/






































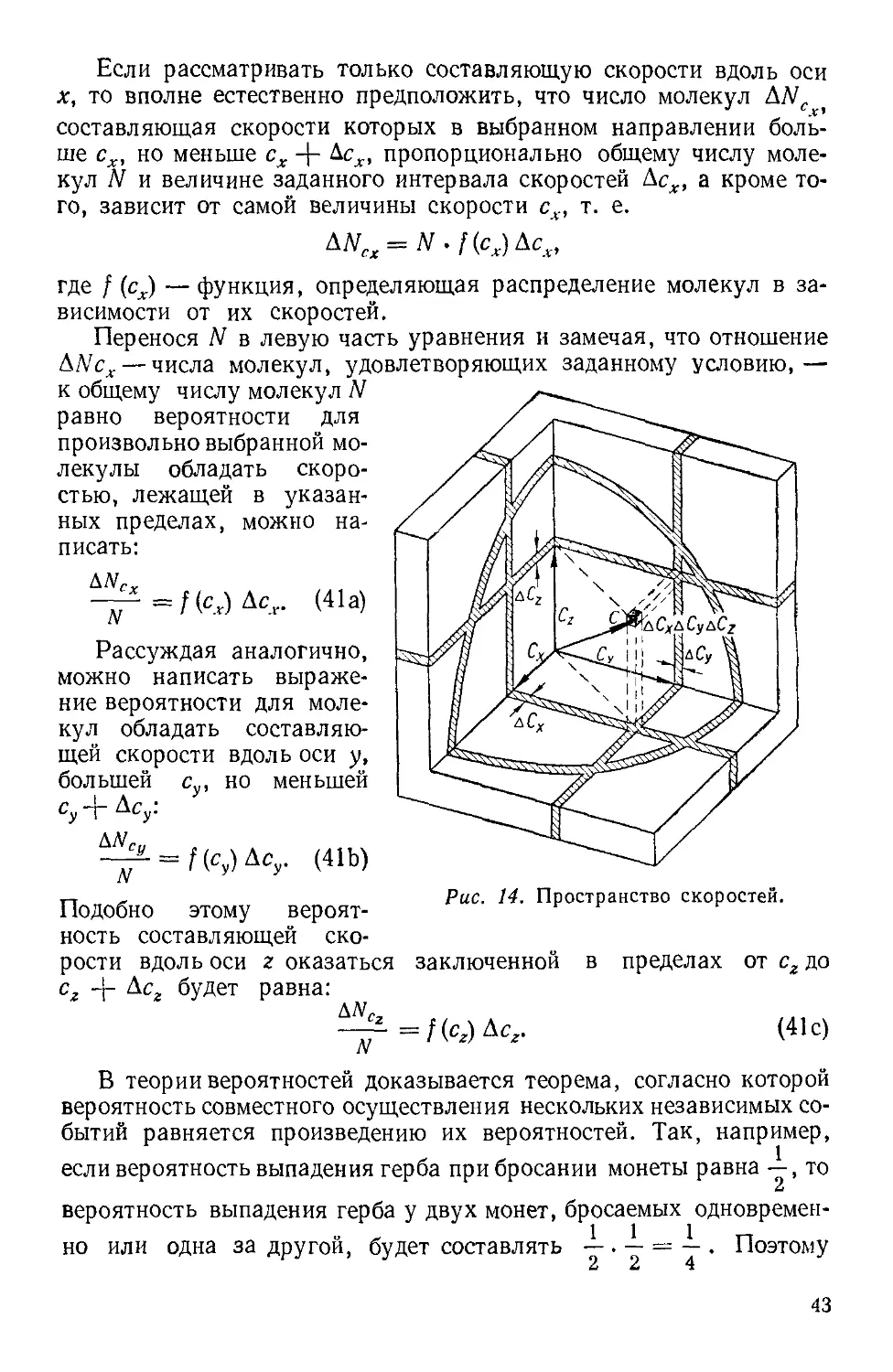





















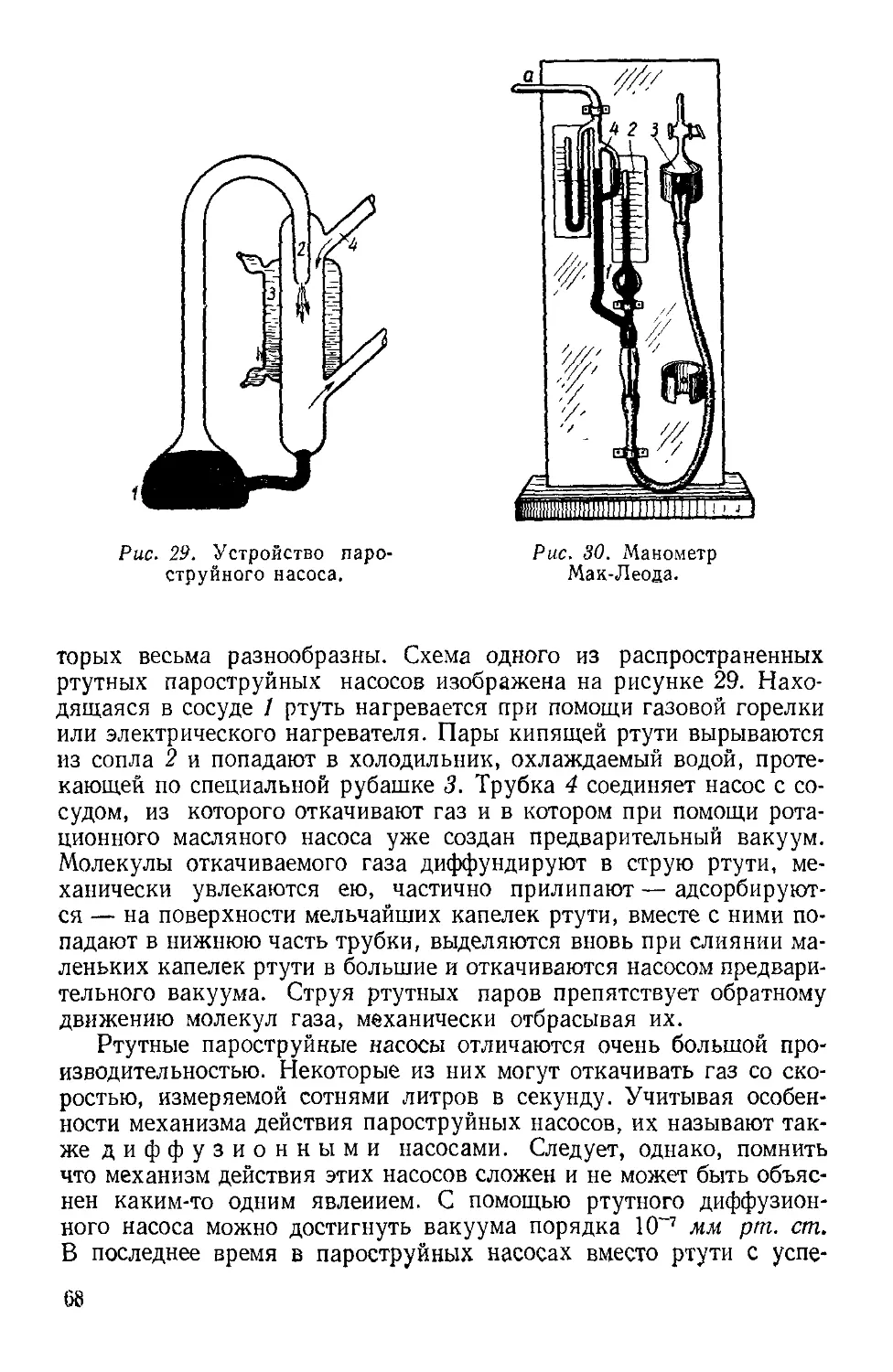








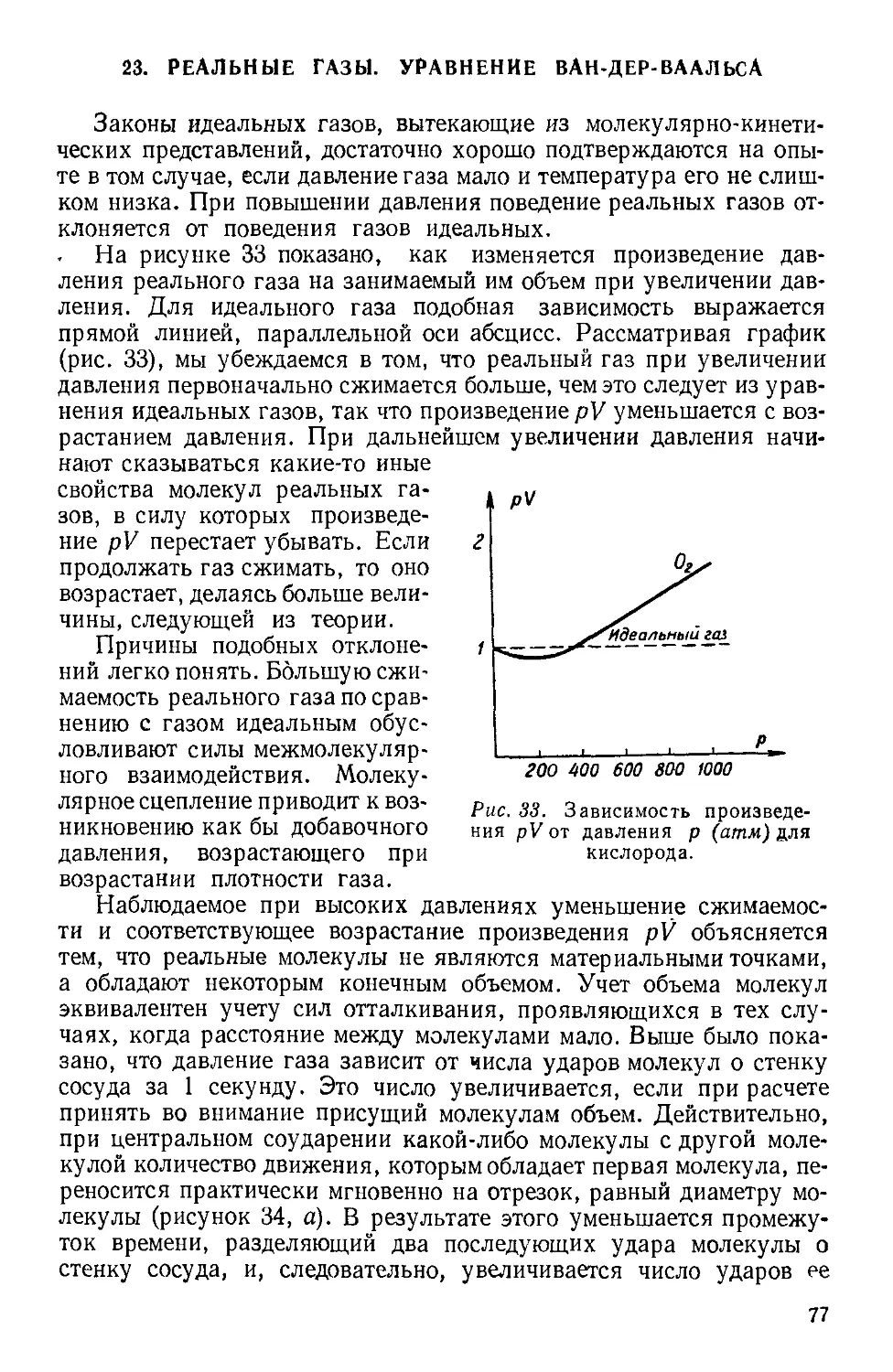





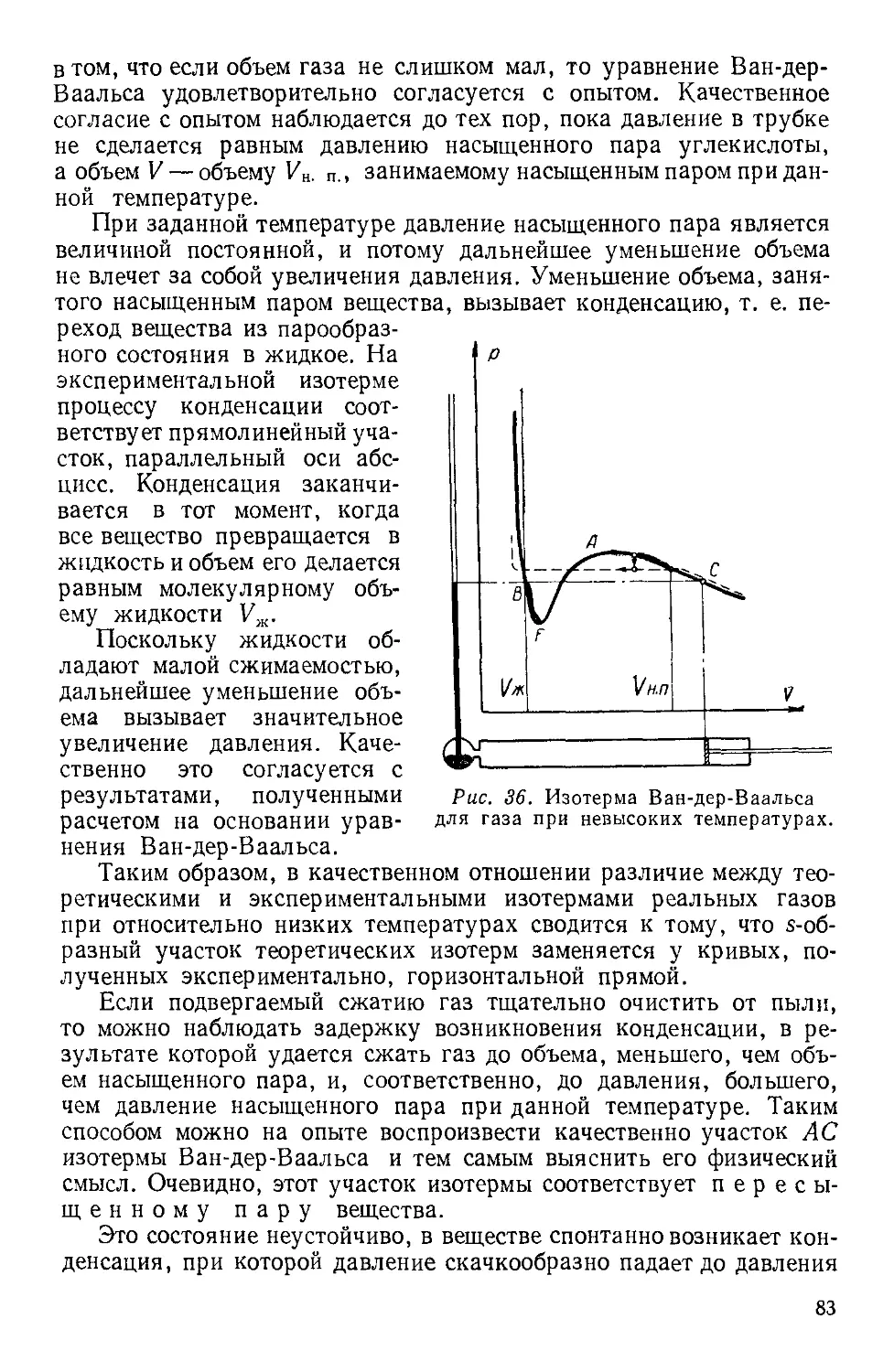

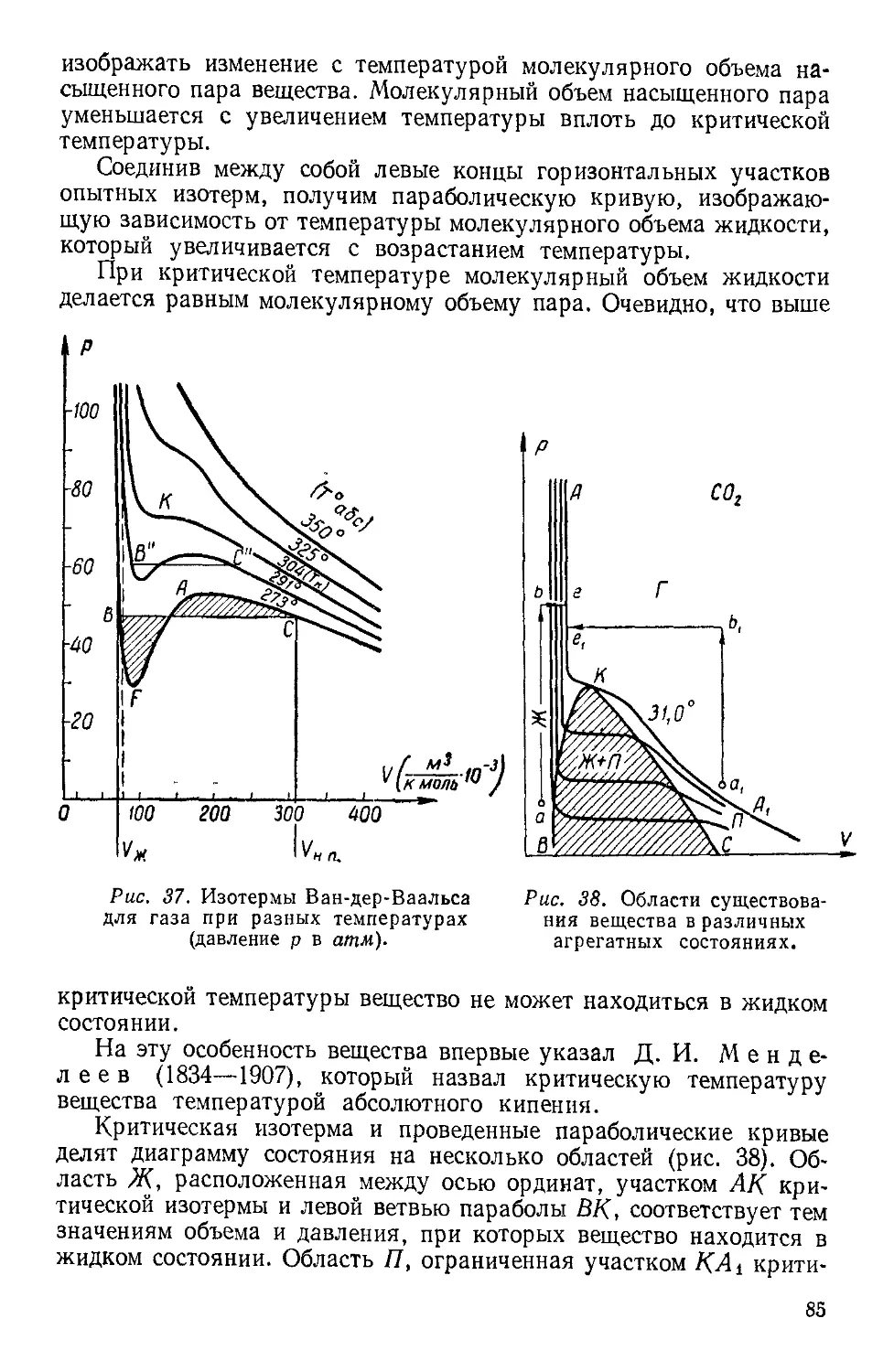








































































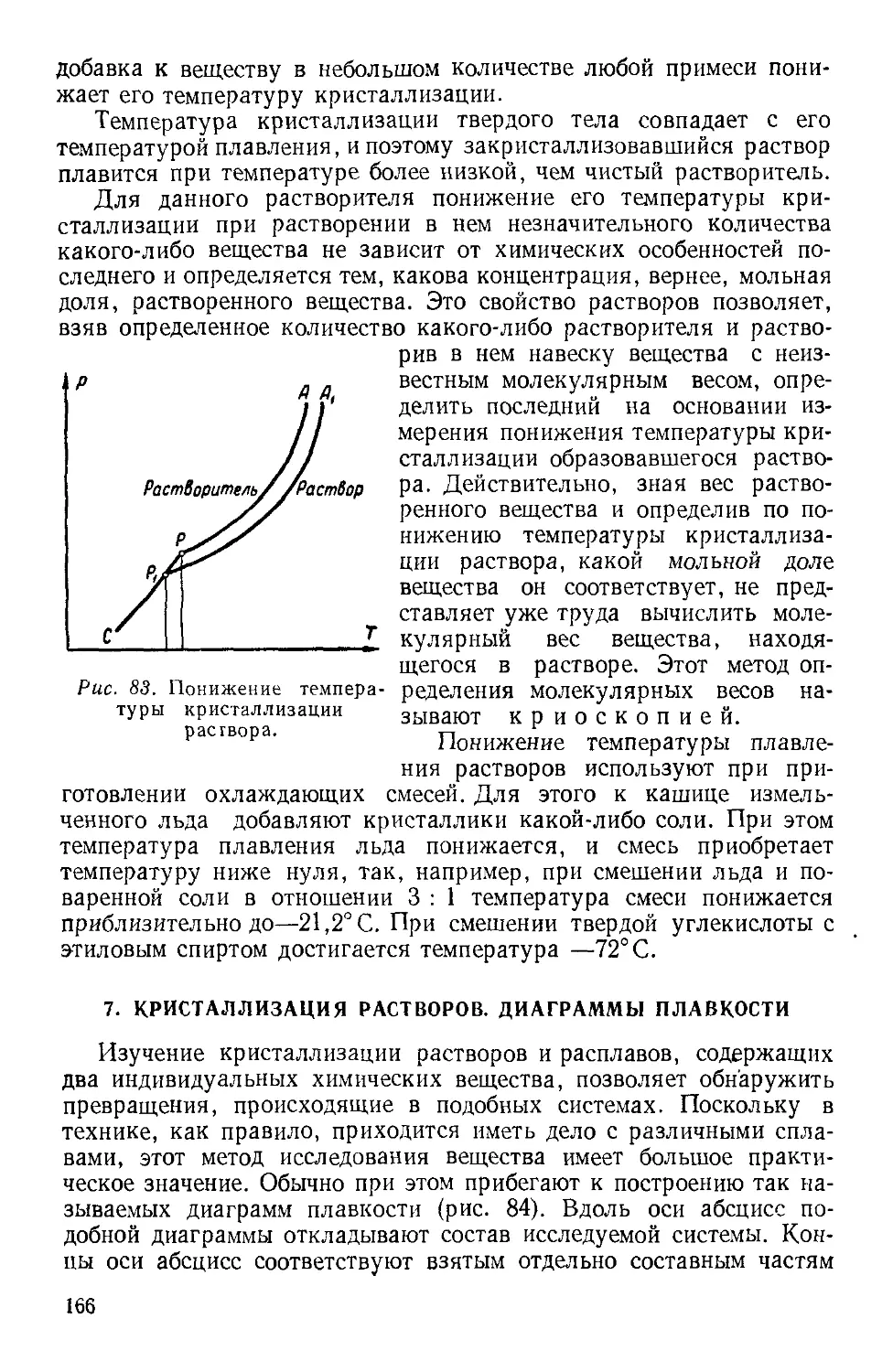































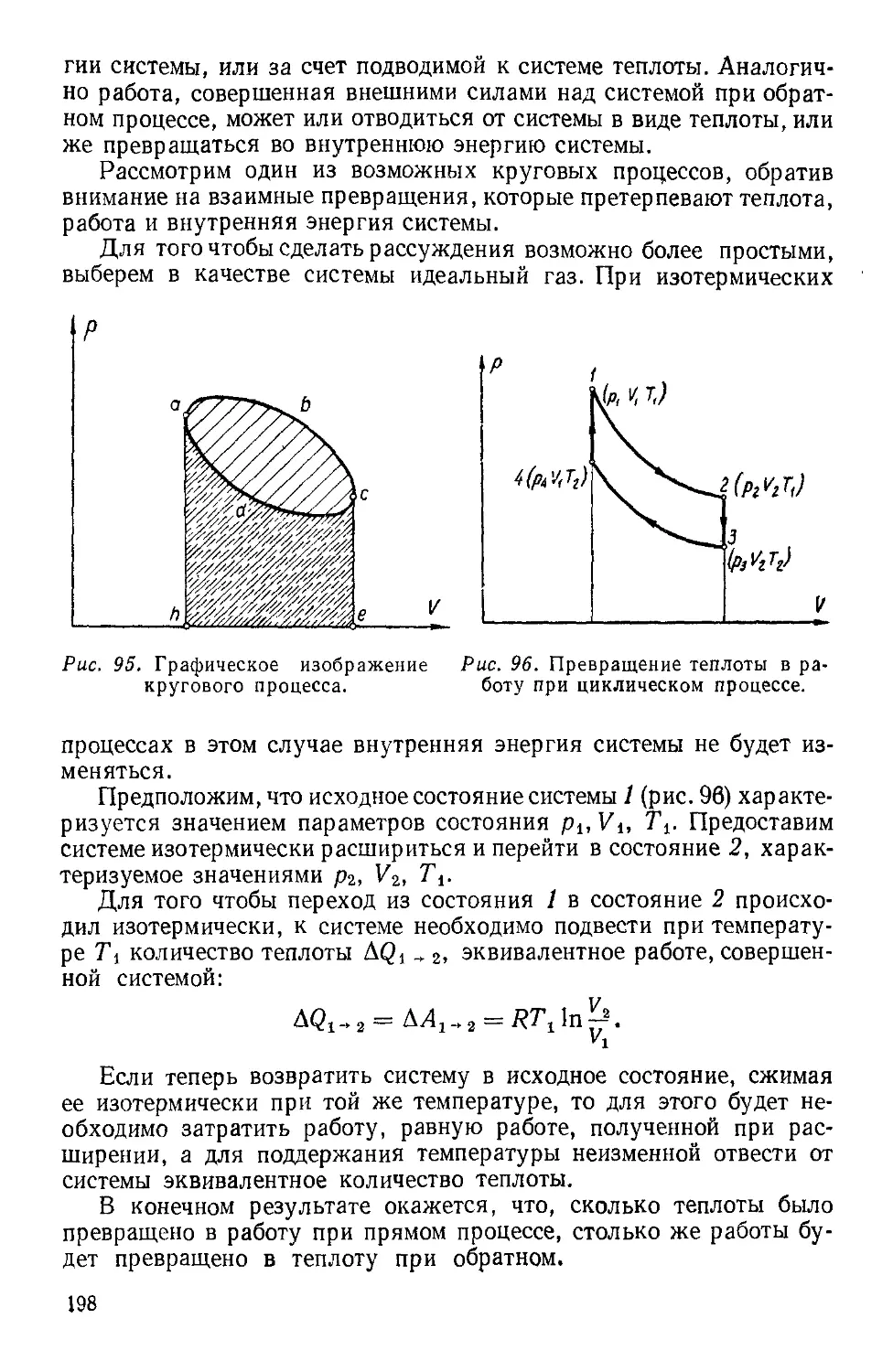





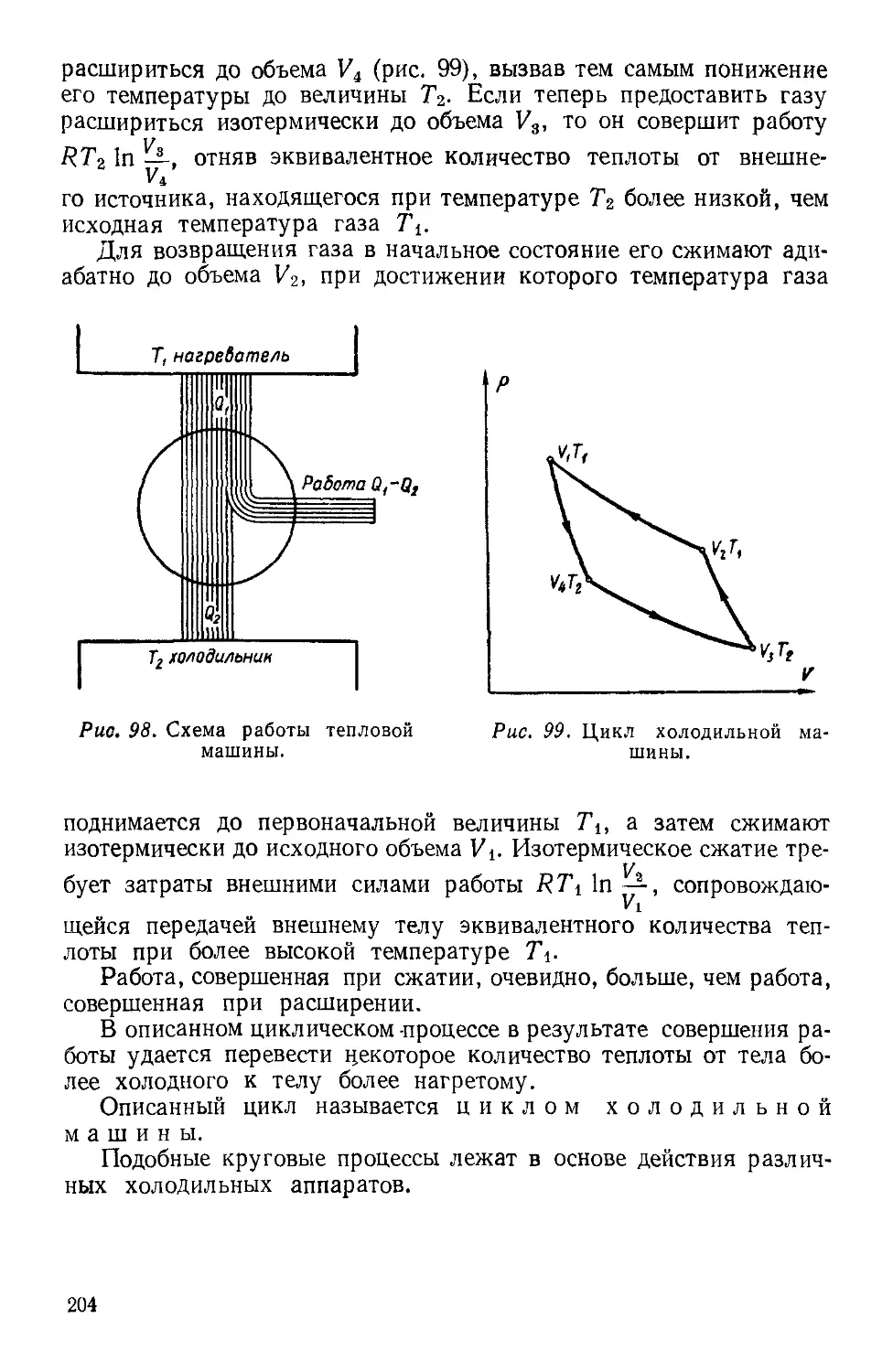
























Текст
Б. Б. КУДРЯВЦЕВ
КУРС
ФИЗИКИ
ТЕПЛОТА
И МОЛЕКУЛЯРНАЯ
ФИЗИКА
Допущено
Министерством
просвещения РСФСР
в качестве учебника
для педагогических
институтов
ИЗДАНИЕ 2-Е
ИЗДАТЕЛЬСТВО «ПРОСВЕЩЕНИЕ»
МОСКВА.1 965
ПРЕДИСЛОВ ИЕ
Во второе издание этой книги внесены лишь незначительные из-
изменения по сравнению с первым. Эти изменения коснулись вопро-
вопроса о применении ультроакустических методов при исследовании га-
газов и жидкостей, вопроса о влиянии дислокаций на механические
свойства тел и о значении надмолекулярных структур для понима-
понимания строения полимеров.
Кроме того, в книге использована Международная система еди-
единиц.
Системой единиц называют совокупность выбранных независимо
основных единиц и образованных на их основе, в соответствии с
физическими законами или зависимостями между величинами (в их
простейшей форме), производных единиц.
В 1960 г. XI Генеральная конференция по мерам и весам при-
приняла Международную систему единиц, которая введена с 1 января
1963 г. в Советском Союзе как предпочтительная. В научных ра-
работах, инженерных расчетах, учебных дисциплинах следует ис-
использовать Международную систему единиц.
Учитывая затруднения, которые могут возникнуть при чтении,
поскольку подавляющее большинство учебных и технических книг
написано с использованием старой системы единиц, ниже приво-
приводится краткая характеристика Международной системы единиц.
Международная система единиц сокращенно обозначается латин-
латинскими буквами S/ (Systeme international) и русскими СИ.
В применявшихся ранее системах единиц CGS и MkGS, для из-
измерения важнейшей для молекулярной физики величины — количе-
количества теплоты, применялась внесистемная единица — калория, для
установления связи которой с другими единицами вводились спе-
специальные переводные коэффициенты. Внесистемные единицы ис-
использовались и для измерения давления {атм кГ/см2, мм рт. cm).
В настоящее время внесистемные единицы могут использоваться
только в каких-либо исключительных случаях.
В основу системы СИ положены шесть основных и две допол-
дополнительные единицы измерений. С помощью этих единиц определя-
югся производные единицы СИ.
ОСНОВНЫЕ ЕДИНИЦЫ СИ
К основным единицам системы СИ (табл. I) относятся:
Метр — единица длины, равная 1650763,73 длины волны в
вакууме излучения, соответствующего переходу между уровнями
2р1а и Ыъ атома криптона 86.
Таблица I
Основные единицы СИ
Величина
Длина
Масса
Время
Сила тока
Термодинамическая тем-
температура
Сила света
Единица
измерения
метр
килограмм
секунда
ампер
градус
Кельвина
свеча
Сокращенные
русскими
буквами
М
кг
сек
а
°К
ев
обозначения
латинскими
буквами
т
kg
s
А
°К
ей
Килограмм — единица массы — представлен массой междуна-
международного прототипа килограмма.
Секунда — 1/31556925,9747 часть тропического года для 1900 г.
января 0 в 12 ч эфемеридного времени.
Ампер — сила неизменяющегося тока, который, проходя по
двум параллельным прямолинейным проводникам бесконечной дли-
длины и ничтожно малого кругового сечения, расположенным на рас-
расстоянии 1 м один от другого в вакууме, вызвал бы между этими про-
проводниками силу взаимодействия, равную 2 • 10~7 единиц силы
Международной системы на каждый метр длины.
Градус Кельвина — единица измерения температуры по тер-
термодинамической температурной шкале, в которой для температуры
тройной точки воды установлено значение 273,16°К (точно).
Свеча — единица силы света, значение которой принимается
таким, чтобы яркость полного излучателя при температуре затвер-
затвердевания платины была равна 60 ев на 1 см2.
ДОПОЛНИТЕЛЬНЫЕ ЕДИНИЦЫ СИ
К дополнительным единицам СИ (табл. II) относятся угловые
единицы — радиан и стерадиан.
Таблица II
Дополнительные единицы СИ
Величина
Плоский угол
Телесный угол
Единица
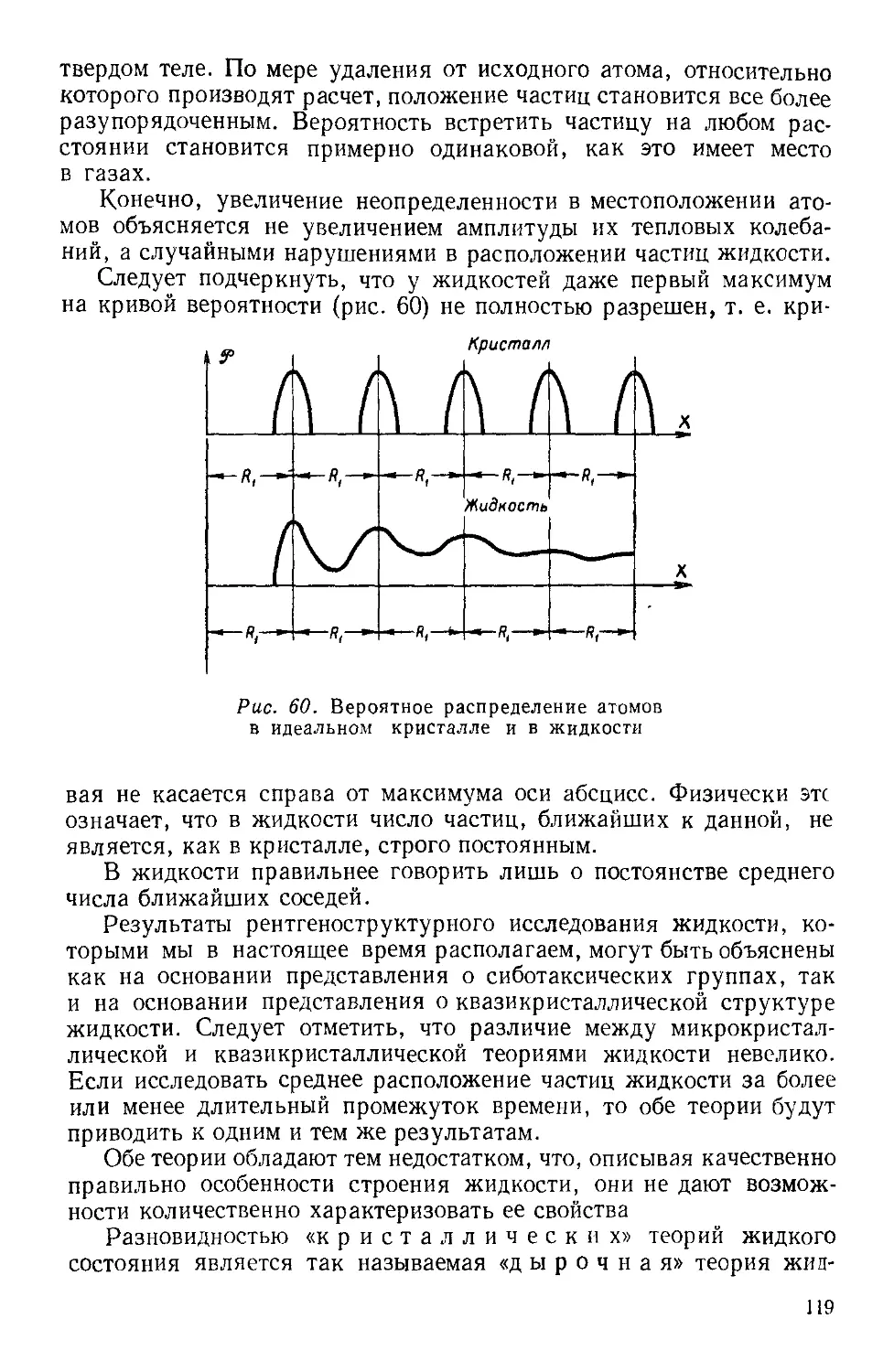
измерения
радиан
стерадиан
Сокращенные обозначения
русскими
буквами
рад
стер
латинскими
буквами
rad
sr
Радиан — угол между двумя радиусами круга, вырезающий
на его окружности дугу, длина которой равна радиусу.
Стерадиан —телесный угол, вершина которого расположена
в центре сферы и который вырезает на поверхности сферы площадь,
равную площади квадрата со стороной, равной радиусу сферы.
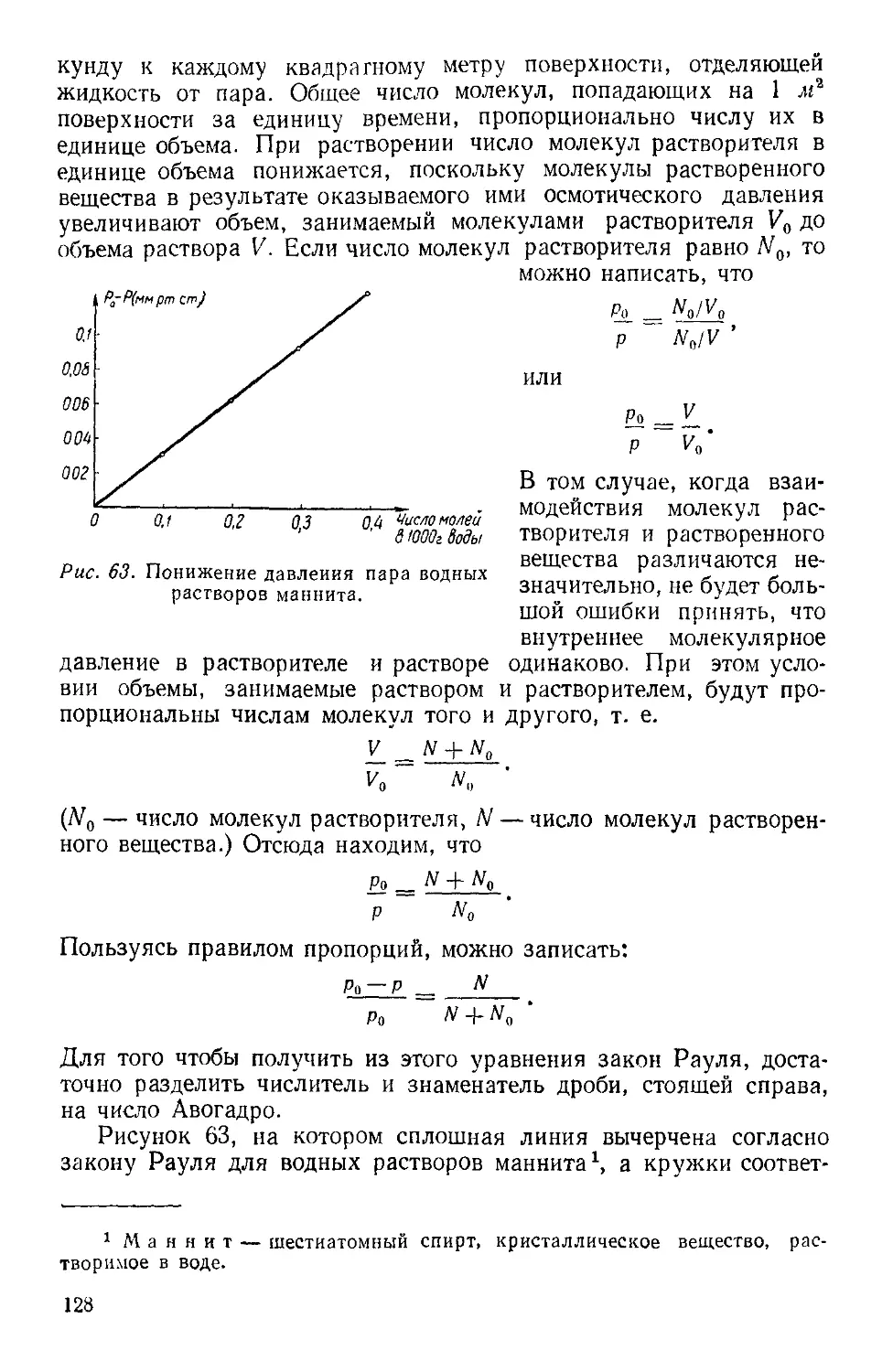
ПРОИЗВОДНЫЕ ЕДИНИЦЫ СИ
Производные единицы СИ можно разделить на следующие груп-
группы в зависимости от области измерений:
1. Единицы пространства и времени.
2. Механические единицы.
3. Электрические и магнитные единицы.
4. Тепловые единицы.
5. Акустические единицы.
6. Световые единицы и единицы энергетической фотометрии.
7. Единицы ионизирующих излучений.
Некоторые производные единицы в системе СИ
Таблица III
Величина
Сила
Работа
Мощность
Давление
Плотность
Единица измерения
1 Ньютон (и) — сила, сообщающая
массе 1 кг ускорение, равное 1 м/сек2
1 джоуль (дж) — работа силы в 1 н
па пути в 1 ж при условии, что сила
действует в направлении перемещения
1 ватт (em) = 1 дж/сек — мощность,
при которой за 1 сек совершается ра-
работа, равная 1 дж
1 н/м2 — равномерно распределенное
давление, при котором на 1 м2 действу-
действует нормально к поверхности сила в 1 н
1 кг/м3— плотность вещества, в 1 ж3
которого содержится масса, равная
1 кг
Переводной коэффициент
1 н = 105 дин
1 н = 0,102кГ
1 дж = 10' эрг
1 дж = 0,102 кГм
1 вт = 10' эрг/сек
1 em = 1,36- Ю-3 л.с.
1 н/м2 = 9,87 • Ю-6 атм
1 н/м2 = 7,50 • 10-3 мм
рт. ст.
1 кг/ж3 = 10~3 г/см3
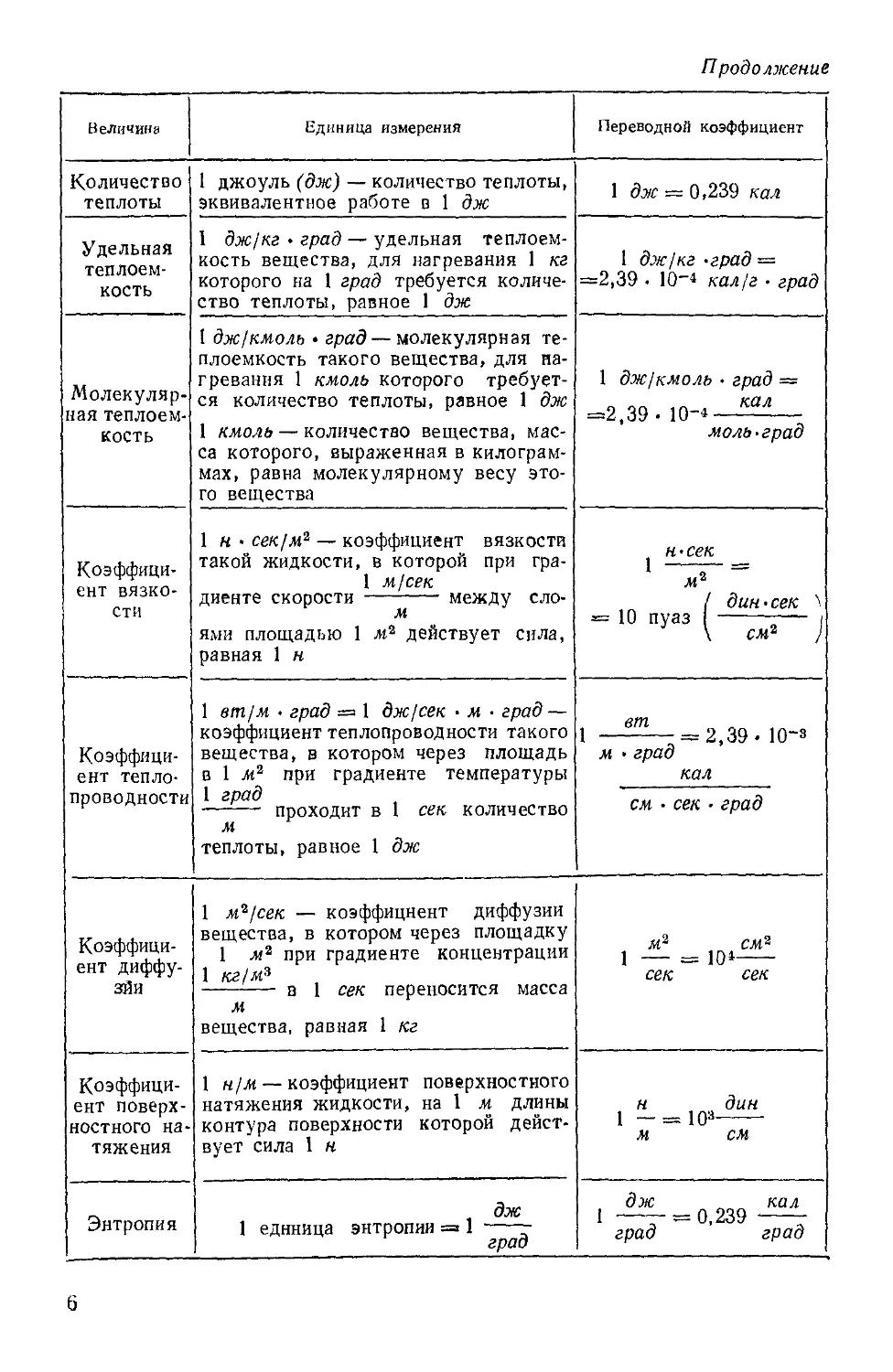
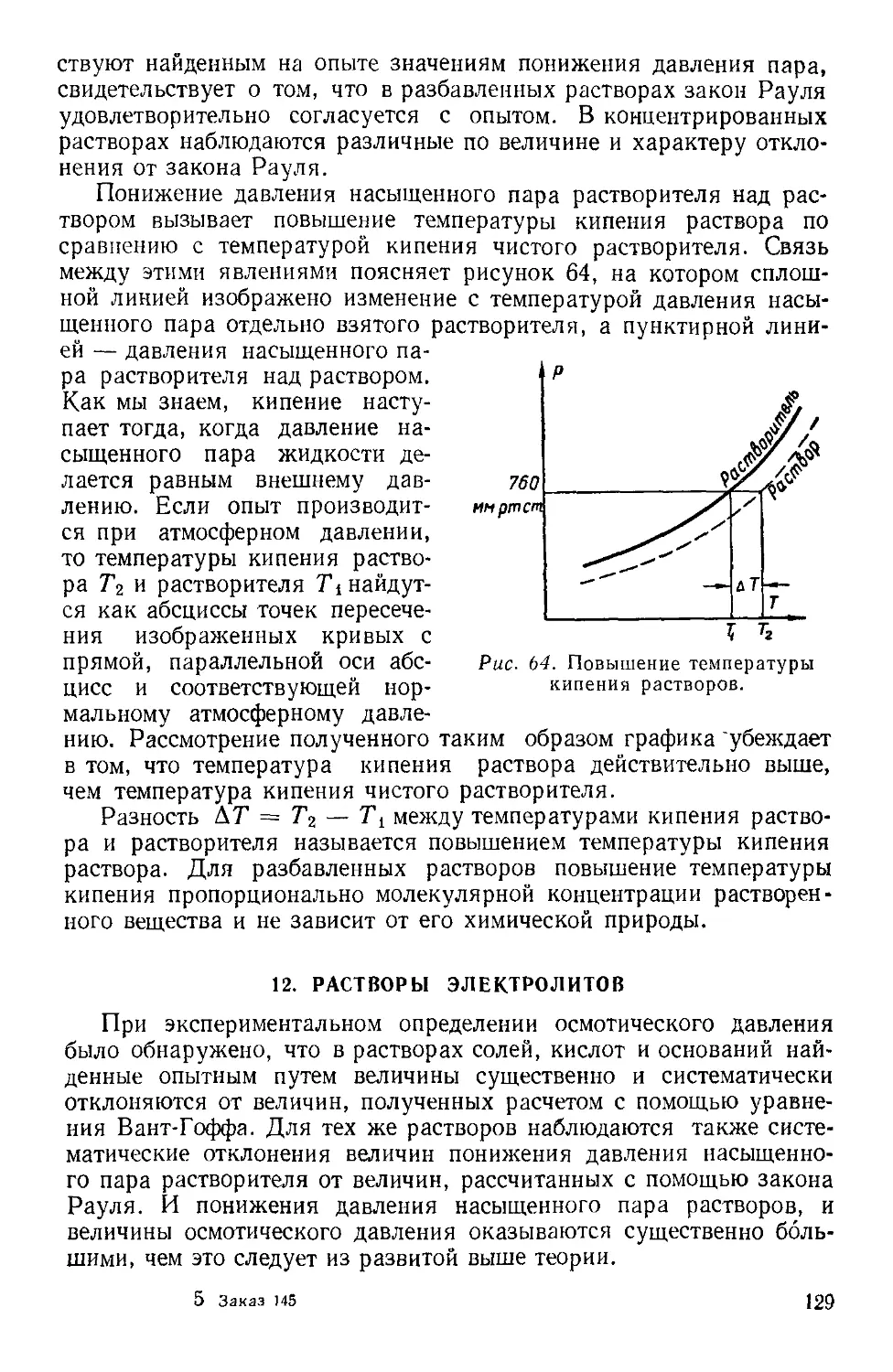
Продолжение
Величина
Количество
теплоты
Удельная
теплоем-
теплоемкость
Молекуляр-
Молекулярная теплоем-
теплоемкость
Коэффици-
Коэффициент вязко-
вязкости
Коэффици-
Коэффициент тепло-
теплопроводности
Коэффици-
Коэффициент диффу-
диффузии
Коэффици-
Коэффициент поверх-
поверхностного на-
натяжения
Энтропия
Единица измерения
1 джоуль (дж) — количество теплоты,
эквивалентное работе в 1 дж
1 дж/кг • град — удельная теплоем-
теплоемкость вещества, для нагревания 1 кг
которого на 1 град требуется количе-
количество теплоты, равное 1 дж
1 дж/кмоль • град — молекулярная те-
теплоемкость такого вещества, для на-
нагревания 1 кмоль которого требует-
требуется количество теплоты, равное 1 дж
1 кмоль — количество вещества, мас-
масса которого, выраженная в килограм-
килограммах, равна молекулярному весу это-
этого вещества
1 н • сек1мг — коэффициент вязкости
такой жидкости, в которой при гра-
1 м/сек
диенте скорости между ело-
м
ями площадью 1 ла действует сила,
равная 1 «
1 вт/м ¦ град = 1 дж/сек • м ¦ град —
коэффициент теплопроводности такого
вещества, в котором через площадь
в 1 м2 при градиенте температуры
1 град
проходит в 1 сек количество
м
теплоты, равное 1 дж
1 м2/сек — коэффициент диффузии
вещества, в котором через площадку
1 м2 при градиенте концентрации
1 кг/м3
в 1 сек переносится масса
м
вещества, равная 1 кг
1 н/м — коэффициент поверхностного
натяжения жидкости, на 1 л длины
контура поверхности которой дейст-
действует сила 1 к
дж
1 единица энтропии = 1
град
Переводной коэффициент
1 дж = 0,239 кал
1 дж/кг -град =
=2,39 • 10-4 кал/г . град
1 дж/кмоль • град —
=2,39-10- КШ
моль.град
Н'Сек
1 ж2 ~
/ дин • сек \
-1Опуаз( „,. ;
1 -^— = 2,39.10-3
м •град
кал
см • сек ¦ град
м? см2
1 — = Ю»
сек сек
iH -х&дин
м см
1 дЖ 0,239 К0Л
град град
В таблице III дается определение и указываются перевод-
переводные коэффициенты для важнейших производных единиц в системе
СИ, которые встречаются в молекулярной физике.
Изменение системы единиц привело к изменению числовых зна-
значений основных физических постоянных. Новые значения основных
констант, встречающихся в молекулярной физике, приведены ни-
ниже:
1. Число молекул в 1 киломоле NA = 6,02 • 1026 кмоль~1.
2. Объем 1 кмоля идеального газа при нормальных условиях
Vo = 22,41 мЧкмоль.
3. Число молекул в единице объема идеального газа при нор-
нормальных условиях п0 = 2,68 • 1025 м~3.
4. Универсальная газовая постоянная /? = 8,31 • №3дж/кмоль-
¦град.
5. Постоянная Больцмана k = 1,38 • 10~23 дж/град.
6. Постоянная Планка h = 6,625 • 1СГ34 дж ¦ сек.
7. Постоянная тяготения у == 6,67 • 10~и м3/кг • сек2.
I. ГАЗООБРАЗНОЕ СОСТОЯНИЕ ВЕЩЕСТВА
1. РАЗВИТИЕ АТОМНЫХ ПРЕДСТАВЛЕНИЙ В НАУКЕ
Представление об атомном строении вещества возникло в глу-
глубокой древности. Наиболее последовательно атомное учение антич-
античной древности было изложено греческим философом и естествоис-
естествоиспытателем Демокритом (V век до нашего летосчисления).
«Обыкновенно мы говорим, — писал он, — о сладком и горьком,
о теплом и холодном, о цвете и запахе, в действительности же су-
существуют атомы и пустое пространство». Различием атомов и осо-
особенностями их движения Демокрит объяснял многие свойства тел,
а также закономерности некоторых явлений, известных из обы-
обыденной жизни. В древности взгляды Демокрита были широко рас-
распространены.
С падением классической цивилизации положение сильно из-
изменилось. Получившая в средние века огромную власть христи-
христианская церковь не признавала материалистического по существу
учения греческих атомистов. Для борьбы с атомизмом привлека-
привлекались и светские власти.
В 1626 году высший суд королевской Франции специальным
декретом запретил распространение учения об атомах под страхом
смертной казни.
Возрождение атомизма связано с возникновением современного
естествознания, которое зарождается в XVII веке. Мысль об атом-
атомном строении вещества высказывает французский философ Пьер
Гассенди A592—1655).
Сторонником атомной теории был известный английский физик
Роберт Бойль A627—1691).
Один из творцов современной физики Исаак Ньютон
A643—1727) считал, что все тела состоят из «имеющих массу, креп-
крепких, непроницаемых, движущихся частичек», т. е. из атомов. Ра-
Работавший в Петербургской Академии наук знаменитый физик Д а-
ниил Бернулли A700—1782) использовал представление
об атомном строении газов для объяснения некоторых их свойств.
Необходимо, однако, отметить, что в XVII—XVIII веках атомные
воззрения не были общепризнанными. Такой крупный ученый, как
Рэне Декарт A596—1650), считал, что основным свойством
материи является ее способность неограниченно делиться, и пото-
потому отказывался признавать атомы. Материальных атомов не при-
признавал и Г. В. Лейбниц A646—1716).
Наиболее последовательно атомные представления в науке
XVIII века были развиты великим русским ученым М. В. Л о-
моносовым A711—1765). Ломоносов считал, что все тела со-
состоят из мельчайших материальных частиц, которые он называл
«элементами». «Элемент, — писал он, — есть часть тела, не
состоящая из каких-либо других меньших и отличающихся от него
тел». На современном языке элементу Ломоносова эквивалентен
атом. Наряду с элементами, как указывал Ломоносов, сущест-
существуют более крупные частицы — корпускулы. «Корпускула
есть собрание элементов, образующее одну малую массу». «Кор-
«Корпускулы однородны, если состоят из одинакового числа одних и тех
же элементов, соединенных одинаковым образом,..». «Корпускулы
разнородны, когда элементы их различны и соединены различным
образом или в различном числе. От этого зависит бесконечное раз-
разнообразие тел».
В наше время вместо слова корпускула говорят моле-
молекула.
Указанное Ломоносовым деление частиц вещества на атомы и
молекулы утвердилось в науке лишь много позднее.
Мельчайшие частицы вещества, согласно Ломоносову, находят-
находятся в движении. У различных тел, как утверждал он .частички «долж-
«должны различаться массою, фигурой, движением».
Особенностями строения атомов и характером их движения Ло-
Ломоносов объясняет многие свойства тел и, что особенно важно, пра-
правильно указывает природу теплоты.
Несмотря на успех, с которым было связано использование в
науке учения об атомах, Ломоносову не удалось сделать это учение
общепризнанным в физике.
Объясняется это тем, что физика в то время не достигла той сте-
степени развития, при которой в ней с необходимостью утверждаются
атомные представления.
Дальнейшие успехи атомной теории некоторое время связыва-
связываются скорее с развитием химии, нежели физики.
В начале XIX века английский химик Д. Дальтон A766—
—1844) на основании атомного учения объясняет наблюдаемые на
опыте особенности химических превращений.
В физике атомная теория прочно утверждается лишь к середи-
середине XIX столетия.
В результате работ ученых разных стран при помощи атомных
представлений удается объяснить многие свойства газов, закономер-
закономерности происходящих в них процессов и, что чрезвычайно важно,
утвердить в науке взгляд на теплоту как на особый вид молекуляр-
молекулярного движения.
Следует подчеркнуть.что победа учения о материальных ато-
атомах была достигнута лишь в результате ожесточенной борьбы. Фи-
Физики-идеалисты отказывались признавать, что вещество в конеч-
конечном счете состоит из материальных атомов. На протяжении всего
XIX века они утверждали, что атомное учение — это только более
или менее удобная гипотеза, а сами атомы — это «плод человечес-
человеческой фантазии»-. Долгое время борьба за атомизм являлась централь-
центральным участком борьбы между материалистическим и идеалистиче-
идеалистическим мировоззрениями в науке. В эту борьбу за прогресс в науке
внесли свой вклад передовые
ученые разных стран. Особенно
много было сделано в этот пе-
период для утверждения, атомного
учения английским физиком
К. Максвеллом A831—
1879), немецким ученым
Р.Клаузиусом A822—1888),
австрийцем Л. Больцманом
A844—1906), польским физиком
М. Смолуховским A872—
1917) и, наконец, французом
Ж- Перреном A870 —
1942). Именное работами Ж.Пер-
рена связано окончательное до-
доказательство реальности мате-
материальных атомов.
Для доказательства реальности атомов и молекул Перрен вос-
воспользовался явлением, которое было открыто в 1826 г. Б р о у-
н о м. Изучая под микроскопом движение соков в растительных
клетках, Броун обнаружил, что взвешенные в растительном соке
мельчайшие твердые частички находятся в непрерывном хаотиче-
хаотическом движении, названном впоследствии его именем. Броуновское
движение не является результатом неравномерного нагрева жидкости
или наличия в ней потоков, вызванных механическими сотрясениями.
Тщательные опыты убедили в том, что броуновское движение
не прекращается ни при каких обстоятельствах. Если, наблюдая в
микроскопе броуновское движение, фиксировать через равные не-
небольшие промежутки времени положение одной из взвешенных в
жидкости частиц, то мы получим характерную ломаную линию, по-
подобную изображенной на рисунке 1. Эта линия не является истинной
траекторией частицы, которая в действительности много сложнее.
Броуновское движение — одна из причин причудливого движения
взвешенных в воздухе пылинок. Движение это хорошо заметно в
солнечном луче, падающем через щель в ставне в затемненную ком-
комнату. Еще знаменитый римский поэт Лукреций Кар
10
Рис. 1. Положения частиц, соверша-
совершающих броуновское движение, отме-
отмечаемые через равные промежутки
времени.
(ок. 99 — 55 гг. до нашего летосчисления) указывал, что это при-
причудливое движение вызвано ударами атомов вещества.
Если тело, находящееся в жидкости, достаточно велико, то на
единицу его поверхности за некоторый небольшой промежуток вре-
времени приходится в среднем одно и то же количество ударов моле-
молекул. Это приводит к тому, что, несмотря на беспорядочность движе-
движения молекул, их удары взаимно компенсируются и тело испыты-
испытывает со всех сторон равномерное давление.
При уменьшении размеров тела подобная компенсация нару-
нарушается: в отдельные мгновения в частицу небольших размеров мо-
может ударяться большее количество молекул, движущихся в каком-
то направлении, чем количество молекул, движущихся в противо-
противоположном направлении. Может случиться и так, что эти количест-
количества молекул будут примерно одинаковыми, но будут существенно
разниться их скорости. В обоих случаях переданные молекулами
импульсы оказываются нескомпенсированными и частица устрем-
устремляется в направлении результирующего вектора количества дви-
движения. В следующее мгновение нескомпенсированными окажутся
удары молекул, имеющих иное направление, и соответственно
изменится направление движения частицы.
Таким образом, под влиянием беспорядочных ударов молекул
взвешенные в жидкости частицы будут двигаться по очень сложной
траектории, характерной для броуновского движения.
Перрен приготовил взвесь мельчайших зернышек гуммигута и
мастики в воде и изучил свойства полученных эмульсий. Оказалось,
что даже sa длительное время не все частицы эмульсии оседают на
дно сосуда, многие остаются взвешенными в жидкости, распреде-
распределяясь так, что в нижних слоях жидкости в каждом кубическом сан-
сантиметре находится большее количество взвешенных частиц, чем в
слоях, расположенных выше. Подсчитывая с помощью микроскопа
число частиц в поле зрения прибора на разной высоте от дна сосуда
и находя средние значения полученных величин, Перрен убедитель-
убедительно доказал, что распределение взвешенных частиц по высоте под-
подчиняется той же закономерности, что и распределение частиц воз-
воздуха в земной атмосфере. Далее, наблюдение движений частиц убе-
убедило его в том, что броуновское движение — это «молекулярное
движение так же точно, как инфракрасный свет, есть в такой же ме-
мере свет, как и свет ультрафиолетовый». Многочисленными расчета-
расчетами, основанными на наблюдении особенностей движения частиц
эмульсий, Перрен подтвердил реальность существования молекул и
атомов и с полным основанием написал в заключение своего ис-
исследования, что «отныне уже будет трудно защищать разумны-
разумными аргументами враждебное отношение к молекулярным гипоте-
гипотезам».
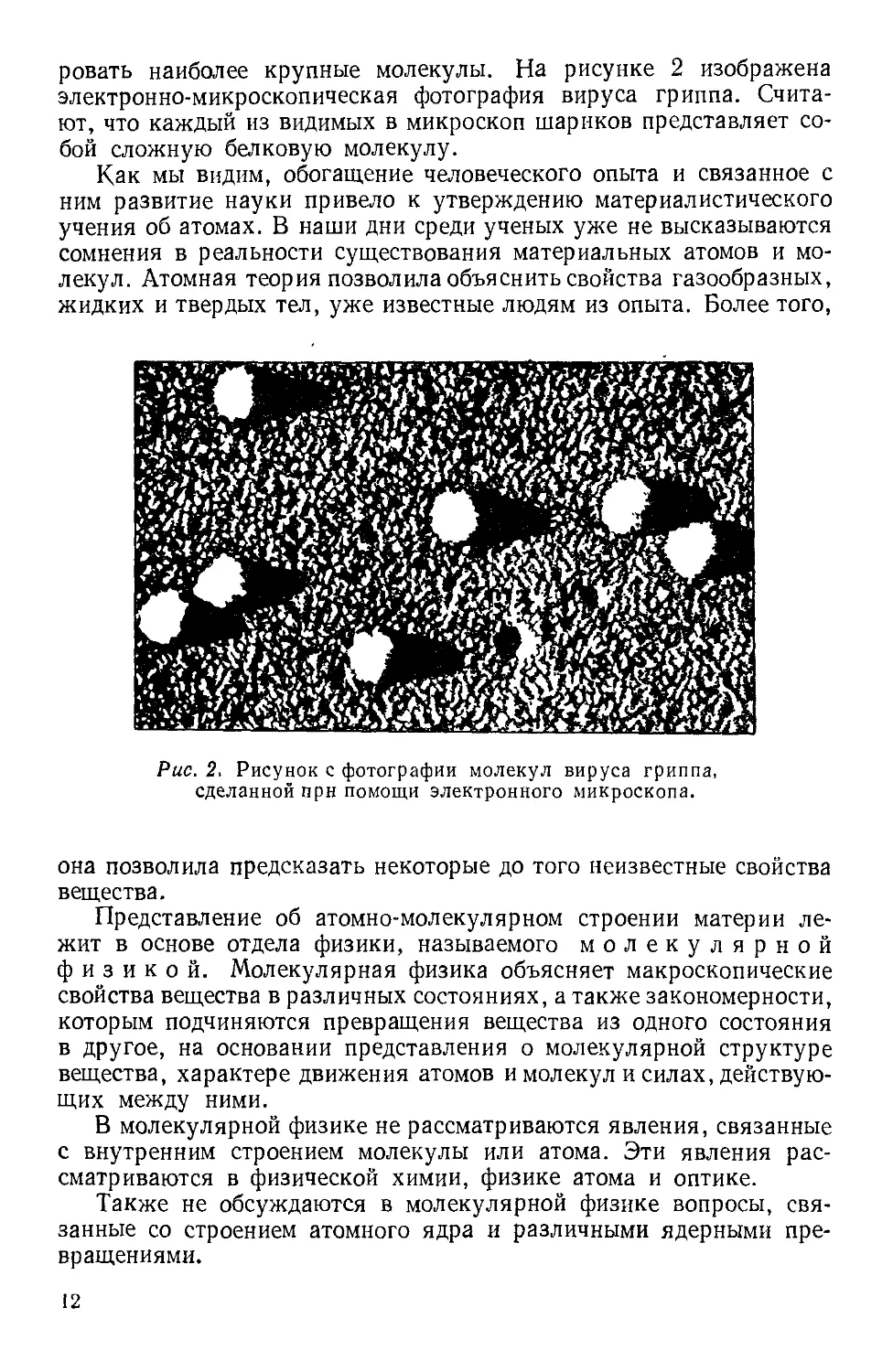
В настоящее время существуют способы прямого наблюдения
молекул. Подобную возможность открыло изобретение электрон-
электронного микроскопа. С помощью этого прибора удалось сфотографи-
п
ровать наиболее крупные молекулы. На рисунке 2 изображена
электронно-микроскопическая фотография вируса гриппа. Счита-
Считают, что каждый из видимых в микроскоп шариков представляет со-
собой сложную белковую молекулу.
Как мы видим, обогащение человеческого опыта и связанное с
ним развитие науки привело к утверждению материалистического
учения об атомах. В наши дни среди ученых уже не высказываются
сомнения в реальности существования материальных атомов и мо-
молекул. Атомная теория позволила объяснить свойства газообразных,
жидких и твердых тел, уже известные людям из опыта. Более того,
Рис. 2. Рисунок с фотографии молекул вируса гриппа,
сделанной при помощи электронного микроскопа.
она позволила предсказать некоторые до того неизвестные свойства
вещества.
Представление об атомно-молекулярном строении материи ле-
лежит в основе отдела физики, называемого молекулярной
физикой. Молекулярная физика объясняет макроскопические
свойства вещества в различных состояниях, а также закономерности,
которым подчиняются превращения вещества из одного состояния
в другое, на основании представления о молекулярной структуре
вещества, характере движения атомов и молекул и силах, действую-
действующих между ними.
В молекулярной физике не рассматриваются явления, связанные
с внутренним строением молекулы или атома. Эти явления рас-
рассматриваются в физической химии, физике атома и оптике.
Также не обсуждаются в молекулярной физике вопросы, свя-
связанные со строением атомного ядра и различными ядерными пре-
превращениями.
12
2. ПОНЯТИЕ О СОСТОЯНИИ ВЕЩЕСТВА
Повседневный опыт учит нас тому, что одно и то же вещество в
зависимости от его состояния может обладать различными свойст-
свойствами. Так, например, физические свойства сильно сжатого газа от-
отличаются от свойств того же газа, взятого при нормальном давле-
давлении, так же как свойства газа, нагретого до высокой температуры,
отличаются от свойств того же газа, охлажденного до низкой тем-
температуры.
Учитывая это обстоятельство, состояние вещества характеризуют
специальными физическими величинами, называемыми парамет-
параметрами состояния. Наиболее часто в качестве параметров состоя-
состояния выбирают три величины, а именно: объем V, занимаемый не-
некоторой массой вещества, температуру t, которую вещество имеет,
и давление р, которое оно оказывает. Если изменяется один из
параметров состояния, например объем, или давление, или темпе-
температура, то изменяется и состояние вещества. Таким образом, со-
состояние вещества определяется совокупностью значений
параметров состояния.
Опыт убеждает в том, что параметры состояния: объем, давле-
давление и температура вещества — не являются независимыми друг от
друга, но связаны между собой определенной функциональной за-
зависимостью
V,t)^O A)
Другими словами, это означает, что для некоторой массы веще-
вещества заданным значениям каких-либо двух параметров состояния
соответствует вполне определенное значение третьего параметра.
Так, например, если один моль1 вещества занимает некоторый объем,
то при заданной температуре его состояние будет характеризовать-
характеризоваться совершенно определенным значением давления. Написанное вы-
выше уравнение называют уравнением состояния. Наи-
Наиболее простой вид имеет уравнение состояния газообразных ве-
веществ.
3. ЗАКОН БОЙЛЯ — МАРИОТТА
Некоторые простые закономерности, связывающие между собой
параметры состояния газообразного вещества, были эмпирически
открыты сравнительно давно. В середине XVII века англичанин
Р. Б о й л ь и француз Э. Мариотт A620—1684) независимо
друг от друга открыли закон, которому подчиняется изменение дав-
давления газа р при изменении объема V, если при этом температура t
остается постоянной. Согласно закону, носящему их имя, при
1 Молем, или грамм-молекулой, вещества называют число граммов ве-
вещества, равное его молекулярному весу, аналогично киломолем, или кило-
килограмм-молекулой вещества называют число килограммов вещества, равное его
молекулярному весу.
13
постоянной температуре для данной массы, газа
произведение объема, занимаемого газом, на его дав-
давление является постоянной величиной, т. е.
рУ = Const. B)
Графически в системе координат р, V эта зависимость изобра-
изображается равнобочными гиперболами (рис. 3).
Поскольку в этом случае изменения состояния происходят при
постоянной температуре, кривые, изображающие графически закон
Бойля—Мариотта, называют изотермами. Следует заметить,
что вид изотерм зависит от выбранной системы координат; так, на-
Т,<Т2<Т3
Рис. 3. Зависимость дав-
давления газа от занимае-
занимаемого им объема при по-
постоянной температуре в
системе координат р, V.
T,<Tt<T}
Рис. 4. Зависимость дав-
давления газа от объема в
1
системе координат р, —.
пример, если условиться откладывать вдоль оси ординат давление
газа, а вдоль оси абсцисс — величину, обратную объему,— , то изо-
изотермы Бойля—Мариотта будут изображаться прямыми линиями
(рис. 4), а не гиперболами.
4. ЗАКОН ГЕЙ-ЛЮССАКА
Зависимость объема газа от его температуры в том случае, если
газ нагревается или охлаждается при постоянном давлении, была
найдена эмпирически французским физиком Гей-Люссаком.
A778-1850).
Согласно закону Гей-Люссака при изменении температуры лю-
любого газа на 1° стоградусной шкалы при постоянном давлении объем
газа V изменяется «<х — часть объема Vo, занимаемого им при нуле
градусов. В более общей форме можно сказать, что при постоянном
14
давлении существует линейная зависимость между объемом газа и
его температурой /, т. е.
У = У0A+а0. C)
Величину а называют термическим коэффициен-
коэффициентом объемного расширения газа. Согласно ска-
сказанному выше а для всех газов равно —, или 0,00367.
27 о
Кривые, изображающие графически изменение объема некоторой
массы газа с температурой при неизменном давлении, называют
изобарами.
В системе координат t, V (рис. 5) изобары Гей-Люссака — пря-
прямые линии, наклоненные к оси температур под таким углом, что при
продолжении их в область отрицательных температур они для всех
-гЪ'~ о f
Рис. 5. Зависимость объема газа
от температуры при постоянном
давлении в системе координат
U V.
-273°
0
t"
Рис. 6. Зависимость давления газа
от температуры при постоянном объ-
объеме в системе координат t, p.
газов пересекают ось абсцисс в одной точке, расположенной на 273°
ниже нуля обычной стоградусной шкалы температур.
Аналогичная зависимость была открыта при измерении давления
газа р в том случае, когда изменяется его температура, а объем оста-
остается постоянным. Как оказалось, для всех газов при повышении тем-
температуры на Г стоградусной шкалы давление р возрастает на —
L> I О
часть того значения Ра, которое оно имеет при нуле градусов, т. е.
Р = РоA+РО- D)
Величину р называют термическим коэффициен-
коэффициентом давления. Для всех газов р* = —.
Графически зависимость давления газа от температуры при не-
неизменном объеме выражается в системе координат р, t прямыми
линиями (рис. 6), называемыми изохорами. В выбранной
системе координат изохоры различных газов при продолжении их в
область отрицательных температур, так же как и изобары, пересе-
пересекают ось абсцисс в одной точке, расположенной на 273° ниже нуля
стоградусной шкалы.
15
Анализ приведенных выше эмпирических закономерностей пока-
показывает, что их можно формулировать много проще, если, не изменяя
величину градуса стоградусной температурной шкалы, проводить
отсчет температур не от обычного нуля градусов стоградусного тер-
термометра, а от точки, расположенной на 273° ниже. Очевидно, что
температура Т, измеренная по этой новой шкале, будет на 273° боль-
больше обычной температуры t, т. е.
7 = 273° + *, ,,
t = T — 273°. К}
Подставляя в уравнения C) и D) вместо t величину (Г — 273)°
и вместо ос и р" их значение —, получим:
273 /яч
т (о)
Р = А>273'
Нуль новой температурной шкалы, расположенный на 273° ни-
ниже нуля обычного стоградусного термометра, называется абсо-
абсолютным нулем температуры, а отсчитанная по новой шкале
температура Т — абсолютной температурой1.
Замечая, что Уои р0 обозначают объем и давление газа при нуле
градусов, а также, что по абсолютной шкале нулю стоградусной
шкалы будет соответствовать температура То = 273°, можно пере-
переписать уравнения F) в следующем виде:
v = vo^ и Р = Ро^-
1 о 'о
Таким образом, при неизменном давлении для данной массы га-
газа справедливо соотношение:
I = Ул = Const, G)
* ' о
а при неизменном объеме — соотношение:
? = ??- Const. (8)
5. ОБЪЕДИНЕННЫЙ ГАЗОВЫЙ ЗАКОН
На практике часто приходится иметь дело с такими изменениями
состояния газа, при которых одновременно изменяются его давление,
объем и температура. Взаимосвязь параметров состояния в этом
случае может быть получена на основании приведенных выше эмпи-
1 Физический смысл величины, называемой абсолютной температурой,
будет подробно рассмотрен ниже.
16
рических закономерностей, справедливых при условии, что один
из параметров состояния искусственно поддерживается постоян-
постоянным.
Предположим, что имеется какое-то количество газа т в неко-
некотором исходном состоянии, характеризуемом следующими значе-
значениями параметров состояния: объем газа Vo, давление р0 и темпе-
температура То. Предположим далее, что состояние газа изменяется, и
конечное состояние характеризуется теперь уже иными значениями
параметров состояния, а именно: Vu plt 7\. Для того чтобы найти
связь между параметрами состояния, допустим, что переход газа
из исходного состояния в конечное происходит в два этапа: перво-
первоначально изменяется только давление газа от значения р0 до значе-
значения рг, а температура остается неизменной, равной То. В результа-
результате указанного изменения давления объем газа изменяется от значе-
значения Vo до значения VJ, которое, поскольку процесс происходит при
постоянной температуре, можно найти, воспользовавшись законом
Бойля—Мариотта:
Р<Уо = РУ\. (9)
В течение второго этапа будем поддерживать неизменным дав-
давление газа plt а температуру заставим изменяться от значения То
до значения Тх. В конце второго этапа давление газа будет рг, тем-
температура 7\, а объем его по необходимости сделается равным Vv
Поскольку второй этап протекает при постоянном давлении, для
определения конечного объема можно воспользоваться законом
Гей-Люссака (уравнение 7):
То
A0)
Подставляя в последнее уравнение значение объема V'x из урав-
уравнения (9), получим:
' oPl * 1 /1 1 \
рЛ = л^ A1)
То Т1
Очевидно, что написанная закономерность будет справедлива для
любого состояния данного количества газа, и поэтому найденному
уравнению можно придать более простую форму:
у = Const, A2)
Р V
где Const = -^~- — некоторая постоянная для данной массы газа
величина.
Таким образом, мы пришли к выводу, что для данной мас-
массы газа произведение давления на объем, занимаемый
17
газом (pV), деленное на абсолютную температуру газа
(Т), остается постоянной величиной при любых изме-
изменениях состояния.
Уравнение A2) выражает связь между параметрами состояния
газообразного вещества и является поэтому уравнением состояния
для газов, подчиняющихся законам Бойля — Мариотта и Гей-Люс-
сака. Технические расчеты часто проводят применительно к 1 кг ве-
вещества. В этом случае постоянную величину Е±Аг стоящую в пра-
вой части уравнения A2), обозначают буквой В и называют удель-
удельной газовой постоянной, а само уравнение записы-
записывают в следующей форме:
pV = ВТ. A3)
Численное значение удельной газовой постоянной зависит от
того, в каких единицах выражаются давление и объем газа. Значе-
Значения В для некоторых веществ приведены в таблице 1.
Таблица 1
Величины удельных газовых постоянных для некоторых
веществ
Вещество
Воздух . . ,
Водород . .
Кислород .
Азот . . .
Углекислота
Метан ...
Пары воды .
Молекулярный
вес
291
2
32
28
44
16
18
Удельная газовая
постоянная
дж
град • кг
2,87 ,
41,5
2,60
2,97
1,89
5,19
4,62
10*
Если условиться относить полученное уравнение не к 1 кг ве-
вещества, а к одной килограмм-молекуле, то его можно записать в еще
более общем виде.
В килограмм-молекуле любого вещества содержится одно и то
же число молекул NА, называемое числом Авогадро и равное NA =
= 6,02 ¦ 1028. Согласно закону, открытому А. Авогадро A776—
1856) при одинаковых температуре и давлении в равных
объемах всех газов содержатся равное число молекул.
Для воздуха приведено значение среднего молекулярного веса.
18
Поэтому при нормальном атмосферном давлении, равном 1,01 X
Х105н/0и\ и температуре 0°С килограмм-молекула любого газа
будет занимать один и тот же объем Vo, равный 22,41 м'л. Таким
образом, очевидно, что величина газовой постоянной
РоУр
То
,рассчи-
,рассчитанная для одной килограмм-молекулы вещества, будет иметь
одно и то же значение для любого газа. Определенную таким обра-
образом газовую постоянную называют универсальной газо-
газовой постояннойи обозначают буквой R.
1,01 • 10» -22,4
273
= 8,ЗЫ03
дж
кмоль • град
Численные значения универсальной газовой постоянной зависят
от единиц, в которых выражены объем и давление газа.
Помимо приведенного выше значения R в Международной си-
системе единиц, часто употребляют значения, приведенные в табли-
таблице 2.
Таблица 2
Численное значение универсальной газовой постоянной R
при выражении давления и объема в различных единицах
Единицы, в кото-
которых измерено
давление
дин
см?
Л1а
am
мм рт. ст.
Единицы, в ко-
которых измерен
объем
см*
Численное значение
8,315. 107
0,848
0,08206
6,237 . 10»
эрг
град • моль
кГм
град ¦ моль
am ¦ л
град • моль
мм рт. ст. см3
град • моль
Универсальная и удельная газовые постоянные связаны между
собой простой зависимостью. Если молекулярный вес вещества М,
а удельная газовая постоянная В, то универсальная газовая по-
постоянная находится из уравнения
R = MB. A4)
Уравнение, объединяющее законы Бойля —Мариотта и Гей-Люс-
сака, было получено впервые французским инженером Клапей-
Клапейроном A799—1864) и носит его имя.
1Э
Обычно уравнение Клапейрона1 записывают для одного кило-
моля вещества в следующей форме:
pV = RT. A5)
Если расчет производится для т килограмм вещества, имеющего
молекулярный вес М, то правую часть уравнения A5) следует умно-
умножить на число киломолей газа, равное —•, и, таким образом, оно при-
м
мет вид
A6)
6. ПОНЯТИЕ ОБ ИДЕАЛЬНОМ ГАЗЕ
При знакомстве со сформулированными выше эмпирическими
газовыми законами естественно возникает вопрос о том, насколько
точно они соответствуют нашим современным знаниям свойств га-
газов. Подобное сравнение теории с опытом более наглядно в тех
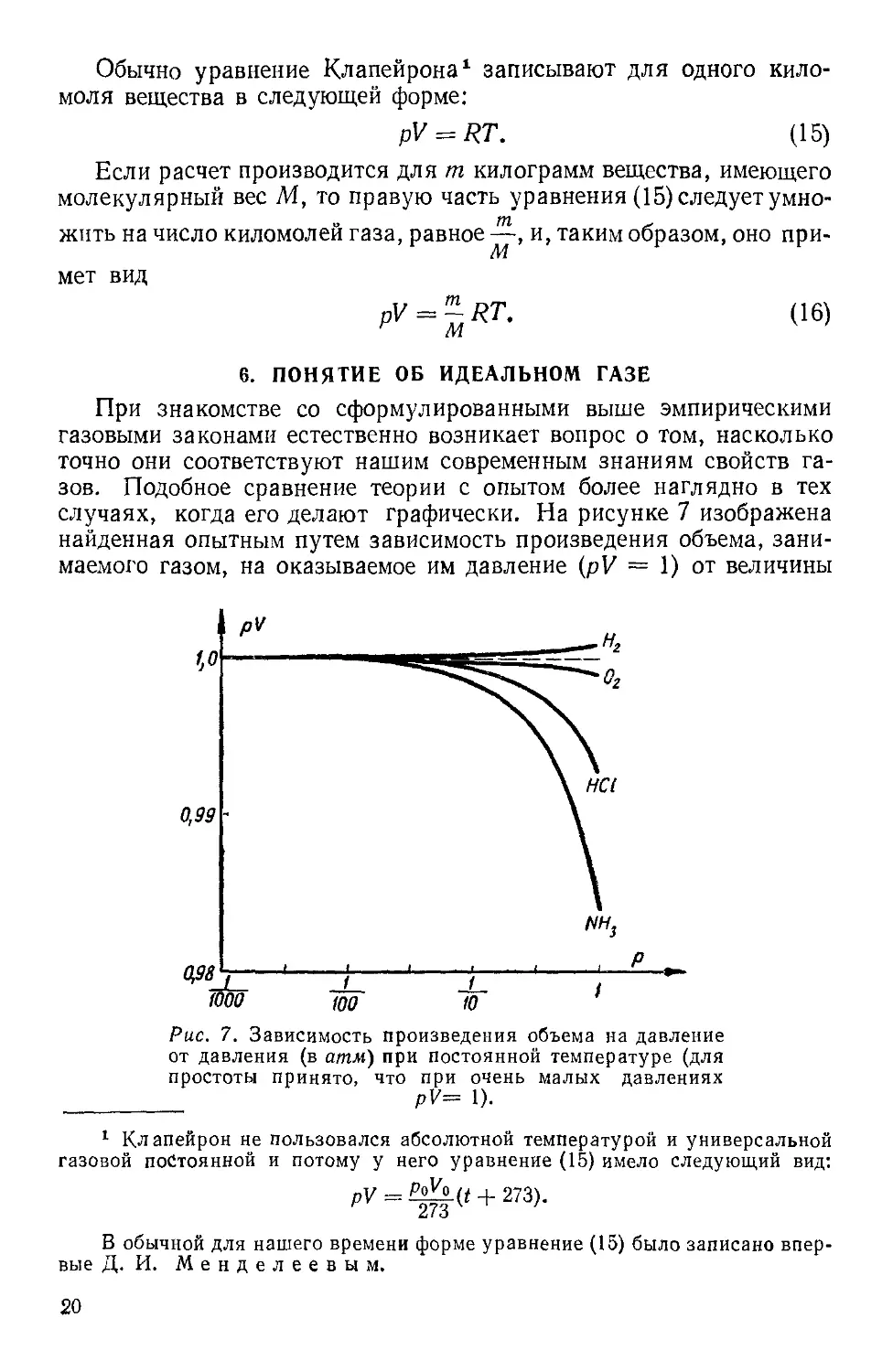
случаях, когда его делают графически. На рисунке 7 изображена
найденная опытным путем зависимость произведения объема, зани-
занимаемого газом, на оказываемое им давление (pV = 1) от величины
W i-
1000
Рис. 7. Зависимость произведения объема на давление
от давления (в атм) при постоянной температуре (для
простоты принято, что при очень малых давлениях
pV= 1).
1 Клапейрон не пользовался абсолютной температурой и универсальной
газовой постоянной и потому у него уравнение A5) имело следующий вид:
«т/ Ром) а 1 t}irx\
РУ--27Т(/ + 273)-
В обычной для нашего времени форме уравнение A5) было записано впер-
впервые Д. И. Менделеевым.
20
давления р, если изменения последнего происходят при постоянной
температуре1. Согласно закону Бойля — Мариотта в этом случае
произведение pV должно было бы оставаться постоянным, в то вре-
время как на опыте оно изменяется по-разному для разных газов.
Таблица 3
Значения термического коэффициента объемного
расширения а и термического коэффициента давления J3
для воздуха при давлении 1,3 • 105 я/д2 и разных температурах
а. 10е
р. ю«
Интервал температуры
0-50°
3676
3675
0—100°
3674
3675
0—150°
3673
3674
0—200°
3672
3674
В то же время весьма важно отметить, что для всех газов
при понижении давления величина pV стремится к од-
одному и тому же постоянному значению из этого следует, что
закон Бойля — Мариотта будет правильно описывать их поведение,
если только газы взять при достаточно малых давлениях.
]
0,00375
0,00370
(W036610
0,00365
а
С1г
у/
_ ¦
—————
—-нг
. р
0,5 1,0 1,5
Рис. 8. Зависимость коэффициента термического
расширения газа от давления (в атм).
Сходные отклонения наблюдаются и при опытной проверке за-
закона Гей-Люссака, как это явствует из данных, приведенных в таб-
таблице 3; термические коэффициенты объемного расширения а и тер-
термические коэффициенты давления р* у различных газов имеют раз-
разные, хотя и близкие, значения.
Если сопоставить термические коэффициенты объемного рас-
расширения а, измеренные при разных давлениях (рис. 8), то выявится,
1 Для того чтобы охватить больший интервал давлений на графике,
вдоль оси абсцисс давление отложено в логарифмическом масштабе.
21
что а изменяется при изменении давления и что это изменение раз-
различно для различных газов.
Большое значение при этом имеет то обстоятельство, что при
понижении давления термические коэффициенты объ-
объемного расширения различных газов стремятся к
одинаковой для всех газов величине, близкой к той, кото-
которая фигурирует в законе Гей-Люссака.
Весьма наглядное сопоставление эмпирических газовых законов
с опытом можно осуществить, определив экспериментально для
одной килограмм-молекулы какого-либо газа значения произве-
произведения pV при постоянной температуре и разных давлениях, как это
делалось раньше, и выполнив несколько серий подобных измерений
при разных абсолютных температурах Т. Это позволит изобразить
dV
графически зависимость отношения—от давления. Если бы пове-
поведение газа в точности следовало уравнению Клапейрона, то вычис-
вычисленное на основе эксперимента отношение — должно было бы иметь
одно и то же значение при всех температурах и давлениях, равное
по величине универсальной газовой постоянной R. На рисунке 9
изображены результаты подобного опыта, в котором в качестве из-
изучаемого газа выбрана углекислота. Рассмотрение рисунка убеждает
в том, что уравнение Клапейрона при относительно больших да-
давлениях "неправильно описывает поведение углекислоты. Однако
S3! 4,
60°С
, Р
200
400
600 800
pV
Рио. 9. Зависимость отношения —от давления (в атм) для СО2,
тот же рисунок свидетельствует о том, что при понижении давления
pV
отношение — при всех температурах стремится к одному и тому же
значению, совпадающему со значением универсальной газовой по-
постоянной. Эти особенности поведения газов указывают на то, что
сформулированные выше простые эмпирические законы будут спра-
справедливы для любого газа, если только последний находится при
22
достаточно низком давлении и если его температура не слишком
низка. Таким образом возникло представление об идеальном
газовом состоянии, находясь в котором газ в точности
следует законам Бойля — Мариотта и Гей-Люссака или, что то же
самое, уравнению Клапейрона.
Каждый газ может находиться в идеальном газовом состоянии,
которое для разных газов и разных температур будет достигаться
при разных давлениях.
В дальнейшем идеальным газом будет называться такой гипо-
гипотетический газ, поведение которого при всех давлениях и темпе-
температурах в точности следует уравнению Клапейрона.
7. ОСНОВНЫЕ ПОЛОЖЕНИЯ КИНЕТИЧЕСКОЙ ТЕОРИИ ГАЗОВ
Атомно-молекулярные представления дают возможность объяс-
объяснить многие свойства газов и, в частности, установить связь, су-
существующую между объемом, занимаемым газом, его температурой
и давлением, т. е. найти уравнение состояния газа. Это удается
сделать в результате детального анализа поведения отдельных
молекул. При подобном анализе предполагается, что движение ча-
частиц вещества — молекул и атомов — подчиняется законам клас-
классической механики. Созданная таким образом теория называется
молекулярно-кинетической теорией газов.
Молекулярно-кинетическая теория газов основывается на не-
небольшом числе весьма общих положений, важнейшие из которых
указаны ниже:
1. Вещество состоит из мельчайших частиц, атомов
или молекул, находящихся в непрерывном движении.
2. В любом, даже очень малом, объеме газа, к кото-
которому еще применимы выводы молекулярно-кинетической
теории, число молекул очень велико. В обычных условиях
второе предположение полностью выполняется. При нуле градусов
и атмосферном давлении, или, как говорят, при нормальных усло-
условиях, в одном кубическом миллиметре воздуха или какого-либо
другого газа содержится около 3 • 1016молекул, так что в объеме,
составляющем одну миллионную часть кубического миллиметра,
содержится еще около 3 • 1О10 молекул. Этот пример показывает,
что даже в объеме, который можно рассматривать как дифферен-
дифференциально малый по сравнению с объемом сосуда, содержащего газ,
находится очень большое количество молекул.
3. Размеры молекул малы по сравнению с рассто-
расстояниями между ними. Простой расчет показывает, что и это
предположение выполняется, если только газ не сильно сжат.
В самом деле, в настоящее время известно, что диаметры молекул
большинства газов заключены в пределах от 2 • 10~10 до 3 • 10~10
метра, в то же время при нормальных условиях расстояние
23
между молекулами в газе составляет приблизительно 3 • 10~9
метра, т. е. в десять раз превосходит молекулярный диаметр.
4. Молекулы взаимодействуют со своими соседями
только в момент соударения, в остальное же время
силами взаимодействия между ними можно пренебречь.
Соударения молекул со стенками сосуда, так же как
и между собой, являются абсолютно упругими, т. е. при
соударениях кинетическая энергия не превращается в другие виды
энергии. Справедливость последних предположений подтвердить
не так легко, как это было в случае первых двух. Как косвенное
подтверждение их справедливости можно рассматривать согласие
с опытом выводов теории, построенной на их основании.
5. При отсутствии внешних сил молекулы газа рас-
распределяются равномерно по всему объему, занятому
газом, и поэтому для любого, даже дифференциально малого,
объема газа dV число заключенных в нем молекул dN можно опре-
определить, умножив соответствующий объем на молекулярную плот-
плотность, т. е. на число молекул п0 в 1 кубическом метре:
dN = nodV.
6. Направления скоростей молекул распределены хао-
хаотично, т. е. в газе отсутствуют какие-либо избирательные на-
направления движения молекул. Другими словами, все направления
движения молекул равновероятны. По абсолютной ве-
величине скорости движения молекул могут изменяться
от величин бесконечно малых до величин бес-
бесконечно больших. Первая часть последнего предположе-
предположения вытекает непосредственно из гипотезы молекулярного хаоса
(беспорядочного движения молекул) и не нуждается в особом об-
обсуждении. В отношении же второй части следует сказать, что стро-
строго говоря, скорость движения молекулы не может быть бесконечно
велика, поскольку подобная молекула должна была бы обладать
бесконечно большой кинетической энергией, что противоречит за-
закону сохранения энергии. Однако при обсуждении ряда вопросов,
как это будет ясно из дальнейшего, характер распределения ско-
скоростей молекул по абсолютной величине не будет играть по суще-
существу никакой роли. Позднее вопрос о распределении молекулярных
скоростей будет рассмотрен детально.
8. ОСНОВНОЕ УРАВНЕНИЕ КИНЕТИЧЕСКОЙ ТЕОРИИ ГАЗОВ
Основное уравнение кинетической теории газов можно получить
различными способами, отличающимися большей или меньшей стро-
строгостью рассуждений. Поскольку окончательный результат во всех
случаях оказывается одним и тем же, ниже приводится возможно
более простой вывод этого важного уравнения. Предположим, что
имеется сосуд в форме куба с ребром а, содержащий один киломоль
24
с,
газа. Далее допустим, что все молекулы—шарики, имеющие одну
и ту же массу т, а число их NA — очень велико. Малость размеров
молекул позволяет пренебречь соударениями их между собой и учи-
учитывать лишь соударения их со стенками сосуда. Конечно, последнее
предположение справедливо только в том случае, если газ занима-
занимает достаточно большой объем, или, что то же самое, если давление
газа невелико.
Влияние, которое оказывают молекулярные соударения, позднее
будет рассмотрено специально. Будем считать дальше, что в отно-
отношении молекулярных ско-
скоростей сь сг, с3, ..., cNa
справедливы предположе-
предположения, сделанные в преды-
предыдущем параграфе.
При своем движении
молекулы будут непрерыв-
непрерывно ударяться о стенки со-
сосуда. Следующие очень
быстро один за другим
молекулярные удары бу-
будут усредняться и, как
указал в 1738 г. академик
Санкт-Петербургской Ака-
Академии наук Д. Бернул-
ли, создавать постоянную
силу, действующую на стенки сосуда. Для того чтобы опре-
определить величину этой силы, рассмотрим соударение со стенкой
какой-либо произвольно выбранной молекулы. Разложим скорость
движения этой молекулы с4 на три взаимно перпендикулярные со-
составляющие: cix, ciy, Ciz, которые направим вдоль трех ребер куба,
пересекающихся в одной из его вершин. Если считать, что стенки
содержащего газ сосуда абсолютно гладкие, то при упругом ударе
молекулы-шарика об одну из стенок будет изменяться, как это до-
доказывается в механике, только нормальная к данной стенке со-
составляющая скорости. Две другие составляющие скорости будут
при ударе оставаться без изменения. Допустим, что в некоторый
момент времени рассматриваемая молекула ударилась о правую
грань куба, соответствующую (см. рис. 10) в выбранной системе
координат плоскости YZ. При ударе нормальная составляющая
скорости cix, сохранив неизменной свою величину, изменит напра-
направление на обратное, т. е. после удара эта составляющая скорости
движения молекулы будет равна — cix. С изменением скорости
молекулы неразрывно связано изменение присущего ей количества
движения, составляющее в данном случае
Рис. 10.
тс1х — (— тс1х) = 2тс1х.
25
Отразившись от стенки, рассматриваемая молекула после боль-
большего или меньшего числа соударений с различными гранями куба
ударится о левую грань, противоположную первой. До этого мо-
момента составляющая скорости cix изменяться не будет, поскольку
предполагается, что между собой молекулы не соударяются, а из
граней куба нормальны к этой составляющей скорости только пра-
правая и левая грани и поэтому только при соударении с ними она
может измениться.
После соударения с левой гранью куба составляющая скорости
(— cix) изменит свое направление на обратное, т. е. сделается
равной -f- с1х. Спустя некоторый промежуток времени рассматри-
рассматриваемая молекула вновь ударится о правую грань куба. Время tit
разделяющее два последующих удара молекулы об одну и ту же
грань, легко определить. Для этого необходимо разделить путь,
пройденный молекулой между этими соударениями и измеренный
вдоль данной оси координат, на соответствующую составляющую
скорости движе»ия молекулы. В рассматриваемом случае путь мо-
молекулы равен 2а, так что время между двумя последующими ударами
найдется из соотношения
2а
г\ = —•
с1г
Зная время, разделяющее два последующих удара молекулы
об одну и ту же грань, можно определить, сколько раз за 1 се-
секунду ударится данная молекула о рассматриваемую грань куба.
Это число ударов пх будет равно:
„ _ 1 _ cLx
Аи = — — —•.
Поскольку при каждом ударе данной молекулы изменение ко-
количества движения составляет 2mcix, общее изменение количества
движения за 1 секунду будет:
2а а
Приведенные выше рассуждения можно повторить применительно
к любой из NA молекул, заключенных в рассматриваемом кубе.
Общее изменение количества движения за 1 секунду, происхо-
происходящее в результате соударения с данной гранью всех заполняющих
куб молекул, найдется суммированием изменений количеств дви-
движений, обусловленных соударениями отдельных молекул, т. е.
тс2. тс\
\ млх
а ' а ' " ' " ' а
Для дальнейших выводов следует учесть, что при разложении
векторов молекулярных скоростей на составляющие вдоль трех
26
произвольно выбранных осей координат должны выполняться сле-
следующие условия:
2 2 i 2 | 2
С\ = С\х-\-С\у-\-С\г,
2 2 I 2 , 2
Суммируя написанные уравнения, получим:
NA NA N А NA
Условие хаотичности молекулярного движения требует, чтобы
при любом расположении координатных осей выполнялось равен-
равенство:
NA NA NA
V 2 V 2 V 2
7, Qa z=z ?л ^iy ^^ Z^ Cizt
1 1 1
или
NA NA NA NA
Это дает возможность преобразовать уравнение A7), выражающее
изменение количества движения, следующим образом:
Na о N л о
. mctx 1 ^ '"с?
2_if _I
a ~~ 3
-W 2
Из механики известно, что изменение количества движения чис-
численно равно импульсу силы
FM = Amv.
Поскольку в приведенных выше рассуждениях подсчитано изме-
изменение количества движения, происходящее за промежуток вре-
времени At, равный одной секунде, то очевидно, что в данном случае
изменения количества движения, происходящие при ударе молекул
о грань куба, численно равны силе F, действующей со стороны
газа на эту грань, т. е.
27
При характеристике свойств газов обычно говорят не о силе,
действующей на стенки сосуда, а о давлении газа р. Сила F, дей-
действующая на грань куба площадью S, связана с давлением газа
простым соотношением: р = ~.
О
В рассматриваемом примере 5 = а2 и, следовательно,
Но а3 = V, т. е. объему, занимаемому газом, так что окончательно
для расчета получаем соотношение:
NA,nu?,
A9)
Приведенные выше рассуждения показывают, каким образом
в результате рассмотрения поведения отдельных молекул можно
сделать заключение о физической сущности макроскопического
свойства газа — его давлении.
Проанализируем более внимательно полученное выражение для
давления газа. Легко заметить, что стоящая в правой части уравне-
уравнения A9) в числителе сумма представляет собой общую кинетиче-
кинетическую энергию молекул газа
Поскольку в знаменателе стоит объем, занимаемый газом, то
очевидно, что вся дробь будет представлять кинетическую энергию
молекул, заключенных в единице объема газа, т. е. плотность
кинетической энергии молекул.
Таким образом, мы приходим к важному выводу о том, что
давление газа измеряется плотностью кинетической
энергии движущихся молекул.
Все молекулы газа имеют одинаковую массу т, значение кото-
которой можно вынести в выражении для кинетической энергии за знак
суммирования. В этом случае общая кинетическая энергия молекул
запишется в виде
Е^^[с2г + с1 + ... + с%А1 B0)
Разделив и умножив правую часть выражения B0) на число моле-
молекул газа NA, получим:
28
NA.m
—
2 NA
Дробь
представляет собой средний квадрат скорости движения молекул
с2. Корень квадратный из этой величины называют средней
квадратичной скоростью движения моле-
молекул:
NA
Пользуясь величиной средней квадратичной скорости, можно
выразить кинетическую энергию молекул газа следующим соотно-
соотношением:
Ek = NA.f. B2)
Выражение для давления газа теперь будет:
р = -^т^. B3)
у 3 V '
Замечая, что дробь — представляет число по молекул в единице
объема газа, можно записать выражение для давления газа в сле-
следующем виде:
р^уПо/пА B4)
Это уравнение обычно называют основным уравнением
кинетической теории газов.
Заметим, что число молекул п0, находящихся при нормальных
условиях в одном кубическом метре газа, равно 2,68 • 1025.
В дальнейшем наряду со средней квадратичной скоростью нам
придется пользоваться и средней арифметической
скоростью, определяемой соотношением:
с=С1 + С2+- + С^ . B5)
N
Следует обратить внимание на то, что квадрат средней квад-
квадратичной скорости с2 отличается от квадрата средней арифмети-
арифметической скорости (сJ. Для того чтобы убедиться в этом, решим сле-
29
дующую задачу: имеется 10 молекул, две из которых движутся со
скоростью 1 м/сек, четыре—со скоростью 2 м/сек, три—со скоростью
3 м/сек и одна — со скоростью 5 м/сек. Требуется определить квад-
квадрат средней арифметической скорости (сJ и квадрат средней квад-
квадратичной скорости сг.
Согласно определению
24 22+ 22+ 22+ 32+ 32+
,_\2 /
[с ) = I
1+1+2 + 2 + 2 + 2
= 7 0 к
=5,76 л/сек
Мы видим, таким образом, что величины с2 и (сJ существенно
различаются.
Если требуется на основании среднего значения молекулярной
скорости найти величину кинетической энергии какой-то совокуп-
совокупности молекул, то для получения правильного ответа необходимо
пользоваться средней квадратичной скоростью, а не средней ариф-
арифметической.
Если в уравнении B3) перенести объем газа У в левую часть,
то оно приобретет следующий вид:
pV = i- NAmc*. B6)
О
Сопоставив написанное выражение с уравнением Клапейрона
и замечая, что левые части этих равенств одинаковы, можно на-
написать
NAm^ RT. B7)
О
Перенеся Л^л в правую часть уравнения, получим:
3 NA
По своему физическому смыслу отношение —¦ представляет
NA
собой универсальную газовую постоянную, отнесенную к одной
молекуле. Эта величина называется постоянной Боль-
ц м а н a k., В системе СИ постоянная Больцмана k равна
1,38- 10~23 дж!град.
Полученное уравнение позволяет сделать чрезвычайно важный вы-
вывод о том, что средняя кинетическая энергия молекул газа
зависит только от температуры газа.
^l = ±kT B8)
2 2 '
30
Этот вывод раскрывает физический смысл абсолютной темпера-
температуры.
Из уравнения B8) следует, что абсолютная температура
является мерой средней кинетической энергии молеку-
молекулярного движения. По мере приближения к абсолютному нулю
средняя кинетическая энергия теплового движения молекул умень-
уменьшается 1.
Следует также обратить внимание на то, что согласно уравнению
B8) при одинаковой абсолютной температуре кинетические энер-
энергии, обусловленные перемещением молекул, будут одинаковыми
даже у молекул, сильно различающихся своей массой. Про-
Процесс выравнивания температуры, который имеет место при сопри-
соприкосновении или смешении холодного газа с газом нагретым, пред-
представляет по существу процесс выравнивания средних кинетиче-
кинетических энергий поступательного движения молекул.
Уравнение B7) позволяет рассчитать среднюю квадратичную ско-
скорость движения молекул любого газа при разных температурах.
Заметив, что произведение массы молекулы т на число АвогадроЛ/^
равняется массе одного киломоля газа, т. е. молекулярному весу ве-
вещества М, можно записать выражение для вычисления средней
квадратичной скорости газа У с2 в следующем виде:
=V
W
B9)
или, подставляя численное значение универсальной газовой посто-
постоянной,
Р= 158
j/J м/сек. C0)
Таким образом, средняя квадратичная скорость поступатель-
поступательного движения молекул прямо пропорциональна квадратному
корню из отношения абсолютной температуры к молекулярному
весу.
Сходная зависимость существует и для средней арифметиче-
1 Обычно возникает вопрос о том, как будет обстоять дело при достиже-
достижении абсолютного нуля. Не следует ли из уравнения 28, что при абсолютном
нуле вовсе прекратится всякое движение частиц вещества? Такое заключе-
заключение было бы неправильным. Предположения, указанные в начале этого па-
параграфа, не выполняются при низких температурах, а следовательно, теря-
теряют силу выводы, делаемые на их основании. Кроме того, частицы вещества
не только перемещаются, но совершают и другие виды движения, в частно-
частности атомы, входящие в состав молекул, колеблются. Колебательное движе-
движение частиц не прекращается и при абсолютном нуле. Каждый атом обладает
при абсолютном нуле температуры некоторой энергией колебательного дви-
движения, не зависящей от температуры и называемой нулевой энергией.
31
ской скорости движения молекул с. В этом случае справедливо со-
соотношение:
уЩ CD
или
с= 145 l/L м/сек. C2)
V М
Пример 1. Вычислить среднюю квадратичную и среднюю арифметиче-
арифметическую скорости движения молекул водорода и атомов парообразной ртути
при температуре 300° по абсолютной шкале. Молекулярный вес водорода 2,
атомный вес ртути 200.
Решение.
Для водорода: Y^ = 158 1/ 522. = 1928 м/сек.
145 l/^i. = 1659 м/сек.
Для ртути:/^ = 158 Ъ^— = 193 м/сек,
A
с = 145 у ^L = 177 м/сек.
Пример 2. Какой вывод придется сделать, если при вычислении давления
газа (стр. 25) предположить, что грань куба, о которую ударяются молекулы,
удаляется от приближающихся молекул со скоростью и, измеренной по от-
отношению к той же неподвижной системе координат, по отношению к которой
измерена скорость молекулы.
Решение. При расчете скорости движения молекулы при соударении
со стенкой следует принимать во внимание не абсолютное значение скорости
движения молекулы, а относительное ее значение, измеренное с учетом дви-
движения стенки. Таким образом, поскольку и молекула и стенка движутся в
одном направлении, относительная скорость движения молекулы до удара
будет (с\х — и). После удара скорость движения молекулы по отношению
стенки должна сохранить свою величину, но изменить знак на обратный. По-
Поскольку же после удара стенка и рассматриваемая молекула движутся в
противоположных направлениях, то в системе координат, в которой измере-
измерены скорости с1х и и, это означает добавочное уменьшение скорости движения
молекулы на величину, равную скорости движения стенки и.
Таким образом, в той системе координат, в которой до соударения со
стенкой скорость движения молекулы была равна с1х, после соударения она
составит только (с1х — 2ц), и, следовательно, кинетическая энергия молекулы
при ударе о движущуюся стенку будет изменяться, в рассмотренном случае —
уменьшаться. Уменьшение кинетической энергии в условиях данной задачи
равно:
те?
с\х
V~ 2 '
Очевидно, что если энергия молекулярного движения не пополняется
извне, то в результате соударений с движущейся стенкой величина средней
кинетической энергии молекул газа уменьшается, т. е. газ охлаждается.
32
9. НЕКОТОРЫЕ СЛЕДСТВИЯ ИЗ ОСНОВНОГО УРАВНЕНИЯ
КИНЕТИЧЕСКОЙ ТЕОРИИ ГАЗОВ
Если в сосуде объемом V находится смесь нескольких газов,
то общая кинетическая энергия молекул газовой смеси равна сум-
сумме кинетических энергий молекул отдельных компонентов смеси,
где Eki, Ek2, Екз и т. д. — кинетические энергии молекул первого,
второго, третьего и т. д. газов, входящих в состав смеси.
Написанное равенство не изменится, если обе его части помно-
2 1 „
жить на одну и ту же величину, равную— • —. Проделав эту опера-
о V
цию, найдем:
з v з v з v з v
Проанализируем физический смысл полученного равенства.
2 F
Выражение -, стоящее в левой части уравнения, согласно выво-
выводам предыдущего параграфа, представляет собой общее давление
газовой смеси р.
2 Ek
Первый член правой части уравнения соответствует тому
давлению ри которое оказывал бы первый газ, входящий в состав
газовой смеси, если бы он один занимал весь объем V. Это давле-
давление называют парциальным давлением первого ком-
компонента газовой смеси. Второй член правой части уравнения, оче-
очевидно, соответствует парциальному давлению второго компонента
газовой смеси, третий — парциальному давлению третьего компо-
компонента смеси и т. д. Физический смысл полученного уравнения
заключается, таким образом, в утверждении, что общее дав-
давление, оказываемое на стенки сосуда смесью нескольких
газов, равно сумме парциальных давлений отдельных
компонентов смеси. Этот важный физический закон был ог-
крыт эмпирически Д. Дальтоном и носит его имя.
Закон Дальтона вытекает непосредственно из основных пред-
представлений кинетической теории газов. Действительно, выше пред-
предполагалось, что молекулы газа движутся, не соударяясь друг с
другом. При этом условии молекулы какого-либо газа в смеси уда-
ударяются о стенки сосуда и создают давление независимо от присут-
присутствия других газов в сосуде. Очевидно, что общее давление газовой
смеси будет при этом равно сумме парциальных давлений компо-
компонентов смеси.
Молекулярно-кинетическое истолкование возникновения давле-
давления газа позволяет объяснить и весомость газов. Вес газа возни-
возникает в результате того, что о верхнюю и нижнюю стенки сосуда
2 Заказ 145 gj
молекулы ударяются, двигаясь с различной скоростью. Для до-
доказательства этого возвратимся еще раз к кубическому сосуду, ко-
которым пользовались при выводе основного уравнения кинетичес-
кинетической теории газов (рис. 10).
Предположим, что какая-либо молекула ударилась о верхнюю
стенку куба, двигаясь со скоростью сУ1. При последующем движе-
движении вниз скорость молекулы под действием силы тяготения возрас-
возрастет. Если время движения рассматриваемой молекулы от верхней
грани куба до нижней tit то при соударении с нижней гранью ее ско-
скорость c'5l будет
где g — ускорение силы тяжести.
Изменение количества движения при ударах молекулы о верх-
верхнюю и нижнюю грани куба будут соответственно равны: 2тс1у при
ударе о верхнюю грань и 2т (сн + gti) при ударе о нижнюю грань.
Время движения молекулы вверх равно времени движения ее
вниз, и поэтому промежуток времени между двумя последующими
ударами рассматриваемой молекулы об одну и ту же грань {t) бу-
будет равен удвоенному времени движения молекулы сверху вниз,
т. е. 2ti. В этом случае число ударов молекулы о стенку за 1 сек
равно —, а изменения количества движения за то же время на верх-
верхней и нижней гранях составит соответственно: 2тсу и 2т (cyi -f-
1 2h
+?^i) — Разность этих величин будет численно равна направлен-
ной вниз избыточной силе, возникающей в результате ударов рас-
рассматриваемой молекулы:
/Ч = 2т (с -f gtj — — 2тсу . i- = mgm
Как мы видим, эта избыточная сила не зависит от скорости дви-
движения молекулы, а зависит лишь от ее массы. Поэтому для вычисле-
вычисления суммарной избыточной силы, действующей на нижнюю грань
куба со стороны всех молекул, достаточно умножить найденную
величину на число молекул NA, т. е.
F = NAmg = Mg.
Здесь М = NАт — общая масса газа. Таким образом, избыточная
сила, действующая на нижнюю грань куба и возникающая в резуль-
результате изменения скоростей движения молекул под действием поля
земного тяготения, и обусловливает вес газа.
34
один квадратный метр
10. РАСПРЕДЕЛЕНИЕ МОЛЕКУЛ ГАЗА В ПОЛЕ ЗЕМНОГО
ТЯГОТЕНИЯ
Действие силы тяжести приводит не только к возникновению
избыточного давления на дно сосуда содержащего газ, но и к оп-
определенному распределению молекулярной плотности по высоте
газового столба. Одновременно с измерением плотности изменяется
и давление газа, измеряемое барометром.
Для нахождения закона, которому подчиняется изменение плот-
плотности в свободном столбе газа, поддерживаемом при постоян-
постоянной температуре, выделим мысленно столб
газа с основанием в
(рис. 11).
Давление газа у основания столба обозна-
обозначим р0 и предположим, что в сечении столба,
расположенном на высоте h, давление газа
будет р. Слои газа, расположенные ниже
выделенного сечения, не оказывают на него
давления. Однако эти слои давят на воздух,
расположенный под ними, и поэтому давление
газа у основания столба будет больше, чем
на высоте h. Для вычисления зависимости
плотности газа от высоты проведем второе
сечение, расположенное на дифференциально
малую величину dh выше первого. Давление
газа во втором сечении будет на величину dp
меньше, чем в первом сечении, т. е. будет
равно р — dp. Уменьшение давления, очевид-
очевидно равно весу столбика газа, заключенного между первым и вторым
сечениями. Если обозначить число молекул в единице объема газа
на высоте h символом nh, а массу молекулы т, то вес столбика газа,
заключенного между указанными сечениями, будет равен nhmgdh,
где g — ускорение силы тяжести.
Как сказано выше, именно на эту величину уменьшается дав-
давление в столбе газа при переходе от первого сечения ко второму,
т. е.
dp——nhmgdh. C4)
Воспользовавшись уравнением Клапейрона, можно написать
для единицы объема газа на высоте h соотношение:
Рис. 11.
где v — число молей газа в единице объема, равное отношению чис-
числа молекул nh к числу Авогадро NА. Учитывая сказанное, получим:
35
Дифференцируя, найдем:
dp = ^dnh. C5)
Приравнивая правые части уравнений C4) и C5), запишем:
или, преобразуя,
RT
-nhmgdh= —dnh,
dnh _ NAmg ^
nh RT
Замечая, что произведение mNA представляет массу килограмм-мо-
килограмм-молекулы, равную молекулярному весу М газа, и интегрируя, най-
найдем:
In nh = -h-\- Const.
л RT i
Для определения постоянной интегрирования заметим, что при
h = О, nh— n0 и, следовательно, Const = In nn.
Таким образом, искомое уравнение принимает вид:
или
Шh C7)
Уравнение C7) показывает, как изменяется с высотой, в ре-
результате действия силы тяжести, количество молекул в единице
объема покоящегося столба газа, имеющего по всему протяжению
одинаковую температуру.
Умножив обе части уравнения C7) на массу отдельной моле-
молекулы т и замечая, что произведения пот и nhm соответствуют мас-
массам газа в одном кубическом метре, т. е. плотностям газа у основа-
основания столба (р0) и на высоте h (рй), получим уравнение, передаю-
передающее изменение плотности газа с высотой:
= ~ Wh C8)
Аналогичная зависимость будет справедлива и для изменения
с высотой давления газа. Именно так должно было бы изменяться
давление воздуха в земной атмосфере, если бы температура в стол-
столбе воздуха была неизменной.
Уравнение, позволяющее рассчитывать изменение давления
воздуха с высотой, было получено впервые французским матема-
математиком и физиком Лапласом A749—1827) и называется б а-
рометрической формулой Лапласа.
Зо
Уравнению C7) можно придать более общую форму, если обра-
обратить внимание на то, что стоящее в показателе степени произведение
Mgh равно изменению потенциальной энергии АЕр одного киломо-
ля газа при перемещении его от уровня, на котором в кубическом
метре содержится п0 частиц, до уровня, на котором в кубическом
метре — nh частиц. Учитывая сказанное, уравнение C7) можно
записать в следующем виде:
ДЕр
RT ,
C9)
Таким образом, разница молекулярных плотностей в столбе
покоящегося газа зависит от различия потенциальных энергий мо-
молекул, находящихся на разной высоте. Можно формулировать как
общее правило: если имеются две области, отличающиеся одна от
другой тем, что потенциальные энергии молекул в них различны,
и если возможен взаимный переход молекул из одной области в дру-
другую, то при равновесии в этих областях будут различны и
плотности вещества. Соотношение в плотностях в этом случае
можно подсчитать, воспользовавшись уравнением C9).
П. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ ЧИСЛА АВОГАДРО
Замечательные работы Ж. Перрена, сыгравшие исключитель-
исключительную роль в деле утверждения молекулярных представлений, свя-
связаны с использованием полученной выше барометрической фор-
формулы. Основная идея опытов Перрена сводилась к предположению,
что законы молекулярно-кинетической теории определяют поведе-
поведение не только атомов и молекул, но и гораздо более крупных час-
частиц, состоящих из многих тысяч молекул. Исходя из весьма общих
соображений, которые здесь не будут рассматриваться, можно пред-
предполагать, что средние кинетические энергии очень мелких частиц,
совершающих броуновское движение в жидкости, совпадают со
средними кинетическими энергиями молекул газа, если только
температура жидкости и температура газа одинаковы. Точно так
же распределение по высоте частиц, взвешенных в жидкости, под-
подчиняется тому же закону, что и распределение по высоте молекул
газа. Подобный вывод очень важен, поскольку на основании его
возможна количественная проверка закона распределения. Про-
Проверку можно осуществить путем непосредственного подсчета с по-
помощью микроскопа количества взвешенных частиц, находящихся
в жидкости на разной высоте.
Уравнение C6) распределения частиц по высоте
удобно в этом случае переписать, разделив числитель и знаменатель
дроби, стоящей в правой части уравнения, на число Аво1адроЛ'л.
37
м
При этом следует заметить, что отношение^- соответствует массе
RT 2
частицы т, а отношение^- равно j средней кинетической энергии
частицы ц0 [сравните уравнение B8)] .
Вводя эти обозначения, получим:
In "ft — . msh
п0 2_-'
Если теперь опытным путем определить количества частиц л4
и гц, соответствующие двум различным значениям h, то можно бу-
будет написать:
In —• = и In —- = .
п0 г_- п0 2^_
Вычитая из первого уравнения второе, найдем:
D0)
nh> 2 _
I"» _
Из этого соотношения можно определить ы0, если только знать мас-
массу частицы т.
При всей простоте и ясности основной идеи опыты Перрена бы-
были связаны с преодолением больших трудностей. В качестве объ-
объекта исследования им были выбраны водные эмульсии мастики и
гуммигута, которые подвергались центрифугированию для полу-
получения эмульсий, состоящих из зернышек одинакового размера.
Размер зернышек, которые считались шариками, определялся по
скорости их оседания. За движением отдельного зернышка следить
было невозможно и потому наблюдалась скорость оседания верх-
верхней границы эмульсии, т. е. средняя скорость оседания многих ты-
тысяч зернышек. Зная плотность эмульгированного вещества и оп-
определяя размеры зернышек эмульсии, можно было вычислить их
массы. Далее необходимо было определить числа nhi, и nhr С этой
целью к предметному стеклышку для микроскопических наблюде-
наблюдений Перрен приклеил второе стекло с просверленным в нем круг-
круглым отверстием, так что образовалась цилиндрическая прозрачная
кювета. Поместив в кювету каплю эмульсии и закрыв для предот-
, вращения испарения кювету покровным стеклышком, можно было
с помощью микроскопа наблюдать зернышки эмульсии. Если вос-
воспользоваться объективом с небольшой глубиной поля зрения, то в
микроскопе будут видны только зернышки, расположенные в очень
тонком слое жидкости. Практически в этих опытах можно сосчитать
лишь небольшое количество зернышек, поскольку их число непре-
непрерывно меняется. Для преодоления этого затруднения в фокальной
38
плоскости окуляра помещался непрозрачный экран с маленьким
круглымотверстием. Благодаря этому поле зрения микроскопа силь-
сильно уменьшалось, и наблюдатель мог сразу определить, сколько
зернышек в данный момент находится в поле зрения (рис. 12).
Повторяя подобные наблюдения через правильные промежутки
времени, записывая наблюдаемые числа зерен и усредняя полу-
полученные данные, Перрен показал, что среднее
число зерен на данном уровне стремится к не-
некоторому определенному пределу, соответствую-
соответствующему плотности эмульсии на этом уровне. Для
того чтобы проиллюстрировать трудоемкость
этих опытов, можно указать, что для получе-
получения точного результата необходимо было про-
производить несколько тысяч измерений.
Определив с желаемой степенью точности
плотность эмульсии на некотором уровне hx,
Перрен перемещал микроскоп в вертикальном
направлении и измерял плотность эмульсии
на втором уровне /г2. Тщательно выполненные
измерения показали, что распределение зерны-
зернышек эмульсии по высоте подчиняется баромет-
барометрической формуле (уравнение 37).
Воспользовавшись найденными значениями
плотности зерен эмульсии на разных уровнях,
Перрен вычислил среднюю кинетическую энер-
энергию частицы и0 и, воспользовавшись соотноше-
соотношением Рис- 12- Распре-
Распределение зерен
эмульсии.
определил экспериментально число Авогадро NA.
Найденные таким образом значения числа Авогадро колебались
от 6 • 1023 до 7 • 1028.
Число Авогадро можно определить различными способами.
В своей работе Перрен сопоставляет найденные им величины со
значениями числа Авогадро, определенными двенадцатью другими
методами, и подчеркивает, что самые разнообразные методы опреде-
определения NА приводят к весьма близким величинам, чего не могло бы
быть, если бы основные положения молекулярно-кинетической тео-
теории не отражали бы правильно явления, объективно существующие
в природе.
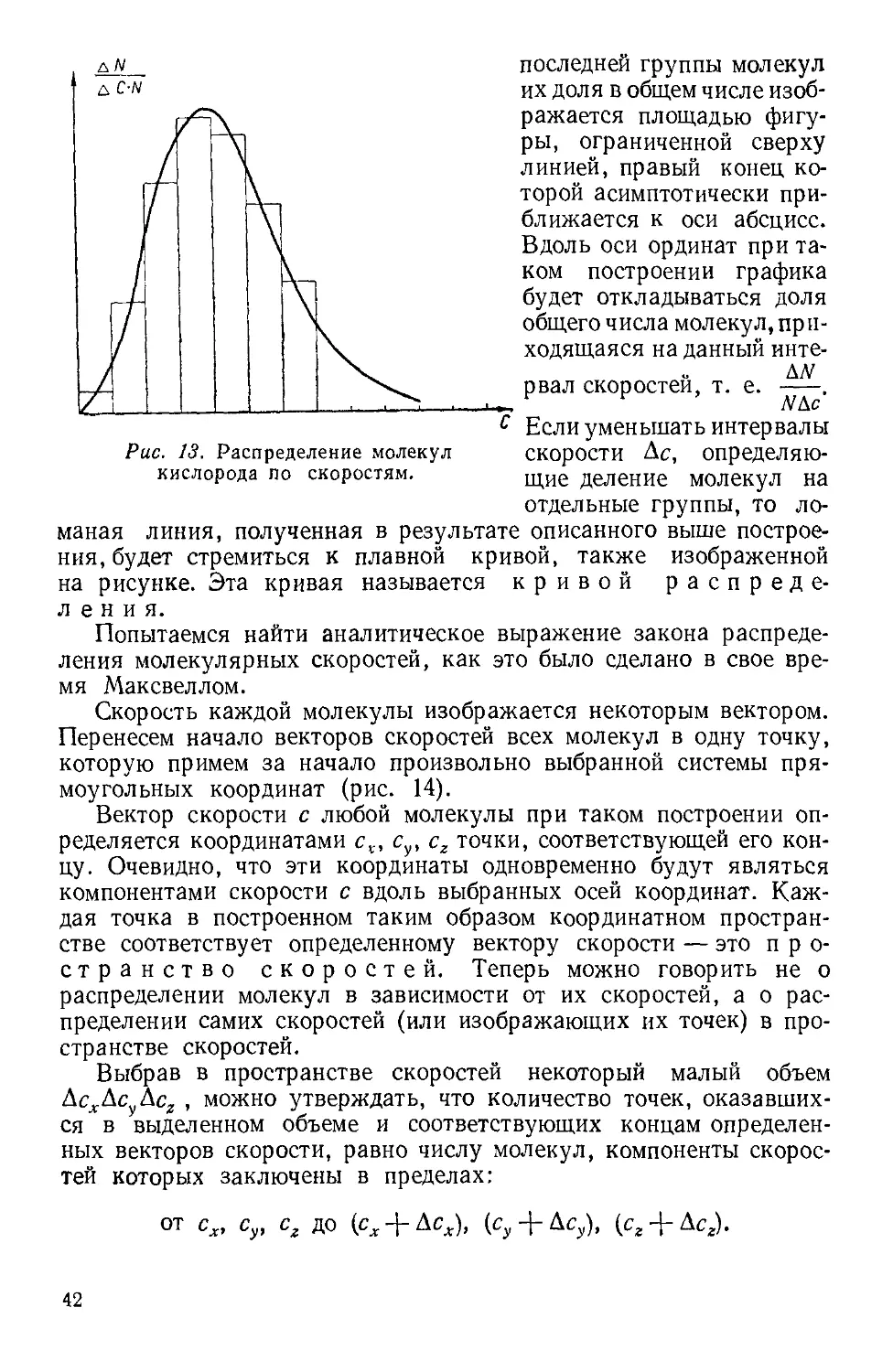
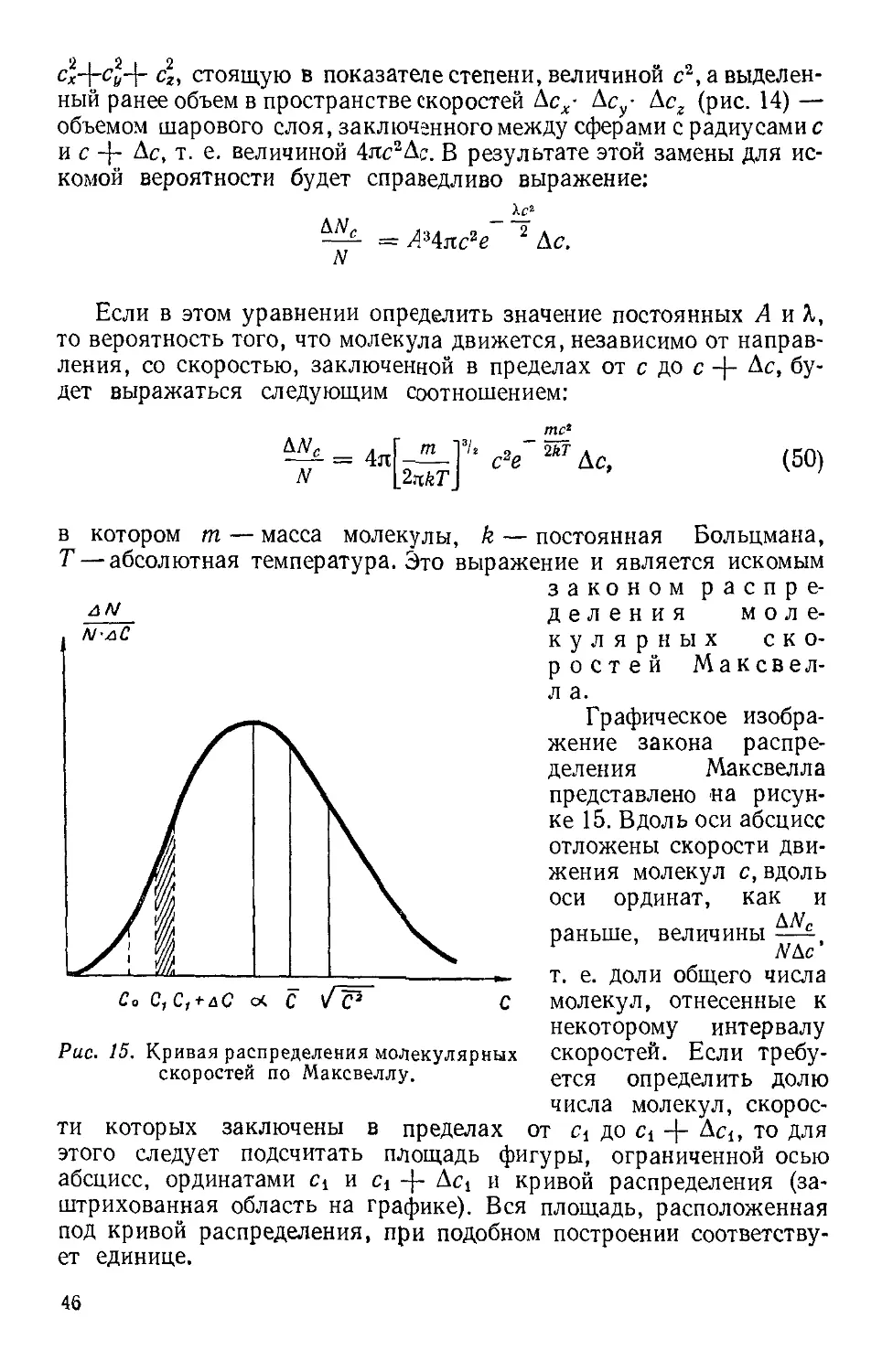
12. ЗАКОН РАСПРЕДЕЛЕНИЯ СКОРОСТЕЙ
В предыдущих рассуждениях принималось, что молекулы газа
могут двигаться с самыми различными скоростями, начиная с очень
малых и кончая бесконечно большими. Как распределяются при
39
этом молекулы в зависимости от их скоростей: сколько молекул
движется быстро и сколько медленно — не обсуждалось. Это оп-
оправдывалось тем, что при подсчете давления распределение моле-
молекулярных скоростей не играет никакой роли. Величина давления
определяется кинетической энергией молекул в единице объема вне
зависимости от того, как эта кинетическая энергия делится между
отдельными молекулами.
Хаотичность молекулярного движения может вообще породить
сомнение в возможности решить задачу о распределении скоростей
молекул. Но, как мы убедимся ниже, подобную задачу решить мож-
можно, и впервые это было сделано К. Максвеллом, нашедшим
закон распределения молекул по скорос-
скоростям. Строгое обоснование закона распределения принадлежит
Л. Больцману, который показал, что, каким бы ни было в на-
начальный момент распределение скоростей молекул, оно с неиз-
неизбежностью переходит в распределение, следующее из закона Макс-
Максвелла, если только температуру газа поддерживать неизменной
достаточно долго.
Конечно, хаотичный характер молекулярного движения накла-
накладывает свой отпечаток на закон распределения молекул по ско-
скоростям и его формулировку. Нельзя, например, ставить вопрос о
том, сколько молекул в данном объеме газа в какой-либо момент
времени движется со скоростью, в точности равной сх. В силу слу-
случайного, беспорядочного характера движения молекул даже при
очень большом числе их в рассматриваемом объеме газа может не
оказаться ни одной молекулы, удовлетворяющей этому условию.
По указанной причине в дальнейшем всегда будет говориться о
числе молекул, скорости которых заключены в некотором интер-
интервале скоростей, скажем о числе молекул, скорости которых боль-
больше некоторой величины сь но меньше величины ct -j- Дс4.
Кроме того, нельзя гарантировать, что указанное число моле-
молекул будет во всех случаях точно соответствовать действительности.
В данном случае следует говорить о вероятности встре-
встретить в газе данное число интересующих нас молекул. При этом не-
необходимо иметь в виду, что в отличие от того, как это имеет место
в обыденной жизни, когда «вероятность события» определяется на
основании субъективной оценки каких-то известных данному че-
человеку факторов, так что вероятность одного и того же события мо-
может оцениваться различными людьми по-разному, научное опре-
определение вероятности объективно и выражается числом, не завися-
зависящим от того, кто его определяет. Позднее определение вероятности
будет рассмотрено более подробно. Здесь мы отметим лишь, что ес-
если газ содержит большое количество молекул, то вычисленная с
помощью закона Максвелла вероятность встретить молекулу, ско-
скорость которой удовлетворяет определенным условиям, с боль-
большой точностью совпадает с долей подобных молекул среди молекул
газа. Выше отмечалось, что на практике приходится иметь дело с
40
количествами вещества, содержащими огромное число молекул, и
поэтому при решении большинства практических задач молекуляр-
молекулярной физики законы теории вероятности выполняются с той стро-
строгостью, которая обычна для физических законов.
Для пояснения физической сущности закона распределения
молекулярных скоростей рассмотрим следующий мысленный опыт.
Предположим, что удалось сконструировать прибор, позволяющий
не только непосредственно наблюдать хаотическое движение мо-
молекул, но и определять их скорости. Допустим, что с помощью это-
этого прибора наблюдают поведение молекул кислорода, температу-
температура которого нуль градусов. Как указано выше, не имеет смысла
пытаться определять точные значения скоростей отдельных моле-
молекул. Наблюдаемые молекулы распределяются в зависимости от их
скоростей по отдельным группам. Предположим, что все молеку-
молекулы делят так, что в первую группу относят те, скорости которых
меньше 100 м/сек, во вторую — те, у которых скорости заключены
в пределах от 100 до 200 м/сек, в третью — от 200 до 300 м/сек и т. д.
вплоть до молекул, движущихся со скоростями от 600 до 700 м/сек,
после чего все остальные молекулы собирают в одну общую груп-
группу. Если подобный опыт проделать несколько раз с различным, но
достаточно большим количеством молекул, определяя всякий раз
не просто количества молекул в указанных выше отдельных груп-
группах, а долю, которую составляют эти молекулы в общем числе мо-
молекул, скорости которых были измерены, то мы заметим, что для
каждой из групп найденная таким образом доля остается неизмен-
неизменной.
В таблице 4 приведены результаты проверки подобного мыслен-
мысленного опыта.
Как можно убедиться, рассматривая
таблицу, молекул с очень большими и
очень малыми скоростями немного. По-
Подавляющее большинство молекул дви-
движется со скоростями, заключенными в
сравнительно узких пределах.
Еще более наглядно графическое
изображение описанного опыта. На ри-
рисунке 13 вдоль оси абсцисс отложены
скорости движения молекул. В соответ-
соответствии с опытом ось абсцисс разделена на
отдельные интервалы по 100 м/сек вплоть
до 700 м/сек. Доли числа молекул —,
скорости которых лежат в указанных
интервалах скоростей, изображаются на
графике площадями соответствующих
прямоугольников, имеющих основанием
заданный интервал скоростей Дс. Для
Таблица 4
Распределение молекул
кислорода по скоростям
при 0° С
Интервал скоро-
скоростей в м/сек
Менее 100
100—200
200—300
300—400
400—500
500—600
600—700
Более 700
Доля мо-
молекул в %
1,4
8,1
16,7
21,5
20,3
16,1
9,2
7,7
41
Рис. 13. Распределение молекул
кислорода по скоростям.
последней группы молекул
их доля в общем числе изоб-
изображается площадью фигу-
фигуры, ограниченной сверху
линией, правый конец ко-
которой асимптотически при-
приближается к оси абсцисс.
Вдоль оси ординат при та-
таком построении графика
будет откладываться доля
общего числа молекул, при-
приходящаяся на данный инте-
рвал скоростей, т. е. .
Если уменьшать интервалы
скорости Ас, определяю-
определяющие деление молекул на
отдельные группы, то ло-
ломаная линия, полученная в результате описанного выше построе-
построения, будет стремиться к плавной кривой, также изображенной
на рисунке. Эта кривая называется кривой распреде-
распределения.
Попытаемся найти аналитическое выражение закона распреде-
распределения молекулярных скоростей, как это было сделано в свое вре-
время Максвеллом.
Скорость каждой молекулы изображается некоторым вектором.
Перенесем начало векторов скоростей всех молекул в одну точку,
которую примем за начало произвольно выбранной системы пря-
прямоугольных координат (рис. 14).
Вектор скорости с любой молекулы при таком построении оп-
определяется координатами сх, су, сг точки, соответствующей его кон-
концу. Очевидно, что эти координаты одновременно будут являться
компонентами скорости с вдоль выбранных осей координат. Каж-
Каждая точка в построенном таким образом координатном простран-
пространстве соответствует определенному вектору скорости — это про-
пространство скоростей. Теперь можно говорить не о
распределении молекул в зависимости от их скоростей, а о рас-
распределении самих скоростей (или изображающих их точек) в про-
пространстве скоростей.
Выбрав в пространстве скоростей некоторый малый объем
AcxAcyAcz , можно утверждать, что количество точек, оказавших-
оказавшихся в выделенном объеме и соответствующих концам определен-
определенных векторов скорости, равно числу молекул, компоненты скорос-
скоростей которых заключены в пределах:
от сх, с„, сг до (сх^Асх), (с -{-Ас), (сг + Дсг).
42
Если рассматривать только составляющую скорости вдоль оси
х, то вполне естественно предположить, что число молекул ANC
составляющая скорости которых в выбранном направлении боль-
больше сх, но меньше сх -\- &сх, пропорционально общему числу моле-
молекул N и величине заданного интервала скоростей &сх, а кроме то-
того, зависит от самой величины скорости сх, т. е.
где / (сх) — функция, определяющая распределение молекул в за-
зависимости от их скоростей.
Перенося N в левую часть уравнения и замечая, что отношение
ANcx — числа молекул, удовлетворяющих заданному условию,—
к общему числу молекул Л/
равно вероятности для
произвольно выбранной мо-
молекулы обладать скоро-
скоростью, лежащей в указан-
указанных пределах, можно на-
написать:
= f(cx)Acx. D1a)
N
Рассуждая аналогично,
можно написать выраже-
выражение вероятности для моле-
молекул обладать составляю-
составляющей скорости вдоль оси у,
большей су, но меньшей
су+Асу:
~- = f(cv)Acv. D1b)
Подобно этому вероят-
вероятность составляющей ско-
скорости вдоль оси 2 оказаться
cz -j- Acz будет равна:
N
Рис. 14. Пространство скоростей,
заключенной в пределах от cz до
= f(cz)Acz. D1с)
В теории вероятностей доказывается теорема, согласно которой
вероятность совместного осуществления нескольких независимых со-
событий равняется произведению их вероятностей. Так, например,
если вероятность выпадения герба при бросании монеты равна —, то
вероятность выпадения герба у двух монет, бросаемых одновремен-
одновременно или одна за другой, будет составлять —. — = — . Поэтому
43
ДДГ
можно предположить, что вероятность1 —? для молекулы обладать
скоростью, компоненты которой заключены в пределах от сх, су,
сг до (сх-\- Асх), (су-\- Асу), {cz-\- Асг), найдется перемножением
написанных выше вероятностей D1а, 41Ь и 41с), т. е.
^ = / (сх) Асх . f (Су) Асу . / (сг) Acz. D2)
Предположим, что нижний предел скорости с постоянен, в
этом случае должно выполняться условие:
с2 + 4+Сг = Const,
cxdcx + cydcy -f czdcz = 0. D3)
Допустим дальше, что неизменным остается и интервал скорос-
скорости, т. е. элементарный объем в пространстве скоростей:
Асх ' Асу • Асг = Const.
При выполнении этих двух предположений должна оставаться
неизменной и вероятность —~ того, что молекула обладает ско-
скоростью, удовлетворяющей сформулированным выше требованиям.
Если это так, то можно написать:
0 и d [Асх ¦ Асу • Асг] - 0.
Подставляя в первое равенство значение —- из уравнения D2)
и принимая во внимание второе равенство, получим:
f (Су) f (сг) Г (сх) dcx + f {cz) f (cx) Г {Cy) dcy +
+ f(cx)f{Cy)r(cz)dcz = O. D4)
Разделив полученное уравнение на произведение функций
f(c*)f{cy)f{cz),
найдем
х^ /(с„) у^ f(cz) г V ;
Умножив выражение D3) на произвольную величину Я и сложив
с уравнением D5), можно, сгруппировав члены в соответствии с
индексами у с, написать:
1 Строго говоря, это предположение не точно, однако расчет, выполнен-
выполненный математически строго, приводит к тем же результатам.
44
В силу произвольности величин dcx, dcy и dcz написанное урав-
уравнение может выполняться только в том случае, если каждый из
стоящих в скобках двучленов порознь равен нулю, т. е.
П?«)| Хс 0; Г (су) _|_ ъс = Q; 11Шл-1с -о, D7)
/(<>) ^ * /(с) ^ у /(г) ^ г ;
заметив, что f'(cx) = —
Последние уравнения легко решаются. Обозначив f(cx) = у и
етив,
получим:
J- . EL _|_ Кг = О,
у dcx
или
— — АСМСГ,
У
откуда
у = Ае~~,
где А — постоянная интегрирования.
Таким образом, искомое выражение вероятности —°-х того,
что скорость молекулы в направлении оси х заключена в пределах
от сх до сх-\~ Асх, будет равна:
Очевидно, что аналогичные выражения можно получить для вероят-
вероятности того, что скорость молекулы вдоль оси у заключена в преде-
пределах от су до су + ^су> и Для вероятности того, что скорость моле-
молекулы в направлении оси z заключена в пределах от сг до сг-\- Асг.
Вероятность —- того, что компоненты скорости молекулы вдоль
N
трех осей координат одновременно заключены в пределах от сх,
су, сг до сх-\- Асх, Cy-j- Асу, сг-\- Асг, найдется, как принято было
выше, перемножением соответствующих вероятностей, т. е.
~ =А3е AcxAcyAcz. D9)
Часто для расчета представляет интерес только абсолютная ве-
величина скорости, но не направление ее в пространстве, или, другими
словами, не значения компонентов скорости сх, су, cz и соот-
соответствующих интервалов Асх, Лсу, Асг, а лишь величина с и неко-
некоторый интервал ее Ас. В этом случае следует заменить сумму
45
с'х-\-си-\- сг, стоящую в показателе степени, величиной с2, а выделен-
выделенный ранее объем в пространстве скоростей Асх- Асу Acz (рис. 14) —
объемом шарового слоя, заключенного между сферами с радиусами с
и с -f- Ac, т. е. величиной 4яс2Лс. В результате этой замены для ис-
искомой вероятности будет справедливо выражение:
N
= АЧпс2е 2 Ас.
Если в этом уравнении определить значение постоянных А и К,
то вероятность того, что молекула движется, независимо от направ-
направления, со скоростью, заключенной в пределах от с до с -f- Ac, бу-
будет выражаться следующим соотношением:
шсг
ш
Ас,
E0)
в котором т — масса молекулы, k — постоянная Больцмана,
Т — абсолютная температура. Это выражение и является искомым
законом распре-
распределения моле-
молекулярных ско-
скоростей Максвел-
л а.
Графическое изобра-
изображение закона распре-
распределения Максвелла
представлено на рисун-
рисунке 15. Вдоль оси абсцисс
отложены скорости дви-
движения молекул с,вдоль
оси ординат, как и
раньше, величины —-,
NAc
т. е. доли общего числа
с0 с,с, + йС с* с (/с5 с молекул, отнесенные к
некоторому интервалу
скоростей. Если требу-
требуется определить долю
числа молекул, скорос-
скорости которых заключены в пределах от сх до с4 -f- Ac и то для
этого следует подсчитать площадь фигуры, ограниченной осью
абсцисс, ординатами d и с4 -\- Дс4 и кривой распределения (за-
(заштрихованная область на графике). Вся площадь, расположенная
под кривой распределения, при подобном построении соответству-
соответствует единице.
Рис. 15. Кривая распределения молекулярных
скоростей по Максвеллу.
46
При практическом применении закона Максвелла иногда бы-
бывает необходимо определить долю числа молекул, скорости которых
превосходят некоторое определенное значение с0. Искомая вели-
величина изображается на графике площадью фигуры, расположенной
справа от ординаты, соответствующей скорости с0, и ограниченной
кривой распределения и осью абсцисс.
Характер кривой распределения молекулярных скоростей ука-
указывает на то, что существует некоторая скорость молекулярного
движения, отличающаяся тем, что в ее окрестности на данный не-
небольшой интервал скоростей приходится большее число молекул,
чем в окрестности любой другой скорости. Эта скорость называется
наивероятнейшей скоростью движения мо-
молекул. Наивероятнейшей скорости соответствует максимум на
кривой распределения.
Вычисление наивероятнейшей скорости а производится по фор-
формуле:
а=уЩ, E1)
или
а= 129 Л ft Ml сек.
Если условиться измерять скорости движения молекул не в
метрах в секунду, а в относительных единицах, равных отношению
с
скорости движения молекулы к наивероятнейшей скорости: и ——,
то закон распределения Максвелла запишется гораздо проще, а
AN
именно: доля числа молекул — , относительные скорости кото-
которых заключены в интервале от и до и -(- Аи, выражается уравне-
уравнением:
— = -Лт и2е~иг Аи. E2)
Л' Уп
Для сравнения на графике, изображающем закон распределения
(рис. 15), указаны_ значения средней арифметической с и средней
квадратичной Vc* скоростей.
На распределение молекулярных скоростей большое влияние
оказывает температура. Наивероятнейшая скорость движения мо-
молекул, как это ясно из уравнения 51, пропорциональна корню квад-
квадратному из абсолютной температуры. Следовательно, при повы-
повышении температуры максимум на кривой распределения смешается
вдоль оси абсцисс вправо. Поскольку же площадь, ограниченная
кривой распределения и осью абсцисс, должна при этом оставать-
оставаться неизменной, то очевидно, что величина ординаты, соответствую-
соответствующей максимуму, уменьшается—кривая делается более пологой.
47
На рисунке 16 сплошная кривая соответствует абсолютной темпе-
температуре 273°, а пунктирная — абсолютной температуре 373°.
Изменение кривой распределения молекулярных скоростей при
изменении температуры объясняет влияние температуры на многие
физико-химические процессы. Особенно велико влияние темпера-
температуры на те процессы, при которых определяющим является коли-
количество молекул со скоростями, превосходящими некоторую опре-
С (м/сек)
1000
1300
Рис. 16. Изменение распределения молекулярных
скоростей при изменении температуры в кислороде
(по оси ординат отложена величина
iV-Дс
деленную величину. Как явствует из графика, число подобных мо-
молекул быстро возрастает с ростом температуры. Именно это возрас-
возрастание объясняет во многих случаях ускорение химических реак-
реакций при нагревании реагирующей смеси.
13. ЭКСПЕРИМЕНТАЛЬНАЯ ПРОВЕРКА ЗАКОНА РАСПРЕДЕЛЕНИЯ
МАКСВЕЛЛА
Экспериментально закон распределения молекулярных скоро-
скоростей был проверен Штерном. Прибор, предназначенный для
этой цели, состоял из двух жестко соединенных между собой коак-
коаксиальных цилиндров (рис. 17). Вдоль оси цилиндров натягивалась
покрытая серебром платиновая проволока. Внутренний цилиндр
имел узкую щель, расположенную вдоль образующей цилиндра.
Пространство внутри цилиндров откачивалось до высокого ваку-
вакуума. По платиновой проволоке пропускался электрический ток,
в результате чего проволока раскалялась. При этом с ее поверх-
поверхности испарялись атомы серебра, которые, не встречая на своем
пути никаких частиц, двигались прямолинейно, пока не ударя-
48
лись о стенки внутреннего цилиндра и не
осаждались на них, образуя налет серебра.
В том месте, где помещалась щель, атомы
серебра проникали в пространство между
цилиндрами и осаждались на внутренней
поверхности внешнего цилиндра.
Таким образом, возникало как бы изоб-
ражениещели, подобное изображениям, по-
получаемым в малярных работах при помощи
трафарета. Описанный прибор приводил- Рис_ 17 Схема опыта
ся во вращательное движение вокруг оси Штерна,
цилиндров с угловой скоростью со.
Во вращающемся приборе серебряный налет оказывался распо-
расположенным не против щели, а несколько смещенным в сторону. Это
смещение возникало в результате того, что за время tit в течение
которого прямолинейно движущиеся атомы серебра перемещались
от щели до стенки наружного цилиндра, прибор успевал повер-
повернуться на угол ср! = (о^. Если бы все атомы двигались с одина-
одинаковой скоростью с1(то изображение щели смещалось бы, не изме-
изменяя своей формы, на некоторое расстояние Asi, измеряя которое,
можно было бы определить скорость движения атомов.
Действительно, если радиусы внешнего и внутреннего цилин-
цилиндров соответственно равны R и г, то
Величина смещения изображения щели:
или
д _ aR(R-r)
с, •
откуда
wtf (R — г)
C==
На опыте наблюдается не только смещение изображения щели,
но и расплывание его, обусловленное различием скоростей движе-
движения атомов серебра. Атомы, движущиеся быстро, смещаются мень-
меньше, чем атомы, движущиеся медленно. В таком простом оформле-
оформлении опыта можно было определить только смещение для середины
изображения щели и вычислить среднюю скорость движения ато-
атомов. Результат проверки удовлетворительно согласовывался с вы-
выводами кинетической теории газов.
В последующие годы опыты Штерна многократно повторялись
и усовершенствовались. В наиболее точных измерениях использова-
49
Рис. 18. Опытное исследование рас-
распределения молекул по скоростям.
лось наблюдение изменения под
действием силы тяжести траекто-
траектории свободно летящих в гори-
горизонтальном направлении ато-
атомов.
В специальной электрической
печи, помещенной в хорошо эва-
эвакуированный сосуд (остаточное
давление 1СИ мм рт. ст., или
1,33- КНя/л2),нагревался цезий.
Струя паров цезия проходила
через отверстие О (рис. 18). С по-
помощью щели в экранеS из струи
паров вырезался узкий парал-
параллельный пучок частиц, направ-
направлявшийся на расположенную горизонтально накаленную вольфра-
вольфрамовую проволочку D. При попадании на раскаленную вольфра-
вольфрамовую проволочку атомы цезия вновь испарялись, но теперь, в
силу высокой температуры, в виде ионов. Таким образом, воз-
возникали электрические заряды, количество которых было равно ко-
количеству атомов, попавших на проволочку. Возникшие ионы со-
собирались с помощью заряженного отрицательного электрода. По
величине ионизационного тока можно было определить количество
атомов цезия, попадавших на проволочку за 1 секунду.
В отсутствие силы тяжести траектории атомов были бы прямо-
молинейны. В действительности они являлись параболами, тем бо-
более отличающимися от прямой, чем медленнее движется атом. Зная
то место {D',D") на плоскости РР, в которое попадает движущий-
движущийся атом, можно рассчитать его траекторию и вычислить ско-
скорость. Перемещая вольфрамовую проволочку в вертикальном нап-
направлении, удалось определить число атомов, соответствующее
различным траекториям, а следовательно, и скоростям движе-
движения атомов,и проверить за-
закон Максвелла.
Результаты этих опытов /
изображены на рисунке 19.
Вдоль оси ординат отложе-
отложены значения ионизационно-
ионизационного тока /, вдоль оси абс-
абсцисс — расстояния d про-
проволочки по вертикали от
точки D. Сплошная кривая
рассчитана теоретически, в
предположении, что скорос-
скорости движения атомов подчи-
подчиняются закону Максвелла.
Точки на графике получе-
Рис. 19. Сопоставление опытных данных о
распределении молекул по скоростям с за-
законом Максвелла.
50
ны экспериментально. Следует отметить хорошее совпадение тео-
теоретически рассчитанной кривой с данными опыта.
14. ЧИСЛО СОУДАРЕНИЙ И ДЛИНА СВОБОДНОГО ПРОБЕГА
МОЛЕКУЛЫ
Большие скорости движения молекул, которые не только
следуют из теоретических соображений, но и подтверждаются
специально поставленными опытами, заставляют задуматься о при-
причинах малых скоростей таких процессов, в которых, как казалось
бы, и наблюдается непосредственно движение молекул, например
процесс распространения пахучего вещества в воздухе. Это ка-
кажущееся противоречие объясняется тем, что каждая молекула при
своем движении претерпевает очень большое количество соударе-
соударений с другими молекулами, в результате чего ее траектория при-
приобретает характерный вид изломанной линии (рис. 20).
Прямолинейные участки траектории /1( /2, /3 1п> проходимые
молекулой между двумя последовательными соударениями, назы-
называются свободными пробегами молекулы. Средняя ариф-
арифметическая величина свободных пробегов называется средним
свободным пробегом /:
E3)
где п — число соударений.
Число свободных пробегов за какой-либо промежуток времени
совпадает с числом соударений молекулы за то же время. Если за
одну секунду молекула испытала z соударений, то длина ее тра-
траектории, численно равная средней скорости ее движения, будет со-
состоять из г свободных пробегов. Числа соударений, испытываемых мо-
молекулой за 1 секунду, могут быть раз-
различными для различных молекул, одна-
однако можно ввести представление о сред-
среднем числе соударений г, численно рав-
равном отношению средней скорости дви-
движения молекулы с к среднему свобод-
свободному пробегу /:
Т
E4)
От величины среднего свободно-
свободного пробега зависят многие свойства
газов.
Рис. 20. Траектория движения
молекулы.
51
Для вычисления среднего свободного пробега молекулы пред-
предположим, что все молекулы газа, за исключением одной, неподвиж-
неподвижны и распределены равномерно по всему объему. Будем считать
далее, что скорость движущейся молекулы совпадает со средней
скоростью с молекулярного движения для данного газа. Двигаясь,
молекула соударяется с другими молекулами всякий раз, когда
она приближается к ним настолько, что расстояние между их цент-
центрами делается равным молекулярному диаметру ст.
Опишем вокруг движущейся молекулы сферу радиусом, рав-
равным молекулярному диаметру, и назовем ее сферой огра-
ограждения молекулы. Всякий раз, когда движущаяся молекула
сближается с какой-либо другой молекулой настолько, что центр
последней оказывается на поверхности сферы ограждения, проис-
происходит соударение молекул. При движении молекулы сфера огра-
ограждения вырезает в пространстве цилиндр с основанием па2. Если
молекула движется в течение 1 секунды, то высота этого цилиндра
равна средней скорости движения молекулы с, а объем, вырезан-
вырезанный сферой ограждения, составляет V = па2с.
Очевидно, что соударения будут происходить всякий раз, когда
центр встречной молекулы будет находиться внутри цилиндра, вы-
вырезанного сферой ограждения. Следовательно, для определения
среднего числа соударений z достаточно подсчитать число молекул
газа, центры которых находятся внутри указанного цилиндра. Это
число равно произведению объема цилиндра V на количество мо-
молекул газа в единице объема п0. Таким образом, среднее число со-
соударений молекулы за одну секунду будет равно:
г— п0ло2с. E5)
При получении этого соотношения все молекулы газа, кроме од-
одной, считались неподвижными. Более строгая теория показывает,
что при учете движения всех молекул и при условии, что скорости
молекулярного движения распределены согласно закону Макс-
Максвелла, среднее число соударений молекулы за 1 секунду будет не-
несколько больше и может быть подсчитано по уравнению:
г = V~2n0no2c. E6)
Пример. Подсчитать среднее число соударений за 1 сек, молекулы кисло-
кислорода при нормальном атмосферном давлении и температуре 0° С.
Решение.
Диаметр молекулы кислорода о* = 2,9 • Ю0 м. При нормальных усло-
условиях одна килограмм-молекула кислорода занимает объем 22,4 м3 • В 1 м3
6,02 • 1026
находится ———-— молекул. Средняя арифметическая скорость движения
молекулы кислорода при 0° С:
Г273
;=145|/,
-ZL = 423 м/сек.
32
52
Среднее число соударений молекулы кислорода за 1 сек при этих услови-
условиях равно:
с no, in26
г = Y 2 • 3,14 B,9.10-ю)з.423 = 4,29.10".
Зная среднее число соударений молекулы, можно определить
средний свободный пробег молекулы:
^E7)
г У 2 п0яо2
Пример. Подсчитать средний свободный пробег молекулы азота, если
газ находится при нормальных условиях. Диаметр молекулы азога
З.ЫО0 м.
Решение. Число молекул в 1 ж3:
6,02.1026
«0 =
22,4
Средний свободный пробег молекулы азота:
Г= ,-6,02.10™ =8,7.10-»*.
"/2— 3,Н.C,Ы0-1°J
Заменяя в уравнении Клапейрона универсальную газовую по-
постоянную R произведением числа Авогадро NА на постоянную Больц-
мана k, можно написать:
pV = NAkT,
или
Но отношение — равно числу молекул в одном кубическом метре
п0 и, следовательно,
р = nokT. E8)
Определяя из этого уравнения п0 и подставляя это значение в урав-
уравнение E7), получим:
7^—
V2 яа2р
E9)
Таким образом, при постоянной температуре средний свобод-
свободный пробег молекулы обратно пропорционален давлению.
При постоянной молекулярной плотности, или, что то же са-
самое, при постоянном объеме газа, средний свободный пробег моле-
молекулы не должен зависеть от температуры.
Независимость при постоянной молекулярной плотности сред-
среднего свободного пробега молекул от температуры является резуль-
53
татом того, что во всех предыдущих рассуждениях не принималось
во внимание взаимодействие между молекулами. Учет молекуляр-
молекулярных взаимодействий приводит к заключению, что при возрастании
температуры средний свободный пробег молекул должен несколь-
несколько увеличиваться. Если обозначить величину среднего свободного
пробега молекул, рассчитанного без учета молекулярного взаимо-
взаимодействия, и, следовательно, независящую от температуры /со, то ре-
реальная величина среднего свободного пробега 1Т при температуре
Т может быть подсчитана с помощью уравнения:
?='-гТс' (б0)
где С — постоянная Сезерленда, зависящая от интенсив-
интенсивности молекулярных взаимодействий. Следует, однако, заметить,
что учет взаимодействия молекул, проявляющегося на расстояниях,
больших по сравнению с молекулярными диаметрами, в значитель-
значительной степени лишает понятие свободного пробега молекул той оп-
определенности и ясности, которые без этого ему присущи. Действи-
Действительно, при наличии взаимодействия на значительных расстояниях
траектории сближающихся молекул будут плавно изменяться,
благодаря чему границы свободного пробега делаются неопреде-
неопределенными.
15. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ ДЛИНЫ СРЕДНЕГО
СВОБОДНОГО ПРОБЕГА МОЛЕКУЛ
Для проверки правильности теоретического расчета длины сред-
среднего свободного пробега молекул используются молекулярные
пучки, подобныетем, о которых говорилось в опытах Штерна (§13).
В предназначенном для этой цели приборе (рис. 21)
создается столь высокий вакуум, что молекулы испа-
испаряющегося вещества А вылетая в виде пучка, через
отверстие О в экране, установленном на их пути,
движутся без соударений до противоположной стен-
стенки прибора, на которой и оседают, создавая налет,
воспроизводящий по форме отверстие.
На расположенных слегка в стороне от траекто-
S,
не возникает,
молекулы на
Рис. 21.
Определение
среднего
свободного
пробега
молекул.
рии пучка экранах Su S2, S3 налета
поскольку прямолинейно движущиеся
них не попадают.
Если ввести в сосуд некоторое количество молекул
газа, то траектория частиц, образующих молекуляр-
молекулярный пучок, будет изменяться в результате соударе-
соударений с молекулами введенного газа.
Это сказывается в появлении налета на боковых
экранах. Чем больше давление газа, тем короче дли-
длина среднего свободного пробега молекул и тем ближе
54
к отверстию в экране возникает налет частиц, образующих молеку-
молекулярный пучок. Изменяя давление газа, можно оценить длину среднего
свободного пробега и проверить выводы молекулярно-кинетической
теории. Подобные опыты подтвердили удовлетворительное согла-
согласие теории с опытом.
16. ЯВЛЕНИЯ ПЕРЕНОСА. ВЯЗКОСТЬ ГАЗОВ
Представление о среднем свободном пробеге молекул играет
большую роль при молекулярно-кинетическом объяснении меха-
механизма многих физических явлений. Особенно важно это физическое
понятие для раскрытия сущности таких явлений, как внутрен-
внутреннее трение, или вязкость, теплопроводность
и диффузия газов.
Как будет показано ниже, все эти три явления связаны с про-
процессами переноса. В случае вязкости — с переносом количе-
количества движения, в случае теплопроводности — кинети-
кинетической энергии, в случае диффузии — м а с с ы мо-
молекул, обладающих тем или иным физическим или химическим
качеством.
В силу общности молекулярного механизма этих явлении их
объединяют в одну группу, называя явлениями переноса.
Рассмотрим молекулярный механизм вязкости газов. Предпо-
Предположим, что в газе (рис. 22) параллельно неподвижной пластинке аа
движется с постоянной '
скоростью расположенная А
выше нее пластинка ЪЬ.
В результате движения
верхней пластинки прихо-
приходят в движение слои газа,
находящиеся между ней и
нижней пластинкой. Ско-
Скорость этого упорядоченно-
упорядоченного движения газа убывает
по мере удаления от верх-
верхней, т. е. движущейся, у
пластинки. Опыт показы- /
вает, что для равномерно- "у
го движения пластинки к Рис 22.
ней должна быть приложе-
приложена некоторая сила F, называемая силой вязкости. Согласно ^за-
^закону, открытому эмпирически Ньютоном, сила вязкости, дейст-
действующая на пластинку с поверхностью S, может быть подсчитана
по уравнению:
F = i\S~, F1)
Дг
в котором градиент скорости ^ соответствует измене-
55
нию скорости v упорядоченного движения газа на расстоянии в
единицу длины в направлении, перпендикулярном направлению
движения газа, а коэффициент ц, называемый коэ ффициен-
том вязкости, или коэффициентом внутрен-
внутреннего трения зависит от свойств газа.
Для выяснения механизма возникновения вязкости выделим в
газе мысленно площадку в 1 м2, параллельную пластинкам аа и bb
и расположенную между ними.
В результате беспорядочного теплового движения молекул че-
через выделенную площадку за 1 секунду будет проходить в направ-
направлении сверху вниз некоторое количество молекул п.
Поскольку плотность газа остается неизменной, то очевидно,
что этот переход компенсируется встречным переходом такого же
количества молекул газа снизу вверх. В то же время молекулы,
проходящие через площадку в направлении сверху вниз, движут-
движутся упорядоченно слева направо со скоростью vt 4- Д^ (смотрите
рисунок 23) большей, чем скорость упорядоченного движения
yt — Д^1 молекул встречного потока, направленного снизу
вверх.
Если обозначить массу молекулы т, то молекулы, движущиеся
сверху вниз, пронесут за 1 секунду через рассматриваемую площад-
площадку количество движения пт (vi -\- Aui), обусловленное упорядочен-
упорядоченным движением газа, а молекулы, движущиеся навстречу, — ко-
количество движения пт (vi — Afj).
Таким образом, в выделенной площадке за одну секунду будет
происходить изменение количества движения, равное разности
пт (ух -\- Аух) — пт (ух — Аих) = 2пт Avv
Изменение количества движения за 1 секунду численно равно дей-
действующей силе. В данном случае это будет сила вязкости. Таким
образом, сила вязкости возникает как следствие перехода молекул
при их беспорядочном тепловом движении из слоев газа, движущих-
движущихся медленно, в слои газа, движущиеся быстро.
Для уяснения механизма этого явления можно представить се-
себе два расположенных рядом, нагруженных зерном транспортера,
движущихся в одну сторону с различными скоростями, снабжен-
снабженных специальным механизмом, перебрасывающим непрерывно ка-
какое-то количество зерна с быстрого транспортера на медленный и
равное ему количество зерна во встречном направлении. Первый
поток зерна, ударяясь о медленный транспортер и отдавая прине-
принесенное избыточное количество движения, увеличивает скорость
движения медленного транспортера, в то время как встречный по-
поток тормозит движение быстрого транспортера. Результат подоб-
подобного переноса будет таким же, как если бы между транспортерами
существовало трение, обусловленное механическим зацеплением
неровностей на трущихся поверхностях.
56
Для теоретического вычисления коэффициента вязкости сделаем
следующие предположения: 1) поскольку движение молекул хао-
хаотично и все направления движения равновероятны, будем считать,
что в выбранной нами (см. рис. 23) системе прямоугольных коор-
координат одна треть молекул движется вдоль оси х, одна треть — вдоль
оси у и одна треть — вдоль оси г; 2) все молекулы движутся с од-
одной и той же скоростью, равной средней скорости движения мо-
молекул с, и имеют один и тот
же свободный пробег, равный
среднему свободному пробе-
пробегу I.
Сила трения, действую-
действующая на площадку S ~ \ м2,
равна изменению количества
движения, происходящему на
этой площадке за 1 секун-
секунду, т. е.
F = 2nmAv1. F2)
Число молекул п, прохо-
проходящих через площадку в ка-
каком-либо одном направлении
за 1 секунду, может быть
подсчитано следующим обра-
образом: построим прямой парал-
параллелепипед с основанием S =
= 1 ж2 (рис. 23) и высотой,
равной средней скорости дви-
движения молекул с. Очевидно,
что молекулы, расположенные на верхнем основании параллеле-
параллелепипеда и движущиеся вниз, за 1 секунду как раз достигнут нижне-
нижнего основания. Таким образом, все молекулы, находящиеся в этом
параллелепипеде и движущиеся вниз, за 1 секунду пройдут че-
через выделенную нами площадку.
Если в каждом кубическом метре газа содержится п0 молекул,
то в рассматриваемом параллелепипеде их будет пос. Из этого числа
вдоль оси координат, совпадающей с высотой параллелепипеда,
движется одна треть молекул, т. е. —. Можно считать, что из это-
О
го числа половина молекул движется снизу вверх, а половина —
сверху вниз, и, следовательно, искомое число молекул п будет рав-
равно М .
6
Для определения разности скоростей упорядоченного движе-
движения молекул Auj обратимся к чертежу на рисунке 23. Из рассмот-
Рис. 23. Расчет коэффициента вязкости
газов.
57
рения подобных треугольников abc и ABC следует, что величина
Ai>i равна произведению
~ • ас.
Здесь ас — то расстояние, с которого молекулы без соударения до-
доходят до площадки S. Поскольку выше было предположено, что
все молекулы имеют один и тот же свободный пробег /, то очевидно,
что именно с этого расстояния и будут доходить молекулы до пло-
площадки 5 без соударений, т. е.
Аг
Подставляя значения п и Aci в выражение для силы вязкости
получим:
kv-T. F3)
Сравнивая полученное уравнение F3) с эмпирическим законом
Ньютона F1), можно написать:
г) = - потсТ.
О
Произведение пот равно плотности газа р, и, следовательно, для
коэффициента вязкости справедливо выражение1:
Л = {рсЛ F4)
Средний свободный пробег / обратно пропорционален давле-
давлению, а плотность газа р прямо пропорциональна давлению, поэто-
поэтому произведение р/ не зависит от давления. Не зависит от дав-
давления и средняя скорость движения молекул. Очевидно, не дол-
должен зависеть от давления и коэффициент вязкости г\.
Таблица 5, в которой приведены значения коэффициента вяз-
вязкости для углекислоты СО2 при разных давлениях, убеждает в том,
что теория удовлетворительно согласуется с опытом.
Независимость коэффициента вязкости от давления является
следствием особенностей механизма этого явления. Действительно,
уменьшение давления газа в два раза уменьшает вдЕое число моле-
молекул, проходящих за 1 секунду через рассматриваемую площадку,
однако одновременно это изменение давления в два раза увеличи-
увеличивает расстояние, на котором происходит последнее соударение мо-
молекул перед попаданием их на площадку, а следовательно, в два
раза увеличивает разность скоростей упорядоченного движения.
1 Хотя при выводе уравнения F4) были сделаны заведомо неточные пред-
предположения, однако полученный результат совпадает с тем, к которому при-
приводят более строгие рассуждения.
58
В результате, как это само собой очевидно, изменение количества
движения оказывается независящим от давления.
Вторая особенность вязкости газов, также объясняемая моле-
кулярно-кинетической теорией этого явления, заключается в уве-
увеличении вязкости с ростом температуры, в том случае, если нагре-
нагревание производить при постоянной плотности. При неизменной
плотности изменение температуры не влияет на средний свободный
Таблица 5
Вязкость углекислоты при
разных давлениях
Таблица 6
Зависимость вязкости
воздуха от температуры
Давление мм
рт. ст.
760
380
20
2
0,6
Коэфф. вязко-
вязкости н-сек/м2
14,9.10-»
14,9 »
14,8»
14,7 »
13,8 »
Температура
в ° С
—104,0
— 31,6
± 0,0
54
229
357
Коэфф
сти «•
11,3
15,3
17,1
19,6
26,3
31,5
вязко-
сек/м2
• ю-6
»
»
»
»
»
пробег, так что изменение коэффициента вязкости обусловлено
только изменением средней скорости молекулярного движения,
возрастающей с ростом температуры. Это на первый взгляд неожи-
неожиданное заключение хорошо согласуется с опытом, в чем убеждают
данные, приведенные в таблице 6.
17. ТЕПЛОПРОВОДНОСТЬ ГАЗОВ
При соприкосновении двух газов, имеющих разную температуру,
газ с более высокой температурой охлаждается, а газ с более низкой
температурой нагревается. Обычно основную роль в выравнивании
температуры играет перемешивание газов, или кон-
конвекция, обусловленная возникновением в газе потоков, имею-
имеющих разную плотность.
Можно, однако, осуществить опыт таким образом, что в газе
будет отсутствовать конвекция, но в то же время будет существо-
существовать постоянный градиент температуры, обусловливающий посто-
постоянный поток теплоты.
В этом случае перенос теплоты осуществляется в результате
беспорядочного теплового движения молекул, которые, проходя
через мысленно выделенную площадку 5 (рис. 24), переносят в од-
одном направлении в среднем большее количество кинетической энер-
59
гии, чем молекулы, движущиеся им навстречу. Подобный механизм
передачи теплоты называют теплопроводностью.
Подсчитаем количество теплоты AQ, перенесенное в силу теп-
теплопроводности через мысленно выделенную в газе площадку, име-
имеющую площадь S = 1 ж2, за 1 секунду, если градиент температуры
Л Т*
равен—. Расчет основан на тех же предположениях, которые бы-
Дг
ли сделаны при вычислении коэффициента вязкости газа.
Если число молекул газа в одном кубическом метре п0 и средняя
скорость молекулярного движения с, то количество молекул, про-
прошедших за 1 секунду через рассматриваемую площадку в одном
Рис. 24. Расчет коэффициента теплопроводности газов.
из двух встречных направлений, будет, как это показано в преды-
предыдущем параграфе,—. Строго говоря, средние скорости молекуляр-
6
ного движения для слоев газа с разной температурой будут слегка
различаться, однако это различие компенсируется сопутствующим
ему различием в количестве молекул п0 в одном кубическом метре
газа.
Обозначая массу молекулы т, найдем, что масса газа, перене-
перенесенная за 1 секунду через площадку 5 = 1 л*2, будет равна —2-, или
6
— , где р — плотность газа.
6
Последнее соударение молекулы перед прохождением через
площадку будет происходить, как и в случае вязкости газа, на рас-
расстоянии среднего свободного пробега /, так что если температура
газа в том слое, где находится площадка, равна 7\, то температура
60
более нагретого газа, поступающего на площадку, будет Tt-\-— /,
Дг
Л Т
а более холодного газа Г t 7.
Дг
Обозначив удельную теплоемкость газа Cv, можно для количе-
количества теплоты AQb перенесенного в результате беспорядочного теп-
теплового движения молекул за 1 секунду через площадку S в одном
направлении, написать выражение:
д<21 сJt.+
1 6 v \ J ' Дг
а для количества теплоты AQ2. перенесенного во встречном направ-
направлении, выражение:
- 6 С^/х Дг/ J.
Таким образом, в результате беспорядочного теплового дви-
движения молекул возникает непрерывный поток теплоты через рас-
рассматриваемую площадку. За 1 секунду через площадку S = 1 м2
проходит количество теплоты AQ, равное:
AQ = AQ, - AQ2 = f Cv- 2 ^7. F5)
Еще до возникновения молекулярно-кинетической трактовки
явления теплопроводности французский математик и физик Ф у-
р ь е A768—1830) нашел, что количество теплоты, проходящее че-
через площадку в 1 мг за 1 секунду, пропорционально градиенту тем-
температуры, т. е.
Коэффициент пропорциональности К называют коэффи-
коэффициентом теплопроводности. Сравнивая уравнения
F5) и F6), можно расшифровать значение коэффициента теплопро-
теплопроводности газа
/С = 1рсСД F7)
О
Коэффициент теплопроводности связан простой зависимостью с
коэффициентом вязкости. Из уравнений F4) и F7) следует, что
К = Т1С„.
В такой простой форме это соотношение плохо согласуется с опы-
опытом. Однако более строгие рассуждения показывают, что выводы
молекулярно-кинетической теории правильно описывают наблю-
наблюдаемое соотношение коэффициентов вязкости и теплопроводности.
61
Анализ уравнения F7) приводит к заключению, что коэффици-
коэффициент теплопроводности газа не должен зависеть от давления. Экс-
Экспериментальная проверка подтверждает этот вывод. Коэффициент
теплопроводности сохраняет постоянное значение вплоть до весь-
весьма малых давлений, при которых уже основные положения теории
оказываются неприменимыми, и величина К начинает изменяться
при изменении давления.
18. ДИФФУЗИЯ ГАЗОВ
Одним из наиболее наглядных примеров явлений переноса слу-
служит диффузия газов. Диффузией газов называют процесс
взаимного проникновения двух соприкасающихся газов, обуслов-
обусловленный тепловым движением молекул.
Если процесс диффузии происходит достаточно длительное вре-
время, то устанавливается стационарное состояние, при котором кон-
концентрация [с], т. е. количество одного из диффундирующих газов в
единице объема, плавно изменяется вдоль направления, в котором
происходит диффузия. В газе возникает постоянный градиент
концентрации, равный -Ю- (рис. 25) и аналогичный гра-
Дг
диенту скорости упорядоченного движения — или градиенту тем-
Аг
AT
пературы , которые приходилось учитывать при рассмотрении
Az
вязкости и теплопроводности газов.
Может показаться, что для объяснения особенностей процесса
диффузии газов достаточно механически повторить рассуждения,
использованные при рассмотрении вязкости и теплопроводности.
Однако это неверно. В явлении вязкости и теплопроводности при-
приходилось иметь дело с градиентом какого-либо свойства молекул,
в то время как сами молекулы оставались идентичными. В случае
же диффузии двух различных газов молекулы их различаются мас-
массой, скоростью движения, размерами, свободным пробегом и т. д.
Эти особенности диффузии не позволяют в этом случае перенести
без изменений схему рассуждений, использованных в предыдущих
параграфах.
Можно рассмотреть, однако, частный случай диффузии, при ко-
котором эти возражения отпадают, это случай самодиффузии,
т.е. диффузионного распространения газа в самом себе. На практике
самодиффузию можно реализовать, воспользовавшись радиоак-
радиоактивными изотопами какого-либо элемента и превратив с их помо-
помощью часть молекул соответствующего газа в меченые молекулы.
Все физические свойства меченых и немеченых молекул, за исклю-
исключением способности первых к радиоактивному распаду, будут оди-
одинаковы. Это сильно облегчит рассмотрение диффузии.
62
Число молекул, проходящих сверху вниз (рис. 25) через пло-
площадку в 1 м2 за 1 секунду, как и в случае вязкости, равно—, где
б
по — число молекул в 1 м3, а с — средняя скорость молекулярно-
молекулярного движения. Последнее соударение перед попаданием на рассмат-
рассматриваемую площадку, определяющее перемешивание и, следователь-
следовательно, состав поступающего на площадку газа, молекулы испытыва-
испытывают на расстоянии среднего свободного пробега / от площадки. Обоз-
Обозначим долю меченых молекул в слое газа, совпадающем с площад-
площадкой, через [Со ], количество по-
подобных молекул в 1 м3 будет
равно по[с'о ]. При принятых
обозначениях число меченых мо-
молекул на расстоянии / с од-
одной стороны площадки будет
«о
Дг
/ }, а с другой
стороны ее — пи , ^
Таким образом, за 1 секунду
через рассматриваемую площад-
площадку в одном направлении прой-
пройдет в результате беспорядочно-
беспорядочного движения количество мече-
меченых молекул:
а во встречном направлении —
количество меченых молекул:
Рис. 25. Расчет коэффициента
диффузии газов.
В результате того, что количества меченых молекул, проходя-
проходящих через площадку во встречных направлениях неодинаковы,
возникает поток подобных молекул, при котором за 1 секунду через
площадку в 1 м2 проходит число меченых молекул Дл, определяе-
определяемое соотношением:
An = Д«! — Ап2 =
пос~Т
F8)
3 Дг
Если умножить обе части написанного равенства на массу мо-
молекулы т, то произведение тАп = AM будет равно числу кило-
килограмм меченого газа, прошедшего за одну секунду через рассмат-
рассматриваемую площадку.
63
Стоящее в правой части уравнения выражение тпй&[с'о ] можно
тогда записать, перенося знак А, так: Атпо[с'0 }. Произведение
по[с\\ равно числу меченых молекул в 1 м3; умножение этого числа
на массу молекулы, т. е. тпо[со'], дает концентрацию радиоактивно-
радиоактивного газа [с']. Таким образом, мы приходим к заключению, что выра-
ктпо-[с'о J
жение !L_равно градиенту концентрации радиоактивного газа
Дг
—i^J . Вводя эти обозначения, можно переписать уравнение F8) в
Дг
следующем виде:
-сТ^^. F9)
Согласно найденному эмпирически закону (закон Фика) коли-
количество вещества ДМ, диффундирующего за 1 секунду через пло-
площадку в 1 л2, пропорционально градиенту концентрации, т. е.
АМ= D ^. G0)
Дг
Входящий в это уравнение коэффициент D называют коэффи-
коэффициентом диффузии. В случае самодиффузии коэффи-
коэффициент D определяется уравнением:
D = jc/~ G1)
При постоянной плотности газа средний свободный пробег / моле-
молекул не зависит от температуры, а средняя скорость движения мо-
молекул с пропорциональна корню квадратному из абсолютной тем-
температуры. Поэтому коэффициент диффузии, а следовательно, и ско-
скорость диффузии при постоянной плотности возрастают пропорцио-
пропорционально VT. При постоянной температуре, но изменяющемся дав-
давлении средняя скорость молекулярного движения остается постоян-
постоянной, в то время как средний свободный пробег изменяется обратно
пропорционально давлению. В силу этого коэффициент диффузии
при постоянной температуре изменяется обратно пропорционально
давлению.
Все три коэффициента: вязкости, теплопроводности и диффузии
(уравнения 64, 67 и 71) — зависят от величины среднего свободного
пробега молекул. Поэтому, определив экспериментально какой-либо
из этих коэффициентов, можно рассчитать величину среднего сво-
свободного пробега молекулы. Зная же средний свободный пробег мо-
молекулы и соответствующее значение плотности газа, можно, вос-
воспользовавшись уравнением E7), вычислить диаметр молекулы газа.
Этим методом были определены диаметры многих молекул.
Найденные этим методом величины называют газокинетическими
диаметрами молекул. В таблице 7 сопоставлены некоторые вели-
64
чины, характеризующие молекулы газов, рассчитанные на основа-
основании кинетической теории газов.
Таблица 7
Молекулярно-кинетические свойства газов при температуре 20° С
и давлении 1,01 • I05«/jh2
Газ
Водород
Гелий
Азот
Кислород
Аргон
Углекис-
Углекислота
Молекуляр-
Молекулярный вес
2,016
4,003
28,016
32,000
39,944
44,010
Средняя
арифметиче-
арифметическая ско-
скорость
в м /сек
1756,7
1246,6
471,25
440,94
394,66
375,99
/-i^tilitlln С мед
него свобод-
свободного пробега
в м
1116- Ю-1"
1753 »
592 »
534 »
622 »
389 »
Диаметр
молекулы
в м
2,40-Ю-10
1,90 »
3,15 »
2,98 »
2,88 г
3,34 »
Среднее чис-
число соударе-
соударений молеку-
молекулы
в 1 сек
15,74-10''
7,14 »
7,97 »
6,96 i>
6,35 »
9,67 »
Рассуждения, использованные при обсуждении механизма явле-
явлений переноса, оправдываются только в том случае, если давление
газа не понижается ниже некоторой определенной при данных кон-
конкретных условиях величины. Например, коэффициент вязкости газа
можно рассчитывать, пользуясь полученными выше формулами,
только в том случае, если расстояние между движущейся и непод-
неподвижной пластинками значительно превосходит средний свободный
пробег молекул газа. Уменьшение этого расстояния до величины
свободного пробега молекулы, или даже меньшей, приведет к тому,
что молекулы, приобретая некоторое количество движения при
ударе об одну из пластинок, будут передавать его, не испытывая
соударений с другими молекулами, непосредственно второй плас-
пластинке. Количество движения, переданное при этих условиях мо-
молекулами газа за 1 секунду, будет пропорционально числу моле-
молекул в единице объема. При этих условиях уменьшение давления
должно уменьшать коэффициент вязкости, который в предыдущих
рассуждениях, когда расстояние между пластинками было значи-
значительным, не зависел от давления.
При не слишком низких давлениях измеренный на опыте коэф-
коэффициент теплопроводности в согласии с кинетическои"теорией газов
не зависит от давления газа. Когда изучается теплопроводность
газа, следует иметь в виду, что обычно передача теплоты осущест-
осуществляется газами, заключенными между двумя стенками, имеющими
разные температуры. Понижая давление газа, можно настолько уве-
увеличить свободный пробег молекул, что они будут пробегать путь
от одной стенки до другой, не испытывая на этом пути соударений.
При этих условиях полученное выше выражение для коэффициен-
коэффициента теплопроводности оказывается неверным. Ударяясь о более нагре-
3 Заказ 145
65
тую стенку, молекулы приобретают большую по сравнению со сред-
средней кинетическую энергию, которую и передают непосредственно
более холодной стенке. Дальнейшее понижение давления приводит
к уменьшению числа молекул, осуществляющих этот процесс, и,
следовательно, к уменьшению коэффициента теплопроводности.
Это обстоятельство используется при устройстве так
называемых сосудов Дьюара, предназначенных для
хранения сжиженных газов.
Сосуды Дьюара (рис. 26) имеют двойные стенки,
в промежутке между которыми создан высокий ва-
вакуум. Благодаря этому теплопроводность сосудов
Дьюара ничтожно мала, передача теплоты в них
осуществляется в основном путем теплоизлучения,
для уменьшения которого стеклянные стенки сосудов
покрывают тонким зеркальным слоем серебра. Сосу-
Сосуды Дьюара небольших размеров используются при
Рис. 26. устройстве термосов, предназначенных для
Сосуд сохранения жидких пищевых продуктов в нагретом
ьюара. или охлаЖденном состоянии.
19. ВАКУУМ. ПОЛУЧЕНИЕ И ИЗМЕРЕНИЕ ВАКУУМА
Состояние разрежения газа, при котором молекулярные соуда-
соударения или вовсе отсутствуют, или же являются немногочисленными
по сравнению с числом соударений молекул со стенками сосуда,
называется вакуумом. Очевидно, что состояние вакуума бу-
будет достигаться в зависимости от размеров сосуда при раз-
разных давлениях. В обычных физических приборах вакуум дости-
достигается при давлениях порядка 10~3 — 10~4 мм рт. ст., или 0,133—
—0,013 н/м\
Однако в узких капиллярных трубках диаметром 0,1 — 0,01 мм
он достигается уже при давлениях порядка от 0,7 до 7 мм рт. ст.,
или 93,1 —931 н!м%. Получение высокого вакуума имеет большой
теоретический и практический интерес. Изучение поведения газов
в состоянии высокого разрежения позволяет лучше понять их свой-
свойства. Практическое значение высокого вакуума связано с тем, что
большинство электронных приборов — радиолампы, телевизионные
трубки и т. д. — требуют для нормальной работы создания в них
высокого вакуума.
Для получения высокого вакуума давление в откачиваемом со-
сосуде первоначально понижается с помощью так называемых насо-
насосов предварительного вакуума. В качестве насосов предваритель-
предварительного вакуума чаще всего используются ротационные мас-
масляные насосы. Насос состоит из металлического цилиндра /
(рис. 27), внутри которого вращается эксцентрически расположен-
расположенный цилиндрический ротор 2. В верхней части поверхность ротора
плотно прилегает к внутренней поверхности цилиндра (на рисунке
66
для наглядности между цилиндрами изображен зазор). В специаль-
специальную прорезь ротора входят лопасти 3, прижимаемые пружиной к
внутренней поверхности цилиндра.
При вращении ротора одна из лопастей, создавая разрежение,
засасывает газ из откачиваемого сосуда в полость насоса, после че-
чего вторая лопасть, захватив наполнившую насос порцию газа, вы-
выталкивает ее через выходное отверстие 4. Эту операцию лопасти
насоса проделывают поочередно. Выходное отверстие снабжено
клапаном 5, препятствующим обратному поступлению воздуха в
полость насоса. Для лучшей герметичности и смазки вращающихся
частей насос заключают в кожух, наполненный маслом. С помощью
ротационного масляного насоса можно создать разрежение, при ко-
Рис. 27. Устройство ротаци-
ротационного масляного насоса.
Рис. 28.
Устройство
молекулярного
насоса.
тором остаточное давление откачиваемого газа составляет около
0,01 мм рт. ст., или 1,33 н1м2 (отдельные образцы насоса могут обес-
обеспечивать остаточное давление до 0,005 мм рт. ст.).
Для получения более высокого вакуума применяют моле-
молекулярные насосы. В металлическом цилиндре молеку-
молекулярного насоса (рис. 28), с хорошо обработанной внутренней по-
поверхностью, быстро вращается второй цилиндр. За исключением
небольшого участка / — 2, зазор между обоими цилиндрами очень
мал (порядка 0,03 мм). Молекулы откачиваемого газа попадают че-
через входное отверстие / в расширенную часть зазора, ударяются о
быстро движущуюся поверхность внутреннего цилиндра, приобре-
приобретают дополнительную скорость в направлении вращения и увлека-
увлекаются к выходному отверстию 2. Насос работает производительно
только в том случае, если в сосуде, из которого откачивается газ,
предварительно уже создано разрежение (форвакуум). Молекуляр-
Молекулярный насос позволяет получить остаточное давление до 10""' мм рт. ст.,
или 1,33 • \0"° н/м2.
Наибольшее распространение для получения высокого вакуума
имеют различные пароструйные насосы, конструкции ко-
3*
67
Рис. 29, Устройство паро-
пароструйного насоса.
Рас. SO. Манометр
Мак-Леоаа.
торых весьма разнообразны. Схема одного из распространенных
ртутных пароструйных насосов изображена на рисунке 29. Нахо-
Находящаяся в сосуде / ртуть нагревается при помощи газовой горелки
или электрического нагревателя. Пары кипящей ртути вырываются
из сопла 2 и попадают в холодильник, охлаждаемый водой, проте-
протекающей по специальной рубашке 3. Трубка 4 соединяет насос с со-
сосудом, из которого откачивают газ и в котором при помощи рота-
ротационного масляного насоса уже создан предварительный вакуум.
Молекулы откачиваемого газа диффундируют в струю ртути, ме-
механически увлекаются ею, частично прилипают — адсорбируют-
адсорбируются — на поверхности мельчайших капелек ртути, вместе с ними по-
попадают в нижнюю часть трубки, выделяются вновь при слиянии ма-
маленьких капелек ртути в большие и откачиваются насосом предвари-
предварительного вакуума. Струя ртутных паров препятствует обратному
движению молекул газа, механически отбрасывая их.
Ртутные пароструйные насосы отличаются очень большой про-
производительностью. Некоторые из них могут откачивать газ со ско-
скоростью, измеряемой сотнями литров в секунду. Учитывая особен-
особенности механизма действия пароструйных насосов, их называют так-
также диффузионными насосами. Следует, однако, помнить
что механизм действия этих насосов сложен и не может быть объяс-
объяснен каким-то одним явлением. С помощью ртутного диффузион-
диффузионного насоса можно достигнуть вакуума порядка 10~7 мм рпг. ст.
В последнее время в пароструйных насосах вместо ртути с успе-
68
хом применяют органические жидкости с малой упругостью пара
(парафины, дибутилфталат и др.).
Для изменения высокого вакуума обычные манометры не при-
пригодны. Широкое применение для этой цели находит манометр
М а к-Л е о д а (рис. 30), действие которого основывается на уве-
увеличении измеряемого давления в результате уменьшения объема,
занимаемого некоторой порцией газа. Основная часть этого при-
прибора — стеклянный баллон 1, снабженный в верхней части узким
капилляром 2, а снизу — отводной трубкой, соединяющей манометр
с сосудом, в котором измеряют давление. Давления газа в баллоне /
и в сосуде, из которого откачивают газ, в момент измерения оди-
одинаковы. Для измерения давления поднимают вверх наполненный
ртутью сосуд 3, соединенный с баллоном / резиновой трубкой,
и отделяют с помощью ртути, поднимающейся вверх по отводной
трубке, то количество газа, которое при измеряемом давлении рх
занимало объем баллона У. Продолжая поднимать дальше сосуд 3,
сжимают отделенное таким образом количество газа, переводя
его в капилляр 2, до объема v. При изменении объема газа темпе-
температура его остается постоянной, а давление возрастает до величи-
величины р, которую можно измерить по разности уровней ртути в капил-
капиллярах 2 и 4. Согласно Закону Бойля —Мариотта можно написать:
РХУ = ро.
откуда следует:
Если объем баллона / много больше объема капилляра, то даже при
малом измеряемом давлении рх величина р будет достаточно велика
для того, чтобы ее измерить.
Малые давления измеряются еще при помощи так называе-
называемых радиационных манометров. В радиационных
манометрах используется зависимость теплопроводности разрежен-
разреженного газа от давления. В определенном интервале давлений теп-
теплопроводность газа изменяется при изменении давления прак-
практически линейно. В продолговатой стеклянной колбочке, соединен-
соединенной с сосудом, в котором измеряется давление, на специальной
ножке укрепляется тонкая платиновая нить, нагреваемая элект-
электрическим током. Температура нити зависит от соотношения меж-
между скоростью, с которой нить нагревается, и скоростью, с кото-
которой происходит ее охлаждение. Чем выше давление газа в кол-
колбочке, тем больше его теплопроводность и тем быстрее остывает
нить, а следовательно, тем ниже ее температура. Электрическое
сопротивление платиновой нити зависит от ее температуры, и
поэтому измерение температуры заменяют измерением сопротив-
сопротивления нити, которое легко выполнить весьма точно. Перед при-
применением радиационный манометр калибруют.
69
20. ВНУТРЕННЯЯ ЭНЕРГИЯ ИДЕАЛЬНОГО ГАЗА
Газ состоит из движущихся с различными скоростями молекул,
обладающих вследствие движения определенной кинетической энер-
энергией. Полная энергия, которой обладают в совокупности все моле-
молекулы газа, называется внутренней энергией газа.
В общем случае внутренняя энергия газа представляет сумму
всех видов энергии, которыми обладают молекулы, включая сюда
и потенциальную энергию молекулярных взаимодействий, если та-
таковые имеют место. В идеальном газе молекулярные взаимодействия
отсутствуют, и поэтому внутренняя энергия его равна суммарной
кинетической энергии молекул.
Выше (§ 8) было получено выражение для кинетической энергии
молекул, обусловленной их перемещением в пространстве. В слу-
случае молекул конечных размеров это движение соответствует по-
поступательному движению. Поступательное движение твердого тела
может быть разложено на три независимых движения, происходя-
происходящих вдоль трех независимых осей координат. По предложению
Максвелла эти независимые движения называют степенями
свободы движения молекул. Заметим, что число степеней сво-
свободы совпадает с числом независимых координат, которые необ-
необходимо ввести для того, чтобы определить положение тела, в данном
случае молекулы, в пространстве.
Любая молекула обладает тремя степенями свободы поступа-
поступательного движения.
В классической кинетической теории молекулы, состоящие из
одного атома, принимались за идеально гладкие твердые шарики,
у которых при соударениях вращательного движения возникнуть
не может. На этом основании предполагалось, что одноатомные мо-
молекулы не обладают степенями свободы вращательного движения.
Хотя в настоящее время представление об атомах сильно отлича-
отличается от этой простой модели, однако заключение о том, что при со-
соударениях атомов не возникает вращательного движения, сохра-
сохраняет свою силу.
Одноатомные молекулы обладают только тремя
степенями свободы поступательного движения.
У двухатомной молекулы к трем степеням свободы поступатель-
поступательного движения следовало бы добавить три степени свободы враща-
вращательного движения (соответствующие трем взаимно перпендику-
перпендикулярным осям вращения). Однако одну из осей вращения всегда мож-
можно совместить с осью молекулы. Вращение вокруг этой оси равно-
равносильно вращению отдельных атомов, поэтому его не учитывают.
Таким образом, всем двухатомным молекулам, а также тем много-
многоатомным молекулам, атомы которых расположены вдоль прямой,
следует приписать две степени свободы вращательного движения.
Общее число степеней свободы двухатомной моле-
молекулы равно пяти.
70
Это число совпадает с числом независимых координат, необхо-
необходимых для определения положения двухатомной молекулы в про-
пространстве. Действительно, для определения положения двухатом-
двухатомной молекулы в пространстве необходимо и достаточно указать ко-
координаты ее центра тяжести хи yit г4 (рис. 31) и два угла ф и со.
Для многоатомной молекулы с нелинейным расположением ато-
атомов сохраняются все три степени свободы вращательного движения,
и поэтому общее число степеней свободы, обусловленных поступа-
поступательным и вращательным движением молекулы, равно шести.
В приведенном подсчете числа степеней свободы принималось,
что атомы в молекулах закреплены неподвижно и не могут коле-
колебаться друг относительно дру-
друга. Опыт показывает, что при
комнатной температуре для
двухатомных газов это пред-
предположение оправдывается.
Средняя кинетическая энер-
энергия поступательного движе-
движения любой молекулы (урав-
(уравнение 28) согласно молеку-
лярно-кинетической теории
является линейной функцией
абсолютной температуры:
— = -kT.
Иис. 31. Число степеней свободы
двухатомной молекулы.
Эту величину можно разложить на три составляющие соответст-
соответственно трем произвольно выбранным координатным осям, или, как
можно теперь сказать, соответственно трем степеням свободы по-
поступательного движения.
Хаотичность молекулярного движения требует, чтобы ни одно
из направлений не было бы преимущественным в отношении дви-
движения, а это равносильно требованию равного распределения ки-
кинетической энергии, связанной с перемещением молекул, между
тремя степенями свободы поступательного движения. Максвелл
обобщил эту закономерность в знаменитом принципе рав-
равного распределения энергии, согласно которому в систе-
системе, состоящей из большого числа частиц, кинетичес-
кинетическая энергия распределяется в среднем поровну между
различными степенями свободы движения частиц.
На три степени свободы поступательного движения молекулы
приходится в среднем кинетическая энергия, равная -kT, следо-
вательно, на одну степень свободы будет приходиться:
Ek = - kT.
*« 2
G2)
71
Такое же количество кинетической энергии будет приходиться при
температуре Т на любую другую степень свободы движения моле-
молекулы.
Таким образом, если молекула газа обладает i степенями свобо-
свободы, то средняя кинетическая энергия ее будет равна:
ьт
Ek = i~. G3)
* 2 v ;
Умножив среднюю кинетическую энергию молекулы на число Аво-
гадро NA, получим кинетическую энергию одного киломоля газа,
которая в случае идеального газа будет равна его внутренней энер-
энергии U. Замечая, что NAk = R, можно написать:
С/ = г —. G4)
2 v
21. ТЕПЛОЕМКОСТЬ ИДЕАЛЬНОГО ГАЗА
Теплоемкостью тела называют физическую величину,
измеряемую количеством теплоты, которое необходимо сообщить
телу для того, чтобы повысить его температуру на один градус.
Если при этом масса тела равна одному килограмму, теплоемкость
называют удельной теплоемкостью, а если одной кило-
килограмм-молекуле, то — молекулярной теплоемкостью.
Количество теплоты, необходимое для нагревания тела на один
градус, как это позднее будет рассмотрено подробно, зависит от ус-
условий, при которых происходит нагревание. Так, например, если
нагревать газ на 1° в цилиндрическом сосуде, закрытом подвижным
поршнем, потребуется иное количество теплоты, чем в том случае,
если при нагревании поршень закрепить неподвижно. Для того
чтобы теплоемкость выражалась определенным числом, необходи-
необходимо производить нагревание всегда при определенных, наперед за-
заданных условиях.
Если условиться нагревать газ при неизменном объеме, то все
подведенное к газу количество теплоты будет идти на увеличение
внутренней энергии газа. Определенная при этих условиях величи-
величина теплоемкости называется теплоемкостью газа при
постоянном объеме — Cv.
Можно, однако, нагревать газ, поддерживая при этом постоян-
постоянное давление. В этом случае подведенная к газу теплота частично
расходуется на увеличение внутренней энергии, которая у идеаль-
идеального газа зависит только от температуры и числа степеней свободы,
а частично — на совершение внешней работы, поскольку при на-
нагревании в этих условиях газ будет расширяться и совершать ра-
работу, связанную с преодолением внешнего давления. Очевидно, что
теплоемкость, измеренная при постоянном давлении, будет больше,
нем теплоемкость, измеренная при постоянном объеме. Найденную
72
таким образом величину называют теплоемкостью газа
при постоянном давлении — Ср.
Молекулярная теплоемкость газа при постоянном объеме Cv свя-
связана простой зависимостью с числом степеней свободы движения
молекул. Из приведенного выше определения следует, что эта ве-
величина численно равна приросту внутренней энергии газа при по-
повышении его температуры на 1 градус. Как показывает, однако, опыт,
теплоемкость газа зависит от температуры, и поэтому более пра-
правильно определить теплоемкость Cv как первую производную от
внутренней энергии газа U по температуре, взятую при постоянном
объемен е.
Cv - М,-
Подставляя в уравнение G5) значение внутренней энергии для од-
одного киломоля газа — уравнение G4), найдем, что молекулярная
теплоемкость идеального газа при постоянном объеме может быть
записана в виде следующего простого выражения:
G6)
Таким образом, молекулярная теплоемкость идеаль-
идеального газа, измеренная при постоянном объеме, опреде-
определяется числом степеней свободы движения молекул
газа.
Учитывая число степеней свободы, присущее молекулам различ-
различной сложности (§ 20), можно ожидать, что для газа с одноатомными
молекулами молекулярная теплоемкость при постоянном объеме
будет равна Cv = — = 12,46 • 103 дж/кмоль • град, а для газа с
двухатомными молекулами Cv — — = 20,77 • 103 дж/кмоль ¦ град.
Z
Для газа с трехатомными нелинейными молекулами величина Cv =
= 3R, или 24,93 дж/кмоль ¦ град.
Таблица 8
Молекулярные теплоемкости некоторых газов, измеренные при постоянном
объеме и при разных температурах
73
Данные, приведенные в таблице 8, убеждают в том, что молеку-
лярно-кинетическая теория позволяет рассчитывать теплоемкость
газов в удовлетворительном согласии с опытом. Одновременно эти
данные позволяют заметить, хотя и незначительные, но имеющие
большое значение для теории расхождения между опытом и выво-
выводами изложенной выше теории.
Бросается в глаза, во-первых, то, что величины молекулярных
теплоемкостей Cv газов с одинаковым (по изложенной теории) стро-
строением молекул заметно различаются, в то время как теоретически
следовало ожидать.что эти величины будут одинаковыми.
Кроме того, у двухатомных газов величина Cv с повышением тем-
температуры существенно возрастает, стремясь при высокой температуре
приблизительно к 29,3 103 дж/кмоль ¦ град.
Казалось бы, это возрастание можно объяснить возникновением
при более высоких температурах новых степеней свободы движения
молекул. В самом деле, с повышением температуры возрастают
средние скорости движения молекул, а следовательно, возрастает
и интенсивность их взаимодействия при соударениях. Естественно
предположить, что в этих условиях при соударениях молекул ато-
атомы, входящие в их состав, смещаются друг относительно друга, в
результате чего возникают колебательные движения и связанные с
ними новые степени свободы.
Несомненно, это явление имеет место. Однако при таком объяс-
объяснении зависимости величины Cv от температуры возникают новые
затруднения.
Согласно представлению о связи, существующей между степеня-
степенями свободы и теплоемкостью, возникновение каждой новой степени
свободы должно сопровождаться скачкообразным возрастанием мо-
Г)
лекулярной теплоемкости на — дж/кмоль ¦ град, в то время как
на опыте наблюдается плавное изменение ее по мере возрастания
температуры.
Это затруднение в молекулярно-кинетическом объяснении те-
теплоемкости удалось разрешить лишь в результате изменения ос-
основных представлений о механизме передачи энергии. При этом
пришлось отказаться от характерного для классической кинети-
кинетической теории предположения о возможности передачи любого сколь
угодно малого количества энергии. Оказалось необходимым при-
признать, что энергия, так же как и вещество, имеет дискретную при-
природу. Передача энергии вращательного и колебательного движе-
движения молекул возможна только определенными порциями. Эти пор-
порции энергии называются квантами.
Кванты энергии могут быть различными по величине. Энергия
вращательного движения молекулы передается квантами, величина
которых мала по сравнению со средней энергией, приходящейся
на одну степень свободы движения молекул при комнатной темпе-
ратуре, и поэтому квантовая природа энергии, связанной с враще-
вращением молекул, в этих условиях не сказывается.
Иное положение наблюдается в случае колебательного движе-
движения молекул. Кванты, которыми передается энергия колебатель-
колебательного движения молекул, гораздо больше квантов энергии враща-
вращательного движения. При комнатной температуре только в очень ред-
редких случаях соударяющиеся молекулы обладают энергией, доста-
достаточной для возбуждения колебательного состояния двухатомной
молекулы. Число молекул, обладающих энергией, достаточной для
возбуждения при их соударении колебаний атомов, входящих в сос-
состав молекул, увеличивается с ростом температуры. Естественно,
что с увеличением температуры увеличивается и количество моле-
молекул, атомы которых колеблются, т. е. таких молекул, у которых не-
необходимо учитывать наряду со степенями свободы поступательного
и вращательного движений также степени свободы колебательного
движения.
Кванты энергии колебательного движения атомов в молекулах
двухатомных газов значительно больше, чем у сложных молекул,
из которых состоят пары различных органических соединений. Это
обстоятельство объясняет, почему при комнатной температуре теп-
теплоемкости Cv двухатомных газов близки к — R дж/кмоль • град,
т. е. колебательные движения атомов в них не возникают, в то вре-
время как при тех же условиях у газов со сложными молекулами теп-
теплоемкости существенно больше: 3R дж/кмоль • град. При расчете
теплоемкости сложных молекул необходимо учитывать особенности
колебательного движения атомов.
22. СООТНОШЕНИЕ МЕЖДУ ТЕПЛОЕМКОСТЬЮ ГАЗА
ПРИ ПОСТОЯННОМ ОБЪЕМЕ И ТЕПЛОЕМКОСТЬЮ ГАЗА
ПРИ ПОСТОЯННОМ ДАВЛЕНИИ
Теплоемкость газа Ср, измеренная при постоянном давлении,
превосходит теплоемкость газа при постоянном объеме Cv на вели-
величину, равную работе А, совершаемой газом, расширяющимся при
нагревании, т. е.
Cp = Cv + A.
Для вычисления величины работы А рассмотрим один киломоль газа,
заключенный при температуре Т в цилиндре, закрытом невесомым
поршнем (рис. 32). Площадь основания цилиндра S, расстояние
между поршнем и дном цилиндра h, так что объем, занимаемый газом,
y = Sft.
Внешнее давление на поршень р равно давлению газа в цилиндре,
связанному с объемом и температурой газа уравнением Клапейро-
Клапейрона:
pV =- RT.
75
Допустим, что газ нагревается при постоянном давлении от тем-
температуры Т до температуры Т -\- Г. Объем, занимаемый газом, при
этом увеличится от величины V до V + А У, а поршень переместит-
переместится на расстояние Ah. Перемещение поршня связано с преодолени-
преодолением силы F, равной произведению pS, и с совершением работы А, ко-
которую можно подсчитать, воспользовавшись соотношением:
А = FAh, или А = pSAh = pAV.
Объем газа, нагретого до температуры Т -\- Г, связан с давле-
давлением, так же как и до нагревания, уравнением Клапейрона,
т.е.
или, раскрывая скобки,
Сократив величину pV, стоящую в левой части
уравнения, с равной ей величиной RT, стоящей
в правой части уравнения, найдем:
pAV = R. G8)
Полученное уравнение поясняет физический
смысл универсальной газовой постоянной, ко-
рис д2 торая, как это теперь ясно, численно равна ра-
работе, совершаемой одним киломолем газа, расши-
расширяющимся при постоянном давлении при нагревании на 1 градус.
Молекулярная теплоемкость идеального газа при постоянном дав-
давлении Ср связана с молекулярной теплоемкостью того же газа
при постоянном объеме Cv соотношением:
CP = CV + R. G9)
Во многих процессах важную роль играет отношение тепло-
теплоемкости Ср к теплоемкости Cv, обозначаемое буквой у:
??
С/
(80)
Очевидно,
Liv kjv
iR
Заменяя в написанном уравнении Cv == — , получим,
2»
76
23. РЕАЛЬНЫЕ ГАЗЫ. УРАВНЕНИЕ ВАН-ДЕР-ВААЛЬСА
pV
Законы идеальных газов, вытекающие из молекулярно-кинети-
ческих представлений, достаточно хорошо подтверждаются на опы-
опыте в том случае, если давление газа мало и температура его не слиш-
слишком низка. При повышении давления поведение реальных газов от-
отклоняется от поведения газов идеальных.
На рисунке 33 показано, как изменяется произведение дав-
давления реального газа на занимаемый им объем при увеличении дав-
давления. Для идеального газа подобная зависимость выражается
прямой линией, параллельной оси абсцисс. Рассматривая график
(рис. 33), мы убеждаемся в том, что реальный газ при увеличении
давления первоначально сжимается больше, чем это следует из урав-
уравнения идеальных газов, так что произведение рV уменьшается с воз-
возрастанием давления. При дальнейшем увеличении давления начи-
начинают сказываться какие-то иные
свойства молекул реальных га-
газов, в силу которых произведе-
произведение pV перестает убывать. Если
продолжать газ сжимать, то оно
возрастает, делаясь больше вели-
величины, следующей из теории.
Причины подобных отклоне-
отклонений легко понять. Большую сжи-
сжимаемость реального газа по срав-
сравнению с газом идеальным обус-
обусловливают силы межмолекуляр-
межмолекулярного взаимодействия. Молеку-
Молекулярное сцепление приводит к воз-
возникновению как бы добавочного
давления, возрастающего при
возрастании плотности газа.
Наблюдаемое при высоких давлениях уменьшение сжимаемос-
сжимаемости и соответствующее возрастание произведения pV объясняется
тем, что реальные молекулы не являются материальными точками,
а обладают некоторым конечным объемом. Учет объема молекул
эквивалентен учету сил отталкивания, проявляющихся в тех слу-
случаях, когда расстояние между молекулами мало. Выше было пока-
показано, что давление газа зависит от числа ударов молекул о стенку
сосуда за 1 секунду. Это число увеличивается, если при расчете
принять во внимание присущий молекулам объем. Действительно,
при центральном соударении какой-либо молекулы с другой моле-
молекулой количество движения, которым обладает первая молекула, пе-
переносится практически мгновенно на отрезок, равный диаметру мо-
молекулы (рисунок 34, а). В результате этого уменьшается промежу-
промежуток времени, разделяющий два последующих удара молекулы о
стенку сосуда, и, следовательно, увеличивается число ударов ее
200 400 600 800 1000
Рис. 33. Зависимость произведе-
произведения pV от давления р (атм) для
кислорода.
77
об эту стенку за одну секунду. По мере увеличения давления воз-
возрастает плотность газа, а вместе с ней возрастает влияние собст-
собственного объема молекул — газ оказывает большее сопротивление
сжатию, чем это следует из уравнения идеальных газов (уравнения
Клапейрона).
Следует заметить, что на необ-
необходимость учитывать при расчете
давления сильно сжатого газа соб-
собственный объем молекул указывал
еще Ломоносов.
Уравнение состояния реально-
реального газа, в котором приняты во
внимание обе причины, вызываю-
вызывающие отклонения его свойств от
свойств идеальных газов, было
предложено впервые голландским
физиком Ван-дер-Ваал ь-
сом A837—1923) и носит его имя.
Уравнение Ван-дер-Ваальса отли-
отличается от уравнения Клапейрона
наличием двух поправочных чле-
членов, один из которых учитыва-
учитывает влияние собственного объема
молекул, а другой — влияние сил
молекулярного притяжения.
Для того чтобы учесть влияние
собственного объема молекул, об-
обратим внимание на то, что при
неограниченном увеличении давле-
ния объем идеального газа V = —
р
стремится к нулю, а объем ре-
реального газа при тех же условиях — к некоторой предельной ве-
величине Ь. Следовательно, для того чтобы в уравнении состояния
идеальных газов учесть влияние собственного объема молекул, не-
необходимо заменить в нем величину V разностью (V — Ь). Таким
образом, уравнение состояния, учитывающее собственный объем
молекул, будет иметь вид:
(81)
Рис. 34. Влияние собственного
объема молекул на давление
газа.
Теоретический расчет величины Ъ сложен. В простейшем случае,
если считать молекулы газа твердыми шариками с радиусом г и
предположить при этом, что в газе происходят только двойные со-
соударения молекул, то можно показать, что Ь равно учетверенному
78
собственному объему молекул, т. е.
6 = 4#„|яг\ (82)
где NА — число Авогадро.
Для того чтобы пояснить, почему берется учетверенный собст-
собственный объем молекул, рассмотрим более внимательно передачу
количества движения при молекулярных соударениях. При цент-
центральном ударе (рис. 34, а) импульс молекулы практически мгновен-
мгновенно передается на расстояние, равное диаметру молекулы 2л, что
эквивалентно вычитанию из объема, занятого газом, объема Vol,
равного N А-п [2г]3 = 8N д-лг3, т.е. восьмикратного объема мо-
4 о
лекул. Однако наряду с центральным ударом молекула будет ис-
испытывать всевозможные соударения (рис. 34, б, в, г) вплоть до сколь-
скользящего удара (рис. 34, г), при котором никакой мгновенной передачи
импульса в направлении движения происходить не будет и, следо-
следовательно, при котором поправка к объему равна нулю, т. е. V02 = 0.
Приравнивая Ь среднему значению поправки -01 ~*~ 02 получим
соотношение (82).
Если газ сжат настолько сильно, что в нем наряду с двойными
соударениями наблюдаются тройные и более сложные соударения
молекул, величина поправочного члена, учитывающего влияние
собственного объема молекул, будет иной.
Величина поправки к давлению, обусловленная молекулярным
притяжением, строго говоря, должна зависеть от плотности газа и
от закона, согласно которому изменяется сила, действующая меж-
между молекулами при изменении расстояния между ними. Однако при
выводе уравнения Ван-дер-Ваальса последняя зависимость не при-
принимается во внимание. Это оказывается возможным сделать, если,
учитывая подвижность молекул, предположить, что все положения
какой-либо молекулы в мысленно выделенном небольшом объеме
газа равновероятны. В этом случае взаимодействие любой молеку-
молекулы в одном из выделенных объемов с молекулами такого же сосед-
соседнего объема будет равно средней величине взаимодействия ее при
всех возможных положениях, занимаемых ею относительно сосед-
соседнего объема. Сказанное будет справедливо для всех молекул как в
первом, так и во втором объеме. Если теперь мысленно провести
плоскость внутри газа (рис. 35), то молекулы, находящиеся в малом
объеме справа от этой плоскости, будут притягиваться молекулами,
находящимися в таком же объеме слева от нее, с одинаковой для
всех молекул средней силой. Необходимо отметить, что, строго го-
говоря, это предположение может быть оправдано только в случае, ес-
если оно относится к разреженному газу.
На рисунке 35, а в каждом из рассматриваемых объемов нахо-
находится по одной молекуле. При увеличении плотности газа в два
раза число молекул в каждом из объемов удваивается (рисунок 35, б),
79
а сила, с которой один объем притягивает другой учетверяется.
При увеличении плотности газа в три раза (рисунок 35, в) сила при-
притяжения увеличится в 9 раз.
Из рассмотренного примера следует, что обусловленное моле-
молекулярным притяжением добавочное, или как его называют, внут-
внутреннее давление рг в реальных газах возрастает прямо пропорцио-
пропорционально квадрату плотности газа или, другими словами, обратно
пропорционально квадрату молеку-
молекулярного объема V, т. е.
где а — постоянная величина, раз-
различная для различных газов, зави-
зависящая от интенсивности молекуляр-
молекулярных взаимодействий.
Внутреннее давление не фиксиру-
фиксируется манометром и потому должно до-
добавляться к манометрическому дав-
давлению.
Учтя, таким образом, влияние на
поведение газов собственного объема
молекул и сил, действующих между
ними, Ван-дер -В аальс предложил за-
записать уравнение состояния реальных
газов в следующем виде:
-b) = RT. (83)
Таблица 9
О-
-О
Рис. 35. Учет молекуляр-
молекулярных взаимодействий.
Сравнение наблюдаемых на опыте величин давления Н2 и СО2
при 100°С с рассчитанными при помощи уравнения идеальных
газов и уравнения Ван-дер-Ваальса
р опытя.
к/ж2
50,5.105
75,7 »
101 »
Водород На
Ур-ние иде-
идеального газа
49,2.10s
73,0
95,9
Ур-ние Ван-
-дер-Ваальса
50,7
76,2
101,8
Углекислота СО2
Ур-ние иде-
идеального газа
57,6
93,2
134,8
Ур-ние Ван-
дер-ВааЛьса
50
74
96,7
В таблице 9 сопоставлены результаты расчета свойств газа с
помощью уравнений Клапейрона и Ван-дер-Ваальса с данными
опыта.
Как можно убедиться, при повышении давления уравнение
Ван-дер-Ваальса лучше передает свойства реального газа, нежели
уравнение Клапейрона.
80
Внутреннее давление —, играющее роль поправочного члена,
когда внешнее давление невелико, увеличивается при возрастании
последнего и может при определенных условиях существенно вли-
влиять на свойства газов.
В приведенной выше форме уравнение Ван-дер-Ваальса (83)
относится к одному киломолю газа, так что входящая в него вели-
величина объема газа — это молекулярный объем. При желании исполь-
использовать написанное уравнение применительно к т килограммам ве-
вещества необходимо помнить, что значения постоянных а и b в этом
случае будут иными.
Если молекулярный вес газа М и если желательно сохранить
значения постоянных а и Ъ, определенных для одного киломоля
вещества, то уравнение Ван-дер-Ваальса следует записывать так:
М
М
(84)
В этом уравнении V — объем, занимаемый т килограммами газа.
Величины а и b для некоторых газов приведены в таблице 10.
Таблица 10
Постоянные в уравнении Ван-дер-Ваальса а и b
для некоторых газов
Вещество
Не
н2
N2
о2
со2
н2о
О Н'МЧКМОЛЬ*
0,035.10а
0,248 »
1,37 »
1,39 »
3,65 »
5,52 »
Ь M^jKMOAb
23,9-Ю-з
26,7 »
38,6 »
31,9 »
42,8 »
30,4 »
В заключение следует подчеркнуть приближенный характер урав-
уравнения Ван-дер-Ваальса, которое, хотя и лучше передает поведение
реальных газов, чем уравнение Клапейрона, все же мало пригодно
для точных расчетов.
Вычисленные с помощью уравнения Ван-дер-Ваальса значения
давления газа достаточно точно совпадают с опытом лишь при от-
относительно высоких температурах и только в некотором интервале
81
давлений. Этим объясняются непрерывные поиски более совершен-
совершенных уравнений, которых в настоящее время известно более 150.
При малых давлениях и соответственно больших значениях мо-
молекулярного объема V можно пренебречь в уравнении Ван-дер-
Ваальса поправкой —, в силу ее малости по сравнению с р, и поп-
поправкой b — по сравнению с молекулярным объемом V. Уравнение
Ван-дер-Ваальса при этих условиях переходит в уравнение идеаль-
идеальных газов.
24. ИССЛЕДОВАНИЕ УРАВНЕНИЯ ВАН-ДЕР-ВААЛЬСА
Уравнение Ван-дер-Ваальса — алгебраическое уравнение третьей
степени относительно объема. Раскрыв скобки и расположив члены
уравнения Ван-дер-Ваальса по убывающим степеням объема, мож-
можно записать последнее в следующем виде:
V*— /б+— \ V2-\--V — -=.0. (85)
\ р I Р Р
Особенности этого уравнения наиболее наглядно выявляются при
графическом изображении полученной с его помощью зависимости
давления газа от занимаемого им объема.
На рисунке 36 изображена подобная зависимость для того слу-
случая, когда температура газа не слишком высока. Вдоль оси абсцисс
отложен объем, вдоль оси ординат — давление газа. Кривая, кото-
которая изображена на графике, рассчитана при постоянной температу-
температуре и, следовательно, является изотермой. Для того чтобы проанали-
проанализировать физический смысл отдельных участков полученной изо-
изотермы, рассмотрим мысленный опыт, схема которого изображена
на том же рисунке. Параллельно оси абсцисс расположена толсто-
толстостенная стеклянная трубка, наполненная углекислотой и закрытая
с правого конца поршнем. К левому концу трубки прикреплен ртут-
ртутный манометр. Положение поршня непосредственно указывает объ-
объем, занимаемый газом. Давление газа регистрируется по положению
уровня ртути в манометре.
Пересечение прямых, одна из которых параллельна оси абсцисс
на уровне ртути в манометре, а вторая — параллельна оси ординат и
указывает положение поршня в трубке, наполненной газом, проис-
происходит в точке С, изображающей на расположенной выше диаграмме
определенное состояние газа. Перемещая поршень и отмечая указан-
указанным способом состояния газа, можно с помощью подобного прибо-
прибора выяснить, насколько правильно передает уравнение Ван-дер-
Ваальса поведение реального газа.
Результаты подобного опыта изображены на рисунке 36 пунк-
пунктирной линией. Сопоставляя рассчитанную теоретически кривую
BFAC с кривой, построенной на основании опыта, мы убеждаемся
62
о
втом, что если объем газа не слишком мал, то уравнение Ван-дер-
Ваальса удовлетворительно согласуется с опытом. Качественное
согласие с опытом наблюдается до тех пор, пока давление в трубке
не сделается равным давлению насыщенного пара углекислоты,
а объем V — объему VH. „., занимаемому насыщенным паром при дан-
данной температуре.
При заданной температуре давление насыщенного пара является
величиной постоянной, и потому дальнейшее уменьшение объема
не влечет за собой увеличения давления. Уменьшение объема, заня-
занятого насыщенным паром вещества, вызывает конденсацию, т. е. пе-
переход вещества из парообраз-
парообразного состояния в жидкое. На
экспериментальной изотерме
процессу конденсации соот-
соответствует прямолинейный уча-
участок, параллельный оси абс-
абсцисс. Конденсация заканчи-
заканчивается в тот момент, когда
все вещество превращается в
жидкость и объем его делается
равным молекулярному объ-
объему жидкости Уж.
Поскольку жидкости об-
обладают малой сжимаемостью,
дальнейшее уменьшение объ-
объема вызывает значительное
увеличение давления. Каче-
Качественно это согласуется с
Рис. 36. Изотерма Ван-дер-Ваальса
для газа при невысоких температурах.
результатами, полученными
расчетом на основании урав-
уравнения Ван-дер-Ваальса.
Таким образом, в качественном отношении различие между тео-
теоретическими и экспериментальными изотермами реальных газов
при относительно низких температурах сводится к тому, что s-об-
разный участок теоретических изотерм заменяется у кривых, по-
полученных экспериментально, горизонтальной прямой.
Если подвергаемый сжатию газ тщательно очистить от пыли,
то можно наблюдать задержку возникновения конденсации, в ре-
результате которой удается сжать газ до объема, меньшего, чем объ-
объем насыщенного пара, и, соответственно, до давления, большего,
чем давление насыщенного пара при данной температуре. Таким
способом можно на опыте воспроизвести качественно участок АС
изотермы Ван-дер-Ваальса и тем самым выяснить его физический
смысл. Очевидно, этот участок изотермы соответствует пересы-
пересыщенному пару вещества.
Это состояние неустойчиво, в веществе спонтанно возникает кон-
конденсация, при которой давление скачкообразно падает до давления
83
насыщенного пара (подобный процесс на рисунке 36 изображен
стрелкой).
Точно так же при изотермическом увеличении объема сжатой
жидкости экспериментально можно наблюдать задержку образо-
образования паровой фазы, несмотря на то что объем, занимаемый жид-
жидкостью, превышает ее молекулярный объем.
Давление в системе при этом будет ниже соответствующего дан-
данной температуре давления насыщенного пара жидкости. Задержка
возникновения паровой фазы позволяет экспериментально воспро-
воспроизвести в качественном согласии с теорией участок BF изотермы
Ван-дер-Ваальса. Эта часть теоретической изотермы соответствует
неустойчивому состоянию растянутой жидкости. Иног-
Иногда это состояние называют состоянием перегретой жидкости
в том смысле, что при данном значении молекулярного объема и тем-
температуры часть жидкости должна была бы находиться в парообраз-
парообразном состоянии, в то время как на опыте паровая фаза отсутствует.
Участок AF изотермы Ван-дер-Ваальса соответствует совер-
совершенно неустойчивому состоянию вещества, при котором давление
в системе должно было бы уменьшаться при уменьшении объема
системы и расти при его увеличении. На опыте это состояние не ре-
реализуется.
Таким образом, уравнение Ван-дер-Ваальса качественно пра-
правильно передает изменение давления вещества при изменении объ-
объема как в случае пара, так и в случае жидкости.
Уравнение предсказывает наличие неустойчивых состояний,
а именно — состояния перегретого пара и состояния растянутой
жидкости. Однако теория Ван-дер-Ваальса не учитывает возмож-
возможности одновременного присутствия вещества в парообразном и жид-
жидком состояниях.
Если изобразить на графике изотермы Ван-дер-Ваальса, рас-
рассчитанные теоретически при разных значениях температуры, то по-
получится семейство кривых, подобное изображенному на рисунке 37.
Как можно убедиться, при повышении температуры s-образный
участок изотермы Ван-дер-Ваальса уменьшается, другими словами,
уменьшается разница между молекулярными объемами насыщен-
насыщенного пара и жидкости. При некоторой температуре Tk, называемой
критической температурой, s-образный участок изотермы
исчезает. На соответствующей изотерме, которую называют кри-
критической, вместо s-образного участка наблюдается точка переги-
перегиба К, которая называется критической точкой. При
более высоких температурах изотермы Ван-дер-Ваальса напоминают
изотермы идеальных газов.
Экспериментально поведение реальных газов было изучено Эн д-
рьюсом A813—1885), найденные им изотермы изображены на
рисунке 38.
Если соединить между собой правые концы горизонтальных
участков экспериментальных изотерм, то полученная кривая будет
84
изображать изменение с температурой молекулярного объема на-
насыщенного пара вещества. Молекулярный объем насыщенного пара
уменьшается с увеличением температуры вплоть до критической
температуры.
Соединив между собой левые концы горизонтальных участков
опытных изотерм, получим параболическую кривую, изображаю-
изображающую зависимость от температуры молекулярного объема жидкости,
который увеличивается с возрастанием температуры.
При критической температуре молекулярный объем жидкости
делается равным молекулярному объему пара. Очевидно, что выше
Р
р
ь
0
а
fl
Д
г
Ж
U///////
wfrf}/',
тж.
Г
ш
W/A
сог
bt
о,
^ у
Рис. 37. Изотермы Ван-дер-Ваальса
для газа при разных температурах
(давление р в атм).
Рис. 38. Области существова-
ния вещества в различных
агрегатных состояниях.
критической температуры вещество не может находиться в жидком
состоянии.
На эту особенность вещества впервые указал Д. И. Менде-
Менделеев A834—1907), который назвал критическую температуру
вещества температурой абсолютного кипения.
Критическая изотерма и проведенные параболические кривые
делят диаграмму состояния на несколько областей (рис. 38). Об-
Область Ж, расположенная между осью ординат, участком АК кри-
критической изотермы и левой ветвью параболы ВК, соответствует тем
значениям объема и давления, при которых вещество находится в
жидком состоянии. Область П, ограниченная участком /041 крити-
85
ческой изотермы и правой ветвью параболы КС, выделяет значения
давления и объема, соответствующие веществу в парообразном сос-
состоянии. Выше критической изотермы расположена область Г —
вещества в газообразном состоянии. Наконец, область, ограничен-
ограниченная осью абсцисс и параболой ВКС, соответствует одновременному
присутствию вещества в жидком и парообразном состояниях. Это
область, в которой вещество находится одновременно в двух раз-
различных агрегатных состояниях, или гетерогенная об-
область.
Таким образом, сжатие вещества при температуре ниже крити-
критической вызывает при определенном давлении его конденсацию, в то
время как любое сжатие вещества при температурах выше крити-
критической не вызывает конденсации. При температурах выше крити-
критической, увеличивая давление, нельзя превратить газообразное ве-
вещество в жидкое.
Теория Ван-дер-Ваальса указывает на возможность непрерыв-
непрерывного перехода вещества из жидкого состояния в газообразное без
распадения вещества на две фазы. Непрерывный переход из жидкого
состояния в газообразное изображен на рисунке 38 стрелками. От-
Отрезки ab и be изображают нагрев вещества до критической темпе-
температуры. Давление в системе при этом возрастает выше критическо-
критического давления. Дальнейшее изменение состояния вещества может
заключаться или в изотермическом расширении, при котором дав-
давление уменьшается до критического и жидкость в критической точ-
точке превращается в пар, или же в изобарном нагревании, при кото-
котором вещество превращается в газ при давлении выше критического.
Естественно, что возможен и обратный непрерывный, т. е. не
сопровождающийся распадением вещества на две фазы, переход
из газообразного состояния в жидкое. Этот переход показан на диа-
диаграмме стрелками аф^ и Ь
25. ОПРЕДЕЛЕНИЕ КРИТИЧЕСКИХ ПАРАМЕТРОВ ВЕЩЕСТВА
Соответствующие критической точке значения давления pk,
объема V k и температуры Tk называют: критическим дав-
давлением, критическим объемом и критической
температурой вещества. Критические параметры pk,
Vk и Tk являются индивидуальными характеристиками вещества,
зависящими от свойств его молекул.
Уравнение Ван-дер-Ваальса позволяет установить однозначную
связь между входящими в это уравнение постоянными а и Ь и кри-
критическими параметрами вещества.
Как всякое уравнение 3-й степени, уравнение Ван-дер-Ваальса
можно представить в виде произведения:
(V_V1)(V-V2)(V-V3) = 0, (86)
в котором Vit Уч и V3 являются тремя корнями уравнения.
86
При критической температуре Vi = V2 = V3 = Vk и, следова-
следовательно:
(V — Vkf = О, или V3 — 3V2Vk + ZVV\ —V3k =0. (87)
Это уравнение должно быть тождественно с уравнением Ван-дер-
Ваальса, написанным для критической изотермы:
\ УЯ + ?У—-=0. (88)
Pk I Pk Pk
Написанные уравнения будут тождественными в том случае,
если будут равны коэффициенты у членов, содержащих одинаковые
степени V.
Из этого условия следуют соотношения:
Pk Pk Pk
пользуясь которыми, можно найти следующие зависимости между
критическими параметрами вещества и соответствующими значе-
значениями постоянных в уравнении Ван-дер-Ваальса:
Т %а . Т/ <1U. _ _ а 1ОГ\\
ИЛИ * 27Rb H* 27Ь2
3 3 Тк
Если подставить найденные значения постоянных а, Ь и R в урав-
уравнение Ван-дер-Ваальса и условиться измерять давление, объем и
температуру для каждого вещества отношением этих величин к их
критическим значениям, а именно:
~~ Pk ' ^ Vk' ~ Tk'
то уравнение состояния примет следующий вид:
- 1) = 8т, (92)
т. е. оно не будет содержать произвольных постоянных1.
Определенные, как это сделано выше, величины давления, объ-
объема, температуры называют: приведенным давлением—я, приве-
приведенным объемом — юн приведенной температурой — т.
Уравнение Ван-дер-Ваальса, в котором используются приведен-
приведенные величины, называется приведенным уравнением
Ван-дер-Ваальса. Если два или несколько веществ нахо-
1 Написанное уравнение содержит определяемые опытным путем постоян-
постоянные вскрытой форме. Этими постоянными являются критические па-
параметры вещества.
87
дятся в состояниях, при которых приведенные значения двух ка-
каких-либо параметров состояния у них одинаковы, то одинаковы
приведенные значения и третьего параметра. В этом случае говорят,
что вещества находятся в соответственных состоя-
состояниях.
26. ПРИРОДА СИЛ МОЛЕКУЛЯРНОГО ПРИТЯЖЕНИЯ
Силы молекулярного притяжения имеют сложную природу, хо-
хотя в конечном счете все они сводятся к взаимодействию электричес-
электрических зарядов.
Как известно, атомы, входящие в состав молекул, состоят из
положительно заряженных ядер, окруженных большим или мень-
меньшим количеством быстро вращающихся электронов. Хотя в сумме
положительные заряды атомных ядер в молекулах равны суммар-
суммарному отрицательному заряду электронов,
т. е. в целом молекулы электрически
нейтральны, однако пространственно поло-
+ у -^ + жительные и отрицательные заряды не
Н >*>. __ejr4. H совпадают.
В настоящее время достоверно извест-
известно, что существуют молекулы, у которых
Рис. 39. Строение электрические заряды распределены не-
немолекулы воды. равномерно. У подобной молекулы в
одной части оказываются преобладающи-
преобладающими положительные заряды, а в другой — отрицательные. Два
равных разноименных электрических заряда, разделенные некото-
некоторым расстоянием, образуют электрический диполь.
Поэтому молекулы с несимметричным распределением электричес-
электрических зарядов называют дипольными, или полярными, молеку-
молекулами. Примером полярной молекулы может служить молекула во-
воды (рис. 39). Та часть молекулы, в которой расположен атом кисло-
кислорода, заряжена преимущественно отрицательно.
Дипольные молекулы всегда стремятся ориентироваться друг
относительно друга так, чтобы против положительного полюса од-
одной молекулы располагался бы отрицательный полюс другой. Это
обусловлено электрическим взаимодействием разноименных полю-
полюсов. Несмотря на то что тепловое движение молекул нарушает по-
подобную ориентацию, все же она является преимущественной, в ре-
результате чего между дипольными молекулами возникают силы при-
притяжения. Поскольку этот вид взаимодействия молекул связан с их
взаимной ориентацией, его называют ориентационным
взаимодействием, или ориентационным эф-
эффектом. Ориентационный эффект — одно из слагаемых ван-
дер-ваальсовых сил.
Однако это взаимодействие не может во всех случаях объяснить
молекулярное притяжение. Наряду с полярными молекулами су-
ществуют молекулы с симметричным расположением электричес-
электрических зарядов, не имеющие дипольного момента. Такие молекулы на-
называют неполярными. Неполярны молекулы двухатомных
газов: азота, кислорода, водорода, молекулы углекислоты, пары
бензола и многие другие.
Симметричность электрических зарядов в неполярных моле-
молекулах легко нарушается, если подобная молекула 'оказывается
вблизи полярной молекулы или иона.
В этих условиях у неполярной молекулы возникает дипольный
момент, называемый индуцированным дипольным
моментом. Взаимодействие индуцированных дипольных мо-
моментов, возникших в молекулах, вызывает в свою очередь возник-
возникновение молекулярного притяжения. Это взаимодействие называют
индукционным эффектом. Индукционный эффект не-
незначителен и не может объяснить наблюдаемую на опыте величину
ван-дер-ваальсовых сил.
За редким исключением молекул с большим дипольным момен-
моментом, основной причиной возникновения молекулярного притяжения
являются взаимодействия так называемых мгновенных, или
осциллирующих диполей.
Входящие в состав молекулы положительно заряженные ядра
атомов и отрицательно заряженные электроны совершают своего
рода колебания друг относительно друга. Таким образом, мгновен-
мгновенная картина даже неполярной молекулы обнаруживает различные
смещения ядер и электронов. Величина этого смещения изменяется
во времени так, что молекулы вне зависимости от того, полярны они
или нет, ведут себя как периодически возникающие (осциллирую-
(осциллирующие) диполи. При сближении молекул мгновенные диполи вызыва-
вызывают появление в соседних молекулах согласованно изменяющихся
индуцированных диполей, взаимодействие с которыми и приводит
к возникновению молекулярного притяжения.
Таблица 11
Соотношение между различными составляющими
молекулярных сил (в %)
Вещество
Вода
Аммиак
Хлористый водород . .
Метан
Ориента-
ционный
эффект
77
45
15
Индук-
Индукционный
эффект
4
5
4
Диспер-
Дисперсионный
эффект
19
50
81
100
89
Те же колебания электрических зарядов в молекулах вызыва-
вызывают различное преломление лучей света с разной длиной волны, на-
называемое дисперсией и проявляющееся в возникновении оптичес-
оптического спектра. По этой причине силы, обусловленные взаимодейст-
взаимодействием мгновенных диполей, называют дисперсионными
силами. В реальных газах с неполярными молекулами ван-дер-
ваальсовые силы практически полностью обусловлены дисперси-
дисперсионным взаимодействием.
В таблице 11 приведены относительные величины ориентацион-
ного, индукционного и дисперсионного взаимодействия для неко-
некоторых молекул.
27. ВНУТРЕННЯЯ ЭНЕРГИЯ РЕАЛЬНОГО ГАЗА
Внутренняя энергия идеального газа, поскольку в нем отсут-
отсутствуют молекулярные взаимодействия, представляет собой кинети-
кинетическую энергию движения молекул, которая зависит от температу-
температуры газа, но не зависит от занимаемого газом объема.
В реальных газах нельзя пренебрегать взаимодействием моле-
молекул, и потому внутренняя энергия реального газа находится сум-
суммированием кинетической энергии движения молекул Ek и потен-
потенциальной энергии их взаимодействия Ер:
^реатьн. газа == *-"ft ~Т~ рш
Потенциальная энергия молекулярного взаимодействия зависит от
взаимного расположения молекул и потому должна изменяться при
изменении объема газа.
Если исключить обмен энергией между газом и внешней сре-
средой, то сумма присущей молекулам газа кинетической и потенци-
потенциальной энергий должна оставаться постоянной и, следовательно,
изменение одного из видов энергии должно компенсироваться про-
противоположным изменением второго вида энергии:
Если учитывать только притяжение молекул, то их потенциаль-
потенциальная энергия должна возрастать при удалении молекул друг от дру-
друга, т. е. расширение реального газа при этих условиях должно со-
сопровождаться уменьшением кинетической энергии его молекул.
Поскольку же мерой средней кинетической энергии молекул газа
служит его абсолютная температура, то при расширении газа, мо-
молекулы которого притягиваются друг к другу, температура его
должна понижаться. Впервые подобный опыт удалось осуществить
совместно Д. Джоулю A818—1889) и В. Томсону A824—
1907). В опыте Джоуля — Томсона для обнаружения изменения
внутренней энергии реального газа при изменении объема газ за-
заставляют расширяться через пористую перегородку (рис. 40), уста-
установленную в трубке, соединяющей сосуды А и В. Специальные на-
90
д
Рис. 40. Схемы опыта
Томсона.
в
Джоуля —
сосы поддерживают в этих
сосудах постоянные давле-
давления: в сосуде А — давле-
давление ри а в сосуде В — мень-
меньшее давление р%. По обе
стороны от пористой пере-
перегородки помещают термо-
термометры.
Как показали опыты,
большинство газов, расши-
расширяясь при комнатной тем-
температуре и не очень больших давлениях, охлаждается.
Исключение составляет водород, который при этих условиях на-
нагревается.
Изменение температуры, сопровождающее расширение реаль-
реального газа, получило название эффекта Джоуля — Том-
Томсона. Охлаждение газа при расширении называют положитель-
положительным эффектом Джоуля — Томсона, нагревание — отрицательным.
Для того чтобы понять существование эффекта Джоуля—Том-
Джоуля—Томсона разного знака, обратимся к графику, поясняющему отклоне-
отклонение поведения реального газа от газа идеального (рис. 41).
Сплошными линиями на графике изображено изменение отноше-
отношения произведения давления газа на занимаемый им объем к аб-
абсолютной температуре при изменении давления для трех различ-
различных температур Ти Т2, Т3, удовлетворяющих условию 7Л1<Г2<713.
В случае идеального газа (пунктирная прямая) отношение —
остается постоянным.
Если в опыте Джоуля — Томсона предоставить газу расши-
расширяться от давления рх до некоторого меньшего давления (р4 —
— Ар), то в зависимости от температуры, при которой производит-
производится этот опыт, отклонение в
поведении реального газа
pV от газа идеального будет
~f~ ' различно. При более низкой
температуре Т1 отношение —-
меньше, чем для идеального
газа, и, следовательно, пре-
преобладает влияние сил притя-
притяжения. При расширении га-
газа в этом случае молекулы
будут совершать работу про-
против сил притяжения за счет
присущей им кинетической
энергии. В результате при
расширении газ будет охлаж-
"Идеальный
газ
Р,
Рис. 41.
91
даться, т. е. будет наблюдаться положительный эффект Джо-
Джоуля—Томсона.
При более высокой температуре Т3, как это явствует из чертежа,
преобладающее значение имеют силы отталкивания, учитываемые
в уравнении Ван-дер-Ваальса поправочным членом Ь. Эти силы бу-
будут совершать работу при расширении реального газа и тем увели-
увеличивать кинетическую энергию молекул. В этом случае при расши-
расширении реального газа будет наблюдаться нагревание, т. е. отрица-
отрицательный эффект Джоуля—Томсона.
При некоторой промежуточной температуре Тг влияние сил при-
притяжения в точности компенсируется влиянием сил отталкивания и
реальный газ ведет себя как газ идеальный, т. е. его расширение
не сопровождается изменением температуры.
Таким образом, при плавном изменении температуры от значе-
значения 7\ до значения Т3 знак эффекта Джоуля—Томсона изменяется
с положительного на отрицательный. Это происходит при темпе-
температуре, называемой температурой инверсии.
Для кислорода температура инверсии -f-790°C, для углекислоты
-f-1800°C, однако для водорода температура инверсии —73°С. Водо-
Водород будет охлаждаться при расширении только в том случае, если
его температура ниже —73°С, в противном же случае при расшире-
расширении он нагревается.
Эффект Джоуля—Томсона находит себе важное применение в
технике при сжижении газов.
28. СЖИЖЕНИЕ ГАЗОВ
Анализ уравнения Ван-дер-Ваальса показывает, что все веще-
вещества при понижении их температуры ниже критической могут быть
превращены в жидкость простым увеличением давления.
В таблице 12 приведены критические температуры и давления
некоторых веществ.
Таблица 12
Критические температуры и давления некоторых веществ
Вещество
Вода
Бензол
Хлор
Углекислота
Кислород
Азот
Водород
Гелий
Критическая
температура
374,0
288,5
144,0
31,1
—118
—147,1
—239,9
-267,9
Критическое
давление н/м>
220.105
47,7 »
77,1 »
73,9 »
50,3 »
33,9 »
13,0 »
2,29»
92
Впервые получить газообразное вещество в жидком состоянии
удалось М. Фарадею A791—1867), превратившему в 1823 году
газ хлор в жидкость. Легко превращается в жидкость углекислота.
Гораздо труднее сжижаются газы с низкой критической темпера-
температурой: азот, кислород, водород, гелий.
В конце XIX века было предложено использовать для сжижения
газов эффект Джоуля—Томсона.
Эта мысль была высказана независимо друг от друга в Англии
Дьюаром ив Германии Линде.
Понижение температуры, обусловленное эффектом Джоуля—
Томсона, обычно невелико. При нуле градусов и средних давлениях
воздух, например, охлаждается всего на 0,26 гра-
градуса при уменьшении давления на 105 н/м2. Однако
при температуре —100°С и давлении 137 • 105 н/м2
воздух, расширяясь до давления 105 н/м2, охлаж-
охлаждается на 93° градуса.
В машине Линде, предложенной для получе-
получения жидкою воздуха (рис. 42), последний в газо-
газообразном виде сжимается компрессором до давле-
давления порядка 107 н/м2. После охлаждения в обыч-
обычном холодильнике сжатый воздух поступает в зме-
змеевик /, состоящий из нескольких труб, располо-
расположенных одна внутри другой. Проходя по внут-
внутренней трубе, воздух подвергается дросселирова-
дросселированию, т. е. внезапному расширению, в результате
которого его температура понижается. Охлажден-
Охлажденный воздух направляется по внешней трубе навст-
навстречу воздуху, идущему на дросселирование. В ре-
результате теплообмена температура воздуха, иду-
идущего по внутренней трубе, понижается. Подоб-
Подобное устройство машины обеспечивает непрерыв-
непрерывное понижение температуры воздуха, поступающего на дроссели-
дросселирование, до тех пор, пока внезапное расширение при дросселиро-
дросселировании и связанное с ним понижение температуры не вызовут его
конденсацию.
Жидкий воздух собирается в специальном приемнике, представ-
представляющем собой большой дьюаровский сосуд B, рис. 42).
Для сжижения водорода в машине Линде его необходимо пред-
предварительно охладить жидким воздухом ниже температуры инверсии.
Особенно трудно сжижается гелий, имеющий самую низкую
критическую температуру —267,9°С и температуру инверсии —258°С.
Заставляя жидкий гелий кипеть при пониженном давлении,
удается достичь температуры, всего на несколько десятых долей
градуса отличающейся от абсолютного нуля. Для дальнейшего
понижения температуры в кипящий гелий помещают кусок желе-
железа и намагничивают его в магнитном поле. Если теперь быстро прек-
прекратить действие магнитного поля, то железо размагнитится. Раз-
Рис. 42.
Схема
машины
Линде.
93
магничивание железа связано с затратой энергии, которая черпа-
черпается из окружающей среды в форме теплоты. Таким способом уда-
удалось достигнуть температур, отличающихся от абсолютного нуля
на тысячные доли градуса.
Понижение температуры, необходимое для сжижения газа, мож-
можно осуществить также, заставляя охлаждаемый газ совершать ра-
работу не только против внутренних сил, обусловленных молекуляр-
молекулярным сцеплением, но и против внешних сил. Охлаждение в этом слу-
случае происходит более эффективно. Машины, предназначенные для
этой цели, называют детандерами. Поршневые детандеры
напоминают своим устройством паровые машины с той разницей,
что в них пар заменен сжатым газом.
Советский ученый П. Л. К а п и ц а сконструировал детандер,
напоминающий своим устройством турбину. Ротор турбодетандера
Капицы делает около 40 000 об/мин.
Работа, совершаемая воздухом в турбодетандере, вызывает столь
интенсивное охлаждение его, что часть воздуха сжижается. При
одинаковой производительности турбодетандер в несколько раз
меньше поршневого детандера.
II. ЖИДКОЕ СОСТОЯНИЕ ВЕЩЕСТВА
1. СРАВНЕНИЕ СВОЙСТВ ЖИДКОСТЕЙ СО СВОЙСТВАМИ
ГАЗОВ И ТВЕРДЫХ ТЕЛ
Жидкости по своим свойствам занимают промежуточное поло-
положение между газами и твердыми телами. Вряд ли имеет смысл
обсуждать вопрос о том, к чему ближе свойства жидкостей: к свой-
свойствам ли соответствующего пара или твердого тела. Ответ на этот
вопрос будет зависеть от того, при какой температуре и давлении
производят это сравнение.
Если производить сравнение при температуре, близкой к темпе-
температуре кристаллизации вещества, то жидкость будет иметь больше
сходства с твердым телом.
Действительно, при плавлении твердого тела объем изменяется
незначительно. Для большинства веществ это изменение составляет
около 10%. Следовательно, при плавлении не может произойти
больших изменений в расположении молекул вещества, бывшего
до того твердым телом. К этому необходимо добавить, что тепло-
теплоемкость жидкости при температуре плавления близка к теплоем-
теплоемкости соответствующего твердого тела, что свидетельствует о сход-
сходстве в характере движения частиц вещества в этих двух состояниях.
Поскольку расположение частиц материи и их движение при плав-
плавлении твердого тела изменяются незначительно, то очевидно, что
в жидкостях при этих условиях должны сохраняться некоторые чер-
черты упорядоченного расположения частиц, характерные для твердого
тела.
В настоящее время имеются экспериментальные доказательства
наличия частичной упорядоченности в расположении молекул жид-
жидкости при температурах, близких к температуре кристаллизации.
Сближает жидкости с твердыми телами также их способность
сопротивляться растяжению, если только предупредить образова-
образование пара. Для того чтобы разорвать жидкость без образования па-
паровой фазы, подчас необходимы большие усилия. Жидкость может
существовать в растянутом состоянии, о котором говорилось уже
при обсуждении уравнения Ван-дер-Ваальса.
95
Следует, однако, отметить, что наряду с чертами сходства с твер-
твердыми телами жидкости обладают одновременно физическими свой-
свойствами, отличными от свойств твердых тел. Жидкости изотропны, т. е
их физические свойства не зависят от направления, в то время как
свойства кристаллических твердых тел зависят от направления.
Кроме того, жидкости отличаются от твердых тел текучестью.
Частицы, образующие жидкость, могут легко перемещаться одна
относительно другой. Можно представить себе некую идеальную,
крайне текучую жидкость, для которой модуль сдвига будет равен
нулю, т. е. у которой перемещение частиц не будет требовать ника-
никакого усилия1.
Как сказано выше, свойства жидкостей зависят от температуры,
и отмеченные черты сходства жидкостей с твердыми телами с по-
повышением температуры постепенно уступают место все более уси-
усиливающемуся сходству жидкости с соответствующим паром. Если
изучать свойства жидкости, находящейся в равновесии с паром при
температурах, близких к критическим, то придется отметить,
что при этих условиях сходными оказываются уже плотности жид-
жидкости и пара, а не плотности жидкости и твердого тела. В данном
случае на первый план выступают черты сходства жидкого и паро-
парообразного состояний вещества.
Исторически первоначально большее значение придавалось
сходству между жидкостью и паром. Жидкость рассматривалась
как сильно сжатый газ.
В последнее время иногда наблюдается обратная тенденция:
подчеркивается преимущественно сходство жидкости с твердыми
телами. Взятые в своих крайних формах, обе тенденции неверны.
Жидкое состояние — это промежуточное состояние, в котором
свойства вещества по мере возрастания температуры переходят
от свойств твердого тела к свойствам соответствующего пара, что
неразрывно связано с соответствующим изменением молекулярного
строения.
Из сказанного выше становится ясной трудность исчерпываю-
исчерпывающего определения понятия жидкого состояния.
В дальнейшем изложении мы будем считать, что жидкость отли-
отличается от газов тем, что ей всегда можно предоставить объем, до-
достаточно большой для того, чтобы она занимала бы его не полностью,
в то время как газ или пар всегда занимают полностью предостав-
предоставленный им объем.
Таким образом, жидкость, как и твердое тело, будет отличать-
отличаться от пара или газа наличием свободной поверхности.
1 Следует отметить, что способность реальных жидкостей к пластическим
деформациям, а следовательно, и текучесть зависят от скорости, с которой
жидкость подвергается механическому воздействию. Если по струе текущей
жидкости ударить металлическим прутом достаточно быстро, струя разла-
разламывается на отдельные кусочки подобно тому, как раскалывается хрупкое
твердое тело.
96
Примем дальше, что от твердого тела жидкость отличается под-
подвижностью своих частиц относительно друг друга, т. е. способно-
способностью течь.
2. МОЛЕКУЛЯРНОЕ ДАВЛЕНИЕ
При не слишком высоких температурах молекулярные объемы
жидкостей значительно меньше молекулярных объемов газов или
паров. Это означает, что молекулы жидкостей расположены гораз-
гораздо ближе друг к другу, нежели молекулы паров, а следовательно,
силы молекулярного притяжения в жидкостях значительно больше,
чем в парах.
В уравнении Ван-дер-Ваальса взаимодействие молекул учиты-
учитывается поправкой к давлению, равной отношению характерной для
данного вещества постоянной величины а к квадрату молекуляр-
молекулярного объема V, т. е. — .
Уравнение Ван-дер-Ваальса в первом приближении применимо
и к жидкостям, и потому, пользуясь им, можно оценить величину
поправки к внешнему давлению, которую необходимо ввести в том
случае, если вещество находится в жидком состоянии.
Молекулярный объем воды при 4°С составляет 18 • \0Г3м3.
Постоянная а для водяного пара — 5,52 • 105 н • ж4 /кмоль. По-
Поправка к давлению, следующая из уравнения Ван-дер-Ваальса для
воды в жидком виде, будет:
J&lOLn .W н/м\
Hi V2 A8 • 10-яJ
Не следует переоценивать значение сделанного расчета. Полу-
Полученная цифра указывает лишь на то, что взаимодействие молекул
в жидкостях эквивалентно очень большому внешнему давлению.
Экспериментальная оценка молекулярного притяжения в жидкостях
действительно подтверждает, что последнее эквивалентно давлению
в сотни, а иногда, вероятно, и в тысячи атмосфер.
Поправку к внешнему давлению, эквивалентную действию сил
притяжения, называют внутренним, или молекуляр-
молекулярным давлением жидкости.
Необходимо подчеркнуть отличие молекулярного давления от
вычисленного ранее для газов термического давления, обуслов-
обусловленного тепловым движением молекул. Молекулярное давление —
это не давление в том смысле, как оно понималось в учении о га-
газах, а скорее условное обозначение величины, учитывающей вли-
влияние сил молекулярного взаимодействия на объем, занимаемый
веществом.
Физически силы молекулярного взаимодействия, а следова-
следовательно, и то свойство жидкости, которое называют молекулярным
давлением, проявляются, если пытаться увеличить или уменьшить
4 Заказ 145 97
объем, занимаемый жидкостью, т. е. сблизить или удалить молекулы
вещества друг от друга.
В жидкости, которая занимает равновесный объем, молекулы
колеблются около положений, в которых силы притяжения между
ними в точности уравновешены силами отталкивания.
Состояние частиц в жидкости можно до некоторой степени уяс-
уяснить на примере грубо приближенной механической модели, если
представить себе две совершающие колебания частицы, соединенные
упругой пружиной. Жесткость пружины может быть весьма велика,
однако в положении равновесия силы притяжения и отталкивания
взаимно уравновешиваются, пружина практически не действует на
частицы, которые она соединяет. Наличие связи между частицами
проявляется лишь в том случае, если последние сближать или уда-
удалять друг от друга.
Большая величина молекулярного давления объясняет многие
свойства жидкостей. Так, например, поскольку молекулярное при-
притяжение эквивалентно большому внешнему давлению, т. е. жидко-
жидкости при атмосферном давлении уже как бы сильно сжаты силами,
действующими между их частицами, естественно, что изменение
внешнего давления оказывает малое влияние на объем жидкостей.
Другими словами, сжимаемость жидкостей мала.
3. ПОВЕРХНОСТНЫЕ СВОЙСТВА ЖИДКОСТЕЙ
Характерное для жидкости наличие поверхности, при прохож-
прохождении через которую плотность вещества скачкообразно изменя-
изменяется, определяет целый ряд свойств жидкости. Молекулы, рас-
расположенные в слое, непосредственно прилегающем к поверхности
жидкости, находятся в состоянии, отличном от состояния молекул
в ее толще. Это различие обусловлено особенностями молекуляр-
молекулярного взаимодействия. Молекулярные силы действуют на сравнитель-
сравнительно коротких расстояниях, порядка 10~9 метра. Если вокруг центра
молекулы описать сферу радиусом, равным расстоянию, на кото-
которое распространяется действие молекулярных сил, мы получим
то, что называют сферой молекулярного действия.
Частицы, находящиеся в толще жидкости, окружены со всех
сторон подобными же частицами, равномерно заполняющими их
сферы молекулярного действия. В этом случае молекулярные силы,
действующие на какую-либо частицу, будучи направлены симмет-
симметрично во все стороны, взаимно компенсируются, так что влияние
их на поведение частицы не сказывается (рис. 43).
В ином положении находятся частицы, расположенные в по-
поверхностном слое. Если для простоты представить поверхност-
поверхностный слой жидкости в виде геометрической плоскости, то частицы,
расположенные на поверхности, будут взаимодействовать, с одной
стороны, с большим количеством подобных им частиц, расположен-
расположенных в сфере молекулярного действия, обращенной в сторону жид-
98
кости, а с другой стороны — с небольшим числом частиц, располо-
расположенных в той половине сферы, которая находится в веществе в па-
парообразном состоянии. Это приводит к тому, что силовое поле моле-
молекулы, расположенной в поверхностном слое, оказывается ненасыщен-
ненасыщенным с той стороны, которая обращена к парообразной фазе.
Если мысленно представить себе процесс перехода одной из мо-
молекул жидкости из толщи на поверхность, то очевидно, что при этом
часть молекулярных связей, существовавших тогда, когда молеку-
молекула была окружена со всех сторон подобными ей, должна быть ра-
разорвана.
Из сказанного следует, что для перемещения молекулы из
толщи жидкости на поверхность необходимо затратить работу.
В первом приближении
можно считать, что при
этом процессе разрывается
ровно половина связей
между рассматриваемой
частицей и окружающими
ее частицами, и, следова-
следовательно, работа, необходи-
необходимая для перевода молеку-
молекулы из толщи жидкости на
поверхность, равна ПОЛО- Рис- 43- Взаимодействие между
вине той работы, которая молекулами жидкости.
необходима для перевода
молекулы из жидкости в паровую фазу. Естественно поэтому,
что всякое увеличение поверхности жидкости требует затраты
определенной работы, поскольку при этом необходимо перевести
какое-то количество молекул жидкости в поверхностный слой.
Лежащие в поверхностном слое частицы притягиваются не
только частицами, расположенными ниже их, но и своими соседями.
В результате этого в поверхностном слое возникает сила, вели-
величина которой зависит от интенсивности взаимодействия частиц
данной жидкости. Для количественной оценки этой силы ее от-
относят к единице длины линии, расположенной в поверхности.
Найденную таким способом величину называют коэффициен-
коэффициентом поверхностного натяжения а, или, иногда,
просто поверхностным натяжением. Размерность
коэффициента поверхностного натяжения н/м. Величина поверх-
поверхностного натяжения не зависит от того, будет ли линия, проведен-
проведенная на поверхности, кривой или прямой. Подобная зависимость
возникает лишь тогда, когда радиус кривизны этой линии делается
сравнимым с радиусом сферы молекулярного действия.
Иногда поверхностную пленку жидкости уподобливают рези-
резиновой пленке. Следует подчеркнуть, что это сходство в значитель-
значительной мере внешнее. Если увеличивать поверхность резиновой плен-
пленки, растягивая ее, то по мере растяжения увеличивается необхо-
4* 99
димая для этого сила. При увеличении же жидкостной пленки сила,
необходимая для этого, не зависит от растяжения пленки. Это объ-
объясняется тем, что при растяжении жидкой пленки расстояния меж-
между частицами жидкости остаются неизменными, а увеличение по-
поверхности достигается в результате перехода молекул из толщи
жидкости в поверхностный слой. Увеличение поверхности в этом
случае скорее напоминает не растяжение резиновой пленки, а сма-
сматывание ткани с валика, при котором натяжение ткани остается не-
неизменным.
Если проволочку, согнутую в виде буквы П, на ножки которой
надета подвижная перекладинка (рис. 44), опустить в мыльную во-
воду, а затем вынуть, то на ней возникнет
мыльная пленка. Поверхность возникшей
пленки можно увеличивать или уменьшать,
перемещая подвижную перекладинку. Обозна-
Обозначив длину перекладинки /, а коэффициент
поверхностного натяжения жидкости а, най-
найдем, что сила, необходимая для перемеще-
перемещения перекладинки, будет равна:
F = 2la. A)
Рис. 44. Опреде-
Определение поверхност-
поверхностного натяжения.
Возникновение множителя 2 в правой час-
части уравнения объясняется тем, что мыльная
пленка, удерживающая перекладинку, имеет
две поверхности. Приложив внешнюю силу,
равную F, можно заставить перекладинку переместиться на рас-
расстояние Ah. Очевидно, внешняя сила совершит при этом работу
АЛ, равную:
АЛ = 2laAh. B)
В результате перемещения перекладинки поверхность жидкости
возрастет на величину
AS = 21 Ah. C)
Сопоставление уравнений B) и C) убеждает в том, что работа,
необходимая для увеличения поверхности, связана простым со-
соотношением с поверхностным натяжением жидкости:
АЛ = aAS. {4)
Написанное соотношение часто используется для определения
поверхностного натяжения. На основании уравнения D) можно
написать:
а =
AS
т. е, коэффициент поверхностного натяжения численно
равен работе, необходимой для образования \ м2 Ho-
Hoвой поверхности жидкости. В этом случае коэффициент по-
поверхностного натяжения жидкости имеет размерность дж/м2.
С работой против сил поверхностного натяжения приходится
считаться при распылении жидкости.
Пример. Определить работу, необходимую для дробления 1 г воды на
шарики радиусом 10~б см, т. е. практически для превращения 1 г воды в ту-
туман.
Коэффициент поверхностного натяжения воды а = 73 • 10~3 н/м, плот-
плотность воды р = 103 кг/м3.
4 4
Решение. Объем капельки V —— kR3, ее масса то =pV = р—я/?3. Чис-
3 3
ло образовавшихся из 1 г воды капелек равно N = = .Поверх-
ность капельки So = 4яЯ2. Суммарная поверхность всех капелек1:
Работа дробления воды:
А = aS = 73 • Ю-3 • 30 « 2 дж.
Таким образом, для дробления воды потребуется работа, равная приблизи-
приблизительно 2 джоулям.
Поскольку возникновение новой поверхности связано с совер-
совершением внешними силами определенной работы, то естественно,
что поверхность жидкости обладает энергией. Энергия, в которую
превращается работа, затраченная на образование поверхности
жидкости, называется свободной энергией поверх-
поверхности.
Свободной энергией называют ту часть общей энергии тела,
которая может быть превращена в работу, если процесс протекает
при постоянной температуре.
В механических процессах, при которых температура не играет
роли, свободной энергии соответствует потенциальная энергия.
Одним из важнейших законов природы является закон, согласно
которому в любой системе при постоянной температу-
температуре устойчивое равновесие наблюдается тогда, когда
свободная энергия системы минимальна. Этот за-
закон называют принципом минимума свободной
энергии, а в механике — принципом минимума
потенциальной энергии. Принцип минимума свобод-
свободной энергии объясняет многие явления, знакомые нам из повседнев-
повседневной жизни.
1 Первоначальная поверхность водяного шарика массой 1 г составляла
около 5' 10~4 hi2, поэтому она в расчете не учитывается.
101
Рис. 45.
Всем хорошо известно, что ма-
маленькие капельки ртути имеют фор-
форму шарика. Эта форма обусловле-
обусловлена тем, что среди различных ге-
геометрических тел при одном и том
же объеме шар обладает наимень-
наименьшей поверхностью. Поэтому капель-
капелька ртути, имеющая форму шара,
будет обладать наименьшей свободной поверхностной энергией по
сравнению с капельками ртути той же массы, но другой формы.
Если присмотреться к капелькам ртути больших размеров
(рис. 45), лежащим на твердой поверхности, то мы заметим, что их
форма отличается от сферической. Это объясняется тем, что свобод-
свободная энергия F капельки, находящейся в поле земного тяготения,
складывается из двух слагаемых:
F = aS -f- tngh.
Первое слагаемое обусловлено наличием поверхности, а второе —
действием силы тяготения. Минимальной должна быть сумма
обоих слагаемых. При увеличении объема капельки ее масса воз-
возрастает быстрее, чем поверхность. Так, например, при увеличении
объема сферической капли в 8 раз ее масса возрастает также в
8 раз, а поверхность — только в 4 раза. Поэтому при увеличении
размеров капли второе слагаемое в написанной выше сумме, про-
пропорциональное массе капельки, приобретает большее значение,
чем первое. В то же время величина потенциальной энергии тяго-
тяготения пропорциональна высоте h, на которой находится центр
тяжести капли. Поэтому для уменьшения потенциальной энергии
тяготения капля должна была бы растечься по поверхности. Этого
не происходит, поскольку растекание капли вызвало бы значитель-
значительное возрастание ее свободной поверхностной энергии.
Реальная форма капли находится из условия минимального значе-
значения суммы обоих составляющих свободной энер-
энергии.
Стремлением свободной энергии к мини-
минимуму объясняется своеобразная форма мыль-
Рис. 46. Форма мыльных пленок на проволочных
каркасах.
ных пленок (рис. 46), возникающих на проволочных каркасах,
погруженных в мыльный раствор и вынутых затем из него.
Поверхностное натяжение жидкости зависит от температуры.
Для сравнительно небольших интервалов температур поверхност-
поверхностное натяжение обычно уменьшается линейно с возрастанием тем-
температуры. В критической точке, когда плотность жидкости и пара
одинакова, поверхностное натяжение жидкости равно нулю.
Поверхностное натяжение существует не только на границе
жидкости с паровой фазой, но и на границе двух несмешивающих-
ся жидкостей, или жидкости и твердого тела. Поверхностное натя-
натяжение на границе двух несмешивающихся жидкостей может суще-
существенно отличаться от поверхностного натяжения каждой из них
на границе с ее насыщенным паром.
То же самое справедливо для поверхностного натяжения на гра-
границе жидкость — твердое тело.
4. ЯВЛЕНИЯ НА ГРАНИЦЕ ЖИДКОСТИ С ТВЕРДЫМ ТЕЛОМ
При соприкосновении жидкости с поверхностью твердого тела
различают два случая: когда жидкость смачивает твердое тело и
когда жидкость не смачивает твердое тело.
Чистая стеклянная палочка, опущенная в воду, смачивается
водой. Вынутая из воды стеклянная палочка несет на конце каплю
воды. Та же палочка, опущенная в ртуть, не смачивается ртутью.
На вынутой из ртути палочке ртути не остается.
Понять, почему одни жидкости смачивают, а другие не смачи-
смачивают данное твердое тело, помогает рисунок 47.
Представим себе, что твердое тело граничит в точке А с гори-
горизонтальной плоской поверхностью жидкости. На молекулу
жидкости в точке А действуют три силы: Д, направленная горизон-
горизонтально и определяемая поверхностным
натяжением а23 жидкости на границе
жидкость — пар; /2, направленная вер-
вертикально вниз и определяемая поверх-
поверхностным натяжением ai2 на границе
твердое тело — жидкость, и /3, направ-
направленная вертикально вверх и определяе-
определяемая поверхностным натяжением сс]3 на
границе твердое тело — пар.
Смачивание или несмачивание по-
поверхности твердого тела будет зависеть
от соотношения сил /3 и /2, т. е. от соотно-
соотношения поверхностных натяжений твердо-
твердого тела на границе с паром жидкости а1з
и с самой жидкостью а12. теЛо — жидкость.
/,
Пар
^=Е2 =
103
Если а1з > а12, то молекула, находившаяся в точке А, начнет
двигаться вверх, в результате чего поверхность жидкости сделает-
сделается изогнутой и силы, действующие на молекулу, будут теперь на-
направлены так, как это изображено на рисунке 48.
Равновесие наступит, когда будет выполняться условие:
«13 = «12 + «23 COS д. F)
Угол б, образованный касательной к поверхности жидкости в той
точке, где жидкость граничит с твердым телом и поверхностью твер-
твердого тела, называют краевым углом.
Следует подчеркнуть, что краевой угол всегда измеряют со
стороны жидкости. У жидкостей, смачивающих твердое тело, крае-
краевой угол острый. Равновес-
Равновесная величина краевого угла оп-
определяется из соотношения:
Рис. 48. Форма
границы твер-
твердое тело—жид-
тело—жидкость, смачи-
смачивающая твер-
твердое тело.
Рис. 49. Форма
границы твер-
твердое тело —жид-
—жидкость, не сма-
смачивающая твер-
твердое тело.
Рис. 50. Форма капель жидкости,
лежащих на поверхности твердого
тела. Слева — капля жидкости, не
смачивающей поверхность, справа —
смачивающей.
В том случае, когда соотношение поверхностных натяжений на
границе твердого тела с жидкостью и паром оказывается обратным
рассмотренному, т. е. когда о13<а12, поверхность жидкости при-
приобретает кривизну противоположного знака (рис. 49).
Условием равновесия будет равенство:
«i2 = «i3 + a23cosA80° —б). G)
Равновесное значение краевого угла определяется из соотношения:
cos б = — °»-°» .
Краевой угол в случае жидкостей, не смачивающих твердое тело,
тупой.
На плоской поверхности твердого тела капли жидкости, сма-
смачивающей и не смачивающей твердое тело, приобретают характер-
характерную форму (рис. 50).
У жидкости, полностью смачивающей твердое тело, краевой угол
равен нулю, а у жидкости, полностью не смачивающей твердое
тело — 180°.
104
Смачивание или несмачивание жидкостью твердого тела опре-
определяет поведение жидкости в узких трубках — капиллярах.
Если жидкость смачивает стенки капиллярной трубки, возникает
вогнутый мениск. Сила поверхностного натяжения /, действующая
по периметру жидкой поверхности в капилляре радиуса R и рав-
равная произведению коэффициента поверхностного натяжения на
периметр сечения капилляра (/ = a2nR), стремится сократить по-
поверхность жидкости. Стремление поверхности жидкости сокра-
сократиться вызывает в свою очередь отклонение величины краевого
угла б от равновесного значения, опре-
определяемого написанными соотношениями.
Для достижения равновесного значения
краевого угла граница жидкости с твер-
твердым телом перемещается по капилляру
вверх, увлекая при этом за собой стол-
столбик жидкости (рис. 51). Подъем жид-
жидкости в капилляре вызывает новое из-
изменение краевого угла, которое при-
приводит к дальнейшему перемещению жид-
жидкости вверх по капилляру, прекращаю-
прекращающемуся тогда, когда составляющая по-
поверхностного натяжения, тянущая столб
жидкости вверх:
f = /cos б = 2nRa cos б,
уравновешивается тяжестью столба mg, ~
т. е.
2nRa cos б = mg, Рцс 5} Подъеы жидкости
„ в капилляре,
где т — масса столбика жидкости,
g — ускорение силы тяжести.
Поскольку масса столбика жидкости т равна его плотности р,
умноженной на объем V = nR^h, где h — высота столбика, можно
написать:
2nRa cos б = nR2hpg.
Написанное соотношение позволяет рассчитать подъем жидко-
жидкости в капилляре по сравнению с уровнем жидкости в широком со-
сосуде:
А =
cos б.
(8)
Таким образом, смачивающая капиллярную трубку жидкость
будет подниматься по ней тем выше, чем уже трубка. В противо-
противоположность описанному явлению уровень жидкости, не смачи-
смачивающей стенки капилляра, будет располагаться в нем ниже, чем
в широком сосуде.
105
Капиллярные явления играют большую роль в жизни растений.
Они во многом определяют водный режим почв. Например, в удер-
удержании влаги выше уровня грунтовых вод важная роль принадле-
принадлежит капиллярному поднятию воды.
С поверхностными явлениями на границе «твердое тело — жид-
жидкость» связано открытое Б. В. Дерягиным с сотрудниками раскли-
расклинивающее действие тонкого слоя жидкости, проникающего в зазор
между поверхностями двух твердых тел. Расклинивающий эффект
возникает в результате молекулярного притяжения и наглядно до-
доказывает существование на границе твердого тела слоя жидкости
с аномальными свойствами, толщина которого превосходит намного
молекулярные размеры.
Расклинивающее действие жидких пленок может объяснить об-
облегчение раскалывания твердого тела в присутствии жидкости.
Если с помощью бритвы не полностью отщепить от куска слюды
тоненький листочек, а затем, не вынимая бритвы, ввести в обра-
образовавшийся зазор каплю воды, то можно наблюдать быстрое пере-
перемещение границы, отделяющей отколотый листочек от основного
куска слюды, и углубление образовавшегося раскола.
5. ДАВЛЕНИЕ ПОД ИЗОГНУТОЙ ПОВЕРХНОСТЬЮ ЖИДКОСТИ
Натяжение, существующее в поверхностном слое, вызывает в
том случае, если поверхность жидкости изогнута, возникновение
давления, называемого лапласовским, поскольку Лаплас впер-
впервые обратил на него внимание.
Лапласовское давление р, как это ясно из рассмотрения рисун-
рисунка 52, направлено внутрь жидкости, если поверхность жидкости
выпуклая, и в сторону паровой фазы, если поверхность — вог-
вогнутая.
С целью определения лапласовского давления подсчитаем ра-
работу, необходимую для дифференциально малого увеличения
объема мыльного пузыря.
Свободная энергия F поверхности мыльного пузыря радиуса R
равна, согласно сказанному выше, произведению площади повер-
поверхности пузыря на коэффи-
коэффициент поверхностного натя-
натяжения а. Учитывая, что
оболочка пузыря имеет две
поверхности (внешнюю и
внутреннюю) площадью по
AvcR2 каждая, можно напи-
написать:
Рис. 52. Возникновение лапласовского
давления.
F = 8л#2а.
(9)
106
Пленка мыльного пузыря оказывает давление р на заключен-
заключенный в пузыре воздух, и поэтому при увеличении радиуса пузыря
на элементарно малую величину dR внешние силы должны совер-
совершить работу против этого давления, равную dA = pdV. Эта работа
затрачивается на увеличение свободной энергии dF поверхности
пузыря, которая возрастает при увеличении его радиуса.
Приравнивая увеличение свободной энергии работе, совершен-
совершенной внешними силами, dA — dF и подставляя вместо объема V его
значение, равное для шара-л^3, а вместо свободной энергии F
О
ее значение из уравнения (9), найдем:
pinR4R = l6nRadR,
или
,-*. am
Если рассматривать не мыльный пузырь, а пузырек, возникший
в толще жидкости, то, поскольку последний обладает только од-
одной поверхностью, избыточное давление в нем будет в половину
меньше подсчитанного, т. е.
Полученные соотношения подтверждают, что лапласовское
давление обусловлено кривизной поверхности. В случае плоской
поверхности, для которой радиус кривизны бесконечно велик,
лапласовское давление обращается в нуль. Следует обратить вни-
внимание также на то, что по мере увеличения радиуса пузырька лап-
лапласовское давление уменьшается, а не увеличивается, как это на-
наблюдается в том случае, когда избыточное давление обусловлено
упругостью пленки (резиновый мяч).
В пузырьках, размеры которых не очень малы, лапласовское
давление незначительно по сравнению с атмосферным и его можно
обычно не учитывать. При уменьшении же радиуса пузырька оно
возрастает и может достигать очень больших величин. Так, напри-
например, в возникшем в воде пузырьке радиусом 10~э м лапласовское
давление составит около 15 • 107 н/м2.
В технике приходится считаться с тем, что при различных
процессах в жидкости могут возникать наполненные паром пу-
пузырьки. Это наблюдается при быстром движении жидкости в трубе,
при распространении в ней интенсивных звуковых и ультразвуковых
волн, при пропускании в жидкость через узкое сопло перегретого
пара жидкости и т. п. Явления возникновения пузырьков в жидко-
жидкости называют кавитацией, а пузырьки — кавитационными
пузырьками. Кавитация возникает, в частности, при вращении
гребных винтов кораблей.
107
Сжатие и полное исчезновение кавитационных пузырьков сопро-
сопровождается возникновением огромных давлений, измеряемых ты-
тысячами атмосфер. Эти давления являются одной из причин свое-
своеобразных явлений, наблюдаемых при кавитации. Так, например,
кавитация может вызывать разрушение гребных винтов кораблей,
дробить большие молекулы органических соединений, вызывать
химические превращения, свечение воды и т. д.
Если поверхность жидкости имеет кривизну, отличную от сфери-
сферической, лапласовское давление может быть подсчитано при помощи
уравнения:
'НУ- A2)
в котором Ri и R2 — радиусы
кривизны двух произвольно про-
проведенных взаимно перпендикуляр-
перпендикулярных нормальных сечений.
Для того чтобы уяснить значе-
значение последнего уравнения, подсчи-
подсчитаем лапласовское давление в жид-
жидкости, удерживающейся в силу
капиллярности между двумя парал-
параллельными пластинками (рис. 53).
Проведем два взаимно перпенди-
перпендикулярных нормальных сечения
одно из которых проходит вдоль
образующей цилиндрической по-
Поскольку радиус кривизны, соответ-
сечению, бесконечно велик, —
Рис. 53. Поведение жидкости,
заключенной между двумя па-
параллельными плоскими пластин-
пластинками.
верхности
ствующий
жидкости,
последнему
то — = 0.
Очевидно, в рассматриваемом случае лапласовское давление будет
равно:
В описанном опыте стремление поверхности жидкости к сокра-
сокращению будет стягивать пластинки, если жидкость их смачивает,
и расталкивать, если не смачивает.
Пример. Требуется определить давление, которое может расплющить
каплю ртути весом 0,02 Г между тонкими стеклянными пластинками в лепеш-
лепешку радиусом 0,9 см. Плотность ртути р = 13 600 кг/м3. Поверхностное натя-
натяжение а = 0,5 н/м.
Решение. Объем капли ртути-
2-10-5
13,6-103
¦= 1,47 - 10~а ж3,
где m — масса ртути.
108
Площадь одного из оснований лепешки:
S = я/?2 = 3,14(9-Ю-3J = 2,54.10"* ле2.
Расстояние между пластинами:
V 1,47-10~9
Радиус кривизны края ртутной лепешки:
„ h 5,8-10-е
Я = у= —- =2,9.10-" м.
Давление, необходимое для того чтобы расплющить ртутную капельку
в лепешку:
6. ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
В случае растворов поверхностные свойства существенно за-
зависят от того, понижает ли растворенное вещество поверхностное
натяжение растворителя или же, наоборот, повышает. Если рас-
растворенное вещество понижает поверхностное натяжение раствори-
растворителя, то в силу стремления свободной энергии жидкости к мини-
минимуму , растворенное вещество будет по возможности полнее пере-
переходить в поверхностный слой. Очевидно, такое вещество будет
плохо растворяться в жидкости, собираясь преимущественно в
поверхностном слое. Подобные вещества называют поверх-
поверхностно-активными. Первые порции поверхностно-актив-
поверхностно-активного вещества практически целиком собираются в поверхностном
слое. Накопление их в этом слое продолжается до тех пор, пока вся
поверхность жидкости не окажется занятой молекулами поверх-
поверхностно-активного вещества.
Наоборот, вещества, повышающие поверхностное натяжение
растворителя, будут преимущественно собираться в толще жидкости,
а поверхностный слой будет состоять практически из чистого раство-
растворителя. Эти вещества называют поверхностно-неактив-
поверхностно-неактивными.
По отношению к воде поверхностно-активными веществами яв-
являются многие органические соединения. Молекулы этих веществ
велики, и различные части молекул могут по-разному взаимодейст-
взаимодействовать с водой. Сказанное легко проследить на примере различных
жирных кислот. Молекулы их состоят из более или менее длинной
цепочки углеродных атомов, оканчивающейся с одной стороны так
называемой кислотной группой, способной при растворении отде-
отделять положительно заряженный атом водорода, а с другой стороны—
атомом углерода, соединенным с тремя атомами водорода.
109
На рисунке 54 изображены структурные формулы уксусной и
нормальной валериановой кислот. Если так называемые кислотные
группы — C<f в правой части цепочек заменить группами
\<ЭН
н
I
—С—Н , такими же, какие стоят в молекулах слева, то образу-
Н
ются соединения, называемые в химии парафинами.
Парафины нерастворимы в воде. Растворимость жирных кислот
обусловлена наличием кислотной группы — С<(
ЮН
Если углеводородная цепь в кислоте невелика—кислота хо-
хорошо растворяется в воде. Кислоты с большой углеводородной цепью
в воде практически нерастворимы. Бу-
I дучи добавлены к воде, молекулы этих ве-
' />^ ществ собираются в поверхностном слое.
н С Сч ]^ак показали опыты, молекулы
I он поверхностно-активных веществ распо-
н лагаются в поверхностном слое упо-
упорядочение Для того чтобы убедиться
в этом, было определено весовое колп-
i ^ у ^ чество различных кислот, которое необ-
I I I i jr ходимо для того, чтобы покрыть сплошь
I ^ ^ С^ поверхность воды слоем кислоты толщи-
' ' ОН ной в 1 молекулу. Подобные опыты
были проделаны для кислот, начиная
Рис. 54. Структурные фор- от пальмитиновой, содержащей угле-
мулы жирных кислот. родную цепочку из 15 атомов угле-
углерода (Ci6Hsl COOH) до церотиновой,
в которой углеводородная цепь содержит уже 25 атомов
углерода (С25Н51СООН). Зная молекулярные веса соответствую-
соответствующих соединений, было нетрудно вычислить, сколько молекул со-
соответствующего поверхностно-активного вещества располагается
на одном квадратном сантиметре мономолекулярного слоя, и опре-
определить площадь на поверхности, занимаемую одной молекулой.
При этом оказалось, что для всех кислот эта площадь приблизи-
приблизительно одна и та же и равна 21 • 10~20 ж2. Такой результат опытов
можно объяснить, только предположив, что молекулы указанных
соединений располагаются в поверхностном слое наподобие частоко-
частокола или хлебных колосьев на поле. Погруженной в воду оказывается
/
при этом кислотная группа — С\оН, которая и занимает в
по
поверхностном слое указанную выше площадь. Углеводородная це-
цепочка оказывается торчащей из воды, поэтому она не влияет на раз-
размеры площади, занимаемой молекулой на поверхности воды (рис. 55).
Поверхностные явления вообще и поверхностно-активные ве-
вещества, в частности, играют важную роль в технике. Поверхностно-
активными веществами являются различные мыла. Мыла увеличи-
увеличивают смачиваемость водой загрязненной поверхности и облегчают
удаление частиц загрязнения с поверхности твердого тела. Кроме
того, мыла удерживают частицы загрязнения, когда последние на-
находятся уже в водной среде.
Особенно большое значение для техники имеет использование
поверхностных явлений в процессе обогащения руд, называемом
Мономолвиулярный слой
Рис. 55. Молекулы поверхностно- Рис. 56. Схема
активных веществ в поверхностном флотации,
слое.
флотацией. Как правило, кусочки руды плохо смачиваются
водой, но хорошо смачиваются жирными органическими вещест-
веществами. Напротив, пустая порода, всегда встречающаяся вместе с
рудой, хорошо смачивается водой и плохо маслами. Если загру-
загрузить раздробленную руду вместе с породой в воду, в которой в
виде мельчайших капелек находятся специальные масла, то кусоч-
кусочки породы окажутся смоченными водой, а кусочки руды — маслом.
Интенсивно вспенивая образовавшуюся массу и отделяя часть, ухо-
уходящую вместе с пеной от той, которая падает на дно сосуда, можно
значительно обогатить руду содержащимися в ней ценными мине-
минералами. Обогащение достигается в результате того, что присутствую-
присутствующие в пене пузырьки воздуха (рис. 56) прилипают к маслянистому
слою, обволакивающему кусочки руды, и уносят их вместе с пеной
на поверхность. К смоченным водой кусочкам пустой породы, удель-
удельный вес которой обычно меньше удельного веса руды, воздушные
пузырьки не прилипают, и пустая порода падает на дно.
Изменяя флотореагент, как называют добавленное для флотации
вещество, можно не только обогащать руду ценными материалами,
Hi
но и производить разделение руд, если одновременно вместе с по-
породой присутствуют несколько различных минералов. В этом слу-
случае первоначально вместе с пеной удаляется один из минералов,
а второй уходит вместе с пустой породой и выделяется при повторе-
повторении флотационного процесса с применением иного флотореагента.
7. ИСПАРЕНИЕ ЖИДКОСТЕЙ. НАСЫЩЕННЫЙ ПАР
И ЕГО СВОЙСТВА
Расположенные в поверхностном слое жидкости молекулы не
остаются в нем неограниченно долго. Тепловое движение вызыва-
вызывает непрерывную замену их молекулами, расположенными в более
глубоких слоях жидкости, с которыми они как бы меняются местами.
При подобном перемещении может оказаться, что молекула, дви-
движущаяся из глубины жидкости к поверхности, будет обладать боль-
большой скоростью. В этом случае можно было бы ожидать, что мо-
молекула обязательно покинет жидкость и перейдет в паровую фазу.
Такое предположение, однако, неверно, поскольку при этом
не учитывается, что удаление молекул из поверхностного слоя свя-
связано с работой испарения — Л11СП, которую необходимо совер-
совершить для преодоления сил молекулярного притяжения. Работа по
преодолению молекулярного сцепления совершается за счет кине-
кинетической энергии движущейся молекулы, определяемой однозначно
ее скоростью. Решающее значение при переходе молекулы в паро-
паровую фазу имеет не вообще величина скорости, а только ее составляю-
составляющая сп в направлении, перпендикулярном к поверхности. Молеку-
Молекула покинет жидкость в том случае, если выполняется неравенство:
тс2„
A3)
где т —масса молекулы.
Поскольку при испарении жидкости работа против сил моле-
молекулярного сцепления совершается за счет энергии теплового дви-
движения ее молекул, то температура жидкости понижается. Для
поддержания температуры испаряющейся жидкости постоянной к
ней необходимо подводить теплоту. Эта теплота не вызывает повы-
повышения температуры и потому ее называют внутренней скры-
скрытой теплотой испарения, или просто внутрен-
внутренней теплотой испарения (обозначается — ^). По-
Помимо работы, обусловленной преодолением сил молекулярного
сцепления, при испарении жидкости необходимо также совершить
работу, связанную с увеличением объема, занимаемого веществом.
Количественно эта работа равна произведению давления пара жид-
жидкости на разность объемов вещества в парообразном и жидком со-
состояниях:
112
Эквивалентное количество теплоты называют внешней теп-
теплотой испарения.
Сумму внутренней и внешней теплот испарения
Ь*=К + Р{УП.-УЖ.) A4)
называют общей теплотой испарения, а часто просто
теплотой испарения жидкости.
Количество теплоты, необходимое для изотермического испа-
испарения 1 кг жидкости, называют удельной теплотой ис-
испарения, а количество теплоты, необходимое для изотермиче-
изотермического испарения 1 киломоля жидкости, — молекулярной
теплотой испарения.
Очевидно, что неравенство A3), определяющее возможность
перехода молекулы из жидкости в область пара, будет выполнять-
выполняться не для каждой молекулы. Если теплота испарения велика, то
только немногие молекулы окажутся обладающими составляющей
скорости сп, достаточной для того, чтобы молекула покинула жид-
жидкость и перешла бы в паровую фазу. Поэтому из общего числа мо-
молекул, которые за 1 секунду подходят к квадратному метру поверх-
поверхности раздела жидкости и пара со стороны жидкости, только неболь-
небольшая часть будет переходить в паровую фазу. Количество молекул,
переходящих за 1 секунду из жидкости в пар на 1 м? поверхности
их раздела, определяет интенсивность испарения жидкости. _
Для молекул, подходящих к поверхности раздела со стороны
паровой фазы, указанного ограничения не существует. При при-
приближении молекул пара к границе, разделяющей пар и жидкость,
они попадают в ненасыщенное силовое поле, существующее вблизи
поверхности жидкости, и их движение в некоторой степени должно
напоминать движение тяжелого тела вблизи земной поверхности.
Поэтому, если только поверхность жидкости не загрязнена, слу-
случаи отражения молекул пара от поверхности жидкости будут
крайне редкими. Можно принять, и это, вероятно, близко к истине,
что все молекулы пара, которые ударяются о незагрязненную поверх-
поверхность жидкости, переходят в жидкую фазу.
Таким образом, интенсивность конденсации пара, измеряемая
числом молекул, переходящих на 1 ж2 поверхности раздела за 1
секунду из паровой фазы в жидкую, по существу определяется чис-
числом соударений молекул пара с указанной поверхностью за 1 се-
секунду.
Известно, что в закрытом сосуде жидкость при достаточном ее
количестве испаряется не целиком. В определенный для данной
температуры момент процесс испарения жидкости компенсируется
процессом конденсации пара. Возникает состояние динамиче-
динамического равновесия, при котором макроскопические коли-
количества жидкости и пара остаются неизменными.
Условием динамического равновесия является равенство ско-
скоростей взаимно компенсирующихся процессов. В рассматриваемом
ИЗ
случае число молекул жидкости, отрывающихся за 1 секунду от
1 квадратного метра поверхности жидкости и переходящих в пар,
должно быть равно числу молекул пара, переходящих за то же вре-
время на той же поверхности в жидкость. Из сказанного выше совершен-
совершенно ясно, что подобное равенство может выполняться только в том
случае, если за единицу времени к данному участку поверхности
раздела фаз со стороны жидкости будет подходить гораздо больше
молекул, чем со стороны пара. Это обусловлено тем, что молекулы
пара все без остатка будут переходить в жидкость, а из жидкости
переходить в пар будет только небольшая часть из числа молекул,
оказавшихся на поверхности.
При одинаковой средней скорости молекулярного движения
указанное условие может выполняться только в том случае, если
плотность жидкости будет гораздо больше плотности пара. Такое
соотношение в плотностях и наблюдается в действительности при
температурах, далеких от критической. Пар, находящийся в рав-
равновесии с жидкостью, называется насыщенным.
Для каждой жидкости, находящейся при некоторой темпера-
температуре в равновесии с собственным паром, число ежесекундно испа-
испаряющихся молекул является постоянной величиной, а следова-
следовательно, постоянным является и число молекул, переходящих из
пара в жидкость. В свою очередь количество конденсирующихся
молекул зависит от плотности, а следовательно, и давления пара,
и поэтому можно утверждать, что при постоянной темпера-
температуре давление насыщенного пара для данной жид-
жидкости—величина постоянная, независящая от объема,
им занимаемого. Изменение объема, занимаемого паровой фазой,
вызывает или дальнейшее испарение жидкости, если объем увели-
увеличивается, или же частичную конденсацию пара, если объем умень-
цается.
Следует также отметить, что давление насыщенного пара не
зависит от величины поверхности, с которой происходит испарение.
Величина поверхности может влиять только на время, необходи-
необходимое для достижения равновесного состояния.
Наоборот, температура сильно влияет на давление насыщенного
пара. Объясняется это тем, что скорость теплового движения мо-
молекул жидкости зависит от температуры и число молекул, скорости
которых превышают необходимую для испарения предельную вели-
величину, быстро возрастает с ростом температуры. Кроме того, с ро-
ростом температуры возрастает число молекул, подходящих к по-
поверхности за 1 секунду. Оба эти обстоятельства увеличивают коли-
количество ежесекундно испаряющихся молекул, а следовательно,
и равновесное значение числа молекул пара, конденсирующихся
за то же время. Графически зависимость давления насыщенного
пара от температуры для различных жидкостей выражается кривы-
кривыми параболического вида (рис. 57), обрывающимися в критической
точке.
114
Таким образом, критическое давление — это максимальное дав-
давление насыщенного пара данной жидкости.
В заключение следует подчеркнуть, что испарение жидкости
происходит при всех температурах. При низких температурах испа-
испарение происходит только со свободной поверхности жидкости. Ког-
Когда при постепенном повышении температуры давление насы-
насыщенного пара жидкости сделается равным атмосферному давлению,
пузырьки пара начнут образовываться в толще жидкости. Процесс
испарения, сопровождающийся образованием пузырей пара внутри
жидкости, низывается кипением. Температура кипения жидкос-
жидкости зависит от внешнего давления:
с понижением внешнего дав-
давления температура кипения
понижается.
Образование пузырьков пара
внутри жидкости часто бывает
затруднено. Одна из причин это-
этого — большое лапласовское давле-
давление в возникающих в толще жид-
жидкости микроскопических пузырь-
пузырьках. Процесс возникновения пу-
пузырьков пара облегчается присут-
присутствием в жидкости растворенных
газов. С повышением температуры
растворимость газов в жидкости
уменьшается и они выделяются
в виде мельчайших пузырьков га-
газа, которые играют роль центров
возникновения пузырьков пара. При отсутствии растворенных га-
газов жидкости легко перегреваются выше температуры кипения и
процесс кипения приобретает бурный характер.
Иногда для предотвращения перегрева всей жидкости в ней
искусственно создают небольшие области перегрева, в которых
возникают микроскопические пузырьки пара, вызывающие кипение.
Рис. 57. Зависимость давления
насыщенного пара жидкости
от температуры.
8. МОЛЕКУЛЯРНОЕ СТРОЕНИЕ ЖИДКОСТЕЙ
В современной молекулярно-кинетической теории материи раз-
различные агрегатные состояния вещества связывают с различной сте-
степенью упорядоченности в расположении его частиц. Для газообраз-
газообразного состояния характерно полностью беспорядочное, хаотичное
расположение молекул. В противоположность этому в идеальном
кристалле частицы расположены в строгом порядке, распространяю-
распространяющемся на весь кристалл. Правильное расположение частиц в кри-
кристаллических твердых телах подтверждается экспериментально
опытами по рассеянию рентгеновских лучей кристаллами.
115
N№
Этими опытами удалось, например, установить, что атомы в
ряде кристаллов образуют так называемую центрированную ку-
кубическую кристаллическую решетку (рис. 58, а). Атомы, нахо-
находящиеся в узлах такой кристаллической решетки, расположены
на вполне определенных расстояниях от произвольно выбранного
атома (О — на рис. 58). Рассматриваемая кристаллическая решет-
решетка характеризуется тем, что на расстоянии ОА от выбранного атома
находятся 8 атомов, на расстоянии ОВ — 6 атомов и т. д.
Указанное пространственное распределение атомов в решетке
можно изобразить графически, отложив на оси абсцисс расстоя-
расстояния R, а на оси ординат — величи-
величину N (R), равную числу атомов
N, находящихся на одном квадрат-
квадратном сантиметре сферической по-
поверхности с радиусом R, описан-
описанной вокруг атома О, выбранного
за начало отсчета.
График, построенный по это-
этому принципу, показан на рисун-
рисунке 58, б.
Рентгенографический метод по-
позволяет на основании результа-
результатов опытов рассчитывать и стро-
строить аналогичные графики для всех
исследуемых веществ.
Применение этого метода к
изучению строения ' простейших
(атомных) жидкостей при темпе-
температурах, близких к температуре
их кристаллизации, привело к
установлению факта, чрезвычайно
важного для теории жидкого со-
состояния. Оказалось, что при этих
условиях в жидкости в значительной степени сохраняется упоря-
упорядоченность в расположении частиц, характерная для кристалла.
Рентгенограммы атомных жидкостей напоминают рентгенограммы,
полученные для порошкообразных кристаллических тел. Подобны-
Подобными же опытами было обнаружено, что с повышением температуры
эта упорядоченность уменьшается, расположение частиц жидкости
приближается к расположению, свойственному частицам газов.
Для объяснения результатов этих опытов было предложено
несколько теорий. По одной из них жидкость состоит из субми-
субмикроскопических кристалликов, разделенных тонкими пленками ве-
вещества в аморфном состоянии, характеризуемом беспорядочным рас-
расположением частиц. Субмикроскопические кристаллики назвали
сиботаксическими областями. В отличие от реаль-
реальных кристалликов сиботаксические области очерчены не резко, они
8 Ю
R
Рис. 58 Строение кристалличе-
кристаллической решетки и зависимость чис-
числа соседних атомов в решетке от
расстояния, выраженного в анг-
ангстремах.
116
плавно переходят в области неупорядоченного расположения, частиц.
Кроме того, сиботаксические области непостоянны, они непрерывно
разрушаются и возникают вновь. Наличие областей упорядочен-
упорядоченного расположения частиц приводит к тому, что у большинства
молекул жидкости соседние с ними частицы располагаются в опре-
определенном, характерном для данной жидкости порядке. Однако
благодаря хаотической ориентации отдельных сиботаксических
групп в отношении друг друга упорядоченное расположение мо-
молекул распространяется только на ближайших к данной молекуле
а
Рис. 59. Сравнение строения идеального кристалла и жидкости.
соседей. На расстоянии трех-четырех молекулярных диаметров
упорядоченность уменьшается столь сильно, что теряет смысл
говорить о правильном порядке в расположении частиц вещества.
В настоящее время считается общепризнанным, что жидкости
свойственна упорядоченность ближнего порядка в расположении
ее частиц в отличие от кристаллов, которые характеризуются упо-
упорядоченностью дальнего порядка.
Различие в строении кристаллического тела и жидкости схема-
схематично показано на рисунке 59. Слева на рисунке изображено строе-
строение идеального гипотетического кристалла. Его структурные ча-
частицы в любом месте кристалла занимают строго определенное
положение относительно друг друга. Однако в жидкостях (на
рисунке — справа), в окрестности произвольно выбранной моле-
молекулы О, соседние молекулы могут иметь расположение, как весьма
близкое к кристаллическому (направление ОВ), так и отличное от
него (направление ОС). Во всяком случае, в жидкости наблюдается
почти «кристаллическое» расположение соседних молекул («ближ-
(«ближний порядок») и нарушение строгого порядка в расположении даль-
дальних молекул (отсутствие «дальнего порядка»).
Следует также обратить внимание на то, что на рассматривае-
рассматриваемом рисунке число частиц, расположенных упорядоченно (рис. 59, а)
117
одинаково с числом частиц, которые расположены неупорядоченно
(рис. 59, б). Сравнение соответствующих площадей убеждает в том,
что при характерном для жидкости неупорядоченном расположении
частиц она занимает больший объем, чем при упорядоченном,
кр иста л л ическом.
Результаты рентгеноструктурного исследования жидкостей мож-
можно объяснить также, исходя из представления о квазикри-
квазикристаллической структуре жидкости. Для того чтобы
пояснить это, обратимся к расположению атомов в идеальном кри-
кристалле. Если мысленно выбрать какой-либо атом в таком кристалле
и постараться определить, какова вероятность встретить соседний
атом на расстоянии R от первого, то при отсутствии теплового дви-
движения искомая вероятность равнялась бы нулю на расстояниях,
меньших расстояния Ri, при котором она делалась бы равной еди-
единице1. Это означает, что в данном направлении соседний атом всег-
всегда встречался бы на одном и том же расстоянии от исходного.
На расстояниях, больших Ru но меньших 2RU искомая вероят-
вероятность вновь равнялась бы нулю, а на расстоянии 2?>4 — единице.
Такое положение повторялось бы на всем протяжении кристалла:
вероятность встретить атом равнялась бы единице для всех рассто-
расстояний, кратных R±.
Тепловое колебательное движение атомов в кристалле приводит
к тому, что вероятность встретить соседний атом будет не равна ну-
нулю также и на расстояниях, незначительно отличающихся от 7?4.
В одном случае соседний атом, совершая колебания, слегка при-
приблизится к тому, от которого ведется отсчет, а в другом случае —
удалится. Графически изменение вероятности встретить атом в за-
зависимости от расстояния между ним и атомом, выбранным за нача-
начало отсчета, изображается характерной кривой (верхняя часть рисун-
рисунка 60).
Отличительной чертой графика является постоянство ширины от-
отдельных колоколообразных участков кривой. Именно это постоян-
постоянство указывает на сохранение упорядоченности на всем протяжении
кристалла.
В жидкости наблюдается иная картина (рис. 60, внизу). Каче-
Качественно вероятность Р встретить атом на каком-либо расстоянии
от исходного атома изменяется подобно тому, как это имеет место
в кристалле. Однако в этом случае только первый колоколообраз-
ный участок кривой выражен в виде четкого максимума. Последую-
Последующие колоколообразные участки, расширяясь, взаимно перекрыва-
перекрываются, так что максимумы на кривой сравнительно быстро исчезают.
Таким образом, расположение близких друг к другу частиц
в жидкости напоминает расположение частиц в кристаллическом
1 В теории вероятностей принято, что достоверное (обязательно проис-
происходящее) событие имеет вероятность, равную единице.
118
твердом теле. По мере удаления от исходного атома, относительно
которого производят расчет, положение частиц становится все более
разупорядоченным. Вероятность встретить частицу на любом рас-
расстоянии становится примерно одинаковой, как это имеет место
в газах.
Конечно, увеличение неопределенности в местоположении ато-
атомов объясняется не увеличением амплитуды их тепловых колеба-
колебаний, а случайными нарушениями в расположении частиц жидкости.
Следует подчеркнуть, что у жидкостей даже первый максимум
на кривой вероятности (рис. 60) не полностью разрешен, т. е. кри-
Кристалл
LUUUU\
Рис. 60. Вероятное распределение атомов
в идеальном кристалле и в жидкости
вая не касается справа от максимума оси абсцисс. Физически этс
означает, что в жидкости число частиц, ближайших к данной, не
является, как в кристалле, строго постоянным.
В жидкости правильнее говорить лишь о постоянстве среднего
числа ближайших соседей.
Результаты рентгеноструктурного исследования жидкости, ко-
которыми мы в настоящее время располагаем, могут быть объяснены
как на основании представления о сиботаксических группах, так
и на основании представления о квазикристаллической структуре
жидкости. Следует отметить, что различие между микрокристал-
микрокристаллической и квазикристаллической теориями жидкости невелико.
Если исследовать среднее расположение частиц жидкости за более
или менее длительный промежуток времени, то обе теории будут
приводить к одним и тем же результатам.
Обе теории обладают тем недостатком, что, описывая качественно
правильно особенности строения жидкости, они не дают возмож-
возможности количественно характеризовать ее свойства
Разновидностью «кристаллических» теорий жидкого
состояния является так называемая «д ы р о ч н а я» теория жип-
119
кости. Согласно этой теории жидкость уподобляется кристаллу, в
котором большое количество атомов оказывается смещенным из
присущих им равновесных положений. При смещении атома из рав-
равновесного положения остается как бы свободное место, которое и
называется «дыркой».
Согласно теории «дырки» в жидкости — это более или менее
расширенные промежутки между молекулами, возникающие спон-
спонтанно, расширяющиеся, а затем сжимающиеся и вновь исчезающие.
Уравнение состояния в «дырочной» теории жидкости имеет,
согласно Я-И. Френкелю, следующий вид:
_ и_
V—V0=Ne kTAV.
Здесь V — молярный объем жидкости при температуре Т; Уо —
минимальный объем, который может занимать жидкость; U—энер-
U—энергия образования дырки; k — постоянная Больцмана; N — число
Авогадро; AV — минимальный объем дырки.
Как уже неоднократно подчеркивалось, по мере увеличения тем-
температуры сходство жидкостей с твердыми телами уменьшается и
возрастает сходство их с соответствующими газами. Поэтому не уди-
удивительно, что при объяснении свойств жидкостей наряду с рассмот-
рассмотренными выше «кристаллическими» моделями жидкости широкое
распространение приобрели теории, в которых жидкость уподобля-
уподобляется сильно сжатому газу. В этих теориях большую роль играет
представление о свободном объеме жидкости1,
определить который точно затруднительно. Существующие в на-
настоящее время методы вычисления свободного объема жидкости
являются грубо приближенными и приводят, как правило, к вели-
величинам, расходящимся между собой.
Из теорий свободного объема наиболее разработана так назы-
называемая «ячеечная» теория жидкости.
Благодаря тому что молекулы жидкости расположены близко
друг к другу, каждую из них можно рассматривать как заключен-
заключенную в ячейку, стенки которой образуют ее ближайшие соседи.
Молекулы могут меняться местами, так что молекула, находя-
находящаяся в центре мысленно выделенной ячейки, может спустя неко-
некоторое время перейти в соседнюю ячейку. Однако подобные мигра-
миграции частиц происходят сравнительно редко, и большую часть вре-
времени молекула проводит внутри данной ячейки.
Движение молекулы в ячейке происходит в силовом поле, обра-
образованном ее ближайшими соседями, число которых для простых
жидкостей полагают равным 12.
1 Свободным объемом жидкости называется разность между объемом
жидкости при данных условиях и объемом, который она занимала бы при иде-
идеально упорядоченном плотном расположении ее частиц, соответствующем аб-
абсолютному нулю температуры.
120
Поскольку данная теория применима к жидкостям, находя-
находящимся при высоких температурах, когда влияние структуры веще-
вещества практически не сказывается, можно считать силовое поле,
в котором происходит движение частицы, сферически симметричным.
Принимая далее определенную форму зависимости потенциаль-
потенциальной энергии молекулярного взаимодействия от расстояния между
частицами и делая ряд упрощающих предположений, можно найти
выражение для потенциальной энергии <р частицы, находящейся
в элементарной ячейке. Обычно этому выражению придают следую-
следующий вид:
-4 В
т уп ут'
где V — объем сферической ячейки, приходящейся на одну части-
частицу, а п, т, А и В — постоянные.
Уравнение состояния жидкости в этом случае можно будет
записать в следующей форме:
kT dm
п = .
и V 6V
Здесь р — давление, k — постоянная Больцмана и Т — темпера-
температура. Подставляя в последнее выражение значение ф, удается выра-
выразить количественно многие физико-химические характеристики ин-
индивидуальных жидкостей. Так, например, пользуясь ячеечной те-
теорией жидкости, можно вычислить критические параметры различ-
различных простых веществ. Рассчитанные значения критической темпе-
температуры в случае простейших газов оказались равными по абсолют-
абсолютной шкале для водорода 41°, неона 47°, азота 128° и аргона 160°,
экспериментальные же значения соответственно равны 33°, 44°, 126°
и 150° К. В приведенном примере согласие величин, рассчитанных
теоретически, с величинами, найденными на опыте, вполне удовлет-
удовлетворительное.
Необходимо, однако, отметить, что написанное выше выраже-
выражение для давления, строго говоря, справедливо для реального газа,
а не для жидкости, и потому ожидать очень хорошего согласия
теории с опытом нет оснований. Несмотря на это замечание, теория
свободного объема имеет свои достоинства, среди которых следует
отметить простоту использованных физических моделей и возмож-
возможность количественного сравнения теории с опытом.
Ячеечная теория дает возможность относительно просто объ-
объяснить свойства жидкостей и рассчитать в первом приближении
некоторые их характеристики.
Теоретически более строгой является статистическая
теория жидкости. В этой теории основную роль играют
две физические величины. Первая из этих величин называется р а-
д паль ной функцией распределения, вторая —
меж молекулярным потенциалом. Радиальная
121
функция распределения определяет вероятность встретить произ-
произвольно выбранную пару частиц в жидкости на некотором заданном
расстоянии, заключенном в пределах от R до R -f- dR. Межмолеку-
Межмолекулярный потенциал определяет взаимодействие молекул жидкости.
Знание этих двух величин позволяет написать теоретически строго
уравнения состояния и энергии жидкости и выразить количествен-
количественно ее различные физико-химические характеристики.
Радиальную функцию распределения для ряда жидкостей мож-
можно определить экспериментально на основании данных рентгено-
структурного анализа. Однако значительные трудности в определе-
определении и расчете межмолекулярного потенциала для конкретных жид-
жидкостей заставляют решать полученные уравнения приближенно.
Указанное обстоятельство затрудняет количественное сопостав-
сопоставление статистической теории жидкости с опытом. Нельзя, однако,
забывать, что эта теория качественно правильно предсказывает
многие свойства жидкостей и присущие им закономерности.
Именно в возможности правильно предвидеть различные свой-
свойства вещества заключается одно из преимуществ статистической те-
теории жидкого состояния.
В будущем, когда будет найдено теоретически строгое выражение
для межмолекулярного потенциала и преодолены вычислительные
затруднения, статистическая теория позволит лучше понять особен-
особенности жидкого состояния вещества.
9. ВЯЗКОСТЬ ЖИДКОСТЕЙ
Одной из важных физических характеристик жидкости является
ее вязкость. Вязкость жидкости очень сильно зависит от темпе-
температуры. При изменениях температуры, которые сравнительно не-
нетрудно осуществить на опыте, вязкость жидкости может изме-
изменяться в миллионы раз. При понижении температуры вязкость неко-
некоторых жидкостей настолько возрастает, что жидкости теряют ха-
характеризующую их способность течь, превращаясь в аморфные
твердые тела. В настоящее время существует несколько теорий
вязкости жидкостей. Наличие нескольких теорий указывает на то,
что ни одна из них не свободна от недостатков.
Одним из наиболее удачных выражений, связывающих вязкость
жидкостей с другими их свойствами, следует признать уравнение
Бачинского A877—1944).
Бачинским было найдено, что для очень большого количества
жидкостей коэффициент вязкости ч\ связан простой зависимостью
с удельным объемом жидкости V:
Входящие в это уравнение постоянные Сию имеют физический
смысл, который легко расшифровать. Постоянная со — это предель-
122
ный объем, обусловленный несжимаемостью молекул вещества.
В знаменателе в правой части уравнения Бачинского стоит, таким
образом, свободный объем жидкости. Постоянная С учитывает силы
молекулярного взаимодействия частиц жидкости. Она связана про-
простой зависимостью с постоянной а в уравнении Ван-дер-Ваальса.
Следует подчеркнуть, что для идеальных газов коэффициент вяз-
вязкости рассчитывается без учета действия межмолекулярных сил, по-
поскольку вязкость идеальных газов обусловлена только хаотиче-
хаотическим движением молекул. В противоположность этому для жидко-
жидкостей на первый план выступает учет силового взаимодействия
молекул.
Для многих жидкостей урав-
уравнение Бачинского хорошо согла-
согласуется с опытом (см. рис. 61).
В уравнении Бачинского зави-
зависимость вязкости от температуры
выражена в неявной форме: из
величин, стоящих в правой части
уравнения, от температуры за-
зависит удельный объем жидкос-
жидкости V.
В настоящее время для ра-
расчета коэффициента вязкости жид-
жидкости предложено большое коли-
количество уравнений, различающихся
в деталях, но в то же время имею-
имеющих много общего. Обычно коэф-
коэффициент вязкости представляют
произведением:
Рис. 61. Связь между текучестью
1
жидкости— , отложенной вдоль
¦
оси абсцисс, и ее удельным объе-
объемом V. Сплошная линия рассчи-
рассчитана по уравнению Бачинского,
точки нанесены на основании
опытных данных.
г] = Ае
RT
A6)
в котором сомножитель А в одних уравнениях — постоянная вели-
величина, а в других — более или менее сложная функция абсолютной
температуры Т, R — универсальная газовая постоянная и, наконец,
<Р (U) — величина, определяемая силами межмолекулярного взаи-
взаимодействия.
Присутствие температуры в показателе степени в уравнении A6)
объясняет большое влияние ее на величину коэффициента вязкости.
С молекулярно-кинетической точки зрения процесс течения
жидкости представляет последовательный переход молекул жидко-
жидкости из одного равновесного положения в соседнее. Эти положения
разделены энергетическими барьерами, которые молекула должна
преодолеть при своем движении. Согласно одной из распространен-
распространенных теорий вязкости возникновение силы вязкости в жидкости
связано с указанным преодолением потенциального барьера.
Сходная картина наблюдается при диффузии различных веществ
в жидком состоянии.
Различие в механизме возникновения вязкости в газах и жидко-
жидкостях приводит к тому, что связь между коэффициентами вязкости
и диффузии в этих состояниях вещества различна.
В газах коэффициент вязкости и коэффициент диффузии уве-
увеличиваются при возрастании температуры, в жидкостях же при
увеличении температуры коэффициент диффузии увеличивается,
а коэффициент вязкости уменьшается. Соотношение между коэф-
коэффициентом вязкости жидкости г\ и коэффициентом диффузии D ча-
часто выражают уравнением Стокса — Эйнштейна:
kT
в котором Г — абсолютная температура, k — постоянная Больц-
мана и R —• радиус диффундирующей молекулы, принимаемой за
шарик небольших размеров.
Пользуясь современными представлениями о природе жидкости,
можно показать, что из написанного соотношения следует близость
коэффициентов диффузии у жидкостей, молекулы которых не
очень сильно различаются своими размерами.
Опыт показывает, однако, что коэффициенты диффузии в ра-
растворах близки по величине и у таких веществ, молекулы кото-
которых очень сильно различаются. Так, например, в водных растворах
при 18° коэффициенты диффузии радиоактивного газа радона,
кислорода, соляной кислоты и сахара соответственно равны: 2,33;
1,61; 1,39 и 0,34 смУсутки. Мало зависят обычно коэффициенты диф-
диффузии и от природы растворителя. Молекулярно-кинетическая
теория материи в настоящее время не может объяснить всех особен-
особенностей процесса диффузии в жидкостях.
10. РАСТВОРЫ. ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
Многие твердые тела способны растворяться в жидкостях. При
растворении вещества его молекулы равномерно распределяются
среди молекул растворителя.
Как правило, процесс растворения не представляет собой про-
простого молекулярного раздробления растворяемого вещества. Обычно
растворение сопровождается энергичным физическим и химическим
взаимодействием молекул растворителя с молекулами растворен-
растворенного вещества. Весьма часто молекулы растворенного вещества
оказываются окруженными молекулами растворителя, образую-
образующими вокруг них более или менее устойчивую оболочку. В еод-
ных растворах эти оболочки называют гидратными оболоч-
оболочками, в неводных — сольватными.
124
Д. И. Менделеев впервые указал на необходимость учитывать
при растворении вещества химические взаимодействия. Хотя даль-
дальнейшее развитие учения о растворах и подтвердило правильность
мысли Менделеева о наличии химических взаимодействий между
молекулами веществ при растворении, однако не у всех раство-
растворов это взаимодействие существенно влияет на свойства растворов.
Во многих растворах взаимодействие между молекулами раство-
растворенного вещества и растворителя мало отличается от взаимодей-
взаимодействия молекул этих веществ, взятых порознь. Подобные растворы
должны напоминать по своим свойствам смеси газов.
Если раствор разбавленный, т. е. если в единице объема раство-
растворителя содержится небольшое количество растворенного вещества,
то молекулы последнего оказываются отстоящими далеко друг от
друга и особенности их взаимодействия никак не проявляются.
Влияние молекул растворителя на движение молекул растворен-
растворенного вещества взаимно компенсируется, так что его оказывается
возможным не учитывать. Растворенное вещество в разбавленном
растворе ведет себя подобно идеальному газу.
Мысль о сходстве свойств растворенного вещества со свой-
свойствами идеального газа была высказана голландским физиком
Вант-Гоффом A852—1911). Развивая мысль об аналогии
между смесью газов и жидким раствором, Вант-Гофф пришел к
выводу о том, что растворенное вещество должно оказывать давле-
давление, подобное парциальному давлению газа, добавленного в неболь-
небольшом количестве к какому-либо другому газу. Это давление л было
названо осмотическим.
Если раствор, содержащий т килограмм растворенного веще-
вещества, молекулярный вес которого М, занимает объем V, то, соглас-
согласно закону Вант-Гофф а, осмотическое давление можно
подсчитать по аналогии с давлением идеального газа при помощи
уравнения:
я = —Я7\ A7)
MV ' у '
в котором R — универсальная газовая постоянная, Т — абсолют-
абсолютная температура. Величина -^— , численно равная числу киломо-
лей растворенного вещества в \ м3 раствора, называется молярной
концентрацией с, поэтому закон Вант-Гоффа часто записывают в
следующей форме:
n = cRT. A8)
Таким образом, осмотическое давление пропорционально
молярной концентрации растворенного вещества и аб-
абсолютной температуре раствора.
Естественно возникает вопрос, как обнаружить проявления
осмотического давления.
125
Рис. 62. Обнаружение осмотическо-
осмотического давления.
Жидкость находится, с одной стороны, под действием внеш-
внешнего давления р, а с друюй стороны — под действием внутрен-
внутреннего давления pt, обусловленного силами молекулярного притя-
притяжения. Осмотическое давление, оказываемое частицами растворен-
растворенного вещества, должно несколько увеличить объем жидкости,
поскольку оно направлено противоположно внешнему и внутрен-
внутреннему давлениям, сжимающим жидкость. Обнаружить, однако, это
в разбавленных растворах, для которых справедливы сформулиро-
сформулированные выше простые закономерности, практически невозможно,
поскольку внутреннее давление по абсолютной величине несоиз-
несоизмеримо больше осмотическо1 о
давления и полностью маски-
маскирует его действие.
Для обнаружения осмоти-
осмотического давления прибегают к
использованию свойств полу-
полупроницаемых перегородок, спо-
способных свободно пропускать
растворитель, но задерживаю-
задерживающих частицы растворенного ве-
вещества. Для точных измерений
полупроницаемые перегородки
обычно приготавливаются сле-
следующим образом: пористый со-
сосуд из неглазурованного фарфора погружают в сосуд больших
размеров, наполненный раствором медного купороса. Внутрь
пористого сосуда наливают раствор железистосинеродистого ка-
калия. Растворенные соли диффундируют в поры фарфорового сосу-
сосуда и, встречаясь, реагируют, образуя пленку железистосинероди-
стой меди, пропускающей воду, но не пропускающей молекулы
многих растворенных веществ, например сахара.
Осмотическое давление в растворе, помещенном во внутренний
сосуд прибора, изображенного на рисунке 62, уравновешивается
внешним и внутренним давлениями, действующими на жидкость.
Если заменить дно сосуда, содержащего раствор, полупроницаемой
перегородкой, то растворитель из внешнего сосуда сможет свобод-
свободно поступать во внутренний. Осмотическое давление на поверхность
раствора окажется неуравновешенным, и поверхность начнет переме-
перемещаться вверх, как бы втягивая при этом растворитель во внут-
внутренний сосуд.
Движение жидкости будет продолжаться до тех пор, пока воз-
возникшая разница уровней жидкости во внешнем и внутреннем сосу-
сосудах не уравновесит осмотическое давление. Результаты экспери-
экспериментального определения осмотического давления (таблица № 13)
подтверждают справедливость представлений Вант-Гоффа. Осмо-
Осмотические явления играют большую роль в жизнедеятельности
растительных и животных организмов. Оболочки клеток, из кото-
Таблица 13
Осмотическое давление сахарозы при 0° С
Концентрация
в кмолях/м*
0,1
0,2
0,3
0,4
0,5
Наблюдаемое
осмотическое
давление лоп
н'м2
2,48 • 106
4,77 »
7,16 »
9,53 »
12,0 »
Осмотическое
давление, рас-
рассчитанное те-
теоретически я теор
Н/Л12
2,26 • 105
4,52 »
6,78 »
9,03 »
11,3 »
яоп
лтеОр
1,10
1,06
1,06
1,06
1,07
рых состоят живые организмы, являются полупроницаемыми пе-
перегородками, и многие виды важного для организма обмена веществ
совершаются в результате осмотического проникновения жидко-
жидкости через полупроницаемую перегородку.
11. ПОНИЖЕНИЕ ДАВЛЕНИЯ НАСЫЩЕННОГО ПАРА
РАСТВОРИТЕЛЯ НАД РАСТВОРОМ
При растворении нелетучих веществ давление пара раствори-
растворителя уменьшается так, что давление пара над поверхностью рас-
раствора р оказывается меньше, чем измеренное при той же темпера-
температуре давление пара над поверхностью чистого растворителя р0.
Разность р0 — р называют понижением давления насыщенного
пара. Величина понижения давления насыщенного пара зависит
от концентрации растворенного вещества.
Опытным путем Раулем A830 —1901) было найдено, что
для разбавленных растворов относительное понижение да-
давления насыщенного пара растворителя над раствором
равно мольной доле растворенного вещества:
Ро —
Ро
A9)
Здесь v — число молей растворенного вещества и v0 —число молей
растворителя. Написанное соотношение обычно называют зако-
законом Рауля.
Таким образом, относительное понижение давления насыщен-
насыщенного пара оказывается независящим от химической природы рас-
растворенного вещества, а зависящим только от отношения числа мо-
молекул растворенного вещества к общему числу молекул в рас-
растворе.
Для объяснения закона Рауля о молекулярно-кинетической
точки зрения вспомним, что при данной температуре в паровую
фазу при испарении переходит определенная доля от общего числа
молекул жидкости, подходящих из более глубоких слоев за 1 се-
127
кунду к каждому квадратному метру поверхности, отделяющей
жидкость от пара. Общее число молекул, попадающих на 1 м%
поверхности за единицу времени, пропорционально числу их в
единице объема. При растворении число молекул растворителя в
единице объема понижается, поскольку молекулы растворенного
вещества в результате оказываемого ими осмотического давления
увеличивают объем, занимаемый молекулами растворителя Vo до
объема раствора V. Если число молекул растворителя равно No, то
можно написать, что
Pa'P(MMPmcmJ у п N IV
Р0_ ivfl/ v о
Р AyV
или
?» = !.
Р V»'
В том случае, когда взаи-
взаимодействия молекул рас-
растворителя и растворенного
вещества различаются не-
незначительно, не будет боль-
большой ошибки принять, что
внутреннее молекулярное
давление в растворителе и растворе одинаково. При этом усло-
условии объемы, занимаемые раствором и растворителем, будут про-
пропорциональны числам молекул того и другого, т. е.
V _ N + No
пл Число мо пей
' дЮООгдады
Рис. 63. Понижение давления пара водных
растворов маннита.
(No — число молекул растворителя, /V —число молекул растворен-
растворенного вещества.) Отсюда находим, что
Пользуясь правилом пропорций, можно записать:
Ро — Р _ N
Ро N-{-Na
Для того чтобы получить из этого уравнения закон Рауля, доста-
достаточно разделить числитель и знаменатель дроби, стоящей справа,
на число Авогадро.
Рисунок 63, на котором сплошная линия вычерчена согласно
закону Рауля для водных растворов маннита1, а кружки соответ-
1 М а н н и т
творимое в воде.
шестиатомный спирт, кристаллическое вещество, рас-
128
ствуют наиденным на опыте значениям понижения давления пара,
свидетельствует о том, что в разбавленных растворах закон Рауля
удовлетворительно согласуется с опытом. В концентрированных
растворах наблюдаются различные по величине и характеру откло-
отклонения от закона Рауля.
Понижение давления насыщенного пара растворителя над рас-
раствором вызывает повышение температуры кипения раствора по
сравнению с температурой кипения чистого растворителя. Связь
между этими явлениями поясняет рисунок 64, на котором сплош-
сплошной линией изображено изменение с температурой давления насы-
насыщенного пара отдельно взятого растворителя, а пунктирной лини-
линией — давления насыщенного па-
пара растворителя над раствором.
Как мы знаем, кипение насту-
наступает тогда, когда давление на-
насыщенного пара жидкости де-
делается равным внешнему дав-
давлению. Если опыт производит-
производится при атмосферном давлении,
то температуры кипения раство-
раствора Т2 и растворителя Т\ найдут-
найдутся как абсциссы точек пересече-
пересечения изображенных кривых с
прямой, параллельной оси абс-
абсцисс и соответствующей нор-
нормальному атмосферному давле-
давлению. Рассмотрение полученного таким образом графика убеждает
в том, что температура кипения раствора действительно выше,
чем температура кипения чистого растворителя.
Разность AT = Тг — Ту между температурами кипения раство-
раствора и растворителя называется повышением температуры кипения
раствора. Для разбавленных растворов повышение температуры
кипения пропорционально молекулярной концентрации растворен-
растворенного вещества и не зависит от его химической природы.
мирт cm
Рис. Ь4. Повышение температуры
кипения растворов.
12. РАСТВОРЫ ЭЛЕКТРОЛИТОВ
При экспериментальном определении осмотического давления
было обнаружено, что в растворах солей, кислот и оснований най-
найденные опытным путем величины существенно и систематически
отклоняются от величин, полученных расчетом с помощью уравне-
уравнения ВантТоффа. Для тех же растворов наблюдаются также систе-
систематические отклонения величин понижения давления насыщенно-
насыщенного пара растворителя от величин, рассчитанных с помощью закона
Рауля. И понижения давления насыщенного пара растворов, и
величины осмотического давления оказываются существенно боль-
большими, чем это следует из развитой выше теории.
5 Заказ 145
129
Наблюдаемые отклонения можно было бы объяснить, если пред-
предположить, что при растворении количество молекул соли или кис-
кислоты в силу каких-то процессов возрастает. Для дальнейшего раз-
развития теории растворов было чрезвычайно важно то обстоятель-
обстоятельство, что указанные систематические отклонения от законов
Вант-Гоффа и Рауля наблюдались в растворах, способных прово-
проводить электрический ток.
Вещества, растворы которых проводят электрический ток, на-
называют электролитами.
В 80-х годах прошлого столетия шведский ученый С в а н т е
Аррениус A859—1927) высказал предположение о том, что
наблюдаемые в растворах электролитов отклонения от законов,
которым подчиняются растворы, не проводящие электрический
ток, объясняются диссоциацией, т. е. распадом молекул
электролитов при растворении.
Мысль о том, что молекулы электролита при растворении рас-
распадаются на части, несущие электрические заряды противопо-
противоположного знака, была высказана еще в начале прошлого века литов-
литовским ученым Ф. X. Гротгусом A785—1822). Однако плодо-
плодотворное развитие гипотезы электролитической диссоциации нераз-
неразрывно связано с работами Аррениуса.
При растворении молекулы электролита распадаются на и о н ы.
В случае кислот ионами являются положительно заряженный ион
водорода и отрицательно заряженный ион кислотного остатка.
Так, например, диссоциация азотной кислоты протекает по следую-
следующей схеме:
Соли диссоциируют с образованием положительно заряженного
иона металла и отрицательно заряженного иона кислотного остатка:
Наконец, основания (щелочи) диссоциируют с образованием поло-
положительно заряженного иона металла и отрицательно заряженного
гидроксильного радикала — ОН. В качестве примера укажем на
диссоциацию едкого натрия:
Диссоциация электролитов объясняется тем, что электроны, осу-
осуществляющие химическую связь атомов в молекуле электролита,
оттянуты преимущественно к определенным атомам рассматрива-
рассматриваемой молекулы.
При растворении в воде важную роль играет большая диэлек-
диэлектрическая постоянная воды, уменьшающая силу электрического вза-
взаимодействия заряженных частиц примерно в 80 раз. Кроме того,
в самой молекуле воды, как указывалось выше, центры противо-
130
положных по знаку электрических зарядов не совпадают: молекула
воды полярна.
В результате этих особенностей воды при растворении в ней
электролита силы связи, удерживающие вместе противоположно
заряженные части молекул электролита, ослабляются, а сами моле-
молекулы энергично взаимодействуют с молекулами воды. Все это
приводит в конечном счете к тому, что молекула электролита дис-
диссоциирует с образованием противоположно заряженных ионов.
Большую роль растворителя в процессе электролитической дис-
диссоциации подчеркивает тот факт, что одно и то же вещество может
быть электролитом при растворении в одном растворителе и неэлек-
неэлектролитом — при растворении в другом. В качестве примера можно
привести хлористый водород, который при растворении в воде обра-
образует раствор соляной кислоты, хорошо проводящий электрический
ток, а при растворении в бензоле остается в недиссоциированном
состоянии, так что раствор не проводит электрического тока.
Некоторые соли, например NaCl, KBr, LiCl и т. д., диссоцииро-
диссоциированы уже в твердом состоянии. Кристаллы этих веществ состоят
не из атомов, а из ионов. При растворении подобных солей проис-
происходит просто разъединение ионов, удерживаемых в кристалле
силами электростатического притяжения. Естественно, что эти соли
диссоциированы полностью.
Электролиты, диссоциирующие полностью, называют силь-
сильными электролитамив отличие от частично диссоцииро-
диссоциированных электролитов, которые называют слабыми.
Теория электролитической диссоциации позволила объяснить
многие свойства растворов электролитов.
Однако не всегда наблюдается хорошее количественное согла-
согласие теории с опытом. Это особенно справедливо в отношении теории
сильных электролитов.
Теории сильных электролитов посвящено в настоящее время
большое количество исследований, в которых делаются попытки
учесть как взаимодействие ионов между собой, так и взаимодей-
взаимодействие ионов с молекулами растворителя.
13. ПРИМЕНЕНИЕ УЛЬТРААКУСТИЧЕСКИХ МЕТОДОВ
К ИССЛЕДОВАНИЮ ГАЗОВ И ЖИДКОСТЕЙ
В последние годы для исследования газов и жидкостей успешно
применяются ультразвуковые колебания (ультразвуки).
Ультразвуками называют распространяющиеся в материальной
среде упругие волны, частоты колебаний которых превышают
20 000 гц и которые не воспринимаются человеческим ухом.
Изучение особенностей распространения ультразвуков в газах
и жидкостях дало много ценных сведений об их свойствах.
При использовании ультраакустических методов изучения
свойств вещества источниками ультразвука, как правило, являются
пьезоэлектрические генераторы.
5- 131
Если из кристалла кварца вырезать пластинку в форме парал-
параллелепипеда, ориентируя его грани определенным образом по отно-
отношению осей кристалла, то при сжатии пластинки на противопо-
противоположных гранях ее возникают разноименные электрические заряды.
Возникновение электрических зарядов в результате давления на-
называют пьезоэлектрическим эффектом. Пьезоэлек-
Пьезоэлектрическим эффектом обладают кристаллы кварца, сегнетовой
соли, турмалина, дигидрофосфата аммония и некоторых других
веществ.
Пьезоэлектрический эффект обратим. Достаточно покрыть
противоположные грани пьезоэлектрической пластинки тонким
слоем металла и подвести к
ним переменное электриче-
электрическое напряжение, как пла-
пластинка начнет в такт с изме-
изменением электрического на-
напряжения изменять свою фор-
форму. Грани пластинки будут
совершать при этом поршне-
образные движения, создавая
в окружающем веществе про-
продольную упругую волну с
частотой, соответствующей ча-
частоте электрических колеба-
колебаний. Источником переменного
электрического напряжения
является обычный ламповый
генератор (рис. 65, а).
По сравнению со слыши-
слышимыми звуками ультразвуки обладают тем преимуществом, что
скорость их распространения, а также коэффициент поглощения
их в веществе можно измерить с большой точностью.
Для этой цели часто используется прибор, называемый уль-
ультразвуковым интерферометром (рис. 65, б).
В этом приборе на некотором расстоянии от колеблющейся квар-
кварцевой пластинки / и параллельно ей располагают плавно перемещаю-
перемещающийся рефлектор 2. Достигшая рефлектора ультразвуковая волна
отражается от него и падает вновь на излучающую кварцевую пла-
пластинку. В результате отражения от рефлектора в пространстве меж-
между ним и кварцевой плчстинкой фактически распространяются две
встречные волны одинаковой частоты. При плавном перемещении
рефлектора вследствие сложения этих волн периодически возникают
стоячие волны.
Возникновение стоячих волн сказывается на режиме колеба-
колебаний кварцевой пластинки, что в свою очередь влияет на величины,
характеризующие колебательную систем} в целом, и, в частности,
на величину анодного тока в генераторе.
Рис. 65. Схема ультраакустических
измерений.
132
Если записывать величину анодного тока / при перемещении
рефлектора, то наблюдается кривая, изображенная на рисунке 65, в.
Расстояние между положениями рефлектора, соответствующее
двум соседним максимумам величины анодного тока, равняется
половине длины звуковой волны (к/2) в веществе, в котором распро-
распространяется звук.
Измерив расстояние / между двумя положениями рефлектора,
соответствующее п максимумам анодного тока, и зная частоту ко-
колебаний генератора /, можно весьма точно определить скорость
звука с с помощью формулы:
с- ^f
Уменьшение разницы между максимальной и минимальной вели-
величинами анодного тока по мере удаления рефлектора от колеблю-
колеблющейся кварцевой пластинки, которое ясно видно на рисунке 65,
вызвано поглощением звука в веществе, в котором он распростра-
распространяется. Это позволяет с помощью кривых, аналогичных изобра-
изображенной на рисунке 65, определять коэффициент поглощения зву-
звука различными веществами. Скорость распространения звука с
связана с физико-химическими свойствами вещества уравнением:
Cv \SVJT
в котором р — давление, V — удельный объем, Ср и Cv — теплоем-
теплоемкости вещества соответственно при постоянном давлении и объеме.
Давление, как мы уже знаем, связано с объемом и темпера-
температурой вещества уравнением состояния, и поэтому, пользуясь напи-
написанной зависимостью и данными о скоростях звука, можно проверять
различные представления о строении вещества, в частности оце-
оценивать свободный объем жидкостей, определять постоянные, вхо-
входящие в уравнение состояния, и т. д.
Экспериментальное определение теплоемкости жидкости при
постоянном объеме затруднительно. Акустические измерения дают
возможность косвенного определения Cv, которым в наше время
широко пользуются.
Если звук распространяется в газе, к которому можно приме-
применить уравнение Клапейрона, то для скорости звука будет справед-
справедливо более простое выражение:
С —г~.
где р — плотность газа, R — универсальная газовая постоянная.
При применении точных ультраакустических методов измере-
измерения скорости звука обнаружилось, что в многоатомных газах
последняя зависит от частоты звука, т. е. наблюдается диспер-
дисперсия звука. Из написанного выше уравнения, выражающего
скорость звука, следует, что дисперсия звука будет наблюдаться
133
в том случае, если при изменении частоты звуковых колебаний
будет изменяться теплоемкость газа Cv. Изменение теплоемкости
при изменении частоты звуковых колебаний тесно связано с про-
процессами распределения акустической энергии между различными
степенями свободы движения молекул многоатомного газа.
Первоначально вся акустическая энергия приходится на сте-
степени свободы, соответствующие поступательному движению моле-
молекул. Спустя некоторое время часть этой энергии перераспреде-
перераспределяется в соответствии с принци-
принципом равного распределения энер-
энергии на степени свободы, свя-
связанные с вращательным и ко-
колебательным движениями мно-
многоатомных молекул.
Если непрерывно увеличи-
увеличивать частоту звуковой волны,
то рано или поздно будет до-
достигнута такая частота, при ко-
которой за время полного коле-
колебания энергия не будет успе-
успевать распределяться равновес-
равновесно между различными степе-
степенями свободы движения моле-
молекул. Молекулы как бы поте-
потеряют какую-то часть степеней
с'
/¦
1
1
Г*
, Поглощение
fg
fg
а
и
J
Рис. 66. Дисперсия звука
в углекислоте.
свободы, а так как теплоем-
теплоемкость газа Cv непосредственно зависит от числа степеней свобо-
свободы, то очевидно, что при достаточно большой частоте звуковых
колебаний Сг, должно уменьшиться, а скорость звука, как это
следует из написанного выше выражения, —возрасти.
На рисунке 66 изображена найденная эмпирически зависимость
квадрата скорости звука с в углекислоте от логарифма частоты ы.
Как можно убедиться, опыт полностью подтверждает теоретические
предположения. При относительно малых частотах скорость зву-
звука не зависит от частоты и имеет величину с0. При высоких частотах
скорость звука опять же не зависит от частоты, но имеет теперь
большее значение с». Между этими предельными значениями ле-
лежит область дисперсии, в которой скорость звука изменяется при
изменении частоты. Одновременно с изменением скорости в области
дисперсии резко возрастает поглощение звука.
В области дисперсии звука наблюдается аномальное поглоще-
поглощение звуковой энергии. Если изобразить графически зависимость
коэффициента поглощения звука, рассчитанного на одну длину
волны, от частоты колебаний, то в области дисперсии эта зависимость
имеет вид характерной колоколообразной кривой, максимум кото-
которой определяет положение дисперсионной области.
Частота, при которой наблюдается дисперсия, зависит от ско-
134
роста, с которой происходит перераспределение энергии между
различными степенями свободы движения молекул.
Как показал опыт, задержка в установлении равновесного рас-
распределения энергии связана с возбуждением молекулярных колеба-
колебаний, которые возникают далеко не при каждом соударении молекул.
При добавке к исследуемому газу небольшого количества какого-
либо другого частота, соответствующая центру дисперсионной об-
области, изменяется, потому что вероятность возбуждения колеба-
колебательных состояний молекулы основного газа при ее соударениях
в сильной степени зависит от особенностей той молекулы, с которой
происходит соударение. Таким образом, изучая дисперсию звука,
можно определить среднее число соударений, которые должна пре-
претерпеть молекула для того, чтобы входящие в ее состав атомы на-
начали бы колебаться, оценить эффективность соударений между раз-
различными молекулами в отношении возбуждения колебательного
состояния и попытаться увязать результаты этих измерений с осо-
особенностями строения молекул.
В жидкостях также наблюдаются дисперсионные явления, од-
однако в этом случае изменение скорости, как правило, мало, и обыч-
обычно предпочитают изучать особенности поглощения звука.
Ультразвуковые методы успешно используются для изучения
механизма и скорости диссоциации электролитов, изучения сжимае-
сжимаемости индивидуальных жидкостей и растворов, особенностей взаи-
взаимодействия компонентов жидких смесей и т. д.
Измерение скоростей ультразвука используется при устройстве
анализаторов, предназначенных для контроля состава различных га-
газообразных и жидких смесей, а также ультраакустических расходо-
расходомеров, определяющих количество жидкости, протекающей по трубе.
Изучение затуханий колебаний стержня, погруженного в иссле-
исследуемую жидкость, дает возможность определить вязкость последней.
На этом принципе сконструированы ультразвуковые вискозиметры.
Большие возможности открывает использование ультраакусти-
ультраакустических методов при исследовании критического состояния вещества,
определении теплоемкостей вещества и его других характеристик
в критической области.
В жидкостях, температура которых далека от критической,
скорость звука в основном определяется межмолекулярным взаимо-
взаимодействием, для изучения которого и можно использовать ультра-
ультраакустические измерения.
Так, например, зависимость потенциальной энергии <р жидкос-
жидкости от объема часто выражают приближенным уравнением, содержа-
содержащим два гиперболических числа:
А В
~ уп ут'
в котором пит — постоянные, а Л и В — величины, независящие от
объема, но изменяющиеся с температурой, V — молекулярный объем.
135
На основании теоретических соображений можно принять т = 6.
В этом случае измерения скоростей звука можно использовать для
нахождения величины п.
В растворах на скорость звука оказывает влияние возникнове-
возникновение гидратных или сольватных оболочек, характеризующихся более
интенсивным взаимодействием молекул. Естественно поэтому было
применить акустические измерения для изучения процессов гидрата-
гидратации различных веществ. Этим способом удается получить данные,
необходимые для понимания свойств растворов.
Аномальное поглощение звука, обусловленное нарушением рав-
равновесного распределения энергии между различными степенями сво-
свободы, как это описано выше, у многоатомных газов, может наблю-
наблюдаться и в жидкостях. Однако у жидкостей оно, как правило, на-
наблюдается при гораздо более высоких частотах, порядка 10е герц,
т. е. в области так называемых гиперзвуков.
Дисперсионные явления, наблюдаемые в жидкостях при распро-
распространении ультразвуков, вызваны нарушением равновесия между
присутствующими в них различными видами молекул.
Молекулы многих жидкостей образуют более или менее прочные
комплексы. Это явление называют ассоциацией жидко-
жидкости. Примером ассоциированной жидкости может служить уксус-
уксусная кислота, часть молекул которой соединена попарно, образуя
так называемые д и м е р ы. Между одиночными молекулами уксус-
уксусной кислоты и димерами существует динамическое равновесие,
другими словами, за какой-либо промежуток времени некоторое
количество димеров распадается, но этот распад компенсируется
возникновением равного количества новых димеров.
Акустические измерения позволяют всесторонне охарактери-
охарактеризовать реакцию возникновения димеров уксусной кислоты, а также
подобные реакции в других жидкостях.
Молекулы некоторых органических жидкостей могут сущест-
существовать в двух формах, подобно тому, как это показано ниже для
молекулы акрилового альдегида:
Н2С = С —Н НаС = С —Н
О = С —Н *~ Н —С = О.
Подобные молекулы называются поворотными изоме-
изомерами, поскольку одна из них может быть получена из другой
поворотом части последней на 180°. Между поворотными изомерами
в жидкости также существует динамическое равновесие, которое
с успехом изучается ультраакустическими методами.
Ультразвуковые методы применяются также для изучения струк-
структуры молекул жидкостей, химических взаимодействий в различ-
различных жидких смесях, непрерывного контроля за плотностью текущей
жидкости и т. д.
136
III. ТВЕРДОЕ СОСТОЯНИЕ ВЕЩЕСТВА
1. КРИСТАЛЛИЧЕСКИЕ И АМОРФНЫЕ ТВЕРДЫЕ ТЕЛА
Тела, которые в обыденной жизни называют твердыми, отли-
отличаются от жидкостей отсутствием способности течь. Не обладая
текучестью, твердые тела сохраняют неизменной свою форму. Жид-
Жидкости же и газы принимают форму сосуда, в котором они находятся.
Как мы увидим в дальнейшем, с молекулярной точки зрения приве-
приведенное выше определение неточно.
Разнообразные твердые тела, с которыми человеку приходится
иметь дело в своей практической деятельности, можно разделить
на две группы, существенно различающиеся по своим свойствам.
Одну группу образуют тела кристаллические, другую —
аморфные.
Одной из основных особенностей кристаллических тел является
анизотропия, т. е. зависимость свойств макроскопически од-
однородного тела от направления. Если исследовать такие характе-
характеристики физических свойств тела, как показатель преломления
световых лучей, коэффициент теплопроводности, модуль упруго-
упругости и т. п., то окажется, что в кристалле в зависимости от направле-
направления эти величины могут иметь разное значение. Иными словами,
в различных направлениях кристалл обладает различной теплопро-
теплопроводностью, различной упругостью, различной способностью прелом-
преломлять световые лучи.
Анизотропия — характерная особенность кристаллов. Аморфные
тела не обладают анизотропией. Если исследовать различные физи-
физические свойства стекла, которое является типичным представителем
аморфного тела, то окажется, что во всех направлениях его свойства
одинаковы.
Независимость физических свойств тела от направления назы-
называют изотропностью. Аморфные тела изотропны. Изотроп-
Изотропность аморфных твердых тел является проявлением сходства их
молекулярной структуры с молекулярной структурой жидкостей.
Можно считать, что твердые аморфные тела — это жидкости, вяз-
137
кость которых при понижении температуры настолько возросла,
что они потеряли способность течь, сохранив при этом молекуляр-
молекулярную структуру, характерную для жидкости.
Следует указать на то, что подавляющее большинство материалов,
используемых человеком в его практической деятельности, изотроп-
изотропны. Это и понятно, поскольку, как правило, практическое исполь-
использование анизотропных материалов затруднительно. В то же время
большая часть материалов, применяемых в технике и строительном
деле, таких, например, как железо, медь, цинк, гранит и т. д.,
является кристаллическими, а следовательно, анизотропными ве-
веществами. Использование этих материалов возможно тем не менее
в силу того, что анизотропия характерна только для отдельных
больших кристаллов, так называемых монокристаллов.
Технические же материалы представляют собой тела, образованные
соединенными вместе маленькими кристалликами. Такие тела назы-
называют поликристаллическими. Чем меньше кристалли-
кристаллики, образующие поликристаллическое тело, тем более оно однород-
однородно и одновременно более изотропно. В тех случаях, когда необхо-
необходима высокая изотропность материала, он должен быть возможно
более мелкокристаллическим.
Бывают, однако, случаи, когда желательно получить отдель-
отдельный монокристалл больших размеров. Для этой цели разработаны
специальные методы выращивания больших кристаллов, позволяю-
позволяющие получать монокристаллы с линейными размерами в десятки
сантиметров.
Многие тела (сера, глицерин, сахар и т. п.) могут существовать
как в кристаллической, так и в аморфной, или, как часто говорят,
в стеклообразной, форме.
Если расплавленную и нагретую до температуры около 350°
серу быстро охладить, вылив ее в холодную воду, она превращается
не в обычную кристаллическую серу, а в аморфную пластическую.
Свойства аморфной серы не похожи на свойства серы кристалличе-
кристаллической. Кристаллическая сера хрупка, аморфная же пластическая
сера легко изменяет свою форму при сравнительно небольшом дав-
давлении, напоминая в этом отношении пластилин.
Как правило, аморфная форма твердого тела менее устойчива,
чем кристаллическая. Аморфные тела самопроизвольно, хотя и очень
медленно, превращаются в кристаллические. В случае стекла этот
процесс сопровождается появлением микроскопических трещин
и называется расстекловыванием.
Следует, однако, отметить, что процесс кристаллизации аморф-
аморфного тела может протекать столь медленно, что аморфное состояние
будет практически полностью устойчиво. Так, например, янтарь,
являющийся аморфным телом, так же как и другие ископаемые
смолы, образовался десятки миллионов лет назад, однако в нем не
обнаруживается никаких признаков кристаллизации, и он может
рассматриваться как полностью устойчивое аморфное вещество.
138
Различие между аморфными и кристаллическими телами про-
проявляется при переходе их в жидкое состояние, т. е. при плавлении.
Для уяснения этого различия проделаем следующий опыт: будем
нагревать мелко измельченное твердое тело и наблюдать происхо-
происходящие при этом изменения температуры, отмечая через определен-
определенные промежутки времени показания прибора, измеряющего темпе-
температуру, и откладывая эти величины в качестве ординат Т на графике
(рис. 67), на котором вдоль оси абсцисс отложено время нагрева t.
Если соединить между собой найденные таким образом точки, то
в случае кристаллического тела возникнет кривая, изображенная
на графике сплошной линией.
I7"
шское
Рис. 67. Плавление твердого кристалличе-
кристаллического тела и аморфного тела.
Первоначально нагрев твердого тела сопровождается повыше-
повышением его температуры, возрастающей приблизительно прямо про-
пропорционально времени нагрева (отрезок АВ). При достижении тем-
температуры плавления дальнейший рост температуры прекращается,
несмотря на нагревание тела (отрезок ВС). Подводимое тепло в этом
случае расходуется на плавление кристалла (теплота плавления).
После того как все твердое тело расплавится (точка С), температура
при дальнейшем подводе теплоты начинает вновь возрастать (отре-
(отрезок CD).
Иная картина наблюдается при нагревании аморфного твердого
тела. В этом случае отсутствует резко выраженная температура
плавления. На соответствующей кривой (пунктирная кривая на
рис. 67) наблюдается только изменение скорости возрастания тем-
температуры. В некотором, не резко ограниченном интервале темпе-
температур, соответствующем области размягчения аморфного тела, ско-
скорость возрастания температуры уменьшается. Вязкость аморфного
твердого тела в области размягчения падает и тело превращается
из твердого в жидкое.
139
Кроме указанных выше особенностей, кристаллы отличаются
от аморфных тел наличием правильной формы. Иногда правильная
форма кристаллического тела бывает искажена особыми условиями
роста кристаллов или же механической обработкой. Однако наличие
правильной формы кристаллов у кристаллических твердых тел всег-
всегда можно установить по характеру их излома. Излом кристалличе-
кристаллического тела имеет шероховатую поверхность, образованную кристал-
кристалликами правильной формы больших или меньших размеров. Излом
же аморфного тела имеет гладкую поверхность с характерными ост-
острыми краями, хорошо знакомыми по излому стекла.
Внешняя форма отдельных кристалликов может сильно изме-
изменяться в зависимости от условий роста кристалла. Последнее осо-
особенно бросается в глаза при рассматривании причудливых форм
снежинок.
Для учения о кристаллах чрезвычайно важно то, что вне зависи-
зависимости от внешней формы кристалла углы между его гранями имеют
всегда постоянную, определенную для данного типа кристалла ве-
величину. В тех случаях, когда рост кристалла не искажен какими-
либо внешними причинами, его форма определяется принципом ми-
минимума свободной поверхностной энергии. При этом следует пом-
помнить, что различные грани кристалла характеризуются различной
поверхностной энергией, и при росте кристалла грани с малой по-
поверхностной энергией получают наибольшее развитие. В целом же
равновесная форма кристалла такова, что его суммарная поверхно-
поверхностная энергия минимальна.
2. МОЛЕКУЛЯРНОЕ СТРОЕНИЕ КРИСТАЛЛОВ
Физические свойства кристаллических твердых тел обусловле-
обусловлены особенностями их молекулярного строения. Частицы, образую-
образующие кристаллы, расположены в пространстве упорядоченно — в уз-
узлах кристаллической решетки. В основе кристаллической решетки
лежит элементарная кристаллическая ячейка с характерным для
данной решетки расположением атомов. Весь кристалл можно по-
построить, перемещая элементарные кристаллические ячейки парал-
параллельно самим себе, до некоторой степени подобно тому, как уклады-
укладывают кирпичи при постройке какого-либо сооружения. Мысль о
правильном расположении частиц, образующих кристалл, была
высказана еще в конце XVIII столетия и развита более подробно
в 1848 г. Б р а в э.
В начале XX столетия для изучения строения кристаллов были
использованы рентгеновские лучи. Если направить на грань кри-
кристалла пучок параллельных рентгеновских лучей под углом б к его
поверхности (рис. 68), то отражение (рассеяние) рентгеновских лу-
лучей будет происходить не только на атомах, расположенных в пло-
плоскости, совпадающей с поверхностью, но и на атомах, расположен-
расположенных в следующей плоскости, отстоящей на расстоянии d от первой.
J40
В зависимости от угла б и от расстояния d лучи, отраженные от
первой и второй плоскостей, будут или усиливать, или ослаблять
друг друга (В. Г. и В. Л. Брэгги, Ю. В. Буль ф). Сходные
закономерности наблюдаются и при сквозном просвечивании кри-
кристалла рентгеновскими лучами (М. Л а у з).
Измеряя углы, при которых наблюдается усиление рентгенов-
рентгеновских лучей при рассеянии их кристаллами, можно определить рас-
расстояния между плоскостями, в которых расположены атомы в кри-
кристаллах, и, таким образом, экспериментально установить, какая
кристаллическая решетка свойственна тому или иному веществу.
Строго научную систематику возможных пространственных
расположений частиц в кристаллах разработал Е. С. Федоров
Рис. 68. Рассеяние рентгеновских лучей кристаллами.
A853—1919). Основываясь на признаках симметрии, удалось по-
показать, что возможно существование 230 различных «пространст-
«пространственных групп», в которые располагаются частицы, образующие
кристаллы. Этими частицами могут быть ионы, т. е. атомы, несущие
электрические заряды, нейтральные атомы и, наконец, молекулы.
Соответственно кристаллические решетки разделяются на ион-
ионные, атомные и молекулярные.
Если считать ионы или атомы, образующие кристалл, шариками
и если предположить, что все шарики имеют одинаковый радиус,
то задача построения кристаллической решетки сводится к задаче
о наиболее плотной упаковке шариков, т. е. о таком их расположе-
расположении, при котором объем, не занятый шариками, был бы минималь-
минимальным. Оказывается, одинаковые шарики можно расположить наи-
наиболее плотно двумя различными способами.
Предположим, что мы мысленно выделим в кристаллической ре-
решетке некоторый слой плотно упакованных шариков-атомов (рис.69).
Для наиболее плотной упаковки всего кристалла в целом шарики
в слоях, расположенных сверху и снизу по отношению к выделенному
слою, должны располагаться в промежутках между шариками в
этом слое. Зачерним тушью, как это сделано на рисунке, положения
шариков в слое, прилегающем к выделенному слою снизу. В этом слу-
случае шарики в слое, лежащем сверху над рассматриваемым, можно
141
Рис. 69. Плотная упаковка ша-
шаров одинакового размера
Рис. 70. Гексагональная плотная
упаковка.
поместить или над лунками, зачерненными тушью, или же над сво-
свободными лунками. В первом случае перпендикуляр, опущенный
из центра шарика, расположенного в верхнем слое, попадает в центр
шарика, отделенного от него одним слоем шаров. Такое расположе-
расположение атомов соответствует гексагональной плотной упаков-
упаковке (рис. 70).
Как сказано выше, можно расположить шарики иначе. Поме-
Поместим шарики верхнего слоя в незачерненные лунки, т. е. в те лун-
лунки, против которых не имеется шариков в слое, расположенном ни-
ниже выделенного слоя, и по такому же принципу будем располагать
шарики в последующих слоях. В этом случае центры шариков обра-
образуют кубическую гранецентрированную ре-
решетку (рис. 71). Перпендикуляр, опущенный из центра одного
из шариков, будет встречать центр шарика, расположенного ниже,
только через два слоя, так что расположение шариков-атомов в
Рис. 71. Кубическая гранецен-
трированная упаковка.
Рис, 72. Упаковка ионов
в кристалле хлористого натрия.
142
первом слое будет совпадать с расположением их в четвертом, рас-
расположение во втором слое — с расположением в пятом и т. д.
Каждый шарик будет соприкасаться при этой упаковке с 12
другими шариками. Замечательно, что плотность упаковки в обо-
обоих случаях одна и та же: шарики-атомы занимают 74% общего
объема кристалла.
В тех случаях, когда радиусы атомов, образующих кристалл,
различаются существенно один от другого, более плотная упаковка
достигается при ином расположении частиц. В качестве примера
можно указать кристалл поваренной соли, химическая формула
которой NaCl (рис. 72). При соотношении радиусов шариков, кото-
Рис. 73. Простейшие кристаллические решетки: а — куби-
кубическая, б — кубическая гранецентрированная; в — гексаго-
гексагональная.
рое соответствует радиусам ионов Na+ и СП, более выгодным в от-
отношении плотности упаковки оказывается расположение атомов
в простой кубической решетке.
Некоторые виды простейших кристаллических решеток изобра-
изображены на рисунке 73, на котором маленькие шарики соответствуют
положению центров частиц, образующих кристалл. Существуют,
однако, и такие кристаллы, в которых образующие их частицы рас-
расположены не так плотно, как при гексагональном или кубическом
гранецентрированном расположении. Характерными в этом отно-
отношении являются кристаллы льда, в которых частицы упакованы рых-
рыхло. Рыхлость структуры льда объясняет уменьшение объема, зани-
занимаемого некоторой массой льда, при его плавлении. Как известно,
плотность воды при температуре плавления больше плотности льда.
Строение кристалла зависит от его химического состава, от свойств
образующих его частиц и в первую очередь от их размеров. Это поз-
позволяет, исходя из химической формулы вещества, делать заключение
о строении соответствующих кристаллов. Однако известны случаи,
когда одно и то же вещество может образовывать кристаллы с раз-
различной структурой. Так, например, углерод может образовывать
кристаллы графита или кристаллы алмаза.
Возникновение той или иной кристаллической структуры зави-
зависит от условий кристаллизации. Так, например, при обычных усло-
143
Рис. 74. Строение кристалла хлористого
натрия.
виях углерод всегда кристаллизуется в форме графита. Однако, ис-
используя давление до 10' атм или 1010 н1м2 и температуру около 2000°,
удалось заставить углерод кристаллизоваться в форме алмаза.
Таким способом в настоящее время в промышленности получают
искусственные алмазы. Применяя высокие давления и температуры,
удалось получить кристаллы, не встречающиеся в природе. В ка-
качестве таковых можно указать на кристаллы нитрида бора, возникаю-
возникающие в том случае, если повысить давление приблизительно до
65 • 108 н/м2 и температу-
температуру до 1500°С. Полученный
при этом материал — бора-
зон — по твердости и химиче-
химической стойкости превосходит
алмаз.
У многих веществ кри-
кристаллическая структура из-
изменяется при изменении
внешних условий, например
температуры. Это явление на-
называется полиморфиз-
полиморфизмом.
Правильное расположение
частиц в кристаллической ре-
решетке обусловливает анизот-
анизотропию свойств кристаллов, о которой говорилось выше. Для ил-
иллюстрации этого рассмотрим механические свойства кристалла
хлористого натрия (NaCl). Если кристалл NaCl (рис.74) рассечь
плоскостью abed, то на образовавшихся вновь гранях будет
расположено равное количество положительно и отрицатель-
отрицательно заряженных частиц, и поэтому силы электростатического при-
притяжения между этими плоскостями будут невелики — вдоль этих
плоскостей кристалл будет легко раскалываться.
Если же попытаться отделить одну из вершин кристалличе-
кристаллического кубика по плоскости efh, то на одной из возникших граней
окажется три положительно заряженные частицы, а на второй —
три отрицательно заряженные частицы. Силы электростатического
притяжения в этом случае велики, и вдоль этих плоскостей кри-
кристалл расколоть значительно труднее.
Плоскости, по отношению которых прочность кристаллов ослаб-
ослаблена, называются плоскостями спайности. Наличие
плоскостей спайности обусловливает специфический характер из-
излома кристаллического тела.
Далее, если выбрать в кристалле направление вдоль линии ab,
то в этом направлении расположены попеременно положительно
и отрицательно заряженные частицы, а в направлении вдоль ли-
линии ef — только частицы одного знака. Естественно, что свойства
кристалла в направлении ab отличаются от его свойств в направлении
И4
ef. Таким образом, упорядоченное расположение разнородных частиц
в кристалле приводит к зависимости его свойств от направления,
т. е. к анизотропии.
В отношении молекулярного строения идеальное кристалличе-
кристаллическое тело является как бы противоположностью идеальному газу.
В то время как молекулы идеального газа распределены хаотично,
т. е. в их распределении полностью отсутствует всякая упорядочен-
упорядоченность, частицы, образующие идеальный кристалл, распределены
• Si О-О °
Рис. 75. Расположение частиц в гипотетическом двухмерном
кристалле (б) и таком же стекле (о).
в совершенном порядке. В отличие от жидкости или аморфного твер-
твердого тела упорядоченность в распределении частиц в кристалле рас-
распространяется не только на ближайших соседей какой-либо произ-
произвольно выбранной частицы, но простирается на все тело. Уяснить
себе это различие помогает рисунок 75, на котором изображено рас-
расположение частиц в гипотетических двухмерном кристалле и стекле.
Следует, однако, отметить, что в реальных кристаллах строго
упорядоченное расположение частиц в узлах кристаллической ре-
решетки нарушается, в ней возникают дефекты. В результатемаксвел-
ловского распределения скоростей хаотического теплового движения
частиц некоторые из них приобретают достаточное количество энер-
энергии для того, чтобы перейти из узла кристаллической решетки в меж-
междоузлие. При этом в кристаллической решетке возникают свободный
узел — «вакансия» и частица, смещенная из своего равновесного
положения, — «атом в междоузлии». Чем выше температура кри-
кристалла, тем больше в его кристаллической решетке «вакансий»
и соответственно «атомов в междоузлиях».
Кроме наличия указанных дефектов, кристаллическая решетка
145
D
в
реального кристалла отличается от рассмотренных идеальных схем
прксутствием в ней атомов посторонних веществ — примесей. При
этом атомы примесей могут или замещать атомы основного вещества
в кристаллической решетке (примесь замещения), искажая струк-
структуру последней в результате различия размеров атомов основного
вещества и примеси, или же внедряться в кристаллическую решетку,
раздвигая образующие ее атомы (примесь внедрения).
Многие свойства кристаллов, и в первую очередь их электриче-
электрические и механические характеристики, весьма чувствительны к нали-
наличию в кристаллической решетке
дефектов, а также присутствию в
ней даже незначительных коли-
количеств примесей.
Как показали исследования по-
последних лет, механические свой-
свойства кристаллических тел очень
сильно зависят от наличия в послед-
последних особого вида дефектов, на-
называемых дислокациями.В случае
так называемой краевой дислока-
дислокации в кристалле имеется область,
содержащая как бы одну лиш-
лишнюю атомную плоскость (рис. 76).
Можно себе представить, что подоб-
подобный дефект возникает в резуль-
результате смещения атомных плоскостей в некоторой части кристалла
(верхняя часть рисунка 76) так, что одна из плоскостей (DB) оказы-
оказывается расположенной в промежутке между атомными плоскостями
в другой части кристалла (сравните верхнюю и нижнюю части
рисунка 76). Линия, проведенная в точке В перпендикулярно пло-
плоскости чертежа, называется линией дислокации, а об-
область кристалла вдоль этой линии называется краевой дисло-
дислокацией.
Если подвергнуть кристалл действию сдвигового напряжения
определенной величины, в нем возникает скольжение одной атомной
плоскости относительно другой. Напряжение, которое для этого
необходимо, можно рассчитать теоретически. Как показывает опыт,
у реальных кристаллов скольжение возникает при напряжениях,
в тысячи раз меньших, чем это следует из теории. Такое расхожде-
расхождение теории и опыта обусловлено наличием дислокаций. Действи-
Действительно, при создании напряжения, вызывающего сдвиг верхней ча-
части кристалла на рисунке 76 вправо, движение краевой дислокации
будет очень облегчено благодаря тому, что атомы, расположенные
вдоль линии дислокации, уже смещены из своих равновесных поло-
положений. При перемещении дислокации происходит последователь-
последовательное смещение атомов в отличие от одновременного смещения, ха-
характерного для идеального кристалла.
Рис. 76. Краевая дислока-
дислокация в кристалле, совер-
совершенном во всех прочих от-
отношениях. Линия дислока-
дислокации перпендикулярна пло-
плоскости чертежа в точке В.
146
В поликристаллических телах, состоящих из беспорядочно ориен-
ориентированных кристаллических зерен, направления легкого скольже-
скольжения, обусловленного дислокациями в отдельных кристалликах,
не совпадают, и поэтому поликристаллические образцы деформи-
деформируются не так легко, как реальные монокристаллы.
Дислокации облегчают не только сдвиговую деформацию крис-
кристаллов, но и деформацию кристаллов при растяжении. При растя-
растяжении большинства кристаллов, используемых в технике, наблюда-
наблюдается скольжение отдельных кристаллических плоскостей, которое
можно обнаружить экспериментально и которое тесно связано с на-
наличием дислокаций.
Правильность высказанных предположений о роли дислокаций
в определении механических свойств кристаллов наглядно подтвер-
подтвердилась, когда были выращены маленькие металлические кристаллы,
свободные от дислокаций. Прочность таких кристаллов приближа-
приближалась к рассчитанной теоретически прочности идеальных кристаллов.
3. ПОЛИМЕРЫ
В последние годы в практической деятельности человека исклю-
исключительное значение приобрели аморфные твердые тела, объединя-
объединяемые под общим названием полимеры. Твердые полимеры воз-
возникают из соответствующих жидкостей — мономеров — не
в результате понижения температуры, как это имеет место у обыч-
обычных аморфных тел, а в результате химического соединения молекул
мономера.
В молекулах некоторых органических соединений атомы углеро-
углерода соединены между собой двойными химическими связями. При
определенных условиях двойные связи могут раскрываться и от-
отдельные молекулы соединяться друг с другом, образуя гигант-
гигантские макромолекулы.
Такой процесс называют полимеризацией, исходные
молекулы — мономерными молекулами и возникаю-
возникающий продукт — полимером.
В качестве примера можно привести реакцию образования поли-
полиэтилена с молекулярным весом 140 000. Молекулы этого вещества
представляют цепочки, состоящие каждая из 5000 отдельных моле-
молекул этилена СН2 = СН2, соединенных между собой за счет раскрыв-
раскрывшихся двойных связей. Число мономерных молекул р, входящих в
состав молекулы полимера, называют степенью полиме-
полимеризации.
При небольших значениях р, порядка 10—20, полимеризация
приводит к возникновению вязких жидкостей, которые при даль-
дальнейшей полимеризации, когда макромолекулы будут уже содержать
более ста исходных мономерных молекул, превращаются в твердые
аморфные тела — всем хорошо известные пластические массы, кау-
чуки, органические стекла и т. п.
147
Молекулярные веса твердых полимеров очень велики — от десят-
десятков тысяч до миллиона.
В зависимости от свойств исходной молекулы при полимериза-
полимеризации могут возникать или линейные макромолекулы в виде гигант-
гигантских молекулярных нитей, или же разветвленные пространственные
полимеры.
Механические свойства вещества, состоящего из линейных поли-
полимерных молекул, отличаются от свойств вещества, образовавшегося
в результате пространственной полимеризации.
В некоторых случаях при полимеризации первоначально воз-
возникают линейные полимеры, которые соответствующей обработкой
превращаются в пространственные. В этом процессе между цепочко-
образными макромолекулами возникают «молекулярные мостики»,
соединяющие между собой отдельные полимерные молекулы. Важ-
Важнейшим примером подобной реакции является вулканизация сырого
каучука, в результате которой получается резина.
Наряду с реакцией полимеризации для получения высокомоле-
высокомолекулярных соединений используется реакция поликонденса-
поликонденсации. При поликонденсации не обязательно наличие в исходных
мономерных молекулах двойных связей. В этом случае объединение
отдельных молекул в одну макромолекулу происходит в результате
реакции, сопровождающейся отщеплением от исходных молекул
атомов или групп атомов, способных к химическому взаимодейст-
взаимодействию. Возникающие при этом ненасыщенные химические связи обес-
обеспечивают соединение исходных молекул в макромолекулы.
Так, если в молекуле А имеются подвижные атомы водорода Н,
а в молекуле Б —две гидроксильные группы ОН, то реакция поли-
поликонденсации может протекать по следующей схеме:
Н — А— Н + НО — В — ОН-Н — А — В — ОН + Н2О
Н —А —В —ОН+Н —А —Н-Н —А —В —А —Н + Н2О
Н — А — В — А — Н + НО — В — ОН- Н—А—В-А—В—ОН+Н2О
Н-[А-В]„-ОН
В результате поликонденсации возникает, как мы видим, поли-
полимерная молекула с общей формулой Н [А — В]п ОН. С помощью
поликонденсации получается очень большое количество разно-
разнообразных высокомолекулярных веществ.
Полимерные вещества с линейными макромолекулами способны,
так же как и другие аморфные тела, переходить в кристаллическое
состояние. Однако, в то время как низкомолекулярные аморфные
тела способны полностью переходить в кристаллическую форму,
у полимеров большие размеры молекул не позволяют им располо-
148
житься с той высокой степенью упорядоченности, которая харак-
характерна для кристаллического тела. В этом случае наряду с областями,
в которых вещество находится в кристаллическом состоянии,
всегда присутствует аморфное вещество. Протяженность областей
кристаллического состояния много меньше длины вытянутых макро-
макромолекул обычных полимерных веществ.
В области кристаллизации нити отдельных макромолекул распо-
располагаются параллельно друг другу. За границами же этой области
нитеобразные молекулы располагаются беспорядочно, образуя амор-
аморфное вещество. Некоторая часть из этих молекул на большем или
меньшем расстоянии от первой кристаллической области может
вновь оказаться в области упорядоченного расположения молекул,
т. е. в области кристаллического состояния вещества. Таким об-
образом, длинная нитеобразная макромолекула на своем протяже-
протяжении может несколько раз входить в состав кристаллических об-
областей.
В качестве примера можно указать, что области кристаллич-
кристалличности целлюлозы имеют линейные размеры, обычно не превышающие
600 А AА — 1 ангстрем = 10~10 м), в то время как длина цепочко-
образной молекулы имеет порядок десятков тысяч ангстрем.
Полимерные вещества, обладающие хорошо развитой простран-
пространственной структурой, вовсе не кристаллизуются, поскольку в этом
случае связи, возникающие между отдельными макромолекулами,
не позволяют им расположиться упорядоченно.
Не кристаллизуются также так называемые нерегулярные
полимеры, в которых нарушено монотонное чередование звеньев
в макромолекулярной цепи.
В последние годы изложенные выше считавшиеся общепризнан-
общепризнанными представления о строении кристаллических и аморфных по-
полимеров существенно видоизменяются. Предполагается, что основ-
основной структурной единицей в полимерах является «пачка» полимер-
полимерных цепей, по своей структуре напоминающая область с упорядочен-
упорядоченно расположенными молекулами в обычной жидкости. Различие
между ними в основном сводится к разнице во временах существо-
существования. Большие размеры макромолекул и соответственно меньшая
их подвижность приводят к тому, что пачка полимерных молекул
существует в среднем гораздо дольше, чем область упорядоченного
расположения молекул в обычных жидкостях.
Образующие пачку нитеобразные молекулы смещены друг отно-
относительно друга, и потому длина пачки много больше длины отдель-
отдельной макромолекулы. По толщине пачка соответствует толшрче
нескольких макромолекул.
Пачки возникают в аморфных полимерах и являются промежу-
промежуточной стадией образования кристаллических областей.
У регулярных полимеров, с монотонным чередованием отдель-
отдельных звеньев в молекулярной цепи, при определенных условиях в
149
пачке возникает упорядоченное расположение уже не самих моле-
молекулярных цепей, а отдельных их звеньев, т. е. происходит кристал-
кристаллизация. Пачки полимерных цепей — это простейшая форма над-
надмолекулярной структуры, из которой могут строиться более слож-
сложные структуры, завершающиеся возникновением единичных кри-
кристаллов.
Растягивание полимера выпрямляет его молекулы и тем способ-
способствует их упорядоченному расположению. Если нагретый полимер
растянуть, а затем быстро охладить, то можно получить полимер-
полимерное вещество с ориентированным расположением молекул.
Одной из отличительных особенностей полимеров является их
высокая эластичность.
При растяжении обычных твердых тел упругая деформация
составляет несколько процентов от первоначальной длины. Поли-
Полимерные материалы также могут упруго деформироваться, и величи-
величина упругой деформации у них так же невелика, как и у низкомоле-
низкомолекулярных твердых тел. В то же время вещества, подобные резине,
можно обратимо растянуть настолько, что их длина будет в 6—10
раз превосходить первоначальную. Эту деформацию называют в ы-
сокоэласт и ческой деформацией.
Высокая эластичность полимеров проявляется только в некото-
некотором определенном интервале температур, ниже которого поли-
полимерное вещество делается твердым и хрупким, а выше — пласти-
пластичным.
Высокая эластичность присуща всем полимерным веществам.
Полимеры с сетчатой пространственной структурой, у которых вы-
высокая эластичность проявляется при комнатных и более низких тем-
температурах, называются резинами.
Область температур, в которой проявляется высокая эластич-
эластичность данного полимера, зависит также от скорости, с которой про-
производится механическое воздействие. Если на полимер действует
периодически изменяющаяся сила, то область высокой эластично-
эластичности зависит от частоты изменения силы. Так, например, при обыч-
обычном статическом измерении упругих свойств резины период воздей-
воздействия силы на образец составляет приблизительно 5 минут, в то
время как резиновая покрышка шины автомобиля, движущегося
со скоростью 60 км/час, подвергается действию силы, изменяющей-
изменяющейся с периодом около 0,01 сек, т. е. в 30 000 раз меньшим. В послед-
последнем случае материал делается хрупким при температуре, примерно
на 30 градусов более высокой, чем в первом. Поэтому не удиви-
удивительно, что какой-либо сорт резины может обладать хорошей элас-
эластичностью при одних условиях работы и вести себя как хрупкий
материал при других.
При объяснении высокоэластической деформации необходимо
помнить о следующих особенностях этого явления:
1. Высокоэластическая деформация обратима, т. е. остаточные,
деформации в образце могут отсутствовать.
150
2. Высокоэластическая деформация по своей величине в тысячи,
а иногда и в десятки тысяч раз больше упругой деформации.
3. Как показывает опыт, при высокоэластической деформации
полимеров объем образца не изменяется, а следовательно, не из-
изменяется потенциальная энергия взаимодействия молекул поли-
полимера.
Высокоэластическая деформация обусловлена особенностями
молекулярного строения полимеров. Наибольшим распростране-
распространением в настоящее время пользуется так называемая кинетическая
теория упругости полимеров, согласно которой высокая эластич-
эластичность ряда пластических материалов обусловлена тепловым движе-
движением их молекул. Согласно этой теории сами по себе цепочки поли-
полимерных молекул не проявляют упругость.
Отдельные звенья макромолекулярной цепи соединены между
собой как бы шарнирами, поворот которых сопряжен с преодоле-
преодолением не очень больших энергетическах барьеров. В результате
этого макромолекула может изменять свою форму без нарушения
химических связей. Такие изменения формы молекулы называют
конформационными превращениями. Тепловое
движение звеньев полимерной молекулы делает маловероятной та-
такую конформацию ее, при которой молекула оказывается вытяну-
вытянутой в виде прямолинейной цепочки. При обычных условиях полимер-
полимерные молекулы имеют неправильную изогнутую форму, приближаю-
приближающуюся в пределе к шарообразному клубку. При растяжении поли-
полимерного образца макромолекулы принудительно распрямляются
и ориентируются относительно друг друга. Если устранить растя-
растягивающую силу, то тепловое движение нарушит вынужденную де-
деформацией образца и несвойственную при данных условиях моле-
молекулам полимера их распрямленную форму — молекулы вновь при-
примут ту конформацию, которая наиболее вероятна при температуре
опыта. Образец приобретает первоначальную форму.
Кинетическая теория высокоэластической деформации объяс-
объясняет многие особенности этого явления, которые были установлены
эмпирически, и позволяет понять, почему это свойство присуще
только полимерным веществам.
Так, например, при увеличении температуры возрастает интен-
интенсивность теплового движения молекул, и потому согласно теории
должна возрастать упругость высокомолекулярных материалов.
Это явление действительно наблюдается на опыте.
У низкомолекулярных тел, упругость которых обусловлена сила-
силами, действующими между частицами тела, наблюдается противопо-
противоположная зависимость упругости от температуры. Кинетическая те-
теория позволяет также объяснить основные особенности зависимо-
зависимости высокоэластической деформации полимеров от времени действия
силы. Дальнейшее развитие молекулярной теории высокополимер-
высокополимерных веществ имеет большое значение ввиду чрезвычайной практи-
практической ценности этих материалов.
151
Реакции возникновения полимеров сравнительно легко регули-
регулируются, в результате чего в настоящее время химики могут созда-
создавать полимеры с различной длиной молекулярной цепи, менять по
своему усмотрению отдельные звенья в этой цепи, наращивать или
«сшивать» отдельные цепи и получать в результате этого вещества
с самыми различными свойствами.
Новых синтетических материалов создано так много, что затруд-
затруднительно даже перечислить их. Среди полимеров можно встретить:
строительные материалы, имитаторы различных ценных пород дере-
дерева; органические стекла; заменители металлов в машиностроении
и приборостроении; заменители кожи; искусственные волокна, нам-
намного превосходящие по своим свойствам естественные; различные
лаки и клеи; материалы, из которых изготавливаются корпуса су-
судов, автомобилей и самолетов, и т. д. Применение полимеров поз-
позволяет решать многие сложные задачи, с которыми связано разви-
развитие современной техники.
4. СИЛЫ, ДЕЙСТВУЮЩИЕ МЕЖДУ ЧАСТИЦАМИ
В ТВЕРДЫХ ТЕЛАХ
Все твердые тела, одни в большей, другие в меньшей степени
сопротивляются разрыву, следовательно, между частицами, образу-
образующими твердое тело, действуют силы притяжения, проявляющиеся
при попытке увеличить расстояние между частицами. Силы притя-
притяжения между частицами возрастают при их сближении. С другой
стороны, частицы, образующие твердое тело, не сближаются неогра-
неограниченно. Для каждой кристаллической решетки существуют совер-
совершенно определенные для данных температуры и давления расстояния
между частицами, расположенными в ее узлах. Это можно объяс-
объяснить, предположив, что при сближении частиц начинают проявляться
силы отталкивания, возрастающие по мере уменьшения расстояния
между частицами. Равновесное состояние кристалла соответствует
такому расположению частиц, при котором силы притяжения меж-
между частицами, образующими его, уравновешиваются силами от-
отталкивания.
Опыт показывает, что силы притяжения между атомами и моле-
молекулами проявляются при таких расстояниях, при которых силы
отталкивания еще не сказываются. Поэтому совершенно естествен-
естественно, что для того.чтобы силы отталкивания могли бы уравновесить
силы притяжения, они должны возрастать при уменьшении расстоя-
расстояния между частицами быстрее, чем силы притяжения. Характер из-
изменения сил притяжения при изменении расстояния между части-
частицами зависит от физической природы этих сил. Как показывает те-
теория, зависимость силы притяжения Fa между двумя частицами от'
расстояния г между ними может быть выражена уравнением:
152
в котором b — постоянная, а показатель степени т зависит от фи-
физической природы сил притяжения. Силы притяжения и обуслов-
обусловленная ими потенциальная энергия записываются с отрицательным
знаком, а силы отталкивания и соответствующая им потенциаль-
потенциальная энергия — с положительным.
Для зависимости сил отталкивания FR от расстояния современ-
современная теория дает весьма сложное выражение:
p, B)
в котором р — постоянная величина, зависящая от природы взаимо-
взаимодействующих частиц, а Р (г) — многочлен, содержащий положитель-
положительные и отрицательные степени рас-
расстояния между частицами — г. Слож-
Сложность теоретически строгого выраже-
выражения приводит к тому, что на прак-
практике избегают им пользоваться и
выражают зависимость сил отталки-
отталкивания от расстояния приближенно
верным выражением:
В КОТОРОМ аип~ ПОСТОЯННЫе. между ^частицамивтве^дых
Для ТОГО чтобы СИЛЫ отталкива- телах: / — сила притяжения;
ния возрастали при сближении час- // — сила отталкивания;
тиц быстрее, чем силы притяжения, I!I ~ результирующая сила,
необходимо, чтобы показатель сте-
степени п в члене, учитывающем отталкивание, был больше показате-
показателя степени т в члене, учитывающем притяжение.
Если изобразить графически изменение сил, действующих меж-
между частицами при их сближении, откладывая вдоль оси абсцисс
расстояние между частицами, вдоль положительного направления
оси ординат — величину силы отталкивания, а вдоль отрицатель-
отрицательного— величину силы притяжения, то соответствующий график бу-
будет подобен графику, изображенному на рисунке 77.
Результирующая кривая, находимая суммированием кривых,
передающих изменение с расстоянием сил притяжения и отталкива-
отталкивания, изображена на рисунке сплошной линией. Изменение ре-
результирующей силы, действующей между частицами, при измене-
изменении расстояния между ними выражается уравнением:
rn+x
D)
Легко убедиться, что при определенном расстоянии го между
частицами силы отталкивания компенсируют силы притяжения.
153
Естественно, что при равновесии неподвижные частицы будут
находиться на расстоянии г0 одна от другой. При увеличении этого
расстояния преобладающими делаются силы притяжения — ча-
частицы сопротивляются разрыву. Наоборот, попытки сблизить ча-
частицы приводят к преобладанию сил отталкивания — частицы ока-
оказывают сопротивление сжатию.
С силами, действующими между частицами, связана соответ-
соответствующая потенциальная энергия. Можно считать, что суммарная
потенциальная энергия взаимодействия двух частиц складывается
из потенциальной энергии, обусловленной притяжением частиц, и
потенциальной энергии, обусловленной их отталкиванием. Неслож-
Несложно показать, что если закон изменения с расстоянием сил, действую-
действующих между частицами, выражается уравнением D), то их потен-
потенциальная энергия Ер будет изменяться при изменении расстоя-
расстояния согласно уравнению:
F А В (Ъ
в котором А и В — постоянные.
В последнее время в качестве приближенно верного выражения
для энергии потенциального взаимодействия частиц часто применя-
применяют уравнение E), в котором полагают п — 12 и т = 6.
Обозначая ?¦„ — энергию, соответствующую минимуму на кри-
кривой потенциальной энергии, можно придать уравнению для энер-
энергии взаимодействия частиц более простую форму:
Здесь г0 — расстояние между частицами, при котором потенциаль-
потенциальная энергия их взаимодействия минимальна.
Графически зависимость энергии взаимодействия частиц от рас-
расстояния между ними изображена на рисунке 78 сплошной линией.
Для сравнения на том же рисунке пунктирной линией изображена
зависимость от расстояния результирующей силы, действующей
между частицами. Вдоль оси ординат на этом рисунке отложены
для одной кривой энергия, которой обладают частицы, для другой
кривой — сила, действующая между ними.
Если частицы неподвижны, т. е. если их кинетическая энергия
равна нулю, то они находятся на расстоянии г0, соответствующем
минимуму на кривой потенциальной энергии. Как мы знаем, в дей-
действительности частицы материи никогда не находятся в покое. Даже
при абсолютном нуле температуры частицы твердого тела совер-
совершают колебания и обладают поэтому некоторой так называемой ну-
нулевой энергией.
Частица, обладающая, помимо потенциальной энергии, некото-
некоторой кинетической энергией Ek, будет находиться не в точке О
154
F.E
(рис. 78), а несколько выше (точка 0,). Расстояние, измеренное
вдоль оси ординат между точкой О и уровнем ааи на котором на-
находится частица, равно ее кинетической энергии. Частица колеб-
колеблется, причем движение ее ограничено отрезком ааи на концах ко-
которого ее кинетическая энергия делается равной: при движении
в одну сторону — потенциальной энергии притяжения, а при дви-
движении в другую сторону — по-
потенциальной энергии отталкива-
отталкивания. Особенности молекулярных
колебаний связаны с тем, что по-
потенциальная яма, ограничивающая
движение частицы, несимметрична
относительно линии 00': левая
ветвь кривой возрастает более кру-
круто, чем правая.
В результате несимметрично-
несимметричности кривой потенциальной энергии
частица при своем движении впра-
вправо смещается на большее расстоя-
расстояние, чем при движении влево, так
что среднее положение, занимае-
занимаемое частицей при колебаниях, не
совпадает с линией 00' (линия,
соответствующая среднему поло-
положению частицы, изображена на
рисунке 78 пунктиром). Кине-
Кинетическая энергия молекулярного
движения пропорциональна абсо-
абсолютной температуре. Чем больше
температура, тем выше приподня-
приподнята на графике частица по сравне-
сравнению со своим наинизшим энергетическим положением и тем более
смещено ее среднее положение вправо от линии 00', соответствующей
положению частицы при абсолютном нуле. Несимметричность кри-
кривой, выражающей взаимодействие частиц, обусловливает расшире-
расширение твердых тел при нагревании. Если бы частицы твердого тела
совершали чисто гармонические колебания, или, другими словами,
если бы рассматриваемая кривая была симметрична, твердые тела
не должны были бы расширяться при нагревании.
Как уже было сказано, в узлах кристаллической решетки могут
находиться ионы, атомы или молекулы. Соответственно различают
ионные, атомные и молекулярные кристаллические решетки.
Ионные решетки свойственны кристаллам большинства неоргани-
неорганических солей: NaCl, KC1, LiJ и т. д.
В подобных решетках ионы одного знака окружены ионами про-
противоположного знака (см. рис. 79). Силы притяжения, удерживаю-
Рас. 78. Зависимость энергии
взаимодействия частиц
от расстояния между ними.
155
щие частицы кристалла в ионных кристаллических решетках, —
это силы электростатического притяжения разноименно заряжен-
заряженных ионов. Потенциальная энергия притяжения двух разноименных
зарядов -{-q и —q, находящихся на расстоянии г0 один от другого,
выражается уравнением:
?, = -?. F)
Показатель степени п у члена, учитывающего потенциальную
энергию, обусловленную силами отталкивания (уравнение 5), тео-
теоретически определить нель-
нельзя. Изменяя значения посто-
постоянных, входящих в уравне-
уравнение, выражающее потенци-
потенциальную энергию двух частиц
E), можно получить удов-
удовлетворительные результаты
при разных значениях п. Час-
Часто полагают п = 9. В этом
случае для потенциальной
энергии двух ионов можно
написать:
п А о2
G)
Рис. 79. Кристаллическая решетка
хлористого цезия.
'о
Хотя молекулярные взаи-
взаимодействия распространяются
только на очень короткие расстояния, так что в основном необходи-
необходимо учитывать взаимодействие частицы с ее ближайшими соседями, все
же полностью игнорировать присутствие в кристаллической решет-
решетке частиц, расположенных за ближайшим окружением, нельзя.
Для учета взаимодействия с более удаленными частицами, а также
геометрических особенностей кристаллической решетки в уравне-
уравнение для потенциальной энергии вводятся специальные коэффици-
коэффициенты, так называемые постоянные Маделунгааир. С учетом
постоянных Маделунга уравнение для потенциальной энергии двух
ионов принимает вид:
? =pA_ai! (8)
Поскольку силы отталкивания убывают с расстоянием много
быстрее сил притяжения, постоянную Маделунга р можно прирав-
приравнять числу ближайших частиц, окружающих данную, т. е. не учи-
учитывать при расчете энергии, обусловленной силами отталкивания;
взаимодействия частицы с ее более далекими соседями.
У твердых тел, кристаллическая решетка которых состоит из
атомов, частицы удерживаются силами, имеющими ту же природу,
156
что и силы, удерживающие атомы в молекулах химических соеди-
соединений.
Теория химической связи излагается в квантовой механике
и здесь можно лишь пояснить, что связь эта возникает в резуль-
результате изменения движения электронов в атомах, образующих при
сближении химическое соединение.
Атомные решетки имеют: алмаз, SiC, A1N, ВеО и т. д.
Химическая связь очень прочна, и потому атомные кристаллы
отличаются высокой температурой плавления, большой твердостью,
исключительно малой летучестью.
В тех случаях, когда в узлах кристаллической решетки рас-
расположены молекулы, последние удерживаются ван-дер-ваальсовыми
силами, подобными тем, которые действуют между молекулами
реальных газов. Эти силы невелики по сравнению с силами, дей-
действующими в атомных и ионных кристаллических решетках. По-
Поэтому молекулярные кристаллы отличаются сравнительно низкой
температурой плавления и малой механической прочностью. Моле-
Молекулярные кристаллические решетки свойственны большинству орга-
органических соединений.
Особую группу кристаллических решеток составляют кристал-
кристаллические решетки металлов. В узлах кристаллических решеток ме-
металлов находятся ионы соответствующего элемента. В отличие от
ионных решеток кристаллов неорганических солей все ионы, распо-
расположенные в узлах кристаллической решетки металла, имеют за-
заряды одного знака.
Ионизация возникает в данном случае в результате того, что
некоторое количество электронов у каждого атома приобретает
способность относительно свободно переходить от одного атома
к другому. Эти электроны оказываются как бы принадлежащими
одновременно всем атомам в металле. Они называются свобод-
свободными электронами. Свободные электроны, перемещаю-
перемещающиеся внутри кристаллической решетки металла, до некоторой
степени напоминают молекулы газа (электронный газ). Взаимодей-
Взаимодействие положительно заряженных ионов со свободными электронами
обеспечивает прочность кристаллической решетки металлов. Нали-
Наличие свободных электронов сообщает металлам способность прово-
проводить электрический ток. Движущиеся с большой скоростью электро-
электроны обеспечивают высокую теплопроводность металлов.
Силы, действующие между частицами, образующими твердое тело
проявляются при изменении расстояния между ними, т. е. при сжа-
сжатии или растяжении тела. В простейших случаях измерения сжи-
сжимаемости позволяют определить значения постоянных, знание ко-
которых необходимо для расчета сил, действующих между частицами
твердого тела. Располагая данными о структуре твердого тела и зная
зависимость от расстояния сил, действующих между его частицами,
можно определить механическую прочность различных твердых
тел.
157
Опыт убеждает в том, что прочность твердых тел, найденная
экспериментально, всегда много меньше рассчитанной теорети-
теоретически.
Низкая механическая прочность реальных твердых тел объяс-
объясняется, как указано выше, наличием дислокаций, а кроме того,
присутствием на поверхности тел микроскопических трещин, уве-
увеличивающихся при растяжении образца и приводящих к его раз-
разрыву.
А. Ф. Иоффе показал, что если кристалл NaCl погрузить в
воду и путем растворения уничтожить имевшиеся на его поверхно-
поверхности трещины, то прочность кристалла на разрыв повышается от 0,4
до 30 и даже до 150 кГ/мм2. Подобным образом возрастает прочность
на разрыв стеклянных нитей при протравливании их плавиковой
кислотой. В данном случае повышение прочности у более тонких
нитей объясняется тем, что их поверхность меньше и на ней менее
вероятно присутствие трещин.
Часто возникает вопрос: почему, если разломать твердое-тело
пополам, а затем прочно прижать друг к другу образовавшиеся
половинки, они не соединяются вместе и не образуют вновь первона-
первоначального тела? Причин для этого несколько.
Во-первых, при разламывании тела поверхности разлома изме-
изменяются, и практически невозможно привести соприкасавшиеся
до разлома точки обеих поверхностей в их первоначальное поло-
положение.
Во-вторых, на образовавшихся при разломе поверхностях воз-
возникает тонкая пленка воздуха, относительно прочно удерживаемая
поверхностями. Воздушная пленка препятствует плотному кон-
контакту соединяемых поверхностей.
В последние годы применяется новый способ соединения метал-
металлических изделий — холодная сварка с помощью ультразвуковых
колебаний. В этом методе соединяемые металлические поверхности
прижимаются одна к другой под сравнительно небольшим давле-
давлением, а затем ту часть детали, в которой осуществляется' сварка,
заставляют совершать ультразвуковые колебания в плоскости, сов-
совпадающей с плоскостью свариваемых поверхностей. Как предпола-
предполагают, при этом свариваемые поверхности деталей очищаются и воз-
возникает прочное соединение металла.
5. ТЕПЛОЕМКОСТЬ ТВЕРДЫХ ТЕЛ
Образующие твердое тело частицы находятся в непрерывном
тепловом движении — они колеблются около положений равнове-
равновесия, совпадающих с узлами кристаллической решетки. Естественно,
что каждая частица взаимодействует с окружающими ее частицами,
и поэтому колебания всех частиц являются связанными между со-
собой. Однако, как показывает сопоставление выводов теории с
опытом, при достаточно высокой температуре эта связь невелика,
158
и можно считать, что каждая частица колеблется независимо от
своих соседей.
Внутренняя энергия, приходящаяся на одну степень свободы
движения атома или молекулы и0, пропорциональна абсолютной
температуре:
kT
U
здесь k — постоянная Больцмана, Т — абсолютная температура.
Если атомы вещества обладают i степенями свободы, то внутрен-
внутренняя энергия U одного килограмм-атома выражается соотношением:
U = - kTNA.
2 л
Поскольку N а — число Авогадро, a NAk = R, то
U = ±RT.
Положение атома (как материальной точки) в пространстве оп-
определяется тремя координатами, следовательно, колеблющийся атом
обладает тремя степенями свободы движения.
При подобном подсчете числа степеней свободы необходимо,
однако, принять во внимание, что колеблющаяся материальная
точка обладает одновременно кинетической и потенциальной энер-
энергией, и поэтому при вычислении внутренней энергии колеблющегося
атома наряду со степенями свободы, приходящимися на кинети-
кинетическую энергию, следует учитывать равное количество степеней
свободы, приходящихся на долю потенциальной энергии. В резуль-
результате этого колеблющийся атом оказывается обладающим не тремя
степенями свободы, а шестью.
Внутренняя энергия килограмм-атома кристаллического твер-
твердого тела, таким образом, равна:
U = SRT. (9)
Поскольку твердые тела обладают малым коэффициентом тер-
термического расширения и, следовательно, мало увеличиваются в
объеме при нагревании, для них часто не различают теплоемкость
при постоянном объеме от теплоемкости при постоянном давле-
давлении, а говорят просто о теплоемкости твердого тела С, которая
численно равна первой производной от внутренней энергии тела
по температуре:
г г dU
Подставляя в выражение для теплоемкости значение внутренней
энергии твердого тела, найдем:
С = 3R. A0)
159
Таким образом, при достаточно высокой температуре
атомная теплоемкость всех твердых тел не зависит
от температуры и равна 3R.
Этот закон был открыт эмпирически еще в XIX столетии Д го-
голо н г о м A785—1838) и Пт и A791—1820) и носит их имя (за-
(закон Дюлонга и Пти). Величины, приведенные в таблице 14, убежда-
убеждают в том, что во многих случаях закон Дюлонга и Пти удовлетвори-
удовлетворительно выполняется, и, следовательно, для указанных в таблице ве-
веществ колебания атомов уже при комнатной температуре можно счи-
считать независимыми. В то же время имеются и такие вещества, как,
например, алмаз или бор, для которых измеренное при комнатной
температуре значение темплоемкости существенно отличается от 3R.
Таблица 14
Теплоемкость твердых тел
Вещество
Алюминий
Железо
Медь
Кадмий
Алмаз
Бор
Кремний
Атомная
емкость
3,07
3,18
2,95
3,05
0,68
1,26
2,34 •
тепло-
R
R
R
R
R
R
R
Таблица 15
Молекулярная теплоемкость
химических соединений
в твердом состоянии
Соединение
РЬО
КС1
AgCl
РЬС12
ВаС12
CaCl.,
Молекулярная
теплоемкость
5,78 •
6,19 ¦
6,26 •
9,51 •
9,3 •
9,1 •
R
R
R
R
R
R
Для этих веществ комнатная температура, очевидно, недостаточ-
недостаточно высока для того, чтобы считать колебания атомов независимыми.
В случае кристаллов солей или каких-либо других химических
соединений обычно определяется не атомная, а молекулярная тепло-
теплоемкость.
Если по-прежнему считать, что кристалл построен из атомов
или ионов, колеблющихся независимо друг от друга, то, очевидно,
число частиц, движение которых необходимо учитывать при' вы-
вычислении внутренней энергии одного килограмм-моля вещества,
будет равно общему числу атомов в одном киломоле вещества или
числу Авогадро, помноженному на число атомов в молекуле. Это
объясняет найденное эмпирически правило, согласно которому
молекулярная теплоемкость твердого соединения рав-
равна сумме атомных теплоемкостей элементов, входя-
входящих в состав соединения.
В случае твердых соединений, состоящих из двух атомов, на<-
пример КС1, РЬО и т. д., молекулярная теплоемкость согласно этому
правилу должна равняться 6R, а для твердых соединений, состоя-
состоящих из трех атомов, например СаС12, РЬС12 и т. д., соответствен-
соответственно 9R. В таблице 15 приведены величины молекулярных теплоем-
костей некоторых соединений, подтверждающие сформулирован-
сформулированное правило.
Как показывает опыт, постоянство теплоемкости твердых тел
нарушается при понижении температуры. Теплоемкости твердых
тел уменьшаются при понижении температуры, стремясь к нулю
при приближении температуры к абсолютному нулю.
Удовлетворительно объяснить зависимость теплоемкости твер-
твердых тел от температуры удалось Д е б а ю на основе теории кван-
квантов. Теория квантов, развитая М. Планком A858—1947), исхо-
исходит из представления о том, что энергия, так же как и вещество,
имеет дискретную (прерывную) природу. Энергия может переда-
передаваться от одних тел другим не сколь угодно малыми порциями,
а лишь определенными дискретными количествами — квантами.
Величина кванта энергии колебательного движения е зависит
от частоты колебаний v и определяется выражением:
e = Av, A1)
в котором h — так называемая универсальная посто-
постоянная Планка, равная F,6254 ± 0,0002) • 10~34 дж ¦ сек.
Квантовая природа энергии проявляется в тепловых явлениях
тогда, когда энергия теплового движения атомов или молекул,
приходящаяся на одну степень свободы — , делается сравнимой с
величиной кванта энергии. Пока же величина кванта энергии очень
kT
мала по сравнению с величиной — , квантовую природу энергии
можно не учитывать. Частота колебаний частиц в кристаллической
решетке имеет порядок 10п секГ1, так что соответствующий квант
имеет величину, равную 6,6 • 10~34 • 1012 дж = 6,6 • 10~22 дж.
Таким образом, квантовую природу энергии необходимо учиты-
учитывать при температурах Т, при которых выполняется неравенство:
— <6,6. Ю~22,
или
Исходя из квантовой теории, Дебай нашел, что при темпера-
температурах, близких к абсолютному нулю, внутренняя энергия кристал-
кристаллического твердого тела пропорциональна четвертой степени тем-
температуры:
U = aT\ A2)
где а — постоянная величина.
6 Заказ 145 jgj
Соответственно для теплоемкости кристаллического твердого
тела им найден закон, называемый законом кубов Дебая:
С — — = 4аГ3. A3)
На рисунке 80 изображено определенное экспериментально из-
изменение теплоемкости твердого тела при изменении температуры.
Как мы видим, при сравнительно высоких температурах теплоем-
теплоемкость твердого тела не зависит от температуры. Это область при-
применимости закона Дюлонга и Пти. При температурах, прилегающих
к абсолютному нулю, наб-
наблюдается пропорциональ-
пропорциональность теплоемкости третьей
степени температуры —это
область, в которой выпол-
выполняется закон Дебая. Меж-
Между этими областями лежит
промежуточная область,
для которой количествен-
количественную связь между теплоем-
теплоемкостью и температурой по-
пока установить не удалось.
Как показали опытные
исследования, не для всех
твердых кристаллических
тел вблизи абсолютного
нуля температуры тепло-
Рис. 80. Изменение теплоемкости твердых
тел при изменении температуры.
емкость изменяется про-
пропорционально кубу температуры. Существуют твердые вещества,
теплоемкость которых при низких температурах пропорциональ-
пропорциональна у одних тел — квадрату, а у других—первой степени тем-
температуры. Эти особенности изменения теплоемкости с температу-
температурой были объяснены В. В. Тарасовым, указавшим на то,
что степень, в которой температура входит в закон Дебая, связа-
связана с особенностями строения кристаллической решетки.
Если частицы в кристаллической решетке связаны примерно
одинаково прочно во всех трех измерениях, выполняется закон
кубов Дебая. Для кристаллов, в которых соседние частицы, распо-
расположенные в определенных плоскостях, связаны много прочнее, чем
соседние частицы, расположенные в двух соприкасающихся плоско-
плоскостях, теплоемкость при низких температурах будет пропорциональ-
пропорциональна второй степени температуры. Это справедливо для графита, слю-
слюды и т. п. Наконец, если кристалл состоит из прочно связанных це-
цепочек частиц, причем сами цепочки соединены между собой слабо,
теплоемкость пропорциональна первой степени температуры. Та-
Такая зависимость наблюдается, например, для плавленого кварца
и ряда других твердых тел.
162
6. СУБЛИМАЦИЯ, ПЛАВЛЕНИЕ И КРИСТАЛЛИЗАЦИЯ
ТВЕРДЫХ ТЕЛ
Скорости теплового движения частиц твердых тел распределены
при не слишком низких температурах согласно закону Максвелла.
Это означает, что в кристалле конечных размеров всегда будет
присутствовать большее или меньшее число частиц, обладающих
скоростью, существенно превосходящей характерное для данной
температуры среднее значение скорости. Естественно, что в тех слу-
случаях, когда силы, удерживающие частицы в кристалле, не очень
велики, может оказаться, что энергия, которой обладает какая-
либо частица, превосходит работу, необходимую для отрыва ча-
частицы от поверхности кристалла. Частицы, входящие в состав кри-
кристалла, могут в этом случае отрываться от поверхности твердого
тела и переходить в окружающее кристалл пространство. Повсед-
Повседневный опыт убеждает нас в том, что подобный процесс действитель-
действительно происходит. Вспомните резкий запах, свойственный таким твер-
твердым веществам, как нафталин, камфора, многим твердым органиче-
органическим соединениям. Запах этих веществ обусловлен непосредствен-
непосредственным переходом вещества из твердого состояния в газообразное.
Этот процесс называют сублимацией, или возгонкой
твердого вещества.
Таким образом, над поверхностью твердого тела устанавливается
характерное для данного вещества давление насыщенного пара.
Давление насыщенного пара над поверхностью твердого тела
зависит от температуры: чем выше температура, тем больше дав-
давление насыщенного пара.
Если повышать температуру кристаллического твердого тела,
то при достижении определенной величины, характерной для дан-
данного вещества, твердое тело плавится. Однако нагрев твердого
тела до температуры плавления еще не вызывает превращения его
в жидкость. Для этого необходимо сообщить твердому телу опреде-
определенное количество теплоты, затрачиваемое на разрушение кристал-
кристаллической решетки. Эта теплота называется теплотой плавле-
плавления твердого тела.
В том случае, когда твердому телу, нагретому до температуры
плавления, сообщается количество теплоты, недостаточное для пол-
полного превращения его в жидкость, какая-то часть вещества остается
в кристаллической форме и может неограниченно долго сосущест-
сосуществовать с жидкостью. При определенных условиях это равновесие не
нарушается и в том случае, если одновременно вещество присутству-
присутствует в парообразном состоянии.
Поскольку переход в парообразное состояние возможен как из
жидкого, так и из твердого состояния, подобное равновесие будет
наблюдаться (см. рис. 81) только в том случае, если давления насы-
насыщенного пара над твердым и жидким веществами одинаковы. Если
такого равенства нет и если, предположим, давление пара над жил-
6* 163
костью больше, чем над твердым телом, — жидкость будет испарять-
испаряться, а пар, конденсируясь, превращаться втвердоетело. Если давле-
давление пара больше над твердым телом, будет протекать противополож-
противоположный процесс.
Зависимость давления насыщенного пара жидкости и твердого
тела от температуры изображается графически кривыми, пересекаю-
пересекающимися при температуре плавления (рис. 82).
При понижении температуры перегретого пара жидкости он при
некоторой температуре становится насыщенным. Эта температура
Пар
Газообразная,
фаза
Рис. 81. Равновесие в случае присут-
присутствия вещества одновременно в трех
агрегатных состояниях.
Рис 82. Диаграмма состоя-
состояния вблизи тройной точки.
будет температурой конденсации. При отводе теп-
теплоты вещество из парообразного состояния превращается при этой
температуре в жидкость.
При дальнейшем охлаждении жидкость при некоторой темпе-
температуре кристаллизируется, т. е. превращается в твердое тело. Эта
температура называется температурой кристаллизации.
Температура кристаллизации зависит от давления.
Если соединить температуры кристаллизации, соответствующие
различным давлениям, то на графике (рис. 82), изображающем рав-
равновесие между различными состояниями вещества, возникнет ли-
линия РВ, соответствующая равновесию между веществом, находя-
находящимся в твердом и жидком состояниях. Эта линия начинается в
точке пересечения кривых давления насыщенных паров жидкости
и твердого тела.
Точка Р, в которой пересекаются три указанные кривые и,
следовательно, в которой в равновесии находится вещество в трех
состояниях, называется тройной точкой.
Каждое вещество имеет только одну тройную точку.
Давление, под которым находится вещество в тройной точке,
представляет собой давление насыщенных паров, и не следует сме-
смешивать это давление о атмосферным. Так, например, тройная точ-
ка воды характеризуется давлением 610,5 н/м2 и температурой
0,0098° С.
Наклон кривой РВ, изображающей линию равновесия между
твердым и жидким состояниями вещества, зависит от изменения
объема твердого тела при плавлении. Если при плавлении объем ве-
вещества увеличивается, т. е. если при температуре плавления плот-
плотность жидкости меньше плотности твердого тела, то при повышении
давления температура плавления повышается. Это наблюдается у
подавляющего большинства веществ.
У некоторых веществ (вода, висмут, чугун и др.) при плавлении
объем, занятый веществом, уменьшается, и плотность жидкости ока-
оказывается больше плотности соответствующего твердого тела. У этих
тел температура плавления понижается при увеличении давления.
В тех случаях, когда температура плавления вещества воз-
возрастает с увеличением давления, возможно, создав достаточно вы-
высокое давление, получить вещество в твердом состоянии при темпе-
температуре выше критической. Так, например, критическая темпера-
температура углекислоты +31,1° С, однако при давлении 11,77 • 108 н!м2
углекислота будет твердой при температуре +93° С.
Кристаллизация жидкостей, как правило, требует для своего
возникновения присутствия кристаллических зародышей. Такими
зародышами могут являться не только кристаллики образующегося
вещества, но и посторонние примеси, пыль, сажа и т. п. При пол-
полном отсутствии зародышевых центров кристаллизации возникнове-
возникновение в жидкости микроскопических кристалликов затруднено и
происходит только при переохлаждении жидкости, т. е.
при понижении ее температуры ниже температуры кристаллизации.
Некоторые жидкости, если их тщательно очистить от посторонних
примесей, могущих служить центрами кристаллизации, теряют
вообще способность кристаллизоваться. При понижении темпера-
температуры они переохлаждаются и в дальнейшем затвердевают, стано-
становясь твердыми аморфными веществами.
Характерными особенностями обладает явление кристаллизации
из раствора.
Понижение давления насыщенного пара над раствором по
сравнению с давлением пара над чистым растворителем приводит
к тому, что температура кристаллизации раствора ниже, чем тем-
температура кристаллизации чистого растворителя.
Для того чтобы пояснить причину этого, проведем на графике,
изображающем равновесие между различными агрегатными состоя-
состояниями вещества (рис. 83), кривую P^Ai—давления пара над
раствором. При кристаллизации разбавленного раствора в виде
твердой фазы выпадает чистый растворитель. Равновесие между
жидкой и твердой фазами наступает при равенстве давлений пара
над твердым и жидким веществом, т. е. при пересечении кри-
кривых PC и PiAi и, следовательно, при температуре, меньшей тем-
температуры кристаллизации чистого растворителя. Таким образом,
165
добавка к веществу в небольшом количестве любой примеси пони-
понижает его температуру кристаллизации.
Температура кристаллизации твердого тела совпадает с его
температурой плавления, и поэтому закристаллизовавшийся раствор
плавится при температуре более низкой, чем чистый растворитель.
Для данного растворителя понижение его температуры кри-
кристаллизации при растворении в нем незначительного количества
какого-либо вещества не зависит от химических особенностей по-
последнего и определяется тем, какова концентрация, вернее, мольная
доля, растворенного вещества. Это свойство растворов позволяет,
взяв определенное количество какого-либо растворителя и раство-
растворив в нем навеску вещества с неиз-
неизвестным молекулярным весом, опре-
определить последний на основании из-
измерения понижения температуры кри-
кристаллизации образовавшегося раство-
раствора. Действительно, зная вес раство-
растворенного вещества и определив по по-
понижению температуры кристаллиза-
кристаллизации раствора, какой мольной доле
вещества он соответствует, не пред-
представляет уже труда вычислить моле-
молекулярный вес вещества, находя-
Растворитель/ /Раствор
щегося в растворе. Этот метод оп-
Рис. 83. Понижение темпера- ределения молекулярных весов на-
натуры кристаллизации зывают криоскопией.
раствора. Понижение температуры плавле-
плавления растворов используют при при-
приготовлении охлаждающих смесей. Для этого к кашице измель-
измельченного льда добавляют кристаллики какой-либо соли. При этом
температура плавления льда понижается, и смесь приобретает
температуру ниже нуля, так, например, при смешении льда и по-
поваренной соли в отношении 3 : 1 температура смеси понижается
приблизительно до—21,2° С. При смешении твердой углекислоты с
этиловым спиртом достигается температура —72° С.
7. КРИСТАЛЛИЗАЦИЯ РАСТВОРОВ. ДИАГРАММЫ ПЛАВКОСТИ
Изучение кристаллизации растворов и расплавов, содержащих
два индивидуальных химических вещества, позволяет обнаружить
превращения, происходящие в подобных системах. Поскольку в
технике, как правило, приходится иметь дело с различными спла-
сплавами, этот метод исследования вещества имеет большое практи-
практическое значение. Обычно при этом прибегают к построению так на-
называемых диаграмм плавкости (рис. 84). Вдоль оси абсцисс по-
подобной диаграммы откладывают состав исследуемой системы. Кон-
Концы оси абсцисс соответствуют взятым отдельно составным частям
166
системы. Вдоль оси ординат откладывают температуры кристаллиза-
кристаллизации, или, что то же самое, температуры плавления смесей соответ-
соответствующего состава. Предположим, желательно исследовать сплавы
свинца с сурьмой. Чистый свинец кристаллизуется при температу-
температуре 326° С, сурьма—при температуре 632° С.Если к расплавленному
свинцу добавить сурьмы, то, несмотря на более высокую температуру
кристаллизации сурьмы, расплав свинца и сурьмы начнет кристал-
кристаллизоваться при температуре более низкой, чем температура кри-
кристаллизации чистого свинца. Так, расплав, содержащий 10% сурьмы,
начнет кристаллизоваться при температуре 250° С,которая ниже тем-
температуры кристаллизации отдельно взятого свинца. Образующееся
при этом твердое вещество
будет содержать только кри-
кристаллики чистого свинца, бла-
благодаря чему содержание сурь-
сурьмы в жидком сплаве будет по-
повышаться. Увеличение содер-
содержания сурьмы в расплаве
вызовет дальнейшее пониже-
понижение температуры кристалли-
кристаллизации.
Таким образом, по мере
протекания процесса кристал-
кристаллизации температура кристал-
кристаллизации плавно изменяется,
следуя кривой АЕ.В точке Е
понижение температуры кри-
кристаллизации прекращается, и
весь расплав затвердевает.
Очевидно, что при составе расплава, соответствующем точке Е,
одновременно кристаллизуются свинец и сурьма. Возникший сли-
слиток состоит из мелких кристалликов свинца и сурьмы. Точка Е
называется эвтектической точкой, а сплав, состав ко-
которого соответствует точке Е, называется эвтектикой.
При охлаждении расплавов, составы которых соответствуют
точкам, расположенным справа от эвтектической точки, кристалли-
кристаллизуется чистая сурьма, жидкий расплав обогащается свинцом и точка
кристаллизации понижается. Возникает правая ветвь диаграммы
плавкости. Обе ветви пересекаются в эвтектической точке.
В расплаве, имеющем состав эвтектики, при охлаждении не
возникает кристаллизации до тех пор, пока температура не пони-
понизится до величины, соответствующей эвтектической точке. По до-
достижении этой температуры расплав затвердевает, как индивиду-
индивидуальное вещество.
В том случае, когда составные части А и В расплава образуют
химическое соединение АВ, диаграмма плавкости имеет вид, изобра-
изображенный на рисунке 85.
Свинец 20
40
60
80
Рис. 84. Диаграмма плавкости системы
свинец — сурьма.
167
Эта диаграмма представляет как бы сочетание двух простых диа-
диаграмм, подобных рассмотренной выше, у которых имеется общая
составная часть АВ.
При охлаждении расплава с составом, отвечающим точке а,
кристаллизация начнется с выпадения кристалликов вещества А.
Содержание вещества А в расплаве будет уменьшаться, а темпе-
температура расплава понижаться вплоть до температуры, соответствую-
соответствующей эвтектической точке ?\. При этой температуре одновременно
с выпадением кристалликов вещества А начнут выпадать кристал-
кристаллики соединения АВ.
Рис. 85. Диаграмма плавкости
системы, составные части которой
образуют соединение.
Рис. 86. Диаграмма плавкости
системы никель — медь.
В случае расплава с составом, отвечающим точке Ь, первона-
первоначально при охлаждении будут выпадать кристаллики соединения
АВ, а состав расплава будет приближаться к эвтектике.
Сходная картина наблюдается при охлаждении расплавов, со-
составы которых на диаграмме плавкости заключены между точ-
точками АВ и В.
Затвердевание расплава с составом аг начинается с выпадения
кристалликов соединения АВ и заканчивается кристаллизацией
эвтектики, состоящей из кристаллов соединения АВ и кристаллов
вещества В. При кристаллизации расплава с составом Ьх первона-
первоначально выпадают кристаллы вещества В, а заканчивается затвер-
затвердевание кристаллизацией эвтектики, состоящей из кристаллов ве-
вещества В и кристаллов соединения АВ.
Особый вид имеют диаграммы плавкости таких двойных сплавов,
у которых атомы одного из веществ, входящих в состав сплава, мо-
могут заменять атомы второго вещества в соответствующей ему кри-
кристаллической решетке, и обратно — атомы второго вещества могут
заменять атомы в кристаллической решетке первого вещества.
Диаграмма плавкости подобной системы (рис. 86) не похожа на
рассмотренные выше.
168
При температурах, превосходящих 1702° по абсолютной шкале,
жидкий никель смешивается в любых пропорциях с медью. Если
расплав с составом, отвечающим на графике точке а, охладить
до 1500° К, он начнет кристаллизоваться, причем выпадающие кри-
кристаллики представляют собой твердый раствор меди в никеле, с
составом, более богатым никелем, чем исходный расплав.
Для нахождения состава образующихся кристаллов следует
провести из точки, соответствующей началу кристаллизации, пря-
прямую, паралельную оси абсцисс, до пересечения ее с кривой, огра-
ограничивающей снизу чечевицеобразнуюфигуру, изображенную на
диаграмме плавкости, и опустить из этой точки перпендикуляр на
ось абсцисс, как это показано стрелками на рисунке 86. По мере
кристаллизации незатвердевшая жидкость будет при этом обога-
обогащаться медью, а температура кристаллизации — понижаться.
Процесс кристаллизации окончится, когда температура кристал-
кристаллизации понизится до величины 1415° К,при которой состав выпа-
выпадающих кристалликов совпадает с составом исходного расплава.
8. АДСОРБЦИЯ
Твердые тела обладают способностью удерживать и уплотнять
на своей поверхности газообразные или растворенные вещества.
Явление уплотнения газообразного или растворенного вещества
на поверхности твердого тела называют адсорбцией, а веще-
вещество, на поверхности которого происходит адсорбция, — адсор-
адсорбентом.
Адсорбция вещества обусловлена тем, что силовое поле частиц
твердого тела, расположенных у поверхности, является ненасы-
ненасыщенным, поэтому молекулы газа или растворенного вещества при-
притягиваются к поверхности твердого тела. В простейшем случае
можно представить себе, что поверхность твердого тела несет элек-
электрические заряды. Эти заряды могут быть или одного знака, или
их знаки могут чередоваться. Электрические заряды поверхности
взаимодействуют с электрическими зарядами в молекулах газов
или растворенных веществ и притягивают их. Таким образом, на
поверхности твердого тела возникает уплотненный слой ориенти-
ориентированных молекул.
Особенно большое значение имеет адсорбция электрически за-
заряженных частиц — ионов — из растворов. Изучение процесса
адсорбции позволило приготовить адсорбенты, избирательно сорби-
сорбирующие ионы определенного знака.
В некоторых случаях при адсорбции наблюдается процесс ион-
ионного обмена, при котором из раствора сорбируются, например,
положительно заряженные ионы металлов, заменяясь поступаю-
поступающими из адсорбента ионами водорода. Такие адсорбенты называют
катионитами. Существуют также адсорбенты, избирательно погло-
поглощающие отрицательно заряженные ионы — анионы — различных
169
солей, обменивая их на гидроксильный ион ОН. Эти адсорбенты
называют а н и о п и т а м и. Заставляя воду, содержащую раство-
растворенные соли, проходить последовательно два сосуда, или, как их
называют, ионнообменные колонны, содержащие со-
соответственно катионит и анионит, можно осуществить замену в пер-
первой колонне иона металла на ион водорода, а во второй — аниона
кислотного остатка на гидроксильный анион. В результате присут-
присутствовавшая в воде соль будет поглощена и заменена соответствую-
соответствующим количеством воды. Этот способ позволяет получить воду, пол-
полностью свободную от примеси солей, не прибегая к перегонке,
и имеет поэтому большое практическое значение.
В общем случае молекулярное силовое поле у поверхности
твердого тела более сложно, чем описанное выше простое электро-
электростатическое поле.
Интенсивность силового поля у различных участков поверх-
поверхности неодинакова. Наиболее активные участки поверхности осо-
особенно энергично взаимодействуют с адсорбируемыми молекулами.
В этом случае иногда наблюдается химическое соединение адсор-
адсорбируемых молекул с атомами адсорбента, сопровождающееся выде-
выделением значительного количества теплоты.
Подобная адсорбция называется хемосорбцией.
Следует заметить, что при адсорбции вещества на активных
участках поверхности, даже в тех случаях, когда не возникает устой-
устойчивого химического соединения, химические свойства адсорбирован-
адсорбированной молекулы существенно изменяются. Этим объясняется то, что
в присутствии адсорбентов протекают некоторые химические реак-
реакции, не наблюдаемые в их отсутствии.
Поскольку процесс адсорбции протекает на поверхности твер-
твердого тела, то в качестве адсорбентов обычно используются тела,
отличающиеся большой пористостью — угли, гель1 кремниевой кис-
кислоты, различные специальные смолы. Пористость адсорбционного
угля повышается специальной обработкой, называемой актива-
активацией угля.
Истинная поверхность приготовленного таким образом активи-
активированного угля очень велика. У специальных сортов угля она дости-
достигает от 500 до 1000 квадратных метров на грамм угля.
В некоторых случаях в узких порах угля может происходить
даже конденсация адсорбируемых паров. Не приходится поэтому
удивляться большим количествам вещества, которые адсорбирует
уголь. Можно указать, например, что одна тонна специально при-
приготовленного угля поглощает до двухсот килограммов паров
бензина.
При повышении температуры количество адсорбированного
вещества уменьшается. Как говорят, при нагревании происходит
ско
170
1 Гель — тиердое или студнеобразное тело, представляющее собой
шгулировавший коллоидный раствор.
десорбция вещества. Поэтому в тех случаях, когда необхо-
необходимо удалить адсорбированные каким-либо телом газы, это тело
нагревают, одновременно откачивая десорбирующееся вещество.
Явлением адсорбции пользуются для достижения вакуума. Так,
например, при производстве осветительных ламп на нить накали-
накаливания наносят слой красного фосфора. Когда нить накаливается,
фосфор испаряется и, соединившись с присутствующими парами
воды и кислородом, осаждается на внутренней поверхности стек-
стеклянного баллончика. Возникший налет поглощает остаточные газы.
При производстве радиоламп для достижения высокого вакуума
обычно применяют бариевый газопоглотитель, наносимый на внут-
внутреннюю поверхность колбочки лампы. Этот газопоглотитель адсор-
адсорбирует азот, кислород, водород, углекислый газ и окись углерода.
В различных областях техники приходится иметь дело с сильно
летучими веществами. Для того чтобы предотвратить потери цен-
ценных продуктов, вызываемые их летучестью, используются ад-
адсорбционные уловители. Подобные аппараты используются также
для улавливания вредных летучих веществ, могущих загрязнить
воздух.
Когда в первую мировую войну были применены отравляющие
вещества: хлор, фосген и другие, для защиты от них было исполь-
использовано явление адсорбции. Русские ученые Н. Д. Зелинский
иН.А. Шилов создали угольный противогаз, в котором отравля-
отравляющие вещества поглощались слоем активированного угля. Сход-
Сходные противогазы применяются и в настоящее время.
Явление адсорбции используется в технике также с целью очист-
очистки растворов от различных загрязнений. Так, например, растворы,
из которых ведется кристаллизация сахара, бывают окрашены.
Для получения привычного для нас бесцветного сахара сахарный
сироп подвергают очистке, фильтруя его через слои активирован-
активированного угля.
Твердые тела могут не только адсорбировать газы, т. е. уплотнять
их на своей поверхности, но и растворять газы, подобно тому как
это имеет место у жидкостей. Растворение газов твердыми телами
называют окклюзией. Некоторые металлы способны окклю-
окклюдировать большое количество газов. Так, например, палладий раст-
растворяет такое количество водорода, которое при нормальном давле-
давлении занимает объем, приблизительно в тысячу раз превосходящий
объем палладия.
При повышении температуры способность твердого тела окклю-
окклюдировать газы возрастает.
IV. ФИЗИЧЕСКИЕ ОСНОВЫ ТЕРМОДИНАМИКИ
1. МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИЙ МЕТОД, МЕТОД
СТАТИСТИЧЕСКОЙ МЕХАНИКИ И ТЕРМОДИНАМИЧЕСКИЙ МЕТОД
При объяснении свойств вещества, при отыскании законов, ко-
которым подчиняются различные превращения его, можно пользо-
пользоваться одним из трех методов, применяемых в настоящее время в
физике. Эти методы построения физической теории называют:
молекулярно-кинетическим методом, ме-
методом статистической механики и термоди-
термодинамическим методом.
Молекулярно-кинетический метод, который использовался в
предыдущих главах, основывается на модельном представлении о
строении вещества. Принимается, что вещество состоит из атомов
и молекул, находящихся в непрерывном движении. Для объясне-
объяснения каких-либо свойств вещества детально анализируется поведе-
поведение отдельно взятой молекулы. При этом анализе предполагается,
что движение атомов и молекул подчиняется законам классической
механики. Выводы, полученные применительно к отдельной моле-
молекуле, переносятся в дальнейшем на совокупность большого количе-
количества молекул. Именно таким образом находилось выше молекуляр-
но-кинетическое выражение для давления газа, теплоемкости, коэф-
коэффициента внутреннего трения и т. п.
Преимуществом этого метода является ясная картина механизма
рассматриваемого явления, проникновение непосредственно в сущ-
сущность различных процессов природы. Эта особенность молеку-
лярно-кинетического метода обусловливает и его основной недо-
недостаток — приближенный характер полученных соотношений и их
чрезвычайное усложнение при попытке более строгого рассмотре-
рассмотрения вопроса.
Более общим является метод статистической механики. В ста-
статистической механике не рассматривается поведение отдельной мо-
молекулы. В этом случае исследователь имеет дело с большой совокуп-
совокупностью частиц, движение каждой из которых подчиняется законам
классической механики, но одновременно является событием слу-
172
чайным в том смысле, что скорости движения и соударения молекул
подчиняются законам теории вероятностей. Выводы статистической
механики неприменимы к совокупностям небольшого числа молекул.
В этом случае будут наблюдаться отклонения, возрастающие по
мере уменьшения числа молекул. В статистической механике,
хотя и в более общей форме, чем в молекулярно-кинетическом мето-
методе, анализируется механизм изучаемых явлений.
Термодинамический метод в отличие от первых двух полностью
исключает рассмотрение механизмов изучаемых процессов. В ка-
качестве основы термодинамических выводов используются наиболее
общие законы природы, которые называют началами.
Начала термодинамики не обосновываются логически. Они яв-
являются обобщением человеческого опыта и подтверждаются прак-
практической деятельностью человека. Используя методы математиче-
математического анализа, можно, опираясь на основные законы термодина-
термодинамики, получить очень большое количество соотношений между раз-
различными свойствами вещества. Например, соотношение между теп-
теплоемкостью вещества при постоянном давлении Ср и теплоемкостью
вещества при постоянном объеме Соили соотношение между теплоем-
теплоемкостью вещества и давлением и т. п. Эти соотношения будут столь
же точны, сколь точны основные законы термодинамики.
Однако термодинамическим выводам свойственны и существен-
существенные недостатки: термодинамический метод позволяет установить
только соотношения между различными свойствами вещества, но
не дает возможности количественно охарактеризовать эти свойства.
Так, например, точно устанавливая соотношение между теплоем-
костями Ср и Cv, термодинамический метод не дает возможности
определить величину теплоемкости Ср или Cv, Для вычисления
теплоемкости необходимо прибегнуть к молекулярно-кинетической
теории.
Кроме того, термодинамический метод не дает возможности най-
найти уравнение состояния, знание которого необходимо для большин-
большинства расчетов.
Правда, в практической деятельности этот недостаток можно
преодолеть, используя определенные экспериментально значения
одних свойств вещества, для того чтобы с помощью строгих термо-
термодинамических соотношений найти значения других свойств веще-
вещества. Этим объясняется широкое использование термодинамиче-
термодинамических расчетов в различных отраслях техники.
2. ПРЕДМЕТ ТЕРМОДИНАМИКИ. ОСНОВНЫЕ ТЕРМОДИНАМИЧЕСКИЕ
понятия.
Исторически термодинамика возникла как раздел физики, изу-
изучающий соотношение между теплотой и работой.
В наше время область термодинамики гораздо шире. По суще-
существу термодинамика изучает те же явления,которые рассматривают -
173
ся в молекулярно-кинетической теории: диффузию, кристаллиза-
кристаллизацию, поверхностные явления, плавление твердых тел,испарение
жидкостей и т. д.
Термодинамическое рассмотрение этих вопросов, однако, суще-
существенно отличается от молекулярно-кинетического. В термодина-
термодинамике все явления рассматриваются не с точки зрения их механизма,
а с точки зрения энергетических соотношений, имеющих место при
этих явлениях.
Таким образом, термодинамика рассматривает самые
разнообразные физические явления с единой точки зре-
зрения: с точки зрения тех превращений энергии, кото-
которыми эти явления сопровождаются.
Для того чтобы пояснить соотношение между кинетическим и
энергетическим подходами к изучению явления, рассмотрим реше-
решение одной из простейших задач механики. Предположим, что тя-
тяжелое тело массой т расположено на высоте h над уровнем моря.
Требуется определить, какую скорость приобретет тело на уровне
моря, падая свободно.
Эту задачу можно решить в рамках кинематики, для этого сле-
следует заметить, что искомая скорость v равна произведению ускоре-
ускорения свободно падающего тела g на время падения t, т. е.
За время падения t тело пройдет путь h, который согласно законам
кинематики найдем из уравнения:
Пользуясь последним соотношением, можно определить время
падения:
и, подставив наиденное значение в выражение для скорости падаю-
падающего тела на уровне моря, получим:
v = g j/1* = V2gh-
Ту же задачу можно решить энергетически. При падении тела
имеет место превращение потенциальной энергии поднятого тела
mgh в кинетическую энергию движущегося тела —. Приравнивая
кинетическую энергию, приобретенную телом, той потенциальной
энергии, которой оно обладало, найдем:
174
или
v = V2gh.
Таким образом, энергетическое рассмотрение вопроса приводит к
тому же самому результату, что и решение задачи с помощью зако-
законов кинематики.
В термодинамике используется специальная терминология, с
которой необходимо познакомиться.
Термодинамической системой или телом
называют вещество, занимающее определенный объем. Объем, заня-
занятый системой, может быть отделен от окружающего пространства
или материальной границей, в том числе жесткими или эластичными
стенками, или же термодинамическая система может быть выделена
мысленно.
Обычно рассматривается определенное состояние системы,
которое характеризуется физическими признаками, имеющими объек-
объективную меру, такими, например, как плотность, температура, дав-
давление.
Эти признаки называются параметрами состояния,
потому что совокупность значений их определяет состояние си-
системы.
Если состояние системы, предоставленной самой себе, т. е. при
отсутствии непрерывного воздействия извне, остается неизменным
неограниченно долго и если при незначительном воздействии из-
извне система, выйдя под влиянием этого воздействия из первоначаль-
первоначального состояния, после прекращения воздействия вновь возвращает-
возвращается в прежнее состояние, то говорят, что система находится в состоя-
состоянии устойчивого равновесия.
От равновесных состояний следует отличать стационар-
стационарные состояния, которые остаются неизменными только в ус-
условиях непрерывного воздействия на систему.
В качестве примера равновесного состояния можно указать на
состояние равновесия при диссоциации. Так, например, в парообраз-
парообразном состоянии йодистый водород частично диссоциирует на йод и
водород:
При данных температуре и давлении в результате диссоциации
определенного количества йодистого водорода в системе образуются
определенные количества йода и водорода.
Изменение температуры смещает равновесие: в одном случае
увеличивается количество продуктов диссоциации, в другом слу-
случае, наоборот, продукты диссоциации соединяются и увеличивается
количество недиссоциированного HJ.
При восстановлении первоначального значения температуры вос-
восстанавливается и первоначальное соотношение между количествами
продуктов диссоциации и недиссоциированного HJ.
175
1(р, К V
Примером стационарного состояния может служить состояние,
возникающее в сосуде с жидкостью, в которую погружена непре-
непрерывно работающая мешалка- При этом в жидкости возникает оп-
определенное распределение давлений и скоростей, сохраняющееся
неизменным до тех пор, пока остаются неизменными условия работы
мешалки, но нарушающееся как только прекратится работа мешалки,
т. е. исчезнет внешнее воздействие.
В дальнейшем все рассуждения, если нет специального указания,
будут относиться только к равновесным состоя-
состояниям.
Если изменяется один из параметров состояния системы, то
изменяется и ее состояние.
Параметров состояния может быть много, однако чаще всего
в качестве их выбирают плотность р, давление р и температуру Т
или же молекулярный объем V, дав-
давление р и температуру Т.
В этом случае для графического
изображения состояния системы не-
необходима пространственная диаграм-
диаграмма. Каждая точка подобной диаграм-
диаграммы соответствует определенному со-
состоянию системы. Так, например,
точка с координатами ри Vu Tt
(рис. 87) соответствует первому со-
состоянию системы, а точка с коорди-
координатами р2, V2, T2 — второму состоя-
состоянию системы.
Очевидно, что линия, соединяю-
соединяющая эти две точки, будет соот-
соответствовать определенному процессу перехода системы из состо-
состояния / в состояние 2. В зависимости от характера этой линии си-
система при переходе из состояния / в состояние 2 будет проходить
через различные промежуточные состояния, т. е. процессы перехода
могут быть различными.
Если в системе протекает процесс, т. е. изменяется ее состояние,
то естественно, что в системе осуществляется целый ряд неравновес-
неравновесных состояний.
Можно, однако, представить себе такой процесс, в котором каждое
последующее состояние бесконечно мало отличается от предыдущего,
так что для любого произвольно выбранного малого промежутка
времени состояние системы можно считать равновесным. Процесс
в этом случае будет представлять собой как бы последовательность
равновесных состояний. Такой процесс называют равновесным
процессом.
Очевидно, что все равновесные процессы протекают бесконечно
медленно. На диаграммах состояния равновесные процессы изобра-
изображаются непрерывными кривыми.
Рис. 87. Графическое изобра-
изображение состояния системы.
176
Все реальные процессы, строго говоря, неравновесные процес-
процессы. К равновесным процессам приближаются процессы, протекаю-
протекающие очень медленно.
В случае равновесных процессов термодинамические законо-
закономерности значительно проще, чем в случае неравновесных процес-
процессов. Все выводы в дальнейшем, если только нет специального указа-
указания, будут относиться именно к равновесным
процессам.
Отличительной особенностью равновесных процессов является
их обратимость: изменяя соответствующим образом параметры
состояния, можно заставить равновесный процесс протекать в об-
обратном направлении так, что система возвратится в исходное поло-
положение, причем в окружающих телах не произойдет никаких изме-
изменений.
Всякий неравновесный процесс необратим. Это не означает,
что систему, состояние которой изменилось в результате необра-
необратимого процесса, нельзя возвратить в исходное состояние. Воз-
Возвратить систему в исходное состояние возможно и в случае не-
необратимого процесса, однако при этом процесс, обратный прямому,
будет вызывать не только возвращение системы в исходное состоя-
состояние, но и определенные изменения в окружающих телах.
Типичным примером необратимого процесса является расшире-
расширение газа в вакуум. Предположим, имеется цилиндр, разделенный
пополам перегородкой, с одной стороны которой находится газ,
а с другой — вакуум. Если убрать перегородку, газ расширится и
заполнит весь цилиндр. Этот процесс будет необратимым, хотя,
вдвигая поршень, можно вновь собрать газ в одной половине ци-
цилиндра, в то время как в другой будет вакуум. Дело в том, что не-
необходимое для этой цели движение поршня потребует изменение
состояния окружающих тел, которое сохранится при возвращении
системы в исходное состояние.
Трудность построения пространственных диаграмм, подобных
изображенной на рисунке 87, приводит к тому, что часто пользуются
плоскими кривыми, возникающими при пересечении пространствен-
пространственных диаграмм соответствующими плоскостями.
Так, например, проводя сечение, параллельное плоскости pV
(рис. 87), получим диаграмму состояний системы, характеризую-
характеризующихся одной и той же температурой. Кривые, проведенные на этой
диаграмме, будут соответствовать различным изотермиче-
изотермическим процессам.
Однако pV диаграммы вовсе не обязательно подразумевают изо-
изотермические изменения состояния. На подобных диаграммах можно
изобразить зависимость между давлением и молекулярным объемом
при любых процессах.
В кинетической теории газов уже говорилось, что для реальных
физических систем параметры состояния не являются независимыми
переменными, их связывает соотношение, называемое у р а в н е-
177
нием состояния,
в следующей форме:
которое для общности можно записать
F{p, V, 7) = 0.
Термодинамика не позволяет определить уравнение состояния в
явном виде. Для этого приходится прибегать к молекулярно-ки-
нетической теории. Простейшим уравнением состояния является
уравнение состояния идеальных газов:
pV = RT.
В термодинамических рассуждениях важную роль играют вели-
величины, однозначно определяемые параметрами состояния. Эти вели-
величины называются функция-
функциями состояния. Важнейшей
функцией состояния является
внутренняя энергия
системы U.
Внутренней энергией си-
системы называют общий
запас энергии, которым
обладает термодинамиче-
термодинамическая система.
Предположим, термодинами-
термодинамическая система находится в со-
состоянии /, характеризуемом па-
Рис. 88. Работа, совершенная газом раметрами состояния рь Кь TV
при изменении его состояния. Внутренняя энергия может
иметь в этом случае только
одно-единственное значение Ut.
Переведем рассматриваемую систему из состояния / в состояние 2,
характеризуемое параметрами состояния р2, V2, T2. В этом состоянии
система будет обладать иным, но опять же единственно возможным
значением внутренней энергии ?/2. Естественно, что разница во внут-
внутренних энергиях при переходе системы из первого состояния во вто-
второе будет иметь одно и то же значение
вне зависимости от того, каким путем совершается переход из одно-
одного состояния в другое.
Это справедливо для всех функций состояния, т. е. для лю-
любой функции состояния изменение ее при переходе сис-
системы из одного состояния в другое не зависит от пу-
пути перехода.
Не все величины, встречающиеся в термодинамике, являются
функциями состояния.
Рассмотрим, например, играющую очень большую роль в термо-
динамике физическую величину, называемую работ ой. В качестве
рассматриваемой системы выберем один киломольгаза, занимающие
178
при давлении pt и температуре Tj объем Vt. На диаграмме pV (рис.88)
состояние системы изображается точкой 1. Предоставим газу расши-
расшириться до объема V2, причем давление и температура при этом примут
значение р2 и Т2. Новому состоянию системы на диаграмме соответ-
соответствует точка 2. Процесс расширения изображается кривой аЬс.
Подсчитаем работу при элементарном изменении объема dV.
Давление в этом случае можно считать постоянным, равным р,
так что для искомой величины работы dA будет справедливо выра-
выражение:
dA = pdV. A)
Работа расширения при переходе системы из состояния / в состоя-
состояние 2 найдется интегрированием написанного выражения в пределах
от Vi до V2:
B)
Геометрически работа А{ ^2 изображается площадью abode, заштри-
заштрихованной на графике сплошными линиями.
Если бы условия расширения газа были бы иными, был бы иным
и характер кривой abc; так, например, можно осуществить расши-
расширение газа таким образом, что процесс изменения состояния будет
изображаться на графике кривой ahc. Работе расширения в этом
случае соответствует площадь ahcde, заштрихованная пунктирной
штриховкой. Из рассмотрения чертежа становится понятным,
что работа, совершаемая при переходе системы из одного состоя-
состояния в другое, зависит от пути перехода и потому не является
функцией состояния.
Поскольку работа не является функцией состояния, не имеет
смысла говорить о количестве работы в какой-либо системе. Дей-
Действительно, если выбрать некоторое состояние системы за стан-
стандартное, приняв, что в этом состоянии количество работы в систе-
системе равно нулю, то количество работы в системе в любом другом со-
состоянии должно быть равно работе, которую надо совершить при пе-
переводе системы из стандартного состояния в рассматриваемое.
В зависимости от пути перехода эта работа, как пояснено выше,
может иметь самые разнообразные значения, и потому представле-
представление о количестве работы в системе не имеет физического смысла.
Особенности физической величины, называемой работой, приводят
к тому, что выражение для дифференциально малой работы dA от-
отличается по своему смыслу от того, который вкладывается в поня-
понятие дифференциала какой-либо функции. Действительно, в математи-
математическом анализе символом dy обозначают бесконечно малое изменение
некоторой функции у, в случае же работы величина dA означает
просто бесконечно малую работу, поскольку самой величине А нель-
нельзя приписать однозначного значения. Эту особенность мы будем в
дальнейшем отмечать штрихом при знаке дифференцирования d'A.
179
3. ФИЗИЧЕСКИЙ СМЫСЛ ПОНЯТИЯ «ТЕПЛОТА»
Второй физической величиной, играющей в термодинамике ис-
исключительно большую роль и не являющейся в то же время функ-
функцией состояния, является теплота.
Поьятие о теплоте возникло очень давно, первоначально на ос-
основании чисто физиологических ощущений человека. Именно на
основании физиологических ощущений человек делил все тела на
горячие и холодные, определяя степень нагретости тела по непо-
непосредственному ощущению.
Оценка температуры тела с помощью наших органов чувств
не только не точна, но может приводить к качественно неверным
заключениям при сравнении температур тел.
Поэтому были разработаны различные способы объективного
определения температуры. Большинство методов измерения темпе-
температуры основывается на представлении о выравнивании темпера-
температур тел, приведенных в соприкосновение в условиях изоляции от
внешних воздействий. При этом подразумевается, что с телами не
происходит никаких химических изменений и что они находятся
в соприкосновении достаточно долгое время.
Для объяснения тепловых явлений в XVIII веке было выдвинуто
представление об особого рода невесомой «тепловой мате-
р и и», или «теплород е». Согласно этой теории считалось,
что чем больше в теле теплорода, тем выше его температура.
Для этой теории совершенно естественным было представление
о количестве теплоты AQ как о количестве тепловой материи, содер-
содержащейся в том или ином теле. Процесс выравнивания температуры
объяснялся как процесс перехода некоторого количества «теплоро-
«теплорода» от одного тела к другому. Температуре в этом случае соответ-
соответствовал как бы уровень теплорода в данном теле.
Таким образом, одно и то же количество тепловой материи со-
сообщало различным телам, обладающим разной «емкостью» по от-
отношению к теплороду, различную температуру. Так возникло пред-
представление о теплоемкости как о величине, численно равной коли-
количеству теплоты, необходимому для повышения температуры тела на
один градус. Теория теплорода сыграла в свое время положитель-
положительную роль в развитии физики, однако она была ошибочна. В том же
XVIII веке некоторыми физиками, и особенно последовательно
М. В. Ломоносовым, высказывалась мысль о том, что тепло-
теплота представляет собой особого рода движение материи. «Очень
хорошо известно, — писал в 1745 году Ломоносов, — что теплота
возбуждается движением: от взаимного трения руки согреваются,
дерево загорается пламенем, при ударе кремня об огниво появ-
появляются искры, железо накаливается докрасна от проковывания
частыми и сильными ударами, а если их прекратить, то теплота
уменьшается и произведенный огонь тухнет». На основании цело-
целого ряда остроумных соображений Ломоносов приходит к заклю-
180
V
чению, что «теплота состоит во внутреннем движении
материи». В 1798 году Румфорд, наблюдая сверление пушеч-
пушечных стволов, высказал мысль о том, что выделяющаяся при этом
теплота возникает в результате совершения работы, с которой свя-
связано сверление металла. Немногим позднее Дэви A799) доказал
экспериментально, что теплота может возникать в результате
совершения работы (лед плавится при трении одного его куска о
другой).
В конце первой половины XIX века Джоуль определяет ме-
механический эквивалент теплоты: для получения одной кило-
килокалории необходимо за-
затратить 427 килограммо-
килограммометров работы.
Эти успехи механической
теории теплоты привели к то-
тому, что в середине XIX века
теория теплорода была оконча-
окончательно отвергнута.
Однако терминология, свой-
свойственная теории теплорода,
сохранилась в науке до наших
дней. До сих пор мы говорим:
поток тепла, количество тепло-
теплоты, теплоемкость и т. п. Эти
термины корнями своими ухо-
уходят в теорию теплорода.
В действительности поня-
понятие о количестве теплоты в
теле не имеет физического
смысла, точно так же как не имеет физического смысла понятие
о количестве работы в теле.
Рассмотрим один киломоль газа, занимающий при температу-
температуре Ti и давлении pt объем Vt.
На диаграмме р, V (рис. 89) состояние системы изобразится
точкой /. Сообщим системе количество теплоты AQ, необходимое
для того, чтобы ее температура повысилась на заданную величину
AT и окончательная температура сделалась бы равной Т4+ AT.
В зависимости от давления один киломоль газа может занимать при
температуре 7\+AT различные объемы. На диаграмме с координа-
координатами pV точки, соответствующие возможным конечным состояниям
рассматриваемой системы, образуют изотерму ab.
Вертикально проведенная прямая /—2 соответствует нагреванию
системы при постоянном объеме, для которого требуется количе-
количество теплоты AQ,^ 2 = С^ЛТ, где Cv— молекулярная теплоем-
теплоемкость газа при постоянном объеме.
Горизонтальная прямая /—3 соответствует нагреванию газа
при постоянном давлении, для которого необходимо иное коли-
181
Рис. 89. Характер процесса нагре-
нагревания определяет количество тепло-
теплоты, необходимое для повышения
температуры системы на определен-
определенное число градусов.
чество теплоты AQ(^3 = СрАТ, где Ср — молекулярная теплоем-
теплоемкость газа при постоянном давлении.
Как было показано выше, теплоемкость газа при постоянном
давлении отличается от теплоемкости газа при постоянном объеме.
Даже если ограничиться этими случаями как предельными, то,
поскольку между точками 2 и 3 на изотерме ab расположено беско-
бесконечное число промежуточных точек, очевидно, что веществу при-
присуще бесконечно большое количество теплоемкостей,
величины которых зависят от характера нагрева.
Поскольку теплоемкость вещества изменяется при изменении
условий нагревания, переход системы из какого-либо состояния,
характеризуемого значениями параметров состояния рь V\, Tit в
иное состояние, характеризуемое значениями параметров р2, V2,
Т2, требует в зависимости от пути перехода разного количества
теплоты. Так, например, можно нагреть вещество до температуры
Т2 при неизменном объеме, а затем, поддерживая температуру по-
постоянной и изменяя объем, довести его до величины У2 при давле-
давлении р2. Тот же переход можно осуществить, нагрев первоначально
вещество при неизменном давлении до температуры Г2, а затем, под-
поддерживая опять же температуру постоянной и изменяя давление,
довести его до величины р2, соответствующей объему V2- Количе-
Количества теплоты, необходимые для нагревания вещества в первом и вто-
втором случаях, будут различны.
Если принять, как это было сделано при обсуждении особен-
особенностей физической величины, называемой работой, некоторое со-
состояние системы за стандартна и условиться, что в этом состоянии
количество теплоты в системе равно нулю, то количество теплоты
в данной системе в каком-либо другом состоянии будет равно тому
количеству теплоты, которое необходимо сообщить ей для перехода
из стандартного в рассматриваемое состояние. Это количество тепло-
теплоты в зависимости от пути перехода может иметь самые различные зна-
значения. Поэтому, так же как и в случае работы, не имеет смысла го-
говорить о количестве теплоты, которой обладает система.
Точно так же дифференциально малое количество теплоты
означает не бесконечно малое изменение величины Q, присущей
системе, а просто бесконечно малое количество теплоты dQ, сооб-
сообщаемое системе или взятое от нее. Для того чтобы подчеркнуть это
в случае дифференциально малого количества теплоты, у знака диф-
дифференцирования ставят знак штрих, т. е. пишут d'Q.
4. ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
В основе термодинамики лежат два основных закона, или на-
начала, термодинамики. Эти законы обобщают многовековой опыт
человека, они не следуют из других законов, открытых челове-
человеком, но сами служат для формулировки различных закономерностей.
182
Первое начало термодинамики — это закон сохране-
сохранения и превращения энергии: при разнообразных процес-
процессах, протекающих в природе, энергия не возникает из
ничего и не уничтожается, но превращается лишь из
одних видов в другие.
Исторически в формулировке первого начала термодинамики
важную роль сыграли неудачные попытки человека построить
машину, которая производила бы работу, не потребляя эквивалент-
эквивалентного количества энергии. Машина, производящая работу без по-
потребления эквивалентного количества энергии, называется веч-
вечным двигателем первого рода, или — по-латыни—
перпетуум мобиле первого рода. Поэтому первое
начало термодинамики часто формулируют в виде указания о невоз-
невозможности построения подобной машины, а именно записывают его
в виде следующего утверждения: невозможно построить
перпетуум мобиле первого рода.
В 1748 г. в письме к знаменитому математику Л. Эйлеру
М. В. Ломоносов писал: «Все перемены, в натуре случающиеся,
такого суть состояния, что сколько чего у одного тела отнимет-
отнимется, столько присовокупится к другому. Так, ежели где убудет
несколько материи, то умножится в другом месте, сколько часов
положит кто на бдение, столько же сну отнимет. Сей все-
всеобщий естественный закон простирается и в самые
правила движения: ибо тело, движущее своей силой
другое, столько же оныя у себя теряет, сколько
сообщает другому, которое от него движение полу-
получает».
Этот закон Ломоносов применял для объяснения физических
явлений. Так, например, желая объяснить охлаждение воды, наблю-
наблюдаемое при растворении некоторых солей, он писал: «Когда какое-
либо тело ускоряет движение другого, то сообщает ему часть дви-
движения, но сообщить часть движения оно не может иначе, как теряя
точно такую же часть. Потому частицы воды, ускоряя вращатель-
вращательное движение частиц соли (при растворении.—Б. К-), теряют часть
своего вращательного движения. А так как последнее — причина
теплоты, то нисколько не удивительно, что вода охлаждается при
растворении соли».
Эти мысли Ломоносова показывают, что он был первым, для
кого был ясен один из основных законов природы и кто сформули-
сформулировал этот закон, конечно, с теми ограничениями, которые неизбеж-
неизбежно накладывает на формулировку различных закономерностей
историческое развитие науки.
Через сто лет, в 1842 г., Р. М а й е р A814—1878) обобщил на-
накопленные наукой данные о взаимной превращаемости теплоты и
работы в принципе, которому он придал следующую формулиров-
формулировку: «При всех химических и физических про-
процессах заданная сила остается постоянной
183
величиной»1. Работы Майера, однако, не убедили его совре-
современников.
Утверждение в науке закона сохранения энергии связано с
работами Г. Гельмгольца A821—1894). Гельмгольц не толь-
только сформулировал A847) закон сохранения энергии в математи-
математической форме, но показал те большие возможности, которые откры-
открывает применение этого важнейшего принципа естествознания в
науке.
Следует отметить, что всеобъемлющее значение первого начала
термодинамики было впервые раскрыто Энгельсом, великим
материалистом и диалектиком. Диалектический материализм утвер-
утверждает, что движение2 является формой существования материи.
«Любая форма движения, — писал Энгельс, — оказалась способ-
способной и вынужденной превращаться в любую форму движения. Дойдя
до этой формы, закон (сохранения и превращения энергии. — Б. К.)
достиг своего последнего выражения. Посредством новых открытий
мы можем доставить ему новые подтверждения, дать ему новое бо-
более богатое содержание. Но к самому закону, как он здесь выражен,
мы не можем прибавить более ничего» (Ф.Энгельс, Диалектика
природы).
Для того чтобы записать первое начало термодинамики в мате-
математической форме, рассмотрим, как собственно происходит изме-
изменение внутренней энергии системы. Для наглядности предположим,
что рассматриваемая система — идеальный газ.
Внутренняя энергия газа будет повышаться при его нагревании,
т. е. когда мы сообщаем газу какое-то количество теплоты. Таким
образом, подвод к системе или отвод от нее теплоты
является одним из способов изменения внутренней
энергии системы.
Вторым способом изменения внутренней энергии
системы является совершение системой некоторой
работы, или же совершение работы над системой.
Действительно, если совершить работу, быстро сжав газ, он
нагреется, т. е. его внутренняя энергия возрастет. Наоборот, ее-,
ли предоставить газу расширяться, не подводя к нему теплоты,
газ будет охлаждаться, т. е. его внутренняя энергия будет убы-
убывать.
Передача теплоты и совершение работы — это два
процесса в результате которых и только благодаря
которым изменяется внутренняя энергия системы.
Учитывая сказанное об изменении внутренней энергии системы,
можно записать закон сохранения энергии, приравняв изменение
внутренней энергии системы dU разности между количеством теп-
1 Во времена Р. Майера термина «энергия» еще не существовало. В сво-
своих работах вместо термина «энергия» он употребляет термин «сила».
а Под движением понимается всякое изменение.
184
лоты, сообщенной системе d'Q, и работой d'A, совершенной си-
системой:
dU = d'Q—d'A. C)
При этой записи считаются положительными все три величины:
1) возрастание внутренней энергии системы; 2) теплота, сообщен-
сообщенная системе; 3) работа, совершенная системой.
Записывая первое начало термодинамики в дифференциальной
форме, следует подчеркнуть, что слева в уравнении имеем полный
дифференциал внутренней энергии. Каждая из величин, стоящих
справа от знака равенства, d'Q и d'A, взятая в отдельности, не яв-
является полным дифференциалом, а величины Q и А не являются
функциями состояния.
Поэтому при принятом определении энергии тепло-
теплоту нельзя считать энергией.
5. МЕТОДЫ НАХОЖДЕНИЯ ТЕРМОДИНАМИЧЕСКИХ
СООТНОШЕНИЙ
Термодинамические выводы, как это было сказано выше, делают-
делаются не в результате обсуждения механизма рассматриваемого явле-
явления или каких-либо модельных представлений, а в результате при-
применения основных законов термодинамики и правил математиче-
математического анализа. Для того чтобы уяснить способы нахождения тер-
термодинамических соотношений, рассмотрим некоторые конкретные
примеры.
Переменными в термодинамике являются термодинамические
параметры состояния, т. е. обычно — давление, объем, температура
(р, V, Т), и функции состояния системы. Параметры состояния си-
системы связаны между собой уравнением состояния.
Взятая изолированно термодинамика не дает возможности на-
написать уравнение состояния в такой форме, которая позволяла
бы находить для реальных веществ значение одного из параметров
состояния на основании значений двух других. Поэтому уравнение
состояния следует записать в общей форме, как указание на связь,
существующую между параметрами состояния:
F(p> V, Г) = 0.
Таким образом, каждый из параметров состояния оказывается
функцией двух других:
p = /1(V, Г); V = /,(/,, Т); Т = /3 (р, V).
Этим функциональным зависимостям соответствует большое коли-
количество частных производных, имеющих определенный физический
смысл. Так, например, частная производная от объема по темпе-
температуре при постоянном давлении входит как составная часть в вы-
185
ражение термического коэффициента объемного расширения а
вещества:
1 ldV\
а = — •— 1 .
V \ дТ I _
В свою очередь частная производная от объема по давлению при
постоянной температуре играет такую же роль в выражении для
коэффициента изотермической сжимаемости pV вещества:
ldV\
Как доказывается в математическом анализе, если некоторая вели-
величина z является функцией двух переменных х и у, т. е.
z = f(x, у),
и между различными частными производными существуют опре-
определенные соотношения. Так, например, доказывается, что
дх}у jdx\
дг!у
—1-
Эти и подобные им соотношения широко используются в тер-
термодинамике. Для того чтобы показать, как это делается, найдем
выражения для коэффициента термического расширения идеального
газа и газа, подчиняющегося уравнению Ван-дер-Ваальса.
Согласно определению термический коэффициент объемного
расширения равен:
В случае идеального газа:
дТ)р
Следовательно,
У« Р ЛГо То
1 Индекс Т у коэффициента рг показывает, что величина Эг^ыла опреде-
определена при постоянной температуре.
186
Для газа, подчиняющегося уравнению Ван-дер-Ваальса, можно,
вместо того чтобы находить производную от объема по темпера-
температуре, воспользоваться для определения а соотношением1, выте-
вытекающим из уравнений D) и E), а именно:
dV\ _ \дГ'у
\dVJT
Давление реального газа согласно теории Ван-дер-Ваальса вы-
выражается соотношением:
= RT a
Р ~~ V — b l/з '
воспользовавшись которым, можно написать:
(др_\ _ R
\dTlv V—b'
1др_\ _^ RT , 2а
[dVlr (V — ftJ ~^~ V3'
Подставляя найденные значения производных в уравнение F),
получим для коэффициента термического расширения газа, подчи-
подчиняющегося уравнению Ван-дер-Ваальса, выражение:
а
V ' дТ )р RTV3 - 2a(V - bJ '
Приведенные примеры подчеркивают несколько формальный
характер нахождения термодинамических соотношений.
1 Для получения уравнения F) приравняем х = р, у = V'. z = Т. Со-
Соотношение D) примет при этом вид:
1
\dpjv (др\
\dTJv
а соотношение E) соответственно:
( ()
KdVJT\dTjp\dp]v
Из последнего соотношения находим:
1др_\ /ЭГ
\dvjr \dp
\
dpjv
(дТ\
Подставляя в уравнение (Ь) значение I — из уравнения (а), получим
уравнение F).
187
Кроме того, эти примеры показывают, что изолированно взятая
термодинамика позволяет получать лишь соотношения между раз-
различными частными производными. Окончательные выражения, свя-
связывающие искомую величину с параметрами состояния, находятся
в результате использования того или иного уравнения состояния
и существенно зависят от вида последнего. Эти окончательные
соотношения уже не обладают строгостью термодинамических урав-
уравнений, поскольку они в скрытой форме содержат приближенно
верные предположения, сделанные при нахождении уравнения
состояния. Весьма часто при этом применение даже сравнительно
простого уравнения состояния, каким является уравнение Ван-дер-
Ваальса, приводит в конечном счете к сложным выражениям.
Следует еще раз подчеркнуть, что для практических целей
указанные ограничения не столь существенны, поскольку всегда
возможно экспериментально определить значения одних частных
производных и, пользуясь ими, рассчитать термодинамически строго
значения других производных.
В качестве переменных в термодинамике используются не только
три параметра состояния, но, как указано выше, и ряд функций
состояния, поэтому число частных производных и соответственно
соотношений между ними очень велико. Термодинамика — раздел
физики, наиболее богатый математическими соотношениями. Учи-
Учитывая это, термодинамические соотношения для удобства исполь-
использования сведены в специальные таблицы.
6. АДИАБАТНЫЕ ПРОЦЕССЫ. УРАВНЕНИЕ ПУАССОНА
В целом ряде физических явлений приходится сталкиваться
с процессами, протекающими в термодинамической системе практи-
практически почти без теплообмена с окружающей средой.
Процессы, протекающие без теплообмена, называются адиа-
адиабатными.
Для того чтобы какой-либо процесс протекал адиабатно, система
должна быть отделена от окружающей среды теплонепроницае-
теплонепроницаемыми стенками. Поскольку подобных стенок в природе не встре-
встречается, не встречается в природе и полностью адиабатных процес-
процессов. Чем лучше теплоизоляция системы, тем ближе процесс к ади-
адиабатному. Так как процесс теплообмена требует некоторого времени,
то к адиабатным процессам приближаются все процессы, протекаю-
протекающие достаточно быстро.
При адиабатном сжатии или расширении газа зависимость меж-
между давлением и объемом отличается от обратной пропорционально-
пропорциональности, следующей из закона Бойля — Мариотта, справедливого в
случае изотермических изменений состояния газа. Для отыскания
этой зависимости воспользуемся первым началом термодинамики:
188
В случае адиабатных процессов теплообмен отсутствует и, следо-
следовательно,
dU=*— dA,
вательно,
так что
или
т. е. работа, совершаемая системой при адиабатных
изменениях состояния, сопровождается эквивалент-
ным изменением внутренней энергии системы. Поскольку
в этом случае работа однозначно связана с состоянием системы, выра-
выражение dA является полным дифференциалом. Элементарная работа
при любом расширении или сжатии газа выражается уравнением
dA = pdV.
Если система представляет собой идеальный газ, то изменение
внутренней энергии dU связано с изменением температуры си-
системы dT простой зависимостью:
аи = Cv • dl.
Подставляя эти значения dA и dU в выражение первого начала,
получим:
pdV=—CV'dT.
Для того чтобы уменьшить число переменных, исключим из напи-
написанного выражения давление газа, воспользовавшись для этого
уравнением состояния идеальных газов, согласно которому
RT
в результате найдем:
Разделим переменные и перенесем Cv в левую часть уравнения:
R_ dV = _dT
Cv ' V ~ T •
Интегрируя в пределах от начальных значений объема и темпера-
температуры Vx и 7\ до конечных — V2 и Т2, получим:
-1п-2 = 1п-\
или, подведя постоянный сомножитель под знак логарифма,
189
Если равны логарифмы каких-либо величин, то равны и сами вели-
величины, и поэтому в написанном соотношении знаки логарифмов мож-
можно отбросить:
V*icv _
или
Как было показано в кинетической теории газов (см. стр. 69)
и, следовательно,
1 = ^-1
Отношение теплоемкости вещества при постоянном давлении
Q
к его теплоемкости при постоянном объеме —? обозначают обычно
п
греческой буквой у, следовательно, — = y — 1. Используя это
обозначение, можно записать полученное соотношение в форме:
Г^!1 = T2Vl~l = Const. (8)
Таким образом, при адиабатных изменениях состояния
для данной массы газа произведение температуры на
объем в степени (к — 1) остается постоянной величиной.
При адиабатном расширении идеального газа его температура
понижается и, наоборот, при адиабатном сжатии — повышается.
Если в уравнении (8) заменить температуру ее выражением через
объем и давление, воспользовавшись для этого уравнением идеаль-
идеальных газов
Т _pV
R '
то получится соотношение между давлением и объемом, имеющее
место при адиабатном изменении состояния:
R
Перенеся универсальную газовую постоянную R из левой части
уравнения в правую и замечая, что произведение R • Const остает-
остается постоянной величиной, можно написать:
p)/i =Const. (9)
190
Аналогично, подставляя в уравнение (8) значение объема,
выраженное через соответствующие значения давления и темпера-
температуры,
р
найдем третью форму уравнения, справедливого для адиабатных
изменений состояния идеального газа:
Р1
или
=Const,
=Const.
Р Т
A0)
\ Изотерма
\ Адиабата
Уравнения, описывающие адиабатные изменения состояния, на-
называют уравнениями Пуассона.
Сопоставим изотермические и адиабатные изменения состояния
идеального газа.
Предположим, что исходное состояние газа, характеризуемое
значениями параметров ри Vit Ть изображается на диаграмме с
координатами р, V точкой 1.
При изотермическом сжатии или
расширении газа точка, изоб-
изображающая состояние системы
(рис. 90), будет двигаться вдоль
соответствующей изотермы, пред-
представляющей с геометрической
точки зрения равнобочную ги-
гиперболу pV = Const.
При адиабатном сжатии и
расширении, как это следует из
уравнения (9), давление изменя-
изменяется быстрее, чем при изотерми-
изотермическом, так что соответствую-
соответствующие точки, изображающие со-
состояние системы, образуют кри-
кривую, расположенную более круто, чем изотерма. Эта кривая
называется адиабатой.
С молекулярной точки зрения большая крутизна адиабаты
по сравнению с изотермой вполне понятна. При изотермическом
сжатии давление возрастает (при расширении — уменьшается)
только в результате изменения молекулярной плотности и соот-
соответственно числа ударов молекул о стенки сосуда. Среднее измене-
изменение количества движения при ударе молекулы о стенку при этом
остается постоянным. В случае же адиабатных изменений объема
сжатие сопровождается повышением температуры, а расширение —
понижением. Поэтому при сжатии число соударений возрастает не
Рис. 90. Изотермическое и адиа-
адиабатное изменение состояния газа.
191
только в результате увеличения молекулярной плотности, но и в
результате увеличения средней скорости молекулярного движения.
Кроме того, при этом возрастает среднее изменение количества дви-
движения, происходящее при соударениях молекулы со стенкой. Все
это, вместе взятое, приводит к тому, что при адиабатном сжатии на-
наблюдается большее возрастание давления, чем при эквивалентном
изотермическом.
При адиабатном расширении в результате понижения темпера-
температуры средняя скорость движения молекул уменьшается. Это вызы-
вызывает большее по сравнению с наблюдаемым при изотермическом рас-
I Адиабата
\ ^ Изотермы
'Изотерма
Рис. 91. Через любую точку ади-
адиабаты можно провести изотерму.
Рис. 92. Через любую точку изо-
изотермы проходит самостоятельная
адиабата.
ширении уменьшение числа ударов молекул за одну секунду о стен-
стенку сосуда, сопровождающееся уменьшением среднего изменения
количества движения при отдельном ударе. Естественно, что при ади-
адиабатном расширении газа его давление уменьшается больше, чем
при эквивалентном изотермическом расширении.
Следует подчеркнуть что каждая точка адиабаты имеет темпе-
температуру, отличную от других, и потому через каждую точку адиаба-
адиабаты можно провести соответствующую изотерму (рис. 91).
Аналогично, через каждую точку изотермы можно провести ади-
адиабату (рис. 92).
В системе координат р, V адиабаты всегда распола-
располагаются круче, чем изотермы.
7. ПРИМЕНЕНИЕ ПЕРВОГО НАЧАЛА ТЕРМОДИНАМИКИ
К НЕКОТОРЫМ ПРОЦЕССАМ В ГАЗАХ
Рассмотрим некоторые примеры применения первого начала тер-
термодинамики к процессам изменения состояния идеального газа.
Одним из простейших процессов изменения состояния газа
является процесс, протекающий при неизменном объеме. Подобные
192
процессы называют изохорными. Поскольку при изохорном
процессе объем V остается постоянным,™ dV — 0, и, следовательно,
работа dA = pdV равна нулю. Первое начало термодинамики за-
запишется в этом случае в виде соотношения:
dU = dQ.
Количество теплоты, которое необходимо сообщить системе для
того, чтобы при постоянном объеме повысить ее температуру на
величину dT, можно выразить, если известна теплоемкость веще-
вещества при постоянном объеме Cv, уравнением:
dQ = CvdT,
и, следовательно, '
dU = CvdT,
или1
Теплоемкость Cv идеального одноатомного газа не зависит от темпе-
температуры, и потому для внутренней энергии идеального одноатомного
газа будет справедливо выражение:
совпадающее с полученным ранее в молекулярно-кинетической
теории.
Таким образом, при изохорном изменении состояния газа
вся подведенная к системе теплота идет на увеличение
внутренней энергии системы.
Иное положение наблюдается при изотермических
изменениях состояния. В этом случае внутренняя энергия идеаль-
идеального газа остается неизменной:
U = Const
Уравнение первого начала при изотермическом изменении со-
состояния газа запишется в следующей форме:
d'Q — d'A = 0,
или
d'Q = d'A.
1 У идеального газа внутренняя энергия зависит только от температуры,
поэтому в данном случае можно воспользоваться обычными значками диффе-
дифференциалов, не указывая на то, что процесс протекает при постоянном объеме.
7 Заказ 145 J93
Из этого выражения следует, что при изотермическом
процессе все подведенное к системе количество тепло-
теплоты превращается в работу.
Для того чтобы подсчитать работу, совершаемую газом, рас-
расширяющимся изотермически от объема F4 до объема V2, необхо-
необходимо проинтегрировать в этих пределах выражение для элемен-
элементарной работы, учтя при этом связь, существующую между объемом
идеального газа и давлением:
RT
Графически вычисленная работа выражается на диаграмме с коор-
координатами р, V (рис. 93) площадью abed, заштрихованной на гра-
графике.
Из полученного уравнения следует, что работа, совершаемая
при изотермическом расширении, зависит только от соотношения
начального и конечного объемов газа, но не от их абсолютных
величин. Следует иметь в виду, что написанное уравнение позео-
ляет рассчитать работу изотермического расширения одного моля
идеального газа.
Работа, совершаемая при расширении п молей газа, будет в п
раз больше, т. е.
-2.
Вместо отношения объемов V2/Vt можно воспользоваться рав-
равным ему обратным отношением давлений:
18
Рг
Расширение газа может происходить при таких условиях, что
неизменным будет оставаться давление газа. Процессы, протекаю-
протекающие при неизменном давлении, называют изобарными. Вы-
Выражение для работы при изобарном расширении особенно просто.
Поскольку давление при этом остается постоянным, при интегриро-
интегрировании выражения для элементарной работы оно выносится за знак
интеграла:
194
Очевидно, что для того чтобы давление газа сохранялось в про-
процессе расширения неизменным, необходимо при этом повышать
его температуру. Повышение температуры газа вызывает в свою
очередь увеличение внутренней энергии, так что в уравнении пер-
первого начала термодинамики сохраняются все слагаемые. При изо-
изобарном расширении количество подведенной к газу теплоты больше
совершаемой работы на величину, равную возрастанию внутренней
энергии:
На диаграмме р, V (рис. 94) работа изобарного расширения
газа изображается площадью прямоугольника abed.
00
Рис. 93. Графическое изображение
работы при изотермическом расшире-
расширении газа.
Рис. 94. Графическое изображе-
изображение работы при изобарном рас-
расширении газа.
При адиабатном процессе, как это выяснено выше, работа
совершается за счет изменения внутренней энергии газа. Если
температура адиабатно расширяющегося газа уменьшается от зна-
значения 7\ до значения Т2, то одновременно и его внутренняя энер-
энергия уменьшается от величины CvTi до величины CvTz, и, следо-
следовательно, газ совершает работу, равную:
При желании можно выразить работу адиабатно расширяю-
расширяющегося газа через соответствующие изменения объема и давле-
давления.
Предположим, что газ, исходное состояние которого опреде-
определяется значениями параметров состояния рь Vit Tit адиабатно рас-
расширяется, причем конечное состояние его характеризуется значе-
значениями параметров р2, V2, 1\.
195
Совершенная при этом работа, как и раньше, будет выражаться
интегралом:
Однако в этом случае объем и давление газа связаны уравнением
Пуассона:
pV = Const = С,
откуда
С
Подставляя это значение р в уравнение, выражающее работу, и
интегрируя, найдем:
но
Заменяя постоянную С в выражении для работы, совершаемой ади-
абатно расширяющимся газом, получим:
-г
Рассмотренные выше закономерности справедливы применитель-
применительно к идеальным газам. В случае реальных газов соответствующие
закономерности усложняются.
Внутренняя энергия ван-дер-ваальсового газа зависит не только
от температуры, но и от объема. Для того чтобы найти эту зави-
зависимость, рассмотрим процесс изотермического изменения объема
реального газа. При изотермическом изменении состояния газа та
часть внутренней энергии, которая обусловлена молекулярным дви-
движением, остается неизменной, поскольку не изменяется средняя
скорость движения молекул.
Как показано выше, для этого к системе необходимо подвести
в случае расширения или от системы отвести в случае сжатия коли-
количество теплоты, эквивалентное совершенной работе.
Если газ подчиняется уравнению Ван-дер-Ваальса, работа d'A,
обусловленная элементарным изменением объема, запишется в виде
следующего соотношения:
или
196
Первый член в правой части написанного выражения — pdV —
соответствует работе против внешнего давления — это внешняя
работа. Второй член в правой части уравнения dV — пред-
представляет собой работу, совершенную против сил молекулярного
притяжения, и ее эквивалентом является изменение энергии взаимо-
взаимодействия молекул. Этот член соответствует потенциальной соста-
составляющей внутренней энергии реального газа.
Изменение внутренней энергии реального газа при неизотерми-
неизотермическом процессе складывается, таким образом, из изменения кине-
кинетической энергии теплового движения молекул и изменения потен-
потенциальной энергии их взаимодействия:
^реальн. газа = dUp + dUk,
ИЛИ
аза= ^2 dV -f CvdT.
В различных термодинамических соотношениях, определяющих
свойства реальных газов, необходимо учитывать наличие потен-
потенциальной энергии молекулярного взаимодействия.
8. КРУГОВЫЕ ПРОЦЕССЫ
В термодинамических рассуждениях большое значение имеет
рассмотрение различных круговых процессов.
Круговым процессом, или циклом называется такая
последовательность превращений, в результате кото-
которой система, выйдя из какого-либо исходного состоя-
состояния, вновь в него возвращается.
На диаграмме состояния круговой процесс изображается зам-
замкнутой кривой.
При изображении кругового процесса на графике с координа-
координатами р, V (рис. 95) он, естественно, распадается на процесс расши-
расширения системы abc и на процесс сжатия cda.
Процесс расширения системы связан с совершением ею работы,
ь то время как сжатие системы вызывается работой внешних сил
над системой.
На графике (рис. 95) работа, совершаемая системой при расшире-
расширении, численно равна площади фигуры abceh, а работа, совершаемая
внешними силами, возвращающими систему в исходное состояние,—
площади фигуры adceh. Разность этих площадей, равная площади
фигуры abed, соответствует разнице между работой, полученной
при расширении системы Л^г. и работой, совершенной для воз-
возвращения системы в исходное состояние Л2-»1.
Согласно первому началу термодинамики работа при расши-
расширении системы может совершаться или за счет внутренней энер-
197
гии системы, или за счет подводимой к системе теплоты. Аналогич-
Аналогично работа, совершенная внешними силами над системой при обрат-
обратном процессе, может или отводиться от системы в виде теплоты, или
же превращаться во внутреннюю энергию системы.
Рассмотрим один из возможных круговых процессов, обратив
внимание на взаимные превращения, которые претерпевают теплота,
работа и внутренняя энергия системы.
Для того чтобы сделать рассуждения возможно более простыми,
выберем в качестве системы идеальный газ. При изотермических
Рис. 95. Графическое изображение Рис. 96. Превращение теплоты в ра-
кругового процесса. боту при циклическом процессе.
процессах в этом случае внутренняя энергия системы не будет из-
изменяться.
Предположим, что исходное состояние системы / (рис. 96) характе-
характеризуется значением параметров состояния ри Vi, 7\. Предоставим
системе изотермически расшириться и перейти в состояние 2, харак-
характеризуемое значениями р2, V2, 7\.
Для того чтобы переход из состояния 1 в состояние 2 происхо-
происходил изотермически, к системе необходимо подвести при температу-
температуре 7\ количество теплоты AQi _> 2, эквивалентное работе, совершен-
совершенной системой:
V,
Если теперь возвратить систему в исходное состояние, сжимая
ее изотермически при той же температуре, то для этого будет не-
необходимо затратить работу, равную работе, полученной при рас-
расширении, а для поддержания температуры неизменной отвести от
системы эквивалентное количество теплоты.
В конечном результате окажется, что, сколько теплоты было
превращено в работу при прямом процессе, столько же работы бу-
будет превращено в теплоту при обратном.
198
Можно, однако, поступить иначе.
Предположим, например, что по достижении системой состоя-
состояния 2 мы понизим ее температуру до величины Т2, сохранив объем
неизменным. Давление в системе при этом должно уменьшиться
до величины р3. Система перейдет в некоторое состояние 3, харак-
характеризуемое параметрами ps, V2, Т2.
Переход из состояния 2 в 3 потребует отвода от системы коли-
количества теплоты AQ2-3- равного
Если теперь сжать газ изотермически до первоначального объ-
объема, то система перейдет в состояние 4, характеризуемое парамет-
параметрами р4, Vu Т2.
Для этого перехода потребуется затратить работу АЛ 3_>4 и для
поддержания неизменной температуры отвести от системы эквива-
эквивалентное количество теплоты:
AQ3->4 = AA,~4 = Wnb.
Как мы видим, на сжатие газа затрачена меньшая работа, чем
была получена при его расширении.
Рассмотренный процесс, однако, не является еще циклическим.
Для замыкания цикла необходимо газ нагреть при постоянном объ-
объеме, повысив его температуру до исходной. При этом к системе не-
необходимо подвести количество теплоты AQ4_i, равное
В результате нагревания цикл замкнется, система возвратится в ис-
исходное состояние с параметрами ри V1( 7\.
Проанализируем рассмотренный круговой процесс.
Прежде всего обратим внимание на то, что количество теплоты
AQ2_3> отнятое от системы при переходе ее из состояния 2 в сос-
состояние 3, равно количеству теплоты AQ4-i. сообщенному системе
при переходе ее из состояния 4 в 1, и поэтому эти количества теп-
теплоты взаимно сокращаются.
Таким образом, следует рассмотреть только изотермическое
расширение системы при переходе ее из состояния / в состояние 2
и изотермическое сжатие при переходе от состояния 3 к 4.
При изотермическом расширении системы к ней при температу-
температуре 7\ подводится количество теплоты AQi_»2. и система соверша-
совершает работу АЛ 1..2. эквивалентную этому количеству теплоты.
При изотермическом сжатии системы внешние силы совершают
работу АЛ3 .4, и от системы при более низкой температуре отво-
отводится эквивалентное количество теплоты AQ3^4.
Разность работ АЛ^г— АЛ3-,4 представляет работу данного
циклического процесса. Эта работа выражается на графике пло-
площадью фигуры, ограниченной контуром цикла. Она эквивалентна
199
разности количеств теплоты, подведенной при расширении и отве-
отведенной при сжатии системы.
С практической точки зрения интересны циклические процессы,
сопровождающиеся превращениями теплоты в работу.
В рассмотренном циклическом процессе от некоторого источ-
источника забиралось количество теплоты AQw2 при температуре Tt
и подводилось к рассматриваемой системе. Часть подведенной к
системе теплоты превращалась в работу, а остаток отводился от
системы опять же в форме теплоты AQ3-4> но ПРИ более низкой
температуре Тг. Такое положение является общим.
При любом циклическом процессе, предназначенном для пре-
превращения теплоты в работу, к системе подводится некоторое коли-
количество энергии в форме теплоты при более высокой температуре,
часть подведенной энергии превращается в работу, а часть отво-
отводится опять же в форме теплоты, но при более низкой температуре.
Циклический процесс, при котором к системе подводилось бы не-
некоторое количество теплоты и все без остатка превращалось бы в
работу, вообще невозможен.
Для характеристики эффективности циклического процесса в
отношении превращения теплоты в работу вводится физическая ве-
величина, называемая коэффициентом пол езного дей ст-
вия цикла, т], равная отношению работы А = А1^2—Л3,4, прак-
практически используемой в данном циклическом процессе, к работе,
которую можно было бы получить при превращении в нее всего ко-
количества теплоты, подведенного к системе:
или, учитывая эквивалентность между теплотой и работой,
Превращения теплоты в работу, подобные описанным выше,
используются в различных тепловых машинах. Чем ближе коэф-
коэффициент полезного действия цикла к единице, тем более совершен-
совершенна с термодинамической точки зрения данная машина. Наиболее
совершенным в отношении коэффициента полезного действия яв-
является циклический процесс, рассмотренный впервые французским
физиком Сади Карно A796—1832) и носящий его имя.
9. ЦИКЛ КАРНО
Цикл Карно (рис. 97) состоит из двух изотерм аЬ и dc и двух
адиабат be и ad. Для того чтобы упростить вычисления, предпо-
предположим, что рассматриваемая система — один киломоль идеального
газа, хотя полученный вывод будет справедлив для любой системы.
Допустим, что начальное A) состояние системы определяется
значениями параметров состояния рь Vu 7\.
200
Предоставим газу возможность расшириться изотермически при
температуре Ti до объема V2. Первоначальное давление газа при
этом уменьшится до величины р2, и система перейдет в состояние 2,
характеризуемое величинами р?, V2, 7\.
Расширяясь изотермически, газ совершит работу Ai^2, равную
Для поддержания температуры неизменной при изотермичес-
изотермическом расширении к системе необходимо подвести количество теп-
теплоты Qi, эквивалентное совершенной при этом работе:
Если теперь предоставить газу расшириться адиабатно от объ-
объема V2 до объема V3, то температура газа понизится до величины 72,
а давление — до величины р3.
Система перейдет в новое состоя-
состояние 3, характеризуемое величи-
величинами р3, V3, T2. Работа Л2_»3,
которую при этом переходе со-
совершит система, будет эквива-
эквивалентна уменьшению внутренней
энергии системы:
2-з — W К11— 1 %)•
Для возвращения системы в ис-
исходное состояние подвергнем
3(p3V3Tz)
Рис. 97. Цикл Карно.
газ, находящийся в результате
адиабатного расширения при бо-
более низкой температуре Т2, изо-
изотермическому сжатию до объема У4. Давление в системе возрастет
при этом до величины р4. Изотермическое сжатие газа потребует
работы Лз-4' равной
Для того чтобы сжатие газа происходило изотермически, от не-
него необходимо отвести количество теплоты Q2. эквивалентное со-
совершенной работе:
Значение объема Vt выбирается с таким расчетом, чтобы замы-
замыкание цикла, которое связано с повышением температуры до ве-
величины Гь можно было бы осуществить адиабатным сжатием газа
от объема У4 до начального объема W
В результате адиабатного сжатия температура газа и его дав-
давление принимают первоначальные значения 7\ и рь и система воз-
возвращается в исходное состояние.
201
Сжатие газа, соответствующее последней ветви цикла Карно,
потребует работы А^и эквивалентной возрастанию внутренней
энергии системы:
• -»1 = Cv (Т1 Т 2).
Работа, совершенная системой при расширении, равна сумме
Работа же, совершенная для возвращения системы в исходное состоя-
состояние, — сумме
Разность между работой, совершенной при расширении системы, и
работой, совершенной при ее сжатии, равна работе, которую мож-
можно использовать при циклическом изменении состояния системы:
RT, In Ь + С„ G\ - Т2) - RT2 In ?-¦ - С, G\ - Т2).
Как явствует из написанного выше выражения и как это сле-
следует непосредственно из физических соображений, работа, совер-
совершенная системой при адиабатном расширении, и работа, совершен-
совершенная при адиабатном сжатии, взаимно сокращаются, так что работа,
которую можно использовать в результате проведения цикла Карно,
равна разности
Коэффициент полезного действия цикла Карно выражается от-
отношением:
Для того чтобы упростить это выражение, заметим, что объемы V2
и V3, так же как объемы V4 и Vu лежат попарно на соответствую-
соответствующих адиабатах, и потому согласно уравнению Пуассона (уравне-
нение 8), для них справедливы соотношения:
Разделив первое равенство почленно на второе, получим:
202
Извлекая из обеих частей равенства корень (f — 1) степени, найдем:
V,
Учитывая это равенство, можно заменить в выражении для г\ ве-
величину In
получить:
личину In — равной величиной In — и, сократив дробь на R In -?,
A4)
Таким образом, коэффициент полезного действия цикла
Карно равен отношению разности между абсолютной
температурой Ти при которой происходит изотерми-
изотермическое расширение газа, и абсолютной температурой
изотермического сжатия газа Т2, к абсолютной тем-
температуре изотермического расширения газа 7\.
В термодинамике доказывается, что коэффициент полезного
действия цикла Карно не зависит от природы вещества, с помощью
которого он осуществляется, а зависит лишь от величины темпе-
температур, при которых теплота сообщается системе и отводится от нее.
Кроме того, при заданных значениях температур Т4 и Т2 цикл Кар-
Карно имеет наибольший по сравнению с другими циклами коэффи-
коэффициент полезного действия.
Для того чтобы повысить коэффициент полезного действия цик-
цикла Карно, следует увеличить температуру Tit при которой тепло-
теплота сообщается системе, и уменьшить температуру Тг, при которой
она отдается системой. Коэффициент полезного действия цикла
Карно стал бы равен 1, если температуру Т2 удалось бы понизить
до абсолютного нуля.
В результате цикла тепловой машины некоторое количество
теплоты Qi забирается при температуре Т\ от какого-либо внеш-
внешнего по отношению к рассматриваемой системе тела, обычно назы-
называемого нагревателем. Часть этой теплоты превращается
в работу, а часть Q2 отдается по-прежнему в виде теплоты, но при
более низкой температуре Г2, второму внешнему по отношению к
системе телу, называемому холодильником. Схематичес-
Схематически этот процесс изображен на рисунке 98.
Воспользовавшись этой терминологией, можно сказать, что
коэффициент полезного действия цикла Карно равен
отношению разности температур нагревателя и хо-
холодильника к абсолютной температуре нагревателя.
При обсуждении особенностей цикла Карно исключительно
большое значение имеет то обстоятельство, что процессы изотер-
изотермического и адиабатного сжатия и расширения, из которых этот
цикл состоит, обратимы. Поэтому можно первоначально предоста-
предоставить газу, занимающему при температуре 7\ объем Vи адиабатно
203
расшириться до объема Vi (рис. 99), вызвав тем самым понижение
его температуры до величины Тг. Если теперь предоставить газу
расшириться изотермически до объема V3, то он совершит работу
RT2 In —, отняв эквивалентное количество теплоты от внешне-
внешнего источника, находящегося при температуре Т2 более низкой, чем
исходная температура газа 7\.
Для возвращения газа в начальное состояние его сжимают ади-
абатно до объема V2, при достижении которого температура газа
Работа Q,-Q
Т2 холодильник
Рис. 98. Схема работы тепловой
машины.
Рис. 99. Цикл холодильной ма-
машины.
поднимается до первоначальной величины Tit а затем сжимают
изотермически до исходного объема Vt. Изотермическое сжатие тре-
требует затраты внешними силами работы Л7\ In —, сопровождаю-
сопровождающейся передачей внешнему телу эквивалентного количества теп-
теплоты при более высокой температуре 7\.
Работа, совершенная при сжатии, очевидно, больше, чем работа,
совершенная при расширении.
В описанном циклическом -процессе в результате совершения ра-
работы удается перевести некоторое количество теплоты от тела бо-
более холодного к телу более нагретому.
Описанный цикл называется циклом холодильной
машины.
Подобные круговые процессы лежат в основе действия различ-
различных холодильных аппаратов.
204
10. ИСПОЛЬЗОВАНИЕ ЦИКЛИЧЕСКИХ ПРОЦЕССОВ
В ТЕРМОДИНАМИЧЕСКИХ РАССУЖДЕНИЯХ
Как уже указывалось выше, коэффициент полезного действия
цикла Карно имеет одно и то же значение для любого вещества и
зависит только от температур, при которых осуществляются изо-
изотермические расширение и сжатие. Больше того, при изменениях
параметров состояния, т. е. давления, объема и температуры, в цик-
л°, Карно может происходить изменение агрегатного состояния ве-
вещества, например испарение жидкости или конденсация пара, и
все же коэффициент полезного действия будет определяться уравне-
уравнением A4), полученным в предыду-
предыдущем параграфе.
Эта особенность цикла Карно
позволяет использовать его для
нахождения важных термодинами-
термодинамических соотношений.
В качестве примера получим
этим способом уравнение Кла-
Клапейрона — Клаузиуса,
устанавливающее связь между дав-
давлением насыщенного пара жидко-
жидкости и температурой.
Пусть рассматриваемая систе-
система состоит из жидкости, находя-
находящейся при температуре Т в рав-
равновесии с собственным паром, дав-
давление которого при этой темпе-
температуре р н. п.
Обозначим молекулярный объем жидкости при температуре Т
через VM, а молекулярный объем пара — VH. п.
На диаграмме р, V (рис. 100) начальное состояние системы изоб-
изображается точкой а. Предоставим системе возможность изотерми-
изотермически расшириться и перейти в состояние, изображенное на диа-
диаграмме точкой Ь. Поскольку система состоит из жидкости, находя-
находящейся в равновесии с собственным паром, изотермическое расши-
расширение происходит при постоянном давлении pHi п и сопровождается
испарением п киломолеи жидкости.
В жидком состоянии п киломолеи вещества занимают объем п Уж,
а в виде насыщенного пара они занимают объем п VHi „. В результа-
результате возрастание объема при переходе системы из точки а в точку Ь
составит п (VH. п —Уж).
Для того чтобы испарение жидкости происходило изотермичес-
изотермически, к системе необходимо подвести количество теплоты, равное про-
произведению числа киломолеи испаряющейся жидкости п на моле-
молекулярную теплоту испарения X, т. е. пК.
Рис. 100. К выводу уравнения
Клапейрона — Клаузиуса
методом циклов.
205
Из состояния, соответствующего точке Ь, система переводится
аднабатно, в результате дифференциально малого увеличения объ-
объема, в состояние, соответствующее точке с, где температура на ве-
величину dT меньше исходной. Давление насыщенного пара умень-
уменьшается соответственно на величину йрКш „.
Уменьшая объем, систему изотермически переводят из состоя-
состояния с в состояние, соответствующее точке d, которое определяет-
определяется тем условием, чтобы адиабатное сжатие системы до исходного
объема сопровождалось подъемом температуры до первоначально-
первоначального значения Т.
Изотермическое сжатие с -* d происходит при постоянном да-
давлении, т. е. изобарно, и сопровождается конденсацией некоторого
количества пара.
Выражение для коэффициента полезного действия рассматри-
рассматриваемого дифференциального цикла Карно можно записать в виде
соотношения: .
а А аГ
пХ ~~~Т'
в котором d'A соответствует количеству теплоты, превращенному
в результате циклического изменения состояния системы в работу,
а произведение пХ — количеству теплоты, подведенному к системе
при изотермическом расширении. Графически (рис. 100) работа
d'A изображается площадью фигуры abed, равной произведению
одного из оснований (ab или cd) на высоту, которая, как это ясно
из графика, определяется изменением давления насыщенного па-
ра dpH.n.
Поскольку основание рассматриваемой фигуры аЪ равно раз-
разности объемов, занимаемых п киломолями вещества в жидком и паро-
парообразном состояниях, т. е. п (VH. п — Vx), то для работы, совер-
совершенной веществом в течение описанного цикла, будет справедливо
выражение:
d'A(V
Воспользовавшись полученным выражением, можно записать
коэффициент полезного действия рассматриваемого циклического
процесса в форме следующего соотношения:
пХ Т '
или, преобразуя его в более обычную форму, получим:
dT T(VH.n-Vx)
Последнее соотношение и называют уравнением Кла-
Клапейрона— Клаузиус а.
Воспользовавшись уравнением Клапейрона—Клаузиуса, удоб-
удобно сравнить термодинамический и молекулярно-кинетический ме-
методы решения одной из физико-химических проблем.
206
Предположим, требуется найти зависимость между упругостью
насыщенного пара жидкости и температурой. В дифференциаль-
дифференциальной форме эта зависимость дается совершенно строго уравнением'
Клапейрона—Клаузиуса, однако в такой форме это уравнение не
удобно для практического применения. Интегрирование получен-
полученного уравнения связано, как это сделается ясным ниже, с целым
рядом приближенно верных предположений.
При температурах, далеких от критической, молекулярный объ-
объем жидкости Уж много меньше молекулярного объема насыщенного
пара VH, п1, поэтому в разности, стоящей в правой части уравнения
A5), молекулярным объемом жидкости можно пренебречь и запи-
записать уравнение Клапейрона — Клаузиуса в форме:
Если теперь предположить, что при небольших давлениях свойства
насыщенного пара подобны свойствам идеального газа и для ве-
величины молекулярного объема пара VH. п справедливо выражение:
RT
V -
' н.п —
Ри.п
то уравнение Клапейрона—Клаузиуса примет вид:
dT RT2
или
A6)
Предположив далее, что теплота испарения К не зависит от темпе-
температуры, можно проинтегрировать написанное уравнение и найти
для давления насыщенного пара следующее соотношение:
In р„.п=-^г+ Const, A7)
или
где А — постоянная.
Но, как показывает опыт, теплота испарения зависит от темпе-
температуры. Это является одной из причин плохого согласия с опы-
опытом уравнения A7). Приняв в качестве первого приближения, что
теплота испарения линейно убывает с температурой:
А. = Ко — аТ,
1 При 20° С объем одного моля воды VM = 18,036-10~6 м3, объем од-
одного моля насыщенного водяного пара VH<n = 1,04 ж3.
207
где а — постоянная величина, и подставив это значение К в урав-
уравнение A6), найдем:
Рн.п \ RT*
RT )
dT.
Интегрирование последнего уравнения приводит к зависимости
давления насыщенного пара от температуры, лучше согласующей-
согласующейся с опытом, а именно:
Здесь А и В — постоянные, значения которых находятся из опыта.
Отказываясь от сделанных выше приближенно верных предпо-
предположений, можно получить более строгие и одновременно более слож-
сложные решения данной физико-химической задачи.
Ту же проблему можно решить молекулярно-кинетическим ме-
методом. Для этой цели будем считать, что средние скорости дви-
движения молекул жидкости и пара одинаковы, а следовательно,
одинаковы и средние кинетические энергии молекул жидкости и
находящегося с ней в равновесии пара.
Поскольку переход жидкости в пар требует затраты энергии в
форме теплоты, называемой теплотой испарения, то
очевидно, что потенциальные энергии молекул вещества в жидком и
парообразном состоянии различаются между собой. Разница в по-
потенциальной энергии АЯр, молекул одного киломоля вещества в жид-
жидком и парообразном состояниях в первом приближении равна м о-
лекулярной теплоте испарения X. В кинетической
теории было показано, что если молекулы вещества могут нахо-
находиться в одной из двух областей, переход между которыми связан
с изменением потенциальной энергии на величину АЕр, то при рав-
равновесии молекулярные плотности вещества в этих двух областях
будут различны. Если обозначить числа молекул в одном кубичес-
кубическом метре в первой и во второй области соответственно nt и л2) то
можно написать (сравните стр. 37):
АЕр
где кЕр — изменение потенциальной энергии одного киломоля веще-
вещества при переходе его из второй области в первую, R — универ-
универсальная газовая постоянная и Г — абсолютная температура.
Применяя это уравнение к случаю равновесия между жидко-
жидкостью и паром, найдем:
..-*/". A9)
Здесь пп и пж — числа молекул в 1 м3 соответственно пара и жид-
жидкости.
208
Число молекул в одном кубическом метре жидкости пж срав-
сравнительно мало зависит от температуры и в первом приближении,
при небольших изменениях температуры, может считаться посто-
постоянным.
Предположив, что пар подчиняется закону идеальных газов,
найдем далее, что число молекул пара в одном кубическом метре пп
связано с давлением pHi п и температурой Т соотношением: '
в котором k — постоянная Больцмана.
Подставляя из последнего уравнения значение пп в уравнение
A9), получим:
х
Рн.п _ ~ RT
кТ
или х
Логарифмирование этого уравнения приводит к выражению:
Inрн.п =- ~ + In T + Const. B0)
л/
Последнее уравнение качественно совпадает с уравнением A8),
полученным выше термодинамическим методом.
Некоторое отличие уравнения, полученного молекулярно-кине-
тическим методом от аналогичного уравнения, полученного термо-
термодинамически, обусловлено тем, что в обоих случаях для получения
окончательного выражения пришлось использовать лишь прибли-
приближенно верные предположения.
11. ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
Выше мы познакомились с термодинамическим методом решения
различных физических задач. Все рассуждения при этом основы-
основывались на использовании одного из основных законов природы:
закона сохранения и превращения энергии, или первого начала
термодинамики.
Как показал человеческий опыт, при всей важности этого за-
закона, его, однако, недостаточно для того, чтобы объяснить свое-
своеобразие протекания различных явлений в природе. Для того чтобы
убедиться в этом, рассмотрим первое начало термодинамики и след-
следствия, вытекающие из него, с несколько иной точки зрения, чем это
делалось выше. Математически первое начало термодинамики вы-
выражается уравнением:
dU = d'Q — d'A,
физический смысл которого сводится к утверждению, что измене-
изменение внутренней энергии системы возможно или в результате совер-
209
шения работы, или в результате передачи некоторого количества
теплоты. Чрезвычайно важно то, что написанное уравнение исчер-
исчерпывает все возможные способы изменения внутренней энергии
системы: внутренняя энергия системы может изменять-
изменяться только в результате совершения работы или пе-
передани некоторого количества теплоты.
Обратим теперь внимание на то обстоятельство, что оба ука-
указанных способа изменения внутренней энергии системы подразу-
подразумевают взаимодействие ее с какими-то телами, не входящими в рас-
рассматриваемую систему. Работа совершается или внешними силами,
т. е. силами, действующими на систему со стороны каких-либо не
входящих в нее тел, или, наоборот, системой, преодолевающей дей-
действие этих внешних сил.
Точно так же количество теплоты, необходимое для изменения
внутренней энергии системы, передается последней или от каких-
либо тел, не входящих в нее, или от самой системы этим телам.
Необходимость для изменения внутренней энергии системы
взаимодействия ее с телами, не входящими в нее, приводит к тому,
что в изолированной системе, т. е. в системе, включаю-
включающей все взаимодействующие тела, внутренняя энергия
остается неизменной. Учитывая сказанное, первое начало тер-
термодинамики иногда так и формулируют, утверждая, что внут-
внутренняя энергия изолированной системы постоянна,
или, что то же самое, в изолированной системе dU = 0.
В различных термодинамических системах можно представить
себе мысленно самые разнообразные процессы. Первое начало тер-
термодинамики позволяет выбрать из этого многообразия процессы,
протекание которых с точки зрения энергетических соотношений
принципиально возможно.
Предположим, например, что рассматриваемая система состоит
из двух порций mi и тг одной и той же жидкости, имеющих соот-
соответственно температуры /4 и t2. При сливании этих порций жид-
жидкости в условиях изоляции от взаимодействия с какими-либо дру-
другими телами для всей смеси устанавливается некоторая общая тем-
температура t. Опираясь на первое начало термодинамики, можно ут-
утверждать, что конечная температура всей смеси не может быть боль-
больше температуры более теплой из смешиваемых порций жидкости.
Процесс, приводящий к подобному результату, не допускается пер-
первым началом термодинамики. Более того, на том же основании мож-
можно утверждать, что в случае действительно изолированной системы
возможны только такие процессы, при которых выполняется сле-
следующее равенство:
Ю А + m2^a = (mi + mz) *• B!)
Огромное значение первого начала термодинамики заключает-
заключается именно в том, что оно указывает, каким образом выбрать из бес-
бесконечного количества процессов, которые человек может себе пред-
210
ставить, те процессы, протекание которых, вообще говоря, воз-
возможно.
Однако, помогая выделить возможные процессы, первое начало
термодинамики не дает основания для дальнейшего различия между
ними: с точки зрения первого начала термодинамики все отобран-
отобранные процессы одинаково возможны.
Для того чтобы уяснить эту особенность, возвратимся к при-
приведенному выше примеру. При смешении двух порций жидкос-
жидкости с разной температурой с точки зрения первого начала термо-
термодинамики возможен любой процесс, в результате которого тем-
температура смеси примет значение t, соответствующее уравне-
уравнению B1).
Однако с точки зрения первого начала термодинамики вполне
возможен и процесс, обратный рассмотренному: первое начало тер-
термодинамики допускает возможность того, что жидкость, масса ко-
которой mi-\-m.2, имеющая повсюду одинаковую температуру t, са-
самопроизвольно разделится на две части т^ и т2 с различными тем-
температурами ti и t2, если только эти температуры удовлетворяют
уравнению B1). Первое начало термодинамики не допускает лишь
изменения внутренней энергии изолированной системы, но никак
не ограничивает перераспределение внутренней энергии внутри
данной изолированной системы.
В то же время опыт учит человека тому, что в природе наблю-
наблюдается иное положение.
Хорошо известно, что при смешении нескольких порций жид
кости с разными температурами смесь всегда приобретает некото-
некоторую температуру, общую для всей жидкости. Также хорошо из-
известно из опыта, что без воздействия извне в жидкости, имев-
имевшей повсюду одинаковую температуру, никогда не возникает раз-
разность температур, обусловленная самопроизвольным переходом
некоторого количества теплоты от одной части жидкости к
другой.
Точно так же, при смешении водного раствора какой-либо соли
с чистой водой всегда наблюдается диффузия растворенного веще-
вещества, приводящая к выравниванию концентрации раствора во всей
жидкости, и никогда не наблюдается, чтобы растворенное в какой-
либо жидкости вещество самопроизвольно собралось бы в одной ее
части, в то время как во второй оказался бы чистый растворитель,
хотя этот процесс и не противоречит первому началу термодина-
термодинамики.
Наконец, можно постоянно наблюдать самопроизвольное пре-
превращение механической работы в теплоту. Так, например, можно
заставить скользить тяжелый брусок по наклонной плоскости,
(рис. 101), причем вся работа, совершаемая силой тяжести, будет
благодаря трению превращаться в теплоту. В результате трения
температура бруска и наклонной плоскости слегка возрастет, а внут-
внутренняя энергия системы останется постоянной.
211
В то же время, сколько бы ни ожидать, не удается наблюдать
самопроизвольного охлаждения бруска и наклонной плоскости,
в результате которого брусок сам начал бы двигаться вверх по на-
наклонной плоскости, хотя этот процесс может также протекать при
неизменной внутренней энергии системы.
Таким образом, возможные с точки зрения первого начала тер-
термодинамики процессы оказываются неравноценными в отношении
их протекания в том смысле, что, как показывает опыт, в изоли-
изолированной системе одни из этих процессов протекают, а другие не
протекают.
На различие таких процессов и указывается вторым основным
законом, или вторым началом, термодинамики.
Второе начало термодинамики ут-
утверждает, что существует функция
состояния, называемая энтро-
энтропией, которая обладает тем свой-
свойством, что при всех реальных про-
процессах, протекающих в изолирован-
изолированной системе, она возрастает.
Таким образом, второму началу
термодинамики можно придать сле-
Рис- 101- дующую формулировку: в изоли-
изолированной системе возможны
только такие процессы, при которых энтропия си-
системы возрастает.
Часто второе начало термодинамики формулируют несколько
иначе, например Кельвин формулировал этот закон в форме утверж-
утверждения, что невозможен процесс, единственным резуль-
результатом которого было бы получение от какого-либо
тела теплоты и превращение ее в эквивалентное коли-
количество работы.
Клаузиус предложил записать второе начало термодинамики
как утверждение невозможности самопроизвольного пере-
перехода теплоты от более холодного тела к телу более
теплому. Эти формулировки второго начала, так же как и еще
несколько формулировок, встречающихся в литературе, приводят
в конечном счете к одним и тем же выводам, и в этом отношении
равноценны.
Формулировка, приведенная в качестве первой, отличается тем,
что в ней более ясно выступает общность второго начала термо-
термодинамики.
Согласно второму началу термодинамики, для того чтобы отве-
ответить на вопрос, возможно ли в изолированной системе то или иное
превращение, необходимо рассчитать приращение энтропии при
этом превращении, и если это приращение окажется положитель-
положительным, то рассматриваемое превращение возможно, так как в резуль-
результате его энтропия изолированной системы возрастает. Те же про-
212
цессы, при которых приращение энтропии оказывается отрицатель-
отрицательным, в изолированной системе невозможны, поскольку при подоб-
подобных процессах энтропия изолированной системы должна убывать.
Количественно в термодинамике определяется не энтропия, а
разность энтропии, соответствующая какому-либо изменению со-
состояния системы. Новая функция состояния — энтропия — обоз-
обозначается буквой S, и согласно определению
dS = d^, B2)
или
2
^^r- B3)
Дифференциальное изменение энтропии dS определяется, таким
образом, отношением дифференциально малого количества теп-
теплоты, полученного или отданного системой, к температуре, при кото-
которой происходит процесс. Для гого чтобы пояснить, как использу-
используются формулы B2) и B3), рассмотрим некоторые примеры.
1. Подсчитаем изменение энтропии при плавлении 1 кмоля A8 кг)
льда. Удельная теплота плавления льда 3,35 105 дж/кг. Плавление
льда происходит при постоянной температуре 273° К, и поэтому в
уравнении B3) Т выносится за знак интеграла jd'Q, который в
данном случае будет равен количеству теплоты, необходимому для
плавления одного киломоля льда.
Таким образом:
SO 3,35 ¦ 105 • 18 П Л in, л , *ч
2 _ s1 = —: = 2,2 . 104 дж/град.
Lit О
2. Один киломоль идеального газа занимает при давлении рг и
температуре 7\ объем VY Определим изменение энтропии при равно-
равновесном переходе газа в состояние, характеризуемое параметрами
состояния р2, V2, Т2.
Запишем первое начало термодинамики:
dU = d'Q — pdV.
В случае идеального газа dU = Cv dT и р = —. Подставив эти
значения в уравнение первого начала, запишем его в виде:
T —.
Разделив это уравнение на Г и приняв во внимание определение
энтропии (уравнение 22), получим:
213
Интегрируя уравнение в пределах от Vu T4 до V2, T2, найдем ис-
искомое решение:
2 1 v "г1! уГ
Читатель может самостоятельно записать приращение энтропии
идеального газа при том же изменении состояния как функцию па-
параметров состояния р и Т или р и V.
Рассмотрим простейшие примеры применения второго начала
термодинамики для определения возможности протекания некото-
некоторых процессов в изолированных термодинамических системах.
Предположим, что в соприкосно-
соприкосновение приведены два весьма боль-
больших куска железа, один при темпе-
температуре 300° К, а второй при 400° К
(рис. 102). Подсчитаем изменение энт-
энтропии, которое будет иметь место
при переходе 100 дж теплоты один
раз от более теплого куска к более
холодному, а второй раз — в обрат-
обратном направлении.
Будем считать, что куски настоль-
настолько велики, что при получении или
потере 100 дж изменением темпера-
температуры можно пренебречь. Когда теплота переходит от тела более
теплого к телу более холодному, общее изменение энтропии в си-
системе составит:
Рис. 102. К расчету изме-
изменения энтропии при тепло-
теплообмене.
Знак минус ставится в том случае, когда теплота отдается телом,
и плюс, когда тело получает некоторое количество теплоты.
В случае, когда теплота переходит от тела более холодного
к телу более теплому, общее изменение энтропии системы составит:
Э =- й дж/зрад-
Таким образом, переход теплоты от тела более нагретого к телу
более холодному сопровождается положительным приращением
энтропии, и, следовательно, этот процесс в изолированной системе
возможен. Наоборот, переход теплоты от более холодного тела к
телу более теплому сопровождается отрицательным приращением
энтропии, и, следовательно, в изолированной системе такой про-
процесс невозможен.
В качестве второго примера рассмотрим изменение энтропии
при изменении объема идеального газа. Изменение энтропии в этом
случае выражается формулой:
214
Если изменение объема происходит изотермически: Tt = Тг
и In -2 - 0, то
Т
т. е. изменение энтропии будет всегда положительно, когда конеч-
конечный объем больше начального. Другими словами, идеальный газ,
представляющий собой изолированную систему, будет самопроиз-
самопроизвольно расширяться, стремясь занять весь предоставленный ему
объем.
Выше были рассмотрены наиболее элементарные примеры приме-
применения второго начала для определения направления возможного
процесса. Однако этот закон позволяет определить направление
и более сложных процессов. Кроме того, он дает возможность пред-
предопределить, при каких именно условиях данный процесс будет
протекать в желательном направлении.
12. СТАТИСТИЧЕСКИЙ СМЫСЛ ВТОРОГО НАЧАЛА
ТЕРМОДИНАМИКИ
Второе начало термодинамики как физическая закономерность
существенно отличается от первого начала. Первое начало термо-
термодинамики, или закон сохранения энергии, выполняется одинаково
строго как в случае применения его к телам макроскопических раз-
размеров, состоящих из огромного множества молекул, так и к телам,
содержащим всего несколько молекул, и даже к отдельной молекуле
или атому.
Второе начало термодинамики в этом отношении резко отли-
отличается от первого начала. Как показывает опыт, этот закон при-
применим только к телам, содержащим конечное, но очень большое
количество молекул. В самом деле, второе начало термодинамики
утверждает, что в теле с одинаковой температурой и плотностью
самопроизвольно никогда не возникает разность температур или раз-
разность плотностей. В то же время мы знаем из опыта, что конденса-
конденсация пара в облаках происходит в виде мельчайших капелек в облас-
областях, в которых самопроизвольно возникает более низкая темпе-
температура. Следовательно, в случае микрообъектов второе начало
термодинамики не только может нарушаться, но и постоянно нару-
нарушается. Эти особенности второго начала обусловлены статистичес-
статистической природой этого закона и объясняются особенностями строения
материи.
Вещество состоит из движущихся хаотически атомов или моле-
молекул. Поэтому нельзя предсказать точно, когда данная молекула
встретится с другой молекулой и какую скорость она приобрета-
приобретает после соударения. Учитывая это, говорят, что соударения мо-
215
лекул — явление случайное и результат его предвычислить точно
невозможно. Однако в цепи большого числа подобных случайных
событий наблюдается, как мы убедимся, определенная закономер-
закономерность.
Для пояснения этой мысли рассмотрим простейший пример слу-
случайного события — бросание игральной кости, имеющей шесть гра-
граней с цифрами от 1 до 6. Предположим, предлагается бросить 600 раз
подряд игральную кость и определить, сколько раз при этом вы-
выпадет шестерка.
Допустим далее, что, проделав этот опыт, обнаружим, что ше-
шестерка выпала 510 раз. Опыт повторили, бросив кость снова 600 раз,
и оказалось, что в этом случае шестерка выпала 490 раз. Когда тот
же опыт проделали в третий раз, то нашли, что из 600 бросаний
500 раз выпала шестерка. Можно быть твердо уверенным в том, что
познакомившись с результатами этих опытов, любой здравомыс-
здравомыслящий человек совершенно справедливо скажет, что эта кость не-
неправильной формы.
Но почему же можно сделать такое заключение?
Ведь выпадение той или иной грани при бросании игральной
кости — явление случайное, и наперед результат его предсказать
нельзя. Объясняется это тем, что при повторении действительно
случайного события должен выполняться совершенно определен-
определенный закон, грубо нарушавшийся в приведенном примере. Чтобы
наглядно показать это, рассмотрим некоторые закономерности, ко-
которым подчиняются случайные события.
Первоначально на том же конкретном примере с бросанием иг-
игральной кости введем понятие оматематической веро-
вероятности некоторого «события», в рассмотренном примере —
выпадения шестерки. Кость имеет шесть граней, причем выпадение
каждой из них одинаково возможно: говорят, что всего имеется 6
одинаково возможных случаев. Шестерка имеется только на одной
грани. Нас интересует событие, при котором выпадает именно шес-
шестерка, таким образом, благоприятствует рассматриваемому «собы-
«событию» только один случай. Если разделить число случаев, благо-
благоприятных рассматриваемому событию, на общее число случаев, воз-
возможных при данном событии, то мы получим величину, называе-
называемую вероятностью Р данного события.
р Число случаев, благоприятных событию
Общее число возможных случаев
В рассматриваемом примере вероятность выпадения шестерки
В теории вероятностей показывается, что для случайных
явлений справедлив закон: при достаточно
216
большом числе испытаний отношение чис-
числа испытаний, при котором наблюдается
какое-либо событие, к общему числу испы-
испытаний стремится к пределу, совпадающему
с вероятностью данного события.
Таким образом, в примере с бросанием игральной кости отно-
отношение числа бросаний, при которых выпала шестерка, к общему
числу бросаний должно при увеличении числа бросаний стремиться
для нормальной кости к 1/6. В описанном же опыте это отношение
составляло 51/60, 49/60, 50/60, т. е. резко отличалось от соответст-
соответствующей вероятности. И именно то, что найденная величина не стре-
стремилась при увеличении числа испытаний к вероятности данного
события, убеждает в том, что в данном случае выпадение шестерки
не являлось случайным событием — кость, очевидно, была непра-
неправильной формы.
Необходимо, однако, оговориться, что указанный закон спра-
справедлив лишь в случае большого числа испытаний и не имеет, напри-
например, силы при пяти, шести, двадцати бросаниях кости.
Законы, которым подчиняются совокупности случайных собы-
событий, называются законами больших чисел или ста-
статистическими законами в отличие от законов дина-
динамических, одинаково справедливых как в отношении сово-
совокупности событий, так и в отношении любого отдельно взятого со-
события.
Не следует рассматривать статистические закономерности как
закономерности, стоящие по рангу ниже закономерностей динами-
динамических. Причднные связи, имеющие место в природе, не могут быть
ограничены только рамками динамических законов, но включают
также и законы статистические. Применительно к телам макроско-
макроскопических размеров статистические закономерности молекулярной
физики выполняются столь же строго, как и закономерности дина-
динамические.
Возникает вопрос: каким образом можно увязать соображения
о вероятности различных событий с изучением свойств и поведения
вещества, которыми занимается молекулярная физика и термоди-
термодинамика?
Основным понятием термодинамики является понятие о состоя-
состоянии вещества. Выше говорилось, что состояние вещества опреде-
определяется совокупностью значений параметров состояния, в простей-
простейшем случае: давления, объема, температуры. И давление, и темпе-
температура являются макроскопическими характеристиками вещества,
возникающими в результате усреднения в одном случае большого
числа молекулярных ударов, а в другом — кинетической энергии
молекулярного движения.
Распределение молекул в пространстве, так же как и распреде-
распределение их скоростей, — явления случайные. При данных физичес-
физических условиях, например при данной внутренней энергии, данном
21?
объеме, данном взаимодействии между молекулами, то или иное
распределение молекул в пространстве, так же как и распределение
их скоростей, характеризуется определенной вероятностью.
Определение термодинамической веро-
вероятности отличается от определения веро-
вероятности математической. Для того чтобы пояснить,
как определяется термодинамическая вероятность, рассмотрим
один из простейших примеров. Предположим, в некотором сосуде
(рис. 103) имеется 6 молекул, которые для удобства обозначим бук-
©
©
©
©
©
©
Рис. 103. Мысленный опыт для вычисления вероятности
состояния.
вами а, Ь, с, d, e, /. Разделим мысленно сосуд на две равные полови-
половины и рассмотрим три «состояния» газа. Первое состояние соответ-
соответствует тому случаю, когда все шесть молекул находятся в одной по-
половине сосуда, а вторая — свободна от молекул. Второе, — когда
в одной половине находится одна молекула, а во второй — пять,
и третье, — при котором в каждой половине сосуда присутствуют
по три молекулы.
Каждое термодинамическое состояние можно рассматривать
или с макроскопической, или с микроскопи-
микроскопической точки зрения. С макроскопической точки зрения не име-
имеет значения, какие именно молекулы находятся в одной половине
сосуда и какие в другой, важно только их количество. С микроско-
микроскопической же точки зрения замена какой-либо молекулы в одной из
половин сосуда на молекулу из другой половины сосуда приводит
уже к новому состоянию. Таким образом, данное макроскопическое
состояние системы может осуществляться различным количеством
микроскопических состояний. Поясним это на приведенном выше
примере.
Первое состояние можно осуществить с микроскопической точ-
точки зрения одним-единственным способом, а именно: поместив все
218
из математики, равно г = С6 = --^-^ = 20. Состояние, при кото-
шесть молекул в одну половину сосуда и оставив вторую половину
пустой.
Второе состояние можно осуществить уже несколькими спосо-
способами. Действительно, предположим, что, для того чтобы осущест-
осуществить второе состояние, в одну из половин сосуда помещена моле-
молекула а, а остальные пять — в другую половину. С макроскопичес-
макроскопической точки зрения это состояние будет неразличимо от тех состояний,
при которых вместо молекулы а в данной половине сосуда находит-
находится молекула Ь, с, d, e или /.
Таким образом, в этом случае одно и то же макроскопическое
состояние может быть осуществлено уже, с помощью 6 состояний,
различных с микроскопической точки зрения. Говорят, что дан-
данному макросостоянию соответствует шесть микросостояний. Естест-
Естественно, что если попадание той или иной молекулы в данную поло-
половину сосуда — явление случайное, то второе макросостояние бу-
будет более вероятно по сравнению с первым.
Еще больше число микросостояний, с помощью которых можно
осуществить третье макросостояние. Число их можно определить,
если подсчитать число сочетаний из 6 по 3, которое, как известно
„з е-5.4
1-2-3 '
ром в каждой половине сосуда находится по три молекулы, оче-
очевидно, более вероятно, чем то состояние, при котором в одной по-
половине находилась одна молекула, а во второй — пять.
Термодинамической вероятностью W какого-либо
состояния называют число микросостояний, с по-
помощью которых может быть осуществлено данное
макросостояние. В настоящее время разработаны способы
расчета термодинамической вероятности различных состояний.
Для газа, на который не действуют внешние силы, наиболее ве-
вероятным состоянием является равномерное распределение молекул
по всему объему. В противоположность этому для газа, находяще-
находящегося в поле тяготения, наиболее вероятным будет распределение
молекул, вытекающее из барометрической формулы.
Точно так же, если исключить внешние воздействия и выждать
достаточно долгое время, то в газе установится наиболее вероят-
вероятное распределение молекулярных скоростей, каковым является
максвелловское распределение.
Представление о термодинамической вероятности состояний
позволяет понять особенности второго начала термодинамики. Как
показал Л. Больцман, функция состояния, называемая энтропиейS,
однозначно связана с термодинамической вероятностью состояния,
а именно энтропия пропорциональна логариф-
логарифму вероятности состояния:
S = k In W. B4)
Коэффициент пропорциональности k — постоянная Больцмана.
219
При статистическом истолковании энтропии становится понят-
понятным, почему в изолированной системе самопроизвольно протекают
только такие процессы, при которых энтропия системы возрастает.
Возрастание энтропии означает возрастание вероятности состояния
системы, и второе начало термодинамики приобретает очень про-
простой смысл утверждения, что самопроизвольно изолирован-
изолированная система может переходить только от состояний
менее вероятных к состояниям более вероятным. Также,
очевидно, что изолированная термодинамическая система, энтро-
энтропия которой достигла максимально возможной при данных значе-
значениях параметров состояния величины, будет находиться в с о с т о-
янии устойчивого равновесия.
Естественно, что этот закон имеет силу только для совокупнос-
совокупностей конечного, но достаточно большого числа молекул. Непра-
Неправильно пытаться применять второе начало термодинамики к пяти,
десяти, двадцати молекулам.
Таким образом, в нарушение второго начала вочень не-
небольших объемах газа или жидкости можно встретить само-
самопроизвольные повышения или понижения температуры или плот-
плотности, обусловленные случайным характером молекулярного дви-
движения. Статистическая природа второго начала термодинамики не
только не исключает возможности подобных отклонений величины
параметров состояния от средних значений, но даже предсказывает
их неизбежность. Такого рода случайные отклонения значений па-
параметров состояния от средних значений называют флуктуаци-
я м и. Флуктуации играют важную роль во многих физических
явлениях. Так, например, привычный для нас синий цвет неба возни-
возникает в результате рассеяния солнечных лучей на случайных флук-
туациях плотности воздуха в земной атмосфере. Если бы подобных
флуктуации не существовало, небо представлялось бы нам иссиня-
черным и даже днем на нем можно было бы видеть не только солнце,
но и звезды. Флуктуации плотности, непрерывно возникающие в
атмосфере, рассеивают солнечные лучи. Наиболее интенсивно рас-
рассеиваются лучи с короткой длиной волны, т. е. сине-фиолетовые.
Именно эти лучи и создают окраску неба.
Особенно сильно развиты флуктуации в том случае, если веще-
вещество находится вблизи критической точки. Флуктуации плотности
обусловливают в этом случае так называемую критическую опале-
сценцию. В результате критической опалесценции вещество в кри-
критической области делается непрозрачным.
Статистический характер второго начала термодинамики следует
иметь в виду при всех применениях этого важнейшего закона при-
природы. Забвение этого легко приводит к ложным с научной течки
зрения выводам.
220
13. КРИТИКА ТАК НАЗЫВАЕМОЙ «ТЕОРИИ ТЕПЛОВОЙ СМЕРТИ
ВСЕЛЕННОЙ»
Примером неправильного применения второго начала термоди-
термодинамики может служить так называемая «теория тепловой смерти
Вселенной», особенно оживленно обсуждавшаяся в конце прошлого
столетия.
Неправильно распространяя на Вселенную второе начало тер-
термодинамики, сторонники этой теории рассуждали приблизительно
так: все виды энергии способны самопроизвольно превращаться в
теплоту, а теплота самопроизвольно переходит от тел с большей
температурой к телам с меньшей температурой.
В природе непрерывно происходит процесс выравнивания суще-
существующих разностей температур. Рано или поздно во всей Вселен-
Вселенной выравняются температура и давление. Вселенная достигнет
состояния термодинамического равновесия, при котором исклю-
исключается возможность каких-либо процессов, — это будет состояние
тепловой смерти Вселенной.
Поскольку подобное состояние еще не достигнуто, следователь-
следовательно, Вселенная не существует вечно, она была создана какое-то ко-
количество лет тому назад. Таким образом, делался вывод о возник-
возникновении (сотворении) мира.
Дальше ученые-идеалисты делали вывод о том, что для созда-
создания Вселенной необходима была деятельность сознательного су-
существа. Необходимость творца Вселенной аргументировалась так:
поскольку Вселенная развивается, переходя от состояний с мень-
меньшей энтропией к состояниям с большей энтропией, следовательно,
она переходит от состояний менее вероятных к состояниям более
вероятным. Двигаясь назад в глубь веков, мы встречаемся, согласно
этим рассуждениям, с состояниями все менее и менее вероятными
и, наконец, в пределе достигаем состояния, вероятность которого
исчезающе мала.
Подобное состояние не может возникнуть случайно, но оно мо-
может быть создано сознательным существом, так же как практичес-
практически не может нормальная игральная кость выпасть миллион раз
подряд вверх шестеркой, но может быть таким образом сознательно
положена человеком.
Подобные ошибочные с точки зрения физики, но имеющие ви-
видимость научной теории рассуждения представляли широкое по-
поле деятельности различным представителям идеалистической фи-
философии.
Фундаментальная ошибка всех этих рассуждений заключалась
в том, что опытный закон, установленный применительно к систе-
системе, содержащей конечное число частиц, необоснованно переносился
на бесконечно протяженную систему. Подобного рода ошибки отно-
относятся к основным логическим ошибкам. Ведь ни у кого не возни-
возникает сомнения в невозможности применения второго начала термо-
221
динамики к системам, содержащим небольшое число частиц,
а ведь столь же ошибочно применение этого закона и к систе-
системам с бесконечно большим числом частиц. Крометого, второе начало
не учитывает целый ряд превращений, которые могут играть боль-
большую роль во Вселенной. Мы еще мало знаем о закономерностях вза-
взаимодействия элементарных частиц с полем, да и сами элементарные
частицы, вероятно, не являются элементарными в обычном смысле
этого слова, а имеют структуру, которую в скором времени при-
придется учитывать.
Функция состояния, называемая энтропией, определяющая рав-
равновесие изолированных систем с конечным числом частиц, имеет,
вероятно, иной смысл, когда говорят о космических системах, пони-
понимая под последними даже не всю Вселенную, а отдельные звездные
скопления.
Неправильно механически переносить наши обычные предста-
представления, возникшие в результате наблюдения поведения макроскопи-
макроскопических тел, на явления, разыгрывающиеся как в микромире, так и
в космосе.
Жидкость, находящаяся достаточно долго в идеальном термо-
термостате, достигает состояния термодинамического равновесия и может
рассматриваться как пребывающая в состоянии тепловой смерти.
В масштабах же микромира в подобной жидкости непрерывно про-
происходят разнообразные процессы: сложные молекулы диссоциируют;
электроны, вращающиеся вокруг атомных ядер в молекулах, изме-
изменяют свое первоначальное движение; присутствующие продукты
диссоциации вновь соединяются с образованием недиссоциирован-
ных продуктов и т. д. С точки зрения атома подобное состояние ни-
никак нельзя рассматривать как состояние, неизменное во времени.
Столь же неправильно переносить наши представления и на косми-
космические системы. Неправильно считать, что Вселенная когда-то воз-
возникла и затем развивалась, стремясь к равновесному состоянию.
Вселенная — вечна. Астрономические исследования последних
лет дают основания предполагать, что звездные миры продолжают
возникать и в наше время. Вселенная, в которой мы живем, непре-
непрерывно обновляется.
Есть все основания предполагать, что во Вселенной, наряду с
процессами выравнивания температур, имеют место процессы, при-
приводящие к значительной концентрации энергии и к возникновению
областей с очень высокой температурой.
Физик должен постоянно помнить об относительном характере
законов науки, о том, что физическая теория является лишь «приб-
«приблизительной копией с объективной реальности» (Ленин В. И.,
«Материализм и эмпириокритицизм»), и не допускать необоснован-
необоснованной экстраполяции физических законов.
ОГ ЛАВ ЛЕНИЕ
Предисловие •.... 3
I. ГАЗООБРАЗНОЕ СОСТОЯНИЕ ВЕЩЕСТВА
1. Развитие атомных представлений в науке 8
2. Понятие о состоянии вещества 13
3. Закон Бойля—Мариотта —
4. Закон Гей-Люссака Н
5. Объединенный газовый закон 16
6. Понятие об идеальном газе 20
7. Основные положения кинетической теории га^ов . , 23
8. Основное уравнение кинетической теории газов 24
9. Некоторые следствия из основного уравнения кинетической тео-
теории газов 33
10. Распределение молекул газа в поле земного тяготения 35
11. Экспериментальное определение числа Авогадро 37
12. Закон распределения скоростей 39
13. Экспериментальная проверка закона распределения Максвелла . . 48
14. Число соударений и длина свободного пробега молекулы .... 51
15. Экспериментальное определение длины среднего свободного про-
пробега молекул 54
16 Явления переноса. Вязкость газов 55
17. Теплопроводность газов 59
18. Диффузия газов 62
19. Вакуум. Получение и измерение вакуума 66
20. Внутренняя энергия идеального газа 70
21. Теплоемкость идеального газа 72
22. Соотношение между теплоемкостью газа при постоянном объеме
и теплоемкостью газа при постоянном давлении 75
23. Реальные газы. Уравнение Ван-дер-Ваальса 77
24. Исследование уравнения Ван-дер-Ваальса 82
25. Определение критических параметров вещества 86
26. Природа сил молекулярного притяжения 88
27. Внутренняя энергия реального газа 90
28. Сжижение газов 92
II. ЖИДКОЕ СОСТОЯНИЕ ВЕЩЕСТВА
1. Сравнение свойств жидкостей со свойствами газов и твердых тел. 95
2. Молекулярное давление 97
3. Поверхностные свойства жидкостей 98
4. Явления на границе жидкости с твердым телом 103
5. Давление под изогнутой поверхностью жидкости 106
6. Поверхностно-активные вещества , 109
7. Испарение жидкостей. Насыщенный пар и его свойства 112
8. Молекулярное строение жидкостей 115
9. Вязкость жидкостей 122
223
Ю.''Растворы. Осмотическое давление 124
11».-Понижение давления насыщенного пара растворителя над раст-
раствором 127
12. Растворы электролитов 129
13. Применение ультраакустических методов к исследованию газов
и жидкостей 131
III. ТВЕРДОЕ СОСТОЯНИЕ ВЕЩЕСТВА
1. Кристаллические и аморфные твердые тела . ......... 137
2 1Чолекулярное строение кристаллов 140
3. Полимеры 147
4. Силы, действующие между частицами в твердых телах 152
5. Теплоемкость твердых тел 158
6 Сублимация, плавление и кристаллизация твердых тел 163
7*. Кристаллизация растворов. Диаграммы плавкости 166
8. Адсорбция 169
IV. ФИЗИЧЕСКИЕ ОСНОВЫ ТЕРМОДИНАМИКИ
1. Молекулярно-кинетический метод, метод статистической механи-
механики и термодинамический метод 172
2 Предмет термодинамики. Основные термодинамические понятия . 173
3. Физический смысл понятия «теплота» 180
4. Первое начало термодинамики '. 182
5. Методы нахождения термодинамических соотношений 185
6. Адиабатные процессы. Уравнение Пуассона 188
7. Применение первого начала термодинамики к некоторым про-
процессам в газах 192
8. Круговые процессы 197
9. Цикл Карно 200
10. Использование циклических процессов в термодинамических рас-
рассуждениях 205
11. Второе начало термодинамики 209
12. Статистический смысл второго начала термодинамики 215
13. Критика так называемой «теории тепловой смерти Вселенной» . 221
Борис Борисович Кудрявцев
КУРС ФИЗИКИ
Теплота и молекулярная физика
Редактор Т В. Михалкевич Художественный редактор В. С. Эрденко. Технический редак-
редактор 7\ А. Семейкина. Корректор М В Голубева
* V
*
Сдано в набор 2/IX 1964 г. Подписано к печати 5/II 1965 г. бОХЭО'/и- Печ л. 14. Уч.-
изд. л 13,39. Тираж 30 тыс. экз. А00054. Т. п. 1965 г №36/255.
* *
Издательство «Просвещение» Государственного комитета Совета Министров РСФСР по пе-
печати Москва, 3-й проезд Марьиной рощи, 41
Саратовский полиграфический комбинат Росглавполиграфпрома Государственного комитета
Совета Министров РСФСР по печати, г. Саратов, ул. Чернышевского, 59.
Заказ № 145. Цена без переплета 40 коп., переплет 15 коп.