники, тобто розчинники з величиною рК,. що зви-
чайно перевищує 15, які не мають виражених кислот-
но-основних властивостей, такі як кетони, нітрили,
нітрометан.
У ряді випадків для титрування застосовують суміші
неводних розчинників, один із яких є апротонним
(бензол, хлороформ та ін.). Присутність апротонного
розчинника зменшує іонний добуток середовища (К,),
що може сприяти поліпшенню умов титрування.
Метод неводного титрування застосовують також д»
кількісного визначення речовин, які містять кар-
бонільну групу, мають слабо виражені кислотно-ос
новні властивості й не піддаються прямому титрувавИ
ню в неводних розчинниках. У цьому випадку про»
дять реакцію оксимування у неводних розчинникахИ
здійснюють кількісне визначення карбоніловмісних,
речовин шляхом титрування надлишку реагенту. Ме-
тод застосовують для кількісного визначення різних
класів органічних сполук: альдегідів, кетонів, хро-|
монів, стероїдних гормонів, глікозидів та ін.
Титрування в неводних середовищах може бути прове-
дене як з індикаторами, так і потенціометрично з вико-|
ристанням як індикаторного скляного або інших, обо-'
ротних до протона електродів. Як електрод порівняння
звичайно використовують хлорсрібний електрод.
При проведенні потенціометричного титрування у не-
водних середовищах електролітичний міст або елек-
трод порівняння заповнюють розчинами калію хлори-1
ду або ЛІТІЮ хлориду у ВІДПОВІДНИХ неводних розчин- і
никах.
При титруванні в основних розчинниках належить!
вживати заходів для захисту титрованого розчину і ти
транту від вуглецю діоксиду, який міститься в повітрі. І
Титрування краще проводити в атмосфері інертного >
газу (азоту, гелію).
У Табл. 1 наведені найбільш часто застосовувані не-
водні розчинники, індикатори і титранти.
Таблиця І
Розчинники, індикатори і титранти, найчастіше застосовувані у неводному титруванні
Розчинники Індикатори 1 Титранти
Кислі Оцтова і мурашина кислоти, оцтовий ангідрид та їх суміші з іншими розчинниками Кристалічний фіолетовий, Судан III, тропеолін 00, метиловий фіолетовий, нейтральний червоний, малахітовий зелений, диметиламіноазобензол Розчин хлорної кислоти в оцтовій кислоті або нітрометані
Основні Диметилформамід, піридин, етилендіамін Тимоловий синій, бромтимоловий синій, а-нафтолбензеїн, о-нітроанілін Розчини натрію гідроксиду, калію гідроксиду, натрію мети лату, літію метил ату, тетраетиламонію гідроксиду в метанолі або в суміші метанолу і бензолу
Диференціюючі Ацетон, діоксан, нітрометан, метил етил кетон, метанол, ізопропанол, трет-бутанол, ди метилсул ьфоксид Метиловий оранжевий, тимоловий синій, нейтральний червоний, метиловий червоний, бромтимоловий синій Розчини кислоти хлористоводневої у метанолі або у гліколевих сумішах; розчини кислоти хлорної у нітрометані. у метанолі або у гліколевих сумішах; розчини, застосовувані при титруванні в основних розчинниках
52
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
Таблиця 2
Величини рК, різних розчинників (рК^ = при температурі від 20 °С до 25 °С
Розчинник рКі
1 Сірчана кислота 3.62
2 Мурашина кислота 6.10
3 Оцтова кислота 14.4
4 Оцтовий ангідрид 14.5
5 Етилендіамін 15.3
б Етиленгліколь 15.6
7 Формамід 16.7
8 Метанол 16.7
9 Пропіленгліколь 16.8
10 Діетиленгліколь 17.5
11 Етанол 19.1
12 Масляна кислота 19.2
13 н-Бутанол 20.1
14 Метилцелозольв 20.7
15 Ізопропанол 22.0
16 Диметилацетамід 23.9
17 Нітрометан 24.0
18 N - Метилпіролідон 24.2
19 Піридин 24.2
20 Диметилсульфоксид 97 % (*) 24.5
21 Диметилформамід 25.3
22 Метилбутилкетон 25.3
23 Сульфолан 25.5
24 Метил етилкетон 25.7
25 Ацетон 25.9
26 Ацетон 90 % (*) 20.3
27 трет-Бутанол 26.8
28 Пропіленкарбонат 29.2
29 Рідкий аміак 29.8
ЗО Ацетон ітрил 32.2
31 Диметилсульфоксид 33.3
(*) - другий компонент - вода
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
53
2.2. Фізичні та фізико-хімічні методи
Величини рКЛ кислот у різній
Кислота Розчинник
Вода Метанол Спирт 95 % Бутанол Ізопропанол трет-Бутанол Етиленгліколь Пропіленгліколь Метилцелозольв Ацетон Ацетон 90 % Метилізобутилкетон Метилетилкетон Формамід
Хлорна 3.94 2.90 2.20
Сірчана 1.44 3.42 5.48
Азотна 0.2 3.17 3.75 4.66
Хлористоводнева 0.8 1.05 1.95 3.10 5.50 8.90 2.47 8.30
Бромистоводнева 0.2 2.0 5.0
п-Толуолсульфонова 3.82
Оцтова 4.75 9.70 10.41 10.35 11.35 14.27 8.32 9.10 11.10 12.55 10.27 16.6 б.9і;
Монохлоронтова 2.86 7.80 8.51 8.50 9.23 12.24 6.05 9.10 11.20 7.60 15.4 4.50 1
Дихлороцтова 1.31 6.30 7.14 7.30 7.80 10.27 4.50 10.20 10.26
Трихлороцтова 0.70 4.90 5.70 6.30 5.90 8.20 8.86 1.46 1
Фенілоцтова 4.31 8.06 8.78 6.57 1
Мурашина 3.75 9.15 8.82 9.70 16.70 5.74 1
Бензойна 4.20 9.52 10.13 10.24 15.10 8.16 8.83 10.70 11.95 9.70 16.6 6.36 1
п-Нітробензойна 3.40 8.40 8.87 9.10 9.60 12.04 10.59 8.09 5.88 її
м- Н ітробензой на 3.46 8.30 9.0 9.15 9.20 10.66 8.03 5.40 1
п-Амінобензойна 4.92
3,5-Динітробензойна 2.80 8.31 10.60 9.63 18.80 9.78
Саліцилова 2.89 7.90 8.60 7.73 9.53 8.90 9.53 7.22 13.0 4.73
Ацетилсаліцилова 3.50 16.30
Нікотинова 4.73 16.60 15.0
Барбітурова 4.01
Винна 3.03 7.40
Лимонна 3.10 10.1
Фенол 9.89 14.2 19.08 22.3
Резорцин 9.20
о-Нітрофеиол 7.17 13.82 10.75
п-Нітрофенол 17.15 11.0 11.0 11.19 14.48 13.52 10.93 8.53
м-Нітрофенол 8.30 12.6
2,4-Динітрофенол 4.02 7.85 8.21 8.36 10.68 8.766 6.45 9.31 4.52
2,5-Динітрофенол 5.22 8.98 8.10 5.94
2,6-Динітрофенол 3.71 7.63 6.77 17.60 4.17
3,5-Динітрофенол 6.70 1083 13.40
Пікринова 0.80 4.80 3.93 4.50 3.70 3.65 3.17 2.18 11.0 3.70 1.33
Барбітал 7.43 12.69 16.8 19.0
Фенобарбітал 7.21 19.20 13.30
Сульфадимезин 7.51 19.60 18.70
Сульфадиметоксин 5.90
Сульфамеразин 7.10
Норсульфазол 6.80
Сульгін 13.00
Метилурацил 9.70
Фторурацил 7.98
Тіоурацил 7.82
Цинхофен 6.22
Рутин 8.96 10.34 13.15
Кверцетин 8.21 9.18 14.41
Лікуразид 9.49 12.30 14.34
Ізосаліпурпозид 7.67 11.66 12.90
Неодикумарин 4.37 5.71 7.37
Зоокумарин 4.70 9.63 7.81
54 ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
оо 22 £ 5 О> = о й оо ро 4^ ь | 8 § <о д 2 Е § О 3: р р5 5 О § о Диметилформаміл
чО О> 04 N4 о 113.28 |11 63 ІЗ р Р О [10.98 о о 04 О 4^ 40 N4 ь Ь 16.40 | § де 3 р р 11.10 1 Г 09'1! ОО о 12.60 | Диметилсульфоксид
| 11.28 І 18.05 1 11.22 й о О 40 діді і з! 15.62 | 1 ЕЕ71 >О 04 О ос р к) о Диметилсульфоксид 97 %
1 06'9 о 1 9Г8 N4 04 О Диметилацетамід
1 9.90 | 16.45 | 14.87 N4 к) 4^ 1 19.71 1 ЕГ91 1 19.67 | 17.09 | о 16.55 | 1 18'81 7.77 | И'9 Пропіленкарбоиат
| 23.4 І—— о | 20.50 04 ОС | 17.50 04 О 1 20.80 м N4 О N5 04 04 О 04 О 1 06'91 19.29 | 18.70 | N4 р О 15.80 | ОО до О N4 М О о Зі 6.20 | 4.60 | 40 О Ацетонітрил
І 10.50 І 19.40 04 О чо | 15.90 N4 р О 1 21.40 N4 р О 17.60 | 17.60 | 1 09'61 ю р о ОГЯ о 20.50 | 0Г8 8.80 | о 2.23 | Нітрометан о £ 3
08’9 | N4 Ю ю 3 о І 12.60 04 О р о 10.50 | 12.30 | N4 О 8.30 | 10.90 | 13.30 | 4.08 | 41-метилпіролідон = І а 7!
І 3.65 9£'9 4^ 3 ос N4 р 04 О р о | 16.20 р. 04 оо 04 > Р6'£ 08'6 8.84 | —- 2.68 | 4.36 | о 4.30 1 к) Піридин
N4 687 2.37 | 2.27 | Рідкий аміак
Ю ’^і о 5.10 | 4.25 | О Оцтова кислота
N4 04 0.34 | 0.89 і 0.58 і 0.28 | Мурашина кислота
N4 N 40 О 1ь> к> Масляна кислота
Ю ОС 40 О ос о ос к> о 4.90 | 06'0 Оцтовий ангідрид
2.2. Фізичні та фізико-хімічні методи
2.2. Фізичні та фізико-хімічні методи
Величини рКА основ у різні
Основа Розчинник
Вода Метанол Етанол 95% Етилен- гліколь Пропілснг- ліколь Метилце- лозольв ХІ-метил- піролідон Мурашина кислота Оцтова кислота Оцтовий ангідрид
Тетраметил гуан іди н 13.60
Гуанозин 12.40 15.0
Піперидин 11.20 11.0 12.51 12.52 11.71 10.40 10.1
Диетиламін 10.90 12.20 9.20 5.19 10 10
Бензиламін °.62 11.30
Цитидин 9.80 13.20 _
Аденін 9.90 13.70
Трибутиламін 9.85 10.10
Триетиламін 10.70 11Д5 8.70 10.20 11.50 _
диметиламін 10.60, 100
н-Бутиламін 10.60
Аденозин 10.55 14.90
Дифеніл гуанідин 10.10 13.60 15.2.6 8.90 10.0
Аміак 9.30 11.70 10 10
Анілін 4.58 6 10 5.70 6.12 6 12 5 49 8.60
Диметиланілін 5.10 4.50 4.40 9.93
Діетиланілін 6.52 10.20 10.60 1
п-Хлоранілін 4.00 4.66 9.27
м-Хлоранілін 3.52 4 35 9.25
о-Нітроанілін 0.29 6.95
м-Нітроанілін 2.50 4.25 6.70
п-Нітроанілін 1.02 1.29 1.01
Піридин 5.15 5.54 4.30 5.92 5.69 5.50 10.0 9.90
п-Толуїдин 5.07 6 60 6.30
м-Толуїдин 4.71 3 20 5 90
а-Піколін 5.95 6.24 5.54 6.38 6.09
а-Нафтиламін 3.92 5.66 5.10 5.05 9.60
Дифе ніл амін 0.90 3.18 7.45
Анетамш 0.48 6.75 8.6(М
Ацетоксим 1 & .8.42
Тіосечовина 0.% 7.75
Ефедрин 9.70 11.29 8 90
Атропін 4.60 11.3.0
Цитозин 9-40 12 50
Новокаїн 8.80 11.30
Промедол 8.40 11.30 1
Морфолін 8.70 10.14 9 19
Тебаїн 8.30
Димедрол 8.20
Платифілін 8.10 11.50
Кодеїн 8.00 8.60 11.40 9.38 5.11 10.80
Морфін 7.80 8.60 9.51 5.08
Гідразин 8.11 11.10
Хінін 8.00 5.23 11.50
Бруцин 7.96
Етилморфін 7.90 11.10
Спазмолітин 7.70 11.60
Пілокарпін 6.80 10.90
Резерпін 6.60 9.54
Наркотин 6.20 7 20
Гідроксиламін 5.97 9.00
Папаверин 5.90 6.92 7.25 10.80
Тифен 6.30 11.30
Уротропін (гексаметилентетрамін) 4.90 9 70 700
Хінолін 4.80 4 58 5.39 5 27 9 86
Амідопірин 4.80 9.13
Дибазол 4.20 9.00
Акрідин 4.11
Теофілін 2.60 6.60
Фенацетин 2.20 6.00
Антипірин 1.51 8.35 9.60
Кофеїн 0.60 5.17 6.30
Теобромін 0.10 5.26 6.10
Сечовина 0.20 4.67 7 65 9.36
Ацетанілід 0.40 6.80 7.30
56
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
о оо о - £ у» 2 її її ЇЇ ЇЇ її 5 £ Г 12.24 ! Ацетон
І 6.04 3.20 1 6.20 1 9.80 1 13.40 1 8.30 ь* о 8ГП о 14.40 6.38 1 р ос 6.94 | 1 07/. 6.20 | 12.40 1 13.44 | 13.48 і Метил- етилкетон
Н чО О о 8.40 | 9.63 | р 40 О 20.30 1 Метил- ізобутил кетон
181 і Пг'і? 2.75 П р к> о О 1 66'6 11.08 1 Формамід Розчі
р о р Ь' О о о і 8.75 ос о І 8.30 О 1 15.18 1 8.20 [ 8.60 чО ІЛ О О 4.36 1 9.45 і 8.90 і 10.50 і р о 9.25 0101 р о 13.65 І Диметил- формаміл ИІІІІИК
і 16.61 о 197 о 10.50 | І 0ГІІ чО О 10.50 І 13.20 І Диметил- сульфо- ксид
чС5 р чО О 11.92 ; 12.08 І 11.15 | 10.56 і 15.90 1 О о 17.91 ? 18.69 1 18.53 І Пропілен- карбонат
о 1 15.80 5-24_ чО О і 180 9171 р 00 6.62 І І П7 7.99 1 11 04 І £0'6 р О 17.20 і 16.86 І 17.96 І 18.35 [ 17.77 17.95 1 18.22 І Нітро- метан
і 9ПІ 19.33 І 10.56 і 16.46 І чО О 18.26 І 18.73 І 18.46 1 18.10 І 18.70 | 18.92 | 23.3 | Ацето- нітрил
2.2. Фізичні та фізико-хімічні методи
2.2. Фізичні та фізико-хімічні методи
ВАЛІДАНІЯ АНАЛІТИЧНИХ
МЕТОДИК І ВИПРОБУВАНЬ*'
У статті описуються процедури, застосовувані для
валідації методик і випробувань2, які включаються до
монографій Державної Фармакопеї України і ана-
літичної нормативної документації на лікарські засо-
би і допоміжні речовини (окремі статті). Оскільки до
окремих статей включаються різні інструментальні й
неінструментальні випробування (ідентифікація, кон-
троль домішок, кількісне визначення і т.п.), вимоги до
валідації випробування залежать від його типу і засто-
совуваного аналітичного методу.
А. ТЕРМІНИ І ВИЗНАЧЕННЯ,
ВИКОРИСТОВУВАНІ ПРИ ВАЛІДАЦІЇ
АНАЛІТИЧНИХ МЕТОДИК
1. Вступ
Валідація аналітичної методики — це експеримен-
тальний доказ того, що методика придатна для роз-
в’язання поставлених завдань.
У даному розділі розглядаються характеристики
аналітичних методик (випробувань), які підлягають
Валідаційні характеристики методик, застосовував
для цілей ідентифікації, контролю домішок і кільки
ного визначення, наведені у Табл. 1.
2. Аналітичні випробування і методики, які підлягають І
валідації
У статті розглядаєт вся проведення валідації для таки
випробувань:
— випробувань на ідентифікацію;
— кількісних випробувань для визначення домішок
— випробувань на граничний вміст3 для контролі
домішок;
— кількісних випробувань для визначення діючоїрі-
човини та інших компонентів (наприклад, коь
сервантів) у субстанціях і готових лікарські
засобах.
Усі аналітичні методики і випробування, які входяті
до монографії і аналітичної нормативної докумен-
тації, мають бути валідовані. Однак для валідації дея-
ких випробувань, наприклад, таких як "Розчинення1
або "Розмір часток", можуть бути потрібними інш
валідаційні процедури, не описані у загальній статті.
валідації (далі "валідаційні характеристики").
Таблиця
Валідаційні характеристики, які розглядаються для різних випробувань і методик
Типи аналітичних методик
Характеристики Ідентифікація Випробування на домішки Кількісне визначення
Кількісні Граничні Розчинення, визначення лише вмісту, активності
Правильність - + - +
Точність: Збіжність Внутрішньолабораторна точність 4- +* - + +*
Специфічність ** + + + +
Межа виявлення - + 1
Межа кількісного визначення - + - -
Лінійність - + - +
Діапазон застосування - + - +
«-» — характеристика звичайно не досліджується;
«+» — характеристика звичайно досліджується;
«*» — у тих випадках, коли проводиться дослідження відтворюваності, дослідження внутрішньолабораторної точності не вимагається;
«**» — недолік специфічності випробування можна компенсувати іншим (іншими) додатковим випробуванням (див. п. 4.2 );
«***» — може бути потрібним у деяких випадках (коли межа визначення і нормована межа вмісту визначуваної домішки близькі).
Гармонізовано з "Керівництвом щодо розробки монографій" Європейської Фармакопеї ("ТесЬпісаі Сінісіе Гот Фе ЕІаЬогаііоп оГтопе
бгаріїь. 2пб ЕФ. РНАКМЕЄКОРА, МотетЬег 1996").
г Випробування — це аналітична методика, описана в окремій статті, у сукупності з вимогами до одержуваних за нею результатів. Ре
зультатом проведення випробуваня є відповідь на питання, відповідає чи ні даний лікарський засіб вимогам окремої статті.
Випробування на граничний вміст домішок — це такі випробування, які регламентують вміст домішок не вище встановленого рівня
58
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
СТИСЛА ХАРАКТЕРИСТИКА ВИПРОБУВАНЬ
Випробування на ідентифікацію призначені для під-
інердження наявності аналізованої речовини у зразку.
Звичайно це досягається шляхом порівняння якихось
п іастивостей (наприклад, спектральних характерис-
тик. хроматографічної поведінки, хімічної реакційної
Ьатності і т.п.) випробовуваного і стандартного зраз-
ків.
Випробування, призначені для контролю домішок, мо-
жуть бути як кількісними, так і граничними. Призна-
чення обох випробувань — охарактеризувати чистоту
ірззка. Для валідації кількісних і граничних випробу-
вань необхідні різні валідаційні характеристики.
Кількісне визначення призначене для визначення
аналізованої речовини у зразку. Така сама валідаційна
процедура може бути застосована до методики
кількісного визначення, пов’язаної з іншим випробу-
ванням (наприклад, у випробуванні "Розчинення").
3. Валідаційні характеристики і вимоги
Набір досліджуваних валідаційних характеристик за-
лежить від призначення аналітичної методики. Типові
валідаційні характеристики:
- правильність;
точність;
- збіжність;
- внутрішньолабораторна точність;
- специфічність;
- межа виявлення;
межа кількісного визначення;
- лінійність;
- діапазон застосування.
Цей перелік треба розглядати як типовий для зазначе-
них випробувань (аналітичних методик). Як правило,
на стадії розробки методики вивчається також
валідаційна характеристика "робасність".
Повторне проведення валідації може бути потрібним
І таких випадках:
- зміна у синтезі лікарської субстанції;
- зміна у складі готового лікарського засобу;
- зміна в аналітичній методиці.
Об’єм проведення повторної валідації визначається
специфікою зміни. Повторна валідація може бути
потрібного і в інших випадках.
4. Словник
4.1. Аналітична методика (апаїуіісаі ргоседиге) — це
спосіб проведення аналізу, тобто детальний виклад
усіх операцій, необхідних для виконання випробуван-
ня. Вона включає в себе опис підготовки випробову-
ваних зразків, стандартів, реактивів; опис використо-
вуваного обладнання із зазначенням параметрів; умо-
ви одержання калібрувальних кривих; використання
розрахункових формул і т п.
4.2. Специфічність (зресі/ісііу) — здатність однозначно
оцінювати аналізовану речовину в присутності інших
компонентів, які можуть бути присутніми у зразку. Це
можуть бути домішки, продукти розкладу, допоміжні
речовини і т.п.
Недолік специфічності випробування може бути ком-
пенсований іншим (іншими) додатковим випробуван-
ням.
Специфічність для різних типів випробувань означає
таке:
Ідентифікація — доказ того, що ідентифіковано саме
аналізовану речовину.
Випробування на домішки — доказ того, що кожне ви-
пробування на домішки дозволяє однозначно характе-
ризувати вмістдомішоку зразку (наприклад, випробу-
вання "Супровідні домішки", "Важкі метали", "Залиш-
кові кількості органічних розчинників" і т.п.).
Кількісне визначення (вміст або активність) — доказ
того, що методика дозволяє точно і правильно встано-
вити вміст або активність аналізованої речовини у
зразку.
4.3. Правильність (ассигасу, ігиепезз) характеризує
ступінь відповідності між відомим справжнім значен-
ням або довідковою величиною і значенням, одержа-
ним за даною методикою.
4.4. Точність (ргесізіоп) аналітичної методики виражає
ступінь близькості (або ступінь розкиду) результатів
для серії вимірів, виконаних за даною методикою на
різних пробах одного і того самого однорідного зраз-
ка. Точність може розглядатися на трьох рівнях:
збіжність, внутрішньолабораторна точність і відтво-
рюваність.
Точність необхідно вивчати на вірогідно однорідних
зразках Однак, якщо однорідний зразок одержати не-
можливо, можна використовувати його розчин або
модельні суміші.
Точність аналітичної методики звичайно характеризу-
ють відхиленням, стандартним відхиленням або віднос-
ним стандартним відхиленням для серії вимірювань.
4.4.1. Збіжність (гереаіаЬПііу) характеризує точність
методики при її виконанні в одних і тих самих умовах
(зокрема, одним і тим самим аналітиком або групою
аналітиків) протягом невеликого проміжку часу.
4.4.2. Внутрішньолабораторна точність (іпіетедіаіе
ргесімоп) характеризує вплив внутрішньолаборатор-
них варіацій: різні дні, різні аналітики, різне облад-
нання і т.п.
4.4.3. Відтворюваність (гергосІисіЬіІііу) характеризує
точність міжлабораторного експерименту.
4.5. Межа виявлення для конкретної аналітичної мето-
дики являє собою мінімальну кількість аналізованої
речовини у зразку, яка може бути виявлена (при цьому
не обов’язково має бути визначене точне значення).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
59
2.2. Фізичні та фізико-хімічні методи
4.6. Межа кількісного визначення для аналітичної ме-
тодики являє собою мінімальну кількість аналізованої
речовини у зразку, яка може бути кількісно визначена
з потрібною правильністю і точністю. Межа кіль-
кісного визначення є валідаційною характеристикою
методик кількісного визначення малих концентрацій
речовин у зразку і розглядається в основному при ви-
значенні домішок і/або продуктів розкладання.
4.7. Лінійність (Ипеагііу) — це здатність методики (у
межах діапазону застосування) давати величини, пря-
мо пропорційні концентрації (кількості) аналізованої
речовини у зразку.
4.8. Діапазоном застосування (ганує) аналітичної ме-
тодики є інтервал між мінімальною і максимальною
концентраціями (кількостями) аналізованої речовини
у зразку (включаючи ці концентрації), для якого пока-
зано, що аналітична методика має потрібну точність,
правильність і лінійність.
4.9. Робасність (гоЬиМпекк) —• це здатність аналітичної
методики не зазнавати впливу малих задаваних (кон-
трольованих) аналітиком змін в умовах виконання ме-
тодики. Робасність є показником надійності методики
при її використанні у зазначених умовах.
В. ПРОВЕДЕННЯ ВАЛІДАЦІЇ АНАЛІТИЧНИХ
МЕТОДИК
1. Вступ
Головним завданням валідації аналітичної методики є
експериментальний доказ того, що дана методика
придатна для досягнення тих цілей, для яких вона
призначена. У звіт з валідації мають бути включені всі
дані, одержані у процесі валідації, і використані для
розрахунків формули з відповідним їхнім обговорен-
ням.
Підходи до проведення валідації методик аналізу
біологічних і біотехнологічних препаратів можуть бу-
ти іншими, ніж зазначено у даній статті.
При проведенні валідації необхідно використовувати
лише стандартні зразки з відомими характеристика-
ми, підтвердженими документально. Необхідний
ступінь їхньої чистоти залежить від завдань, які
розв’язуються при їхньому використанні.
Послідовність розгляду валідаційних характеристик
відбиває процес, за яким може розроблятися і валіду-
ватися аналітична методика. Однак доцільно планува-
ти експеримент так, щоб відповідні валідаційні ха-
рактеристики вивчалися одночасно, наприклад: спе-
цифічність, лінійність, діапазон застосування, пра-
вильність і точність.
2. Специфічність
Дослідження специфічності проводиться при
валідації випробувань на ідентифікацію, контроль
домішок і кількісне визначення. Спосіб підтверджене
ня специфічності залежить від завдань, для розв’язаі
ня яких призначена аналітична методика.
У тих випадках, коли методика недостатньо спе-
цифічна, застосовують поєднання двох або більше
аналітичних методик для досягнення необхідною
рівня вибірності.
2.1. Ідентифікація. Випробування на ідентифікації!
мають забезпечувати можливість розрізняти сполуки
близької будови, які можуть бути присутніми у зразю
разом із визначуваним компонентом. Вибірність не
тодики може бути підтверджена одержанням позитиь-
них результатів (можливо, шляхом порівняння з відо-
мим стандартним зразком) для зразків, які містять
визначуваний компонент, і негативних результатів,
одержаних для зразків, які не містять його. Для
підтвердження відсутності хибнопозитивних резуль-
татів випробування на ідентифікацію може бути пе-
ревірене для речовин з близькою будовою або су-
провідних аналізованій речовині. Вибір потенційно
заважаючих проведенню випробування речовин маї
бути обгрунтований.
2.2. Кількісне визначення і випробування на домішки.!
При валідації хроматографічних методик для підтвер-
дження специфічності мають використовуватися ха- 4
рактерні хроматограми із зазначенням індивідуальним
речовин. Аналогічний підхід використовують і діл
інших методів розділення.
Для хроматографічних методик ступінь розділення
має бути досліджений для відповідних концентрацій]
речовин. Для підтвердження специфічності може бути
використаний ступінь розділення двох речовин, які
найбільш близько елююються.
У разі використання неспецифічного методу кіль-
кісного визначення необхідно застосовувати додат-
кові аналітичні методики і підтверджувати спе- І
цифічність усього комплексу методик. Наприклад, як-
що кількісне визначення проводиться титриметрич-1
ним методом, його можна доповнити відповідним ви-1
пробуванням на домішки.
Для кількісного визначення і для випробувань на
домішки застосовують однакові підходи, описанії
нижче.
2.2.1. Зразки домішок наявні. Для методу кількісного
визначення необхідно підтвердити вибірність визна-І
чення аналізованої речовини у присутності домішок
і/або інших компонентів зразка. Це можна зробити
внесенням до зразка (субстанції або лікарського засо-
бу) домішок і/або інших компонентів зразка у
відповідній концентрації і наступним доказом того
що це не відбилося на одержуваному результаті (шля-
хом порівняння результатів, одержаних на вихідному і
забрудненому зразках).
Для випробувань на чистоту підтвердження вибірності
проводять шляхом забруднення субстанції або готово-
го лікарського засобу відповідними кількостями
домішок і доказу розділення цих домішок як одна від
одної, так і від інших компонентів зразка.
60
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
2.2.2. Зразки домішок відсутні. У випадку, якщо зразки
домішок або продуктів розкладу відсутні, підтвер-
дження специфічності проводять шляхом порівняння
результатів аналізу зразків, що містять домішки або
продукти розкладу, пропонованою методикою й
іншою арбітражною методикою. Як остання може бу-
ти використана фармакопейна методика або інша
валідована методика. Цей підхід передбачає попе-
реднє забруднення зразка продуктами розкладу шля-
хом витримування його у стресових умовах: вплив
світла, тепла, вологості, гідролізу, окиснення і т.п.
При валідації кількісного визначення треба порівняти
результати аналізів, одержаних з використанням ме-
тодики, що валідується, і арбітражної методики.
При валідації випробування на чистоту треба порівня-
ти результати визначення домішок, одержані з вико-
ристанням методики, що валідується, і арбітражної
методики
Для доказу того, що пік аналізованої речовини
відповідає лише одному компоненту, використовують
тести на чистоту піків, наприклад, із використанням
діодно-матричного детектування, мас-спсктрометрії
і т.п.
3. Лінійність
Лінійна залежність має бути досліджена у межах діапа-
зону застосування аналітичної методики. Вона може
бути підтверджена безпосередньо на субстанції (шля-
хом розведення вихідного розчину) або, для лікарсь-
ких засобів, на модельних сумішах із використанням
відповідної процедури.
За одержаними даними будують графік залежності
сигналу як функції концентрації або кількості визна-
чуваного компонента і візуально оцінюють його
лінійність. Якщо лінійна залежність спостерігається,
результати обробляють підхожим статистичним мето-
дом, наприклад, методом найменших квадратів. У де-
яких випадках для одержання лінійності дані треба
піддати попередньому математичному перетворенню.
Мають бути визначені й подані: коефіцієнт кореляції,
точка перетину з віссю ординат, тангенс кута нахилу
прямої і залишкова сума квадратів відхилень, а також
графік з усіма експериментальними даними. Для
оцінки лінійності можуть знадобитися відхилення ек-
спериментальних даних від прямої.
Деякі аналітичні методики, наприклад, імуно-
аналітичні, не показують лінійності ні за яких матема-
тичних перетворений. У таких випадках аналітичний
відклик має бути описаний підхожою функцією кон-
центрації аналізованої речовини у зразку.
Для підтвердження лінійності використовують не
менше п’яти концентрацій. Інші підходи мають бути
обгрунтовані.
4. Діапазон застосування
Діапазон застосування методики залежить від її при-
значення і визначається при вивченні лінійності. У
межах діапазону застосування методика має забезпе-
чувати потрібну лінійність, правильність і точність.
Мінімальні допустимі діапазони застосування мето-
дик:
— Для кількісного визначення лікарських суб-
станцій або лікарських форм — від 80% до 120% від
номінального вмісту.
— Для однорідності дозування — від 70 % до 1 ЗО % від
номінального вмісту, якщо для випробування не
потрібний більш широкий інтервал (наприклад, для
дозованих аерозолей).
Для випробувань на розчинення — ±20 % (абсо-
лютних) від нормованої величини вивільнення. На-
приклад, якщо при контролі вивільнення пролонгова-
них лікарських засобів нормована величина вивіль-
нення становить від 20 % за першу годину і до 90 % за
24 год, діапазон застосування має бути від 0 % до
110 % від номінального вмісту.
— Для визначення домішок — від концентрації, у
якій домішка звичайно виявляється, до 120 % від нор-
мованого вмісту.
Для домішок, які мають надзвичайно сильну дію або
мають токсичний або непередбачуваний фармако-
логічний ефект, межа детектування/кіл вкісного ви-
значення має відповідати тому рівню концентрації, на
якому ці домішки мають контролюватися.
Примітка: при проведенні валідації випробування на
домішки безпосередньо у процесі розробки методики
необхідно визначити діапазон застосування, всере-
дині якого знаходиться гадана межа нормування
домішок.
У випадку, коли кількісне визначення і випробування
на домішки виконуються сумісно як одне випробу-
вання, і використовується лише стандарт основної ре-
човини, відповідний його номінальному вмісту, то
діапазон застосування має охоплювати діапазон кон-
центрації від нормованого вмісту домішки до 120% від
номінального вмісту основної речовини.
5. Правильність
Правильність вивчають у межах діапазону застосуван-
ня аналітичної методики.
5.1. Кількісне визначення
5.1.1. Субстанції. Можуть використовуватися такі
способи визначення правильності:
— застосування аналітичної методики до зразка з
відомим ступенем чистоти, наприклад, до стандартно-
го зразка;
— порівняння результатів аналізу, одержаних із ви-
користанням методики, що валідується, і арбітражно-
го методу, правильність і точність якого відомі (вико-
ристання незалежного методу, див. п. 2.2.);
— висновок про правильність можна зробити після
того, як установлені точність, лінійність і спе-
цифічність.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
5.1.2. Готові лікарські засоби. Можуть використовува-
тися такі способи визначення правильності:
— застосування методики до штучних домішок, до
яких були додані відомі кількості аналізованої речо-
вини;
— у випадку, коли неможливо одержати зразки усіх
компонентів лікарського засобу, можливе застосуван-
ня методу добавок або арбітражної методики, пра-
вильність якої доведена (див. п. 2.2.);
— висновок про правильність можна зробити після
того, як установлені точність, лінійність і спе-
цифічність.
5.2. Домішки (кількісний вміст). Правильність вивча-
ють на зразках (субстанції або готового лікарського за-
собу) з доданою відомою кількістю домішок.
Якщо домішки або продукти розкладання недоступні,
застосовують арбітражний метод (див. п. 2.2.). Якщо
домішки невідомі, чутливість їх визначення може бути
визнаною за таку, що дорівнює чутливості визначення
субстанції. У випадку, якщо чутливість визначення
субстанції і домішки істотно відрізняється, вводять
коефіцієнт перерахунку.
Має бути зазначений конкретний спосіб нормування
вмісту домішок, наприклад, у масових відсотках, у
відсотках відносно площі піка головного компонента
або ін.
5.3. Подання даних. Правильність оцінюють не менш
як для дев'яти визначень для трьох різних концен-
трацій, охоплюючих увесь діапазон застосування, тоб-
то три концентрації і три визначення для кожної
концентрації. Визначення мають включати всі стадії
методики.
Правильність виражають у відсотках знайденого зна-
чення від уведеного або як різницю між середнім і
справжнім значенням з урахуванням відповідних
довірчих інтервалів.
6. Точність
Валідаційна характеристика "точність" вивчається для
методик кількісного визначення.
6.1. Збіжність. Збіжність вивчають, виконуючи:
— не менше дев’яти визначень, охоплюючих діапа-
зон застосування методики (три концентрації/три по-
втори) або
— не менше шести визначень для зразків із вмістом
аналізованої речовини, близьким до номінального.
6.2. Внутрішньолабораторна точність. Установлюють
вплив випадкових факторів на точність аналітичної
методики, що валідується. Типовими досліджуваними
факторами є різні дні, різні аналітики, різне облад-
нання і т.п. При вивченні впливу різних факторів най-
краще використовувати планування експерименту.
6.3. Відтворюваність. Відтворюваність оцінюють шля-
хом проведення міжлабораторних досліджень. Від-
творюваність має бути вивчена при включенні мето-
дики до Фармакопеї.
6.4. Подання даних. При вивченні точності мають по-
даватися: стандартне відхилення, відносне стандартні
відхилення і довірчий інтервал.
7. Межа виявлення
Залежно від того, є методика інструментальною І
неінструментальною, можливі різні підходи ДЛЯ Вк
значення межі виявлення. Використовуються та
підходи.
7.1. Візуальна оцінка. Візуальну оцінку використав}-
ють як для неінструментальних. так і для інструмен-
тальних методів.
Межу виявлення установлюють шляхом аналізу
зразків з відомими концентраціями аналізованої речо-
вини і оцінкою мінімального вмісту, за якого аналізо-
вана речовина надійно визначається.
7.2. Відношення Сигнал/Шум. Цей підхід підхожиіі
тільки до тих методів, для яких спостерігається шуі
базової лінії. Для визначення відношення сигнал/шуь
порівнюють величини сигналів, одержані для кон
трольного досліду і для зразків з низькими концепт
раціями аналізованої речовини. На підставі одержав
них даних установлюють мінімальну концентрацію,І
для якої величина відношення сигнал/шум становить
звичайно від трьох до двох.
7.3. Використання калібрувальної прямої і стандартного
відхилення аналітичного сигналу. Межа виявлення
(МВ) може бути виражена як:
МВ = З.Зо/8, (1)
де:
о — стандартне відхилення сигналу,
8 — тангенс кута нахилу калібрувальної прямої.
Значення тангенса кута нахилу калібрувальної прямо
обчислюють з калібрувальної прямої для аналізована]
речовини. Оцінка стандартного відхилення сигналу
може бути проведена багатьма способами, наприклад,
наступними.
7.3.1. Використання стандартного відхилення сигналу
для контрольного досліду. Вимірюють розмір аналітич-
ного сигналу для необхідного числа зразків, що не
містять аналізованої речовини, і обчислюють стан-
дартне відхилення.
7.3.2. Використання калібрувальної прямої. Одержують
калібрувальну пряму для зразків з вмістом аналізова-
ної речовини, близьким до межі виявлення, і обчис-
люють її параметри. Як стандарт не відхилення 8 у
формулі (1) може бути використане стандартне відхи-
лення вільного члена лінійної залежності.
7.4. Подання даних. Подають значення межі виявлен-
ня із зазначенням способу, використаного для його
визначення. Якшо визначення межі виявлення грун-
тується на відношенні сигнал/шум, подають відпо-
відні хроматограми.
62
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
Якшо значення межі виявлення одержано шляхом об-
ннлень або екстраполяцій, його оцінка має бути під-
тверджена аналізом необхідного числа зразків з вмістом
лімлізованої речовини, близьким до межі виявлення.
8. Межа кількісного визначення
Можливі декілька підходів для визначення межі
Кількісного визначення, які залежать від того, є мето-
лика інструментальною чи неінструментальною. Мо-
жуть бути використані такі підходи:
8.1. Візуальна оцінка. Візуальну оцінку використову-
ють як для неінструментальних, так і для інстру-
ментальних методів.
Межу кількісного визначення установлюють шляхом
аналізу зразків з відомими концентраціями аналізова-
ної речовини і оцінкою мінімального вмісту, за якого
аналізована речовина визначається кількісно з
потрібного правильністю і точністю.
8.2. Віїношення Сигнал/Шум. Цей підхід застосовний
інше до тих методів, для яких спостерігається шум ба-
лвої лінії (див. п. 7.2.). Для визначення відношення
сигнал/шум порівнюють величини сигналів, одержані
для холостого досліду і для зразків з низькими кон-
центраціями аналізованої речовини. На підставі одер-
жаних даних установлюють мінімальну концентрацію,
для якої величина відношення сигнал/шум становить
близько 10:1.
8.3. Використання калібрувальної прямої і стандартного
відхилення сигналу
МВ=10-сг/8, (2)
лс.
о стандартне відхилення сигналу,
8 — тангенс кута нахилу калібрувальної прямої.
Значення тангенса кута нахилу калібрувальної прямої
може бути визначене з калібрувальної прямої для
аналізованої речовини. Оцінка стандартного відхи-
лення сигналу може бути проведена багатьма способа-
ми. наприклад, наступними.
8.3.1. Використання стандартного відхилення сигналу
оля контрольного досліду. Вимірюють величину
аналітичного сигналу для необхідного числа зразків,
шо не містять аналізованої речовини, і обчислюють
стандартне відхилення.
8.3.2. Використання калібрувальної прямої. Одержують
калібрувальну пряму для зразків із вмістом аналізова-
ної речовини, близьким до межі виявлення, і обчис-
люють її параметри. Як стандартне відхилення о у
формулі (2) може бути використане стандартне відхи-
лення вільного члена лінійної залежності.
8.4. Падання даних. Подають значення межі виявлен-
ня із зазначенням способу, використаного для його
визначення. Значення межі визначення має бути
підтверджене аналізом необхідного числа зразків із
вмістом аналізованої речовини, близьким до межі
виявлення.
9. Робасність
Оцінку робасності проводять на стадії розробки мето-
дики з урахуванням типу методики, що вивчається. Ця
оцінка має довести надійність результатів аналізу при
невеликих змінах параметрів методики.
Якщо на результати аналізу впливають умови його
проведення, ці умови мають бути стандартизовані, і до
тексту методики вносять відповідні застереження.
Типові приклади параметрів, які вивчаються:
— стійкість у часі аналітичних розчинів;
— час екстракції.
У випадку рідинної хроматографії:
— рН рухомої фази;
— склад рухомої фази;
— колонки (різні серії і/або постачальники);
— температура;
— швидкість рухомої фази.
У випадку газової хроматографії:
— колонки (різні серії і/або постачальники);
— температура;
— швидкість газу-носія.
10. Перевірка придатності хроматографічної системи
Перевірка придатності системи є складовою частиною
багатьох аналітичних методик. Цей тест грунтується на
уявленні про те, що обладнання, електроніка, аналітичні
операції і аналізовані зразки становлять єдину систему,
яку можна досліджувати як ціле. Параметри, які уво-
дяться до тесту "Перевірка придатності хроматографічної
системи", залежать від використовуваного методу аналізу
й обгрунтовуються дослідженнями з робасності.
С. ВАЛІДАЦІЯ АНАЛІТИЧНИХ МЕТОДИК -
ОСОБЛИВОСТІ її ЗАСТОСУВАННЯ
ДО МЕТОДІВ, ВИКОРИСТОВУВАНИХ
У ФАРМАКОПЕЇ
1. Оптичне обертання (2.2.7)
1.1. Вступ. Обирають розчинник, який дозволяє одер-
жувати максимально можливий кут обертання.
Досліджують стабільність кута обертання випробову-
ваного розчину протягом не менше 2 год. Якщо не-
обхідно, зазначають, що розчин використовують
свіжоприготованим. У необхідних випадках зазнача-
ють час досягнення стабільного значення кута обер-
тання.
Там, де можливо, використовують О-лінію натрію.
1.2. Ідентифікація. Якщо випробовувана речовина яв-
ляє собою енантіомер, для ідентифікації використо-
вують питомий показник оптичного обертання.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
63
2.2. Фізичні та фізико-хімічні методи
Якщо питомий показник оптичного обертання вико-
ристовують лише для цілей ідентифікації, його зна-
чення можна не перераховувати на суху речовину. Рег-
ламентовані межі величини питомого показника ма-
ють враховувати допустимі межі кількісного вмісту
аналізованої речовини і чистоту зразків різного похо
дження. які витримують вимоги відповідної моно-
графії.
Якщо питомий показник оптичного обертання вико-
ристовується також і для контролю чистоти енан-
тіомерів, випробування розділу "Ідентифікація" може
містити посилання: витримує вимоги випробування
"Питоме оптичне обертання".
1.3. Випробування. Питомий показник оптичного
обертання (в окремих випадках кут обертання) може
бути використаний для підтвердження оптичної чис-
тоти енантіомера. Цей метод менш чутливий за метол
рідинної хроматографії (РХ). У випадку, коли вимірю-
вання питомого оптичного обертання призначене для
того, щоб нормувати вміст одного з енантіомерів. не-
обхідно показати, що в умовах методики аналізований
енантіомер має достатню величину оптичного обер-
тання, щоб бути виявленим. Результат визначення по-
дають у перерахунку на суху речовину. Там. де це мож-
ливо, подають дані про вплив потенційних домішок.
Межі питомого показника оптичного обертання уста-
новлюють із урахуванням можливого вмісту домішок.
За відсутності інформації про оптичне обертання
домішок звичайно установлюють межі відхилення
±5 % від середнього значення, одержаного на зразках,
що відповідають усім іншим вимогам окремої статті.
Там. де це можливо, випробовують зразки різного
походження. Корисно також випробовувати зразки з
термінами придатності, близькими до граничного.
Вимірювання оптичного обертання може використо-
вуватися для підтвердження того, що субстанція є ра-
цематом. У таких випадках звичайно установлюють
межі від (- 0.10)° до (+ 0.10)°.
1.4. Кількісне визначення. Оптичне обертання може
використовуватися для кількісного визначення суб-
станції. При цьому необхідно використовувати стан-
дартний зразок з відомою оптичною чистотою.
2. Абсорбційна спектрофотометрія в ультрафіолетовій і
видимій областях спектра (2.2.25)
Має бути доведена придатність вибраних умов визна-
чення, таких як використовувані розчинники та їх
якість, рН розчину і т.п.
Звичайно ультрафіолетова спектрофотометрія має об-
межену специфічність, яку можна підвищити викори-
станням першої і другої похідної спектра.
2.1. Ідентифікація. Ультрафіолетова спектрофото-
метрія сама по собі рідко використовується для іден-
тифікації. Коли цей метод включають у набір випро-
бувань для ідентифікації, необхідно вивчити його спе-
цифічність шляхом порівняння спектрів аналізованої
речовини зі спектрами подібних сполук. Спе-
цифічність методу можна підвищити, якщо викорис-
товувати не абсолютні значення оптичних густин, а
спектральні відношення.
2.2. Випробування на граничний вміст домішок. Якщо
ультрафіолетова спектрофотометрія використо-
вується у випробуваннях на допустимі межі вмісту
домішок, треба показати, що аналізовані домішки да-
ють достатній внесок у вимірювану оптичну густину
За вибраної довжини хвилі має бути встановлена оп-
тична густина, відповідна нормованій концентрації
аналізованої домішки.
2.3. Кількісне визначення. Якшо ультрафіолетова спек-
трофотометрія використовується для кількісного ви-
значення, треба оцінити вплив домішок на світлопо-
глинання. При кількісному визначенні не рекомен-
дується використовувати питомий показник погли-
нання. Якшо питомий показник поглинання все ж за-
стосовується, його значення треба установлювати нг
підставі міжлаборагорного дослідження, використо-
вуючи серії з відомою чистотою. Чистота цих зразків
має оцінюватися з використанням різних методів, які
включають як методи розділення, так і абсолютні ме-
тоди (які не вимагають використання стандартного
зразка).
3. Неінсі рументальні випробування на чистоту і
граничний вміст домішок
3.1. Зовнішній вигляд розчину (2.2.1 і 2.2.2). Це візу-
альні випробування, призначені для оцінки загально
чистоти субстанції і засновані на порівнянні забар-
влення (або опалесценції) випробовуваного розчину
серії еталонів. Часто невідомо, які домішки і в якії
концентрації зумовлюють забарвлення або опалес-
ценцію. У цьому випадку валідація грунтується ні
зіставленні даних, одержаних для різних серій, пода-
них виробником (або виробниками). Якщо домішкі
відомі й доступні, валідацію цього методу проводячі
шляхом порівняння збільш досконалим методом.
3.2. Кислотність і лужність. Це неспецифічне випробу
вання є однією з характеристик чистоти зразка і вико
ристовується для контролю протеолітичних домішок.
3.3. Випробування на граничний вміст аніонів і катіо
нів (2.4). Придатність цих випробувань треба довесті
шляхом використання методу добавок і/або порівняв
ня з іншими, більш досконалими методами.
Сульфатна зола (2.4.14). Це випробування призначені
для визначення суми катіонів металів, присутніх вор
ганічних субстанціях, але не підхоже для неорганічна:
солей органічних сполук. Звичайно межа не має пере
вищувати 0.1 %. Цей метод не потребує валідашї.
Важкі метали (2.4.8). Застосовують різні способі
проведення цього випробування. Звичайно меж;
вмісту важких металів становить 0.001 % (10 ррт) аб
0.002 % (20 ррт), але іноді й 0.0005 % (5 ррт), шозна
ходиться поблизу межі виявлення.
64
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ
2.2. Фізичні та фізико-хімічні методи
Д ь випгіаііії випробування на важкі метали аналізу-
ють випробовуваний зразок і зразок, спеціально за-
жинений свинцем у відповідній концентрації. За-
барв ення випробовуваного зразка має бути меншим,
і забрудненого зразка таким, що дорівнює або
перевищує забарвлення еталона.
і ряду методик, що вимагають спалювання зразка,
Існує небезпека втрат деяких важких металів (таких
як, наприклад, ртуть і свинець у присутності хло-
ридів). У такому разі, коли це можливо, контро-
>ють вміст важких металів методом атомно-аб-
сорбційної спектрометрії або іншим інструменталь-
ні”.! методом.
Якшо відомо, що при синтезі субстанції використо-
внться каталізатор, наприклад, паладій, нікель або
іншії, їх вміст доцільно контролювати колориметрич-
ними або інструментальними методами (наприклад,
ломно-абсорбційна спектрометрія і т.п.)
Ктьорові реакції або реакції осадження. Для окремих
Канонів і аніонів описані граничні випробування, за-
лизані на візуальному порівнянні забарвлення або
опалесценції. При цьому необхідно довести, що:
- забарвлення або опалесценція для нормованих
концентрацій виразно видні;
- знайдена концентрація доданого іона однакова як
ця випробовуваного розчину, так і для розчину
порівняння (як візуально, так і за допомогою методів,
.основаних на вимірюванні поглинання);
- значення оптичної густини розчинів, що містять
50 г<, 100 % і 150 % аналізованої домішки від нормова-
іоїконцентрації, мають значуще відрізнятися;
визначення домішки на рівні нормованої концен-
трації проводять не менше шести разів і обчислюють
сіандартне відхилення. Знайдена концентрація має
сіановити не менше 80 % від уведеної, а відносне
стандартне відхилення — не більше 20 %.
Доцільно провести порівняння результатів гранично-
го випробування з результатами кількісного визначен-
ня з використанням незалежного методу, наприклад,
помно-абсорбційної спектрофотометрії для катіонів
або іонної хроматографії для аніонів. Результати,
одержані двома методами, мають бути близькими.
4. Атомно-абсорбційна спектрометрія (2.2.23)
Метод атомно-абсорбційної спектрометрії (ААС) за-
стосовують для випробувань щодо визначення вмісту
окремих елементів, присутніх у зразку.
4.1. Специфічність. Специфічність даного методу ви-
значається тим, шо атоми аналізованого елемента по-
глинають характеристичне випромінювання віл дже-
рела з строго дискретними довжинами хвиль,
відповідними даному елементу. Однак можливі пере-
шкоди внаслідок як оптичних, так і хімічних ефектів.
Перед початком валідації необхідно виявити такі пе-
решкоди і, якщо можливо, зменшити їх вплив шляхом
використання відповідних засобів.
Ці перешкоди можуть призвести до систематичної по-
хибки при використанні методу прямого калібрування
або до зміни чутливості методу. Основним джерелом
похибки в методі ААС є похибки, пов’язані з проце-
сом калібрування і заважаючим впливом матриці.
4.2. Калібрування. Використання лінійної моделі
калібрування описано у статті 2.2.23 "Атомно-аб-
сорбційна спектрометрія ". Для доведення придатності
лінійної регресійної моделі рекомендується вико-
ристовувати не менше п’яти калібрувальних розчинів.
У деяких випадках можливе використання пара-
болічної моделі калібрування. При цьому також вико-
ристовують не менше п’яти калібрувальних розчинів.
Рекомендується використовувати концентрації
калібрувальних розчинів з рівномірним розподілом
всередині діапазону застосування.
Для кожної концентрації рекомендується виконувати
не менше п’яти вимірювань.
Проблеми з калібруванням часто можуть виявлятися
візуально. Однак калібрувальні графіки самі по собі не
можна використовувати як доказ придатності методу
калібрування. Калібрувальний графік подають у тако-
му вигляді:
а) На графіку відкладають виміряні оптичні густи-
ни як функції концентрацій і будують криву, що опи-
сує цю калібрувальну функцію, разом з її довірчими
інтервалами. Експериментальні точки мають знаходи-
тися у межах довірчого інтервалу побудованої кривої.
Ь) На графіку відкладають залишкові відхилення
(різниці між виміряними й обчисленими за калібру-
вальним графіком оптичними густинами) як функції
концентрації. Ці різниці мають розподілятися навко-
ло осі абспис випадковим чином.
У деяких випадках розкид значень сигналу зростає із
зростанням концентрації, що може бути виявлено з
графіка залишкових відхилень або статистичними ме-
тодами. При цьому найбільша точність може бути до-
сягнута при використанні калібрування з ваговими
множниками. Може бути застосована як лінійна, так і
квадратична вагова функція.
У випадку використання вагової моделі будується
графік зважених різниць (тобто різниць, помножених
на ваги) як функції концентрації у такий спосіб:
а) На графіку відкладають виміряні оптичні густи-
ни як зважені функції концентрацій і будують криву,
що описує цю калібрувальну функцію разом з її
довірчими інтервалами.
Ь) На графіку відкладають зважені залишкові
відхилення (тобто зважені різниці між виміряними й
обчисленими за калібрувальним графіком оптичними
густинами) як функції концентрації.
Необхідно показати, що модель достатньо точно опи-
сує експериментальні дані.
4.3. Ефекти матриці. Якщо для одержання калібру-
вальної функції використовують метод калібрувальної
кривої, необхідно показати, що чутливість для розчи-
ну аналізованого зразка і калібрувальних розчинів од-
накова.
Якшо застосовується калібрування у вигляді прямої
лінії, відмінності у чутливості можуть бути виявлені
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
65
зико-хімічні методи
2.2. Фізшг
----------
Якщо пит^
ристовую^
чення м^/Ь^
ламентс
ють вг,
аналіз. V
ДЖСНу.
граб.
Як'
Р’
калібрувальної прямої,
донних розчинів, і роз-
// стандартної добавки до
Точність оцінки нахилів
дід числа і розподілу точок
л\ побудови обох регресійних
х використовувати достатнє чис-
Тяти) і вибирати концентрації пе-
діапазону калібрування.
м для можливості використання мето-
іьної кривої є незначимість розходжень
ержаних прямих за критерієм Стьюдента.
зходження істотні, використовують метод
[тгних добавок.
*.4. Межа виявлення і межа кількісного визначення
(метод заснований на стандартному відхиленні сигналу
контрольного досліду див.7.3.1 і 8.3.1, розділ В). Вико-
нують контрольні досліди, для яких найкраще вико-
ристовувати розчини "плацебо”, які містять усі компо-
ненти зразка, за винятком визначуваного. Якщо такі
контрольні досліди виконати неможливо, допустимо
використовувати холості розчини, що містять усі реа-
генти і приготовані так само, як і випробовуваний роз-
чин.
Для випробувань на граничний вміст домішок -1
обхідно враховувати таке:
Виявлення. Необхідно уникати використання особ
вих обприскуючих реагентів, якщо у методиці не в |
користовується стандарт нормованої домішки.
Межа виявлення. Якщо використовують кількіс:
інструментальну методику, для визначення межі вігЛ
лення використовують підходи, описані у пунЛ
7 розділу В. Якщо використовують візуальну метол
ку, необхідно показати, що виявляється кількісні
відповідна зазначуваній межі виявлення.
Коефіцієнт перерахунку. Якщо домішки доступні
необхідно показати, що чутливості визначене
домішки і основної речовини близькі. Для випроМ
вань на граничний вміст домішок відмінності у чутли-І
востях можуть бути показані шляхом порівняння мзі
виявлення.
Межа кількісного визначення, лінійність, діапазоні
збіжність. Ці дані необхідно подавати при викорис-
танні інструментальної кількісної ТШХ.
5.1.2. Рідинна хроматографія (2.2.29)
5. Розділювальні методи
ІДЕНТИФІКАЦІЯ
5.1. Хроматографічні методи. Різноманітні хромато-
графічні методи можуть використовуватися для іден-
тифікації, контролю домішок і кількісного визначен-
ня. Нижче описані особливості валідації даних ме-
тодів.
5.1.1. Тонкошарова хроматографія (2.2.27)
Специфічність. Для тестів ідентифікації звичайно не
можна домогтися специфічності, використовуючи
тільки тонкошарову хроматографію (ТШХ) саму по
собі, однак достатня вибірність може бути досягнута
при поєднанні ТШХ з іншими методами. Якщо для
граничного випробування вибірність недостатня, ви-
користовують додаткове(і) випробування для контро-
лю домішки (домішок), зона якої не була відділена від
інших зон. Необхідно довести вибірність сукупності
використовуваних методик. Для випробувань іден-
тифікації поліпшення вибірності може бути досягнуте
при використанні обприскування реактивом, який
дозволяє розрізняти близькі сполуки за кольором.
Стаціонарна фаза. Необхідно показати, що дане ви-
пробування придатне для проведення аналізу на плас-
тинках одного типу, але різного походження.
Тест "Перевірка придатності хроматографічної систе-
ми". Такий тесі звичайно проводиться для підтвер-
дження розділення двох сполук, які близько елюю-
ються, однією з яких є аналізована речовина. Не-
обхідно довести, що розділення вибраних сполук га-
рантує придатність системи для досягнення поставле-
них цілей.
Специфічність. Для випробувань ідентифікації звичайЯ
но не можна домогтися специфічності, використову- І
ючи лише рідинну хроматографію саму но собі, п роті І
достатня вибірність може бути досягнута прі 1
поєднанні рідинної хроматографії з іншими метода-!
ми. Вибірність має бути показана для часів утримував-1
ня, відносних часів утримування або для коефіцієнтів!
ємності для аналізованої речовини і близьких за було-1
вою речовин. Ці дані необхідно подавати для
декількох стаціонарних фаз одного типу.
ВИЗНАЧЕННЯ НА ГРАНИЧНИЙ ВМІСТ ДОМІШОК
Специфічність. Вибірність розділення. Має бути по-
казано розділення відомих і можливих домішок з ос-
новною речовиною і, якщо можливо, між собою. Спе-
цифічність може бути підтверджена при використанні
для детектування мас-спектрометра. Домішки, які не
відділяються від основної речовини, мають контролю-
ватися іншим методом. Необхідно подавати дані про
час утримування, відносний час утримування або ко-
ефіцієнт ємності для основної речовини і домішок. Ці
дані мають подаватися для декількох стаціонарних фаз
одного типу.
Вибірність детектуючої системи. Вибір детектора і
умов детектування має бути обгрунтований. Спе-
цифічність може бути підтверджена, наприклад, при
використанні для детектування мас-спектрометра.
Коефіцієнт перерахунку. Якщо домішки доступні, не-
обхідно показати, що чутливості визначення домішки
і основної речовини близькі (при використанні УФ-
детектування — за вибраної довжини хвилі детекту-
66
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.2. Фізичні та фізико-хімічні методи
жим. це необхідно показати і для інших типів детек-
ІГОрііі - наприклад, рефрактометричного або кондук-
... грішного). Якшо відношення чутливостей відо-
мім домішки і основної речовини виходить за межі
1)5-1 2 і якшо допустима межа вмісту цієї домішки
і 0.1 г., необхідно використовувати коефіцієнт
Ирервхунку або стандарт нормованої домішки як
нинішній стандарт.
Межа виявлення або кількісного визначення. Ці межі
м.іють визначатися для методу зовнішнього стандарту
при використанні розведень випробовуваної суб-
станції або при використанні стандарту домішки. Як-
шо пік домішки виходить у безпосередній близькості
щ піка субстанції (особливо якщо безпосередньо за
ним), межа виявлення або кількісного визначення має
і "іновлюватися за цією домішкою. Метод, описаний
і пункті 7 розділу В, придатний для обчислювання
’і ші зазначених меж
Стабільність. Необхідно подавати дані, що підтвер-
• кують термін придатності випробовуваного розчину
і розчину порівняння. Також мають подаватися дані
іро стабільність рухомої фази.
Ступінь витягу. При використанні екстракції необхідно
іивчити ступінь витягу відомих і доступних домішок за
оптимальних умов. Необхідно подати дані, які підтвер-
чують, шо екстракція забезпечує достатню точність.
Одержання похідних. Якщо використовують перед- або
піс.іяколонкове одержання похідних, необхідно уста-
। увити оптимальні умови реакції (час, температура та
ін) і дослідити стабільність одержаних похідних.
Тест "Перевірка придатності хроматографічної систе-
ми". Як описано для ТШХ. Використання співвідно-
шення сигнал/шум вимагається лише тоді, коли межа
визначення і нормована межа вмісту домішки близькі
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Специфічність. Це бажана, але не основна вимога. Як-
шо метод не специфічний, то можливість його вико-
ристання забезпечується низьким рівнем вмісту
домішок, які контролюються іншим випробуванням.
Тест "Перевірка придатності хроматографічної систе-
ми". Як описано для ТШХ.
5.3.1. Газова хроматографія (2.2.29).
ІДЕНТИФІКАЦІЯ
Специфічність. Як описано для рідинної хроматографії.
ВИПРОБУВАННЯ НА ГРАНИЧНИЙ ВМІСТ ДОМІШОК
Специфічність. Як описано для рідинної хроматографії.
Коефіцієнт перерахунку. Як описано для рідинної хро-
матографії. Мають подаватися коефіцієнти перера-
хунку нормованої домішки відносно основної речови-
ни. Це особливо важливо при використанні селектив-
них детекторів, таких як детектор з електронного за-
хвату і т.п.
Межі виявлення і кількісного визначення. Як описано
для рідинної хроматографії.
Стабільність. Як описано для рідинної хроматографії.
Одержання похідних. Як описано для рідинної хрома-
тографії.
Внутрішній стандарт. Необхідно показати, шо за вибра-
них умов пік внутрішнього стандарту не перекриваєть-
ся з піками можливих домішок або основної речовини.
Ступінь витягу. Як описано для рідинної хроматографії.
ТЕСТ "ПЕРЕВІРКА ПРИДАТНОСТІ ХРОМАТОГРАФІЧНОЇ
СИСТЕМИ"
Нижче наводяться деякі особливості, які необхідно
враховувати для даного тесту.
Відношення Сигнал/Шум. Звичайно визначають для
сигналів, що дорівнюють або дещо більших за межу
виявлення і межу кількісного визначення
Ступінь розділення. Визначають для піка аналізованої
речовини і найближчого піка домішки або для піка ана-
лізованої речовини і піка внутрішнього стандарту. Якщо
коефіцієнт асиметрії відрізняється від прийнятого діапа-
зону (0.8-1.2), доцільно нормувати його межі (2.2.29).
Це особливо важливо для тих випадків, коли використо-
вуються набивні колонки або пік нормованої домішки
елююється безпосередньо за піком основної речовини.
Там, де можливо, підтверджують виконання випробу-
вання з використанням колонок подібного типу.
Метод аналізу рівноважної парової фази. Цей метол за-
стосовують для випробування легколетких речовин.
Необхідно показати, що вибрані температура і час по-
переднього нагрівання посудин з випробовуваним
зразком забезпечують установлення рівноваги. Треба
вивчити вплив матриці. Валідація методу полягає в
проведенні повторних випробувань зразка. При цьому
після кожного вводу проби газова фаза у посудині
заміщується і посудини знову врівноважуються перед
новим вводом газової фази. За одержаними даними
будують графік залежності логарифмів площ піків
аналізованої речовини від числа екстракцій і розрахо-
вують його характеристики. Близькість коефіцієнта
кореляції одержаної залежності до 1 0 засвідчує пра-
вильність вибору умов випробування. Ефекти впливу
матриці можна усунути при використанні методу
стандартних добавок.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Специфічність. Як описано для рідинної хромато-
графії.
Тест "Перевірка придатності хроматографічної систе-
ми". Як описано для рідинної хроматографії.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
67
2.3. Ідентифікація
2.3. ІДЕНТИФІКАЦІЯ
2.3.1. РЕАКЦІЇ ІДЕНТИФІКАЦІЇ НА ІОНИ
І ФУНКЦІОНАЛЬНІ ГРУПИ
АЛКАЛОЇДИ
Декілька міліграмів або зазначену в окремій статті
кількість випробовуваної субстанції розчиняють у
5 мл води Р, додають кислоту хлористоводневу розведе-
ну Р до кислої реакції розчину (2.2.4), потім 1 мл роз-
чину калію йодвісмутату Р; відразу утворюється оран-
жево-червоний осад.
АЛЮМІНІЙ
Близько 15 мг випробовуваної субстанції розчиняють
у 2 мл води Р. До одержаного розчину або 2 мл розчи-
ну, зазначеного в окремій статті, додають близько 0.5 мл
кислоти хлористоводневої розведеної Р і близько 0.5 мл
реактиву тіоацетаміду Р; осад не утворюється. Потім
додають краплями розчин натрію гідроксиду розведе-
ний Р; утворюється гелеподібний білий осад, який
розчиняється при наступному додаванні розчину
натрію гідроксиду розведеного Р. До одержаного роз-
чину поступово додають розчин амонію хлориду Р; зно-
ву утворюється гелеподібний білий осад.
АМІНИ АРОМАТИЧНІ ПЕРВИННІ
Випробовуваний розчин, зазначений в окремій статті,
підкислюють кислотою хлористоводневою розведеною Р,
додають 0.2 мл розчину натрію нітриту Р і через 1-2 хв
додають 1 мл розчину ф-нафтолу Р; з’являється інтен-
сивне оранжеве або червоне забарвлення і, як прави-
ло, утворюється осад такого самого кольору.
АМОНІЮ СОЛІ
До випробовуваного розчину, зазначеного в окремій
статті, додають 0.2 г магнію оксиду Р. Крізь рідину про-
пускають струмінь повітря і газ, шо виходить, спрямо-
вують у суміш 1 мл 0.1 М розчину кислоти хлористо-
водневої і 0.05 мл розчину метилового червоного Р; за-
барвлення індикатора переходить у жовте. Потім до-
дають 1 мл свіжоприготованого розчину 100 г/л
натрію кобальтинітриту Р; утворюється жовтий осад.
АМОНІЮ СОЛІ Й СОЛІ ЛЕТКИХ ОСНОВ
Близько 20 мг випробовуваної субстанції розчиняють
у 2 мл води Р. До одержаного розчину або до 2 мл роз-
чину, зазначеного в окремій статті, додають 2 мл роз-
чину натрію гідроксиду розведеного Р. При нагріванні
розчину виділяються пари, які виявляються за запа-
хом і лужною реакцією (2.2.4).
АРСЕН кр
.ір
а) 5 мл випробовуваного розчину нагрівають на і .р
дяній бані з рівним об’ємом реактиву гіпофосфіту ш
утворюється коричневий осад. ш
___________________________________________і п<
Ь) Арсен(ПІ) (арсеніти). До 0.3 мл розчину, що міспи н
випробовувану субстанцію в кількості, еквіваленти ।
близько 30 мг арсеніт-іона (АТ)3), додають 0.5 Ц
кислоти хлористоводневої розведеної Р і 0.1 мл роччл к
натрію сульфіду Р; утворюється жовтий осад, нерш .-
чинний у кислоті хлористоводневій концентрованійі |
розчинний у розчині аміаку Р. Ик
с) Арсен(У) (арсенати). До 0.3 мл розчину, що містиі
випробовувану субстанцію в кількості, еквівалентні
близько 1 мг арсенат-іона (АЮд-), додають по 1N
розчину 100 г/л амонію хлориду Р, розчину аміаку!
розчину 100 г/л магнію сульфату Р; утворюється біли
кристалічний осад, розчинний у кислоті хлористою
невій розведеній Р (відмінність від арсенітів).
АЦЕТАТИ
а) Випробовувану субстанцію нагрівають з рівно
кількістю кислоти щавлевої Р; виділяється кислотної
това, яка виявляється за запахом і кислою реакцій
(2.2.4).
Ь) Близько ЗО мг випробовуваної субстанції розчиня-
ють у 3 мл води Р. До одержаного розчину або до 3 ш
розчину, зазначеного в окремій статті, послідовно до-
дають 0.25 мл розчину лантану нітрату Р, 0.1 м?
0.05 Мрозчину йоду і 0.05 мл розчину аміаку розведено-
го Р2. Суміш обережно нагрівають до кипіння; протя-
гом декількох хвилин утворюється синій осад або з’яв-
ляється синє забарвлення.
___________________________________________л
с) До 2 мл нейтрального розчину, що містить випробо-
вувану субстанцію в кількості, еквівалентній близько
20-60 мг ацетат-іона (СН3СОО‘), додають 0.2 мл роз-
чину 30 г/л заліза(ІІІ) хлориду Р; з’являється червоно-
буре забарвлення, яке зникає при додаванні кислот
мінеральних розведених.
сі) 2 мл розчину, що містить випробовувану субстанцію
в кількості, еквівалентній близько 20-60 мг ацетат-іона
(СН3СОО~), нагрівають з рівною кількістю кислоти
сірчаної концентрованої Р і 0.5 мл 96 % спирту Р; утво-
рюється етилацетат, який виявляється за запахом.
АЦЕТИЛ
Близько 15 мг або зазначену в окремій статті кількість
випробовуваної субстанції поміщають у пробірку за-
вдовжки близько 180 мм і зовнішнім діаметром 18 мм
і додають 0.15 мл кислоти фосфорної Р. Пробірку за-
68
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.3. Ідентифікація
фіімють пробкою, крізь яку пропущено невеличку
цюбірку завдовжки близько 100 мм і зовнішнім діаме-
ірон 10 мм, яка містить воду Р і виконує роль холо-
пі іьника. На зовнішню поверхню меншої пробірки
мішають 1 краплю розчину лантану нітрату Р. Як-
ію субстанція відносно легко гідролізується, пристрій
помішають на 5 хв у водяну баню, потім виймають
меншу пробірку. Для субстанцій, які важко гідролізу-
єшся, суміш повільно нагрівають на відкритому по-
,м'ї до кипіння. Краплю знімають, змішують на фар-
оровііі пластинці з 0.05 мл 0.01 М розчину йоду. На
* рай краплі наносять 0.05 мл розчину аміаку розведено-
>Р2:через 1-2 хв у місці з’єднання двох крапель з’яв-
іиється синє забарвлення, яке посилюється і збері-
гаїться протягом короткого проміжку часу.
БАРБІТУРАТИ (ЗА ВИНЯТКОМ КІ-ЗАМ1ЩЕНИХ)
Близько 5 мг випробовуваної субстанції розчиняють
у З мл метанолу Р, додають 0.1 мл розчину, що містить
1001 л кобальту нітрату Рі 100 г/л кальцію хлориду Р,
перемішують і додають при струшуванні 0.1 мл розчи-
<іг натрію гідроксиду розведеного Р; з’являється фіоле-
во-синє забарвлення і утворюється осад.
БЕНЗОАТИ
аі До 1 мл розчину, зазначеного в окремій статті, дода-
ють 0.5 мл розчину заліза(ІН) хлориду РІ; утворюється
бпдо-жовтий осад, розчинний в ефірі Р.
Ь) 0.2 г випробовуваної субстанції, якщо необхідно,
здрібненої, поміщають у пробірку, змочують 0.2 мл або
0.3 мл кислоти сірчаної Р. обережно нагрівають дно
пробірки; на внутрішніх стінках пробірки з’являється
білий наліт.
о 0.5 г випробовуваної субстанції розчиняють у 10 мл
води Р. До одержаного розчину або до 10 мл розчину,
«значеного в окремій статті, додають 0.5 мл кислоти
.лористоводневої Р; утворюєт ься осад, який після пе-
рекристалізації з теплої води Р і висушування у ваку-
умі має температуру плавлення (2.2.14) від 120 °С
до 124 °С
БРОМІДИ
а) Наважку випробовуваної субстанції, еквівалентну
близько 3 мгбромід-іона (Вг“), розчиняють у 2 мл во-
ди Р. Одержаний розчин або 2 мл розчину, зазна-
ченого в окремій статті, підкислюють кислотою азот-
ною розведеною Р, додають 0.4 мл розчину срібла нітра-
ту РІ, перемішують і відстоюють; утворюється світло-
жовтий сирнистий осад. Осад відокремлюють центри-
фугуванням і промивають трьома порціями води Р
по 1 мл кожна. Ці операції проводять швидко у захи-
щеному від яскравого світла місці, при цьому допус-
кається, щоб рідина над осадом не була уповні прозо-
рою. Одержаний осад суспендують у 2 мл води Р і до-
дають 1.5 мл розчину аміаку Р; осад повільно розчи-
няється.
Ь) Наважку випробовуваної субстанції, еквівалентну
близько 5 мг бромід-іона /Вг-), або кількість суб-
станції, зазначену в окремій статті, поміщають у неве-
лику пробірку, додають 0.25 мл води Р, близько 75 мг
свинцю(ІР) оксиду Р, 0.25 мл кислоти оцтової Рі обе-
режно струшують. Верхню внутрішню частину
пробірки висушують за допомогою фільтрувального
паперу і залишають на 5 хв. Смужку фільтрувального
паперу необхідного розміру імпрегнують, уміщуючи її
край у краплю розчину фуксину знебарвленого Р і не-
гайно поміщають імпрегновану частину в пробірку.
Протягом 10 с біля нижнього краю фільтрувального
паперу з’являється фіолетове забарвлення, яке чітко
відрізняється від червоного забарвлення фуксину, що
спостерігається у верхній імпрегнованій частині
смужки паперу.
______________________________________________N
с) До 1 мл розчину, що містить випробовувану суб-
станцію у кількості, еквівалентній близько 2-30 мг
бромід-іона (Вг~), додають 1 мл кислоти хлористовод-
невої розведеної Р, 0.5 мл розчину (свіжоприготованого)
50 г/л хлораміну Р, 1 мл хлороформу Р і збовтують; хло-
роформний шар набуває жовто-бурого забарвлення.
ВІСМУТ
а) 0.5 г випробовуваної субстанції розчиняють у 10 мл
кислоти хлористоводневої розведеної Р. Одержаний
розчин або 10 мл розчину, зазначеного в окремій
статті, кип’ятять протягом 1 хв, охолоджують і, якщо
необхідно, фільтрують До 1 мл одержаного розчину
додають 20 мл води Р; утворюється білий або світло-
жовтий осад, колір якого після додавання від 0.05 мл
до 0.1 мл розчину натрію сульфіду Р змінюється на ко-
ричневий.
Ь) Близько 45 мг випробовуваної субстанції розчиня-
ють у 10 мл кислоти азотної розведеної Р. Одержаний
розчин або 10 мл розчину, зазначеного в окремій
статті, кип’ятять протягом 1 хв, охолоджують і, якщо
необхідно, фільтрують. До 5 мл одержаного розчину
додають 2 мл розчину 100 г/л тіосечовини Р; з’яв-
ляється жовтувато-оранжеве забарвлення або утво-
рюється оранжевий осад. Потім додають 4 мл розчину
25 г/л натрію фториду Р; розчин не знебарвлюється
протягом 30 хв.
ЗАЛІЗО
а) Наважку випробовуваної субстанції, еквівалентну
близько 10 мгзаліза-юна (Ее2+), розчиняють в 1 мл во-
ди Р. До одержаного розчину або до 1 мл розчину, за-
значеного в окремій статті, додають 1 мл розчину калію
фериціаніду Р; утворюється синій осад, нерозчинний
при додаванні кислоти хлористоводневої розведеної Р.
9
Ь) Наважку випробовуваної субстанції, еквівалентну
близько 1 мг заліза-іона (Ее3+), розчиняють у 30 мл во-
ди Р. До одержаного розчину або до 3 мл розчину, за-
значеного в окремій статті, додають 1 мл кислоти хло-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
69
2.3. Ідентифікація
ристоводневої розведеної Р і 1 мл розчину калію
тіоціанату Р; з’являється червоне забарвлення.
Відбирають дві порції одержаного розчину по 1 мл
кожна. До однієї порції додають 5 мл спирту ізоаміло-
вого Рабо 5 мл ефіру Р, струшують і залишають до роз-
шарування; органічний шар набуває рожевого за-
барвлення. До другої порції додають 2 мл розчину
ртуті(П) хлориду Р; червоне забарвлення розчину
зникає.
с) Наважку випробовуваної субстанції, еквівалентну
не менше 1 мгзаліза-іона (Ее3+), розчиняють у 1 мл во-
ди Р. До одержаного розчину або до 1 мл розчину, за-
значеного в окремій статті, додають 1 мл розчину калію
фероціаніду Р; утворюється синій осад, який не розчи-
няється при додаванні 5 мл кислоти хлористоводневої
розведеної Р.
ЙОДИДИ
а) Наважку випробовуваної субстанції, еквівалентну
близько 4 мг йодид-іона (1 ), розчиняють у 2 мл води Р.
Одержаний розчин або 2 мл розчину, зазначеного
в окремій статті, підкислюють кислотою азотною роз-
веденою Р, додають 0.4 мл розчину срібла нітрату Р1,
перемішують і відстоюють до утворення світло-жовто-
го сирнистого осаду. Осад відокремлюють центрифу-
гуванням і промивають трьома порціями води Р по
1 мл кожна. Цю операцію проводять швидко в захи-
щеному від яскравого світла місці, при цьому допус-
кається, щоб рідина над осадом не була уповні прозо-
рою. Осад суспендують у 2 мл води Р і додають 1.5 мл
аміаку Р; осад не розчиняється.
Ь) До 0.2 мл розчину випробовуваної субстанції, що
містить близько 5 мг йодид-іона (Г) в 1 мл, або до
0.2 мл розчину, зазначеного в окремій статті, додають
0.5 мл кислоти сірчаної розведеної Р, 0.1 мл розчину
калію діхромату Р, 2 мл води Р, 2 мл хлороформу Р,
струшують протягом кількох секунд і залишають до
розшарування; хлороформний шар набуває фіолето-
вого або фіолетово-червоного забарвлення.
КАЛІЙ
а) 0.1 г випробовуваної субстанції розчиняють у 2 мл
води Р. До одержаного розчину або до 2 мл розчину, за-
значеного в окремій статті, додають 1 мл розчину
натрію карбонату Р і нагрівають; осад не утво-
рюється. До гарячого розчину додають 0.05 мл розчину
натрію сульфіду Р; осад не утворюється. Розчин охо-
лоджують у льодяній воді, додають 2 мл розчину
150 г/л кислоти винної Р і відстоюють; утворюється
білий кристалічний осад.
Ь) Близько 40 мг випробовуваної субстанції розчиня-
ють в 1 мл води Р. До одержаного розчину або до 1 мл
розчину, зазначеного в окремій статті, додають 1 мл
кислоти оцтової розведеної Р і 1 мл свіжоприготовано-
го розчину 100 г/л натрію кобальтинітриту Р; відразу
утворюється жовтий або оранжево-жовтий осад.
с) Сіль калію, внесена у безбарвне полум’я, забарвлю
його у фіолетовий колір або при розгляданні мере
синє скло — у пурпурово-червоний.
КАЛЬЦІЙ
а) До 0.2 мл нейтрального розчину, шо містить випро-
бовувану субстанцію в кількості, еквівалентній близь-
ко 0.2 мг кальцій-іона (Са2+) в 1 мл, або до 0.2 мл роз- |
чину, зазначеного в окремій статті, додають 0.5 мл роз
чину 2 г/л гліоксальгідроксіанілу Р у спирті Р, 0.2 м.'
розчину натрію гідроксиду розведеного Р і 0.2 мл роз
чину натрію карбонату Р. Суміш струшують з 1 м
або 2 мл хлороформу Р і додають від 1 мл до 2 м І
води Р; хлороформний шар набуває червоного забар-1
влення.
Ь) Близько 20 мг або зазначену в окремій статті кіль-
кість випробовуваної субстанції розчиняють у 5 мл кис-1
лоти оцтової Р. До одержаного розчину додають 0.5 мл
розчину калію фероціаніду Р: розчин залишається про-1
зорим. До розчину додають близько 50 мг амоніюхлорі1
ду Р; утворюється білий кристалічний осад.
_____________________________________________А
с) До 1 мл розчину, що містить випробовувану суб
станцію у кількості 2-20 мг кальцій-іона (Са2+), дода-
ють 1 мл розчину 40 г/л амонію оксалату Р: утво-
рюється білий осад, нерозчинний у кислоті оцтовій
розведеній Р \ розчині аміаку Р, розчинний у розведе-
них мінеральних кислотах.
сі) Сіль кальцію, змочена кислотою хлористоводневою Р
і внесена у безбарвне полум’я, забарвлює його в оран-
жево-червоний колір.
КАРБОНАТИ Й ГІДРОКАРБОНАТИ
а) 0.1 г випробовуваної субстанції поміщають і
пробірку і суспендують у 2 мл води Р. До одержаної су-
спензії або до 2 мл суспензії, зазначеної в окремій
статті, додають 3 мл кислоти оцтової розведеної Р.
Пробірку відразу закривають притертою пробкою зі
скляною трубкою, двічі вигнутою під прямим кутом;
спостерігається бурхливе виділення бульбашок газу
без кольору і запаху. Пробірку обережно нагрівають!
пропускають газ, що виділяється, крізь 5 мл розчину
барію гідроксиду Р; утворюється білий осад, що розчи-
няється при додаванні надлишку кислоти хлористо-
водневої Р1.
_____________________________________________N
Ь) 0.2 г випробовуваної субстанції розчиняють у 2 мл
води Р. До одержаного розчину додають 0.5 мл насиче-
ного розчину магнію сульфату Р; утворюється білий
осад (відмінність від гідрокарбонатів, розчини яких
утворюють осад лише при кип’ятінні суміші).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.3. Ідентифікація
, Примітка. Для одержання насиченого розчину маг-
ьфату до 100 г магнію сульфату Р додають
,(11 мл води Р і залишають на 24 год при частому збов-
I жуванні. Розчин фільтрують.
сі '1.2 г випробовуваної субстанції розчиняють у 2 мл
а Р. До одержаного розчину додають 0.05 мл розчи-
молфталеїну Р; з’являється червоне забарвлення
(цимінність від гідрокарбонатів, розчини яких зали-
ваються безбарвними).
КСАНТИНИ
До кількох міліграмів або до зазначеної в окремій
Статті кількості випробовуваної субстанції додають
НІ \п розчину водню пероксиду концентрованого Р,
II ’ мл кислоти хлористоводневої розведеної Р і упарю-
ють на водяній бані до одержання сухого жовтувато-
червоного залишку. До залишку додають 0.1 мл аміаку
рамеденого Р2; колір залишку змінюється на червоно-
фппетовий.
ЛАКТАТИ
Наважку випробовуваної субстанції, еквівалентну
близько 5 мг кислоти молочної, розчиняють у 5 мл во-
іи Р. До одержаного розчину або до 5 мл розчину, за-
значеного в окремій статті, додають 1 мл бромної во-
Лі Р, 0.5 мл кислоти сірчаної розведеної Р і нагрівають
на водяній бані, періодично перемішуючи скляною
паличкою до знебарвлювання розчину. До розчину до-
аають 4 г амонію сульфату Р і перемішують, додають
краплями, не перемішуючи, 0.2 мл розчину 100 г/л
натрію нітропрусиду Ру кислоті сірчаній розведеній Р,
обережно додають, також не перемішуючи, 1 мл роз-
чину аміаку концентрованого Р\ відстоюють протягом
Ч хв; на межі двох рідин утворюється темно-зелене
кільце.
МАГНІЙ
Близько 15 мг випробовуваної субстанції розчиняють у
2 мл води Р. До одержаного розчину або до 2 мл розчи-
ну зазначеного в окремій статті, додають 1 мл розчину
аміаку розведеного Р1; утворюється білий осад, що роз-
чиняється при додаванні 1 мл розчину амонію хлориду Р.
До одержаного розчину додають 1 мл розчину динатрію
Ідрофосфату Р; утворюється білий кристалічний осад.
НАТРІЙ
аі 0.1 г випробовуваної субстанції розчиняють у 2 мл
води Р. До одержаного розчину або до 2 мл розчину,
^значеного в окремій статті, додають 2 мл розчину
150 г/л калію карбонату Р і нагрівають до кипіння;
осад не утворюється. До розчину додають 4 мл розчину
калію піроантимонату Р і нагрівають до кипіння,
потім охолоджують у льодяній воді і, якщо необхідно,
потирають внутрішні стінки пробірки скляною палич-
кою; утворюється густий осад білого кольору.
Ь) Наважку випробовуваної субстанції, еквівалентну
близько 2 мг натрій-іона (N3+), розчиняють у 0.5 мл
води Р. До одержаного розчину або до 0.5 мл роз-
чину, зазначеного в окремій статті, додають 1.5 мл
метоксифенілоцтової кислоти реактиву Р, охолод-
жують у льодяній воді протягом 30 хв; утворюється
об’ємний білий кристалічний осад. Суміш поміща-
ють у воду при температурі 20 °С і перемішують
протягом 5 хв; осад не зникає. До суміші додають
1 мл розчину аміаку розведеного Р1; осад цілком
розчиняється. До одержаного розчину додають
1 мл розчину амонію карбонату Р; осад не утво-
рюється.
____________________________________________N
с) Сіль натрію, змочена кислотою хлористоводневою Р
і внесена в безбарвне полум’я, забарвлює його у жов-
тий колір.
НІТРАТИ
а) Наважку порошку субстанції, еквівалентну близько
1 мг нітрат-іона (N05), або кількість, зазначену в ок-
ремій статті, додають до суміші 0.1 мл нітробензолу Р\
0.2 мл кислоти сірчаної Рї через 5 хв охолоджують у
льодяній воді. Продовжуючи охолодження, повільно
при перемішуванні додають 5 мл води Р, 5 мл розчину
натрію гідроксиду концентрованого Р, 5 мл ацетону Р,
збовтують і відстоюють; верхній шар набуває темно-
фіолетового забарвлення.
____________________________________________N
Ь) Нітрати. Розчин, що містить випробовувану суб-
станцію у кількості, еквівалентній близько 2 мг нітрат-
іона (N0^), не знебарвлює розчин 1 г/л калію перман-
ганату Р, підкислений кислотою сірчаною розведе-
ною Р (відмінність від нітритів).
с) Нітрити. Кілька кристалів антипірину розчиня-
ють у фарфоровій чашці в 0.1 мл кислоти хлористо-
водневої розведеної Р, додають 0.1 мл розчину, що
містить близько 1 мг нітрит-іона (N0^); з’являється
зелене забарвлення (відмінність від нітратів).
РТУТЬ
а) Близько 0.1 мл розчину випробовуваної субстанції
поміщають на ретельно очищену поверхню мідної
фольги; з’являється темно-сіра пляма, яка при нати-
ранні стає блискучою. Фольгу висушують і нагрівають
у пробірці; пляма зникає.
Ь) До розчину, зазначеного в окремій статті, додають
розчин натрію гідроксиду розведений Р до сильнолуж-
ної реакції середовища (2.2.4); утворюється густий
осад жовтого кольору (солі ртуті).
____________________________________________N
с) До 1 мл розчину, що містить випробовувану
субстанцію у кількості, еквівалентній 10-30 мг
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
71
2.? ітифікація
.і£2+). додають обережно краплями
йодиду Р; утворюється червоний осад,
у надлишку цього реактиву.
САЛІЦИЛАТИ
а) До І мл розчину, зазначеного в окремій статті, дода-
ють 0.5 мл розчину заліза(ІІІ) хлориду РІ; з’являється
фіолетове забарвлення, яке не зникає після додавання
0.1 мл кислоти оцтової Р.
Ь) 0.5 г випробовуваної субстанції розчиняють у 10 мл
води Р. До одержаного розчину або до 10 мл розчину,
зазначеного в окремій статті, додають 0.5 мл кислоти
хлористоводневої Р. Одержаний осад після перекрис-
талізації з гарячої води Р і висушений у вакуумі має
температуру плавлення (2.2.14) від 156 °С до 161 °С.
СВИНЕЦЬ
а) 0.1 г випробовуваної субстанції розчиняють в 1 мл
кислоти оцтової Р. До одержаного розчину або до 1 мл
розчину, зазначеного в окремій статті, додають 2 мл
розчину калію хромату Р; утворюєті.ся жовтий осад,
що розчиняється при додаванні 2 мл розчину натрію
гідроксиду концентрованого Р.
Ь) 50 мг випробовуваної субстанції розчиняють у 1 мл
кислоти оцтової Р. До одержаного розчину або до 1 мл
розчину, зазначеного в окремій статті, додають 10 мл
води Р і 0.2 мл розчину калію йодиду Р; утворюється
жовтий осад. Суміш кип’ятять протягом 1-2 хв; осад
розчиняється. Розчину дають охолонути; знову утво-
рюється осад у вигляді блискучих жовтих пластинок.
СРІБЛО
Близько 10 мг випробовуваної субстанції розчиняють
у 10 мл води Р. До одержаного розчину або до 10 мл
розчину, зазначеного в окремій статті, додають 0 3 мл
кислоти хлористоводневої РІ; утворюється білий сир-
нистий осад, який розчиняється при додаванні 3 мл
розчину аміаку розведеного РІ.
СИЛІКАТИ
Кількість випробовуваної субстанції, зазначену в ок-
ремій статті, змішують у свинцевому або платиновому
тиглі за допомогою мідного дроту із близько 10 мг
натрію фториду Р і декількома краплями кислоти
сірчаної Р до утворення суспензії. Тигель накривають
тонкою прозорою пластиковою пластинкою з вися-
чою краплею води Р і обережно нагрівають; через ко-
роткий проміжок часу навколо краплі води з’яв-
ляється біле кільце.
СУЛЬФАТИ
а) Близько 45 мг випробовуваної субстанції розчиня-
ють у 5 мл води Р. До одержаного розчину або до 5 мл
розчину, зазначеного в окремій статті, додають І і
кислоти хлористоводневої розведеної Р і 1 мл розчі
барію хлориду РІ; утворюється білий осад.
Ь) До суспензії, одержаної в результаті реакції (а),м
дають 0.1 мл 0.05 М розчину йоду; жовте забарвлен,
йоду не зникає (відмінність від сульфітів і ДИТІОНІТІі
але знебарвлюється при додаванні краплями розчіц
олова хлориду Р (відмінність від йодатів). Суми
кип’ятять; осад не знебарвлюється (відмінність відІ
ленатів і вольфраматів).
СУЛЬФІТИ
а) До 2 мл розчину, що містить випробовувану су
станцію у кількості, еквівалентній 10-30 мг сульфі ‘
іона (8О3 ), додають 2 мл кислоти хлористоводнесЛ
розведеної Р і струшують; поступово виділяєш
сірчистий газ, що виявляється за характерним різкг
запахом.
Ь) До зазначеного в окремій статті розчину, що містин
сульфіт-іон (8О3), додають 0.1 мл 0.05 М розчину ію
ду; реактив знебарвлюється.
СУРМА
Близько 10 мг випробовуваної субстанції розчиняюпі
при нагріванні у розчині 0.5 г калію-натрію тартрі
ту Ру 10 мл води Рі охолоджують. До 2 мл одержаної
го розчину або до 2 мл розчину, зазначеного в окремі
статті, додають краплями розчин натрію сульфіду Р
утворюється оранжево-червоний осад, який розчи!
няється при додаванні розчину натрію гідроксиду ро
веденого Р.
ТАРТРАТИ
а) Близько 15 мг випробовуваної субстанції розчини]
ють у 5 мл води Р. До одержаного розчину або до 5 мл
розчину, зазначеного в окремій статті, додають 0.05 N .
розчину 10 г/л заліза(П) сульфату Р і 0.05 мл /юзчіад
водню пероксиду розведеного Р; з’являється нестійке]
жовте забарвлення. Після знебарвлення розчину с
нього додають краплями розчин натрію гідроксиду роз-і
ведений Р; з’являється інтенсивне синє забарвлення.
Ь) До 0.1 мл розчину, що містить випробовувану суб-
станцію, у кількості еквівалентній близько 15 мг/мі
кислоти винної, або до 0.1 мл розчину, зазначеного г
окремій статті, додають 0.1 мл розчину 100 г/л кам
броміду Р, 0.1 мл розчину 20 г/л резорцину Р, 3 мл кис-
лоти сірчаної Рі нагрівають на водяній бані від 5 хв»
10 хв; з’являється темно-синє забарвлення. Розчин
охолоджують і вливають у воду Р; забарвлення розчи-
ну змінюється на червоне.
72
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.3. Ідентифікація
» фосфати (Ортофосфати)
рДс 5 мл розчину, зазначеного в окремій статті, якщо
щцбмлно, нейтралізованого, додають 5 мл розчину
ірігіш нітрату РІ; утворюється жовтий осад, колір
Нмп> не змінюється при кип’ятінні і який розчи-
ніться при додаванні розчину аміаку Р.
►і До і мл розчину, зазначеного в окремій статті, дода-
ній- 2 мл молібденованадієвого реактиву Р\ перемішу-
ють: з'являється жовте забарвлення.
ХЛОРИДИ
й Наважку випробовуваної субстанції, еквівалентну
(ііінько 2 мг хлорид-іона (СІ ), розчиняють у 2 мл во-
вн Р Одержаний розчин або 2 мл розчину, зазначено-
го в окремій статті, підкислюють кислотою азотною
^жденою Р, додають 0.4 мл розчину срібла нітра-
су РІ, перемішують і відстоюють; утворюється білий
пінистий осад, який центрифугують і промивають
трьома порціями води Рпо 1 мл кожна. Цю операцію
вводять швидко в захищеному від яскравого світла
місці, при цьому допускається, щоб рідина над осадом
нї біла уповні прозорою. Осад суспендують у 2 мл во-
лі Рі додають 1.5 мл розчину аміаку Р; осад швидко
рої'іпняється; допускається наявність декількох круп-
ній часток, які розчиняються повільно.
М Наважку випробовуваної субстанції, еквівалентну
близько 15 мг хлорид-іона (СІ), або кількість, зазна-
чені в окремій статті, поміщають у пробірку, додають
н 2 г калію діхромату Р і 1 мл кислоти сірчаної Р. Біля
«ліпного отвору пробірки поміщають фільтрувальний
напір, просякнутий 0.1 мл розчину дифенілкарбазиду Р
шри цьому просякнутий папір не має стикатися з
и’іію біхроматом); папір забарвлюється у фіолетово-
и'рвоний колір.
ЦИТРАТИ
а) Наважку випробовуваної субстанції, еквівалентну
близько 50 мг кислоти лимонної, розчиняють у 5 мл
води Р. До одержаного розчину або до 5 мл розчину, за-
значеного в окремій статті, додають 0.5 мл кислоти
сірчаної Р\ 1 мл розчину калію перманганату Р. Розчин
нагрівають до знебарвлення, додають 0.5 мл розчину
100 г/л натрію нітропрусиду Р в кислоті сірчаній розве-
деній Р, 4 г кислоти сульфамінової Р. До суміші дода-
ють розчин аміаку концентрований Р до лужної реакції
середовища, додаючи його краплями до повного роз-
чинення кислоти сульфамінової. Додавання надлиш-
ку розчину аміаку концентрованого Р призводить до
появи фіолетового забарвлення, яке переходить у
фіолетово-синє.
____________________________________________N
Ь) До 1 мл нейтрального розчину, що містить випробо-
вувану субстанцію в кількості, еквівалентній 2-10 мг
цитрат-іона, додають 1 мл розчину 200 г/л кальцію
хлориду Р; розчин залишається прозорим; при
кип’ятінні розчину утворюється білий осад, розчин-
ний у кислоті хлористоводневій розведеній Р.
с) До кількості субстанції, еквівалентній 1-2 мг цит-
рат-іона, додають 0.5 мл ангідриду оцтового Р і нагрі-
вають; через 20-40 с з’являється червоне забарвлення.
ЕФІРИ СКЛАДНІ
До близько 30 мг або до зазначеної в окремій статті
кількості випробовуваної субстанції додають 0.5 мл
розчину 70 г/л гідроксиламіну гідрохлориду Р у мета-
нолі Р, 0.5 мл розчину 100 г/л калію гідроксиду Р в
спирті Р, нагрівають при збовтуванні і охолоджують.
Одержаний розчин підкислюють кислотою хлористо-
водневою розведеною Р, додають 0.2 мл розчину
заліза(ІІІ) хлориду РІ, розведеного у 10 разів; з’яв-
ляється синювато-червоне або червоне забарвлення.
ЦИНК
ііОІ г випробовуваної субстанції розчиняють у 5 мл
Р. До одержаного розчину або до 5 мл розчину, за-
кінченого в окремій статті, додають 0.2 мл розчину
натрію гідроксиду концентрованого Р; утворюється
білий осад. Потім додають ше 2 мл розчину натрію
гідроксиду концентрованого Р; осад розчиняється. До
-держаного розчину додають 10 мл розчину амонію
•лориду Р; розчин залишається прозорим. До розчину
'мають 0.1 мл розчину натрію сульфіду Р; утворюється
1'11111 пластівчастий осад.
N
Ьі До 2 мл розчину, що містить випробовувану
спхланцію у кількості, еквівалентній 5-20 мг цинк-
Іона (2п2+), додають 0.5 мл розчину калію фероціаніду Р;
'створюється білий осад, нерозчинний у кислоті хлори-
стоводневій розведеній Р.
2.3.2. ІДЕНТИФІКАЦІЯ ЖИРНИХ ОЛІЙ
МЕТОДОМ ТОНКОШАРОВОЇ
ХРОМАТОГРАФІЇ
Визначення проводять методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар ок-
тадецилсилільний силікагель для високоефективної
тонкошарової хроматографії.
Випробовуваний розчин. Якщо немає інших зазначень
в окремій статті, близько 20 мг (краплю) жирної олії
розчиняють у 3 мл метиленхлориду Р.
Розчин порівняння. Близько 20 мг (краплю) олії куку-
рудзяної Р розчиняють у 3 мл метиленхлориду Р.
На лінію старту хроматографічної пластинки окремо
наносять по 1 мкл випробовуваного розчину і розчину
порівняння. Пластинку двічі хроматографують на
відстань 0.5 см, використовуючи як рухому фазу ефір Р,
і двічі хроматографують на відстань 8 см, використо-
вуючи як рухому фазу суміш розчинників: метилен-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
73
2.3. Ідентифікація
хлорид Р — кислота оцтова льодяна Р — ацетон Р
(20:40:50). Потім пластинку сушать на повітрі, обпри-
скують розчином 100 г/л кислоти фосфорномолібдено-
вої Ру спирті Р, нагрівають при температурі 120 °С
протягом 3 хв і переглядають при денному світлі. Ти-
пова хроматограма для ідентифікації жирних олій на-
ведена на Рис. 2.3.2.-1.
2.3.3. ІДЕНТИФІКАЦІЯ ФЕНОТІАЗИН1В
МЕТОДОМ ТОНКОШАРОВОЇ
ХРОМАТОГРАФІЇ
Визначення проводять методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар
кізельгур с Р.
Пластинку імпрегнують, помістивши в закриту каме-
ру з необхідною кількістю імпрегнувальної суміші, що
містить 10 % (об/об) феноксіетанолу Ру розчині 50 г/л
поліетиленгліколю 300 Р в ацетоні Р, у такий спосіб,
щоб вона була занурена в імпрегнувальну суміш при-
близно на 5мм. Коли фронт розчинників пройде 17 см
від нижнього краю пластинки, її виймають з камери і
відразу використовують для хроматографування. Хро-
матографування проводять у тому самому напрямку,
що й імпрегнування.
Випробовуваний розчин. 20 мг випробовуваної суб-
станції розчиняють у хлороформі Р'\ доводять до 10 мл
тим самим розчинником.
Розчин порівняння. 20 мг відповідного фармакопейно-
го стандартного зразка (ФСЗ) розчиняють у хлоро-
формі Р і доводять тим самим розчинником до 10 мл І
На лінію старту' хроматографічної пластинки окреме
наносять по 2 мкл випробовуваного розчину і розчин!
порівняння. Пластинку поміщають у камеру із суміш
шю: петролейний ефір Р- діетиламін Р(5():І), насте І
ну феноксіетанолом Р (тобто, від 3 мл до 4 мл фенокЛ
етанолу Р додають до вищезазначеної суміші розчині
ників, збовтують до утворення стійкої каламутності
декантують і використовують верхній шар, який мох
бути каламутним). Хроматографування проводять!
темному місці. Коли фронт розчинників пройде 15СЯ]
від лінії старту, пластинку виймають з камери
поміщають в УФ-світло за довжини хвилі 365 нм і ч<
рез декілька хвилин проводять оцінку хроматограми. І
На хроматограмі випробовуваного розчину мас виям
лятися пляма на рівні плями на хроматограмі розчин»
порівняння, яка відповідає їй за кольором флуор&н
ценції і розміром. Потім пластинку обприскують роз-
чином 10 % (об/об) кислоти сірчаної Ру спирті Р. Н
хроматограмі випробовуваного розчину має виявляти
ся пляма на рівні плями на хроматограмі розчин»!
порівняння, яка відповідає їй за забарвленням та його
стабільністю протягом не менше 20 хв.
2.3.4. ВИЗНАЧЕННЯ ЗАПАХУ
Від 0.5 г до 2.0 г випробовуваної субстанції розподіл*
ють тонким шаром на годинниковому склі діаметром
від 6 см до 8 см; через 15 хв визначають запах або при-
ходять до висновку про його відсутність.
12 34 5678 9 10
Рисунок 2.3.2.-1. Типова хроматограма для ідентифікації жирних олій
1 — арахісова олія; 2 — оливкова олія; 3 — кунжутна олія; 4 — кукурудзяна олія; 5 — мигдальна олія:
6 — соєва олія; 7 — соняшникова олія; 8 — рапсова олія; 9 — рапсова олія (яка не містить ерукової кислоти):
10 — олія зародків пшениці.
74
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
2.4. ВИПРОБУВАННЯ НА
ГРАНИЧНИЙ ВМІСТ
ДОМІШОК
2.4.1. АМОНІЮ СОЛІ
Д А застосовують, якщо немає інших зазначень в
окремій статті.
МЕТОДА
Кількість випробовуваної речовини, зазначену в ок-
реиііі статті, поміщають у пробірку, розчиняють у
|4уд води Р, якщо необхідно, підлужують розчином
ктрію гідроксиду розведеним Р і доводять об’єм роз-
імну водою Рао 15 мл. Додають 0.3 мл розчину калію
тетрайодмеркурату лужного Р.
Як еталон використовують розчин, одержаний до-
данням до 10 мл еталонного розчину амонію
і І ррт Р5 мл води Р і 0.3 мл розчину калію тет-
ршодмеркурату лужного Р. Пробірки закривають.
Через5 хв жовте забарвлення випробовуваного розчи-
ііу мас бути не інтенсивнішим за забарвлення еталона.
МЕТОД В
Кількість ретельно розтертої випробовуваної речови-
ми зазначену в окремій статті, поміщають у посудину
Місткістю 25 мл, споряджену поліетиленовою проб-
кою. і розчиняють або суспендують в 1 мл води Р. До-
тають 0 ЗО г магнію оксиду важкого Р, поміщають
) пі судину під пробку смужку срібно-марганцевого па-
перу Р, змочену кількома краплями води Р таким чи-
ном, щоб відрізок паперу розміром (5 х 5) мм знахо-
дься нижче нижнього краю пробки, після чого посу-
шну негайно закривають пробкою. Перемішують
вміст посудини коловими рухами, не допускаючи по-
плання бризок на папір, і витримують у водяному
термостаті при температурі 40 °С протягом ЗО хв.
Пзралельно за цих самих умов готують еталон. До за-
вченої в окремій статті кількості еталонного розчину
имонію (І ррт ЇЇН4) Р додають 1 мл води Р, 0.30 г
щігнію оксиду важкого Р і далі чинять, якз випробову-
нзнпм розчином.
Сіре забарвлення срібно-марганцевого паперу Р, одер-
жане у досліді з випробовуваним розчином, має бути
не інтенсивнішим за забарвлення срібно-марганцевого
паперу Р, одержане в досліді з еталоном.
І N
МЕТОД С
Застосовують для зразків, що містять лужноземельні
та важкі метали.
Кількість випробовуваної речовини, зазначену в окремій
статті, поміщають у пробірку, розчиняють у мінімально-
му об’ємі води Р, додають при охолодженні 2 мл розчину
натрію гідроксиду розведеного Р і 2 мл розчину натрію
карбонату Р. Розчин розводять водою Р до зазначеної в
окремій статті концентрації, збовтують і фільтрують.
10 мл одержаного фільтрату поміщають у пробірку, до-
водять об’єм розчину водою Рао 15 мл і додають 0.3 мл
розчину калію тетрайодмеркурату лужного Р.
Як еталон використовують суміш 10 мл еталонного
розчину амонію (1 ррт МН4) Р, 5 мл води Рі 0.3 мл роз-
чину калію тетрайодмеркурату лужного Р. Пробірки
закривають.
Через 5 хв жовте забарвлення випробовуваного розчи-
ну має бути не інтенсивнішим за забарвлення еталона.
МЕТОД Б
Застосовують для зразків, що містять більше 300ррт
домішки заліза.
Кількість випробовуваної речовини, зазначену в ок-
ремій статті, поміщають у пробірку, розчиняють у
10 мл води Р, долають 2 краплі розчину натрію гідрокси-
ду розведеного Р і 3 мл розчину 200 г/л калію-натрію
тартрату Р. Ретельно переміщують, доводять об’єм
розчину водою Рдо 15 мл і додають 0 3 мл розчину калію
тетрайодмеркурату лужного Р. Пробірку закривають.
Паралельно за цих самих умов готують еталон, вико-
ристовуючи замість 10 мл випробовуваного розчину
10 мл еталонного розчину амонію (/ ррт N11^ ) Р.
Через 5 хв жовте забарвлення випробовуваного розчи-
ну має бути не інтенсивнішим за забарвлення еталона.
2.4.2. АРСЕН
МЕТОД А
Прилад (див. Рис. 2.4.2.-1) складається з конічної кол-
би місткістю 100 мл, закритої скляною притертою
пробкою, крізь яку проходить скляна трубка за-
вдовжки близько 200 мм із внутрішнім діаметром 5 мм.
Нижня частина трубки витягнута до внутрішнього
діаметра 1.0 мм; на відстані 15 мм від кінчика трубки
розташований бічний отвір діаметром від 2 мм до 3 мм.
Трубка поміщена таким чином, щоб бічний отвір зна-
ходився мінімум на 3 мм нижче нижньої поверхні
пробки. Верхній кінець трубки повинен мати цілком
плоску притерту поверхню, розташовану під прямим
кутом до осі трубки. Друга скляна трубка завдовжки
30 мм з таким самим внутрішнім діаметром і такою са-
мою плоскою притертою поверхнею поміщається
зверху першої трубки і щільно прикріплюється до неї
двома пружинами.
Нижню трубку нещільно заповнюють 50-60 мг свинце-
во-ацетатної вати Р або поміщають невеликий ват-
ний тампон і скручену трубочкою смужку свинцево-
ацетатного паперу Р масою 50-60 мг. Між плоскими
поверхнями трубок поміщають шматочок ртутно-
бромідного паперу Р такого розміру, щоб закрити отвір
трубки (15 х 15) мм.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
75
2.4. Випробування на граничний вміст домішок
Зазначену кількість випробовуваної речовини по-
міщають у конічну колбу і розчиняють у 25 мл води Рабо
зазначений об’єм випробовуваного розчину поміша-
ють у конічну колбу, доводять об’єм розчину водою Р
до 25 мл. Додають 15 мл кислоти хлористоводневої Р,
0.1 мл розчину олова(II) хлориду Р, 5 мл розчину калію
йодиду Р, залишають на 15 хв і потім додають 5 г цинку
активованого Р. Негайно сполучають дві частини при-
ладу, колбу поміщають у водяну баню, температура
якої підтримується такою, щоб забезпечити рівно-
мірне виділення газу.
Паралельно за цих самих умов проводять дослід з ета-
лоном, що складається з 1 мл еталонного розчину арсе-
ну (І ррт Аз) Р і 24 мл води Р.
Не менш як через 2 год забарвлення ртутно-бромід-
ного паперу, одержане в досліді з випробовуваним
розчином, має бути не інтенсивнішим за забарвлення
ртутно-бромідного паперу, одержане в досліді з етало-
ном.
МЕТОД В
Кількість випробовуваної речовини, зазначену в ок-
ремій статті, поміщають у пробірку, що містить 4 мл
кислоти хлористоводневої Р і близько 5 мг калію йоди-
ду Р, і додають 3 мл реактиву гіпофосфіту Р. Суміш
нагрівають на водяній бані протягом 15 хв, час від ча-
су струшуючи.
Паралельно за цих самих умов готують еталон, вико-
ристовуючи замість випробовуваної речовини 0.5 мл
еталонного розчину арсену (10 ррт Аз) Р.
Після нагрівання на водяній бані забарвлення випро
бовуваного розчину має бути не інтенсивнішим за за-
барвлення еталона.
__________________________________________Я
МЕТОДА
Температура водяної бані не має перевищувати 40°С
Рисунок 2.4.2.-1. Прилад для випробування
на арсен (метод А)
Розміри зазначені у міліметрах
МЕТОД В
Метод В застосовують у випадку визначення поряд
арсеном селену і телуру, а також при визначенні арсе-
ну в зразках, що містять сурму, вісмут, ртуть і срібло,і
також сульфіди і сульфіти, та в деяких інших випад-
ках.
2.4.3. КАЛЬЦІЙ
При приготуванні всіх розчинів, застосовуваних у дано-
му випробуванні, має використовуватися вода дисти-
льована Р.
До 0.2 мл еталонного розчину кальцію спиртової
(100 ррт Са) Р додають 1 мл розчину амонію океані
ту Р. Через 1 хв додають суміш 1 мл кислоти оцтово
розведеної Рі 15 мл розчину, що містить зазначену!
окремій статті кількість випробовуваної речовини,
струшують.
Паралельно за цих самих умов готують еталон, вико-
ристовуючи суміш 1 мл кислоти оцтової розведеної Р,
10 мл еталонного розчину кальцію водного (10ррт Са) Рі
5 мл води дистильованої Р.
Через 15 хв опалесценція випробовуваного розчинук
має перевищувати опалесценцію еталона.
2.4.4. ХЛОРИДИ
До 15 мл розчину, зазначеного в окремій статті, долі
ють 1 мл кислоти азотної розведеної Р і виливають
суміш за один раз у пробірку, що містить 1 мл розчив)
срібла нітрату Р2.
Паралельно за цих самих умов готують еталон, вико
ристовуючи замість 15 мл випробовуваного розчині
10 мл еталонного розчину хлориду (5ррт СІ) Р і 5 мл ва-
ди Р. Пробірки поміщають у захищене від світла місце,
76
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 200і
2.4. Випробування на граничний вміст домішок
Ч<Тсі5 хв пробірки переглядають на чорному фоні го-
гчі.интально(перпендикулярно до осі пробірок). Опа-
цепція випробовуваного розчину не має перевищу-
л и опалесценцію еталона.
2.4.5. ФТОРИДИ
Кь ькість випробовуваної речовини, зазначену в ок-
; іііі статті. 0.1 г піску Р, промитого кислотою, і 20 мл
. ііші рівних об’ємів кислоти сірчаної Р і води Р
помішають у внутрішню пробірку приладу; зображе-
но на Рис. 2.4.5.-1. Оболонку, заповнену тетрахюр-
стіном Р, нагрівають до температури кипіння тетра-
оретану (146 °С). Приєднують генератор водяної па-
і і відганяють вміст пробірки з перегрітою водяною
рою, збираючи відгін у мірну колбу місткістю
Кін мл, яка містить 0.3 мл 0.1М розчину натрію гідрок-
> 10 І мл розчину фенолфталеїну Р. Об’єм розчину в
пробірці під час відгону має бути постійним (20 мл).
Підтримують лужну реакцію вмісту мірної колби,
,„шо необхідно, додаючи краплями 0.1 М розчин
мрію гідроксиду. Доводять об’єм розчину водою Рро
г значки і перемішують (випробовуваний розчин).
Рисунок 2.4.5.-1. Прилад для випробування
на фториди
Розміри зазначені у міліметрах
Паралельно за цих самих умов готують еталон, вико-
ристовуючи замість випробовуваної речовини 5 мл
еталонного розчину фториду (10 ррт Р) Р.
У циліндр із скляною притертою пробкою поміщають
20 мл випробовуваного розчину, в другий такий самий
циліндр — 20 мл еталона, потім у кожний циліндр до-
дають по 5 мл реактиву амінометилалізариндіоцтової
кислоти Р.
Через 20 хв синє забарвлення випробовуваного розчи-
ну, що з’явилося замість первісного червоного, має бу-
ти не інтенсивнішим за забарвлення еталона
2.4.6. МАГНІЙ
До 10 мл розчину випробовуваної речовини, пригото-
ваного, як зазначено в окремій статті, додають 0 1 г ди-
натрію тетраборату Р. Визначають рН розчину і,
якщо необхідно, доводять до рН 8.8-9.2, використову-
ючи кислоту хлористоводневу розведену Р або' розчин
натрію гідроксиду розведений Р. Розчин поміщають у
ділильну лійку, струшують протягом 1 хв з двома пор-
ціями, по 5 мл кожна, розчину 1 г/л гідроксихіноліну Р
у хлороформі Р, залишають до розшарування і відкида-
ють органічний шар. До водного шару додають
0.4 мл бутиламіну Р \ 0.1 мл триетаноламіну Р. Визна-
чають рН розчину і, якщо необхідно, доводять до
рН 10.5-І 1.5. Додають 4 мл розчину 1 г/л гідрок-
сихіноліну Р у хлороформі Р, струшують протягом 1 хв,
залишають до розшарування, нижній шар відбирають
і використовують для випробування.
Паралельно за цих самих умов готують еталон, вико-
ристовуючи замість 10 мл розчину випробовуваної ре-
човини суміш 1 мл еталонного розчину магнію
(10 ррт Му) Рта 9 мл води Р.
Забарвлення випробовуваного розчину має бути не
інтенсивнішим за забарвлення еталона.
2.4.7. МАГНІЙ І ЛУЖНОЗЕМЕЛЬНІ МЕТАЛИ
До 200 мл води Рдодають 0.1 г гідроксиламіну гідрохло-
риду Р, 10 мл аміачного буферного розчину рН 10.0 Р,
1 мл 0.1 Мрозчину цинку сульфату і близько 15 мг інди-
каторної суміші протравного чорного 11 Р. Нагрівають
до температури близько 40 °С. Одержаний розчин ти-
трують 0.01 М розчином натрію едетату до переходу
забарвлення розчину від фіолетового до синього. До
одержаного розчину додають зазначену в окремій
статті кількість випробовуваної речовини, розчинену
в 100 мл води Р, або використовують зазначений в ок-
ремій статті розчин. Якщо забарвлення розчину стає
фіолетовим, знову титрують до переходу забарвлення
розчину до синього.
Об’єм 0.01 Мрозчину натрію едетату, витрачений на
друге титрування, не має перевищувати об’єм титран-
ту, зазначений в окремій статті.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
77
2.4. Випробування на граничний вміст домішок
2.4.8. ВАЖКІ МЕТАЛИ
МЕТОДА
До 12 мл водного розчину, зазначеного в окремій
статті, додають 2 мл буферного розчину рН 3.5 Р і пе-
ремішують. Одержану суміш додають до 1.2 мл реак-
тиву тіоацетаміду Рі негайно перемішують.
Паралельно за тих самих умов готують еталон, вико-
ристовуючи замість 12 мл випробовуваного розчину
суміш 10 мл еталонного розчину свинцю (1 ррт або
2ррт РЬ) Р, як зазначено в окремій статті, і 2 мл ви-
пробовуваного розчину.
Готують холостий розчин, використовуючи суміш
10 мл води Р і 2 мл випробовуваного розчину.
Порівняно з холостим розчином еталон повинен мати
світло-коричневе забарвлення.
Через 2 хв коричневе забарвлення випробовуваного
розчину має бути не інтенсивнішим за забарвлення
еталона.
МЕТОД В
Випробовувану речовину розчиняють в органічному
розчиннику з мінімально необхідною кількістю води
(наприклад, діоксан або ацетон з додаванням 15 % води).
До 12 мл розчину, зазначеного в окремій статті, дода-
ють 2 мл буферного розчину рН 3.5 Р і перемішують.
Одержану суміш додають до 1 2 мл реактиву тіоаце-
таміду Рі негайно перемішують.
Паралельно за тих самих умов готують еталон, вико-
ристовуючи замість 12 мл випробовуваного розчину
суміш 10 мл еталонного розчину свинцю (І ррт або
2ррт РЬ), як зазначено в окремій статті, і 2 мл випро-
бовуваного розчину. Еталонний розчин свинцю (І ррт
або 2 ррт РЬ) готують шляхом розведення еталонного
розчину свинцю (100ррт РЬ) Ррозчинником, який за-
стосовується для розчинення випробовуваної речови-
ни.
Готують холостий розчин, використовуючи суміш
10 мл розчинника, застосовуваного для розчинення
випробовуваної речовини, і 2 мл випробовуваного
розчину. Порівняно з холостим розчином еталон
повинен мати світло-коричневе забарвлення.
Через 2 хв коричневе забарвлення випробовуваного
розчину має бути не інтенсивнішим за забарвлення
еталона.
МЕТОД С
У фарфоровий або кварцовий тигель поміщають 4 мл
розчину 250 г/л магнію сульфату Р у кислоті сірчаній
розведеній Р і додають кількість випробовуваної речо-
вини, зазначену в окремій статті (не більше 2 г). Пе-
ремішують тонкою скляною паличкою і обережно
нагрівають. Якщо суміш рідка, обережно упарюють на
водяній бані до сухого залишку, потім поступово
нагрівають до обвуглювання і продовжують нагрівання
до одержання майже білого або в крайньому разі а-
ватого залишку. Спалювання проводять при темпе;
турі не більше 800 °С. Залишають до охолоджені
потім залишок у тиглі змочують кількома крапляІ
кислоти сірчаної розведеної Р. Упарюють до сухою
лишку, знов спалюють і залишають до охолодженні
Загальний час спалювання не має перевищувати 2 пЛ
Залишок з тигля кількісно переносять у пробіркудвоу
порціями кислоти хлористоводневої розведеної Р, і в
5 мл кожна. Додають 0.1 мл розчину фенолфталеїну Л
потім підлужують розчином аміаку концентрованім М
до появи рожевого забарвлення. Охолоджують, доЛ
ють кислоту оцтову льодяну /'до знебарвлення розчи .
і додають ще 0.5 мл кислоти оцтової льодяної Р. Якщ
необхідно, фільтрують і промивають фільтр водою ш
Доводять об’єм розчину водою Р до 20 мл і переміні) І
ють. 12 мл одержаного розчину поміщають у пробірк
додають 2 мл буферного розчину рН 3.5 Рі перемішую»І
Одержану суміш додають до 1.2 мл реактиву іпіоащ
таміду Рі негайно перемішують.
Паралельно готують еталон, використовуючи 4 к.
розчину 250 г/л магнію сульфату Ру кислоті сірчанп 1
розведеній Р і зазначений об’єм еталонного розчину,І
свинцю (10ррт РЬ) Р. Спалюють за тих самих умов, ц» І
і випробовувану речовину, кількісно переносять кис І
лотою хлористоводневою, додають розчин аміак; і]
потім кислоту оцтову. Доводять об’єм розчину Д"
20 мл водою Р і перемішують. До 10 мл одержаною]
розчину додають 2 мл розчину, одержаного при об-'
робці випробовуваної речовини, і 2 мл буферного роз-
чину рН 3.5 Р і перемішують. Одержану суміш додаючі
до 1.2 мл реактиву тіоацетаміду Р і негайно пе-
ремішують.
Готують холостий розчин, використовуючи суміш
10 мл води Рі 2 мл розчину, одержаного при обробні
випробовуваної речовини. Порівняно з холостим роз-
чином еталон повинен мати світло-коричневе забарв-
лення
Через 2 хв коричневе забарвлення випробовуваного
розчину має бути не інтенсивнішим за забарвлення
еталона.
МЕТОД О
Кількість випробовуваної речовини, зазначену в ок-
ремій статті, поміщають у фарфорофий або кварцовий
тигель і старанно змішують із 0.5 г магнію оксиду РІ.
Спалюють при слабкому червоному жару (близько
600 °С) до утворення однорідного залишку білого або
сірувато-білого кольору. Якщо після 30 хв спалювання
суміш залишається забарвленою, тигель залишають до
охолодження, вміст перемішують тонкою скляною
паличкою і повторюють спалювання. Якщо не-
обхідно, операцію повторюють. Нагрівають при тем-
пературі 800 °С близько 1 год. Залишок з тигля
кількісно переносять у пробірку двома порціями по
5 мл суміші рівних об’ємів розчину кислоти хлористо-
водневої РІ і води Р. Додають 0.1 мл розчину фенолфта-
леїну Р, потім підлужують розчином аміаку концентро-
ваним Р до появи рожевого забарвлення. Охолоджу-
ють, підкислюють кислотою оцтовою льодяною Р до
знебарвлення розчину і додають ще 0.5 мл кислотиоц-
78
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
тової льодяної Р. Якщо необхідно, фільтрують і проми-
вають фільтр водою Р. Доводять об’єм розчину водою Р
ю 20 мл і перемішують. 12 мл одержаного розчину
помішають у пробірку, додають 2 мл буферного розчину
рН 3.5 Р і перемішують. Одержану суміш додають до
1.2 мл реактиву тіоацетаміду Р і негайно перемі-
шують.
Паралельно готують еталон таким чином. До 0.5 г
магнію оксиду РІ додають зазначений в окремій статті
об'єм еталонного розчину свинцю (10 ррт РЬ) Р. Вису-
шують у сушильній шафі при температурі від 100 °С до
105 °С. потім спалюють за тих самих умов, що й випро-
бовувану речовину, кількісно переносять кислотою
оористоводневою, додають розчин аміаку і потім кис-
юту оцтову. Доводять об’єм розчину до 20 мл водою Р і
перемішують. До 10 мл одержаного розчину додають
2 мл розчину, одержаного при обробці випробовуваної
речовини, і 2 мл буферного розчину рН 3.5 Р '\ перемішу-
ють. Одержану суміш додають до 1.2 мл тіоаце-
тамідногореактиву Рі негайно перемішують.
Готують холостий розчин, використовуючи суміш
10 мл води Р і 2 мл випробовуваного розчину.
Порівняно з холостим розчином еталон повинен мати
світло-коричневе забарвлення.
Через 2 хв коричневе забарвлення випробовуваного
розчину має бути не інтенсивнішим за забарвлення
еталона.
МЕТОД Е
Кількість випробовуваної речовини, зазначену в ок-
ремій статті, розчиняють у ЗО мл або у зазначеній
кількості води Р.
У тримач пристрою для стерильного фільтрування,
на підкладку якого поміщено мембранний фільтр
(розмір пор - 3 мкм), а поверх нього — передфільтр
(Рис. 2.4.8.-1), установлюють шприц місткістю 50 мл
без поршня.
Випробовуваний розчин поміщають у шприц і уводять
поршень, розвиваючи такий тиск, щоб профільтру-
вався увесь розчин. Відкривають тримач і виймають
передфільтр таким чином, щоб мембранний фільтр не
забруднився домішками. У противному разі його
заміняють іншим мембранним фільтром і операцію
повторюють за тих самих умов.
До фільтрату або до зазначеного в окремій статті
об’єму фільтрату додають 2 мл буферного розчину
рН 3.5 Р і перемішують. Одержану суміш додають до
1.2 мл реактиву тіоацетаміду Р і перемішують, зали-
шають на 10 хв і знову фільтрують, як описано вище,
але розташування фільтрів змінюють таким чином,
щоб рідина проходила спочатку крізь мембранний
фільтр, а потім - крізь передфільтр (Рис. 2.4.8.-1). Тиск
поршня шприца має бути помірним і постійним для
забезпечення повільного і рівномірного фільтрування.
Після закінчення фільтрування тримач відкривають,
мембранний фільтр виймають і висушують за допомо-
гою фільтрувального паперу.
Еталон готують аналогічно, використовуючи зазначе-
ний в окремій статті об’єм еталонного розчину свинцю
(1 ррт РЬ) Р.
Забарвлення мембранного фільтра, одержане в досліді
з випробовуваним розчином, має бути не інтен-
сивнішим за забарвлення мембранного фільтра, одер-
жане у досліді з еталоном.
МЕТОД Е
Кількість випробовуваної речовини, зазначену в ок-
ремій статті, поміщають у чисту суху колбу К’єльдаля
місткістю 100 мл (у разі інтенсивного ціноутворення
треба застосувати колбу місткістю 300 мл). Колбу
Рисунок 2.4.8.-1. Прилад для випробування на солі важких металів (метод Е)
Розміри зазначені у міліметрах
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
79
2.4. Випробування на граничний вміст домішок
закріплюють під кутом 45° і додають суміш 8 мл кисло-
ти сірчаної Р і 10 мл кислоти азотної Ру кількості,
достатній для повного змочування випробовуваної ре-
човини. Обережно нагрівають до початку реакції,
потім дають реакції стихнути і додають додаткові
порції тієї самої суміші кислот, нагріваючи після кож-
ного додавання. Операцію повторюють, доки об’єм
доданої суміші кислот не досягне 18 мл. Посилюють
нагрівання і обережно кип’ятять до потемніння роз-
чину. Охолоджують, додають 2 мл кислоти азотної Р і
знову нагрівають до потемніння розчину. Продовжу-
ють додавання кислоти азотної Рз наступним нагрі-
ванням, доки розчин не перестане темніти, потім
сильно нагрівають до появи густих білих парів. Охоло-
джують, обережно додають 5 мл води Р, кип’ятять з
обережностями до появи густих білих парів і продов-
жують нагрівання до одержання залишку об’ємом від
2 мл до 3 мл. Охолоджують, обережно додають 5 мл во-
ди Р і визначають забарвлення розчину. Якщо розчин
має жовте забарвлення, додають краплями 1 мл розчи-
ну водню пероксиду концентрованого Р і знову нагрі-
вають до появи густих білих парів і продовжують
нагрівання до одержання залишку об’ємом від 2 мл до
З мл. Якщо забарвлення розчину все ще залишається
жовтим, повторюють додавання 5 мл води Рі 1 мл роз-
чину водню пероксиду концентрованого Р до знебарв-
лення розчину. Охолоджують, обережно доводять
об’єм розчину водою Рц,о 25 мл.
Доводять рН розчину до 3.0-4.0 розчином аміаку кон-
центрованим РІ (при наближенні до зазначеного зна-
чення рН можна застосовувати розчин аміаку розведе-
ний РІ), використовуючи як зовнішній індикатор
індикаторний папір, що діє у вузькому інтервалі рН,
потім доводять об’єм розчину водою Р до 40 мл, пе-
ремішують і додають 2 мл буферного розчину рН 3.5 Р.
Одержану суміш додають до 1.2 мл тіоацетамідного
реактиву Р і негайно перемішують. Доводять об’єм
розчину водою Рро 50 мл і перемішують.
Паралельно за цих самих умов готують еталон, вико-
ристовуючи замість випробовуваної речовини зазна-
чений в окремій статті об’єм еталонного розчину свин-
цю (10 ррт РЬ) Р.
Через 2 хв коричневе забарвлення випробовуваного
розчину має бути не інтенсивнішим за забарвлення
еталона.
2.4.9. ЗАЛІЗО
Кількість випробовуваної речовини, зазначену в ок-
ремій статті, розчиняют ь у воді Р, доводять об’єм роз-
чину водою Рдо 10 мл і перемішують або використову-
ють 10 мл розчину, зазначеного в окремій статті Дода-
ють 2 мл розчину 200 г/л кислоти лимонної Р і 0.1 мл
кислоти тіогліколевої Р. Перемішують, підлужують
розчином аміаку Р, доводять об’єм розчину водою Р до
20 мл
Паралельно за цих самих умов готують еталон, використо-
вуючи 10 мл еталонного розчину заліза( ІII) (1 ррт Ге) Р
Через 5 хв рожеве забарвлення випробовуваного ро
ну має бути не інтенсивнішим за забарвлення еталс
2.4.10. СВИНЕЦЬ У ЦУКРАХ
Визначення свинцю проводять методом атомно- і
сорбційної спектрометрії (2.2.23, метод 2).
Випробовуваний розчин. 20.0 г випробовуваної речовеИ
ни розчиняють у суміші рівних об’ємів кислоти оцп
воїрозведеної Рі води Р, доводять об’єм розчину ції Д
самою сумішшю розчинників до 100.0 мл і переміцЯ
ють. Додають 2.0 мл насиченого розчину (близьЛ
10 г/л) амонію піролідиндитіокарбамату Рі 10.0 мліЛ
тилізобутилкетону Р і струшують протягом ЗО с) Л
хищеному від яскравого світла місці. Залишають
розшарування. Для випробування використовую^
шар метил ізобутил кетону.
Розчини порівняння. Готують три розчини порівнянні
аналогічно до випробовуваного розчину, але з додавані
ням до 20.0 г випробовуваної речовини відповідно 0.5 м.
1.0 мл і 1.5 мл еталонного розчину свинцю (10ррт РЬ) Р. І
Установлюють нульову точку на приладі, ви користі
вуючи метилізобутилкетон Р, оброблений аналогічні
до випробовуваного розчину, але без додавання ви
пробовуваної речовини. Вимірюють поглинання
довжини хвилі 283.3 нм, використовуючи як джере.
випромінювання лампу з порожнистим свинцевий
катодом і повітряно-ацетиленове полум’я.
Випробовувана речовина має містити не більше
0.5 ррт свинцю, якщо немає інших зазначень в ок
ремій статті.
2.4.11. ФОСФАТИ
До 100 мл приготованого і, якщо необхідно, нейт-
ралізованого, як зазначено в окремій статті, розчин
додають 4 мл сульфомолібденового реактиву Рі |
Струшують і додають 0.1 мл розчину олова) ! І) хлори-
ду РІ.
Паралельно за цих самих умов готують еталон, вико-
ристовуючи замість 100 мл розчину випробовуваної
речовини суміш 2 мл еталонного розчину фосфату
(5ррт РО4) Рі 98 мл води Р. Через 10 хв порівнюють
забарвлення 20 мл кожного розчину.
Забарвлення випробовуваного розчину має бути не
інтенсивнішим за забарвлення еталона.
2.4.12. КАЛІЙ
До 10 мл розчину, приготованого, як зазначено в ок-
ремій статті, додають 2 мл свіжоприготованого розчи-
ну 10 г/л натрію тетрафенілборату Р.
Паралельно за цих самих умов готують еталон, викори-
стовуючи замість 10 мл розчину випробовуваної речо-
вини суміш 5 мл еталонного розчину калію (20ррт К) Р
і 5 мл води Р.
80
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
Через 5 хв опалесценція випробовуваного розчину не
чає перевищувати опалесценцію еталона.
вання до постійної маси, якшо немає інших зазначень
в окремій статті.
2.4.13. СУЛЬФАТИ
При приготуванні усіх розчинів, застосовуваних у да-
ному випробуванні, має використовуватися вода дис-
тильована Р.
До 1.5 мл еталонного розчину сульфату (10 ррт 5О4 ) РІ
додають 1 мл розчину 250 г/л барію хлориду Р. Струшу-
ють і залишають на 1 хв, потім додають 15 мл випробо-
вуваного розчину, приготованого, як зазначено в ок-
ремій статті, і 0.5 мл кислоти оцтової Р.
Паралельно за цих самих умов готують еталон, вико-
ристовуючи замість випробовуваного розчину 15 мл
еталонного розчину сульфату (10 ррт 8О4 ) Р.
Через 5 хв опалесценція випробовуваного розчину не
має перевищувати опалесценцію еталона.
2.4.14. СУЛЬФАТНА ЗОЛА
Цей метод застосовують, якщо немає інших зазначень
в окремій статті.
Фарфоровий, кварцовий або платиновий тигель
нагрівають при червоному жару протягом ЗО хв, охо-
лоджують в ексикаторі і зважують. Випробовувану ре-
човину поміщають у тигель і додають 2 мл кислоти
сірчаної розведеної Р, нагрівають спочатку на водяній
бані, потім обережно на полум’ї. Потім температуру
поступово збільшують до 600 °С і продовжують спа-
лювання до зникнення темних часток. Тигель залиша-
ють до охолодження, додають декілька крапель кисло-
ти сірчаної розведеної Р, нагрівають і спалюють, як
описано вище, потім знову охолоджують. Додають
декілька крапель розчину амонію карбонату Р. Упарю-
ють і обережно спалюють, охолоджують в ексикаторі,
зважують і повторюють спалювання по 15 хв до
постійної маси.
________________________________________N
МЕТОДА
Близько 1 г (точна наважка) або зазначену в окремій
статті кількість випробовуваної речовини поміщають
у попередньо прожарений і зважений фарфоровий,
кварцовий або платиновий тигель, змочують 1 мл кис-
лоти сірчаної Р, обережно нагрівають на полум’ї або
на піщаній бані до видалення пари кислоти сірчаної і
прожарюють при температурі (600+25) °С до зникнен-
ня темних часток. Після закінчення спалювання ти-
гель охолоджують в ексикаторі, зважують і обчислю-
ють вміст зольного залишку у випробовуваній речо-
вині. Якщо вміст золи перевищує межу, зазначену в
окремій статті, залишок знову змочують 1 мл кислоти
сірчаної Р, нагрівають і спалюють, як описано вище, і
знову обчислюють вміст золи. Продовжують спалю-
МЕТОД В
Близько 1 г (точна наважка) або зазначену в окремій
статті кількість випробовуваної речовини поміщають
у попередньо прожарений і зважений фарфоровий,
кварцовий або платиновий тигель. Обережно нагріва-
ють на полум’ї або на піщаній бані до повного обвуг-
лювання речовини, охолоджують і, якщо немає інших
зазначень в окремій статті, змочують залишок 1 мл
кислоти сірчаної Р, обережно нагрівають до видалення
пари кислоти сірчаної і спалюють при температурі
(800±25) °С до зникнення темних часток. Після закін-
чення спалювання тигель охолоджують в ексикаторі,
зважують і обчислюють вміст зольного залишку у ви-
пробовуваній речовині. Якщо вміст золи перевищує
межу, зазначену в окремій статті, залишок знову змо-
чують 1 мл кислоти сірчаної Р, нагрівають і спалюють,
як описано вище, і знову обчислюють вміст золи.
Продовжують спалювання до постійної маси, якщо
немає інших зазначень в окремій статті.
2.4.15. НІКЕЛЬ У ПОЛЮЛАХ
Визначення нікелю проводять методом атомно-аб-
сорбційної спектрометрії (2.2.23, метод 2).
Випробовуваний розчин. 20.0 г випробовуваної речови-
ни розчиняють у суміші рівних об’ємів кислоти оцто-
вої розведеної Р і води Р, доводять об’єм розчину цією
самою сумішшю розчинників до 100.0 мл і перемішу-
ють. Додають 2.0 мл насиченого розчину (близько
10 г/л) амонію піролідиндитіокарбамату Р\ 10.0 мл ме-
тилізобутилкетону Р і струшують протягом ЗО с у за-
хищеному від яскравого світла місці. Залишають до
розшарування. Для випробування використовують
шар метилізобутилкетону.
Розчини порівняння. Готують три розчини порівняння
аналогічно до випробовуваного розчину, але з дода-
ванням до 20.0 г випробовуваної речовини відповідно
0.5 мл, 1.0 мл і 1.5 мл еталонного розчину нікелю
(10 ррт N1) Р.
Установлюють нульову точку на приладі, використо-
вуючи метилізобутилкетон Р, оброблений аналогічно
до випробовуваного розчину, але без додавання ви-
пробовуваної речовини. Вимірюють поглинання за
довжини хвилі 232.0 нм, використовуючи як джерело
випромінювання лампу з порожнистим нікелевим ка-
тодом і повітряно-ацетиленове полум’я.
Випробовувана речовина має містити не більше 1 ррт
нікелю, якщо немає інших зазначень в окремій статті.
2.4.16. ЗАГАЛЬНА ЗОЛА
Фарфоровий, кварцовий або платиновий тигель
нагрівають при червоному жару протягом 30 хв, охо-
лоджують в ексикаторі і зважують. Якщо немає інших
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
81
2.4. Випробування на граничний вміст домішок
зазначень в окремій статті, 1.00 г випробовуваної ре-
човини або здрібненої на порошок лікарської рослин-
ної сировини поміщають у тигель і рівномірно роз-
поділяють по дну тигля. Висушують при температурі
від 100 °С до 105 °С протягом 1 год і потім спалюють
до постійної маси у муфельній печі при температурі
(600±25) °С, охолоджуючи тигель в ексикаторі після
кожного спалювання. Протягом усієї процедури у
тиглі не повинно з’являтися полум'я. Якщо після три-
валого спалювання зола все ще містить темні частки,
вміст тигля кількісно переносять гарячою водою на
беззольний фільтр і спалюють залишок на фільтрі ра-
зом з фільтрувальним папером. Фільтрат об’єднують із
золою, обережно упарюють до сухого залишку і спа-
люють до постійної маси.
2.4.17. АЛЮМІНІЙ
Розчин випробовуваної речовини, приготований, як
зазначено в окремій статті, поміщають у ділильну
лійку, струшують з двома порціями, по 20 мл кожна,
розчину 5 г/л гідроксихіноліну Ру хлороформі Р, потім
з 10 мл цього самого розчину. Хлороформні шари
відділяють і збирають у мірну колбу місткістю 50.0 мл
Доводять об’єм розчину хлороформом Рдо познач-
ки і перемішують (випробовуваний розчин).
Еталон готують аналогічно, використовуючи зазначе-
ний в окремій статті розчин порівняння.
Холостий розчин готують аналогічно, використовую-
чи зазначений в окремій статті розчин
Вимірюють інтенсивність флуоресценції (2.2.21) ви-
пробовуваного розчину (І/), еталона (12) і холостого
розчину (7р, використовуючи збуджуюче випроміню-
вання за довжини хвилі 392 нм і вторинний фільтр із
смугою пропускання, що має максимум за довжини
хвилі 518 нм, або монохроматор, установлений на
пропускання цієї довжини хвилі.
Флуоресценція (1ГІз) випробовуваного розчину не
має перевищувати флуоресценцію еталона (І2~Із).
2.4.18. ВІЛЬНИЙ ФОРМАЛЬДЕГІД
Метод А застосовують, якщо немає інших зазначень в
окремій статті. Метод В застосовують для вакцин, в
яких для нейтралізації надмірного формальдегіду ви-
користовують натрію метабісульфіт.
Паралельно за цих самих умов готують еталон, впкс-Е
ристовуючи замість розведеної вакцини 1 мл /дазвди.
формальдегіду Р, розведеного до вмісту формальдегід
(СН2О) 20 мкг/мл.
Пробірки переглядають уздовж їх вертикальних осе,
Забарвлення випробовуваного розчину має бути ж
інтенсивнішим за забарвлення еталона.
МЕТОД В
У пробірку поміщають 0.5 мл випробовуваної вакци І
ни, розведеної у співвідношенні Ідо 100, додають 5 м І
розчину 0.5 г/л метилбензотіазолонгідразону г/фохк-і
раду Р і 0.05 мл полісорбату 80 Р, пробірку закривають!
струшують і залишають на 60 хв. Потім додають 1 м |
реактиву заліза(ПІ) хлориду і сульфамінової кислоти ’
і залишають на 15 хв.
Паралельно за цих самих умов готують еталон, викг
ристовуючи замість 0.5 мл розведеної випробовуванії
вакцини 0.5 мл розчину формальдегіду Р, розведеним
до вмісту формальдегіду (СН2О) 5 мкг/мл.
Вимірюють оптичну густину (2.2.25) одержаних ро
чинів на спектрофотометрі за довжини хвилі 628 нм
використовуючи як компенсаційний розчин ХОЛОСТИі
розчин.
Оптична густина випробовуваного розчину не маєш-1
ревищувати оптичну густину еталона.
Якщо випробовувана вакцина являє собою емульсіиі
водну фазу відділяють таким способом. До вакцини!
додають рівний об’єм ізопропілміристапіу Р і не
ремішують. До трьох об’ємів одержаної суміші дода-
ють два об’єми 1 Мрозчину кислоти хлористоводневая
три об’єми хлороформу Рі чотири об’єми розчину9г/,!
натрію хлориду Р. Ретельно перемішують і центри фі-
гу юті, з прискоренням 15 000 § протягом 60 хв. Водний
шар відбирають і вимірюють його об’єм. Водний шар
використовують для випробування на формальдегід
як зазначено вище.
Обчислюють концентрацію формальдегіду в еталоні
з урахуванням розведення вакцини в процесі розді-
лення шарів і готують еталон з відповідною конпен
рацією.
Якщо описана процедура не дозволяє досягти розді-
лення шарів, додають 100 г/л полісорбату 20 Рдороь
чину 9 г/л натрію хлориду Р і повторюють процедуру,
центрифугуючи з прискоренням 22 500 у.
МЕТОДА
Вакцини, використовувані для людини, розводять у
10 разів. Бактеріальні токсоїди, застосовувані у вете-
ринарії, розводять у 25 разів.
1 мл випробовуваної вакцини, розведеної, як зазначе-
но вище, поміщають у пробірку і додають 4 мл води Рі
5 мл реактиву ацетилацетону РІ. Пробірку поміща-
ють у водяну баню з температурою 40 °С на 40 хв.
2.4.19. ЛУЖНІ ДОМІНІКИ У ЖИРНИХ ОЛІЯХ
У пробірку поміщають 10 мл свіжоперегнаного ацето-
ну Р і 0.3 мл води Р, додають 0.05 мл розчину 0.4 г/д
бромфеполового синього Ру спирті Р; якщо необхідно,
розчин нейтралізують 0.01 Мрозчином кислоти хлори-
стоводневої або 0.01 М розчином натрію гідроксиду,
потім додають 10 мл випробовуваної олії, струшуютьі
залишають до розділення шарів.
82
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
зміни забарвлення верхнього шару на жовте має
гни витрачено не більше 0.1 мл 0.01 Мрозчину кисло-
ти аористоводневої.
2.4.20. АНТИОКСИДАНТИ У ЖИРНИХ ОЛІЯХ
Ііиздобування проводять методом тонкошарової хро-
члографії (2.2.27), використовуючи як тонкий шар
ипікаге\ь с Р. Перед застосуванням пластинки вису-
гютьпри температурі 130 °С протягом 2 год.
"Лшробовуваний розчин (а). 20 г випробовуваної речо-
№и, взятої з центра зразка, або 20 г олії поміщають
. їли.іьн) лійку, додають 50 мл петролейного ефіру Р
енергійно струшують з двома порціями по 30 мл ме-
г.ягау (75 % об/об). Після чіткого розділення шарів
кні, метанольні, шари об’єднують і випарюють при
ширеному тиску і якомога більш низькій температурі
..гмосфсрі азоту. Залишок розчиняють у 5 мл хлоро-
'.гиу, вільного від етанолу Р. Зберігають у добре заку-
пній посудині.
°;иробовуваний розчин (Ь). Шар петролейного ефіру
верхній шар), одержаний при приготуванні випробо-
аного розчину (а), обережно упарюють до сухого
залишку. Додають 0.5 г пірогалолу Р, розчиненого у
п мл етанолу Р, потім 15 мл свіжоприготованого
рі мину 330 г/л натрію гідроксиду Р і кип’ятять із зво-
тм холодильником протягом 30 хв. Охолоджують,
лі.іають250 мл води Рі витягують неомилювані речо-
і гни трьома порціями по 100 мл петролейного ефіру Р.
Ію'еднані витяги петролейного ефіру промивають во-
гіою Рпо відсутності лужної реакції середовища і упа-
юють до сухого залишку. Залишок розчиняють у 5 мл
Проформу, вільного від етанолу Р. Зберігають у
ьно закупореній посудині.
А. НЕПОЛ1ПДРОКС1АНТИОКСИДАНТИ
ГИ.стику поміщають у хроматографічну камеру з
иороформом, вільним від етанолу, Р. Коли фронт роз-
чинника пройде близько 12 см від нижнього краю
тастинки. пластинку виймають з камери, висушують
протягом 20 хв на повітрі, потім протягом 20 хв в екси-
каторі під вакуумом.
На лінію старту хроматографічної пластинки, підгото-
ваної, як зазначено вище, у стартову точку № 1
(Рис. 2.4.20.-1) наносять випробовуваний розчин (а) у
вигляді плями діаметром не більше 5 мм. Об’єм ви-
пробовуваного розчину (а), що наноситься, залежить
під його концентрації і становить звичайно від 2 мкл
.о 10 мкл. У стартові точки № 2 і № 3 (Рис. 2.4.20.-1)
наносять по 2 мкл забарвленого розчину, шо містить
по 0.1 г/л диметилового жовтого Р, Судану червоно-
ЮЄ Р та індофенолового синього Ру бензолі Р. На плас-
тинці позначають довжину пробігу (10 см) у двох на-
прямках. Першим разом пластинку хроматографують
\ камері з хлороформом, вільним від етанолу Р. Плас-
тинку висушують на повітрі протягом 10 хв, потім по-
вертають на 90° і хроматографують у камері з бензо-
юи Р. Пластинку висушують на повітрі протягом 5 хв
і обприскують розчином 200 г/л кислоти фосфорно-
молібденової Р в етанолі Р до постійного забарвлення
пластинки у жовтий колір. Через 2 хв починають про-
являтися сині плями. Протягом перших 5-10 хв плас-
тинку обробляють парами аміаку, доки фон не стане
чисто білим. На пластинці залишаються сині, світло-
фіолетові або зеленуваті плями.
Хроматограму оцінюють, порівнюючи з Рис. 2.4.20,-1.
Якщо на лінії старту виявляються сині плями, прово-
дять розділення й ідентифікацію полігідроксіантиок-
сидантів (метод В).
Рисунок 2.4.20.-1. Типові хроматограми
антиоксидантів (методи А і С)
а = Стартова точка № 1 + пляма галатів + пляма
нордигідрогваярепіової кислоти
Ь = Стартова точка № 2
с = Стартова точка № З
І = Гваякова смола
2 = 3-( 1,1-Диметилетил)-4-метоксифенол
З = 2-( 1,1-Диметилепшл)-4-метоксифенол
4 = 2,2,5, 7,8-Пентаметил-6-хроманол
5 = Тетраетилтіурам дисульфід
6 = а- Токоферол
7 = Дибутилгідроксіанізол
8 = Бутилгідроксітолуол
А = жовтий
В = червоний
С = синій
Метод А: суцільна лінія
Метод С: пунктирна лінія
В. ПОЛІ ГІДРОКСІ АНТИОКСИДАНТИ
Налінію старту хроматографічної пластинки наносять
1 мкл, 2 мкл, 4 мкл і 6 мкл випробовуваного розчи-
ну (а), а також від 1 мкл до 2 мкл забарвленого розчи-
ну, приготованого, як зазначено в методі А. Пластин-
ку поміщають у камеру із сумішшю розчинників: кис-
лота оцтова льодяна Р - бензол Р - петролейний ефір Р
(30:60:60). Коли фронт розчинників пройде близько
13 см від лінії старту, пластинку виймають із камери.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
83
2.4. Випробування на граничний вміст домішок
висушують на повітрі, обприскують розчином 200 г/л
кислоти фосфорномолібденової Р в етанолі Р і продов-
жують проявлення за методикою, описаною для іден-
тифікації неполігідроксіантиоксидантів.
Полігідроксіантиоксиданти ідентифікують, порівню-
ючи положення плям на хроматограмі випробовува-
ного розчину по відношенню до плям на хроматограмі
забарвленого розчину (див. Рис. 2.4.20.-2).
Рисунок 2.4.20.-2. Типові хромотограми
полігідроксіантиоксидантів (Метод В)
1 = Забарвлений розчин
2 = Бутилований гідроксіинізол
З = Гваякова смола
4 = Нордигідрогваярепюва кислота
5 = Мепіилгалат
6 = Етилгалат
7 = Пропілгалат
8 = Октилгалат
9 = Додецилгалат
А = жовтий
В = червоний
С = синій
С. АНТИОКСИДАНТИ, ЩО НЕ ВИТЯГАЮТЬСЯ
МЕТАНОЛОМ
Випробування проводять методом тонкошарової хро-
матографії на іншій пластинці. Хроматографування
проводять за методикою, описаною для неполігідроксі-
антиоксидантів, використовуючи замість випробову-
ваного розчину (а) випробовуваний розчин (Ь). Плас-
тинку обприскують розчином 10 г/л дихлорхінон-
хлоріміду Рв етанолі Р. Плями виявляються протягом
15 хв. Хроматограму оцінюють, порівнюючи з
Рис. 2.4.20.-1. Зони, позначені пунктиром, відповіда-
ють а-токоферолу і бутилованому гідрокситолуолу.
Плями р- і у-токоферолу знаходяться у зоні, відповід-
ній 2-( 1.1-диметилетил)-4-метоксифенолу.
Якщо у випробовуваній речовині в істотній кількості
присутній бутилований гідрокситолуол, його визнача-
ють за методом А.
2.4.21. СТОРОННІ ОЛІЇ У ЖИРНИХ ОЛІЯХ
МЕТОДОМ ТОНКОШАРОВОЇ
ХРОМАТОГРАФІЇ
Випробування проводять методом тонкошарової хр
матографії (2.2.27), використовуючи як тонкий шс*
кізельгур б Р. Пластинку імпрегнують, помістивши!
камеру, яка містить суміш вазелінового масла Ріпп
релейного ефіру Р( 10:90) у такій кількості, щоб нижнії
край пластинки занурився на 5 мм нижче поверхів
рідини. Коли суміш для імпрегнування підніметься
менш як на 12 см від нижнього краю пластинки.її
виймають із камери і дають розчиннику випарити»»
протягом 5 хв. Наступне хроматографування ііроиі
дять у тому самому напрямку, іцо й імпрегнування. І
Приготування суміші жирних кислот. До 2 г олії дощ-
ють 30 мл 0.5 М розчину калію гідроксиду спиртоеоЛ
і нагрівають із зворотним холодильником протягом
45 хв. Додають 50 мл води Р, залишають до охолоджен-
ня, переносять у ділильну лійку і струшують з трьом» 1
порціями ефіру Р, по 50 мл кожна. Ефірні шарі
відділяють, водний шар підкислюють кислотою хіори- І
стоводневою Р і знову струшують з трьома порція» І
ефіру Р, по 50 мл кожна. Усі ефірні шари об’єднують І
і струшують з трьома порціями води Р, по 10 мл кожні. І
відкидаючи промивні води. Об’єднані ефірні шарив* І
сушують над натрію сульфатом безводним Рі фільтр
ють. Потім ефір випарюють на водяній бані, а із з* з
лишку готують випробовуваний розчин або розчин]
порівняння. Жирні кислоти також можна витягти І і
розчину, одержаного в ході визначення неомилювант
речовин.
Випробовуваний розчин. 40 мг суміші жирних кислот |
одержаної з випробовуваної олії, розчиняють у 4 м.
хлороформу Р.
Розчин порівняння. 40 мг суміші жирних кислот, одср-ч
жаноїз суміші 19 об’ємів кукурудзяної олії Р і 1 об’єм?
рапсової олії Р, розчиняють у 4 мл хлороформу Р.
На лінію старту хроматографічної пластинки наносять
по 3 мкл кожного розчину. Поміщають пластинку в
камеру з сумішшю розчинників: вода Р - кислота оц-
това льодяна Р (10:90). Коли фронт розчинники
пройде 8 см від лінії старту, пластинку виймають із ка-
мери, висушують при температурі 110 °С протягом
10 хв і залишають до охолодження. Потім, якщо нема
інших зазначень в окремій статті, пластинку поміша-
ють у закриту щільно припасованою кришкою хрома-
тографічну камеру, попередньо насичену парами йод,
(на дно камери у випарювальній чашці помішений
йод Р). Через деякий час проявляються коричневі аби
жовтувато-коричневі плями. Пластинку виймають
із камери і залишають на кілька хвилин. Коли корич-
невий фон зникне, пластинку обприскують розчином
крохмалю Р. Сині плями, що з’являються, можуть на-
бувати коричневого забарвлення при висушуванні
і знову стають синіми після обприскування водою Р.
На хроматограмі випробовуваного розчину завжди
мають виявлятися пляма з В/ близько 0.5 (олеїнова
кислота) і пляма з Ау близько 0.65 (лінолева кислота),
відповідні плямам на хроматограмі розчину порівнян-
ня. На хроматограмах деяких олій може виявлятися
84
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
ЯшзЛ, близько 0.75 (ліноленова кислота). Шляхом
Порівняння плям на хроматограмі випробовуваного
доу з плямами на хроматограмі розчину порівнян-
ні переконуються у відсутності на хроматограмі ви-
!|пч'б чуваного розчину плями з Яу близько 0.25 (еру-
іпш кислота).
2.4.22. СТОРОННІ ОЛІЇ У ЖИРНИХ ОЛІЯХ
МЕТОДОМ ГАЗОВОЇ ХРОМАТОГРАФІЇ
™ бхвання на сторонні олії проводять шляхом пе-
реведення жирних кислот, що містяться у випробову-
КГНїй олії, у метилові ефіри.
МЕТОДА
Ц/іі метод незастосовний для олій, що містять гліце-
► жирних кислот з епокси-, гідроксіепокси-, цикло-
ві овими або циклопропеніловими групами, а також
чіп олій, у складі яких велика частина жирних кислот
чш довжину ланцюга менше восьми атомів вуглецю, і
ІП шій з кислотним числом більше 2.0.
.'апробування проводять методом газової хромато-
щефії (2.2.28).
Лмробовуваний розчин. Випробовувану олію висушу-
іль перед метилуванням, якщо це зазначено в ок-
ремій статі 1.0 г олії поміщають у круглодонну колбу
МІСТКІСТЮ 25 мл зі шліфом, споряджену зворотним хо-
Йильником і газовідвідною трубкою. У колбу дода-
<і;ь 10 мл метанолу безводного Р, 0.2 мл розчину 60 г/л
цігію гідроксиду Р у метанолі Р, приєднують зворот-
ним холодильник і, пропускаючи крізь суміш азот Р із
швидкістю близько 50 мл/хв, струшують і нагрівають
Ю кипіння. Коли розчин стане прозорим (звичайно
через 10 хв), продовжують нагрівання протягом на-
сипних 5 хв. Потім колбу охолоджують під проточ-
ною водою і вміст переносять у ділильну лійку. Кол-
бі промивають 5 мл гептану Р, переносять змиви у ту
с.ім\ ділильну лійку і струшують. Додають 10 мл роз-
чині' 200 г/л натрію хлориду Р і енергійно струшують.
Залишають до розшарування, потім переносять ор-
і знімний шар у посудину, що містить натрію сульфат
безводний Р, і через деякий час фільтрують.
Розчин порівняння (а). Готують 0.50 г суміші речовин, за-
нотовуваних для калібрування (каліброваної суміші)
складу, наведеного в одній з Табл. 2.4.22, як зазначено в
окремій статті (якщо в окремій статті не зазначений
певний розчин, готують суміш складу, наведеного в
Табл. 2.4.22.-1). Суміш розчиняють у гептані Р і дово-
дять об’єм розчину тим самим розчинником до 50.0 мл.
Розчин порівняння (Ь). 1.0 мл розчину порівняння (а)
доводять гептаном Рао 10.0 мл.
Хроматографують на газовому хроматографі з по-
іуменево-іонізаційним детектором за таких умов:
- колонка скляна або з нержавіючої сталі завдовжки
від 2 м до 3 м і внутрішнім діаметром від 2 мм до
4 мм, заповнена діатомітом для газової хромато-
графії Рз розміром часток від 125 мкм до 200 мкм,
з нанесеним від 5 % до 15 % поліетиленглікольсук-
цинатом Р або поліетиленглікольадипінатом Р;
— газ-носій азот для хроматографії Р;
— швидкість газу-носія 25 мл/хв;
— температура колонки 180 °С;
— температура блока вводу проб і детектора 200 °С.
Якщо необхідно або якщо зазначено в окремій статті,
температуру колонки збільшують від 120 °С до 200 °С
із швидкістю 5 °С за хв.
Хроматографувати можливо також на газовому хрома-
гографі з полуменево-іонізаційним детектором за та-
ких умов:
— колонка капілярна скляна або кварцова зав-
довжки від 10 м до 30 м і внутрішнім діаметром від
0.2 мм до 0.8 мм, внутрішня поверхня якої вкрита
шаром полі[(ціанопропіл)(метил)][(феніл)(метил)]
силоксану Рабо макроголу 20 000 Р з товщиною ша-
ру від 0.1 мкм до 0.5 мкм або іншою підхожою не-
рухомою фазою;
— газ-носій гелій для хроматографії Р або водень для
хроматографії Р;
— швидкість газу-носія 1.3 мл/хв (для колонки з
внутрішнім діаметром 0.32 мм);
— поділ потоку 1:100 або менше у залежності від
внутрішнього діаметра застосовуваної колонки (у
разі використання колонки з внутрішнім діамет-
ром 0.32 мм поділ потоку має становити 1:50);
— температура колонки від 160 °С до 200 °С, у залеж-
ності від довжини колонки і використовуваної не-
рухомої фази (для колонки завдовжки 30 м, вкритої
шаром макроголу 20 000 Р, температура має стано-
вити 200 °С);
— температура блока вводу проб і детектора 250 °С.
Якщо необхідно або якщо зазначено в окремій статті,
температуру колонки збільшують від 170 °С до 230 °С
із швидкістю З °С за хв (для колонки, вкритої шаром
макроголу 20 000 Р).
Хроматографують 0.5 мкл розчину порівняння (а).
Чутливість системи регулюють таким чином, щоб ви-
сота основного піка на одержаній хроматограмі стано-
вила від 50 % до 70 % шкали реєструючого пристрою.
Визначають часи утримування жирних кислот, що
входять до складу калібрувальної суміші. Хроматогра-
фують 1 мкл розчину порівняння (Ь) і розраховують
відношення сигнал/шум для піка, відповідного ме-
тилміристату.
Хроматографують від 0.5 мкл до 1.0 мкл випробовува-
ного розчину. Час хроматографування має бути у 2.5
рази більше часу утримування метилолеату. Хромато-
граму оцінюють, як описано нижче.
При використанні калібрувальних сумішей № 1 або
№ 3 хроматографічна система вважається придатною,
якщо виконуються такі умови:
— на хроматограмі розчину порівняння (а) число тео-
ретичних тарілок (п) (2.2.28), обчислене за піком,
відповідним метилстеарату, становить не менше
2000 для набивної колонки і не менше 30 000 для
капілярної колонки;
— на хроматограмі розчину порівняння (а) ко-
ефіцієнт розділення /АД (2.2.28) піків, відповідних
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
85
2.4. Випробування на граничний вміст домішок
метилолеату і метилстеарату, становить не менше
1.25 для набивної колонки і не менше 1.8 для
капілярної колонки;
— на хроматограмі розчину порівняння (Ь) відно-
шення сигнал/шум (2.2.28) для піка метилміриста-
ту становить не менше 5
При використанні калібрувальної суміші № 2 хрома-
тографічна система вважається придатною, якщо ви-
конуються такі умови:
— на хроматограмі розчину порівняння (а) число те-
оретичних тарілок (н) (2.2.28), обчислене за піком,
відповідним метилкапрату, становить не менше
1500 для набивної колонки і не менше
15 000 для капілярної колонки;
— на хроматограмі розчину порівняння (а) ко-
ефіцієнт розділення (К^) (2.2.28) піків, відповідних
метилкаприлату і метилкапрату, становить не мен-
ше 2 для набивної колонки і не менше 4 для
капілярної колонки;
— на хроматограмі розчину порівняння (Ь) відношен-
ня сигнал/шум (2.2.28) для піка метилкапроату
становить не менше 5.
ОЦІНКА ХРОМАТОГРАМ
Треба уникати умов хроматографування, шо можуть
дати нерозділені піки (наявність компонентів із неве-
ликою відмінністю між часами утримування, напри-
клад, ліноленова і арахідонова кислоти).
Якісний аналіз. Будують калібрувальну криву, викори-
стовуючи хроматограми розчину порівняння (а) і дані
Таблиць 2.4.22.
а) Для хроматограм, одержаних в ізотермічному ре-
жимі, обчислюють логарифми зведених часів утриму-
вання як функцію еквівалента числа атомів вуглецю
у жирних кислотах. Калібрувальна крива насичених
кислот являє собою пряму лінію. Логарифми зведених
часів утримування ненасичених кислот розташовані
на цій лінії як точки, відповідні нецілим значенням
"еквівалента довжини ланцюга". Ідентифікацію ком-
понентів жирних кислот випробовуваної олії прово-
дять, розраховуючи логарифми зведених часів утриму-
вання піків, одержаних на хроматограмі випробовува-
ного розчину, і установлюючи за калібрувальною кри-
вою "еквіваленти довжини ланцюга".
б) Для хроматограм, одержаних з використанням
лінійного градієнта температури, визначають часи ут-
римування, що знаходяться у залежності від числа
атомів вуглецю в жирних кислотах, і ідентифікують
жирні кислоти, що входять до складу випробовуваної
олії, шляхом порівняння з калібрувальною кривою.
Кількісний аналіз. Звичайно використовують метод
внутрішньої нормалізації; при цьому суму площ усіх
піків на хроматограмі, крім піків, що відносяться до
розчинника, беруть за 100 %. Рекомендується застосу-
вання електронного інтегратора. Вміст кожного компо-
нента обчислюють як відношення площі відповідного
піка до суми площ усіх піків. Піки, площа яких стано-
вить менше 0.05 % від суми площ усіх піків, не врші
ють, якщо немає інших зазначень в окремій статті
У певних випадках, тобто при наявності жирних й
лот з 12 або менше атомами вуглецю, в окремій стаї
має бути зазначений поправочний коефіцієнт для і,
ретворсння площ піків у відсотки (м/м).
МЕТОД В
Цей метод незастосовний для олій, що містять глй.
риди жирних кислот з епокси-, гідрокс іепокси-,
пропіловими і циклопропеніловими групами і для олі;
кислотним числом більше 2.0.
Випробовуваний розчин. 0.100 г випробовуваної ре*
вини поміщають у нентрифугову пробірку з кришко
що закручується, розчиняють у 1 мл гептану Р і 1V
димепіилкарбонату Р і енергійно перемішують пя
помірному нагріванні (від 50 °С до 60 °С). До шетеп
лого розчину додають 1 мл розчину 12 г/л натрію Р ,
метанолі безводному Р, приготованого з необхідної’!
обережністю, і енергійно перемішують протягом 5 та
Додають 3 мл води дистильованої Р і енергійно пе ।
ремішують протягом 30 с. Центрифугують протятої;
15 хв з прискоренням 1500 Хроматографують 1 мк
органічного шару.
Розчини порівняння і оцінка хроматограм. Якщо в о»
ремій статті немає інших зазначень, діють, як зазнач! !
но у методі А.
Хроматографують на газовому хроматографі з по-І
луменево-іонізаційним детектором за таких умов:
— кварцова колонка завдовжки 30 м, з внутрішнічі
діаметром 0.25 мм, покрита шаром макрогол
20 000 Рз товщиною шару 0.25 мкм;
— газ-носій гелій для хроматографії Р;
— швидкість газу-носія 0.9 мл/хв;
— поділ потоку 1:100;
— температура колонки і швидкість підняття темпе-
ратури — за такою програмою:
Час (хв) Темпера- тура! С) Швид- кість ПІДНЯТТЯ темпера- тури (“ С/хв) Примітки !
Колонка 0-15 100 - Ізотермічний режим
15-36 100—225 10 Лінійний градієнт тем- ператури
36-61 225 - Ізотермічний режим
Блок вводу проб 250
Детектор 250
МЕТОД С
Цей метод незастосовний для олій, які містять гліце-
риди жирних кислот з епокси-, гідроперекисними, аль-
86
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
Приготування суміші речовин, застосовуваних для калібрування *
Табл. 2.4.22,-1
Склад (у відсотках м/м)
Речовина для калібрування Еквівалент довжини ланцюга** Ізотермічний режим Лінійний градієнт температури
(1) (2)
Метилмурат Р 12.0 12.0 5 10
Меішміристат Р 14.0 14.0 5 15
Меїтипальмітат Р 16.0 16.0 10 15
Мети ктеарат Р 18.0 18.0 20 20
Меітчарахідат Р 20.0 20.0 40 20
М.тчолеат Р 18.6 18.3 20 20
Табл. 2.4.22.-2
Приготування суміші речовин, застосовуваних для калібрування *
І- Склад (у відсотках м/м)
Речовина для калібрування Еквівалент довжини ланцюга** Ізотермічний режим Лінійний градієнт температури
(1) (2)
Метиікапроат Р 6.0 6.0 5 10
Мети ікаприлат Р 8.0 8.0 5 35
Мети ікапрат Р 10.0 10.0 10 35
Метииаурат Р 12.0 12.0 20 10
Меттміристат Р 14.0 14.0 40 10
Табл. 2.4.22.-З
Приготування суміші речовин, застосовуваних для калібрування *
Склад (у відсотках м/м)
Речовина для калібрування Еквівалент довжини ланцюга** Ізотермічний режим Лінійний градієнт температури
(1) (2)
Мети.іміристат Р 14.0 14.0 5 15
Мети тальмітат Р 16.0 16.0 10 15
Мети істеарат Р 18.0 18.0 15 20
Мети іарахідат Р 20.0 20.0 20 15
Метило.іеат Р 18.6 18.3 20 15
Меішиейкозаноат Р 20.2 20.2 10 10
Мети ібегенат Р 22.0 22.0 10 5
Меітшігноцерат Р 24.0 24.0 10 5
Для ГХ із застосуванням капілярної колонки і поділом потоку рекомендується додавати до суміші речовин, застосовуваних для калібру-
і.-ння. компоненти з більшою довжиною ланцюга.
"Ці значення, обчислені з використанням калібрувальної кривої, подані як приклад для колонки, заповненої поліетиленглікольсукцина-
Р( 1) і макроголом 20 000 Р (2).
дегідними, кетоновими, циклопропіловими і циклопро-
пеніювими групами і спряженими поліненасиченими і
ацетиленовими компонентами через часткове або по-
вне руйнування цих груп.
Випробовуваний розчин. 0.10 г випробовуваної речовини
поміщають у конічну колбу місткістю 25 мл, розчиня-
ють у 2 мл розчину 20 г/л натрію гідроксиду Р у мета-
нолі Р і кип'ятять із зворотним холодильником протя-
гом ЗО хв. Потім через холодильник додають 2.0 мл роз-
чину бору фториду в метанолі Р і кип’ятять ще ЗО хв,
після чого додають через холодильник 4.0 мл гептану Р
і кип’ятять 5 хв. Охолоджують, додають 10.0 мл розчину
натрію хлориду насиченого Р, струшують протягом 15 с
і додають такий об’єм розчину натрію хлориду насичено-
го Р, щоб верхній шар піднявся до шийки колби. Відби-
рають 2.0 мл верхнього шару, поміщають у ділильну
лійку, промивають трьома порціями води Р, по 2 мл
кожна, і висушують над натрію сульфатом безводним Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
87
2.4. Випробування на граничний вміст домішок
Розчини порівняння, хроматографічна методика і
оцінка хроматограм. Якщо в окремій статті немає
інших зазначень, діють, як зазначено у методі А.
_________________________________________N
МЕТОДА
Допускається застосування інших методик переведен-
ня жирних кислот, що містяться у випробовуваній
олії, у метилові ефіри, зазначених в окремих статтях.
ОЦІНКА ХРОМАТОГРАМ
Якісний аналіз, а) Допускається ідентифікація жирних
кислот випробовуваної олії шляхом порівняння часів
утримування піків на хроматограмі випробовуваного
розчину з часами утримування піків на хроматограмі
розчину порівняння або на стандартній хроматограмі,
зазначеній в окремій статті.
Зведений час утримування — різниця між часом утри-
мування піка речовини і часом утримування речови-
ни, що не сорбується (за умов визначення).
2.4.23. СТЕРИНИ У ЖИРНИХ ОЛІЯХ
ВІДОКРЕМЛЕННЯ СТЕРИНОВОЇ ФРАКЦІЇ
Одержують неомилювані речовини жирної ОЛІЇ 1 потім
відокремлюють стеринову фракцію методом тонкоша-
рової хроматографії (2.2.27). використовуючи як тон-
кий шар силікагель б Р завтовшки від 0.3 мм до 0.5 мм.
Випробовуваний розчин (а). У колбу місткістю 150 мл,
споряджену зворотним холодильником, поміщають
розчин 2 г/л бетуліну Р у метиленхлориді Р у такому
об’ємі, щоб кількість бетуліну відповідала близько
10 % вмісту стеринів у зразку, взятому для визначення
(тобто у випадку оливкової олії додають 500 мкл. у ви-
падку інших рослинних олій — 1500 мкл розчину бе-
туліну). Упарюють до сухого залишку під азотом Р.
Якщо в окремій статті визначається лише вміст
індивідуальних стеринів у відсотках до стеринової
фракції, розчин бетуліну можна не додавати.
У колбу поміщають 5.00 г випробовуваної речовини.
Додають 50 мл 2 М розчину калію гідроксиду спиртово-
го Р \ нагрівають на водяній бані протягом 1 год, часто
перемішуючи вміст колби коловими рухами. Охолод-
жують до температури нижче 25 °С і кількісно перено-
сять вміст колби у ділильну лійку, що містить 100 мл
води Р. Обережно струшують з трьома порціями ефіру,
вільного від пероксидів Р, по 100 мл кожна. Ефірні шари
збирають в іншу ділильну лійку, що містить 40 мл
води Р, злегка струшують протягом декількох хвилин,
залишають до розшарування і відкидають водний шар
Ефірний шар струшують з кількома порціями води Р,
по 40 мл кожна, доки водний шар не припинить дава-
ти лужну реакцію з фенолфталеїном. Ефірний шар
кількісно переносять у колбу, висушену до постійної
маси, промиваючи ділильну лійку ефіром, вільним від
пероксидів Р. Ефір випарюють з необхідними обере. І
ностями, до залишку додають 6 мл ацетону Р. Обере
но видаляють розчинник під струменем азоту Р. Вис і
шують до постійної маси при температурі від 100 °С до
105 °С, охолоджують в ексикаторі і зважують. Залиш» і
розчиняють у мінімальному об’ємі метиленхлориду Р. І
Випробовуваний розчин (Ь). У колбу місткістю 150 м1 ~
споряджену зворотним холодильником, помішати
5.00 г рапсової олії Р і далі діють, як зазначено дія ви
пробовуваного розчину (а), починаючи зі слів "до,1.
ють 50 мл 2 М розчину калію гідроксиду спиртового Р І
Випробовуваний розчин (с). У колбу місткістю 150 г 11
споряджену зворотним холодильником, поміщаюі. І
5.00 г соняшникової олії Р і далі діють, як зазначеної» І
випробовуваного розчину (а), починаючи зі слів"м
дають 50 мл 2 М розчину калію гідроксиду спиртові |
го Р".
Розчин порівняння. 25 мг холестерину Р і 10 мг а. ।
туліну Р розчиняють у 1 мл метиленхлориду Р.
Для кожного випробовуваного розчину використо»-І
ють окрему пластинку. На кожну з пластинок наїю-1
сять смугою розміром (20 х 3) мм 20 мкл розчиі |
порівняння і смугою розміром (40 х 3) мм 400 мкл ви І
пробовуваного розчину (а) або випробовуваного рої ]
чину (Ь), або випробовуваного розчину (с) відповідно
Пластинки поміщають у камеру із сумішшю розчин-1
ників ефір Р - гексан Р (35:65). Коли фронт розчин
ників пройде 18 см від лінії старту, пластинки вийма
ють із камери, висушують у струмені азоту Р, обпри-
скують розчином 2 г/л дихлорфлуоресцеїну Р в ста-1
нолі Рі переглядають в ультрафіолетовому світлі задо-І
вжини хвилі 254 нм. На хроматограмі розчину порі
вняння виявляються смуги, відповідні холестерину,]
бетуліну. На хроматограмах випробовуваних розчині)
виявляються смуги з близькими значеннями
відповідні стеринам. З хроматограми кожного випро-
бовуваного розчину знімають область покриття, і
якій розташовані смуги стеринів, а також додатком
зони на 2-3 мм вище і нижче видимих зон розчин,
порівняння, і поміщають нарізно у три колй
місткістю по 50 мл кожна. У кожну колбу додають по
15 мл теплого метиленхлориду Р \ струшують. Кожний
розчин фільтрують крізь скляний фільтр (40) аС.
підхожий фільтрувальний папір і промивають кожнім
фільтр трьома порціями метиленхлориду Р, по 15 мі
кожна. Об’єднані фільтрати і змиви поміщають у грі
колби, висушені до постійної маси, упарюють до сухо-
го залишку під струменем азоту Рі зважують.
ВИЗНАЧЕННЯ СТЕРИНІВ
Визначення стеринів проводять методом газової хро-
матографії (2.2.28).
Усі операції проводять, захищаючи розчини і реактиві
від вологи, розчини готують безпосередньо перед заїж-
суванням.
Випробовуваний розчин. До виділених з випробовува-
ної речовини методом тонкошарової хроматографії
стеринів додають свіжоприготовану суміш: хпортрв-
мепіилсилан Р - гексаметилдисилазан Р - піридин беї-
88
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
Мж.щ/’11:3:9) (0.02 мл суміші на міліграм залишку).
ЙСрежно струшують до повного розчинення залишку
І Впіиають в ексикаторі над фосфору(У) оксидом Р на
’шв. Якшо необхідно, центрифугують і використову-
падосадову рідину
Л'їми порівняння (а). До дев’яти частин стеринів,
її । .ених з рапсової олії Р методом тонкошарової хро-
мі,лграфії, додають одну частину холестерину Р. До
иержаної суміші додають свіжоприготовану суміш:
І триметилсилан Р- гексаметилдисилазан Р - піри-
Ьі безводний Р (1:3:9) (0.02 мл суміші на міліграм
Ніші стеринів) Обережно струшують до повного
розчинення залишку і залишають в ексикаторі над фо-
И оксидом Дна ЗО хв. Якщо необхідно, центри-
пють і використовують надосадову рідину.
ї-нин порівняння (Ь). До стеринів, виділених з соняш-
>в()ї олії Р методом тонкошарової хроматографії,
мають свіжоприготовану суміш: хлортриметилси-
Р - гексаметилдисилазан Р- піридин безводний Р
11'3:9) (0.02 мл суміші на міліграм суміші стеринів),
ііісрежно струшують до повного розчинення за-
„шку і залишають у ексикаторі над фосфору(У) окси-
о.кРна 30 хв. Якщо необхідно, центрифугують і ви-
црристовують надосадову рідину.
Хроматографують на газовому хроматографі з по-
іуменево-іонізаційним детектором за таких умов:
кварцова колонка завдовжки від 20 м до ЗО м і
внутрішнім діаметром від 0.25 мм до 0.32 мм, по-
крита полі[метил(95)феніл(5)]силоксаном Р або
по.ііІмеітсі(94)феніл(5)вініл( 1)]силоксаном Рз тов-
щиною шару 0.25 мкм;
- глз-носій водень для хроматографії Р (лінійна
швидкість від 30 см/с до 50 см/с) або гелій для хро-
матографії Р (лінійна швидкість від 20 см/с до
35 см/с). Лінійну швидкість вимірюють таким спо-
собом: за умов визначення стеринів уводять від
1 мкл до 3 мкл метану або пропану. Вимірюют ь час
у секундах, необхідний для проходження газу крізь
колонку від моменту вводу до появи піка (Ім).
Лінійну швидкість обчислюють за формулою
де £ — довжина колонки у сантиметрах;
— поділ потоку 1:50 або 1:100;
— температура колонки 260 °С;
— температура блока вводу проб 280 °С;
— температура детектора 290 °С.
Хроматографують за зазначених умов по 1 мкл кожно-
го розчину.
На хроматограмі розчину порівняння (а) виявляються
чотири основних піки, відповідних холестерину, браси-
кастерину, кампестерину і р-ситостерину: на хромато-
грамі розчину порівняння (Ь) виявляються чотири ос-
новних піки, відповідних кампестерину, стигмастерину,
Р-ситостерину і Д7-сигмастенолу. Часи утримування сте-
ринів відносно Р-ситостерину подані у Табл. 2.4.23.-1.
Пік внутрішнього стандарту (бетулін) має бути чітко
відділений від піків визначуваних стеринів.
Ідентифікують піки, виявлені на хроматограмі випро-
бовуваного розчину, і обчислюють відсотковий вміст
кожного стерину в стериновій фракції випробовуваної
речовини за формулою:
-£-•100 ,
О
де:
А - площа піка, відповідного визначуваному сте-
рину;
5- сума площ усіх піків, відповідних компонен-
там, зазначеним у Табл. 2.4.23.-1.
Якщо це зазначено в окремій статті, обчислюють
кількість кожного стерину, у міліграмах, що міститься
Таблиця 2.4.23.-1
Часи утримування стеринів у відношенні до часу утримування (3-ситостерину для двох різних колонок
Полі [метил(95)-фсніл (5)] - силоксан Полі[метил(94)-феніл(5) вініл( 1)]силоксан
Холестерин 0.63 0.67
Браспкастерин 0.71 0.73
24-Метиленхолестерин 0.80 0.82
Кампестерин 0.81 0.83
Кампестанол 0.82 0.85
Стигмастерин 0.87 0.88
Д7-Кампестерин 0.92 0.93
А5.23 Стигмастадієнол 0.95 0.95
Клеростерин 0.96 0.96
Ц-Сітостерин 1 1
Снтостанол 1.02 1.02
Д5-Авенастерин 1.03 1.03
‘5.24-Стигмастадієнол 1.08 1.08
А7-Стигмастенол 1.12 1.12
д’-Авенастерин 1.16 1.16
Бстулін 1.4 1.6
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
89
2.4. Випробування на граничний вміст домішок
у 100 г випробовуваної речовини, за формулою:
А • т5 • 100
4- т ’
де:
А — площа піка, відповідного визначуваному ком-
поненту;
— площа піка, відповідного бетуліну;
т — маса наважки зразка випробовуваної речови-
ни, у грамах;
т$ — маса доданого бетуліну Р, у міліграмах.
2.4.25. ЗАЛИШКОВІ КІЛЬКОСТІ
ЕТИЛЕНОКСИДУ І ДІОКСАНУ
Випробування призначене для визначення вмісту за-
лишкових кількостей етиленоксиду і діоксану в речо-
винах, розчинних у воді або диметилацетаміді. Для ре-
човин, нерозчинних або недостатньо розчинних у цих
розчинниках, приготування розчину випробовуваної
речовини і умови визначення методом парофазної га-
зової хроматографії зазначають в окремій статті.
Випробування проводять методом парофазної газової
хроматографії (2.2.28).
А. Для речовин, розчинних або таких, що змішуються
з водою, застосовують таку методику.
Випробовуваний розчин. 1.00 г випробовуваної речови-
ни поміщають у контейнер місткістю 10 мл (у залеж-
ності від умов проведення випробування можуть за-
стосовуватися контейнери іншого об’єму) і додають
1.0 мл води Р. Контейнер закривають, вміст перемішу-
ють до одержання однорідного розчину і витримують
протягом 45 хв при температурі 70 °С.
Розчин порівняння (а). 1.00 г випробовуваної речовини
поміщають у такий самий контейнер місткістю 10 мл,
додають 0.50 мл розчину етиленоксиду РЗ і 0.50 мл роз-
чину діоксану РІ. Контейнер закривають, вміст пе-
ремішують до одержання однорідного розчину і вит-
римують протягом 45 хв при температурі 70 °С.
Розчин порівняння (Ь). 0.50 мл розчину етиленоксиду РЗ
поміщають у контейнер місткістю 10 мл, додають
0.1 мл свіжоприготованого розчину 10 мг/л ацеталь-
дегіду Р і 0.1 мл розчину діоксану РІ. Контейнер закри-
вають, вміст перемішують до одержання однорідного
розчину і витримують протягом 45 хв при температурі
70 °С.
В. Для речовин, розчинних або таких, що змішуються
з диметилацетамідом, застосовують таку методику.
Випробовуваний розчин. 1.00 г випробовуваної речови-
ни помішають у контейнер місткістю 10 мл (у залеж-
ності від умов проведення випробування можуть за-
стосовуватися контейнер іншого об’єму), додають
1.0 мл диметилацетаміду Р і 0.20 мл води Р.
Контейнер закривають, вміст перемішують до одер-
жання однорідного розчину і витримують протягом 45
хв при температурі 90 °С.
Розчин порівняння (а). 1.00 г випробовуваної речовини
поміщають у контейнер місткістю 10 мл і додають
1.0 мл диметилацетаміду Р, 0 10 мл розчину діокса-
ну Р і 0.10 мл розчину етиленоксиду Р2. Контейнер.-'
кривають, вміст перемішують до одержання о*
норідного розчину і витримують протягом 45 ХВ 11
температурі 90 °С.
Розчин порівняння (Ь). 0.10 мл розчину етиленоксиди
поміщають у контейнер місткістю 10 мл, дода-<
0.1 мл свіжоприготованого розчину 10 мг/л ацепц
дегіду Рі 0.1 мл розчину діоксану Р. Контейнер закрі
ють вміст перемішують до одержання однорідна
розчину і витримують протягом 45 хв при темпера?®
90 °С.
Можуть бути використані такі умови підготовки іук
дення рівноважної парової фази:
— температура зрівноважування 70 °С (90 °С для р Л
чинів у диметилацетаміді);
— час зрівноважування 45 хв;
— температура блока вводу газової проби 75®
(150 °С для розчинів у диметилацетаміді);
— газ-носій — гелій для хроматографії Р;
— час напускання газу-носія у контейнер з аналі
ваною пробою 1 хв;
— час вводу газової проби 12 с.
Хроматографують на газовому хроматографі з пі
луменево-іонізаційним детектором за таких умов: І
— колонка капілярна скляна або кварцова з
вдовжки 30 м і внутрішнім діаметром 0.32 м
внутрішня поверхня якої покрита шаром шй
мепіилсилоксану Р завтовшки 1.0 мкм;
— газ-носій — гелій для хроматографії Рабо сзотіЯ
хроматографії Р;
— лінійна швидкість газу-носія близько 20 см/с; І
— поділ потоку 1:20;
— підтримують температуру колонки 50 °С протяг
5 хв, потім підвищують до 180 °С із швидкісЯ
5 °С/хв, потім — до температури 230 СС Л
швидкістю 30 °С/хв і підтримують таку темпераг 1
ру протягом 5 хв;
— температура блока вводу проб 150 °С;
— температура детектора 250 °С.
Хроматографують підхожий об’єм, наприклад, 1.0кЛ
проби газової фази над розчином порівняння (Ь).Чуі
ливість системи регулюють таким чином, щоб висот
піків етиленоксиду і ацетальдегіду на одержанії! хро
матограмі становили не менше 15 % усієї шкал
реєструючого пристрою.
Хроматографічна система вважається придатною, я
що виконуються такі умови:
— коефіцієнт розділення піків ацетальдегіду і етилі
ноксиду становить не менше 2.0;
— відношення сигнал/шум для піка діоксану стан
вить не менше 5.
Хроматографують по черзі підхожі об’єми, наприклад
по 1.0 мл (або такий самий об’єм, як для розчин
порівняння (Ь)), проб газових фаз над випробовуй-
ним розчином і розчином порівняння (а), одержую1)
по три хроматограми для кожної з проб. Розміри пліь
піків етиленоксиду і діоксану на хроматограмі випри
бовуваного розчину мають становити не більше полі
вини величин площ відповідних піків на хроматогя
розчину порівняння (а) (1 ррт етиленоксиду і 50 рр
діоксану).
90
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
ПЕРЕВІРКА ТОЧНОСТІ
Для кожної пари введень обчислюють різницю площ
йків етиленоксиду і діоксану на хроматограмі розчи-
И'порівняння (а) і на хроматограмі випробовуваного
резчину.
Хроматографічна система вважається придатною, як-
аю виконуються такі умови:
- відносне стандартне відхилення, розраховане для
трьох значень різниці площ піків етиленоксиду,
становить не більше 15 %;
- відносне стандартне відхилення, розраховане для
трьох значень різниці площ піків діоксану, стано-
вить не більше 10 %.
Якшо наважки випробовуваної речовини, взяті для
приготування випробовуваного розчину і розчину
порівняння (а), відрізнялися від 1.00 г більше ніж на
(1.5 Ч, необхідно внести відповідні поправки.
Вміст етиленоксиду у мільйонних частинах (ррт) об-
числюють за формулою:
АТС
(Лк-Л/Т) - (Лу-Л/К)
№'
Лт — площа піка етиленоксиду на хроматограмі ви-
пробовуваного розчину;
Лк — площа піка етиленоксиду на хроматограмі роз-
чину порівняння (а);
маса наважки випробовуваної речовини, взята
для приготування випробовуваного розчину, у
грамах;
А/к — маса наважки випробовуваної речовини, взята
для приготування розчину порівняння, у гра-
мах;
С — кількість етиленоксиду, доданого до розчину
порівняння (а), у мікрограмах.
Вміст діоксану в мільйонних частинах (ррт) обчислю-
ють за формулою:
ИГС
(Лк-Л/Т) - (£)у- Л/к)
де:
Д — площа піка діоксану на хроматограмі випробо-
вуваного розчину;
£)к — площа піка діоксану на хроматограмі розчину
порівняння (а);
С — кількість діоксану, доданого до розчину
порівняння (а), у мікрограмах.
2.4.26. М,М-ДИМЕТИЛАНІЛІН
МЕТОДА
Випробування проводять методом газової хромато-
графії (2.2.28), використовуючи як внутрішній стан-
дарт 8,8-діетилані.іін Р.
Розчин внутрішнього стандарту. 50 мг N,N-610010-
.іані.ііну Р розчиняють у 4 мл 0.1 М розчину кислоти
хлористоводневої і доводять об’єм розчину водою Рдо
50 мл. 1 мл одержаного розчину розводять водою Рдо
100 мл.
Випробовуваний розчин. У колбу з притертою скляною
пробкою помішають 0.50 г випробовуваної речовини і
розчиняють у 30.0 мл води Р. потім додають 1.0 мл роз-
чину внутрішнього стандарту і термостатують розчин
при температурі від 26 °С до 28 °С. Додають 1.0 мл роз-
чину натрію гідроксиду концентрованого Р і перемішу-
ють до повного розчинення. Додають 2.0 мл триметил-
пентану Р. струшують протягом 2 хв і залишають до
розшарування. Використовують верхній шар.
Розчин порівняння. 50.0 мг 14,14-диметиланшну Р роз-
чиняють у 4.0 мл 0.1 М розчину кислоти хлористовод-
невої і доводять об’єм розчину водою Р до 50.0 мл.
1.0 мл одержаного розчину розводять водою Р до
100.0 мл. 1.0 мл цього розчину розводять водою Рдо
30.0 мл. Додають 1.0 мл розчину внутрішнього стан-
дарту і 1.0 мл розчину натрію гідроксиду концентрова-
ного Р. Додають 2.0 мл триметилпентану Р, струшу-
ють протягом 2 хв і залишають до розшарування. Ви-
користовують верхній шар.
Поперемінно хроматографують по 1 мкл випробову-
ваного розчину і розчину порівняння на газовому хро-
матографі з полуменево-іонізаційним детектором за
таких умов:
— капілярна кварцова колонка завдовжки 25 м і
внутрішнім діаметром 0.32 мм, внутрішня поверх-
ня якої покрита шаром модифікованого поліме-
тилфенілсилоксану Рзавтовшки 0.52 мкм;
— газ-носій — гелій для хроматографії Р;
— поділ потоку 1:20;
— тиск на вході в колонку 50 кПа, об’ємна швидкість
газу-носія, що скидається, 20 мл/хв;
— кварцова вставка у випарник, заповнена шаром
діатоміту для газової хроматографії Р з нанесеним
полі(диметил)силоксаном Ру кількості 10 % (м/м)
завтовшки 1 см;
— підтримують температуру колонки 150 °С протя-
гом 5 хв, потім температуру підвищують до 275 °С
із швидкістю 20 °С за хв і підтримують таку темпе-
ратуру протягом 3 хв;
— температура блока вводу проб 220 °С;
— температура детектора 300 °С.
Час утримування 19,19-диметиланіліну Р становить
близько 3.6 хв, ІЧ,8і-діетиланіліну Р— близько 5.0 хв.
МЕТОД В
Випробування проводять методом газової хромато-
графії (2.2.28), використовуючи як внутрішній стан-
дарт нафталін Р.
Розчин внутрішнього стандарту. 50 мг нафталіну Р
розчиняють у циклогексані Р і доводять об’єм розчину
цим самим розчинником до 50 мл. 5 мл одержаного
розчину розводять циклогексаном Рдо 100 мл.
Випробовуваний розчин. У пробірку з притертою скля-
ною пробкою поміщають 1.00 г випробовуваної речо-
вини, додають 5 мл 1 М розчину натрію гідроксиду і
1.0 мл розчину внутрішнього стандарту. Пробірку за-
ікККОПЕЯ УКРАЇНИ 2001
91
2.4. Випробування на граничний вміст домішок
кривають і енергійно струшують протягом І хв. Якщо
необхідно, центрифугують і використовують верхній
шар.
Розчин порівняння. До 50.0 мг N,Ві-диметиланіліну Р
додають 2 мл кислоти хлористоводневої Р і 20 мл во-
ди Р. Струшують до розчинення і доводять об’єм роз-
чину водою Р до 50.0 мл. 5.0 мл одержаного розчину
розводять водою Рдо 250.0 мл. 1.0 мл одержаного роз-
чину поміщають у пробірку з скляною притертою
пробкою, додають 5 мл 1 М розчину натрію гідроксиду
і 1.0 мл розчину внутрішнього стандарту. Пробірку за-
кривають і енергійно струшують протягом 1 хв. Якщо
необхідно, центрифугують і використовують верхній
шар.
Хроматографують по 1 мкл випробовуваного розчину
і розчину порівняння на газовому хроматографі з по-
луменево-іонізаційним детектором за таких умов:
— скляна колонка завдовжки 2 м і внутрішнім діаме-
тром 2 мм, заповнена діатомітом силанізованим
для газової хроматографії Рз нанесеним у кількості
З % (м/м) поліметилфенілсилоксаном Р;
— газ-носій — азот для хроматографії Р;
— швидкість газу-носія ЗО мл/хв;
— температура колонки 120 °С;
— температура блока вводу проб і детектора 150 °С.
2.4.27. НІКЕЛЬ У ГІДРОГЕНІЗОВАНИХ
РОСЛИННИХ ОЛІЯХ
Визначення нікелю проводять методом атомно-аб-
сорбційної спектрометрії (2.2.23. метод 1).
При застосуванні закритих реакційних посудин високо-
го тиску і мікрохвильового лабораторного обладнання
необхідно суворо дотримуватися інструкцій з техніки
безпеки і користування обладнанням, що додаються ви-
робником.
Випробовуваний розчин. 0.100 г випробовуваної речо-
вини поміщають у підхожу герметичну реакційну по-
судину з фторполімеру або кварцового скла, стійку до
високого тиску, додають 6.0 мл кислоти азотної Р і
2.0 мл розчину водню пероксиду концентрованого Р.
Паралельно за пих самих умов готують холостий розчин.
Закриті посудини витримують у лабораторній мікро-
хвильовій печі за відповідною програмою, наприклад,
протягом 10 хв — за потужності печі 250 Вт; протя-
гом 5 хв — 600 Вт; протягом 6 хв — 400 Вт; протягом
7 хв — 250 Вт. Після охолодження посудини відкрива-
ють, їх вміст кількісно переносять у мірні колби
місткістю 25.0 мл, доводять об’єм розчинів водою Рао
позначки і перемішують.
Калібрувальні розчини. До 10.0 мл еталонного розчину
нікелю (10 ррт N1) Р додають 1.0 мл кислоти азотної Р,
2.0 мл розчину водню пероксиду концентрованого Р і до-
водять об’єм розчину до 20.0 мл водою Р. 20 мкл, 50 мкл,
100 мкл і 150 мкл одержаного розчину вносять,
відповідно, у чотири мірні колби місткістю 25.0 мл. У
кожну колбу додають 6.0 мл кислоти азотної Р, дово-
дять об’єм розчинів водою Рао позначки і перемішують.
Одержані калібрувальні розчини містять відповідн 01
4 нг/мл, 10 нг/мл, 20 нг/мл і ЗО нг/мл (ррЬ) нікелю. ’ Р
Холостий розчин. 6.0 мл кислоти азотної Р помішаю,
у мірну колбу місткістю 25.0 мл, доводять об’єм розчи |
ну водою Рао позначки і перемішують.
Методика. Готують суміші одного об’єму розчин. І»
5.0 г/л магнію нітрату Р і двох об’ємів холостого рої-І
чину, випробовуваного розчину, калібрувальних ро>-1
чинів і нульового розчину, відповідно. Вимірюють по-
глинання розчинів за довжини хвилі 232.0 нм на
підхожому атомно-абсорбційному спектрометрі
графітовою піччю, оснащеному системою компсн-1
сації фону Зеємана, піролітично покритою трубкою її
платформою і лампою з порожнистим нікелевим ка-
тодом. Температура висушування має становити І
100 °С протягом 10 с після 10 с розігріву, температура
озолення — 1400 °С протягом 10 с після 20 с розігріву,
температура атомізації — 2500 °С протягом 5 с. Ну- |
льову точку приладу установлюють за холостим розчи- І
ном. За одержаними показаннями будують калібру-
вальну криву. За калібрувальною кривою визначають
концентрації випробовуваного і холостого розчинів.У І
разі, якщо значення абсорбції випробовуваного роз-
чину виходить за межі калібрувальної кривої, розчин
розводять холостим розчином (фактор розведення Д І
Вміст N1 у зразку, у мкг/г (ррт), обчислюють за фор- І
мулою: ’
І
т • 40
де; І
с — концентрація випробовуваного розчину, знай- І
дена з калібрувального графіка з урахуванням
холостого розчину, у нанограмах у мілілітрі;
/ — фактор розведення;
т — маса наважки, взята для приготування випро-
бовуваного розчину, у грамах.
2.4.28. 2-ЕТИЛГЕКСАНОВА КИСЛОТА
Випробування проводять методом газової хромато-
графії (2.2.28). використовуючи як внутрішній стан-
дарт кислоту 3-циклогексилпропіонову Р.
Розчин внутрішнього стандарту. 100 мг кислоти
3-циклогексилпропіонової Ррозчиняють у циклогексані Р
і доводять об’єм розчину тим самим розчинником до
100 мл.
Випробовуваний розчин. 0.300 г випробовуваної речо-
вини розчиняють у 4.0 мл розчину (33 % об/об) кисло-
ти хлористоводневої Р. Енергійно струшують по 1 хв з
двома порціями розчину внутрішнього стандарту, по
1.0 мл кожна. Залишають до розшарування, якщо не-
обхідно, центрифугують Для випробування викорис-
товують об’єднані верхні шари.
Розчин порівняння. 75.0 мг кислоти 2-етилгексанової Р
розчиняють у розчині внутрішнього стандарту і дово-
дять об’єм розчину тим самим розчинником до
50.0 мл. До 1.0 мл одержаного розчину додають 4.0 мл
розчину (33 % об/об) кислоти хлористоводневої Р і
92
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.4. Випробування на граничний вміст домішок
Вісрііино струшують протягом І хв. Залишають до
Іиімр)вання, якщо необхідно, центрифугують. До
1 Нижнього шару додають 1.0 мл розчину внутрішнього
лиі іарту і енергійно струшують протягом І хв. Зали-
ло розшарування, якщо необхідно, центрифу-
г.ють Для випробування використовують об’єднані
. чнішари.
і Хроматографують по І мкл випробовуваного розчину
' «розчину порівняння на газовому хроматографі з по-
.іуммево-іонізаційним детектором за таких умов:
- капілярна кварцова колонка завдовжки 10 м і
трішнім діаметром 0.53 мм, покрита шаром ма-
кроголу 20 000 2-нітротерефталату Р завтовшки
10 мкм;
- газ-носііі — гелій для хроматографії Р;
- швидкість газу-носія 10 мл/хв;
- шмпература колонки і швидкість підняття тем-
ператури — за такою програмою:
Час (хв) Темпера- тура(°С) Швид- кість підняття темпера- тури (°С/хв) Примітки
колонка 0-2 40 - Ізотермічний режим
2-7.3 40—200 зо Лінійний градієнт тем- ператури
7.3-10.3 200 - Ізотермічний режим
Бюк % проб 200
Детектор 300
Хроматографічна система вважається придатною, як-
п о коефіцієнт розділення піків 2-етилгексанової кис-
лоти (перший пік) і внутрішнього стандарту становить
це менше 2.0.
Вміст 2-етилгексанової кислоти, у відсотках, обчис-
люють за формулою:
да:
плоша піка 2-етилгексанової кислоти на хро-
матограмі випробовуваного розчину;
Лк площа піка 2-етилгексанової кислоти на хро-
матограмі розчину порівняння;
/т - плоша піка внутрішнього стандарту на хрома-
тограмі випробовуваного розчину;
/ плоша піка внутрішнього стандарту на хрома-
тограмі розчину порівняння;
тТ — маса наважки випробовуваної речовини, взята
для приготування випробовуваного розчину, у
грамах;
— маса наважки 2-етилгексанової кислоти, взята
для приготування розчину порівняння, у гра-
мах.
______________________________________________N
2.4.290. ЦИНК
До 10.0 мл розчину випробовуваної речовини, приго-
тованого, як зазначено в окремій статті, додають
2.0 мл розчину кислоти хлористоводневої РІ і 0.2 мл
розчину калію фероціаніду Р.
Паралельно готують еталон з використанням замість
випробовуваного розчину 10 мл еталонного розчину
цинк-іона (5 ррт 7п), який готують шляхом розведен-
ня водою Р у 1000 разів еталонного розчину цинку
(5 мг/мл Хп) Р.
Через 10 хв опалесценція випробовуваного розчину не
має перевищувати опалесценцію еталона.
Примітка. У разі появи у випробовуваному розчині
синього забарвлення, шо заважає нефелометричному
порівнянню, треба заздалегідь відділити залізо. Для
цього до розчину випробовуваної речовини, нагрітого
до кипіння, додають розчин аміаку розведений РІ до
появи виразного запаху і суміш фільтрують. Об’єм
фільтрату доводять водою Р до необхідної концент-
рації і використовують для випробування на цинк за
описаною вище методикою.
2.4.300. РЕЧОВИНИ, ЩО ЛЕГКО
ОБВУГЛЮЮТЬСЯ
Якщо в окремій статті немає інших зазначень, кіль-
кість випробовуваної речовини, зазначену в окремій
статті (тонко здрібненої, якшо вона знаходиться у
твердій фазі), невеликими порціями помішають у
пробірку з безбарвного скла, стійкого до дії кислоти
сірчаної, що містить зазначену в окремій статті
кількість сірчаної кислоти (95.0±0.5) %, (об/об). Про-
бірку попередньо промивають кислотою сірчаною Р,
потім водою Р і висушують.
Суміш перемішують скляною паличкою до повного
розчинення випробовуваної речовини, залишають на
15 хв, якшо немає інших зазначень в окремій статті, і
порівнюють забарвлення одержаного розчину із за-
барвленням зазначеного в окремій статті еталона,
помішеного в аналогічну пробірку з безбарвного скла.
Примітка. Для одержання кислоти сірчаної
((95.0±0.5) %, (об/об)) сірчану кислоту Р додають до
певного об’єму води Р, необхідного для досягнення за-
значеної концентрації.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
93
2.5. Методи кількісного визначення
2.5. МЕТОДИ КІЛЬКІСНОГО
ВИЗНАЧЕННЯ
2.5.1. КИСЛОТНЕ ЧИСЛО
Кислотним числом ІА називають кількість калію
гідроксиду, у міліграмах, необхідну для нейтралізації
вільних кислот, що містяться в 1 г випробовуваної ре-
човини.
Близько 10.00 г або зазначену в окремій статті наваж-
ку речовини (г) розчиняють у 50 мл суміші рівних
об’ємів спирту Р і ефіру Р, попередньо нейтралізова-
ної 0.1 М розчином калію гідроксиду, якщо немає
інших зазначень в окремій статті, використовуючи як
індикатор 0.5 мл розчину фенолфталеїну РІ. Після роз-
чинення випробовуваної речовини одержаний розчин
титрують 0.1М розчином калію гідроксиду до появи ро-
жевого забарвлення, яке не зникає протягом 15 с.
Кислотне число (ІА) обчислюють за формулою:
5.610л
‘а т ’
де:
л — кількість 0.1 М розчину калію гідроксиду,
витрачена на титрування, у мілілітрах;
5.610 — кількість калію гідроксиду, шо відповідає
1 мл 0.1 М розчину калію гідроксиду, у мілі-
грамах;
т — маса наважки речовини, у грамах.
_______________________________________________N
Якщо випробовувана речовина не розчиняється у
суміші розчинників, до колби приєднують зворотний
холодильник і злегка нагрівають на теплій водяній бані
при постійному перемішуванні до розчинення речови-
ни. Потім додають 0.5 мл розчину фенолфталеїну РІ і
титрують 0.1 Мрозчином калію гідроксиду до появи ро-
жевого забарвлення, яке не зникає протягом 15 с.
Якщо об’єм 0.1 М розчину калію гідроксиду, не-
обхідний для титрування, менше 2 мл, відповідним
способом збільшують масу наважки випробовуваної
речовини або використовують більш розведений тит-
рант (в останньому випадку вносять відповідні зміни у
формулу розрахунку).
Якщо випробовувана речовина з метою консервації
була насичена вуглецю діоксидом, перед зважуванням
її витримують у випарювальній чашці протягом 24 год
у вакуум-ексикаторі.
2.5.2. ЕФІРНЕ ЧИСЛО
Ефірним числом 1Е називають кількість калію гідро-
ксиду, у міліграмах, необхідну для омилення ефірів,
що містяться в 1 г випробовуваної речовини.
Ефірне число (1^ обчислюють за формулою:
де:
— число омилення:
1А — кислотне число.
2.5.3. ГІДРОКСИЛЬНЕ ЧИСЛО
Гідроксильним числом 10Н називають кількість мія
грамів калію гідроксиду, еквівалентну кількості кисло
ти, що зв’язується при ацилюванні 1 г речовини.
МЕТОДА
Наважку речовини, відповідно до Табл. 2.5.3.-ІЇ
поміщають у круглодонну колбу з шліфом місткістю
150 мл, якщо немає інших зазначень в окремій статті.!
Додають об’єм розчину оцтового ангідриду РІ, зазначе-
ний у Табл. 2.5.3.-1.
Таблиця 2.5.3.-1
Передбачуване значення Іон Наважка речо- вини, у грамах Об’єм розчину оцтового ангід- риду РІ, у мілі- літрах
10- 100 20 5.0
100-150 1.5 5.0
150- 200 1.0 5.0
200 - 250 0.75 5.0
250 - 300 0.60 або 1.20 5.0 або 10.0
300 - 350 1.0 10.0
350 - 700 0.75 15.0
700 - 950 0.5 15.0
До колби приєднують повітряний холодильник, помі-
щають на киплячу водяну баню, підтримуючи рівень
води у бані на 2.5 см више рівня рідини у колбі, й
нагрівають протягом 1 год. Потім через верхній кінець
повітряного холодильника додають 5 мл води Р. Якшо
розчин каламутнішає, до нього додають піридин Р до
зникнення каламуті, відмічаючи витрачений об’єм.
Колбу струшують, поміщають у киплячу водяну баню
на 10 хв. Потім колбу виймають з водяної бані й охоло-
джують до кімнатної температури. Повітряний холо-
дильник і стінки колби промивають 5 мл спирту Р, по-
передньо нейтралізованого з використанням розчину
фенолфталеїну РІ. Одержаний розчин титрують
0.5 М розчином калію гідроксиду спиртовим, використо-
вуючи як індикатор 0.2 мл розчину фенолфталеїну РІ.
Паралельно проводять контрольний дослід.
Гідроксильне число обчислюють за формулою:
28.05(л-, - п.)
Іон = -------------- + ’
де:
П/ — об’єм 0.5 М розчину калію гідроксиду спирто-
вого, витрачений на титрування випробовува-
ної речовини, у мілілітрах;
94
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.5. Методи кількісного визначення
— об’єм 0.5 Мрозчину калію гідроксиду спирто-
вого, витрачений на титрування в контроль-
ному досліді, у мілілітрах;
т — маса наважки речовини, у грамах;
28 05 — кількість калію гідроксиду, що відповідає
1 мл 0.5 М розчину калію гідроксиду, у мілі-
грамах;
Ц — кислотне число.
МЕТОД Б
Наважку речовини поміщають у суху конічну колбу з
притертою пробкою місткістю 5 мл і додають 2.0 мл
реактиву пропіонового ангідриду Р. Колбу закривають,
обережно струшують до розчинення речовини і зали-
шають на 2 год, якщо немає інших зазначень в окре-
мій статті. Виймають пробку, колбу з її вмістом помі-
шають у конічну колбу з широким горлом місткістю
500 мл, яка містить 25.0 мл розчину 9 г/л аніліну Р у
циклогексані Р і ЗО мл кислоти оцтової льодяної Р. От-
риманий розчин перемішують, залишають на 5 хв, до-
дають 0.05 мл розчину кристалічного фіолетового Р і
титрують 0.1 Мрозчином кислоти хлорної до появи яс-
краво-зеленого забарвлення.
Паралельно проводять контрольний дослід.
Гідроксильне число обчислюють за формулою:
5.610(И| - и2)
!он= т ’
де:
И/ — об’єм 0.1 М розчину кислоти хлорної, ви-
трачений на титрування випробовуваної ре-
човини, у мілілітрах;
п2 — об’єм 0.1 М розчину кислоти хлорної, ви-
трачений на титрування в контрольному
досліді, у мілілітрах;
т — маса наважки речовини, у грамах;
5.610 — кількість калію гідроксиду, що відповідає 1 мл
0.1 М розчину калію гідроксиду, у міліграмах;
Вміст води, присутньої в речовині, визначають
напівмікрометодом (2.5.12).
Перерахування гідроксильного числа проводять за
формулою: /он = (знайдене значення гідроксильного
числа речовини) — 31.1 у,
де:
у - вміст води в речовині, у відсотках.
2.5.4. ЙОДНЕ ЧИСЛО
Йодним числом // називають кількість галогену в пе-
рерахунку на йод, у грамах, необхідну для зв’язування
100 г випробовуваної речовини за описаних умов.
Наважку речовини (відповідно до Табл. 2.5.4,-1, якщо
немає інших зазначень в окремій статті) поміщають у
колбу з притертою пробкою місткістю 250 мл, попе-
редньо висушену або промиту кислотою оцтовою льо-
дяною Р, розчиняють у 15 мл хлороформу Р, якщо не-
має інших зазначень в окремій статті. До одержаного
розчину повільно додають 25.0 мл розчину йоду бро-
міду Р.
Таблиця 2.5.4,-1
Передбачуване значення її Маса наважки речовини, у грамах
менше 20 1.0
від 20 до 60 від 0.5 до 0.25
від 60 до 100 від 0.25 до 0.15
більше 100 від 0.15 до 0.10
Колбу закривають пробкою і витримують у темному
місці при частому перемішуванні протягом ЗО хв, як-
що немає інших зазначень в окремій статті. Додають
10 мл розчину 100 г/л калію йодиду Р, 100 мл води Рі
титрують 0.1 М розчином натрію тіосульфату при
інтенсивному перемішуванні до світло-жовтого за-
барвлення, потім додають 5 мл розчину крохмалю Р і
титрують 0.1 М розчином натрію тіосульфату крап-
лями до знебарвлення.
Паралельно проводять контрольний дослід.
Йодне число ///) обчислюють за формулою:
1.269(л2 -
т
де:
п/ — кількість 0.1 Мрозчину натрію тіосульфату, ви-
трачена на титрування випробовуваної
речовини, у мілілітрах;
п2 — кількість 0.1 Мрозчину натрію тіосульфату, ви-
трачена на титрування в контрольному досліді,
у мілілітрах;
т — маса наважки речовини, у грамах.
______________________________________________N
Допускається проводити визначення йодного числа за
такою методикою.
Наважку випробовуваної речовини (відповідно до
Табл. 2.5.4,-1) поміщають у суху колбу з притертою
пробкою місткістю 250 мл, розчиняють у 3 мл ефіру Р,
якщо немає інших зазначень в окремій статті,
повільно додають 20.0 мл 0.1Мрозчину йоду хлориду Р.
Колбу закривають пробкою, змоченою розчином
10 г/л калію йодиду Р, і витримують у темному місці
при частому перемішуванні протягом 1 год, якщо не-
має інших зазначень в окремій статті. Додають 10 мл
розчину 10 г/л калію йодиду Р, 50 мл води Рі титрують
0.1 М розчином натрію тіосульфату при постійному,
інтенсивному перемішуванні до світло-жовтого за-
барвлення, потім додають 3 мл ефіру Р, інтенсивно пе-
ремішують, додають 5 мл розчину крохмалю Р і титру-
ють 0.1 М розчином натрію тіосульфату краплями до
знебарвлення.
Паралельно проводять контрольний дослід.
При аналізі твердих жирів наважку розчиняють у 6 мл
ефіру Р, додають 20.0 мл 0.1 Мрозчину йоду хлориду Рк.
Подальше визначення проводять, як зазначено вище.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
95
2.5. Методи кількісного визначення
Приготування 0.1 М розчину йоду хлориду Рл. 11.06 г
калію йодиду Р і 7.10 г калію йодату Р поміщають у
колбу з притертою пробкою, додають 50 мл води Р і
50 мл кислоти хлористоводневої концентрованої Р, за-
кривають пробкою і струшують до повного розчинен-
ня йоду, що утворюється при реакції. Розчин перено-
сять у ділильну лійку і збовтують з 10 мл хлороформу Р.
Якщо хлороформний шар забарвлюється у фіолетовий
колір, додають при енергійному струшуванні крап-
лями розчин 10 г/л калію йодату Р до знебарвлення
хлороформного шару. Якщо хлороформний шар зали-
шається безбарвним, то додають розчин 10 г/л калію
йодиду Р краплями до появи блідо-рожевого забарв-
лення. Після відстоювання водний шар зливають у
мірну колбу місткістю 1000 мл і доводять об’єм розчи-
ну водою до позначки. Одержаний розчин повинен
мати світло-жовте забарвлення.
МЕТОД Б
Випробування проводять у захищеному від світла місці.
Наважку речовини, відповідно до Табл. 2.5.5.-1, помії
щають у конічну колбу з притертою скляною проб-
кою, додають 50 мл суміші: триметилпентан Р—кис
лота оцтова льодяна Р(2:3).
Таблиця 2.5.5.-1
Передбачуване значення переписного числа І Іаважки речовини, у грамах
від 0 до 12 від 2.00 до 5.00
від 12 до 20 від 1.20 до 2.00
від 20 до ЗО від 0.80 до 1.20
від 30 до 50 від 0.500 до 0.800
від 50 до 90 від 0.300 до 0.500
2.5.5. ПЕРЕКИСНЕ ЧИСЛО
Перекисним числом Ір називають кількість мілі-
еквівалентів активного кисню, відповідну кількості
перекисів, що містяться у 1000 г випробовуваної речо-
вини, яку визначають методами, наведеними нижче.
Якщо немає зазначень в окремій статті, використову-
ють метод А. Заміна методу А на метод Б у цьому ви-
падку вимагає проведення валідації.
МЕТОД А
Близько 5.00 г (точна наважка) речовини поміщають у
конічну колбу з притертою скляною пробкою
місткістю 250 мл, додають 30 мл суміші: хлороформ Р -
кислота оцтова льодяна Р (2:3). Колбу струшують до
розчинення речовини, додають 0.5 мл насиченого роз-
чину калію йодиду Р, перемішують протягом 1 хв і до-
дають 30 мл води Р. Одержаний розчин титрують
0.01 Мрозчином натрію тіосульфату, повільно дода-
ючи титрант при безперервному перемішуванні май-
же до повного зникнення жовтого забарвлення.
Потім додають 5 мл розчину крохмалю Р і продовжу-
ють титрувати, інтенсивно перемішуючи до знебарв-
лення розчину.
Паралельно проводять контрольний дослід.
Об’єм 0.01 М розчину натрію тіосульфату, витраче-
ний на титрування в контрольному досліді, не має пе-
ревищувати 0.1 мл.
Перекисне число обчислюють за формулою:
де:
/і/ — об’єм 0.01 Мрозчину натрію тіосульфату, вит-
рачений на титрування випробовуваної речови-
ни, у мілілітрах;
п2 — об’єм 0.01 М розчину натрію тіосульфату, вит-
рачений на титрування в контрольному досліді,
у мілілітрах:
т — маса наважки речовини, у грамах.
Колбу закривають пробкою і вміст перемішують до І
розчинення речовини. До одержаного розчину мірною І
піпеткою додають 0.5 мл насиченого розчину калію йо-
диду Р. Колбу закривають пробкою, залишають на
1 хв±1 с, ретельно струшуючи розчин не менше трьох
разів, додають 30 мл води Р і титрують 0.1 М розчином
натрію тіосульфату, додаючи титрант поступово при
постійному та інтенсивному перемішуванні майже до
повного зникнення жовтого забарвлення йоду. Потім
додають близько 0.5 мл розчину крохмалю РІ і продов-
жують титрування при постійному інтенсивному пе-
ремішуванні поблизу точки еквівалентності з метою
повного вивільнення йоду з шару органічного розчин-
ника. Розчин натрію тіосульфату додають краплями до
зникнення синього забарвлення.
Якщо об’єм 0.1Мрозчину натрію тіосульфату, витра-
чений на титрування, менше 0.5 мл, визначення по-
вторюють з 0.01 М розчином натрію тіосульфату при
постійному та інтенсивному перемішуванні.
Примітка. Для перекисних чисел близько 70 і більше
спостерігається затримка знебарвлення індикатора
крохмалю від 15 с до 30 с, що зумовлено здатністю три-
метилпентану спливати на поверхню водної фази. Тому
необхідний час для змішування розчинника з водним
титрантом для повного вивільнення йоду. Для перекис-
них чисел менше 15 рекомендують використовувати
0.01 М розчин натрію тіосульфату. З метою запобігти
розшарування фаз і домогтися швидкого вивільнення
йоду до реакційної суміші допускається додавання не-
великої кількості (0.5 % - 1 % (м/м)) емульгатора з ви-
соким гідрофільно-ліпофільним балансом (ГЛБ).
Паралельно проводять контрольний дослід
Якщо об’єм титранту, витрачений на титрування у
контрольному досліді, перевищує 0.1 мл, випробуван-
ня повторюють із свіжоприготованими реактивами.
Перекисне число обчислюють за формулою:
1000(И, - го)с
т
де:
с — концентрація розчину натрію тіосульфату, у мо-
лях на літр;
96
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.5. Методи кількісного визначення
І,- об’єм розчину натрію тіосульфату, витрачений
на титрування випробовуваної речовини, у мі-
лілітрах;
1,— об’єм розчину натрію тіосульфату, витрачений
на титрування контрольного досліду, у мілі-
літрах;
т — маса наважки речовини, у грамах.
2.5.6. ЧИСЛО ОМИЛЕННЯ
Числом омилення /4 називають кількість калію гідро-
ксиду, у міліграмах, необхідну для нейтралізації
вільних кислот і омилення складних ефірів, що
\ стяться в 1 г випробовуваної речовини.
Наважку речовини (відповідно до Табл. 2.5.6.-1, якщо
немає інших зазначень в окремій статті) поміщають у
колбу з боросилікатного скла місткістю 250 мл, спо-
ряджену зворотним холодильником.
Таблиця 2.5.6.-1
Передбачуване значення числа омилення Маса наважки речовини, у грамах
від Зло 10 від 12 до 15
від 10 до 40 від 8 до 12
від 40 до 60 від 5 до 8
від 60 до 100 від 3 до 5
від 100 до 200 від 2.5 до 3
від 200 до 300 від 1 до 2
від 300 до 400 від 0.5 до 1
Додають 25.0 мл 0.5 М розчину калію гідроксиду спир-
тового і декілька скляних кульок. До колби приєдну-
ють іворотний холодильник і нагрівають на киплячій
водяній бані протягом ЗО хв, якщо немає інших зазна-
чень в окремій статті. Додають 1 мл розчину фенолфта-
іеїну РІ і гарячий розчин відразу титрують 0.5 Мроз-
чином кислоти хлористоводневої.
Паралельно проводять контрольний дослід.
Число омилення (І.) обчислюють за формулою:
28.05(и2 - п^)
т
де:
п і — об’єм 0.5 Мрозчину кислоти хлористоводне-
вої, витрачений на титрування випробову-
ваної речовини, у мілілітрах;
— об’єм 0.5 Мрозчину кислоти хлористоводне-
вої, витрачений на титрування в контроль-
ному досліді, у мілілітрах;
т - маса наважки випробовуваної речовини, у
грамах;
28 05 — кількість калію гідроксиду, відповідна 1 мл
0.5 М розчину кислоти хлористоводневої, у
міліграмах.
_____________________________________________N
Якшо випробовувана речовина з метою консервації
була насичена вуглецю діоксидом, перед зважуванням
її витримують у випарювальній чашпі протягом 24 год
у вакуум-ексикаторі.
2.5.7. НЕОМИЛЮВАНІ РЕЧОВИНИ
Термін "неомилювані речовини" застосовується до ре-
човин нелетких при температурі від 100 °С до 105 °С,
які екстрагуються органічним розчинником з випро-
бовуваного зразка після його омилення. Вміст неоми-
люваних речовин обчислюють у відсотках (м/м).
Треба використовувати скляний посуд із шліфами без
змащування.
Наважку випробовуваної речовини, зазначену в ок-
ремій статті, помішають у колбу місткістю 250 мл,
споряджену зворотним холодильником. Додають
50 мл 2 М розчину калію гідроксиду спиртового і
нагрівають на водяній бані протягом 1 год, періодич-
но перемішуючи коловими рухами. Потім охолоджу-
ють до температури нижче 25 °С і вміст колби за до-
помогою 100 мл води Р переносять у ділильну лійку.
Одержаний розчин обережно струшують з трьома
порціями ефіру, вільного від пероксидів, Р по 100 мл
кожна. Усі ефірні витяги збирають в іншу ділильну
лійку, в яку попередньо помішають 40 мл води Р, обе-
режно струшують протягом декількох хвилин і зали-
шають до повного розділення шарів, після чого
відкидають водний шар. Ефірний шар промивають
двома порціями води Р, по 40 мл кожна. Потім ре-
тельно відмивають по черзі 40 мл розчину 30 г/л
калію гідроксиду Рі 40 мл води Р, повторюючи дану
процедуру три рази. Потім ефірний шар відмивають
водою Р порціями по 40 мл до відсутності лужної ре-
акції у водному шарі по фенолфталеїну. Ефірний
шар кількісно переносять у доведену до постійної
маси колбу за допомогою ефіру, вільного від перок-
сидів, Р.
Ефір відганяють з відповідною обережністю і до за-
лишку додають 6 мл ацетону Р. Розчинник ретельно
видаляють у струмені повітря. Залишок у колбі вису-
шують при температурі від 100 °С до 105 °С до по-
стійної маси, охолоджують в ексикаторі і зважують.
Вміст неомилюваних речовин, у відсотках, обчислю-
ють за формулою:
100 -а „
Неомилювані речовини =-----—---% ’
де:
а — маса залишку, у грамах;
т — маса наважки речовини, у грамах.
Залишок розчиняють у 20 мл спирту Р, попередньо
нейтралізованого за розчином фенолфталеїну Р, і тит-
рують 0.1 М розчином натрію гідроксиду етанольним
Якщо витрачений об’єм 0.1 М розчину натрію гідро-
ксиду етанольного перевищує 0.2 мл, розділення двох
шарів було неповним; при цьому зважений залишок
не може розглядатися як "неомилювані речовини". У
цьому випадку випробування треба повторити.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
97
2.5. Методи кількісного визначення
2.5.9. ВИЗНАЧЕННЯ АЗОТУ ПІСЛЯ
МІНЕРАЛІЗАЦІЇ СІРЧАНОЮ КИСЛОТОЮ
НАПІВМІКРОМЕТОД
Наважку випробовуваної речовини, що містить близь-
ко 2 мг азоту, поміщають у колбу для спалювання, до-
дають 4 г здрібненої суміші, що складається зі 100 г
калію сульфату Р, 5 г міді сульфату Р\2.5ь селену Р, і
три скляні кульки. Додають 5 мл кислоти сірчаної Рта-
ким чином, щоб вона змивала всі частки, що прилип-
ли до шийки колби, і стікала по стінках колби. Вміст
колби перемішують коловими рухами. Щоб уникнути
великих втрат сірчаної кислоти, шийку колби закри-
вають нещільно, наприклад, скляною грушоподібною
пробкою з коротким запаяним відростком. Колбу
нагрівають, поступово доводячи до кипіння з конден-
сацією пари сірчаної кислоти у шийці колби; при цьо-
му необхідно стежити за тим, щоб верхня частина кол-
би не перегрівалася. Нагрівання продовжують протя-
гом ЗО хв, якщо немає інших зазначень в окремій
статті. Охолоджують, розчиняють твердий залишок,
додаючи до суміші обережно 25 мл води Р, знову охо-
лоджують і приєднують до приладу для перегонки з
водяною парою. Додають ЗО мл розчину натрію гідрок-
сиду концентрованого Рі негайно починають перегон-
ку, пропускаючи пару крізь суміш. Близько 40 мл
відгону збирають у приймач, що містить 20.0 мл
0.01 Мрозчину кислоти хлористоводневої і достатню
кількість води Р для того, щоб кінець холодильника
був занурений. Наприкінці перегонки приймач опус-
кають таким чином, щоб кінець холодильника знахо-
дився над поверхнею рідини. Не слід допускати, шоб
на зовнішній поверхні холодильника залишалася
рідина із вмісту приймача. Відгін титрують 0.01 Мроз-
чином натрію гідроксиду, використовуючи як індика-
тор змішаний розчин метилового червоного Р.
Випробування повторюють, використовуючи замість
випробовуваної речовини 50 мг глюкози Р.
0.01401(л2 - и,)
Вміст азоту = ------—--------% ’
де:
т — маса наважки випробовуваної речовини, у гра-
мах;
пІ — об’єм 0.01 Мрозчину натрію гідроксиду, витра-
чений на титрування розчину, одержаного
після спалювання випробовуваної речовини, у
мілілітрах;
п2 — об’єм 0.01 Мрозчину натрію гідроксиду, витра-
чений на титрування розчину, одержаного
після спалювання глюкози, у мілілітрах.
____________________________________________N
Після закінчення відгонки кінець холодильника про-
мивають зовні невеликою кількістю води, збираючи
промивну воду у той самий приймач.
Відгін титрують до переходу забарвлення з червоно-
фіолетового у зелене.
2.5.11. КОМПЛЕКСОМЕТРИЧНЕ
ТИТРУВАННЯ
Алюміній. 20.0 мл розчину, зазначеного в окремій
статті, поміщають у конічну колбу місткістю 500 мл,
додають 25.0 мл 0.1 Мрозчину натрію едетату і 10 мл
суміші рівних об’ємів розчину 155 г/л амонію ацета-
ту Рі кислоти оцтової розведеної Р, кип’ятять протя-
гом 2 хв і охолоджують. Додають 50 мл етанолу Р, 3 мл
свіжоприготованого розчину 0.25 г/л дитизону Р в
етанолі Р і надлишок натрію едетату титрують
0.1 М розчином цинку сульфату до переходу зеленува-
то-синього забарвлення розчину в червонувато-фіоле-
тове.
1 мл 0.1 М розчину натрію едетату відповідає
2.698 мг А1.
Вісмут. Розчин випробовуваної речовини у кислоті
азотній Р, зазначений в окремій статті, поміщають у
конічну колбу місткістю 500 мл. Доводять об’єм роз-
чину водою Рдо 250 мл і, якщо немає інших зазначень
в окремій статті, додають краплями, при перемішу-
ванні, розчин аміаку концентрований Р до появи
каламуті. Потім додають 0.5 мл кислоти азотної Р,
нагрівають до температури близько 70 °С до зникнен-
ня каламуті, додають близько 50 мг індикаторної суміші
ксиленолового оранжевого Р і титрують 0.1 М розчином
натрію едетату до переходу блідо-рожево-фіолетово-
го забарвлення розчину в жовте.
1 мл 0.1 М розчину натрію едетату відповідає
20.90 мг Ві.
Кальцій. Розчин, зазначений в окремій статті, поміша-
ють у конічну колбу місткістю 500 мл. Доводять об’єм
розчину водою Р до 300 мл, додають 6.0 мл розчину
натрію гідроксиду концентрованого Р, близько 15 мг
індикаторної суміші кальконкарбоновоїкислоти Рі ти-
трують 0.1 М розчином натрію едетату до переходу
фіолетового забарвлення розчину в синє.
1 мл 0.1 М розчину натрію едетату відповідає
4.008 мг Са.
Магній. Розчин, зазначений в окремій статті, поміща-
ють у конічну колбу місткістю 500 мл. Доводять об’єм
розчину водою Р до 300 мл, додають 10 мл аміачного
буферного розчину рН 10.0 Рі близько 50 мг індикатор-
ної суміші протравного чорного 11 Р. Розчин нагріва-
ють до температури близько 40 °С і титрують при цій
температурі 0.1 М розчином натрію едетату до пере-
ходу фіолетового забарвлення розчину в синє.
1 мл 0.1 М розчину натрію едетату відповідає
2.431 мг Мр.
Свинець. Розчин, зазначений в окремій статті, поміща-
ють у конічну колбу місткістю 500 мл. Доводять об’єм
розчину водою Рао 200 мл, додають близько 50 мг інди-
каторної суміші ксиленолового оранжевого Р, а потім
гексаметилентетрамін Р до появи фіолетово-рожево-
го забарвлення розчину. Титрують 0.1 М розчином
натрію едетату до переходу фіолетово-рожевого за-
барвлення розчину в жовте.
98
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.5. Методи кількісного визначення
І мл 0.1 М розчину натрію едетату відповідає
20.72 мг РЬ.
Цинк. Розчин, зазначений в окремій статті, поміща-
ють у конічну колбу місткістю 500 мл, об’єм розчину
доводять водою Р до 200 мл, додають близько 50 мг
індикаторної суміші ксиленолового оранжевого Р, а
потім гексаметилентетрамін Р до появи фіолетово-
рожевого забарвлення розчину. Після цього додають
ще 2 г гексаметилентетраміну Р і титрують 0.1 Мроз-
чином натрію едетату до переходу фіолетово-рожево-
го забарвлення у жовте.
1 мл 0.1 М розчину натрію едетату відповідає
6.54 мг 2п.
_____________________________________________N
Комплексометричне титрування — це метод титру-
вання, заснований на реакціях комплексоутворення.
Комплексонометричне титрування як окремий випа-
док комплексометричного титрування засноване на
реакції комплексоутворення катіонів металів з ком-
плексонами — амінополікарбоновими кислотами та їх
солями. Утворювані комплексні сполуки називають
комплексонатами.
Дія комплексонометричного титрування як титрант
застосовують звичайно динатрієву сіль етилендіамін-
тетраоцтової кислоти, відому під назвами: натрію еде-
тат,грилон Б, комплексон ПІ, хелатон III та ін.
Натрію едетат утворює з катіонами багатовалентних ме-
талів стійкі й добре розчинні у воді комплексонати у
стехіометричному співвідношенні 1 1 і використо-
вується для кількісного визначення алюмінію, вісмуту,
кальцію, свинцю, магнію та цинку в лікарських засобах.
Індикатори, застосовувані для візуального визначення
кінцевої точки титрування, називаються металоінди-
каторами. Вони є органічними барвниками і мають
властивість змінювати забарвлення при утворенні
комплексних сполук з катіонами металів. Мета-
лоіндикатори для комплексонометрії підбирають та-
ким чином, щоб їх взаємодія з катіонами визначува-
них металів була оборотною і стійкість їхніх ком-
плексів була значно меншою стійкості комплексо-
натів, утворюваних у процесі титрування.
Пряме титрування розчинами натрію едетату проводять
так: до аналізованого розчину, якщо необхідно, нейт-
ралізованого, додають буферний розчин для створення
потрібного значення рН, потім додають металоіндика-
тор. У процесі титрування розчином трилону Б забарв-
лення розчину в точці еквівалентності змінюється від
забарвлення комплексу металоіндикатора з титрова-
ним катіоном металу до забарвлення вільного мета-
лоіндикатора.
При зворотному титруванні до випробовуваного роз-
чину додають натрію едетат і його надлишок відтитро-
вують при певному значенні рН у присутності
відповідного металоіндикатора розчинами солей цин-
ку, магнію, свинцю та ін.
Допускається використання як титранту 0.05 Мрозчи-
ну натрію едетату, що має бути зазначено в окремій
статті.
Допускається проводити визначення алюмінію за та-
кою методикою.
Алюміній. Точну наважку випробовуваної речовини,
еквівалентну 0.02-0.03 г алюмінію, розчиняють у 2 мл
1 М розчину кислоти хлористоводневої і 50 мл води Р.
Додають 50 мл 0.05 Мрозчину натрію едетату і нейт-
ралізують 1 Мрозчином натрію гідроксиду, використо-
вуючи як індикатор метиловии червоний Р. Розчин
нагрівають до кипіння і витримують на киплячій во-
дяній бані протягом 10 хв, охолоджують, додають
0.05 г індикаторної суміші ксиленолового оранжевого Р,
5 г гексаметилентетраміну Р і титрують надлишок
натрію едетату 0.05 М розчином свинцю нітрату до
блідо-рожево-фіолетового забарвлення.
1 мл 0.05 М розчину натрію едетату відповідає
0.001349 г алюмінію.
2.5.12. ВИЗНАЧЕННЯ ВОДИ
НАІПВМІКРОМЕТОДОМ
(метод К.Фішера)
Для титрування використовують посудину місткістю
близько 60 мл, споряджену двома платиновими елект-
родами, трубкою для підведення азоту, трубкою, за-
повненою осушувальним агентом, і пробкою, в яку
вставляється кінчик бюретки. Випробовувану речови-
ну вносять у посудину через трубку, розташовану з
протилежного боку відносно трубки-осушувача, і
таку, що закривається притертою пробкою. Пе-
ремішування розчину в процесі титрування здійсню-
ють за допомогою магнітної мішалки або шляхом про-
дування висушеного азоту через розчин.
Кінцеву точку титрування визначають амперометрич-
но. Електрична схема складається з потенціометра з
опором близько 2000 Ом, підключеного до джерела
постійного струму з напругою 1.5 В; він забезпечує не-
обхідну різницю потенціалів. Різниця потенціалів
відрегульована таким чином, щоб через платинові
електроди, сполучені послідовно з мікроампермет-
ром, проходив невеликий початковий струм. При до-
даванні реактиву стрілка мікроамперметра відхи-
ляється, але відразу ж повертається до вихідного поло-
ження. У кінці реакції одержуване відхилення має бу-
ти незмінним не менше 30 с.
Иодсірчистии реактив Рвикористовують після визна-
чення його титру за водою (4.1.1). Використовувані
розчини і реактиви мають бути безводними, і
необхідно вжити застережних заходів до запобігання
дії на них атмосферної вологи. Иодсірчистииреактив Р
необхідно захищати від дії світла і бажано берегти в
ємності, спорядженій автоматичною бюреткою.
Иодсірчисті реактиви, що є в продажу, часто відрізня-
ються за складом від иодсірчистого реактиву Р: піридин
замінений у них на інші основи. Використання таких ре-
активів має бути попередньо валідоване з метою під-
твердження у кожному конкретному випадку стехіо-
метрії та відсутності несумісності між випробовува-
ною речовиною і реактивом (1.1. Загальні зауваження).
Якщо немає інших зазначень в окремій статті, вико-
ристовують метод А.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
99
2.5. Методи кількісного визначення
МЕТОД А
Близько 20 мл метанолу безводного Рабо розчинника,
зазначеного в окремій статті, поміщають у посудину
для титрування і титрують йодсірчистим реактивом Р,
визначаючи кінцеву точку титрування амперометрич-
но. Зазначену кількість випробовуваної речовини
швидко поміщають у посудину для титрування. Суміш
перемішують протягом 1 хв і знову титрують йодсірчи-
стим реактивом Р, визначаючи кінцеву точку титру-
вання амперометрично.
МЕТОД В
Близько 10 мл метанолу безводного Р або розчинника,
зазначеного в окремій статті, поміщають у посудину для
титрування і титрують йодсірчистим реактивом Р, ви-
значаючи кінцеву точку титрування амперометрично.
Потім швидко вносять у посудину для титрування за-
значену кількість випробовуваної речовини і точно
відміряний об’єм йодсірчистого реактиву Р, узятий з
надлишком приблизно на 1 мл або об’єм, зазначений
в окремій статті. Посудину закривають пробкою, ви-
тримують у захищеному від світла місці протягом 1 хв
або протягом часу, зазначеного в окремій статті,
періодично перемішуючи вміст посудини. Надлишок
йодсірчистого реактиву Р титрують до початкового
значення сили струму, використовуючи метанол без-
водний Рабо розчинник, зазначений в окремій статті,
до якого було додано точно відому кількість води Р,
еквівалентну близько 2.5 г/л.
___________________________________________N
Дозволяється використання йодсірчистого реактиву
(реактиву К Фішера), що випускається підприємства-
ми України відповідно до вимог нормативної доку-
ментації, затвердженої уповноваженими державним,!
органами.
Реактив К.Фішера являє собою розчин сірки ліоксиду
йоду і піридину в метанолі. Взаємодія реактиву з во-
дою відбувається стехіометрично за рівнянням:
І2 + 8О2 + Н2О + ЗС5Н51У —* 2С5Н5М-НІ+С5Н5И8О3
С5Н5М8О3 + СН3ОН—~С5Н5М-Н8О4СН3
Реактив К.Фішера зазначеного вище складу не засто-
совний для аналізу сполук, які реагують з одним або
кількома компонентами реактиву, як, наприклад, ас-
корбінова кислота, меркаптани, сульфіди, гідрокар-
бонати і карбонати лужних металів, альдегіди, кетони
та ін.
При визначенні води у твердих речовинах, нерозчин-
них у метанолі, тонко здрібнену наважку речовини
збовтують із метанолом, після чого титрують реакти-
вом К.Фішера. Деякі речовини або суміші можна роз-
чиняти у безводних оцтовій кислоті, хлороформі,
піридині та інших розчинниках.
Кінцеву точку титрування допускається визначати!
візуально за зміною забарвлення титрованої рідини
від жовтого до червонувато-коричневого за умови за-
безпечення необхідної точності. При цьому необхідно
проводити контрольний дослід.
100
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
2.6. БІОЛОГІЧНІ ВИПРОБУВАННЯ
2.6.1. СТЕРИЛЬНІСТЬ
Випробування проводять для контролю субстанцій, го-
тових лікарських засобів або виробів, які відповідно до
вимог Фармакопеї мають бути стерильними. Однак
задовільний результат аналізу означає лише те, що в
умовах випробування у зразку не були виявлені
життєздатні мікроорганізми. Для доказу стериль-
ності серії необхідне виконання додаткових умов, які
уговорюються в розділі "Інструкція щодо застосуван-
ня випробування на стерильність".
ПОПЕРЕДЖЕННЯ МІКРОБНОГО ЗАБРУДНЕННЯ
Випробування на стерильність проводять за асептич-
них умов, використовуючи, наприклад, ламінар-бокс
масу А, розташований у чистому приміщенні класу В,
або ізолятор. Заходи, що вживаються для попереджен-
ня мікробного забруднення, не мають чинити впливу
на мікроорганізми, які можуть бути виявлені у зразку
в результаті випробування. Умови проведення випро-
бування треба регулярно контролювати шляхом
аналізу проб, відібраних відповідним чином у робочій
зоні, або проведення інших контрольних заходів, ви-
кладених у відповідних Директивах Європейського
Співтовариства і примітках до керівних матеріалів з
СМР.
ЖИВИЛЬНІ СЕРЕДОВИЩА
Живильні середовища для випробування можуть готу-
ватися, як описано нижче. Допускається використан-
ня сухих живильних середовищ за умови, що після приго-
тування вони відповідають вимогам за ростовими вла-
стивостями.
Для проведення випробування на стерильність мо-
жуть бути використані живильні середовища, наведені
нижче. Рідке тіогліколеве середовище призначене на-
самперед для вирощування анаеробних бактерій, од-
нак може бути також використане для виділення ае-
робних бактерій. Соєво-казеїнове середовище при-
значене насамперед для вирощування аеробних бак-
терій. однак може бути також використане для
виділення грибів. Допускається використання інших
живильних середовищ за умови, що буде доведена
їхня здатність підтримувати ріст широкого спектра
мікроорганізмів.
Рідке тіогліколеве середовище
Е-Цистин 0.5 г
Гранульований агар
(вміст вологи не більше 15 %) 0.75 г
Натрію хлорид 2.5 г
Глюкози моногідрат 5.5 г
Дріжджовий екстракт (водорозчинний) 5.0 г
Панкреатичний гідролізат казеїну 15.0 г
Натрію тіогліколят або 0.5 г
Кислота тіогліколева 0.3 мл
Розчин резазурину натрію (1:1000),
свіжоприготований 1.0 мл
Вода Р 1000 мл
рН після стерилізації 7.1±0.2
Змішують Е-цистин, агар, натрію хлорид, глюкозу, во-
дорозчинний дріжджовий екстракт і панкреатичний
гідролізат казеїну з водою Р і нагрівають до розчинен-
ня інгредієнтів. До одержаного розчину додають
натрію тіогліколят або кислоту тіогліколеву і пе-
ремішують до розчинення. Якщо необхідно, додають
1 М розчин натрію гідроксиду так, щоб значення рН
живильного середовища після стерилізації становило
7.1±0.2. Якшо необхідно, розчин нагрівають, не дово-
дячи до кипіння, а потім фільтрують гарячим крізь
зволожений паперовий фільтр. Додають розчин реза-
зурину натрію і перемішують. Живильне середовище
розливають у контейнери, які забезпечують таке спів-
відношення площі поверхні живильного середовища
до його глибини, щоб на кінець періоду інкубації
зміна кольору середовища, що засвідчує про
збільшення концентрації кисню, спостерігалася не
більш як у верхній третині середовища. Стерилізують
у паровому стерилізаторі, застосовуючи валідований
метод. Якшо середовище не призначене для негайно-
го використання, його зберігають при температурі від
2 °С до 25 °С у стерильному повітронепроникному
контейнері. Якщо необхідно, середовище регенеру-
ють безпосередньо перед використанням, наприклад,
шляхом нагрівання на водяній бані протягом 20 хв і
наступного швидкого охолодження. При цьому не-
обхідно вжити заходів шодо попередження контакту
нестерильного повітря з живильним середовищем.
Соєво-казеїнове живильне середовище
Панкреатичний гідролізат казеїну 17.0 г
Папаїновий гідролізат соєвої муки 3.0 г
Натрію хлорид 5.0 г
Дикалію гідрофосфат 2.5 г
Глюкози моногідрат 2.5 г
Вода Р 1 000 мл
рН після стерилізації 7.3±0.2
Інгредієнти складу змішують із водою Р і злегка
нагрівають до розчинення. Охолоджують розчин до
кімнатної температури. Якшо необхідно, додають
1 М розчин натрію гідроксиду так. шоб значення рН
живильного середовища після стерилізації становило
7.3±0.2. Якщо необхідно, фільтрують для одержання
прозорого середовища, розливають у підхожі посуди-
ни і стерилізують у паровому стерилізаторі, застосову-
ючи валідований метод. Якщо середовище не призна-
чене для негайного використання, його зберігають
при температурі від 2 °С до 25 °С у стерильному герме-
тичному контейнері.
Незалежно від того, чи використовують при випробу-
ванні на стерильність живильні середовища, описані
више, чи середовища іншого складу, усі вони мають
задовольняти наведені нижче вимоги щодо стериль-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
101
2.6. Біологічні випробування
пості та ростових властивостей. Випробування на
відповідність живильних середовищ зазначеним ви-
могам проводять за наведеними нижче методиками
перед випробуванням лікарського засобу на сте-
рильність або одночасно з ним.
Стерильність. Декілька контейнерів з живильним се-
редовищем інкубують при температурах, зазначених у
Табл. 2.6.1.-1 протягом 14 діб. Після закінчення
періоду інкубації не має спостерігатися ріст мікроор-
ганізмів.
Ростові властивості. Випробування для аеробів, ана-
еробів і грибів. Порції випробовуваних живильних се-
редовищ інокулюють невеликою кількістю
(10-100 колонієутворюючих одиниць(КУО)) тест-
мікроорганізмів. При випробуванні рідкого тіогліко-
левого середовища використовують не менше одного
виду аеробних і не менше одного виду анаеробних
бактерій. Для соєво-казеїнового середовища викорис-
товують не менше одного виду грибів і одного виду ае-
робних бактерій. Одну порцію середовища інокулю-
ють одним тест-мікроорганізмом. Рекомендовані
тест-мікроорганізми наведені у Табл. 2.6.1.-1. Інкубу-
ють за умов, зазначених у Табл. 2.6.1.-1, не більше
трьох діб (для бактерій) і не більше п’яти діб (для
грибів).
Робочу культуру, використовувану при випробуванні,
одержують таким чином, щоб кількість пересівань,
зроблених від вихідного тест-штаму, не перевищувала
п’яти.
У тому випадку, коли після закінчення періоду інку-
бації у випробовуваному живильному середовищі спос-
терігається виразно видимий ріст мікроорганізмів, йо-
го вважають придатним для подальшого використання.
Якщо після закінчення періоду інкубації у посівах, шо
містять лікарський засіб, спостерігається виразно ви-
Таблиня 2.6.1.-!
Тест-мікроорганізми, рекомендовані для контролю ростових властивостей живильних
середовищ і для перевірки придатності методики
ПЕРЕВІРКА ПРИДАТНОСТІ МЕТОДИКИ
ВИПРОБУВАННЯ
При перевірці придатності методики випробування
тіогліколевому середовищі використовують ненелц
кількість, (10-100 КУО) анаеробних і аеробних б»
терій, наведених у Табл. 2.6.1.-1. Для соєво-казеїн»
го середовища використовують гриби, зазначені і
Табл. 2.6.1.-1. Проводять випробування відповідної
методики, описаної нижче у розділі "Випробувані
лікарського засобу на стерильність", але з такив
змінами.
Випробування методом мембранної фільтрації. Вмісі І
контейнера(ів) з випробовуваним зразком пропуск,
ють крізь мембранний фільтр, мембранний фільрі
відмивають, додавши до останньої порції промив»
рідини невелику кількість (10-100 КУО) відповідно™
тест-мікроорганізму.
Випробування методом прямого висівання. Вміст кон-
тейнера(ів) з випробовуваним зразком (для кетгуту}»
інших шовних матеріалів для ветеринарії — нитки
вносять у живильне середовище, потім інокулююп
середовище невеликою кількістю (10-100 КУО
відповідного тест-мікроорганізму.
Незалежно від методу випробування проводять кон
трольний дослід, як описано вище у розділі "Живильні
середовища. Ростові властивості. Випробування дл.
аеробів, анаеробів і грибів". Усі посіви інкубують при
температурах, зазначених у Табл. 2.6.1.-1. Тривалість
інкубації має становити не більше трьох діб (для бак-
терій) і не більше п’яти діб (для грибів).
Тест-мікроорганізми Умови інкубації
Видова назва Рекомендовані штами Температура (°С) Максимальна тривалість
Аеробні бактерії Д ія всіх аеробів
Зіаріїуіососси^ аигеиз АТСС 6538 СІР4.83 КСТС 10788 КС1МВ9518 30-35 три доби
Васіїїиз зиЬііІії АТСС 6633 СІР 52.62 МСІМВ8054
Рзешіопюпах аегикіпоха АТСС 9027 КІСІМВ8626 СІР82.118
Анаеробні бактерії Для всіх анаеробів
СІоМг 'иііит зрого^епез АТСС 19404 СІР79.3 30-35 три доби
Гриби Для всіх грибів
Сапсіісіа аІЬісапз АТСС 10231 ІР 48.72 АТСС 2091 1Р 1180.79 20-25 п’ять діб
Аг>рег§і11из пі§ег АТСС 16404
102
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
німий ріст тест-мікроорганізмів, що не поступається
□ інтенсивністю контролю, вважають, що випробову-
кнмй лікарський засіб або не має антимікробної ак-
иеності за умов випробування на стерильність, або
антимікробна активність повністю нейтралізується. У
пиальшому випробування на стерильність проводять
без зміни методики.
Якщо після закінчення періоду інкубації в посівах, що
містять лікарський засіб, виразно видимий ріст тест-
иікроорганізму відсутній або поступається за інтен-
сивністю контролю, вважають, що антимікробна ак-
тивність лікарського засобу за умов випробування на
стерильність не була повністю нейтралізована. Зміню-
ють умови випробування так, щоб повністю нейт-
ралізувати антимікробну активність, і повторюють пе-
ревірку придатності методики.
Перевірку придатності методики проводять:
і) при випробуванні на стерильність нового
лікарського засобу;
) іавжди, у тому випадку, коли вносять зміни в
умови проведення випробування на стерильність.
Допускається проводити перевірку придатності мето-
іики одночасно з випробуванням лікарського засобу
на стерильність. При цьому облік результатів випро-
бування на стерильність проводять у тому випадку, як-
що підтверджена придатність використовуваної мето-
дики.
ВИПРОБУВАННЯ ЛІКАРСЬКОГО ЗАСОБУ
НА СТЕРИЛЬНІСТЬ
Випробування проводять, використовуючи метод
мембранної фільтрації або метод прямого висівання.
Незалежно від методу випробування проводять
відповідний негативний контрольний дослід, викори-
стовуючи зразки, стерильність яких була доведена
раніше. Метод мембранної фільтрації треба викорис-
товувати в усіх випадках, коли природа випробовува-
ного лікарською засобу це дозволяє — для випробу-
вання лікарських засобів у вигляді водних розчинів,
шо піддаються фільтрації; лікарських засобів, що
змішуються або розчиняються у водних розчинниках,
або оліях, які не мають антимікробної активності за
умов випробування.
Випробування методом мембранної фільтрації. Викори-
стовують мембранні фільтри з номінальним розміром
пор не більше 0.45 мкм, здатні ефективно затримувати
мікроорганізми. Для водних, розведених спиртових
розчинів та розчинів в оліях, наприклад, використову-
ють целюлозно-нітратні мембранні фільтри, а для
концентрованих спиртових розчинів — целюлозно-
ацетатні мембранні фільтри. Якщо необхідно, для де-
яких лікарських засобів, наприклад, для антибіотиків,
можуть бути використані спеціальні мембранні
фільтри.
Використовують мембранні фільтри діаметром близь-
ко 50 мм. При використанні мембранних фільтрів
іншого діаметра об’єм розчинника і промивної рідини
змінюють відповідним чином. Фільтраційну установ-
ку і мембранні фільтри стерилізують підхожим спосо-
бом. Конструкція фільтраційної установки має забез-
печувати асептичні умови при внесенні й фільтрації
лікарського засобу, а також при видаленні мембранно-
го фільтра для перенесення його у живильне середови-
ще або має дозволяти проводити інкубацію посівів
безпосередньо в самій установці після додавання в неї
живильного середовища.
Водні розчини. Перед початком випробування реко-
мендується пропустити крізь мембранний фільтр не-
велику кількість підхожого стерильного розчинника,
наприклад, нейтрального розчину 1 г/л м’ясного або
казеїнового пептону з рН 7.1±0.2. Розчинник може
містити відповідні нейтралізатори і/або відповідні
інактиватори. наприклад, при випробуванні ан-
тибіотиків.
Увесь вміст контейнера(ів) з випробовуваним зразком
переносять на мембранний фільтр або мембранні
фільтри. Якщо необхідно, попередньо доводять об’єм
зразка до 100 мл відповідним стерильним розчинни-
ком, однак у будь-якому випадку кількість лікарсько-
го засобу, взята для аналізу, має бути не менше зазна-
ченої у Табл. 2.6.1.-2. Негайно фільтрують. Якщо
лікарський засіб має антимікробну активність за умов
випробування, мембранний фільтр відмивають не
менше трьох разів, пропускаючи крізь нього при кож-
ному відмиванні той самий об’єм вибраної стерильної
промивної рідини, що і при перевірці придатності ме-
тодики. Після відмивання мембранний фільтр пере-
носять у живильне середовище або розрізають його,
дотримуючись правил асептики, на дві рівні частини,
кожну з яких поміщають у відповідне живильне сере-
довище. Використовують ті самі кількості живильного
середовища, що і при перевірці придатності методики.
При використанні установки закритого типу живиль-
не середовище вносять безпосередньо в установку. Як-
що немає інших зазначень в окремій статті, посіви
інкубують не менше 14 діб при температурі від ЗО °С до
35 °С (живильні середовища, призначені переважно
для виявлення бактерій) і при температурі від 20 °С до
25 °С (живильні середовища, призначені переважно
для виявлення грибів).
Тверді розчинні речовини. Кількість лікарського засобу,
взята для висівання на кожне живильне середовище,
має бути не менше зазначеної у Табл. 2.6.1.-2. Зразок
розчиняють у підхожому розчиннику, наприклад, ней-
тральному розчині 1 г/л м’ясного або казеїнового пеп-
тону, і проводять випробування, як описано для вод-
них розчинів, використовуючи тип мембранних
фільтрів відповідно до вибраного розчинника.
Олії і розчини в оліях. Кількість лікарського засобу, взя-
та для висівання на кожне живильне середовище, має
бути не менше зазначеної у Табл. 2.6.1.-2. Олії і розчи-
ни олій з досить малою в’язкістю допускається філь-
трувати крізь сухий мембранний фільтр без поперед-
нього розведення В’язкі олії, якщо необхідно, розчи-
няють у підхожому стерильному розчиннику, напри-
клад, в ізопропілміристаті, який не має антимікробної
активності за умов випробування. Дають можливість
олії проникнути в пори мембранного фільтра само-
пливом, а потім фільтрують, використовуючи тиск,
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
103
2.6. Біологічні випробування
або вакуум. Мембранний фільтр відмивають не менше
трьох разів, пропускаючи крізь нього щоразу близько
100 мл відповідної стерильної промивної рідини, на-
приклад, розчину 1 г/л м’ясного або казеїнового пеп-
тону, що містить 10 г/л полісорбату-80 або підхожу
кількість іншого емульгатора, шо не має антимікроб-
ної активності за умов випробування. Мембранний
фільтр або мембранні фільтри поміщають у живильне
середовище або живильні середовища, або вносять
живильне середовище або живильні середовища у
фільтраційну установку й інкубують, як описано для
водних розчинів.
Мазі і креми. Кількість лікарського засобу, взята для
висівання на кожне живильне середовище, має бути
не менше зазначеної у Табл. 2.6.1.-2. Мазі на жировій
основі та емульсії типу вода/олія допускається розво-
дити ізопропілміристатом у співвідношенні 1:100, як
описано вище, використовуючи, якщо необхідно,
нагрівання до температури не більше 40 °С. У винят-
кових випадках допускається нагрівання до темпера-
тури не більше 44 °С. Фільтрацію проводять негайно з
максимально можливою швидкістю. Далі випробу-
вання проводять, як описано вище для олій і розчинів
в оліях.
Випробування методом прямого висівання. Випробо-
вуваний лікарський засіб вносять безпосередньо у
живильне середовище у кількості, зазначеній у
Табл. 2.6.1.-2. Якщо немає інших зазначень в окремій
статті, кількість лікарського засобу має становити не
більше 10 % від об’єму живильного середовища.
У тому випадку, коли лікарський засіб має ан-
тимікробну активність за умов випробування, її нейт-
ралізують шляхом додавання підхожих нейтралі.,
торів або збільшення об’єму живильного сери
виша. Якщо для висівання необхідно використати в®
лику кількість зразка, краще застосовувати концсиі
роване живильне середовище, приготоване з урах»
ванням наступного розведення. Там, де це можім®
концентроване живильне середовище рекомендуєтьЛ
додавати безпосередньо в контейнер з лікарським за
собом.
Рідини, що містять олії. У живильне середовище по-
передньо додають 10 г/л полісорбату або підхож
кількість іншого емульгатора, що не має антимікрої і
ної активності за умов випробування.
Мазі і креми. Лікарський засіб емульгують у розв-|
денні 1:10 за допомогою підхожого емульгатора в
підхожому стерильному розчиннику, наприклад, ней-
тральному розчині 1 г/л м’ясного або казеїнового неп-1
тону. Одержану емульсію вносять у живильне середо-1
више, яке не містить емульгатора.
Якщо немає інших зазначень в окремій статті, посіви|
інкубують не менше 14 діб при температурах, зазначе-
них у Табл. 2.6.1.-1. Посіви переглядають кілька разіву'
період інкубації. Посіви олійних лікарських засоби,
щодня акуратно струшують. Однак у тому випадку, ко-
ли тіогліколеве або аналогічне йому середовище за-
стосовується для виявлення анаеробних мікроор-
ганізмів, струшування або перемішування має бути
зведене до мінімуму для того, щоб не порушувати ана-
еробних умов.
Кетгут та інші шовні матеріали для ветеринарії. Кіль-
кість зразка, взята для висівання на кожне живиль-
Таблиця 2.6.1.-2
Кількість готового лікарського засобу, необхідна для випробування на стерильність
Тйп готового лікарського засобу Кількість готового лікарського засобу в одному контейнері Мінімальна кількість зразка, яку необхідно використовувати для висівання на кожне живильне середовище, якщо немає інших зазначень в окремій статті
Парентеральні лікарські засоби Рідини - менше 1 мл - 1 мл і більше Порошки - менше 50 мг - 50 мг і більше, але не більше 300 мг - 300 мг і більше Увесь вміст кожного контейнера Половина вмісту кожного контейнера, але не більше 20 мл Увесь вміст кожного контейнера Половина вмісту кожного контейнера 150 мг
Офтальмологічні та інші неін’єкційні лікарські засоби Водні розчини Інші готові лікарські засоби, розчинні у воді або ізопропілміристаті Нерозчинні готові лікарські засоби креми і мазі, що піддаються суспендуванню або емульгуванню Увесь вміст кожного контейнера, але не менше 2.5 мл Увесь вміст кожного контейнера, але не менше 0.25 г Увесь вміст кожного контейнера, але не менше 0.25 г
Кетгут та інші шовні ма- теріали для ветеринарії Три відрізки нитки (по 30 см кожний)
104
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
І середовище, має бути не менше зазначеної у
І іГ . 2.6.1.-2, Розкривають упаковку, дотримуючись
правил асептики, і переносять по три відрізка,
ші< рані від початку, середини і кінця нитки, у кожне
< івильне середовище. Довжина кожного відрізка має
вповити ЗО см При випробуванні матеріалів у ка-
лій упаковці використовують усю нитку. Кожний
шрізок нитки вносять у відповідне живильне середо-
пгде. Живильне середовище має цілком покривати
випробовуваний матеріал (використовують від
мі до 150 мл живильного середовища).
іБЛІКІ ІНТЕРПРЕТАЦІЯ РЕЗУЛЬТАТІВ
П сіви переглядають періодично під час і після
пкінчення інкубаційного періоду, відмічаючи на-
ївність візуально виявлюваного росту мікроор-
інізмів Якщо випробовуваний зразок викликає по-
тіння живильного середовища, що робить немож-
впм візуальний облік, то через 14 діб після початку
інкубації з кожної посудини переносять певну
ніькість середовища в посудини з тим самим свіжим
Вівильним середовищем. Продовжують інкубацію
віілних і повторних посівів. Загальний час інкубації
'Цс становити не менш як (14+7) діб від початку ви-
пробування.
Лікарський засіб витримує випробування на сте-
ильність, якщо при візуальному обліку не вияв-
імєгься ріст мікроорганізмів. При наявності росту
мікроорганізмів вважають, що лікарський засіб не ви-
тримує випробування на стерильність, якщо не дове-
гна невірогідність результатів випробування, викли-
чна причинами, не пов’язаними з випробовуваним
іікарським засобом. Результати випробування можуть
бути визнані невірогідними, якщо виконується одна
то кілька з умов, наведених нижче:
і) одержані незадовільні результати мікробіологі-
чного контролю навколишнього середовища в
ході проведення випробування на стерильність;
Ь) виявлені помилки, допущені в ході випробування;
с) виявлений ріст мікроорганізмів у негативному
контролі;
ф після ідентифікації мікроорганізмів, виділених з
лікарського засобу, однозначно визнано, що при-
чиною виникнення росту нього виду або видів є
матеріали і/або технічні прийоми, використані
при випробуванні на стерильність.
Якшо результати випробування визнані невірогідни-
ми. його повторюють на тій самій кількості зразків,
шо й початкове.
Якщо в результаті повторного випробування не було
виявлено росту мікроорганізмів, вважають, що
лікарський засіб витримує випробування на
стерильність. Якщо в результаті повторного ви-
пробування було виявлено ріст мікроорганізмів,
лікарський засіб не витримує випробування на сте-
рильність.
КОНТРОЛЬ СТЕРИЛЬНОСТІ
ПАРЕНТЕРАЛЬНИХ, ОФТАЛЬМОЛОГІЧНИХ
І ІНШИХ НЕІН’ЄКЦІЙНИХ ГОТОВИХ
ЛІКАРСЬКИХ ЗАСОБІВ, ДО ЯКИХ СТАВЛЯТЬСЯ
ВИМОГИ ЩОДО СТЕРИЛЬНОСТІ
При випробуванні методом мембранної фільтрації ви-
користовують, якщо це можливо, увесь вміст контейне-
ра, але не менше кількості, зазначеної у Табл. 2.6.1.-2.
Якщо необхідно, доводять об’єм зразка до 100 мл
відповідним стерильним розчинником, наприклад,
нейтральним розчином 1 г/л м’ясного або казеїнового
пептону. Якщо немає інших зазначень в окремій
статті, загальний об’єм, що пропускається крізь один
мембранний фільтр, не має перевищувати 1000 мл.
При випробуванні методом прямого висівання вико-
ристовують зразок у кількостях, зазначених у
Табл. 2.6.1.-2. Із кожного контейнера проводять
висівання на середовище для виявлення бактерій і на
середовище для виявлення грибів. У тому випадку, як-
що об’єм або кількість лікарського засобу в одному
контейнері недостатня для проведення випробування,
використовують вміст двох і більше контейнерів для
висівання на різні живильні середовища.
Якщо об’єм вмісту одного контейнера перевищує
100 мл, використовують метод мембранної фільтрації,
якшо немає інших зазначень в окремій статті.
ІНСТРУКЦІЯ ЩОДО ЗАСТОСУВАННЯ
ВИПРОБУВАННЯ НА СТЕРИЛЬНІСТЬ
Наведена методика випробування на стерильність так
само, як і всі інші фармакопейні методики, призначе-
на для того, щоб дати змогу незалежному контролюю-
чому компетентному органу встановити, чи відповідає
конкретний лікарський засіб вимогам Фармакопеї.
Виробник не зобов’язаний дотримуватися наведених
методик, він може вносити до них зміни або викорис-
товувати інші, якщо гарантує, що при випробуванні
описаними вище офіційними методами його про-
дукція відповідатиме вимогам Фармакопеї.
Рекомендації виробникам. Рівень надійності, з яким за
задовільним результатом випробування на сте-
рильність (відсутність нестерильних контейнерів у
зразку) можна зробити висновок про якість усієї серії,
залежить від однорідності серії, умов виробництва і
прийнятого порядку відбору проб. У даній статті під
серією розуміють сукупність герметично закупорених
контейнерів, вироблених таким чином, що під час ви-
робничого процесу ризик мікробного забруднення був
однаковий для кожного з них.
У випадку, коли лікарський засіб піддається процедурі
кінцевої стерилізації, результати автоматичного кон-
тролю обгрунтованих з точки зору біології фізичних
параметрів, що підтверджують дотримання встановле-
них режимів при стерилізації серії, характеризують її
стерильність з більшою надійністю за результати ви-
пробування на стерильність. Умови, в яких допусти-
мий параметричний випуск серії, наведені у статті
"Методи приготування стерильних продуктів" (5.1.1).
Для підтвердження дотримання асептичних умов у
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
105
2.6. Біологічні випробування
процесі виробництва може бути використаний метод
наповнення живильними середовищами. Однак ви-
пробування на стерильність є єдиним аналітичним
методом, що дозволяє оцінити стерильність лікарсь-
кого засобу, виготовленого в асептичних умовах, і,
крім того, у будь-якому випадку це єдиний аналітич-
ний метод, що дозволяє оцінити стерильність зразка
лікарського засобу при проведенні експертизи.
При проведенні випробування на стерильність мож-
ливість виявлення мікроорганізмів прямо про-
порційна їх кількості у випробовуваному зразку і зале-
жить від здатності цих мікроорганізмів давати види-
мий ріст на живильних середовищах за умов випробу-
вання. Ймовірність виявлення мікроорганізмів мала
при дуже низькому рівні забруднення лікарського за-
собу, навіть при рівномірному мікробному забруд-
ненні серії. Інтерпретація результатів випробування
на стерильність грунтується на тому припущенні, що
одержувані результати були б ідентичні для кожного
контейнера, що входить до складу серії. Оскільки ви-
пробування кожного контейнера провести неможли-
во, необхідно розробити план відбору проб. При ви-
пробуванні продукції, виготовленої за асептичних
умов, рекомендується відбирати контейнери, напов-
нені на початку і в кінці серії, а також після впливу
істотних перешкод.
У Табл. 2.6.1.-З наведені рекомендовані мінімальні
кількості зразків для аналізу залежно від кількості
контейнерів у серії. При складанні плану відбору проб
необхідно враховувати об’єм лікарського засобу в
йому контейнері, метод стерилізації, а також іі
фактори, які можуть чинити вплив на стерильні
лікарського засобу.
ЖИВИЛЬНІ СЕРЕДОВИЩА
Для проведення випробування на стерильність моя
жуть бути також використані живильні середовищі
наведені нижче. Рідке тіогліколеве середовище при *
значене для вирощування бактерій, середовище Саб\
ро — для вирощування грибів.
Рідке тіогліколеве середовище
Цистин (цистеїн) 0.75' І
Агар 0.75г
Натрію хлорид 2.5» І
Глюкоза 5Л|
Дріжджовий екстракт (10 %) 5.01
Панкреатичний гідролізат казеїну 150
Натрію тіогліколят або 0.5
кислота тіогліколева 0.3 м.
Розчин резазурину натрію 1:1000
свіжоприготований 1.0 щ І
Вода очищена 1 000вд І
рН після стерилізації 7.0±0.2
Таблиця 2.6.1.-.
Рекомендовані мінімальні кількості контейнерів для аналізу
Кількість контейнерів у серії Мінімальна кількість контейнерів, необхідна для випробування на стерильність на кожному живильному середовищі*
Парентеральні готові лікарські засоби - Не більше 100 контейнерів - Більше 100, але не більше 500 контейнерів - Більше 500 контейнерів 10 % від серії, але не менше 4 контейнерів 10 контейнерів 2 %, але не більше 20 контейнерів
Офтальмологічні та інші неін ’єкціині готові лікарські засоби - Не більше 200 контейнерів - Більше 200 контейнерів - У тому випадку, якщо готовий лікарський засіб випускається в однодозових контейнерах, міні- мальну кількість контейнерів, необхідну для випробування, визначають, як описано вище для парентеральних готових лікарських засобів 5 % від серії, але не менше 2 контейнерів 10 контейнерів
Кетгут та інші шовні матеріали для ветеринарії 2 % від серії, але не менше 5 і не більше 20 контейнерів
Порошки в упаковці "іп Ьиік" - Менше 4 контейнерів - Більше 4, але не більше 50 контейнерів - Більше 50 контейнерів Кожний контейнер 20 % від серії, але не менше 4 контейнерів 2 % від серії, але не менше 10 контейнерів
* Якщо вміст одного контейнера є достатнім для висівання на двох середовищах, то в цій колонці наведена кількість контейнерів,
необхідних для випробування на стерильність на двох живильних середовищах.
106
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
Рідке середовище Сабуро
Пептон ферментативний
Гіюкоза
Вода очищена
10г
40 г
1 000 мл
рН після стерилізації 5.6±0.2
Обидва середовища стерилізують у паровому стери-
Ііеторі при температурі 121 °С протягом 15 хв.
РОЗЧИННИКИ І ПРОМИВНІ РІДИНИ
„ля розчинення лікарських засобів і відмивання мем-
інних фільтрів можуть бути використані розчинни-
ки, які не виявляють антимікробної активності за
•.мов випробування. Рекомендовані розчинники і
промивні рідини наведені нижче.
Розчин 9 г/л натрію хлориду
Рідина № 1
Пептон ферментативний 1 г
ІІода очищена і 000 мл
рН після стерилізації 7.1 ±0.2
Рідина № 2
Пептон ферментативний 5 г
Полісорбат-80 10 г
Вода очищена 1 000 мл
рН після стерилізації 7.1+0.2
Стерилізують у паровому стерилізаторі, як описано
зишедля живильних середовищ. Час стерилізації ви-
значають при валідації методу стерилізації залежно від
•Гему рідин, які стерилізуються.
ТЕСТ-МІКРООРГАНІЗМИ
При перевірці придатності методики і для контролю
ростових властивостей живильних середовищ можуть
бути також використані такі штами тест-мікроор-
ганізмів
- додатково для тіогліколевого середовиїца — тест-
мікроорганізм ЕзсИегісИіа соїі АТСС 25922;
- замість тест-мікроорганізму Сапсіісіа аІЬісапк
АТСС 10231 (ІР48. 72, АТСС 2091, ІР 1180. 79) мо-
же бути використаний тест-мікроорганізм
Сашііда аІЬісапх N0+0 885-653;
- замість тест-мікроорганізму Сіозігісііит зрогозепез
АТСС 19404 (СІР 79.3) може бути використаний
тест-мікроорганізм Сіо^ігісііит 8рого%епе8 № 272.
Допускається використання інших штамів мікроор-
ганізмів за узгодженням з компетентним уповноваже-
ним органом.
При випробуванні використовують 18-24-годинні
культури бактерій і 48-годинні культури грибів.
Рекомендовані інактиватори: полісорбат-80, лецитин,
лзачектин; п-амінобензойна кислота для сульфаніла-
мідів; псніциліназа або р-лактамаза для пеніцилінів
або цефалоспоринів.
2.6.8. ПІРОГЕНИ
Випробування на пірогени полягає у вимірюванні
підвищення температури тіла, спричиненого у кро-
ликів внутрішньовенним введенням стерильного роз-
чину випробовуваного лікарського засобу.
Добір тварин. Використовують здорових статевозрілих
кроликів обох статей масою не менше 1.5 кг, які діста-
ють повноцінне і збалансоване харчування без ан-
тибіотиків і не втрачають масу тіла протягом тижня,
що передує випробуванню. Кролик не може бути ви-
користаний у випробуванні на пірогени:
а) якщо він був використаний у випробуванні на
пірогени з негативним результатом протягом по-
передніх трьох діб;
б) якщо він був використаний у попередні три тижні
у випробуванні на пірогени, в якому зразок не ви-
тримав випробування.
Приміщення для тварин. Кроликів утримують нарізно в
тихому приміщенні з підхожою постійною температу-
рою. Напередодні увечері й до повного завершення
випробування кроликів позбавляють їжі; під час ви-
пробування позбавляють води. Випробування прово-
дять у тихій кімнаті, де немає ризику збудження тва-
рин і в якій температура не відрізняється більш як на
З °С від температури в житлових приміщеннях для
кроликів або в яких кроликів утримували принаймні
протягом 18 год перед випробуванням.
Матеріали. Скляний посуд, шприци та голки. Ретельно
миють увесь скляний посуд, шприци та голки водою
для ін’єкцій і прогрівають гарячим повітрям при тем-
пературі 250 °С протягом 30 хв або при температурі
200 °С протягом 1 год.
Фіксуючі клітки. Фіксуючі клітки для кроликів, тем-
пературу яких вимірюють за допомогою електричного
пристрою, мають бути виготовлені таким чином, щоб
тварини фіксувалися лише за допомогою вільно
закріплених на шиї хомутів; тулуб залишається
відносно вільним, щоб кролики могли сидіти в зви-
чайному положенні. їх не утримують за допомогою
ременів або інших подібних методів, які можуть спри-
чинити шкоду тварині. Тварин поміщають у клітки не
менш як за 1 год до першого вимірювання температу-
ри і залишають у них протягом випробування.
Термометри. Використовують термометр або елект-
ричний пристрій, що визначають температуру з
точністю до 0.1 °С, і уводять у пряму кишку кролика
на глибину близько 5 см. Глибина уведення пристрою
постійна у кожного кролика в кожному випробуванні.
При використанні електричного пристрою його мож-
на залишити у цьому положенні протягом усього ви-
пробування.
Попереднє випробування. Після добору тварин, за
один-три дні до випробування зразка, тим тваринам,
яких не використовували протягом попередніх
двох тижнів, уводять внутрішньовенно 10 мл на кіло-
грам маси тілаапірогенний 9 г/л розчин натрію хлори-
ду, підігрітий до температури 38.5 °С. Реєструють тем-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
107
2.6. Біологічні випробування
пературу тварин, починаючи за 90 хв до ін’єкції і про-
довжуючи протягом 3 год після ін’єкції розчину. Будь-
яка тварина, у якої виявляється зміна температури
більш як на 0.6 °С, не використовується в основному
випробуванні.
Основне випробування. Випробування проводять, ви-
користовуючи групу із трьох кроликів.
Підготовка і введення випробовуваного зразка. Перед
введенням випробовуваний розчин нагрівають при-
близно до температури 38.5 °С. Випробовуваний зра-
зок може бути розчинений або розведений в апіроген-
ному розчині 9 г/л натрію хлориду або іншому розчин-
нику, зазначеному в окремій статті. Розчин повільно
уводять у крайову вену вуха кожного кролика протя-
гом не більше 4 хв, якщо немає інших зазначень в ок-
ремій статті. Кількість випробовуваного зразка, що
вводиться, залежить від його властивостей і зазна-
чається в окремій статті. Об’єм, що вводиться, має бу-
ти не менш як 0.5 мл і не більш як 10 мл на кілограм
маси тіла.
Визначення вихідної і максимальної температур.
"Вихідна температура" кожного кролика — це середнє
значення із двох показань температур, зареєстрованих
у кролика з інтервалом ЗО хв протягом 40 хв, що безпо-
середньо передують введенню випробовуваного зраз-
ка. "Максимальна температура" кожного кролика є
найвищою зареєстрованою температурою даного кро-
лика протягом 3 год після ін’єкції. Температуру кож-
ного кролика реєструють з інтервалом не більше ЗО хв,
починаючи не раніш як за 90 хв перед введенням ви-
пробовуваного зразка і продовжуючи протягом 3 год
після уведення. Різницю між максимальною темпера-
турою і вихідною температурою для кожного кролика
беруть за його реакцію. Якщо ця різниця від’ємна, ре-
зультат оцінюють як нульову реакцію.
Кроликів, у яких при визначенні вихідної температу-
ри виявляють відмінності між двома послідовними
температурними показниками більш як на 0.2 °С, ви-
лучають із випробування. У будь-якому випробуванні
використовують лише тих кроликів, вихідні темпера-
тури яких не різняться між собою більш як на 1 °С.
Кроликів, що мають вихідну температуру вище 39.8 °С
або нижче 38.0 °С, вилучають із випробування.
Інтерпретація результатів. Випробування проводять
спочатку на групі із трьох кроликів, якщо необхідно,
далі повторюють на наступних групах із трьох кро-
ликів до загальної кількості чотири групи, залежно від
одержаних результатів. Якщо сумарна реакція у
першій групі не перевищує значення, наведеного у
другій колонці Табл. 2.6.8.-1, зразок витримує випро-
бування. Якщо сумарна реакція перевищує значення,
наведене у другій колонці таблиці, але не перевищує
значення, наведеного в третій колонці таблиці, випро-
бування повторюють, як зазначено вище. Якщо су-
марна реакція перевищує значення, наведене в третій
колонці таблиці, зразок не витримує випробування.
Кроликів, використаних раніше у випробуванні на
пірогени, у якому середнє підвищення температури
перевищило 1.2 °С, вилучають із випробування.
Таблиця 2.6.8.•
Кількість кроликів Зразок витримує випробування, якщо сумарна реакція не перевищує Зразок не витримує 1 випробування, якщо сумарна реакція перевищує
3 1.15 °С 2.65 С
6 2.80 °С 4.30 °С
9 4.45 °С 5.95 °С
12 6.60 °С 6.60 °С
____________________________________________и
У тому випадку, коли виробництво лікарского засобу ні
проводиться відповідно до вимог належної виробнича]
практики (НВП; СМР), установлених у Європейсьтц
Співтоваристві, до даного лікарського засобу стсв-
ляться такі вимоги до випробування на пірогени1.
Випробування на пірогени виконують з метою обме-1
жити небезпеку пропасної реакції пацієнтів ні 1
ін’єкційне введення ліків і гарантувати їхню відпо
відну якість і безпеку.
Випробування полягає у вимірюванні рівня підви- і
щення температури тіла кроликів після ін’єкційного І
введення стерильного розчину лікарського засобу.
Матеріали. Скляний посуд, шприци та голки. Скляний І
посуд, шприци та голки мають бути ретельно вимиті І
водою для ін’єкцій і депірогенізовані гарячим І
повітрям при температурі 250 °С протягом 30 хв або І
при температурі 200 °С протягом І год.
Розчинники. Вода та інші розчинники, які використо-І
вують для промивання зазначених пристроїв або для І
розчинення випробовуваних зразків, мають бути І
апірогенними і стерильними.
Термометри. Використовують термометри, що мають!
точність вимірювання ± 0.1 °С і дозволяють зареєстру-1
вати максимальні показання протягом не більше 5 хв. ]
Для стандартизації умов випробування можуть бути І
використані багатоканальні пірогентестери, які дозво-
ляють здійснювати автоматизований моніторинг тем-
ператури одночасно на великій кількості тварин ]
стандартних для кожної умовах.
Добір тварин. Умови утримування. Використовують
здорових статевозрілих кроликів обох статей, масою
тіла не менше 1.5 кг. не альбіносів. Протягом тижня,
що передує випробуванню, кролики не мають втрача-
ти у масі.
Одного і того самого кролика використовують для ви-
пробування не раніш як через дві доби, якщо уведений
йому в попередньому випробуванні зразок був визна-
' У даних вимогах використані матеріали Фармакопеї США — із
дозволу Фармакопеї США.
108
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
пій апірогенним, і не раніш як через два тижні, якщо
попередньому випробуванні на пірогени зареєстро-
ию підвищення його температури на 0.6 °С і більше.
Кроликів, яких вперше використовують для випробу-
ань на пірогени, попередньо готують до процедури
апробування, витримуючи їх протягом не більше
.емиліб і здійснюючи з ними усі підготовчі процедури
(огляд, зважування, вимірювання температури) згідно
зрозділами "Добір тварин. Умови утримування" і "Ме-
тодика випробувань", за винятком ін’єкції.
Порядок повторного використання кроликів, на яких
випробовували зразки, що мають антигенні власти-
вості, має бути зазначений в окремій статті.
Кроликів утримують нарізно в стандартних клітках на
повноцінному збалансованому харчуванні, в ізольова-
ному від шуму та інших збуджувальних впливів
приміщенні при температурі від 20 °С до 23 °С. Відхи-
лення від вибраної температури мають бути не більше
±3°С.
Методика випробування. Випробування проводять по-
тайно На І етапі його виконують на трьох кроликах.
Якщо необхідний II етап, його виконують на п’ятьох
кроликах. Увечері напередодні випробування у тварин
забирають корм і не дають його до повного завершення
випробування, у перебігу якого їм не дають також воду.
Не менш як за 18 год до випробування відібраних кро-
ликів переводять до окремого ізольованого тихого
приміщення, призначеного лише для випробування
на пірогени і в якому умови подібні до тих, в яких тва-
рини утримувалися до цього.
Визначення вихідної і максимальної температур. Не
більш як за ЗО хв до ін’єкції випробовуваного зразка
вимірюють "вихідну температуру", що є базовою при
визначенні будь-якого підвищення температури у да-
ного кролика, викликаного ін’єкцією випробовувано-
го зразка. Вихідна температура кроликів в одній групі
має розрізнятися не більш як на ±1 °С і має бути не
виїде 39.8 °С.
“Максимальна температура” — це найвища темпера-
турауданого кролика протягом 3 год після ін’єкції ви-
пробовуваного зразка. Різницю між вихідною і макси-
мальною температурою кожного кролика враховують
як "максимальне підвищення температури".
Підготовка і введення випробовуваного зразка. Реєст-
рація температури. Кількість одноразово відібраної
проби випробовуваного зразка має бути достатньою
для проведення випробування на вісьмох кроликах.
Випробовуваний зразок вводять без додаткового роз-
ведення, якщо немає інших зазначень в окремій
статті. Сухі лікарські засоби для парентерального за-
стосування вводять у концентраціях і розчинниках,
передбачених інструкцією для медичного застосуван-
ня, якщо немає інших зазначень в окремій статті.
Розчин випробовуваного зразка, нагрітого перед
ін'єкцією до температури (37±2) °С, вводять не
пізніше як через ЗО хв після вимірювання вихідної
температури у крайову вену вуха.
Розчин вводять в об’ємі від 0.5 мл до 10 мл на 1 кг ма-
си тіла повільно протягом від 2 хв до 4 хв, якщо в ок-
ремій статті немає іншіх зазначень про швидкість або
шляхи введення (підшкірно, внутрішньом’язово,
внутрішньочеревно). Доза випробовуваного зразка за-
лежить від його біологічної активності і зазначається в
окремій статті.
Температурний датчик вводять у пряму кишку на гли-
бину від 7 см до 9 см, і ця глибина має бути незмінною
протягом усього випробування. Температуру тіла кро-
ликів реєструють кожні ЗО хв протягом 3 год після
ін’єкції випробовуваного зразка, визначаючи у кож-
ного кролика максимальне підвищення температури.
При використанні пірогентестерів температурний
датчик залишають у прямій кишці на весь час випро-
бування. При цьому кроликів утримують у фіксуючих
клітках за допомогою м’яко закріплених навколо шиї
хомутів, щоб не завдати їм болю і дозволити тулубу
знаходитися у фізіологічному положенні протягом
усього випробування. Кроликів поміщають у ці клітки
не менш як на 1 год до першого вимірювання темпе-
ратури і залишають в них протягом усього випробу-
вання.
Інтерпретація результатів. Будь-яке зниження темпе-
ратури вважають нульовим підвищенням. Якщо в
жодного із трьох кроликів не відмічено максимально-
го підвищення температури на 0.5 °С і більше, випро-
бовуваний зразок відповідає вимогам щодо відсут-
ності пірогенів. Якщо хоча б у одного кролика відміче-
но максимальне підвищення температури на 0.5 °С і
більше, випробування продовжують, використовуючи
п’ять інших кроликів.
Якщо не більш як у трьох із вісьмох кроликів відміче-
ні індивідуальні максимальні підвищення температу-
ри на 0.5 °С і більше і якщо сума вісьмох індивідуаль-
них максимальних підвищень температури не переви-
щує 3.3 °С, випробовуваний зразок відповідає вимо-
гам на відсутність пірогенів.
2.6.9. АНОМАЛЬНА ТОКСИЧНІСТЬ
ЗАГАЛЬНЕ ВИПРОБУВАННЯ
Кількість випробовуваного лікарського засобу, зазна-
чену в окремій статті, розчинену в 0.5 мл води для
ін ’єкціи Рабо у стерильному розчині 9 г/л натрію хло-
риду. вводять внутрішньовенно кожній із п’яти здоро-
вих мишей масою від 17 гдо 22 г. Розчин вводять про-
тягом інтервалу часу від 15 с до ЗО с, якщо немає інших
зазначень в окремій статті.
Зразок витримує випробування, якщо жодна з мишей
не гине в межах 24 год або протягом часу, зазначеного
в окремій статті. Якщо більш як одна тварина гине,
зразок не проходить випробування. У випадку заги-
белі однієї тварини випробування повторюють. Зра-
зок витримує випробування, якщо жодна з тварин у
другій групі не гине в межах зазначеного інтервалу ча-
су.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
109
2.6. Біологічні випробування
ІМУННІ СИРОВАТКИ І ВАКЦИНИ
ДЛЯ людини
Якщо немає інших зазначень в окремій статті, вводять
внутрішньочеревно одну людську дозу, але не більше
1 мл кожній із п’яти здорових мишей масою від 17 гдо
22 г. Людську дозу зазначають на етикетці випробову-
ваного лікарського засобу або у супровідному доку-
менті. Спостерігають за тваринами протягом семи діб.
Зразок витримує випробування, якщо у жодної з тва-
рин не виявляються ознаки інтоксикації. Якщо більш
як одна тварина гине, зразок не відповідає вимогам.
Якщо одна з тварин виявляє ознаки інтоксикації або
гине, випробування повторюють. Зразок витримує ви-
пробування, якщо жодна з тварин у другій групі не ви-
являє ознак інтоксикації або не гине в межах зазначе-
ного часу.
Випробування також має бути проведене на двох здо-
рових морських свинках масою від 250 г до 350 г. Вво-
дять внутрішньочеревно кожній тварині одну людську
дозу випробовуваного лікарського засобу, але не більше
5 мл. Людську дозу зазначають на етикетці лікарського
засобу або у супровідному документі. Спостерігають за
тваринами протягом семи діб. Зразок витримує випро-
бування, якщо жодна з тварин не виявляє ознак інток-
сикації. У випадку загибелі більш як однієї тварини
зразок не відповідає вимогам. Якщо одна з тварин ви-
являє ознаки інтоксикації або гине, випробування по-
вторюють. Зразок витримує випробування, якщо жод-
на з тварин у другій групі не виявляє ознак інтоксикації
і не гине протягом зазначеного часу.
__________________________________________N
Випробування на аномальну токсичність виконують із
метою виключення небезпеки підвищення рівня ток-
сичності лікарського засобу, якщо в його складі при
виробництві або зберіганні відбулися зміни, неперед-
бачені регламентом виробництва або окремою статею.
ЗАГАЛЬНЕ ВИПРОБУВАННЯ
Випробування проводять на білих мишах, на яких
раніше не проводили ніяких випробувань і яких утри-
мують у стандартних умовах на повноцінному збалан-
сованому харчуванні.
Розчинні лікарські засоби вводять внутрішньовенно,
якщо немає інших зазначень в окремій статті. Лікарсь-
кі засоби у вигляді суспензій уводять внутрішньочерев-
но в об’ємі не більше 1 мл зі швидкістю 0.1 мл/с.
Зразок витримує випробування, якшо протягом 24 год
після ін’єкції або в межах часу, зазначеного в окремій
статті, жодна із п’ятьох мишей не загине і не більш як
у однієї миші спостерігаються ознаки інтоксикації, ха-
рактер і вияв яких перевищують допустимий рівень,
зазначений в окремій статті. Якщо гине одна із п’ятьох
мишей або симптоми нерегламентованої інтоксикації
виявляються більш як у однієї із п’ятьох мишей, ви-
пробування повторюють ще на п’яти мишах. Зразок
витримує випробування, якщо жодна з тварин у другій
групі не гине.
2.6.11. ДЕПРЕСОРНІ РЕЧОВИНИ
Випробування проводять на кішці, маса тіла якої не
більше 2 кг, наркотизованій хлоралозою або барбітД
ратами для підтримання постійного кров’яного тиску
Випробування проводять за умов, які дозволяють зь-
побігти зниженню температури тіла тварини і пілтри-1
мувати її так, щоб ректальна температура зберігалася)!
фізіологічних межах. У трахею вводять дихальну труб-И
ку. У загальну сонну артерію уводять канюлю, запов-
нену гепаринізованим розчином 9 г/л натрію хлориду І
і сполучають її з пристроєм, що забезпечує тривалу І
реєстрацію кров’яного тиску. У стегнову вену вводяті-І
другу канюлю, заповнену гепаринізованим розчином г
9 г/л натрію хлориду, крізь яку можна вводити розчи-
ни гістаміну і випробовуваного зразка лікарського за І
собу.
Визначають чутливість тварин до гістаміну задопомо- І
гою внутрішньовенних ін’єкцій через рівні інтервали] І
розчину гістаміну Р у дозах, відповідних 0.1 мкг і І
0.15 мкг гістаміну основи на кілограм маси тіла. По-
вторюють меншу дозу не менше трьох разів. Другу і 11
наступні ін’єкції вводять не раніш як через І хв після
повернення кров’яного тиску до рівня, який був без-
посередньо перед попередньою ін’єкцією. Тварину
використовують у випробуванні лише в тому випадку; І
якщо одержано зниження кров’яного тиску, яке чітко І
реєструється, постійне для меншої дози, і якшо вели- І
ка доза викликає більш високу реакцію.
Розчиняють випробовуваний зразок у достатній
кількості розчину 9 г/л натрію хлориду або іншого
розчинника, як зазначено в окремій статті. Вводять
внутрішньовенно на кілограм маси тіла 1 мл розчину
гістаміну Р. потім — дві послідовні ін’єкції розчину
випробовуваного лікарського засобу в дозі, зазначеній
в окремій статті, і, нарешті, І мл розчину гістаміну Р.
Другу, третю і четверту ін’єкції роблять не раніш як че-
рез 1 хв після повернення кров’яного тиску до рівня,
на якому він був безпосередньо перед попередньою
ін’єкцією. Повторюють ні серії ін’єкцій двічі і завер-
шують випробування уведенням 1.5 мл розчину
гістаміну Р на кілограм маси тіла.
Випробування вважають не дійсним, якщо реакція на
1.5 мл розчину гістаміну Р не перевищує реакцію на
1 мл. Зразок не витримує випробування, якщо середнє
значення реакцій на його введення вище за середнє
значення реакцій на 1 мл розчину гістаміну на кіло
грам маси тіла або якщо будь-яка одна його доза ви-
кликала більш високу депресорну реакцію, ніж завер-
шальна доза розчину гістаміну. Тварин не використо-
вують повторно у випробуванні на депресорні речови-
ни, якщо будь-яка одна його доза викликає більш ви-
соку депресорну реакцію, ніж завершальна доза роз-
чину гістаміну, або якщо реакція на велику дозу
гістаміну, введену після випробовуваного зразка, ниж-
ча за середню реакцію на менші дози попередньо уве-
деного гістаміну.
Випробування на депресорні речовини виконують із
метою виключення небезпеки зниження артеріально-
110
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
л иску (депресорної реакції) після введення лікарев-
ого засобу, якщо в його складі при виробництві вия-
інлися речовини, які мають депресорну дію.
Найбільш небезпечні щодо цього лікарські засоби, які
сержгють із тканин тварин і людини або шляхом
ч.кробіологічного синтезу, бо при цьому до них мо-
«угьбути внесені високоактивні депресорні речовини
іаринного походження — гістамін, серотонін, бра-
іикінін та ін.
Ігя наркотнзування кішки допускається використан-
ня уретану.
2.6.12. ВИПРОБУВАННЯ МІКРОБІОЛОГІЧНОЇ
ЧИСТОТИ НЕСТЕРИЛЬНИХЛІКАРСЬКИХ
ЗАСОБІВ (ВИЗНАЧЕННЯ ЗАГАЛЬНОГО
ЧИСЛА ЖИТТЄЗДАТНИХ АЕРОБНИХ
МІКРООРГАНІЗМІВ)
Випробування, описані нижче, дозволяють здійсню-
їзтиі кількісне визначення мезофільних бактерій та
грибів, здатних рости за аеробних умов.
Випробування призначені насамперед для того, щоб
визначити, чи задовольняє субстанція вимогам щодо
мікробіологічної чистоти, наведеним в окремій статті.
Якщо випробування проводять із цією метою, вико-
нують зазначення, наведені нижче, включаючи
кі іькість зразків, шо відбирають для аналізу, та інтер-
претацію одержаних результатів. Наведені нижче ме-
юдики можуть бути також використані при випробу-
ванні ефективності антимікробних консервантів
(5.1.3) відповідно до Фармакопеї. Крім того, їх можна
застосовувати при визначенні якості сировини і гото-
вих лікарських засобів відповідно до рекомендацій,
викладених у статті "Мікробіологічна чистота лікарсь-
ких засобів" (5.1.4). У цьому випадку, наприклад, при
визначенні виробником якості сировини і/або гото-
вих лікарських засобів або при проведенні перевірки
придатності вибраної методики порядок проведення
випробування, в тому числі кількість зразків, що
відбираються для аналізу, і інтерпретацію одержаних
результатів виробник установлює за погодженням із
компетентним уповноваженим органом.
Випробування проводять за умов, що дозволяють за-
побігти випадковому мікробному забрудненню випро-
бовуваного зразка. Заходи, що вживаються для запобі
гання мікробному забрудненню випробовуваного
зразка, не мають впливати на мікроорганізми, які мо-
жуть бути виявлені при проведенні випробування. У
випадку, якщо випробовуваний лікарський засіб вияв-
ляє антимікробну активність, вона має бути підхожим
чином нейтралізована. При використанні з цією ме-
тою інактиваторів треба підтвердити їхню нейтралізу-
ючу ефективність і нешкідливість для мікроорганізмів.
Визначення загального числа аеробних мікроор-
ганізмів проводять методом мембранної фільтрації або
методом висівання на чашки Петрі, як описано у
даній статті.
Метод найбільш імовірного числа (НІЧ) призначений
для використання втих випадках, коли неможливо за-
стосувати інші методи. При виборі методу випробу-
вання необхідно враховувати природу випробовувано-
го лікарського засобу та очікувану кількість мікроор-
ганізмів. Треба належним чином провести перевірку
придатності вибраної методики.
Якщо мета проведення випробування пов’язана із
статтями 5.1.3 або 5.1.4, треба використовувати метод
глибинного або поверхневого висівання на чашки
Петрі або метод мембранної фільтрації.
Підготовка зразка
Порядок відбору проб. Відбір проб лікарського засобу
треба проводити в суворо визначеному порядку. Поря-
док відбору проб залежить від розмірів серії, ступеня
небезпеки для здоров’я, пов’язаної з мікробним за-
брудненням лікарського засобу вище допустимого
рівня, характеристиками лікарського засобу й очікува-
ним рівнем мікробного забруднення. Якщо немає
інших зазначень в окремій статті, для випробування
використовують зразки по 10 г або 10 мл субстанції або
готового лікарського засобу, відібрані з дотриманням
застережних заходів, як зазначено вище. Зразки відби-
рають у випадковому порядку з упаковок іп Ьиік або з
контейнерів з готовим лікарським засобом. Якщо не-
обхідно, при відборі однієї проби змішують вміст
кількох контейнерів, для того щоб одержати зразок не-
обхідної маси або об’єму (залежно від природи випро-
бовуваної субстанції або готового лікарського засобу).
Прикладом плану відбору проб є трирівневий порядок
пробовідбору, використовуваний для лікарських за-
собів, у яких велика ймовірність нерівномірного роз-
поділу мікроорганізмів. У цьому випадку від кожної
серії відбирають п’ять зразків, кожний з яких випро-
бовують окремо. При цьому припускають, що в ре-
зультаті випробування можуть бути виявлені зразки,
що характеризуються трьома рівнями мікробного за-
бруднення: (і) — підхожі зразки, тобто зразки, що
містять менш як т колонієутворюючих одиниць
(КУО) у грамі або мілілітрі, де т — гранично допусти-
ма кількість мікроорганізмів, установлена відповід-
ною окремою статтею; (іі) — граничні зразки, тобто
зразки, що містять у грамі або мілілітрі більше т, але
менш як 10/77 КУО; (Ні) — браковані зразки, тобто
зразки, що містять більше 1 От КУО у грамі або
мілілітрі.
Лікарські засоби, розчинні у воді. 10 г або 10 мл випро-
бовуваного лікарського засобу розчиняють або роз-
бавляють буферним розчином із натрію хлоридом і
пептоном рН 7.0 або іншою підхожою рідиною. Як
правило, використовують розведення 1:10. Якщо не-
обхідно, допускається використання інших розведень
відповідно до властивостей лікарського засобу або до
вимог щодо чутливості методики. Якщо відомо, що
лікарський засіб виявляє антимікробну активність, до
розчинника може бути доданий інактиватор. Якщо
необхідно, доводять рН розчину приблизно до 7 і готу-
ють подальші десятикратні серійні розведення, вико-
ристовуючи той самий розчинник.
Нерозчинні у воді лікарські засоби, що не містять
жирів. 10 г або 10 мл випробовуваного лікарського за-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
111
2.6. Біологічні випробування
собу суспендують у буферному розчині з натрію хло-
ридом і пептоном рН 7.0 або в іншій підхожій рідині.
Як правило, готують суспензію у розведенні 1:10.
Якщо необхідно, допускається збільшення об’єму
розчинника відповідно до властивостей лікарського
засобу. Для суспендування погано змочуваних суб-
станцій до буферного розчину може бути додана
підхожа поверхнево-активна речовина, наприклад,
полісорбат-80 у концентрації 1 г/л буферного розчину.
У тому випадку, коли відомо, що лікарський засіб
виявляє антимікробну активність, до розчинника мо-
же бути доданий інактиватор. Якщо необхідно, дово-
дять рН розчину приблизно до 7 і готують подальші
десятикратні серійні розведення, використовуючи той
самий розчинник.
Лікарські засоби, що містять жири. 10 г або 10 мл ви-
пробовуваного лікарського засобу гомогенізують із
стерильним полісорбатом-80 або іншою підхожою
стерильною поверхнево-активною речовиною в
кількості, що не перевищує половину маси випробо-
вуваного зразка. Якщо необхідно, допускається попе-
реднє підігрівання поверхнево-активної речовини до
температури не більше 40 °С. У виняткових випадках
допускається нагрівання до температури не більше
45 °С. Ретельно перемішують, якщо необхідно,
підтримуючи температуру за допомогою водяної бані
або термостата. Додають буферний розчин з натрію
хлоридом і пептоном рН 7.0, попередньо підігрітий до
відповідної температури, у кількості, необхідній для
одержання розведення вихідного зразка 1:10. Ретельно
перемішують, підтримуючи температуру протягом
мінімального часу, необхідного для утворення
емульсії, але у будь-якому разі не більше ЗО хв. По-
дальші серійні десятикратні розведення готують, ви-
користовуючи буферний розчин із натрію хлоридом і
пептоном рН 7.0, що містить підхожу концентрацію
стерильного полісорбату-80 або іншої стерильної по-
верхнево-активної речовини.
Трансдермальні пластирі. Десять пластирів звільняють
від захисного покриття (знімних прокладок) за допо-
могою стерильного пінцета й поміщають у стерильні
пластмасові або скляні лотки клейкою поверхнею дого-
ри. Якщо необхідно, клейку поверхню покривають сте-
рильною марлею (або сіткою з полімерного моново-
локна типу тканинного фільтра) і переносять десять
пластирів у посудину, що містить не менше
500 мл буферного розчину з натрію хлоридом і пепто-
ном рН 7.0 і підхожі інактиватори, наприклад, полісор-
бат-80 і/або лецитин. Енергійно струшують не менше
30 хв (зразок А). Іншу групу з десяти пластирів готують,
як описано вище, помішають у посудину, що містить не
менше 500 мл рідкого живильного середовища Б, і
енергійно струшують не менше ЗО хв (зразок В).
Випробування зразка
Метод мембранної фільтрації. Використовують мемб-
ранні фільтри з розміром пор не більше 0.45 мкм,
здатні ефективно затримувати мікроорганізми. Ма-
теріал мембранного фільтра треба вибирати таким чи-
ном, щоб компоненти випробовуваного лікарського
засобу не впливали на його ефективність. Целюлозно- г
нітратні фільтри, наприклад, можуть бути використані і
для водних, розведених спиртових розчинів і розчини,
в оліях, а целюлозно-ацетатні фільтри — для концент-1
рованих спиртових розчинів. Конструкція фільтра
ційної установки має дозволяти проводити перене-
сення мембранного фільтра на живильне середовище.
Підхожу кількість зразка, підготовану, як описано ; І
розділі "Підготовка зразка" (відповідну 1 г випробову-
ваного лікарського засобу або меншій його кількості, І
якщо очікується високий ступінь мікробної забрудне- І
ності), переносять на кожний із двох мембранних |
фільтрів і негайно фільтрують. Кожний мембранний І
фільтр відмивають трьома порціями, близько 100 мл І
кожна, підхожої рідини, наприклад, буферного розчи- І
ну з натрію хлоридом і пептоном рН 7.0. До промивної І
рідини можуть бути додані поверхнево-активні речо-
вини, наприклад, полісорбат-80 або інактиватори. До-1
пускається використовувати для відмивання мемб-
ранних фільтрів менше трьох порцій промивної ріди- І
ни за умови перевірки придатності методики. Мемб- І
ранний фільтр, призначений переважно для підрахун-
ку бактерій, помішають на поверхню відповідного гу- І
стого живильного середовища, наприклад, середови-
ща В, а інший, призначений переважно для підрахун-
ку грибів, — на поверхню відповідного густого жи-
вильного середовища, наприклад, середовища С.
Чашки з середовищем В інкубують при температурі
від ЗО °С до 35 °С, чашки з середовищем С — від 20 °С
до 25 °С протягом п’яти діб, якщо вірогідні результати
випробування не будуть одержані за більш короткий
час. Відбирають чашки, кількість колоній на яких не
перевищує 100, і визначають число колонієутворю-
ючих одиниць у грамі або мілілітрі лікарського засобу.
При випробуванні транедермальних пластирів 50 мл
зразка А пропускають крізь кожний із двох стерильних
мембранних фільтрів. Один мембранний фільтр
поміщають на поверхню густого живильного середо-
вища В для визначення загального числа бактерій,
інший мембранний фільтр — на поверхню густого жи-
вильного середовища С для визначення загального
числа грибів.
Метод висівання на чашки
а. Метод глибинного висівання. У кожну чашку Петрі
діаметром 9 см вносять 1 мл випробовуваного зразка,
підготованого, як описано вище у розділі "Підготовка
зразка", і від 15 мл до 20 мл розплавленого густого жи-
вильного середовища для вирощування бактерій (на-
приклад, середовище В) або від 15 мл до 20 мл роз-
плавленого густого живильного середовища для виро-
щування грибів (наприклад, середовище С). Темпера-
тура живильного середовища має становити не більше
45 °С. При використанні чашок Петрі більшого діаме-
тра відповідно збільшують кількість живильного сере-
довища. Для кожного розведення використовують не
менше двох чашок Петрі з кожним живильним сере-
довищем. Посіви інкубують при температурі від ЗО °С
до 35 °С (від 20 °С до 25 °С для грибів) протягом п’яти
діб, якщо вірогідні результати випробування не будуть
одержані за коротший час. Відбирають чашки,
112
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
кіповіані одному розведенню, для якого кількість ко-
лій на одній чаїнці Петрі не перевищує 300 (100 ко-
ші для грибів). Обчислюють середнє арифметичне
мічення числа колоній і визначають число ко-
лйеутворюючих одиниць у грамі або мілілітрі.
в Метод поверхневого висівання. У чашки Петрі діаме-
рам 9 см вносять від 15 мл до 20 мл розплавленого гу-
догі живильного середовища для вирощування бак-
іерій (наприклад, середовище В) або розплавленого
'стого живильного середовища для вирощування
>ибів (наприклад, середовище С) із температурою
,іилко45 °С і дають середовищу застигнути. При ви-
іристанні чашок Петрі більшого діаметра відповідно
йільшують об’єм живильного середовища. Підсушу-
ють чашки, наприклад, у ламінарному струмені сте-
-ільного повітря або у термостаті. Точно відміряний
юем зразка (не менше 0.1 мл), підготованого, як опи-
. іно в розділі "Підготовка зразка", розподіляють по
поверхні живильного середовища. Для кожного роз-
велення використовують не менше двох чашок Петрі з
кожним живильним середовищем. Інкубацію посівів і
визначення числа колонієутворюючих одиниць у
тікарському засобі проводять, як описано вище для
етодх глибинного висівання.
Метод найбільш імовірного числа. Метод найбільш
ймовірного числа (НІЧ) поступається щодо точності
метолам мембранної фільтрації і висівання на чашки
Петрі. Результати визначення числа грибів, одержані
даним методом, як правило, невірогідні. У зв’язку з
цим використання методу НІЧ допускається лише для
визначення загального числа бактерій у тих випадках,
коли неможливо застосувати інші методи. Якщо в ок-
ремій статті передбачено застосування методу НІЧ,
визначення проводять, як описано нижче.
Готують серію не менш як трьох послідовних десяти-
кратних розведень випробовуваного лікарського засо-
бу, як описано в розділі "Підготовка зразка". Від кож-
ного розведення відбирають три проби по 1 габо 1 мл,
які вносять у три пробірки, що містять від 9 мл до
10 мл підхожого рідкого живильного середовища (на-
приклад, живильного середовища А). Якщо не-
обхідно, в живильне середовище може бути додана
підхожа поверхнево-активна речовина, наприклад,
полісорбат-80 або інактиватор. Таким чином, для
трьох розведень використовують дев’ять пробірок. Усі
пробірки інкубують при температурі від ЗО °С до 35 °С
протягом п’яти діб. Для кожного розведення познача-
ють кількість пробірок, у яких спостерігається ріст
мікроорганізмів. У тому разі, якщо у зв’язку з приро-
дою лікарського засобу візуальний облік результатів
утруднений або сумнівний, проводять пересівання на
те саме рідке живильне середовище або на підхоже гу-
сте живильне середовище (наприклад, середовище В),
інкубують від 18 год до 24 год при тій самій темпера-
турі й враховують одержані результати. Визнача-
ють найбільш імовірну кількість бактерій у грамі
або мілілітрі випробовуваного лікарського засобу за
Табл. 2.6 12.-1.
Таблиця 2.6.12.-1
Найбільш ймовірне число бактерій
Три пробірки для кожного розведення
Кількість пробірок, в яких спостерігається ріст мікроорганізмів НІЧ в 1 г Категорія* 95 % довірчі інтервали
0.1г 0.01г 0.001г 1 2
0 0 0 <3 - -
0 1 0 3 X <1 17
1 0 0 3 X 1 21
1 0 1 7 X 2 27
1 1 0 7 X 2 28
1 2 0 11 X 4 35
2 0 0 9 X 2 38
2 0 1 14 X 5 48
2 1 0 15 X 5 50
2 1 1 20 X 8 61
2 2 0 21 X 8 63
3 0 0 23 X 7 129
3 0 1 38 X 10 180
3 1 0 43 X 20 210
3 1 1 75 X 20 280
3 2 0 93 X ЗО 390
3 2 1 150 X 50 510
3 2 2 210 X 80 640
3 3 0 240 X 100 1400
3 3 1 460 X 200 2400
3 3 2 1100 X 300 4800
3 3 3 >1100 - -
* Категорія І: результати, одержувані в 95 % випадків. Категорія 2: менш імовірні результати, одержувані лише у 4 % випадків. Ці резуль-
мти не треба використовувати при прийнятті важливих рішень. Результати, менш імовірні, ніж категорія 2, не наведені бо завжди
невірогідні.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
113
2.6. Біологічні випробування
Ростові властивості живильних середовищ
і перевірка придатності методик випробування
Тест-штами бактерій вирощують кожний окремо на
підхожому рідкому живильному середовищі (напри-
клад, середовищі А) при температурі від ЗО °С до 35 °С
від 18 год до 24 год. Тест-штами грибів вирощують кож-
ний окремо на підхожому густому живильному середо-
вищі (наприклад, середовищі С без додавання ан-
тибіотика). Сапдіда аІЬісапз інкубують при температурі
від 20 °С до 25 °С протягом 48 год, Азрег%іІІиз піуег —
при температурі від 20 °С до 25 °С протягом семи діб.
Маріїуіососсиь аигеих
ЕзсИегісИіа соїі
ВасіІІиз хиЬііІіх
Сапсіісіа аІЬісапз
Л$рег§іІІи8 пі$ег
АТСС 6538 (N01М В 9518,
С1Р4.83)
АТСС 8739 (N01М В 8545,
С1Р53. 126)
АТСС 6633 (N01М В 8054,
СІ Р 52.62)
АТСС 10231 (МСРР 3179,
ІР 48.72)
АТСС 16404 (1М1 149007,
1Р 1431. 83)
У буферному розчині з натрію хлоридом і пептоном
рН 7.0 готують робочі суспензії тест-мікроорганізмів,
які містять близько 100 колонієутворюючих одиниць у
мілілітрі. Суспензію кожного мікроорганізму окремо в
присутності й у відсутності випробовуваного зразка
використовують для перевірки придатності методики
визначення загального числа життєздатних аеробних
мікроорганізмів При використанні методу мембран-
ної фільтрації і методу висівання на чашки результати,
одержані при підрахунку кожного з тест-мікроор-
ганізмів у присутності й у відсутності випробовувано-
го зразка, мають відрізнятися не більш як у 5 разів.
При використанні методу найбільш імовірного числа
результат, одержаний при підрахунку КУО тест-мікро-
організму, має знаходитися всередині 95 % довірчого
інтервалу результату, одержаного в присутності випро-
бовуваного зразка. Для перевірки стерильності жи-
вильного середовища і розчинника, а також дотри-
мання асептичних умов випробування проводять, ви-
користовуючи стерильний буферний розчин із натрію
хлоридом і пептоном рН 7.0 замість випробовуваного
зразка. При цьому не має спостерігатися ріст мікроор-
ганізмів.
найбільш імовірного числа визначають загальне чис-
ло бактерій.
Якщо допустимі межі вмісту мікроорганізмів визна-1
чені в окремій статті, результати інтерпретують так: І
ІО2 мікроорганізмів — максимально допустима ме-1
жа: 5x102;
10' мікроорганізмів — максимально допустима ме-1
жа: 5x10’ і т.д.
При використанні, наприклад, трирівневого план) І
відбору проб чинять так.
Визначають загальне число мікроорганізмів окремо
для кожного з п’яти зразків. Вважають, що субстанція
або готовий лікарський засіб задовольняє вимоги що-1
до мікробіологічної чистоти, якщо виконуються такі І
умови: (/) — загальне число мікроорганізмів, визначе-
не для кожного випробовуваного зразка, не переви-1
щує допустимі межі більш як у 10 разів (відсутні бра-1
ковані зразки), та (й) — не більш як для двох випробо-
вуваних зразків загальне число мікроорганізмів, ви-
значене в результаті випробування, знаходиться в
інтервалі між допустимою межею і її десятикратним
значенням (тобто не більше двох граничних зразків).
Застосування трирівневого плану відбору проб має бу-
ти передбачено в окремій статті.
Рекомендовані розчини і живильні середовища наве-
дені в статті 2.6.13.
________________________________________________N
При випробуванні ефективності антимікробних кон-
сервантів (5.1.3) може бути також використаний метод
двошарового висівання на чашки Петрі.
При визначенні якості лікарських засобів відповідно
до рекомендацій, викладених у статті "Мікробіологічна
чистота лікарських засобів" (5.1.4), може бути також
використаний метод двошарового висівання на чашки
Петрі при наявності відповідних зазначень в окремій
статті.
Для визначення мікробіологічної чистоти допус-
кається використання автоматизованих методів за
умови, що в результаті перевірки придатності було до-
ведено, що вони дають еквівалентні результати
порівняно з методами, описаними вище.
Інтерпретація результатів
Підготовка зразка
Загальне число бактерій визначають, виходячи із се-
реднього значення числа колонієутворюючих оди-
ниць, які виросли на густому живильному середови-
щі В. Загальне число грибів визначають, виходячи із
середнього значення числа колонієутворюючих оди-
ниць, вирощених на густому живильному середови-
щі С. Загальне число життєздатних аеробних мікро-
організмів знаходять як суму загального числа бак-
терій і загального числа грибів, визначених, як описа-
но вище. Якщо установлено, шо на живильному сере-
довищі для вирощування бактерій і живильному сере-
довищі для вирощування грибів спостерігається ріст
одних і тих самих мікроорганізмів, необхідно внести
відповідні поправки. При використанні методу
Порядок відбору проб. Якщо немає інших зазначень в
окремій статті, від кожної серії лікарського засобу
відбирають середню пробу, яка складається з рівних ра-
зових проб, взятих не менш як із 10 різних контейнерів.
Випробування зразка
Метод мембранної фільтрації. Якщо немає інших за-
значень в окремій статті, після закінчення фільтрації
мембранні фільтри промивають 1-5 порціями під-
хожої промивної рідини.
При випробуванні лікарського засобу, нерозчинного у
воді й такого, що утворює суспензії (таблетки, порош-
114
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
І >іітаін.) проводять фільтрацію надосадової рідини
іііо використовують попередню фільтрацію крізь пе-
I Ьфільтр, вміщуючи його у фільтраційну установку
ред мембранним фільтром.
Метод висівання на чашки
. Метод двошарового висівання. У чашки Петрі діаме-
ром 9 см вносять від 15 мл до 20 мл розплавленого гу-
дого живильного середовища для вирощування бак-
ерій (наприклад, середовища № 1) або розплавлено-
> густого живильного середовища для вирощування
грибів (наприклад, середовища № 2) із температурою
•ід 45 °С до 50 °С і дають середовищу застигнути. При
«користанні чашок Петрі більшого діаметра
відповідно збільшують об’єм живильного середовища.
І мл зразка, підготованого, як описано в розділі
"Підготовка зразка", вносять у пробірку, що містить
близько 4 мл розплавленого і охолодженого до темпс-
' гатури не більше 45 °С густого живильного середови-
ща № 1 або густого живильного середовища № 2.
Швидко перемішують вміст пробірки і переносять у
чашку Петрі, підготовану, як описано вище, що
містить густе живильне середовище № 1 або № 2,
відповідно. Швидким погойдуванням чашки Петрі
рівномірно розподіляють верхній шар живильного се-
редовища. Для кожного розведення використовують
не менше двох чашок Петрі з кожним живильним се-
редовищем. Після застигання середовища чашки пе-
ревертають й інкубують при температурі від ЗО °С до
35 °С (від 20 °С до 25 °С для грибів) протягом п’яти
діб, якшо вірогідні результати випробування не будуть
одержані за коротший час. Відбирають чашки,
відповідні одному розведенню, для якого кількість ко-
йоній на одній чашці Пет рі не перевищує 300 (100 ко-
лоній для грибів). Обчислюють середнє арифметичне
значення числа колоній і визначають число ко-
тоніеутворюючих одиниць у грамі або мілілітрі.
Ростові властивості живильних середовищ
і перевірка придатності методик випробування
При перевірці придатності методик випробування і
для контролю ростових властивостей живильних серс-
। ловиш можуть бути також використані такі штами
тест -м ікроор га н і зм і в.
Замістьтест-мікроорганізму ЕьсІїегісИіа соїі АТСС 8739
(МС1МВ 8545, С1Р 53.126) може бути використаний
тест-мікроорганізм ЕхсИегісИіа соїі АТСС 25922.
Замістьтест-мікроорганізму Васіїїиз яиЬііІія АТСС 6633
(КС1МВ 8054, С1Р 52.62) може бути використа-
ний тест-мікроорганізм Васіїїиа сегеих АТСС 10702
(МСТС 8035).
Замість тест-мікроорганізму Сапсіісіа аІЬісапх АТСС
10231 (МСРЕ 3179, 1Р 48.72) може бути використаний
тест-мікроорганізм Сапсіісіа аІЬісапх МСТС 885-653.
Допускається використання інших штамів мікроор-
ганізмів за погодженням з компетентним уповноваже-
ним органом.
Для вирощування тест-штамів бактерій може бути ви-
користане рідке живильне середовище № 1, для виро-
щування тест-штамів грибів може бути використане
густе живильне середовище № 2.
Для приготування робочих суспензій тест-мікроор-
ганізмів може бути використаний розчин 9 г/л натрію
хлориду.
Ростові властивості. Невелику кількість (близько
100 КУО) суспензії кожного мікроорганізму окремо
вносять у відповідні живильні середовища (напри-
клад, тест-штами бактерій вносять у живильне середо-
вище № 1, тест-штами грибів — у живильне середови-
ще № 2). Посіви інкубують за умов, зазначених вище,
протягом 48 год (середовище № 1) і 72 год (середови-
ще № 2). Середовище вважають підхожим, якщо після
закінчення періоду інкубації спостерігається ріст
мікроорганізмів.
Інтерпретація результатів
Якщо при випробуванні зразка методом мембранної
фільтрації при розведенні лікарського засобу 1:10 не
виявлений ріст мікроорганізмів на мембранних фільт-
рах, результати позначають таким чином: "В 1 г (мл)
лікарського засобу бактерії (гриби) не виявлені".
У тому випадку, коли при випробуванні зразка мето-
дом висівання на чашки при розведенні лікарського
засобу 1:10 не виявлений ріст мікроорганізмів на
чашках Петрі, результати позначають таким чином:
"В 1 г (мл) лікарського засобу менше 10 бактерій
(грибів)".
Якщо виробництво лікарського засобу не проводиться
відповідно до вимог належної виробничої практики
(НВП; СМР), встановлених в Європейському Співто-
варистві, і допустимі межі вмісту мікроорганізмів ви-
значені в окремій статті, результати інтерпретують та-
ким чином
102 мікроорганізмів — максимально допустима ме-
жа: 1х102;
10’ мікроорганізмів — максимально допустима ме-
жа: 1x10’ і т.п.
2.6.13. ВИПРОБУВАННЯ МІКРОБІОЛОГІЧНОЇ
ЧИСТОТИ НЕСТЕРИЛЬНИХ ЛІКАРСЬКИХ
ЗАСОБІВ (ВИПРОБУВАННЯ НА ОКРЕМІ
ВИДИ МІКРООРГАНІЗМІВ)
Методики, викладені в загальній статті, допускають
використання селективних живильних середовищ, що
не дозволяють виділяти мікроорганізми зі сублеталь-
ними ушкодженнями. Якщо для забезпечення якості
лікарського засобу потрібно виявляти мікроорганізми
зі сублетальними ушкодженнями, треба передбачити
процедуру відновлення їхньої життєздатності перед
використанням селективних живильних середовищ.
Якшо випробовуваний лікарський засіб виявляє ан-
тимікробну активність, вона має бути підхожим спо-
собом нейтралізована.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
115
2.6. Біологічні випробування
Ентеробактерії і деякі інші грамнегативні бактерії
Випробування призначене для виявлення бактерій
род. ЕпіегоЬасїегіасеае, однак дозволяє також виявити
інші мікроорганізми (наприклад, Аеготопах, Раеисіо-
топаз).
Виявлення бактерій. Готують випробовуваний зразок,
як описано у статті 2.6.12, використовуючи рідке жи-
вильне середовище □ замість буферного розчину з
натрію хлоридом і пептоном рН 7.0, гомогенізують й
інкубують при температурі від 35 °С до 37 °С протягом
часу, достатнього для відновлення життєздатності
мікроорганізмів, але не достатнього для збільшення
їхньої кількості (як правило, 2 год, але не більше
5 год). Струшують посудину і переносять її вміст (го-
могенізат А) в об’ємі, відповідному 1 г або 1 мл
лікарського засобу в 100 мл нагромаджувального сере-
довища Е. Посіви інкубують при температурі від 35 °С
до 37 °С від 18 год до 48 год. Після закінчення періоду
інкубації роблять пересівання на чашки з густим жи-
вильним середовищем Е Інкубують при температурі
від 35 °С до 37 °С від 18 год до 24 год. Лікарський засіб
витримує випробування, якщо ріст колоній грамнега-
тивних бактерій не виявляється.
Кількісна оцінка. Гомогенізат А і/або його розведення,
що містять відповідно 0.1 г, 0.01 г і 0.001 г (або 0.1 мл,
0.01 мл і 0.001 мл) випробовуваного лікарського засо-
бу, вносять у відповідні об’єми рідкого нагромаджу-
вального середовища Е. Інкубують при температурі
від 35 °С до 37 °С від 24 год до 48 год. Після закінчен-
ня періоду інкубації з кожної посудини роблять пе-
ресівання на чашку з густим живильним середови-
щем Е так, щоб одержати ріст ізольованих колоній.
Інкубують при температурі від 35 °С до 37 °С від 18 год
до 24 год. Наявність росту добре розвинутих червоних
або червонястих колоній грамнегативних бактерій
означає позитивний результат випробування. Позна-
чають найменшу кількість лікарського засобу, що дає
позитивний результат, і найбільшу кількість лікарсь-
кого засобу, що дає негативний результат. Визначають
імовірне число бактерій за Табл. 2.6.13.-1.
Таблиця 2.6.13.-1
Результат, одержаний для кожної кількості лікарського засобу
0.1г або 0.1 мл 0.01 г або 0.01 мл 0.001 габо 0.001 мл Імовірне число бактерій у грамі або мілілітрі лікарського засобу
+ + + більше 103
+ + - менше 103, але більше 102
+ - - менше 102, але більше 10
- - - менше 10
При випробуванні трансдермальних пластирів 50 мл
зразка В пропускають крізь стерильний мембранний
фільтр, як описано у статті 2.6.12, помішають мемб-
ранний фільтр у 100 мл рідкого нагромаджувального
середовища Е й інкубують при температурі від 35 °С до
37 °С від 18 год до 24 год. Після закінчення періоду
інкубації роблять пересівання на поверхню густо
живильного середовища Е для виявлення ентсробаЛ
терій та інших грамнегативних мікроорганізмів.
Еас/іегісіїіа соїі
10 мл випробовуваного зразка, підготованого, якого
сано в статті 2.6.12, або його кількість, відповідну!
або І мл лікарського засобу, вносять у 100 мл рідкої
живильного середовища А, гомогенізують й інкубуюіь *
при температурі від 35 °С до 37 °С від 18 год до 48 год І
Після закінчення періоду інкубації струшують посу-іґ
дину, переносять 1 мл її вмісту в 100 мл рідкого жи-
вильного середовища С й інкубують при температурі
від 43 °С до 45 °С від 18 год до 24 год. Роблять пе
ресівання на чашки з густим живильним середови-1
щем Н й інкубують при температурі від 35 °С до 37 с( І
від 18 год до ТІ год. Ріст червоних не слизових колоній 1
грамнегативних паличок указує на можливість забруд І
нення лікарського засобу Е. соїі. У цьому випадку про
водять підхожі додаткові біохімічні тести, наприклад .
тест на індол. Лікарський засіб витримує випробуван-1
ня, якщо на густому живильному середовищі Н не ви-1
явлений ріст описаних вище колоній або додаткові І
біохімічні тести дали негативний результат.
Еаїтопеїіа
Готують випробовуваний зразок, як описано в стані
2.6.12, використовуючи рідке живильне середовище А
замість буферного розчину з натрію хлоридом і пепто-І
ном рН 7.0, гомогенізують й інкубують при темпера-
турі від 35 °С до 37 °С від 18 год до 24 год. 1 мл одержа-
ної нагромаджувальної культури вносять у 10 мл І
рідкого живильного середовища 1 й інкубують при
температурі від 41 °С до 43 °С від 18 год до 24 год. Роб-1
лять пересівання не менш як на два з трьох живильних
середовища: густе живильне середовище 3, густе жи-
вильне середовище К і густе живильне середовище І.
Інкубують при температурі від 35 °С до 37 °С від 18 гол
до 72 год. Наявність на живильних середовищах ха-
рактерного росту, описаного нижче, указує на мож-
ливість забруднення лікарського засобу Еаїтопеїіа.
— густе живильне середовище 1: добре розвинуті
безбарвні колонії;
— густе живильне середовище К: добре розвинуті
червоні або червоні з чорним центром колонії;
— густе живильне середовище І_: дрібні прозорі
безбарвні, рожеві або каламутно-білі колонії, як
правило, оточені рожевою або червоною зоною.
Декілька підозрілих колоній, кожну окремо, пересіва-
ють на поверхню і вглиб густого живильного середо-
вища ІМ у пробірках. Роблять попередній висновок
про забруднення лікарського засобу 8аІтопеІІа, якщо
після закінчення періоду інкубації спостерігається
зміна кольору живильного середовища М із червоного
на жовтий (у глибині агару, але не на його поверхні),
шо супроводжується, як правило, газоутворенням.
При цьому може спостерігатися утворення сірковод-
ню в глибині агару. Остаточний висновок роблять
після проведення відповідних біохімічних або серо-
логічних тестів. Лікарський засіб витримує випробу-
116
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
І іня, якшо на густих живильних середовищах не ви-
іі іениіі ріст описаних вище колоній або додаткові
боммічні та серологічні тести дали негативний ре-
| штат.
Рші(Іопюпа)і аеги^іпоха
! мл випробовуваного зразка, підготованого, як опи-
г.ілО у статті 2.6.12, або його кількість, що відповідає
І * ібо 1 мл лікарського засобу, внося ть у 100 мл рідко-
... живильного середовища А. гомогенізують й інкубу-
гь при температурі від 35 °С до 37 °С від 18 год до
-• год. Роблять пересівання на чашку з густим жи-
ьним середовищем N й інкубую і ь при температурі
135 °С до 37 °С від 18 год до 72 год. Лікарський засіб
гримує випробування, якщо ріст мікроорганізмів на
•двіньному середовищі не виявляється. Якщо вияв-
, ний ріст грамнегативних паличок, роблять пе-
ргсівання різних за морфологічними ознаками ізо-
: ованих колоній на рідке живильне середовище А й
(кубують при температурі від 41 °С до 43 °С від 18 год
24 год. Лікарський засіб витримує випробування,
юно при температурі від 41 °С до 43 °С не спос-
ірігаеться ріст мікроорганізмів.
При випробуванні транедермальних пластирів 50 мл
(різка А пропускають крізь стерильний мембранний
фільтр, як описано у статті 2.6.12, поміщають мемб-
р нний фільтр у 100 мл рідкого живильного середови-
іАиінкубуютьпри температурі від 35 °С до 37 °С від
год до 48 год. Після закінчення періоду інкубації
облять пересівання на поверхню густого живильного
середовища N.
Зіаріїуіососсіії аигеих
10 мл випробовуваного зразка, підготованого, як опи-
ано у статті 2.6.12, або його кількість, що відповідає
1 габо 1 мл лікарського засобу, вносять у 10 мл рідко-
го живильного середовища А, гомогенізують й інкубу-
ють при температурі від 35 °С до 37 °С від 18 год до
48 год. Роблять пересівання на чашку з густим жи-
вильним середовищем О й інкубують при температурі
від 35 "С до 37 °С від 18 год до 72 год. Ріст чорних ко-
лоній грампозитивних коків, оточених прозорою зо-
ною, указує на наявність 5. аигеиа, що може бути
підтверджено відповідними біохімічними тестами, на-
приклад, тестами на коагулазу і дезоксирибонуклеазу.
Лікарський засіб витримує випробування, якщо на гу-
стому живильному середовищі О не виявлений ріст
описанихвише колоній або додаткові біохімічні тести
дали негативний результат.
При випробуванні транедермальних пластирів 50 мл
зразка А пропускають крізь стерильний мембранний
фільтр, як описано у статті 2.6.12, поміщають мемб-
ранний фільтр у 100 мл рідкого живильного середови-
шаАй інкубують при температурі від 35 °С до 37 °С від
18 год до 48 год. Після закінчення періоду інкубації
роблять пересівання на поверхню густого живильного
середовища О.
Ростові й селективні властивості живильних середовищ
і перевірка придатності методик випробування
Випробування, описані нижче, треба проводити що-
найменше для кожної партії сухого живильного сере-
довища.
Тест-мікроорганізми, наведені нижче, вирощують
кожний окремо у пробірках з підхожим живильним
середовищем, наприклад, зазначеним нижче, при
температурі від ЗО °С до 35 °С від 18 год до 24 год:
ЗіарИуІососсиз аигеиь АТСС 6538 (ІЧС1М В 9518,
С1Р 4.83): рідке живильне
середовище А,
Рьеисіотопах аеги^іпо^а АТСС 9027 (МС1МВ 8626,
С1Р82. 118) рідке живильне
середовище А,
Еаскегіскіа соїі АТСС 8739 (Г4С1МВ 8545,
СІР 53. 126): рідке живильне
середовище А,
Заїтопеїіа іуркітигіит рекомендований номер
штаму не наводиться (може
бути використаний штам не
патогенний для людини,
наприклад, Заїтопеііа аЬопу
1ЧІСТС 6017, СІР 80.39): рідке
живильне середовище А.
Готують робочу суспензію кожного тест-мікроор-
ганізму, що містить близько 1000 життєздатних клітин
у 1 мл, використовуючи буферний розчин з натрію
хлоридом і пептоном рН 7.0. Змішують рівні об’єми
кожної суспензії. 0.4 мл одержаної суміші (близько 100
мікроорганізмів кожного виду) вносять у живильне
середовище і проводять випробування на наявність
5. аигеиз, Р. аепурпоха, Е. соїі і 8аІтопеІІае у присут-
ності й відсутності випробовуваного зразка. Для кож-
ного тест-мікроорганізму має бути одержаний пози-
тивний результат випробування.
СІохігШіа
Випробування, описані нижче, проводять в особливих
випадках. Перший метод призначений для випробу-
вання на відсутність патогенних клостридій у тих ви-
падках, коли їхня наявність у лікарському засобі не
допускається. Такі лікарські засоби, як правило,
містять низьке загальне число мікроорганізмів. Дру-
гий метод являє собою напівкількісне випробування
на Сіоаігісііит рег/гіпуеп^ і призначений для лікарських
засобів, у яких кількість мікроорганізмів даного виду є
критерієм якості.
1. Випробування на Сіоіїгісііа. Готують випробовуваний
зразок, як описано у статті 2.6.12. Відбирають дві рівні
порції, відповідні 1 г або 1 мл випробовуваного
лікарського засобу. Одну порцію прогрівають при тем-
пературі 80 °С протягом 10 хв і швидко охолоджують.
Іншу порцію не прогрівають. 10 мл кожної з гомо-
генізованих порцій переносять у дві посудини
(38 х 200 мм) або інші підхожі посудини, що містять
100 мл живильного середовища Р. Інкубують за ана-
еробних умов при температурі від 35 °С до 37 °С про-
тягом 48 год. Після закінчення періоду інкубації роб-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
117
2.6. Біологічні випробування
лять пересівання з кожної посудини в живильне сере-
довище <2 з гентаміцином й інкубують за анаеробних
умов при температурі від 35 °С до 37 °С протягом
48 год. Лікарський засіб витримує випробування, як-
що після закінчення періоду інкубації не спос-
терігається ріст мікроорганізмів.
При наявності росту мікроорганізмів роблять пе-
ресівання кожної окремої колонії на живильне середо-
вище ф без гентаміцину й інкубують як за аеробних, так
і за анаеробних умов. Наявність лише за анаеробних
умов росту грампозитивних паличок (з ендоспорами
або без них), що дають негативну реакцію на каталазу,
свідчить про наявність Сіомгісііит зрр. Якщо необхідно,
порівнюють культуральні ознаки мікроорганізмів, що
виросли на двох чашках, і проводять тест на каталазу,
щоб виключити аеробні й факультативно анаеробні
мікроорганізми ВасіИиз арр., що дають позитивну ре-
акцію на каталазу. Тест на каталазу проводять шляхом
додавання краплі розчину водню пероксиду розведеного Р
безпосередньо до окремої колонії, що виросла на густо-
му живильному середовищі, або до мікробної маси на
предметному склі. Утворення бульбашок газу свідчить
про позитивну реакцію на каталазу.
2. Підрахунок СІоМгісІіит рег/гіпуеп^. Готують випробо-
вуваний зразок, як описано у статті 2.6.12, і розводять
у співвідношенні 1:100 і 1:1000 буферним розчином із
натрію хлоридом і пептоном рН 7.0. Визначають
найбільш імовірне число бактерій, як описано вище в
статті 2.6.12, використовуючи живильне середови-
ще К у пробірках або інших підхожих посудинах із ма-
ленькими трубками Дюрама. Змішують при мінімаль-
ному струшуванні й інкубують при температурі від
45.5 °С до 46.5 °С від 24 год до 48 год. Наявність чор-
ного забарвлення в посудинах внаслідок утворення
заліза сульфіду і сильне газоутворення в трубках Дю-
рама (не менше 1/10 об’єму трубки) свідчить про на-
явність С. реі/гіпуепь. Визначають найбільш імовірне
число С. рег/і-іпуеп^за Табл. 2.6.12.-1.
Контроль. Використовують такі тест-штами:
Для методу 1: СІоНгідіит зрого^епех, наприклад,
АТСС 19404 (КІСТС 532) або СІ Р 79.3.
Для методу 2: СІовІгіВіит рег/гіпуупу наприклад,
АТСС 13124 (ІЧС1МВ 6125, МСТС 8237, СІР 103 409).
Якшо необхідно, використовують разом з С. врогодепех
для контролю селективності живильних середовищ і
анаеробних умов.
Розділ, наведений нижче, носить довідковий і рекомен-
даційний характер і не є обоє язковою частиною Фар-
макопеї.
РЕКОМЕНДОВАНІ РОЗЧИНИ
1 ЖИВИЛЬНІ СЕРЕДОВИЩА
Наведені нижче розчини і живильні середовища да-
ють задовільні результати при визначенні
мікробіологічної чистоти відповідно до Фармакопеї.
Допускається використання інших живильних сере-
довищ, що не відрізняються за ростовими і селектив-
ними властивостями щодо тих мікроорганізмів,
виділення яких вони призначені
Буферний розчин із натрію хлоридом і пептоном рН 7.0
Калію дипдрофосфат 3.6 г еквівалем 0.067 N
Динатрію гідрофосфат дигідрат 7.2 г еквівалеи
0.067Ц
Натрію хлорид 4.3 г
Пептон (м’ясний 1.0 г
або казеїновий)
Вода очищена 1 000 мл
До розчину можуть бути додані поверхне во-активь
речовини або інактиватори, наприклад, полісор-
бат-80: від 1 г/л до 10 г/л.
Стерилізують у паровому стерилізаторі при темпера-
турі 121 °С протягом 15 хв.
Рідке живильне середовище А (соєво-казеїновий
бульйон)
Панкреатичний гідролізат казеїну 17.0г
Папаїновий гідролізат соєвих бобів 3.0г
Натрію хлорид 5.01
Дикалію гідрофосфат 2.5 г
Ілюкози моногідрат 2.51
Вода очищена 1 000 мл
Установлюють рН середовища таким чином, шоб
після стерилізації його значення становило 7.3±0.2.
Стерилізують у паровому стерилізаторі при темпера-
турі 121 °С протягом 15 хв.
Густе живильне середовище В (соєво-казеїновий агар)
Панкреатичний гідролізат казеїну 15.0 г
Папаїновий гідролізат соєвих бобів 5.0 г
Натрію хлорид 5.0 г
Агар 15.0 г
Вода очищена 1 000 мл
Установлюють рН середовища таким чином, щоб
після стерилізації його значення становило 7.3+0.2
Стерилізують у паровому стерилізаторі при темпера-
турі 121 °С протягом 15 хв.
Густе живильне середовище С (агар Сабуро із глюкозою
й антибіотиками)
Пептони (м’ясний або казеїновий)
Ілюкози моногідрат
Агар
Вода очищена
10.0 г
40.0 г
15.0 г
1 000 мл
Установлюють рН середовища таким чином, щоб
після стерилізації його значення становило 5.6±0.2.
Стерилізують у паровому стерилізаторі при темпера-
турі 121 °С протягом 15 хв. Безпосередньо перед вико-
ристанням до живильного середовища додають сте-
рильні розчини антибіотиків з розрахунку 0.10 г бен-
118
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
м пеніциліну натрію і 0.10 г тетрацикліну на літр се-
Ьмвиша або перед стерилізацією додають хлорам-
б<і ілл із розрахунку 50 мг на літр живильного сере-
дина.
Иж живильне середовище В (лактозний бульйон)
Яловичини екстракт
Плнкреатичний гідролізат желатину
Ьггози моногідрат
Вша очищена
кгановлюють рН середовища таким чином, щоб
після стерилізації його значення становило 6.9+0.2.
Стерилізують у паровому стерилізаторі при темпера-
гюі 121 С протягом 15 хв і охолоджують негайно
аоя стерилізації.
Рідхе нагромаджувальне середовище Е (нагромаджу-
ваний бульйон Мозеля для бактерій род. ЕпІегоЬас-
нгіасеае)
Панкреатичний гідролізат желатину 10.0 г
Июкози моногідрат 5.0 г
Бичача жовч зневоднена 20.0 г
Калію дигідрофосфат 3.0 г
□инатрію гідрофосфат дигідрат 8.0 г
Брильянтовий зелений 15 мг
В на очищена 1 000 мл
Установлюють рН середовища таким чином, щоб
після прогрівання його значення становило 7.2±0.2.
Прогрівають при температурі 100 °С протягом ЗО хв і
негайно охолоджують.
Густе живильне середовище Г (агар з жовчю, глюкозою,
кристалічним фіолетовим і нейтральним червоним)
Дріжджовий екстракт 3.0 г
Панкреатичний гідролізат желатину 7.0 г
Солі жовчних кислот 1.5 г
Лактози моногідрат 10.0 г
Натрію хлорид 5.0 г
І.іюкози моногідрат 10.0г
Агар 15.0 г
Нейтральний червоний ЗО мг
Кристалічний фіолетовий 2 мг
Вода очищена 1 000 мл
Установлюють рН середовища таким чином, щоб
після нагрівання його значення становило 7.4±0.2.
Нагрівають до кипіння. Нагрівання в паровому сте-
рилізаторі не допускається.
Рідке живильне середовище С (бульйон Мак-Конкі)
Панкреатичний гідролізат желатину 20.0 г
Лактози моногідрат 10.0 г
Бичача жовч зневоднена 5.0 г
Бромкрезоловий пурпуровий 10 мг
Вода очищена 1 000 мл
Встановлюють рН середовища таким чином, щоб
після стерилізації його значення становило 7.3±0.2.
Стерилізують у паровому стерилізаторі при темпера-
турі 121 °С протягом 15 хв.
3.0 г
5.0 г
5.0 г
1 000 мл
Густе живильне середовище Н (агар Мак-Конкі)
Панкреатичний гідролізат желатину 17.0 г
Пептони (м’ясний і казеїновий) 3.0 г
Лактози моногідрат 10.0 г
Натрію хлорид 5.0 г
Солі жовчних кислот 1.5 г
Агар 13.5 г
Нейтральний червоний 30.0 мг
Кристалічний фіолетовий 1 мг
Вода очищена 1 000 мл
Установлюють рН середовища таким чином, щоб
після стерилізації його значення становило 7.1 ±0.2.
Кип’ятять протягом 1 хв при постійному струшуванні,
потім стерилізують у паровому стерилізаторі при тем-
пературі 121 °С протягом 15 хв.
Рідке живильне середовище ] (тетратіонатно-жовчний бульйон з брильянтовим зеленим)
Пептон 8.6 г
Бичача жовч зневоднена 8.0 г
Натрію хлорид 6.4 г
Кальцію карбонат 20.0 г
Калію тетратіонат 20.0 г
Брильянтовий зелений 70 мг
Вода очищена 1 000 мл
Установлюють рН середовища таким чином, щоб
після стерилізації його значення становило 7.0±0.2.
Нагрівають до моменту закипання. Повторне на-
грівання не допускається.
Густе живильне середовище Л (дезоксихолатний
цитратний агар)
Яловичий екстракт 10.0 г
М’ясний пептон 10.0 г
Лактози моногідрат 10.0 г
Натрію цитрат 20.0 г
Заліза(Ш) цитрат 1.0 г
Натрію дезоксихолат 5.0 г
Агар 13.5 г
Нейтральний червоний 20 мг
Вода очищена 1 000 мл
Установлюють рН середовища таким чином, щоб
після нагрівання його значення становило 7.3±0.2.
Повільно нагрівають до кипіння і кип’ятять протягом
1 хв. Охолоджують до температури 50 °С і розливають
у чашки Петрі. Нагрівання в паровому стерилізаторі
не допускається.
Густе живильне середовище К (дезоксихолатний агар з
ксилозою і лізином)
Ксилоза 3.5 г
Б-Лізин 5.0 г
Лактози моногідрат 7.5 г
Сахароза 7.5 г
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
119
2.6. Біологічні випробування
Натрію хлорид 5.0 г
Дріжджовий екстракт 3.0 г
Феноловий червоний 80 мг
Агар 13.5 г
Натрію дезоксихолат 2.5 г
Натрію тіосульфат 6.8 г
Заліза(ІП) амонію цитрат 0.8 г
Вода очищена 1 000 мл
Густе живильне середовище N (цетримідний агар)
Панкреатичний гідролізат желатину 20.0
Магнію хлорид 1.4
Калію сульфат 10.01
Цстримід 0.3 '
Агар 13.6г
Вода очищена 1 000 мі
Гліцерин 10.0 ш
Встановлюють рН середовища таким чином, шоб
після нагрівання його значення становило 7.4±0.2.
Нагрівають до моменту закипання. Охолоджують до
температури 50 °С і розливають у чашки Петрі.
Нагрівання в паровому стерилізаторі не допус-
кається.
Густе живильне середовище Ь (агар із сахарозою, лак-
тозою, брильянтовим зеленим і феноловим червоним)
Пептони (м’ясний і казеїновий) 10.0 г
Дріжджовий екстракт 3.0 г
Натрію хлорид 5.0 г
Лактози моногідрат 10.0 г
Сахароза 10.0 г
Агар 20.0 г
Феноловий червоний 80 мг
Брильянтовий зелений 12.5 мг
Вода очищена 1 000 мл
Нагрівають до кипіння і кип’ятять протягом 1 хв. Ус-
тановлюють рН середовища таким чином, щоб після
стерилізації його значення становило 6.9+0.2. Сте-
рилізують безпосередньо перед використанням у па-
ровому стерилізаторі при температурі 121 °С протягом
15 хв. Охолоджують до температури 50 °С і розливають
у чашки Петрі.
Густе живильне середовище М (трицукровий агар із
залізом)
Яловичний екстракт 3.0 г
Дріжджовий екстракт 3.0 г
Пептони (казеїновий і яловичний) 20.0 г
Натрію хлорид 5.0 г
Лактози моногідрат 1.0 г
Сахароза 10.0 г
Глюкози моногідрат 1.0 г
Заліза(ІП) амонію цитрат 0.3 г
Натрію тіосульфат 0.3 г
Феноловий червоний 25 мг
Агар 12.0 г
Вода очищена 1 000 мл
Нагрівають до кипіння і кип’ятять протягом 1 хв при
струшуванні. Установлюють рН середовища таким чи-
ном, щоб після стерилізації його значення становило
7.4+0.2. Розливають середовище в пробірки так, шоб
заповнити одну третину пробірки за висотою, сте-
рилізують у паровому стерилізаторі при температурі
121 °С протягом 15 хв. Дають середовищу застигнути
таким чином, шоб одержати в одній пробірці
стовпчик живильного середовища і скошену поверх-
ню.
Нагрівають до кипіння і кип’ятять протягом 1 хв при
струшуванні. Установлюють рН середовища таким чи-
ном, щоб після стерилізації його значення становило
7.2±0.2. Стерилізують у паровому стерилізаторі при
температурі 121 °С протягом 15 хв.
Густе живильне середовище О (агар Байєрд-Паркера)
Панкреатичний гідролізат казеїну 10.0 г
Яловичний екстракт 5.0 г
Дріжджовий екстракт 1.0г
Літію хлорид 5.0 г
Агар 20.0 г
Іліцин 12.0г
Натрію піруват 10.0 г
Вода очищена 950 мл
Нагрівають до кипіння і кип’ятять протягом 1 хв при
струшуванні. Установлюють рН середовища таким чи-
ном, щоб після стерилізації його значення становило
6.8±0.2. Стерилізують у паровому стерилізаторі при
температурі 121 °С протягом 15 хв. Охолоджують до
температури від 45 °С до 50 °С і додають 10 мл сте-
рильного розчину 10 г/л калію телуриту і 50 мд
емульсії яєчного жовтка.
Живильне середовище Р (збагачене середос а це для
клостридій)
Яловичний екстракт 10.0 г
Пептон 10.0 г
Дріжджовий екстракт 3.0 г
Розчинний крохмаль 1.0 г
Глюкози моногідрат 5.0 г
Цистеїну гідрохлорид 0.5 г
Натрію хлорид 5.0 г
Натрію ацетат 3.0 г
Агар 0.5 г
Вода очищена 1 000 мл
Замочують агар і розчиняють його, нагріваючи до
кипіння при безперервному перемішуванні. Якщо не-
обхідно, установлюють рН середовища таким чином,
щоб після стерилізації його значення становило
близько 6.8. Стерилізують у паровому стерилізаторі
при температурі 121 °С протягом 15 хв.
Живильне середовище О (колумбійський агар)
Панкреатичний гідролізат казеїну 10.0 г
Пептичний гідролізат м’яса 5.0 г
Панкреатичний гідролізат серця 3.0 г
Дріжджовий екстракт 5.0 г
Кукурудзяний крохмаль 1.0 г
Натрію хлорид 5.0 г
120
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
Агар (у відповідності до
ГСіеутворюючих властивостей)
Втаї очищена
від 10.0 г
до 15.0 г
1 000 мл
Збочують агар і розчиняють його, нагріваючи до
кипіння при безперервному перемішуванні. Якщо не-
обхідно, установлюють рН середовища таким чином,
щоб після стерилізації його значення становило
’ 3+0.2. Стерилізують у паровому стерилізаторі при
іемпературі 121 °С протягом 15 хв. Дають охолонути
зо температури від 45 °С до 50 °С, додають, якщо не-
обхідно, 20 мг гентаміцину сульфату, в перерахунку на
геитаміцина основу, і розливають у чашки Петрі.
Живильне середовище К (лактозно-сульфітне середови-
ще)
Панкреатичний гідролізат казеїну 5.0 г
Дріжджовий екстракт 2.5 г
Натрію хлорид 2.5 г
Лактози моногідрат 10.0 г
Цистеїну гідрохлорид 0.3 г
Вела очищена 1 000 мл
Розчиняють інгредієнти складу, установлюють рН
’ 1+0.1, розливають по 8 мл у пробірки розміром
16 мм на 160 мм з маленькими трубками Дюрама. Сте-
рилізують у паровому стерилізаторі при температурі
121 С протягом 15 хв і зберігають при температурі
4“С.
Перед використанням середовище прогрівають на во-
дяній бані протягом 5 хв і охолоджують. Додають у
кожну пробірку 0.5 мл розчину 12 г/л натрію ме-
яабісульфіту Р і 0.5 мл розчину 10 г/л заліза(Ш)
амонію цитрату. Обидва розчини треба попередньо
пропустити крізь мембранні фільтри (розмір пор:
0.45 мкм). Розчини готують безпосередньо перед ви-
користанням.
НЕЙТРАЛІЗАТОРИ
Нейтралізатори використовують для нейтралізації ан-
тимікробної активності лікарських засобів. Нейт-
ралізатори можуть бути доданими до буферного роз-
чину і натрію хлоридом і пептоном рН 7.0, краще пе-
ред стерилізацією. При використанні нейтралізаторів
мас бути доведена їх нейтралізуюча ефективність і
нешкідливість для мікроорганізмів.
Типова нейтралізуюча рідина має такий склад:
Полісорбат-80 ЗО г
Лецитин (яєчний) 3 г
Гістидину гідрохлорид 1 г
Пептон (м’ясний або казеїновий) 1 г
Натрію хлорид 4.3 г
Калію ди гідрофосфат 3.6 г
Динатрію гідрофосфат дигідрат 7.2 г
Вода очищена 1 000 мл
Стерилізують у паровому стерилізаторі при темпера-
турі 121 °С протягом 15 хв.
Якщо розчин не має достатньої нейтралізуючої здат-
ності, збільшують концентрацію полісорбату-80 або
лецитину або використовують нейтралізатори, наве-
дені у Табл. 2.6.13.-2.
Якщо виробництво лікарського засобу не проводиться
відповідно до вимог належної виробничої практики
(НВП; СМР), установлених в Європейському Співто-
варистві, можуть бути також використані методи-
ки, описані нижче.
Ентеробактерїї і деякі інші
грамнегативні бактерії
Випробування використовують для виявлення бак-
терій, що належать до род. ЕпіегоЬасіегіасеае. а також
деяких інших грамнегативних бактерій. Випробуван-
ня проводять методом прямого висівання або методом
мембранної фільтрації.
Виявлення бактерій
Метод прямого висівання. Готують випробовуваний
зразок, як описано у статті 2.6.12. Підготований зра-
зок у кількості, відповідній 1 габо 1 мл лікарського за-
собу, вносять у 100 мл живильного середовища № З,
гомогенізують і інкубують при температурі від 35 °С до
37 °С від 18 год до 24 год. Після закінчення періоду
інкубації роблять пересівання на чашки Петрі з густи-
ми живильними середовищами № 4 і № 5. Посіви
інкубують при температурі від 35 °С до 37 °С від 24 год
до 48 год. Наявність на живильних середовищах ха-
Таблиця 2.6.13.-2
Антимікробні інактиватори, що додаються до буферного розчину з натрію хлоридом і пептоном рН 7.0
Тип антимікробного агента Інактиватор Концентрація Примітки
Феноли Натрію лаурилсульфат Полісорбат-80 і лецитин Яєчний жовток 4 г/л ЗО г/л і 3 г/л від 5 мл/л до 50 мл/л Додають після стерилізації буферного розчину з натрію хлоридом і пептоном рН 7.0
Ртутно-органічні сполуки Натрію тіогліколят від 0.5 г/л до 5 г/л
Галогени Натрію тіосульфат 5 г/л
Четвертинні амонієві сполуки Яєчний жовток від 5 мл/л до 50 мл/л Додають після стерилізації буферного розчину з натрію хлоридом і пептоном рН 7.0
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
121
2.6. Біологічні випробування
рактерного росту грамнегативних паличок, описаного
нижче, вказує на забруднення лікарського засобу бак-
теріями род. ЕпіегоЬасіегіасеае та деякими іншими
грам негати вн и ми бактер і я м и:
— густе живильне середовище № 4: круглі малинові
з металевим блиском або без нього колонії;
рожеві, безбарвні, блискучі опуклі колонії діамет-
ром від 2 мм до 4 мм;
— густе живильне середовище № 5: чорні з метале-
вим блиском колонії, ділянки середовища під
якими забарвлені в чорний колір; зеленувато-
бурі, ясно-зелені, бурі колонії.
Для ідентифікації бактерій род. ЕпіегоЬасіегіасеае
підозрілі колонії, кожну окремо, пересівають на сере-
довище № 1, скошене в пробірках, і інкубують при
температурі від 35 °С до 37 °С від 18 год до 24 год.
Після закінчення періоду інкубації підтверджують чи-
стоту кожної культури і проводять тест на наявність
цитохромоксидази. Культури, що дали негативну ре-
акцію на цитохромоксидазу, пересівають, кожну окре-
мо, у дві пробірки з рідким живильним середовищем
№ 6 і одну пробірку з рідким живильним середовищем
№ 7. Після висівання в одну із двох пробірок із сере-
довищем № 6 вносять близько 0.5 мл стерильного ва-
зелінового масла.
Усі посіви інкубують при температурі від 35 °С до
37 °С від 18 год до 24 год. У випадку зміни кольору се-
редовища № 6 із червоного на жовтий і, як правило,
наявності газоутворення в пробірках з вазеліновим
маслом і без нього роблять висновок про ферментацію
глюкози. Про наявність нітритів на середовищі № 7
судять із появи червоного забарвлення при внесенні у
середовище реактиву Грісса. Лікарський засіб витри-
мує випробування, якщо на живильних середовищах
не виявлений ріст грамнегативних неспороутворюю-
чих паличок, що дають негативну оксидазну реакцію,
ферментують глюкозу з утворенням кислоти (або кис-
лоти і газу) і відновлюють нітрати у нітрити.
Метод мембранної фільтрації. 10 мл зразка, підготова-
ного, як описано у статті 2.6.12, переносять на мемб-
ранний фільтр і негайно фільтрують. Якщо немає
інших зазначень в окремій статті, кожний мембран-
ний фільтр відмивають 1-5 порціями по 100 мл
підхожої промивної рідини. Мембранний фільтр
поміщають у 100 мл рідкого живильного середовища
№ 3. Далі випробування проводять, як описано для
методу прямого висівання.
При випробуванні трансдермальних пластирів десять
пластирів підготовляють, як описано у статті 2 6.12,
поміщають у посудину, що містить не менше 500 мл
рідкого живильного середовища № 11, і енергійно
струшують не менше ЗО хв. 50 мл підготованого зразка
пропускають крізь стерильний мембранний фільтр, як
описано у статті 2.6.12, і поміщають мембранний
фільтр у 100 мл рідкого живильного середовища № 3.
Далі випробування проводять, як описано для методу
прямого висівання.
Тест на наявність цитохромоксидази. Смужку фільт-
рувального паперу змочують реактивом на цитохро-
моксидазу і наносять скляною паличкою чисту добову
культуру випробовуваних бактерій, що виросли на се
редовищі № 1. Синє забарвлення, що з’являється че-
рез 2-5 хв, свідчить про позитивну реакцію на пито-
хромоксидазу.
Кількісна оцінка
Метод прямого висівання. Готують випробовуваний
зразок, як описано у статті 2.6.12, використовуючи
рідке живильне середовище № 11 замість буферного]
розчину з натрію хлоридом і пептоном рН 7.0, гомо-|
генізують і інкубують при температурі від 35 °С до
37 °С протягом часу, достатнього для відновлення
життєздатності мікроорганізмів, але не достатнього
для збільшення їх кількості (як правило, 2 год, але не
більше 5 год). Струшують посудину і переносять її
вміст (гомогснізат А) і/або його розведення, шо
містять відповідно 0.1 г, 0.01 г і 0.001 г (або 0.1 мл,
0.01 мл і 0.001 мл) випробовуваного лікарського засо-
бу, у відповідні об’єми рідкого живильного середови-
ща № 3. Інкубують при температурі від 35 °С до 37 °С
від 24 год до 48 год. Після закінчення періоду інкубації
з кожної посудини роблять пересівання на чашку з гу-
стим живильним середовищем № 4 так, щоб одержати
ріст ізольованих колоній. Інкубують при температурі
від 35 °С до 37 °С від 18 год до 24 год. Позитивний ре-
зультат випробування встановлюють при наявності на
середовищі № 4 росту типових колоній грамнегатив-
них паличок, приналежність яких до род. Епіего-
Ьасіегіасеае підтверджують біохімічними тестами,
описаними у розділі "Ентеробактерії і деякі грам-
негативні бактерії Виявлення бактерій". Позначають
найменшу кількість лікарського засобу, що дає пози-
тивний результат, і найбільшу кількість лікарського
засобу, що дає негативний результат. Визначають
імовірне число бактерій за Табл. 2.6.13.-1.
Метод мембранної фільтрації. Підготовку зразка і
процедуру фільтрації проводять, як описано у роздиі
"Ентеробактерії і деякі грамнегативні бактерії. Вияв-
лення бактерій. Метод мембранної фільтрації". Після
закінчення фільтрації мембранний фільтр поміщають
на поверхню густого живильного середовища №4 у
чашці Петрі і інкубують при температурі від 35 °С
до 37 °С від 24 год до 48 год. Посіви переглядають че-
рез 24 год і остаточно через 48 год. При наявності на
мембранних фільтрах росту типових колоній
грамнегативних паличок, приналежність яких до
род. ЕпіегоЬасіегіасеае підтверджують біохімічними
тестами, описаними вище, підраховують їх число і та-
ким чином визначають число бактерій род. ЕпіегоЬас-
Гегіасеае в 1 г лікарського засобу.
ЕясИегісИіа соїі
Випробування проводять методом прямого висівання
або методом мембранної фільтрації.
Виявлення бактерій
Метод прямого висівання. 10 мл зразка, підготованого,
як описано в розділі "Ентеробактерії і деякі грамне-
122
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
іїійвні бактерії. Кількісна оцінка. Метод прямого
Иківання", вносять у 100 мл рідкого живильного сере-
щвчша № 3 і інкубують при температурі від 35 °С до
37 С від 18 год до 24 год. Після закінчення інкубації
роблять пересівання на густе живильне середовище
44 і інкубують при температурі від 35 °С до
У 0 від 18 год до 24 год. Ріст характерних малинових
колоній з металевим блиском або без нього або роже-
вів колоній діаметром від 2 мм до 4 мм указує на мож-
ливість забруднення лікарського засобу Е. соїі. У цьо-
му випадку роблять пересівання підозрілих колоній,
мжноі окремо, на середовище № 1 і інкубують при
температурі від 35 °С до 37 °С від 18 год до 24 год. Ко-
мнії, шо виросли на середовищі № 1, мікроскопують
і при виявленні в мазках лише грамнегативних пали-
чок проводять тест на цитохромоксидазу. У випадку
негативної реакції проводять додаткові біохімічні тес-
ти на утилізацію цитрату і на індол.
Для біохімічної ідентифікації мікроорганізмів можуть
бути використані готові тест-системи.
Якщо на живильних середовищах виявлений ріст
тгамнегативних неспороутворюючих паличок, що не
мають ферменту цитохромоксидази, не утилізують
цитрат і утворюють індол, вважають, що лікарський
засіб забруднений Е. соїі.
Метод мембранної фільтрації. Підготовку і фільтрацію
іразка проводять, як описано у розділі "Ентеробак-
герії. Виявлення бактерій. Метод мембранної
фільтрації". Після закінчення інкубації роблять пе-
ресівання з середовища № 3 на середовище № 4. Далі
випробування проводять, як описано для методу пря-
мого висівання.
Тест на утилізацію цитрату. Роблять пересівання на
густе живильне середовище № 14 і інкубують при тем-
пературі від 35 °С до 37 °С від 24 год до 48 год. При на-
явності бактеріального росту утилізацію цитрату вста-
новлюють за зміною кольору середовища із зеленого
на синій.
Тест на індол. Роблять пересівання на рідке живильне
середовище № 15. Посіви інкубують при температурі
від 35 °С до 37 °С від 24 год до 48 год. При наявності
бактеріального росту наявність індолу встановлюють
за появою червоного забарвлення при внесенні у сере-
довище реактиву Ковача або Ерліха.
Кількісна оцінка
Випробування проводять методом прямого висівання
або методом мембранної фільтрації, як описано ви-
ще в розділі "Ентеробактерії. Кількісна оцінка". Ріст
Е. соїі на середовищі № 4 підтверджують, використо-
вуючи методики, наведені вище у розділі "Еьсіїсгісіїіа
соїі. Виявлення бактерій".
Каїтопеїіа
випробування проводять методом прямого
висівання
аоо
методом мембранної
фільтрації.
Метод прямого висівання. 10 мл зразка, підготовано-
го, як описано в розділі "Ентеробактерії і деякі грам-
негативні бактерії. Кількісна оцінка", вносять у 100 мл
рідкого живильного середовища № 3 і інкубують при
температурі від 35 °С до 37 °С від 18 год до 24 год.
Після закінчення інкубації роблять пересівання 1 мл з
середовища № 3 у 10 мл рідкого живильного середо-
вища № 12 і інкубують при температурі від 35 °С до
37 °С від 16 год до 18 год. Після закінчення періоду
інкубації роблять пересівання із середовища № 12 на
поверхню густого живильного середовища № 5 у чаш-
ках Петрі і інкубують при температурі від 35 °С до
37 °С від 24 год до 48 год. Ріст чорних колоній із харак-
терним металевим блиском, під якими ділянка сере-
довища профарбовується у чорний колір, або світлих
зеленуватих колоній указує на можливість забруднен-
ня лікарського засобу Еаїтопеїіа. У цьому випадку
роблять пересівання підозрілих колоній, кожної окре-
мо. у пробірки зі скошеним густим живильним сере-
довищем № 1 і інкубують при температурі від 35 °С до
37 °С від 18 год до 24 год. Колонії, що виросли на се-
редовищі № 1. мікроскопують і при виявленні у маз-
ках лише грамнегативних паличок проводять тест на
цитохромоксидазу. У випадку негативної реакції роб-
лять пересівання на густе живильне середовище № 13,
наносячи невелику кількість культури петлею спочат-
ку на скошену частину живильного середовища, а
потім уколом у стовпчик, і інкубують при температурі
від 35 °С до 37 °С від 18 год до 24 год. Якщо після
закінчення періоду інкубації спостерігається зміна ко-
льору середовища № 13 із червоного на жовтий (у гли-
бині агару, але не на його поверхні), що супровод-
жується, як правило, утворенням сірководню в гли-
бині агару (наявність чорного забарвлення), вважа-
ють, що лікарський засіб забруднений Ваїтопеїіа.
Для біохімічної ідентифікації мікроорганізмів можуть
бути використані тест-системи.
Метод мембранної фільтрації. Підготовку і фільтрацію
зразка проводять, як описано у розділі "Ентеробак-
терії і деякі грамнегативні бактерії. Виявлення бак-
терій. Метод мембранної фільтрації". Після закінчен-
ня періоду інкубації роблять пересівання з середови-
ща № 3 на середовище № 5. Далі випробування про-
водять, як описано для методу прямого висівання.
Еіаріїуіососсіні аигеиа
Випробування проводять методом прямого висівання
або методом мембранної фільтрації.
Метод прямого висівання. Готують випробовуваний
зразок, як описано у статті 2.6.12. Підготований зра-
зок у кількості, відповідній 1 габо 1 мл лікарського за-
собу, вносять у 100 мл живильного середовища № 8,
гомогенізують і інкубують при температурі від 35 °С
до 37 °С від 18 год до 24 год. Після закінчення періоду
інкубації роблять пересівання на чашку з густим жи-
вильним середовищем № 10 і інкубують при темпера-
турі від 35 °С до 37 °С від 24 год до 48 год. Ріст золота-
во-жовтих колоній, оточених жовтими зонами (що
свідчить про ферментацію маніту), указує на мож-
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
2001
123
2.6. Біологічні випробування
ливість забруднення лікарського засобу 5. аигеих. У
цьому випадку роблять пересівання підозрілих ко-
лоній, кожної окремо, у пробірки зі скошеним густим
живильним середовишем № 1 і інкубують при темпе-
ратурі від 35 °С до 37 °С від 18 год до 24 год. Колонії,
що виросли на середовищі № 1, мікроскопують і при
виявленні у мазках тільки грампозитивних коків, роз-
ташованих гронами, проводять тест на плазмокоагу-
лазу.
Для біохімічної ідентифікації мікроорганізмів можуть
бути використані готові тест-системи.
Лікарський засіб витримує випробування, якщо на
живильних середовищах не виявлений ріст грампози-
тивних коків, що ферментують маніт і дають позитив-
ну реакцію плазмокоагуляції.
Метод мембранної фільтрації. 10 мл зразка, підготова-
ного, як описано у статті 2.6.12, переносять на мемб-
ранний фільтр і негайно фільтрують. Якщо немає
інших зазначень в окремій статті, кожний мембран-
ний фільтр відмивають 1-5 порціями по 100 мл
підхожої промивної рідини. Мембранний фільтр
поміщають у 100 мл рідкого живильного середовища
№ 8. Далі випробування проводять, як описано для
методу прямого висівання.
При випробуванні трансдермальних пластирів 50 мл
зразка А пропускають крізь стерильний мембранний
фільтр, як описано у статті 2.6.12, поміщають мемб-
ранний фільтр у 100 мл рідкого живильного середови-
ща № 8 і інкубують при температурі від 35 °С до 37 °С
від 18 год до 48 год. Після закінчення періоду інкубації
роблять пересівання на поверхню густого живильного
середовища № 10. Далі випробування проводять, як
описано для методу прямого висівання.
Тест на плазмокоагулазу (реакція плазмокоагуляції).
Кров, узяту стерильним шприцом із серця кролика,
поміщають у 5 % стерильний розчин натрію цитрату,
відбирають плазму, розводять у співвідношенні 1:5
стерильним розчином 9 г/л натрію хлориду і розлива-
ють по 0.5 мл у стерильні пробірки. У кожну пробірку
поміщають 1 петлю чистої добової культури стафіло-
кока, що виросла на середовищі № 1, і інкубують при
температурі від ЗО °С до 35 °С від 4 год до 6 год. Якщо
протягом цього часу не спостерігається згортання
плазми, реакцію плазмокоагуляції вважають негатив-
ною. Одночасно з випробуванням проводять два кон-
трольних досліди: 1) контроль розчину плазми,
2) контроль культури стафілокока, що дає позитивну
реакцію на плазмокоагулазу.
Допускається використовувати суху кролячу цитратну
плазму промислового виробництва, яку готують
згідно з інструкцією щодо застосування.
Раеисіотопаз аеги%іпояа
Випробування проводять методом прямого висівання
або методом мембранної фільтрації.
Метод прямого висівання. Готують випробовуваний
зразок, як описано у статті 2.6.12. Підготований зра-
зок у кількості, відповідній 1 габо 1 мл лікарською®
собу, вносять у 100 мл живильного середовища N
гомогенізують і інкубують при температурі від 351 «І
37 °С від 18 год до 24 год. Після закінчення ііеряа
інкубації роблять пересівання на чашку з густим і І
вильним середовищем № 9 і інкубують при темпері
турі від 35 °С до 37 °С від 24 год до 48 год. Ріст зеле
ватих, як правило, флуоресціюючих колоній, б.чак
них в ультрафіолетовому світлі (що свідчить про!
явність пігменту піоціаніну), указує на можливість!
бруднення лікарського засобу Р. аегиріпоха. У щд-
випадку роблять пересівання підозрілих колоні
кожної окремо, в пробірки із скошеним густим»
вильним середовищем № 1 й інкубують при темпер»
турі від 35 °С до 37 °С від 18 год до 24 год. Колонії,В(
виросли на середовищі № 1, мікроскопують і при в.
явленні у мазках тільки грамнегативних паличок
водять тест на цитохромоксидазу.
Для біохімічної ідентифікації мікроорганізмів можу
бути використані готові тест-системи.
Лікарський засіб витримує випробування, якшо ні
живильних середовищах не виявлений ріст грамнет-
тивних паличок, що утворюють синьо-зеленік
пігмент піоціанін і дають позитивну реакцію на та
хромоксидазу.
Метод мембранної фільтрації. 10 мл зразка, підгото»!
ного, як описано у статті 2.6.12, переносять на мемС
ранний фільтр і негайно фільтрують. Якщо не\м
інших зазначень в окремій статті, кожний мембран-
ний фільтр відмивають 1-5 порціями по 100 уд
підхожої промивної рідини. Мембранний фільт
поміщають у 100 мл рідкого живильного середовищі
№ 8. Далі випробування проводять, як описано дії
методу прямого висівання.
При випробуванні трансдермальних пластирів 50 м
зразка А пропускають крізь стерильний мембраншп
фільтр, як описано у статті 2.6.12, поміщають мемЬ
ранний фільтр у 100 мл рідкого живильного середовн
ща № 8 і інкубують при температурі від 35 °С до 371
від 18 год до 48 год. Після закінчення періоду інкубац)
роблять пересівання на поверхню густого живильнок
середовища № 9. Далі випробування проводять, яі
описано для методу прямого висівання.
Ростові властивості живильних середовищ
і перевірка придатності методик випробування
При перевірці придатності методик випробування
для контролю ростових властивостей живильних сері
довищ замість тест-мікроорганізму ЕхНегісІйа сс
АТСС 8739 (МС1МВ 8545, СІР 53.126) може бути викс
ристаний тест-мікроорганізм ЕзсИегісИіа соїі АТС
25922.
Для вирощування тест-мікроорганізмів допускаєт
використовувати рідке живильне середовище № 1.
Для приготування робочих суспензій тест-мікро
ганізмів допускається використовувати розчин 9
натрію хлориду.
124
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ
2.6. Біологічні випробування
І'пускається проводити процедуру перевірки придат-
? «кгі методик випробування одночасно з випробуван-
" лікарського засобу на мікробіологічну чистоту.
цьому облік результатів випробування проводять
му випадку, якщо підтверджена придатність вико-
I рєіовуваної методики.
РЕКОМЕНДОВАНІ РОЗЧИНИ І ЖИВИЛЬНІ
СЕРЕДОВИЩА
Нижче наведені склади розчинів і живильних серело-
пйш, рекомендованих для проведення випробування
ІГ.Імікробіологічну чистоту.
Лслускаеться використання сухих живильних середо-
япцтого самого або аналогічного складу, які випуска-
ться промисловістю.
Густе живильне середовище № 1 (м’ясо-пептонний
агар)
Допускається використання густого живильного сере-
овища № І замість густого живильного середовища В.
Пептон ферментативний сухиіі 10.0 г
Натрію хлорид 5.0 г
Еіюкоза 1.0 г
Згар мікробіологічний 13.0 г
М’ясна вода (1:2) 1 000 мл
До м'ясної води додають пептон і натрію хлорид, роз-
чиняють при нагріванні, вносять глюкозу, установлю-
ють рН таким чином, щоб після стерилізації його зна-
чення становило 7.3± 0.2, кип’ятять протягом 1 хв, до-
дать замочений заздалегідь агар, нагрівають до по-
вного його розплавлення і фільтрують крізь ватно-
марлевий фільтр. Стерилізують у паровому стериліза-
торі при температурі 121 °С протягом 15 хв.
Рідке живильне середовище № 1
Допускається використання рідкого живильного сере-
довища № 1 замість рідкого живильного середовища А.
Пептон ферментативний сухий 10.0 г
Натрію хлорид 5.0 г
Еіюкоза 1.0 г
М’ясна вода 1 000 мл
До м'ясної води додають пептон і натрію хлорид, роз-
чиняють при нагріванні, вносять глюкозу, установлю-
ють рН таким чином, щоб після стерилізації його зна-
чення становило 7.3+0.2, кип’ятять протягом 1 хв.
Фільтрують крізь паперовий фільтр. Стерилізують у
паровому стерилізаторі при температурі 121 °С протя-
гом 15 хв.
Густе живильне середовище № 2 (агар Сабуро)
Допускається використання густого живильного сере-
довища № 2 замість густого живильного середовища С.
Пептон ферментативний сухий 10.0 г
Глюкоза 40.0 г
Агар мікробіологічний
Вода очищена
13.0 г
1 000 мл
Інгредієнти складу розчиняють у воді, установлюють
рН таким чином, щоб після стерилізації його значен-
ня становило 5.6±0.2, додають замочений заздалегідь
агар, нагрівають до повного його розплавлення і
фільтрують крізь ватно-марлевий фільтр. Стерилізу-
ють у паровому стерилізаторі при температурі 121 °С
протягом 15 хв Після стерилізації додають 0.10 г бен-
зилпеніциліну і 0.10 г тетрацикліну на 1 л середовища
або перед стерилізацією додають хлорамфенікол із
розрахунку 50 мг на 1 л живильного середовища.
Рідке живильне середовище № 3 (середовище збагачен-
ня для бактерій род. ЕпіегоЬасіегіасеае)
Пептон ферментативний сухий 10.0 г
Динатрію гідрофосфат 7.5 г
Калію ди гідрофосфат 2.5 г
Глюкоза 10.0 г
Феноловий червоний 0.08 г
Малахітовий зелений 0. 015 г
М’ясна вода 1 000 мл
Інгредієнти складу, крім глюкози та індикаторів роз-
чиняють у м’ясній воді при нагріванні, потім вносять
глюкозу, додають 8 мл 1 % розчину фенолового черво-
ного і 3 мл 0.5 % розчину малахітового зеленого, уста-
новлюють рН таким чином, щоб після стерилізації йо-
го значення становило 7.3+0.2, кип’ятять І хв,
фільтрують крізь паперовий фільтр. Стерилізують у
паровому стерилізаторі при температурі 121 °С протя-
гом 15 хв.
Густе живильне середовище № 4 (агар Ендо)
Живильний агар сухий 26.5 г
ЕКДА 1.22 г
Динатрію гідрофосфат 0.48 г
Натрію сульфіт безводний 0.83 г
Натрію карбонат 0.03 г
Лактоза 10.7 г
Фуксин основний 0.23 г
Вода очищена 1 000 мл
Установлюють рН таким чином, щоб після нагрівання
його значення становило 7.3±0.2. Нагрівають до по-
вного розплавлення агару і кип’ятять від 2 хв до 3 хв.
Фільтрують крізь ватно-марлевий фільтр, потім
нагрівають до моменту закипання. Охолоджують до
температури від 45 °С до 50 °С і розливають у чашки
Петрі.
Густе живильне середовище № 5 (вісмутсульфіт агар)
Панкреатичний гідролізат м’яса 10 1 г
Динатрію гідрофосфат безводний 3.68 г
Натрію хлорид 2.6 г
Натрію карбонат 0.65 г
Вісмуту цитрат 2.38 г
Заліза(ІІ) амонію сульфат 0.97 г
О-глюкоза 3.9 г
Агар мікробіологічний 15.0 г
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
125
2.6. Біологічні випробування
Брильянтовий зелений
Вода очищена
0.028 г
1 000 мл
Агар мікробіологічний
Вода очищена
15.0
1 000 м
Установлюють рН таким чином, щоб після нагрівання
його значення становило 7.6±0.2. Нагрівають до по-
вного розплавлення агару і кип’ятять від 3 хв до 5 хв.
Охолоджують до температури від 45 °С до 50 °С і роз-
ливають у чашки Петрі.
Рідке живильне середовище № 6 (для визначення фер-
ментації глюкози)
Пептон ферментативний сухий 10.0 г
Натрію хлорид 5.0 г
Глюкоза 40.0 г
Феноловий червоний 0.08 г
М’ясна вода 1 000 мл
Пептон і натрію хлорид розчиняють у м’ясній воді при
нагріванні, вносять глюкозу, додають 8 мл 1 % розчи-
ну фенолового червоного, встановлюють рН таким
чином, щоб після стерилізації його значення
становило 7.2+0.2, кип’ятять 1 хв, фільтрують крізь
паперовий фільтр і розливають по 4-5 мл у пробірки з
поплавцями. Стерилізують у паровому стерилізаторі
при температурі 121 °С протягом 15 хв. Після закін-
чення стерилізації середовище швидко охолоджують.
Рідке живильне середовище № 7 (для визначення
відновлення нітратів у нітрити)
Пептон ферментативний сухий
Натрію хлорид
Калію нітрат
Вода очищена
5.0 г
5.0 г
1.5 г
1 000 мл
Інгредієнти складу розчиняють у воді при нагріванні,
встановлюють рН таким чином, щоб після сте-
рилізації його значення становило 7.2±0.2, кип’ятять
І хв, фільтрують крізь паперовий фільтр, розливають у
пробірки по 4-5 мл. Стерилізують у паровому сте-
рилізаторі при температурі 121 °С протягом 15 хв.
Рідке живильне середовище № 8 (для вирощування
РхеиЛотопая аегиріпояа і 8іарІіуІососсия аигеия)
Пептон ферментативний сухий
Натрію хлорид
Дикалію гідрофосфат
Глюкоза
Вода очищена
10.0 г
5.0 г
2.5 г
2.5 г
1 000 мл
Інгредієнти складу, крім глюкози розчиняють у воді
при нагріванні, вносять глюкозу, встановлюють рН та-
ким чином, щоб після стерилізації його значення ста-
новило 7.3+0.2, кип’ятять 1 хв, фільтрують крізь папе-
ровий фільтр. Стерилізують у паровому стерилізаторі
при температурі 121 °С протягом 15 хв.
Густе живильне середовище № 9 (для виявлення
пігменту піоціаніну)
Пептон ферментативний сухий 20.0 г
Магнію хлорид безводний 1.4 г
Калію сульфат безводний 10.0 г
Гліцерин 10.0 г
Інгредієнти складу, крім гліцерину, розчиняють у вол.І
і залишають на 15 хв. Потім вносять гліцерин, ретелі
но перемішують, розчиняють при нагріванні, встанов-
люють рН таким чином, щоб після стерилізації його
значення становило 7.2±0.2, кип’ятять 1 хв, додають'
замочений заздалегідь агар, нагрівають до повного йо-
го розплавлення, фільтрують крізь ватно-марлсшіг
фільтр. Стерилізують у паровому стерилізаторі прь
температурі 121 °С протягом 15 хв.
Густе живильне середовище № 10 (для ідентифікації
Ьіаріїуіососсия аигеия}
Пептон ферментативний сухий 10.0г
Натрію хлорид 75.0г
Маніт 10.0г
Феноловий червоний О.О25г
Агар мікробіологічний 15.0г
Вода очищена 1 000 мл
Інгредієнти складу розчиняють у воді, вносять 2.5 ш І
1 % розчину фенолового червоного, установлюютьрН І
таким чином, шоб після стерилізації його значення
становило 7.4±0.2, кип’ятять 1 хв, додають замочений
заздалегідь агар, нагрівають до повного його розплав-
лення, фільтрують крізь ватно-марлевий фільтр. Сте-
рилізують у паровому стерилізаторі при температурі
121 °С протягом 15 хв.
Рідке живильне середовище № 11 (лактозний бульйон
для попереднього збагачення бактерій род. ЕпІегоЬаае-
гіасеае)
Пептон ферментативний сухий
Лактоза
Вода очищена
8.0 г
5.0 г
1000 мл
Установлюють рН таким чином, щоб після сте-
рилізації його значення становило 6.9+0.1. Стерилізу-
ють у паровому стерилізаторі при температурі 121 °С
протягом 15 хв.
Рідке живильне середовище № 12 (селенітове середо-
вище сухе для виділення Ьаїтопеїіа)
Панкреатичний гідролізат казеїну 5.5 г
Лактоза 4.2 г
Динатрію фосфат 6.3 г
Натрію гідроселеніт (без телуру) 4.2 г
Вода очищена 1000 мл
Інгредієнти складу вносять у воду очищену, установ-
люють рН таким чином, щоб після стерилізації його
значення становило 7.5+0.2. Нагрівають до моменту
закипання і розливають у стерильні пробірки.
Густе живильне середовище № 13 (трицукровий агар із
залізом для ідентифікації Заїтопеїіа)
М’ясний екстракт
Дріжджовий екстракт
Пептон ферментативний сухий
3.0 г
3.0 г
15.0 г
126
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
П?. І.ОЗОПСПТОН 5.0 г
[ »1ю.шза 10.0 г
Сшроза 10.0 г
Гтоза 1.0 г
ки. а(І1І) сульфат 0.2 г
і і грію хлорид 5.0 г
Н*трію тіосульфат 0.3 г
Л толовий червоний 0.024 г
пар мікробіологічний 15.0 г
Вааа очищена 1 000 мл
шанов.'іюють рН таким чином, щоб після сте-
-ілізації його значення становило 7.2±0.1. Розлива-
ють у пробірки по 5-7 мл. Стерилізують у паровому
.іерилізаторі при температурі 121 °С протягом 15 хв.
Охолоджують так, щоб одержати стовпчик середови-
ща висотою від 2 см до 2.5 см і скошену поверхню.
Густе живильне середовище № 14 (для виявлення фер-
ментації цитрату)
Натрію хлорид
Магнію сульфат
\монію дигідрофосфат
Дикалію гідрофосфат
Натрію цитрат
Бромтимоловий синій
\гар мікробіологічний
Вода очищена
5.0 г
0.2 г
1.0 г
1.0 г
3.0 г
0.08 г
20.0 г
1 000 мл
Усі інгредієнти складу, крім агару і бромтимолового
синього, поміщають у посудину місткістю 1500 мл,
розчиняють у 500 мл свіжоприготованої очищеної во-
їн, додають агар, доводять до 1000 мл свіжоприготова-
ною водою очищеною і нагрівають до розплавлення
агару. Установлюють рН таким чином, щоб після сте-
рилізації його значення становило 7.2±0.1. додають
40 мл 0.2 % водного розчину бромтимолового синьо-
ііі, перемішують і розливають у пробірки по 5-7 мл.
Стерилізують у паровому стерилізаторі при темпера-
гурі 121 °С протягом 15 хв. Охолоджують так, щоб
іержати скошену поверхню.
Різке живильне середовище № 15 (бульйон Хотгінгера
для визначення індолу)
Використовують готове живильне середовище.
Реактив Ковача
Спирт аміловий або ізоаміловий 75 мл
п-Диметиламінобензальдегід 5 г
Кислота хлористоводнева концентрована 25 мл
Наважку п-диметиламінобензальдегіду розчиняють у
спирті аміловому або ізоаміловому при нагріванні на
водяній бані при температурі від 50 °С до 55 °С, охоло-
джують і повільно додають кислоту хлористоводневу
концентровану. Розчин зберігають у захищеному від
світла місці. Реактив має бути жовтого кольору.
Реактив Ерліха
96 % спирт
п-Диметиламінобензальдегід
95 мл
1 г
Кислота хлористоводнева концентрована 20 мл
Наважку п-диметиламінобензальдегіду розчиняють у
96 % спирті й повільно додають кислоту хлористовод-
неву концентровану. Розчин зберігають у захищеному
від світла місці.
Феноловий червоний - 1 % розчин
Феноловий червоний 1.0 г
0.1 М розчин натрію гідроксиду 28.2 мл
Вода очищена до 100 мл
Наважку фенолового червоного розтирають у ступці,
додаючи невеликими порціями 0.1 М розчин натрію
гідроксиду. Одержаний розчин переносять у мірну
колбу місткістю 100 мл і доводять об’єм розчину до
позначки водою очищеною. Зберігають у флаконі
нейтрального світлозахисного скла при температурі
від 4 °С до 10 °С.
Малахітовий зелений - 0.5 % розчин
Малахітовий зелений 0.5 г
Вода очищена до 100 мл
Наважку малахітового зеленого переносять у скляний
флакон, додають гарячу стерильну воду очищену,
поміщають на добу в термостат при температурі від
35 °С до 37 °С, періодично струшуючи.
Реактив Грісса
Розчин № 1:0.5 г кислоти сульфанілової розчиняють у
ЗО мл кислоти оцтової льодяної, додають 100 мл води
очищеної. Розчин придатний протягом місяця.
Розчин №2: 0.1 г 1-нафтіламіну розчиняють у 100 мл
киплячої води очищеної, охолоджують і додають 30 мл
кислоти оцтової льодяної. Розчин придатний протя-
гом семи діб.
Перед використанням зміщують рівні об’єми розчинів
№ 1 і № 2.
Реактив на нитохромоксидазу
Розчин № 1: 1 % спиртовий розчин а-нафтолу.
Розчин № 2: 1 % водний розчин М.ІМ-диметил-п-
фенілендіаміну дигідрохлориду.
Розчини придатні протягом 14 діб при зберіганні при
температурі від 4 °С до 10 °С у флаконах нейтрального
світлозахисного скла.
Перед використанням змішують розчини № 1 і № 2 у
співвідношенні 2 З
2.6.14. БАКТЕРІАЛЬНІ ЕНДОТОКСИНИ
Дана стаття описує п’ять методів, призначених для
визначення відповідності концентрації бактеріальних
ендотоксинів у лікарському засобі до вимог Фармако-
пеї.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
127
2.6. Біологічні випробування
При проведенні випробування на бактеріальні ендо-
токсини (ЛАЛ-тесту) використовують лізат амебо-
цитів мечохвоста Еітиіив роїуркетив. Додавання роз-
чину, що містить ендотоксини, до розчину лізату при-
зводить до появи каламутності, осаджування або геле-
утворення суміші. Швидкість реакції залежить від
концентрації ендотоксинів, рН і температури. Для ре-
акції необхідна наявність певних двовалентних
катіонів, ферментної системи, що забезпечує утворен-
ня тромбу, і білка, здатного до утворення тромбу, які
містяться у лізаті. Концентрацію ендотоксинів можна
також розраховувати за концентрацією барвника, що
вивільнився, у реакції лізису хромогенного пептиду в
розчині лізату після активації його ендотоксинами.
Описано такі п’ять методів:
Метод А — метод гелеутворення: граничне випробу-
вання;
Метод В — напівкількісний метод гелеутворення;
Метод С — турбідиметричний кінетичний метод;
Метод О — кінетичний метод з використанням хро-
могенного пептиду;
Метод Е — метод кінцевої точки з використанням
хромогенного пептиду.
Якщо немає інших зазначень в окремій статті, вико-
ристовують метод А, який пройшов валідацію для да-
ного лікарського засобу. У супротивному випадку ви-
пробування виконують методом, зазначеним в ок-
ремій статті.
Окремі статті установлюють вимоги щодо бактеріаль-
них ендотоксинів, виражені граничною концент-
рацією ендотоксинів; зразок відповідає вимогам, як-
що вміст у ньому ендотоксинів не перевищує гранич-
ної концентрації. Відповідність цій вимозі можна про-
демонструвати, тільки показавши, що концентрація
ендотоксинів у зразку менша за граничну концент-
рацію ендотоксинів.
Випробування проводять за методикою, що дозволяє
уникнути мікробного забруднення. Перед проведен-
ням випробування на ендотоксини для випробовува-
ного зразка необхідно підтвердити:
— що використовуване обладнання не адсорбує ен-
дотоксини;
— величину лямбда (X) використовуваного лізату
амебоцитів; величина X визначається як заявлена
на етикетці чутливість лізату амебоцитів (методи
А і В) або найнижча концентрація ендотоксину,
використовувана для одержання стандартної
кривої (кількісні методи);
— відсутність факторів, що заважають проведенню
ЛАЛ-тесту.
Якщо необхідно, проводять обробку обладнання з ме-
тою видалення ендотоксинів.
Якщо в окремій статті на випробовуваний зразок не-
має інших зазначень, у п’яти описаних методах - від
методу А до методу Е - застосовують одні й ті самі кри-
терії. Термін "пробірка" включає і будь-яку іншу посу-
дину, наприклад, таку як планшета для мікротитру-
вання.
У випробуванні використовують такі реактиви і стан-
дартний препарат.
Ендотоксин БСП (біологічний стандартний претЛ
рат)1. Ендотоксин БСП калібрований у Міжнародне
Одиницях (МО) у порівнянні з Міжнародним Ста,
дартом.
Лізат амебоцитів Ьітиіив. Продукт має бути приведД
ний відповідно до вимог компетентного уповноваженії
го органу країни-виробника. Його готують, дотримуй
чись зазначень інструкції на даний лізат. Для кожне
партії лізату підтверджують заявлену на етикетці чув]
ливість (?.), що виражена у МО ендотоксину на мілілітр
Вода ЛАЛ. Вода є придатною, якщо вона дає негатив,]
ний результат у випробуванні на ендотоксини для вн-|
пробовуваного зразка. Вона може бути приготована!
триразовою перегонкою води в апараті, споряджено-!
му ефективним пристроєм для запобігання попадання!
часток ззовні, або іншими приладдями, що дозволяв
ють одержати воду потрібної якості.
0.1 М розчин кислоти хлористоводневої ЛАЛ і 0.1 1ИІ
розчин натрію гідроксиду ЛАЛ. Реактиви готують з
кислоти хлористоводневої Р і натрію гідроксиду Р, І
відповідно, використовуючи воду ЛАЛ.
Кожний з реактивів є придатним, якщо після доведем-1
ня рН до 6.0-8.0 він дає негативний результат за умові
проведення випробування.
Якщо немає інших зазначень, розчини і розведення, ви-
користовувані при проведенні випробування, готують!
використанням води ЛАЛ.
МЕТОДИ ГЕЛЕУТВОРЕННЯ
Методи А і В грунтуються на утворенні стійкого гелю
у розчині, що містить бактеріальні ендотоксини, після
його змішування й інкубування з ЛАЛ-реактивом.
Обидва методи вимагають підтвердження чутливості
лізату, зазначеної виробником відповідно до зазначень
розділу "Чутливість лізату", і випробування на на-
явність заважаючих факторів відповідно до зазначень
розділу "Заважаючі фактори".
Відмінність методів А і В полягає в такому: за мето-
дом А перевіряють, чи справді два розчини випробо-
вуваного зразка, приготовані паралельно, містять
менше ендотоксинів за граничну концентрацію ендо-
токсинів, зазначену у відповідній окремій статті; за
методом В концентрацію ендотоксинів у випробову-
ваному зразку визначають напівкількісно, і середнє
геометричне величин концентрацій ендотоксинів мас
бути менше за граничну концентрацію ендотоксинів,
зазначену в окремій статті.
Наступні розділи "Методика", "Чутливість лізату" і
"Заважаючі фактори" застосовні як до методу А, такі
до методу В.
Методика. Об'єм лізату, відповідний вибраній посу-
дині (наприклад, пробірці або предметному склу),
'Під біологічним стандартним препаратом ендоксину мають на
увазі стандартний препарат, кількісний аналіз якого проведений у
порівнянні з Міжнародним Стандартом ВООЗ, а його активність
виражена в Міжнародних Одиницях (МО) ендотоксину на мілілітр.
128
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
»>ІІІ . іютьу кожну з необхідної кількості таких посу-
К шо зберігаються при температурі (37±1) °С. Че-
І|г» певні проміжки часу, що дозволяють урахувати
пшиїи результат, додають у кожну посудину рівні
ній випробовуваного розчину і негайно обережно
Шілють із лізатом. Інкубують суміш, не допускаючи
м'г ті і зводячи до мінімуму втрату води при випа-
рншні протягом постійного інтервалу часу, вибрано-
іл . попередньому експерименті (звичайно — від
З* їв то 60 хв), і враховують результати. Позитивним
(н гльтатом вважають утворення стійкого гелю, що не
^кується при обережному перевертанні посудини,
що такий гель не утворюється, результат вважають
«і ітивним.
^гливість лізату. Готують не менше чотирьох пара-
цчіВііх рядів дворазових розведень розчинів ендо-
швкину БСП шія одержання концентрацій 2Х, X, 1/27.
11/4?, де X- зазначена на етикетці чутливість викори-
гппііваного лізату. Принаймні заключне розведення
<одного ряду має давати негативний результат.
Дхлджують, як описано у розділі "Методика", розве-
зення і негативний контрольний розчин, що скла-
Ьться з води ЛАЛ. Обчислюють середнє логарифмів
н,(нижчої концентрації ендотоксину в кожному ряду
Ьпсіень, для якої виявлено позитивний результат.
Антилогарифм цього середнього дає розрахункову
чутливість лізату. Якщо остання не відрізняється від
ашаченої на етикетці чутливості більш як у два рази,
її зичену на етикетці чутливість вважають підтверд-
*еною і використовують у перебігу всіх випробувань,
иійснюваних із використанням цього лізату.
|Заважаючі фактори. Величина рН розчинів має знахо-
дитись в інтервалі, зазначеному виробником лізату.
Іжічайно цей інтервал рН становить 6.0-8.0. При не-
ічїх дності коригування рН до розчину перед внесен-
ням лізату додають 0.1 Мрозчин кислоти хлористовод-
тюї ЛАЛ, 0.1 М розчин натрію гідроксиду ЛАЛ або
підхожий буферний розчин.
Чинять відповідно до розділу "Чутливість лізату", але
дія приготування розведень стандарту ендотокси-
н\БСПвикористовують випробовувані зразки, в яких
цотоксини не виявлені. Ці зразки використовують у
розведенні, що не перевищує максимально допусти-
мого розведення (МДР), яке обчислюють за форму-
лою
МЛР =
Гранична концентрація ендотоксинів
Чутливість лізату
іе кожну з величин виражають у МО ендотоксину на
мілілітр.
Якщо гранична концентрація ендотоксину в окремій
статті виражена в МО ендотоксину на міліграм
лікарського засобу або на одиницю активності, гра-
ничну концентрацію ендотоксину множать на кон-
центрацію лікарського засобу у випробовуваному роз-
чині (у міліграмах на мілілітр або в одиницях актив-
ності на мілілітр). Якщо необхідно, описану операцію
множення застосовують до розчину випробовуваного
іразка, приготованого відповідно до зазначень на ети-
кетці лікарського засобу.
Якщо чутливість лізату, визначена у присутності ви-
пробовуваного зразка, відрізняється від чутливості,
визначеної за відсутності випробовуваного зразка не
більш як у два рази, останній не містить факторів, що
заважають проведенню ЛАЛ-тесту, і може бути про-
аналізований без наступної обробки. У противному
разі випробовуваний зразок діє як інгібітор або акти-
ватор, і заважаючі фактори усувають за допомогою
відповідної обробки, наприклад, розведення,
фільтрації, нейтралізації, діалізу або додавання речо-
вин, які витісняють адсорбовані ендотоксини. Засто-
сування більш чутливого лізату дозволяє використо-
вувати більш розведений розчин випробовуваного
зразка, і це може допомогти в усуненні заважаючих
факторів.
Якщо випробовуваний зразок не відповідає вимогам
випробування у розведенні, меншому за максимально
допустиме розведення, випробування повторюють,
використовуючи максимально допустиме розведення.
Якщо заважаючий фактор проходить крізь фільтр із
номінальною межею розділення, відповідною
відносній молекулярній масі від 10 000 до 20 000, може
бути використана ультрафільтрація, наприклад, із за-
стосуванням асиметричних мембранних фільтрів із
триацетату целюлози. Фільтри мають бути обов’язко-
во перевірені на наявність компонентів, які виклика-
ють псевдопозитивні результати випробувань. Зали-
шок на фільтрі, що містить ендотоксини, промивають
водою ЛАЛ або підхожим буферним розчином і визна-
чають ендотоксини у воді ЛАЛ або у буферному роз-
чині. Для кожного випробовуваного зразка визнача-
ють об’єм, необхідний для проведення випробування,
і кінцевий об’єм, використовуваний для виявлення
ендотоксинів.
Для того, щоб установити, що вибраний метод оброб-
ки ефективно усуває заважаючі фактори, не видаляю-
чи ендотоксинів, повторюють випробування на на-
явність заважаючих факторів, використовуючи ви-
пробовуваний зразок, до якого доданий ендотоксин
БСП і який потім обробляють вибраним методом.
МЕТОД А - МЕТОД ГЕЛЕУТВОРЕННЯ:
ГРАНИЧНЕ ВИПРОБУВАННЯ
Ендотоксини у випробовуваному зразку. Виконують ви-
пробування з використанням двох паралельних проб
відповідно до розділу "Методика" у розведенні, що не
перевищує максимально допустиме розведення ви-
пробовуваного зразка, у якому, якщо необхідно, усува-
ють заважаючі фактори.
Одночасно досліджують негативний контроль, що
складається з води ЛАЛ, і два позитивних контроля,
кожний з яких містить ендотоксин БСП у концент-
рації, відповідній подвоєному значенню чутливості
лізату (2Х), і один з яких містить випробовуваний зра-
зок (якщо необхідно, оброблений для усунення зава-
жаючих факторів після додавання ендотоксину БСП) у
концентрації, використовуваній у перебігу випробу-
вання. Випробування дійсне, якщо негативний і обид-
ва позитивних контролі дають відповідні результати.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
129
2.6. Біологічні випробування
Зразок відповідає вимогам випробування, якщо в
кожній із двох паралельних проб одержані негативні
результати. Зразок не відповідає вимогам випробуван-
ня, якщо в кожній із двох паралельних проб одержані
позитивні результати. Якщо в одній із проб одержаний
позитивний результат, а в другій — негативний, ви-
пробування повторюють.
МЕТОД В - НАПІВКІЛЬКІСНИЙ МЕТОД
ГЕЛЕУТВОРЕННЯ
Ендотоксини у випробовуваному зразку. Якщо не-
обхідно, у випробовуваному зразку усувають заважа-
ючі фактори. Готують такі розчини:
а) два незалежні паралельні ряди розчинів із чотирь-
ох пробірок, шо містять випробовуваний зразок у
концентраціях 1, 1/2, 1/4 і 1/8 у відношенні до
розведення, для якого доведена відсутність зава-
жаючих факторів. Для одержання розведень вико-
ристовують воду ЛАЛ;
Ь) два паралельні ряди розчинів з чотирьох пробірок,
що містять ендотоксин БСП у концентраціях 2/,
1/2/ 1/4/. Для одержання розведень використо-
вують воду ЛАЛ;
с) два незалежні паралельні розчини, що містять ви-
пробовуваний зразок у розведенні, для якого дове-
дена відсутність заважаючих факторів, і ендоток-
син БСП у концентрації 2/
б) воду ЛАЛ як негативний контроль.
Проводять кількісне визначення відповідно до розділу
"Чутливість лізату". Випробування дійсне у випадку
дотримання таких трьох умов:
— результат, одержаний для води ЛАЛ (б), є негатив-
ним;
— результати, одержані для розчинів (с), є позитив-
ними;
— середнє геометричне значень концентрацій ендо-
токсинів, одержане для розчинів (Ь), знаходиться
у межах від 1/2/ до 2/.
Визначають для кожного ряду розчинів (а) найнижчу
концентрацію зразка, що дає позитивний результат, і,
отже, що містить / МО ендотоксину на мл. Якщо ко-
ефіцієнт розведення випробовуваного зразка, для
якого доведено відсутність заважаючих факторів, ста-
новив ді і цей зразок був розведений далі з ко-
ефіцієнтом д2з одержанням найнижчої концентрації,
що дає позитивний результат, добуток X х ф х с12 доз-
воляє одержати число МО ендотоксину на мілілітр
вихідного розчину випробовуваного зразка. Обчислю-
ють середнє геометричне двох одержаних величин для
цих двох рядів розчинів (а). Зразок витримує випробу-
вання, якщо середнє геометричне значень концент-
рації ендотоксинів менше за граничну концентрацію
ендотоксину, зазначену у відповідній окремій статті.
КІНЕТИЧНІ МЕТОДИ
Яку турбідиметричному кінетичному методі (метод С),
так і в кінетичному методі з використанням хромоген-
ного пептиду (метод О) використовують лінійну рег-
ресію логарифма відклику по відношенню до лога-
рифма концентрації ендотоксинів. Деталі метолу опА Я
сані у розділі "Методика" відповідно для кожногом:-. н
тоду окремо; розділи "Перевірка надійності критерії! ч
для стандартної кривої", "Заважаючі фактори" і "Енд • г
токсини у випробовуваному зразку” належать якії
турбідиметричного кінетичного методу, так і до кіне
тичного методу з використанням хромогенного пеп-С 1
тиду. '
МЕТОД С - ТУРБІДИМЕТРИЧНИЙ
КІНЕТИЧНИЙ МЕТОД В
Методика. Вимірюють час реакції, необхідний дії І
розвитку певного ступеня каламутності розчину; ти
якого додано лізат, із використанням підхожого при В
ладу. Концентрація ендотоксинів у розчині може бути
визначена з логарифма часу реакції за допомого
калібрувальної кривої, побудованої відповідно до І
опису у розділі "Перевірка надійності критеріїв для
стандартної кривої".
Ступінь зміни каламутності у лінійній частині криво
регресії може бути також використаний для вимірю |
вання концентрації ендотоксинів.
МЕТОД О - КІНЕТИЧНИЙ МЕТОД
ХРОМОГЕННОГО ПЕПТИДУ
Методика. Вимірюють час реакції, необхідний для
розвитку певної інтенсивності забарвлення післі,
вивільнення барвника з підхожого хромогенного пеп-
тиду за допомогою комплексу ендотоксин-лізат із ви-
користанням спектрофотометра за певної довжини
хвилі. Концентрація ендотоксинів у розчині може бу-
ти визначена з логарифма часу реакції за допомогою
калібрувального графіка, побудованого відповідно до
опису у розділі "Перевірка надійності критеріїв дія
стандартної кривої".
МЕТОД С І МЕТОД О
Перевірка надійності критеріїв для стандартної кривої.
Необхідна у випадках, коли використовується нова
партія лізату або змінюється будь-яка інша умова, шо
могла б вплинути на результати випробування.
Готують не менше двох незалежних рядів, кожний з
яких складається як мінімум із чотирьох концентрацій
ендотоксину БСП, що знаходяться у межах потрібного
інтервалу. Не рекомендується застосовувати концент-
рації, що виходять за інтервал, зазначений виробни-
ком. Використовують принаймні одну концентрацію
на одиницю за логарифмічною шкалою і негативний
контроль — воду ЛАЛ. У кожну пробірку додають од-
накові об'єми лізату і при методі О відповідний об’єм
хромогенного пептиду. Вимірюють час реакції, як за-
значено вище.
Будують графік логарифма часу реакції як функції ло-
гарифма концентрації ендотоксинів і аналізують рег-
ресію логарифма часу реакції у відношенні до лога-
рифма концентрації ендотоксинів, використовуючи
стандартні методи аналізу (метод найменших квад-
ратів).
130
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
.і'..я регресії для інтервалу концентрацій ендотокси-
<о. означеного виробником лізату, повинна мати зна-
ним нахил і значущу лінійність при рівні значу-
чкгі 95 %>.
ийииіачають кількість логарифмічних рівновіддалених
інюнаентрапій, для яких крива регресії є лінійною. Як-
Ші ця кількість дорівнює трьом (7Ч, Х2 і А3), викорис-
івзютьдругу концентрацію (Х2) як ХП1 при проведенні
випробування на наявність заважаючих факторів і ви-
•ирібування на наявність ендотоксинів у випробовува-
му зразку. Якщо ця кількість становить чотири або
п'ять, якХП1 для зазначених цілей використовують тре-
тю концентрацію (А3).
[ Ні додаток до цих вимог необхідно виконати будь-яку
ж ну вимогу або виконати будь-які інші випробуван-
ні зазначені виробником лізату.
Заважаючі фактори. Готують чотири незалежні пара-
и'.іьні розчини, що містять стандарт ендотоксину в
-.онцентрації А™, і випробовуваний зразок, коефіцієнт
розведення якого обчислюють за формулою:
Гранична концентрація ендотоксину
Значення рН розчинів мають знаходитися в інтервалі,
означеному виробником лізату. Це звичайно дося-
дється при використанні зразка зі значенням рН в
інтервалі від 6.0 до 8.0. Якщо Того необхідно відкори-
вати, до розчину перед внесенням лізату додають
І/ Мрозчин кислоти хлористоводневоїЛАЛ, 0.1 Мроз-
чин натрію гідроксиду ЛАЛ або підхожий буферний
розчин.
Виконують кількісне визначення для цих чотирьох
паралельних розчинів і обчислюють середню концен-
трацію ендотоксинів як антилогарифм середньої лога-
рифмічної концентрації ендотоксинів.
Якшо середня концентрація ендотоксинів становить
не менше 50 % від Хт, випробовуваний зразок не
містить факторів, що заважають активності лізату за
змов проведення випробування; ці зразки можуть бу-
ти випробувані без наступної обробки з метою вида-
тення заважаючих факторів.
Якшо середня концентрація ендотоксинів менша за
50% від Ащ, заважаючі фактори треба видалити, як
описано для методу А.
Якшо середня концентрація ендотоксинів перевищує
найвищу концентрацію у лінійній частині кривої рег-
ресії, випробування повторюють при більш високому
розведенні випробовуваного зразка, яке обчислюють
за формулою:
Гранична концентрація ендотоксину
де^ОиЛ ?-т
Ендотоксини у випробовуваному зразку. Якщо не-
обхідно, у випробовуваному зразку усувають заважа-
ючі фактори. Значення рН розчинів має знаходитися в
інтервалі, зазначеному виробником лізату. Звичайно
цей інтервал рН становить від 6.0 до 8.0. Для коригу-
вання значення рН до розчину перед внесенням ліза-
ту додають 0.1 М розчин кислоти хлористоводне-
вої ЛАЛ, О.1 М розчин натрію гідроксиду ЛАЛ або
підхожий буферний розчин.
Готують такі розчини:
а) два незалежні паралельні розчини випробовува-
ного зразка у розведенні, для якого доведена
відсутність заважаючих факторів;
Ь) два незалежні паралельні розчини, що містять ен-
дотоксин БСПу концентрації Ат або Ат.. і випро-
бовуваний зразок у розведенні, описаному для
розчинів (а);
с) два незалежні паралельні розчини у трьох лога-
рифмічно рівновіддалених концентраціях ендо-
токсину БСП, що перекривають лінійну частину
кривої регресії;
сі) воду ЛАЛ як негативний контроль.
Виконують кількісне визначення, як описано у розділі
"Перевірка надійності критеріїв для стандартної кри-
вої". Обчислюють концентрацію ендотоксинів у кож-
ному з паралельних розчинів (а) і (Ь), використовуючи
криву регресії, одержану для контрольних розчинів
(С).
Випробування дійсне, якщо виконані такі три умови:
— результат для негативного контролю (б) не пере-
вищує межі для контрольної величини, одержаної
при перевірці чутливості лізату;
— результати для контрольної серії (с) відповідають
вимогам, зазначеним у розділі "Перевірка надій-
ності критеріїв для стандартної кривої";
— вміст ендотоксину, обчислений на основі середньо-
го геометричного величин концентрації! ендоток-
синів у розчинах (Ь) після віднімання середнього
геометричного величин концентрацій ендоток-
синів у розчинах (а), становить більше 50 % і мен-
ше 200 %. Обчислюють відсоток вмісту шляхом
ділення результату віднімання на >.п1 або Ат- і по-
множують результат на 100.
Зразок витримує випробування, якщо концентрація
ендотоксинів у кожному із двох розчинів (а) становить
менше Ат або Ат. МО ендотоксину на мілілітр. Якщо
концентрація ендотоксинів в одному із двох розчинів
нижче, а в другому — вище цієї межі, випробування
повторюють. Зразок витримує випробування, якщо
обидва розчини (а) відповідають зазначеній межі.
МЕТОД Е - МЕТОД КІНЦЕВОЇ ТОЧКИ З
ВИКОРИСТАННЯМ ХРОМОГЕННОГО ПЕПТИДУ
Методика. Вимірюють концентрацію барвника, який
вивільнився з підхожого хромогенного пептиду за до-
помогою розчину, що містить ендотоксин, інкубова-
ного з лізатом і хромогенним пептидом, використову-
ючи спектрофотометр за певної довжини хвилі. Кон-
центрація ендотоксинів у розчині може бути розрахо-
вана з величини оптичної густини за вибраної довжи-
ни хвилі за допомогою калібрувального графіка, побу-
дованого відповідно до опису у розділі "Перевірка
надійності критеріїв для стандартної кривої".
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
131
2.6. Біологічні випробування
Перевірка надійності критеріїв для стандартної кривої.
Проводиться у випадках, коли використовується нова
партія лізату або змінюється будь-яка інша умова, яка
могла б вплинути на результати випробування.
Готують чотири незалежні ряди розведень ендотокси-
ну БСП у воді ЛАЛ, які перекривають інтервал, зазна-
чений виробником лізату. Використовують холостий
реактив, приготований згідно з інструкцією виробни-
ка лізату.
Зазначений об’єм лізату і хромогенного пептиду вно-
сять у кожну пробірку і інкубують протягом часу, за-
значеного виробником лізату.
Зупиняють реакцію і вимірюють оптичну густину за
певної довжини хвилі.
Будують графік залежності оптичної густини для кож-
ної концентрації чотирьох паралельних рядів від кон-
центрації ендотоксинів і аналізують регресію оптичної
густини у відношенні до концентрації, використовую-
чи стандартні методи аналізу (метод найменших квад-
ратів).
Лінія регресії для інтервалу концентрацій ендоток-
синів, зазначеного виробником лізату, повинна мати
значущий нахил і значущу лінійність при рівні значу-
щості 95 %.
Визначають концентрацію ендотоксинів ХП1, що є се-
реднім арифметичним найвищої (Х^) і нижчої (^) кон-
центрацій ендотоксинів, для яких крива регресії є
лінійною; при цьому всі величини виражають у МО
ендотоксинів на мілілітр.
На додаток до цих вимог необхідно виконати будь-яку
іншу вимогу або виконати будь-які інші випробуван-
ня, зазначені виробником лізату.
Заважаючі фактори. Готують чотири незалежні пара-
лельні розчини, що містять ендотоксин БСП у кон-
центрації Хт , і випробовуваний зразок, коефіцієнт
розведення для якого обчислюють за формулою:
Гранична концентрація ендотоксину
Лп
Значення рН розчинів має знаходитися в інтервалі, за-
значеному виробником лізату. Це звичайно дося-
гається при використанні зразка з рН в інтервалі від
6.0 до 8.0. Для коригування значення рН до розчину
перед внесенням лізату додають ОЛ М розчин кислоти
хлористоводневої ЛАЛ, ОЛ М розчин натрію гідроксиду
ЛАЛ або підхожий буферний розчин.
Виконують кількісне визначення для цих чотирьох
паралельних розчинів і обчислюють середню концен-
трацію ендотоксинів.
Якщо середня концентрація ендотоксинів становить
не менше 50 % і не більше 200 % від А,т, випробовува-
ний зразок не містить заважаючих факторів і може бу-
ти досліджений без наступного їх усунення.
Якщо середня концентрація ендотоксинів менше
50 % або більше 200 % від А,т, заважаючі фактори тре-
ба усунути, як описано у методі А.
Якщо середня концентрація ендотоксинів перевииИ
найвищу концентрацію у лінійній частині кривоїр ♦
ресії, випробування повторюють при більш високої
розведенні випробовуваного зразка, яке обчислюю»
за формулою:
Гранична концентрація ендотоксину
Л-т'
де Х| < 2.т' <
Ендотоксини у випробовуваному зразку. Якщо ні
обхідно, у випробовуваному зразку усувають заважЛ
ючі фактори. Значення рН розчинів має знаходитися1*
інтервалі, зазначеному виробником лізату. Звичайне
цей інтервал рН становить від 6.0 до 8.0. Для корит
вання рН перед додаванням лізату додають ОЛ МряЯ
чин кислоти хлористоводневої ЛАЛ, 0.1 М розвдіїї
натрію гідроксиду ЛАЛ або підхожий буферний роЛ
чин.
Готують такі розчини:
а) два незалежні паралельні розчини випробовув»
ного зразка у розведенні, для якого доведені
відсутність заважаючих факторів;
Ь) два незалежні паралельні розчини, що містять
ендотоксин БСП у концентрації Хт або А.т., і ви-
пробовуваний зразок у розведенні, описаному дл»
розчинів (а);
с) два паралельні розчини ендотоксину БСП у кон-
центрації і два паралельні розчини ендоток-
сину БСП у концентрації Х|ї
сі) воду ЛАЛ як негативний контроль.
Виконують кількісне визначення, як описано у розділі
"Перевірка надійності критеріїв для стандартної кри-1
вої". Визначають оптичну густину після інкубування
кожного з паралельних розчинів (а). (Ь), (с) і (сі). Ви-
користовують оптичну густину розчинів (с) і (ф для І
побудови лінії регресії і обчислюють концентрації ен-
дотоксину розчинів (а) і (Ь).
Випробування дійсне, якщо виконані такі три умови:
— результат у негативному контролі (сі) не переви-
щує величини у контрольній (холостій) пробі.!
одержаній при контролі чутливості лізату;
— результати у контрольних розчинах (с) відповіда-
ють калібрувальному графіку, використовуваному
при контролі чутливості лізату;
— вміст ендотоксину, обчислений на основі серед-
нього арифметичного величин концентрацій ен-
дотоксину в розчинах (Ь) після віднімання серед-
нього арифметичного величин концентрацій ен-
дотоксину в розчинах (а), становить більше 50%
і менше 200 %. Обчислюють відсоток вмісту шля-
хом ділення результату віднімання на ХП1 або ).т
і помножують результат на 100.
Зразок витримує випробування, якщо концентрація
ендотоксинів кожного з двох розчинів (а) становить
менше Хт або МО ендотоксину на мілілітр. Якщо
концентрація ендотоксинів в одному з двох розчинів
нижче, а в другому — вище цієї межі, випробування
повторюють. Зразок витримує випробування, якщо
обидва розчини (а) відповідають зазначеній межі.
132
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
|*купний розділ наведено тільки для інформації і як
..мицтво; він не є обов’язковим при проведенні ви-
цЙут на бактеріальні ендотоксини згідно з цією
І акопеєю.
ВИПРОБУВАННЯ НА
МКТЕРІАЛЬНІ ЕНДОТОКСИНИ.
РЕКОМЕНДАЦІЇ
І ВСТУП
Ігютоксини, джерелом яких є грамнегативні мікро-
ііргінізми, є найбільш розповсюдженою причиною
іі[чгенних токсичних реакцій при забрудненні ними
мрських засобів; їхня пірогенна активність набагато
л«ша за активність більшості інших пірогенних речо-
иин. Ці ендотоксини є ліпополісахаридами. Незважа-
вчи на те, що існує незначна кількість пірогенів іншої
(і іічної природи, які мають різну структуру, звичайно
оме відсутність бактеріальних ендотоксинів у
перському засобі має на увазі відсутність пірогенних
хипонентів.
Ндавність ендотоксинів у лікарському засобі може ма-
імватися факторами, що викривлюють реакцію між
ендотоксинами і лізатом амебоцитів. Отже, при ба-
жанні замінити передбачене окремою статтею випро-
бування на пірогени на кроликах випробуванням на
оіктеріальні ендотоксини необхідно довести, що це
апробування може бути здійснене для даного
іікарського засобу; це може спричинити процедуру
усунення заважаючих факторів.
Перш ніж розглядати можливість використання ви-
їробування на бактеріальні ендотоксини стосовно до
конкретного лікарського засобу, необхідно одержати
гаку інформацію.
11. Установити придатність матеріалів, використову-
юнихдля проведення випробування. Має бути гаран-
тована відсутність ендотоксинів у воді ЛАЛ та інших
.«активах; необхідно перевірити заявлену виробни-
ком чутливість лізату амебоцитів.
1.2. Оскільки випробовуваний лікарський засіб може
бути перешкодою для результатів випробування, чут-
тивість лізату визначають у присутності та у відсут-
ності цього лікарського засобу. Між двома значення-
ми чутливості не має бути суттєвої різниці.
У випробуванні на бактеріальні ендотоксини мають
зазначатися методи усунення заважаючих факторів
(див. "Методи гелеутворення"); при наявності заважа-
ючих факторів треба провести інше випробування
після того, як такий метод буде застосований для пе-
того, чи справді усунені перешкоди.
Якщо лікарський засіб не витримує випробування
(позитивний результат), дозволяється провести по-
вторне випробування. Уявна невідповідність лікарсь-
кого засобу вимогам випробування може бути викли-
кана помилками у приготуванні чи розведенні або
іншим випадковим забрудненням, внесеним у процесі
проведення випробування.
Даний розділ пояснює причини вимог випробування
на бактеріальні ендотоксини і стосується одержання
та інтерпретації результатів.
Заміна випробування на пірогени на кроликах ЛАЛ-
тестом фактично означає використання альтернатив-
ного методу аналізу і, отже, потребує валідації; у дано-
му розділі наведено деякі зазначення про те, як треба
чинити.
У статті на лікарський засіб зазначається основний
метод проведення випробування на бактеріальні ен-
дотоксини. Якщо конкретний метод не зазначений як
основний, використовують метод А. Якщо передба-
чається використати метод, відмінний від основного,
необхідно довести, що цей метод є придатним для да-
ного лікарського засобу й дає результати, які узгоджу-
ються з одержаними за основним методом (див. також
п. 11 "Заміна випробування на пірогени на кроликах").
Хоча випробування на бактеріальні ендотоксини пе-
редбачає використання як джерела лізату вид Ілтиіиз
роїурИетиа, активний лізат можна одержати також із
близькоспоріднених видів, таких, які належать до
Таскуріеия. Термін "лізат амебоцитів" використаний у
даній статті для позначення лізату амебоцитів (ЛАЛ-
реактиву), що пройшов валідацію, незалежно від його
біологічного походження.
2. МЕТОД
Додавання ендотоксинів до лізату амебоцитів може
призвести до появи каламутності, осадження або
утворення гелю; як кінцеву точку при проведенні ви-
пробування на бактеріальні ендотоксини методами А і
В використовують лише гелеутворення. Перевагою у
цьому випадку є простота вирішення того, чи витри-
мав зразок лікарського засобу випробування на основі
наявності або відсутності гелеутворення, видимого
неозброєним оком. Кількісні методи, описані як ме-
тоди С, О, Е, були розроблені пізніше; для їх виконан-
ня необхідна більша кількість обладнання, але їх ,гт-
ше автоматизувати для цілей регулярних випробувань
великих кількостей зразків одного і того самого
лікарського засобу.
Ендотоксини можуть адсорбуватися на поверхні
пробірок і піпеток, виготовлених із деяких видів плас-
тика або типів скла. Можуть виникнути перешкоди,
зумовлені вивільненням речовин із пластичних ма-
теріалів. Використовувані матеріали треба перевіряти;
наступні партії пробірок або піпеток можуть трохи
відрізнятися за складом і, отже, рекомендується по-
вторювати такі випробування, починаючи працювати
з новими партіями матеріалів.
Результат випробування на пірогени на кроликах за-
лежить від дози пірогену, результат випробування на
бактеріальні ендотоксини залежить від концентрації
ендотоксину в реакційній суміші. Рішення викорис-
тати випробування на бактеріальні ендотоксини як
граничного випробування має на увазі, по-перше,
що для лікарських засобів, які підлягають випробу-
ФАРМАКОПЕЯ УКРАЇНИ 2001
133
2.6. Біологічні випробування
ванню, треба визначити граничну концентрацію ен-
дотоксинів і, по-друге, необхідно знати, чи переви-
щує концентрація ендотоксинів у випробовуваному
зразку цю порогову концентрацію, чи її значення
нижче за цю величину. Кількісні методи С, В, Е роб-
лять можливим визначення концентрації ендоток-
синів у випробовуваному зразку, але при проведенні
контролю якості за рутинною методикою заключне
питання полягає у тому, чи перевищує ця концент-
рація певну межу.
При встановленні граничної концентрації ендотокси-
ну для випробовуваного зразка треба приділити на-
лежну увагу дозі випробовуваного лікарського засобу
для людини. Мета цього полягає в тому, щоб гаранту-
вати, що доти, доки концентрація ендотоксинів у
зразку залишається нижче цієї межі, навіть макси-
мальна доза лікарського засобу, уведена зазначеним
шляхом за годину, не буде містити такої кількості ен-
дотоксинів, яка викликає токсичну реакцію.
Якщо концентрація ендотоксинів у зразку дорівнює
граничній величині, як і в тому випадку, коли концен-
трація ендотоксинів значно вище цієї величини,
відбувається гелеутворення, і зразок не проходить ви-
пробування, оскільки характер випробування "усе або
нічого" робить неможливим розмежувати концент-
рацію, яка точно дорівнює граничній концентрації, і
більш високу концентрацію. Лише у тому випадку, ко-
ли гелеутворення не спостерігається, можна зробити
висновок, що концентрація ендотоксинів нижче гра-
ничної концентрації.
Для лікарських засобів у твердому стані цю граничну
концентрацію ендотоксину на одиницю маси або на
одиницю активності лікарського засобу треба переве-
сти у концентрацію ендотоксинів на мілілітр розчину,
який підлягає випробуванню, оскільки випробування
може бути проведене лише для розчину. Випадок, ко-
ли лікарські засоби вже перебувають у рідкому стані
(наприклад, розчини для внутрішньовенних вливань),
буде обговорений нижче.
Для визначення граничної концентрації ендотоксину
в МО ендотоксину на одиницю маси або на одиницю
активності необхідно визначити такі величини:
М — максимальна доза лікарського засобу для до-
рослого в одиницях маси (або одиницях актив-
ності) на кілограм маси тіла за годину при вве-
денні лікарського засобу зазначеним шляхом.
При встановленні максимальної дози для до-
рослого як масу тіла дорослої людини беруть
величину 70 кг. Педіатричну дозу на кілограм
маси тіла за годину треба використовувати, як-
що вона вища за відповідну максимальну дозу
для дорослого.
К — максимальна доза ендотоксину в МО ендоток-
сину на кілограм маси тіла за годину, яку
пацієнт може одержати при введенні лікарсь-
кого засобу зазначеним шляхом без будь-якого
несприятливого ефекту (Табл. 2.6.14.-1).
Припустимо, що є розчин для проведення випробу-
вання, що містить с мг (або одиниць активності)
лікарського засобу на мілілітр. Тоді об’єм М/с мл -
об’єм, що містить максимальну дозу М. Якщо!
об’єм містить К МО ендотоксину, випробування .
давати позитивний результат.
Отже, гранична концентрація ендотоксинів (ГКЕї
МО ендотоксину на мілілітр, що еквівалентна ц
ничній концентрації ендотоксину на міліграм або
одиницю активності лікарського засобу у твердо
стані, дорівнює:
де:
К — максимально допустима доза ендотоксину
МО на кілограм маси тіла за годину;
с — концентрація розчину в міліграмах або одини -
цях активності на мілілітр;
М — максимальна доза лікарського засобу в мікро- і
грамах або одиницях активності на кілограмз<|
годину.
Для рідких лікарських засобів максимальну дозушх*
дорослого на кілограм маси тіла за годину виражаю» В
у мілілітрах. Наведений вище вираз для граничні
концентрації ендотоксину застосовний і до таки І
лікарських засобів, за умови, що Мзаміняють величи-
ною максимальної дози в мілілітрах на кілограм мас; ।
тіла за годину, асе її величиною.
Раніше граничну концентрацію ендотоксину визнала-1
ли як "Максимально допустиму концентрацію ендо-
токсину" або "МДКЕ". Однак, на практиці зразок, шс
містить точно МДКЕ ендотоксину, не витримав би ви-
пробування так само, як і зразок, шо містить більше
ендотоксину. Єдиний шлях гарантувати, що МДКЕ1.1
зразку не перевищена, полягає в тому, щоб продемон-1
струвати, що концентрація ендотоксинів у зразку мен-
ша за МДКЕ; отже, більш логічно використовувати
термін "гранична концентрація ендотоксинів" (або
ГКЕ) як концентрації ендотоксинів, що не має бути
досягнута.
Гранична концентрація ендотоксинів залежить від
лікарського засобу і наводиться в окремих статтях.
Значення Л'наведені в Табл. 2.6.14.-1.
Таблиця 2.6.14.-І
Шлях уведення К МО ендотоксину на кілограм маси тіла за годину
Внутрішньовенно 5.0
Внутрішньовенно, для радіофармацевтичних лікарських засобів 2.5
Інтратекально 0.2
Яке розведення лікарського засобу треба використо-
вувати у випробуванні для того, щоб бути впевненими
у тому, що негативний результат випробування
свідчить про те, що концентрація ендотоксинів у ньо-
му нижче ГКЕ, а позитивний результат означає, що
134
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
викачається принаймні, ГКЕ? Ступінь розведення у
ому випадку залежить від ГКЕ і чутливості лізату;
іін називається "Максимально допустимим розведен-
ням (МДР), і його величину можна одержати розра-
хунковим шляхом за формулою:
Ж
і, - заявлена виробником чутливість лізату в МО
ендотоксину на мілілітр.
Якшо величина максимально допустимого розведен-
ня не є цілим числом, для рутинних цілей можна ви-
користовувати найближче ціле число, менше МДР
цію означає, розчин лікарського засобу розводять у
меншому ступені за МДР). У такому випадку негатив-
ний результат випробування показує, що концент-
,тшія ендотоксинів у зразку нижче граничної величи-
ні!. А проте, якщо концентрація ендотоксинів у зразку
ми проведенні випробування нижче ГКЕ, але достат-
ньо висока для того, щоб реакція з лізатом призвела до
утворення гелю, випробування за цих умов може бути
позитивним. Отже, якщо випробування з таким "зруч-
нім" коефіцієнтом розведення є позитивним, зразок
треба розбавити до МДР і повторити випробування. У
випадку будь яких сумнівів треба використовувати
МДР.
Не підкреслює важливість підтвердження чутливості
лізату.
Розведення до 41.67 мл необхідне також у тих випад-
ках, коли випробування виконують для перевірки
сумнівних результатів.
3. СТАНДАРТНИЙ ПРЕПАРАТ
Як стандартний препарат використовують ендоток-
син БСП. Його кількісний аналіз проведений
порівняно з Міжнародним стандартом ВООЗ, а його
активність виражена у Міжнародних Одиницях (МО)
ендотоксину на мілілітр. Міжнародна Одиниця ендо-
токсину визначається як специфічна активність пев-
ної маси Міжнародного Стандарту.
Для рутинних цілей може бути використаний інший
стандартний препарат ендотоксину, за умови, що про-
ведений його кількісний аналіз порівняно з Міжна-
родним Стандартом ендотоксину або ендоток-
сином БСПї його активність виражена в Міжнародних
Одиницях ендотоксину.
Флакон стандарту ендотоксину звичайно містить
більше речовини, ніж необхідно для одного випробу-
вання. Не виявлено втрати активності стандарту ендо-
токсину, якшо його ампули розкриті у камері з
ламінарним потоком і зберігалися при температурі
4 °С протягом періоду до двох тижнів закриті під-
хожим матеріалом після розкриття. Проте, рекомен-
дується перевіряти активність стандарту ендотоксину,
якшо передбачається тривале використання відкритих
флаконів.
Приклад
Треба провести випробування для розчину 50 мг/мл
натрію фенітоїну, призначеного для внутрішньовен-
ного уведення. Визначають МДР, задаючи такі зна-
ення змінних:
Л/ — максимальна доза для людини становить 15 мг
на кілограм маси тіла за годину;
с — 50 мг/мл;
К — 5 МО ендотоксину на кілограм маси тіла за
годину;
і. — 0.4 МО ендотоксину на мілілітр.
ГКЕ і МДР у випробовуваному розчині становлять:
5-50
15
МДР =
ГКЕ
X
5-50 1
------------= 41.67
15 0.4
Для рутинних випробувань цього лікарського засобу
може бути доцільним розбавити 1 мл випробовувано-
го розчину до 20 мл (величина МДР/2, заокруглена до
більш низького цілого числа). Однак якщо при цьому
випробування дасть позитивний результат, необхідно
розвести 1 мл до 41.67 мл і повторити випробування.
4. ВОДА ЛАЛ
Визначення відсутності ендотоксину в цьому реактиві
у випробуванні на пірогени на кроликах заперечене з
практичних і теоретичних причин:
— кролик не має чутливості, достатньої для того,
щоб визначити ендотоксин у воді ЛАЛ, призна-
ченій для проведення випробувань для зразків з
дуже низькою граничною концентрацією ендо-
токсинів;
— внаслідок відносно низької точності температур-
ної реакції у кроликів потрібно було б багато по-
вторних випробувань;
— терміни "пірогени" і "ендотоксини" позначають
групи речовин, які не повністю співпадають одна
з одною.
При описі випробування на бактеріальні ендотоксини
показано, що для приготування води ЛАЛ можуть бути
використані методи, відмінні від потрійної перегонки.
Із хорошими результатами було використано метод
зворотного осмосу. Можна віддати перевагу дистилю-
ванню води більше трьох разів. Який би метод не ви-
користовувався, одержаний реактив має бути вільним
від визначуваних ендотоксинів.
5. рН СУМІШІ
Оптимальне гелеутворення суміші у випробуванні на
бактеріальні ендотоксини спостерігається при
рН 6.0-7.5. Однак додавання лізату до зразка може
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
135
2.6. Біологічні випробування
призвести до зниження рН. Щоб бути певним, 1ЦО
рН суміші не нижче 6.0, треба переконатися у тому,
що рН зразка, який підлягає випробуванню, не мен-
ше 6.5.
6. ВАЛІДАЦІЯ ЛІЗАТУ
При приготуванні розчинів лізату важливо дотримува-
тись інструкції виробника.
Позитивні коефіцієнти розведення у гелеутворюючих
методах А і В переводять у логарифми. Причина цього
полягає в тому, що, якщо накреслити криву розподілу
за частотою цих логарифмічних величин, вона зви-
чайно наближається до кривої нормального розподілу
набагато ближче за розподіл за частотою самих ко-
ефіцієнтів розведення; фактично вони настільки
подібні, що допускається використання нормального
розподілу за частотою як математичної моделі й об-
числення меж, взятих за основу порівняння, за допо-
могою /-критерію Стьюдента.
При застосуванні кінетичних методів Є і О викорис-
товують логарифм концентрації ендотоксинів,
оскільки можна використовувати модель лінійної рег-
ресії логарифма часу реакції відносно до логарифма
концентрації ендотоксинів. При застосуванні хромо-
генного методу кінцевої точки Е використовують іншу
модель. У цьому випадку результат (оптична густина
барвника, шо вивільнився) можна розглядати як
лінійну функцію концентрації ендотоксинів у викори-
стовуваному інтервалі концентрацій.
7. ПОПЕРЕДНЄ ВИПРОБУВАННЯ
НА ЗАВАЖАЮЧІ ФАКТОРИ
Деякі лікарські засоби не можна піддавати випробу-
ванню на наявність ендотоксинів безпосередньо, бо
вони не змішуються з реактивами, не можуть бути до-
ведені до рН 6.5-7.5 або уповільнюють або активують
утворення гелю. Отже, потрібне проведення поперед-
нього випробування для перевірки наявності заважа-
ючих факторів. Якщо вони виявлені, необхідно проде-
монструвати, що процедура їхнього видалення є ефек-
тивною.
Мета попереднього випробування — перевірити
нульову гіпотезу про те, що чутливість лізату у при-
сутності випробовуваного зразка не відрізняється
значно від його чутливості у відсутності зразка. У ме-
тодах А і В використовують простий критерій: нульо-
ва гіпотеза приймається, якщо чутливість лізату у
присутності зразка становить принаймні 0.5 чутли-
вості самого лізату і не більше як удвічі перевищує
цю величину.
Класичним підходом було б обчислення середніх зна-
чень логарифма коефіцієнта розведення для чутли-
вості у відсутності і у присутності зразка й перевірка
різниці між двома середніми значеннями за допомо-
гою /-критерій Стьюдента.
Випробування на заважаючі фактори у гелеутворк
чих методах А і В вимагає використання зра
лікарського засобу, в якому ендотоксини не виявлі
Це являє теоретичну проблему, якщо необхідно
пробовувати зовсім новий лікарський засіб, і
кількісних методів С, О і Е передбачений інн
підхід.
8. УСУНЕННЯ ЗАВАЖАЮЧИХ ФАКТОРІВ
Способи усунення заважаючих факторів не маї
призводити до підвищення або зниження кількі
ендотоксину у випробовуваному зразку (наприкл
до зниження в результаті адсорбції). Для перевір
цього до випробовуваного зразка додають відс
кількість ендотоксину і потім після усунення завж
ючих факторів вимірюють кількість виявленого єн
токсину.
Методи С і И. Якщо властивості випробовуваного
лікарського засобу справляють заважаючу дію, яку- не
можна усунути класичними методами, можлива побу-
дова стандартної кривої для аналогічного лікарського
засобу, звільненого від ендотоксинів за допомогою на-
лежної обробки або розведення. Потім проводять ви-
пробування на ендотоксини порівняно з цією стан-
дартною кривою.
Установлено, що у багатьох випадках достатньою е
ультрафільтрація крізь асиметричні мембранні
фільтри з триацетату целюлози, описана у випробу-
ванні на бактеріальні ендотоксини. Фільтри маюті
відповідним чином пройти валідацію, оскільки іноді
похідні целюлози (р-О-глюкани) можуть викликати
помилково позитивні результати.
Установлено, що полісульфонові фільтри є непри-
датними і при їх використанні були одержані помнп-
ково позитивні результати.
9. МЕТА ПРОВЕДЕННЯ КОНТРОЛЬНИХ
ВИПРОБУВАНЬ
Метою позитивного контролю, що містить воду ЛАЛі
стандартний препарат ендотоксину з концентрацією,
шо у два рази перевищує зазначену на етикетці чут-
ливість лізату, є підтвердження активності лізату при
проведенні випробування за передбачених для цього
умов. Метою негативного контролю є підтвердження
відсутності визначуваної концентрації ендотоксину у
воді ЛАЛ.
Другий позитивний контроль, що містить випробову-
ваний зразок у концентрації, використовуваній у ви-
пробуванні, призначений для того, щоб показати
відсутність інгібуючих факторів за умов проведення
випробування.
10. ОБЛІК ТА ІНТЕРПРЕТАЦІЯ РЕЗУЛЬТАТІВ
Як зазначено вище, випробування з використанням
лізату амебоцитів використовують як граничне випро-
бування, і вибір межі залежить від описаних факторів.
Незначні кількості ендотоксинів у воді ЛАЛ або у
136
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.6. Біологічні випробування
ібць-якому іншому реактиві або матеріалі, впливу
><чго піддається ЛАЛ-реактив при проведенні випро-
міння, можуть не визначатися доти, доки вони не
..дніть межі чутливості лізату. Однак вони можуть
щвишувати кількість ендотоксину в розчині, що
ч діть випробовуваний зразок, до величини, що лед-
е перевищує межу чутливості, і викликати позитивну
ІКЦІЮ.
ІКТОГО, що це відбудеться, може бути знижений
шіяхом перевірки води ЛАЛта інших реактивів за до-
нйогою найбільш чутливого з усіх наявних лізатів або
іуцнаймні більш чутливого за лізат, використовува-
н.ипри випробуванні зразка. Навіть у цьому випадку
гкшк такого "помилково позитивного результату" не
можна повністю виключити. Однак треба розуміти.
у цьому відношенні методика випробування є
"спечною шодо невдачі", на відміну від методики,
юпускає випробування, що дає помилково нега-
гивний результат, шо може призвести до випуску не-
юброякісного лікарського засобу, небезпечного для
цоров'я пацієнта.
.1 ЗАМІНА ВИПРОБУВАННЯ НА ПІРОГЕНИ НА
КРОЛИКАХ НА ВИПРОБУВАННЯ НА
ЕНДОТОКСИНИ
Окремі статті на лікарські засоби, призначені для па-
рентерального застосування, що можуть містити ток-
.ичні кількості бактеріальних ендотоксинів, вимага-
ють проведення або випробування на пірогени на
кроликах, або випробування на бактеріальні ендо-
токсини. Якщо зазначене проведення випробування
на бактеріальні ендотоксини і жоден із п'яти методів
(від А до Е), описаних у даній статті, не зазначений,
•дя такого лікарського засобу вважається дійсним
граничне випробування за методом А. Якщо
.значено про один з інших методів (від В до Е), тоді
йме він є дійсним для даного лікарського засобу.
Заміна випробування на пірогени на кроликах ви-
пробуванням на бактеріальні ендотоксини або заміна
існуючого методу випробування на бактеріальні ен-
дотоксини іншим методом має розглядатися як вико-
ристання альтернативного методу при заміні фарма-
копейного випробування, як описано у розділі /. "За-
ший зауваження
"Випробування та методики кількісного визначення,
наведені у Фармакопеї, є офіційними методиками,
проте за узгодженням із компетентними уповноваже-
ними органами можуть використовуватися й інші ме-
тодики, за умови, що ці методики дають результати,
які відповідають фармакопейним методикам. У ви-
падку сумнівів або розбіжностей вирішальною є фар-
макопепна методика".
Як методичні рекомендації для валідації методу ви-
пробування на бактеріальні ендотоксини, відмінного
від зазначеного в окремій статті, пропонуються такі.
II.І. Методики, матеріали і реактиви, використову-
вані згідно з даним методом, мають пройти валідацію,
як описано для даного випробування.
11.2. Наявність заважаючих факторів (і якщо не-
обхідно спосіб їхнього видалення) треба випробовува-
ти на зразках, відібраних принаймні з трьох виробни-
чих серій. Треба мати на увазі, що для методів О і Е, в
яких використовують хромогенний пептид, потрібні
реактиви, що не застосовуються для методів А, В або
С, і, отже, відповідність методів А, В або С вимогам
відносно заважаючих факторів не можна екстраполю-
вати на метод О або метод Е без додаткового випробу-
вання.
1ЕЗ. Якщо є в наявності зразки з виробничих серій,
шо дають позитивний результат при випробуванні ме-
тодами, передбаченими в окремій статті Фармакопеї,
їх треба випробовувати також і методом, призначеним
для використання як альтернативним. Якщо таких
зразків немає, порівняння альтернативного методу з
методом, передбаченим окремою статтею Фармакопеї
для тих самих зразків, є марним.
12. ВАЛІДАЦІЯ ВИПРОБУВАННЯ ДЛЯ НОВИХ
ЛІКАРСЬКИХ ЗАСОБІВ
Рекомендації, описані в п.п. 11.1 і 11.2, треба застосо-
вувати до всіх нових лікарських засобів, призначених
для парентерального застосування, і таких, що підля-
гають випробуванню на наявність бактеріальних ен-
дотоксинів, відповідно до вимог Фармакопеї.
13. НОВІ ЛІКАРСЬКІ ЗАСОБИ, ШО ВПЛИВАЮТЬ
НА ТЕМПЕРАТУРУ ТІЛА
Якщо на стадії розробки показано, що новий лікарсь-
кий засіб може впливати на температуру тіла, то для
доказу відсутності бактеріальних ендотоксинів можна
використовувати один із методів (від А до Е) Особли
во корисну інформацію може надати кількісний метод
(В, С, Е) або Е). У такому випадку чинять відповідно
до вказівок п.п. 11.1 і 11.2. Однак, якщо є ознаки за-
бруднення зразка пірогенними речовинами, що не є
ендотоксинами, необхідно одержати інформацію на
основі більш широких випробувань.
__________________________________________N
3. СТАНДАРТНИЙ ПРЕПАРАТ
— Як ендотоксин БСПдопускається також викорис-
товувати стандартний препарат ендотоксину, калібро-
ваний у Ендотоксинових Одиницях (ЕО або ЕО)
порівняно зі Стандартним Препаратом Ендотоксину
США (К8Е) (1 ЕО (ЕО) дорівнює 1 МО)). Чутливість
використовуваного лізату також має бути виражена в
Ендотоксинових Одиницях (ЕО або ЕО), а всі описані
вище розрахункові величини також мають бути вира-
жені в Ендотоксинових Одиницях.
— Рекомендується, щоб ендотоксин БСП і лізат, ви-
користовувані у випробуванні, були виготовлені од-
ним виробником. При цьому обов'язкова наявність
сертифіката аналізу стандартного препарату
порівняно з Міжнародним стандартом ВООЗ або зі
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
137
2.6. Біологічні випробування
Стандартним Препаратом Ендотоксину США з вико-
ристанням даного лізату.
— Допускається використання інших валідованих
процедур калібрування ендотоксину БСП.
— Допускається використання готових реакти-
вів (0.1 М розчину кислоти хлористоводневої ЛАЛ і
0.1 Мрозчину натрію гідроксиду ЛАЛ) із маркування^
"Вільний від ендотоксинів". Кожний із реактивів в
придатним, якщо після доведення рН до 6.О-8.0 ві
дає негативний результат за умов проведення випр»І
бувай ня.
138
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ
2.7. Біологічні методи кількісного визначення
1.1. БІОЛОГІЧНІ МЕТОДИ
КІЛЬКІСНОГО ВИЗНАЧЕННЯ
112. КІЛЬКІСНЕ ВИЗНАЧЕННЯ
АНТИБІОТИКІВ МІКРОБІОЛОГІЧНИМ
МЕТОДОМ
Активність антибіотиків визначають шляхом по-
ГІЬіЯННЯ ступеня пригнічення росту чутливих мікро-
нізмів у результаті дії випробовуваного антибіоти-
тандартного зразка у відомих концентраціях.
тіндаріні зразки, використовувані для кількісного
| ачення. являють собою речовини, активність
точно встановлена відповідно до міжнародного
ІВКіартного препарату.
•. ькісне визначення треба проводити таким чином,
а >5 мати можливість підтвердити придатність мате-
і і.ичної моделі, на якій грунтується розрахунок ак-
тивності. При використанні моделі паралельних ліній
.і лінії, що характеризують залежність "логарифм до-
» відповідь (або перетворена відповідь)" для випро-
буваного і стандартного зразків, мають бути пара-
. ьні; залежність має бути лінійною у всьому діапа-
«іі концентрацій, використовуваних при розрахун-
ки. Виконання цих умов треба підтвердити для зада-
і.тго рівня ймовірності, звичайно Р = 0.05. Допус-
кався використання інших моделей, наприклад, мо-
лі співвідношення коефіцієнтів регресії, за умови,
. чх придатність обгрунтована.
Лхшо немає інших зазначень в окремій статті, при
А якісному визначенні активності довірчі інтервали
іР = 0.05) мають становити не менше 95 % і не більше
1115 "Ь від установленої активності.
«, зькісне визначення проводять, використовуючи
метод А або метод В.
\. МЕТОД ДИФУЗІЇ
Живильне середовище, рекомендоване ДЛЯ КІЛЬКІСНО-
ГО визначення, розплавляють і вносять в нього при
(дповідній температурі, наприклад, від 48 °С до 50 °С
іля вегетативних форм, певну кількість суспензії
мікроорганізмів, чутливих до даного антибіотика.
Кількість суспензії має бути такою, шоб забезпечувати
ііткі, підхожого діаметра зони пригнічення росту
тест-мікроорганізму для всіх концентрацій антибіоти-
к.:, використовуваних при кількісному визначенні.
Негайно після внесення тест-мікроорганізму живиль-
не середовище розливають у чашки Петрі або великі
прямокутні чашки так, щоб у них утворився од-
норідний шар завтовшки від 2 мм до 5 мм. Допус-
кається розливати середовище у два шари, з яких іно-
кульовано лише верхній. Чашки т реба зберігати таким
чином, щоб до їх використання не спостерігався ріст
або загибель мікроорганізмів і щоб на момент викори-
стання поверхня живильного середовища була сухою.
Використовуючи зазначені в Табл. 2.7.2.-1 розчин-
ник і буферний розчин, готують розчини стандартно-
го зразка з відомими концентраціями і розчини ви-
пробовуваного антибіотика, передбачувані концент-
рації яких не мають істотних відмінностей від відпо-
відних концентрацій стандартного зразка. Розчини
наносять на поверхню середовища, використовуючи
стерильні циліндри з фарфору, нержавіючої сталі або
іншого підхожого матеріалу, або вносять розчини в
лунки, підготовані в густому живильному середовищі.
У всі циліндри або лунки вносять рівні об’єми роз-
чинів. Допускається використовувати стерильні диски
з фільтрувального паперу підхожої якості, які просо-
чують розчином стандартного зразка і випробовува-
ного лікарського засобу і помішають на поверхню жи-
вильного середовища.
Щоб мати можливість оцінити придатність методики
кількісного визначення, треба використовувати не
менше трьох доз стандартного зразка і трьох відпо-
відних доз випробовуваного антибіотика, шо мають
здогадно ту саму активність. Бажано, щоб дози скла-
дали геометричну прогресію. При проведенні
постійних кількісних визначень, для яких лінійність
системи була продемонстрована на достатньо великій
кількості тридозових визначень, допускається вико-
ристовувати дводозовий варіант за узгодженням з
компетентним уповноваженим органом. Однак в усіх
спірних випадках треба проводити кількісне визна-
чення тридозовим методом, як описано вище.
На кожній чашці Петрі або прямокутній чашці розчи-
ни розміщають відповідно до статистично прийнятно-
го плану. Виняток становлять маленькі чашки Петрі,
на яких неможливо розмістити більше шести роз-
чинів. Розчини випробовуваного антибіотика і
стандартного зразка чергують таким чином, щоб ви-
ключити взаємодію більш концентрованих розчинів.
Чашки інкубують при підхожій температурі близько
18 год. Для зменшення впливу різниці у часі між вне-
сенням розчинів і для уточнення лінії регресії реко-
мендується використовувати попередню дифузію при
кімнатній температурі або при температурі близько
4 °С тривалістю від 1 год до 4 год.
Вимірюють діаметри круглих зон пригнічення росту з
точністю не менше 0.1 мм або їх площі з відповідною
точністю і розраховують активність, використовуючи
підхожі статистичні методи.
Число повторностей для однієї дози при кожному ви-
значенні має бути достатнім для забезпечення потріб-
ної точності. Може бути проведено кілька визначень,
результати яких об’єднують при статистичній обробці
для досягнення потрібної точності й підтвердження
того, що активність випробовуваного антибіотика не
нижча за допустиму мінімальну активність.
В ТУРБІДИМЕТРИЧНИЙ МЕТОД
У підхоже живильне середовище вносять суспензію
вибраних мікроорганізмів, чутливість яких до випро-
бовуваного антибіотика така, що забезпечує достатньо
сильне пригнічення їх росту за умов проведення ви-
пробування. Використовують певну кількість сус-
пензії, яка підбирається таким чином, щоб одержати
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
139
2.7. Біологічні методи кількісного визначення
Кількісне визначення методом дифузії
Таблиця 2.711
Антибіотик Стандартний зразок Розчинник для приготування основного розчину Буферний розчин (рН) Тест-мікроорга- нізм Живильне середо- вище і остаточне значення рН (±0.1) Температурі інкубації 1
Амфотерн- ими В Амфотери- цину в Фсз Диметил- формамід Р рН 10.5 (0.2 М) Бассішготусез сеге\Біае АТСС 9763 ІР 1432-83 Г-рН 6.1 35 °С-37 ТІ
Бацитрацину цинкова сіль Бацитрацину цинкової солі ФСЗ 0.01 М розчин кислоти хлористоводневої рН 7.0 (0.05 М) МІСГОСОССН8 Да\и<> МСТС 7743 СІР 53.160 АТСС 10240 А - рН 7.0 35 °С-39‘СІ
Блеоміиину сульфат Блеоміцину с£ль^)ату Вода Р рН 6.8 (0.1 М) МусоЬасіегіит хте%таііх АТСС 607 С - рН 7.0 35 °С-37 °С 1
Ванкоміиину гідрохлорид Ванкоміцину гід^охлориду Вода Р рН 8.0 Васіііи.ч .чиІМПіх МСТС 8236 СІР 52.62 АТСС 6633 А-рН8.0 37 °С-39‘СІ
Гентаміцину сульфат Гентаміцину с^ль^іату Вода Р РН 8.0 (0.05 М) ВасіІІиз ритіїи.ч МСТС 8241 СІР 76.18 Біаріїуіососси^ еріаегтідіі МСІВ 8853 С1Р68.21 АТСС 12228 А-рН 7.9 А-рН7.9 35 °С-39‘СІ 35 °С-39‘СІ
Дигідростреп- томіцину сульфат Дигідро- стреп- томіцину с^ль^іату Вода Р рН 8.0 (0.05 М) ВасНІи.ч .чиЬіііі.ч МСТС 8236 СІР 1.83 ВасіІІи.ч .чиїлііі.ч МСТС 10400 СІР 52.62 АТСС 6633 А - рН 7.9 А - рН 7.9 30°С-37‘С І 30°С-37‘С 1
Канаміцину моносульфат Канаміцину сульфат кислий Канаміцину моно- с^льї^ату Вода Р рН 8.0 (0.05 М) Васіііи.ч мМІБ МСТС 10400 СІР 52.62 АТСС 6633 Біаріїуіососсиз аигеих МСТС 7447 СІР 53.156 АТСС 6538 Р А-рН 7.9 А-рН7.9 30 °С-37 °С 35 °С-39 °С
Колістину сульфат Колістинме- тансульфонат натрію Колістину с^ль^іату Колістинме- тансульфо- натрію Вода Р рН 6.0 (0.05 М) Вогдеіеііа Ьгопсіїіаерііса МСТС 8344 СІР53.157 АТСС 4617 Ехскегісіїіа соїі МС1В8879 СІР 54.127 АТСС 10536 В-рН 7.3 В-рН 7.3 35 °С-39‘С 35 °С-39‘С
Неоміцину сульфат Неоміцину с^ль^мту Вода Р рН 8.0 (0.05 М) Васіїїиа ритіїи.ч МСТС 8241 СІР 76.18 Васіїїих хиЬііІіх МСТС 10400 СІР 52.62 АТСС 6633 Е - рН 7.9 Е-рН 7.9 ЗО °С-37 °С ЗО °С-37 °С
140
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.7. Біологічні методи кількісного визначення
Продовження таблиці 2.7.2,-1
1 Антибіотик Стандартний зразок Розчинник для приготування основного розчину Буферний розчин (рН) Тест-мікроорга- нізм Живильне середо- вище і остаточне значення рН (±0.1) Температура інкубації
| ’Н; • іміцину Ввмміаг Нетшміцину с^ль^ату Вода Р рН 8.0±0.1 ВіарИуІососсиз аигеиз АТСС 6538 Р СІР 53.156 А-рН 7.9 32 °С - 35 °С
ВНштин Ністатину ФСЗ Диметил- формамід Р рНб.О (0.05 М), що містить 5 % (об/об) диметил- формаміду Р Сапсіісіа ігорісаіік СІР 1433-83 МС¥С 1393 ЗассИаготусез сегех’імае МС¥С 87 СІР 1432-83 АТСС 9763 Р-рН 6.0 Е - рН 6.0 ЗО °С - 37 °С 30 °С - 32 °С
1 Ііліміксину 1 Ііуіьфат Поліміксину В с^ль^ату Вода Р рН 6.0 (0.05 М) Вагсіеіеііа ЬгопсИізерііса МСТС 8344 СІР 53.157 АТСС 4617 В - рН 7.3 35 °С - 39 °С
Рифаміцину ітрієва сіль Рифаміцину натрієвої солі ФСЗ Метанол Р рН 7.0 (0.05 М) Місгососсиз /Іасил МСТС 8340 СІР 53.45 АТСС 9341 А рН 6.6 35 °С - 39 °С
і іііраміцин Сріраміцину Метанол Р рН 8.0 (0.05 М) Васіїїиз міЬіПіз МСТС 10400 СІР 52.62 АТСС 6633 А-рН 7.9 ЗО °С - 32 °С
стрептомі- цину сульфат і Стрепто- міцину ср'їьрату Вода Р рН 8.0 (0.05 М) Васіїїик зиЬШіз МСТС 8236 СІР 1.83 ВасіПиз 8иЬШіз МСТС 10400 СІР 52.62 АТСС 6633 А-рН7.9 А - рН 7.9 30 °С - 37 °С ЗО °С - 37 °С
Тилозин для ветеринарії Тилозину тар- тратдля ветеринарії Тилозину ФСЗ 2.5 % розчин об/об метанолу Ру 0.1 М фосфатному буфер- ному розчині рН 7.0 Р Суміш метанолу Р і 0.1 М фосфатного буферного розчину рН 7.0 Р у співвід- ношенні 40:60 (об/об) МІСГОСОССЦ8 Да\'и8 МСТС 8340 СІР 53.45 АТСС 9341 А - рН 8.0 32 °С - 35 °С
Тобраміцин Тобраміцину Вода Р рН 8.0 (0.05 М) Васіїїиз зиЬШіз МСТС 10400 СІР 52.62 АТСС 6633 А - рН 7.9 ЗО °С - 37 °С
Фраміцетину сульфат Фраміцетину срт^ату Вода Р рН 8.0 (0.05 М) Васіїїиь 8иЬііІІ8 МСТС 10400 СІР 52.62 АТСС 6633 Васіїїик ритіїиз МСТС 8241 СІР 76.18 Е-рН 7.9 Е-рН7.9 ЗО °С - 37 °С ЗО °С - 37 °С
Еритроміцину естолат Еритроміцину стилсукцинат Еритроміцину стеарат Еритроміцину Метанол Р (відповідно до зазначень в окремій статті) Метанол Р рН 8.0 (0.05 М) Васіїїик ритіїиз МСТС 8241 СІР 76 18 Васіїїиз яиЬііПз МСТС 10400 СІР 52.62 АТСС 6633 А-рН 7.9 А - рН 7.9 ЗО °С - 37 °С ЗО °С - 37 °С
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
141
2.7. Біологічні методи кількісного визначення
легко вимірювану каламутність після закінчення інку-
баційного періоду тривалістю близько 4 год.
Середовише використовують негайно після внесення
в нього мікроорганізмів.
Використовуючи зазначені в Табл.2.7.2.-2 розчинник
і буферний розчин, готують розчини стандартного
зразка з відомими концентраціями і розчини випро-
бовуваного антибіотика, передбачувані концентрації
яких не мають істотних відмінностей від відповідних
концентрацій стандартного зразка. Для того щоб мати
можливість оцінити придатність методики кількісно-
го визначення, треба використовувати не менше трьох
доз стандартного зразка і трьох відповідних доз випро-
бовуваного антибіотика, що мають здогадно ту саму
активність. Бажано, щоб дози становили геометричну
прогресію. При цьому може знадобитися з великого
числа доз вибрати три послідовні дози, для яких за-
лежність "логарифм дози — відповідь" є лінійною.
Відповідні дози використовують для стандартного
зразка і випробовуваного антибіотика.
Рівні об’єми кожного з розчинів вносять в однакові
пробірки і додають у кожну пробірку рівні об’єми іно-
кульованого середовища (наприклад, 1 мл розчину
і 9 мл середовища).
У той самий час готують дві контрольні пробірки з
інокульованим середовищем, що не містить ан-
тибіотика, в одну з яких негайно вносять 0.5 мл фор-
мальдегіду Р. Ці пробірки використовують для настро-
ювання оптичного приладу, за допомогою якого про-
водять вимірювання.
Усі пробірки розташовують у випадковому порядку у
вигляді латинського квадрата або випадкового блока
і поміщають на водяну баню або інший підхожий
пристрій, що дозволяє швидко довести пробірки до
необхідної температури інкубації. Пробірки витриму-
ють при цій температурі від 3 год до 4 год, забезпечую-
чи однорідність температури і рівний час інкубації для
кожної пробірки.
Після закінчення періоду інкубації зупиняють ріст
мікроорганізмів, додаючи в кожну з пробірок 0.5 мл
формальдегіду Р, або шляхом теплової обробки і за до-
помогою підхожого оптичного приладу вимірюють
каламутність вмісту пробірок з точністю до третьої
значущої цифри. Допускається використовувати ме-
тод, що дозволяє робити вимірювання каламутності
вмісту кожної пробірки після закінчення строго ви-
значеного інкубаційного періоду, однакового для всіх
пробірок.
Розраховують активність, використовуючи відповідні
статистичні методи.
Залежність "логарифм дози — відповідь (перетворена
або неперетворена)”, часто виявляється лінійною ли-
ше в дуже обмеженому діапазоні концентрацій. При
розрахунках активності треба використовувати лише
цей інтервал, який має включати в себе принаймні три
послідовні дози, що необхідно для перевірки ліній-
ності. При проведенні постійних кількісних визна-
чень, для яких лінійність системи була продемонстро-
вана на достатньо великій кількості тридозових визна-
чень, допускається використовувати дводозовий
варіант за узгодженням з компетентним уповноваже-И
ним органом. Однак в усіх спірних випадках треба І
проводити кількісне визначення тридозовим мето-1
дом, як описано вище.
Число повторностей для однієї дози при кожному І
визначенні має бути достатнім для забезпечення І
потрібної точності. Може бути проведено кілька І
визначень, результати яких об’єднують при статис-
тичній обробці для досягнення потрібної точності й
підтвердження того, шо активність антибіотика не І
нижча за потрібну допустиму мінімальну активність. І
Наступний розділ має довідковий і рекомендаційний ха-
рактер і не є обов’язковою частиною загального методу. І
РЕКОМЕНДОВАНІ ТЕСТ-МІКРООРГАНІЗМИ
Нижче наведені рекомендовані тест-мікроорганізми
та умови їх використання. Допускається використову-
вати інші мікроорганізми, якщо доведена їхня чут-
ливість до випробовуваного антибіотика, застосовую-1
чи підхожі живильні середовища, підхожі умови інку-
бації і значення рН. Концентрації використовуваних!
розчинів випробовуваного антибіотика і стандартного
зразка треба підбирати таким чином, щоб за умов про-
ведення випробування залежність між логарифмом
концентрації і відповіддю була лінійною.
Приготування інокулята
Васіїіиз сегеих гаг. тусоісіех; Васіїїих хиЬйІіх; Васіїїія
ритіїих. Суспензії спор мікроорганізмів готують таким
чином.
Мікроорганізми вирощують при температурі від 35 °С
до 37 °С протягом семи діб на поверхні підхожого жи-
вильного середовища, що містить 0.001 г/л Марган-
цю^!) сульфату Р. Мікроорганізми, що виросли на
поверхні живильного середовища і знаходяться пере-
важно у формі спор, змивають за допомогою стериль-
ної води Р. Одержану суспензію спор прогрівають про-
тягом ЗО хв при температурі 70 °С і розводять до
підхожої концентрації спор, звичайно від ЮхІО6 до
ІООхІО6 спор в 1 мл. Суспензія спор може зберігатися
протягом тривалого часу при температурі не
вище 4 °С.
Може бути використаний інший спосіб приготування
суспензії спор. Мікроорганізми вирощують на середо-
вищі С при температурі 26 °С від чотирьох діб до
шести діб, потім, дотримуючи правил асептики, дода-
ють 0.001 г/л марганцю)II) сульфату Р і продовжують
інкубацію протягом 48 год. Мікроскопічно підтвер-
джують утворення спор у достатній кількості (близько
80 %) і центрифугують суспензію. Одержаний осад по-
вторно суспендують у стерильній воді Р, створюючи
концентрацію від 10x106 до 100x106 спор в
1 мл, і прогрівають протягом ЗО хв при температурі
70 °С. Суспензію треба зберігати при температурі не
вище 4 °С.
Вогсіеіеііа Ьгопсйіхерйса. Тест-мікроорганізми вирощу-
ють на поверхні живильного середовища В при темпе-
ратурі від 35 °С до 37 °С від 16 год до 18 год. Мікроор-
142
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.7. Біологічні методи кількісного визначення
Таблиця 2.7.2.-2
Кількісне визначення турбідиметрични.м методом
Антибіотик Стандартний зразок Розчинник для приготування основного розчину Буферний розчин (рН) Тест-мікроорга- нізм Живильне середо- вище і остаточне значення рН (±0.1) Температура інкубації
| Вннкоміцину пірохлорид Ванкоміцину гідрохлориду Вода Р рН 8.0 МарИуІососсих аигеиа АТСС 6538 Р СІР 53.156 С - рН 7.0 37 °С-39 °С
1 Гентаміцину [ мьфат Гентаміцину срль^іату Вода Р рН 7.0 МарИуІососсих аигеих ГЧСТС 7447 СІР 53.156 АТСС 6538 Р С-рН 7.0 35 °С - 37 °С
Граміцидин Граміцидину Метанол Р рН 7.0 (для запобі- гання адсор- бції при розведенні може бути додана по- верхнево- активна ре- човина, на- приклад, 0.1 мг/мл полісорбату- 80Р) Мгеріососсих /аесаііа АТСС 10541 МарИуіососсия аигеих АТСС 6538 Р С-рН7.0 35 °С - 37 °С
Дигідростреп- юміцину сульфат Дигідро- стреп- томіцину с^ль^ату Вода Р рН 8.0 КІеЬзіеІІа рпеитопіае ТЧСТС 7427 СІР 53.153 АТСС 10031 С-рН7.0 35 °С - 37 °С
Канаміцину моносульфат Канаміцину сульфат кислий Канаміцину моносульфату ФСЗ Вода Р рН 8.0 МарИуІососсиа аигеих ТЧСТС 7447 СІР 53.156 АТСС 6538 Р С-рН7.0 35 °С - 37 °С
Колістину сульфат Колістинме- тансульфонат натрію Колістину срушрату Колістинме- тансульфо- нат§ натрію Вода Р рН 7.0 ЕасіїегісИіа соїі ТЧСІВ 8666 СІР 2.83 АТСС 9637 С-рН7.0 35 °С - 37 °С
Неоміцину сульфат Неоміцину срлмрату Вода Р рН 8.0 МарИуІососсиа аигеих ТЧСТС 7447 СІР 53.156 АТСС 6538 Р С - рН 7.0 35 °С - 37 °С
Рифаміцину натрієва сіль Рифаміцину натрієвої солі ФСЗ Метанол Р рН 7.0 Езсіїегісійа соїі ТЧСІВ 8879 СІР 54.127 АТСС 10536 С - рН 7.0 35 °С - 37 °С
Спірамінин Спіраміцину ФСЗ Метанол Р рН 7.0 Маріїуіососсиз аигеиз ТЧСТС 7447 СІР 53.156 АТСС 6538 Р С-рН7.0 35 °С - 37 °С
Стрептомі- цину сульфат Стрептомі- ціну сульфату ФСЗ Вода Р рН 8.0 КІеЬмеІІа рпеитопіае ТЧСТС 7427 СІР 53.153 АТСС 10031 С - рН 7.0 35 °С - 37 °С
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
143
2.7. Біологічні методи кількісного визначення
Продовження таблиці 2.7.2.
Антибіотик Стандартний зразок Розчинник для приготування основного розчину Буферний розчин (рН) Тест-мікроорга- нізм Живильне середо- вище і остаточне значення рН (±0.1) Температура інкубації
Тилозин для ветеринарії Тилозину тар- трат для ве- теринарії Тилозину ФСЗ 2.5 % (об/об) розчин метанолу Ру 0.1 М фосфатному буферному розчині рН7.0Р рН 7.0 Зіаріїуіососсиз аигеиз ТЧСТС 6571 АТСС 9144 СІР 53.154 С-рН7.0 37 °С
Тобраміцин Тобраміцину ФСЗ Вода Р рН 7.0 Зіаріїуіососсих аигеиз ТЧСТС 7447 АТСС 6538 Р СІР 53.156 АТСС 9144 С - рН 7.0 35 °С -37 °С І
Фраміцетину сульфат Фраміцетину сульфату ФСЗ Вода Р рН 8.0 Зіаріїуіососсиз аигеия ГЧСТС 7447 СІР 53.156 АТСС 6538 Р С - рН 7.0 35 °С-37 °С
Еритроміцину естолат Еритроміцину етилсукцинат Еритроміцину стеарат Еритроміцину ФСЗ Метанол Р (відповідно до зазначень окремої статті) Метанол Р рН 8.0 КІеЬмеІІа рпеитопіае ТЧСТС 7427 СІР 53.153 АТСС 10031 Зіаріїуіососсиь аигеих ТЧСТС 7447 СІР 53.156 АТСС 6538 Р П - рН 7.0 С-рН 7.0 35 °С-37 °С II 35 °С-37 °С
ганізми, що виросли на поверхні живильного середо-
вища, змивають стерильною водою Р і розводять до
одержання підхожої каламутності.
(МарИуІососсих аигеиз; КІеЬаіеІІа рпешпопіае; ЕхсВегіМа
соїі; Місгососсиз/істиз; 8іарИуІососсиз ерідегтісііа. Готу-
ють, як описано вище для В. ЬгопсИікерііса, використо-
вуючи середовище А і підбираючи каламутність, що
забезпечує задовільну залежність "доза - відповідь"
при проведенні турбідиметричного кількісного визна-
чення або дає чіткі зони пригнічення росту підхожого
діаметра при проведенні кількісного визначення ме-
тодом дифузії.
ЗассИаготусез сегеуімае; Сапсіісіа ікорісаііх. Мікроор-
ганізми вирощують на поверхні середовища Е при
температурі від ЗО °С до 37 °С протягом 24 год. Мікро-
організми, що виросли на поверхні середовища, зми-
вають стерильним розчином 9 г/л натрію хлориду Р і
розводять до підхожої каламутності тим самим розчи-
ном.
го для кількісного визначення блеоміцину сульфату,
6.4 г калію дигідрофосфату Р\ 18.9 г динатрію гідрофо-
сфату Р розчиняють у воді Рі доводять об’єм до 1000 мл.
Таблиця 2.7.2.-3
рн Об’єм 0.2 М розчину натрію гідроксиду, у мілілітрах
5.8 3.72
6.0 5.70
6.2 8.60
6.4 12.60
6.6 17.80
6.8 23.65
7.0 29.63
7.2 35.00
7.4 39.50
7.6 42.80
7.8 45.20
8.0 46.80
БУФЕРНІ РОЗЧИНИ
Буферні розчини із значеннями рН від 5.8 до 8.0 готу-
ють шляхом зміщування 50.0 мл 0.2 Мрозчину калію
дигідрофосфату із зазначеною у Табл. 2.7.2.-З
кількістю 0.2 М розчину натрію гідроксиду. Об’єм до-
водять до 200.0 мл свіжоприготованою методом дис-
тиляції водою Р.
Буферні розчини використовують для кількісного
визначення всіх антибіотиків, перелічених у
Табл. 2.7.2.-1, за винятком блеоміцину сульфату. Для
приготування буферного розчину (рН 6.8), необхідно-
ЖИВИЛЬНІ СЕРЕДОВИЩА
Для кількісного визначення антибіотиків використо-
вують живильні середовища, наведені нижче, або
еквівалентні їм.
Живильне середовище А
Пептон....................................6 г
Панкреатичний гідролізат казеїну..............4 г
Яловичий екстракт............................1.5 г
144
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.7. Біологічні методи кількісного визначення
йріжджовий екстракт.....................................З г
Г юкози моногідрат......................................1 г
^ар..............................................15 г
Кта...........................................до 1000 мл
Живильне середовище В
Панкреатичний гідролізат казеїну.......................17 г
Папаїновий гідролізат соєвих бобів......................З г
Натрію хлорид ..........................................5 г
Дихазію гідрофосфат...................................2.5 г
Глюкози моногідрат....................................2.5 г
Агар...................................................15 г
Полісорбат-80..........................................10 г
Вода..........................................до 1000 мл
Полісорбат-80 додають до гарячого розчину решти
інгредієнтів після кип’ятіння і безпосередньо перед
введенням розчину до потрібного об’єму.
Живильне середовище С
Пептон..............................................6 г
Яловичий екстракт.................................1.5 г
Дріжджовий екстракт.................................З г
Натрію хлорид.....................................3.5 г
Глюкози моногідрат..................................І г
Дикалію гідрофосфат..............................3.68 г
Натрію гідрофосфат...............................1.32 г
Вода.......................................до 1000 мл
Живильне середовище В
Екстракт серця.................................1.5 г
Дріжджовий екстракт...............................1.5 г
Казеі'н-пептон......................................5 г
Еіюкози моногідрат..................................1 г
Натрію хлорид.....................................3.5 г
Дикалію гідрофосфат..............................3.68 г
Натрію гідрофосфат...............................1.32 г
Натрію нітрат.......................................2 г
Вода.......................................до 1000 мл
Живильне середовище Е
Пептон.............................................5 г
М'ясний екстракт...................................З г
Динатрію гідрофосфат додекагідрат...............26.9 г
Агар...............................................10 г
Вода.......................................до 1000 мл
Динатрію гідрофосфат додають у вигляді стерильного
розчину після стерилізації середовища.
Живильне середовище Е
Пептон...........................................9.4 г
Дріжджовий екстракт..............................4.7 г
Яловичий екстракт................................2.4 г
Натрію хлорид...................................30.0 г
Ечюкози моногідрат..............................10.0 г
Агар............................................23.5 г
Вода......................................до 1000 мл
Живильне середовище 6
Гліцерин..........................................10 г
Пептон............................................10 г
М’ясний екстракт...............................10 г
Натрію хлорид...................................З г
Агар...........................................15 г
Вода....................................до 1000 мл
рН після стерилізації 7.0±0.1
______________________________________________N
Активність антибіотиків виражають в одиницях дії
(ОД) або мікрограмах. Для більшості антибіотиків 1 ОД
відповідає 1 мкг активної речовини.
А. МЕТОД ДИФУЗІЇ
Кількісне визначення антибіотиків методом дифузії
може також проводитися за умов, зазначених у
Табл. 2.7.2.-1.
Живильні середовища, що рекомендуються для
кількісного визначення, розплавляють і розливають у
чашки Петрі в один або два шари. Для нижнього шару
використовують неінокульовані живильні середови-
ща, для верхнього або одного шару — густе живильне
середовище, інокульоване тест-мікроорганізмом.
Температура розплавленого живильного середовища,
в яке вносять тест-мікроорганізми, має становити від
48 °С до 50 °С для вегетативних клітин і від 65 °С до
70 °С при використанні суспензії спор.
Основні розчини стандартних і випробовуваних
зразків готують у стерильних розчинниках. Якшо не-
має інших зазначень в окремій статті, концентрація
основного розчину має становити близько 1 мг/мл.
Робочі розчини стандартного зразка і випробовувано-
го антибіотика готують шляхом розведення основного
розчину стерильним розчинником. Концентрації ро-
бочих розчинів, шо містять малу, середню і велику до-
зи, мають становити геометричну прогресію. Спів-
відношення, що рекомендується — 1:2:4. Рекомендо-
вана концентрація робочих розчинів, які містять се-
редню дозу (контрольна концентрація), наведена у
Таб.2.7.2-4. Допускається, якщо необхідно, зміна кон-
трольної концентрації за умови, що залежність "лога-
рифм дози — відповідь" буде лінійною у використову-
ваному діапазоні концентрацій.
При використанні для чашок Петрі кількісного визна-
чення на кожній чашці розміщають шість циліндрів
або готують шість лунок. Діаметри усіх циліндрів або
лунок мають відрізнятися не більш як на ± 0.1 мм.
Робочі розчини стандартного зразка і випробовувано-
го антибіотика вносять у циліндри або лунки однієї
чашки Петрі так, щоб розчини з однаковими концен-
траціями не межували один з одним. При внесенні
розчинів спочатку вносять усі розчини в лунки або
циліндри однієї чашки Петрі, потім вносять розчини в
лунки або циліндри наступної чашки і т.д.
Кількість чашок Петрі, використовуваних в одному
визначенні, має бути достатньою для забезпечення
статистичної вірогідності результатів, але не менше
шести чашок.
Якщо немає інших зазначень в окремій статті, три-
валість інкубації має становити від 16 год до 18 год.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
145
2.7. Біологічні методи кількісного визначення
Таблиця 2.7.2.-і
Рекомендовані умови кількісного визначення антибіотиків методом дифузії
Антибіотик/ Стандартний зразок Розчинник для приготування основного розчину/Термін придатності основного розчину стандартного зразка при температурі від 4 °С до 10 °С Розчинник для приготування робочих розчинів3; контрольна концентрація Тест- мікр оор ганізм; висівна доза Живильне середо- вище4 і остаточне значення рН (±0.1) Кількість І живильного 1 середовища на 1 1 чашку Петрі (мл)1!
Нижній шар2 Верхній шар Нижній шар Верхній шар
Амфотери- цин В/ Амфотери- цину В ФСЗ Диметилсульфоксид/ 1 доба Буферний розчин рН 7.0, термін придатності при кімнатній темпе- ратурі не більше ЗОхв; 1 мкг/мл Сапсіісіа ШіІВ Л1А-01; 3 мл робочої зависі на 100 мл середовища - № 11 + 0.1 % глюкози - 15 1 _ 1
Геліоміцин/ Геліоміцину ФСЗ 0.1 М розчин натрію гідроксиду до концетрації геліоміцину 1 мг в 5 мл/ 10 діб 0.1 М розчин натрію гідроксиду; 4 мкг/мл Васіїїиз хиЬііііх АТСС 6633; 108 спор/мл - № 12 - 1 15
Гентаміцину сульфат/ Гентаміцину сульфату ФСЗ Буферний розчин рН 8.0/ 14 діб Буферний розчин рН 8.0; 2 мкг/мл Васіїїиз ритііиз ТЧСТС 8241; 5Т07 спор/мл № 6 № 6 20 5
Граміцидин С/ Граміцидину С ФСЗ 96 % спирт 1 30 діб Вода очищена; 100 мкг/мл Васіїїиз сегеиз \'аг. тусоісіех 537; 8-Ю6 спор/мл - № 9 - 10
Д актиномі- цин/ Дактиномі- цину ФСЗ Вода очищена (витри- мують при температурі від 4 °С до 10 °С до по- вного розчинення)/ 15 діб Буферний розчин рН 8.0; 4 мкг/мл Васіїіиі сегеиз гаг. тусоісіех НВ; 2-Ю7 спор/мл - № 10 - 10
Дигідростреп- томіцину сульфат/ Дигідростреп - томіцину сульфату ФСЗ Буферний розчин рН 6.2/ 30 діб Буферний розчин рН 8.0; 2 мкг/мл ВасіИих сегеиз уаг. тусоісіех 537; 2-Ю7 спор/мл - №6 - 10
Канаміцин/ Канаміцину моносульфату ФСЗ Вода очищена/ 30 діб Буферний розчин рН 8.0; 10 мкг/мл Васіііиз ритіїих МСТС 8241; 4-107 спор/мл №5 рН 7.9 №5 рН 7.9 15 5
Карміноміцин/ Карміноміцину гідрохлориду ФСЗ Вода очищена/ 10 діб Буферний розчин рН8.0; 15 мкг/мл Васіїїиз сегеиз саг. тусоісіех 537; 108 спор/мл - №3 рН7.9 - 15
146
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.7. Біологічні методи кількісного визначення
Продовження таблиці 2.7.2.-4
1 Антибіотик/ Стандартний зразок Розчинник для приготування основного розчину/Термін придатності основного розчину стандартного зразка при температурі від 4 °С до 10 °С Розчинник для приготування робочих розчинів3; контрольна концентрація Тест-мікроорганізм; висівна доза Живильне середо- вище4 і остаточне значення рН (±0.1) Кількість живильного середовища на 1 чашку Петрі (мл)1
Нижній шар2 Верхній шар Нижній шар Верхній шар
Леворин/ Леворину ФСЗ Диметилсульфоксид/ 3 доби Диметилсульфо- ксид до концент- рації леворину 100 мкг/мл, потім у буферному розчині рН 7.0. Термін придатності при кімнатній температурі не більше 30 хв; 0.5 мкг/мл Сапсіісіа иііііх Л1А-01;/ від 2 до 2.5 мл робочої зависі на 100 мл середовища - № 11 + 1 % глюкози - 15
Метациклін/ Метацикліну гідрохлориду ФСЗ 0.01 М розчин кислоти хлористоводневої/ 7 діб Буферний розчин рН 6.0; 0.5 мкг/мл Васіїїил виЬіїїів \’аг. Л2/ 3-Ю7 спор/мл - №4 + 1 % глюкози - 15
Мікогептин/ Мікогептину ФСЗ Диметилсульфоксид/ 3 лоби (не більше 4-5 год при кімнатній температурі) Диметилсу- льфоксид до концентрації мікогептину 50 мкг/мл, потім буферний розчин рН 8.0 (термін придатності при кімнатній температурі не більше 30 хв); 1 мкг/мл Сапсіісіа иіііів Л1А-01; від 2.5 мл до 3.0 мл робочої зависі на 100 мл сереловиша - № 11 + 1 % глюкози - 15
Мономіцин/ Мономіцину ФСЗ Вода очищена/ 30 ліб Розчин калію хлориду 3 %; 2 мкг/мл Васіїїил сегеив гаг. тусоісіел 537; 2-Ю7 спор/мл - № 6 - 10
Неоміцину сульфат/ Неоміцину сульфату ФСЗ Буферний розчин рН 8.0/ 30 діб Буферний розчин рН 8.0; 4 мкг/мл Васіїїив сегеиа гаг. тусоісіех 537; 2-Ю7 спор/мл № 6 №6 20 5
Ністатин/ Ністатину ФСЗ Диметилформамід/ 3 доби Буферний розчин рН 6.0; 20 ОД/мл Сапсіісіа иіііів ЛІА-01; від 3 мл до 3.5 мл робочої зависі на 100 мл середовища - № 11 + 1 % глюкози - 15
Оксацилін/ Оксациліну натрієвої солі ФСЗ Буферний розчин рН 7.0/ Здоби Буферний розчин рН 7.0; 4 мкг/мл Зіаркуіососсил аигеиз АТСС 6538 Р; 4-Ю7клітин на 1 мл №8 рН 6.9 №5 рН 6.9 + 0.1 % глюкози 10 5
Олеандоміцин/ Олеандоміцину фосфату ФСЗ Буферний розчин рН 6.2/ 7 діб Буферний розчин рН 8.0; 4 мкг/мл Васіїїив сегеив гаг. тусоісіех НВ; і 2-Ю7 спор/мл - №6 - 10
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
147
2.7. Біологічні методи кількісного визначення
Продовження таблиці 1.1.1.-'-
Антибіотик/ Стандартний зразок Розчинник для приготування основного розчину/Термін придатності основного розчину стандартного зразка при температурі від 4 °С до 10 °С Розчинник для приготування робочих розчинів3; контрольна концентрація Тест-мікроорганізм; висівна доза Живильне середо- вище4 і остаточне значення рН (±0.1) Кількість живильного середовища на 1 чашку Петрі (мл)1! |
Нижній шар2 Верхній шар Нижній шар Верхній шар 11
Оливоміцин/ Оливоміцину кислоти ФСЗ Стандартний зразок у 96 % спирті до концентрації оливоміцину 5 мг/мл, потім буферний розчин рН 7.0; випробовуваний зразок у воді очищеній/ 14 діб Буферний розчин рН 7.0; 2 мкг/мл Восіїїих зиЬіїііз АТСС 6633; 107 спор/мл №8 рН 6.9 № 13 10 5 1
Поліміксин В/ Поліміксину В сульфату ФСЗ Буферний розчин рН 6.0/ 14 діб Буферний розчин рН 6.0; 100 ОД/мл ВогФеїеІІа ЬгопсЬЕерііса АТСС 4617; від 4-Ю7 до 6-107 клітин на 1 мл - №7 - 10
Поліміксин М/ Поліміксину М сульфату ФСЗ Буферний розчин рН 6.0/ 14 діб Буферний розчин рН 6.0; 100 ОД/мл Вогсіекіїа ЬгопсЬЕерііса АТСС 4617; від 4-Ю7 до 6-Ю7 клітин на 1 мл - №7 - 10
Рубоміцин/ Рубоміцину гідрохлориду ФСЗ Вода очищена/ 10 діб Буферний розчин рН 8.0; 20 мкг/мл Васіїїиз сегеиь ьаг. тусоісіех 537; 107 спор/мл - №3 рН 7.9 - 15
Сизоміцин/ Сизоміцину сульфату ФСЗ Буферний розчин рН 8.0/ 14 діб Буферний розчин рН8.0; 2 мкг/мл Васіїїиа ритіїиз МСТС 8241; 5-Ю7 спор/мл № 8 рН 7.9 №5 рН 7.9 20 5
Стрептоміцин/ Стрептоміцину сульфату ФСЗ Буферний розчин рН 6.2/ 30 діб Буферний розчин рН 8.0; 2 мкг/мл Васіїїиз сегеиз уаі: тусоісіех 537; 2-Ю7 спор/мл - № 6 - 10
Феноксиме- тилпеніцилін/ Феноксиме- тилпеніциліну ФСЗ Буферний розчин рН 7.0/ 7 діб Буферний розчин рН 7.0; 1 ОД/мл Зіаріїуіососсиз аигеи8 АТСС 6538 Р/ 4-Ю7 клітин на 1 мл № 8 рН 6.9 №5 рН 6.9 + 0.1 % глюкози 10 5
Флориміцин/ Флориміцину сульфату ФСЗ Вода очищена/ 7 діб Буферний розчин рН 8.0; 10 мкг/мл Васіііиз зиЬііІЕ АТСС 6633/ 2-Ю7 спор/мл - №5 рН 7.9 - 10
Еритроміцин/ Еритроміцину ФСЗ 1 мл 96 % спирту на 10 мг наважки, потім буферний розчин рН 8.0 до концентрації еритроміціну 1000 мкг/мл/ 7 діб Буферний розчин рН 7.0/ 2 мкг/мл Васіїїиз сегеиз уаг.тусоісіех НВ/ 2-107спор/мл - №6 - 10
Примітки:
1. Об’єми живильних середовищ зазначені для чашок Петрі діаметром 100 мм і варіанта методу дифузії з використанням циліндрів
При використанні лунок допускається збільшення об’єму середовища для нижнього або одного шару.
2. " - " — використовують один шар живильного середовища.
3. Буферні розчини наведені у Табл. 2.7.2.-3.
4. Глюкозу додають у розплавлене живильне середовище у вигляді стерильного 40 % розчину.
148
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.7. Біологічні методи кількісного визначення
ЕКОМЕНДОВАН1 тест-мікроорганізми
Приготування інокулята
Учіеіеііа ЬгопсІїіїерГіса. Для вирощування тест-мікро-
рганізму може бути використане живильне середови-
К№ 1 (рН 7.0-7.2). Тривалість інкубації на середо-
іші № 1 становить від ЗО год до 36 год.
Іаріїуіососсиз аигеиз. Для вирощування тест-мікроор-
пнізмуможе бути використане живильне середовище
Ч 1 (рН 7.0-7.2). Тривалість інкубації на середовищі
' І становить від 18 год до 20 год.
ВйсіІІизсегеихуаг. тусоісіех; Васіїїииритіїих. приго-
вання суспензії спор може бути використаний
спосіб, наведений нижче. Тест-мікроорганізми виро-
пують на живильному середовищі № 1. Значення рН
ередовища для вирощування В. сегеих гаг. тусоісіеа
МіВ.ритіІизЬІСТС 8241 має становити від 7.0до7.2,
«ачення рН середовища для вирощування В. сегеих
аг. тусоісіез 537 - від 7.8 до 8.0. Посіви інкубують при
температурі від 35 °С до 37 °С від 18 год до 20 год.
Мікроорганізми, що виросли на поверхні живильного
ередовища, змивають за допомогою стерильного роз-
ину 9 г/л натрію хлориду (від 5 мл до 10 мл) і засіва-
ть декілька матраців із 300 мл живильного сере-
іовиша№ 3 зі скошеною поверхнею. Значення рН се-
редовища для вирощування В. сегеих гап тусоісіех 537 і
В.ритіїих МСТС 8241 має становити від 6.0 до 6.2, зна-
чення рН середовища для вирощування В. сегеиз \аг.
тусоісіез НВ — від 7.8 до 8.0. Посіви інкубують при
.ліпературі від 35 °С до 37 °С від п'яти діб до семи діб.
У процесі вирощування проводять мікроскопічний
контроль культури. Кількість спор, спостережувана
при мікроскопії, має становити не менше 80 % від за-
шіьної кількості клітин. Культуру, що містить достат-
ню кількість спор, змивають із поверхні живильного
середовища стерильною водою очищеною. Одержану
суспензію спор прогрівають при температурі від 60 °С
до 70 °С протягом ЗО хв. Потім промивають стериль-
ною водою очищеною при центрифугуванні до одер-
жання прозорої надосадової рідини. Промиту завись
спор знову прогрівають протягом 30 хв при темпера-
турі від 60 "С до 70 °С. Завись спор зберігають у скля-
них пробірках при температурі від 4 °С до 10 °С і вико-
ристовують, доки інтенсивність росту і чіткість зон
при кількісному визначенні задовольняють постав-
лені вимоги.
Саяій/аиіїЛ$Л1А-01. Тест-мікроорганізми вирощують
у пробірках зі скошеним живильним середовищем № 2
протягом 48 год при температурі від 29 °С до 31 °С.
Після закінчення періоду інкубації культуру змивають
з поверхні живильного середовища стерильним роз-
чином 9 г/л натрію хлориду і засівають матрац зі ско-
шеною поверхнею. Посіви інкубують протягом 48 год,
після закінчення періоду інкубації змивають 50 мл
стерильного розчину 9 г/л натрію хлориду. Готують ро-
бочу завись, каламутність якої має бути такою, щоб
при розведенні її у ЗО разів стерильним розчином 9 г/л
натрію хлориду оптична густина становила від 0.22 до
0.23. Для визначення каламутності суспензії викорис-
товують нефелометр із нейтральним світлофільтром і
кювети з товщиною шару 3 мм. Робоча завись може
зберігатися протягом ЗО діб.
ЖИВИЛЬНІ СЕРЕДОВИЩА
Для кількісного визначення антибіотиків, відповідно
до рекомендацій Табл. 2.7.2.-4, можуть бути викорис-
тані живильні середовища, наведені нижче, або їм
еквівалентні.
Середовище № 1
М’ясо-пептонний бульйон..................1000 мл
Агар.........................................20 г
Середовище № 2
М’ясо-пептонний бульйон..................1000 мл
Агар.........................................17 г
Глюкоза......................................10 г
Натрію хлорид.................................5 г
Значення рН середовища після стерилізації має стано-
вити від 7.0 до 7.2.
Середовище № З
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок.................................. 5 г
Агар.........................................25 г
Вода очищена...........................до 1000 мл
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмів
без оболонок може бути використаний панкреатич-
ний гідролізат м’яса (за Хоттінгером) глибокого роз-
щеплення Середовище має містити від ЗО мг% до 35 мг%
амінного азоту
Середовище № 4
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок...................................5 г
Агар......................................10-15 г
Вода очищена...........................до 1000 мл
Значення рН середовища після стерилізації має стано-
вити від 6 8 до 7.0.
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмів
без оболонок може бути використаний панкреатичний
гідролізат м’яса (за Хоттінгером) глибокого розщеплен-
ня. Середовище має містити від 130 мг% до 140 мг%
амінного азоту
Середовище № 5
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок....................................5 г
Агар........................................10-12 г
Динатрію гідрофосфат............................З г
Вода очищена............................до 1000 мл
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмів
без оболонок може бути використаний панкреатичний
гідролізат м’яса (за Хоттінгером) глибокого розщеплен-
ня. Середовище має містити від 130 мг% до 140 мг%
амінного азоту.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
149
2.7. Біологічні методи кількісного визначення
Середовище № 6
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок....................................5 г
Агар...........................................15 г
Динатрію гідрофосфат............................З г
Вода очищена............................до 1000 мл
Значення рН середовища після стерилізації має стано-
вити від 7.8 до 8.0.
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмів
без оболонок може бути використаний панкреатичний
гідролізат м’яса (за Хотгінгером) глибокого розщеплен-
ня. Середовище має містити від ЗО мг% до 35 мг%
амінного азоту.
Середовище № 7
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок....................................5 г
Агар...........................................15 г
Калію дигідрофосфат............................25 г
Вода очищена...........................до 1000 мл
Значення рН середовища після стерилізації має стано-
вити від 7.0 до 7.2.
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмів
без оболонок може бути використаний панкреатичний
гідролізат м’яса (за Хоттінгером) глибокого розщеплен-
ня. Середовище має містити від 130 мг% до 140 мг%
амінного азоту.
Середовище № 8
Агар......................................15-20 г
Динатрію гідрофосфат...........................З г
Вода очищена...........................до 1000 мл
Середовище № 9
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок...................................5 г
Агар..........................................20 г
Калію хлорид..................................20 г
Вода очищена...........................до 1000 мл
Значення рН середовища після стерилізації має стано-
вити від 7.0 до 7.2.
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмів
без оболонок може бути використаний панкреатич-
ний гідролізат м’яса (за Хотгінгером) глибокого розщеп-
лення. Середовище має містити від 30 мг% до 35 мг%
амінного азоту.
Середовище № 10
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок................................5 г
Агар........................................15 г
Вода очищена..........................до 1000 мл
Значення рН середовища після стерилізації має стано-
вити від 7.8 до 8.0.
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмі;
без оболонок може бути використаний панкреатич-
ний гідролізат м’яса (за Хоттінгером) глибокого розшем
лення. Середовище має містити від ЗО мг% до 35 мЛ
амінного азоту.
Середовище № 11
Агар......................................18-20
Динатрію гідрофосфат.........................5г
Натрію хлорид...............................20г
Калію хлорид................................20г
Амонію цитрат..................................5 г
Вода очищена..............................1000 мл
Значення рН середовища після стерилізації має стано-
вити від 5.8 до 6.0.
Середовище № 12
Агар..........................................18 г
Глюкоза........................................5 г
Динатрію гідрофосфат..........................15 г
Сечовина.......................................2 г
Амонію хлорид................................2г
Вода очище на..........................до 1000 мі
Значення рН середовища після стерилізації має стано-
вити від 7.8 до 8.0.
Середовище № 13
Ферментативний гідролізат біомаси мікроорганізмів
без оболонок................................ 51
Агар.......................................15-201
Глюкоза.......................................51
Натрію хлорид.................................ЗО і
Вода очище на...........................до 1000 мл
Значення рН середовища після стерилізації має стане
вити від 6.8 до 7.0.
При приготуванні живильного середовища замість
ферментативного гідролізату біомаси мікроорганізмів
без оболонок може бути використаний панкреатичний
гідролізат м’яса (за Хоттінгером) глибокого розщеплен
ня. Середовище має містити від 130 мг% до 140 мг%
амінного азоту.
При приготуванні живильних середовищ на основі су
хого ферментативного гідролізату мікроорганізмів без
оболонок інгредієнти складу вносять у воду очищену,
додають замочений заздалегідь агар і нагрівають до
повного його розплавлення. Установлюють рН таким
чином, щоб після стерилізації його значення станови
ло величину, зазначену вище для кожного живильного
середовища і у Табл. 2.7.2.-4. Якщо необхідно
фільтрують крізь ватно-марлевий фільтр. Стерилізу
ють у паровому стерилізаторі при температурі 121 °С
протягом 15 хв.
Примітка. Кількість агару в живильних середовищах
зазначена для випробування з використанням
циліндрів. При проведенні випробування з викорис
танням лунок кількість агару збільшують до 20-25
Допускається зміна кількості агару в середовищах за
лежно від його якості.
г.
150
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
2001
2.9. Фармако-технологі
апробування
2.9. ФАРМАКО-ТЕХНОЛОГІЧНІ
ВИПРОБУВАННЯ
2.9.1. РОЗПАДАННЯ ТАБЛЕТОК І КАПСУЛ
Випробування на розпадання дозволяє визначити, чи
розпадаються таблетки або капсули в межах визначе-
ного часу, якшо вони помішені в рідке середовише в
експериментальних умовах, зазначених нижче.
Вважають, що зразки розпалися, якщо на сітці:
з) немає залишку;
в) є залишок, який складається з м’якої маси, що не
має відчутно твердого ядра, котре не змочується;
і є лише фрагменти покриття (таблетки), або лише
фрагменти оболонки на сітці, або, якщо були ви-
користані диски, фрагменти оболонки, які при-
липли до нижньої поверхні диска (капсули).
Обладнання. Головна частина обладнання (див.
Рис 29 1 -1) складається з жорсткого кошика із
Сітчастим дном-підставкою (кошик), яка підтримує
шість циліндричних скляних трубочок завдовжки
(77.5+2.5) мм з внутрішнім діаметром 21.5 мм і товщи-
ною стінки близько 2 мм. Кожна трубка має
циліндричний диск діаметром (20.7±0 15) мм і товщи-
ною (9.5±0.15) мм, виготовлений з прозорої пластмаси
з густиною від 1.18 до 1.20. У кожному диску просверд-
лені п’ять отворів діаметром 2 мм, один з них розташо-
ваний в центрі диска, інші чотири — рівномірно по ко-
лу радіусом 6 мм від центра диска. На бічній поверхні
диска вирізані чотири рівновіддалені одна від одної
виїмки так, що на верхній поверхні диска вони мають
ширину 9.5 мм і глибину 2.55 мм, по нижній поверхні
мають форму квадрата зі стороною 1.6 мм. Скляні
трубки втримуються вертикально зверху і знизу двома
накладними прозорими пластмасовими пластинами
діаметром 90 мм, завтовшки 6 мм із шістьма отворами.
Отвори рівновіддалені від центра пластини і знахо-
дяться на рівній відстані один від одного. До нижньої
поверхні нижньої пластини прикріплено сітку з не-
ржавіючого сталевого дроту' діаметром 0.635 мм, з
розміром отворів 2.00 мм. Пластини утримуються жор-
стко на відстані 77.5 мм одна відносно іншої верти-
кальними металевими стрижнями по колу. Ще один
металевий стрижень прикріплений до центра верхньої
пластини що дозволяє прикріпити кошик до ме-
ханічного пристрою, який може піднімати та опускати
його плавно із постійною частотою в межах 28-32 цик-
лі іа хвилину на відстань від 50 мм до 60 мм.
Кошик поміщають у рідину, зазначену у відповідних
загальних та окремих статтях, у підхожій посудині, пе-
реважно в склянці місткістю 1 л. Об’єм рідини має бу-
ти таким, шоб, коли кошик знаходиться в крайньому
верхньому положенні, сітка має бути як мінімум на
15 мм нижче поверхні рідини; коли ж кошик знахо-
диться в найнижчому положенні, сітка має бути на
25 мм вище дна посудини, а верхні відкриті кінці
скляних трубок — над поверхнею рідини. Температуру
рідини від 36 °С до 38 °С підтримують за допомогою
підхожого пристрою
Конструкція кошика може змі'
держання зазначених вище вив
та дротяної сітки.
йтися за умови до-
Іія скляних трубок
2
21.5
шш
-
Рисунок 2.9.1.-1
Розміри зазначені у міліметрах
Методика. У кожну з шести трубок поміщають одну
таблетку або капсулу і, якщо зазначено, поміщають
диск; опускають кошик у посудину з рідиною, зазна-
ченою в загальних та окремих статтях. Вмикають при-
лад, по закінченні зазначеного часу кошик виймають і
досліджують стан таблеток або капсул.
Препарат витримав випробування, якщо всі таблетки
або капсули розпалися.
_____________________________________________N
Обладнання. Допускається підтримувати температуру
рідини від 35 °С до 39 °С.
Допускається використовувати сітку з нержавіючого
сталевого дроту, яку прикріпляють до нижньої по-
верхні нижньої пластини, з розміром отворів 0.50 мм.
2.9.2. РОЗПАДАННЯ СУПОЗИТОРІЇВ І ПЕСАРІЇВ
Випробування на розпадання дозволяє визначити,
розм’якшуються чи розпадаються ректальні або
вагінальні супозиторії чи капсули або вагінальні таб-
летки у межах установленого часу, якщо вони по-
міщені у рідке середовище в експериментальних умо-
вах, зазначених нижче.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
151
2.9. Фармако-технологічні випробування
Вважають, що зразки розпалися, якщо:
а) спостерігається повне розчинення;
в) компоненти супозиторія розділилися: розплавлені
жирові речовини зібралися на поверхні рідини, не-
розчинні часточки осіли на дно, а розчинні компо-
ненти розчинилися; залежно від складу й способу
приготування компоненти можна розподілити за
одним або кількома з вищезазначених шляхів;
с) розм’якшення зразка супроводжується помітною
зміною форми, без повного розділення компо-
нентів; розм’якшенням вважається також відсут-
ність у супозиторія твердого ядра, що чинить опір
тискові скляної палички;
б) спостерігається розрив желатинової оболонки рек-
тальної або вагінальної капсули, що дозволяє
вивільнюватися її вмісту;
е) на перфорованому диску не залишилося осаду або
осад, що залишився, складається лише з м’якої чи
піноподібної маси, яка не має твердого ядра, шо
чинить опір тискові скляної палички (вагінальні
таблетки).
Обладнання. Прилад (див. Рис. 2.9.2,-1) складається з
прозорого скляного або пластмасового порожнього
циліндра з відповідною товщиною стінок, всередині
якого за допомогою трьох тримачів закріплено мета-
левий пристрій. Пристрій являє собою два перфоро-
ваних диски з нержавіючого металу, закріплених на
відстані близько ЗО мм один від одного. Діаметр дисків
майже дорівнює внутрішньому діаметру циліндра, і в
кожному диску є 39 отворів діаметром 4 мм.
Рисунок 2.9.2-1. Прилад для визначення розпадання
ректальних і вагінальних супозиторіїв і капсул
Розміри зазначені у міліметрах
Випробування проводять, використовуючи три там
прилади, кожний з яких містить окремий зразок
Кожний прилад поміщають у посудину з термостату
ючим пристроєм, місткістю не менше 4 л, заповнену
водою з температурою від 36 °С до 37 °С, якщо немає
інших зазначень в окремій статті.
Посудина споряджена повільно рухомою мішалкою і
пристроєм, який підтримує прилад у вертикальному
положенні не менш як на 90 мм нижче від поверхи
води і дає змогу перевертати його на 180°, не виймаю-
чи з води.
Три прилади можна також помістити разом в одну по-
судину місткістю не менше 12 л.
Методика. Випробовують три супозиторія або капсу-
ли. Кожен зразок поміщають на нижній диск прист-
рою, встановлюють пристрій у циліндр приладу і
закріплюють його. Поміщають прилад у посудину і
водою і починають випробування. Прилади перевер-
тають кожні 10 хв.
Після закінчення часу, зазначеного в загальній або ок-
ремій статті, досліджують зразки.
Препарат витримує випробування, якщо всі зразки
розпалися.
МЕТОДИКА ВИПРОБУВАННЯ
ДЛЯ ВАГ1 ПАЛЬНИХ ТАБЛЕТОК
Застосовують описаний вище прилад, установлений
на тримачах (див. Рис. 2.9.2.-2). Прилад помішають)
лабораторну склянку підхожого діаметра, шо містить
воду з температурою від 36 °С до 37 °С. Поверхня води
має бути трохи нижче верхнього перфорованого дис-
ка. Потім за допомогою піпетки додають воду з темпе-
ратурою від 36 °С до 37 °С доти, доки перфорацію дис-
ка покриватиме лише однорідна плівка води.
Випробовують три вагінальні таблетки. Кожну окремо
помішають на верхній диск пристрою, накривають
прилад скляною пластинкою, щоб підтримати
відповідні умови вологості.
Після закінчення часу, зазначеного у загальній або ок-
ремій статті, досліджують зразки.
Препарат витримує випробування, якщо всі зразки
розпалися.
152
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
Рисунок 2.9.2.-2
4 скляна пластинка О — вода
В - вагінальна таблетка Е — склянка
С - поверхня води
2.9.3. ТЕСТ "РОЗЧИНЕННЯ" ДЛЯ ТВЕРДИХ
ДОЗОВАНИХ ФОРМ
ЗАГАЛЬНІ ПОЛОЖЕННЯ
Даний тест використовується для визначення ступеня
розчинення діючих речовин твердих дозованих форм
(наприклад, таблетки, капсули і супозиторії).
Для проведення тесту можливе використання приладу
злопаттю-мішалкою, кошиком або, в спеціальних ви-
падках, із проточною кюветою, якщо немає інших за-
значень в окремій статті. У кожному конкретному ви-
падку застосування тесту "Розчинення" має бути за-
значене таке:
- використовуваний прилад; у тих випадках, коли
застосовується прилад із проточною кюветою, має
бути зазначений також тип проточної кювети
(див. Рис. 2.9.3.-4/5/6);
- склад, об’єм і температура середовища розчи-
нення;
— швидкість обертання або швидкість протікання
середовища розчинення;
- час, метод і об’єм випробовуваного розчину, що
відбирається, або умови для неперервного контро-
лю;
- метод аналізу;
- кількість або кількості діючих речовин, які мають
розчинитися протягом зазначеного часу.
Обладнання. Вибір використовуваного приладу зале-
жить від фізико-хімічних характеристик дозованої
форми. Усі частини приладу, що можуть вступати в
контакт із препаратом або середовищем розчинення,
мають бути хімічно інертними, не адсорбувати, не ре-
агувати або якимось іншим чином спотворювати ре-
зультати тесту. Усі металеві частини приладу, які мо-
жуть вступати в контакт із препаратом або середови-
щем розчинення, мають бути виготовлені з відповід-
ної нержавіючої сталі або вкриті відповідним матері-
алом для того, щоб ці частини не взаємодіяли чи яки-
мось іншим чином не спотворювали результати тесту.
Прилад має бути сконструйований так, щоб звести до
мінімуму будь-які коливання і вібрацію, зумовлені
проточною системою або елементом, що плавно обер-
тається.
Бажано використовувати прилад, який дозволяє спо-
стерігати за випробовуваним препаратом і мішалкою
під час проведення тесту "Розчинення".
Прилад із лопаттю. Прилад (див Рис. 2.9.3.-1) вклю-
чає:
— циліндричну посудину з боросилікатного скла або
іншого підхожого прозорого матеріалу з напівсфе-
ричним дном і номінальним об’ємом 1000 мл;
кришку, яка уповільнює випаровування; у кришці
має бути центральний отвір для осі мішалки й інші
отвори для термометра та пристроїв, які викорис-
товують для вибирання рідини;
— мішалку, що складається з вертикального вала, на
кінець якого прикріплена лопать у формі частини
круга, відрізаного двома паралельними хордами;
лопать має проходити крізь діаметр вала таким чи-
ном, щоб нижня частина лопаті знаходилася
врівень з нижньою частиною вала; вал має розта-
шовуватися так, щоб його вісь була на відстані не
більше 2 мм від осі посудини, а нижня частина ло-
паті була на висоті (25±2) мм від внутрішньої по-
верхні дна посудини; верхня частина вала має
приєднуватися до мотора, спорядженого регуля-
тором швидкості; мішалка має обертатися плавно,
без помітного хитання;
— водяну баню, що підтримує постійну температуру
середовища розчинення (37.0±0.5) °С.
Рисунок 2.9 3.-1. Прилад з лопаттю
Розміри зазначені у м! нметрах
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
153
2.9. Фармако-технологічні випробування
Прилад із кошиком. Прилад (див. Рис. 2.9.3.-2) вклю-
чає:
— посудину, ідентичну описаній вище посудині для
приладу з лопаттю;
— мішалку, шо складається з вертикального вала, до
нижньої частини якого прикріплений циліндрич-
ний кошик, що складається з двох частин: верхня
частина з отвором діаметром 2 мм має бути прива-
реною до вала і спорядженою трьома пружними
затискачами або іншим підхожим пристосуван-
ням, що дозволяє віддаляти нижню частину коши-
ка для введення випробовуваного препарату і
міцно утримувати нижню частину концентрично з
віссю посудини під час обертання; нижня частина
кошика являє собою зварену у вигляді циліндра
оболонку з вузьким обідком листового металу
зверху і знизу; якщо немає інших зазначень в ок-
ремій статті, оболонка складається з дроту діамет-
ром 0.254 мм і площею отворів 0.381 мм2; кошик із
золотим покриттям завтовшки 2.5 мкм можна ви-
користовувати для проведення випробувань у роз-
веденому кислотному середовищі; дно кошика
має бути на висоті (25+2) мм від внутрішньої по-
верхні дна посудини; верхня частина вала має
приєднуватися до мотора, спорядженого регуля-
тором швидкості; мішалка має обертатися плавно,
без помітних коливань;
— водяну баню, що підтримує постійну температуру
середовища розчинення (37.0±0.5) °С.
Рисунок 2.9.3.-2. Прилад із кошиком
Розміри зазначені у міліметрах
Проточний прилад. Прилад (див. Рис. 2.9.3.-3) вклю-
чає.
— резервуар для середовища розчинення;
— насос, який прокачує середовище розчинени
вгору через проточну кювету;
— проточну кювету (див. Рис. 2.9.3.-4/5/6) з прозорого
матеріалу, установлену вертикально, із фільтруючою
системою, що запобігає втраті часток, які не розчи
нилися.
Резервуар для Насос
середовища
розчинення
Проточна кювета
з фільтром, яка
термостатично
контролюється
Посудина для
збирання аналі-
зованої проби
Рисунок 2 9.3.-3. Проточний прилад
Рисунок 2.9.3.-4. Проточна кювета
Розміри зазначені у міліметрах
154
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
Рисунок 2.9.3.-5. Проточна кювета
Розміри зазначені у міліметрах
Проточна кювета, показана на Рис. 2.9.3.-6, спеціаль-
но призначена для ліпофільних твердих дозованих
форм, таких як супозиторії та м’які капсули. Вона
складається з трьох прозорих частин, які вставляють-
ся одна в одну. Нижня частина (1) зроблена з двох спо-
лучених камер, приєднаних до пристрою переповнен-
ня.
Середовище розчинення проходить камерою А і
піднімається вгору. Рух потоку в камері В спрямова-
ний вниз, потім до маленької капілярної трубки, що
веде вгору до фільтруючого пристрою. Середня части-
на (2) кювети має порожнину, призначену для збиран-
ня ліпофільних допоміжних речовин, які спливають у
середовищі розчинення. Металева решітка служить
грубим фільтром. У верхній частині (3) є місце, куди
поміщається фільтр із паперу, скловолокна або целю-
лози;
- водяну баню, що підтримує постійну температуру
середовища розчинення (37.0±0.5) °С.
Рисунок 2.9.3.-6. Проточна кювета
Розміри зазначені у міліметрах
Середовище розчинення. Якщо середовищем розчинення
є буферний розчин, його рН установлюється з точністю
до ±0.05 від зазначеного значення. Перед проведенням
випробування із середовища розчинення виводяться
розчинені гази, бо вони можуть викликати утворення
бульбашок, які істотно впливають на результати.
МЕТОДИКА
Прилади з лопаттю і кошиком
Поміщають зазначений об’єм середовища розчинення у
посудину, збирають прилад, нагрівають середовище роз-
чинення до (37.0 ± 0.5) °С і видаляють термометр.
Поміщають одну одиницю випробовуваного препара-
ту в прилад. Для приладу з лопаттю: перед початком
обертання лопаті помішають препарат на дно посуди-
ни; тверді дозовані форми, що при цьому можуть
спливати, поміщають на дно посудини горизонтально
за допомогою підхожого пристрою, наприклад, дроту
або скляної спіралі.
Для приладу з кошиком: препарат поміщають у сухий
кошик, який опускають у відповідне положення перед
початком обертання.
Слід ужити заходів для недопущення наявності буль-
башок повітря на поверхні препарату. Обертання ло-
паті або кошика із зазначеною швидкістю (±4 %) по-
чинають негайно.
Проточний прилад
Для кювет, поданих на Рис. 2.9.3.-4/5. Щоб захистити
вхід до камери, призначений для рідини, на дно кону-
са поміщають одну кульку діаметром 5 мм (±0.5 мм),
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
155
2.9. Фармако-технологічні випробування
а далі — скляні кульки необхідного розміру, краще
діаметром 1 мм (±0.1 мм). За допомогою спеціального
тримача поміщають одну одиницю випробовуваного
препарату до кювети на/або всередині одержаного ша-
ру скляних кульок. Збирають фільтруючу голівку.
Нагрівають середовище розчинення до температури
(37.0±0.5) °С. Використовуючи насос, пропускають із
зазначеною швидкістю (±5 %) середовище розчинен-
ня крізь дно кювети для одержання відповідного непе-
рервного потоку через відкритий або закритий ланцюг
Для кювети, поданої на Рис. 2.9.3. 6 Поміщають одну
одиницю випробовуваного препарату в камеру А. За-
кривають кювету разом із підготовленим фільтруючим
пристроєм. На початку випробування в камері А вида-
ляють повітря через маленькій отвір, з’єднаний із
фільтруючим пристроєм. Нагрівають середовище роз-
чинення до відповідної температури, беручи до уваги
температуру плавлення препарату Використовуючи
підхожий насос, пропускають із зазначеною швид-
кістю (±5 %) нагріте середовище розчинення крізь
дно кювети, одержуючи неперервний потік через
відкритий або закритий ланцюг. Камера В заповню-
ється середовищем розчинення, коли середовище
розчинення почне переливатися через край, повітря
почне виходити через капіляр.
Препарат поширюється у середовищі розчинення
відповідно до своїх фізико-хімічних властивостей. В
обгрунтованих і дозволених випадках випробуванню
можуть піддаватися значущі частини супозиторіїв ве-
ликого розміру.
ВІДБІР ПРОБ І ОЦІНКА РЕЗУЛЬТАТІВ
У випадках використання приладу з лопаттю або ко-
шиком за означений час або із зазначеними інтерва-
лами, або неперервно здійснюють відбір зазначеного
об’єму чи об’ємів з ділянки посередині між поверхнею
середовища розчинення і верхньою частиною кошика
або лопаті на відстані не ближче 10 мм від стінки посу-
дини. У випадку використання приладу із проточною
кюветою відбір проб завжди проводять біля вихідного
отвору кювети, незалежно від того, відкритий ланцюг
чи закритий.
Треба компенсувати відібраний об’єм рідини додаван-
ням такого самого об’єму середовища розчинення або
відповідними змінами у розрахунках, виключаючи ті ви-
падки, коли використовуються неперервні виміри при
проведенні випробувань із лопаттю або кошиком (ві-
дібрана рідина при цьому повертається назад до посуди-
ни), або коли відбирається лише одна порція рідини.
Відібрану рідину фільтрують, використовуючи інерт-
ний фільтр із відповідним розміром пор, який не ви-
кликає значної адсорбції активного компонента з роз-
чину і не містить таких речовин, які екстрагуються се-
редовищем розчинення і могли б впливати на резуль-
тати зазначеного аналітичного методу. Аналіз фільтра-
ту проводять методом, зазначеним в окремій статті.
Кількість діючої речовини, що розчинилася упродовж
зазначеного часу, виражається у відсотках від вмісту
зазначеного у розділі "Склад".
_____________________________________________А
ЗАГАЛЬНІ ПОЛОЖЕННЯ'
Якщо немає інших зазначень в окремій статті, прове-
дення тесту "Розчинення" не є обов’язковим для жу-
вальних таблеток, полівітамінних препаратів та к І
інших випадках, для яких обгрунтовано нсінформа-
тивність даного тесту.
Під ступенем розчинення твердої дозованої форми
розуміють кількість діючої речовини, у відсотках, від
вмісту, зазначеного в розділі "Склад", яка в умова*. І
описаних в окремій статті, має перейти у розчин.
Як середовище розчинення можуть використовувати-
ся вода Р, 0.1 М розчин кислоти хлористоводневої, фос-
фатні буферні розчини з рН від 6.8 до 7.6 та інші водні
розчинники. Неводні розчинники у середовищах роз-
чинення використовують у виняткових випадках, і їхні
застосування вимагає додаткового обгрунтування.
Звичайними середовищами розчинення є вода Рабо
0.1М розчин кислоти хлористоводневої. Для кишково-
розчинних твердих дозованих форм і форм із заданим
ступенем вивільнення умови проведення тесту "Роз-
чинення" зазначають в окремій статті.
Перед проведенням випробування із середовища розчи-
нення видаляють розчинені гази, наприклад, фільтру-
ванням під вакуумом або обробкою ультразвуком.
Звичайний об’єм середовища розчинення — 900 -
1000 мл, температура середовища розчинення -
(37.0±0.5) °С.
Під час використання приладу з лопаттю або з коти- І
ком швидкість обертання становить звичайно 50 об/хв І
для лопаті і 100 об/хв — для кошика.
Звичайно у прилади для проведення тесту "Розчинен-
ня" поміщають одну одиницю випробовуваного пре
парату, однак можливе вміщення і кількох одиниць
одночасно. У цьому випадку при проведенні оцінки
результатів дана сукупність одиниць розглядається як І
одна одиниця випробовуваного препарату з відповід- І
ними змінами у розрахунках.
Обладнання. Вибір використовуваного приладу зале-
жить від фізико-хімічних характеристик твердої дозо-
ваної форми. Найбільш поширеними є прилади з ко-
шиком і лопаттю. Проточний прилад звичайно
доцільно застосовувати в тому випадку, коли діючі ре- І
човини досліджуваного препарату погано розчинніІ
волі й водних середовищах розчинення.
Бажано застосовувати прилад із зазначеними техніч-
ними параметрами, однак, якщо необхідно, в них мо-
жуть бути внесені обгрунтовані зміни.
МЕТОДИКА
Прилади з лопаттю і кошиком. Поміщають зазначений
об’єм середовища розчинення у посудину, збирають
прилад, нагрівають середовище розчинення дотемпе-
156
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
(тури (37.0±0.5) °С і видаляють термометр у тих ви-
Ьшх, коли це необхідно.
ІДБІР ПРОБ 1 ОЦІНКА РЕЗУЛЬТАТІВ
Уін. випадках, коли регламентується ступінь розчи-
неная лише за один проміжок часу, тест може бути
! Іркдениіі і за коротший час. Якщо ж регламен-
ться ступінь розчинення за два або більше
нжків часу, то відбір проб має здійснюватися без
риппнення роботи приладу за суворо обумовлений
з точністю (+2 %).
"“зводять паралельно дослідження розчинення для
іік-.ти одиниць випробовуваного препарату. Якщо не-
чл інших зазначень в окремій статті, для кожної оди-
ішні випробовуваного препарату за 45 хв у розчин має
ерейти не менше 75 % і не більше 115 % діючої речо-
ини від її вмісту, зазначеного в розділі "Склад". Якшо
аз одиниць випробовуваного препарату не
іповідає цій вимозі, проводять дослідження розчи-
нна ше шести одиниць випробовуваного препарату,
кідодаткові шість одиниць випробовуваного препа-
щгу мають відповідати вищезазначеній вимозі.
1 разі використання в тесті "Розчинення" сукупності
зіниць, яка вважається однією одиницею випробо-
ілзаного препарату, проводять паралельно дослід-
. ння розчинення для шести таких одиниць.
Одержані результати перераховують на одну одиницю
іозованого лікарського засобу. Якщо немає інших
означень в окремій статті, для кожної одиниці
аіпробовуваного препарату за 45 хв до розчину має
перейти не менше 75% і не більше 115% діючої
речовини від її вмісту, зазначеного у розділі "Склад".
Додаткові випробування в даному випадку не
проводять.
У разі застосування тесту "Розчинення" для твердих
юзованих форм із кількома діючими речовинами
можлива регламентація ступеня розчинення лише
однієї з діючих речовин, якщо вона (регламентація)
идовольняє вищезазначені вимоги, і за умови, шо ре-
шта діючих речовин має більш високий ступінь розчи-
Таблиця 2.9.5.-1
Лікарська форма Середня маса Припустиме відхилення, %
Таблетки (без оболонки і покриті плівковою оболонкою) 80 мг і менше 10
Більше 80 мг, але менше 250 мг 7.5
250 мг і більше 5
Капсули, гранули (без покриття, однодозові) і порошки (однодозові) Менше 300 мг 10
300 мг і більше 7.5
Стерильні порошки для парентерального застосування * (однодозові) Більше 40 мг 10
Супозиторії і песарії Для всіх випадків 5
‘Якщо середня маса стерильного порошку для парентерального за-
стосування дорівнює 40 мг і менше, препарат підлягає випробуван-
ню на однорідність вмісту діючої речовини в одиниці дозованого
лікарського засобу (2.9.6) і не підлягає випробуванню на од-
норідність маси.
Для капсул і стерильних порошків для парентерально-
го застосування випробування проводять, як описано
нижче.
КАПСУЛИ
Зважують нерозпаковану капсулу. Потім розпакову-
ють капсулу в такий спосіб, щоб не була втрачена
будь-яка частина оболонки, і видаляють якомога
повніше її вміст. Якщо капсули з м’якою оболонкою,
промивають оболонку ефіром або іншим підхожим
розчинником і залишають на повітрі до видалення за-
паху розчинника. Потім зважують оболонку. За різни-
цею зважувань розраховують масу вмісту капсули. По-
вторюють процедуру з іншими 19 капсулами.
1.9.5. ОДНОРІДНІСТЬ МАСИ ДЛЯ ОДИНИЦІ
ДОЗОВАНОГО ЛІКАРСЬКОГО ЗАСОБУ
!0 одиниць дозованого лікарського засобу або вміст
.ожного з 20 контейнерів, у випадку однодозових лі-
карських засобів в індивідуальних контейнерах, відби-
иють за статистично обгрунтованою схемою, зважують
:сжну окремо і розраховують середню масу. Лікарський
асіб витримав випробування, якщо не більше двох
ндивідуальних мас відхиляються від середньої маси на
«личину, яка перевищує значення, зазначене у
Габл. 2 9.5.-1. При цьому жодна індивідуальна маса не
іає відхилятися від середньої маси на величину, що у
іварази перевищує значення, зазначене вТабл. 2.9.5.-1.
СТЕРИЛЬНІ ПОРОШКИ
ДЛЯ ПАРЕНТЕРАЛЬНОГО ЗАСТОСУВАННЯ
Видаляють паперову етикетку з поверхні контейнера.
Контейнер миють і сушать. Потім контейнер роз-
кривають і зразу зважують. Обережним постукуван-
ням звільняють якомога повніше контейнер від вмісту,
обполіскують його, якщо необхідно, водою Рі 96%
спиртом Р і сушать при температурі від 100 °С до
105 °С протягом 1 год або, якшо природа контейнера
не дозволяє використовувати нагрівання при такій
температурі, сушать при більш низькій температурі до
постійної маси. Після цього охолоджують в ексикаторі
і зважують. За різницею зважувань розраховують масу
вмісту контейнера. Повторюють процедуру з іншими
19 контейнерами.
ФАРМАКОПЕЯ УКРАЇНИ 2001
157
2.9. Фармако-технологічні випробування
___________________________________N
Випробування "Однорідність маси для одиниці дозова-
ного лікарського засобу" не застосовують у тих випадках,
коли для всіх діючих речовин дозованого лікарського
засобу проводять випробування "Однорідність вмісту
діючої речовини в одиниці дозованого лікарського засобу"
(2.9.6), якщо немає інших зазначень в окремій статті.
2.9.6. ОДНОРІДНІСТЬ ВМІСТУ ДІЮЧОЇ
РЕЧОВИНИ В ОДИНИЦІ ДОЗОВАНОГО
ЛІКАРСЬКОГО ЗАСОБУ
Випробування на однорідність вмісту діючої речови-
ни в одиниці дозованого лікарського засобу грун-
тується на кількісному визначенні вмісту в індивіду-
альних однодозових одиницях препарату з метою
з’ясування, чи знаходиться цей вміст у межах, вста-
новлених стосовно середнього вмісту у випробову-
ваному зразку.
Таке випробування не проводиться для полівітамін-
них лікарських засобів і для лікарських засобів, що
містять мікроелементи, якщо немає інших зазначень
в окремій статті.
Метод. Використовуючи підхожу аналітичну методи-
ку, визначають вміст діючої речовини в кожній
з ІОдозованиходинипьлікарськогозасобу, відібраних
за статистично обгрунтованою схемою.
Застосовують критерії тестів А, В або С, як зазначено
у статті для випробовуваної дозованої форми.
ТЕСТ А
Таблетки, порошки для парентерального застосування,
суспензії для ін’єкцій. Препарат витримує випробуван-
ня, якщо вміст у кожній його однодозовій одиниці пе-
ребуває в межах 85 — 115 % від середнього вмісту.
Препарат не витримує випробування, якщо вміст у
більш як одній одиниці виходить за вищезазначені
межі або якщо вміст хоча б в одній одиниці виходить
за межі 75 — 125 % від середнього вмісту.
Якщо вміст в одній одиниці препарату виходить за
межі 85 — 115%, але перебуває у межах 75 — 125 %,
визначають вміст у кожній з 20 додаткових однодозо-
вих одиниць препарату, відібраних за статистично об-
грунтованою схемою. Препарат витримує випробу-
вання, якщо вміст не більш як в одній з проаналізова-
них ЗО одиниць виходить за межі 85 — 115 % і в жодній
одиниці не виходить за межі 75 — 125 % від середньо-
го вмісту.
ТЕСТ В
Капсули, порошки не для парентерального застосування,
гранули, супозиторії, песарії. Препарат витримує випро-
бування, якщо вміст не більш як в одній одиниці вихо-
дить за межі 85 — 115 % і в жодній одиниці не виходить
за межі 75 — 125 % від середнього вмісту в препараті.
Препарат не витримує випробування, якщо вміст більш І
як у трьох одиницях виходить за межі 85 — 115 % від се-
реднього вмісту або якщо хоча б в одній одиниці вихо-
дить за межі 75 — 125 % від середнього вмісту.
Я кщо вміст у двох або трьох одиницях препарату вихо-
дить за межі 85 — 115 %, але знаходиться у межах
75— 125 %, визначають вмісту кожній з20додаткових
однодозових одиниць препарату, відібраних за статис-
тично обгрунтованою схемою. Препарат витримує ви-
пробування, якщо середній вміст не більш як у трьохз
проаналізованих ЗО одиниць виходить за межі 85 -
115 % і в жодній одиниці не виходить за межі 75-
125 % від середнього вмісту.
ТЕСТС
Трансдермальні пластирі. Препарат витримує випробу-
вання, якщо середній вміст у 10 однодозових одини-
цях знаходиться у межах 90 — 110 % від вмісту, зазна-
ченого у розділі "Склад", і якщо вміст у кожній з
10 одиниць знаходиться у межах 75 — 125 % від серед-
нього вмісту.
____________________________________________А
У тому випадку, коли виробництво дозованого
лікарського засобу не проводиться відповідно до ви-
мог належної виробничої практики (НВП, СМР),
встановлених у Європейському Співтоваристві, дода-
ного лікарського засобу ставляться такі вимоги щодо
однорідності вмісту1.
ЗАГАЛЬНІ ПОЛОЖЕННЯ
Однорідність вмісту діючої речовини (ОВДР) в одиниці
дозованого лікарського засобу являє собою характери-
стику розподілу вмісту діючої речовини (речовин) за
одиницями даного лікарського засобу (таблетки, кап-
сули, супозиторії, ліофілізовані лікарські засоби і сте-
рильні розсипки лікарських засобів у однодозових кон-
тейнерах, гранули в однодозових контейнерах і т. д.).
Вимоги даної статті поширюються на дозовані лі-
карські засоби, що містять одну і більше діючих речо-
вин, якщо немає інших зазначень в окремій статті.
Вимоги даної статті не поширюються на дозовані
полівітамінні лікарські засоби і на лікарські засоби
які містять мікроелементи, якщо немає інших
зазначень в окремій статті
Для дозуючих аерозолей вимоги ОВДР мають бути на-
ведені в окремій статті.
Для визначення ОВДР треба застосовувати один з
двох методів:
1) метод прямого визначення;
2) розрахунково-ваговий метод.
Визначення ОВДР методом прямого визначення до-
пускається для всіх дозованих лікарських засобів, які
підлягають випробуванню. Використання даного ме-
1 У даних вимогах використані матеріали Фармакопеї США — з
дозволу Фармакопеї США.
158
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
толу обов’язкове у таких випадках:
- для таблеток, покритих та не покритих оболон-
кою, твердих і м’яких капсул, що містять діючу ре-
човину в кількості 50 мг і менше;
- для супозиторіїв і песаріїв;
- для дозованих транедермальних пластирів;
- для суспензій в однодозових контейнерах або
м'яких капсулах;
- для гранул і стерильних розсипок лікарських за-
собів в однодозових контейнерах, які містять інші
діючі й допоміжні речовини у кількості менше
50 % за масою.
Визначення ОВДР розрахунково-ваговим методом
допускається у таких випадках:
- для м’яких капсул з рідким вмістом;
- для гранул та стерильних розсипок лікарських за-
собів у однодозових контейнерах, які не містять
інших діючих або допоміжних речовин або містять
інші діючі й допоміжні речовини в кількості більш
як 50 % за масою (наприклад, стерильні розсипки
антибіотиків);
- для ліофілізованих лікарських засобів в однодозо-
вих контейнерах як таких, шо містять, так і таких,
що не містять інших діючих і допоміжних речовин
(наприклад, для ліофілізованих ін’єкційних
лікарських засобів).
МЕТОД ПРЯМОГО ВИЗНАЧЕННЯ
Від серії дозованого лікарського засобу, що підлягає
випробуванню, відбирають за статистично обгрунто-
ваною схемою пробу в кількості ЗО одиниць. З них у
довільному порядку відбирають 10 одиниць.
У кожній з 10 відібраних одиниць визначають кіль-
кісний вміст діючої речовини за методикою, зазначе-
ною в окремій статті.
РОЗРАХУНКОВО-ВАГОВИЙ МЕТОД
Від серії дозованого лікарського засобу, що підлягає
випробуванню, відбирають за статистично обгрунто-
ваною схемою пробу в кількості ЗО одиниць, які
досліджуються за нижченаведеними методиками з
точністю зважування 0.0002 г.
М’які капсули. Зважують точно кожну з 10 відібраних
неушкоджених капсул, ретельно стежачи за їхньою
цілісністю. Розрізають капсули за допомогою підхо-
жого сухого і чистого різального інструмента, напри-
клад, ножиць або скальпеля, і вимивають вміст підхо-
жим розчинником, який добре розчиняє вміст, але не
розчиняє оболонку капсули. Дають можливість роз-
чиннику випаритися при кімнатній температурі з по-
верхні оболонок. Зважують точно кожну з оболонок і
розраховують масу вмісту. За результатами кількісного
визначення, проведеного відповідно до окремої
статі і, розраховують вміст діючої речовини в кожній з
капсул, вважаючи розподіл діючої речовини за масою
вмісту капсул однорідним.
Гранули в однодозових контейнерах. Зважують точно
кожен з 10 відібраних однодозових контейнерів. Роз-
кривають, ретельно видаляють вміст кожного контей-
нера, зважують точно кожний порожній контейнер і
розраховують масу вмісту. За результатами кількісного
визначення, проведеного відповідно до окремої
статт і, розраховують вміст діючої речовини в кожному
з контейнерів, вважаючи розподіл діючої речовини за
масою гранул однорідним.
Ліофілізовані та стерильні розсипки лікарських засобів у
однодозових контейнерах. Тест проводиться так само,
як зазначено више в розділі “Гранули в однодозових
контейнерах”.
РОЗРАХУНОК ВІДНОСНОГО СТАНДАРТНОГО
ВІДХИЛЕННЯ
1 «
х = ~іг
де:
К8О — відносне стандартне відхилення (стан-
дартне відхилення, виражене у відсотках
до середнього результату);
X — середній результат одиничного визначен-
ня;
п — число випробовуваних дозованих оди-
ниць лікарського засобу;
X), х2 ...х„ — індивідуальні значення, одержані для
випробовуваних дозованих одиниць.
КРИТЕРІЇ
Якщо спеціально не зазначено в окремій статті, засто-
совують такі критерії:
(А) Для симетричних меж вмісту діючої речовини або в
тому випадку, коли півсума верхньої та нижньої меж
вмісту діючої речовини менша за зазначену в розділі
"Склад".
Пресовані таблетки (покриті оболонкою і без), супози-
торії, песарії, суспензії в однодозових контейнерах, гра-
нули, ліофілізовані та стерильні розсипки лікарських за-
собів у однодозових контейнерах. Якщо інші критерії не
зазначені в окремій статті, вимоги ОВДР вважаються
виконаними за умови, що вміст діючої речовини у
кожній з 10 одиниць, визначений за Розрахунково-ва-
говим методом або Методом прямого визначення, зна-
ходиться в межах 85.0 — 115.0 % від зазначеного в роз-
ділі "Склад”, а Відносне стандартне відхилення не пере-
вищує 6.0 %.
Якщо хоча б в одній одиниці дозованого лікарського
засобу вміст діючої речовини виходить за межі
85.0 — 115.0 %, але при цьому в усіх одиницях знахо-
диться в межах 75.0 — 125.0 % від зазначеного у розділі
"Склад”, або якщо Відносне стандартне відхилення пе-
ревищує 6.0 %, або порушені одночасно обидві умови,
слід піддати аналізу додаткові 20 одиниць дозованого
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
159
2.9. Фармако-технологічні випробування
лікарського засобу. Вимоги ОВДР вважаються вико-
наними, якщо не більш як в одній одиниці з ЗО вміст
діючої речовини виходить за межі 85.0 — 115.0 % і при
цьому в усіх одиницях знаходиться у межах
75.0 — 125.0 %, а Відносне стандартне відхилення для
ЗО одиниць не перевищує 7.8 %.
Капсули, трансдермальні пластирі і формовані (зокрема,
тритурапійні)таблетки. Якшо інші критерії не зазначені
в окремій статті, вимоги ОВДР вважаються виконани-
ми за умови, що не менш як у дев’яти з 10 одиниць до-
зованого лікарського засобу вміст діючої речовини
знаходиться в межах 85.0 — 115.0 % від зазначеного в
розділі "Склад" і при цьому в жодній з одиниць не ви-
ходить за межі 75.0 — 125.0 %, а Відносне стандартне
відхилення для 10 одиниць не перевищує 6.0 %.
Якщо в двох або трьох одиницях вміст діючої речови-
ни виходить за межі 85.0 — 115.0 %, але не виходить за
межі 75.0 — 125.0 %, або якщо Відносне стандартне
відхилення перевищує 6.0 %, або якщо одночасно по-
рушуються обидві умови, слід піддати аналізу додат-
кові 20 одиниць дозованого лікарського засобу. Вимо-
ги ОВДР вважаються виконаними, якщо не більш як у
трьох одиницях з 30 вміст діючої речовини виходить за
межі 85.0 — 115.0 % від зазначеного у розділі "Склад" і
при цьому жодна з одиниць не виходить за межі
75.0 — 125.0 %, а Відносне стандартне відхилення для
30 одиниць не перевищує 7.8 %.
(Б) Якщо півсума верхньої і нижньої меж кількісного
вмісту діючої речовини більша за зазначену в розділі
"Склад".
(1) Якщо фактично знайдене середнє значення
кількісного вмісту діючої речовини для випробо-
вуваних одиниць дозованого лікарського засобу мен-
ше або дорівнює зазначеному в розділі "Склад", вимо-
ги такі самі, як у випадку (А).
(2) Якщо фактично знайдене середнє значення
кількісного вмісту діючої речовини для випробо-
вуваних одиниць дозованого лікарського засобу
більше або дорівнює півсумі верхньої і нижньої меж
кількісного вмісту діючої речовини, вимоги ОВДР
такі самі, як у випадку (А), за тим винятком, що слова
"зазначене в розділі "Склад" замінюються словами
"півсума верхньої і нижньої меж кількісного вмісту
діючої речовини".
(3) Якщо фактично знайдене середнє значення
кількісного вмісту діючої речовини для випробо- І
вуваних одиниць дозованого лікарського засобу лежить
в інтервалі між зазначеним у розділі "Склад" і півсумою
верхньої і нижньої меж кількісного вмісту діючої речо
вини, вимоги ОВДР такі самі, як у випадку (А), за тим
винятком, що слова "зазначене в розділі "Склад"
замінюються словами "середнє значення для ви-
пробовуваних одиниць дозованого лікарського засобу" |
У всіх інших випадках, не описаних у розділах А і Б, І
додаткові визначення не проводяться, і лікарський за- І
сіб вважається таким, що не витримав випробування.
2.9.7. СТИРАНІСТЬ ТАБЛЕТОК БЕЗ ОБОЛОНКИ
Випробування дозволяє визначити стираність табле-
ток без оболонки за певних умов, тобто ушкодження
поверхні таблеток під дією механічного удару або сти-
рання.
Обладнання. Використовують барабан із внутрішнім
діаметром близько 286 мм і завглибшки близько
39 мм, виготовлений із прозорого синтетичного
полімеру; внутрішні поверхні барабана мають бути
відполіровані й не мають електризуватись (див.
Рис. 2.9.7.-1). Одна сторона барабана знімна. При
кожному оберті барабана таблетки приводяться в рух
за допомогою зігнутої лопаті, розташованої між цент-
ром барабана і його зовнішньою стінкою. Барабан
прикріплюється до горизонтальної осі пристрою, шо
забезпечує швидкість обертання приблизно 25 об/хв.
Отже, при кожному оберті барабана таблетки пада-
ють, перевертаючись або ковзаючи, з висоти приблиз-
но 130 мм на стінку барабана або одна на одну.
0286
Рисунок 2.9.7.-1. Прилад для визначення стираності таблеток
Розміри зазначені у міліметрах
160
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
Методика. При масі однієї таблетки менше 0.65 гдля
випробування беруть 20 таблеток; при масі однієї таб-
летки більше 0.65 г — 10 таблеток. Таблетки поміща-
ють на сито номер 1000 і ретельно видаляють пил за
допомогою стисненого повітря або м’якого пензлика.
Таблетки зважують (точна наважка) і поміщають у ба-
рабан. Після 100 обертів барабана таблетки виймають
ізнов ретельно видаляють пил. Якщо на жодній з таб-
леток немає ознак сколювання або тріщин, таблетки
зважують з точністю до міліграма.
Звичайно випробування проводять один раз. Якщо
одержані результати викликають сумнів або втрата в
масі перевищує 1 %, випробування повторюють ще
двічі й обчислюють середнє з трьох вимірів. Якщо не-
має інших зазначень в окремій статті, втрата в масі має
бути не більше 1 % від сумарної маси випробовуваних
таблеток.
При випробуванні таблеток з діаметром 13 мм і більше
для одержання відтворюваних результатів може ви-
никнути потреба відрегулювати барабан таким чином,
шоб таблетки, що лежать поруч, не упиралися одна в
одну і мали можливості падати вільно. Звичайно до-
статньо установити вісь під кутом 10° до основи.
Подання результатів. Стираність виражають втратою в
масі, обчисленою у відсотках від вихідної маси випро-
бовуваних таблеток.
Необхідно зазначати число таблеток, взятих для ви-
пробування.
0200
Рисунок 2.9.7.-2. Прилад для визначення
стираності таблеток
Розміри зазначені у міліметрах
____________________________________________N
Обладнання. Допускається використання приладу
(див. Рис 2.9.7.-2), що складається з барабана діамет-
ром близько 200 мм і завглибшки близько 38 мм;
внутрішні поверхні барабана, виготовленого із
прозорого синтетичного полімеру, мають бути відпо-
ліровані й не мають електризуватись. Одна сторона
барабана знімна. По внутрішньому периметру стінки
розташовані 12 лопатей (35 мм х 35 мм) під кутом 20°
до дотичної барабана, які при його обертанні надають
руху таблеткам. Барабан прикріплюється до горизон-
тальної осі пристрою, що забезпечує швидкість обер-
тання приблизно 20 об/хв. Прилад споряджений го-
динником, який автоматично вимикає пристрій, коли
заданий час випробування закінчується.
Методика. Якщо хоча б в одній з 10 випробовуваних
таблеток виявляють тріщини або сколювання, випро-
бування проводять додатково на 20 таблетках.
Якщо хоча б в одній з 20 випробовуваних таблеток ви-
являють тріщини або сколювання, серія таблеток не
витримує випробування на стираність.
Подання результатів. Стираність виражають втратою в
масі, обчисленою у відсотках від вихідної маси ви-
пробовуваних таблеток.
2.9.8. СТІЙКІСТЬ ТАБЛЕТОК
ДО РОЗДАВЛЮВАННЯ
Випробування дозволяє визначити стійкість табле-
ток до роздавлювання за певних умов шляхом вимі-
рювання сили, необхідної для руйнування таблеток.
Обладнання. Прилад являє собою два розташовані
один проти одного затискачі, один із яких може пе-
ремішуватися в напрямку до другого. Площини по-
верхонь затискачів перпендикулярні напрямку пряму-
вання. Здавлюючі поверхні затискачів мають бути
плоскими і перевищувати за розміром зону контакту з
таблеткою. Прилад калібрують із використанням сис-
теми, що забезпечує точність 1 Н (Ньютон).
Методика. Таблетку поміщають між затискачами, бе-
ручи до уваги її форму, а також розділювальну лінію і
напис, якшо вони є. Для всіх вимірів таблетка має бу-
ти орієнтованою однаково стосовно напрямку сили,
що прикладається. Виміри проводять для 10 таблеток.
Перед кожним виміром ретельно видаляють усі фраг-
менти попередньої таблетки.
Ця методика не застосовна при використанні пов-
ністю автоматизованого приладу.
Подання результатів. Необхідно зазначати середнє,
мінімальне і максимальне значення виміряної сили в
ньютонах.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
161
2.9. Фармако-технологічні випробування
Також зазначають тип використаного приладу і, якщо
необхідно, орієнтацію таблеток.
_____________________________________________N
Обладнання. Допускається використання приладу з
затискачами, що можуть переміщуватися із постійною
швидкістю у напрямі один до одного. Прилад має за-
безпечувати припинення здавлювання при будь-яко-
му порушенні цілісності таблетки.
Методика. Таблетку поміщають між затискачами на
ребро, якщо немає інших зазначень в окремій статті.
Таблетки повинні мати стійкість до роздавлювання не
нижче значень, наведених у Табл. 2.9.8.-1, якщо в ок-
ремій статті немає інших зазначень:
Таблиця 2.9.8,-1
Діаметр, мм Стійкість до роздавлювання, Н
6 10
7 20
8 25
9 зо
10 зо
11 40
12 50
13 50
Для таблеток, призначених для здрібнювання або роз-
жовування, в окремій статті зазначають верхню межу
стійкості до роздавлювання.
ня маси порошку, що пройшов крізь сито (сита), дозі-1
гальної маси випробовуваного порошку, у відсотка!
(м/м).
Якщо зазначено один номер сита, то не менше 97 я
маси порошку має проходити крізь зазначене ситої
якщо немає інших зазначень в окремій статті.
Для визначення здрібненості порошку збирають сита,
порошок повністю просіюють і зважують кожної
фракцію.
______________________________________________л
В окремій статті зазначають наважку порошку, час і
умови просіювання.
2.9.13. ВИЗНАЧЕННЯ РОЗМІРУ ЧАСТОК
ПОРОШКІВ МЕТОДОМ МІКРОСКОПІЇ І
Відповідну кількість порошку (наприклад, від 10 мгло
100 мг) суспендують у 10.0 мл підхожої рідини, у якій
порошок не розчиняється, додаючи, якщо необхідної
речовину, що покращує змочуваність часток порошку
Порцію гомогенної суспензії поміщають у підхожу
лічильну чашку і переглядають під мікроскопом пло-
щу, відповідну не менше 10 мкг випробовуваного по-
рошку. Підраховують усі частки, які мають розміри,
що виходять за межі зазначеного інтервалу. Допустиму
кількість часток, що виходить за межі інтервалу, зазна-
чають в окремій статті.
2.9.12. СИТОВИЙ АНАЛІЗ
Здрібненість порошку може бути виражена розмірами
отворів сит відповідно до Табл. 2.1.4.-1.
Здрібненість порошку визначають просіюванням
крізь сита з певними номерами і виражають поданими
нижче термінами.
Грубий порошок. Не менше 95 % маси порошку має
проходити крізь сито номер 1400 і не більше 40 % ма-
си порошку — крізь сито номер 355.
Середньо-дрібний порошок. Не менше 95 % маси по-
рошку має проходити крізь сито номер 355 і не більше
40 % маси порошку — крізь сито номер 180.
Дрібний порошок. Не менше 95 % маси порошку має
проходити крізь сито номер 180 і не більше 40 % маси
порошку — крізь сито номер 125.
Дуже дрібний порошок. Не менше 95 % маси порошку
має проходити крізь сито номер 125 і не більше
40 % маси порошку — крізь сито номер 90.
Якщо такі терміни не можуть бути використані,
здрібненість порошку виражають у вигляді відношен-
2.9.15. НАСИПНИЙ ОБ’ЄМ
Випробування дозволяє визначити за заданих умов
насипний об’єм та насипну густину матеріалу, шо
складається з твердих часток (наприклад, порошків,
гранул), до усадки, здатність матеріалу до усадки, а та-
кож його об’єм і густину після усадки.
Обладнання. Прилад (див. Рис. 2.9.15.-1) складається з
таких частин:
— струшувальний пристрій, що забезпечує 250±15
зіскоків циліндра за хвилину з висоти (3±0.2) мм;
підставка для градуйованого циліндра, спорядже-
на тримачем, має масу (450±5) г;
— градуйований циліндр місткістю 250 мл (ціна
поділки — 2 мл); маса циліндра — (220±40) г.
Методика. У сухий циліндр поміщають без ущільнен-
ня 100.0 г (т — маса наважки, у грамах) випробовува-
ного матеріалу. Якщо це неможливо, беруть наважку
випробовуваного матеріалу, що має насипний об’єму
діапазоні 50 мл — 250 мл; цю наважку зазначають у
звіті. Закріплюють циліндр на підставці й фіксують
насипний об’єм до усадки Уо. Проводять 10 , 500, 1250
зіскоків циліндра і фіксують об’єми УІ0, У500, УП50
з точністю до найближчої позначки. Якщо різниця
МІЖ У500 І У]250 перевищує 2 мл, проводять ще
1250 зіскоків циліндра.
162
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
51
Підставка
я циліндра
Градуйований
циліндр
Ексцентрик
..і тни
Цей розмір має бути таким,
щоб зазор (1) задовольняв
вимоги специфікацій і щоб
при цьому в нижній точці
ексцентрика підставка ци-
ліндра вільно сідала б на
верхнє ковадло
Рисунок 2.9.16.-1. Лійка і насадка
Насадку виготовляють з нержавіючої
кислототривкої сталі (У4А, СгИі)
Розміри зазначені у міліметрах
Рисунок 2.9.15.-1
Подання результатів
а) Об’єми:
- насипний об’єм — об’єм до усадки: Уо, мл;
- об’єм після усадки: И/250, мл або У2500, мл:
Ь) Здатність до усадки: різниця об’ємів УІ0, мл
- Ут, мл:
с) Густини:
- насипна густина — густина до усадки: т/У0, г/мл;
- густина після усадки: т/УІ250, г/мл або т/У2500, г/мл.
______________________________________________N
Обладнання. Допускається використання інших при-
ладів.
2.9.16. ПЛИННІСТЬ
Випробування дозволяє визначити здатність ма-
теріалів, що складаються зтвердих часток (наприклад,
порошків, гранул), текти у вертикальному напрямку
за заданих умов.
Обладнання. У залежності від плинності випробовува-
них матеріалів використовують лійки без вихідного
ствола з різними розмірами вихідних отворів і лійки з
вихідним стволом. Кути цих лійок різні. Типові лійки
показані на Рис. 2.9.16.-1 і 2.9.16.-2. Лійка підтри-
мується у вертикальному положенні за допомогою
спеціального пристрою. Вся конструкція має бути за-
хищена від вібрацій (метод нерухомої лійки).
Рисунок 2.9.16.-2
Розміри зазначені у міліметрах
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
163
2.9. Фармако-технологічні випробування
Насадка Діаметр (<У вихідного отвору, мм
1 10 ±0.01
2 15 ±0.01
3 25 ±0.01
Методика. У суху лійку, вихідний отвір якої закритий
тим чи іншим способом, помішають без ущільнення
наважку випробовуваного матеріалу, взяту з точністю
0.5 %. Кількість випробовуваного матеріалу залежить
від його насипного об'єму і від використовуваного
приладу. Відкривають вихідний отвір і визначають час,
необхідний для повного витікання зразка з лійки.
Проводять три виміри.
Подання результатів. Плинність виражають у секундах
і десятих частках секунди, віднесених до 100 г зразка.
Результати залежать від умов зберігання випробовува-
ного матеріалу.
Результати можуть бути подані таким чином:
а) як середнє значення одержаних результатів за умо-
ви, що жоден із результатів не відхиляється від се-
реднього більш як на (±10 %);
Ь) у вигляді діапазону значень, якшо окремі результа-
ти відхиляються від середнього значення більш як
на (±10 %);
с) у вигляді графіка залежності маси від часу витікання,
б) якщо зразок повністю не витік із лійки, зазначають
нескінченний час.
________________________________________________N
Метод нерухомої лійки. Методика. Кількість випробо-
вуваного матеріалу має займати об’єм не менше
90 % об’єму лійки. Використовують лійку, подану на
Рис. 2.9.16.-1 з насадкою 1. Відкривають вихідний
отвір і визначають час, необхідний для повного
витікання зразка з лійки. Якшо наважка випробовува-
ного матеріалу рівномірно висипається не менш як за
25 с, рекомендується використовувати лійку, подану на
Рис. 2 9.16.-2. шо може бути виготовлена також ізскла
Якшо наважка випробовуваного матеріалу не виси-
пається рівномірно з лійки (Рис. 2.9.16.-1) з насад-
кою 1, послідовно випробовують плинність, викорис-
товуючи лійку з насадкою 2 або 3.
Подання результатів. Необхідно зазначати лійку і на-
садку, використовувані при визначенні плинності.
Метод лійки з вібропристроєм. Допускається проводи-
ти визначення плинності з використанням лійки з
вібропристроєм (Рис. 2.9.16.-3), шо забезпечує амплі-
туду коливань від 0.04 мм до 0.1 мм за частоти 50 Гц.
Конструкція має забезпечувати стійкість приладу при
вібрації.
Методика. У суху лійку, вихідний отвір якої закритий
заслінкою, поміщають без ущільнення наважку ви-
пробовуваного матеріалу з точністю 0.25 г. Вмикають
вібропристрій і через 20 с відкривають заслінку.
Визначають час, необхідний для повного виті]
зразка з лійки. Проводять три виміри.
Рисунок 2 9.16.-3. Лійка і насадка
Розміри зазначені у міліметрах
Подання результатів. Результати подають, як зазначено
для методу нерухомої лійки. Необхідно зазначати ме-
тод і розмір отвору лійки, використовуваної при
визначенні плинності.
2.9.17. ОБ’ЄМ, ЩО ВИТЯГАЄТЬСЯ
Вимоги даної статті поширюються на ін’єкційні та
інфузійні лікарські засоби для парентерального засто-
сування.
ІН’ЄКЦІЙНІ ЛІКАРСЬКІ ЗАСОБИ
Однодозові препарати можуть випускатися в однодо-
зових контейнерах, картриджах або попередньо на-
повнених шприцах.
Однодозові контейнери
Об’єм ін’єкційного розчину в однодозовому контей-
нері має бути достатнім для гарантованого добування
номінальної дози. Цей об’єм більший за номінальний,
що може бути небезпечним у випадку введення всього
вмісту.
Відповідність вимозі до об’єму, що витягається, гаран-
тована шляхом одержання об’єму наповнення біль-
шого за номінальний об’єм на величину, визначувану
характеристиками препарату.
Суспензії та емульсії необхідно струшувати перед
відбором вмісту й перед визначенням густини.
Масляні або в’язкі препарати, якшо необхідно, нагріва-
ють і ретельно струшують безпосередньо перед відбо-
ром вмісту. Перед визначенням вміст охолоджують.
Контейнери з номінальним об’ємом менше 5мл
Відбирають шість контейнерів, п’ять для проведення
164
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
апробування і один для обполіскування використо-
вуваних шприца й голки.
Вибирають шприц місткістю не більший за подвійний
змірюваний об’єм і насаджують підхожу голку. На-
тірають у шприц невеликий об’єм випробовуваного
.н’єкційного лікарського засобу з контейнера, при-
значеного для обполіскування, і виливають рідину з
шприца, тримаючи його вертикально голкою догори
пи вилучення повітря.
Витягають максимально можливий об’єм вмісту одного
п’яти контейнерів, використовуваних для випробуван-
ня, видаляють бульбашки повітря і переносять цей
об’єм, уникаючи випорожнення голки, у сухий зваже-
ній контейнер; зважують і визначають масу вмісту. Про-
цедуру повторюють з чотирма іншими контейнерами.
Визначення густини проводять при тій самій темпера-
турі, шо і визначення об’єму, що витягається. Об’єм,
шо витягається, розраховують діленням маси вмісту
кожного контейнера на густину препарату.
Препарат витримує випробування на об’єм, що витя-
гається, якщо об’єм, розрахований для кожного з
п’яти контейнерів, не менший за номінальний.
Контейнери з номінальним об’ємом 5мл і більше
Відбирають шість контейнерів, п’ять для проведення
випробування і один для обполіскування використо-
вуваних шприца й голки.
Вибирають шприц місткістю не більший за подвійний
вимірюваний об’єм і насаджують підхожу голку. На-
бирають у шприц невеликий об’єм випробовуваного
ін’єкційного лікарського засобу з контейнера, при-
значеного для обполіскування, і виливають рідину з
шприца, утримуючи його вертикально голкою догори
дія вилучення повітря.
Витягають максимально можливий об’єм вмісту одно-
гоз п’яти контейнерів, використовуваних для випро-
бування, видаляють бульбашки і переносять цей
об’єм, уникаючи випорожнення голки, у сухий
мірний циліндр такої місткості, шоб вимірюваний
об’єм заповнив не менше 40 % номінального об’єму
циліндра. Вимірюють об’єм, що витягається. Проце-
іуру повторюють з чотирма іншими контейнерами.
Препарат витримує випробування на об’єм, що витя-
гається, якщо об’єм, виміряний у кожному з п’яти
контейнерів, не менший за номінальний.
картриджі й попередньо наповнені шприци
Об'єм ін’єкційного лікарського засобу, який міститься
у картриджі або попередньо наповненому шприці, має
бути достатнім для відбору номінальної дози.
Суспензії та емульсії необхідно струшувати перед
відбором вмісту і перед визначенням густини.
Масляні або в’язкі препарати, якщо необхідно, нагріва-
ють і ретельно струшують безпосередньо перед відбором
вмісту. Перед визначенням вміст охолоджують.
Відбирають п’ять контейнерів. Якщо треба, до кон-
тейнера приєднують приладдя (голку, поршень,
шприц), необхідне для його використання, і перено-
сять увесь вміст, уникаючи випорожнення голки, у су-
хий зважений контейнер шляхом повільного і
постійного тиску на поршень. Зважують і визначають
масу вмісту.
Процедуру повторюють ще з чотирма іншими контей-
нерами.
Визначення густини проводять при тій самій темпера-
турі, що і визначення об’єму, шо витягається. Об’єм,
що витягається, розраховують діленням маси вмісту
кожного контейнера на густину препарату.
Препарат витримує випробування на об’єм, що витя-
гається, якщо розрахований об’єм для кожного з п’яти
контейнерів не менший за номінальний.
ВНУТРІШНЬОВЕННІ ІНФУЗІЙН1 ЛІКАРЬСКІ
ЗАСОБИ
Відбирають один контейнер. Переносять вміст у сухий
мірний циліндр такої місткості, шоб визначуваний
об’єм заповнив не менше 40 % номінального об’єму
циліндра. Вимірюють об’єм, що витягається.
Об’єм, шо витягається, має бути не меншим за
номінальний об’єм, зазначений на контейнері.
___________________________________________N
Кожний контейнер для ін’єкційних лікарських
засобів наповнюють об’ємом, що перевищує но-
мінальний. Надлишковий об’єм рекомендовано у
Табл. 2.9.17.-1.
Таблиця 2.9.17.-1
Номінальний об'єм (мл) Надлишковий об'єм (мл)
Для рухомих рідин Для в'язких рідин
0.5 0.10 0.12
1.0 0.10 0.15
2.0 0.15 0.25
5.0 0.30 0.50
10.0 0.50 0.70
20.0 0.60 0.90
30.0 0.80 1.20
50.0 і більше 2 % 3 %
2.9.19. МЕХАНІЧНІ ВКЛЮЧЕННЯ:
НЕВИДИМІ ЧАСТКИ
Механічні включення ін'єкційних і внутрішньовен-
них інфузійних розчинів — це побічні рухомі нероз-
чинні частки, за винятком бульбашок газу, випадково
присутніх у розчинах.
Лікарські засоби, які потребують випробування на ме-
ханічні включення, і вимоги щодо вмісту механічних
включень зазначені у відповідній окремій статті.
Обладнання. Для контролю використовують прилад, за-
снований на принципі світлоблокування, який дозволяє
автоматично вимірювати кількість і розмір часток.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
165
2.9. Фармако-технологічні випробування
Прилад калібрують, використовуючи дисперсійні за-
висі ФСЗ сферичних часток розміром від 5 мкм до
25 мкм. Стандартні частки дисперговані у воді Р,
вільній від часток. Необхідно уникати агрегації часток
дисперсійної фази.
Загальні зазначення. Контроль необхідно здійснювати
в умовах, що обмежують попадання механічних вклю-
чень, найкраще в зоні ламінарного потоку повітря.
Ретельно промивають скляний посуд і використовуване
фільтраційне обладнання (крім мембранних фільтрів)
теплим розчином миючого засобу з наступним обпо-
ліскуванням необхідною кількістю води для видалення
слідів миючого засобу. Безпосередньо перед випробу-
ванням обполіскують обладнання від верху до низу,
зовні і потім усередині водою, вільною від часток, Р.
Необхідно виключити виникнення повітряних буль-
башок у випробовуваному зразку, особливо під час пе-
ренесення проби до посудини, у якій буде проводити-
ся вимірювання.
Для того, щоб переконатися в якості очистки облад-
нання, скляного посуду і чистоті води, необхідно ви-
конати таке випробування. Визначають наявність ме-
ханічних включень у п’яти пробах води, вільної від
часток, Р(кожна по 5 мл), згідно з методикою, описа-
ною нижче. Якщо у 25 мл для об’єднаних п’яти проб
число часток розміром у 10 мкм і більше перевищує
25, прийняті запобіжні заходи для проведення випро-
бувань недостатні. Обробку повторюють доти, доки
обладнання, скляний посуд і вода не стануть придат-
ними для проведення контролю.
Методика. Перемішують вміст зразка, повільно і без-
перервно перевертаючи контейнер п’ять разів. Якщо
необхідно, обережно видаляють етикетки і ковпачки.
Зовнішні поверхні контейнера, який розкривають,
очищають струменем води, вільної від часток, Рі роз-
кривають контейнер, уникаючи внесення будь-якого
забруднення. Для видалення бульбашок повітря дають
відстоятися розчину протягом 2 хв.
Відбирають чотири проби, не менше 5 мл кожна, і ви-
значають кількість часток з розмірами, що дорівню-
ють або перевищують значення, зазначені у відповід-
ному нормативному документі. Виключають резуль-
тат, одержаний для першої проби, і обраховують се-
редню кількість часток у випробовуваному зразку.
______________________________________________N
Додаткові умови проведення випробування і норми
контролю лікарських засобів для парентерального за-
стосування на наявність механічних включень зазна-
чені у відповідному нормативному документі.
2.9.20. МЕХАНІЧНІ ВКЛЮЧЕННЯ:
ВИДИМІ ЧАСТКИ
Механічні включення ін’єкційних і внутрішньовен-
них інфузійних розчинів — це побічні рухомі нероз-
чинні частки, за винятком бульбашок газу, випадково
присутніх у розчинах.
Дане випробування призначене для візуальної оцінки
якості парентеральних розчинів за наявністю видимих
часток. Можуть бути використані інші валідовам
методи.
Обладнання. Обладнання (див. Рис. 2.9.20.-1) скла!
дається з пристрою для перегляду, що включає:
— матовий чорний екран підхожого розміру, якиі
знаходиться у вертикальному положенні;
— білий матовий екран відповідного розміру, що зна-
ходиться у вертикальному положенні, поруч з чор-|
ним екраном;
— плафон, що переміщується і споряджений підхо-І
жим затемненим джерелом денного світла з під-
хожим відбивачем (освітлювач може складатися з
двох флуоресцентних ламп по 13 Вт кожна, зав-
довжки 525 мм). Інтенсивність освітлення уточім
контролю має бути від 2000 люкс до 3750 люкс; для |
кольорових скляних і пластмасових контейнерів
віддають перевагу значенням, близьким до
верхньої межі.
Рисунок 2.9.20,-1. Обладнання для виявлення
видимих часток
Методика. Прибирають наклеєні етикетки з контей-
нера, миють його зовні і сушать. Плавно обертають
або перевертають контейнер, уникаючи утворення
повітряних бульбашок, і переглядають приблизно
протягом 5 с перед білим екраном. Повторюють цю
процедуру перед чорним екраном. Відзначають на-
явність будь-яких часток.
______________________________________________А
Додаткові умови проведення випробування і норми
контролю лікарських засобів для парентерального за-
стосування на наявність механічних включень зазна-
чені у відповідному нормативному документі.
2.9.21. МЕХАНІЧНІ ВКЛЮЧЕННЯ:
МЕТОД МІКРОСКОПІЇ
Механічні включення ін’єкційних і внутрішньовен-
них інфузійних розчинів — це сторонні рухомі нероз-
чинні частки, за винятком бульбашок газу, випадково
присутніх у розчинах.
Дане випробування призначене для встановлення
природи часток, які можуть бути присутніми у роз-
чині, і для визначення їхніх характеристик. Воно може
166
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
2.9. Фармако-технологічні випробування
вказувати на можливе джерело забруднення. Ця
інформація дозволить виробникові вжити заходів для
ипобігання забруднень.
Загальні зазначення. Усі процедури виконують у зоні
ламінарного потоку повітря.
Підготовка матеріалів. Ретельно миють скляний по-
суд і використовуване фільтраційне обладнання, крім
мембранних фільтрів, теплим розчином миючого за-
собу і обполіскують великою кількістю води Рао вида-
лення слідів миючого засобу. Безпосередньо перед ви-
пробуванням обполіскують зазначене вище обладнан-
ня від верху до низу, зовні і потім усередині
водою, вільною від часток, Р.
Обладнання. Використовують вакуум-фільтраційну
систему, виконану із скла або нержавіючої сталі, об-
ладнану мембранним фільтром з нанесеною сіткою.
Фільтр має відповідну пористість і колір.
Використовують бінокулярний мікроскоп, що скла-
дається з:
- ахроматичного об’єктива із збільшенням хІО;
- окулярів із збільшенням хІО, з яких, принаймні,
один споряджений сіткою, що дозволяє точно
вимірювати частки розміром у 10 мкм і більше;
- системи освітлення для спостереження з падаю-
чим або відбитим світлом;
- об’єкт-мікрометр для калібрування сітки окуляра.
Методика. Блок мембранного фільтра і фільтраційного
апарата. Обполіскують лійку і основу фільтротримача
водою, вільною від часток, Р; попередньо вимитим
пінцетом беруть мембранний фільтр із сіткою; для ви-
далення всіх часток обполіскують обидва боки фільтра
водою, вільною від часток, Р, тримаючи фільтр у вер-
тикальному положенні й повільно спрямовуючи стру-
мінь води з боку в бік, починаючи зверху і донизу.
Промитий фільтр поміщають сіткою вгору на основу
фільтротримача вакуум-фільтраційного апарата і цен-
трують. Установлюють фільтраційну лійку на основу
фільтротримача без зсуву її по стороні сітки фільтра.
Обертають зібраний блок, обполіскуючи внутрішню
частину лійки і поверхню фільтра струменем води,
вільної від часток, Р протягом близько 15 с. Приєдну-
ють блок до посудини для фільтрату.
Фільтрація випробовуваного зразка. 25 мл випробову-
ваного ретельно перемішаного препарату переносять у
фільтраційний апарат. Дають відстоятися розчину при-
близно 1 хв. Проводять вакуум-фільтрацію. Повільно
відключають вакуум і обполіскують внутрішні стінки
лійки струменем води, вільної від часток, Р. Не до-
пускається спрямовувати струмінь води на поверхню
фільтра. Дають можливість відстоятися розчину і
фільтрують змиви під вакуумом. Після завершення
фільтрації вакуум вимикають не зразу, щоб підсушити
фільтр. Акуратно видаляють лійку; сталевим пінцетом,
попередньо відмитим струменем води, вільної від
часток, Р переносять фільтр у чисту чашку Петрі. На-
кривають чашку чистою кришкою так, щоб вона була
трохи відкрита. Поміщають чашку в зону ламінарного
потоку повітря для підсушки фільтра, уникаючи при
цьому утворення згинів. Потім помішають чашку
Петрі на стіл мікроскопа і підраховують частки на
фільтрі, як описано нижче.
Підготовка контрольної проби. Готують контрольну
пробу так само, як і випробовуваний зразок. Кон-
трольну пробу проводять з 25 мл води, вільної від час-
ток, Р пропущеної безпосередньо через лійку вакуум-
фільтраційної системи.
Калібрування мікроскопа. Калібрують сітку окуляра
мікроскопа, використовуючи об’єкт-мікрометр.
Визначення. Можна використовувати як візуальний
підрахунок, так і електронний пристрій реєстрації ча-
сток. Досліджують усю поверхню фільтра при
збільшенні хІОО і освітленні падаючим або відбитим
світлом. Підраховують частки з розмірами 10 мкм і
більше і класифікують їх за попередньо вибраними
діапазонами розмірів.
Для кожного діапазону з кількості часток у випробову-
ваному зразку віднімають кількість часток того самого
діапазону у контрольній пробі. Якщо у контрольній
пробі виявляють більш як п’ять часток розміром
25 мкм і більше, умови проведення випробування вва-
жаються незадовільними, і випробування повторю-
ють.
Оцінка результатів. Якщо можливо, установлюють
природу виявлених часток і визначають їхні характе-
ристики. Якщо необхідно, число виявлених часток
реєструють.
_______________________________________________N
Додаткові умови проведення випробування і норми
контролю лікарських засобів для парентерального за-
стосування на наявність механічних включень зазна-
чені у відповідному нормативному документі.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
167
4.1.1. Реактиви
4. РЕАКТИВИ
11. РЕАКТИВИ, ЕТАЛОННІ
РОЗЧИНИ, БУФЕРНІ
РОЗЧИНИ
Якщо найменування реактиву або розчину реактиву су-
проводжується літерою Р та виділене курсивом, це
точить, що реактив внесений до нижченаведеного пе-
кїіку. Специфікації, наведені для реактивів, не гаран-
тують можливості використання останніх як лікарсь-
ких засобів.
В описі кожного реактиву є семизначний код, виділений
курсивом (приміром, 1002501). Цей номер залишається
«змінним для кожного реактиву за будь-яких подаль-
ших переглядів переліку. Він може бути використаний
дія ідентифікації реактиву та, приміром, при обліку
та складуванні реактивів. Опис може також містити
помер СИетісаІ АЬзігасі Зегеісе ВеЦхІгу (СА5), який лег-
ко впізнати за характерним позначенням, наприклад,
9002-93-1.
Леякл з реактивів, внесених до переліку, є токсичними і
при роботі з ними необхідно дотримуватись відповідних
застережних заходів.
Водні розчини реактивів готують із використанням во-
ди Р. Якщо розчин реактиву описано висловом типу
"розчин 10 г/л кислоти хлористоводневої", розчин готу-
ють відповідним розведенням водою Р більш концентро-
ваного розчину реактиву, наведеного у цьому ж розділі.
Розчини реактивів, використовувані для випробувань
на граничний вміст барію, кальцію та сульфати, готу-
ють із використанням води дистильованої Р. Якщо роз-
чинник не вказано, мається на увазі водний розчин.
Реактиви й розчини реактивів мають зберігатися у
щііьно закупорених контейнерах.
4.1.1. РЕАКТИВИ
Агароза для електрофорезу. 1002000. [9012-36-6].
Нейтральний лінійний полісахарид, головний компо-
нент якого одержують із агару.
Порошок білого або майже білого кольору. Практично
не розчинна в холодній воді, дуже мало розчинна в га-
рячій воді.
Агароза для хроматографії. 1001800. [9012-36-6].
4% суспензія у воді Р набухлих гранул діаметром від
60 мкм до 140 мкм. Використовують в гель-хромато-
графії для розділення білків із молекулярними масами
від 6 х 104 до 20 х 106 та полісахаридів із молекулярни-
ми масами від 3 х 10і до 5 х ІО6.
.Агароза поперечно-зшита для хроматографії. 1001900.
[61970-08-91.
Одержують із агарози реакцією з 2,3-дибромпропано-
.юм у сильно лужному середовищі.
4 % суспензія у воді Р набухлих гранул діаметром від
60 мкм до 140 мкм. Використовують в гель-хромато-
графії для розділення білків із молекулярними масами
від 6 х 10’ до 20 х 106 та полісахаридів із молекулярни-
ми масами від 3 х 10і до 5 х 106.
Агароза поперечно-зшита для хроматографії РІ.
1001901. [65099-79-8].
Одержують з агарози реакцією з 2,3-дибромпропа-
нолом у сильно лужному середовищі.
4 % суспензія у воді Р набухлих гранул діаметром
від 60 мкм до 140 мкм. Використовують в гель-
хроматографії для розділення білків із молекуляр-
ними масами від 7 х 104 до 40 х 106 та полісахаридів
із молекулярними масами від 1 х 10’ до 2 х 107.
Агароза-ДЕАЕ для іонообмінної хроматографії.
1002100. [57407-08-6].
Поперечно-зшита агароза, із заміщеними діети-
ламіноетильними групами, у вигляді кулястих гранул.
Агароза/поперечно-зшитий поліакриламід. 1002200.
Агароза в поперечно-зшитій поліакриламідній мат-
риці. Використовують для розділення глобулярних
білків із молекулярними масами від 2 х 104 до 35 х ІО4.
Аденозин. С10НІЗМ5О4. (М.м. 267.2). 1001600. [58-61-7].
6-Аміно-9-[3-О-рибофуранозил-9//-пурин.
Кристалічний порошок білого кольору. Мало розчинний
у воді, практично не розчинний в ацетоні, 96 % спирті та
ефірі. Розчиняється у розведених розчинах кислот.
Температура плавлення: близько 234 °С.
Адипінова кислота. С6Н1ПО4. (М.м. 146.1). 1095600.
[124-04-91.
Кристали у вигляді призм. Легко розчинна в метанолі,
розчинна в ацетоні, практично не розчинна в петро-
лейному ефірі.
Температура плавлення: близько 152 °С.
Азометан Н. С17Н12ККаО882. (М.м. 445.4). 1008700.
[5941-07-1]. Натрію 4-гідрокси-5-(2-гідроксибен-
зиліденаміно)-2,7-нафталіндисульфонат кислий.
Азометину Н розчин. 1008701.
0.45 г азометину Н Р та 1 г кислоти аскорбінової Р
розчиняють у воді Рпри слабкому нагріванні й до-
водять об'єм розчину тим самим розчинником до
100 мл
Азот. N2. (М.м. 28.01). 1059300. [7727-37-9].
Азот промитий та висушений.
АзотРІ. 1059400.
Містить не менше 99.999 % (об/об) N2.
Вуглецю монооксид: не більше 5 ррт.
Кисень: не більше 5 ррт.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
169
4.1.1. Реактиви
Азот, вільний від кисню. 1059600.
Азот Р очищують від кисню пропусканням крізь роз-
чин пірогалолу лужний Р.
Азот для хроматографії. 1059500.
Містить не менше 99.95 % (об/об) N2-
Азотна кислота. НМ)3. (М.м. 63.0). 1058400. [7697-37-2].
Містить не менше 63.0 % (м/м) і не більше 70.0 %
(м/м) НЬЮ3.
Прозора, безбарвна або майже безбарвна рідина,
змішується із водою.
^2о : від 1-384 до 1.416.
Розчин 10 г/л є сильною кислотою й дає реакцію на
нітрати (2.3.1).
Прозорість (2.2.1). Кислота азотна має бути прозорою.
Кольоровість (2.2.2, метод II). Забарвлення кислоти
азотної має бути не інтенсивнішим за еталон ¥6.
Хлориди (2.4.4). Не більше 0.00005 % (0.5 ррт). До 5 г
кислоти азотної додають 10 мл води Рта 0.3 мл розчину
срібла нітрату Р2, витримують протягом 2 хв у захи-
щеному від світла місці. Одержаний розчин має вит-
римувати випробування на хлориди. Еталон готують із
використанням суміші 13 мл води Р, 0.5 мл кислоти
азотної Р, 0.5 мл еталонного розчину хлориду
(5 ррт СІ) Р та 0.3 мл розчину срібла нітрату Р2.
Сульфати (2.4.13). Не більше 0.0002 % (2 ррт). До 10 г
кислоти азотної додають 0.2 г натрію карбонату Р і ви-
парюють насухо; залишок розчиняють у 15 мл води дис-
тильованої Р. Одержаний розчин має витримувати ви-
пробування на сульфати. Еталон готують із викорис-
танням 2 мл еталонного розчину сульфату
(10ррт 500 Рта 13 мл води дистильованої Р.
Арсен (2.4.2, метод А). Не більше 0.000002 % (0.02
ррт). До 50 г кислоти азотної додають 0.5 мл кислоти
сірчаної Р і обережно нагрівають до появи білих парів;
до залишку додають 1 мл розчину 100 г/л гідрокси-
ламіну гідрохлориду Р і доводять водою Рцо об'єму 2 мл.
Одержаний розчин має витримувати випробування на
арсен. Еталон готують із використанням 1.0 мл ета-
лонного розчину арсену (1 ррт Ах) Р.
Важкі метали (2.4.8, метод А). Не більше 0.0002 %
(2 ррт). 10 мл розчину, приготованого для випробу-
вання на залізо, доводять водою Р до об'єму 20 мл.
12 мл одержаного розчину мають витримувати
випробування на важкі метали. Еталон готують із ви-
користанням еталонного розчину свинцю (2 ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.0001 % (1 ррт). Осад, одер-
жаний при випробуванні на сульфатну золу, розчиня-
ють в 1 мл кислоти хлористоводневої розведеної Р і до-
водять об'єм розчину водою Рцо 50 мл. 5 мл одержано-
го розчину доводять водою Р до об'єму 10 мл.
Одержаний розчин має витримувати випробування на
залізо.
Сульфатна зола. Не більше 0.001 %. 100 г кислоти
азотної обережно випарюють насухо; залишок змочу-
ють кількома краплями кислоти сірчаної Р і нагріва-
ють до блідо-червоного кольору.
Кількісне визначення. До 1.50 г кислоти азотної доЛ
ють близько 50 мл води Р і титрують / М розчин1.
натрію гідроксиду, використовуючи як індика»
0.1 мл розчину метилового червоного Р.
1 мл / М розчину натрію гідроксиду відповідає 63.0 Л
НЬ4О3.
Зберігають у захищеному від світла місці.
Азотна кислота, вільна від свинцю. 1058403. І
Кислота азотна /’ мас витримувати таке додати»!
ве випробування.
До 100 г кислоти азотної Р додають 0.1 г /адтри
карбонату безводного Р і випарюють насухо; залі-І
шок розчиняють у воді Р при слабкому нагрівані г
й доводять об'єм розчину тим самим розчинникоиі
до 50.0 мл. Вміст свинцю визначають методом атом І
но-абсорбційної спектрометрії (2.2.23, метод /І) І
Інтенсивність поглинання вимірюють за довжини
хвилі 283.3 нм або 217.0 нм, використовуючи ж
джерело випромінювання лампу з порожниста»
свинцевим катодом та повітряно-ацетиленове по-
лум'я. Не більше 0.00001 % (0.1 ррт РЬ).
Азотна кислота, вільна від свинцю й кадмію.
1058401.
Кислота азотна Р має витримувати такі додаткові
випробування.
Випробовуваний розчин. До 100 г кислоти азотної І
додають 0.1 г натрію карбонату безводного Р, ви-
парюють насухо; залишок розчиняють у воді Рпри
слабкому нагріванні й доводять об'єм розчинутим
самим розчинником до 50.0 мл.
Кадмій. Не більше 0.00001 % (0.1 ррт Сф. Вміст
кадмію визначають методом атомно-абсорбційної
спектрометрії (2.2.23, метод 11). Інтенсивність по-
глинання вимірюють за довжини хвилі 228.8 нм,
використовуючи як джерело випромінювання лам-
пу з порожнистим кадмієвим катодом та повітряно-
ацетиленове або повітряно-пропанове полум'я.
Свинець. Не більше 0.00001 % (0.1 ррт РЬ). Вміст
свинцю визначають методом атомно-абсорб-
шйної спектрометрії (2.2.23, метод 11). Інтен-
сивність поглинання вимірюють за довжини хвилі
283.3 нм або 217.0 нм, використовуючи лампу із
порожнистим свинцевим катодом та повітряно-
ацетиленове полум'я.
Азотна кислота розведена. 1058402.
Містить близько 125 г/л НМО3 (М.м. 63.0).
20 г кислоти азотної Р доводять водою Рао об'єму
100 мл.
Азотна кислота димляча. 1058500. [52583-42-3].
Прозора рідина, жовтавого кольору, яка димить на
повітрі.
г/20 : близько 1.5.
170
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Азогу закис. И2О. (М.м. 44.01). 1108500.
Містить не менше 99.99 % (об/об) К2О.
коту монооксид: не більше 1 ррт.
Вуглецю монооксид: не більше 1 ррт.
Азоіу монооксид. N0. (М.м. 30.01). 1108300.
Містить не менше 98.0 % (об/об) N0.
Акриламід. С3Н5МО. (М.м. 71.1). 1001500. [79-06-1].
Пропенамід.
Безбарвні або білого кольору пластівці або кристалічний
порошок білого або майже білого кольору. Дуже легко
розчинний у воді й метанолі, легко розчинний в етанолі.
Температура плавлення: близько 84 °С.
Акриламід-бісакриламіду (29:1) ЗО % розчин.
1001501.
290 г акриламіду Р і 10 г метиленбісакриламіду Р
розчиняють в 1 л води Р і фільтрують.
Акриламід-бісакриламіду (36.5:1) ЗО % розчин.
1001502.
292 г акриламіду Р і 8 г метиленбісакриламіду Р
розчиняють в І л води Р '\ фільтрують.
Акрилова кислота. С3Н4О2. (М.м. 72.1). [79-10-7].
Проп-2-енова кислота. Вінілмурашина кислота.
Містить не менше 99 % С3Н4О2. Стабілізована 0.02 %
розчином монометилового ефіру гідрохінону.
їдка рідина. Змішується з водою й 96 % спиртом. Лег-
ко полімеризується у присутності кисню.
$: близько 1.05.
1$ близько 1.421.
Температура кипіння: близько 141 °С.
Температура плавлення: від 12 °С до 15 °С.
Аіанін. 1102900.156-41-7]. Див. статтю Аланін.
0-Аіанін. 1004500. [107-95-9]. Див. З-Амінопропіонова
кислота Р.
Аіеуринова кислота. СІ6Н32О5. (М.м. 304.4). 1095700.
|533-87-9]. (9К8,1057Д-9,10,16-Тригілроксигексадека-
нова кислота.
Порошок білого кольору, маслянистий на дотик. Роз-
чинна в метанолі.
Температура плавлення: близько 101 °С.
Алізарин 8. СІ4Н7КаО78,Н2О. (М.м. 360.3). 1002600.
[130-22-3]. Показник Шульца № 1145. Кольоровий
індекс № 58005. Натрію 1,2-дигідроксіантрахінон-З-
сульфонат моногідрат. Натрію 3,4-дигідрокси-9,10-ді-
оксо-9,10-дигідроантрацен-2-сульфонат моногідрат.
Порошок оранжево-жовтого кольору. Легко розчин-
ний у воді й 96 % спирті.
Алізарину 8 розчин. 1002601.
Розчин 1 г/л.
Випробування на чутливість. Реактив змінює за-
барвлення від жовтого до оранжево-червоного
при встановленні титру 0.05 М розчину барію пер-
хлорату (4.2.2).
Зміна забарвлення. Від жовтого до фіолетового в інтер-
валі рН 3.7-5.2.
Альбумін бичачий. 1002300. [9048-46-8].
Альбумін бичачий сироватковий. Містить близько
96 % білка.
Порошок від білого до світлого жовтувато-коричнево-
го кольору.
Вода (2.5.12). Не більше 3.0 %. Визначення проводять
із 0.800 г альбуміну бичачого.
Альбумін бичачий, використовуваний при кількісному
визначенні Тетракозактиду, має бути апірогенним, не
має проявляти протеолітичну активність при визна-
ченні відповідними методами, наприклад, при викорис-
танні хромогенного субстрату, і не повинен мати кор-
тикостероїдну активність, що визначається вимірю-
ванням флуоресценції, як зазначено в статті Тетрако-
зактид у кількісному визначенні біологічної актив-
ності.
Альбуміну людського розчин. 1002400. [9048-46-8]. Див.
статтю Альбуміну людського розчин.
Альбуміну людського розчин РІ. 1002401.
Альбуміну людського розчин Р розводять розчином
9 г/л натрію хлориду Р до концентрації білка 1 г/л.
Доводять рН розчину до 3.5-4.5 кислотою оцтовою
льодяною Р.
Альдегіддегідрогеназа. 1103000. Фермент, одержаний із
хлібопекарських дріжджів, окиснює ацетальдепд до
кислоти оцтової в присутності нікотинамід-аденіну
динуклеотиду, солей калію та тіолів при рН 8.0.
Альдегіддегідрогенази розчин. 1103001. Кількість
альдегіддегідрогенази Р, яка відповідає 70 одини-
цям. розчиняють у воді Р і доводять об'єм розчину
тим самим розчинником до 10 мл. Розчин
стабільний протягом 8 год при температурі 4 °С.
Алюміній. АІ. (А.м. 26.98). 1118200. [7429-90-5].
М'який, ковкий метал білого з блакитнуватим
відтінком кольору у вигляді брусків, листів, порошку,
стрічки або дроту. На повітрі утворюється окисна
плівка, яка захищає метал від корозії.
Аналітичної чистоти.
Алюмінію-калію сульфат. 1003000. [7784-24-9|. Див.
статтю Галуни.
Алюмінію нітрат. А1(МО3)3,9Н2О. (М.м. 375 1). 1002800.
[7784-27-2]. Алюмінію нітрат нонагідрат.
гом'лш-ід ґНдрм АІ^ППРЯ УКРАЇНИ 7001
171
4.1.1. Реактиви
Кристали, що розпливаються на повітрі. Дуже легко
розчинний у воді й 96 % спирті, дуже мало розчинний
в ацетоні.
Зберігають у повітронепроникному контейнері.
Алюмінію оксид безводний. 1002000. [1344-28-1].
Алюмінію оксид, що складається з у-Л12О3, зневодне-
ний та активований нагрівом. Розмір часток від
75 мкм до 150 мкм.
Алюмінію оксид основний. 1118300. Алюмінію оксид
безводний Росновної форми, придатний для хромато-
графічних колонок.
рН (2.3.3). Від 9 до 10. Вимірюють рН суспензії, одер-
жаної струшуванням 1 г з 10 мл води, вільної від вугле-
цю діоксиду, Р протягом 5 хв.
Алюмінію оксид нейтральний. А12О3. (М.м. 102.0).
1118400.
Гранульований порошок білого кольору.
Обмінна ємність. 1.00 г прокаїну гідрохлориду Р розчи-
няють у спирті (90 %, об/об) Рта доводять об'єм роз-
чину тим самим розчинником до 100 мл. 20.0 мл одер-
жаного розчину й 5.0 г випробовуваного реактиву
поміщають у колбу місткістю 100 мл із притертою
скляною пробкою, відстоюють протягом 15 хв,
періодично струшуючи, й фільтрують. До 10.0 мл
фільтрату додають 10 мл води Р, 0.05 мл розчину бром-
фенолового синього РІ і титрують 0.1 Мрозчином кисло-
ти хлористоводневої по одержання зеленого забарв-
лення розчину. Забарвлення розчину має змінитися
при додаванні не більше 1.4 мл 0.1 М розчину кислоти
хлористоводневої.
Водорозчинні речовини. Не більше 0.2 %. Використову-
ють хроматографічну колонку із внутрішнім діамет-
ром 1 см, завдовжки 40 см, нижній кінець якої звуже-
ний до діаметра від 2 мм до 3 мм і має пористий скля-
ний фільтр (100) або бавовняний тампон вище звуже-
ноїчастини. Колонку заповнюють 10.0 г випробовува-
ного реактиву й 25 мл води Р, елююють водою Р до
одержання 20 мл прозорого елюату, який випарюють
та сушать при температурі 150 °С; маса залишку не має
перевищувати 20 мг.
Розчин 8. Залишок, одержаний у випробуванні "Водо-
розчинні речовини", розчиняють при нагріванні у
воді Р, фільтрують та доводять об'єм фільтрату водою Р
до 100 мл.
Хлориди (2 4 4). Не більше 0 05 %. 1 мл розчину 8 дово-
дять водою Р до об'єму 15 мл. Одержаний розчин має
витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.1 %>. 1 мл розчину 8 до-
водять водою Рдо об'єму 15 мл. Одержаний розчин має
витримувати випробування на сульфати Еталон готу-
ють із використанням 10 мл еталонного розчину суль-
фату (10 ррт 80) Р.
Хроматографічна розділювальна здатність. Хромато-
графічну колонку, описану в випробуванні "Водороз-
чинні речовини", заповнюють випробовуваним реак-
тивом до висоти 5 см. Крізь колонку пропускають 5 мл
розчину азобензолу Р та 5 мл метоксіазобензолу Я
потім 20 мл суміші розчинників бензол Р-петролейіш
ефір Р(1:4). У верхній частині колонки утворюєтьД
шар яскраво-жовтого кольору метоксіазобензолузпЖ
товшки від 3 мм до 5 мм, а нижче від нього спи
терігається шар азобензолу блідо-жовтого кольо,
завтовшки 2 см.
Алюмінію хлорид. А1С13,6Н2О. (М.м. 241.4). /0027ЙВ
[7784-13-6]. Алюмінію хлорид гексагідрат.
Містить не менше 98.0 % А1С13,6Н2О.
Кристалічний порошок від білого до злегка жовтаво
кольору, гігроскопічний. Легко розчинний у водії І
96 % спирті, розчинний в ефірі.
Зберігають у повітронепроникному контейнері.
Алюмінію хлориду реактив. 1002702.
2.0 г алюмінію хлориду Р розчиняють у 100 мі >
5 % (об/об) розчину кислоти оцтової льодяної Р\ і
метанолі Р.
Алюмінію хлориду розчин. 1002701.
65.0 г алюмінію хлориду Р розчиняють у воді Рі до-1
водять об'єм розчину тим самим розчинником до І
100 мл. Додають 0.5 г вугілля активованого Р, пе- І
ремішують протягом 10 хв та фільтрують. До
фільтрату при безперервному перемішуванні дода- І
ють достатню кількість розчину 10 г/л натрію
гідроксиду Р (близько 60 мл) до одержання рН
близько 1.5.
Аміаку розчин концентрований. 1004700. Див. статтю
Аміаку розчин концентрований.
Аміаку розчин. 1004701.
Містить не менше 170 г/л і не більше 180 г/л МН3
(М.м. 17 03).
67 г розчину аміаку концентрованого Р доводять во-
дою Рпо об'єму 100 мл.
: від 0.931 до 0.934.
Аміаку розчин Р, використовуваний у випробу-
ванні на граничний вміст заліза, має задовольняти
таку додаткову вимогу: 5 мл розчину аміаку Рвипа-
рюють на водяній бані насухо. До сухого залишку
додають 10 мл води Р, 2 мл розчину 200 г/л кисло-
ти лимонної Р, 0.1 мл кислоти тіогліколевої Р та
розчину аміаку Рпо лужної реакції, доводять об'єм
одержаного розчину водою Р до 20 мл. Розчин не
має набувати рожевого забарвлення.
Зберігають при температурі нижче 20 °С, захища-
ючи від вуглецю діоксиду.
Аміаку розчин розведений РІ. 1004702.
Містить не менше 100 г/л і не більше 104 г/л 1МН3
(М.м. 17.03).
41 г розчину аміаку концентрованого Рдоводять во-
дою Рпо об'єму 100 мл.
172
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Аміаку розчин розведений Р2. 1004703.
Містить не менше 33 г/л і не більше 35 г/л ЬІН3
(М.м. 17.03).
14 г розчину аміаку концентрованого Р доводять во-
дою Рро об'єму 100 мл.
Аміаку розчин розведений РЗ. 1004704.
Містить не менше 1.6 г/л і не більше 1.8 г/л 1ЧН3
(Мм. 17.03).
0.7 г розчину аміаку концентрованого Р доводять
водою Р до об’єму 100 мл.
иіаку розчин концентрований РІ. 1004800.
Іістить не менше 32.0 % (м/м) N143 (М.м. 17.03).
розора, безбарвна рідина.
]0° : від 0.883 до 0.889.
ількісне визначення. 50.0 мл 1 Мрозчину кислоти хло-
істоводневої помішають у колбу з притертою проб-
ою, точно зважують, додають 2 мл розчину аміаку
онцентрованого і знов зважують. Титрують 1 М роз-
міном натрію гідроксиду, використовуючи як індика-
ор 0.5 мл змішаного розчину метилового червоного Р.
т 1 М розчину кислоти хлористоводневої відповідає
7.03 мг N143.
берігають при температурі не вище 20 °С, захищаючи
ід вуглецю діоксиду.
імідо-чорний 10В. С22Н14^№2О982. (М.м. 617).
003100. [1064-48-8]. Показник Шульца № 299. Ко-
ьоровий індекс № 20470. Динатрію 5-аміно-4-гідрок-
и-6-[(4-нітрофеніл)азо]-3-(фенілазо)нафталін-2,7-
мсульфонат.
Іорошок від темно-коричневого до чорного кольору.
Іомірно розчинний у воді, розчинний у 96 % спирті.
Амідо-чорного 10В розчин. 1003101.
Розчин 5 г/л амідо-чорного 10В Ру суміші розчин-
ників кислота оцтова Р- метанол Р (10:90).
х-Амілаза. 1100800. 1,4-а-О-Глюкан-глюканогідрола-
я.(ЕС3.2/./).
Порошок від білого ло світло-коричневого кольору.
а-Амілази розчин. 1100801.
Розчин а-амілази Рз активністю 800 ФАО (фран-
ко-американських одиниць)/г.
Аміноазобензол. С12Н1^3. (М.м. 197.2). 1003200.
[60-09-3]. Кольоровий індекс № 11000. 4-(Феніл-
азо)анілін.
Голчасті кристали коричнювато-жовтого з блакитну-
ватим відтінком кольору. Мало розчинний у воді, лег-
ко розчинний у 96 % спирті й ефірі.
Температура плавлення: близько 128 °С.
4-Амінобензойна кислота. С7Н^О2. (М.м. 137.1).
1003300. [150-13-0].
Кристалічний порошок білого кольору. Мало розчин-
на у воді, легко розчинна в 96 % спирті, практично не
розчинна в петролейному ефірі.
Температура плавлення: близько 187 °С.
Хроматографія. Визначення проводять, як зазначено
у статті Прокаїну гідрохлорид', на хроматограмі має ви-
являтися лише одна основна пляма.
Зберігають у захищеному від світла місці.
4-Амінобензойної кислоти розчин. 1003301.
1 г кислоти 4-амінобензойної Р розчиняють у
суміші 18 мл кислоти оцтової безводної Р, 20 мл во-
ди Р чаї мл кислоти фосфорної Р.
Безпосередньо перед використанням одержаний
розчин змішують із ацетоном Р(2:3).
2-Амінобензойна кислота. С7Н^О2. (М.м. 137.1).
1003400. [118-92-3]. Антранілова кислота.
Кристалічний порошок від білого до блідо-жовтого
кольору. Помірно розчинна у холодній воді, легко роз-
чинна в гарячій воді, 96 % спирті, ефірі й гліцерині.
Розчини в 96 % спирті або ефірі та особливо в гліце-
рині виявляють фіолетову флуоресценцію.
Температура плавлення: близько 145 °С.
Амінобутанол. С4Н,^О. (М.м. 89.1). 1003500.
[5856-63-3]. 2-Амінобутанол.
Масляниста рідина. Змішується з водою, розчинний у
96 % спирті.
<72о : близько 0.94.
: близько 1.453.
Температура кипіння: близько 180 °С.
6-Аміногексанова кислота. С6Н,^О2. (М.м. 131.2).
1103100. [60-32-2]. 6-Амінокапронова кислота.
Безбарвні кристали. Легко розчинна у волі, помірно
розчинна в метанолі, практично не розчинна в етанолі.
Температура плавлення: близько 205 °С.
Аміногідроксинафталінсульфонова кислота.
С10Н^О48. (М.м. 239.3). 1112400. [116-63-2].
4-Аміно-З-гідроксинафталін- 1-сульфонова кислота.
Голчасті кристали білого або сірого кольору, під дією
світла набувають рожевого кольору, особливо якщо
вологі. Практично не розчинна в воді, 96 % спирті й
ефірі, розчинна у розчинах гідроксидів лужних ме-
талів та гарячих розчинах натрію метабісульфіту.
Зберігають у захищеному від світла місці.
Аміногідроксинафталінсульфонової кислоти розчин.
1112401.
Змішують 5.0 г натрію сульфіту безводного Р, 94.3 г
натрію гідросульфіту Р і 0.7 г кислоти аміногідро-
ксинафталінсульфонової Р. 1.5 г одержаної суміші
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
173
4.1.1. Реактиви
розчиняють у воді Р і доводять об'єм розчину тим
самим розчинником до 10.0 мл.
Термін придатності розчину 1 доба.
Аміногіпурова кислота. С9Н|0М2О3. (М.м. 194.2).
1003700. [61-78-9]. (4-Амінобензамідо)оцтова кислота.
Порошок білого або майже білого кольору. Помірно
розчинна у воді, розчинна у 96 % спирті, дуже мало
розчинна в ефірі.
Температура плавлення: близько 200 °С.
Аміногіпурової кислоти реактив. 1003701.
З г кислоти фталевої Р\ 0.3 г кислоти аміногіпуро-
вої Р розчиняють у 96 % спирті Р і доводять об'єм
розчину тим самим розчинником до 100 мл.
Амінометилалізариндіоцтова кислота.
С19Н15МО8.2Н2О. (М.м. 421.4). 1003900. [3952-78-1].
2,2'-[(3,4-дигідроксіантрахінон-3-іл)метиленнітри-
ло]діоитової кислоти дигідрат.
Дрібнокристалічний порошок від світлого коричню-
вато-жовтого до оранжево-коричневого кольору.
Практично не розчинна у воді, розчинна в розчинах
гідроксидів лужних металів.
Температура плавлення: близько 185 °С.
Втрата в масі при висушуванні (2.2.32). Не більше
10.0 %. Визначення проводять із 1.000 г.
Амінометилалізариндіоцтової кислоти реактив.
1003901.
Розчин І. 0.36 г церію(ІІІ) нітрату Р розчиняють у
воді Р і доводять об'єм розчину тим самим розчин-
ником до 50 мл.
Розчин II. 0.7 г кислоти амінометилалізариндіоцто-
вої Рсуспендують у 50 мл води Р, додають до розчи-
нення близько 0.25 мл розчину аміаку концентрова-
ного Р, потім додають 0.25 мл кислоти оцтової льо-
дяної Р і доводять об'єм розчину водою Рао 100 мл.
Розчин III. 6 г натрію ацетату Р розчиняють у
50 мл води Р, додають 11.5 мл кислоти оцтової льо-
дяної Рі доводять об'єм розчину водою Рао 100 мл.
До 33 мл ацетону Р додають 6.8 мл розчину 111,
1.0 мл розчину 11, 1.0 мл розчину 1 і доводять об'єм
одержаного розчину водою Рао 50 мл.
Випробування на чутливість. До 1.0 мл еталонного
розчину фториду (10 ррт Р) Р додають 19.0 мл во-
ди Р і 5.0 мл реактиву амінометилалізариндіоцто-
вої кислоти. Через 20 хв має з'явитися блакитне за-
барвлення.
Термін придатності розчину 5 діб.
Амінометилалізариндіоцтової кислоти розчин.
1003902.
0.192 г кислоти амінометилалізариндіоцтової Р роз-
чиняють у 6 мл свіжоприготованого 1 М розчину
натрію гідроксиду, додають 750 мл води Р, 25 мл сук-
цинатного буферного розчину рН 4.6 Р та по краплях
0.5 М розчин кислоти хлористоводневої до зміни за-
барвлення розчину від фіолетово-червоного до жов-
того (рН від 4.5 до 5), потім додають 100 мл ацето-
ну Р і доводять об'єм розчину водою Рао 1000 мл.
Амінонітробензофенон. С13Н10М2О3. (М.м. 242.2).
1004000. [1775-95-7]. 2-Аміно-5-нітробензофенон.
Кристалічний порошок жовтого кольору.
Практично
не розчинний у воді, розчинний у тетрагідрофурані,
мало розчинний у метанолі.
Температура плавлення: близько 160 °С.
А\%м : від 690 до 720. Визначення проводять за довжи-
ни хвилі 233 нм, використовуючи розчин 0.01 г/л іш
танолі Р.
Амінопіразолон. СцН|3М3О. (М.м. 203.2). 1004600.
[83-07-8]. 4-Аміно-2,3-диметил-1-фенілпіразолін-5-он
Голчасті кристали або порошок світло-жовтого кольо
ру. Помірно розчинний у воді, легко розчинний у 96 Я
спирті, мало розчинний в ефірі.
Температура плавлення: близько 108 °С.
Амінопіразолону розчин. 1004601.
Розчин 1 г/л в буферному розчинірН 9.0 Р.
Амінополіефір.
[23978-09-8].
С18Н36М2О6. (М.м. 376.5).
біцикло[8,8,8]гексакозан.
1112500.
4,7,13,16,21,24-гексаокса-1,10-діаза
Температура плавлення: від 70 °С до 73 °С.
3-Амінопропанол. С3Н9ЮО. (М.м. 75.1). 1004400.
[156-87-6]. З-Амінопропан-1-ол.
Прозора, безбарвна, в'язка рідина.
<72о : близько 0.99.
Ир : близько 1.461.
Температура плавлення: близько 11 °С.
З-Амінопропіонова кислота. С3Н7ЮО2.
1004500. [107-95-9]. Р-Аланін.
Містить не менше 99 % С3Н7МО2.
Кристалічний
порошок білого кольору.
(М.м. 89.1)
Легко
на у воді, мало розчинна в 96 % спирті,
розчинна в ацетоні й ефірі.
Температура плавлення:
данням.
близько
розчин
практично
200 °С,
із
розкла
4-Амінофенол. С6Н7ЮО. (М.м. 109.1). 1004300.
[123-30-8].
Кристалічний
порошок білого
барвлений,
під дією повітря
кольору
й світла
або
дещо за
набуває
Помірно розчинний у воді, розчинний в етанолі.
Температура плавлення: близько
данням.
186 °С,
Зберігають у захищеному від світла місці.
Амінохлорбензофенон. СІЗНІ0С1МО.
кольору.
із
розкла
(М.м. 231.7)
1003600. [719-59-5]. 2-Аміно-5- хлорбензофенон.
174
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
2001
4.1.1. Реактиви
Кристалічний порошок жовтого кольору. Практично
нерозчинний у воді, легко розчинний в ацетоні, роз-
чинний у 96 % спирті.
Температура плавлення: близько 97 °С.
Хроматографія. Визначення проводять, як зазначено в
статті Хлордіазепоксиду гідрохлорид, використовуючи
5 мкл розчину 0.5 г/л в метанолі Р, на хроматограмі має
виявлятися лише одна основна пляма з К. близько 0.9.
Зберігають у захищеному від світла місці.
Амоксициліну тригідрат. 1103400. Див. статтю Амокси-
циііну тригідрат.
.Амоніюацетат. С2Н7МО2. (М.м. 77.1). 1004900. [631-61-8].
Безбарвні кристали, які дуже легко розпливаються на
повітрі Дуже легко розчинний у воді й 96 % спирті.
Зберігають у повітронепроникному контейнері.
Амонію ацетату розчин. 1004901.
150 г амонію ацетату Ррозчиняють у воді Р, дода-
ють 3 мл кислоти оцтової льодяної Р та доводять
об'єм розчину водою Рао 1000 мл.
Термін придатності 7 діб.
Амонію ванадат. МН4УО3. (М.м. 117.0). 1006800.
[7803-55-6]. Амонію триоксованадат(У).
Кристалічний порошок від білого до жовтавого кольо-
ру. Мало розчинний у воді, розчинний у розчині аміаку
розведеному РІ.
Амонію ванадату розчин. 1006801.
1.2 г амонію ванадату Ррозчиняють у 95 мл води Р
і доводять об'єм розчину кислотою сірчаною Р до
100 мл.
Амонію гідрокарбонат. МН4НСО3. (М.м. 79.1). 1005500.
11066-33-7|.
Містить не менше 99 % N Н4НСО3.
Амонію дигідрофосфат. (МН4)Н2РО4. (М.м. 115.0).
1005400. [7722-76-1]. Амонію фосфат однозамішений.
Кристалічний порошок білого кольору або безбарвні
кристали Легко розчинний у воді
рН(2.2.3). Близько 4.2. Вимірюють рН розчину 23 г/л.
(1 /?)-(-)-Амонію 10- камфоросульфонат. С10Ні9МО48.
(Мм. 249.3). 1103200.
Містить не менше 97.0 % (1 Д)-(-)-амонію 10-камфо-
росульфонату.
[«Іо : -18°±2°. Визначення проводять, використовую-
чи розчин 50 г/л у воді Р.
Амонію карбонат. 1005200. [506-87-6]. Суміш амонію
гідрокарбонату (1ЧН4НСО3, М.м. 79.1) та амонію кар-
бамату (МН2СОО1ЧН4, М.м. 78.1) в різних кількісних
співвідношеннях.
Напівпрозора маса білого кольору. Повільно розчин-
ний приблизно в чотирьох частинах води. Розкла-
дається в киплячій воді
Амонію карбонат у вільному стані виділяє не менше
30 % (м/м) МН3 (М.м. 17.03).
Кількісне визначення. 2.00 г амонію карбонату розчи-
няють у 25 мл води Р, повільно додають 50.0 мл
1 М розчину кислоти хлористоводневої і титрують
1 М розчином натрію гідроксиду, використовуючи як
індикатор 0.1 мл розчину метилового оранжевого Р.
1 мл 1 М розчину кислоти хлористоводневої відповідає
17.03 мг 1^Н3.
Зберігають при температуре не вище 20 °С.
Амонію карбонату розчин. 1005201.
Розчин 158 г/л.
Амонію молібдат. (МН4)6Мо7О24,4Н2О. (М.м. 1236).
1005700. [12054-85-2].
Безбарвні кристали або кристали від жовтавого до зе-
ленуватого кольору. Розчинний у воді, практично не
розчинний у 96 % спирті.
Амонію молібдату реактив. 1005701.
Послідовно змішують по 1 об'єму розчину 25 г/л
амонію молібдату Р. розчину 100 г/л кислоти ас-
корбінової Рта розчину 294.5 г/л (Н28О4) кислоти
сірчаної Р, потім додають 2 об'єми води Р.
Термін придатності 1 доба.
Амонію молібдату реактив РІ. 1005706.
Змішують 10 мл розчину 60 г/л динатрію арсена-
ту Р, 50 мл розчину амонію молібдату Р, 90 мл кис-
лоти сірчаної розведеної Р і доводять об'єм розчину
водою Рпо 200 мл.
Суміш витримують при температурі 37 °С протягом
24 год.
Зберігають у флаконах оранжевого скла.
Амонію молібдату розчин. 1005702.
Розчин 100 г/л.
Амонію молібдату розчин Р2. 1005703.
5.0 г амоніюмолібдату Р розчиняють при нагріванні
в 30 мл води Р, потім охолоджують і доводять рН до
7.0 розчином аміаку розведеним Р2, об’єм одержано-
го розчину доводять водою Рдо 50 мл.
Амонію молібдату розчин РЗ. 1005704.
Розчин /,5 г амонію молібдату Р розчиняють при
нагріванні у 20 мл води Р.
Розчин II. Змішують 150 мл 96 % спирту Різ 150 мл
води Р. При охолодженні додають 100 мл кислоти
сірчаної Р.
Безпосередньо перед використанням до розчину І
додають розчин П у співвідношенні 20:80.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
175
4.1.1. Реактиви
Амонію молібдату розчин Р4. 1005705.
1.0 г амонію молібдату Р розчиняють у воді Р, до-
водять тим самим розчинником до об'єму 40 мл,
додають 3 мл кислоти хлористоводневої Р, 5 мл
кислоти хлорної Р і доводять об'єм розчину ацето-
ном Рао 100 мл.
Зберігають у захищеному від світла місці.
Термін придатності 1 міс.
Амонію молібдату розчин Р5. 1005706.
1.0 г амонію молібдату Р розчиняють у 40.0 мл
15 % (об/об) розчину кислоти сірчаної Р.
Термін придатності 1 доба.
Амонію нітрат. МН4МО3. (Мм 80.0). 1005800.
[6484-52-2].
Кристалічний порошок білого кольору або безбарвні
кристали. Гігроскопічний, дуже легко розчинний у
воді, легко розчинний у метанолі, розчинний у 96 %
спирті.
Зберігають у повітронепроникному контейнері
Амонію нітрат РІ. 1005801.
Має задовольняти вимоги для амонію нітрату Рта
витримувати такі додаткові випробування.
Кислотність (2.2.4). Розчин повинен мати слабо
кислу реакцію.
Хлориди (2.4.4). Не більше 0.01 % (100 ррт). 0.50 г
мають витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.015 % (150 ррт).
1.0 г має витримувати випробування на сульфа-
ти.
Сульфатна зола (2.4.14). Не більше 0.05 %. Визна-
чення проводять із 1.0 г.
Амонію оксалат. С2Н8М2О4,Н2О. (М.м. 142.1). 1005900.
[6009-70-7].
Безбарвні кристали. Розчинний у воді.
Амонію оксалату розчин. 1005901.
Розчин 40 г/л.
Амонію персульфат. (МН4)2$2О8. (М.м. 228.2). 1006000
[7727-54-0].
Кристалічний порошок або гранули білого кольору.
Легко розчинний у воді.
Амоніюпіролілинлитіокарбамат. С5Н12М282. (М.м. 164.3).
1006200. [5108-96-3]. Амонію 1-піролідинілдитіофор-
міат.
Кристалічний порошок від білого до світло-жовтого
кольору. Помірно розчинний у воді, дуже мало роз-
чинний у 96 % спирті.
Зберігають у контейнері, який містить невелику
кількість амонію карбонату в полотняному мішечку.
Амонію рейнекат. КІН4[Сг(КС8)4(?<Нз)2І,Н2О.
(М.м. 354.4). 1006300. [13573-16-5]. Амонію діамінтет-
ракіс(ізотіоціанато)хромат( ПІ) моногідрат.
Порошок або кристали червоного кольору. Помірно
розчинний у холодній воді, розчинний у гарячій воді й
96 % спирті.
Амонію рейнекату розчин. 1006301.
Розчин 10 г/л.
Готують безпосередньо перед використанням.
Амоніюсульфамат. МН2$ОзМН4. (М.м. 114.1). 1006400.
[7773-06-0].
Кристалічний порошок білого кольору або безбарвні
кристали. Гігроскопічний, дуже легко розчинний у
воді, мало розчинний у 96 % спирті.
Температура плавлення: близько 130 °С.
Зберігають у повітронепроникному контейнері.
Амонію сульфат. (NN4)2804. (М.м. 132.1). 1006500.
[7783-20-2].
Безбарвні кристали або гранули білого кольору Дуже
легко розчинний у воді, практично не розчинний в
ацетоні й 96 % спирті.
рН(2.2.3). Від 4.5 до 6.0. Вимірюють рН розчину 50 г/л
у воді, вільній від вуглецю діоксиду, Р.
Сульфатна зола (2.4.14). Не більше 0.1 %.
Амонію тіоціанат. NН48СN. (М.м. 76.1). 1006700.
[1762-95-4].
Безбарвні кристали, які розпливаються на повітрі Ду-
же легко розчинний у воді, розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Амонію тіоціанату розчин. 1006701.
Розчин 76 г/л.
Амонію форміат. €^N02. (М.м. 63.1). 1112600.
[540-69-2].
Кристали, що розпливаються, або гранули. Дуже лег-
ко розчинний у воді, розчинний у 96 % спирті.
Температура плавлення: від 119 °С до 121 °С.
Зберігають у повітронепроникному контейнері.
Амонію фосфат. ^Н4)2НРО4. (М.м. 132.1). 1006100.
[7783-28-0]. Діамонію гідрофосфат.
Кристали або гранули білого кольору. Гігроскопічний,
дуже легко розчинний у воді, практично не розчинний
у 96 % спирті.
рН (2.2.3). Близько 8. Вимірюють рН розчину 200 г/л.
Зберігають у повітронепроникному контейнері.
Амонію хлорид. 1005300. 112125-02-9]. Див. статтю
Амонію хлорид.
176
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Амонію хлориду розчин. 1005301.
Розчин 107 г/л.
Амонію цитрат. С6НИК2О7. (М.м. 226.2). 1103300.
|3012-65- 5]. Діамонію гідроцитрат.
Кристалічний порошок білого кольору або безбарвні
кристали. Легко розчинний у воді, мало розчинний у
96 % спирті.
рН (2.2.3). Близько 4.3. Вимірюють рН розчину
22.6 г/л
Амонію церію(ІУ) нітрат. (МН4)2Се(МО3)6. (М.м. 548.2).
1005000.116774-21-3|.
Кристалічний порошок оранжево-жовтого кольору
або прозорі кристали оранжевого кольору. Розчинний
уводі.
Амонію церію(ІУ) сульфат. (МН4)4Се($О4)4,2Н2О.
(АЬ/.бЗЗ). 1005100. |10378-47-9].
Кристалічний порошок або кристали оранжево-жов-
того кольору Повільно розчинний у воді.
Анетол. С10НІ2О. (М.м. 148.2). 1006900. [4180-23-8].
1-Метокси-4-(пропен-1 -іл)бензол.
Кристалічна маса білого кольору при температурі до
20-21 °С, при температурі вище 23 °С - рідина. Прак-
тично не розчинний у воді, легко розчинний в етанолі,
ефірі, етилацетаті й петролейному ефірі.
Яр : близько 1.56.
Температура кипіння: близько 230 °С.
Анетол, використовуваний у газовій хроматографії,
.має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28) в умовах, зазначених у статті Олія
анісова, використовуючи анетол як випробовуваний
розчин.
Площа основного піка, який відповідає транс-ането-
лу, із часом утримування близько 41 хв, має бути не
менше 99.0 % суми площ усіх піків.
цис-Анетол. СІ()НІ2О. (М.м. 148.2). 1007000. (7)-1-Ме-
токси-4-(пропеніл-1)бензол.
Кристалічна маса білого кольору при температурі до
20-21 °С, при температурі више 23 °С - рідина. Прак-
тично не розчинний у воді, легко розчинний в етанолі,
розчинний в ефірі, етилацетаті й петролейному ефірі.
Лр : близько 1.56.
Температура кипіння: близько 230 °С.
цис-Анетол, використовуваний у газовій хромато-
графії, має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28) в умовах, зазначених у статті Олія
анісова, використовуючи цпс-анетол як випробовува-
ний розчин.
Площа основного піка має бути не менше 92.0 % суми
площ усіх піків.
л-Анізидин. С7Н9МО. (М.м. 123.2). 1103500. [104-94-9].
4-Метоксіанілін
Кристали білого кольору. Помірно розчинний у воді,
розчинний в етанолі.
Містить не менше 97.0 % С7НУКІО.
Викликає подразнення шкіри; сенсибілізатор.
Зберігають у захищеному від світла місці при темпера-
турі від 0 °С до 4 °С.
При зберіганні /і-анізидин темнішає внаслідок окис-
нення. Окиснений л-анізидин може бути відновлений
і знебарвлений таким чином: 20 г п-анізидину Ррозчи-
няють у 500 мл води Рпри температурі 75 °С, додають
1 г натрію сульфіту Рта 10 г вугілля активованого Р,
перемішують протягом 5 хв і фільтрують. Одержаний
фільтрат охолоджують і відстоюють при температурі
близько 0 °С не менше 4 год, потім фільтрують, одер-
жані кристали промивають невеликою кількістю во-
ди Р, охолодженої до температури 0 °С, й сушать у ва-
куумі над фосфору) V) оксидом Р.
Анілін. С6Н7М. (Мм 93.1) 1007100. [62-53-3]. Бен-
золамін.
Безбарвна або жовтавого кольору рідина. Розчинний у
воді, змішується з 96 % спиртом та ефіром.
<72о : близько 1 02.
Температура кипіння: від 183 °С до 186 °С.
Зберігають у захищеному від світла місці.
Аніонообмінна смола. 1007200.
Смола у хлоридній формі, яка містить четвертинні
амонієві групи [СН2КГ(СН3)3], приєднані до полімерної
решітки, шо складається з полістиролу поперечно-зши-
гого 2 % дивінілбензолу. Випускають у вигляді гранул,
розмір яких має бути зазначений в окремих статтях.
Смолу промивають на скляному фільтрі (40) 1 М роз-
чином натрію гідроксиду до негативної реакції на хло-
риди у промивному розчині, потім промивають во-
дою Р до одержання нейтральної реакції у промивній
воді. Суспендують у свіжоприготованій воді, вільній від
аміаку, Р. і захищають від вуглецю діоксиду.
Аніонообмінна смола снльноосновна. 1026600.
Гелеподібна смола в ОН-формі, яка містить четвер-
тинні амонієві групи [СН21Ч (СН3)3, тип 1 ], приєднані
до полімерної решітки, що складається з полістиролу
поперечно-зшитого 8 % дивінілбензолу.
Прозорі гранули коричневого кольору.
Розмір часток: від 0.2 мм до 1.0 мм.
Вміст вологи: близько 50 %.
Повна обмінна ємність: не менше 1.2 мекв/мл.
Аніонообмінна смола снльноосновна для хроматографії.
1112700.
Смола з четвертинними амонієвими групами,
приєднаними до решітки латексу поперечно-зшитого
дивініл бензолом.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
177
4.1.1. Реактиви
Анісовий альдегід. С8Н8О2. (М.м. 136.1). 1007300.
1123-11-5]. 4-Метоксибензальдегід.
Масляниста рідина. Дуже мало розчинний у воді,
змішується з 96 % спиртом та ефіром.
Температура кипіння: близько 248 °С.
Анісовий альдегід, використовуваний у газовій хрома-
тографії, має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28) в умовах, зазначених у статті Олія
анісова, використовуючи анісовий альдегід як випро-
бовуваний розчин.
Площа основного піка має бути не менше 99.0 % суми
площ усіх піків.
Анісового альдегіду розчин. 1007301.
Послідовно змішують 0.5 мл анісового альдегіду Р,
10 мл кислоти оцтової льодяної Р, 85 мл мета-
нолу Р та 5 мл кислоти сірчаної Р.
Анісового альдегіду розчин РІ. 1007302.
10 мл анісового альдегіду Р змішують із 90 мл 96 %
спирту Р, додають 10 мл кислоти сірчаної Р і пе-
ремішують.
Аноліт для ізоелектрофокусування рН від 3 до 5 (0.1 М
розчин кислоти глутамінової й 0.5 М розчин кислоти фо-
сфорної). 1112800.
До розчину 14.71 г кислоти глутамінової Ру воді Рао-
дають 33 мл кислоти фосфорної Рі доводять об'єм роз-
чину водою Рао 1000 мл.
Антимоніду калію тартрат. С4Н4КО78Ь, 1/2Н2О.
(М.м. 333.9). 1007600. Калію аква[тартрато-
(4-)-О/,С)2,О-?]-антимоніат(111) гемігідрат.
Гранульований порошок білого кольору або прозорі
безбарвні кристали. Розчинний у воді й гліцерині, лег-
ко розчинний у киплячій воді, практично не розчин-
ний у 96 % спирті. Водний розчин має слабко кислу
реакцію.
Антитромбін 111. 1007800. [90170-80-2].
Антитромбін 111 виділяють із людської плазми хрома-
тографічно з використанням гепарин-агарозної ко-
лонки.
Питома активність має бути не менше 6 МО/мг.
Антитромбіну III розчин РІ. 1007801.
Антитромбін 111 Р обробляють, як зазначено ви-
робником. і розводять буферним розчином
трис(гідроксиметил)амінометану-натрію хлориду
р//7.4 Рдо активності 1 МО/мл.
Антитромбіну III розчин Р2. 1007802.
Антитромбін 111 Р обробляють, як зазначено ви-
робником, і розводять буферним розчином
трис(гідроксиметил)амінометану-натрію хлориду
рН 7.4 Рао активності 0.5 МО/мл.
Антрацен. СІ4Н10. (М.м. 178.2). 1007400. 1120-12-7].
Кристалічний порошок білого кольору. Практично нД
розчинний у воді, мало розчинний у хлороформі.
Температура плавлення: близько 218 °С.
Антрон. С14НІ0О. (М.м. 194.2). 1007500. [90-44-8].
9-(10//)-Антраценон.
Кристалічний порошок світло-жовтого кольору.
Температура плавлення: близько 155 °С.
Апігенін. С15НІ0О5. (М.м. 270.2). 1095800. [520-36-5].І
4',5,7-Тригідроксифлавон.
Легкий порошок жовтуватого кольору. Практично не І
розчинний у воді, помірно розчинний у 96 % спирті. І
Температура плавлення: близько 310 °С, із розкла-І
данням.
Хроматографія. Визначення проводять, як зазначеної
в статті Квітки римської ромашки, використовуючи!
10 мкл розчину 0.25 г/л у метанолі Р. На верхній тре-1
тині хроматограми має виявлятися основна пляма із І
жовтувато-зеленою флуоресценцією.
Апігенін-7-глюкозид. С21Н20О10. (М.м. 432.6). 1095900. І
Легкий порошок жовтуватого кольору. Практично
не розчинний у воді, помірно розчинний у 96 Й
спирті.
Температура плавлення: від 198 °С до 201 °С.
Хроматографія. Визначення проводять, як зазначено
в статті Квітки римської ромашки, використовуючи
10 мкл розчину 0.25 г/л у метанолі Р. На середній тре-
тині хроматограми має виявлятися основна пляма із
жовтуватою флуоресценцією.
Апротинін. 1007900. [9087-70-1]. Див. статтю Апротинін.
Арабіноза. С5Н10О5. (М.м. 150.1). 1008000. [87-72-9].
І_-(+)-Арабіноза.
Кристалічний порошок білого кольору. Легко розчин-
на у воді.
[«]□ : від +103° до +105°. Визначення проводять, ви-
користовуючи розчин 50 г/л у воді Р, яка містить
близько 0.05 % МН3.
Арбутин. С12Н16О7. (М.м. 272.3). 1008100. [497-76-7].
Арбутозид. 4-Гідроксифеніл-р-О-глюкопіранозид.
Дрібні, блискучі голчасті кристали білого кольору.
Легко розчинний у воді, дуже легко розчинний у га-
рячій воді, розчинний у 96 % спирті, практично не
розчинний в ефірі.
[«]□ : близько -64°. Визначення проводять, викорис-
товуючи розчин 20 г/л.
Температура плавлення: близько 200 °С.
Хроматографія. Визначення проводять, як зазначено
в статті Листя мучниці', на хроматограмі має бути лише
одна основна пляма.
178
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Аргінін. 1103600. [74-79-31- Див. статтю Аргінін.
Аргон. Аг. (А.м. 39.95). 1008200. 17440-37-1].
Містить не менше 99.995 % (об/об) Аг.
Арсену(ІП) оксид. А$2О3. (М.м. 197.8) 1008300.
11327-53-3]. Арсенистий ангідрид. Діарсену триоксид.
Кристалічний порошок або біла маса. Мало розчин-
ний у воді, розчинний у киплячій воді.
Аскорбінова кислота. 1008400. 150-81-7]. Див. статтю
Кислота аскорбінова.
Аскорбінової кислоти розчин. 1008401.
50 мг кислоти аскорбінової Р розчиняють в 0.5 мл
води Р і доводять об'єм розчину диметилформ-
амідом Рао 50 мл.
Е-Аспартил-Б-фенілаланін. С|3НІ6М2О5. (М.м. 280.3).
1008500. [13433-09-5]. (5)-3-Аміно-А-[(5)-1-карбок-
си-2-фенілетил]бурштинова кислота.
Порошок білого кольору.
Температура плавлення: близько 210 °С, із розкла-
данням.
Ацеталь. СЬНІ4О2. (М.м. 118.2). 1112300. [105-57-7].
Аиетальдегідудіетилацеталь. 1,1-Діетоксіетан.
Прозора, безбарвна, летка рідина. Змішується із во-
дою й 96 % спиртом.
</$: близько 0.824.
«0: близько 1.382.
Температура кипіння: близько 103 °С.
Ацетальдегід. С2Н4О. (М.м. 44.1). 1000200. [75-07-0].
Етаналь.
Прозора, безбарвна, займиста рідина. Змішується з
водою й 96 % спиртом.
(/*: близько 0.788.
Лр1 близько 1.332.
Температура кипіння: близько 21 °С.
Ацетилхлорид. С2Н3С1О. (Мм. 78.5). 1000800.175-36-51.
Прозора, безбарвна, займиста рідина. Розкладається у
воді й 96 % спирті, змішується з етил єн хлоридом.
: близько 1.10.
Температурні межі перегонки (2.2.11). Від 49 °С до
53 °С; має переганятись не менше 95 %.
А-Ацетил-с-капролактам. СХН13МО2. (М.м. 155.2).
1102700. 11888-91-1]. А-Ацетилгексан-6-лактам.
Безбарвна рідина. Змішується з етанолом.
і/]”: близько 1.100.
: близько 1.489.
Температура кипіння: близько 135 °С.
Анетилацетамід. С4Н7МО2. (М.м. 101.1). 1102600.
[5977-14-0]. З-Оксобутанамід.
Температура плавлення: від 53 °С до 56 °С.
Ацетилацетон. С5Н8О2. (М.м. 100.1). 1000900. [123-54-6].
2,4-Пентандіон.
Безбарвна або жовтавого кольору, легко займиста
рідина. Легко розчинний у воді, змішується з ацето-
ном, 96 % спиртом, ефіром та кислотою оцтовою льо-
дяною.
/ід : віл 1.452 до 1.453.
Температура кипіння: від 138 °С до 140 °С.
Ацетилацетону реактив РІ. 1000901.
До 100 мл розчину амонію ацетату Р додають
0.2 мл ацетилацетону Р.
Ацетилевгенол. СІ2Н|4О3. (М.м. 206.2). 1100700.
[93-28-7]. 2-Метокси-4-(2-пропеніл)фенілацетат.
Масляниста рідина жовтого кольору. Легко розчинний
у 96 % спирті й ефірі, практично не розчинний у
воді.
и® : близько 1 521.
Температура кипіння: від 281 °С до 282 °С.
Ацетилевгенол, використовуваний у газовій хромато-
графії, має витримувати таке додаткове випробуван-
ня.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено в статті Олія гвоздич-
на, використовуючи ацетилевгенол як випробовува-
ний розчин.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків.
А-Ацетилнеурамінова кислота. СцН|9МО9. (М.м. 309.3).
1001100. [131-48-6]. О-Сіалова кислота.
Голчасті кристали білого кольору. Розчинна у воді й
метанолі, мало розчинна в 96 % спирті, практично не
розчинна в ацетоні й ефірі.
[«]□ : близько -36°. Визначення проводять, викорис-
товуючи розчин 10 г/л.
Температура плавлення: близько 186 °С, із розкла-
данням.
Ацетилтирозину етиловий ефір. С|3НІ7МО4,Н2О.
(М.м. 269.3). 1001200. [36546-50-6|. А-Ацетил-(.-тиро-
зину етиловий ефір моногідрат. Етил-(5)-2-ацетамідо-
3-(4-гідроксифеніл)пропіонат моногідрат.
Кристалічний порошок білого кольору; придатний
для кількісного визначення хімотрипсину.
[«!□ : від +21° до +25°. Визначення проводять, вико-
ристовуючи розчин 10 г/л у 96 % спирті Р.
А^м : від 60 до 68. Визначення проводять за довжини
хвилі 278 нм, у 96 % спирті Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
179
4.1.1. Реактиви
Ацетилтирозину етилового ефіру 0.2 М розчин.
1001201.
0.54 г ацетилтирозину етилового ефіру Р розчиня-
ють у 96 % спирті Р і доводять об'єм розчину тим
самим розчинником до 10.0 мл.
Л-Ацетилтриптофан. С13Н14М2О3. (М.м. 246.3).
1102800. [1218-34-4]. 2-Ацетиламіно-3-(індол-3-
іл)пропіонова кислота.
Порошок білого або майже білого кольору або без-
барвні кристали. Мало розчинний у воді, розчинний у
розведених розчинах гідроксидів лужних металів.
Температура плавлення: близько 205 °С.
Кількісне визначення. 10.0 мг розчиняють в суміші роз-
чинників ацетонітрил Р - вода Р (10:90) й доводять
об'єм розчину тією самою сумішшю до 100.0 мл. Виз-
начення проводять, як зазначено в статті Триптофан у
випробуванні "1,1'-Етиліденбіс(триптофан) та інші
супровідні домішки".
Площа основного піка має бути не менше 99.0 % суми
площ усіх піків.
Ацетилхоліну хлорид. С7Н16С1МО2. (М.м. 181.7).
1001000. [60-31-1].
Кристалічний порошок. Дуже легко розчинний у хо-
лодній воді й 96 % спирті, практично не розчинний в
ефірі; розкладається в гарячій воді й розчинах лугів.
Зберігають при температурі -20 °С.
Ацетон. 1000600. [67-64-1]. Див. статтю Ацетон.
Ацетонітрил. С2Н31\1. (М.м. 41.05). 1000700. [75-05-8].
Метил ціан ід. Етан нітрил.
Прозора, безбарвна рідина. Змішується з водою, аце-
тоном, ефіром і метанолом.
<72о : близько 0.78.
Лр : близько 1.344.
Розчин 100 г/л ацетонітрилу має нейтральну реакцію
за лакмусовим папером.
Температурні межі перегонки (2.2.11). Від 80 °С до
82 °С; має переганятись не менше 95 %.
Ацетонітрил, використовуваний для спектрофото-
метрії, має задовольняти таку додаткову вимогу.
Мінімальне пропускання (2.2.25). 98 %>. Визначення
проводять в області довжин хвиль від 255 нм до 420 нм.
використовуючи як компенсаційну рідину воду Р.
Ацетонітрил для хроматографії. 1000701. Див. Аце-
тонітрил Р.
Ацетонітрил, використовуваний у хроматографії,
має задовольняти такі додаткові вимоги.
Мінімальне пропускання (2.2.25). 98 %. Визначення
проводять за довжини хвилі 240 нм, використову-
ючи як компенсаційну рідину воду Р.
Мінімальна чистота (2.2.28). 99.8 %.
Ацетонітрил для хроматографії РІ. 1000702.
Має задовольняти вимоги для ацетонітрилу Рі
такі додаткові вимоги.
Містить не менше 99.9 % С2Н31Ч.
Оптична густина (2.2.25). Не більше 0.10.1
Вимірюють за довжини хвилі 200 нм, використо-|
вуючи як компенсаційну рідину воду Р.
Барбалоїн. С21Н22О9,Н2О. (М.м. 436.4). 1008800.
[1415-73-2]. Алоїн. 1,8-Дигідрокси-З-гідроксиметил-1
10-р-Б-глюкопіранозил-10//-антрацен-9-он.
Кристалічний порошок або голчасті кристали віджов- І
того до темно-жовтого кольору, темнішає під дією І
повітря й світла. Помірно розчинний у воді й 96$ І
спирті, розчинний в ацетоні, розчинах аміаку й
гідроксидів лужних металів, дуже мало розчинний в
ефірі.
А\^м : близько 192 - за довжини хвилі 269 нм;
близько 226 - за довжини хвилі 296.5 нм;
близько 259 - за довжини хвилі 354 нм.
Визначення проводять, використовуючи як розчин-
ник метанол Р, у перерахунку на безводну речовину.
Хроматографія. Визначення проводять, як зазначено
в статті Кора крушини', на хроматограмі має виявля-
тись лише одна основна пляма.
Барбітал. 1008900. [57-44-3]. Див. статтю Барбітал.
Барбітал-натрій. С8НцМ2№О3. (М.м. 206.2). 1009000.
[144-02-5].
Містить не менше 98.0 % 5,5-діетил-1 //,3/7,5Н-
піримідин-2,4,6-триону натрію.
Кристалічний порошок білого кольору або безбарвні
кристали. Легко розчинний у воді, мало розчинний у
96 % спирті, практично не розчинний в ефірі.
Барбітурова кислота. С4Н4М2О3. (М.м. 128.1). 1009100.
[67-52-7 ]. 1//,3//,5//-Піримідин-2,4,6-трион.
Порошок білого або майже білого кольору. Мало роз-
чинна у воді, легко розчинна в киплячій воді й розве-
дених кислотах.
Температура плавлення: близько 253 °С.
Барію гідроксид. Ва(ОН)2,8Н2О. (М.м. 315.5). 1009400.
[12230-71-6]. Барію дигідроксид.
Безбарвні кристали. Розчинний у воді.
Барію гідроксиду розчин. 1009401.
Розчин 47.3 г/л.
Барію карбонат. ВаСО3. (М.м. 197.3). 1009200. [513-77-9].
Порошок білого кольору або розсипчаста маса. Прак-
тично не розчинний у воді.
Барію сульфат. 1009500. [7727-43-7]. Див. статтю Барію
сульфат.
180
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Барію хлорид. ВаС12,2Н2О. {М.м. 244.3). 1009300.
|10326-27-9]. Барію дихлорид.
Безбарвні кристали. Легко розчинний у воді, мало
розчинний у 96 % спирті.
Барію хлориду розчин РІ. 1009301.
Розчин 61 г/л.
Барію хлориду розчин Р2. 1009302.
Розчин 36.5 г/л.
Бензальдегід. С7Н6О. {М.м. 106.1). 1009600. [100-52-7].
Безбарвна або жовтавого кольору рідина. Мало роз-
чинний у воді, змішується з 96 % спиртом і ефіром.
ф®: близько 1.05.
Ир1: близько 1.545.
Температурні межі перегонки (2.2.11). Від 177 °С до
180 °С; має переганятись не менше 95 %.
Зберігають у захищеному від світла місці.
Бензетонію хлорид. С27Н42С1НО2,Н2О. {М.м. 466.1).
1009900. [121-54-0]. Бензилдиметил[2-[2-[4-(1,1,3,3-
тетраметилбутил)феноксі]етоксі]етил]амонію хлорид
моногідрат.
Дрібний порошок білого кольору або безбарвні крис-
тали. Розчинний у воді й 96 % спирті, мало розчинний
в ефірі.
Температура плавлення: близько 163 °С.
Зберігають у захищеному від світла місці.
Бензил. С14Н10О2. {М.м. 210.2). 1117800. [134-81-6].
Дифеніл етандіон.
Кристалічний порошок жовтуватого кольору. Не роз-
чинний у воді, розчинний у 96 % спирті, етилацетаті й
толуолі.
Температура плавлення: 95 °С.
Бензилбензоат. 1010800. [120-51-4]. Див. статтю Бен-
зиібензоат.
Бензилкоричний ефір. С16Н14О2. {М.м. 238.3). 1010900.
[103-41-3]. Бензил-З-фенілпроп-2-еноат. Бензилпи-
намат.
Безбарвні або жовтуватого кольору кристали. Прак-
тично не розчинний у воді, розчинний у 96 % спирті й
ефірі.
Температура плавлення: близько 39 °С.
Бензиловий спирт. 1010700. [100-51-6]. Див. статтю
Спирт бензиловий.
Бензилпеніциліну натрієва сіль. 1011000. [69-57-8]. Див.
статтю Бензилпеніциліну натрієва сіль.
2-Бензилпіридин. С12НцГ\. {М.м. 169.2). 1112900.
[101-82-6].
Містить не менше 98.0 % С12НИ1Ч.
Рідина жовтого кольору.
Температура плавлення: від 13 °С до 16 °С.
Бензоїларгініну етилового ефіру гідрохлорид.
С15Н23С1М4О3. {М.м. 342.8). 1010500. [2645-08-1].
А-Бензоїл-Е-аргінін етилового ефіру гідрохлорид.
Етил(5)-2-бензамідо-5-гуанідиновалерату гідрохло-
рид.
Кристалічний порошок білого кольору. Дуже легко роз-
чинний у воді й етанолі, практично не розчинний в ефірі.
|о(]р : від-15° до-18°. Визначення проводять, викори-
стовуючи розчин 10 г/л.
Температура плавлення: близько 129 °С.
А\%м : від 310 до 340. Визначення проводять за довжи-
ни хвилі 227 нм, використовуючи розчин 0.01 г/л.
Л-Бензоїл-Ь-проліл-Ь-фенілаланіл-Ь-аргініну 4-нітро-
анілідапетат. С^Н^К/О^. {М.м. 703). 1010600.
Бензоїлхлорид. С7Н5С1О. {М.м. 140.6). 1010400.
[98-88-4].
Безбарвна, сльозоточива рідина. Розчинний в ефірі,
розкладається у воді й 96 % спирті.
<72о : близько 1.21.
Температура кипіння: близько 197 °С.
Бензоїн. С14Н12О2. {М.м. 212.3). 1010200. [579-44-2].
2-Гідрокси-1,2-дифенілетанон.
Кристали злегка жовтавого кольору. Дуже мало роз-
чинний у воді, легко розчинний в ацетоні, розчин-
ний у гарячому 96 % спирті, помірно розчинний в
ефірі.
Температура плавлення: близько 137 °С.
Бензойна кислота. 1010100. [65-85-0]. Див. статтю Кис-
лота бензойна.
Бензол. С6Н6. {М.м. 78.1). 1009800. [71-43-2].
Прозора, безбарвна, займиста рідина. Практично не
розчинний у воді, змішується з 96 % спиртом і ефіром.
Температура кипіння: близько 80 °С.
Бензофенон. С13Н10О. {М.м. 182.2). 1010300. [119-61-9].
Дифенілметанон.
Кристали у вигляді призм. Практично не розчинний у
воді, легко розчинний у 96 % спирті й ефірі.
Температура плавлення: близько 48 °С.
1,4-Бензохінон. С6Н4О2. {Мм. 108.1). 1118500. [106-51-4].
Циклогекса-2,5-дієн-1,4-діон.
Містить не менше 98.0 % С6Н4О2.
Бергаптен. С12Н8О4. {М.м. 216.2). 1103700. [484-20-8].
5-Метоксипсорален.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
181
4.1.1. Реактиви
Безбарвні кристали. Практично не розчинний у воді,
помірно розчинний у 96 % спирті, мало розчинний у
кислоті оцтовій льодяній.
Температура плавлення: близько 188 °С.
Бетулін. С30Н50О2. (М.м. 442.7). 1011100. [473-98-3].
Луп-20(39)-ен-3р,28-діол.
Кристалічний порошок білого кольору.
Температура плавлення: від 248 °С до 251 °С.
Бісбензімід. С25Н27С13М()О,5Н2О. (Мм. 624). 1103800.
[23491 -44-3]. 4-[5-|5-(4-Метилпіперазин-1 -іл)бен-
зімідазол-2-іл]бензімідазол-2-іл]фенолу тригідрохло-
рид пентагідрат.
Бісбензіміду вихідний розчин. 1103801.
5 мг бісбензіміду Р розчиняють у воді Р і доводять
об'єм розчину тим самим розчинником до 100 мл.
Зберігають в темному місці.
Бісбензіміду робочий розчин. 1103802.
Безпосередньо перед використанням 100 мкл
вихідного розчину бісбензіміду Р доводять фізіоло-
гічним фосфатно-буферним розчином рН 7.4 Р до
об'єму 100 мл.
Біурет. С2Н5М3О2. (Мм. 103.1). 1011600. [108-19-0].
Кристали білого кольору, гігроскопічні. Розчинний у
воді, помірно розчинний у 96 % спирті, дуже мало роз-
чинний в ефірі.
Температура плавлення: від 188 °С до 190 °С, із роз-
кладанням.
Зберігають у повітронепроникному контейнері.
Біуретовий реактив. 1011601.
1.5 г міді(ІІ) сульфату Р і 6.0 г калію-натрію тар-
трату Р розчиняють у 500 мл води Р, додають
300 мл розчину 100 г/л натрію гідроксиду Р,
вільного від карбонатів, доводять об'єм розчину
тим самим розчинником до 1000 мл і перемішу-
ють.
Біфеніл-4-ол. С12НІ0О. (М.м. 170.2). 1011300. [90-43-7|.
4-Фенілфенол.
Кристалічний порошок білого кольору. Практично не
розчинний у воді.
Температура плавлення: від 164 °С до 167 °С.
ВКР індикатора розчин. 1013000.
0.1 г бромтимолового синього Р, 20 мг метилового чер-
воного Рта 0.2 г фенолфталеїну Р розчиняють у 96 %
спирті Р, доводять об'єм розчину тим самим розчин-
ником до 100 мл і фільтрують.
Борна кислота. 1011800. [ 10043-35-3]. Див. статтю Кис-
лота борна.
Борнеол. С10НІ8О. (М.м. 154.3). 1011900. [507-70-0].
ендо-1,7,7-Триметилбіцикло[2.2.1 ]гептан-2-ол.
Безбарвні кристали. Легко сублімується, практично
не розчинний у воді, легко розчинний у 96 % спирті,
ефірі й петролейному ефірі.
Температура плавлення: близько 208 °С.
Хроматографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель с Р. На лінію старту хромато-
графічної пластинки наносять 10 мкл розчину 1 г/л в
толуолі Р. Хроматографують у хлороформі Р. Коли
фронт розчинника пройде 10 см від лінії старту, плас-
тинку виймають із камери, сушать на повітрі й обпри-
скують розчином анісового альдегіду Р, витрачаючи
10 мл на пластинку площею 200 мм2, сушать при тем-
пературі від 100 °С до 105 °С протягом 10 хв. На хро-
матограмі має виявлятись лише одна основна пляма.
Борнілацетат. СІ2Н20О2. (Мм. 196.3). 1012000. [5655-61-8].
ендо-1,7,7-Триметилбіцикло[2.2.1 ]гепт-2-ил ацетат.
Безбарвні кристали або безбарвна рідина. Дуже мало
розчинний у воді, розчинний у 96 % спирті й ефірі.
Температура плавлення: близько 28 °С.
Хроматографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель с Р. На лінію старту хромато-
графічної пластинки наносять 10 мкл розчину 2 г/л в
толуолі Р. Хроматографують у хлороформі Р. Коли
фронт розчинника пройде 10 см, пластинку виймають
із камери, сушать на повітрі й обприскують розчином
анісового альдегіду Р, витрачаючи 10 мл на пластинку
площею 200 мм2, сушать при температурі від 100 °С до
105 °С протягом 10 хв. На хроматограмі має виявля-
тись лише одна основна пляма.
Бору фторид. ВЕ3. (М.м. 67.8). 1012100. [7637-07-2].
Бору трифторид.
Безбарвний газ.
Бору фториду розчин у метанолі. 1012101.
Розчин 140 г/л бору фториду Ру метанолі Р.
Бору хлорид. ВС13. (М.м. 117.2). 1112000. [10294-34-5].
Бору трихлорид.
Безбарвний газ. Бурхливо реагує із водою. Викорис-
товують у вигляді розчинів у підхожих розчинниках
(2-хлоретанол, метиленхлорид, гексан, гептан, мета-
нол).
Токсичний, спричиняє корозію.
Температура кипіння: близько 12.6 °С.
: близько 1.420.
Бору хлориду розчин у метанолі. 1112001.
Розчин 120 г/л у метанолі Р.
Зберігають у захищеному від світла місці при тем-
пературі -20 °С, переважно в ампулах.
182
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.±.±. реактиви
Брильянтовий синій. 1012200. [6104-59-2]. Див. Кислот-
ний синій 83 Р.
Бром. Вг2. (Мм. 159.8). 1012400. [7726-95-6].
Димляча рідина коричнювато-червоного кольору. Ма-
лорозчинний у воді, розчинний у 96 % спирті й ефірі.
: близько 3.1.
Бромна вода. 1012402.
З мл брому Р струшують із 100 мл води Р до наси-
чення.
Зберігають над надлишком брому Ру захищеному
від світла місці.
Бромна вода РІ. 1012403.
0.5 мл брому Рструшують із 100 мл води Р
Зберігають у захищеному від світла місці.
Термін придатності 7 діб.
Брому розчин. 1012401.
ЗО г брому Р і 30 г калію броміду Р розчиняють у
воді Рі доводять об'єм розчину тим самим розчин-
ником до 100 мл.
5-Бром-2'-деоксіуридин. С9НцВг\2О5. (М.м. 307.1).
1012500. [59-14-3]. 5-Бром-1-(2-деокси-р-О-е/ш/и/ю-
пентофуранозил)-1 /7,3//-піримідин-2,4-діон.
Температура плавлення: близько 194 °С.
Хроматографія. Визначення проводять, як зазначено
в статті Йодоксіуридин\ наносять 5 мкл розчину
0.25 г/л; на одержаній хроматограмі має виявлятись
лише одна основна пляма.
Бромелайни. 1012300. [37189-34-7].
Концентрат протеолітичних ферментів, одержаний із
Анапа! сотози! Мегг.
Порошок тьмяно-жовтого кольору.
Активність. 1 г бромелайнів має вивільнювати близь-
ко 1.2 камінного азоту із розчину желатину Р протягом
20хв при температурі 45 °С і рН 4.5.
Бромелайнів розчин. 1012301.
Розчин 10 г/л бромелайнів Ру суміші розчинників
фосфатний буферний розчин рН 5.5 Р- розчин 9 г/л
натрію хлориду Р (1.9).
Бромистоводнева кислота ЗО % розчин. 1098700.
[10035-10-6].
ЗО % розчин кислоти бромистоводневої в кислоті оц-
товій льодяній Р.
Перед розкриттям вміст обережно дегазують.
Бромистоводнева кислота розведена. 1098701.
5.0 мл ЗО % розчину кислоти бромистоводневої Р
поміщають у флакони з темного скла, укупорюють
в атмосфері аргону Р поліетиленовими пробками й
зберігають у захищеному від світла місці. Безпосе-
редньо перед використанням додають 5.0 мл кис-
лоти оцтової льодяної Р і перемішують.
Зберігають у темному місці.
Бромистоводнева кислота 47 % розчин. 1118900.
47 % (м/м) розчин кислоти бромистоводневої у воді Р.
Бромистоводнева кислота розведена РІ. 1118901.
Містить 7.9 г/л НВг.
16.81 г 47 % розчину кислоти бромистоводневої Р
розчиняють у воді Р і доводять об'єм розчину тим
самим розчинником до 1000 мл.
Бромкрезоловий зелений. С2іН|4Вг4О58. (М.м. 698).
1012600. [76-60-8|. 3',3",5',5"-Тетрабром-л/-крезол-
сульфонфталеїн. 4,4'-(5//-2,1-Бензоксатіол-3-іліден)-
біс[2.6-дибром-3-метилфенол]5.5-діоксид.
Порошок білого з коричнюватим відтінком кольору.
Мало розчинний у воді, розчинний у 96 % спирті й
розведених розчинах гідроксидів лужних металів.
Бромкрезолового зеленого розчин. 1012601.
50 мг бромкрезолового зеленого Р розчиняють в
0.72 мл 0.1 М розчину натрію гідроксиду й 20 мл
96 % спирту Р, доводять об'єм розчину водою Рдо
100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0.2 мл роз-
чину бромкрезолового зеленого, з'являється синє
забарвлення, яке має перейти у жовте при дода-
ванні не більше 0.2 мл 0.02 Мрозчину кислоти хло-
ристоводневої.
Зміна забарвлення. Від жовтого до синього в інтер-
валі рН 3.6-5.2.
Бромкрезолового зеленого і метилового червоного
розчин. 1012602.
0.15г бромкрезолового зеленого Р і 0.1 г метилового
червоного Р розчиняють у 180 мл етанолу Р і дово-
дять об'єм розчину водою Рдо 200 мл.
Бромкрезоловий пурпуровий. С2іН|6Вг2О58. (М.м. 540.2).
1012700. [115-40-2]. 3'.3"-Дибром-о-крезолсульфон-
фталеїн. 4 4'-(3//-2,1-Бензоксатіол-3-іліден)біс-[2-
бром-6-метилфенол]5,5-діоксид.
Порошок рожевуватого кольору Практично не роз-
чинний у воді, розчинний у 96 % спирті й розведених
розчинах гідроксидів лужних металів.
Бромкрезолового пурпурового розчин. 1012701.
50 мг бромкрезолового пурпурового Р розчиняють в
0.92 мл 0.1 М розчину натрію гідроксиду й 20 мл
96 % спирту Р, доводять об'єм розчину водою Р до
100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0.2 мл
розчину бромкрезолового пурпурового й 0.05 мл
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
183
4.1.1. Реактиви
0.02 М розчину натрію гідроксиду, з'являється
синювато-фіолетове забарвлення, яке має пе-
рейти у жовте при додаванні не більше 0.2 мл
0.02 М розчину кислоти хлористоводневої.
Зміна забарвлення. Від жовтого до синювато-
фіолетового в інтервалі рН 5.2-6.8.
Бромтимоловий синій. С27Н2ЯВГ2О58. (М.м. 624).
1012900. [76-59-5]. 3',3"-Дибромтимолсульфонфта-
леїн. 4,4'-(3//-2,1-Бензоксатіол-3-іліден)біс(2-бром-
6-ізопропіл-3-метилфенол)5,5-діоксид.
Порошок від червонувато-рожевого до коричнювато-
го кольору. Практично не розчинний у воді, розчин-
ний у 96 % спирті й розведених розчинах гідроксидів
лужних металів.
Бромтимолового синього розчин РІ. 1012901.
50 мг бромтимолового синього Р розчиняють в
суміші 4 мл 0.02 М розчину натрію гідроксиду й
20 мл 96 % спирту Р, доводять об'єм розчину во-
дою Рдо 100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0.3 мл роз-
чину бромтимолового синього; з'являється жовте
забарвлення, яке має перейти у синє при дода-
ванні не більше 0.1 мл 0.02 М розчину натрію
гідроксиду.
Зміна забарвлення. Від жовтого до синього в інтер-
валі рН 5.8-7.4.
Бромтимолового синього розчин Р2. 1012902.
Розчин 10 г/л в диметилформаміді Р.
Бромтимолового синього розчин РЗ. 1012903.
До 0.1 г бромтимолового синього Р додають 3.2 мл
0.05 М розчину натрію гідроксиду й 5 мл спирту
(90 %, об/об) Р, нагрівають до розчинення, одер-
жаний розчин охолоджують і доводять спиртом
(90 %, об/об) Рдо об'єму 250 мл.
Бромфеноловий синій. Сі9Ні0Вг4О58. (М.м. 670).
1012800. [115-39-9]. 3',3",5',5"-Тетрабромфенолсуль-
фонфталеїн. 4,4'-(3//-2,1-Бензоксатіол-3-іліден)біс
(2,6-дибромфенол)5,5-діоксид.
Порошок світлого оранжево-жовтого кольору. Дуже ма-
ло розчинний у воді, мало розчинний у 96 % спирті, лег-
ко розчинний у розчинах гідроксидів лужних металів.
Зміна забарвлення. Від жовтого до синювато-І
фіолетового в інтервалі рН 2.8-4.4.
Бромфенолового синього розчин РІ. 1012802.
50 мг бромфенолового синього Ррозчиняють при обе-
режному нагріванні у 3.73 мл 0.02 М розчину натрім
гідроксиду й доводять водою Рдо об'єму 100 мл.
Бромфенолового синього розчин Р2. 1012803.
0.2 г бромфенолового синього Р розчиняють при
нагріванні у 3 мл 0.1 М розчину натрію гідроксиду
й 10 мл 96 % спирту Р, одержаний розчин охолод-
жують і доводять 96 % спиртом Рдо об'єму 100 мл.
Бруцин. С2зН261Ч2О4,2Н2О. (М.м. 430.5). 1013100.
[357-57-3]. 10,11-Диметоксистрихнін.
Безбарвні кристали. Мало розчинний у воді, легко
розчинний у 96 % спирті й ефірі.
Температура плавлення: близько 178 °С.
Бурштинова кислота. С4Н6О4. (М.м. 118.1). 1085600.
[110-15-6]. Бутандикарбонова кислота.
Кристалічний
порошок або безбарвні
кольору. Розчинна у воді й 96 % спирті.
кристали
білого
Температура плавлення: від 184 °С до 187 °С.
Бутанол. С4Н10О. (М.м. 74.1). 1013200. [71-36-3]. и-Бу-
танол. 1-Бутанол.
Прозора, безбарвна рідина.
Змішується
з 96%
спир-
том.
: близько 0.81.
Температура кипіння: від 116 °С до 119 °С.
2-Бутанол РІ. С4Н|0О. (М.м. 74.1).
втор-Бутиловий спирт.
Містить не менше 99.0 % С4Н10О.
1013301.
[78-92-2]
Прозора, безбарвна рідина. Розчинний
змішується з 96 % спиртом і ефіром.
г/20 : близько 0.81.
Температурні межі перегонки (2.2.11). Від
100 °С; має переганятись не менше 95 %.
Кількісне визначення. Проводять
матографії, як зазначено в статті
у ВОДІ
99 °С до
методом газової хро
Спирт ізопропіловии
Бромфенолового синього розчин. 1012801.
0.1 г бромфенолового синього Р розчиняють в суміші
1.5 мл 0.1 М розчину натрію гідроксиду й 20 мл
96 % спирту Р, доводять об'єм розчину водою Р до
100 мл.
Випробування на чутливість. До 20 мл води, вільної
від вуглецю діоксиду, Р додають 0.05 мл розчину
бромфенолового синього і 0.05 мл 0.1 М розчи-
ну кислоти хлористоводневої; з'являється жовте
забарвлення, яке має перейти у синю-
вато-фіолетове при додаванні не більше 0.1 мл
0.1 М розчину натрію гідроксиду.
Бутиламін. С4НІ(М. (М.м. 73.1). 1013600. [109-73-9].
1-Бутанамін.
Переганяють і використовують протягом 1 міс.
Безбарвна рідина. Змішується з водою, 96
ефіром.
: близько 1.401.
Температура кипіння: близько 78 °С.
трет- Бутиламін.
тилетиламін Р.
1100900. [75-64-9]. Див.
спиртом
1,1-Диме
184
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
2001
4.1.1. Реактиви
Бутилацетат. С6Н12О2. (Мм. 116.2). 1013400. [123-86-4].
Прозора, безбарвна, займиста рідина. Мало розчин-
ний у воді, змішується з 96 % спиртом та ефіром.
4,: близько 0.88.
Лр : близько 1.395.
Температурні межі перегонки (2.2.11). Від 123 °С до
126 °С; має переганятись не менше 95 %.
Бутилацетат РІ. 1013401.
Прозора, безбарвна, займиста рідина. Мало роз-
чинний у воді, змішується з 96 % спиртом та
ефіром.
: близько 0.883.
п$ : близько 1.395.
Бутанол. Не більше 0.2 %. Визначення проводять
методом газової хроматографії.
н-Бутилформіат. Не більше 0.1 %. Визначення
проводять методом газової хроматографії.
н-Бутилпропіонат. Не більше 0.1 %. Визначення
проводять методом газової хроматографії.
Вода. Не більше 0.1 %.
Кількісне визначення. Не менше 99.5 % С6Н)2О2.
Визначення проводять методом газової хромато-
графії.
Бутилборна кислота. С4НцВО2. (Мм. 101.9). 1013700.
14426-47-5].
Містить не менше 98 % С4НИВО2.
Температура плавлення: від 90 °С до 92 °С.
трем-Бутилгідроксипероксид. С4Н)0О2. (Мм. 90.1).
1118000. [75-91-2]. 1,1-Диметилетилгідроксипероксид.
Займиста рідина. Розчинний в органічних розчинниках.
4°: 0.898.
и™: 1.401.
Температура плавлення: 35 °С.
Бутилгідрокситолуол. 1013800. [128-37-0]. Див. статтю
Бутилгідрокситолуол.
трет-Бутилметиловий ефір. /013900. [1634-04-4]. Див.
/, 1-Диметилетилметиловий ефір Р.
Бутиловий ефір парагідроксибензойної кислоти.
1103900. [94-26-8]. Див. статтю Бутиловий ефір пара-
гідроксибензойної кислоти.
Бутильований гідрокситолуол. 1013800. [128-37-0]. Див.
Бутилгідрокситолуол Р.
Бутиролактон. С4Н6О2. (Мм. 86.1). 1104000. [96-48-0].
Дигідро-2(3/Д-фуранон. у-Бутиролактон.
Масляниста рідина. Змішується з водою, розчинний у
метанолі й ефірі.
: близько 1.435.
Температура кипіння: близько 204 °С.
Вазелін. 1062100.
Напіврідка суміш вуглеводнів, одержана з нафти й
знебарвлена. Практично не розчинний у воді й 96 %
спирті, розчинний в ефірі й петролейному ефірі РІ;
розчин іноді виявляє слабку опалесценцію.
Вазелінове масло. 1062000. [8042-47-5]. Див. статтю
Вазелінове масло.
Валеріанова кислота. С5Н|0О2. (Мм. 102.1). 1095200.
[109-52-4]. Пентанова кислота.
Безбарвна рідина. Розчинна у воді, легко розчинна в
96 % спирті й ефірі.
г/2о : близько 0.94.
: близько 1.409.
Температура кипіння: близько 186 °С.
Ванадію(У) оксид. У2О5. (М.м. 181.9). 1034000.
[1314-62-1]. Ванадієвий ангідрид. Диванадію пентоксид.
Містить не менше 98.5 % У2О5.
Порошок від жовто-коричневого до оранжевувато-ко-
ричневого кольору. Мало розчинний у воді, розчин-
ний у концентрованих мінеральних кислотах і розчи-
нах гідроксидів лужних металів з утворенням солей.
Прозорість (2.2.1). 1 г ванадію(У) оксиду нагрівають з
10 мл кислоти сірчаної Р протягом 30 хв, охолоджують
і доводять об'єм розчину тією самою кислотою до
10 мл. Розчин має бути прозорим.
Чутливість до водню пероксиду. 1.0 мл розчину, приго-
тованого для випробування на прозорість, обережно
доводять водою Р до об'єму 50.0 мл (випробовуваний
розчин). До 0.5 мл випробовуваного розчину додають
0.1 мл розчину 0.1 г/л (Н2О2) водню пероксиду Р. Одер-
жаний розчин повинен мати чітко виражене оранжеве
забарвлення порівняно із забарвленням контрольного
розчину, приготованого з 0.5 мл випробовуваного роз-
чину і 0.1 мл води Р. Оранжеве забарвлення випробо-
вуваного розчину має перейти в оранжево-жовте після
додавання 0.4 мл розчину 0.1 г/л (Н2О2) водню перок-
сиду.
Втрата в масі після прожарювання. Не більше 1.0 %.
Визначення проводять із 1.00 г при температурі
700 °С.
Кількісне визначення. 0.200 г ванадію(У) оксиду розчи-
няють при нагріванні в 20 мл 70 % (м/м) розчину кис-
лоти сірчаної Р, додають 100 мл води Р і 0.02 Мрозчи-
ну калію перманганату до утворення червонуватого за-
барвлення. Надлишок калію перманганату знебарв-
люють додаванням розчину 30 г/л натрію нітриту Р,
потім додають 5 г сечовини Рі 80 мл 70 % (м/м) розчи-
ну кислоти сірчаної Р, охолоджують і негайно титру-
ють 0.1 Мрозчином заліза сульфату до появи зеленува-
то-червоного забарвлення, використовуючи як інди-
катор 0.1 мл фероїну Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
185
4.1.1. Реактиви
1 мл 0.1 Мрозчину заліза сульфату відповідає 9.095 мг
^О5.
Ванадію(У) оксиду розчин у кислоті сірчаній.
1034001.
0.2 г ванадію(1/) оксиду Р розчиняють у 4 мл кисло-
ти сірчаної Р і доводять об'єм розчину водою Р до
100 мл.
Ванілін. 1095300. [121-33-5]. Див. статтю Ванілін.
Ваніліну реактив. 1095301.
До 100 мл розчину 10 г/л ваніліну Ру 96 % спирті Р
обережно по краплях додають 2 мл кислоти сірча-
ної Р.
Термін придатності 2 доби.
Ваніліну розчин у кислоті фосфорній. 1095302.
1.0 г ваніліну Р розчиняють у 25 мл 96 % спир-
ту Р, додають 25 мл води Р і 35 мл кислоти фос-
форної Р.
Винна кислота. 1087200. [87-69-4]. Див. статтю Кисло-
та винна.
Відновлююча суміш. 1074700.
Для одержання однорідної суміші послідовно змішують
попередньо подрібнені реактиви: 20 мг калію броміду Р,
0.5 г гідразину сульфату Р і 5 г натрію хлориду Р.
Вінілацетат. С4Н6О2. (М.м. 86.10). 1111800. [108-05-4].
</20 : близько 0.930.
Температура кипіння: близько 72 °С
2-Вінілпіридин. С7Н7М. (М.м. 105.1). 1102200. [100-69-6].
Рідина жовтого кольору. Змішується з водою.
: близько 0.97
: близько 1.549.
1-Вінілпіролідин-2-он. С6Н9МО. (Мм. 111.1). 1111900.
[88-12-0].
Містить не менше 99.0 % С6Н9МО.
Прозора, безбарвна рідина
Вода (2.5.12, напівмікромепюд). Не більше 0.1 %. Виз-
начення проводять із 2.5 г, використовуючи як роз-
чинник суміш 50 мл метанолу безводного Рі 10 мл бу-
тиролактону Р
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28).
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізапійним детектором за таких
умов:
— колонка кварцова капілярна розміром 30 м х 0.5 мм,
покрита шаром макроголу 20 000 Р завтовшки
1.0 мкм;
— газ-носій гелій для хроматографії Р;
— температура блока вводу проби 190 °С;
— температуру колонки програмують таким чином І
витримують температуру 80 °С протягом 1 хв, по- І
тім піднімають до 190 °С із швидкістю 10 °Сза.хв І
і витримують температуру 190 °С протягом 15 хв. І
Хроматографують 0.3 мкл випробовуваної речовини, І
регулюючи швидкість потоку газу-носія таким чи-
ном, щоб час утримування піка, який відповідає
1-вінілпіролідин-2-ону, становив близько 17 хв. Вміст
С6Н9МО визначають методом внутрішньої нор-
малізації.
Вінілполімер октадецилсилільний для хроматографії.
1121600.
Сферичні частки (5 мкм) сополімеру вінілового спир-
ту, зв'язаного октадеиилсиланом.
Містить вуглецю 17 %.
Вінілхлорид. С2Н3С1. (М.м. 62.5). 1095400. [75-01-4|.
Безбарвний газ. Мало розчинний в органічних роз-
чинниках.
Вісмуту нітрат основний. [4ВіМОз(ОН)2,ВЮ(ОН)|.
(М.м. 1462). 1011500. [1304-85-4].
Порошок білого кольору. Практично не розчинний у
воді.
Вісмуту нітрат основний РІ. 1011501
Містить не менше 71.5 % і не більше 74.0 % вісму-
ту (Ві), і не менше 14.5 %, але не більше 16.5%
нітрату, в перерахунку на азоту(У) оксид (N20/)
Вісмуту нітрату основного розчин. 1011502.
5 г вісмуту нітрату основного РІ розчиняють у
суміші 8.4 мл кислоти азотної Рта 50 мл води Р.
доводять об'єм розчину водою Р до 250 мл і
фільтрують, якщо необхідно.
Кислотність. До 10 мл додають 0 05 мл розчину
метилового оранжевого Р; забарвлення розчину
має змінитися при додаванні від 5.0 мл до 6.25 мл
1 М розчину натрію гідроксиду.
Вода. 1095500. [7732-18-5]. Див. статтю Вода очищена.
Вода, вільна від аміаку. 1095501. [7732-18-5].
До 100 мл води Р додають 0.1 мл кислоти сірча-
ної Р, переганяють, використовуючи прилад для
визначення Температурних меж перегонки
(2.2 11). відкидають перші 10 мл і збирають на-
ступні 50 мл.
Вода, вільна від вуглецю діоксиду. 1095502. [7732-18-5|.
Воду Р кип'ятять протягом кількох хвилин, охоло-
джують.
Зберігають, захищаючи від атмосферного впливу.
Вода, вільна від нітратів. 1095506. [7732-18-5].
До 100 мл води Р додають кілька міліграмів калію
перманганату Р та барію гідроксиду Р, переганя-
186
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
ють, використовуючи прилад для визначення Тем-
пературних меж перегонки (2.2.11), відкидають
перші 10 мл і збирають наступні 50 мл.
Вода, вільна від часток. 1095507. [7732-18-5].
Воду Р фільтрують крізь мембранний фільтр із
розміром пор 0.22 мкм.
Вода дистильована. 1095504. [7732-18-5].
Вода Р, одержана шляхом перегонки.
Вода для ін'єкцій. 1095505. [7732-18-5]. Див. статтю
Вода для ін'єкцій.
Вода для хроматографії. 1095503. [7732-18-5].
Деіонізована вода Р, яка має опір не менше
0.18 Мом-м.
Водень для хроматографії. Н2. (М.м. 2.016). 1043700.
11333-74-0].
Містить не менше 99.95 % (об/об) Н2.
Водню пероксиду розчин концентрований. 1043900.
17722-84-1]. Див. статтю Розчин водню пероксиду (ЗО %).
Водню пероксиду розчин розведений. 1043800. [7722-84-1].
Див. статтю Розчин водню пероксиду розведений (3 %).
Вугілля активоване. 1017800. [64365-11-3]. Див. статтю
Вугілля активоване.
Вугілля графітизоване для хроматографії. 1015900.
Вуглецеві ланцюжки з довжиною ланцюга більше за
Сч; розмір часток від 400 мкм до 850 мкм.
Густина: 0.72.
Площа поверхні: 10 м2/г.
Не використовують при температурі вище 400 °С.
Вуглеводні з низьким тиском пари (тип Ь). 1049400.
Масляниста маса. Розчинні у бензолі й толуолі.
Вутлецю діоксид. 1015600. [124-38-9]. Див. статтю Вуг-
іецю діоксид.
Вутлецю діоксид РІ. СО2. (М.м. 44.01). 1015700.
Містить не менше 99.995 % (об/об) СО2.
Вуглецю монооксид: менше 5 ррт.
Кисень: менше 25 ррт.
Вутлецю дисульфід. С82. (М.м. 76.1). 1015800. [75-15-0].
Сірковуглець.
Безбарвна або жовтуватого кольору займиста рідина.
Практично не розчинний у воді, змішується з етано-
лом та ефіром.
(і® : близько 1.26.
Температура кипіння: від 46 °С до 47 °С.
Вуглецю монооксид. СО. (М.м. 28.01). 1016000.
[630-08-0].
Містить не менше 99.97 % (об/об) СО.
Вутлецю тетрахлорид. СС14. (М.м. 153.8). 1016100.
[56-23-5]. Тетрахлорметан. Вуглець чотирихлористий.
Прозора, безбарвна рідина. Практично не розчинний
у воді, змішується з 96 % спиртом.
с$ : від 1.595 до 1.598.
Температура кипіння: від 76 °С до 77 °С.
Галактоза. С6Н12О6. (М.м. 180.2). 1039700. [59-23-4].
О-(+)-Галактоза.
Кристалічний порошок білого кольору. Легко розчин-
на у воді.
[а][) : від +79° до +81°. Визначення проводять, вико-
ристовуючи розчин 100 г/л у воді Р, яка містить близь-
ко 0.05 % КН3.
Галова кислота. С7НЙО5,Н2О. (М.м. 188.1). 1039800.
[5995-86-8]. 3,4,5-Тригідроксибензойна кислота мо-
ногідрат.
Кристалічний порошок або довгі голчасті кристали,
безбарвні або жовтавого кольору. Розчинна у воді, лег-
ко розчинна в гарячій воді, 96 % спирті й гліцерині,
мало розчинна в ефірі.
Галова кислота втрачає кристалізаційну воду при тем-
пературі 120 °С і плавиться при температурі близько
260 °С із розкладанням.
Хроматографія. Визначення проводять, як зазначено
в статті Листя мучниці; на хроматограмі має виявля-
тись лише одна основна пляма.
Гарпагозид. С^НзоОц. (М.м. 494.5). 1098600.
Кристалічний порошок білого кольору, дуже гігро-
скопічний. Розчинний у воді й 96 % спирті.
Температура плавлення: від 117 °С до 121 °С.
Зберігають у повітронепроникному контейнері.
Гвайазулен. С15Н|8. (М.м. 198.3). 1041500. [489-84-9].
1,4-Диметил-7-ізопропілазулен.
Кристали темно-синього кольору або рідина синього
кольору. Дуже мало розчинний у воді, змішується з
жирними й ефірними оліями й вазеліновим маслом,
помірно розчинний у 96 % спирті, розчинний у роз-
чині 500 г/л кислоти сірчаної та 80 % (м/м) кислоті
фосфорній з утворенням безбарвного розчину
Температура плавлення: близько 30 °С.
Зберігають у захищеному від світла й повітря місці.
Гваякова смола. 1041400.
Смола, одержана з серцевини дерева Сиаіасит офісі-
паїе Т. і Сиаіасит хапсСит Ь.
Тверді, гладкі фрагменти червонувато-коричневого або
зеленувато-коричнюватого кольору, блищать на зламі.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
187
4.1.1. Реактиви
Гексадиметрину бромід. (С|3Н30Вг2М2)п. 1042300.
[28728-55-4]. 1,5-Диметил-1,5-діазаундекаметилену
поліметобромід. Полі( 1,1,5,5-тетраметил-1,5-азонію-
ундекаметилен дибромід).
Аморфний порошок білого кольору, гігроскопічний.
Розчинний у воді.
Зберігають у повітронепроникному контейнері.
Гексакозан. С2бН54. (Мм. 366.7). 1042200. [630-01-3].
Безбарвні або білого кольору пластівці.
Температура плавлення: близько 57 °С.
2,2’,2",6,6',6"-Гекса(1,1-диметилетил)-4,4',4"-[(2,4,6-
триметил-1,3,5-бензонітрил)трисметилен]трифенол.
С54Н7ХО3. (Мм. 775). 1042Ю0. 2,2',2",6,6',6"-Гекса-
/л/)Є/л-бутил-4,4’,4"-| (2,4,6-три метил- 1,3,5-бен-
зонітрил)трисметилен]трифенол.
Кристалічний порошок. Практично не розчинний у воді,
розчинний в ацетоні, мало розчинний у 96 % спирті.
Температура плавлення: близько 244 °С.
Гексаметилдисилазан. С6Н|ЧК’8і2. (Мм. 161.4). 1042400.
[999-97-3].
Прозора, безбарвна рідина.
^0° : близько 0.78.
: близько 1.408.
Температура кипіння: близько 125 °С.
Зберігають у повітронепроникному контейнері.
Гексаметилентетрамін. С6Н|21Ч4. (Мм. 140.2). 1042500.
[100-97-0]. Гексамін. 1,3,5,7-Тетра-азатрицик-
ло[3.3.1.13-7] декан.
Безбарвний кристалічний порошок. Дуже легко роз-
чинний у воді.
Гексан. С6Н)4. (М.м. 86.2). 1042600. [110-54-3].
Безбарвна, займиста рідина. Практично не розчинний
у воді, змішується з етанолом та ефіром.
^0° : ВІД 0.659 до 0.663.
: від 1.375 до 1.376.
Температурні межі перегонки (2.2.11). Від 67 °С до
69 °С; має переганятись не менше 95 %.
Гексан, використовуваний в спектрофотометрії, має
витримувати таке випробування.
Мінімальне пропускання (2.2.25)'. 97 %. Визначення
проводять в області довжин хвиль від 260 нм до 420 нм,
використовуючи як компенсаційну рідину воду Р.
Гексиламін. С6Н|51Ч. (Мм. 101.2). 1042700. [111-26-2].
Гексанамін.
Безбарвна рідина. Мало розчинний у воді, розчинний
у 96 % спирті та ефірі.
(/2о : близько 0.766.
: близько 1.418.
Температура кипіння: від 127 °С до 131 °С.
Гелій для хроматографії. Не. (А.м. 4.003). 10418011
[7440-59-7].
Містить не менше 99.995 % (об/об) Не.
Гемоглобін. 1041700. [9008-02-0].
Азот. Від 15 % до 16 %.
Залізо. Від 0.2 % до 0.3 %.
Втрата в масі при висушуванні (2.2.32). Не більше 2 ?«.
Сульфатна зола (2.4.14). Не більше 1.5 %.
Гемоглобіну розчин. 1041701.
2 г гемоглобіну Р поміщають у стакан місткістю
250 мл, додають 75 мл кислоти хлористовод-1
невої розведеної Р2 й перемішують до повного
розчинення. Доводять рН (2.2.3) 1 М розчи-
ном кислоти хлористоводневої до 1.6±0.1. Одер-і
жаний розчин переносять у колбу місткістю
100 мл з допомогою розчину кислоти хлористо-
водневої розведеної Р2 й додають 25 мг тіомер-\
салю Р.
Термін придатності 1 доба.
Зберігають при температурі 5±3 °С; перед викори-
станням знов доводять рН до 1.6.
Гепарин. 1041900. [9041-08-1]. Див. статтю Гепарин
натрію.
Гептан. С7Н16. (Мм. 100.2). 1042000. [142-82-5].
Безбарвна, займиста рідина. Практично не розчинний
у воді, змішується з етанолом та ефіром.
<72о : від 0.683 до 0.686.
: від 1.387 до 1.388.
Температурні межі перегонки (2.2.11). Від 97 °С до
98 °С; має переганятись не менше 95 %.
Гераніолу ацетат. С|2Н20О2. (М.м. 196.3). 1106500.
[105-87-3]. (£)-3,7-Диметилокта-2,6-дієн-1-іл ацетат.
Безбарвна або слабко жовтого кольору рідина, із слаб-
ким запахом троянди й лаванди.
: від 0.896 до 0.913.
: близько 1.463.
Температура кипіння25: близько 138 °С.
Гераніолу ацетат, використовуваний в газовій хрома-
тографії, має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено в статті Олія квіток
померанця, використовуючи гераніолу ацетат як ви-
пробовуваний розчин.
Площа основного піка має бути не менше 99.0 % суми
площ усіх піків.
188
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Гіалуронідази розріджувач. 1043300.
0.140 г желатину гідролізованого Р розчиняють при
температурі 37 °С у 200 мл суміші рівних об'ємів фос-
фатного буферного розчину рН 6.4 Р і води Р.
Термін придатності 2 год.
Гідразину сульфат. Н61Ч2О48. (М.м. 130.1). 1043400.
110034-93-2].
Безбарвні кристали. Помірно розчинний у холодній
воді, розчинний у гарячій воді (50 °С) й легко розчин-
ний в киплячій воді, практично не розчинний у
% % спирті.
Арсен (2.4.2, метод А). Не більше 0.0001 % (1 ррт).
1.0 г має витримувати випробування на арсен.
Сульфатна зола (2.4.14). Не більше 0.1 %.
Гідрокортизону ацетат. 1098800. [50-03-3]. Див. статтю
Гідрокортизону ацетат.
Гідроксиламіну гідрохлорид. 1\ІН4СЮ. (М.м. 69.5).
1044300. [5470-11-1].
Кристалічний порошок білого кольору. Дуже легко
розчинний у воді, розчинний у 96 % спирті.
Гідроксиламіну розчин лужний. 1044302.
Безпосередньо перед використанням змішують
рівні об'єми розчину 139 г/л гідроксиламіну
гідрохлориду Р і розчину 150 г/л натрію гідрокси-
ду Р.
Гідроксиламіну розчин лужний РІ. 1044303.
Розчин А. 12.5 г гідроксиламіну гідрохлориду Р роз-
чиняють у метанолі Р і доводять об'єм розчину
тим самим розчинником до 100 мл.
Розчин В. 12.5 г натрію гідроксиду Р розчиняють у
метанолі Р і доводять об'єм розчину тим самим
розчинником до 100 мл.
Безпосередньо перед використанням змішують
рівні об'єми розчинів АйВ.
Гідроксиламіну гідрохлориду розчин Р2. 1044304.
2.5 г гідроксиламіну гідрохлориду Р розчиняють у
4.5 мл гарячої води Р, додають 40 мл 96 % спирту Р,
0.4 мл розчину бромфенолового синього Р2 та до-
статню кількість 0.5 М розчину калію гідроксиду
спиртового до одержання зеленувато-жовтого за-
барвлення, доводять об'єм розчину 96 % спиртом Р
до 50.0 мл.
Гідроксиламіну розчин спиртовий. 1044301.
3.5 г гідроксиламіну гідрохлориду Р розчиняють у
95 мл спирту (60 %, об/об) Р, додають 0.5 мл роз-
чину 2 г/л метилового оранжевого Р у спирті
(60 %, об/об) Р і достатню кількість 0.5 М розчину
калію гідроксиду в спирті (60 %, об/об) до одер-
жання чіткого жовтого забарвлення розчину, дово-
дять спиртом (60 %, об/об) Рдо об'єму 100 мл.
4-Гідроксибензойна кислота. С7Н6О3. (М.м. 138.1).
1106700. [99-96-7].
Кристалічний порошок. Дуже мало розчинна у воді,
дуже легко розчинна в 96 % спирті, розчинна в ацетоні
та ефірі.
Температура плавлення: від 214 °С до 215 °С.
Гідроксиметилфурфурол. С6Н6О3. (М.м. 126.1). 1044400.
[67-47-0]. 5- Гідроксиметилфурфурол.
Голчасті кристали. Легко розчинний у воді, ацетоні й
96 % спирті, розчинний в ефірі.
Температура плавлення: близько 32 °С.
Гідроксинафтолового синього натрієва сіль.
С20Н1^^а3О1І83. (М.м. 620). 1044500. [63451-35-4].
Тринатрію 2,2'-дигідроксі-1,1'-азонафталін-3',4,6'-
трисульфонат.
12-Гідроксистеаринова кислота. С18Н36О3. (М.м. 300.5).
1099000. [106-14-9]. 12-Гідроксіоктадеканова кислота.
Порошок білого кольору.
Температура плавлення: від 71 °С до 74 °С.
Гідроксихінолін. С9Н71ЧО. (М.м. 145.2). 1044600.
[148-24-3]. 8-Гідроксихінолін. Хінолін-8-ол.
Кристалічний порошок білого або жовтавого кольору.
Мало розчинний у воді, легко розчинний в ацетоні.
96 % спирті й розведених мінеральних кислотах.
Температура плавлення: близько 75 °С.
Сульфатна зола (2.4.14). Не більше 0.05 %.
4-Гідроксіізофталева кислота. С8Н6О5. (М.м. 182.1).
1106900. [636-46-4]. 4-Гідроксибензол-1,3-дикарбо-
нова кислота.
Голчасті або у вигляді пластинок кристали. Дуже мало
розчинна у воді, легко розчинна в 96 % спирті й ефірі.
Температура плавлення: близько 314 °С, із розкла-
данням.
5-Гідроксіурацил. С4Н41Ч2О3. (М.м. 128.1). 1044700.
[496-76-4]. Ізобарбітурова кислота. Піримідин-2,4,5-
триол.
Кристалічний порошок білого кольору.
Температура плавлення: близько 310 °С, із розкла-
данням.
Хроматографія. Визначення проводять, як зазначено
в статті Фторурацшг, на хроматограмі має виявлятись
лише одна основна пляма з Л близько 0.3.
Зберігають у повітронепроникному контейнері.
Гідрохінон. С6НЙО2. (М.м. 110.1). 1044100. [123-31-9].
Бензол-1,4-діол.
Безбарвні або білого кольору, голчасті, дрібні криста-
ли, що темнішають під дією повітря й світла. Розчин-
ний у воді, 96 % спирті й ефірі.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
189
4.1.1. Реактиви
Температура плавлення: близько 173 °С.
Зберігають у захищеному від світла й повітря місці.
Гіосцинаміну сульфат. 1044900. [620-61-1]. Див. статтю
Гіосцинаміну сульфат.
Гіосцину гідробромід. 1044800. [114-49-8]. Див. статтю
Гіосцину гідробромід.
Гіперозид. С2ІН2()О|2. (М.м. 464.4). 1045000. 2-(3,4-
Дигідроксифеніл)-3-р-О-галактопіранозилокси-5,7-
дигідроксихромен-4-он.
Голчасті кристали світло-жовтого кольору. Розчиннии
у метанолі.
[«Іо : -8.3°. Визначення проводять, використовуючи
розчин 2 г/л у піридині Р.
Температура плавлення: близько 240 °С, із розкла-
данням.
Розчин у метанолі Р має два максимуми поглинання
(2.2.25) за довжин хвиль 259 нм та 364 нм.
Гіпоксантин. С5Н4ІМ4О. (Л/.ж. 136.1). 1045300. [68-94-0].
1//-Пурин-6-он.
Кристалічний порошок білого кольору. Дуже мало
розчинний у воді, помірно розчинний у киплячій воді,
розчинний у розведених кислотах і розведених розчи-
нах гідроксидів лужних металів, розкладається, не
плавлячись, при температурі близько 150 °С.
Хроматографія. Визначення проводять, як зазначено
в статті Меркаптопурин', на хроматограмі має виявля-
тись лише одна основна пляма.
Гіпофосфіту реактив. 1045200.
10 г натрію гіпофосфіту Р розчиняють при слабкому
нагріванні в 20 мл води Р і доводять об'єм розчину кис-
лотою хлористоводневою Рао 100 мл, відстоюють і де-
кантують або фільтрують крізь скловату.
Гістаміну дигідрохлорид. 1042800. [56-92-8]. Див. стат-
тю Гістаміну дигідрохлорид.
Гістаміну фосфат. Ю42900. [23297-93-0]. Див. статтю
Гістаміну фосфат.
Гістаміну розчин. ]042901.
Розчин 9 г/л натрію хлориду Р, який містить
0.1 мкг/мл гістаміну фосфату або гістаміну
дигідрохлориду в перерахунку на гістамін-основу.
Гістидину гідрохлорид моногідрат. С6Н10С1М3О2,Н2О.
(М.м. 209.6). 1043000. [123333-71-1]. (Л5)-2-Аміно-3-
(імідазол-4-іл)пропіонової кислоти гідрохлорид мо-
ногідрат.
Безбарвні кристали або кристалічний порошок. Роз-
чинний у воді.
Температура плавлення: близько 250 °С, із розкла-
данням.
Хроматографія. Визначення проводять, як зазначся В
в статті Гістаміну дигідрохлорид', на хроматограмі ма. И
виявлятись лише одна основна пляма.
Гітоксин. С41Н64О|4. (М.м. 781). 1040200. [4562-36-1]І
Глікозид Рфіїаііа ригригеа £. Зр-(О-2,6-Дидеокси4-И
0-/ш6о-гексопіранозил-(1—>4)-О-2,6-дидеокси-р-0-И
/ш6о-гексопіранозил-(1—>4)-2,6-дидеокси-р-В-р«бов
гексопіранозилокси)-14,1бр-дигідрокси-5р,14(3-кард-И
20(22)-енолід.
Кристалічний порошок білого кольору. Практично не І
розчинний у воді й більшості органічних розчинників І
розчинний у піридині.
[а]р : від +20° до +24°. Визначення проводять, вико 1
ристовуючи розчин 5 г/л у суміші розчинників хюро-1
форм Р- метанол Р (50:50).
Хроматографія. Визначення проводять, як зазначено І
в статті Листя наперстянки', на хроматограмі має ви-1
являтись лише одна основна пляма.
Гліколева кислота. С2Н4О3. (М.м. 76.0). 1040800.
[79-14-1]. 2-Гідроксіоцтова кислота.
Кристали. Розчинна у воді, ацетоні, 96 % спирті, ефірі
й метанолі.
Температура плавлення: близько 80 °С.
Гліоксальгідроксіаніл. С|4НІ2М2О2. (М.м. 240.3).
1041000. [1149-16-2]. Гліоксальбіс(2-гідроксіаніл).
Кристали білого кольору. Розчинний у гарячому
96 % спирті.
Температура плавлення: близько 200 °С.
Гліоксалю розчин. 1098400. [107-22-2].
Містить близько 40 % (м/м) гліоксалю.
Кількісне визначення. 1.000 г розчину гліоксалю
поміщають в колбу із притертою скляною пробкою,
додають 20 мл розчину 70 г/л гідроксиламіну гідрохіо-
риду Рта 50 мл води Р, витримують протягом 30 хв і ти-
трують 1 Мрозчином натрію гідроксиду до переходу за-
барвлення розчину від червоного до зеленого, викори-
стовуючи як індикатор 1 мл змішаного розчину мети-
лового червоного Р. Паралельно проводять контроль-
ний дослід.
1 мл 1 М розчину натрію гідроксиду відповідає 29.02 мг
гліоксалю (С2Н2О2).
Гліцерин. 1040500. [56-81-5]. Див. статтю Гліцерин.
Гліцерин (85 %). 1040600. Див. статтю Гліцерин (85 %).
Гліцин. 1040700. [56-40-6]. Див. статтю Гліцин.
Гліцирретинова кислота. С30Н46О4. (М.м. 470.7).
1040900. [471-53-4]. Гліцирретинова кислота. 12,13-Ди-
дегідро-Зр-гідроксі-11-оксоолеан-30-ова кислота.
Суміш а- й р-гліцирретинових кислот, в якій р-ізомер
переважає.
190
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Порошок від білого до жовтувато-коричневатого ко-
ьору. Практично не розчинна у воді, розчинна в ета-
золі й кислоті оцтовій льодяній,
|р : від +145° до +155°. Визначення проводять, ви-
користовуючи розчин 10.0 г/л в етанолі Р.
ро.шітографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель 6Е254 Р, суспензію якого готу-
іть, використовуючи 0.25 % (об/об) розчин кислоти
шфорної Р. На хроматографічну пластинку наносять
5мкл розчину 5 г/л кислоти гліцирретинової в суміші
рівних об смів хлороформу Р та метанолу Р. Хромато-
пафують в суміші розчинників метанол Р-хлоро-
чорм Р (5:95). Коли фронт розчинників пройде 10 см,
хромагограму переглядають в УФ-світлі за довжини
виді 254 нм. На хроматограмі має виявлятись темна
пляма (Яу близько 0.3), відповідна кислоті р-гліцирре-
тиновій, та менша пляма (Яу близько 0.5), відповідна
кислоті а-гліцирретиновій. Пластинку обприскують
розчином анісового альдегіду Р і нагрівають при темпе-
ратурі від 100 °С до 105 °С протягом 10 хв. Обидві пля-
ми мають бути забарвлені в синювато-фіолетовий
колір; між ними дозволяєтся наявність меншої плями
синювато-фіолетового кольору.
Глутамінова кислота. 1040400. [56-86-0]. Див. статтю
Кислота глутамінова.
Глутаровий альдегід. С5Н8О2. (М.м. 100.1). 1098300.
[ІІ1-30-8].
Масляниста рідина. Розчиннии у воді.
; близько 1.434
Температура кипіння: близько 188 °С.
Глюкоза. 1025700. [50-99-7]. Див. статтю Глюкоза без-
водна.
Гіюкозаміну гідрохлорид. С6НІ4С1МО5. (М.м. 215.6).
1040300. [66-84-2]. О-Глюкозаміну гідрохлорид.
Кристали Розчинний у воді, практично не розчинний
в ефірі.
|а]р : +100°, яке знижується до +47.5° через 30 хв.
Визначення проводять, використовуючи розчин
100 г/л у воді Р.
Б-Глюкуронова кислота. С6Н,0О7. (М.м. 194 І).
1119700 [6556-12-3].
Містить не менше 96.0 % С6Н|0О7, у перерахунку на
суху речовину, висушену в вакуумі (2.2.32)
Розчинна у воді й 96 % спирті.
Виявляє мутаротацію: [а]р : +11.7°->+36.3°.
Кількісне визначення. 0.150 г розчиняють при пе-
ремішуванні в метанолі безводному Р і титрують
0.1 М розчином тетрабутиламонію гідроксиду по-
тенціометрично (2.2.20), захищаючи розчин від дії
вуглецю діоксиду повітря під час розчинення й титру-
вання.
1 мл 0.1 М розчину тетрабутиламонію гідроксиду
відповідає 19.41 мгС6Н10О7.
Гольмію(ІІІ) оксид. Но2О3. (М.м. 377.9). 1043100.
[12055-62-8]. Дигольмію триоксид.
Порошок жовтуватого кольору. Практично не розчин-
ний у воді
Гольмію перхлорату розчин. 1043101.
Розчин 40 г/л гольмію! 111) оксиду Р у розчині
141 г/л (НС1О4) кислоти хлорної Р.
Гуанідину гідрохлорид. СН51\І3НС1. (М.м. 95.5).
1098500. [50-01-1].
Кристалічний порошок. Легко розчинний у воді й
96 % спирті.
Гуанін. С5Н5М5О. (М.м. 151.1). 1041600. [73-40-5].
2-Аміно-1,7-дигідро-6/7-пурин-6 он.
Аморфний порошок білого кольору. Практично не
розчинний у воді, мало розчинний у 96 % спирті, роз-
чинний у розчинах аміаку й розведених розчинах
гідроксидів лужних металів.
Гуміарабік. 1000100. Див. статтю Гуміарабік.
Гуміарабіку розчин. 1000101.
100 г гуміарабіку Р розчиняють у 1000 мл води Р
при перемішуванні механічною мішалкою протя-
гом 2 год. Центрифугують із прискоренням близь-
ко 2000 £ протягом 30 хв до одержання прозорого
розчину.
Зберігають в поліетиленових контейнерах міст-
кістю близько 250 мл при температурі від 0 °С до
- 20 °С.
Дантрон. С)4Н8О4. (М.м. 240.2). 1024500. [117-10-2].
1,8-Дигідроксіантрахін-9(10//)-он.
Кристалічний порошок оранжевого кольору.
Температура плавлення: близько 195 °С.
Дейтерію оксид. ’Н2О. (М.м. 20.03). 1025300. [7789-20-0|.
Вода дейтерована.
Ступінь дейтерування не менше 99.7 %.
: близько 1.11.
: близько 1.328.
Температура кипіння: близько 101 °С.
Дейтерована кислота оцтова. С2?Н4О2. (М.м. 64.1).
1101100 [1186-52-3]. Тетрадейтерооцтова кислота.
Оцтова-</? кислота-Д
Ступінь дейтерування не менше 99.7 %.
с/2о : близько 1.12.
: близько 1.368.
Температура кипіння: 115 °С.
Температура плавлення. 16 °С.
4.1.1. Реактиви
Дейтерований ацетон. С32Н6О. (М.м. 64.1). 1024900.
|666-52-4]. Ацетон-Д6. (2Н6)-Ацетон.
Ступінь дейтерування не менше 99.5 %.
Прозора, безбарвна рідина. Змішується з водою, ди-
метилформамідом, етанолом, ефіром та метанолом.
Д2о близько 0.87
: близько 1.357.
Температура кипіння: близько 55 °С.
Вода й дейтерію оксид: не більше 0.1 %.
Дейтерований диметисульфоксид. С22Н6О8. (М.м. 84.2).
1025100. [2206-27-1]. (2Н6)-Диметилсульфоксид. Ди-
метилсульфоксид-</6.
Ступінь дейтерування не менше 99.8 %.
В'язка, практично безбарвна, сильно гігроскопічна
рідина. Розчинний у воді, ацетоні, етанолі та ефірі.
: близько 1.18.
Температура плавлення: близько 20 °С.
Вода й дейтерію оксид: не більше 0.1 %.
Зберігають у повітронепроникному контейнері.
Дейтерований метанол. С2Н4О. (М.м. 36.1). 1025200.
[811 98-3]. (2Н4)-Метанол. Метанол-Д4.
Ступінь дейтерування не менше 99.8 %.
Прозора, безбарвна рідина. Змішується з водою, 96 %
спиртом і метиленхлоридом.
Д2о : близько 0.888.
: близько 1.326.
Температура кипіння: 65.4 °С.
Дейтерований хлороформ. С2НС13. (М.м. 120.4).
1025000. [865-49-6]. (’Н)-Хлороформ. Хлороформ-Д
Ступінь дейтерування не менше 99.7 %.
Прозора, безбарвна рідина. Практично не розчинний
у воді, змішується з ацетоном, 96 % спиртом та
ефіром. Може бути стабілізований срібною фольгою.
г/2о : близько 1.51.
: близько 1.445.
Температура кипіння: близько 60 °С.
Вода й дейтерію оксид: не більше 0.05 %.
Декан. С10Н22. (М.м. 142.3). 1024600.1124-18-5].
Безбарвна рідина. Практично не розчинний у воді.
: близько 1.411.
Температура кипіння: близько 174 °С.
Деканол. С|0Н22О. (М.м. 158.3). 1024700. [112-30-1].
н-Дециловий спирт.
192
В'язка рідина, яка твердіє при температурі 6 °С. Прак-
тично не розчинний у воді, розчинний у 96 % спиртій
ефірі.
Но : близько 1.436.
Температура кипіння: близько 230 °С.
Декстран 2000 синій. 1011700. [9049-32-5].
Готують із декстрану, який має середню молекулярну ма-
су 2 х 10', введенням поліциклічного хромофору; то на-
дає речовині синього кольору. Ступінь заміщення 0.017.
Висушують при заморожуванні. Швидко й повністю
розчиняється у воді Рта водних солевих розчинах.
Розчин 1 г/л у фосфатному буферному розчині рН 7 Р
має максимум поглинання (2.2.25) за довжини хвилі
280 нм.
Декстран поперечно-зшитий для хроматографії Р2.
1025500.
Гранули кулястої форми, придатні для розділення
пептидів і білків із молекулярними масами від 15 х 10!
до 30 х 10’. Сухі гранули мають діаметр від 20 мкм до
80 мкм.
Декстран поперечно-зшитий для хроматографії РЗ.
1025600.
Гранули кулястої форми, придатні для розділення
пептидів та білків із молекулярними масами від 4 х 10і
до 15 х 104. Сухі гранули мають діаметр від 40 мкм до
120 мкм.
Декстроза. 1025700. [50-99-7]. Див. Глюкоза Р.
2'-Деоксіуридин. С9Н|2М2О5. (М.м. 228.2). 1024800.
[951-78-0]. 1-(2-Деокси-р-0-ерптро-пентофурано-
зил)-1 //,3//-піримідин-2,4-діон.
Температура плавлення: близько 165 °С.
Хроматографія. Визначення проводять, як зазначено
в статті Йодоксіуридин; наносять 5 мкл розчину
0.25 г/л; на хроматограмі має виявлятись лише одна
основна пляма.
Дибензил. С14Н14. (М.м. 182.3). 1011200. [103-29-7].
1,2-Дифенілетан.
Кристалічний порошок білого кольору. Практично не
розчинний у воді, дуже легко розчинний у метилен-
хлориді, легко розчинний в ацетоні, розчинний у
96 % спирті.
Температура плавлення: від 50 °С до 53 °С.
Дибузиловий ефір. С8Н|8О. (М.м. 130.2). 1026700.
[142-96-1].
Безбарвна, займиста рідина. Практично не розчинний
у воді, змішується з етанолом та ефіром.
Д2о : близько 0.77.
Др : близько 1.399.
Не переганяють, якщо дибутиловий ефір не витримує
випробування на пероксиди.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
іПероксиди. 8 мл розчину крохмалю з калію йодидом Р
домішають у циліндр із притертою скляною пробкою,
істкістю 12 мл і діаметром близько 1.5 см, повністю
заповнюють випробовуваним ефіром, закривають
іробкою та перемішують. Витримують у темному
місці протягом ЗО хв; не має з'являтись забарвлення.
Назва й концентрація будь-якого доданого стабіліза-
тора мають бути зазначені на етикетці.
Дибутилфталат. СІ6Н22О4. (М.м. 278.3). 1026800.
[84-74-2]. Дибутилбензол-1,2-дикарбоксилат.
Прозора, безбарвна або слабко забарвлена масляниста
рідина. Дуже мало розчинний у воді, змішується з аце-
тоном, 96 % спиртом та ефіром.
: від 1.043 до 1.048.
«о : від 1.490 до 1.495.
10,11-Дигідрокарбамазепін. С15Н14ЬІ2О. (М.м. 238.3).
1028900. [3564-73-6]. 10.11-Дигідро-5/7-дибензо-
|/\/]азепін-5-карбоксамш.
Температура плавлення: від 205 °С до 210 °С.
Дигідрок инафталін. 1029000. [132-86-5]. Див. 1,3-Ди-
гідроксинафталін Р.
1,3-Дигідроксинафталін. С|()Н8О2. (М.м. 160.2).
1029000. [132-86-5]. Нафталін-1,3-діол.
Кристалічний порошок, звичайно коричнювато-фіоле-
тового кольору. Легко розчинний у воді й 96 % спирті.
Температура плавлення: близько 125 °С.
2,7-Дигідроксинафталін. С10Н8О2. (М.м. 160.2).
1029100. [582-17-2]. Нафталін-2,7-діол.
Голчасті кристали. Розчинний у воді, 96 % спирті й
ефірі.
Температура плавлення: близько 190 °С.
2,7-Дигідроксинафталіну розчин. 1029101.
10 мг 2,7-дигідроксинафталіну Р розчиняють у
100 мл кислоти сірчаної Р і витримують до знебарв-
лення.
Термін придатності 2 доби.
Дигітоксин. 1028800. [71-63-6]. Див. статтю Дигіток-
син.
Дигітонін. С56Н92О29. (М.м. 1229). 1028700. [11024-24-1].
ЗР-[0-р-о-Глюкопіранозил-(1->3)-0-р-о-галак-
топіранозил-(1->2)-0-[р-о-ксилопіранозил-(1->3)]-
0-р-ц-галактопіранозил-(1->4)-О-р-о-галактопіра-
нозилокси[-(25А)-5а-спіростан-2а,15р-діол.
Кристали. Практично не розчинний у воді, помірно
розчинний в етанолі, мало розчинний у 96 % спирті,
практично не розчинний в ефірі.
Дидодецил-3,3'-тіодипропіонат. С30Н58О48. (М.м.
514.8). 1027700. [123-28-4].
Кристалічний порошок білого кольору. Практично не
розчинний у воді, легко розчинний в ацетоні й петро-
лейному ефірі, мало розчинний у 96 % спирті.
Температура плавлення: близько 39 °С.
Ди(2-етилгексил)фталат. С24Н38О4. (М.м. 390.5).
1028100. Ди(2-етилгексил)бензол-1,2-дикарбоксилат.
Прозора, масляниста рідина. Практично не розчин-
ний у воді, розчинний в органічних розчинниках.
б?2о : близько 0.98.
: близько 1.486.
В'язкість (2.2.9). Близько 80 мПа-с.
Дикалію гідрофосфат. К2НРО4. (М.м. 174.2). 1033000.
[7758-11-4].
Кристалічний порошок білого кольору, гігроско-
пічний. Дуже легко розчинний у воді, мало розчинний
у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Дикарбоксидину гідрохлорид. С2()Н26С12М2О6.
(М.м. 461.3). 1026900. [56455-90-4]. 4,4'-[(4,4'-Діаміно-
дифеніл-3,3'-діїл)діокси]дибутанової кислоти дигідро-
хлорид.
Диметикон. 1105400. [9006-65-9]. Див. статтю Димети-
кон.
Диметиламінобензальдегід. С9НИКО. (М.м. 149.2).
1029800. [100-10-7]. 4-Диметиламінобензальдегід.
Кристали білого або жовтувато-білого кольору. Роз-
чинний у 96 % спирті й розведених кислотах.
Температура плавлення: близько 74 °С.
Диметиламінобензальдегіду розчин РІ. 1029801.
0.2 г диметиламінобензальдегіду Р розчиняють у
20 мл 96 % спирту Р, додають 0.5 мл кислоти хло-
ристоводневої Р, одержаний розчин струшують із
вугіллям активованим Рі фільтрують. Забарвлення
розчину не має бути інтенсивнішим за забарвлен-
ня розчину йоду РЗ.
Готують безпосередньо перед використанням.
Диметиламінобензальдегіду розчин Р2. 1029802.
0.2 г диметиламінобензальдегіду Р розчиняють без
нагрівання в суміші 4.5 мл води Р і 5.5 мл кислоти
хлористоводневої Р.
Готують безпосередньо перед використанням.
Диметиламінобензальдегіду розчин Р6. 1029803.
0.125 г диметиламінобензальдегіду Р розчиняють в
охолодженій суміші 35 мл води Р і 65 мл кислоти
сірчаної Р, додають 0.1 мл розчину 50 г/л
заліза) 111) хлориду Р.
Перед використанням витримують 24 год у захи-
щеному від світла місці.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
193
4.1.1. Реактиви
Зберігають при кімнатній температурі 7 діб; у хо-
лодильнику - протягом кількох місяців.
Диметиламінобензальдегіду розчин Р7. 1029804.
1.0 г диметиламінобензальдегіду Р розчиняють у
50 мл кислоти хлористоводневої Р і додають 50 мл
96 % спирту Р.
Зберігають у захищеному від світла місці.
Термін придатності 1 міс.
Диметиламінобензальдегіду розчин Р8. 1029805.
0.25 г диметиламінобензальдегіду Р розчиняють в
суміші 5 г кислоти фосфорної Р, 45 г води Р і 50 г
кислоти оцтової безводної Р.
Готують безпосередньо перед використанням.
4-Диметиламінокоричний альдегід. СИНІЗМО.
(М.м. 175.2). 1029900. [6203-18-5]. 3-(4-Диметиламіно-
феніл)проп-2-еналь. 4-Диметиламіноциннамальдегід.
Кристали або порошок від оранжевого до оранжево-
коричневого кольору. Чутливий до світла.
Температура плавлення: близько 138 °С.
4-Диметиламінокоричного альдегіду розчин.
1029901.
2 г 4-диметиламінокоричного альдегіду Р розчиня-
ють у суміші 100 мл кислоти хлористоводневої РІ і
100 мл етанолу Р. Зберігають у прохолодному
місці. Безпосередньо перед використанням роз-
чин розводять етанолом Ру 4 рази.
Зберігають у прохолодному місці.
Диметиламінонафталінсульфонілхлорид.
Сі2Ні2С1ЬІО28. (М.м. 269.8). 1030000. [605-65-2].
5-Диметиламіно-1 -нафталінсульфонілхлорид.
Кристалічний порошок жовтого кольору. Мало роз-
чинний у воді, розчинний у метанолі.
Температура плавлення: близько 70 °С.
Зберігають у прохолодному місці.
Диметиланілін. С8НцМ. (М.м. 121.2). 1030100. [121-69-7].
А'.А'-Димстиланілін.
Прозора, масляниста рідина. Свіжоперегнаний - май-
же безбарвний, при зберіганні темнішає до червонува-
то-коричневого кольору. Практично не розчинний у
воді, легко розчинний у 96 % спирті й ефірі.
: близько 1.558.
Температурні межі перегонки (2.2.11). Від 192 °С до
194 °С; має переганятись не менше 95 %.
ТУ^ТУ-Диметиланілін. 1030100. [121-69-7]. Див. Диме-
тиланілін Р.
2,6-Диметиланілін. С8НцМ. (М.м. 121.2). 1030200.
[87-62-7]. 2,6-Ксилідин.
Безбарвна рідина. Помірно розчинний у воді, розчин-1
ний у 96 % спирті.
б?2о : близько 0.98.
2,3-Диметиланілін. С8НцМ. (М.м. 121.2). 1105300.
[87-59-2]. 2,3-Ксилідин.
Рідина жовтуватого кольору. Помірно розчинний а
воді, розчинний у 96 % спирті.
: від 0.993 до 0.995.
: близько 1.569.
Температура кипіння: близько 224 °С.
Диметилацетамід. С4Н9МО. (М.м. 87.1). 1029700.
[127-19-5]. Л'.Л'-Димстилапсіамід.
Містить не менше 99.5 % С4НЧМО.
Безбарвна рідина. Змішується з водою й більшістю ор-
ганічних розчинників.
б?2о : близько 0.94.
:близько 1.437.
Температура кипіння: близько 165°С.
Диметилгліоксим. С4НЯМ2О2. (М.м. 116.1). 1030400.
[95-45-4]. 2,3-Бутандіондіоксим.
Кристалічний порошок білого кольору або безбарвні
кристали. Практично не розчинний у холодній воді,
дуже мало розчинний у киплячій воді, розчинний у
96 % спирті й ефірі.
Температура плавлення: близько 240 °С, із розкла-
данням.
Сульфатна зола (2.4.14). Не більше 0.05 %.
Диметилдециламін. С,2Н27М. (М.м. 185.4). 1113500.
[1120-24-7]. А'.А-диметилдециламін.
Містить не менше 98.0 % (м/м) СІ2Н27М.
Температура кипіння: близько 234 °С.
1,1-Диметилетиламін. С4НцМ. (М.м. 73.1). 1100900.
[75-64-9]. 2-Аміно-2-метилпропан. л/ре/н-Бутиламін.
Рідина. Змішується з 96 % спиртом.
сІ2о : близько 0.694.
: близько 1.378.
Температура кипіння: близько 46 °С.
1,1-Диметилетилметиловий ефір. С5НІ2О. (М.м. 88.1).
1013900. [1634-04-4]. трет-Бутилметиловий ефір.
Безбарвна, прозора, займиста рідина.
: близько 1.376.
Мінімальне пропускання (2.2.25) визначають, викорис-
товуючи як компенсаційну рідину воду Р:
не менше 50 % за довжини хвилі 240 нм,
не менше 80 % за довжини хвилі 255 нм,
не менше 98 % за довжини хвилі 280 нм.
194
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Диметиловий жовтий. С|4Н|5К3. (М.м. 225.3). 1029600.
[60-11-7]. Показник Шульца № 28. Індекс кольоро-
вості № 11020. 4-(Диметиламіно)азобензол. Метило-
вий жовтий.
Дрібні кристали жовтого кольору або пластівці жовто-
го чи оранжевого кольору. Практично не розчинний у
воді, дуже мало розчинний у 96 % спирті.
Хроматографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель с Р. На хроматографічну плас-
тинку наносять 10 мкл розчину 0.1 г/л у метиленхло-
риді Рі хроматографують у цьому самому розчиннику,
фронт розчинника має пройти не менше 10 см; на хро-
матограмі має виявлятись лише одна основна пляма.
Зберігають у повітронепроникному контейнері.
Диметилового жовтого й орацетового синього розчин.
1118700.
10 мг диметилового жовтого Р і 10 мг орацетового си-
нього В Р розчиняють у 300 мл метиленхлориду Р.
Л,Л-Диметилоктиламін. С10Н23Т4. (М.м. 157.3).
1030500. [7378-99-6]. Октилдиметиламін.
Безбарвна рідина.
Дщ близько 0.765.
: близько 1.424.
Температура кипіння: близько 195 °С.
1,3-Диметил-2-імідозолідинон. С5НІ(ІК2О. (М.м. 114.2).
[80-73-9]. А',М-Диметилетиленсечовина.
«р : близько 1.4720.
Температура кипіння: близько 224 °С.
Диметилпіперазин. С6НІ4М2. (М.м. 114.2). 1030700.
[106-58-1]. 1,4-Диметилпіперазин.
Безбарвна рідина. Змішується з водою й 96 % спир-
том.
: близько 0.85.
По : близько 1.446.
Температура кипіння: близько 131 °С.
Диметилстеариламід. С20Н4,МО. (М.м. 311.6). 1030800.
/V, А'-Диметилстеариламід.
Тверда маса білого або майже білого кольору. Розчин-
ний у більшості органічних розчинників, в ацетоні
включно.
Температура плавлення: близько 51 °С.
Диметилсульфоксид. С2Н6О8. (М.м. 78.1). 1029500.
[67-68-5].
Прозора, безбарвна, масляниста, гігроскопічна ріди-
на. Змішується з водою й 96 % спиртом.
і/™ : близько 1.10.
Температура кипіння: близько 189 °С.
Вода (2.5.12). Не більше 10 г/л.
Диметилсульфоксид, використовуваний у спектрофото-
метрії, має задовольняти вимоги для диметилсульфокси-
ду Р, але з іншим вмістом води, який наведено нижче, і,
крім того, має задовольняти такі додаткові вимоги.
Мінімальне пропускання (2.2.25) визначають, викорис-
товуючи як компенсаційну рідину воду Р.
10 % за довжини хвилі 262 нм,
35 % за довжини хвилі 270 нм,
70 % за довжини хвилі 290 нм,
98 % за довжини хвилі 340 нм і більше.
Вода (2.5.12). Не більше 0.2 % (м/м).
Зберігають у повітронепроникному контейнері.
Диметилсульфон. С2Н6О28. (М.м. 94.1). 1030900.
[67-71-0].
Кристалічний порошок білого кольору. Легко розчин-
ний у воді, розчинний в ацетоні й 96 % спирті.
Температура плавлення: від 108 °С до 110 °С.
Диметилтетрадеииламін. СІ6Н35М. (М.м. 241.5).
1031000. Л',Л'-Диметилтетрадециламін.
Містить не менше 98.0 % (м/м) і не більше 101.0 %
(м/м) СІ6Н35М.
Прозора або майже прозора, безбарвна або жовтувато-
го кольору рідина. Практично не розчинний у воді,
змішується з ацетоном, 96 % спиртом і метанолом.
г/2д : близько 0.80.
Температура кипіння: близько 260 °С.
Вода (2.5.12). Не більше 0.3 % (м/м).
Кількісне визначення. 0.200 г розчиняють в 10 мл 96 %
спирту Р і титрують 0.1 М розчином кислоти хлористо-
водневої до появи червоного забарвлення розчину, ви-
користовуючи як індикатор 0.1 мл розчину метилового
червоного Р.
1 мл 0.1 М розчину кислоти хлористоводневої
відповідає 24.15 мгСІ6Н35М.
2,6-Диметилфенол. С8НІ0О. (М.м. 122.2). 1030600.
[576-26-1].
Безбарвні голчасті кристали. Мало розчинний у воді,
легко розчинний у 96 % спирті й ефірі.
Температура кипіння: близько 203 °С.
Температура плавлення: від 46 °С до 48 °С.
3,4-Диметилфенол. С^НщО. (М.м. 122.2). 1098100.
[95-65-8].
Кристали білого або майже білого кольору. Мало роз-
чинний у воді, легко розчинний у 96 % спирті.
Температура кипіння: близько 226 °С.
Температура плавлення: від 25 °С до 27 °С.
Диметилформамід. С3Н7МО. (М.м. 73.1). 1030300.
[68-12-2].
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
195
4.1.1. Реактиви
Прозора, безбарвна рідина. Змішується з водою й 96 %
спиртом.
і/20 : від 0.949 до 0.952.
Температура кипіння: близько 153 °С.
Вода (2.5.12). Не більше 0.1 %.
Диметилформаміду діетилацеталь. С7Н,7МО2.
(М.м. 147.2) 1113600. [1188-33-6]. А,А-димстилфор-
маміду діетилацеталь.
: близько 1.40.
Температура кипіння: від 128 °С до 130 °С.
Диметоксипропан. С5Н,2О2. (М.м. 104.1). 1105200.
[77-76-9]. 2,2-Диметоксипропан.
Безбарвна рідина. Розкладається під дією вологого
повітря або води.
т/2о : близько 0.847.
: близько 1.378.
Температура кипіння: близько 83 °С.
Димідію бромід. С20НІ8ВгМ3. (М.м. 380.3). 1031100.
[518-67-2]. 3,8-Діаміно-5-метил-6-фенілфенантри-
дінію бромід.
Кристали темно-червоного кольору. Мало розчинний
у воді при температурі 20 °С, помірно розчинний у
воді при температурі 60 °С і 96 % спирті, практично не
розчинний в ефірі.
Димідію броміду й сульфанового синього змішаний
розчин. 1031101.
Окремо розчиняють 0.5 г димідію броміду Рі 0.25 г
сульфанового синього Ру ЗО мл гарячій суміші роз-
чинників етанол Р - вода Р (1:9) і перемішують.
Обидва розчини змішують і доводять об'єм розчи-
ну тією самою сумішшю розчинників до 250 мл.
20 мл одержаного розчину змішують із 20 мл
14.0 % (об/об) розчину кислоти сірчаної Р, попе-
редньо розведеної приблизно 250 мл води Р, дово-
дять водою Рдо об'єму 500 мл.
Зберігають у захищеному від світла місці.
Динатрію арсенат. 1\Іа2НА8О4,7Н2О. (М.м. 312.0).
1102500. [10048-95-0]. Динатрію гідроарсенат геп-
тагідрат.
Кристали, які звітрюються на повітрі. Легко розчинний
у воді, розчинний у гліцерині, мало розчинний у 96 %
спирті. Водний розчин має лужну реакцію за лакмусом.
г/Іо : близько 1.87.
Температура плавлення: близько 57 °С (при швидкому
нагріванні).
Динатрію гідрофосфат. 1033300. [10039-32-4]. Див.
статтю Динатрію фосфат додекагідрат.
Динатрію гідрофосфату розчин. 1033301.
Розчин 90 г/л.
Динатрію гідрофосфат безводний. ІМа2НРО4.
(М.м. 142.0). 1033400. [7558-79-4|.
Динатрію гідрофосфат дигідрат. /033500. [10028-24-7]. |
Див. статтю Динатрію фосфат дигідрат.
Динатрію гідроцитрат. С6Н6№207,1 5Н2О. (М.м. 263.1). (
1033200. [144-33-2]. Натрію цитрат кислий. Динатрію І
2-гідроксипропан-1,2,3-трикарбоксилат кислий сес- І
квігідрат.
Порошок білого кольору. Розчинний менш ніжу 2ча-
стинах води, практично не розчинний у 96 % спирті. ’
Динатрію тетраборат. 1033600. [1330-43-4]. Див. статтю 1
Бура.
Бури розчин. 1033601.
9.55 г динатрію тетраборату Р розчиняють у
кислоті сірчаній Р при нагріванні на водяній бані
й доводять об'єм розчину тією самою кислотою
до 1 л.
Динітробензоїлхлорид. С7Н3С1М2О5. (М.м. 230.6).
1031400. [99-33-2]. 3,5-Динітробснзоїлхлорид.
Кристалічний порошок світло-жовтого кольору або
безбарвні кристали.
Температура плавлення близько 68 °С.
Динітробензойна кислота. С7Н4М2О6. (М.м. 212.1).
1031300. [99-34-3]. 3,5-Динітробснзойна кислота.
Кристали майже безбарвні. Мало розчинна у воді, ду-
же легко розчинна у 96 % спирті.
Температура плавлення: близько 206 °С
Динітробензойної кислоти розчин. 1031301.
Розчин 20 г/л у 96 % спирті Р.
Динітробензол. С6Н4М2О4. (М.м. 168.1). 1031200.
|528-29-0]. 1,3-Динітробензол.
Кристалічний порошок або кристали жовтуватого ко-
льору. Практично не розчинний у воді, мало розчин-
ний у 96 % спирті.
Температура плавлення: близько 90 °С.
Динітробензолу розчин. 1031201.
Розчин 10 г/л у 96 % спирті Р.
Динітрофенілгідразин. С6Н61Ч4О4. (М.м. 198.1).
1031500. [119-26-6]. 2,4-Динітрофенілгідразин.
Кристали червонувато-оранжевого кольору. Дуже ма-
ло розчинний у воді, мало розчинний у 96 % спирті.
Температура плавлення: близько 203 °С (метод мит-
тєвого плавлення).
Динітрофенілгідразину оцтово-хлористоводневий
розчин. 1031501.
0 2г динітрофенілгідразину Р розчиняють у 20 мл
метанолу Р, додають 80 мл суміші рівних об'ємів
196
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
кислоти оцтової Р і кислоти хлористоводневої РІ і
перемішують.
Готують безпосередньо перед використанням.
Динітрофенілгідразину хлористоводневий розчин.
1031502.
0 50 г динітрофенілгідразину Р розчиняють при
нагріванні в кислоті хлористоводневій розведеній Р,
доводять об'єм розчину тим самим розчинником
до 100 мл, охолоджують і фільтрують.
Готують безпосередньо перед використанням.
Динонілфталат. С26Н42О4 (М.м. 418.6). 1031600.
128553-12-0].
Безбарвна або світло-жовтого кольору в'язка рідина.
</$: від 0.97 до 0.98.
: від 1.482 до 1.489.
Кислотність. 5.0 г струшують із 25 мл води Р протягом
1 хв. Після розділення шарів відокремлюють водний
шар, додають 0.1 мл розчину фенолфталеїну Р, забарв-
лення розчину має змінитися при додаванні не більше
0.3 мл 0.1 Мрозчину натрію гідроксиду (0.05 % в пере-
рахунку на кислоту фталеву).
Вода (2.5.12). Не більше 0.1 %.
Дитизон. С|3Н|2і\48. (М.м. 256.3). 1033900. [60-10-6].
1,5-Дифенілтіокарбазон.
Порошок синювато-чорного, або коричнювато-чор-
ного, або чорного кольору. Практично не розчинний у
воді, розчинний у 96 % спирті.
Зберігають у захищеному від світла місці.
Дитизону розчин. 1033901.
Розчин 0.5 г/л у хлороформі Р.
Готують безпосередньо перед використанням.
Дитизону розчин Р2. 1033903.
40.0 мг дитизону Р розчиняють у хлороформі Р і до-
водять об'єм розчину тим самим розчинником до
1000.0 мл. 30.0 мл одержаного розчину доводять
хлороформом Рдо об'єму 100.0 мл.
Встановлення титру. Кількість ртуті(П) хлори-
ду Р, еквівалентну 0.1354 г Н^С12, розчиняють у
суміші рівних об'ємів кислоти сірчаної розведеної Р
і води Р і доводять об'єм розчину тією самою
сумішшю розчинників до 100.0 мл. 2.0 мл одержа-
ного розчину доводять сумішшю рівних об'ємів
кислоти сірчаної розведеної Р і води Р до об'єму
100.0 мл (розчин містить 20 ррт Н§). 1.0 мл одер-
жаного розчину помішають у ділильну лійку, дода-
ють 50 мл кислоти сірчаної розведеної Р, 140 мл во-
ди Р і 10 мл розчину 200 г/л гідроксиламіну гідро-
хюриду Р. Титрують приготованим розчином ди-
тизону; після кожного додавання титранту суміш
струшують 20 разів, наприкінці титрування суміш
залишають для розділення шарів, потім відкида-
ють хлороформний шар і продовжують титрувати
до одержання синювато-зеленого забарвлення.
Кількість ртуті (£) в міліграмах, еквівалентну
вмісту дитизону в одному мілілітрі розчину, обчис-
люють за формулою: £= 20/У, де V - об'єм розчину
дитизону, витрачений на титрування, в мілілітрах.
Дитизон РІ. С,3НІ21М48. (М.м. 256.3). 1105500. |60-10-6].
1,5-Дифенілтіокарбазон.
Містить не менше 98.0 % С13Н|2ІМ48.
Порошок синювато-чорного, або коричнювато-чор-
ного, або чорного кольору. Практично не розчинний у
воді, розчинний у 96 % спирті.
Зберігають у захищеному від світла місці.
5,5'-Дитіобіс(2-нітробензойна кислота). СІ4Н8ІМ2О882.
(М.м. 396.4). 1097300. [69-78-3]. З-Карбокси-4-нітро-
фенілдисульфід. Реактив Ельмана. ОТІМ В.
Порошок жовтого кольору. Помірно розчинна у 96 %
спирті.
Температура плавлення: близько 242 °С.
Дитіол. С7Н882. (М.м. 156.3). 1033800. [496-74-2]. То-
луол-3,4-дитіол. 4-Метилбензол-1,2-дитіол.
Кристали білого кольору, гігроскопічні. Розчинний у
метанолі й розчинах гідроксидів лужних металів.
Температура плавлення: близько ЗО °С.
Зберігають у повітронепроникному контейнері.
Дитіолу реактив. 1033801.
До 1 г дитіолу Р додають 2 мл кислоти тіогліколе-
вої Р і доводять розчином 20 г/л натрію гідрокси-
ду Рдо об'єму 250 мл.
Готують безпосередньо перед використанням.
Дитіотреїтол. С4Ні0О282. (М.м. 154.2). 1098200.
[27565-41-9]. трео-1,4-Димеркаптобутан-2,3-діол.
Голчасті, слабо гігроскопічні кристали. Легко розчин-
ний у воді, ацетоні й етанолі.
Зберігають у повітронепроникному контейнері.
Дифеніламін. СІ2Н,,(8. (М.м. 169.2). 1032100. [122-39-4].
Кристали білого кольору. Мало розчинний у воді, роз-
чинний у 96 % спирті.
Температура плавлення: близько 55 °С.
Зберігають у захищеному від світла місці.
Дифеніламіну розчин. 1032101.
Розчин 1 г/л у кислоті сірчаній Р.
Зберігають у захищеному від світла місці.
Дифеніламіну розчин РІ. 1032102.
Розчин 10 г/л у кислоті сірчаній Р. Розчин має бу-
ти безбарвним.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
197
4.1.1. Реактиви
Дифеніламіну розчин Р2. 1032103.
1 г дифеніламіну Р розчиняють у 100 мл кислоти
оцтової льодяної Р і додають 2.75 мл кислоти сірча-
ної Р.
Розчин використовують негайно.
Дифенілантрацен. С26НІ8. (Мм. 330.4) 1032200.
[1499-10-1]. 9.10-Дифенілантрацен.
Кристалічний порошок від жовтуватого до жовтого
кольору. Практично не розчинний у воді, легко роз-
чинний в ефірі.
Температура плавлення близько 248 °С.
Дифенілбензидин. С24Н2о192- (М.м. 336.4). 1032300.
[531-91-9]. /У./У-Дифенілбензидин. /V,/У’-Дифсніл-
біфеніл-4,4'-діамін.
Кристалічний порошок білого або білого із сіруватим
відтінком кольору. Практично не розчинний у воді,
мало розчинний в ацетоні й 96 % спирті.
Температура плавлення; близько 248 °С.
Нітрати. 8 мг розчиняють в охолодженій суміші 5 мл
води Р і 45 мл кислоти сірчаної, вільної від азоту, Р;
одержаний розчин має бути безбарвним або злегка
блакитнуватого кольору.
Сульфатна зола (2.4.14). Не більше 0.1 %.
Зберігають у захищеному від світла місці
Дифенілборної кислоти аміноетиловий ефір.
С14Н|6ВІ\Ю. (М.м. 225.1). 1032400. [524-95-8].
Кристалічний порошок білого або жовтавого кольору.
Практично не розчинний у воді, розчинний у 96 %
спирті.
Температура плавлення: близько 193 °С.
Дифенілкарбазид. СІЗН,4М4О. (М.м. 242.3). 1032500.
[ 140-22-7]. 1,5-Дифенілкарбондигідразид
Кристалічний порошок білого кольору, поступово
рожевіє на повітрі. Дуже мало розчинний у воді, роз-
чинний в ацетоні, 96 % спирті іі кислоті оцтовій льо-
дяній.
Температура плавлення: близько 170 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %.
Зберігають у захищеному від світла місці.
Дифенілкарбазиду розчин. 1032501.
0.2 г дифенілкарбазиду Л1 розчиняють у 10 мл кисло-
ти оцтової льодяної Р і доводять об'єм розчину
етанолом Рдо 100 мл.
Готують безпосередньо перед використанням.
Дифенілкарбазон. С)3Н12М4О. (М.м. 240.3). 1032600.
[538-62-5]. 1,5-Дифенілкарбазон.
Кристалічний порошок оранжево-жовтого кольору.
Практично не розчинний у воді, дуже легко розчин-
ний у 96 % спирті.
Температура плавлення: близько 157 °С, із розкла-
данням.
Дифенілкарбазон-ртутний реактив. 1032601.
Розчин /.0.1 г дифенілкарбазону Р розчиняють в
етанолі Р і доводять об'єм розчину тим самим роз-
чинником до 50 мл.
Розчин 11. І г ртуті(П) хлориду Р розчиняють в
етанолі Рі доводять об'єм розчину тим самим роз-
чинником до 50 мл.
Змішують рівні об’єми розчинів 1 і II.
Дифенілоксазол. С|5Н]^О. (М.м. 221.3). 1032700.
[92-71-7]. 2,5-Дифенілоксазол.
Порошок білого кольору. Практично не розчинний) І
воді, розчинний у метанолі, помірно розчинний І
діоксані й кислоті оцтовій льодяній.
Температура плавлення: близько 70 °С.
: близько 1260. Визначення проводять за довжини
хвилі 305 нм, використовуючи як розчинник метанол Р.
Дифенілоксазол, використовуваний для рідинної сцин-
тиляції, має бути відповідного ступеня чистоти.
Дифенілфеніленоксиду полімер. 1032800. Полімер
2,6-дифеніл-н-феніленоксиду.
Пористі кульки білого або майже білого кольору,
розмір яких зазначають у випробуваннях, де він вико-
ристовується.
Дихлорбензол. С6Н4С12. (М.м. 147.0). 1027100. [95-50-1].
1,2- Дихлорбензол.
Безбарвна масляниста рідина. Практично не розчин-
ний у воді, розчинний в етанолі й ефірі.
г/2о • близько 1.31.
Температура кипіння: близько 180 °С.
Дихлорофос. С4Н7С12О4Р. (М.м. 221). 1101200.162-73-7].
2,2-Дихлорвінілдиметилфосфат.
Рідина від безбарвного до коричнювато-жовтого ко-
льору. Розчинний у воді, змішується з більшістю ор-
ганічних розчинників.
: близько 1.452.
Дихлороцтова кислота. С2Н2СІ2О2. (М.м. 128.9)
/027000. [79-43-6].
Безбарвна рідина. Змішується з водою, 96 % спиртом
та ефіром.
г/2о близько 1.566.
«р : близько 1.466.
Температура кипіння: близько 193 °С.
Дихлороцтової кислоти розчин. 1027001.
67 мл кислоти дихлороцтової Р доводять водою Р
до об'єму 300 мл і нейтралізують розчином аміаку Р
за синім лакмусовим папером Р. Охолоджують, до-
198
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
дають 33 мл кислоти дихлороцтової Р і доводять
водою Рдо об'єму 600 мл.
Дихлорфеноліндофенолу натрієва сіль.
СІ2Н6С12М\ІаО2,2Н2О. {М.м. 326.1). 1027300. [620-45-1].
Натрію 2,6-дихлор-Л'-(4-гідроксифеніл)-1,4-бензо-
хінонмоноіміну дигідрат. 2,6-Дихлорфеноліндофено-
лят натрію.
Порошок темно-зеленого кольору. Легко розчинна у
воді й етанолі. Водний розчин має темно-синє забарв-
лення, яке при підкислюванні розчину переходить у
рожеве.
Дихлорфеноліндофенолу титрований розчин.
1027301.
50.0 мг дихлорфеноліндофенолу натрієвої солі Р
розчиняють у 100.0 мл води Р і фільтрують.
Встановлення титру. 20.0 мг кислоти аскорбіно-
вої Ррозчиняють у 10 мл свіжоприготованого роз-
чину 200 г/л кислоти метафосфорної Р і доводять
об'єм розчину водою Рдо 250.0 мл. 5.0 мл одержа-
ного розчину швидко титрують приготованим
розчином дихлорфеноліндолфенолу, з мікробю-
ретки із ціною поділки 0.01 мл, до появи рожевого
забарвлення, яке не зникає протягом 10 с; час ти-
трування має бути не більшим 2 хв. Розчин ди-
хлорфеноліндолфенолу розводять водою Р до
одержання розчину, 1 мл якого відповідає 0.1 мг
кислоти аскорбінової (С6Н8О6).
Термін придатності 3 доби. Титр установлюють
безпосередньо перед використанням.
Дихлорфлуоресцеїн. С20НІ()С12О5. {М.м. 401.2). 1027200.
[76-54-0]. 2,7-Дихлорфлуоресцеїн. 2-(2,7-Дихлор-6-
гідрокси-3-оксо-3//-ксантен-9-іл)бензойна кислота.
Порошок від жовтувато-коричневого до жовто-оранже-
вого кольору. Мало розчинний у воді, легко розчинний
у 96 % спирті й розведених розчинах гідроксидів лужних
металів із утворенням розчину з жовтувато-зеленою
флуоресценцією, практично не розчинний в ефірі.
Дихлорхінонхлорімід. С6Н2С13МО. {М.м. 210.4).
1027400. [101-38-2]. 2,6-Дихлор-7У-хлор-1,4-бензо-
хінонмоноімін.
Кристалічний порошок від світло-жовтого до зелену-
вато-жовтого кольору. Практично не розчинний у
воді, розчинний у 96 % спирті й розведених розчинах
гідроксидів лужних металів.
Температура плавлення: близько 66 °С.
Дициклогексиламін. С,2Н23Т4. {М.м. 181.3). 1027500.
|101-83-7]. Л^А-Дициклогексиламін.
Безбарвна рідина. Помірно розчинний у воді, змі-
шується зі звичайними органічними розчинниками.
«о : близько 1.484.
Температура кипіння: близько 256 °С.
Температура тверднення (2.2.18). Від 0 °С до 1 °С.
Дициклогсксилссчовина. СІЗН24І\І2О. {М.м. 224.4).
1027600. [2387-23-7]. 1,3-Дициклогексилсечовина.
Кристалічний порошок білого кольору.
Температура плавлення: близько 232 °С.
Діазобензолсульфонової кислоти розчин РІ. 10265000.
0.9 г кислоти сульфанілової Р розчиняють в суміші
ЗО мл кислоти хлористоводневої розведеної Р та 70 мл
води Р. До 3 мл одержаного розчину додають 3 мл роз-
чину 50 г/л натрію нітриту Р, охолоджують в льо-
дяній бані протягом 5 хв, потім додають 12 мл розчину
натрію нітриту, знову охолоджують і доводять водою Р
до об'єму 100 мл. Реактив поміщають у льодяну баню.
Готують безпосередньо перед використанням, витри-
муючи в льодяній бані протягом 15 хв.
3,3'-Діамінобензидину тетрагідрохлорид.
СІ2НІ8С141\І4,2Н2О. {М.м. 396.1). 1098000. [7411-49-6].
3,3',4,4'-Дифенілтетрамін.
Порошок майже білого або трохи рожевого кольору.
Розчинний у воді.
Температура плавлення: близько 280 °С, із розкла-
данням.
Діатоміт. 1025900. [91053-39-3].
Дрібний гранульований порошок білого або майже
білого кольору, одержаний із крем'янистих оболонок
скам’янілих діатомових водоростей або їх уламків.
Практично не розчинний у воді, 96 % спирті й ефірі.
Ідентифікують з допомогою мікроскопа при збіль-
шенні х 500.
Діатоміт для газової хроматографії. 1026000.
Дрібний гранульований порошок білого або майже
білого кольору, одержаний із крем'янистих оболонок
скам’янілих діатомових водоростей або їх уламків.
Практично не розчинний у воді, 96 % спирті й ефірі.
Ідентифікують з допомогою мікроскопа при збіль-
шенні х 500; очищують шляхом обробки кислотою
хлористоводневою Р і промиванням водою Р.
Розмір часток. Не більше 5 % має залишатись на ситі
№ 180 і не більше 10 % має проходити крізьсито № 125.
Діатоміт для газової хроматографії РІ. 1026100.
Дрібний гранульований порошок білого або майже
білого кольору, одержаний із крем'янистих оболонок
скам'янілих діатомових водоростей або їх уламків.
Практично не розчинний у воді, 96 % спирті й ефірі.
Ідентифікують з допомогою мікроскопа при збіль-
шенні х 500; очищують шляхом обробки кислотою
хлористоводневою Р і промиванням водою Р.
Розмір часток. Не більше 5 % має залишатись на ситі
№ 250 і не більше 10 % має проходити крізьсито № 180.
Діатоміт для газової хроматографії Р2. 1026200.
Дрібний гранульований порошок білого або майже
білого кольору, із питомою площею поверхні близько
0.5 м2/г, одержаний із крем'янистих оболонок скам'я-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
199
4.1.1. Реактиви
нілих діатомових водоростей або їх уламків. Практич-
но не розчинний у воді, 96 % спирті й ефірі. Іден-
тифікують з допомогою мікроскопа при збільшенні
х 500; очищують шляхом обробки кислотою хлористо-
водневою Р і промиванням водою Р.
Розмір часток. Не більше 5 % має залишатись на ситі
№ 180. Не більше 10 % має проходити крізь сито
№ 125.
Діатоміт силанізований для газової хроматографії.
1026300.
Діатоміт для газової хроматографії Р, силанізований
диметилдихлорсиланом або іншими підхожими си-
ланізуючими реагентами.
Діатоміт силанізований РІ для газової хроматографії Р.
1026400.
Одержують із подрібненої червоної вогнетривкої цег-
ли і силанізують диметилдихлорсиланом або іншими
підхожими силанізуючими реагентами. Очищують
шляхом обробки кислотою хлористоводневою Р і про-
миванням водою Р.
Діетаноламін. С4НцМО2. (М.м. 105.1). 1027800.
[111-42-2]. 2,2'-1мінобісетанол.
Прозора, в'язка рідина трохи жовтуватого кольору або
кристали, які розпливаються на повітрі, плавляться
при температурі близько 28 °С. Дуже легко розчинний
у воді, ацетоні й метанолі.
г/2о : близько 1.09.
рН (2.2.3). Від 10.0 до 11.5. Вимірюють рН розчину
50 г/л.
Діетаноламін, використовуваний у випробуванні на
лужну фосфатазу, має задовольняти таку додаткову
вимогу.
Етаноламін. Не більше 1.0 %. Визначення проводять
методом газової хроматографії (2.2.28), використову-
ючи як внутрішній стандарт пропаноламін Р.
Розчин внутрішнього стандарту. 1.00 г пропано-
ламіну Ррозчиняють в ацетоні Р і доводять об'єм роз-
чину тим самим розчинником до 10.0 мл.
Випробовуваний розчин (а). 5.00 г діетиламіну розчиня-
ють в ацетоні Р і доводять об'єм розчину тим самим
розчинником до 10.0 мл.
Випробовуваний розчин (Ь). 5.00 г діетиламіну розчиня-
ють в ацетоні Р, додають 1.0 мл розчину внутрішнього
стандарту й доводять об'єм розчину тим самим роз-
чинником до 10.0 мл.
Розчини порівняння. 0.50 г етаноламіну Ррозчиняють в
ацетоні Р і доводять об'єм розчину тим самим розчин-
ником до 10.0 мл. До 0.5 мл, 1.0 мл та 2.0 мл одержано-
го розчину додають по 1.0 мл розчину внутрішнього
стандарту й доводять об'єм кожного розчину ацето-
ном Р до 10.0 мл.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором.
Хроматографують по 1.0 мкл кожного випробовувано- II
го розчину й по 1.0 мкл кожного розчину порівнянні
за таких умов:
— колонка розміром 1 м х 4 мм, заповнена исиіин
ром дифенілфеніленоксиду Різ розміром часток вц
180 мкм до 250 мкм,
— газ-носій азот для хроматографії Р,
— швидкість газу-носія 40 мл/хв;
— температуру колонки підтримують при 125 °С
протягом 3 хв; потім підвищують температуру до
300 °С із швидкістю 12 °С/хв;
— температура блока вводу проб 250 °С;
— температура детектора 280 °С.
Зберігають у повітронепроникному контейнері.
Діетиламін. С4Нц1\І. (М.м. 73.1). 1028000. [109-89-7].
Прозора, безбарвна, займиста рідина. Має сильно
лужну реакцію, змішується з водою і 96 % спиртом
іїу) : близько 0.71.
Температура кипіння: близько 55 °С.
Діетиламіноетилдекстран. 1028200.
Аніонообмінна смола у формі гідрохлориду.
Порошок, який утворює з водою гель.
А^А'-Діетиланілін. СІ0НІ5М. (М.м. 149.2). 1028400.
191-66-7].
г/2о близько 0.938.
Температура кипіння: близько 217 °С.
Температура плавлення: близько -38 °С.
Діетиленгліколь. С4Н,0О3. (М.м. 106.1). 1028300.
[111-46-6]. 2,2'-Оксидіетанол.
Містить не менше 99.5 % (м/м) С4НІ0О3.
Прозора, безбарвна, гігроскопічна рідина. Змішується
з водою, ацетоном і 96 % спиртом.
г/20 : близько 1.118.
: близько 1.447.
Температура кипіння: від 244 °С до 246 °С.
Зберігають у повітронепроникному контейнері.
А^А/-Діетилетан-1,2-діамін. 1028500. [100-36-7]. Див.
1\',І\!-діетилетилендіамін Р.
А,Л-Діетилетиленліамін. С6НІ61Ч2 (М.м. 116.2).
1028500. [100-36-7].
Містить не менше 98.0 % С6НІ6ІМ2.
Трохи масляниста рідина, безбарвна або жовтавого
кольору, із сильним запахом аміаку. Справляє подраз-
ливу дію на шкіру, очі та слизові оболонки.
0$ : 0.827.
Температура кипіння: від 145 °С до 147 °С.
Вода (2.5.12). Не більше 1.0 %. Визначення проводять
із 0.500 г.
200
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Шетилфенілендіамінусульфат. С|0Н|8М2О4$.(Мм. 262.3).
1028600 |6283-63-2]. N,79'- Діетил-л-фенілендіаміну
сульфат. А,А-Діетилбензол-1,4-діаміну сульфат.
Порошок білого або жовтавого кольору. Розчинний у
воді.
Температура плавлення: близько 185 °С. із розкла-
данням.
Зберігають у захищеному від світла місці.
Діетилфенілевдіаміну сульфату розчин. 1028601.
До 250 мл води Р додають 2 мл кислоти сірчаної Р
та 25 мл 0.02 М розчину натрію едетату. В одержа-
ному розчині розчиняють 1.1г діетилфенілендіамі-
ну сульфату Рі доводять водою Рцо об’єму 1000 мл.
Використовують лише безбарвний розчин.
Зберігають у прохолодному, захищеному від світла
місці.
Термін придатності 1 міс.
Діетокситетрагідрофуран. С8НІ6О3. (М.м. 160.2).
1027900. [3320-90-9]. 2,5-Діетокситетрагідрофуран.
Суміш цис і транс ізомерів.
Прозора, безбарвна або жовтавого кольору рідина.
Практично не розчинний у воді, розчинний у
96% спирті, ефірі й більшості інших органічних роз-
чинників.
4і? : близько 0.98.
: близько 1.418.
Діізобутилкетон. СЧН|8О. (М.м. 142.2). 1029200.
[108-83-8].
Прозора, безбарвна рідина. Мало розчинний у воді,
змішується з більшістю органічних розчинників.
я® : близько 1.414.
Температура кипіння: близько 168 °С.
Діізопропіловий ефір. С6НІ4О. (М.м. 102.2). 1029300.
[108-20-3].
Прозора, безбарвна рідина Дуже мало розчинний у
воді, змішується з 96 % спиртом та ефіром.
4“: від 0.723 до 0.728.
Температура кипіння: від 67 °С до 69 °С.
Не переганяють, якщо діізопропіловий ефір не витри-
мує випробування на пероксиди.
Пероксиди. 8 мл розчину крохмалю з калію йодидом Р
поміщають у циліндр із притертою скляною пробкою
місткістю 12 мл і діаметром близько 1.5 см, повністю
заповнюють випробовуваним ефіром, закривають
пробкою та перемішують. Витримують у темному місці
протягом ЗО хв; не має з'являтись забарвлення.
Назва й концентрація будь-якого доданого стабіліза-
тора мають бути зазначені на етикетці.
Зберігають у захищеному від світла місці.
Діоксан. С4Н8О2. (М.м. 88.1). 1032000. [123-91-1].
1,4-Діоксан.
Прозора, безбарвна рідина. Змішується з водою й
більшістю органічних розчинників.
; близько 1.03.
Температура тверднення (2.2.18). Від 9 °С до 11 °С.
Вода (2.5.12). Не більше 0.5 %.
Не переганяють, якщо діоксан не витримує випробу-
вання на пероксиди.
Пероксиди. 8 мл розчину крохмалю з калію йодидом Р помі-
щають у циліндр із притертою скляною пробкою міст-
кістю 12 мл і діаметром близько 1.5 см, заповнюють пов-
ністю діоксаном та перемішують. Витримують у темному
місці протягом ЗО хв; не має з'являтись забарвлення.
Діоксан, використовуваний для рідинної сцинтиляції,
має бути відповідного ступеня чистоти.
Діоксану вихідний розчин. 1032001.
1.00 г діоксану Р розчиняють у воді Р і доводять
об'єм розчину тим самим розчинником до
100.0 мл. 5.0 мл одержаного розчину доводять во-
дою Рао об'єму 50.0 мл (1.0 мг/мл).
Діоксану розчин. 1032002.
50.0 мл вихідного розчину діоксану Р доводять во-
дою Рао об'єму 100.0 мл (0.5 мг/мл діоксану).
Діоксану розчин РІ. 1032003.
10.0 мл розчину діоксану Р доводять водою Р до
об'єму 50.0 мл (0.1 мг/мл діоксану).
Ді(октаденил)-3,3'-тіодипропіонат. С42Н82О48.
(М.м. 683). 1031900. [693-36-7].
Кристалічний порошок білого кольору. Практично не
розчинний у воді, легко розчинний у метиленхлориді,
помірно розчинний в ацетоні, 96 % спирті й петролей-
ному ефірі.
Температура плавлення: від 58 °С до 67 °С.
2,2'-Ді(октадецилокси)-5,5'-спіробі( 1,3,2-діоксафос-
форинан). С4|Н82О6Р2. (М.м. 733). 1031800.
Тверда воскоподібна речовина білого кольору. Прак-
тично не розчинний у воді, розчинний у розчинах
гідрокарбонатів.
Температура плавлення: від 40 °С до 70 °С.
Діоктадецилдисульфід. С36Н7482. (М.м. 571.1). 1031700.
[1844-09-3].
Порошок білого кольору. Практично не розчинний у
воді.
Температура плавлення: від 53 °С до 58 °С.
Докузат натрію. 1034100. [577-11-7]. Див. статтю Доку-
зат натрію.
Дотриаконтан. С32Н66. (М.м. 450.9). 1034200. [544-85-4].
н-Дотриаконтан.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
201
4.1.1. Реактиви
Пластинки білого кольору. Практично не розчинний у
воді, помірно розчинний у гексані, мало розчинний в
ефірі.
Температура плавлення: близько 69 °С.
Домішки. Не більше 0.1 % домішок із часом утри-
мування, характерним для а-токоферолу ацетату; виз-
начення проводять методом газової хроматографії, як
зазначено в статті а- Токоферолу ацетат.
Евгенол. Сі0НІ2О2. (М.м. 164.2). 1037000. [97-53-0].
4-Аліл-2-метоксифенол.
Безбарвна або блідо-жовтого кольору масляниста
рідина, під дією повітря й світла темнішає та стає
більш в'язкою. Практично не розчинний у воді,
змішується з 96 % спиртом, ефіром і жирними та
ефірними оліями.
^20 близько 1.07.
Температура кипіння: близько 250 °С.
Евгенол, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено в статті Олія гвоздична,
використовуючи евгенол як випробовуваний розчин.
Плоша основного піка має бути не менше 98.0 % суми
площ усіх піків.
Зберігають у захищеному від світла місці.
Еметину дигідрохлорид. 1034300. [316-42-7]. Див. стат-
тю Еметину гідрохлорид пентагідрат.
Емодин. СІ5Н10О5. (М.м. 270.2). 1034400.
[518-82-1 ]. 1,3,8-Тригідрокси-6-метилантрахінон.
Голчасті кристали оранжево-червоного кольору.
Практично не розчинний у воді, мало розчинний в
ефірі, розчинний у 96 % спирті й розчинах гідроксидів
лужних металів.
Хроматографія. Визначення проводять, як зазначено
в статті Корені ревеню-, на хроматограмі має виявлятись
лише одна основна пляма.
Еритритол. С4НІ0О4. (М.м. 122.1). 1113800. [149-32-6].
(А*,5*)-Бутан-1,2,3,4-тетрол. мезо- Еритритол.
Кристали у вигляді тетрагональних призм. Дуже легко
розчинний у воді, розчинний у піридині, мало розчин-
ний у 96 % спирті.
Температура плавлення: близько 121.5 °С.
Ерукамід. С22Н43МО. (М.м. 337.6). 1034500. [112-84-5].
(7)-Докоз-13-еноамід.
Порошок жовтуватого або білого кольору або гранули.
Практично не розчинний у воді, дуже легко розчин-
ний у метиленхлориді, розчинний в етанолі.
Температура плавлення: близько 70 °С.
Ескулін. Сі5Ні6О9,1.5Н2О. (М.м. 367.3). 1119400.
[531-75-9]. 6-([3-О-Глюкопіранозилокси)-7-гідрокси-
2/7-хромен-2-он.
Порошок білого або майже білого кольору або бо
барвні кристали. Помірно розчинний у воді (І
96 % спирті, легко розчинний у гарячій воді й гарячиІ
му 96 % спирті.
Хроматографія (2.2.27). Визначення проводять, як ц-
значено в статті Корені елеутерокока', на хроматограйИ
має виявлятись лише одна основна пляма.
Естрагол. СІ0НІ2О. (М.м. 148.2). 1034700. [140-67-0]. І
1-Метокси-4-проп-2-енілбензол.
Рідина. Змішується з 96 % спиртом.
: близько 1.52.
Температура кипіння: близько 216 °С.
Естрагол, використовуваний у газовій хроматографії 1
має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро- 1
матографії (2.2.28) за умов, зазначених у статті О.іи І
анісова, використовуючи естрагол як випробовуванні! І
розчин.
Площа основного піка має бути не менше 98.0 % суми І
площ усіх піків.
17а-Естрадіол. С|8Н24О2. (М.м. 272.4). 1034600.
[57-91-01.
Кристалічний порошок білого або майже білого ко-
льору або безбарвні кристали.
Температура плавлення: від 178 °С до 179 °С.
Есцин. 1001700. [11072-93-8].
Суміш споріднених сапонінів, одержаних із зерен!
Ае.чсиіи.ч ИірросаМапит Ь.
Дуже дрібний аморфний порошок майже білого або
дещо червонуватого або жовтуватого кольору.
Хроматографія. Визначення проводять, як зазначено
в статті Корені сенеги, але використовують 20 мкл роз-
чину. Після обприскування хроматограми розчином
анісового альдегіду Р і нагрівання на хроматограмі ви-
пробовуваного розчину має виявлятись основна пля-
ма з Аублизько 0.4.
Етанол. С2Н6О. (М.м. 46.07). 1034800. [64-17-5]. Див.
статтю Етанол безводний.
Етанол РІ. 1034801.
Має задовольняти вимоги для етанолу Рі таку до-
даткову вимогу.
Метанол. Не більше 0.005 % (об/об). Визначають
методом газової хроматографії (2.2.28).
Випробовуваний розчин. Випробовуваний етанол.
Розчин порівняння. 0.50 мл метанолу безводного Р
доводять випробовуваним етанолом до об'єму
100.0 мл. 1.0 мл одержаного розчину доводять ви-
пробовуваним етанолом до об'єму 100.0 мл.
Хроматографування проводять із використанням
полуменево-іонізаційного детектора в таких умо-
вах:
202
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
— колонка скляна розміром 2 м х 2 мм, заповнена
сополімером етилвінілбензол-дивінілбензолу Р із
розміром часток від 75 мкм до 100 мкм,
— газ-носій азот для хроматографії Р;
— швидкість потоку ЗО мл/хв;
— температура колонки 130 °С;
— температура інжектора при вводі 150 °С;
— температура детектора 200 °С.
Вводять по 1 мкл випробовуваного розчину і роз-
чину порівняння, по черзі, тричі. Після кожного
хроматографування нагрівають колонку до темпе-
ратури 230 °С протягом 8 хв. Інтегрують пік мета-
нолу.
Вміст метанолу (X), у відсотках, обчислюють за фор-
мулою:
а • Ь
с -Ь
вміст метанолу в розчині порівняння, у відсот-
ках (об/об);
площа піка метанолу на хроматоірамі випробо-
вуваного розчину;
с — площа піка метанолу на хроматограмі розчину
порівняння.
Етаноламін. С2Н7ХЮ. (М.м. 61.1). Ю34900. [141-43-5].
2-Аміноетанол.
Прозора, безбарвна, в'язка, гігроскопічна рідина.
Змішується з водою й метанолом, помірно розчинний
в ефірі.
(/і®: близько 1.04.
Ир : близько 1.454.
Температура плавлення: близько 11 °С.
Зберігають у повітронепроникному контейнері.
Етилакрилат. С5Н8О2. (М.м. 100.1). 1035400. [140-88-5].
Етилпрон-2-еноат.
Безбарвна рідина.
</2о '• близько 0.924.
: близько 1.406.
Температура кипіння: близько 99 °С.
Температура плавлення: близько -71 °С.
Етилацетат. С4Н8О2. (М.м. 88.1). 1035300. [141-78-6].
Прозора, безбарвна рідина. Розчинний у воді, змі-
шується з 96 % спиртом.
: від 0.901 до 0.904.
Температура кипіння: від 76 °С до 78 °С.
Етилацетат оброблений. 1035301.
200 г кислоти сульфамінової Р диспергують в етил-
ацетаті Р і доводять тим самим розчинником
до об'єму 1000 мл. Одержану суспензію перемішу-
ють протягом 3 діб і фільтрують крізь паперовий
фільтр.
Термін придатності 1 міс.
Етилбензол. С8Н10. (М.м. 106.2). 1035800. [100-41-4].
Містить не менше 99.5 % (м/м) С8Н10, визначають ме-
тодом газової хроматографії.
Прозора, безбарвна рідина. Практично не розчинний
у воді, розчинний в ацетоні й 96 % спирті.
е/2о : близько 0.87
: близько 1.496.
Температура кипіння: близько 135 °С.
2-Етилгексан-1,3-діол. С8Н18О2. (М.м. 146.2). 1105900.
[94-96-2].
Дещо масляниста рідина. Розчинний в етанолі, 2-про-
панолі, пропіленгліколі та олії рициновій.
</2о : близько 0.942.
Яр* : близько 1.451.
Температура кипіння: близько 244 °С.
2-Етилгексанова кислота. С8Н,6О2. (М.м. 144.2).
1036600. [149-57-5]. 2-Етилкапронова кислота.
Безбарвна рідина.
еІ2о : близько 0.91.
Ир : близько 1.425.
Супровідні домішки. Визначення проводять мето-
дом газової хроматографії (2.2.28). Хроматогра-
фують 1 мкл розчину, приготованого таким чином:
суспендують 0.2 г 2-етилгексанової кислоти у 5 мл
води Р, додають 3 мл кислоти хлористоводневої роз-
веденої Р і 5 мл гексану Р, струшують протягом
1 хв, після розділення шарів використовують верхній
шар. Хроматографують за умов, описаних для
2-етилгексанової кислоти у статті Амоксицилін
натрію.
Сума площ піків, крім основного піка й піка розчинни-
ка, не має перевищувати 2.5 % площі основного піка.
Етиленбіс[3,3-ди(3-(1,1-диметилетил)-4-гідрокси-
феніл)бутират]. С50Н66О8. (М.м. 795). 1035900.
[32509-66-3]. Етиленбіс[3,3-ди(3-/ире/и-бутил-4-
гідроксифеніл)бутират].
Кристалічний порошок. Практично не розчинний у
воді й петролейному ефірі, дуже легко розчинний в
ацетоні, ефірі й метанолі.
Температура плавлення: близько 165 °С.
Етиленбіс[3,3-ди(3-/ире/л-бутил-4-гідроксифеніл)бути-
рат]. 1035900. [32509-66-3]. Див. Етиленбіс[3,3-ди(3-
(1,1-диметилетил)-4-гідроксифеніл)бутират! Р.
Етиленгліколь. С2Н6О2. (М.м. 62.1). 1036100. [107-21-1].
Етан-1,2-діол.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
203
4.1.1. Реактиви
Безбарвна, дещо в'язка гігроскопічна рідина. Змішується
з водою й 96 % спиртом, мало розчинний в ефірі.
3%) від 1.113 до 1.115.
: близько 1.432.
Температура плавлення: близько -12 °С.
Температура кипіння: близько 198 °С.
Кислотність. До 10 мл додають 20 мл води Рі 1 мл роз-
чину фенолфталеїну Р; забарвлення розчину має
змінитися до рожевого при додаванні не більше
0.15 мл 0.02 М розчину натрію гідроксиду.
Вода (2.5.12). Не більше 0.2 %.
Етиленгліколюмоноетиловийефір. С4Н1(|О2. (М.м 90 1).
1036200. [110-80-5]. 2-Етоксіетанол.
Прозора, безбарвна рідина. Змішується з водою, аце-
тоном, 96 % спиртом і ефіром.
^2о : близько 0.93.
: близько 1.406.
Температура кипіння: близько 135 °С.
Етиленгліколю монометиловий ефір. С3НЯО2.
(М.м. 76.1). 1036300. [109-86-4]. 2-Метоксіетанол.
Прозора, безбарвна рідина. Змішується з водою, аце-
тоном, 96 % спиртом і ефіром.
^20 ' близько 0.97.
: близько 1.403.
Температура кипіння: близько 125 °С.
Етилендіамін. С2Н^2 (М.м. 60.1). 1036500. 1107-15-3].
Етан-1,2-діамін.
Прозора, безбарвна, димляча рідина; має сильно луж-
ну реакцію. Змішується з водою і 96 % спиртом, мало
розчинний в ефірі.
Температура кипіння: близько 116 °С.
(Етилендинітрил)тетраоцтова кислота. С10Н|6М2О8.
(М.м. 292.2). 1105800. [60-00-4]. А,А-1,2-етан-
діїлбіс[У-(карбоксиметил)гліцин]. Едетова кислота.
Кристалічний порошок білого кольору. Дуже мало
розчинний у воді.
Температура плавлення: близько 250 °С, із розкла-
данням.
Етиленоксид. С2Н4О. (М.м. 44.05). 1036400. |75-21-8|
Оксиран.
Безбарвний, займистий газ. Дуже легко розчинний у
воді й етанолі.
Температура зрідження, близько 12 °С
Етиленоксиду вихідний розчин. 1036401.
Усі операції, які проводяться у ході приготування
розчинів, виконують у витяжній шафі. Захищають
руки й обличчя, надягаючи поліетиленові захисні р\'
кавички й підхожу маску для обличчя.
Розчини зберігають у повітронепроникному кон-
тейнері, в холодильнику при температурі від 4 "Сдс
8 "С. Усі випробування проводять тричі.
До сухої чистої тест-пробірки, охолодженої і
суміші з 1 частини натрію хлориду Р і 3 частин
подрібненого льоду, повільно вводять потік газо-
подібного етиленоксиду Р, дозволяючи конденсу
ватись на внутрішній стінці тест-пробірки. З до-
помогою скляного шприца, попередньо охолод-
женого до температури 10 °С, поміщають близько
300 мкл (що відповідає приблизно 0.25 г етиле-
ноксиду) рідкого етиленоксиду Ру 50 мл макрого-
лу 200 РІ. Визначають абсорбовану кількість ети-
леноксиду зважуванням до і після абсорбції (Мео)
Розводять макроголом 200 РІ до об'єму 100.0 мл.
Перед використанням ретельно перемішують.
Кількісне визначення. До 10 мл суспензії 500 г/д
магнію хлориду Р в етанолі Р додають 20.0 мл
0.1 М розчину кислоти хлористоводневої в спирті
Колбу закривають пробкою, збовтують до одер-
жання насиченого розчину і для досягнення
рівноваги витримують протягом ночі. 5.00 г
вихідного розчину 2.5 г/л етиленоксиду Р поміща-
ють у колбу, зважують, витримують протягом ЗОхв
і титрують 0.1 М розчином калію гідроксиду спир-
товим потенціометрично (2.2.20).
Проводять контрольний дослід, використовуючи
замість вихідного розчину етиленоксиду таку саму
кількість макроголу 200 РІ.
Вміст етиленоксиду (X), у міліграмах в одному
грамі, обчислюють за формулою:
(К0-Иі)-/- 4,404
де:
Уо і V, — об’єм 0.1 М розчину калію гідроксиду
спиртового, витрачений на титрування
контрольного й випробовуваного роз-
чину, відповідно;
ґ — поправковий коефіцієнт до моляр-
ності 0.1 М розчину калію гідроксиду
спиртового;
т — маса випробовуваного зразка, у грамах.
Етиленоксиду розчин. 1036402.
Зважують кількість охолодженого вихідного розчи-
ну етиленоксиду Р, яка відповідає 2.5 мг етиленок-
сиду, в охолодженій колбі й доводять макроголом
200 РІ до 50.0 г, ретельно перемішують. 2.5 г одер-
жаного розчину доводять макроголом 200 РІ до
об’єму 25.0 мл (5 мкг етиленоксиду в 1 г розчину).
Готують безпосередньо перед використанням.
Етиленоксиду розчин РІ. 1036403.
1.0 мл (точна наважка) охолодженого вихідного роз-
чину етиленоксиду Рдоводятьмакроголом 200 Ріло
204
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
об’єму 50.0 мл і ретельно перемішують. 2.5 г одер-
жаного розчину доводять макроголом 200 РІ до
об’єму 25.0 мл. Вміст етиленоксиду, в ррт, обчис-
люють із об’єму, визначеного зважуванням, беру-
чи густину макроголу 200 РІ, що дорівнює 1.127.
Готують безпосередньо перед використанням.
Етиленоксиду розчин Р2. 1036404.
1.00 г охолодженого вихідного розчину етиленокси-
ду Р (що відповідає 2.5 мг етиленоксиду) поміща-
ють у попередньо зважену колбу, яка містить 40.0 г
охолодженого макроголу 200 РІ, і перемішують.
Визначають точну масу й розводять до розрахун-
кової маси таким чином, щоб одержати розчин,
який містить 50 мкг етиленоксиду в 1 г розчину.
Зважують 10.00 г, поміщають у колбу, яка містить
близько 30 мл води Р, перемішують і доводять во-
дою Рао об’єму 50.0 мл (10 мкг/мл етиленоксиду).
Готують безпосередньо перед використанням.
Етиленоксиду розчин РЗ. 1036405.
10.0 мл розчину етиленоксиду Р2 доводять водою Р
до об’єму 50.0 мл (2 мкг/мл етиленоксиду).
Готують безпосередньо перед використанням.
Етиленхлорид. С2Н4С12. (М.м. 99.0). 1036000. [107-06-2].
1.2-Дихлоретан.
Прозора, безбарвна рідина. Розчинний приблизно у
120 частинах води й 2 частинах 96 % спирту,
змішується з ефіром.
<$}: близько 1.25.
Температурні межі перегонки (2.2.11). Від 82 °С до
84 °С; має переганятись не менше 95 %.
І,Г-Етиліденбіс(триптофан). С24Н26М4О4. (М.м. 434.5).
1106000. [132685-02-0]. 3,3’-[Етиліденбіс(1/7-індол-
1,3-діїл)]біс[(25)-2-амінопропіонова кислота].
Містить не менше 98.0 % С24Н261М4О4.
Кристалічний порошок білого або майже білого ко-
льору. Мало розчинний у воді, дуже мало розчинний у
96 % спирті, практично не розчинний в ефірі.
Температура плавлення: близько 223 °С, із розкла-
данням.
Кількісне визначення. Проводять, як зазначено в статті
Триптофан у розділі “1,Г-Етилїденбіс(триптофан) та
інші супровідні речовини”.
Площа основного піка на хроматограмі розчину
порівняння (а) має бути не менше 98.0 % суми плош
усіх піків.
А-Етилмалеїмід. С6Н7МО2. (М.м. 125.1). 1036700.
[128-53-0]. І-Етил-1 //-пірол-2,5-діон.
Безбарвні кристали. Помірно розчинний у воді, легко
розчинний у 96 % спирті.
Температура плавлення: від 41 °С до 45 °С.
Зберігають при температурі від 2 °С до 8 °С.
2-Етил-2-метилбурштинова кислота. С7Н12О4.
(М.м. 160.2). 1036800. [631-31-2]. 2-Етил-2-метилбу-
тандикарбонова кислота.
Температура плавлення: від 104 °С до 107 °С.
Етилметилкетон. 1054100. [78-93-3]. Див. Метилетил-
кетон Р.
Етилпарагідроксибензоат. 1035700. [120-47-8]. Див.
статтю Етилпарагідроксибензоат.
Етилформіат. С3Н6О2. (М.м. 74.1). 1035600. [109-94-4].
Етилметаноат.
Прозора, безбарвна займиста рідина. Легко розчин-
ний у воді, змішується з 96 % спиртом і ефіром
с/20 : близько 0.919.
: близько 1.36.
Температура кипіння: близько 54 °С.
Етилціаноацетат. С5Н7МО2. (М.м. 113.1). 1035500.
[105-56-6].
Безбарвна або світло-жовтого кольору рідина. Мало
розчинний у воді, змішується з 96 % спиртом і ефіром.
Температура кипіння: від 205 °С до 209 °С, із розкла-
данням.
Етоксихризоїдину гідрохлорид. С14Н|7С1М4О.
(М.м. 292.8). 1035200 [2313-87-3]. 4-[(4-Етоксифе-
н іл )діазе н і л ] фен іле н -1,3 -діам і ну гі дрохл орид.
Порошок червонуватого кольору. Розчинний у 96 %
спирті.
Етоксихризоїдину розчин. 1035201.
Розчин 1 г/л у 96 % спирті Р.
Випробування на чутливість. До суміші 5 мл кисло-
ти хлористоводневої розведеної Р і 0.05 мл розчину
етоксихризоїдину додають 0.05 мл 0.0167Мрозчину
бромід-бромату. Забарвлення розчину має зміни-
тися від червоного до світло-жовтого протягом 2 хв.
Еуглобуліни бичачі. 1037100.
Використовують свіжу бичачу кров, зібрану в розчин
антикоагулянту (наприклад, розчин натрію цитрату).
Відкидають будь-яку гемолізовану кров. Центрифугу-
ють із прискоренням від 1500 § до 1800 § при темпера-
турі від 15 °С до 20 °С для одержання супернатанту
плазми з низьким вмістом тромбоцитів.
До 1 л плазми бичачої додають 75 г барію сульфату Р,
струшують протягом 30 хв, потім центрифугують із
прискоренням від 1500 8 до 1800 8 при температурі від
15 °С до 20 °С і відокремлюють прозору надосадову
рідину. Додають 10 мл розчину 0.2 мг/мл апротиніну Р
і струшують до змішування. У контейнер із мінімаль-
ною місткістю 30 л у камері з температурою 4 °С
поміщають 25 л води дистильованої Р, охолодженої до
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
205
4.1.1. Реактиви
температури 4 °С, додають близько 500 г твердого вуг-
лецю діоксиду й негайно, при перемішуванні, додають
надосадову рідину, одержану із плазми. Утворюється
білий осад. Для осадження витримують при темпера
турі 4 °С від 10 год до 15 год. Прозору надосадову ріди-
ну відокремлюють з допомогою сифона. Збирають
осад центрифугуванням при температурі 4 °С. Сус-
пендують осад механічним диспергуванням у 500 мл
води дистильованої Р при температурі 4 °С, збовтують
протягом 5 хв і відокремлюють осад центрифугуван-
ням при температурі 4 °С. Осад механічно диспергу-
ють у 60 мл розчину, який містить 9 г/л натрію хлори-
ду Рі 0.9 г/л натрію цитрату Р, доводять рН до 7.2-7.4
розчином 10 г/л натрію гідроксиду Р і фільтрують крізь
скляний фільтр. Одержаний осад подрібнюють у
ступці, фільтр і ступку промивають 40 мл розчину,
який містить 9 г/л натрію хлориду Р і 0.9 г/л натрію
цитрату Р, доводять тим самим розчином до об’єму
100 мл і ліофілізують. Звичайно вихід становить від 6 г
до 8 г еуглобулінів із 1 л плазми бичачої.
Випробування на придатність. Готують розчин, вико-
ристовуючи фосфатний буферний розчин рН 7.4 Р,
який містить 30 г/л альбуміну бичачого Р.
У тест-пробірку діаметром 8 мм, поміщену у водяну
баню при температурі 37 °С, вносять 0.2 мл розчину
порівняння урокінази, який містить 100 МО/мл, і
0.1 мл розчину тромбіну людського Р, який містить
20 МО/мл. Швидко вводять 0.5 мл розчину, який міс-
тить 10 мг еуглобулінів бичачих у мілілітрі. Має утво-
ритися щільний згусток за час менше 10 с. Відмічають
час, який минув між додаванням розчину еуглобулінів
бичачих і руйнуванням згустка. Час лізису не має пе-
ревищувати 15 хв.
Зберігають у сухому місці при температурі 4 °С.
Термін придатності 1 рік.
Еуглобуліни людські. 1037200.
Для приготування використовують свіжу людську
кров, зібрану в розчин антикоагулянту (наприклад,
розчин натрію цитрату), або людську кров для перели-
вання, зібрану в пластмасові контейнери для крові, із
терміном зберігання, який тільки-но скінчився.
Відкидають будь-яку гемолізовану кров. Центрифугу-
ють із прискоренням від 1500 § до 1800 § при темпера-
турі 15 °С для одержання супернатанту плазми з низь-
ким вмістом тромбоцитів. Можна змішувати плазми,
одержані з крові однієї групи.
До 1 л плазми додають 75 г барію сульфату Р, збовту-
ють протягом 30 хв, потім центрифугують при темпе-
ратурі 15 °С із прискоренням не менш як 1500 & і відо-
кремлюють прозору надосадову рідину. Додають 10 мл
розчину 0.2 мг/мл апротиніну Р і струшують до змішу-
вання. У контейнер з мінімальною місткістю 30 л у ка-
мері з температурою 4 °С поміщають 25 л води дисти-
льованої Р, охолодженої до температури 4 °С, додають
близько 500 г твердого вуглецю діоксиду й негайно до-
дають, при перемішуванні, надосадову рідину, одержа-
ну із плазми; утворюється білий осад. Залишають для
осадження при температурі 4 °С на 10 - 15 год. Видаля-
ють прозору надосадову рідину з допомогою сифону-
вання. Збирають осад центрифугуванням при темпе-1
ратурі 4 °С. Суспендують осад механічним диспергу- І
ванням у 500 мл води дистильованої Р при температурі І
4 °С, збовтують протягом 5 хв і відокремлюють осад І
центрифугуванням при температурі 4 °С. Механічно І
диспергують осад у 60 мл розчину, який містить 9 г/л 1
натрію хлориду Р і 0.9 г/л натрію цитрату Р, і добо- і
дять рН до 7.2-7.4 розчином натрію гідроксиду Р
Фільтрують крізь скляний фільтр. Одержаний осад 1
подрібнюють з допомогою придатного інструмента. І
Промивають фільтр та інструмент 40 мл розчину, який
містить 9 г/л натрію хлориду Рі 0.9 г/л натрію цитра-
ту Р, і розводять до об’єму 100 мл тим самим розчи-
ном. Ліофілізують. Вихід звичайно становить від 6 гдо
8 г еуглобулінів з 1 л плазми людської.
Випробування на придатність. Для цього випробуван-
ня готують розчин, використовуючи фосфатний бу-
ферний розчин рН 7.2 Р, який містить 30 г/л альбуміну
бичачого Р. У тест-пробірку діаметром 8 мм, поміщену
до водяної бані при температурі 37 °С, вносять 0.1 мл
розчину порівняння стрептокінази, який містить
10 МО/мл стрептокіназної активності, та 0.1 мл роз-
чину тромбіну людського Р, який містить 20 МО/мл.
Швидко додають 1 мл розчину, який містить 10 мг/мл
еуглобулінів людських. Має утворитися щільний згус-
ток за час менший 10 с. Відмічають час, який минув
між додаванням розчину еуглобулінів людських і руй-
нуванням згустка. Час лізису не має перевищувати
15 хв.
Зберігають у повітронепроникному контейнері при
температурі 4 °С.
Термін придатності 1 рік.
Ефір. С4НІ0О. (М.м. 74.1). 1035000. 160-29-7].
Прозора, безбарвна, летка й дуже рухлива, легко зай-
миста рідина. Гігроскопічний, розчинний у воді,
змішується з 96 % спиртом.
г?2о ' від 0.713 до 0.715.
Температура кипіння: від 34 °С до 35 °С.
Не переганяють, якщо ефір не витримує випробування
на пероксиди.
Пероксиди. 8 мл розчину крохмалю з калію йодидом Р
поміщають у циліндр із притертою скляною проб-
кою місткістю 12 мл і діаметром близько 1.5 см.
Об’єм циліндра заповнюють повністю випробовуваним
ефіром, перемішують і витримують у темно-
му місці протягом ЗОхв; не має з’являтись забарвлення.
Назва й концентрація будь-якого доданого стабіліза-
тора мають бути зазначені на етикетці.
Зберігають у повітронепроникному контейнері, за-
хищеному від світла місці, при температурі не ви-
ще 15 °С.
Ефір, вільний від пероксидів. 1035)00. Див. статтю Ефір
для наркозу.
Желатин. 1040000. [9000-70-8]. Див. статтю Желатин.
206
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
[Желатин гідролізований. 1040100.
50 г желатину Р розчиняють у 1000 мл води Р. Оброб-
ляють насиченою парою в автоклаві при температурі
121 °С протягом 90 хв і ліофілізують.
Заліза(ІІІ) амонію сульфат. ЕеІ'І Н4(8О4)2,12Н2О.
(Мм. 482.2). 1037700. [7783-83-7]. Заліза амонію ди-
сульфат додекагідрат.
Кристали блідо-фіолетового кольору, які вицвітають
на повітрі. Дуже легко розчинний у воді, практично не
розчинний у 96 % спирті.
Заліза(ІІІ) амонію сульфату розчин Р2. 1037702.
Розчин 100 г/л.
Перед використанням фільтрують, якщо необхідно.
Заліза(ІП) амонію сульфату розчин Р5. 1037704.
Струшують 30.0 г заліза(ПІ) амонію сульфату Р із
40 мл кислоти азотної Р і доводять об’єм розчину
водою Рао 100 мл. Якщо розчин каламутний, його
центрифугують або фільтрують.
Зберігають у захищеному від світла місці.
Заліза(Ш) амонію сульфату розчин Р6. 1037705.
20 г заліза(ІП) амонію сульфату Р розчиняють у
75 мл води Р, додають 10 мл 2.8 % (об/об) розчину
кислоти сірчаної Р і доводять об’єм розчину во-
дою Рцо 100 мл.
Заліза(Ш) нітрат. Ее(МО3)3,9Н2О. (М.м. 404). 1106100.
|7782-61-8|.
Містить не менше 99.0 % (м/м) Ее(ГМО3)3,9Н2О.
Кристали або кристалічна маса світло-рожевого ко-
льору. Дуже легко розчинний у воді.
Ві на кислота: не більше 0.3 % (у вигляді НГМО3).
Заліза(ІІІ) сульфат. Ее2(8О4)3,хН2О. 1037900.
[10028-22-5]. Заліза трисульфат водний.
Порошок жовтувато-білого кольору, сильно гігро-
скопічний, розкладається на повітрі. Мало розчинний
уводі й 96 % спирті.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла місці.
Заліза(ІІІ) хлорид. ГеС13,6Н2О. (М.м. 270.3). 1037800.
[10025-77-1]. Заліза трихлорид гексагідрат.
Кристалічна маса жовто-оранжевого або коричневого
кольору, яка розпливається на повітрі. Дуже легко роз-
чинний у воді, розчинний у 96 % спирті та ефірі. Під
дією світла заліза(Ш) хлорид і його розчини частково
відновлюються.
Зберігають у повітронепроникному контейнері.
Заліза(ІІІ) хлориду розчин РІ. 1037801.
Розчин 105 г/л.
Заліза(Ш) хлориду розчин Р2. 1037802.
Розчин 13 г/л.
Заліза(ІІІ) хлориду розчин РЗ. 1037803.
2.0 г заліза(ПІ) хлориду Р розчиняють в етанолі Р
і доводять об’єм розчину тим самим розчинником
до 100.0 мл.
Заліза(Ш) хлориду й сульфамінової кислоти реак-
тив. 1037804.
Розчин містить 10 г/л заліза(ПІ) хлориду Р і 16 г/л
кислоти сульфамінової Р.
Заліза(ІІ) амонію сульфат. Ее(1ЧН4)2(8О4)2,6Н2О.
(М.м. 392.2). 1038200. [7783-85-9]. Заліза діамонію ди-
сульфат гексагідрат.
Кристали блідого блакитнувато-зеленуватого кольору
або гранули. Легко розчинний у воді, практично не
розчинний у 96 % спирті.
Зберігають у захищеному від світла місці.
Заліза(ІІ) сульфат. 1038300. [7782-63-0]. Див. статтю
Заліза сульфат.
Заліза(П) сульфату розчин Р2. 1038301.
0.45 г заліза(П) сульфату Р розчиняють у 50 мл
0.1 М розчину кислоти хлористоводневої та дово-
дять об’єм розчину водою, вільною від вуглецю діок-
сиду, Р до 100 мл.
Готують безпосередньо перед використанням.
Заліза саліцилату розчин. 1046700.
0.1 г заліза(III) амонію сульфату Р розчиняють у суміші
2 мл кислоти сірчаної розведеної Р і 48 мл води Р, дово-
дять об’єм розчину водою Р до 100 мл. До одержаного
розчину додають 50 мл розчину 11.5 г/л натрію саліци-
лату Р. 10 мл кислоти оцтової розведеної Р, 80 мл роз-
чину 136 г/л натрію ацетату Р і доводять об’єм розчи-
ну водою Рдо 500 мл.
Готують безпосередньо перед використанням.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла місці.
Залізо. Ге. (А.м. 55.85). 1046600. [7439-89-6].
Порошок сірого кольору або дріт. Розчиняється в роз-
ведених мінеральних кислотах.
Замінник тромбоцитів. 1066400.
До 0.5-1 г фосфоліпідів Р додають 20 мл ацетону Р і
струшують протягом 2 год, потім центрифугують про-
тягом 2 хв і зливають рідину з осаду. Залишок сушать у
вакуумі (водяний насос), додають 20 мл хлороформу Р,
струшують протягом 2 год і фільтрують під вакуумом;
одержаний залишок суспендують у 5-10 мл розчину
9 г/л натрію хлориду Р.
Для використання у кількісному визначенні факто-
ра IX готують розведену суспензію в розчині 9 г/л
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
207
4.1.1. Реактиви
натрію хлориду Ртаким чином, щоб різниця часу коа-
гуляції між послідовними розведеннями суспензій
БСП становила близько 10 с.
Зберігають розведені суспензії при температурі -ЗО °С.
Термін придатності 6 тижнів.
Ізатин. С8Н5МО2. (М.м. 147.1). 1046800. [91-56-5].
1ндолін-2,3-діон.
Дрібні кристали жовтувато-червоного кольору. Ма-
ло розчинний у воді, розчинний у гарячій воді,
96 % спирті й ефірі, розчинний у розчинах гідроксидів
лужних металів з утворенням фіолетового забарвлен-
ня, яке при стоянні переходить у жовте.
Температура плавлення: близько 200 °С, із частковою
сублімацією.
Сульфатна зола (2.4.14). Не більше 0.2 %.
Ізатину реактив. 1046801.
6 мг заліза(ІІІ) сульфату Р розчиняють у 8 мл во-
ди Р, додають при перемішуванні 50 мл кислоти
сірчаної Р; до одержаного розчину додають 6 мг
ізатину Рі перемішують до розчинення.
Розчин має бути світло-жовтого кольору, але не
повинен мати оранжевий або червоний колір.
Ізоаміловий спирт. С5Н12О. (М.м. 88.1). 1046900.
[123-51-3]. З-Метилбутан-1-ол.
Безбарвна рідина. Мало розчинний у воді, змішується
з 96 % спиртом і ефіром.
Температура кипіння, близько 130 °С.
Ізоандростерон. С]9Н30О2. (М.м. 290.4). 1107100.
[481-29-8]. Епіандростерон. 3(3-Гідрокси-5а-андрос-
тан-17-он.
Порошок білого кольору. Практично не розчинний у
воді, розчинний в органічних розчинниках.
Температура плавлення: від 172 °С до 174 °С.
[«]□ : +88°. Визначення проводять, використовуючи
розчин 20 г/л у метанолі Р.
АА (2.2.41)'. 14.24 х 10і. Визначення проводять за до-
вжини хвилі 304 нм, використовуючи розчин 1.25 г/л.
Ізоментол. С,0Н20О. (М.м. 156.3). 1047000. [23283-97-8].
(+)-1зоментол: (15,2/?,5/?)-2-ізопропіл-5-метилцик-
логексанол.
(+)-Ізоментол: суміш рівних частин (15,18,58)- та
(1 /?,25,55)-2-ізопропіл-5-метилциклогексанолу.
Безбарвні кристали. Практично не розчинний у воді,
дуже легко розчинний у 96 % спирті та ефірі.
[«]□: (+)-Ізоментол: близько +24°. Визначення про-
водять, використовуючи розчин 100 г/л у 96 % спир-
ті Р.
Температура кипіння (+)-1зоментолу\ близько 218 °С.
Температура кипіння (+)-1зоментолу: близько 218 °С.
Температура плавлення (+)-Ізоментолу. близько
80 °С.
Температура плавлення (+)-Ізоментолу. близької
53 °С.
(+)-Ізоментон. С10Н18О. (М.м. 154.2). 1047100.
(1/?)-^пс-л-Ментан-3-он. (1/?)-^пс-2-Ізопропіл-5-ме- ,
тилциклогексанон.
Містить різні кількості ментону.
Безбарвна рідина. Дуже мало розчинний у воді, роз-
чинний у 96 % спирті та ефірі.
б/?}) : близько 0.904.
: близько 1.453.
[«]□ : близько +93.2°.
Ізоментон, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28) за умов, зазначених у статті Олія
м'яти перцевої, з використанням (+)-ізоментону як
випробовуваного розчину.
Площа головного піка має бути не менше 80.0 % суми
площ усіх піків
Ізопропіламін. С3Н9Ь1. (М.м. 59.1). 1119800. (75-31-0].
Пропан-2-амін.
Безбарвна, сильно летка, займиста рідина.
: близько 1.374.
Температура кипіння: від 32 °С до 34 °С.
Ізопропілміристат. 1047200. [110-27-0]. Див. статтю
Ізопропілміристат.
4-Ізопропілфенол. С9Н12О. (М.м. 136.2). Ю47300.
[99-89-8].
Містить не менше 98 % С9НІ2О.
Температура кипіння: близько 212 °С
Температура плавлення: від 59 °С до 61 °С.
Імідазол. С3Н4ІМ2. (М.м. 68.1). 1045400 [288-32-4].
Кристалічний порошок білого кольору; розчинний у
воді й 96 % спирті.
Температура плавлення: близько 90 °С.
Імінодибензил.СІ4НІЗК.(Л/.лг. 195 3). 1045500. [494-19-9].
10,11-Дигідродибенз|6,/]азегйн
Кристалічний порошок блідо-жовтого кольору. Прак
тично не розчинний у воді, легко розчинний в ацетоні.
Температура плавлення: близько 106 °С.
Індигокармін. С16Н8М2ІМа2О8$2. (М.м. 466.3). Ю45600.
[860-22-0]. Показник Шульца № 1309. Індекс кольо-
ровості № 73015. Динатрію 3,3'-діоксо-2,2'-бісін-
доліден-5,5-дисульфонат. Е 132.
Звичайно містить натрію хлорид.
Порошок від синього до фіолетово-синього кольору
або гранули синього кольору з мідним блиском
208
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Помірно розчинний у воді, практично не розчинний у
96% спирті. Осаджується з водного розчину натрію
оридом.
Індигокарміну розчин. 1045601.
0.2 г індигокарміну Р розчиняють у суміші 10 мл
кислоти хлористоводневої Р і 990 мл розчину
200 г/л кислоти сірчаної, вільної від азоту, Р.
Розчин має витримувати таке випробування:
10 мл одержаного розчину додають до розчину
1.0 мг калію нітрату Ру 10 мл води Р, негайно до-
дають 20 мл кислоти сірчаної, вільної від азоту, Р і
нагрівають до кипіння. Синє забарвлення розчину
має зникнути протягом 1 хв.
Індигокарміну розчин РІ. 1045602.
4 г індигокарміну Р розчиняють у воді Р, додаючи
воду окремими порціями до об’єму 900 мл, потім
додають 2 мл кислоти сірчаної Р і доводять об’єм
розчину водою Рао 1000 мл.
Встановлення титру. 10.0 мл еталонного розчину
нітрату (100 ррт N0) Р поміщають у конічну
колбу із широким горлом місткістю 100 мл, дода-
ють 10 мл води Р, 0.05 мл розчину індигокарміну РІ
і негайно додають (за один раз, але обережно)
30 мл кислоти сірчаної Р. Одержаний розчин не-
гайно титрують приготовленим розчином індиго-
карміну Р1 до одержання стабільного синього за-
барвлення.
Кількість мілілітрів (п), витрачена на титрування,
відповідає 1 мг МО3.
Індометацин. 1101500. [53-86-1]. Див. статтю Індоме-
тацин.
Індофеноловий синій. С18Н|6]М2О. (М.м. 276.3).
1045700. [132-31-0]. Показник Шульца № 939. Індекс
кольоровості № 49700. А-[4-(Диметиламіно)феніл)]-
1,4-нафтохінонмоноімін.
Порошок фіолетово-чорного кольору. Практично не
розчинний у воді.
Хроматографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель с Р На хроматографічну плас-
тинку наносять 10 мкл розчину 0.1 г/л у метиленхло-
риді Р і хроматографують у цьому ж розчиннику.
Фронт розчинника має пройти не менше 10 см. На
хроматограмі має виявлятись лише одна основна пля-
ма. Допускається пляма на старті.
Іонообмінна смола сильнокислотна. 1085400.
Смола у протонованій формі з групами кислоти суль-
фонової, приєднаними до решітки, що складається з
полістиролу, поперечно зшитого 8 % дивінілбензолу.
Випускають у вигляді гранул кулястої форми; якщо
немає інших зазначень, розмір часток становить від
0.3 мм до 1.2 мм.
Ємність. Від 4.5 ммоль/г до 5 ммоль/г, при вмісті води
від 50 % до 60 %.
Приготування колонки. Якщо немає інших зазначень,
використовують трубку із вплавленим всередину дис-
ком із пористого скла, завдовжки 400 мм, внутрішнім
діаметром 20 мм і висотою заповнення близько
200 мм. Смолу попередньо змішують з водою Р, одер-
жану завись уводять у трубку, не допускаючи утворен-
ня бульбашок повітря між частками. Під час роботи
рідина не має опускатися нижче поверхні смоли.
Якщо смола у протонованій формі, промивають во-
дою Рдоти, поки для нейтралізації 50 мл знадобиться
не більше 0 05 мл 0.1 Мрозчину натрію гідроксиду, ви-
користовуючи як індикатор 0.1 мл розчину метилового
оранжевого Р. Якшо смола у натрієвій формі або є по-
треба в її регенерації, крізь колонку повільно пропус-
кають близько 100 мл суміші рівних об’ємів кислоти
хлористоводневої РІ і води Р, а потім промивають во-
дою Р, як описано вище.
Йод. /045800. [7553-56-2]. Див. статтю Йод.
Йодкрохмальний папір. 1085101.
Смужки фільтрувального паперу занурюють у
100 мл розчину крохмалю, вільного від йоду, Р, який
містить 0 1 г калію йодату Р Надлишок рідини ви-
даляють. Сушать у захищеному від світла місці.
Йоду розчин РІ. 1045801.
До 10.0 мл 0.05 Мрозчину йоду додають 0.6 г калію
йодиду Р і доводять об’єм розчину водою Р до
100.0 мл.
Готують безпосередньо перед використанням.
Йоду розчин Р2. 1045802.
До 10.0 мл 0.05 М розчину йоду додають 0.6 г калію
йодиду Р і доводять об’єм розчину водою Р до
1000.0 мл.
Готують безпосередньо перед використанням.
Йоду розчин РЗ. 1045803.
2.0 мл розчину йоду Р1 доводять водою Рро об’єму
100.0 мл.
Готують безпосередньо перед використанням.
Йоду розчин Р4. 1045806.
14 г йоду Р розчиняють у 100 мл розчину 400 г/л
калію йодиду Р, додають 1 мл кислоти хлористо-
водневої розведеної Р і доводять об’єм розчину во-
дою Рао 1000 мл.
Зберігають у захищеному від світла місці.
Йоду розчин спиртовий. 1045804.
Розчин 10 г/л у 96 % спирті Р.
Зберігають у захищеному від світла місці.
Йоду розчин у хлороформі. 1045805.
Розчин 5 г/л у хлороформі Р.
Зберігають у захищеному від світла місці
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
209
4.1.1. Реактиви
2-Йодбензойна кислота. С7Н5Ю2. (М.м. 248.0).
1046100. [88-67-5].
Кристалічний порошок від білого до світло-жовтого
кольору. Мало розчинна у воді, розчинна в 96 %
спирті.
Температура плавлення: близько 160 °С.
Хроматографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар целюлозу для хроматографії Р254 Р. Налінію
старту хроматографічної пластинки наносять 20 мкл
розчину кислоти 2-йодбензойної, приготованого роз-
чиненням 40 мг у 4 мл 0.1 Мрозчину натрію гідрокси-
ду й розведенням водою Р до об’єму 10 мл. Хромато-
графують, використовуючи як рухому фазу верхній
шар, одержаний при струшуванні суміші розчинників
вода Р-кислота оцтова льодяна Р-толуол Р (20:40:40).
Коли фронт розчинників пройде 12 см, пластинку
переглядають в УФ-світлі за довжини хвилі 254 нм. На
хроматограмі має виявлятись лише одна основна пля-
ма.
Йоду бромід. ІВг. (М.м. 206.8). 1045900. [7789-33-5].
Кристали від синювато-чорного до коричнювато-чор-
ного кольору. Легко розчинний у воді, 96 % спирті,
ефірі й кислоті оцтовій льодяній.
Температура кипіння: близько 116 °С.
Температура плавлення: близько 40 °С.
Зберігають у прохолодному, захищеному від світла місці.
Йоду броміду розчин. 1045901
20 г йоду броміду Р розчиняють у кислоті оцтовій
льодяній Р і доводять об’єм розчину тим самим
розчинником до 1000 мл.
Зберігають у захищеному від світла місці.
Йоду(¥) оксид перекристалізований. І2О5. (М.м. 333.8).
1046000. [12029-98-0]. Дийод пентоксид. Йодний
ангідрид.
Містить не менше 99.5 % І2О5.
Кристалічний порошок білого кольору або гранули від
білого до сірувато-білого кольору. Гігроскопічний, ду-
же легко розчинний у воді з утворенням НІО3.
Стабільність при нагріванні. 2 г, попередньо витри-
мані при температурі 200 °С протягом 1 год, розчиня-
ють у 50 мл води Р. Розчин має бути безбарвним.
Кількісне визначення. 0.100 г йоду(У) оксиду перекрис-
талізованого розчиняють у 50 мл води Р, додають 3 г
калію йодиду Рі 10 мл кислоти хлористоводневої розве-
деної Р. Титрують вивільнений йод 0.1 М розчином
натрію тіосульфату, використовуючи як індикатор
1 мл розчину крохмалю Р.
1 мл 0.1 М розчину натрію тіосульфату відповідає
2.782 мг І2О5.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла місці.
2-Йодгіпурова кислота. С<2НДМО3,2Н2О. (М.м. 341.1).
1046200. [147-58-0]. 2-(2-Иодбензамідо)оцтова кислота
Кристалічний порошок білого або майже білого коль-
ору. Помірно розчинна у воді.
Температура плавлення: близько 170 °С.
Вода (2.5.12). Від 9 % до 13 %. Визначення проводять
із 1.000 г.
Хроматографія. Визначення проводять методом тонко-
шарової хроматографії (2.2.27), використовуючи як
тонкий шар целюлозу для хроматографії Г2І4 Р. На лінію
старту хроматографічної пластинки наносять 20 мкл
розчину кислоти 2-йодгіпурової, приготованого розчи-
ненням 40 мг у 4 мл 0.1 М розчину натрію гідроксиду і
розведенням водою Р до об’єму 10 мл. Хроматографу-
ють, використовуючи як рухому фазу верхній шар, ।
одержаний при струшуванні суміші розчинників во-
да Р-кислота оцтова льодяна Р-толуол Р (20:40:40). Ко- '
ли фронт розчинників пройде 12 см, пластинку пере-
глядають в УФ-світлі за довжини хвилі 254 нм. На хро-
матограмі має виявлятись лише одна основна пляма.
Йодетан. С2Н51. (М.м. 155.9). 1099100. [75-03-6].
Рідина від безбарвного до злегка жовтуватого коль-
ору, під дією повітря й світла темнішає. Змішується з
96 % спиртом і більшістю органічних розчинників.
е/2о : близько 1.95.
: близько 1.513.
Температура кипіння: близько 72 °С.
Зберігають у повітронепроникному контейнері.
Йодистоводнева кислота. НІ. (М.м. 127.9). 1098900.
[10034-85-2].
Кислоту йодистоводневу переганяють над червоним
фосфором, пропускаючи під час перегонки вуглецю
діоксид Р або азот Р. Використовують безбарвну або
майже безбарвну, киплячу при сталій температурі,
суміш (від 55 % до 58 % НІ), яка переганяється при
температурі від 126 °С до 127 °С.
Кислоту поміщають у невеликі флакони зі скла корич-
невого кольору, попередньо продуті вуглецю діокси-
дом Р або азотом Р, зі скляними пробками, гермети-
зують парафіном.
Зберігають у захищеному від світла місці.
Йодна кислота. Н5Ю6. (М.м. 227.9). 1108900.
[10450-60-9].
Кристали. Легко розчинна у воді, розчинна в 96%
спирті.
Температура плавлення: близько 122 °С.
Йодної та оцтової кислоти розчин. 1063000.
0.446 г натрію перйодату Р розчиняють у 2.5 мл розчи-
ну 25 % (об/об) кислоти сірчаної Р і доводять об’єм
розчину кислотою оцтовою льодяною Рдо 100.0 мл.
Йодоцтова кислота. С2Н3ІО2. (М.м. 185.9). 1107000.
[64-69-7].
210
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Безбарвні або білого кольору кристали. Розчинна у
воді й 96 % спирті.
Температура плавлення: від 82 °С до 83 °С.
Йодплатинату реактив. 1046300.
До 3 мі розчину 100 г/л кислоти хлорплатинової Р дода-
ють 97 мл води Р і 100 мл розчину 60 г/л калію йодиду Р.
[ Зберігають у захищеному від світла місці.
І «сірчистий реактив. 1046400.
Пристрій для приготування реактиву, який скла-
I дається з круглодонної колби місткістю 3000-4000 мл із
трьома вхідними отворами для мішалки, термометра й
трубки, заповненої осушувачем, має бути закритим і
сухим у процесі підготовки. У колбу поміщають 700 мл
піридину безводного Р і 700 мл монометалевого ефіру
етиленгліколю Р, додають при постійному перемішу-
ванні 220 г ретельно подрібненого йоду Р, попередньо
висушеного над фосфору(У) оксидом Р. Перемішуван-
ня продовжують до повного розчинення йоду (близько
ЗОхв), потім охолоджують колбу до температури -10 °С
і швидко додають при постійному перемішуванні 190 г
сірки діоксиду Р. Температура реакційної суміші не має
перевищувати 30 °С. Охолоджують.
Встановлення титру. Близько 20 мл метанолу безвод-
ного Р поміщають у посудину для титрування й титру-
ють приготованим йодсірчистим реактивом (2.5.12,
визначення води). Додають точно зважену достатню
кількість води Р і повторюють визначення води. Об-
числюють кількість води, в міліграмах, яка відповідає
1 мл йодсірчистого реактиву.
1 мл йодсірчистого реактиву відповідає як мінімум
3.5 мг води.
Необхідно вжити застережних заходів для запобігання
дії на розчини атмосферної вологи. Титр встановлю-
ють безпосередньо перед використанням.
Зберігають у сухому контейнері.
5-Йодурацил. С4Н3ІМ2О2. (М.м. 238.0). 1046500.
|696-07-1]. 5-Йод-1/7,3/7-піримідин-2,4-діон.
Температура плавлення: близько 276 °С, із розкла-
данням.
Хроматографія. Визначення проводять, як зазначено
в статті Йодоксіуридин, на хроматографічну пластинку
наносять 5 мкл розчину 0.25 г/л; на одержаній хрома-
тограмі має бути лише одна основна пляма.
Кадмій. Ссі. (А.м. 112.4). 1014100. [10108-64-2].
Блискучий метал сріблясто-білого кольору. Практич-
но не розчинний у воді, легко розчинний у кислоті
азотній та гарячій кислоті хлористоводневій.
Казеїн. 1016600. [9000-71-9].
Суміш споріднених фосфопротеїнів, одержаних із мо-
лока.
Аморфний порошок або гранули білого кольору. Дуже
мало розчинний у воді й неполярних органічних роз-
чинниках, розчинний у кислоті хлористоводневій
концентрованій із утворенням блідо-фіолетового за-
барвлення. Утворює солі з кислотами та основами.
Ізоелектрична точка казеїну знаходиться при значенні
рН близько 4.7. Лужні розчини мають ліве обертання
плошини поляризації.
Калію бікарбонат. 1069900. [298-14-6]. Див. Калію
гідрокарбонат Р.
Калію бікарбонату розчин насичений, метанольний.
1069901. Див. Калію гідрокарбонату розчин насиче-
ний, метанольний Р.
Калію бромат. КВгО3. (М.м. 167.0). 1068700. [7758-01-2].
Кристали або гранульований порошок білого кольору.
Розчинний у воді, мало розчинний у 96 % спирті.
Калію бромід. 1068800. [7758-02-3]. Див. статтю Калію
бромід.
Калію бромід, використовуваний в інфрачервоній аб-
сорбційній спектрофотометрії (2.2.24), має витриму-
вати таке додаткове випробування.
ІЧ-спектр диска калію броміду, завтовшки 2 мм, попе-
редньо висушеного при температурі 250 °С протягом
1 год, повинен мати практично рівну базову лінію в
інтервалі довжин хвиль від 4000 см_і до 620 см1. Не по-
винен мати максимумів із поглинанням більше 0.02
над базовою лінією, за винятком максимумів для води
за довжин хвиль 3440 см 1 і 1630 см1.
Калію гідрокарбонат. КНСО3. (М.м. 100.1). 1069900.
[298-14-6]. Калію бікарбонат.
Прозорі, безбарвні кристали. Легко розчинний у воді,
практично не розчинний у 96 % спирті.
Калію гідрокарбонату розчин насичений, метаноль-
ний. 1069901.
0.1 г калію гідрокарбонату Р розчиняють у 0.4 мл
води Р при нагріванні на водяній бані, додають
25 мл метанолу Р і перемішують коловими руха-
ми, продовжуючи нагрівання до розчинення.
Готують безпосередньо перед використанням.
Калію гідроксид. 1070300. [1310-58-3]. Див. статтю
Калію гідроксид.
Калію гідроксиду 2 М розчин спиртовий. 1070301.
12 г калію гідроксиду Р розчиняють у 10 мл води Р і
доводять об’єм розчину 96 % спиртом Рао 100 мл.
Калію гідроксиду 0.5 М розчин спиртовий
(10 %, об/об). 1070302.
28 г калію гідроксиду Р розчиняють у 100 мл
96 % спирту Р і доводять об’єм розчину водою Рао
1000 мл.
Калію гідроксиду розчин спиртовий. Ю70303.
З г калію гідроксиду Р розчиняють у 5 мл води Р і
доводять об’єм розчину 96 % спиртом, вільним від
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
211
4.1.1. Реактиви
альдегідів, Рао 100 мл. Декантують прозорий роз-
чин. Розчин має бути майже безбарвним.
Калію гідроксиду розчин спиртовий РІ. 1070304.
6.6 г калію гідроксиду Р розчиняють у 50 мл води Р
і доводять об’єм розчину етанолом Рдо 1000 мл.
Калію гідросульфат. КН8О4. Калію бісульфат.
(Л/.ж. 136.2). 1070100. [7646-93-7].
Прозорі, безбарвні, гігроскопічні кристали. Легко роз-
чинний у воді з утворенням сильнокислого розчину.
Зберігають у повітронепроникному контейнері.
Калію гідротартрат. С4Н5КО6. (М.м. 188.2). 1070200.
[868-14-4]. Калію гідро(2/?,3/?)-2,3-дигідроксибутан-
1,4-діоат.
Кристалічний порошок білого кольору або безбарвні,
трохи матові кристали. Мало розчинний у воді, розчин-
ний у киплячій воді, дуже мало розчинний у 96 %
спирті.
Калію гідрофталат. С8Н5КО4. (М.м. 204.2). 1070000.
[877-24-7]. Калію гідробензол-1,2-дикарбоксилат.
Кристали білого кольору. Розчинний у воді, мало роз-
чинний у 96 % спирті.
Калію гідрофталату 0.2 М розчин. 1070001.
Розчин калію гідрофталату Р містить 40.84 г, у
перерахунку на С8Н5КО4, у 1000.0 мл.
Калію дигідрофосфат. 1069600. [7778-77-0]. Див. стат-
тю Калію дигідрофосфат.
Калію дигідрофосфату 0.2 М розчин. 1069601.
Розчин калію дигідрофосфату Р містить 27.22 г, у
перерахунку на КН2РО4, у 1000.0 мл.
Калію дихромат. К2Сг2О7. (М.м. 294.2). 1069500.
[7778-50-9]. Дикалію дихромат. Калію біхромат.
Калію дихромат, використовуваний для калібровки
спектрофотометрів (2.2.25), має містити не менше
99.9 % К2Сг2О7, у перерахунку на суху речовину, вису-
шену при температурі 130 °С.
Кристали оранжево-червоного кольору. Розчинний у
воді, практично не розчинний у 96 % спирті.
Кількісне визначення. 1.000 г калію дихромату розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 250.0 мл. 50.0 мл одержаного розчину
поміщають у колбу місткістю 500 мл, додають свіжо-
приготований розчин, який складається з 4 г калію йо-
диду Р,2г натрію гідрокарбонату Р і 6 мл кислоти хло-
ристоводневої Р у 100 мл води Р. Колбу закривають
пробкою, витримують у захищеному від світла місці
протягом 5 хв і титрують 0.1 М розчином натрію
тіосульфату, використовуючи як індикатор 1 мл роз-
чину крохмалю, вільного від йоду, Р.
1 мл 0.1 М розчину натрію тіосульфату відповідає
4.903 мг К2Сг2О7.
Калію дихромату розчин. 1069501.
Розчин 106 г/л.
Калію дихромату розчин РІ. 1069502.
Розчин 5 г/л.
Калію йодат. КІО3. (М.м. 214.0). 1070400. [7758-05-6].
Кристалічний порошок білого кольору. Розчинний у
воді.
Калію йодид. 1070500. [7681-11-0]. Див. статтю Калію
йодид.
Калію йодиду розчин. 1070502.
Розчин 166 г/л.
Калію йодиду йодований розчин. 1070503.
2 г йоду Р і 4 г калію йодиду Р розчиняють у 10 мл
води Р, після повного розчинення доводять об’єм
розчину водою Рао 100 мл.
Калію йодиду насичений розчин. 1070504.
Насичений розчин калію йодиду Ру воді, вільній від
вуглецю діоксиду, Р має містити нерозчинені кри-
стали.
0.5 мл насиченого розчину калію йодиду змішують
із 30 мл суміші хлороформ Р - кислота оцтова Р
(2:3), додають 0.1 мл розчину крохмалю Р; якщо
з’являється синє забарвлення, то воно має зник-
нути при додаванні 0.05 мл 0.1 М розчину натрію
тіосульфату.
Зберігають у захищеному від світла місці.
Калію йодовісмутату розчин. 1070600.
До 0.85 г вісмуту нітрату основного Р додають 40 мл
води Р, 10 мл кислоти оцтової льодяної Р і 20 мл розчи-
ну 400 г/л калію йодиду Р.
Калію йодовісмутату розчин РІ. 1070601.
100 г кислоти винної Ррозчиняють у 400 мл води Р,
додають 8.5 г вісмуту нітрату основного Р, стру-
шують протягом 1 год, додають 200 мл розчину
400 г/л калію йодиду Р та енергійно струшують.
Витримують 24 год і фільтрують.
Зберігають у захищеному від світла місці.
Калію йодовісмутату розчин Р2. 1070602.
Вихідний розчин. Суспендують 1.7 г вісмуту ніт-
рату основного Р і 20 г кислоти винної Р у 40 мл
води Р. До суспензії додають 40 мл розчину 400 г/л
калію йодиду Р, струшують протягом 1 год і
фільтрують.
Термін придатності розчину кілька днів, при
зберіганні у флаконах оранжевого скла.
Розчин для обприскування. Безпосередньо перед
використанням змішують 5 мл вихідного розчину
із 15 мл води Р.
212
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Калію йодовісмутату розчин розведений. 1070603.
100 г кислоти винної Р розчиняють у 500 мл води Р
і додають 50 мл розчину калію йодовісмутату Рі.
Зберігають у захищеному від світла місці.
Калію карбонат. К2СО3. (М.м. 138.2). 1068900. [584-08-7].
Дикалію карбонат.
Гранульований порошок білого кольору, гігро-
скопічний. Дуже легко розчинний у воді, практично
не розчинний в етанолі.
Зберігають у повітронепроникному контейнері.
Калію-натрію тартрат. С4Н4ККаО6,4Н2О. (М.м. 282.2).
1083500. [6381-59-5].
Безбарвні призматичні кристали. Дуже легко розчин-
ний у воді.
Калію нітрат. К1ЧО3. (М.м. 101.1). 1070700. [7757-79-1].
Безбарвні кристали. Дуже легко розчинний у волі.
Каліюперйодат. К1О4. (М.м. 230.0). 1070800. [7790-21-8].
Кристалічний порошок білого кольору або безбарвні
кристали. Розчинний у воді.
Калію фериперйодату розчин. 1070801.
1 V каліюперйодату Ррозчиняють у 5 мл свіжопри-
готованого розчину 120 г/л калію гідроксиду Р, до-
дають 20 мл води Р \ 1.5 мл розчину заліза(ІП) хло-
риду РІ, доводять свіжоприготованим розчином
120 г/л калію гідроксиду Рдо об’єму 50 мл.
Калію перманганат. 1070900. [7722-64-7]. Див. статтю
Калію перманганат.
Калію перманганату розчин у кислоті фосфорній.
1070901.
З г калію перманганату Р розчиняють у суміші
15 мл кислоти фосфорної Р і 70 мл води Р, доводять
об’єм розчину водою Рцо 100 мл.
Калію перманганату розчин. 1070902.
Розчин ЗО г/л.
Калію перренат. ККеО4. (М.м. 289.3). 1071000.
[10466-65-6].
Кристалічний порошок білого кольору. Розчинний у
воді, мало розчинний у 96 % спирті, метанолі й
пропіленгліколі.
Калію персульфат. К282О8. (М.м. 270.3). 1071100.
[7727-21-1]. Дикалію пероксидисульфат.
Безбарвні кристали або кристалічний порошок білого
кольору. Помірно розчинний у воді, практично не роз-
чинний у 96 % спирті. Водні розчини розкладаються
при кімнатній температурі, швидше - при нагріванні.
Зберігають у прохолодному місці.
Калію піроантимонат. К8Ь(ОН)6. (М.м. 262.9). 1071300.
[12208-13-8]. Калію гексагідроксоантимоніат.
Кристали або кристалічний порошок білого кольору.
Помірно розчинний у воді.
Калію піроантимонату розчин. 1071301.
2 г калію піроантимонату ^розчиняють у 95 мл га-
рячої води Р, швидко охолоджують, додають роз-
чин, який містить 2.5 г калію гідроксиду Ру 50 мл
води Р, і 1 мл розчину натрію гідроксиду розведено
го Р. Витримують протягом 24 год, фільтрують і
доводять водою Р до об’єму 150 мл
Калію плюмбіту розчин. 1071200.
1 .7 г свинцю(ІІ) ацетату Р. 3.4 г калію цитрату Рі 50 г
калію гідроксиду Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 100 мл.
Калію сульфат. К28О4. (М.м. 174.3). 1033100. [7778-80-5].
Дикалію сульфат.
Безбарвні кристали. Розчинний у воді.
Калію тартрат. С4Н4К2О6,1/2Н2О. (М.м. 235.3).
1071400. [921-53-9]. Дикалію (2Р,ЗР)-2,3-дигідрокси-
бутан-1,4-дикарбоксилат гемігідрат.
Гранульований порошок або кристали білого кольору.
Дуже легко розчинний у воді, дуже мало розчинний у
96 % спирті.
Калію тетрайодомеркурату розчин. 1071500.
1. 35 г ртуті(П) хлориду Р розчиняють у 50 мл води Р,
додають 5 г калію йодиду Р і доводять об’єм розчину
водою Рро 100 мл.
Калію тетрайодомеркурату лужний розчин. 1071600.
11 г калію йодиду Р і 15 г ртуті(П) йодиду Р розчиня-
ють у воді Р, доводять об’єм розчину тим самим роз-
чинником до 100 мл. Безпосередньо перед викорис-
танням одержаний розчин змішують з розчином
250 г/л натрію гідроксиду Р (1:1).
Калію тетраоксалат. С4Н3КО8,2Н2О. (М.м. 254.2).
1071700. [6100-20-5].
Кристалічний порошок білого кольору. Помірно роз-
чинний у воді, розчинний у киплячій воді, мало роз-
чинний у 96 % спирті.
Калію тіоціанат. К8СК. (М.м. 97.2). 1071800. [333-20-0].
Безбарвні кристали, які розпливаються на повітрі. Ду-
же легко розчинний у воді й 96 % спирті.
Зберігають у повітронепроникному контейнері.
Калію тіоціанату розчин. 1071801.
Розчин 97 г/л.
Калію фериціанід. К3|Ее(С1М)ь]. (М.м. 329.3). 1069700.
[13746-66 2]. Калію гексаціаноферат(ІП).
Кристали червоного кольору. Легко розчинний \ воді.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
213
4.1.1. Реактиви
Калію фериціаніду розчин. 1069701.
Промивають 5 г калію фериціаніду Р невеликою
кількістю води Р, розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 100 мл.
Готують безпосередньо перед використанням.
Калію фероціанід. К4[Ре(С1Ч)6],ЗН2О. (Мм. 422.4).
1069800. [14459-95-1]. Калію гексаціаноферат(П).
Прозорі кристали жовтого кольору. Легко розчинний
у воді, практично не розчинний у 96 % спирті.
Калію фероціаніду розчин. 1069801.
Розчин 53 г/л.
Калію хлорат. КС1О3. (Мм. 122.6). 1069000. [3811-04-9].
Порошок або гранули, або кристали білого кольору.
Розчинний у воді.
Калію хлорид. 1069100. [7447-40-7]. Див. статтю Калію
хлорид.
Калію хлорид, використовуваний для інфрачервоної аб-
сорбційної спектрофотометрії (2.2.24), має задоволь-
няти таку додаткову вимогу.
ІЧ-спектр диска калію хлориду, завтовшки 2 мм,
попередньо висушеного при температурі 250 °С
протягом 1 год, повинен мати практично рів-
ну базову лінію в інтервалі довжин хвиль від
4000 см ' до 620 см '. Не повинен мати максимумів із
поглинанням більше 0.02 над базовою лінією, за ви-
нятком максимумів для води за довжин хвиль
3440 см'1 і 1630 см '.
Калію хлориду 0.1 М розчин. 1069101.
Розчин каліюхлориду Рмістить 7.46 г, у перерахун-
ку на КС1, у 1000.0 мл.
Калію хромат. К2СгО4. (М.м. 194.2). 1069200. [7789-00-6].
Дикалію хромат.
Кристали жовтого кольору. Легко розчинний у воді.
Калію хромату розчин. 1069201.
Розчин 50 г/л.
Калію цитрат. 1069300. [6100-05-6]. Див. статтю Калію
цитрат.
Калію ціанід. КСКІ. (М.м. 65.1). 1069400. [151-50-8].
Кристалічний порошок або маса, або гранули білого
кольору. Легко розчинний у воді, мало розчинний у
96 % спирті.
Калію ціаніду розчин. 1069401.
Розчин 100 г/л.
Кальконкарбонова кислота. С21Н14ЬІ2О78,ЗН2О. (М.м.
492.5). 1015300. [3737-95-9]. 2-Гідрокси-1-(2-гідрокси-4-
сульфо-1-нафтілазо)нафталін-3-карбонова кислота.
Порошок коричнювато-чорного кольору. Мало роз-І
чинна у воді, дуже мало розчинна в ацетоні й 96% І
спирті, помірно розчинна в розведених розчинаї І
натрію гідроксиду.
Кальконкарбонової кислоти індикаторна суміш
1015301.
Змішують одну частину кальконкарбонової кислої І
ти Р із 99 частинами натрію хлориду Р.
Випробування на чутливість. 50 мг індикатор-1
ної суміші кальконкарбонової кислоти розчиняють І
у суміші 2 мл розчину натрію гідроксиду концентро-
ваного Р і 100 мл води Р; з’являється блакитне за-
барвлення, яке має перейти у фіолетове при дода-
ванні 1 мл розчину 10 г/л магнію сульфату Рі 0.1 мл
розчину 1.5 г/л кальцію хлориду Р; при додаванні!
0.15 мл 0.01 М розчину натрію едетату знов з’яв-
ляється блакитне забарвлення.
Кальцію гідроксид. Са(ОН)2. (М.м. 74.1). 1015000.
[1305-62-0]. Кальцію дигідроксид.
Порошок білого кольору. Майже повністю розчинний
у 600 частинах води.
Кальцію гідроксиду розчин. 1015001.
Свіжоприготований насичений розчин.
Кальцію карбонат. 1014500. [471-34-1]. Див. статтю
Кальцію карбонат.
Кальцію карбонат РІ. 1014501.
Має задовольняти вимоги для кальцію карбонату Р
і таку додаткову вимогу.
Хлориди (2.4.4). Не більше 50 ррт.
Кальцію лактат. 1015100. [41372-22-9]. Див. статтю
Кальцію лактат пентагідрат.
Кальцію сульфат. Са$О4,1/2Н2О. (М.м. 145.1). 1015200.
[10034-76-1]. Кальцію сульфат гемігідрат.
Порошок білого кольору. Розчинний приблизно у
1500 частинах води, практично не розчинний у 96%
спирті. При змішуванні з водою, маса якої дорівнює
половині маси кальцію сульфату, порошок швидко
твердіє, перетворюючись на тверду пористу масу.
Кальцію сульфату розчин. 1015201.
5 г кальцію сульфату Р збовтують із 100 мл води Р
протягом 1 год і фільтрують.
Кальцію хлорид. 1014600. [10035-04-8]. Див. статтю
Кальцію хлорид.
Кальцію хлориду розчин. 1014601.
Розчин 73.5 г/л.
Кальцію хлориду 0.01 М розчин. 1014602.
0.147 г кальцію хлориду Р розчиняють у воді Р і до-
водять об’єм розчину тим самим розчинником до
100.0 мл.
214
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Кальцію хлориду 0.02 М розчин. 1014603.
2.94 г кальцію хлориду Р розчиняють у 900 мл во-
ди Р, встановлюють рН розчину в межах від 6.0 до
6.2 і доводять об’єм розчину водою Рдо 1000.0 мл.
Зберігають при температурі від 2 °С до 8 °С.
Кальцію хлорид РІ. СаС12,4Н2О. (М.м. 183.1). 1014700.
Кальцію хлорид тетрагідрат.
Містить не більше 0.05 ррт Ре.
Кальцію хлорид безводний. СаС12. (М.м. 111.0).
1014800. [10043-52-4].
Містить не менше 98.0 % СаС12, у перерахунку на суху
речовину.
Гранули білого кольору, які розпливаються на повітрі.
Дуже легко розчинний у воді, легко розчинний у 96 %
спирті й метанолі.
Втрата в масі при висушуванні (2.2.32). Не більше
5.0%. Визначення проводять у сушильній шафі при
температурі 200 °С.
Зберігають у повітронепроникному контейнері, захи-
щаючи від дії вологи.
Камедь бобів ріжкового дерева. /104500. Подрібнений
ендосперм фруктових кісточок Сегаіопіа мііциа Ь.
ТаиЬ.
Порошок білого кольору, який містить від 70 % до
80 % розчинної у воді смоли, що складається в основ-
ному з галактоманоглікону.
Камфора. 1113000. [76-22-2]. Див. статтю Камфора ра-
цемічна.
Камфора, використовувана в газовій хроматографії,
мас витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хрома-
тографії (2.2.28), як зазначено в статті Олія лавандова.
Випробовуваний розчин 10 г/л розчин випробовуваної
субстанції у гексані Р.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків, за винятком піка гексану.
(15)-(+)10-Камфоросульфонова кислота. С10Н16О43.
(М.м. 232.3). 1104100. [3144-16-9]. (1А,4А)-(+)-2-Ок-
со-10-борненсульфонова кислота. |(15)-7,7-Диметил-
2-оксобіцикло[2.2.1] гептан-1-іл]метансульфонова
кислота. Кислота Рейхлера
Кристали у вигляді призм. Гігроскопічна, розчинна у
воді.
Містить не менше 99.0 % (15)-(+)10-камфоросульфо-
нової кислоти.
Температура плавлення: близько 194 °С, із розкла-
данням.
[а]і, : +20°±1°. Визначення проводять, використову-
ючи розчин 43 г/л у воді Р.
ДА (2.2.41)-. 10.2 х 10’. Визначення проводять за до-
вжини хвилі 290.5 нм, використовуючи розчин 1.0 г/л.
Каолін легкий. 1047400. 11332-58-7].
Очищений природний алюмосилікат гідратований.
Містить підхожий диспергатор.
Легкий порошок білого кольору, який не містить твер-
дих спечених часток, маслянистий на дотик. Практич-
но не розчинний у воді й мінеральних кислотах.
Великі частки. Не більше 0.5 %. 5.0 г каоліну поміща-
ють у циліндр із притертою скляною пробкою за-
вдовжки близько 160 мм і діаметром 35 мм, додають
60 мл розчину 10 г/л натрію пірофосфату Р, енергійно
струшують та відстоюють протягом 5 хв. З допомогою
піпетки відбирають 50 мл рідини на рівні близько 5 см
нижче поверхні й відкидають. До рідини, що залиши-
лася, додають 50 мл води Р, струшують, відстоюють
протягом 5 хв і видаляють 50 мл, як описано вище. Цю
операцію повторюють доти, поки не буде видалено за-
галом 400 мл. Переносять суспензію, яка залишилася,
у фарфорову чашку, випарюють на водяній бані насу-
хо й сушать до постійної маси при температурі від
100 °С до 105 °С. Маса залишку має бути не більше
25 мг.
Дрібні частки 5.0 г каоліну диспергують у 250 мл во-
ди Р при енергійному струшуванні протягом 2 хв і не-
гайно виливають у скляний циліндр діаметром 50 мм
З допомогою піпетки відбирають 20 мл, помішають у
фарфорову чашку, випарюють на водяній бані насухо
й сушать до постійної маси при температурі від 100 °С
до 105 °С. Залишок суспензії відстоюють при темпера-
турі 20 °С протягом 4 год і з допомогою піпетки вида-
ляють 20 мл на рівні точно 5 см нижче поверхні, не
скаламучуючи осаду. Залишок поміщають у фарфоро-
ву чашку, випарюють насухо і сушать до постійної ма-
си при температурі від 100 °С до 105 °С. Маса другого
залишку має бути не менше 70 % від маси першого за-
лишку.
Каприловий спирт. 1024700. Див. Деканол Р.
Карбазол. С12Н9М. (Мм. 167.2). 1015400. [86-74-8].
Дибензопірол.
Кристали. Практично не розчинний у воді, легко роз-
чинний в ацетоні, мало розчинний в етанолі.
Температура плавлення: близько 245 °С.
Карбомер. 1015500. [9007-20-9].
Поперечно-зшитий полімер акрилової кислоти, після
висушування при температурі 80 °С протягом 1 год
містить велику кількість карбоксильних груп (СО2Н,
від 56 % до 68 %).
Середня молекулярна маса близько 3 х 106.
рН (2.2.3). Близько 3. Вимірюють рН суспензії 10 г/л.
Карбофенотіон. СИН16С1О2Р83. (М.м. 342.9). 1016200.
[786-19-6]. О,О-Діетил-5-[[(4-хлорфеніл)тіо]метил]-
фосфордитіоат.
Рідина жовтуватого кольору. Практично не розчинний
у воді, змішується з органічними розчинниками.
<745 : близько 1.27.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
215
4.1.1. Реактиви
Карвакрол. С10Н14О. (М.м. 150.2). 1016400. [499-75-2].
5-Ізопропіл-2-метилфенол.
Рідина коричнюватого кольору. Практично не розчин-
ний у воді, дуже легко розчинний у 96 % спирті й ефірі.
г?2о : близько 0.975.
Ир : близько 1.523.
Температура кипіння: близько 237 °С.
Карвакрол, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено в статті Олія м’яти
перцевої.
Випробовуваний розчин. 0.1 г розчиняють у 10 мл аце-
тону Р.
На хроматограмі випробовуваного розчину площа ос-
новного піка має бути не менше 95.0 % суми площ усіх
піків, за винятком піка ацетону.
Карвон. СІ0Н|4О. (М.м. 150.2). 1016500. [2244-16-8].
/57-и-Мента-6,8-дієн-2-он. (+)-2-Метил-5-(1-метил-
етеніл)циклогекс-2-енон.
Рідина. Практично не розчинний у воді, змішується з
96 % спиртом.
г?20 : близько 0.965.
: близько 1.500.
[а]р : близько +6Г.
Температура кипіння: близько 230 °С.
Карвон, використовуваний в газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено в статті Олія м’яти
перцевої, використовуючи карвон як випробовуваний
розчин.
Площа головного піка має бути не менше 98.0 % суми
площ усіх піків.
Катехін. С15Н14О6,хН2О. (М.м. 290.3 для безводного).
1119000. [154-23-4]. (+)-(2/?,35)-2-(3,4-Дигідрокси-
феніл)-3,4-дигідро-2/7-хромен-3,5,7-триол. Катехол.
Ціаніданол. Ціанідол.
Катіонообмінна смола сильна (кальцієва форма).
1104600.
Смола у кальцієвій формі з групами сульфонової кис-
лоти, приєднаними до решітки полімеру, який скла-
дається із полістиролу поперечно-зшитого 8 % ди-
вінілбензолу. Розмір часток зазначають після назви
реактиву в окремих статтях.
Катіоніт слабоосновний. 1096000. Див. Катіоніт слаб-
кий Р.
Катіоніт слабкий. 1096000.
Смола поліметакрилова, слабо кисла, яка містить кар-
боксильні групи у протонованій формі.
Розмір часток: від 75 мкм до 160 мкм.
Використовувані межі рН: від 5 до 14.
Максимальна температура використання 120 °С.
Катіонообмінна смола. 1016700.
Смола у протонованій формі з групами сульфонової І
кислоти, приєднаними до решітки полімеру, який І
складається з полістиролу поперечно-зшитого 8 Й 1
дивінілбензолу. Випускають у вигляді гранул, розмір]
яких зазначають після назви реактиву у випробуваніІ
нях, в яких він використовується.
Катіонообмінна смола РІ. 1121900.
Смола у протонованій формі з групами сульфоно-
вої кислоти, приєднаними до решітки полімеру,
який складається із полістиролу поперечно-зши- І
того 4 % дивінілбензолу. Випускають у вигляді гра-
нул, розмір яких зазначають після назви реактивуу
випробуваннях, в яких він використовується.
Католіт для ізолектрофокусування рН від 3 до 5 (0.1 М
розчин Д-аланіну). /113100.
8.9 г Р-аланіну Р розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 1000 мл.
Кетостеариловий спирт. 1017500. [67762-27-0]. Див.
статтю Спирт кетостеариловий.
Кисень. О2. (М.м. 32.00). 1108800.
Містить не менше 99.99 % (об/об) О2.
Азот і аргон: не більше 100 ррт.
Вуглецю діоксид: не більше 10 ррт.
Вуглецю монооксид: не більше 5 ррт.
Кислотний синій 83. С45Н441\І31\ІаО782. (М.м. 826).
1012200. [6104-59-2]. Кольоровий індекс № 42660.
Брильянтовий синій Р. Кумасі брильянтовий си-
ній Р 250.
Порошок коричневого кольору. Не розчинний у
холодній воді, мало розчинний у киплячій воді й ета-
нолі, розчинний у кислоті сірчаній, кислоті оцтовій
льодяній і розведених розчинах гідроксидів лужних
металів.
Кумасі фарбувальний розчин. 1012201.
Розчин 1.25 г/л кислотного синього 83 Р у суміші
розчинників кислота оцтова льодяна Р - мета-
нол Р - вода Р( 1:4:5). Фільтрують.
Знебарвлюючий розчин. 1012202.
Суміш розчинників кислота оцтова льодяна Р-ме-
танол Р-вода Р( 1:4:5).
Кислотний синій 90. С47Н48КІ3І\аО782. (М.м. 854).
1001300. [6104-58-1]. Кольоровий індекс № 42655.
Натрій [4-][4-[(4-етоксифеніл)аміно]феніл][|4-(етил)-
(3-сульфонатобензил)аміно]феніл]метилен]циклогек-
са-2,5-дієн-1 -іл ідей] (етил )-(3-сул ьфонато-бен-
зил)амоній.
216
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Порошок темно-коричневого кольору з фіолетовим
ілиском і з украпленими частками, які мають ме-
талічний блиск. Розчинний у воді й етанолі.
Втрата в масі при висушуванні (2.2.32). Не більше
:.О %. 0.500 г сушать у сушильній шафі при темпера-
турі від 100 °С до 105 °С.
А : більше 500, у перерахунку на суху речовину. Ви-
значення проводять за довжини хвилі 577 нм, вико-
ристовуючи розчин 0.01 г/л у буферному розчині
рН 7.0.
Кислотний синій 92. С26Н16М3Ма3О10$3. (М.м. 696).
1001400. [3861-73-2]. Кольоровий індекс № 13390. Ку-
масі блакитний. Аназолен-натрій. Тринатрію 8-гідрок-
си-4'-(феніламіно)азонафталін-3,5',6-трисульфонат.
Кристали темно-синього кольору. Мало розчинний у
96 % спирті, розчинний у воді, ацетоні й моноетило-
вому ефірі етиленгліколю.
Кислотного синього 92 розчин. 1001401.
0.5 г кислотного синього 92 Р розчиняють у суміші
10 мл кислоти оцтової льодяної Р, 45 мл 96 % спир-
ту Р і 45 мл води Р.
ельгурс. 1047600.
Складається з кізельгуру, обробленого кислотою хло-
ристоводневою і кальцинованого додаванням близько
15 % кальцію сульфату гемігідрату.
Дрібний порошок сірувато-білого кольору; при розти-
ранні з водою сірий колір стає більш вираженим. Се-
редній розмір часток від 10 мкм до 4(1 мкм.
Камцію сульфат. Визначення проводять методом,
зазначеним для силікагелю б Р.
рН(2.2.3). Від 7 до 8. Вимірюють рН суспензії, одержа-
ної струшуванням 1 г із 10 мл води, вільної від вуглецю
діоксиду, Р протягом 5 хв.
Хроматографічна розділювальна здатність. Визначен-
ня проводять методом тонкошарової хроматографії
(1.227). Пластинки готують, використовуючи завись
кізельгуру о із розчином 2.7 г/л натрію ацетату Р. На
лінію старту хроматографічної пластинки наносять
5 мкл розчину, який містить по 0.1 г/л лактози, саха-
рози, глюкози й фруктози у піридині Р. Хроматографу-
ютьу системі розчинників вода Р - 2-пропанол Р-ети-
іацетат Р (12:23:65). Час прохождення фронту роз-
чинників на відстань 14 см близько 40 хв. Пластинку
сушать на повітрі, обприскують розчином анісового
альдегіду Р, витрачаючи близько 10 мл, і нагрівають
при температурі від 100 °С до 105 °С протягом 5 хв. На
хроматограмі мають виявлятися чотири чітких, добре
розділених без “хвостів”, плями.
Кізельїур для хроматографії. 1047500.
Легкий порошок білого або жовтувато-білого кольору.
Практично не розчинний у воді, розведених кислотах
і органічних розчинниках.
Швидкість фільтрації. Використовують хромато-
графічну колонку розміром 0.25 м х 10 мм із пластин-
кою з пористого скла (100) і двома позначками на ви-
соті 0.10 м і 0.20 м над пластинкою. Колонку заповню-
ють випробовуваною речовиною до першої позначки,
а до другої позначки заповнюють водою Р. Коли перші
краплі починають витікати з колонки, знов заповню-
ють до другої позначки водою Р і вимірюють час
витікання з колонки перших 5 мл води. Швидкість по-
току має бути не менше 1 мл/хв.
Кольоровість (2.2.2, метод І). Елюат, одержаний при
випробуванні на швидкість фільтрації, має бути без-
барвним.
Кислотність або лужність. До 1.00 г додають 10 мл во-
ди Р, енергійно збовтують і витримують протягом 5 хв.
Суспензію фільтрують крізь фільтр, попередньо про-
митий гарячою водою Рао нейтральної реакції в про-
мивній воді. До 2.0 мл фільтрату додають 0.05 мл роз-
чину метилового червоного Р; розчин повинен мати
жовте забарвлення. До 2.0 мл фільтрату додають
0.05 мл розчину фенолфталеїну Р1\ дозволяється слаб-
ко рожеве забарвлення розчину.
Водорозчинні речовини. 10.0 г поміщають у хрома-
тографічну колонку розміром 0.25 м х 10 мм, елюю-
ють водою Р, збираючи перші 20 мл елюату, випа-
рюють насухо, залишок сушать при температурі
від 100 °С до 105 °С. Маса залишку має бути не більше
10 мг.
Залізо (2.4.9). Не більше 0.02 % (200 ррт). До 0.50 г до-
дають 10 мл суміші рівних об’ємів кислоти хлористо-
водневої РІ і води Р, енергійно струшують, витриму-
ють протягом 5 хв і фільтрують. 1 мл фільтрату має ви-
тримувати випробування на залізо.
Втрата в масі після прожарювання. Не більше 0.5 %.
Під час прожарювання (600 °С) речовина не повинна
мати коричневе або чорне забарвлення.
Клобетазолу пропіонат. С25Н32С1ЕО5. (М.м. 467.0).
1097700. [25122-46-7]. 21-Хлор-9-фтор-1 ір.17-
ди гідрокси-1бр-метилпрегна-1,4-дієн-3,20-діон-17-
пропіонат.
Кристалічний порошок білого кольору. Не розчинний
у воді, розчинний у 96 % спирті й ацетоні.
[а]р : близько +104°. Визначення проводять у діок-
сані.
Температура плавлення: близько 196 °С.
Кобальту нітрат. Со(1МО3)2,6Н2О. (М.м. 291.0). 1021700.
[10026-22-9].
Дрібні кристали гранатового кольору. Дуже легко роз-
чинний у воді.
Кобальту хлорид. СоС12,6Н2О. (М.м. 237.9). 1021600.
[7791-13-1].
Кристалічний порошок червоного кольору або крис-
тали темно-червоного кольору. Дуже легко розчинний
у воді, розчинний у 96 % спирті.
Кодеїн. 1021800. [6059-47-8]. Див. статтю Кодеїн.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
217
4.1.1. Реактиви
Кодеїну фосфат. 1021900. [52-28-8]. Див. статтю Ко-
деїну фосфат гемігідрат.
Конго червоний. С32Н22М6І\Іа2О6$2- (М.м. 697) 1022000.
[573-58-0). Показник Шульца № 360. Кольоровий
індекс № 22120. Динатрію (біфеніл-4,4'-дїіл-біс-2,2’-
азо)біс( 1 -амінонафталін-4-сульфонат).
Порошок коричнювато-червоного кольору. Розчин-
ний у воді.
Конго червоного розчин. 1022001.
0.1 г конго червоного Р розчиняють у суміші 20 мл
96 % спирту Рі води Рі доводять об’єм розчину во-
дою Рао 100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0 2 мл роз-
чину конго червоного і 0.3 мл 0.1 М розчину
натрію гідроксиду, з’являється синє забарвлення,
яке має перейти у рожеве при додаванні не більше
0.3 мл 0.1 М розчину натрію гідроксиду.
Зміна забарвлення. Від синього до рожевого в
інтервалі рН 3.0-5.0.
Конго червоного папір. 1022002.
Смужки фільтрувального паперу занурюють на
кілька хвилин у розчин конго червоного Р. Висушу-
ють.
Коричний альдегід. С9Н8О. (Мм. 132.1). 1020700.
[104-55-2]. З-Фенілпропеналь.
Масляниста рідина від жовтуватого до зеленувато-
жовтого кольору. Мало розчинний у воді, дуже легко
розчинний у 96 % спирті й ефірі
г/20 • від 1.048 до 1.051.
п2$ : близько 1.620.
Зберігають у прохолодному, захищеному від світла місці.
Кортизону ацетат. 1097800. [50-04-4]. Див. статтю Кор-
тизону ацетат.
Кофеїн. 1014400. [58-08-2]. Див. статтю Кофеїн.
Кофейна кислота. С9Н8О4. (Мм. 180.2). 1014300.
[331-39-5]. (£)-3-(3,4-Дипдроксифеніл)пропюнова
кислота.
Кристали або пластинки білого або майже білого ко-
льору. Легко розчинна у гарячій воді й 96 % спирті,
помірно розчинна у холодній воді.
Температура плавлення: близько 225 °С, із розкла-
данням.
Свіжоприготований розчин з рН 7.6 має два максимуми
поглинання (2.2.25) за довжин хвиль 293 нм і 329 нм.
Крезол. С7Н8О (М.м. 108.1). 1022700. [95-48-7]. о-Кре-
зол. 2-Метилфенол.
Кристали або переохолоджена рідина, яка темнішає
на світлі й повітрі. Змішується з етанолом і ефіром,
розчинний приблизно у 50 частинах води й розминав
гідроксидів лужних металів.
<72() : близько 1.05.
: від 1.540 до 1.550.
Температура кипіння: близько 190 °С.
Температура тверднення (2.2.18). Не нижче 30.5 °С. І
Залишок після випарювання. Не більше 0.1 % (м/м> І
Випарюють на водяній бані й сушать у сушильнім І
шафі при температурі від 100 °С до 105 °С.
Зберігають у захищеному від кисню, світла й вологи І
місці, перед використанням переганяють.
Крезоловий червоний. С2|Н18О5Б. (М.м. 382.4),
1022800 [1733-12-6]. Крезолсульфонфталеїн
4,4’-(3/7-2,1-Бензоксатіол-3-іліден)біс(2-метилфе І
нол)8,8-діоксид.
Кристалічний порошок червонувато-коричневого ко- І
льору. Мало розчинний у волі, розчинний у 96 % спирті
й розведених розчинах гідроксидів лужних металів.
Крезолового червоного розчин. 1022801.
0.1 г крезолового червоного Р розчиняють в суміші
2.65 мл 0.1 М розчину натрію гідроксиду й 20 мл
96 % спирту Р, доводять об’єм розчину водою /'до
100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0.1 мл роз-
чину крезолового червоного і 0.15 мл 0.02 Мрозчи-
ну натрію гідроксиду, з’являється пурпурно-чер-
воне забарвлення, яке має перейти у жовте при до-
даванні не більше 0.15 мл 0.02 М розчину кислоти
хлористоводневої.
Зміна забарвлення. Від жовтого до червоного в
інтервалі рН 7.0-8.6.
лг-Крезоловий пурпуровий. С21Н18О58. (М.м. 382.44).
1121700. [2303-01-7]. м-Крезолсульфонфталеїн.
Кристалічний порошок оливково-зеленого кольору
Мало розчинний у воді, розчинний у 96 % спирті, кис-
лоті оцтовій льодяній та метанолі.
м-Крезолового пурпурового розчин. 1121701.
0.1 г м-крезолового пурпурового Р розчиняють у
13 мл 0.01 Мрозчину натрію гідроксиду Р, доводять
об’єм розчину водою Рдо 100 мл і перемішують.
Зміна забарвлення. Від червоного до жовтого в
інтервалі рН 1.2-2.8.
Від жовтого до фіолетового в інтервалі рН 7.4-9.0.
Кремневольфрамова кислота. Н48і\УІ2О40,хН2О.
1078000. [11130-20-4].
Кристали білого або жовтувато-білого кольору, які
розпливаються на повітрі. Дуже легко розчинна у воді
й 96 % спирті.
Зберігають у повітронепроникному контейнері.
218
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Кристалічний фіолетовий. С25Н30С11М3. (М.м. 408.0).
1022900. [548-62-9]. Показник Шульца№ 78. Кольоро-
вий індекс № 42555. Гексаметилпарарозаніліну хлорид.
Кристали або порошок темно-зеленого кольору. Роз-
чинний у воді й 96 % спирті.
Кристалічного фіолетового розчин. 1022901.
0.5 г кристалічного фіолетового Р розчиняють у
кислоті оцтовій безводній Р і доводять об’єм роз-
чину тим самим розчинником до 100 мл.
Випробування на чутливість. До 50 мл кислоти оц-
тової безводної Р додають 0.1 мл розчину крис-
талічного фіолетового; з’являється блакитнувато-
фіолетове забарвлення, яке має перейти у блакит-
нувато-зелене при додаванні 0.1 мл 0.1 М розчину
кислоти хлорної.
Крохмаль розчинний. 1085100. [9005-84-9].
Порошок білого кольору.
Готують розчин 20 г/л у гарячій воді Р. Розчин повинен
мати слабку опалесценцію і залишатись рідким при
охолодженні.
Крохмалю розчин. 1085103.
1.0 г крохмалю розчинного Р розтирають на поро-
шок із 5 мл води Р, суміш повільно при постійному
перемішуванні вливають у 100 мл киплячої води Р,
яка містить 10 мг ртуті(ІІ) йодиду Р.
При використанні реактиву кожного разу прово-
дять випробування на чутливість.
Випробування на чутливість. Суміш, яка скла-
дається із 1 мл розчину крохмалю, 20 мл води Р,
близько 50 мг калію йодиду Рі 0.05 мл розчину йо-
ду РІ, повинна мати синє забарвлення.
Крохмалю розчин РІ. 1085105.
1 г крохмалю розчинного Р змішують із невеликою
кількістю холодної води Р. Одержану суміш дода-
ютьдо 200 мл киплячої води Р, додають 250 мг кис-
лоти саліцилової Р, кип’ятять протягом 3 хв і не-
гайно охолоджують.
Термін придатності від 2 до 3 тижнів при збері-
ганні розчину при температурі від 4 °С до 10 °С.
Свіжий розчин крохмалю готують у тому випадку,
коли в точці еквівалентності перехід забарвлення
від синього до безбарвного не різкий.
Випробування на чутливість. До 2 мл розчину крох-
малю РІ додають 20 мл води Р, близько 50 мг калію
йодиду Рі 0.05 мл розчину йоду Р1\ одержаний роз-
чин повинен мати синє забарвлення.
Крохмалю розчин, вільний від йоду. 1085104.
Готують розчин, як зазначено для розчину крохма-
лю Р, але без ртуті(П) йодиду.
Готують безпосередньо перед використанням.
Крохмалю розчин із калію йодидом. 1070501.
0.75 г калію йодиду Р розчиняють у 100 мл води Р,
нагрівають до кипіння і додають при перемішу-
ванні розчин 0.5 г крохмалю розчинного Р у 35 мл
води Р. Кип’ятять протягом 2 хв і охолоджують.
Випробування на чутливість. Суміш, яка скла-
дається із 15 мл розчину крохмалю із калію йоди-
дом, 0.05 мл кислоти оцтової льодяної Р і 0.3 мл
розчину йоду Р2, повинна мати синє забарвлення.
Ксантгідрол. С13Н10О2. (М.м. 198.2). 1096100. [90-46-0].
9-Ксантенол.
Містить не менше 90.0 % С13Н10О2.
Порошок від білого до світло-жовтого кольору. Дуже
мало розчинний у воді, розчинний у 96 % спирті, ефірі
й кислоті оцтовій льодяній.
Доступний також у вигляді розчину, який містить від
90 г/л до 110 г/л ксантгідролу в метанолі Р.
Температура плавлення: близько 123 °С.
Кількісне визначення. 0.300 г ксантгідролу поміщають у
колбу місткістю 250 мл, розчиняють у 3 мл метанолу Р
або використовують 3.0 мл розчину. Додають 50 мл
кислоти оцтової льодяної Р і по краплях при струшу-
ванні 25 мл розчину 20 г/л сечовини Р. Відстоюють
12 год, потім фільтрують крізь скляний фільтр (16).
Осад на фільтрі промивають 20 мл 96 % спирту Р, су-
шать у сушильній шафі при температурі від 100 °С до
105 °С і зважують.
1 г осаду відповідає 0.9429 г ксантгідролу.
Зберігають у захищеному від світла місці. Якщо вико-
ристовують метанольний розчин, то зберігають у не-
великих герметично закритих ампулах і якщо не-
обхідно, перед використанням фільтрують.
Ксантгідрол РІ. 1096101.
Має задовольняти вимоги для ксантгідролу Р і та-
ку додаткову вимогу.
Містить не менше 98.0 % С13Н10О2.
Ксантгідролу розчин. 1096102.
До 100 мл кислоти оцтової безводної Р додають
0.1 мл розчину 100 г/л ксантгідролу Р у мета-
нолі Р, 1 мл кислоти хлористоводневої Р і витриму-
ють 24 год.
Ксиленоловий оранжевий. С31Н2ХМ2№4О138. (Мм. 761).
1096300. [3618-43-7]. Тетранатрію 3,3'-(ЗЯ-2,1-бен-
зоксатіол-3-іліден)біс[(6-гідрокси-5-метил-3,1-фені-
лен)метиленімінобісацетат]5,5-діоксид.
Кристалічний порошок червонувато-коричневого ко-
льору. Розчинний у воді.
Ксиленолового оранжевого індикаторна суміш.
1096301.
Розтирають на порошок 1 частину ксиленолового
оранжевого Р із 99 частинами калію нітрату Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
219
4.1.1. Реактиви
Випробування на чутливість. До 50 мл води Рдода-
ють 1 мл кислоти оцтової розведеної Р, 50 мг інди-
каторної суміші ксиленолового оранжевого й
0.05 мл розчину свинцю(ІІ) нітрату Р. Додають
гексаметилентетрамін Рдоти, поки забарвлення
розчину не зміниться від жовтого до фіолетово-
червоного; після додавання 0.1 мл 0 1 М розчину
натрію едетату забарвлення розчину має зміни-
тися на жовте.
Ксилоза. С5Н10О5. (М.м. 150.1). 1096400. [58-86-6].
В- Ксилоза.
Кристалічний порошок білого кольору або безбарвні
голчасті кристали. Дуже легко розчинна у воді, роз-
чинна у гарячому 96 % спирті.
[а]р : близько +20°. Визначення проводять, викорис-
товуючи розчин 100 г/л, через 10 год після його приго-
тування.
Ксилол. С8Н10. (М.м. 106.2). 1096200. [1330-20-7].
Суміш ізомерів. Прозора, безбарвна, займиста ріди-
на. Практично не розчинний у воді, змішується з
96 % спиртом і ефіром
<У2о : близько 0.867.
п™ : близько 1.497.
Температура кипіння: близько 138 °С.
о-Ксилол. С8Н10. (Мм. 106.2). //00600. [95-47-6].
1,2-Диметилбензол.
Прозора, безбарвна, займиста рідина. Практично не
розчинний у воді, змішується з 96 % спиртом та
ефіром.
<72® : близько 0.881.
: близько 1.505.
Температура кипіння: близько 144 °С.
Температура плавлення: близько -25 °С.
м-Ксилол. С8Н)0. (М.м. 106.2). 1117700. [108-38-3].
1,3-Диметил бензол.
Прозора, безбарвна, займиста рідина. Практично не
розчинний у воді, змішується з 96 % спиртом та ефіром.
<72о : близько 0.884
: близько 1.497.
Температура кипіння: близько 139 °С.
Температура плавлення: близько -47 °С.
Кукурудзяна олія. 1050400.
Жирна олія, одержана із зародків 7еа тауя £. вичав-
люванням або екстракцією.
Прозора рідина від світло-жовтого до золотисто-жов-
того кольору. Практично не розчинна в 96 % спирті,
змішується з ефіром і петролейним ефіром.
Йодне число (2.5.4). Від 103 до 128.
Перекисне число (2.5.5). Не більше 5.
Число омилювання (2.5.6). Від 187 до 195.
Ідентифікація. Визначення проводять методом тон-
кошарової хроматографії (2.3.2). Одержана хроматог-
рама має співпадати з хроматограмою для кукурудзя-
ної олії, наведеної на Рис. 2.3.2.-1.
Кумасі синій. 1001400. [3861-73-2]. Див. Кислотний
синій 92 Р.
Кумасі синього розчин. 1001401. Див. Кислотного
синього 92 розчин Р.
Куркумін. С21Н20О6. (М.м. 368.4). 1023500. [458-37-7].
1,7-Біс(4-гідрокси-3-метоксифеніл)гепта-1,6-дієн-
3,5-дюн.
Кристалічний порошок оранжево-коричневого кольо-
ру. Практично не розчинний у воді, розчинний у кис-
лоті оцтовій льодяній, практично не розчинний в ефірі.
Температура плавлення: близько 183 °С.
Лавандолол. С10Н18О. (М.м. 154.2). 1114100. [6544-40-7].
2-1зопропеніл-5-мстилгекс-4-ен- 1-ол.
Масляниста рідина з характерним запахом.
г/2о : близько 0.875.
: близько 1.407.
| «]□ : близько -10.2°.
Температура кипіння13: близько 94 °С.
Лавандолол, використовуваний в газовій хромато-
графії, має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28) за умов, зазначених у статті Олія
лавандова.
Випробовуваний розчин. Випробовувана субстанція.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків.
Лавандололу ацетат. СІ2Н20О2. (М.м. 196.3). 1114200.
[50373-59-6]. 2-1зопропеніл-5-метилгекс-4-ен-І-ілу
ацетат.
Безбарвна рідина з характерним запахом.
<У2о : близько 0.911.
И0 : близько 1.454.
Температура кипіння І3: від 106 °С до 107 °С.
Лавандололу ацетат, використовуваний в газовій хро-
матографії, має витримувати таке додаткове випро-
бування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28) за умов, зазначених у статті Ой-
лавандова.
Випробовуваний розчин. Випробовувана субстанція.
Площа основного піка має бути не менше 93.0 % суми
площ усіх піків.
220
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
І Лакмус. 1049300. 11393-92-6]. Показник Шульца
| V 1386.
Фрагменти синьо-фіолетового кольору, одержані з різ-
них видів Косеїіа, Ресапога або інших лишайників. Роз-
чинний у волі, практично не розчинний у 96 % спирті.
Зміна забарвлення. Від червоного до синього в інтер-
валі рН 5-8.
Лакмусовий папір синій. 1049301.
10 частин грубо здрібненого лакмусу Р кип’ятять із
100 частинами 96 % спирту Р протягом І год.
Спирт декантують, до залишку додають суміш із
45 частин 96 % спирту Р і 55 частин води Р. Через
2 дні прозору рідину декантують, просочують
смужки фільтрувального паперу й сушать.
Випробування на чутливість. Смужку фільтруваль-
ного паперу розміром 10 х 60 мм занурюють у
суміш 10 мл 0.02 М розчину кислоти хлористовод-
невої та 90 мл води Р. При струшуванні папір має
набути червоного забарвлення протягом 45 с.
Лакмусовий папір червоний. 1049302.
До синього екстракту лакмусу додають по краплях
кислоту хлористоводневу розведену Р до переходу
синього забарвлення у червоне. Смужки фільтру-
вального паперу просочують одержаним розчи-
ном і сушать.
Випробування на чутливість. Смужку фільтруваль-
ного паперу розміром 10 х 60 мм занурюють у
суміш 10 мл 0.02 М розчину натрію гідроксиду й
90 мл води Р. При струшуванні папір має набути
синього забарвлення протягом 45 с.
Лактобіонова кислота. С12Н22О12. (М.м. 358.3).
1101600. [96-82-2].
Кристалічний порошок білого кольору. Легко розчин-
на у воді, практично не розчинна в 96 % спирті.
Температура плавлення' близько 115 °С.
Лактоза. 1047900. [5989-81-1]. Див. статтю Лактоза.
Лантану(ПІ) нітрат. Еа(КІО3)з,6Н2О. (М.м. 433.0).
1048000. [10277-43-7]. Лантану тринітрат гексагідрат
Безбарвні кристали, які розпливаються на повітрі.
Легко розчинний у воді.
Зберігають у повітронепроникному контейнері.
Лантану(ІІІ) нітрату розчин. 1048001.
Розчин 50 г/л.
Лантану(ІІІ) оксид. Ба2О3. (М.м. 325.8). 1114000.
[1312-81-8]. Лантану триоксид.
Аморфний порошок майже білого кольору. Практич-
но не розчинний у воді, розчинний у розведених міне-
ральних кислотах, поглинає вуглецю діоксид із
повітря.
Кальцій. Не більше 5 ррт.
Л антану(Ш) хлориду розчин. / /14001.
До 58.65 г лантану(ІІІ) оксиду Р повільно додають
100 мл кислоти хлористоводневої Р, нагрівають до
кипіння, охолоджують і доводять об’єм розчину во-
дою Рао 1000.0 мл.
Лауриловий спирт. СІ2Н26О. (М.м. 186.3). 1119900.
1-Додеканол.
г/2о : близько 0.820.
Температура кипіння: від 24 °С до 27 °С.
Лейцин. 1048500. [61-90-5]. Див. статтю Лейцин.
Лимонен. СІ0Н|6. (М.м. 136.2). 1048600. [5989-27-5].
О-Лимонен. (+)-л-Мента-1,8-дієн. (А)-4-Ізопро-
пеніл-1 -метил циклогекс-1 -єн.
Безбарвна рідина. Практично не розчинний у воді,
розчинний у 96 % спирті.
г/2о : близько 0.84.
По : від 1.471 до 1.474.
| а] □ : від +96° до +106°.
Температура кипіння: від 175 °С до 177 °С.
Лимонен, використовуваний в газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія м’яти
перцевої, використовуючи лимонен як випробовува-
ний розчин.
Площа основного піка має бути не менше 99.0 % суми
площ усіх піків.
Лимонна кислота. 1021000. [5949-29-1]. Див. статтю
Кислота лимонна моногідрат.
При використанні у випробуванні на залізо кислота ли-
монна має витримувати таке додаткове випробування.
0.5 г кислоти лимонної розчиняють у 10 мл води Р, до-
дають 0.1 мл кислоти тіогліколевої Р, перемішують,
додають розчин аміаку Рао лужної реакції та доводять
об’єм одержаного розчину водою Рао 20 мл. Розчин не
має забарвлюватись у рожевий колір.
Лимонна кислота безводна. 1021200. [77-92-9]. Див.
статтю Кислота лимонна безводна.
Лимонна олія. 1!01700. Див. статтю Олія лимонна.
Линалілу ацетат. СІ2Н20О2. (М.м. 196.3). 1107200.
[115-95-7]. (Я5)-1,5-Диметил-1-вінілгекс-4-еніл аце-
тат.
Безбарвна або злегка жовта рідина із сильним запахом
бергамоту й лаванди.
с/25 : від 0.895 до 0.912.
: від 1.448 до 1.451.
Температура кипіння: близько 215 °С.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
221
4.1.1. Реактиви
Линалілу ацетат, використовуваний в газовій хрома-
тографії, має витримувати таке додаткове випробу-
вання.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено в статті Олія квіток
померанцю, використовуючи линалілу ацетат як ви-
пробовуваний розчин.
Площа основного піка має бути не менше 95.0 % суми
площ усіх піків.
Линалол. С10Н18О. (М.м. 154.2). 1048700. [78-70-6].
(Р5)-3,7-Диметилокта-1,6-дієн-3-ол
Суміш двох стереоізомерів (ликареолу й коріандролу)
Рідина. Практично не розчинний у воді, розчинний в
ефірі.
</2о : близько 0.860.
: близько 1.462.
Температура кипіння: близько 200 °С.
Линалол, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія анісова.
використовуючи линалол як випробовуваний розчин.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків.
Літій. □. (А.м. 6.94). 1048800. [7439-93-2].
М’який метал, на свіжому зрізі сріблясто-сірого ко-
льору, при контакті з повітрям швидко стає тьмяним.
Бурхливо реагує з водою з утворенням водню й розчи-
ну літію гідроксиду; розчинний у метанолі з утворен-
ням водню й розчину літію метоксиду; практично не
розчинний в ефірі й петролейному ефірі.
Зберігають під петролейним ефіром або рідким па-
рафіном.
Літію гідроксид. І_іОН,Н2О. (М.м. 41.96). 1049100.
[1310-66-3]. Літію гідроксид моногідрат.
Гранульований порошок білого кольору. Є сильним
лугом, швидко поглинає воду й вуглецю діоксид, роз-
чинний у воді, помірно розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Літію карбонат. Бі2СО3. (М.м. 73.9). 1048900.1554-13-2].
Дилітію карбонат.
Легкий порошок білого кольору. Помірно розчинний у
воді, дуже мало розчинний у 96 % спирті. Насичений роз-
чин при температурі 20 °С містить близько 13 г/л Бі2СО3.
Літію метаборат безводний. 1_іВО2. (М.м. 49.75).
1120000. [13453-69-5].
Літію сульфат. Бі28О4,Н2О. (М.м. 128.0). 1049200.
[10102-25-7]. Дилітію сульфат моногідрат.
Безбарвні кристали. Легко розчинний у воді, практич-
но не розчинний у 96 % спирті.
Літію хлорид. БіСІ. (М.м. 42.39). 1049000. [7447-41-8]. І
Кристалічний порошок або гранули, або кубічні кри- І
стали; розпливається на повітрі, легко розчинний і
воді, розчинний в ацетоні й 96 % спирті. Водні розчи- '
ни мають нейтральну або слабку лужну реакцію.
Зберігають у повітронепроникному контейнері.
Магній. М§. (А.м. 24.30). 1049500. [7439-95-4].
Стрічка, або стружка, або дріт сріблясто-білого кольо-
ру, або порошок сірого кольору.
Магнію ацетат. С4Н6МеО4,4Н2О (М.м. 214.5). 1049600
[ 16674-78-5]. Магнію діацетат тетрагідрат.
Безбарвні кристали, які розпливаються на повітрі.
Легко розчинний у воді й 96 % спирті.
Зберігають у повітронепроникному контейнері.
Магнію нітрат. Ме(МО3)2,6Н2О. (М.м. 256.4). 1049800.
[13446-18-9]. Магнію нітрат гексагідрат.
Безбарвні прозорі кристали, які розпливаються на
повітрі. Дуже легко розчинний у воді, легко розчин-
ний у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Магнію нітрату розчин. 1049801.
17.3 г магнію нітрату Р розчиняють при обереж-
ному нагріванні у 5 мл води Р, додають 80 мл
96 % спирту Р, охолоджують і доводять об’єм роз-
чину тим самим розчинником до 100.0 мл.
Магнію оксид. 1049900. |1309-48-4|. Див. статтю
Магнію оксид легкий.
Магнію оксид РІ. 1049901.
Має задовольняти вимоги для магнію оксиду Різ
такими змінами.
Арсен (2.4.2, метод А). Не більше 0.0002 % (2 ррт)
0.5 г магнію оксиду розчиняють в суміші 5 мл во-
ди Р і 5 мл кислоти хлористоводневої РІ. Одержа-
ний розчин має витримувати випробування на ар-
сен.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 0.75 г магнію оксиду розчиняють у сумі-
ші 3 мл води Р і 7 мл кислоти хлористоводневої РІ,
додають 0.05 мл розчину фенолфталеїну Р і розчину
аміаку концентрованого Рао одержання рожевого
забарвлення. Надлишок аміаку нейтралізу-
ють кислотою оцтовою льодяною Р, додають
0.5 мл надлишку кислоти й доводять водою Р до
об’єму 15 мл і фільтрують, якщо необхідно. 12 мл
розчину мають витримувати випробування на
важкі метали. Еталон готують, використовуючи
5 мл еталонного розчину свинцю (1 ррт РЬ) Рі 5 мл
води Р.
Залізо (2.4.9). Не більше 0.005 % (50 ррт). 0.2 г
магнію оксиду розчиняють у 6 мл кислоти хлорис-
товодневої розведеної Р і доводять об’єм розчину
водою Рао 10 мл. Одержаний розчин має витриму-
вати випробування на залізо.
222
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Магнію оксид важкий. 1050000. [1309-48-4]. Див. стат-
но Магнію оксид важкий.
Магнію сульфат. 1050200. [10034-99-8]. Див. статтю
Магнію сульфат.
Магнію хлорид. 1049700. [7791-18-6]. Див. статтю
Магнію хлорид гексагідрат.
Макрогол 200. 1099200. [25322-68-3]. Поліетилен-
чіколь 200.
Прозора, безбарвна або майже безбарвна, в’язка ріди-
на. Легко розчинний в ацетоні та етанолі, практично
не розчинний в ефірі й жирних оліях
: близько 1.127.
4 : близько 1.450.
Макрогол 200 РІ. 1099201.
500 мл макроголу 200 Р помішають у круглодонну
колбу місткістю 1000 мл, відганяють леткі речови-
ни при температурі 60 °С протягом 6 год, викори-
стовуючи ротаційний випарник і вакуум від
1.5 кПа до 2.5 кПа.
Макрогол 300. 1067100. [25322-68-3]. Поліетилен-
гліколь 300. Див. статтю Макроголи
Макрогол 400. 1067200. [25322-68-3]. Поліетилен-
г.ііколь 400. Див. статтю Макроголи.
Макрогол 1000. 1067300. [25322-68-3]. Поліетилен-
гліколь 1000. Див. статтю Макроголи.
Макрогол 1500. 1067400. [25322-68-3]. Поліетилен-
гліколь 1500. Див. статтю Макроголи.
Макрогол 20 000. 1067600. Поліетиленгліколь 20 000.
Див. статтю Макроголи.
Макрогол 20 000 2-нітротерсфталат. 1067601.
Поліетиленгліколь 20 000 2 нітрозерефталат.
Макрогол 20 000 Р модифікований обробкою кис-
лотою 2-нітротерефталевою.
Тверда воскоподібна маса білого або майже білого
кольору. Розчинний в ацетоні.
Малахітовий зелений. С23Н25С1М2. (М.м. 364.9).
1050500. [123333-61 9] Показник Шульца№ 754. Ко-
льоровий індекс № 42000. [4-[[4-(Диметиламіно)-
феніл (феніл метилен | циклогекса-2,5-дієн-І-іліден]-
диметиламонію хлорид.
Кристали зеленого кольору з металічним блиском.
Дуже легко розчинний у воді з утворенням розчину
синювато-зеленого кольору, розчинний у 96 % спирті
й метанолі.
Розчин 0 01 г/л у 96 % спирті Рмає максимум погли-
нання (2.2 25) за довжини хвилі 617 нм.
Малахітового зеленого розчин. 1050501.
Розчин 5 г/л у кислоті оцтовій безводній Р.
Малеїнова кислота. 1050600. [110 16-7|. Див. статтю
Кислота малеїнова.
Малеїновий ангідрид. С4Н2О3. (М.м. 98.1). 1050700.
[108-31-6]. Бутендіоновий ангідрид. 2,5-Фурандюн.
Кристали білого кольору. Розчинний у воді з утворен-
ням кислоти малеїнової, дуже легко розчинний в аце-
тоні та етилацетаті, легко розчинний у толуолі, роз-
чинний у 96 % спирті з утворенням ефіру, дуже мало
розчинний у петролейному ефірі.
Температура плавлення: близько 52 °С.
Будь-який залишок, не розчинний у толуолі, не має
перевищувати 5 % (кислота малеїнова).
Малеїнового ангідриду розчин. 1050701.
5 г малеїнового ангідриду Ррозчиняють у толуолі Р
і доводять об’єм розчину тим самим розчинником
до 100 мл.
Термін придатності 1 міс; розчин фільтрують у ви-
падку помутніння.
Маніт. 1051000. [69-65-8]. Див. статтю Маніт.
Маноза. С6НІ2О6. (М.м. 180.2). 1051100. [3458-28-4].
О-(+)-Маноза.
Кристалічний або дрібнокристалічний порошок біло-
го кольору. Дуже легко розчинна у воді, мало розчин-
на в етанолі.
[а|о : від + 13.7° до +14.7°. Визначення проводять, ви-
користовуючи розчин 200 г/л у воді Р, яка містить
близько 0.05 % МН3.
Температура плавлення: близько 132 °С, із розкла-
данням.
Марганцю(П) сульфат. Мп8О4,Н2О. (М.м. 169.0).
1050900. [10034-96-5]. Марганцю сульфат моногідрат.
Кристалічний порошок або кристали блідо-рожевого
кольору. Легко розчинний у воді, практично не роз-
чинний у 96 % спирті.
Втрата в масі після прожарювання. Від 10.0 % до
12.0 %. Визначення проводять із 1.000 г при темпера-
турі 500 °С.
Масляна кислота. С4Н8О2. (М.м. 88.1). 1014000.
[107-92-6]. Бутанова кислота.
Містить не менше 99.0 % С4Н8О2.
Масляниста рідина. Змішується з водою, 96 % спир-
том.
г/2о ' близько 0.96
: близько 1.398.
Температура кипіння: близько 163 °С
Мезитилоксид. С6Н)()О (М.м. 98.1). 1120100. [141-79-7].
Метилпент-З-ен-2-он.
Безбарвна масляниста рідина. Розчинний у 30 частинах
води, змішується з більшістю органічних розчинників.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
223
4.1.1. Реактиви
(/20 близько 0.858.
Температура кипіння: від 129 °С до 130 °С.
Меклозину гідрохлорид. 1051200. [1104-22-9]. Див.
статтю Меклозину гідрохлорид.
Меламін. С3Н61Ч6. (М.м. 126.1). 1051300. [108-78-1].
1,3,5-Триазин-2,4,6-триамін.
Аморфний порошок білого кольору. Дуже мало роз-
чинний у воді й 96 % спирті.
Менадіон. 1051400. [58-27-5]. Див. статтю Менадіон.
Ментилацетат. С|2Н22О2. {М.м. 198.3). 1051800.
[16409-45-3]. 2-1зопропіл-5-метилциклогексилацетат.
Безбарвна рідина. Мало розчинний у воді, змішується
з 96 % спиртом і ефіром.
г/2о : близько 0.92
: близько 1.447.
Температура кипіння: близько 225 °С.
Ментилацетат, використовуваний у газовій хромато-
графії, має витримувати таке додаткове випробуван-
ня.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія м ’яти
перцевої, використовуючи ментилацетат як випробо-
вуваний розчин.
Площа основного піка має бути не менше 98 0 % від
суми площ усіх піків.
Ментол. 1051600. [2216-51-5]. Див. статті Левоментол
та Ментол рацемічний.
Ментол, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Ментол ра-
цемічний у випробуванні «Супровідні домішки», вико-
ристовуючи ментол як випробовуваний розчин.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків, за винятком піка розчинника.
Ментон. С1()Н,8О. {М.м. 154.2). 1051700. [14073-97-3].
(25,5Д)-2-Ізопропіл-5-метилциклогексанон.
(-)-щ/л7//с-л-Ментан-3-он.
Містить різні кількості ізоментону.
Безбарвна рідина. Дуже мало розчинний у воді, дуже
легко розчинний у 96 % спирті й ефірі.
г/2о : близько 0.897.
По : близько 1.450.
Ментон, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія м’яти
перцевої, використовуючи ментон як випробовуваний
розчин.
Площа основного піка має бути не менше 90.0 % су»-1
площ усіх піків.
Ментофуран. С1()Н14О. (М.м. 150.2). 1051500.
[17957-94-7]. 3,9-Епокси-м-мента-3.8-дієн. 3,6-Диме-І
тил -4,5,6,7-тетрагідробензофуран.
Рідина злегка синюватого кольору. Дуже мало розчин-
ний у воді, розчинний у 96 % спирті.
д™ : близько 0.965.
: близько 1.480.
[а]р : близько +93°.
Температура кипіння: 196 °С.
Ментофуран, використовуваний у газовій хромато-
графії, має витримувати таке додаткове випробуван-1
ня.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія мятиі
перцевої, використовуючи ментофуран як випробову-
ваний розчин.
Площа основного піка має бути не менше 97.0 % від
суми площ усіх піків.
2-Меркаптоетанол. С2Н6О8. (М.м. 78.1). 1099300.
[60-24-2].
Рідина. Змішується з водою.
(У2о : близько 1.116.
Температура кипіння: близько 157 °С.
Меркаптопурин. 1051900. [6112-76-1]. Див. статтю
Меркаптопурин.
Метакрилова кислота. С4Н6О2. (М.м. 86.1). 1101800.
[79-41-4]. 2-Метилпроп-2-енова кислота.
Безбарвна рідина.
: близько 1.431.
Температура кипіння: близько 160 °С.
Температура плавлення: близько 16 °С.
Метаніловий жовтий. С18Н141Ч3№038. (М.м. 375.4).
1052900. [587-98-4]. Показник Шульца№ 169. Кольо-
ровий індекс № 13065. Натрію 3-[4-(феніламі-
но)фенілазо]бензолсульфонат.
Порошок коричнювато-жовтого кольору. Розчинний
у воді й 96 % спирті, дуже мало розчинний в ефірі.
Метанілового жовтого розчин. 1052901.
Розчин 1 г/л у метанолі Р.
Випробування на чутливість. До 50 мл кислоти оц
тової безводної Рдодають 0.1 мл розчину метаніло-
вого жовтого; з’являється рожевувато-червоне за-
барвлення, яке має перейти у фіолетове при дода-
ванні 0.05 мл 0.1 Мрозчину кислоти хлорної.
Зміна забарвлення. Від червоного до оранжево-
жовтого в інтервалі рН 1.2-2.3.
224
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
ІНетанол. СН4О. (Мм. 32.04). 1053200. |67-56-1 ].
Прозора, безбарвна, займиста рідина. Змішується з
•одою й 96 % спиртом.
: від 0.791 до 0.793.
Температура кипіння: від 64 °С до 65 °С.
Метанол РІ. 1053201.
Має задовольняти вимоги для метанолу Р\ витри-
мувати таке додаткове випробування.
Мінімальне пропускання (2.2.25) визначають, ви-
користовуючи як компенсаційну рідину воду Р
20 % за довжини хвилі 210 нм,
50 % за довжини хвилі 220 нм,
75 % за довжини хвилі 230 нм,
95 % за довжини хвилі 250 нм,
98 % за довжини хвилі 260 нм і більше.
Метанол Р2. 1053202.
Метанол Р2, використовуваний у рідинній хрома-
тографії, має задовольняти таку вимогу.
Містить не менше 99.8 % СН4О (М.м. 32.04).
Оптична густина (2.2.25). Не більше 0.17.
Вимірюють за довжини хвилі 225 нм, використо-
вуючи як компенсаційну рідину воду Р.
Метанол підкислений. 1053203.
1.0 мл кислоти хлористоводневої РІ доводять ме-
танолом Рдо об’єму 100.0 мл.
Метанол безводний. 1053400. [67-56-1].
1000 мл метанолу Робробляють 5 гмагнію Р. Якщо не-
обхідно. ініціюють реакцію, додаючи 0.1 мл розчину
ртуті(П) хлориду Р. Після припинення виділення га-
зу рідину переганяють, відгін збирають у сухий кон-
тейнер і захищають від вологи.
Вода (2.5.12). Не більше 0.3 г/л.
Метанол, вільний від альдегідів. 1053300.
25 г йоду Р розчиняють в 1 л метанолу Р, одержаний
розчин додають при постійному помішуванні до
400 мл 1 М розчину натрію гідроксиду, потім додають
150 мл води Р і залишають на 16 год. Фільтрують і
кип’ятять зі зворотним холодильником до зникнення
запаху йодоформу. Розчин переганяють фракційною
перегонкою.
Містить не більше 0.001 % альдегідів і кетонів.
Метансульфонова кислота. СН4О38. (М.м. 96.1).
1053100. [75-75-2].
Прозора, безбарвна рідина, яка твердіє при темпера-
турі близько 20 °С. Змішується з водою, мало розчин-
на у толуолі, практично не розчинна у гексані.
: близько 1.48.
: близько 1.430.
Метафосфорна кислота. (НРО3)Х. 1053000. [37267-86-0].
Склоподібні грудочки або палички, які містять деяку
кількість натрію метафосфату. Гігроскопічна, дуже
легко розчинна у воді.
Нітрати. 1.0 г кип’ятять із 10 мл води Р, охолоджують,
додають 1 мл розчину індигокарміну Р, 10 мл кислоти
сірчаної, вільної від азоту, Р і нагрівають до кипіння.
Синє забарвлення не має повністю зникнути.
Відновлюючі речовини. Не більше 0.01 %, у перера-
хунку на Н3РО3. 35.0 г розчиняють у 50 мл води Р,
додають 5 мл розчину 200 г/л кислоти сірчаної Р,
50 мг калію броміду Р і 5.0 мл 0.02 М розчину калію
бромату й нагрівають на водяній бані протягом
ЗО хв; охолоджують, додають 0.5 г калію йодиду Р і
титрують йод, що виділився, 0.1 М розчином нат-
рію тіосульфату, використовуючи як індикатор
1 мл розчину крохмалю Р. Проводять контрольний
дослід.
1 мл 0.02 Мрозчину калію бромату відповідає 4.10 мг
Н3РО3.
Зберігають у повітронепроникному контейнері.
4-Метиламінофенолу сульфат. С14Н2(№068.
(М.м. 344.4). 1053800. [55-55-0].
Безбарвні кристали. Дуже легко розчинний у воді, ма-
ло розчинний у 96 % спирті, практично не розчинний
в ефірі.
Температура плавлення: близько 260 °С.
Метилантранілат. С8Н9ЬЮ2. (М.м. 151.2). 1107300.
[134-20-3]. Метил-2-амінобензоат.
Безбарвні кристали або рідина від безбарвного до жов-
туватого кольору. Розчинний у воді, легко розчинний
у 96 % спирті та ефірі.
Температура плавлення: від 24 °С до 25 °С.
Температура кипіння: від 134 °С до 136 °С.
Метилантранілат, використовуваний у газовій хрома-
тографії, має витримувати таке додаткове випробу-
вання.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено в статті Олія квіток
померанця, використовуючи метилантранілат як ви-
пробовуваний розчин.
Площа основного піка має бути не менше 95.0 % від
суми площ усіх піків.
Метиларахідат. С21Н42О2. (М.м. 326.6). 1053900.
[1120-28-1]. Метилейкозаноат.
Містить не менше 98.0 % С21Н42О2. Визначення про-
водять методом газової хроматографії (2.4.22).
Кристалічна маса від білого до жовтого кольору. Роз-
чинний у 96 % спирті й петролейному ефірі
Температура плавлення: близько 46 °С.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
225
4.1.1. Реактиви
Метилацетат. С3Н6О2. (М.м. 74.1). 1053700. [79-20-9].
Прозора, безбарвна рідина. Розчинний у воді, змі-
шується з 96 % спиртом.
с/20 : близько 0.933.
: близько 1.361.
Температура кипіння: від 56 °С до 58 °С.
Метилбегенат. С23Н46О2. (М.м. 354.6). 1107500.
[929-77-1]. Метилдокозаноат.
Температура плавлення: від 54 °С до 55 °С.
Метил бензотіазолонгідразону гідрохлорид.
С8Н10С1К38,Н2О. (М.м. 233.7). 1055300. [38894-11-0].
3-Метилбензотіазол-2(3//)-он гідразону гідрохлорид
моногідрат.
Кристалічний порошок майже білого або жовтуватого
кольору.
Температура плавлення: близько 270 °С.
Випробування на придатність для визначення аль-
дегідів. До 2 мл метанолу, вільного від альдегідів, Рао-
дають 60 мкл розчину 1 г/л пропіонового альдегіду Р у
метанолі, вільному від альдегідів, Р і 5 мл розчину 4 г/л
метилбензотіазолонгідразону гідрохлориду, змішують
і залишають на ЗО хв. Готують контрольний розчин,
який не містить пропіонового альдегіду. До випробо-
вуваного й контрольного розчинів додають по 25.0 мл
розчину 2 г/л заліза(ІІІ) хлориду Р, доводять об’єм
кожного розчину ацетоном Р до 100.0 мл і перемішу-
ють. Оптична густина (2.2.25) випробовуваного роз-
чину, виміряна за довжини хвилі 660 нм із викорис-
танням контрольного розчину як компенсаційної
рідини, має бути не менше 0.62.
2-Метилбут-2-ен. С5Н10. (М.м. 70.1). 1055400. [513-35-9].
Дуже легко займиста рідина. Практично не розчинний
у воді, змішується з 96 % спиртом та ефіром.
Температура кипіння: від 37.5 °С до 38.5 °С.
2-Метилбутан. С5Н12. (М.м. 72.2). 1099500. [78-78-4].
Ізопентан.
Містить не менше 99.5 % С5НІ2.
Безбарвна, легко займиста рідина.
б/2о : близько 0.621.
«о : близько 1.354.
Температура кипіння: близько 29 °С.
Вода (2.5.12). Не більше 0.02 %.
Залишок після випарювання. Не більше 0.0003 %.
Мінімальне пропускання (2.2.25) визначають, викорис-
товуючи як компенсаційну рідину воду Р.
50 % за довжини хвилі 210 нм,
85 % за довжини хвилі 220 нм,
98 % за довжини хвилі 240 нм і більше.
Метилдеканоат. СцН22О2. (М.м. 186.3). 1054000.
[110-42-9]. Метил-н-деканоат.
Містить не менше 99.0 % СцН22О2.
Прозора, безбарвна або жовтого кольору рідина. Роз-
чинний у петролейному ефірі.
: від 0.871 до 0.876.
: від 1.425 до 1.426.
Сторонні домішки. Визначення проводять методом га-
зової хроматографії (2.2.28), хроматографуючи рівні
об’єми кожного із таких розчинів речовин: (1) розчин
0.02 г/л метилдеканоату у вуглеці дисульфіду Р, (11)
розчин 2 г/л метилдеканоату у вуглеці дисульфіду Р,
(111) вуглецю дисульфід Р. Хроматографують за умов І
випробування на бутилгідрокситолуол, зазначених )
статті Ланолін.
На хроматограмі розчину (11) сума площ усіх піків,
окрім основного піка й піка розчинника, має бути
меншою за площу основного піка на хроматограмі
розчину (1).
З-О-Метилдопаміну гідрохлорид. С9Н14С1МО2.
(М.м. 203.7). 1055600. [1477-68-5]. 4-(2-Аміноетил)-
2-метоксифенолу гідрохлорид.
Температура плавлення: від 213 °С до 215 °С.
Хроматографія. Визначення проводять, як зазначено
у статті Допаміну гідрохлорид', наносять 10 мкл розчину
0.075 г/л у метанолі Р. На хроматограмі має бути лише
одна основна пляма.
4-О-Метилдопаміну гідрохлорид, С9НІ4С1ТЮ2.
(М.м. 203.7). 1055700. [645-33-0]. 5-(2-Аміноетил)-2-
метоксифенолу гідрохлорид.
Температура плавлення: від 207 °С до 208 °С.
Хроматографія. Визначення проводять, як зазначено
у статті Допаміну гідрохлорид', наносять 10 мкл розчину
0.075 г/л у метанолі Р. На хроматограмі має бути лише
одна основна пляма.
2-Метил-5-нітроімідазол. С4Н5К3О2. (М.м. 127.1).
1056100. [88054-22-2].
Порошок від білого до світло-жовтого кольору.
Температура плавлення: від 252 °С до 254 °С.
Метилейкозеноат. С20Н38О2. (М.м. 310.5). 1120500. Ме-
тил цис-11 -ейкозеноат.
Метиленбісакриламід. С7Н10М2О2. (М.м. 154.2).
1056000. [110-26-9]. А.А'-Метиленбіспропенамід.
Дуже дрібний порошок білого або майже білого ко-
льору. Мало розчинний у воді, розчинний у 96 %
спирті.
Температура плавлення: 300 °С, із розкладанням.
Метиленовий синій. С16Н18С1ІМ38.хН2О. (М.м. 319.9,
для безводного). 1055800. [7220-79-3]. Показник
Шульца № 1038. Кольоровий індекс № 52015. 3,7-Ди-
метиламінофенотіазину-5 хлорид.
Існує в різних гідратованих формах і може містити до
22 % води.
226
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кристалічний порошок темно-зеленого або бронзово-
го кольору. Легко розчинний у воді, розчинний у 96 %
і’пирті
Метиленхлорид. СН2С12. {М.м. 84.9). 1055900. [75-09-2].
Диктор метан.
Безбарвна рідина. Помірно розчинний у воді, змішу-
ться з 96 % спиртом та ефіром.
Температура кипіння: від 39 °С до 42 °С.
Метиленхлорид, використовуваний у флуориметрії,
мас витримувати таке додаткове випробування.
Флуоресценція. При опроміненні світлом із довжиною
хвилі 365 нм поглинання (2.2.21), виміряне за довжи-
ни хвилі 460 нм у кюветі із товщиною шару І см, не
має бути інтенсивнішим за поглинання розчину, який
містить 0.002 ррт хініну Р у 0.5 М розчині кислоти
сірчаної, виміряної за тих самих умов.
Метиленхлорид підкислений. 1055901.
До 100 мл метиленхлориду Р додають 10 мл кисло-
ти хлористоводневої Р, струшують. Після
розділення шарів використовують нижній шар.
Метилетилкетон. С4НХО. {М.м. 72.1). 1054100. [78-93-3].
Етилметилкетон. 2-Бутанон.
Прозора, безбарвна, займиста рідина. Дуже легко роз-
чинний у воді, змішується з 96 % спиртом і ефіром.
: близько 0 81
Температура кипіння: від 79 °С до 80 °С.
Метилізобутилкетон. С6Н|2О. (М.м. 100.2). 1054300.
[108-10-1]. 4-Метил-2-пентанон.
Прозора, безбарвна рідина. Мало розчинний у воді,
змішується із більшістю органічних розчинників.
И[) близько 0.80.
Температура кипіння: близько 115 °С.
Температурні межі перегонки (2.2.11). Переганяють
100 мл. Інтервал температури перегонки не має пе-
ревищувати 4 0 °С; має переганятись від 1 мл до
95 мл.
Залишок після випарювання. Не більше 0.01 %. Випа-
рюють на водяній бані, залишок сушать при темпера-
турі від 100 °С до 105 °С.
Метилізобутилкетон РІ. 1054301.
50 мл свіжоперегнаного метилізобутилкетону Р
струшують із 0.5 мл кислоти хлористоводневої РІ
протягом 1 хв. Після розділення шарів нижній шар
відкидають.
Готують безпосередньо перед використанням.
Метилкапрат. 1054000. Див. Метилдеканоат Р.
Метилкаприлат. С9НІ8О2. {М.м. 158.2). 1120400.
[111-11-5]. Метилоктаноат.
№ : близько 0 876.
«р : близько 1.417.
Температура кипіння від 193 °С до 194 °С.
Метилкапроат. С7Н14О2. (М.м. 130.2). 1120300. [106-70-7].
Метилгексаноат.
</2о : близько 0.885.
«р : близько 1.405.
Температура кипіння: від 150 °С до 151 °С.
Метиллаурат. С13Н26О2. (М.м. 214.4). 1054400. [111-82-0].
М етилдодеканоат.
Містить не менше 98.0 % СИН26О2. Визначення про-
водять методом газової хроматографії (2.4.22).
Безбарвна або жовтого кольору рідина. Розчинний у
96 % спирті й петролейному ефірі.
с/2§ : близько 0.87.
п$ : близько 1.431.
Температура плавлення: близько 5 °С.
Метиллігноперат. С25Н50О2. (М.м. 382.7). 1120600.
[557-59-51. Метилтетракозаноат.
Пластівці.
Температура плавлення: близько 58 °С.
Метиллінолсат. С19Н34О2. (М.м. 294.5). 1120700.
[112-63-0]. Метил-цис,цис-9,12-октадекадієноат.
с/2о : близько 0.888.
Лр близько 1.466
Температура кипіння: віл 207 °С до 208 °С.
Метиллінолснат. С19Н32О2. (М.м. 292.5). 1120800.
[ЗО 1 -00-8]. Метил-цис,цис,цис-9,12,15-октадекатриє-
ноат.
^20 : близько 0.901.
Лр : близько 1.471.
Температура кипіння: близько 207 °С.
Метилмаргарат. С18Н36О2. (М.м. 284.5). 1120900.
[1731-92-6]. Метилгептадеканоат.
Температура плавлення: від 32 °С до 34 °С.
Метилметакрилат. С5Н8О2. (М.м. 100.1). 1054500.
[80-62-6]. Метил-2-метилпроп-2-еноат.
Безбарвна рідина
Лр : близько 1.414.
Температура кипіння: близько 100 °С.
Температура плавлення: близько -48 °С.
Містить підхожий стабілізуючий реагент.
Метилміристат. С15Н30О2. (М.м. 242.4). 1054600.
[ 124-10-7]. Метилтетрадеканоат.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
227
4.1.1. Реактиви
Містить не менше 98.0 % СІ5Н30О2. Визначення про-
водять методом газової хроматографії (2.4.22).
Безбарвна або жовтавого кольору рідина. Розчинний у
96 % спирті й петролейному ефірі.
г/2о • близько 0.87.
: близько 1.437.
Температура плавлення: близько 20 °С.
Метиловий зелений. С26Н33С12М3. (М.м. 458.5).
1054200. [7114-03-6]. Показник Шульца № 788.
Кольоровий індекс № 42585. 4-[[4-(Диметиламіно)-
феніл][4-(диметилімініо)циклогекса-2,5-дієніліден]-
метилфеніл]триметиламонію дихлорид.
Порошок зеленого кольору. Розчинний у воді, роз-
чинний у кислоті сірчаній із утворенням жовтого за-
барвлення, яке переходить у зелене при розведенні во-
дою.
Метилового зеленого-йодомеркуратний папір.
1054201.
Тонкі смужки підхожого фільтрувального паперу
занурюють у розчин 40 г/л метилового зеленого Р,
сушать на повітрі, потім занурюють їх на І год у
розчин, який містить 140 г/л калію йодиду Р і
200 г/л ртуті(Н) йодиду’ Р. Смужки промивають
водою дистильованою Рдоти, поки промивні води
не стануть практично безбарвними, й сушать на
повітрі.
Зберігають у захищеному від світла місці.
Термін придатності 2 доби.
Метиловий оранжевий. С14Н14К3ІМаО38. (М.м. 327.3).
1054800. [547-58-0]. Показник Шульца № 176. Кольо-
ровий індекс № 13025. Натрію 4’-(диметиламіно)азо-
бензол-4-сульфонат.
Кристалічний поровюк оранжево-жовтого кольору.
Мало розчинний у воді, практично не розчинний у
96 % спирті.
Метилового оранжевого змішаний розчин. 1054801.
20 мг метилового оранжевого Р і 0.1 г бромкрезоло-
вого зеленого Р розчиняють в 1 мл 0.2 М розчину
натрію гідроксиду і доводять об’єм розчину водою Р
до 100 мл.
Зміна забарвлення. Від оранжевого до жовтаво-зе-
леного в інтервалі рН 3.0-4.4.
Метилового оранжевого розчин. 1054802.
0.1 г метилового оранжевого Р розчиняють у 80 мл
води Р і доводять об’єм розчину 96 % спиртом Рао
100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0.1 мл роз-
чину метилового оранжевого; з’являється жовте
забарвлення, яке має перейти у червоне при дода-
ванні не більше 0.1 мл 0.1 Мрозчину кислоти хло-
ристоводневої.
Зміна забарвлення. Від червоного до жовтого і
інтервалі рН 3.0-4.4.
Метиловий червоний. С15Н15ЬЇ3О2. (М.м. 269.3)
1055100. [493-52-7]. Показник Шульца № 250. Кольо
ровий індекс № 13020. 2-(4-Диметиламінофені.іа-
зо)бензойна кислота.
Порошок темно-червоного кольору або кристали
фіолетового кольору. Практично не розчинний у воді,
розчинний у 96 % спирті.
Метилового червоного змішаний розчин. 1055101.
0.1 г метилового червоного Р і 50 мг метиленового І
синього Р розчиняють у 100 мл 96 % спирту Р.
Зміна забарвлення. Від червоно-фіолетового до зе-
леного в інтервалі рН 5.2-5.6.
Метилового червоного розчин. 1055102.
50 мг розчиняють в суміші 1.86 мл 0.1 М розчину
натрію гідроксиду та 50 мл 96 % спирту Р, дово-
дять об’єм розчину водою Рдо 100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0.1 мл роз-
чину метилового червоного і 0.05 мл 0.02 Мрозчи-
ну кислоти хлористоводневої; з’являється червоне
забарвлення, яке має перейти у жовте при дода-
ванні не більше 0.1 мл 0.02 М розчину натрію
гідроксиду.
Зміна забарвлення. Від червоного до жовтого в
інтервалі рН 4.4-6.0.
Метилолеат. С19Н36О2. (М.м. 296.4). 1054700. [112-62-9].
Метил-С2)-октадек-9-еноат.
Містить не менше 98.0 % С19Н36О2. Визначення про-
водять методом газової хроматографії (2.4.22).
Безбарвна або жовтавого кольору рідина. Розчинний у
96 % спирті й петролейному ефірі.
с/2о : близько 0.88.
: близько 1.452.
Метилпальмітат. С17Н34О2. (М.м. ТК}.5(. 1054900.
[112-39-0]. Метилгексадеканоат.
Містить не менше 98.0 % С17Н34О2. Визначення про-
водять методом газової хроматографії (2.4.22).
Кристалічна маса білого або жовтого кольору. Розчин-
ний у 96 % спирті іі петролейному ефірі.
Температура плавлення: близько ЗО °С.
Метилпальмітолеат. С,7Н32О2. (М.м. 268.4). 1121000.
[1120-25-8]. Метил-цис-9-гексадекеноат.
с/2(() : близько 0.876.
«р : близько 1.451.
Метилпарагідроксибензоат. 1055000. [99-76-3]. Див.
статтю Метилпарагідроксибензоат.
228
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
4-Метилпентан-2-ол. С6НІ4О. (М.м. 102.2). 1114300.
1108-11-2].
Прозора, безбарвна, летка рідина.
(/4°: близько 0 802.
близько 1411.
Температура кипіння: близько 132 °С.
Метилпіперазин. С5Н12М2. (М.м. 100.2). 1056300.
[74879-18-8]. 1-Метилпіперазин.
Безбарвна рідина. Змішується з водою й 96 % спиртом.
і/2,: близько 0 90.
гір : близько 1 466.
Температура кипіння: близько 138 °С.
4-(4-Метилпіперидино)пірилин. СИН16К2- (М.м. 176.3).
1114400. [80965-30-6|.
Прозора рідина.
Ир : близько 1.565.
2-Метилпропанол. С4Н,0О. (М.м. 74.1). 1056400.
|78-83-1[. Ізобутиловий спирт. 2-Метилпропан-1-ол.
Прозора, безбарвна рідина. Розчинний у воді, змі-
шується з 96 % спиртом та ефіром.
: близько 0 80.
Яр : від 1.397 до 1.399.
Температура кипіння: близько 107 °С.
Температурні межі перегонки (2.2.11). Від 107 °С до
109 °С; має переганятися не менше 96 %.
2-Метил-2-пропанол. С4НИ)О. (М.м. 74.1). 1056500.
|75-65-О]. 1,1-Диметилетиловий спирт. Треді-Бутило-
вий спирт.
Прозора, безбарвна рідина або кристалічна маса. Роз-
чинний у воді, змішується з 96 % спиртом та ефіром.
Температура тверднення (2.2.18). близько 25 °С.
Температурні межі перегонки (2.2.11). Від 81 °С до
83 °С; має переганятися не менше 95 %.
Метилстеараг. С19Н38О2. (М.м. 298.5). 1055200.
[112-61-8|. Метилоктадеканоат.
Містить не менше 98.0 % С19Н38О2. Визначення про-
водять методом газової хроматографії (2.4.22).
Кристалічна маса білого або жовтого кольору. Розчин-
ний у 96 % спирті й петролейному ефірі.
Температура плавлення: близько 38 °С.
Метилтридеканоат. СІ4Н28О2. (М.м. 228.4). 1121100.
[1731-88-0].
Безбарвна або жовтава рідина. Розчинний у 96 %
спирті й петролейному ефірі.
<$} : близько 0.86.
Яр : близько 1.441.
Температура плавлення: близько 6 °С.
Метилтрикозаноат. С24Н48О2. (М.м. 368.6). 1111500.
[2433-97-8]. Метиловий ефір трикозанової кислоти.
Містить не менше 99 0 % С24Н48О2.
Кристали білого кольору. Практично не розчинний у
воді, розчинний у гексані.
Температура плавлення: від 55 °С до 56 °С.
Метилфснілоксазолілбензол. С2()Н2(1К2О2. (М.м. 392.5).
1056200. [3073-87-8]. 1,4-Біс[2-(4-метил-5-феніл)ок-
сазоліл]бензол.
Дрібний порошок зеленувато-жовтого кольору із си-
ньою флуоресценцією або дрібні кристали. Розчин-
ний у 96 % спирті, помірно розчинний у ксилолі.
Температура плавлення: близько 233 °С.
Метилфенілоксазолілбензол, використовуваний для
рідинної сцинтиляції, має бути відповідного ступеня
чистоти.
Метилцелюлоза 450. 1055500. [9004-67-5]. Див. статтю
Метилцелюлоза.
Номінальна в’язкість: 450 мПа-с.
Метилцинамат. С10НІ0О2. (М.м. 162.2). 1099400.
[103-26-4].
Безбарвні кристали. Практично не розчинний у воді,
розчинний у 96 % спирті та ефірі.
: близько 1.56.
Температура кипіння: близько 260 °С.
Температура плавлення: від 34 °С до 36 °С.
Б-Метіошн. 1053500. [63-68-3]. Див. стагтю Метіонін.
Метоксифенілоцтова кислота. С9Н(0О3. (М.м. 166.2).
1053600. [7021-09-2]. (Я5)-2-Метокси-2-фенілоцтова
кислота.
Кристалічний порошок білого кольору або кристали
білого або майже білого кольору. Помірно розчинна у
воді, легко розчинна в 96 % спирті та ефірі.
Температура плавлення: близько 70 °С.
Зберігають у прохолодному місці.
Метоксифенілоцтової кислоти реактив. 1053601.
2.7 г кислоти метоксифенілоцтової Р розчиняють
у 6 мл розчину тетраметиламонію гідроксиду Р і
додають 20 мл етанолу Р.
Зберігають у поліетиленовому контейнері.
(7?5)-Метотрексат. 1120200.
Містить не менше 96.0 % С2оН22І\805
Температура плавлення: близько 195 °С.
Міді(П) ацетат. С4Н6СиО4,Н2О. (М.м. 199.7). 1022200.
[142-71-2].
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
229
4.1.1. Реактиви
Кристали або порошок блакитнувато-зеленого кольо-
ру. Легко розчинний у киплячій воді, розчинний у воді
й 96 % спирті, мало розчинний в ефірі й гліцерині
(85%).
Міді едетату розчин. 1022300.
До 2 мл розчину 20 г/л міді(II) ацетату Р додають 2 мл
0.1Мрозчину натрію едетату й доводять об’єм розчи-
ну водою Рр.о 50 мл.
Міді(ІІ) нітрат. Си(МО3)2,ЗН2О. (М.м. 241.6). 1022400.
[10031-43-3]. Міді динітрат тригідрат.
Кристали синього кольору. Гігроскопічний, дуже лег-
ко розчинний у воді, водний розчин має сильнокислу
реакцію, легко розчинний у 96 % спирті и кислоті
азотній розведеній.
Зберігають у повітронепроникному контейнері.
Міді(П) сульфат. СиБО4,5Н7О. (М.м. 249.7). 1022500.
[7758-99-81.
Порошок або кристали синього кольору Повільно
звітрюється на повітрі, дуже легко розчинний у воді,
мало розчинний у 96 % спирті.
Міді(ІІ) сульфату розчин. 1022501.
Розчин 125 г/л.
Міді тетрааміакату аміачний розчин. 1022600.
34.5 гміді(П) сульфату /'розчиняють у 100 мл води Р,
додають при перемішуванні по краплях розчин аміаку
концентрований Р до розчинення утвореного осаду.
Підтримуючи температуру не вище 20 °С, при безпе-
рервному струшуванні додають по краплях ЗО мл роз-
чину натрію гідроксиду концентрованого Р. Фільтру-
ють крізь скляний фільтр (40), промивають водою Рао
одержання прозорого фільтрату. Струшують із 200 мл
розчину аміаку концентрованого Р і фільтрують крізь
скляний фільтр, потім знов фільтрують, щоб зменши-
ти осад до мінімуму.
Міді(ІІ) хлорид. СііС12,2Н2О. (М.м. 170.5). 1023000.
[10125-13 0|. Міді хлорид дигідрат.
Порошок або кристали зеленувато-блакитного кольо-
ру, які розпливаються на повітрі, звітрюються в сухо-
му повітрі. Легко розчинний у воді, 96 % спирті й ме-
танолі, помірно розчинний в ацетоні, мало розчинний
в ефірі.
Зберігають у повітронепроникному контейнері.
Мідно-тартратний розчин. 1023300.
Розчин І. 34.6 г міді(ІІ) сульфату Р розчиняють у воді Р,
доводять об’єм розчину тим самим розчинником до
500 мл.
Розчин 11. 173 г калію натрію тартрату Р і 50 г
натрію гідроксиду Р розчиняють у 400 мл води Р.
Нагрівають до кипіння, охолоджують, доводять об’єм
одержаного розчину водою, вільною від вуглецю діокси-
ду. Рао 500 мл.
Безпосередньо перед використанням змішують рівні
об’єми розчинів 1 та II.
Мідно-тартратний розчин Р2. 1023302.
Змішують 1 мл розчину, який містить 5 г
міді(П) сульфату Рі 10 г/л калію тартрату Р,
50 мл розчину натрію карбонату РІ.
Готують безпосередньо перед використанням
Мідно-тартратний розчин РЗ. 1023303.
Змішують рівні об’єми розчину 10 г/л МІдІ(11)суПг
фату Р \ розчину 20 г/л натрію тартрату Р.
До 1.0 мл одержаного розчину додають 50 мл раз
чину натрію карбонату РІ
Готують безпосередньо перед використанням.
Мідно-тартратний розчин Р4. 1023304.
Розчин 1. Розчин 150 г/л міді(П) сульфату Р.
Розчин 11. 2.5 г натрію карбонату безводного Р,
2.5 г калію-натрію тартрату Р, 2.0 г натрію гідро
карбонату Рі 20.0 г натрію сульфату безводного!
розчиняють у воді Р, доводять об’єм одержаної!
розчину тим самим розчинником до 100 мл.
Безпосередньо перед використанням змішують
розчини І та 11 у співвідношенні 1:25.
Мідно-цитратний розчин. 1023100.
25 г міді(II) сульфату Р, 50 г кислоти лимонної Р і 144 г
натрію карбонату безводного Р розчиняють у воді Р\
доводять об’єм розчину тим самим розчинником до
1000 мл.
Мідно-цитратний розчин РІ. 1023200.
25 г міді(II) сульфату Р, 50 г кислоти лимонної Р і 144 г
натрію карбонату безводного Р розчиняють у воді Рі
доводять об’єм розчину тим самим розчинником до
1000 мл (випробовуваний розчин)
Розчин коригують, аби він задовольняв такі вимоги:
а) До 25.0 мл випробовуваного розчину додають 3 г
калію йодиду Р, потім обережно невеликими порціями
додають 25 мл 25 % (м/м) розчину кислоти сірчаної Рі
титрують 0.1 М розчином натрію тіосульфату, вико-
ристовуючи як індикатор 0.5 мл розчину крохмалю Р.
який додають наприкінці титрування.
На титрування має бути витрачено від 24.5 мл до
25.5 мл Д / М розчину натрію тіосульфату.
Ь) 10.0 мл випробовуваного розчину доводять водою Р
до об’єму 100.0 мл і перемішують. До 10.0 мл одержа-
ного розчину додають 25.0 мл 0.1 М розчину кислоти
хлористоводневої, нагрівають на водяній бані протя-
гом І год, охолоджують, доводять водою Р до вихідно-
го об’єму й титрують 0.1 М розчином натрію гідрокси-
ду, використовуючи як індикатор 0.1 мл розчину фе-
нолфталеїну РІ.
На титрування має бути витрачено від 5.7 мл до 6.3 мл
0.1М розчину натрію гідроксиду.
230
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
с) 10.0 мл випробовуваного розчину доводять водою Р
до об’єму 100.0 мл і перемішують. 10.0 мл одержаного
розчину титрують 0.1 М розчином кислоти хлористо-
водневої, використовуючи як індикатор 0.1 мл розчину
фенолфталеїну РІ.
На титрування має бути витрачено від 6.0 мл до 7.5 мл
0.1 Мрозчину кислоти хлористоводневої.
Мідь. Си. (А.м. 63.55). 1022100. [7440-50-8].
Фольга очищена, стружка, дріт або металевий поро-
шок електролітичної чистоти.
Міозмін. С9Н10М2. (М.м. 146.2). 1121200. [532-12-7].
3-(4,5-Дигідро-3//-пірол-2-іл)піридин.
Безбарвні кристали.
Температура плавлення: близько 45 °С.
Міристиловий спирт. С14Н30О. (М.м. 214.4). 11121300.
1-Тетрадеканол.
її™: близько 0.823.
Температура плавлення: віл 38 °С до 40 °С.
Міристицин. СцН12О3. (М.м. 192.2). 1099600. [607-91-0].
5-Аліл-1-метокси-2,3-метилендіоксибензол. 4-Ме-
токси-6-(проп-2-еніл)-1,3-бензодіоксол.
Безбарвна масляниста рідина. Практично не розчин-
ний у воді, мало розчинний в етанолі, розчинний в
ефірі, змішується з толуолом і ксилолом.
($}: близько 1.144.
«о : близько 1.540.
Температура кипіння: від 276 °С до 277 °С.
Температура плавлення: близько 173 °С.
Хроматографія. Визначення проводять, як зазначено
у статті Олія бадьянова; на одержаній хроматограмі мас
виявлятися лише одна основна пляма.
Зберігають у прохолодному, захищеному від світла місці.
0-Мірцен. С10Н16. (Мм. 136.2). 1114500. [123-35-3].
7-Метил-З-метиленокта-1,6-дієн.
Масляниста рідина із приємним запахом. Практич-
но не розчинний у воді, змішується з 96 % спир-
том, розчинний в ефірі та кислоті оцтовій льодя-
ній, розчинний у розчинах гідроксидів лужних ме-
талів.
: близько 0.794.
«о : близько 1.470.
(і-Мірцен, використовуваний у газовій хромато-
графії, має витримувати таке додаткове випробуван-
ня.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія м’яти
перцевої.
Випробовуваний розчин. Випробовувана субстанція.
Площа основного піка має бути не менше 90.0 % від
суми площ усіх піків.
Міцний синій В, сіль. С14Н12С12М4О2. (М.м. 339.2).
1037400. [84633-94-3]. Показник Шульца № 490. Ко-
льоровий індекс № 37235. 3,3'-Диметокси(біфеніл)-
4,4'-бісдіазонію дихлорид.
Порошок темно-зеленого кольору. Розчинний у воді.
Стабілізований цинку хлоридом.
Зберігають у повітронепроникному контейнері при
температурі від 2 °С до 8 °С.
Міцний червоний В, сіль. С17Н13М3О9$2. (М.м. 467.4).
1037500. [56315-29-8]. Показник Шульца № 155. Ко-
льоровий індекс № 37125. 2-Метокси-4-нітробензол-
діазонію кислий нафталін-1,5-дисульфонат.
Порошок оранжево-жовтого кольору. Розчинний у
воді, мало розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла місці при температурі від 2 °С до 8 °С.
Молекулярне сито. 1056600.
Молекулярне сито складається з натрію алюмосиліка-
ту. Має вигляд кульок із розмірами пор 0.4 нм і діаме-
тром 2 мм.
Молібденованадієвий реактив. 1056700.
У стакані місткістю 150 мл змішують розтерті на поро-
шок 4 г амонію молібдату Р і 0.1 г амонію ванадату Р,
додають 70 мл води Р і перемішують скляною палич-
кою до розчинення. Через кілька хвилин має утвори-
тися прозорий розчин, до якого додають 20 мл кисло-
ти азотної Р і доводять об’єм розчину водою Р до
100 мл.
Молочна кислота. 1047800. [50-21-5]. Див. статтю Кис-
лота лактонова.
Молочної кислоти реактив. 1047801.
Розчин А. До 60 мл кислоти молочної Р додають
45 мл розчину кислоти молочної Р, насиченого без
нагрівання Суданом червоним С Р\ попередньо від-
фільтрованого. Кислота молочна насичується
повільно без нагрівання, тому завжди необхідний
надлишок барвника.
Розчин В. Готують 10 мл насиченого розчину
аніліну Рї фільтрують.
Розчин С. 75 мг калію йодиду Ррозчиняють у воді Р
і доводять об’єм розчину тим самим розчинником
до 70 мл. До одержаного розчину додають 10 мл
96 % спирту Р й 0.1 г йоду Р, струшують.
Змішують розчини АйВ, додають розчин С.
Морфіну гідрохлорид. 1056900. Див. статтю Морфіну
гідрохлорид.
Морфолін. С4Н9МО. (Мм. 87.1). 1057000. [110-91-8].
Тетрагідро-1,4-оксазин.
Безбарвна, гігроскопічна, займиста рідина. Розчин-
ний у воді й 96 % спирті.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
231
4.1.1. Реактиви
с/20 близько 1.01.
Температурні межі перегонки (2.2.11). Від 126 °С до
130 °С; має переганятися не менше 95 %.
Зберігають у повітронепроникному контейнері.
Мурашина кислота безводна. СН2О2. {М.м. 46.03).
1039300. [64-18-6].
Містить не менше 98.0 % (м/м) СН2О2.
Безбарвна рідина. Спричиняє корозію, змішується з
водою й 96 % спиртом.
г/2§ : близько 1.22.
Кількісне визначення. 10 мл води Р поміщають у
конічну колбу, точно зважують, швидко додають
близько 1 мл кислоти мурашиної безводної та знов
зважують. Додають 50 мл води Р і титрують 1 М розчи-
ном натрію гідроксиду, використовуючи як індикатор
0.5 мл розчину фенолфталеїну Р.
1 мл 1 Мрозчину натрію гідроксиду відповідає 46.03 мг
СН2О2.
Натрій. N3. {А.м. 22.99). 1078500. [7440-23-5].
Метал, на свіжому зрізі має блискучу сріблясто-сіру по-
верхню. На повітрі швидко тьмяніє та повністю окис-
нюється до натрію оксиду, який перетворюється на
натрію карбонат. Бурхливо реагує з водою з утворенням
водню й натрію гідроксиду; розчинний у безводному
метанолі з утворенням водню й натрію метилату; прак-
тично не розчинний в ефірі й петролейному ефірі.
Зберігають у петролейному ефірі або рідкому парафіні
(приміром, гас).
Натрію азид. 1Ча1Ч3. {М.м. 65.0). 1078900. [26628-22-8].
Кристалічний порошок або кристали білого кольору.
Легко розчинний у воді, мало розчинний у 96 %
спирті, практично не розчинний в ефірі.
Натрію арсеніту розчин. 1008301.
0.50 г арсену(ПІ) оксиду Р розчиняють у 5 мл розчину
натрію гідроксиду розведеного Р, додають 2.0 г натрію
гідрокарбонату Рі доводять об’єм розчину водою Рао
100.0 мл.
Натрію аскорбату розчин. 1078800. [134-03-2].
3.5 г кислоти аскорбінової Р розчиняють у 20 мл
1 М розчину натрію гідроксиду.
Готують безпосередньо перед використанням.
Натрію ацетат. 1078600. [6131-90-4]. Див. статтю
Натрію ацетат.
Натрію ацетат безводний. С2Н31^аО2. {М.м. 82.0).
1078700. [127-09-3].
Безбарвні кристали або гранули. Дуже легко розчин-
ний у воді, помірно розчинний у 96 % спирті.
Втрата в масі при висушуванні (2.2.32). Не більше
2.0 %. Визначення проводять при температурі від
100 °С до 105 °С.
Натрію бікарбонат. 1081300. [144-55-8]. Див. Дотрімі
гідрокарбонат Р.
Натрію вісмутат. N38105. {М.м. 280.0). 1079000.
[12232-99-4].
Містить не менше 85.0 % N38103.
Порошок жовтого або жовтаво-коричневого кольорі
Повільно розкладається під дією вологи або висока!
температури, практично не розчинний у холодній воді
Кількісне визначення. 0.200 г суспензують у 10 мл роз-
чину 200 г/л калію йодиду Р, долають 20 мл кислоти]
сірчаної розведеної Р і титрують 0.1 М розчином натрію І
тіосульфату до одержання оранжевого забарвлення. І
використовуючи як індикатор 1 мл розчину крохма-
лю Р.
1 мл 0.1 М розчину натрію тіосульфату відповідає І
14.00 мг N38105.
Натрію бутансульфонат. С4Н9№038. {М.м. 160.2). |
1115600. [2386-54-1].
Кристалічний порошок білого кольору. Розчинний у
воді.
Температура плавлення: понад 300 °С.
Натрію вольфрамат. №2\УО4.2Н2О. {М.м. 329.9)
1084700. [10213-10-2]. Динатрію вольфрамат дигідрат.
Кристалічний порошок білого кольору або безбарвні
кристали. Легко розчинний у воді з утворенням прозо-
рого розчину, практично не розчинний у 96 % спирті
Натрію гексансульфонат. С6Н13№038. {М.м. 188.2).
1081200. [2832-45-3].
Порошок білого або майже білого кольору. Легко роз
чинний у воді.
Натрію гептансульфонат. С7Н15№О38. {М.м. 202.3).
1081000. [22767-50-6].
Кристалічна маса білого або майже білого кольору.
Легко розчинний у воді, розчинний у метанолі.
Натрію гептансульфонат моногідрат.
С7Н15№О38,Н2О. {М.м. 220.3). 1081100.
Містить не менше 96 % С7Н15№038 у перерахунку на
безводну речовину.
Кристалічний порошок білого кольору. Розчинний у
воді, дуже мало розчинний в етанолі, практично не
розчинний в ефірі.
Вода (2.5.12). Не більше 8 %. Визначення проводять із
0.300 г.
Кількісне визначення. 0.150 г розчиняють у 50 мл кисло-
ти оцтової безводної Р і титрують 0.1 М розчином кис-
лоти хлорної потенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної відповідає 20.22 мг
С7Н15№038.
Натрію гідрокарбонат. 1081300. [144-55-8]. Див. статтю
Натрію гідрокарбонат.
232
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Натрію гідрокарбонату розчин. 1081301.
Розчин 42 г/л.
Натрію гідроксид. 1081400. [1310-73 2]. Див. статтю
Натрію гідроксид.
Натрію гідроксиду розчин. 1081401.
20.0 г натрію гідроксиду Ррозчиниють у воді Р і до-
водять об’єм розчину тим самим розчинником
до 100.0 мл. Концентрацію розчину визнача-
ють титруванням / М розчином кислоти хлористо-
водневої, використовуючи як індикатор розчин ме-
тилового оранжевого Р, якщо необхідно, розчин
зміцнюють або розводять до концентрації 200 г/л.
Натрію гідроксиду розчин розведений. /081402.
8.5 г натрію гідроксиду Р розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
100 мл.
Натрію гідроксиду метанольний розчин. 1081403.
40 мг натрію гідроксиду Р розчиняють у 50 мл во-
ди Р, одержаний розчин охолоджують і додають
50 мл метанолу Р.
Натрію гідроксиду розчин концентрований. 1081404.
42 г натрію гідроксиду Р розчиняють у воді Р і до-
водять об’єм розчину тим самим розчинником до
100 мл.
Натрію гідросульфіт. N301038. {М.м. 104.1). 1115700
[7631-90-5]. Натрію бісульфіт.
Кристалічний порошок білого кольору. Легко розчин-
ний у воді, помірно розчинний у 96 % спирті.
На повітрі частково втрачає сірки діоксид і поступово
окиснюється до сульфату.
Натрію гіпоброміту розчин. 1081500.
20 мл розчину натрію гідроксиду концентрованого Р і
500 мл води Р змішують на льодяній бані, додають
5 мл розчину брому Р і обережно перемішують до роз-
чинення.
Готують безпосередньо перед використанням.
Натрію гіпофосфіт. №Н2РО2,Н2О. {М.м. 106.0).
1081700. [10039-56-2]. Натрію фосфінат моногідрат.
Кристалічний порошок битого кольору або безбарвні
кристали. Гігроскопічний, легко розчинний у воді,
розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Натрію гіпохлориту розчин концентрований. 1081600.
Містить не менше 25 г/л і не більше 30 г/л активного
хлору.
Рідина жовтавого кольору, має лужну реакцію.
Кількісне визначення. У колбу з 50 мл води Р послідо-
вно поміщають 1 г калію йодиду Р і 12.5 мл кислоти
оцтової розведеної Р. 10.0 мл розчину концентрова-
ного натрію гіпохлориту доводять водою Р до об’єму
100.0 мл. 10.0 мл одержаного розчину додають у колбу
з реактивами та титрують 0.1 М розчином натрію
тіосульфату, використовуючи як індикатор 1 мл роз-
чину крохмалю Р
1 мл 0.1 М розчину натрію тіосульфату відповідає
3.546 мг активного хлору.
Зберігають у захищеному від світла місці.
Натрію глюкуронат. С6Н9НаО7,Н2О. {М.м. 234.1).
1080900. Натрію О-глюкуронат моногідрат
[а]^1 : близько +21.5°. Визначення проводять, вико-
ристовуючи розчин 20 г/л.
Натрію декансульфонат. С10Н
21 НаО38. (М.м. 244.3).
1079800. [13419-61-9].
Кристалічний порошок або пластівці білого чи майже
білого кольору. Легко розчинний у воді, розчинний у
метанолі.
Натрію дезоксирибонуклеат. 1079900. [73049-39-5].
(Близько 85 % має молекулярну масу 2x107 або
більше).
Волокниста речовина білого кольору; одержують із
тимуса теляти.
Випробування на придатність. 10 мг розчиняють в
імідазольному буферному розчині рН 6.5 Р і доводять
об’єм розчину тим самим буферним розчином до
10.0 мл (розчинА). 2.0 мл розчину А доводять іміда-
зольним буферним розчином рН 6 5 Рдо об’єму 50.0 мл.
Оптична густина (2.2.25) одержаного розчину, виміря-
на задовжини хвилі 260 нм, має становити від 0.4до0.8.
До 0.5 мл розчину А додають 0.5 мл імідазольного бу-
ферного розчину рН 6.5 Р, 3 мл розчину 25 г/л (НС1О4)
кислоти хлорної, утворюється осад, який центрифугу-
ють. Вимірюють оптичну густину надосадової рідини
задовжини хвилі 260 нм, використовуючи як компен-
саційну рідину суміш, що складається з 1 мл імідазоль-
ного буферного розчину рН 6.5 Р і 3 мл розчину 25 г/л
(НС1О4) кислоти хлорної. Оптична густина має бути
не більше 0.3.
У кожну з двох пробірок поміщають по 0.5 мл розчи-
ну А і 0.5 мл розчину порівняння зразка стрептодорна-
зи, який містить 10 МО/мл, в імідазольному буферному
розчині рН 6.5 Р. В одну пробірку негай но додають 3 мл
розчину 25 г/л (НС1О4) кислоти хлорної, утворюється
осад, який центрифугують і збирають надосадову ріди-
ну (а). Другу пробірку нагрівають при температурі
37 °С протягом 15 хв, додають 3 мл розчину 25 г/л
(НС1О4) кислоти хлорної, центрифугують і збирають
надосадову рідину (Ь). Вимірюють оптичну густину на-
досадової рідини (Ь) задовжини хвилі 260 нм, викори-
стовуючи як компенсаційний розчин надосадову ріди-
ну (а). Оптична густина має бути не менше 0.15.
Натрію дигідрофосфат. 1080100. [10028-24-7]. Див.
статтю Натрію дигідрофосфат дигідрат.
Натрію дигідрофосфат безводний. КаН2РО4.
{М.м. 120.0). 1080200. [7558-80-7].
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
233
4.1.1. Реактиви
Порошок білого кольору, гігроскопічний.
Зберігають у повітронепроникному контейнері.
Натрію дигідрофосфат моногідрат. №Н2РО4,Н2О.
(Мм. 138.0) 1080300. [10049-21 5].
Кристали або гранули білого кольору, які злегка
розпливаються на повітрі. Легко розчинний у воді,
практично не розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Натрію дитіоніт. Ма282О4. (М.м. 174.1). 1080400.
[7775-14-6].
Кристалічний порошок білого або сірувато-білого ко-
льору; на повітрі окислюється. Дуже легко розчинний
у воді, мало розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Натрію діетилдитіокарбамат. С5Н ИІКГ\'а82,ЗН2О.
(Мм. 225.3). 1080000. [20624-25-3]
Безбарвні або білого кольору кристали. Легко розчин-
ний у воді, розчинний у 96 % спирті. Водний розчин
безбарвний.
Натрію додецилсульфат. 1080500. [151-21-3]. Див. стат-
тю Натрію лаурилсульфат. за винятком вмісту, який
має бути не менше 99.0 %.
Буферний робочий розчин для електрофорезу в сис-
темі натрію додецилсульфат-поліакриламілний гель
(8П8-РА6Е). 1114900.
151.4 г трис(гідроксиметил)амінометану Р, 721.0 г
гліцину Р і 50.0 г натрію лаурилсульфату Р розчи-
няють у воді Р і доводять тим самим розчинником
до об’єму 5000 мл. Безпосередньо перед викорис-
танням розводять водою Р у 10 разів і перемішу-
ють.
рН (2.2.3) одержаного розчину має бути від 8.1 до 8.8.
Буферний зразковий розчин (концентрований) для
електрофорезу в системі натрію додецилсульфат -
поліакриламідний гель (8Н8-РАСЕ). /115000.
1.89 г трис(гідроксиметил)амінометану Р, 5.0 г
натрію лаурилсульфату Р, 50 мг бромфенолового
синього Р і 25.0 мл гліцерину Ррозчиняють у 100 мл
води Р. Доводять рН розчину до 6.8 кислотою хло-
ристоводневою Р і доводять водою Р до об’єму
125 мл.
Буферний зразковий розчин (концентрований) для
електрофорезу в системі натрію додецилсульфат -
поліакриламідний гель (8Б8-РАСЕ) для відновних
умов. 1122100.
3.78 г трис(гідроксиметил)амінометану Р, 10.0 г
натрію додецилсульфату Р, 100 мг бромфенолового
синього Р150.0 мл гліцерину Р розчиняють у 200 мл
води Р. До одержаного розчину додають 25.0 мл
2-меркаптоетанолу Р і доводять рН (2.2.3) розчи-
ну до 6.8 кислотою хлористоводневою Р і доводять
водою Рао об’єму 250.0 мл.
Альтернативно як відновлююча речовина замість!
2-меркаптоетанолу може бути використаний!
дитіотреїтол. У цьому разі зразковий буферний!
розчин готують таким чином: 3.78 г трисїгідрокЛ
симетил)амінометану Р, 10.0 г натрію <)о<)еі(ш-
сульфату Р, 100 мг бромфенолового синього Р'
50.0 мл гліцерину Р розчиняють у 200 мл води Р.
Доводять рН (2.2.3) розчину до 6.8 кислотоюхіо-1
ристоводневою Р і розводять водою Р до об’ємі І
250.0 мл. Безпосередньо перед використанням
додають дитіотреїтол Рао кінцевої концентрації
100 мМ.
Натрію едетат. 1080600. [6381-92-6]. Див. статтю Ди-
натрію едетат.
Натрію йодид. 1081800. [7681-82-5]. Див. статтю І
Натрію йодид.
Натрію карбонат. 1079200. |6132-02-1]. Див. статтю
Натрію карбонату декагідрат.
Натрію карбонат безводний. №2СО3. (Мм. 106.0).
1079300. [497-19-8]. Динатрію карбонат.
Порошок білого кольору, гігроскопічний. Легко роз-
чинний у воді.
Втрата в масі при висушуванні при температурі близь-
ко 300 °С має бути не більше 1 %.
Зберігають у повітронепроникному контейнері.
Натрію карбонату розчин. 1079301.
Розчин 106 г/л натрію карбонату безводного Р.
Натрію карбонату розчин РІ. 1079302.
Розчин 20 г/л натрію карбонату безводного Р}
0.1 М розчині натрію гідроксиду.
Натрію кобальтинітрит. №3|Со(МО2)6]. (М.м. 403.9).
1079700. [ 13600-98-11. Тринатрію гексанітрокобаль-
тат(ІІІ).
Порошок оранжево-жовтого кольору. Легко розчин-
ний у воді, мало розчинний у 96 % спирті.
Натрію кобальтинітриту розчин. 1079701.
Розчин 100 г/л.
Готують безпосередньо перед використанням.
Натрію лаурилсульфат. 1081900. [151 21-3]. Див. стат-
тю Натрію лаурилсульфат.
Натрію метабісульфіт. 1082000. [7681-57-4]. Див. стат-
тю Натрію метабісульфіт.
Натрію метансульфонат. СН38О3№. (М.м. 118.1).
1082100. [2386-57-4].
Кристалічний порошок білого кольору, гігро-
скопічний.
Зберігають у повітронепроникному контейнері.
234
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Натрію молібдат. Ка2МоО4,2Н2О. (М.м. 242.0).
1082200. [10102-40-6]. Динатрію молібдат дигідрат.
Кристалічний порошок білого кольору або безбарвні
кристали. Легко розчинний у воді.
Натрію нафіохінонсульфонат. С10Н5№О5$. (М.м. 260.2).
1082300. |521-24-4]. Натрію 1,2-нафтохінон-4-суль-
фонат.
Кристалічний порошок від жовтого до оранжево-жов-
того кольору. Легко розчинний у воді, практично не
розчинний у 96 % спирті.
Нагрію нітрат. КаГ\'О3. (М.м. 85.0). 1082400.17631 -99-4].
Порошок або гранули білого кольору або безбарвні,
прозорі кристали, які розпливаються на повітрі. Легко
розчинний у воді, мало розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері
Натрію нітрит. NаNО2. (М.м. 69.0). 1082500. [7632-00-0].
Містить не менше 97.0 % 1МаМО2.
Гранульований порошок білого кольору або кристалічний
порошок жовтавого кольору. Легко розчинний у воді.
Натрію нітриту розчин. 108250!.
Розчин 100 г/л.
Готують безпосередньо перед використанням.
Натрію нітропрусид. Ка2[Ее(СМ)5(1\ІО)],2Н2О.
(Мм. 298.0). 1082600. [13755-38-9]. Натрію пен-
таціано-нітрозилферат( III) дигідрат.
Порошок або кристали червонувато-коричневого
кольору Легко розчинний у воді, мало розчинний у
96 % спирті.
Натрію оксалат. С2Ка2О4. (М.м. 134.0). 1082900. [62-76-0].
Кристалічний порошок білого кольору. Розчинний у
воді, практично не розчинний у 96 % спирті та ефірі.
Натрію октансульфонат. С8Н17№О38. (М.м. 216.3).
1082700. [5324-84-5].
Містить не менше 98.0 % С8Н,7КаО38
Кристалічний порошок або пластівці білого або май-
же білого кольору. Легко розчинний у воді, розчинний
у метанолі.
Оптична густина (2.2.25). Оптична густина розчину
54 г/л за довжини хвилі 200 нм має бути не більше
0.10, а за довжини хвилі 250 нм - не більше 0.01.
Натрію октилсульфат. С8Н)7КаО4$. (М.м. 232.3).
1082800. |142-31-4].
Кристалічний порошок або пластівці білого або май-
же білого кольору. Легко розчинний у воді, розчинний
у метанолі.
Натрію пентансульфонат. С5НцКаО38. (М.м. 174.2).
1083000. |22767-49-3].
Тверда кристалічна речовина білого кольору. Розчин-
ний у воді.
Натрію перйодат. К'аІО4. (М.м. 213.9). 1083200.
[7790-28-5]. Натрію метаперйодат.
Містить не менше 99.0 % №ІО4.
Кристалічний порошок або кристали білого кольору.
Розчинний у воді й мінеральних кислотах.
Натрію перйодату розчин. 1083201.
1.07 г натрію перйодату Ррозчиняють у воді Р. до-
дають 5 мл кислоти сірчаної розведеної Р і доводять
об’єм розчину водою Рдо 100.0 мл.
Готують безпосередньо перед використанням.
Натрію перхлорат. Г\'аСЮ4,Н2О. (М.м. 140.5). 1083100.
[7791-07-3].
Містить не менше 99.0 % КаС1О4,Н2О.
Кристали білого кольору, які розпливаються на
повітрі. Дуже легко розчинний у воді.
Зберігають у щільно закритому контейнері.
Натрію пікрату лужний розчин. 1083300.
Змішують 20 мл розчину кислоти пікринової Р і 10 мл
розчину 50 г/л натрію гідроксиду Р, доводять об’єм
розчину водою Рдо 100 мл.
Термін придатності 2 доби з моменту приготування.
Натрію пірофосфат. Ка4Р2О7,10Н2О. (М.м. 446.1).
1083600. [13472-36-1]. Тетранатрію дифосфат де-
ка гідрат.
Безбарвні кристали, які злегка звітрюються. Легко
розчинний у воді.
Натрію родизонат. С6Ка2О6. (М.м. 214.0). 1122300.
[523-21 -7]. [(3,4,5,6-тетраоксоциклогекс-1 -єн-1,2-
ілен)діокси]динатрій.
Кристали фіолетового кольору. Розчинний у воді з ут-
воренням оранжево-жовтого розчину. Розчини не-
стабільні, їх готують у день використання.
Натрію саліцилат. 1083700. [54-21-7]. Див. статтю
Натрію саліцилат.
Натрію сульфат безводний. 1083800. [7757-82-6|.
Прожарений при температурі від 600 °С до 700 °С
натрію сульфат безводний має задовольняти вимоги,
зазначені у статті Натрію сульфат безводний.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. Визначення проводять при температурі 130 °С.
Натрію сульфід. №28,9Н2О. (М.м. 240.2). 1083900.
[1313-84-4]. Динатрію сульфід нонагідрат.
Безбарвні кристали, які швидко жовтіють, а також
розпливаються на повітрі. Дуже легко розчинний у воді
Зберігають у повітронепроникному контейнері.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
235
4.1.1. Реактиви
Натрію сульфіду розчин. 1083901.
12 г натрію сульфіду Р розчиняють при нагріванні в
45 мл суміші розчинників вода Р-гліцерин (85 %) Р
(10:29), потім охолоджують і доводять об’єм розчи-
ну тією самою сумішшю розчинників до 100 мл.
Розчин має бути безбарвним.
Натрію сульфіт. 1084000. [27610-45-3|. Див. статтю
Натрію сульфіт гептагідрат.
Натрію сульфіт безводний. 1084100. [7757-83-7]. Див.
статтю Натрію сульфіт безводний.
Натрію тартрат. (М.м. 230.1).
1084200. [6106-24-7]. Динатрію (2Я,ЗЯ)-2,3-дигідро-
ксибутандіоат дигідрат.
Кристали або гранули білого кольору. Дуже легко роз-
чинний у воді, практично не розчинний у 96 % спирті.
Натрію тетрадейтеродиметилсилапентаноат.
С6Н92Н4№О28і. (М.м. 172.3). 1084300. Т8Р. Натрію
(2,2.3.3-тетрадейтеро)-4,4-диметил-4-силапентаноат.
Ступінь дейтерування не менше 99 %>.
Кристалічний порошок білого кольору. Легко розчин-
ний у воді, етанолі й метанолі.
Температура плавлення: близько 300 °С.
Вода й дейтерію оксид: не більше 0.5 %.
Натрію тетрафенілборат. №В(С6Н5)4. (М.м. 342.2).
1084400. [143-66-8].
Об’ємний порошок білого або дещо жовтуватого ко-
льору. Легко розчинний у воді й ацетоні.
Натрію тетрафенілборату розчин. 1084401.
Розчин 10 г/л.
Якщо необхідно, перед використанням фільтрують.
Термін придатності 7 діб
Натрію тіогліколят. С2Н3№О28. (М.м. 114.1). 1084500.
[367-51-1]. Натрію меркаптоацетат.
Гранульований порошок або кристали білого кольору.
Гігроскопічний, легко розчинний у воді й метанолі,
мало розчинний у 96 % спирті.
Зберігають у повітронепроникному контейнері.
Натрію тіосульфат. 1084600. [10102-17-7]. Див. статтю
Натрію тіосульфат.
Натрію флуоресцеїнат. С20Н1()^а2О5. (М.м. 376.3).
1080700. [518-47-8]. Показник Шульца№ 880. Кольо-
ровий індекс № 45350. Флуоресцеїн натрію. Динатрію
2-(3-оксо-6-оксидо-3//-ксантен-9-іл)бензоат.
Порошок оранжево-червоного кольору. Легко роз-
чинний у воді. Водні розчини мають інтенсивну жов-
тувато-зелену флуоресценцію.
Кристалічний порошок або гранули білого кольор
які розпливаються. Розчинний у воді й гліцерині, Ма-
ло розчинний у 96 % спирті.
Температура плавлення: близько 253 °С.
Натрію фосфат додекагідрат. №3РО4,12Н2О.
(М.м. 380.1). 1094300. [10101-89-0]. Тринатрію фосфа:
додекагідрат.
Безбарвні або білого кольору кристали. Легко розчин-
ний у воді.
Натрію фторид. 1080800. [7681-49-4]. Див. статтю
Натрію фторид.
Натрію хлорид. 1079500. [7647 14-5]. Див. статтю
Натрію хлорид.
Натрію хлориду розчин. 1079502.
Розчин 20 % (м/м).
Натрію хлориду насичений розчин. 1079503.
1 частину натрію хлориду Р змішують із 2 частина-
ми води Р, періодично струшують і відстоюють.
Перед використанням розчин декантують і
фільтрують, якшо необхідно.
Натрію цетостеарилсульфат. 1079400. Див. статтю
Натрію цетостеарилсульфат.
Натрію цитрат. 1079600. [6132-04-3]. Див. статтю
Натрію цитрат.
Нафталін. С10Н8. (М.м. 128.2). 1057100. [91-20-3].
Кристали білого кольору. Практично не розчинний)
воді, легко розчинний в ефірі, розчинний у 96 Й
спирті.
Температура плавлення: близько 80°С.
Нафталін, використовуваний для рідинної сцинтиляції
має бути відповідного ступеня чистоти.
Нафтарзон. С16НиА8М2№2О1082. (М.м. 576.3)
1121400. [132-33-2]. Торин. Динатрію 4-[(2-арсоно
феніл)азо|-3-гідроксинафталін-2,7-дисульфонат.
Порошок червоного кольору. Розчинний у воді.
Нафтарзону розчин. 1121401.
Розчин 0.58 г/л.
Випробування на чутливість. До 50 мл 96 % спир
ту Р додають 20 мл води Р, 1 мл 0.05 М розчин
кислоти сірчаної за 1 мл розчину нафтарзону й ти
трують 0.025 М розчином барію перхлорату до
реходу забарвлення розчину від оранжево-жовт
до оранжево-рожевого.
Зберігають у захищеному від світла місці
Термін зберігання 7 діб.
Натрію форміат. СНГ\аО2. (М.м. 68.0). 1122200
[141-53-7].
Нафтиламін. С10Н9М. (М.м. 143.2). 1057700. [134-32-
1-Нафтиламін.
4.1.1. Реактиви
Кристалічний порошок білого кольору, під дією світла
й повітря рожевіє. Мало розчинний у воді, легко роз-
чинний у 96 % спирті й ефірі.
Температура плавлення: близько 51 °С.
Зберігають у захищеному від світла місці.
Нафтилетилендіаміну дигідрохлорид. С12Н16С12К2.
(Мм. 259.2). 1057800. [1465-25-4]. А-(1-Нафтил)ети-
лендіаміну дигідрохлорид. Може містити крис-
талізаційний метанол.
Порошок білого або жовтувато-білого кольору. Роз-
чинний у воді, мало розчинний у 96 % спирті.
а-Нафтол. С10Н8О. (М.м. 144.2). 1057300. [90-15-3].
1-Нафтол.
Кристалічний порошок білого кольору або безбарвні
або білого кольору кристали, які темніють під впли-
вом світла. Мало розчинний у воді, легко розчинний у
96 % спирті та ефірі.
Температура плавлення: близько 95 °С.
Зберігають у захищеному від світла місці.
а-Нафтолу розчин. 1057301.
0.10 г а-нафтолу Р розчиняють у 3 мл розчину
150 г/л натрію гідроксиду Р і доводять об’єм роз-
чину водою Рао 100 мл.
Готують безпосередньо перед використанням.
0-Нафтол. С10Н8О. (М.м. 144.2). 1057400. [135-19-3].
2-Нафтол.
Пластинки або кристали білого або слабко рожевого
кольору. Дуже мало розчинний у воді, дуже легко роз-
чинний у 96 % спирті.
Температура плавлення: близько 122 °С.
Зберігають у захищеному від світла місці.
/З-Нафтолу розчин. 1057401.
5 г свіжоперекристалізованого р-нафтолу Р роз-
чиняють у 40 мл розчину натрію гідроксиду розве-
деного Р і доводять об’єм розчину водою Р до
100 мл.
Готують безпосередньо перед використанням.
Д-Нафтолу розчин РІ. 1057402.
3 .0 мг р-нафтолу Р розчиняють у 50 мл кислоти
сірчаної Р і доводять об’єм розчину тією самою
кислотою до 100.0 мл.
Готують безпосередньо перед використанням.
Нафтолбензеїн. С27Н20О3. (М.м. 392.5). 1057600.
|6948-88-5]. а-Нафтолбензеїн. Фенілбіс(4-гідрокси-
нафтил)метанол.
Порошок коричнювато-червоного кольору або блис-
кучі кристали коричнювато-чорного кольору. Прак-
тично не розчинний у воді, розчинний у 96 % спирті й
кислоті оцтовій льодяній.
Нафтолбензеїну розчин. 1057601.
Розчин 2 г/л у кислоті оцтовій безводній Р.
Випробування на чутливість. До 50 мл кислоти оц-
тової льодяної Р додають 0.25 мл розчину нафтол-
бензеїну; з’являється коричнювато-жовте забарв-
лення, яке має перейти у зелене при додаванні не
більше 0.05 мл 0.1 Мрозчину кислоти хлорної.
Нерилацетат. С12Н20О2. (М.м. 196.3). 1108000. [141-12-8].
(2)-3,7-Диметилокта-2.6-дієнілацетат.
Безбарвна, масляниста рідина.
г/2у : близько 0.907.
«о : близько 1.460.
Температура кипіння25: 134 °С.
Нерилацетат, використовуваний у газовій хромато-
графії, має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія квіток
померанця, використовуючи нерилацетат як випробо-
вуваний розчин.
Площа основного піка має бути не менше 93.0 % суми
площ усіх піків.
транс-Неролідол. С15Н26О. (М.м. 222.4). Н07900.
[40716-66-3]. 3,7,11 -Триметилдодека-1,6,10-триєн-3-ол.
Рідина слабко жовтого кольору з легким запахом лілії
або конвалії. Практично не розчинний у воді й гліце-
рині, змішується з 96 % спиртом.
г/20 : близько 0.876.
: близько 1.479.
Температура кипіння12: від 145 °С до 146 °С.
транс-Неролідол, використовуваний у газовій хрома-
тографії, має витримувати таке додаткове випробу-
вання.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія квіток
померанця, використовуючи транс-неролідол як ви-
пробовуваний розчин.
Площа основного піка має бути не менше 90.0 % суми
площ усіх піків.
Нікель-алюмінієвий сплав. 1058100.
Містить від 48 % до 52 % алюмінію (А1, А.м. 26.98) і від
48 % до 52 % нікелю (N1, А.м. 58.70).
Перед використанням подрібнюють до дрібного по-
рошку (180).
Практично не розчинний у воді, розчинний у міне-
ральних кислотах.
Нікель-алюмінієвий сплав, вільний від галогенів.
1118100.
Містить від 48 % до 52 % алюмінію (А1, А.м. 26.98) і від
48 % до 52 % нікелю (N1, А.м. 58.70).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
237
4.1.1. Реактиви
Тонкий порошок сірого кольору. Практично не роз-
чинний у воді, розчинний у мінеральних кислотах з
утворенням солей.
Хлориди. Не більше 0.001 %(10 ррт). 2.00 г розчиня-
ють у 40 мл кислоти азотної Р, розчин упарюють май-
же насухо. Залишок розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 20.0 мл.
Розчин розливають порівну в дві пробірки. У кожну
пробірку додають по 1.0 мл 0.1 М розчину срібла нітра-
ту й через 15 хв фільтрують. До одержаного фільтрату
однієї пробірки додають 0.25 мл розчину 40 мкг/мл
(СІ) натрію хлориду (еталонний розчин). Через 5 хв
порівнюють опалесценцію випробовуваного розчину
з еталонним розчином. Випробовуваний розчин має
витримувати випробування на хлориди.
Нікелю сульфат. Мі$О4,7Н2О {М.м. 280.9). 1058000.
[10101-98-1]. Нікелю сульфат гептагідрат.
Кристалічний порошок або кристали зеленого кольо-
ру. Легко розчинний у воді, мало розчинний у 96 %
спирті.
Нікелю хлорид. МіС12. {М.м. 129.6). 1057900. [7718-54-9].
Нікелю хлорид безводний.
Кристалічний порошок жовтого кольору. Дуже легко
розчинний у воді, розчинний у 96 % спирті. Сублі-
мується у відсутності повітря й легко абсорбує аміак.
Водний розчин має кислу реакцію.
Нікотинамід-аденіну динуклеотид. С2|Н27М7О|4Р2.
{М.м. 663). 1108100. [53-84-9]. І^АО .
Порошок білого кольору, сильно гігроскопічний. Лег-
ко розчинний у воді.
Нікотинамід-аденіну динуклеотиду розчин. 1108101
40 мг нікотинамід-аденіну динуклеотиду Р розчи-
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Готують безпосередньо перед використанням.
Нільський синій А. С20Н2^3О58. {М.м. 415.5). 1058200.
[3625-57-8]. Показник Шульца № 1029. Кольоровий
індекс №51180. 5-Аміно-9-(діетиламіно)бензо[а]фе-
ноксазинілію кислий сульфат.
Кристалічний порошок зеленого кольору із бронзо-
вим блиском. Помірно розчинний у 96 % спирті, кис-
лоті оцтовій льодяній та піридині.
Розчин 0.005 г/л у спирті (50 %, об/об) Р має макси-
мум поглинання (2.2.25) за довжини хвилі 640 нм.
Нільського синього А розчин. 1058201.
Розчин 10 г/л у кислоті оцтовій безводній Р.
Випробування на чутливість. До 50 мл кислоти оц-
тової безводної Р додають 0.25 мл розчину
нільського синього А; з’являється блакитне за-
барвлення. яке має перейти у синьо-зелене при
додаванні 0.1 мл 0.1 Мрозчину кислоти хлорної.
Зміна забарвлення. Від синього до червоного в
інтервалі рН 9.0-13.0.
Нінгідрин. СУН4О?,Н2О. {М.м. 178.1). 1058300. [485-47-2|. І
1,2,3-Індантрион моногідрат.
Кристалічний порошок білого або злегка жовтого ко-
льору. Розчинний у воді й 96 % спирті, мало розчин-
ний в ефірі.
Зберігають у захищеному від світла місці.
Нінгідрину і олова(ІІ) хлориду реактив. 1058301.
0.2 г нінгідрину Р розчиняють у 4 мл гарячої води Р,
додають 5 мл розчину 1.6 г/л олова(П) хлориду Р,
залишають на 30 хв, фільтрують і зберігають при
температурі від 2 °С до 8 °С.
Безпосередньо перед використанням до 2.5 мл
одержаного розчину додають 5 мл води Р і 45 мл
2-пропанолу Р.
Нінгідрину і олова(П) хлориду реактив РІ. 1058302.
4 г нінгідрину Р розчиняють у 100 мл моноетилоео-
го ефіру етиленгліколю Р. Обережно струшують
з 1 г катіонообмінної смоли Р (від 300 мкм до
840 мкм) і фільтрують (розчин А). 0.16 г олова(ІІ)
хюриду Р розчиняють у 100 мл буферного розчину
рН 5.5 Р (розчин В).
Безпосередньо перед використанням змішують
рівні об’єми розчинів АйВ.
Нінгідрину розчин. 1058303.
Розчин 2 г/л нінгідрину Р у суміші розчинників
кислота оцтова розведена Р - бутанол Р(5:95).
Нінгідрину розчин РІ. 1058304.
1.0 г нінгідрину Р розчиняють у 50 мл 96 % спир-
ту Р і додають 10 мл кислоти оцтової льодяної Р
Нінгідрину розчин Р2. 1058305.
З г нінгідрину Р розчиняють у 100 мл розчину
45.5 г/л натрію метабісульфіту Р.
Нінгідрину розчин РЗ. 1058306.
Розчин 4 г/л нінгідрину Р у суміші розчинників
кислота оцтова безводна Р-бутанол 7і (5:95).
Нітроанілін. С6Н6М2О2. (Мм. 138.1). 1058600. [100-01-6|.
4-Нітроанілін.
Кристалічний порошок яскраво-жовтого кольору. Ду-
же мало розчинний у воді, помірно розчинний у кип-
лячій воді, розчинний у 96 % спирті та ефірі, утворює
водорозчинні солі із сильними мінеральними кисло-
тами.
Температура плавлення: близько 147 °С.
Нітробензальдегід. С7Н51\Ю3. {М.м. 151.1). 1058700.
[552-89-6]. 2-Нітробензальдегід.
Голчасті кристали жовтого кольору. Мало розчинний у
воді, легко розчинний у 96 % спирті, розчинний в
ефірі, сублімується парою.
Температура плавлення: близько 42 °С.
238
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Нітробензальдегідний папір. 1058701.
0.2 г нітробензальдегіду Р розчиняють у 10 мл розчи-
ну 200 г/л натрію гідроксиду Р. Термін придатності
розчину 1 год. У одержаний розчин занурюють ниж-
ню половину смужки із повільно фільтруючого па-
перу завдовжки 10 см і завширшки 0.8 -1 см. Надли-
шок реактиву видаляють, промокаючи смужку між
двома аркушами фільтрувального паперу.
Використовують протягом кількох хвилин після
приготування.
Нітробензальдегіду розчин. 1058702.
0.12 г порошку нітробензальдегіду Р додають до
10 мл розчину натрію гідроксиду розведеного Р,
струшують протягом 10 хв і фільтрують.
Готують безпосередньо перед використанням.
Нітробензилхлорид. С7Н6С1МО2. (М.м. 171.6). 1059000.
1100-14-1 ]. 4- Н ігробе н зилхлорид.
Кристали світло-жовтого кольору. Спричиняє сльозо-
течу. Практично не розчинний у воді, дуже легко роз-
чинний у 96 % спирті та ефірі.
Нітробензоїлхлорид. С7Н4С1Т9О3. (М.м. 185.6).
1058900. [122-04-3] 4-Нітробензоїлхлорид.
Кристали або кристалічна маса жовтого кольору, яка
розпливається на повітрі. Розчинний у розчині натрію
гідроксиду з утворенням жовтувато-оранжевого за-
барвлення.
Температура плавлення: близько 72 °С.
Нітробензол. С6Н5КО2. (ЛО/. 123.1). 1058800. [98-95-3].
Безбарвна або жовтавого кольору рідина. Практично
не розчинний у воді, змішується з 96 % спиртом і
ефіром.
Температура кипіння: близько 211 °С.
Динітробензол. До 0.1 мл нітробензолу додають 5 мл
ацетону Р, 5 мл води Р і 5 мл розчину натрію гідрокси-
ду концентрованого Р і струшують; після розділення
шарів верхній шар має бути майже безбарвний.
4-(4-Нітробензил)піридин. С12Н10М2О2. (М.м. 214.2).
1101900. [1083-48-3].
Порошок жовтого кольору.
Температура плавлення: близько 70 °С.
Нітрованадомолібденовий реактив. 1060100. Див.
Нітромолібденованадієвий реактив Р
Нітроетан. С2Н5МО2. (М.м. 75.1). 1059200. [79-24-3].
Прозора, безбарвна, масляниста рідина.
Температура кипіння: близько 114 °С.
Нітрозодипропіламін. С6Н14М2О. (М.м. 130.2). 1099900.
[621-64-7]. Дипропілнітрозамін.
Рідина. Розчинний в етанолі, ефірі та концентрованих
кислотах.
: близько 0.915.
Температура кипіння: близько 78 °С.
Ступінь чистоти підходить для визначення хемі-
люмінесценції.
Нітрозодипропіламіну розчин. 1099901.
Вводять 78.62 г етанолу Р, проколюючи ін’єк-
ційною голкою пробку посудини, яка містить
нітрозодипропіламін Р, розводять етанолом Р у
співвідношенні 1:100 і помішають по 0.5 мл у кон-
тейнери з обтиснутими кришками.
Зберігають у захищеному від світла місці при тем-
пературі 5 °С.
Нітрометан. СН3НО2. (М.м. 61.0). 1059700. [75-52-5].
Прозора, безбарвна, масляниста рідина. Мало роз-
чинний у воді, змішується з 96 % спиртом та ефіром.
г/2о : від 1.132 до 1.134.
п2° : від 1.381 до 1.383.
Температурні межі перегонки (2.2.11). Від 100 °С до
103 °С; має переганятись не менше 95 %.
Нітромолібденованадієвий реактив. 1060100.
Розчин І. 10 г амонію молібдату Р розчиняють у воді Р.
додають 1 мл розчину аміаку Р і доводять об’єм розчи-
ну водою Рао 100 мл.
Розчин II. 2.5 г амонію ванадату Р розчиняють у га-
рячій воді Р, додають 14 мл кислоти азотної Р і дово-
дять об’єм розчину водою Рао 500 мл
До 96 мл кислоти азотної Р додають 100 мл розчину 1
і 100 мл розчину 11 і доводять об’єм розчину водою Рао
500 мл.
Нітротетразолієвий синій. С40Н30С12ЇМ|0О6. (М.м. 818).
1060000. [298-83-9]. 3,3'-(3,3'-Диметокси-4,4'-ди-
фенілен)ди[2-(4-нітрофеніл)-5-феніл-2//-тетразолію]
дихлорид. л-Нітротетразолієвий синій.
Кристали. Розчинний у метанолі з утворенням прозо-
рого розчину жовтого кольору.
Температура плавлення: близько 189 °С, із розкла-
данням.
Нітрофурантоїн. 1099700. [67-20-9]. Див. статтю
Нітрофурантоїн.
(5-Нітро-2-фурил)метилену діацетат. С9Н9ІЧО7.
(М.м. 243.2). 1099800. [92-55-7]. Нітрофурфуролу
діацетат. 5-Нітрофурфурилідену діацетат.
Кристали жовтого кольору.
Температура плавлення: близько 90 °С.
Нітрохромовий реактив. /059100.
0.7 г калію дихромату Р розчиняють у кислоті
азотній Р і доводять об’єм розчину тією самою кисло-
тою до 100 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
239
4.1.1. Реактиви
Нордазепам. С^НцСІ^О. (Мм. 270.7). 1060200.
[340-57-8]. 7-Хлор-2,3-дигідро-5-феніл-1//-1,4-бен-
зодіазепін-2-он.
Кристалічний порошок білого або світло-жовтого ко-
льору. Практично не розчинний у воді, мало розчин-
ний у 96 % спирті.
Температура плавлення: близько 216 °С.
В,Ь-Норлейцин. С6Н13КІО2. (М.м. 131.2). 1060300.
[616-06-8]. (/?5)-2-Аміногексанова кислота. Амінока-
пронова кислота.
Блискучі кристали. Помірно розчинний у воді, роз-
чинний у кислотах.
Норпсевдоефедрину гідрохлорид. С9Н |4С1К'О.
(М.м. 187.7). 1060400. [53643-20-2]. (1Я,2Я)- або
(15,25)-2-Аміно-1-фенілпропанолу гідрохлорид.
Кристалічний порошок. Розчинний у воді.
Температура плавлення: від 180 °С до 181 °С.
Носкапіну гідрохлорид. 1060500. [912-60-7]. Див. стат-
тю Носкапіну гідрохлорид.
Октадецил[3-[3,5-біс(1,1-диметилетил)-4-гідрокси-
феніл]пропіонат]. С35Н62Оз. (М.м. 530.9). 1060600.
[2082-79-3]. Октадецил-3-(3,5-ди-шреш-бутил-4-
гідрокс и фе н іл) п роп і о нат.
Кристалічний порошок білого або жовтавого кольо-
ру. Практично не розчинний у воді, дуже легко роз-
чинний в ацетоні й гексані, мало розчинний у мета-
нолі.
Температура плавлення: від 49 °С до 55 °С.
Октанол. С8НІ8О. (М.м. 130.2). 1060700. [111-87-5].
1-Октанол. Каприловий спирт.
Безбарвна рідина. Не розчинний у воді, змішується з
96 % спиртом та ефіром.
^20 : близько 0.828.
Температура кипіння: близько 195 °С.
З-Октанон. С8Н16О. (М.м. 128.2). 1114600. [106-68-3].
Етилпентилкетон.
Безбарвна рідина з характерним запахом.
^20 : близько 0.822.
: близько 1.415.
Температура кипіння: близько 167 °С.
З-Октанон, використовуваний у газовій хромато-
графії, має витримувати таке додаткове випробуван-
ня:
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28). як зазначено у статті Олія лавандо-
ва.
Випробовуваний розчин. Випробовувана речовина.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків.
Октоксинол 10. С34Н62Оц (середня). (М.м. 647) І
1060800. [9002-93-1]. а-[4-(1,1,3,3-Тетраметилбу-1
тил )фе н іл ] -а>- гідрокси пол і(оксіетиле н).
Прозора, в’язка рідина світло-жовтого кольору. Змі- І
шується з водою, ацетоном і 96 % спиртом, розчинний І
у толуолі.
Зберігають у повітронепроникному контейнері.
Олеамід. С18Н35МО. (М.м. 281.5). 1060900. (7)- Окта- ]
дек-9-еноамід.
Порошок або гранули від білого до жовтуватого ко- І
льору. Практично не розчинний у воді, дуже легке І
розчинний у метиленхлориді, розчинний в етанолі.
Температура плавлення: близько 80 °С.
Оливкова олія. 1061000. [8001-25-0]. Див. статтю Олія
оливкова рафінована.
Олова(ІІ) хлорид. 8пС12,2Н2О. (М.м. 225.6). 1085000. І
[10025-69-1]. Олова дихлорид дигідрат.
Містить не менше 97.0 % 8пС12,2Н2О.
Безбарвні кристали. Дуже легко розчинний у воді, легко
розчинний у 96 % спирті, кислоті оцтовій льодяній, кис-
лоті хлористоводневій розведеній та концентрованій.
Кількісне визначення. 0.500 г поміщають у колбу з при-
тертою скляною пробкою, розчиняють у 15 мл кисло
ти хлористоводневої Р, додають 10 мл води Р і 5 мл хло-
роформу Р. Швидко титрують 0.05 М розчином калію
йодату до знебарвлення хлороформного шару.
1 мл 0.05 Мрозчину калію йодату відповідає 22.56 мг
8пС12,2Н2О.
Олова(ІІ) хлориду розчин. 1085001.
20 г олова Р нагрівають із 85 мл кислоти хлористо-
водневої Р до припинення виділення водню, охо-
лоджують.
Зберігають розчин над надлишком олова Р, захи-
щаючи від повітря.
Олова(ІІ) хлориду розчин РІ. 1085002.
Безпосередньо перед використанням розчин оло-
ва( 11) хлориду Р розводять кислотою хлористовод-
невою розведеною Р (1:10).
Олова(ІІ) хлориду розчин Р2. 1085003.
До 8 г олова(ІІ) хлориду Р додають 100 мл 20%
(об/об) розчину кислоти хлористоводневої Р, стру-
шують до розчинення, якщо необхідно, нагріва-
ють на водяній бані при температурі 50 °С і пропу-
скають азот Р протягом 15 хв.
Готують безпосередньо перед використанням.
Олово. 8п. (А.м. 118.7). 1090800. [7440-31-5].
Гранули сріблясто-білого кольору. Розчинне в кислоті
хлористоводневій з виділенням водню.
240
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Арсен (2.4.2, метод А). Не більше 0.001 % (10 ррт).
0.1 гадає витримувати випробування на арсен.
Орацетовий синій 2К. С20Н|4М2О2. (М.м. 314.3).
1061100. [4395-65-7]. Кольоровий індекс № 61110.
І-Лміно-4-(феніламіно)антрацен-9,10-діон.
Температура плавлення: близько 194 °С.
Орацетовий синій В. 1118600. Суміш 1-метиламіно-4-
анілінантрахінону (С2)Н16М2О2; М.м. 328.4) та
І-аміно-4-анілінантрахінону (С20Н14ІЧ2О2; М.м. 314.3).
Порошок синьо-фіолетового кольору. Практично не
розчинний у воді, легко розчинний в ацетоні й кислоті
оцтовій безводній.
Орцин.С7НХО2,Н2О. (М.м. 142.2). 1108700. [6153-39-5].
5-Метилбензол-1,3-діол моногідрат.
Кристалічний порошок, чутливий до світла.
Температура кипіння: близько 290 °С.
Температура плавлення: від 58 °С до 61 °С.
Осмію(УІІІ) оксид. О$О4. (М.м. 254.2). 1061200.
|20816-12-О]. Осмію тетраоксид.
Голчасті кристали світло-жовтого кольору або крис-
талічна маса жовтого кольору. Гігроскопічний, чутли-
вий до світла, розчинний у воді, 96 % спирті та ефірі.
Зберігають у повітронепроникному контейнері.
Осмію(УШ) оксиду розчин. 1061201.
Розчин 2.5 г/л у 0.05 Мрозчині кислоти сірчаної.
Оцтовий ангідрид. С4Н6О3. (М.М. 102.1). 1000500.
[108-24-7|.
Містить не менше 97.0 % (м/м) С4Н6О3.
Прозора безбарвна рідина.
Температура кипіння: від 136 °С до 142 °С.
Кількісне визначення. 2.00 г помішають у скляну колбу
з притертою пробкою, розчиняють у 50.0 мл 1 Мроз-
чину натрію гідроксиду, кип’ятять зі зворотним холо-
дильником протягом 1 год і титрують / М розчином
кислоти хлористоводневої, використовуючи як інди-
катор 0.5 мл розчину фенолфталеїну Р. Обчислюють
кількість мілілітрів 1 Мрозчину натрію гідроксиду, ви-
траченого на титрування 1 г (л,).
2.00 г поміщають у скляну колбу з притертою проб-
кою, розчиняють у 20 мл циклогексану Р, охолоджують
на льоду, додають охолоджену суміш 10 мл аніліну Р і
20 мл циклогексану Р, кип’ятять зі зворотним холо-
дильником протягом 1 год, додають 50.0 мл / Мрозчи-
ну натрію гідроксиду, перемішують і титрують 1 Мроз-
чином кислоти хлористоводневої, використовуючи як
індикатор 0.5 мл розчину фенолфталеїну Р. Обчислю-
ють кількість мілілітрів 1 Мрозчину натрію гідроксиду,
витраченого на титрування 1 г(л2).
Вміст С4Н6О3, у відсотках, обчислюють за формулою:
10.2(И] - л2)
Оцтового ангідриду розчин РІ. 1000501.
25.0 мл оцтового ангідриду Р розчиняють у безвод-
ному піридині Рі доводять тим самим розчинником
до об’єму 100.0 мл.
Зберігають, захищаючи від світла й повітря.
Оцтового ангідриду — кислоти сірчаної розчин.
1000502.
Обережно змішують 5 мл оцтового ангідриду Р і
5 мл кислоти сірчаної Р. Одержану суміш додають
при охолодженні по краплях до 50 мл етанолу Р.
Готують безпосередньо перед використанням.
Оцтова кислота безводна. С2Н4О2. (М.м. 60.1). 1000300.
[64-19-7].
Містить не менше 99.6 % (м/м) С2Н4О2.
Безбарвна рідина або білі блискучі папоротеподібні
кристали. Легко змішується або розчиняється у воді,
96 % спирті, ефірі, гліцерині (85 %) та більшості жир-
них та ефірних олій.
г/2о : від 1.052 до 1.053.
Температура кипіння: від 117 °С до 119 °С.
Розчин 100 г/л є сильною кислотою (2.2.4).
Розчин 5 г/л кислоти оцтової, нейтралізованої розчи-
ном аміаку розведеним Р2, дає реакцію (Ь) на ацетати
(2.3.1).
Температура тверднення (2.2.18). Не нижче 15.8 °С.
Вода (2.5.12). Не більше 0.4 %. Якшо вміст води пере-
вищує 0.4 %, додають обчислену кількість оцтового
ангідриду Р.
Зберігають у захищеному від світла місці.
Оцтова кислота льодяна. С2Н4О2. (М.м. 60.1). 1000400.
[64-19-7].
Містить не менше 98.0 % (м/м) С2Н4О2.
г/2у : від 1.052 до 1.053.
Температура кипіння- від 117 °С до 119 °С.
Розчин 100 г/л є сильною кислотою. Розчин 5 г/л кис-
лоти оцтової, нейтралізований розчином аміаку розве-
деним Р2, дає реакцію (Ь) на ацетати (2.3.1).
Кількісне визначення. 5.00 г кислоти оцтової льодяної
розводять водою Рао об’єму 100.0 мл. 25.0 мл одержа-
ного розчину титрують 7 Мрозчином натрію гідрокси-
ду, використовуючи як індикатор 0.5 мл розчину фе-
нолфталеїну Р.
1 мл / М розчину натрію гідроксиду відповідає 60.1 мг
С2Н4О2.
Оцтова кислота. 1000401.
Містить не менше 290 г/л і не більше 310 г/л
С2Н4О2 (М.м. 60.1).
30 г кислоти оцтової льодяної Р доводять водою Р
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
241
4.1.1. Реактиви
до об’єму 100 мл.
Оцтова кислота розведена. 1000402.
Містить не менше 115 г/л і не більше 125 г/л
С2Н4О2 (Мм. 60.1).
12 г кислоти оцтової льодяної Р доводять водою Р
до об’єму 100 мл.
Паладій. Реї. (А.м. 106.4). 1114700. [7440-05-3].
Метал сірувато-білого кольору. Розчинний у кислоті
хлористоводневій.
Паладію хлорид. Рс1С12. (М.м. 177.3). 1061500. [7647-10-1].
Кристали червоного кольору.
Температура плавлення: від 678 °С до 680 °С.
Паладію хлориду розчин. 1061501.
1 г паладію хлориду Р розчиняють у 10 мл теплої
кислоти хлористоводневої Р, одержаний розчин
доводять сумішшю рівних об’ємів кислоти хлорис-
товодневої розведеної Р і води Р до об’єму 250 мл.
Безпосередньо перед використанням розчин роз-
водять двома об’ємами води Р
Пальмітинова кислота. С!6Н32О2. (М.м. 256.4).
1061600. [57-10-3]. Гексадеканова кислота.
Кристалічні лусочки білого кольору. Практично не
розчинна у воді, легко розчинна в гарячому 96 %
спирті та ефірі.
Температура плавлення: близько 63 °С.
Хроматографія. Визначення проводять, як зазначено
у статті Хюрамфеніколу пальмітат; на одержаній хро-
матограмі має виявлятися лише одна основна пляма.
Панкреатину порошок. 1061700 Див. статтю Панкреа-
тину порошок.
Папаверину гідрохлорид. 1061800. [61-25-6]. Див. стат-
тю Папаверину гідрохлорид.
Парарозаніліну гідрохлорид. С19Н18С^3. (М.м. 323 8).
1062200. [569-61-9|. Показник Шульца № 779. Кольо-
ровий індекс № 42500. 4-[Біс(4-амінофеніл)мети-
лен]циклогекса-2,5-дієнімінію хлорид.
Кристалічний порошок синювато-червоного кольору.
Мало розчинний у воді, розчинний в етанолі, прак-
тично не розчинний в ефірі. Розчини у воді й етанолі
мають інтенсивне червоне забарвлення, розчини в
кислоті сірчаній та кислоті хлористоводневій мають
жовте забарвлення.
Температура плавлення: близько 270 °С, із розкла-
данням.
Парарозаніліну знебарвлений розчин. 1062201.
0.1 г парарозаніліну гідрохлориду Р поміщають у
колбу з притертою скляною пробкою, додають
60 мл води Р і розчину 1.0 г натрію сульфіту без-
водного Р, або розчину 2.0 г натрію сульфіту Р, або
розчину 0.75 г натрію метабісульфппу Ру 10 мл во-
ди Р, потім повільно при перемішуванні додають І
6 мл кислоти хлористоводневої розведеної Р. за- І
кривають колбу пробкою і продовжують пе-
ремішування до розчинення, об’єм одержаного І
розчину доводять водою Рпо 100 мл.
Розчин використовують через 12 год після приго-
тування.
Зберігають у захищеному від світла місці.
Парацетамол. 1061900. [103-90-2]. Див. статтю Пара-
цетамол.
Парацетамол, вільний від 4-аміпофенолу. 1061901.
Парацетамол Рперекристалізовують із води Рісу-
шать у вакуумі при температурі 70 °С; процедуру
повторюють доти, поки парацетамол не буде вит-
римувати таке випробування: 5 г висушеного па-
рацетамолу розчиняють у суміші рівних об’ємів
метанолу Р і води Р і доводять об’єм розчину тією
самою сумішшю розчинників до 100 мл. Додають
1 мл свіжоприготованого розчину, який містить
10 г/л натрію нітропрусиду Р і 10 г/л натрію кар-
бонату безводного Р, перемішують і витримують
протягом 30 хв у захищеному від світла місці. Не
має з’являтися синє або зелене забарвлення.
Пеніцилінази розчин. 1062800.
10 г казеїну гідролізату, 2.72 г калію дигідрофосфату Р
і 5.88 г натрію цитрату Р розчиняють у 200 мл води Р.
доводять рН до 7.2 розчином 200 г/л натрію гідрокси-
ду Р і доводять водою Рпо об’єму 1000 мл.
0.41 г магнію сульфату Р розчиняють у 5 мл води Р, до-
дають 1 мл розчину 1.6 г/л заліза(II) амонію сульфату Р
і доводять об’єм розчину водою Рпо 10 мл.
Стерилізують обидва розчини нагріванням в авто-
клаві, охолоджують, змішують, розподіляють тонкими
шарами у конічних колбах і культивують із Васіїїів
сегеиз (КСТС 9946). Витримують колби при темпера-
турі від 18 °С до 37 °С до очевидних ознак росту, а
потім витримують при температурі від 35 °С до 37 °С
протягом 16 год, постійнно струшуючи для забезпе-
чення максимальної аерації. Центрифугують, надоса-
дову рідину стерилізують методом мембранної
фільтрації. 1.0 мл розчину пеніцилінази містить не
менше 0.4 мікрокатал (шо відповідає гідролізу не мен-
ше 500 мг бензилпеніциліну до бензилпеніцилінової
кислоти за годину) при температурі 30 °С і рН 7 за
умови, що концентрація бензилпеніциліну не опус-
кається нижче рівня, необхідного для ферментного
насичення.
Константа Міхаеліса для пеніцилінази за бензил-
пеніциліном у розчині пеніцилінази становить близь-
ко 12 мкг/мл.
Стерильність (2.6.1). Має витримувати випробування
на стерильність.
Зберігають при температурі від 0 °С до 2 °С і використо-
вують протягом 2-3 діб. Ліофілізований препарат
зберігають у запаяних ампулах протягом кількох місяців.
242
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Пентаеритритилтетракіс[3-(3,5-ди(1,1-диметилетил)-
4-гідроксифеніл)проп іонат]. СузНюіР 12- (М.м. 1178).
1062400. [6683-19-8]. Пентаеритритилтетракіс[3-(3,5-
ди-т/х,т-бутил-4-гідроксифеніл)пропіонат]. 2,2'-біс-
(Гідроксиметил)пропан-1,3-діолтетракіс[3-[3,5-
ди( 1.1 -диметилетил)-4-гідроксифеніл] 1 пропіонат.
Кристалічний порошок від білого до злегка жовтого
кольору. Практично не розчинний у воді, дуже легко
розчинний в ацетоні, розчинний у метанолі, мало роз-
чинний у гексані.
Температура плавлення: від 110 °С до 125 °С.
а-форма: від 120 °С до 125 °С.
Р-форма: від 110 °С до 115 °С.
Пентан. С5НІ2. (М.м. 72.2). 1062500.1109-66-0].
Прозора, безбарвна, займиста рідина. Дуже мало роз-
чинний у воді, змішується з ацетоном, етанолом і
ефіром.
: близько 0.63.
: близько 1.359.
Температура кипіння: близько 36 °С.
Пентан, використовуваний у спектрофотометрії, мас
витримувати таке додаткове випробування.
Мінімальне пропускання (2.2.25) визначають, викорис-
товуючи як компенсаційну рідину воду Р:
20 % за довжини хвилі 200 нм,
50% задовжини хвилі 210 нм,
85 % за довжини хвилі 220 нм,
93 % задовжини хвилі 230 нм,
98 % задовжини хвилі 240 нм.
Пентанол. С5Н,2О. (М.м. 88.1). 1062600. [71-41-0].
1-Пентанол. н-Аміловий спирт.
Безбарвна рідина. Помірно розчинний у воді,
змішується з 96 % спиртом і ефіром.
Лр : близько 1.410.
Температура кипіння: близько 137 °С.
/преш-Пентиловий спирт. С5НІ2О. (М.м. 88.1). 1062700.
|75-85-4]. мреш-Аміловий спирт. 2-Метил-2-бутанол.
Летка займиста рідина. Легко розчинний у воді,
змішується з 96 % спиртом, ефіром і гліцерином.
4о: близько 0.81.
Температурні межі перегонки (2.2.11). Від 100 °С до
104 °С; має переганятись не менше 95 %.
Зберігають у захищеному від світла місці.
Пепсину порошок. 1062800. [9001-75-6]. Див. статтю
Пепсину порошок.
Петролейний ефір. 1063100. [8032-32-4].
Прозора, безбарвна, займиста рідина, не флуоресціює.
Практично не розчинний у воді, змішується з 96 %
спиртом.
е/2о : від 0.661 до 0.664.
Температурні межі перегонки. (2.2.11). Від 50 °С до
70 °С.
Петролейний ефір РІ. 1063101.
Має задовольняти вимоги для петролейного
ефіру Р із такими змінами:
е/2о : від 0.630 до 0.656.
Температурні межі перегонки (2.2.11). Від 40 °С до
60 °С.
Не має мутнішати при температурі 0 °С.
Петролейний ефір Р2. 1063102.
Має задовольняти вимоги для петролейного
ефіру Різ такими змінами:
г/2р : від 0.620 до 0.630.
Температурні межі перегонки (2.2.11). Від 30 °С до
40 °С.
Не має мутнішати при температурі 0 °С.
Петролейний ефір РЗ. 1063103.
Має задовольняти вимоги для петролейного
ефіру Р із такими змінами:
Д2о : від 0.659 до 0.671.
Температурні межі перегонки (2.2.11). Від 40 °С до
80 °С.
Пікринова кислота. С6Нз1\І3О7. (М.м. 229.1). 1065800.
[88-89-1]. 2,4,6-Тринітрофенол.
Призми або пластинки жовтого кольору. Розчинна у
воді й 96 % спирті.
Зберігають зволоженою водою Р.
Пікринової кислот и розчин. 1065801.
Розчин 10 г/л.
Пікринової кислоти розчин РІ. 1065802.
До 100 мл насиченого розчину кислоти пікрино-
вої Р додають 0.25 мл розчину натрію гідроксиду
концентрованого Р.
Д-Пінен. СІ0Н16. (М.м. 136.2). 1109000. [19902-08-0].
6.6-Диметил-2-метиленбіцикло[3.1.1]гептан.
Безбарвна, масляниста рідина із запахом, який нага-
дує скипидар. Практично не розчинний у воді,
змішується з 96 % спиртом і ефіром.
</2о : близько 0.867.
п$ : близько 1.474.
Температура кипіння: від 155 °С до 156 °С.
Р-Пінен, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія квіток
пі-пм/Апії* т«пллАі/г\пга х/і/раїніЛ ЗПП1
243
4.1.1. Реактиви
померанця, використовуючи Д-пінен як випробовува-
ний розчин.
Площа основного піка має бути не менше 99.0 % суми
площ усіх шків.
Піперазину гідрат. 1065900. [142-63-2|. Див. статтю
Піперазину гідрат.
Піперидин. С5НИМ. (М.м. 85.2). 1066000. 1110-89-4].
Гексагідропіридин.
Безбарвна або злегка жовтуватого кольору рідина, має
лужну реакцію. Змішується з водою, 96 % спиртом,
ефіром і петролейним ефіром.
Температура кипіння: близько 106 °С.
Пірид-2-иламін. С5Н6М2. (М.м. 94.1). 1073400. [504-29-0].
2-Амінопіридин.
Великі кристали. Розчинний у воді, 96 % спирті та
ефірі.
Температура кипіння: близько 210 °С.
Температура плавлення: близько 58 °С.
Піридилазонафтол. СІ5НцКІ3О. (М.м. 249.3). 1073500.
[85-85-8]. 1-(2-Піридилазо)-2-нафтол.
Порошок цегляно-червоного кольору. Практично не
розчинний у воді, розчинний у 96 % спирті, метанолі
й гарячих розведених розчинах гідроксидів лужних
металів.
Температура плавлення: близько 138 °С.
Піридилазонафтолу розчин. 1073501.
Розчин 1 г/л в етанолі Р.
Випробування на чутливість. До 50 мл води Р дода-
ють 10 мл ацетатного буферного розчину рН 4.4 Р,
0.10 мл 0 02 М розчину натрію едетату і 0.25 мл роз-
чину піридилазонафтолу; після додавання 0.15 мл
розчину 5 г/л міді(П) сульфату Р забарвлення має
змінитися від світло-жовтого до фіолетового
Піридин. С5Н51Ч. (Мм. 79.1). 1073200. [110-86-1].
Прозора, безбарвна, гігроскопічна рідина. Змішується
з водою й 96 % спиртом.
Температура кипіння: близько 115 °С.
Зберігають у повітронепроникному контейнері.
Піридин безводний. 1073300. [ 110-86-1 ].
Піридин Рсушать над натрію карбонатом безводним Р,
фільтрують! переганяють.
Вода (2.5.12) Не більше 0.01 % (м/м).
Піровиноградна кислота. С3Н4О3. (М.м. 88.1). 1109300.
[127-17-3]. 2-Оксопропанова кислота.
Рідина жовтуватого кольору. Змішується з водою, ета-
нолом і ефіром.
б?2о : близько 1.267.
Лр : близько 1.413.
Температура кипіння: близько 165 °С.
Пірогалол. С6Н6О3. (М.м. 126 1) 1073700 [87-66-1]
Бензол-1,2,3-триол
Кристали білого кольору, під дією повітря та світла ко-
ричневіють. Дуже легко розчинний у воді, 96 % спирті
та ефірі, мало розчинний у сірковуглеці. Під дією
повітря водні розчини, а ше швидше лужні розчини
набувають коричневого забарвлення внаслідок аб-
сорбції кисню.
Температура плавлення: близько 131 °С.
Зберігають у захищеному від світла місці.
Пірогалолу лужний розчин. 1073701.
0.5 г пірогалолу Р розчиняють у 2 мл води, вільної
від вуглецю діоксиду, Р.
12 г калію гідроксиду Р розчиняють у 8 мл води,
вільної від вуглецю діоксиду, Р.
Безпосередньо перед використанням змішують
обидва розчини.
Пірокатехін. С6Н6О2 (М.м. 110 1) 1073600 |120-80-9|.
Бензол-1,2-діол.
Безбарвні або слабко жовтого кольору кристали. Роз-
чинний у воді, ацетоні, 96 % спирті та ефірі.
Температура плавлення: близько 102 °С.
Зберігають у захищеному від світла місці.
Пісок. 1075800.
Крупинки кремнію діоксиду білого або злегка сірува-
того кольору з розміром часток від 150 мкм до 300 мкм.
Плазма зі зниженим вмістом тромбоцитів. 1066100.
45 мл людської крові відбирають пластмасовим шпри-
цем місткістю 50 мл, який містить 5 мл стерильного
розчину 38 г/л натрію цитрату Р, і негайно центри-
фугують із прискоренням 1500 з при температурі 4 °С
протягом 30 хв. Відбирають із допомогою пластмасо-
вого шприца верхні 2/3 шару плазми, що сплив, і не-
гайно центрифугують із прискоренням 3500 § при тем-
пературі 4 °С протягом 30 хв. Відбирають верхні 2/3
шару рідини і швидко заморожують її у необхідній
кількості пластмасових пробірок при температурі
< 40 °С. Використовують пластмасове устаткування
або устаткування, оброблене силіконом.
Плазми субстрат. 1066200.
Плазму відокремлюють від людської або бичачоі
крові, збирають у розчин 38 г/л натрію цитрату Р
об’єм якого становить 1/9 об’єму плазми, або в роз-
чин, що містить 20 г/л динатрію гідроцитрату Р і
25 г/л глюкози Р, об’єм якого становить 2/7 об’єму
плазми. У першому випадку субстрат готують у день
збору крові; у другому випадку субстрат готують про-
тягом 2 днів від дня збору крові.
Зберігають при температурі -20 °С.
244
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Плазми субстрат РІ. 1066201.
Для взяття і обробки крові використовують во-
довідштовхуюче устаткування (виготовлене із
підхожих пластмас або скла, обробленого силіко-
ном).
Необхідний об’єм крові збирають від кожної з не
менш ніж п’яти овець. Достатнім об’ємом є відбір
285 мл крові у 15 мл розчину антикоагулянту, але й
менший об’єм може бути зібрано. Кров беруть у
живої тварини або під час забою, використовуючи
голку, приєднану до підхожої канюлі з довжиною,
достатньою для досягнення дна посудини для збо-
ру. Відкидають перші кілька мілілітрів і збирають
лише кров, яка вільно тече. Кров збирають у до-
статню кількість розчину антикоагулянту, який
містить 8.7 г натрію цитрату Рі 4 мг апротиніну Р
у 100 мл води Р. Співвідношення крові й розчину
антикоагулянту має бути 19:1. Під час збору і одра-
зу після нього кров злегка перемішують, не допус-
каючи спінювання. Після закінчення збору посу-
дину закривають і охолоджують до температури
від 10 °С до 15 °С. Після охолодження вміст усіх
колб об’єднують, за винятком тих, у яких спо-
стерігається очевидний гемоліз або утворення
згустків, і зберігають зібрану кров при температурі
від 10 °С до 15 °С.
Якнайшвидше, у межах 4 год після збору, об’єдна-
ну кров центрифугують із прискоренням від 1000 §
до 2000 § при температурі від 10 °С до 15 °С протя-
гом 30 хв. Відокремлюють надосадову рідину і
центрифугують із прискоренням 5000 § протягом
30 хв. (Якщо необхідно, для одержання прозорої
плазми можна центрифугувати з більшим приско-
ренням, наприклад, із прискоренням 20000 £ про-
тягом 30 хв, але фільтрація при цьому не припус-
тима). Відокремлюють надосадову рідину, негайно
ретельно перемішують і поміщають субстрат плаз-
ми у невеликі контейнери із пробками, порціями,
достатніми для проведення повного кількісного
визначення гепарину (приміром, від 10 мл до
ЗО мл). Одразу ж швидко охолоджують до темпе-
ратури нижче -70 °С (наприклад, занурюючи кон-
тейнери у рідкий азот) і зберігають при темпера-
турі не вище -30 °С.
Плазма придатна як субстрат плазми для
кількісного визначення гепарину, якщо за умов
кількісного визначення вона забезпечує час утво-
рення згустку, відповідний використовуваному
методу визначення, і забезпечує одержання кру-
тих логарифмічних кривих доза - відгук.
Перед використанням необхідну порцію плазми
розморожують на водяній бані при температурі
37 °С, обережно перемішуючи до повного розмо-
рожений. Розморожену плазму утримують при
температурі від 10 °С до 20 °С і негайно викорис-
товують.
Якщо необхідно, розморожений субстрат плазми
злегка центрифугують, але не фільтрують.
Плазми субстрат Р2. 1066202.
Готують із людської крові, яка містить менше 1 %
звичайної кількості фактора ЇХ. Збирають кров у
розчин 38 г/л натрію цитрату Р, об’єм якого ста-
новить 1/9 об’єму плазми.
Зберігають у невеликих кількостях у пластмасових
контейнерах при температурі -30 °С або нижче.
Плазми субстрат із недостатнім вмістом фактора V.
1066300.
Переважно використовують плазму, одержану від до-
нора із природженою недостатністю, або готують її та-
ким чином: відокремлюють плазму від людської крові,
зібраної в розчин 13.4 г/л натрію оксалату Р, об’єм
якого становить 1/10 об’єму крові. Культивують при
температурі 37 °С від 24 год до 36 год. Час згортання,
визначений за методом, описаним для розчину факто-
ра Vзгортання крові Р, має бути від 70 с до 100 с. Як-
що час згортання менше 70 с, то культивують знову від
12 год до 24 год.
Зберігають у невеликих кількостях при температурі
-20 °С або нижче.
Плазміноген людський. 1109100. [9001-91-6].
Речовина, наявна в крові, яка може бути активована
до плазміну, ферменту, шо здійснює лізис фібрину в
згустках крові.
Повідон. Полівінілпіролідон. 1068500. [9003-39-8]. Див.
статтю Повідон.
Полі(диметил)(дифеніл)силоксан. 1066900. ОВ-5, 8Е52.
Містить 95 % метильних груп і 5 % фенільних груп.
Нерухома фаза для газової хроматографії.
Полі(диметил)(дифеніл)(дивініл)силоксан. /100000.
8Е54.
Містить 94 % метильних груп, 5 % фенільних груп і
1 % вінільних груп.
Нерухома фаза для газової хроматографії.
Полі(диметил)силоксан. 1066800.
Каучук силіконовий (метил). Органосиліконовий
полімер, який має вигляд напіврідкої безбарвної смоли.
Характеристична в’язкість, визначена, як зазначено
нижче, має бути близько 115 мл-г'.
1.5 г, 1 г і 0.3 г полі(диметил)силоксану зважують із
точністю до 0.1 мг у мірних колбах місткістю 100 мл,
додають від 40 мл до 50 мл толуолу Р, струшують до
розчинення і доводять об’єм розчину тим самим роз-
чинником до 100.0 мл. Визначають в’язкість (2.2.9)
кожного розчину і в’язкість толуолу Р за тих самих
умов. Концентрацію кожного розчину зменшують
удвічі, розводячи толуолом Р, і визначають в’язкість
одержаних розчинів.
с — концентрація, г/100 мл;
Г/ — час витікання випробовуваного розчину;
і2 — час витікання толуолу;
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
245
4.1.1. Реактиви
77/ — в’язкість випробовуваного розчину, у мПа-с:
т]2 — в’язкість толуолу, в мПа-с;
<7/ — відносна густина випробовуваного розчину;
сі2 — відносна густина толуолу.
Для одержання значень відносної густини використо-
вують такі дані:
Концентрація (с), г/100 мл Відносна густина (б//)
0-0.5 1.000
0.5 - 1.25 1.001
1.25 - 2.20 1.002
2.20 - 2.75 1.003
2.75 - 3.20 1.004
3.20 - 3.75 1.005
3.75 - 4.50 1.006
Питому в’язкість (т7„ит) визначають за рівнянням:
= Пі - П2 =
П2 (2^2
Приведену в’язкість (г]пі>) визначають за рівнянням:
Л/Ш/П
П,;/? — £
Поліефірний гідроксильований гель для хроматографії
1067000.
Гель із невеликим розміром часток, який мі*і
гідрофільну поверхню до гідроксильних груп. Маєме-І
жу ексклюзії за декстраном із молекулярною масою І
від 2 х 10’ до 2.5 х 106.
Поліметилфенілсилоксан. 1067900.
Містить 50 % метальних груп і 50 % фенільних груп І
(Середня молекулярна маса 4000).
Дуже в’язка рідина (в’язкість близько 1300 мПа-с).
Нерухома фаза для газової хроматографії.
: близько 1.09.
Лр : близько 1.540.
Полі[метил(95)феніл(5)]силоксан. 1068000.
Див. Полі(диметил)(дифеніл)силоксан Р.
Полі[метил(94)феніл(5)вініл(1)]силоксан. 1068100.
Див. Полі(диметил)(дифеніл)(дивініл)силоксан Р.
Поліоксіетильована рицинова олія. 1068200.
Рідина світло-жовтого кольору, стає прозорою при
температурі близько 26 °С.
Полісорбат 20. 1068300. [9005-64-5]. Див. статтю
Полісорбат 20.
Характеристичну в’язкість (77) одержують екстрапо-
ляцією попереднього рівняння до с = 0. Для цього
проводять криву т]пит/с або 1о§ чіпит/<-' як функцію с.
Екстраполяцією до с = 0 одержують 77. Характеристич-
ну в’язкість виражають у мл/г, тому одержане значен-
ня необхідно помножити на 100.
Інфрачервоний спектр поглинання (2.2.24), одержа-
ний нанесенням речовини, якщо необхідно дисперго-
ваної у кількох краплях вуглецю тетрахлориду Р, на
диск натрію хлориду, не повинен мати поглинання за
довжини хвилі 3053 см-1, яке відповідає вінільним гру-
пам.
Втрата в масі при висушуванні (2.2.32). Не більше
2.0 %. Визначення проводять із 1.000 г, сушать у ваку-
умі при температурі 350 °С протягом 15 хв. Не більше
0.8 %, визначення проводять із 2.000 г, сушать при
температурі 200 °С протягом 2 год.
Поліетиленглікольадипінат. (С8Н12О4)П. [М.м. (172.2)п].
1067700.
Воскоподібна маса білого кольору. Практично не роз-
чинний у воді, розчинний у хлороформі.
Температура плавлення: близько 43 °С.
Поліетиленглікольсукцинат. (С6Н8О4)П. [М.м. (144.1)п].
1067800.
Кристалічний порошок білого кольору. Практично не
розчинний у воді, розчинний у хлороформі.
Температура плавлення: близько 102 °С.
Полісорбат 80. Твін-80. 1068400. [9005-65-6]. Див.
статтю Полісорбат 80.
Полістирол 900-1000. 1112200. [9003-53-6].
Органічний стандарт, використовуваний для калібру-
вання в газовій хроматографії.
Ми: близько 950.
Ми/Мп: 1.10.
Полі(ціанопропіл)силоксан. 1066700.
Полісилоксан, заміщений на 100 % ціанопропільними
групами.
Полі [ (ціанопропіл)(феніл)] [диметил]силоксан. 1114800.
Містить 6 % ціанопропілфенільних груп і 94 % диме-
тильних груп.
Нерухома фаза для газової хроматографії.
Полі(ціанопропіл)(7)(феніл)(7)(метил)(86)силоксан.
1109200.
Полісилоксан, заміщений на 7 % ціанопропільними
групами, на 7 % фенільними групами і на 86 % диме-
тильними групами.
Нерухома фаза для газової хроматографії.
Полі(ціанопропіл)(фенілметил)силоксан. 1066600.
Містить 90 % ціанопропільних груп і 10 % фенілме-
тильних груп.
246
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Нерухома фаза для газової хроматографії.
Полі [(ціанопропіл)метил фені лмети лей локсан].
1066500. Див. Полі[(ціанопропіл)(метил)][(феніл)(ме-
іпи.і)]силоксан Р.
Полі[(ціанопропіл)(метил)][(феніл)(метил)]силоксан.
1066500.
Містить 25 % ціанопропільних груп, 25 % фенільних
груп і 50 % метальних груп. (Середня молекулярна
маса - 8000).
Дуже в’язка рідина (в’язкість близько 9000 мПа-с).
(/§ : близько 1.10.
: близько 1.502.
Прокаїну гідрохлорид. 1109400. Див. статтю Прокаїну
гідрохлорид.
В-Проліл-Б-фенілаланіл-Е-аргінін-4-нітроаніліду
дигідрохлорид. С26Н36С12МХО5. (М.м. 612). 1072800.
Пропанол. С3НХО. (М.м. 60.1). 1072000. [71-23-8].
1-Пропанол.
Прозора, безбарвна рідина. Змішується з водою й 96 %
спиртом.
від 0.802 до 0.806.
Температура кипіння: близько 97.2 °С.
Температурні межі перегонки (2.2.11). Від 96 °С до
99 °С; має переганятись не менше 95 %.
2-Пропанол. С3НХО. (Мм. 60.1). 1072100. [67-63-0|.
Ізопропіловий спирт.
Прозора, безбарвна, займиста рідина. Змішується з
водою й 96 % спиртом.
<1$ : близько 0.785.
Температура кипіння: від 81 °С до 83 °С.
2-Пропанол РІ. 1072101.
Має задовольняти вимоги для 2-пропанолу Р ї такі
додаткові вимоги:
«0 : близько 1.378.
Вода (2.5.12). Не більше 0.05 %. Визначення про-
водять із 10 г.
Мінімальне пропускання (2.2.25) визначають, ви-
користовуючи як компенсаційну рідину воду Р:
25 % за довжини хвилі 210 нм,
55 % за довжини хвилі 220 нм,
75 % за довжини хвилі 230 нм,
95 % за довжини хвилі 250 нм,
98 % за довжини хвилі 260 нм.
Пропаноламін. С3Н9МО. (М.м. 75.1). 1072200. [156-87-6].
З-Аміно-1-пропанол.
Прозора, безбарвна, в’язка рідина.
б/2о : близько 0.99.
«р : близько 1.461.
Температура плавлення: близько 11 °С.
Пропілацетат. С5Н|0О2. (М.м. 102.1). 1072600.1109-60-4].
і/2о : близько 0.888.
Температура кипіння: близько 102 °С.
Температура плавлення: близько -95 °С.
Пропіленгліколь. 1072900. [57-55-6]. Див. статтю
Пропіленгліколь.
Пропіленоксид. С3Н6О. (М.м. 58.1). 1121800.
Безбарвна рідина. Змішується з 96 % спиртом.
Пропілпарагідроксибензоат. 1072700. [94-13-3]. Див.
статтю Пропілпарагідроксибензоат.
Пропіонова кислота. С3Н6О2. (М.м. 74.1). 1072400.
[79-09-4].
Масляниста рідина. Розчинна в 96 % спирті та ефірі,
змішується з водою.
і/2о : близько 0.993.
«р : близько 1.387.
Температура кипіння: близько 141 °С.
Температура плавлення: близько -21 °С.
Пропіоновий альдегід. С3Н6О. (М.м. 58.1). 1072300.
[123-38-6|. Пропаналь.
Рідина. Легко розчинний у воді, змішується з 96 %
спиртом і ефіром.
і/2о : близько 0.81.
«о1 : близько 1.365.
Температура кипіння: близько 49 °С.
Температура плавлення: близько -81 °С.
Пропіоновий ангідрид. С6Н10О3. (М.м. 130.1). 1072500.
[123-62-6].
Прозора, безбарвна рідина. Розчинний у 96 % спирті
та ефірі.
і/2о : близько 1.01.
Температура кипіння: близько 167 °С.
Пропіонового ангідриду реактив. 1072501.
1 г кислоти толуолсульфонової Р розчиняють у
30 мл кислоти оцтової льодяної Р і додають 5 мл
пропіонового ангідриду Р.
Використовують через 15 хв після приготування.
Термін придатності 1 доба.
Протаміну сульфат. 1073000. [53597-25-4 (сальмін)
9007-31-2 (клупеїн)]. Див. статтю Протаміну сульфат.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
247
4.1.1. Реактиви
Протеаза 8(арііу1ососсия аигеїія штам У8. 1115800.
[66676-43-5]. Тип XVIІ-В.
Мікробіологічний позаклітинний протеолітичний
фермент. Ліофілізований порошок містить від 500 оди-
ниць до 1000 одиниць в 1 мг розчину.
Протравний чорний 11. С20Н|2М3№О78. (М.м. 461.4).
1056800. [1787-61-7]. Показник Шульца № 241. Ко-
льоровий індекс № 14645. Натрію 2-гідрокси-1-[(1-
гідроксинафт-2-іл)азо]-6-нітронафталін-4-сульфонат.
Еріохром чорний.
Порошок коричнювато-чорного кольору. Розчинний
у воді й 96 % спирті.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла місці.
Протравного чорного 11 індикаторна суміш. 1056801.
1 г протравного чорного II Р змішують із 99 г
натрію хлориду Р.
Випробування на чутливість. 50 мг індикаторної
суміші розчиняють у 100 мл води Р, з’являється ко-
ричнювато-фіолетове забарвлення, яке має пе-
рейти у синє при додаванні 0.3 мл розчину аміаку
розведеного РІ. При подальшому додаванні 0.1 мл
розчину 10 г/л магнію сульфату Р забарвлення має
змінитися на фіолетове
Зберігають у повітронепроникному контейнері,
захищеному від світла місці.
Пулегон. С|0НІ6О. (М.м. 152.2). 1073100. [89-82-7|.
(Я)-2-1зопропіліден-5-метилциклогексанон.
(+)-н-Мент-4-ен-3-он.
Безбарвна, масляниста рідина. Практично не розчин-
ний у воді, змішується з 96 % спиртом і ефіром.
г/^5 : близько 0.936.
Нц : від 1.485 до 1.489.
[а]р : віл +19.5° до +22.5°.
Температура кипіння: від 222 °С до 224 °С.
Пулегон, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія м’яти
перцевої, використовуючи пулегон як випробовуваний
розчин
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків.
Рамноза. С6Н12О5,Н2О. (М.м. 182.2). 1074900.
[6155-35-7]. Б-(+)-Рамноза. 6-Деокси-Б-маноза.
Кристалічний порошок білого кольору. Легко розчин-
на у воді.
[а]р : від +7.8° до +8.3°. Визначення проводять, вико-
ристовуючи розчин 50 г/л у воді Р, яка містить близь-
ко 0.05 % МН3.
Рапонтицин. С21Н24О9. (М.м. 420.4). 1075000. [ 155-58-8|.
3-Гідрокси-5-[2-(3-гідрокси-4-метоксифеніі)-
етеніл]феніл-Р-О-глюкопіранозид.
Кристалічний порошок жовтувато-сірого кольору І
Розчинний у 96 % спирті й метанолі.
Хроматографія. Визначення проводять, як зазначенії І
у статті Корінь ревеню', на одержаній хроматограмі має І
виявлятися лише одна основна пляма.
Рапсова олія. 1074600.
Жирна олія, одержана вичавлюванням із зерен різних
сортів Вгазмеа парих £.
Фракція жирних кислот містить від 40 % до 55 % кис- |
лоти ерукової.
Прозора рідина від жовтого до земно-жовтого кольо-
ру. Практично не розчинна у 96 % спирті, змішується
з ефіром і петролейним ефіром.
Йодне число (2.5.4). Від 94 до 120.
Число омилення (2.5.6). Від 168 до 181.
Перекисне число (2.5.5). Не більше 5.
Кислота ерукова. Визначення проводять, як зазначено
у випробуванні на сторонні жирні кислоти методом
тонкошарової хроматографії (2.4.21), використовую-
чи такі розчини:
Розчин А. Розчиняють 20 мг суміші жирних кислоту
4 мл хлороформу Р.
Розчин В. 2.0 мл розчину А доводять хлороформом Рдо
об’єму 50 мл.
На хроматограмі розчину А має виявлятися п’ять
чітких плям. Пляма із найнижчим значенням /?у
близько 0.25 має бути найбільш інтенсивною або
однією з найінтенсивніших і має відповідати кислоті
еруковій. На хроматограмі розчину В має бути чітко
видно пляму, яка відповідає кислоті еруковій.
Резорцин. 1074800. [ 108-46-3]. Див. статтю Резорцин.
Резорцину реактив. 1074801.
До 80 мл кислоти хлористоводневої Р додають
10 мл розчину 20 г/л резорцину Р, 0.25 мл розчину
25 г/л міді(ІІ) сульфату Р і доводять водою Р до
об’єму 100.0 мл.
Використовують через 4 год після приготування.
Зберігають при температурі від 2 °С до 8 °С.
Термін придатності 7 діб
Рибоза. С5Н10О5. (М.м. 150.1). 1109600. [50-69-1].
О-Рибоза
Розчинна у воді, мало розчинна в 96 % спирті.
Температура плавлення: від 88 °С до 92 °С.
Рицинолеїнова кислота. С18Н34О3. (М.м. 298.5).
1100100. [141-22-0]. 12-Гідроксіолеїнова кислота.
В’язка рідина від жовтого до жовтувато-коричневого
кольору. Містить суміш жирних кислот, одержаних
248
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
гідролізом олії рицинової. Практично не розчинна у
воді, дуже легко розчинна в етанолі, розчинна в
ефірі.
Л’5: близько 0.942.
: близько 1.472.
Температура плавлення: близько 285 °С, із розкла-
данням.
Родамін В. С28Н31С11\2О3. (М.м. 479.0). 1075100
[81-88-9]. Показник Шульца № 864. Кольоровий
індекс № 45170. [9-(2-Карбоксифеніл)-6-(діети-
ламіно)-3//-ксантен-3-іліден]діетиламонію хлорид.
Кристали зеленого кольору або порошок червонува-
то-фіолетового кольору. Дуже легко розчинний у воді
іі 96 % спирті.
Ртуть. Не. (А.м. 200.6). 1052800. [7439-97-6|.
Рідина сріблясто-білого кольору, яка розсипається на
сферичні краплі, що не залишають металевого сліду
при терті об папір.
г/іо : близько 13.5.
Температура кипіння: близько 357 °С.
Ртуті(П) нітрату розчин. 1052801.
З мл ртуті Робережно розчиняють у 27 мл кисло-
ти азотної димлячої Р, одержаний розчин розво-
дять водою Ррівним об’ємом.
Зберігають у захищеному від світла місці.
Термін придатності 2 міс.
Ртуті(П) ацетат. С4Н(1НеО4. (М.м. 318.7). 1052000.
[1600-27-7]. Ртуті діацетат.
Кристали білого кольору. Дуже легко розчинний у
воді, розчинний у 96 % спирті.
Ртуті(ІІ) ацетату розчин. 1052001.
3.19 г ртуті(П) ацетату Р розчиняють у кислоті
оцтовій безводній Р, доводять об’єм розчину тією
самою кислотою до 100 мл. Якщо необхідно,
одержанні! розчин нейтралізують 0.1 М розчином
кислоти хлорної, використовуючи як індикатор
0.05 мл розчину кристалічного фіолетового Р.
Ртуті(ІІ) бромід. Н§Вг2. (М.м. 360.4). 1052100. [7789-47-1].
Ртуті дибромід.
Кристали або кристалічний порошок білого або
світло-жовтого кольору. Мало розчинний у воді, роз-
чинний у 96 % спирті.
Ртутно-бромідний папір. 1052101.
У прямокутну чашку поміщають розчин 50 г/л
ртуті(П) броміду Р в етанолі Р, занурюють у роз-
чин шматочки білого фільтрувального паперу з гу-
стиною 80 г/м2 (швидкість фільтрування, яка
дорівнює часу фільтрування, вираженому в секун-
дах, при фільтруванні 100 мл води при температурі
20 °С крізь фільтр з поверхнею 10 см’ і постійному
тиску 6,7 кПа: від 40 с до 60 с), розміром 1.5 х 20 см,
складені удвічі. Папір підвішують на неметалеву
нитку, дозволяючи стікти надлишку рідини, су-
шать у захищеному від світла місці. Відрізають по
1 см із кожного кінця кожної смужки і нарізають
решту паперу на квадратики зі стороною 1.5 см або
диски діаметром 1.5 см.
Зберігають у контейнері зі скляною пробкою, за-
горнутому в чорний папір.
Ртуті(ІІ)йодид. Н(Д2. (М.м. 454.4). 1052300. [7774-29-0].
Ртуті дийодид.
Щільний кристалічний порошок яскраво-червоного
кольору. Мало розчинний у воді, помірно розчинний в
ацетоні, 96 % спирті та ефірі, розчинний у надлишку
розчину калію йодиду Р.
Зберігають у захищеному від світла місці.
Ртуті(ІІ) нітрат. Н£(І8Ю3)2,Н2О. (М.м. 342.6). 1052400.
[7782-86-7]. Ртуті динітрат моногідрат.
Безбарвні або злегка забарвлені кристали. Гігро-
скопічний, розчинний у воді в присутності невеликої
кількості кислоти азотної.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла міст.
Ртуті(П) оксид. ІфО. (М.м. 216.6). 1052500. [21908-53-2].
Ртуті оксид жовтий. Ртуті оксид.
Порошок від жовтого до оранжево-жовтого кольору.
Практично не розчинний у воді й 96 % спирті.
Зберігають у захищеному від світла місці.
Ртуті(ІІ) сульфату розчин. 1052600. [7783-35-91.
1 г ртуті(ІІ) оксиду Р розчиняють у суміші 20 мл во-
ди Рї 4 мл кислоти сірчаної Р.
Ртуті(ІІ) тіоціанат. Н£?(БСМ)2. (М.м. 316.7). 1052700.
[592-85-8|. Ртуті ди(тіоціанат). Ртуті роданід.
Кристалічний порошок білого кольору. Дуже мало
розчинний у воді, мало розчинний у 96 % спирті та
ефірі, розчинний у розчинах натрію хлориду.
Ртуті(ІІ) тіоціанату розчин. 1052701.
0.3 г ртуті(П) тіоціанату Р розчиняють в ета-
нолі Рі доводять об’єм розчину тим самим розчин-
ником до 100 мл.
Термін придатності 7 діб
Ртуті(ІІ) хлорид. 1052200. [7487-94-7|. Див. статтю
Ртуті хлорид.
Ртуті(ІІ) хлориду розчин. 1052201.
Розчин 54 г/л.
Рутеній червоний.
[(МН3)5КііОКи(МН3)4ОКи(МН3)5|С16,4Н2О. (М.м. 858).
1075200. [11103-72-31.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
249
4.1.1. Реактиви
Порошок коричнювато-червоного кольору. Розчин-
ний у воді.
Рутенію червоного розчин. 1075201.
Розчин 0.8 г/л у розчині свинцю(ІІ) ацетату Р.
Рутин. С27Н30О16,ЗН2О. (Мм. 665). 1075300. [153-18-4].
Рутозид. 3-(О-6-Деокси-а-Ь-манопіранозил-(1—>6)-
Р-О-глюкопіранозилокси)-2-(3,4-дигідроксифеніл)-
5,7-дигідрокси-4//-хромен-4-он.
Кристалічний порошок жовтого кольору, темнішає
під світлом. Дуже мало розчинний у воді, розчинний
приблизно у 400 частинах киплячої води, мало роз-
чинний у 96 % спирті, практично не розчинний в
ефірі, розчинний у розчинах гідроксидів лужних ме-
талів та аміаку.
Температура плавлення: близько 210 °С, із розкла-
данням.
Розчин у 96 % спирті Р має два максимуми поглинан-
ня (2.2.25) за довжин хвиль 259 нм і 362 нм.
Зберігають у захищеному від світла місці.
Сабінен. С10Н16. (Мм. 136.2). 1109700. |2009-00-9|.
Туй-4(10)-єн. 4-Метилен-1-ізопропілбіцикло[3.1.0]ге-
ксан.
Безбарвна масляниста рідина.
г/2| : близько 0.843.
Ир : близько 1.468.
Температура кипіння: від 163 °С до 165 °С.
Сабінен, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування:
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія квіток
померанця, використовуючи сабінен як випробовува-
ний розчин.
Площа основного піка має бути не менше 99.0 % суми
площ усіх піків.
Саліцилова кислота. 1075600. [69-72-7]. Див. статтю
Кислота саліцилова.
Саліциловий альдегід. С7Н6О2. (М.м. 122.1). 1075400.
[90-02-8]. 2-Гідроксибензальдегід.
Прозора, безбарвна, масляниста рідина.
г/2о : близько 1.167.
п& : близько 1.574.
Температура кипіння: близько 196 °С.
Температура плавлення: близько -7 °С.
Саліцилового альдегіду азин. С14Н12М2О2. (Мм. 240.3).
1075500. 2,2'-Азинодиметилдифенол.
0.30 г гідразину сульфату Р розчиняють у 5 мл води Р,
додають 1 мл кислоти оцтової льодяної Рі 2 мл свіжо-
приготованого 20 % (об/об) розчину саліцилового аль-
дегіду Р у 2-пропанолі Р. Перемішують, витримують до
утворення жовтого осаду, потім струшують із двома І
порціями метиленхлориду Р по 15 мл кожна. Об’єд- І
нані органічні витяжки, висушені над натрію сульфа-1
том безводним Р, декантують або фільтрують і випа- І
рюють насухо. Осад перекристалізовують при охолод- 1
женні з суміші розчинників метанол Р - толуол Р
(40:60). Кристали сушать у вакуумі.
Температура плавлення: близько 213 °С.
Хроматографія. Визначення проводять, як зазначено і
у статті Повидон у випробуванні на гідразин; на одер- І
жаній хроматограмі має виявлятися лише одна основ- і
на пляма.
Сантонін. С|5Н|5О3. (Мм. 246.3). 1122000. (-)-а-Сан-
тонін. 3,5а.9-Триметил-За,5,5а,9Ь-тетрагідро-3//,4її-
нафто] 1,2]-фуран-2,8-діон.
Безбарвні блискучі кристали, які жовтіють під дією
світла. Дуже мало розчинний у воді, легко розчинний
у гарячому 96 % спирті, помірно розчинний в етанолі.
Температура плавлення: від 174 °С до 176 °С.
[а]І5 :-173° в етанолі.
Хроматографія. Визначення проводять, як зазначено
у випробуванні Ідентифікація С у статті Квітки
арніки; на хроматограмі, одержаній із 10 мкл розчину,
має виявлятися темна пляма з /^близько 0.5. Хрома-
тограму обприскують розчином анісового альдегіду Р,
нагрівають при температурі 105 °С протягом 5-10 хв.
На хроматограмі при денному світлі спостерігається
пляма спочатку жовтого кольору, яка потім швидко
набуває фіолетово-червоного кольору.
Сахароза. 1085700. [57-50-1]. Див. статтю Сахароза.
Якщо сахарозу використовують для перевірки поляри-
метра, її зберігають у сухому вигляді в запаяній ампулі.
Свинцю(ІІ) ацетат. С4Н6О4РЬ,ЗН2О. (М.м. 379.3).
1048100. [6080-56-4]. Свинцю діацетат.
Безбарвні кристали, які звітрюються на повітрі. Легко
розчинний у воді, розчинний у 96 % спирті.
Свинцево-ацетатна вата. 1048101.
Гігроскопічну вату занурюють у суміш розчинників
кислота оцтова розведена Р - розчин свинцю(П) аце-
тату Р (1:10). Не віджимаючи вати, видаляють
надлишок рідини, потім поміщають її на кілька
шарів фільтрувального паперу й сушать на повітрі.
Зберігають у повітронепроникному контейнері.
Свинцево-ацетатний папір. 1048102.
Фільтрувальний папір, густина якого 80 г/м2, зану-
рюють у суміш кислота оцтова розведена Р-розчин
свинцю(П) ацетату Р (1:10), потім папір вийма-
ють, сушать і нарізають на смужки розміром
15 мм х 40 мм.
Свинцю(П) ацетату розчин. 1048103.
Розчин 95 г/л у воді, вільній від вуглецю діоксиду, Р.
250
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Свинцю(ІІ) ацетату основного розчин. 1048400
Ц335-32-6]. Свинцевий оцет.
Містить не менше 16.7 % (м/м) і не більше 17.4 %
(м/м) РЬ (А.м. 207.2) у вигляді сполуки, яка приблизно
відповідає формулі С8Н14О10РЬ3.
40.0 г свинцю(П) ацетату Ррозчиняють у 90 мл води,
вільної від вуглецю діоксиду, Р. Доводять рН розчину до
7.5 розчином натрію гідроксиду концентрованим Р,
центрифугують і використовують прозорий, безбарв-
ний розчин над осадом.
При зберіганні, в добре закритому контейнері, розчин
мас залишатись прозорим.
Свиншо(ІУ) оксид. РЬО2. (М.м. 239.2). 1048200.
[1309-60-0]. Свинцю діоксид.
Порошок темно-коричневого кольору, що виділяє ки-
сень при нагріванні. Практично не розчинний у воді,
розчинний у кислоті хлористоводневій з виділенням
хлору, розчинний у кислоті азотній розведеній у при-
сутності пероксиду водню, щавлевої кислоти або
інших відновлюючих реагентів, розчинний у гарячих
концентрованих розчинах гідроксидів лужних металів.
Свинцю(ІІ) нітрат. РЬ(КЮ3)2. (М.м. 331.2). 1048300.
[10099-74-8]. Свинцю динітрат.
Кристалічний порошок білого кольору або безбарвні
кристали. Легко розчинний у воді.
Свинцю(П) нітрату розчин. 1048301.
Розчин 33 г/л.
Селен. 8е. (А.м. 79.0). 1075900. [7782-49-2].
Порошок або гранули від коричнювато-червоного до
чорного кольору. Практично не розчинний у воді й
96 % спирті, розчинний у кислоті азотній.
Температура плавлення: близько 220 °С.
Селеніста кислота. Н28еО3. (М.м. 129.0). 1100200.
[7783-00-8].
Кристали, які розпливаються на повітрі. Легко роз-
чинна у воді.
Зберігають у повітронепроникному контейнері.
Серин. 1076000. |56-45-І]. Див. статтю Серин.
Сечовина. 1095000. [57-13-6]. Див. статтю Сечовина.
Сіалова кислота. 1001100. [131-48-6]. Див. !9-ацетил-
неурамінова кислота Р.
Силікагель с. 1076300. [112926-00-8].
Містить близько 13 % кальцію сульфату гемігідрату.
Дрібний гомогенний порошок білого кольору, з
розміром часток близько 15 мкм.
Кальцію сульфат. 0.25 г поміщають у колбу з притер-
тою скляною пробкою, додають 3 мл кислоти хлорис-
товодневої розведеної Р і 100 мл води Р, енергійно
збовтують протягом ЗО хв, фільтрують крізь скляний
фшьтр і промивають залишок. Фільтрат і промивні во-
ди об’єднують і проводять визначення вмісту кальцію
методом комплекси метрі! (2.5.11).
1 мл 0.1 Мрозчину натрію едетату відповідає 14.51 мг
Са8О4,1/2 Н2О.
рН (2.2.3). Близько 7. Вимірюють рН суспензії, одер-
жаної збовтуванням 1 г із 10 мл води, вільної від вугле-
цю діоксиду, Р протягом 5 хв.
Силікагель ск254. 1076400. [112926-00-8].
Містить близько 13 % кальцію сульфату гемігідрату і
близько 1.5 % флуоресцентного індикатора, який має
оптимальну інтенсивність поглинання за довжини
хвилі 254 нм.
Дрібний гомогенний порошок білого кольору з
розміром часток близько 15 мкм.
Кальцію сульфат. Визначення проводять метолом, за-
значеним для силікагелю б Р.
рН (2.2.3). Має задовольняти вимоги для силікагелю с Р.
Флуоресценція. Визначення проводять методом тонко-
шарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель с Р. На хроматографічну плас-
тинку наносять у десять крапок послідовно зростаючі
об’єми від 1 мкл до 10 мкл розчину 1 г/л кислоти бен-
зойної Р в суміші розчинників кислота мурашина без-
водна Р- 2-пропанол Р( 10:90). Хроматографують у тій
самій суміші розчинників. Коли фронт розчинників
пройде 10 см, пластинку сушать і переглядають в УФ-
світлі за довжини хвилі 254 нм. На верхній третині
хроматограми на флуоресціюючому фоні мають вияв-
лятись темні плями кислоти бензойної, починаючи
від 2 мкг і більше.
Силікагель ОВ для хіральних розділень. 1110300.
Силікагель для хроматографії дуже дрібний, тонко
здрібнений із розміром часток 5 мкм, покритий таким
продуктом заміщення:
Силікагель н. 1076500. [112926-00-8].
Дрібний гомогенний порошок білого кольору з
розміром часток близько 15 мкм.
рН (2.2.3). Має задовольняти вимоги для силікагелю с Р.
Силікагель н силанізований. 1076600.
Приготування тонкого шару. Див. силікагель нр254 си-
ланізований Р.
Дрібний гомогенний порошок білого кольору; після
струшування з водою спливає на поверхню внаслідок
гідрофобних властивостей.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
251
4.1.1. Реактиви
Хроматографічнарозділювальна здатність. Має витри-
мувати випробування для силікагелю нг2,4силанізовано-
го Р.
Силікагель НР254. 1076700.
Містить близько 1.5 % флуоресцентного індикатора,
який має оптимальну інтенсивність поглинання задо-
вжини хвилі 254 нм.
Дрібний гомогенний порошок білого кольору з
розміром часток близько 15 мкм.
рН (2.2.3). Має задовольняти вимогу для силікагелю с Р
Флуоресценція. Має задовольняти вимогу для силікаге-
лю сг254 Р.
Силікагель нр254 силанізований. 1076800.
Містить близько 1.5 % флуоресцентного індикатора,
який має оптимальну інтенсивність поглинання задо-
вжини хвилі 254 нм
Дрібний гомогенний порошок білого кольору; після
струшування з водою спливає на поверхню внаслідок
гідрофобних властивостей.
Приготування тонкого шару. ЗО г енергійно струшують
із 60 мл суміші розчинників метанол Р - вода Р (1:2)
протягом 2 хв. Ретельно очищені пластинки покрива-
ють шаром завтовшки 0.25 мм, використовуючи
пристрій для нанесення. Пластинки з покриттям су-
шать на повітрі, потім нагрівають у сушильній шафі
при температурі від 100 °С до 105 °С протягом 30 хв.
Хроматографічна розділювальна здатність. У конічну
колбу місткістю 250 мл поміщають по 0.1 г метиллау-
рату Р, метилміристату Р, метилпальмітату Р і ме-
тилстеарату Р, додають 40 мл розчину калію гідрокси-
ду спиртового Р і нагрівають зі зворотним холодиль-
ником на водяній бані протягом 1 год. Охолоджують,
переносять розчин у ділильну лійку з допомогою
100 мл води Р, підкислюють (рН від 2 до 3) кислотою
хлористоводневою розведеною Р і струшують із трьома
порціями хлороформу Р по 10 мл кожна. Об’єднані
хлороформні витяжки сушать над натрію сульфатом
безводним Р, фільтрують і випарюють насухо на во-
дяній бані. Залишок розчиняють у 50 мл хлороформу Р
Проводять визначення методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар
силікагель НР254 силанізований. На хроматографічну
пластинку наносять у три крапки по 10 мкл розчину в
хлороформі і хроматографують у системі розчинників
кислота оцтова льодяна Р - вода Р - діоксан Р
(10:25:65). Коли фронт розчинників пройде 14 см,
пластинку сушать при температурі 120 °С протягом
ЗОхв, охолоджують, обприскують розчином 35 г/л
кислоти фосфорномолібденової Р у 2-пропанолі Р і
нагрівають при температурі 150 °С до появи плям.
Пластинку обробляють парою аміаку до одержання
білого фону. На хроматограмах мають виявлятись чо-
тири чітко розділені добре виражені плями.
Силікагель для ексклюзійної хроматографії. 1077900.
Силікагель дуже тонко здрібнений з розміром часток
10 мкм і дуже гідрофільною поверхнею. Середній
діаметр пор близько 30 нм. Він сумісний із водними
розчинами з рН від 2 до 8 і органічними розчинника-1
ми. Придатний для розділення протеїнів із молеку.пяр- І
ними масами від 1x10’ до ЗхЮ5.
Силікагель для хроматографії. 1076900.
Силікагель дуже тонко здрібнений з розміром часток І
від 3 мкм до 10 мкм. Розмір часток зазначають після І
назви сорбенту у випробуваннях, в яких він викорис- І
товується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель модифікований амілазою для хроматографії.
1109800.
Силікагель дуже тонко здрібнений з розміром часток
10 мкм і поверхнею, хімічно модифікованою аміла-
зою. Розмір часток зазначають після назви сорбенту)
випробуваннях, в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель амінопропілметилсилільний для хромато-
графії. 1102400.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною аміноиропілметилсилільними групами. Розмір
часток зазначають після назви сорбенту у випробуван-
нях, в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель амінопропілсилільний для хроматографії.
1077000.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною амінопропілсилільними групами. Розмір часток
зазначають після назви сорбенту у випробуваннях, в
яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель безводний. 1076100. [112926-00-8].
Аморфна кремнієва кислота, частково зневоднена й
полімеризована, яка поглинає при температурі 20 °С
близько 30 % води відносно своєї маси. Містить як
індикатор кобальту хлорид. Практично не розчинний у
воді, частково розчинний у розчинах натрію гідроксиду.
Силікагель бутилсилільний для хроматографії. 1076200.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною бутилсилільними групами. Розмір часток зазна-
чають після назви сорбенту у випробуваннях, в яких
він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
252
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Кремнію діоксид сфероїдальним: ЗО нм.
Об’єм мор: 0.6 см’/г.
Питома площа поверхні: 80 м2/г.
Силікагель гексилсилільний для хроматографії.
1077100.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною гексилсилільними групами. Розмір часток зазна-
чають після назви сорбенту у випробуваннях, в яких
він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель гідрофільний для хроматографії. 1077200.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм, поверхня якого модифікована з
метою надання гідрофільних властивостей. Розмір ча-
сток зазначають після назви сорбенту у випробуван-
нях, в яких він використовується.
Силікагель диметилоктадецилсилільний для хромато-
графії. 1115100.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною диметилоктадецилсилільними групами. Розмір
часток зазначають після назви сорбенту у випробуван-
нях, в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті. Розмір час-
ток нерегулярний.
Питома площа поверхні: 300 м2/г.
Силікагель діольний для хроматографії. /110000.
Сферичні частки кремнію діоксиду із прищепленими
дигідроксипропільними групами. Розмір пор 10 нм.
Силікагель нітрильний для хроматографії. 1077300.
Силікагель дуже тонко здрібнений з поверхнею,
хімічно модифікованою ціанопропілсилільними гру-
пами. Розмір часток зазначають після назви сорбенту
у випробуваннях, в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді, 96 % спирті та ефірі.
Силікагель нітрильний для хроматографії РІ. 1077400.
Силікагель дуже тонко здрібнений, який складається з
пористих сферичних часток, із хімічно зв’язаними
нітрильними групами. Розмір часток зазначають після
назви сорбенту у випробуваннях, в яких він викорис-
товується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді. 96 % спирті та ефірі.
Силікагель нітрильний для хроматографії Р2. 1077400.
Силікагель надчистий, з поверхнею, хімічно модифі-
кованою ціанопропілсилільними групами. Містить
менше 20 ррт металів. Розмір часток зазначають після
назви сорбенту у випробуваннях, в яких він викорис-
товується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді, 96 % спирті та ефірі.
Силікагель октадеканоїламінопропілсилільний для хро-
матографії. 1115200.
Силікагель тонко здрібнений з розміром часток від
З мкм до 10 мкм і поверхнею, хімічно модифікованою
амінопропілсилільними групами, ацильованими ок-
тадеканоїл-групами. Розмір часток зазначають після
назви сорбенту у випробуваннях, в яких він викорис-
товується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель октадецилсилільний для хроматографії.
1077500.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною октадецилсилільними групами. Розмір часток за-
значають після назви сорбенту у випробуваннях, в
яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель октадецилсилільний для хроматографії РІ.
1110100.
Силікагель надчистий, дуже тонко здрібний із
розміром пор близько 10 нм і поверхнею, хімічно мо-
дифікованою октадецилсилільними групами (вміст
вуглецю 19 %).
Вміст металів має бути менше 20 ррт.
Силікагель октадецилсилільний для хроматографії Р2.
1115300.
Силікагель надчистий, дуже тонко здрібнений із
розміром пор 15 нм і поверхнею, хімічно модифікова-
ною октадецилсилільними групами (вміст вугле-
цю 20 %), оптимізований для аналізу поліциклічних
ароматичних вуглеводнів. Розмір часток зазначають
після назви сорбенту у випробуваннях, в яких він ви-
користовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель октадецилсилільний деактивований відносно
основ для хроматографії. 1077600.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм; перед уведенням октадецил-
силільних груп його попередньо обробляють шляхом
ретельного промивання і гідролізу більшості поверх-
невих силоксанових містків для зведення до мінімуму
взаємодії з основними компонентами. Розмір часток
зазначають після назви сорбенту у випробуваннях, в
яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
253
4.1.1. Реактиви
Силікагель октадепилсилільний, деактивований віднос-
но основ, ендкепований для хроматографії. 1108600.
Силікагель дуже тонко здрібнений з розміром часток
від З мкм до 10 мкм і розміром пор близько 10 нм,
містить близько 16 % вуглецю. Перед введенням окта-
децилсилільних груп його попередньо обробляють
шляхом ретельного промивання й гідролізу більшості
поверхневих силоксанових містків. Для подальшого
зведення до мінімуму будь-якої взаємодії з основними
сполуками ретельно ендкепують для усунення біль-
шості силанольних груп, шо залишилися. Розмір час-
ток зазначають після назви сорбенту у випробуваннях,
в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель октадецилсилільний ендкепований для хро-
матографії. 1115400.
Силікагель дуже тонко здрібнений з розміром часток від
З мкм до 10 мкм і поверхнею, хімічно модифікованою
октадецилсилільними групами. Для зведення до мініму-
му взаємодії з основними сполуками ретельно ендкепу-
ють для усунення більшості силанольних груп, що зали-
шилися. Розмір часток зазначають після назви сорбенту
у випробуваннях, в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель октилсилільний для хроматографії. 1077700.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною октилсилільними групами. Розмір часток зазна-
чають після назви сорбенту у випробуваннях, в яких
він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель октилсилільний для хроматографії РІ.
1077701.
Силікагель дуже тонко здрібнений з розміром час-
ток від 3 мкм до 10 мкм і поверхнею, хімічно мо-
дифікованою октилсилільними й метальними
групами. Розмір часток зазначають після назви
сорбенту у випробуваннях, в яких він використо-
вується.
Дрібний гомогенний порошок білого кольору.
Практично не розчинний у воді й 96 % спирті.
Силікагель октилсилільний для хроматографії Р2.
1077702.
Силікагель надчистий, дуже тонко здрібнений з
розміром пор 10 нм і поверхнею, хімічно мо-
дифікованою октилсилільними групами (містить
19 % вуглецю). Містить менше 20 ррт металів.
Силікагель октилсилільний ендкепований для хромато-
графії. 1119600.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною октилсилільними групами. Для зведення до
мінімуму взаємодії з основними сполуками ретельно
ендкепують для усунення більшості силанольних
груп, що залишилися. Розмір часток зазначають після
назви сорбенту у випробуваннях, в яких він викорис-
товується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді і 96 % спирті.
Силікагель триметилсилільний для хроматографії.
1115500.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною триметилсилільними групами. Розмір часток за-
значають після назви сорбенту у випробуваннях, в
яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Силікагель для хроматографії, сильний аніоніт. 1077800.
Силікагель дуже тонко здрібнений з розміром часток
від 3 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною групами четвертинного амонію. Розмір часток за-
значають після назви сорбенту у випробуваннях, в
яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
рН: використовувані межі від 2 до 8.
Силікагель фенілсилільний для хроматографії. 1110200.
Силікагель дуже тонко здрібнений з розміром часток
від 5 мкм до 10 мкм і поверхнею, хімічно модифікова-
ною фенільними групами.
Силікагель фенілсилільний для хроматографії РІ.
1075700.
Силікагель тонко здрібнений з розміром часток 5 мкм
і поверхнею, хімічно модифікованою фенільними гру-
пами. Розмір часток зазначають після назви сорбенту
у випробуваннях, в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді, 96 % спирті й метиленхло-
риді.
Кремнію діоксид сфероїдальний: 8 нм.
Питома площа поверхні: 180м2/г.
Вміст вуглецю: 5.5 %.
Силікагель ціаносилільний для хроматографії. /109900.
Силікагель дуже тонко здрібнений з поверхнею,
хімічно модифікованою ціаносилільними групами.
Розмір часток зазначають після назви сорбенту у ви-
пробуваннях, в яких він використовується.
Дрібний гомогенний порошок білого кольору. Прак-
тично не розчинний у воді й 96 % спирті.
Синенсетин. С20Н20О7. (Мм. 372). 1110500. [2306-27-6].
З',4',5,6,7-Пентаметоксифлавон.
254
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Сірка. 1110800. Див. статтю Сірка для зовнішнього за-
стосування.
Сірки діоксид. 8О2. (М.м. 64.1). 1086700. [7446-09-5].
Сірчистий ангідрид.
Безбарвний газ. При стисненні перетворюється на
безбарвну рідину.
Сірки діоксид РІ. 8О2. (М.м. 64.1). 1110900.
Містить не менше 99.9 % (об/об) 8О2.
Сірководень. Н28. (М.м. 34.08). 1044000. [7783-06-4].
Газ; мало розчинний у воді.
Сірководень РІ. Н28. (М.м. 34.08). 1106600.
Містить не менше 99.7 % (об/об) Н28.
Сірчана кислота. Н28О4. (М.м. 98.1). 1086800. [7664-93-9].
Містить не менше 95.0 % (м/м) і не більше 97.0 %
(м/м) Н28О4.
Безбарвна, їдка, маслянистої консистенції, дуже
гігроскопічна рідина. Змішується з водою й 96 %
спиртом з інтенсивним виділенням тепла.
4": від 1.834 до 1.837.
Розчин 10 г/л є сильною кислотою й дає реакцію на
сульфати (2.3.1).
Прозорість (2.2.1). Кислота сірчана має бути прозо-
рою.
Кольоровість (2.2.2, метод 11). Кислота сірчана має
бути безбарвною.
Речовини, здатні окиснюватися. Обережно при охоло-
дженні 20 г кислоти сірчаної додають до 40 мл води Р,
додають 0.5 мл 0.002 М розчину калію перманганату,
фіолетове забарвлення має зберігатись не менше 5 хв.
Хлориди. Не більше 0.00005 % (0.5 ррт). Обережно при
охолодженні 10 г кислоти сірчаної додають до 10 мл во-
ди Р, охолоджують і доводять об’єм розчину тим самим
розчинником до 20 мл. Додають 0.5 мл розчину срібла
нітрату Р2\ витримують протягом 2 хв у захищеному
від світла місці. Одержаний розчин має витримувати
випробування на хлориди. Еталон готують із викорис-
танням 1 мл еталонного розчину хлориду (5 ррт СІ) Р,
19 мл води Рі 0.5 мл розчину срібла нітрату Р2.
Нітрати. Не більше 0.00005 % (0.5 ррт). Обережно
при охолодженні 50 г або 27.2 мл кислоти сірчаної до-
дають до 15 мл води Р, потім додають 0.2 мл свіжопри-
готованого розчину 50 г/л бруцину Ру кислоті оцтовій
льодяній Р. Через 5 хв забарвлення одержаного розчи-
ну має бути не інтенсивнішим за забарвлення еталон-
ного розчину, приготованого аналогічно до випробо-
вуваного із використанням 12.5 мл води Р, 50 г кисло-
ти сірчаної, вільної від азоту, Р, 2.5 мл еталонного роз-
чину нітрату (10 ррт 6'Оф Р і 0.2 мл розчину 50 г/л
бруцину Ру кислоті оцтовій льодяній Р.
Амоній. Не більше 0.0002 % (2 ррт). Обережно при
охолодженні 2.5 г кислоти сірчаної додають до води Р,
доводять об’єм розчину тим самим розчинником до
20 мл, охолоджують і по краплях додають 10 мл розчи-
ну 200 г/л натрію гідроксиду Р і 1 мл лужного розчину
калію тетрайодомеркурату Р; забарвлення розчину
має бути не інтенсивнішим за забарвлення еталонно-
го розчину, приготованого з використанням 5 мл ета-
лонного розчину амонію (1 ррт ИН4) Р, 15 мл води Р,
10 мл розчину 200 г/л натрію гідроксиду Рі 1 мл луж-
ного розчину калію тетрайодомеркурату Р.
Арсен (2.4.2, метод А). Не більше 0.000002 %
(0.02 ррт). Обережно при охолодженні до 50 г кисло-
ти сірчаної додають 3 мл кислоти азотної Р, обережно
упарюють до об’єму 10 мл, охолоджують, до одержа-
ного залишку додають 20 мл води Р і упарюють до
об’єму 5 мл. Розчин має витримувати випробування
на арсен. Еталон готують із використанням 1.0 мл
еталонного розчину арсену (1 ррт Аз) Р.
Важкі метали (2.4.8, метод А). Не більше 0.0002 %
(2 ррт). 10 мл розчину, приготованого для випробу-
вання на залізо, доводять водою Р до об’єму 20 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (2ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.0001 % (1 ррт). Зольний за-
лишок, одержаний при визначенні залишку після
прожарювання, розчиняють при слабкому нагріванні
у 1 мл кислоти хлористоводневої розведеної Р і дово-
дять об’єм розчину водою Рао 50.0 мл. 5 мл одержано-
го розчину доводять водою Рао об’єму 10 мл. Розчин
має витримувати випробування на залізо.
Залишок після прожарювання. Не більше 0.001 %. Ви-
значення проводять із 100 г кислоти сірчаної шляхом
обережного випарювання у невеликому тиглі над
відкритим полум’ям і нагрівання залишку до червоно-
го жару.
Кількісне визначення. У колбу з притертою скляною
пробкою помішають 30 мл води Р, точно зважують, до-
дають 0.8 мл кислоти сірчаної, охолоджують і знову
зважують. Титрують / М розчином натрію гідроксиду,
використовуючи як індикатор 0.1 мл розчину метило-
вого червоного Р.
1 мл 1 Мрозчину натрію гідроксиду відповідає 49.04 мг
Н28О4.
Зберігають у контейнері з притертою скляною пробкою,
виготовленому із скла або іншого інертного матеріалу.
Сірчана кислота, вільна від азоту. 1086806.
Має задовольняти вимоги для кислоти сірчаної Рі
витримувати таке додаткове випробування.
Нітрати. До 5 мл води Р обережно додають 45 мл
кислоти сірчаної, охолоджують до температури
40 °С і додають 8 мг дифенілбензидину Р; одержа-
ний розчин має бути блідо-рожевого або злегка
блідо-блакитного кольору.
Сірчаної кислоти 2.5 М розчин спиртовий. 1086801.
Обережно при постійному охолодженні і пе-
ремішуванні 14 мл кислоти сірчаної Р додають до
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
255
4.1.1. Реактиви
60 мл етанолу Р, охолоджують і доводять об’єм
розчину етанолом Рдо 100 мл.
Готують безпосередньо перед використанням.
Сірчаної кислоти 0.25 М розчин спиртовий.
1086802.
10 мл 2.5 М спиртового розчину кислоти сірчаної Р
доводять етанолом Рдо об’єму 100 мл.
Готують безпосередньо перед використанням.
Сірчаної кислоти розчин спиртовий. 1086803.
Обережно при постійному охолодженні й пе-
ремішуванні 20 мл кислоти сірчаної Р додають до
60 мл 96 % спирту Р, охолоджують і доводять
об’єм розчину 96 % спиртом Рдо 100 мл.
Готують безпосередньо перед використанням.
Сірчана кислота розведена. 1086804.
Містить 98 г/л Н28О4.
5.5 мл кислоти сірчаної Р додають до 60 мл води Р,
охолоджують і доводять об’єм розчину тим самим
розчинником до 100 мл.
Кількісне визначення. У колбу з притертою скля-
ною пробкою поміщають 30 мл води Р, додають
10.0 мл кислоти сірчаної розведеної і титрують
1 М розчином натрію гідроксиду, використову-
ючи як індикатор 0.1 мл розчину метилового черво-
ного Р.
1 мл 1 М розчину натрію гідроксиду відповідає
49.04 мг Н28О4.
Сквалан. С30Н62. (М.м. 422.8). 1084900. [111-01-3].
2,6,10,15,19,23-Гексаметилтетракозан.
Безбарвна масляниста рідина. Легко розчинний в
ефірі й жирних оліях, мало розчинний в ацетоні, 96 %
спирті, кислоті оцтовій льодяній і метанолі.
: від 0.811 до 0.813.
п% : від 1.451 до 1.453.
Смола іонообмінна сильнокислотна. 1085400. Див. Іоно-
обмінна смола сильнокислотна Р.
Соняшникова олія. 1086900. Жирна олія, одержана ви-
чавлюванням із зерен Неііапііша аппиих А.
Прозора рідина світло-жовтого кольору.
с/Ц : близько 0.92.
Гідроксильне число (2.5.3). Від 14 до 16.
Йодне число (2.5.4). Від 125 до 136.
Число омилення (2.5.6). Від 188 до 194.
Сополімер етилвінілбензол-дивінілбензолу. 1036900.
Тверді, пористі гранули кулястої форми з поперечно-
зшитого полімеру. Існують різні марки з різними
розмірами гранул. Розмір гранул зазначають у випро-
буваннях, в яких він використовується.
Сополімер етилвінілбензол-дивінілбензолу РІ.
1036901.
Тверді, пористі гранули кулястої форми з попе-
речно-зшитого полімеру, з номінальною питомою
площею поверхні від 500 м2/г до 600 м2/г і се-
реднім розміром пор 7.5 нм. Існують різні марки з
різними розмірами гранул. Розмір гранул зазнача-
ють у випробуваннях, в яких він використо
вується.
Сополімер стирол-дивінілбензолу. 1085500.
Тверді, пористі гранули із поперечно-зшитого поліме-
ру. Існують різні марки з різними розмірами гранул.
Розмір гранул зазначають після назви реактиву у ви-
пробуваннях, в яких він використовується.
Сорбіт. 1084800. [50-70-4|. Див. статтю Сорбіт.
96% спирт. С2Н6О. (М.м. 46.07). 1002500. [64-17-5].
Див. статтю 96 % спирт етиловии.
96 % спирт, вільний від альдегідів. 1002501.
1200 мл 96 % спирту Р змішують із 5 мл розчину
400 г/л срібла нітрату Рі 10 мл охолодженого роз-
чину 500 г/л калію гідроксиду Р. струшують, відсто-
юють протягом кількох днів і фільтрують.
Фільтрат переганяють безпосередньо перед вико-
ристанням.
Спирт (X відсотків, об/об). 1002502.
Для одержання розчину, вміст спирту у якому
відповідає величині X, змішують відповідні об’єми
води Р і 96 % спирту Р, враховуючи ефекти нагрі-
вання і зменшення об’єму, які супроводжують
приготування такої суміші.
Срібла діетилдитіокарбамат. С5Н10А§М82. (М.м. 256.1).
1110400. [1470-61-7].
Порошок від блідо-жовтого до сірувато-жовтого ко-
льору. Практично не розчинний у воді, розчинний у
піридині.
Готують таким чином. 1.7 г срібла нітрату Р розчиня-
ють у 100 мл води Р. Окремо розчиняють 2.3 г натрію
діетилдитіокарбамату Ру 100 мл води Р. Обидва роз-
чини охолоджують до температури 10 °С. потім їх
змішують і при перемішуванні збирають осад жовтого
кольору на скляному фільтрі, промивають 200 мл хо-
лодної води Рі сушать у вакуумі протягом 2-3 год.
Срібла діетилдитіокарбамат не повинен змінювати за-
барвлення або мати сильний запах.
Срібла нітрат. 1078300. [7761-88-8]. Див. статтю Срібла
нітрат.
Срібла нітрату реактив. 1078305.
До суміші 3 мл розчину аміаку концентрованого Р і
40 мл 1 М розчину натрію гідроксиду додають по
краплях при перемішуванні 8 мл розчину 200 г/л
срібла нітрату Р і доводять об’єм розчину водою Р
до 200 мл.
256
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Срібла нітрату розчин РІ. 1078301.
Розчин 42.5 г/л.
Зберігають у захищеному від світла місці.
Срібла нітрату розчин Р2. 1078302.
Розчин 17 г/л.
Зберігають у захищеному від світла місці.
Срібла нітрату аміачний розчин. 1078303.
2.5 г срібла нітрату Р розчиняють у 80 мл води Р,
по краплях додають розчин аміаку розведений Ріпо
розчинення осаду і доводять об’єм розчину во-
дою Рао 100 мл.
Готують безпосередньо перед використанням.
Срібла нітрату розчин у піридині. 1078304.
Розчин 85 г/л у піридині Р.
Зберігають у захищеному від світла місці.
Срібла оксид. А£2О. (Мм. 231.7). 1078400. [20667-12-3].
Порошок коричнювато-чорного кольору. Практично
нерозчинний у воді й 96 % спирті, легко розчинний у
розведеній кислоті азотній і розчинах аміаку.
Зберігають у захищеному від світла місці.
Срібно-марганцевий папір. 1078200.
Смужки повільно фільтруючого паперу занурюють у
розчин, який містить 8.5 г/л марганцю(П) сульфату Р
і 8.5 г/л срібла нітрату Р. Вигримують протягом
кількох хвилин, сушать над фосфору(У) оксидом Р, за-
хищаючи від дії парів кислот та лугів.
Стеаринова кислота. С|ХН36О. (М.м. 284.5). 1085200.
|57-11-4|. Октадеканова кислота.
Порошок або пластівці білого кольору. Масляниста на
дотик, практично не розчинна у воді, розчинна в гаря-
чому 96 % спирті та ефірі.
Температура плавлення: близько 70 °С.
Стрептоміцину сульфат. 1085300. [3810-74-0]. Див.
статтю Стрептоміцину сульфат.
Стронцію карбонат. 8гСО3. (М.м. 147.6). 1122700.
[1633-05-2].
Кристалічний порошок білого кольору.
Містить не менше 99.5 % 8гСО3.
Судан червоний 6. С17Н14М2О2. (М.м. 278.3). 1085800.
Показник Шульца № 149. Кольоровий індекс
№ 12150. Розчинний Червоний 1. 1-[(2-Метокси-
феніл)азо|нафталін-2-ол.
Порошок червонувато-коричневого кольору. Прак-
тично не розчинний у волі.
Хроматографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель с Р. На хроматографічну плас-
тинку наносять 10 мкл розчину 0,1 г/л у метиленхло-
риді Р і хроматографують у тому ж розчиннику. До-
вжина пробігу розчинника від лінії старту близько
10 см. На одержаній хроматограмі має виявлятись ли-
ше одна основна пляма.
Судан оранжевий. СІ6Н12М2О. (М.м. 248.3). 1110700.
[842-07-9|. Кольоровий індекс № 12055. 1-(Феніла-
зо)нафталін-2-ол. Судан І.
Порошок оранжево-червоного кольору. Практично не
розчинний у воді, розчинний у метиленхлориді.
Температура плавлення: близько 131 °С.
Сульфамінова кислота. Н3КО38. (М.м. 97.1). 1085900.
[5329-14-6].
Кристалічний порошок або кристали білого кольору.
Легко розчинна у воді, помірно розчинна в ацетоні,
96 % спирті й метанолі, практично не розчинна в
ефірі.
Температура плавлення: близько 205 °С, із розкла-
данням.
Сульфаніламід. С6Н8М2О2$. (М.м. 172.2). 1086100.
[63-74-1]. 4-Амінобензолсульфонамід.
Порошок білого кольору. Мало розчинний у воді, легко
розчинний у киплячій воді, ацетоні, розведених кисло-
тах і розчинах гідроксидів лужних металів, помірно роз-
чинний у 96 % спирті, ефірі та петролейному ефірі.
Температура плавлення: близько 165 °С.
Сульфанілова кислота. С6Н7МО3$. (М.м. 173.2).
1086200. [121-57-3]. 4-Амінобензолсульфонова кислота.
Безбарвні кристали. Помірно розчинна у воді, прак-
тично не розчинна в 96 % спирті.
Сульфановий синій. С27Н3|М2№О682. (М.м. 566.6).
1086000. [129-17-9]. Показник Шульца № 769. Кольо-
ровий індекс № 42045. Дисульфін синій. Кислотний
синій 1. Синій У8. Натрію [[[4-(діетиламіно)фе-
ніл](2,4-дисульфонатофеніл)метилен|циклогекса-
2,5-дієн-1 -іліден]діетиламоній.
Порошок фіолетового кольору. Розчинний у воді. Роз-
ведені розчини мають синє забарвлення, яке перехо-
дить у жовте при додаванні кислоти хлористоводневої
концентрованої.
Сульфатіазол. С9Н9М3О282. (М.м. 255.3). 1086300.
[72-14-01. 4-Аміно-А-(тіазол-2-іл)бензолсульфонамід.
Порошок або кристали білого чи жовтувато-білого ко-
льору. Дуже мало розчинний у воді, розчинний в аце-
тоні, мало розчинний у 96 % спирті. Розчиняється в
розведених мінеральних кислотах, розчинах гідро-
ксидів і карбонатів лужних металів.
Температура плавлення: близько 200 °С.
Сульфомолібденовий реактив Р2. 1086400.
Близько 50 мг амонію молібдату Ррозчиняють у 10 мл
кислоти сірчаної Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
257
4.1.1. Реактиви
Сульфомолібденовий реактив РЗ. 1086500.
2.5 г амонію молібдату Р розчиняють при нагріванні у
20 мл води Р. 28 мл кислоти сірчаної Р розводять
водою Рао об’єму 50 мл, потім охолоджують. Обидва
розчини змішують і доводять об'єм розчину водою Рдо
100 мл.
Зберігають у поліетиленовому контейнері.
Сульфосаліциловакислота. С7Н6О6$,2Н2О. (М.м. 254.2).
1086600. [5965-83-3]. 2-Гідрокси-5-сульфобензойна
кислота.
Кристалічний порошок або кристали білого кольору. Ду-
же легко розчинна у воді й 96 % спирті, розчинна в ефірі.
Температура плавлення: близько 109 °С.
Сурми(ПІ)хлорид. 5>ЬС13. (М.м. 228.1). 1007700.
110025-91-9]. Сурми трихлорид.
суспензії еритроцитів доводять сумішшю розчинників
фосфатний буферний розчин рН 7.2 Р — розчин 9 г/л
натрію хлориду /*(1:9) до об’єму 100 мл.
Тагатоза. С6Ні2О6. (М.м. 180.16). 1111000. [87-81-0].
п-ліксо- Гексулоза.
Порошок білого кольору.
[«]□ : -2.3°. Визначення проводять, використовуючи
розчин 21.9 г/л у воді Р.
Температура плавлення: від 134 °С до 135 °С.
Талію сульфат. Т12$О4. (М.м. 504.8). 1089100.
[7446-18-6]. Диталію сульфат.
Ромбоподібні призми білого кольору. Мало розчин-
ний у воді, практично не розчинний у 96 % спирті.
Тальк. 1087000. [14807-96-6]. Див. статтю Тальк.
Безбарвні кристали або прозора кристалічна маса.
Гігроскопічний, легко розчинний в етанолі. Сурми
хлорид гідролізується водою.
Зберігають у повітронепроникному контейнері, захи-
щають від вологи.
Сурми(ІП) хлориду розчин. 1007701.
30 г сурми(ПІ) хлориду /"швидко промивають двома
порціями хлороформу, вільного від етанолу, Р по
15 мл кожна; відкидають промивні розчини і негай-
но промиті кристали розчиняють при слабкому на-
гріванні у 100 мл хлороформу, вільного від етанолу, Р.
Зберігають розчин над кількома грамами натрію
сульфату безводного Р.
Сурми(ІІІ) хлориду розчин РІ. 1007702
Розчин 1. 110 г сурми(ІП) хлориду Р розчиняють у
400 мл етиленхлориду Р, додають 2 г алюмінію ок-
сиду безводного Р, перемішують і фільтрують крізь
скляний фільтр (40). Доводять об’єм фільтрату
етиленхлоридом Р до 500.0 мл і перемішують. Оп-
тична густина (2.2.25) одержаного розчину,
виміряна за довжини хвилі 500 нм у кюветі з тов-
щиною шару 2 см, не має перевищувати 0.07.
Розчин II. У витяжній шафі змішують 100 мл
свіжоперегнаного ацетилхлориду Р і 400 мл ети-
ленхлориду Р.
Зберігають у прохолодному місці.
Змішують 90 мл розчину І і 10 мл розчину II.
Зберігають у флаконах оранжевого скла з притер-
тою пробкою. Термін придатності 7 діб. Реактив
непридатний при появі забарвлення.
Суспензія еритроцитів кролика. 1074500.
1.6 % (об/об) суспензію еритроцитів кролика готують
таким чином: видаляють фібрин із 15 мл свіжовідібра-
ної крові кролика, струшуючи зі скляними кульками,
потім центрифугують із прискоренням 2000 £ протя-
гом 10 хв і промивають еритроцити трьома порціями
розчину 9 г/л натрію хлориду Р, по 30 мл кожна. 1.6 мл
Танінова кислота. 1087100. [ 1401-55-4]. Дубильна кис-
лота.
Блискучі лусочки або аморфний порошок від жовту-
ватого до світло-коричневого кольору. Дуже легко
розчинна у воді, легко розчинна у 96 % спирті, роз-
чинна в анетоні, практично не розчинна в ефірі.
Зберігають у захищеному від світла місці.
Теофілін. 1089300. [58-55-9]. Див. статтю Теофілін.
/-Терпінен. С10НІ6. (Мм. 136.2). 1115900. [99-85-4].
1 -Ізопропіл-4-метилциклогекса-1,4-дієн.
Масляниста рідина.
Д}5 : близько 0.850.
: від 1.474 до 1.475.
Температура кипіння: від 183 °С до 186 °С.
/-Терпінен,
використовуваний у
газовій
має витримувати таке випробування.
хроматографії
Кількісне визначення. П роводять методом газової хро
матографії (2.2.28), як зазначено в статті
перцевої.
Олія
м яти
Випробовуваний розчин. Випробовувана речовина.
Плоша основного піка має бути
площ усіх піків.
не
менше 93.0
% суми
Терпінен-4-ол. С|0НІХО. (М.м. 154.2).
1116000.
4-Метил-1 -(1 -метилетил)циклогекс-3-ен-1 -ОЛ.
/?-Мент-1-ен-4-ол.
Безбарвна масляниста рідина.
б/2о : близько 0.934.
: близько 1.477.
Температура кипіння: від 209 °С до 212 °С.
Терпінен-4-ол,
використовуваний у
[562-74-3]
газовій
графії, має витримувати таке випробування.
Кількісне визначення.
Проводять методом
хромато
газової
хро
матографії (2.2.28), як зазначено в статті
дова.
Олія
лаван
258
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ
2001
4.1.1. Реактиви
Випробовуваний розчин. Випробовувана речовина.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків.
а-Терпінеол. СІ0НІ8О. (М.м. 154.2). 1087300. |98-55-5].
(й5)-2-(4-Метилциклогекс-3-еніл)-2-пропанол.
Безбарвні кристали. Практично не розчинний у воді,
розчинний у 96 % спирті та ефірі.
</20: близько 0.935.
1$: близько 1.483.
[аїр : близько 92.5°.
Температура плавлення: близько 35 °С.
Може містити від 1 % до 3 % /1-терпінеолу.
а-Терпінеол, використовуваний у газовій хромато-
графії, має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія анісова.
Випробовуваний розчин. 100 г/л у гексані Р.
Площа основного піка має бути не менше 97.0 % суми
площ усіх піків, за винятком піка гексану.
Тестостерон. 1116100. [58-22-0]. Див. статтю Тесто-
стерон.
Тестостерону пропіонат. 1087400. [57-85-2]. Див. статтю
Тестостерону пропіонат.
Тетрабутиламонію бромід. СІ6Н36ВгМ. (М.м. 322.4).
1087500. [1643-19-2].
Кристали білого або майже білого кольору.
Температура плавлення: від 102 °С до 104 °С.
Тетрабутиламонію гідроксид. С16Н37МО,30Н2О.
(М.м. 800). 1087800. [2052-49-5].
Містить не менше 98.0 % С16Н37ТМО,30Н2О.
Кристали білого або майже білого кольору. Розчинний
у воді.
Кількісне визначення. 1.000 г розчиняють у 100 мл во-
ди Рі негайно титрують 0.1Мрозчином кислоти хлори-
стоводневої потенціометрично (2.2.20). Паралельно
проводять контрольний дослід.
1 мл 0.1 М розчину кислоти хлористоводневої
відповідає 80.0 мг С16Н37МО,30Н2О.
Тетрабутиламонію гідроксиду розчин (104 г/л).
1087801. [2052-49-5].
Розчин, який містить 104 г/л С16Н37МО (М.м. 259.5),
приготований розведенням реактиву відповідного
ступеня чистоти.
Тетрабутиламонію гідроксиду розчин (400 г/л).
1087802. [2052-49-5].
Розчин, який містить 400 г/л С16Н37МО
(М.м. 259.5), приготований розведенням реактиву
відповідного ступеня чистоти.
Тетрабутиламонію гідросульфат. С16Н37™О48.
(М.м. 339.5). 1087700. [32503-27-8].
Кристалічний порошок або безбарвні кристали. Легко
розчинний у воді й метанолі.
Температура плавлення: від 169 °С до 173 °С.
Оптична густина (2.2.25). Не більше 0.05. Вимірюють
оптичну густину розчину 50 г/л в області довжин
хвиль від 240 нм до 300 нм.
Тетрабутиламонію дигідрофосфат. СІ6Н38ІМО4Р.
уМ.м. 339.5). 1087600. [5574-97-0].
Порошок білого кольору, гігроскопічний.
рН (2.2.3). Близько 7.5. Вимірюють рН розчину
170 г/л.
Оптична густина (2.2.25). Близько 0.10. Вимірюють оп-
тичну густину розчину 170 г/л задовжини хвилі 210 нм.
Зберігають у повітронепроникному контейнері.
Тетрабутиламонію йодид. С16Н361М. (М.м. 369.4).
1087900. [311-28-4].
Містить не менше 98.0 % СІ6Н36ІМ.
Кристалічний порошок або кристали білого кольору
або дещо забарвлені. Розчинний у 96 % спирті.
Сульфатна зола (2.4.14). Не більше 0.02 %.
Кількісне визначення. 1.200 г розчиняють у 30 мл во-
ди Р, додають 50.0 мл 0.1 М розчину срібла нітрату і
5 мл кислоти азотної розведеної Р. Титрують надли-
шок срібла нітрату 0.1 Мрозчином амонію тіоціанату,
використовуючи як індикатор 2 мл розчину заліза(Ш)
амонію сульфату Р2.
1 мл 0.1М розчину срібла нітрату відповідає 36.94 мг
С16Н36І1Ч.
Тетрагептиламонію бромід. С28Н60ВгМ. (М.м. 490.7).
1088400. [4368- 51-8].
Кристалічний порошок або кристали білого кольору
або дещо забарвлені.
Температура плавлення: від 89 °С до 91 °С.
Тетрагептиламонію гідросульфат. С24Н53МО48.
(Мм. 451.8) 1116300. [32503-34-7].
Температура плавлення: від 100 °С до 102 °С.
Тетрагідрофуран. С4Н8О. (Мм. ТІЛ). 1088500. [109-99-9].
Тетраметиленоксид.
Прозора, безбарвна, займиста рідина. Змішується з
водою, 96 % спиртом і ефіром.
б/2о : близько 0.89.
Не переганяють, якщо тетрагідрофуран не витримує
випробування на пероксиди.
Пероксиди. 8 мл розчину крохмалю з калію йодидом Р
поміщають у циліндр із притертою пробкою місткістю
12 мл і діаметром близько 1.5 см, заповнюють пов-
ністю тетрагідрофураном, потім перемішують і витри-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
259
4.1.1. Реактиви
мують у темному місці протягом ЗО хв: не має з’явля-
тись забарвлення.
Тетрагідрофуран, використовуваний у спектрофото-
метри, має витримувати таке додаткове випробуван-
ня.
Мінімальне пропускання (2.2.25). Визначення прово-
дять, використовуючи як компенсаційну рідину во-
ду Р:
20 % за довжини хвилі 255 нм,
80 % за довжини хвилі 270 нм,
98 % за довжини хвилі 310 нм.
Тетрадекан. С14Н30. (М.м. 198.4). 1088200. [629-59-4].
//-Тетрадекан.
Містить не менше 99.5 % (м/м) С14Н30.
Безбарвна рідина.
^20 : близько 0.76.
п^ : близько 1.429.
Температура кипіння: близько 252 °С.
Температура плавлення: близько -5 °С.
Тетрадециламонію бромід. С40Н84ВгМ. (М.м. 659).
1088300. [14937-42-9]. Тетракис(децил)амонію бромід.
Кристалічний порошок або кристали білого кольору
або дещо забарвлені.
Температура плавлення: від 88 °С до 89 °С.
Тетраетиламонію гідроксиду розчин. С8Н21МО.
(М.м. 147.3). 1100300. [77-98-5].
Розчин 200 г/л; безбарвна рідина, є сильним лугом.
^2о : близько 1.01.
: близько 1.372.
Кваліфікація — для "ВЕРХ".
Тетраетиламонію гідросульфат. С8Н2ІКО48. (М.м. 227.3).
1116200. [16873-13-5].
Гігроскопічний порошок.
Температура плавлення: близько 245 °С.
Тетраетиленпентамін. С8Н23Н5. (М.м. 189.3). 1102000.
[ 112-57-2]. 3,6,9-Триазаундекан-1,11 -діамін.
Безбарвна рідина. Розчинний в ацетоні.
: близько 1.506.
Зберігають у сухому й прохолодному місці.
Тетразолієвий синій. С40Н32С12Н8О2. (М.м. 728).
1089000. [1871-22-3]. 3,3'-(3,3'-Диметокси[1,Г-
біфеніл]-4,4'-діїл)біс[2,5-дифеніл-2//-тетразолій]ди-
хлорид.
Кристали жовтого кольору. Мало розчинний у воді,
легко розчинний у 96 % спирті й метанолі, практично
не розчинний в ацетоні та ефірі.
Температура плавлення: близько 245 °С, із розкла-
данням.
Тетраметиламонію гідроксид. С4НІЗМО,5Н2О.
(М.м. 181.2). 1122800. Тетраметиламонію гідроксид
пентагідрат.
Кваліфікація — для "ВЕРХ".
Тетраметиламонію гідроксиду розчин. 1088600. [75-59-2].
Містить не менше 10.0 % (м/м) С4Н13МО (М.м. 91.2).
Прозора, безбарвна або дещо забарвлена рідина.
Змішується з водою й 96 % спиртом.
Кількісне визначення. До 1.000 г додають 50 мл води Р\
титрують 0.05Мрозчином кислоти сірчаної, використо-
вуючи як індикатор 0.1 мл розчину метилового червоно
го Р.
1 мл 0.05 Мрозчину кислоти сірчаної відповідає 9.12 мг
С4Н13МО.
Тетраметиламонію гідроксиду розчин розведений.
1088601.
10 мл розчину тетраметиламонію гідроксиду Р до-
водять 96 % спиртом, вільним від альдегідів, Рпо
об’єму 100 мл.
Готують безпосередньо перед використанням.
Тетраметиламонію гідросульфат. С4Н13Т9О48. (Мм. 171.2).
11 і6400. [80526-82-5].
Гігроскопічний порошок.
Температура плавлення: близько 295 °С.
Тетраметиламонію хлорид. С4НІ2С1КІ. (М.м. 109.6).
1100400. [75-57-0].
Безбарвні кристали. Розчинний у воді й 96 % спирті.
Температура плавлення: близько 300 °С, із розкла-
данням.
Тетраметилдіамінодифенілметан. СІ7Н22^. (М.м. 254.4).
1088700. [101-61-1]. 4,4'-Метиленбіс-(Л,,Л,-диметил-
анілін).
Кристали від білого до блакитнувато-білого кольору
або листочки. Практично не розчинний у воді, мало
розчинний у 96 % спирті, розчинний у мінеральних
кислотах, легко розчинний в ефірі.
Температура плавлення: близько 90 °С.
Тетраметилдіамінодифенілметану реактив. 1088701.
Розчин А. 2.5 г тетраметилдіамінодифенілметану Р
розчиняють у 10 мл кислоти оцтової льодяної Р і
додають 50 мл води Р.
Розчин В. 5 г калію йодиду Р розчиняють у 100 мл
води Р.
Розчин С. 0.30 г нінгідрину Р розчиняють у 10 мл
кислоти оцтової льодяної Р і додають 90 мл води Р.
Розчини А й В змішують, до одержаного розчину
додають 1.5 мл розчину С.
Тетраметилетилендіамін. С6Н16]Ч2. (М.м. 116.2).
1088800. [110-18-9]. ТУ,/V,А'',/V-Тетраметилетилен-
діамін.
260
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Безбарвна рідина. Змішується з водою, 96 % спиртом
та ефіром.
/?2о: близько 0.78.
и®: близько 1.418.
Температура кипіння: близько 121 °С.
Тетраметилсилан. С4НІ2$і. {М.м. 88.2). 1088900. [75-76-3].
ТМ$.
Прозора, безбарвна рідина. Дуже мало розчинний у
воді, розчинний в ацетоні й 96 % спирті.
/?2о: близько 0.64.
я® : близько 1.358.
Температура кипіння: близько 26 °С.
Тетраметилсилан, використовуваний у спектроскопії
ядерного магнітного резонансу, має витримувати таке
додаткове випробування.
Успектрі ЯМР приблизно 10 % (об/об) розчину тетра-
метилсилану в дейтерованому хлороформі Р інтен-
сивність будь-якого стороннього сигналу, за винятком
тих, які відповідають обертанню бічних зв’язків, і хло-
роформу, не має перевищувати інтенсивності бічних
ліній С-13, розташованих на відстані 59.1 Гц по обидва
боки основного сигналу тетраметилсилану.
Тетрахлоретан. С2Н2С14. {М.м. 167.9). 1088000. [79-34-5].
1,1,2,2-Тетрахлоретан.
Прозора, безбарвна рідина. Мало розчинний у воді,
змішується з 96 % спиртом та ефіром.
^2о близько 1.59.
я®: близько 1.495.
Температурні межі перегонки (2.2.11). Від 145 °С до
147 °С; має переганятись не менше 95 %.
Тимін. С5Н61\І2О2. (М.м. 126.1). 1090400. [65-71-4].
5-Метилпіримідин-2,4( 1 //,3//)-діон.
Короткі голчасті кристали або пластинки. Мало роз-
чинний у холодній воді, розчинний у гарячій воді,
розчинний у розведених розчинах гідроксидів лужних
металів.
Тимол. 1090500. [89-83-8]. Див. статтю Тимол.
Тимол, використовуваний у газовій хроматографії, мас
витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28), як зазначено у статті Олія м’яти
перцевої.
Випробовуваний розчин. 0.1 г у 10 мл ацетону Р.
Площа основного піка має бути не менше 95.0 % суми
площ усіх піків, за винятком піка ацетону.
Тимоловий синій. С27Н30О5$. (М.м. 466.6). 1090600.
[76-61-9]. Тимолсульфонфталеїн. 4,4'-(3//-2,1-Бен-
зоксатіол-3-іліден)біс[2-ізопропіл-5-метилфенол]-
5,5-діоксид.
Кристалічний порошок від коричнювато-зеленого до
зеленувато-синього кольору. Мало розчинний у воді,
розчинний у 96 % спирті й розведених розчинах гідро-
ксидів лужних металів.
Тимолового синього розчин. 1090601.
0.1 г тимолового синього Р розчиняють у суміші
2.15 мл 0.1 М розчину натрію гідроксиду і 20 мл
96 % спирту Р, доводять об’єм розчину водою Рао
100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р додають 0.1 мл роз-
чину тимолового синього й 0.2 мл 0.02 Мрозчину
натрію гідроксиду, з’являється синє забарвлення,
яке має перейти у жовте при додаванні не більше
0.1 мл 0.02 М розчину кислоти хлористоводневої.
Зміна забарвлення. Від червоного до жовтого в
інтервалі рН 1.2-2.8. Від оливково-зеленого до си-
нього в інтервалі рН 8.0-9.6.
Тимолфталеїн. С28Н30О4. (М.м. 430.5). 1090700.
[125-20-2]. 3,3-Біс(4-гідрокси-5-ізопропіл-2-метил-
фе н іл) - З Н- ізобе нзофуран -1 -он.
Порошок від білого до жовтувато-білого кольору. Прак-
тично не розчинний у воді, розчинний у 96 % спирті й
розведених розчинах гідроксидів лужних металів.
Тимолфталеїну розчин. 1090701.
Розчин 1 г/л у 96 % спирті Р.
Випробування на чутливість. До 100 мл води, вільної
від вуглецю діоксиду, Р додають 0.2 мл розчину ти-
молфталеїну, розчин безбарвний; при додаванні не
більше 0.05 мл 0.1 Мрозчину натрію гідроксиду має
з’явитися синє забарвлення розчину.
Зміна забарвлення. Від безбарвного до синього в
інтервалі рН 9.3-10.5.
Тирамін. С8НИМО. (М.м. 137.2). 1117600. [51-67-2].
4-(2-Аміноетил)фенол.
Кристали. Мало розчинний у воді, розчинний у гаря-
чому етанолі.
Температура плавлення: від 164 °С до 165 °С.
Тирозин. С9НИМО3. (М.м. 181.2). 1094800. [60-18-4].
2-Аміно-3-(4-гідроксифеніл)пропіонова кислота.
Кристалічний порошок або кристали білого кольору
або безбарвні кристали. Мало розчинний у воді, прак-
тично не розчинний в ацетоні, етанолі та ефірі, роз-
чинний у кислоті хлористоводневій розведеній і роз-
чинах гідроксидів лужних металів.
Хроматографія. Визначення проводять, як зазначено
у статті Леводопа-, на одержаній хроматограмі має ви-
являтись лише одна основна пляма.
Титан. Ті. (А.м. 47.88). 1091000. [7440-32-6].
Містить не менше 99 % Ті.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
261
4.1.1. Реактиви
Металевий порошок або тонкий дріт, діаметром не
більше 0.5 мм, або губка.
Температура плавлення: близько 1668 °С.
Густина: близько 4.507 г/см3.
Титановий жовтий. С28НІ9Н5Ма2О684. (М.м. 696).
1090900. [1829-00-1]. Показник Шульца № 280. Ко-
льоровий індекс № 19540. Тіазоловий жовтий. Ди-
натрію 2,2'-[( 1-триазен-1,3-діїл)ди-4,1 -фенілен]біс-
[6-м етилбен зотіазол - 7 - сул ьфо нат].
Порошок жовтувато-коричневого кольору. Легко роз-
чинний у воді й 96 % спирті.
Титанового жовтого папір. 1090901.
Смужки фільтрувального паперу занурюють у роз-
чин титанового жовтого Р, витримують кілька
хвилин і сушать при кімнатній температурі.
Титанового жовтого розчин. 1090902.
Розчин 0.5 г/л.
Випробування на чутливість. До 10 мл води Рдода-
ють 0.1 мл розчину титанового жовтого, 0.2 мл
еталонного розчину магнію (10 ррт М%) Р і 1.0 мл
1 Мрозчину натрію гідроксиду. Одержаний розчин
порівнюють із еталонним розчином, приготова-
ним тим самим способом, за винятком магнію;
має спостерігатись чітке рожеве забарвлення роз-
чину.
Титану діоксид. 1117900. 113463-67-7]. Див. статтю Ти-
тану діоксид.
Титану(ІІІ) хлорид. ТіС13. (М.м. 154.3). 1091200.
[7705-07-9]. Титану трихлорид.
Кристали червонувато-фіолетового кольору, які
розпливаються на повітрі. Розчинний у воді й 96 %
спирті, практично не розчинний в ефірі.
Температура плавлення: близько 440 °С.
Зберігають у повітронепроникному контейнері.
Титану(ІІІ) хлориду розчин. 1091201.
Розчин 150 г/л у розчині 100 г/л (НСІ) кислоти
хлористоводневої.
е/2о : близько 1.19.
Титану(ІІІ) хлорид і кислоти сірчаної реактив.
1091202.
20 мл розчину титану(НІ) хлориду Р обережно
змішують із 13 мл кислоти сірчаної Р, додають до-
статню кількість розчину водню пероксиду концен-
трованого Р до одержання жовтого забарвлення,
нагрівають до початку виділення білих парів і охо-
лоджують. Розводять водою Р, повторюють випа-
рювання й додавання води Р до одержання без-
барвного розчину, доводять об’єм розчину водою Р
до 100 мл.
Тіамазол. С4Н6М2$. (М.м. 114.2). 1089400. [60-56-0].
М еті м азол. 1 - М етил -1Н-1 м ідазол - 2 -ті ол.
Кристалічний порошок білого або майже білого ко
льору. Легко розчинний у воді, розчинний у 961
спирті й метиленхлориді, помірно розчинний в
ефірі.
Температура плавлення: близько 145 °С.
2-(2-Тієніл)оцтова кислота. С6Н6О28. (М.м. 142.1).
1089500. [1918-77-0].
Порошок коричневого кольору.
Температура плавлення: близько 65 °С.
Тіоацетамід. С2Н5М$. (М.м. 75.1). 1089600. [62-55-5].
Кристалічний порошок або безбарвні кристали. Легко
розчинний у воді й 96 % спирті.
Температура плавлення: близько 113 °С.
Тіоацетаміду розчин. 1089602.
Розчин 40 г/л.
Тіоацетаміду реактив. 1089601.
До 0.2 мл розчину тіоацетаміду Р додають 1 мл
суміші 5 мл води Р, 15 мл / М розчину натрію
гідроксиду і 20 мл гліцерину (85 %) Р, нагрівають на
водяній бані протягом 20 с.
Готують безпосередньо перед використанням.
Тіобарбітурова кислота. С4Н4М2О28. (М.м. 144.2).
1111200. [504-17-6]. 4,6-Дигідрокси-2-сульфаніл-
піримідин.
Тіогліколева кислота. С2Н4О28. (М.м. 92.1). 1089700.
[68-11-1]. 2-Меркаптооцтова кислота.
Безбарвна рідина. Змішується з водою, розчинна в
96 % спирті.
Тіомсрсал. С9Н9Н£МаО28. (М.м. 404.8). 1089800.
[54-64-8]. Натрію меркуротіолат. Натрію 2-[(етилмер-
куріо)тіо [бензоат.
Легкий кристалічний порошок жовтувато-білого ко-
льору. Дуже легко розчинний у воді, легко розчинний
у 96 % спирті, практично не розчинний в ефірі.
Тіосечовина. СН4Тч2$. (М.м. 76.1). 1089900. [62-56-6].
Кристалічний порошок або кристали білого кольору
Розчинна у воді й 96 % спирті.
Температура плавлення: близько 178 °С.
Тозиларгініну метилового ефіру гідрохлорид.
С14Н23С1М4О48. (М.м. 378.9). 1092000. [1784-03-8].
/У-Тозил-Ь-аргінін метилового ефіру гідрохлорид.
Етил-(5)-5-гуанідино-2-(4-метилбензолсульфон-
амідо)валерату гідрохлорид.
[се] □ : від -12° до -16°. Визначення проводять, викори-
стовуючи розчин 40 г/л.
Температура плавлення: близько 145 °С.
262
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Тозиларгініну метилового ефіру гідрохлориду розчин.
1092001.
До 98.5 мг тозиларгініну метилового ефіру гідрохло-
риду Р додають 5 мл буферного розчину трис(гідрок-
симетил)амінометану рН 8.1 Р, струшують до роз-
чинення, додають 2.5 мл змішаного розчину мети-
лового червоного Р і доводять об’єм розчину водою Р
до 25.0 мл.
Тозил-лізил-хлорметану гідрохлорид. С|4Н22СІ2^Оз8>.
(Мм. 369.3). 1092100. [4238-41-9]. А-Тозил-Е-лізил-
хлорметану гідрохлорид. (35)-7-Аміно-1-хлор-3-(4-
метилбензолсульфонамідо)гептан-2-он гідрохлорид.
[а]™ : від -7° до -9°. Визначення проводять, викорис-
товуючи розчин 20 г/л.
Температура плавлення: близько 155 °С, із розкла-
данням.
: від 310 до 340. Визначення проводять за довжи-
ни хвилі 230 нм, використовуючи як компенсаційну
рідину воду Р.
ІГозилфенілаланілхлорметан. С|7Н1ХС1МО3$. (Мм. 351.9).
1092200. |402-71-1]. А-Тозил-Ь-фенілаланілхлорметан.
[а]р : від -85° до -89°. Визначення проводять, викори-
стовуючи розчин 10 г/л у 96 % спирті Р.
Температура плавлення: близько 105 °С.
: від 290 до 320. Визначення проводять за довжи-
ни хвилі 228.5 нм у 96 % спирті Р.
о-Толідин. С14Н16№>. (М.м. 212.3). 1123000. [119-93-7].
3,3'-Диметилбензидин.
Містить не менше 97.0 % С|4Н|6М2.
Кристалічний порошок світло-коричневого кольору.
Температура плавлення: близько 130 °С.
о-Толідину розчин. 1123001.
0.16 г о-толідину Р розчиняють у 30.0 мл кислоти
оцтової льодяної Р, додають 1.0 г калію іюдиду Р
і доводять об’єм розчину водою Рро 500.0 мл.
о-Толуїдин. С7Н9М. (М.м. 107.2). 1091700. [95-53-4].
2-Метиланілін.
Рідина блідо-жовтого кольору, під дією повітря й світла
стає червонувато-коричневою. Мало розчинний у
воді, розчинний у 96 % спирті й розведених кислотах.
(/з”: близько 1.01.
: близько 1.569.
Температура кипіння: близько 200 °С.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла місці.
и-Толуїдин. С7Н9№ (М.м. 107.2). 1091800. [106-49-0].
4-Метиланілін.
Блискучі пластинки або пластівці. Мало розчинний у
воді, легко розчинний в ацетоні й 96 % спирті, роз-
чинний в ефірі.
Температура плавлення: близько 44 °С.
Толуїдиновий синій. С15Н16С1К38. (М.м. 305.8).
1091900. [92-31-9]. Показник Шульца №1041. Кольо-
ровий індекс № 52040. Толуїдиновий синій О. З-Амі-
но-7-диметиламіно-2-метилфенотіазину-5 хлорид.
Порошок темно-зеленого кольору. Розчинний у воді,
мало розчинний у 96 % спирті.
Толуол. С7Н8. (М.м. 92.1). 1091300. [108-88-3]. Метил-
бензол.
Прозора, безбарвна, займиста рідина. Дуже мало роз-
чинний у воді, змішується з 96 % спиртом.
с/2о : від 0.865 до 0.870.
Температура кипіння: близько 110 °С.
Толуол, вільний від сірки. 1091301.
Має задовольняти вимоги для толуолу Р і таку до-
даткову вимогу.
Сірковмісні сполуки. До 10 мл толуолу додають 1 мл
етанолу Р, 3 мл розчину калію плюмбіту Р
і кип’ятять зі зворотним холодильником протягом
15 хв. Через 5 хв водний шар не має потемніти.
Речовини, споріднені тіофену. 2 мл толуолу струшу-
ють із 5 мл реактиву ізатину Р протягом 5 хв і за-
лишають на 15 хв; у нижньому шарі не має спос-
терігатись синє забарвлення.
о-Толуолсульфонамід. С7Н9№О28. (М.м. 171.2). 1091400.
[88-19-7]. 2-Метилбензолсульфонамід.
Кристалічний порошок білого кольору. Мало розчин-
ний у воді та ефірі, розчинний у 96 % спирті й розчи-
нах гідроксидів лужних металів.
Температура плавлення: близько 156 °С.
л-Толуолсульфонамід. 1091500. Див. Толуолсульфонамід Р.
Толуолсульфонамід. С7Н9МО28. (М.м. 171.2). 1091500.
[70-55-3]. 4-Метилбензолсульфонамід. л-Толуолсуль-
фонамід.
Кристалічний порошок білого кольору. Мало розчин-
ний у воді та ефірі, розчинний у 96 % спирті й розчи-
нах гідроксидів лужних металів.
Температура плавлення: близько 136 °С.
Хроматографія. Визначення проводять, як зазначено
в статті Толбутамід', на одержаній хроматограмі має
виявлятись лише одна основна пляма.
Толуолсульфонова кислота. С7Н8О3$,Н2О. (М.м. 190.2).
1091600. [6192-52-5]. 4-Метилбензолсульфонова кислота.
Містить не менше 87.0 % С7Н8О3$.
Кристалічний порошок або кристали білого кольору.
Легко розчинна у воді, розчинна в 96 % спирті та ефірі.
Трагакант. 1092300. [9000-65-1]. Див. статтю Трагакант.
Треонін. 1090000. [72-19-5]. Див. статтю Треонін.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
263
4.1.1. Реактиви
Триамцинолон. С2|Н27РО6. (М.м. 394.4). 1111300.
[124-94-7]. 9-Фтор-11 р, 16а, 17,21-тетрагідроксипрег-
на-1,4-дієн-3,20-діон.
Кристалічний порошок.
Температура плавлення: від 262 °С до 263 °С.
Триацетин. С9Н14О6. (Мм. 218.2). 1092400. [102-76-1].
Пропан-1,2,3-триїптриацетат.
Майже прозора, безбарвна або жовтуватого кольору
рідина. Розчинний у воді, змішується з 96 % спиртом
та ефіром.
с/2о : близько 1.16.
Лр : близько 1.43.
Температура кипіння: близько 260 °С.
Триетаноламін. С6Н15]\ІО3. (М.м. 149.2). 1092900.
[102-71 -6]. 2,2',2"-Нітрилотриетанол.
Безбарвна, в’язка, дуже гігроскопічна рідина, під дією
повітря й світла набуває коричневого забарвлення.
Змішується з водою, ацетоном, 96 % спиртом, гліце-
рином (85 %) і метанолом.
г/2о : близько 1.13.
Зберігають у повітронепроникному контейнері, захи-
щеному від світла місці.
Триетиламін. С6Н|5М. (Мм. 101.2). 1093000. [121-44-8].
А^А-Діетилетанамін.
Безбарвна рідина. Мало розчинний у воді при темпера-
турі нижче 18.7 °С, змішується з 96 % спиртом та ефіром.
б/2о : близько 0.727.
Лр : близько 1.401.
Температура кипіння: близько 90 °С.
Триетилендіамін. С6Н12М2. (М.м. 112.2). 1093100.
1,4-Діазабіпикло[2.2.2]октан.
Кристали, дуже гігроскопічні. Легко сублімується при
кімнатній температурі. Легко розчиняється у воді,
ацетоні та етанолі.
Температура кипіння: близько 174 °С.
Температура плавлення: близько 158 °С.
Зберігають у повітронепроникному контейнері.
Трикозан. С23Н48. (Мм. 324.6). 1092800. [638-67-5].
Кристали білого кольору. Практично не розчинний у
воді, розчинний в ефірі й гексані.
Лр : близько 1.447.
Температура плавлення: близько 48 °С.
Триметилпентан. С8Н18. (Мм. 114.2). 1093400. [540-84-1].
Ізо-октан. 2,2,4-Триметилпентан.
Безбарвна, займиста рідина. Практично не розчинний
у воді, розчинний в етанолі.
^2о : від 0.691 до 0.696.
Лр : від 1.391 до 1.393.
Температурні межі перегонки (2.2.11). Від 98 °С до І
100 °С; має переганятись не менше 95 %.
Триметилпентан, використовуваний у спектрофото-9
метрії, має витримувати таке додаткове випробування. І
Мінімальне пропускання (2.2.25): 98 %. Визначення
проводять в області довжин хвиль від 250 нм до 420 нм.
використовуючи як компенсаційну рідину воду Р.
Триметилпентан РІ. 1093401.
Має задовольняти вимоги для триметилпентану Р
із таким зміненням.
Оптична густина (2.2.25). Не більше 0.07. Визна-
чення проводять в області довжин хвиль від 220 нм
до 360 нм, використовуючи як компенсаційну
рідину воду Р.
ТУ-Триметилсилілімідазол. С6Н|2]\І2Зі. (М.м. 140.3).
1100500. [18156-74-6]. 1-Триметилсилілімідазол.
Безбарвна, гігроскопічна рідина.
г/2о : близько 0.96.
Лр : близько 1.48.
Зберігають у повітронепроникному контейнері.
ТУ, О-біс(Триметилсиліл)ацетамід. С8 Н21N О3і2.
(М.м. 203.4). 1093600. [10416-59-8].
Безбарвна рідина.
б/2о : близько 0.83.
Трипсин. 1094500. [9002-07-7].
Протеолітичний фермент, одержаний активацією
трипсиногену, вилученого із підшлункової залози би-
ка (Вох іаигих Т.).
Кристалічний або аморфний порошок білого кольору.
Помірно розчинний у воді.
Трипсин д ля пептидного картування. 1094600. [9002-07-7].
Трипсин високої чистоти, оброблений з метою підви-
щення хімотрипсинової активності.
Триптофан. С||Н12М2О2. (Мм. 204.2). 1094700. [73-22-3].
Кристалічний порошок від білого до жовтувато-білого
кольору або безбарвні кристали. Мало розчинний у
воді, дуже мало розчинний у 96 % спирті, практично
не розчинний в ефірі.
[а]р : близько -30°. Визначення проводять, викорис-
товуючи розчин 10 г/л.
1,3,5-Трис[3,5-ди(1,1-диметилетил)-4-гідроксибензил]-
1,3,5-триазин-2,4,6(1//,3//,5Д)-трион.
(Мм. 784.1). 1094000. [27676-62-6].
Кристалічний порошок білого кольору.
Температура плавлення: від 218 °С до 222 °С.
264
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Трис[2,4-ди(1,1-диметилетил)феніл] фосфіт. С42Н63О3Р.
(Ют. 647). 1094100. [31570-04-4].
Порошок білого кольору.
Температура плавлення: від 182 °С до 186 °С.
Трис(гідроксиметил)амінометан. 1094200. |77-86-1].
Див. статтю Трометамін.
Трис(гідроксимстил)амінометану розчин. 1094201.
Розчин трис(гідроксиметил)амінометану Р міс-
тить еквівалент 24.22 гС4НцМО3у 1000.0 мл.
Трисціаноетоксипропан. С12Н7М3О3. (М.м. 251.3).
1093900. 1,2.3-Трис(2-ціаноетокси)пропан.
В’язка рідина коричнево-жовтого кольору. Розчинний
у метанолі.
Використовують як нерухому фазу в газовій хромато-
графії.
$: близько 1.11.
В’язкість (2.2.9). Близько 172 мПа-с.
Трифенілметанол. С19Н16О. (М.м. 260.3). 1093700.
|76-84-6]. Трифенілкарбінол.
Безбарвні кристали. Практично не розчинний у воді,
легко розчинний у 96 % спирті.
Трифенілтетразолію хлорид. С19Н15СІК4. (М.м. 334.8).
1093800. [298-96-4]. 2,3.5-Трифеніл-2//-тетразолію
хлорид.
Містить не менше 98.0 % С,9Н |5СІГ\4.
Порошок блідо-жовтого або тьмяно-жовтого кольору
Розчинний у воді, ацетоні й 96 % спирті, практично не
розчинний в ефірі.
Температура плавлення: близько 240 °С, із розкла-
данням.
Кількісне визначення. 1.000 г розчиняють у суміші 5 мл
кислоти азотної розведеної Р і 45 мл води Р, додають
50.0 мл 0.1 М розчину срібла нітрату й нагрівають до
кипіння. Охолоджують, додають 3 мл дибутилфтала-
ту Р. енергійно струшують і титрують 0.1 М розчином
амонію тіоціанату, використовуючи як індикатор 2 мл
розчину заліза)III) амонію сульфату Р2.
1 мл 0.1 М розчину срібла нітрату відповідає 33.48 мг
С19НІ5С1М4.
Зберігають у захищеному від світла місці.
Трифенілтетразолію хлориду розчин. 1093801.
Розчин 5 г/л у 96 % спирті, вільному від альдегідів, Р.
Зберігають у захищеному від світла місці.
Трифтороцтова кислота. С7НЕ3О2. (М.м. 114.0).
1093200 [76-05-1].
Містить не менше 99 % С2НР3О2.
Рідина, змішується з ацетоном, 96 % спиртом та
ефіром.
</2о близько 1.53.
Температура кипіння: близько 72 °С.
Використовують кваліфікацію, придатну для секвен-
тації протеїнів.
Зберігають у повітронепроникному контейнері.
Трифтороцтовий ангідрид. С4Р6О3. (М.м. 210.0).
1093300. 1407-25-0].
Безбарвна рідина.
е/2о : близько 1.5.
1,1,1-Трихлоретан. С2Н3С13. (М.м. 133.4). 1092600.
[71-55-6]. Метилхлороформ.
Незаймиста рідина. Практично не розчинний у воді,
розчинний в ацетоні, ефірі й метанолі.
г/29 : близько 1.34.
: близько 1.438.
Температура кипіння: близько 74 °С.
Трихлоретилен. С2НС13. (М.м. 131.4). 1102100. [79-01-6].
Безбарвна рідина. Практично не розчинний у воді,
змішується з 96 % спиртом та ефіром.
г/2о : близько 1.46.
: близько 1.477.
Трихлортрифторетан. С2С13Р3. (М.м. 187.4). 1092700.
[76-13-1]. 1,1,2-Трихлор-1,2,2-трифторетан.
Безбарвна, летка рідина. Практично не розчинний у
воді, змішується з ацетоном та ефіром.
е/2о : близько 1.58.
Температурні межі перегонки (2.2.11). Від 47 °С до
48 °С; має переганятись не менше 98 %.
Трихлороцтова кислота. С2НС13О2. (М.м. 163.4).
1092500. [76-03-91.
Безбарвні кристали або кристалічна маса. Дуже легко
розпливається на повітрі, дуже легко розчинна у воді й
96 % спирті.
Зберігають у повітронепроникному контейнері.
Трихлороцтової кислоти розчин. 1092501.
40.0 г трихлороцтової кислоти Р розчиняють у
воді Р і доводять об’єм розвинутим самим розчин-
ником до 1000.0 мл. Концентрацію визначають ти-
труванням 0.1 М розчином натрію гідроксиду та,
якщо необхідно, доводять до концентрації
(40±1) г/л.
Тромбін бичачий. 1090200. [9002-04-4].
Ферментний препарат, одержаний із бичачої плазми,
який перетворює фібриноген на фібрин.
Порошок жовтувато-білого кольору.
Зберігають при температурі не вище 0 °С.
ІПЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
265
4.1.1. Реактиви
Тромбін людський. 1090100. [9002-04-41
Сухий тромбін людський. Ферментний препарат, який
перетворює фібриноген людський на фібрин. Одержу-
ють із людської рідкої плазми шляхом осадження
підхожими солями та органічними розчинниками в
умовах контролю рН, іонної сили й температури.
Порошок жовтувато-білого кольору. Легко розчинний
у розчині 9 г/л натрію хлориду з утворенням каламут-
ного блідо-жовтого забарвлення.
Зберігають у скляних контейнерах, закупорених в ат-
мосфері азоту, при температурі не вище 25 °С.
Тромбіну людського розчин. 1090101.
Тромбінлюдський Р відновлюють, як зазначено ви-
робником, і розводять буферним розчином
трис(гідроксиметил)амінометану-натрію хлори-
ду рН 7.4 Рао концентрації 5 МО/мл.
Тромбопластину реактив. 1090300.
1 5 г мозку бичачого, висушеного ацетоном, Р, екстра-
гують 60 мл води Р при температурі 50 °С протягом
15 хв, центрифугують при 1500 об/хв протягом 2 хв і
декантують надосадову рідину. Екстракт зберігає ак-
тивність протягом кількох діб при зберіганні в холо-
дильнику. Може містити 3 г/л крезолу Р як ан-
тимікробного консерванта.
Туйон. С|0Н16О. (М.м. 152.2). 1116500. [546-80-5].
4- Метил-1-(1 -метил етил )біцикло[3.1.0]гексан-3-он.
Безбарвна або майже безбарвна рідина. Практично не
розчинний у воді, розчинний у 96 % спирті та бага-
тьох інших органічних розчинниках.
</20 близько 0.925.
: близько 1.455.
[а|р : близько -15°.
Температура кипіння: близько 86 °С.
ТШХ пластинка із шаром силікагелю. 1116700.
Підкладка зі скла, металу або пластика, покрита ша-
ром силікагелю з підхожею товщиною і розміром час-
ток (звичайно від 2 мкм до 10 мкм для пластин із
дрібним розміром часток (Високоефективна тонко-
шарова хроматографія (ВЕТШХ) і від 5 мкм до 40 мкм
для звичайних ТШХ пластин). Якщо необхідно,
розмір часток зазначають після назви сорбенту у ви-
пробуваннях, де він використовується.
Сорбент може містити зв’язуючу органічну речовину.
Хроматографічна розділювальна здатність. На плас-
тинку наносять необхідний об’єм розчину для визна-
чення придатності ТШХ пластинок Р (10 мкл для зви-
чайної пластинки й від 1 мкл до 2 мкл для пластинки
із дрібним розміром часток). Хроматографують у сис-
темі розчинників метанол Р-толуол Р (20:80). Коли
фронт розчинників пройде дві третини довжини плас-
тинки, вона вважається придатною, якщо на ній вид-
но чотири чітко розділені плями:
пляма бромкрезолового зеленого з /?^не більше 0.15 І
пляма метилового оранжевого з Яу-у межах від 0.1 до0.25,1
пляма метилового червоного з у межах від 0.35 до
0.55,
пляма Судану червоного С з Я^у межах від 0.75 до 0.98 І
Розчин для визначення придатності ТШХ пласти-
нок. 1116600.
Змішують по 1.0 мл розчину 0.5 г/л Судану червоно-1
го С Р у толуолі Р, свіжоприготованого розчину
0.5 г/л метилового оранжевого Р в етанолі Р, роз-
чину 0.5 г/л бромкрезолового зеленого в ацетоні Р,
розчину 0.25 г/л метилового червоного Р в аце-
тоні Р і доводять об’єм одержаного розчину аце-
тоном Рао 10.0 мл.
ТШХ пластинка із шаром силікагелю г254. 1116800.
Має задовольняти вимоги для ТШХ пластинки із ша-
ром силікагелю Р із такими змінами
Містить флуоресцентний індикатор з інтенсивністю
поглинання за довжини хвилі 254 нм.
Гасіння флуоресценції. На пластинку наносять у п’ять
крапок послідовно зростаючі об’єми від 1 мкл до
10 мкл для звичайної ТШХ пластинки й від 0.2 мкл до
2 мкл для ВЕТШХ пластинки розчину 1 г/л кислоти
бензойної Ру суміші розчинників етанол Р - циклогек-
сан Р (15:85). Хроматографують у тій самій суміші роз-
чинників. Коли фронт розчинників пройде половину
довжини пластинки, її виймають із камери й сушать до
випаровування розчинників. Пластинку переглядають
в УФ-світлі за довжини хвилі 254 нм. На звичайних
ТШХ пластинках кислота бензойна має виявлятись у
вигляді темних плям на флуоресціюючому фоні при-
близно на середині хроматограми для нанесених
кількостей 2 мкг і більше. На ВЕТШХ пластинках кис-
лота бензойна має виявлятись у вигляді темних плям
на флуоресціюючому фоні приблизно на середині хро-
матограми для нанесених кількостей 0.2 мкг і більше.
ТШХ пластинка із шаром силікагелю с. 1116900.
Має задовольняти вимоги для ТШХ пластинки із ша-
ром силікагелю Р із таким зміненням.
Містить кальцію сульфат гемігідрат (гіпс) як зв’язую-
чу речовину.
ТШХ пластинка із шаром силікагелю сг254. 1117000.
Має задовольняти вимоги для ТШХ пластинки із ша-
ром силікагелю Р із такими змінами.
Містить кальцію сульфат гемігідрат (гіпс) як зв’язую-
чу речовину.
Гасіння флуоресценції. Має задовольняти вимоги для
ТШХ пластинки із шаром силікагелю Р254 Р.
ТШХ пластинка із шаром силікагелю силанізованого.
1117100.
Підкладка зі скла, металу або пластика, покрита ша-
ром силікагелю силанізованого з підхожою товщиною
266
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
й розміром часток (звичайно від 2 мкм до 10 мкм для
пластин із дрібним розміром часток. Високоефектив-
на тонкошарова хроматографія (ВЕТШХ) і від 5 мкм
до 40 мкм для звичайних ТШХ пластин). Якщо не-
обхідно, розмір часток зазначають після назви сорбен-
туу випробуваннях, де він використовується.
Сорбент може містити зв’язуючу органічну речовину.
Хроматографічна розділювальна здатність По 0.1 г
жтиллаурату Р, метилміристату Р, метилпальміта-
ту Р і метилстеарату Р поміщають у конічну колбу
місткістю 250 мл, додають 40 мл розчину калію гідрок-
сиду спиртового Р\ нагрівають зі зворотним холодиль-
ником на водяній бані протягом 1 год Охолоджують,
розчин поміщають у ділильну лійку з допомогою
100 мл води Р. підкислюють кислотою хлористоводне-
вою розведеною Рао рН від 2 до 3 і струшують з трьома
порціями метиленхлориду Р, по 10 мл кожна. Об’єд-
нані метиленхлориди! витяжки сушать над натрію
сульфатом безводним Р, фільтрують і випарюють насу-
хо на водяній бані. Сухий залишок розчиняють у 50 мл
метиленхлориду Р (випробовуваний розчин). Визна-
чення проводять методом ТШХ (2.2.27), використову-
ючи ТШХ пластинку із шаром силікагелю силанізова-
ного Р. На пластинку наносять у три крапки не-
обхідний об’єм випробовуваного розчину (близько
10 мкл для звичайної ТШХ пластинки й від 1 мкл до
2 мкл для ВЕТШХ пластинки з дрібним розміром час-
ток). Хроматографують у системі розчинників кисло-
та оцтова льодяна Р-вода Р-діоксан Р( 10:25:65). Коли
фронт розчинників пройде дві третини довжини плас-
тинки, її виймають із камери й сушать при температурі
120 °С протягом 30 хв. Пластинку охолоджують, об-
прискують розчином 35 г/л кислоти фосфорномолібде-
нової Р у 2-пропанолі Р і нагрівають при температурі
150 °С до появи плям. Потім пластинку обробляють
парами аміаку до одержання фону білого кольору.
Пластинка вважається придатною, якщо на ній видно
чотири чітко розділені плями.
ТШХ пластинка із шаром силікагелю силанізованого Е2м-
1117200.
Має задовольняти вимоги для ТШХ пластинки із ша-
ром силікагелю силанізованого Різ таким зміненням.
Містить флуоресцентний індикатор з інтенсивністю
поглинання за довжини хвилі 254 нм.
Уридан. С9НІ2М2О6. (М.м. 244.2). 1095100. [58-96-8].
І -]3-о- Рибофуранозилурацил.
Кристалічний порошок білого або майже білого ко-
льору. Розчинний у воді.
Температура плавлення: близько 165 °С.
Фактора V згортання крові розчин. 1021400.
Розчин фактора Vзгортання крові може бути пригото-
ваний таким чином (або будь-яким іншим способом,
який виключає фактор VIII).
Готують реактив фактора V із свіжооксалатованої плаз-
ми бичачої фракціонуванням при температурі 4 °С з
насиченим розчином амонію сульфату Р, приготова-
ним при температурі 4 °С. Відокремлюють фракцію,
яка осаджується в інтервалі насичення від 38 % до 50 %
і містить фактор V, без істотного забруднення фактором
VIII. Амонію сульфат видаляють діалізом і розводять
розчином 9 г/л натрію хлориду Рдо одержання розчину,
який містить від 10 % до 20 % кількості фактора V, на-
явного у звичайній свіжій плазмі крові людини.
Визначення вмісту фактора V. Готують два розведення
препарату фактора V в імідазольному буферному роз-
чині рН 7.3 Р, які містять один об’єм препарату
відповідно у 10 й 20 об’ємах буферного розчину. Кож-
ний розчин випробовують таким чином: змішують
О. 1 мл субстрату плазми без фактора V Р, 0.1 мл ви-
пробовуваного розчину, 0.1 мл реактиву тромбоплас-
тину Р і 0.1 мл розчину 3.5 г/л кальцію хлориду Р і
вимірюють час згортання крові, тобто інтервал між мо-
ментом додавання розчину кальцію хлориду й першою
ознакою утворення фібрину, яку можна спостерігати
візуально або з допомогою відповідних приладів.
Таким самим чином визначають час згортання крові
(два паралельних визначення) чотирьох розчинів зви-
чайної плазми крові людини в імідазольному буферно-
му розчинірН 7.3 Р, які містять відповідно 1 об’єм у де-
сяти (еквівалент 100 % фактора V), 1 об’єм у 50 (20 %),
1 об’єм у 100 (10 %) та 1 об’єм у 1000 (1 %). Викорис-
товуючи двобічний логарифмічний папір, наносять
середнє значення часу згортання крові для кожного
розчину плазми людини від еквівалента відсоткового
вмісту фактора V, у відсотках, і з допомогою інтерпо-
ляції визначають вміст фактора V, у відсотках, для двох
розведених розчинів фактора V. Середнє значення
двох результатів дає вміст фактора V, у відсотках, у ви-
пробовуваному розчині.
Зберігають розчин у замороженому стані при темпера-
турі не вище -20 °С
Фактор коагуляції Ха бичачий. 1037300. |9002-05-5].
Напівочищений препарат одержують із рідкої бичачої
плазми; його можна одержати також за допомогою ак-
тивації зимогену фактора X з допомогою підхожого ак-
тиватора, такого як отрута гадюки Руссела.
Зберігають ліофілізований препарат при температурі
-20 °С, заморожений розчин зберігають при темпера-
турі не вище -20 °С.
Фактора Ха бичачого розчин. 1037301.
Відновлюють відповідно до зазначень виробника
й розводять буферним розчином трис(гідроксиме-
тил)амінометану-натрію хлориду рН 7.4 Р.
Зміна оптичної густини не має перевищувати 0.15-
0.20 за хв. Вимірювання проводять за довжини
хвилі 405 нм (2.2.25), використовуючи як компен-
саційну рідину буферний розчин трис(гідроксиме-
тил)амінометану- натрію хлориду рН 7.4 Р.
Феназон. 1063400. [60-80-0]. Див. статтю Феназон.
Фенантрен. СІ4Н|0. (М.м. 178.2). 1063200. [85-01-8].
Кристали білого кольору. Практично не розчинний у
воді, легко розчинний в ефірі, помірно розчинний у
96 % спирті.
Температура плавлення: близько 100 °С.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
267
4.1.1. Реактиви
Фенантроліну гідрохлорид. С12Н9С^2,Н2О. (М.м. 234.7).
1063300. [3829-86-5]. 1,10-Фенантроліну гідрохлорид
моногідрат.
Порошок білого або майже білого кольору. Легко роз-
чинний у воді, розчинний у 96 % спирті.
Температура плавлення: близько 215 °С, із розкла-
данням.
Фенілаланін. 1064100. [63-91-2]. Див. статтю Феніл-
аланін.
Фенілгідразину гідрохлорид. С6Н9С1М2. (М.м. 144.6).
1064500. [59-88-1].
Кристалічний порошок білого або майже білого
кольору, під дією повітря набуває коричневого забарв-
лення. Розчинний у воді й 96 % спирті
Температура плавлення: близько 245 °С, із розкла-
данням.
Зберігають у захищеному від світла місці.
Фенілгідразину гідрохлориду розчин. 1064501.
0.9 г фенілгідразину гідрохлориду Р розчиняють у
50 мл води Р, знебарвлюють вугіллям активова-
ним Р і фільтрують. До фильтрату додають ЗО мл
кислоти хлористоводневої Р і доводять об’єм роз-
чину водою Р до 250 мл.
Фенілгідразину розчин у кислоті сірчаній. 1064502.
65 мг фенілгідразину гідрохлориду Р, попередньо
перекристалізованого із спирту (85 %, об/об) Р,
розчиняють у суміші розчинників вода Р - кислота
сірчана Р (80:170) і доводять тією самою сумішшю
розчинників до об’єму 100 мл.
Готують безпосередньо перед використанням.
а-Фенілгліцин. С8Н9МО2. (М.м. 151.2). 1064300.
[2835-06-5]. (А5)-2-Аміно-2-фенілоцтова кислота.
л-Фенілендіаміну дигідрохлорид. С6Н |0С12М2.
(М.м. 181.1). 1064200. [615-28-1]. 1,4-Діамінобензолу
дигідрохлорид.
Кристалічний порошок або кристали білого кольору
або злегка забарвлені. На повітрі червоніє. Легко роз-
чинний у воді, мало розчинний у 96 % спирті та ефірі.
Фенілізотіоціанат. С7Н51М$. (М.м. 135 2). 1121500.
[іда-72-0].
Рідина. Не розчинний у воді, розчинний у 96 % спирті.
с/29 : близько 1.13.
Ир : близько 1.65.
Температура кипіння: близько 221 °С
Температура плавлення: близько -21 °С.
Феноксибензаміну гідрохлорид. С18Н23С12ЬІО.
(М.м. 340.3). /063900. А-(2-Хлоретил)-А-(1-метил-2-
феноксіетил)бензиламіну гідрохлорид.
Містить від 97.0 % до 103.0 % С|8Н23С12МО, у перер,
хунку на суху речовину.
Кристалічний порошок білого або майже білого к
льору. Помірно розчинний у воді, легко розчинник,
96 % спирті.
Температура плавлення: близько 138 °С.
Втрата в масі при висушуванні (2.2.32). Не більш,
0.5 %. Сушать над фосфору(Р) оксидом Р при тисю
який не перевищує 670 Па, протягом 24 год.
Кількісне визначення. 0.500 г розчиняють у 50.0 млхи-
роформу, вільного від етанолу, Р і екстрагують трьош
порціями 0.01Мрозчину кислоти хлористоводневої,»
20 мл кожна. Кислотний шар відкидають, а хлоро-
формний шар фільтрують крізь вату. 5.0 мл одержано-1
го фільтрату доводять хлороформом, вільним від етано- і
лу, Р до об’єму 500.0 мл. Вимірюють оптичну густин,
одержаного розчину в закритій кюветі в максимуміл
довжини хвилі 272 нм.
Обчислюють вміст С|8Н23С12МО. беручи питомий по-
казник поглинання рівним 56.3.
Зберігають у захищеному від світла місці.
Феноксіоцтова кислота. С8Н8О3. (М.м. 152.1). 1063800 І
[122-59-8]. 2-Феноксіетанова кислота.
Кристали майже білого кольору. Помірно розчинна у
воді, легко розчинна в 96 % спирті, ефірі й кислоті оц-
товій льодяній.
Температура плавлення: близько 98 °С.
Хроматографія. Визначення проводять, як зазначено
в статті Феноксиметилпеніцилін', на одержаній хрома-
тограмі має виявлятись лише одна основна пляма.
Фсноксіетанол. С8Н|(ІО2. (М.м. 138.2). 1064000.
[122-99-6]. 2-Феноксіетанол.
Прозора, безбарвна масляниста рідина. Малорозчин-
ний у воді, легко розчинний у 96 % спирті та ефірі.
с/29 : близько 1.11.
: близько 1.537.
Температура тверднення (2.2.18). Не нижче 12 °С.
Фенол. 1063500. [108-95-2]. Див. статтю Фенол.
Феноловий червоний. 1063600. [143-74-8]. Див. статтю
Фенолсульфонфталеїн.
Фенолового червоного розчин. 1063601.
0.1 г фенолового червоного Р розчиняють у суміші
2.82 мл 0.1 М розчину натрію гідроксиду і 20 мл
96 % спирту Р, доводять об’єм розчину водою Рао
100 мл.
Випробування на чутливість. До 100 мл води,
вільної від вуглецю діоксиду, Р, додають 0.1 мл роз-
чину фенолового червоного; з’являється жовте за-
барвлення, яке має перейти у червонувато-фіоле-
тове при додаванні не більше 0.1 мл 0.02 Мрозчи-
ну натрію гідроксиду.
268
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Зміна забарвлення. Від жовтого до червонувато-
фіолетового в інтервалі рН 6.8-8.4.
Фенолового червоного розчин Р2. 1063603.
Розчин І. 33 мг фенолового червоного Р розчиняють
у 1.5 мл розчину натрію гідроксиду розведеного Р і
доводять об’єм розчину водою Т до 100 мл.
Розчин II. 25 мг амонію сульфату Р розчиняють у
235 мл води Р, додають 105 мл розчину натрію
гідроксиду розведеного Р і 135 мл кислоти оцтової
розведеної Р.
Розчин 11 змішують із 25 мл розчину І. Якщо не-
обхідно, доводять рН розчину до 4.7.
Фенолового червоного розчин РЗ. 1063604.
Розчин /. 33 мг фенолового червоного Р розчиняють
у 1.5 мл розчину натрію гідроксиду розведеного Рі
доводять об’єм розчину водою Рро 50 мл.
Розчин II. 50 мг амонію сульфату Р розчиняють у
235 мл води Р; додають 105 мл розчину натрію
гідроксиду розведеного Р і 135 мл кислоти оцтової
розведеної Р.
Розчин II змішують із 25 мл розчину 1. Якщо не-
обхідно, доводять рН розчину до 4.7.
Фенолфталеїн. С20Н14О4. (Мм. 318.3). 1063700. [77-09-8].
3,3-Біс(4-гідроксифеніл)-3//-ізобензофуран-1 -он.
Порошок від білого до жовтувато-білого кольору
Практично не розчинний у воді, розчинний у
96 % спирті.
Фенолфталеїну розчин. 1063702.
0.1 г фенолфталеїну Р розчиняють у 80 мл
96 % спирту Р і доводять об’єм розчину водою Р
до 100 мл.
Випробування на чутливість. До 100 мл води, віль-
ної від вуглецю діоксиду, Р додають 0.1 мл розчину
фенолфталеїну; при додаванні не більше 0.2 мл
0.02 М розчину натрію гідроксиду забарвлення
розчину має змінитися від безбарвного до роже-
вого.
Зміна забарвлення. Від безбарвного до яскраво-ро-
жевого в інтервалі рН 8.2-10.0.
Фенолфталеїну розчин РІ. 1063703.
Розчин 10 г/л у 96 % спирті Р
Фенхон. С10Н16О. (Мм. 152.2). 1037600. [7787-20-4].
1,3,3-Триметилбіцикло[2.2.1]гептан-2-он.
Масляниста рідина. Змішується з 96 % спиртом та
ефіром, практично не розчинний у воді.
Яр : близько 1.46.
Температура кипіння 15мм: близько 66 °С.
Фенхон, використовуваний у газовій хроматографії,
має витримувати таке випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28) за умов, зазначених у статті Фен-
хель гіркий, використовуючи фенхон як випробовува-
ний розчин.
Площа основного піка не має бути менше 98 0 % за-
гальної площі всіх піків.
Фероїн. 1038100. [14634-91-4].
0.7 г заліза(Н) сульфату Р і 1.76 г фенантроліну гідро-
хлориду Р розчиняють у 70 мл води Р і доводять об’єм
розчину тим самим розчинником до 100 мл.
Випробування на чутливість. До 50 мл кислоти сірчаної
розведеної Р додають 0.15 мл розчину осмію(УІП) окси-
ду Рі 0.1 мл фероїну. Після додавання 0.1 мл 0.1 Мроз-
чину амонію церію нітрату забарвлення розчину має
змінитися від червоного до блакитного.
Фероцифен. С26Н|6РсМ6. (М.м. 468.3). 1038000.
[ 14768-11-7]. Диціанобіс(1,10-фенантролін)залізо(ІІ).
Кристалічний порошок фіолетово-бронзового кольо-
ру. Практично не розчинний у воді й 96 % спирті.
Зберігають у сухому захищеному від світла місці.
Фібрин синій. 1101400.
1.5 г фібрину змішують із ЗО мл розчину 5 г/л індиго-
карміну Р в 1 % (об/об) розчині кислоти хлористовод-
невої розведеної Р, суміш нагрівають до температури
80 °С і витримують при такій температурі близько 30 хв
при перемішуванні, охолоджують і фільтрують. Осад
ретельно промивають, ресуспендуючи в 1 % (об/об)
розчин кислоти хлористоводневої розведеної Р і пе-
ремішуючи близько 30 хв, фільтрують. Осад промива-
ють тричі, сушать при температурі 50 °С і подрібнюють.
Фібрин конго червоний. 1038400.
Промитий фібрин нарізають на маленькі шматочки і
залишають на ніч у розчині 20 г/л конго червоного Ру
спирті (90 %, об/об) Р і фільтрують; фібрин промива-
ють водою Р і зберігають під ефіром Р.
Фібриноген. 1038500. [9001-32-5]. Див. статтю Фібри-
ноген людський ліофілізований.
Флороглюцин. С6Н6О3,2Н2О. (М.м. 162.1). 1064600.
[6099-90-7]. Бензол-1,3,5-триол.
Кристали білого або жовтуватого кольору. Мало роз-
чинний у воді, розчинний у 96 % спирті.
Температура плавлення: близько 223 °С (метод мит-
тєвого плавлення).
Флороглюцину розчин. 1064601.
До 1 мл розчину 100 г/л флороглюцину Р у 96%
спирті Р додають 9 мл кислоти хлористоводневої Р.
Зберігають у захищеному від світла місці.
Флуорантен. С16Н10. (М.м. 202.3). 1038600. [206-44-0].
1,2-( 1,8-Нафтилен)бензол. 1,2-Бензаценафтен.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
269
4.1.1. Реактиви
Кристали від жовтого до жовтувато-коричневого кольору.
Температура кипіння: близько 384 °С.
Температура плавлення: від 105 °С до 110 °С.
Флуоресцеїн. С2оН|205. (М.м. 332.3). 1106300.
[2321-07-5]. З',6'-Дигідроксиспіро[ізобензофуран-
1(3//),9'-[9//]ксантен]-3-он.
Порошок оранжево-червоного кольору. Практично не
розчинний у воді, розчинний у теплому 96 % спирті,
практично не розчинний в ефірі, розчинний у розчи-
нах гідроксидів лужних металів У розчині флуорес-
цеїн виявляє зелену флуоресценцію.
Температура плавлення: близько 315 °С.
Флуоресцсїн-спряжена сироватка проти сказу. 1038700.
Імуноглобулінова фракція з високим рівнем антитіл
проти сказу, приготована із сироватки підхожих тва-
рин, імунізованих інактивованим вірусом сказу; іму-
ноглобулін спряжений із флуореспеїн-ізотіоціана-
том.
Флуфенамінова кислота. СІ4НІ0Р3МО2. (М.м. 281.2).
1106200. [530-78-9]. 2-[[3-(Трифторметил)феніл|-
аміно]бензойна кислота.
Кристалічний порошок або голчасті кристали блідо-
жовтого кольору. Практично не розчинна у воді, легко
розчинна в 96 % спирті.
Температура плавлення: від 132 °С до 135 °С.
Фолієва кислота. 1039000. [75708-92-8]. Див. статтю
Кислота фолієва.
Формальдегід. 1039100. [50-00-0]. Див. Розчин фор-
мальдегіду Р.
Формальдегіду розчин. 1039101 Див. статтю Розчин
формальдегіду (35 %).
Формальдегіду розчин у сірчаній кислоті. 1086805.
2 мл розчину формальдегіду Р змішують зі 100 мл
кислоти сірчаної Р.
Формамід. СН3МО. (М.м. 45.0). 1039200 [75-12-7].
Прозора, безбарвна, масляниста, гігроскопічна ріди-
на. Змішується з водою й 96 % спиртом. Гідролі-
зується водою.
Температура кипіння: близько 103 °С. Визначення
проводять під тиском 2 кПа.
Зберігають у повітронепроникному контейнері.
Формамід оброблений. 1039201.
1.0 г кислоти сульфамінової Р диспергують у 20.0 мл
формаміду Р, який містить 5 % (об/об) води Р.
Формамід РІ. 1039202.
Має задовольняти вимоги для формаміду Р і ви-
тримувати таке додаткове випробування.
Вода (2.5.12). Не більше 0.1 %, визначення прово-1
дя ть з рівним об’ємом метанолу безводного Р
Фосфоліпіди. 1064800.
Певну кількість мозку людського або бичачого, добре
відокремленого від кровоносних судин, промивають! І
розріджують у підхожому пристрої. Від 1000 гдо 1300г І
розрідженої речовини зважують, вимірюють об’єм І
(V мл), потім екстрагують трьома порціями ацетону Р. ]
по 4У мл кожна, й фільтрують при зниженому тиску;І
одержаний залишок сушать при температурі 37 °С ]
протягом 18 год; потім залишок екстрагують сумішшю
розчинників петролейний ефір Р2 - петролейний
ефір РІ (2:3), двома порціями, кожна по 2\ мл.
фільтруючи кожен екстракт крізь фільтрувальний
папір, попередньо зволожений тією самою сумішшю
розчинників. Об’єднані витяжки випарюють насухо
при температурі 45 °С піл тиском не більше 670 Па. За-
лишок розчиняють у 0.2У мл ефіру Р і витримують при
температурі 4 °С до утворення осаду. Прозору надоса-
дову рідину центрифугують, випарюють під низьким
тиском до об’єму 100 мл на кілограм розрідженої речо-
вини і зважують. Витримують при температурі 4 °С до
утворення осаду (від 12 год до 24 год) і центрифугують.
До прозорої надосадової рідини додають ацетон РІ
кількості, яка у п’ять разів перевищує її об’єм, центри-
фугують і відкидають надосадову рідину. Осад сушать.
Зберігають в ексикаторі під вакуумом, захищеному від
світла місці.
Фосфору(У) оксид. Р2О5. (М.м. 141 9) 1032900.
[1314-56-3]. Дифосфору пентоксид. Фосфорний
ангідрид.
Аморфний порошок білого кольору, який розпливається
на повітрі. З водою утворює гідрати з виділенням тепла.
Зберігають у повітронепроникному контейнері.
Фосфорна кислота. 1065100. |7664-38-2]. Див. статтю
Кислота фосфорна концентрована
Фосфорна кислота розведена. 1065101. Див. статтю
Кислота фосфорна розведена.
Фосфорновольфрамової кислоти розчин. 1065200.
До 10 г натрію вольфрамату Р додають 8 мл кислоти
фосфорної Р і 75 мл води Р, нагрівають зі зворотним хо-
лодильником протягом 3 год, охолоджують і доводять
об’єм розчину водою Рдо 100 мл.
Фосфорномолібденова кислота. 12МоО3,Н3РО4,хН20.
1064900. [5 \429-74-4].
Дрібні кристали оранжево-жовтого кольору. Легко
розчинна у воді, розчинна в 96 % спирті та ефірі.
Фосфорномолібденової кислоти розчин. 1064901.
4 г фосфорномолібденової кислоти Р розчиняють у
воді Р, доводять об’єм розчину тим самим розчин
ником до 40 мл Обережно при охолодженні дода
ють 60 мл кислоти сірчаної Р.
Готують безпосередньо перед використанням.
270
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Фосфорномолібденово-вольфрамовий реактив. 1065000.
100 г натрію вольфрамату Р і 25 г натрію молібдату Р
розчиняють у 700 мл води Р, додають 100 мл кислоти
хлористоводневої Р і 50 мл кислоти фосфорної Р.
Суміш нагрівають у скляній колбі зі зворотним холо-
іиіьником протягом 10 год, додають 150 г літію суль-
фату Р, 50 мл води Р і кілька краплин брому Р.
Кип’ятять до видалення надлишку брому (15 хв), охо-
лоджують, доводять об’єм розчину водою Рао 1000 мл
і фільтрують. Реактив повинен мати жовте забарвлен-
ня. Реактив не придатний для використання, якшо
набуває зеленого відтінку, але може бути регенерова-
ний шляхом кип’ятіння з кількома краплями брому Р.
Надлишок брому обов’язково видаляють кип’я-
тінням.
Зберігають при температурі від 2 °С до 8 °С.
Фосфорномолібденово-вольфрамовий реактив роз-
ведений. 1065001.
Змішують фосфорномолібденово-вольфрамовии ре-
актив Р із водою Р (1:2).
Фруктоза. 1106400. [57-48-7]. Див. статтю Фруктоза.
Фталазин. С8Н6М2. (М.м. 130.1). 1065400. [253-52-1].
Кристали блідо-жовтого кольору. Легко розчинний у
воді, розчинний в етанолі, етилацетаті й метанолі,
помірно розчинний в ефірі.
Температура плавлення: від 89 °С до 92 °С.
Фталева кислота. С8Н6О4. (М.м. 166.1). 1065600.
[88-99-3]. Бензол-1,2-дикарбонова кислота.
Кристалічний порошок білого кольору. Розчинна в га-
рячій воді й 96 % спирті.
Фталевий альдегід. С8Н6О2. (М.м. 134.1). 1065300.
|643-79-8]. Бензол-1,2-дикарбоксальдегід.
Кристалічний порошок жовтого кольору.
Температура плавлення: близько 55 °С.
Зберігають у захищеному від світла місці, без доступу
повітря.
Фталевого альдегіду реактив. 1065301.
2А~1 г кислоти борної Р розчиняють у 75 мл води Р,
доводять рН до 10.4 розчином 450 г/л калію гідро-
ксиду Р і доводять водою Рдо об’єму 100 мл.
1.0 г фталевого альдегіду Р розчиняють у 5 мл мета-
нолу Р, додають 95 мл приготованого розчину кис-
лоти борної і 2 мл кислоти тіогліколевої Р і дово-
дять рН до 10.4 розчином 450 г/л калію гідроксиду Р.
Зберігають у захищеному від світла місці.
Термін придатності 3 доби.
Фталевий ангідрид. С8Н4О3. (М.м. 148.1). 1065700.
[85-44-9]. Ізобензофуран-1,3-діон.
Містить не менше 99.9 % С8Н4О3.
Пластівці білого кольору.
Температура плавлення: від 130 °С до 132 °С.
Кількісне визначення. 2.000 г розчиняють у 100 мл во-
ди Р, кип’ятять зі зворотним холодильником протягом
30 хв, охолоджують і титрують 1 М розчином натрію
гідроксиду, використовуючи як індикатор розчин фе-
нолфталеїну Р.
1 мл 1 Мрозчину натрію гідроксиду відповідає 74.05 мг
С8Н4О3.
Фталевого ангідриду розчин. 1065701.
42 г фталевого ангідриду Р розчиняють у 300 мл
піридину безводного Р і витримують протягом
16 год.
Зберігають у захищеному від світла місці.
Термін придатності 7 діб.
Фталеїновий пурпуровий. С32Н32М2О|2,хН2О.
(М.м. 637, для безводного). 1065500. ]2411-89-4]. Ме-
тилфталеїн. 2,2',2",2'"-[о-Крезолфталеїн-3’,3"-біс(ме-
тиленнітрило)]тетраоцтова кислота. (1,3-Дигідро-3-
оксо-ізобензофуран-1-іліден)біс[(6-гідроксі-5-метил-
3,1-фенілен)біс(метиленіміно)діоптова кислота].
Порошок від жовтувато-білого до коричнюватого ко-
льору. Практично не розчинний у воді, розчинний у
96 % спирті. Реактив надходить у продаж у вигляді
натрієвої солі: порошок від жовтувато-білого до ко-
ричнюватого кольору; розчинний у воді, практично не
розчинний у 96 % спирті.
Випробування на чутливість. 10 мг розчиняють в 1 мл
розчину аміаку концентрованого Р і доводять об’єм
розчину водою Рао 100 мл. До 5 мл одержаного розчи-
ну додають 95 мл води Р, 4 мл розчину аміаку концент-
рованого Р, 50 мл 96 % спирту Р і 0.1 мл 0.1 Мрозчину
барію хлориду, з’являється синьо-фіолетове забарв-
лення розчину, яке має зникнути після додавання
0.15 мл 0.1 М розчину натрію едетату.
2-Фтор-2-деокси-В-глюкоза. С6НцЕО5. (М.м. 182.2).
1113900. [86783-82-6].
Кристалічний порошок.
Температура плавлення: від 174 °С до 176 °С.
1-Фтор-2-нітро-4-(трифторметил)бензол. С7Н3Е4МО2.
(М.м. 209.1). 1038900. [367-86-2].
Температура плавлення: близько 197 °С.
Фтординітробензол. С6Н3ЕМ2О4. (М.м. 186.1). 1038800.
[70-34-8]. 1-Фтор-2,4-динїтробензол.
Кристали блідо-жовтого кольору. Розчинний в ефірі й
пропіленгліколі.
Температура плавлення: близько 29 °С.
Фтористоводнева кислота. НЕ (М.м. 20.01). 1043600.
[7664-39-3].
Містить не менше 40.0 % (м/м) НЕ.
Прозора, безбарвна рідина.
Залишок після прожарювання. Не більше 0.05 % (м/м).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
271
4.1.1. Реактиви
Кислоту фтористоводневу випарюють у платиновому
тиглі, залишок обережно прожарюють до постійної
маси.
Кількісне визначення. В точно зважену колбу зі скля-
ною притертою пробкою, яка містить 50.0 мл 7 Мроз-
чину натрію гідроксиду, поміщають 2 г кислоти фтори-
стоводневої та зважують. Титрують 0.5 М розчином
кислоти сірчаної, використовуючи як індикатор 0.5 мл
розчину фенолфталеїну Р.
1 мл І Мрозчину натрію гідроксиду відповідає 20.01 мг
НЕ.
Зберігають у поліетиленовому контейнері.
Фукоза. С6Н|2О5. (М.м. 164.2). 1039500. [6696-41-9].
6-Деокси-Е-галактоза.
Порошок білого кольору. Розчинна у воді й 96 %
спирті.
[а] р : близько -76°. Визначення проводять у розчині
90 г/л через 24 год після приготування.
Температура плавлення: близько 140 °С.
Фуксин основний. 1039400. [632-99-5]. Суміш ро-
заніліну гідрохлориду (С2(ІН2()С1Гу3; М.м. 337.9; кольо-
ровий індекс № 42510; показник Шульца № 780) і па-
ро-розаніліну гідрохлориду (С19Н18С1Кз; М.м. 323.8;
кольоровий індекс № 42500; показник Шульца
№ 779).
При необхідності очищують таким чином: 1 г фуксину
основного розчиняють у 250 мл кислоти хлористовод-
невої розведеної Р, витримують протягом 2 год при
кімнатній температурі, фільтрують, одержаний
фільтрат нейтралізують розчином натрію гідроксиду
розведеним Рі додають його надлишок від 1 мл до 2 мл.
Фільтрують крізь скляний фільтр (40), осад промива-
ють водою Р, розчиняють у 70 мл метанолу Р, поперед-
ньо нагрітого до кипіння, й додають 300 мл води Р при
температурі 80 °С. Охолоджують і фільтрують; криста-
ли сушать у вакуумі.
Кристали із зеленувато-бронзовим блиском. Розчин-
ний у воді й 96 % спирті.
Зберігають у захищеному від світла місці.
Фуксину знебарвлений розчин. 1039401.
0.1 г фуксину основного Р розчиняють у 60 мл во-
ди Р, додають розчин, який містить 1 г натрію
сульфіту безводного Р або 2 г натрію сульфіту Р у
10 мл води Р. Повільно, при постійному пере-
мішуванні додають 2 мл кислоти хлористоводне-
вої Р, доводять об’єм розчину водою Рдо 100 мл.
Витримують у захищеному від світла місці не мен-
ше 12 год, знебарвлюють вугіллям активованим Рі
фільтрують. Якщо розчин мутнішає, його фільтру-
ють перед використанням. Якщо при витриму-
ванні розчину з’являється фіолетове забарвлення,
його знову знебарвлюють вугіллям активованим Р.
Випробування на чутливість. До 1.0 мл додають
1.0 мл води Р і 0.1 мл 96 % спирту, вільного від аль-
дегідів, Р. Додають 0.2 мл розчину, який містить
0.1 г/л формальдегіду (СН2О, М.м. 30.0). Протя- І
гом 5 хв має з’явитися світло-рожеве забарвлення І
розчину.
Зберігають у захищеному від світла місці.
Фуксину знебарвлений розчин РІ. 1039402.
До 1 г фуксину основного Р додають 100 мл води Р, І
нагрівають до температури 50 °С і охолоджують
періодично перемішуючи. Витримують протягом І
48 год, перемішують і фільтрують. До 4 мл фільтра-1
ту додають 6 мл кислоти хлористоводневої Р, пе- І
ремішують і доводять об’єм розчину водою Рао
100 мл.
Розчин використовують через 1 год після приготу-1
вання.
Фурфурол. С5Н4О2. (М.м. 96.1). 1039600. [98-01-1].
2-Фуральдегід. 2-Фуранкарбальдегід.
Прозора, масляниста рідина, безбарвна або коричню-
вато-жовтого кольору. Змішується з 11 частинами во-
ди, змішується з 96 % спиртом і ефіром.
б/2о : від 1.155 до 1.161.
Температурні межі перегонки (2.2.11). Від 159 °С до
163 °С; має переганятись не менше 95 %.
Зберігають у темному місці.
Хінальдиновий червоний. С21Н23ІМ2. (М.м. 430.3).
1073800. [117-92-0]. 2-[2-[4-(Диметиламіно)феніл|-
етеніл|-1-етилхіноліну йодид.
Порошок темного синювато-чорного кольору. Помір-
но розчинний у воді, легко розчинний у 96 % спирті.
Хінальдинового червоного розчин. 1073801.
0.1 г хінальдинового червоного Р розчиняють у ме-
танолі Рі доводять об’єм розчину тим самим роз-
чинником до 100 мл.
Зміна забарвлення. Від безбарвного до червоного в
інтервалі рН 1.4-3.2.
Хінгідрон. С|2Н|0О4. (М.м. 218.2). 1073900. [106-34-3].
Еквімолекулярна сполука 1,4-бензохінону й гідрохінону.
Блискучі кристали або кристалічний порошок темно-
зеленого кольору. Мало розчинний у воді, помірно
розчинний у гарячій воді, розчинний у 96 % спирті,
розчині аміаку концентрованого та ефірі.
Температура плавлення: близько 170 °С.
Хінідин. С20Н24М2О2. (М.м. 324.4). 1074000. [56-54-2].
(5)-(6-Метоксихінол-4-іл)[(2/?,4А,57?)-5-вінілхіну-
клідин-2-іл|метанол.
Кристалічний порошок білого кольору. Дуже мало
розчинний у воді, помірно розчинний у 96 % спирті,
мало розчинний в ефірі й метанолі.
[а|р : близько +260°. Визначення проводять, викори-
стовуючи розчин 10 г/л в етанолі Р.
272
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Температура плавлення: близько 172 °С.
Зберігають у захищеному від світла місці.
Хінідину сульфат. 1109500. [6591-63-5]. Див. статтю
Хінідину сульфат.
Хінін. С20Н24М2О2. (Мм. 324.4). 1074100. [130-95-0].
(Я)-(6-Метоксихінол-4-іл)| (25,45,5 Я)-5-він іл-
хінуклілин-2-іл]метанол.
Мікрокристалічний порошок білого кольору. Дуже
мало розчинний у воді, мало розчинний у киплячій
воді, дуже легко розчинний в етанолі, розчинний в
ефірі.
[а]р : близько -167°. Визначення проводять, викори-
стовуючи розчин 10 г/л в етанолі Р.
Температура плавлення: близько 175 °С.
Зберігають у захищеному від світла місці.
Хініну гідрохлорид. 1074200. [6119-47-7]. Див. статтю
Хініну гідрохлорид.
Хініну сульфат. 1074300. |6119-70-6]. Див. статтю Хініну
сульфат.
2-Хлор-4-нітроанілін. С6Н5СІЇ\2О2. (М.м. 172.6).
1018800. [121-87-9].
Кристалічний порошок жовтого кольору. Легко роз-
чинний у метанолі.
Температура плавлення: близько 107 °С.
Зберігають у захищеному від світла місці.
Хлоральгідрат. 1017900. [302-17-0]. Див. статтю Хлор-
альгідрат.
Хлоральгідрату розчин. 1017901.
Розчин 80 г у 20 мл води Р.
Хлорамін. 1018000. [127-65-1]. Див. статтю Хюрамін.
Хлораміну розчин. 1018001.
Розчин 20 г/л.
Готують безпосередньо перед використанням.
Хлораміну розчин РІ. 1018002.
Розчин 0.1 г/л хлораміну Р.
Готують безпосередньо перед використанням.
Хлораміну розчин Р2. 1018003.
Розчин 0.2 г/л.
Готують безпосередньо перед використанням.
Хлоранілін. С6Н6С11\'. (М.м. 127.6). 1018300. [106-47-8].
4-Хлоранілін.
Кристали. Розчинний у гарячій воді, легко розчинний
у 96 % спирті та ефірі.
Температура плавлення: близько 71 °С.
Хлорацетанілід. С8Н8С1МО. (М.м. 169.6). 1018100.
[539-03-7]. 4’-Хлорацетанілід.
Кристалічний порошок. Практично не розчинний у
воді, розчинний у 96 % спирті.
Температура плавлення: близько 178 °С.
4-Хлорбензолсульфонамід. С6Н6С1\СТ$. (Мм. 191.6).
1097400. [98-64-6].
Порошок білого кольору.
Температура плавлення: близько 145 °С.
Хлорбутанол. 1018400. [57-15-8]. Див. статтю Хлорбу-
танол безводний.
2-Хлоретанол. С2Н5С1О. (М.м. 80.5). 1097500.1107-07-3].
Безбарвна рідина. Розчинний в 96 % спирті.
: близько 1.197.
: близько 1.442.
Температура кипіння: близько 130 °С.
Температура плавлення: близько -89 °С.
2-Хлоретанолу розчин. 1097501.
125 мг 2-хлоретанолу /"розчиняють у 2-пропанолі Р
і доводять об’єм розчину тим самим розчинником
до 50 мл. 5 мл одержаного розчину доводять 2-про-
панолом Рцо об’єму 50 мл.
(2-Хлоретил[діетиламіну гідрохлорид. С6Н15С12ГЧ.
(М.м. 172.1). 1018500. 1869-24-91.
Кристалічний порошок білого кольору. Дуже легко
розчинний у воді й метанолі, легко розчинний у мети-
ленхлориді, практично не розчинний у гексані.
Температура плавлення: близько 211 °С.
Хлористоводнева кислота. 1043500. [7647-01-0]. Див.
статтю Кислота хлористоводнева концентрована.
Хлористоводнева кислота РІ. 1043501.
Містить 250 г/л НСІ.
70 г кислоти хлористоводневої Р доводять водою Р
до об’єму 100 мл.
Хлористоводнева кислота бромована. 1043507.
До 100 мл кислоти хлористоводневої Р додають
1 мл розчину брому Р.
Хлористоводнева кислота розведена. 1043503
Містить 73 г/л НСІ.
20 г кислоти хлористоводневої Р доводять водою Р
до об’єму 100 мл.
Хлористоводнева кислота розведена РІ. 1043504.
Містить 0.37 г/л НСІ.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
273
4.1.1. Реактиви
1.0 мл кислоти хлористоводневої розведеної Р дово-
дять водою Рдо об’єму 200 мл.
Хлористоводнева кислота розведена Р2. 1043505.
ЗО мл ІМ розчину кислоти хлористоводневої дово-
дять водою Р до об’єму 1000 мл, рН розчину
1.6±0.1.
Розчин хлористоводневої кислоти у спирті. 1043506.
5.0 мл 1 М розчину кислоти хлористоводневої дово-
дять 96 % спиртом Рдо об’єму 500.0 мл.
Хлористоводнева кислота, вільна від свинцю. 104359?.
Має задовольняти вимоги для кислоти хлористоводне-
вої Р і витримувати таке додаткове випробування.
Свинець (2.2.22, метод І). Не більше 20 ррт, визна-
чення проводять методом атомно-емісійної спектро-
метрії.
Випробовуваний розчин. 200 г кислоти хлористоводне-
вої поміщають у кварцовий тигель, випарюють майже
насухо, до одержаного залишку додають 5 мл кислоти
азотної, приготованої дистиляцією кислоти азотної Р
при температурі нижче температури кипіння, і випа-
рюють насухо. До одержаного залишку додають 5 мл
кислоти азотної, приготованої дистиляцією кислоти
азотної Р при температурі нижче температури
кипіння.
Розчини порівняння. Готують розчини порівняння, ви-
користовуючи еталонний розчин свинцю (0.1 ррт РЬ) Р,
розведений кислотою азотною, приготованою дисти-
ляцією кислоти азотної Р при температурі нижче тем-
ператури кипіння.
Вимірюють інтенсивність емісії за довжини хвилі
220.35 нм.
Хлорна кислота. НС1О4. (М.м. 100.5). 1062900.
[7601-90-3].
Містить не менше 70.0 % (м/м) і не більше 73.0 %
(м/м) НС1О4.
Прозора, безбарвна рідина. Легко змішується з водою.
: близько 1.7.
Кількісне визначення. 2.50 г кислоти хлорної додають
до 50 мл води Рі титрують 1 Мрозчином натрію гідрок-
сиду, використовуючи як індикатор 0.1 мл розчину ме-
тилового червоного Р.
1 мл 1 Мрозчину натрію гідроксиду відповідає 100.5 мг
НС1О4.
Хлорної кислоти розчин. 1062901.
8.5 мл кислоти хлорної Р доводять водою Р до
об’єму 100 мл.
Хлорогенова кислота. С16Н|8О9. (М.м. 354.3). 1104700.
[327-97-9]. (15,ЗА,4Я, 5 Я)-3-[(3,4-Дигідроксицина-
моїл)окси]-1,4,5-тригідроксициклогексанкарбонова
кислота.
Кристалічний порошок білого кольору. Розчинна у
киплячій воді, ацетоні й етанолі.
Температура плавлення: близько 208 °С.
[«][) : близько - 35.2°.
Хроматографія. Визначення проводять, як зазначек І
в статті Сухий екстракт листя беладонни, стандарт,.' І
зовании за умов, описаних в "Ідентифікації А"; на хр. І
матограмі має виявлятись лише одна пляма.
Хлороформ. СНС13. (М.м. 119.4). 1018600. [67-66-3]
Трихлорметан.
Прозора, безбарвна рідина. Мало розчинний у воді
змішується з 96 % спиртом.
: від 1-475 до 1.481.
Температура кипіння; близько 60 °С.
Хлороформ містить від 0.4 % (м/м) до 1.0 % (м/м) ета-
нолу.
Етанол. 1.00 г хлороформу поміщають у колбу з при-
тертою скляною пробкою, додають 15.0 мл нітрохро-
мового реактиву Р, закривають колбу, енергійно стру-
шують протягом 2 хв і витримують протягом 15 хв. До-
дають 100 мл води Рі 5 мл розчину 200 г/л калію йоди-
ду Р. Через 2 хв титрують 0.1 М розчином натрію
тіосульфату до одержання світло-зеленого забарв-
лення, використовуючи як індикатор 1 мл розчин}
крохмалю Р. Проводять контрольний дослід.
Вміст етанолу (X), у відсотках, обчислюють за форму-
лою:
(п2 - л?1)0.115
де:
П| — об’єм 0.1 М розчину натрію тіосульфату, ви-
трачений на титрування випробовуваного роз-
чину, в мілілітрах;
п2 — об’єм 0.1 М розчину натрію тіосульфату, ви-
трачений на титрування контрольного досліду;
в мілілітрах;
т — маса наважки хлороформу, в грамах.
Хлороформ, вільний від етанолу. 1018602.
200 мл хлороформу Р промивають водою Р, струшу-
ючи з чотирма порціями по 100 мл кожна. Сушать
над 20 г натрію сульфату безводного Р протягом
24 год. Фільтрат переганяють над 10 г натрію суль-
фату безводного Р, відкидаючи перші 20 мл відго-
ну.
Готують безпосередньо перед використанням.
Хлороформ підкислений. 1018601.
До 100 мл хлороформу Р додають 10 мл кислоти
хлористоводневої Р. струшують, відстоюють і
розділяють два шари.
Хлороформ, стабілізований аміленом. СНС13. (М.м. 119.4).
1018700.
Прозора, безбарвна рідина. Мало розчинний у воді,
змішується з 96 % спиртом.
274
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
Вода. Не більше 0.05 %.
Залишок після випарювання. Не більше 0.001 %.
Мінімальне пропускання (2.2.25) визначають, викорис-
товуючи як компенсаційну рідину воду Р.
не менше 50 % за довжини хвилі 255 нм,
не менше 80 % за довжини хвилі 260 нм,
не менше 98 % за довжини хвилі 300 нм.
Кількісне визначення. Не менше 99.8 % СНС13. Визна-
чення проводять методом газової хроматографії.
Хлороцтова кислота. С2Н3С1О2. (М.м. 94.5). 1018200.
[79-11-8].
Безбарвні або білого кольору кристали, які розплива-
ються на повітрі. Дуже легко розчинна у воді, розчин-
на в 96 % спирті та ефірі.
Зберігають у повітронепроникному контейнері.
Хлорплатинова кислота. Н2С16Рі,6Н2О. (М.м. 517.9).
1019000. [18497-13-7]. Іексахлорплатинова(ІУ) кислота.
Містить не менше 37.0 % (м/м) платини (А.м. 195.1).
Кристали або кристалічна маса коричнювато-черво-
ного кольору. Дуже легко розчинна у воді, розчинна в
% % спирті.
Кількісне визначення. 0.200 г кислоти хлорплатинової
прожарюють при температурі 900 °С до постійної ма-
си і зважують залишок (платини).
Зберігають у захищеному від світла місці.
3-Хлорпропан-1,2-діол. С3Н7С1О2. (М.м. 110.5).
1097600. [96-24-2].
Безбарвна рідина. Розчинний у воді, 96 % спирті та
ефірі.
: близько 1.322.
Ир : близько 1.480.
Температура кипіння: близько 213 °С.
5-Хлорсаліцилова кислота. С7Н5СЮ3. (М.м. 172.6).
1019100. [321-14-2].
Кристалічний порошок білого або майже білого
кольору. Розчинна у метанолі.
Температура плавлення: близько 173 °С.
Хлортіазид. 1112100. [58-94-6|. Див. статтю
Хлортіазид.
Хлортриметилсилан. С3Н9С18і. (М.м. 108.6). 1019300.
[75-77-4].
Прозора, безбарвна рідина, шо димить на повітрі.
• близько 0.86
Ир : близько 1.388.
Температура кипіння: близько 57 °С.
Хлорфенол. С6Н5С1О. (М.м. 128.6). 1018900. [106-48-9].
4-Хлорфенол.
Безбарвні або майже безбарвні кристали. Мало роз-
чинний у воді, дуже легко розчинний у 96 % спирті,
ефірі й розчинах гідроксидів лужних металів
Температура плавлення: близько 42 °С.
Холестерин. 1019400. [57-88-5]. Див. статтю Холестерин.
Холіну хлорид. С5НІ4С1МО. (М.м. 139 6). 1019500.
]67-48-1 ]. (2- Гідроксиетил)три метиламонію хлорид.
Кристали, які розпливаються на повітрі. Дуже легко
розчинний у воді й 96 % спирті.
Хроматографія. Визначення проводять, як зазначено
в статті Суксаметонію хлорид, використовуючи 5 мкл
розчину 0.2 г/л у метанолі Р, на хроматограмі має ви-
являтись лише одна основна пляма.
Зберігають у повітронепроникному контейнері.
Хромазурол 8. С23Н)3С12№а3О98. (М.м. 605). 1019600.
[1667-99-8]. Показник Шульца №841. Кольоровий
індекс № 43825. Тринатрію 5-[(3-карбоксилато-5-ме-
тил-4-оксоциклогекса-2,5-дієн-1-іліден)(2,6-дихлор-3-
сульфонатофеніл)метил]-2-гідрокси-3-метилбензоат.
Порошок коричнювато-чорного кольору. Розчинний
у воді, мало розчинний у 96 % спирті.
Хромова суміш. 1019700.
Насичений розчин хрому(УІ) оксиду Р у кислоті
сірчаній Р.
Хромотроп II В. СІ6Н9КІ3№2ОІ0$2. (М.м. 513.4).
1020200. [548-80-1]. Показник Шульца № 67. Кольо-
ровий індекс № 16575. Динатрію 4,5-дигідрокси-3-(4-
нітрофенілазо)нафталін-2,7-дисульфонат.
Порошок червонувато-коричневого кольору. Розчин-
ний у воді з утворенням жовтувато-червоного розчину,
практично не розчинний у 96 % спирті.
Хромотропу II В розчин. 1020201.
Розчин 0.05 г/л у кислоті сірчаній Р.
Хромотропова кислота. СІ0Н8О882. (М.м. 320.3).
1119100. [148-25-4]. 4,5-Дигідрокси-2,7-нафталінди-
сульфонова кислота.
Голчасті кристали білого кольору. Розчинна у воді.
Температура плавлення: близько 300 °С.
Хромотропової кислоти розчин.
0.50 г кислоти хромотропової Р розчиняють при-
близно у 80 мл води Р і доводять тим самим роз-
чинником до об’єму 100 мл.
Термін придатності розчину 24 год.
Хромотропової кислоти натрієва сіль.
С10Н6Ма2О882,2Н2О. (Мм. 400.3). 1020300. [5808-22-0].
Показник Шульца № 1136. Динатрію 1,8-дигідрокси-
нафталін-3,6-дисульфонат дигідрат.
Порошок жовтувато-білого кольору. Розчинна у воді,
практично не розчинна в 96 % спирті.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
275
4.1.1. Реактиви
Хромоформний субстрат РІ. 1020000.
А-а-бензилоксикарбоніл О-аргініл-Е-гліцил-Е-аргі-
нін-4-нітроаніліду дигідрохлорид розчиняють у воді Р
до одержання 0.003 М розчину. Перед використанням
розводять буферним розчином трис(гідроксиме-
тил)амінометану-ЕПТА рН 8.4 Р до одержання
0.0005 М розчину.
Хромоформний субстрат Р2. 1020100.
О-фенілаланіл-Е-піпеколіл-Е-аргінін-4-нітроаніліду
дигідрохлорид розчиняють у воді Р до одержання
0.003 М розчину. Перед використанням розводять бу-
ферним розчином трис(гідроксиметил)амінометану-
ЕПТА рН 8.4 Рао одержання 0.0005 М розчину.
Хрому(ІІІ) трихлорид гексагідрат. [Сг(Н2О)4С12 |С1,2Н2О.
(М.м. 266.5). 1104800. [10060-12-5].
Кристалічний порошок темно-зеленого кольору,
гігроскопічний.
Зберігають у сухому місці, захищаючи від дії окис-
ників.
Хрому(УІ)оксид. СгО3. (М.м. 100.0). 1019900. [1333-82-0].
Хрому триоксид.
Голчасті кристали або гранули темного коричнювато-
червоного кольору, які розпливаються на повітрі. Ду-
же легко розчинний у воді
Зберігають у повітронепроникному контейнері.
Хрому-калію сульфат. СгК(8О4)2,12Н2О. (М.м. 499.4).
1019800. [7788-99-0]. Хромові галуни.
Великі кристали від фіолетово-червоного до чорного
кольору. Легко розчинний у воді, практично не роз-
чинний у 96 % спирті.
Цезію хлорид. СкСІ. (М.м. 168.4). 1014200. [7647-17-8].
Порошок білого кольору Дуже легко розчинний у
воді, легко розчинний у метанолі, практично не роз-
чинний в ацетоні.
Целюлоза для хроматографії. 1016800. [9004-34-6].
Дрібний гомогенний порошок білого кольору із се-
реднім розміром часток менше ЗО мкм.
Приготування тонкого шару. 15 г суспендують у 100 мл
води Р і гомогенізують на електромішалці протягом
60 с. Ретельно очищені пластини покривають шаром
завтовшки 0.1 мм, використовуючи прилад для нане-
сення. Сушать на повітрі.
Целюлоза для хроматографії РІ. 1016900.
Мікрокристалічна целюлоза. Дрібний гомогенний по-
рошок білого кольору із середнім розміром часток
менше ЗО мкм.
Приготування тонкого шару. 25 г суспендують у 90 мл
води Р і гомогенізують на електромішалці протягом
60 с. Ретельно очищені пластини покривають шаром
завтовшки 0.1 мм, використовуючи прилад для нане-
сення. Сушать на повітрі.
Целюлоза для хроматографії г254. 1017000.
Мікрокристалічна целюлоза е254. Тонкий гомогенний |
порошок білого кольору із середнім розміром часток
менше 30 мкм, який містить флуоресцентний індика- 1
тор із інтенсивністю поглинання за довжини хвилі
254 нм.
Приготування тонкого шару. 25 г суспендують і І
100 мл води Рі гомогенізують на електромішалці про-
тягом 60 с. Ретельно очищені пластини покривають
шаром завтовшки 0.1 мм, використовуючи прилад
для нанесення. Сушать на повітрі.
Церію(ІП) нітрат. Се(МО3)3,6Н2О. (М.м. 434.3).
1017400. [10294-41-4|. Церію тринітрат гексагідрат.
Кристалічний порошок від безбарвного до ледь жов-
того кольору. Легко розчинний у воді й 96 % спирті.
Церію(ІУ) сульфат. Се(8О4)2,4Н2О. (М.м. 404.3).
1017300. [123333-60-8]. Церій сірчанокислий.
Кристалічний порошок або кристали жовтого або
оранжево-жовтого кольору. Дуже мало розчинний у
воді, повільно розчинний у розведених кислотах.
Цетилтриметиламонію бромід. С1УН42ВгК. (М.м. 364.5).
1017700. [57-09-0]. Цстримонію бромід. /У-Гексаде-
цил-А,А, А-триметиламонію бромід.
Кристалічний порошок білого кольору. Розчинний у
воді, легко розчинний у 96 % спирті.
Температура плавлення: близько 240 °С.
Цетрімід. 1017600. [8044-71-1]. Див. статтю Цетрімід.
Цефаліну реактив. 1017200.
До 0.5 - 1 г мозку бичачого, висушеного в ацетоні Р, до-
дають 20 мл ацетону Р і залишають на 2 год. Центри-
фугують із прискоренням 500 § протягом 2 хв, рідину
над осадом зливають. Залишок сушать при зниженому
тиску, потім додают ь 20 мл хлороформу Р і залишають
на 2 год, часто збовтуючи. Фільтрують або центрифу-
гують і випарюють хлороформ при зниженому тиску.
Залишок суспендують у 5 - 10 мл розчину 9 г/л натрію
хлориду Р.
Розчинники, використовувані для приготування реак-
тиву, мають містити підхожий антиоксидант, на-
приклад, 0.02 г/л бутил нова ного гідроксианізолу.
Зберігають у замороженому або висушеному при тем-
пературі не вище 0 °С, ліофілізованому вигляді.
Термін придатності 3 міс.
Цефаліну дигідрохлорид. С28Н40С12М9О4,7Н2О.
(М.м. 666). 1017100. [5884-43-5]. (Я)-1-[(25,ЗЯ,11Ь5)-
3-етпл-1,3,4,6,7,11 Ь-гексагідро-9,10-диметокси-2//-
бензо[а]хінолізин-2-ілметил]-1,2,3,4-тетрагідро-7-
метоксіізохінолін-6-ол дигідрохлорид гептагідрат.
Кристалічний порошок від білого до жовтуватого ко-
льору. Легко розчинний у воді, розчинний в ацетоні й
96 % спирті.
[«][) : близько +25°. Визначення проводять, викорис-
товуючи розчин 20 г/л.
276
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.1. Реактиви
ІЦиклогексан. С6Ні2. (Мм. 84.2). 1023900. [110- 82-7].
Прозора, безбарвна займиста рідина. Практично не роз-
чинний у воді, змішується з органічними розчинниками.
: близько 0.78.
Температура кипіння: близько 80.5 °С.
Циклогексан, використовуваний у спектрофотометрії,
мас витримувати таке додаткове випробування.
Мінімальне пропускання (2.2.25) визначають, викорис-
товуючи як компенсаційну рідину воду Р:
45 % за довжини хвилі 220 нм,
70 % за довжини хвилі 235 нм,
90% за довжини хвилі 240 нм,
98 % за довжини хвилі 250 нм.
Циклогексан РІ. 1023901.
Має задовольняти вимоги для циклогексану Р і та-
ку додаткову вимогу.
Інтенсивність поглинання, виміряна за довжини
хвилі 460 нм (при опромінюванні пучком світла з
довжиною хвилі 365 нм), не має бути інтен-
сивнішою за поглинання розчину 0.002 ррт хіні-
ну Ру 0.05 Мрозчині кислоти сірчаної.
Циклогексиламін. С6НІ31\'. (М.м. 99.2). 1024000.
1108-91-8].
Безбарвна рідина. Розчинний у воді, змішується з
найпоширенішими розчинниками.
: близько 1.460.
Температура кипіння: від 134 °С до 135 °С.
Циклогексилендинітрилтетраоцтова кислота.
С14Н22^О8,Н2О. (М.м. 364.4). 1024100. транс-Цикло-
гексилен-1 ,2-динітрило-А,Ц,А’-тетраоптова кис-
лота.
Кристалічний порошок білого кольору.
Температура плавлення: близько 204 °С.
З-Циклогексилпропіонова кислота. С9Н16О2.
(М.м. 156.2). 1119200. [701-97-3].
Безбарвна рідина.
</2о : близько 0.998.
и® : близько 1.4648.
Температура кипіння: близько 130 °С.
Цинеол. С10НІ8О. (М.м. 154.3). 1020600. [470-82-6].
1,8-Цинеол. Евкаліптол. 1,8-Епокси-и-ментан.
Безбарвна рідина. Практично не розчинний у воді.
Змішується з етанолом і ефіром.
і/® : від 0.922 до 0.927.
«р : від 1.456 до 1.459.
Температура тверднення (2.2.18). Від 0 °С до 1 °С.
Температурні межі перегонки (2.2.11). Від 174 °С до
177 °С.
Фенол. 1 г струшують із 20 мл води Р. Після розділення
шарів до 10 мл водного шару додають 0.1 мл розчину
заліза(111) хлориду РІ. Розчин не має набувати фіоле-
тового кольору.
Терпентинова олія. 1 г цинеолу розчиняють у 5 мл
спирту (90 %, об/об) Р, по краплях додають свіжопри-
готовану бромну воду Р. Для одержання жовтого за-
барвлення, яке не зникає протягом ЗО хв, має бути ви-
трачено не більше 0.5 мл.
Залишок після випарювання. Не більше 0.05 %. До
10.0 мл додають 25 мл води Р, випарюють на водяній
бані, залишок сушать до постійної маси при темпера-
турі від 100 °С до 105 °С.
Цинеол, використовуваний у газовій хроматографії,
має витримувати таке додаткове випробування.
Кількісне визначення. Проводять методом газової хро-
матографії (2.2.28). як зазначено в статті Олія м’яти
перцевої, використовуючи цинеол як випробовуваний
розчин.
Площа основного піка має бути не менше 98.0 % суми
площ усіх піків.
Цинк. 7.п. (А.м. 65.4). 1096500. [7440-66-6].
Містить не менше 99-5 % 2п.
Циліндри, або гранули, або кульки сріблясто-білого
кольору або стружка із синім блиском.
Арсен (2.4.2, метод А). Не більше 0.00002 % (0.2 ррт).
5.0 г цинку розчиняють у суміші 15 мл кислоти хлори-
стоводневої Р і 25 мл води Р; одержаний розчин має
витримувати випробування на арсен.
Цинк активований. 1096501.
Цинк у вигляді циліндрів або кульок поміщають у
конічну колбу, додають достатню кількість 50 ррт
кислоти хлорплатинової Р, щоб повністю покрити
метал, через 10 хв метал промивають водою, вида-
ляють воду й негайно сушать.
Арсен. До 5 г цинку активованого додають 15 мл
кислоти хлористоводневої Р, 25 мл води Р, 0.1 мл
розчину олова(II) хлориду Р і 5 мл розчину калію йо-
диду Р. Далі діють, як зазначено у випробуванні на
арсен (2.4.2, метод А). На ртутно-бромідному па-
пері Р не має спостерігатись забарвлення.
Активність. Повторюють випробування на арсен,
використовуючи ті самі реактиви, додають розчин,
який містить 1 мкг арсену. На ртутно-бромідному
папері /"з’являється помітне забарвлення.
Цинку ацетат. (С2Н3О2)27п,2Н2О. (М.м. 219.5).
1102300. [5970-45-6]. Цинку ацетат дигідрат.
Блискучі кристали білого кольору, злегка звітрюються
на повітрі. Легко розчинний у воді, розчинний у 96 %
спирті.
При температурі 100 °С втрачає кристалізаційну воду.
г/2о : близько 1.735.
Температура плавлення: близько 237 °С.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
277
4.1.1. Реактиви
Цинку ацетату розчин. /102301.
54.9 г цинку ацетату Р розчиняють у суміші 600 мл
води Рі 150 мл кислоти оцтовоїльодяної Р. При пе-
ремішуванні додають 150 мл розчину аміаку кон-
центрованого Р, охолоджують і доводять рН до 6.4
розчином аміаку Р, доводять об’єм розчину во-
дою Рдо 1 л.
Цинку оксид. 1096700. [1314-13-2]. Див. статтю Цинку
оксид.
Цинку порошок. 7.п. (А.м. 65.4). 1096800. [7440-66-6].
Містить не менше 90.0 % 7п (А.м. 65.4).
Дуже дрібний порошок сірого кольору. Розчинний у
кислоті хлористоводневій розведеній Р.
Цинку сульфат. 1097000. [7446-20-0]. Див. статтю Цин-
ку сульфат.
Цинку хлорид. 1096600. [7646-85-7]. Див. статтю Цин-
ку хлорид.
Цинку хлориду розчин в кислоті мурашиній. 1096601.
20 г цинку хлориду Р розчиняють у 80 г розчину
850 г/л кислоти мурашиної безводної Р.
Цинку хлориду розчин йодований. 1096602
20 г цинку .хлориду Рі 6.5 г калію йодиду Ррозчиня-
ють у 10.5 мл води Р, додають 0.5 г йоду Рі струшу-
ють протягом 15 хв. Якщо необхідно, фільтрують.
Зберігають у захищеному від світла місці.
Цинку йодиду і крохмалю розчин. 1096502.
До розчину 2 г цинку хлориду Ру 10 мл води Рдодають
0.4 г крохмалю розчинного Р і нагрівають до розчинен-
ня крохмалю. Після охолодження до кімнатної темпе-
ратури додають 1.0 мл безбарвного розчину, який
містить0.10 г цинку Ру вигляді ошурок і 0.2 г йоду Ру
воді Р, доводять об’єм розчину водою Р до 100 мл,
фільтрують.
Зберігають у захищеному від світла місці.
Випробування на чутливість. 0.05 мл розчину натрію
нітриту Р доводять водою Р до об’єму 50 мл. До 5 мл
одержаного розчину додають 0.1 мл кислоти сірчаної
розведеної Р і 0.05 мл приготованого розчину цинку
йодиду і крохмалю та змішують; розчин забарв-
люється у синій колір.
Ципхонідин. С|9Н22І^2О. (М.м. 294.4). 1020400.
[485-71-2]. (Я)-(Хінол-4-іл)[(25,45',5Р)-5-вінілхіну-
клідин-2-іл] метанол.
Кристалічний порошок білого кольору. Дуже мало
розчинний у воді й петролейному ефірі, помірно роз-
чинний у 96 % спирті, мало розчинний в ефірі.
[а]р : від -105° до -110°. Визначення проводять, вико-
ристовуючи розчин 50 г/л у 96 % спирті Р.
Температура плавлення: близько 208 °С, із розкла-
данням.
Зберігають у захищеному від світла місці.
Цинхонін. С19Н22І\12О. (М.м. 294.4). 1020500. [118-10-51 І
(5')-(Хінол-4-іл)[(2Л,45,5Р)-5-вінілхінуклідин-1 І
іл] метанол.
Кристалічний порошок білого кольору. Мало розчин-1
ний у воді, помірно розчинний у 96 % спирті й мета-1
полі, мало розчинний в ефірі.
[«Іо* : від +225° до +230°. Визначення проводять, ви-1
користовуючи розчин 50 г/л у 96 % спирті Р.
Температура плавлення: близько 263 °С.
Зберігають у захищеному від світла місці.
Цирконілу нітрат. Основна сіль, склад якої приблизно
відповідає формулі 7гО(МО3)2,2Н2О. 1097200.
114985-18-3].
Кристалічний порошок або кристали білого кольору
Гігроскопічний, розчинний у воді. Водний розчин
прозорий або злегка опалесціюючий.
Зберігають у повітронепроникному контейнері.
Цирконілу ніт рату розчин. 1097201.
Розчин 1 г/л у суміші розчинників вода Р- кисло-
та хлористоводнева Р (40:60).
Цирконілу хлорид. Основна сіль, склад якої приблизно
відповідає формулі 7гС12О,8Н2О. 1097100 [15461-27-5|.
Містить не менше 96.0 % ХгС12О,8Н2О.
Кристалічний порошок або кристали білого або майже
білого кольору. Легко розчинний у воді й 96 % спирті.
Кількісне визначення. 0.600 г розчиняють у суміші 5 мл
кислоти азотної Р і 50 мл води Р, додають 50.0 мл
0.1М розчину срібла нітрату, 3 мл розчину дибутш-
фталату Р, збовтують і титрують 0.1 М розчином
амонію тіоціанату до червонувато-жовтого забарв-
лення, використовуючи як індикатор 2 мл розчину
заліза(Ш) амонію сульфату Р2.
1 мл 0.1 М розчину срібла нітрату відповідає 16.11 мг
7гС12О, 8Н2О.
Ь-Цистеїн. С3Н7КО28. (М.м. 121.1). 1024200. [52-90-4].
Порошок. Легко розчинний у воді, 96 % спирті й кис-
лоті оптовій, практично не розчинний в ацетоні.
Цистеїну гідрохлорид. 1024300. 17048-04-6]. Див. стат-
тю Цистеїну гідрохлорид моногідрат.
Ь-Цистин. С6НІ21\І2О482. (М.м. 240.3). 1024400. [56-89-3].
Кристалічний порошок білого кольору Практично не
розчинний у воді й 96 % спирті, розчиняється у розве-
дених розчинах гідроксидів лужних металів. Розкла-
дається при температурі 250 °С.
[а]о : від -218° до -224°. Визначення проводять в
1 М розчині кислоти хлористоводневої.
Цитраль. С10Н16О. (Мм. 152.2). 1020800. [5392-40-5].
Суміш (2£)- та (27)-3,7-Диметилокта-2,6-дієналю.
278
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.2. Еталонні розчини для випробувань на граничний вміст домішок
І \іина світло-жовтого кольору. Практично не розчинна
Ігводі, змішується з 96 % спиртом, ефіром і гліцерином.
І Ірматографія. Визначення проводять методом тон-
кміарової хроматографії (2.2.27), використовуючи як
такий шар силікагель ск254 Р. На хроматографічну
І «тинку наносять 10 мкл розчину 1 г/л у толуолі Р.
І Хроматографують, використовуючи систему розчин-
Віків етилацетат Р— толуол Р (15:85). Коли фронт
І і' ічинників пройде 15 см, пластинку виймають із ка-
I пери й сушать на повітрі. Переглядають в УФ-світлі за
дожпип хвилі 254 нм. На одержаній хроматограмі має
І виявлятись лише одна основна пляма.
І Цигрована плазма кролика. 1020900.
У кролика, який не приймав їжі протягом 12 год,
І «вбирають кров внутрішньосерцевою пункцією, ви-
I корпстовуючи пластиковий шприц із голкою № 1, що
містить відповідний об’єм розчину 38 г/л натрію цит-
I хіту Р, так, шоб кінцеве співвідношення об’ємів роз-
іпну натрію цитрату й крові становило 1:9. Відокрем-
I дюють плазму центрифугуванням при прискоренні від
І500 р до 1800 § і температурі від 15 °С до 20 °С протя-
гом ЗО хв.
Зберігають при температурі від 0 °С до 6 °С.
Термін придатності 4 год з момента відбору крові.
Цитроптен. СцНі0О4. (М.м. 206.2). 1021300. [487-06-9].
Ліметтин. 5,7-Диметокси-2//-1-бензопіран-2-он.
Голчасті кристали. Практично не розчинний у воді,
ефірі й петролейному ефірі, легко розчинний в аце-
тоні й 96 % спирті.
Температура плавлення: близько 145 °С.
Хроматографія. Визначення проводять методом тон-
кошарової хроматографії (2.2.27), використовуючи як
тонкий шар силікагель сг254 Р- На хроматографічну
пластинку наносять 10 мкл розчину 1 г/л у толуолі Р.
Хроматографують, використовуючи систему розчин-
ників етилацетат Р — толуол Р (15:85). Коли фронт
розчинників пройде 15 см, пластинку виймають із ка-
мери й сушать на повітрі. Переглядають в УФ-світлі за
довжини хвилі 254 нм. На одержаній хроматограмі має
виявлятись лише одна основна пляма.
Ціаноброміду розчин. 1023700. [506-68-3].
До води бромної Р додають по краплях при охолод-
женні 0.1 М розчин амонію тіоціанату до зникнення
жовтого забарвлення.
Готують безпосередньо перед використанням.
Ціаногуанідин. С2Н4\4. (М.м. 84.1). 1023800. [461-58-5].
Диціандіамід. 1-Ціаногуанідин.
Кристалічний порошок білого кольору. Помірно роз-
чинний у воді й 96 % спирті, практично не розчинний
в ефірі й метиленхлориді.
Температура плавлення: близько 210 °С.
Ціанокобаламін. 1023600. [68-19-9]. Див. статтю
Ціанокобаламін.
Ціанооцтова кислота. С3Н3І\1О2. (М.м. 85.1). 1097900.
[372- 09-8].
Гігроскопічні кристали білого або жовтувато-білого
кольору. Розчинна у воді.
Зберігають у повітронепроникному контейнері.
Шлунковий штучний сік. 1039900.
2.0 г натрію хлориду Р і 3.2 г пепсину порошку Р розчи-
няють у воді Р, додають 80 мл 1 М розчину кислоти хло-
ристоводневої та доводять об’єм розчину водою Р до
’.ООО мл.
Щавлева кислота. С2Н2О4,2Н2О. (М.м. 126.1). 1061400.
[6153-56-6]. Етандикарбонова кислота дигідрат.
Кристали білого кольору. Розчинна у воді, легко роз-
чинна в 96 % спирті.
Щавлевої кислоти і сірчаної кислоти розчин.
1061401.
Розчин 50 г/л кислоти щавлевої Р в охолодженій
суміші рівних об’ємів кислоти сірчаної Р і води Р.
4.1.2. ЕТАЛОННІ РОЗЧИНИ ДЛЯ ВИПРОБУВАНЬ
НА ГРАНИЧНИЙ ВМІСТ ДОМІШОК
Алюмінію еталонний розчин (200 ррт А1). 5000200.
0.352 г алюмінію-калію сульфату Р, у перерахунку на
А1К(8О4)2,12Н2О, розчиняють у воді Р, додають 10 мл
кислоти сірчаної розведеної Р і доводять об’єм розчину
водою Рдо 100.0 мл.
Алюмінію еталонний розчин (100 ррт А1). 5000203.
8.947 г алюмінію хлориду Р розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл.
Розчин розводять водою Р у 10 разів безпосередньо
перед використанням.
Алюмінію еталонний розчин (10 ррт А1). 5000201.
1.39 г алюмінію нітрату Р, у перерахунку на
А1(МО3)3,9Н2О, розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 100.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
Алюмінію еталонний розчин (2 ррт А1). 5000202.
0.352 г алюмінію-калію сульфату Р, у перерахунку на
А1К(8О4), 12Н2О, розчиняють у 10 мл кислоти сірчаної
розведеної Р і доводять об’єм розчину водою Р до
100.0 мл.
Розчин розводять водою Р в 100 разів безпосередньо
перед використанням.
Амонію еталонний розчин (100 ррт І\Н4). 5000300.
0.741 г амонію хлориду Р, у перерахунку на 1ЧН4С1, роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
279
4.1.2. Еталонні розчини для випробувань на граничний вміст домішок
10 мл одержаного розчину доводять водою Рао об’єму
25 мл безпосередньо перед використанням.
Амонію еталонний розчин (2.5 ррт ІЧН4). 5000301.
0.741 г амонію хлориду Р, у перерахунку на N Н4С1, роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
Розчин розводять водою Р у 100 разів безпосередньо
перед використанням.
Амонію еталонний розчин (1 ррт N114). 5000302.
Еталонний розчин амонію (2.5 ррт ГН4) ? розводять
водою Р у 2.5 рази безпосередньо перед викорис-
танням.
Арсену еталонний розчин (10 ррт Ах). 5000500.
0.330 г арсену(ПІ) оксиду Р. у перерахунку на Ах2О3,
розчиняють у 5 мл розчину натрію гідроксиду розведе-
ного Р і доводять об’єм розчину водою Рао 250.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
Арсену еталонний розчин (1 ррт Ах). 5000501.
Еталонний розчин арсену (10 ррт Аз) Р розводять во-
дою Ру 10 разів безпосередньо перед використанням.
Арсену еталонний розчин (0.1 ррт Ах). 5000502.
Еталонний розчин арсену (1 ррт Аз) Р розводять во-
дою Ру 10 разів безпосередньо перед використанням.
Ацетальдегіду еталонний розчин (100 ррт С2Н4О).
5000100.
1.0 г ацетальдегіду Ррозчиняють у 2-нропанолі Р і до-
водять об’єм розчину тим самим розчинником до
100.0 мл. 5.0 мл одержаного розчину доводять 2-про-
панолом Р до об’єму 500.0 мл.
Готують безпосередньо перед використанням.
Ацетальдегіду еталонний розчин (100 ррт С2П4О) РІ.
5000101.
1.0 г ацетальдегіду Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 100.0 мл.
5.0 мл одержаного розчину доводять водою Р до об’єму
500.0 мл.
Готують безпосередньо перед використанням.
Барію еталонний розчин (50 ррт Ва). 5000600.
0.178 г барію хлориду Р, у перерахунку на ВаС12,2Н2О,
розчиняють у воді дистильованій Р і доводять об’єм
розчину тим самим розчинником до 100 0 мл.
Розчин розводять у 20 разів водою дистильованою Р
безпосередньо перед використанням.
Ванадію еталонний розчин (1 г/л УЗ- 5003300.
0.230 г амонію ванадату Р, у перерахунку на N Н4\'О3,
розчиняють у воді Р і доводять тим самим розчинни-
ком до об’єму 100 0 мл.
Гліоксалю еталонний розчин (20 ррт С2Н2О2). 500ЛАІ
У мірній колбі місткістю 100 мл зважують кільки І
розчину гліоксалю Р, яка відповідає 0.200 г С2Н?0і. І
доводять об’єм розчину етанолом Рао позначки.
Розчин розводять етанолом Ру 100 разів безпосерег І
ньо перед використанням.
Заліза еталонний розчин (0.1 % Ре). 5001605.
0.100 г Ге розчиняють у мінімгільно необхідній кілька
суміші рівних об’ємів кислоти хлористоводневої Р іво- П
ди Р, доводять об’єм розчину водою Рао 100.0 мл.
Заліза еталонний розчин (250 ррт Ре). 5001606.
4.840 г заліза(ПІ) хлориду Р розчиняють у розчині І
150 г/л кислоти хлористоводневої Р і доводять об’єм І
розчину тією самою кислотою до 100.0 мл.
Розчин розводять водою Ру 40 разів безпосередньо пе-
ред використанням.
Заліза еталонний розчин (20 ррт Ре). 5001600.
0.863 г заліза(Ш) амонію сульфату Р, у перерахунку на
ЕеМН4(8О4)2,12Н3О, розчиняють в 25 мл кислоти
сірчаної розведеної Р і доводять об’єм розчину водою!
до 500.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Заліза еталонний розчин (10 ррт Ре). 5001601.
7.022 г заліза)II) амонію сульфату Р, у перерахунку на
Ее(МН4)2(8О4)2,6Н2О, розчиняють у 25 мл кислоти
сірчаної розведеної Р і доводять об’єм розчину водою Р
до 1000.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
Заліза еталонний розчин (8 ррт Ре). 5001602.
80 мг заліза Р розчиняють у 50 мл розчину 220 г/л
(НС1) кислоти хлористоводневої Р і доводять об’єм
розчину водою Рао 1000.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Заліза еталонний розчин (2 ррт Ре). 5001603.
Еталонний розчин заліза (20 ррт Ре) Р розводять во-
дою Ру 10 разів безпосередньо перед використанням.
Заліза еталонний розчин (1 ррт Ре). 5001604.
Еталонний розчин заліза (20 ррт Ге) Р розводять во-
дою Ру 20 разів.
Йодиду еталонний розчин (10 ррт І). 5003800.
0.131 г калію йодиду Р, у перерахунку на К1, розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 100.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
280
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.2. Єталонні розчини для випробувань на граничний вміст домішок
кадмію еталонний розчин (0.1 % Сії). 5000700.
1.100 г кадмію Р, у перерахунку на Ссі. розчиняють у
.іінімально необхідній кількості суміші рівних об’ємів
кислоти хлористоводневої Р і води Р, доводять об єм
розчину 1 % (об/об) розчином кислоти хлористовод-
невої Рас 100.0 мл.
Кадмію еталонний розчин (10 ррт). 5000701.
Еталонний розчин кадмію (0.1 % Ссі) Р розводять 1 %
(об/об) розчином кислоти хлористоводневої Р у 100
разів безпосередньо перед використанням.
Калію еталонний розчин (100 ррт К). 5002400.
0.446 г калію сульфату Р, у перерахунку на К28О4. роз-
чиняють у води Р і доводять об’єм розчину тим самим
розчинником до 100.0 мл.
Розчин розводять водою Ру 20 разів безпосередньо пе-
ред використанням.
Калію еталонний розчин (20 ррт К). 5002401.
Еталонний розчин калію (100 ррт К) Р розводять во-
дою Ру 5 разів безпосередньо перед використанням.
Кальцію еталонний розчин (400 ррт Са). 5000800.
1.000 г кальцію карбонату Р, у перерахунку на СаСО3,
розчиняють у 23 мл / М розчину кислоти хлористовод-
невої і доводять об’єм розчину водою дистильованою Р
до 100.0 мл.
Розчин розводять водою дистильованою Р у 10 разів
безпосередньо перед використанням.
Кальцію еталонний розчин (100 ррт Са). 5000801.
1.624 г кальцію карбонату Р, у перерахунку на СаСО3,
розчиняють у 3 мл кислоти оцтової Р і доводять об’єм
розчину водою дистильованою Рао 250.0 мл.
Розчин розводять водою дистильованою Ру 10 разів
безпосередньо перед використанням.
Кальцію еталонний розчин (100 ррт Са) РІ. 5000804.
2.769 г кальцію хлориду безводного Р, у перерахунку на
СаС12, розчиняють у кислоті хлористоводневій розве-
деній Р і доводять об’єм розчину тим самим розчинни-
ком до 1000.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням
Кальцію еталонний розчин спиртовий (100 ррт Са).
5000802.
2.50 г кальцію карбонату Р, у перерахунку на СаСО3,
розчиняють у 12 мл кислоти оцтової Р і доводять
об’єм розчину водою дистильованою Рао 1000 0 мл.
Розчин розводять 96 % спиртом Ру 10 разів безпосе-
редньо перед використанням.
Кальцію еталонний розчин (10 ррт Са). 5000803.
0.624 г кальцію крбонату Р, у перерахунку на СаСО3,
розчиняють у 3 мл кислоти оцтової Р і доводять об’єм
розчину водою дистильованою Рао 250.0 мл.
Розчин розводять у 100 разів водою дистильованою Р
безпосередньо перед використанням.
Магнію еталонний розчин (100 ррт М§). 5001800.
1.010 г магнію сульфату Р. у перерахунку на
М§8О4,7Н2О. розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 100.0 мл.
Розчин розводять водою Р у 10 разів безпосередньо
перед використанням.
Магнію еталонний розчин (10 ррт М§). 5001801.
Еталонний розчин магнію (100ррт М§) Ррозводять во-
дою Ру 10 разів безпосередньо перед використанням.
Магнію еталонний розчин (10 ррт М§) РІ. 5001802.
8.365 г магнію хлориду Р розчиняють у кислоті хлорис-
товодневій розведеній Р і доводять об’єм розчину тим
самим розчинником до 1000.0 мл.
Розчин розводять водою Р у 100 разів безпосередньо
перед використанням.
Міді еталонний розчин (0.1 % Си). 5001100.
0.393 г міді(П) сульфату Р, у перерахунку на
Си8О4,5Н2О, розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 100.0 мл.
Міді еталонний розчин (10 ррт Сн). 5001101.
Еталонний розчин міді (0.1 % Си) Р розводять водою Р
у 100 разів безпосередньо перед використанням.
Міді еталонний розчин (0.1 ррт Си). 5001102.
Еталонний розчин міді (10ррт Си) Р розводять водою Р
у 100 разів безпосередньо перед використанням.
Натрію еталонний розчин (200 ррт N3). 5002700.
0.509 г натрію хлориду Р. у перерахунку на ІМаСІ, роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 100.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Натрію еталонний розчин (50 ррт N3). 5002701.
Еталонний розчин натрію (200ррт N0) Р розводять во-
дою Ру 4 рази.
Нікелю еталонний розчин (10 ррт N1). 5002000.
4.78 г нікелю сульфату Р. у перерахунку на
Мі$О4,7Н2О, розчиняють у воді Рідоводять об’ємроз-
чину тим самим розчинником до 1000.0 мл
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
Нікелю еталонний розчин (0.2 ррт N1). 5002002.
Еталонний розчин нікелю (10ррт N1) Р розводять во-
дою Р у 50 разів безпосередньо перед використан-
ням.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
281
4.1.2. Еталонні розчини для випробувань на граничний вміст домішок
Нікелю еталонний розчин (0.1 ррт Мі). 5002001.
Еталонний розчин нікелю (10 ррт N1) Р розводять во-
дою Ру 100 разів безпосередньо перед використанням.
Нітрату еталонний розчин (100 ррпі МО3). 5002100.
0.815 г калію нітрату Р, у перерахунку на КМО3, роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 500.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Нітрату еталонний розчин (10 ррт МО3). 500210!.
Еталонний розчин нітрату (100 ррт N0^) Р розводять
водою Ру 10 разів безпосередньо перед використан-
ням.
Ні трату еталонний розчин (2 ррт МО3). 5002102.
Еталонний розчин нітрату (10 ррт N0^) Р розводять
водою Ру 5 разів безпосередньо перед використанням.
Олова еталонний розчин (5 ррт 8п). 5003100.
0.500 г олова Р, у перерахунку на 8п, розчиняють у
суміші 5 мл води Рі 25 мл кислоти хлористоводневої Р,
доводять об’єм розчину водою Рпо 1000.0 мл.
Розчин розводять розчином 2.5 % (об/об) кислоти
хлористоводневої Р у 100 разів безпосередньо перед
використанням.
Олова еталонний розчин (0.1 ррт 8п). 5003101.
Еталонний розчин олова (5 ррт 5>п) Р розводять во-
дою Ру 50 разів безпосередньо перед використанням.
Паладію еталонний розчин (500 ррт Рті). 5003600.
50.0 мг паладію Р розчиняють у 9 мл кислоти хлорис-
товодневої Р і доводять об’єм розчину водою Р до
100.0 мл.
Паладію еталонний розчин (20 ррт Реї). 5003602.
0.333 г паладію хлориду Р розчиняють у 2 мл теплої
кислоти хлористоводневої Р і доводять об’єм розчину
сумішшю рівних об’ємів кислоти хлористоводневої
розведеної Рі води Рцо 1000.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Паладію еталонний розчин (0.5 ррт Р<1). 5003601.
Еталонний розчин паладію (500 ррт Реї) Р розводять
сумішшю 0.3 об’єму кислоти азотної Р і 99.7 об’єму
води Р.
Платини еталонний розчин (ЗО ррт Рі). 5002300
80 мг кислоти хлорплатинової Ррозчиняють в 1 Мроз-
чині кислоти хлористоводневої та доводять об’єм роз-
чину тим самим розчинником до 100.0 мл.
Розчин розводять 1 М розчином кислоти хлористовод-
невоїу 10 разів безпосередньо перед використанням.
Ртуті еталонний розчин (1000 ррт Н§). 5001900.
1.354 г ртуті(П) хлориду Р, у перерахунку на Н§С І
розчиняють у 50 мл кислоти азотної розведеної І
водять об’єм розчину водою Рдо 1000.0 мл.
Свинцю еталонний розчин (0.1 % РЬ). 5001700.
0.400 г свинцю(П) нітрату Р, у перерахунку 11
РЬ(І\ІО3)2, розчиняють у воді Р і доводять тим самії" І
розчинником до об’єму 250.0 мл.
Свинцю еталонний розчин (100 ррт РЬ). 5001701.
Еталонний розчин свинцю (0.1% РЬ) Р розводять во-1
дою Р у 10 разів безпосередньо перед використанням. І
Свинцю еталонний розчин (10 ррт РЬ). 5001702.
Еталонний розчин свинцю (100 ррт РЬ) Р розводять во-
дою Ру 10 разів безпосередньо перед використанням.
Свинцю еталонний розчин (10 ррт РЬ) РІ. 5001706.
0.160 г свинцю) 11) нітрату Р розчиняють у 100 мл во-
ди Р, додають 1 мл кислоти азотної, вільної від свин-
цю, Р і доводять об’єм розчину водою Рао 1000.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Свинцю еталонний розчин (2 ррт РЬ). 5001703.
Еталонний розчин свинцю (10 ррт РЬ) Р розводять во-
дою Ру 5 разів безпосередньо перед використанням.
Свинцю еталонний розчин (1 ррт РЬ). 5001704.
Еталонний розчин свинцю (10 ррт РЬ) Р розводять во-
дою Ру 10 разів безпосередньо перед використанням.
Свинцю еталонний розчин (0.1 ррт РЬ). 5001705.
Еталонний розчин свинцю (1 ррт РЬ) Р розводять во-
дою Ру 10 разів безпосередньо перед використанням.
Селену еталонний розчин (100 ррт 8е). 5002500.
0.100 г селену Р розчиняють у 2 мл кислоти азотноїР,
випарюють насухо. Залишок розчиняють у 2 мл води Р
і випарюють насухо; цю операцію повторюють тричі
Залишок розчиняють у 50 мл кислоти хлористоводне-
вої розведеної Р і доводять об’єм розчину тією самою
кислотою до 1000.0 мл.
Селену еталонний розчин (1 ррт 8е). 5002501.
6.54 мг кислоти селенистої Р. у перерахунку на
Н28еО3, розчиняють у води Р і доводять об’єм розчину
тим самим розчинником до 100.0 мл.
Розчин розводять водою Ру 40 разів безпосередньо пе-
ред використанням.
Срібла еталонний розчин (5 ррт А§). 5002600.
0.790 г срібла нітрату Р, у перерахунку на А§МО3, роз-
чиняють у води Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
282
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.2. Еталонні розчини для випробувань на граничний вміст домішок
Стронцію еталонний розчин (1.0 % 8г). 5003900.
1.6849 г стронцію карбонату Р. у перерахунку на
$гСО3, покривають водою Р; додають кислоти хлорис-
товодневої Рдо розчинення і припинення бульбашок
газу, фільтрують і розводять водою Рдо 100.0 мл.
Сульфату еталонний розчин (100 ррпі 8О4). 5002802.
0.181 г калію сульфату Р. у перерахунку на К28О4, роз-
чиняють у воді дистильованій Р і доводять об’єм
розчину тим самим розчинником до 100.0 мл.
Розчин розводять водою дистильованою Р у 10 разів
безпосередньо перед використанням.
Сульфату еталонний розчин (10 ррт 8О4). 5002800.
0.181 г калію сульфату Р, у перерахунку на К2$О4, роз-
чиняють у воді дистильованій Р і доводять об’єм
розчину тим самим розчинником до 100.0 мл.
Розчин розводять водою дистильованою Ру 100 разів
безпосередньо перед використанням.
Сульфату еталонний розчин (10 ррт 8О4) РІ. 5002801.
0.181 г калію сульфату Р, у перерахунку на К2$О4, роз-
чиняють у спирті (ЗО %, об/об) Р і доводять об’єм роз-
чину тим самим розчинником до 100.0 мл.
Розчин розводять спиртом (ЗО %, об/об) Ру 100 разів
безпосередньо перед використанням.
Сульфіту еталонний розчин (1.5 ррт 8О2). 5002900.
0.152 г натрію метабісульфіту Р, у перерахунку на
№282О5, розчиняють у воді Рідоводять об’єм розчину
тим самим розчинником до 100.0 мл.
5 0 мл одержаного розчину доводять водою Рдо об’єму
100 мл (розчин 1). До 3 мл розчину 1 долають 4.0 мл
0.1 Мрозчину натрію гідроксиду і доводять об’єм роз-
чину водою Рао 100.0 мл.
Сурми еталонний розчин (1 ррт 5Ь). 5000400.
0.274 г антимоніду калію тартрату Р, у перерахунку на
С4Н4КО78Ь,1/2Н2О, розчиняють у 20 мл кислоти хло-
ристоводневої РІ і доводять прозорий розчин водою Р
до об’єму 100.0 мл. До 10.0 мл одержаного розчину до-
дають 200 мл кислоти хлористоводневої РІ і дово-
дять об’єм розчину водою Р до 1000.0 мл (розчин 1).
До 100.0 мл розчину 1 додають 300 мл кислоти хло-
ристоводневої РІ і доводять об’єм розчину водою Р
до 1000.0 мл.
Розведені розчини готують безпосередньо перед вико-
ристанням.
Талію еталонний розчин (10 ррт ТІ). 5003000.
0.1235 г талію сульфату Р, у перерахунку на Т12$О4,
розчиняють у розчині 9 г/л натрію хлориду Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл. 10.0 мл одержаного розчину доводять роз-
чином 9 г/л натрію хлориду Рдо об’єму 100.0 мл.
Титану еталонний розчин (100 ррт Ті). 5003200.
100.0 мг титану Р розчиняють, якщо необхідно, при
нагріванні у 100 мл кислоти хлористоводневої Р. роз-
веденої водою Рдо об’єму 150 мл; дають охолонути й
доводять водою Рдо об’єму 1000 мл
Фероціаніду еталонний розчин (100 ррт Ее(СМ)6).
5001200.
0.20 г калію фероціаніду Р, у перерахунку на
К4Ее(СМ)6,ЗН2О, розчиняють у воді Р, доводять об’єм
розчину тим самим розчинником до 100 0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Фериціаніду еталонний розчин (50 ррт Ге(СМ)6).
5001300.
0.78 г калію фериціаніду Р, у перерахунку на
К3Ее(СМ)6, розчиняють у воді Р, доводять об’єм роз-
чину тим самим розчинником до 100.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
Фториду еталонний розчин (10 ррт Г). 5001400.
0.442 г натрію фториду Р, у перерахунку на ІЧаЕ, попе-
редньо висушеного при температурі 300 °С протягом
12 год, розчиняють у воді Р і доводять об’єм розчину
тим самим розчинником до 1000.0 мл (1 мл = 0.2 мг Е).
Зберігають у поліетиленовому контейнері.
Розчин розводять водою Ру 20 разів безпосередньо пе-
ред використанням.
Фториду еталонний розчин (1 ррт Г). 5001401.
Еталонний розчин фториду (10 ррт Е) Р розводять во-
дою Ру 10 разів безпосередньо перед використанням.
Формальдегіду еталонний розчин (5 ррт СН2О).
5001500.
1.0 г розчину формальдегіду Р, у перерахунку на СН2О,
розводять водою Рдо 1 л.
Розчин розводять водою Р у 200 разів безпосередньо
перед використанням.
Фосфату еталонний розчин (200 ррт РО4). 5004200.
0.286 г калію дигідрофосфату Р, у перерахунку на
КН2РО4, розчиняють у воді Р, доводять об’єм розчину
тим самим розчинником до 1000.0 мл.
Фосфату еталонний розчин (5 ррт РО4). 5002200.
0.716 г калію дигідрофосфату Р, у перерахунку на
КН2РО4, розчиняють у воді Р, доводять об’єм розчину
тим самим розчинником до 1000.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
Хлориду еталонний розчин (8 ррт СІ). 5000900.
1.32 г натрію хлориду Р, у перерахунку на ІМаСІ, розчи-
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
Розчин розводять водою Ру 100 разів безпосередньо
перед використанням.
ЛРРЖАІЗНА ФАРМАКОПЕЯ УКРАЇНИ 2001
283
4.1.3. Буферні розчини
Хлориду еталонний розчин (5 ррт СІ). 5000901.
0.824 г натрію хлориду Р, в перерахунку на ІМаСІ, роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
Розчин розводять водою Р у 100 разів безпосередньо
перед використанням.
Хрому еталонний розчин (0.1 % Сг). 5001002.
2.83 г калію дихромату Р, в перерахунку на К2Сг2О7,
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 1000.0 мл.
Хрому еталонний розчин (100 ррт Сг). 5001000.
0.283 г калію дихромату Р, у перерахунку на К2Сг2О7,
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 1000.0 мл.
Хрому еталонний розчин (0.1 ррт Сг). 5001001.
Еталонний розчин хрому (100 ррт Сг) Р розводять во-
дою Ру 1000 разів безпосередньо перед використанням.
Цинку еталонний розчин (5 мг/мл Хп). 5003400.
3.15 г цинку оксиду Р розчиняють у 15 мл кислоти хло-
ристоводневої Рі доводять об’єм розчину водою Рдо
500.0 мл.
Цинку еталонний розчин (100 ррт Хп). 5003401.
0.440 г цинку сульфату Р, у перерахунку на
7п$О4,7Н2О, розчиняють в 1 мл кислоти оцтової Р і
доводять об’єм розчину водою Рдо 100.0 мл.
Розчин розводять водою Ру 10 разів безпосередньо пе-
ред використанням.
Цинку еталонний розчин (10 ррт Хп). 5003402.
Еталонний розчин цинку (100 ррт Хп) Р розводять у 10
разів водою Рбезпосередньо перед використанням.
Цинку еталонний розчин (5 ррт Хп). 5003403.
Еталонний розчин цинку (100 ррт Хп) Р розводять во-
дою Ру 20 разів безпосередньо перед використанням.
Цирконію еталонний розчин (1 г/л Хг). 5003500.
0.293 г цирконілу нітрату Р, у перерахунку на
7гО(НО3)2,2Н2О, розчиняють у суміші 2 об’ємів кис-
лоти хлористоводневої Р і 8 об’ємів води Р і доводять
об’єм розчину тією самою сумішшю розчинників до
100.0 мл.
4.1.3. БУФЕРНІ РОЗЧИНИ
Забуферений ацетоновий розчин. 4000100.
8.15 г натрію ацетату Р і 42 г натрію хлориду Р розчи-
няють у воді Р, додають 68 мл 0.1 М розчину кислоти
хлористоводневої, 150 мл ацетону Р і доводять об’єм
розчину водою Рдо 500 мл.
Буферний розчин рН 2.0. 4000200.
6.57 г калію хлориду Р розчиняють у воді Р, додають І
119.0 мл 0.1М розчину кислоти хлористоводневої тань
водять об’єм розчину водою Рдо 1000.0 мл.
Фосфатний буферний розчин рН 2.0. 4007900.
8.95 г динатрію гідрофосфату Р і 3.40 г калію дигідро
фосфату Р розчиняють у воді Р, доводять об’єм розчи-
ну тим самим розчинником до 1000.0 мл. Якщо не-
обхідно, доводять рН (2.2.3) кислотою фосфорною Р.
Сульфатний буферний розчин рН 2.0. 4008900.
132.1 г амонію сульфату Р розчиняють у воді Р, дово-
дять об'єм розчину тим самим розчинником до
500.0 мл (розчин І).
Обережно при постійному охолодженні й перемішу-
ванні 14 мл кислоти сірчаної Р додають до приблизно
400 мл води Р; охолоджують і доводять об’єм розчину
водою Рдо 500.0 мл (розчин II).
Змішують рівні об’єми розчинів І і II. Якщо не-
обхідно, доводять рН (2.2.3).
Буферний розчин рН 2.5. 4000300.
100 г калію дигідрофосфату Р розчиняють у 800 мл во-
ди Р, встановлюють рН (2.2.3) 2.5 з допомогою кисло-
ти хлористоводневої Р і доводять об’єм розчину во-
дою Рдо 1000.0 мл.
Буферний розчин рН 2.5 РІ. 4000400.
До 4.9 г кислоти фосфорної розведеної Рдодають 250 мп
води Р, встановлюють рН (2.2.3) з допомогою розчину
натрію гідроксиду розведеного Р і доводять об’єм роз-
чину водою Рдо 500.0 мл.
Буферний розчин рН 3.0. 4008000.
21.0 г кислоти лимонної Ррозчиняють у 200 мл 1Мроз-
чину натрію гідроксиду й доводять об’єм розчину во-
дою Рдо 1000 мл.
40.3 мл одержаного розчину доводять 0.1 М розчином
кислоти хлористоводневої до об’ єму 100.0 мл.
Фосфатний буферний розчин рН 3.0. 4000500.
0.7 мл кислоти фосфорної Р змішують із 100 мл води Р
і доводять об’єм розчину тим самим розчинником до
900 мл. Встановлюють рН (2.2.3) 3.0 з допомогою роз-
чину натрію гідроксиду концентрованого Р і доводять
об’єм розчину водою Рдо 1000 мл.
Фосфатний буферний розчин рН 3.0 РІ. 4010000.
3.40 г калію дигідрофосфату Р розчиняють у 900 мл во-
ди Р. Встановлюють рН (2.2.3) 3.0 з допомогою кисю-
ти фосфорної Р і доводять об’єм розчину водою Р до
1000.0 мл.
Фосфатний буферний розчин рН 3.2. 4008100.
До 900 мл розчину 4 г/л натрію дигідрофосфату Рдо-
дають 100 мл розчину 2.5 г/л кислоти фосфорної Р. Як-
що необхідно, доводять рН (2.2.3).
284
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.3. Буферні розчини
Фосфатний буферний розчин рН 3.2 РІ. 4008500.
Встановлюють рН (2.2.3) 3.2 розчину 35.8 г/л ди-
натрію гідрофосфату Р з допомогою кислоти фосфор-
ної розведеної Р.
100.0 мл розчину доводять водою Рдо об’єму 2000.0 мл.
Буферний розчин рН 3.5. 4000600.
25.0 г амонію ацетату Р розчиняють у 25 мл води Р,
додають 38.0 мл кислоти хлористоводневої РІ. Якщо
необхідно, встановлюють рН (2.2.3) з допомогою кис-
лоти хлористоводневої розведеної Р або розчину аміаку
розведеного Р і доводять об’єм розчину водою Р до
100.0 мл.
Фосфатний буферний розчин рН 3.5. 4000700.
68.0 г калію дигідрофосфату Р розчиняють у воді Р, дово-
дять об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлюють рН (2.2.3) кислотою фосфорною Р.
Буферний розчин рН 3.6. 4000800.
До 250.0 мл 0.2 Мрозчину калію гідрофталату Рдода-
ють 11.94 мл 0.2 Мрозчину кислоти хлористоводневої і
доводять об’єм розчину водою Рдо 1000.0 мл.
Буферний розчин рН 3.7. 4000900.
До 15.0 мл кислоти оцтової Р додають 60 мл 96 %
спирту Рі 20 мл води Р; встановлюють рН (2.2.3) 3.7 з
допомогою розчину аміаку Рі доводять об’єм розчину
водою Рдо 100.0 мл.
Забуферений розчин міді сульфату рН 4.0. 4001000.
0.25 сміді(ІІ) сульфату Рі 4.5 г амонію ацетату Р роз-
чиняють у кислоті оцтовій розведеній Р і доводять
об’єм розчину тим самим розчинником до 100.0 мл.
Ацетатний буферний розчин рН 4.4. 4001100.
136 г натрію ацетату Р і 77 г амонію ацетату Р роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл, потім додають 250.0 мл
кислоти оцтової льодяної Р і перемішують.
Фталатний буферний розчин рН 4.4. 4001200.
2.042 г калію гідрофталату Р розчиняють у 50 мл во-
ди Р, додають 7.5 мл 0.2 Мрозчину натрію гідроксиду й
доводять об’єм розчину водою Рдо 200.0 мл.
0.05 М фосфатний буферний розчин рН 4.5. 4009000.
6.80 г калію дигідрофосфату Р розчиняють у 1000.0 мл
води Р.
рН (2.2.3) розчину має становити 4.5.
Ацетатний буферний розчин рН 4.5. 4010100.
63 г натрію ацетату безводного Р розчиняють у воді Р.
додають 90 мл кислоти оцтової Р, встановлюють рН
(2.2.3) 4.5 і доводять об’єм розчину водою Рдо 1000 мл.
Ацетатний буферний розчин рН 4.6. 4001400.
5.4 г натрію ацетату Р розчиняють у 50 мл води Р, до-
дають 2.4 г кислоти оцтової льодяної Р, доводять во-
дою Р до об’єму 100.0 мл. Якщо необхідно, доводять
рН (2.2.3).
Сукцинатний буферний розчин рН 4.6. 4001500.
11.8 г кислоти бурштинової Р розчиняють у суміші
600 мл води Р і 82 мл 1 М розчину натрію гідроксиду й
доводять об’єм розчину водою Рдо 1000.0 мл.
Ацетатний буферний розчин рН 4.7. 4001600.
136.1 г натрію ацетату Р розчиняють у 500 мл води Р.
250 мл одержаного розчину змішують із 250 мл кислоти
оцтової розведеної Р. Струшують двічі зі свіжопригото-
ваним, відфільтрованим розчином 0.1 г/л дитизону Ру
хлороформі Р. Струшують із вуглецю тетрахлоридом Р
до знебарвлення екстракту. Водний шар фільтрують для
видалення слідів вуглецю тетрахлориду.
Буферний (ацетатний) розчин рН 5.0. 4009100.
До 120 мл розчину 6 г/л кислоти оцтовоїльодяної Рдо-
дають 100 мл 0.1 М розчину калію гідроксиду і приблиз-
но 250 мл води Р, перемішують. Встановлюють рН 5.0
з допомогою розчину 6 г/л кислоти оцтової Р або
0.1 Мрозчину калію гідроксиду й доводять об’єм одер-
жаного розчину водою Рдо 1000.0 мл.
Буферний розчин рН 5.2. 4001700.
1.02 г калію гідрофталату Р розчиняють у 30.0 мл
0.1 Мрозчину натрію гідроксиду й доводять об’єм роз-
чину водою Рдо 100.0 мл.
Буферний розчин рН 5.5. 4001800.
54.4 г натрію ацетату Р розчиняють у 50 мл води Р,
якщо необхідно, нагрівають до температури 35 °С.
Після охолодження повільно додають 10 мл кислоти
оцтової безводної Р, перемішують і доводять об’єм
розчину водою Рдо 100.0 мл.
Ацетатно-едетатний буферний розчин рН 5.5. 4001900.
250 г амонію ацетату Рі 15 г натрію едетату Ррозчи-
няють у 400 мл води Рі додають 125 мл кислоти оцто-
вої льодяної Р.
Фосфатний буферний розчин рН 5.5. 4002000.
Розчині. 13.61 г калію дигідрофосфату Р розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 1000.0 мл.
Розчин 11. 35.81 г динатрію гідрофосфату Р розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 1000.0 мл.
Змішують 96.4 мл розчину І і 3.6 мл розчину II.
Фосфатно-цитратний буферний розчин рН 5.5. 4008700.
Змішують 56.85 мл розчину 28.4 г/л динатрію гідрофо-
сфату безводного Рі 43.15 мл розчину 21 г/л кислоти
лимонної Р.
Фосфатний буферний розчин рН 5.8. 4002100.
1.19г динатрію гідрофосфату дигідрату Рі 8.25 г калію
дигідрофосфату Ррозчиняють у воді Рі доводять об’єм
розчину тим самим розчинником до 1000.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
285
4.1.3. Буферні розчини
Ацетатний буферний розчин рН 6.0. 4002200.
100 г амонію ацетату Р розчиняють у 300 мл води Р,
додають 4.1 мл кислоти оцтової льодяної Р. Якщо не-
обхідно, встановлюють рН (2.2.3) з допомогою розчи-
ну аміаку Р або кислоти оцтової Р і доводять об’єм
розчину водою Рдо 500.0 мл.
Діетиламонію фосфату буферний розчин рН 6.0.
4002300.
68 мл кислоти фосфорної Лдоводять водою Рдо об’єму
500 мл. До 25 мл одержаного розчину додають 450 мл
води Рі 6 мл діетиламіну Р, якщо необхідно, встанов-
люють рН (2.2.3) 6±0.05 з допомогою діетиламіну Р
або кислоти фосфорної Р і доводять об’єм розчину во-
дою Рдо 500.0 мл.
Фосфатний буферний розчин рН 6.0. 4002400.
Змішують 63.2 мл розчину 71.5 г/л динатрію гідрофос-
фату Р і 36.8 мл розчину 21 г/л кислоти лимонної Р.
Фосфатний буферний розчин рН 6.0 РІ. 4002500.
6.8 г натрію дигідрофосфату Р розчиняють у воді Р і
доводять об’єм розчину водою Р до 1000.0 мл. Дово-
дять рН (2.2.3) розчином натрію гідроксиду концентро-
ваним Р.
Фосфатний буферний розчин рН 6.0 Р2. 4002600.
До 250.0 мл 0.2 Мрозчину калію дигідрофосфату Рдо-
дають 28.5 мл 0.2 М розчину натрію гідроксиду й дово-
дять об’єм розчину водою Рдо 1000.0 мл.
Фосфатний буферний розчин рН 6.4. 4002700.
1.79 г динатрію гідрофосфату Р, 1.36 г калію дигідро-
фосфату Р і 7.02 г натрію хлориду Р розчиняють у
воді Р і доводять об'єм розчину тим самим розчинни-
ком до 1000.0 мл.
Фосфатний буферний розчин рН 6.4 РІ. 4002800.
2.5 г динатрію гідрофосфату Р, 2.5 г натрію дигідро-
фосфату Р і 8.2 г натрію хлориду Р розчиняють у
950 мл води Р. Якщо необхідно, встановлюють рН
(2.2.3) 6.4 з допомогою І М розчину натрію гідроксиду
або 7 М розчину кислоти хлористоводневої й доводять
об’єм розчину водою Рдо 1000.0 мл.
0.5 М фталатний буферний розчин рН 6.4. 4009200.
100 г калію гідрофталату Ррозчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл. Якщо необхідно, доводять рН (2.2.3) розчи-
ном натрію гідроксиду концентрованим Р.
Буферний розчин рН 6.5. 4002900.
60.5 г динатрію гідрофосфату Р і 46 г калію дигідро-
фосфату Р розчиняють у воді Р, додають 100 мл
0.02 Мрозчину натрію едетату, 20 мг ртуті(П) хлори-
ду Р і доводять об’єм розчину водою Рдо 1000.0 мл.
Імідазольний буферний розчин рН 6.5. 4003000.
6.81 г імідазолу Рі 1.23 г магнію сульфату Р розчиня-
ють у 752 мл 0.1 М розчину кислоти хлористоводневої.
Якщо необхідно, встановлюють рН (2.2.3) і доводять!
об’єм розчину водою Рдо 1000.0 мл.
Фосфатний буферний розчин рН 6.6. 4003100.
До 250.0 мл 0.2 Мрозчину калію дигідрофосфату Р до-
дають 89.0 мл 0.2 М розчину натрію гідроксиду і дово-
дять об’єм розчину водою Рдо 1000.0 мл.
Фосфатний забуферений фізіологічний розчин рН 6.8.
4003200.
1.0 г калію дигідрофосфату Р, 2.0 г дикалію гідрофосфа-1
ту Р і 8.5 г натрію хлориду Р розчиняють у 900 мл во-
ди Р. Якщо необхідно, встановлюють рН (2.2.3) і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл.
Фосфатний буферний розчин рН 6.8. 4003300.
Зміщують 77.3 мл розчину 71.5 г/л динатрію гідрофос-
фату Р і 22.7 мл розчину 21 г/л кислоти лимонної Р.
Фосфатний буферний розчин рН 6.8 РІ. 4003400.
До 51.0 мл розчину 27.2 г/л калію дигідрофосфату Р
додають 49.0 мл розчину 71.6 г/л динатрію гідрофос-
фату Р, якщо необхідно, доводять рН (2.2.3).
Зберігають при температурі від 2 °С до 8 °С.
1 М трис-гідрохлориду буферний розчин рН 6.8.
4009300.
60.6 г трис(гідроксиметил)амінометану Р розчиняють
у 400 мл води Р, встановлюють рН (2.2.3) з допомогою
кислоти хлористоводневої Р і доводять об’єм розчину
водою Рдо 500.0 мл.
Буферний розчин рН 7.0. 4003500.
До 1000 мл розчину, який містить 18 г/л динатрію
гідрофосфату Р і 23 г/л натрію хлориду Р, додають для
встановлення рН (2.2.3) достатню кількість (близько
280 мл) розчину, що містить 7.8 г/л натрію дигідро-
фосфату Р і 23 г/л натрію хлориду Р. Розчиняють в
одержаному розчині необхідну кількість натрію азиду Р
до одержання розчину 0.2 г/л.
Малеатний буферний розчин рН 7.0. 4003600.
10.0 г натрію хлориду Р, 6.06 г трис(гідроксиме-
тил)амінометану Р і 4.90 г малеїнового ангідриду Рроз-
чиняють у 900 мл води Р. Встановлюють рН (2.2.3) з
допомогою розчину 170 г/л натрію гідроксиду Лі дово-
дять об’єм розчину водою Рдо 1000.0 мл.
Зберігають при температурі від 2 °С до 8 °С.
Термін придатності 3 доби.
Фосфатний буферний розчин рН 7.0. 4003700.
Змішують 82.4 мл розчину 71.5 г/л динатрію гідрофос-
фату Р із 17.6 мл розчину 21 г/л кислоти лимонної Р.
0.025 М фосфатний буферний розчин рН 7.0. 4009400.
Змішують 1 об’єм 0.063 М фосфатного буферного роз-
чину рН 7.0 Різ 1.5 об’ємами води Р.
286
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.3. Буферні розчини
0.063 М фосфатний буферний розчин рН 7.0. 4009500.
5.18 г динатрію гідрофосфату безводного Р і 3.65 г
натрію дигідрофосфату моногідрату Р розчиняють у
950 мл води Р, встановлюють рН (2.2.3) з допомогою
кислоти фосфорної Р і доводять об’єм розчину водою Р
до 1000.0 мл.
0.067 М фосфатний буферний розчин рН 7.0. 4003800.
Розчин /. 0.908 г калію дигідрофосфату Ррозчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 100.0 мл.
Розчин II. 2.38 г динатрію гідрофосфату Ррозчиняють
уводі Рі доводять об’єм розчину тим самим розчинни-
ком до 100.0 мл.
Змішують 38.9 мл розчину І і 61.1 мл розчину II та, як-
що необхідно, доводять рН (2.2.3).
0.1 М фосфатний буферний розчин рН 7.0. 4008200.
1.361 г калію дигідрофосфату Р розчиняють у воді Р і
доводять об’єм розчину тим самим розчинником до
100.0 мл. Доводять рН (2.2.3) розчином 35 г/л ди-
натрію гідрофосфату Р.
0.03 М фосфатний буферний розчин рН 7.0. 4010300.
5.2 г дикалію гідрофосфату Р розчиняють у 900 мл во-
ди для хроматографії Р. Встановлюють рН 7.0±0.1 з
допомогою кислоти фосфорної Р і доводять об’єм роз-
чину водою для хроматографії Рдо 1000 мл.
Фосфатний буферний розчин рН 7.0 РІ. 4003900.
Змішують 250.0 мл 0.2 М розчину калію дигідрофосфа-
ту Рі 148.2 мл розчину 8 г/л натрію гідроксиду Р, як-
шо необхідно, встановлюють рН (2.2.3) і доводять
об’єм розчину водою Рао 1000.0 мл.
Фосфатний буферний розчин рН 7.0 Р2. 4004000.
Змішують 50.0 мл розчину 136 г/л калію дигідрофосфа-
ту Рі 29.5 мл 1 М розчину натрію гідроксиду, доводять
об’єм розчину водою Рао 100.0 мл і встановлюють рН
(2.2.3) 7.0±0.1.
Фосфатний буферний розчин рН 7.0 РЗ. 4008600.
5 г калію дигідрофосфату Р і 11 г дикалію гідрофосфа-
ту Р розчиняють у 900 мл води Р. Встановлюють рН
(2 2.3) 7.0 з допомогою кислоти фосфорної розведеної Р
або розчину натрію гідроксиду розведеного Р, доводять
об'єм розчину водою Рао 1000 мл і перемішують.
Фосфатний буферний розчин рН 7.0 Р4. 4010200.
28.4 г динатрію гідрофосфату безводного Р і 18.2 г
копіюдигідрофосфату Ррозчиняютьу воді Р і доводять,
об’єм розчину тим самим розчинником до 500 мл.
Буферний розчин рН 7.2. 4004100.
До 250.0 мл 0.2 Мрозчину калію дигідрофосфату Р до-
дають 175.0 мл 0.2 Мрозчину натрію гідроксиду. Дово-
дять об’єм розчину водою Рдо 1000.0 мл та, якщо не-
обхідно, доводять рН (2.2.3).
Фосфатний буферний розчин рН 7.2. 4004200.
Змішують 87.0 мл розчину 71.5 г/л динатрію гідрофос-
фату Р і 13.0 мл розчину 21 г/л кислоти лимонної Р.
Забуферений сольовий розчин рН 7.2. 4004300.
8.0 г натрію хлориду Р, 0.2 г калію хлориду Р, 0.1 г
кальцію хлориду безводного Р, 0.1 г магнію хлориду Р,
3.18 г динатрію гідрофосфату Раа 0.2 г калію дигідро-
фосфату Р розчиняють у воді Р і доводять об’єм
розчину водою Рао 1000.0 мл.
Фосфатно-альбуміновий забуферений фізіологічний роз-
чин рН 7.2. 4004400.
10.75 г динатрію гідрофосфату Р, 7.6 г натрію хлори-
ду Р і 10 г альбуміну бичачого Р розчиняють у воді Р і
доводять тим самим розчинником до об’єму 1000.0 мл.
Безпосередньо перед використанням встановлюють
рН (2.2.3) з допомогою розчину натрію гідроксиду роз-
веденого Рабо кислоти фосфорної розведеної Р.
Фосфатно-альбуміновий забуферений фізіологічний роз-
чин рН 7.2 РІ. 4009600.
10.75 г динатрію гідрофосфату Р, 7.6 г натрію хлори-
ду Рі 1 г альбуміну бичачого Ррозчиняють у воді Рі до-
водять тим самим розчинником до об’єму 1000.0 мл.
Безпосередньо перед використанням доводять рН
(2.2.3) розчином натрію гідроксиду розведеним Р або
кислотою фосфорною розведеною Р.
Імідазольний буферний розчин рН 7.3. 4004500.
3.4 г імідазолу Рі 5.8 г натрію хлориду Р розчиняють у
воді Р, додають 18.6 мл / Мрозчину кислоти хлористо-
водневої, доводять об’єм розчину водою Рао 1000.0 мл
та, якщо необхідно, доводять рН (2.2.3).
Буферний розчин рН 7.4. 4004600.
0.6 г калію дигідрофосфату Р, 6.4 г динатрію гідрофо-
сфату Р і 5.85 г натрію хлориду Р розчиняють у
воді Р, доводять об’єм розчину тим самим розчинни-
ком до 1000.0 мл та, якщо необхідно, доводять
рН (2.2.3).
Барбітал-буферний розчин рН 7.4. 4004700.
Змішують 50 мл розчину, який містить 19.44 г/л
натрію ацетату Р і 29.46 г/л барбітал-натрію Р у
воді Р, і 50.5 мл 0.1 М розчину кислоти хлористоводне-
вої, додають 20 мл розчину 85 г/л натрію хлориду Р і
доводять об’єм розчину водою Рао 250 мл.
Фосфатний буферний розчин рН 7.4. 4004800.
До 393.4 мл 0.1 М розчину натрію гідроксиду додають
250.0 мл 0.2 М розчину калію дигідрофосфату Р.
Трис(гідроксиметил)амінометан-натрію хлорид буфер-
ний розчин рН 7.4. 4004900.
6.08 г трис(гідроксиметил)амінометану Р і 8.77 г
натрію хлориду Ррозчиняють у 500 мл води дистильо-
ваної Р, додають 10.0 г альбуміну бичачого Р. Встанов-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
287
4.1.3. Буферні розчини
люють рН (2.2.3) з допомогою кислоти хлористоводне-
вої Р і доводять об’єм розчину водою дистильованою Р
до 1000.0 мл.
Фосфатний забуферений фізіологічний розчин рН 7.4.
4005000.
2.38 г динатрію гідрофосфату Р, 0.19 г калію
дигідрофосфату Р і 8.0 г натрію хлориду Р розчиняють
у воді Р, доводять об’єм розчину тим самим розчин-
ником до 1000.0 мл та, якщо необхідно, доводять
рН (2.2.3).
Боратний буферний розчин рН 7.5. 4005200.
2.5 г натрію хлориду Р, 2.85 г динатрію тетраборату Р
і 10.5 г кислоти борної Р розчиняють у воді Р, доводять
об’єм тим самим розчинником до 1000.0 мл та, якщо
необхідно, доводять рН (2.2.3).
Зберігають при температурі від 2 °С до 8 °С.
Буферний (НЕРЕ8) розчин рН 7.5. 4009700.
2.38 г кислоти 2-[4-(2-гідроксіетил)піперазин-1-
іл]етансульфонової Р розчиняють у приблизно 90 мл
води Р, встановлюють рН 7.5 з допомогою розчину
натрію гідроксиду Р і доводять об’єм розчину водою Р
до 100 мл.
0.33 М фосфатний буферний розчин рН 7.5. 4005300.
Розчин І. 119.31 г динатрію гідрофосфату Р розчиня-
ють у воді Р і доводять об’єм тим самим розчинником
до 1000.0 мл.
Розчин 7/. 45.36 г калію дигідрофосфату Р розчиняють
у воді Рі доводять об’єм розчину тим самим розчинни-
ком до 1000.0 мл
Змішують 85 мл розчину І і 15 мл розчину II та, якщо
необхідно, доводять рН (2.2.3).
0.2 М фосфатний буферний розчин рН 7.5. 4005400.
27.22 г калію дигідрофосфату Р розчиняють у 930 мл
води Р, встановлюють рН (2.2.3) 7.5 з допомогою роз-
чину 300 г/л калію гідроксиду Р і доводять об’єм розчи-
ну водою Рао 1000 0 мл.
Трис(гідроксиметил)амінометан-буферний розчин рН 7.5.
4005500.
7.27 г трис(гідроксиметил)амінометану Р і 5.27 г
натрію хлориду Р розчиняють у воді Р. якщо не-
обхідно, встановлюють рН (2.2.3) і доводять об’єм
розчину водою Рцо 1000.0 мл.
0.05 М трис-гідрохлориду буферний розчин рН 7.5.
4005600.
6.057 г трис(гідроксиметил)амінометану Р розчиня-
ють у воді Р, якщо необхідно, встановлюють рН (2.2.3)
з допомогою кислоти хлористоводневої Р і доводять
об’єм розчину водою Рао 1000.0 мл.
Натрію цитрату буферний розчин рН 7.8 (0.034 М роз-
чин натрію цитрату і 0.101 М розчин натрію хлориду).
4009800.
10.0г натрію цитрату Р і 5.90 г натрію хлориду Р роз- І
чиняють у 900 мл води Р, встановлюють рН (2.2.3) з І
допомогою кислоти хлористоводневої Р і доводять І
об’єм розчину водою Рао 1000 мл.
Буферний розчин рН 8.0. 4005900.
До 50.0 мл 0.2 Мрозчину калію дигідрофосфату Рпола-
ють 46.8 мл 0.2 Мрозчину натрію гідроксиду і доізодять
об’єм розчину водою Рао 200.0 мл.
Буферний розчин рН 8.0 РІ. 4010400.
20 г дикалію гідрофосфату /'розчиняють у 900 мл води Р.
встановлюють рН (2.2.3) з допомогою кислоти фосфор-
ної Р і доводять об’єм розчину водою Рао 1000 мл.
0.0015 М боратний буферний розчин рН 8.0. 4006000.
0.572 г динатрію тетраборату Рі 2.94 г кальцію хюри-
ду Р розчиняють у 800 мл води Р. Встановлюють рН
(2.2.3) з допомогою 7 Мрозчину кислоти хлориспювод-
невоїта доводять об’єм розчину водою Рао 1000.0 мл.
1 М фосфатний буферний розчин рН 8.0. 4007800.
136.1 г калію дигідрофосфату Р розчиняють у воді Р,
встановлюють рН (2.2.3) з допомогою 1 М розчину
натрію гідроксиду і доводять об’єм розчину водою Рдо
1000.0 мл.
0.1 М фосфатний буферний розчин рН 8.0. 4008400.
0.523 г калію дигідрофосфіату Р і 16.73 г дикалію гідро-
фосфату Р розчиняють у воді Р і доводять об’єм роз-
чину тим самим розчинником до 1000.0 мл.
0.02 М фосфатний буферний розчин рН 8.0. 4006100.
До 50.0 мл 0.2 М розчину калію дигідрофосфіату 7'дода-
ють 46.8 мл 0.2 Мрозчину натрію гідроксиду й доводять
об’єм розчину водою Рао 500.0 мл.
Трис(гідроксиметил)амінометан-буферний розчин рН 8.1.
4006200.
0.294 г кальцію хлориду Р розчиняють у 40 мл розчину
трис(гідроксиметил)амінометану Р встановлюють
рН (2.2.3) з допомогою 1 Мрозчину кислоти хлористо-
водневої і доводять об’єм розчину водою Рао 100.0 мл.
Трис-гліцин-буферний розчин рН 8.3. 4006300.
6.0 г трис(гідроксиметил)амінометану Р і 28.8 г гліци-
ну Р розчиняють у воді Р і доводять об’єм розчину тим
самим розчинником до 1000.0 мл.
Безпосередньо перед використанням до 1 об’єму дода-
ють 10 об’ємів води Р.
Барбітал-буферний розчин рН 8.4. 4006400.
8.25 г барбітал-натрію Р розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл.
Трис-ЕБТА-В8А буферний розчин рН 8.4. 4006500.
6.1 г трис(гідроксиметил)амінометану Р, 2.8 г натрію
едетату Р, 10.2 г натрію хлориду Р і 10 г альбуміну би-
288
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.1.3. Буферні розчини
чачого Ррозчиняють у воді Р, встановлюють рН (2.2.3)
Я.4 з допомогою / Мрозчину кислоти хлористоводневої
таловодять об’єм розчину водою Рдо 1000 0 мл.
Трис(гідроксиметил)амінометан-ЕОТА буферний розчин
рН 8.4. 4006600.
5.12 г натрію хлориду Р, 3.03 г трис(гідроксиме-
тил)амінометану Рі 1.40 г натрію едетату Ррозчиня-
ють у 250 мл води дистильованої Р, встановлюють рН
(2.2.3) 8.4 з допомогою кислоти хлористоводневої Р і
доводять об’єм розчину водою дистильованою Р до
500.0 мл.
Трис-ацетатний буферний розчин рН 8.5. 4006700.
0.294 г кальцію хлориду Р і 12.11 г трис(гідроксиме-
тил)амінометану Р розчиняють у воді Р, встановлю-
ють рН (2.2.3) з допомогою кислоти оцтової Р і дово-
дять об’єм розчину водою Рдо 1000.0 мл.
Барбітал-буферний розчин рН 8.6 РІ. 4006900.
1.38 г барбіталу Р, 8.76 г барбітал-натрію Р і 0.38 г
кальцію лактату Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
1.5 М трис-гідрохлориду буферний розчин рН 8.8.
4009900.
90.8 г трис(гідроксиметил)амінометану Р розчиняють
у 400 мл води Р, встановлюють рН (2.2.3) з допомогою
кислоти хлористоводневої Р і доводять об’єм розчину
водою Рдо 500.0 мл.
Буферний (фосфатний) розчин рН 9.0. 4008300.
1.74 г калію дигідрофосфату Ррозчиняють у 80 мл во-
ди Р, якшо необхідно, встановлюють рН (2.2.3) з допо-
могою / М розчину калію гідроксиду й доводять об’єм
розчину водою Рдо 100.0 мл.
Буферний розчин рН 9.0. 4007000.
Розчин 1. 6.18 і кислоти борної Р розчиняють у 0.1 М
розчині калію хлориду Р і доводять об’єм розчину тим
самим розчинником до 1000.0 мл.
Розчин 11. 0.1 М розчин натрію гідроксиду.
Змішують 1000.0 мл розчину І і 420.0 мл розчину II.
Буферний розчин рН 9.0 РІ. 4007100.
6.20 кислоти борної Р розчиняють у 500 мл води Р,
встановлюють рН (2.2.3) з допомогою 1 М розчину
натрію гідроксиду (близько 41.5 мл) і доводять об’єм
розчину водою Рдо 1000.0 мл.
Аміачний буферний розчин рН 9.5. 4007200.
33.5 г амонію хлориду Ррозчиняють у 150 мл води Р, до-
дають 42.0 мл розчину аміаку концентрованого Р і до-
водять об’єм розчину водою Рдо 250.0 мл.
Зберігають у поліетиленовому контейнері.
Аміачний буферний розчин рН 10.0. 4007300.
5.4 г амонію хлориду Р розчиняють у 20 мл води Р, до-
дають 35.0 мл розчину аміаку Р і доводять об’єм розчи-
ну водою Рдо 100.0 мл.
Діетаноламін-буферний розчин рН 10.0. 4007500.
96 4 г діетаноламіну Р розчиняють у воді Р, доводять
об’єм розчину тим самим розчинником до 400 мл, до-
дають 0.5 мл розчину 186 г/л магнію хлориду Р, вста-
новлюють рН (2.2.3) з допомогою / Мрозчину кислоти
хлористоводневої ії доводять об’єм розчину водою Рдо
500.0 мл.
Буферний розчин рН 10.9. 4007600.
6.75 г амонію хлориду Р розчиняють у розчині аміаку Р
і доводять об’єм розчину тим самим розчинником до
100.0 мл.
Буфер для регулювання іонної сили. 4007700.
58.5 г натрію хлориду Р, 57.0 мл кислоти оцтової льо-
дяної Р, 61.5 г натрію ацетату Р і 5.0 г циклогексилен-
динітрилтетраоцтової кислоти Ррозчиняютьу воді Р
і доводять об’єм розчину тим самим розчинником до
500.0 мл. Встановлюють рН до 5.0-5.5 з допомогою
розчину 335 г/л натрію гідроксиду Р і доводять об’єм
розчину водою дистильованою Рдо 1000.0 мл.
Буфер для регулювання іонної сили РІ. 4008800.
Розчин (а). 210 г кислоти лимонної Р розчиняють у
400 мл води дистильованої Р, встановлюють рН (2.2.3)
7.0 з допомогою розчину аміаку концентрованого Р і
доводять об’єм розчину водою дистильованою Р до
1000.0 мл.
Розчин (Ь). 132 г амонію фосфату Р розчиняють у воді
дистильованій Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
Розчин (с). До суспензії 292 г (етилендинітрил)тетра-
оцтової кислоти Р приблизно у 500 мл води дистильо-
ваної Рдодають близько 200 мл розчину аміаку концен-
трованого Рдо розчинення. Встановлюють рН (2.2.3)
до 6-7 з допомогою розчину аміаку концентрованого Рі
доводять об’єм розчину водою дистильованою Р до
1000.0 мл.
Змішують рівні об’єми розчинів (а), (Ь) і (с) та дово-
дять рН до 7.5 розчином аміаку концентрованим Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
289
4.2. Реактиви, титровані розчини для об’ємного аналізу
4.2. РЕАКТИВИ, ТИТРОВАНІ
РОЗЧИНИ ДЛЯ ОБ’ЄМНОГО
АНАЛІЗУ
4.2.1. ВИХІДНІ СТАНДАРТНІ РЕЧОВИНИ ДЛЯ
ТИТРОВАНИХ РОЗЧИНІВ
Вихідні стандартні речовини для встановлення титру
титрованих розчинів позначають буквами РО і готу-
ють таким чином.
Арсену(ПІ) оксид. Аб2О3. (М.м. 197.8). 2000100.
[1327-53-3]
Арсену(ПІ) оксид Рсублімують.
Зберігають над силікагелем безводним Р.
Калію бромат. КВгО3. (М.м. 167.0). 2000300. [7758-01-2].
Калію бромат Р перекристалізовують із киплячої во-
ди Р. Кристали збирають і сушать до постійної маси
при температурі 180 °С.
Калію гідрофталат. С8Н5КО4. (М.м. 204.2). 2000400.
[877-24-7].
Калію гідрофталат Рперекристалізовують із киплячої
води Р. Кристали збирають при температурі вище
35 °С і сушать до постійної маси при температурі
ПО °С.
Кислота бензойна. С7Н6О2. (М.м. 122.1). 2000200.
[65-85-0].
Кислоту бензойну Рсублімують.
Натрію карбонат. 1\Іа2СО3. (М.м. 106.0). 2000500.
[497-19-81.
Насичений розчин натрію карбонату Р фільтрують
при кімнатній температурі. Крізь фільтрат повільно
пропускають потік вуглецю діоксиду Рпри постійному
охолодженні й перемішуванні Через 2 год осад збира-
ють на скляному фільтрі, промивають фільтр льодя-
ною водою Р, насиченою вуглецю діоксидом. Сушать
при температурі від 100 °С до 105 °С і прожарюють до
постійної маси при температурі від 270 °С до 300 °С,
періодично перемішуючи.
Натрію хлорид. ІМаСІ. (М.м. 58 44) 2000600. [7647-14-5].
До 1 об’єму насиченого розчину натрію хлориду Р до-
дають 2 об’єми кислоти хлористоводневої Р. Одержані
кристали збирають і промивають кислотою хлористо-
водневою РІ, яку видаляють нагріванням на водяній
бані. Прожарюють до постійної маси при температурі
300 °С.
Сульфанілова кислота. С6Н7!\ІО38. (М.м. 173.2).
2000700. [121-57-3].
Кислоту сульфанілову Р перекристалізовують із кипля-
чої води Р, фільтрують і сушать до постійної маси при
температурі від 100 °С до 105 °С.
Цинк. 7п. (А.м. 65.4). 2000800. [7440-66-6].
Використовують цинк із вмістом не менше 99.9 % 2п. І
4.2.2. ТИТРОВАНІ РОЗЧИНИ
Титровані розчини мають готуватися відповідно до
звичайних вимог хімічного аналізу. Перевіряють
точність використовуваного обладнання для того,
щоб пересвідчитись у його придатності для передбачу-
ваного використання.
Концентрацію титрованих розчинів виражають їх по-
лярністю. Молярність виражає кількість речовини,
розчиненої в 1 л розчину, у вигляді КІЛЬКОСТІ молів.
Розчин, який містить х молей речовини у одному
літрі, позначають лМ розчином.
Концентрація титрованих розчинів не має різнитися
від зазначеної більш як на 10 %. Молярність титрова-
них розчинів визначають із точністю 0.2 %.
Титровані розчини стандартизують описаними нижче
методами. Якщо титрований розчин використовують
у кількісному аналізі, в якому кінцеву точку визнача-
ють електрохімічним методом (приміром, амперо-
метрії або потенціометрії), то розчин стандартизують
тим самим методом. Склад середовища, в якому стан-
дартизують титрований розчин, має бути таким са-
мим, як і той, у якому розчин буде використано.
Розчини, більше розведені за описані нижче,
одержують розведенням останніх водою, вільною від
вуглецю діоксиду, Р. Поправкові коефіцієнти одержа-
них розчинів такі самі, як і у вихідних розчинів. Роз-
чини з молярністю нижче 0.1 М готують безпосеред-
ньо перед використанням
1 М розчин азотної кислоти. 3003600.
96.6 г кислоти азотної Р доводять водою Р до об’єму
1000.0 мл.
Встановлення титру. 1 000 г натрію карбонату РО
розчиняють у 50 мл води Р, додають 0.1 мл розчину ме-
тилового оранжевого Р '\ титрують приготованим роз-
чином кислоти азотної до появи червонувато-жовтого
забарвлення розчину; кип’ятять протягом 2 хв, розчин
знову набуває жовтого забарвлення, охолоджують і
продовжують титрування до появи червонувато-жов-
того забарвлення.
1 мл 1 М розчину кислоти азотної відповідає 53.00 мг
ІЧа2СО3.
0.1 М розчин амонію тіоціанату. 3000500.
7.612 г амонію тіоціанату Ррозчиняють у воді Рі до-
водять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. До 20.0 мл 0.1 М розчину срібла
нітрату додають 25 мл води Р, 2 мл кислоти азотної
розведеної Р, 2 мл розчину заліза(ПІ) амонію сульфа-
ту Р2 Ї титрують приготованим розчином амонію тіо-
ціанату до появи червонувато-жовтого забарвлення.
290
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.2. Реактиви, титровані розчини для об’ємного аналізу
0.1 М розчин амонію церію нітрату. 3000100.
Розчин, який містить 56 мл кислоти сірчаної Р і 54.82 г
амонію церію(ІУ) нітрату Р, збовтують протягом 2 хв,
додають послідовно п’ять порцій води Р по 100 мл
кожна, перемішуючи після кожного додавання. Дово-
дять об’єм прозорого розчину водою Р до 1000.0 мл.
Титр одержаного розчину встановлюють через 10 діб.
Встановлення титру. 80.0 мг арсену(ПІ) оксиду РО
розчиняють при обережному нагріванні у 15 мл
02 М розчину натрію гідроксиду. До прозорого розчи-
ну додають 50 мл кислоти сірчаної розведеної Р, 0.15 мл
розчину 2.5 г/л осмію(УІІІ) оксиду Ру кислоті сірчаній
розведеній Лі 0.1 мл фероїну Р; одержаний розчин тит-
рують приготованим розчином амонію церію нітрату
до зникнення червоного забарвлення. Поблизу точки
еквівалентності титрують повільно.
1 мл 0.1 М розчину амонію церію нітрату відповідає
4 946 мг Аз2О3.
Зберігають у захищеному від світла місці.
0.01 М розчин амонію церію нітрату. 3000200.
До 100.0 мл 0.1 М розчину амонію церію нітрату дода-
ють при охолодженні 30 мл кислоти сірчаної Р і дово-
дять об’єм розчину водою Р до 1000.0 мл.
0.1 М розчин амонію церію сульфату. 3000300.
65.0 г амонію(ІУ) церію сульфату Р розчиняють у
суміші 500 мл води Р'\ 30 мл кислоти сірчаної Р; охоло-
джують і доводять об’єм розчину водою Рдо 1000.0 мл.
Встановлення титру. 80.0 мг арсену(Ш) оксиду РО
розчиняють при обережному нагріванні у 15 мл
0.2 М розчину натрію гідроксиду. До одержаного про-
зорого розчину додають 50 мл кислоти сірчаної розве-
деної Р, 0.15 мл розчину 2.5 г/л осмію(УІІІ) оксиду Ру
кислоті сірчаній розведеній Р, 0.1 мл фероїну Р і титру-
ють приготованим розчином амонію церію сульфату
до зникнення червоного забарвлення. Поблизу точки
еквівалентності титрують повільно.
1 мл 0.1 М розчину амонію церію сульфату відповідає
4.946 мг Ак2О3.
0.01 М розчин амонію церію сульфату. 3000400.
До 100.0 млО.ІМ розчину амонію церію сульфату до.да-
ють при охолодженні 30 мл кислоти сірчаної Р і дово-
дять об’єм розчину водою Р до 1000.0 мл.
0.1 М розчин барію хлориду. 3000600.
24.4 г барію хлориду Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлення титру. До 10.0 мл приготованого роз-
чину барію хлориду додають 60 мл води Р, 3 мл розчи-
ну аміаку концентрованого Р, 0.5-1 мг фталеїнового
пурпурового Р і титрують 0.1 М розчином натрію еде-
тату. Коли забарвлення розчину почне слабшати, до-
дають 50 мл 96 % спирту Р і продовжують титрування
до зникнення синювато-фіолетового забарвлення.
0.05 М розчин барію перхлорату. 3000700.
15.8 г барію гідроксиду Р розчиняють у суміші 75 мл во-
ди Р \7 5 мл кислоти хлорної Р, доводять рН розчину
до 3 кислотою хлорною Р і фільтрують, якщо не-
обхідно Додають 150 мл 96 % спирту Р. розводять во-
дою Р до об’єму 250 мл і доводять об’єм розчину бу-
ферним розчином рН 3.7 Рдо 1000.0 мл.
Встановлення титру. До 5.0 мл 0.05 М розчину кисло-
ти сірчаної додають 5 мл води Р, 50 мл буферного роз-
чину рН 3.7 Р, 0.5 мл розчину алізарину 5 Р і титрують
приготованим розчином барію перхлорату до появи
оранжево-червоного забарвлення
Титр установлюють безпосередньо перед використан-
ням.
0.025 М розчин барію перхлорату. 3009600.
500.0 мл 0.05 М розчину барію перхлорату доводять бу-
ферним розчином рН 3.7 Рдо об’єму 1000.0 мл.
0.004 М розчин бензетонію хлориду. 3000900.
1.792 г бензетонію хлориду Р, попередньо висушеного
до постійної маси при температурі від 100 °С до 105 °С,
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 1000.0 мл.
Встановлення титру. Обчислюють молярність розчи-
ну, виходячи із вмісту С27Н42С1НО2 у висушеному бен
зетонію хлориді, визначеного таким чином. 0.350 г ви-
сушеної речовини розчиняють у 30 мл кислоти оцтової
безводної Р, додають 6 мл розчину ртуті(ІІ) ацетату Р
і титрують 0.1 Мрозчином кислоти хлорної, використо-
вуючи як індикатор 0.05 мл розчину кристалічного
фіолетового Р. Паралельно проводять контрольний
дослід.
І мл 0.1 М розчину кислоти хлорної відповідає 44.81 мг
С27 Н 42С1N О2.
0.0167 М розчин бромід-бромату. 3001000.
2.7835 г калію бромату РО і 13 г калію броміду Ррозчи-
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
0.1 М розчин заліза амонію сульфату. 3001300.
50.0 г заліза(Ш) амонію сульфату Р розчиняють у
суміші 6 мл кислоти сірчаної Р і 300 мл води Р і дово-
дять об’єм розчину водою Рдо 1000.0 мл.
Встановлення титру. До 25.0 мл приготованого роз-
чину заліза амонію сульфату додають 3 мл кислоти
хлористоводневої Р, 2 г калію иодиду Р і через 10 хв ти-
трують 0.1 М розчином натрію тіосульфату, викорис-
товуючи як індикатор 1 мл розчину крохмалю Р.
1 мл 0.1 М розчину натрію тіосульфату відповідає
48.22 мг ЕеМН4(!8О4)2,12Н2О.
0.1 М розчин заліза сульфату. 3001400.
27.80 г заліза(/1) сульфату Р розчиняють у 500 мл кис-
лоти сірчаної розведеної Р і доводять об’єм розчину во-
дою Рдо 1000.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
291
4.2. Реактиви, титровані розчини для об’ємного аналізу
Встановлення титру. До 25.0 мл приготованого роз-
чину заліза сульфату додають 3 мл кислоти фосфор-
ної Р і негайно титрують 0.02 М розчином калію пер-
манганату.
Титр установлюють безпосередньо перед використан-
ням.
0.5 М розчин йоду. 3009400.
127 г йоду Р і 200 г калію йодиду Ррозчиняють у воді Р
і доводять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. 400.0 мг арсену(Ш) оксиду РО
розчиняють у суміші 10 мл розчину натрію гідроксиду
розведеного Р і 10 мл води Р, додають 10 мл кислоти
хлористоводневої розведеної Р. З г натрію гідрокарбо-
нату Рі титрують приготованим розчином йоду, вико-
ристовуючи як індикатор 1 мл розчину крохмалю Р.
1 мл 0.5 М розчину йоду відповідає 49.46 мг Ак2О3.
Зберігають у захищеному від світла місці.
0.05 М розчин йоду. 3002700.
12.7 г йоду Рі 20 г калію йодиду Ррозчиняють у воді Рі
доводять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. 80.0 мг арсену(ІІІ) оксиду РО
розчиняють у суміші 10 мл розчину натрію гідроксиду
розведеного Р і 10 мл води Р, додають 10 мл кислоти
хлористоводневої розведеної Р, 3 г натрію гідрокарбо-
нату Рі титрують приготованим розчином йоду, вико-
ристовуючи як індикатор 1 мл розчину крохмалю Р.
1 мл 0.05 Мрозчину йоду відповідає 4.946 мг Аз2О3.
Зберігають у захищеному від світла місці.
0.01 М розчин йоду. 3002900.
0.3 г калію йодиду Р розчиняють у 20.0 мл 0.05 Мроз-
чину йоду і доводять об’єм розчину водою Рдо 100.0 мл.
0.033 М розчин калію бромату. 3004200.
5.5670 г калію бромату РО розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
0.02 М розчин калію бромату. 3004300.
3.340 г калію бромату РО розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл.
0.0167 М розчин калію бромату. 3004400.
Готують розведенням 0.033 Мрозчину калію бромату.
0.083 М розчин калію бромату. 3004500.
Готують розведенням 0.033 М розчину калію бромату.
1 М розчин калію гідроксиду. 3009100.
60 г калію гідроксиду Р розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 1000.0 мл.
Встановлення титру. 20.0 мл приготованого розчині І
калію гідроксиду титрують / Мрозчином кислоти хло-
ристоводневої, використовуючи як індикатор 0.5 мл '
розчину фенолфталеїну Р.
0.1 М розчин калію гідроксиду. 3004800.
6 г калію гідроксиду Ррозчиняють у воді, вільній від вуг-
лецю діоксиду, Р і доводять об’єм розчину тим самим
розчинником до 1000.0 мл.
Встановлення титру. 20.0 мл приготованого розчину
калію гідроксиду титрують 0.1 М розчином кислоти
хлористоводневої, використовуючи як індикатор
0.5 мл розчину фенолфталеїну Р.
0.5 М розчин калію гідроксиду в спирті (60 %, об/об).
3004900.
З г калію гідроксиду Р розчиняють у спирті (60 %,
об/об), вільному від альдегідів, Р і доводять об’єм роз-
чину тим самим розчинником до 100.0 мл.
Встановлення титру. 20.0 мл приготованого розчину
калію гідроксиду в спирті (60 %, об/об) титрують
0.5 М розчином кислоти хлористоводневої, використо-
вуючи як індикатор 0.5 мл розчину фенолфталеїну Р.
0.5 М розчин калію гідроксиду спиртовий. 3005000.
З г калію гідроксиду Р розчиняють у 5 мл води Р і дово-
дять об’єм розчину 96 % спиртом, вільним від аль-
дегідів, Рдо 100.0 мл.
Встановлення титру. 20.0 мл приготованого розчину
калію гідроксиду спиртового титрують 0.5 Мрозчином
кислоти хлористоводневої, використовуючи як інди-
катор 0.5 мл розчину фенолфталеїну Р.
0.1 М розчин калію гідроксиду спиртовий. 3005100.
20 мл 0.5 М розчину калію гідроксиду спиртового дово-
дять 96 % спиртом, вільним від альдегідів, Р до об’єму
100.0 мл.
0.01 М розчин калію гідроксиду спиртовий. 3009000.
2.0 мл 0.5 Мрозчину калію гідроксиду спиртового дово-
дять 96 % спиртом, вільним від альдегідів, Р до об’єму
100.0 мл.
0.1 М розчин калію гідрофталату. 3004700.
Близько 800 мл кислоти оцтової безводної Р поміща-
ють у конічну колбу, додають 20.42 г калію гідрофтала-
ту РО і нагрівають на водяній бані до розчинення, за-
хищаючи від дії вологи. Охолоджують до температури
20 °С і доводять об’єм розчину кислотою оцтовою без-
водною Рдо 1000.0 мл.
0.0167 М розчин калію дихромату. 3004600.
4.90 г калію дихромату Р розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. До 20.0 мл приготованого роз-
чину калію дихромату додають 1 г калію йодиду Р, 7 мл
кислоти хлористоводневої розведеної Р, 250 мл води Р і
292
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.2. Реактиви, титровані розчини для об’ємного аналізу
титрують 0.1 М розчином натрію тіосульфату до пере-
ходу забарвлення розчину від синього до світло-зеле-
ного, використовуючи як індикатор 3 мл розчину крох-
малю Р.
0.05 М розчин калію йодату. 3005200.
10.70 г калію йодату Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлення титру. 25.0 мл приготованого розчину
калію йодату доводять водою Р до об’єму 100.0 мл. До
20.0 мл одержаного розчину додають 2 г калію йоди-
ду Р. 10 мл кислоти сірчаної розведеної Р і титрують
0.1 М розчином натрію тіосульфату, використовуючи
як індикатор 1 мл розчину крохмалю Р, який додають
наприкінці титрування
0.001 М розчин калію йодиду. 3009200.
10.0 мл розчину 166 г/л калію йодиду Рдоводять водою Р
до об’єму 100.0 мл. 5 мл одержаного розчину доводять
водою Рдо об’єму 500.0 мл.
0.02 М розчин калію перманганату. 3005300.
3.2 г калію перманганату Р розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл; одержаний розчин нагрівають на водяній
бані протягом 1 год, охолоджують і фільтрують крізь
скляний фільтр.
Встановлення титру. До 20.0 мл приготованого роз-
чину калію перманганату додають 2 г калію йодиду Р,
10 мл кислоти сірчаної розведеної Р і титрують
0.1 Мрозчином натрію тіосульфату, використовуючи
як індикатор 1 мл розчину крохмалю Р, який додають
наприкінці титрування.
Титр установлюють безпосередньо перед використан-
ням.
Зберігають у захищеному від світла місці.
0.1 М розчин літію метилату. 3003300.
0.694 г літію Р розчиняють у 150 мл метанолу безвод-
ного Р і доводять об’єм розчину толуолом Р до
1000.0 мл.
Встановлення титру. До 10 мл диметилформаміду Р
додають 0.05 мл розчину 3 г/л тимолового синього Р у
метанолі Р і титрують приготованим розчином літію
метилату до одержання синього забарвлення розчину.
Негайно додають 0.200 г кислоти бензойної РО. пе-
ремішують до розчинення й титрують приготованим
розчином літію метилату до повторного одержання
синього забарвлення розчину. Під час титрування роз-
чин захищають від атмосферного вуглецю діоксиду.
Титр розчину літію метилату встановлюють за об’ємом
титранту, витраченим у повторному титруванні.
Титр установлюють безпосередньо перед використан-
ням
1 мл 0.1 М розчину літію метилату відповідає 12.21 мг
СН6О2.
0.1 М розчин магнію хлориду. 3003400.
20.33 г магнію хлориду Ррозчиняють у воді Рі доводять
об’єм розчину тим самим розчинником до 1000 0 мл.
Встановлення титру. Проводять визначення магнію
методом комплексометрії (2.5.11).
1 М лужний розчин міді-етилендіаміну. 3008700.
Молярне відношення етилендіаміну до міді становить
2.00±0.04.
0.02 М розчин міді сульфату. 3001200.
5.0гміді(П) сульфату Ррозчиняютьу воді Рі доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлення титру. До 20.0 мл приготованого розчи-
ну міді сульфату додають 2 г натрію ацетату Р, 0.1 мл
розчину піридилазонафтолу Р і титрують 0.02 М розчи-
ном натрію едетату до змінення забарвлення від фіо-
летово синього до яскраво-зеленого. Поблизу точки
еквівалентності титрують повільно.
0.1 М розчин натрію арсеніту. 3005800.
4.946 г арсену! 111) оксиду РО, у перерахунку на Аз2О3,
розчиняють у суміші 20 мл розчину натрію гідроксиду
концентрованого Р і 20 мл води Р, доводять об’єм роз-
чину водою Р до 400.0 мл і нейтралізують кислотою
хлористоводневою розведеною Р за лакмусовим папе-
ром Р. Розчиняють в одержаному розчині 2 г натрію
гідрокарбонату Рі доводять об’єм розчину водою Рдо
500.0 мл.
1 М розчин натрію гідроксиду. 3006300.
42 г натрію гідроксиду Р розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 1000.0 мл.
Встановлення титру. 20.0 мл приготованого розчину
натрію гідроксиду титрують 1 М розчином кислоти
хлористоводневої, використовуючи індикатор, зазна-
чений у методиці кількісного визначення з викорис-
танням І М розчину натрію гідроксиду.
Якщо необхідне використання розчину натрію гідро-
ксиду, вільного від альдегідів, його готують таким чи-
ном. Розчиняють натрію гідроксид Ру воді Рдо одер-
жання розчину від 400 г/л до 600 г/л і залишають сто-
яти. Прозору рідину над осадом зливають, захищаючи
від дії вуглецю діоксиду, розводять водою, вільною від
вуглецю діоксиду, Р до необхідної молярності. Розчин
має витримувати таке випробування. Титрують 20.0 мл
розчину кислоти хлористоводневої тієї самої моляр-
ності, що й приготований розчин натрію гідроксиду,
використовуючи як індикатор 0.5 мл розчину фенол-
фталеїну Р. У точці еквівалентності додають невелику
кількість кислоти, необхідну для зникнення рожевого
забарвлення; концентрують розчин до 20 мл кип’я-
тінням; під час кип’ятіння додають кислоту в
кількості, необхідній для зникнення рожевого забарв-
лення, яке не має поновлюватися при тривалому
кип’ятінні. Об’єм витраченої кислоти не має переви-
щувати 0.1 мл.
0.1 М розчин натрію гідроксиду. 3006600.
100.0 мл 1 М розчину натрію гідроксиду доводять во-
дою, вільною від вуглецю діоксиду, Р до об’єму
1000.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
293
4.2. Реактиви, титровані розчини для об’ємного аналізу
Встановлення титру. Проводять титрування, як
зазначено для 1 Мрозчину натрію гідроксиду, викори-
стовуючи 0.1 М розчин кислоти хлористоводневої.
0.1 М розчин натрію гідроксиду етанольний. 3007000.
До 250 мл етанолу Р додають 3.3 г розчину натрію
гідроксиду концентрованого Р.
Встановлення титру. 0.200 г кислоти бензойної РО
розчиняють у 2 мл води Рі 10 мл 96 % спирту Р і тит-
рують приготованим розчином натрію гідроксиду ета-
нольним, використовуючи як індикатор 0.2 мл розчи-
ну тимолфталеїну Р.
Титр установлюють безпосередньо перед використан-
ням.
1 мл 0.1 М розчину натрію гідроксиду етанольного
відповідає 12.21 мгС7Н6О2.
0.1 М розчин натрію едетату. 3005900.
37.5 г натрію едетату Р розчиняють у 500 мл води Р,
додають 100 мл 1 М розчину натрію гідроксиду й дово-
дять об’єм розчину водою Рпо 1000.0 мл.
Встановлення титру. 0.120 г цинку РО розчиняють у
4 мл кислоти хлористоводневої РІ і додають 0.1 мл
бромної води Р; видаляють надлишок брому кип’ятін-
ням, додають розчин натрію гідроксиду розведений Рпо
слабкокислої або нейтральної реакції та проводять
кількісне визначення пинку методом комплексометрії
(2.5.11).
1 мл 0.1 М розчину натрію едетату відповідає 6.54 мг
7п.
Зберігають у поліетиленовому контейнері.
0.02 М розчин натрію едетату. 3006000.
7.444 г натрію едетату Р розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. 0.100 г цинку РО розчиняють у
4 мл кислоти хлористоводневої РІ і додають 0.1 мл
бромної води Р', видаляють надлишок брому кип’ятін-
ням. Переносять розчин у мірну колбу й доводять
об’єм розчину водою Рпо 100.0 мл. 25.0 мл одержано-
го розчину поміщають у конічну колбу місткістю
500 мл і доводять водою Р до об’єму 200 мл. Додають
близько 50 мг індикаторної суміші ксиленолового оран-
жевого Р і гексаметилентетраміну Р до одержання
фіолетово-рожевого забарвлення розчину. Додають 2 г
гексаметилентетраміну Р у надлишку. Титрують при-
готованим розчином натрію едетату до переходу
фіолетово-рожевого забарвлення у жовте.
1 мл 0.02 Мрозчину натрію едетату відповідає 1.308 мг
7п.
0.1 М розчин натрію метилату. 3007100.
175 мл метанолу безводного Р охолоджують у льодяній
воді і додають невеликими порціями близько 2.5 г
щойно нарізаного натрію Р; коли метал розчиниться,
доводять толуолом Рпо об’єму 1000.0 мл.
Встановлення титру. До 10 мл диметилформамід)1 Р
додають 0.05 мл розчину 3 г/л тимолового синього Ру
метанолі Р і титрують приготованим розчином натрію
метилату до одержання синього забарвлення розчину.
Негайно додають 0.200 г кислоти бензойної РО, пе-
ремішують до розчинення й титрують приготованим
розчином натрію метилату до повторного одержання
синього забарвлення розчину. Під час титрування роз-
чин захищають від атмосферного вуглецю діоксиду,
Титр розчину натрію метилату встановлюють за
об’ємом титранту, витраченого у повторному титру- ]
ванні.
Титр установлюють безпосередньо перед використан-
ням.
1 мл 0.1 М розчину натрію метилату відповідає
12.21 мг С7Н6О2.
0.1 М розчин натрію нітриту. 3007200.
7.5 г натрію нітриту Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлення титру. 0.300 г кислоти сульфанілової Р0
розчиняють у 50 мл кислоти хлористоводневої розведе-
ної Р і проводять визначення первинної ароматичної
аміногрупи (2.5.8) електрометрично, використовуючи
приготований розчин натрію нітриту.
Титр установлюють безпосередньо перед використан-
ням.
1 мл 0.1 М розчину натрію нітриту відповідає 17.32 мг
С6Н7МО38.
0.1 М розчин натрію перйодату. 3009500.
21.4г натрію перйодату Р розчиняють у 500 мл води Р
і доводять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. 20.0 мл приготованого розчину
натрію перйодату поміщають у колбу з притертою
пробкою, додають 5 мл кислоти хлорної Р, колбу за-
кривають пробкою й перемішують. Доводять
рН (2.2.3) до 6.4 насиченим розчином натрію гідро-
карбонату Р. Додають 10 мл розчину калію йодиду Р,
закривають пробкою, перемішують, витримують 2 хв і
титрують 0.025 М розчином натрію арсеніту до одер-
жання ледь жовтого забарвлення, потім додають 2 мл
розчину крохмалю Р і титрують до знебарвлення розчи-
ну.
0.1 М розчин натрію тіосульфату. 3007300.
25 г натрію тіосульфату Р і 0.2 г натрію карбонату Р
розчиняють у воді, вільній від вуглецю діоксиду, Р і до-
водять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. До 10.0 мл 0.033 Мрозчину калію
бромату додають 40 мл води Р, 10 мл розчину калію йо-
диду Р, 5 мл кислоти хлористоводневої РІ і титрують
приготованим розчином натрію тіосульфату, викорис-
товуючи як індикатор 1 мл розчину крохмалю Р, який
додають наприкінці титрування.
294
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
4.2. Реактиви, титровані розчини для об’ємного аналізу
0.1 М розчин оцтової кислоти. 3008900.
6.0 г кислоти оцтової льодяної Р доводять водою Р до
об’єму 1000.0 мл.
Встановлення титру. До 25.0 мл приготованого роз-
чину кислоти оцтової додають 0.5 мл розчину фенол-
фталеїну Р\ титрують 0.ІМ розчином натрію гідрокси-
ду
0.02 М розчин ріуті нітрату. 3003500.
6.85 гртуті(ІІ) нітрату Р розчиняють у 20 мл 1Мроз-
чину кислоти азотної та доводять об’єм розчину во-
дою Рао 1000.0 мл.
Встановлення титру. 15.0 мг натрію хлориду РО розчи-
няютьу 50 мл води Рі титрують приготованим розчином
ртуті нітрату потенціометрично (2.2.20), використовую-
чи як індикаторний електрод платиновий або ртутний, а
як електрод порівняння — ртуть-сульфатний.
1 мл 0.02 М розчину ртуті нітрату відповідає 2.338 мг
№С1.
0.5 М розчин сірчаної кислоти. 3007800.
28 мл кислоти сірчаної Р змішують із водою Р і дово-
дять об’єм розчину тим самим розчинником до
1000.0 мл.
Встановлення титру. 1.000 г натрію карбонату РО
розчиняють у 50 мл води Р, додають 0.1 мл розчину ме-
тилового оранжевого Р і титрують приготованим роз-
чином кислоти сірчаної до появи червонувато-жовтого
забарвлення. Кип’ятять близько 2 хв; розчин знову на-
буває жовтого забарвлення, охолоджують і титрують
знову до повторної появи червонувато-жовтого за-
барвлення.
1 мл 0.5 Мрозчину кислоти сірчаної відповідає 53.00 мг
№2СО3.
0.05 М розчин сірчаної кислоти. 3008000.
100.0 мл 0.5 М розчину кислоти сірчаної доводять во-
дою Рдо об’єму 1000.0 мл.
Встановлення титру. Визначення проводять, як за-
значено для 0.5 М розчину кислоти сірчаної, викорис-
товуючи 0.100 г натрію карбонату РО, розчиненого у
20 мл води Р.
1 мл 0.05 Мрозчину кислоти сірчаної відповідає 5.30 мг
Ка2СО3.
0.1 М розчин свинцю нітрату. 3003100.
33 г свинцю нітрату Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлення титру. До 20.0 мл приготованого роз-
чину свинцю нітрату додають 300 мл води Р і прово-
дять визначення свинцю методом комплексометрії
(2.5.11).
0.1 М розчин срібла нітрату. 3005600.
17.0 г срібла нітрату Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлення титру. 0.100 г натрію хлориду РО роз-
чиняють у 30.0 мл води Р і титрують приготованим
розчином срібла нітрату потенціометрично (2.2.20).
1 мл 0.1 М розчину срібла нітрату відповідає 5.844 мг
№С1.
Зберігають у захищеному від світла місці.
0.001 М розчин срібла нітрату. 3009300.
5.0 мл 0.1 М розчину срібла нітрату доводять водою Р
до об’єму 500.0 мл.
0.1 М розчин тетрабутиламонію гідроксиду. 3008300.
40 г тетрабутиламонію йодиду Р розчиняють у 90 мл
метанолу безводного Р, додають 20 г тонко здрібнено-
го срібла оксиду Р і енергійно струшують протягом
1 год. Центрифугують кілька мілілітрів суміші й про-
водять випробування рідини над осадом на йодиди.
При одержанні позитивної реакції додатково додають
2 г срібла оксиду Р і струшують протягом наступних
30 хв; цю процедуру повторюють доти, поки рідина не
буде вільною від йодидів, суміш фільтрують крізь
тонкий скляний фільтр і промивають реакційну посу-
дину й фільтр трьома порціями толуолу Р по 50 мл
кожна. До одержаного фільтрату додають промивний
толуол і доводять толуолом Р до об’єму 1000.0 мл.
Крізь розчин пропускають сухий азот, вільний від вуг-
лецю діоксиду, протягом 5 хв.
Встановлення титру. До 10 мл диметилформаміду Р
додають 0.05 мл розчину 3 г/л тимолового синього Ру
метанолі Р і титрують приготованим розчином тетра-
бутиламонію гідроксиду до одержання чистого си-
нього забарвлення розчину. Негайно додають 0.200 г
кислоти бензойної РО, перемішують до розчинення й
титрують приготованим розчином тетрабутиламонію
гідроксиду знову до одержання чистого синього за-
барвлення розчину. Титр розчину тетрабутиламонію
гідроксиду встановлюють за об’ємом титранту, витра-
ченого у повторному титруванні.
Титр установлюють безпосередньо перед використан-
ням.
1 мл 0.1 М розчину тетрабутиламонію гідроксиду
відповідає 12.21 мгС7Н6О2.
0.1 М розчин тетрабутиламонію гідроксиду у 2-пропа-
нолі. 3008400.
Готують, як зазначено для 0.1 Мрозчину тетрабутил-
амонію гідроксиду, використовуючи як розчинник
2-пропанол Р замість толуолу Р; титр установлюють,
як зазначено для 0.1 М розчину тетрабутиламонію
гідроксиду.
6 М розчин хлористоводневої кислоти. 3001500.
618.0 г кислоти хлористоводневої Р доводять водою Р
до об’єму 1000.0 мл.
З М розчин хлористоводневої кислоти. 3001600.
309.0 г кислоти хлористоводневої Р доводять водою Р
до об’єму 1000.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
295
4.2. Реактиви, титровані розчини для об'ємного аналізу
2 М розчин хлористоводневої кислоти. 3001700.
206.0 г кислоти хлористоводневої Р доводять водою Р
до об’єму 1000.0 мл.
1 М розчин хлористоводневої кислоти. 3001800.
103.0 г кислоти хлористоводневої Р доводять водою Р
до об’єму 1000.0 мл.
Встановлення титру. 1.000 г натрію карбонату РО
розчиняють у 50 мл води Р, додають 0.1 мл розчину ме-
тилового оранжевого Р '\ титрують приготованим роз-
чином кислоти хлористоводневої до появи червонува-
то-жовтого забарвлення. Кип’ятять протягом 2 хв;
розчин знову набуває жовтого забарвлення, охолод-
жують і продовжують титрування до появи червонува-
то-жовтого забарвлення.
1 мл 1 М розчину кислоти хлористоводневої відповідає
53.00 мг №2(203.
0.1 М розчин хлористоводневої кислоти. 3002100.
100.0 мл 1 М розчину кислоти хлористоводневої дово-
дять водою Рао об’єму 1000.0 мл.
Встановлення титру. Проводять титрування, як за-
значено для 1 М розчину кислоти хлористоводневої,
використовуючи 0.100 г натрію карбонату РО, розчи-
неного у 20 мл води Р.
1 мл 0.1 М розчину кислоти хлористоводневої
відповідає 5.30 мг №2СО3.
0.1 М розчин хлористоводневої кислоти у спирті.
3008800.
9.0 мл кислоти хлористоводневої Р доводять
96 % спиртом, вільним від альдегідів, Р до об’єму
1000.0 мл.
0.1 М розчин хлорної кислоти. 3003900.
8.5 мл кислоти хлорної Р поміщають у мірну колбу, яка
містить близько 900 мл кислоти оцтової льодяної Р, і
перемішують. Додають 30 мл оцтового ангідриду Р, до-
водять об’єм розчину кислотою оцтовою льодяною Р
до 1000.0 мл, перемішують і залишають на 24 год.
Визначають вміст води (2.5.12) без додавання метано-
лу та, якщо необхідно, доводять вміст води від 0.1 % до
0.2 % додаванням оцтового ангідриду Р або води Р. За-
лишають на 24 год.
Встановлення титру. 0.350 г калію гідрофталату РО
розчиняють у 50 мл кислоти оцтової безводної Р, як-
що необхідно, обережно нагріваючи. Охолоджують,
захищаючи від повітря, і титрують приготованим
розчином кислоти хлорної, використовуючи як інди-
катор 0.05 мл розчину кристалічного фіолетового Р.
Вимірюють температуру розчину хлорної кислоти під
час титрування. Якщо температура, при якій прово-
диться кількісне визначення, відрізняється від тем-
ператури, при якій було встановлено титр 0.1 Мроз-
чину кислоти хлорної, тоді об’єм (Ус), необхідний для
кількісного визначення, обчислюють за формулою:
Ус = V [1 + (/] - /2)0.00111,
де:
і1 — температура, при якій установлюють титр;
і2 — температура, при якій проводять кількісне
визначення;
V — об’єм, витрачений на титрування фактично, у
мілілітрах.
1 мл 0.1 М розчину кислоти хлорної відповідає 20.42 мг
С8Н5КО4.
0.05 М розчин хлорної кислоти. 3004000.
50.0 мл 0.1 М розчину кислоти хлорної доводять кисло-
тою оцтовою безводною Рао об’єму 100.0 мл.
0.1 М розчин церію сульфату. 3001100.
40.4 г церію(ІР) сульфату Р розчиняють у суміші
500 мл води Р і 50 мл кислоти сірчаної Р; охолоджують
і доводять об’єм розчину водою Рао 1000.0 мл.
Встановлення титру. До 25.0 мл приготованого роз-
чину церію сульфату додають 2.0 г калію йодиду Р.
150 мл води Р і негайно титрують 0.1 М розчином
натрію тіосульфату, використовуючи як індикатор
1 мл розчину крохмалю Р.
0.05 М розчин цинку хлориду. 3008500.
6.82 г цинку хлориду Р, зваженого із відповідними за-
побіжними заходами, розчиняють у воді Р. Якщо не-
обхідно, по краплях додають кислоту хлористоводневу
розведену Р до зникнення опалесценції та доводять
об’єм розчину водою Рао 1000.0 мл.
Встановлення титру. До 20.0 мл приготованого роз-
чину цинку хлориду додають 5 мл кислоти оцтової
розведеної Р і проводять визначення цинку методом
комплексометрії (2.5.11).
0.1 М розчин цинку сульфату. 3008600.
29 г цинку сульфату Р розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 1000.0 мл.
Встановлення титру. До 20.0 мл приготованого роз-
чину цинку сульфату додають 5 мл кислоти оцтової
розведеної Р і проводять визначення цинку методом
комплексометрії (2.5.11).
296
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Загальні тексти
5. ЗАГАЛЬНІ ТЕКСТИ
5.1.1. МЕТОДИ ПРИГОТУВАННЯ СТЕРИЛЬНИХ
ПРОДУКТІВ
Під стерильністю розуміють відсутність життєздатних
мікроорганізмів. Стерильність не може бути гаранто-
вана випробуванням на стерильність; вона має забез-
печуватися належним чином валідованим процесом
виробництва. Важливо, щоб при вивченні впливу ви-
браної процедури стерилізації на продукт (включаючи
контейнер і закупорювальні засоби) було підтвердже-
но її ефективність, а також збереження цілісності про-
дукту. Процедура стерилізації має бути валідована до її
застосування на практиці. Рекомендується вибирати
контейнер, що дає змогу використовувати оптималь-
ний режим стерилізації. Найменше порушення валідо-
ваного процесу стерилізації спричиняє ризик одер-
жання нестерильного або неякісного продукту. По-
вторну валідацію варто проводити щоразу при вне-
сенні змін у процедуру стерилізації, у тому числі при
зміні об’єму продукту, стерилізованого за один раз.
При проектуванні виробничого процесу треба врахо-
вувати принципи належної виробничої практики (ви-
кладені, наприклад, в Інструкції Європейського
Співтовариства з СМР), у тому числі:
— залучення кваліфікованого персоналу, що має
належну підготовку;
— використання відповідних приміщень;
- використання підхожого виробничого обладнан-
ня, яке легко очищається і стерилізується;
- вживання адекватних заходів для зведення до
мінімуму біозабруднень перед стерилізацією;
- використання валідованих процедур на всіх кри-
тичних стадіях виробництва;
моніторинг навколишнього середовища і прове-
дення контролю на проміжних стадіях.
Необхідні заходи для зведення до мінімуму біозабруд-
нень перед стерилізацією включають використання
інгредієнтів з низьким рівнем мікробного забруднен-
ня. Для інгредієнтів, які внаслідок свого походження,
природи або методу приготування зазнають мікробно-
го забруднення, можуть бути рекомендовані
мікробіологічний моніторинг і установлення допусти-
мих меж мікробного забруднення.
Методи, описані нижче, застосовні головним чином для
інактивації або усунення бактерій, дріжджових і
плісеневих грибів. Для продуктів тваринного походжен-
ня або одержаних від людини, а також у тих випадках,
коли такі продукти використовуються в процесі ви-
робництва, при валідації виробничого процесу треба до-
вести, що використовувані методи придатні для інак-
тивації або усунення відповідних вірусних забруднень.
Рекомендації з цих питань наведені, наприклад, у
відповідних "Інструктивних указаннях Європейського
Співтовариства ".
У всіх випадках, коли це можливо, стерилізацію про-
дукту треба проводити у контейнері (кінцева сте-
рилізація). При використанні повністю валідованого
методу кінцевої стерилізації — стерилізації парою, су-
хим жаром або іонізуючим випромінюванням — за уз-
годженням із відповідним компетентним уповноваже-
ним органом допускається параметричний випуск
серії стерильного продукту, при якому висновок про
якість серії приймається на підставі даних про процес
стерилізації, а не результатів випробування на сте-
рильність.
У тих випадках, коли неможливо здійснити кінцеву
стерилізацію, використовують фільтрацію крізь
фільтри, здатні затримувати бактерії, або виробництво
за асептичних умов. По можливості при цьому треба
проводити додаткову обробку продукту (наприклад,
нагрівання) у контейнері. У всіх випадках контейнер і
закупорювальні засоби мають забезпечувати збере-
ження стерильності продукту протягом терміну при-
датності.
СТУПІНЬ НАДІЙНОСТІ СТЕРИЛІЗАЦІЇ (СНС)
У тих випадках, коли це можливо, для методів, наведе-
них нижче, зазначають "ступінь надійності сте-
рилізації" (СНС). Неможливо гарантувати або довес-
ти, що стерильність досягнута для кожної одиниці з
великої кількості одиниць, підданих стерилізації.
Процес інактивації мікроорганізмів фізичними або
хімічними методами описується експоненціальним
законом, отже, завжди існує ймовірність виживання
мікроорганізмів у процесі стерилізації. У конкретному
випадку ця ймовірність визначається кількістю, ви-
дом і стійкістю мікроорганізмів, а також середови-
щем, в якому мікроорганізми знаходяться у процесі
обробки. Під СНС процесу стерилізації розуміють
ступінь надійності, з якою може бути гарантовано сте-
рильність усіх одиниць у серії. Для конкретного про-
цесу СНС визначають як можливість наявності несте-
рильної одиниці у сукупності одиниць, підданих сте-
рилізації. Наприклад, СНС=106 означає, що у
підданій стерилізації серії готового продукту об’ємом
10' одиниць існує ймовірність виявити не більше од-
ного життєздатного мікроорганізму. СНС процесу
стерилізації для конкретного продукту встановлюють
у ході належного процесу валідації.
МЕТОДИ І УМОВИ СТЕРИЛІЗАЦІЇ
Стерилізація може бути проведена одним з методів,
описаних нижче. Допускається модифікувати або
комбінувати ці методи за умови проведення валідації
вибраної процедури стерилізації як щодо її ефектив-
ності, так і щодо збереження цілісності продукту, у то-
му числі контейнера і закупорювальних засобів.
При застосуванні будь-якого з методів стерилізації
треба проводити моніторинг на критичних стадіях
процесу з метою підтвердити, що необхідні умови сте
рилізації забезпечуються для всієї серії протягом усьо-
го процесу стерилізації. Цю вимогу треба виконувати в
усіх випадках, у тому числі при використанні стан-
дартних умов.
ПГРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
297
Загальні тексти
Кінцева стерилізація. При проведенні кінцевої сте-
рилізації треба враховувати неоднорідність фізичних і
хімічних (якшо вони мають відношення до пронесу
стерилізації) умов всередині стерилізаційної камери.
Для кожної конфігурації завантаження контейнерів
певного типу або розміру треба визначити зону сте-
рилізаційної камери, найменш доступну для сте-
рилізуючого агента (наприклад, зону у паровому сте-
рилізаторі з найменшою температурою). Для підтвер-
дження того, що кожне із завантажень буде оброблене
належним чином, треба також визначити можливу
найменшу летальність мікроорганізмів у ході циклу
стерилізації і підтвердити відтворюваність процесу
стерилізації.
Після остаточного вибору режиму кінцевої сте-
рилізації у повсякденній практиці треба при можли-
вості поповнювати інформацію про хід процесу сте-
рилізації шляхом моніторингу і реєстрації підхожим
способом фізичних і хімічних (якщо вони мають
відношення до процесу стерилізації) умов, у яких зна-
ходиться стерилізований матеріал у стерилізаційній
камері у ході кожного циклу стерилізації.
Парова стерилізація (автоклавуваїшя). Найкраще, як-
що можливо, проводити стерилізацію насиченою па-
рою під тиском, особливо при стерилізації готових
лікарських засобів у вигляді водних розчинів. Стан-
дартні умови при такому методі кінцевої стерилізації
готових лікарських засобів у вигляді водних розчинів
такі: прогрівання при температурі не менше 121 °С
протягом 15 хв. Допускається використовувати інші
поєднання часу і температури, якщо достатньо пере-
конливо доведено, що вибраний режим при рутинно-
му застосуванні забезпечує необхідний і відтворюва-
ний рівень летальності мікроорганізмів у рамках уста-
новлених допусків. Використовувані процедури і за-
побіжні заходи мають забезпечувати СНС не більше
10 й. Рекомендації щодо проведення валідації з вико-
ристанням концепції Ео (5.1.5).
У ході процесу стерилізації треба реєструвати фізичні
умови (температуру і тиск) у камері парового сте-
рилізатора. Температуру, як правило, вимірюють за
допомогою термочутливих елементів, які поміщають у
контрольні контейнери. Додаткові термоелементи
поміщають у зоні завантаженої камери з найменшою
температурою (визначеною попередньо). У ході кож-
ного циклу стерилізації треба реєструвати умови сте-
рилізації, наприклад, у вигляді температурних діаграм
або іншим підхожим способом.
Якщо передбачена біологічна оцінка, її проводять з
використанням підхожих біологічних індикаторів
| (5-1.2).
Сухожарова стерилізація. При такому методі кінце-
вої стерилізації стандартними умовами є: прогріван-
ня при температурі не менше 160 °С протягом не
менше 2 год. Допускається використовувати інші
|поєднання часу і температури, якщо достатньо
переконливо доведено, що вибраний режим при
|рутинному застосуванні забезпечує необхідний
і відтворюваний рівень летальності мікроорганіз-
мів у рамках установлених допусків. Використову-
,298
вані процедури і запобіжні заходи мають забез-
печувати СНС не більше 10'.
Сухожарову стерилізацію здійснюють у сухожаровй
шафі, оснащеній пристроєм для примусової цирку-
ляції повітря, або за допомогою іншого обладнання,
спеціально призначеного для цих цілей. Стерилізатор
треба завантажувати таким чином, щоб забезпечити
однорідність температури у межах усього завантажен-
ня. Температуру у стерилізаторі вимірюють, як прави-
ло, за допомогою термочутливих елементів, поміше-
них у контрольні контейнери, і додаткових термоеле-
ментів. розташованих у зоні завантаженого стериліза-
тора з найменшою температурою (визначеною попе-
редньо). У ході кожного циклу стерилізації реєструють
температуру підхожим способом.
Якщо передбачена біологічна оцінка, її проводять з
використанням підхожих біологічних індикаторів
(5.1.2). 1
Сухожарову стерилізацію при температурі більше
220 °С проводять для стерилізації скляного посуду і
усунення пірогенів. У цьому випадку замість викорис-
тання біологічних індикаторів має бути доведене
зменшення кількості термостійких ендотоксинів на
три порядки (5.1.2).
Радіаційна стерилізація. Цей вид стерилізації здійсню-
ють шляхом опромінення продукту іонізуючим ви-
промінюванням. Це може бути або гама-випроміню-
вання, джерелом якого є підхожий радіоактивний ізо-
топ (наприклад, кобальт-60), або пучок електронів,
прискорених за допомогою підхожого прискорювача.
У деяких країнах є інструктивні документи, що регла-
ментують використання іонізуючого випромінювання
для стерилізації, наприклад, відповідні "Інструктивні
указання Європейського Співтовариства".
Для цього методу кінцевої стерилізації стандартна
увібрана доза становить 25 кГр. Можуть бути викорис-
тані інші дози, якшо достатньо переконливо доведе-
но, що вибраний режим при рутинному застосуванні
забезпечує необхідний і відтворюваний рівень леталь-
ності мікроорганізмів у рамках установлених допусків.
Використовувані процедури і запобіжні заходи мають
забезпечувати СНС не більше 10 й.
У процесі стерилізації треба постійно здійснювати
вимірювання увібраного продуктом випромінювання
за допомогою установлених дозиметричних методів,
що не залежать від дози. Дозиметри треба калібрувати
по відношенню до стандартного джерела на еталонній
радіаційній установці безпосередньо після одержання
від постачальника, а потім через певні проміжки часу,
шо не перевищують одного року.
Якщо передбачена біологічна оцінка, її проводять із
використанням підхожих біологічних індикаторів
(5.1.2.).
Газова стерилізація. Цей метод стерилізації допус-
кається використовувати лише у тому випадку, коли
не можуть бути застосовані інші методи. При цьому
має бути забезпечене проникнення газу і вологи у про-
дукт, шо стерилізується, а також подальше видалення
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Загальні тексти
газу способом, що дозволяє знизити концентрацію га-
зу і продуктів його перетворень у продукті, який сте-
рилізується, до рівня, що не викликає токсичних
ефектів при використанні продукту. Спосіб видалення
газу має бути попередньо обгрунтований. Відповідні
рекомендації у випадку використання оксиду етилену
наведені, наприклад, у відповідних "Інструкційних
указаннях Європейського Співтовариства".
Там, де це можливо, при проведенні стерилізації треба
вимірювати і реєструвати концентрацію газу, відносну
вологість, температуру і тривалість процесу. Вимірю-
вання треба проводити у тих зонах, де умови стери-
іізації досягаються найгірше, що встановлюють у ході
валідаиії.
Дія кожного завантаження визначають ефективність
стерилізації з використанням підхожого біологічного
індикатора (5.1.2).
Перед випуском кожної серії треба проводити кон-
троль стерильності на відповідній кількості зразків
(2.6.1).
Фільтрація. Деякі продукти, що не підлягають
кінцевій стерилізації, можуть бути піддані фільтрації
крізь фільтри, придатність яких попередньо доведена
шляхом мікробіологічних випробувань із викори-
станням підхожих тест-мікроорганізмів, напри-
клад, суспензії Раеисіопюпах (іітіпиіа (АТСС 19146,
МС1МВ 11091 або СІР 103020). При випробуванні ре-
комендується використовувати концентрацію мікро-
організмів не менше 10’ КУО/см2 активної поверхні
фільтру. Суспензію мікроорганізмів рекомендується
готувати у триптонно-соєвому бульйоні. Після прохо-
дження крізь фільтр бульйон збирають, дотримую-
чись правила асептики, й інкубують за аеробних умов
при температурі 32 °С. При виробництві таких про-
дуктів вимагаються спеціальні запобіжні заходи. Ор-
ганізація процесу виробництва і стан навколишнього
середовища мають забезпечувати мінімальний ризик
мікробного забруднення, їх треба регулярно піддавати
належному моніторингу. Обладнання, контейнери,
закупорювальні засоби і, по можливості, інгредієнти
треба піддавати належній стерилізації. Рекомен-
дується проводити фільтрацію безпосередньо перед
стадією наповнення контейнерів. Операції, які ідуть
за стадією фільтрації, необхідно проводити за асеп-
тичних умов.
Розчини пропускають крізь мембранні фільтри, здатні
утримувати бактерії, з номінальним розміром пор не
більше 0.22 мкм, допускається використовувати
фільтри інших типів, що не поступаються ефек-
тивністю. Треба здійснювати належні заходи для за-
побігання втратам розчиненої речовини в результаті ад-
сорбції на фільтрі, а також вивільнення утриманих
фільтром забруднень. Треба враховувати рівень біоза-
бруднень до початку фільтрації, пропускну здатність
фільтра, об’єм серії і тривалість фільтрації. Термін ви-
користання фільтра не має перевищувати часу, встанов-
леного при валідації для даного фільтра у поєднанні з
конкретним фільтрованим лікарським засобом.
Цілісність готового до застосування стерилізуючого
фільтра перевіряють до і після використання за допо-
могою випробувань, відповідних типу фільтра і стадії
перевірки, наприклад, випробуванням точки буль-
башки, витримуваного тиску або швидкості дифузії.
У зв’язку з тим, що при проведенні стерилізуючої
фільтрації існують додаткові потенційні фактори ри-
зику у порівнянні з іншими методами стерилізації, ре-
комендується проводити попередню фільтрацію крізь
утримуючий бактерії фільтр у тих випадках, коли
низький рівень біозабруднень не може бути забезпече-
ний іншими засобами.
ВИРОБНИЦТВО ЗА АСЕПТИЧНИХ УМОВ
Метою виробництва за асептичних умов є збереження
стерильності продукту, виготовленого з компонентів,
кожний з яких був попередньо простерилізований од-
ним з методів, описаних вище. Це досягається шляхом
використання умов і обладнання, призначених для за-
побігання мікробному забрудненню. За асептичних
умов можуть здійснюватися такі стадії виробничого
процесу, як наповнення контейнерів і закупорювання,
змішування інгредієнтів з наступним асептичним на-
повненням і асептичним закупорюванням.
Для збереження стерильності інгредієнтів складу і го-
тового лікарського засобу в ході виробничого процесу
особливу увагу треба приділяти:
— стану навколишнього середовища;
— персоналу;
— критичним поверхням;
— стерилізації контейнерів/закупорювальних засобів
і передавальним процедурам;
— граничному термінові зберігання лікарського засо-
бу до наповнення контейнера.
Валідація пронесу включає належну перевірку всього
переліченого вище, а також систематичний контроль
за допомогою імітаційних тестів з використанням жи-
вильного середовища, яке інкубують і досліджують на
наявність мікробного забруднення (тести наповнення
живильними середовищами). На додаток до цього пе-
ред випуском кожної серії будь-якого лікарського за-
собу, простерилізованого методом фільтрації/або ви-
готовленого за асептичних умов, треба проводити ви-
пробування стерильності на відповідній кількості
зразків (2.6.1).
5.1.2. БІОЛОГІЧНІ ІНДИКАТОРИ СТЕРИЛІЗАЦІЇ
Біологічні індикатори — це стандартизовані препара-
ти певних мікроорганізмів, використовувані для
оцінки ефективності процедури стерилізації.
Біологічний індикатор, як правило, являє собою по-
пуляцію спор бактерій, нанесених на інертний носій,
наприклад, смужку фільтрувального паперу, скляну
пластинку або пластикову пробірку. Інокульований
носій ізолюють таким чином, щоб запобігти його по-
шкодженню або забрудненню, але водночас забезпе-
чити контакт стерилізуючого агента з мікроорганізма-
ми. Суспензії спор можуть знаходитися у герметично
закритих ампулах. Біологічні індикатори готують та-
ким чином, щоб існувала можливість їх зберігання за
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
299
І
Загальні тексти
певних умов, при цьому має бути зазначений термін
придатності.
Допускається безпосередньо інокулювати рідкий про-
дукт, який підлягає стерилізації, або рідкий продукт,
аналогічний стерилізованому, мікроорганізмами того
самого виду, який використовується для виробництва
біологічних індикаторів. У цьому випадку має бути до-
ведено, що лікарський засіб не чинить інгібуючої дії
на спори, особливо на їх проростання.
Для біологічного індикатора зазначають такі характе-
ристики: вид бактерій, використовуваних як еталонні
мікроорганізми, номер штаму у вихідній колекції,
число життєздатних спор, що припадають на носій і
величину О. Величина Б — це значення параметра
стерилізації (тривалість або увібрана доза), що забез-
печує зниження числа життєздатних клітин мікроор-
ганізмів до 10 % від їх початкового числа. Величина О
має значення для строго визначених експерименталь-
них умов стерилізації. Біологічний індикатор має
містити тільки зазначені мікроорганізми. Допус-
кається використання біологічних індикаторів, які
містять більше одного виду бактерій на одному носії.
Має бути зазначена інформація про необхідне жи-
вильне середовище і умови інкубації.
Індикатори рекомендується розміщувати у зонах, най-
менш доступних для стерилізуючого агента. Ці зони
визначають емпірично або на підставі попередніх
фізичних вимірювань, якщо такі можливі. Після за-
вершення дії стерилізуючого агента носії спор перено-
сять у живильне середовище, дотримуючись правила
асептики для запобігання мікробному забрудненню у
процесі випробування. Допускається використання
індикаторів, у яких ампула з живильним середовищем
помішена безпосередньо в упаковці, що захищає іно-
кульований носій.
Вибір тест-мікроорганізму для біологічних індика-
торів здійснюють із урахуванням таких вимог:
— стійкість тест-штаму стосовно конкретного ме-
тоду стерилізації має бути велика у порівнянні зі
стійкістю усіх патогенних мікроорганізмів та
мікроорганізмів, які можуть забруднювати продукт;
— тест-штам має бути непатогенним;
— тест-штам має легко культивуватися.
Якщо після інкубації спостерігається ріст підданих
стерилізації еталонних мікроорганізмів, це свідчить
про незадовільно проведену процедуру стерилізації.
1. Парова стерилізація. Біологічні індикатори, призна-
чені для контролю стерилізації парою, рекомен-
дується використовувати при валідації циклів сте-
рилізації. Рекомендується використовувати спори
ВасіІІи$ Меагоікегторкіїиз (наприклад, АТСС 7953,
МСТС 10007, МСІМВ 8157 або СІР 52.81). Число
життєздатних спор має перевищувати 5x10' на носій.
Величина Б при температурі 121 °С має становити
більше 1.5 хв. При обробці біологічного індикатора
парою при температурі (121 ± 1) °С протягом 6 хв має
спостерігатися збереження життєздатних спор, а об-
робка при тій самій температурі протягом 15 хв має
призводити до повної загибелі еталонних тест-мікро-
організмів.
2. Сухожарова стерилізація. Для приготування біо-
логічних індикаторів рекомендуються спори ВасіІІих
зиЬііІіз (наприклад, єаг. пі$ег АТСС 9372, МСІМВ 8058
або СІР 77.18). Число життєздатних спор має переви-
щувати їх 105 на носій. Величина Б при температурі
160 °С становить близько 5-10 хв. Для стерилізації і
депірогенізації скляного обладнання часто викорис-
товують сухий жар при температурі більше 220 °С. У
цьому випадку замість використання біологічних
індикаторів може бути доведене зниження кількості
термостійких бактеріальних ендотоксинів на три по-
рядки.
3. Радіаційна стерилізація. Біологічні індикатори мо-
жуть використовуватися для моніторингу поточних
операцій як додаткова можливість оцінки ефектив-
ності установленої дози випромінювання, особливо у
випадку стерилізації прискореними електронами. Ре-
комендуються спори ВасіІІиз риті/их (наприклад,
АТСС 27.142, МСТС 10327, МСІМВ 10692 або СІР
77.25). Число життєздатних спор має становити
більше 1 х Ю7 спор на носій. Величина Б має станови-
ти більше 1.9 кГр. Потрібно переконатися, що після
опромінення біологічного індикатора дозою 25 кГр
(мінімальна увібрана доза) ріст еталонних мікроор-
ганізмів не спостерігається.
4. Газова стерилізація. Використання біологічних інди-
каторів необхідне при проведенні усіх процедур газо-
вої стерилізації: як при валідації циклів стерилізації,
так і при проведенні рутинних операцій. Число
життєздатних спор має становити більше 5x10' на
носій. У випадку застосування водню пероксиду і кис-
лоти пероцтової рекомендується використову-
вати спори Васіїїиз Меагоікегторкіїиз (наприклад,
АТСС 7953, МСТС 10007, МСІМВ 8157 або СІР 52 81),
при застосуванні оксиду етилену і формальдегіду ре-
комендується використовувати спори ВасіІІиз яіМІів
(наприклад, єаг. пі^ег АТСС 9372, МСІМВ 8058 або
СІР 77.18). Для кожної конкретної процедури мають
бути відомі параметри стійкості тест-мікроорганізму.
Наприклад, при використанні оксиду етилену величи-
на Б складає більше 2 5 хв для циклу стерилізації з
такими параметрами: концентрація оксиду етилену
600 мг/л, температура 54 °С і відносна вологість 60 %.
Потрібно переконатися, що після 60-хвилинного цик-
лу стерилізації із зазначеними параметрами не спос-
терігається ріст еталонних мікроорганізмів, тоді як
після 15-хвилинного циклу при більш низькій темпе-
ратурі (600 мг/л, 30°С, 60 %) життєздатні спори
зберігаються. Біологічний індикатор має дозволяти
виявити недостатню вологість у стерилізаторі й про-
дукті, щоб гарантувати інактивацію зневоднених
мікроорганізмів. Життєздатність спор має зберігатися
при впливі на індикатор оксиду етилену (600 г/л.
54 °С, 60 хв) без зволоження.
300
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Загальні тексти
5.1.3. ЕФЕКТИВНІСТЬ АНТИМІКРОБНИХ
КОНСЕРВАНТІВ
Якшо готовий лікарський засіб сам по собі не має до-
статньої антимікробної активності, до його складу мо-
жуть бути введені антимікробні консерванти, що
особливо важливо для лікарських засобів у вигляді
водних розчинів. Оскільки мікробне забруднення мо-
же викликати інфікування пацієнта або псування го-
тового лікарського засобу, антимікробні консерванти
призначені для запобігання розмноженню мікроор-
ганізмів або обмеження мікробної забрудненості гото-
вого лікарського засобу в процесі зберігання і застосу-
вання, особливо у випадку використання багатолезо-
вих конейнерів. Антимікробні консерванти не мають
використовуватися як альтернатива належній вироб-
ничій практиці.
Ефективність антимікробного консерванта може по-
силюватися або послаблюватися в результаті взаємодії
здіючою речовиною або іншими компонентами гото-
вого лікарського засобу, а також із пакувальними або
закупорювальними матеріалами. Тому протягом тер-
міну придатності треба контролювати антимікробну
активність готового лікарського засобу, що збері-
гається у контейнері, з метою доказу того, що вона не
знижується у процесі зберігання. Для такого контро-
лю можуть бути використані зразки, добуті з контей-
нера безпосередньо перед випробуванням.
На стадії розробки лікарського засобу треба довести,
що антимікробна активність самого лікарського засо-
бу або при необхідності лікарського засобу з добавкою
підхожого консерванта(ів) забезпечує належний за-
хист від небажаних ефектів, які можуть бути результа-
том мікробного забруднення лікарського засобу або
розмноження в ньому мікроорганізмів у процесі
зберігання і використання.
Випробування ефективності антимікробних консер-
вантів може бути проведене, як описано нижче. Дане
випробування не призначене для рутинного контролю.
ВИПРОБУВАННЯ ЕФЕКТИВНОСТІ
АНТИМІКРОБНИХ КОНСЕРВАНТІВ
Для проведення випробування у готовий лікарський
засіб, що знаходиться по можливості у контейнері,
вносять зазначену нижче кількість підхожих тест-
мікроорганізмів і зберігають інокульовані зразки при
зазначеній нижче температурі. Через певні проміжки
часу з інокульованих зразків відбирають проби і ви-
значають у них число мікроорганізмів
Ефективність консервантів у готовому лікарському за-
собі вважають задовільною, якщо за умов проведення
випробування, при зберіганні інокульованих зразків
при заданій температурі, протягом зазначених
проміжків часу спостерігається значне зменшення або
не спостерігається збільшення, залежно від вимог до
готового лікарського засобу, числа мікроорганізмів.
Критерії оцінки, що показують зменшення числа
мікроорганізмів за певний період часу, залежать від
потрібного ступеня захисту готових лікарських засобів
(див. Табл. 5.1.3,-1/2/3).
Тест-мікроорганізми
Рзеисіотопаз аеги%іпо8а АТСС 9027; N01МВ 8626; СІР 82.118.
ЕіарИуІососсиа аигеиз АТСС 6538; N010 10788; N0^16 9518; СІР4.83.
Сапсіісіа аІЬісапя АТСС 10231; NСРЕ3179; ІР 48.72.
А$рег§Ши8 пі$ег АТСС 16404; ІМІ 149007; ІР 1431.83.
При випробуванні використовують монокультури пе-
релічених мікроорганізмів. У разі необхідності вико-
ристовують також мікроорганізми, які можуть являти
ймовірне мікробне забруднення готового лікарського
засобу. Наприклад, при випробуванні усіх готових
лікарських засобів для орального застосування реко-
мендується використовувати ЕхсИегісИіа соїі (АТСС
8739; МСІМВ 8545; СІР 53.126), а при випробуванні
готових лікарських засобів для орального застосуван-
ня, що містять високі концентрації цукру, — Ху§о$ас-
сііаготусез гоихіі (ХС¥С 381: ІР 2021.92).
Приготування інокуляту. При підготовці до проведення
випробування свіжовирощену вихідну культуру кож-
ного із зазначених тест-мікроорганізмів висівають на
поверхню густого живильного середовища В (2.6.12) у
випадку вирощування бактерій або густого живильно-
го середовища С без додавання антибіотиків (2.6.12) у
випадку вирощування грибів. Культури бактерій інку-
бують при температурі від ЗО °С до 35 °С від 18 год до
24 год; культуру С. аІЬісапз інкубують при температурі
від 20 °С до 25 °С протягом 48 год; культуру А. пц>ег —
при температурі від 20 °С до 25 °С протягом одного
тижня або до одержання добре розвинутих спор. Для
досягнення мікроорганізмом його оптимального ста-
ну можуть бути потрібні пересівання, однак рекомен-
дується обмежитися мінімально можливим їх числом.
Для приготування суспензій бактеріальних культур і
культури С. аІЬісапз мікробну масу змивають із по-
верхні живильного середовища стерильною суспенду-
ючою рідиною, що містить 9 г/л натрію хлориду Р і
1 г/л пептону, і переносять у підхожу посудину. Потім
за допомогою тієї самої рідини доводять вміст мікро-
організмів до 108 у мілілітрі. Для приготування сус-
пензії культури А. пірег використовують стерильну су-
спендуючу рідину, що містить 9 г/л натрію хлориду Р і
0.5 г/л полісорбату-80 Р, і за її допомогою доводять
вміст спор до 108 у мілілітрі.
Із кожної суспензії негайно відбирають підхожий зра-
зок і визначають число колонієутворюючих одиниць у
1 мл кожної суспензії методом прямого висівання на
чашки або методом мембранної фільтрації (2.6.12).
Одержане значення править для визначення числа
життєздатних мікроорганізмів в інокуляті і вихідного
числа мікроорганізмів при проведенні випробування.
Інокулят треба використовувати негайно після приго-
тування.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
301
Загальні тексти
МЕТОДИКА
Для підрахунку життєздатних мікроорганізмів у іноку-
льованих зразках використовують те саме густе жи-
вильне середовище, яке було використане для почат-
кового вирощування відповідних мікроорганізмів.
Кожний контейнер із випробовуваним лікарським за-
собом інокулюють суспензією, що містить один з тест-
мікроорганізмів, забезпечуючи мікробне навантажен-
ня від 10' КУО до 10" КУО у мілілітрі або грамі лікарсь-
кого засобу. Об’єм інокуляту має складати не більше
1 % від об’єму зразка. Ретельно перемішують для за-
безпечення рівномірного розподілу мікроорганізмів у
зразку.
Інокульовані зразки лікарського засобу витримують
при температурі від 20 °С до 25 °С у захищеному від
світла місці. Із кожного випробовуваного зразка
відбирають проби (звичайно 1 мл або 1 г) безпосеред-
ньо після інокуляції і через зазначені інтервали часу
(Табл. 5.1.3-1/2/3) визначають число життєздатних
мікроорганізмів методом висівання на чашки або ме-
тодом мембранної фільтрації (2.6.12). Антимікробну
активність готового лікарського засобу треба усунути
шляхом розведення, фільтрації або за допомогою
підхожого інактиватора. При використанні процедури
розведення треба брати до уваги зниження чутливості
методу при визначенні малого числа життєздатних
мікроорганізмів. При використанні інактиватора тре-
ба підтвердити шляхом контрольних дослідів, шо його
присутність не впливає на життєздатність мікроор-
ганізмів. Треба підтвердити придатність методики для
доказу потрібного зниження числа життєздатних
мікроорганізмів.
КРИТЕРІЇ ОЦІНКИ ЕФЕКТИВНОСТІ
АНТИМІКРОБНИХ КОНСЕРВАНТІВ
У Табл. 5.1.3.-1/2/3 подані критерії оцінки анти-
мікробної активності у вигляді логарифма зменшення
числа життєздатних мікроорганізмів.
Таблиця 5.1.3.-1
Парентеральні і офтальмологічні лікарські засоби
зменшення
6 год 24 год 7 діб 14 діб 28 діб
Бактерії А 2 3 - - НВ*
В - 1 3 - НЗ**
Гриби А - - 2 - НЗ
В - - - 1 НЗ
*НВ — мікроорганізми не виявляються;
**НЗ — не спостерігається збільшення числа мікро-
організмів.
Критерій А відповідає рекомендованій ефективності.
Я кщо обгрунтовано, що критерій А не може бути до-
сягнутий, наприклад, із причини підвищеного ризику
несприятливих впливів, лікарський засіб має задо-
вольняти критерій В.
Таблиця 5.1.3.-2
Лікарські засоби для місцевого застосування
зменшення
2 доби 7 діб 14 діб 28 діб
Бактерії А 2 3 - НЗ
В - - 3 НЗ
Гриби А - - 2 НЗ
В - - 1 НЗ
Критерій А відповідає рекомендованій ефективності.
Якшо обгрунтовано, що критерій А не може бути до-
сягнутий, наприклад, із причини підвищеного ризику
несприятливих впливів, лікарський засіб має задо-
вольняти критерій В.
Таблиця 5.1.3.-3
Лікарські засоби для орального застосування
зменшення
14 діб 28 діб
Бактерії 3 НЗ
Гриби 1 НЗ
Наведені критерії відповідають рекомендованій ефек-
тивності.
_________________________________А
ВИПРОБУВАННЯ ЕФЕКТИВНОСТІ
АНТИМІКРОБНИХ КОНСЕРВАНТІВ
Тест-мікроорганізми
При випробуванні ефективності антимікробних кон-
сервантів можуть бути також використані такі штами
тест-мікроорганізмів:
— Замість тест-мікроорганізму Езсіїегісіїіа соїі
АТСС 8739 (МС1МВ 8545; СІР 53.126) може бути
використаний тест-мікроорганізм ЕзсИегісІйа соїі
АТСС 25922.
— Замість тест-мікроорганізму Сапсіісіа аІЬісапх
АТСС 10231(]\ІСРЕ 3179; 1Р 48.72) може бути вико-
ристаний тест-мікроорганізм Сапсіісіа аІЬісат
КІСТС 885-653.
Допускається використання інших штамів мікроор-
ганізмів за узгодженням із компетентним уповнова-
женим органом.
Для вирощування тест-штамів бактерій може бути ви-
користане густе живильне середовище № 1, для виро-
щування тест-штамів грибів — густе живильне середо-
вище № 2 без додавання антибіотиків (2.6. ІЗ).
МЕТОДИКА
Для забезпечення однорідного розподілу мікроор-
ганізмів у зразку допускається попереднє підігрівання
лікарського засобу до температури не більше 45 °С.
302
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Загальні тексти
5.1.4. МІКРОБІОЛОГІЧНА ЧИСТОТА
ЛІКАРСЬКИХ ЗАСОБІВ
Заний розділ носить довідковий і рекомендаційний ха-
рактер і не є обов’язковою частиною Фармакопеї.
При виробництві, пакуванні, зберіганні і розповсюд-
женні готових лікарських засобів мають бути вжиті
відповідні заходи для забезпечення їх мікробіологічної
чистоти. До готових лікарських засобів рекомен-
дується ставити вимоги, наведені нижче.
КАТЕГОРІЯ 1
Готові лікарські засоби, до яких ставляться вимоги що-
до стерильності у відповідних загальних статтях на
іікарські форми та інші готові лікарські засоби, марко-
вані як стерильні.
- Мають витримувати випробування на стерильність
(2.6.1).
КАТЕГОРІЯ 2
Готові лікарські засоби для місцевого застосування і за-
стосування у респіраторному тракті, за винятком
тих, до яких ставляться вимоги щодо стерильності, і
трансдермальні пластирі.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 102 мікроорганізмів (ае-
робних бактерій і грибів сумарно) у грамі,
мілілітрі або на один пластир (включаючи клейкий
шар і основу).
Трансдермальні пластирі: відсутність ентеробак-
терій і деяких інших грамнегативних бактерій на
одному пластирі (включаючи клейкий шар і осно-
ву). Інші готові лікарські засоби: не більше 10і ен-
теробактерій і деяких інших грамнегативних бак-
терій у грамі або мілілітрі (2.6.13).
— Відсутність Рхеидотопаз аепщіпояа в 1 г, 1 мл або на
одному пластирі (включаючи клейкий шар і осно-
ву) (2.6.13).
— Відсутність ЕіарИуІососсих аигеих в 1 г, 1 мл або на
одному пластирі (включаючи клейкий шар і осно-
ву) (2.6.13).
КАТЕГОРІЯ З
А. Готові лікарські засоби для орального застосування і
ректального введення.
- Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше ІО3 бактерій і не більше
102 грибів у грамі або мілілітрі.
— Відсут ність ЕхсйегіМа соїі в 1 габо 1 мл (2.6.13).
В. Готові лікарські засоби для орального застосування,
до складу яких входять субстанції і допоміжні речови-
ни природного (тваринного, рослинного або мінерально-
го) походження, для яких попередня антимікробна об-
робка неможлива і щодо яких компетентний уповнова-
жений орган допускає мікробне забруднення більше 103
життєздатних мікроорганізмів у грамі або мілілітрі.
За винятком рослинних лікарських засобів, що нале-
жать до категорії 4.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 104 бактерій і не більше
102 грибів у грамі або мілілітрі.
— Не більше 102 ентеробактерій і деяких інших грам-
негативних бактерій у грамі або мілілітрі (2.6.13).
— Відсутність Еаїтопеїіа у 10 г або 10 мл (2.6.13).
— Відсутність ЕзсГіегісГііа соїі в 1 габо 1 мл (2.6.13).
— Відсутність ЕїарНуїососсих аигеих в 1 г або 1 мл
(2.6.13).
КАТЕГОРІЯ 4
Лікарські засоби, що складаються тільки з рослинних
компонентів, одного або кількох (цілісних, здрібнених,
розтертих на порошок).
А. Рослинні лікарські засоби, до яких перед вживанням
додають киплячу воду.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 107 бактерій і не більше
105 грибів у грамі або мілілітрі.
— Не більше 102 ЕхсГіегісйіа соїі у грамі або мілілітрі
(2.6.13), із використанням підхожих розведень.
В. Рослинні лікарські засоби, до яких перед вживанням
не додають киплячу воду.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 105 бактерій і не більше
104 грибів у грамі або мілілітрі.
— Не більше 103 ентеробактерій і деяких інших грам-
негативних бактерій у грамі або мілілітрі (2.6.13).
— Відсутність Ехсйегісйіа соїі в 1 г або 1 мл (2.6.13).
— Відсутність Еаїтопеїіа у 10 г або 10 мл (2.6. ІЗ).
_________________________________________________N
У тому випадку, коли виробництво лікарського засобу
не проводиться відповідно до вимог належної виробни-
чої практики (ГІВП, СМР), встановлених у Європей-
ському Співтоваристві, до даного лікарського засобу
мають ставитися такі вимоги щодо мікробіологічної
чистоти.
КАТЕГОРІЯ 1
Готові лікарські засоби, до яких ставляться вимоги що-
до стерильності відповідно до загальних статей на
лікарські форми (парентеральні: для ін’єкцій, інфузій
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
303
Загальні тексти
та ін.; офтальмологічні; для введення у порожнини
тіла, де в нормальному стані відсутні мікроорганізми),
й інші готові лікарські засоби, марковані як стерильні.
— Мають витримувати випробування на стерильність
(2.6.1).
КАТЕГОРІЯ 2
Готові лікарські засоби для місцевого, трансдермально-
го, інтравагінального застосування; для введення у по-
рожнини вуха, носа і застосування у ротовій порож-
нині; готові лікарські засоби для інгаляції; за винятком
тих, до яких ставляться вимоги щодо стерильності.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6 /2): не більше 102 мікроорганізмів (бак-
терій і грибів сумарно) у грамі, у мілілітрі або на
один пластир (включаючи клейкий шар і основу).
— Відсутність ентеробактерій і деяких інших грамне-
гати в них бактерій в 1 г, 1 мл або на одному пластирі
(включаючи клейкий шар і основу) (2.6.13).
— Відсутність Рзеисіотопаз аегиДпоза в 1 г, 1 мл або на
одному пластирі (включаючи клейкий шар і осно-
ву) (2.6. ІЗ).
— Відсутність Зіаріїуіососсш аигеиз в 1 г, 1 мл або на
одному пластирі (включаючи клейкий шар і осно-
ву) (2.6.13).
КАТЕГОРІЯ З
А. Готові лікарські засоби для орального застосування і
ректального введення.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 103 бактерій і не більше
102 грибів у грамі або мілілітрі.
— Відсутність бактерій род. ЕпіегоЬасіегіасеае в 1 г
або 1 мл (2.6.13).
— Відсутність Ряеисіопюпаз аегиДпоза в 1 г або І мл
(2.6.13).
— Відсутність ЗіарИуІососсш аигеиз в 1 г або 1 мл
(2.6.13).
В. Готові лікарські засоби для орального застосування,
до складу яких входить сировина природного (тварин-
ного, рослинного або мінерального) походження, для
якої попередня антимікробна обробка неможлива, і що-
до яких компетентний уповноважений орган допускає
мікробне забруднення сировини більше 103 життєздат-
них мікроорганізмів у грамі або мілілітрі. За винятком
рослинних лікарських засобів, що належать до кате-
горії 4.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 104 бактерій і не більше
І О2 грибів у грамі або мілілітрі.
— Не більше 102 ентеробактерій і деяких інших грам
негативних бактерій у грамі або мілілітрі (2.6.13).
— Відсутність ЗаІтопеІІа у 10 г або 10 мл (2.6.13).
— Відсутність ЕзсІїегісНіа соїі в 1 габо 1 мл (2.6.13).
— Відсутність Зіаріїуіососси.', аигеиз в 1 г або 1 мл
(2.6.13).
— Відсутність Рзеидотопаз аетДпоза в 1 г або 1 мл
(2.6.13).
КАТЕГОРІЯ 4
Лікарські засоби, що складаються лише з рослинних
компонентів, одного або декількох (цілісних, здрібнених,
розтертих на порошок), рослинні збори.
А. Рослинні лікарські засоби, до яких перед вживанням
додають киплячу воду.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 107 бактерій і не більше
105 грибів у грамі або мілілітрі.
— Не більше 102 ЕзсИегісИіа соїі у грамі або мілілітрі
(2.6.13).
В. Рослинні лікарські засоби, до яких перед вживанням
не додають киплячу воду.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12). Не більше 105 бактерій і не більше
104 грибів у грамі або мілілітрі.
— Не більше 103 ентеробактерій і деяких інших грам-
негативних бактерій у грамі або мілілітрі (2.6. ІЗ).
— Відсутність Езсіїегісіїіа соїі в 1 габо 1 мл (2.6.13)
— Відсутність Заїтопеїіа у 10 г або 10 мл (2.6.13).
Для того, щоб забезпечити потрібну мікробіологічну
чистоту готових лікарських засобів, використовувані
при їх виробництві субстанції і допоміжні речовини ма-
ють відповідати наведеним нижче вимогам щодо
мікробіологічної чистоти.
КАТЕГОРІЯ 1
Субстанції і допоміжні речовини для виробництва сте-
рильних готових лікарських засобів, що не піддаються
стерилізації.
— Мають витримувати випробування на стерильність
(2.6.1).
КАТЕГОРІЯ 2
Субстанції і допоміжні речовини для виробництва сте-
рильних готових лікарських засобів, що піддаються
стерилізації; готових лікарських засобів для місцевого,
трансдермального, інтравагінального застосування,
для введення у порожнини вуха, носа і застосування у
304
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Загальні тексти
ротовій порожнині; готових лікарських засобів для
інгаляції.
- Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 102 мікроорганізмів
(бактерій і грибів сумарно) у грамі або мілілітрі.
- Відсутність ентеробактерій і деяких інших грамне-
гативних бактерій в 1 габо 1 мл (2.6.13).
- Відсутність Рхеисіотопак аепц’іпоза в 1 г або 1 мл
(2.6.13).
- Відсутність ЕїарНуїососсиз аигеиз в 1 г або І мл
(2.6.13).
КАТЕГОРІЯ З
А. Субстанції і допоміжні речовини для виробництва
готових лікарських засобів для орального застосування
і ректального введення.
- Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 103 бактерій і не більше
102 грибів у грамі або мілілітрі.
- Відсутність бактерій род. ЕпіегоЬасїегіасеае
в 1 г або І мл (2.6.13).
— Відсутність Рлеисіотопак аеги%іпо$а в 1 г або 1 мл
(2.6.13).
— Відсутність ЕіарИуІососсиз аигеиз в 1 г або 1 мл
(2.6.13).
В. Субстанції і допоміжні речовини природного (тва-
ринного, рослинного або мінерального) походження, для
яких попередня антимікробна обробка неможлива і що-
до яких компетентний уповноважений орган допускає
мікробне забруднення більше 103 життєздатних мікро-
організмів у грамі або мілілітрі.
— Загальне число життєздатних аеробних мікроор-
ганізмів (2.6.12): не більше 104 бактерій і не більше
102 грибів у грамі або мілілітрі.
— Не більше 102 ентеробактерій і деяких інших грам-
негативних бактерій у грамі або мілілітрі (2.6.13).
— Відсутність За/топеііа у 10 г або 10 мл (2.6.13).
— Відсутність ЕзсИегісйіа соїі в І г або І мл (2.6.13).
— Відсутність Зіарйуіососсиз аигеиз в 1 г або 1 мл
(2.6.13).
— Відсутність Рзеисіотопаз аеги§іпоза в 1 г або 1 мл
(2.6.13).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
305
Залишкові кількості органічних розчинників"
5.4. ЗАЛИШКОВІ КІЛЬКОСТІ
ОРГАНІЧНИХ РОЗЧИННИКІВ14
Дана стаття поширюється на субстанції та готові
лікарські засоби, у процесі виробництва яких викори-
стовуються органічні розчинники, й установлює межі
граничного вмісту залишкових кількостей органічних
розчинників, що найчастіше застосовуються.
Аналітичний нормативний документ (АНД) на такі
лікарські засоби має містити контроль залишкових
кількостей органічних розчинників.
Відсутність в АНД такого розділу або відсутність у
розділі методики визначення будь-якого з органічних
розчинників, що використовувалися при виробництві
лікарського засобу, має бути обгрунтована в кожному
окремому випадку.
Виробник (заявник) може включати до АНД й менш
жорсткі вимоги до вмісту залишкових кількостей ор-
ганічних розчинників, але в такому разі він зобов’яза-
ний обгрунтувати їх з фармакологічної та токсико-
логічної точок зору, а також з точки зору стабільності
субстанції чи готового лікарського засобу. Таке об-
грунтування необхідне і тоді, коли в АНД регламенту-
ються межі гранічного вмісту залишкових кількостей
органічних розчинників, не перелічених у Табл. 5.4.-1.
Граничний вміст залишкових кількостей органічних
розчинників у субстанціях і готових лікарських засобах
має відповідати величинам, наведеним у Табл. 5.4.-1.
Методи контролю. Контроль наявності залишковім І
кількостей органічних розчинників може проводити-1
ся будь-якими валідованими методами. Найприи- І
нятнішим є метод газової хроматографії (ГХ). Прг
цьому визначення проводять методом порівняння і І
стандартним зразком (звичайно у варіанті абсолютно-
го градуювання)
Типові методики контролю залишкових кількостей
органічних розчинників методом ГХ наведені у додат-
ках. Враховуючи те, що в конкретній субстанції або
готовому лікарському засобі можуть бути присутні не
всі розчинники одночасно, до АНД можуть бути вне-
сені необхідні зміни.
Вимоги до хроматографа. Для визначення залишкових
кількостей органічних розчинників можуть застосову-
ватися будь-які серійні газові хроматографії з детекто-
рами іонізації полум’я, захвату електронів, постійної
швидкості рекомбінації та ін., що відповідають вимо-
гам АНД виробника та показали позитивний результат
при проведенні тесту "Перевірка придатності хромато-
графічної системи".
Загальні вимоги до придатності хроматографічної систе-
ми. Хроматографічна система вважається придатною
якщо виконуються такі умови:
— відносне стандартне відхилення хроматографічно-
го сигналу (у варіанті абсолютного градуювання -
висоти або площі піка у варіанті внутрішнього
стандарту — відношення висот або площ піків)
розраховане з шести повторних хроматограм стан-
Граничний вміст залишкових кількостей органічних розчинників
у субстанціях і готових лікарських засобах
Таблиця 5.4.-І
Назва Граничний вміст, % Назва Граничний вміст, %
Етиленоксид 0.001 Ацетон 0.1
Вуглецю тетрахлорид 0.001 Диметилформамід 0.1
Ацетонітрил 0.005 Етилацетат 0.1
Хлороформ 0.005 Ефір діетиловий 0.1
Бензол 0.01 Кислота оцтова 0.1
1,4-Діоксан 0.01 2-Метоксиетанол 0.1
Етиленхлорид 0.01 Метанол 0.1
Трихлоретилен 0.01 Толуол 0.1
Метиленхлорид 0.05 Формамід 0.1
2-Метилпіридин 0.05 Пентан. гексан. гептан 0.2
2-Метилпропанол 0.05
Піридин 0.05 2-Пропанол 0.5
1-Бутанол 0.05 Етанол 1.0
306
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Залишкові кількості органічних розчинників"
дартного зразка, не перевищує величин, зазначе-
них в окремій статті;
- піки всіх аналізованих компонентів випробовува-
ного розчину мають розділятися з найближчими
сусідніми піками (відповідні коефіцієнти
розділення (//,) наводяться в АНД).
Вимоги до розчинників. Розчинники, що використову-
ються для аналізу, не мають давати на хроматограмах
піків, які перекриваються з піками аналізованих ком-
понентів.
Як один із розчинників може бути використана вільна
вії органічних речовин вода.
Вимоги до хімічного посуду. З метою уникнення ад-
сорбції малих концентрацій аналізованих розчин-
ників посуд, використовуваний для пробопідготовки,
мас бути дезактивований, наприклад, силанізований.
ДОДАТОК 1*
ВИЗНАЧЕННЯ ХЛОРОФОРМУ, БЕНЗОЛУ,
ТРИХЛОРЕТИЛЕНУ, 1,4-ДІОКСАНУ
ТА МЕТИЛЕНХЛОРИДУ
МЕТОДИКА і
Вимоги до хроматографії. Газовий хроматограф із про-
грамуванням температури, без дільника потоку, спо-
ряджений:
- полуменево-іонізаційним детектором;
- колонкою кварцовою капілярною розміром
ЗО м х 0.53 мм, покритою шаром хімічно прищеп-
леного полі[метил(95)феніл(5)]силоксану Р з тов-
щиною шару 5 мкм;
— передколонкою кварцовою розміром 5 м х 0.53 мм,
дезактивованою полі[метил(95)феніл(5)]силокса-
ном Р.
Випробовуваний розчин. 0.100 г препарату поміщають у
конічну колбу місткістю 10 мл, розчиняють в 5 мл во-
ди Р(або іншого розчинника, зазначеного в окремій
статті) і перемішують.
Розчин робочого стандартного зразка (РСЗ 1). У
конічну колбу місткістю 20 мл послідовно поміщають
2.3 мл (2 г) 1,4-діоксану Р, 2.2 мл (2 г) бензолу Р,
1.36 мл (2 г) трихлоретилену Р, 0.67 мл (1 г) хлорофор
яу Р та 7.6 мл (10 г) метиленхлориду Р. Суміш пе-
ремішують.
14 мкл одержаної суміші поміщають у мірну колбу
місткістю 1000 мл, яка містить 500 мл води Р(або іншо-
го розчинника, зазначеного в окремій статті), доводять
об’єм розчину тим самим розчинником до позначки й
перемішують. В 1 мл одержаного розчину РСЗ 1 міс-
титься 1 мкг хлороформу, 2 мкг бензолу, 2 мкг трихло-
ретилену, 2 мкг 1,4-діоксану і 10 мкг метиленхлориду.
Розчин використовують свіжоприготованим.
Перевірка придатності хроматографічної системи. Для
перевірки придатності хроматографічної системи пе-
ред початком аналізу хроматографують 1 мкл розчину
РСЗ 1, одержуючи не менше шести хроматограм за та-
ких умов:
— температура колонки, що програмується: 35°С —
5 хв, приріст температури 8°С/хв до 175 °С, потім
приріст температури 35°С/хв до 260°С, при 260 °С
витримують не менше 16 хв;
— температура блока вводу проб 70°С;
— температура детектора 260°С;
— газ-носій гелій для хроматографії Р;
— лінійна швидкість газу-носія 35 см/с.
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— відносне стандартне відхилення, розраховане для
площ піків хлороформу, або бензолу, або трихлор-
етилену. або 1,4-діоксану, або метиленхлориду,
становить не більше 15 %;
— коефіцієнти розділення (ЯД, розраховані для піків
хлороформу, або бензолу, або трихлоретилену, або
1,4-діоксану, або метиленхлориду, із найближчими
сусідніми піками становлять не менше 1.
Визначення. По 1 мкл випробовуваного розчину і роз-
чину РСЗ 1 поперемінно хроматографують, одержую-
чи не менше п’яти хроматограм за зазначених вище
умов.
Вміст хлороформу, або бензолу, або трихлоретилену,
або 1,4-діоксану, або метиленхлориду (А) у препараті,
у відсотках, обчислюють за формулою:
де:
5, — середнє значення площ піків хлороформу, або
бензолу, або трихлоретилену, або 1,4-діоксану,
або метиленхлориду, розраховане з хроматограм
випробовуваного розчину;
5П — середнє значення площ піків хлороформу, або
бензолу, або трихлоретилену, або 1,4-діоксану,
або метиленхлориду, розраховане з хроматограм
розчину РСЗ 1;
С — вміст в 1 мл розчину РСЗ 1 хлороформу (1 мкг),
або бензолу (2 мкг), або трихлоретилену (2 мкг),
або 1,4-діоксану (2 мкг), або метиленхлориду
(10 мкг).
МЕТОДИКА 2
Вимоги до хроматографа. Газовий хроматограф з про-
грамуванням температури, без дільника потоку, спо-
ряджений:
— полуменево-іонізаційним детектором;
— колонкою кварцовою капілярною розміром
30 м х 0.53 мм, покритою шаром полі[(ціанпропіл)
(феніл)][диметил]силоксану Р з товщиною шару
З мкм;
— передколонкою кварцовою розміром 5 м х 0.53 мм,
дезактивованою поліІметил(95)феніл(5)]силокса-
ном Р.
Випробовуваний розчин. 0.100 г препарату поміщають у
флакон місткістю 20 мл, споряджений прокладкою та
металевим обтискним ковпачком, додають 5 мл води Р
(або іншого розчинника, зазначеного в окремій статті),
1 г натрію сульфату безводного Р\ обтискують ковпачок.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
307
Залишкові кількості органічних розчинників"
Закупорений флакон нагрівають при температурі
80 °С протягом 60 хв, якщо немає інших зазначень в
окремій статті.
Розчин РСЗ 2. 5 мл розчину РСЗ 1 (Методика 1) помі-
щають у флакон місткістю 20 мл, споряджений про-
кладкою та металевим обтискним ковпачком, який
містить 1 г натрію сульфату безводного Р, і обтискують
ковпачок. Закупорений флакон нагрівають при тем-
пературі 80 °С протягом 60 хв.
Відбір газової фази здійснюють негайно.
Перевірка придатності хроматографічної системи. Для
перевірки придатності хроматографічної системи пе-
ред початком аналізу хроматографують 1 мкл розчину
РСЗ 1, одержуючи не менше шести хроматограм за та-
ких умов:
— температура колонки, що програмується: 40 °С —
20 хв, потім швидке підвищення температури до
240 °С, при 240 °С витримують 5 хв;
— температура блока вводу проб 140 °С,
— температура детектора 260 °С;
— газ-носій гелій для хроматографії Р;
— лінійна швидкість газу-носія 35 см/с.
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— відносне стандартне відхилення, розраховане для
площ піків хлороформу, або бензолу, або грихлор-
етилену, або 1,4-діоксану, або метиленхлориду
становить не більше 15 %;
— коефіцієнти розділення (Ту, розраховані для піків
хлороформу, або бензолу, або трихлоретилену, або
1,4-діоксану, або метиленхлориду, із найближчими
сусідніми піками становлять не менше 3.
Випробування. По 1 мл газової фази випробовуваного
розчину і розчину РСЗ 2 поперемінно хроматографу-
ють, одержуючи не менше п’яти хроматограм за зазна-
чених вище умов.
Вміст хлороформу, або бензолу, або трихлоретилену,
або 1,4-діоксану, або метиленхлориду (А) у препараті, у
відсотках, обчислюють за формулою [1] (Методика 1).
МЕТОДИКА З
Вимоги до хроматографа — аналогічно до Методики 2.
Випробовуваний розчин, розчин РСЗ 3 (РСЗ 1), перевірка
придатності хроматографічної системи, умови визна-
чення та розрахункова формула — аналогічно до Ме-
тодики 1.
ДОДАТОК 2
ВИЗНАЧЕННЯ ВУГЛЕЦЮ ТЕТРАХЛОРИДУ,
ХЛОРОФОРМУ, МЕТИЛЕНХЛОРИДУ
ТА ЕТАНОЛУ
Вимоги до хроматографа. Газовий хроматограф, споря-
джений:
— детектором захвату електронів (ДЗЕ) або детекто-
ром постійної швидкості рекомбінації (ДПР) -
для визначення вуглецю тетрахлориду, хлорофор-
му, метиленхлориду;
— полуменево-іонізаційним детектором (ДІП) — для
визначення етанолу;
— колонкою розміром 1.6 м х 3 мм, заповненою сор-
бентом із розміром часток 0.125 — 0.160 мм (на-
приклад, Сепарон ВО-19 фірми Теььек Еісі (Чехія)
або аналогічний).
Випробовуваний розчин. 1.00 г препарату помішають у
мірну колбу місткістю 5 мл, додають 1.0 мл розчину
внутрішнього стандарту та 2.5 мл диметилформаміду
очищеного (або іншого розчинника, зазначеного в
окремій статті), обережно перемішують до повного
розчинення препарату, доводять об’єм розчину тим
самим розчинником до позначки і перемішують.
Розчин внутрішнього стандарту. 20 мл диметилформ-
аміду очищеного поміщають у мірну колбу місткістю
50 мл, додають 0.2 мл етиленхлориду Р, доводять об’єм
розчину до позначки диметилформамідом очищеним і
перемішують.
Розчин зберігають у захищеному від світла місці при
температурі не вище 5 °С.
Термін придатності розчину 14 діб.
Приготування диметилформаміду очищеного. 800 мл
диметилформаміду Р, 20 мл бензолу Рта 10 мл води Р
помішають у круглодонну колбу місткістю 1000 мл.
Суміш переганяють при атмосферному тиску до до-
сягнення температури пари від 152 °С до 155 °С
(близько 80 мл відгону), потім охолоджують до темпе-
ратури 50 °С, знижують тиск до 100 мм рт.ст. та відга-
няють основну фракцію.
Зберігають диметилформамід очищений у захищено
му від світла місці при температурі не вище 10 °С.
Термін придатності 4 міс.
Розчин РСЗ 4 25 мл диметилформаміду очищеного
поміщають у мірну колбу місткістю 100 мл, додають
0.025 мл (0.04 г) вуглецю тетрахлориду Р, 0.134 мл
(0.20 г) хлороформу Р, 1.36 мл (2 г) метиленхлориду Р, до-
водять об’єм одержаного розчину диметилформамідом
очищеним до позначки і перемішують (розчин А).
Розчин А зберігають у захищеному від світла місці при
температурі не вище 10 °С.
Термін придатності розчину А 3 доби.
10 мл диметилформаміду очищеного поміщають у
мірну колбу місткістю 50 мл, додають 0.127 мл (0.10 г)
етанолу Р, 0.25 мл розчину А, 1.0 мл розчину
внутрішнього стандарту, доводять об’єм розчину диме-
тилформамідом очищеним до позначки і перемішують.
308
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Залишкові кількості органічних розчинників"
•озчин використовують свіжоприготованим.
І Перевірка придатності хроматографічної системи. Для
|і юевірки придатності хроматографічної системи пе-
ня початком аналізу хроматографують 2 мкл розчину
РСЗ4, одержуючи не менше шести хроматограм за та-
яхумов’
ямпсратура:
- колонки — 185 °С;
• блока вводу проб — 200 °С;
в-носій:
- для ДЗЕ та ДПР — відповідно до інструкції хрома-
тографа;
- для ДІП — аргон Рабо азот для хроматографії Р;
подання газу-носія — 45 мл/хв.
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
- відносне стандартне відхилення, розраховане для
площ піків вуглецю тетрахлориду або хлороформу,
становить не більше 15 %; а для метиленхлориду
або етанолу та внутрішнього стандарту (етилен-
хлориду) — не більше 5 %;
- коефіцієнт розділення (Р) будь-якої пари піків
визначуваних компонентів, етиленхлориду та ди-
метилформаміду становить не менше 2.
Визначення. По 2 мкл випробовуваного розчину та роз-
чину РСЗ 4 поперемінно хроматографують, одержуючи
не менше п’яти хроматограм за зазначених вище умов.
Вміст вуглецю тетрахлориду, або хлороформу, або ме-
тиленхлориду (А) в препараті, у відсотках, обчислю-
ють за формулою:
ЬС
Х=~Т~'^ [2]
де:
Ь — середнє значення відношення площ піків вугле-
цю тетрахлориду, або хлороформу, або метилен-
хлориду до площ піків внутрішнього стандарту
(етиленхлориду), розраховане з хроматограм ви-
пробовуваного розчину;
Ь51 —середнє значення відношення плош піків вугле-
цю тетрахлориду, або хлороформу, або метилен-
хлориду до площ піків внутрішнього стандарту
(етиленхлориду), розраховане з хроматограм роз-
чину РСЗ 4;
С — концентрація в розчині РСЗ 4 вуглецю тетрахло-
риду (0.0002 %), хлороформу (0.001 %) та мети-
ленхлориду (0.01 %).
Вміст етанолу (X) у препараті, у відсотках, обчислю-
ють за формулою:
5. • С
Х= -------------5, [3]
ло
де:
— середнє значення площ піків етанолу, розрахо-
ване з хроматограм випробовуваного розчину;
$о —середнє значення площ піків етанолу, розрахо-
ване з хроматограм розчину РСЗ 4;
С— концентрація у розчині РСЗ 4 етанолу (0.2 %).
ДОДАТОК З*
ВИЗНАЧЕННЯ МЕТИЛЕНХЛОРИДУ
В ТАБЛЕТКАХ, ПОКРИТИХ ОБОЛОНКОЮ
Вимоги до хроматографа. Газовий хроматограф з про-
грамуванням температури, споряджений:
— полуменево-іонізаційним детектором;
— колонкою скляною розміром 1.8 м х 2 мм, запов-
неною графітизованим вуглецем із мінімальною
поверхнею 12 м2/г, який містить 0.2 % нанесеного
макроголу 1500.
Випробовуваний розчин. 1.00 г таблеток поміщають у
конічну колбу з притертою пробкою місткістю 50 мл,
додають 20 мл води Р, поміщають колбу в ультразвуко-
ву баню і витримують до повного розпадання табле-
ток, після чого центрифугують розчин. 2 мл одержа-
ного розчину поміщають у флакон місткістю 20 мл,
споряджений прокладкою та металевим обтискним
ковпачком, і закупорюють. Флакон поміщають на
20 хв у водяну баню з температурою 85 °С.
Розчин РСЗметиленхлориду. 3.8 мкл (5 мг) метиленхло-
риду Рпоміщають у мірну колбу місткістю 1000 мл, яка
містить 500 мл води Р, доводять об’єм розчину водою Р
до мітки і перемішують.
Розчин використовують свіжоприготованим.
Розчин РСЗ 5. Розчин готують так само, як і випробо-
вуваний, але замість 20 мл води Р використовують
20 мл розчину РСЗ метиленхлориду.
Відбір газової фази здійснюють негайно.
Приготування зразка для перевірки придатності хро-
матографічної системи. 3.1 мкл (2.5 мг) 96 % спирту Р
змішують з 500 мл розчину РСЗ метиленхлориду.
Розчин використовують свіжоприготованим.
2 мл одержаного розчину поміщають у флакон міст-
кістю 20 мл, споряджений прокладкою та металевим
обтискним ковпачком, і закупорюють. Флакон помі-
щають на 20 хв у водяну баню з температурою 85 °С.
Відбір газової фази здійснюють негайно.
Перевірка придатності хроматографічної системи.
1 мл газової фази зразка для перевірки придатності
хроматографічної системи хроматографують, одержу-
ючи не менше шести хроматограм за таких умов:
— температура колонки, що програмується: 80 °С —
2 хв, приріст температури 30 °С/хв до 150 °С, при
150 °С витримують 5 хв;
— температура блока вводу проб 200 °С;
— температура детектора 200 °С;
— газ-носій азот для хроматографії Р;
— подання газу-носія 20 мл/хв.
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— відносне стандартне відхилення, розраховане для
площі піка метиленхлориду, становить не біль-
ше 10 %;
При розробці методик (Додатки 1 і 3) використані або адаптовані
матеріали Фармакопеї США. Із дозволу Фармакопейної
Конвенції США, Інк.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
309
Залишкові кількості органічних розчинників"
— коефіцієнт розділення піків (7?,), розрахований для
піків метиленхлориду та етанолу, має бути не мен-
ше 1.5;
— відносний час утримування піка етанолу по відно-
шенню до піка метиленхлориду становить близь-
ко 0.8.
Випробування. По 1 мл газової фази випробовуваного
розчину і розчину РСЗ 5 поперемінно хроматографу-
ють, одержуючи не менше п’яти хроматограм за зазна-
чених вище умов.
Вміст метиленхлориду (X) у 1 г препарату, у мікрогра-
мах, обчислюють за формулою:
5, • 5 • 20
^0“ ^1
де:
5] — середнє значення площ піків метиленхлорид
розраховане з хроматограм випробовуваного,
розчину;
50 — середнє значення площ піків метиленхлориду
розраховане з хроматограм розчину РСЗ 5.
Вміст метиленхлориду в таблетках, покритих оболон-
кою, має бути таким, щоб його добова доза для
пацієнта не перевищувала 500 мкг.
310
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Аланін
АЛАНІН
Аіапіпит
1ШШ
Н3С^,СО2Н
н мн2
ЧН7МО2 М.м. 89.1
кланін містить не менше 98.5 % і не більше 101.0 %
!5)-2-амінопропанової кислоти, у перерахунку на суху
речовину.
ВИРОБНИЦТВО
субстанція одержана в результаті процесу, шо
ає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації”.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Легко розчинний у воді Р, дуже мало роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, Ц.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ аланіну.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
В. 0.5 г субстанції розчиняють у суміші 1 мл води Р,
0.5 мл розчину 100 г/л натрію нітриту Р\ 0.25 мл роз-
чину кислоти хлористоводневої РІ, збовтують; виділя-
ються бульбашки газу. До одержаного розчину дода-
ють 2 мл розчину натрію гідроксиду розведеного Р і
відразу 0.25 мл розчину калію йодиду йодованого Р, ви-
тримують протягом 30 хв; утворюється жовтий осад із
характерним запахом.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді дистильо-
ваній Рі доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). 10 мл розчину 8 доводять
водою Р до об’єму 20 мл. Одержаний розчин має бути
прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування "Про-
зорість розчину", має бути не інтенсивнішим за еталон
В¥6.
Питоме оптичне обертання (2.2.7). Від +13.5° до +15.5°,
у перерахунку на суху речовину. 2.50 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рао об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ аланіну розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчину (Ь)
доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ аланіну і 10 мг ФСЗ
гліцину розчиняють у воді Р і доводять об’єм розчину
тим самим розчинником до 25 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг аланіну і 2 мкг гліцину)
розчину порівняння (с). Пластинку сушать на повітрі
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
313
Аміаку розчин концентрований
й поміщають у камеру із сумішшю розчинників кисло-
та оцтова льодяна Р - вода Р - бутанол Р (20:20:60).
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й
обприскують розчином нінгідрину Р. Пластинку
нагрівають при температурі від 100 °С до 105 °С протя-
гом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт) 5 мл роз-
чину 8 доводять водою Рдо об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0 03 % (300 ррпг). 10 мл
розчину 8 доводять водою дистильованою Рдо об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
помішають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р і
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв. Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (100 ррт Ь'Н4) Р,
0.5 мл води Рі 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваним розчином має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 20 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
80.0 мг субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової безвод-
ної Р і титрують 0.1 Мрозчином кислоти хлорної до пе-
реходу забарвлення від коричнювато-жовтого до зеле-
ного, використовуючи як індикатор 0.1 мл розчину
нафтолбензеїну Р.
1 мл 0.1 М розчину кислоти хлорної відповідає 8.91 мг
С3Н7МО2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
____________________________________________А
рН (2.2.3). Від 5.5 до 7.0. Вимірюють рН розчину 8.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентераль-
них лікарських засобів без подальшої процедури вида-
лення пірогенів, вона має витримувати випробування
"Пірогени” (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
АМІАКУ РОЗЧИН
КОНЦЕНТРОВАНИЙ
Аттопіае зокДіо сопсспітаїа
АММОІЧІА 8ОИ ПОХ, СОМ БМРА ТЕІ)
Аміаку розчин концентрований містить не менше 25.0 %
і не більше 30.0 % (м/м) аміаку (1\ІН3; М.м. 17.03).
ВЛАСТИВОСТІ
Опис. Прозора, безбарвна, дуже лужна рідина.
Розчинність. Зміщується з водою Р і 96 % спиртом Р
ІДЕНТИФІКАЦІЯ
А. Відносна густина (2.2.5). Від 0.892 до 0.910.
314
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Амоксициліну тригідрат
В. Субстанція повинна мати сильнолужну реакцію
(2.2.4).
С. До 0.5 мл субстанції додають 5 мл води Р, пропуска-
ють повітря і одержану газову суміш спрямовують на
поверхню розчину, що містить 1 мл 0.ІМ кислоти хло-
ристоводневої і 0.05 мл розчину метилового червоно-
го Р; червоне забарвлення розчину переходить у жов-
те. До одержаного розчину додають 1 мл розчину
натрію кобальтинітриту Р; утворюється жовтий осад.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8.220 мл субстанції упарюють майже насухо на
водяній бані, охолоджують, додають І мл кислоти оц-
тової розведеної Р і доводять об’єм розчину водою дис-
тильованою Рпо 20 мл
Прозорість розчину (2.2.1). До 2 мл субстанції додають
8мл води Р. Одержаний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин, приго-
тований для випробування "Прозорість розчину", має
бути безбарвним.
Сухий залишок. 50 мл субстанції упарюють на водяній
бані і висушують при температурі від 100 °С до 105 °С
протягом 1 год Маса сухого залишку не має переви-
щувати 1 мг (0.02 г/л)
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
50.0 мл 7 Мрозчину кислоти хлористоводневої поміща-
ють у колбу зі скляною притертою пробкою, точно
зважують, додають 2 мл субстанції і повторно зважу-
ють. Одержаний розчин титрують 1 М розчином
натрію гідроксиду до переходу забарвлення від черво-
ного до жовтого, використовуючи як індикатор 0.1 мл
розчину метилового червоного Р.
1 мл 7 М розчину кислоти хлористоводневої відповідає
17.03 мг1\Н3.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, при температурі
не вище 20 °С.
Речовини, що окислюються. До 100 мл кислоти сірчаної
розведеної Р при охолодженні обережно додають
8.8 мл субстанції і 0.75 мл 0.002 Мрозчину калію пер-
манганату. Одержаний розчин витримують протягом
5 хв; розчин має залишатися слабко-рожевим.
АМОКСИЦИЛІНУ ТРИГІДРАТ
Піридин і супровідні домішки. Оптична густина (2.2.25)
субстанції, вимірювана за довжини хвилі 252 нм, має
бути не більше 0.06 (0.0002 % (2 ррт), у перерахунку
на піридин). Як компенсаційний розчин використо-
вують воду Р.
АтохісіПіпит Ігіїїусігісшп
АМОХІСІШМ ТКІШОКАТЕ
Карбонати. Не більше 0.006 % (60 ррт). 10 мл суб-
станції помішають у пробірку зі скляною притертою
пробкою, додають 10 мл розчину кальцію гідроксиду Р.
Відразу закривають пробкою і перемішують. Опалес-
ценція одержаного розчину не має перевищувати опа-
лесценцію еталона, приготованого аналогічно до ви-
пробовуваного розчину з використанням 10 мл розчи-
ну 0.1 г/л натрію карбонату безводного Р.
Хлориди (2.4.4). Не більше 0.0001 % (1 ррт). 5 мл роз-
чину 8 доводять водою Рдо об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.0005 % (5 ррт). З мл
розчину 8 доводять водою дистильованою Рдо об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Важкі метали (2.4.8, метод А). Не більше 0.0001 %
(1 ррт). 4 мл розчину 8 доводять водою Рдо об’єму
20 мл. 12 мл одержаного розчину мають витримувати
випробування на важкі метали. Еталон готують із ви-
користанням еталонного розчину свинцю (2 ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.000025 % (0 25 ррт). 4 мл
розчину 8 доводять водою Рдо 10 мл Одержаний роз-
чин має витримувати випробування на залізо.
, ЗН2О
с16н19гм3о58,зн2о
М.м. 419.4
Амоксициліну тригідрат містить не менше 95.0 % і не
більше 100.5 % (25,5Р,6Р)-6-[[(2/?)-2-аміно-2-(4-
гідроксифеніл)ацетил]аміно]-3,3-диметил-7-оксо-4-
тіа-1-азабіцикло[3.2.0]гептан-2-карбонової кислоти, у
перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Мало розчинний у воді Рі 96 % спирті Р,
практично не розчинний в ефірі Рі жирних оліях.
(Розчиняється в розведених розчинах мінеральних
кислот і гідроксидів лужних металів)
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
315
Амоксициліну тригідрат
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А.
Друга ідентифікація: В, С.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ амоксициліну
тригідрату.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель н силанізований Р.
Випробовуваний розчин. 25 мг субстанції розчиняють у
розчині натрію гідрокарбонату Р і доводять об’єм роз-
чину тим самим розчинником до 10 мл.
Розчин порівняння (а). 25 мг ФСЗ амоксициліну три-
гідрату розчиняють у розчині натрію гідрокарбонату Р
і доводять об’єм розчину тим самим розчинником до
10 мл.
Розчин порівняння (Ь). 25 мг ФСЗ амоксициліну
тригідрату і 25 мг ФСЗ ампіциліну тригідрату розчи-
няють у розчині натрію гідрокарбонату Р і доводять
об’єм розчину тим самим розчинником до 10 мл.
Налінію старту хроматографічної пластинки наносять
1 мкл (2.5 мкг) випробовуваного розчину, 1 мкл
(2.5 мкг) розчину порівняння (а) і 1 мкл (2.5 мкг амо-
ксициліну тригідрату і 2.5 мкг ампіциліну тригідрату)
розчину порівняння (Ь). Пластинку поміщають у ка-
меру із сумішшю розчинників ацетон Р — розчин
154 г/л амонію ацетату Р, рН якого доведено до 5.0
кислотою оцтовою льодяною Р, (10:90). Коли фронт
розчинників пройде 15 см від лінії старту, пластинку
виймають із камери, сушать на повітрі, витримують у
парі йоду до появи плям і переглядають при денному
світлі.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна ій за
розміром і забарвленням.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
С. Близько 2 мг субстанції поміщають у пробірку зав-
вишки близько 150 мм і діаметром 15 мм і змочують
0.05 мл води Р. Потім додають 2 мл розчину формаль-
дегіду в кислоті сірчаній Р і перемішують обертальни-
ми рухами; одержаний розчин має бути практично
безбарвним. Пробірку нагрівають у водяній бані про-
тягом 1 хв; поступово з’являється темно-жовте забарв-
лення.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 0.100 г субстанції за допомогою ультразвуку
або обережно нагріваючи розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 50.0 мл.
Прозорість розчину (2.2.1). 1.0 г субстанції розчиняють І
у 10 мл 0.5 М розчину кислоти хлористоводневої. Окре- 1
мо 1.0 г субстанції розчиняють у 10 мл розчину аміаку І
розведеного Р2. Одержані розчини відразу після приго
тування за ступенем каламутності не мають переви-
щувати еталон 11.
рН (2.2.3). Від 3.5 до 5.5. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від +290° до +315с,
у перерахунку на безводну речовину. Визначення про-
водять. використовуючи розчин 8.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29) в умовах, описаних у
розділі "Кількісне визначення", підбираючи співвідно-
шення рухомих фаз А: В і чутливість реєструючого при-
строю, як зазначено у розділі "Кількісне визначення".
У вибраних умовах в ізократичному режимі хромато-
графують розчин порівняння (б).
У цих самих умовах хроматографують свіжопригото-
ваний випробовуваний розчин (Ь). Відразу після вихо-
ду піка амоксициліну починають хроматографування
в режимі лінійного градієнта так, щоб через 25 хв після
початку градієнта співвідношення рухомих фаз А:В
становило 0:100. Продовжують хроматографування
протягом 15 хв із використанням рухомої фази В.
Потім протягом 15 хв урівноважують колонку рухо-
мою фазою початкового складу. Як контрольний
дослід хроматографують рухому фазу А в умовах, за-
значених для розчину порівняння (б).
На хроматограмі випробовуваного розчину (Ь) площа
будь-якого піка, крім основного і піків, що відповіда-
ють пікам на хроматограмі контрольного досліду, не
має перевищувати площу основного піка на хромато-
грамі розчину порівняння (б) (1 %).
Диметиланілін. Не більше 0.002 % (20 ррт). Визначен-
ня проводять методом газової хроматографії (2.2.28),
використовуючи нафталін Ряк внутрішній стандарт.
Розчин внутрішнього стандарту. 50.0 мг нафталіну Р
розчиняють у циклогексані Р і доводять об’єм розчину
тим самим розчинником до 50.0 мл. 5.0 мл одержаного
розчину доводять циклогексаном Рдр об’єму 100.0 мл.
Випробовуваний розчин. 1.00 г субстанції поміщають у
пробірку із притертою скляною пробкою, додають
5 мл 7 М розчину натрію гідроксиду і 1.0 мл розчину
внутрішнього стандарту. Пробірку закривають й ін-
тенсивно струшують протягом 1 хв. Якщо необхідно,
центрифугують і використовують верхній шар.
Розчин порівняння. До 50.0 мг диметиланіліну Р дода-
ють 2 мл кислоти хлористоводневої Р і 20 мл води Р,
струшують до повного розчинення і доводять об’єм
розчину водою Рдо 50.0 мл. 5.0 мл одержаного розчи-
ну доводять водою Рао об’єму 250.0 мл. 1.0 мл одержа-
ного розчину поміщають у пробірку із притертою
скляною пробкою, додають 5 мл 1 М розчину натрію
гідроксиду й 1.0 мл розчину внутрішнього стандарту.
Пробірку закривають й інтенсивно струшують протя-
гом 1 хв. Якщо необхідно, центрифугують і викорис-
товують верхній шар.
316
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Амоксициліну тригідрат
Хроматографування проводять на газовому хромато-
графія полуменево-іонізаційним детектором за таких
иов:
- колонка скляна розміром 2 м х 0.2 мм. заповнена
діатомітом силанізованим для газової хромато-
графії Р, з нанесеним 3 % (м/м) поліметилфенілси-
локсаном Р;
- газ-носій азот для хроматографії Р;
- швидкість газу-носія ЗО мл/хв;
- температура колонки 120 °С;
- температура детектора і блока вводу проб 150 °С.
Поперемінно хроматографують 1 мкл випробовувано-
го розчину і 1 мкл розчину порівняння.
Вода (2.5.12). Не менше 11.5 % і не більше 14.5 %. Ви-
значення проводять із 0.100 г субстанції напівмікро-
методом
Сульфатна зола (2.4.14). Не більше 1.0 %. Визначення
проводять із 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять методом рідинної хромато-
графії (2.2.29).
Випробовуваний розчин (а). ЗО 0 мг субстанції розчиня-
ють у рухомій фазі А і доводять об’єм розчину тією са-
мою рухомою фазою до 50.0 мл.
Випробовуваний розчин (Ь). 30.0 мг субстанції розчиня-
ють у рухомій фазі А і доводять об’єм розчину тією са-
мою рухомою фазою до 20.0 мл.
Розчин порівняння (а). 30.0 мг ФСЗ амоксициліну
тригідрату розчиняють у рухомій фазі А і доводять
об єм розчину тією самою рухомою фазою до 50.0 мл.
Розчин порівняння (Ь). 4.0 мг ФСЗ цефадроксилу розчи-
няють у рухомій фазі А і доводять об’єм розчину тією
самою рухомою фазою до 50.0 мл. До 5.0 мл одержано-
го розчину додають 5.0 мл розчину порівняння (а) і до-
водять об’єм розчину рухомою фазою А до 100.0 мл.
Розчин порівняння (с). 1.0 мл розчггну порівняння (а)
доводять рухомою фазою А до об’єму 20.0 мл. 1.0 мл
одержаного розчину доводять рухомою фазою А до
об’єму 50.0 мл.
Розчин порівняння (сі). 2.0 мл розчину порівняння (а)
доводять рухомою фазою А до об’єму 20.0 мл. 5.0 мл
одержаного розчину доводять рухомою фазою А до
об’єму 20.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
- колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октадецилсилільним для хро-
матографії Рз розміром часток 5 мкм;
— швидкість рухомої фази 1.0 мл/хв;
- рухома фаза А: ацетонітрил Р — буферний розчин
рН 5.0(1:99);
— рухома фаза В: ацетонітрил Р — буферний розчин
рН 5.0 (20:80);
— буферний розчин рН 5.0: до 250 мл 0.2 М розчину
калію дигідрофосфату додають розчин натрію
гідроксиду розведеного Рдо рН 5.0, доводять об’єм
розчину водою Рао 1000.0 мл;
— детектування за довжини хвилі 254 нм;
— об’єм проби, що уводиться, 50 мкл.
Урівноважують колонку рухомою фазою зі співвідно-
шенням рухомих фазА:В (92:8).
Хроматографують розчин порівняння (Ь). Хромато-
графічна система вважається придатною, якщо вико-
нуються такі умови:
— коефіцієнт розділення двох основних піків має
бути не менше 2.0;
— коефіцієнт ємності для першого піка (амоксици-
лін) має бути від 1.3 до 2.5
Якщо необхідно, коригують співвідношення рухомих
фаз А: В.
Хроматографують розчин порівняння (с). Регулюють
систему таким чином, щоб відношення сигнал-шум
для основного піка становило не менше 3.
Хроматографують розчин порівняння (а) шість разів.
Хроматографічна система вважається придатною, як-
шо відносне стандартне відхилення для площі основ-
ного піка не перевищує 1.0 %.
Поперемінно хроматографують випробовуваний роз-
чин (а) і розчин порівняння (а).
ЗБЕРІГАННЯ
У повітронепроникному контейнері.
ДОМІШКИ
А. (25,5/?,6/?)-6-аміно-3,3-диметил-7-оксо-4-тіа-1-
азабіцикло[3.2.0]гептан-2-карбонова кислота (6-амі-
нопеніциланова кислота),
В. (25',5/(,6/?)-6-[[(25)-2-аміно-2-(4-гідроксифеніл)-
ацетил]аміно]-3,3-диметил-7-оксо-4-тіа-1-азабіцик-
ло[3.2.0]гептан-2-карбонова кислота (Е-амоксипи-
лін),
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
317
Амоксициліну тригідрат
С. (45)-2-[5-(4-гідроксифеніл)-3,6-діоксопіперазин-
2-іл]-5,5-диметилтіазолідин-4-карбонова кислота
(амоксицилін дикетопіперазини),
тил]аміно]-3,3-диметил-7-оксо-4-тіа-1-азабшик-
ло[3.2.0]гептан-2-карбонова кислота (Ь-(4-гідрокси-
феніл)гліциламоксицилін),
Н. (2Я)-2-[(2,2-диметилпропаноїл)аміно]-2-(4-гідро-
ксифеніл)оцтова кислота,
І. (2Я)-2-аміно-2-(4-гідроксифеніл)оцтова кислота.
Б. (45)-2-[[[(2Я)-2-аміно-2-(4-гідроксифеніл)апе-
тил]аміно]карбоксиметил]-5,5-диметилтіазолідин-4-
карбонова кислота (пеніпилоїнові кислоти амокси-
циліну).
Е. (2Я5,45)-2-[[[(2Я)-2-аміно-2-(4-гідроксифеніл)-
ацетил]аміно]метил]-5,5-диметилтіазолідин-4-карбо-
нова кислота (пенілоїнові кислоти амоксициліну).
Г. З - (4- гідроксифеніл) п ірази н - 2-ол,
6. (25,5Л,6/?)-6-[[(2Л)-2-[[(2Л)-2-аміно-2-(4-гідро-
ксифеніл)ацетил]аміно]-2-(4-гідроксифеніл)аце-
X Олігомери амоксициліну і пеніцилоїнової кислоти
амоксициліну,
К. Олігомери пеніцилоїнової кислоти амоксициліну.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
318
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Аргінін
АРГІНІН
Аг§іпіпит
об’ємів розчину натрію гіпохлориту концентрованого Р
і води Р; з’являється червоне забарвлення.
ВИПРОБУВАННЯ НАЧИСТОТУ
ШІКІКЕ
С6Н14К4О2
М.м. 174.2
Аргінін містить не менше 98.5 % і не більше 101.0 %
5)-2-аміно-5-гуанідинопентанової кислоти, у пере-
рахунку на суху речовину.
ВИРОБНИЦТВО
Якшо субстанція одержана в результаті процесу, шо
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Легко розчинний у воді Р, дуже мало роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Пруга ідентифікація: А, В, Б, Е.
А. Субстанція має відповідати вимогам щодо питомо-
гооптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Розчин 8, приготований, як зазначено у розділі
"Випробування на чистоту", повинен мати сильно-
лужну реакцію (2.2.4).
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ аргініну.
Б. На хроматограмі випробовуваного розчину (Ь), одер-
жаній при випробуванні "Речовини, виявлювані нінгід-
рином”, має виявлятися основна пляма на рівні основної
плями на хроматограмі розчину порівняння (а), відпо-
відна їй за розміром і забарвленням.
Е. Близько 25 мг субстанції розчиняють у 2 мл води Р.
Додають 1 мл розчину а-нафтолу Рі 2 мл суміші рівних
Розчин 8. 2.5 г субстанції розчиняють у воді дистильо-
ваній Рі доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
Питоме оптичне обертання (2.2.7). Від +25.5° до +28.5°,
у перерахунку на суху речовину. 2.00 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27),
використовуючи ТШХ пластинки із шаром силікаге-
лю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у кислоті хлористоводневій розведеній Р і доводять
об’єм розчину тією самою кислотою до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рпо об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ аргініну розчиняють
у 0.1 М розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рпо об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ аргініну і 10 мг ФСЗ
лізину гідрохлориду розчиняють у 0.1 М розчині кисло-
ти хлористоводневої і повопять об’єм розчину тією са-
мою кислотою до 25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг аргініну і 2 мкг лізину
гідрохлориду) розчину порівняння (с), сушать на
повітрі й поміщають у камеру із сумішшю розчин-
ників розчин аміаку концентрований Р - 2-пропанол Р
(30:70). Коли фронт розчинників пройде 15 см від лінії
старту, пластинку виймають із камери, сушать при
температурі від 100 °С до 105 °С до зникнення запаху
аміаку й обприскують розчином нінгідрину Р. Пластин-
ку нагрівають при температурі від 100 °С до 105 °С
протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
319
Аргініну гідрохлорид
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). До 5 мл
розчину 8 додають 0.5 мл кислоти азотної розведеної Р
і доводять водою Рао об’єму 15 мл. Одержаний розчин
має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). До 10 мл
розчину 8 додають 1.7 мл кислоти хлористоводневої
розведеної Р і доводять водою дистильованою Р до
об’єму 15 мл. Одержаний розчин має витримувати ви-
пробування на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, шо складається із двох годинникових сте-
кол діаметром 60 мм, сполучених по краях. На вну-
трішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р'\ ре-
тельно розтирають скляною паличкою. Нижнє годин-
никове скло відразу накривають верхнім і нагрівають
при температурі 40 °С протягом 15 хв. Паралельно в
аналогічних умовах готують еталон, використовуючи
0.1 мл еталонного розчину амонію (100 ррт Б'Н) Р, 0.5 мл
води Р і 0.30 г магнію оксиду важкого Р. Лакмусовий
папір над випробовуваним розчином має бути забарв-
лений у синій колір не інтенсивніше, ніж над еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 20 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у 50 мл води Р і титрують
0.1 М розчином кислоти хлористоводневої до переходу
забарвлення від зеленого до фіолетово-червоного, ви-
користовуючи як індикатор 0.2 мл розчину метилового
червоного змішаного Р.
1 мл 0.1 М розчину кислоти хлористоводневої
відповідає 17.42 мг С6Ні4М4О2.
ЗБЕРІГАННЯ
У шільно закупореному контейнері, у захищеному ви
світла місці.
_____________________________________________а
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
АРГІНІНУ ГІДРОХЛОРИД
Аг§іпіпі Ііусігосіїїогісіїип
АКСІМПЕ НУВКОСНЕОКЮЕ
С6Н15СЖ4О2
М.м. 210.7
Аргініну гідрохлорид містить не менше 98.5 % і не
більше 101.0 % (5)-2-аміно-5-гуанідинопентанової
кислоти гідрохлориду, у перерахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу фер-
ментації, що включає стадії ферментації, вона має ви-
тримувати вимоги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Легко розчинний у воді Р, дуже мало роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, Е.
Друга ідентифікація: А, С, В, Е.
320
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Аргініну гідрохлорид
А.Субстанція має відповідати вимогам щодо питомо-
ооптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ аргініну гідрохлориду.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином”, має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
В. Близько 25 мг субстанції розчиняюі ь у 2 мл води Р.
Долають 1 мл розчину а-нафтолу Рі 2 мл суміші рівних
об’ємів розчину натрію гіпохлориту концентрованого Р
іводи Р; з’являється червоне забарвлення.
Е. Близько 20 мг субстанції дають реакцію (а) на хло-
риди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді дистильо-
ваній Рі доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон ВУ6.
Питоме оптичне обертання (2.2.7). Від +21.0° до +23.5°,
у перерахунку на суху речовину 2.00 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рро об’єму 50 мл.
Розчин порівняння (а) 10 мг ФСЗ аргініну гідрохлориду
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ аргініну гідрохлориду і
10 мг ФСЗлізину гідрохлориду розчиняють у воді Рі до-
водять об’єм розчину тим самим розчинником до
25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а). 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг аргініну гідрохлориду і
2 мкг лізину гідрохлориду) розчину порівняння (с).
Пластинку сушать на повітрі й поміщають у камеру із
сумішшю розчинників розчин аміаку концентрова-
ний Р - 2-пропанол Р (30:70). Коли фронт розчинників
пройде 15 см від лінії старту, пластинку виймають із
камери, сушать при температурі від 100 °С до 105 °С до
зникнення запаху аміаку й обприскують розчином
нінгідрину Р. Пластинку нагрівають при температурі
від 100 °С до 105 °С протягом 15 хв.
На хроматоірамі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 10 мл
розчину 8 доводять водою дистильованою Рдо об’єму
15 мл. Розчин має витримувати випробування на суль-
фати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, шо складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р і
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв. Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (100 ррт ХН4) Р,
0.5 мл води Р і 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваним розчином має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкепюном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Рі дово-
дять об’єм розчину тим самим розчинником до 20 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
321
Атенолол
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.180 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають ЗО мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної до пе-
реходу забарвлення від коричнювато-жовтого до зеле-
ного, використовуючи як індикатор 0.1 мл розчину
нафтолбензеїну Р.
1 мл 0.1 Мрозчину кислоти хлорної відповідає 21.07 мг
С6Н15СН\4О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________N
рН (2.2.3). Від 4.5 до 6.5. 10 мл розчину 8 доводять во-
дою дистильованою Рдо об’єму 20 мл.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якшо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
АТЕНОЛОЛ
Аїепоіоіит
ІДЕНТИФЩКАЦ1Я
Перша ідентифікація: С.
Друга ідентифікація: А, В, Б.
А. Температура плавлення (2.2.14). Від 152 °С до 155 °С.
В. 0.100 г субстанції розчиняють у метанолі Р і дово-
дять об’єм розчину тим самим розчинником до 100 мл.
10.0 мл одержаного розчину доводять метанолом Рао
об’єму 100 мл. Ультрафіолетовий спектр поглинання
(2.2.25) одержаного розчину в області від 230 нм до
350 нм повинен мати два максимуми за довжин хвиль
275 нм і 282 нм. Відношення оптичної густини в мак-
симумі за довжини хвилі 275 нм до оптичної густини в
максимумі за довжини хвилі 282 нм має бути від 1.15
до 1.20.
С. Інфрачервоний спектр поглинання (2.2.24) суб
станції має відповідати спектру ФСЗ атенололу.
Б. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель вг254 Р.
Випробовуваний розчин. 10 мг субстанції розчиняють у
1 мл метанолу Р.
Розчин порівняння. 10 мг ФСЗ атенололу розчиняють у
1 мл метанолу Р.
Налінію старту хроматографічної пластинки наносять
10 мкл (100 мкг) випробовуваного розчину і 10 мкл
(100 мкг) розчину порівняння. Пластинку поміщають
у камеру із сумішшю розчинників розчин аміаку кон-
центрований РІ - метанол Р (1:99). Коли фронт роз-
чинників пройде 15 см від лінії старту, пластинку вий-
мають із камери, сушать на повітрі й переглядають в
УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром.
АТЕИОІОІ
ОН
І
(СН3)2СН-МН-СН2-СН-СН2-О
сн2-с—мн2
С|4Н22М2О3
М.м. 266.3
Атенолол містить не менше 99.0 % і не більше 101.0 %
(ЯУ)-4-[2-(гідрокси-3-ізопропіламінопропокси)фе-
ніл]ацетаміду, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Білий або майже білий порошок.
Розчинність. Помірно розчинний у воді Р, розчинний в
етанолі Р, мало розчинний у метиленхлориді Р, прак-
тично не розчинний в ефірі Р.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 0.10 г субстанції розчиняють у воді Рі дово-
дять об’єм розчину тим самим розчинником до 10 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон 6 шка-
ли найбільш підхожого кольору.
Оптичне обертання (2.2.7). Від +0.10° до -0.10°, у пере-
рахунку на суху речовину. Визначення проводять для
розчину 8.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин (а). 50.0 мг субстанції розчиня-
ють у 20 мл рухомої фази і доводять об’єм розчину ру-
хомою фазою до 25.0 мл.
322
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Атропіну сульфат
Випробовуваний розчин (Ь). 50.0 мг субс танції розчиня-
ють у 0.1 мл диметилсульфоксиду Р, якшо необхідно,
злегка нагріваючи у водяній бані протягом декількох
секунд, і доводять об’єм розчину рухомою фазою до
25.0 мл.
Розчин порівняння (а). 0.5 мл випробовуваного розчи-
нена) доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (Ь). 50.0 мг ФСЗ атенололу для
валідації колонки розчиняють у 0.1 мл диметилсуль-
фоксиду Р, якщо необхідно, злегка нагріваючи у во-
дяній бані протягом декількох секунд, і доводять
об’єм розчину рухомою фазою до 25.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
- колонка з нержавіючої сталі розміром 0.15 м х 4.6 мм,
заповнена силікагелем для хроматографії октаде-
цилсилільним Рз розміром часток 5 мкм;
- рухома фаза: 1.0 г натрію октансульфонату РіОАс
тетрабутиламонію гідросульфату Р розчиняють у
1 л суміші тетрагідрофуран Р - метанол Р - розчин
3.4 г/л калію дигідрофосфату Р (20:180:800) і дово-
дять рН кислотою ортофосфорною Рдо 3.0;
- швидкість рухомої фази 1.0 мл/хв;
- детектування за довжини хвилі 226 нм.
Урівноважують колонку при швидкості рухомої фази
1.0 мл/хв протягом близько 30 хв.
Хроматографують 10 мкл розчину порівняння (а).
Чутливість системи регулюють таким чином, щоб ви-
сота основного піка становила не менше 50 % шкали
реєструючого пристрою.
Хроматографують 10 мкл розчину порівняння (Ь).
Одержана хроматограма має бути аналогічною до
зразка хроматограми, що додається до ФСЗ атенололу
для валідації колонки: пік біс-ефіру виходить першим і
відокремлюється від піка третинного аміну, що зви-
чайно виходить піком із двома максимумами. Якщо
необхідно, регулюють вміст натрію октансульфонату в
рухомій фазі. Збільшення вмісту натрію октансульфо-
нату збільшує час утримування третинного аміну.
Поперемінно хроматографують 10 мкл випробовува-
ного розчину (а) і 10 мкл розчину порівняння (а). Час
хроматографування має бути в 4 рази більше часу ут-
римування основного піка. На хроматограмі випробо-
вуваного розчину (а) площа будь-якого піка, крім ос-
новного, не має перевищувати половини площі ос-
новного піка на хроматограмі розчину порівняння (а)
(0.25 %); сума площ усіх піків, крім основного, не має
перевищувати площу основного піка на хроматограмі
розчину порівняння (а) (0.5 %). Не враховують піки,
площі яких становлять менше 0.1 площі основного
піка на хроматограмі розчину порівняння (а).
Якшо субстанція містить більше 0.15 % біс-ефіру,
її придатність треба підтверджувати повторним хрома-
тографуванням 10 мкл випробовуваного розчину (Ь).
Хлориди (2.4.4). Не більше 0.1 %. 50 мг субстанції роз-
чиняють у суміші 1 мл кислоти азотної розведеної Р і
15 мл води Р. Одержаний розчин має втримувати ви-
пробування на хлориди без подальшого додавання
кислоти азотної розведеної Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.200 г субстанції розчиняють у 80 мл кислоти оцтової
льодяної Р і титрують 0.1 М розчином кислоти хлорної
потенціометрично (2.2.20).
1 мл 0.1 Мрозчину кислоти хлорної відповідає 26.63 мг
С|4Н22^О3.
ДОМІШКИ
А. 2-(4-гідроксифеніл)ацетамід,
В. 2-[4-(2,3-дигідроксипропокси)феніл]ацетамід,
С. 2-[4-(2,3-епоксипропокси)феніл]апетамід,
В. 2-[4-(2-гідрокси-3-хлорпропокси)феніл]ацетамід,
Е. 4,4'-(2-гідроксипропан-1,3-дїїлдіокси)біс(феніла-
цетамід),
Г. 4,4'-[А-ізопропіл-3,3'-імінобіс(2-гідроксипропо-
кси)]біс(фенілацетамід),
С. 2-[4-(2-гідрокси-3-ізопропіламінопропокси)фе-
ніл]оі ітова кислота,
Н. 2-[4-(2-гідрокси-3-ізопропіламінопропокси)фе-
ніл]ацетонітрил.
_______________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
АТРОПІНУ СУЛЬФАТ
Аїгоріпі 8и1Га8
АТКОРШЕ ЕЦЬРНАТЕ
С34Н48^ОІ08,Н2О
М. м. 695
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
323
Атропіну сульфат
Атропіну сульфат містить не менше 99.0 % і не більше
101.0 % біс(1Л,Зг,55)-3-[(А5)-(3-гідрокси-2-феніл-
пропіоніл)окси]-8-метил-8-азабіцикло[3.2.1]октану
сульфату, у перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Дуже легко розчинний у воді Р, легко роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р
(Плавиться при температурі близько 190 °С із розкла-
данням. Визначення проводять із субстанції, висуше-
ної при температурі 135 °С протягом 15 хв.)
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, Е.
Друга ідентифікація: С, В, Е, Г.
А. Водний розчин субстанції, приготований для ви-
пробування "Оптичне обертання", як зазначено в
розділі "Випробування на чистоту", майже не має оп-
тичного обертання.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ атропіну сульфа-
ту.
С. Близько 50 мг субстанції розчиняють у 5 мл води Рі
додають 5 мл розчину кислоти пікринової Р. Температу-
ра плавлення (2.2.14) осаду, промитого водою Рі вису-
шеного при температурі від 100 °С до 105 °С протягом
2 год, має бути від 174 °С до 179 °С.
Ц. До близько 1 мг субстанції додають 0.2 мл кислоти
азотної димлячої Рі упарюють насухо на водяній бані.
Зал ишок розчиня ють у 2 мл ацетону Р і додають 0.1 мл
розчину 30 г/л калію гідроксиду Р у метанолі Р; з’яв-
ляється фіолетове забарвлення.
Е. Субстанція лає реакції на сульфати (2.3.1).
Е. Субстанція дає реакцію на алкалоїди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
рН (2.2.3). Від 4.5 до 6.2. 0.6 г субстанції розчиняють у
воді, вільній від вуглецю діоксиду, Р і доводять об’єм
розчину тим самим розчинником до 30 мл.
Оптичне обертання (2.2.7). Від -0.50° до +0.05°. 2.50 г
субстанції розчиняють у воді Р, доводять об’єм розчи-
ну тим самим розчинником до 25.0 мл і вимірюють кут
оптичного обертання одержаного розчину в кюветі з
товщиною шару 2 дм.
Сторонні алкалоїди і продукти розкладання. Визначення
проводять методом тонкошарової хроматографії
(2.2.27), використовуючи як тонкий шар силікагель сР.
Випробовуваний розчин. 0.2 г субстанції розчиняють)
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Розчин порівняння (а). 1 мл випробовуваного розчину
доводять метанолом Р до об’єму 100 мл.
Розчин порівняння (Ь). 5 мл розчину порівняння (а) до-
водять метанолом Рдо об’єму 10 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (200 мкг) випробовуваного розчину, 10 мкл
(2 мкг) розчину порівняння (а) і 10 мкл (1 мкг) розчи-
ну порівняння (Ь). Пластинку поміщають у камеру із
сумішшю розчинників ацетон Р - вода Р - розчин
аміаку концентрований Р (90:7:3). Коли фронт роз-
чинників пройде 10 см від лінії старту, пластинку вий-
мають із камери, сушать при температурі від 100 °С до
105 °С протягом 15 хв, охолоджують і обприскують
розчином калію йодовісмутату розведеним Р до появи
плям.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (а)
(1.0 %), і тільки одна пляма може бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Апоатропін. Не більше 0.5 %. 0.10 г субстанції розчиня-
ють у 0.01 Мрозчині кислоти хлористоводневої і дово-
дять об’єм розчину тим самим розчинником до
100.0 мл. Вимірюють оптичну густину (2.2.25) одержа-
ного розчину за довжини хвилі 245 нм. Питомий по-
казник поглинання має бути не більше 4.0, у перера-
хунку на безводну речовину.
Вода (2.5.12). Від 2.0 % до 4.0 %. Визначення прово-
дять з 0.50 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.500 г субстанції розчиняють, якщо необхідно, при
нагріванні, у 30 мл кислоти оцтової безводної Р. Роз-
чин охолоджують і титрують 0.1 М розчином кислоти
хлорної потенціометрично (2.2.20).
1 мл 0.1 М розчин кислоти хлорної відповідає 67.68 мг
0з4Ч48^2О]|)8.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
324
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Ацетилцистеїн
АЦЕТИЛ ЦИСТЕЇН
Асеіуїсузіеіпит
ОЛ7.СГ57Ї7АЕ
С5Н9МО38
М.м. 163.2
Ацетилцистеїн містить не менше 98.0 % і не більше
101.0 % (Л)-2-ацетамідо-3-меркаптопропанової кис-
тоти, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Легко розчинний у воді Р \ 96 % спирті Р,
практично не розчинний у метиленхлориді Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: А, В, О, Е.
А. Субстанція має відповідати вимогам шодо питомо-
гооптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Температура плавлення (2.2.14). Віл 104 °С до 110 °С
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках із калію бромідом Р, має
відповідати спектру ФСЗ ацетилцистеїну.
0. На хроматограмі випробовуваного розчину (Ь).
одержаній при випробуванні "Супровідні домішки",
час утримування і плоша основного піка мають
відповідати часу утримування і площі основного піка
на хроматограмі розчину порівняння (Ь).
Е. До 0.5 мл розчину 8, приготованого, як зазначено в
розділі "Випробування на чистоту", додають 0.05 мл
розчину 50 г/л натрію нітропрусиду Р \ 0.05 мл розчи-
ну аміаку концентрованого Р; з’являється темно-фіо-
летове забарвлення.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 20 мл.
Прозорість розчину (2.2.1) Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 2.0 до 2.8. До 2 мл розчину 8 додають
8 мл води, вільної від вуглецю діоксиду, Р і перемішу-
ють.
Питоме оптичне обертання (2.2.7). Від +21.0° до +27.0°,
у перерахунку на суху речовину. До 1.25 г субстанції
додають І мл розчину 10 г/л натрію едетату Р, пе-
ремішують, додають 7.5 мл розчину 40 г/л натрію
гідроксиду Р, перемішують до розчинення субстанції і
доводять об’єм одержаного розчину фосфатним бу-
ферним розчином рН 7.0 Р2ро 25.0 мл
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29). Всі розчини, крім ви-
пробовуваного розчину (с), готують безпосередньо пе-
ред використанням.
Випробовуваний розчин (а). 0.80 г субстанції суспенду-
ють у 1 мл / М розчину кислоти хлористоводневої і до-
водять об’єм розчину водою Рдо 100 0 мл.
Випробовуваний розчин (Ь). 5.0 мл випробовуваного
розчину (а) доводять водою Рдо об’єму 100.0 мл. 5.0 мл
одержаного розчину доводять водою Р до об’єму
50.0 мл.
Випробовуваний розчин (с). Використовують випробо-
вуваний розчин (а) після зберігання не менше 1 год.
Розчин порівняння (а). 4.0 мг ФСЗ ацетилцистеїну,
4.0 мг Ь-цистину Р, 4.0 мг В-цистеїну Р, 4.0 мг ФСЗ
домішки С ацетилцистеїну і 4.0 мг ФСЗ домішки І)
ацетилцистеїну суспендують у 1 мл 1 М розчину кисло-
ти хлористоводневої і доводять об’єм розчину водою Р
до 100.0 мл.
Розчин порівняння (Ь). 4.0 мг ФСЗ ацетилцистеїну сус-
пендують у 1 мл 1М розчину кислоти хлористоводневої
і доводять об’єм розчину водою Рдо 100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4 мм,
заповнена силікагелем октадецилсилільним для
хроматографії Р з розміром часток 5 мкм;
— рухома фаза: ацетонітрил Р - вода Р (3 97); рН
одержаної суміші доводять до 3.0 кислотою фос-
форною концентрованою Р;
— швидкість рухомої фази 1.0 мл/хв;
детектування за довжини хвилі 220 нм.
При хроматографуванні за зазначених умов часи утри-
мування піків мають бути: Е-цистину - близько
2.2 хв, Б-цистеїну - близько 2.4 хв; 2-метил-2-тіазолін-
4-карбонової кислоти, що утворюється у випробову-
ваному розчині (с) - близько 3.3 хв; ацетилцистеїну -
близько 6.4 хв; домішки С ацетилцистеїну - близько
12 хв, домішки В ацетилцистеїну - близько 14 хв.
Хроматографують 20 мкл розчину порівняння (а).
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
325
Ацетилцистеїн
— коефіцієнт розділення піків Б-цистину і Б-цис-
теїну становить не менше 1.5;
— коефіцієнт розділення піків домішки С ацетилци-
стеїну і домішки О ацетилцистеїну становить не
менше 2.0.
Як контрольний дослід хроматографують 20 мкл
0.01 Мрозчину кислоти хлористоводневої.
Поперемінно хроматографують у зазначених умовах,
одержуючи не менше трьох хроматограм, 20 мкл роз-
чину порівняння (а), 20 мкл розчину порівняння (Ь),
20 мкл випробовуваного розчину (а), 20 мкл випробо-
вуваного розчину (Ь) і 20 мкл випробовуваного розчи-
ну (с). Час хроматографування має бути в 5 разів
більше часу утримування піка ацетилцистеїну і стано-
вити близько 30 хв.
Із хроматограми випробовуваного розчину (а) обчис-
люють вміст, у відсотках, конкретно зазначуваних до-
мішок і неназиваних домішок, використовуючи такі
формули:
конкретно зазначувана домішка =
Я] • т2100
А2 пі\
неназивана домішка =
Ау ту 100
Аутх
де:
А, — площа піка зазначуваної домішки (Е-цистин,
Е-цистеїн, домішка С ацетилцистеїну і доміш-
ка О ацетилцистеїну) на хроматограмі випро-
бовуваного розчину (а);
А2 — площа піка зазначуваної домішки (Е-цистин,
Е-цистеїн, домішка С ацетилцистеїну і доміш-
ка О ацетилцистеїну) на хроматограмі розчину
порівняння (а);
А3 — площа піка неназиваної домішки на хромато-
грамі випробовуваного розчину (а);
А4 — площа піка ацетилцистеїну на хроматограмі
розчину порівняння (Ь);
тх — маса наважки субстанції, узята для приготу-
вання випробовуваного розчину (а);
т2 — маса наважки зазначуваної домішки, узята для
приготування розчину порівняння (а);
т2 — маса наважки ацетилцистеїну, узята для при-
готування розчину порівняння (Ь).
Вміст кожної зазначуваної домішки і кожної ненази-
ваної домішки не має перевищувати 0.5 %. Сумарний
вміст зазначуваних і неназиваних домішок не має пе-
ревищувати 0.5 %. Не враховують пік розчинника, пік
2-метил-2-тіазолін-4-карбонової кислоти з часом ут-
римування близько 3.3 хв і будь-який пік, площа яко-
го становить менше 0.1 площі основного піка на хро-
матограмі розчину порівняння (Ь).
Важкі метали (2.4.8, метод С). Не більше 0.001 V
(10 ррт). 2.0 г субстанції мають витримувати випрой
вання на важкі метали. Еталон готують із ви кориті
ням 2 мл еталонного розчину свинцю (10ррт РЬ) Р.
П,инк. Не більше 0.001 % (10 ррт). Визначення про» 1
дять методом атомно-абсорбційної спектрометрії І
(2.2.23, метод II).
Випробовуваний розчин. 1.00 г субстанції розчиняють, І
0.001 М розчині кислоти хлористоводневої і доводять І
об’єм розчину тією самою кислотою до 50.0 мл.
Розчини порівняння. Готують розведенням етамннол І
розчину цинку (5мг/мл 7п) Р 0.001 Мрозчином кисяоїЛ 1
хлористоводневої.
Вимірюють поглинання одержаних розчинів за до-
вжини хвилі 213.8 нм, використовуючи якджерелови-
промінювання лампу з порожнистим цинковим като-
дом, повітряно-ацетиленове полум’я і метод корекції
неселективного поглинання.
Втрата в масі при висушуванні (2.2.32). Не більше
1.0 %. 1.000 г субстанції сушать у вакуумі при темпера-
турі 70 °С протягом 3 год.
Сульфатна зола (2.4.14). Не більше 0.2 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.140 г субстанції розчиняють у 60 мл води Р, додають
10 мл кислоти хлористоводневої розведеної Р, охолод-
жують на льодяній бані, додають 10 мл розчину калію
йодиду і титрують 0.05 М розчином йоду, використову-
ючи як індикатор 1 мл розчину крохмалю Р.
1 мл 0.05 М розчину йоду відповідає 16.32 мг
С5Н9КО38.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. Е-цистин,
В. Е-цистеїн,
С. А.А-діацетил-Е-цистин,
326
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Ацетилцистеїн
н
Б. \,5-діацстил-Е-цистеїн.
N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
327
Бензилпеніциліну калієва сіль
БЕНЗИЛПЕНІЦИЛІНУ КАЛІЄВА
СІЛЬ
ВепгуІрепісіПіпит каїісит
ШІУІРЕМСИиН Р0ТА88ШМ
С16Ні7К\2О48
М.м. 372.5
Бензилпеніциліну калієва сіль містить не менше
96.0 % і не більше 101.0 % калію (25,5Л,6Л)-3,3-диме-
тил-7-оксо-6-[(фенілацетил)аміно]-4-тіа-1-азабіцик-
ло[3.2.0]гептан-2-карбоксилату, у перерахунку на суху
речовину. Субстанція продукується певними штамами
РепісіПіитпоіаіит або спорідненими організмами, або
одержується будь-якими іншими способами.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Дуже легко розчинний у воді Р. практично
не розчинний у жирних оліях і вазеліновому маслі Р.
Розчин порівняння (а). 25 мг ФСЗ бензилпеніциліну
калієвої солі розчиняють у 5 мл води Р.
Розчин порівняння (Ь). 25 мг ФСЗ бензилпеніциліну
калієвої солі і 25 мг ФСЗ феноксиметилпеніциліну
калієвої солі розчиняють у 5 мл води Р.
На лінію старту хроматографічної пластинки наносять
1 мкл (5 мкг) випробовуваного розчину, 1 мкл
(5 мкг) розчину порівняння (а), 1 мкл (5 мкг бензил-
пеніциліну калієвої солі і 5 мкг феноксиметил-
пеніциліну калієвої солі) розчину порівняння (Ь).
Пластинку помішають у камеру із сумішшю розчин-
ників ацетон Р- розчин 154 г/л амонію ацетату Р, рН
якого доведено до 5.0 кислотою оцтовою льодяною Р,
(30:70). Коли фронт розчинників пройде 15 см від лінії
старту, пластинку виймають із камери, сушать на
повітрі й витримують у парі йоду до проявлення плям.
Хроматограму переглядають при денному світлі.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром і забарвленням.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
С. Близько 2 мг субстанції помішають у пробірку зав-
вишки близько 150 мм і діаметром 15 мм і змочують
0.05 мл води Р. Потім додають 2 мл розчину формаль-
дегіду в кислоті сірчаній Р і перемішують оберталь-
ними рухами: одержаний розчин має бути практично
безбарвним. Пробірку нагрівають у водяній бані
протягом і хв; поступово з’являється червонувато-ко-
ричневе забарвлення.
С. Субстанція дає реакцію (а) на калій (2.3.1).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, Б.
Друга ідентифікація: В, С, В.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ бензилпеніциліну
калієвої солі.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель н силанізований Р.
Випробовуваний розчин. 25 мг субстанції розчиняють у
5 мл води Р.
ВИПРОБУВАННЯ НАЧИСТОТУ
рН (2.2.3). Від 5.5 до 7.5. 2.0 г субстанції розчиняють у
воді, вільній від вуглецю діоксиду, Р і доводять об’єм
розчину тим самим розчинником до 20 мл.
Питоме оптичне обертання (2.2.7). Від +270с до +300°, у
перерахунку на суху речовину. 0.500 г субстанції розчи-
няють у воді, вільній від вуглецю діоксиду, Р і доводять
об’єм розчину тим самим розчинником до 25.0 мл.
Оптична густина (2.2.25). 94.0 мг субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 50.0 мл. Вимірюють оптичну густину
одержаного розчину за довжин хвиль 325 нм, 280 нм і
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
329
Бензилпеніциліну калієва сіль
264 нм (максимум), розбавивши розчин, якщо не-
обхідно, для виміру задовжини хвилі 264 нм.
Оптична густина за довжин хвиль 325 нм і 280 нм не
має перевищувати 0.10; а за довжини хвилі 264 нм
(максимум) має бути від 0.80 до 0.88, у перерахунку на
нерозведений (1.88 г/л) розчин.
Перевіряють розділювальну здатність приладу
(2.2.25); відношення оптичних густин має бути не
менше 1.7.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29), як описано у розділі
"Кількісне визначення".
20 мкл розчину порівняння (б) хроматографують в ізо-
кратичному режимі, використовуючи обрану рухому
фазу. 20 мкл випробовуваного розчину (Ь) хроматогра-
фують в ізократичному режимі. Відразу після виходу
піка бензилпеніциліну починають такий лінійний
градієнт:
Час (хв) Рухома фаза А (% об/об) Рухома фаза В (% об/об) Примітки
0-20 70—-0 зо—►ЮО ЛІНІЙНИЙ градієнт
20-35 0 100 ізократичний режим
35-50 70 зо установлення рівноваги
Як контрольний дослід хроматографують воду Р в
умовах, зазначених для випробовуваного розчину (Ь).
На хроматограмі випробовуваного розчину (Ь) площа
будь-якого піка, крім основного, не має перевищува-
ти площу основного піка на хроматограмі розчину
порівняння (б) (1 %).
Втрата в масі при висушуванні (2.2.32). Не більше 1.0 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Стерильність (2.6.1). Якщо субстанція призначена для
виробництва парентеральних лікарських засобів без
подальшої процедури стерилізації, вона має витриму-
вати випробування на стерильність.
Пірогени (2.6.8). Якшо субстанція призначена для ви-
робництва парентеральних лікарських засобів без по-
дальшої процедури видалення пірогенів, вона має ви-
тримувати випробування на пірогени. Уводять на 1 кг
маси кролика 1 мл розчину, що містить 1.5 мг суб-
станції у 1 мл води для ін’єкцій Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять методом рідинної хромато-
графії (2.2.29).
Випробовуваний розчин (а). 50.0 мг субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 50.0 мл.
Випробовуваний розчин (Ь). Розчин готують безносе- >
редньо перед використанням. 80.0 мг субстанції розчи- і
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 20.0 мл.
Розчин порівняння (а). 50.0 мг ФСЗ бензилпеніциліну
натрієвої солі розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 50.0 мл.
Розчин порівняння (Ь). 10 мг ФСЗ бензилпеніциліну
натрієвої солі і 10 мг ФСЗ кислоти фенілоцтовоїроз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 50 мл.
Розчин порівняння (с). 1.0 мл розчину порівняння (а)
доводять водою Рао об’єму 20.0 мл. 1.0 мл одержаного
розчину доводять водою Рао об’єму 50.0 мл.
Розчин порівняння (сі). 4.0 мл розчину порівняння (а)
доводять водою Рао об’єму 100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка розміром 0.25 м х 4.6 мм, заповнена
силікагелем октадецилсилільним для хромато-
графії Р, з розміром часток 5 мкм;
— рухома фаза А: розчин 68 г/л калію дигідрофосфа-
ту Р, рН якого доведено до 3.5 розчином 500 г/л
кислоти фосфорної розведеної Р - метанол Р
вода Р( 10:30:60);
— рухома фаза В: розчин 68 г/л калію дигідрофосфа-
ту Р, рН якого доведено до 3.5 розчином 500 г/л
кислоти фосфорної розведеної Р - вода Р - мета-
нол Р( 10:40:50);
— швидкість рухомої фази 1.0 мл/хв;
— детектування за довжини хвилі 225 нм.
Урівноважують колонку рухомою фазою у співвідно-
шенні рухомих фаз А:В (70:30).
Хроматографують 20 мкл розчину порівняння (Ь).
Хроматографічна система вважається придатною, як-
шо виконуються такі умови:
— коефіцієнт розділення двох основних піків мас
бути не менше 6.0 (якщо необхідно, коригують
співвідношення рухомих фаз А:В);
— коефіцієнт ємності для другого піка (бензилпені-
цилін), має бути від 4.0 до 6.0.
Хроматографують 20 мкл розчину порівняння (с). Си-
стему регулюють таким чином, шоб відношення сиг-
нал/шум становило не менше 3.
Хроматографують 20 мкл розчину порівняння (а)
шість разів. Хроматографічна система вважається
придатною, якшо відносне стандартне відхилення для
площі основного піка не перевищує 1.0 %.
Вміст бензилпеніциліну калієвої солі, у відсотках,
розраховують шляхом помноження відсоткового
вмісту бензилпеніциліну натрієвої солі на 1.045.
ЗБЕРІГАННЯ
У повітронепроникному контейнері. Якщо субстанція
стерильна, зберігають у стерильному повітронепро-
никному контейнері з контролем першого розкриття.
330
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Бензилпеніциліну натрієва сіль
МАРКУВАННЯ
У необхідних випадках зазначають:
- субстанція стерильна;
- субстанція апірогенна.
ДОМІШКИ
і епімср при С*
Е. (2А5,45)-2-[((фенілацетил)аміно]метил]-5,5-диме-
тилтіазолідин-4-карбонова кислота (пенілоїнові кис-
лоти бензилпеніциліну).
А. (2£5А,6Я)-6-аміно-3,3-диметил-7-оксо-4-тіа-1 -
азабіцикло[3.2.0|гептан-2-карбонова кислота (6-амі-
нопеніциланова кислота),
В.фенілоцтова кислота,
ВИПРОБУВАННЯ НА ЧИСТОТУ
Прозорість розчину (2.2.1). 0.30 г субстанції розчиня-
ють у воді Р\ доводять об’єм розчину тим самим роз-
чинником до 10 мл. Одержаний розчин має бути про-
зорим.
Кольоровість розчину (2.2.2, метод 11). Розчин, приго-
тований для випробування "Прозорість розчину", має
бути не інтенсивнішим за еталон ¥7.
Аномальна токсичність (2.6.9). Якщо субстанція при-
значена для виробництва парентеральних лікарських
засобів, вона має витримувати випробування на ано-
мальну токсичність.
С. (25',5/(,6/?)-6-[[(4-гідроксифеніл)ацетил]аміно|-
3,3-диметил-7-оксо-4-тіа-1-азабіцикло13.2.0]гептан-
2-карбонова кислота,
БЕНЗИЛПЕНІЦИЛІНУ
НАТРІЄВА СІЛЬ
ВепхуІрепісіПіпит паігісит
НЕІ\7ЛІРЕІ\ІСіи.1І\ 9ОІНІМ
В. (35,7/?,7аЯ)-5-бензил-2,2-диметил-2,3,7,7а-тет-
рагідроімідазо[5,1-£]тіазол-3,7-дикарбонова кислота
(пенілова кислота бензилпеніциліну),
о.
Н СО2№
,сн
н н
СІ6Н17^\аО48
н
м-
сн
М.м. 356.4
Е. (45’)-2-[карбокси[(фенілацетил)аміно]метил|-5,5-
диметилтіазолідин-4-карбонова кислота (пеніцило-
їнові кислоти бензилпеніциліну),
Бензилпеніциліну натрієва сіль містить не менше 96.0 %
і не більше 101.0 % натрію (25,5/?,6Я)-3.3-диметил-7-
оксо-6-[ (феніл ацетил )аміно]-4-тіа-1 -азабіцик-
ло|3.2.0|гептан-2-карбоксилату, у перерахунку на суху
речовину. Субстанція продукується певними штамами
РепісіИіит поіаіит або спорідненими організмами, або
одержується будь-якими іншими способами.
N
з
з
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
331
Бензилпеніциліну натрієва сіль
ВИРОБНИЦТВО
Якшо при одержанні субстанції використовують тех-
нологію, за якої можлива наявність у ній залишкових
кількостей 2-етилгексанової кислоти, субстанція має
витримувати таке випробування:
2-Етилгексанова кислота. Не більше 0.5 % (м/м). Ви-
значення проводять методом газової хроматографії
(2.2.28), використовуючи відповідну валідовану мето-
дику.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Дуже легко розчинний у воді Р, практично
не розчинний у жирних оліях і вазеліновому маслі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: В, С, В.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ бензилпеніциліну
натрієвої солі.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель н силанізований Р.
Випробовуваний розчин. 23 мг субстанції розчиняють у
5 мл води Р.
Розчин порівняння (а). 25 мг ФСЗ бензилпеніциліну
натрієвої солі розчиняють у 5 мл води Р.
Розчин порівняння (Ь). 25 мг ФСЗ бензилпеніциліну
натрієвої солі і 25 мг ФСЗ феноксиметилпеніциліну
калієвої солі розчиняють у 5 мл води Р.
Налінію старту хроматографічної пластинки наносять
1 мкл (5 мкг) випробовуваного розчину, 1 мкл (5 мкг)
розчину порівняння (а), 1 мкл (5 мкг бензил-
пеніциліну натрієвої солі і 5 мкг феноксиметил-
пеніциліну калієвої солі) розчину порівняння (Ь).
Пластинку помішають у камеру із сумішшю розчин-
ників ацетон Р- розчин 154 г/л амонію ацетату Р, рН
якого доведено до 5.0 кислотою оцтовою льодяною Р,
(30:70). Коли фронт розчинників пройде 15 см від лінії
старту, пластинку виймають із камери, сушать на
повітрі й витримують у парі йоду до проявлення плям.
Хроматограму переглядають при денному світлі.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром і забарвленням.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
С. Близько 2 мг субстанції поміщають у пробірку заіг
вишки близько 150 мм і діаметром 15 мм і змочую™
0.05 мл води Р. Потім додають 2 мл розчину формаль-
дегіду в кислоті сірчаній Р і перемішують обертальними
рухами; одержаний розчин має бути практично безбарві
ним. Пробірку нагрівають у водяній бані 1 хв; поступо-
во з’являється червонувато-коричневе забарвлення.
Ц. Субстанція дає реакцію (а) на натрій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
рН (2.2.3). Від 5.5 до 7.5. 2.0 г субстанції розчиняють,
воді, вільній від вуглецю діоксиду, Р і доводять об’ец
розчину тим самим розчинником до 20 мл.
Питоме оптичне обертання (2.2.7). Від +285° до +311)',
у перерахунку на суху речовину. 0.500 г субстанції
розчиняють у воді, вільній від вуглецю діоксиду, Р і
доводять об’єм розчину тим самим розчинником
до 25.0 мл.
Оптична густина (2.2.25). 90.0 мг субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 50.0 мл. Вимірюють оптичну густин)
одержаного розчину за довжин хвиль 325 нм, 280 нм і
264 нм (максимум), розбавивши розчин, якщо не-
обхідно, для виміру за довжини хвилі 264 нм.
Оптична густина за довжин хвиль 325 нм і 280 нм має
бути не менше 0.10; а за довжини хвилі 264 нм (макси-
мум) має бути від 0.80 до 0.88, у перерахунку на нероз-
ведений (1.80 г/л) розчин.
Перевіряють розділювальну здатність приладу
(2.2.25); відношення оптичних густин має бути не
менше 1.7.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29), як описано у розділі
"Кількісне визначення".
20 мкл розчину порівняння (б) хроматографуютьвізо-
кратичному режимі, використовуючи обрану рухом;
фазу. 20 мкл випробовуваного розчину (Ь) хроматогра-
фують в ізократичному режимі. Відразу після виход)
піка бензилпеніциліну починають такий
градієнт:
Час (хв) Рухома фаза А (% об/об) Рухома фаза В (% об/об) Примітки
0-20 70—►О зо—►ЮО ЛІНІЙНИЙ градієнт;
20-35 0 100 ізократичний режим;
35-50 70 зо установлення рівноваги
Як контрольний дослід хроматографують воду Р і
умовах, зазначених для випробовуваного розчину (Ь).
На хроматограмі випробовуваного розчину (Ь) площ:
будь-якого піка, крім основного, не має перевищува
ти площу основного піка на хроматограмі розчин;
порівняння (б) (1 %).
332
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ
Бензилпеніциліну натрієва сіль
Втрата в масі при висушуванні (2.2.32). Не більше
1.0%. 1 000 г субстанції сушать при температурі від
ІОО°Сдо 105 °С.
Стерильність (2.6.1). Якщо субстанція призначена для
виробництва парентеральних лікарських засобів без
подальшої процедури стерилізації, вона має витриму-
вати випробування на стерильність.
Пірогени (2.6.8). Якшо субстанція призначена для ви-
робництва парентеральних лікарських засобів без по-
дальшої процедури видалення пірогенів, вона має ви-
тримувати випробування на пірогени. Уводять на 1 кг
маси кролика 1 мл розчину, шо містить 1.5 мг суб-
станції у 1 мл води для ін’єкцій Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять методом рідинної хромато-
графії (2.2.29).
Випробовуваний розчин (а). 50.0 мг субстанції розчиня-
ють у воді Рі доводять об’єм розчину тим самим роз-
чинником до 50.0 мл.
Випробовуваний розчин (Ь). Розчин готують безпосе-
редньо перед використанням. 80.0 мг субстанції розчи-
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 20.0 мл.
Розчин порівняння (а). 50.0 мг ФСЗ бензилпеніциліну
натрієвої солі розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 50.0 мл.
Розчин порівняння (Ь). 10 мг ФСЗ бензилпеніциліну
натрієвої солі і 10 мг ФСЗ кислоти фенілоцтової роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 50 мл.
Розчин порівняння (с). 1.0 мл розчину порівняння (а)
доводять водою Рдо об’єму 20.0 мл. 1.0 мл одержаного
розчину доводять водою Рдо об’єму 50.0 мл.
Розчин порівняння (сі). 4.0 мл розчину порівняння (а)
доводять водою Рдо об’єму 100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка розміром 0.25 м х 4.6 мм. заповнена силі-
кагелем октадецилсилільним для хроматографії Р,
з розміром часток 5 мкм;
— рухома фаза А: розчин 68 г/л калію дигідрофосфа-
ту Р, рН якого доведено до 3.5 розчином 500 г/л
кислоти фосфорної розведеної Р - метанол Р -
вода Р (10-30:60);
— рухома фаза В: розчин 68 г/л калію дигідрофосфа-
ту Р, рН якого доведено до 3.5 розчином 500 г/л
кислоти фосфорної розведеної Р - вода Р - мета-
нол Р( 10:40:50);
— швидкість рухомої фази 1 мл/хв;
— детектування за довжини хвилі 225 нм.
Урівноважують колонку рухомою фазою у співвідно-
шенні рухомих фаз А:В (70:30).
Хроматографують 20 мкл розчину порівняння (Ь).
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— коефіцієнт розділення двох основних піків має
бути не менше 6.0 (якшо необхідно, коригують
співвідношення рухомих фаз А:В);
— коефіцієнт ємності для другого піка (бензилпені-
цилін), має бути від 4.0 до 6.0.
Хроматографують 20 мкл розчину порівняння (с).
Хроматографічну систему регулюють таким чином,
щоб відношення сигнал/шум становило не менше 3.
Хроматографують 20 мкл розчину порівняння (а)
шість разів. Хроматографічна система вважається при-
датною, якшо відносне стандартне відхилення для
площі основного піка не перевищує 1.0 %.
Поперемінно хроматографують випробовуваний роз-
чин (а) і розчин порівняння (а).
ЗБЕРІГАННЯ
У повітронепроникному контейнері. Якщо субстанція
стерильна, зберігають у стерильному повітронепро-
никному контейнері з контролем першого розкриття.
МАРКУВАННЯ
У необхідних випадках зазначають:
— субстанція стерильна;
— субстанція апірогенна.
ДОМІШКИ
А. (25,5/(,6А)-6-аміно-3,3-диметил-7-оксо-4-тіа-1-
азабіцикло[3.2.0|гептан-2-карбонова кислота (6-амі-
нопеніниланова кислота),
В. фенілоцтова кислота.
С. (25'.5А,6А)-6-|[(4-гідроксифеніл)ацетил]аміно]-
3,3-диметил-7-оксо-4-тіа-1-азабіцикло[3.2.0]гептан-
2-карбонова кислота,
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
333
Бісакодил
ВІ8АСОПУС
БІСАКОДИЛ
Візасодуіит
Б. (38,7К.,7аВ.)-5-бензил-2,2-диметил-2,3,7,7а-тет-
рагідроімідазо[5,1 -Л]тіазол-3,7-дикарбонова кислота
(пенілова кислота бензилпеніциліну),
Е. (45)-2-[карбокси[(фенілацетил)аміно]метил]-5,5-
диметилтіазолідин-4-карбонова кислота (пеніци-
лоїнові кислоти бензилпеніциліну),
М.м. 361.4
Е. (2Я5,45)-2-1[(фенілацетил)аміно]метил|-5,5-диме-
тилтіазолідин-4-карбонова кислота (пенілоїнові кис-
лоти бензилпеніциліну).
Бісакодил містить не менше 98.0 % і не більше 101.0 %
4,4'-(2-піридилметилен)дифеніл діацетату, у перера-
хунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Практично не розчинний у воді Р, розчин-
ний в ацетоні Р, помірно розчинний у 96 % спирті Р.
мало розчинний в ефірі Р.
(Розчиняється в розведених мінеральних кислотах).
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 0.30 г субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл. Одержаний розчин має бути про-
зорим.
Кольоровість розчину (2.2.2, метод II). Розчин, приго-
тований для випробування "Прозорість розчину", має
бути не інтенсивнішим за еталон ¥7.
Аномальна токсичність (2.6.9). Якщо субстанція при-
значена для виробництва парентеральних лікарських
засобів, вона має витримувати випробування на ано-
мальну токсичність.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: С.
Друга ідентифікація: А, В, П.
А. Температура плавлення (2.2.14). Від 131 °С до
135 °С.
В. 10.0 мг субстанції розчиняють у розчині 6 г/л калію
гідроксиду Ру метанолі Р, доводять об’єм розвинутим
самим розчинником до 100.0 мл. 10.0 мл одержано-
го розчину доводять тим самим розчинником до
об’єму 100.0 мл. Ультрафіолетовий спектр поглинання
(2.2.25) одержаного розчину в області від 220 нм до
350 нм повинен мати максимум за довжини хвилі
248 нм і плече за довжини хвилі близько 290 нм. Пито-
мий показник поглинання в максимумі має бути від
632 до 672.
С. Інфрачервоний спектр поглинання (2.2.24) субстанції
має відповідати спектру ФСЗ бісакодилу. У випадку
334
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Бромгексину гідрохлорид
різниці одержаних спектрів окремо розчиняють суб-
танцію і ФСЗ бісакодилу у хлороформі Р, упарюють на-
охо і повторно записують спектри одержаних залишків.
В.Хроматограму, одержану при випробуванні "Супро-
відні домішки", обприскують сумішшю рівних об’ємів
9.05 М розчину йоду і кислоти сірчаної розведеної Р,
потім переглядають при денному світлі.
На хроматограмі випробовуваного розчину (Ь) має ви-
івлятися основна пляма на рівні основної плями на
іроматограмі розчину порівняння (а), відповідна їй за
розміром.
ВИПРОБУВАННЯ НА ЧИСТОТУ
істотність або лужність. До 1.0 г субстанції додають
20 мл води, вільної від вуглецю діоксиду, Р, струшують,
нагрівають до кипіння, охолоджують і фільтрують. До
(держаного розчину додають 0.2 мл 0.01 М розчину
натрію гідроксиду і 0.1 мл розчину метилового червоно-
го Р: з’являється жовте забарвлення, шо переходить у
червоне забарвлення при додаванні не більше 0.4 мл
0.01 М розчину кислоти хлористоводневої.
(Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель сг254 Р.
Випробовуваний розчин (а). 0.20 г субстанції розчиня-
ють в ацетоні Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять ацетоном Рдо об’єму 10 мл.
Розчин порівняння (а). 20 мг ФСЗ бісакодилу розчиня-
ють в ацетоні Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Розчин порівняння (Ь). 1 мл випробовуваного розчину (а)
доводять ацетоном Рдо об’єму 100 мл.
Розчин порівняння (с). 5 мл розчину порівняння (Ь) до-
водять ацетоном Рро об’єму 10 мл.
Налінію старту хроматографічної пластинки наносять
10 мкл (200 мкг) випробовуваного розчину (а), 10 мкл
(20 мкг) випробовуваного розчину (Ь), 10 мкл (20 мкг)
розчину порівняння (а), 10 мкл (2 мкг) розчину
порівняння (Ь) і 10 мкл (1 мкг) розчину порівняння (с).
Пластинку поміщають у камеру із сумішшю розчин-
ників ксилол Р - метилетилкетон Р (50:50). Коли
фронт розчинників пройде 10 см від лінії старту, плас-
тинку виймають із камери, сушать на повітрі, якщо
необхідно, нагрівають до температури від 100 °С до
105 °С і переглядають в УФ-світлі за довжини хвилі
254 нм.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
а пляму на хроматограмі розчину порівняння (Ь)
(1.0 %), і тільки одна пляма може бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (с)
(0.5 %).
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 0.500 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.300 г субстанції розчиняють у 60 мл кислоти оцтової
безводної Р і титрують 0.1 М розчином кислоти хлорної
потенціометрично (2.2.20).
1 мл 0.1 Мрозчину кислоти хлорної відповідає 36.14 мг
С22Н19М)4.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
БРОМГЕКСИНУ ГІДРОХЛОРИД
ВготИехіпі ИудгосИІогідит
ВКОМНЕХІКЕ НУВКОСНЕОКЮЕ
, неї
СІ4112| Вг2СТ\2
М.м. 412.6
Бромгексину гідрохлорид містить не менше 98.5 % і не
більше 101.5 % А-(2-аміно-3,5-дибромфенілметил)-
А-метилциклогексиламіну гідрохлориду, у перерахун-
ку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Дуже мало розчинний у воді Р, мало роз-
чиннийу 96% спирті Р і метиленхлориді Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
335
Бупренорфіну гідрохлорид
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, Е.
Друга ідентифікація: В, С, В, Е.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ бромгексину
гідрохлориду. У випадку різниці одержаних спектрів
окремо розчиняють субстанцію і ФСЗ бромгексину
гідрохлориду в мінімальному об’ємі метанолу Р, упа-
рюють насухо на водяній бані й повторно записують
спектри одержаних залишків.
В. Хроматограму, одержану при випробуванні "Су-
провідні домішки", переглядають в УФ-світлі за дов-
жини хвилі 254 нм. На хроматограмі випробовуваного
розчину (Ь) має виявлятися основна пляма на рівні ос-
новної плями на хроматограмі розчину порівняння (с),
відповідна їй за розміром.
С. Близько 25 мг субстанції розчиняють у суміші 1 мл
кислоти сірчаної розведеної Рі 50 мл води Р. До одержа-
ного розчину додають 2 мл метиленхлориду Р, 5 мл
розчину хлораміну Р і струшують; нижній шар забарв-
люється в коричнево-жовтий колір
В. Близько 1 мг субстанції розчиняють у 3 мл 0.1 М
розчину кислоти хлористоводневої. Одержаний розчин
дає реакцію на первинні ароматичні аміни (2.3.1).
Е. Близько 20 мг субстанції розчиняють в 1 мл мета-
нолу Р і додають 1 мл води Р. Одержаний розчин дає
реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель сг254 Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у метанолі Р і доводять об’єм розчину тим самим
розчинником до 5 мл.
Випробовуваний розчин (Ь). 1.0 мл випробовуваного
розчину (а) доводять метанолом Рао об’єму 10 мл.
Розчин порівняння (а). 0.5 мл випробовуваного розчи-
ну (Ь) доводять метанолом Рдо об’єму 20 мл.
Розчин порівняння (Ь). 7.5 мл розчину порівняння (а)
доводять метанолом Рао об’єму 10 мл.
Розчин порівняння (с). 20 мг ФСЗ бромгексину гідрохло-
риду розчиняють у метанолі Р і доводять об’єм розчи-
ну тим самим розчинником до 10 мл.
Налінію старту хроматографічної пластинки наносять
20 мкл (400 мкг) випробовуваного розчину (а), 20 мкл
(40 мкг) випробовуваного розчину (Ь), 20 мкл (1 мкг)
розчину порівняння (а), 20 мкл (0.75 мкг) розчину
порівняння (Ь) і 20 мкл (40 мкг) розчину порівняння (с).
Пластинку поміщають у камеру із сумішшю розчин-
ників кислота оцтова льодяна Р - вода Р - бутанол Р
(17:17:66). Коли фронт розчинників пройде 15 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й переглядають в УФ-світлі за довжини хвип
254 нм.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (а) (0.25 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) чітко видна пляма.
Втрата в масі при висушуванні (2.2.32). Не більше
1.0 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.300 г субстанції розчиняють у 70 мл 96 % спирту Р.
додають 1 мл 0.1 М розчину кислоти хлористоводневої
і титрують 0.1 М розчином натрію гідроксиду по-
тенціометрично (2.2.20). У розрахунок беруть об’єм
титранту між двома стрибками потенціалів на кривій
титрування.
1 мл 0.1 М розчину натрію гідроксиду
41.26 мгСІ4Н21Вг2СІМ2.
відповідає
ЗБЕРІГАННЯ
У захищеному від світла місці.
Залишкові кількості органічних розчинників. Суб
станція має витримувати вимоги статті (5.4).
БУПРЕНОРФІНУ ГІДРОХЛОРИД
Виргепогрізіпі Иудгосізіогідит
всрвековрніке нткосньоківЕ
, неї
С29Н42СІХО4
М. м. 504.
Бупренорфіну гідрохлорид містить не менше 98.5 %
не більше
101.5 %
(25)-2-
[17-(циклопропілметил)
4,5а-епокси-3-гідрокси-6-метокси-6а,14-етано-
14а
336
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
2001
Бупренорфіну гідрохлорид
«рфінан-7а-іл]-3,3-диметил-бутан-2-олу гідрохло-
?вду, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору
Розчинність. Помірно розчинний у воді Р, легко роз-
чинний у метанолі Р, розчинний у 96 % спирті Р,
практично не розчинний у циклогексані Р.
ІДЕНТИФІКАЦІЯ
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати еталонному спектру ДФУ бу-
пренорфіну гідрохлориду.
В. З мл розчину 8, приготованого, як зазначено в
розділі "Випробування на чистоту", дають реакцію (а)
на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8.0.250 г субстанції розчиняють у 5.0 мл мета-
нолу Р при постійному перемішуванні й доводять
об'єм розчину водою, вільною від вуглецю діоксиду, Р
до 25.0 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод П). Розчин 8 має
бути безбарвним.
Кислотність або лужність. До 10.0 мл розчину 8 дода-
ють 0.05 мл розчину метилового червоного Р\ забарв-
лення розчину має змінитися при додаванні не більше
0.2 мл 0.02 М розчину натрію гідроксиду або 0.02 М роз-
чину кислоти хлористоводневої.
Питоме оптичне обертання (2.2.7). Від -92° до -98°, у
перерахунку на суху речовину. 0.100 г субстанції роз-
чиняють у метанолі Р і доводять об’єм розчину тим са-
мим розчинником до 10.0 мл.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 25.0 мг субстанції розчиняють
у рухомій фазі й доводять об’єм розчину рухомою фа-
зою до 10.0 мл.
Розчин порівняння (а). 5 мг субстанції розчиняють у
2.0 мл метанолу Р, додають 0.25 мл 2 Мрозчину кисло-
ти хлористоводневої.
Розчин порівняння (Ь). 0.5 мл випробовуваного розчину
доводять рухомою фазою до об’єму 200.0 мл.
Розчин порівняння (с). 0.65 мл випробовуваного розчи-
ну доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (Д). 4.0 мл розчину порівняння (Ь)
доводять рухомою фазою до об’єму 10.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октадецилсилільним для
хроматографії Р з розміром часток 5 мкм;
— рухома фаза: розчин 10 г/л амонію ацетату Р - ме-
танол Р( 10:60);
— швидкість рухомої фази 1 мл/хв;
— детектування за довжини хвилі 288 нм;
— температура колонки 40 °С.
Хроматографують 20 мкл розчину порівняння (а).
Швидкість рухомої фази регулюють так, щоб час утри-
мування піка бупренорфіну становив близько 15 хв.
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— на одержаній хроматограмі має бути два піки;
— відносний час утримування першого піка віднос-
но другого піка (бупренорфін) має бути 0.93.
Поперемінно хроматографують по 20 мкл випробову-
ваного розчину і розчинів порівняння (Ь), (с) і (сі). Час
хроматографування має бути в 2.5 рази більше часу
утримування основного піка.
На хроматограмі випробовуваного розчину площа
будь-якого піка, крім основного, не має перевищува-
ти площу основного піка на хроматограмі розчину
порівняння (Ь) (0.25 %); сума площ усіх піків, крім ос-
новного, не має перевищувати площу основного піка
на хроматограмі розчину порівняння (с) (0.65 %). Не
враховують піки, площа яких менша площі основного
піка на хроматограмі розчину порівняння (<5).
Втрата в масі при висушуванні (2.2.32). Не більше
1.0%. 1.000 г субстанції сушать при температурі від
115 °С до 120 °С.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.400 г субстанції розчиняють у 40 мл кислоти оцтової
безводної Р, додають 10 мл оцтового ангідриду Р і тит-
рують 0.1 М розчином кислоти хлорної потенціомет-
рично (2.2.20).
І мл 0.1 М розчину кислоти хлорної відповідає 50.41 мг
С29Н42СІ.\О4.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
337
Бупренорфіну гідрохлорид
А. К1 = Н. К2 = СН2- СН2-СН= СН2 : (25)-2-[17-(бут-
3-еніл)-4,5а-епокси-3-гідрокси-6-метокси-6а,14-ета-
но-14а-морфінан-7а-іл]-3,3-диметилбутан-2-ол,
В. К1 = Н, К2 = Н : (25)-2-[4,5а-епокси-3-гідрокси-6-
метокси-6а,14-етано-14а-морфінан-7а-іл]-3,3-диме-
тилбутан-2-ол,
С. НІ = СН3, К2 = СК : 4,5а-епокси-7а-[( 15)-1 -гід-
рокси- 1,2,2-триметилпропіл1-3,6-диметокси-6а, 14-
етано-14а-морфінан-17-карбонітрил.
________________________________________________А
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
338
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Валін
ВАЛІН
Уаііпшп
УАЬІІЇЕ
С5Н,^О2
М.м. 117.1
Валін містить не менше 98.5 % і не більше 101.0 % (5)-
2-аміно-З-метилбутанової кислоти, у'перерахунку на
суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Розчинний у воді Р. дуже мало розчинний
в 96 % спирті Р, практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перта ідентифікація: А, В.
Друга ідентифікація А, С.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ валіну.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді Р і дово-
дять об'єм розчину тим самим розчинником до 100 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
Питоме оптичне обертання (2.2.7). Від +26.5° до +29.0°,
у перерахунку на суху речовину. 2.00 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об'єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0 10 г субстанції розчиня-
ють у кислоті хлористоводневій розведеній Рі доводять
об'єм розчину тією самою кислотою до 10 мл
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рдо об'єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ валіну розчиняють у
0.1 М розчині кислоти хлористоводневої і доводять
об'єм розчину тією самою кислотою до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рдо об'єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ валіну і 10 мг ФСЗ
фенілаланіну розчиняють у 0 1 М розчині кислоти хло-
ристоводневої і доводять об'єм розчину тією самою
кислотою до 25 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг валіну і 2 мкг феніла-
ланіну) розчину порівняння (с). Пластинку сушать на
повітрі й поміщають у камеру із сумішшю розчин-
ників кислота оцтова льодяна Р - вода Р - бутанол Р
(20:20:60). Коли фронт розчинників пройде 15 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й обприскують розчином нінгідрину Р. Плас-
тинку нагрівають при температурі від 100 °С до 105 °С
протягом 15 хв.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
339
Ванілін
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 10 мл роз-
чину 8 доводять водою Р до об'єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють у воді дистильованій Р і дово-
дять об'єм розчину тим самим розчинником до 15 мл.
Одержаний розчин має витримувати випробування на
сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм. змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють або суспенду-
ють у 0.5 мл води Р. До одержаного розчину або сус-
пензії додають 0.30 г магнію оксиду важкого Р і ретель-
но розтирають скляною паличкою. Нижнє годинни-
кове скло відразу накривають верхнім і нагрівають при
температурі 40 °С протягом 15 хв. Паралельно в анало-
гічних умовах готують еталон, використовуючи 0.1 мл
еталонного розчину амонію (100ррт ПН4) Р, 0.5 мл во-
ди Р і 0.30 г магнію оксиду важкого Р. Лакмусовий
папір над розчином або суспензією має бути забарвле-
ний у синій колір не інтенсивніше, ніж над еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тил ізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р
Втрата вмасі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної до пе-
реходу забарвлення від коричнювато-жовтого до зеле-
ного, використовуючи як індикатор 0.1 мл розчину
нафтолбензеїну Р.
1 мл 0.1 М розчину кислоти хлорної відповідає 11.71 мг
С5НиКО2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
________________________________________
рН (2.2.3). Від 5.5 до 7.0. Вимірюють рН розчину 8
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6 14).
ВАНІЛІН
УапіШгшт
УАМІІШ
С8Н8О3
М.м. 152.1
Ванілін містить не менше 99 0 % і не більше 101.0 % 4-
гідрокси-3-метоксибензальдегіду, у перерахунку на
суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок або голчасті кристали
білого або білого з жовтавим відтінком кольору.
Розчинність. Мало розчинний у воді Р, легко розчин-
ний у 96% спирті Р\ метанолі Р, розчинний в ефірі Р.
(Розчиняється в розведених розчинах гідроксидів
лужних металів).
340
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Ванілін
ІДЕНТИФІКАЦІЯ
І Перша ідентифікація: В.
Друга ідентифікація: А, С, В.
А. Температура плавлення (2.2.14). Від 81 °С до 84 °С.
В, Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ ваніліну.
С. Хроматограму, одержану при випробуванні "Су-
провідні домішки", після обприскування перегляда-
ють при денному світлі. На хроматограмі випробову-
ваного розчину (Ь) має виявлятися основна пляма на
рівні основної плями на хроматограмі розчину
порівняння (а), відповідна їй за розміром і забарвлен-
ням.
1). До 5 мл насиченого розчину субстанції додають
0.2 мл розчину заліза(Ш) хлориду РІ, з'являється синє
забарвлення. Одержаний розчин нагрівають до темпе-
ратури 80 °С; з'являється коричневе забарвлення; при
охолодженні утворюється білий осад.
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 1.0 г субстанції розчиняють
у 96 % спирті Р і доводять об'єм розчину тим самим
розчинником до 20 мл. Одержаний розчин має бути
прозорим.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину, приготованого для випробування "Прозорість
розчину", має бути не інтенсивнішим за еталон В6.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель се254 Р.
Випробовуваний розчин (а). 0.1 г субстанції розчиняють
у метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 5 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять метанолом Рдо об'єму 10 мл.
Розчин порівняння (а). 10 мг ФСЗ ваніліну розчиняють
у метанолі Р і доводять об'єм розчину тим самим роз-
чинником до 5 мл.
Розчин порівняння (Ь). 0.5 мл випробовуваного роз-
чину (а) доводять метанолом Р до об'єму 100 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (100 мкг) випробовуваного розчину (а), 5 мкл
(10 мкг) випробовуваного розчину (Ь), 5 мкл (10 мкг)
розчину порівняння (а) і 5 мкл (0.5 мкг) розчину
порівняння (Ь). Пластинку поміщають у ненасичену
камеру із сумішшю розчинників кислота оцтова без-
водна Р-метанол Р - метиленхлорид Р (0.5:1:98.5). Ко-
ли фронт розчинників пройде 10 см від лінії старту,
пластинку виймають із камери, сушать у струмені хо-
лодного повітря і переглядають в УФ-світлі за до-
вжини хвилі 254 нм.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Одержану хроматограму обприскують динітро-
фенілгідразину оцтово-хлористоводневим розчином Р.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Реакція з кислотою сірчаною. 50 мг субстанції розчиня-
ють у 5 мл кислоти сірчаної Р. Через 5 хв забарвлення
одержаного розчину має бути не інтенсивнішим за за-
барвлення суміші 4.9 мл жовтого основного розчину і
0.1 мл червоного основного розчину або суміші 4.9 мл
жовтого основного розчину і 0.1 мл блакитного основ-
ного розчину (2.2.2, метод І).
Втрата в масі при висушуванні (2.2.32). Не більше 1.0 %.
1.000 г субстанції сушать в ексикаторі протягом 4 год.
Сульфатна зола (2.4.14). Не більше 0.05 %. Визначення
проводять із 2.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.120 г субстанції розчиняють у 20 мл 96 % спирту Р,
додають 60 мл води, вільної від вуглецю діоксиду, Р і ти-
трують 0.1 М розчином натрію гідроксиду потенціо-
метрично (2.2.20).
1 мл 0.1 М розчину натрію гідроксиду відповідає
15.21 мгС8Н8О3.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
л. пч , ./лгігл х/і/илїиіл злої
341
Верапамілу гідрохлорид
ВЕРАПАМІЛУ ГІДРОХЛОРИД
Уегаратіїі кусігосіїїогісіит
УЕКАРАМІЕ НтКОСНЮКЮЕ
Верапамілу гідрохлорид містить не менше 99.0 % і не
більше 101.0 % (2/?5)-2-(3,4-диметоксифеніл)-5-[[2-
(3,4-диметоксифеніл)етил](метил)аміно]-2-(1-метил-
етил)пентаннітрилу гідрохлориду, у перерахунку на су-
ху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору.
Розчинність. Розчинний у воді Р, помірно розчинний у
96 % спирті Р, легко розчинний у метанолі Р.
(Плавиться при температурі близько 144 °С).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, В.
Друга ідентифікація: А, С, В.
А. 20.0 мг субстанції розчиняють у 0.01 Мрозчині кис-
лоти хлористоводневої і доводять об’єм розчину тим
самим розчинником до 100.0 мл. 5.0 мл одержаного
розчину доводять 0.01 Мрозчином кислоти хлористо-
водневої що об’єму 50.0 мл. Ультрафіолетовий спектр
поглинання (2.2.25) одержаного розчину в області від
210 нм до 340 нм повинен мати два максимуми за дов-
жин хвиль 229 нм і 278 нм і плече за довжини хвилі
близько 282 нм. Відношення оптичної густини в мак-
симумі задовжини хвилі 278 нм до оптичної густини в
максимумі за довжини хвилі 229 нм має бути від 0.35
до 0.39.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ верапамілу гідрохлориду.
С. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
підхожий силікагель із флуоресцентним індикатором з
оптимальною інтенсивністю поглинання за довжини
хвилі 254 нм.
Випробовуваний розчин. 10 мг субстанції розчиняють/
метиленхлориді Р і доводять об'єм розчину тим самил
розчинником до 5 мл.
Розчин порівняння (а). 20 мг ФСЗ верапамілу гідрохю-1
риду розчиняють у метиленхлориді Р і доводять об'ємі
розчину тим самим розчинником до 10 мл.
Розчин порівняння (Ь). 5 мг ФСЗпапаверину гідрохюри-
ду розчиняють у розчині порівняння (а) і доводять 1
об'єм розчину тим самим розчинником до 5 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (10 мкг) випробовуваного розчину, 5 мкл
(10 мкг) розчину порівняння (а), 5 мкл (5 мкг папаве- 1
рину гідрохлориду і 10 мкг верапамілу гідрохлориду) І
розчину порівняння (Ь). Пластинку помішають у ка-
меру із сумішшю розчинників діетиламін Р - цикло-
гексан Р( 15:85). Коли фронт розчинників пройде 15см
від лінії старту, пластинку виймають із камери, сушать |
на повітрі і переглядають в УФ-світлі задовжини хвилі
254 нм.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
В. Субстанція дає реакцію (Ь) на хлориди (2.3.1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 1.0 г субстанції, обережно нагріваючи, роз-
чиняють у 20.0 мл води, вільної від вуглецю діоксиду, Р.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод 11). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 4.5 до 6.0. Вимірюють рН розчину 8.
Оптичне обертання (2.2.7). Від -0.10° до +0.10°. Визна-
чення проводять, використовуючи розчин 8.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 25.0 мг субстанції розчиняють
у рухомій фазі складу першого ізократичного ступеня
і доводять об’єм розчину тією самою рухомою фазою
до 10.0 мл.
Розчин порівняння (а). 5 мг ФСЗ верапамілу гідрохюри-
ду, 5 мг ФСЗ домішки 1 верапамілу і 5 мг ФСЗдомішки М
верапамілу розчиняють у рухомій фазі складу першого
ізократичного ступеня і доводять об’єм розчину тією
самою рухомою фазою до 20 мл. І мл одержаного роз-
чину доводять рухомою фазою складу першого ізокра-
тичного ступеня до об’єму 10 мл.
342
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Верапамілу гідрохлорид
Розчин порівняння (Ь). 1.0 мл випробовуваного розчину
доводять рухомою фазою складу першого ізократич-
ного ступеня до 100.0 мл. 1.0 мл одержаного розчину
доводять рухомою фазою складу першого ізократич-
ного ступеня до об’єму 10.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
- колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
іаповнена силікагелем октадеканоїламінопропілси-
чільним для хроматографії Рз розміром часток 5 мкм;
- рухома фаза А: розчин 6.97 г/л калію гідрофосфа-
ту Р, рН якого доведено до 7.20 кислотою фосфор-
ною Р;
- рухома фаза В. ацетонітрил Р;
- швидкість рухомої фази 1.5 мл/хв у подвійному
ізократичному режимі за таких умов:
Час (хв) Рухома фаза А (% об/об) Рухома фаза В (% об/об) Примітки
0-22 63 37 перший ізократичний ступінь
22-27 63—35 37—*б5 перехід до другого ізократичного ступеня
27-35 35 65 другий ізократичний ступінь
35-36 35—► 63 65—*37 перехід до рухомої фази складу першого ізократичного ступеня
36-50 63 37 установлення рівноваги
— детектування за довжини хвилі 278 нм.
Колонку врівноважують рухомою фазою складу пер-
шого ізократичного ступеня близько 60 хв.
Хроматографують 10 мкл розчину порівняння (а). При
хроматографуванні за зазначених умов часи утриму-
вання піків мають бути: верапамілу - близько 16 хв;
домішки 1 верапамілу - близько 21 хв; домішки М ве-
рапамілу. шо елююється у вигляді подвійного піка, —
близько 32 хв. Хроматографічна система вважається
придатною, якщо коефіцієнт розділення піків вера-
памілу і домішки 1 верапамілу становить не менше 5 і
домішка М верапамілу елююється з колонки.
Хроматографують 10 мкл розчину порівняння (Ь).
Чутливість системи регулюють таким чином, щоб ви-
сота основного піка становила не менше 15 % шкали
реєструючого пристрою.
Поперемінно хроматографують 10 мкл випробовува-
ного розчину і 10 мкл розчину порівняння (Ь). На хро
матограмі випробовуваного розчину площа будь-яко-
го піка, крім основного, не має перевищувати площу
основного піка на хроматограмі розчину порівнян-
ня (Ь) (0.1 %); сума площ усіх піків, крім основного, не
має перевищувати 3 площі основного піка на хромато-
грамі розчину порівняння (Ь) (0.3 %). Не враховують
піки, площа яких становить менше 0.1 площі основ
ного піка на хроматограмі розчину порівняння (Ь).
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 1 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.400 г субстанції розчиняють у 50 мл етанолу Р, дода-
ють 5 0 мл 0.01 М розчину кислоти хлористоводневої і
титрують 0.1 Мрозчином натрію гідроксиду потенціо-
метрично (2.2.20). У розрахунок беруть об'єм титранту
між двома стрибками потенціалів на кривій титруван-
ня.
1 мл 0.1 М розчину натрію гідроксиду відповідає
49.11 мг С27Н39С1М2О4.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. А,А-біс[2-(3,4-диметоксифеніл)етил]-А,А-диме-
тилпропан-1,3-діамін,
В. 2-(3,4-диметоксифеніл)-А,-метилетанамін,
С. 2-(3,4-диметоксифеніл)-А,А-диметилетанамін,
В. 3-хлор-/У-[2-(3,4-диметоксифеніл)етил|-А-метил-
пропан-1-амін,
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
343
Верапамілу гідрохлорид
Е. Аг-СН2ОН: (3.4-диметоксифеніл)метанол,
і енантіомер
Г. (2А5)-2-(3,4-диметоксифеніл)-5-(метиламіно)-2-
(І-метилетил)пентаннітрил,
С. Аг-СНО: 3,4-диметоксибензальдегід,
і енантіомер
Н. (2/?.У)-5-[[2-(3,4-диметоксифеніл)етил|( метил)
аміно]-2-(3,4-диметоксифеніл)-2-етилпентан нітрил,
і енантіомер
І. (2/?5)-2-(3,4-диметоксифеніл)-2-[2-||2-(3,4-диме-
токсифен іл)етил|(метил)аміно|етил |-3-метилбутан-
нітрил
і енантіомер
Л. (2/?5)-2-(3,4-диметоксифеніл)-5-[[2-(3,4 диметок-
сифен іл)етил]аміно|-2-( 1 -метилетил)пентан нітрил
(А-норверапаміл),
Аг
і енантіомер
К. (2/?5)-2-(3.4-диметоксифеніл)-3-метилбутанніт-
рил.
Аг
Е. 1-(3,4-диметоксифеніл) 2-метилпропан-1-он,
М. 5,5'-[]2-(3,4-диметоксифеніл)етил]іміно]біс[2-
(3,4-диметоксифеніл)-2-(1-метил етил)пентаннітрил|,
N. 5,5'-(метиліміно)біс[2-(3,4-диметоксифеніл)-2-(І-
метилетил)пентаннітрил],
і енантіомер
О. (2/?5)-2-(3,4-диметоксифеніл)-5-[2-[2-(3.4-диме-
токсифенілїетил | (метил )а міно]-2-пропіл пентан-
нітрил.
N0 СМ
Аг-^^^^^^Аг
Н3С СН3 Н3С СН3
Р. 2,6-біс(3,4-диметоксифеніл)-2,6-біс( 1-метилетил)
гептан-1,7-динітрил.
_________________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.400 г субстанції розчиняють в 40 мл оцтового
ангідриду Р і титрують 0.1 М розчином кислоти хіорної
до появи жовтого забарвлення, використовуючи як
індикатор 0.1 мл розчину кристалічного фіолетового Р.
1 мл 0 1 М розчину кислоти хлорної відповідає 49 11 мг
С27Н(9СІМ2О4
344
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Галоперидол
ГАЛОПЕРИДОЛ
Наїорегідоіит
Галоперидол містить не менше 99.0 % і не більше
101.0 % 4-[4-(4-хлорфеніл)-4-гідроксипіперидин-1-
іл]-1-(4-фторфеніл)бутан-1-ону, у перерахунку на суху
речовину.
ВЛАСТИВОСТІ
Опис. Порошок білого або майже білого кольору.
Розчинність. Практично не розчинний у воді Р, мало
розчинний у 96 % спирті Р, у метанолі Р і метиленхло-
риді Р
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, Е.
Друга ідентифікація: А, С, О, Е.
А. Температура плавлення (2.2.14). Від 150 °С до 153 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ галоперидолу.
С. Визначення проводять методом тонкошарової хро-
матографії (2 2.27), використовуючи як тонкий шар
підхожий силікагель октадецилсилільний.
Випробовуваний розчин. 10 мг субстанції розчиняють у
метанолі Р і доводять об'єм розчину тим самим роз-
чинником до 10 мл.
Розчин порівняння (а). 10 мг ФСЗ галоперидолу розчи-
няють у метанолі Р і доводять об'єм розчину тим са-
мим розчинником до 10 мл.
Розчин порівняння (Ь). 10 мг ФСЗ галоперидолу і 10 мг
ФСЗ бромперидолу розчиняють у метанолі Рі доводять
об'єм розчину тим самим розчинником до 10 мл.
Налінію старту хроматографічної пластинки наносять
1 мкл (1 мкг) випробовуваного розчину, 1 мкл (1 мкг)
розчину порівняння (а) і 1 мкл (1 мкг галоперидолу і
1 мкг бромперидолу) розчину порівняння (Ь). Плас-
тинку поміщають у камеру із сумішшю розчинників
тетрагідрофуран Р - метанол Р- розчин 58 г/л натрію
хлориду Р (10:45:45). Коли фронт розчинників пройде
15 см від лінії старту; пластинку виймають із камери,
сушать на повітрі протягом 15 хв і переглядають в
УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
плями, що можуть бути не цілком розділені.
Ц. Близько 10 мг субстанції розчиняють у 5 мл етано-
лу Р, додають 0.5 мл розчину динітробензолу Р і 0 5 мл
2 Мрозчину калію гідроксиду спиртового Р; з’являється
фіолетове забарвлення, що через 20 хв переходить у
коричнювато-червоне.
Е. 0.1 г субстанції поміщають у фарфоровий тигель,
додають 0.5 г натрію карбонату безводного Р. нагріва-
ють над відкритим полум’ям протягом 10 хв і охолод-
жують. Потім до одержаного залишку додають 5 мл
кислоти азотної розведеної Р і фільтрують. До І мл
фільтрату додають 1 мл води Р. Одержаний розчин дає
реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Прозорість розчину (2.2.1). 0.2 г субстанції розчиняють
у 20 мл розчину 1 % (об/об) кислоти молочної Р. Одер-
жаний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування "Прозорість
розчину", має бути не інтенсивнішим за еталон ¥7.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 0.100 г субстанції розчиняють
у метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 10.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
345
Галоперидол
Розчин порівняння (а). 5.0 мг ФСЗ галоперидолу і 2.5 мг
ФСЗ бромперидолу розчиняють у метанолі Р і доводять
об’єм розчину тим самим розчинником до 50.0 мл.
Розчин порівняння (Ь). 5.0 мл випробовуваного розчину
доводять метанолом Рдо об’єму 100.0 мл. 1.0 мл одержа-
ного розчину доводять метанолом Рдо об’єму 10.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка розміром 0.1 м х 4.6 мм, заповнена силіка-
гелем октадецилсилільним, деактивованим віднос-
но основ, для хроматографії Р, з розміром час-
ток 3 мкм;
— швидкість рухомої фази 1.5 мл/хв;
— рухома фаза А; розчин 17 г/л тепірабутиламонію
гідроксиду сульфату Р;
— рухома фаза В: ацетонітрил Р;
— використовують таку програму градієнта:
Час (хв) Рухома фаза А (% об/об) Рухома фаза В (% об/об) Примітки
0- 15 90—«-50 10—-50 ЛІНІЙНИЙ градієнт
15-20 50 50 ізократичний режим
20-25 90 10 переключення на початковий склад рухомої фази
25 = 0 90 10 повторний старт градієнта
— детектування за довжини хвилі 230 нм.
ДОМІШКИ
Колонку врівноважують ацетонітрилом Р протягом
не менше 30 хв і потім урівноважують рухомою фазою
початкового складу протягом не менше 5 хв.
Хроматографують 10 мкл розчину порівняння (Ь).
Чутливість системи регулюють таким чином, щоб ви-
сота основного піка становила не менше 50 % шкали
реєструючого пристрою.
Хроматографують 10 мкл розчину порівняння (а). При
хроматографуванні за зазначених умов часи утриму-
вання піків мають бути: галоперидолу - близько 5.5 хв
і бромперидолу - близько 6 хв. Хроматографічна сис-
тема вважається придатною, якщо коефіцієнт
розділення піків галоперидолу і бромперидолу стано-
вить не менше 3.0. Якщо необхідно, регулюють вміст
ацетонітрилу в рухомій фазі або час програмування
для рухомої фази лінійного градієнта.
Як контрольний дослід хроматографують 10 мкл ме-
танолу Р.
Поперемінно хроматографують 10 мкл випробовува-
ного розчину і 10 мкл розчину порівняння (Ь).
На хроматограмі випробовуваного розчину площа будь-
якого піка, крім основного, не має перевищувати площу
основного піка на хроматограмі розчину порівняння (Ь)
(0.5 %); сума площ усіх піків, крім основного, не має пе-
ревищувати 2 площі основного піка на хроматограмі
розчину порівняння (Ь) (1 %). Не враховують піки, пло-
ща яких становить менше 0.1 площі основного піка на
хроматограмі розчину порівняння (Ь).
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять у платиновому тиглі з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.300 г субстанції розчиняють у 50 мл суміші кислота
оцтова льодяна Р - метилетилкетон Р (1:7) і титрують
0.1 М розчином кислоти хлорної, використовуючи як
індикатор 0.2 мл розчину нафтолбензеїну Р.
1 мл 0.1 М розчину кислоти хлорної відповідає 37.59 мг
С2ІН23С1Е]\О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
А. 1 -(4-фторфеніл)-4-(4-гідрокси-4-фенілпіперидин-
1-іл)бутан-1-он,
В. 4-[4-(4-хлорфеніл)-4-гідроксипіперидин-1-іл]-1-
(2-фторфеніл)бутан-1-он,
С. 4-|4-(4-хлорфеніл)-4-гідроксипіперидин-1-іл]-1
(3-етил-4-фторфеніл)бутан-1-он.
346
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гентаміцину сульфат
В. 4-[4-(4-хлорфеніл)-4-гідроксипіперидин- 1-іл]-1 -
|4-|4-(4-хлорфеніл)-4-гідроксипіперидин-1 -
іл]феніл]бутан-1-он,
Е. 4-[4-(4'-хлорбіфеніл-4-іл)-4-гідроксипіперидин-1 -
іл]-1-(4-фторфеніл)бутан-і-он,
Е. 4-[4-(3'-хлорбіфеніл-4-іл)-4-гідроксипіперидин-1-
іл]-І-(4-фторфеніл)бутан-1-он.
________________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ГЕНТАМІЦИНУ СУЛЬФАТ
Оепіатусіпі 8и1Га8
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель б Р.
Випробовуваний розчин. 25 мг субстанції розчиняють у
воді Рі доводять об’єм розчину тим самим розчинни-
ком до 5 мл.
Розчин порівняння. 25 мг ФСЗ гентаміцину сульфату
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 5 мл.
Налінію старту хроматографічної пластинки наносять
10 мкл (50 мкг) випробовуваного розчину і 10 мкл
(50 мкг) розчину порівняння. Пластинку помішають у
камеру з рухомою фазою і хроматографують. Як рухо-
му фазу використовують нижній шар суміші рівних
об’ємів розчинників метанол Р - хлороформ Р - розчин
аміаку концентрований Р. Коли фронт розчинників
пройде 15 см віл лінії старту, пластинку виймають із
камери, сушать на повітрі, обприскують розчином
нінгідрину РІ і витримують при температурі 110 °С
протягом 5 хв.
На хроматограмі випробовуваного розчину мають ви-
являтися три основні плями на рівні трьох основних
плям на хроматограмі розчину порівняння, що відпо-
відають їм за розміром і забарвленням.
С. На хроматограмі випробовуваного розчину, одер-
жаній при випробуванні "Компонентний склад", ма-
ють виявлятися чотири основні піки, що мають ті ж
часи утримування, що і чотири основні піки на хрома-
тограмі розчину порівняння.
В. Субстанція дає реакцію (а) на сульфати (2.3.1).
СЕВ'ТАМУСІІЇ ЗВІР НАТЕ
Гентаміцину сульфат являє собою суміш сульфатів ан-
тибіотиків, продукованих Місготопохрога ригригеа.
Антимікробна активність субстанції має бути не мен-
ше 590 ОД/мг, у перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Порошок білого або майже білого кольору.
Розчинність. Легко розчинний у воді Р, практично не
розчинний у 96 % спирті Р і ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: С, Б.
Пруга ідентифікація: А, В, В.
А. Близько 10 мг субстанції розчиняють у 1 мл води Рі
додають 5 мл розчину 400 г/л кислоти сірчаної Р.
Нагрівають на водяній бані протягом 100 хв, охолод-
жують і доводять об’єм розчину водою Рао 25 мл. Уль-
трафіолетовий спектр поглинання (2.2.25) одержано-
го розчину в області довжин хвиль від 240 нм до 330 нм
не повинен мати жодного максимуму.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 0.8 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 20 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон 6 шка-
ли найбільш підхожого кольору.
рН (2.2.3). Від 3.5 до 5.5. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від +107° до +121 °,
у перерахунку на безводну речовину. 2.5 г субстанції
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 25.0 мл.
Метанол. Не більше 1.0% (м/м). Визначення прово-
дять методом газової хроматографії (2.2.28), викорис-
товуючи пропанол Ряк внутрішній стандарт.
Розчин внутрішнього стандарту. 2.5 мл пропанолу Р
доводять водою Рао об’єму 500.0 мл.
Випробовуваний розчин (а). 0.50 г субстанції розчиня-
ють у 2.0 мл води Р.
правил гидрмдкопря УКРАЇНИ 2001
347
Гентаміцину сульфат
Випробовуваний розчин (Ь). 0.50 г субстанції розчиня-
ють у 1.0 мл розчину внутрішнього стандарту і
1.0 мл води Р.
Розчин порівняння До 1.25 мл метанолу Р додають
1.25 мл пропанолу Р і доводять об’єм розчину водою Р
до 500.0 мл.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
— колонка розміром 1.5-2.0 м х 2-4 мм, заповнена
сополімером етилвінілбензол-дивінілбензолу Р з
розміром часток 150-180 мкм;
— газ-носій азот для хроматографії Р;
— швидкість газу-носія від 30 мл/хв до 40 мл/хв;
— температура колонки від 120 °С до 140 °С;
— температура детектора і блока вводу проб не мен-
ше ніж на 50 °С вище температури колонки.
Поперемінно хроматографують обрані об’єми випро-
бовуваних розчинів і розчину порівняння. Вміст мета-
нолу в субстанції обчислюють, використовуючи зна-
чення його густини при температурі 20 °С, що дорі-
внює 0.792 г/мл.
Компонентний склад. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 0.10 г субстанції розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 100.0 мл. До 10.0 мл одержаного розчину дода-
ють 5 мл метанолу Р і 4 мл реактиву фталевого аль-
дегіду Р, перемішують і доводять об’єм розчину мета-
нолом Рцо 25.0 мл. Нагрівають у водяній бані при тем-
пературі 60 °С протягом 15 хв і охолоджують до
кімнатної температури. Якщо розчин не використо-
вується відразу, його охолоджують до температури
0 °С і використовують протягом 4 год.
Розчин порівняння. Приготування проводять, як опи-
сано для випробовуваного розчину, використовуючи
0 10 г ФСЗ гентаміцину сульфату замість субстанції.
Хроматографування проводять на рідинному хро-
матографі за таких умов:
— колонка з нержавіючої сталі розміром
0.10-0.125 м х 4.6-5.0 мм, заповнена силікагелем
октадецилсилільним для хроматографії Р з розмі-
ром часток 5 мкм;
— рухома фаза: розчин 5.5 г натрію гептансульфона-
ту Р у суміші 50 мл кислоти оцтової льодяної Р,
250 мл води Р і 700 мл метанолу Р;
— швидкість рухомої фази 1.5 мл/хв;
— спектрофотометричний детектор із проточною
кюветою об’ємом близько 10 мкл; детектування за
довжини хвилі 330 нм (для детектора з перемін-
ною довжиною хвилі) або 340 нм (для детектора з
фільтрами).
На хроматограмах, одержаних у зазначених умовах,
час утримування компонента С2 має бути в межах від
10 хв до 20 хв, і мають виявлятися добре розділені піки
з відносними часами утримування близько 0.13 (реак-
тив), близько 0.27 (компонент С,), близько 0.65 (ком-
понент С|а), близько 0.85 (компонент С2а) і близько
1.00 (компонент С2).
Хроматографують 5 мкл розчину порівняння. Хрома-
тографічна система вважається придатною, якщо ко-
ефіцієнт розділення (2.2.29) піків С2а і С2 становить ні
менше 1.3. Якшо необхідно, регулюють вміст метано-
лу в рухомій фазі.
Хроматографують 5 мкл розчину порівняння. Чут
ливість реєструючого пристрою і об’єм проби, ЩО вво-
диться, регулюють таким чином, щоб висота піка,
відповідного компоненту Є,, становила близько 758-
шкали реєструючого пристрою. На хроматограмі прово-
дять горизонтальну базову лінію, починаючи з точки, шо
передує піку реактиву. Для кожного компонента вимі-
рюють висоту піка відносно проведеної базової лінії.
Фактор відгуку /?х для кожного компонента обчислю-
ють за формулою:
де:
— відоме співвідношення компонентів для ком-
понента Сху ФСЗ гентаміцину сульфату;
Нх — висота піка, що відповідає компоненту Сх, на
хроматограмі розчину порівняння.
Хроматографують 5 мкл випробовуваного розчину.
Чутливість реєструючого пристрою і об’єм проби, ІЦО
вводиться, регулюють таким чином, щоб висота піка,
відповідного компоненту С], становила близько 75 %
шкали реєструючого пристрою. На хроматограмі про-
водять горизонтальну базову лінію, починаючи з точ-
ки, що передує піку реактиву. Для кожного компонен-
та вимірюють висоту піка відносно проведеної базової
лінії. Відносний вміст компонентів Сь С1а, С2іС2ав
субстанції обчислюють за формулою:
Сх(%) = ------;-----------~
Я. Н. Н. РР
/?І /?|а /?2 ^2а
100 ,
де:
Н'х — висота піка, що відповідає компоненту Сх, на
хроматограмі випробовуваного розчину.
Відносний вміст компонентів має знаходитися в таких
межах: С, - від 25.0 % до 50.0 %; С,а - від 10.0 % до
35.0 %; суми С2 і С2а - від 25.0 % до 55.0 %.
Вода (2.5.12). Не більше 15.0%. Визначення прово-
дять з 0.300 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 1.0 %. Визначення
проводять з 0 50 г субстанції.
Сульфати. Від 32.0 % до 35.0 % сульфату ($О4). у пере-
рахунку на безводну речовину. 0.250 г субстанції роз-
чиняють у 100 мл води дистильованої Р і доводять рН
розчину до 11 розчином аміаку концентрованим Р. До
одержаного розчину додають 10.0 мл 0.1 М розчину
барію хлориду, близько 0.5 мг фталеїнового пурпуро-
вого Р і титрують 0.1 М розчином натрію едетату.
348
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гістидин
додаючи 50 мл 96 % спирту Р, коли забарвлення роз-
чину почне змінюватися, і продовжують титрування
до зникнення фіолетово-блакитного забарвлення.
І мл 0.1 М розчину барію хлориду відповідає 9.606 мг
сульфату (5>О4).
Стерильність (2.6.1). Якщо субстанція призначена для
виробництва парентеральних або офтальмологічних
лікарських засобів без подальшої процедури сте-
рилізації, вона має витримувати випробування на
стерильність.
Бактеріальні ендотоксини (2.6.14). Не більше 1.67 МО у
1 мг, якщо субстанція призначена для виробництва
парентеральних лікарських засобів без подальшої
процедури видалення бактеріальних ендотоксинів.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять мікробіологічним методом
(2.7.2).
витримувати випробування на пірогени. Уводять на
І кг маси кролика 1 мл розчину, що містить 10 мг ген-
таміцину в 1 мл води для ін’єкцій Р.
Аномальна токсичність (2.6.9). Якщо субстанція при-
значена для виробництва парентеральних лікарських
засобів, вона має витримувати випробування на ано-
мальну токсичність. Уводять кожній миші протягом
ЗО с 0.5 мг гентаміцину в 0.5 мл води для ін’єкцій Р.
Термін спостереження 48 год.
Депресорні речовини (2.6.11). Якщо субстанція при-
значена для виробництва парентеральних лікарських
засобів, вона має витримувати випробування на де-
пресорні речовини.
МАРКУВАННЯ
При проведенні випробування "Пірогени" замість
субстанція вільна від бактеріальних ендотоксинів"
зазначають:
— субстанція апірогенна.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці. Якщо субстанція стерильна, зберігають у
стерильному, повітронепроникному контейнері з кон-
тролем першого розкриття.
МАРКУВАННЯ
У необхідних випадках зазначають:
- субстанція стерильна;
- субстанція вільна від бактеріальних ендотоксинів.
____________________________________________N
НІ8ТЮІІЧЕ
ГІСТИДИН
Ні81іс1іпит
с6н9іч3о2
М.м. 155.2
Гістидин містить не менше 98.5 % і не більше 101.0 %
(5)-2-аміно-3-(імідазол-4-іл)пропанової кислоти, у
перерахунку на суху речовину.
ВИРОБНИЦТВО
Гентаміцину сульфат являє собою суміш сульфатів ан-
тибіотиків — гентаміцинів С,, С1а С2 і С2а. 1 ОД
відповідає 1 мкг гентаміцину.
Опис. Гігроскопічний.
Замість випробування "Бактеріальні ендотоксини"допу-
скається застосування випробування "Пірогени"(2.6.8).
Пірогени (2.6.8). Якщо субстанція призначена для ви-
робництва парентеральних лікарських засобів без
подальшої процедури видалення пірогенів, вона має
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Розчинний у воді Р, дуже мало розчинний
у 96 % спирті Р, практично не розчинний в ефірі Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
349
Гістидин
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, Б.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗгістидину. У випадку різниці одержаних спектрів
окремо розчиняють субстанцію і ФСЗ гістидину в
мінімальному об’ємі води Р, упарюють насухо при
температурі 60 °С і повторно записують спектри одер-
жаних залишків.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлюва-
ні нінгідрином", має виявлятися основна пляма на
рівні основної плями на хроматограмі розчину порів-
няння (а), відповідна їй за розміром і забарвленням.
Б. 0.1 г субстанції розчиняють у 7 мл води Р і додають
З мл розчину 200 г/л натрію гідроксиду Р. 50 мг кисло-
ти сульфанілової Р розчиняють у суміші 0.1 мл кисло-
ти хлористоводневої Р і 10 мл води Р і додають 0.1 мл
розчину натрію нітриту Р. Другий розчин додають до
першого і перемішують; з’являється оранжево-черво-
не забарвлення.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді дистильо-
ваній Р, нагріваючи у водяній бані, і доводять об’єм
розчину тим самим розчинником до 50 мл.
Прозорість розчину (2.2.!). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
Питоме оптичне обертання (2.2.7). Від +11.8° до +12.8°,
у перерахунку на суху речовину. 2.75 г субстанції роз-
чиняють у 12 мл кислоти хлористоводневої РІ і дово-
дять об’єм розчину водою Рдо 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2 2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рао об’єму 50 мл.
Розчин порівняння (а) 10 мг ФСЗ гістидину розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ гістидину і 10 мг ФСЗ
проліну розчиняють у воді Р і доводять об’єм розчину
тим самим розчинником до 25 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг) роз-
чину порівняння (а). 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг гістидину і 2 мкг проліну)
розчину порівняння (с). Пластинку сушать на повітрі і
поміщають у камеру із сумішшю розчинників кислота
оцтова льодяна Р - вода Р - бутанол Р (20:20:60). Коли
фронт розчинників пройде 15 см від лінії старту, плас-
тинку виймають із камери, сушать на повітрі й обпри-
скують розчином нінгідрину Р. Пластинку нагрівають
при температурі від 100 °С до 105 °С протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 5 мл роз-
чину 8 доводять водою Рао об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 10 мл
розчину 8 доводять водою дистильованою Рао об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовляють
комірку, шо складається із двох годинникових стекол
діаметром 60 мм, сполучених по краях. На внутрішню
поверхню верхнього годинникового скла поміщають
шматочок червоного лакмусового паперу Р розміром
(5 х 5) мм, змочений декількома краплями води Р. 50 мг
здрібненої субстанції помішають на нижнє годиннико-
ве скло і розчиняють у 0.5 мл води Р До розчину дода-
ють 0.30 г магнію оксиду важкого Р і ретельно розтира-
ють скляною паличкою. Нижнє годинникове скло
відразу накривають верхнім і нагрівають при темпера-
турі 40 °С протягом 15 хв. Паралельно в аналогічних
умовах готують еталон, використовуючи 0.1 мл еталон-
ного розчину амонію (100 ррт ПН4) Р, 0.5 мл води Р і
0.30 г магнію оксиду важкого Р. Лакмусовий папір над
випробовуваним розчином має бути забарвлений у
синій колір не інтенсивніше, ніж над еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три разиж-
тилізобутилкепюном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод О) Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у суміші 3 мл
розчину кислоти хлористоводневої розведеної Р і 15 мл
води Р, якщо необхідно слабко нагріваючи, доводять
350
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гістидину гідрохлорид моногідрат
іб'єм розчину водою Р до 20 мл. 12 мл одержаного
зозчину мають витримувати випробування на важкі
ііетали. Еталон готують із використанням еталонного
розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2 32) Не більше
0.5%. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
Гістидину гідрохлорид моногідрат містить не менше
98.5 % і не більше 101.0 % (5)-2-аміно-3-(імідазол-4-
іл)пропанової кислоти гідрохлориду, у перерахунку на
суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.130 г субстанції розчиняють у 50 мл води Р '\ титрують
0.1 М розчином кислоти хлористоводневої потенціоме-
трично (2.2.20).
1 мл 0.1 М розчину кислоти хлористоводневої відпо-
відає 15.52 мг С6Н^3О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________N
рН (2.2.3). Від 7.0 до 8.5. 10 мл розчину 8 доводять во-
дою дистильованою Рдо об’єму 20 мл.
Залишкові кількості органічних розчинників. Субстан-
ція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
“Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
ГІСТИДИНУ ГІДРОХЛОРИД
МОНОГІДРАТ
НІ8ііс1іпі Иусігосізіогісіит топоііусігісит
НІЇПйІІУЕ ітЖОСІП.ОКПіЕ М()М)П}1)КАТЕ
С6Н10СІ^О2,Н2О
М.м. 209.6
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Легко розчинний у воді Р, мало розчинний
у 96 % спирті Р, практично не розчиннии в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, С, Г.
Друга ідентифікація: А, В, В, Е, Е.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Субстанція має відповідати вимогам щодо рН, за-
значеним у розділі "Випробування на чистоту".
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ гістидину гідрохлориду моногідрату
В. На хроматограмі випробовуваного розчину (Ь), одер-
жаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні ос-
новної плями на хроматограмі розчину порівняння (а),
відповідна їй за розміром і забарвленням.
Е. 0 1 г субстанції розчиняють у 7 мл води Р і додають
З мл розчину 200 г/л натрію гідроксиду Р 50 мг кисло-
ти сульфанілової Р розчиняють у суміші 0 1 мл кисло-
ти хлористоводневої Р \ 10 мл води Р і додають 0.1 мл
розчину натрію нітриту Р. Другий розчин додають до
першого і перемішують; з’являється оранжево-черво-
не забарвлення.
Е. Близько 20 мг субстанції дають реакцію (а) на хло-
риди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р, приготованій із води дистильова-
ної Р, і доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
351
Гістидину гідрохлорид моногідрат
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
рН (2.2.3). Від 3.0 до 5.0. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від +9.2° до +10.6°,
у перерахунку на суху речовину. 2.75 г субстанції роз-
чиняють у 12 мл кислоти хлористоводневої РІ і дово-
дять об’єм розчину водою Рао 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рао об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ гістидину гідрохлори-
ду моногідрату розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рар об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗгістидину гідрохлори-
ду моногідрату і Юмг ФСЗ проліну розчиняють у воді Р
і доводять об’єм розчину тим самим розчинником до
25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг гістидину гідрохлориду
моногідрату і 2 мкг проліну) розчину порівняння (с).
Пластинку сушать на повітрі і поміщають у камеру із
сумішшю розчинників кислота оцтова льодяна Р - во-
да Р - бутанол Р (20:20:60). Коли фронт розчинників
пройде 15 см від лінії старту, пластинку виймають із
камери, сушать на повітрі й обприскують розчином
нінгідрину Р. Пластинку нагрівають при температурі
від 100 °С до 105 °С протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якшо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 10 мл
розчину 8 доводять водою дистильованою Р до об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Рі
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв. Паралель-
но в аналогічних умовах готують еталон, використо-
вуючи 0.1 мл еталонного розчину амонію (100 ррт
ХН4) Р, 0.5 мл води Р і 0.30 г магнію оксиду важкого Р.
Лакмусовий папір над випробовуваним розчином має
бути забарвлений у синій колір не інтенсивніше, ніж
над еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 20 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Від 7.0 % до
10.0 %>. 1.000 г субстанції сушать при температурі від
145 °С до 150 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.160 г субстанції розчиняють у 50 мл води, вільної від
вуглецю діоксиду, Р і титрують 0.1 М розчином натрію
гідроксиду потенціометрично (2.2.20).
1 мл 0.1 М розчину натрію гідроксиду відповідає
19.16 мг С6Н10С1М3О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
____________________________________________N
Залишкові кількості органічних розчинників. Субстан-
ція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентераль-
них лікарських засобів без подальшої процедури вида-
лення пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
352
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гліцерин
ГЛІЦЕРИН
Сіусегоіит
опсекоь
он
С3Н8О3 М.м. 92.1
Гліцерин містить не менше 98.0 % і не більше 101.0 %
пропан-1,2,3-тріолу, у перерахунку на безводну речо-
вину.
ВЛАСТИВОСТІ
Опис. Сиропоподібна, масляниста на дотик, безбарв-
на або майже безбарвна, прозора рідина. Дуже гігро-
скопічна.
Розчинність. Змішується з водою Р і 96% спиртом Р,
малорозчинний в ацетоні Р, практично не розчинний
в ефірі Р, жирних і ефірних оліях.
ІДЕНТИФІКАЦІЯ
Перта ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Субстанція має відповідати вимогам щодо показни-
ка заломлення, зазначеним у розділі "Випробування
на чистоту".
В. До 5 мл субстанції додають 1 мл води Рі обережно
перемішують. Інфрачервоний спектр поглинання
(2.2.24) одержаного розчину має відповідати еталон-
ному спектру ДФУ гліцерину (85 %).
С. 1 мл субстанції змішують з 0.5 мл кислоти азот-
ної Р і нашаровують 0.5 мл розчину калію дихромату Р.
На межі двох шарів рідини утворюється блакитне
кільце; блакитне забарвлення не має переходити в
нижній шар протягом 10 хв.
0.1 мл субстанції й 2 г калію гідросульфату Р нагріва-
ють у випарювальній чашці; з’являються випари (ак-
ролеїн), що викликають почорніння фільтрувального
паперу, змоченого розчином калію тетрайодомеркура-
ту лужним Р.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 100.0 г субстанції доводять водою, вільною від
вуглецю діоксиду, Рдо об’єму 200.0 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод І/). 10 мл розчину 8
доводять водою Рдо об’єму 25 мл. Одержаний розчин
має бути безбарвним.
Кислотність або лужність. До 50 мл розчину 8 додают ь
0.5 мл розчину фенолфталеїну Р; розчин безбарвний.
Рожеве забарвлення розчину має з’явитися при дода-
ванні не більше 0.2 мл 0. 1 Мрозчину натрію гідроксиду.
Показник заломлення (2.2.6). Від 1.470 до 1 475.
Альдегіди. Не більше 0.0005 % (5 ррт). 7.5 мл розчину 8
поміщають у колбу із притертою скляною пробкою,
додають 7.5 мл води Р і 1.0 мл розчину парарозаніліну
знебарвленого Р. Колбу закривають пробкою, пе-
ремішують і залишають при температурі (25±1)°С
протягом 1 год. Оптична густина (2.2.25) одержаного
розчину, виміряна за довжини хвилі 552 нм, не має пе-
ревищувати оптичну густину еталона, приготованого
аналогічно до випробовуваного розчину із викорис-
танням 7.5 мл еталонного розчину формальдегіду
(5 ррт СН2О) Р
Результати аналізу вважаються вірогідними, якщо ета-
лон має рожеве забарвлення.
Ефіри. До розчину субстанції, одержаного після про-
ведення випробування "Кислотність або лужність",
додають 0. ] Мрозчин натрію гідроксиду до 10.0 мл (за-
гальний об’єм з урахуванням кількості, витраченої у
випробуванні "Кислотність або лужність"). Одержа-
ний розчин кип'ятять протягом 5 хв зі зворотним хо-
лодильником, охолоджують, додають 0.5 мл розчину
фенолфталеїну Р і титрують 0.1 М розчином кислоти
хлористоводневої; забарвлення розчину має змінитися
при додаванні не менше 8.0 мл 0.1 Мрозчину кислоти
хлористоводневої.
Домішка А та супровідні домішки. Визначення прово-
дять методом газової хроматографії (2.2.28).
Випробовуваний розчин. 10.0 мл розчину 8 доводять во-
дою Рдо об’єму 100.0 мл.
Розчин порівняння (а). 1.000 г діетиленгліколю Р розчи-
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 100.0 мл.
Розчин порівняння (Ь). 1.0 мл розчину порівняння (а)
доводять випробовуваним розчином до об’єму 10.0 мл.
1.0 мл одержаного розчину доводять випробовуваним
розчином до об’єму 20.0 мл.
Розчин порівняння (с). 1.0 мл випробовуваного розчину
змішують з 5.0 мл розчину порівняння (а) і доводять
об’єм розчину водою Рдо 100.0 мл. 1.0 мл одержаного
розчину доводять водою Рдо об’єму 10.0 мл.
Розчин порівняння (сі). 5.0 мл розчину порівняння (а)
доводять водою Рдо об’єму 100.0 мл.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
— колонка кварцова капілярна розміром
ЗО м х 0.53 мм, покрита сумішшю поперечно-зши-
того поліціанопропілфенілсилоксану (6 %) і полі-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
353
Гліцерин
диметилсилоксану (94 %), з товщиною шару
З мкм;
— температура колонки, що програмується: 100 °С у
момент уведення проби, приріст температури
7.5 °С/хв до 220 °С, при 220 °С витримують протя-
гом 4 хв;
— температура блока вводу проб і детектора 220 °С і
250 °С, відповідно;
— газ-носій гелій для хроматографії Р;
— лінійна швидкість газу-носія 38 см/с;
— поділ потоку 10:1.
Хроматографують 0.5 мкл розчину порівняння (с).
Чутливість системи регулюють таким чином, щоб ви-
сота піка домішки А становила не менше 50 % шкали
реєструючого пристрою. При хроматографуванні за
зазначених умов послідовність виходу піків має бути
такою: домішка А, гліцерин.
Хроматографічна система вважається придатною, як-
що коефіцієнт розділення піків домішки А і гліцерину
на хроматограмі розчину порівняння (с) становить не
менше 7.0.
Поперемінно хроматографують 0.5 мкл випробовува-
ного розчину, 0.5 мкл розчину порівняння (Ь) і 0.5 мкл
розчину порівняння (0). На хроматограмі випробовува-
ного розчину площа піка домішки А не має перевищу-
вати половини площі відповідного піка на хроматогра-
мі розчину порівняння (Ь) (0.1 %); площа будь-якого
піка, крім піка домішки А і піка з часом утримування
менше часу утримування гліцерину, не має перевищу-
вати половини площі піка домішки А на хроматограмі
розчину порівняння (Ь) (0.1 %); сума площ усіх піків із
часом утримування більше часу утримування гліцерину
не має перевищувати 2.5 площі піка домішки А на хро-
матограмі розчину порівняння (Ь) (0.5 %). Не врахову-
ють піки, площа яких становить менше 0.05 площі піка
домішки А на хроматограмі розчину порівняння (0).
Галогенпохідні. Не більше 0.0035 % (35 ррт). До 10 мл
розчину 8 додають 1 мл розчину натрію гідроксиду роз-
веденого Р, 5 мл води Р і 50 мг нікель-алюмінієвого
сплаву, вільного від галогенів, Р. Одержаний розчин
нагрівають на водяній бані протягом 10 хв, охолоджу-
ють і фільтрують. Колбу і фільтр промивають водою Р
до одержання 25 мл фільтрату. До 5 мл фільтрату дода-
ють 4 мл 96 % спирту Р, 2.5 мл води Р, 0.5 мл кислоти
азотної Р, 0.05 мл розчину срібла нітрату Р2, пе-
ремішують і витримують протягом 2 хв. Опалесценція
одержаного розчину не має перевищувати опалес-
ценцію еталона, приготованого одночасно з випробо-
вуваним розчином із 7.0 мл еталонного розчину хлори-
ду (5 ррт СІ) Р. 4 мл 96% спирту Р, 0.5 мл води Р,
0.5 мл кислоти азотної Р і 0.05 мл розчину срібла
нітрату Р2.
Цукри. До 10 мл розчину 8 додають 1 мл кислоти
сірчаної розведеної Р і нагрівають на водяній бані про-
тягом 5 хв. До одержаного розчину додають 3 мл
вільного від карбонатів розчину натрію гідроксиду роз-
веденого Р (приготованого, як зазначено для вільного
від карбонатів / М розчину натрію гідроксиду (4.2.2)).
перемішують і краплями додають 1 мл свіжопригото-
ваного розчину міді(ІІ) сульфату Р; розчин має бути
прозорим і блакитним. Одержаний розчин продовжу- |
ють нагрівати на водяній бані протягом 5 хв, розчин
має залишитися блакитним і не має утворюватися
осад.
Хлориди (2.4.4). Не більше 0.001 % (10 ррт). 1 мл роз-
чину 8 доводять водою Р до об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Еталон готують із використанням 1 мл еталонногороз
чину хлориду (5 ррт СІ) Р, який доводять водою Рао
об’єму 15 мл.
Важкі метали (2.4.8, метод А). Не більше 0.0005%
(5 ррт). 8 мл розчину 8 доводять водою Р до об’єму
20 мл. 12 мл одержаного розчину мають витримувати
випробування на важкі метали. Еталон готують із ви-
користанням еталонного розчину свинцю (1 ррт РЬ) Р.
Вода (2.5.12). Не більше 2.0 %. Визначення проводять
з 1.500 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.01 %. Визначення
проводять з 5.0 г субстанції після упарювання і спалю-
вання.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.1000 г субстанції ретельно перемішують з 45 мл во-
ди Р, додають 25.0 мл розчину 21.4 г/л натрію перйода-
ту Р, витримують протягом 15 хв у захищеному від
світла місці, додають 5.0 мл розчину 500 г/л ети-
ленгліколю Р і витримують протягом 20 хв у захищено-
му від світла місці. Одержаний розчин титрують 0.1 М
розчином натрію гідроксиду, використовуючи як інди-
катор 0.5 мл розчину фенолфталеїну Р.
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину натрію гідроксиду відповідає 9.21 мг
С3Н8О3.
ЗБЕРІГАННЯ
У повітронепроникному контейнері.
ДОМІШКИ
А. діетиленгліколь,
В. етиленгліколь,
С. пропіленгліколь.
__________________________________________________/V
Речовини, що містять аміни. 10 мл розчину 8 і І мл роз-
чину 2.5 г/л нінгідрину Р поміщають у колбу і витриму-
354
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гліцерин (85 %)
ють у водяній бані протягом 5 хв, потім розчин пере-
ливають у пробірку; розчин не повинен мати фіолето-
вого відтінку, припустиме лише жовтаве забарвлення.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ГЛІЦЕРИН (85 %)
Сіусегоіиш (85 рег сепіит)
СІУСЕКОЬ (85 РЕВ СЕN7)
Гліцерин (85 %) - водний розчин, містить не менше
83.5% (м/м) і не більше 88.5 % (м/м) пропан-1,2,3-
тріолу (С3Н8О3, М.м. 92.10).
ВЛАСТИВОСТІ
Опис. Сиропоподібна, масляниста на дотик, безбарв-
на або майже безбарвна, прозора рідина. Дуже гігро-
скопічна.
Розчинність. Змішується з водою Р і 96% спиртом Р.
малорозчинний в ацетоні Р, практично не розчинний
в ефірі Р, жирних і ефірних оліях.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація. А, В.
Друга ідентифікація: А, С, В.
А. Субстанція має відповідати вимогам шодо показни-
ка заломлення, зазначеним у розділі "Випробування
на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати еталонному спектру ДФУ
гліцерину (85 %).
С. 1 мл субстанції змішують з 0.5 мл кислоти азот-
ної Р і нашаровують 0.5 мл розчину калію дихромату Р.
На межі двох шарів рідини утворюється блакитне
кільце: блакитне забарвлення не має переходити в
нижній шар протягом 10 хв.
В. 1 мл субстанції й 2 г калію гідросульфату Р нагріва-
ють у випарювальній чашці; з’являється пара (акро-
леїн), що викликає почорніння фільтрувального папе-
ру, змоченого розчином калію тетрайодомеркурату
лужним Р.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 117.6г субстанції доводять водою, вільною від
вуглецю діоксиду, Рао об’єму 200.0 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). 10 мл розчину 8
доводять водою Рдо об’єму 25 мл. Одержаний розчин
має бути безбарвним.
Кислотність або лужність. До 50 мл розчину 8 додають
0.5 мл розчину фенолфталеїну Р; розчин безбарвний.
Рожеве забарвлення розчину має з’явитися при дода-
ванні не більше 0.2 мл 0.1 Мрозчину натрію гідроксиду.
Показник заломлення (2.2.6). Від 1.449 до 1.455
Альдегіди. Не більше 0.0005 % (5 ррт). 7.5 мл розчину 8
поміщають у колбу із притертою скляною пробкою,
додають 7.5 мл води Р і 1.0 мл розчину парарозаніліну
знебарвленого Р. Колбу закривають пробкою, пе-
ремішують і залишають при температурі (25±1)°С
протягом 1 год. Оптична густина (2.2.25) одержаного
розчину, виміряна за довжини хвилі 552 нм, не має пе-
ревищувати оптичну густину еталона, приготованого
аналогічно до випробовуваного розчину із викорис-
танням 7.5 мл еталонного розчину формальдегіду (5ррт
СН2О) Р.
Результати аналізу вважаються вірогідними, якщо ета-
лон має рожеве забарвлення.
Ефіри. До розчину субстанції, одержаного після про-
ведення випробування "Кислотність або лужність",
додають 0.1 М розчин натрію гідроксиду до 10 0 мл (за-
гальний об’єм з урахуванням кількості, витраченої у
випробуванні "Кислотність або лужність"). Одержа-
ний розчин кип’ятять протягом 5 хв зі зворотним хо-
лодильником, охолоджують, додають 0.5 мл розчину
фенолфталеїну Р і титрують 0.1 М розчином кислоти
хлористоводневої; забарвлення розчину має змінитися
при додаванні не менше 8.0 мл 0.1 Мрозчину кислоти
хлористоводневої.
Домішка А та супровідні домішки. Визначення прово-
дять методом газової хроматографії (2.2.28).
Випробовуваний розчин. 10.0 мл розчину 8 доводять во-
дою Ра.о об’єму 100.0 мл.
Розчин порівняння (а). 1.000 г диетиленгліколю Ррозчи-
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 100.0 мл.
Розчин порівняння (Ь). 1.0 мл розчину порівняння (а)
доводять випробовуваним розчином до об’єму 10.0 мл.
1.0 мл одержаного розчину доводять випробовуваним
розчином до об’єму 20.0 мл.
Розчин порівняння (с). 1.0 мл випробовуваного розчину
змішують з 5.0 мл розчину порівняння (а) і доводять
об’єм розчину водою Рдо 100.0 мл. 1.0 мл одержаного
розчину доводять водою Рдо об’єму 10.0 мл.
Розчин порівняння (сі). 5.0 мл розчину порівняння (а)
доводять водою Рдо об’єму 100.0 мл.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
— колонка кварцова капілярна розміром
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
355
Гліцерин (85 %)
ЗО м х 0.53 мм, покрита сумішшю поперечно-
зшитого поліціанопропілфенілсилоксану (6 %) і
полідиметилсилоксану (94 %), з товщиною шару
З мкм;
— температура колонки, що програмується: 100 °С у
момент уведення проби, приріст температури
7.5 °С/хв до 220 °С, при 220 °С витримують протя-
гом 4 хв;
— температура блока вводу проб і детектора 220 °С і
250 °С, відповідно;
— газ-носій гелій для хроматографії Р;
— лінійна швидкість газу-носія 38 см/с;
— поділ потоку 10:1.
Хроматографують 0.5 мкл розчину порівняння (с).
Чутливість системи регулюють таким чином, щоб ви-
сота піка домішки А становила не менше 50 % шкали
реєструючого пристрою. При хроматографуванні за
зазначених умов послідовність виходу піків має бути
такою: домішка А, гліцерин.
Хроматографічна система вважається придатною, як-
що коефіцієнт розділення піків домішки А і гліцерину
на хроматограмі розчину порівняння (с) становить не
менше 7.0.
Поперемінно хроматографують 0.5 мкл випробовува-
ного розчину, 0.5 мкл розчину порівняння (Ь) і 0.5 мкл
розчину порівняння (сі). На хроматограмі випробову-
ваного розчину площа піка домішки А не має переви-
щувати половини плоші відповідного піка на хромато-
грамі розчину порівняння (Ь) (0.1 %); площа будь-
якого піка, крім піка домішки А і піка з часом утриму-
вання менше часу утримування гліцерину, не має пе-
ревищувати половини площі піка домішки А на хро-
матограмі розчину порівняння (Ь) (0.1 %); сума площ
усіх піків із часом утримування більше часу утриму-
вання гліцерину не має перевищувати 2.5 площі піка
домішки А на хроматограмі розчину порівняння (Ь)
(0.5 %). Не враховують піки, площа яких становить
менше 0.05 площі піка домішки А на хроматограмі
розчину порівняння (б).
Галогенпохідні. Не більше 0.003 % (30 ррт). До 10 мл
розчину Здолають 1 мл розчину натрію гідроксиду роз-
веденого Р, 5 мл води Р і 50 мг нікель-алюмішєвого
сплаву, вільного від галогенів, Р. Одержаний розчин
нагрівають на водяній бані протягом 10 хв, охолоджу-
ють і фільтрують. Колбу і фільтр промивають водою Р
до одержання 25 мл фільтрату. До 5 мл фільтрату дода-
ють 4 мл 96 % спирту Р, 2.5 мл води Р, 0.5 мл кислоти
азотної Р, 0.05 мл розчину срібла нітрату Р2, пе-
ремішують і витримують протягом 2 хв. Опалесценія
одержаного розчину не має перевищувати опалес-
ценію еталона, приготованого одночасно з випробо-
вуваним розчином із 7.0 мл еталонного розчину хлори-
ду (5 ррт СІ) Р, 4 мл 96 % спирту Р, 0.5 мл води Р,
0.5 мл кислоти азотної Р і 0.05 мл розчину срібла
нітрату Р2.
Цукри. До 10 мл розчину 8 додають 1 мл кислоти
сірчаної розведеної Рі нагрівають на водяній бані про-
тягом 5 хв. До одержаного розчину додають 3 мл
вільного від карбонатів розчину натрію гідроксиду роз-
веденого Р (приготованого, як зазначено для вільного
від карбонатів / М розчину натрію гідроксиду (4.2.2)),
перемішують і краплями додають 1 мл свіжопригото-
ваного розчину міді(ІІ) сульфату Р; розчин має бути
прозорим і блакитним. Одержаний розчин продовжу-
ють нагрівати на водяній бані протягом 5 хв; розчин
має залишитися блакитним і не має утворюватися
осад.
Хлориди (2.4.4). Не більше 0.001 % (10 ррт). 1 мл роз-
чину 8 доводять водою Рдо об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Еталон готують із використанням 1 мл еталонного роз-
чину хлориду (5 ррт СІ) Р, який доводять водою Рдо
об’єму 15 мл.
Важкі метали (2.4.8, метод А). Не більше 0.0005%
(5 ррт). 8 мл розчину 8 доводять водою Р до об’єму
20 мл. 12 мл одержаного розчину мають витримувати
випробування на важкі метали. Еталон готують із ви-
користанням еталонного розчину свинцю (1 ррт РЬ) Р.
Вода (2.5.12). Від 12.0 % до 16.0 %. Визначення прово-
дять з 0.200 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.01 %. Визначення
проводять з 5.0 г субстанції після упарювання і спалю-
вання.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.1000 г субстанції ретельно перемішують з 45 мл во-
ди Р, додають 25.0 мл розчину 21.4 г/л натрію перйода-
ту Р, витримують протягом 15 хв у захищеному від
світла місці, додають 5.0 мл розчину 500 г/л ети-
ленгліколю Р і витримують протягом 20 хв у захищено-
му від світла місці. Одержаний розчин титрують 0.1 М
розчином натрію гідроксиду, використовуючи як інди-
катор 0.5 мл розчину фенолфталеїну Р.
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину натрію гідроксиду відповідає 9.21 мг
С3Н,О3.
ЗБЕРІГАННЯ
У повітронепроникному контейнері.
ДОМІШКИ
А. діетиленгліколь,
В. етиленгліколь,
С. пропіленгліколь.
356
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гліцерину тринітрату розчин
ГЛІЦЕРИНУ ТРИНІТРАТУ
РОЗЧИН
Оіісегоіі ігіпіїгаіІ8 зоїиііо
СИ’СЕКП ТРШІТКАТЕ 8ОШТІОХ
С,Н5І\3О9 М.м. 227.1
Епіиерину тринітрату розчин є етанольним розчином
гліцерину тринітрату із вмістом не менше 9.85 г/л і не
більше 101.0 г/л пропан-1,2,3-тріолу тринітрату. Суб-
станція має містити не менше 98.5 % і не більше
101.0% від зазначеного на етикетці вмісту гліцерину
тринітрату.
ВЛАСТИВОСТІ
Опис. Гліцерину тринітрату розчин являє собою про-
зору, безбарвну або світло-жовтого кольору рідину.
(Чистий гліцерину тринітрат являє собою безбарвну
рідину).
Розчинність. Гліцерину тринітрату розчин змішується
з ацетоном Р і етанолом Р.
(Чистий гліцерину тринітрат легко розчинний в ета-
нолі Р, змішується з ацетоном Р і не змішується з во-
дою Р)
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, Б.
Друга ідентифікація: В, С, Б.
Для розведення гліцерину тринітрату використовують
тільки абсолютний етанол, інакше з розчину можуть
осаджуватися краплі чистого гліцерину тринітрату.
Залишки і розчини, одержані після випробувань "Іден-
тифікація" і "Випробування на чистоту", нагрівають
на водяній бані протягом 5 хв із розчином натрію гідро-
ксиду розведеним Р.
А. 50 мкл розчину 10 г/л субстанції, якшо необхідно,
попередньо розведеного етанолом Р, поміщають на
диск калію броміду Р і упарюють розчинник у вакуумі.
Інфрачервоний спектр поглинання (2.2.24) субстанції
має відповідати еталонному спектру ДФУ гліцерину
тринітрату.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи ТШХ пластинки
із шаром силікагелю с Р.
Випробовуваний розчин. Кількість субстанції, від-
повідну 50 мг гліцерину тринітрату, розчиняють в аце-
тоні Р і доводять об’єм розчину тим самим розчинни-
ком до 100 мл.
Розчин порівняння. 0.05 мл ФСЗ розчину гліцерину
тринітрату доводять ацетоном Р до об’єму 1 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (2.5 мкг) випробовуваного розчину і 5 мкл роз-
чину порівняння. Пластинку поміщають у камеру із
сумішшю розчинників етилацетат Р - толуол Р
(20:80). Коли фронт розчинників пройде 10 см від лінії
старту; пластинку виймають із камери, сушать на
повітрі й обприскують свіжоприготованим розчином
крохмалю з калію йодидом Р. Витримують пластинку в
УФ-світлі за довжини хвилі 254 нм протягом 15 хв і
переглядають при денному світлі
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром і забарвленням.
С. До кількості субстанції, відповідної 10 мг гліцерину
тринітрату, додають 10 мл ефіру, вільного від пероксиду, Р.
Розчин упарюють насухо в струмені азоту при темпе-
ратурі не більше 40 СС. Одержаний залишок дає ре-
акцію на нітрати (2.3.1).
В. Субстанція має витримувати вимоги, описані у
розділі "Кількісне визначення".
ВИПРОБУВАННЯ НАЧИСТОТУ
Для розведення гліцерину тринітрату використовують
тільки абсолютний етанол, інакше з розчину можуть
осаджуватися крапельки чистого гліцерину тринітра-
ту.
Залишки і розчини, одержані після випробувань "Іден-
тифікація" і "Випробування на чистоту", нагрівають
на водяній бані протягом 5 хв із розчином натрію гід-
роксиду розведеним Р.
Кольоровість розчину (2.2.2, метод II). Якщо не-
обхідно, попередньо розводять субстанцію етано-
лом Р до одержання розчину із вмістом гліцерину
тринітрату 10 г/л. Забарвлення одержаного розчину
має бути не інтенсивнішим за еталон ¥7.
Неорганічні нітрати. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин. Якщо необхідно, розводять
субстанцію етанолом Р до одержання розчину із
вмістом гліцерину тринітрату 10 г/л.
Розчин порівняння. 5 мг калію нітрату Ррозчиняють у
1 мл води Р і доводять об’єм розчину 96 % спиртом Р
до 100 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (100 мкг) випробовуваного розчину і 10 мкл
(0.5 мкг) розчину порівняння. Пластинку поміщають
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
357
Гліцерину тринітрату розчин
у камеру із сумішшю розчинників кислота оцтова
льодяна Р- ацетон Р- толуол Р( 15:30:60). Коли фронт
розчинників пройде 15 см від лінії старту, пластинку
виймають із камери, ретельно сушать у струмені
повітря до повного зникнення запаху кислоти оцтової
і рясно обприскують свіжоприготованим розчином
крохмалю з калію йодидом Р. Пластинку витримують в
УФ-світлі за довжини хвилі 254 нм протягом 15 хв і
переглядають при денному світлі.
На хроматограмі випробовуваного розчину пляма,
відповідна нітрат-іону, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 % від
вмісту гліцерину тринітрату, у перерахунку на калію
нітрат).
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. Кількість субстанції, від-
повідну 2 мг гліцерину тринітрату, розчиняють у ру-
хомій фазі і доводять об’єм розчину рухомою фазою до
20.0 мл.
Розчин порівняння (а). 0.10 г ФСЗ розчину гліцерину
тринітрату розчиняють у рухомій фазі, додають кіль-
кість еквівалентну 1.0 мг ФСЗ пентаеритритилу тет-
ранітрату розведеного і доводять об’єм розчину рухо-
мою фазою до 100.0 мл. Одержаний розчин витриму-
ють в ультразвуковій бані і фільтрують, якщо
необхідно.
Розчин порівняння (Ь). 1.0 мл випробовуваного розчину
доводять рухомою фазою до об’єму 100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октадецилсилільним для
хроматографії Р з розміром часток 5 мкм;
— швидкість рухомої фази 1 мл/хв;
— рухома фаза: ацетонітрил Р - вода Р (1:1);
— детектування за довжини хвилі 210 нм.
Поперемінно хроматографують 20 мкл розчину
порівняння (а) і 20 мкл розчину порівняння (Ь). Чут-
ливість системи регулюють таким чином, шоб висота
основного піка на хроматограмі розчину порівнян-
ня (Ь) становила не менше 70 % шкали реєструючого
пристрою. Хроматографічна система вважається при-
датною, якщо на хроматограмі розчину порівняння (а)
коефіцієнт розділення піків гліцерину тринітрату і
пентаеритритилу тетранітрату становить не менше 2.0.
Хроматографують 20 мкл випробовуваного розчину.
Час хроматографування має бути в 3 рази більше часу
утримування основного піка.
На хроматограмі випробовуваного розчину площа
будь-якого піка, крім основного, не має перевищува-
ти площу основного піка на хроматограмі розчину
порівняння (Ь) (1 %, у перерахунку на гліцерину
тринітрат); сума площ усіх піків, крім основного, не
має перевищувати 3 площі основного піка на хромато-
грамі розчину порівняння (Ь) (3 %, у перерахунку на
гліцерину тринітрат). Не враховують піки, площа яких
становить менше 0.1 площі основного піка на хрома-
тограмі розчину порівняння (Ь).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
До кількості субстанції, відповідної 60.0 мг гліцерин)
тринітрату, додають 80 мл піридину Р і титрують 0.1 М
розчином тетрабутиламонію гідроксиду потенціомет-
рично (2.2.20).
І мл 0.1 М розчину тетрабутиламонію гідроксиду
відповідає 7.57 мг С3Н5^О<).
ЗБЕРІГАННЯ
Розведені 10 г/л розчини зберігають у захищеному від
світла місці, при температурі від 2 °С до 15 °С. Більш
концентровані розчини зберігають у захищеному від
світла місці, при температурі від 15 °С до 20 °С.
МАРКУВАННЯ
На етикетці зазначають вміст гліцерину тринітрату.
ДОМІШКИ
А. неорганічні нітрати,
/К2
О
/О. Л
41 43
В. К1 = N02, К2 = Н, ВЗ = Н : 1-нітрат пропан-1,2.3-
тріолу,
С. В1 = Н, В2 = N02, КЗ = Н : 2-нітрат пропан-1,2,3-
тріолу,
В. К1 = N02, ^2 = N02, КЗ = Н : 1,2-динітрат пропан-
1,2,3-тріолу,
Е. В1 = N02, К2 = Н, КЗ = N02: 1,3-Динітрат про-
пан- 1,2,3-тріолу.
_______________________________________________________N
РОЗЧИН НІТРОГЛІЦЕРИНУ
Еоіиїіо пііго^іусегіпі
358
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гліцин
ГЛІЦИН
Оіусіпит
сіусіке
н2м соон
с2н^о2
М.м. 75.1
Гліцин містить не менше 98.5 % і не більше 101.0 %
2-аміноетанової кислоти, у перерахунку на суху речо-
вину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору.
Розчинність. Легко розчинний у воді Р, мало розчин-
ний у 96 % спирті Р, практично не розчинний в
ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: В, С.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках з 1 мг субстанції і 0.4 г
калію броміду Р, має відповідати спектру ФСЗ гліцину.
У випадку різниці одержаних спектрів окремо
розчиняють субстанцію і ФСЗ гліцину в мінімальному
об’ємі спирту (60% об/об) Р, упарюють насухо і по-
вторно записують спектри одержаних залишків.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин. 25 мг субстанції розчиняють у
воді Рі доводять об’єм розчину тим самим розчинни-
ком до 10 мл.
Розчин порівняння. 25 мг ФСЗ гліцину розчиняють у
воді Рі доводять об’єм розчину тим самим розчинни-
ком до 10 мл.
На лінію старту хроматографічної пластинки наносять
2 мкл (5 мкг) випробовуваного розчину і 2 мкл (5 мкг)
розчину порівняння. Пластинку поміщають у камеру
із сумішшю розчинників розчин аміаку концентрова-
ний Р - пропанол Р (30:70). Коли фронт розчинників
пройде 15 см від лінії старту, пластинку виймають із
камери, сушать при температурі від 100 °С до 105 °С
протягом 10 хв і обприскують розчином 2 г/л нінгідри-
ну Р в суміші кислота оцтова розведена Р - бутанол Р
(5:95). Потім пластинку нагрівають при температурі
від 100 °С до 105 °С протягом 2 хв.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром і забарвленням.
С. 50 мг субстанції розчиняють у 5 мл води Р. До одер-
жаного розчину додають 1 мл розчину натрію гіпохло-
риту концентрованого Р, кип’ятять протягом 2 хв, до-
дають 1 мл кислоти хлористоводневої Р і кип’ятять
протягом 4-5 хв. Потім додають 2 мл кислоти хлорис-
товодневої Р і 1 мл розчину 20 г/л резорцину Р,
кип’ятять протягом І хв і охолоджують. Додають
10 мл води Рі перемішують. До 5 мл одержаного роз-
чину додають 6 мл розчину натрію гідроксиду розведе-
ного Р; розчин забарвлюється у фіолетовий колір із
зеленувато-жовтою флуоресценцією. Через кілька
хвилин забарвлення розчину переходить в оранжеве,
потім у жовте, а інтенсивна флуоресценція зали-
шається.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон ¥7.
рН (2.2.3). Від 5.9 до 6.4. 10 мл розчину 8 доводять
водою, вільною від вуглецю діоксиду, Р до об’єму
20 мл.
Хлориди (2.4.4). Не більше 0.0075 % (75 ррт). 0.67 г
субстанції розчиняють у воді Р і доводять об’єм розчи-
ну тим самим розчинником до 15 мл. Одержаний роз-
чин має витримувати випробування на хлориди.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С протягом 2 год.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
70 .0 мг субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової льодя-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
359
Глюкоза безводна
ної Р і відразу титрують 0.1 М розчином кислоти хлор-
ної потенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної відповідає 7.51 мг
С2Н^О2.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентераль-
них лікарських засобів без подальшої процедури ви-
далення пірогенів, вона має витримувати випробу-
вання "Пірогени" (2.6.8) або "Бактеріальні ендоток-
сини" (2.6. 14).
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
ГЛЮКОЗА БЕЗВОДНА
Оіпсозит апкудгісит
СІ ІСО8Е АМі}1)і«)18
с6н|2о6
М.м. 180.2
Глюкоза безводна являє собою О-(+)-глюкопіранозу.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору із солод-
ким смаком.
Розчинність. Легко розчинна у воді Р, помірно розчин-
на у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
А. Субстанція має відповідати вимогам щодо пи-
томого оптичного обертання, зазначеним у розділі
"Випробування на чистоту".
В. Визначення проводять методом тонкошарової хро- І
матографії (2.2.27), використовуючи як тонкий шар І
силікагель с Р.
Випробовуваний розчин. 10 мг субстанції розчиняютьу
суміші вода Р - метанол Р (2:3) і доводять об’єм розчи-
ну тією самою сумішшю розчинників до 20 мл.
Розчин порівняння (а). 10 мг ФСЗ глюкози розчиняють
у суміші вода Р - метанол Р (2:3) і доводять об’єм роз-
чину тією самою сумішшю розчинників до 20 мл.
Розчин порівняння (Ь). По 10 мг ФСЗ фруктози. ФСЗ
глюкози, ФСЗ лактози і ФСЗ сахарози розчиняють у
суміші вода Р - метанол Р (2:3) і доводять об’єм розчи-
ну тією самою сумішшю розчинників до 20 мл.
Налінію старту хроматографічної пластинки наносять
2 мкл (1 мкг) випробовуваного розчину, 2 мкл (І мкг)
розчину порівняння (а) і 2 мкл (по 1 мкг фруктози,
глюкози, лактози і сахарози) розчину порівняння (Ь) і
ретельно сушать. Пластинку поміщають у камеру із
сумішшю розчинників вода Р - метанол Р - кислота
оцтова безводна Р - етиленхлорид Р (10:15:25:50).
Точно відмірюють об’єми компонентів зазначеної
суміші, тому що невеликий надлишок води
призводить до помутніння. Коли фронт розчинників
пройде 15 см від лінії старту, пластинку виймають із
камери, сушать у струмені теплого повітря і відразу
повторно хроматографують зі свіжою рухомою фазою.
Пластинку сушать у струмені теплого повітря і
рівномірно обприскують розчином 0.5 г тимолу Р в
суміші 5 мл кислоти сірчаної Р і 95 мл 96% спирту Р.
Пластинку нагрівають при температурі 130 °С протя-
гом 10 хв.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром і забарвленням.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються чо-
тири чітко розділені плями.
С. 0.1 г субстанції розчиняють у 10 мл води Р, додають
З мл розчину мідно-тартратного Р і нагрівають; утво-
рюється червоний осад.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 10.0 г субстанції розчиняють у воді дисти-
льованій Р і доводять об’єм розчину тим самим роз-
чинником до 100 мл.
Прозорість розчину (2.2.1). 10.0 г субстанції розчиня-
ютьу 15 мл води Р. Одержаний розчин має бути прозо-
рим і без запаху.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину, приготованого для випробування "Прозорість
розчину", має бути не інтенсивнішим за еталон В¥7.
Кислотність або лужність. 6.0 г субстанції розчиняють у
25 мл води, вільної від вуглецю діоксиду, Р, додають
0.3 мл розчину фенолфталеїну Р; розчин безбарвний.
360
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Глюкоза безводна
Рожеве забарвлення розчину має з’явитися при дода-
ванні не більше 0.15 мл 0.1М розчину натрію гідроксиду.
Питоме оптичне обертання (2.2.7). Від +52.5° до +53.3°,
у перерахунку на безводну речовину. 10.0 г субстанції
розчиняють у 80 мл води Р, додають 0.2 мл розчину
аміаку розведеного РІ, витримують протягом 30 хв і
доводять об’єм розчину водою Рао 100.0 мл.
Сторонні цукри, розчинний крохмаль, декстрини. 1.0 г
субстанції розчиняють при кипінні в 30 мл спирту
(90 % об/об) Ріл охолоджують; розчин має залишатися
прозорим.
Сульфіти. Не більше 0.0015 % (15 ррт 8О2). 5.0 г суб-
станції розчиняють у 40 мл води Р, додають 2.0 мл
0.1 Мрозчину натрію гідроксиду і доводять об’єм роз-
чину водою Рдо 50.0 мл. До 10.0 мл одержаного розчи-
ну додають 1 мл розчину 310 г/л кислоти хлористовод-
невої Р, 2.0 мл розчину фуксину знебарвленого РІ і
2.0 мл розчину 0.5 % (об/об) формальдегіду Р. Одержа-
ний розчин витримують протягом 30 хв і вимірюють
оптичну густину (2.2.25) у максимумі за довжини
хвилі 583 нм.
Еталон готують таким чином: 76 мг натрію мета-
бісульфіту Р розчиняють у воді Р і доводять об’єм роз-
чину тим самим розчинником до 50.0 мл 5.0 мл одер-
жаного розчину доводять водою Р до об’єму 100.0 мл.
До 3.0 мл розчину додають 4.0 мл 0.1М розчину натрію
гідроксиду і доводять об’єм розчину водою Р до
100.0 мл. До 10.0 мл одержаного розчину відразу дода-
ють 1 мл розчину 310 г/л кислоти хлористоводневої Р,
2.0 мл розчину фуксину знебарвленого РІ і 2.0 мл розчи-
ну 0.5 % (об/об) формальдегіду Р. Розчин витримують
протягом ЗО хв і вимірюють оптичну густину в макси-
мумі за довжини хвилі 583 нм.
Як компенсаційний розчин для обох вимірів викорис-
товують розчин, приготований аналогічно до еталону,
із використанням 10 мл води Р, починаючи зі слів
"...додають 1 мл розчину 310 г/л кислоти хлористовод-
невої Р...".
Оптична густина випробовуваного розчину не має пе-
ревищувати оптичну густину еталона.
Хлориди (2.4.4). Не більше 0.0125 % (125 ррт). 4 мл роз-
чину 8 доводять водою Р до об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0 02 % (200 ррт) 7.5 мл
розчину 8 доводять водою дистильованою Р до об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Арсен (2.4.2, метод А). Не більше 0.0001 % (1 ррт)
1.0 г субстанції мають витримувати випробування на
арсен.
Барій. До 10 мл розчину 8 додають 1 мл кислоти сірча-
ної розведеної Р. Опалесценція одержаного розчину
відразу після приготування і через 1 год не має переви-
щувати опалесценцію суміші 1 мл води дистильова-
ної Р\ 10 мл розчину 8.
Кальцій (2.4.3). Не більше 0.02 % (200 ррт). 5 мл роз-
чину 8 доводять водою дистильованою Р до об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на кальцій.
Свинець у цукрах (2.4.10). Не більше 0.00005 %
(0.5 ррт). Субстанція має витримувати випробування
на свинець у цукрах.
Вода (2.5.12). Не більше 1.0 %. Визначення проводять
з 0.50 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14) Не більше 0.1 %. 5.0 г суб-
станції розчиняють у 5 мл води Р, додають 2 мл кисло-
ти сірчаної Р, упарюють насухо на водяній бані і про-
жарюють до постійної маси. У випадку неповного озо-
лення повторюють нагрівання з кислотою сірчаною Р.
Пірогени (2.6.8). Якщо субстанція призначена для ви-
робництва парентеральних лікарських засобів вели-
ких об’ємів, вона має витримувати випробування на
пірогени. Уводять на 1 кг маси кролика 10 мл розчину,
що містить 50 мг субстанції в 1 мл води для ін ’єкціи Р.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
МАРКУВАННЯ
У необхідних випадках зазначають:
— субстанція апірогенна.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
361
Діазепам
ВІЛ2ЕРАМ
ДІАЗЕПАМ
Оіахерашит
розчином 5 г/л кислоти сірчаної Р у метанолі Р до
об’єму 100.0 мл. Ультрафіолетовий спектр поглинання
(2.2.25) одержаного розчину в області від 325 нм до
400 нм повинен мати максимум за довжини хвилі
366 нм. Питомий показник поглинання в максимумі
має бути від 140 до 155.
С. Близько 10 мг субстанції розчиняють у 3 мл кисло-
ти сірчаної Р; розчин при перегляді в УФ-світлі за до-
вжини хвилі 365 нм виявляє зеленувато-жовту флуо-
ресценцію.
В. 80 мг субстанції поміщають у фарфоровий тигель,
додають 0.3 г натрію карбонату безводного Р і нагріва-
ють на відкритому полум’ї протягом 10 хв. Після охо-
лодження одержаний залишок розчиняють у 5 мл кис-
лоти азотної розведеної Р і фільтрують. До 1 мл
фільтрату додають 1 мл води Р; розчин дає реакцію (а)
на хлориди (2.3.1).
С16НІЗСІМ2О М.м. 284.7
Діазепам містить не менше 99.0 % і не більше 101.0 %
7-хлор-1 -метил-5-феніл-2,3-дигідро-1Н-1,4-бензо-
діазепін-2-ону, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
ксіпьору.
Розчинність. Дуже мало розчинний у воді Р, розчин-
ний у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
А. Температура плавлення (2.2.14). Від 131 °С до 135 °С.
В. Розчини готують у захищеному від яскравого світла
місці и вимірюють оптичне поглинання розчинів відразу
після приготування.
25 мг субстанції розчиняють у розчині 5 г/л кислоти
сірчаної Ру метанолі Р і доводять об’єм розчину тим
самим розчинником до 250.0 мл (розчин А). 5.0 мл
розчину А доводять розчином 5 г/л кислоти сірчаної Р
у метанолі Р до об’єму 100.0 мл. Ультрафіолетовий
спектр поглинання (2.2.25) одержаного розчину в об-
ласті від 230 нм до 330 нм повинен мати два максиму-
ми за довжин хвиль 242 нм і 285 нм. Питомий показ-
ник поглинання в максимумі за довжини хвилі 242 нм
має бути близько 1020. 25.0 мл розчину А доводять
ВИПРОБУВАННЯ НАЧИСТОТУ
Супровідні Домініки і продукти розкладу. Випробування
проводять у захищеному від яскравого світла місці.
Визначення проводять методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар
силікагель СР2І4 Р.
Випробовуваний розчин. 1.0 г субстанції розчиняють в
ацетоні Р\доводять об’єм розвинутим самим розчин-
ником до 10 мл. Розчин готують безпосередньо перед
використанням.
Розчин порівняння. 1 мл випробовуваного розчину до-
водять ацетоном Рдо об’єму 100 мл. 1 мл одержаного
розчину доводять ацетоном Рпо об’єму 10 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (0.5 мг) випробовуваного розчину і 5 мкл
(0.5 мкг) розчину порівняння. Пластинку поміщають
у камеру із сумішшю рівних об’ємів етилацетату Р і
гексану Р. Коли фронт розчинників пройде 12 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й переглядають в УФ-світлі за довжини хвилі
254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.1 %).
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 4 мл еталонного розчину свинцю (10 ррт РЬ) Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
363
Динатрію фосфат дигідрат
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %. 1.000 г субстанції сушать у вакуумі при температурі 60 °С протягом 4 год. Розчинність. Розчинний у воді Р, практично не роз- чинний у 96 % спирті Р.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення проводять з 1.0 г субстанції. ІДЕНТИФІКАЦІЯ А. Розчин 8, приготований, як зазначено в розділі "Випробування на чистоту", має слабколужну реакцію
КІЛЬКІСНЕ ВИЗНАЧЕННЯ (2.2.4).
0.500 г субстанції розчиняють у 50 мл оцтового ангід- риду Р і титрують 0.1 М розчином кислоти хлорної до жовтувато-зеленого забарвлення, використовуючи як індикатор 0.3 мл розчину нільського синього А Р. В. Субстанція має витримувати випробування "Втрата в масі при висушуванні”, як зазначено в розділі "Ви- пробування на чистоту".
І мл 0.1 М розчину кислоти хлорної відповідає 28.47 мг СІЙН|(СІХ2О. С. Розчин 8 дає реакцію (Ь) на фосфати (2.3.1). Ц. Розчин 8 дає реакцію (а) на натрій (2.3.1).
ЗБЕРІГАННЯ ВИПРОБУВАННЯ НАЧИСТОТУ
У щільно закупореному контейнері, у захищеному від світла місці. Розчин 8. 5.0 г субстанції розчиняють у воді дистильо- ваній Рі доводять об’єм розчину тим самим розчиннії-
N ком до 100 мл. Прозорість розчину (2.2 1) Розчин 8 має бути прозорим.
СИБАЗОН Кольоровість розчину (2.2.2, метод 11). Розчин 8 має
БіЬаіопит бути безбарвним.
Залишкові кількості органічних розчинників. Суб- станція має витримувати вимоги статті (5.4). Відновлюючі речовини. До 5 мл розчину 8 додають 5 мл кислоти сірчаної розведеної Р, 0.25 мл 0.02 М розчину калію перманганату і нагрівають на водяній бані про- тягом 5 хв; розчин має зберегти слабко-червоне за-
КІЛЬКІСНЕ ВИЗНАЧЕННЯ барвлення.
Допускається титрування в присутності індикатора роз- чину кристалічного фіолетового Р до зеленого забарв- лення. Паралельно проводять контрольний дослід. Натрію дигідрофосфат. Розраховують співвідношення (А) із кількостей мілілітрів 1 М розчину кислоти хлори- стоводневої (25 мл) і 1 М розчину натрію гідроксиду («і і п2, мл), витрачених при кількісному визначенні, за формулою:
ДИНАТРІЮ ФОСФАТ ДИГІДРАТ щ - 25 А ” 25 - л, |
Піпаїгіі рИозрИаз сііЬусІгісиз Одержане співвідношення не має перевищувати 0 025.
ОІ80ОШМРІІО8РНАТЕ РІНЮКАТЕ Хлориди (2.4.4). Не більше 0.04 % (400 ррт). До 2.5 мл розчину 8 додають 10 мл кислоти азотної розведеної Р і доводять водою Рпо об’єму 15 мл. Одержаний розчин
ІЧа2НРО4,2Н2О М.м. 178.0 має витримувати випробування на хлориди.
Динатрію фосфат дигідрат містить не менше 98.0 % і не більше 101.0 % На2НРО4, у перерахунку на суху ре- човину. Сульфати (2.4.13). Не більше 0.1 %. До 3 мл розчину 8 додають 2 мл кислоти хлористоводневої розведеної Р і доводять водою дистильованою Р до об’єму 15 мл. Одержаний розчин має витримувати випробування на сульфати.
ВЛАСТИВОСТІ Арсен (2.4.2, метод А). Не більше 0.0004 % (4 ррт).
Опис. Порошок білого або майже білого кольору або безбарвні кристали. 5 мл розчину 8 мають витримувати випробування на арсен.
364
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Динатрію фосфат додекагідрат
Важкі метали (2.4.8, метод А). Не більше 0.002 %
(20 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.004 % (40 ррт). 5 мл розчи-
ну 8 доводять водою Р до об’єму 10 мл. Одержаний
розчин має витримувати випробування на залізо.
Втрата в масі при висушуванні (2.2.32). Від 19.5 % до
21.0 %. 1.000 г субстанції сушать при температурі
130 °С.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
2.000 г субстанції (т, г) розчиняють у 50 мл води Р, до-
дають 25.0 мл 1 М розчину кислоти хлористоводневої і
титрують / М розчином натрію гідроксиду потенціо-
метрично (2.2.20) до першого стрибка потенціалів на
кривій титрування («р мл). Продовжують титрування
до другого стрибка потенціалів на кривій титрування
(п2 — загальний об’єм / М розчину натрію гідроксиду,
витраченого при титруванні, мл).
Вміст №2НРО4 (А), у відсотках, обчислюють за фор-
мулою:
1420-(25 - и,)
№•(100 - Д)
де:
(І — втрата в масі при висушуванні, у відсотках.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
_______________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДИНАТРІЮ ФОСФАТ
ДОДЕКАГІДРАТ
Оіпаїгіі рИоврЬаБ с1ос1еса1іус1гіси8
Р 1301)11 М РН08РНА1ЕТЮРЕСАНТРКА ТЕ
Ха2НРО4,12Н2О
М.м. 358.1
Динатрію фосфат додекагідрат містить не менше
98.0 % і не більше 101.0 % №2НРО4, у перерахунку на
безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристали безбарвні, прозорі, легко вивітрюються
Розчинність. Дуже легко розчинний у воді Р, практич-
но не розчинний у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
А. Розчин 8, приготований, як зазначено в розділі
"Випробування на чистоту", дає реакції на фосфати
(2.3.1).
В. Розчин 8 дає реакції на натрій (2.3. І).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 5.0 г субстанції розчиняють у воді дистильо-
ваній Р і доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
Відновлюючі речовини. До 5 мл розчину 8 додають 5 мл
кислоти сірчаної розведеної Р, 0.25 мл 0 02 М розчину
калію перманганату і нагрівають на водяній бані про-
тягом 5 хв; розчин має зберегти слабко-червоне за-
барвлення.
Натрій дигідрофосфат. Розраховують співвідношення
(А) із кількостей мілілітрів 1 Мрозчину кислоти хлори-
стоводневої (25 мл) і / М розчину натрію гідроксиду
(и, і п2, мл), витрачених при кількісному визначенні,
за формулою:
«2 25
Одержане співвідношення не має перевищувати 0.025.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). До 2.5 мл
розчину 8 додають 10 мл кислоти азотної розведеної Р
і доводять водою Рцо об’єму 15 мл. Одержаний розчин
має витримувати випробування на хлориди.
Сульфати (2.4. ІЗ). Не більше 0.05 % (500 ррт). До 3 мл
розчину 8 додають 2 мл кислоти хлористоводневої роз-
веденої Рі доводять водою дистильованою Рдо об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Арсен (2.4 2, метод А). Не більше 0.0002 % (2 ррт)
5 мл розчину 8 мають витримувати випробування на
арсен.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р
ПГРМСДРНД (ПДРМДКГ1ПРЯ УКРАЇНИ 2001
365
Дисульфірам
Залізо (2.4.9). Не більше 0.002 % (20 ррт). 5 мл розчи-
ну 8 доводять водою Р до об’єму 10 мл. Одержаний
розчин має витримувати випробування на залізо.
Вода (2.5.12). Від 57.0 % до 61.0 %. Визначення прово-
дять з 0.100 г субстанції напівмікрометодом, викорис-
товуючи як розчинник суміш метанол Р - диметил-
формамід Р( 10:40).
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Практично не розчинний у воді Р, легко
розчинний у метиленхлориді Р, розчинний в ефірі Р.
помірно розчинний у 96 % спирті Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
4.00 г субстанції (т, г) розчиняють у 25 мл води Р, до-
дають 25.0 мл ЇМ розчину кислоти хлористоводневе! і
титрують 1 Мрозчином натрію гідроксиду потенціоме-
трично (2.2.20) до першого стрибка потенціалів на
кривій титрування (пх, мл). Продовжують титрування
до другого стрибка потенціалів на кривій титрування
(«2 — загальний об’єм 1 М розчину натрію гідроксиду,
витраченого при титруванні, мл).
Вміст №2НРО4 (X), у відсотках, обчислюють за фор-
мулою:
1420-(25 - п.)
V— ______________1_
/и(100 - сі)
де:
сі — втрата в масі при висушуванні, у відсотках.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДИСУЛЬФІРАМ
ВізиІГігатит
ШМІНКАМ
5 8
н5с2 II II с2н5
/М— с— 8— 8— С—N
Н5С2 С2Н5
СІ0Н2(^284
М.м. 296.5
Дисульфірам містить не менше 98.5 % і не більше
101.0 % біс(діетилтіокарбамоїл) дисульфіду, у перера-
хунку на суху речовину.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Температура плавлення (2.2. !4). Від 70 °С до 73 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ дисульфіраму.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Супровідні домішки",
має виявлятися основна пляма на рівні основної пля-
ми на хроматограмі розчину порівняння (а), відповід-
на їй за розміром.
В. Близько 10 мг субстанції розчиняють у 10 мл мета-
нолу Р і додають 2 мл розчину 0.5 г/л міді(П) хлориду Р
у метанолі Р; з’являється жовте забарвлення, шо пере-
ходить у зеленувато-жовте.
ВИПРОБУВАННЯ НАЧИСТОТУ
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель із флуоресцентним інди-
катором, з оптимальною інтенсивністю поглинання за
довжини хвилі 254 нм.
Випробовуваний розчин (а). 0.20 г субстанції розчиня-
ють в етилацетаті Р і доводять об’єм розчину тим са-
мим розчинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять етилацетатом Рдо об’єму 10 мл.
Розчин порівняння (а). 10 мг ФСЗ дисульфіраму розчи-
няють в етилацетаті Р і доводять об’єм розчину тим
самим розчинником до 5 мл.
Розчин порівняння (Ь). 1 мл випробовуваного розчину (Ь)
доводять етилацетатом Рго об’єму 20 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (200 мкг) випробовуваного розчину (а), 10 мкл
(20 мкг) випробовуваного розчину (Ь). 10 мкл (20 мкг)
розчину порівняння (а), 10 мкл (10 мкг) розчину
порівняння (Ь). Пластинку помішають у камеру із
сумішшю розчинників бутилацетат Р - гексан Р
(30:70). Коли фронт розчинників пройде 15 см від лінії
старту, пластинку виймають із камери, сушать на повітрі
й переглядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
366
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Допаміну гідрохлорид
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Діетилдитіокарбамат. Не більше 0.015 % (150 ррт).
0.20 г субстанції розчиняють у 10 мл ефіру, вільного від
пероксидів, Р, додають 5 мл буферного розчину рН 8.0 Р
і ретельно збовтують. Верхній шар відкидають, а
нижній шар промивають 10 мл ефіру, вільного від пе-
роксидів, Р. До нижнього шару додають 0.2 мл розчи-
ну 4 г/л міді(ІІ) сульфату Р, 5 мл циклогексану Р і
збовтують. Жовте забарвлення нижнього шару
одержаного розчину має бути не інтенсивнішим за
забарвлення еталону, приготованого аналогічно до
випробовуваного розчину з використанням 0.2 мл
свіжоприготованого розчину 0.15 г/л натрію діетил-
дитіокарбамату Р.
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать у вакуумі при температурі
50 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.450 г субстанції розчиняють у 80 мл ацетону Р, дода-
ють 20 мл розчину 20 г/л калію нітрату Р і титрують
0.1 М розчином срібла нітрату потенціометрично
(2.2.20), використовуючи срібний і хлорсрібний елек-
троди, сполучені електролітичним містком, заповне-
ним насиченим розчином калію нітрату Р.
1 мл 0.1 М розчину срібла нітрату відповідає 59.3 мг
с10н2(№84.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДОМІШКИ
5 5
Н5С2 Ц Ц с2н5
/ЬІ — с— 5— с —
Н5С2 С2Н5
А. сульфірам,
Н5С2 х,8
—С\
НдС2 5
В. діетилдитіокарбамат.
_______________________________________N
ТЕТУРАМ
Теіигатит
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДОПАМІНУ ГІДРОХЛОРИД
Воратіпі ЬусІгосЬІогісІит
ИОРАМІМ: 1І\І)К()(ІІІ.ОКІІ)Е
, неї
С8НІ2С1ІЧО2
М.м. 189.6
Допаміну гідрохлорид містить не менше 98.0 % і не
більше 102.0 % 4-(2-аміноетил)бензол-1,2-діол гідро-
хлориду, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Легко розчинний у воді Р, розчинний у
96 % спирті Р, помірно розчинний в ацетоні Р\мети-
ленхлориді Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, Е.
Друга ідентифікація: А, С, В, Е.
А. 40.0 мг субстанції розчиняють у 0.1 Мрозчині кислоти
хлористоводневої і доводять об’єм розчину тією самою
кислотою до 100.0 мл. 10.0 мл розчину доводять 0.1 М
розчином кислоти хлористоводневої до об’єму 100.0 мл.
Ультрафіолетовий спектр поглинання (2.2.25) одержа-
ного розчину в області від 230 нм до 350 нм повинен ма-
ти максимум за довжини хвилі 280 нм. Питомий показ-
ник поглинання в максимумі має бути від 136 до 150.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках із калію хлоридом Р, має
відповідати спектру ФСЗ допаміну гідрохлориду.
С. Близько 5 мг субстанції розчиняють у суміші 5 мл
/ М розчину кислоти хлористоводневої і 5 мл води Р. До
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
367
Допаміну гідрохлорид
одержаного розчину додають 0.1 мл розчину натрію
нітрату Р, що містить 100 г/л амонію молібдату Р;
з’являється жовте забарвлення, яке при додаванні роз-
чину натрію гідроксиду концентрованого Р переходить
у червоне.
В. Близько 2 мг субстанції розчиняють у 2 мл води Р, до-
дають 0.2 мл розчину заліза(Ш) хлориду Р2; з’являється
зелене забарвлення, яке при додаванні 0.1 г гексамети-
лентетраміну Р переходить у синьо-фіолетове.
Е. Субстанція дає реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 0.4 г субстанції розчиняють
у воді Рі доводять об’єм розчину тим самим розчинни-
ком до 10 мл. Одержаний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування "Про-
зорість розчину", має бути не інтенсивнішим за еталон
В6 або ¥6.
Кислотність або лужність. 0.5 г субстанції розчиняють
у воді, вільній від вуглецю діоксиду, Р і доводять об’єм
розчину тим самим розчинником до 10 мл. додають
0.1 мл розчину метиленового червоного Р і 0.75 мл
0.01 М розчину натрію гідроксиду; з’являється жовте
забарвлення, яке переходить у червоне при додаванні
1.5 мл 0.0! М розчину кислоти хлористоводневої.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель с Р.
Випробовуваний розчин. 0.15 г субстанції розчиняють у
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 5 мл.
Розчин порівняння (а). 7.5 мг 4-О-метилдопаміну
гідрохлориду Р розчиняють у метанолі Р і доводять
об’єм розчину тим самим розчинником до 100 мл.
Розчин порівняння (Ь). 7.5 мг З-О-метилдопаміну
гідрохлориду Р і 7.5 мг 4-О-метилдопаміну гідрохлори-
ду Р розчиняють у метанолі Р і доводять об’єм розчи-
ну тим самим розчинником до 100 мл.
Налінію старту хроматографічної пластинки наносять
10 мкл (300 мкг) випробовуваного розчину, 10 мкл
(0.75 мкг) розчину порівняння (а), 10 мкл (0.75 мкг 3-
О-метилдопаміну гідрохлориду і 0.75 мкг 4-О-метил-
допаміну гідрохлориду) розчину порівняння (Ь). Пла-
стинку поміщають у камеру із сумішшю розчинників
кислота мурашина безводна Р - вода Р - метанол Р -
хлороформ Р (2:7:36:52). Коли фронт розчинників
пройде 15 см від лінії старту, пластинку виймають із
камери, сушать на повітрі протягом 15 хв і рівномірно,
рясно обприскують свіжоприготованою сумішшю
рівних об’ємів розчину калію фериціаніду Р і розчину
заліза(Ш) хлориду РІ.
На хроматограмі випробовуваного розчину будь-яка
пляма з Кг більшим за Кг основної плями не має бути
інтенсивнішою за пляму на хроматограмі розчину
порівняння (а) (0.25 %).
Результати аналізу вважаються вірогідними, якшо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції мат витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С протягом 2 год.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Щоб уникнути перегріву реакційной суміші, титрують
при постійному перемішуванні і припиняють титру-
вання відразу після досягнення точки еквівалентності.
0.1500 г субстанції розчиняють у 10 мл кислоти мура-
шиної безводної Р, додають 50 мл оцтового ангідриду Р
і титрують 0.1 М розчином кислоти хлорної по-
тенціометрично (2.2.20).
І мл 0.1 М розчину кислоти хюрної відповідає 18.96 мг
С8НІ2СІМ)2.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. 5-(2-аміноетил)-2-метоксифенол (4-О-метилдопа-
мін).
_______________________________________________ТУ
ДОФАМІН
ОорИатіпит
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
368
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Еконазолу нітрат
ЕКОНАЗОЛУ НІТРАТ
Есопа/оіі пкгаз
ЕС05А701.Е N11 ПАТЕ
Еконазолу нітрат містить не менше 98.5 % і не більше
101.5 % (А5)-І-[2-(4-хлорбензилокси)-2-(2,4-дихлор-
феніл)етил|імідазолу нітрату, у перерахунку на суху
речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Дуже мало розчинний у воді Р, розчин-
ний у метанолі Р, помірно розчинний у метиленхло-
риді Р, мало розчинний у 96 % спирті Р, практично не
розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: А, В, Б, Е.
А. Температура плавлення (2.2.14). Від 161 °С до 166 °С.
В. 40 мг субстанції розчиняють у суміші 0.1 М розчин
кислоти хлористоводневої Р- 2-пропанол Р(1:9) і дово-
дять об’єм розчину тією самою сумішшю розчинників
до 100.0 мл. Ультрафіолетовий спектр поглинання
(2.2.25) одержаного розчину в області від 240 нм до
320 нм повинен мати три максимуми за довжин хвиль
265 нм, 271 нм і 280 нм. Відношення оптичної густини
в максимумі за довжини хвилі 271 нм до оптичної гус-
тини в максимумі за довжини хвилі 280 нм має бути
від 1.55 до 1.70.
Результати аналізу вважаються вірогідними, якщо при
виконанні випробування на розділювальну здатність
(2.2.25) відношення оптичних густин становить не
менше 2.
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ еконазолу нітрату.
В. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Супровідні домішки",
до обприскування пластинки при перегляді в
УФ-світлі задовжини хвилі 254 нм має виявлятися ос-
новна пляма на рівні основної плями на хроматограмі
розчину порівняння (а), відповідна їй за розміром.
Е. Субстанція дає реакцію на нітрати (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 0.50 г субстанції розчиня-
ють у метанолі Р і доводять об’єм розчину тим самим
розчинником до 50 мл. Одержаний розчин має бути
прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування "Прозорість
розчину", має бути не інтенсивнішим за еталон 7 шка-
ли найбільш підхожого кольору.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.21), використовую-
чи як тонкий шар силікагель із флуоресцентним інди-
катором з оптимальною інтенсивністю поглинання за
довжини хвилі 254 нм.
Випробовуваний розчин (а). 0.25 г субстанції розчиня-
ють у суміші розчин аміаку концентрований Р - мета-
нол Р (1:9) і доводять об’єм розчину тією самою
сумішшю розчинників до 5 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять сумішшю розчин аміаку концентро-
ваний Р - метанол Р(1:9) до об’єму 10 мл.
Розчин порівняння (а). 25 мг ФСЗ еконазолу нітрату
розчиняють у суміші розчин аміаку концентрова-
ний Р - метанол Р (1:9) і доводять об’єм розчину тією
самою сумішшю розчинників до 5 мл.
Розчин порівняння (Ь). 2.5 мл випробовуваного розчи-
ну (Ь) розчиняють у суміші розчин аміаку концентро-
ваний Р - метанол Р (1:9) і доводять об’єм розчину
тією самою сумішшю розчинників до 100 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
369
Етилморфіну гідрохлорид
Налінію старту хроматографічної пластинки наносять
10 мкл (500 мкг) випробовуваного розчину (а), 10 мкл
(50 мкг) випробовуваного розчину (Ь), 10 мкл (50 мкг)
розчину порівняння (а) і 10 мкл (0.125 мкг) розчину
порівняння (Ь). Пластинку поміщають у камеру із
сумішшю розчинників розчин аміаку концентрований Р
- толуол Р - діоксан Р (1:40:60). Коли фронт розчин-
ників пройде 10 см від лінії старту, пластинку виймають
із камери, сушать у струмені повітря протягом 15 хв і
переглядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.25 %). Пластинку обприскують розчином калію йо-
довісмутату Р2 і переглядають при денному світлі. На
хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь)
(0.25 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) чітко видна пляма.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5%. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С протягом 2 год.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0. 4000 г субстанції розчиняють у 50 мл кислоти оцто-
вої льодяної Р і титрують 0.1М розчином кислоти хюр-
ноїпотенціометрично (2.2.20).
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину кислоти хлорної відповідає
44.47 мг С18Н16С13ГЧ3О4.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ЕТИЛМОРФІНУ ГІДРОХЛОРИД
ЕШуІтогрИіпі ЬуйгосИІогідит
ЕТНУЬМОКРНІПЕ ІІ\І)ІЮ(1ІІ.()КІІ)Е
С19Н24СІ\О„2Н2О
М.м. 385.9
Етилморфіну гідрохлорид містить не менше 99.0% і
не більше 100.5 % (5Д,65)-4.5-епокси-3-етокси-А-ме-
тилморфін-7-ен-6-олу гідрохлориду, у перерахунку на
безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Розчинний у воді Рі 96 % спирті Р, прак-
тично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: В, С, В.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати еталонному спектру ДФУ
етилморфіну гідрохлориду.
В. 0.5 г субстанції поміщають у пробірку, розчиняють
у 6 мл води Р і додають 15 мл 0.1 М розчину натрію
гідроксиду. Стінку пробірки потирають скляною па-
личкою; утворюється білий кристалічний осад, який
збирають, промивають водою Р і розчиняють у 20 мл
води Р, нагрітої до температури 80 °С. Одержаний роз-
чин фільтрують і охолоджують на льодяній бані; утво-
рюються кристали, температура плавлення (2.2.14)
яких після висушування у вакуумі протягом 12 год має
бути від 85 °С до 89 °С.
С. До близько 10 мг субстанції додають 1 мл кислоти
сірчаної Р, 0.05 мл розчину заліза(ІП) хюриду Р2 і
нагрівають на водяній бані; з’являється блакитне за-
барвлення. До одержаного розчину додають 0.05 мл
кислоти азотної Р; з’являється червоне забарвлення.
В. Розчин 8, приготований, як зазначено у розділі
"Випробування на чистоту", дає реакцію (а) на хлори-
ди (2.3.1).
370
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Етил олеат
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 0.500 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 25.0 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
рН (2.2.3). Від 4.3 до 5.7. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від -102° до -105°, у
перерахунку на суху речовину. Визначення проводять,
використовуючи розчин 8.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель С Р.
Випробовуваний розчин. 0.25 г субстанції розчиняють у
суміші рівних об’ємів 96 % спирту Рі води Рі доводять
об’єм розчину тією самою сумішшю розчинників до
10 мл.
Розчин порівняння. 0.5 мл випробовуваного розчину до-
водять сумішшю рівних об’ємів 96 % спирту Рі води Р
до об’єму 100 мл.
Налінію старту хроматографічної пластинки наносять
10 мкл (250 мкг) випробовуваного розчину і 10 мкл
(1.25 мкг) розчину порівняння. Пластинку сушать на
повітрі й поміщають у камеру із сумішшю розчин-
ників розчин аміаку концентрований Р- ацетон Р- 96%
спирт Р - толуол Р (2.5:32.5:35:35). Коли фронт роз-
чинників пройде 15 см від лінії старту, пластинку вий-
мають із камери, сушать у струмені повітря й обприс-
кують розчином калію йодовісмутату розведеним Р.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %).
Вода (2.5.12). Від 8.0 % до 10.0 %. Визначення прово-
дять з 0.250 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.1 %>. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.300 г субстанції розчиняюі ь у ЗО мл кислоти оцтової
безводної Р, додають 20 мл оцтового ангідриду Р, 5 мл
розчину ртуті( 11) ацетату Р і титрують 0.1 М розчи-
ном кислоти хлорної потенціометрично (2.2.20).
І мл 0.1 М розчину кислоти хлорної відповідає
34.99 мг СІ9Н24СІІ\О3.
ЗБЕРІГАННЯ
У шільно закупореному контейнері, у захищеному від
світла місці.
___________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги етапі (5.4).
ЕТИЛОЛЕАТ
ЕіЬуІіз оіеаз
РИПІ. ОЬЕАТЕ
Етилолеат є сумішшю, що складається з етилових
ефірів жирних кислот, переважно кислоти олеїнової.
Можливі добавки підхожих антиоксидантів.
ВИРОБНИЦТВО
При виробництві етилолеату з тканин ссавців або з ре-
човин, одержаних від теплокровних тварин, стан здо-
ров’я тварин, використовуваних для виділення етило-
леату, має відповідати вимогам, що ставляться до тва-
рин, придатних для вживання людиною. Ці вимоги
встановлюються компетентними уповноваженими
органами. Крім того, тканини цих тварин не мають
містити ніяких небезпечних речовин і матеріалів, за-
значених спеціальними міжнародними або відповід-
ними національними законодавствами.
ВЛАСТИВОСТІ
Опис. Рідина прозора, безбарвна або світло-жовтого
кольору.
Розчинність. Практично не розчинний у воді Р,
змішується з 96 % спиртом Р, метиленхлоридом Р і пе-
тролейним ефіром РІ.
ІДЕНТИФІКАЦІЯ
А. Субстанція має витримувати вимоги випробування
"Відносна густина", зазначені в розділі "Випробування
на чистоту".
В. Субстанція має витримувати вимоги випробування
"Число омилення", зазначені в розділі "Випробування
на чистоту".
С. Субстанція має витримувати вимоги випробування
"Кислота олеїнова", зазначені в розділі "Випробування
на чистоту".
ВИПРОБУВАННЯ НАЧИСТОТУ
Відносна густина (2.2.5). Від 0.866 до 0.874.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
371
Етилолеат
Кислотне число (2.5.1). Не більше 0.5. Визначення
проводять з 10.0 г субстанції.
Йодне число (2.5.4). Від 75 до 90.
Перекисне число (2.5.5). Не більше 10.0.
Число омилення (2.5.6). Від 177 до 188. Визначення
проводять із 2.0 г субстанції.
Кислота олеїнова. Проводять випробування для визна-
чення сторонніх олій у жирних оліях методом газової
хроматографії (2.4.22). Фракція жирних кислот у суб-
станції має містити не менше 60 % кислоти олеїнової.
Вода (2.5.12). Не більше 1.0 %. Визначення проводять
з 1.00 г субстанції напівмікрометодом.
Загальна зола (2.4.16). Не більше 0.1 %. Визначення
проводять із 2.0 г субстанції.
ЗБЕРІГАННЯ
У захищеному від світла місці.
МАРКУВАННЯ
На етикетці зазначають застосування, назву і концен-
трацію доданих антиоксидантів.
___________________________________________А
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
372
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Ізолейцин
ІЗОЛЕЙЦИН
коїеисіпит
ІЇОЬЕІ/СтЕ
н мн2
С6НП1\Ю2
М.м. 131.2
Ізолейцин містить не менше 98.5 % і не більше 101.0 %
(25,35)-2-аміно-3-метилпентанової кислоти, у пере-
рахунку на суху речовину.
ВИРОБНИЦТВО
Якшо субстанція одержана в результаті процесу, шо
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок або пластинки білого
або майже білого кольору.
Розчинність. Помірно розчинний у воді Р, мало роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р.
(Розчиняється в розведених мінеральних кислотах і
розведених розчинах гідроксидів лужних металів).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Пруга ідентифікація: А, В, 1).
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту”.
В. 0.5 г субстанції розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 25 мл
Одержаний розчин обертає площину поляризації пра-
воруч.
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ ізолейцину.
Ц. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.І). 0.5 г субстанції розчиняють
уІМ розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 10 мл. Одержа-
ний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод П). Забарвлення
розчину, приготованого для випробування "Про-
зорість розчину", має бути не інтенсивнішим за еталон
В¥6.
Питоме оптичне обертання (2.2.7). Від +39.0° до +42.0°,
у перерахунку на суху речовину. 1.00 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27),
використовуючи ТШХ пластинки із шаром силікаге-
лю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у 0.1 М розчині кислоти хлористоводневої і дово-
дять об’єм розчину тією самою кислотою до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рао об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ ізолейцину розчиня-
ють у 0.1 М розчині кислоти хлористоводневої і дово-
дять об’єм розчину тією самою кислотою до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ ізолейцину і 10 мг
ФСЗ валіну розчиняють у 0.1 Мрозчині кислоти хлори-
стоводневої і доводять об’єм розчину тією самою кис-
лотою до 25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг ізолейцину і 2 мкг валіну)
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
373
Ізосорбіду динітрат розведений
розчину порівняння (с). Пластинку сушать на повітрі
й поміщають у камеру із сумішшю розчинників кисло-
та оцтова льодяна Р - вода Р - бутанол Р (20:20:60).
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й
обприскують розчином нінгідрину Р. Пластинку
нагрівають при температурі від 100 °С до 105 °С протя-
гом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 0.25 г суб-
станції розчиняють у воді Р і доводять об’єм розчину
тим самим розчинником до 15 мл. Одержаний розчин
має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють у 3 мл кислоти хлористоводне-
вої розведеної Р і доводять об’єм розчину водою дисти-
льованою Рдо 15 мл. Одержаний розчин має витриму-
вати випробування на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, шо складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють або суспенду-
ють у 0.5 мл води Р. До одержаного розчину або сус-
пензії додають 0.30 г магнію оксиду важкого Рі ретель-
но розтирають скляною паличкою. Нижнє годинни-
кове скло відразу накривають верхнім і нагрівають при
температурі 40 °С протягом 15 хв. Паралельно в ана-
логічних умовах готують еталон, використовуючи
0.1 мл еталонного розчину амонію (100 ррт І8Н4) Р,
0.5 мл води Рі 0.30 г магнію оксиду важкого Р Лакму-
совий папір над розчином або суспензією має бути за-
барвлений у синій колір не інтенсивніше, ніж над ета-
лоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8. метод О). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначеная
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної до пе-
реходу забарвлення від коричнювато-жовтого до зеле-
ного, використовуючи як індикатор 0.1 мл розчину
нафтолбензеїну Р.
1 мл 0.1 Мрозчину кислоти хюрної відповідає 13.12 мг
С6Н13М)2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері
світла місці.
захищеному
У
рН (2.2.3). Від 5.5 до 7.0.
1.0
г субстанції
розчиняють у
воді Р і доводять об’єм розчину тим самим розчиним
ком до 100 мл.
Залишкові кількості органічних розчинників. Суботам
ція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб
станція призначена для
виробництва
лікарських засобів без подальшої процедури видален
ня пірогенів, вона має
"Пірогени" (2.6.8) або
(2.6.14).
парентеральних
витримувати випробування
"Бактеріальні ендотоксини
ІЗОСОРБІДУ ДИНІТРАТ
РОЗВЕДЕНИЙ
ібобогЬісіі діпіігаз с!і1иШ$
І8080КВЮЕ ОІШТКАТЕ, ІНИТЕІ)
МО2
І
с6н8и2о8
М.м. 236.1
Ізосорбіду динітрат розведений є сухою сумішшю ізо
сорбіду динітрату
"Лактози
моногідрату"
або
374
ДЕРЖАВНА ФАРМАКОПЕЯ
УКРАЇНИ
2001
Ізосорбіду динітрат розведений
"Маніту". Містить не менше 95.0 % і не більше 105.0 %
(м/м) від зазначеного на етикетці вмісту 1,4:3,6-
діангідро- О- гл юкитолу 2,5 -ди н ітрату.
ЗАПОБІЖИ/ ЗАХОДИ, нерозведений ізосорбіду ди-
нітрат під впливом удару або при нагріванні може
вибухати. Мають бути вжиті необхідні запобіжні за-
ходи і треба працювати лише з дуже невеликими його
кількостями.
ВЛАСТИВОСТІ
Опис. Нерозведений ізосорбіду динітрат являє собою
дрібний кристалічний порошок білого кольору.
Розчинність. Розчинність ізосорбіду динітрату розве-
деного залежить від розріджувача і його концентрації.
(Нерозведений ізосорбіду динітрат дуже мало розчин-
ний у воді Р, дуже легко розчинний в ацетоні Р,
помірно розчинний у 96 % спирті Р.)
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С, В.
Пруга ідентифікація В, С, В.
А. Інфрачервоний спектр поглинання (2.2.24) залиш-
ку, одержаного у випробуванні Б, у дисках має відпо-
відати спектру залишку, одержаного аналогічно до
субстанції, з використанням ФСЗ ізосорбіду динітра-
ту.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. Наважку субстанції, шо
відповідає 10 мг ізосорбіду динітрату, струшують з
10 мл 96 % спирту Р протягом 5 хв і фільтрують.
Розчин порівняння. Наважку ФСЗ ізосорбіду динітра-
ту, шо відповідає 10 мг ізосорбіду динітрату, струшу-
ють з 10 мл 96 % спирту Р протягом 5 хв і фільтрують.
Налінію старту хроматографічної пластинки наносять
10 мкл (10 мкг) випробовуваного розчину і 10 мкл
(10 мкг) розчину порівняння. Пластинку поміщають у
камеру із сумішшю розчинників метанол Р - мети-
ленхлорид Р (5:95). Коли фронт розчинників пройде
15 см від лінії старту, пластинку виймають із камери,
сушать на повітрі й обприскують свіжоприготованим
розчином крохмалю з калію йодидом Р. Витримують
пластинку в УФ-світлі за довжини хвилі 254 нм протя-
гом 15 хв і переглядають при денному світлі.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром і забарвленням.
С. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. Наважку субстанції, що відпо-
відає 0.10 г лактози або маніту, струшують з 10 мл во-
ди Р'\ фільтрують, якшо необхідно.
Розчин порівняння (а). 0.10 г лактози Р розчиняють у
воді Р \ доводять об’єм розчину тим самим розчинни-
ком до 10 мл.
Розчин порівняння (Ь). 0.10 г маніту Р розчиняють у
воді Р\ доводять об’єм розчину тим самим розчинни-
ком до 10 мл.
Розчин порівняння (с). Змішують рівні об’єми розчинів
порівняння (а) і (Ь).
Налінію старту хроматографічної пластинки наносять
І мкл (10 мкг) випробовуваного розчину, 1 мкл
(10 мкг) розчину порівняння (а), 1 мкл (10 мкг) розчи-
ну порівняння (Ь) і 1 мкл (5 мкг лактози і 5 мкг маніту)
розчину порівняння (с) і ретельно сушать. Пластинку
поміщають у камеру із сумішшю розчинників
вода Р - метанол Р - кислота оцтова безводна Р - ети-
ленхлорид Р (10:15:25:50). Точно відмірюють об’єми
компонентів зазначеної суміші, тому що невеликий
надлишок води призводить до помутніння. Коли
фронт розчинників пройде 15 см від лінії старту, плас-
тинку виймають із камери, сушать у струмені теплого
повітря і відразу повторно хроматографують зі свіжою
рухомою фазою. Коли фронт розчинників пройде
15 см від лінії старту, пластинку виймають із камери,
сушать у струмені теплого повітря, обприскують роз-
чином кислоти 4-амінобензойної Р і сушать у струмені
холодного повітря до зникнення запаху ацетону. Пла-
стинку витримують при температурі 100 °С протягом
15 хв, охолоджують й обприскують розчином 2 г/л
натрію перйодату Р. Пластинку сушать у струмені хо-
лодного повітря і знову витримують при температурі
100 °С протягом 15 хв.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), якщо розріджува-
чем є лактоза, або на рівні основної плями на хрома-
тограмі розчину порівняння (Ь), якщо розріджувачем
є маніт, відповідна їй за розміром і забарвленням.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Б. Наважку субстанції, що відповідає 25 мг ізосорбіду
динітрату, струшують з 10 мл ацетону Рпротягом 5 хв,
одержаний розчин фільтрують, упарюють насухо при
температурі не більше 40 °С і сушать залишок над фо-
сфоруй) оксидом Рпри тиску не більше 0.7 кПа про-
тягом 16 год. Температура плавлення (2.2.14) залишку
має бути від 69 °С до 72 °С.
ВИПРОБУВАННЯ НАЧИСТОТУ
Неорганічні нітрати. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель н Р.
Випробовуваний розчин. Наважку субстанції, що
відповідає 0.10 г ізосорбіду динітрату, струшують з
5 мл 96 % спирту Рі фільтрують.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
375
Ізосорбіду динітрат розведений
Розчин порівняння. 10 мг калію нітрату Р розчиняють
у 1 мл води Р і доводять об’єм розчину 96 % спиртом Р
до 100 мл.
На лінію старту хроматографічної пластинки нано-
сять 10 мкл (100 мкг) випробовуваного розчину і
10 мкл (1 мкг) розчину порівняння. Пластинку
поміщають у камеру із сумішшю розчинників кисло-
та оцтова льодяна Р - ацетон Р - толуол Р (15:30:60).
Коли фронт розчинників пройде 15 см від лінії стар-
ту, пластинку виймають із камери, ретельно сушать у
струмені повітря до повного зникнення запаху кисло-
ти оцтової і рясно обприскують пластинку свіжопри-
готованим розчином крохмалю з калію йодидом Р. Пла-
стинку витримують в УФ-світлі за довжини хвилі
254 нм протягом 15 хв і переглядають при денному
світлі.
На хроматограмі випробовуваного розчину пляма,
відповідна нітрат-іону, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %, у
перерахунку на калію нітрат).
Ізосорбіду 5-нітрат та ізосорбіду 2-нітрат. Визначення
проводять методом рідинної хроматографії (2.2.29) за
умов, описаних у розділі "Кількісне визначення",
змінюючи таку умову:
— детектування задовжини хвилі 210-215 нм.
При хроматографуванні за зазначених умов часи утри-
мування піків мають бути: ізосорбіду динітрату —
близько 5 хв; ізосорбіду 2-нітрату — близько 8 хв; ізо-
сорбіду 5-нітрату — близько 11 хв.
Хроматографують 10 мкл розчину порівняння (с).
Чутливість системи регулюють таким чином, щоб ви-
сота основного піка становила не менше 20 % шкали
реєструючого пристрою.
Хроматографують 10 мкл розчину порівняння (е).
Хроматографічна система вважається придатною, як-
що на хроматограмі розчину порівняння (е) ко-
ефіцієнт розділення піків ізосорбіду динітрату й ізо-
сорбіду 2-нітрату становить не менше 6.0.
Поперемінно хроматографують по 10 мкл випро-
бовуваного розчину (а), розчину порівняння (с) і роз-
чину порівняння (0). На хроматограмі випробовува-
ного розчину (а) площа піка ізосорбіду 2-нітрату не
має перевищувати площу основного піка на хромато-
грамі розчину порівняння (с) (0.5 %); площа піка ізо-
сорбіду 5-нітрату не має перевищувати площу основ-
ного піка на хроматограмі розчину порівняння (єі)
(0 5%).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять методом рідинної хромато-
графії (2.2.29).
Випробовуваний розчин (а). До наважки субстанції,
відповідної 25.0 мг ізосорбіду динітрату, додають 20 мл
рухомої є])ази. витримують в ультразвуковій бані про-
тягом 15 хв і доводять об’єм розчину рухомою фазою
до 25.0 мл. Одержаний розчин фільтрують крізь підхо-
жий мембранний фільтр.
Випробовуваний розчин (Ь). 1.0 мл випробовуваного
розчину (а) доводять рухомою фазою до об’єму
10.0 мл.
Розчин порівняння (а). До наважки ФСЗ ізосорбіду
динітрату, відповідної 25.0 мг ізосорбіду динітрату,
додають 20 мл рухомої фази, витримують в ультразву-
ковій бані протягом 15 хв і доводять об’єм розчину ру-
хомою фазою до 25.0 мл. Одержаний розчин фільтру-
ють крізь підхожий мембранний фільтр.
Розчин порівняння (Ь). 1.0 мл розчину порівняння (а)
доводять рухомою фазою до об’єму 10.0 мл.
Розчин порівняння (с). 10.0 мг ФСЗ ізосорбіду 2-нітра-
ту розчиняють у рухомій фазі й доводять об’єм розчи
ну рухомою фазою до 10.0 мл. 0.1 мл одержаного роз-1
чину доводять рухомою фазою до об’єму 20.0 мл.
Розчин порівняння (сі). 10.0 мг ФСЗ ізосорбіду мо-
нонітрату розчиняють у рухомій фазі й доводять
об’єм розчину рухомою фазою до 10.0 мл. 0.1 мл одер-
жаного розчину доводять рухомою фазою до об’єму
20.0 мл
Розчин порівняння (е). 5 мг ФСЗ ізосорбіду 2-нітрату
розчиняють у рухомій фазі й доводять об’єм розчину
рухомою фазою до 10 мл. До 1 мл одержаного розчину
додають 0.5 мл розчину порівняння (а) і доводять
об’єм розчину рухомою фазою до 10 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм.
заповнена силікагелем амінопропілметилсилільния
для хроматографії Р з розміром часток 10 мкм;
— рухома фаза: етанол Р- триметилпентан Р( 15:85);
— швидкість рухомої фази 1 мл/хв;
— детектування задовжини хвилі 230 нм.
Хроматографують 20 мкл розчину порівняння (Ь).
Чутливість системи регулюють таким чином, щоб ви-
сота основного піка становила не менше 50 % шкали
реєструючого пристрою
Якщо площі піків на двох послідовних хроматограмах
різняться не більше як на 1.0 %, хроматографують роз-
чин порівняння (Ь) ще чотири рази і розраховують
відносне стандартне відхилення із шести хроматограм.
Хроматографічна система вважається придатною, як-
шо відносне стандартне відхилення для основного
піка не перевищує 2.0 %.
Поперемінно хроматографують 20 мкл випробовува-
ного рочину і 20 мкл розчину порівняння (Ь).
Вміст ізосорбіду динітрату обчислюють у відсотках від
кількості, зазначеної на етикетці.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
МАРКУВАННЯ
На етикетці зазначають вміст ізосорбіду динітрату, у
відсотках.
376
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Ізосорбіду динітрат розведений
ДОМІШКИ
А. неорганічні нітрати,
В. ізосорбіду 2-нітрат,
С. ізосорбіду мононітрат (ізосорбіду 5-нітрат).
_________________________________________________N
Залишкові кількості органічних розчинників. Субстан-
ція має витримувати вимоги статті (5.4).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
377
Кальцію глюконат
КАЛЬЦІЮ ГЛЮКОНАТ
Саісіі §1исопа$
САІСІІІМ СІ.ІСОІХАТЕ
он он н он
НОН2С—С—С—С—С—С00' Са2+,Н2О
н н ОН н
_ 2
С,2Н22СаОІ4,Н2О
М.м. 448.4
Кальцію глюконат містить не менше 98 5 % і не
більше 102.0 % кальцію О-глюконату моногідрату.
ВЛАСТИВОСТІ
Опис. Кристалічний або гранульований порошок
білого кольору.
Розчинність. Помірно розчинний у воді Р, легко роз-
чинний у киплячій воді Р.
ІДЕНТИФІКАЦІЯ
А. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. 20 мг субстанції розчиняють у
І мл води Р, якщо необхідно, нагріваючи на водяній
бані при температурі 60 °С.
Розчин порівняння. 20 мг ФСЗ кальцію глюконату роз-
чиняють у 1 мл води Р, якщо необхідно, нагріваючи на
водяній бані при температурі 60 °С
Налінію старту хроматографічної пластинки наносять
5 мкл (100 мкг) випробовуваного розчину і 5 мкл
(100 мкг) розчину порівняння. Пластинку поміща-
ють у камеру із сумішшю розчинників розчин аміаку
концентрований Р - етилацетат Р - вода Р - 96 %
спирт Р( 10:10:30:50). Коли фронт розчинників прой-
де 10 см від лінії старту, пластинку виймають із каме-
ри, сушать при температурі 100 °С протягом 20 хв.
Потім пластинку охолоджують й обприскують розчи-
ном 50 г/л калію дихромату Р у розчині 40 % (м/м)
кислоти сірчаної Р. Через 5 хв хроматограму перегля-
дають при денному світлі.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром і забарвленням.
В. Розчин 8, приготований, як зазначено в розділі "Ви-
пробування начистоту", дає реакцію на кальцій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.0 г субстанції розчиняють у воді Р, нагрітій
до температури 60 °С, і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину 8 при температурі 60 °С має бути не інтен-
сивнішим за еталон ¥6.
Прозорість розчину (2.2.1). Розчин 8 після охолоджен-
ня за ступенем каламутності не має перевищувати ета-
лон II.
Органічні домішки і кислота борна 0.5 г субстанції
поміщають у фарфорову чашку, попередньо промиту
кислотою сірчаною Р, і помішають у льодяну баню.
Додають 2 мл охолодженої кислоти сірчаної Р і пе-
ремішують, не має з’являтися жовте або коричневе за-
барвлення. До одержаного розчину додають 1 мл роз-
чину хромотропу 11 В Р; з’являється фіолетове забарв-
лення, що не переходить у темно-синє. Забарвлення
одержаної суміші має бути не інтенсівнішим за
забарвлення суміші І мл розчину хромотропу II В Рі
2 мл охолодженої кислоти сірчаної Р.
Сахароза і відновлюючі цукри. 0.5 г субстанції розчиня-
ють у суміші 2 мл кислоти хлористоводневої РГ\ 10 мл
води Р. Кип’ятять протягом 5 хв, охолоджують, дода-
ють 10 мл розчину натрію карбонату Р і витримують
протягом 10 хв. Потім доводять об’єм розчину водою Р
до 25 мл і фільтрують До 5 мл фільтрату додають 2 мл
розчину мідно-тартратного Р і кип’ятять протягом
1 хв, одержаний розчин витримують протягом 2 хв; не
має утворюватися червоний осад.
Хлориди (2.4 4). Не більше 0.02 % (200 ррт) 12 5 мл роз-
чину 8 доводять водою Р до об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.01 % (100 ррт). 10.0 г
субстанції розчиняють при нагріванні в суміші 10 мл
кислоти оцтової Р і 90 мл води дистильованої Р. 15 мл
одержаного розчину мають витримувати випробуван-
ня на сульфати.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
379
Кальцію глюконат для ін’єкцій
Важкі метали (2.4.8, метод О). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Субстанцію поступово і обе-
режно нагрівають до повного перетворення в білу ма-
су і прожарюють. Еталон готують із використанням
2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Магній і лужні метали. 1.00 г субстанції розчиняють у
100 мл киплячої води Р, додають 10 мл розчину амонію
хлориду Р, 1 мл розчину аміаку Рі краплями 50 мл га-
рячого розчину амонію оксалату Р. Одержаний розчин
витримують протягом 4 год, потім доводять об’єм роз-
чину водою Рло 200 мл і фільтрують. 100 мл фільтрату
упарюють насухо і прожарюють. Маса залишку не має
перевищувати 2 мг (0.4 %).
Мікробіологічна чистота (2.6.12). Визначення прово-
дять методом прямого висівання. У 1 г субстанції до-
пускається наявність не більше 103 мікроорганізмів
(бактерій і грибів сумарно).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.8000 г субстанції розчиняють у 20 мл гарячої води Р,
охолоджують і доводять об’єм розчину водою Р до
300 мл. Визначення кальцію проводять методом ком-
плексометричного титрування (2.5.11).
1 мл 0.1 Мрозчину натрію едетату відповідає 44.84 мг
С12Н22СаО14,Н2О.
_________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КАЛЬЦІЮ ГЛЮКОНАТ ДЛЯ
ІН’ЄКЦІЙ
Саісіі £Іисопа8 ас! іпіесїаЬіІе
( АІ.СІІ М 67. (СОНАТІ ІОН ІТ4ІЕСТКЖ
Са
СО2
Н
НО
Н
Н
ОН
Н
ОН
ОН
СН2ОН
_2
, Н2О
С12Н22СаОІ4.Н20
М.м. 448.4
Кальцію глюконат для ін’єкцій містить не менше
99.0 % і не більше 101.0 % кальцію О-глюконату мо-
ногідрату.
ВЛАСТИВОСТІ
Опис. Кристалічний або гранульований порошок
білого кольору.
Розчинність. Помірно розчинний у воді Р, легко роз-
чинний у киплячій воді Р.
ІДЕНТИФІКАЦІЯ
А. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. 20 мг субстанції розчиняють у
І мл води Р, якщо необхідно, нагріваючи на водяній
бані при температурі 60 °С.
Розчин порівняння. 20 мг ФСЗ кальцію глюконату роз-
чиняють у 1 мл води Р. якщо необхідно, нагріваючи на
водяній бані при температурі 60 °С.
Налінію старту хроматографічної пластинки наносять
5 мкл (100 мкг) випробовуваного розчину і 5 мкл
(100 мкг) розчину порівняння. Пластинку поміща-
ють у камеру із сумішшю розчинників розчин аміаку
концентрований Р - етилацетат Р - вода Р - 96%
спирт Р (10:10:30:50). Коли фронт розчинників прой-
де 10 см від лінії старту, пластинку виймають із каме-
ри, сушать при температурі 100 °С протягом 20 хв.
Потім пластинку охолоджують й обприскують розчи-
ном 50 г/л калію дихромату Р у розчині 40 % (м/м)
кислоти сірчаної Р. Через 5 хв хроматограму перегля-
дають при денному світлі.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром і забарвленням.
В. Близько 20 мг субстанції дають реакцію (Ь) на
кальцій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. До 10.0 г субстанції додають 90 мл киплячої
води дистильованої Р, кип’ятять при перемішуванні
протягом не більше 10 с до повного розчинення, і до-
водять об’єм розчину тим самим розчинником то
100.0 мл.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 при температурі 60 °С має бути не інтен-
сивнішим за еталон В7.
Прозорість розчину (2.2.1). Розчин 8 після охолоджен-
ня за ступенем каламутності не має перевищувати ета-
лон II.
рН (2.2.3). Від 6.4 до 8.3. 1.0 г субстанції, нагріваючи
на водяній бані, розчиняють у 20 мл води, вільної від
вуглецю діоксиду. Р.
Органічні домішки і кислота борна. 0.5 г субстанції
поміщають у фарфорову чашку, попередньо промиту
380
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кальцію глюконат для ін’єкцій
кислотою сірчаною Р, і поміщають у льодяну баню.
Додають 2 мл охолодженої кислоти сірчаної Р і пе-
ремішують; не має з’являтися жовте або коричневе за-
барвлення. До одержаного розчину додають 1 мл роз-
чину хромотропу // В Р; з’являється фіолетове забарв-
лення, що не переходить у темно-синє. Забарвлення
одержаної суміші має бути не інтенсивнішим за
забарвлення суміші 1 мл розчину хромотропу II В Р і
2 мл охолодженої кислоти сірчаної Р.
Оксалати. Не більше 0.01 % (100 ррт). Визначення
проводять методом рідинної хроматографії (2.2.29).
Випробовуваний розчин. 1.00 г субстанції розчиняють у
воді для хроматографії Р і доводять об’єм розчину тим
самим розчинником до 100.0 мл.
Розчин порівняння. 1.00 г субстанції розчиняють у воді
для хроматографії Р, додають 0.5 мл розчину 0.152 г/л
натрію оксалату Р у воді для хроматографії Р і дово-
дять об’єм розчину тим самим розчинником до
100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з кондуктометричним детектором за таких
умов:
— захисна колонка розміром 30 мм х 4 мм, заповне-
на підхожою сильною аніонообмінною смолою з
розміром часток від 30 мкм до 50 мкм;
- дві колонки, кожна розміром 0.25 м х 4 мм, запов-
нені підхожою сильною аніонообмінною смолою
з розміром часток від ЗО мкм до 50 мкм;
— мікромембранна аніон-заглушуюча колонка,
з’єднана послідовно з передколонкою і аналітич-
ними колонками; аніон-заглушуюча колонка об-
ладнана пристроєм, що дозволяє пропускати роз-
чин для регенерації заглушуючої колонки в на-
прямку, протилежному напрямку прямування ру-
хомої фази зі швидкістю 4 мл/хв;
— рухома фаза: 0.212 г натрію карбонату безводно-
го Р і 63 мг натрію гідрокарбонату Р розчиняють у
воді для хроматографії Р і доводять об’єм розчину
тим самим розчинником до 1000.0 мл;
— швидкість рухомої фази 2 мл/хв;
— заглушуючий регенеруючий розчин: розчин
1.23 г/л кислоти сірчаної Р у воді для хромато-
графії Р.
Хроматографують 50 мкл розчину порівняння п’ять
разів. Хроматографічна система вважається придат-
ною, якщо відносне стандартне відхилення для площі
піка оксалату не перевищує 2.0 %.
Поперемінно хроматографують 50 мкл випробовува-
ного розчину і 50 мкл розчину порівняння, одержую-
чи не менше трьох хроматограм.
Вміст оксалату (А), у ррт, обчислюють за формулою:
• 50
х~ Х.Г
де:
$т — площа піка оксалату на хроматограмі випро-
бовуваного розчину;
8К — площа піка оксалату на хроматограмі розчину
порівняння.
Сахароза і відновлюючі цукри. 0.5 г субстанції розчи-
няють у суміші 2 мл кислоти хлористоводневої РІ і
10 мл води Р. Кип'ятять протягом 5 хв, охолоджують,
додають 10 мл розчину натрію карбонату Рі витриму-
ють протягом 10 хв. Потім доводять об’єм розчину во-
дою Рдо 25 мл і фільтрують. До 5 мл фільтрату дода-
ють 2 мл розчину мідно-тартратного Р і кип’ятять
протягом 1 хв, одержаний розчин витримують протя-
гом 2 хв: не має утворюватися червоний осад.
Хлориди (2.4.4). Не більше 0.005 % (50 ррт). До 10 мл
попередньо профільтрованого розчину 8 додають 5 мл
води Р. Одержаний розчин має витримувати випробу-
вання на хлориди.
Фосфати (2.4.11). Не більше 0.01 % (100 ррт). 1 мл
розчину 8 доводять водою Рдо об’єму 100 мл. Одержа-
ний розчин має витримувати випробування на фосфа-
ти.
Сульфати (2.4.13). Не більше 0.005 % (50 ррт). 15 мл
попередньо профільтрованого розчину 8 мають ви-
тримувати випробування на сульфати. Еталон готують
із використанням 7.5 мл еталонного розчину сульфату
(10ррт 504) Рі 7.5 мл води дистильованої Р.
Залізо. Не більше 0.0005 % (5 ррт Ее). Визначення
проводять методом атомно-абсорбційної спектро-
метрії (2.2.23, метод І).
Випробовуваний розчин. 2.0 г субстанції поміщають у
колбу з політетрафторетилену місткістю 100 мл, дода-
ють 5 мл кислоти азотної Р, кип’ятять, упарюючи
майже насухо. Додають 1 мл розчину водню пероксиду
концентрованого Р і знову упарюють майже насухо.
Обробку розчином водню пероксиду повторюють до
одержання прозорого розчину. Одержаний розчин за
допомогою 2 мл кислоти азотної Р переносять у мірну
колбу місткістю 25 мл і доводять об’єм розчину кисло-
тою хлористоводневою розведеною Р до позначки.
Компенсаційний розчин готують аналогічно до ви-
пробовуваного розчину, використовуючи 0.65 г каль-
цію хлориду РІ замість субстанції.
Розчини порівняння. Готують розведенням еталонного
розчину заліза (20 ррт Ре) Р кислотою хлористоводне-
вою розведеною Р.
Вимірюють поглинання одержаних розчинів за дов-
жини хвилі 248.3 нм, використовуючи як джерело ви-
промінювання лампу з порожнистим залізним като-
дом і повітряно-ацетиленове полум’я. Урахування не-
селективного поглинання проводять за допомогою
дейтерієвої лампи.
Магній і лужні метали. До 0.50 г субстанції додають
суміш 1.0 мл кислоти оцтової розведеної Р і 10.0 мл во-
ди Р і швидко кип’ятять при перемішуванні до повно-
го розчинення. До киплячого розчину додають 5.0 мл
розчину амонію оксалату Р і витримують протягом
6 год. Потім фільтрують крізь скляний фільтр (1.6) у
фарфоровий тигель, обережно упарюють насухо і про-
жарюють. Маса залишку не має перевищувати 2 мг
(0.4 %).
ЛЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
381
Камфора рацемічна
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Бактеріальні ендотоксини (2.6.14). Не більше 167 МО
у 1 г
Мікробіологічна чистота (2.6.12). Визначення прово-
дять методом прямого висівання. У 1 г субстанції до-
пускається наявність не більше 102 мікроорганізмів
(бактерій і грибів сумарно). Не допускається наявність
Е^скегісИіа соїі, Рзеидотопах аегиріпо.'іа і Біаркуіососсиз
аигеих (2.6.13).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.350 г субстанції розчиняють у 20 мл гарячої води Р,
охолоджують і доводять об’єм розчину водою Р до
300 мл. Визначення кальцію проводять методом ком-
плексометричного титрування (2.5.11), використову-
ючи 50 мг індикаторної суміші кислоти кальконкарбо-
нової Р.
1 мл 0.1 Мрозчину натрію едетату відповідає 44.84 мг
СІ2Н22СаО14,Н2О.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Замість випробування "Бактеріальні ендотоксини " до
пускається застосування випробування "Пірогени"
(2.6.8).
КАМФОРА РАЦЕМІЧНА
Сатріїога гасетіса
САМРНОК, ПАСЕ МІС
і енантіомер
СщНібО
М.м. 152.2
Камфора рацемічна являє собою (1Я5,45Л)-1,7,7-три-
метилбіцикло[2.2.1]гептан-2-он.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок або пухка кристалічна
маса білого кольору, легко летка навіть при кімнатній
температурі.
Розчинність. Мало розчинна у воді Р, дуже легко роз-
чинна в 96 % спирті Р, ефірі Р і петролейному ефірі Р
Легко розчинна в жирних оліях, дуже мало розчинна;
гліцерині Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: А, В, И.
А. Субстанція має відповідати вимогам щодо оптич-
ного обертання, зазначеним у розділі "Випробування
на чистоту".
В. Температура плавлення (2.2.14). Від 172 °С до 180 °С.
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у суспензії з вазеліновим маслом Р,
має відповідати спектру ФСЗ камфори рацемічної.
1). 1.0 г субстанції розчиняють у 30 мл метанолу Р, до-
дають 1.0 г гідроксиламіну гідрохлориду Рі 1.0 г натрію
ацетату безводного Р. Кип’ятять суміш зі зворотним
холодильником протягом 2 год, охолоджують і дода-
ють 100 мл води Р; утворюється осад. Одержану суміш
фільтрують, промивають 10 мл води Р і перекрис-
талізовують із 10 мл суміші 96 % спирт Р- вода Р(4:6).
Температура плавлення (2.2.14) висушених у вакуумі
кристалів має бути від 118 °С до 121 °С.
ВИПРОБУВАННЯ НАЧИСТОТУ
Зважування проводять швидко.
Розчин 8. 2.50 г субстанції розчиняють у 10 мл 96 %
спирту Р і доводять об’єм розчину тим самим розчин-
ником до 25.0 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод П). Розчин 8 маї
бути безбарвним.
Кислотність або лужність. 1.0 г субстанції розчиняють
у 10 мл 96 % спирту Р і додають 0.1 мл розчину фенол-
фталеїну Рі; розчин безбарвний. Забарвлення розчи-
ну має змінитися при додаванні не більше 0.2 мл 0.1 М
розчину натрію гідроксиду.
Оптичне обертання (2.2.1). Від +0.15° до -0.15°. Визна-
чення проводять, використовуючи розчин 8.
Супровідні домішки. Визначення проводять методом
газової хроматографії (2.2.28).
Випробовуваний розчин. 50 мг субстанції розчиняють у
гексані Р і доводять об’єм розчину тим самим розчин-
ником до 50.0 мл.
382
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Канаміцину моносульфат
Розчин порівняння (а). 50 мг субстанції і 50 мг борніла-
цетату /"розчиняють у гексані Рі доводять об’єм роз-
чину тим самим розчинником до 50.0 мл.
Розчин порівняння (Ь). 1.0 мл випробовуваного розчину
доводять гексаном Рао об’єму 200.0 мл.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
- колонка розміром 2 м х 2 мм, заповнена діатомі-
том для газової хроматографії Р з нанесе-
ним шаром 10 % (м/м) макроголу 20 000 Р:
- газ-носій азот для хроматографії Р;
- швидкість газу-носія 30 мл/хв;
- температура колонки 130 °С;
- температура детектора і блока вводу проб 200 °С.
Поперемінно хроматографують 1 мкл випробовувано-
го розчину, 1 мкл розчину порівняння (а) і 1 мкл роз-
чину порівняння (Ь). Чутливість системи регулюють
таким чином, щоб висота піка, одержаного на хрома-
тограмі випробовуваного розчину, становила не мен-
ше 80 % шкали реєструючого пристрою. Час хромато-
графування має бути у 3 рази більше часу утримуван-
ня камфори.
Хроматографічна система вважається придатною, як-
шо виконуються такі умови:
- коефіцієнт розділення піків камфори і борнілаце-
тату на хроматограмі розчину порівняння (а) ста-
новить не менше 1.5;
- відношення сигнал/шум, розраховане для основ-
ного піка на хроматограмі розчину порівняння (Ь),
становить не менше 5.
На хроматограмі випробовуваного розчину сума площ
усіх піків, крім основного, не має перевищувати 4 %
площі основного піка; площа жодного піка, крім ос-
новного, не має перевищувати 2 % площі основного
піка Не враховуються піки, плоша яких менше площі
основного піка на хроматограмі розчину порівнян-
ня (Ь).
Галогени. Не більше 0.01 % (100 ррт). 1.0 г субстанції
поміщають у колбу для перегонки і розчиняють у
10 мл 2-пропанолу Р, додають 1.5 мл розчину натрію
гідроксиду розведеного Р і 50 мг нікель-алюмінієвого
сплаву Р, нагрівають на водяній бані до упарювання
2-пропанолу Р. Охолоджують і додають 5 мл води Р.
Одержаний розчин перемішують і фільтрують крізь
вологий фільтр, попередньо промитий водою Р до
відсутності реакції на хлориди. Об’єм фільтрату дово-
дять водою Рао 10.0 мл. До 5.0 мл одержаного розчину
краплями додають кислоту азотну Р до розчинення
осаду, що утворюється, потім доводять водою Р до
об’єму 15 мл. Одержаний розчин має витримувати ви-
пробування на хлориди (2.4.4).
Вода. 1 г субстанції розчиняють у 10 мл петролейного
ефіру Р. Одержаний розчин має бути прозорим (2.2.1).
Залишок після сублімації. Не більше 0.05 %. 2.0 г суб-
станції упарюють на водяній бані і висушують при
температурі від 100 °С до 105 °С протягом 1 год. Маса
залишку не має перевищувати 1 мг.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
КАНАМІЦИНУ МОНОСУЛЬФАТ
Капатусіпі топозиІГаз
КАЬАМЕСІК МОРЮ81ЛРНАТЕ
С18Н38і\4Оі58,Н2О
М.м. 601
Канаміцину моносульфат О-З-аміно-З-деоксі-а-О-
глюкопіранозил-(1-»6)-0-[6-аміно-6-деоксі-а-0-
глюкопіранозил-( 1->4)]-2-деоксі-О-стрептаміну
сульфат - антибіотик, продукований певними штама-
ми Зїгерїотусех капатусебсиа. Антимікробна ак-
тивність має бути не менше 750 ОД/мг, у перерахунку
на суху речовину.
ВИРОБНИЦТВО
Спосіб виробництва субстанції має виключити або
звести до мінімуму вміст речовин, що знижують
кров’яний тиск. Субстанція має витримувати таке ви-
пробування:
Аномальна токсичність (2.6.9). Уволять кожній миші
0.5 мл розчину, що містить 2 мг канаміцину в 1 мл во-
ди для ін ’єкціи Р.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
383
Канаміцину моносульфат
Розчинність. Розчинний у близько восьми частинах
води Р, практично не розчинний в ацетоні Р.
96 % спирті Р і ефірі Р.
ІДЕНТИФІКАЦІЯ
А. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи пластинку, по
криту сумішшю з товщиною шару 0.75 мм. Суміш
одержують таким чином: 0.3 г карбомеру Рзмішують із
240 мл води Р, витримують, помірно перемішуючи-,
протягом 1 год, доводять рН одержаної суміші до 7.0,
додаючи порціями розчин натрію гідроксиду розведе-
ного Р, постійно перемішуючи, потім додають ЗО г
силікагелю н Р.
Пластинку нагрівають при температурі 110 °С протя-
гом І год, охолоджують і відразу використовують.
Випробовуваний розчин. 10 мг субстанції розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 10 мл.
Розчин порівняння (а). 10 мг ФСЗ канаміцину моносуль-
фату розчиняють у воді Р і доводять об’єм розчину
тим самим розчинником до 10 мл.
Розчин порівняння (Ь). 10 мг ФСЗ канаміцину моносуль-
фату, 10 мг ФСЗ неоміцину сульфату і 10 мг ФСЗ
стрептоміцину сульфату розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 10 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (10 мкг) випробовуваного розчину, 10 мкл
(10 мкг) розчину порівняння (а) і 10 мкл (10 мкг ка-
наміцину моносульфату, 10 мкг неоміцину сульфату і
10 мкг стрептоміцину сульфату) розчину порівняння (Ь).
Пластинку поміщають у камеру із розчином 70 г/л
калію дигідрофосфату Р. Коли фронт розчину пройде
12 см від лінії старту, пластинку виймають із камери,
сушать у струмені теплого повітря й обприскують
сумішшю рівних об’ємів розчину 2 г/л дигідроксинаф-
таліну Р у 96 % спирті Р і розчину 460 г/л кислоти
сірчаної Р Пластинку нагрівають при температурі
150 °С протягом від 5 хв до 10 хв.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром і забарвленням.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються
три чітко розділені плями.
В. 0.5 г субстанції розчиняють у 10 мл води Р і додають
10 мл розчину кислоти пікринової Р. Якщо необхідно,
прискорюють кристалізацію потиранням скляною па-
личкою стінку пробірки і витримують до утворення
кристалів. Кристали збирають, промивають 20 мл во-
ди Р, фільтрують і висушують при температурі
100 °С. Температура плавлення (2.2.14) кристалів має
бути близько 235 °С із розкладанням.
С. Близько 50 мг субстанції розчиняють у 2 мл води Р,
додають 1 мл розчину 10 г/л нінгідрину Рі нагрівають
протягом декількох хвилин на водяній бані; поступово
з’являється фіолетове забарвлення.
В. Субстанція дає реакції на сульфати (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 0.20 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 20.0 мл.
рН (2.2.3). Від 6.5 до 8.5. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від +112° до +123°,
у перерахунку на суху речовину. Визначення прово-
дять, використовуючи розчин 8.
Канаміцип В. Визначення проводять методом тонко-
шарової хроматографії (2.2.27), використовуючи пла-
стинки, приготовані, як зазначено у випробуванні А
розділу "Ідентифікація". Пластинку нагрівають при
температурі 110 °С протягом 1 год, охолоджують і
відразу використовують.
Випробовуваний розчин. 0.1 г субстанції розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 20 мл.
Розчин порівняння. 4 мг ФСЗ канаміцину В сульфату
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 20 мл.
На лінію старту хроматографічної пластинки наносять
4 мкл (20 мкг) випробовуваного розчину і 4 мкл
(0.8 мкг) розчину порівняння. Пластинку поміщають
у камеру з розчином 70 г/л калію дигідрофосфату Р
Коли фронт розчину пройде 12 см від лінії старту, пла-
стинку виймають із камери, сушать у струмені теплого
повітря й обприскують реактивом нінгідрину і оло-
ва(ЇІ) хлориду Р. Пластинку нагрівають при темпера-
турі 110 °С протягом 15 хв.
На хроматограмі випробовуваного розчину пляма, що
відповідає канаміцину В, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння.
Втрата в масі при висушуванні (2.2.32). Не більше
1.5 %. 1.00 г субстанції сушать при температурі 60 °С і
залишковому тиску не більше 670 Па протягом 3 год.
Сульфатна зола (2.4 14) Не більше 0.5 %. Визначення
проводять з 1.0 г субстанції.
Сульфати. Від 15.0 % до 17 0 % сульфату (8О4), у пере-
рахунку на суху речовину. 0.250 г субстанції розчиня-
ють у 100 мл води Р і доводять рН розчину до 11 розчи-
ном аміаку концентрованим Р. До одержаного розчину
додають 10.0 мл 0.1 М розчину барію хлориду, близько
0.5 мг фталеїнового пурпурного Р і нітрують 0.1 Мроз-
чином натрію едетату, додаючи 50 мл 96 % спирту Р,
коли забарвлення розчину почне змінюватися, і про-
довжують титрування до зникнення фіолетово-бла-
китного забарвлення
1 мл 0.1 М розчину барію хлориду відповідає 9.606 мг
сульфату (8О4).
384
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Каптоприл
Стерильність (2.6.1). Якщо субстанція призначена для
виробництва парентеральних лікарських засобів без
подальшої процедури стерилізації, вона має витриму-
вати випробування на стерильність.
Пірогени (2.6.8). Якщо субстанція призначена для ви-
робництва парентеральних лікарських засобів без
подальшої процедури видалення пірогенів, вона має
витримувати випробування на пірогени. Уводять на
1 кг маси кролика 1 мл розчину, що містить 10 мг ка-
наміцину в 1 мл води для ін ’єкцій Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять мікробіологічним методом
(2.7.2).
ЗБЕРІГАННЯ
У щільно закупореному контейнері. Якщо субстанція
стерильна, зберігають у стерильному контейнері з
контролем першого розкриття.
МАРКУВАННЯ
У необхідних випадках зазначають:
— субстанція стерильна;
— субстанція апірогенна.
____________________________________________N
Вступна частина. 1 ОД відповідає 1 мкг канаміцину.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Депресорні речовини (2.6.11). Якщо субстанція призначе-
на для виробництва парентеральних лікарських засобів,
вона має витримувати випробування на депресорні ре-
човини. Уводять на 1 кг маси кішки 1 мл розчину, що
містить 4 мг канаміцину в 1 мл води для ін ’єкцій Р.
КАПТОПРИЛ
Саріоргіїит
САРТОРКИ.
С9Н15МО38
М.м. 217.3
Каптоприл містить не менше 98.0 % і не більше
101.5 % (2А)-1-[(2У)-3-меркапто-2-метилпропаноїл]-
піролідин-2-карбонової кислоти, у перерахунку на су-
ху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Легко розчинний у воді Р, метилен-
хлориді Р і метанолі Р.
(Розчиняється в розведених розчинах гідроксидів
лужних металів).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В.
Друга ідентифікація: А, С, О.
А. Температура плавлення (2.2.14). Від 105 °С до 108 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ каптоприлу.
С. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. 10 мг субстанції розчиняють у
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 2 мл.
Розчин порівняння. 10 мг ФСЗ каптоприлу розчиняють
у метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 2 мл.
Налінію старту хроматографічної пластинки наносять
2 мкл (10 мкг) випробовуваного розчину і 2 мкл
(10 мкг) розчину порівняння. Пластинку помішають у
камеру із сумішшю розчинників кислота оцтова льо-
дяна Р- толуол Р(1:3). Коли фронт розчинників прой-
де 10 см від лінії старту, пластинку виймають із каме-
ри, сушать у струмені холодного повітря й обприску-
ють розчином 20 г/л 5.5'-дитіобіс(2-нітробензойної
кислоти) Р у метанолі Р, рН якого доведено до 8 роз-
чином аміаку розведеним Р.
На хроматограмі випробовуваного розчину має вияв-
лятись основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром і забарвленням.
Ц. Близько 20 мг субстанції розчиняють у 2 мл води Р.
До одержаного розчину додають 0.5 мл 0.05 М розчину
йоду, розчин відразу знебарвлюється.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 0.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 25.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
385
Каптоприл
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 2.0 до 2.6. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від -156° до -161°, у
перерахунку на суху речовину. Визначення проводять,
використовуючи розчин 8.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 50 мг субстанції розчиняють у
рухомій фазі і доводять об’єм розчину рухомою фазою
до 100.0 мл.
Розчин порівняння (а). 2.0 мл випробовуваного розчину
доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (Ь). 10 мг субстанції розчиняють у
рухомій фазі, додають 1 мл 0.05 Мрозчину йоду і дово-
дять об’єм розчину рухомою фазою до 100.0 мл.
10.0 мл одержаного розчину доводять рухомою фазою
до об’єму 100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.125 м х 4 мм,
заповнена силікагелем октилсилільним для хрома-
тографії Рз розміром часток 5 мкм;
— рухома фаза: кислота фосфорна концентрована Р -
метанол Р - вода Р (0.05:50:50);
— швидкість рухомої фази 1 мл/хв;
— детектування за довжини хвилі 220 нм.
Хроматографують 20 мкл розчину порівняння (а).
Чутливість системи регулюють таким чином, щоб ви-
сота основного піка становила не менше 40 % шкали
реєструючого пристрою.
Хроматографують 20 мкл розчину порівняння (Ь).
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— на хроматограмі мають бути три піки;
— коефіцієнт розділення для останніх двох піків має
бути не менше 2.0.
Поперемінно хроматографують 20 мкл випробовува-
ного розчину і 20 мкл розчину порівняння (а). Час
хроматографування має бути у 3 рази більше часу ут-
римування основного піка на хроматограмі випробо-
вуваного розчину.
На хроматограмі випробовуваного розчину площа будь-
якого піка, крім основного, не має перевищувати поло-
вини площі основного піка на хроматограмі розчину
порівняння (а) (1.0 %); сума площ усіх піків, крім ос-
новного, не має перевищувати площу основного піка на
хроматограмі розчину порівняння (а) (2.0 %). Не врахо-
вують піки з часом утримування менше 1.4 хв і піки,
площа яких становить менше 0.1 площі основного піка
на хроматограмі розчину порівняння (а).
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
1.0 %. 1.000 г субстанції сушать під високим вакуумом
протягом 3 год при температурі 60 °С.
Сульфатна зола (2.4.14). Не більше 0.2 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у 30 мл води Рі титрують
0.05 М розчином йоду потенціометрично (2.2.20), ви-
користовуючи комбінований платиновий електрод.
1 мл 0.05 Мрозчину йоду відповідає 21.73 мг С9НІ5МО38.
ЗБЕРІГАННЯ
У повітронепроникному контейнері.
ДОМІШКИ
А. (25.2’5)-1, Г-[3.3'-дитіобіс[(25)-2-метилпропано-
їл]]біспіролідин-2-карбонова кислота (каптоприл-ди-
сульфід).
_________________________________________________/V
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КЕТАМІНУ ГІДРОХЛОРИД
Кеїатіпі Ізудгоскіогідит
КЕТАМ 15 Е НЕОКОСНЮКЮЕ
, НСІ і енантіомер
С13Н17С12М)
М.м. 274.2
386
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кетаміну гідрохлорид
Кетаміну гідрохлорид містить не менше 99.0 % і не
більше 101.0 % (Я5)-2-(2-хлорфеніл)-2-(метила-
міно)циклогексанону гідрохлориду.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору.
Розчинність. Легко розчинний у воді Р і метанолі Р,
розчинний у 96 % спирті Р.
(Плавиться при температурі близько 260 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати еталонному спектру ДФУ ке-
таміну гідрохлориду.
В. Субстанція дає реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 5.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 25 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод 11). Розчин 8 має
бути безбарвним.
рН(225>. Від3.5до4.1. 10 мл розчину 8 доводять во-
дою, вільною від вуглецю діоксиду, Рдо об’єму 20 мл.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 50.0 мг субстанції розчиняють
у рухомій фазі і доводять об’єм розчину рухомою фа-
зою до 50.0 мл.
Розчин порівняння (а). 25.0 мг ФСЗ домішки А кетаміну
розчиняють у рухомій фазі і доводять об’єм розчину
рухомою фазою до 50.0 мл (якщо необхідно, викорис-
товують ультразвук). До 1.0 мл одержаного розчину
додають 0.5 мл випробовуваного розчину і доводять
об’єм розчину рухомою фазою до 100.0 мл. Розчин го-
тують безпосередньо перед використанням.
Розчин порівняння (Ь). 1.0 мл випробовуваного розчину
доводять рухомою фазою до об’єму 10.0 мл. 1.0 мл
одержаного розчину' доводять рухомою фазою до
об’єму 20.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.125 м х 4.0 мм
і передколонка з нержавіючої сталі розміром
4 мм х 4.0 мм, заповнені силікагелем октадецил-
силільним для хроматографії Р, з розміром часток
5 мкм;
— рухома фаза: 0.95 г натрію гексансульфонату Р
розчиняють у 1 л суміші ацетонітрил Р - вода Р
(25:75), потім додають 4 мл кислоти оцтової льодя-
ної Р;
— швидкість рухомої фази 1.0 мл/хв;
— детектування за довжини хвилі 215 нм.
Поперемінно хроматографують 20 мкл випробовува-
ного розчину, 20 мкл розчину порівняння (а) і 20 мкл
розчину порівняння (Ь). Час хроматографування має
бути в 10 разів більше часу утримування кетаміну.
Хроматографічна система вважається придатною, як-
що на хроматограмі розчину порівняння (а) ко-
ефіцієнт розділення піків домішки А кетаміну і ке-
таміну становить не менше 1.5. Якшо необхідно, регу-
люють співвідношення води й ацетонітрилу в рухомій
фазі.
На хроматограмі випробовуваного розчину сума площ
усіх піків, крім основного, не має перевищувати пло-
щу основного піка на хроматограмі розчину порівнян-
ня (Ь) (0.5 %). Не враховують піки, плоша яких стано-
вить менше 0.2 площі основного піка на хроматограмі
розчину порівняння (Ь).
Важкі метали (2.4.8, метод А). Не більше 0.002 %
(20 ррт). 10 мл розчину 8 доводять водою Рло об’єму
20 мл. 12 мл одержаного розчину мають витримувати
випробування на важкі метали. Еталон готують із ви-
користанням еталонного розчину свинцю (2 ррт РЬ) Р.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.200 г субстанції розчиняють у 50 мл метанолу Р, до-
дають 1.0 мл 0.1 М розчину кислоти хлористоводневої і
титрують 0.1М розчином натрію гідроксиду по-
тенціометрично (2.2.20). У розрахунок беруть об’єм
титранту між двома стрибками потенціалів на кривій
титрування.
1 мл 0.1 М розчину натрію гідроксиду відповідає
27.42 мгС13Н17СІ^О.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
А. 1-[(2-хлорфеніл)(метиліміно)метил|циклопентанол,
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
387
Кислота аскорбінова
і енантіомер
В. (7?5)-2-(2-хлорфеніл)-2-гідроксициклогексанон,
С. 2-хлорфеніл-1-гідроксициклопентил кетон.
______________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.200 г субстанції розчиняють у 1 мл кислоти мураши-
ної безводної Р, додають 20 мл кислоти оцтової безвод-
ної Р, 5 мл розчину ртуті(ІІ) ацетату Р і титрують
0.1 М розчином кислоти хіорної до синьо-зеленого за-
барвлення, використовуючи як індикатор 0.1 мл роз-
чину кристалічного фіолетового Р.
1 мл й/ Мрозчину кислоти хіорної відповідає 27.42 мг
С13Н17СІ2М).
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже біло-
го кольору або безбарвні кристали, шо змінюють ко-
лір під впливом повітря і вологи.
Розчинність. Легко розчинна у воді Р, розчинна у 96 %
спирті Р. практично не розчинна в ефірі Р.
(Плавиться при температурі близько 190 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, С.
Друга ідентифікація: А, С, В.
А. 0.10 г субстанції розчиняють у воді Р\ відразу дово-
дять об’єм розчину тим самим розчинником до
100.0 мл. До 10 мл 0.1 Мрозчину кислоти хлористовод-
невої аоааюзь 1.0 мл одержаного розчину і доводять
водою Рдо об’єму 100.0 мл. Вимірюють оптичну густи-
ну (2.2.25) одержаного розчину в максимумі за довжи-
ни хвилі 243 нм відразу після приготування розчину.
Питомий показник поглинання в максимумі має бути
від 545 до 585.
В. Інфрачервоний спектр поглинання (2.2.24) 1 мг
субстанції, одержаний у дисках, має відповідати спек-
тру ФСЗ кислоти аскорбінової.
С. рН (2.2.3) розчину 8, приготованого, як зазначено у
розділі "Випробування на чистоту", має бути від 2.1
до 2.6.
В. До 1 мл розчину 8 додають 0.2 мл кислоти азотної
розведеної Р і 0.2 мл розчину срібла нітрату Р2; утво-
рюється сірий осад.
КИСЛОТА АСКОРБІНОВА
Асідит а8согЬісит
А5СОКВІС АСИ)
с6н8о6
М.м. 176.1
Кислота аскорбінова містить не менше 99.0 % і не
більше 100.5 % (А)-5-[(5)-1,2-дигідроксіетил]-3,4-ди-
гідрокси-5Я-фуран-2-ону.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 20 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон ВУ7.
Питоме оптичне обертання (2.2.7). Від +20.5° до +21.5 .
2.50 г субстанції розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 25.0 мл.
Кислота щавлева. Не більше 0.2 %. 0.25 г субстанції
розчиняють у 5 мл води Р, нейтралізують розчином
натрію гідроксиду розведеним Р за червоним лакмусо-
вим папером Р, додають 1 мл кислоти оцтової розведе-
ної Р і 0.5 мл розчину кальцію хлориду Р (випробовува-
ний розчин).
70 мг кислоти щавлевої Р розчиняють у воді Рї дово-
дять об’єм розчину тим самим розчинником до 500 мл.
388
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кислота аспарагінова
До 5 мл одержаного розчину додають 1 мл кислоти оц-
тової розведеної Р і 0.5 мл розчину кальцію хлориду Р
(розчин порівняння).
Розчини витримують протягом 1 год. Опалесценція
випробовуваного розчину не має перевищувати опа-
лесценцію розчину порівняння.
Мідь. Не більше 0.0005 % (5 ррт). Визначення прово-
дять методом атомно-абсорбційної спектрометрії
(2.2.23, метод І).
Випробовуваний розчин. 2.0 г субстанції розчиняють у
0.1 Мрозчині кислоти азотної і доводять об’єм розчи-
ну тією самою кислотою до 25.0 мл.
Розчини порівняння Готують розведенням еталонного
розчину міді (10 ррт Си) Р 0.1 М розчином кислоти
азотної по концентрації 0.2 ррт. 0.4 ррт і 0.6 ррт Си.
Вимірюють поглинання одержаних розчинів за до-
вжини хвилі 324.8 нм, використовуючи як джерело ви-
промінювання лампу з порожнистим мідним катодом
і повітряно-ацетиленове полум’я. Настроюють реєст-
руючий пристрій на нульове значення, використову-
ючи 0.1 М розчин кислоти азотної.
Залізо. Не більше 0.0002 % (2 ррт). Визначення про-
водять методом атомно-абсорбційної спектрометрії
(2.2.23, метод І).
Випробовуваний розчин. 5.0 г субстанції розчиняють у
0.1 Мрозчині кислоти азотної і доводять об’єм розчи-
ну тією самою кислотою до 25.0 мл.
Розчини порівняння. Готують розведенням еталонного
розчину заліза (20 ррт Ре) Р 0.1 М розчином кислоти
азотної по концентрації 0.2 ррт. 0.4 ррт і 0.6 ррт Ге.
Вимірюють поглинання одержаних розчинів за до-
вжини хвилі 248.3 нм, використовуючи як джерело ви-
промінювання лампу із порожнистим залізним като-
дом і повітряно-ацетиленове полум’я. Настроюють
реєструючий пристрій на нульове значення, викорис-
товуючи 0.1 М розчин кислоти азотної.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 1 5 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 15 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у суміші 10 мл кислоти
сірчаної розведеної Рі 80 мл води, вільної від вуглецю
діоксиду, Р Додають 1 мл розчину крохмалю Р і тит-
рують 0.05 М розчином йоду по одержання стійкого
синьо-фіолетового забарвлення.
1 мл 0.05 Мрозчину йоду відповідає 8.81 мг С6Н8О6.
ЗБЕРІГАННЯ
У щільно закупореному неметалевому контейнері, у
захищеному від світла місці.
____________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КИСЛОТА АСПАРАГІНОВА
Ас і сіп ш аБрагїісит
ЛРІ’ЛКТІС АСИ)
СО2Н
но2с
н гчн2
С4Н7М)4 М.м. 133.1
Кислота аспарагінова містить не менше 98.5 % і не
більше 101.5 % (А)-2-аміно-бутандикарбонової кисло-
ти, у перерахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Мало розчинна у воді Р, практично не
розчинна у 96 % спирті Р і ефірі Р.
(Розчиняється в розведених розчинах гідроксидів
лужних металів і розведених мінеральних кислотах).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: А. В. В.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Суспензія 1 г субстанції в 10 мл води Р повинна ма-
ти сильнокислу реакцію (2.2.4).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
389
Кислота аспарагінова
С. Інфрачервоний спектр поглинання (2.224) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ кислоти аспарагінової.
В. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні ос-
новної плями на хроматограмі розчину порівняння (а),
відповідна їй за розміром і забарвленням.
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.І). 0.5 г субстанції розчиняють
уІМ розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 10 мл. Одержа-
ний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування "Прозорість
розчину", має бути не інтенсивнішим за еталон В¥6.
Питоме оптичне обертання (2.2.7/ Від +24.0° до +26.0°,
у перерахунку на суху речовину. 2.000 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у 2 мл розчину аміаку Р і доводять об’єм розчину
водою Рдо 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рдо об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ кислоти аспарагіно-
вої розчиняють у 2 мл розчину аміаку розведеного РІ і
доводять об’єм розчину водою Рдо 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчину (Ь)
доводять водою Рдо об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ кислоти аспарагінової
і 10 мг ФСЗ кислоти глутамінової розчиняють у
2 мл розчину аміаку розведеного Рі і доводять об’єм
розчину водою Рдо 25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг кислоти аспарагінової і
2 мкг кислоти глутамінової) розчину порівняння (с).
Пластинку сушать на повітрі і помішають у камеру із
сумішшю розчинників кислота оцтова льодяна Р - во-
да Р - бутанол Р (20:20:60). Коли фронт розчинників
пройде 15 см від лінії старту, пластинку виймають із
камери, сушать на повітрі й обприскують розчином
нінгідрину Р. Пластинку нагрівають при температурі
від 100 °С до 105 °С протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 0.25 гсуб-
станції розчиняють у 3 мл кислоти азотної розведе-
ної Рі доводять об’єм розчину водою Рдо 15 мл. Одер-
жаний розчин має витримувати випробування на хло-
риди без подальшого додавання розчину кислоти
азотної розведеної Р.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють у 4 мл кислоти хлористоводне-
вої Р і доводять об’єм розчину водою дистильованою Р
до 15 мл. Через 30 хв одержаний розчин має витриму-
вати випробування на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції помішають на
нижнє годинникове скло і суспендують у 0.5 мл
води Р. До одержаної суспензії додають 0.30 г магнію
оксиду важкого Р і ретельно розтирають скляною па-
личкою. Нижнє годинникове скло відразу накривають
верхнім і нагрівають при температурі 40 °С протягом
15 хв. Паралельно в аналогічних умовах готують ета-
лон, використовуючи 0.1 мл еталонного розчину
амонію (100 ррт ХП4) Р, 0.5 мл води Р і 0.30 г магнію
оксиду важкого Р. Лакмусовий папір над випробову-
ваною суспензією має бути забарвлений у синій колір
не інтенсивніше, ніж над еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод И). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1 000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять із 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0 100 г субстанції, якщо необхідно, злегка нагріва-
ючи, розчиняють у 50 мл води, вільної від вуглецю діок-
сиду, Р, охолоджують і титрують 0.1 М розчином
натрію гідроксиду до переходу забарвлення розчину
390
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кислота ацетилсаліцилова
від жовтого до синього, використовуючи як індикатор
0.1 мл розчину бромтимолового синього Р І.
1 мл 0.1 М розчину натрію гідроксиду відповідає
13.31 мгС4Н^О4.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
____________________________________________А
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів. вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції' має відповідати спектру ФСЗ кислоти ацетил-
саліцилової.
В. До 0.2 г субстанції додають 4 мл розчину натрію
гідроксиду розведеного Р. кип’ятять протягом 3 хв, охо-
лоджують і додають 5 мл кислоти сірчаної розведеної Р\
випадає кристалічний осад. Одержану суміш фільтру-
ють, осад промивають, потім сушать при температурі
від 100 °С до 105 °С. Температура плавлення (2.2.14)
одержаного залишку має бути від 156 °С до 161 °С.
С. У пробірці змішують 0.1 г субстанції з 0.5 г кальцію
гідроксиду Р. Суміш нагрівають і в парах, що з’яв-
ляються, витримують шматочок фільтрувального па-
перу, просочений 0 05 мл розчину нітробензальдегіду Р\
поступово папір забарвлюється в жовтувато-зелений
або блакитнувато-зелений колір. Потім папір змочу-
ють кислотою хлористоводневою розведеною Р; папір
забарвлюється в блакитний колір.
В. Близько 20 мг осаду, одержаного у випробуванні В,
при нагріванні розчиняють у 10 мл води Р ї охолоджу-
ють. Одержаний розчин дає реакцію (а) на саліцилати
(2.3.1).
КИСЛОТА АЦЕТИЛСАЛІЦИЛОВА випробування на чистоту
Асідит асеїуІБаїісуІісит
АСЕТУЬБАБУСІЬІС АСЮ
С)Н8О4
М.м. 180.2
Кислота ацетилсаліцилова містить не менше 99.5 % і
не більше 101.0 % 2-(ацетокси)бензойної кислоти, у
перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Мало розчинна у воді Р, легко розчинна у
96 % спирті Р, розчинна в ефірі Р.
(Плавиться при температурі близько 143 °С (миттєвий
метод)).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: В, С, В.
Прозорість розчину (2.2.1). 1.0 г субстанції розчиняють
у 9 мл 96 % спирту Р. Одержаний розчин має бути
прозорим.
Кольоровість розчину (2.2.2, метод 11). Розчин, приго-
тований, як зазначено у випробуванні "Прозорість
розчину", має бути безбарвним.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29) Розчини готують без-
посередньо перед використанням
Випробовуваний розчин. 0.10 г субстанції розчиняють в
ацетонітрилі для хроматографії Р і доводять об’єм
розчину тим самим розчинником до 10.0 мл.
Розчин порівняння (а). 50.0 мг кислоти саліцилової Р
розчиняють у рухомій фазі і доводять об’єм розчину
рухомою фазою до 50.0 мл. 1.0 мл одержаного розчину
доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (Ь). 10.0 мг кислоти саліцилової Р
розчиняють у рухомій фазі і доводять об’єм розчину
рухомою фазою до 10.0 мл. До 1.0 мл одержаного роз-
чину додають 0.2 мл випробовуваного розчину і дово-
дять рухомою фазою до об’єму 100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октадецилсилільним для
хроматографії Р з розміром часток 5 мкм;
— рухома фаза: кислота фосфорна Р - ацетонітрил
для хроматографії Р - вода Р (2:400:600);
— швидкість рухомої фази 1 мл/хв;
— детектування за довжини хвилі 237 нм.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
391
Кислота борна
Хроматографують 10 мкл випробовуваного розчину,
10 мкл розчину порівняння (а) і 10 мкл розчину
порівняння (Ь). Час хроматографування випробовува-
ного розчину має бути в 7 разів більше часу утриму-
вання кислоти ацетилсаліцилової.
Хроматографічна система вважається придатною, як-
що на хроматограмі розчину порівняння (Ь) ко-
ефіцієнт розділення двох основних піків становить не
менше 6.0.
На хроматограмі випробовуваного розчину площа
будь-якого піка, крім основного, не має перевищува-
ти площу основного піка на хроматограмі розчину
порівняння (а) (0.1 %); сума площ усіх піків не має пе-
ревищувати 2.5 площі основного піка на хроматограмі
розчину порівняння (а) (0 25 %). Не враховують піки,
плоша яких становить менше 0.25 площі основного
піка на хроматограмі розчину порівняння (а).
Важкі метали (2.4.8, метод В). Не більше 0.002 %
(20 ррт). 1.0 г субстанції розчиняють у 12 мл ацето-
ну Р ї доводять об’єм розчину водою Р до 20 мл. 12 мл
одержаного розчину мають витримувати випробуван-
ня на важкі метали. Еталон готують із використанням
еталонного розчину свинцю (1 ррт РЬ), одержаного
шляхом розведення еталонного розчину свинцю
(100ррт РЬ) Р сумішшю вода Р - ацетон Р(6.9)
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %>. 1.000 г субстанції сушать у вакуумі.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
1.000 г субстанції поміщають у колбу із притертою
скляною пробкою, розчиняють у 10 мл 96 % спирту Р
і додають 50.0 мл 0.5 М розчину натрію гідроксиду.
Колбу закривають і витримують протягом 1 год. Одер-
жаний розчин титрують 0.5 М розчином кислоти хло-
ристоводневої, використовуючи як індикатор 0.2 мл
розчину фенолфталеїну Р.
Паралельно проводять контрольний дослід.
1 мл 0.5 М розчину натрію гідроксиду відповідає
45.04 мг С9Н8О4.
ЗБЕРІГАННЯ
У повітронепроникному контейнері.
В. 4-гідроксибензен-1,3-дикарбоксильна кислота
(4-гідроксіізофталева кислота),
С. 2-гідроксибензойна кислота (саліцилова кислота),
В. 2-[[2-(ацетокси)бензоїл]окси]бензойна кислота
(ацетилсаліцилсаліцилова кислота),
Е.2-[(2-гідроксибензо'іл)окси]бензойна кислота (салі-
цилсаліцилова кислота),
Г. 2-(ацетокси)бензоєвий ангідрид (ацетилсаліцило-
вий ангідрид).
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КИСЛОТА БОРНА
Асідит Ьогісит
домішки
ІЮКІС АСИ)
Н3ВО3
М.м. 61.8
А. 4-гідроксибензойна кислота,
Кислота борна містить не менше 99.0 % і не більше
100.5 % Н3ВО3.
392
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кислота глутамінова
ВЛАСТИВОСТІ
Опис. Кристалічний порошок або кристали білого ко-
льору, або безбарвні, блискучі, жирні на дотик пластин-
ки.
Розчинність. Розчинна у воді Р, 96 % спирті Р, легко
розчинна у киплячій воді Р і гліцерині (85 %) Р
КИСЛОТА ГЛУТАМІНОВА
Асідит §1иІатісит
сштаміс лею
ІДЕНТИФІКАЦІЯ
Н МН2
А. 0.1 г субстанції, обережно нагріваючи, розчиняють
у 5 мл метанолу Р, додають 0.1 мл кислоти сірчаної Р і
розчин підпалюють; полум’я має зелену облямівку. С5НЧІ\О4
М.м. 147.1
В. Розчин 8, приготований, як зазначено у розділі
"Випробування на чистоту", є кислотою (2.2.4).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 3.3 г субстанції розчиняють у 80 мл киплячої
води дистильованої Р, охолоджують і доводять об’єм
розчину водою, вільною від вуглецю діоксиду, Р до
100 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 3.8 до 4.8. Вимірюють рН розчину 8.
Розчинність у спирті. 1.0 г субстанції розчиняють у
10 мл киплячого 96 % спирту Р Одержаний розчин за
ступенем каламутності не має перевищувати еталон II
(2.2.1) і має бути безбарвним <2 2.2, метод II).
Органічні речовини. Субстанція не має темні ги при
прожарюванні до блідо-червоною забарвлення.
Сульфати (2.4.13). Не більше 0.045 % (450 ррт). 10 мл
розчину 8 доводять водою дистильованою Рдо об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Важкі метали (2.4.8, метод А). Не більше 0.0015 %
(15 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням суміші 2.5 мл еталонного розчину свинцю
(2ррт РЬ) Р і 7.5 мл води Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
1.000 г субстанції розчиняють при нагріванні в 100 мл
води Р, що містить 15 гманіту Р, і титрують 1 Мрозчи-
ном натрію гідроксиду до появи рожевого забарвлен-
ня. використовуючи як індикатор 0.5 мл розчину фе-
нолфталеїну Р.
1 мл ЇМ розчину натрію гідроксиду відповідає 61.8 мг
Н3ВО3.
Кислота глутамінова містить не менше 98.5 % і не
більше 100.5% (5)-2-амінопентан-І,5-дикарбонової
кислоти, у перерахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Легко розчинна в киплячій воді Р, мало
розчинна в холодній воді Р, практично не розчинна в
кислоті оцтовій Р, ацетоні Р, 96 % спирті Р і ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, 1).
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Ви-
пробування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ кислоти глутамінової. У випадку різниці одержа-
них спектрів окремо розчиняють субстанцію і ФСЗ
кислоти глутамінової в мінімальному об’ємі води Р,
упарюють насухо при температурі 60 °С і повторно за-
писують спектри одержаних залишків.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівняння (а),
відповідна їй за розміром і забарвленням.
Ц. До 2.0 мл розчину 8, приготованого, як зазначено в
розділі "Випробування на чистоту", додають 0.1 мл
розчину фенолфталеїну Р, від 3.0 мл до 3.5 мл / М роз-
чину натрію гідроксиду до появи червоного забарвлен-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
393
Кислота глутамінова
ня. Потім додають суміш 3 мл розчину формальдегіду Р,
З мл води, вільної від вуглецю діоксиду, Р і 0.1 мл розчи-
ну фенолфталеїну Р, до якої попередньо доданий
1 М розчин натрію гідроксиду до появи рожевого за-
барвлення; розчин знебарвлюється. До одержаного
розчину додають / М розчин натрію гідроксиду до по-
яви червоного забарвлення. Загальний об’єм витраче-
ного 1Мрозчину натрію гідроксиду має бути від 4.0 мл
до 4.7 мл.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 5.00 г субстанції при слабкому нагріванні
розчиняють у 1 М розчині кислоти хлористоводневої і
доводять об’єм розчину тією самою кислотою до 50.0 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
Питоме оптичне обертання (2.2.7). Від +30.5° до +32.5°,
у перерахунку на суху речовину. Визначення прово-
дять, використовуючи розчин 8.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у 5 мл розчину аміаку розведеного Р2 і доводять
об’єм розчину водою Рао 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рао об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ кислоти глутамінової
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ кислоти глутамінової
і 10 мг ФСЗ кислоти аспарагінової розчиняють у воді Р
і доводять об’єм розчину тим самим розчинником до
25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь). 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг кислоти глутамінової і
2 мкг кислоти аспарагінової) розчину порівняння (с).
Пластинку сушать на повітрі протягом 15 хв і поміща-
ють у камеру із сумішшю розчинників кислота оцто-
ва льодяна Р - вода Р - бутанол Р (20:20:60). Коли
фронт розчинників пройде 15 см від лінії старту, плас-
тинку виймають із камери, сушать на повітрі й обпри-
скують розчином нінгідрину Р. Пластинку нагрівають
при температурі від 100 °С до 105 °С протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 0.25 г суб- |
станції розчиняють у 3 мл кислоти азотної розведе-1
ної Рі доводять об’єм розчину водою Рао 15 мл. Одержа-
ний розчин має витримувати випробування на хлориди
без подальшого додавання кислоти азотної розведеної Р.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 5 мл
розчину 8 доводять водою дистильованою Рао об'єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
помішають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і суспендують у 0.5 мл води Р.
До суспензії додають 0.30 г магнію оксиду важкого Рі
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв. Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (100ррт ХН^) Р,
0.5 мл води Р і 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваною суспензією має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хюристоводневоїрозведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод В). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.130 г субстанції при слабкому нагріванні розчиня-
ють у 50 мл води, вільної від вуглецю діоксиду, Р, охоло-
джують і титрують 0.1 М розчином натрію гідроксиду
до переходу жовтого забарвлення в блакитне, викори-
стовуючи як індикатор 0.1 мл розчину бромтимолового
синього РІ.
394
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кислота лимонна безводна
І мл 0.1 М розчину натрію гідроксиду відповідає
14.71 мгС5Н9ІЧО4.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________N
рН (2.2.3). Від 3.1 до 3.7. 10 мл розчину 8 доводять во-
дою дистильованою Рао об’єму 20 мл.
Залишкові кількості органічних розчинників.Субстанція
має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без процедури видалення піро-
генів, вона має витримувати випробування "Пірогени"
(2.6.8) або "Бактеріальні ендотоксини " (2.6.14).
КИСЛОТА ЛИМОННА БЕЗВОДНА
Асідит сіігісит апЬусІгісит
СШІСАСІ1), ЛМІМЖОІА,
СН2—СОгН
НО---С---СО2Н
І
СН2-СО2Н
С6Н8О7 М.м. 192.1
Кислота лимонна безводна містить не менше 99.5 % і
не більше 101.0% 2-гідроксипропан-1,2,3-трикарбо-
нової кислоти, у перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали, або гранули.
Розчинність. Дуже легко розчинна у воді Р. легко роз-
чинна у 96 % спирті Р, помірно розчинна в ефірі Р.
(Плавиться при температурі близько 153 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, Е.
Друга ідентифікація: А, С, Б, Е.
А. 1 г субстанції розчиняють у 10 мл води Р. Одержа-
ний розчин повинен мати сильнокислу реакцію
(2.2.4).
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, висушеної при температурі від 100 °С до
105 °С протягом 24 год, має відповідати спектру ФСЗ
кислоти лимонної безводної, висушеного за тих самих
умов.
С. Близько 5 мг субстанції додають до суміші 1 мл оц-
тового ангідриду Р і 3 мл піридину Р; з’являється чер-
воне забарвлення.
Б. 0.5 г субстанції розчиняють у 5 мл води Р, нейт-
ралізують 1 М розчином натрію гідроксиду (близько
7 мл), додають 10 мл розчину кальцію хлориду Р і
нагрівають до кипіння; утворюється білий осад.
Е. Субстанція має відповідати вимогам випробування
"Вода", зазначеним у розділі "Випробування на чистоту".
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2 2. /). 2 0 г субстанції розчиняють
у воді Рі доводять об’єм розчину тим самим розчинни-
ком до 10 мл. Одержаний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод /І). Забарвлення
розчину, приготованого, як зазначено у випробуванні
"Прозорість розчину", має бути не інтенсивнішим за
еталони ¥7, В¥7 або С¥7.
Речовини, шо легко обвуглюються. 1.0 г субстанції
поміщають у чисту пробірку, додають 10 мл кислоти
сірчаної Р і одержану суміш відразу нагрівають у во-
дяній бані при температурі (90±І) °С протягом 60 хв.
Відразу швидко охолоджують. Забарвлення одержано-
го розчину має бути не інтенсивнішим за забарвлення
суміші І мл червоного основного розчину і 9 мл жов-
того основного розчину (2.2.2, метод і).
Кислота щавлева. Не більше 0.035 % (350 ррт), у пере-
рахунку на безводну кислоту щавлеву. 0.80 г субстанції
розчиняють у 4 мл води Р, додають 3 мл кислоти хло-
ристоводневої Р і 1 г цинку Р у гранулах. Кип’ятять
протягом 1 хв і витримують протягом 2 хв. Надосадову
рідину переносять у пробірку, що містить 0.25 мл роз-
чину 10 г/л фенілгідразину гідрохюриду Рі нагрівають
до кипіння. Одержаний розчин швидко охолоджують,
переносять у мірний циліндр і додають рівний об’єм
кислоти хлористоводневої Р і 0.25 мл розчину 50 г/л
калію ферищаніду Р Струшують і витримують протя-
гом 30 хв; рожеве забарвлення розчину має бути не
інтенсивнішим за еталон, приготований аналогічно до
випробовуваного розчину з використанням 4 мл роз-
чину 0.1 г/л кислоти щавлевої Р.
Сульфати (2 4.13) Не більше 0.015 % (150 ррт). 1.0 г
субстанції розчиняють у воді дистильованій Р і дово-
дять об’єм розчину тим самим розчинником до 15 мл.
Одержаний розчин має витримувати випробування на
сульфати.
ЛГРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
395
Кислота нікотинова
Важкі метали (2.4.8, метод А). Не більше 0.001 % (10 ррт). До 5.0 г субстанції окремими порціями дода- ють 39 мл розчину натрію гідроксиду розведеного Р до розчинення і доводять об’єм розчину водою дистильо- ваною Рао 50 мл. 12 мл одержаного розчину мають ви- тримувати випробування на важкі метали. Еталон го- тують із використанням еталонного розчину свинцю (1 ррт РЬ) Р. КИСЛОТА НІКОТИНОВА Асідит пісоїіпісит МСОТШІС АСЮ
Вода (2.5.12). Не більше 1.0 %. Визначення проводять із 2.000 г субстанції напівмікрометодом. N
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення проводять з 1.0 г субстанції. ^^соон
Бактеріальні ендотоксини (2.6.14). Не більше 0.5 МО у 1 мг, якщо субстанція призначена для виробництва парентеральних лікарських засобів без подальшої процедури видалення бактеріальних ендотоксинів. С6Н5М)2 М.м. 123.1 Кислота нікотинова містить не менше 99.5 % і не більше 100.5 % піридин-3-карбонової кислоти, у пере-
Алюміній (2.4.17). Якщо субстанція призначена для виробництва розчинів для діалізу, вона має витриму- вати випробування на алюміній. Не більше 0.00002 % (0.2 ррт). рахунку на суху речовину. ВЛАСТИВОСТІ
20 г субстанції розчиняють у 100 мл води Р і додають 10 мл ацетатного буферного розчину рН 6.0 Р. Одержа- ний розчин має витримувати випробування на алюміній. Як еталон використовують суміш 2 мл ета- лонного розчину алюмінію (2 ррт АІ) Р, 10 мл ацетат- ного буферного розчину рН 6.0 Рі 98 мл води Р. Як холо- стий розчин використовують суміш 10 мл ацетатного буферного розчину рН 6.0 Р ї 100 мл води Р. Опис. Кристалічний порошок білого кольору. Розчинність. Розчинна у киплячій воді Р\ киплячому 96 % спирті Р, помірно розчинна у воді Р, практично не розчинна в ефірі Р. (Розчиняється в розведених розчинах гідроксидів і карбонатів лужних металів) ІДЕНТИФІКАЦІЯ
КІЛЬКІСНЕ ВИЗНАЧЕННЯ Перша ідентифікація: А, В.
0.550 г субстанції розчиняють у 50 мл води Р і титрують / М розчином натрію гідроксиду, використовуючи як індикатор 0.5 мл розчину фенолфталеїну Р. 1 мл 1 Мрозчину натрію гідроксиду відповідає 64 03 мг С6Н8О7. Друга ідентифікація: А, С. А. Температура плавлення (2.2.14). Від 234 °С до 240 °С. В. Інфрачервоний спектр поглинання (2.2.24) субстанції має відповідати спектру ФСЗ кислоти нікотинової. С. Близько 10 мг субстанції розчиняють у ІОмлвоФ/Р.
ЗБЕРІГАННЯ До 2 мл одержаного розчину додають 2 мл розчину ціаноброміду Р і 3 мл розчину 25 г/л аніліну Рї струшу-
У щільно закупореному контейнері. ють; з’являється жовте забарвлення.
МАРКУВАННЯ ВИПРОБУВАННЯ НАЧИСТОТУ
У необхідних випадках зазначають: Супровідні домішки. Визначення проводять методом
— субстанція вільна від бактеріальних ендотоксинів, — субстанція призначена для виробництва розчинів для діалізу. тонкошарової хроматографії (2.2.27), використовую- чи як тонкий шар силікагель сг254 Р. Випробовуваний розчин 0.5 г субстанції розчиняють у
N воді Р, якщо необхідно, злегка нагріваючи, і доводять об’єм розчину тим самим розчинником до 25 мл.
Залишкові кількості органічних розчинників. Суб- станція має витримувати вимоги статті (5.4). Розчин порівняння. 0.5 мл випробовуваного розчину доводять водою Рао об’єму 100 мл. Налінію старту хроматографічної пластинки наносять
Арсен (2.4.2, метод А). Не більше 0.0001 % (1 ррт). 1 г субстанції має витримувати випробування на арсен. 5 мкл (100 мкг) випробовуваного розчину і 5 мкл (0.5 мкг) розчину порівняння. Пластинку поміщають
396 ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кислота сорбінова
у камеру із сумішшю розчинників вода Р - кислота му-
рашина безводна Р - пропанол Р (5:10:85). Коли фронт
розчинників пройде 15 см від лінії старту, пластинку
виймають із камери, сушать при температурі від 100 °С
до 105 °С протягом 10 хв і переглядають в УФ-світлі за
довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %).
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 0.25 г суб-
станції розчиняють у воді Р, нагріваючи на водяній
бані, і доводять об’єм розчину тим самим розчинни-
ком до 15 мл. Одержаний розчин має витримувати ви-
пробування на хлориди.
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
1.0%. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С протягом 1 год.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КИСЛОТА СОРБІНОВА
Асідит 8ОгЬісит
8ОКВІС АСІй
С6Н8О2
М.м. 112.1
Кислота сорбінова містить не менше 99.0 % і не
більше 101.0 % (Е,£)-гекса-2.4-дієнової кислоти, у пе-
рерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Мало розчинна у воді Р, легко розчинна у
96 % спирті Р і ефірі Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у 50 мл води Рі титрують
0.1 Мрозчином натрію гідроксиду до одержання роже-
вого забарвлення, використовуючи як індикатор
0.25 мл розчину фенолфталеїну Р.
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину натрію гідроксиду відповідає
12 31 мгС6Н5ГМО2.
ЗБЕРІГАННЯ
У захищеному від світла місці.
__________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: А, В, І).
А. Температура плавлення (2.2.14). Від 132 °С до 136 °С.
В. 50.0 мг субстанції розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 250.0 мл.
2.0 мл одержаного розчину доводять 0.1 М розчином
кислоти хлористоводневої до 200.0 мл. Ультрафіолето-
вий спектр поглинання (2.2.25) одержаного розчину в
області від 230 нм до 350 нм повинен мати максимум
за довжини хвилі 264 нм. Питомий показник погли-
нання у максимумі має бути від 2150 до 2550.
С. Інфрачервоний спектр поглинання (2.2.24) субстанції
має відповідати спектру ФСЗ кислоти сорбінової.
В. 0.2 г субстанції розчиняють у 2 мл 96 % спирту Р і
додають 0.2 мл води бромноїР, розчин знебарвлюється.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.25 г субстанції розчиняють у 96 % спирті Р і
доводять об’єм розчину тим самим розчинником до 25 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
Альдегіди. Не більше 0.15 %, у перерахунку на С2Н4О.
1.0 г субстанції розчиняють у суміші 30 мл води Р і
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
397
Клонідину гідрохлорид
50 мл 2-пропанолу Р, доводять рН 0.1 М розчином кис-
лоти хлористоводневої або 0.1 М розчином натрію
гідроксиду до 4 і доводять об’єм розчину водою Р до
100 мл. До 10 мл одержаного розчину додають 1 мл
розчину фуксину знебарвленого Р і витримують протя-
гом ЗО хв. Забарвлення розчину має бути не інтен-
сивнішим за еталон, приготований аналогічно до ви-
пробовуваного розчину додаванням 1 мл розчину фук-
сину знебарвленого Р до суміші 1.5 мл еталонного роз-
чину ацетальдегіду (100ррт С2Н4О) Р, 4 мл 2-пропано-
лу Р і 4.5 мл води Р.
Важкі метали (2.4.8, метод В). Не більше 0.001 %
(10 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням 5 мл еталонного розчину свинцю (1 ррт РЬ) Р і
5 мл 96 % спирту Р.
Вода (2.5.12). Не більше 1.0 %. Визначення проводять
із 2.000 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0 2%. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.1000 г субстанції розчиняють у 20 мл 96 % спирту Р і
титрують 0.1 М розчином натрію гідроксиду до рожево-
го забарвлення, використовуючи як індикатор 0.2 мл
розчину фенолфталеїну Р.
1 мл 0.1 М розчину натрію гідроксиду відповідає
11.21 мгС6Н8О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КЛОНІДИНУ ГІДРОХЛОРИД
Сіопідіпі кудгосіїїогісіит
сіохпзіхе НУОКОСНЮКЮЕ
С9НІ()СІ3Г\3
М.м. 266.6
Клонідину гідрохлорид містить не менше 98.5 % і не
більше 101.0% 2-[(2,6-дихлорфеніл)аміно]-2-іміда-
золіну гідрохлориду, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Розчинний у воді Р і 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, О.
Друга ідентифікація: А, С, Б.
А. 30 0 мг субстанції розчиняють у 0 01 Мрозчині кисло-
ти хлористоводневої і об’єм розчину тим самим
розчинником до 100.0 мл. Ультрафіолетовий спектр по-
глинання (2.2.25) одержаного розчину в області від
245 нм до 350 нм повинен мати два максимуми за до-
вжин хвиль 272 нм і 279 нм і плече за довжини хвилі
близько 265 нм. Питомий показник поглинання в мак-
симумах має бути близько 18 і близько 16 відповідно.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ клонідину гідро-
хлориду.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Супровідні домішки",
має виявлятися основна пляма на рівні основної пля-
ми на хроматограмі розчину порівняння (а),
відповідна їй за розміром і забарвленням.
Б. Субстанція дає реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.25 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 25 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим
Кольоровість розчину (2.2.2, метод П). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон ¥7.
рН (2.2.3). Від 4.0 до 5.0. Вимірюють рН розчину 8.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель в Р.
Випробовуваний розчин (а). 0.1 г субстанції розчиняють
у метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять метанолом Р до об’єму 10 мл.
Розчин порівняння (а). 10 мг ФСЗ клонідину гідрохлори-
ду розчиняють у метанолі Р і доводять об’єм розчину
тим самим розчинником до 10 мл.
398
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кофеїн
Розчин порівняння (Ь). 1 мл випробовуваного розчину (а)
доводять метанолом Рао об’єму 10 мл. 5 мл одержано-
го розчину доводять метанолом Рао об’єму 100 мл.
Налінію старту хроматографічної пластинки наносять
10 мкл (100 мкг) випробовуваного розчину (а), 10 мкл
(10 мкг) випробовуваного розчину (Ь), 10 мкл
(100 мкг) розчину порівняння (а) і 10 мкл (0.5 мкг)
розчину порівняння (Ь). Струшують у ділильній лійці
суміш розчинників кислота оцтова льодяна Р - бута-
нол Р - вода Р (10:40'50) і витримують до розшаруван-
ня. Верхній шар фільтрують і використовують
фільтрат як рухому фазу.
Пластинку поміщають у камеру із рухомою фазою
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й
обприскують розчином калію йодовісмутату Р2. Плас-
тинку сушать на повітрі протягом 1 год і знову обпри-
скують спочатку розчином калію йодовісмутату Р2, а
потім відразу — розчином 50 г/л натрію нітриту Р.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Втрата в масі при висушуванні (2.2.32). Не більше
0 5%. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.200 г субстанції розчиняють у 70 мл 96 % спирту Р і
титрують 0.1 М розчином натрію гідроксиду етаноль-
ним потенціометрично (2.2.20).
1 мл 0.1 М розчину натрію гідроксиду етанольного
відповідає 26.66 мг С9Н10С1з1Ч3.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
__________________________________________/V
КЛОФЕЛІН
СІорИеІіпит
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КОФЕЇН
СоГГеіпит
САЕЕЕІПЕ
СвНіо^Ог
М.м. 194.2
Кофеїн містить не менше 98.5 % і не більше 101.5 %
1,3,7-триметил-3,7-дигідро-1//-пурин-2,6-діону, у пе-
рерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок або шовковисті криста-
ли білого кольору; легко сублімуються.
Розчинність. Помірно розчинний у воді Р, легко роз-
чинний у киплячій воді Р, мало розчинний в етанолі Р
і еф ір і Р.
(Розчиняється у концентрованих розчинах лужних
бензоатів або саліцилатів).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, Е.
Друга ідентифікація: А, С, Б, Е, Г.
А. Температура плавлення (2.2.14). Вщ 234 °С до 239 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ кофеїну.
С. До 2 мл насиченого розчину субстанції додають
0.05 мл розчину калію йодиду йодованого Р; розчин за-
лишається прозорим. До одержаного розчи ну додають
0 І мл кислоти хлористоводневої розведеної Р; утво-
рюється коричневий осад, який розчиняється при
нейтралізації розчином натрію гідроксиду розведеним Р.
О. Близько 10 мг субстанції поміщають у пробірку із
притертою скляною пробкою, розчиняють у 0.25 мл
суміші 0.5 мл ацетилацетону Р і 5 мл розчину натрію
гідроксиду розведеного Р. Одержаний розчин нагріва-
ють у водяній бані при температурі 80 °С протягом
7 хв, охолоджують і додають 0.5 мл розчину димети-
ламінобензальдегіду Р2. Ще раз нагрівають у водяній
бані при температурі 80 °С протягом 7 хв, охолоджу-
ють і додають 10 мл води Р; з’являється інтенсивне
синє забарвлення.
пгпм/лппл ал пм а і/гіпса х/іголїіділ топі
399
Кофеїну моногідрат
Е. Субстанція мас витримувати вимоги випробування
"Втрата в масі при висушуванні", зазначені у розділі
"Випробування на чистоту".
Г. Субстанція дає реакцію на ксантини (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 0 5 г субстанції розчиняють при нагріванні в
50 мл води, вільної від вуглецю діоксиду, Р, приготова-
ної із води дистильованої Р, охолоджують і доводять
об’єм розчину тим самим розчинником до 50 мл.
Прозорість розчину (2.2.1) Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод 11). Розчин 8 має
бути безбарвним .
Кислотність. До 10 мл розчину 8 додають 0.05 мл роз-
чину бромтимолового синього РІ; з’являється зелене
або жовте забарвлення, що переходить у синє при до-
даванні не більше 0.2 мл 0.01 Мрозчину натрію гідрок-
сиду.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2 27), використовую-
чи як тонкий шар силікагель ср254 Р
Випробовуваний розчин. 0.2 г субстанції розчиняють у
суміші розчинниківл/е/панол Р-хлороформ Р(4:6) і до-
водять об’єм розчину тією самою сумішшю розчин-
ників до 10 мл.
Розчин порівняння. 0.5 мл випробовуваного розчину
доводять сумішшю розчинників метанол Р - хлоро-
форм Р (4:6) до об’єму 100 мл.
Налінію старту хроматографічної пластинки наносять
10 мкл (200 мкг) випробовуваного розчину і 10 мкл
(1 мкг) розчину порівняння. Пластинку поміщають у
камеру із сумішшю розчинників розчин аміаку концен-
трований Р - ацетон Р - хлороформ Р - бутанол Р
(10:30:30:40). Коли фронт розчинників пройде 15 см
від лінії старту, пластинку виймають із камери, сушать
на повітрі і переглядають в УФ-світлі за довжини хвилі
254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %).
Сульфати (2.4.13). Не більше 0.05 % (500 ррт). 15 мл
розчину 8 мають витримувати випробування на суль-
фати. Еталон готують із використанням суміші 7.5 мл
еталонного розчину сульфату (10 ррт 80^ Р і 7.5 мл
води дистильованої Р.
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С протягом 1 год.
Сульфатна зола (2.4 14). Не більше 0.1 % Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.170 г субстанції розчиняють при нагріванні в 5 мл
кислоти оцтової безводної Р. Охолоджують, додають
10 мл оцтового ангідриду Р, 20 мл толуолу Р\ титрують
0.1 М розчином кислоти хлорної потенціометрично
(2.2.20).
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину кислоти хіорної відповідає 19.42 мг
С8Н10^О2.
_____________________________________________N
Речовини, що легко обвуглюються. 0.5 г субстанції роз-
чиняють у 5 мл кислоти сірчаної Р. Одержаний розчин
має бути прозорим і безбарвним (2.2.2, метод 1).
Залишкові кількості органічних розчинників. Суб
станція має витримувати вимоги статті (5.4).
ЗБЕРІГАННЯ
У сухому, захищеному від світла місці.
КОФЕЇНУ МОНОГІДРАТ
Соїїеіпигп топокудгісит
САННІМ МОбіОНУИКАТЕ
С8НіиК4О2,Н2О М.м. 212.2
Кофеїну моногідрат містить не менше 98.5 % і не
більше 101.5 % 1,3,7-триметил-3,7-дигідро-7/7-пу-
рин-2,6-діону, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Субстанція витримує вимоги, описані в статті "Кофеїн".
ІДЕНТИФІКАЦІЯ
Субстанція має витримувати випробування розділу
"Ідентифікація", описані в статті "Кофеїн"; перед про-
веденням випробувань А і В субстанцію сушать при
температурі від 100 °С до 105 °С протягом 1 год.
Перша ідентифікація: А, В, Е.
Друга ідентифікація: А, С, В, Е, Г.
400
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Кофеїну моногідрат
ВИПРОБУВАННЯ НАЧИСТОТУ
Субстанція має витримувати вимоги розділу "Випро-
бування на чистоту", описані в статті "Кофеїн", з ураху-
ванням таких змін.
Втрата в масі при висушуванні (2.2.32). Від 5.0 % до
9.0%. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С протягом 1 год.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Кількісне визначення проводять, як описано в статті
"Кофеїн", використовуючи попередньо висушену при
температурі від 100 °С до 105 °С протягом 1 год суб-
станцію.
1 мл 0.1 М розчину кислоти хлорної відповідає 19.42 мг
С8НкМО2.
____________________________________________N
ЗБЕРІГАННЯ
У сухому, захищеному від світла місці
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
401
Леицин
ЛЕЙЦИН
кеисіпит
ІЕОСІПЕ
Н3С^^х/СО2Н
І А
сн3 н мн2
С6НІ31\О2
М.м. 131.2
Лейцин містить не менше 98.5 % і не більше 101.0 %
(5)-2-аміно-4-метилпентанової кислоти, у перерахун-
ку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Блискучі пластинки або кристалічний порошок
білого або майже білого кольору.
Розчинність. Помірно розчинний у воді Р, практично
не розчинний у 96 % спирті Р і ефірі Р.
(Розчиняється в розведених мінеральних кислотах і
розведених розчинах гідроксидів лужних металів).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: А, В, Ц.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. 0 50 г субстанції розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 25 мл.
Одержаний розчин обертає площину поляризації
ліворуч.
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ лейцину
Ц. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 0.5 г субстанції розчиняють
у 1 М розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 10 мл. Одержа-
ний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину, приготованого для випробування "Прозорість
розчину", має бути не інтенсивнішим за еталон В¥6.
Питоме оптичне обертання (2.2.7). Від +14.5° до +16.5°,
у перерахунку на суху речовину. 1.00 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у 0.1 М розчині кислоти хлористоводневої і дово-
дять об’єм розчину тією самою кислотою до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рао об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ лейцину розчиняють
у 0.1 М розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчину (Ь)
доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ лейцину і 10 мг ФСЗ
валіну розчиняють у 0.1 Мрозчині кислоти хлористо-
водневої і доводять об’єм розчину тією самою кисло-
тою до 25 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг лейцину і 2 мкг валіну)
розчину порівняння (с). Пластинку сушать на повітрі
і поміщають у камеру із сумішшю розчинників кисло-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
403
Лізину гідрохлорид
та оцтова льодяна Р - вода Р - бутанол Р (20:20:60).
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й
обприскують розчином нінгідрину Р. Пластинку
нагрівають при температурі від 100 °С до 105 °С протя-
гом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 0.25 г суб-
станції розчиняють у воді Р і доводять об’єм розчину
тим самим розчинником до 15 мл. Одержаний розчин
має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють у 3 мл кислоти хлористоводне-
вої розведеної Р і доводять об’єм розчину водою дисти-
льованою Рао 15 мл. Одержаний розчин має витриму-
вати випробування на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовляють
комірку, що складається із двох годинникових стекол
діаметром 60 мм, сполучених по краях. На внутрішню
поверхню верхнього годинникового скла поміщають
шматочок червоного лакмусового паперу Р розміром
(5 х 5) мм, змочений декількома краплями води Р. 50 мг
здрібненої субстанції поміщають на нижнє годиннико-
ве скло і розчиняють або суспендують у 0.5 мл води Р.
До одержаного розчину або суспензії додають 0.30 г
магнію оксиду важкого Р і ретельно розтирають скля-
ною паличкою. Нижнє годинникове скло відразу на-
кривають верхнім і нагрівають при температурі 40 °С
протягом 15 хв. Паралельно в аналогічних умовах готу-
ють еталон, використовуючи 0.1 мл еталонного розчину
амонію (100ррт НП4) Р, 0.5 мл води Р і 0.30 гмагнію ок-
сиду важкого Р. Лакмусовий папір над розчином або
суспензією має бути забарвлений у синій колір не
інтенсивніше, ніж над еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилїзобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів долають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод О). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають ЗО мл кислоти оцтової безвод-
ної Р і титрують 0.1М розчином кислоти хюрної до пе-
реходу забарвлення від коричнювато-жовтого до зеле-
ного, використовуючи як індикатор 0.1 мл розчину
нафтолбензеїну Р.
1 мл 0.1 Мрозчину кислоти хюрної відповідає 13.12 мг
С6Н13М)2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
____________________________________________N
рН (2.2.3). Від 5.5. до 7.0. 1.0 г субстанції розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 100 мл.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якшо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
ЛІЗИНУ ГІДРОХЛОРИД
Ьувіпі Иудгосізіогідит
/1Л7А/ НУ1Ж0СНШКІВЕ
С6Н15С11У2О2
М.м. 182.7
Лізину гідрохлорид містить не менше 98.5 % і не біль-
ше 101.0% (5)-2,6-діаміногексанової кислоти гідро-
хлориду, у перерахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
404
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лізину гідрохлорид
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Легко розчинний у воді Р, мало розчинний
у 96 % спирті Р, практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, Е.
Друга ідентифікація: А, С, Ц, Е.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗлізину гідрохлориду. У випадку різниці одержаних
спектрів окремо розчиняють субстанцію і ФСЗ лізину
гідрохлориду в мінімальному об’ємі води Р, упарюють
насухо при температурі 60 °С і повторно записують
спектри одержаних залишків.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порів-
няння (а), відповідна їй за розміром і забарвленням.
І). До 0.1 мл розчину 8, приготованого, як зазначено
в розділі "Випробування на чистоту", додають 2 мл
води Р і 1 мл розчину 50 г/л кислоти фосфорномолібде-
нової Р; утворюється жовтувато-білий осад.
Е. До 0.1 мл розчину 8 додають 2 мл води Р Одержа-
ний розчин дає реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 5.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р, приготованій із води дистильованої
Р, і доводять об’єм розчину тим самим розчинником
до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення роз-
чину 8 має бути не інтенсивнішим за еталон В7 або О¥7.
Питоме оптичне обертання (2.2.7). Від +21.0° до +22.5°,
у перерахунку на суху речовину. 2.00 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тією самою кислотою до 25.0 мл
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХпластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину доводять водою Рдо об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ лізину гідрохлориду
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчину (Ь)
доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ лізину гідрохлориду і
10 мг ФСЗ аргініну розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг лізину гідрохлориду і
2 мкг аргініну) розчину порівняння (с). Пластинку су-
шать на повітрі й поміщають у камеру із сумішшю роз-
чинників розчин аміаку концентрований Р - 2-пропа-
нол Р (30:70). Коли фронт розчинників пройде 15 см
від лінії старту, пластинку виймають із камери, сушать
при температурі від 100 °С до 105 °С до зникнення за-
паху аміаку й обприскують розчином нінгідрину Р Пла-
стинку нагрівають при температурі від 100 °С до 105 °С
протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 5 мл
розчину 8 доводять водою дистильованою Рао об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р і
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв. Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (К)Оррт ХН4) Р,
0.5 мл води Р і 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваним розчином має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.003 % (30 ррт). 0.33 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Рі струшують протягом 3 хв.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
405
Лінкоміцину гідрохлорид
Одержаний водний розчин має витримувати випробу-
вання на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у 5 мл кислоти мураши-
ної безводної Р, додають 50 мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної по-
тенціометрично (2.2.20).
1 мл 0.1 Мрозчину кислоти хлорної відповідає 18.27 мг
С6Н15СІК2О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
_____________________________________________N
рН (2.2.3). Від 5.0 до 6.0. 10 мл розчину 8 доводять во-
дою, вільною від вуглецю діоксиду, Рдо об’єму 20 мл.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
ЛІНКОМІЦИНУ ГІДРОХЛОРИД
Гупсотусіпі Иусігосіїїогісіит
Ь¥ПСОМ¥СІП ІІ\1)ІЮСІІІ.()КІІ)Е
С18Н35СІК2О68,Н2О
М.м. 461.0
Лінкоміцину гідрохлорид складається в основному і
моногідрату метил-6,8-дидеокси-6-[(2А,4Я)-І-метил-
4-пропілпіролідин-2-карбоксамідо]-1-тіо-0-еритро-
а-О-галакто-октопіранозиду гідрохлориду - антибіо-
тик, продукований Бігеріотусех Ипсоїпехіх гаг. Ііпсоїпе-
або одержаний будь-якими іншими способами.
Субстанція містить не менше 82.5 % і не більше 93.0%
лінкоміцину (С1ііН3418І2Об8), у перерахунку на безвод-
ну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Дуже легко розчинний у воді Р, мало роз-
чинний у 96 % спирті Р, дуже мало розчинний в аце-
тоні Р, практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, П.
Друга ідентифікація: В, С, О.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ лінкоміцину
гідрохлориду.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. 10 мг субстанції розчиняють у
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Розчин порівняння (а). 10 мг ФСЗлінкоміцину гідрохю-
риду розчиняють у метанолі Р і доводять об’єм розчи-
ну тим самим розчинником до 10 мл.
Розчин порівняння (Ь). 10 мг ФСЗлінкоміцину гідрохло-
406
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лінкоміцину гідрохлорид
риду і 10 мг ФСЗ кліндаміцину гідрохлориду розчиня-
ють у метанолі Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (5 мкг) випробовуваного розчину, 5 мкл (5 мкг)
розчину порівняння (а) і 5 мкл (5 мкг лінкоміцину
гідрохлориду і 5 мкг кліндаміцину гідрохлориду) роз-
чину порівняння (Ь). Пластинку поміщають у камеру з
рухомою фазою і хроматографують. Як рухому фазу
використовують верхній шар суміші розчинників
2-пропанол Р - розчин 150 г/л амонію ацетату Р, рН
якого попередньо доведено до 9.6 розчином аміаку Р, -
етилацетат Р (20:40:45). Коли фронт розчинників
пройде 15 см від лінії старту, пластинку виймають із
камери, сушать на повітрі й обприскують розчином
1 г/л калію перманганату Р.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром і забарвленням.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
С. Близько 10 мг субстанції розчиняють у 2 мл кисло-
ти хюристоводневої розведеної Рі нагрівають на во-
дяній бані протягом 3 хв, додають 3 мл розчину натрію
карбонату Р і 1 мл розчину 20 г/л натрію нітропруси-
ду Р; поступово з’являється фіолетово-червоне забар-
влення.
В. 0.1 г субстанції розчиняють у воді Рі доводять об’єм
розчину тим самим розчинником до 10 мл. Розчин дає
реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 20 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон ¥6.
рН (2.2.3). Від 3.5 до 5.5. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від +135° до +150°,
у перерахунку на безводну речовину. 1.000 г субстанції
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 25.0 мл.
Лінкоміцин В. На хроматограмі випробовуваного роз-
чину (а), одержаній у розділі "Кількісне визначення",
площа піка лінкоміцину В, що виходить перед піком
лінкоміцину, становить не більше 5 % площі піка
лінкоміцину.
Важкі метали (2.4.8, метод С). Не більше 0.0005 %
(5 ррт ). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 1.0 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Вода (2.5.12). Від 3.1 % до 4.6 %. Визначення прово-
дять з 0.500 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.5 %. Визначення
проводять з 1.0 г субстанції.
Стерильність (2.6.1). Якщо субстанція призначена для
виробництва парентеральних лікарських засобів без
подальшої процедури стерилізації, вона має витриму-
вати випробування на стерильність.
Бактеріальні ендотоксини (2.6.14). Не більше 0.50 МО у
1 мг, якщо субстанція призначена для виробництва
парентеральних лікарських засобів без подальшої
процедури видалення бактеріальних ендотоксинів.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять методом газової хроматографії
(2.2.28), використовуючи як внутрішній стандарт до-
триаконтан Р.
Розчин внутрішнього стандарту. 0.200 г дотриаконта-
ну Р розчиняють у хлороформі Р і доводять об’єм роз-
чину тим самим розчинником до 25.0 мл.
Випробовуваний розчин (а). 0.100 г субстанції розчиня-
ють у розчині 20 г/л імідазолу Р у хлороформі Р і дово-
дять об’єм тим самим розчином до 100.0 мл. Струшу-
ють до повного розчинення. 4.0 мл одержаного розчи-
ну поміщають у пентрифужну пробірку місткістю
15 мл із притертою скляною пробкою, додають 1.0 мл
суміші хлортриметилсилан Р - А,О-біс(триметил-
силіл)ацетамід РІЗ'.ЧО) і обережно перемішують обер-
тальними рухами. Пробірку нещільно закривають
пробкою і витримують при температурі 65 °С протя-
гом 30 хв.
Випробовуваний розчин (Ь). Розчин готують аналогічно
до випробовуваного розчину (а), додаючи перед роз-
чиненням субстанції 10.0 мл розчину внутрішнього
стандарту.
Розчин порівняння. Розчин готують аналогічно до ви-
пробовуваного розчину (а), використовуючи замість
субстанції 0.100 г ФСЗлінкоміцину гідрохюриду і дода-
ючи перед розчиненням ФСЗлінкоміцину гідрохюриду
10.0 мл розчину внутрішнього стандарту.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
— скляна колонка розміром 1.5 м х 3 мм, заповнена
діатомітом силанїзованим для газової хромато-
графії Р, з нанесеним 3 % (м/м) поліметилфеніл-
силоксаном Р;
— газ-носій гелій для хроматографії Р;
— швидкість газу-носія 45 мл/хв;
— температура колонки 260 °С;
— температура блока вводу проб і детектора від
260 °С до 290 °С.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
407
Лінкоміцину гідрохлорид
Поперемінно хроматографують обрані об'єми випро-
бовуваних розчинів і розчину порівняння.
ЗБЕРІГАННЯ
У повітронепроникному контейнері при температурі
не више ЗО °С. Якщо субстанція стерильна, зберігають
у стерильному, повітронепроникному контейнері з
контролем першого розкриття.
МАРКУВАННЯ
У необхідних випадках зазначають:
— субстанція стерильна;
— субстанція вільна від бактеріальних ендотоксинів.
____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Замість випробування "Бактеріальні ендотоксини"до-
пускається застосування випробування "Пірогени"
(2.6.8).
Аномальна токсичність (2.6.9). Якщо субстанція при-
значена для виробництва парентеральних лікарських
засобів, вона має витримувати випробування на ано-
мальну токсичність. Уводять кожній миші протягом
ЗО с 2 мі лінкоміцину у 0 5 мл води для ін’єкцій Р.
Термін спостереження 24 год.
Депресорні речовини (2.6 11). Якщо субстанція при-
значена для виробництва парентеральних лікарських
засобів, вона має витримувати випробування на де-
пресорні речовини.
МАРКУВАННЯ
При проведенні випробування "Пірогени" замість
субстанція вільна від бактеріальних ендотоксинів"
зазначають:
— субстанція апірогенна.
408
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Магнію оксид важкий
МАГНІЮ ОКСИД ВАЖКИЙ
Ма§пе8Іі ох усі шп ропсіего8ит
МАСІЇЕЕ111М ОХІБЕ, НЕАУУ
М§о М.м. 40.30
Магнію оксид важкий містить не менше 98.0 % і не
більше 100.5 % М&О, у перерахунку на прожарену ре-
човину.
ВЛАСТИВОСТІ
Опис. Дрібний порошок білого кольору.
Розчинність. Практично не розчинний у воді Р, у якій
виявляє лужну реакцію за фенолфталеїном.
(Розчиняється в розведених кислотах, у більшості ви-
падків зі слабким виділенням бульбашок газу).
Насипний об’єм. 15 г субстанції займає об’єм близько
ЗО мл.
ІДЕНТИФІКАЦІЯ
Близько 15 мг субстанції розчиняють у 2 мл кислоти
азотної розведеної Р і нейтралізують розчином натрію
гідроксиду розведеним Р. Одержаний розчин дає ре-
акцію на магній (2.3.1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 5.0 г субстанції розчиняють у суміші 70 мл
кислоти оцтової Р і ЗО мл води дистильованої Р,
кип’ятять протягом 2 хв, охолоджують і доводять
об’єм розчину кислотою оцтовою розведеною Р до
100 мл. Одержаний розчин, якшо необхідно, фільтру-
ють крізь попередньо прожарений і зважений фарфо-
ровий або кварцовий фільтр-тигель необхідної порис-
тості для одержання прозорого розчину.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В3.
Розчинні речовини. До 2.00 г субстанції додають 100 мл
води Р, перемішують і кип’ятять протягом 5 хв. Гарячу
суміш фільтрують крізь скляний фільтр (40), охолод-
жують і доводять об’єм розчину водою Р до 100 мл.
50 мл одержаного розчину упарюють насухо і сушать
при температурі від 100 °С до 105 °С. Маса сухого за-
лишку не має перевищувати 20 мг (2.0 %).
Речовини, нерозчинні в кислоті оцтовій. Маса сухого за-
лишку, одержаного при приготуванні розчину 8, про-
митого, висушеного і прожареного при температурі
600 °С, не має перевищувати 5 мг (0.1 %).
Хлориди (2.4.4). Не більше 0.1 %. 1 мл розчину 8 дово-
дять водою Рдо об’єму 15 мл. Одержаний розчин має
витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 1.0 %. 0.3 мл розчину 8
доводять водою дистильованою Р до об’єму 15 мл.
Одержаний розчин має витримувати випробування на
сульфати.
Арсен (2.4.2, метод А). Не більше 0.0004 % (4 ррт).
5 мл розчину 8 мають витримувати випробування на
арсен.
Кальцій (2.4.3). Не більше 1.5 %. 1.3 мл розчину 8 до-
водять водою дистильованою Рдо об’єму 150 мл. 15 мл
одержаного розчину мають витримувати випробуван-
ня на кальцій.
Важкі метали (2.4.8, метод А). Не більше 0.003 %
(ЗО ррт). До 20 мл розчину 8 додають 15 мл кислоти
хлористоводневої РІ, 25 мл метилізобутилкетону Р і
збовтують протягом 2 хв. Після розділення фаз водний
шар упарюють насухо, залишок розчиняють у 1 мл
кислоти оцтової Р і доводять водою Рдо об’єму ЗО мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.07 %. 0.15 г субстанції роз-
чиняють у 5 мл кислоти хлористоводневої розведеної Р
і доводять об’єм розчину водою Рдо 10 мл. 1 мл розчи-
ну доводять водою Рдо об’єму 10 мл. Одержаний роз-
чин має витримувати випробування на залізо.
Втрата в масі при прожарюванні. Не більше 8.0 %. Ви-
значення проводять з 1.00 г субстанції при температурі
900 °С.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.700 г субстанції розчиняють у 20 мл кислоти хлорис-
товодневої розведеної Р і доводять об’єм розчину
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
409
Магнію оксид легкий
водою Р до 100.0 мл. Визначення магнію в 10.0 мл
одержаного розчину проводять методом комплексо-
метричного титрування (2.5.11).
1 мл й 7 Мрозчину натрію едетату відповідає 4.030 мг
м^о.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
МАГНІЮ ОКСИД ЛЕГКИЙ
Ма^пеБІі охудит ієує
МАСМЕ8ІЕМ ОХЮЕ, ЬІСНТ
М&О М.м. 40.30
Магнію оксид легкий містить не менше 98.0 % і не
більше 100.5 % М£О, у перерахунку на прожарену ре-
човину.
ВЛАСТИВОСТІ
Опис. Дрібний аморфний порошок білого кольору.
Розчинність. Практично не розчинний у воді Р, у якій
виявляє лужну реакцію за фенолфталеїном.
(Розчиняється в розведених кислотах, у більшості ви-
падків зі слабким виділенням бульбашок газу).
Насипний об’єм. 15 г субстанції займає об’єм близько
150 мл.
ІДЕНТИФІКАЦІЯ
Близько 15 мг субстанції розчиняють у 2 мл кислоти
азотної розведеної Р і нейтралізують розчином натрію
гідроксиду розведеним Р. Одержаний розчин дає ре-
акцію на магній (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 5.0 г субстанції розчиняють у суміші 70 мл
кислоти оцтової Р і ЗО мл води дистильованої Р, ки-
п’ятять протягом 2 хв, охолоджують і доводять об’єм
розчину кислотою оцтовою розведеною Р до 100 мл.
Одержаний розчин, якщо необхідно, фільтрують крізь
попередньо прожарений і зважений фарфоровий або
кварцовий фільтр-тигель необхідної пористості для
одержання прозорого розчину.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В2.
Розчинні речовини. До 2.00 г субстанції додають 100 мл
води Р, перемішують і кип’ятять протягом 5 хв. Гарячу
суміш фільтрують крізь скляний фільтр (40), охолод-
жують і доводять об’єм розчину водою Р до 100 мл.
50 мл одержаного розчину упарюють насухо і сушать
при температурі від 100 °С до 105 °С. Маса сухого за-
лишку не має перевищувати 20 мг (2.0 %).
Речовини, нерозчинні в кислоті оцтовій. Маса сухого за-
лишку, одержаного при приготуванні розчину 8, про-
митого, висушеного і прожареного при температурі
600 °С, не має перевищувати 5 мг (0.1 %).
Хлориди (2.4.4). Не більше 0.15 %. 0.7 мл розчину 8до-
водять водою Р по об’єму 15 мл. Одержаний розчин
має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 1.0 %. 0.3 мл розчину 8
доводять водою дистильованою Р до об’єму 15 мл.
Одержаний розчин має витримувати випробування на
сульфати.
Арсен (2.4.2, метод А). Не більше 0.0004 % (4 ррт).
5 мл розчину 8 мають витримувати випробування на
арсен.
Кальцій (2.4.3). Не більше 1.5 %. 1.3 мл розчину 8 до-
водять водою дистильованою Рао об’єму 150 мл. 15 мл
одержаного розчину мають витримувати випробуван-
ня на кальцій.
Важкі метали (2.4.8, метод А). Не більше 0.003 %
(ЗО ррт). До 20 мл розчину 8 додають 15 мл кислоти
хлористоводневої РІ, 25 мл метилізобутилкетону Р і
збовтують протягом 2 хв. Після розділення фаз водний
шар упарюють насухо, залишок розчиняють у 1.5 мл
кислоти оцтової Р і доводять водою Рдо об’єму ЗО мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (І ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.1 %. 50 мг субстанції розчи-
няють у 5 мл кислоти хлористоводневої розведеної Р і
доводять об’єм розчину водою Рдо 10 мл. 2 мл розчи-
ну доводять водою Р до об’єму 10 мл. Одержаний роз-
чин має витримувати випробування на залізо.
Втрата в масі при прожарюванні. Не більше 8.0 %>.
Визначення проводять з 1.00 г субстанції при темпера-
турі 900 °С.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.700 г субстанції розчиняють у 20 мл кислоти хлорис-
товодневої розведеної Р і доводять об’єм розчину
водою Р до 100.0 мл. Визначення магнію в 10.0 мл
одержаного розчину проводять методом комплексо-
метричного титрування (2.5.11).
1 мл О.ІМ розчин натрію едетату відповідає 4.030 мг
М§О.
410
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Метилпарагідроксибензоат
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
забарвлюється від жовтого до оранжево-коричнюва-
того кольору, в той час як розчин (а) забарвлюється від
оранжевого до червоного кольору, чітко більш інтен-
сивно за розчин (Ь).
ВИПРОБУВАННЯ НАЧИСТОТУ
МЕТИЛПАРАГІДРОКСИБЕНЗОАТ
МеШуІІБ рагаИудгохуЬепхоаБ
МІ ТИМ. РАКАН¥ВКОХ¥ВЕХХОАТЕ
Метилпарагідроксибензоат містить не менше 99.0 % і
не більше 100.5 % метил-4-гідроксибензоату.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Дуже мало розчинний у воді Р, легко роз-
чинний у 96 % спирті Р і метанолі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Температура плавлення (2.2.14). Від 125 °С до 128 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ метилпа-
рагідроксибензоату.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Супровідні домішки",
має виявлятися основна пляма на рівні основної пля-
ми на хроматограмі розчину порівняння (Ь),
відповідна їй за розміром.
О. Близько 10 мг субстанції поміщають у пробірку, до-
дають 1 мл розчину натрію карбонату Р, кип’ятять
протягом ЗО с і охолоджують (розчин (а)). Близько
10 мг субстанції поміщають в іншу пробірку такого
самого розміру, додають 1 мл розчину натрію карбона-
ту Р і перемішують до часткового розчинення суб-
станції (розчин (Ь)). До розчину (а) і розчину (Ь) одно-
часно додають по 5 мл розчину амінопіразолону Р, 1 мл
розчину калію фериціаніду Р \ перемішують; розчин (Ь)
Розчин 8. 1.0 г субстанції розчиняють у 96 % спирті Р
і доводять об’єм розчину тим самим розчинником
до 10 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
Кислотність. До 2 мл розчину 8 додають 3 мл 96 %
спирту Р, 5 мл води, вільної від вуглецю діоксиду, Р і
0.1 мл розчину бромкрезолового зеленого Р; блакитне за-
барвлення розчину має з’являтися при додаванні не
більше 0.1 мл 0.1 Мрозчину натрію гідроксиду.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар підхожий силікагель октадецил-
силільний із флуоресцентним індикатором з опти-
мальною інтенсивністю поглинання за довжини хвилі
254 нм.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють в ацетоні Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять ацетоном Рдо об’єму 10 мл.
Розчин порівняння (а). 0.5 мл випробовуваного розчи-
ну (а) доводять ацетоном Рао об’єму 100 мл.
Розчин порівняння (Ь). 10 мг ФСЗ метилпарагідрокси-
бензоату розчиняють у ацетоні Р і доводять об’єм роз-
чину тим самим розчинником до 10 мл.
Розчин порівняння (с). 10 мг ФСЗ етилпарагідроксибен-
зоату розчиняють в 1 мл випробовуваного розчину (а)
і доводять об’єм розчину ацетоном Рао 10 мл.
На лінію старту хроматографічної пластинки наносять
2 мкл (20 мкг) випробовуваного розчину (а), 2 мкл
(2 мкг) випробовуваного розчину (Ь), 2 мкл (0.1 мкг)
розчину порівняння (а), 2 мкл (2 мкг) розчину
порівняння (Ь) і 2 мкл (2 мкг етилпарагідроксибензоа-
ту і 2 мкг субстанції) розчину порівняння (с). Плас-
тинку поміщають у камеру із сумішшю розчинників
кислота оцтова льодяна Р - вода Р - метанол Р
(1:30:70). Коли фронт розчинників пройде 15 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й переглядають в УФ-світлі за довжини хвилі
254 нм.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (а) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
411
Метіонін
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
2.000 г субстанції поміщають у термостійку конічну
колбу із притертою скляною пробкою, додають
40 мл / М розчину натрію гідроксиду і обережно
нагрівають зі зворотним холодильником протягом
1 год. Розчин охолоджують, холодильник обполіску-
ють водою Р, приєднуючи промивні води до розчину.
Надлишок натрію гідроксиду титрують 0.5 Мрозчином
кислоти сірчаної потенціометрично (2.2.20), продов-
жуючи титрування до другого стрибка потенціалів на
кривій титрування.
Паралельно проводять контрольний дослід.
1 мл / М розчину натрію гідроксиду відповідає 152.1 мг
СвНвОз.
МЕТІОНІН
МеіИіопіпит
МЕТНІОМИЕ
Н МН2
С5НнМ)28
М.м. 149.2
Метіонін містить не менше 99.0 % і не більше 101.0 %
(5)-2 аміно-4-(метилтіо)бутанової кислоти, у перера-
хунку на суху речовину.
ДОМІШКИ
А. К = Н : 4-гідроксибензойна кислота,
В. К = СН2-СН3: етил-4-гідроксибензоат,
С. К = СН2-СН2-СН3: пропіл-4-гідроксибензоат,
В. К = СН2-СН2-СН2-СН3: бутил-4-гідроксибензоат.
_________________________________________________N
НІПАГІН
Міра^іпит
МЕТНУЕРАКАВЕП
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Розчинний у воді Р. дуже мало розчинний
у 96 % спирті Р, практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ метіоніну.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
В. 0.1 г субстанції і 0.1 г гліцину Р розчиняють у 4.5 мл
розчину натрію гідроксиду розведеного Р, додають 1 мл
розчину 25 г/л натрію нітропрусиду Р. Одержаний
розчин нагрівають при температурі 40 °С протягом
10 хв, охолоджують і додають 2 мл суміші кислота фос-
форна Р - кислота хлористоводнева Р (1:9); поступово
з’являється темно-червоне забарвлення.
412
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Метіонін
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 100 мл.
Прозорість розчину (2.2.І). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 5.5 до 6.5. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від +22.5° до +24.0°,
у перерахунку на суху речовину. 1.00 г субстанції роз-
чиняють у кислоті хлористоводневій РІ і доводять
об’єм розчину тим самим розчинником до 50.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у кислоті хлористоводневій розведеній Р і доводять
об’єм розчину тим самим розчинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рдо об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗметіоніну розчиняють
у розчині 10 г/л кислоти хлористоводневої Р і доводять
об’єм розчину тим самим розчинником до 50 мл.
Розчин порівняння (Ь). 5 мл випробуваного розчину (Ь)
доводять водою Рдо об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗметіоніну і 10 мг ФСЗ
серину розчиняють у розчині 10 г/л кислоти хлористо-
водневої Р і доводять об’єм розчину тим самим роз-
чинником до 25 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь). 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь), 5 мкл (2 мкг метіоніну і 2 мкг серину)
розчину порівняння (с). Пластинку поміщають у ка-
меру із сумішшю розчинників кислота оцтова
льодяна Р - вода Р - бутанол Р (20:20:60). Коли фронт
розчинників пройде 15 см від лінії старту, пластинку
виймають із камери, сушать на повітрі й обприскують
розчином нінгідрину Р. Пластинку нагрівають при тем-
пературі від 100 °С до 105 °С протягом 5 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якшо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). До 10 мл
розчину 8 додають 25 мл води Р, 5 мл кислоти азотної
розведеної Р і 10 мл розчину срібла нітрату Р2 і витри-
мують у захищеному від світла місці протягом 5 хв.
Опалесценція одержаного розчину не має перевищу-
вати опалесценцію еталона, приготованого ана-
логічно до випробовуваного розчину з використанням
10 мл еталонного розчину хлориду (5 ррт СІ) Р.
Пробірки переглядають горизонтально на чорному
фоні.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0 5г
субстанції розчиняють у 3 мл кислоти хлористоводне-
вої розведеної Р і доводять об’єм розчину водою Р до
15 мл. Одержаний розчин маг витримувати випробу-
вання на сульфати.
Амонію солі (2.4.1, метод В). Не більше 0.02%
(200 ррт). 0.10 г субстанції мають витримувати випро-
бування на амонію солі. Еталон готують із використан-
ням 0.2 мл еталонного розчину амонію (100ррт ПН4) Р.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 2 0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0 125 г субстанції розчиняють у 5 мл кислоти мураши-
ної безводної Р, додають ЗО мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної по-
тенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної відповідає 14.92 г
С5Н,^О28.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видалення
пірогенів, вона має витримувати випробування "Піро-
гени" (2.6.8) або "Бактеріальні ендотоксини "(2.6.14).
пспм/лпил ФЛРМЛітПРЯ УКРАЇНИ 7001
413
Налоксону гідрохлорид дигідрат
НАЛОКСОНУ ГІДРОХЛОРИД
ДИГІДРАТ
Наїохопі ИудгосЬІогідит діИусІгісит
ХАІХЇХОХЕ НТОПОСНТОППУЕ ОІНУИКАТЕ
, неї , 2 Н2О
М.м. 399.9
Налоксону гідрохлорид дигідрат містить не менше
98.0 % і не більше 102.0 % 4,5а-епокси-3,14-дигідро-
кси-17-(проп-2-еніл)морфінан-6-ону гідрохлориду, у
перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору. Гігроскопічний.
Розчинність. Легко розчинний у воді Р, розчинний у
96 % спирті Р, практично не розчинний у толуолі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: В, С.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ налоксону гідро-
хлориду дигідрату.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи ТШХ пластинки
із шаром силікагелю с Р.
Випробовуваний розчин. 8 мг субстанції розчиняють у
0 5 мл води Р і доводять об’єм розчину метанолом Рдо
1 мл.
Розчин порівняння. 8 мг ФСЗ налоксону гідрохлориду
дигідрату розчиняють у 0.5 мл води Р і доводять об’єм
розчину метанолом Рдо 1 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (40 мкг) випробовуваного розчину і 5 мкл
(40 мкг) розчину порівняння. Пластинку поміщають у
камеру із сумішшю розчинників, що складається із
5 об’ємів метанолу Р і 95 об'ємів верхнього шару су-
міші розчин аміаку розведений Р2 - бутанол Р (60:100).
Камеру поміщають у захищене від світла місце. Коли
фронт розчинників пройде 10 см від лінії старту, плас-
тинку виймають із камери, сушать у струмені повітря й
обприскують свіжоприготованим розчином 5 г/л калію
фериціаніду Ру розчині заліза) 111) хлориду РІ. Пластин-
ку переглядають при денному світлі.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, відповідна їй за
розміром і забарвленням.
С. Субстанція дає реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 0.50 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 25.0 мл.
Прозорість розчину (2.2. і). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
Кислотність або лужність. До 10.0 мл розчину 8 дода-
ють 0.05 мл розчину метилового червоного Р; забарв-
лення розчину має змінитися при додаванні не більше
0.2 мл 0.02 Мрозчину натрію гідроксиду або 0.02 М роз-
чину кислоти хлористоводневої.
Питоме оптичне обертання (2.2.7). Від -170° до -181°, у
перерахунку на безводну речовину. Визначення про-
водять, використовуючи розчин 8.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 0.125 г субстанції розчиняють
у 0.1 М розчині кислоти хлористоводневої, доводять
об’єм розчину тим самим розчинником до 25.0 мл.
Розчин порівняння (а). 10.0 мг ФСЗ налоксону гідрохло-
риду дигідрату і 10.0 мг ФСЗ домішки А налоксону
гідрохлориду дигідрату розчиняють у 0.1 М розчині
кислоти хлористоводневої, доводять об’єм розчину
тим самим розчинником до 10.0 мл. 1.0 мл одержано
го розчину доводять 0.1 М розчином кислоти хлорис-
товодневої до об’єму 100.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
415
Налоксону гідрохлорид дигідрат
Розчин порівняння (Ь). 1.0 мл випробовуваного розчину
доводять 0.1 М розчином кислоти хлористоводневої до
об’єму 20.0 мл. 1.0 мл одержаного розчину доводять
0.1 М розчином кислоти хлористоводневої до об’єму
10.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром
0.125 м х 4.0 мм, заповнена силікагелем октил-
силільним ендкепованим для хроматографії Р з розмі-
ром часток 5 мкм;
— рухома фаза А: ацетонітрил Р - тетрагідрофу-
ран Р - розчин кислоти октансульфонової
(20:40:940). Розчин кислоти октансульфонової го-
тують таким чином: 1.17 г натрію октансульфона-
ту Ррозчиняють у 1000 мл води Р, рН одержаного
розчину доводять розчином 50 % (об/об) кислоти
фосфорної Рао 2.0 і фільтрують;
— рухома фаза В: ацетонітрил Р - тетрагідрофуран Р
- розчин кислоти октансульфонової (170:40:790);
— швидкість рухомої фази 1.5 мл/хв;
— використовують таку програму градієнта:
Час (хв) Рухома фаза А (% об/об) Рухома фаза В (% об/об) Примітки
0-40 100—0 0—100 ЛІНІЙНИЙ градієнт
40-50 0 100 ізократичний режим
— детектування за довжини хвилі 230 нм;
— температура колонки 40 °С;
— об’єм проби, шо уводиться, 20 мкл.
Хроматографують розчин порівняння (а). Хромато-
графічна система вважається придатною, якщо вико-
нуються такі умови:
— відношення сигнал/шум, розраховане для піка
налоксону, має бути не менше 10;
— коефіцієнт розділення піків домішки А і налок-
сону має бути не менше 4. Якщо необхідно, змі-
нюють хроматографічні умови.
При хроматографуванні за зазначених умов час утри-
мування піка налоксону має бути близько 11 хв;
відносний час утримування домішки А до налоксону
— близько 0.8.
Поперемінно хроматографують випробовуваний роз-
чин і розчин порівняння (Ь). На хроматограмі випро-
бовуваного розчину площа будь-якого піка, крім ос-
новного, не має перевищувати площу основного піка
на хроматограмі розчину порівняння (Ь) (0.5 %): сума
площ усіх піків, крім основного, не має перевищува-
ти 2 площі основного піка на хроматограмі розчину
порівняння (Ь) (1 %). Не враховують піки, площа яких
становить менше 0.1 площі основного піка на хрома-
тограмі розчину порівняння (Ь).
Вода (2.5.12). Від 7.5 % до 11.0 %. Визначення прово-
дять з 0.200 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.2 %. Визначення
проводять з 0.50 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.300 г субстанції розчиняють у 50 мл 96 % спирту Р,
додають 5.0 мл 0.01 Мрозчину кислоти хлористоводне-
вої і титрують 0.1 М розчином натрію гідроксиду ета-
нольним потенціометрично (2.2.20). У розрахунок бе-
руть об’єм титранту між двома стрибками потенціалів
на кривій титрування.
1 мл 0.1 М розчину натрію гідроксиду етанольного
відповідає 36.38 мг С19Н22СЖО4.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. К1 = К2 = КЗ = Н : 4,5а-епокси-3,14-дигідрокси-
морфінан-6-он (нороксиморфон),
В. К1 = КЗ = СН2-СН=СН2, К2 = Н : 4,5а-епокси-14-
гідрокси-17-(проп-2-еніл)-3-(проп-2-енілок-
си)морфінан-6-он (З-О-алілналоксон),
С. К1 = Н, К2 = ОН, КЗ = СН2-СН=СН2: 4,5а-епок-
си-3,10а, 14-тригідрокси-17-(проп-2-еніл)морфінан-
6-он (Юа-гідроксиналоксон),
В. 7,8-дидегідро-4,5а-епокси-3,14-дигідрокси-17-
(проп-2-еніл)морфінан-6-он (7,8-дидегідроналоксон),
Е. 4,5а:4',5'а-діепокси-3,3',14,14'-тетрагідрокси-
17,17'-біс(проп-2-еніл)-2,2’-біморфінаніл-6,6’-діон
(2,2'-бісналоксон).
416
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Напроксен
ТУ
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
НАПРОКСЕН
Б. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках із калію бромідом Р, має
відповідати спектру ФСЗ напроксену.
Е. Близько 2 мг субстанції розчиняють у 2 мл кисло-
та сірчаної Р; з’являється жовте забарвлення. До
одержаного розчину додають 50 мг хлоралгідрату Р і
збовтують до розчинення: розчин забарвлюється в
оранжевий колір, що переходить в оранжево-черво-
ний.
Наргохешіт
8ЛРК0ХЕХ
С14Н14О3
М.м. 230.3
Напроксен містить не менше 98.5 % і не більше
100.5 % (5)-2-(6-метоксинафт-2-іл)пропіонової кис-
лоти, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Практично не розчинний у воді Р, роз-
чинний у 96 % спирті Р і у метанолі Р, помірно роз-
чинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В. Б.
Друга ідентифікація: А, В, С, Е.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Температура плавлення (2.2.14). Від 154 °С до
158 °С.
С. 40.0 мг субстанції розчиняють у метанолі Р і дово-
дять об’єм розчину тим самим розчинником до
100.0 мл. 10.0 мл одержаного розчину доводять мета-
нолом Р до об’єму 100.0 мл. Ультрафіолетовий спектр
поглинання (2.2.25) одержаного розчину в області від
230 нм до 350 нм повинен мати чотири максимуми за
довжин хвиль 262 нм, 271 нм, 316 нм і 331 нм. Питомі
показники поглинання у максимумах мають бути від
216 до 238, від 219 до 241, від 61 до 69 і від 79 до 87,
відповідно.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Прозорість розчину (2.2.!). 1.25 г субстанції розчиня-
ють у метанолі Р і доводять об’єм розчину тим самим
розчинником до 25 мл. Одержаний розчин має бути
прозорим.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину, приготованого для випробування "Прозорість
розчину", має бути не інтенсивнішим за еталон В¥7.
Питоме оптичне обертання (2.2.7). Від +63° до +68.5°, у
перерахунку на суху речовину. 0.500 г субстанції роз-
чиняють у хлороформі Р і доводять об’єм розчину тим
самим розчинником до 25.0 мл.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель Р-
Випробовуваний розчин. 0.25 г субстанції розчиняють у
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 5 мл.
Розчин порівняння. 0.5 мл випробовуваного розчину
доводять метанолом Рло об’єму 100 мл.
На лінію старту хроматографічної пластинки нано-
сять 10 мкл (500 мкг) випробовуваного розчину і
10 мкл (2.5 мкг) розчину порівняння. Пластинку
поміщають у камеру із сумішшю розчинників кисло-
та оцтова льодяна Р - тетрагідрофуран Р - толуол Р
(3:9:90). Коли фронт розчинників пройде 15 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й переглядають в УФ-світлі за довжини хвилі
254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %).
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С протягом 3 год.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
л.члллі/лпга \1/ПАЇ ПІЛ лот
417
Натрію дигідрофосфат дигідрат
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.200 г субстанції розчиняють у суміші 25 мл води Р і
75 мл метанолу Р і титрують 0.1 М розчином натрію
гідроксиду, використовуючи як індикатор 1 мл розчину
фенолфталеїну Р.
1 мл 0.1 М розчину натрію гідроксиду відповідає
23.03 мг СІ4НІ4О3.
ЗБЕРІГАННЯ
У іцільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________ТУ
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
НАТРІЮ ДИГІДРОФОСФАТ
ДИГІДРАТ
Каігіі діИудго§епорИо8р1іа8 сііЬусІгіси8
КОІНІМ ШНЇІЖОСЕХ РНО8РНАТЕ тНПЖАТЕ
1УаН2РО4,2Н2О М.м. 156.0
Натрію дигідрофосфат дигідрат містить не менше
98.0 % і не більше 100.5 % КаН2РО4, у перерахунку на
суху речовину.
ВЛАСТИВОСТІ
Опис. Порошок білого кольору або безбарвні кристали.
Розчинність. Дуже легко розчинний у воді Р, дуже ма-
ло розчинний у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
А. Розчин 8, приготований, як зазначено в розділі
"Випробування на чистоту", має слабкокислу реакцію
(2.2.4).
В. Розчин 8 дає реакцію на фосфати (2.3.1).
С. Розчин 8, попередньо нейтралізований розчином
100 г/л калію гідроксиду Р, дає реакцію (а) на натрій
(2.3.1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 10.0 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р, одержаній із води дистильова-
ної Р, і доводять об’єм розчину тим самим розчинни-
ком до 100 мл.
Прозорість розчину (2.2.!). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 4.2 до 4.5. До 5 мл розчину 8 додають
5 мл води, вільної від вуглецю діоксиду, Р.
Відновлюючі речовини. До 5 мл розчину 8 додають
0.25 мл 0.02 Мрозчину калію перманганату і 5 мл кис-
лоти сірчаної розведеної Р і нагрівають у водяній бані
протягом 5 хв; розчин має зберегти слабко-червоне за-
барвлення.
Хлориди (2.4.4) Не більше 0.02 % (200 ррт). 2.5 мл роз-
чину 8 доводять водою Р до об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). До 5 мл
розчину 8 додають 0.5 мл кислоти хлористоводневої Р
і доводять водою дистильованою Р до об’єму 15 мл.
Одержаний розчин має витримувати випробування на
сульфати.
Арсен (2.4.2, метод А). Не більше 0.0002 % (2 ррт).
0.5 г субстанції мають витримувати випробування на
арсен.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (І ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 10 мл роз-
чину 8 мають витримувати випробування на залізо.
Втрата в масі при висушуванні (2.2.32). Від 21.5% до
24.0 %. 0.50 г субстанції сушать при температурі 130 °С.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
2.500 г субстанції розчиняють у 40 мл води Рі титрують
/ М розчином натрію гідроксиду, вільним від карбо-
натів, потенціометрично (2.2.20).
1 мл / М розчину натрію гідроксиду відповідає 0.120 г
КаН2РО4.
ЗБЕРІГАННЯ
У шільно закупореному контейнері.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4)
418
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Натрію диклофенак
НАТРІЮ ДИКЛОФЕНАК
ОісІоГепасит паїгісит
80БІУМ П1СІ.ОЇЕКЛС
С14Н |оСІ2І\І\а02
М.м. 318.1
Натрію диклофенак містить не менше 99.0 % і не
більше 101.0% натрію 2-[(2,6-дихлорфеніл)аміно|-
феніл)ацетату, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або білого з жов-
тавим відтінком кольору, мало гігроскопічний.
Розчинність. Помірно розчинний у воді Р, легко роз-
чинний у метанолі Р, розчинний у 96 % спирті Р, ма-
ло розчинний в ацетоні Р, практично не розчинний в
ефірі Р.
(Плавиться при температурі близько 280 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, Б.
Друга ідентифікація: В, С, Б-
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ натрію диклофенаку.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель 6Т254 Р.
Випробовуваний розчин. 25 мг субстанції розчиняють в
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 5 мл.
Розчин порівняння (а). 25 мг ФСЗ натрію диклофенаку
розчиняють у метанолі Рі доводять об’єм розчину тим
самим розчинником до 5 мл.
Розчин порівняння (Ь). 10 мг ФСЗ індометацину розчи-
няють у розчині порівняння (а) і доводять об’єм роз-
чину тим самим розчинником до 2 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (25 мкг) випробовуваного розчину, 5 мкл
(25 мкг) розчину порівняння (а) і 5 мкл (25 мкг натрію
диклофенаку і 25 мкг індометапину) розчину
порівняння (Ь). Пластинку помішають у камеру із
сумішшю розчинників розчин аміаку концентрова-
ний Р - метанол Р - етилацетат Р (10:10:80). Коли
фронт розчинників пройде 10 см від лінії старту, плас-
тинку виймають із камери, сушать на повітрі й пере-
глядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
С. Близько 10 мг субстанції розчиняють у 10 мл 96 %
спирту Р. До 1 мл одержаного розчину додають 0.2 мл
свіжоприготованої суміші рівних об’ємів розчину 6 г/л
калію фериціаніду Р і розчину 9 г/л заліза(ПІ) хлори-
ду Р. Розчин витримують протягом 5 хв у захищеному
віл світла місці, додають 3 мл розчину 10 г/л кислоти
хлористоводневої Р і витримують протягом 15 хв у за-
хищеному від світла місці; розчин поступово забарв-
люється у синій колір і утворюється синій осад.
В. 60 мг субстанції розчиняють у 0.5 мл метанолу Р і
додають 0.5 мл води Р. Розчин дає реакцію (Ь) на
натрій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 1.25 г субстанції розчиня-
ють у метанолі Р і доводять об’єм розчину тим самим
розчинником до 25.0 мл. Одержаний розчин має бути
прозорим.
Кольоровість розчину. Оптична густина (2.2.25) розчи-
ну, приготованого для випробування "Прозорість роз-
чину", виміряна за довжини хвилі 440 нм, не має пере-
вищувати 0.05.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 50.0 мг субстанції розчиняють
у метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 50.0 мл.
Розчин порівняння (а). 2.0 мл випробовуваного розчину
доводять метанолом Рдо об’єму 100.0 мл. 1.0 мл одер-
жаного розчину доводять метанолом Р до об’єму
10.0 мл.
Розчин порівняння (Ь). 1.0 мг ФСЗ домішки А диклофе-
наку розчиняють у метанолі Р, додають 1.0 мл випро-
бовуваного розчину і доводять об’єм розчину метано-
лом Рао 200.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октилсилільним ендкепова-
ним для хроматографії Р з розміром часток
5 мкм;
— рухома фаза: суміш рівних об’ємів розчину 1 г/л кис-
пг-пхіл* пп А гьл олл л гонга Х/КРАЇНИ 7001
419
Натрію метабісульфіт
лоти фосфорної Р \ розчину 1.6 г/л натрію дигідро-
фосфату Р, рН якого доведено до 2.5, - метанол Р
(34:66);
— швидкість рухомої фази 1 мл/хв;
— детектування задовжини хвилі 254 нм.
Хроматографують 20 мкл розчину порівняння (Ь).
При хроматографуванні за зазначених умов часи утри-
мування піків мають бути: диклофенаку — близько
25 хв і домішки А диклофенаку — близько 12 хв. Чут-
ливість системи регулюють таким чином, щоб висота
піків становила не менше 50 % шкали реєструючого
пристрою. Час хроматографування має бути у 1.5 рази
більше часу утримування диклофенаку.
Хроматографічна система вважається придатною, як-
шо коефіцієнт розділення піка диклофенаку і піка
домішки А диклофенаку на хроматограмі розчину
порівняння (Ь) становить не менше 6.5.
Поперемінно хроматографують 20 мкл випробовувано-
го розчину і 20 мкл розчину порівняння (а).
На хроматограмі випробовуваного розчину площа
будь-якого піка, крім основного, не має перевищува-
ти площу основного піка на хроматограмі розчину
порівняння (а) (0.2 %); сума площ усіх піків, крім ос-
новного, не має перевищувати 2 5 площі основного
піка на хроматограмі розчину порівняння (а) (0.5 %).
Не враховують піки, площа яких становить менше
0.25 площі основного піка на хроматограмі розчину
порівняння (а).
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С протягом З год.
В. 2-[(2,6-дихлорфеніл)аміно]бензальдегід,
С. 12-[(2,6-дихлорфеніл)аміно]феніл]метанол,
В. 2-[2-[(2-бром-6-хлорфеніл)аміно]феніл]оцтова
кислота.
Е. індолін-2-он.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у 30 мл кислоти оцтової
льодяної Р і титрують 0.1 М розчином кислоти хлорної
потенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної Р відповідає
31.81 міСІ4Н10С12ХХаО2.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. 1-(2,6-дихлорфсніл)індолін-2-он,
НАТРІЮ МЕТАБІСУЛЬФІТ
Наїгіі теїаЬІБНІГіБ
ЬОШІМ МЕТАВІ8ЕЕРНІТЕ
N328305 М.м. 190.1
Натрію метабісульфіт (натрію дисульфіт) містить не
менше 95.0 % і не більше 100.5 % N338205.
420
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Натрію тетраборат
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Легко розчинний у воді Р, мало розчин-
ний у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
А. Субстанція має відповідати вимогам випробування
"рН", зазначеним у розділі "Випробування на чистоту".
В. До 0.4 мл розчину калію йодиду йодованого Р дода-
ють 8 мл води дистильованої Р і 1 мл розчину 8, розве-
деного в 10 разів водою дистильованою Р; одержаний
розчин безбарвний і дає реакцію (а) на сульфати
(2.3.1).
С. Розчин 8 дає реакцію (а) на натрій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 5.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду,Р, приготованій із води дистильова-
ної Р, і доводять тим самим розчинником до 100 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод 11). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 3.5 до 5.0. Вимірюють рН розчину 8.
Тіосульфати. До 5 мл розчину 8 додають 5 мл кислоти
хлористоводневої розведеної Р; розчин має залишатися
прозорим (2.2.1) протягом не менше 15 хв.
Арсен (2.4.2, метод А). Не більше 0.0005 % (5 ррт). У
чашці змішують 0.20 г субстанції з 2 мл води Р і крап-
лями додають 1.5 мл кислоти азотної Р. Суміш упарю-
ють насухо на водяній бані, потім нагрівають на по-
лум’ї до припинення виділення парів. Одержаний за-
лишок розчиняють у 25 мл води Р. Розчин має витри-
мувати випробування на арсен.
Важкі метали (2.4.8, метод А). Не більше 0.002 %
(20 ррт). 40 мл розчину 8 поміщають у кварцовий ти-
гель, додають 10 мл кислоти хлористоводневої Р і упа-
рюють насухо. Одержаний залишок розчиняють у
19 мл води Р і додають 1 мл розчину 40 г/л натрію
фториду Р. 12 мл розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (2 ррт РЬ) Р.
Залізо (2.4.9). Не більше 0.002 % (20 ррт). 10 мл роз-
чину 8 мають витримувати випробування на залізо.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.200 г субстанції розчиняють у 50.0 мл 0.05 М розчину
йоду, додають 5 мл кислоти хлористоводневої розведе-
ної Р і титрують 0.1 М розчином натрію тіосульфату,
використовуючи наприкінці титрування як індикатор
1 мл розчину крохмалю Р.
1 мл 0.05 Мрозчину йоду відповідає 4.753 мг N328205.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
НАТРІЮ ТЕТРАБОРАТ
Богах
БОКАХ
№2В4О7,10Н2О М.м. 381.4
Натрію тетраборат містить не менше 99.0 % і не
більше 103.0 % динатрію тетраборату декагідрату.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору, або без-
барвні кристали, або кристалічна маса. Вивітрюється
на повітрі.
Розчинність. Розчинний у воді Р, дуже легко розчин-
ний у киплячій воді Р. легко розчинний у гліцерині Р.
ІДЕНТИФІКАЦІЯ
А. До 1 мл розчину 8, приготованого, як зазначено в
розділі "Випробування на чистоту", додають 0.1 мл
кислоти сірчаної Р, 5 мл метанолу Р і підпалюють; по-
лум’я має зелену облямівку.
В. До 5 мл розчину 8 додають 0.1 мл розчину фенол-
фталеїну Р; з’являється червоне забарвлення. При до-
даванні 5 мл гліцерину (85 %) Рзабарвлення зникає.
С. Розчин 8 дає реакцію на натрій (2.3.1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8.4.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 100 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
421
Натрію фторид
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 9.0 до 9.6. Вимірюють рН розчину 8.
Сульфати (2.4.13). Не більше 0.005 % (50 ррт). 15 мл
розчину 8 мають витримувати випробування на суль-
фати. Еталон готують із використанням 3 мл еталон-
ного розчину сульфату (10 ррт ЗО4) Р і 12 мл води дис-
тильованої Р.
Амонію солі (2.4.1). Не більше 0.001 % (10 ррт). 6 мл
розчину 8 доводять водою Р до об’єму 14 мл. Одержа-
ний розчин має витримувати випробування на амонію
солі. Еталон готують із використанням 2.5 мл еталон-
ного розчину амонію (1 ррт N11 4) Р і 7.5 мл води Р.
Арсен (2.4.2, метод А). Не більше 0.0005 % (5 ррт).
5 мл розчину 8 мають витримувати випробування на
арсен.
Кальцій (2.4.3). Не більше 0.01 % (100 ррт). 15 мл роз-
чину 8 мають витримувати випробування на кальцій.
Еталон готують із використанням 6 мл еталонного роз-
чину кальцію (10ррт Са) Р і 9 мл води дистильованої Р.
Важкі метали (2.4.8, метод А). Не більше 0.0025 %
(25 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (І ррт РЬ) Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
20 г маніту Р розчиняють у 100 мл води Р, якщо не-
обхідно, нагрівають, охолоджують, додають 0.5 мл роз-
чину фенолфталеїну Р і нейтралізують 0.1 М розчином
натрію гідроксиду до одержання рожевого забарвлен-
ня. До одержаного розчину додають 3.00 г субстанції,
нагрівають до повного розчинення, охолоджують і ти-
трують 1 М розчином натрію гідроксиду до повторної
появи рожевого забарвлення.
1 мл 1 М розчину натрію гідроксиду відповідає 0.1907 г
Ка2В4О7,10Н2О.
ЗБЕРІГАННЯ
У шільно закупореному контейнері.
____________________________________________N
Карбонати і гідрокарбонати. 1 г субстанції розчиняють
у 20 мл води Р. 5 мл одержаного розчину поміщають у
пробірку, додають 1 мл З М розчину кислоти хлористо-
водневої Р; не має спостерігатися виділення бульба-
шок газу.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
НАТРІЮ ФТОРИД
N31111 Ппогісіит
801)! ІМ ПЛОКНЗЕ
М.м. 41.99
Натрію фторид містить не менше 98.5 % і не більше
100.5 % №Р, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Порошок білого кольору або безбарвні кристали.
Розчинність. Розчинний у воді Р, практично не роз
чинний у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
А. До 2 мл розчину 8, приготованого, як зазначено в
розділі "Випробування на чистоту", додають 0.5 мл
розчину кальцію хлориду Р: утворюється білий геле-
подібний осад, що розчиняється при додаванні 5 мл
розчину заліза(III) хлориду РІ.
В. До близько 4 мг субстанції додають суміш 0.1 мл
розчину алізарину 5 Р і 0.1 мл розчину цирконілу нітра-
ту Р і перемішують; забарвлення розчину змінюється
від червоного до жовтого.
С. Розчин 8 дає реакцію (а) на натрій (2.3.1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють без нагрівання у
воді, вільній від вуглецю діоксиду, Р і доводять об’єм
розчину тим самим розчинником до 100 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
Кислотність або лужність. 2.5 г калію нітрату Ррозчи-
няють у 40 мл розчину 8 і доводять об’єм розчину во-
дою, вільною від вуглецю діоксиду, Р до 50 мл. Одержа-
ний розчин охолоджують до температури 0 °С і дода-
ють 0.2 мл розчину фенолфталеїну Р. Якщо розчин без-
барвний, при додаванні не більше 1.0 мл 0.1 М розчину
натрію гідроксиду має з’являтися червоне забарвлення
і зберігатися протягом не менше 15 с. Якщо розчин за-
барвлений у червоний колір, забарвлення розчину має
змінитися при додаванні не більше 0.25 мл 0.1 Мроз-
чину кислоти хлористоводневої.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 10 мл роз-
чину 8 доводять водою Р до об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
422
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Натрію цитрат
Фторосилікати. Нейтралізований розчин, одержаний у
випробуванні "Кислотність або лужність", нагрівають
до кипіння. Гарячий розчин титрують 0.1 М розчином
натрію гідроксиду; червоне забарвлення розчину з’яв-
ляється при додаванні не більше 0.75 мл 0.1 Мрозчину
натрію гідроксиду.
Сульфати (2.4.13). Не більше 0.02 % (200 ррт). 0.25 г
субстанції розчиняють у 10 мл насиченого розчину
кислоти борної Р у воді дистильованій Р, додають 5 мл
води дистильованої Р і 0.6 мл кислоти хлористоводне-
вої РІ. Одержаний розчин має витримувати випробу-
вання на сульфати. Еталон готують, змішуючи 0.6 мл
кислоти хлористоводневої РІ. 5 мл еталонного розчину
сульфату (10 ррт 8О4) Рі 10 мл насиченого розчину
кислоти борної Р у воді дистильованій Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі 130 °С
протягом З год.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
До 80.0 мг субстанції додають суміш 5 мл оцтового
ангідриду Р і 20 мл кислоти оцтової безводної Р,
нагрівають до розчинення. Після охолодження до
одержаної суміші додають 20 мл діоксану Р і титрують
0.1 М розчином кислоти хлорної ар появи зеленого за-
барвлення, використовуючи як індикатор 0.1 мл роз-
чину кристалічного фіолетового Р.
Паралельно проводять контрольний дослід.
1 мл 0.І Мрозчину кислоти хлорної відповідає 4.199 мг
1\аЕ.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
___________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
НАТРІЮ ЦИТРАТ
Наші сіїга$
5001ОМ СІТКАТЕ
С6Н51Уа3О7.2Н2О М.м. 294.1
Натрію цитрат містить не менше 99.0 % і не більше
1010% тринатрію 2-гідроксипропан-1,2,3-трикар-
боксилату, у перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок або гранульовані крис-
тали білого кольору, злегка розпливаються у вологому
повітрі.
Розчинність. Легко розчинний у воді Р, практично не
розчинний у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
А. До 1 мл розчину 8, приготованого, як зазначено в
розділі "Випробування на чистоту", додають 4 мл во-
ди Р. Одержаний розчин дає реакцію на цитрати
(2.3.1).
В. 1 мл розчину 8 дає реакцію (а) на натрій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 10.0 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р, приготованій із води дистильо-
ваної Р, і доводять об’єм розчину до 100 мл тим самим
розчинником.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
Кислотність або лужність. До 10 мл розчину 8 додають
0.1 мл розчину фенолфталеїну Р; забарвлення розчину
має змінитися при додаванні не більше 0.2 мл 0.1 М
розчину кислоти хлористоводневої або 0.1 М розчину
натрію гідроксиду.
Речовини, що легко обвуглюються. До 0.20 г здрібненої
субстанції додають 10 мл кислоти сірчаної Р, нагріва-
ють у водяній бані при температурі (90± 1) °С протя-
гом 60 хв і швидко охолоджують. Забарвлення одержа-
ного розчину має бути не інтенсивнішим за еталон ¥2
або С¥2 (2.2.2, метод II).
Хлориди (2.4.4). Не більше 0.005 % (50 ррт). 10 мл роз-
чину 8 доводять водою Р до об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Оксалати. Не більше 0.03 % (300 ррт). 0.50 г суб-
станції розчиняють у 4 мл води Р, додають 3 мл кисло-
ти хлористоводневої Р, І г цинку Р гранульованого і
нагрівають на водяній бані протягом 1 хв. Витримують
протягом 2 хв, рідину зливають у пробірку, що містить
0.25 мл розчину 10 г/л фенілгідразину хлористоводне-
вого Рі нагрівають до кипіння. Швидко охолоджують,
поміщають у мірний циліндр, додають рівний об’єм
кислоти хлористоводневої Р і 0.25 мл розчину калію фе-
риціаніду Р, збовтують і витримують протягом 30 хв.
Рожеве забарвлення розчину має бути не інтен-
сивнішим за еталон, приготований аналогічно до ви-
пробовуваного розчину із використанням 4 мл розчи-
ну 50 мг/л щавлевої кислоти Р.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
423
Нікотинамід
Сульфати (2.4.13). Не більше 0.015% (150 ррт). До
10 мл розчину 8 додають 2 мл кислоти хлористоводне-
вої РІ і доводять об’єм розчину водою дистильованою
Р до 15 мл. Одержаний розчин має витримувати ви-
пробування на сульфати.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Вода (2.5.12). Від 11.0 % до 13.0 %. Визначення прово-
дять з 0.300 г субстанції напівмікрометодом. Після до-
давання субстанції перед титруванням розчин пе-
ремішують протягом 15 хв.
Пірогени (2.6.8). Якщо субстанція призначена для ви-
робництва парентеральних лікарських засобів вели-
ких об’ємів, вона має витримувати випробування на
пірогени. Уводять на 1 кг маси кролика 10 мл свіжо-
приготованого розчину, що містить 10.0 мг/мл суб-
станції і 7.5 мг вільного від пірогенів кальцію хлориду Р
у воді для ін ’єкцій Р.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у 20 мл кислоти оцтової
безводної Р, нагріваючи до температури близько 50 °С,
залишають до охолодження і титрують 0.1 Мрозчином
кислоти хлорної до появи зеленого забарвлення, вико-
ристовуючи як індикатор 0.25 мл розчину нафтолбен-
зеїну Р.
1 мл 0.1 Мрозчину кислоти хлорної відповідає 8.602 мг
С6Н5Ка3О7.
ЗБЕРІГАННЯ
У повітронепроникному контейнері.
______________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Якщо субстанція призначена для виробництва парен-
теральних лікарських засобів, вона має витримувати
такі випробування:
Арсен (2.4.2, метод А). Не більше 0.0001 % (1 ррт).
10 мл розчину 8 мають витримувати випробування на
арсен.
Кальцій (2.4.3). Не більше 0.03 % (300 ррт). 3.3 мл роз-
чину 8 доводять водою дистильованою Р до об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на кальцій.
НІКОТИНАМІД
Кісоіїпатісіит
ПІСОТШАМЮЕ
С6н6м2о
М.м. 122.1
Нікотинамід містить не менше 99.0 % і не більше
І01.-0 % піридин-3-карбоксаміду, у перерахунку на су-
ху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Легко розчинний у воді Р і етанолі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Температура плавлення (2.2.14). Від 128 °С до 131 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ нікотинаміду.
С. 0.1 г субстанції кип’ятять з 1 мл розчину натрію
гідроксиду розведеного Р; виділяється аміак, шо вияв-
ляється за запахом.
В. 2 мл розчину 8, приготованого, як зазначено в
розділі “Випробування на чистоту”, доводять во-
дою Р до об’єму 100 мл. До 2 мл одержаного розчину
додають 2 мл розчину ціаноброміду Р і 3 мл розчину
25 г/л аніліну Р і струшують; з’являється жовте за-
барвлення.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥7.
рН (2.2.3). Від 6.0 до 7.5. Вимірюють рН розчину 8.
424
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Нітразепам
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин. 0.4 г субстанції розчиняють у
суміші рівних об’ємів 96 % спирт Р - вода Р і доводять
об’єм розчину тією самою сумішшю розчинників до
5.0 мл.
Розчин порівняння. 0.5 мл випробовуваного розчину
доводять сумішшю рівних об’ємів 96 % спирт Р - во-
да Рдо об’єму 200 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (400 мкг) випробовуваного розчину і 5 мкл
(1 мкг) розчину порівняння. Пластинку поміщають у
камеру із сумішшю розчинників вода Р - етанол Р -
хлороформ Р (4:45:48). Коли фронт розчинників прой-
де 10 см від лінії старту, пластинку виймають із каме-
ри, сушать на повітрі й переглядають в УФ-світлі за
довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.25 %).
Важкі метали (2.4.8, метод А). Не більше 0.003 %
(30 ррт). 12 мл розчину 8 доводять водою Рдо об’єму
18 мл. 12 мл одержаного розчину мають витримувати
випробування на важкі метали Еталон готують із ви-
користанням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5%. 1.00 г субстанції сушать у вакуумі протягом
18 год.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у 20 мл кислоти оцтової
безводної Р, якщо необхідно, злегка нагріваючи, дода-
ють 5 мл оцтового ангідриду Р і титрують 0.1М розчи-
ном кислоти хлорної до появи зеленувато-синього за-
барвлення, використовуючи як індикатор розчин кри-
сталічного фіолетового Р.
1 мл 0.1 Мрозчину кислоти хлорної відповідає 12.21 мг
С6Н6^О.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
НІТРАЗЕПАМ
Ніїга/ератит
1УІТКА2ЕРАМ
Нітразепам містить не менше 99.0 % і не більше
ІОі.О % 7-нітро-5-феніл-1,3-дигідро-2//-І,4-бензо-
діазепін-2-ону, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок жовтого кольору.
Розчинність. Практично не розчинний у воді Р, мало
розчинний у 96 % спирті Р, ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С.
Друга ідентифікація: А, В, І), Е.
А. Температура плавлення (2.2.14). Від 226 °С до 230 °С.
В. Розчини готують у захищеному від світла місці й
вимірюють оптичне поглинання розчинів відразу після
приготування.
25.0 мг субстанції розчиняють у розчині 5 г/л кислоти
сірчаної Р у метанолі Р і доводять об’єм розчину тим
самим розчинником до 250.0 мл. 5.0 мл одержаного
розчину доводять розчином 5 г/л кислоти сірчаної Ру
метанолі Р до 100.0 мл. Ультрафіолетовий спектр по-
глинання (2.2.25) одержаного розчину в області від
230 нм до 350 нм повинен мати максимум за довжини
хвилі 280 нм. Питомий показник поглинання у макси-
мумі має бути від 890 до 950.
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ нітразепаму.
Ц. Близько 20 мг субстанції розчиняють у суміші 5 мл
кислоти хлористоводневої Рі 10 мл води Р. Кип’ятять
протягом 5 хв, охолоджують, додають 2 мл розчину
1 г/л натрію нітриту Рі витримують протягом 1 хв. До
одержаного розчину додають 1 мл розчину 5 г/л суль-
фамінової кислоти Р, перемішують, витримують про-
тягом 1 хв і додають 1 мл розчину 1 г/л нафтилети-
лендіаміну дигідрохлориду Р; розчин забарвлюється в
червоний колір.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
425
Нітрофурал
Е. Близько 10 мг субстанції розчиняють, якщо не-
обхідно нагріваючи, у 1 мл метанолу Р, додають
0.05 мл розчину натрію гідроксиду розведеного Р; роз-
чин забарвлюється у червоний колір.
ВИПРОБУВАННЯ НАЧИСТОТУ
Супровідні домішки. Випробування проводять у захище-
ному від світла місці.
Визначення проводять методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар
силікагель ск254 Р.
Випробовуваний розчин. 0.2 г субстанції розчиняють в
ацетоні Р і доводять об’єм розчину тим самим розчин-
ником до 10 мл. Готують розчин безпосередньо перед
використанням.
Розчин порівняння (а). 10 мг амінонітробензофенону Р
розчиняють в ацетоні Р і доводять об’єм розчину тим
самим розчинником до 100 мл. 10 мл одержаного роз-
чину доводять ацетоном Рдо об’єму 50 мл.
Розчин порівняння (Ь). 10 мг ФСЗ домішки А нітразепа-
му розчиняють в ацетоні Р і доводять об’єм розчину
тим самим розчинником до 100 мл. 10 мл одержаного
розчину доводять ацетоном Р до об’єму 50 мл.
Розчин порівняння (с). І мл випробовуваного розчину
доводять ацетоном Рдо 20 мл. 1 мл одержаного розчи-
ну доводять ацетоном Р до об’єму 50 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (200 мкг) випробовуваного розчину, 10 мкл
(0.2 мкг) розчину порівняння (а), 10 мкл (0.2 мкг) роз-
чину порівняння (Ь) і 10 мкл (0.2 мкг) розчину порів-
няння (с). Пластинку помішають у камеру із сумішшю
розчинників етилацетат Р-нітрометан Р(\5:5). Ко-
ли фронт розчинників пройде 12 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й пе-
реглядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину пляма,
відповідна амінонітробензофенону, не має бути інтен-
сивнішою за пляму на хроматограмі розчину
порівняння (а) (0.1 %); пляма, відповідна домішці А
нітразепаму, не має бути інтенсивнішою за пляму на
хроматограмі розчину порівняння (Ь) (0.1 %); будь-
яка пляма, крім основної і плям, відповідних
амінонітробензофенону і домішці А нітразепаму, не
має бути інтенсивнішою за пляму на хроматограмі
розчину порівняння (с) (0.1 %).
Важкі метали (2.4.8, метод О). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5%. 1.00 г субстанції сушать при температурі від
100 °С до 105 °С протягом 4 год.
Сульфатна зола (2.4.14). Не більше 0 1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у 25 мл оцтового
ангідриду Р і титрують О.ІМ розчином кислоти хіорної
потенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хіорної відповідає 28 13 мг
С15Нн^О3.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. 3-аміно-6-нітро-4-фенілхінол-2-он,
В. 2-аміно-5-нітробензофенон.
_________________________________________А
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
НІТРОФУРАЛ
КкгоГигаІит
МІКОІІКАЬ
о
О2Г\І О СН=М—ЬІН—с—мн2
хт
С6Н6ІЧ4О4 М.м. 198.1
Нітрофурал містить не менше 97.0 % і не більше
103.0 % 5-нітро-2-фуральдегіду семикарбазону, у пе-
рерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок жовтого або коричню-
вато-жовтого кольору.
Розчинність. Дуже мало розчинний у воді Р, мало роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація' В.
Друга ідентифікація: А, С, О.
426
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Нітрофурал
А. Випробування проводять у захищеному від яскравого
І світла місці.
Ультрафіолетовий спектр поглинаня (2.2.25) розчину,
приготованого, як зазначено в розділі “Кількісне ви-
значення”, в області від 220 нм до 400 нм повинен ма-
ти два максимуми за довжин хвиль 260 нм і 375 нм.
Відношення оптичної густини в максимумі за довжини
хвилі 375 нм до оптичної густини в максимумі за до-
вжини хвилі 260 нм має бути від 1.15 до 1.30.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції. одержаний у дисках, має відповідати спектру
ФСЗ нітрофуралу.
С. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. 10 мг субстанції розчиняють у
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Розчин порівняння. 10 мг ФСЗнітрофуралу розчиняють
у метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (5 мкг) випробовуваного розчину, 5 мкл (5 мкг)
розчину порівняння. Пластинку поміщають у камеру
із сумішшю розчинників метанол Р - нітрометан Р
(10:90). Коли фронт розчинників пройде 15 см від лінії
старту, пластинку виймають із камери, сушать на
повітрі й обприскують розчином фенілгідразину гідро-
хіориду Р.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння, що відповідає їй за
розміром і забарвленням.
0. Близько 1 мг субстанції розчиняють у 1 мл диме-
тилформаміду Р і додають 0.1 мл розчину калію гідро-
ксиду спиртового Р; з’являється фіолетово-червоне за-
барвлення.
ВИПРОБУВАННЯ НАЧИСТОТУ
рН (2.2.3). Від 5.0 до 7.0. До 1.0 г субстанції додають
100 мл води, вільної від вуглецю діоксиду, Р. збовтують і
фільтрують.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29).
Випробовуваний розчин. 0.10 г субстанції розчиняють у
рухомій фазі й доводять об’єм розчину рухомою фа-
зою до 100.0 мл.
Розчин порівняння (а). 10.0 мг (5-нітро-2-фурил)мети-
лендіацетату Р розчиняють у рухомій фазі й доводять
об’єм розчину рухомою фазою до 20.0 мл. 1.0 мл одер-
жаного розчину доводять рухомою фазою до об’єму
100.0 мл.
Розчин порівняння (Ь). 10 мг ФСЗ нітрофуралу і 10 мг
нітрофурантоїну Р розчиняють у рухомій фазі й дово-
дять об’єм розчину рухомою фазою до 100 мл. 5 мл
одержаного розчину доводять рухомою фазою до
об’єму 100 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октадецилсилільним для
хроматографії Рз розміром часток 5 мкм;
— рухома фаза: ацетонітрил Р - вода Р (40:60);
— швидкість рухомої фази 1 мл/хв;
— детектування за довжини хвилі 310 нм.
Хроматографують 20 мкл розчину порівняння (а).
Чутливість реєструючого пристрою регулюють таким
чином, щоб висота основного піка становила не мен-
ше 50 % шкали реєструючого пристрою.
Хроматографують 20 мкл розчину порівняння (Ь).
Хроматографічна система вважається придатною, як-
що коефіцієнт розділення піків нітрофурантоїну й
нітрофуралу становить не менше 2.0.
Поперемінно хроматографують 20 мкл випробовува-
ного розчину і 20 мкл розчину порівняння (а). Час
хроматографування має бути в 10 разів більше часу
утримування нітрофуралу, шо становить близько 3 хв.
На хроматограмі випробовуваного розчину площа
будь-якого піка, крім основного, не має перевищува-
ти площу основного піка на хроматограмі розчину
порівняння (а) (0.5 %); сума площ усіх піків, крім ос-
новного, не має перевищувати 2 площі основного піка
на хроматограмі розчину порівняння (а) (1.0%). Не
враховують піки, площа яких становить менше 0.05
площі основного піка на хроматограмі розчину порів-
няння (а).
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Випробування проводять у захищеному від яскравого
світла місці.
60.0 мг субстанції розчиняють у 20 мл диметилфор-
маміду Р і доводять об’єм розчину водою Р до 500.0 мл.
5.0 мл одержаного розчину доводять водою Р до об’єму
100.0 мл. Аналогічно готують розчин порівняння із ви-
користанням 60.0 мг ФСЗ нітрофуралу.
Оптичну густину (2.2.25) одержаних розчинів вимірю-
ють у максимумі за довжини хвилі 375 нм.
Вміст С6Н6М4О4 розраховують, виходячи зі значень
оптичних густин і концентрацій розчинів.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
427
Нітрофурал
ДОМІШКИ
А. 5-нітро-2-фуральдегід азин,
ФУРАЦИЛІН
Ригасіїіпит
При проведенні випробування “рН”, зазначеного у
розділі “Випробування на чистоту”, суспензію суб-
станції збовтують протягом !5хв.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
В. (5-нітро-2-фурил)метилен діацетат.
428
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Оксазепам
ОКСАЗЕПАМ
Охахератит
поглинання в максимумі за довжини хвилі 229 нм має
бути від 1220 до 1300.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ оксазепаму.
0ХА2ЕРАМ
М.м. 286.7
Оксазепам містить не менше 98.5 % і не більше 101.0 %
7 хлор-З-гідрокси-5-феніл-1,3-дигідро-2/7-1,4-бен-
зодіазепін-2-ону, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Практично не розчинний у воді Р, мало
розчинний у 96 % спирті Р і метиленхлориді Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, С.
Друга ідентифікація А, С, В.
А. Розчини готують у захищеному від світла місці й
вимірюють оптичне поглинання розчинів відразу після
приготування.
20.0 мг субстанції розчиняють у 96 % спирті Р і дово-
дять об’єм розчину тим самим розчинником до
100.0 мл. 10.0 мл одержаного розчину доводять
96 % спиртом Рао об’єму 50.0 мл (розчин А). 10.0 мл
розчину А доводять 96% спиртом Рао об’єму 100.0 мл
(розчин В). Ультрафіолетовий спектр поглинання
(2.2.25) розчину А в області від 300 нм до 350 нм
повинен мати максимум за довжини хвилі 316 нм.
Ультрафіолетовий спектр поглинання (2.2.25) розчи-
ну В в області від 220 нм до 250 нм повинен мати мак-
симум за довжини хвилі 229 нм. Питомий показник
С. Хроматограми, одержані при випробуванні "Су-
провідні домішки", переглядають в УФ-світлі за до-
вжини хвилі 254 нм. На хроматограмі випробовувано-
го розчину (Ь) має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порів-
няння (а), відповідна їй за розміром.
В. Близько 20 мг субстанції розчиняють у суміші 5 мл
кислоти хлористоводневої Р і 10 мл води Р. Кип’ятять
протягом 5 хв і охолоджують. До одержаного розчину
додають 2 мл розчину 1 г/л натрію нітриту Р і витри-
мують протягом 1 хв. Додають 1 мл розчину 5 г/л кис-
лоти сульфамінової Р, перемішують, витримують про-
тягом 1 хв і додають І мл розчину 1 г/л нафтилети-
лендіаміну дигідрохюриду Р; розчин забарвлюється в
червоний колір.
ВИПРОБУВАННЯ НАЧИСТОТУ
Супровідні домішки. Випробування проводять у захище-
ному від світла місці.
Визначення проводять методом тонкошарової хрома-
тографії (2.2 27), використовуючи як тонкий шар
підхожий силікагель із флуоресцентним індикатором з
оптимальною інтенсивністю поглинання за довжини
хвилі 254 нм. Перед використанням пластинку проми-
вають метанолом Р. Коли фронт розчинника пройде
17 см від лінії старту, пластинку виймають із камери і
сушать на повітрі, потім при температурі від
100 СС до 105 °С протягом 30 хв.
Випробовуваний розчин (а). 50 мг субстанції розчиня-
ють в ацетоні Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Випробовуваний розчин (Ь). 2 мл випробовуваного роз-
чину (а) доводять ацетоном Рао об’єму 10 мл.
Розчин порівняння (а). 10 мі ФСЗ оксазепаму розчиня-
ють в ацетоні Р і доводять об’єм розчину тим самим
розчинником то 10 мл.
Розчин порівняння (Ь). 10 мг ФСЗ оксазепаму і 10 мг
ФСЗ бромазепаму розчиняють в ацетоні Р і доводять
об’єм розчину тим самим розчинником до 10 мл.
Розчин порівняння (с). 1 мл розчину порівняння (Ь) до-
водять ацетоном Рао об’єму 100 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
429
Орнітину гідрохлорид'
Розчин порівняння (сі). 5 мл розчину порівняння (с) до-
водять ацетоном Рао об’єму 10 мл.
На лінію старту хроматографічної пластинки наносять
20 мкл (100 мкг) випробовуваного розчину (а), 20 мкл
(20 мкг) випробовуваного розчину (Ь), 20 мкл (20 мкг)
розчину порівняння (а), 20 мкл (20 мкг оксазепаму і
20 мкг бромазепаму) розчину порівняння (Ь), 20 мкл
(0.2 мкг) розчину порівняння (с), 20 мкл (0.1 мкг) роз-
чину порівняння (б). Пластинку помішають у камеру
із сумішшю розчинників метанол Р - метиленхлорид Р
(10:100). Коли фронт розчинників пройде 15 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й переглядають в УФ-світлі за довжини хвилі
254 нм.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (с)
(0.2 %), і тільки одна пляма може бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (б)
(0.1 %).
Результати аналізу вважаються вірогідними, якшо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С при залишковому тиску не більше 0.7 кПа.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у суміші 10 мл кислоти
оцтової льодяної Р і 90 мл оцтового ангідриду Р і тит-
рують 0.1 М розчином кислоти хлорної потенціомет-
рично (2.2.20).
1 мл 0.1 М розчину кислоти хюрної відповідає 28.67 мг
С15НнС1М2О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
430
ОРНІТИНУ ГІДРОХЛОРИД1"
ОгпііЬіпі ІіудгосЬІогідит
ОКПІТНШЕ ИІІЖОСІН.ОКІІ)!
СО2Н
н2гч , неї
Н ЬІН2
С5Н13СІІУ2О2 М.м. 168.6
Орнітину гідрохлорид містить не менше 99.0 % і не
більше 102.0 % (5)-2,5-діамінопентанової кислоти
гідрохлориду, у перерахунку на суху речовину;
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті “Продукти ферментації”.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Легко розчинний у воді Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, Е.
Друга ідентифікація: А, С, Б, Е.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ орнітину гідрохлориду.
С. Близько 10 мг субстанції розчиняють у 2 мл води Р,
додають 0.1 М розчин натрію гідроксиду до одержання
рН від 6.0 до 7.0, 0.5 мл розчину 2.5 г/л нінгідрину Р ї
нагрівають на водяній бані; протягом 10 хв з’являється
синьо-фіолетове забарвлення.
Б. Близько 10 мг субстанції розчиняють у 2 мл води Р,
додають 1 мл розчину 50 г/л кислоти фосфорно-
молібденової Р; з’являється жовтувато-біла каламуть
або осад.
Е. Субстанція дає реакцію (а) на хлориди (2.3.1)
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Орнітину гідрохлорид1
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 100 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
Питоме оптичне обертання (2.2.7). Від +23.0° до +25.0°,
у перерахунку на суху речовину 1.00 г субстанції роз-
чиняють у розчині 220 г/л кислоти хлористоводневої Р
і доводять об’єм розчину тією самою кислотою до
25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять метолом тонкошарової хроматографії (2.2.27),
використовуючи ТШХ пластинки із шаром силікаге-
лю Р.
Розчин порівняння. 1 мл розчину 8 доводять водою Рро
об’єму 100 мл.
Налінію старту хроматографічної пластинки наносять
2 мкл (50 мкг) розчину 8 і 2 мкл (0.5 мкг) розчину
порівняння. Пластинку сушать на повітрі й поміша-
ють у камеру із сумішшю розчинників вода Р - кисло-
та оцтова льодяна Р - бутанол Р (25:25:50). Коли
фронт розчинників пройде 10 см від лінії старту, плас-
тинку виймають із камери, сушать при температурі від
100 °С до 105 °С протягом 15 хв і обприскують розчи-
ном нінгідрину Р. Нагрівають пластинку при темпера-
турі від 100 °С до 105 °С протягом 10 хв.
На хроматограмі розчину 8 будь-яка пляма, крім ос-
новної. не має бути інтенсивнішою за пляму на хрома-
тограмі розчину порівняння (1.0 %).
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Сульфати (2.4.13). Не більше 0.02 % (200 ррт). 0.75 г
субстанції розчиняють у 15 мл води дистильованої Р.
Одержаний розчин має витримувати випробування на
сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції помішають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р і
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв. Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (100 ррт ПН4) Р,
0.5 мл води Р і 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваним розчином має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкепюном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 20 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
“Пірогени” (2.6.8) або “Бактеріальні ендотоксини”
(2.6.14).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.140 г субстанції розчиняють у 5 мл кислоти мураши-
ної безводної Р, додають 50 мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної по-
тенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної відповідає 16.86 мг
С5Н13СІК2О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
431
Пентоксифілін
ПЕНТОКСИФІЛШ
РепІохіГуПіпіит
РЕМОХІГУІ.І.ІХЕ
С13Н18^О3
М.м. 278.3
Пентоксифілін містить не менше 99.0 % і не більше
101.0 % 3,7-диметил-1-(5-оксогексил)-3,7-дигідро-
1//-пурин-2,6-діону, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Розчинний у воді Р, легко розчинний у ме-
тиленхлориді Р, помірно розчинний у 96 % спирті Р,
дуже мало розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Температура плавлення (2.2.14). Від 103 °С до
107 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ пентоксифіліну.
С. Хроматограму, одержану при випробуванні “Су-
провідні домішки”, переглядають в УФ-світлі за дов-
жини хвилі 254 нм. На хроматограмі випробовуваного
розчину (Ь) має виявлятися основна пляма на рівні ос-
новної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром.
В. Субстанція дає реакцію на ксантини (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р, приготованій із води дистильова-
ної Р, доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). 4 мл розчину 8 доводять
водою Р до об’єму 10 мл. Одержаний розчин має бути
прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування “Прозорість
розчину”, має бути не інтенсивнішим за еталон У7.
Кислотність. До 8 мл розчину 8 додають 12 мл води Р і
0.05 мл розчину бромтимолового синього РІ; з’яв-
ляється зелене або жовте забарвлення, шо переходить
у синє забарвлення при додаванні не більше 0.2 мл
0.01 М розчину натрію гідроксиду.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27). використовуючи
як тонкий шар силікагель СЕ254 Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у метанолі Р і доводять об’єм розчину тим самим
розчинником до 5 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять метанолом Рдо об’єму 10 мл.
Розчин порівняння (а). 20 мг ФСЗ пентоксифіліну роз-
чиняють у метанолі Рі доводять об’єм розчину тим са-
мим розчинником до 10 мл.
Розчин порівняння (Ь). 1 мл випробовуваного розчи-
ну (Ь) доводять метанолом Рло об’єму 50 мл.
Розчин порівняння (с). 20 мг ФСЗпентоксифіліну і 20 мг
ФСЗ теофіліну розчиняють у метанолі Р і доводять
об’єм розчину тим самим розчинником до 10 мл.
На лінію старту хроматографічної пластинки наносять
20 мкл (400 мкг) випробовуваного розчину (а), 20 мкл
(40 мкг) випробовуваного розчину (Ь), 20 мкл (40 мкг)
розчину порівняння (а), 20 мкл (0.8 мкг) розчину
порівняння (Ь), 20 мкл (40 мкг пентоксифіліну і 40 мкг
теофіліну) розчину порівняння (с). Пластинку помі-
щають у камеру із сумішшю розчинників метанол Р -
етилацетат Р(15 85). Коли фронт розчинників прой-
де 10 см від лінії старту, пластинку виймають із каме-
ри, сушать на повітрі і переглядають в УФ-світлі за до-
вжини хвилі 254 нм
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.2 %).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
433
Піперазину адипінат
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). НебільшеО.ОІ % (100ррт). 20 мл роз-
чину 8 поміщають у ділильну лійку, струшують із
двома порціями по 20 мл кожна 2-метилпропанолу Р.
Відбирають 10 мл водного шару і доводять водою Рдо
об’єму 15 мл Одержаний розчин має витримувати ви-
пробування на хлориди.
Сульфати (2.4.13). Не більше 0.02 % (200 ррт). 15 мл
розчину 8 мають витримувати випробування на суль-
фати.
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать над фосфору(У) окси-
дом Р при температурі 60 °С і тиску не більше 700 Па.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.200 г субстанції розчиняють у 5 мл кислоти оцтової
льодяної Р, додають 20 мл оцтового ангідриду Р і тит-
рують 0.1 М розчином кислоти хлорної потенціомет-
рично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної відповідає
27.83 мг С13Н18Х4О3.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. Теобромін.
____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ПІПЕРАЗИНУ АДИПІНАТ
Рірегахіпі адіра8
РІРЕКАШЧЕ АИ1РА ТЕ
ГІН ГІН , НО2С-(СН2)4-СО2Н
СІ0Н20ІУ2О4
М.м. 232.3
Піперазину адипінат містить не менше 98.0 % і не
більше 101.0 % піперазину адипінату; у перерахунку на
безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору.
Розчинність. Розчинний у воді Р, практично не роз-
чинний у 96 % спирті Р.
(Плавиться при температурі близько 250 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А.
Друга ідентифікація В, С.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ піперазину адипінату.
В. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Супровідні домішки",
після обприскування розчинами нінгідрину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром і забарвленням.
С. До 10 мл розчину 8, приготованого, як зазначено в
розділі “Випробування на чистоту”, додають 5 мл кис-
лоти хлористоводневої Р і струшують із трьома
порціями по 10 мл кожна ефіру Р. Ефірні витяги
об’єднують, упарюють насухо, залишок промивають
5 мл води Р і сушать при температурі від 100 °С до
105 °С. Температура плавлення (2.2.14) одержаного за-
лишку має бути від 150 °С до 154 °С.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
434
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Піридоксину гідрохлорид
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В8.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар підхожий силікагель.
Випробовуваний розчин (а). 1.0 г субстанції розчиняють
у 6 мл розчину аміаку концентрованого Р і доводять
об’єм розчину етанолом Р до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять сумішшю етанол Р - розчин аміаку
концентрований Р(2:3) до об'єму 10 мл.
Розчин порівняння (а). 0.1 г ФСЗ піперазину адипінату
розчиняють у суміші етанол Р- розчин аміаку концен-
трований Р(2:3) і доводять об’єм розчину тією самою
сумішшю розчинників до 10 мл.
Розчин порівняння (Ь). 25 мг етилендіаміну Р розчиня-
ють у суміші етанол Р - розчин аміаку концентрова-
ний Р (2:3) і доводять об’єм розчину тією самою
сумішшю розчинників до 100 мл.
Розчин порівняння (с). 25 мг триетилендіаміну /'розчи-
няють у суміші етанол Р - розчин аміаку концентрова-
ний Р (2:3) і доводять об’єм розчину тією самою
сумішшю розчинників до 100 мл.
Розчин порівняння (3). 12.5 мг триетилендіаміну Рроз-
чиняють у 5.0 мл випробовуваного розчину (а) і дово-
дять об’єм розчину сумішшю етанол Р- розчин аміаку
концентрований Р (2:3) до 50 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (500 мкг) випробовуваного розчину (а), 5 мкл
(50 мкг) випробовуваного розчину (Ь), 5 мкл (50 мкг)
розчину порівняння (а), 5 мкл (1.25 мкг) розчину порів-
няння (Ь), 5 мкл (1.25 мкг) розчину порівняння (с),
5 мкл (1.25 мкг триетилендіаміну і 50 мкг піперазину
адипінату) розчину порівняння (сі). Пластинку поміща-
ють у камеру зі свіжоприготованою сумішшю розчин-
ників розчин аміаку концентрований Р - ацетон Р
(20:80). Коли фронт розчинників пройде 15 см від лінії
старту, пластинку виймають із камери, сушать при тем-
пературі 105 °С і обприскують послідовно розчином
З г/л нінгідрину Ру суміші кислота оцтова льодяна Р -
бутанол Р (3:100) і розчином 1.5 г/л нінгідрину Рв ета-
нолі Рі сушать при температурі 105 °С протягом 10 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.25 %).
Пластинку обприскують 0.05 Мрозчином йоду і витри-
мують протягом 10 хв.
На хроматограмі випробовуваного розчину (а) пляма,
відповідна триетилендіаміну, не має бути інтенси-
внішою за пляму на хроматограмі розчину порівнян-
ня (с) (0.25 %). Не враховують пляму на лінії старту.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (сі) виявляються дві
чітко розділені плями.
Важкі метали (2.4.8, метод А). Не більше 0.002 %
(20 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Вода (2.5.12). Не більше 0.5 %. Визначення проводять
з 1.00 г субстанції напівмікрометодом
Сульфатна зола (2 4.14). Не більше 0 І %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції, обережно нагріваючи, розчиняють у
10 мл кислоти оцтової безводної Р і доводять об’єм
розчину тим самим розчинником до 70 мл. Одержа-
ний розчин титрують 0.1 М розчином кислоти хлорної
до переходу забарвлення розчину від коричнювато-
жовтого до зеленого, використовуючи як індикатор
0.25 мл розчину нафтолбензеїну Р.
1 мл 0.1 Мрозчину кислоти хлорної відповідає 11.61 мг
С10Н20ЇЧ2О4.
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
__________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ПІРИДОКСИНУ ГІДРОХЛОРИД
Ругідохіпі Иусігосіїїогісіит
Р\КІІ)()\І5Е НУІ)КОСНІ.ОКІІ)Е
С8Н12СІГ4О3
М.м. 205.6
Піридоксину гідрохлорид містить не менше 99.0 % і не
більше 101.0% (5 гідрокси-6 метилпіридин-3,4-діїл)-
диметанол гідрохлориду. у перерахунку на суху речо-
вину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
435
Піродоксину гідрохлорид
Розчинність. Легко розчинний у воді Р, мало розчинний
у 96 % спирті Р, практично не розчинний в ефірі Р.
(Плавиться при температурі близько 205 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, В.
Друга ідентифікація: А, С, В.
А. 1.0 мл розчину 8, приготованого, як зазначено в
розділі "Випробування на чистоту", доводять 0.1 М
розчином кислоти хлористоводневої до об’єму 50.0 мл
(розчин (а)). 1.0 мл розчину (а) доводять 0.1М розчи-
ном кислоти хлористоводневої до об’єму 100.0 мл. Уль-
трафіолетовий спектр поглинання (2.2.25) одержано-
го розчину в області від 250 нм до 350 нм повинен ма-
ти максимум за довжини хвилі від 288 нм до 296 нм.
Питомий показник поглинання в максимумі має бути
від 425 до 445.
1.0 мл розчину (а) доводять сумішшю рівних об’ємів
0.025 М розчину калію дигідрофосфату і 0.025 М роз-
чину динатрію гідрофосфату (2.2.3) до об’єму 100.0 мл.
Ультрафіолетовий спектр поглинання (2.2.25) одержа-
ного розчину в області від 220 нм до 350 нм повинен
мати максимуми за довжин хвиль від 248 нм до 256 нм
і від 320 нм до 327 нм. Питомі показники поглинання
в максимумах мають бути від 175 до 195 і від 345 до 365,
відповідно.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ піридоксину
гідрохлориду.
С. На хроматограмі випробовуваного розчину (Ь).
одержаній при випробуванні "Супровідні домішки",
має виявлятися основна пляма на рівні основної пля-
ми на хроматограмі розчину порівняння (а),
відповідна їй за розміром і забарвленням.
В. Розчин 8 дає реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.50 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 50.0 мл
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон ¥,.
рН (2.2.3). Від 2.4 до 3.0. Вимірюють рН розчину 8.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи ТШХ пластинки із шаром силікагелю С Р.
Випробовуваний розчин (а). 1.0 г субстанції розчиняють
у воді Рі доводять об’єм розчину тим самим розчинни-
ком до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рдо об’єму 10 мл.
Розчин порівняння (а). 0.10 г ФСЗ піридоксину гідрохю-
риду розчиняють у воді Р і доводять об’єм розчину тим
самим розчинником до 10 мл.
Розчин порівняння (Ь). 2.5 мл розчину порівняння (а)
доводять водою Р до об’єму 100 мл. 1 мл одержаного
розчину доводять водою Рдо об’єму 10 мл.
Налінію старту хроматографічної пластинки наносять
2 мкл (200 мкг) випробовуваного розчину (а), 2 мкл
(20 мкг) випробовуваного розчину (Ь), 2 мкл (20 мкг)
розчину порівняння (а) і 2 мкл (0.5 мкг) розчину
порівняння (Ь). Пластинку поміщають у камеру із
сумішшю розчинників розчин аміаку концентрова-
ний Р - метиленхлорид Р - тетрагідрофуран Р - аце-
тон Р (9:13:13:65). Коли фронт розчинників пройде
15 см від лінії старту, пластинку виймають із камери,
сушать на повітрі й обприскують розчином 50 г/л
натрію карбонату Р у суміші 96 % спирт Р - вода Р
(30:70). Пластинку сушать у струмені теплого повітря
й обприскують розчином 1 г/л дихлорхінонхлоріміду Р
у 96 % спирті Р і відразу переглядають одержану хро-
матограму.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.25 %). Не враховують пляму на лінії старту;
Важкі метали (2.4.8, метод А). Не більше 0.002 %
(20 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у суміші 5.0 мл 0.01 М
розчину кислоти хлористоводневої і 50 мл 96 % спир-
ту Р. Одержаний розчин титрують 0.1 М розчином
натрію гідроксиду Р потенціометрично (2.2.20). У роз-
рахунок беруть об’єм титранту між двома стибками
потенціалів на кривій титрування.
1 мл 0.1 М розчину натрію гідроксиду відповідає
20.56 мг С8Н12С1М)3.
ЗБЕРІГАННЯ
У захищеному від світла місці.
______________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
436
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Прокаїнаміду гідрохлорид
ПРОКАЇНАМІДУ ГІДРОХЛОРИД
Ргосаіпатісіі ИусІгосИїогісІит
РКОСАШАМІРЕ НУРКОСНЕОКПУЕ
СІЗН22СІК,О
М.м. 271.8
Прокаїнаміду гідрохлорид містить не менше 98.0 % і не
більше 101.0 % 4-аміно-А'-[2-(діетиламіно)етил]бен-
заміду гідрохлориду, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або білого з жов-
тавим відтінком кольору, гігроскопічний.
Розчинність. Дуже легко розчинний у воді Р, легко роз-
чинний у 96 % спирті Р, мало розчинний в ацетоні Р,
практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: С, О.
Друга ідентифікація: А, В, О, Е.
А. Температура плавлення (2.2.14). Від 166 °С до 170 °С.
В. 10.0 мг субстанції розчиняють у 0.1 М розчині натрію
гідроксиду і доводять об’єм розчину тим самим розчин-
ником до 100.0 мл. 10.0 мл одержаного розчину доводять
0.1 М розчином натрію гідроксиду до об’єму 100.0 мл.
Ультрафіолетовий спектр поглинання (2.2.25) одержа-
ного розчину в області від 220 нм до 350 нм повинен ма-
ти максимум за довжини хвилі 273 нм. Питомий показ-
ник поглинання в максимумі має бути від 580 до 610.
С. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ прокаїнаміду гід-
рохлориду.
В. 1 мл розчину 8, приготованого, як зазначено в роз-
ділі "Випробування на чистоту", доводять водою Р до
об’єму 5 мл. Одержаний розчин дає реакцію (а) на
хлориди (2.3.1).
Е. 1 мл розчину 8 доводять водою Рдо об’єму 2 мл. 1 мл
одержаного розчину дає реакцію на первинні арома-
тичні аміни (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р, і доводять об’єм розчину тим са-
мим розчинником до 25 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В6.
рН (2.2.3). Від 5.6 до 6.3. Вимірюють рН розчину 8.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель СР254 Р.
Випробовуваний розчин. 0.10 г субстанції розчиняють у
96 % спирті Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Розчин порівняння. 1 мл випробовуваного розчину до-
водять 96 % спиртом Рдо об’єму 200 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину і 5 мкл
(0.25 мкг) розчину порівняння. Пластинку поміщають
у камеру із сумішшю розчинників кислота оцтова льо-
дяна Р - вода Р - бутанол Р( 15:30:60). Коли фронт роз-
чинників пройде 12 см від лінії старту, пластинку вий-
мають із камери, сушать у струмені холодного повітря і
переглядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %).
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.2500 г субстанції розчиняють у 50 мл кислоти хлори-
стоводневої розведеної Р і проводять визначення, як
зазначено в статті "Визначення первинних ароматичних
амінів" (2.5.8).
1 мл 0.1 Мрозчину натрію нітриту відповідає 27.18 мг
С13Н22С11\3О.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці.
_____________________________________________N
НОВОКАЇНАМІД
Ноуосаіпатісіит
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
437
Прокаїну гідрохлорид
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ПРОКАЇНУ ГІДРОХЛОРИД
Ргосаіпі Ьусігосіогісіит
РРОСЛІМ: НУОКОСНЮКЮЕ
/ \ II
Н2ІЧ—£ д—С—О--------С^-СН^С^ , НС!
С13Н2ІСИ\2О2 М.м. 272.8
Прокаїну гідрохлорид містить не менше 99.0 % і не
більше 101.0% 2-діетиламіноетил-4-амінобензоату
гідрохлориду, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
Розчинність. Дуже легко розчинний у воді Р, розчинний
у 96 % спирті Р, практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, Е.
Друга ідентифікація: А, С, Ц, Е, Г.
А. Температура плавлення (2.2.14). Від 154 °С до 158 °С.
В. Інфрачервоний спектр поглинання (2.2.24) субстанції
має відповідати спектру ФСЗ прокаїну гідрохлориду.
С. До близько 5 мг субстанції додають 0.5 мл кислоти
азотної димлячої Р, упарюють насухо на водяній бані,
охолоджують і залишок розчиняють у 5 мл ацетону Р.
До одержаного розчину додають 1 мл 0.1 М розчину
калію гідроксиду спиртового; з’являється тільки ко-
ричнювато-червоне забарвлення.
І). До 0.2 мл розчину 8, приготованого, як зазначено у
розділі "Випробування на чистоту", додають 2 мл во-
ди Р, 0.5 мл кислоти сірчаної розведеної Р і збовтують.
До одержаного розчину додають І мл розчину 1 г/л
калію перманганату Р; забарвлення відразу зникає.
Е. Субстанція дає реакцію (а) на хлориди (2.3.1).
Г. І мл розчину 8 доводять водою Р до об’єму 100 мл.
2 мл одержаного розчину дають реакцію на первинні
ароматичні аміни (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 5.0 до 6.5. 4 мл розчину 8 доводять во-
дою, вільною від вуглецю діоксиду, Рдо об’єму 10 мл.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель ср254 Р.
Випробовуваний розчин. 1.0 г субстанції розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 10 мл.
Розчин порівняння. 50 мг кислоти 4-амінобензойної Р
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 100 мл. 1 мл одержаного розчину
доводять водою Рдо об’єму 10 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (500 мкг) випробовуваного розчину і 5 мкл
(0.25 мкг) розчину порівняння. Пластинку поміщають
у камеру із сумішшю розчинників кислота оцтова
льодяна Р - гексан Р - ефір дибутиловий Р (4:16:80). Ко-
ли фронт розчинників пройде 10 см від лінії старту,
пластинку виймають із камери, сушать при темпера-
турі від 100 °С до 105 °С протягом 10 хв і переглядають
в УФ-світлі задовжини хвилі 254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.05 %).
На хроматограмі випробовуваного розчину основна
пляма має виявлятися на лінії старту.
Важкі метали (2.4.8, метод Е). Не більше 0.0005%
(5 ррт). 1.0 г субстанції розчиняють у воді Р і доводять
об’єм розчину тим самим розчинником до 25.0 мл і
фільтрують. 10 мл фільтрату мають витримувати ви-
пробування на важкі метали. Еталон готують із вико-
ристанням 2 мл еталонного розчину свинцю (1 ррт РЬ ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.00 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.400 г субстанції розчиняють у 50 мл кислоти хлорис-
товодневої розведеної Р і проводять визначення, як за-
значено в статті "Визначення первинних ароматичних
амінів" (2.5.8).
ллє
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Пролін
1 мл 0.1 М розчину натрію нітриту відповідає 27.28 мг
С13Н21СІ1У2О2.
ЗБЕРІГАННЯ
У захищеному від світла місці.
________________________________________N
НОВОКАЇН
Коуосаіпит
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.300 г субстанції розчиняють у 10 мл води Р і 10 мл
кислоти хлористоводневої розведеної Р і далі проводять
визначення, як зазначено в статті ‘‘Визначення первин-
них ароматичних амінів ”(2.5.8). Як індикатор викори-
стовують розчин нейтрального червоного (0.1 мл на
початку і 0.1 мл наприкінці титрування), проводячи
титрування до переходу забарвлення розчину від чер-
воно-фіолетового до синього; або розчин тропеолі-
ну ОО в суміші з метиленовим синім Л* (0.2 мл розчину
тропеоліну ОО і 0.1 мл розчину метиленового синьо-
го), проводячи титрування до переходу забарвлення
від червоно-фіолетового до блакитного.
1 мл 0.1 Мрозчину натрію нітриту відповідає 27.28 мг
СІЗН21С1М2О2.
ПРОЛІН
Ргоііпит
РКОЬІІХЕ
С,Н9І\О2 М.м. 115.1
Пролін містить не менше 98.5 % і не більше 101.0 %
(5)-піролідин-2-карбонової кислоти, у перерахунку на
суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Дуже легко розчинний у воді Р, легко роз-
чинний у 96 % спирті Р, практично не розчинний в
еф ір і Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ проліну.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді дистильо-
ваній Р і доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод П). Розчин 8 має
бути безбарвним.
Питоме оптичне обертання (2.2.7). Від -84.0° до -86.0°,
у перерахунку на суху речовину. 1.00 г субстанції роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у 0.1 М розчині кислоти хлористоводневої і дово-
дять об’єм розчину тією самою кислотою до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рдо об’єму 50 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
439
Пролін
Розчин порівняння (а). 10 мг ФСЗ проліну розчиняють у
0.1 М розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рдо об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ проліну і 10 мг ФСЗ
треоніну розчиняють у 0.1 М розчині кислоти хлорис-
товодневої і доводять об’єм розчину тією самою кис-
лотою до 25 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг проліну і 2 мкг треоніну)
розчину порівняння (с). Пластинку сушать на повітрі
і поміщають у камеру із сумішшю розчинників кисло-
та оцтова льодяна Р - вода Р - бутанол Р (20:20:60).
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й
обприскують розчином нінгідрину Р. Пластинку
нагрівають при температурі від 100 °С до 105 °С протя-
гом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.5 %).
Результати аналізу вважаються вірогідними, якшо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 5 мл роз-
чину 8 доводять водою Рао об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 10 мл
розчину 8 доводять водою дистильованою Р до об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, шо складається із двох годинникових сте-
кол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями во-
ди Р. 50 мг здрібненої субстанції поміщають на нижнє
годинникове скло і розчиняють у 0.5 мл води Р. До роз-
чину додають 0.30 г магнію оксиду важкого Р і ретельно
розтирають скляною паличкою. Нижнє годинникове
скло відразу накривають верхнім і нагрівають при тем-
пературі 40 °С протягом 15 хв. Паралельно в аналогіч-
них умовах готують еталон, використовуючи 0.1 мл
еталонного розчину амонію (100 ррт ІЇН4) Р, 0.5 мл
води Р і 0.30 г магнію оксиду важкого Р. Лакмусовий
папір над випробовуваним розчином має бути забарв-
лений у синій колір не інтенсивніше, ніж над еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Рі дово-
дять об’єм розчину тим самим розчинником до 20 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають ЗО мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної ао пе-
реходу забарвлення від коричнювато-жовтого до зеле-
ного, використовуючи як індикатор 0.1 мл розчину
нафпюлбензеїну Р.
1 мл 0.1 М розчину кислоти хлорної відповідає 11.51 мг
СДІ9\О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
_____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видалення
пірогенів, вона має витримувати випробування "Піро-
гени" (2.6.8) або "Бактеріальні ендотоксини"(2.6.14).
440
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Прометазину гідрохлорид
ПРОМЕТАЗИНУ ГІДРОХЛОРИД
РготеШахіпі ИусІгосШогідит
рзюметнахше нупкосііеокюе
, НСІ і енантіомер
М.м. 320.9
Прометазину гідрохлорид містить не менше 99.0 % і
не більше 101.0 % (2/?5)-А,/У-диметил-1-(10//-феноті-
азин-10-іл)пропан-2-аміну гідрохлориду, у перерахун-
ку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або білого з жов-
тавим відтінком кольору.
Розчинність. Дуже легко розчинний у воді Р, легко роз-
чинний у 96 % спирті Р \ метиленхлориді Р.
(Плавиться при температурі близько 222 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В, О.
Друга ідентифікація: В, С, В.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ прометазину
гідрохлориду.
В. Субстанція має витримувати вимоги випробування
на фенотіазини методом тонкошарової хроматографії
(2.3.3).
С. 0.1 г субстанції розчиняють у 3 мл води Р і додають
краплями 1 мл кислоти азотної Р, випадає осад, що
швидко розчиняється, з’являється червоне забарвлен-
ня, що переходить в оранжеве, а потім у жовте. Одер-
жаний розчин нагрівають до кипіння; з’являється
оранжеве забарвлення й утворюється оранжево-чер-
воний осад.
О. Субстанція дає реакцію (Ь) на хлориди (2.3.1).
ВИПРОБУВАННЯ НА ЧИСТОТУ
рН (2.2.3). Від 4.0 до 5.0 для розчину, виміряного
відразу після приготування. 1.0 г субстанції розчиня-
ють у воді, вільній від вуглецю діоксиду, Р і доводять
об’єм розчину тим самим розчинником до 10 мл.
Супровідні домішки. Випробування проводять у захище-
ному від яскравого світла місці. Розчини готують без-
посередньо перед використанням.
Визначення проводять методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар
підхожий силікагель.
Випробовуваний розчин. 0.20 г субстанції розчиняють у
суміші діетиламін Р - метанол Р (5:95) і доводять
об’єм розчину тією самою сумішшю розчинників до
10 мл.
Розчин порівняння (а). 20 мг ФСЗ ізопрометазину
гідрохлориду розчиняють у суміші діетиламін Р - ме-
танол Р (5:95) і доводять об’єм розчину тією самою
сумішшю розчинників до 100 мл.
Розчин порівняння (Ь). 0.5 мл випробовуваного розчину
доводять сумішшю діетиламін Р - метанол Р(5:95) до
об’єму 100 мл.
Розчин порівняння (с). 0.2 мл випробовуваного розчину
доводять сумішшю діетиламін Р - метанол Р (5:95) до
об’єму 100 мл.
На лінію старту хроматографічної пластинки нано-
сять 10 мкл (200 мкг) випробовуваного розчину,
10 мкл (2 мкг) розчину порівняння (а), 10 мкл (1 мкг)
розчину порівняння (Ь) і 10 мкл (0.4 мкг) розчину
порівняння (с). Пластинку поміщають у ненасичену
камеру із сумішшю розчинників діетиламін Р - аце-
тон Р - циклогексан Р (5:10:85). Коли фронт розчин-
ників пройде 12 см від лінії старту, пластинку вийма-
ють із камери, сушать на повітрі і переглядають в
УФ-світлі за довжини хвилі 254 нм. Не враховують
пляму на лінії старту.
На хроматограмі випробовуваного розчину пляма,
відповідна ізопрометазину гідрохлориду, не має бути
інтенсивнішою за пляму на хроматограмі розчину
порівняння (а) (1 %); будь-яка пляма, крім основної і
плями, відповідної ізопрометазину гідрохлориду, не
має бути інтенсивнішою за пляму на хроматограмі
розчину порівняння (Ь) (0.5 %), і не більше трьох із
цих плям можуть бути інтенсивнішими за пляму на
хроматограмі розчину порівняння (с) (0.2 %).
Важкі метали (2.4.8, метод Е). Не більше 0.001 %
(10 ррт). 1.0 г субстанції розчиняють у 5 мл води Р, до-
дають 5 мл ацетону Р і 5 мл буферного розчину рН 3.5 Р
і фільтрують. Одержаний фільтрат має витримувати
випробування на важкі метали. Еталон готують із
використанням 5 мл еталонного розчину свинцю
(2 ррт РЬ) Р.
Втрата в масі при висушуванні (22.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
441
Пропілпарагідроксибензоат
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у суміші 5.0 мл 0.01 М
розчину кислоти хлористоводневої і 50 мл 96 % спир-
ту Р і титрують 0.1 М розчином натрію гідроксиду по-
тенціометрично (2.2.20). У розрахунок беруть об’єм
титранту між двома стрибками потенціалів на кривій
титрування.
1 мл 0.1 М розчину натрію гідроксиду відповідає
32.09 мг СІ7Н2ІС1М28.
_____________________________________л
ДИПРАЗИН
РіргаДпілт
Залишкові кількості органічних розчинників Суб-
станція має витримувати вимоги статті (5.4).
ЗБЕРІГАННЯ
У шільно закупореному контейнері, у захищеному від
світла місці.
ДОМІШКИ
ПРОПІЛПАРАГІДРОКСИБЕНЗОАТ
Ргору1І8 рагаІіусІгохуЬепхоаБ
РКОРУЬ РАКАНУРКОХУВЕКгОАТЕ
А. фенотіазин,
і енантіомер
В. (2/?5)-А,А-диметил-2-(10//-фенотіазин-10-іл)про-
пан-1-амін (ізопрометазин).
і енантіомер
С. (2/?А)-7У-метил-1 -(10//-фенотіазин- 10-іл)пропан-
2-амін,
і енантіомер
В. (2/?5)-7У,А-диметил-1 -(10//-фенотіазин-10-іл)про-
пан-2-амін 5-оксид.
С10Н|2О3
М.м. 180.2
Пропілпарагідроксибензоат містить не менше 99 0 % і
не більше 100.5 % пропіл-4-гідроксибензоату.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору.
Розчинність. Дуже мало розчинний у воді Р, легко роз-
чинний у 96 % спирті Р і метанолі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Температура плавлення (2.2.14). Від 96 °С до 99 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ пропілпарагідро-
ксибензоату.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Супровідні домішки",
має виявлятися основна пляма на рівні основної пля-
ми на хроматограмі розчину порівняння (Ь), відпо-
відна їй за розміром.
442
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
пропілпарагідроксибензоат
В. Близько 10 мг субстанції поміщають у пробірку, до-
дають 1 мл розчину натрію карбонату Р, кип’ятять
протягом ЗО с і охолоджують (розчин (а)). Близько
10 мг субстанції поміщають в іншу пробірку такого са-
мого розміру, додають 1 мл розчину натрію карбона-
ту Р і перемішують до часткового розчинення суб-
станції (розчин (Ь)). До розчину (а) і розчину (Ь) одно-
часно додають по 5 мл розчину амінопіразолону Р, 1 мл
розчину калію фериціаніду Р \ перемішують; розчин (Ь)
забарвлюється від жовтого до оранжево-коричнюва-
того кольору, а розчин (а) забарвлюється від оранже-
вого до червоного кольору чітко більш інтенсивно за
розчин (Ь).
лінії старту, пластинку виймають із камери, сушать на
повітрі й переглядають в УФ-світлі за довжини хвилі
254 нм.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (а) (0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 1.0 г субстанції розчиняють у 96 % спирті Р
і доводять об’єм розчину тим самим розчинником
до 10 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод 11). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
Кислотність. До 2 мл розчину 8 додають 3 мл 96%
спирту Р, 5 мл води, вільної від вуглецю діоксиду, Р і
0 1 мл розчину бромкрезолового зеленого Р; блакитне за-
барвлення розчину має з’явитися при додаванні не
більше 0.1 мл 0.1М розчину натрію гідроксиду Р.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар підхожий силікагель октадецил-
силільний із флуоресцентним індикатором з опти-
мальною інтенсивністю поглинання за довжини хвилі
254 нм.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють в аиетоні Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять ацетоном Рдо об’єму 10 мл.
Розчин порівняння (а). 0.5 мл випробовуваного розчи-
ну (а) доводять ацетоном Рдо об’єму 100 мл.
Розчин порівняння (Ь). 10 мг ФСЗ пропілпарагідрокси-
бензоату розчиняють в ацетоні Рі доводять об’єм роз-
чину тим самим розчинником до 10 мл.
Розчин порівняння (с). 10 мг ФСЗ етилпарагідроксибен-
зоату розчиняють у 1 мл випробовуваного розчину (а)
і доводять об’єм розчину ацетоном Рао 10 мл.
Налінію старту хроматографічної пластинки наносять
2 мкл (20 мкг) випробовуваного розчину (а), 2 мкл
(2 мкг) випробовуваного розчину (Ь), 2 мкл (0.1 мкг)
розчину порівняння (а), 2 мкл (2 мкг) розчину
порівняння (Ь) і 2 мкл (2 мкг етилпарагідроксибензоа-
ту і 2 мкг субстанції) розчину порівняння (с). Плас-
тинку поміщають у камеру із сумішшю розчинників
кислота оцтова льодяна Р - вода Р - метанол Р
(1:30:70). Коли фронт розчинників пройде 15 см від
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
2,000 г субстанції поміщають у колбу із притертою
скляною пробкою, додають 40.0 мл 1 М розчину
натрію гідроксиду і обережно нагрівають зі зворотним
холодильником протягом 1 год. Розчин охолоджують
до кімнатної температури, холодильник обполіскують
водою Р, приєднуючи промивні води до розчину. Тит-
рують надлишок натрію гідроксиду 0.5 М розчином
кислоти сірчаної потенціометрично (2.2.20), продов-
жуючи титрування до другого стрибка потенціалів на
кривій титрування.
Паралельно проводять контрольний дослід.
І мл / М розчину натрію гідроксиду Р відповідає
180.2 мгС10НІ2О3.
ДОМІШКИ
А. К = Н : 4-гідроксибензойна кислота,
В. К = СН3 : метил-4-гідроксибензоат,
С. К = СН2-СН3: етил-4-гідроксибензоат.
В. К. = СН2-СН2-СН2-СН3: бутил-4-гідроксибензоат.
НІПАЗОЛ
Міраяоїшп
РКОРПРАКАВЕН
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
443
Резорцин
РЕЗОРЦИН
Ке8огсіпокіт
кееоксііуоь
с6н6о2
М.м. 110.1
Резорцин містить не менше 98 5 % і не більше 101.0 %
бензол-1,3-діолу, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок або кристали безбарвні,
або блідо рожево-сірого кольору. Червоніють під
впливом світла і повітря.
Розчинність. Дуже легко розчинний у воді Р \ 96%
спирті Р. легко розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
А. Температура плавлення (2.2.14). Від 109 °С до 112 °С.
В. 0.1 г субстанції розчиняють у 1 мл води Р, додають
1 мл розчину натрію гідроксиду концентрованого Р і
0.1 мл хлороформу Р, нагрівають і охолоджують; з’яв-
ляється інтенсивне темно-червоне забарвлення, що
при додаванні невеликого надлишку кислоти хлорис-
товодневої Р переходить у блідо-жовте забарвлення.
С. Ретельно змішують тонко здрібнені порошки
близько 10 мг субстанції і близько 10 мг калію гідро-
фталату Рао одержання однорідного порошку. Одер-
жану суміш нагрівають на полум’ї до появи оранжево-
жовтого забарвлення. Потім охолоджують, додають
І мл розчину натрію гідроксиду розведеного Р, 10 мл во-
ди Р\збовтують до розчинення. В одержаному розчині
виявляється інтенсивна зелена флуоресценція.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим роїчинником до 25 мл
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В5 або
К5 і не має змінюватися при нагріванні у водяній бані
протягом 5 хв.
Кислотність або лужність. До 10 мл розчину 8 додають
0.05 мл розчину бромфенолового синього Р2, забарвлен-
ня розчину має змінитися при додаванні не більше
0.05 мл 0.1 М розчину кислоти хлористоводневої або
0.1 М розчину натрію гідроксиду.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель с Р.
Випробовуваний розчин. 0.5 г субстанції розчиняють у
метанолі Р і доводять об’єм розчину тим самим роз-
чинником до 10 мл.
Розчин порівняння. 0.1 мл випробовуваного розчину
доводять метанолом Рдо об’єму 20 мл.
На лінію старту хроматографічної пластинки наносять
2 мкл (100 мкг) випробовуваного розчину, 2 мкл
(0.5 мкг) розчину порівняння. Пластинку помішають у
камеру із сумішшю розчинників етилацетат Р - гек-
сан Р (40:60). Коли фронт розчинників пройде 15 см
від лінії старту, пластинку виймають із камери, сушать
на повітрі протягом 15 хв і проявляють парою йоду.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %).
Пірокатехін. До 2 мл розчину 8 додають 1 мл розчину
амоніюмолібдату Р2і перемішують. Жовте забарвлен-
ня випробовуваного розчину має бути не інтен-
сивнішим за забарвлення еталона, приготованого ана-
логічно до випробовуваного розчину із використан-
ням 2 мл розчину 0.1 г/л пірокатехіну Р.
Втрата в масі при висушуванні (2.2.32). Не більше
1.0%. 1.00 г здрібненої субстанції сушать в ексикаторі
протягом 4 год.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1 0 г субстанції.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
445
Рибофлавін
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.500 г субстанції розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до 250.0 мл. 25.0 мл
одержаного розчину поміщають у колбу із притертою
скляною пробкою, додають 1.0 г калію броміду Р,
50.0 мл 0.0167 М розчину калію бромату, 15 мл хлоро-
форму Р і 15.0 мл кислоти хлористоводневої РІ. Колбу
закривають, збовтують і залишають у захищеному від
світла місці протягом 15 хв, періодично збовтуючи.
Потім додають 10 мл розчину 100 г/л калію іюдиду Р,
ретельно перемішують, витримують протягом 5 хв і
титрують 0.1 М розчином натрію тіосульфату, вико-
ристовуючи як індикатор 1 мл розчину крохмалю Р.
1 мл 0.0167 М розчину калію бромату відповідає
1.835 мгС6Н6О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ВЛАСТИВОСТІ
Опис. Кристалічний порошок жовтого або оранжево-
жовтого кольору.
Розчинність. Дуже мало розчинний у воді Р, практично
не розчинний у 96 % спирті Рі ефірі Р.
(Субстанція легше розчинна в розчині 9 г/л натрію
хлориду Р, ніж у воді Р. Розчини розкладаються під
впливом світла, особливо у присутності гідроксидів
лужних металів).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В.
Друга ідентифікація: А. С.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту”.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗрибофлавіну.
С. Близько 1 мг субстанції розчиняють у 100 мл води Р.
Одержаний розчин у світлі, що проходить, має блідо
зеленувато-жовте забарвлення, у відбитому світлі роз-
чин виявляє інтенсивну жовтувато-зелену флуорес-
ценцію, що зникає при додаванні мінеральних кислот
або гідроксидів лужних металів.
РИБОФЛАВІН
КіЬоПауіпит
КІВОРЬАУШЕ
СН2ОН
но—с—н
І
но—с—н
І
СІ7Н20І\4О6
М.м. 376.4
Рибофлавін містить не менше 98.0 % і не більше
101.0 % 7,8-диметил-10-[(25,35,4/?)-2,3,4,5-тетрагід-
роксипентил]-3//,10//-бензоптеридин-2,4-діону, у пе-
рерахунку на суху речовину.
ВИПРОБУВАННЯ НАЧИСТОТУ
Кислотність або лужність. До 0.5 г субстанції додають
25 мл води Р, кип’ятять протягом 2 хв, охолоджують і
фільтрують. До 10 мл фільтрату додають 0.05 мл розчи-
ну фенолфталеїну РІ і 0.4 мл 0.01 М розчину натрію
гідроксиду; з’являється оранжеве забарвлення. До
одержаного розчину додають 0.5 мл 0.01 М розчину
кислоти хлористоводневої; з’являється жовте забарв-
лення, що переходить в оранжеве при додаванні
0.15 мл розчину метилового червоного Р.
Питоме оптичне обертання (2.2.7). Від -115° до -135°, у
перерахунку на суху речовину. 50.0 мг субстанції роз-
чиняють у 0.05 М розчині натрію гідроксиду, вільного
від карбонатів, і доводять об’єм розчину тим самим
розчинником до 10.0 мл. Оптичне обертання одержа-
ного розчину вимірюють не більш як через ЗО хв з мо-
менту розчинення.
Оптична густина (2.2.25). Розчин, приготований для
випробування "Кількісне визначення”, розбавляють
рівним об’ємом води Р. Ультрафіолетовий спектр по-
глинання одержаного розчину має чотири максимуми
за довжини хвиль 223 нм, 267 нм, 373 нм і 444 нм.
Відношення оптичної густини в максимумі за довжи-
ни хвилі 373 нм до оптичної густини в максимумі за
довжини хвилі 267 нм має бути від 0.31 до 0.33, а відно-
шення оптичної густини в максимумі за довжини
хвилі 444 нм до оптичної густини в максимумі за до-
вжини хвилі 267 нм має бути від 0.36 до 0.39.
446
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Рибофлавін
Люміфлавін. 25 мг субстанції струшують з 10 мл хлоро-
форму Р протягом 5 хв і фільтрують. Забарвлення
одержаного фільтрату має бути не інтенсивнішим за
еталон В¥6 (2.2.2, метод II).
Втрата в масі при висушуванні (2.2.32). Не більше
1.5%. 1.00 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г залишку, одержаного при випробу-
ванні "Втрата в масі при висушуванні".
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять при ослабленому освітленні.
65 0 мг субстанції поміщають у мірну колбу коричне-
вого скла місткістю 500 мл і суспендують у 5 мл води Р.
Коли субстанція цілком змочена, додають 5 мл розчи-
ну натрію гідроксиду розведеного Р і перемішують до
повного розчинення. Потім додають 100 мл води Р
і 2.5 мл кислоти оцтової льодяної Р і доводять об’єм
розчину водою Рдо 500.0 мл. 20.0 мл одержаного роз-
чину помішають у мірну колбу коричневого скла
місткістю 200 мл, додають 3.5 мл розчину 14 г/л
натрію ацетату Рі доводять об’єм розчину водою Рдо
200 0 мл.
Оптичну густину (2.2.25) одержаного розчину вимірю-
ють за довжини хвилі 444 нм.
Вміст СІ7Н2()М4О6 обчислюють, використовуючи пи-
томий показник поглинання, що дорівнює 328.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці.
____________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
447
Серин
СЕРИН
Бегіпит
міом
С3Н7М)3
СО2Н
мн2
М.м. 105.1
Серин містить не менше 98.5 % і не більше 101.0 %
(5)-2-аміно-3-гідроксипропанової кислоти, у перера-
хунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Легко розчинний у воді Р, практично не
розчинний у 96 % спирті Р і ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, О.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ серину.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином”, має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
Ц. 1 мл розчину 10 г/л субстанції поміщають у
пробірку, додають 5 мл розчину 20 г/л натрію перйода-
ту Р, закривають скловатою, змоченою водою Р, і
нагрівають на водяній бані протягом 5 хв, збираючи
пари на скловату. Потім скловату переносять в іншу
пробірку, що містить 1 мл розчину 15 г/л хромотропо-
вої кислоти натрієвої солі Р і 3 мл кислоти сірчаної Р,
нагрівають на водяній бані протягом 10 хв; з’являється
фіолетово-червоне забарвлення.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді дистильо-
ваній Р і доводять об’єм розчину тим самим розчинни-
ком до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод П). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В¥6.
Питоме оптичне обертання (2.2.7). Від +14.0° до +16.0°,
у перерахунку на суху речовину. 2.50 г субстанції роз-
чиняють у кислоті хлористоводневій розведеній Р і до-
водять об’єм розчину тією самою кислотою до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у 0.1 М розчині кислоти хлористоводневої і дово-
дять об’єм розчину тією самою кислотою до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рао об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ серину розчиняють у
0.1 М розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ серину і 10 мг ФСЗ
метіоніну розчиняють у 0.1 Мрозчині кислоти хлорис-
товодневої і доводять об’єм розчину тією самою кис-
лотою до 25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
449
Серин
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг серину і 2 мкг метіоніну)
розчину порівняння (с). Пластинку сушать на повітрі
й поміщають у камеру із сумішшю розчинників кисло-
та оцтова льодяна Р - вода Р - бутанол Р (20:20:60).
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й
обприскують розчином нінгідрину Р. Пластинку
нагрівають при температурі від 100 °С до 105 °С протя-
гом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 5 мл роз-
чину 8 доводять водою Рао об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 10 мл
розчину 8 доводять водою дистильованою Р до об’єму
15 мл. Одержаний розчин має витримувати випробу-
вання на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р і
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв. Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (100 ррт МН4) Р,
0.5 мл води Р і 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваним розчином має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 20 мл.
12 мл розчину мають витримувати випробування на
важкі метали. Еталон готують із використанням ета-
лонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної до пе-
реходу забарвлення від коричнювато-жовтого до зеле-
ного, використовуючи як індикатор 0.1 мл розчину
нафтолбензеїну Р.
1 мл 0.1 М розчину кислоти хлорної відповідає
10.51 мгС3Н7МО3.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному віл
світла місці.
_________________________________________ТУ
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
СПИРТ ІЗОПРОПІЛОВИЙ
АІсоИоІ І8оргору1іси8
18ОРКОРУІАІ.СОНОІ.
С3Н8О
М.м. 60.1
Спирт ізопропіловий — пропан-2-ол.
ВЛАСТИВОСТІ
Опис. Безбарвна прозора рідина.
450
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Спирт ізопропіловий
Розчинність. Змішується з водою Р, 96 % спиртом Р і
ефіром Р.
ІДЕНТИФІКАЦІЯ
А. Відносна густина (2.2.5). Від 0.785 до 0.789.
В. Показник заломлення (2.2.6). Від 1.376 до 1.379.
С. До 1 мл субстанції додають 2 мл розчину калію
дихромату Р, 1 мл кислоти сірчаної розведеної Р і
кип’ятять; утворюються пари, що забарвлюють філь-
трувальний папір, просочений розчином нітробензаль-
дегіду Р, у зелений колір. Фільтрувальний папір змочу-
ють кислотою хлористоводневою розведеною Р; забарв-
лення переходить у синє.
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). Субстанція має бути прозо-
рою. 1 мл субстанції доводять водою Рао об’єму 20 мл.
Одержаний розчин через 5 хв має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Субстанція має
бути безбарвною.
Кислотність або лужність. 25 мл субстанції обережно
кип’ятять протягом 5 хв, додають 25 мл води, вільної
від вуглецю діоксиду, Р, витримують до охолодження,
захищаючи від вуглецю діоксиду повітря. До одержа-
ного розчину додають 0.1 мл розчину фенолфталеїну Р;
розчин безбарвний. Слабко-рожеве забарвлення роз-
чину має з’явитися при додаванні не більше 0.6 мл
0.01 Мрозчину натрію гідроксиду Р.
Бензол і супровідні домішки. Визначення проводять
методом газової хроматографії (2.2.28).
Випробовуваний розчин (а). Випробовувана субстанція.
Випробовуваний розчин (Ь). 1.0 мл 2-бутанолу РІ дово-
дять випробовуваним розчином (а) до об’єму 50.0 мл.
5.0 мл одержаного розчину доводять випробовуваним
розчином (а) до об’єму 100.0 мл.
Розчин порівняння (а). 0.5 мл 2-бутанолу РІ і 0.5 мл
пропанолу Р доводять випробовуваним розчином (а)
до об’єму 50.0 мл. 5.0 мл одержаного розчину доводять
випробовуваним розчином (а) до об’єму 50.0 мл.
Розчин порівняння (Ь). 100 мкл бензолу Р доводять ви-
пробовуваним розчином (а) до об’єму 100.0 мл. 0.20 мл
одержаного розчину доводять випробовуваним розчи-
ном (а) до об’єму 100.0 мл.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
— колонка капілярна розміром 30 м х 0.32 мм,
покрита шаром полі[(ціанопропіл)(феніл)][ди-
метил]силоксану Р, із товщиною шару 1.8 мкм;
— газ-носій гелій для хроматографії Р;
— поділ потоку 1:5 із лінійною швидкістю 35 см/с;
— швидкість газу-носія 1.4 мл/хв;
— для піддувки детектора використовують гелій для
хроматографії Р або азот для хроматографії Р.
Використовують таку програму температурного режи-
му:
Час (хв) Темпера- тура('С) Швид- кість підняття темпера- тури СС/хв) Примітки
Колонка 0-12 40 - ізотермічний режим
12-32 40—-240 10 лінійний градієнт
32-42 240 - ізотермічний режим
Блок вводу проб - 280 - -
Детектор - 280 -
Хроматографують 1 мкл розчину порівняння (а). Чут-
ливість системи регулюють таким чином, щоб висота
двох піків, що йдуть за основним піком, становила не
менше 50 % шкали реєструючого пристрою. Хромато-
графічна система вважається придатною, якщо ко-
ефіцієнт розділення першого піка (пропанол) і друго-
го піка (2-бутанол) становить не менше 10.
Хроматографують 1 мкл випробовуваного розчину (Ь).
На хроматограмі площа будь-якого піка, крім основ-
ного і 2-бутанолу, не має перевищувати площу піка
2-бутанолу (0.1 %), і сума площ усіх піків не має пере-
вищувати 3 площі цього піка (0.3 %).
Хроматографують 1 мкл розчину порівняння (Ь). Чут-
ливість системи регулюють таким чином, щоб висота
піка, що іде за основним піком, із часом утримування
близько 10 хв, становила не менше 10 % шкали
реєструючого пристрою.
Хроматографують 1 мкл випробовуваного розчину (а).
На хроматограмі площа піка бензолу не має переви-
щувати половини площі відповідного піка на хромато-
грамі розчину порівняння (Ь) (2 ррт).
Пероксиди. 8 мл розчину крохмалю з калію йодидом Р
поміщають у пробірку із притертою скляною пробкою
місткістю 12 мл і діаметром близько 15 мм, заповню-
ють повністю субстанцією та інтенсивно перемішу-
ють. Витримують у темному місці протягом 30 хв; роз-
чин не має забарвлюватись.
Нелеткий залишок. 100 г субстанції, якщо вона витри-
мала випробування "Пероксиди", упарюють насухо на
водяній бані, потім сушать при температурі від 100 °С
до 105 °С. Маса сухого залишку не має перевищувати
2 мг ( 20 ррт).
Вода (2.5.12). Не більше 0.5 %. Визначення проводять
із 5.0 г субстанції напівмікрометодом.
ЗБЕРІГАННЯ
У захищеному від світла місці.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
451
Спирт ізопропіловий
ДОМІШКИ
А. ацетон,
О. К=Н: етоксіетан (діетиловий ефір),
Е. СН3-ОН: метанол,
ОН
В. бензол,
Е. пропанол-1 ол(н-пропанол).
С. К=СН3:2-(1-метилетокси)пропан (діізопропіловий
ефір),
452
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Твердий жир
твердий жир
Адерз 8ОІіди$
НАПОРАТ
Твердий жир містить суміш тригліцеридів, дигліце-
ридів і моногліцеридів, що можуть бути одержані ете-
рифікацією жирних кислот природного походження
гліцерином або переетерифікацією природних жирів.
Кожний тип твердого жиру характеризується своїми
номінальними значеннями температури плавлення,
гідроксильного числа, числа омилення. Твердий жир
не містить добавок.
ВЛАСТИВОСТІ
Опис. Крихка воскоподібна маса білого або майже
білого кольору. При нагріванні до температури 50 °С
субстанція плавиться з утворенням безбарвної або
слабко-жовтавої рідини.
Розчинність. Практично не розчинний у воді Р, легко
розчинний в ефірі Р, мало розчинний в етанолі Р.
ІДЕНТИФІКАЦІЯ
Визначення проводять методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар
силікагель с Р.
Випробовуваний розчин. 1.0 г субстанції розчиняють в
етиленхлориді Р і доводять об’єм розчину тим самим
розчинником до 10 мл.
На лінію старту хроматографічної пластинки наносять
2 мкл (0.2 мкг) випробовуваного розчину. Пластинку
помішають у камеру із сумішшю розчинників ефір Р -
ети.іенхлорид Р (10:90). Коли фронт розчинників
пройде 12 см від лінії старту, пластинку виймають із
камери, сушать на повітрі й витримують у камері, на-
сиченій парою йоду, до появи плям.
Хроматограму переглядають при денному світлі. На
хроматограмі має виявлятися пляма з К( близько 0.6,
відповідна тригліцеридам (К^ 1), і плями, відповідні
1,3-дигліцеридам (Кч[ 0.5) та 1,2-дигліцеридам (Ву 0.3),
можливе виявлення плями, відповідної 1-моногліце-
ридам (К8І 0.05).
ВИПРОБУВАННЯ НА ЧИСТОТУ
Лужні домішки. 2.00 г субстанції розчиняють у суміші
розчинників, що містить 1.5 мл 96 % спирту Р\ 3.0 мл
ефіру Р, додають 0.05 мл розчину бромфенолового синьо-
го Р; жовте забарвлення має з’явитися при додаванні
не більше 0.15 мл 0.01 М розчину кислоти хлористовод-
невої.
Температура плавлення (2.2.15). Від 30 °С до 45 °С. Тем-
пература плавлення не має відрізнятися більш як на
2 °С від номінального значення. Субстанцію розплав-
ляють, заповнюють капіляр і витримують при темпе-
ратурі нижче 10 °С протягом 24 год.
Кислотне число (2.5.1). Не більше 0.5. 5.0 г субстанції
розчиняють в описаній суміші розчинників.
Гідроксильне число (2.5.3, метод А). Не більше 50. Зна-
чення гідроксильного числа не має відрізнятися від
номінального значення більш як на 5 одиниць. Якщо
номінальне значення гідроксильного числа менше 5,
гідроксильне число має бути не більше 5.
Йодне число (2.5.4). Не більше 3.
Перекисне число (2.5.5). Не більше 3.
Число омилення (2.5.6). Від 210 до 260. Визначення
проводять із 2.0 г субстанції. Число омилення не має
відрізнятися більш як на 5 % від номінального значен-
ня числа омилення.
Неомилювані речовини (2.5.7). Не більше 0.6 %. Визна-
чення проводять із 5.0 г субстанції.
Важкі метали (2.4.8, метод О). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Загальна зола (2.4.16). Не більше 0.05 %. Визначення
проводять із 2.00 г субстанції.
ЗБЕРІГАННЯ
У прохолодному, захищеному від світла місці.
МАРКУВАННЯ
На етикетці зазначають:
— номінальне значення температури плавлення,
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
453
Тіаміну гідробромід"
— номінальне значення гідроксильного числа,
— номінальне значення числа омилення.
______________________________________________N
ВИПРОБУВАННЯ НАЧИСТОТУ
1-Моногліцериди. Не більше 5 %, у перерахунку на мо-
ногліцеростеарат. 10.000 г субстанції поміщають у
ділильну лійку місткістю 100 мл, додають 50 мл хлоро-
форму Р, перемішують до розчинення, потім додають
25 мл розчину 20 г/л кислоти лимонної Р і ретельно
збовтують протягом 1 хв. Після розділу шарів верхній
(водний) шар відокремлюють, нижній (хлороформ-
ний) шар промивають двома порціями по 25 мл кожна
води Р. У разі утворення емульсії хлороформний шар
промивають двома порціями по 25 мл кожна розчину
20 г/л кислоти лимонної Р. Хлороформний шар пере-
носять у мірну колбу місткістю 100 мл, доводять об’єм
розчину хлороформом Р до позначки. 10.0 мл одержа-
ного розчину поміщають у конічну колбу зі скляною
притертою пробкою місткістю 100 мл, додають 10.0 мл
розчину йодної та оцтової кислот Р, закривають проб-
кою, ретельно перемішують і витримують протягом
30 хв у захищеному від світла місці. Потім додають
4 мл розчину калію йодиду Р, колбу закривають проб-
кою, збовтують і точно через 1 хв додають 10 мл води Р.
Йод, що виділився, титрують 0.1 М розчином натрію
тіосульфату до солом’яно-жовтого забарвлення,
після чого додають 2 мл розчину крохмалю Р і продов-
жують титрування при сильному збовтуванні до зне-
барвлення.
Паралельно проводять контрольний дослід з 10.0 мл
хлороформу Рі 10.0 мл розчину йодної та оцтової кис-
лот Р.
Вміст 1-моногліцеридів (А) у субстанції, у перерахун-
ку на моногліцеростеарат, у відсотках, обчислюють за
формулою:
(И2 - К0 А -0.0179 100 100 (Е, - Г0 Х17.9
т 10 т
де:
0.0179 — кількість моногліцеростеарату, шо від по-
відає 1 мл 0.1 М розчину натрію тіосульфа-
ту Р, в грамах;
— об’єм 0.1 Мрозчину натрію тіосульфату Р,
витрачений на титрування випробовувано-
го розчину, у мілілітрах;
И2 — об’єм 0.1М розчину натрію тіосульфату Р,
витрачений на титрування в контрольному
досліді, у мілілітрах;
К — поправочний коефіцієнт до молярності
0.1 М розчину натрію тіосульфату Р;
т — маса наважки субстанції, у грамах.
ТІАМІНУ ПДРОБРОМНГ
Тіііатіпі ЬудгоЬготідит
тніамііїе нуркопкомійБ
, Вг? НВг ,* 1/2 Н20
С12Н18Вг2М4О8, 1/2 Н2О
С12НІ8Вг2М4О8
М.м. 435.2
М.м. 426.2
Тіаміну бромід містить не менше 98.5 % і не більше
101.5 % 3-[(4-аміно-2-метилпіримідин-5-іл)метил]-5-
(2-гідроксіетил)-4-метилтіазолу броміду гідроброміду,
у перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або білого з жов-
тавим відтінком кольору зі специфічним запахом.
Розчинність. Легко розчинний у воді Р, мало розчинний
у 96 % спирті Р, практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, С, В.
Друга ідентифікація: В, С, В.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, попередньо висушеної до постійної маси при
температурі від 100 °С до 105 °С, одержаний у дисках
із калію бромідом Р, має відповідати спектру ФСЗ
тіаміну гідроброміду. У випадку різниці одержаних
спектрів, окремо розчиняють субстанцію і ФСЗ
тіаміну гідроброміду Р, упарюють насухо і повторно
записують спектри одержаних залишків.
В. Близько 20 мг субстанції розчиняють у 10 мл води Р,
додають 1 мл кислоти оцтової розведеної Р і 1.6 мл / М
розчину натрію гідроксиду, нагрівають на водявий бані
протягом 30 хв і охолоджують. До одержаного розчину
додають 5 мл розчину натрію гідроксиду розведеного Р,
10 мл розчину калію фериціаніду Р, 10 мл бутанолу Р
й інтенсивно струшують протягом 2 хв. У верхньому
(спиртовому) шарі спостерігається інтенсивна світло-
блакитна флуоресценція, особливо в УФ-світлі за дов-
жини хвилі 365 нм. Наведене вище випробування по-
вторюють. використовуючи 0.9 мл / Мрозчину натрію
гідроксиду і 0.2 г натрію сульфіту Р замість 1.6 мл ЇМ
розчину натрію гідроксиду; флуоресценція не спосте-
рігається.
С. Субстанція дає реакцію (а) на броміди (2.3.1).
454
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Тіаміну гідробромід"
В. 50 мг субстанції розчиняють у 25 мл води Р. До 5 мл
одержаного розчину додають 1 мл кислоти хлористо-
водневої Р, 1 мл розчину хлораміну Р, І мл хлороформу Р
й інтенсивно струшують протягом 2 хв; у хлороформ-
ному шарі з’являється жовте забарвлення
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 25 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозо-
рим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон 5 шка-
ли найбільш підхожого кольору.
рН (2.2.3). Від 2 7 до 3.4. 3.0 г субстанції розчиняють у
50 мл води, вільної від вуглецю діоксиду, Р.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29). Випробування прово-
дять, захищаючи розчини від дії прямих сонячних про-
менів.
Випробовуваний розчин. 50.0 мг субстанції розчиняють
у рухомій фазі і доводять об’єм розчину рухомою фа-
зою до 10.0 мл. Розчин готують безпосередньо перед
використанням.
Розчин порівняння (а). 20.0 мг ФСЗ 2-метил-4-аміно-5-
оксиметилпіримідину розчиняють у рухомій фазі і до-
водять об’єм розчину рухомою фазою до 10.0 мл.
Розчин порівняння (Ь). 0.5 мл розчину порівняння (а)
доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (с). 20.0 мг ФСЗ 4-метил-5-р-
оксіетилтіазолу розчиняють у рухомій фазі і доводять
об’єм розчину рухомою фазою до 10.0 мл.
Розчин порівняння (сі). 0.5 мл розчину порівняння (с)
доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (е). 50.0 мг ФСЗ тіаміну гідро-
броміду розчиняють у рухомій фазі і доводять об’єм
розчину рухомою фазою до 100.0 мл. До 1.0 мл одер-
жаного розчину додають 0.5 мл розчину порівнян-
ня (а) і 0.5 мл розчину порівняння (с) і доводять об’єм
розчину рухомою фазою до 100.0 мл.
Розчин порівняння (/). 250.0 мг ФСЗ тіаміну гідро-
броміду розчиняють у 25.0 мл розчину порівняння (Ь) і
доводять об’єм розчину розчином порівняння (сі) до
50.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром
0.15 мх 3.9 мм, заповнена октадецилсилільним
силікагелем для хроматографії Рз розміром часток
4 мкм*;
— рухома фаза: 0.60 г натрію гексансульфонату Р
* Підхожими колонками є ІЧоха-Рак С1!{ або БеІІа-Рак С)8
розчиняють у суміші 80 мл ацепюнітрилу Р,
915 мл води Р і 5 мл кислоти оцтової безводної Р;
— швидкість рухомої фази 1 мл/хв;
— температура колонки (30.0±0.1) °С;
— детектування за довжини хвилі 250 нм.
При хроматографуванні за зазначених умов піки вихо-
дять у такому порядку: 4-метил-5-р-оксіетилтіазол;
2-метил-4-аміно-5-оксиметилпіримідин; тіаміну гі-
дробромід.
Хроматографують 5 мкл розчину порівняння (Г). Хро-
матографічна система вважається придатною, якшо
коефіцієнт розділення піків 2-метил-4-аміно-5-окси-
метилпіримідину і тіаміну гідроброміду, 4-МЄТИЛ-5-Р-
оксіетилтіазолу і 2-метил-4-аміно-5-оксиметилпіри-
мідину становить не менше 1.5.
Хроматографують 5 мкл розчину порівняння (е). Хро-
матографічна система вважається придатною, якщо
виконуються такі умови:
— коефіцієнт симетрії, розрахований за піком тіамі-
ну гідроброміду, має бути не більше 1.5;
— ефективність хроматографічної колонки має бути
не менше 1500 теоретичних тарілок.
Поперемінно хроматографують 5 мкл випробовувано-
го розчину і 5 мкл розчину порівняння (е).
На хроматограмі випробовуваного розчину площі
піків 4-метил-5-Р-оксіетилтіазолу і 2-метил-4-аміно-
5-оксиметилпіримідину не мають перевищувати
площі піків 4-метил-5-р-оксіетилтіазолу і 2-метил-4-
аміно-5-оксиметилпіримідину, відповідно, на хрома-
тограмі розчину порівняння (е) (0.2 %). На хромато-
грамі випробовуваного розчину площа будь-якого
піка, крім основного і піків 4-метил-5-Р-оксіетил-
тіазолу і 2-метил-4-аміно-5-оксиметилпіримідину, не
має перевищувати площу піка тіаміну гідроброміду на
хроматограмі розчину порівняння (е) (0.1 %).
Нітрати. До 0 4 мл розчину 8 додають 1.6 мл води Р і
2 мл кислоти сірчаної Р і охолоджують. На одержаний
розчин обережно нашаровують 2 мл свіжоприготова-
ного розчину 80 г/л заліза(ІІ) сульфату Ру воді, вільній
від вуглецю діоксиду, Р; на межі поділу двох шарів не
має утворюватися коричневе кільце.
Сульфати (2.4. ІЗ). Не більше 0.05 % (500 ррт). 5 мл
розчину 8 доводять водою, вільною від вуглецю діокси-
ду, Рцо об’єму 15 мл. Одержаний розчин має витриму-
вати випробування на сульфати.
Важкі метали (2.4.8, метод В) Не більше 0.001 %
(10 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням еталонного розчину свинцю (1 ррт РЬ) Р.
Вода (2.5.12). Не більше 5.0 %. Визначення проводять
з 0.40 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1 0 г субстанції.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
455
Тіаміну гідрохлорид
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у суміші 5 мл кислоти
мурашиної безводної Р, додають 65 мл кислоти оцтової
безводної Р і при перемішуванні 10 мл розчину
ртуті(П) ацетату Р. Одержаний розчин титрують
0.1 М розчином кислоти хлорної потенціометрично
(2.2.20).
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину кислоти хлорної відповідає
21.31 мг С12Н18Вг^4О8.
ЗБЕРІГАННЯ
У повітронепроникному неметалевому контейнері, у
захищеному від світла місці.
ДОМІШКИ
А. 4-метил-5-р-оксіетилтіазол,
В. 2-метил-4-аміно-5-оксиметилпіримідин
ТІАМІНУ ГІДРОХЛОРИД
Тіііатіпі Ііудгосіїїогідит
ПІІАМІІУЕ НУОКОСНЕОКЮЕ
СІ2НІ8СІ2І\4()8
М.м. 337.3
Тіаміну гідрохлорид містить не менше 98.5 % і не
більше 101.5 % ЗД(4-аміно-2-метилпіримідин-5-іл)-
метил]-5-(2-гідроксіетил)-4-метилтіазолу хлориду
гідрохлориду, у перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору або безбарвні кристали.
Розчинність. Легко розчинний у воді Р, розчинний у
гліцерині Р. мало розчинний у 96 % спирті Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А. С.
Друга ідентифікація: В, С.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ тіаміну гідрохло-
риду. У випадку різниці одержаних спектрів окремо
розчиняють субстанцію і ФСЗ тіаміну гідрохлориду у
воді Р, упарюють насухо і повторно записують спектри
одержаних залишків.
В. Близько 20 мг субстанції розчиняють у 10 мл води Р.
додають 1 мл кислоти оцтової розведеної Р і 1.6 мл ЇМ
розчину натрію гідроксиду Р, нагрівають на водяній
бані протягом 30 хв і охолоджують. До одержаного
розчину додають 5 мл розчину натрію гідроксиду розве-
деного Р, 10 мл розчину калію фериціаніду Р, 10 мл бу-
танолу Р й інтенсивно струшують протягом 2 хв. У
верхньому (спиртовому) шарі спостерігається інтен-
сивна світло-блакитна флуоресценція, особливо
помітна в УФ-світлі за довжини хвилі 365 нм Наведе-
не вище випробування повторюють, використовуючи
0.9 мл 1 М розчину натрію гідроксиду і 0.2 г натрію
сульфіту Рзамість 1.6 мл 1 Мрозчину натрію гідрокси-
ду; флуоресценція не спостерігається.
С. Субстанція дає реакцію (а) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р, приготованій із води дистильова-
ної Р, і доводять об’єм розчину тим самим розчинни-
ком до 25 мл.
Прозорість розчину (2.2.1). 2.5 мл розчину 8 доводять
водою Р до об’єму 5 мл. Одержаний розчин має бути
прозорим
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування "Про-
зорість розчину”, має бути не інтенсивнішим за еталон
¥7 або О¥7.
рН (2.2.3). Від 2.7 до 3.3. 2.5 мл розчину 8 доводять во-
дою, вільною від вуглецю діоксиду, Рдо об’єму 10 мл.
Нітрати. До 0.4 мл розчину 8 додають 1.6 мл води Р
і 2 мл кислоти сірчаної Р і охолоджують. На одержаний
456
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Тіаміну гідрохлорид
розчин обережно нашаровують 2 мл свіжоприготова-
ного розчину 80 г/л заліза(ІІ) сульфату Ру воді, віль-
ній від вуглецю діоксиду, Р; на межі поділу двох шарів
не має утворюватися коричневе кільце.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 5 мл
розчину 8 доводять водою Р до об’єму 15 мл. Одержа-
ний розчин має витримувати випробування на суль-
фати.
Важкі метали (2.4.8, метод А). Не більше 0.002 %
(20 ррт). 12 мл розчину 8 мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (2 ррт РЬ) Р.
Вода (2.5.12). Не більше 5.0 %. Визначення проводять
з 0.40 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у суміші 5.0 мл 0.01 М
розчину кислоти хлористоводневої Р і 50 мл 96 % спир-
ту Р. Одержаний розчин титрують 0.1 М розчином
натрію гідроксиду потенціометрично (2.2.20). У
розрахунок беруть об’єм титранту між двома стрибка-
ми потенціалів на кривій титрування.
1 мл 0.1 М розчину натрію гідроксиду відповідає
16.86 мг С12Н18С12^О8.
ЗБЕРІГАННЯ
У шільно закупореному неметалевому контейнері, у
захищеному від світла місці.
____________________________________________N
ВЛАСТИВОСТІ
Опис. Субстанція має специфічний запах.
ВИПРОБУВАННЯ НАЧИСТОТУ
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29). Випробування прово-
дять, захищаючи розчини від дії прямих сонячних про-
менів.
Випробовуваний розчин. 50.0 мг субстанції розчиняють
у рухомій фазі і доводять об’єм розчину рухомою фа-
зою до 10.0 мл. Розчин готують безпосередньо перед
використанням.
Розчин порівняння (а). 20.0 мг ФСЗ 2-метил-4-аміно-5-
оксиметилпіримідину розчиняють у рухомій фазі і до-
водять об’єм розчину рухомою фазою до 10.0 мл.
Розчин порівняння (Ь). 0.5 мл розчину порівняння (а)
доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (с). 20.0 мг ФСЗ 4-метил-5-(3-
оксіетилтіазолу розчиняють у рухомій фазі й доводять
об’єм розчину рухомою фазою до 10.0 мл.
Розчин порівняння (д). 0.5 мл розчину порівняння (с)
доводять рухомою фазою до об’єму 100.0 мл.
Розчин порівняння (е). 50.0 мг ФСЗ тіаміну гідрохюри-
ду розчиняють у рухомій фазі і доводять об’єм розчину
рухомою фазою до 100.0 мл. До 1.0 мл одержаного роз-
чину додають 0.5 мл розчину порівняння (а) і 0.5 мл
розчину порівняння (с) і доводять об’єм розчину рухо-
мою фазою до 100.0 мл.
Розчин порівняння (ф). 250.0 мг ФСЗ тіаміну гідрохлориду
розчиняють у 25.0 мл розчину порівняння (Ь) і доводять
об’єм розчину розчином порівняння (б) до 50.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.15 мм х 3.9 мм,
заповнена силікагелем октадецилсилільним для
хроматографії Рз розміром часток 4 мкм*;
— рухома фаза: 0.60 г натрію гексансульфонату Р
розчиняють у суміші 80 мл ацетонітрилу Р, 915 мл
води Р і 5 мл кислоти оцтової безводної Р;
— швидкість рухомої фази 1 мл/хв;
— температура колонки (30.0±0.1) °С;
— детектування за довжини хвилі 250 нм.
При хроматографуванні за зазначених умов піки вихо-
дять у такому порядку: 4-метил-5-р-оксіетилтіазол;
2-метил-4-аміно-5-оксиметилпіримідин; тіамину гі-
дрохлорид.
Хроматографують 5 мкл розчину порівняння (і). Хро-
матографічна система вважається придатною, якщо
коефіцієнт розділення піків 2-метил-4-аміно-5-окси-
метилпіримідину і тіаміну гідрохлориду, 4-метил-5-р-
оксіетилтіазолу і 2-метил-4-аміно-5-оксиметил-піри-
мідину становить не менше 1.5.
Хроматографують 5 мкл розчину порівняння (е). Хро-
матографічна система вважається придатною, якщо
виконуються такі умови:
— коефіцієнт симетрії, розрахований за піком тіамі-
ну гідрохлориду, має бути не більше 1.5;
— ефективність хроматографічної колонки має бути
не менше 1500 теоретичних тарілок.
Поперемінно хроматографують 5 мкл випробовувано-
го розчину і 5 мкл розчину порівняння (е).
На хроматограмі випробовуваного розчину площі піків
4-метил-5-/5-оксіетилтіазолу і 2-метил-4-аміно-5-ок-
симетилпіримідину не мають перевищувати площі
піків 4-метил-5-/ї-оксіетилтіазолу і 2-метил-4-аміно-
5-оксиметилпіримідину, відповідно, на хроматограмі
розчину порівняння (е) (0.2 %). На хроматограмі ви-
пробовуваного розчину площа будь-якого піка, крім
основного і піків 4-метил-5-Р-оксіетилтіазолу і 2-ме-
тил-4-аміно-5-оксиметилпіримідину, не мають пере-
вищувати площу піка тіаміну гідрохлориду на хрома-
тограмі розчину порівняння (е) (0.1 %).
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
* Підхожими колонками є Моуа-Рак С|8 або Веііа-Рак СІ8.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
457
Тирозин
ДОМІШКИ
А. 4-метил-5-р-оксіетилтіазол,
В. 2-метил-4-аміно-5-оксиметилпіримідин.
ТИРОЗИН
Туговіпиш
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ тирозину.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином”, має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
В. До близько 50 мг субстанції додають 1 мл кислоти
азотної розведеної Р; протягом 15 хв з’являється тем-
но-червоне забарвлення.
Е. Близько ЗО мг субстанції розчиняють у 2 мл розчину
натрію гідроксиду розведеного Р. Додають 3 мл свіжо-
приготованої суміші рівних об’ємів розчину 100 г/л
натрію нітриту Р і розчину 0.5 г кислоти суль-
фанілової Ру суміші 6 мл кислоти хлористоводневої РІ
і 94 мл води Р; з’являється оранжево-червоне забарв-
лення.
ТУКОММ:
С9Н,^О3
М.м. 181.2
Тирозин містить не менше 99.0 % і не більше 101.0 %
(5)-2-аміно-3-(4-гідроксифеніл)пропанової кислоти,
у перерахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати
вимоги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або
безбарвні кристали.
Розчинність. Дуже мало розчинний у воді Р, практично
не розчинний у 96 % спирті Р.
(Розчиняється в розведених мінеральних кислотах і
розведених розчинах гідроксидів лужних металів).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, Ц, Е.
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 0.5 г субстанції розчиняють
у кислоті хлористоводневій розведеній Р і доводять
об’єм розчину тією самою кислотою до 20 мл. Одержа-
ний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод П). Забарвлення
розчину, приготованого для випробування "Про-
зорість розчину”, має бути не інтенсивнішим за еталон
У7.
Питоме оптичне обертання (2.2.7). Від -11.0° до -12.3°.
у перерахунку на суху речовину. 1.25 г субстанції роз-
чиняють у суміші рівних об’ємів кислоти хлористо-
водневої розведеної Рі води Рі доводять об’єм розчину
тією самою сумішшю розчинників до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у розчині аміаку розведеного Р2 і доводять об’єм
розчину водою Рао 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рдо об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ тирозину розчиняють
у 1 мл розчину аміаку розведеного Р2 і доводять об’єм
розчину водою Рдо 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рао об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ тирозину і 10 мг ФСЗ
фенілаланіну розчиняють у 1 мл розчину аміаку розве-
деного Р2 і доводять об’єм розчину водою Рао 25 мл.
458
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Треонін
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг тирозину і 2 мкг феніл-
аланіну) розчину порівняння (с). Пластинку сушать
на повітрі й поміщають у камеру із сумішшю розчин-
ників розчин аміаку концентрований РІ - пропанол Р
(30:70). Коли фронт розчинників пройде 15см від лінії
старту, пластинку виймають із камери, сушать на
повітрі й обприскують розчином нінгідрину Р. Плас-
тинку нагрівають при температурі від 100 °С до 105 °С
протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 0.25 г суб-
станції розчиняють у 3 мл кислоти азотної розведе-
ної Р і доводять об’єм розчину водою Р до 15 мл.
Одержаний розчин має витримувати випробування на
хлориди без подальшого додавання кислоти азотної
розведеної Р.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють при слабкому нагріванні в 5 мл
кислоти хлористоводневої розведеної Р і доводять
об’єм розчину водою дистильованою Р до 15 мл.
Одержаний розчин має витримувати випробування на
сульфати.
Амонію солі (2.4.1, метод В). Не більше 0.02%
(200 ррт). 0.10 г субстанції мають витримувати випро-
бування на амонію солі. Еталон готують із викорис-
танням 0.2 мл еталонного розчину амонію (100 ррт
БН4) Р. Замінюють магнію оксид важкий Р 2.0 мл
розчину натрію гідроксиду концентрованого Р.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
іпилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у 5 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної по-
тенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної відповідає
18.12 мг СцНцМОз-
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени” (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
ТРЕОНІН
Тіігеопіпит
ТНКГ.ОМХБ
н он
X .со2н
Н3С X
н ин2
С4Н<)МО3
М.м. 119.1
Треонін містить не менше 99.0 % і не більше 101.0 %
(25,ЗЯ)-2-аміно-3-гідроксибутанової кислоти, у пере-
рахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
459
Треонін
Розчинність. Розчинний у воді Р, практично не роз-
чинний у 96 % спирті Рі ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту”.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ треоніну.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином”, має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
Ц. 1 мл розчину 2 г/л субстанції змішують з 1 мл роз-
чину 20 г/л натрію перйодату Р, додають 0.2 мл піпе-
ридину Р і 0.1 мл розчину 25 г/л натрію нітропруси-
ду Р; з’являється блакитне забарвлення, яке через
кілька хвилин переходить у жовте.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 100 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод П). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 5.0 до 6.5. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від -27.6° до -29.0°,
у перерахунку на суху речовину. 1.50 г субстанції роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27),
використовуючи ТШХ пластинки із шаром силікаге-
лю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у кислоті хлористоводневій розведеній Р і доводять
об’єм розчину тією самою кислотою до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять водою Рцо об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ треоніну розчиняють
у розчині 1 % (об/об) кислоти хлористоводневої Р і до-
водять об’єм розчину тією самою кислотою до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять водою Рдо об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ треоніну і 10 мг ФСЗ
проліну розчиняють у розчині 1 % (об/об) кислоти хло-
ристоводневої Р і доводять об’єм розчину тією самою
кислотою до 25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг треоніну і 2 мкг проліну)
розчину порівняння (с). Пластинку сушать на повітрі
і поміщають у камеру із сумішшю розчинників кисло-
та оцтова льодяна Р - вода Р - бутанол Р (20 20:60).
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать на повітрі й
обприскують розчином нінгідрину Р. Пластинку
нагрівають при температурі від 100 °С до 105 °С протя-
гом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 10 мл роз-
чину 8 доводять водою Р до об’єму 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
Сульфати (2.4 ІЗ). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють у воді дистильованій Р і дово-
дять об’єм розчину тим самим розчинником до 15 мл.
Одержаний розчин має витримувати випробування на
сульфати.
Амонію солі (2.4.!, метод В). Не більше 0.02%
(200 ррт). 0.10 г субстанції мають витримувати випро-
бування на амонію солі. Еталон готують із викорис-
танням 0.2 мл еталонного розчину амонію (100 ррт
N114) Р.
Залізо (2.4.9) Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній ліііці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
460
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Триптофан
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 5 мл кислоти мураши-
ної безводної Р, додають ЗО мл кислоти оцтової безвод-
ної Р і титрують 0.1 М розчином кислоти хлорної по-
тенціометрично (2.2.20).
1 мл 0.1 М розчину кислоти хлорної відповідає
11.91 мгС4Н9МО3.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
___________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
ТРИПТОФАН
Тгурїоріїапит
М.м. 204.2
не більше
с„нІ2іч2о2
Триптофан містить не менше 98.5 % і
101.0 % (5)-2-аміно-3-(1 /7-індол-3-іл)пропанової кис-
лоти, у перерахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний або аморфний порошок білого
або майже білого кольору.
Розчинність. Помірно розчинний у воді Р, мало роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р.
(Розчиняється в розведених розчинах гідроксидів
лужних металів і мінеральних кислот).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ триптофану.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівнян-
ня (а), відповідна їй за розміром і забарвленням.
В. Близько 20 мг субстанції розчиняють у 10 мл води Р,
додають 5 мл розчину диметиламінобензальдегіду Р6 і
2 мл кислоти хлористоводневої Р1. Одержаний розчин
нагрівають на водяній бані; з’являється фіолетово-
синє забарвлення.
ВИПРОБУВАННЯ НА ЧИСТОТУ
Прозорість розчину (2.2.1). 0.1 г субстанції розчиняють
уІМ розчині кислоти хлористоводневої і доводять
об’єм розчину тією самою кислотою до 10 мл. Одержа-
ний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину, приготованого для випробування "Про-
зорість розчину", має бути не інтенсивнішим за еталон
В¥6.
Питоме оптичне обертання (2.2.7). Від -30.0° до -33.0°,
у перерахунку на суху речовину. 0.25 г субстанції роз-
чиняють у воді Р, якщо необхідно, при нагріванні на
водяній бані і доводять об’єм розчину тим самим роз-
чинником до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27),
використовуючи ТШХ пластинки із шаром силікаге-
лю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у суміші рівних об’ємів кислоти оцтової льодяної Р
і води Р і доводять об’єм розчину тією самою сумішшю
розчинників до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять сумішшю рівних об’ємів кислоти
оцтової льодяної Р і води Рро об'єму 50 мл.
ЛРРЖАВПА ФАРМАКОПЕЯ УКРАЇНИ 2001
461
Триптофан
Розчин порівняння (а). 10 мг ФСЗ триптофану розчи-
няють у суміші рівних об’ємів кислоти оцтової льодя-
ної Р і води Р і доводять об’єм розчину тією самою
сумішшю розчинників до об’єму 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчи-
ну (Ь) доводять сумішшю рівних об’ємів кислоти оц-
тової льодяної Р\ води Рро об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ триптофану і 10 мг
ФСЗ тирозину розчиняють у суміші рівних об’ємів
кислоти оцтової льодяної Р і води Р і доводять об’єм
розчину тією самою сумішшю розчинників до 25 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг триптофану і 2 мкг тиро-
зину) розчину порівняння (с). Пластинку сушать на
повітрі і поміщають у камеру із сумішшю розчинників
кислота оцтова льодяна Р - вода Р - бутанол Р
(20:20:60). Коли фронт розчинників пройде 15 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й обприскують розчином нінгідрину Р. Плас-
тинку нагрівають при температурі від 100 °С до 105 °С
протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
1,Г-Етиліденбіс(триптофан) та інші супровідні домішки.
Визначення проводять методом рідинної хромато-
графії (2.2.29).
Буферний розчин рН 2.3. 3.90 г натрію дигідрофосфа-
ту Р розчиняють у 1000 мл води Р, додають близько
700 мл розчину 2.9 г/л кислоти фосфорної Р і доводять
тим самим розчином до рН 2.3.
Розчини готують безпосередньо перед використанням.
Стандартний розчин. 10.0 мг 19-ацетилтриптофану Р
розчиняють у суміші ацетонітрил Р - вода Р (10:90) і
доводять об’єм розчину тією самою сумішшю розчин-
ників до 100.0 мл. 2.0 мл одержаного розчину доводять
тією самою сумішшю розчинників до об’єму 100.0 мл.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ютьу суміші ацетонітрил Р- вода 73 (10:90) і доводять
об’єм розчину тією самою сумішшю розчинників до
10.0 мл.
Випробовуваний розчин (Ь). 0.10 г субстанції розчиня-
ють у стандартному розчині і доводять об’єм розчину
тим самим розчином до 10.0 мл.
Розчин порівняння (а). 1.0 мг 1, Г-епшліденбіс(трипто-
фану) Р розчиняють у суміші ацетонітрил Р - вода Р
(10:90) і доводять об’єм розчину тією самою сумішшю
розчинників до 100.0 мл.
Розчин порівняння (Ь). 10.0 мл розчину порівняння (а)
доводять стандартним розчином до об’єму 50.0 мл.
Розчин порівняння (с). 10.0 мл розчину порівняння (а)
доводять сумішшю ацетонітрил Р - вода Р (10:90) до
об’єму 50.0 мл.
Розчин порівняння (сі). 0.10 г субстанції розчиняють у
розчині порівняння (с) і доводять об'єм розчину тим
самим розчином до 10.0 мл.
Розчин порівняння (е). 1.0 мл розчину порівняння (с)
доводять сумішшю ацетонітрил Р - вода Р (10:90) до
об’єму 10.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром
0.25 м х 4.6 мм, заповнена силікагелем октадецил-
силільним для хроматографії Р з розміром часток
5 мкм;
— рухома фаза А: ацетонітрил Р- буферний розчин
рН 2.3 (115: 885);
— рухома фаза В: ацетонітрил Р- буферний розчин
рН 2.3 (350 : 650);
— швидкість рухомої фази 0.7 мл/хв;
— використовують таку програму градієнта:
Час (хв) Рухома фаза А (% об/об) Рухома фаза В (% об/об) Примітки
0- 10 100 0 ізократичний режим
10-45 100^0 0-100 лінійний градієнт
45-65 0 100 ізократичний режим
65-66 0^100 100-0 лінійний градієнт
66-80 100 0 установлення рівноваги
— детектування за довжини хвилі 220 нм;
— температура колонки 40 °С.
Поперемінно хроматографують 20 мкл розчину
порівняння (Ь), 20 мкл розчину порівняння (сі) і
20 мкл розчину порівняння (е). При хроматографу-
ванні за зазначених умов час утримування піків має
бути: триптофану — близько 8 хв; А-ацетилтриптофа-
ну — близько 29 хв; 1,Г-етиліденбіс(триптофану) —
близько 34 хв. Чутливість системи регулюють таким
чином, щоб висота піка А-ацетилтриптофану на хро-
матограмі розчину порівняння (Ь) становила не мен-
ше 50 % шкали реєструючого пристрою.
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— на хроматограмі розчину порівняння (Ь)
коефіцієнт розділення піків А-ацетилтриптофа-
ну і 1,1’-етиліденбіс(триптофану) становить не
менше 8.0. Якщо необхідно, змінюють час
градієнтного елюювання. Збільшення тривалості
елюювання рухомою фазою А збільшує час
утримування і призводить до кращого розділення;
— на хроматограмі розчину порівняння (е) відно-
шення сигнал/шум становить не менше 15.
Поперемінно хроматографують 20 мкл випробовува-
ного розчину (а) і 20 мкл випробовуваного розчи-
ну (Ь). Перевіряють, чи з’являється пік із часом утри-
мування, що відповідає А-ацетилтриптофану, на хро-
462
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Триптофан
матограмі випробовуваного розчину (а). При появі
подібного піка на хроматограмі випробовуваного роз-
чину (Ь) коригують площу піка Л'-ацетилтриптофану,
віднімаючи від неї площу піка Л'-ацетилтриптофану,
розраховану із хроматограми випробовуваного розчи-
ну (а). На хроматограмі випробовуваного розчину (Ь)
площа піка 1,1’-етиліденбіс(триптофану) не має пере-
вищувати 0.5 площі основного піка на хроматограмі
розчину порівняння (е) (10 ррт); сума площ усіх піків
із часом утримування, менше часу утримування піка
триптофану, не має перевищувати 0.6 площі піка
Л'-ацетилтриптофану на хроматограмі розчину по-
рівняння (Ь) (100 ррт); сума площ усіх піків із часом
утримування, більше часу утримування піка трипто-
фану, крім піка Л'-ацетилтриптофану і піків, час утри-
мування яких у 1.8 раза більше часу утримування піка
Л'-ацетилтриптофану, не має перевищувати 1.9 площі
піка Л'-ацетилтриптофану на хроматограмі розчину
порівняння (Ь) (300 ррт). Не враховують піки, площа
яких менше 0.02 площі піка Л'-ацетилтриптофану на
хроматограмі розчину порівняння (Ь).
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). 0.25 г суб-
станції розчиняють у 3 мл кислоти азотної розведе-
ної Рї доводять об’єм розчину водою Рдо 15 мл. Одер-
жаний розчин має витримувати випробування на хло-
риди без подальшого додавання розчину кислоти
азотної розведеної Р.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють у суміші кислота хлористовод-
нева розведена Р - вода дистильована Р (5:25) і дово-
дять об’єм розчину тією самою сумішшю розчинників
до 15 мл. Одержаний розчин має витримувати випро-
бування на сульфати.
Амонію солі (2.4.1, метод В). Не більше 0.02%
(200 ррт). 0.10 г субстанції мають витримувати випро-
бування на амонію солі. Еталон готують із викорис-
танням 0.2 мл еталонного розчину амонію (100 ррт
Ь!Н4) Р.
Залізо (2.4.9). Не більше 0.002 % (20 ррт). 0.50 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Р і витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод В). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.150 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової безвод-
ної Р і титрують 0.1Мрозчином кислоти хлорної, вико-
ристовуючи як індикатор 0.1 мл розчину нафтолбен-
зеїну Р.
1 мл 0.1 М розчину кислоти хлорної відповідає
20.42 мгСнН12М2О2.
ЗБЕРІГАННЯ
У шільно закупореному контейнері, у захищеному від
світла місці.
ДОМІШКИ
А. 3,3’-|етиліденбіс( 1 //-індол-1,3-діїл)|біс[(25)-2-
амінопропанова| кислота (1,Г-етиліденбіс(трипто-
фан)),
В. (5)-2-аміно-3-[(ЗА5)-3-гідрокси-2-оксо-2,3-дигід-
ро-1//-індол-3-іл!пропанова кислота (діоксііндоліл-
аланін),
С. (5)-2-аміно-4-(2-амінофеніл)-4-оксобутанова кис-
лота (кінуренін),
НО
И. (5)-2-аміно-3-(5-гідрокси-1 //-індол-3-іл)пропано-
ва кислота (5-гідрокситриптофан),
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
463
Триптофан
Е. (5)-2-аміно-4-[2-(форміламіно)феніл]-4-оксобута-
нова кислота (А'-формілкіиуреиіи),
Н N4-
Г. (5)-2-аміно-3-(феніламіно)пропанова кислота (3-
феніламіноаланін),
(5)-2-аміно-3-[2-[2,3-дигідрокси-1 -(1 //-індол-3-
іл)пропіл]-1 //-індол-3-іл]пропанова кислота.
К. (5)-2-аміно-3-[2-(1//-індол-3-ілметил)-1 //-індол-
3-іл]пропанова кислота.
С. (5)-2-аміно-3-(2-гідрокси-1Я-індол-3-іл)пропано-
ва кислота (2-гідрокситриптофан),
Н. (3/?5)-1,2,3,4-тетрагідро-9//-Р-карболін-3-карбо-
нова кислота,
І. 1 -метил-1,2,3,4-тетрагідро-9Я-Р-карболін-3-карбо-
нова кислота,
Е. 1-( 1//-індол-3-ілметил)-1,2,3,4-тетрагідро-9//-р-
карболін-3-карбонова кислота.
_________________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
464
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Фенілаланін
ФЕНІЛАЛАНІН
Різепуіаіапіппт
РПЕХУЕАІ.іМХЕ
С9НПМО2
М.м. 165.2
Фенілаланін містить не менше 98.5 % і не більше
101.0 % (5)-2-аміно-3-фенілпропанової кислоти, у пе-
рерахунку на суху речовину.
ВИРОБНИЦТВО
Якшо субстанція одержана в результаті процесу, що
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору, або блискучі, білі пластинки.
Розчинність. Помірно розчинний у воді Р, дуже мало
розчинний у 96 % спирті Р, практично не розчинний
в ефірі Р.
(Розчиняється в розведених мінеральних кислотах і
розведених розчинах гідроксидів лужних металів).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ фенілаланіну.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Речовини, виявлювані
нінгідрином", має виявлятися основна пляма на рівні
основної плями на хроматограмі розчину порівняння (а),
відповідна їй за розміром і забарвленням.
Б. До близько 10 мг субстанції додають 0.5 г калію
нітрату Р і 2 мл кислоти сірчаної Р, нагрівають на во-
дяній бані протягом 20 хв, охолоджують, додають 5 мл
розчину 50 г/л гідроксиламіну гідрохлориду Р і витри-
мують у льодяній бані протягом 10 хв. До одержаного
розчину додають 9 мл розчину натрію гідроксиду кон-
центрованого Р; з’являється фіолетово-червоне або
фіолетово-коричневе забарвлення.
ВИПРОБУВАННЯ НАЧИСТОТУ
Прозорість розчину (2.2.1). 0.5 г субстанції розчиняють
уІМ розчині кислоти хлористоводневої і доводять
об’єм розчину тим самим розчинником до 10 мл.
Одержаний розчин має бути прозорим.
Кольоровість розчину (2.2.2, метод П). Забарвлення
розчину, приготованого для випробування "Про-
зорість розчину", має бути не інтенсивнішим за еталон
В¥6.
Питоме оптичне обертання (2.2.7). Від -33.0° до -35.5°,
у перерахунку на суху речовину. 0.50 г субстанції роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи 1111 X пластинки із шаром силікагелю Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у суміші рівних об’ємів кислоти оцтової льодяної Р
і води Р, доводять об’єм розчину тією самою сумішшю
розчинників до 10 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять сумішшю рівних об’ємів кислоти
оцтової льодяної Р і води Рцо об’єму 50 мл.
Розчин порівняння (а). 10 мг ФСЗ фенілаланіну розчи-
няють у суміші рівних об’ємів кислоти оцтової льодя-
ної Р і води Р і доводять об’єм розчину тією самою
сумішшю розчинників до 50 мл.
Розчин порівняння (Ь). 5 мл випробовуваного розчину (Ь)
доводять сумішшю рівних об’ємів кислоти оцтової
льодяної Р і води Рдо об’єму 20 мл.
Розчин порівняння (с). 10 мг ФСЗ фенілаланіну і 10 мг
ФСЗ тирозину розчиняють у суміші рівних об’ємів
кислоти оцтової льодяної Р і води Р і доводять об’єм
розчину тією самою сумішшю розчинників до 25 мл.
Фенол
На лінію старту хроматографічної пластинки наносять
5 мкл (50 мкг) випробовуваного розчину (а), 5 мкл
(1 мкг) випробовуваного розчину (Ь), 5 мкл (1 мкг)
розчину порівняння (а), 5 мкл (0.25 мкг) розчину
порівняння (Ь) і 5 мкл (2 мкг фенілаланіну і 2 мкг ти-
розину) розчину порівняння (с). Пластинку сушать на
повітрі і поміщають у камеру із сумішшю розчин-
ників кислота оцтова льодяна Р - вода Р - бутанол Р
(20:20:60). Коли фронт розчинників пройде 15 см від
лінії старту, пластинку виймають із камери, сушать на
повітрі й обприскують розчином нінгідрину Р. Плас-
тинку нагрівають при температурі від 100 °С до 105 °С
протягом 15 хв.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не мас бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(0.5 %).
Результати аналізу вважаються вірогідними якщо на
хроматограмі розчину порівняння (с) виявляються дві
чітко розділені плями.
Хлориди (2.4.4). Не більше 0.02 %(200 ррт). 0.25 г суб-
станції розчиняють у 3 мл кислоти азотної розведе-
ної Р і доводять об’єм розчину водою Рдо 15 мл. Одер-
жаний розчин має витримувати випробування на
хлориди, без подальшого додавання кислоти азотної
розведеної Р.
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.5 г
субстанції розчиняють у суміші кислота хлористовод-
нева розведена Р - вода дистильована Р (5:25) і дово-
дять об’єм розчину тією самою сумішшю розчинників
до 15 мл. Одержаний розчин має витримувати випро-
бування на сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
помішають шматочок червоного лакмусового паперу Р
розміром (5x5 )мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р і
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40 °С протягом 15 хв Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (100 ррт КП4) Р,
0.5 мл води Р і 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваним розчином має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.001 % (10 ррт). 1.0 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Рі витягають три рази ме-
тилїзобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод В). Не більше 0.001 %
(10 ррт). 2.0 г субстанції мають витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1 000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 3 мл кислоти мураши-
ної безводної Р, додають 30 мл кислоти оцтової безвод-
ної Р і титрують 0.1М розчином кислоти хлорної до пе-
реходу забарвлення від жовтого до зеленого,
використовуючи як індикатор 0.1 мл розчину нафтол-
бензеїну Р.
1 мл 0.1 М розчину кислоти хлорної відповідає
16.52 мгС9Н1^О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці
____________________________________________N
рН (2.2.3). Від 5.3 до 6.3. 0.02 г субстанції розчиняють
при нагріванні у 20 мл води Р і охолоджують.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени" (2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
ФЕНОЛ
РИепоІит
РНЕІЧОЬ
он
с6н6о
М.м. 94.1
466
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
ч»ентог
Фенол містить не менше 99.0 % і не більше 100.5 %
С6Н6О.
ВЛАСТИВОСТІ
Опис. Безбарвні або блідо-рожеві, або блідо-жовтуваті
кристали або кристалічна маса, що розпливається на
повітрі.
Розчинність. Розчинний у воді Р, дуже легко розчин-
ний у 96 % спирті Р, гліцерині Р, метиленхлориді Р.
ІДЕНТИФІКАЦІЯ
А. 0.5 г субстанції розчиняють у 2 мл розчину аміаку
концентрованого Р і доводять об’єм розчину водою Р
до 100 мл. До 2 мл одержаного розчину додають
0.05 мл розчину натрію гіпохлориту концентровано-
го Р; з’являється блакитне забарвлення, шо згодом
стає більш інтенсивним.
В. До 1 мл розчину 8, приготованого як зазначено в
розділі "Випробування на чистоту", додають 10 мл во-
ди Р і 0.1 мл розчину заліза(III) хлориду РІ; з’являється
фіолетове забарвлення, шо зникає при додаванні 5 мл
2-пропанолу Р
С. До 1 мл розчину 8 додають 10 мл води Рі 1 мл бром-
ної води Р; випадає осад блідо-жовтого кольору.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.0 г субстанції розчиняють у воді Р, дово-
дять об’єм розчину тим самим розчинником до 15 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон В6.
Кислотність. До 2 мл розчину 8 додають 0.05 мл розчи-
ну метилового оранжевого Р. Одержаний розчин має
бути жовтим.
Температура тверднення (2.2.18). Не менше 39.5 °С.
Сухий залишок. Не більше 0.05 %. 5.000 г субстанції
упарюють на водяній бані насухо, потім сушать при
температурі від 100 °С до 105 °С протягом 1 год.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
2.000 г субстанції розчиняють у воді Р і доводять об'єм
розчину тим самим розчинником до 1000.0 мл. 25.0 мл
одержаного розчину поміщають у колбу із притертою
скляною пробкою, додають 50.0 мл 0.0167 М розчину
бромід-бромату і 5 мл кислоти хлористоводневої Р, за-
кривають пробкою, витримують протягом 30 хв,
періодично перемішуючи, потім залишають на 15 хв.
Додають 5 мл розчину 200 г/л калію йодиду Р, пе-
ремішують і титрують 0.1 М розчином натрію тіосуль-
фату до появи слабко-жовтого забарвлення. Потім
додають 0.5 мл розчину крохмалю Р, 10 мл хлорофор-
му Р і продовжують титрування, енергійно перемішу-
ючи до повного знебарвлення розчину.
Паралельно проводять контрольний дослід.
І мл 0.0167 М розчину бромід-бромату відповідає
1.569 мгС6Н6О.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці.
____________________________________________N
ВЛАСТИВОСТІ
Опис.На поверхні субстанції допускається наявність
окремих або зрощених голчастих кристалів.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: Ц.
Друга ідентифікація: А, В, С.
В. Інфрачервоний спектр поглинання (2.2.24) розчи-
ну 3 г/л субстанції у вуглеці тетрахлориді для хромато-
графії має відповідати еталонному спектру ДФУ фе-
нолу.
ВИПРОБУВАННЯ НАЧИСТОТУ
Крезоли та інші леткі домішки. Визначення проводять
методом газової хроматографії (2.2.28), використову-
ючи тимол Ряк внутрішній стандарт.
Розчин внутрішнього стандарту. 50.0 мг тимолу Р
розчиняють у метанолі Р і доводять об’єм розчину тим
самим розчинником до 100.0 мл.
Випробовуваний розчин. 1.000 г субстанції розчиняють
у метанолі Р, додають 2.0 мл розчину внутрішнього
стандарту і доводять об’єм розвинутим самим розчин-
ником до 10.0 мл.
Розчин порівняння (а). 50.0 мг о-крезолу Р, 50.0 мг
л/-крезолу, 50.0 мг //-крезолу розчиняють у метанолі Р
і доводять об’єм розчину тим самим розчинником до
50.0 мл.
Розчин порівняння (Ь). До 1.0 мл розчину порівняння
(а) додають 2.0 мл розчину внутрішнього стандарту і
доводять об’єм розчину метанолом Рао 10.0 мл.
Розчин порівняння (с). 50.0 мг субстанції і 50.0 мг
дифенілоксиду розчиняють у метанолі Р і доводять
об’єм розчину тим самим розчинником до 50.0 мл. До
1.0 мл одержаного розчину додають 1.0 мл розчину
порівняння (а), 2.0 мл розчину внутрішнього стандар-
ту і доводять об’єм розчину метанолом Рао 10.0 мл.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
467
Фенол
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
— колонка капілярна кварцова розміром 25 м х 0.35 мм
із полі(диметил)(дифеніл)силоксаном Р, з товщи-
ною шару 0.2 мкм;
— газ-носій гелій для хроматографії Р;
— швидкість газу-носія 1.5 мл/хв;
— температуру колонки програмують: 50 °С протя-
гом 2 хв, підвищення температури зі швидкістю
7 °С/хв до 180 °С, при температурі 180 °С протя-
гом 10 хв;
— температура детектора і блока вводу проб 220 °С;
— час хроматографування має на 10 % перевищувати
час утримування дифенілоксиду на хроматограмі
розчину порівняння (Ь) і становити близько 20 хв.
Поперемінно хроматографують по 2 мкл випробову-
ваного розчину, розчину порівняння (Ь), розчину
порівняння (с), одержуючи не менше п’яти хромато-
грам кожного розчину. Порядок виходу піків на хро-
матограмі розчину порівняння (Ь) має бути: метанол,
о-крезол, сумарний пік п- і ти-крезолів, тимол (внут-
рішній стандарт).
Хроматографічна система вважається придатною, як-
що виконуються такі умови:
— ефективність хроматографічної колонки, розра-
хована за піком о-крезолу із хроматограм розчи-
ну порівняння (с), має бути не менше 6000 теоре-
тичних тарілок;
— коефіцієнт розділення піків фенолу та о-крезолу,
розрахований із хроматограм розчину порівнян-
ня (с), має бути не менше 4.7;
— коефіцієнт розділення піків тимолу (внутрішній
стандарт) і дифенілоксиду, розрахований із хрома-
тограм розчину порівняння (с), має бути не
менше 5.9;
— висота піка о-крезолу на хроматограмі розчину
порівняння (Ь) має бути не менше 30 % шкали
реєструючого пристрою.
Вміст суми крезолів (А), у відсотках, обчислюють за
формулою:
з
й, 10 • 100
_____і______________
Во- пц -50-10
в, ї тОі' 2
і
в0«і
де:
в] — середнє значення відношення суми площ піків
усіх ізомерів крезолу до площі піка внутріш-
нього стандарту, обчислене із хроматограм
випробовуваного розчину;
50 — середнє значення відношення суми площ піків
усіх ізомерів крезолу до площі піка внутріш-
нього стандарту, обчислене із хроматограм
розчину порівняння (Ь);
гщ — маса наважки субстанції, у грамах;
ліОІ — маса наважки о-крезолу, л/-крезолу і я-крезол у,
у грамах.
Сума площ усіх інших додаткових піків на хроматогра-
мах випробовуваного розчину не має перевищувати
площу піка о-крезолу на хроматограмі розчину порів-
няння (Ь) (0.1 %).
Хлориди (2.4.4). Не більше 0.01 %(100 ррт). 10 мл роз-
чину 8 доводять водою Р до об’єму 20 мл. 15 мл одер-
жаного розчину мають витримувати випробування на
хлориди.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 12 мл розчину 100 г/л субстанції у воді Р
мають витримувати випробування на важкі метали.
Еталон готують із використанням еталонного розчину
свинцю (1 ррт РЬ) Р.
Вода (2.5.12). Не більше 0.5 %. Визначення проводять
з 0.800 г субстанції напівмікрометодом.
ПРИМІТКА.
.м-Крезол. С7Н8О. (М.м. 108.1). З-Метилфенол.
Безбарвна або червоного кольору прозора рідина.
Швидко темніє при зберіганні. Не розчинний у воді,
легко розчинний у спиртах, ефірі
: близько 1.03.
по : від 1.5395 до 1.5403
Температура кипіння в межах 200°С - 204 °С
Температура тверднення (2.2.18): не нижче 10.5 °С.
л-Крезол. С7Н8О. (М.м. 108.1/. 4-Метилфенол.
Кристали безбарвні або злегка забарвлені, швидко
темніють на повітрі. Легко розчинний у 96 % спирті.
Температура кипіння (при 760 мм.рт.ст.): в межах
200°С - 202 °С.
Температура тверднення (2.2.18): не нижче 32.5 °С.
Дифенілоксид. СІ2Н10О. (М.м. 170.2/.
Безбарвні кристали. При температурі вище 26.5 °С
рідина з запахом герані.
Практично не розчинний у воді, легко розчинний в
органічних розчинниках. Не розчинний в мінераль-
них кислотах і розчинах гідроксидів лужних металів.
Температура кипіння: в межах 256-259 °С.
Температура тверднення (2.2.18): від 26.5 °С до 28 °С
Вуглецю тетрахлорид для хроматографії. СС14
(М.м. 153.8/.
Безбарвна прозора рідина.
п% : 1.460310.0002.
Вода: не більше 0.05 %.
Вміст суми крезолів у субстанції має бути не більше 0.3 %.
468
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ типі
Фторурацил
ФТОРУРАЦИЛ
Ріиогоигасіїит
Н.ГОііОІВ.КІІ
С4Н3ГМ2О2
М.м. 130.1
Фторурацил містить не менше 98.5 % і не більше
101.0 % 5-фтор-1 //,3/7-піримідин-2,4-діону, у перера-
хунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Помірно розчинний у воді Р, мало роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А.
Друга ідентифікація: В, С.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ фторурацилу.
В. Хроматограми, одержані при випробуванні "Су-
провідні домішки", переглядають в УФ-світлі за довжи-
ни хвилі 254 нм. На хроматограмі випробовуваного
розчину (Ь) має виявлятися основна пляма на рівні ос-
новної плями на хроматограмі розчину порівняння (а),
відповідна їй за розміром
С. 0.5 мл насиченого розчину хрому(УІ) оксиду Ру кис-
лоті сірчаній концентрованій Р нагрівають у тер-
мостійкій пробірці на відкритому полум’ї пальника до
появи білих парів у верхній частині пробірки, при цьо-
му розчин не утворює плівки на стінках пробірки. До
одержаної суміші додають 2 мг субстанції і знову
нагрівають на відкритому полум’ї пальника до
виділення білих парів із верхньої частини пробірки;
при перемішуванні розчин стікає по стінках пробірки
плівкою.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 0.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 50 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення роз-
чину 8 має бути не інтенсивнішим за еталон В¥7 або ¥7.
рН (2.2.3). Від 4.5 до 5.0. Вимірюють рН розчину 8.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель СТ2Я Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у суміші рівних об’ємів метанолу Р і води Рі дово-
дять об’єм розчину тією самою сумішшю розчинників
до 10.0 мл.
Випробовуваний розчин (Ь). 2 мл випробовуваного роз-
чину (а) доводять сумішшю рівних об’ємів метанолу Р
і води Рдо об’єму 10 мл.
Розчин порівняння (а). 20 мг ФСЗ фторурацилу розчи-
няютьусуміші рівних об’ємів, метанолу Рі води Рідо-
водять об’єм розчину тією самою сумішшю розчин-
ників до 10 мл.
Розчин порівняння (Ь). 2.5 мл розчину порівняння (а)
доводять сумішшю рівних об’ємів метанолу Рі води Р
до об’єму 200 мл.
Розчин порівняння (с). 5 мг ФСЗ 5-гідроксіурацилу роз-
чиняють у суміші рівних об’ємів метанолу Р і води Р і
доводять об’єм розчину тією самою сумішшю розчин-
ників до 200 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (100 мкг) випробовуваного розчину (а), 10 мкл
(20 мкг) випробовуваного розчину (Ь). 10 мкл (20 мкг)
розчину порівняння (а), 10 мкл (0.25 мкг) розчину
порівняння (Ь) і 10 мкл (0.25 мкг) розчину порівнян-
ня (с). Пластинку сушать у струмені теплого повітря і
поміщають у камеру із сумішшю розчинників мета-
нол Р - вода Р - етилацетат Р (15:15:70). Коли фронт
розчинників пройде 12 см від лінії старту, пластинку
виймають із камери, сушать на повітрі і переглядають
в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину (а) будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (Ь) (0.25 %).
Пластинку обприскують свіжоприготованим розчи-
ном 5 г/л солі міцного синього В Р і потім 0.1 М розчи-
ном натрію гідроксиду.
На хроматограмі випробовуваного розчину (а) пляма,
відповідна 5-гідроксіурацилу, не має бути інтен-
сивнішою за пляму на хроматограмі розчину
порівняння (с) (0.25 %).
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). Визначення проводять у платиновому тиглі.
1.0 г субстанції має витримувати випробування на
важкі метали. Еталон готують із використанням 2 мл
еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать у вакуумі при темпера-
турі 80 °С протягом 4 год.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
469
Фуросемід
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції у платиновому тиглі.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.1000 г субстанції розчиняють, обережно нагріваючи,
у 80 мл диметилформаміду Р і титрують 0.1 М розчином
тетрабутиламонію гідроксиду, використовуючи як
індикатор 0.25 мл розчину 10 г/л тимолового синього Р
у диметилформаміді Р.
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину тетрабутиламонію гідроксиду
відповідає 13.01 мгС4Н3ЕІЧ2О2.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці.
__________________________________________N
Треба дотримувати обережності при роботі зі фтору-
рацилом. не допускати вдихання часток, попадання на
шкіру.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ФУРОСЕМІД
Ригозетідиш
РОКО8ЕМЮЕ
С12Нц(ІА2О58
М.м. 330.7
Фуросемід містить не менше 98.5 % і не більше
101.0 % 4-хлор-2-(фурфуриламіно)-5-сульфамоїлбен-
зойної кислоти, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Практично не розчинний у воді Р, розчин-
ний в ацетоні Р, помірно розчинний у 96 % спирті Р,
мало розчинний в ефірі Р, практично не розчинний у
метиленхлориді Р.
(Розчиняється в розведених розчинах гідроксидів
лужних металів).
(Плавиться при температурі близько 210 °С із розкла-
данням).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В.
Друга ідентифікація: А, С.
А. 50 мг субстанції розчиняють у розчині 4 г/л натрію
гідроксиду Р і доводять об’єм розчину тим самим роз-
чинником до 100 мл. 1 мл одержаного розчину дово-
дять тим самим розчином 4 г/л натрію гідроксиду Рдо
об’єму 100 мл. Ультрафіолетовий спектр поглинання
(2.2.25) одержаного розчину в області від 220 нм до
350 нм повинен мати три максимуми за довжин хвиль
228 нм, 270 нм і 333 нм. Відношення оптичної густини
в максимумі за довжини хвилі 270 нм до оптичної гус-
тини в максимумі за довжини хвилі 228 нм має бути
від 0.52 до 0.57.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ фуросеміду.
С. Близько 25 мг субстанції розчиняють у 10 мл 96 %
спирту Р. До 5 мл одержаного розчину додають 10 мл
води Р. До 0.2 мл одержаного розчину додають 10 мл
кислоти хлористоводневої розведеної Р і нагрівають зі
зворотним холодильником протягом 15 хв. Одержа-
ний розчин охолоджують, додають 18 мл ЇМ розчину
натрію гідроксиду і 1 мл розчину 5 г/л натрію нітри-
ту Р, витримують протягом 3 хв, додають 2 мл розчину
25 г/л кислоти сульфамінової Р і перемішують. Потім
додають 1 мл розчину 5 г/л нафтилетилендіаміну
дигідрохлориду Р; з’являється фіолетово-червоне за-
барвлення.
ВИПРОБУВАНЯ НАЧИСТОТУ
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29). Розчини готують без-
посередньо перед використанням і захищають від дії
світла.
Випробовуваний розчин. 50.0 мг субстанції розчиняють
у рухомій фазі і доводять об’єм розчину рухомою фа-
зою до 50.0 мл.
Розчин порівняння (а). 20 0 мг ФСЗ домішки А фуро-
семіду розчиняють у рухомій фазі і доводять об’єм роз-
чину рухомою фазою до 20.0 мл.
Розчин порівняння (Ь). 1.0 мл випробовуваного розчину
і 1.0 мл розчину порівняння (а) доводять рухомою фа-
зою до об’єму 20.0 мл. 1.0 мл одержаного розчину до-
водять рухомою фазою до об’єму 20.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
470
ЛРРЖДВНА ФДРМДКОПРЯ Vк'РДЇНР^ ЗПП1
Фуросемід
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октилсилільним для хрома-
тографії Р з розміром часток 5 мкм;
— рухома фаза: 0.2 г калію дигідрофосфату Р і 0.25 г
цетриміду Р розчиняють у 70 мл води Р, доводять
рН розчину до 7.0 розчином аміаку Р і додають
30 мл пропанолу Р;
— швидкість рухомої фази 1 мл/хв;
— детектування за довжини хвилі 238 нм.
Хроматографують 20 мкл розчину порівняння (Ь).
Чутливість системи регулюють таким чином, щоб ви-
соти двох піків, одержаних на хроматограмі розчину
порівняння (Ь), становили не менше 20 % шкали
реєструючого пристрою. Хроматографічна система
вважається придатною, якщо коефіцієнт розділення
першого піка (домішка А фуросеміду) і другого піка
(фуросемід) становить не менше 4.
Хроматографують 20 мкл випробовуваного розчину.
Час хроматографування має бути у 3 рази більше часу
утримування фуросеміду.
На хроматограмі випробовуваного розчину площа
будь-якого піка, крім основного, не має перевищува-
ти площу першого піка на хроматограмі розчину
порівняння (Ь) (0.25 %); сума площ усіх піків, крім ос-
новного, не має перевищувати 2 площі першого піка
на хроматограмі розчину порівняння (Ь) (0.5 %). Не
враховують піки, площа яких становить менше
0.1 площі першого піка на хроматограмі розчину
порівняння (Ь).
Хлориди (2.4.4). Не більше 0.02 % (200 ррт). До 0.5 г
субстанції додають суміш 0.2 мл кислоти азотної Р і
30 мл води Р, струшують протягом 5 хв, витримують
протягом 15 хв і фільтрують. 15 мл одержаного фільтра-
ту мають витримувати випробування на хлориди.
Сульфати (2.4.13) Не більше 0.03 % (300 ррт). До 1.0 г
субстанції додають суміш 0.2 мл кислоти оцтової Р і
30 мл води дистильованої Р, струшують протягом 5 хв,
витримують протягом 15 хв і фільтрують. 15 мл одер-
жаного фільтрату мають витримувати випробування
на сульфати.
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5%. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у 20 мл диметилфор-
маміду Р і титрують 0.1 М розчином натрію гідроксиду,
використовуючи як індикатор 0.2 мл розчину бромти-
молового синього Р2.
Паралельно проводять контрольний дослід.
1 мл 0.1 М розчину натрію гідроксиду відповідає
33.07 мгС^НцСІ^Одй.
ЗБЕРІГАННЯ
У захищеному від світла місці.
ДОМІШКИ
А. 2-хлор-4(фурфуриламіно)-5-сульфамоїлбензойна
кислота,
В. 2,4-дихлор-5-сульфамоїлбензойна кислота,
С. 2-аміно-4-хлор-5-сульфамоїлбензойна кислота,
В. 2,4-біс(фурфуриламіно)-5-сульфамоїлбензойна
кислота.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
471
Хлорамін
ХЛОРАМІН
СШогатіпит
СНЬОПАМІНЕ
сн3
С7Н7СІІ\\аО28,3112О
М.м. 281.7
Хлорамін містить не менше 98.0 % і не більше 103.0 %
натрію /У-хлор-4-метилбензол-сульфоніміду тригідрату.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або білого з жов-
тавим відтінком кольору.
Розчинність. Легко розчинний у воді Р, розчинний у
96 % спирті Р, практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
А. Розчин 8, приготований, як зазначено в розділі "Ви-
пробування на чистоту”, забарвлює червоний лакмусо-
вий папір Ру синій колір, а потім знебарвлює його
В. До 10 мл розчину 8 додають 10 мл розчину водню пе-
роксиду розведеного Р; утворюється білий осад, що
розчиняється при нагріванні. Одержаний гарячий
розчин фільтрують, охолоджують. Білі кристали, що
утворилися, промиті й висушені при температурі від
100 °С до до 105 °С, повинні мати температуру плав-
лення (2.2.14) від 137 °С до 140 °С.
С. 1 г субстанції прожарюють, дотримуючи запобіж-
них заходів від займання. Залишок розчиняють у 10 мл
води Р. Одержаний розчин дає реакцію (а) на хлориди
(2.3.1).
Ц. Розчин, приготований для випробування С, дає ре-
акцію (а) на сульфати (2.3.1).
Е. Розчин, приготований для випробування С, дає ре-
акцію (Ь) на натрій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 1.0 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 20 мл.
Прозорість розчину (2.2.1). Розчин 8 за ступенем кала-
мутності не має перевищувати еталон II.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 8.0 до 10.0. Вимірюють рН розчину 8.
Ортопохідні. До 2 г субстанції додають 10 мл води Р,
перемішують, додають 1 г натрію метабісульфіту Р і
нагрівають до кипіння. Охолоджують до температури
0 °С, швидко фільтрують і промивають трьома пор-
ціями льодяної води Р по 5 мл кожна. Осад, висуше-
ний над фосфору(У) оксидом Р при тиску не більше
600 Па, повинен мати температуру плавлення (2.2.14)
не нижче 134 °С.
Залишок нерозчинний в етанолі. 1.00 г субстанції збов-
тують з 20 мл етанолу Р протягом 30 хв і фільтрують
крізь попередньо зважений фільтр. Залишок на
фільтрі промивають 5 мл етанолу Рі сушать при тем-
пературі від 100 °С до 105 °С. Маса сухого залишку не
має перевищувати 20 мг (2 %).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.125 г субстанції поміщають у колбу із притертою
скляною пробкою, розчиняють у 100 мл води Р, дода-
ють 1 г калію йодиду Рі 5 мл кислоти сірчаної розведе-
ної Р, витримують протягом 3 хв і титрують 0.1 М роз-
чином натрію тіосульфату, використовуючи як інди-
катор 1 мл розчину крохмалю Р.
1 мл 0.1 М розчину натрію тіосульфату відповідає
14.08 мг С7Н7СІ8№О28,ЗН2О.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці, при температурі від 8 °С до 15 °С.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
473
Хлорпромазину гідрохлорид
ХЛОРПРОМАЗИНУ ГІДРОХЛОРИД
СШогрготахіпі НусІгосИІогіскіт
СНЮКРКОМАгШЕ нткоснюкіРЕ
СН2-СН2-СН2-М(СНз)2
^-'17^20^'12^2^
М.м. 355.3
Хлорпромазину гідрохлорид містить не менше 99.0 % і не
більше 101.0 % 2-хлор-10-(3-диметиламінопропіл)фе-
нотіазину гідрохлориду, у перерахунку на суху речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору. Розкладається під впливом повітря і світла.
Розчинність. Дуже легко розчинний у воді Р, легко роз-
чинний у 96 % спирті Р, практично не розчинний в
ефірі Р.
(Плавиться при температурі близько 196 °С).
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В, С, В.
Друга ідентифікація: А, С, В.
А. Розчини готують у захищеному від яскравого світла
місці й вимірюють оптичне поглинання розчинів відразу
після приготування.
50.0 мг субстанції розчиняють у 0.1 Мрозчині кислоти
хлористоводневої} доводять об’єм розчину тією самою
кислотою до 500.0 мл. 5.0 мл одержаного розчину до-
водять 0.1 М розчином кислоти хлористоводневої до
об’єму 100.0 мл. Ультрафіолетовий спектр поглинання
(2.2.25) одержаного розчину в області від 230 нм до
340 нм повинен мати два максимуми за довжин хвиль
254 нм і 306 нм. Питомий показник поглинання в мак-
симумі за довжини хвилі 254 нм має бути від 890
до 960.
В. Інфрачервоний спектр поглинання (2.2.24) розчину
60 г/л субстанції, у метиленхлориді Рпри вимірюванні в
кюветі з товщиною шару 0.1 мм, має відповідати спек-
тру ФСЗ хлорпромазину гідрохлориду.
С. Субстанція має витримувати випробування "Іден-
тифікація фенотіазинів методом тонкошарової хрома-
тографії" (2.3.3).
Б. Субстанція дає реакцію (Ь) на хлориди (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
рН (2.2.3). Від 3.5 до 4.5. 1.0 г субстанції розчиняють у
воді, вільній від вуглецю діоксиду, Р і доводять об’єм
розчину тим самим розчинником до 10 мл. Вимірю-
ють рН свіжоприготованого розчину.
Супровідні домішки. Випробування проводять у захище-
ному від яскравого світла місці.
Визначення проводять методом тонкошарової хрома-
тографії (2.2.27), використовуючи як тонкий шар
силікагель СР254 Р.
Випробовуваний розчин. 0.2 г субстанції розчиняють у
суміші діетиламін Р — метанол Р (5:95) і доводять
об’єм розчину тією самою сумішшю розчинників до
10 мл. Розчин готують безпосередньо перед викорис-
танням.
Розчин порівняння. 1 мл випробовуваного розчину до-
водять сумішшю діетиламін Р — метанол Р (5:95) до
об’єму 200 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (200 мкг) випробовуваного розчину, 10 мкл
(Імкг) розчину порівняння. Пластинку поміщають у
камеру із сумішшю розчинників ацетон Р — діети-
ламін Р — циклогексан Р (10:10:80). Коли фронт роз-
чинників пройде 15 см від лінії старту, пластинку вий-
мають із камери, сушать на повітрі протягом 15 хв і пе-
реглядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (0.5 %).
Не враховують пляму на лінії старту.
Важкі метали (2.4.8, метод С). Не більше 0.001 %
(10 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 1 мл еталонного розчину свинцю (10 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше
0.5 %. 1.000 г субстанції сушать при температурі від
100 °С до 105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.250 г субстанції розчиняють у суміші 5.0 мл 0.01 М
розчину кислоти хлористоводневої і 50 мл 96 % спир-
ту Р і титрують 0.1 М розчином натрію гідроксиду по-
тенціометрично (2.2.20). У розрахунок беруть об’єм
титранту між двома стрибками потенціалів на кривій
титрування.
1 мл 0.1 М розчину натрію гідроксиду відповідає
35.53 мг С17Н20С12М28.
ЗБЕРІГАННЯ
У повітронепроникному контейнері, у захищеному від
світла місці.
ЛІ Л
А ґЬЛПАЛ А ігґЛПРЯ V1гРАЇI^IЛ 9ПП1
Хлорпромазину гідрохлорид
______________________________________N
АМІНАЗИН
Атіпсщпит
Роботу із субстанцією треба проводити під тягою, у
гумових рукавичках. Після закінчення роботи руки
необхідно вимити холодною, злегка підкисленою водою,
без мила.
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
пгрткдннд <Т>ДРМДКОПРЯ УКРАЇНИ 2001
475
Цефалексин
ц
ЦЕФАЛЕКСИН
СеГаїехіпит
СЕЕАЕЕХІП
С16Ні7^О48,Н2О
М.м. 365.4
Цефалексин містить не менше 95.0 % і не більше
101.0 % (6А,77?)-7-[(/?)-2-аміно-2-фенілацетамідо]-3-
метил-8-оксо-5-тіа-1 -азабіцикло[4.2.0]окт-2-ен-2-
карбонової кислоти, у перерахунку на безводну речо-
вину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Розчиняється у воді Р (близько 1 г у
100 мл води), практично не розчинний у 96 % спирті Р
і ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А.
Друга ідентифікація: В, С.
А. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідати спектру ФСЗ цефалексину.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель нр254 силанізований Р.
Випробовуваний розчин. 20 мг субстанції розчиняють у
5.0 мл суміші рівних об’ємів метанолу Рі 0.067 М фо-
сфатного буферного розчину рН 7.0 Р.
Розчин порівняння (а). 20 мг ФСЗ цефалексину розчи-
няють у 5.0 мл суміші рівних об’ємів метанолу Р і
0.067 М фосфатного буферного розчину рН 7.0 Р.
Розчин порівняння (Ь). 20 мг ФСЗ цефалексину і 20 мг
ФСЗ цефрадину розчиняють у 5.0 мл суміші рівних
об’ємів метанолу Р і 0.067 М фосфатного буферного
розчину рН 7.0 Р.
На лінію старту хроматографічної пластинки наносять
1 мкл (4 мкг) випробовуваного розчину, 1 мкл (4 мкг)
розчину порівняння (а), І мкл (4 мкг цефалексину і
4 мкг цефрадину) розчину порівняння (Ь). Пластинку
поміщають у камеру із сумішшю розчинників аце-
тон Р - розчин 154 г/л амонію ацетату Р, рН якого
попередньо доведено до 6.2 кислотою оцтовою Р,
(15:85). Коли фронт розчинників пройде 15 см від лінії
старту, пластинку виймають із камери, сушать і пере-
глядають в УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину має виявля-
тися основна пляма на рівні основної плями на хромато-
грамі розчину порівняння (а), відповідна їй за розміром.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
С. Близько 2 мг субстанції поміщають у пробірку зав-
вишки близько 150 мм і діаметром 15 мм і змочують
0.05 мл води Р. Потім додають 2 мл розчину формаль-
дегіду в кислоті сірчаній Рі перемішують обертальни-
ми рухами, одержаний розчин світло-жовтий. Пробір-
ку нагрівають у водяній бані протягом 1 хв; поступово
з’являється темно-жовте забарвлення
ВИПРОБУВАННЯ НАЧИСТОТУ
рН (2.2.3). Від 4.0 до 5.5. 50 мг субстанції розчиняють
у воді, вільній від вуглецю діоксиду, Р і доводять об’єм
розчину тим самим розчинником до 10 мл.
Питоме оптичне обертання (2.2.7). Від +149° до +158°.
у перерахунку на безводну речовину. 0.125 г субстанції
розчиняють у фталатному буферному розчині рИ 4.4 Р
і доводять об’єм розчину тим самим розчинником до
25.0 мл.
Оптична густина (2.2.25). 50 мг субстанції розчиняють у
воді Р і доводять об’єм розчину тим самим розчинни-
ком до 100.0 мл. Оптична густина одержаного розчину
за довжини хвилі 330 нм не має перевищувати 0.05.
2.0 мл розчину доводять водою Рдо об’єму 50.0 мл. Уль-
трафіолетовий спектр поглинання одержаного розчину
в області від 220 нм до 300 нм повинен мати максимум
за довжини хвилі 262 нм. Питомий показник поглинан-
ня в максимумі за довжини хвилі 262 нм має бути від
220 до 245, у перерахунку на безводну речовину.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
477
Цефалексин
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель с Р. Пластинку імпрегну-
ють розчином 5 % (об/об) тетрадекану Ру гексані Р.
Пластинку сушать до видалення запаху розчинника і
проводять хроматографування в тому ж напрямку, що
й імпрегнування.
Випробовуваний розчин. 0.25 г субстанції розчиняють у
кислоті хлористоводневій розведеній Р і доводять об’єм
розчину тією самою кислотою до 10 мл.
Розчин порівняння (а). 1 мл випробовуваного розчину
доводять кислотою хлористоводневою розведеною Рдо
об’єму 100 мл.
Розчин порівняння (Ь). 25 мг ФСЗ кислоти 7-амінодеза-
цетоксицефалоспоринової розчиняють у кислоті хло-
ристоводневій розведеній Р і доводять об’єм розчину
тією самою кислотою до 10 мл (розчин порівняння Ь').
1 мл одержаного розчину доводять кислотою хлорис-
товодневою розведеною Рдо об’єму 10 мл.
Розчин порівняння (с). 25 мг а-фенілгліцину Р розчиня-
ють у кислоті хлористоводневій розведеній Р і доводять
об’єм розчину тією самою кислотою до 10 мл (розчин
порівняння с'). 1 мл одержаного розчину доводять кис-
лотою хлористоводневою розведеною Р до об’єму
10 мл.
Розчин порівняння (б). 0.25 г субстанції розчиняють у
суміші 1 мл розчину порівняння Ь' і 1 мл розчину
порівняння с' і доводять об’єм розчину кислотою хло-
ристоводневою розведеною Рдо 10 мл.
Налінію старту хроматографічної пластинки наносять
5 мкл (125 мкг) випробовуваного розчину, 5 мкл
(1.25 мкг) розчину порівняння (а). 5 мкл (1.25 мкг)
розчину порівняння (Ь), 5 мкл (1.25 мкг) розчину
порівняння (с) і 5 мкл (125 мкг субстанції, 1.25 мкг
кислоти 7-амінодезацетоксицефалоспоринової і
1.25 мкг а-фенілгліцину) розчину порівняння (<і).
Пластинку поміщають у камеру із сумішшю розчин-
ників ацетон Р— розчин 72 г/л динатрію гідрофосфа-
ту Р — розчин 21 г/л кислоти лимонної Р (3:80:120).
Коли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери і сушать при темпера-
турі 90 °С протягом 3 хв. Гарячу пластинку обприску-
ють розчином 1 г/л нінгідрину Р в рухомій фазі,
нагрівають при температурі 90 °С протягом 15 хв і охо-
лоджують.
На хроматограмі випробовуваного розчину пляма,
відповідна кислоті 7-амінодезацетоксицефалоспори-
новій, не має бути інтенсивнішою за пляму на хрома-
тограмі розчину порівняння (Ь) (1.0 %); пляма,
відповідна а-фенілгліцину (положення якого визна-
чають порівнянням із хроматограмою розчину (сі)), не
має бути інтенсивнішою за пляму на хроматограмі
розчину порівняння (с) (1.0 %); будь-яка пляма, крім
основної і плям, відповідних кислоті 7-амінодезаце-
токсицефалоспориновій і а-фенілгліцину, не має бути
інтенсивнішою за основну пляму на хроматограмі
розчину порівняння (а) (1.0 %).
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (сі) виявляються
три чітко розділені плями.
Диметиланілін. Не більше 0.002 % (20 ррт). Визначен-
ня проводять методом газової хроматографії (2.2.28),
використовуючи нафталін Ряк внутрішній стандарт.
Розчин внутрішнього стандарту. 50.0 мг нафталіну Р
розчиняють у циклогексані Р і доводять об’єм розчину
тим самим розчинником до 50.0 мл. 5.0 мл одержано-
го розчину доводять циклогексаном Р до об’єму
100.0 мл.
Випробовуваний розчин. 1.00 г субстанції поміщають у
пробірку із притертою скляною пробкою, додають
5 мл 7 М розчину натрію гідроксиду і 1.0 мл розчину
внутрішнього стандарту. Пробірку закривають і
енергійно струшують протягом 1 хв. Вміст пробірки,
якщо необхідно, центрифугують і використовують
верхній шар.
Розчин порівняння. До 50.0 мг диметиланіліну Р дода-
ють 2 мл кислоти хлористоводневої Р і 20 мл води Р, пе-
ремішують до розчинення і доводять об’єм розчину
водою Р до 50.0 мл. 5.0 мл одержаного розчину дово-
дять водою Р до об’єму 250.0 мл. 1.0 мл одержаного
розчину поміщають у пробірку із притертою скляною
пробкою, додають 5 мл 7 Мрозчину натрію гідроксиду
і 1.0 мл розчину внутрішнього стандарту. Пробірку за-
кривають пробкою і енергійно струшують протягом
1 хв. Вміст пробірки, якщо необхідно, центрифугують
і використовують верхній шар.
Хроматографування проводять на газовому хромато-
графі з полуменево-іонізаційним детектором за таких
умов:
— колонка скляна розміром 2 м х 2 мм, заповнена
діатомітом силанізованим для газової хромато-
графії Р, з нанесеним 3 % (м/м) поліметилфеніл-
силоксаном Р;
— газ-носій азот для хроматографії Р;
— швидкість газу-носія ЗО мл/хв:
— температура колонки 120 °С;
— температура детектора і блока вводу проб 150 °С.
Поперемінно хроматографують 1 мкл випробовувано-
го розчину і 1 мкл розчину порівняння.
Вода (2.5.12). Від 4.0 % до 8.0 %. Визначення прово-
дять з 0.300 г субстанції напівмікрометодом.
Сульфатна зола (2.4.14). Не більше 0.2 %. Визначення
проводять з 1.0 г субстанції.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять методом рідинної хромато-
графії (2.2.29).
Випробовуваний розчин. 50.0 мг субстанції розчиняють
у воді Рі доводять об’єм розчину тим самим розчинни-
ком до 100.0 мл.
Розчин порівняння (а). 50.0 мг ФСЗ цефалексину розчи-
няють у воді Р і доводять об’єм розчину тим самим
розчинником до 100.0 мл.
Розчин порівняння (Ь). 10 мг ФСЗ цефалексину і 10 мг
ФСЗ цефрадину розчиняють у воді Р і доводять об’єм
розчину тим самим розчинником до об’єму 100.0 мл.
478
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Цефтриаксону натрієва сіль
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка розміром 0.25 м х 4.6 мм, заповнена
силікагелем октадецилсилільним для хромато-
графії Р з розміром часток 5 мкм або 10 мкм;
— рухома фаза: метанол Р - ацетонітрил Р - розчин
13.6 г/л калію дигідрофосфату Р - вода Р
(2:5:10:83);
— швидкість рухомої фази 1.5 мл/хв;
— детектування за довжини хвилі 254 нм;
— об’єм проби, що уводиться, 20 мкл.
Хроматографують розчин порівняння (Ь). Чутливість
системи регулюють таким чином, щоб висота піків
становила не менше 50 % шкали реєструючого при-
строю. Хроматографічна система вважається придат-
ною, якщо коефіцієнт розділення піків цефалексину і
цефрадину становить не менше 4. Якщо необхідно,
регулюють вміст ацетонітрилу в рухомій фазі.
Хроматографують розчин порівняння (а) шість разів.
Хроматографічна система вважається придатною, як-
що відносне стандартне відхилення для площі піка це-
фалексину не перевищує 1.0 %.
Поперемінно хроматографують випробовуваний роз-
чин і розчин порівняння (а). Розраховують вміст це-
фалексину, у відсотках.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці при температурі не більше 30 °С.
____________________________________________/V
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Рекомендується внести такі зміни в проведення
кількісного визначення.
Випробовуваний розчин. 100.0 мг субстанції розчиня-
ють у воді Р і доводять об’єм розчину тим самим роз-
чинником до 100.0 мл.
Розчин порівняння (а). 100.0 мг ФСЗ цефалексину роз-
чиняють у воді Р і доводять об’єм розчину тим самим
розчинником до 100.0 мл.
Хроматографують розчин порівняння (а) шість разів.
Хроматографічна система вважається придатною, як-
що відносне стандартне відхилення для площі піка це-
фалексину не перевищує 0.85 %.
ЦЕФТРИАКСОНУ НАТРІЄВА СІЛЬ
Сеїїгіахопит паїгісит
СЕЕТКІАХОПЕ 5()І)ПМ
СІ8Н,^а2О783,Зі/2Н2О
М. м. 662
Цефтриаксону натрієва сіль містить не менше 96.0 % і
не більше 102.0 % динатрію (2)-(6/?,7/?)-7-[2-(2-аміно-
1,3-тіазол-4-іл)-2-(метоксііміно)ацетамідо|-8-оксо-3-
|(2,5-дигідро-2-метил-6-оксидо-5-оксо-1,2,4-триа-
зин-3-іл)тіометил]-5-тіа-1-азабіцикло[4.2.0]окт-2-ен-
2-карбоксилату, у перерахунку на безводну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок майже білого або білого
з жовтавим відтінком кольору. Слабко гігроскопічний.
Розчинність. Дуже легко розчинний у воді Р, помірно
розчинний у метанолі Р, дуже мало розчинний в ета-
нолі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: В, С, О.
А. Інфрачервоний спектр поглинання субстанції
(2.2.24) має відповідати спектру ФСЗ цефтриаксону
натрієвої солі.
В. Визначення проводять методом тонкошарової хро-
матографії (2.2.27), використовуючи як тонкий шар
силікагель НГ254 силанізований Р.
Випробовуваний розчин. 20 мг субстанції розчиняють у
5 мл суміші рівних об’ємів метанолу Р і 0.067 М фос-
фатного буферного розчину рН 7.0 Р.
Розчин порівняння (а). 20 мг ФСЗ цефтриаксону
натрієвої солі розчиняють у 5 мл суміші рівних об’ємів
метанолу Р і 0.067 М фосфатного буферного розчину
рН 7.0 Р.
Розчин порівняння (Ь). 20 мг ФСЗ цефтриаксону
натрієвої солі і 20 мг ФСЗ цефрадину розчиняють у
5 мл суміші рівних об’ємів метанолу Р і 0.067 М фос-
фатного буферного розчину рН 7.0 Р.
На лінію старту хроматографічної пластинки наносять
1 мкл (4 мкг) випробовуваного розчину, 1 мкл (4 мкг)
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
479
Цефтриаксону натрієва сіль
розчину порівняння (а), 1 мкл (4 мкг цефтриаксону
натрієвої солі і 4 мкг цефрадину) розчину порівняння
(Ь). Пластинку поміщають у камеру із сумішшю роз-
чинників метилацетат Р-розчин 150 г/л амонію аце-
тату, рН якого попередньо доведено до 6.2 кислотою
оцтовою Р, (10:90). Коли фронт розчинників пройде
15 см віл лінії старту, пластинку виймають із камери,
сушать у струмені теплого повітря і переглядають в
УФ-світлі за довжини хвилі 254 нм.
На хроматограмі випробовуваного розчину має вияв-
лятися основна пляма на рівні основної плями на хро-
матограмі розчину порівняння (а), відповідна їй за
розміром.
Результати аналізу вважаються вірогідними, якщо на
хроматограмі розчину порівняння (Ь) виявляються дві
чітко розділені плями.
С. Близько 2 мг субстанції помішають у пробірку зав-
вишки близько 150 мм і діаметром 15 мм і змочують
0.05 мл води Р. Потім додають 2 мл розчину формаль-
дегіду в кислоті сірчаній Р і перемішують обертальни-
ми рухами; одержаний розчин має бути зеленувато-
жовтим. Пробірку нагрівають у водяній бані протягом
1 хв; поступово з’являється жовте забарвлення.
В. Субстанція дає реакцію (а) на натрій (2.3.1).
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.40 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 20.0 мл.
Прозорість розчину (2.2.1). 2 мл розчину 8 доводять во-
дою Р до об’єму 20 мл. Одержаний розчин має бути
прозорим.
Кольоровість розчину (2.2.2). Забарвлення розчину,
приготованого для випробування "Прозорість розчи-
ну", має бути не інтенсивнішим за еталон У5 або ВУ5.
рН (2.2.3). Від 6.0 до 8.0. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від -155° до -170°, у
перерахунку на безводну речовину. 0.250 г субстанції
розчиняють у воді Р і доводять об’єм розчину тим са-
мим розчинником до 25.0 мл.
Супровідні домішки. Визначення проводять методом
рідинної хроматографії (2.2.29) за умов, описаних у
розділі "Кількісне визначення".
Поперемінно хроматографують випробовуваний роз-
чин і розчин порівняння (с). Час хроматографування
має бути в 2 рази більше часу утримування основного
піка. На хроматограмі випробовуваного розчину пло-
ща будь-якого піка, крім основного, не має перевищу-
вати площу основного піка на хроматограмі розчину
порівняння (с) (1.0 %); сума площ усіх піків, крім ос-
новного, не має перевищувати 4 площі основного піка
на хроматограмі розчину порівняння (с) (4.0 %). Не
враховують піки, площа яких менше 10 % площі ос-
новного піка на хроматограмі розчину порівняння (с).
Вода (2.5.12). Від 8.0 % до 11.0 %. Визначення прово-
дять з 0.100 г субстанції напівмікрометодом.
Стерильність (2.6.1). Якщо субстанція призначена для
виробництва парентеральних лікарських засобів без
подальшої процедури стерилізації, вона має витриму-
вати випробування на стерильність.
Бактеріальні ендотоксини (2.6.14). Не більше 0.20 МО в
1 мг, якщо субстанція призначена для виробництва
парентеральних лікарських засобів без подальшої
процедури видалення бактеріальних ендотоксинів.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
Визначення проводять метолом рідинної хромато-
графії (2.2.29).
Випробовуваний розчин. 30.0 мг субстанції розчиняють
у рухомій фазі і доводять об’єм розчину тим самим
розчинником до 100.0 мл.
Розчин порівняння (а). 30.0 мг ФСЗ цефтриаксону
натрієвої солі розчиняють у рухомій фазі й доводять
об’єм розчину тим самим розчинником до 100.0 мл.
Розчин порівняння (Ь). 5.0 мг ФСЗ цефтриаксону
натрієвої солі і 5.0 мг ФСЗ домішки А цефтриаксону
розчиняють у рухомій фазі й доводять об’єм рухомою
фазою до 100.0 мл.
Розчин порівняння (с). 1.0 мл випробовуваного розчину
доводять рухомою фазою до об’єму 100.0 мл.
Хроматографування проводять на рідинному хрома-
тографі з УФ-детектором за таких умов:
— колонка з нержавіючої сталі розміром 0.25 м х 4.6 мм,
заповнена силікагелем октадецилсилільним для
хроматографії Р, з розміром часток 5 мкм;
— рухома фаза: 2.0 г тетрадециламонію броміду Р і
2.0 г тетрагептиламонію броміду Р розчиняють у
суміші 440 мл води Р, 55 мл 0.067 М фосфатного
буферного розчину рН 7.0 Р, 5.0 мл цитратного бу-
ферного розчину рН 5.0 і 500 мл ацетонітрилу Р.
Цитратний буферний розчин рН 5.0 готують та-
ким чином: 20.17 г кислоти лимонної Р розчиня-
ють у 800 мл води Р, доводять рН розчину до 5.0
розчином натрію гідроксиду концентрованим Р
і доводять об’єм одержаного розчину водою Р до
1000.0 мл.
— швидкість рухомої фази 1.5 мл/хв:
— детектування за довжини хвилі 254 нм;
— об’єм проби, що уводиться, 20 мкл.
Хроматографують розчин порівняння (Ь). Чутливість
системи регулюють таким чином, щоб висота піків
становила не менше 50 % шкали реєструючого прист-
рою. Хроматографічна система вважається придат-
ною, якшо коефіцієнт розділення двох основних піків
становить не менше 3.0.
Хроматографують розчин порівняння (а) шість разів.
Хроматографічна система вважається придатною, як-
480
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Циклофосфамід
що відносне стандартне відхилення для плоші піка
цефтриаксону не перевищує 1.0 %.
Поперемінно хроматографують випробовуваний роз-
чин і розчин порівняння (а). Розраховують вміст цеф-
триаксону натрієвої солі у відсотках.
МАРКУВАННЯ
При проведенні випробування "Пірогени" замість
"—субстанція вільна від бактеріальних ендотоксинів"
зазначають:
— субстанція апірогенна.
ЗБЕРІГАННЯ
У щільно закупореному контейнері, у захищеному від
світла місці при температурі не вище ЗО °С. Якщо суб-
станція стерильна, зберігають у стерильному, повітро-
непроникному контейнері з контролем першого
розкриття.
ЦИКЛОФОСФАМІД
Сус1ор1іо8р1іатіс1ит
МАРКУВАННЯ
У необхідних випадках зазначають:
— субстанція стерильна;
— субстанція вільна від бактеріальних ендотоксинів.
ДОМІШКИ
А. динатрій (£)-(6/?,7/?)-7-[(2-аміно-1,3-тіазол-4-
іл)(метоксііміно)ацетамідо]-3-[[2,5-дигідро-2-метил-
6-оксидо-5-оксо-1,2,4-триазин-3-іл)тіо]метил]-8-ок-
со-5-тіа-1-азабіцикло[4.2.0]окт-2-ен-2-карбоксилат
(Е-ізомер),
В. (7)-2-(2-аміно-1,3-тіазол-4-іл)-А-[(5аЕ,6Е)-1,7-ді-
оксо-1,4,6,7-тетрагідро-ЗЯ,5а//-азето(2,1 -й]фуро[3,4-
^][1,3|тіазин-6-іл]-2-(метоксііміно)апетамід,
С. 6-гідрокси-2-метил-3-меркапто-1,2,4-триазин-
5(2Е)-он,
Б. 5-( 1,3-бензотіазол-2-іл)(2)-(2-аміно-1,3-тіазол-4-
іл) (метокс і і м і н о )тіоацетат,
Е. (6А,7/?)-7-аміно-3-[[2,5-дигідро-6-гідрокси-2-ме-
тил-5-оксо-1,2,4-триазин-3-іл)тіо]метил]-8-оксо-5-
тіа-1 -азабіцикло[4.2.0]окт-2-ен-2-карбоксильна кис-
лота.
________________________________________________N
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Замість випробування "Бактеріальні ендотоксини " до-
пускається застосування випробування "Пірогени"
(2.6.8).
Пірогени (2.6.8). Якщо субстанція призначена для ви-
робництва парентеральних лікарських засобів, вона
має витримувати випробування на пірогени. Уволять
на 1 кг маси кролика 1 мл розчину, що містить 40 мг
цефтриаксону в 1 мл води для ін ’єкцііл Р.
Аномальна токсичність (2.6.9). Якщо субстанція при-
значена для виробництва парентеральних лікарських
засобів, вона має витримувати випробування на ано-
мальну токсичність.
СїСЬОРПО8РНАМЮЕ
^°^р^СН2-СН2-СІ
І гмн "'СНг-СНг-СІ
, н2о
С7Н15СІ2М2О2Р,Н2О М.м. 279.1
Циклофосфамід містить не менше 98.0 % і не більше
102.0 % (Е5)-2-[біс(2-хлоретил)аміно]тетрагідро-2//-
1,3,2-оксазафосфорин 2-оксиду, у перерахунку на без-
водну речовину.
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого або майже білого
кольору.
Розчинність. Розчинний у воді Р, легко розчинний у
96 % спирті Р, мало розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: В.
Друга ідентифікація: А, С, Ц.
А. Визначають температуру плавлення (2.2.14) суб-
станції. Змішують рівні кількості субстанції і ФСЗ
циклофосфаміду і визначають температуру плавлення
суміші. Температури плавлення субстанції і суміші
(близько 51 °С) не мають відрізнятися більш як
на 2 °С.
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції має відповідані спектру ФСЗ циклофосфаміду.
С. На хроматограмі випробовуваного розчину (Ь),
одержаній при випробуванні "Супровідні домішки",
має виявлятися основна пляма на рівні основної пля-
ми на хроматограмі розчину порівняння (а), відпо-
відна їй за розміром і забарвленням.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
481
Циклофосфамід
О. 0.1 г субстанції розчиняють у 10 мл води Рі додають
5 мл розчину срібла нітрату Р; розчин залишається
прозорим. Одержаний розчин кип’ятять, утворюється
білий осад, що розчиняється в розчині аміаку концент-
рованому Р і знову утворюється при додаванні кислоти
азотної розведеної Р.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 0.50 г субстанції розчиняють у воді, вільній
від вуглецю діоксиду, Р і доводять об’єм розчину тим
самим розчинником до 25.0 мл.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Забарвлення
розчину 8 має бути не інтенсивнішим за еталон У6.
рН (2.2.3). Від 4.0 до 6.0. Вимірюють рН свіжопригото-
ваного розчину 8.
Супровідні домішки. Визначення проводять методом
тонкошарової хроматографії (2.2.27), використовую-
чи як тонкий шар силікагель с Р.
Випробовуваний розчин (а). 0.10 г субстанції розчиня-
ють у 96 % спирті Р і доводять об’єм розчину тим са-
мим розчинником до 5 мл.
Випробовуваний розчин (Ь). 1 мл випробовуваного роз-
чину (а) доводять 96 % спиртом Рао об’єму 10 мл.
Розчин порівняння (а). 10 мг ФСЗ циклофосфаміду роз-
чиняють у 96 % спирті Рі доводять об’єм розчину тим
самим розчинником до 5 мл.
Розчин порівняння (Ь). 0.1 мл випробовуваного розчи-
ну (а) доводять 96 % спиртом Рдо об’єму 10 мл.
На лінію старту хроматографічної пластинки наносять
10 мкл (200 мкг) випробовуваного розчину (а), 10 мкл
(20 мкг) випробовуваного розчину (Ь), 10 мкл (20 мкг)
розчину порівняння (а) і 10 мкл (2 мкг) розчину
порівняння (Ь). Пластинку поміщають у камеру із
сумішшю розчинників кислота мурашина безводна Р -
ацетон Р- вода Р - метилетилкетон Р (2:4:12:80). Ко-
ли фронт розчинників пройде 15 см від лінії старту,
пластинку виймають із камери, сушать у струмені теп-
лого повітря і нагрівають при температурі 110 °С про-
тягом 10 хв.
На дно хроматографічної камери поміщають випарю-
вальну чашку, що містить розчин 50 г/л калію перман-
ганату Р, і додають рівний об’єм кислоти хлористо-
водневої Р. Гарячу пластинку поміщають у камеру і ви-
тримують у парі хлору протягом 2 хв. Потім пластинку
виймають із камери і витримують у струмені холодно-
го повітря до зникнення надлишку хлору доти, доки
крапля розчину крохмалю із калію йодидом Р, нанесена
нижче лінії старту, буде давати ледве помітне блакитне
забарвлення. Уникають тривалого перебування в
струмені холодного повітря. Пластинку обприскують
розчином крохмалю із калію йодидом Р і витримують
протягом 5 хв для проявлення плям.
На хроматограмі випробовуваного розчину (а) будь-
яка пляма, крім основної, не має бути інтенсивнішою
за пляму на хроматограмі розчину порівняння (Ь)
(1.0 %). Не враховують пляму на лінії старту.
Хлориди (2.4.4). Не більше 0.033 % (330 ррт). 0.15 г
субстанції розчиняють у воді Р і доводять об’єм розчи-
ну тим самим розчинником до 15 мл. Свіжоприготова-
ний розчин має витримувати випробування на хлори-
ди.
Фосфати (2.4.11). Не більше 0.01 % (100 ррт). 0.10 г
субстанції розчиняють у воді Р і доводять об’єм розчи-
ну тим самим розчинником до 100 мл. Одержаний
розчин має витримувати випробування на фосфати.
Важкі метали (2.4.8, метод С). Не більше 0.002 %
(20 ррт). 1.0 г субстанції має витримувати випробу-
вання на важкі метали. Еталон готують із використан-
ням 2 мл еталонного розчину свинцю (10ррт РЬ) Р.
Вода (2.5.12). Від 6.0 % до 7.0 %. Визначення прово-
дять з 0.300 г субстанції напівмікрометодом.
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 50 мл розчину 1 г/л
натрію гідроксиду Р в етиленгліколі Р і кип’ятять зі
зворотним холодильником протягом 30 хв. Розчин
охолоджують, обполіскують холодильник 25 мл во-
ди Р, додають 75 мл 2-пропанолу Р, 15 мл кислоти
азотної розведеної Р, 10.0 мл 0.1 М розчину срібла
нітрату і 2.0 мл розчину заліза(ІІІ) амонію сульфа-
ту Р2 і титрують 0.1 М розчином амонію тіоціанату.
1 мл 0.1 М розчину срібла нітрату відповідає 13.05 мг
ЗБЕРІГАННЯ
У щільно закупореному контейнері.
__________________________________________N
ЦИКЛОФОСФАН
Сусіоркозркап ит
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
482
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Цистеїн
ЦИСТЕЇН*
Сузїеіпит
СЕ8ТЕШЕ
о
нз'х'х'^хон
н' N42
С3П7ХО28 М.м. 121.2
Цистеїн містить не менше 98.0 % і не більше 101.0 %
(/?)-2-аміно-3-меркаптопропанової кислоти, у пере-
рахунку на суху речовину.
ВИРОБНИЦТВО
Якщо субстанція одержана в результаті процесу, шо
включає стадії ферментації, вона має витримувати ви-
моги статті "Продукти ферментації".
ВЛАСТИВОСТІ
Опис. Кристалічний порошок білого кольору або без-
барвні кристали з характерним запахом.
Розчинність. Легко розчинний у воді Р і 96 % спирті Р,
практично не розчинний в ефірі Р.
ІДЕНТИФІКАЦІЯ
Перша ідентифікація: А, В.
Друга ідентифікація: А, С, В, Е.
А. Субстанція має відповідати вимогам щодо питомо-
го оптичного обертання, зазначеним у розділі "Випро-
бування на чистоту".
В. Інфрачервоний спектр поглинання (2.2.24) суб-
станції, одержаний у дисках, має відповідати спектру
ФСЗ цистеїну.
С. Близько 10 мг субстанції розчиняють у 2 мл води Р,
додають 0.1 М розчин кислоти хлористоводневої до рН
близько 4.0, 0.5 мл розчину 2.5 г/л нінгідрину Р,
нагрівають на водяній бані протягом 10 хв; з’яв-
ляється жовте або жовто-коричневе забарвлення.
Одержаний розчин охолоджують до кімнатної темпе-
ратури, додають краплинами від 1 мл до 2 мл 0.1 М
розчину натрію гідроксиду; з’являється червоно-фіоле-
тове забарвлення.
В. Близько 5 мг субстанції розчиняють у 5 мл води Р,
додають 2 мл розчину натрію гідроксиду розведеного Р і
0.2 мл розчину 25 г/л натрію нітропрусиду Р; з’яв-
ляється жовте забарвлення, що протягом 2 хв перехо-
дить у червоне.
Е. 0.10 г субстанції обережно змішують з 1 мл розчину
водню пероксиду концентрованого Р, 0.1 мл розчину
заліза(ІП) хлориду РІ і охолоджують. До одержаного
розчину додають 1 мл кислоти хлористоводневої розве-
деної Р, 5 мл води Р, 1 мл розчину барію хлориду РІ; про-
тягом 3 хв з’являється біла каламуть або осад.
ВИПРОБУВАННЯ НАЧИСТОТУ
Розчин 8. 2.5 г субстанції розчиняють у воді, вільній від
вуглецю діоксиду, Р і доводять об’єм розчину тим са-
мим розчинником до 100 мл. Розчин використовують
свіжоприготованим.
Прозорість розчину (2.2.1). Розчин 8 має бути прозорим.
Кольоровість розчину (2.2.2, метод II). Розчин 8 має
бути безбарвним.
рН (2.2.3). Від 4.5 до 6.0. Вимірюють рН розчину 8.
Питоме оптичне обертання (2.2.7). Від +8.0° до +9.5°, у
перерахунку на суху речовину. 3.00 г субстанції розчи-
няють у розчині 220 г/л кислоти хлористоводневої Р і
доводять об’єм розчину тією самою кислотою до
25.0 мл.
Речовини, виявлювані нінгідрином. Визначення прово-
дять методом тонкошарової хроматографії (2.2.27), ви-
користовуючи ТШХ пластинки із шаром силікагелю Р.
Випробовуваний розчин. 0.10 г субстанції розчиняють у
6 мл розчину 20 г/л 61-етилмалеїміду Р. Одержаний
розчин використовують протягом 15 хв.
Розчин порівняння. 1 мл випробовуваного розчину до-
водять водою Рдо об’єму 100 мл.
На лінію старту хроматографічної пластинки наносять
5 мкл (83 мкг) випробовуваного розчину і 5 мкл
(0.83 мкг) розчину порівняння. Пластинку сушать на
повітрі й поміщають у камеру із сумішшю розчин-
ників вода Р— кислота оцтова льодяна Р— бутанол Р
(25:25:50). Коли фронт розчинників пройде 10 см від
лінії старту, пластинку виймають із камери, сушать
при температурі від 100 °С до 105 °С протягом 15 хв і
обприскують розчином нінгідрину Р. Пластинку
нагрівають при температурі від 100 °С до 105°С протя-
гом 10 хв.
На хроматограмі випробовуваного розчину будь-яка
пляма, крім основної, не має бути інтенсивнішою за
пляму на хроматограмі розчину порівняння (1.0 %).
Залишкові кількості органічних розчинників. Суб-
станція має витримувати вимоги статті (5.4).
Хлориди (2.4.4). Не більше 0.04 % (400 ррт). 5 мл роз-
чину 8 нагрівають до 60 "С і обережно, краплями,
змішують із 5 мл розчину водню пероксиду концентро-
ваного Р, кип’ятять протягом 1 год, охолоджують і до-
водять об’єм розчину водою Р до 15 мл. Одержаний
розчин має витримувати випробування на хлориди.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
483
Цистеїн"
Сульфати (2.4.13). Не більше 0.03 % (300 ррт). 0.50 г
субстанції розчиняють у 15 мл води дистильованої Р.
Одержаний розчин має витримувати випробування на
сульфати.
Амонію солі. Не більше 0.02 % (200 ррт). Підготовля-
ють комірку, що складається із двох годинникових
стекол діаметром 60 мм, сполучених по краях. На
внутрішню поверхню верхнього годинникового скла
поміщають шматочок червоного лакмусового паперу Р
розміром (5 х 5) мм, змочений декількома краплями
води Р. 50 мг здрібненої субстанції поміщають на
нижнє годинникове скло і розчиняють у 0.5 мл води Р.
До розчину додають 0.30 г магнію оксиду важкого Р і
ретельно розтирають скляною паличкою. Нижнє го-
динникове скло відразу накривають верхнім і нагріва-
ють при температурі 40°С протягом 15 хв. Паралельно
в аналогічних умовах готують еталон, використовую-
чи 0.1 мл еталонного розчину амонію (100 ррт N11 4) Р,
0.5 мл води Р і 0.30 г магнію оксиду важкого Р. Лакму-
совий папір над випробовуваним розчином має бути
забарвлений у синій колір не інтенсивніше, ніж над
еталоном.
Залізо (2.4.9). Не більше 0.002 % (20 ррт). 0.5 г суб-
станції в ділильній лійці розчиняють у 10 мл кислоти
хлористоводневої розведеної Рї витягають три рази ме-
тилізобутилкетоном РІ, порціями по 10 мл, струшую-
чи щоразу протягом 3 хв. До об’єднаних органічних
витягів додають 10 мл води Р і струшують протягом
З хв. Одержаний водний розчин має витримувати ви-
пробування на залізо.
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт). 2.0 г субстанції розчиняють у воді Р і дово-
дять об’єм розчину тим самим розчинником до 20 мл
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують із викорис-
танням еталонного розчину свинцю (1 ррт РЬ) Р.
Втрата в масі при висушуванні (2.2.32). Не більше 0.5 %.
1.000 г субстанції сушать при температурі від 100 °С до
105 °С.
Сульфатна зола (2.4.14). Не більше 0.1 %. Визначення
проводять із 1.0 г субстанції.
Пірогени або бактеріальні ендотоксини. Якщо суб-
станція призначена для виробництва парентеральних
лікарських засобів без подальшої процедури видален-
ня пірогенів, вона має витримувати випробування
"Пірогени"(2.6.8) або "Бактеріальні ендотоксини"
(2.6.14).
КІЛЬКІСНЕ ВИЗНАЧЕННЯ
0.100 г субстанції розчиняють у 10.0 мл кислоти хлорис-
товодневої розведеної Р, доводять об’єм розчину водою
Р до 50 мл, охолоджують у льодяній бані, додають 2 г
калію йодиду Р ї 15.0 мл 0.05 Мрозчину йоду. Реакційну
посудину закривають, витримують протягом 15 хв у за-
хищеному від світла місці і титрують 0.1 М розчином
натрію тіосульфату до зникнення забарвлення, вико-
ристовуючи наприкінці титрування 1 мл розчину крох-
малю Р.
Паралельно проводять контрольний дослід.
1 мл 0.05 М розчину йоду відповідає 12.12 мг
С3Н7МО28.
ЗБЕРІГАННЯ
У шільно закупореному контейнері, у захищеному від
світла місці.
484
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
ЗАГАЛЬНІ СТАТТІ
НА ЛІКАРСЬКІ ФОРМИ
ТА СУБСТАНЦІЇ
ВУШНІ ЛІКАРСЬКІ ЗАСОБИ
Аигісиїагіа
ВИЗНАЧЕННЯ
Вушні лікарські засоби являють собою рідкі, м'які або
тверді лікарські засоби, призначені для закапування,
розпилення, вдування або аплікації у слуховий отвір
або для промивання вуха.
Вушні лікарські засоби звичайно містять одну або
більше діючих речовин у підхожому розчиннику. Вони
можуть містити допоміжні речовини, наприклад, для
регулювання тонічності або в’язкості, створення або
стабілізації необхідного значення рН, збільшення роз-
чинності діючих речовин, забезпечення стабільності
або надання відповідних антимікробних властивос-
тей. Допоміжні речовини у використовуваних кон-
центраціях не мають негативно впливати на дію
лікарського засобу і справляти токсичну або небажану
місцеву подразливу дію.
Лікарські засоби, які застосовуються при пораненнях
вуха, особливо при ушкодженні барабанної перетинки
або перед хірургічними операціями, мають бути сте-
рильними, не мають містити антимікробних консер-
вантів і мають постачатися в однодозових контейне-
рах.
Якщо немає інших зазначень, водні вушні лікарські
засоби випускають у багатодозових контейнерах, які
містять підхожий антимікробний консервант у не-
обхідній концентрації, за винятком лікарських за-
собів, які виявляють достатню антимікробну дію.
Контейнери для вушних лікарських засобів мають
відповідати вимогам статеіі "Матеріали, використову-
вані для виробництва контейнерів" (3.1 та підрозділи,)
та "Контейнери"(3 2та підрозділи/
Вушні лікарські засоби можуть бути класифіковані як:
— вушні краплі й аерозолі;
— вушні м’які лікарські засоби;
— вушні порошки;
— вушні промивки;
— вушні тампони.
ВИРОБНИЦТВО
При розробці вушних лікарських засобів, до складу
яких входять антимікробні консерванти, уповноваже-
ному органу мають бути подані дані, шо підтверджу-
ють ефективність вибраних консервантів. Метод ви-
значення і критерії ефективності консервантів мають
відповідати вимогам статті "Ефективність ан-
тимікробних консервантів " (5.1.3).
При виробництві, пакуванні, зберіганні та реалізації
вушних лікарських засобів мають бути вжиті від-
повідні заходи, які забезпечують необхідну мік-
робіологічну чистоту відповідно до вимог статті
"Мікробіологічна чистота лікарських засобів " (5.1.4).
Стерильні вушні лікарські засоби виготовляють з ви-
користанням матеріалів і методів, які забезпечують
стерильність, запобігають забрудненню лікарських за-
собів і розвитку мікроорганізмів, відповідно до вимог
статті "Методи приготування стерильних продуктів"
(5.1.1).
При виробництві вушних лікарських засобів, які
містять дисперговані частки, треба передбачити захо-
ди, що забезпечують необхідний розмір часток та його
контроль.
ВИПРОБУВАННЯ
Стерильність (2.6.1). Якщо на етикетці зазначено, що
препарат стерильний, він має витримувати випробу-
вання на стерильність.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах. Якщо препарат
стерильний, його зберігають у стерильних повітроне-
проникних контейнерах з контролем першого роз-
криття.
МАРКУВАННЯ
На етикетці зазначають:
— назву кожного антимікробного консерванта;
— "стерильно" (якщо необхідно).
Вушні краплі й аерозолі
ВИЗНАЧЕННЯ
Вушні краплі й аерозолі являють собою розчини,
емульсії або суспензії, які містять одну або більше
діючих речовин у підхожих рідинах (наприклад, вода,
гліколі або жирні масла), призначені для введення у
слуховий отвір без виявлення небезпечного тиску на
барабанну перетинку. Вони також можуть бути введе-
ними у слуховий отвір за допомогою турунди, просо-
ченої лікарським засобом.
Емульсії можуть розшаровуватися, однак при збовту-
ванні мають легко перетворюватися на емульсію. Сус-
пензії можуть утворювати осад, що має швидко ресу-
спендуватися при збовтуванні, утворюючи суспензію,
досить стабільну, щоб забезпечити необхідну дозу при
введенні.
Вушні краплі і аерозолі звичайно випускають у багато-
дозових контейнерах, споряджених підхожою насад-
кою. Якшо аерозолі випускають у контейнерах підти-
ском, вони мають відповідати вимогам статті "Лі-
карські засоби, що знаходяться під тиском ".
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
487
Гранули
Вушні м’які лікарські засоби
ВИЗНАЧЕННЯ
Вушні м’які лікарські засоби призначені для введення
у зовнішній слуховий отвір. Якщо необхідно, заклада-
ють або вводять турунду, просочену лікарським засо-
бом.
Вушні м’які лікарські засоби мають відповідати вимо-
гам статті "М’які лікарські засоби для місцевого засто-
сування
їх випускають у контейнерах, споряджених підхожою
насадкою.
Вушні порошки
ВИЗНАЧЕННЯ
Вушні порошки мають відповідати вимогам статті
"Порошки для зовнішнього застосування
їх випускають у контейнерах, споряджених підхожою
насадкою для нанесення або вдування.
Вушні промивки
ВИЗНАЧЕННЯ
Вушні промивки являють собою лікарські засоби,
призначені для очищення зовнішнього слухового от-
вору. Вони звичайно являють собою водні розчини із
значенням рН. відповідним фізіологічним межам.
Вушні тампони
ВИЗНАЧЕННЯ
Вушні тампони призначені для введення у зовнішній
слуховий отвір. Вони мають відповідати вимогам
статті "Тампони медичні
_______________________________________N
Вушні краплі
ВИПРОБУВАННЯ
Вушні краплі, що являють собою розчини, звичайно
контролюють за такими показниками якості: опис,
ідентифікація, прозорість, кольоровість, рН (крім не-
водних і масляних розчинів), супровідні домішки,
об’єм вмісту контейнера, мікробіологічна чистота або
стерильність, кількісне визначення.
Для вушних крапель у вигляді суспензій додатково
контролюють розмір часток і стійкість суспензії.
Для вушних крапель, що являють собою масляні роз-
чини, додатково контролюють кислотне і перекисне
числа.
Для вушних крапель, що містять речовини, які забез-
печують в’язкість, додатково контролюють в’язкість.
Кількісне визначення. Проводять визначення діючих
речовин, антимікробних консервантів, неводних роз-
чинників та інших речовин, зазначених в окремій
статті. Вміст визначуваних речовин виражають у гра-
мах, міліграмах або одиницях дії (ОД) в 1 мл препара-
ту. Вміст діючих речовин має бути від 90 % до 110 % від
вмісту, зазначеного у розділі "Склад”, якщо немає
інших зазначень в окремій статті
ЗБЕРІГАННЯ
У щільно закупорених контейнерах, що забезпечують
стабільність і зручність дозування препарату при за-
стосуванні.
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву й концентрацію діючих речовин і антимік-
робних консервантів;
— необхідний перелік допоміжних речовин;
— умови зберігання та термін придатності;
— умови і термін зберігання після розкриття
ГРАНУЛИ
Огапиіа
Вимоги до гранул, що використовуються для приготу-
вання розчинів або суспензій для орального застосуван-
ня, наведені у статті "Рідкі лікарські засоби для ораль-
ного застосування Вимоги даної статті не поширю-
ються на гранули для ветеринарного застосування, як-
що немає інших зазначень в окремій статті.
ВИЗНАЧЕННЯ
Гранули — лікарська форма, що складається з твердих
сухих, досить міцних агрегатів часток порошку. Грану-
ли призначені для приймання всередину: для ковтан-
ня, розжовування, розчинення, диспергування у воді
або в іншій підхожій рідині перед застосуванням.
Гранули містять одну або більше діючих речовин з на-
повнювачами чи без них. При необхідності викорис-
товують барвники і ароматизуючі добавки, дозволені
до медичного застосування.
Гранули випускають в однодозових або багатодозових
контейнерах. Кожну дозу гранул із багатодозового
контейнера одержують за допомогою відповідного
пристрою для відмірювання запропонованої кіль-
488
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Гранули
кості. При однодозовому фасуванні кожну дозу паку-
ють в індивідуальний контейнер, наприклад, пакетик,
паперовий пакет або банку.
Контейнери для гранул мають відповідати вимогам
статей "Матеріали, що використовуються для вироб-
ництва контейнерів"(3.1 та підрозділи,) та "Контейне-
ри" (3.2та підрозділи^, якщо немає інших зазначень в
окремій статті.
Гранули можуть бути класифіковані як:
— гранули "шипучі";
— гранули, покриті оболонкою;
— гранули кишково-розчинні;
— гранули з модифікованим вивільненням.
ВИРОБНИЦТВО
При виробництві, пакуванні, зберіганні й реалізації
гранул мають бути вжиті відповідні заходи, які забез-
печують необхідну мікробіологічну чистоту відпо-
відно до вимог статті "Мікробіологічна чистота лі-
карських засобів " (5.1.4).
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Гранули в однодозових
контейнерах з вмістом діючої речовини менше 2 мг
або менше 2 % від загальної маси мають витримувати
випробування однорідності вмісту діючої речовини в
одиниці дозованих лікарських засобів (тест В), якщо
немає інших зазначень в окремій статті. Для гранул,
що містять більше однієї діючої речовини, вимоги по-
ширюються лише на ті речовини, вміст яких
відповідає умовам, викладеним вище. Дане випробу-
вання не поширюється на полівітамінні препарати і
препарати, що містять мікроелементи.
Однорідність маси (2.9.5). Гранули в однодозових кон-
тейнерах, за винятком гранул, покритих оболонкою,
мають витримувати випробування однорідності маси
для одиниці дозованого лікарського засобу. Випробу-
вання на однорідність маси не вимагається, якщо ви-
пробування однорідності вмісту передбачено для всіх
діючих речовин.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах або, якщо препа-
рат містить леткі речовини чи вміст необхідно захис-
тити. — у повітронепроникних контейнерах.
Гранули "шипучі"
ВИЗНАЧЕННЯ
Гранули "шипучі" — гранули без оболонки, що голов-
ним чином містять кислоти і карбонати або гідрокар-
бонати. які швидко реагують у присутності води з
виділенням вуглекислого газу Гранули призначені для
розчинення або диспергування у воді перед застосу-
ванням.
ВИПРОБУВАННЯ
Розпадання. Помішають одну дозу гранул "шипучих" у
склянку з 200 мл води Р при температурі від 15 °С до
25 °С; виділяються численні бульбашки газу. Гранули
вважають такими, що розпалися, якщо після припи-
нення виділення газу вони або розчинилися, або дис-
пергувалися у воді. Повторюють процедуру на п’яти
інших дозах. Гранули витримують випробування, як-
що кожна з шести доз розпадається протягом не біль-
ше 5 хв.
ЗБЕРІГАННЯ
У повітронепроникних контейнерах.
Гранули, покриті оболонкою
ВИЗНАЧЕННЯ
Гранули, покриті оболонкою, — це лікарські засоби в
багатолезових контейнерах, які звичайно складаються
з гранул, покритих одним або кількома шарами із
суміші різних допоміжних речовин.
ВИРОБНИЦТВО
Речовини, які використовують для одержання обо-
лонки, звичайно наносять у вигляді розчину або сус-
пензії в умовах, у яких відбувається випаровування
розчинника.
ВИПРОБУВАННЯ
Розчинення. Випробування можна здійснити для
підтвердження відповідного вивільнення діючої речо-
вини або речовин, наприклад, одним із способів, опи-
саних у статті "Тест "Розчинення"для твердих дозова-
них форм " (2.9.3).
Гранули кишково-розчинні
ВИЗНАЧЕННЯ
Гранули кишково-розчинні — це гранули з моди-
фікованим вивільненням, стійкі до шлункового соку і
здатні вивільняти діючі речовини в кишковому соку.
Це досягається покриттям гранул матеріалом, стійким
до шлункового соку, або іншими підхожими способа-
ми.
ВИРОБНИЦТВО
Проводять випробування, яке підтверджує, шо техно-
логія забезпечує необхідне вивільнення діючої речо-
вини або речовин.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
489
Екстракти
Гранули з модифікованим
вивільненням
ВИЗНАЧЕННЯ
Гранули з модифікованим вивільненням — це грану-
ли, покриті оболонкою або без оболонки, одержані з
використанням спеціальних допоміжних речовин або
спеціальних способів, які. окремо чи разом, призна-
чені для регулювання швидкості вивільнення або міс-
ця, де вивільняються діюча речовина або речовини.
ВИРОБНИЦТВО
Проводять випробування, яке підтверджує, що техно-
логія забезпечує необхідне вивільнення діючої речо-
вини або речовин.
________________________________________N
Гранули контролюють за такими показниками якості:
опис, ідентифікація, розмір гранул, втрата в масі при
висушуванні, тальк і аеросил, рН, розпадання, су-
провідні домішки, розчинення, однорідність маси або
однорідність вмісту діючої речовини для гранул в од-
нодозових контейнерах, маса вмісту контейнера для
гранул у багатолезових контейнерах, мікробіологічна
чистота, кількісне визначення.
ВИПРОБУВАННЯ
Розпадання (2.9.]). Визначення проводять з наважки
0.5 г із використанням сітки з розміром отворів 0.5 мм;
гранули мають розпадатися протягом не більше 15 хв,
якщо немає інших зазначень в окремій статті.
Розмір гранул (2.9.12). Визначення проводять
відповідно до вимог статті "Ситовий аналіз". Розмір
гранул має відповідати вимогам, зазначеним в окремій
статті Звичайно він знаходиться у межах від 0.2 мм до
3.0 мм.
Тальк, аеросил. Визначення проводять відповідно до
вимог статті "Таблетки". Вміст тальку та аеросилу не
має перевищувати вимог, зазначених в окремій статті.
Розчинення. Визначення проводять відповідно до ви-
мог статті "Тест "Розчинення" для твердих дозованих
форм"(2.9.3). Для гранул у багатодозових контейнерах
для випробування відбирають шість проб, які містять
одну дозу в кожній пробі.
Якщо проводять випробування за показником
"Розчинення", випробування "Розпадання" не вима-
гається.
Однорідність вмісту. Гранули мають витримувати ви-
моги статті "Однорідність вмісту діючої речовини в оди-
ниці дозованого лікарського засобу" (2.9.6), якщо немає
інших зазначень в окремій статті.
Кількісне визначення. Для визначення вмісту діючих
речовин у гранулах беруть наважку не менш як із 10 г
розтертих гранул, якщо немає інших зазначень в ок-
ремій статті.
Вміст визначуваних речовин виражають у грамах,
міліграмах або в одиницях дії (ОД) в 1 г препарату, як-
що немає інших зазначень в окремій статті.
Для гранул в однодозових контейнерах вміст визначу-
ваних речовин виражають у грамах, міліграмах або в
одиницях дії (ОД) в одній дозі, якщо немає інших за-
значень в окремій статті.
Відхилення вмісту діючих речовин мають становити
не більше (±10 %) від вмісту, зазначеного у розділі
"Склад", якщо немає інших зазначень в окремій статті.
МАРКУВАННЯ
Для гранул в однодозових контейнерах на етикетці за-
значають назву і кількість діючої речовини.
Для гранул у багатодозових контейнерах на етикетці
зазначають назву і кількість діючої речовини в одній
дозі. На етикетці додатково зазначають:
— умови і термін зберігання після розкриття;
— спосіб застосування.
ЗБЕРІГАННЯ
У сухому, захищеному від світла місці, якщо немає
інших зазначень в окремій статті
ЕКСТРАКТИ
Ехігасіа
ВИЗНАЧЕННЯ
Екстракти — це концентровані препарати рідкої,
твердої або густої консистенції, які звичайно одержу-
ють із висушеної рослинної чи тваринної сировини. У
деяких випадках матеріал, що екстрагується, може
піддаватися попередній обробці, наприклад, інакти-
вації ферментів, здрібненню або знежирюванню.
Екстракти виготовляють мацерацією, перколяцією
або іншим придатним валідованим методом, викори-
стовуючи спирт або інший підхожий розчинник.
Після екстрагування непотрібні матеріали, якщо не-
обхідно, видаляють.
ВИРОБНИЦТВО
Метод перколяції. Якщо необхідно, сировину, що
екстрагується, здрібнюють до часток певного розміру,
ретельно змішують з порцією зазначеного екстрагенту
і залишають на необхідний час. Переносять суміш в
490
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Екстракти
екстрактор і повільно перколюють, стежачи за тим,
щоб сировина увесь час була повністю вкрита шаром
відповідного екстрагенту. Залишок можна віджати і
одержану рідину з’єднати з перколятом.
Метод мацерації. Якщо необхідно, сировину, що
екстрагується, здрібнюють до часток певного розміру,
ретельно змішують з порцією зазначеного розчинника
і мацерують у закритому екстракторі необхідний час.
Залишок відділяють від екстракту і, якщо необхідно,
віджимають. В останньому випадку обидві рідини
з’єднують.
Концентрування до бажаної консистенції проводять,
використовуючи придатні методи, звичайно під змен-
шеним тиском і з використанням температури, за якої
руйнування компонентів зводиться до мінімуму. Вміст
залишкових розчинників в екстракті не має переви-
щувати зазначених меж.
Регулювання складу до певного вмісту складових час-
тин проводять, використовуючи підхожий до-
поміжний матеріал або екстракт іншої концентрації з
рослинного або тваринного матеріалу, шо застосо-
вується при виготовленні даного препарату.
Рідкі екстракти
ВИЗНАЧЕННЯ
Рідкі екстракти є препаратами, в яких, як правило, од-
на частина за масою або за об’ємом еквівалентна одній
частині за масою вихідної висушеної лікарської сиро-
вини. Ці препарати стандартизують, якщо необхідно,
так, щоб вони відповідали вимогам щодо вмісту роз-
чинника, діючих речовин або сухого залишку.
Рідкі екстракти можуть бути приготовані описаними
вище методами з використанням лише етанолу певної
концентрації або води або шляхом розчинення густо-
го або сухого екстрактів у якомусь із зазначених роз-
чинників і, якщо необхідно, з наступним фільтру-
ванням. Кожний спосіб, однак, має давати
порівнювані за складом препарати. При зберіганні
можливе утворення невеликого осаду, що допус-
кається за умови відсутності істотної зміни складу.
До рідких екстрактів можуть бути уведені підхожі ан-
тимікробні консерванти.
ВИПРОБУВАННЯ
Відносна густина (2.2.5). Значення відносної густини
рідкого екстракту має відповідати межам, зазначеним
в окремій статті.
Вміст етанолу (2.9.10). Для спиртових рідких екс-
трактів проводять визначення вмісту етанолу. Вміст
етанолу має відповідати межам, зазначеним в окремій
статті.
Метанол і 2-пропанол (2.9.11). У спиртовмісних рідких
екстрактах допускається вміст не більше 0.05 %
(об/об) метанолу і не більше 0.05 % (об/об) 2-пропа-
нолу, якщо немає інших зазначень в окремій статті.
Сухий залишок. Вміст сухого залишку рідкого екстрак-
ту має відповідати межам, зазначеним в окремій
статті.
2.00 г або 2.0 мл екстракту поміщають у плоскодонну
чашку або бюкс діаметром близько 50 мм і заввишки
близько 30 мм. Випарюють насухо на водяній бані і су-
шать у сушильній шафі при температурі від 100 °С до
105 °С протягом 3 год. Охолоджують в ексикаторі над
фосфору(V) оксидом Р і зважують. Результат виража-
ють у вагових відсотках або в грамах на літр.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах, у захищеному від
світла місці.
МАРКУВАННЯ
На етикетці зазначають:
— яка сировина використана — рослинна чи
тваринна:
— де необхідно, зазначають, що використовувалась
свіжа рослинна або тваринна сировина;
— концентрацію спирту, що використовується для
приготування;
— якщо необхідно, вміст спирту у відсотках (об/об) в
екстракті;
— вміст діючих речовин і/або співвідношення
вихідного матеріалу до одержаного рідкого
екстракту;
— назву і концентрацію кожного антимікробного
консерванта.
Густі екстракти
ВИЗНАЧЕННЯ
Густими екстрактами є препарати проміжної консис-
тенції між рідкими і сухими екстрактами. Вони вироб-
ляються шляхом часткового упарювання використо-
вуваного розчинника. Застосовують лише спирт
відповідної концентрації або воду. Густі екс-
тракти звичайно мають сухий залишок не менш як
70 % (за масою). До них можуть бути уведені підхожі
антимікробні консерванти.
ВИПРОБУВАННЯ
Сухий залишок. Густий екстракт за вмістом сухого за-
лишку має відповідати межам, зазначеним у відпо-
відній окремій статті.
2.00 г екстракту поміщають у плоскодонну чашку або
бюкс діаметром близько 50 мм і заввишки близько
30 мм. Випарюють насухо на водяній бані і сушать у
сушильній шафі при температурі від 100 °С до 105 °С
протягом 3 год. Охолоджують в ексикаторі над фосфо-
ЛЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
491
Екстракти
ру(У) оксидом Р і зважують. Результат виражають у ва-
гових відсотках.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах, у захищеному від
світла місці.
МАРКУВАННЯ
На етикетці зазначають:
— яка сировина використана — рослинна чи тварин-
на;
— де необхідно, зазначають, що використовувалася
свіжа рослинна чи тваринна сировина;
— концентрацію спирту, що використовується для
виготовлення, у відсотках (об/об);
— вміст діючих речовин і/або співвідношення
вихідного матеріалу до одержаного густого екс-
тракту;
— назву і концентрацію кожного антимікробного
консерванта.
Сухі екстракти
ВИЗНАЧЕННЯ
Сухі екстракти — це препарати, одержувані видален-
ням розчинника, що використовується для їх приготу-
вання. Сухі екстракти звичайно містять не менше 95 %
сухого залишку за масою. До них можуть додаватися
підхожі допоміжні речовини.
Регулювання складу сухих екстрактів до певного
вмісту складових частин проводять, використовуючи
підхожі допоміжні матеріали або сухий екстракт іншої
концентрації з рослинного або тваринного матеріалу,
що застосовується при виготовленні даного препарату.
Якщо зазначено в окремій статті, проводять визначен-
ня вмісту залишкового розчинника, що використо-
вується для приготування препарату.
ВИПРОБУВАННЯ
Втрата в масі при висушуванні. Значення втрати в масі
при висушуванні сухого екстракту має відповідати ме-
жам, зазначеним в окремій статті.
0.50 г здрібненого на тонкий порошок екстракту
поміщають у плоскодонну чашку або бюкс діаметром
близько 50 мм і заввишки близько ЗО мм і сушать у су-
шильній шафі при температурі від 100 °С до 105 °С
протягом 3 год. Охолоджують в ексикаторі над фосфо-
ру (V) оксидом Рі зважують. Результат виражають у ва-
гових відсотках.
ЗБЕРІГАННЯ
У повітронепроникних контейнерах, у захищеному від
світла місці.
МАРКУВАННЯ
На етикетці зазначають:
— назву і кількість кожного допоміжного матеріалу;
— яка сировина використана — рослинна чи тварин-
на;
— де необхідно, зазначають, що використовувалася
свіжа рослинна чи тваринна сировина;
— назву і концентрацію етанолу у відсотках (об/об)
у розчиннику, використаному для приготування;
— вміст діючих речовин і/або співвідношення
вихідного матеріалу до одержаного сухого екс-
тракту.
___________________________________________N
ВИЗНАЧЕННЯ
Екстракти — це концентровані витяги з лікарської
рослинної або тваринної сировини. Розрізняють рідкі
екстракти (Ехігасіа Пиісіа); густі екстракти (Ехігасіа
хрікьа) — в’язкі маси з втратою в масі при висушуванні
не більше 25 %; сухі екстракти (Ехігасіа хісса) — сипкі
маси з втратою в масі при висушуванні не більше 5 %.
ВИРОБНИЦТВО
Для екстрагування лікарської рослинної сировини за-
стосовують воду, спирт різної концентрації й інші
екстрагенти, іноді з додаванням кислот, лугів, гліце-
рину та ін.
При виготовленні рідких екстрактів з однієї вагової
частини лікарської сировини одержують одну або дві
об’ємні частини екстракту, якшо немає інших зазна-
чень в окремій статті.
Одержані витяги звичайно відстоюють не менше
двох діб при температурі не вище 10 °С до одержання
прозорої рідини і фільтрують.
При виготовленні густих і сухих екстрактів допус-
кається використання або рідких екстрактів, або на-
стойок, які упарюють до необхідної концентрації, як-
що немає інших зазначень в окремій статті.
ВИПРОБУВАННЯ
Екстракти додатково контролюють за такими показ-
никами якості: опис, ідентифікація, важкі метали і
вміст органічних розчинників, мікробіологічна
чистота, кількісне визначення.
У рідких екстрактах додатково визначають об’єм
вмісту контейнера.
У густих і сухих екстрактах додатково визначають
втрату в масі при висушуванні.
У густих і сухих екстрактах, що використовуються як
готові лікарські засоби, додатково визначають масу
вмістуконтейнера.
492
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Капсули
Сухий залишок. Допускається проводити визначення з
5 0 мл рідкого екстракту, який поміщають у зважений
бюкс, випарюють на водяній бані, сушать при темпе-
ратурі від 100 °С до 105 °С протягом 3 год, потім охо-
лоджують в ексикаторі протягом ЗО хв і зважують.
Важкі метали (2.4.8, метод А). НебільшеО.ОІ % (100 ррт).
До 1.0 мл рідкого екстракту або 1.00 г густого чи сухого
екстракту додають 1 мл кислоти сірчаної Р, обережно
спалюють і прожарюють. До одержаного залишку до-
дають при нагріванні 5 мл розчину 615 г/л амонію аце-
тату Р, фільтрують крізь беззольний фільтр, промива-
ють 5 мл води Р і доводять об’єм фільтрату водою Р до
100 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують з викори-
станням еталонного розчину свинцю (1 ррт РЬ) Р.
У препаратах, що містять залізо у кількості 0.05 % і
більше, визначення важких металів проводять після
відокремлення заліза згідно із зазначеннями в окремій
статті.
Кількісне визначення. Вміст визначуваних речовин, які
визначають для рідких екстрактів, виражають у відсот-
ках (м/об), а для густих та сухих — у відсотках (м/м).
ЗБЕРІГАННЯ
У контейнерах, що забезпечують стабільність препа-
рату протягом зазначеного терміну придатності, і, як-
що необхідно, у прохолодному, захищеному від світла
місці. Під час зберігання рідких екстрактів можливе
утворення осаду
МАРКУВАННЯ
Для рідких екстрактів на етикетці додатково зазнача-
ють вміст діючих речовин у відсотках (м/об). а для
густих та сухих — у відсотках (м/м).
КАПСУЛИ
Сар$и1е$
Вимоги цієї статті не обоє ’язкові для лікарських за-
собів у капсулах, призначених до застосування не ораль-
ним, а іншим способом. Вимоги до таких лікарських за-
собів наведені в інших статтях, наприклад, "Лікарські
засоби для ректального застосування" або "Лікарські
засоби для вагінального застосування ",
ВИЗНАЧЕННЯ
Капсули — тверді лікарські засоби з твердою або м’я-
кою оболонкою різної форми і місткості, звичайно
капсула містить одну дозу діючої речовини. Капсули
призначені для орального застосування
Оболонка капсули виготовлена з желатину або інших
речовин. Консистенція оболонки може бути забезпе-
чена шляхом додавання таких речовин, як гліцерин
або сорбіт. До складу оболонки можуть входити такі
допоміжні речовини, як поверхнево-активні речови-
ни, непрозорі наповнювачі, антимікробні консерван-
ти, підсолоджувачі, барвники, дозволені до медичного
застосування, ароматизатори та ін. Поверхня капсул
може бути маркована.
Вміст капсул може бути твердим, рідким або пасто-
подібним. Він складається з однієї або більше діючих
речовин і допоміжних речовин, таких як розчинники,
розріджувачі, змащувальні й розпушувальні речовини
та ін , або без допоміжних речовин. Вміст капсули не
має руйнувати оболонку. Однак оболонка під дією
травних соків має руйнуватися і вивільнювати вміст
капсули.
Там, де це необхідно, контейнери для капсул мають
відповідати вимогам статті "Матеріали, використову-
вані для виробництва контейнерів" (3.1 та підрозділи)
та "Контейнери"(3.2та підрозділи).
Капсули можуть бути класифіковані як:
капсули тверді;
— капсули м’які;
— капсули кишково-розчинні;
— капсули з модифікованим вивільненням.
ВИРОБНИЦТВО
При виробництві, пакуванні, зберіганні і реалізації
капсул мають бути вжиті відповідні заходи, які забез-
печують необхідну мікробіологічну чистоту відпо-
відно до вимог статті "Мікробіологічна чистота лі-
карських засобів" (5.1.4).
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Капсули з вмістом діючої
речовини менше 2 мгабо менше 2 % від маси вмісту ма-
ють витримувати випробування однорідності вмісту
діючої речовини в одиниці дозованого лікарського за-
собу (тест В), якщо немає інших зазначень в окремій
статті. Якшо лікарська форма містить більше однієї
діючої речовини, вимоги поширюються лише на ті ре-
човини, вміст яких відповідає вищезазначеним умовам.
Дане випробування не поширюється на полівітамінні
препарати і препарати, що містять мікроелементи.
Однорідність маси (2.9.5). Капсули мають витримувати
випробування однорідності маси для одиниці дозова-
ного лікарського засобу. Випробування на однорідність
маси не вимагається, якщо випробування однорідності
вмісту передбачено для всіх діючих речовин.
Розчинення. Випробування може бути проведене для
підтвердження відповідного вивільнення діючої речо-
вини або речовин, наприклад, одним із способів, опи-
саних у статті "Тест "Розчинення"для твердих дозова-
них форм" (2.9.3)
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
493
Капсули
Якщо проводять випробування за показником "Розчи-
нення”, випробування "Розпадання” не вимагається.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах, при температурі
не вище ЗО °С.
МАРКУВАННЯ
Зазначають назву всіх антимікробних консервантів,
що входять до складу.
Капсули тверді
ВИЗНАЧЕННЯ
Капсули тверді мають оболонку, що складається з двох
попередньо виготовлених частин циліндричної фор-
ми, один кінець кожної частини заокруглений і закри-
тий, а інший кінець відкритий.
ВИРОБНИЦТВО
Діючу речовину або речовини звичайно у твердому
стані (у вигляді порошку або гранул) засипають в одну
з частин оболонки, яку щільно закривають другою ча-
стиною. Надійність закриття капсули може бути поси-
лена відповідними засобами.
ВИПРОБУВАННЯ
Розпадання. Тверді капсули мають витримувати ви-
пробування на розпадання таблеток або капсул (2.9.1).
Як рідке середовище використовують воду Р. Якшо за-
значено в окремій статті, як рідке середовище може
бути використаний 0.1 М розчин кислоти хлористовод-
невої або штучний шлунковий сік Р. Якщо капсула
спливає на поверхню води, треба використовувати
диск. Прилад вмикають на ЗО хв, якшо немає інших
зазначень в окремій статті, і досліджують стан капсул.
Випробування вважають витриманим, якщо розпали-
ся всі шість капсул
Капсули м'які
ВИЗНАЧЕННЯ
Капсули м’які звичайно мають більш товсту оболонку,
ніж тверді капсули. Оболонка складається з однієї ча-
стини і має різні форми.
ВИРОБНИЦТВО
М’які капсули звичайно виготовляють, заповнюють і
запечатують в одній технологічній стадії, але для екс-
темпорального застосування оболонка може бути ви-
готовлена заздалегідь. Матеріал оболонки може міс-
тити діючу речовину.
Рідини можуть бути поміщеними в капсулу безпосе-
редньо; тверді речовини звичайно розчиняють або
дисперіують у підхожому розчиннику для утворення
розчину або суспензії майже пастоподібної консис-
тенції.
Можлива часткова міграція складових компонентів
вмісту капсули до оболонки і навпаки, зумовлена при-
родою контактуючих матеріалів і поверхонь.
ВИПРОБУВАННЯ
Розпадання. М’які капсули мають витримувати випро-
бування на розпадання таблеток або капсул (2.9.1). Як
рідке середовище використовують воду Р. Якщо зазна-
чено в окремій статті, як рідке середовище може бути
використаний 0.1 М розчин кислоти хлористоводневої
або штучний шлунковий сік Р. У кожну скляну трубку
поміщають диск. Рідкі лікарські речовини, роз-
поділені у м’якій капсулі, можуть обволікати диск; у
такому випадку, або якщо зазначено в окремій статті,
використання дисків виключають. Прилад вмикають
на ЗО хв, якщо немає інших зазначень в окремій статті,
і досліджують стан капсул. Якщо капсули не витрима-
ли випробування внаслідок прилипання до дисків, ви-
пробування повторюють на наступних шести капсулах
без дисків. Випробування вважають витриманим, як-
що розпалися усі шість капсул.
Капсули кишково-розчин ні
ВИЗНАЧЕННЯ
Капсули кишково-розчинні — це капсули з модифіко-
ваним вивільненням, які мають бути стійкими у
шлунковому соку і вивільнювати діючу речовину або
речовини у кишковому соку. Вони можуть бути виго-
товлені шляхом покриття твердих або м’яких капсул
кислотостійкою оболонкою (кишково-розчинні кап-
сули) або ж шляхом заповнення капсул гранулами чи
частками, покритими кислотостійкою оболонкою.
ВИРОБНИЦТВО
Для капсул, заповнених гранулами або частками з
кислотостійкою оболонкою, проводять випробуван-
ня, яке підтверджує відповідне вивільнення діючої ре-
човини або речовин.
ВИПРОБУВАННЯ
Розпадання. Для капсул з кишково-розчинною обо-
лонкою проводять визначення розпадання (2.9. і) з та-
кими змінами. Як рідке середовище використовують
494
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лікарські засоби для вагінального застосування
0.1 М розчин кислоти хлористоводневої і вмикають
прилад на 2 год, якщо немає інших зазначень в ок-
ремій статті, без дисків. Досліджують стан капсул. Час
стійкості у кислому середовищі коливається у залеж-
ності від складу випробовуваних капсул. Звичайно він
складає від 2 до 3 год, але у будь-якому випадку він має
бути не менше 1 год. Жодна з капсул не має виявляти
ознак розпаду або розривів, крізь які можливий вихід
вмісту. Кислоту замінюють фосфатним буферним роз-
чином рН 6.8 Р. Якщо зазначено в окремій статті може
бути використаний буферний розчин рН 6.8 з дода-
ванням порошку панкреатину (наприклад, 0.35 г по-
рошку панкреатину Р на 100 мл буферного розчину). У
кожну скляну трубку поміщають диск. Прилад вмика-
ють на 1 год і досліджують стан капсул. Якщо капсули
не витримали випробування внаслідок прилипання до
дисків, випробування повторюють на наступних шес-
ти капсулах без дисків. Випробування вважають ви-
триманим. якщо розпалися всі шість капсул.
Капсули з модифікованим
вивільненням
ВИЗНАЧЕННЯ
Капсули з модифікованим вивільненням — це тверді
або м’які капсули, які мають у складі вмісту, або обо-
лонки, або в тому і другому одночасно спеціальні до-
поміжні речовини, або виготовлені спеціальними ме-
тодами, що призначені для зміни швидкості або місця
вивільнення діючої речовини або речовин.
ВИРОБНИЦТВО
Проводять випробування, яке підтверджує відповідне
вивільнення діючої речовини або речовин.
______________________________________N
ВИПРОБУВАННЯ
Капсули звичайно контролюють за такими показни
ками якості: опис, ідентифікація, однорідність маси,
однорідність вмісту, супровідні домішки, розпадання,
розчинення, втрата в масі при висушуванні або вода,
мікробіологічна чистота, кількісне визначення.
Для м’яких капсул, вміст яких являє собою масла або
масляні розчини, додатково контролюють кислотне і
перекисне числа.
Ідентифікація. Проводять визначення справжності
всіх діючих речовин і антимікробних консервантів, які
входять до складу препарату.
Однорідність вмісту (2.9.6). Капсули мають витримува-
ти вимоги статті "Однорідність вмісту діючої речовини
в одиниці дозованого лікарського засобу” (2.9.6), якщо
немає інших зазначень в окремій статті.
Кількісне визначення. Для кількісного ви значення ви-
користовують вміст 20 капсул.
Вміст діючої речовини у капсулі. Якщо немає інших за-
значень в окремій статті, відхилення у вмісті діючих
речовин мають становити при дозуванні менше 1 мг
(± 15 %). від 1 мг до 10 мг (± 10 %), від 10 мгдо 100 мг
(± 7.5 %) і від 100 мг й вище (± 5 %).
МАРКУВАННЯ
На етикетці додатково зазначають:
— вміст діючої речовини (речовин) у капсулі;
— термін придатності;
— умови зберігання.
ЛІКАРСЬКІ ЗАСОБИ ДЛЯ
ВАГІНАЛЬНОГО ЗАСТОСУВАННЯ
ВИЗНАЧЕННЯ
Капсули повинні мати гладку поверхню без пошкод-
жень і видимих повітряних і механічних включень.
Тверді капсули залежно від місткості виготовляють
вісьмох розмірів — від 000 (найбільшого розміру) до 5
(найменшого розміру). Номери капсул і відповідна їм
місткість наведені у таблиці нижче.
Номер 000 00 0 1 2 3 4 5
Середня місткість капсули, у мл 1.37 0.95 0.68 0.50 0.37 0.30 0.21 0.13
М’які капсули можуть бути різних розмірів, звичайно
місткістю до 1.5 мл. Оболонка м’яких капсул може бу-
ти жорсткою або еластичною залежно від вмісту плас-
тифікаторів.
Хафгіаііа
ВИЗНАЧЕННЯ
Лікарські засоби для вагінального застосування
(вагінальні лікарські засоби) можуть бути рідкими,
м’якими або твердими і призначені для застосування у
піхву з метою забезпечення місцевої дії. Вони містять
одну або більше діючих речовин у відповідній ОСНОВІ
Контейнери для вагінальних лікарських засобів мають
відповідати вимогам статей "Матеріали, використову-
вані для виробництва контейнерів" (3.1 та підрозділи)
та "Контейнери"(3.2та підрозділи).
Лікарські засоби для вагінального застосування мо-
жуть бути класифіковані як:
— литі песарії (вагінальні супозиторії);
— вагінальні таблетки;
державна Фармакопея України 2001
495
Лікарські засоби для вагінального застосування
— вагінальні капсули;
— вагінальні піни;
— вагінальні тампони.
ВИРОБНИЦТВО
При виробництві, пакуванні, зберіганні і реалізації
лікарських засобів для вагінального застосування мають
бути вжиті відповідні заходи, шо забезпечують необхідну
мікробіологічну чистоту відповідно до вимог статті
"Мікробіологічна чистота лікарських засобів" (5.1.4).
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Якщо немає інших зазна-
чень в окремій статті, тверді однодозові лікарські за-
соби з вмістом діючої речовини менше 2 мг або менше
2 % від загальної маси мають витримувати випробу-
вання однорідності вмісту однодозових препаратів
(тест А — вагінальні таблетки або тест В — литі песарії,
вагінальні капсули). Якщо лікарський засіб містить
більше однієї діючої речовини, вимоги поширюються
лише на ті речовини, які відповідають вищезазначе-
ним умовам.
Однорідність маси (2.9.5). Тверді лікарські засоби в од-
нодозових контейнерах мають витримувати випробу-
вання однорідності маси для одиниці дозованого
лікарського засобу. Випробування однорідності маси
не вимагається, якшо випробування однорідності
вмісту передбачено для всіх діючих речовин.
Розчинення. Для підтвердження відповідного
вивільнення діючої речовини або речовин з твердих
однодозових лікарських засобів випробування може
бути проведене, наприклад, одним із способів, описа-
них у статті "Тест "Розчинення"для твердих дозованих
форм" (2.9.3)
Якщо проводять тест "Розчинення", випробування
"Розпадання" не вимагається.
Литі песарії
Лікарські засоби для вагінального застосування, за
визначенням відповідні литим песаріям, можуть бути
виготовлені також методом пресування. Вони мають
відповідати вимогам, які ставляться долитих песаріїв.
ВИРОБНИЦТВО
Обирають випробування, що підтверджує відповідне
вивільнення діючої речовини або речовин із супози-
торіїв, призначених для модифікованого вивільнення
або пролонгованої місцевої дії.
ВИПРОБУВАННЯ
Розпадання. Якшо песарії не призначені для мо-
дифікованого вивільнення або місцевої пролонгова-
ної дії, вони мають витримувати випробування розпа-
дання супозиторіїв і песаріїв (2.9.2). Стан песаріїв
досліджують через 60 хв, якшо немає інших зазначень
в окремій статті.
ЗБЕРІГАННЯ
У шільно закупорених контейнерах.
Вагінальні таблетки
ВИЗНАЧЕННЯ
Вагінальні таблетки (пресовані песарії) — це тверда
однодозова лікарська форма, загалом подібна до таб-
леток без оболонки або до таблеток, покритих оболон-
кою, за визначенням, наведеним у статті "Таблетки".
ВИРОБНИЦТВО
Обирають випробування, шо підтверджує відповідне
вивільнення діючої речовини або речовин з вагіналь-
них таблеток, призначених для модифікованого ви-
вільнення або пролонгованої місцевої дії.
ВИЗНАЧЕННЯ
ВИПРОБУВАННЯ
Литі песарії (вагінальні супозиторії) — тверді однодо-
зові лікарські засоби. Вони можуть бути різної форми,
звичайно яйцеподібної; за об’ємом і консистенцією
мають відповідати вагінальному застосуванню. За ви-
нятком форми, вони відповідають визначенню для
литих супозиторіїв, наведеному у статті "Лікарські за-
соби для ректального застосування".
Для виготовлення литих песаріїв можуть бути викори-
стані методи і допоміжні речовини, описані для литих
супозиторіїв у статті "Лікарські засоби для ректального
застосування". Діюча речовина або речовини можуть
бути дисперговані або розчинені у простій або
складній основі, яка може бути розчинною і не роз-
чинною. але такою, що плавиться при температурі
тіла або диспергується у воді.
Розпадання. Якшо вагінальні таблетки не призначені
для модифікованого вивільнення або місцевої про-
лонгованої дії, вони мають витримувати випробуван-
ня розпадання супозиторіїв і песаріїв (спеціальний
метод для вагінальних таблеток, 2.9.2). Стан таблеток
досліджують через ЗО хв, якщо немає інших зазначень
в окремій статті.
Вагінальні капсули
ВИЗНАЧЕННЯ
Вагінальні капсули (песарії з оболонкою) — це тверда
однодозова лікарська форма, загалом подібна до
496
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лікарські засоби для парентерального застосування
м’яких капсул, що відрізняється лише формою і
розміром. Вагінальні капсули можуть мати різну фор-
му, звичайно яйцеподібну. Вони мають бути гладкими
і однорідними за зовнішнім виглядом.
ВИРОБНИЦТВО
Обирають випробування, шо підтверджує відповідне
вивільнення діючої речовини або речовин з вагіналь-
них капсул, призначених для модифікованого ви-
вільнення або пролонгованої місцевої дії.
ВИПРОБУВАННЯ
Розпадання. Якшо вагінальні капсули не призначені
для модифікованого вивільнення або місцевої про-
лонгованої дії, вони мають витримувати випробуван-
ня розпадання супозиторіїв і песаріїв (2.9.2) Стан
капсул досліджують через ЗО хв, якшо немає інших за-
значень в окремій статті.
Вагінальні піни
ВИЗНАЧЕННЯ
Вагінальні піни мають відповідати вимогам статті
"Піни медичні".
Вагінальні тампони
ВИЗНАЧЕННЯ
Вагінальні тампони — це тверда однодозова лікарська
форма, призначена для введення у піхву на певний
час.
Вони мають відповідати вимогам статті "Тампони ме-
дичні".
___________________________________________N
ВИПРОБУВАННЯ
Лікарські засоби для вагінального застосування зви-
чайно контролюють за такими показниками якості:
опис, ідентифікація, середня маса і однорідність маси
(для литих песаріїв, вагінальних капсул і таблеток),
однорідність вмісту, розпадання, розчинення, темпе-
ратура плавлення або час повної деформації, супро-
відні домішки, мікробіологічна чистота, кількісне
визначення.
У лікарських засобах для вагінального застосування,
якщо необхідно, додатково контролюють кислотне і
перекисне числа, а також розмір часток.
Середня маса. Визначення середньої маси проводять
для литих песаріїв, вагінальних капсул і таблеток.
Відхилення середньої маси від маси, зазначеної у
розділі "Склад", не має перевищувати (±5 %), якшо
немає інших зазначень в окремій статті. Визначення
середньої маси проводять, як зазначено у статті "Од-
норідність маси для одиниці дозованого лікарського за-
собу" (2.9.5).
Однорідність вмісту (2.9.6). Якщо немає інших зазна-
чень в окремій статті, тверді лікарські засоби в одно-
дозових контейнерах мають витримувати випробуван-
ня однорідності вмісту однодозових препаратів.
Температура плавлення. Для литих песаріїв, виготовле-
них на ліпофільній основі, визначають температуру
плавлення за методом (2.2.15), яка не має перевищу-
вати 37 °С, якшо немає інших зазначень в окремій
статті.
Час повної деформації. Для литих песаріїв визначають
час повної деформації згідно з Додатком 1 до статті
"Лікарські засоби для ректального застосування". До-
пускається використання інших приладів. Час повної
деформації має бути не більше 15 хв, якшо немає
інших зазначень в окремій статті.
Кількісне визначення. Вміст визначуваних речовин ви-
ражають у грамах, міліграмах або в одиницях дії (ОД)
в одиниці дозованого лікарського засобу, якщо немає
інших зазначень в окремій статті.
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву діючої речовини або речовин та їх вміст у
препараті або в одиниці дозованого лікарського
засобу;
— спосіб застосування;
— умови зберігання;
— термін придатності.
ЛІКАРСЬКІ ЗАСОБИ ДЛЯ
ПАРЕНТЕРАЛЬНОГО
ЗАСТОСУВАННЯ
Рагепіегаїіа
Вимоги цієї статті не поширюються на препарати, ви-
готовлені з людської крові, імунологічні препарати,
радіофармацевтичні препарати і протези, які імплан-
туються Спеціальні вимоги можуть бути введені для
препаратів, які використовують у ветеринарії, залеж-
но від виду тварин, для яких вони призначені.
ВИЗНАЧЕННЯ
Лікарські засоби для парентерального застосування
(парентеральні лікарські засоби) — це стерильні
ЛГРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
497
Лікарські засоби для парентерального застосування
лікарські засоби, призначені для введення шляхом
ін’єкцій, інфузій або імплантацій в організм людини
чи тварини.
Для виготовлення лікарських засобів для паренте-
рального застосування використовують допоміжні ре-
човини, наприклад, такі, шо забезпечують ізо-
тонічність препаратів відносно крові, регулюють рН,
покращують розчинність діючих речовин, запобігають
їхньому розкладанню, забезпечують відповідні ан-
тимікробні властивості препарату. Ці речовини у ви-
користовуваних концентраціях не мають негативно
впливати на дію лікарського засобу і не мають викли-
кати токсичність або небажане місцеве подразнення.
Контейнери для лікарських засобів для парентераль-
ного застосування мають бути виготовлені з ма-
теріалів, які достатньо прозорі і дозволяють візуально
переглянути вміст контейнера, за винятком імплан-
татів, якщо немає інших зазначень в окремій статті.
Контейнери для лікарських засобів для парентераль-
ного застосування мають відповідати вимогам статей
"Матеріали, використовувані для виробництва кон-
тейнерів" (3.1 та підрозділи) та "Контейнери" (3.2 та
підрозділи/ якщо немає інших зазначень в окремій
статті. Лікарські засоби для парентерального застосу-
вання випускають у скляних контейнерах (3.2.1) чи в
інших, таких як пластмасові контейнери (3.2.2, 3.2.7і
3.2.9), або у заздалегідь наповнених шприцах, що задо-
вольняють вимоги, викладені у відповідних норматив-
них документах. Герметичність контейнера забезпечу-
ють відповідними способами. Закупорювальні засоби
мають забезпечувати надійну ізоляцію, запобігати до-
ступу мікроорганізмів та інших забруднень і звичайно
дозволяють діставати частину або увесь вміст контей-
нера без видалення закупорювального засобу. Пласти-
кові матеріали або еластомери (3.2.9), призначені для
закупорювання, мають бути достатньо густими і елас-
тичними, щоб при проходженні голки виділялася най-
менша кількість часток. Закупорювальні засоби для
багатодозових контейнерів мають бути достатньо елас-
тичними, щоб забезпечити герметизацію контейнера
при видаленні голки.
Лікарські засоби для парентерального застосування
можуть бути класифіковані як:
— ін’єкційні лікарські засоби;
— внутрішньовенні інфузійні лікарські засоби;
— концентрати для ін’єкційних або внутрішньовен-
них інфузійних лікарських засобів;
— порошки для ін’єкційних або внутрішньовенних
інфузійних лікарських засобів;
— імплантати.
ВИРОБНИЦТВО
Щодо лікарських засобів для парентерального засто-
сування, до складу яких входять антимікробні консер-
ванти, уповноваженому органу мають бути подані
дані, що підтверджують ефективність вибраних кон-
сервантів. Метод визначення і критерії ефективності
консервантів мають відповідати вимогам статті "Ефек-
тивність антимікробних консервантів"(5.1.3).
Лікарські засоби для парентерального застосування
виготовляють з використанням матеріалів і методів,
шо забезпечують стерильність і запобігають забруд-
ненню лікарських препаратів і розвитку в них мікро-
організмів, відповідно до вимог статті "Методи приго-
тування стерильних продуктів"(5.1.1).
Вода, використовувана у виробництві лікарських засо-
бів для парентерального застосування, має відповідати
вимогам, зазначеним у статті "Вода для ін ’єкцій".
ВИПРОБУВАННЯ
Бактеріальні ендотоксини (2.6.14). Випробування на
бактеріальні ендотоксини може замінити випробуван-
ня на пірогени у таких випадках:
— для препаратів, включених до Фармакопеї, якшо
це випробування зазначено у монографії;
— у будь-яких інших випадках, якщо умови і вимоги
випробування зазначені в окремій статті.
Якщо зазначено випробування на бактеріальні ендо-
токсини, випробування на пірогени не проводять, як-
шо відсутні інші зазначення в окремій статті.
Стерильність (2.6.1). Лікарські засоби для паренте-
рального застосування мають витримувати випробу-
вання на стерильність.
ЗБЕРІГАННЯ
У стерильних повітронепроникних контейнерах із
контролем першого розкриття.
МАРКУВАННЯ
На етикетці зазначають назву і концентрацію кожного
антимікробного консерванта.
Ін’єкційні лікарські засоби
ВИЗНАЧЕННЯ
Ін’єкційні лікарські засоби — це стерильні розчини,
емульсії або суспензії. їх готують шляхом розчинення,
емульгування або суспендування діючих і допоміжних
речовин у воді для ін’єкцій, або у запропонованій сте-
рильній неводній рідині, або в суміші цих розчинників.
Розчини для ін’єкцій у відповідних умовах спо-
стерігання мають бути прозорими і практично вільни-
ми від часток.
Емульсії для ін’єкцій не мають виявляти ознак розша-
рування. У суспензіях для ін’єкцій може спостерігати-
ся осад, який має швидко диспергуватися при збовту-
ванні, утворюючи суспензію. Суспензія, шо утворила-
ся, має бути достатньо стабільною для того, шоб забез-
печити необхідну дозу при введенні.
Однодозові препарати. Об’єм ін’єкційного лікарського
засобу в однодозовому контейнері має бути достатнім
для відбору і введення номінальної дози при звичай-
ному методі введення.
498
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лікарські засоби для парентерального застосування
Багатолезові препарати. Багатодозові водні ін’єкційні
лікарські засоби містять відповідний антимікробний
консервант у необхідній концентрації, за винятком
препаратів, що виявляють відповідні антимікробні
властивості. При випуску препарату для парентераль-
ного введення у багатодозовому контейнері необхідно
зазначити запобіжні заходи щодо його введення і
особливо щодо зберігання між відбором доз.
Антимікробні консерванти. Водні препарати, які готу-
ють в асептичних умовах і які не можуть бути піддані
термічній стерилізації, мають містити певні анти-
мікробні консерванти у відповідних концентраціях.
Антимікробні консерванти не застосовують, якщо:
— об’єм, що вводять в одноразовій дозі, перевищує
15 мл, крім випадків, де це доведено і затверджено;
— препарати призначені для внутрішньопорожнин-
них ін’єкцій або інших ін’єкцій, які мають доступ
до спинномозкової рідини, або інтра- чи ретро-
бульбарних ін’єкцій.
Такі препарати випускають в однодозових контейнерах.
ВИРОБНИЦТВО
При виробництві ін’єкційних лікарських засобів, які
містять дисперговані частки, треба передбачити захо-
ди, шо забезпечують необхідний розмір часток та його
контроль.
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Суспензії для ін’єкцій в од-
нодозових контейнерах із вмістом діючої речовини
менше 2 мг або менше 2 % від загальної маси мають
витримувати випробування однорідності вмісту
діючої речовини в одиниці дозованих лікарських за-
собів (тест А), якшо немає інших зазначень в окремій
статті. Для препаратів, що містять більше однієї діючої
речовини, вимоги поширюються лише на ті речовини,
вміст яких відповідає вищезазначеним умовам. Дане
випробування не поширюється на полівітамінні пре-
парати і препарати, що містять мікроелементи.
Пірогени (2.6.8). Якщо об’єм ін’єкційного лікарського
засобу в одній дозі становить 15 мл або більше і випро-
бування на бактеріальні ендотоксини не зазначене в
окремій статті, препарат має витримувати випробуван-
ня на пірогени. Якщо об’єм ін’єкційного розчину в
одній дозі становить менше 15 мл, на етикетці зазначе-
но, що препарат апірогенний, і випробування на бак-
теріальні ендотоксини не описане або неможливе, то
препарат має витримувати випробування на пірогени.
персійним середовищем; вони вільні від пірогенів і
звичайно ізотонічні крові. Вони переважно призна-
чені для застосування у великих об’ємах. Внутрішньо-
венні інфузійні лікарські засоби не містять ніяких ан-
тимікробних консервантів.
Розчини для внутрішньовенних інфузійних лікарсь-
ких засобів оцінюють у відповідних умовах спо-
стерігання, при цьому вони мають бути прозорими і
практично вільними від часток.
Емульсії для внутрішньовенних інфузій не мають ви-
являти ознак розшарування.
ВИРОБНИЦТВО
При виробництві внутрішньовенних інфузійних
лікарських засобів, які містять дисперговані частки,
треба передбачити заходи, що забезпечують не-
обхідний розмір часток та його контроль.
Об’єм внутрішньовенного інфузійного лікарського
засобу в контейнері має бути достатнім для введення
номінальної дози при звичайному методі введення
(2.9 17).
ВИПРОБУВАННЯ
Пірогени (2.6.8). Якшо випробування на бактеріальні
ендотоксини не зазначене в окремій статті, препарат
має витримувати випробування на пірогени, за
відсутності інших зазначень в окремій статті. Уводять
10 мл на кілограм маси тіла кожного кролика, якщо
немає інших зазначень в окремій статті.
Концентрати для ін’єкційних або
внутрішньовенних інфузійних
лікарських засобів
»
ВИЗНАЧЕННЯ
Концентрати для ін’єкційних або внутрішньовенних
інфузійних лікарських засобів являють собою сте-
рильні розчини, призначені для ін’єкцій або інфузій
після розведення. Концентрати розводять до зазначе-
ного об’єму відповідною рідиною перед застосуван-
ням. Після розведення одержаний розчин має
відповідати вимогам, які ставляться до ін’єкційних
або інфузійних лікарських засобів.
Внутрішньовенні інфузійні
лікарські засоби
ВИЗНАЧЕННЯ
Внутрішньовенні інфузійні лікарські засоби — це сте-
рильні водні розчини або емульсії з водою як дис-
ВИПРОБУВАННЯ
Пірогени (2.6.8). Якщо випробування на бактеріальні
ендотоксини не зазначене в окремій статті, препарат
має витримувати випробування на пірогени. описане
для ін’єкційних або внутрішньовенних інфузійних
лікарських засобів, відповідно, після розведення до
визначеного об’єму.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
499
Лікарські засоби для парентерального застосування
Порошки для ін’єкційних або
внутрішньовенних інфузійних
лікарських засобів
ВИЗНАЧЕННЯ
Порошки для ін’єкційних або внутрішньовенних
інфузійних лікарських засобів являють собою тверді
стерильні речовини, поміщені у контейнер. При стру-
шуванні із зазначеним об’ємом відповідної стерильної
рідини вони швидко утворюють або прозорий,
вільний від часток розчин, або однорідну суспензію.
Після розчинення або суспендування вони мають
відповідати вимогам, що ставляться до внутрішньо-
венних інфузійних або ін’єкційних лікарських за-
собів, відповідно.
Ліофільні препарати для парентерального застосуван-
ня розглядають як порошки для ін’єкційних або
внутрішньовенних інфузійних лікарських засобів.
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Порошки для ін’єкційних
або внутрішньовенних інфузійних лікарських засобів
із вмістом діючої речовини менше 2 мг або менше 2 %
від загальної маси або з масою дозованої одиниці, що
дорівнює 40 мг чи менше, мають витримувати випро-
бування однорідності вмісту діючої речовини в оди-
ниці дозованих лікарських засобів (тест А), якщо не-
має інших зазначень в окремій статті. Для препаратів,
до складу яких входить більше однієї діючої речовини,
вимоги поширюються на речовини, вміст яких
відповідає вищезазначеним умовам. Дане випробу-
вання не поширюється на полівітамінні препарати і
препарати, що містять мікроелементи.
Однорідність маси (2.9.5). Порошки для ін’єкційних
або внутрішньовенних інфузійних лікарських засобів
мають витримувати випробування однорідності маси
для одиниці дозованого лікарського засобу. Випробу-
вання однорідності маси не вимагається, якщо випро-
бування однорідності вмісту діючої речовини перед-
бачене для всіх діючих речовин.
Пірогени (2.6.8). Якщо випробування на бактеріальні
ендотоксини не зазначене в окремій статті, препарат
має витримувати випробування на пірогени, описане
для ін’єкційних або внутрішньовенних інфузійних
лікарських засобів, відповідно, після розведення або
суспендування до визначеного об’єму рідини.
Імплантати
ВИЗНАЧЕННЯ
Імплантати являють собою стерильні тверді лікарські
засоби, що мають підхожі для парентеральної імплан-
тації розміри й форми і вивільнюють діючі речовини
протягом тривалого часу. Вони упаковані в індивіду-
альні стерильні контейнери.
______________________________________________N
ВИРОБНИЦТВО
Для виготовлення лікарських засобів для паренте-
рального застосування використовують діючі, допо-
міжні речовини і розчинники, дозволені до медичного
застосування.
Розчинники. Як водні розчинники звичайно застосо-
вують воду для ін’єкцій Р, ізотонічний розчин натрію
хлориду, розчин Рінгера, 5 % розчин глюкози або інші
водні розчини.
Як неводні розчинники звичайно використовують
жирні рослинні олії або інші органічні розчинники.
Якщо немає інших зазначень в окремій статті, рос-
линні олії, призначені для виготовлення ін’єкційних
лікарських засобів, мають відповідати таким вимогам:
кислотне число має бути не більше 0.56, йодне число
має бути від 79 до 137, число омилення має бути від
185 до 200. Рослинні олії мають бути прозорі при
температурі 10 °С і не повинні мати запаху і смаку
згірклості.
Речовини, що не омилюються, у рослинних оліях ви-
значають за такою методикою: доливають 10 мл олії в
колбу, поміщену на киплячу баню, додають 15 мл роз-
чину натрію гідроксиду Р(1:6) і 30 мл 96 % спирту Р,
часом перемішуючи до одержання прозорої суміші.
Переносять розчин до мілкого посуду, випарюють
спирт на киплячій водяній бані, змішують осад з
100 мл води Р. Розчин, що утворюється, має бути про-
зорим. За іншими показниками якості рослинні олії
додатково мають відповідати вимогам, зазначеним у
відповідній окремій статті.
У складі комплексного розчинника можуть бути вико-
ристані спирт етиловий, гліцерин, пропіленгліколь,
макрогол 400, бензилбензоат, бензиловий спирт та ін.
Допоміжні речовини. Як допоміжні речовини викорис-
товують аскорбінову, хлористоводневу, винну, лимон-
ну, оцтову кислоти, натрію карбонат, натрію гідрокар-
бонат, натрію гідроксид, натрію або калію сульфіт,
гідросульфіт або метабісульфіт, натрію тіосульфат,
натрію цитрат, натрію дигідрофосфат і динатрію гідро-
фосфат, натрію хлорид, метилпарагідроксибензоат.
пропілпарагідроксибензоат, ронгаліт, динатрієву сіль
етилендіамінтетраоцтової кислоти, спирт полівініло-
вий, хлоробутанол, крезол, фенол та ін.
Кількість деяких допоміжних речовин, якщо немає
інших зазначень в окремій статті, не має перевищува-
ти таких концентрацій: для речовин, подібних до хло-
робутанолу, крезолу, фенолу — 0.5 %; сірчистого
ангідриду або еквівалентних кількостей сульфіту,
гідросульфіту або метабісульфіту калію або натрію —
0.2 %.
500
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лікарські засоби для парентерального застосування
ВИПРОБУВАННЯ
Рідкі лікарські засоби для парентерального застосу-
вання звичайно контролюють за такими показниками
якості: опис, ідентифікація, прозорість, кольоровість,
рН, супровідні домішки, об’єм, шо витягається, сте-
рильність, пірогени або бактеріальні ендотоксини,
аномальна токсичність, механічні включення, кіль-
кісне визначення.
Для рідких лікарських засобів для парентерального за-
стосування у вигляді в’язких рідин додатково контро-
люють густину.
Для рідких лікарських засобів для парентерального за-
стосування у вигляді суспензій додатково контролюють
розмір часток, однорідність вмісту (у випадку суспензій
в однодозових контейнерах), стійкість суспензій.
У порошках для ін’єкцій або внутрішньовенних ін-
фузій додатково контролюють такі показники якості
час розчинення, втрату в масі при висушуванні або во-
ду, однорідність вмісту або однорідність маси.
Для проведення випробувань "Прозорість", "Кольо-
ровість", "рН" при контролі порошків для ін’єкційних
або внутрішньовенних інфузійних лікарських засобів
використовують розчин лікарського засобу в тому
розчиннику і в тій концентрації, які зазначені в
інструкції для застосування, якшо немає інших зазна-
чень в окремій статті.
Прозорість. Розчини мають бути прозорими (2.2.1) у
порівнянні з водою Рабо відповідним розчинником,
якщо немає інших зазначень в окремій статті.
Кольоровість. Забарвлення лікарських засобів для па-
рентерального застосування визначають шляхом по-
рівняння з еталонами відповідно до вимог статті "Ви-
значення ступеня забарвлення рідин" (2.2.2) або до за-
значень окремої статті.
Об’єм, що витягається. Визначення проводять відпо-
відно до вимог статті "Об’єм, що витягається"(2.9.17).
Однорідність вмісту. Порошки для ін’єкційних або
внутрішньовенних інфузійних лікарських засобів, а
також однодозові суспензії для ін’єкцій мають відпо-
відати вимогам статті "Однорідність вмісту діючої ре-
човини в одиниці дозованого лікарського засобу"(2.9.6),
якщо немає інших зазначень в окремій статті.
Пірогени. Випробуванню на наявність пірогенів мають
підлягати всі лікарські засоби для парентерального
застосування незалежно від дози, об’єму і шляху
введення, використовуваних у клініці.
Випробування на наявність пірогенів проводять
відповідно до вимог статті "Пірогени"(2.6.8) або "Бак-
теріальні ендотоксини" (2.6.14).
Аномальна токсичність. Випробування на аномальну
токсичність проводять відповідно до вимог статті
"Аномальна токсичність " (2.6.9).
Механічні включення. Випробування проводять відповід-
но до вимог статті "Механічні включення"(2.9.19 - 2.9.21).
Стійкість суспензії та інші показники. Суспензії для па-
рентерального застосування після струшування до
одержання однорідної суспензії мають зберігати од-
норідність протягом не менше 5 хв, якщо немає інших
зазначень в окремій статті.
Суспензія має вільно проходити в шприц крізь голку
№ 0840, якщо немає інших зазначень в окремій статті.
Розмір часток для суспензій контролюють за методи-
ками, зазначеними в окремій статті.
Кількісне визначення. Вміст визначуваних речовин в
рідких лікарських засобах для парентерального засто-
сування виражають у грамах або міліграмах в 1 мл
препарату, якщо немає інших зазначень в окремій
статті.
Вміст визначуваних речовин у порошках для ін’єкцій
або внутрішньовенних інфузій в однодозових контей-
нерах виражають у грамах, міліграмах або одиницях дії
(ОД) в одній дозі, якщо немає інших зазначень в ок-
ремій статті.
МАРКУВАННЯ
Ін’єкційні лікарські засоби
На кожному контейнері зазначають назву лікарського
засобу, його концентрацію або активність, об’єм або
масу, номер серії.
На етикетці додатково зазначають:
— назву і концентрацію діючих речовин для багато-
компонентних препаратів;
— спосіб введення;
— стерильно;
— апірогенний чи вільний від ендотоксинів;
— перелік допоміжних речовин;
— термін придатності;
— умови зберігання.
На етикетці лікарських засобів для парентерального
застосування в однодозових контейнерах зазначають,
що будь-яка порція вмісту, що залишилася, не має
більше використовуватися для застосування.
Внутрішньовенні інфузійні
лікарські засоби
На етикетці додатково до маркування, наведеного для
ін’єкційних лікарських засобів, зазначають:
— осмоляльність (осмолярність);
— склад препарату;
— іонний склад препарату в ммоль/л.
Порошки для ін’єкційних
лікарських засобів
На етикетці додатково до маркування, наведеного для
ін’єкційних лікарських засобів, зазначають відомості
про застосування.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
501
Лікарські засоби для ректального застосування
Якщо до порошку для ін’єкцій додається контейнер із
розчинником для приготування парентерального
лікарського засобу, на етикетці контейнера зазнача-
ють склад розчинника.
Концентровані розчини для
приготування ін’єкційних або
внутрішньовенних інфузійних
лікарських засобів
На етикетці додатково до маркування, наведеного для
ін’єкційних лікарських засобів, зазначають, що роз-
чин розводять перед використанням відповідно до
інструкції для застосування
ЛІКАРСЬКІ ЗАСОБИ ДЛЯ
РЕКТАЛЬНОГО ЗАСТОСУВАННЯ
Кесіаііа
ВИЗНАЧЕННЯ
Лікарські засоби для ректального застосування (рек-
тальні лікарські засоби) призначені для введення у
пряму кишку з метою одержання загальної або місце-
вої дії. Вони можуть також бути використані з діагно-
стичною метою.
Контейнери для ректальних лікарських засобів мають
відповідати вимогам статей "Матеріали, використову-
вані для виробництва контейнерів" (3. і та підрозділи)
та "Контейнери"(3.2та підрозділи), якщо немає інших
зазначень в окремій статті.
Лікарські засоби для ректального застосування мо-
жуть бути класифіковані як:
— ректальні супозиторії;
— ректальні капсули;
— ректальні розчини і суспензії;
— порошки і таблетки для приготування ректальних
розчинів або суспензій:
— м’які лікарські засоби для ректального застосу-
вання;
— ректальні піни;
— ректальні тампони.
ВИРОБНИЦТВО
При розробці лікарських засобів для ректального за-
стосування, до складу яких входять антимікробні кон-
серванти, уповноваженому органу мають бути надані
дані, шо підтверджують ефективність вибраних кон-
сервантів. Метод визначення і критерії ефективності
консервантів мають відповідати вимогам статті "Ефек-
тивність антимікробних консервантів"(5.1.3).
При виробництві, пакуванні, зберіганні й реалізації
лікарських засобів для ректального застосування ма-
ють бути вжиті відповідні заходи, шо забезпечують не-
обхідну мікробіологічну чистоту відповідно до вимог
статті "Мікробіологічна чистота лікарських засобів"
(5.1.4).
При виробництві м’яких і рідких лікарських засобів
для ректального застосування, що містять дисперго-
вані частки, належить передбачити заходи, які забез-
печують необхідний розмір часток і його контроль.
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Якшо немає інших зазна-
чень в окремій статті, тверді лікарські засоби в одно-
дозових контейнерах із вмістом діючої речовини мен-
ше 2 мг або менше 2 % від загальної маси мають ви-
тримувати випробування однорідності вмісту діючої
речовини в одиниці дозованого лікарського засобу
(тест А — таблетки або тест В — супозиторії, ректальні
капсули). Якщо лікарська форма містить більше однієї
діючої речовини, вимоги поширюються лише на ті ре-
човини, вміст яких відповідає вищезазначеним умо-
вам.
Однорідність маси (2.9.5). Тверді лікарські засоби в од-
нодозових контейнерах мають витримувати випробу-
вання однорідності маси для одиниці дозованого
лікарського засобу. Випробування однорідності маси
не вимагається, якщо випробування однорідності
вмісту передбачене для всіх діючих речовин.
Розчинення. Для підтвердження відповідного ви-
вільнення діючої речовини або речовин з твердих од-
нодозових препаратів випробування може бути прове-
дене, наприклад, одним із методів, описаних для су-
позиторіїв і м’яких капсул (2.9.3).
Якщо проводять тест за показником "Розчинення",
випробування "Розпадання" не вимагається.
МАРКУВАННЯ
На етикетці зазначають назву кожного антимікробно-
го консерванта.
Ректальні супозиторії
ВИЗНАЧЕННЯ
Супозиторії — тверді однодозові лікарські засоби
Форма, об’єм і консистенція супозиторіїв мають
відповідати ректальному застосуванню.
Вони містять одну або більше діючих речовин, дис-
пергованих або розчинених у простій чи складній ос-
нові, яка може розчинятися, або диспергуватися у
воді, або плавитися при температурі тіла. До складу
супозиторіїв, якщо необхідно, можуть входити до-
поміжні речовини, такі як розріджувачі, адсорбенти,
поверхнево-активні й змащувальні речовини, ан-
тимікробні консерванти, а також барвники, дозволені
до медичного застосування.
502
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лікарські засоби для ректального застосування
ВИРОБНИЦТВО
Супозиторії готують пресуванням або литтям. Якщо
необхідно, діючу речовину або речовини попередньо
здрібнюють і просіюють крізь відповідні сита. Якщо
супозиторії готують литтям, приготовану масу попе-
редньо розплавлюють при нагріванні й розливають у
відповідні форми. Супозиторії тверднуть при охолод-
женні. Щоб забезпечити процес тверднення, додають
такі допоміжні речовини, як тверді жири, макроголи,
олію какао, різні гелеутворюючі суміші, які містять,
наприклад, желатин, воду і гліцерин.
Обирають випробування, що підтверджує відповідне
вивільнення діючої речовини або речовин із супози-
торіїв, призначених для модифікованого вивільнення
або пролонгованої місцевої дії.
ВИПРОБУВАННЯ
Розпадання. Якщо супозиторії не призначені для мо-
дифікованого вивільнення або місцевої пролонгова-
ної дії, вони мають витримувати випробування розпа-
дання супозиторіїв і песаріїв (2.9.2). Якщо немає
інших зазначень в окремій статті, стан супозиторіїв на
жировій основі досліджують через ЗО хв, а супози-
торіїв на гідрофільній основі — через 60 хв.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах.
Ректальні капсули
ВИЗНАЧЕННЯ
Ректальні капсули (супозиторії з оболонкою) — це
тверда однодозова лікарська форма, в основному схо-
жа за визначенням з м’якими капсулами, наведеним у
статті "Капсули". Крім того, вони можуть мати ковзну
оболонку. Вони мають бути гладкими, однорідними за
зовнішнім виглядом і мати подовжену форму.
ВИРОБНИЦТВО
Обирають випробування, що підтверджує відповідне
вивільнення діючої речовини або речовин з ректаль-
них капсул, призначених для модифікованого
вивільнення або пролонгованої місцевої дії.
Ректальні розчини і суспензії
ВИЗНАЧЕННЯ
Ректальні розчини і суспензії (клізми) — рідкі
лікарські засоби, призначені для введення у пряму
кишку з метою одержання загальної або місцевої дії.
Вони можуть також бути використані з діагностичною
метою.
Ректальні розчини і суспензії — однодозові лікарські
засоби, що містять одну або більше діючих речовин,
розчинених або диспергованих у воді, гліцерині, мак-
роголах або в інших підхожих розчинниках. У сус-
пензіях допускається наявність осаду, що легко дис
пергується при збовтуванні з утворенням суспензії,
яка має бути достатньо стабільною для введення
відповідної дози.
Ректальні розчини або суспензії можуть містити до-
поміжні речовини, призначені для забезпечення не-
обхідної в’язкості, забезпечення або стабілізації рН,
для покрашення розчинності діючої речовини (речо-
вин), або для стабілізації лікарського засобу. Ці речо-
вини не мають несприятливо впливати на основну
терапевтичну дію і, у використовуваних концентра-
ціях, не мають справляти надмірну місцеву подразли-
ву дію.
Ректальні розчини і суспензії випускають у контейнерах
об’ємом від 2.5 мл до 2000 мл. Контейнер має бути при-
стосований для введення лікарського засобу в пряму
кишку або забезпечений відповідним аплікатором.
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Ректальні суспензії мають
витримувати таке випробування. Звільняють кожний
контейнер якнайповніше і визначають вміст діючої
речовини для кожного контейнера. Вони мають ви-
тримувати випробування однорідності вмісту (тест В).
Однорідність маси. Ректальні розчини мають витриму-
вати таке випробування. Звільняють кожний із 20 кон-
тейнерів якнайповніше і зважують вміст. Визначають
середню масу вмісту. Для контейнерів з масою вмісту
менше 100 г маса вмісту лише для двох контейнерів
може відхилятися більш як на (±10 %) від середньої
маси, і маса вмісту жодного контейнера не має відхи-
лятися більш як на (±20 %). Для контейнерів з масою
вмісту більше 100 г маса вмісту лише для двох контей-
нерів може відхилятися більш як на (±5 %) від серед-
ньої маси і маса вмісту жодного контейнера не має
відхилятися більш як на (±10 %) від середньої маси.
ВИПРОБУВАННЯ
Розпадання. Якщо капсули не призначені для мо-
дифікованого вивільнення або місцевої пролонгова-
ної дії, вони мають витримувати випробування розпа-
дання супозиторіїв і песаріїв (2.9.2). Стан капсул
досліджують через 30 хв, якшо немає інших зазначень
в окремій статті.
Порошки й таблетки для приготування
ректальних розчинів або суспензій
ВИЗНАЧЕННЯ
Порошки й таблетки, призначені для приготування рек-
тальних розчинів або суспензій, — це однодозові
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
503
Лікарські засоби для ректального застосування
лікарські засоби, шо розчиняють або диспергують у воді
безпосередньо перед застосуванням. Вони можуть
містити допоміжні речовини, які сприяють розчиненню
або диспергуванню і запобігають агрегації часток.
Після розчинення або диспергування вони мають
відповідати вимогам, які ставляться до ректальних
розчинів або суспензій.
ВИПРОБУВАННЯ
Розпадання. Таблетки, призначені для приготування
ректальних розчинів або суспензій, мають розпадати-
ся протягом 3 хв, якщо випробування проводять в
умовах, прийнятих для таблеток і капсул (2.9.1), але з
використанням як середовища води Рпри температурі
від 15 °С до 25 °С.
МАРКУВАННЯ
На етикетці зазначають:
— спосіб приготування ректального розчину або сус-
пензії;
— умови і термін зберігання розчину або суспензії
після приготування.
М’які лікарські засоби для
ректального застосування
ВИЗНАЧЕННЯ
М’які лікарські засоби для ректального застосуван-
ня — це креми, гелі й мазі.
Вони звичайно являють собою однодозові лікарські
засоби у контейнерах, забезпечених відповідним
аплікатором.
М’які лікарські засоби для ректального застосування
мають відповідати вимогам статті "М’які лікарські за-
соби для місцевого застосування "
Ректальні піни
ВИЗНАЧЕННЯ
Ректальні піни мають відповідати вимогам статті
"Піни медичні".
Ректальні тампони
ВИЗНАЧЕННЯ
Ректальні тампони — це тверда однодозова лікарська
форма, призначена для введення у нижню частину
прямої кишки на певний час.
Вони мають відповідати вимогам статті "Тампони ме-
дичні".
___________________________________________N
ВИПРОБУВАННЯ
Однорідність вмісту. Якщо немає інших зазначень в
окремій статті, тверді лікарські засоби в однодозових
контейнерах мають відповідати вимогам статті
"Однорідність вмісту діючої речовини в одиниці дозо-
ваного лікарського засобу"(2.9.6).
Ректальні супозиторії
ВИЗНАЧЕННЯ
Супозиторії можуть мати форму конуса, циліндра із
загостреним кінцем або іншу форму з максимальним
діаметром не більше 1.5 см.
Маса одного супозиторія звичайно знаходиться у ме-
жах від 1 г до 4 г, для дітей — від 0.5 г до 1.5 г.
Супозиторії мають бути однорідними. Однорідність
супозиторія визначають візуально на поздовжньому
зрізі. На зрізі мають бути відсутні вкраплення, допус-
кається наявність повітряного стрижня або лійко-
подібної заглибини.
ВИРОБНИЦТВО
Діючі речовини при необхідності здрібнюють,
просіюють, змішують з основою безпосередньо або
після розчинення чи розтирання з невеликою
кількістю води, гліцерину, вазелінового масла або
іншого підхожого розчинника. Термолабільні речови-
ни додають до напівохололої маси безпосередньо пе-
ред формуванням супозиторіїв.
Як ліпофільні основи для виготовлення супозиторіїв
застосовують масло какао, сплави масла какао з па-
рафіном і гідрогенізованими жирами, рослинні й тва-
ринні гідрогенізовані жири, твердий жир, ланоль,
сплави гідрогенізованих жирів із воском, твердим па-
рафіном та інші основи, дозволені до медичного за-
стосування.
Як гідрофільні основи використовують желатино-
гліцеринові гелі, сплави поліетиленоксидів із різними
молекулярними масами й інші речовини, дозволені до
медичного застосування. Желатино-гліцеринову ос-
нову виготовляють із желатину медичного, гліцерину і
води.
При виготовленні супозиторіїв можуть застосовувати-
ся бутилгідрокситолуол. бутилгідроксианізол, лимон-
на кислота, емульгатори (емульгатор № 1, емульгатор
Т-1), спирти шерстного воску, аеросил й інші до-
поміжні речовини, дозволені до медичного застосу-
вання.
Супозиторії можуть бути одержані методом викачу-
вання. Як сполучну речовину при цьому застосовують
ланолін безводний.
504
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Лікарські засоби для ректального застосування
ВИПРОБУВАННЯ
Лікарські засоби для ректального застосування іви-
чайно контролюють за такими показниками якості:
опис, ідентифікація, середня маса і однорідність маси
(для супозиторіїв і ректальних капсул, таблеток для
приготування ректальних розчинів і суспензій, а та-
кож для ректальних розчинів і суспензій), розпадання
(для супозиторіїв і ректальних капсул, таблеток для
приготування ректальних розчинів і суспензій), од-
норідність вмісту, температура плавлення або час пов-
ної деформації, розчинення, супровідні домішки,
мікробіологічна чистота, кількісне визначення.
У лікарських засобах для ректального застосування
при необхідності додатково контролюють кислотне і
перекисне числа, а також розмір часток.
Середня маса. Визначення середньої маси проводять
для супозиторіїв і ректальних капсул, таблеток для
приготування ректальних розчинів і суспензій, а та-
кож для ректальних розчинів і суспензій. Відхилення
середньої маси від маси, зазначеної у розділі "Склад",
не має перевищувати (±5 %), якшо немає інших
зазначень в окремій статті. Визначення середньої ма-
си проводять, як зазначено у статті "Однорідність маси
для одиниці дозованого лікарського засобу" (2.9.5).
Температура плавлення. Для супозиторіїв, виготовле-
них на ліпофільній основі, визначають температуру
плавлення за методом (2.2.15), яка не має перевищу-
вати 37 °С, якшо немає інших зазначень в окремій
статті.
Час повної деформації. Для супозиторіїв визначають
час повної деформації згідно з Додатком 1. Допус-
кається використання інших приладів. Час повної де-
формації має бути не більше 15 хв, якщо немає інших
зазначень в окремій статті.
Кількісне визначення. Вміст визначуваних речовин ви-
ражають у грамах, міліграмах або в одиницях дії (ОД)
в одиниці дозованого лікарського засобу або в 1 г
лікарського засобу, якщо немає інших зазначень в ок-
ремій статті.
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву діючої речовини (речовин) та її (їх) вміст у
препараті або в одиниці дозованого лікарського
засобу;
— спосіб застосування;
— умови зберігання;
— термін придатності.
ДОДАТОК 1
Визначення часу повної деформації
Визначення часу повної деформації проводять у скля-
ному приладі (див. Рис. 1), шо складається з відкритої
із обох боків скляної трубки з капілярним переходом
(?), скляного штока (в) і металевого стрижня (д) масою
7.5 г і діаметром 2 мм. Трубку (?) з короткого кінця за-
кривають пробкою і заповнюють водою температури
(37 ± 1)°С. Перед початком визначення прилад помі-
щають у посудину з циркулюючою водою при темпе-
ратурі (37 ± 1) °С. Супозиторій, попередньо витрима-
ний на льоду протягом 15 хв, уводять у трубку (?) і
закріплюють за допомогою штока (в), потім негайно
на супозиторії встановлюють металевий стрижень (д) і
вмикають секундомір. Заміряють час від введення су-
позиторія в трубку (?) до появи стрижня (д) унизу зву-
ження трубки. Цей час беруть за час повної дефор-
мації супозиторія.
Рисунок 1. Прилад для визначення часу повної
деформації супозиторіїв
а — скляна посудина; б — термометр, ціна поділки
1 °С; в — скляний шток; ? — скляна трубка для проб;
д — металевий стрижень.
Ректальні розчини і суспензії
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Ректальні суспензії мають
витримувати таке випробування. Звільняють кожний
контейнер якнайповніше і визначають вміст діючої
речовини для кожного контейнера. Вони мають ви-
тримувати випробування однорідності вмісту.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
505
Лікарські засоби, що знаходяться під тиском
ЛІКАРСЬКІ ЗАСОБИ,
ЩО ЗНАХОДЯТЬСЯ ПІД ТИСКОМ
Ргаерагаііопев рЬагтасеиїісаеп
іп уа8І8 сит ргевви
Талі, де це необхідно, до лікарських засобів, що знахо-
дяться під тиском, ставлять додаткові вимоги, зазна-
чені в інших статтях, наприклад, "Лікарські засоби для
інгаляції", "Рідкі лікарські засоби для зовнішнього за-
стосування ", "Порошки для зовнішнього застосування ”,
"Назальні лікарські засоби" та "Вушні лікарські засо-
би".
ВИЗНАЧЕННЯ
Лікарські засоби, що знаходяться під тиском, — це
лікарські засоби, що знаходяться у спеціальних кон-
тейнерах під тиском газу і містять одну або більше
діючих речовин. Дані лікарські засоби при виході з
контейнера при натискуванні на клапан являють со-
бою аерозоль (дисперсію твердих або рідких часток в
газі, розмір яких залежить від призначення лікарсько-
го засобу), рідину або м’яку піну Тиск, необхідний для
виходу лікарського засобу з контейнера, забезпечують
відповідні пропеленти. Лікарські засоби, шо зна-
ходяться під тиском, являють собою розчин, емульсію
або суспензію. Вони призначені для місцевого нане-
сення на шкіру, на слизові оболонки або для інгаляцій.
До складу лікарських засобів можуть входити такі до-
поміжні речовини, як солюбілізатори, суспендуючі
речовини, емульгатори, розчинники, а також ковзні
речовини, що запобігають засмічуванню клапана.
Пропеленти. Пропеленти являють собою зріджені під
тиском гази, стиснуті гази або низькокиплячі рідини.
Зрідженими газами є фторовані вуглеводні й вугле-
водні з низькою молекулярною масою, такі як пропан
або бутан. Стиснуті гази — це вуглецю діоксид, азот
або азоту оксид.
Можуть використовуватися суміші пропелентів для
одержання лікарського засобу з оптимальними влас-
тивостями і заданими характеристиками тиску, дози і
розпилення.
Контейнери. Контейнери мають бути міцними і
стійкими до внутрішнього тиску. Вони можуть бути
виготовлені з металу, скла, пластика або комбінації
цих матеріалів і не мають взаємодіяти з вмістом.
Скляні контейнери повинні мати захисне пластикове
покриття.
Розпилюючі пристрої. Клапан має герметично закри-
вати контейнер у неробочому положенні і забезпечу-
вати необхідну дозу лікарського засобу в процесі ви-
користання. На характеристику розпилення вплива-
ють розміри, кількість і розташування отворів, а також
тип розпилюючого пристрою. Клапани забезпечують
неперервний вихід (клапан неперервної дії) або вида-
ють відміряну кількість лікарського засобу при кож-
ному натискуванні (клапан дозувальної дії).
Матеріали, використовувані для виробництва клапа-
нів, не мають взаємодіяти з вмістом контейнера.
Вимоги, які ставляться до лікарських засобів, що знахо-
дяться під тиском. Лікарські засоби, що знаходяться
під тиском, мають бути оснащені розпилюючим при-
строєм відповідно до його призначення.
При необхідності лікарські засоби мають відповідати
вимогам, які ставляться до пропелентів, розміру час-
ток, дози, одержуваної при одному натискуванні на
дозувальний клапан.
МАРКУВАННЯ
На етикетці зазначають:
— спосіб застосування;
— запобіжні заходи;
— для лікарських засобів, що знаходяться у контей-
нері з клапаном дозувальної дії, зазначають
кількість діючої речовини в одній дозі.
______________________________________________N
ВИПРОБУВАННЯ
Лікарські засоби, що знаходяться під тиском, звичай-
но контролюють за такими показниками якості:
опис, перевірка на герметичність, вимірювання тиску
всередині контейнера, визначення відсотка виходу
вмісту контейнера, ідентифікація, супровідні до-
мішки, мікробіологічна чистота, кількісне визначен-
ня.
Для лікарських засобів, шо знаходяться під тиском,
споряджених дозувальним клапаном, додатково кон-
тролюють середню масу лікарського засобу в одній
дозі і кількість доз, що витягаються з контейнера.
Для лікарських засобів у вигляді суспензій або
емульсій, призначених для загальної дії, що знахо-
дяться під тиском із клапаном дозувальної дії, додат-
ково контролюють однорідність дозування.
Для лікарських засобів, що знаходяться під тиском, у
вигляді суспензій для введення в бронхи й легені до-
датково контролюють розмір часток.
Перевірка контейнера на герметичність. Для перевірки
контейнера на герметичність може бути використана
така методика. Контейнер без ковпачка і розпилювача
або насадки повністю занурюють у водяну баню при
температурі (45 ± 5) °С не менш як на 15 хв і не більш
як на ЗО хв для скляних балонів, покритих пластико-
вим покриттям, та не менш як на 10 хв і не більш як на
20 хв — для металевих балонів. Товщина шару води
над штоком клапана має бути не менше 1 см. Не має
спостерігатися виділення бульбашок газу.
Вимірювання тиску всередині контейнера. Проводять
для лікарських засобів, що знаходяться під тиском, в
яких як пропеленти використовують стиснуті гази.
Перед вимірюванням тиску контейнери витримують
506
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
М’які лікарські засоби для місцевого застосування1'1
при температурі (20 ± 2) °С протягом 1 год. Контроль
тиску проводять за допомогою манометра (клас точ-
ності 2.5). Гранично допустимий тиск у контейнері
при 20 °С має бути не вище 0.8 МПа.
Визначення відсотка виходу вмісту контейнера. Контей-
нер зважують з точністю до 0.01 г (/л,), видаляють
вміст і зважують контейнер (/я4). Вихід вмісту у відсот-
ках (А) обчислюють за формулою:
т, - тл
Х = ----' - х 100 ,
т5
де:
т5 — маса вмісту, зазначена на етикетці.
Випробування проводять при температурі (20 ± 2) °С.
Відсоток виходу вмісту контейнера зазначають в ок-
ремій статті.
Визначення середньої маси лікарського засобу в одній
дозі. Проводять для лікарських засобів, шо знаходять-
ся під тиском, споряджених дозувальним клапаном.
За допомогою розпилювача роблять перші п’ять нати-
скувань і контейнер з розпилювачем зважують з
точністю до 0.01 г (ш2)- Потім роблять точну кількість
натискувань (від 10 до 20) з інтервалом 10-15 с і зважу-
ють (т3). Середню масу однієї дози у грамах (тср) об-
числюють за формулою:
- т3
де:
п — число натискувань, зазначене в окремій
статті.
Випробування проводять при температурі (20 ± 2) °С.
Межі визначення середньої маси лікарського засобу у
дозі зазначають в окремій статті.
Кількість доз лікарського засобу, що втягаються. Про-
водять для лікарських засобів, що знаходяться під ти-
ском, споряджених дозувальним клапаном. Середню
кількість доз (И), що витягаються, в одному контей-
нері обчислюють за формулою:
Визначення розміру часток. Проводять для лікарських
засобів, шо знаходяться під тиском і являють собою
суспензії для введення у бронхи і легені. Випробуван-
ня проводять мікроскопічним методом. Методики
визначення і вимоги до розміру часток мають бути за-
значені в окремій статті. Якщо немає інших зазначень
в окремій статті, кількість часток розміром менше
10 мкм має бути не менше 95 %, при цьому кількість
часток розміром менше 5 мкм має бути не менше 90 %.
Однорідність дозування. Лікарські засоби, що являють
собою дозовані аерозолі у вигляді суспензій або
емульсій, призначені для загальної дії. мають витри-
мувати випробування однорідності дозування, зазна-
чене у статті "Лікарські засоби для інгаляції".
М’ЯКІ ЛІКАРСЬКІ ЗАСОБИ ДЛЯ
МІСЦЕВОГО ЗАСТОСУВАННЯ14
ІІП£иепіа
Вимоги даної статті поширюються на всі м 'які
лікарські засоби, які умовно об’єднуються загальним
поняттям "мазі" ("Іїпуиепіа") і призначені для місцево-
го застосування. Там, де це необхідно, до м 'яких лікар-
ських засобів, призначених для застосування на певних
поверхнях тіла або слизових оболонках, мають бути ви-
сунуті додаткові вимоги у відповідних статтях, таких
як: "Вушні лікарські засоби", "Очні лікарські засоби",
"Назальні лікарські засоби", "Лікарські засоби для рек-
тального застосування" та "Лікарські засоби для
вагінального застосування".
ВИЗНАЧЕННЯ
М’які лікарські засоби для місцевого застосування
призначені для нанесення на шкіру, рани і певні сли-
зові оболонки для місцевої терапевтичної зм’якшу-
вальної або захисної дії або для проникнення лікарсь-
ких речовин крізь шкіру або слизові оболонки. Вони
характеризуються специфічними реологічними влас-
тивостями при встановленій температурі зберігання:
неньютонівським типом течії, певною структурною
в’язкістю, псевдопластичними або пластичними і
тиксотропними властивостями. За зовнішнім вигля-
дом вони мають бути однорідними, крім тих випадків,
коли неоднорідність є характерною особливістю пре-
парату.
М’які лікарські засоби звичайно містять діючі та до-
поміжні речовини. Допоміжні речовини утворюють
просту або складну основу, яку можна виробляти ок-
ремо або одержувати у процесі виготовлення м’якого
лікарського засобу. Діючі речовини мають бути
рівномірно розподілені в основі, яка залежно від її
складу і властивостей може впливати на їх вивільнен-
ня, біодоступність і терапевтичну дію.
М’які лікарські засоби і основи можуть являти собою
одно-, дво- або багатофазові системи і складатися з
природних і/або синтетичних речовин.
Допоміжні речовини, що входять до складу м’яких
лікарських засобів, за функціональним призначенням
можна класифікувати як:
— м’які основи-носїї (вазелін, ланолін та ін.);
— речовини, що підвищують температуру плавлення
і в’язкість (парафін, спермацет, гідрогенізовані
рослинні олії, воски, макроголи з високою моле-
кулярною масою та ін.);
— гідрофобні розчинники (мінеральні та рослинні
олії, ізопропілпальмітат, ізопропілміристат,
поліалкілсилоксани, бензилбензоат та ін.);
— вода і гідрофільні розчинники (етанол та ізопро-
панол, макроголи 200-600, пропіленгліколь,
пропіленкарбонат, гліцерин, диметилсульфоксид
та ін.);
— емульгатори типу масло/вода (м/в) (натрію лау-
рилсульфат, емульгуючий віск (емульгатор № 1),
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
507
М’які лікарські засоби для місцевого застосування"
полісорбати, поліоксиетиленгліколеві ефіри ви-
щих жирних спиртів, цетилпіридинію хлорид, солі
вищих жирних кислот, оксіетильована рицинова
олія, поліоксиетиленгліколеві ефіри стеаринової
кислоти та ін.);
— емульгатори типу вода/масло (в/м) (вищі жирні
спирти, холестерин, спирти шерстного воску,
спени, гліцерилмоноолеат, гліцерилмоностеарат
та ін.),
— гелеутворювачі (карбомери, альгінова кислота та її
солі, похідні целюлози, поліетилен низькомолеку-
лярний, полоксамери або проксаноли, макрого-
ли 1500-8000, бентоніт, каолін, кремнію діоксид
колоїдний, гуміарабік, трагакант, желатин та ін.);
— антимікробні консерванти (бензалконію хлорид,
мірамістин, цетримід, цетилпіридинію хлорид,
солі хлоргексидину, бензойна і сорбінова кислоти
та їх солі, ефіри п-гідроксибензойної кислоти (па-
рабени), спирт бензиловий, крезол, хлоркрезол,
імідосечовина, феноксиетанол, макрогол, етанол
та ін.);
— антиоксиданти (альфа-токоферол, аскорбінова
кислота та її похідні, бутилгідроксіанізол і бу-
тил гідрокситолуол, етилендіамінтетраоцтова
кислота та її солі, лимонна кислота, пропілгалат,
натрію метабісульфіт та ін.);
— солюбілізатори (бета-циклодекстрин, гідрофільні
поверхнево-активні речовини (ПАР) та ін.);
— запашники (ментол, ефірні олії, фенілетанол та
ін.);
— речовини для створення або стабілізації певного
значення рН (лимонна кислота, фосфорнокислі
солі натрію та ін.);
— барвники, коригенти смаку та ін.
Деякі допоміжні речовини, крім того, можуть правити
за зм’якшувальні і зволожувальні добавки, пенетрато-
ри, змочувані та ін. Допоміжні речовини одночасно
можуть виконувати декілька вищеперерахованих
функцій, наприклад, гелеутворювачі, емульгатори й
речовини, що підвищують температуру плавлення і
в’язкість основ, є також стабілізаторами дисперсних
систем. Деякі допоміжні речовини являють собою
суміші різних речовин: ланолін водний, емульгуючий
віск (емульгатор № 1), неіоногенний емульсійний
віск, сплав вазеліну зі спиртами шерстного воску та ін.
М’які лікарські засоби і основи можуть бути кла-
сифіковані за такими ознаками:
— за спорідненістю до води: на гідрофільні і гідро-
фобні (ліпофільні);
— за здатністю абсорбувати воду і механізмом її аб-
сорбції;
— за типом дисперсних систем: на розчини, сплави
(однофазні системи); емульсії типу м/в і в/м, сус-
пензії, колоїдні дисперсії вищих жирних спиртів
або кислот, стабілізовані гідрофільними ПАР
(двофазові системи); множинні емульсії м/в/м і
в/м/в, а також комбіновані системи (багатофазові
системи);
— за реологічними властивостями м’якого лікар-
ського засобу і/або дисперсійного середовища при
установлених температурі зберігання і способі за-
стосування;
— за концентрацією і дисперсним станом до-
поміжних і/або лікарських речовин.
За сукупністю цих ознак м’які лікарські засоби для
місцевого застосування (Цп^иепіа) можуть бути кла-
сифіковані як:
— мазі;
— креми;
— гелі;
— пасти;
— лініменти.
Мазі
Мазі — м’які лікарські засоби для місцевого застосу-
вання, дисперсійне середовище яких при установ-
леній температурі зберігання має неньютонівський
тип течії і високе значення реологічних параметрів.
Гідрофобні мазі
Гідрофобні мазі приготовані, як правило, на вуглевод-
невих основах (вазелін, вазелінове масло, парафін) і
можуть містити інші ліпофільні допоміжні речовини
(рослинні олії, жири тваринного походження, воски,
синтетичні гліцериди і рідкі поліалкілсилоксани). До
їх складу може бути уведена лише незначна кількість
води або водних розчинів. Гідрофобні мазі при засто-
суванні мають оклюзійний (який запобігає контакту з
повітрям) ефект, справляють зм’якшувальну дію, важ-
ко змиваються водою і не змішуються з ексудатом.
Гідрофобні абсорбційні мазі
Гідрофобні абсорбційні мазі при втиранні в шкіру мо-
жуть абсорбувати (емульгувати) ексудат. Основи для
них можуть бути розділені на дві групи:
— гідрофобні основи, що складаються з вуглеводнів і
емульгаторів типу в/м (вазелін і ланолін або спир-
ти шерстного воску), до складу яких може бути
уведена значна кількість води або водних розчинів
з утворенням емульсії типу в/м;
— гідрофобні основи, які є емульсіями типу в/м або
м/в/м (вазелін і водний ланолін); до їх складу шля-
хом емульгування додатково може бути введена
вода або водний розчин.
Гідрофільні мазі
Гідрофільні мазі, як правило, є гіперосмолярними і
при застосуванні можуть абсорбувати значну кількість
ексудату. Основи для них можуть бути розділені на дві
групи:
— водорозчинні основи, що, як правило, містять
гідрофільні неводні розчинники (макрогол 400,
пропіленгліколь та ін.) і досить великі концент-
рації водорозчинних полімерів (макрогол 1500,
проксанол 268 та ін.);
— водозмивні основи, які, крім водорозчинних
полімерів і гідрофільних неводних розчинників,
містять ліпофільні речовини (вищі жирні спирти,
вазелін, вазелінове масло, ланолін, воски та ін.).
508
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
М’які лікарські засоби для місцевого застосування'
Ці основи, як правило, являють собою емульсії типу
м/в і вимагають присутності емульгатора типу м/в.
Креми
Креми — м’які лікарські засоби для місцевого застосу-
вання, що являють собою дво- або багатофазові дис-
персні системи, дисперсійне середовище яких при
установленій температурі зберігання, як правило, має
ньютонівський тип течії і низькі значення реологічних
параметрів.
Гідрофобні креми
Гідрофобні креми приготовані на основі емульсії в/м
або м/в/м, стабілізованої підхожими емульгаторами.
Гідрофільні креми
Гідрофільні креми приготовані на основі емульсії м/в
або в/м/в, стабілізованої підхожими емульгаторами.
До них також зараховують колоїдні дисперсні систе-
ми, шо складаються з диспергованих у воді або у
змішаних водно-гліколевих розчинниках вищих жир-
них спиртів або кислот і стабілізовані гідрофільними
ПАР.
Гелі
Гелі — м’які лікарські засоби для місцевого застосу-
вання, що являють собою одно-, дво- або багатофазові
дисперсні системи з рідким дисперсійним середови-
щем, реологічні властивості яких обумовлені при-
сутністю гелеутворювачів у порівняно невеликих кон-
центраціях. При цьому гелеутворювачі додатково мо-
жуть виконувати роль стабілізаторів дисперсних сис-
тем: суспензій або емульсій; такі гелі можуть назива-
тися відповідно суспензійними гелями або емульгеля-
ми.
Гідрофобні гелі
Гідрофобні гелі (олеогелі) приготовані на основах, що
складаються з гідрофобного розчинника (вазелінове
або рослинне масло та ін.) і ліпофільного гелеутворю-
вача (поліетилен низькомолекулярний, кремнію діок-
сид колоїдний, алюмінієве або цинкове мило та ін.).
Гідрофільні гелі
Гідрофільні гелі (гідрогелі) приготовані на основах,
що складаються з води, гідрофільного змішаного або
неводного розчинника (гліцерин, пропіленгліколь,
етанол, ізопропанол) і гідрофільного гелеутворювача
(карбомери. похідні целюлози, трагакантта ін.).
Пасти
Пасти — м’які лікарські засоби для місцевого застосу-
вання, що являють собою суспензії, які містять значну
кількість (звичайно більше 20 % м/м) твердої дисперс-
ної фази, рівномірно розподіленої в основі. Як основа
для паст можуть бути використані основи для мазей,
кремів і гелів.
Лініменти
Лініменти — м’які лікарські засоби для місцевого за-
стосування, які плавляться при температурі тіла. До
лініментів можуть бути зараховані мазі, креми, гелі та
пасти, що характеризуються цією ознакою.
Спеціальні вимоги
При розробці м’яких лікарських засобів треба подати
до уповноваженого органу обгрунтування вибору
складу і первинної упаковки, а також пояснення ролі
кожної з допоміжних речовин препарату. Основу для
м’яких лікарських засобів треба обирати з урахуван-
ням призначення препарату, його ефективності й
нешкідливості, біодоступності діючих речовин,
сумісності компонентів препарату, реологічних влас-
тивостей, фізико-хімічної, хімічної і мікробіологічної
стабільності протягом терміну придатності.
При розробці, виробництві, пакуванні, зберіганні, ре-
алізації і застосуванні м’яких лікарських засобів ма-
ють бути вжиті відповідні заходи, які забезпечують не-
обхідну мікробіологічну чистоту ВІДПОВІДНО до вимог
статті "Мікробіологічна чистота лікарських засобів"
(5.1.4).
Мікробіологічну чистоту м’яких лікарських засобів
треба забезпечувати за рахунок наявності антимікроб-
ної консервуючої дії компонентів і/або належних умов
виробництва. Ефективність антимікробної консерву-
ючої дії має бути підтверджена експериментально
відповідно до методів і критеріїв оцінки, викладених у
статті "Ефективність антимікробних консервантів"
(5.1.3). Вибір консервантів і антиоксидантів треба
здійснювати з урахуванням таких факторів, як умови
зберігання, частота розпакування, додаткова підго-
товка препарату для застосування Якщо для ефектив-
ності консерванта або стабільності препарату має зна-
чення величина рН, треба установити межі рН і пода-
ти відповідні експериментальні дані до уповноваже-
ного органу^
М’які лікарські засоби для місцевого застосування, при-
значені для нанесення на великі відкриті рани або на ду-
же ушкоджену шкіру, при відсутності в них ефективної
консервуючої дії (5.1.3) мають бути стерильними.
Стерильні м’які лікарські засоби треба виробляти з
використанням вихідної сировини, первинних паку-
вальних матеріалів і за допомогою способів, що забез-
печують стерильність і запобігають забрудненню
мікроорганізмами та їх розмноженню, відповідно до
вимог статті "Методи приготування стерильних
продуктів"(5.1.1). Для таких препаратів необхідно ус-
тановлювати термін зберігання після першого роз-
криття.
При виробництві, зберіганні й реалізації м’яких
лікарських засобів необхідно вжити заходів, які забез-
ЛГРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 7ПП1
^па
М’які лікарські засоби для місцевого застосування”
печують їхню однорідність (рівномірний розподіл
лікарських і допоміжних речовин, відсутність сторон-
ніх включень). Необхідно передбачити процедури, що
контролюють однорідність м'яких лікарських засобів.
При виробництві м’яких лікарських засобів, які
містять дисперговані частки, необхідно передбачити
заходи щодо забезпечення і контролю необхідного
розміру часток, зумовленого призначенням даного
лікарського засобу.
ВИПРОБУВАННЯ
М’які лікарські засоби контролюють за такими показ-
никами якості: опис, ідентифікація, однорідність, ма-
са вмісту контейнера, мікробіологічна чистота,
кількісне визначення.
При необхідності додатково контролюють розмір час-
ток, рН, кислотне і перекисне числа, характерні влас-
тивості основи, супровідні домішки, герметичність
контейнера.
Стерильні м’які лікарські засоби мають витримувати
випробування на стерильність (2.6.1).
В описі методик випробування якості м’яких лікарсь-
ких засобів необхідно зазначати методики відбору
аналізованої проби.
Опис. Контролюють зовнішній вигляд і характерні ор-
ганолептичні властивості. М’які лікарські засоби не
повинні мати згірклого запаху, а також ознак фізичної
нестабільності (агрегація часток, коалесценція, коагу-
ляція, розшарування), якшо немає інших зазначень в
окремій статті.
Ідентифікація. Проводять визначення ідентифікації
всіх діючих речовин і антимікробних консервантів, які
входять до складу препарату.
При необхідності визначають ідентифікацію допоміж-
них речовин.
Однорідність. М’які лікарські засоби мають бути од-
норідними. Однорідність визначають за зовнішнім
виглядом і за методикою, наведеною у Додатку 1.
При необхідності однорідність визначають за кількі-
сним вмістом компонентів при спеціальному відборі
проб, шо дозволяє контролювати рівномірність їх роз-
поділу.
Розмір часток. У м’яких лікарських засобах, що
містять компоненти у вигляді твердої або рідкої дис-
персної фази, контролюють розмір часток, якщо від
нього залежать біодоступність, терапевтична ефек-
тивність і нешкідливість або даний показник регла-
ментується призначенням препарату.
Вимоги, які ставляться до розміру часток, методики
визначення і критерії оцінки, наводять в окремій
статті. Розмір часток у м’яких лікарських засобах ви-
значають методом мікроскопії.
Герметичність контейнера. Для стерильних і при не-
обхідності для нестерильних м’яких лікарських за-
собів проводять визначення герметичності контей-
нера відповідно до методики, викладеної у Додатку 1.
рН (2.2.3). Залежно від типу основи і складу препарату
визначають рН водної витяжки, водного розчину або
самого лікарського засобу. Вимоги, які ставляться до
рН, і методики визначення наводять в окремій статті.
Кислотне число (2.5.1) і Перекисне число (2.5.5). Кон-
тролюють при необхідності у м’яких лікарських засо-
бах, до складу яких входять речовини, здатні до
гідролізу і окиснення. Регламентовані вимоги і мето-
дики визначення наводять в окремій статті.
Кількісне визначення. Проводять кількісне визначення
усіх діючих речовин. Допустиме відхилення вмісту
діючих речовин при їх дозуванні менше 10 % мають
складати (±10 %), при дозуванні 10 % і більше —
(±5 %) від вмісту, зазначеного у розділі "Склад", якщо
немає інших зазначень в окремій статті.
Кількісний вміст визначуваних речовин виражають у
грамах, міліграмах або одиницях дії (ОД) в 1 г препара-
ту, якщо немає інших зазначень в окремій статті. Для
консервантів регламентують і контролюють верхню і
нижню межі вмісту. Для інших допоміжних речовин,
здатних негативно впливати на фізіологічні функції,
контролюють і регламентують верхню межу вмісту. Як-
що допоміжна речовина впливає на біодоступність
діючої речовини, регламентують верхню і нижню межі
вмісту і проводять їх кількісне визначення.
УПАКОВКА
Упаковка для м’якихлікарських засобів має бути інди-
ферентною стосовно препарату; щільно закупорюва-
тися для запобігання контакту вмісту з навколишнім
середовищем; при необхідності вона має бути герме-
тичною і світлонепроникною. Краще використовува-
ти металеві необоротно стискувані туби із внутрішнім
лаковим покриттям, захисною мембраною і латек-
сним кільцем. Можуть бути використані інші види
первинної упаковки, відповідні вищеперерахованим
вимогам.
Контейнер зі стерильними м’якими лікарськими за-
собами має бути герметичним і мати пристрій для
контролю першого розкриття, наприклад, захисну
ме'мбрану.
Контейнер для назальних, вушних, очних, ректальних
і вагінальних м’якихлікарських засобів має бути спо-
ряджений відповідними аплікаторами.
Контейнери для м’якихлікарських засобів мають від-
повідати вимогам статей "Матеріали, використовувані
для виробництва контейнерів" (3.1 та підрозділи,)
та "Контейнери " (3.2 та п ідрозділ и).
МАРКУВАННЯ
Маркування додатково має містити такі відомості:
— назву і кількісний вміст усіх антимікробних кон-
сервантів;
— назву всіх допоміжних речовин;
510
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Назальні лікарські засоби
— "Для місцевого застосування";
— при необхідності термін зберігання після першого
розкриття;
— для стерильних препаратів — зазначення про сте-
рильність.
ЗБЕРІГАННЯ
При температурі не вище +25 °С, якщо немає інших
зазначень в окремій статті; не допускається заморожу-
вання.
ДОДАТОК 1
МЕТОДИКА ВИЗНАЧЕННЯ ОДНОРІДНОСТІ
Беруть чотири проби препарату по 20-30 мг кожна,
поміщають по дві проби на предметне скло, накрива-
ють другим предметним склом і міцно притискують до
утворення плям діаметром близько 2 см.
При розгляді одержаних проб неозброєним оком (на
відстані близько ЗО см від очей) у всіх чотирьох пробах
не мають виявлятися видимі частки, сторонні вклю-
чення і, якщо немає інших зазначень в окремій статті,
ознаки фізичної нестабільності; агрегація і коалес-
ценція часток, коагуляція.
Якщо одна з проб не витримує випробування, ви-
значення проводять додатково ще на восьми пробах.
При цьому вісім додаткових проб мають витримувати
випробування.
НАЗАЛЬНІ ЛІКАРСЬКІ ЗАСОБИ
№$а1іа
ВИЗНАЧЕННЯ
Назальні лікарські засоби являють собою рідкі, м’які
або тверді лікарські засоби, призначені для введення в
носові порожнини з метою одержання загальної або
місцевої дії. Вони містять одну або більше діючих ре-
човин. Назальні лікарські засоби не мають справляти
подразливої та іншої несприятливої дії на слизову но-
са та її волоски. Водні назальні лікарські засоби зви-
чайно ізотонічні.
Назальні лікарські засоби випускають у багатодозових
та однодозових контейнерах, споряджених, якщо це
необхідно, пристроєм, який забезпечує зручність за-
стосування і запобігає забрудненню.
Якщо немає інших зазначень, водні назальні лікарські
засоби випускають у багатодозових контейнерах, які
містять підхожий антимікробний консервант у не-
обхідній концентрації, за винятком лікарських за-
собів, які виявляють достатню антимікробну дію.
Контейнери для назальних лікарських засобів мають
відповідати вимогам статей "Матеріали, використову-
вані для виробництва контейнерів" (3.1 та підрозділи^
та "Контейнери"(3.2та підрозділи/
Назальні лікарські засоби можуть бути класифіковані
як:
— назальні краплі та рідкі аерозолі;
— назальні порошки;
— назальні м’які лікарські засоби;
— назальні промивки;
— назальні палички.
ДОДАТОК 2
МЕТОДИКА ВИЗНАЧЕННЯ ГЕРМЕТИЧНОСТІ
КОНТЕЙНЕРА
Відбирають 10 туб з препаратом і ретельно витирають
їх зовнішні поверхні фільтрувальним папером. Туби
поміщають у горизонтальному положенні на аркуш
фільтрувального паперу і витримують у термостаті при
температурі (60 ± 3) °С протягом 8 год.
На фільтрувальному папері не має бути патьоків пре-
парату з жодної з туб. Якщо патьоки спостерігаються
лише з однієї туби, то випробування проводять додат-
ково ще з 20 тубами. Якщо патьоки спостерігаються
більше як з однієї туби, результати випробування вва-
жають незадовільними.
Результати випробування вважають задовільними, як-
що не спостерігають патьоків з перших 10 туб або спо-
стерігалися патьоки лише для однією з ЗО туб.
ВИРОБНИЦТВО
При розробці назальних лікарських засобів, до складу
яких входять антимікробні консерванти, уповноваже-
ному органу мають бути надані дані, що підтверджу-
ють ефективність вибраних консервантів. Метод ви-
значення і критерії ефективності консервантів мають
відповідати вимогам статті "Ефективність ан-
тимікробних консервантів" (5.1.3).
При виробництві, пакуванні, зберіганні й реалізації
назальних лікарських засобів мають бути вжиті
відповідні заходи, які забезпечують необхідну
мікробіологічну чистоту відповідно до вимог статті
"Мікробіологічна чистота лікарських засобів " (5.1.4).
Стерильні назальні лікарські засоби виготовляють з
використанням матеріалів і методів, які забезпечують
стерильність, запобігають забрудненню лікарських за-
собів і розвитку мікроорганізмів, відповідно до вимог
статті "Методи стерилізації лікарських засобів" (5.1.1).
При виробництві назальних лікарських засобів, які
містять дисперговані частки, треба передбачити захо-
ди, що забезпечують необхідний розмір часток та їхній
контроль.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
511
Назальні лікарські засоби
ВИПРОБУВАННЯ
Стерильність (2.6.1). Якщо на етикетці зазначено, що
препарат стерильний, він має витримувати випробу-
вання на стерильність.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах. Якщо препарат
стерильний, зберігають у стерильних повітронепро-
никних контейнерах з контролем першого розкриття.
МАРКУВАННЯ
На етикетці зазначають:
— назву кожного антимікробного консерванта;
— "стерильно" (якщо необхідно).
Назальні краплі та рідкі аерозолі
ВИЗНАЧЕННЯ
Назальні краплі та рідкі аерозолі являють собою роз-
чини, емульсії або суспензії, призначені для закапу-
вання або упорскування в носові порожнини.
Емульсії можуть розшаровуватися, однак при збовту-
ванні мають легко перетворюватися в емульсію. Сус-
пензії можуть утворювати осад, що має швидко ресус-
пендуватися при збовтуванні, утворюючи суспензію,
досить стабільну, щоб забезпечити необхідну дозу при
введенні.
Назальні краплі звичайно випускають у багатодозових
контейнерах, споряджених підхожою насадкою.
Рідкі назальні аерозолі випускають у контейнерах з
розпилювальним пристроєм або у контейнерах під ти-
ском, споряджених підхожою насадкою, а також дозу-
ючим клапаном або без нього. Якщо аерозолі випуска-
ють у контейнерах під тиском, вони мають відповіда-
ти вимогам статті "Лікарські засоби, що знаходяться
під тиском".
Розмір розпилених краплинок має бути таким, щоб
забезпечувати їх осадження у носовій порожнині.
ВИПРОБУВАННЯ
Якщо немає інших зазначень, назальні краплі, які ви-
пускаються в однодозових контейнерах, та дозовані
аерозолі, призначені для загальноїдії. мають витриму-
вати такі випробування.
Однорідність маси. Назальні краплі, що являють собою
розчини, мають витримувати таке випробування.
Звільняють кожен з 10 контейнерів якнайповніше і
зважують вміст. Визначають середню масу вмісту. Ма-
са вмісту не більше як двох контейнерів може відхиля-
тися від середньої маси більш як на (±10 %), і маса
вмісту жодного контейнера не має відхилятися більш
як на (±20 %) від середньої маси.
Дозовані назальні аерозолі, що являють собою розчи-
ни, мають витримувати таке випробування. Один раз
натискують на клапан і відкидають вивільнений вміст.
Не менш як через 5 с знову натискують на клапан і
відкидають вивільнений вміст. Повторюють цю опе-
рацію ще три рази. Зважують контейнер, один раз на-
тискують на клапан і знову зважують контейнер. За
різницею обчислюють масу вмісту, що вивільнився.
Повторюють цю операцію ще для дев’яти контей-
нерів. Препарат витримує випробування, якщо
індивідуальна маса лише для двох контейнерів відхи-
ляється від середнього значення більш як на (±25 %),
але не більш як на (±35 %).
Однорідність вмісту (2.9.6). Назальні краплі, шо явля-
ють собою суспензії або емульсії, мають витримувати
таке випробування. Звільняють кожний контейнер
якнайповніше і визначають вміст діючої речовини для
кожного контейнера. Вони мають витримувати ви-
пробування однорідності вмісту (тест В).
Однорідність дозування. Дозовані назальні аерозолі,
що являють собою суспензії або емульсії, мають ви-
тримувати таке випробування. Використовують при-
лад, що дозволяє кількісно утримувати дозу після на-
тиснення на розпилюючий пристрій.
Збовтують контейнер протягом 5 с, випускають дозу і
відкидають. Не менш як через 5 с знову збовтують
контейнер протягом 5 с, випускають дозу і відкида-
ють. Повторюють зазначену операцію ще три рази.
Через 2 с натискують на розпилювальний пристрій,
спрямовуючи дозу назального аерозолю у збиральну
посудину. Вміст збиральної посудини шляхом
послідовних промивань об’єднують і визначають
вміст діючої речовини в об’єднаному розчині.
Повторюють вищезазначену операцію ще для дев’яти
контейнерів.
Якщо немає інших зазначень в окремій статті, препа-
рат витримує випробування, якщо вміст діючої речо-
вини в дозі не більш як для одного контейнера не
укладається в межі від 75 % до 125 %, але не виходить
за межі від 65 % до 135 % від середнього значення.
Якщо вміст діючої речовини у дозі для двох або трьох
окремих контейнерів не укладається в межі від 75 % до
125 %, але укладається в межі від 65 % до 135 %, повто-
рюють випробування ще для 20 контейнерів. Препа-
рат задовольняє вимоги, якщо вміст діючої речовини у
дозі не більш як для трьох контейнерів виходить за
межі від 75 % до 125 %. але укладається у межі від 65 %
до 135 % від середнього значення.
Назальні порошки
ВИЗНАЧЕННЯ
Назальні порошки являють собою порошки, призна-
чені для введення в носові порожнини за допомогою
підхожого пристрою. Вони мають відповідати вимо-
гам статті "Порошки для зовнішнього застосування".
512
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Настойки
Розмір часток, які осідають у носових порожнинах,
має підтверджуватися відповідними методами визна-
чення розміру часток.
Назальні м’які лікарські засоби
ВИЗНАЧЕННЯ
Назальні м’які лікарські засоби мають відповідати ви-
могам статті "М’які лікарські засоби для місцевого за-
стосування”. Контейнер повинен мати підхожий
пристрій для нанесення вмісту.
Назальні промивки
ВИЗНАЧЕННЯ
Назальні промивки звичайно являють собою водні
ізотонічні розчини, призначені для очищення носо-
вих порожнин. Назальні промивки, призначені для
застосування при ушкодженнях частин носа або перед
хірургічною операцією, мають бути стерильними.
Назальні палички
ВИЗНАЧЕННЯ
Назальні палички мають задовольняти вимоги статті
"Палички
_________________________________________N
Назальні краплі
ВИПРОБУВАННЯ
Назальні краплі, що являють собою розчини, звичай-
но контролюють за такими показниками якості: опис,
ідентифікація, прозорість, кольоровість, рН (крім не-
водних і масляних розчинів), супровідні домішки,
об’єм вмісту контейнера (для багатодозових контей-
нерів), мікробіологічна чистота або стерильність,
кількісне визначення.
Для назальних крапель, що являють собою масляні
розчини, додатково контролюють кислотне і перекис-
не числа.
Для назальних крапель, що містять речовини, які забез-
печують в'язкість, додатково контролюють в’язкість.
Для назальних крапель, призначених для загальноїдії,
у вигляді розчинів, що випускають в однодозових кон-
тейнерах, додатково контролюють однорідність маси.
Для назальних крапель, призначених для загальноїдії.
у вигляді суспензій або емульсій, що випускають в од-
нодозових контейнерах, додатково контролюють од-
норідність вмісту, розмір часток, стійкість суспензії.
Для назальних рідких дозованих аерозолей, які міс-
тять суспензії або емульсії, додатково контролюють
однорідність дозування.
рН. Визначають для назальних крапель, за винятком
неводних і масляних розчинів. Якщо немає інших за-
значень в окремій статті, значення рН назальних кра
пель має відповідати фізіологічним межам.
Однорідність вмісту. Назальні краплі, шо являють со-
бою суспензії або емульсії, мають відповідати вимогам
статті "Однорідність вмісту діючої речовини в одиниці
дозованого лікарського засобу" (2.9.6), якщо немає ін-
ших зазначень в окремій статті.
Кількісне визначення. Проводять визначення діючих
речовин, антимікробних консервантів, неводних роз-
чинників та інших речовин, зазначених в окремій
статті. Вміст визначуваних речовин виражають у гра-
мах, міліграмах або одиницях дії (ОД) в 1 мл препара-
ту. Вміст діючих речовин має бути від 90 % до 110 % від
вмісту, зазначеного у розділі "Склад", якщо немає
інших зазначень в окремій статті.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах, які забезпечують
стабільність і зручність дозування препарату при за-
стосуванні.
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву і концентрацію діючих речовин і антимік-
робних консервантів;
— необхідний перелік допоміжних речовин;
— умови зберігання і термін придатності:
— термін зберігання після розкриття.
ЗБЕРІГАННЯ
У прохолодному, захищеному від світла місці, якщо
немає інших зазначень в окремій статті.
НАСТОЙКИ
ТіпсШгае
ВИЗНАЧЕННЯ
Настойки — не рідкі препарати, звичайно одержувані
із висушеної рослинної або тваринної сировини. Для
приготування настойок сировина перед екстракцією
може бути піддана попередній обробці, наприклад,
роздрібненню, знежирюванню або інактивації фер-
ментів.
ПРР’З'ДНПД ФАРМАКОПЕЯ УКРАЇНИ 2001
513
Настойки
Настойки виготовляють мацерацією, перколяцією або
іншим підхожим валідованим методом із застосуван-
ням спирту відповідної концентрації.
Настойки можуть бути також виготовлені розчинен-
ням або розведенням екстрактів у спирті відповідної
концентрації.
Настойки звичайно виготовляють, використовуючи
одну частину сировини і 10 частин екстрагенту або
одну частину сировини і п’ять частин екстрагенту На-
стойки звичайно прозорі. Під час зберігання допус-
кається утворення невеликого осаду за умови відсут-
ності суттєвої зміни складу.
ВИРОБНИЦТВО
Метод перколації. Якщо необхідно, сировину
здрібнюють до часток певного розміру і піддають екс-
тракції відповідною кількістю екстрагенту. Сировину
ретельно змішують з порцією зазначеного екс-
трагенту і залишають на необхідний час. Потім пере-
носять в екстрактор і повільно перколюють, стежачи
за тим, щоб сировина була повністю покрита шаром
відповідного екстрагенту. Залишок можна віджати і
одержану рідину можна об’єднати з перколятом.
Метод мацерації. Якщо немає інших зазначень, не-
обхідну кількість підготованого матеріалу екстрагують
змішуванням із відповідною кількістю зазначеного
екстрагенту і мацерують у закритому екстракторі не-
обхідний час. Залишок відділяють від екстрагенту і,
якщо необхідно, віджимають. В останньому випадку
обидві рідини об’єднують.
Приготування із екстрактів. Для приготування насто-
йок розчиненням або розведенням екстрактів викори-
стовують спирт відповідної концентрації. Вміст спир-
ту і діючих речовин або вміст спирту і сухого залишку,
якщо це передбачено, має відповідати таким саме по-
казникам в аналогічних настойках, одержуваних ма-
церацією або перколяцією.
Регулювання складу. Регулювання вмісту діючих речо-
вин, якщо необхідно, проводять додаванням екстра-
генту придатної концентрації або додаванням
відповідної настойки.
ВИПРОБУВАННЯ
Відносна густина (2.2.5). Значення відносної густини
настойки має відповідати межам, зазначеним в ок-
ремій статті.
Вміст етанолу (2.9.10). Вміст етанолу має відповідати
межам, зазначеним в окремій статті.
Метанол і 2-пропанол (2.9.11). Допускається не більше
0.05 % (об/об) метанолу і не більше 0.05 % (об/об)
2-пропанолу, якщо немає інших зазначень в окремій
статті.
Сухий залишок. Вміст сухого залишку настойки має
відповідати межам, зазначеним в окремій статті.
2.00 г або 2.0 мл настойки поміщають у зважений бюкс
діаметром близько 50 мм і заввишки близько ЗО мм,
випарюють насухо на водяній бані й сушать у су-
шильній шафі при температурі від 100 °С до 105 °С
протягом 3 год. Бюкс охолоджують в ексикаторі над
фосфору) V) оксидом Р і зважують. Результат виража-
ють у вагових відсотках або в грамах на літр.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах, у захищеному від
світла місці.
МАРКУВАННЯ
На етикетці зазначають:
— який матеріал використано — рослинний чи тва-
ринний:
— що використовувалася свіжа рослинна або тварин-
на сировина;
— концентрацію спирту, що використовується для
приготування;
— концентрацію спирту в настойці;
— вміст діючої речовини і/або співвідношення
вихідного матеріалу до екстрагенту або вихідного
матеріалу до одержаної настойки.
___________________________________________N
ВИЗНАЧЕННЯ
Настойки — це рідкі спиртові або водно-спиртові ви-
тяги, що одержують звичайно з висушеної або свіжої
рослинної або тваринної сировини без нагрівання і
усунення екстрагенту.
При виготовленні настойок з однієї вагової частини
лікарської рослинної сировини одержують п’ять
об’ємних частин готового продукту, із сильнодіючої
сировини — 10 об’ємних частин готового продукту,
якшо немає інших зазначень в окремій статті.
Одержувані витяги звичайно відстоюють не менше
двох діб при температурі не више 10 °С до одержання
прозорої рідини і фільтрують.
ВИПРОБУВАННЯ
Настойки звичайно контролюють за такими показни-
ками якості: опис, ідентифікація, вміст етанолу або
відносна густина; сухий залишок, важкі метали, об’єм
вмісту контейнера, мікробіологічна чистота, кількісне
визначення.
Сухий залишок. Допускається проводити визначення з
5.0 мл настойки.
514
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Очні лікарські засоби
Важкі метали (2.4.8, метод А). Не більше 0.001 %
(10 ррт), якшо немає інших зазначень в окремій
статті.
5.0 мл настойки випарюють насухо, додають 1 мл кис-
лоти сірчаної Р. обережно спалюють і прожарюють.
До одержаного залишку додають при нагріванні 5 мл
розчину 615 г/л амонію ацетату Р, фільтрують крізь
беззольний фільтр, промивають 5 мл води Р і доводять
об’єм фільтрату водою Рло 50 мл.
12 мл одержаного розчину мають витримувати випро-
бування на важкі метали. Еталон готують з вико-
ристанням еталонного розчину свинцю (/ ррт РЬ) Р.
Кількісне визначення. Вміст визначуваних речовин у
настойках виражають у відсотках (м/об).
ЗБЕРІГАННЯ
У прохолодному, захищеному від світла місці. Під час
зберігання настойок можливе випадання осаду.
ОЧНІ ЛІКАРСЬКІ ЗАСОБИ
Осиїагіа
ВИЗНАЧЕННЯ
Очні лікарські засоби являють собою стерильні рідкі,
м’які або тверді лікарські засоби, призначені для на-
несення на очне яблуко і/або кон’юнктиву чи для вве-
дення до кон’юнктивального мішка.
Контейнери для очних лікарських засобів мають
відповідати вимогам статей "Матеріали, використову-
вані для виробництва контейнерів” (3.1 та підрозділи/
та "Контейнери"(3.2та підрозділи).
Очні лікарські засоби можуть бути класифіковані як:
— очні краплі;
— очні примочки;
— очні м’які лікарські засоби:
— очні вставки.
ВИРОБНИЦТВО
При розробці очних лікарських засобів, до складу
яких входять антимікробні консерванти, уповноваже-
ному органу мають бути подані дані, що підтверджу-
ють ефективність вибраних консервантів. Метод ви-
значення і критерії ефективності консервантів мають
відповідати вимогам статті "Ефективність ан-
тимікробних консервантів" (5.1.3).
Очні лікарські засоби виробляють, використовуючи
матеріали та методи, які забезпечують стерильність і
запобігають забрудненню лікарських засобів і розвит-
ку мікроорганізмів, відповідно до вимог статті "Мето-
ди стерилізації лікарських засобів" (5. /. 1).
При виробництві очних лікарських засобів, які містять
дисперговані частки, слід передбачити заходи, шо за-
безпечують необхідний розмір часток та його контроль.
ВИПРОБУВАННЯ
Стерильність (2.6.1). Очні лікарські засоби мають ви-
тримувати випробування на стерильність. Аплікатори,
що додаються окремо, також мають витримувати ви-
пробування на стерильність. їх виймають з контейне-
ра в асептичних умовах і помішають у посудину з жи-
вильним середовищем до повного занурення. Інку-
бацію посівів і оцінку результатів проводять відпо-
відно до вимог статті "Стерильність".
ЗБЕРІГАННЯ
У стерильних повітронепроникних контейнерах з
контролем першого розкриття, якшо немає інших за-
значень в окремій статті.
МАРКУВАННЯ
На етикетці зазначають назву кожного антимікробно-
го консерванта.
Очні краплі
ВИЗНАЧЕННЯ
Очні краплі являють собою стерильні водні або мас-
ляні розчини або суспензії, які містять одну або
більше діючих речовин, призначених для інстиляції в
око. У деяких випадках для забезпечення стабільності
очних крапель вони можуть випускатися у сухій сте-
рильній формі, яка безпосередньо перед використан-
ням розчиняється або суспендується у запропоно-
ваній стерильній рідині.
Очні краплі можуть містити допоміжні речовини, на-
приклад, для забезпечення необхідної тонічності,
в’язкості, створення або стабілізації необхідного зна-
чення рН, збільшення розчинності діючих речовин,
забезпечення стабільності препарату. Ці речовини у
використовуваних концентраціях не мають негативно
впливати на дію лікарського засобу і чинити надмірне
місцеве подразнення.
Водні препарати, що випускаються у багатодозових
контейнерах, мають містити підхожі антимікробні
консерванти в необхідних концентраціях, за винятком
тих випадків, коли сам препарат виявляє достатню ан-
тимікробну дію. Вибрані антимікробні консерванти
мають бути сумісними з іншими інгредієнтами препа-
рату і зберігати ефективність протягом усього періоду
використання очних крапель.
Якшо очні краплі не містять антимікробних консер-
вантів, то вони мають бути упаковані переважно в од-
нодозові контейнери. Очні краплі, призначені для ви-
користання при хірургічних процедурах, не мають
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
515
Очні лікарські засоби
містити антимікробних консервантів і мають випуска-
тися в однодозових контейнерах.
Очні краплі, шо являють собою розчини, у відпо-
відних умовах нагляду мають бути практично прозо-
рими і практично вільними від часток.
Очні краплі у вигляді суспензій можуть утворювати
осад, який має швидко ресуспендуватися при збовту-
ванні, утворюючи суспензію, що має бути достатньо
стабільною і забезпечувати необхідну дозу при введенні.
Препарати у багатодозових контейнерах випускають у
таких контейнерах, які дозволяють дозувати крап-
лями. Контейнер має містити не більше 10 мл препа-
рату, якщо немає інших зазначень в окремій статті.
ВИПРОБУВАННЯ
Розмір часток. Очні краплі у формі суспензії мають ви-
тримувати таке випробування: певну кількість сус-
пензії вносять до лічильної камери або за допомогою
мікропіпетки наносять на предметне скло і перегляда-
ють під мікроскопом площу, відповідну 10 мкг твердої
фази. Спочатку зразок переглядають при малому
збільшенні (наприклад, х 50), відмічаючи частки з
максимальним розміром більше 25 мкм. Потім
здійснюють вимірювання цих часток при більшому
збільшуванні (наприклад, від х 200 до х 500). Допус-
кається, щоб не більше 20 часток мали максимальний
розмір більше 25 мкм і з них не більше двох часток ма-
ли максимальний розмір більше 50 мкм. Не допуска-
ється наявність часток розміром більше 90 мкм.
центраціях, за винятком тих випадків, коли сам пре-
парат виявляє достатню антимікробну дію. Вибрані
антимікробні консерванти мають бути сумісними з
іншими інгредієнтами препарату і зберігати ефек-
тивність протягом усього періоду використання очних
примочок.
Якщо очні примочки не містять антимікробних кон-
сервантів, вони мають бути упаковані в однодозові
контейнери. Очні примочки, призначені для викорис-
тання при хірургічних процедурах і для надання пер-
шої медичної допомоги, не мають містити ан-
тимікробних консервантів і мають випускатися лише
в контейнерах для одноразового використання.
Очні примочки у відповідних умовах нагляду мають
бути практично прозорими і практично вільними від
часток. Багатодозовий контейнер має містити не
більше 200 мл очної примочки, якщо немає інших за-
значень в окремій статті.
МАРКУВАННЯ
На етикетці зазначають:
— для однодозових контейнерів — вміст має вико-
ристовуватися лише один раз;
— для багатодозових контейнерів — термін зберіган-
ня препарату після розкриття контейнера. Цей
термін не має перевищувати чотирьох тижнів, як-
що немає інших зазначень в окремій статті.
Очні м’які лікарські засоби
МАРКУВАННЯ
На етикетці багатодозових контейнерів зазначають
термін зберігання препарату після розкриття контей-
нера. Цей термін не має перевищувати чотирьох
тижнів, якщо немає інших зазначень в окремій статті.
Очні примочки
ВИЗНАЧЕННЯ
Очні примочки являють собою стерильні водні розчи-
ни, призначені для змочування і промивання очей, а
також для просочування матеріалів, які накладають на
око.
Очні примочки можуть містити допоміжні речовини,
наприклад, для забезпечення необхідної тонічності,
в’язкості, створення або стабілізації необхідного зна-
чення рН, збільшення розчинності діючих речовин,
забезпечення стабільності препарату. Ці речовини у
використовуваних концентраціях не мають негативно
впливати надію лікарського засобу і чинити надмірне
місцеве подразнення.
Водні розчини очних лікарських засобів, які випуска-
ються у багатодозових контейнерах, мають містити
підхожі антимікробні консерванти в необхідних кон-
ВИЗНАЧЕННЯ
Очні м’які лікарські засоби являють собою однорідні
стерильні мазі, креми або гелі, призначені для нане-
сення на кон’юнктиву. Вони містять одну або більше
діючих речовин, розчинених або диспергованих у
підхожій основі.
Очні м’які лікарські засоби мають відповідати вимо-
гам загальної статті "М’які лікарські засоби для місцево-
го застосування". Основа не має подразнювати
кон’юнктиву.
Очні м'які лікарські засоби упаковують у стерильні,
необоротно стискувані, мілкоємні туби, що не пружи-
нять, з умонтованим або доданим наконечником.
Вміст туби має бути не більше 5 г. Туби мають бути
щільно закупорені, щоб запобігати мікробного за-
бруднення. Очні м’які лікарські засоби можуть також
випускатися у спеціально призначених однодозових
контейнерах.
ВИПРОБУВАННЯ
Розмір часток. Очні м’які лікарські засоби, що містять
дисперговані тверді частки, мають витримувати таке
випробування: пробу, яка містить не менше 10 мкг
твердої діючої речовини, обережно наносять тонким
шаром і переглядають під мікроскопом усю площу
516
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Очні лікарські засоби
зразка. Спочатку зразок переглядають при малому
збільшенні (наприклад, х 50), відмічаючи частки з
максимальним розміром більше 25 мкм. Потім
здійснюють вимірювання цих часток при більшому
збільшуванні (наприклад, від х 200 до х 500). Для кож-
ної проби, яка містить 10 мкг твердої діючої речовини,
має бути не більше 20 часток із максимальним
розміром більше 25 мкм, і з них не більше двох часток
із максимальним розміром більше 50 мкм. Не допус-
кається наявність часток із максимальним розміром
більше 90 мкм.
Очні вставки
ВИЗНАЧЕННЯ
Очні вставки являють собою стерильні тверді або
м’які препарати відповідного розміру і форми, при-
значені для вставки у кон’юнктивальний мішок. Вони
звичайно складаються з матриці, в яку включено
діючу речовину, або діюча речовина оточена мембра-
ною, що контролює швидкість вивільнення. Діюча ре-
човина має бути достатньо розчинною у фізіологічній
рідині і вивільнюватися певний період часу.
Кожна очна вставка випускається в індивідуальному
стерильному контейнері.
ВИРОБНИЦТВО
Виробництво очних вставок має забезпечувати не-
обхідне вивільнення діючої речовини.
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). При необхідності визна-
чення проводять відповідно до вимог статті "Од-
норідність вмісту діючої речовини в одиниці дозованого
лікарського засобу"(2.9.6), використовуючи тест А.
МАРКУВАННЯ
На етикетці зазначають:
— загальну кількість діючої речовини в одній
вставці, якщо необхідно;
— дозу, вивільнювану за одиницю часу, якщо необхідно.
_________________________________________N
Очні краплі
ВИРОБНИЦТВО
При виробництві очних крапель застосовують сте-
рильні розчинники: воду очищену, ізотонічні буферні
розчини, масла та ін.
Як стабілізатори, консерванти, пролонгатори та інші
допоміжні речовини використовують: натрію хлорид,
натрію сульфат, натрію нітрат, натрію метабісульфіт,
натрію тіосульфат, натрію дигідрофосфат і динатрію
гідрофосфат, кислоту борну, кислоту сорбінову, ме-
тилпарагідроксибензоат, пропілпарагідроксибензоат.
бензалконію хлорид, похідні целюлози та ін.
Звичайно очні краплі мають бути ізотонічними із
слізною рідиною, відповідною 0.9 % розчину натрію
хлориду. Допускається иробництво розчинів, осмо-
ляльність (осмолярність) яких знаходиться в межах
осмоляльності (осмолярності) 0.6 % — 2 % розчину
натрію хлориду. В окремих випадках очні краплі мо-
жуть мати осмоляльність, більшу за осмоляльність 2 %
розчину натрію хлориду.
Для очних лікарських засобів уповноваженому органу
мають бути подані дані про осмоляльність (осмо-
лярність) препарату.
ВИПРОБУВАННЯ
Очні краплі звичайно контролюють за такими показ-
никами якості, опис, ідентифікація, прозорість,
кольоровість, рН, супровідні домішки, об’єм вмісту
контейнера (для багатодозових контейнерів), сте-
рильність, механічні включення, кількісне визначен-
ня.
Для очних крапель у вигляді масляних розчинів додат-
ково контролюють кислотне і перекисне числа.
Для очних крапель у вигляді суспензій додатково кон-
тролюють розмір часток.
Для очних крапель, що містять речовини, які забезпе-
чують в’язкість, додатково контролюють в’язкість.
РН. Визначають для очних крапель, за винятком мас-
ляних розчинів. Оптимальним значенням рН є 7.4, що
відповідає рН слізної рідини.
Якщо діючі речовини препарату при зазначеному зна-
ченні рН не стабільні або погано розчиняються, то
значення рН може відрізнятися від оптимального і
має знаходитися у межах від 3.5 до 8.5.
Кількісне визначення. Проводять визначення діючих
речовин, антимікробних консервантів, неводних роз-
чинів та інших речовин, зазначених в окремій статті.
Вміст визначуваних речовин зазначають у грамах,
міліграмах або одиницях дії (ОД) в 1 мл препарату.
Вміст діючих речовин має бути від 90 % до 110 % від
вмісту, зазначеного у розділі "Склад", якщо немає
інших зазначень в окремій статті.
УПАКОВКА
Контейнери мають забезпечувати герметичність, сте-
рильність, стабільність і зручні ь дозування препара-
ту при застосуванні.
ПГПЧ/АІША ЖАОМАЕЛПГа Х/ЦРАЇНІЛ золі
517
Піни медичні
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву і концентрацію діючих речовин і антимік-
робних консервантів;
— перелік допоміжних речовин;
— умови зберігання і термін придатності.
ЗБЕРІГАННЯ
У прохолодному, захищеному від світла місці, якщо
немає інших зазначень в окремій статті.
Очні мазі
ВИПРОБУВАННЯ
Очні мазі додатково контролюють за такими показни-
ками якості: маса вмісту контейнера, металеві частки,
герметичність контейнера.
Для очних мазей, основи яких містять тригліцериди
жирних кислот, додатково контролюють кислотне і
перекисне числа.
Металеві частки. Вміст кожної з 10 туб поміщають у 10
чашок Петрі, поверхня яких не містить видимих подря-
пин. Чашки закривають кришками і нагрівають при
температурі 85 °С протягом 2 год до повного розплав-
лення препарату. Чашки Петрі поміщають на стійку
рівну поверхню і охолоджують при кімнатній темпера-
турі до загуснення. Знімають кришки, перевертають
кожну чашку Петрі догори дном і розглядають під
мікроскопом, спорядженим мікрометричною сіткою,
при збільшенні х ЗО. На додаток до звичайного джерела
світла має бути джерело, що знаходиться під кутом 45°
зверху. Досліджують все дно кожної чашки Петрі на на-
явність металевих часток. Варіювання інтенсивності
верхнього джерела світла дозволяє визначити такі част-
ки металу за їхнім характерним відбиванням світла.
Підраховують кількість металевих часток, розмір яких
перевищує 50 мкм.
Вимоги даного випробування вважаються виконани-
ми, якщо кількість таких часток у 10 тубах — не більше
50 і лише в одній тубі допускається більше восьми та-
ких часток.
Якщо ці вимоги не виконані, то випробування прово-
дять додатково із вмістом 20 туб. Вимоги вважаються
виконаними, якщо кількість металевих часток
розміром більше 50 мкм у 30 тубах, не перевищує 150 і
не більш як у трьох тубах допускається більше восьми
таких часток у кожній.
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву і концентрацію діючих речовин і ан-
тимікробних консервантів;
— перелік допоміжних речовин;
— умови зберігання і термін придатності.
ЗБЕРІГАННЯ
У прохолодному, захищеному від світла місці, якщо
немає інших зазначень в окремій статті.
ПІНИ МЕДИЧНІ
Мивсі медісаіі
До пін медичних (пін) можуть бути поставлені додат-
кові вимоги, зазначені в інших статтях, наприклад,
"Лікарські засоби для ректального застосування",
"Лікарські засоби для вагінального застосування" і
"Рідкі лікарські засоби для зовнішнього застосування".
ВИЗНАЧЕННЯ
Піни — лікарська форма, що складається з великого
об’єму газу, диспергованого у рідині. Вони звичайно
містять одну або більше діючих речовин, поверхнево-
активні речовини, які забезпечують утворення піни, а
також інші допоміжні речовини. Піни звичайно при-
значені для нанесення на шкіру або слизові оболонки.
Піни звичайно утворюються безпосередньо під час за-
стосування з рідкого лікарського засобу, шо знахо-
диться у контейнері під тиском. Контейнер має бути
споряджений клапаном і насадкою натискного типу,
пристосованої для застосування піни.
Піни, призначені для застосування на великих відкри-
тих ранах або на дуже ушкодженій шкірі, мають бути
стерильними.
Піни, що випускаються у контейнерах підтиском, ма-
ють задовольняти вимоги статті "Лікарські засоби, що
знаходяться під тиском
ВИРОБНИЦТВО
Стерильні піни виробляють з використанням ма-
теріалів і методів, які забезпечують стерильність і за-
побігають забрудненню лікарських засобів і розвитку
в них мікроорганізмів відповідно до вимог статті "Ме-
тоди приготування стерильних продуктів"(5.1.1).
ВИПРОБУВАННЯ
Відносна густина піни. Контейнер витримують при
температурі близько 25 °С протягом не менше 24 год.
Стежачи за тим, щоб не нагріти контейнер, установ-
люють на насадку натискного типу жорстку трубку за-
вдовжки 70-100 мм і внутрішнім діаметром близько
1 мм. Струшують контейнер для одержання одно-
рідного вмісту, випускають і відкидають від 5 мл до
10 мл піни. Зважують плоскодонну чашку місткістю
близько 60 мл і заввишки близько 35 мм. Поміщають
518
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Порошки для зовнішнього застосування
кінець жорсткої трубки, установленої на насадці нати-
скного типу, в куток чашки, натискують насадку і
рівномірно заповнюють чашку за допомогою колових
рухів. Після того як піна цілком розшириться, згла-
джують її рівень шляхом видалення зайвої піни під-
хожим плоским предметом, наприклад, предметним
склом для мікроскопа. Зважують і визначають масу та-
кого самого об’єму води Р, наповнивши ту саму чашку
водою Р
Відносну густину піни обчислюють за формулою:
т/ е,
де:
т — маса наважки випробовуваного зразка піни, у
грамах;
є — маса наважки об’єму води Р, у грамах.
Проводять три визначення. Жодне значення не має
відхилятися більш як на (±20 %) від середнього зна-
чення.
Час розширення. Прилад (Рис. 1) складається з бюрет-
ки місткістю 50 мл, внутрішнім діаметром 15 мм,
ціною поділки 0 1 мл, спорядженої краном з одним
отвором розміром 4 мм. Позначка, відповідна ЗО мл,
має бути не ближче 210 мм від осі крана. Нижня час-
тина бюретки сполучена за допомогою пластмасової
трубки завдовжки не більше 50 мм і внутрішнім діаме-
тром 4 мм з піноутворювальним контейнером, облад-
наним підхожою для даного сполучення насадкою на-
тискного типу. Контейнер витримують при темпера-
турі близько 25 "С протягом не менше 24 год. Контей-
нер струшують для одержання однорідного вмісту так,
щоб не нагріти його, і відкидають перші 5 — 10 мл
піни. Сполучають насадку і носик бюретки. Натиску-
ють на насадку і за один раз випускають близько 30 мл
піни. Закривають кран і одночасно вмикають хроно-
метр. Спостерігають за об’ємом піни у бюретці, кожні
10 с відмічають збільшення об’єму піни доти, поки не
буде досягнуто максимального об’єму.
Проводять три визначення. Час досягнення макси-
мального об’єму в кожному визначенні не має переви-
щувати 5 хв.
Стерильність (2.6.1). Стерильний препарат має витри-
мувати випробування на стерильність.
МАРКУВАННЯ
На етикетці зазначають "стерильно" (якщо необхідно).
______________________________________N
ВИПРОБУВАННЯ
Піни звичайно контролюють за такими показниками
якості: опис, ідентифікація, маса вмісту контейнера,
супровідні домішки, відносна густина піни, час роз-
ширення, мікробіологічна чистота (стерильність),
кількісне визначення діючих речовин і антимікробних
консервантів.
Піни, що знаходяться у контейнерах під тиском, до-
датково контролюють за такими показниками якості:
перевірка контейнера на герметичність, вимірювання
тиску всередині контейнера, визначення відсотка ви-
ходу вмісту контейнера.
Якщо необхідно, додатково контролюють рН.
ПОРОШКИ ДЛЯ ЗОВНІШНЬОГО
ЗАСТОСУВАННЯ
Риіуегез асі изит сіегтісит
Вимоги даної статті не поширюються на порошки, ви-
користовувані у ветеринарії, якщо немає інших зазна-
чень в окремій статті.
ВИЗНАЧЕННЯ
Порошки для зовнішнього застосування являють со-
бою лікарську форму, що складається з твердих окре-
мих сухих часток різного ступеня здрібненості. По-
рошки для зовнішнього застосування містять одну або
більше діючих речовин з наповнювачами або без них.
При необхідності використовують барвники, дозво-
лені до медичного застосування.
Порошки для зовнішнього застосування випускають у
однодозових або багатодозових контейнерах. Вони не
мають містити агрегатів часток порошку. Порошки
для зовнішнього застосування, призначені для вико-
ристання на великих відкритих ранах або на дуже
ушкодженій шкірі, мають бути стерильні.
Порошки для зовнішнього застосування у багатодозо-
вих контейнерах можуть випускатися у контейнерах із
кришками, що просіюють, або у контейнерах із ме-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
519
Порошки для зовнішнього застосування
ханічним розпилювачем, або у контейнерах під
тиском.
Порошки для зовнішнього застосування, що випуска-
ються у контейнерах під тиском, мають відповідати
вимогам статті "Лікарські засоби, що знаходяться під
тиском
Контейнери для порошків для зовнішнього застосу-
вання мають відповідати вимогам загальних статей
"Матеріали, використовувані для виробництва кон-
тейнерів" (3.1 і підрозділи^ та "Контейнери"(3.2 і під-
розділи/
ВИРОБНИЦТВО
При виробництві, пакуванні, зберіганні та реалізації
порошків для зовнішнього застосування мають бути
вжиті відповідні заходи, які забезпечують необхідну
мікробіологічну чистоту відповідно до вимог статті
"Мікробіологічна чистота лікарських засобів " (5.1.4).
Стерильні порошки для зовнішнього застосування ви-
готовляють з використанням матеріалів і методів, які
забезпечують стерильність, запобігають забрудненню
лікарських засобів і розвитку мікроорганізмів відпо-
відно до вимог статті "Методи стерилізації лікарських
засобів"(5.1. 1).
ВИПРОБУВАННЯ
Здрібненість. Здрібненість порошку для зовнішнього
застосування визначають ситовим аналізом (2.9.12)
або іншим придатним методом.
Однорідність вмісту (2.9.6). Порошки для зовнішнього
застосування в однодозових контейнерах із вмістом
діючої речовини менше 2 мг або менше 2 % від загаль-
ної маси мають витримувати випробування
однорідності вмісту діючої речовини в одиниці дозова-
ного лікарського засобу (тест В), якщо немає інших за-
значень в окремій статті Для порошків для зовнішнію-
го застосування, що містять більше однієї дію-
чої речовини, вимоги поширюються лише на ті ре-
човини, вміст яких відповідає вищезазначеним умовам
Дане випробування не поширюється на полівітамінні
препарати і препарати, що містять мікроелементи.
Однорідність маси (2.9.5). Порошки для зовнішнього за-
стосування в однодозових контейнерах мають ви-
тримувати випробування однорідності маси для одиниці
дозованого лікарського засобу. Випробування одно-
рідності маси не вимагається, якщо випробування од-
норідності вмісту передбачене для всіх діючих речовин.
Стерильність (2.6.1). Якщо на етикетці зазначено, що
препарат стерильний, він має витримувати випробу-
вання на стерильність.
ЗБЕРІГАННЯ
МАРКУВАННЯ
На етикетці зазначають:
— для зовнішнього застосування;
— "стерильно" (якщо необхідно).
_____________________________________________ТУ
ВИПРОБУВАННЯ
Порошки для зовнішнього застосування звичайно
контролюють за такими показниками якості: опис,
ідентифікація, здрібненість, однорідність маси або од-
норідність вмісту діючої речовини для порошків в од-
нодозовому контейнері, маса вмісту контейнера для
порошків у багатодозовому контейнері, втрата в масі
при висушуванні або вода, супровідні домішки,
мікробіологічна чистота (стерильність), кількісне ви-
значення.
Якщо необхідно, порошки контролюють за показни-
ками рН і важкі метали.
Однорідність вмісту. Порошки для зовнішнього за-
стосування в однодозових контейнерах мають ви-
тримувати вимоги статті "Однорідність вмісту дію-
чої речовини в одиниці дозованого лікарського за-
собу" (2.9.6). якщо немає інших зазначень в окремій
статті.
Кількісне визначення. Вміст визначуваних речовин ви-
ражають у грамах, міліграмах або одиницях дії (ОД) в
одному грамі препарату.
Для порошків в однодозових контейнерах вміст
визначуваних речовин виражають у грамах, мілі-
грамах або одиницях дії (ОД) в одній дозі, якщо
немає інших зазначень в окремій статті.
Відхилення у вмісті діючих речовин мають стано-
вити не більше (±10 %) від вмісту, зазначеного в
розділі "Склад", якщо немає інших зазначень в ок-
ремій статті.
МАРКУВАННЯ
Для порошків для зовнішнього застосування в
однодозових контейнерах на етикетці додатково
зазначають:
— назву і кількість діючих речовин;
— термін придатності;
— умови зберігання;
— спосіб застосування.
У щільно закупорених контейнерах.
Порошки для орального застосування
ПОРОШКИ ДЛЯ ОРАЛЬНОГО
ЗАСТОСУВАННЯ
Ріі1уєгє8 асі и$ит регогаїіа
Вимоги до порошків, використовуваних для приготу-
вання розчинів або суспензій для орального застосуван-
ня, наведені в статті "Рідкі лікарські засоби для ораль-
ного застосування". Вимоги даної статті не поширю-
ються на порошки для орального застосування, викори-
стовувані у ветеринарії, якщо немає інших зазначень в
окремій статті.
ВИЗНАЧЕННЯ
Порошки для орального застосування являють собою
лікарську форму, що складається з твердих окремих
сухих часток різного ступеня здрібненості, яка має
властивість сипкості.
Порошки містять одну або більше діючих речовин з
наповнювачами або без них. Якщо необхідно, викори-
стовують барвники, дозволені до медичного за-
стосування, і ароматизатори. Порошки звичай-
но приймають з водою або іншою підхожою рідиною,
їх можна також ковтати безпосередньо. Порошки ви-
пускають в однодозових або багатодозових контейне-
рах.
Контейнери для порошків для орального застосуван-
ня мають відповідати вимогам загальних статей "Ма-
теріали, використовувані для виробництва контей-
нерів" (3.1 та підрозділи) та "Контейнери" (3.2 та
підрозділи). Кожну дозу порошку з багатодозового
контейнера відбирають за допомогою відповідного
пристрою для відмірювання прописаної кількості.
При однодозовому фасуванні кожна доза має бути
упакована в індивідуальний контейнер, наприклад,
пакетик, паперовий пакет або банку.
ВИРОБНИЦТВО
При виробництві, пакуванні, зберіганні й реаліза-
ції порошків для орального застосування має бути
вжито відповідних заходів, що забезпечують не-
обхідну мікробіологічну чистоту відповідно до ви-
мог статті "Мікробіологічна чистота лікарських за-
собів" (5. 1.4).
ВИПРОБУВАННЯ
Здрібненість. Якщо зазначено в окремій статті,
здрібненість порошку для зовнішнього застосування
визначають ситовим аналізом (2.9.12) або іншим при-
датним методом
Однорідність вмісту (2.9.6). Порошки для орального
застосування в однодозових контейнерах із вмістом
діючої речовини менше 2 мг або менше 2 % від загаль-
ної маси мають витримувати випробування од-
норідності вмісту діючої речовини в одиниці дозова-
ного лікарського засобу (тест В), якщо немає інших
зазначень в окремій статті. Для порошків, що містять
більше однієї діючої речовини, вимоги поширюються
лише на ті речовини, вміст яких відповідає вищеза-
значеним умовам. Дане випробування не поши-
рюється на полівітамінні препарати і препарати, що
містять мікроелементи.
Однорідність маси (2.9.5) Порошки для орального за-
стосування в однодозових контейнерах мають витри-
мувати випробування однорідності маси для одиниці
дозованого лікарського засобу. Випробування од-
норідності маси не вимагається, якщо випробування
однорідності вмісту передбачене для всіх діючих речо-
вин.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах або, якщо препа-
рат містить леткі речовини, зберігають у повітроне-
проникних контейнерах.
Порошки "шипучі"
ВИЗНАЧЕННЯ
Порошки "шипучі” — однодозові або багатолезові по-
рошки, що містять, головно, кислоти і карбонати або
гідрокарбонати, що швидко реагують у присутності
води з виділенням вуглецю діоксиду. Порошки "ши-
пучі" призначені для розчинення або диспергування у
воді перед застосуванням.
ЗБЕРІГАННЯ
У повітронепроникних контейнерах.
__________________________________________N
Приготування порошків
"ех іетроге"
Розрізняють порошки: прості, які складаються з однієї
речовини; складні, які складаються з двох і більше ре-
човин.
Складні порошки готують з урахуванням властивос-
тей діючих речовин, наповнювачів та їхніх кількостей.
При наявності в складі складного порошку речовин у
різних кількостях змішування починають з речовин,
що входять у менших кількостях, поступово додаючи
решту речовин.
Отруйні й сильнодіючі речовини у кількостях менше
0.05 г на всю масу, що готується, використовують у ви-
гляді тритурацій — суміші з лактозою та іншими до-
поміжними речовинами, дозволеними до медичного
застосування (1:100 або 1:10).
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
521
Рідкі лікарські засоби для орального застосування
ВИПРОБУВАННЯ
Порошки звичайно контролюють за такими показни-
ками якості: опис, ідентифікація, здрібненість, од-
норідність маси або однорідність вмісту діючої речо-
вини для порошків у однодозовому контейнері, маса
вмісту контейнера для порошків у багатодозовому
контейнері, втрата в масі при висушуванні або вода,
супровідні домішки, мікробіологічна чистота, кіль-
кісне визначення.
Якщо необхідно, порошки контролюють за такими
показниками: час розчинення, рН, важкі метали.
Однорідність вмісту. Порошки мають витримувати ви-
моги статті "Однорідність вмісту діючої речовини в оди-
ниці дозованого лікарського засобу" (2.9.6), якщо немає
інших зазначень в окремій статті.
Кількісне визначення. Вміст визначуваних речовин ви-
ражають у грамах, міліграмах або одиницях дії (ОД) в
одному грамі препарату.
Для порошків в однодозових контейнерах вміст ви-
значуваних речовин виражають у грамах, міліграмах
або одиницях дії (ОД) в одній дозі, якщо немає інших
зазначень в окремій статті.
Відхилення у вмісті діючих речовин мають становити
не більше (±10 %) від вмісту, зазначеного у розділі
"Склад", якщо немає інших зазначень в окремій статті.
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву і кількість діючої речовини;
— термін придатності;
— умови зберігання;
— спосіб застосування.
Додатково зазначають назву і концентрацію усіх ан-
тимікробних консервантів.
РІДКІ ЛІКАРСЬКІ ЗАСОБИ ДЛЯ
ОРАЛЬНОГО ЗАСТОСУВАННЯ
Гіянісіа регогаїіа
Вимоги даної статті не поширюються на рідкі
лікарські засоби для орального застосування, викорис-
товувані у ветеринарії, якщо немає інших зазначень.
ВИЗНАЧЕННЯ
Рідкі лікарські засоби для орального застосування
звичайно являють собою розчини, емульсії або сус-
пензії, що містять одну або більше діючих речовин у
відповідному розчиннику. Деякі лікарські засоби для
орального застосування можуї ь складатися лише з
рідких діючих речовин.
522
Вони призначені для пиття у нерозведеному вигляді
або після розведення. Ці препарати можуть бути також
приготовані перед застосуванням з рідких концент-
ратів або порошків, гранул або таблеток для приготу-
вання оральних розчинів або суспензій за допомогою
відповідного розчинника.
Рідкі лікарські засоби для орального застосування
можуть містити підхожі антимікробні консер-
ванти, антиоксиданти та інші допоміжні речови-
ни, які забезпечують диспергування, суспендування,
а також загусники, емульгатори, речовини, призна-
чені для створення або стабілізації необхідного
значення рН, для забезпечення змочування і роз-
чинності, стабілізатори, ароматизатори, смакові до-
бавки і барвники, дозволені до медичного застосу-
вання.
Емульсії можуть розшаровуватися, однак при збовту-
ванні мають легко відновлювати попередній вигляд.
Суспензії можуть утворювати осад, який має швидко
ресуспендуватися при збовтуванні, утворюючи сус-
пензію, яка має бути достатньо стабільною, шоб забез-
печити необхідну дозу при прийманні.
Рідкі лікарські засоби для орального застосуван-
ня випускають у багатодозових або однодозових кон-
тейнерах. Об’єм однієї дози рідкого лікарського
засобу для орального застосування може становити
5 мл або бути кратним до нього, або дозуватися в
краплях. Кожна доза багатодозового лікарського за-
собу застосовується за допомогою дозуючого прист-
рою, призначеного для вимірювання прописаного
об’єму.
Контейнери для рідких лікарських засобів для ораль-
ного застосування мають відповідати вимогам статті
"Матеріали, використовувані для виробництва кон-
тейнерів" (3.1 та підрозділи) та "Контейнери" (3.2
та підрозділи).
ВИРОБНИЦТВО
Для рідких лікарських засобів, призначених для
орального застосування, до складу яких входять ан-
тимікробні консерванти, уповноваженому органу ма-
ють бути подані дані, шо підтверджують ефективність
вибраних консервантів. Метод визначення і критерії
ефективності консервантів мають відповідати вимо-
гам статті "Ефективність антимікробних консер-
вантів” (5. 1.3).
При виробництві, пакуванні, зберіганні й реалізації
-рідких лікарських засобів для орального застосування
мають бути вжиті відповідні заходи, які забезпечують
необхідну мікробіологічну чистоту відповідно до ви-
мог статті "Мікробіологічна чистота лікарських за-
собів" (5. 1.4).
При виробництві рідких лікарських засобів для ораль-
ного застосування, які містять дисперговані частки,
треба передбачити заходи, які забезпечують необ-
хідний розмір часток та його контроль.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Рідкі лікарські засоби для орального застосування
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Рідкі лікарські засоби у ви-
гляді суспензій в однодозових контейнерах мають ви-
тримувати таке випробування: після збовтування
звільняють кожний контейнер якомога повніше і про-
водять визначення вмісту діючої речовини. Зазначені
суспензії мають витримувати випробування од-
норідності вмісту для однодозових препаратів (тест В).
Однорідність маси (2.9.5). Рідкі лікарські засоби у ви-
гляді емульсій в однодозових контейнерах мають ви-
тримувати таке випробування. Звільняють кожен з
20 контейнерів якомога повніше і зважують вміст
кожного контейнера. Визначають середню масу
вмісту. Маса вмісту не більше двох контейнерів може
відхилятися більш як на (±10 %) від середньої маси, і
маса вмісту жодного контейнера не має відхилятися
більш як на (±20 %).
Доза і однорідність дозування крапель для орального за-
стосування. Кількість крапель, відповідну одній дозі,
помішають у мірний циліндр за допомогою пристрою,
що капає або дозує і входить до комплекту упаковки.
Швидкість капання не має перевищувати двох кра-
пель на секунду. Рідину зважують, додають ще одну
дозу і знову зважують; повторне додавання з наступ-
ним зважуванням проводять доти, доки не буде зваже-
но 10 доз. Визначають середню масу дози. Маса жод-
ної дози не має відхилятися більш як на (±10 %) віл се-
редньої маси. Сумарна маса десяти доз не має
відрізнятися більш як на (±15 %) від номінальної ма-
си 10 доз. Якщо необхідно, вимірюють загальний
об’єм 10 доз. Об’єм не має відрізнятися більш як на
(±15 %) від номінального об’єму 10 доз.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах.
МАРКУВАННЯ
На етикетці зазначають:
— назву всіх антимікробних консервантів;
— для оральних крапель — кількість крапель в одно-
му мілілітрі або в одному грамі препарату, якщо
доза вимірюється краплями.
Порошки і гранули для
оральних розчинів і суспензій
ВИЗНАЧЕННЯ
Порошки і гранули, використовувані для приготуван-
ня розчинів і суспензій для орального застосування, в
основному відповідають визначенням, наведеним в
статтях "Порошки для орального застосування" і "Гра-
нули". Вони можуть містити також допоміжні речови-
ни, які сприяють диспергуванню або розчиненню або
запобігають злипанню.
Після розчинення або суспендування вони мають за-
довольняти вимоги, які ставляться до розчинів і сус-
пензій для орального застосування.
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Порошки і гранули в одно-
дозовому контейнері з вмістом діючої речовини мен-
ше 2 мг або менше 2 % від загальної маси мають ви-
тримувати випробування однорідності вмісту діючої
речовини в одиниці дозованого лікарського засобу
(тест В), якщо немає інших зазначень в окремій статті.
Для порошків і гранул, що містять більше однієї
діючої речовини, вимоги поширюються лише на ті ре-
човини, вміст яких відповідає вищезазначеним умо-
вам. Дане випробування не поширюється на
полівітамінні препарати і препарати, які містять
мікроелементи.
Однорідність маси (2.9.5). Порошки і гранули в одно-
дозових контейнерах мають витримувати випробуван-
ня однорідності маси для одиниці дозованого лікар-
ського засобу. Випробування однорідності маси не ви-
магається, якщо випробування однорідності вмісту
передбачено для всіх діючих речовин.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах.
МАРКУВАННЯ
На етикетці зазначають:
— спосіб приготування розчину або суспензії;
— умови і термін зберігання після приготування.
__________________________________________N
ВИЗНАЧЕННЯ
До рідких лікарських засобів для орального застосу-
вання належать також сиропи, настойки та екстракти
для орального застосування.
ВИПРОБУВАННЯ
Рідкі лікарські засоби для орального застосування
звичайно контролюють за такими показниками
< якості: опис, ідентифікація, рН, супровідні домішки,
об’єм вмісту контейнера, мікробіологічна чистота,
кількісне визначення.
Для в’язких рідких лікарських засобів для орального
застосування додатково контролюють густину і
в’язкість.
Для рідких лікарських засобів для орального застосу-
вання у вигляді суспензій додатково контролюють
стійкість суспензії.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
523
Субстанції"
Кількісне визначення. Вміст визначуваних речовин
звичайно зазначають у грамах або одиницях дії в 1 мл
або в одній дозі препарату.
МАРКУВАННЯ
На етикетці додатково зазначають:
— назву діючих речовин і антимікробних консер-
вантів;
— вміст діючих речовин у дозі або в об’ємі;
— для емульсій і суспензій — час збовтування препа-
рату перед застосуванням.
УПАКОВКА
Упаковка має забезпечувати стабільність і зручність
дозування препарату при застосуванні.
Сиропи
ВИЗНАЧЕННЯ
Густі прозорі рідини, що містять одну або більше дію-
чих речовин, розчинених у концентрованих водних
розчинах сахарози або інших цукрів. Якщо необхідно,
до сиропів додають антимікробні консерванти, анти-
оксиданти, стабілізатори, ароматизатори, смакові до-
бавки та інші допоміжні речовини.
СУБСТАНЦІЇ*
ЗАГАЛЬНІ ПОЛОЖЕННЯ
1. Субстанція — стандартизована біологічно активна
речовина (звичайно одержувана шляхом синтезу) або
стандартизована суміш біологічно активних речовин
(звичайно одержувана з об’єктів тваринного або рос-
линного походження), яка використовується для
виробництва готових лікарських засобів.
2. Вимоги даної статті поширюються, передусім, на
індивідуальні органічні речовини. Для субстанцій, що
являють собою стандартизовану суміш біологічно ак-
тивних речовин тваринного або рослинного поход-
ження, а також неорганічних речовин можливі відхи-
лення відданих вимог або додаткові вимоги, зазначені
в окремих статтях.
3. Вимоги даної статті поширюються також на до-
поміжні речовини, які використовуються для вироб-
ництва готових лікарських засобів.
4. Субстанції звичайно не використовують як готові
лікарські засоби. Підприємство-виробник готового
лікарського засобу проводить контроль якості суб-
станції перед виробництвом готового лікарського за-
собу.
5. Зразки всіх серій субстанцій, використаних для ви-
готовлення готових лікарських засобів, зберігають на
підприємстві щонайменше два роки після випуску го-
тової продукції.
6. Якість субстанції регламентується вимогами
відповідної монографії Державної Фармакопеї
України (ДФУ) і/або аналітичного нормативного
документа (АНД), затвердженого уповноваженим
органом.
7. Усі субстанції, які застосовуються для виготовлення
готових лікарських засобів, мають відповідати вимо-
гам відповідної монографії ДФУ.
8. У тому випадку, коли субстанція даного виробника
має Сертифікат відповідності монографії Європей-
ської Фармакопеї або аналогічний дозвіл уповноваже-
ного органу, її якість може контролюватися безпосе-
редньо відповідною монографією ДФУ.
9. У решті випадків якість субстанцій контролюється
за АНД, затвердженим уповноваженим органом.
Рівень вимог даного АНД має бути не нижчим за ви-
моги відповідної монографії ДФУ.
10. Відповідність вимогам монографії є необхідною,
але не завжди достатньою умовою для остаточного
висновку про якість субстанції будь-якого конкретно-
го виробника. При оцінці якості субстанції необхідно
брати до відома виробництво її відповідно до вимог
належної виробничої практики за відомою техно-
логією, умови реалізації.
11. Вимоги монографії або АНД враховують кон-
кретні технології виробництва субстанцій з певними
профілями домішок. Тому відповідно до вимог даної
монографії можуть контролюватися лише субстанції,
одержані за цими конкретними технологіями, що
має бути підтверджене Сертифікатом відповідності
монографії Європейської Фармакопеї для даної суб-
станції або аналогічним дозволом уповноваженого
органу.
12. Усі зміни, що вносяться у технологію виробництва
субстанції, освоєння старої або розробка нової техно-
логії виробництва іншими виробниками мають супро-
воджуватися поданням до уповноваженого органу да-
них, які підтверджують можливість контролю якості
цієї субстанції за вимогами відповідної монографії або
АНД.
13. У тих випадках, коли зміна технології виробництва
субстанції може призвести до появи домішок, які не
контролюються відповідною монографією, необхідна
інформація має бути подана до уповноваженого орга-
ну. До внесення змін до монографії якість таких суб-
станцій контролюють за АНД, затвердженим уповно-
важеним органом. АНД має бути затверджений також
і в тих випадках, коли для субстанції відсутня моно-
графія ДФУ.
524
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Субстанції
ПОКАЗНИКИ ЯКОСТІ, ЩО ВКЛЮЧАЮТЬСЯ до
АНАЛІТИЧНОГО НОРМАТИВНОГО ДОКУМЕНТА
НА СУБСТАНЦІЇ
АНД на субстанції звичайно містить такі розділи:
1. Вступна частина.
2. Опис.
3. Розчинність.
4. Ідентифікація.
5. Температура плавлення*.
6. Температура кипіння або температурні межі
перегонки*.
7. Температура тверднення*.
8. Відносна густина (густина)*.
9. Питоме оптичне обертання (оптичне обертання)*.
10. Питомий показник поглинання*.
11. Показник заломлення*.
12. В’язкість*.
13. Показники якості розчину:
— Прозорість.
— Кольоровість.
— Кислотність (лужність) або рН.
14. Механічні включення*.
15. Супровідні домішки'
— Конкретно зазначувані домішки.
— Неназивані домішки.
— Сумарний вміст домішок.
16. Залишкові кількості органічних розчинників.
17. Речовини, що легко обвуглюються*.
18. Неорганічні аніони (хлориди, сульфати, нітрати
і т.д.)*.
19. Неорганічні катіони (залізо та ін.)*.
20. Втрата в масі при висушуванні або вода.
21. Загальна зола або сульфатна зола.
22. Важкі метали.
23. Арсен*.
24. Мікробіологічна чистота (або стерильність).
25. Пірогени* (бактеріальні ендотоксини)*.
26. Кількісне визначення.
27. Активність*.
28. Пакування.
29. Маркування.
ЗО. Транспортування.
ЗІ. Зберігання.
32. Термін придатності.
33. Основна фармакологічна дія.
Примітка. Розділи, позначені включають залежно від природи
і призначення субстанції. Допускається введення інших розділів.
Вступна частина. Зазначають межі вмісту основної ре-
човини в субстанції (звичайно в перерахунку на суху
або безводну речовину) або, якщо це неможливо ви-
значити, наводять вимоги за параметрами, пов’язани-
ми з вмістом основної речовини в субстанції. Для
індивідуальних органічних субстанцій наводять
міжнародну непатентовану назву, хімічну назву за но-
менклатурою ШРАС, структурну формулу і брутто-
формулу (для неорганічних субстанцій — молекуляр-
ну формулу), молекулярну масу (для кристалосоль-
ватів для сольватованої і несольватованої молекули).
Зазначають також виробника субстанції та її передба-
чуване застосування.
Опис. Зазначають характеристики фізичного стану і
колір субстанції. Не треба включати опис смаку. У не-
обхідних випадках наводять інформацію про запах і
гігроскопічність.
Допустимий діапазон кольоровості субстанції звичай-
но має бути у межах відтінків, наприклад: "Крис-
талічний порошок білого або білого з жовтуватим
відтінком кольору".
У деяких випадках може бути зазначений чисельний
діапазон розміру часток, а також уведено дослідження
форми кристалів. Такі випробування виносять в ок-
ремі розділи.
Розчинність (1.4) Розчинність субстанції у різних роз-
чинниках розглядають як додаткову характеристику її
ідентифікації і чистоти, тому бажано використовувати
розчинники, які охоплюють широку шкалу поляр-
ності, наприклад: вода, 96 % спирт, ацетон, мети-
ленхлорид, гексан Не рекомендується використання
легкокиплячих та легкозаймистих (наприклад, діети-
ловий ефір) або дуже токсичних (наприклад, бензол)
розчинників.
Ідентифікація. Для ідентифікації субстанцій звичайно
застосовують сполучення інфрачервоної спектро-
скопії, електронної спектрофотометрії або хромато-
графії (газової, рідинної або тонкошарової) з харак-
терними хімічними реакціями. Можливе використан-
ня й інших фізико-хімічних методів.
Температура плавлення (2.2.14 - 2.2.16). Випробування
звичайно застосовують для характеристики твердих
речовин.
Температура кипіння (2.2.12) або Температурні межі пе-
регонки (2.2.11), Відносна густина (густина) (2.2.5),
В’язкість (2.2.8 — 2.2.10), Показник заломлення (2.2.6),
Температура тверднення (2.2.18). Дані випробування
вводять для характеристики рідких речовин.
Питоме оптичне обертання (оптичне обертання) (2.2.7).
Уводять для характеристики оптично активних речо-
вин.
Питомий показник поглинання (2.2.25). Уводять зви-
чайно в тих випадках, коли випробовувана речовина
має максимуми поглинання в області від 220 нм до
800 нм. Даний показник є додатковою характеристи-
кою ідентифікації й чистоти субстанції.
Прозорість розчину (2.2.1), Кольоровість розчину
(2.2.2) Дані випробування обов’язково уводять для
субстанцій, використовуваних для приготування па-
рентеральних, очних, назальних і вушних лікарських
засобів. Для решти лікарських засобів їх бажано уво-
дити для всіх водорозчинних субстанцій. Випробуван-
ня звичайно проводять у водних розчинах субстанцій,
але можливе використання змішаних розчинників або
самої субстанції.
Розчини субстанцій, як правило, мають бути прозори-
ми.
Випробування "Кольоровість розчину" звичайно не
уводять у тому випадку, якщо субстанція забарвлена.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
525
Субстанції”
Дане випробування, при необхідності, можна заміни-
ти регламентацією оптичної густини за певних дов-
жин хвиль.
рН (Кислотність або лужність). Для проведення даного
випробування можуть використовуватися два підходи:
вимірювання рН (2.2.3) або напівкількісне індикатор-
не титрування (кислотність і/або лужність). Випробу-
вання проводять звичайно у водних розчинах суб-
станції, але можливе використання і змішаних роз-
чинників. Допустимий інтервал рН звичайно має бути
не більше 2.
Механічні включення (2.9.19 — 2.9.21). Дане випробу-
вання уводять для контролю якості стерильних суб-
станцій, використовуваних для приготування парен-
теральних і очних лікарських засобів. Перевірку
здійснюють звичайно у тій максимальній концент-
рації, яку використовують у відповідних готових
лікарських засобах.
Супровідні домішки. Дане випробування контролює
технологічні домішки (напівпродукти і побічні про-
дукти), продукти розкладання, а також у деяких ви-
падках сторонні домішки. У межах цього випробуван-
ня звичайно не контролюють неорганічні домішки і
залишкові кількості летких органічних розчинників.
Як правило, усі домішки, концентрація яких переви-
щує 0.1 %, мають бути ідентифіковані. Тест "Супро-
відні домішки" може контролювати конкретно як
зазначувані домішки, так і ті, що не конкретизуються
(неназивані). Інформацію про природу і кількість цих
домішок необхідно подавати до уповноваженого ор-
гану.
Для контролю супровідних домішок можуть застосо-
вуватися різні хроматографічні і спектроскопічні ме-
тоди або їх комбінації, однак більшість субстанцій
контролюють хроматографічними методами.
Сумарний вміст супровідних домішок звичайно не
має перевищувати 2 %.
У АНД має бути зазначено, які домішки контролює
розділ "Супровідні домішки", мають бути наведені їхні
структурні та молекулярні формули і маси. Треба за-
уважити. шо в субстанції можуть виявлятися не лише
ці зазначені домішки, а також можливі слідові
кількості інших супровідних домішок, наведених у до-
датку до АНД. Якщо субстанція містить домішку в
кількості вище 0.1 %, яка не контролюється цим ви-
пробуванням, це може означати, що ця субстанція
одержана за іншою (не дозволеною уповноваженим
органом) технологією, або що використовувана ви-
робником технологія не забезпечує захист від забруд-
нення сторонніми домішками. Така серія субстанції
може використовуватися лише після спеціального
дозволу уповноваженого органу.
Конкретно зазначувані домішки. Даний показник уво-
дять в тому випадку, коли необхідно регламентувати
вміст якихось конкретних, нерідко високотоксичних
домішок. Визначення їх може виноситися в окремий
розділ. У деяких випадках в ролі таких домішок мо-
жуть виступати сторонні домішки (тобто які не є ні
напівпродуктом синтезу, ні продуктом розкладання),
які привносяться у процесі технології. Межі вмісту
домішок, які конкретно зазначаються, необхідно об-
грунтовувати фармакологічними або токсикологічни-
ми даними.
Для контролю конкретно зазначуваних домішок кра-
ще застосовувати хроматографічні метоли з викорис-
танням стандартних зразків домішок.
Неназивані домішки. У тих випадках, коли немає
підстав вважати якісь домішки особливо токсичними,
їхні назви при проведенні випробування звичайно не
зазначають. При застосуванні хроматографічних ме-
тодів для нормування таких домішок допускається ви-
користання випробовуваної субстанції у необхідних
розведеннях. При цьому звичайно нормують вміст як
окремих домішок, так і їхню суму.
Вміст таких домішок необхідно обгрунтовувати фар-
макологічними або токсикологічними даними.
У деяких випадках у межах даного випробування рег-
ламентують також конкретно зазначувані домішки.
Залишкові кількості органічних розчинників (2.4.24,
5.4). Вміст залишкових кількостей органічних розчин-
ників, які використовуються при одержанні суб-
станції, має відповідати вимогам статті "Залишкові
кількості органічних розчинників" (5.4). До уповнова-
женого органу треба подати інформацію про всі роз-
чинники, які використовуються при виробництві суб-
станції, і обгрунтування вибору розчинників, контро-
льованих в АНД. Наявність у субстанції інших роз-
чинників у концентраціях, що перевищують 10 % від
регламентованих статтею (5.4), є ознакою викорис-
тання незареєстрованої технології виробництва суб-
станції. Така серія субстанції може використовуватися
лише після спеціального дозволу уповноваженого ор-
гану.
Речовини, що легко обвуглюються (2.4.30). Дане випро-
бування є неспецифічним тестом на органічні
домішки і уводиться в деяких випадках для підтверд-
ження чистоти субстанції. Випробування проводять з
кислотою сірчаною концентрованою з наступною
оцінкою забарвлення одержаного розчину.
Неорганічні аніони (2.4). У назві даного розділу має бу-
ти конкретно зазначено, який аніон контролюють,
наприклад, "Хлориди". Вибір контрольованих аніонів
визначається технологічним процесом з обгрунтуван-
ням у супровідній документації. При цьому контрольо-
вані аніони можуть бути нетоксичними (наприклад,
хлориди, сульфати та ін.). Регламентація їхнього
вмісту має на меті додатково відрізнити зареєстровану
технологію від незареєстрованої. Межі їхнього вмісту
вимагають необхідного обгрунтування.
Контроль аніонів як домішок не уводять у тому випад-
ку, якшо вони входять до складу субстанції (напри-
клад, речовина є гідрохлоридом або сульфатом).
Неорганічні катіони (2.4). У назві даного розділу має
бути конкретно зазначено, який катіон контролюють.
Цей розділ уводять в тому випадку, коли контроль
526
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Таблетки
і
вмісту конкретних катіонів є суттєвим для якості
субстанції. Межі їхнього вмісту вимагають необ-
хідного обгрунтування.
Контроль катіонів як домішок не вводять в тому ви-
падку, якщо вони входять до складу субстанції (напри-
клад, речовина є натрієвою сіллю).
Випробування Втрата в масі при висушуванні (2.2.32)
або Вода (2.5.12) уводять для контролю вмісту летких
речовин і/або вологи в субстанції. Введення одного з
цих випробувань, як правило, обов’язкове.
Відсутність їх треба пояснювати у пояснювальній за-
писці. Якщо немає інших зазначень в окремій статті й
субстанція не є кристалосольватом, то втрата в масі
при висушуванні або вміст води не має перевищувати
0.5%.
Якщо субстанція є кристалогідратом (кристалосоль-
ватом), то регламентують верхню і нижню межі.
Загальна зола (2.4.16) або Сульфатна зола (2.4.14). Дані
випробування характеризують загальну мінералізацію
субстанції. Як правило, сульфатна або загальна зола
не має перевищувати 0.1 %. Відсутність даного випро-
бування в АНД або підвищений вміст сульфатної або
загальної золи вимагає відповідного обгрунтування.
Важкі метали (2.4.8). Вміст важких металів не має пе-
ревищувати 0.001 %, якщо немає інших зазначень в
। окремій статті.
Арсен (2.4.2). Дане випробування вводять у тому ви-
падку, коли або вихідна сировина може містити арсен,
наприклад, для сировини природного походження,
або можливе забруднення ним у процесі одержання
субстанції. Вміст арсену, як правило, не має переви-
щувати 0.0001 %.
Мікробіологічна чистота (2.6.12, 2.6.13, 5.1.4). Дане ви-
пробування уводять для контролю якості нестериль-
них субстанцій. Мікробіологічна чистота субстанцій
має забезпечувати необхідну мікробіологічну чистоту
готових лікарських засобів.
Відсутність даного випробування в АНД вимагає
відповідного обгрунтування.
Стерильність (2.6.1). Дане випробування уводять для
субстанцій, використовуваних у виробництві готових
стерильних лікарських засобів, які не піддаються про-
цедурі стерилізації.
Пірогени (2.6.8) або Бактеріальні ендотоксини (2.6.14).
Дані випробування проводять у таких випадках:
— якщо субстанція використовується у виробництві
готових лікарських засобів, які вимагають відсут-
ності пірогенів або бактеріальних ендотоксинів,
але при цьому не піддаються відповідній проце-
дурі їх вилучення;
— якщо в маркуванні субстанції є зазначення про
відсутність пірогенів або бактеріальних ендоток-
синів.
Граничні значення і тест-методи установлюються в
окремій статті.
Кількісне визначення. Для кількісного визначення ос-
новної речовини в субстанції бажане використання
прямих методів аналізу. У випадку солей звичайно до-
статньо аналізу лише одного з іонів — краще фармако-
логічно активного. Вміст основної речовини в синте-
тичній субстанції звичайно має бути в межах від 99.0 %
до 101.0 %, якщо не наводиться відповідне обгрунту-
вання. При необхідності визначають біологічну ак-
тивність. Якщо визначення вмісту основної речовини
в субстанції неможливе, проводять визначення таких
кількісних показників, які зв’язані із вмістом основ-
ної речовини у субстанції.
Пакування і зберігання. Упаковка та умови зберігання
мають забезпечувати якість субстанції протягом
терміну придатності.
Маркування. Маркування має містити відомості про
виробника, торгову і міжнародну непатентовану назву
субстанції, умови зберігання, застережні заходи (якщо
вони необхідні), дату виготовлення і термін придат-
ності.
Термін придатності. Термін придатності субстанцій —
це проміжок часу, протягом якого виробник суб-
станції гарантує її відповідність вимогам АНД при до-
триманні умов зберігання. Протягом усього терміну
придатності субстанцію можна використовувати для
виробництва готових лікарських засобів за умови від-
повідності ії вимогам АНД.
ТАБЛЕТКИ
ТаЬиІеПае
Вимоги даної статті не обоє ’язкові для таблетованих
лікарських засобів, призначених до застосування не
оральним, а іншим способом. Вимоги до таких лікар-
ських засобів можуть бути наведеними в інших загаль-
них статтях, наприклад, "Лікарські засоби для рек-
тального застосування" або "Лікарські засоби для
вагінального застосування". Вимоги даної статті не
поширюються на таблетки для ветеринарного засто-
сування, якщо немає інших зазначень.
ВИЗНАЧЕННЯ
Таблетки — тверда лікарська форма, яка містить одну
дозу однієї або більше діючих речовин і одержана пре-
суванням певного об’єму часток. Таблетки призначені
для прийняття всередину. Деякі таблетки ковтають
цілими, деякі — попередньо розжовують, інші ж розчи-
няють або диспергують у воді перед вживанням або за-
лишають у роті, де діюча речовина вивільнюється.
Частки складаються з однієї або більше діючих і таких
допоміжних речовин, які розводять, зв’язують, розпу-
шують, ковзають, зволожують; речовин, здатних
змінити поведінку лікарської форми у травному
тракті, барвників, дозволених до медичного застосу-
вання, і ароматизаторів або без допоміжних речовин.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
527
Таблетки
Таблетки звичайно являють собою цільні правильні,
круглі циліндри, верхня і нижня поверхні яких плоскі
або опуклі, краї поверхонь можуть бути скошені. На
поверхні таблеток можуть бути нанесені штрихи, рис-
ки для поділу, написи та інші позначення. Таблетки
можуть бути покритими оболонкою.
Контейнери для таблеток мають відповідати вимогам
статей "Матеріали, використовувані для виробництва
контейнерів" (3.1 та підрозділив та "Контейнери" (3.2
та підрозділи/ якщо немає інших зазначень в окремій
статті.
Таблетки для приймання всередину можуть бути кла-
сифіковані як:
— таблетки без оболонки;
— таблетки, покриті оболонкою;
— таблетки "шипучі";
— таблетки розчинні;
— таблетки дисперговані;
— таблетки кишково-розчинні;
— таблетки з модифікованим вивільненням;
— таблетки для застосування у ротовій порожнині.
ВИРОБНИЦТВО
Таблетки одержують пресуванням певного об’єму час-
ток або агрегатів часток, одержаних методами грану-
ляції. При виготовленні ядер таблеток мають бути
ужиті відповідні заходи, шо забезпечують необхідну
механічну міцність і стійкість таблеток ло роздавлю-
вання і стирання. Це підтверджується випробування-
ми "Стираність таблеток без оболонки" (2.9.7)
і "Стійкість таблеток до роздавлювання"(2.9.8). Таб-
летки для розжовування виготовляють, забезпечуючи
легке руйнування при жуванні.
При виробництві, пакуванні, зберіганні й реалізації
таблеток мають бути вжиті відповідні заходи, що за-
безпечують необхідну мікробіологічну чистоту
відповідно до вимог статті '"Мікробіологічна чистота
лікарських засобів ” (5.1 4).
ВИПРОБУВАННЯ
Однорідність вмісту (2.9.6). Таблетки із вмістом діючої
речовини менше 2 мг або менше 2 % від маси таблет-
ки мають витримувати випробування однорідності
вмісту діючої речовини в одиниці дозованого лікарсь-
кого засобу (тест А), якщо немає інших зазначень в
окремій статті. Якщо лікарська форма містить більше
однієї діючої речовини, вимоги поширюються лише
на ті речовини, вміст яких відповідає вищезазначеним
умовам. Таке випробування не поширюється на
полівітамінні препарати і препарати, що містять
мікроелементи.
Однорідність маси (2.9.5). Таблетки без оболонки і, як-
що немає інших зазначень в окремій статті, таблетки,
покриті плівковою оболонкою, мають витримувати ви-
пробування однорідності маси для одиниці дозованого
лікарського засобу. Випробування на однорідність маси
не вимагається, якщо випробування однорідності
вмісту передбачене для всіх діючих речовин.
Розчинення. Випробування може бути проведене для
підтвердження відповідного вивільнення діючої речо-
вини або речовин, наприклад, одним із способів, опи-
саних у загальній статті "Тест "Розчинення" для твер-
дих дозованих форм " (2.9.3).
Якщо проводять випробування за показником "Роз-
чинення", випробування "Розпадання" не вима-
гається.
ЗБЕРІГАННЯ
У щільно закупорених контейнерах, шо запобігає роз-
давлюванню і механічним ударам.
Таблетки без оболонки
ВИЗНАЧЕННЯ
Таблетки без оболонки — це одношарові таблетки,
одержані одноразовим пресуванням часток, або бага-
тошарові таблетки, які складаються з концентричних
або паралельних шарів, одержані послідовним пресу-
ванням часток різного складу. Використовувані допо-
міжні речовини спеціально не призначені для вивіль-
нення діючої речовини у шлунково-кишковому тракті.
Таблетки без оболонки відповідають загальному ви-
значенню таблеток. На розламі при розгляданні під
лупою видно ту чи іншу відносно однорідну структуру
(одношарові таблетки) або пошарову структуру (бага-
тошарові таблетки), але не ознаки оболонки.
ВИПРОБУВАННЯ
Розпадання. Таблетки без оболонки мають витримува-
ти випробування на розпадання таблеток або капсул
(2.9.1). Як рідке середовище використовують воду Р. У
кожну скляну трубку поміщають диск. Прилад вмика-
ють на 15 хв, якщо немає інших зазначень в окремій
статті, і досліджують стан таблеток. Якщо таблетки не
витримали випробування внаслідок прилипання їх до
дисків, випробування повторюють на наступних шес-
ти таблетках без дисків.
Випробування вважають витриманим, якщо розпали-
ся усі шість таблеток.
Таблетки для розжовування випробуванню на розпадан-
ня не підлягають.
Таблетки, покриті оболонкою
ВИЗНАЧЕННЯ
Таблетки, покриті оболонкою, — це таблетки, покриті
одним або кількома шарами суміші різних речовин,
таких як натуральні або синтетичні смоли, камеді, же-
латин, неактивні і нерозчинні наповнювачі, цукри,
пластифікатори, поліспирти, воски, барвники, дозво-
лені для медичного застосування, і іноді ароматизато-
528
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Таблетки
ри та діючі речовини. Речовини, використовувані для
покривання таблеток, звичайно наносять у вигляді
розчинів або суспензій в умовах, що дозволяють роз-
чиннику випаритися. Коли оболонка являє собою ду-
же тонке полімерне покриття, таблетки визначають як
таблетки, покриті плівковою оболонкою.
Таблетки, покриті оболонкою, мають гладку поверх-
ню, яка часто забарвлена і може бути відполірованою;
на розламі при розгляданні під лупою видно ядро,
оточене одним або кількома суцільними шарами
різної структури.
ВИПРОБУВАННЯ
Розпадання. Таблетки, покриті оболонкою, за винят-
ком плівкової, мають витримувати випробування
на розпадання таблеток або капсул (2.9.1). Як рідке се-
редовище використовують воду Р У кожну скля-
ну трубку поміщають диск. Прилад вмикають на
60 хв, якщо немає інших зазначень в окремій стат-
ті, і досліджують стан таблеток. Якщо не розпа-
лася хоча б одна з шести таблеток, випробування
повторюють на наступних таблетках, замінивши
воду Ру посудині на 0.1 М розчин кислоти хлористо-
водневої. Випробування вважають витриманим, якщо
усі шість таблеток розпалися у кислому середовищі.
Таблетки, покриті плівковою оболонкою, мають витри-
мувати випробування на розпадання в умовах, прийня-
тих для таблеток без оболонки, при цьому прилад вми-
кають на ЗО хв, якщо інше не зазначено в окремій статті.
Якщо таблетки, покриті оболонкою, або таблетки,
покриті плівковою оболонкою, не витримали випро-
бування внаслідок прилипання таблеток до дисків,
випробування повторюють на наступних шести таб-
летках без дисків.
Випробування вважають витриманим, якщо розпали-
ся усі шість таблеток.
Таблетки для розжовування, покриті оболонкою, ви-
пробуванню на розпадання не підлягають.
Таблетки "шипучі"
ВИЗНАЧЕННЯ
Таблетки "шипучі" — це таблетки без оболонки, ос-
новну масу яких складають кислоти і карбонати або
гідрокарбонати, що швидко реагують у присутності
води з виділенням вуглекислого газу Ці таблетки при-
значені для розчинення або диспергування у воді пе-
ред застосуванням.
ВИПРОБУВАННЯ
Розпадання. Одну таблетку поміщають у склянку, яка
містить 200 мл води Р при температурі від 15 °С до
25 °С; виділяються численні бульбашки газу. Таблетка
вважається такою, що розпалася, якщо після припи-
нення виділення газу навколо таблетки або її фраг-
ментів вона або розчинилася, або диспергувалася у
воді без агломератів часток. Повторюють процедуру
на п’яти інших таблетках.
Випробування вважають витриманим, якщо кожна з
шести таблеток розпалася вищезазначеним способом
протягом 5 хв, якщо немає інших зазначень в окремій
статті.
Таблетки розчинні
ВИЗНАЧЕННЯ
Таблетки розчинні — це таблетки без оболонки або
таблетки, покриті плівковою оболонкою. Ці таблетки
перед застосуванням розчиняють у воді. В одержано-
му розчині допускається легка опалесценція через до-
поміжні речовини, використані при виготовленні
таблеток.
ВИПРОБУВАННЯ
Розпадання. Розчинні таблетки мають розпадатися
протягом 3 хв, перевіряють їх за методикою випробу-
вання на розпадання для таблеток і капсул (2.9.1), ви-
користовуючи як середовище воду Р з температурою
від 15 °С до 25 °С.
Таблетки дисперговані
ВИЗНАЧЕННЯ
Таблетки дисперговані — це таблетки без оболонки
або таблетки, покриті плівковою оболонкою. Перед
застосуванням їх диспергують у воді до утворення го-
могенної суспензії.
ВИПРОБУВАННЯ
Розпадання. Дисперговані таблетки мають розпадати-
ся протягом 3 хв, випробування на розпадання прово-
дять за методикою для таблеток і капсул (2.9.1), вико-
ристовуючи як середовище воду Р з температурою віл
15"Сдо25"С.
Ступінь диспергування. Дві таблетки поміщають у кол-
бу, яка містить 100 мл води Р, і перемішують до повно-
го диспергування. Має утворитися однорідна сус-
пензія. яка проходить крізь сито з номінальним
розміром отворів 710 мкм.
Таблетки кишково-розчинні
ВИЗНАЧЕННЯ
Таблетки кишково-розчинні — таблетки з модифіко-
ваним вивільненням, що мають бути стійкими у
шлунковому соку і вивільнювати діючу речовину або
речовини в кишковому соку. Такі таблетки виготовля-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
529
Таблетки
ють, покриваючи ядра таблеток оболонкою, стійкою
до шлункового соку (таблетки, покриті кишково-роз-
чинною оболонкою), або виготовлюють із гранул або
часток з нанесеною на них раніше оболонкою,
стійкою до шлункового соку.
Таблетки, покриті кишково-розчинною оболонкою,
зараховують до групи таблеток, покритих оболонкою.
ВИРОБНИЦТВО
Для таблеток, приготованих з гранул або часток, за-
здалегідь покритих кишково-розчинною оболонкою,
проводять випробування, яке підтверджує необхідне
вивільнення діючої речовини або речовин.
ВИПРОБУВАННЯ
Розпадання. Для таблеток, покритих кишково-роз-
чинною оболонкою, проводять випробування на роз-
падання (2.9.1) з наступними змінами. Як середовише
використовують 0.1 Мрозчин кислоти хлористоводне-
вої. Прилад вмикають на 2 год, якщо немає інших за-
значень в окремій статті, без дисків і досліджують стан
таблеток. Час стійкості таблеток у кислому середовищі
може бути різним і залежить від складу випробовува-
них таблеток. Звичайно він становить від 2 до 3 год,
але навіть якщо наведені інші зазначення в окремій
статті, час стійкості в кислому середовищі має бути не
меншим 1 год. Жодна з таблеток не має виявляти оз-
нак розпадання (не враховуючи фрагментів покриття)
і мати тріщин, крізь які можливий вихід вмісту. Кис-
1 лоту заміняють фосфатним буферним розчином
рН 6.8 Р\в кожну скляну трубку вносять диск. Прилад
вмикають на 60 хв і досліджують стан таблеток. Якщо
таблетки не витримали випробування внаслідок при-
1 липання до дисків, випробування повторюють на
шести наступних таблетках без дисків.
Випробування вважають витриманим, якщо розпали-
ся всі шість таблеток.
Таблетки з модифікованим
вивільненням
ВИЗНАЧЕННЯ
1 Таблетки з модифікованим вивільненням — таблетки,
покриті оболонкою, або без оболонки, які містять
спеціальні допоміжні речовини або виготовлені
спеціальними способами, які окремо або разом при-
шачені для зміни швидкості або місця вивільнення
ііючої речовини або речовин.
ВИРОБНИЦТВО
Проводять випробування, яке підтверджує відповідне
1 вивільнення діючої речовини або речовин.
| 530
Таблетки для застосування
у ротовій порожнині
ВИЗНАЧЕННЯ
Таблетки для застосування у ротовій порожнині — і
звичайно таблетки без оболонки. Склад забезпеч
повільне вивільнення і місцеву дію діючої речовиг
або речовин або вивільнення і всмоктування діюч
речовини або речовин у певних ділянках рота.
ВИЗНАЧЕННЯ
Для виготовлення таблеток можуть використовувать
ся й інші методи, наприклад, формування. Вимоги д
таких таблеток зазначені в окремих статтях.
Залежно від фізико-хімічних властивостей лікарськи
речовин, їх дозування і методу виготовлення таблето
застосовують такі допоміжні речовини відповідно д
їхнього призначення.
Розріджувачі застосовують для забезпечення не
обхідної маси таблеток, якщо до її складу входить ма
ла кількість діючої речовини або речовин. З метою по
крашення біодоступності важкорозчинних і гідрофоб
них лікарських речовин застосовують в основному во
дорозчинні розріджувачі. До групи розріджувачі)
можна зарахувати аеросил, авіцел, гліцин, лактозу
крохмаль, кальцію гідрофосфат, магнію карбонат
магнію оксид, модифіковані крохмалі, натрію гідро-
карбонат, целюлозу мікрокристалічну. До складу таб-
леток для розжовування звичайно входять маніт.
сорбіт, цукор.
Зв’язувальні речовини застосовують для грануляції і
забезпечення необхідної міцності таблеток при пресу-
ванні, їх додають у вигляді розчинів або в сухому ви-
гляді. До цієї групи можна зарахувати альгінову кисло-
ту та її натрієву сіль, бентоніти, гуміарабік, желатин,
цукор, полівінілпіролідон, природні камеді, трага-
кант, макрогол, метилцелюлозу, карбоксиметилцелю-
лозу, крохмальну патоку, декстрин, полівініловий
спирт. При одержанні таблеток методом прямого пре-
сування як зв’язувальне, як правило, використовують
мікрокристалічну целюлозу.
Розпушувачі застосовують для забезпечення не-
обхідного розпадання таблеток і розчинення діючої
речовини. До цієї групи допоміжних речовин можна
зарахувати крохмаль, хімічно модифіковані крохмаль і
целюлозу, агар-агар, альгінову кислоту і натрієву сіль
альгінової кислоти, аеросил, полісорбат 80, натрію ла-
урилсульфат, метилцелюлозу, натрієву сіль карбо-
ксиметилцелюлози, поперечно-зшитий полівінілпі-
ролідон, мікрокристалічну целюлозу. У шипучих таб-
летках як розпушуючий компонент звичайно викори-
стовують газоутворювальні суміші натрію гідрокарбо-
нату з кислотою винною або лимонною.
Ковзні і змащувальні речовини застосовують для по-
кращення плинності і зменшення прилипання табле-
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
Таблетки
тованих сумішей до пресуючих поверхонь. До них
можна зарахувати такі речовини: крохмаль, аеросил,
тальк, масло какао, стеаринову кислоту та її кальцієву
і магнієву солі. Більшість змащувальних і ковзних ре-
човин гідрофобні. Вони уповільнюють швидкість роз-
падання таблетки і розчинення діючої речовини, тому
не рекомендується перевищувати вміст полісор-
бату 80, кислоти стеаринової, кальцію або магнію сте-
арату більше 1 %, тальку — 3 %, аеросилу — 10 % від
маси таблетки.
Макрогол і натрію лаурилсульфат використовують як
водорозчинні ковзні речовини.
Барвники і коригенти смаку використовують для на-
дання таблеткам необхідного кольору і смаку.
Речовини, використовувані для нанесення оболонки,
поділяються на декілька груп залежно від методу по-
криття.
При нанесенні цукрової оболонки використовують
гуміарабік, желатин, магнію карбонат, крохмаль, олії
рослинні, масло какао, метилцелюлозу, борошно
пшеничне, кальцію стеарат, тальк, натрію альгінат,
кальцію карбонат, магнію оксид, патоку, титану(ІУ)
оксид та ін.
При нанесенні плівкової оболонки звичайно застосо-
вують водорозчинні речовини або дисперговані у воді
речовини, такі як гідроксипропілметилцелюлоза,
метилцелюлоза, гідроксипропілцелюлоза, натрій-кар-
боксиметилцелюлоза, співполімери метакрилової
кислоти та її ефірів, макрогол, полівінілпіролідон
У деяких випадках використовують неводні розчин-
ники.
При нанесенні оболонки пресуванням застосовують
глюкозу, мікрокристалічну целюлозу та інші до-
поміжні речовини.
ВИПРОБУВАННЯ
Таблетки звичайно коніролюють за такими показни-
ками якості: опис, ідентифікація діючих речовин, се-
редня маса таблетки, однорідність маси, стираність,
стійкість до роздавлювання, розпадання, тальк і аеро-
сил, втрата в масі при висушуванні або вода, супро-
відні домішки, однорідність вмісту, залишкові кіль-
кості органічних розчинників (при їх використанні в
технології), розчинення, мікробіологічна чистота,
кількісне визначення діючих речовин.
Середня маса таблетки. Відхилення середньої маси
таблетки від маси, зазначеної у розділі "Склад", не має
перевищувати (±5 %), якщо немає інших зазначень в
окремій статті. Визначення середньої маси таблетки
проводять, як зазначено в статті "Однорідність маси
для одиниці дозованого лікарського засобу " (2.9.5).
Тальк, аеросил. Визначення проводять відповідно до
методики, описаної уДодатку 1. Вміст тальку і аероси-
лу не має перевищувати вимог, зазначених в окремій
статті.
Однорідність вмісту. Таблетки мають витримувати ви-
моги статті "Однорідність вмісту діючої речовини в оди-
ниці дозованого лікарського засобу" (2.9.6), якщо немає
інших зазначень в окремій статті.
Кількісне визначення. Для кількісного визначення бе-
руть наважку порошку розтертих таблеток (не менше
20 штук).
Для таблеток, покритих оболонкою, допускається
проводити випробування з певною кількістю табле-
ток (не менше п’яти), зазначеною в окремій статті.
Вміст визначуваних речовин виражають у грамах,
міліграмах або одиницях дії (ОД) на одну таб-
летку, якщо немає інших зазначень в окремій
статті.
Вміст діючої речовини в таблетці. Відхилення у вмісті
діючих речовин мають становити при дозуванні мен-
ше 1 мг (+15 %), від 1 мгдо 10 мг (±10 %), від 10 мгдо
100 мг (±7.5 %) і вище 100 мг (±5 %) від вмісту, зазна-
ченого у розділі "Склад", якщо немає інших зазначень
в окремій статті.
МАРКУВАННЯ
На етикетці додатково зазначають;
— вміст діючої речовини (речовин) в таблетці;
— термін придатності;
— умови зберігання;
— спосіб застосування, якщо необхідно.
ДОДАТОК 1
ВИЗНАЧЕННЯ ТАЛЬКУ
Близько 1 г (точна наважка) порошку розтертих табле-
ток обробляють у посудині 200 мл теплої води Р, ріди-
ну відфільтровують крізь беззольний фільтр і посуди-
ну ретельно обполіскують водою. Залишок на фільтрі
декілька разів промивають теплою водою Р(по 10 мл)
до відсутності видимого залишку після випарювання
краплі промивної води на годинниковому склі. Фільтр
із залишком висушують, спалюють, прожарюють і
зважують з точністю до 0.0001 г.
Якщо таблетки містять неспалимі або нерозчинні в
теплій воді речовини, то наважку таблеток після
спалювання і прожарювання обробляють при на-
гріванні 30 мл кислоти хлористоводневої розведеної Р,
розчин фільтрують і залишок на фільтрі промива-
ють гарячою водою до відсутності в промивній воді
реакції на хлориди. Фільтр із залишком висушують,
спалюють, прожарюють і зважують з точністю до
0.0001 г.
Визначення аеросилу проводять за тією самою мето-
дикою.
ДЕРЖАВНА ФАРМАКОПЕЯ УКРАЇНИ 2001
531
Державна Фармакопея України
1 -е видання
Розроблено
Державним підприємством "Науково-експертний фармакопейний центр
Підписано до друку 27.07.2001 року. Формат 60x84/8.
Папір офсетний. Гарн. Ньютон. Друк офсетний.
Ум. друк. арк. 64.22.
Тираж 10000 (1-3500) прим.
Видавнича група"РІРЕГ"
(свідоцтво про внесення до державного реєстру видавців серія ДК № 532 від 16.07.2001)
61003, м. Харків, пр. Московський, 10/12
Віддруковано поліграфічним підприємством "РІРЕГ
61003, м. Харків, пр. Московський, 10/12