/



















































































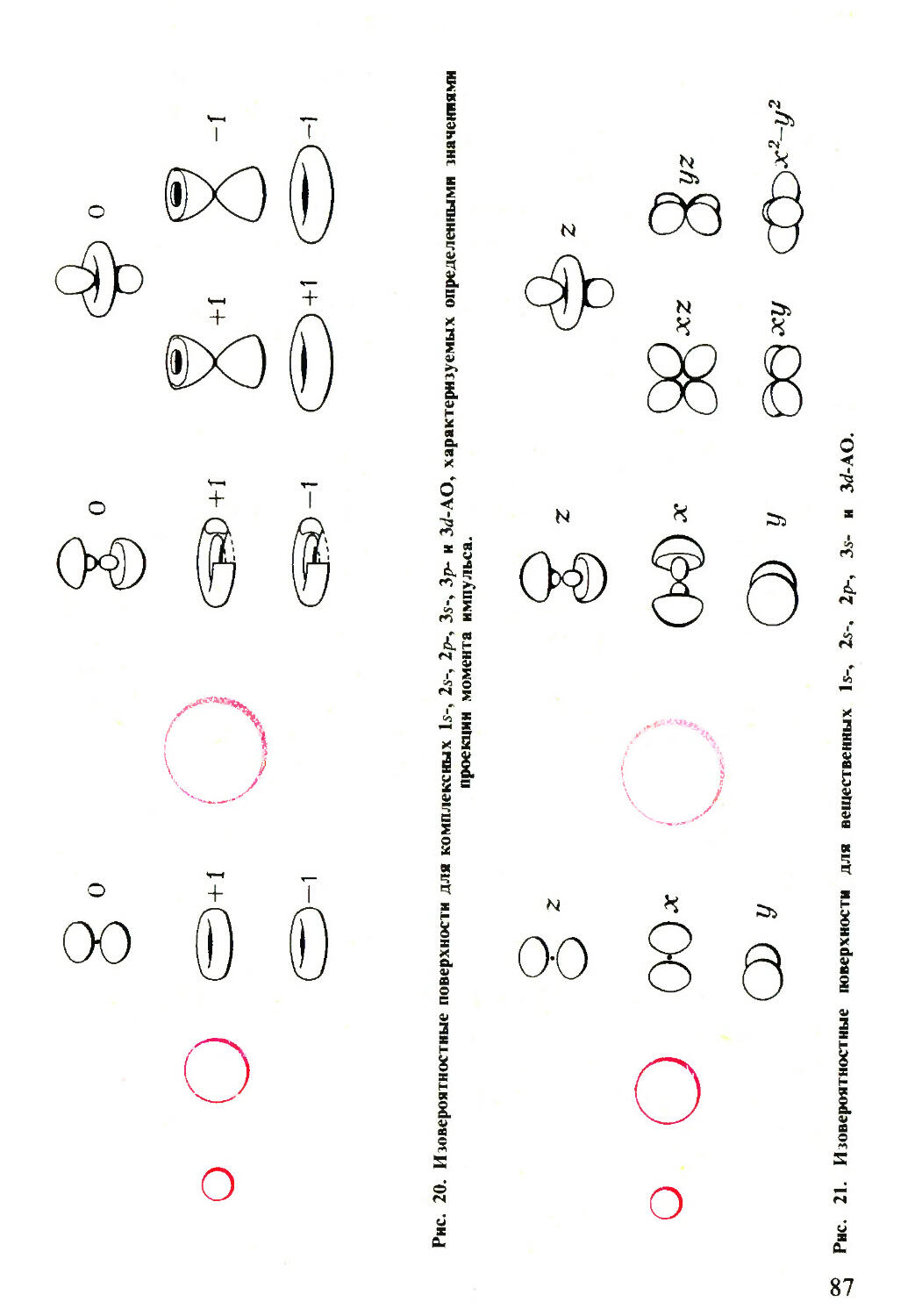

























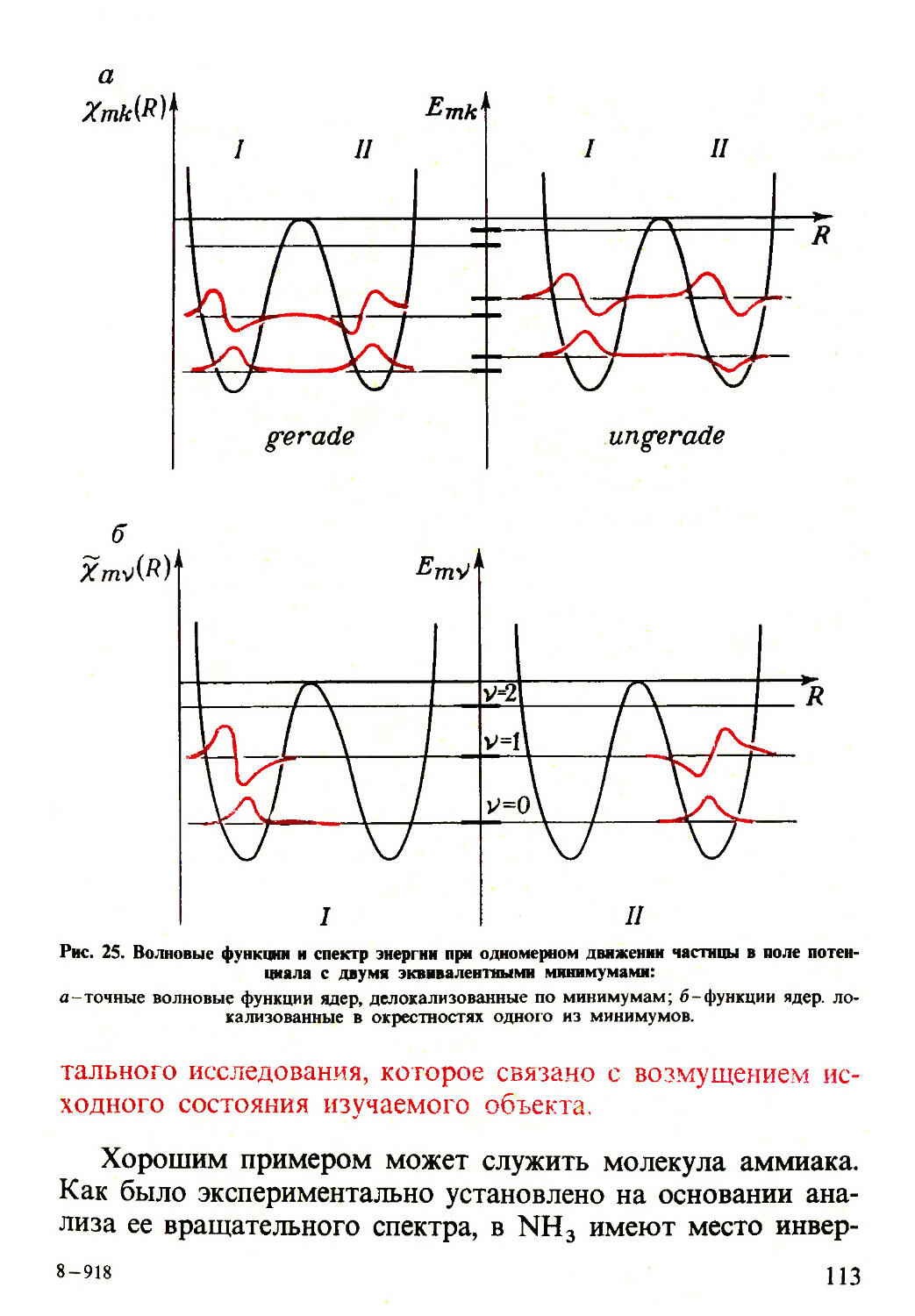




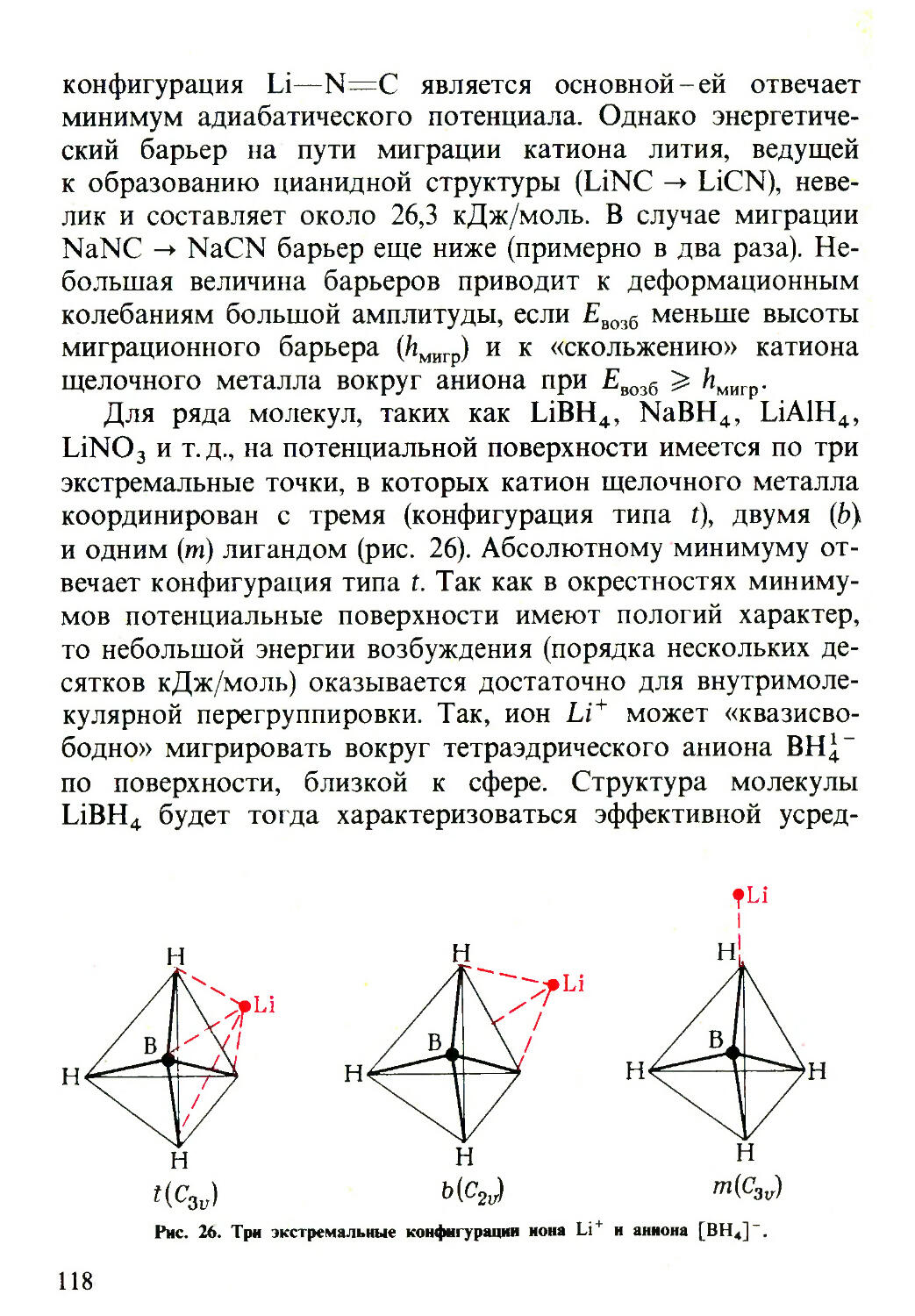
















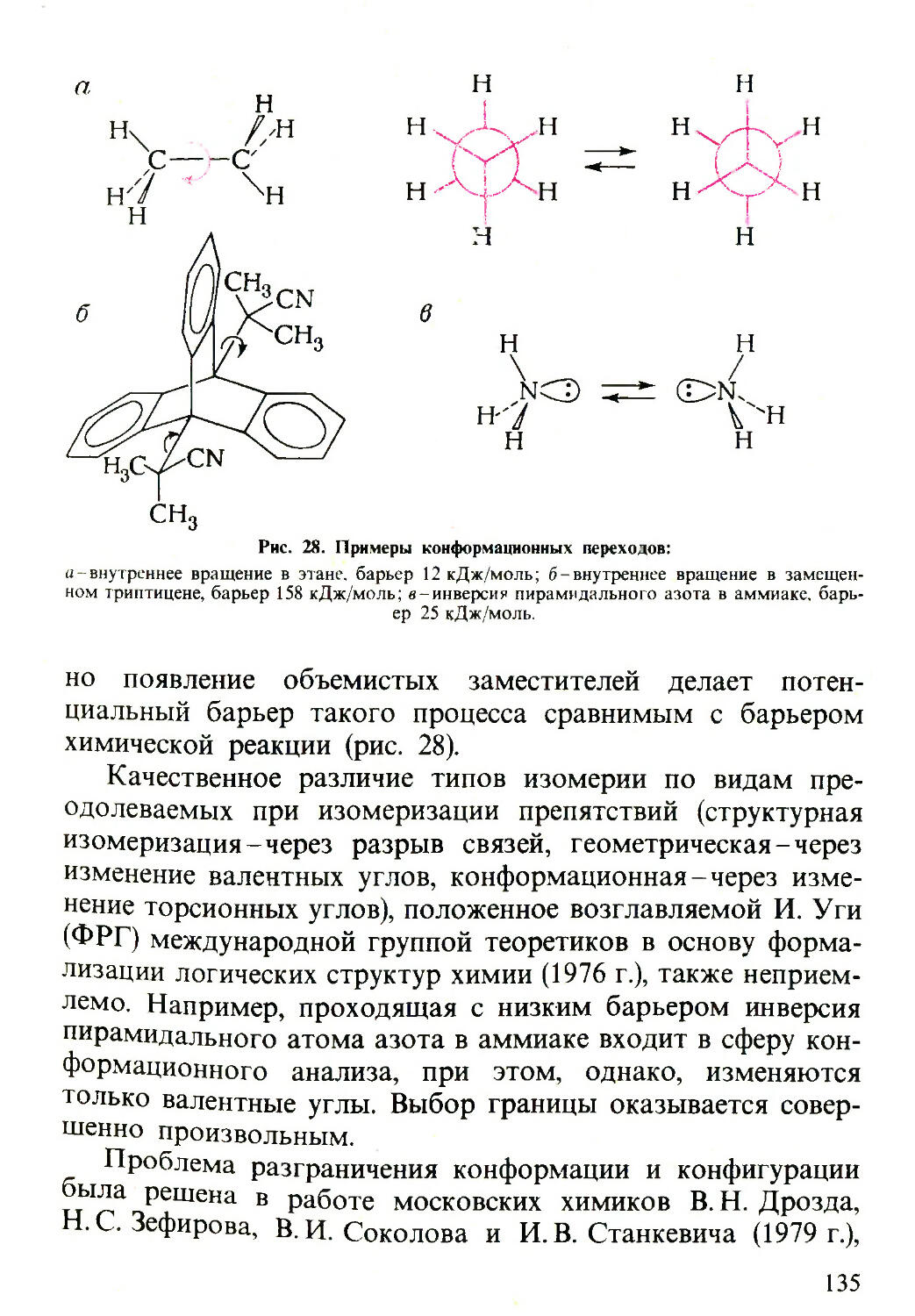








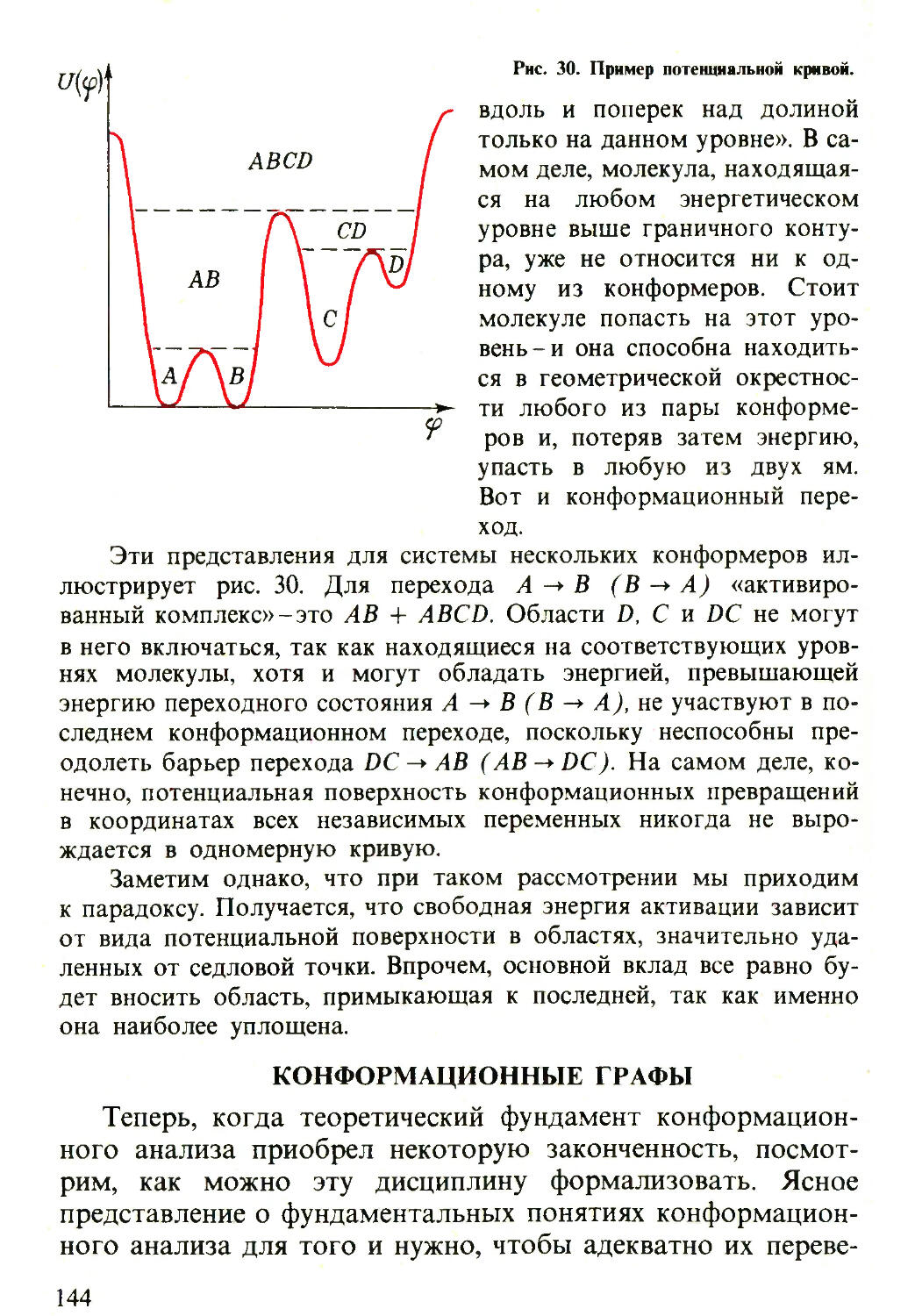











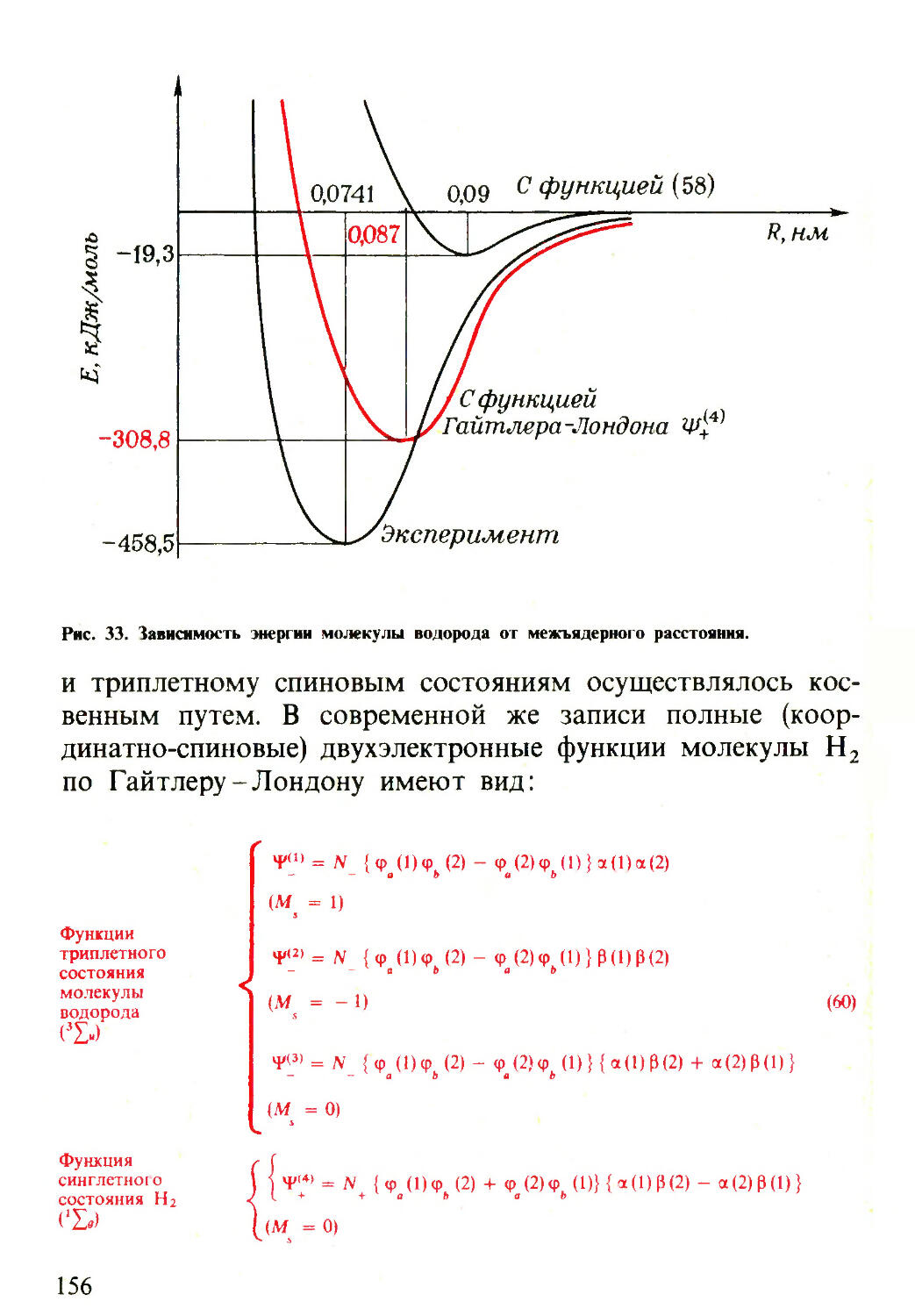












































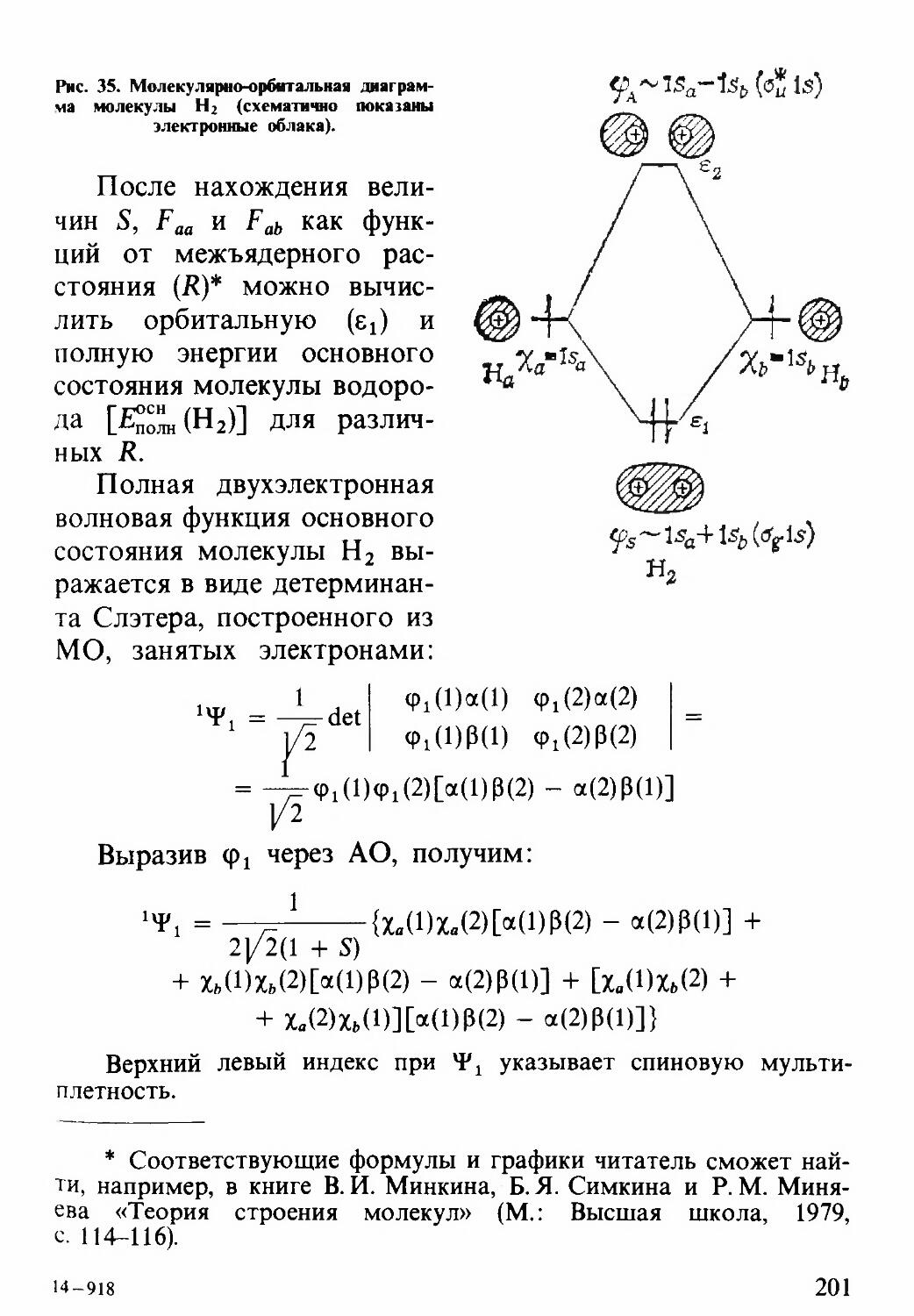





































Текст
т научно-—
=■ ПОПУЛЯРНАЯ
i ЛИТЕРАТУРА
И.С. ДМИТРИЕВ
Электрон
глазами химика
«Ы И.С. ДМИТРИЕВ
ЛИТЕРАТУРА
Электрон
глазами химика
(ОЧЕРКИ О СОВРЕМЕННОЙ КВАНТОВОЙ ХИМИИ)
Ленинград
ХИМИЯ
Ленинградское отделение
1983
541
Д53
УДК 541.2
Дмитриев И. С.
Д53 Электрон глазами химика- JL: Химия,
1983-232 с., ил - (Вопросы современной химии).
Рассмотрены вопросы электронного строения атомов и молекул. В доступной
форме описаны достижения, проблемы и перспективы развития квантовохимических
представлений. Читателю предложен круг современных проблем в области молекуляр¬
ной квантовой химии. Систематически и последовательно изложены основы структур¬
ной теории.
Рассчитана на старшеклассников, студентов, преподавателей, широкий круг хими¬
ков-всех, кто интересуется современными проблемами теоретической химии.
1802000000-101
Д 101-83 541
050(01)-83
Рецензент-канд. хим. наук А. Г. Авраменко
© Издательство «Химия», 1983
СОДЕРЖАНИЕ
От автора 4
Введение. РОЖДЕНИЕ ГЕРОЯ 5
Глава I. ЭЛЕКТРОН-ВОЛНА И ЧАСТИЦА 16
Глава II. ЭЛЕКТРОН В АТОМЕ 77
Глава III. ЧТО ТАКОЕ МОЛЕКУЛЯРНАЯ 98
СТРУКТУРА?
Глава IV. ЭЛЕКТРОН ГЛАЗАМИ ХИМИКА 153
ОТ АВТОРА
По теории строения молекул написано немало хороших
книг, и автору, естественно, не хотелось пересказывать уже
известное. Поэтому значительное место отведено материа¬
лу, несколько нетрадиционному для изданий такого рода.
Это, в частности, касается главы, посвященной молекуляр¬
ной структуре. Вместе с тем, требования систематичности
и последовательности изложения заставляют касаться и ве¬
щей довольно известных. В этих случаях автор стремился
подчеркнуть аспекты, на которые обычно мало обращают
внимания (см., например, раздел, посвященный концепции
гибридизации).
В книге много математических формул. Возможно, од¬
ним читателям это покажется недостатком, другим же, на¬
оборот-достоинством. На наш взгляд, спор о том, сколько
химику математики надо,-отчасти схоластический. Времена,
когда математические потребности химика сводились к ру¬
диментарному владению латинским и греческим алфавита¬
ми, безвозвратно канули в Лету. И потому каждый, кто за¬
хочет по-настоящему разобраться в современной структур¬
ной химии, рано или поздно сталкивается с необходи¬
мостью перейти от сугубо качественных рассуждений к более
строгой теории или, по крайней мере, уяснить, какое физи¬
ко-математическое содержание, какая модель стоит за таки¬
ми широко используемыми ныне понятиями, как орбиталь,
гибридизация, конфигурация и т.д. Мы надеемся, что эта
книжка поможет, хотя бы отчасти, разобраться в этом.
Автор приносит сердечную благодарность рецензенту
к.х.н. А. Г. Авраменко, научному редактору к.х.н. С. Г. Семе¬
нову, к.х.н. С. М. Шевченко, любезно согласившемуся напи¬
сать разделы, посвященные некоторым понятиям современ¬
ной стереохимии.
Природы превращенья шире,
Чем смена дня и ночи в мире
И. ГЕТЕ
Введение
РОЖДЕНИЕ ГЕРОЯ
Мысль о существовании элементарного электрического
заряда возникла еще в XVIII в., т. е. задолго до его экспери¬
ментального открытия. В трудах Б. Франклина, В. Вебера,
О. Моссотти, Г. Дэви и многих других естествоиспытателей
можно найти намеки или прямые указания на возможность
существования «электрического атома». Важным аргумен¬
том в пользу такого предположения послужили открытые
в 1830-х годах М. Фарадеем количественные законы электро¬
лиза, согласно которым для получения 1 г-экв любого веще¬
ства при 100%-ном выходе по току требуется одно и то же
количество электричества. Анализ этого закона привел не¬
мецкого ученого Г. Гельмгольца к идее элементарного элек¬
трического заряда. «Если применить атомистическую гипо¬
тезу к электрическим процессам,-отмечал Гельмгольц
в 1881 г.,-то она в соединении с законом Фарадея приводит
к поразительным следствиям. Если мы допускаем существо¬
вание химических атомов, то мы вынуждены заключить от¬
сюда, что и электричество разделяется на определенные эле¬
ментарные количества, которые играют роль атомов элек¬
тричества».
В этом же году был опубликован доклад английского
физика Дж. Стони (сделанный им еще в 1874 г.) «О физиче¬
ских единицах природы», где автор высказал аналогичные
идеи. Позже, в 1891 г., Стони предложил термин «электрон».
«В каждом химическом атоме,-писал Стони,-может быть
несколько элементарных зарядов. Эти заряды, которые
удобно назвать «электронами», не могут быть отделены от
5
атомов, но они обнаруживаются, когда атомы вступают
в химическое соединение».
В 1897 г. электронная гипотеза получила эксперимен¬
тальное подтверждение в исследованиях Э. Вихерта
и Дж. Дж. Томсона. И с этого времени началось создание раз¬
нообразных электронных моделей атомов и молекул. Одна¬
ко первые модели были гадательными. Положение измени¬
лось только после работ Э. Резерфорда.
«Теперь мы знаем, как выглядит атом»
Летом 1906 г., в Канаде, Резерфорд, который тогда воз¬
главлял физическую лабораторию Макгилльского универси¬
тета, заметил странное явление: узенький пучок альфа-ча¬
стиц, пронизав тонкий слюдяной листок, немного расширил¬
ся. Отчего? Что могло сбить частицы с прямого пути?
Тогда ученый ограничился лишь коротким замечанием: «Та¬
кой результат ясно показывает, что атомы вещества должны
быть средоточием очень интенсивных электрических полей».
Но спустя три года, в Манчестере...
«Он повернулся ко мне и сказал,-вспоминал ученик Ре¬
зерфорда Э. Марсден- Посмотрите-ка, не сможете ли вы
получить некий эффект прямого отражения а-частиц от ме¬
таллической поверхности?» И, может быть неожиданно для
самого Резерфорда, эксперимент подтвердил сделанное
предположение - Гейгеру и Марсдену удалось наблюдать а-
частицы, возвращающиеся назад. Из этого следовало, что
в атоме есть положительно заряженная, массивная сердце¬
вина-ядро, занимающее небольшой объем и отбрасываю¬
щее положительно заряженные а-частицы, если те попадали
в него. В 1911 г. Резерфорд опубликовал статью, в которой
сформулировал концепцию планетарного атома или-другое
наименование,-ядерную модель атома, предупредив, одна¬
ко, читателей, что «вопрос об устойчивости предлагаемого
атома на этой стадии не следует подвергать рассмотре¬
нию... Устойчивость окажется, очевидно, зависящей от тон¬
6
ких деталей структуры атома и движения составляющих его
заряженных частей».
Предупреждение это было ненапрасным. С позиций клас¬
сической физики, атом Резерфорда существовать не мог, так
как электроны, по законам электростатики, не могли нахо¬
диться в покое, а ускоренно двигаясь, они должны были, по
законам электродинамики, излучать энергию и в конце кон¬
цов упасть на ядро.
Таким образом, складывалась весьма запутанная и про¬
тиворечивая ситуация: эксперимент говорил в пользу плане¬
тарной (ядерной) модели атома, тогда как согласно из¬
вестным физическим законам такой атом существовать не
мог. Выход был найден Н. Бором, теория которого опира¬
лась на модель атома, предложенную Резерфордом, эмпири¬
чески установленные закономерности в атомных спектрах
и гипотезу М. Планка. На последней надо остановиться
особо.
Рождение кванта
На протяжении шести лет берлинский профессор Макс
Планк занимался проблемой равновесного электромагнит¬
ного излучения абсолютно черного тела. Он искал единую
формулу распределения энергии в спектре этого излучения.
До него были известны формулы, описывающие два край¬
них случая-испускания длинных и коротких волн. Общее же
решение было неизвестно. После долгих раздумий и колеба¬
ний Планк пришел к выводу, что проблема может быть ре¬
шена, если допустить, что энергия испускается не непрерыв¬
но, а отдельными порциями {квантами), причем энергия
одного кванта (е) равна:
е = hv (1)
где v-частота излучения, a h- некая универсальная постоянная, на¬
званная впоследствии постоянной Планка, h = 6,62559 х
х 10"34 Дж с.
7
Сам Планк говорил о своей гипотезе весьма скромно:
«математический прием», «рабочее предположение» и т.д.
«Это была чисто формальная гипотеза,-писал он Р. Вуду,-и,
по правде говоря, я не ожидал от нее бог весть чего, разве
лишь одного-чтобы любой ценой получился положи¬
тельный результат». Этот результат был доложен Планком
на заседании Немецкого физического общества 14 декабря
1900 г.
«На следующий день утром,-вспоминал он позже,-меня
разыскал мой коллега Рубенс и рассказал, что после заседа¬
ния, глубокой ночью, он сравнил мою формулу с данными
своих измерений и всюду нашел радующее согласие». Ра¬
дующее, впрочем, скорее экспериментатора Рубенса, полу¬
чившего, наконец, желанную формулу, чем теоретика-клас-
сика Планка, воспитанного на принципе «природа не делает
скачков» и отстаивавшего на страницах своей докторской
диссертации 1879 г. мысль о том, что атомистические
взгляды на строение материи приводят к противоречиям.
Но, как бы то ни было, решающий шаг был сделан.
«В раннюю пору моего пребывания в Манчестере, весной
1912 года,-вспоминал Н. Бор,-я пришел к убеждению, что
строение электронного роя в резерфордовском атоме упра¬
вляется квантом действия (постоянной Планка h)».
Постулаты Бора
Первое, что осознал Бор,-эго неприменимость законов
классической физики в микромире. На все возражения, сво¬
дившиеся к выводу о неизбежности падения электрона на
ядро, он отвечал: «Но ведь атом все-таки устойчив!».
Как-то в начале февраля 1913 г. Бор поделился своими
раздумьями о строении атомов со спектроскопистом X. Хан¬
сеном и тот его спросил: «А как твоя теория объясняет
спектральные формулы?» Этот вопрос оказался для Бора
неожиданным. «Я ничего не знал ни о каких спектральных
формулах»,-признавался он впоследствии.
8
В тот же день Бор разыскал в книге И. Штарка «Прин¬
ципы атомной динамики» спектральную формулу Бальмера,
описывающую серию линий в видимой части спектра ато¬
марного водорода:
где v-частота излучения, с-скорость света, п и к - целые числа,
причем п = 2, а к = 3, 4, 5, 6,...; Я-так называемая постоянная
Ридберга (R = 109677,576 см”1).*
«Как только я увидел формулу Бальмера,-вспоминал
Бор,-мне все тут же стало ясно».
Теория атома Бора была основана на следующих посту¬
латах :
I. В изолированном атоме существуют такие состояния
движения электрона, в которых он не излучает энергии. Эти
состояния называются стационарными. Каждое такое со¬
стояние характеризуется определенной энергией Еп, где
и-целое число, нумерующее возможные стационарные со¬
стояния.
II. Переход электрона из одного стационарного состоя¬
ния (с большей энергией Еп) в другое (энергия которого Ек
меньше) сопровождается испусканием кванта монохромати¬
ческого излучения, частота которого определяется следую¬
щим условием:
Еп — Ек — hv (2)
Атом способен поглощать квант излучения, если энергия
этого кванта в точности равна разности энергий каких-либо
двух стационарных состояний атома.
* Мы привели формулу Бальмера в том виде, как ее предло¬
жил записывать в 1890 г. шведский физик И. Ридберг и как ее
впервые увидел Бор.
9
Если теперь перейти к наглядному образу электрона,
движущегося по определенной орбите вокруг ядра, то при¬
веденные постулаты можно сформулировать так:
I. В атоме существуют орбиты, вращаясь по которым
электрон не излучает.
II. Излучение происходит только при перескоке электро¬
на с одной стационарной орбиты на другую.
Здесь следует обратить внимание на совершенно неклас¬
сический характер этих постулатов: с одной стороны, Бор
ввел чуждые классике представления о квантовых скачках
и стационарных состояниях, которые согласно электродина¬
мическим законам никак не могли появиться в системе
«ядро-электрон», а с другой, он нарушил привычную взаи¬
мосвязь между частотой излучения и частотой вращения
движущегося заряда (электрона). В классической физике бы¬
ло установлено, что частота колебаний заряда равна частоте
испускаемого им излучения. В теории же Бора этой связи
просто не было, для процесса излучения совершенно несу¬
щественно, как часто облетает электрон ядро, важна лишь
разность энергий стационарных состояний, между которыми
происходит квантовый скачок.
Для определения размеров стационарных орбит (которые
Бор поначалу рассматривал как круговые) датский физик
предположил, что момент импульса электрона (I) квантует¬
ся, т. е., как и энергия принимает лишь вполне определенные
значения, кратные постоянной Планка:
Для плоской круговой орбиты I = mvr (рис. 1) и тогда:
mvr = п-
2п
(3)
10
Рис. 1. К определению момента количества j
движения ■ классической механике.
Это условие выделяет из всех
мыслимых круговых орбит опре¬
деленные, названные Бором раз¬
решенными или стационарными.
Объединяя это условие с условием
частот (2), а также учитывая из¬
вестное соотношение
Ze2
к
указывающее на равенство центростремительной и кулонов-
ской сил, получаем следующую систему уравнений:
h
mvr = п
2я
mv2 е
г
г г2
Отсюда легко получить значения энергии Е„ (учитывая,
2 2
mir ч
что Е = ) и радиусов г стационарных орбит:
2 г
mZ2e4
П2
(4а)
Z- заряд ядра, для атома водорода Z = 1 и А = —— =
2 К
= 1,054 10-34 Дж с.
А2 ,
г = — п2 (46)
11
Из формулы (4а) следует, что энергетические состояния
водородного атома образуют определенную последователь¬
ность (энергетические уровни). Состояние, отвечающее значе¬
нию п = 1, называется основным или нормальным, все
остальные состояния (п > 1) называются возбужденными.
По мере возрастания п энергетические уровни сближаются,
и так как возможные значения полной энергии в стацио¬
нарных состояниях атома приняты отрицательными, то при
п -► оо, Е„ -► 0, а г„ -► оо. Электрон в этом случае оказывается
удаленным от ядра на бесконечное расстояние и поэтому не
связанным с ним. Энергия, необходимая для удаления элек¬
трона из атома водорода, находящегося в основном состоя¬
нии, равна:
— Е1 = —Ех = 13,6 эВ (или 1313,15 кДж/моль)
Кроме того, из формулы (46) нетрудно найти радиус пер¬
вой боровской орбиты:
А2
г. = —— = 5,281-10"2 нм
те2
Спектр водородного атома
Если излучение возникает при переходах электрона со
всевозможных уровней к на какой-то определенный уровень
п{к > п), то получается серия спектральных линий.
Например, если п = 2, а к = 3, 4, 5, 6, ..., то это-серия
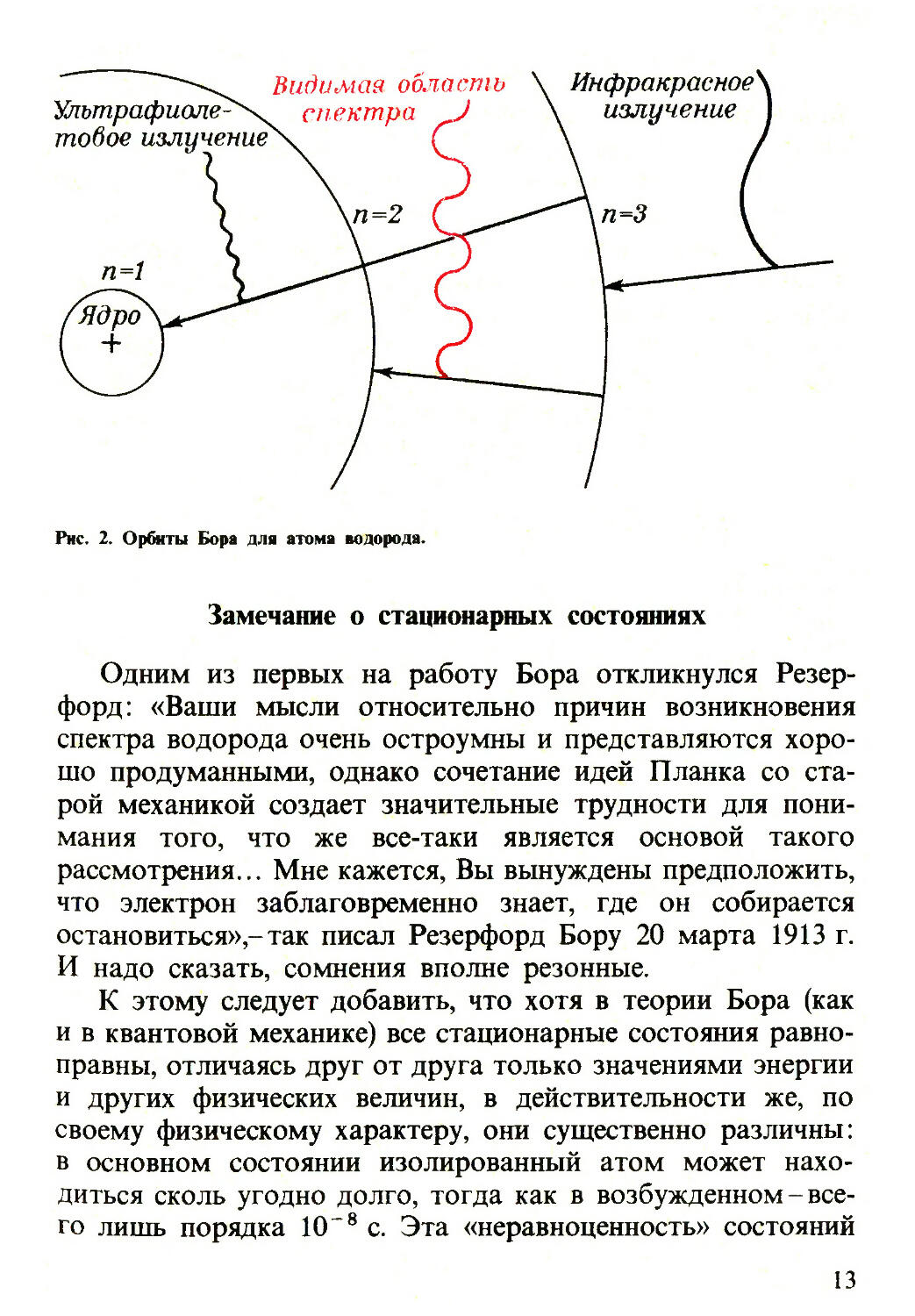
Бальмера (рис. 2).
Отсюда следует формула Бора для частот излучения
те4 ( 1 1 \
атома водорода: v='^r(^2которая позволяет
теоретически определить постоянную Ридберга (R — ■ )
\ 4яЙ3с /
и частоты спектральных линий.
12
Замечание о стационарных состояниях
Одним из первых на работу Бора откликнулся Резер¬
форд: «Ваши мысли относительно причин возникновения
спектра водорода очень остроумны и представляются хоро¬
шо продуманными, однако сочетание идей Планка со ста¬
рой механикой создает значительные трудности для пони¬
мания того, что же все-таки является основой такого
рассмотрения... Мне кажется, Вы вынуждены предположить,
что электрон заблаговременно знает, где он собирается
остановиться»,-так писал Резерфорд Бору 20 марта 1913 г.
И надо сказать, сомнения вполне резонные.
К этому следует добавить, что хотя в теории Бора (как
и в квантовой механике) все стационарные состояния равно¬
правны, отличаясь друг от друга только значениями энергии
и других физических величин, в действительности же, по
своему физическому характеру, они существенно различны:
в основном состоянии изолированный атом может нахо¬
диться сколь угодно долго, тогда как в возбужденном - все¬
го лишь порядка 10- 8 с. Эта «неравноценность» состояний
13
(хотя все они полагаются стационарными) получила свое
объяснение только в квантовой теории поля.
В поисках гармонии
Существенный вклад в развитие теории Бора внес
А. Зоммерфельд, который, «следуя, как когда-то Кеплер при
изучении планетной системы, внутреннему чувству гармо¬
нии», разработал представление об эллиптических орбитах,
введя для них соответствующее условие квантования, кото¬
рое заменило боровскую формулу (3):
f pdq = nh (5)
где р- импульс, а <?-координаты электрона.
Не останавливаясь подробно на теории Зоммерфельда*,
отметим только, что она описывала движение электрона
с помощью двух квантовых чисел-главного (л) и орбиталь¬
ного (/) и, кроме того, учитывала возможность различной
ориентации плоскости орбиты в пространстве. Главное
квантовое число (л) характеризовало при этом диаметр ор¬
биты, орбитальное (/)- степень ее вытянутости и магнитное
квантовое число (w)-ориентацию орбиты в пространстве.
Мы не будем, однако, разбирать этот вопрос досконально,
так как в квантовой механике смысл квантовых чисел иной
(о чем см. гл. II).
Новые трудности
Несомненно, теория Бора - Зоммерфельда явилась круп¬
нейшим достижением физики. Наличие в атомах дискретных
состояний было подтверждено экспериментально в опытах
* Более детальные сведения о ней читатель сможет получить
в книге: М. Борн. Атомная физика. М.: Мир, 1970, с. 135-153.
14
Д. Франка и Г. Герца (1913 г.). Серьезным успехом этой тео¬
рии стало также вычисление постоянной Ридберга для водо¬
родоподобных систем и объяснение структуры их линей¬
чатых спектров. В частности, Бору удалось правильно
объяснить серии спектральных линий иона Не+, до того
приписываемые водороду. Теория Бора-Зоммерфельда
объяснила физическую природу характеристических рентге¬
новских спектров, расщепление спектральных линий в силь¬
ном магнитном поле (так называемый нормальный эффект
Зеемана) и другие явления.
Однако с момента создания этой теории были ясны и ее
недостатки. Так, она приводила к существенным трудностям
при описании электронной структуры многоэлектронных
атомов и молекул, требовала введения явно искусственных
предположений при рассмотрении интенсивностей спек¬
тральных линий и т.д.
По мере возрастания числа трудностей при использова¬
нии теории Бора-Зоммерфельда, становилось очевидным,
что она является лишь переходным этапом на пути созда¬
ния теории микромира и необходимо дальнейшее углубле¬
ние наших представлений о природе вещества.
Природа говорит языком математики
Г. ГАЛИЛЕЙ
Глава I
ЭЛЕКТРОН-ВОЛНА И ЧАСТИЦА
НЕИЗБЕЖНОСТЬ НЕВЕРОЯТНОГО
Электрон - волна ?
Начнем с описания следующего эксперимента. Элек¬
троны определенной энергии, вылетая из источника, пооди¬
ночке проходят через маленькие отверстия в поставленной
на их пути преграде, а затем попадают на фотопластинку
или на люминесцирующий экран, где оставляют след. После
проявления фотопластинки на ней можно увидеть совокуп¬
ность чередующихся светлых и темных колец (рис. 3), т.е.
дифракционную картину, которая представляет собой до¬
вольно сложное физическое явление, включающее как соб¬
ственно дифракцию (т.е. огибание волной препятствия), так
и интерференцию (наложение) волн. Не останавливаясь на
детальном рассмотрении этих явлений, отметим лишь сле¬
дующие моменты:
-и дифракция и интерференция, наблюдаемые в по¬
добных экспериментах с электронами, говорят о нали¬
чии у них (и, вообще,-у микрочастиц) волновых
свойств, ибо только волны способны огибать пре¬
пятствия и налагаться друг на друга в местах встречи;
-даже когда электроны проходят через отверстия по¬
одиночке, т.е. с большим временным интервалом, ре¬
зультирующая дифракционная картина остается такой
же, как и при «массированном обстреле», что говорит
о наличии волновых свойств у каждого отдельного
электрона;
16
-чтобы объяснить дифракцию электронов, необходи¬
мо сопоставить с их движением какую-то волновую
функцию, свойства которой должны определять на¬
блюдаемую дифракционную картину. Но раз есть вол¬
новая функция, то должно быть и волновое уравнение,
решением которого эта функция является.
Для удобства изложения, мы напишем сначала не само
уравнение, а его решение применительно к движению сво¬
бодного электрона.
Формула плоской волны
В силу ряда причин, на которых мы здесь не будем оста¬
навливаться, одномерному свободному движению электрона
в направлении оси х можно сопоставить плоскую монохро¬
матическую волну:
*Р = ,4cos [2я ( vt) ]
А,
X
или 'Р = Asin [2тс (- vt) ]
А,
где и V,-соответственно, амплитуда (с максимальным абсо¬
лютным значением А), длина и частота волны, t- время.
Вводя понятия круговой
частоты со = 2rcv и волнового
— 2п — —
вектора к = —п (где и-еди-
X,
ничный вектор, указываю¬
щий направление перемеще¬
ния плоской волны), приве¬
денные формулы можно пе¬
реписать так:
Рис. 3. Дифракция электронного пучка.
2-918
17
Ч* — Acosikx — cot)
и (6)
¥ = /Isin (kx - oot)
Стоящее под знаком тригонометрической функции выра¬
жение (кх - соt) называют фазой волны и обозначают бук¬
вой ф. Формулы (6) удобно записать в эквивалентной ком¬
плексной форме:
= A (cos ф + i sin ф) = Ае‘9 (7)
где Л-может быть также комплексным.
Математическое пояснение. При написании формулы (7) мы
воспользовались известным соотношением Л. Эйлера
ещ = cos ф + / sin ф (8)
с которым нам еще не раз придется встретиться.
Заметим (это пригодится далее), что функция е'9 периодична
с периодом 2т (п = 0, + 1, ±2,...).
Пока мы сделали только первый шаг к получению
волновой функции свободного электрона-написали форму¬
лу плоской волны (7). Прежде чем сделать следующий
шаг-несколько замечаний физического и исторического
характера.
Интенсивность волны
Когда волны интерферируют, интенсивность результи¬
рующей волны, пропорциональная квадрату модуля ее ам¬
плитуды |'Р|2, зависит от того, совпадают или не совпадают
фазы налагающихся волн. В описанном выше эксперименте
физически наблюдаемой характеристикой является почерне¬
ние фотопластинки, которое зависит от числа электронов,
попавших на единицу ее площади. Это наводит на мысль,
что вероятность попадания электрона в ту или иную точку
18
пластинки пропорциональна |VP|2. Далее мы рассмотрим эту
идею более детально.
В поисках электронных оболочек
Итак, электрон обладает волновыми свойствами. Этот
факт был сначала предсказан выдающимся французским фи¬
зиком JI. де Бройлем в 1924 г., а затем установлен экспери¬
ментально в 1927 г. американцами Дж. Дэвиссоном и
А. Джермером.
J1. де Бройль предположил, что свободно движущемуся
электрону с импульсом р и энергией Е можно сопоставить
волну с волновым вектором к и частотой со*, причем:
- h
р = %к или р = - (9)
А.
и Е = hcо (10)
Эти соотношения де Бройля сыграли выдающуюся роль
в истории создания квантовой физики.
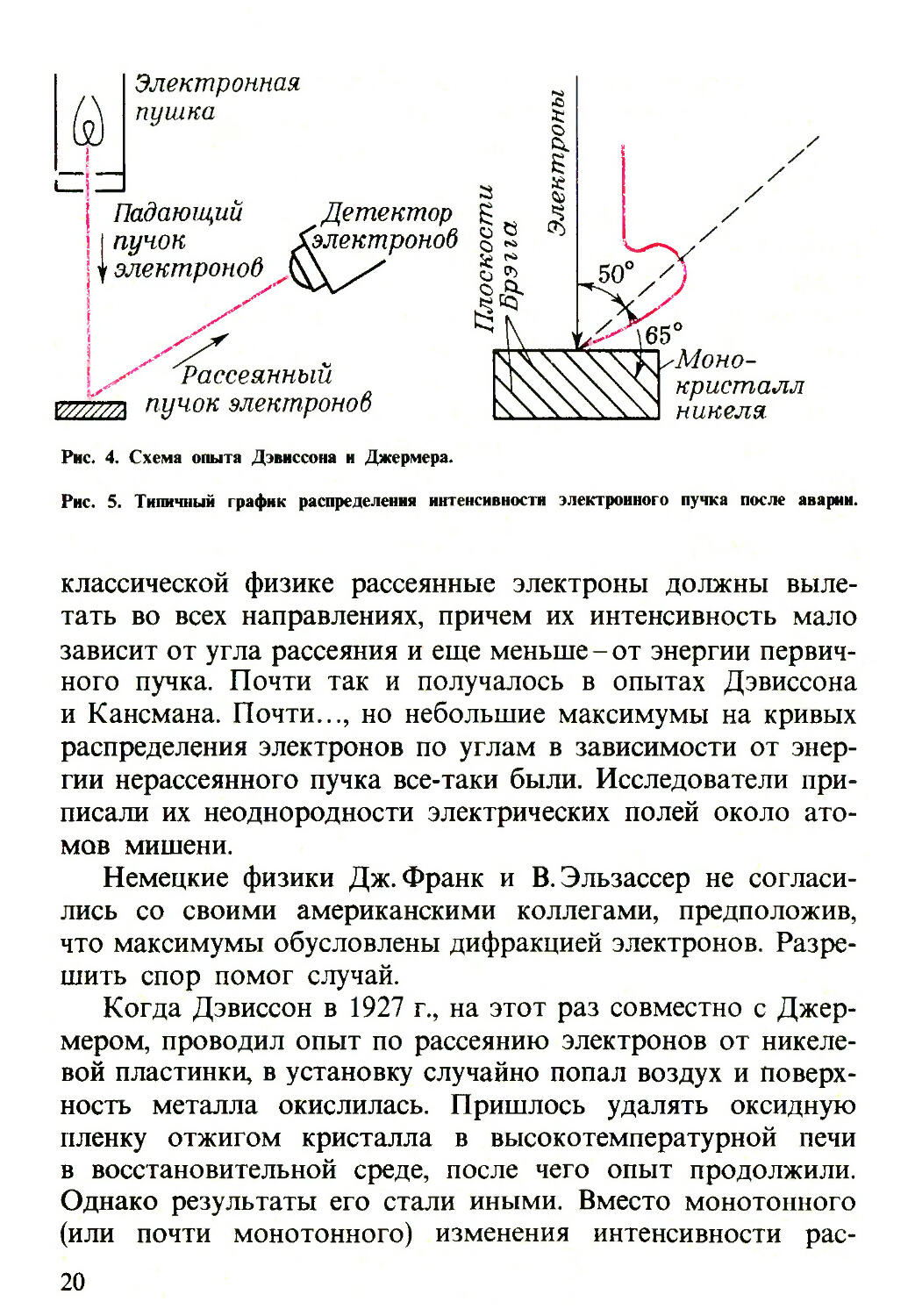
Теперь об эксперименте Дэвиссона и Джермера. Понача¬
лу Дэвиссон искал ... электронные оболочки атомов, а точ¬
нее, изучая отражение электронов от твердых тел, он стре¬
мился «прощупать» конфигурацию электрического поля,
окружающего отдельный атом. В 1923 г. совместно со
своим учеником Г. Кансманом он получил кривые распреде¬
ления рассеянных электронов по углам в зависимости от
скорости первоначального (нерассеянного) пучка. Схема
опыта показана на рис. 4. В этой установке можно было из¬
менять энергию первичного пучка, угол падения на мишень
(поверхность металла) и положение детектора. Согласно
* Мы не рассматриваем здесь причин, побудивших де Бройля
сделать такое предположение. Вопрос этот сложный, и между исто¬
риками физики до сих пор нет единого мнения.
2*
19
Электронная
пушка
\ Падающий
! I пун
| эле
Детектор S
Электронов §
! Рассеянный
У7/////Л пучок электронов
Рис. 4. Схема опыта Дэвиссона и Джермера.
Рис. 5. Типичный график распределения интенсивности электронного пучка после аварии.
классической физике рассеянные электроны должны выле¬
тать во всех направлениях, причем их интенсивность мало
зависит от угла рассеяния и еще меньше - от энергии первич¬
ного пучка. Почти так и получалось в опытах Дэвиссона
и Кансмана. Почти..., но небольшие максимумы на кривых
распределения электронов по углам в зависимости от энер¬
гии нерассеянного пучка все-таки были. Исследователи при¬
писали их неоднородности электрических полей около ато¬
мов мишени.
Немецкие физики Дж. Франк и В.Эльзассер не согласи¬
лись со своими американскими коллегами, предположив,
что максимумы обусловлены дифракцией электронов. Разре¬
шить спор помог случай.
Когда Дэвиссон в 1927 г., на этот раз совместно с Джер-
мером, проводил опыт по рассеянию электронов от никеле¬
вой пластинки, в установку случайно попал воздух и поверх¬
ность металла окислилась. Пришлось удалять оксидную
пленку отжигом кристалла в высокотемпературной печи
в восстановительной среде, после чего опыт продолжили.
Однако результаты его стали иными. Вместо монотонного
(или почти монотонного) изменения интенсивности рас¬
20
сеянных электронов от угла наблюдались ярко выраженные
максимумы и минимумы, положение которых зависело от
энергии электронов. На рис. 5 показан один из таких
максимумов.
Причина столь резкого изменения картины рассеяния по¬
сле аварии состояла в образовании в результате отжига мо¬
нокристаллов никеля, которые служили своего рода дифрак¬
ционными решетками. Если де Бройль прав и электрон
обладает волновыми свойствами, то картина рассеяния дол¬
жна напоминать рентгенограмму Лауэ. А рассчитывать
рентгенограммы к тому времени уже умели, формула Брэг¬
га была известна. Так, для случая, представленного на рис. 5,
угол а между плоскостями Брэгга и направлением макси¬
мального рассеяния электронов составляет 65°.Измеренное
рентгенографическим методом расстояние а между плоско¬
стями в монокристалле Ni равно 0,091 нм. Уравнение Брэг¬
га, описывающее положение максимумов при дифракции,
имеет вид: nA, = 2asina (п-целое число). Принимая п = 1
и подставляя экспериментальные значения а и а, получаем
для А,:
X - 2-0,091 -8т65° = 0,165 нм
Формула де Бройля
h h h
Р ™ “ 1/2т£„н
дает:
6,63-ю-34
А- = —= 0,166 НМ
|/2-9,1 -10-31 -8,64-10“18
что превосходно согласуется с экспериментом.
Впоследствии аналогичные результаты были получены
Г. Томсоном (1928 г.) и в 1930-х гг.-многими другими физи¬
ками. По словам Э. Шредингера: «Некоторые исследователи
21
(Дэвиссон и Джермер и молодой Томсон (сын Дж. Дж. Том¬
сона-Я. Д.)) приступили к выполнению опыта, за который
еще несколько лет назад их бы поместили в психиатриче¬
скую больницу для наблюдения за их душевным состоя¬
нием. Но они добились успеха!» В итоге, наличие у электро¬
на волновых свойств стало твердо установленным экспери¬
ментальным фактом.
Что же такое электрон?
Но не будем торопиться объявлять электрон волной.
Обратимся вновь к описанному выше опыту, точнее, к его
заключительной части. Каждый электрон, попадая на реги¬
стрирующую пластинку, вызывает почернение только
в одном определенном месте ее поверхности, т.е. в одном
«зерне» фотослоя, что указывает на корпускулярную приро¬
ду электрона.
Выходит, что в одних условиях,-скажем, проходя через
дифракционную решетку,-электрон ведет себя как волна,
тогда как в других,-взаимодействуя с фотопластинкой,-как
частица (корпускула).
Сэр У. Брэгг-старший подытожил ситуацию так: по поне¬
дельникам, средам и пятницам электрон ведет себя как вол¬
на, по вторникам, четвергам и субботам-как частица, в вос¬
кресенье же он отдыхает.
Впоследствии, как мы увидим, были предложены и дру¬
гие, не менее глубокие обобщения.
С позиций классической физики указанная двойствен¬
ность в поведении электрона кажется совершенно невероят¬
ной, ибо речь идет о совмещении в одном и том же объекте
несовместимых свойств. Действительно, даже беглое сравне¬
ние волн и частиц выявляет их коренные различия. Волна
способна огибать препятствия, частица-нет, волна занимает
все доступное ей пространство, частица-локализована, т.е.
в каждый момент времени находится в определенном месте
(«точке») пространства и т.д. и т.д. Впрочем ...?!
22
Идея королевского звездочета
Впрочем, некая формальная общность между частицами
и волнами все-таки есть. Она была впервые выявлена заме¬
чательным ирландским математиком, механиком и астроно¬
мом сэром У. Р. Гамильтоном в 1833 г. Его идея, изложен¬
ная в работе «Об общем методе выражения путей света
и планет с помощью коэффициентов некоторой характери¬
стической функции», состояла в следующем.
Изложение классической механики начинается обычно
с законов Ньютона. Но можно начать и «с другого конца»,
а именно, с формулировки весьма общего утверждения, име¬
нуемого принципом наименьшего действия. Согласно этому
принципу реальному движению механической системы (в от¬
личие от всех других ее мыслимых движений) отвечает эк¬
стремальное (а для достаточно малого промежутка времени
At = t2 — ^-минимальное) значение интеграла, называемо¬
го действием
S = I Lit
^1
где L- некоторая функция координат, скоростей и, вообще говоря,
времени, именуемая функцией Лагранжа.
Как показал Гамильтон, любой величине в механике от¬
вечает аналогичная ей величина в геометрической оптике.
Так, распространение плоской волны можно представить
как перемещение в пространстве поверхности постоянной
фазы: ф = const. В то же время движению системы тождест¬
венных материальных точек вдоль пучка траекторий можно
сопоставить перемещение в пространстве некоторой поверх¬
ности постоянного действия: S = const.
Аналогия «фаза-действие» может быть продолжена, тог¬
да «подобными» окажутся такие физические величины, как
23
8S dtp
dt ’ dt
энергия и частота, а также импульс и волновой вектор
со = —
~к = Vtp
Р = VS;
где V («набла-оператор»)-символ, введенный У.Гамильтоном:
v = + +
(об операторах см. далее).
Обнаруженная Гамильтоном «оптико-механическая ана¬
логия», без малого 100 лет не привлекала к себе практически
никакого внимания. Полученные английским ученым анали¬
тические результаты были затем использованы К. Якоби
в теоретической механике и X. Брюнсом в оптике (теория эй¬
конала). Аналогия оказалась разъятой, на нее никто, кроме,
может быть, проницательного Ф. Клейна, в XIX в. и в нача¬
ле XX столетия не обращал внимания. Только де Бройль су¬
мел понять ее значение для физики микромира. Именно глу¬
бокий анализ оптико-механической аналогии Гамильтона,
в совокупности с другими идеями, привел его к гипотезе
о «волне-частице», т. е. к мысли о двойственной корпуску¬
лярно-волновой природе микрообъектов.
Не лезьте электрону в душу
Что же такое электрон? Как устроена эта частица, совме¬
щающая в себе несовместимое? Вопрос этот до сих пор не
получил ответа. В 1920-х гг., на заре квантовой механики,
физики поставили перед собой другую задачу - построить
механику микромира, т. е. найти законы, определяющие дви¬
* Подобие здесь сводится к сходству соответствующих фор¬
мул, физический же смысл фигурирующих в них величин и их раз¬
мерности, разумеется, различны.
24
жение электрона в различных условиях, не прибегая к моде¬
лям, описывающим его внутреннюю структуру.
Итак, дано: микрообъект с отрицательным зарядом
и определенной массой, совмещающий в себе каким-то
образом свойства волны и частицы. Спрашивается: каковы
особенности физического и математического описания дви¬
жения такого микрообъекга?
Одним из важнейших физических принципов такого опи¬
сания является принцип неопределенности.
Закон о невозможных измерениях
В классической механике (и, вообще, в классической фи¬
зике) предполагалось, что может быть достигнуто сколь
угодно малое воздействие измерительного прибора на изу¬
чаемый с его помощью объект, так же как и сколь угодно
малая длительность измерения. Экспериментатор может как
бы «подглядеть» явление, не нарушая его естественного хо¬
да. Иными словами, физический процесс рассматривался как
нечто, происходящее само по себе, без воздействия изме¬
ряющего прибора. «Основные абстракции, используемые
классической физикой,-писал акад. В. А. Фок,-сводятся
к предположениям об абсолютном характере физических
процессов (в смысле их независимости от условий наблюде¬
ния) и возможности сколь угодно детального (в пределе-
исчерпывающе точного и всестороннего) их описания».
В квантовой механике дело обстоит иначе. С увеличе¬
нием точности измерения воздействие прибора на микроси¬
стему увеличивается и измерение одной физической вели¬
чины вносит неконтролируемые изменения в численные
значения некоторых других величин. Ка пример, если для из¬
мерения координаты электрона использовать рассеяние све¬
товых квантов - фотонов (т.е. «освещать» элек трон), то по¬
грешность такого измерения будет иметь порядок длины
25
волны света: Ах ~ X. При этом чем меньше X, тем выше
точность измерения. Однако с уменьшением длины световой
2л-к
волны, одновременно, растет импульс фотона р - —-—, ко-
А
торый частично будет передаваться электрону при столкно¬
вении с ним кванта света.
Тем самым, в численное значение импульса электрона
вносится неконтролируемое изменение Арх порядка импуль¬
са фотона. Можно показать, что величины Ах и Арх и дру¬
гие одноименные компоненты связаны при этом следующим
образом:
AxApx:j.‘-h; AyAp,2>h; AzApt > h (Н)
Эти соотношения называются соотношениями неопреде¬
ленностей В. Гейзенберга.
В итоге условия, благоприятные для точного измерения
координаты электрона (малая длина волны), оказываются
несовместимыми с условиями, необходимыми для точного
измерения его импульса (малое значение энергии кванта све¬
та) и наоборот.
Невозможность точного одновременного измерения двух
физических величин есть результат того, что электрон (как
и любая другая микрочастица) по самой своей двойственной
природе не допускает одновременной локализации в коор¬
динатном и в импульсном пространстве.
Движение без траектории
Из сказанного следует, что движение электрона не может
быть описано с помощью понятия о траектории. В самом
деле, чтобы начертить траекторию частицы, надо знать
в каждый момент времени ее положение в пространстве (г)
и скорость (у) или импульс (р = mv). Но как раз это в к ван¬
26
товой механике невозможно. С квантовомеханических пози¬
ций говорить об электронных орбитах в атомах или молеку¬
лах, как это делалось в теории Бора, не имеет никакого
смысла. Кстати, сам Бор часто вспоминал, как в 1950-х гг.
к нему после лекции подошел студент и спросил: «Неужели
действительно были такие идиоты, которые думали, что
электрон вращается по орбите?»
ВЕРОЯТНОСТНОЕ БУДУЩЕЕ ЭЛЕКТРОНА
Сначала решение - потом уравнение
Обратимся вновь к формуле (7) и преобразуем ее с уче¬
том соотношений де Бройля (т. е. фактически учтем данные
эксперимента):
Полученное выражение описывает волновой процесс, свя¬
занный с движением свободного электрона в направлении
оси х. Теперь следует найти уравнение, которому отвечает
такая плоская волна. Вообще говоря, эта задача не имеет
однозначного решения. Однако можно показать, что при¬
емлемым с физической точки зрения оказывается уравнение
вида:
vp = Ае1^ (Р* - Et)
(12)
2т дх2
(13)
То, что (12) есть решение (13) легко убедиться подстанов¬
кой. В более общем случае трехмерного движения электрона
в некотором поле U (х, у, z) уравнение (13) принимает вид:
Это уравнение лежит в основе квантовой механики и ее
приложений. Оно называется временным уравнением Шре-
дингера (1926 г.).
Не следует думать, что мы изложили вкратце путь «вы¬
вода» этого уравнения из законов классической физики
и формул де Бройля. Такой вывод невозможен, ибо кванто¬
вая механика-более общая теория и справедливость уравне¬
ния Шредингера доказывается его соответствием колоссаль¬
ному фактическому материалу квантовой физики, а также
его «внутренним совершенством», т. е. согласованностью
с общими физическими представлениями. Выше мы привели
лишь некоторые «наводящие рассуждения». Теперь немного
об истории открытия этого уравнения.
«Пенные гребни» на волнах излучения
Положение, сложившееся в физике накануне появления
работы Шредингера, без преувеличения можно назвать от¬
чаянным. «Физика теперь снова зашла в тупик, во всяком
случае для меня,-писал в мае 1925 г. В. Паули,-она стала
слишком трудной, и я предпочел бы быть комиком в кино».
Многие ученые догадывались, что волны де Бройля-это
решения какого-то неизвестного пока уравнения. Но лишь
австрийскому ученому Э. Шредингеру удалось его найти.
Эрвин Шредингер (1887-1961)-был не только выдаю¬
щимся физиком и математиком, он любил поэзию и сам пи¬
сал стихи, переводил Гомера на английский и провансаль¬
ских поэтов на немецкий язык, пробовал свои силы как
скульптор, а его работа «Что такое жизнь? (С точки зрения
физика)» получила всемирную известность. Воспитанный
в духе классической физики, испытавший сильное влияние
идей и личности J1. Больцмана, Шредингер с трудом при¬
нимал идеи Бора о квантовых скачках.
«К современной теории атома,-вспоминал австрийский
физик в 1929 г.,-я приближался очень медленно. Ее внут¬
ренние противоречия звучат как пронзительные диссонансы
28
Рис. 6. Только целое число волн де Бройля
может уложиться в орбиту Бора.
по сравнению с чистой, не¬
умолимо ясной последова¬
тельностью мысли Больцма¬
на. Было время, когда я пря¬
мо-таки готов был обратить¬
ся в бегство... Известное об¬
легчение впервые доставила
мне идея де Бройля об элек¬
тронных волнах...».
С концепцией де Бройля Шредингер познакомился бла¬
годаря статье А. Эйнштейна о квантовой теории газов
(1925 г.). «Можно полагать,-писал Эйнштейн,-что каждому
движению соответствует волновое поле... Это волновое по¬
ле-пока еще неизвестной физической природы-в принципе
должно оказывать свое влияние на движение... Думаю, что
речь здесь идет не только о простой аналогии». Под влия¬
нием этой статьи Эйнштейна Шредингер пишет летом
1925 г., т. е. всего за полгода до открытия своего волнового
уравнения, работу «К эйнштейновской теории газа», кото¬
рую заканчивает такими словами: «...Все это означает ни¬
что иное, как принятие всерьез волновой теории де Брой¬
ля-Эйнштейна движущихся частиц, согласно которой эти
частицы представляются в виде некоторых «пенных греб¬
ней» (Schaumkamm) на фоне образующих их волн излуче¬
ния».
Кроме идеи о волновой природе материи, Шредингера
привлекла в работе де Бройля оригинальная интерпретация
квантовых условий Бора-Зоммерфельда (5). По де Бройлю
устойчивыми будут лишь те орбиты, в которых укладывает¬
ся целое число волн (рис. 6). Иными словами, длина устой¬
чивой орбиты (Г) должна быть целым кратным длины волны
электрона: I = пк (где п-целое). Тогда, подставляя в (5)
формулу де Бройля (9), получаем: jl/Xdq = п
29
Это уравнение натолкнуло Шредингера на идею сформу¬
лировать теорию атома как математическую задачу на собс¬
твенные значения.
Математическое пояснение. Подобные задачи часто встре¬
чаются в математической физике. Приведем в качестве простейше¬
го примера уравнение свободных колебаний однородной струны,
закрепленной на концах:
д2и (х, t) д2и (х, г)
dt2 ~ й дх2 ( }
где и(х, Г)-смещение точек струны в момент времени t от положе¬
ния равновесия (рис. 7), а-постоянная величина. Так как струна за¬
креплена с обоих концов, то значения функции и (х, t) в точках х =
= 0 и х = I будет равно нулю:
и\х = 0= 0 и и\х = 1= 0 (16)
Эти условия называются граничными. Кроме того, имеются
определенные начальные условия, но в дальнейшем они нам не
понадобятся.
Будем искать частные решения уравнения (15) в виде
и (х, t) = X (х) T(t) (17)
т.е. методом разделения переменных.
Подставляя (17) в (15), получим:
д2Т д2Х
х д Т _ агтд Х 1 / dt2 \ ( дх>
dt дх а \ Т / \ X
В этом равенстве левая часть зависит только от t, а правая —
только от х. Такое возможно лишь когда обе части равенства не
зависят ни от х, ни от t, а равны некой постоянной, которую мы
Рис. 7. К задаче о колебании струны, закреп¬
ленной с обоих концов.
х=0 х=1
30
обозначим через — b. Тогда можно записать:
д2Т
—т + а2ЬТ=0 (18а)
д2Х
—-г + ЬХ = 0 (186)
дх
Найдем нетривиальные (т. е. ненулевые) решения уравнения
(186), для которого граничные условия (16) примут следующий вид:
XUo=XU,= 0 (19)
В итоге мы приходим к такой задаче: найти значения парамет¬
ра Ь, при которых существуют нетривиальные решения уравнения
(186), удовлетворяющие граничным условиям (19). Эти значения па¬
раметра b называются собственными, а соответствующие им реше¬
ния -собственными функциями уравнения (186).
Можно показать (мы это здесь опускаем), что нетривиальные
решения возможны только при Ь)0; тогда общее решение уравне¬
ния (186) имеет вид:
X (х) = Cjcos (]/b-x) + C2sin (1/b-х)
Приняв во внимание граничные условия (19), получаем:
Х(0) = СГ1 + С2-0 = Сх = О
и
X (/) = С2 ■ sin (|/b ■ Г) = О
Отсюда:
sin(]/&•/) = 0 и 1\ГЪ = кп (к = 0, +1, + 2,...)
Следовательно, нетривиальные решения задачи возможны
лишь при
т. е. только при некоторых, «избранных», значениях этого парамет¬
ра.
31
Аналогия с квантованными орбитами, в которых может
уместиться лишь целое число волн де Бройля, напрашивает¬
ся сама собой. Конечно, уравнение (15) не похоже на уравне¬
ние (14)-разные порядки производной по времени. Но важ¬
но другое-идея рассмотреть задачу о движении электрона
в атоме как математическую задачу на определение соб¬
ственных значений и собственных функций некоторого диф¬
ференциального уравнения. Оставалось найти это уравнение.
Исторические статьи
С задачами на собственные значения Шредингер подроб¬
но познакомился на лекциях Фрица Хазенерля-талантливо¬
го австрийского физика, трагически погибшего во время
первой мировой войны. Получая Нобелевскую премию,
Шредингер сказал: «Если б Хазенерль не был убит таким
молодым, он стоял бы сейчас на этом месте». Исключитель¬
но важную роль в работе Шредингера сыграла дружеская
помощь выдающегося немецкого математика Германа Вей¬
ля. Почти во всех основных статьях Шредингера
1926-1927 гг. можно встретить слова благодарности Вейлю
за ценные консультации при разработке методов решения
и за сообщение о современном состоянии соответствующих
математических проблем.
К лету 1925 г. почва для открытия волнового уравнения
была подготовлена. «В то время,-вспоминал в 1964 г. П. Де¬
бай,-Шредингер получил мою кафедру в университете Цю¬
риха, а я был в Техническом университете, и у нас был со¬
вместный семинар. Мы говорили о теории де Бройля
и пришли к выводу, что не понимаем ее... Поэтому я попро¬
сил Шредингера устроить для нас специальный коллоквиум.
Он начал его готовить. Между его выступлением и его пу¬
бликациями прошло всего лишь несколько месяцев».
Первая из цикла статей под общим заглавием: «Кванто¬
вание как задача о собственных значениях» поступила в ре¬
дакцию «Annalen der Physik» 27 января 1926 г. В ней было
32
дано так называемое стационарное уравнение Шредингера
(см. далее). Последняя, четвертая, публикация цикла посту¬
пила в редакцию 21 июня 1926 г., в ней содержится приве¬
денное выше временное уравнение.
“ ... what does psi really mean?”
В свое время среди физиков ходила эпиграмма на Шре¬
дингера, составленная на английском и немецком языках
и приписываемая Э.Хюккелю:
Gar Manches rechnet Erwin schon Erwin with his psi can do
Mit seiner Wellenfunktion Calculations quite a few
Nur wissen mocht'man gerne wohl But one thing has not been seen
Was man sich dabei vorstelfn soil? Just what does psi really mean?*
Действительно, вопрос о физическом смысле ^-функции
стал после открытия уравнения Шредингера одним из цен¬
тральных. Вокруг него долго велись горячие споры. Сам Шре¬
дингер связывал с волновой функцией некоторое непрерывное
распределение электрического заряда, представляя электрон
как «размазанную» в пространстве заряженную массу. «Он ду¬
мал,-писал о Шредингере М. Борн,-что осуществил возврат
к классическому мышлению. Он рассматривал электрон не как
частицу, но как некоторое распределение плотности, которое
давалось квадратом модуля его волновой функции | |2. Он
считал, что следует полностью отказаться от идеи частиц
и квантовых скачков и никогда не сомневался в правильности
этого убеждения. Я, напротив, имел возможность каждоднев¬
но убеждаться в плодотворности концепции частиц, наблюдая
за блестящими опытами Дж. Франка по атомным и молеку¬
лярным столкновениям, и был убежден, что частицы нельзя
просто упразднить. Следовало найти путь к объединению
частиц и волн. Я видел связующее звено в идее вероятности...»
* Перевод стишка: Эрвин может с помощью своей пси-функции
многое вычислить. Одно лишь неясно-что же такое пси-функция?
3-918
33
И еще одна цитата, хорошо передающая суть споров вокруг
проблемы физической интерпретации математического аппа¬
рата квантовой механики. В лекции «Современное состояние
атомной физики», прочитанной в Гамбургском университете
в феврале 1927 г. немецкий физик А. Зоммерфельд так характе¬
ризовал ситуацию в квантовой теории:«... В трехмерном про¬
странстве электрон нельзя локализовать. Это подчеркивает
Гейзенберг, а Шредингер иллюстрирует это, «размазывая» за¬
ряд электрона в сплошную пространственную массу. Лично
я не верю в этот размазанный, растекающийся электрон уже
потому, что вне атома корпускулярно концентрированные
электроны, обладающие большой скоростью, с несомнен¬
ностью могут быть установлены экспериментом. С другой сто¬
роны, неоспоримый факт, что сплошные плотности Шрединге-
ра при расчете физических и химических действий атома
оказывают неоценимую помощь и в этом смысле реальны
в большей степени, нежели точечно локализованный электрон
старой теории. Весьма возможно, что сплошную плотность за¬
ряда и связанный с нею сплошной ток заряда в теории Шредин-
гера мы должны понимать статистически в смысле нескольких
важных работ Борна...»
Согласно интерпретации Борна квадрат модуля волно¬
вой функции | *Р (х, у, z, t)\2 определяет вероятность обнару¬
жения электрона в момент времени t в «точке» пространства
с координатами (х, у, г). Иными, более точными, словами,
выражение | У (х, у, z, t)\2dV есть вероятность локализации
электрона к моменту времени t в элементе объема dV
окрестности точки (х, у, z). Тогда | ¥ (х, у, z, t) |2 следует по¬
нимать как плотность вероятности локализации электрона
в окрестности точки (х, у, z) к моменту времени t.
Возвращаясь к рассмотренным в начале главы дифрак¬
ционным экспериментам, можно сказать, что почернение фо¬
топластинки, пропорциональное числу электронов, попадаю¬
щих на единицу ее площади, определяется квадратом модуля
34
волновой функции (квадратом модуля амплитуды волны де
Бройля).
В итоге физически наблюдаемой величиной оказывается
не сама Т-функция, но | |2, поэтому возможное наличие
в выражении для ¥ мнимой единицы не приводит к проти¬
воречиям в теории.
Поле, описываемое функцией ¥(х, у, z),-ero иногда на¬
зывают дебройлевским в отличие от электромагнитного
и гравитационного полей-не силовое. Если бы оно описы¬
вало, например, карточную игру, то в результате прохожде¬
ния такой «волны удачи» по поверхности Земли в один мо¬
мент времени вероятность выигрыша для данного игрока,
скажем в Лас-Вегасе, могла быть много больше, чем в Мон-
те-Карло, тогда как в следующий момент-наоборот
и т.д.
С позиций физика-классика
Заметим, что в классической физике, в тех случаях, когда
условия задачи были не полностью известны и по не¬
известным параметрам проводилось усреднение, также ис¬
пользовались вероятностные представления. Но там они
оказывались как бы внешними по отношению к физическим
законам. В сознание теоретика-классика прочно вошла
мысль о первичности однозначно предсказывающих законо¬
мерностей по отношению к вероятностным, ибо последние
характеризуют наше незнание и, как следствие, неполноту
формулировки задачи. Вероятности в классической физике
считались принципиально устранимыми благодаря более
тонким опытам и более точным измерениям.
Облако вероятности
Часто вероятность локализации электрона в пространстве
изображают наглядно с помощью множества точек, похожего
на облако (рис. 8). Чем больше точек в некоторой области, т. е.
чем гуще облако, тем большую часть времени проводит там
з*
35
Рис. 8. Электронное облако.
электрон. Там же, где вероятность
застать его меньше, точечное облако
разреженней.
Физики и химики часто употреб¬
ляют выражение «электронное обла¬
ко», «распределение электронной
плотности» и т.п. Мы тоже будем
их использовать. Однако следует помнить, что электронное
облако-это не наглядный образ самого электрона, «разма¬
занного» в пространстве, а наглядное изображение распреде¬
ления вероятности его возможной локализации в различных
пространственных областях, т. е., в конечном счете, электрон¬
ное облако характеризует состояние движения электро¬
на.
Для лучшего понимания сказанного приведем такую ана¬
логию. Допустим, перед нами очень много фотографий
одного из участников футбольного матча, скажем, вратаря.
Используя эти фотографии (или кинопленку, или, наконец,
визуальное наблюдение), можно нанести на чертеж все точ¬
ки футбольного поля, где во время матча в разные моменты
застал вратаря объектив фотографа. Ясно, что наибольшее
число отметок придется на участок поля, непосредственно
примыкающий к воротам,-там вратарь бывает чаще, там
больше поэтому вероятность его обнаружения. Полученное
таким способом «вратарное облако», конечно не будет обра¬
зом самого вратаря, а будет характеризовать его движение
по футбольному полю во время матча. Правда, в отличие
от электрона, вратарь-макрообъект и его движение можно
было бы представить и по-другому, начертив, к примеру,
траекторию его перемещений и избежав, тем самым, введе¬
ния вероятностных представлений. Для электрона же такой
альтернативы нет, его движение можно описать только
в терминах вероятности и потенциальной возможности.
36
Ограничения на ^-функцию
Обычно на \)/-функцию накладывают ряд ограничений
(стандартных условий). Волновая функция в области ее
определения должна быть:
- однозначна,
-непрерывна и непрерывно дифференцируема,
-конечна,
-нормирована.
Последнее условие означает следующее. Поскольку ве¬
роятность обнаружения электрона где-либо в доступной для
него области пространства, равная сумме вероятностей его
обнаружения во всех точках этой области, есть 1 (ибо это-
вероятность достоверного события, «обшарив» всю область
определения волновой функции, мы электрон обязательно
найдем), то:
J| ф(х, у,z, t)\zdV = 1 (20)
Приведенное выражение называется условием нормировки
волновой функции. Так как координаты x,y,z изменяются
непрерывно, то вместо суммы в формуле (20) стоит инте¬
грал.
Кроме того, в зависимости от особенностей системы
(например, ее симметрии, топологических свойств и т.д.)
на соответствующие волновые функции могут наклады¬
ваться дополнительные условия сверх указанных стандарт¬
ных условий.
Еще раз подчеркнем, что понятие волновой функции-
центральное в квантовой механике, и не только потому, что
она описывает распределение электронной плотности в про¬
странстве, но и потому, что она содержит в себе всю воз¬
можную, допускающую опытную проверку информацию
о состоянии микросистемы (атома, молекулы, иона и т.д.)
и позволяет вычислить вероятности возможных результатов
любых измерений.
37
Наложение состояний
Одним из основных принципов квантовой механики
является принцип суперпозиции (наложения) состояний. Суть
его заключается в следующем. Допустим, некоторая система
может находиться и в состоянии \|/х, в котором какая-то фи¬
зическая величина L имеет определенное значение Ll и в со¬
стоянии \|/2, в котором L = L2. Тогда существует состояние,
в котором измерение величины L приводит либо к значению
Lu либо-к L2: \|/ = + С2\|/2, где С* и С2-числа, во¬
обще говоря, комплексные.
Разумеется, число налагающихся состояний может быть
и больше двух: \|/х, v|/2, ..., \|/„. В этом случае можно гово¬
рить о существовании суперпозиции этих состояний:
'I' = I cj/m
т = 1
Измерение величины L в состоянии \|/ даст одно из зна¬
чений Lu L2, L3, ..., L„.
ГАРМОНИЯ ФИЗИКИ И МАТЕМАТИКИ
Квантовая механика-наука в высшей степени математи¬
зированная, поэтому для понимания дальнейшего материа¬
ла книги необходимо, хотя бы в общих чертах, познако¬
миться с ее математическим аппаратом, чему и будет
посвящен настоящий раздел.
Что такое оператор?
В январе 1926 г. М. Борн и Н. Винер высказали идею,
развитую впоследствии К. Эккартом, Дж. фон Нейманом
и другими исследователями и составляющую ныне одну из
38
основ формализма квантовой теории. Суть этой идеи со¬
стояла в сопоставлении каждой классической физической ве¬
личине некоторого оператора, обладающего определенными
свойствами.
«В сотрудничестве с Норбертом Винером,-вспоминал
Борн,-я пытался распространить теорию непрерывного
энергетического спектра на случай более общих систем (сво¬
бодные частицы) с прерывным спектром; мы развили опера¬
торное исчисление, которое очень близко примыкало к шре-
дингеровскому методу, в то время (зима 1925-1926 г., когда
Борн читал лекции в Массачусетском технологическом ин¬
ституте-И. Д.) нам не известному».
Математическое пояснение. Оператором называют прави¬
ло, с помощью которого одной функции ф может быть сопоставле¬
на другая f Символически это записывают так:
f=L ф
Оператор обозначается буквой со «шляпкой».
Например, функции х2 можно сопоставить функцию 2х с по¬
мощью оператора дифференцирования ——:
dx
2х = -у~(х2)
dx
Если для оператора L и функции v|/ (\j/ ф 0) выполняется соотно¬
шение
1\|/ = 1л|/ (21)
где L-некоторое число, вообще говоря, комплексное, то v|f назы¬
вается^ собственной функцией, а L-собственным значением опера¬
тора L. Так, рассмотренное выше уравнение (186) можно записать
в операторной форме:
ЬХ{х) = - ЬХ{х)
t д*
где L = —-.
дх2
Совокупность собственных значений оператора называют его
спектром.
39
Свойства квантовомеханических операторов
Математический аппарат квантовой механики построен
таким образом, что экспериментально наблюдаемыми зна¬
чениями физической величины могут быть только соб¬
ственные значения уравнения (21), а волновыми функциями
системы-только фигурирующие в этом уравнении соб¬
ственные функции оператора L. Чтобы это условие выпол¬
нялось, L должен обладать определенными свойствами,
а именно он должен быть линейным и самосопряженным
(эрмитовым).
Математическое пояснение, а) Линейным называется
оператор, удовлетворяющий следующему условию:
L(Citi + ^2^2) — CjLv|/j + C2Lv|/2
где С, и С2 - вещественные или комплексные числа.
Требование линейности связано с принципом суперпозиции,
б) И операторы и их собственные функции могут быть_.-ком-
плексными, т.е. включать в себя мнимую единицу i = ]/ — 1. Но
физические величины вещественны, и потому им должны соответ¬
ствовать только операторы с вещественными собственными значе¬
ниями. Это налагает на операторы дополнительное условие-они
должны быть самосопряженными или-другое название -эрмитовы¬
ми. Оператор называется эрмитовым, если выполняется следующее
соотношение:
В приведенном равенстве одновременно с добавлением к сим¬
волу оператора знака комплексного сопряжения (*), меняются ме¬
стами функции \|f и ср*, одна из них (\|/) «уходит» из-под символа
оператора, а другая (<р*) встает на ее место.
Напоминаем: знак (*) показывает, что в соответствующей ма¬
тематической величине (числе, функции, операторе и т.д.) надо вез¬
де изменить знак перед мнимой единицей. Для вещественной вели¬
чины L имеет место соотношение: If = L.
Рассмотрим некоторые примеры. Нетрудно убедиться в неэр-
d
митовости оператора дифференцирования . Действительно, если
dx
40
vj/ (x) и ф (x) обращаются в нуль на бесконечности, то, пользуясь из¬
вестной формулой интегрирования по частям (Judv = uv — fvdu),
которая в данном случае дает:
4-00 +00 +00+00 +00
d С
<р* v]fdx — ф*^\|/ = ф*у|/
dx
т оо
^Ф* = — v|/—-—(p*dx
-00 — 00
получаем: | ф* ~ \J/dx Ф | \|/ ~q>*dx.
Однако этот недостаток оператора дифференцирования легко
исправить, умножив его на мнимую единицу. Мы предоставляем
. d
читателю самостоятельно доказать эрмитовость оператора г~-.
В дальнейшем выражения вида { q>*L\\fdV и |ф*\|idV мы часто
будем записывать в более компактной форме, предложенной
П. Дираком: <ф|Ь|\|/> и <ф|\|/>. Если функция характеризуется ка¬
ким-либо индексом, то вА скобках Дирака указывают только этот
индекс, например: |ф*1хрт^К = <rc|L|m>. Условие эрмитовости
принимает тогда вид:
<ф|£|\|/> = «1|/|£|ф»* ИЛИ <n|L|m> = (<m | L | п»*
Эрмитовы операторы обладают следующими важными свой¬
ствами :
1) Их собственные значения вещественны; в самом деле:
t'bn = LA
откуда <и|L|n> = L„<n|n> = (<n|L|«>)* = L*<n|n> и L„ = L*
2) Их собственные функции \J/n и v|/m, относящиеся к различным
собственным значениям L„ и Lm (Ln Ф Lm), ортогональны:
(m | п} = J i|/*i|/ndF = 0 (m ф n)
Это также легко доказать. Действительно, если Li]/„ = Lnvj/„ и
U>m = bmxj/m, то
<m|L|n> = Ln (m I n> = (<n|L|m>)* = Lm«n|m»* = Lm<m|«>
откуда (L„ — Lm)<mjn> = 0 и, при условии Ь„ФЬт, <m|n> = О
41
Спектр оператора
Итак, квантовомеханические операторы физических вели¬
чин должны быть линейными и эрмитовыми. Оба требова¬
ния-физические по своему происхождению. Допустим, что
спектр оператора дискретен и, решая уравнение вида (21),
мы получаем набор соответствующих собственных функций
и собственных значений. При этом возможны два случая:
либо каждому собственному значению Ln отвечает одна
собственная функция \J/„, так что
Ь- Ь %■■■■
I i t
Lt, L2,..., Ln, ...
либо некоторым (или всем) собственным значениям отвеча¬
ет больше одной собственной функции:
Ъ’ ?г tn-vf„tn+,’-’tn+r-
I t I (
Ln Lz, ... ,Ln_j, Ln, Ln+I ,
В последнем случае говорят, что собственное значение
f-кратно вырождено.
Собственные функции \|/„, \|/я+1, ..., \)/и+/, соответствую¬
щие вырожденному собственному значению L„, могут быть
взаимно неортогональны. Но из них можно составить раз¬
нообразные линейные комбинации
fln'l'n + ая+1^л+1 + ••• + ая+/\|/л+/ = фп
которые также будут собственными функциями L, относя¬
щимися к т о му же собственному значению L„, причем систе¬
ма функций фп может быть сделана ортогональной.
42
От классической механики к квантовой
До сих пор мы вели общий разговор о том, какими,
должны быть операторы физических величин в квантовой
механике. Теперь поговорим о том, каковы же они на самом
деле, т.е. каков их конкретный вид.
В квантовой механике существуют определенныел прави¬
ла сопоставления линейного эрмитового оператора L физи¬
ческой величине L, имеющей классический аналог, т. е.
являющейся функцией классических переменных -хг и рк (ко¬
ординат и компонент импульса). Если оставить в стороне
некоторые «тонкости», то в простейшем виде эти правила
сводятся к следующему.
Классическое выражение для данной физической вели¬
чины записывают в канонической или (другое название) га¬
мильтоновой форме, где переменными служат координаты
и импульсы. Например, выраженная таким образом полная
энергия материальной точки, движущейся в потенциальном
поле U(x,y,z,t),-функция Гамильтона-имеет вид:
Н = —(р2х + Ру + р2) + U (х, у, z, f) (22)
2m
Для получения из классической функции Гамильтона
квантовомеханического оператора полной энергии частицы
(гамильтониана) нужно канонические переменные заменить
на операторы: х -> х, у -> у, z -> £ и рх -» рх и т. д. Таким об¬
разом, для построения нужного оператора L надо знать
прежде всего операторы координат и проекций импульса.
Операторы координат
Эти операторы самые простые-они представляют собой
умножение функции, на которую они действуют, на эту
координату
xv(/ (х, у, z) = xvj/ (х, у, z) И т. д.
43
или, короче:
х — х, у — у, z — z (23)
Оператор физической величины, являющейся функцией
только от координат, например оператор потенциальной
энергии U(x,y,z), есть также оператор умножения: U —
= U (х, у, z).
Оператор импульса
Декартовым компонентам импульса в квантовой механи¬
ке отвечают операторы:
8 8 д
Рх = - ik ; Ру = - Й И Рг - - Й (24)
ОХ су CZ
Компоненты оператора импульса образуют вектор:
* / д _ 8 ... 3 _\
Р = — / + ~^-j + ) — ~ ^ (25)
Оператор энергии
Теперь можно определить явный вид некоторых операто¬
ров. Начнем с оператора энергии (гамильтониана). Под¬
ставляя в классическую функцию Гамильтона (22) выражения
(23) и (24), получаем:
h2 ( д2 д2 д2 \
H=-^{-^ + W + ^) + Uix-y’2,=
= - v2 + U(x,y,z) (26)
2т
где У2-так называемый оператор Лапласа (лапласиан), который,
( h2 \
будучи умноженным на I ), представляет собой оператор ки-
v - е
нетической энергии частицы: Ткин = V2.
2т
44
Гамильтониан играет в квантовой механике особую
роль, ибо он практически определяет свойства системы.
В дальнейшем мы встретимся с ним не раз.
Момент импульса
Теперь обратимся к оператору момента импульса элек¬
трона. В классической механике момент импульса М опре¬
деляется как векторное произведение следующего вида:
Математическое пояснение. Существуют различные
способы перемножения векторов, из которых наиболее распростра¬
нены следующие два:
а) Скалярное произведение векторов А и В определяется так:
где ф-угол_между перемножаемыми векторами.
Если АВ = 0, то А и В называют ортогональными друг другу,
б) При перемножешга векторов по второму способу получает^
ся_не скаляр, а вектор С, перпендикулярный плоскости векторов А
и В и имеющий величину | С | ==J А_\ | B^sin ф. Направление С тако¬
во, что совокупность векторов А, В и С образует правую систему
координат:
Полученное_произведение называется векторным и обозначает¬
ся символами [А х В], [ЛВ] или А х В. Векторное произведение
можно записать через компоненты:
Обратите внимание на циклическую перестановку (чередова¬
ние) индексов. х
М = [г х р]
АВ = АХВХ + АуВу + AZBZ = |Л||В|со8ф
С
Площадь параллелограмма равна:
| С | = | А 11 В | sin ф
Нетрудно убедиться, что векторное произведение обладает
свойством антикоммутативности:
[А х В] = - [В х А]
В квантовой механике оператор момента импульса опре¬
деляется следующей формулой
М=\?хр]
или, в компонентах:
-й(,А_2£);
- - й(2"^ ~ х^); Й:= “ ''*(х¥ “ У^)
Часто оператор проекции момента на ось квантования
z удобно выразить не в декартовых, а в так называемых
сферических координатах (рис. 9). Тогда он после весьма
громоздких преобразований принимает простой вид:
М2= - Ш— (27)
Зф
В то же время вид операторов Мх и Му в этих координа¬
тах, наоборот, усложняется. А
Отвечающие оператору М2 собственные функции и соб¬
ственные значения находят из уравнения
<ЗФ (г, 0, ф)
М Ф(г, 0, ф) = — ih = М„Ф(г, 0, ф)
5ф
решение которого можно записать следующим образом:
м
Ф(г,0,ф) = С(г,0)е~^(р
где С (г, 0)-произвольная функция от г и 0.
46
Рис. 9. Так связаны между собой сфериче¬
ские и декартовы координаты (d V- элемент
объема):
О г < оо; 0 ^ 0 < к; 0 ^ ф ^ 2я; х =
= rsinOcostp; у = rsin0sm(p; г = гcos0;
dV = dxdydz = r2 sin f) dr dQdq.
Для выполнения условий
однозначности Ф(г, 0, ср) не¬
обходимо, чтобы
Ф (г, 0, ф) = Ф (г, 0, ф + 2п)
т. е.
м м
ег-~ Ф = Ф + 2л)
откуда:
м
1—Г~2п
е h =1
Это условие удовлетворяется, если MJk- целое [см. по¬
яснение к формуле (8)], т.е.:
Мг = mh (28)
где т = 0, +1, ± 2, ...
Собственные функции оператора М2, отвечающие его соб¬
ственным значениям (28) и условию нормировки
2я
J Ф*Фт^ф = 1
о
имеют вид:
Фт(Ф) -
У 2п
Уравнения, определяющие собственные функции и соб¬
ственные значения оператора квадрата момента М2, имеют
47
более сложный вид, поэтому мы ограничимся тем, что при¬
ведем только конечный результат;
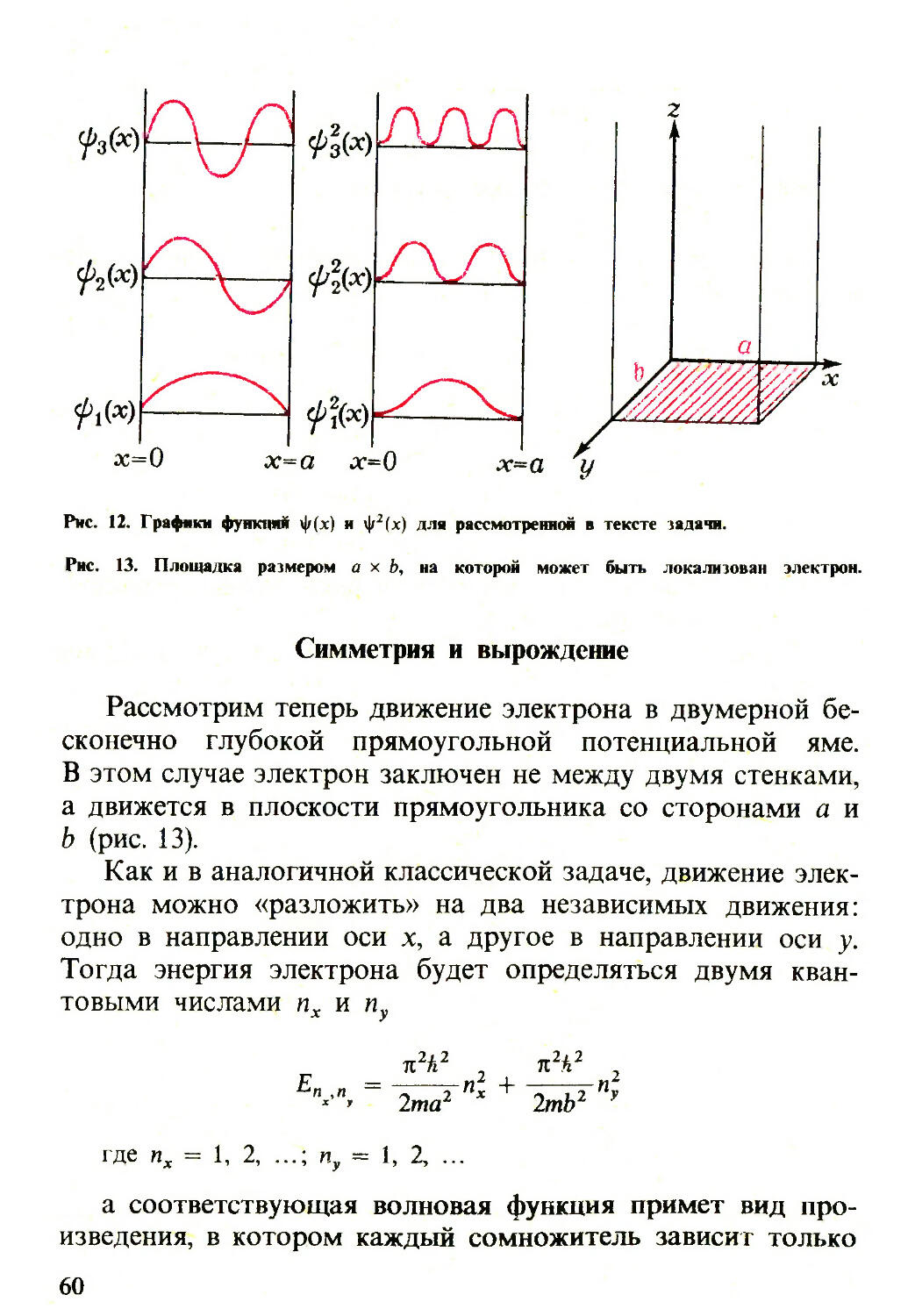
Собственные значения М2: М2 =■■■/(/ +1)А2 (/ — 0, 1, 2, ...)
Вид сомножителя @im (0) мы здесь не конкретизируем,
ввиду его сложности, заметим только, что функции Ylm(Q> Ф)>
называемые сферическими или шаровыми, удовлетворяют
условиям конечности и однозначности при целых положи¬
тельных значениях / ^ \т\. Кроме того, сферические функ¬
ции будут не только собственными функциями оператора
~ 'У А
М2, но и Mz, причем имеет место (21 + 1)-кратное вырожде¬
ние, т. е. каждому значению квадрата момента импульса (ка¬
ждому I) отвечает (21 + 1) различных собственных функций
Иными словами, при фиксированном значении момента
импульса его проекция на произвольную ось квантования
z может принимать (21 + 1) значений, так как в силу условия
/ ^ \т\, число т изменяется в интервале от — / до + /.
Хотя мы не стали выписывать общее математическое вы¬
ражение сферических функций, для лучшего понимания
дальнейшего полезно привести формулы нескольких первых
Yim (0, ф) (все функции нормированы):
Собственные функции М2: У(гй(0,<р) = &im(Q)eim4>
I = 0, т = 0 : У00
]/4п
I = 1, т = 0 : У10
т = ±1 : Yh±l = + i
48
I = 2, m = 0 : Y20 = - /-^-(3cos20 - 1)
! 16n
m = ±1 : У2-+1 = ±
m = + 2 : У2, ± 2 = ~
sin2 де'
На этом мы закончим рассказ о важнейших квантовоме¬
ханических операторах и обратимся вновь к общей теории.
От перестановки сомножителей
Намеченное выше соответствие между физическими ос¬
новами квантовой механики и теорией линейных операторов
будет неполным, если не рассмотреть вопрос о том, как на
языке операторов формируется критерий возможности
одновременного измерения двух физических величин.
Чтобы ответить на этот вопрос, следует обратить внима¬
ние на одну очень важную особенность операторов. Допу-
d
стим, мы имеем два оператора А = sin и В = дейст¬
вующих на функцию х2. Нетрудно убедиться в том, что
результат действия произведения этих операторов будет за¬
висеть от порядка сомножителей*, в самом деле:
АВ(х2) = sin-—(х2)
dx
sin 2х
но ВА(х2) = —— sin(x2) — 2xcosx2
dx
* Везде предполагается, что операторы Л и В определены на
одном и том же множестве функций.
4-918
49
В то же время произведение операторов В — ~ и С =
= С (оператор умножения на постоянную величину), дей¬
ствуя на ту же функцию х2, будет давать один и тот же ре¬
зультат независимо от порядка операторов-сомножителей:
ВС (х2) = -4—Сх2 = 2Сх
dx
СВ(х2) = С~х2 = 2Сх
ах
Поэтому говорят, что операторы могут быть коммути¬
рующими или некоммутирующими. Два оператора А и В
называются коммутирующими, если для любой ограничен¬
ной функции \|/ выполняется равенство: АЩ = ВА\\/, т.е.
АВ = В А, и некоммутирующими в случае АВ Ф В А.
Оператор [Л2? — J5.4] называется коммутатором и обо¬
значается символом [А, В].
Вернемся теперь к поставленному выше вопросу. Для то¬
го, чтобы две величины А и В могли иметь определенные
значения в некотором состоянии, описываемом волновой
функцией \|/„ (х,у, z), эта волновая функция^ очевидно, должна
быть собственной функцией операторов А и В, т. е. должны
иметь место два уравнения:
Мп = Ах\>п И = в%
Можно доказать, что это условие будет выполняться
только в случае коммутации операторов А и В: [А, В] = 0.
Таким образом, если две квантовомеханические вели¬
чины имеют одновременно определенные значения, то отве¬
чающие им операторы коммутируют, и наоборот, если двум
физическим величинам отвечают коммутирующие опера¬
торы, то эти величины будут одновременно иметь опреде¬
ленные значения. Если же операторы не коммутируют, то
соответствующие физические величины либо не могут иметь
50
одновременно определенных значений, либо имеют их толь¬
ко в особых случаях (далее мы приведем пример такого осо¬
бого случая).
Еще раз подчеркнем: коммутирующие операторы имеют
общую систему собственных функций, тогда как системы
собственных функций некоммутирующих операторов раз¬
личны (совпадать могут лишь отдельные функции).
Правда, в случае вырождения отнюдь не любая соб¬
ственная функция одного из коммутирующих операторов
обязательно будет собственной функцией другого. Но при
этом всегда можно путем составления линейных комбина¬
ций построить общую систему собственных функций двух
коммутирующих операторов (к этому мы еще вернемся).
Теперь для лучшего понимания сказанного приведем не¬
сколько примеров.
Как найти коммутатор?
Нетрудно доказать, что одноименные координата и про¬
екция импульса не могут иметь одновременно определенных
значений, так как отвечающие этим величинам квантовоме¬
ханические операторы не коммутируют:
[х,рх]Н>(х) = (хрх-рхх)\|/(х)= - ^x^--£-x^\|f(*) = й\|/(х)
[х, /У = /А Ф о
Аналогично:
[£ Ру] = &Р-А = й Ф О
Приведем еще несколько важных примеров коммута¬
ционных соотношений:
[М2, MJ = [М2, Му] = [М2, MJ = О
[Мд,Му] = ihMz; [Му,Мг] = ШМХ; [Mz. Мх] = ihMy
4* 51
Таким образом, проекции момента импульса Мх, Му и
Мг не могут иметь одновременно определенные значения.
Исключением является случай, когда момент импульса ра¬
вен нулю (соответственно, и / = 0), так как при этом Мх =
= Му = Mz = 0. Это и есть упомянутый выше «особый
случай».
В то же время квадрат момента импульса и одна из его
проекций (скажем, Mz) могут иметь одновременно опреде¬
ленные значения. Поэтому-то, говоря о понятии момента
импульса в квантовой механике, мы рассматривали только
собственные функции двух операторов: М2 и Mz.
Важное неравенство
Итак, мы выяснили условия, при которых возможно
одновременное измерение двух физических величин. Теперь
обратимся к случаю, когда такое измерение невозможно,
так как соответствующие операторы не коммутируют. Вы¬
ясним, как связаны между собой неопределенности значений
одновременно неизмеримых величин А и В.
За меру «разброса» величины около среднего значения
примем ее дисперсию, которая определяется средним квадра¬
том отклонения этой величины от ее среднего значения, т. е.:
ДЛ2 = (А - Af = А2 - (А)2
Как было показано Вейлем и Шредингером, для физиче¬
ских величин А и В, которым отвечают некоммутирующие
операторы А и В, имеют место следующие соотношения:
[А, В] = (АВ - В А) = iC
и TiA2 АВ2 ^ —С2
4
Последняя формула устанавливает минимально возмож¬
ное значение произведения неопределенностей величин
А и В.
52
Для частного случая (А = х, В = рх и С = К) получаем:
Лх2 • Apl ^ —h2
х 4
Это полностью согласуется с приведенным ранее соотно¬
шением неопределенностей Гейзенберга.
Математические ожидания
Важнейшей формулой квантовой механики является фор¬
мула, определяющая средние значения или, как еще говорят,
математические ожидания физических величин.
Если состояние микросистемы описывается волновой
функцией \Jjfp, являющейся одной из собственных функций
оператора L, то в этом состоянии физическая величина L
имеет определенное значение L„, которое мы и должны по¬
лучить экспериментально.
Когда же волновая функция \|/ неА совпадает ни с одной
из собственных функций оператора L, то в этом состоянии
величина L не имеет определенного значения. При измере¬
ниях, проводимых над системой, может с определенной ве¬
роятностью получиться любое собственное значение L„.
Допустим, мы имеем большое число совершенно одина¬
ковых экземпляров системы, находящихся в одном и том же
состоянии, описываемом функцией v|/ (например, число Аво-
гадро атомов водорода). Производя измерение величины L
в каждой системе, мы получим набор значений этой вели¬
чины и определим ее среднее значение (математическое ожи¬
дание) L в состоянии \|/, которое выражается следующей
формулой:
L = 129)
Если ^-собственная функция оператора L (т.е. Ь\|/ =
= Ljty), то L= fy*LtydV = Lraj\J/*vMF = Lm, т.е. величина L
имеет вполне определенное значение (дисперсия AL2 = 0),
равное одному из собственных значений оператора L.
5?
Записывая формулу (29), мы полагали, что функция
\|/ нормирована на единицу. В противном случае вид (29) не¬
сколько усложняется:
- I" vjI*IMV
L = - — (30)
| v|;*vjidV
Заметим, что в квантовой механике все средние значения
физических величин могут быть заданы точно, но... не все
одновременно.
Законы сохранения в квантовой механике
Завершая рассказ о свойствах квантовомеханических опе¬
раторов, обратимся к вопросу о законах сохранения. В клас¬
сической механике есть такой термин: интеграл движения.
Им обозначают физические величины, сохраняющие при
движении постоянное значение, определяемое начальными
условиями. Есть такие величины и в квантовой механике, их
средние значения в любом состоянии не изменяются с тече¬
нием времени.
Чтобы квантовомеханическая физическая величина L бы¬
ла интегралом движения и для нее был бы справедлив со¬
ответствующий закон сохранения, необходимо, чтобы опе¬
ратор L удовлетворял двум условиям:
а) не зависел явно от времени и
б) коммутировал с гамильтонианом ([Я, L] = 0).
Рассмотрим один простой пример. Гамильтониан сво¬
бодного электрона (U(х, у, z) = 0) имеет вид:
h1 ( Ъ2 Ъ2 82 \ 1 А, я,
н ~ “ ьЛ. а? + w +1?) = ~ + р’+ р’]
Очевидно, что
[Я, рх] = [.Н,ру] = [Я,р2] = 0
ссли свободный электрон в какой-то начальный момент
54
времени находился в состоянии с определенным импульсом,
то это значение импульса будет сохраняться во времени.
Стационарное состояние
Для правильного понимания многих квантовомеханиче¬
ских вопросов необходимо особо остановиться на понятии
стационарного состояния.
Рассмотрим систему, у которой оператор Гамильтона не
зависит явно от времени. В этом случае волновое уравнение
Шредингера (14) допускает разделение переменных:
(х, у, z,t) = \|/ (х, у. Z) fit)
Тогда, учитывая формулы (14) и (26), получаем:
d44x,y,z,t) df(t) *
ih—— = $\|/(х, V,z)—:— = НУ (x,y,z,t) =
dt 'dr
= /(1)Щ(х,у, z) =f{{) £v|f (x, y, z)
Отсюда:
= (31)
dt
и
H\\i (x, y, z) = £\|/ (x, y, z) (32)
Последнее уравнение называется стационарным уравне¬
нием Шредингера. Его решения \|/ (х, у, z) соответствуют со¬
стояниям системы, в которых энергия имеет определенное
значение; такие состояния называются стационарными.
Решением уравнения (31) будет экспоненциальная функ¬
ция: f (t) — e{~i/A)Et.
Из сказанного следует, что волновая функция стационар¬
ного состояния имеет вид:
¥ (х, у, z, t) = \J/ (х, у, z) е( ~ ф) Et (33)
55
Таким образом, стационарным в квантовой механике бу¬
дет не такое состояние, которое вообще не зависит от вре¬
мени, но то, которое зависит от него по приведенному выше
закону (33).
Важная особенность стационарного состояния состоит
в том, что в нем математическое ожидание любой физиче¬
ской величины, оператор которой не зависит явно от време¬
ни, постоянно.
В самом деле:
L = fЧ>* (х, у, z, t) U¥ (х, y,z, t)dV =
= J e(mEtif*(x,y,z) Le(~ i/A)£tv|i(x,y,z)dV =
_ e{i/b)Ete( i/A)Etb\f(x,y,z)dV = Jv|/*(x, y, z) Ia|/(x, y, z)dV
Нетрудно показать, что распределение электронной
плотности в стационарном состоянии также не зависит от
времени:
I ¥ (х, У, z, t) |2 = | v|/(x, у, z) I2
Отсюда, кстати, следует, что всякие утверждения о само¬
произвольном изменении характера ее распределения в изо¬
лированной молекуле,-например, переход бензола из со¬
стояния Q в состояние и обратно,-ошибочны
с позиций квантовой механики.
ЭЛЕКТРОН НА ДНЕ КОЛОДЦА
Посмотрим теперь, как изложенный выше аппарат кван¬
товой механики используется в конкретных задачах. В каче¬
стве примера обратимся к одномерному движению электро¬
на в прямоугольной потенциальной яме ширины а
с бесконечно высокими стенками (рис. 10). Эта простая зада¬
ча (вполне доступная даже тем читателям, чья математиче¬
ская подготовка ограниченна) вместе с тем очень полезна,
56
так как позволяет проиллюстрировать ряд важных кванто¬
вомеханических концепций.
Откуда возникает квантование энергии?
Итак, в нашем случае электрон движется вдоль оси х
в обоих направлениях (вперед и назад), но по условию зада¬
чи он не может находиться вне ямы, где U = оо, и потому
его волновая функция \|/(х) = 0 при х < 0 и х > а. Найдем
выражение для функции \|/ (х) и энергии электрона внутри
ямы, т.е. между точками 0 и а (область II на рис. 10). Урав¬
нение Шредингера для этой области, если принять в ней
U(х) = 0, имеет вид:
*2 = ЕШ (34)
2т dx2
с граничными условиями \|/(0) = \|/(а) = 0
II!
п~1 п=2 п=3 п
Рис. 10. Одномерное движете электрона по дну потенциального «колодца».
Рис. 11. 'Энергетические уровни, получаемые при решении задачи о движении электрона
на дне «колодца».
57
Вводя обозначение к2 = 2mE/h2, получаем:
+ = о
Общее решение такого дифференциального уравнения
хорошо известно:
v|/(x) = Asinkx + В cos/сх (35)
В формуле (35) коэффициенты Л и В не равны нулю одновре¬
менно-в противном случае получается тривиальное решение.
Теперь учтем граничные условия:
а) v|/(0) = A sin к ■ 0 + В cos к • 0 = В = О
б) \J/(а) = A sin ка — О
Таким образом, коэффициент В оказался равным нулю
и поэтому, если нас интересуют только нетривиальные ре¬
шения, следует положить А Ф 0. Тогда sin ка = 0, откуда
ка = ия (где и-целое) и
2тЕ п2п2
к- = —= ——
h а'
ИЛИ
K2h2
Е■ - W-" (36)
Из формулы (36) видно, что энергия электрона может
принимать только определенные значения, т.е. квантуется.
Эти значения-собственные значения уравнения (34)-обра¬
зуют систему энергетических уровней (рис. 11), нумеруемых
квантовым числом п. Квантование энергии не было заложе¬
но в условие задачи, а появилось в процессе ее решения
вследствие учета граничных условий, которые, в свою оче¬
редь, вытекают из физических ограничений, налагаемых на
движение.
58
Нахождение волновой функции
Теперь мы можем записать выражение для собственных
функций уравнения (34):
\|/„(х) = Л sin
или, подставляя сюда (36),
\|/„(х) = Л sin
2тЕп
h2
кпх
Эта функция, как нетрудно убедиться, удовлетворяет
всем стандартным условиям. При этом, в силу условия
нормировки:
J|\Mx)|2dx = 42Jsin
откуда
и
/2 . кпх
■ и а
т -
На рис. 12 приведены первые три волновые функции
и соответствующие им плотности вероятности для электро¬
на, движущегося в прямоугольной потенциальной яме с бе¬
сконечно высокими стенками.
Легко видеть, что вероятность локализации электрона
в окрестности некоторой точки внутри ямы может суще¬
ственно изменяться при переходе от одного состояния
к другому. Этот вывод имеет весьма общий характер. Так,
например, распределение электронной плотности в основ¬
ном и возбужденных состояниях молекулы различно.
59
2
а
fx(x)
х=0
х=а ar=0
х=а у
Рис. 12. Графики функций ф(х) и \|/2(х) для рассмотренной в тексте задачи.
Рис. 13. Площадка размером а х Ъ, на которой может быть локализован электрон.
Рассмотрим теперь движение электрона в двумерной бе¬
сконечно глубокой прямоугольной потенциальной яме.
В этом случае электрон заключен не между двумя стенками,
а движется в плоскости прямоугольника со сторонами а и
b (рис. 13).
Как и в аналогичной классической задаче, движение элек¬
трона можно «разложить» на два независимых движения:
одно в направлении оси х, а другое в направлении оси у.
Тогда энергия электрона будет определяться двумя кван¬
товыми числами пх и пу
где пх = 1, 2, пу = 1, 2, ...
а соответствующая волновая функция примет вид про¬
изведения, в котором каждый сомножитель зависит только
Симметрия и вырождение
60
от одной независимой переменной:
VW, ,п (*> У) = МЖ (У)
X у X у
Рассмотрим два состояния системы: пх — 1, пу = 2 и
пх = 2, пу = 1.
Если а ф Ь, то каждому состоянию отвечает свое значе¬
ние энергии:
Состояние Энергия
пх= и Пу — 2 Е12 =
пх = 2, п= 1 Е21 =
пЧ2 ,
(Л
+
22 '
2т '
U2
~ьТ,
пЧ2 ,
( 22
+
I2'
2т '
V а2
'Ьг
Таким образом, Е12 Ф E2i, и, следовательно, вырожде¬
ние отсутствует (рис. 14, /).
Когда же а = Ь, то в обоих состояниях энергия системы
одинакова
кЧ2
Е12 - *21 = ^~2та2 (37)
/
пу=1 пу=2
II
пу=1 пу=2 пу=[
Рис. 14. Взаимосвязь симметрии поля, в котором движется электрон и структуры вырождения
энергетических уровней.
61
или, в общем случае:
£".Л “ 1ЙГ(”2‘ + "’2)
Таким образом, всем комбинациям чисел пх и пу, даю¬
щим одно и то же значение суммы (и* + и,) будет отвечать
одно и то же значение энергии, т. е. будет иметь место выро¬
ждение (рис. 14, П).
Итак, увеличение симметрии системы (в данном случае,
переход от прямоугольника, ограничивающего область дви¬
жения электрона, к квадрату) приводит к вырождению по
энергии некоторых состояний [см. (37)]. Связь между симме¬
трией системы и структурой вырождения энергетических
уровней является одной из глубоких идей квантовой механи¬
ки *.
ЭЛЕКТРОННОЕ ВЕРЕТЕНО
Четвертая координата
Уравнение Шредингера описывает состояния электрона,
движущегося в трехмерном пространстве. При этом требо¬
вания теории относительности никак не учитываются. Если
же их учесть, то уравнение Шредингера следует заменить
другим, релятивистским уравнением Дирака, из которого
непосредственно вытекает существование у электрона соб¬
ственного момента импульса, а следовательно, и собствен¬
ного магнитного момента. Собственный момент электрона
(S) называют также спиновым (от английского глагола to
spin-прясть, плести, крутить(ся), вертеть!ся)) или просто
спином.
* Подробнее см. И. С. Дмитриев. Симметрия в мире молекул.
Л.: Химия, 1976.
62
Спин-это внутренняя степень свободы электрона, имею¬
щая сугубо квантовый характер. При переходе к классиче¬
ской механике спин обращается в нуль и в этом смысле он
не имеет классического аналога.
Правда, иногда с целью наглядной иллюстрации этого
понятия электрон уподобляют вращающемуся вокруг своей
оси заряженному шарику. Но надо признать, что это крайне
грубая аналогия, ибо, как мы видели, электрон даже отда¬
ленно не напоминает шарик. Поэтому в дальнейшем мы
этой аналогией пользоваться не будем.
Хотя понятие о спине строго следует из релятивистской
квантовой теории, его можно ввести и в нерелятивистское
рассмотрение как величину, необходимую для правильного
описания ряда опытных фактов (тонкой структуры атомных
спектров, аномального эффекта Зеемана и т.д.). Такую тео¬
рию частиц со спином называют феноменологической или
полуэмпиртеской. Познакомимся с некоторыми положения¬
ми этой теории.
Момент орбитальный и момент собственный
Математические выражения для квадрата спинового мо¬
мента импульса электрона (S2) и его проекции на ось кван¬
тования z(Sz) полностью аналогичны выражениям для ква¬
драта орбитального момента М2 и его проекции М2:
М2 = 1(1 + 1 )h2
S2 = s(s + \)h2
(38)
М. = mh
Sz = mh
(т = 0, ± 1, ..., ± /)
63
Рис. 15. Две возможные проекции спинового
момента электрона на ось квантования z.
Однако квантовое число спи¬
нового момента (s) в от¬
личие от I может прини¬
мать лишь одно значение
1
s = — и тогда:
s=MF0*=J?*<з9)
Итак, спиновый момент электрона или, короче, его спин
равен ({/3/2) к. Иногда об электроне говорят как о частице
с полуцелым спином, имея в виду значения квантового чис¬
ла s, а не величину собственного момента электрона S.
Особо следует сказать о часто встречающихся формули-
1 1
ровках: «спин электрона может быть равен + — или - —»
или «электрон со спином вверх и вниз». В обоих случаях
речь идет не о спине электрона-момент не может быть от¬
рицательным [(см. формулы (38) и (39)], а о значениях кван¬
тового числа ms, характеризующего проекцию спинового мо¬
мента электрона на ось квантования (рис. 15).
Алгебра спинов
Так как понятие о спине не имеет классического аналога,
то получить выражение для оператора спинового момента
импульса электрона так, как это мы делали раньше, т. е. ис¬
ходя из канонической классической формулы для данной
физической величины, невозможно. Однако было установле¬
но, что коммутационные соотношения для операторов
квадрата собственного момента импульса электрона S2
64
и его проекций Sx, Sy и Sz аналогичны приведенным выше
соотношениям для операторов М2, Мх, Му и Мг, т.е.:
Р = s2x + s2 + s2
[S„ S,,] = ihS2; [Sr S.] = ihSx; [S., Sx] = ihSy
[P,Sx] = lP,Sy] = \S2,Sz] = 0
Коммутирующим операторам S2 и Sz отвечают только
две собственные функции (соответственно двум возможным
значениям проекции спинового момента). Эти функции обо¬
значаются символами а и Р и удовлетворяют следующим
соотношениям:
1
Sz а = msh а = Н ha
Szp = mshfi = - yAp
S2 а = s(s + l)h2a = ~2\^2 + *у^2<х =
р$ = Адгр
Р 4 к
При этом спиновые функции аи|3 ортогональны и нор¬
мированы на единицу.
Магнитные моменты
С механическим моментом электрона (как орбитальным,
так и спиновым) связаны соответствующие магнитные мо¬
менты. Если воспользоваться классической моделью, то ве¬
личина орбитального магнитного момента цорб, отвечающе¬
го движению электрона со скоростью v по круговой орбите
5-918 65
радиуса г, равна
е
Ц°рб - - ~^М = ~УМ
е
где у = гиромагнитное отношение, т.е. отношение величины
2 тс
магнитного момента к механическому.
Поскольку М = |//(/ + 1)А, то
^орб = ~ I/ф + *)~2~
еЬ. _
Величина —— = = 9,2731 10 21 эрг/г с представ-
Ъпс
ляет собой ахомную единицу магнитного момента, называе¬
мую магнетоном Бора.
По аналогии с орбитальным моментом вводится спи¬
новый магнитный момент электрона цсп:
Цеп ~ Усп^
е
При этом у = —, что в два раза больше гиромагнит-
тс
ного отношения для цорб.
Учитывая, что S = j/s(s + 1)А (где 5 = получаем:
Взаимодействие магнитных полей, создаваемых орби¬
тальным и спиновым моментами электрона, называется
спин-орбиталъным взаимодействием. Его строгое рассмотре¬
ние возможно только в рамках релятивистской квантовой
теории.
66
КОГДА ЭЛЕКТРОНОВ МНОГО...
До сих пор речь шла в основном о квантовомеханиче¬
ском описании состояний одной частицы (точнее, одного
электрона). Если же электронов в системе много (в данном
случае «много» означает два и больше), то в поведении та¬
кой системы появляются свои особенности, которые нельзя
вывести из других, сформулированных ранее положений
квантовой механики, и потому эти особенности следует рас¬
сматривать как новые и самостоятельные квантовомехани¬
ческие постулаты. В чем же их суть?
Теоретик считает мух
Рассмотрим для начала систему, состоящую из двух
электронов. Допустим, что в некоторый момент времени t0
координаты этих электронов заданы точно и мы можем ска¬
зать, что, скажем, в окрестности точки (хх, z{) находится
первый электрон, а в окрестности точки (х2, у2» z2)~ второй.
В то же время, согласно соотношению неопределенностей
Гейзенберга, мы ничего не можем сказать об импульсах то¬
го и другого электрона в момент t0. Последнее означает,
что электроны могут двигаться с любыми скоростями и
в любых направлениях. Но тогда, по прошествии некото¬
рого времени мы сможем найти их в любом месте про¬
странства, т.е. области локализации электронов перекры¬
ваются. На рис. 16 условно показано «расплывание» волно¬
вой функции электронов. Заштрихованная область отвечает
большей вероятности нахождения в ней одного из электро¬
нов. Естественно, обнаружив электрон в этой области, мы
никаким способом не сможем установить, какой же это
электрон-«1» или «2». Таким образом, в квантовой механи¬
ке нельзя указать, в каком месте пространства в данный мо¬
мент времени находится каждый из электронов N-электрон-
ной системы. Одинаковость микрочастиц в квантовой
механике имеет, как мы видим, гораздо более глубокую
5*
67
природу, чем одинаковость
классических частиц. В клас¬
сической механике всегда
можно (по крайней мере в
принципе!) определить инди¬
видуальную траекторию каж¬
дого из множества одинако¬
вых объектов (например,
бильярдных шаров), для чего
достаточно либо как-то эти
Рис. 16. Иллюстрация невозможности разли¬
чить одинаковые микрочастицы по состоя¬
нию их движения.
объекты пометить, либо внимательно следить за движением
каждого из них. Достаточно наглядным примером может
служить наблюдение за полетом нескольких мух. Стоит не¬
много отвлечься, «потерять» траектории их движения, и
сказать, где какая муха уже нельзя. В квантовой механике
траектория «потеряна» с самого начала, ибо мы в принципе
не можем пользоваться этим понятием при описании движе¬
ния микрочастиц (о чем уже шла речь выше), и потому в
квантовой механике одинаковые частицы оказываются тож¬
дественными и принципиально неразличимыми.
Сказанное позволяет сформулировать следующий посту¬
лат, называемый принципом тождественности микрочастиц.
В системе, состоящей из N тождественных частиц, осу¬
ществляются лишь такие состояния, которые не меняются
при перестановке любых двух таких частиц. При этом под
перестановкой понимают «обмен» как пространственными
координатами, так и спиновыми переменными г'-й и у'-й
частиц, т.е. (г;, стг) ?± (rp Oj).
Здесь спиновая переменная ст принимает два значения,
соответствующие двум возможным проекциям спинового
момента электрона на ось z.
Одним из первых, кто указал на важность понятия
тождественных частиц для квантовой механики, был В. Гей¬
68
зенберг, который уже в начале 1926 г. писал: «Характерная
черта атомных систем состоит в том, что они включают
в себя части-электроны,-которые одинаковы и подвержены
действию одинаковых сил».
Оператор перестановки
Прежде чем перейти к дальнейшему обсуждению особен¬
ностей многоэлектронных систем, остановимся на физиче¬
ском смысле многоэлектронной волновой функции, описы¬
вающей такие системы. N-Электронная волновая функция
зависит от 4N переменных, так как каждый электрон харак¬
теризуется тремя пространственными координатами г =
= г (х, у, z) и одной спиновой (а):
vN) (40)
Квадрат модуля этой функции определяет вероятность
одновременной локализации электронов с определенными
значениями проекции спина в окрестностях определенных
точек пространства.
Из принципа тождественности следует, что гамильто¬
ниан /V-частичной системы не изменяется при перестановке
частиц, понимаемой в указанном выше смысле. Эту переста¬
новку можно описать математически посредством специаль¬
ного оператора Pik, определив его следующим равенством:
■ ;ri,ai; ...;rk,ok- ...;rN,oN) =
(41)
- 4(r1,ol;...;rk,Gk;...;ri,oi;..:;rN,oN)
Как видим, действие оператора перестановки Pik сводит¬
ся к замене (гь ог() (rk, ак). В простейшем случае двухча¬
стичной системы равенство (41) принимает вид:
Обычно для краткости совокупность пространственных
и спиновых координат /-й частицы обозначают одной бук¬
вой, например х,- = (r;; а;), а иногда используют и более
компактную запись, указывая лишь условный номер ча¬
стицы: *Р(1,2,..., N). В дальнейшем мы будем пользоваться
обоими вариантами.
Принцип антисимметрии
В силу инвариантности гамильтониана Л'-электронной
системы относительно перестановки электронов должно вы¬
полняться следующее соотношение
PikH = HPik
т. е. операторы Pik и Я-коммутируют друг с другом.
Это, в свою очередь, означает, что для них существуют
общие собственные функции ¥:
= £¥ (42а)
Pik4> = W (426)
где X- собственное значение оператора перестановки.
Теперь найдем X,. Ясно, что двукратное действие опера¬
тора Pik на волновую функцию Ч^х^хз, -..,xN) ничего не из¬
меняет, так как в итоге электроны i и к «возвращаются на
свои места»:
Р^¥ (xl,x2,...,xi,...,xk,..., xN) = Pik¥ {xl,x2,...,xk,...,xi,...,xN) =
= '¥{xl,x2,...,xi,...,xk,...,xN) (43)
С другой стороны, если учесть (426), этот же результат
можно записать иначе:
AW*!» *2, •••’*!’ •••>**’ • ■.%) =
70
= X2xF(xj,х2, ..,X;,...,хк,...,xN)
(44)
Сравнивая (43) и (44), получаем: X2 = 1, откуда:
Xj = 1 и Х,2 = — 1
Какое из полученных значений X следует выбрать?
В нерелятивистской квантовой теории постулируется (а
в более общей теории доказывается), что система частиц
с полуцелыми спинами,-т. е. система фермионов, к числу ко¬
торых относятся электроны,-описываете я волновой функ¬
цией, меняющей знак при перестановке координат и спи¬
новых переменных любой пары электронов, например х,- и
-V
Т(Х!,Х2, ...;x„...,xt,...,xN) =
= - 'Р(х1,х2,хк,х„ xN) (45)
Этот принцип называется принципом антисимметрии или
принципом Паули.
ЧТО ТАКОЕ ОРБИТАЛЬ?
Обратимся вновь к волновой функции N-электронной си¬
стемы (40). Эта функция, как следует из сказанного выше,
описывает состояние всей такой системы в целом. В связи
с этим возникает вопрос: можно ли каждому отдельному
электрону такой системы приписать свою волновую функ¬
цию?
Один из многих
Строго говоря, ответ должен быть отрицательным, и вот
почему. В квантовой механике состояния далеко не каждого
микрообъекта можно описать с помощью волновой функ¬
ции. Если, например, два электрона находятся на значитель¬
ном расстоянии друг от друга и потому практически
могут рассматриваться как невзаимодействующие, то тогда
каждому из них можно сопоставить свою волновую функ¬
71
цию. При наличии же взаимодействия между частицами вол¬
новой функцией описывается лишь Система в целом, но не
каждая из составляющих ее частиц. Иными словами, поня¬
тие состояния отдельной частицы в последнем случае теряет
смысл.
Так обстоит дело при строгом подходе. Но практически,
точно решить уравнение Шредингера для систем с двумя
и более электронами до сих пор не удалось. Поэтому для
описания атомных и молекулярных многоэлектронных си¬
стем приходится обращаться к тем или иным приближе¬
ниям, из которых наиболее распространенным является
одноэлектронное или (другое название)-орбитальное. В его
основе лежит представление о существовании индиви¬
дуальных состояний каждого электрона, которые пред¬
ставляют собой стационарные состояния движения электро¬
на в некотором эффективном поле, создаваемом ядром (или
ядрами) и всеми остальными электронами. Эти стацио¬
нарные состояния описываются соответствующими одно¬
электронными функциями. Иными словами, в рамках такого
приближения каждый электрон многоэлектронной системы
полагается как бы независимым. Одноэлектронная функ¬
ция называется орбиталью, если она зависит только от про¬
странственных координат, или спин-орбиталью, если наряду
с тремя пространственными включает также спиновую пере¬
менную.
Детерминант Слэтера
Теперь рассмотрим такой вопрос: каким образом много¬
электронная волновая функция выражается через одноэлек¬
тронные (т. е. через спин-орбитали)?
Обратимся для примера к двухэлектронной системе.
Учитывая вероятностную трактовку волновой функции,
а также принимая одноэлектронное приближение и вспоми¬
ная, что вероятность одновременного наступления двух неза¬
висимых событий равна произведению вероятностей каждого
из них, можно, казалось бы, представить двухэлектронную
функцию как произведение спин-орбиталей
¥(хх,х2) = (46)
В этом случае первый электрон находится в состоянии
vJ/j, а второй-в состоянии v|/2. Однако, в силу принципа то¬
ждественности, мы можем записать и другое равенство:
¥(х1?х2) = \|/i(x2)v|/2(x1) (47)
Оно отвечает ситуации, когда первый электрон находит¬
ся в состоянии \J/2, а второй -vJ/j.
Кроме того, ни (46), ни (47) не удовлетворяют принципу
антисимметрии (точнее, они не обладают вообще никакой
симметрией относительно перестановки электронов). Этот
недостаток можно устранить, если составить такую линей¬
ную комбинацию из функций-произведений (46) и (47):
ч^х^хг) = A [\|/j (хj)v)/2 (х2) - vl/1(x2)v)/2(x1)] (48)
Убедимся в антисимметричности этого выражения:
^(хрхг) = A (Xj) v|/2 (х2) - vMx2)v|/2(Xi)] =
= - A [v|/ j (x2)vl/2(xx) - v|/,(x1)\|/2(x2)] = - Ч^х^х^
Антисимметричную двухэлектронную функцию (48), со¬
ставленную из спин-орбиталей 1|/х и \|/2, можно также пред¬
ставить в виде детерминанта*:
(Xi) v|M*2)
\|/2(Xi) \1/2(х2)
где нормирующий множитель А равен 1 /|/2-
* О детерминантах см. И. С. Дмитриев. Молекулы без химиче¬
ских связей. JL: Химия, 1980, с. 11.
73
В более общем случае TV-электронной системы ее волно¬
вая функция выражается через ортогональные и нормиро¬
ванные спин-орбитали следующим образом:
1/лп
det
*F(Xj ,х2,...,х.у) =
'M*i) ^Лхг)
... vj/^Xiv)
... \|/2(xN)
\|/2(x'i) v|/2(x2)
>M*l) *M*2) ... fv(*v)
(49)
Этот детерминант называется детерминантом Слэтера
исокращенно записывается так: det|\|/ 1(x1)\j/2(x2)...v|/w(xjv)|.
Такой способ построения многоэлектронной волновой
функции системы из спин-орбиталей обеспечивает выполне¬
ние принципа антисимметрии, так как при перестановке
двух столбцов (что соответствует перестановке двух элек¬
тронов) детерминант, как известно, меняет знак. Из (49) так¬
же следует, что если среди номеров состояний окажутся два
одинаковых (что соответствует равенству двух строк), весь
детерминант тождественно обратится в нуль.
Таким образом, в одном и том же состоянии (т.е. на
одной и той же спин-орбитали) не может находиться более
одного электрона. Последнее утверждение составляет содер¬
жание принципа Паули, сформулированного в рамках одно¬
электронного приближения.
Если пренебречь спин-орбитальным взаимодействием,
имеющим гораздо меньшую по сравнению с электростати¬
ческими эффектами величину, то каждую спин-орбиталь
можно представить в виде произведения пространственной
и спиновой частей:
\|/ (х) = ф (г) < или
Ь
Два электрона системы, заселяющие орбитали и отли¬
чающиеся только спиновыми характеристиками, называют¬
74
ся спаренными*, а ЛГ-электронная система, состоящая толь¬
ко из спаренных электронов, называется системой
с замкнутыми оболочками. В такой системе число электро¬
нов четное и детерминант Слэтера в этом случае принимает
вид:
Wi
CTj,... ',rN,aN)
=
q>i(ri)a(l)
Ф1(г2)а(2)...
4>i(r2k)ct(2k)
<Pi(ri)P(l)
<Pi (гг) P (2)...
Ф1 (r2fc) Р(2/с)
Ф2(г, )a(l)
Ф2(г2)а(2)...
Фг(Ыа(2 к)
Фг(п)Р(1)
Ф2(г2)Р(2)...
Фг(г2/с)Р(2/с)
4>*(ri)P(l)
ф!с(г2)Р(2)-
<Рк(Г2к) P(2fc)
(50)
Отсюда, в частности, следует что на одной орбитали не
может находиться более двух электронов, причем спины
этих электронов должны быть антипараллельными (это на¬
иболее распространенная формулировка принципа Паули).
В зависимости от того, идет речь об атомной или о мо¬
лекулярной системе, говорят об атомных или о молеку¬
лярных орбиталях или спин-орбиталях (обозначают их-АО
и МО или АСО и МСО).
Метод Хартри-Фока
В основе этого метода лежит одноэлектронное приближение,
т. е. считается, что каждый электрон движется независимо в некото¬
ром усредненном поле ядра (ядер) и остальных электронов. Одно¬
электронные функции (орбитали) находятся из решения уравнений
Харгри-Фока:
F ф,- = 8;ф
(51)
где F-оператор, называемый фокианом, е(-орбитальные энергии,
индекс i нумерует заполненные электронами орбитали (мы рассма¬
триваем невозбужденные состояния системы, полный спин которой
равен нулю).
* Еще раз подчеркнем, что в силу тождественности электро¬
нов, нельзя сказать, какой из них имеет «спин вверх», а какой-
«спин вниз».
75
Решения системы уравнений (51) определяют набор «наилуч¬
ших» (в рамках одноэлектронного приближения) орбиталей ф; для
основного состояния многоэлектронной системы с замкнутой обо¬
лочкой и соответствующих им собственных значений фокиана. По¬
следние играют роль «орбитальных энергий» и служат разумным
обобщением понятия энергии отдельной независимой частицы. Сам
фокиан можно представить как сумму двух операторов:
тор электронного взаимодействия.
Для системы с заполненной электронной оболочкой полная
энергия определяется следующим равенством:
где Jjj-кулоновский и K{j-обменный интегралы, описывающие по¬
парное взаимодействие электронов:
При этом первый представляет собой среднюю энергию электро¬
статического отталкивания электронов, находящихся на орбиталях
Ф; и фj, второй же появляется вследствие учета принципа антисим¬
метрии.
Система Хартри-Фока (51) является системой нелинейных ин-
тегродифференциальных уравнений. Нелинейность уравнений озна¬
чает, что их решения фг есть собственные функции оператора F, ко¬
торый, в свою очередь, определяется через эти орбитали ф;. Эта
особенность уравнений Хартри-Фока позволяет решать их мето¬
дом итераций. Однако мы не будем останавливаться здесь на вы¬
числительной стороне дела.
F = Hi + G
где Яд-оператор энергии электрона в поле ядра (ядер), a G-onepa-
к к к
i = 1 i = 1 j = 1
,2 ГГ <рНгМ(г2)^у^у2
JJ Г12
фГЫфД^) ф* (г2) ф; (r2)dv1 dV2
И в сфере звезд, и в облике планет
На атомы Вселенная крошится,
Все связи рвутся, все в куски дробится,
Основы расшатались, и сейчас
Все стало относительным для нас.
Д. ДОНН
Глава II
ЭЛЕКТРОН В АТОМЕ
В этой главе мы остановимся на кратком описании
атомных состояний. Теории строения атома посвящена об¬
ширная литература от сверхпопулярной брошюры издатель¬
ства «Малыш» до солидных научных монографий*. Поэто¬
му здесь мы коснемся в первую очередь тех вопросов, на
которых обычно мало акцентируют внимание.
ОРБИТАЛЬНАЯ МОДЕЛЬ АТОМА
Квантовые числа
Начнем с рассмотрения электронных состояний атома
водорода. Заметим, что задача эта представляет собой при¬
мер одной из немногих квантовомеханических задач, имею¬
щих точное аналитическое решение, что обусловлено воз¬
можностью разделения переменных в сферической системе
координат (г, 0, ф). Иными словами, волновая функция (или
АО-здесь эти понятия совпадают) \|/(г, 0, ср), описывающая
движение единственного электрона водородного атома, мо¬
жет быть представлена в виде произведения:
= адеие) ф»(ф) (52)
* Хорошим примером популярного и в то же время коррект¬
ного изложения этой темы может служить книга: Л. Пономарев.
По ту сторону кванта. М.: Молодая гвардия, 1972.
77
Нижние индексы в формуле (52) показывают, что АО ха¬
рактеризуется тремя квантовыми числами: п, I и т (неко¬
торые из них нам уже знакомы).
Квантовое число /, целое и неотрицательное, определяет
орбитальный момент импульса электрона, точнее, его ква¬
драт: /(/ + 1)А2.
Квантовое число т. целое и не превышающее по абсо¬
лютной величине 1(\т\ ^ Г), представляет проекцию орби¬
тального момента импульса на произвольно выбранную ось
квантования z.
И, наконец, главное квантовое число п нумерует орби¬
тальные энергии 8П в порядке их возрастания.
Для обозначения АО используются строчные латинские
буквы: I = О, 1, 2, 3, 4, ...
s р d f д ...
Использование первых четырех букв исторически обусло¬
влено и происходит от названия спектральных линий (sharp,
principal, diffuse, fundamental), остальные следуют в алфавит¬
ном порядке. Перед этими буквами указывается главное
квантовое число, например: Is, 2р, 4/и т.д. Квантовое число
т может указываться в виде нижнего индекса: 2р0, 2р+1,
3d±1, 3d±2 и т.д. В зависимости от абсолютной величины
числа т различают орбитали типа ст(|т| = 0), л(|т| = 1),
5(|т| = 2) и т.д.
Скрытая симметрия водородного атома
Характерным для атома водорода является то, что энер¬
гия е„ не зависит ни от числа /, ни от т и определяется толь¬
ко главным квантовым числом п:
те4 1
е" = ~
Иными словами, энергетический спектр атома водорода
вырожден по квантовым числам, / и т. Кратность вырожде¬
78
ния энергетических уровней зависит от симметрии системы*.
Атомные системы характеризуются сферической симме¬
трией, которая является их геометрической симметрией. Эта
«шарообразность» атома водорода и приводит к тому, что
энергия не зависит от магнитного числа т.
Математическое дополнение. Группа пространственной
симметрии атома водорода О (3) является прямым произведением
группы ортогональных унимодулярных преобразований трехмерно¬
го координатного пространства SO (3) на группу инверсии про¬
странства относительно начала координат С„ т.е. 0(3) = S0(3) х С,.
Координатные волновые функции электрона при этом класси¬
фицируются по (21 + 1)-мерным неприводимым представлениям
(НП) группы SO (3), что соответствует заданию квантовых чисел /
и и, и по одномерным НП группы С(, что соответствует заданию
четности состояния. Квантовое число /, определяющее величину
квадрата орбитального момента импульса, нумерует НП D"1
группы О (3). Каждому значению I соответствует (21 + 1) различных
значений т, т.е. (21 + 1) равно размерности НП D(l).** Таким обра¬
зом, с помощью группы О (3) можно объяснить вырождение энер¬
гетического спектра атома водорода только по числу т.
Но почему энергия не зависит от /? Не связано ли это
с наличием некоторой симметрии, отражающей более глу¬
бокие и потому менее очевидные физические характеристи¬
ки, чем пространственные?
Действительно, как было показано акад. В. А. Фоком
в 1935 г., полная группа симметрии атома Н, объясняющая
оба типа вырождения (по т и по I), есть группа вращений
четырехмерного шара О (4). Для того чтобы связать теорию
атома водорода с симметрией четырехмерного шара, Фок
записал уравнение Шредингера не в обычном виде, а
в особых, введенных им координатах, зависящих от компо¬
нент импульса электрона, причем число таких координат
(размерность пространства Фока) равно четырем.
* См. гл. I, раздел «Электрон на дне колодца».
197 ** ^м' нашУ КНИГУ «Симметрия в мире молекул». Л.: Химия,
79
Математическое дополнение. В классической механике
а
доказывается, что при движении частицы в поле вида U — — (где
г
а-некая постоянная), частным случаем которого служит кулонов-
ское поле, имеет место закон сохранения, специфический именно
для этого поля. Сохраняющейся величиной оказывается при этом
так называемый вектор Лапласа-Рунге-Ленца:
_ г 1 ,
А = — Н [М х р]
г та
В квантовой механике этому вектору по описанным в первой
главе правилам сопоставляется оператор А, коммутирующий с га¬
мильтонианом. Если ввести вместо операторов М и А два вспомо¬
гательных оператора К и N, определяемых равенствами:
К = ~-(М + А); N=j-(M-A)
и найти для их компонент коммутационные соотношения, то не¬
трудно будет получить следующие собственные значения операто¬
ров Ж2 и N2:
Х2\|/ = ц(ц -|- 1)\J/; N2\\i = v(v + l)v|f
где (ц, v = 0, 72, 1, 172, 2, ...).
Можно также показать, что квантовые числа ц и v задают НП
групп 0(4), которые можно поэтому обозначить символом (ц, v).
Однако для описания атома водорода приемлемы далеко не все
представления (ц, v). Из условия М А = 0 следует, что К2 = N2, т. е.
ц = v. Следовательно, необходимо принимать во внимание только
НП (ц, ц).
Гамильтониан атома водорода можно выразить через опера¬
торы К2 и N2, тогда его собственные значения, т. е. возможные зна¬
чения энергии атома, равны:
те4 1
£ц = ~ 2к2 (2ц + \)2
Числа (2\i + 1) = п-целые (п = 1, 2, 3, ...), поэтому:
те4 1
Замечание о многоэлектронных атомах
Мы видим, что описание строения атома водорода дале¬
ко не простое дело. Для многоэлектронных атомов пробле¬
ма еще более усложняется. Как правило, в этом случае
одноэлектронное приближение используется в рамках моде¬
ли центральносимметричного поля, т. е. считается, что элек¬
трон взаимодействует с ядром по некоторому закону U(r).
Это позволяет произвести разделение переменных г, 0, ср
и при рассмотрении многоэлектронных атомов. Но точное
аналитическое выражение для радиальных функций Rnl(r)
при этом, к сожалению, не получается. Эти функции опреде¬
ляются путем решения уравнений Хартри-Фока.
Кроме того, орбитальные энергии в этом случае зависят
уже от двух квантовых чисел п и /, причем п нумерует еп1
с фиксированными / в порядке их возрастания целыми чис¬
лами, начиная с (/ + 1).
Радиальная зависимость АО в многоэлектронных атомах
может быть довольно сложной, но их узловая структура
подобна узловой структуре орбиталей водородного атома:
функция Rnt(r) характеризуется (п - I - 1) узлом, т.е. обра¬
щается в нуль (п — / — 1) раз при конечном значении г > 0.
После этих замечаний обратимся к вопросу о графиче¬
ском представлении атомных орбиталей.
КАК НАРИСОВАТЬ АТОМНУЮ ОРБИТАЛЬ?
Как уже отмечалось, АО в одноэлектронном приближе¬
нии с использованием модели центрально симметричного
поля представляют в виде произведения:
1|/„1тМ,ф) = Rnl(r) У/т(0, ф) = я»0,т(е)Фт(ф)
где Ylm(Q, ф)-уже знакомые нам сферические функции.
Поэтому в дальнейшем мы рассмотрим три случая. tpa-
фического представления АО: для радиальных функций
6-918
81
R„i{r), для сферических Ylm(Q, ф) и так называемые изовероят-
ностные поверхности. Начнем по порядку.
Графическое представление радиальных функций
Для графического представления радиальных функций
используется либо график самой функции Д„,(г), либо гра¬
фик соответствующей ей плотности вероятности локализа¬
ции электрона на расстоянии г от атомного ядра:
рni(r) = r2$ S ф)|2 QdQd(p = г2Д„2г(г)
о о
причем функция р„г(г) нормирована на единицу:
J Р Jr)dr = 1
о
Следует отметить, что в соответствии с условием норми¬
ровки сферических функций интегрирование по углам 0 и
Ф не приводит к появлению множителя 4п, который иногда
ошибочно включается в выражение для р„,(г).
Примеры графического представления радиальных функ¬
ций приведены на рис. 17.
Графическое представление угловой зависимости
атомных орбиталей
Для графического представления сферических функций
П.(9.Ф) = @/т(0)Фт(ф)
используются полярные диаграммы, т.е. графики функций
г(в) - |П.(в,ф)|2
в сферической системе координат.
82
z
I =0
m= 0
2
M = 1
m= 0
г =1
QOm=±I
Рис. 17. Графическое представление радиальной электронной плотности p„i(г) = r2Rh(r) для
некоторых состояний атома водорода.
Рис. 18. Полярные диаграммы для s-, р- и ^-состояний атома водорода.
Полярная диаграмма описывает распределение вероятно¬
сти локализации электрона по направлениям, заданным
углами 0 и ф. Легко видеть, что полярные диаграммы ак¬
сиально симметричны, если атомные орбитали характери¬
зуются определенными значениями квантового числа т, так
как в этом случае их зависимость от угла ф должна иметь
вид:
На рис. 18 приведены сечения полярных диаграмм пло¬
скостью yz. Полные поверхности получаются вращением их
вокруг ОСИ Z.
6*
83
Изовероятностные поверхности
Соответствующее атомным орбиталям распределение
плотности вероятности локализации электрона в определен¬
ной точке трехмерного пространства может характеризо¬
ваться семейством изовероятностных поверхностей (или по¬
верхностей равной вероятности), определяемых уравнением
К^Дф)!2 = с
где С-некоторая константа.
В частности, распределение электронной плотности, со¬
ответствующее ns-орбитали, описывается одной или не¬
сколькими (в зависимости от значения главного квантового
числа п и конкретного значения константы С) концентриче¬
скими сферами: для Is-одна сфера радиуса гДС), для
2s-либо одна сфера радиуса гх(С) (С2 < С ^ CJ, либо две
сферы радиусов ^(С) и г2(С) (С — С2), либо три сферы ра¬
диусов (С), г2 (С) и гз (С) (О < С < С2) (рис. 19).
В качестве других примеров на рис. 20 приведены изове-
роятностные поверхности для Is-, 2s-, 2р-, 3s-, Зр- и З^-орби-
талей атома водорода.
Рис. 19. К определению иювероятносгных поверхностей для Zs-AO.
84
Орбитали комплексные и вещественные
Атомные орбитали могут иметь как комплексную, так
и вещественную форму. До сих пор мы рассматривали ком¬
плексные АО, характеризующиеся определенными значения¬
ми проекции орбитального момента импульса. Однако
в квантовой химии часто используют вещественные комби¬
нации таких орбиталей, определяемые по формулам:
Ф*./,+ц = -"F"!>*/..и + (“
V 2
4Vi,-n =
Г-
Здесь индекс ц = |т| уже не имеет смысла проекции мо¬
мента импульса. К сожалению, на это обстоятельство не
всегда обращают внимание. Во многих учебниках состояние
электрона в атоме характеризуется квантовыми числами п,
I и т, а для иллюстрации приводятся графические изображе¬
ния вещественных АО.
Если выразить ±ц через декартовы координаты
(x,y,z), то каждая из этих функций окажется пропорциональ¬
ной некоторому полиному от х, у и z, который обычно
указывается при ф вместо индекса ц.
Легко убедиться, что между комплексными и веще¬
ственными атомными орбиталями существует соответствие:
Фnl,m
%1,+ц
Mo
%l, -Ц
ns
ns
т
—
nPz
—
пр+1
nPx
—
ПРу
nd0
—
ndz2
—
nd±i
ndxz
—
ndyz
nd± 2
ndx2 _ y2
ndXy
85
Поверхности, представляющие вещественные АО ±|1,
могут не обладать аксиальной симметрией (рис. 21).
Ввиду неоднозначности выбора АО могут возникнуть
следующие два вопроса:
-какие АО, комплексные или вещественные, целесообраз¬
нее использовать при рассмотрении электронной структуры
атома?
-каково реальное пространственное распределение элек¬
тронной плотности в атоме, получаемое в одноэлектронном
приближении?
Какие АО выбрать?
В связи с первым вопросом следует отметить, что если
рассматривается изолированный атом, то оба типа АО-и
комплексные, и вещественные-могут использоваться в рав¬
ной мере. (Для расчетов молекул по методу МО ЛКАО-см.
далее-более удобными оказываются вещественные АО).
Необходимо, однако, иметь в виду, что для получения
какой-либо информации об электронной структуре атома
нужно оказать на него определенное «воздействие», напри¬
мер поместить в аксиальное магнитное поле. В этом случае
следует предпочесть комплексные АО, которые, в отличие
от вещественных АО, будут собственными функциями воз¬
мущенного гамильтониана.
Теорема Унзольда
Ответ на второй вопрос впервые был дан А. Унзольдом.
Он показал, что поскольку заселенность всех АО, с данными
п и I, но различными т, статистически равновероятна, то ус¬
редненная по различным т плотность вероятности сфериче¬
ски симметрична
У 14W(Г. 0, Ф) |2 * R?1 (Г) X ! У‘п(Ь V'112 “ '~VtT " R”l(r)
/ч v *- I m ‘5 - i
86
N Н
а»
0Q
+ I
•И
* с
/
о +"
00
о
о
* * г»
00 8 0
О
г
87
21. Изовероятностные поверхности для вещественных Is-, 2s-, 2р-, 3s- и 3d-AO.
в то время как симметрия отдельной атомной орбитали мо¬
жет быть иной (например, изображаться диаграммой в виде
«гантели»). Тем самым Унзольд разграничил понятия сим¬
метрии АО и симметрии распределения электронной плот¬
ности в атоме.
Гибридные АО
Более общим, чем преобразование комплексных-АО в ве¬
щественные является преобразование гибридизации. Под ги¬
бридизацией АО понимается их преобразование А, смеши¬
вающее АО не только с одинаковыми, но и с различными
квантовыми числами I момента импульса:
'Ы'О =
I т
где (1,т) нумерует строки, а (п, к) - столбцы матрицы унитарного
преобразования А.
Иногда гибридизацию понимают в более узком смысле, ис¬
ключая из приведенного выше преобразования АО с одним и тем
же /, но различными ж. Подробнее на концепции гибридизации мы
остановимся далее.
Электронные термы и конфигурации
Рассмотрим теперь, что представляют собой энергетиче¬
ские уровни многоэлектронного атома. Слэтеровский де¬
терминант, составленный из спин-орбиталей,' является N-
электронной функцией, удовлетворяющей принципу Паули
и соответствующей определенным проекциям /V-элек¬
тронных орбитального и спинового моментов, опреде¬
ляемых квантовыми числами ML и Ms. Однако однодетерми-
нантная волновая функция необязательно будет собствен¬
ной для операторов квадрата полного орбитального
и полного спинового моментов. Собственные функции этих
операторов представляются линейными комбинациями де¬
терминантов Слэтера, соответствующих одним и тем же
N N
значениям квантовых чисел ML = £ ш(0 и ms(i)
i = i г=1
в пределах выбранной конфигурации.
Под электронной конфигурацией атома понимается
определенное распределение электронов по «/-оболочкам:
/
К = (nll1)Vi(n2l2y2...(nflf)v', причем £ vp = ЛГ
Р ~ 1
Каждая пр1р-оболочка представляет набор 2(2/р + 1)
спин-орбиталей, из которых vp заселены, т. е. включены в де¬
терминант Слэтера. Эти vp спин-орбитали можно выбрать
из (пр/р)-оболочки C2p(2/p+i) способами. Следовательно, кон-
/
фигурации К соответствует J~[ Cv2\2i +1) однодерминант-
р = 1
ных функций, причем их число определяется фактически лишь
незамкнутыми оболочками, для которых vp < 2(2Ip + 1).
Например, для конфигурации ls22s22p2 атома углерода
можно построить С\ = 15 детерминантов. Из них можно
составить далее 15 линейных комбинаций, соответствующих
определенным значениям квантовых чисел L и S и образую¬
щих атомные термы.
Термом называется совокупность многоэлектронных
функций определенной конфигурации, характеризующаяся
общими для всех функций терма значениями квантовых чи¬
сел полных орбитального и спинового моментов (L и S). От¬
дельные волновые функции терма различаются по кван¬
товым числам проекций указанных моментов (Мг и Ms).
Если не принимать во внимание взаимодействие орби¬
тального и спинового моментов, то все волновые функции
терма отвечают одному и тому же (2L+ 1)(2S + 1)-кратно
вырожденному энергетическому уровню атома. Спин-орби-
тальное взаимодействие приводит к расщеплению этого вы¬
рожденного уровня на уровни тонкой структуры, характе¬
ризуемые квантовым числом полного спин-орбитального
момента J. Поправка на спин-орбитальное взаимодействие
89
определяется приближенным выражением
№KLSj = уArisW + 1) - L(L + 1) - S(S + 1)]
из которого следует правило Ланде для константы спин-ор-
битального взаимодействия:
akls = (&ekls,j ~ &EKLS,J- l)/J
Легко убедиться, что
Ed J + DA.Eklsj = О
j
т. е. энергия терма равна средневзвешенному значению энер¬
гетических уровней тонкой структуры:
Х(2 J + i)ekls,j
j? _ _£
KLS (2 L + 1)(2S + 1)
Согласно правилам Хунда, энергия EKLS j будет наимень¬
шей, если: 1) квантовое число S максимально; 2) при равных
S максимально квантовое число L; 3) при равных S и L кван¬
товое число J максимально при AKLS < 0 и минимально при
А-KLS > О-
В качестве примера использования правил Хунда рас¬
смотрим структуру энергетических уровней атома углерода
для конфигурации \s22s22p2 (рис. 22). Из пятнадцати одно-
терминантных шестиэлектронных функций этой конфигура¬
ции можно составить девять функций терма 3Р (L = 1 и
S = 1), пять функций терма lD (L= 2 и 5 = 0) и единствен¬
ную функцию терма 1S (L= 0 и S = 0). Наименьшей
энергии отвечает терм 3Р, обладающий максимальной муль-
типлетностью по спину. За ним следует терм XD, поскольку
он характеризуется большим значением квантового числа L,
чем терм lS, при равной спиновой мультиплетности.
90
Рис. 22. Энергетические уровни атома углерода.
Спин-орбитальное взаимодействие приводит к расщепле¬
нию лишь терма ЪР, так как для остальных термов полный
спиновый момент равен нулю (а мультиплетность- единице).
Для терма 3Р константа А > 0 и, следовательно, уровни
тонкой структуры этого терма возрастают в последователь¬
ности 3Р0, 3Pt, 3Р2, где нижний индекс указывает значения
квантового числа J.
Строго говоря, орбитальные энергии &п1 различны для
разных термов одной конфигурации. Согласно расчету
Э. Клементи атомным орбиталям lsz2s22p2-конфигурации
углерода в зависимости от терма соответствуют энергии enJ
(эВ):
C:ls22s22p2
3 р
lD
ls
2 Р
-11,70
- 10,34
-8,43
2s
- 19,32
-19,59
-20,13
Is
- 308,31
- 308,85
- 309,93
91
Таким образом, расстояние между энергетическими уров¬
нями 2s- и 2р-АО при переходе от терма 3Р к терму 1S уве¬
личивается почти на 4,3 эВ (414,9 кДж/моль).
В большей степени орбитальные энергии зависят от
атомной конфигурации. Эту зависимость можно показать на
примере рассмотренной выше Ь22з22р2-конфигурации и воз¬
бужденных ls22s2p3 и Ь’22р4-конфигураций атома углерода.
Из множества термов, соответствующих этим конфигура¬
циям, выберем термы 3Р и 1D (в эВ):
С* : ls22sl2p3
3Р
С* : ls22p4
3 р
'D
2 р
-9,80
— 8,98
2 р
-1,09
-0,27
2 s
- 22,86
— 16,87
—
—
—
Is
- 308,85
- 309,39
Is
- 294,14
- 294,41
Под полной электронной энергией атомной конфигура¬
ции следует понимать средневзвешенное значение энергии ее
термов:
£I(2L+ 1X2S + 1)EKIS
F _ L S _ _
K~ ZK2L+ 1)(2S + 1)
L S
= EEI(2L + l)(2S+l)(2J + l)£J£i.s/ {\ c;-(2,i + 1|
L S J P~1
Было бы ошибкой отождествлять энергию конфигурации
с суммой орбитальных энергий
/
■^орб - ILj Vp£p
р = 1
Эта величина, как и орбитальные энергии, определяется
не только конфигурацией, но и термом атомного состояния.
92
Кроме того, Еорб составляет лишь часть, причем меньшую
часть, полной электронной энергии термов.
По мере увеличения заряда атомного ядра погрешности,
связанные с пренебрежением одноэлектронным спин-орби-
тальным взаимодействием, увеличиваются, и приходится
учитывать расщепление каждой (и/) -оболочки на две под-
оболочки, различающиеся новым спин-орбитальным кван¬
товым числом /': _ , {'=/4-1
J ; J L + 2
£nl(
Ч 7 1
£ш/г; j 2
При этом атомные спин-орбитали уже не могут быть
представлены как произведение орбитали и спиновой функ¬
ции (а или Р) и конфигурация атома характеризуется распре¬
делением электронов по (п//)-оболочкам:
(Ир^р)р ~*{nplpjp) ’’{ftplpjp)р •> Vp "Ь vp = ^р
Многоэлектронные волновые функции, соответствующие
уровням тонкой структуры, строятся в этом приближении,
называемом приближением j — j-связи, непосредственно из
детерминантов «расщепленной» конфигурации.
Схему j — ;-связи иллюстрирует пример атома свинца,
основная конфигурация которого (...6s26p2) аналогична ос¬
новной конфигурации атома углерода (...2s22p2), но суще¬
ственно отличается от последней структурой энергетических
уровней (рис. 23).
Следует подчеркнуть, что выбор квантовых чисел, опре¬
деляющих состояние атома, зависит от того, в каком при¬
ближении мы его рассматриваем. Так, без учета спин-орби-
тального взаимодействия состояния атома характеризуются
квантовыми числами Lh S. Однако при учете этого взаимо¬
действия уже нельзя говорить о сохранении орбитального
и спинового моментов по отдельности, и соответствующие
им квантовые числа L и S не будут более «хорошими» кван-
93
ш
Рис. 23. Энергетические уровни атома свинца.
товыми числами. Вместо них следует использовать кванто¬
вое число J, характеризующее полный спин-орбитальный
момент импульса, который в этом приближении будет со¬
храняться. В то же время, если спин-орбитальное расщепле¬
ние энергетических уровней достаточно мало, можно уста¬
новить соответствие между уровнями тонкой структуры
и определяемыми в более грубом приближении энергетиче¬
скими уровнями термов. Точно так же для тяжелых атомов
квантовое число I, характеризующее одноэлектронный орби¬
тальный момент импульса, перестает служить «хорошим»
квантовым числом, лишь только мы учитываем спин-орби¬
тальное взаимодействие на одноэлектронном уровне рас¬
смотрения.
Периодический закон и квантовая механика
Объяснение физического смысла Периодического закона
представляет собой одно из важнейших достижений кванто¬
вой механики в химии. Для понимания природы периодич¬
ности необходимо иметь в виду следующее:
94
-сходство физико-химических свойств атомов обус¬
ловлено сходством их электронных конфигураций,
причем в первую очередь важно сходство в распреде¬
лении электронов по внешним, валентным АО (сравни¬
те С: ls22s22p2; Si: ls2ls22p63s23p2 и т.д.);
- заполнение АО происходит в порядке возрастания
их энергий, что ориентировочно может быть выражено
следующей последовательностью:
Is < 2s < 2р < 3s < Зр < As ~ 3d < Ар < 5s ~ Ad < 5р <
< 6s ~ 5d ~ 4/ < 6р < 7s < ...
Эта последовательность справедлива только для
нейтральных многоэлектронных атомов, находящихся
в основном состоянии;
-заполнение АО происходит в соответствии
с принципом Паули, согласно которому каждая АО
(каждая ячейка) может быть заселена не более чем
двумя электронами, причем в случае двукратно засе¬
ленной орбитали электроны должны иметь противопо¬
ложные спины.
Здесь уместно еще раз остановиться на терминологии.
Электроны, обладающие одним и тем же значением п, обра¬
зуют электронные слои (слои с п = 1, 2, 3, 4, ... часто обо¬
значают буквами К, L, М, N, ...). Слои, в свою очередь, по¬
строены из оболочек, заполненных электронами с одина¬
ковым значением числа / (их еще называют nl-оболочками).
Каждый n-й период открывается элементом, у которого
начинает заполняться АО с главным квантовым числом п,
т.е. п-й слой, что всегда соответствует ns-оболочке. Таким
образом, каждый период открывается щелочным металлом
с валентной электронной конфигурацией: ns1. Завершается
п-й период элементами, у которых наружные оболочки пол¬
ностью заселены. В первом периоде это соответствует кон¬
фигурации Is2 (Не), во всех остальных -ns2np6.
Кроме того, элементы делят на переходные и непере¬
ходные. Последние образуют главные подгруппы, а первые-
95
дополнительные. К непереходным относят ns- и пр-эле¬
менты, т.е. элементы, у которых заполняются ns- и пр-АО.
Переходными называются элементы, в атомах которых про¬
исходит заполнение {п - 1 )d или (п — 2)/ оболочек. Приме¬
ром d-элементов могут служить З^-элементы 4-го периода
(от Sc до Zn). К /-элементам относятся лантаноиды (запол¬
няется 4f оболочка) и актиноиды (заполняется 5/ оболочка).
Номер группы, к которой относится химический элемент,
равен числу наружных электронов его атома. Под наружны¬
ми электронами понимают у элементов главных подгрупп-
электроны, заселяющие оболочки «поверх» конфигурации
благородных газов, у элементов дополнительных под¬
групп - «поверх» оболочки из десяти (п — 1 )d электронов.
Каверзный вопрос
Как правило, порядок заполнения электронных nl-оболо¬
чек по мере увеличения атомного номера элемента (Is, 2s,
2р, 3s, 3р, 4s, 3d, Ар, ...) объясняется тем, что орбитальные
энергии в многоэлектронном атоме возрастают в той же по¬
следовательности. Так, например, «опережающее» заполне¬
ние 4s-AO в атомах К и Са по сравнению с 3d-AO связы¬
вают с тем, что £4s < e3d. Но тогда встает вопрос: почему
e4s < e3d ? Обычно ответ сводится к тому, что преимущество
4.S-AO обусловлено наличием трех «внутренних» локальных
максимумов, которые обеспечивают их большее проникно¬
вение в остов по сравнению с 3d-AO, не имеющими таких
максимумов.
Однако это объяснение нельзя признать удачным. Во-
первых, разница в узловой структуре орбиталей одинаковой
симметрии сама по себе еще не гарантирует определенного
соотношения их энергий. Во-вторых (и это самое важное!),
появление локальных максимумов, обусловленных ортого¬
нальностью 4s-AO к s-орбиталям остова, следует рассматри¬
вать скорее как проявление эффекта «выталкивания» этих
орбиталей из остова. Не будь условий ортогональности, 4s-
96
орбиталь «провалилась» бы в остов, превратившись в безуз-
ловую Is-АО, имеющую только один большой максимум на
ядре. Следует также заметить, что учет условий ортогональ¬
ности возможен и при использовании безузловых 45-орбита¬
лей, но с соответствующей заменой потенциала эффективно¬
го поля, действующего на описываемые этой орбиталью
электроны, псевдопотенциалом, который отличается от ис¬
ходного некоторой положительной добавкой. Иными слова¬
ми, условия ортогональности должны приводить к увеличе¬
нию орбитальных энергий.
На самом деле, порядок заполнения орбиталей обусло¬
влен не отношением их энергий, а требованием минимума
полной энергии атома, которая, как отмечалось выше, от¬
лична от суммы одноэлектронных энергий. Более того, сами
энергии орбиталей зависят от выбора конфигураций, т. е. от
порядка их заполнения.
Приходится признать, что порядок заполнения АО, опре¬
деляющий структуру Периодической системы, пока еще не
нашел удовлетворительного объяснения.
В ней уж нет ничего от коровы,
одна белизна лишь.
ОВИДИЙ
Глава III
ЧТО ТАКОЕ МОЛЕКУЛЯРНАЯ СТРУКТУРА?
Понятие о молекулярной структуре возникло более ста
лет тому назад. Но до сих пор не утихает полемика по по¬
воду того, что же следует понимать под структурой моле¬
кулы и, вообще, сколько у нее структур. Ниже мы коснемся
этой важной и трудной темы подробнее.
ИЕРАРХИЯ ПРИБЛИЖЕНИЙ
С позиций квантовой механики
С позиций квантовой механики молекула представляет
собой систему электронов и атомных ядер. Поэтому состоя¬
ния изолированной молекулы должны описываться вол¬
новыми функциями вида:
T(r1; CTi; г2, а2 fN, aN; Rt, R2, ..., RK)
(53)
где r; и ст4- соответственно, пространственные и спиновые коорди¬
наты электронов, Ку-координаты ядер, ЛГ-число электронов,
К -число ядер.
Строго говоря, приведенная волновая функция должна
была бы зависеть и от спиновых переменных отдельных
ядер, однако мы здесь не будем учитывать такую зависи¬
мость.
98
Физический смысл волновой функции (53) состоит в том,
что квадрат ее модуля определяет вероятность одновремен¬
ной локализации первого электрона в окрестности точки (гь
стО конфигурационного (координатно-спинового) пространс¬
тва, второго электрона-в окрестности точки (г2, ст2) этого
пространства и т.д., в то время как первое ядро находится
в окрестности точки Rlt второе-в окрестности R2 и т.д. Су¬
щественным обстоятельством при этом оказывается прин¬
ципиальная неразличимость всех электронов молекулы,
в силу чего целесообразно ввести функции электронной
плотности, определяющие вероятность локализации любого
из N электронов системы в окрестности точки г, независимо
от значения их спиновых переменных и координат ядер.
х '¥{rbcs1;...-,r'hGi;...,rN,oN;R1,Rn)d3rrd(7l ...d3rf_ t х
х d<3i _ 1daid3ri + {dai+ 5 ... d3 rNdaNd3 Rx ...d3~RK
где N к К, соответственно-числа электронов и ядер в молекуле.
Аналогичным образом могут быть определены функции
распределения плотности локализации ядер или, как они бу¬
дут называться в дальнейшем-функции ядерной плотности:
х *Р(г,, а,; ...; ry, cry; Rit ..., R'h ..., Rfc)d3 rxdGx . ..d3 rNdaA\ x
При этом ядра, в отличие от электронов, считаются раз¬
личными, даже если они обладают одинаковыми зарядами,
массами и находятся в одинаковых «внутренних» состоя¬
ниях (т.е. не являются изотопами или ядерными изомера¬
ми). Такое допущение вполне оправдано в силу относитель-
X d-3 /?!... d:s Rj _ ! d3 Rj + i ... d3RK
но большой массы ядер
и малости
7*
99
квантовомеханических эффектов, связанных с неразличи¬
мостью одинаковых ядер.
Очевидно, и это следует подчеркнуть особо, - вследствие
однородности и изотропности пространства для изолирован¬
ной молекулы вероятность найти любой электрон или любое
ядро в окрестности любой точки внутримолекулярного про¬
странства одинакова, т, е. величины рв,(г) и р} в дей¬
ствительности от г и Д. не зависят. Поэтому, в строгой
квантовомеханической теории нет аналога классического по¬
нятия молекулярной структуры (как бы мы ни понимали
этот термин). Слова «в строгой теории» означают,, что речь
идет о состояниях молекулы, описываемых собственными
функциями (53) ее полного нерелятивистского гамильтониа¬
на, вид которого приводится ниже.
Иными словами, представления о химической связи ме¬
жду атомами, о геометрии молекулы, ее симметрии и топо¬
логии и многие другие имеют смысл только в рамках опре¬
деленных приближений, вообще говоря, не вытекающих
из основных (или, как часто говорят, первых) принципов
квантовой механики*. В свою очередь, выбор приближения
определяется не только характером постановки решаемой
задачи, особенностями рассматриваемой системы, а также
соображениями физического и математического порядка, но
учитывает (чаще всего, неявно) весь рациональный опыт ис¬
торического развития данной предметной области, причем
последний фактор не менее важен, чем все остальные.
В самом деле, что заставляет теоретиков, занимающихся
изучением строения молекул, немало сил тратить на обсуж¬
дение проблем локализации молекулярных орбиталей, выбо¬
ра оптимального анализа заселенностей и т. д. ? Ведь в прин¬
ципе расчет можно провести, используя делокализованные
(канонические) молекулярные орбитали, или же ограничить¬
* При этом сама квантовая механика является приближенной
теорией по отношению, скажем, к квантовой электродинамике.
100
ся одноцентровым базисом, в результате чего при стандарт¬
ном анализе заселенностей вся электронная плотность ока¬
жется отнесенной к одному атому молекулы. Однако
в обоих случаях результаты расчетов не удается интерпрети¬
ровать в рамках традиционных химических представлений,
т. е. в терминах химических связей, неподеленных элек¬
тронных пар и т. д. И дело не только в необходимости учета
старых, давно известных фактов типа аддитивности и транс-
ферабельности многих молекулярных свойств, дело еще
в стремлении согласовать квантовомеханическое описание
с определенным исторически сложившимся стилем химиче¬
ского мышления. Но мы слишком забежали вперед, вернем¬
ся к нашей теме и посмотрим, как в квантовой химии «ро¬
ждается» понятие молекулярной структуры.
Адиабатическое приближение
Первый шаг на пути к квантовомеханическому аналогу
классического понятия молекулярной структуры состоит
в отделении поступательного (трансляционного) и враща¬
тельного движений молекулы как целого от внутримолеку¬
лярных движений. Это осуществляется посредством перехо¬
да от неподвижной (лабораторной) системы координат
к координатам центра тяжести молекулярной системы и
к относительным координатам*. Не останавливаясь на ма¬
тематической стороне дела, заметим, что отделение поступа¬
тельного движения приводит к радиально-неоднородному
распределению электронной и ядерной плотности в молеку¬
ле, а отделение вращения обусловливает угловую неодно¬
родность этого распределения.
В итоге структура химического соединения будет (после
отделения указанных движений) определяться функциями pei
и р uc.
* Детальное изложение этого вопроса читатель сможет найти
в книге: Д.И. Блохинцев. Основы квантовой механики. М.: Наука,
1976, с. 445-448, 655, 656.
101
Далее, как правило, используют так называемое адиаба¬
тическое (от греческого «адиабатос»-замкнутый, непрохо¬
димый) приближение или-другое название-приближение
Борна - Оппенгеймера (1927 г.). Остановимся на нем подроб¬
нее.
Модельная задача
Начнем с простой модельной задачи о движении ча¬
стицы в потенциальном ящике с бесконечно высокими стен¬
ками, которую мы рассматривали в главе I. Но на этот раз
несколько видоизменим ее условия: пусть существует внеш¬
няя сила, меняющая размер ящика по закону
а = а0 + ах sin юt
где to - циклическая частота, связанная с периодом движения стенки
(Г) соотношением со = 2п/Т, а-размер ящика.
Допустим также, что скорость изменения ширины ящика
а много меньше скорости движения частицы в нем. Тогда за
малый интервал времени At« Т величина а практически не
изменится, тогда как частица успеет несколько раз «пробе¬
жать» от одной стенки до другой! Ясно, что это возможно,
если время х одного пробега расстояния а0 будет много
меньше Т (т « Т).
Из сказанного следует, что во время At (т < At < Т) ча¬
стица будет находиться в квазистационарном состоянии, так
как за это время размер ящика, а потому и потенциальная
энергия, практически не изменяется и можно считать, что
частица будет оставаться в данный временной промежуток
на некотором энергетическом уровне Еп, не меняя квантово¬
го числа п, хотя величина Еп будет медленно меняться во
времени по закону:
Иными словами, состояние движения частицы, описывае¬
мое волновой функцией
будет как бы «подстраиваться» под медленное изменение
внешних условий. В таких случаях говорят, что движение
имеет адиабатический характер.
В соответствие с соотношением неопределенности, мож¬
но записать
Следовательно, расстояние а0 частица пройдет за время
Тогда приведенное выше условие х« Т примет вид
тенциальном ящике.
Итак, если неравенство (54) выполнено, то можно гово¬
рить об адиабатическом («подстраивающемся») движении,
если же частота внешнего поля со сравнима с собственной
Ч> (х, t) =
{sin (х ]/2mE„ (t) /И)} е ~l,h JE"(t)dt
mva0 ~ h
откуда:
h
v
ma0
х
v h
ИЛИ
CO «со,
'p
(54)
где G)p
2nk
-^2—характеристическая частота движения частицы в по-
частотой сор (случай резонанса), то состояние движения ча¬
стицы изменяется и она переходит на другой энергетический
уровень.
Критерий адиабатичности
Можно ли применить подобные рассуждения к молеку¬
лам? Да, можно, причем двояко. Во-первых, из спектроско¬
пии известно, что характеристические частоты электронов
в молекулярных системах лежат в видимой и ультрафиоле¬
товой областях спектра, тогда как частоты колебаний
ядер-в инфракрасной области, так что (озе//соЫие1) ~ 100
и критерий адиабатичности для молекул выполняется (прав¬
да, как мы увидим далее,-не всегда). Во-вторых, слоистое
строение электронных оболочек атомов и молекул позво¬
ляет разделить электроны на группы в зависимости от ско¬
рости их движения, так как периоды движения оптических
(валентных) электронов и электронов остова существенно
различаются. В настоящее время адиабатическое разделение
быстрых и медленных электронов применяется главным
образом в теории атомов, и мы о нем в дальнейшем гово¬
рить не будем, сосредоточив внимание на адиабатическом
разделении электронных и ядерных движений.
Историческая справка
Это разделение широко используется в квантовой химии
и в молекулярной спектроскопии. Исторически оно проводи¬
лось еще до появления квантовой механики. Первая попыт¬
ка обосновать адиабатическое приближение принадлежала
Борну и Гейзенбергу (1924 г.), но она оказалась неудачной,
так как неправильно был выбран параметр малости, по ко¬
торому производилось разложение энергии молекулы. Вто¬
рая попытка (Борн и Оппенгеймер, 1927 г.) удалась, в ре¬
зультате чего полуинтуитивные рассуждения химиков
и спектроскопистов получили квантовомеханическое обосно¬
104
вание. В дальнейшем разработкой этого вопроса занима¬
лись как сам Борн, так и многие другие авторы.
Не вдаваясь в математические детали, рассмотрим ос¬
новные особенности приближения Борна - Оппенгеймера.
Уравнение для электронов
Запишем полный нерелятивистский гамильтониан моле¬
кулы следующим образом:
Н = f Nucl (R) + Tel(f) + U (r, R)
где TNuc'(i?) и Tel (г),- соответственно, операторы кинетической
энергии ядер и электронов, U (F, R)- оператор потенциальной энер¬
гии молекулы, включающей энергию притяжения электронов
к ядрам, а также энергии межэлектронного и межъядерного
отталкивания.
Уравнение Шредингера для молекулярной системы при¬
мет тогда вид:
Я¥(г, R) = ЕЧ' (г, R)
где Е- полная энергия молекулы, a R и г-совокупности ядерных
и электронных координат (спиновые переменные не учтены).
Зависимость волновой функции от двух наборов динами¬
ческих переменных (R и г) выражает тот факт, что движения
электронов и ядер в молекуле строго говоря, связаны между
собой. Но, как уже отмечалось, этой связью часто пренебре¬
гают, полагая, что указанные виды внутримолекулярных
движений можно разделить, т. е. считать их как бы незави¬
симыми. Тогда подсистемы ядер и электронов будут харак¬
теризоваться каждая своей волновой функцией и своим
уравнением Шредингера. Для электронной оболочки моле¬
кулы последнее может быть записано следующим образом:
Не1Фт(г\Щ = em(R)<bm{r\R) (55)
где Hel — Tel + U(г | К)-электронный гамильтониан.
105
Собственные значения em(R) и собственные функции
Фт (г | R) этого уравнения характеризуются набором кван¬
товых чисел электронного состояния (т) и зависят от
ядерных координат не как от динамических переменных,
а как от параметров, поскольку от них как от параметров
зависит Hel. По этой причине ядерные координаты в форму¬
ле (55) отделены от координат электронов вертикальной
чертой.
Таким образом, для каждого фиксированного R, т.е. для
каждой фиксированной ядерной конфигурации, собственная
функция Фт(г|Я) гамильтониана Hel описывает состояние
движения электронов в поле неподвижных^ ядер. Собст¬
венные значения гамильтониана Hel, т.е. em{R), называются
электронными термами молекулы. Каждый электронный
терм представляет собой энергетическую гиперповерхность
в ЗК-мерном пространстве ядерных координат.
Уравнение для ядер
Подсистема ядер, при заданном электронном состоянии,
в рассматриваемом приближении описывается волновыми
функциями %mk(R), которые характеризуются совокупностью
квантовых чисел к ядерных состояний и являются собст¬
венными функциями гамильтониана #Nucl:
= [rNucl + £<К)])и(Ю = Ex,k(R) (56)
где выражение
< (К) = zJK) + (f IR) ТШс1Фт 0r \R)d2f
называется^адиабатическим потенциалом. Как правило, в ка¬
честве &^{R) используют электронный терм молекулы, т.е.
tf(R) = em(R).
106
Полная волновая функция молекулы \J/m*(r, R) в адиаба¬
тическом приближении вычисляется в виде произведения
ядерной и электронной функций:
Утк{г, R) = 1тМФт(г\т (57)
При квантовомеханическом рассмотрении молекулы
в приближении Борна - Оппенгеймера сначала решается элек¬
тронное уравнение (55), а затем полученные значения элек¬
тронных термов em (R) используются для исследования дина¬
мики ядер, описываемой уравнением (56).
Коварная теорема
Без преувеличения можно сказать, что большая часть
квантовохимических работ выполняется в адиабатическом
приближении. Однако, как показывает детальный математи¬
ческий анализ, при наличии электронного вырождения это
приближение оказывается неприменимым. В этом случае
адиабатический потенциал теряет смысл потенциальной
энергии ядер (и, соответственно, поверхности потенциальной
энергии), становясь формальным понятием. Вместе с ним те¬
ряют физический смысл и отдельные состояния вырожден¬
ного электронного терма, которые полностью смешиваются
с колебаниями ядер и образуют качественно новые состоя¬
ния, называемые вибронными или электронно-ядерными.
Кроме того, при конфигурации ядер Q° = {Qi, Q*k}*, от¬
вечающей наличию электронного вырождения существуют
некоторые особенности в поведении адиабатического потен¬
циала, описываемые теоремой Яна-Теллера. Содержание
этой теоремы, которую мы здесь приводим в формулировке
И. Б. Берсукера, состоит в следующем.
* Здесь -совокупность координат i-ro ядра, но координат не
декартовых, а нормальных, использование которых позволяет запи¬
сать энергию колебаний в наиболее простом виде (см. нашу книгу
«Симметрия в мире молекул». J1.: Химия, 1976).
107
Рис. 24. Адиабатический потенциал вблизи точки
вырождения.
Пусть адиабатический по¬
тенциал , Qk) нелиней¬
ной симметричной молекулы,
являющийся формальным ре¬
шением электронного уравне¬
ния Шредингера, имеет несколь¬
ко пересекающихся в точке Q°
ветвей. (Для примера, на рис. 24
представлен случай двукратно¬
го вырождения, т. е. когда
двум электронным состояниям Фх и Ф2 нелинейной сим¬
метричной молекулы отвечают в точке Q0 одинаковые
значения Ead, т.е. имеет место пересечение ветвей адиаба¬
тического потенциала). Тогда в этой точке потенциал не
имеет минимума. Иными словами, для нелинейной сим¬
метричной многоатомной системы в случае электронного
вырождения всегда найдутся такие ядерные смещения,
для которых (8s/dQ)Q0 Ф 0.
При этом оказывается, что получаемое основное виброн-
ное состояние обладает той же кратностью вырождения, той
же мультиплетностью и теми же прочими сопоставимыми
характеристиками, что и исходный электронный терм при
максимально симметричной конфигурации ядер.
Поэтому отсутствие минимума адиабатического потен¬
циала вблизи точки вырождения, вообще говоря, нельзя ин¬
терпретировать как условие обязательного самопроизвольно¬
го искажения исходной симметричной ядерной конфигура¬
ции и перехода к новой, менее симметричной и потому
лишенной исходного вырождения.
Короче говоря, вблизи точки вырождения адиабатиче¬
ский потенциал минимума не имеет, но изменится ли от
108
этого ядерная конфигурация, сказать заранее, без рассмотре¬
ния уравнения Шредингера для подсистемы ядер, нельзя.
С точки зрения химика
Вернемся, однако, к приближению Борна - Оппенгеймера.
Для химика его значение чрезвычайно велико, так как оно
привносит в теорию строения молекул широкий круг фунда¬
ментальных понятий. Прежде всего сюда относятся практи¬
чески все стереохимические понятия и представления (длина
химической связи, угол между связями, конформация, кон¬
фигурация, симметрия ядерного полиэдра и т.д.), а также
понятия многомерной поверхности потенциальной энергии
и потенциальной кривой и многие, многие другие, которые
вне рамок адиабатического приближения теряют смысл.
Более того, это приближение позволяет обосновать расг
смотрение химической связи как «электронного» явления
и сформулировать критерий ее образования на языке потен¬
циальных поверхностей (или кривых), т.е. позволяет утвер¬
ждать, что основному состоянию химически связанных ато¬
мов соответствует минимум на потенциальной поверхности
(кривой).
Тяга к интерпретации явлений в терминах понятий, орга¬
нически связанных с адиабатическим приближением, у хими¬
ка столь велика (ибо тяга эта закалена и укоренена в более
чем вековой традиции химического мышления), что даже
в случаях его неприменимости наблюдается тенденция со¬
хранить хоть в какой-то мере традиционный стиль химиче¬
ского теоретизирования. Однако открытия современной хи¬
мии неумолимо ломают старые каноны.
МОЛЕКУЛЫ ЖЕСТКИЕ И НЕЖЕСТКИЕ
Структура химического соединения в значительной сте¬
пени определяется особенностями потенциальной поверх¬
ности, которая может иметь множество минимумов с раз¬
109
личными характеристиками. Рассмотрим несколько наибо¬
лее важных случаев.
Изомеры и квантовая механика
Если минимумы на потенциальной поверхности доста¬
точно глубоки и неэквивалентны, т. е. различаются по
глубине и (или) по форме, то каждая волновая функция ядер
%mk{R) будет локализована в окрестности одного из них
и молекулярная структура будет характеризоваться коор¬
динатами этого минимума (Rmin). Каков химический смысл
этого утверждения? Он состоит в том, что каждому мини¬
муму на поверхности потенциальной энергии отвечает опре¬
деленная ядерная конфигурация, при этом конфигурации
с наиболее глубоким минимумом будет соответствовать ос¬
новное состояние ядерной подсистемы молекулы, тогда как
остальным конфигурациям-возбужденные состояния этой
же молекулы. Таким образом, квантовомеханически мы
имеем одну и ту же молекулярную систему в различных
ядерных состояниях, каждому из которых отвечает свое рас¬
пределение электронной плотности и своя совокупность
свойств. Но в силу того, что переход из одной конфигура¬
ции в другую сильно затруднен (из-за большой глубины
и неэквивалентности минимумов) химик воспринимает
ядерные состояния, локализованные в окрестностях каждой
такой «потенциальной бездны», как разные химические
соединения с одинаковым брутто-составом и называет их
изомерами.
Например, взаимные превращения так называемых ва¬
лентных изомеров бензола
01 (!) о й и т-л
бензол фульвен бензвален призман
110
могут осуществляться фотохимически, тогда как термиче¬
ские превращения невозможны вследствие высоких потен¬
циальных барьеров между минимумами, а туннельные пере¬
ходы-вследствие неэквивалентности последних, а также
из-за их большой высоты и ширины.
В настоящее время понятие изомерии заметно усложни¬
лось. Возросло, по сравнению с прошлым веком, число раз¬
личных видов изомерии (особенно, стереоизомерии). Кроме
того, нет единого мнения относительно правомерности ис¬
пользования этого понятия в случаях, когда вещества неста¬
бильны (например, циклооктатетраен, [4.2.0]-бициклоокта-
триен и т.д.), т.е. когда, в отличие от рассмотренной выше
ситуации, разделяющие минимумы барьеры невысоки. Это,
в свою очередь, требует уточнения выражения «глубокий
минимум». По мнению некоторых исследователей (Э. Илие-
ла, В. Прелога, К. Мислоу и др.), разумно рассматривать
одинаковые по составу соединения как изомеры, если барье¬
ры между соответствующими минимумами выше кТ (т. е.
выше 2,5 кДж/моль при 25°С).
Аммиак наизнанку
Теперь обратимся к потенциальным поверхностям, мини¬
мумы которых глубоки^ но эквивалентны. В этом случае
волновые функции %mk(R) будут делокализованы по таким
минимумам. В простейшем случае эту ситуацию можно
смоделировать, рассмотрев одномерное движение частицы
в поле потенциала с двумя эквивалентными минимумами.
Впервые такая задача была поставлена Ф. Хундом в мае
1927 г. Он показал возможность существования энергетиче¬
ски равноценных ядерных конфигураций многоатомных мо¬
лекул, причем время перехода из одной конфигурации в дру¬
гую может, по его словам, «иметь порядок от атомных до
космических величин» в зависимости от высоты барьера.
Впоследствии аналогичные задачи рассматривались многи¬
ми физиками в связи с разнообразными проблемами-
111
А. Нордхеймом (1927 г.) при изучении термоэлектронной
эмиссии, Р. Оппенгеймером (1927 г.) при исследовании пове¬
дения атома водорода во внешнем электрическом поле и,
наиболее известный пример, Г. Герни, Э. Кондоном и
Г. А. Гамовым (1928 г.) в теории а-распада атомных ядер.
Не вдаваясь в обсуждение математической стороны дела,
отметим лишь наиболее существенные качественные ас¬
пекты проблемы Хунда. Самой важной чертой энергетиче¬
ского спектра, получаемого при рассмотрении этой про¬
блемы, является его «парный» (дублетный) характер, по¬
скольку соответствующие волновые функции могут быть
как симметричны, так и антисимметричны относительно
плоскости, проходящей через начало координат и перпенди¬
кулярной оси R (рис. 25а). Иными словами, спектр энергии
включает пары колебательных энергетических уровней Е(Л1 и
E(“l (v- колебательное квантовое число), расстояние между
которыми (AEmv) мало по сравнению с расстоянием между
уровнями одной симметрии-д (от нем. gerade-четный) и
и (ungerade - нечетный).
Если условие малости A£mv выполнено, то каждая пара
функций Xml и Xml может быть преобразована в две эквива¬
лентные функции
xJ,(R) = 1/1/2 [х“ (R) + хКОО]
и
Й, («) = 1/1/2 К (R) - A (R)]
каждая из которых локализована в окрестности одного из
минимумов (I или И) (рис. 256) и характеризуется средней
энергией колебательного уровня v:
к, = уК + ед
В этой ситуации структуру химического соединения нель¬
зя рассматривать независимо от методов его эксперимен-
112
а
б
Рис. 25. Волновые функции и спектр энергии при одномерном движении частицы в поле потен-
■01 ала с двумя эквивалентными минимумами:
а-точные волновые функции ядер, делокализованные по минимумам; б-функции ядер, ло¬
кализованные в окрестностях одного из минимумов.
тального исследования, которое связано с возмущением ис¬
ходного состояния изучаемого объекта.
Хорошим примером может служить молекула аммиака.
Как было экспериментально установлено на основании ана¬
лиза ее вращательного спектра, в NH3 имеют место инвер¬
8-918
ИЗ
сионные колебания, т.е. молекула как бы «выворачивается
наизнанку». Определенный из микроволновых спектров
барьер инверсии оказался равным & 24,3 кДж/моль. Какой
«предстанет» эта молекула перед исследователем, зависит
от метода ее изучения.
Если энергия возмущения, характерная для данного ме¬
тода, меньше энергии расщепления колебательных уровней,
то наблюдается делокализация ядерной плотности по экви¬
валентным минимумам. Молекула аммиака в этом случае
будет иметь симметрию D3h (тригональная бипирамида)
и нулевой дипольный момент.
Если же энергия возмущения имеет тот же порядок что и
AEmv или больше этой величины, то ядра локализуются
в окрестности одного из минимумов. Тогда для аммиака
экспериментально наблюдается структура с симметрией
тригональной пирамиды (C3l)) и с отличным от нуля ди-
польным моментом.
Кстати, о симметрии...
Из сказанного ясно, что для классификации состояний
инверсионно-нежестких молекул пользоваться обычными
точечными группами симметрии основной конфигурации
уже нельзя. Например, группа C3v не годится для описания
колебательно-вращательных состояний молекулы аммиака,
так как не учитывает возможность инверсионной переста¬
новки ядер.
В 1963 г. английский ученый Лонге-Хиггинс предложил
использовать для таких случаев перестановочные группы*.
Для молекулы такая группа включает следующие операции
симметрии:
* О перестановочных группах см. гл. VII нашей книги «Моле¬
кулы без химических связей». В более полном виде теория таких
групп изложена в книге И. Г. Каплана «Симметрия многоэлек¬
тронных систем». М.: Наука, 1969.
114
Е- идентичная операция
{(123), (132)}-циклические перестановки к
атомов водорода
{(12), (13), (23)}-парные перестановки 2 з
атомов водорода
Е*- инверсия ядер относительно центра массы молекулы
и две сложные («комбинированные») операции
{(123)*, (132)*}-циклическая перестановка плюс инверсия
и
{(12)* (13)*, (23)*}-парные перестановки с последующей
инверсией.
По теореме Кэли любая конечная группа G порядка п
изоморфна подгруппе группы перестановок Р„, а в ряде
частных случаев G может быть изоморфна и самой группе
Рп. Так, например, описанная выше группа перестановок
изоморфна точечной группе D3h (Е = Е, (123) = С3, (12) = С2,
Е* = стА, (12)* = а„, (123)* = 53 и состояния молекулы ам¬
миака можно классифицировать по неприводимым предста¬
влениям этой группы.
Величина барьеров
Несколько слов следует сказать о влиянии природы ли¬
гандов и центрального атома на инверсионную нежесткость.
Как показали исследования, замещение атомов водорода
в аммиаке на более электроотрицательные лиганды, имею¬
щие неподеленные электронные пары, например галогены,
—NH2 (и вообще —NR2), —OR, приводит к заметному уве¬
личению инверсионного барьера. Так, последовательное за¬
мещение водорода на фтор (NH3 -> NH2F -> NHF2 -+ NF3)
сопровождается резким возрастанием барьера
с 24,3 кДж/моль (NH3) до ^84, ^ 176 и 314 кДж/моль. На¬
оборот, электроположительные заместители понижают
барьер, стабилизируя плоскую структуру, как это имеет ме¬
сто в соединениях NH2BH2, NH2Li, NH2BeH и т.д.
8»
115
Что касается влияния природы центрального атома на
величину барьера инверсии, то для молекул состава АХ3 ин¬
версия наиболее отчетливо проявляется, когда А-атом эле¬
мента главной подгруппы V группы или изоэлектронный
с ним ион (NH3, РН3, СН3 , НэО+ и т.д.).
Короткая жизнь энергетического уровня
Расщепление колебательного уровня (AEJ можно связать
со временем его жизни т„. Произведение этих величин имеет
порядок постоянной Планка: т„АEv ~ к.
В частном случае двух эквивалентных минимумов
и одной колебательной (инверсионной) координаты (модель¬
ная задача) можно показать, что вероятность Dv прохожде¬
ния системы из одной потенциальной ямы в другую равна:
где v„- частота колебаний в одной из потенциальных ям.
Тогда «частота туннелирования» ядер сквозь потен¬
циальный барьер определяется как произведение Dvvv, а ве¬
личина, обратная этому произведению, будет характеризо¬
вать время жизни колебательного уровня v:
1 1 2h
1 \DV гсД Ev AEV
откуда xAEv = 2h.
Энергия возмущения, вызываемого «вмешательством»
прибора при экспериментальном изучении структуры хими¬
ческого соединения, также может быть связана со временем
взаимодействия «молекула-прибор» т', причем различные
экспериментальные методы характеризуются разными зна¬
чениями этой величины:
116
Метод т\ с
Электронография 10 “ 20
Нейтронография 10"18
Рентгенография 10"18
УФ-спектроскопия 10-15-10-14
ИК- и КР-спектроскопия 10~13
ЭПР 10 "МО" 8
ЯМР 10-МО"9
Исследование молекулярных 10"6
пучков
Для сравнения укажем «времена жизни» т (в с) неко¬
торых локализованных состояний при v = 0:
NH3 2,5-10“11 ND3 2,7-10-10
РН3 10 ~3 AsH3 10
РС15 104 PF5 10"5
Из этих данных видно, в частности, что для молекулы
аммиака электронографическим методом определяется
структура C3v, а метод ЯМР или исследование молеку¬
лярных пучков свидетельствуют о более высокой симметрии
D3h. В первом случае молекула NH3 обладает ненулевым
постоянным дипольным моментом, во втором случае он
отсутствует.
Овраги и мелкие минимумы
Когда молекулярная система обладает потенциальной
поверхностью со множеством минимумов, разделенных
малыми и легко преодолимыми барьерами, то ее структура
уже не может характеризоваться ядерной^ конфигурацией,
так как плотность распределения ядер Pj(Rj) в этом случае
существенно делокализована. Атомы или фрагменты таких
молекул постоянно мигрируют из одной внутримолекуляр¬
ной области в другую на расстояния порядка длины хими¬
ческой связи и более.
В качестве примера приведем молекулу цианида лития.
Расчет и эксперимент показали, что линейная изоцианидная
117
конфигурация Li—N=C является основной-ей отвечает
минимум адиабатического потенциала. Однако энергетиче¬
ский барьер на пути миграции катиона лития, ведущей
к образованию цианидной структуры (LiNC -»• LiCN), неве¬
лик и составляет около 26,3 кДж/моль. В случае миграции
NaNC ->• NaCN барьер еще ниже (примерно в два раза). Не¬
большая величина барьеров приводит к деформационным
колебаниям большой амплитуды, если £возб меньше высоты
миграционного барьера (/iMHrp) и к «скольжению» катиона
щелочного металла вокруг аниона при £возб ^ ^МИГр-
Для ряда молекул, таких как LiBH4, NaBH4, LiAlH4,
LiN03 и т.д., на потенциальной поверхности имеется по три
экстремальные точки, в которых катион щелочного металла
координирован с тремя (конфигурация типа t), двумя (Ь)
и одним (т) лигандом (рис. 26). Абсолютному минимуму от¬
вечает конфигурация типа t. Так как в окрестностях миниму¬
мов потенциальные поверхности имеют пологий характер,
то небольшой энергии возбуждения (порядка нескольких де¬
сятков кДж/моль) оказывается достаточно для внутримоле¬
кулярной перегруппировки. Так, ион Li+ может «квазисво-
бодно» мигрировать вокруг тетраэдрического аниона ВН4_
по поверхности, близкой к сфере. Структура молекулы
LiBH4 будет тогда характеризоваться эффективной усред-
Н Н
t{C3u) ь(<у
Рис. 26. Три экстремальные конфигурации иона Li +
и
тЫ
аниона [ВН4]~.
118
Рис. 27. Схематическое изображение структуры молекулы LMX4
а- глобально нежесткой; б-жесткой
ненной конфигурацией с симметрией Td, что наглядно изо¬
бражено на рис. 27 (взятом из работы О. П. Чаркина).
Подобных «структурных формул» классическая химия не
знала. Не знала их и квантовая химия до начала 60-х гг.,
и только изучение нежестких молекул заставило пересмо¬
треть традиционное понимание термина «молекулярная
структура».
Кстати, о формулах!
Надо сказать, что привычные, школьные, структурные
(или графические) формулы соединений далеко не всегда
оказываются адекватными, даже если оставить в стороне
вопрос о структурной нежесткости молекул. Приведем один
пример. Как следует записать структурные формулы ни¬
трата и сульфата таллия (I)?
Если задать такой вопрос школьнику, он начертит для
TINO3 следующую графическую формулу
а для T12S04: Т1—
Т1—
Однако, как показывают электронографические исследо¬
вания, в парах нитрата таллия сосуществуют три структуры
причем первая преобладает (71%). Для сульфата реализуется
также неклассическая структура:
Как видим, порядок связи атомов может быть разным.
Итак, понятие молекулярной структуры далеко не одно¬
значно и включает в себя и топологическое понимание
структуры, восходящее к А. М. Бутлерову , и электронную
структуру соединения, и его ядерную структуру.
Однако наше рассмотрение последней будет неполным,
если мы не остановимся на таких стереохимических поня¬
тиях, как конформация, конфигурация и др.. Они часто
встречаются в литературе, но смысл их не всегда ясен.
ЕЩЕ РАЗ О ГЕОМЕТРИИ ЯДЕРНОГО ОСТОВА*
Чтобы правильно ориентироваться в современных спо¬
рах об интерпретации терминов «конформация», «конфор-
мер», «конформационный переход» и др. (о чем мы порассу¬
ждаем позже), необходимо представлять их генезис.
* Этот и следующий разделы написаны канд. хим. наук
С. М. Шевченко.
Т1
О
120
«Откуда есть пошла...»
Симптоматично, что сформировавшийся на протяжении
жизни одного поколения конформационный анализ стал уже
обрастать легендами. Наиболее странная и романтическая
из них связывает появление термина «конформация» с лите¬
ратурными пристрастиями выдающегося английского хими¬
ка У. Н. Хоуорта.
В. И. Соколов в монографии «Введение в теоретическую
стереохимию» (1979 г.) пишет: «Английское слово
conformation ввел Хоуорт в 1927 г. ...Впервые оно зареги¬
стрировано в поэзии Эдгара По». В журнале «Химия
и жизнь» (№ 1 за 1978 г.) 3. Е. Гельман уточняет, что слово
это использовано Э. По не в поэзии, а в прозе а именно
в рассказе «Береника», появившемся в 1874 г. Оба автора
ссылаются на весьма авторитетный источник-М. Стейси,
одного из ближайших учеников и коллег Хоуорта. Однако
в статье Стейси, представляющей собой запись инаугура¬
ционной лекции по случаю получения первой Хоуортовской
мемориальной медали Химического общества, сказано бук¬
вально следующее: «Доктор Р. С. Типсон сообщил мне сле¬
дующую цитату из «Береники» Э. А. По (приводится цита¬
та-С. Ш.). Хотя Хоуорт часто цитировал поэтов, например
Мильтона, Роберта Бриджеса и т.д., я сомневаюсь, взял ли
он слово из этого источника, но, конечно, он всегда размы¬
шлял о размерах своих ныне хорошо изученных кольчатых
систем, и он был очарован широкими различиями в устой¬
чивости колец, и вообще нет сомнения, что он мог думать
в трех измерениях». Кроме того, 3. Е. Гельман, хотя и связы¬
вает термин «конформация» с Э. По, справедливо отмечает,
что (вопреки мнению В. И. Соколова) слово
conformation - вовсе не неологизм великого романтика, оно
гораздо более давнего происхождения. «Словарь английско¬
го языка на исторических принципах» Мэррея (1901 г.)
указывает на 1511 г. как на дату первого появления этого
слова в письменном источнике. Оно встречается и, скажем,
121
у Бена Джонсона (1637 г.), не менее Э. По популярного в ан¬
глоязычных странах. Таким образом, ответ на вопрос, «от¬
куда пришло в химию слово конформация?» однозначен: из
литературного английского языка.
Зачем нужны новые слова?
Но важнее другое: почему именно это слово Хоуорт,
произвел, если можно так выразиться, в термины, причем
столь удачно, что он в конечном счете прижился? И вообще,
почему потребовался новый термин помимо обычного сло¬
ва «форма»? Кстати, в русском переводе знаменитой книги
Хоуорта «Строение сахаров» именно «форма» фигурирует
на месте оригинальной «конформации».
Здесь важно не допустить распространенной в отече¬
ственной литературе ошибки: обсуждая иноязычный термин,
имеющий в русском языке хождение в среде специалистов,
авторы иногда забывают, что собственно научным терми¬
ном, который должен обозначать лишь строго определенное
понятие, он является лишь для нас. Для носителей того язы¬
ка, из которого он пришел, он-слово, смысл которого «ин¬
туитивно ясен». В такой ситуации мы нередко находимся
в более выгодном положении, чем носители языка, в кото¬
ром термин впервые возник, поскольку для них данное сло¬
во обладает не единственным значением, но более или менее
широким спектром значений. В результате может возник¬
нуть двусмысленная ситуация, когда разные исследователи
одинаково называют разные вещи. В полной мере такая не¬
приятная возможность осуществилась и в случае понятия
«конформация».
По нашему мнению, введение в химический обиход слова
conformation главным образом было инспирировано тем,
что спектр его значений много уже, нежели у гораздо более
старого и более распространенного слова form. Слово form
в английском языке, равно как и «форма» в русском, обни¬
мает столь широкий круг явлений, что не может рассматри¬
122
ваться как термин; при его использовании смысл явления
может оказаться затемненным. Когда же потребовалось
подобрать понятие, описывающее не всю совокупность
форм, а определенного вида формы молекул, и пришло на
помощь понятие conformation, одно (из трех) значение кото¬
рого как нельзя более подходило к существу описываемого
явления: способ, которым вещи придана форма посред¬
ством определенного расположения ее частей; форма, зави¬
сящая от расположения частей; структура, организация. От¬
метим еще, что слово conformation издавна использовалось
в анатомии, означая там взаимное расположение частей че¬
ловеческого тела. Оттуда, кстати, оно и попало в рассказ
Э. По «Береника», где возникает в соответствующем контек¬
сте.
Конформация и констелляция
Представляют определенный интерес причины, по которым
быстро распространившийся в 1950-е гг. термин «конформация» не
прижился поначалу в публикациях на немецком языке, где до по¬
следнего времени как правило использовался эквивалентный тер¬
мин «констелляция» (Konstellation), предложенный Ф. Эбелем
в 1932 г. Насколько нам известно, единственное внятное объясне¬
ние, чем констелляция лучше конформации, принадлежит В. Хюкке-
лю (1956 г.). Оно заключается в том, что термин «конформация»
вызывает ассоциацию с конформными отображениями в математи¬
ке, не имеющими ничего общего со стереохимией. А термин «кон¬
стелляция» напоминает о планетных констелляциях и, якобы, пред¬
почтительнее, потому что разнообразие последних обязано своим
существованием вращательному движению. Но необходимо отме¬
тить, что подобные «жесткие» ассоциации могут возникнуть только
у немца. Латинские слова conformatio и constellatio перешли в со¬
временные европейские языки, по-разному трансформировав свое
значение. По-немецки konform означает «единообразный, одинако¬
вый, конформный», либо «согласно, соответственно», a Konstella¬
tion-«положение звезд (в определенный момент), стечение обстоя¬
тельств, положение дел». Так что, действительно, использование
слова Konformation в интересующем нас смысле должно
123
было вызывать у носителя немецкого языка внутренний про¬
тест, поскольку связано с совсем иными ассоциациями, нежели
слово conformation у англичанина. По своему смыслу немецкое сло¬
во (Constellation заметно ближе к английскому conformation. В то
же время по-английски слово constellation имеет строго фиксиро¬
ванное значение «созвездие» без необходимых оттенков и поэтому
не могло привиться в англоязычной химической литературе. Во
французском языке значения слов conformation и constellation те же,
что и в английском.
Молекула становится реальностью
В конце 20-х гг. нашего века появились первые эксперимен¬
тальные данные по геометрии насыщенных циклов. Молекула
окончательно «обрела плоть и кровь», перестав быть до некоторой
степени умозрительной конструкцией. Стало ясно, что молекулу
как таковую можно измерить, и наиболее прозорливые химики по¬
чувствовали большие перспективы, таящиеся в такой возможности,
предугадывая связь важных физико-химических свойств с тонкими
особенностями стереохимии соединения. Хоуорт одним из первых
стал широко использовать молекулярные модели-пространствен¬
ные конструкции из шариков и стержней, дающие верное предста¬
вление о взаимном расположении частей молекулы. Необычайная
простота таких приспособлений не помешала им оказать (и оказы¬
вать по сей день) большую помощь химикам-органикам.
В своей монографии «Строение сахаров» (1929 г.) Хоуорт писал
именно о «конформациях моделей», которые, в его понимании,
должны были отражать реально наблюдаемую геометрию струк¬
турных единиц сахаров. Термин «конформация» должен был слу¬
жить целям отграничения и объединения определенного вида
структурной информации. Ныне мы ясно понимаем, что необычай¬
ная важность для химии тонких структурных эффектов, относящих¬
ся к сфере конформационного анализа, сделала вполне оправ¬
данным введение особого термина для их обозначения. Однако
к моменту выхода монографии Хоуорта появились лишь первые
разрозненные данные (рентгеноструктурного анализа и химических
исследований), подтвердившие теорию неплоских циклов Заксе -
Мора. Поэтому обобщения и вывод о перспективности этой теории
представляются авторам поздней монографии «Конформационный
анализ» (Э. Илиел и др., 1965 г.) «несколько преждевременными».
124
В действительности, заслуга Хоуорта и заключается в том, что он
указал на перспективность конформационных исследований. Хотя
это смелое предвидение впоследствии полностью оправдалось, не¬
достаток «предмета обобщения», т.е. работ, рассматривающих
форму молекул, привел к тому, что само обобщение, то есть поня¬
тие конформации, показалось ненужным и не появлялось в литера¬
туре в течение двадцати лет. Такая же судьба постигла и термин
«констелляция», введенный Эбелем в его главе фундаментального
коллективного труда «Stereochemie» в связи с обсуждением кресло¬
видной и ваннообразной форм циклогексана, а также гош- и транс¬
изомеров дизамещенных этанов. Он не ссылался на книгу англи¬
чанина, однако в его работе приведены ссылки на оригинальные
статьи Хоуорта того же периода. Поэтому можно предполагать,
что Эбель был знаком с подходом Хоуорта к строению углеводов.
Основное значение публикации Эбеля заключается в том, что в ней
была четко высказана мысль о связи конформационной (констелля-
ционной) изомерии с внутримолекулярным движением, а именно-с
внутренним вращением вокруг ординарной связи углерод-углерод.
Физики и... химики
В течение последующего двадцатилетия, когда работы, отно¬
симые ныне к сфере конформационного анализа, появлялись «по
капле», существовал определенный разрыв между структурными
исследованиями и исследованиями внутреннего вращения. Скорее
всего, это можно объяснить тем, что результаты первых предста¬
вляли тогда интерес прежде всего для химиков (хотя и не сразу
входили в их обиход), тогда как последние оставались «внутренним
делом» физики. Исследования внутреннего вращения развивались,
в связи с появлением новых для того времени спектральных мето¬
дов, интенсивно, и уже в 1944 г. был опубликован важнейший обзор
М. В. Волькенштейна в «Успехах химии»-обзор, содержащий ос¬
новы учения о ротационной изомерии. Развитая на основе этого
учения поворотно-изомерная модель оказалась весьма плодотвор¬
ной в физике макромолекул, однако связь работ по внутреннему
вращению с химией не выглядела очевидной и химики, по суще¬
ству, мало интересовались этими работами. Невидимая стена, еще
разделявшая в те годы физику и органическую химию, мешала
и физикам получить правильное представление о ценности их ра¬
бот. Публикации Хоуорта и Эбеля и даже послевоенные статьи, со¬
125
держащие конформационный подход и терминологию, были, по-
видимому, физикам неизвестны, и поэтому термин «конформация»
не сразу проник в физическую литературу.
Второе рождение
После второй мировой войны объем стереохимической инфор¬
мации стал резко расти. Теперь уже количество экспериментальных
данных настоятельно требовало обобщения, единой концепции. По¬
этому и был заново введен в литературу старый термин, оказав¬
шийся по своему смщслу идеально подходящим к новому времени.
Второе рождение термин «конформация» обрел в 1950 г. в статье
будущего Нобелевского лауреата Д. Бартона, в которой без каких-
либо ссылок дается следующее определение: «Слово конформация
используется для того, чтобы обозначать различающиеся ненапря¬
женные расположения в пространстве набора валентно связанных
атомов.» По нашему мнению, отсутствие ссылки на Хоуорта
здесь-не случайный огрех, поскольку она отсутствует и в более по¬
здней публикации Бартона. При этом в последней есть ссылка на
статью В. Прелога, в которой в аналогичном смысле используется
термин «констелляция»: «Как констелляции мы определим те
формы молекул, которые проистекают из свободного вращения во¬
круг ординарных связей, например кресло и ванна циклогексана».
В свою очередь, в этой обзорной работе Прелог не ссылается ни на
Эбеля, ни, тем более, на Хоуорта.
О пользе неформального общения
Интересно здесь то, что по своему духу определение
англичанина Бартона явно восходит к Хоуорту, а определе¬
ние Прелога, для которого родным языком был немецкий,
к Эбелю. Вышеизложенное позволяет предположить, что
слова «конформация» и «констелляция» все-таки существо¬
вали в научном обиходе между 1929 и 1950 гг., но-лишь
в устной речи, причем происхождение их было забыто. Они
не проникали в публикации, поскольку, как уже было сказа¬
но, до конца 40-х гг. объем и, казалось, значение структур¬
нохимических исследований не требовали особой термино¬
126
логии. Ведь введение терминологии-это признак обособле¬
ния какого-то раздела науки, а прежде, чем обособиться, он
должен сложиться. Так, еще в 1948 г. Бартон использовал
слова «форма» и «конфигурация» там, где в 1950 г. он уже
применял слово «конформация».
Как известно, непосредственное общение специалистов
чрезвычайно важно для плодотворных исследований, осо¬
бенно в новых областях. Так что есть основания считать,
что вклад Хоуорта и Эбеля заключается не только в том,
что они несколько опередили свое время, но и в том, что их
взгляды оказали непосредственное влияние на подход к во¬
просам стереохимии следующего поколения ученых.
Если теоретические концепции слишком опережают свое
время, они, как правило, остаются незамеченными и не
оказывают прямого влияния на развитие науки. Если они
в точности соответствуют достигнутому уровню познания,
их роль-лишь роль стартовой площадки для последующего
взлета. Наиболее удачным является промежуточный ва¬
риант, при котором взгляды, высказанные «с небольшим
опережением», определяют направление последующих изы¬
сканий и подход к новым явлениям.
Термин «конформация», по нашему мнению, имеет кон¬
цептуальный характер, поскольку, как постепенно выясни¬
лось, с ним связан определенный подход к стереохимии,
приведший к возникновению целой группы производных по¬
нятий (конформер, конформационный переход, конформа¬
ционный эффект и т.д.) и к выделению конформационного
анализа в самостоятельную область химии.
Неформальное общение осуществляется преимуществен¬
но в пределах одного коллектива исследователей, знаком¬
ство же со взглядами прочих групп, в особенности, если
речь идет о группах зарубежных, происходит, как правило,
по публикациям или официальным выступлениям. Поэтому,
вероятно, до появления работ 1950 г. слова «конформация»
и «констелляция» использовались, соответственно, в англо-
и немецкоязычных группах независимо. После этих публика¬
127
ций названные термины стали широко использоваться в пе¬
чатных работах, и уже к середине 1950-х гг. оформились
принципы конформационного анализа, главным из которых
является связь физических и химических свойств соединения
с предпочтительной конформацией его молекулы. Подготов¬
ка основополагающего обзора «Принципы конформацион¬
ного анализа» потребовала от Д. Бартона и Р.Куксона
изучения истории вопроса, и только тогда было восстано¬
влено авторство Хоуорта и Эбеля. В этом обзоре утвержда¬
лась эквивалентность терминов «конформация» (в англий¬
ской химической литературе), «констелляция» (в немецкой
химической литературе) и «вращательный изомер» (в лите¬
ратуре по химической физике). С тех пор конформационная
изомерия стала прочно ассоциироваться с внутренним вра¬
щением и низкими потенциальными барьерами. Слово «кон¬
стелляция» постепенно исчезло из научного обихода (по¬
скольку его использование имело смысл лишь для ограни¬
ченного круга исследователей).
Торжество конформационного анализа
Одновременный выход в 1965 г. монографий «Конфор-
мационный анализ», о которой мы уже упоминали, и «Кон¬
формационная теория» М. Ханака ознаменовал окончатель¬
ное становление конформационного анализа. Традиции,
зачастую не воспринимаемые сознательно, нередко опреде¬
ляют использование теоретических концепций и терминоло¬
гии. Термин «конформация» появился в органической хи¬
мии, и поэтому конформационный анализ рассматривался
главным образом как раздел последней. Однако после раз¬
вития их органиками конформационные представления пере¬
шли и в другие разделы химии. Термин «конформация» стал
широко использоваться и в химической физике. Подобное
проникновение очень характерно для науки второй поло¬
вины XX в., отличительной чертой которой стала взаимо¬
связь и пересечение различных областей знания. Однако сле¬
128
дует отметить, что собственно конформационному анализу
взаимодействие с химической физикой сослужило в одном
отношении дурную службу. Как известно, химики нередко
склонны к излишнему пиетету по отношению к работам фи¬
зиков. Многочисленные публикации по внутреннему враще¬
нию, появившиеся во время становления конформационного
анализа, повлияли на восприятие конформационной изоме¬
рии, причем установилась обусловленная психологическими
причинами традиция связывать понятие конформации ис¬
ключительно с внутренним вращением, сводить конформа-
ционную изомерию к ротационной. Только в последние
годы получил распространение более общий подход к поня¬
тию конформации, и стало ясно, насколько широк круг во¬
просов, попадающих в сферу конформационного анализа.
Термины и уровень познания
Как известно, значение понятия не является непо¬
движным, оно изменяется с течением времени, соответствуя
состоянию культуры (в широком смысле). Но в оптималь¬
ном случае в каждый данный момент времени значение
термина должно быть фиксировано. Иначе, строго говоря,
его нельзя и называть термином. К сожалению, в тех слу¬
чаях, когда в разряд терминов попадают слова, смысл ко¬
торых «интуитивно ясен», нередко наблюдается путаница
и различия в их понимании.
Говоря о первом появлении слова «конформация» в хи¬
мии, В. И. Соколов в своей монографии пишет: «Термином
«конформация» стали характеризовать относительное рас¬
положение атомов в молекуле в данный момент времени,
моментальный портрет молекулы...» Эти слова-типичный
пример осовременивания истории. К моменту написания
«Строения сахаров» еще не было ясного представления о ха¬
рактере внутримолекулярных движений. Поэтому Хоуорт
писал именно о «форме моделей». Первая интерпретация
термина «конформация» соответствовала достигнутому
9-918
129
к моменту его появления уровню знания и означала не про¬
извольное, а оптимальное расположение атомов в простран¬
стве. Эбель уже связывал возможность появления раз¬
личных констелляций молекулы с внутренним вращением,
и в его представлении констелляция означала произвольное
взаиморасположение атомов, возникающее в результате
вращения вокруг ординарной связи углерод-углерод.
У начала неразберихи
Цитированные выше «определения» терминов «конфор¬
мация» и «констелляция» были, собственно, не определения¬
ми, а пояснениями «химического» значения слова. Но с той
поры и началась неразбериха с интерпретацией понятия
«конформация», под которым каждая группа исследователей
понимала нечто свое и зачастую нечто, не имеющее физиче¬
ского смысла, неразберихи, апофеозом которой стали «Пра¬
вила ИЮПАК» 1974 г. (раздел Е: стереохимия). Выше от¬
мечалось, что в психологическом плане такая ситуация была
спровоцирована использованием в качестве научного терми¬
на употреблявшегося ранее слова, смысл которого «очеви¬
ден», а вместе с тем спектр значений достаточно широк.
Бросая взгляд на изменение представлений о конформа¬
ции с 1950 г. по настоящее время, отметим, что основное
различие во взглядах касалось двух вопросов: а) отвечает ли
конформации только оптимальное расположение атомов
в пространстве (минимум потенциальной энергии) или
любое мгновенное расположение; б) каким образом отгра¬
ничить конформационную изомерию от других ее видов (в
частности, от конфигурационной). В 1950 г. Бартон писал
о «ненапряженных расположениях в пространстве», т. е., ка¬
залось бы, склонялся к варианту оптимального расположе¬
ния. Однако, по существу, ненапряженная система-кон¬
струкция условная, и поэтому его определение было
двусмысленным и неработоспособным. В последующей
публикации 1953 г. Бартон уточнил: «... расположения
130
в пространстве атомов молекулы, которые свободны от
углового напряжения» (это уточнение ничего не измени¬
ло)-и тут же указал в качестве примера на конформации
этана, возможное число которых бесконечно. Значит, кон¬
формации в его понимании отвечало произвольное мгновен¬
ное расположение атомов, что подтверждается и указанием
на тождественность терминов «конформация» и «констелля¬
ция» (в определении Прелога). Первые определения Бартона
представляли странный гибрид альтернативных взглядов на
понятие конформации. В то же время Прелог определенно
называл констелляцией произвольное расположение атомов,
однако включал в сферу действия понятия лишь ротацион¬
ную изомерию. Близким по смыслу и непротиворечивым
было и несколько более позднее определение У. Клайна
(1954 г.): «Термин «конформация» обозначает различные
расположения в пространстве атомов в единственной клас¬
сической органической структуре (конфигурации), располо¬
жения, получающиеся в результате вращения или искривле¬
ния (но не разрыва) связей». К моменту подготовки
фундаментальной работы 1956 г. Бартон, казалось, устранил
противоречия в своих взглядах и в качестве «одного из на¬
иболее общих определений» дал следующее: «Конформации
молекул - такие расположения атомов молекулы в простран¬
стве, которые не совпадают друг с другом». Это определе¬
ние не связывает конформационный анализ с конкретным
видом внутримолекулярного движения. Косвенное ограниче¬
ние сферы действия понятия содержится лишь в слове «мо¬
лекула», и, таким образом, все упирается именно в опреде¬
ление молекулы. Заметим, что, хотя Бартон, очевидно, об
этом не думал, при таком определении все виды геометри¬
ческой и даже оптическая изомерия становятся частными
случаями конформационной. Бартон и Куксон справедливо
отмечали, что при этом определении «все молекулы будут,
теоретически, иметь бесконечное число конформаций». Они
все же связывали конформационную изомерию с преимуще¬
ственно низкими потенциальными барьерами.
9'
131
Конформации-это изомеры?
Следует отметить, что, будучи химиками-органиками,
Бартон и Куксон не отдавали себе отчета в том, что данное
ими определение не позволяет рассматривать конформации
молекулы как изомеры. Они писали о «барьерах между раз¬
личными конформациями» и о том, что термины «конфор¬
мация», «констелляция» и «вращательный изомер» синони¬
мичны. В то же время ясно (это было ясно и в 1956 г.), что
как изомеры могут рассматриваться лишь конформации, от¬
вечающие минимумам поверхности потенциальной энергии
молекулы, ибо только для них имеется теоретическая воз¬
можность существования в качестве индивидуального соеди¬
нения, только они являются принципиально наблюдаемыми.
Отметим, что в литературе по химической физике термин
«вращательный изомер» всегда использовался не в смысле
определения конформации Бартона и Куксона, а лишь
в приложении к устойчивым состояниям. Действительным
синонимом термина «вращательный изомер» термин «кон¬
формация» стал в опубликованном также в 1956 г. обзоре
У. Добена и К. Питцера: «Под конформацией подразуме¬
вается пространственное расположение атомов в молекуле,
которое может возникнуть при вращении вокруг ординарной
связи и способно к существованию».
В соответствии с такой точкой зрения М. Ханак (1965 г.)
предлагал использовать термин «конформация» только
в отношении структур, отвечающих минимуму адиабатиче¬
ского потенциала, а прочие метастабильные структуры на¬
зывать формами. Однако взгляд на конформацию как на
изомер, в конечном счете, не получил поддержки. Вероятно,
это связано с тем, что конформационный анализ на раннем
этапе его развития в представлении химиков ассоциировался
с внутренним вращением. Поэтому использование термина
«конформация» в смысле «изомер» выглядело ненужным
дублированием термина «ротационный изомер». Впрочем,
термин «конформационный изомер», или «конформер»,
132
вскоре также получил широкое хождение, причем под кон-
формером понималась стабильная конформация из беско¬
нечного множества расположений атомов, возникающих из-
за вращения вокруг ординарных связей.
В литературе на русском языке термин «конформация»
возник в значении мгновенного расположения атомов:
«Конформациями называют различные пространственные
формы молекулы, способные переходить друг в друга путем
вращения атомов без разрыва межатомных связей, в отли¬
чие от конфигураций, взаимопревращения которых воз¬
можны лишь при разрыве связей» (И. Н. Назаров, JI. Д. Бер¬
гельсон, 1957 г.).
Конформации и внутреннее вращение
Одновременный выход в 1965 г. двух монографий
(Э. Илиела и др. и М. Ханака) продемонстрировал как
огромные успехи экспериментальных методов, так и ограни¬
ченность теоретических представлений, лежащих в основе
новой научной дисциплины. В обеих книгах, составивших
фундамент подготовки следующего поколения химиков-ор-
гаников, конформации понимаются как различающиеся рас¬
положения атомов в молекуле, получаемые при вращении
вокруг простых связей без разрыва последних. Пожалуй,
можно сказать, что внутреннее вращение преследовало кон¬
формационный анализ, как злой рок. Уже было хорошо из¬
вестно, что геометрия молекулы может меняться в результа¬
те, например, проходящей с преодолением низкого барьера
инверсии пирамидального гетероатома. Подобные процессы
единодушно относили к сфере конформационного анализа.
И тем не менее в «Правилах ИЮПАК» 1974 г. (раздел Е:
стереохимия) была зафиксирована следующая «расшири¬
тельная» трактовка:
«Существуют различные взгляды относительно точного
определения термина «конформация», (а) Классическая ин¬
терпретация: конформации молекулы определенной конфи¬
133
гурации-это различные расположения ее атомов в про¬
странстве, которые отличаются друг от друга только
в результате вращения вокруг ординарных связей. (Ь) Это
определение обычно расширяют, включая и вращение во¬
круг ст-связей или связей промежуточного порядка между
первым и вторым, (с) Третья точка зрения состоит в даль¬
нейшем расширении определения с тем, чтобы включить
также вращение вокруг связей произвольного порядка,
в том числе двойных связей.
Молекулы, различающиеся по конформации, опреде¬
ляются как конформационные изомеры.» (Е-1.7).
«Молекула в конформации, в которую ее атомы само¬
произвольно возвращаются после малых смещений, опреде¬
ляется как конформер» (Е-5.1).
Подобная трактовка, разумеется, неприемлема, посколь¬
ку связывает общее понятие конформации молекулы
с частным видом внутримолекулярных движений. Кроме то¬
го, понятия «конформера» и «конформационного изомера»
здесь определены различно, хотя в химической литературе
первый термин всегда рассматривался как синоним второго,
от которого он и произошел.
Сложности, связанные со строгим определением термина
«конформация», послужили предметом специального обсуж¬
дения в приложении к «Правилам ИЮПАК». Основным не¬
достатком публикации ИЮПАК является непонимание ее
авторами того обстоятельства, что произвольные метаста-
билъные состояния молекулы, вообще говоря, нельзя рассма¬
тривать как изомеры. Принципиальным вопросом, по суще¬
ству, поставившим в тупик авторов Правил, является
разграничение конформационной и иных видов простран¬
ственной изомерии, различие между конформацией и конфи¬
гурацией. Современные экспериментальные данные не по¬
зволяют провести такое разграничение, как это нередко
делалось раньше, по высотам потенциальных барьеров, раз¬
деляющих изомеры. Скажем, внутреннее вращение вокруг ор¬
динарной связи относят к сфере конформационного анализа,
134
сн3
Рнс. 28. Примеры конформационных переходов:
а внутреннее вращение в этане, барьер 12 кД ж/моль; б-внутреннее вращение в замещен¬
ном триптицене, барьер 158 кДж/моль; в инверсия пирамидального азота в аммиаке, барь¬
ер 25 кДж/моль.
но появление объемистых заместителей делает потен¬
циальный барьер такого процесса сравнимым с барьером
химической реакции (рис. 28).
Качественное различие типов изомерии по видам пре¬
одолеваемых при изомеризации препятствий (структурная
изомеризация-через разрыв связей, геометрическая-через
изменение валентных углов, конформационная-через изме¬
нение торсионных углов), положенное возглавляемой И. Уги
(ФРГ) международной группой теоретиков в основу форма¬
лизации логических структур химии (1976 г.), также неприем¬
лемо. Например, проходящая с низким барьером инверсия
пирамидального атома азота в аммиаке входит в сферу кон¬
формационного анализа, при этом, однако, изменяются
только валентные углы. Выбор границы оказывается совер¬
шенно произвольным.
Проблема разграничения конформации и конфигурации
™ решена в работе московских химиков В. Н. Дрозда,
Н. С. Зефирова, В. И. Соколова и И. В. Станкевича (1979 г.),
где строго определено понятие стереохимической конфи¬
гурации как имеющее смысл лишь для хиральных систем. Ес¬
ли это определение станет общепринятым, формально все
виды пространственной изомерии, кроме оптической, могут
включаться в сферу конформационной изомерии (выделение
структурной изомерии связано уже с определением понятия
молекулы).
Подобная расширительная трактовка получает в послед¬
ние годы все большее распространение. В отношении само¬
го понятия конформации сегодня также победила макси¬
мально широкая трактовка, и конформация толкуется как
произвольное расположение ядерного остова молекулы
в пространстве в данный момент времени. Такой подход,
думается, наиболее удобен (особенно ясно это видно в дина¬
мической стереохимии), логичен и непротиворечив, и данная
интерпретация сохранится, по всей видимости, долгое вре¬
мя. Однако не следует забывать, что при этом определении
различные конформации молекулы не могут рассматривать¬
ся как изомеры, а переход между произвольными конформа¬
циями не является конформационной изомеризацией, анало¬
гичной структурной изомеризации.
Конформация - «ненаблюдаемая величина» ?
Получается, что конформация (в том смысле, в котором
это слово ныне употребляется)-до известной степени услов¬
ная конструкция. В принципе ей не всегда можно сопоста¬
вить экспериментальные данные. В то же время возникнове¬
ние представлений о конформационной изомерии всегда
рассматривалось как расширение классических представле¬
ний о последней. Было бы противоестественным такое рас¬
ширение, в результате которого первоначальное понятие по¬
теряло смысл.
Все же конформационную изомерию следует рассматри¬
вать, как частный случай изомерии, и при образовании кон¬
цептуальных понятий целесообразно здесь, как и при рас¬
136
смотрении иных видов изомерии, исходить из наблюдаемых
величин. Таковыми являются геометрические параметры изо¬
меров, их относительные энергии и величины барьеров
перехода одного изомера в другой. Кроме того, необходимо
исходить из известного правила, диктуемого принципом не¬
определенности: «Анализ химических систем требует моде¬
лей, которые представляют определенные классы эквива¬
лентности состояний, а не сами состояния» (И. Уги и др.,
1976 г.). Очевидно, что конформации представляют собой
именно сами состояния, но вовсе не классы. Эксперимен¬
тальные данные могут быть сопоставлены лишь тем кон¬
формациям, которые относительно стабильны в физическом
смысле, т.е. являются конформерами в смысле приведенно¬
го выше правила ИЮПАК Е-5.1.
Поэтому требуется четкое разграничение понятий кон¬
формации как произвольной мгновенной структуры и кон-
формера как изомера, генетически связанного с многообра¬
зием возможных конформаций данной молекулы.
Нередко встречающееся смешение понятий конформации
и конформера (пример тому-правило ИЮПАК Е-1.7), недо¬
пустимо по двум причинам. Во-первых, оно приводит к вве¬
дению «изомеров», по физическому смыслу таковыми не
являющихся. Во-вторых, используются два разных термина
для обозначения одного и того же объекта, что предста¬
вляет собой засорение научного языка.
Топология ППЭ
Как уже говорилось, в адиабатическом приближении ка¬
ждому электронному состоянию молекулы соответствует
единственная поверхность потенциальной энергии (ППЭ)
в координатах ядер. По существу, конформационный анализ
можно представить как анализ топологических особенно¬
стей многомерной ППЭ. Устойчивым конформациям отве¬
чают локальные минимумы адиабатического потенциала
137
?2
Рис. 29. Пример конформационной карты:
линии эквипотенциалов, ПНЭ; а, Ь- точки
минимумов; с-седловая точк ; фь
ф2 - независимые переменные.
АВ
В
У*1 деленном механизме реакции
(в данном случае -конформа-
глубиной не менее двух кван¬
тов колебаний (для каждой
степени свободы) в миниму¬
ме. Оптимальные пути пере¬
хода между ними лежат че¬
рез седловые точки первого
порядка, отвечающие пере¬
ходным состояниям. Обыч¬
ное представление об опре-
ционного перехода) означает непременное существование
конечного числа определенных оптимальных путей (в идеа¬
ле - единственного пути), соединяющих любую пару мини¬
мумов.
В 1970 г. японский теоретик (теперь Нобелевский лауреат)
К. Фукуи ввел понятие о пути наименьшей энергии (ПНЭ) —
кривой, связующей пару минимумов, проходящей через
переходное состояние и ортогональной ко всем пересе¬
каемым контурам эквипотенциалов (рис. 29). Понятно, что
статистически возможны переходы по любым маршрутам,
но большинство переходов осуществляется по путям, близ¬
ким ПНЭ.
ППЭ дает полную информацию о конформационных
возможностях системы. Отметим, что если для химических
реакций расчет ППЭ чрезвычайно трудоемок, то для кон¬
формационных превращений использование метода атом-
атомных потенциалов зачастую делает эту задачу вполне
разрешимой. Однако трудность наглядного представления
многомерных поверхностей делает их бесполезными для ин¬
терпретации конформационного поведения сложных систем.
Широкое распространение получили лишь сечения этих ги¬
перповерхностей -конформационные карты (пример-
138
-рис. 29). Кроме того, ППЭ и ПНЭ характеристичны, лишь
будучи рассчитанными в «канонической» (определяемой
с учетом масс атомов) системе координат, выбор которой
сложен. А вот стационарные точки, в частности, седловые
и точки минимума, не зависят от избранной системы коор¬
динат. Экспериментально определяемые геометрические
и энергетические характеристики конформационных изоме¬
ров, а также величины барьеров переходов между ними
можно связать со стационарными точками, отвлекаясь от
иных особенностей ППЭ. Поэтому информация о ППЭ за¬
частую избыточна; практически значимое описание системы
конформеров следует строить, исходя из понятия стацио¬
нарных точек и приписывая этим точкам некоторые инте¬
гральные характеристики.
Конформер и конформация
Понятие конформера связано с понятием оптимальной
конформации, но не сводится к нему. По мнению некоторых
химиков, изомерия может рассматриваться как теоретиче¬
ская концепция только для молекул, находящихся в ос¬
новных состояниях. Однако в таком случае понятие конфор-
мационной изомерии теряет смысл, например при комнат¬
ной температуре, когда некоторые ненулевые уровни вну¬
треннего вращения заселены. Подобный подход противоре¬
чит и цитированному выше правилу классов эквивалентно¬
сти. Поэтому разумно связать понятие конформера не
только с точкой минимума ППЭ, но и с окрестностью этой
точки.
Под конформером следует понимать набор конформа¬
ций, которые образуют в пространстве независимых геоме¬
трических переменных непрерывную область, включающую
координаты минимума ППЭ, и лежат по энергии ниже на-
инизшей седловой точки первого порядка, примыкающей
к названной области.
139
Эти области определены однозначно вследствие одно¬
значности и непрерывности функции адиабатического потен¬
циала. Случай двух конформеров (А и В) иллюстрирует
рис. 29. Такой подход позволяет распространить концепцию
конформационной изомерии на молекулы, находящиеся на
ненулевых вращательных и колебательных уровнях. Кон-
формеру отвечают усредненные (с учетом заселенностей
энергетических уровней) геометрические и энергетические ха¬
рактеристики, наблюдаемые в эксперименте. Разумеется, ес¬
ли существенно заселены уровни, лежащие выше переваль¬
ной точки, пара конформеров превращается в один (АВ на
рис. 29). Это происходит при достаточно высоких темпера¬
турах. Э. Илиел (1977 г.) указывал, что трудно определить
изомеры безотносительно к температуре; рассматривать два
минимума как отвечающие паре изомеров можно в том
и только том случае, когда барьер между ними превышает
кТ (к-постоянная Больцмана, Г-абсолютная температура).
Рассмотрение может быть распространено на произвольное
число конформеров, если для каждого из них проводить
граничный контур от наинизшей седловой точки.
Какое понятие «главнее»?
Именно понятие конформера, лежащее в русле тради¬
ционных представлений об изомерии, представляется клю¬
чевым в конформационном анализе. Поэтому целесообразно
определить конформационный переход лишь как переход
между конформерами (но не между произвольными конфор¬
мациями). Однако нередко конформационный переход опре¬
деляется как непрерывный процесс изменения внутренних
координат ядерного остова молекулы, протекающий без на¬
рушения ее целостности. В таком определении конформа¬
ционный переход непосредственно связывается с понятием
конформации. В силу того, что конформация в определен¬
ном смысле «ненаблюдаема», «переход» остается искус¬
ственной конструкцией, которую, в общем случае, нельзя со¬
140
поставить с экспериментальными данными, например, пара¬
метрами активации. Кроме того, приведенное определение
конформационного перехода противоречит и сложившимся
представлениям, ибо тогда конформационным переходом
можно считать, скажем, валентное колебание С—Н метана.
Забавно, что обсуждение определений термина «кон¬
формация» в приложении к «Правилам ИЮПАК» 1974 г.
кончалось фразой: «Обнадеживает, однако, что никакие оп¬
ределения в этой области не включали (пока) колебания
атомов, для которых во всех случаях рассматриваются толь¬
ко усредненные по времени положения». Вот уж, действи¬
тельно, нет предела дерзаниям ученых.
Различным электронным состояниям отвечают свои
энергетические гиперповерхности, поэтому понятие конфор-
мационной изомерии в данном определении теряет смысл
при рассмотрении набора электронных состояний в совокуп¬
ности, но сохраняет его для каждого из состояний, взятых
в отдельности. То есть, если понятие конформации имеет
смысл как для адиабатических, так и для неадиабатических
систем, то понятие конформера имеет смысл только для си¬
стем адиабатических. Именно поэтому в динамической сте¬
реохимии-разделе этой науки, изучающем изменение гео¬
метрии молекул в ходе химических реакций,-уместно
пользоваться лишь термином конформация и говорить об
изменении конформации, а не о конформационном переходе.
Хотя, конечно, ничто нам не мешает рассматривать систему
взаимодействующих молекул как одну псевдомолекулу
и обсуждать ее конформеры. Другое дело, что в большинст¬
ве случаев это нецелесообразно.
Конформационная кинетика
Конформеры, их относительные энергии-это конформа¬
ционная термодинамика. Попробуем теперь разобраться с конфор-
мационной кинетикой. Скорость конформационного перехода—
вещь вполне реальная, измеряемая. Определив зависимость кон¬
141
станты скорости К от температуры Т, мы найдем свободную
энергию активации AF , поскольку
Л F ^
кТ _
К = х—-е кт
п
где и-трансмиссионный коэффициент (см. следующий раздел).
Но AFФ = F* - F°, где F° — свободная энергия конформе-
ра (понятие, физический смысл которого мы уяснили), a F —
— свободная энергия переходного состояния (называемого часто
активированным комплексом). Но что такое переходное состоя¬
ние? Обычно ему сопоставляют седловую точку на ПНЭ. Одна¬
ко РФ = иФ — ТБФ, где UФ и S , соответственно, энергия
и энтропия активированного комплекса. Энергия-это понятно.
Но как можно сопоставить точке энтропию? Перевальная точка
отвечает конформации (для любой из которых SBHyTp = 0), а не
конформеру, состоянию, а не классу состояний. В то же время
экспериментальное значение энтропии активации не совпадает
с значением энтропии исходного конформера, взятым с обратным
знаком.
Поиски связей
Как же прийти к энтропии средней энергии U* и сво¬
бодной энергии FФ активированного комплекса, исходя из ППЭ?
Это-интегральные характеристики в том смысле, что они выводят¬
ся из статистического интеграла Z в пространстве N независимых
переменных {ср1,ф2, •••, Фдг}:
Фи Ф12 Фш ч
р р г 4>iV'
Z = — ... \ е кт dq> 1 ...dq>N
Ф01 ф02 ФОЛ?
(С-нормирующий множитель)
Фи Фш
Щ(рь ■■■, Фуу)
и = ^2 I I ^(фь •••> Ф^)е кт d9l...dvN
1_
Фо1 ФО N
142
S = к\п Z + U/T; F = — к Tin Z = U - TS
Для конформеров «все честно»-потенциальная функция из¬
вестна, пределы интегрирования тоже, считай и сравнивай... Но
классическая термодинамика описывает лишь равновесные си¬
стемы. Переходное состояние таковым не является. В кинетике
обычно закрывают на это глаза, используя «квазитермодинамиче-
ский подход»: постулируют, что тем же способом можно описать
и активированный комплекс. Все бы хорошо, ведь теоретически
ППЭ должна давать нам полное описание системы, да вот как
быть с пределами интегрирования? Для конформеров они ясны: от
минимума до граничного контура. Часто говорят, что энтропию
активации можно сопоставить с формой сечения ППЭ гиперпло¬
скостью, перпендикулярной ПНЭ в точке перевала. Действительно,
вероятность того, что молекула, обладая энергией, превышающей
энергию активации, преодолевает барьер, должна зависеть от
формы такого сечения. Но нетрудно заметить, что размерность
пространства, в котором проводится интегрирование, при таком
подходе уменьшается на единицу. Энтропия уменьшается вслед¬
ствие «изъятия» одной степени свободы, и активированный ком¬
плекс по физическому смыслу не является аналогом исходного
конформера.
Чтобы активированный комплекс обрел объем, имел ту же раз¬
мерность в фазовом пространстве, что и конформер, рассматри¬
вают седловую точку с окрестностью. Но какой? Произвол в выбо¬
ре окрестности вносит неопределенность в сравниваемые инте¬
гральные характеристики исходного конформера и активированно¬
го комплекса. Мы уже указывали, что конформационный анализ -
это анализ топологии ППЭ. Следовательно, при выборе пределов
интегрирования необходимо исходить именно из свойств самой
ППЭ, а не вырезать из нее наиболее приглянувшиеся куски.
Активированный комплекс без конца и края?
Тогда логичным было бы рассматривать как активированный
комплекс конформационного перехода набор конформаций, по
энергии лежащих на уровне и выше наинизшей перевальной точки.
Здесь уместно процитировать замечательную книгу М. Дьюара
и Р. Догерти «Теория ВМО в органической химии» (М.: Мир, 1977,
с. 263): «Реакция ведет себя, как птица, которая может парить
143
Рис. 30. Пример потенциальной кривой.
вдоль и поперек над долиной
только на данном уровне». В са¬
мом деле, молекула, находящая¬
ся на любом энергетическом
уровне выше граничного конту¬
ра, уже не относится ни к од¬
ному из конформеров. Стоит
молекуле попасть на этот уро¬
вень -и она способна находить¬
ся в геометрической окрестнос¬
ти любого из пары конформе¬
ров и, потеряв затем энергию,
упасть в любую из двух ям.
Вот и конформационный пере¬
ход.
Эти представления для системы нескольких конформеров ил¬
люстрирует рис. 30. Для перехода А -*■ В (В -у А) «активиро¬
ванный комплекс»-это АВ + ABCD. Области D, С и DC не могут
в него включаться, так как находящиеся на соответствующих уров¬
нях молекулы, хотя и могут обладать энергией, превышающей
энергию переходного состояния А -> В (В -> А), не участвуют в по¬
следнем конформационном переходе, поскольку неспособны пре¬
одолеть барьер перехода DC -* АВ (АВ -> DC). На самом деле, ко¬
нечно, потенциальная поверхность конформационных превращений
в координатах всех независимых переменных никогда не выро¬
ждается в одномерную кривую.
Заметим однако, что при таком рассмотрении мы приходим
к парадоксу. Получается, что свободная энергия активации зависит
от вида потенциальной поверхности в областях, значительно уда¬
ленных от седловой точки. Впрочем, основной вклад все равно бу¬
дет вносить область, примыкающая к последней, так как именно
она наиболее уплощена.
КОНФОРМАЦИОННЫЕ ГРАФЫ
Теперь, когда теоретический фундамент конформацион-
ного анализа приобрел некоторую законченность, посмот¬
рим, как можно эту дисциплину формализовать. Ясное
представление о фундаментальных понятиях конформацион-
ного анализа для того и нужно, чтобы адекватно их переве-
144
сги на язык математики. Ведь давно известно: нечетко задав
условия, посредством любого математического аппарата по¬
лучишь ерунду. Для чего нужна формализация? Химики сей¬
час все большую часть своей работы стараются переложить
на ЭВМ, а уж с ней-то в терминах «интуитивно ясных»
представлений не пообщаешься.
Лучше всего для описания конформационных возможно¬
стей молекулы подходит аппарат теории графов. На самой
этой теории мы здесь останавливаться не будем, поскольку
и сама она, и применение ее в химии обсуждались в
брошюре «Молекулы без химических связей» (JL: «Химия»,
1980).
Как представить непредставимое?
Мы уже говорили, что многомерную ППЭ саму по себе
нельзя представить наглядно. Чтобы заменить ее на¬
глядным двумерным объектом, И. В. Кривошей (1969 г.)
ввел понятие дерева химической реакции (гомоморфное ото¬
бражение ППЭ на двумерный граф-дерево). Однако эта
конструкция, по определению не допускающая циклических
подграфов, недостаточно отражает топологию ППЭ, напри¬
мер маскируя симметричные ПНЭ. В то же время она со¬
держит обычно не представляющую интереса и потому из¬
быточную информацию о максимумах на ППЭ.
Более удачно использование в конформационном анали¬
зе графа конформационных взаимопревращений, впервые
появившегося в работе И. Уги и др. (1976 г). Существенным
недостатком этой работы, однако, является связь определе¬
ния графа с конкретным частным видом внутримолекуляр¬
ного движения-внутренним вращеним. Рис. 28 уже проде¬
монстрировал нам, что сфера конформационного анализа
отнюдь не ограничивается ротационной изомерией.
По нашему мнению, нецелесообразно при внесении пред¬
ставлений теории графов в стереохимию различать a priori
виды внутримолекулярных движений, стереоизомеры и кон-
10-918
145
формеры, вводя отдельно конфигурационный и конформа¬
ционный графы, как это сделано в вышеназванной работе
Уги и др. В принципе ППЭ может быть построена в про¬
странстве независимых переменных для произвольной си¬
стемы ядер. Тогда ее топологические особенности будут от¬
ражать все многообразие изомеров заданного состава.
Введение понятия пространственной изомерии означает раз¬
деление такой полной ППЭ на области, соответствующие
структурным изомерам, и рассмотрение топологических
особенностей ППЭ внутри этих областей. Химическое со¬
единение следует представлять конструкцией, отражающей
как топологию молекулы (порядок связей), так и возможные
превращения в рамках заданной топологии. Описание топо¬
логии молекулы посредством молекулярных графов, по су¬
ществу являющихся математическим эквивалентом струк¬
турных формул, хорошо разработано. Будучи дополненным
представлением о стереохимической конфигурации как свой¬
стве хиральных молекул, оно оставляет прочие виды изоме¬
рии конформационному анализу. В таком случае геометри¬
ческая изомерия становится частным случаем конформа-
ционной, что естественно, поскольку разница между ними
чисто количественная. Соотношение понятий конформации
и стереохимической конфигурации детально проанализиро¬
вано в работе группы московских теоретиков (1979 г.), на ко¬
торую мы уже ссылались.
Определение конформационного графа
Мы введем максимально общее определение конформа¬
ционного графа как гомоморфного отображения многомер¬
ной ППЭ на не более чем трехмерный и потому наглядный
граф, отражающий основные топологические особенности
гиперповерхности. Конформационным графом (КГ) мы на¬
зовем такой граф, вершины которого однозначно соответ¬
ствуют локальным минимумам ППЭ, а ребра-соединяю¬
щим эти минимумы ПНЭ. Нетрудно заметить, что ло¬
146
кальные минимумы ППЭ и конформеры, а также ПНЭ
и конформационные переходы попарно находятся в отноше¬
нии однозначного соответствия. Тогда КГ может быть изо¬
морфно задан через фундаментальные понятия конформе¬
ра и конформационного перехода. В этом случае
в матричном виде КГ задается с учетом окрестностей ста¬
ционарных точек ППЭ, так что вершины его отвечают кон-
формерам, а ребра -конформационным переходам. Предста¬
вление об определенном механизме химической реакции,
частным случаем которой является конформационный пере¬
ход, означает непременное существование конечного числа
ПНЭ, соединяющих минимумы. Поэтому КГ конечный,
связный и не имеет петель. Пара вершин соединена ребром
в том и только в том случае, когда между отвечающей этой
паре вершин парой конформеров возможен непосред¬
ственный конформационный переход.
Немного математики
Как известно, графы представляют матрицами смежно¬
сти. Простейший способ задания неориентированного
КГ-через матрицу смежности А, такую, что элемент ее
ai} = 1, если ребро ij существует, и = 0, если такое ребро
отсутствует (рис. 31). Очевидно, что . Матрица А дает
лишь качественную картину взаимосвязи конформеров. Бо¬
лее точное описание можно провести, представив КГ как
ориентированный и введя матрицу длин дуг (А) такую, что
ее элемент аи = exp( — AF^j/kT), где Д/^-барьер перехода
i-ro конформера в j-й (рис. 31). При отсутствии ребра ij на
соответствующем неориентированном графе dy = а= 0.
В общем случае ai} Ф ап, и матрица А содержит информа¬
цию о разностях энергий конформеров AF-ф AF^ = AFfj —
~ Таким образом, она полностью характеризует как
кинетические, так и термодинамические свойства системы
конформеров. С использованием матрицы длин дуг легко
провести исследование такой системы, например найти вре¬
мя установления равновесия при изменении температуры,
ю* 147
0
1
0
0
I
0
1
0
0
I
0
I
0
0
1
0
Рис. 31. Пример конформаиионной карты, соответствующего ей конформационного графа и от¬
вечающей такому графу матрицы смежности А.
Трансмиссионный коэффициент
Рассмотрение КГ позволяет оценить трансмиссионный
коэффициент к конформационного перехода (i -*■ j). Значение
этого коэффициента связано с числом возможных маршру¬
тов (i -у j), а также с числом промежуточных вершин и вет¬
влений на каждом из таких маршрутов. Оно отражает ве¬
роятность того, что, пойдя по ПНЭ, молекула, обладающая
достаточной энергией, перейдет из одной ямы в другую. По¬
лагают, что если переход-это просто перевал через одну
седловину, то х = 1.
Например, конформационная динамика циклогексана мо¬
жет быть представлена графом, изображенным на рис. 32, а.
Молекула циклогексана, преодолев седловую точку, прини¬
мает последовательность разделенных невысоким барьером
твист-форм. Заметим, что с методической точки зрения КГ
(рис. 32, а) лучше иллюстрирует поведение циклогексана,
чем обычно используемый в учебных пособиях график
(рис. 32,6), который создает такое впечатление, будто при
переходе (кресло) -*■ (кресло)' молекула обязательно при¬
нимает все шесть вырожденных meucm-форм.
148
Вероятность перехода из meuem-формы в прежнее и ин¬
вертированное кресло одинакова, поэтому х = 0,5. Для
маршрута, проходящего через п минимумов, разделен¬
ных равными барьерами, х = (0,5)". Для неравных барье¬
ров эта формула дает нижнюю оценку; точное значе¬
ние х находится с помощью матрицы Л. При наличии раз¬
ветвлений у промежуточных вершин х уменьшается, при
увеличении числа возможных маршрутов (i -*■возрастает*.
Тот случай, когда непосредственный конформационный пе¬
реход (i -*■ j) возможен по двум энергетически эквивалент¬
ным путям (энантиомерные переходные состояния) отве¬
чает кратному ребру КГ, и соответствующие элементы ма¬
трицы А, имеющей смысл матрицы констант скоростей
одностадийных конформационных переходов, должны быть
домножены на 2.
Матрица длин дуг А конформационного графа пол¬
ностью характеризует конформационную «жесткость» си¬
стемы. Введя понятие конформационного расстояния R(j на
матрице А, равного числу ребер на кратчайшем маршруте ij,
можно охарактеризовать и структурную близость конфор-
меров i и j. Применение хорошо разработанных методов
теории графов в формализованном конформационном ана¬
лизе, вероятно, перспективно.
Кресло Твист
Кресло q
Координата реакции
Рее. 32. а Минимальный конформационный граф циклогексана (i -кресловидные, I -твист-
конформерыХ 6 крива* инверсии циклогексааа.
149
«Никто не обнимет необъятного»
Следует отметить, что общий КГ, описывающий все
многообразие конформационных превращений системы,
в практической работе встречается редко, лишь при
рассмотрении небольших молекул. Чаще представляют ин¬
терес только определенные виды конформеров и конформа¬
ционных переходов. В этом случае строится подграф КГ, от¬
ражающий часть свойств ППЭ. Такой подграф получается
из общего исключением ряда вершин и ребер. Он может от¬
вечать части ППЭ или ППЭ, рассчитанной в зависимости от
ограниченного числа независимых переменных, либо объе¬
динению нескольких конформеров в один условный. Кроме
того, учет высоко лежащих локальных минимумов и высо¬
кобарьерных путей перехода при наличии низкобарьерных
путей зачастую не оправдан. Введя верхнюю границу Fmax
по энергии для конформеров, можно упростить КГ. В этом
случае, однако, надо учитывать влияние на трансмис¬
сионный коэффициент высоко лежащих минимумов, отве¬
чающих вершинам степени выше 1.
Чтобы более наглядно представить характер конформа-
ционной динамики, исключим лишние ребра КГ, введя сле¬
дующую конструкцию. Минимальным конформационным
графом (МКГ) назовем конформационный граф, любая пара
вершин которого соединена ребром в том и только том слу¬
чае, когда конформационный переход (по представленному
этим ребром пути) происходит с преодолением наименьшего
барьера из возможных для всех маршрутов, соединяющих
данные вершины. Очевидно, что подобная конструкция един¬
ственна. В случае энантиомерных переходных состояний со¬
храняются кратные ребра. Пометив вершины графа, можно
ввести на нем отношения эквивалентности. Удобство пред¬
ложенной конструкции заключается в том, что в большин¬
стве случаев она отвечает реально наблюдаемым процессам.
Так, например, понижение температуры мало сказывается
на виде спектра ЯМР обменивающейся системы А В, пока
150
не начнется «вымораживание» самого быстрого из процес¬
сов, обеспечивающих взаимопревращения А и В.
МКГ дает картину оптимальных конформационных
переходов в системе конформеров. Заметим, что наличие ци¬
клов на МКГ, если не учитывать возможность случайного
вырождения, отражает свойства симметрии молекулы.
В случае систем, проявляющих свойство хиральности,
возможны две ситуации. Если все конформеры, предста¬
вленные на МКГ, хиральны, последний может быть разбит
на два эквивалентных подграфа-дерева разрезом ребер, со¬
ответствующих взаимопревращениям пар энантиомеров. Ес¬
ли система содержит как хиральные, так и ахиральные кон¬
формеры, подобное разбиение проводится разрезом таких
же ребер и удвоением вершин, соответствующих ахи-
ральным конформерам. Такие разбиения и представляют со¬
бой учет стереохимической конфигурации при представле¬
нии молекул в рамках теории графов. Введя верхнюю
границу F^ах по высоте барьеров, можно разделить МКГ на
подграфы, соответствующие геометрическим изомерам.
Еще одна модификация
Конструкция МКГ принципиально важна, однако прак¬
тически нередко удобнее использовать расширенный
МКГ (РМКГ), в котором сохраняются все ребра, отвечаю¬
щие конформационным переходам с барьерами, не превы¬
шающими минимальный на заданную величину AF^ax. Кон-
формационные переходы в ряде случаев могут протекать
в сопоставимой степени по нескольким маршрутам (в част¬
ности, низкобарьерный путь по сложному маршруту может
стать «невыгодным» из-за уменьшения х), что и отражает
конструкция РМКГ. Заметим, что существование циклов на
этом графе уже не связано жестко с симметрией молекулы.
Установление механизма конформационной изомериза¬
ции эквивалентно построению МКГ. Конформационные
графы характеризуют молекулу в обоих аспектах ее конфор-
151
мационного поведения-и термодинамическом, и кинетиче¬
ском. Если при задании КГ указываются и векторы коорди¬
нат в пространстве независимых переменных, отвечающие
его вершинам, то химическое соединение может быть адек¬
ватно представлено совокупностью молекулярного и кон¬
формационного графов с учетом стереохимической конфигу¬
рации. Отметим, наконец, что при расчете ППЭ построение
и анализ конформационных графов могут быть переданы
ЭВМ, с тем чтобы конечная обработанная информация по¬
ступала в наглядной форме.
Теории-те же сети; только тот, кто
забрасывает, тот и может поймать.
Ф. ФОН Г АРДЕН БЕРГ (НОВ АЛИС)
Глава IV
ЭЛЕКТРОН ГЛАЗАМИ ХИМИКА
Теперь более детально рассмотрим вопрос об электрон¬
ном строении химических соединений. Начнем с историче¬
ских замечаний.
ТАЙНА ВАЛЕНТНОГО ШТРИХА
Первые шаги квантовой химии
С появлением квантовой механики возникло представле¬
ние о непрерывном характере распределения электронного
заряда в атомах, молекулах и твердых телах, что коренным
образом отличало «новую» квантовую теорию, построен¬
ную в 1925-1926 гг., от «старой» теории Н. Бора (1913 г.). Ве¬
роятностная интерпретация волновой функции уточнила это
представление, показав, что фактически речь идет о не¬
прерывном (за исключением особых точек) распределении
плотности вероятности пространственной локализации элек¬
трона.
Важность этого представления для теории химической
связи трудно переоценить. Не случайно поэтому, немецкие
ученые В. Гайтлер и Ф. Лондон свою известную статью
1927 г. «Взаимодействие нейтральных атомов и гомеополяр-
ная связь с точки зрения квантовой механики», с которой
берет начало современная квантовая химия, начали словами:
«Взаимодействие между нейтральными атомами до сих пор
представляло большие трудности для теоретического рас¬
153
смотрения. Развитие квантовой механики дало для разра¬
ботки этой проблемы совершенно новую точку зрения: пре¬
жде всего в новой «модели» распределение заряда пол¬
ностью отлично от модели Бора, что уже влечет за собой
совершенно новое соотношение сил (Kraftespiel) между ней¬
тральными атомами».
Опираясь на математический аппарат квантовой механи¬
ки и на ее физические представления, Гайтлеру и Лондону
удалось решить проблему, стоявшую перед естествознанием
в течение многих веков: в чем причина химического связы¬
вания (или агрегирования, или сродства, или связи и т. д.-в
разное время терминология была различной)? Какова физи¬
ческая реальность, стоящая за символикой валентных штри¬
хов классической химии?
Молекула Н2 с позиций квантовой механики
Рассматривая задачу об изменении энергии двух электро-
нейтральных атомов водорода, находящихся в основном со¬
стоянии, когда их ядра сближаются до конечного расстоя¬
ния R, Гайтлер и Лондон полагали, что атомы в молекуле
в какой-то мере сохраняют свою индивидуальность и меж¬
атомное взаимодействие следует рассматривать как возму¬
щение.
Когда атомы На и Нь пространственно удалены друг от
друга, их можно считать невзаимодействующими и элек¬
тронное состояние каждого из них следует описывать вол¬
новыми функциями фв(1) и фь (2) изолированных атомов На
и Н6:
Ф^1) = Ц(1) = ~=re~ral ф„(2) = lsb(2) = ~^=ге~гь2
уп ]/п
где цифры «1» и «2»-обозначают условные номера электро¬
нов атомов На и Нь, га1 и га2- расстояния электрон - ядро.
154
Если невзаимодействующие атомы рассматривать как
единую систему, то произведение соответствующих им соб¬
ственных функций представляет собой собственную функ¬
цию этой системы. При этом можно построить две двух¬
электронные функции:
Фа (1) фь (2)-первый электрон около ядра А, второй-около ядра В
Фа (2) cpj, (1)-первый электрон около В, второй-около А
(58)
В силу неразличимости электронов первая функция —
произведение ничуть «не хуже» второй, и той и другой отве¬
чает одна и та же энергия, численно равная удвоенной энер¬
гии атома водорода. Таким образом, система двух атомов
водорода, считающихся невзаимодействующими, оказывает¬
ся двукратно вырожденной.
Вычисление с функцией вида (58) энергии такой системы
в предположении, что межатомное взаимодействие на ко¬
нечных R, близких по значению к равновесному, невелико
и может рассматриваться как возмущение, дает неглубокий
минимум на кривой E-R (рис. 33) в точке R0 = 0,09 нм. Ве¬
личина минимума (т. е. энергия диссоциации /)(Н2)) соста¬
вляет всего лишь 19,3 кДж/моль (эксперимент дает R0 =
= 0,07413 нм и D(H2) = 458,5 кДж/моль). Разумеется, такой
результат нельзя считать удачным даже при самых непритя¬
зательных требованиях к расчету, и поэтому следует по¬
строить более приемлемые двухэлектронные функции. Таки¬
ми функциями могут быть линейные комбинации вида:
Ч> + = ЛГ + [Фо(1)Фь(2) + фД2)фь(1)] (59а)
= N_[ Фа(1)фь (2) - Ф„(2)фь (1) ] (596)
где N+ и N_-нормировочные множители.
Эти функции, однако, не учитывают электронных спинов.
Ко времени написания статьи Гайтлера и Лондона принцип
Паули еще не был сформулирован в общем виде и отнесе¬
ние функций *F + и 'Р_, соответственно, к синглетному
155
Рис. 33. Зависимость энергии молекулы водорода от межъядерного расстояния.
и триплетному спиновым состояниям осуществлялось кос¬
венным путем. В современной же записи полные (коор¬
динатно-спиновые) двухэлектронные функции молекулы Н2
по Гайтлеру-Лондону имеют вид:
Функции
триплетного
состояния
молекулы
водорода
(Т-)
Функция
синглетного
СОСТОЯНИЯ Н2
СЕ.)
Ф? = {*.(»)<р„ (2) - Ф.(2)фь(1)}а(1)в(2)
(М5 = 1)
Ч*2’ = N { Фа (1) Фй (2) - <р (2)<рб(1)}р(1)р(2)
(JW = -1)
Ч,(3) = N_ { фо (1) фк (2) - V (2) Ф6 (1)} { а (1) Р (2) + а (2) р (1) ]
(М = 0)
(60)
Ч**» = N+ {Фо(1)Ф(1 (2) + фа(2)фь(1)}{а(1)р(2) - а(2)р(1)}
(М = 0)
156
Далее мы вернемся к этим функциям, а сейчас обратимся
к энергетическим характеристикам молекулы водорода, по¬
лучаемым в методе Гайтлера - Лондона.
Математические формулы...
Гамильтониан молекулы Н2 в атомных единицах можно
записать в следующем виде, сгруппировав слагаемые наибо¬
лее удобным для дальнейших рассуждений образом:
1 11 liiii
н = vf vj + +
2 ги. 2 г2ь г1Ь г2й г, 2 Rjr
Y ' V ' 4 1 у J
На-гамиль- Иь гамиль- Я'-гамильтониан, учитывающий
тониан тониан взаимодействия, характерные
атома Н„ атома Нь для молекулы
Теперь можно вычислить энергию синглетного и трип-
летного состояний с учетом орто-нормированности функ¬
ций фа и фь и спиновых функций а и (3:
В результате для энергии молекулы получается следую¬
щее выражение
где 2ЕН- энергия двух изолированных атомов водорода, находя¬
щихся в основном состоянии;
это интеграл перекрывания орбиталей ф0 и фь, центриро¬
ванных на разных атомах На и Нь:
±НЧ±d\dV2 _ <Ч,±|Я|'{,±>
(4>±т±1(^иг = <Ч'±|Т4>
Е +
Q ± А
1 ± S2
(61)
(62 а)
Q = - 2 >ь(2)Фь(2)
dVxdV2 (626)
-это кулоновский интеграл и
157
А = -2S
Ф0(1)Фь(1)
dVt +
фа(1)фь(1)фа (2)ФЬ (2)
dVydV2
(62 в)
-это обменный интеграл.
В приведенных интегральных выражениях спиновые функ¬
ции а и (3 в силу их орто-нормированности отсутствуют.
Если бы расчет производился с простейшими функциями-
произведениями (58), то формула (61) приняла бы вид
и минимум на потенциальной кривой был бы обусловлен тем,
что первый интеграл в Q при не очень больших R отрицателен
и по абсолютной величине превосходит второй, так что в це¬
лом Q получается отрицательной величиной. Но энергия хи¬
мической связи при таком грубом расчете составляет, как мы
видели, лишь 10% от экспериментальной величины, тогда как
расчет по методу Гайтлера - Лондона дает: R0 = 0,0869 нм и
Ещш — — 303,8 кДж/моль (рис. 33).
В работе Гайтлера и Лондона в аналитическом виде были
выражены только кулоновский интеграл и интеграл перекры¬
вания, для А авторы ограничились оценкой. Впоследствии
японский физик Й. Сугиура вычислил интеграл А. Но и до
расчета Сугиуры было уже ясно, что по порядку величины
Ra и Emin совпадают с экспериментальными, чего не давала
ни одна доквантовомеханическая теория ковалентной связи.
Каков же физический смысл интегралов в формуле (61)?
Обратимся сначала к кулоновскому интегралу. Подынте¬
гральное выражение первого слагаемого в выражении для Q
Е — 2ЕН + Q
... и их физический смысл
158
Рис. 34. К физическому смыслу кулоновско- rir, ,т,
го интеграла. фъУЧЩ
1
представляет собой энергию d<Jia=fa^)dV1
взаимодействия заряда dqla =
= ф о (1) d V j, соответствую¬
щего орбитали фа, с ядром b
(рис. 34), а сам интеграл
Фа2(1)
dV1
^2a = ^a(2)d^
2a
^26=Уь(2)с/^
характеризует энергию взаимодействия зарядового распре¬
деления, отвечающего орбитали фа, с ядром Ь. Аналогичный
смысл будут иметь интегралы
Фа2(2)
dVv
Фь
dV, и
Фь2( 2)
появляющиеся в процессе вывода формулы (61). Все они
представляют взаимодействия ядер На и Нь с «чужими» (т. е.
первоначально им не принадлежавшими) электронами (см.
рис. 34)*. Так как все написанные выше четыре интеграла
численно равны друг другу в силу симметрии задачи и не¬
различимости электронов, то в первом слагаемом интеграла
Q должен был бы появиться коэффициент 4, но после сокра¬
щений в ходе математических выкладок остается коэффици¬
ент 2.
По нашему мнению, читателю, особенно начинающему, было
бы крайне полезно проделать эти выкладки самостоятельно, в виде
упражнения.
* Интегралы, описывающие взаимодействия ядро-«свой»
электрон, «ушли» в выражение для Ен.
159
Рассуждения, аналогичные вышеприведенным, показы¬
вают, что второе слагаемое в Q характеризует энергию вза¬
имодействия двух зарядовых распределений, обусловленных
орбиталями фа и фь (см. рис. 34).
Так как в рассматриваемом варианте метода Гайтлера —
Лондона ионные члены не учитываются (т. е. «разные»
электроны постоянно находятся на разных орбиталях), то
и интегралов типа
Теперь перейдем к обменному интегралу А. Его смысл
раскрыть труднее. Математически он возникает при выводе
формулы (61) в силу: а) неразличимости электронов и свя¬
занной с ней необходимостью учитывать в выражении для
два возможных способа «равновесного», т.е. (без ионных
членов) распределения электронов по орбиталям ф0 и фь:
фа(1)фь(2) и фа (2) фь(1) и б) в силу учета принципа Паули.
Название этого интеграла как раз и отражает то обстоя¬
тельство, что второе слагаемое в нем содержит координаты
обоих электронов и их «переставленные» координаты. От¬
сюда видно, что термин «обменный», строго говоря, следо¬
вало бы применять только к этому второму слагаемому
в выражении для А, но по сложившейся традиции обменным
интегралом называют всю приведенную выше сумму (62Ь).
При значительных межъядерных расстояниях R величина
А становится малой и обменной энергией можно прене¬
бречь. Напротив, при R, близких к равновесному, величина
А становится большой и отрицательной, обусловливая
около 90% энергии химической связи в синглетном (основ¬
ном) состоянии молекулы водорода. Для триплетного же со¬
здесь не появляется.
Что такое обмен?
160
стояния при всех R наблюдается отталкивание атомов, так
как А входит в (61) с обратным знаком.
Естественно, что эти обстоятельства на заре квантовой
химии послужили причиной истолкования природы химиче¬
ской связи как проявления некоего особого неклассического
«обменного взаимодействия». При этом термин «обмен» по¬
нимали двояко:
-с одной стороны, как отражение того, что при образо¬
вании молекулы водорода из двух атомов имеется конечная
вероятность обнаружения около ядра атома На электрона,
принадлежавшего первоначально (при R -> оо) атому Нь,
и наоборот,
-с другой стороны, под обменом зачастую подразумева¬
ли некий реальный, периодический процесс, происходящий
с некоторой частотой v = (£+ — E_)/h.
Иными словами, считалось возможным дать квантово¬
механической теории химической связи псевдоклассическую
интерпретацию с помощью представления о синхронном
перескоке электронов от атома к атому.
Такая трактовка обменного интеграла в первые годы
развития квантовой химии получила широкое распростране¬
ние, однако она противоречит как принципу неразличимости
электронов, так и самой постановке задачи,-ведь с самого
начала речь шла о стационарных состояниях молекулы
водорода.
В действительности, понятие обмена отражает перерас¬
пределение электронной плотности, получаемое в нулевом
приближении теории возмущений по межэлектронному
взаимодействию вследствие учета перестановочной симме¬
трии волновой функции молекулы.
Говоря об обменном интеграле и связанных с ним эф¬
фектах, следует отметить ту существенную роль, которую
в них играет перекрывание орбиталей <рв и <рь, т.е. интеграл
5. Действительно, при нулевом значении этого интеграла
П-918
161
(ортогональные орбитали) обменный интеграл сводится
к двухэлектронному
f ф.окщф.скс) и <ab|j_|f)o>
J г г
'12 '12
который является положительным.
Но тогда оказывается, что энергетический уровень три-
плетного состояния Е_ лежит ниже синглетного
£_ = 2£H + Q-<ab| — \Ьа} < Е+ = 2£н + Q + <аЬ\ — | Ьа>
г12 г12
что противоречит эксперименту.
Лишь существенное перекрывание атомных орбиталей
обеспечивает большое и отрицательное значение обменного
интеграла и связывающий характер основного (синглетного)
состояния молекулы Н2 в методе Гайтлера-Лондона.
Первый интеграл в выражении для А имеет большое по
абсолютной величине (большее, чем <ab |—^—| Ьа~)) и отрица-
г 12
тельное значение и описывает энергию электростатического
притяжения между атомными ядрами и зарядом перекрыва¬
ния в области химической связи.
Суммарное электронное облако при сближении атомов
усиливается в пространстве между ядрами благодаря «нало¬
жению» орбиталей, что оказывает стягивающее действие на
ядра атомов и тем самым «цементирует» молекулу. Такова
картина в синглетном состоянии.
В «триплете» же перекрываются орбитали с различными
знаками, что ослабляет плотность электронного облака
между ядрами и приводит к отталкиванию атомов.
В 1933 г. Джеймс и Кулидж провели расчет молекулы во¬
дорода, не выражая двухэлектронную функцию ¥ через
атомные орбитали фа и фь, а непосредственно включив в нее
величину г12 и используя сфероидальные координаты. При
162
этом получились очень хорошие результаты. Разумеется,
в их расчетах не фигурировали ни кулоновский, ни об¬
менный интегралы. Это показывает, что одна и та же функ¬
ция, описывающая состояние многоэлектронной системы,
может быть представлена различным образом, соответ¬
ственно чему существует и неоднозначность в разложении
энергии на составные части и неоднозначность выбора поня¬
тий, в терминах которых описывают многоэлектронную
систему. Но из факта, что понятие обмена связано с опреде¬
ленными приближениями (и в ряде методов, например в ме¬
тоде Джеймса-Кулиджа, не используется), не следует делать
вывод, будто оно не отражает никакой физической или хи¬
мической реальности. Всякое конкретное понятие ограничено
определенной моделью и преходяще, как и последняя. Но
в области своей применимости, оно по-своему отражает те
или иные грани объективной реальности. Так, понятие обме¬
на выражает тот факт, что при образовании молекулы
электроны, принадлежавшие ранее одним атомам, могут на¬
ходиться в околоядерном пространстве других; указывает
на важную роль заряда перекрывания для образования хи¬
мической связи и учитывает перестановочную симметрию
системы тождественных микрочастиц (электронов).
Иными словами, понятие обмена отражает такие важные
для понимания природы химической связи явления, как кол¬
лективизация (обобществление) электронов и перестройку
электронных оболочек взаимодействующих атомов.
Заметим, что для объяснения природы химической связи
не пришлось вводить никаких новых типов динамических
взаимодействий. Между образующими молекулу частицами
действуют только известные из классической физики элек¬
тростатические (кулоновские) силы притяжения и отталкива¬
ния. Новизна, привнесенная квантовой механикой, состоит
в ином, по сравнению с классикой, способе описания движе¬
ния частиц (о чем мы уже писали выше) и в учете особого
вида «несиловых» (по выражению В. А. Фока) взаимодей¬
ствий, выражаемых принципом Паули.
и*
163
Завершая разговор о работе Гайтлера и Лондона, сле¬
дует остановиться на одном недоразумении, связанном с об¬
суждаемым здесь кругом вопросов.
Память о спине
Оно состоит в переоценке роли спаривания спинов элек¬
тронов, которое рассматривается как наиболее глубокая
и важная причина образования химической связи. Между
тем, непосредственного участия в образовании молекулы
спины электронов не принимают и взаимодействия, благо¬
даря которым атомы объединяются в молекулы, имеют чи¬
сто электростатическую природу.
Вопрос о роли спина в теории многоэлектронных систем
не нов, он возник уже в конце 1920-х гг. Суть проблемы со¬
стояла в том, что гамильтониан такой системы (например,
молекулы) в нерелятивистском приближении не зависит от
ее полного спина (5) и, казалось бы, его собственные значе¬
ния (т.е. значения энергии) также не должны зависеть от S.
Между тем, как мы уже видели на примере молекулы водо¬
рода, наблюдаемые в действительности значения энергии су¬
щественно зависят от того, в каком спиновом состоянии на¬
ходится многоэлектронная система. Это противоречие было
формально разрешено в принципе антисимметрии, согласно
которому, напоминаем, iV-электронная волновая функция
должна быть антисимметричной относительно перестановки
переменных любой пары электронов. При этом в число
переменных, наряду с тремя пространственными, скажем,
декартовыми, координатами, обязательно должны входить
спиновые переменные (а) электронов.
Таким образом, из множества собственных функций
бесспинового гамильтониана принцип антисимметрии
(принцип Паули) отбирает лишь некоторые, удовлетворяю¬
щие указанному выше условию. Все прочие решения от¬
брасываются как не имеющие физического смысла.
164
Наличие переменных ст обеспечивает наиболее простую
формулировку принципа Паули. Однако она не является
единственно возможной. Более того, введение спиновых пе¬
ременных в волновую функцию кажется несколько искус¬
ственным, что наводит на мысль о возможности иной
формулировки принципа, в которой спиновые переменные
отдельных электронов не фигурировали бы явно. Впервые
в общем виде правильные условия симметрии для коорди¬
натных волновых функций были получены в 1940 г. В. А. Фо¬
ком. В 1960-70-х гг. в работах И. Г. Каплана, Ф. Матсена
и других авторов была разработана так называемая «бес-
спиновая» схема квантовой химии, физически эквивалентная
обычной, но в которой свойства симметрии волновой функ¬
ции выражаются с помощью групп перестановок. Уровни
энергии многоэлектронной системы при этом характери¬
зуются перестановочной симметрией соответствующих им
координатных волновых функций, вид которых несет в себе
как бы «память о спине».
Спаривание спинов, таким образом, оказывается лишь
своего рода «мнемоническим правилом» или, по выражению
Ван Флека, «индикатором» образования химической связи
в рамках модели Гайтлера - Лондона, но не объяснением
природы этого явления.
После объяснения на основе квантовой механики при¬
роды химической связи в молекуле водорода были предпри¬
няты многочисленные попытки, с одной стороны, улучшить
метод Гайтлера-Лондона, а с другой,-распространить его
на другие, более сложные молекулы, что привело в итоге
к созданию метода валентных связей (ВС).
МЕТОД ВС
Алгебра химических связей
Основная идея этого метода заключается в предположе¬
нии, что атомы в молекуле сохраняют в известной мере
свою индивидуальность. А если выражаться более точным
165
языком, то индивидуальность в значительной степени сохра¬
няют атомные орбитали, набор которых математически
представляет данный атом.
Таким образом, многоэлектронная волновая функция ме¬
тода ВС строится из орбиталей отдельных атомов.
Казалось бы, все просто и ясно. Но на деле такое по¬
строение оказывается чрезвычайно громоздким. Поэтому
мы начнем с иллюстрирующего примера, в качестве которо¬
го выберем молекулу гидрида бериллия ВеН2, а уже потом
обратимся к рассмотрению общей процедуры метода.
Основному состоянию атома Be отвечает электронная
конфигурация ls22s2. В химических соединениях бериллий
двухвалентен, поэтому его валентному состоянию обычно
сопоставляют конфигурацию ls22s12p1 (о понятии валентно¬
го состояния см. далее). Тогда в образовании химических
связей в молекуле ВеН2 будут участвовать четыре ва¬
лентных АО: cpj = 2s и ф2 = 2рх АО атома бериллия и
Ф3 = lsa и ф4 = lsb АО атомов водорода*.
Совокупность всех орбиталей {ф15 ф2, ф3, ф4} называется
конфигурацией, а каждый из поднаборов ГВе = {ф1? ф2},
Гн = {фз} и Гн = {ф4}~ валентной конфигурацией соответ¬
ствующих атомов.
Полная волновая функция молекулы должна включать
координатную и спиновую части. В простейшем случае (т. е.
без учета принципа неразличимости электронов) координат¬
ную волновую функцию молекулы можно записать в виде
произведения АО:
ф(1>2д4) =tMbB &£>. &Е
^Ве Гн Гн
а Ь
* Здесь следовало бы взять в качестве cpj- и <р2-гибридные АО
бериллия, но для целей настоящего раздела это необязательно, по¬
скольку мы здесь не претендуем на точное описание геометрии
молекулы.
166
Затем Ф (1,2,3,4) следует домножить на спиновую функ¬
цию молекулы 0(1,2,3,4), построенную из одноэлектронных
спиновых функций аир. Вот тут-то и начинаются сложно¬
сти. Точнее, не сложность,-ибо с чисто математической точ¬
ки зрения, мы имеем дело с обычной алгеброй,-но услож¬
ненность, «тяжеловесность» алгебраических конструкций.
Чтобы построить 0(1,2,3,4) надо сначала из каждой
пары функций а (0 и (3 (/) составить N/2 двухэлектронных
функций* у (i,j) = [a (j) Р (/) — а (/) Р (0 ]/|/2 описывающих
спаривание одноэлектронных спиновых моментов i-ro и j-го
электронов. Но разбиение молекулярных электронов на
пары (у) можно осуществить множеством способов. В на¬
шем случае вариантов разбиения три:
у(1,3) = [а(1)р(3)-а(3)Р(1)]/|/2
Y (2,4) = [а (2) р (4) — а (4) Р (2)] |/2
y(l,4) = [a(l)P(4)-a(4)P(l)]j/2
У (2,3) = [а (2) Р (3) — а (3) Р (2)] f]/l
у (1,2) = [а(1)Р(2) - а (2) P(l)] /J/2
у (3,4) = [а (3) р (4) - а(4)Р(3)]/|/2
Справа приведена графическая иллюстрация попарного
разбиения четырех электронов. Каждому варианту разбиения
сопоставляют функцию - произведение:
©I = у (1,3) у (2,4); ©„ = у (1,4) у (2,3); ©iii = У (1,2) у (3,4)
называемую структурой ВС.
* N- число принятых в рассмотрение электронов. В нашем
случае N = 4. Учет полного числа электронов, ничего не изменив
принципиально, неоправданно усложнил бы выкладки.
167
Непосредственным вычислением нетрудно убедиться, что
@ш = ©I - ©и
т.е. из трех структур только две будут линейно-независимы¬
ми. Какие же из них выбрать в качестве базисных? С мате¬
матической точки зрения, это безразлично, а вот с химиче¬
ской-предпочтение следует отдать структурам 0j и 0П, так
как только в них атомы бериллия и водорода оказываются
связанными.
Тогда полная волновая функция молекулы ВеН2 примет
вид
где коэффициенты СЛ и Сп определяются минимизацией
полной энергии молекулы.
Особо следует отметить, что в выражении (63) для каж¬
дого слагаемого С,Ф0, и СпФ0п между парой атомных ор¬
биталей {фг, ф;} и функцией у (г, j) имеется определенное со¬
ответствие, поэтому АО ф; и ф; называют спаренными
в данной структуре ВС.
Спаривание орбиталей можно представить графически.
Для выбранных нами базисных структур гидрида бериллия
соответствующие диаграммы примут вид:
Здесь штриховой линией обведены точки, изображаю¬
щие орбитали ф, и ф2 атома Be, Стягивая эти точки в одну,
мы переходим от диаграмм, выражающих спаривание орби¬
талей, к диаграммам, отображающим связи атомов. В дан¬
ном случае структуры 1 и II приводят к одной и той же диа¬
грамме атомных связей, которая идентична классической
'Р0 = 0(1,2,3,4)(Cj@i + СП0П)
(63)
4
\
168
структурной формуле молекулы ВеН2:
/Н
Ве(
ХН
Однако такое случается не всегда. Наиболее известным
примером невозможности передать электронное строение
соединения одной схемой связей атомов (т.е. одной класси¬
ческой структурной формулой) является молекула бензола.
А как же антисимметрия?
Предвижу напоминание читателя: «Все это хорошо, но
4*0 не удовлетворяет принципу антисимметрии!» Да, это так
и потому ее надо антисимметризовать, т.е. подействовать
на нее оператором антисимметризации А:
441,2,3,4)= Л {Ф (1,2, 3,4) £ СК0Х(1,2,3,4)} (64)
X = I, II
Заметим, что процедура антисимметризации сохраняет
указанное выше соответствие (ф{, ф7} у(i, j), т.е. и при
учете принципа Паули понятие спаривания электронов (а
точнее, атомных орбиталей) сохраняется. Выше орбитальное
спаривание было показано наглядно в виде диаграмм, ко¬
торые называют диаграммами Румера. Здесь, правда, надо
сделать оговорку...
Схемы Румера и таблицы Юнга
Как было показано Гайтлером и Румером, число линей¬
но-независимых многоэлектронных спиновых функций 0Х,
полученных методом спиновых спариваний п валентных
орбиталей, составляет:
и!/[(и/2)!(и/2 4- 1)!]
В нашем случае п = 4, и число линейно-независимых структур рав¬
но двум (4!/2!3! — 21
169
Ю. Б. Румером был предложен также графический метод
построения линейно-независимых наборов 0Х, согласно ко¬
торому каждой одноэлектронной функции сопоставляется
точка окружности или иной выпуклой кривой на плоскости
и затем эти точки попарно соединяются отрезками прямых
линий; каждый отрезок изображает двухэлектронную спино¬
вую функцию у (i,j). Многоэлектронные спиновые функции,
отвечающие диаграммам Румера с непересекающимися
штрихами, образуют линейно-независимый базис для разло¬
жения (64), а остальные будут тогда их линейными комби¬
нациями.
Т.е., по Румеру, нам бы следовало выбрать для моле¬
кулы ВеНг структуры ©п и ©ш. Выходит-мы ошиблись?!
Нет, просто Румер предложил хороший и-для небольших
и-удобный способ выбора независимых структур. Но спо¬
соб этот-не единственно возможный.
Обычно правило Румера иллюстрируется диаграммами
для л-электронной системы бензола
3 3 3 3 3
2f 2
li ^5 1
6 6 6 6 6
I и III IV v
которым отвечает пять линейно-независимых структур ©и
(х = 1,11,V)
Но есть и другой способ построения структур ВС
с помощью так называемых стандартных таблиц Юнга.
Каждая такая таблица содержит два столбца номеров АО
в возрастающем сверху вниз и слева направо порядке
и отвечает определенной диаграмме Румера (возможно,
с пересекающимися отрезками прямых) и определенной
структуре 0К. Так, для тс-электронной системы бензола
П
170
стандартные таблицы Юнга имеют вид:
f
2
3
4
5
б
1
3
2
4
5
6
f
2
3
5
4
6
1
3
2
5
4
6
1
4
2
5
3
6
I II III IV V
Им соответствуют обобщенные диаграммы Румера
3 3 3 3 3
а этим диаграммам-пять спиновых функций @х, образую¬
щих линейно-независимый набор, отличный от полученного
по правилу Румера. Такие наборы мы будем называть
юнговскими.
Для молекулы ВеНг мы также можем написать два неза¬
висимых набора ©х: румеровский
1« *2
4* *3
0ш
1* «2
4* *3
и юнговский
'\/2
1
3
2
4
1
2
3
4
171
Мы видим, таким образом, что выбор базисных структур
в методе ВС неоднозначен. Кроме того, заранее не ясно, ка¬
кой из методов получения таких структур приводит к разум¬
ным с химической точки зрения результатам. Для моле¬
кулы бензола химически предпочтителен румеровский базис,
состоящий из двух структур Кекуле и трех структур Дью¬
ара.
Сложной и до сих пор окончательно не решенной проб¬
лемой является определение веса каждой структуры, ко¬
торый зависит как от коэффициентов Сх, так и от перекры¬
вания соответствующих многоэлектронных функций.
Прост ли метод ВС?
Все сказанное говорит против широко распространенно¬
го мнения о простоте и наглядности метода ВС. Чтобы еще
усилить это сомнение, обратимся к вопросу об определении
кратности химической связи в рассматриваемом методе.
Для примера возьмем молекулу бензола, ограничившись
только структурами Кекуле:
О О
0, ©н
Кратность связи в таких случаях может быть представле¬
на следующей формулой:
клв = I», I I С (65)
х аеА ЬеВ
их) _ [ 1 -если орбитали фа и фь спарены в структуре и
где /с„(, — ^
(О-в противном случае
сои-вес структуры х.
Веса двух изображенных выше структур молекулы бензо¬
ла равны, так как ©j и ©п эквивалентны по симметрии, т.е.
172
со* = — (х = I, II). Тогда, с учетом а-электронной системы,
которую описывают в приближении полного спаривания
единственной структурой, кратность связи двух соседних
атомов углерода равна:
Действительно, как показывает эксперимент, все связи
в бензольном кольце полностью выравнены и имеют длину
0,140 нм, промежуточную между длиной этановой (0,154 нм)
и этиленовой (0,134 нм) связей.
При учете всех пяти румеровских структур кратности
связей углерод-углерод несколько уменьшаются, но возни¬
кают ненулевые порядки связей противостоящих атомов
углерода: CjQ, С3С6 и С2С5.
Если же перейти к юнговскому набору базисных струк¬
тур, то связи соседних атомов С2С3, С4С5 и CiC6 оказы¬
ваются одинарными, тогда как кратности связей QC2, С3С4
и С5С6 больше единицы. Подобных примеров можно при¬
вести немало.
Мы видим, что в методе ВС далеко не всегда возможно
с достаточной степенью точности представить электронное
строение молекулы одной структурой (т.е. в приближении
полного спаривания). В наибольшей степени это относится
к л-электронным системам. Мы уже приводили в качестве
примера молекулу бензола. Другим примером может слу¬
жить нитрат-анион. Ему также можно сопоставить несколь¬
ко способов спаривания л-орбиталей:
*с,с, + 1 = 0,5-2 + 0,5-1 - 1,5
Резонанс, структуры и диаграммы Румера
:0:
173
По сложившейся традиции в таких случаях говорят о ре¬
зонансе или «наложении» (суперпозиции) структур.
Когда молекуле нельзя сопоставить одну схему спарива¬
ния, ее электронное строение может быть передано набором
таких схем. Реальное же распределение электронной плотно¬
сти оказывается усредненным по всем линейно-независимым
структурам с учетом веса каждой из них. Набор соответ¬
ствующих структур называется резонансным гибридом, а са¬
ми структуры -каноническими или резонансными.
Заметим, что ни одна из резонансных структур сама по
себе не отвечает реально существующему состоянию моле¬
кулы и не передает ее истинного строения. Развитая в рам¬
ках метода ВС концепция резонанса заменила классическое
представление молекулы одной структурной формулой на¬
бором схем спаривания, дающим более полное и правиль¬
ное представление о распределении электронной плотности.
Однако не следует всегда отождествлять приведенные
выше графические изображения структур ВС с классически¬
ми структурными формулами.
В диаграммах Румера штрихи, повторяем, характери¬
зуют связи отдельных орбиталей (причем, не обязательно
атомных), тогда как химические структурные формулы ото¬
бражают межатомные связи различной кратности. Далее,
классические структурные формулы определяют индиви¬
дуальные химические соединения с определенными свой¬
ствами и с определенным распределением валентностей ато¬
мов по химическим связям. Вещества, отвечающие разным
структурным формулам, обладают разными ядерными кон¬
фигурациями, т.е. различным расположением атомов в про¬
странстве. Диаграммы Румера определяют базис для описа¬
ния состояний электронной подсистемы молекулы при
фиксированной и одинаковой для всех диаграмм ядерной
конфигурации, т.е. все диаграммы соответствуют одному
и тому же соединению.
174
К спорам о резонансе
Пожалуй, ни одна квантовохимическая концепция не вы¬
зывала столь острой дискуссии, как концепция резонанса.
Основной вопрос этих дискуссий состоял в следующем:
можно ли отдельным резонансным структурам сопоставить
реальное химическое соединение? Не происходят ли по¬
стоянные быстрые самопроизвольные переходы от одного
соединения, описываемого одной резонансной структурой,
к другому, отвечающему другой такой структуре? Сейчас
кажется невероятным, что для ответа на эти вопросы соби¬
рались конференции, печатались статьи, монографии и т.д.
Но как бы там ни было, «проблема» была поставлена
и требовала своего разрешения. Начнем с терминологии.
Термин «резонансные структуры» можно применять только
в отношении эквивалентных структур метода ВС. Нельзя,
к примеру, называть резонансными структуры бутадиена
НС—СН _ НС=СН
Н2С СН2 Н2С—СН2
I II
так как первая структура может использоваться в качестве
структурной формулы соединения, а вторая-нет, поскольку
вес ее пренебрежимо мал. Действительно, длина ординарной
связи в такой структуре будет меньше длины двойной, что
противоречит известным эмпирическим закономерностям.
О резонансе и резонансных структурах имеет смысл гово¬
рить, когда соответствующие этим структурам квантовоме¬
ханические средние значения энергии равны или близки. При
этом надо иметь в виду, что эти значения электронной энер¬
гии определяются при фиксированной и тождественной для
всех структур конфигурации атомных ядер методами кван¬
товой химии. Они не имеют непосредственного физического
или химического смысла и не измеряются эксперименталь¬
но.
175
Было бы грубой ошибкой связывать резонанс с какими-
либо колебаниями, осциляциями, пульсациями или флуктуа¬
циями в молекулах. Такие псевдоклассические представле¬
ния, имеющие сомнительную ценность в отношении элек¬
тронной системы молекулы, совершенно неправильны
в отношении атомных ядер, которые на данном уровне рас¬
смотрения (электронная задача в адиабатическом приближе¬
нии) следует считать неподвижными.
Иногда резонанс путают с таутомерией. Так, структуры
разумеется, не являются резонансными. Они различаются не
только распределением тг-электронной плотности, но и по¬
ложением атома водорода: в кето-форме он связан с углеро¬
дом, в енольной форме-с кислородом. Геометрические ха¬
рактеристики этих форм, конечно, различны. В подобном
случае следует говорить не о резонансе, а о таутомерии.
В отличие от резонансных структур, таутомеры-это реаль¬
но существующие соединения, которые обычно можно отде¬
лить друг от друга. Так, кетонная и енольная формы нахо¬
дятся в состоянии подвижного равновесия. В приведенном
выше примере в равновесной таутомерной смеси преобла¬
дает кетонная форма, поскольку она на 58,8 кДж/моль ста¬
бильнее енольной, но обе формы являются, в отличие от ре¬
зонансных структур, реальными, экспериментально наблю¬
даемыми соединениями.
Часто с резонансом связывают понятие об «электронных
изомерах». Последние определяют как соединения, характе¬
ризуемые одной и той же ядерной конфигурацией, но раз¬
личным распределением электронной плотности. Такое
представление также ошибочно. Различным распределениям
электронной плотности отвечают разные ядерные конфигу¬
кетон (ацетон)
енол (1-пропен-2-ол)
176
рации и наоборот, о чем мы уже писали в предыдущей главе.
Следовательно, электронным изомерам неизбежно должны
соответствовать различные ядерные конфигурации, и это по¬
нятие в итоге сводится к обычному понятию изомерии.
Тогда возникает вопрос: какие же стороны объективной
реальности отражает понятие резонанса?
Необходимость учета нескольких резонансных структур
связана прежде всего с тем, что не всегда оказывается воз¬
можным приписать химическую связь отдельным парам
атомов и, следовательно, соединение нельзя охарактеризо¬
вать классической структурной формулой, которая не проти¬
воречила бы его свойствам. В этом случае химическая связь
делокализована между тремя и большим числом атомов.
Такой делокализации и соответствует резонанс ковалентных
структур.
Кроме того, в соединениях с локализованными двухцен¬
тровыми связями последние часто оказываются поляризо¬
ванными. Для отражения полярности связи следует учиты¬
вать так называемый ионно-ковалентный резонанс. Напри¬
мер, для молекулы N20:
В некоторых случаях, без учета резонанса структур,
в рамках метода ВС может получаться качественно непра¬
вильное описание электронной структуры молекулы. Так,
для бензола ни одна из двух классических формул Кекуле не
отражает реальной симметрии молекулы, а также ее физиче¬
ских и химических свойств. Другой пример-диоксид углеро¬
да С02. Длина связи углерод - кислород в нем равна
0,115 нм, тогда как длина «нормальной» двойной связи
С=0 (в кетонах) равна 0,122 нм, а расчетная длина тройной
связи С=0-0,110нм. Т.е. связь углерод - кислород в С02
оказалась промежуточной между двойной и тройной, что
можно объяснить в терминах концепции резонанса:
е ф
N—N=0
® ©
и N=N—О
12-918
177
Возвращаясь к вопросу о реальности резонансных струк¬
тур, укажем на такую аналогию (которая, впрочем, может
рассматриваться больше, чем просто формальное сходство
ситуаций). При решении физических задач часто приходится
разлагать какой-то вектор, которому отвечает вполне реаль¬
ная, экспериментально измеримая физическая величина, на
компоненты. Сделать это можно, вообще говоря, разными
способами. Обычно выбирают наиболее удобное, адекват¬
ное симметрии задачи и выбору системы координат, разло¬
жение. При этом далеко не всегда компоненты удается сопо¬
ставить с измеримыми физическими величинами, да это и не
требуется. Аналогично, в методе ВС-полная волновая функ¬
ция разлагается на «компоненты», каждой из которых отве¬
чает определенная схема спаривания орбиталей. Те схемы,
которые входят в разложение с наибольшим весом, обычно
включают в резонансный набор структур ВС.
Достоинства непопулярного метода
Исторически метод ВС был первым широко распростра¬
ненным методом квантовой химии в 30-х гг. Затем, в по¬
слевоенные годы его начал вытеснять метод молекулярных
орбиталей (МО), о котором речь пойдет ниже. После перио¬
да абсолютного господства в квантовой химии теории МО,
у исследователей, примерно с 1960 г., наблюдается все более
возрастающий интерес к методу ВС. По словам Р. Мак-Ви¬
ни, сказанным им в 1969 г., «Метод ВС как метод построе¬
ния достаточно хороших молекулярных электронных вол¬
новых функций сильно дискредитировали за последние 20
лет, и теперь его обычно рассматривают просто как полуэм-
пирическую схему... Вместе с тем, следует подчеркнуть, что
на его основе можно развить математически совершенно
строгую теорию, которая с успехом может использоваться
для проведения неэмпирических расчетов. Метод валентных
связей заслуживает большего внимания, чем обычно ему
уделяют».
178
Усиление интереса к указанному методу в настоящее вре¬
мя в значительной степени объясняется прогрессом ЭВМ.
Последнее обстоятельство позволяет производить количе¬
ственные ab initio расчеты многоатомных молекул, хоть
и более сложным в вычислительном отношении, но зато
и более точно описывающим энергетику и механизм хими¬
ческих процессов, методом ВС. При этом в наши дни не
только проводится его последовательная численная реализа¬
ция в расчетах конкретных, постепенно усложняющихся мо¬
лекулярных систем, но также ведется дальнейшее совершен¬
ствование формализма. В частности У. Годдардом был
разработан обобщенный метод ВС, который был с успехом
применен как для расчета и качественного рассмотрения от¬
дельных соединений, так и для анализа механизмов химиче¬
ских реакций.
Завершая рассказ о методе ВС, остановимся на понятиях
валентности и валентного состояния атома в рамках этого
метода.
Что такое валентность в методе ВС?
В соответствии с принятыми химическими представле¬
ниями валентность атома А можно определить как сумму
кратностей связей, образуемых этим атомом. Тогда, исполь¬
зуя формулу (65), можно записать такое определение в мате¬
матическом виде:
*4 = I кАВ= I X«n’ = L 2>«»Г
В(#А) В(#А) и аеАЬеВ хае А
где величина — 0 или 1 и представляет со-
В(#А)ЬеВ
бой вклад атомной орбитали аеА в валентность атома А.
Проиллюстрируем это определение на примере моле¬
кулы бензола. В качестве базисных структур ограничимся
12* 179
двумя структурами Кекуле. Тогда:
vc, = [®i(*i2(*) + М2 И) + ®п (М2 (я) + М2 И] +
+ 1>1(Мб(л) + МбН) + °>п(Мб(я) + /с1б(ст))]
Значки я и а соответствуют спариванию я-или ст-АО
бензола. Так как a>j = соц = 0,5, то (без учета связи СН):
VCt = 0,5 (1 + 1) + 0,5 (0 + 1) + 0,5 (0+1)4- 0,5 (1 + 1) = 3
Учитывая, что кратность связи СН равна 1
KCiH=0,5 1 +0,5-1 = 1
получаем полную валентность атома углерода в бензоле
у поли = 4
что соответствует классическому значению.
Можно показать, что величина 1^с) не зависит от х для
синглетных состояний, если каждому атому сопоставлять
единственную валентную конфигурацию, так что верхний
индекс в приведенном обозначении можно убрать. Кроме
того, сумма £сох равна единице. Тогда
X
к = I к
ае А
т.е. валентность атома А равна числу неспаренных орбита-
лей в его валентной конфигурации ГА, т. е. спин-валентности
А, и не зависит ни от выбора базисных структур, ни от веса
каждой из них. В этом смысле валентность является инва¬
риантом атома в молекуле.
Для того, чтобы этот вывод привести в соответствие
с эмпирическими правилами валентности атомов, было
введено...
180
..понятие валентного состояния
Рассмотрим содержание этого понятия на примере со¬
единений углерода, в подавляющем большинстве которых
атом С образует четыре ковалентные связи, хотя в валент¬
ной конфигурации основного состояния он имеет только две
неспаренные орбитали (два неспаренных электрона, как ча¬
ще говорят).
Если, однако, затратив некоторую энергию, возбудить
углеродный атом так, чтобы один из электронов 2s-AO
перешел бы на свободную 2р-орбиталь, то число неспа¬
ренных электронов станет равным 4.
Но перевод атома в валентное состояние не сводится
только к его возбуждению (промотированию). Следует
учесть также неопределенность в ориентации спинов неспа¬
ренных электронов, участвующих в образовании химических
связей. А если говорить точнее, то необходимо принять во
внимание, что волновая функция валентного состояния ато¬
ма не является собственной функцией операторов квадрата
полного спина атома (S2) и его проекции на ось квантования
2 (SJ- равно как она не является и собственной функцией
операторов квадрата полного орбитального момента коли¬
чества движения (L2) и его проекции (!*).
Таким образом, валентное состояние атома не есть ка¬
кое-либо его стационарное состояние (основное или возбуж¬
денное), и потому оно не будет спектроскопически наблю¬
даемым.
В итоге, переход атома углерода в валентное состояние
можно условно представить следующей схемой, учитываю¬
щей как энергию возбуждения атома (Евозб), так и энергию,
181
связанную с переориентацией спинов (£сп):
С; ts!2s*2p2 с.тз_: ,si2st2pe an-W»
щрш [Триш
НЕ Щ]
С*ал: is22sl2p3
иР^ш
ОБ
Отсутствие стрелок у символов электронов в правой части
схемы указывает на неопределенность в ориентации их спинов.
Как видим, вклад Ет в суммарную затрату энергии на
переход атома в валентное состояние весьма заметен и соиз¬
мерим с £возб, Кроме того, орбитали атома в молекуле мо¬
гут быть так или иначе гибридизованы, чему также соответ¬
ствует определенная энергия Егаб_. Однако ее вклад как
правило не превышает 20-60 кДж/моль, и им часто прене¬
брегают.
Результаты расчетов рядов С -* СО -*■ С02 и т. п. по¬
казывают, что возбуждение 2s -+ 2р претерпевает заметная
часть электронной плотности
(С) /sW2;
о2—(С+0,35О~°'35)ls22s1'7s2j
\ 41 f
,>^iC'™o;0AS)iАЛ,
1 t t
-0.22
-0,13
- 0,94
1 +0,40 j
так что в первом приближении можно пользоваться
моделью, принимающей перенос одного электрона с 2s-
на 2р-АО.
Энергетические затраты на перевод атома в валентное
состояние (£мл) компенсируются энергией, выделяющейся
при образовании химических связей. Разумеется, если £вал
очень велика, то она не может окупиться образованием до¬
полнительных связей. Так, у атомов Си, Ag и Ай Е^, отве¬
182
чающая переходу (п — 1 )d10nsl -> (и — 1 )d9nslnpl, довольно
значительна и максимальна у серебра. Возможно, с этим об¬
стоятельством связан тот факт, что для Ag наиболее харак¬
терно одновалентное состояние, тогда как для Си и Аи-
двух- и трехвалентные.
После драки...
В заключение разговора о понятии валентного состояния ато¬
ма небольшое отступление полемического характера. Как-то авто¬
ру этих строк случилось написать для журнала «Химия в школе»
статью о химической связи, где среди прочих вопросов рассматри¬
валось и понятие валентного состояния. В редакционном ответе от
имени рецензента было сказано: «Утверждение о хаотическом рас¬
пределении ориентаций спинов не имеет смысла: валентное состоя¬
ние атома-это не какое-то реальное состояние, а лишь тот нуль
отсчета, от которого можно по тем или иным соображениям дви¬
гаться при построении качественной теории. Большого смысла
в этом представлении нет. К тому же в значительной степени оно
устарело».
Мы привели здесь это курьезное замечание потому, что подоб¬
ное мнение среди химиков стало почему-то распространенным. Ва¬
лентное состояние атома-не просто некий «нуль отсчета». Оно бы¬
ло введено в теорию ВС с целью распространить ее на случаи,
когда число неспаренных электронов в основном состоянии атома
меньше числа образуемых им двухэлектронных двухцентровых свя¬
зей. Вместе с тем, это понятие используется и в методе молеку¬
лярных орбиталей, в рамках которого оно обычно понимается как
эффективная электронная конфигурация с дробными заселенностя¬
ми АО и эффективными зарядами, что позволяет учесть как промо-
тирование электронов с одних АО на другие, так и их перенос от
атома к атому при образовании химических связей (см. приве¬
денный выше пример для ряда С - СО - С02). И используется
это понятие в обоих методах не только для построения качествен¬
ной теории, но и при квантовомеханических расчетах*.
* Детально этот вопрос изложен в главах 3 и 4 книги
О. П. Маркина «Стабильность и структура газообразных неоргани¬
ческих молекул, радикалов и ионов» (М.: Наука, 1980), из которой,
между прочим, ясно видно, что понятие валентного состояния от¬
нюдь не устарело.
183
Теперь о «нереальности» валентного состояния. Оно нереально
лишь в том смысле, что не является, как уже отмечалось, спектро¬
скопически наблюдаемым. Но оно отражает вполне реальное физи¬
ческое явление-перераспределение электронной плотности при
переходе от изолированных атомов к «атомам в молекуле», сопро¬
вождаемое расширением валентных возможностей атомов.
Не каждое используемое в теории понятие имеет своего, как
говорят методологи, «референта» («представителя») в объективной
реальности. Примерами таких понятий в квантовой химии могут
служить резонансные структуры, обменное взаимодействие, пере¬
ходное состояние в химической кинетике, которое, кстати, тоже
спектроскопически ненаблюдаемо, и т. д. Но отсюда не следует, что
такие понятия «не имеют большого смысла» и должны быть из¬
гнаны из теории.
О гибридизации-позже!
На этом мы закончим основной разговор о методе ВС.
Обычно в литературе при изложении этого метода рассма¬
тривают еще понятие о гибридизации АО, которое истори¬
чески действительно возникло и сформировалось в рамках
теории валентных связей. Однако гибридизационные пред¬
ставления столь же широко используются и в теории МО.
Учитывая, что в рамках именно этой последней теории не¬
давно были получены нетривиальные результаты, касаю¬
щиеся гибридизации АО, мы обсудим эту концепцию после
рассказа о методе МО. Итак...
МЕТОД МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
В первой главе мы уже рассмотрели понятие о молеку¬
лярных орбиталях и спин-орбиталях в связи с обсуждением
одноэлектронного приближения и метода ССП (метода Хар-
три-Фока). Здесь мы остановимся на теории МО более де¬
тально. Начнем с вопроса о способе представления молеку¬
лярных орбиталей.
184
Как найти МО?
Задача нахождения МО многоатомной системы в общем
виде довольно сложна и потому для ее практического реше¬
ния необходимо ввести дополнительные упрощения. Чаще
всего (уотя и не всегда) используют так называемое прибли¬
жение МОЛКАО, в рамках которого каждую МО предста¬
вляют в виде ограниченного разложения по атомным орби¬
талям всех атомов молекулы, т. е. в виде линейной
комбинации АО:
м
Ф< = £ (66)
Ц=1
г де %м - атомные орбитали входящих в молекулу атомов,
си{-коэффициенты разложения (вообще говоря, комплексные), опре¬
деляющие вид МО.
Набор используемых АО называют базисным набором
или просто базисом. Базисные функции предполагаются
нормированными, но не обязательно ортогональными (если
две АО центрированы на соседних атомах, то они могут
быть и неортогональны).
Аппроксимация МО в форме ЛКАО, по существу, пред¬
ставляет собой математическое выражение в рамках метода
МО и на языке этого метода физической идеи о молекуле
как системе взаимодействующих атомов (идее, восходящей
к началу XIX в., к работам Дальтона, Берцелиуса и других
ученых-атомистов). Действительно, на электрон, находящий¬
ся вблизи какого-либо атома, действует поле, обусловленное
главным образом именно этим атомом. Следовательно,
волновая функция в данной, околоатомной, области должна
быть близка соответствующей АО. Если МО известны, то
с их помощью можно определить физико-химические свой¬
ства системы. Разумеется, на деле все не так просто, как
в общем изложении.
185
Уравнения Рутана
Итак, при заданном атомно-орбитальном базисе надо
найти МО. Для этого нужно знать коэффициенты сЦ1-.
В 1951 г. К. Рутан получил систему нелинейных уравнений
для определения этих коэффициентов и молекулярно-орби¬
тальных энергий, носящую ныне его имя. Не останавливаясь
на выводе этих уравнений, приведем только их вид для слу¬
чая молекулярных систем с замкнутыми оболочками, когда
каждая МО занята двумя электронами с противоположны¬
ми спиновыми моментами и полный спин такой системы
равен нулю.
В дальнейшем, подчеркнем, мы будем иметь в виду именно
этот случай.
Запишем гамильтониан молекулы в виде:
N 1 Л 1
Ям„л = 2Х + т1—
it Z к,I kl
(кф О
1 Z
где hk = — -гг VI — £ ——-одноэлектронный гамильтониан к-го
2 гак
а а"
электрона, движущегося в поле ядерного остова и потому назы¬
ваемый оператором остова.
Второе слагаемое характеризует межэлектронное взаи¬
модействие. В одноэлектронном приближении, где предпо¬
лагается, что электрон движется в поле ядер и эффективном
поле остальных электронов, член, содержащий оператор 1 /гк1
заменяется эффективным потенциалом, зависящим только
от координат рассматриваемого электрона.
Теперь можно записать уравнения Рутана для молеку¬
лярной системы с замкнутой оболочкой, содержащей
N электронов, попарно занимающих N/2 молекулярных
орбиталей:
м
I (iV - = 0 (ц = 1,2,..,, М; i = 1,2,...,N/2) (67)
V — 1
186
где
SMV = №„XvdV = <^|V>
N/2 M M
fMv = <HA1V> + I L I C^Ca;[2<^HVCT> - <^MCTV>]
j= 1 X = 1 <j = 1
<ц№> a Jx;(l)«*,(l)iF1
это матричный элемент оператора остова;
<(Л|»1«т> а Яй(1))5(2)—X,mX.(2)dV,dV2
<^iov> S я*;(1)йр)—хлихладмп
Г12
это матричные элементы межэлектронного кулоновского взаимо¬
действия; ц, А,, ст и v-индексы АО; i-нумерует МО и их энергии е,.
Запишем эти уравнения в более компактной матричной
форме:
FC = SCe
(68)
где F- матрица с элементами F^v, называемая матрицей Фока,
а элементами матриц С и S будут, соответственно, коэффициенты
разложения (66) и интегралы перекрывания базисных АО; е-имеет
диагональную форму:
е =
О
о
'■'Н/2
Часто уравнения Рутана записывают в несколько ином
по сравнению с (67) виде, вводя следующие обозначения
где
Ку = <H*IV>
187
ММ 1
Gmv = I I f\e[<^Hvo> ~ -7<ИИСТУ>]
Л=1о=Х 1
N/2
Л» = 2 Z C*jCaj
i= i
или, в матричной форме:
F = h + G
А-матрица остова, G- матрица электронного взаимодействия.
Совокупность величин Ри образует так называемую ма¬
трицу порядков связей.
Важным дополнительным условием служит условие ор-
то-нормированности МО:
S<f,9,dV= I Z C'S^C,, = 8„
X=lo = l
Система уравнений Рутана имеет нетривиальные реше¬
ния, если ее детерминант равен нулю, т.е.:
det|F - eS| =
| ~ е F12 - eSi2 — |
= det | F2i — eS^t ^22 — e ••• ^2м ~ e^2M | (^®)
I ?М1 ~ Fm2 - e$M2 — Рмм — 8^лш I
Трудности решения
Строго говоря, получение точных решений уравнений
(68) предполагает бесконечный базис функций, т.е. требует
решения бесконечной системы уравнений. Но, как показал
Рутан и как подтверждает обширная расчетная практика,
удовлетворительного приближения можно достичь и при ко¬
188
нечном базисе АО. При этом многое зависит от выбора ба¬
зиса-его размеров и качества. Расширяя базисный набор
путем добавления новых линейно-независимых функций,
можно достичь такой ситуации, когда вычисляемые характе¬
ристики системы (орбитальные энергии, наборы коэффи¬
циентов cxj и т. д.) окажутся «нечувствительными» к дальней¬
шему расширению базиса. В этом случае говорят
о достижении хартри-фоковского предела. Предельный ба¬
зисный набор АО дает очень точные результаты, почти та¬
кие же, как при численном интегрировании уравнений Хар-
три-Фока*. Однако увеличение числа АО в базисе сопрово¬
ждается существенным возрастанием вычислительных труд¬
ностей. Поэтому в реальных расчетах, особенно сложных
многоатомных систем, используют базисы укороченные по
сравнению с предельными.
Характерной особенностью уравнений (68) является их
нелинейность относительно искомых коэффициентов cxj, так
как от этих коэффициентов зависят элементы матрицы Фо¬
ка. Поэтому для решения системы Рутана используют ме¬
тод последовательных приближений. Начинают с того, что
предварительно в заданном базисе вычисляют матрицы S,
h и интегралы, входящие в G. Особые трудности при этом
связаны с вычислением так называемых многоцентровых
интегралов типа:
<ИйИ _ ГГ <(l>g(Z)Z.(l)X.(2) iViiy2'
где входящие в подынтегральное выражение АО могут быть
центрированы на двух, трех и четырех атомах. Чтобы избе¬
жать трудоемких вычислений трех- и четырехцентровых ин¬
тегралов часто используют различные дополнительные при¬
ближения, на которых мы здесь не будем останавливаться.
* Предельный хартри-фоковский базисный набор даже для
Атомов элементов II периода включает все АО вплоть до ва¬
кантных в основном состоянии этих атомов 3d- и ^-орбиталей.
189
После расчета интегралов можно приступать к итерацион¬
ной процедуре.
Допустим, исходя из физических условий задачи, удалось
выбрать некоторые разумные значения коэффициентов с(х®}
для всех МО. Подставляя их в выражение для Fцу, вычис¬
ляют элементы исходной матрицы Фока F$. Теперь систе¬
му (68) можно решать как линейную относительно коэффи¬
циентов, если предварительно из секулярного уравнения (70)
найти орбитальные энергии е|0). В результате получают
новый набор коэффициентов с[)\ с помощью которого со¬
ставляют новую матрицу Фока с элементами F™. Затем из
секулярного уравнения находят орбитальные энергии е{1)
и далее, используя величины и ej1* снова решают систе¬
му Рутана, что дает набор коэффициентов с$, и т.д. до со¬
гласования, т.е. до тех пор, пока вновь вычисленные значе¬
ния не совпадут с требуемой точностью со значениями,
полученными в предыдущей итерации.
Замечательное свойство уравнений Рутана
Уравнения Рутана обладают одним замечательным свой¬
ством-они инвариантны относительно унитарных преобра¬
зований базиса АО. Вот что это означает. Запишем формулу
(66) в матричном виде:
Ф = ХС
где ф = (ф1,Ф2,...,Фа^/2) и х = (Xi»X2»-»Xm)-
Исходный базисный набор выразим теперь через АО
другого, штрихованного, базиса с помощью унитарной
матрицы
X = x'rf (71)
Математическое пояснение. Напомним, что матрица
U называется унитарной, если выполняется соотношение I/+ U =
= UU* — I, в котором U + -эрмитово сопряженная матрица, т.е.
U+ = U*, где тильдой обозначено транспонирование, а звездоч¬
190
кой-комплексное сопряжение. Если элементы матрицы U веще¬
ственные, то UU = UU = I, т.е. в этом случае матрица U-ортого¬
нальна (/-везде единичная матрица).
Можно показать, что вид уравнений Рутана, а также ве¬
личины полной (£полн) и орбитальных (е;) энергий при уни¬
тарном преобразовании базиса не изменяются.
Преобразование (71) может сводиться к следующему:
-замене АО одного и того же атома с одинаковыми п и
/ их линейной комбинацией (примером может служить сме¬
шение 2рх~, 2ру- и 2pz-kO при вращении локальной системы
координат, связанной с данным атомом);
-смешению различных АО рассматриваемого атома
(преобразование гибридизации);
-смешению АО разных атомов, что приводит к получе¬
нию неатомных базисов, как, скажем, в случае трансформа¬
ции АО в групповые орбитали, преобразующиеся по непри¬
водимым представлениям (НП) точечной группы симметрии
молекулы.
При переходе к симметризованному базису матрица
(F — Se) разбивается на блоки, каждый из которых отвечает
определенному неприводимому представлению:
где верхние индексы в скобках нумеруют НП.
Блочно-диагональный вид принимает и матрица поряд¬
ков связей Р.
191
В поисках базиса
Теперь несколько замечаний о выборе базиса. В принци¬
пе МО можно представить в виде разложения по раз¬
личным наборам функций. Но это-в принципе! Практиче¬
ски же необходимо, чтобы базис был оптимальный, т.е.
чтобы при сравнительно небольшом числе входящих в него
функций, он позволял бы получать достаточно точные ре¬
зультаты. В итоге, идея приближения MOJIKAO оказалась
одной из наилучших. Но здесь следует иметь в виду одно
важное обстоятельство: те АО, которые используются в мо¬
лекулярных расчетах,-это не орбитали, полученные при чис¬
ленном интегрировании уравнений Хартри-Фока для ато¬
мов. Последние заданы не в аналитическом, а в табличном
виде, и поэтому крайне неудобны для молекулярных расче¬
тов. Это значит, что необходимо каким-то образом аппрок¬
симировать АО. Последние, как правило, представляют
В В1Ше: Хы. = №!»1ы,(в,ф)
где N- нормировочный множитель.
Основная задача при построении базиса сводится к ап¬
проксимации радиальной части АО (Rnl(r)), что можно сде¬
лать по-разному, например, представить ее в виде:
К,(г) =
j
(21 + 1 ~|l/2 J _гг
(2п^\—J ’r"J е слэйтеровская орбиталь;
rij-главное квантовое число; (^-орбитальная экспонента, задаю¬
щая расстояние максимума электронной плотности от центра ато¬
ма и характеризующая экранирующее влияние других электронов.
Другой вариант аппроксимации АО состоит в использо¬
вании так называемых гауссовых функций:
Rni(r) = I CjN/’e
j
192
- a,r2
Элементарная функция гауссового типа (ФГТ) отличает¬
ся от функции слэйтеровского типа (ФСТ) прежде всего ква¬
дратичной зависимостью от г в экспоненте. При этом, если
ФСТ хорошо передают хартри-фоковские АО вблизи ядра
и при г -> оо, то ФГТ в этих областях имеют неправильную
асимптотику и их надо в 2-5 раз больше для представления
одной и той же Rnt(r), чем в разложении по ФСТ. Но тем не
менее, эти недостатки гауссового базиса окупаются быстро¬
той вычисления соответствующих интегралов.
Не вдаваясь далее в тонкости расчетных методов, отме¬
тим только, что базисные наборы могут быть как мини¬
мальными, так и расширенными, в зависимости от того,
включены в них только АО, заполненные в основных со¬
стояниях атомов, или же к ним добавлены вакантные АО
с более высокими квантовыми числами. Кроме того, расши¬
рение базиса может быть связано с увеличением числа функ¬
ций, аппроксимирующих данную АО.
В последнее время, в связи с усовершенствованием вы¬
числительной техники и методики, расчеты с расширенными
базисами постепенно становятся все более доступными. Но
следует особо подчеркнуть необходимость сбалансирован¬
ности базиса. Это значит, что базисные наборы для каждого
атома должны иметь одинаковую полноту. Так, например,
для молекулы РО базисы (spd)P + (spd)Q и (sp)P + {sp)Q будут
сбалансированными (при этом первый-расширенный, а вто¬
рой-минимальный), тогда как базисы (spd)P + (sp)Q и
(sp)p + (ярсОо-несбалансированы. В последних двух случаях
наблюдается искусственное перераспределение электронной
плотности к атому, представленному более «полным» обра¬
зом.
Пороки метода МО
Так как метод Хартри - Фока - Рутана приближенный, то,
естественно, получаемые в его рамках значения физических
величин отличаются от экспериментальных. Вот некоторые
13-918
193
примеры. Энергия диссоциации молекулы Н2 по методу МО
в зависимости от способа расчета оказывается равной от
255,7 до 350 кДж/моль, что в любом случае заметно ниже
экспериментальной величины (458,5 кДж/моль). Для моле¬
кулы кислорода соответствующие значения равны
136 кДж/моль (теория) и 496 кДж/моль (эксп.).* А молекулы
F2 по Хартри-Фоку вообще существовать не должно. Кро¬
ме того, метод молекулярных орбиталей приводит к непра¬
вильным волновым функциям системы при бесконечном
разведении ядер, т. е. имеет неправильный диссоциативный
предел.
Эти недостатки метода связаны с тем, что в нем не
учитывается корреляция электронов.
Электронная корреляция
Электроны, вследствие действующего между ними куло-
новского отталкивания, будут «избегать» друг друга, так
что вероятность одновременного нахождения двух электро¬
нов в одной точке пространства равна нулю. Это значит,
что движение электронов определенным образом скоррели¬
ровано.
Иногда говорят, что каждый электрон окружен ку-
лоновской дыркой, т. е. в его окрестности не должен нахо¬
диться никакой другой электрон.
Кроме кулоновской (динамической) корреляции суще¬
ствует еще корреляция электронов с параллельными спина¬
ми, которая называется фермиевской (или статической)
и которая обусловлена принципом Паули.
Метод Хартри-Фока учитывает лишь последний тип
корреляции (дырку Ферми), движение же электронов с анти-
* Кстати, метод ВС дает для Н2 энергию диссоциации
388 кДж/моль (расчет Вейнбаума, 1933 г.) и 495 кДж/моль для 02-
194
параллельными спинами в обычной теории ССП * не скор¬
релировано, и такие электроны могут с заметной вероят¬
ностью одновременно находиться в одной и той же точке
пространства. Действительно, вероятность W^(r') нахожде¬
ния электрона со спином (3 в окрестности точки г при усло¬
вии одновременного пребывания электрона со спином а
N
в окрестности точки г равна ——— (г') (iV-число элек¬
тронов в системе) и не зависит от положения последнего
электрона (со спином а). Иными словами, движение обоих
электронов в модели Хартри-Фока статистически независи¬
мо. Так, для гомонуклеарных двухатомных молекул вероят¬
ность того, что один электрон находится около одного ядра,
а другой-около другого, равна вероятности одновременной
локализации электронов около одного из ядер. В итоге, это
приводит к неверному поведению Л/-электронной волновой
функции молекулы Ч'охф пРи увеличении межъядерного рас¬
стояния Яик диссоциации молекулы при R -> оо на атомы
не в основных, а в возбужденных или ионизированных со¬
стояниях. Поэтому при рассмотрении энергий диссоциации,
активационных барьеров и других величин, характеризую¬
щих реакционную способность молекул, ограниченный ме¬
тод Хартри-Фока оказывается неудовлетворительным.
Таким образом, результаты расчетов, выполненных по
методу ОХФ, отличаются от результатов, полученных из
точного решения нерелятивистского уравнения Шредингера
(разумеется, такие решения далеко не всегда известны). Эти
отличия связывают с корреляционными эффектами, подра¬
* Под «обычной» теорией самосогласованного поля (ССП) мы
подразумеваем так называемый ограниченный метод Хартри-Фока
(ОХФ), в рамках которого поведение каждых двух спаренных элек¬
тронов может быть описано одной и той же пространственной ор¬
биталью, так что соответствующие МСО имеют вид: ф,а и (р;|3.
В неограниченном методе Хартри-Фока (НХФ) это ограничение
снято и используются различные орбитали для разных
спинов.
13*
195
зумевая под этим кулоновскую корреляцию. Так, электрон¬
ная энергия, вычисленная в приближении ОХФ, отличается
от истинной Еэл (которая может быть найдена эксперимен¬
тально) на величину £корр:
^корр — £эл ~ ^ОХФ
Для систем с не очень большим числом электронов
в расчетах с расширенным многоэкспоненциальным базисом
АО ЕОХФ составляет 99-99,9% Еэл. Однако радоваться этому
обстоятельству приходится не всегда, ибо, несмотря на
большую относительную точность расчета Еохф, энергия
диссоциации молекулы (De) определяется в ограниченном
методе Хартри-Фока с большой абсолютной ошибкой
(вплоть до 200% от истинного значения), а иногда и с не¬
верным знаком (как, например, для молекулы F2). Это не¬
удивительно, поскольку энергия диссоциации (энергия свя¬
зи)-наименее удобная для квантовохимического расчета
величина. Ведь она получается в виде малой разности двух
больших величин - полной энергии молекулы и полной энер¬
гии исходных атомов (или фрагментов).
Кроме того, сравнивать рассчитанные величины энергий
связи с экспериментом иногда крайне затруднительно и по
другим причинам.
Иногда не совсем ясно, что следует принять за фраг¬
менты соединения,-атомы, ионы, нейтральные лиганды
и т.д. Например, в одном расчете энергия связи металл-ли¬
ганд для соединения Ni(CO)4 получилась равной
360 кДж/моль [по отношению к атому Ni(JS) и ней¬
тральным лигандам], тогда как в другом-она составляет
604 кДж/моль [но уже по отношению к Ni(3F)]; экспери¬
мент же дает 587 кДж/моль (для газовой фазы).
Учет энергии корреляции можно осуществить различны¬
ми методами, например, методом конфигурационного взаи¬
модействия (КВ). В этом методе полная волновая функция
записывается в виде линейной комбинации детерминантов
196
Слэтера, каждый из которых характеризует различные спо¬
собы размещения электронов по орбиталям. Однако вычис¬
ления £корр методом КВ достаточно сложны: для учета
40-60% этой энергии приходится рассматривать до тысячи
возбужденных конфигураций.
Для практических приложений метода МО, особенно
в молекулярной спектроскопии, важно сопоставление рас¬
считанных энергий возбуждения и потенциалов ионизации
молекул с опытными значениями. Обе указанные величины,
строго говоря, должны определяться как разность полных
энергий возбужденной молекулы или ее положительно заря¬
женного иона и ее основного состояния. Если в основном
состоянии молекула обладает заполненной оболочкой (S =
= 0), то при возбуждении или ионизации ее оболочка стано¬
вится открытой (S ф 0), а для таких систем уравнения ССП
MOJ1KAO значительно усложняются*.
Указанные трудности обусловили появление прибли¬
женных методов оценки энергий электронных переходов
и потенциалов ионизации молекул. Чаще всего используется
упрощающее предположение, согласно которому в процес¬
сах возбуждения и ионизации все молекулярные орбитали,
как заполненные, так и вакантные (виртуальные), найденные
из расчета основного состояния, не успевают реорганизо¬
ваться, т.е. остаются как бы «замороженными». Тогда воз¬
буждение молекулы можно рассматривать как одноэлек¬
тронный переход с заполненной МО ф; на виртуальную ф,-.
Энергия перехода имеет вид:
* Расчеты ССП возбужденных и ионизированных состояний
сложных соединений были выполнены только в последние 5 лет
и то, как правило, без учета КВ, хотя оно для этих состояний осо¬
бенно важно.
Теорема Купмэнса
2К -для синглетного воз-
4 бужденного состояния
0 для триплета
(72)
197
где Sj и - орбитальные энергии соответствующих МО, a Jtj и
Ку-двухэлектронные интегралы между МО фг и ср,, описывающие
попарное взаимодействие электронов:
=^mm»^dVidy2 (куло„овск„й)
Ки = JJШШШщу, (обменный,
Формулу (72) можно распространить на процесс иониза¬
ции молекулы, рассматривая его как переход с МО ср, в не¬
связанное состояние фю с энергией — 0, учитывая, что
при этом Ji00 = Kio0 = 0:
Ii = AEi^„=-ei (73)
Таким образом, орбитальный потенциал ионизации мо¬
лекулы с замкнутыми оболочками равен (в приближении за¬
мороженных МО) взятой с обратным знаком одноэлектрон¬
ной энергии соответствующей МО.
Это утверждение известно под названием теоремы Куп-
мэнса (1933 г.).
Следует иметь в виду, что под действием электронного
удара или кванта света электрон может уйти в принципе
с любого уровня 8( молекулы, так что получается набор раз¬
личных потенциалов ионизации.
В приближении «замороженных» МО орбитальные энер¬
гии имеют определенный физический смысл, и соотношение
(73) позволяет сопоставлять найденные из опыта потен¬
циалы ионизации или их последовательность с расчетными.
Иногда согласованность имеет место если не в численных
значениях, то по крайней мере в их относительном порядке.
Но чаще всего этого нет: во многих соединениях перестрой¬
ка (или, как еще говорят, релаксация) МО происходит и при
ионизации, и при электронных возбуждениях.
198
МЕТОД МО В ДЕЙСТВИИ
После изложения основных идей и уравнений метода
МО обратимся к конкретным примерам его использования
при исследовании электронного строения молекул. Начнем
с молекулы водорода в основном состоянии.
Молекула Н2 по методу МО
Воспользуемся минимальным базисом АО, т.е. будем
строить МО ф молекулы Н2 из ls-орбиталей водородных
атомов На и Нь:
Ф = СаХа + Сь1ь
где Ха = {1[\/п)е~г° И Хь = (1/|/тг)е_Гь.
Система уравнений Рутана примет тогда следующий
вид:
ca(Faa - е) + cb(Fab - 8SJ = О
(74)
cjfba ~ eSJ + ch(Fbb - е) = О
Далее, в силу симметрии задачи, можно записать:
Fab = ^ъа 5 Faa = Fbb; Sab = Sba = S (7 5)
Как уже отмечалось выше, уравнения Рутана имеют не¬
тривиальные решения только, если детерминант системы
(74) равен нулю, т.е.
det
откуда
(Faa - е) (Fah - eS)
(Fab-eS) (Faa- в)
= 0 (76)
(Faa - б)2 = (Fab - 8S)2
Решая это квадратное (относительно е) секулярное урав¬
нение, находим выражения для орбитальных энергий:
199
г
(7?а)
1 - S
(776)
Подставляя затем (77 а) и (776) в систему (74), получаем
следующие соотношения для коэффициентов:
са = сь для ег; са=-сь для е2
Впрочем, к этому результату можно было придти также из со¬
ображений симметрии.
Отсюда, учитывая условие нормировки МО, находим их
окончательный вид:
Фа = -7==^=:—(х„ — Хё) = Фл - МО, антисимметричная относи-
|/ ^ ( 1 ~ Ь)
Ввиду сравнительной простоты задачи, мы обошлись без ите¬
рационной процедуры, точнее, уже первая итерация ведет к согла¬
сованию.
Величина приблизительно равна орбитальной энер¬
гии изолированного атома водорода. Тогда, исходя из фор¬
мул (77 а) и (776), можно сказать, что при образовании моле¬
кулы Н2 атомные уровни энергии расщепляются на
молекулярные, лежащие на энергетической шкале выше
и ниже исходных атомных (рис. 35). При этом «подъем»
уровня е2 больше «падения» ех. Так как в молекуле водоро¬
да всего два электрона, то они будут в основном состоянии
занимать МО, энергия которой ниже, т.е. (ps. Эта орбиталь
называется связывающей, а орбиталь (рА-антисвязывающей
(разрыхляющей). Далее мы дадим более точные определе¬
ния связывающих и антисвязывающих МО.
Ф, =
]/2(1 + S)
(Ха + /О,) = Ф5- МО, симметричная относительно
перестановки ядер
гельно перестановки ядер
200
Рис. 35. Молекулярно-орбитальная диаграм¬
ма молекулы Н2 (схематично показаны
электронные облака).
После нахождения вели¬
чин S, Faa и Fab как функ¬
ций от межъядерного рас¬
стояния (R)* можно вычис¬
лить орбитальную (ej и
полную энергии основного
состояния молекулы водоро¬
да KL(H2)] Для различ¬
ных R.
Полная двухэлектронная
волновая функция основного
состояния молекулы Н2 вы¬
ражается в виде детерминан¬
та Слэтера, построенного из
МО, занятых электронами:
1
lvF, = —^det
f2
<pA~-LSa-%(o*is)
fs-lsa^lsb(6^s)
Ho
V~2
Ф1 (l)a(l) Ф1 (2) a(2)
<Pi(l)P(D Ф1(2)Р(2)
a(2)P(l)]
ф1(1)ф1(2)[а(1)Р(2)
Выразив через АО, получим:
lvFi = —7^ {XeU)Xe(2)[a(l)P(2) - a(2)P(l)] +
2/2(1 + S)
+ Хь(1)Хь(2)[а(1)Р(2) - a(2)p(l)] + [хЛПХьР) +
+ Xa&)Хь(1)][a(1)P(2) - a(2)P(l)]}
Верхний левый индекс при указывает спиновую мульти-
плетность.
* Соответствующие формулы и графики читатель сможет най¬
ти, например, в книге В. И. Минкина, Б. Я. Симкина и Р. М. Миня¬
ева «Теория строения молекул» (М.: Высшая школа, 1979,
с. 114-116).
14-918
201
Но последнее слагаемое в фигурных скобках есть не что
иное, как функция Гайтлера - Лондона для основного (син-
глетного) состояния молекулы Н2 [ср. с формулой (60)*].
Первые же два слагаемых отвечают ионным структурам
Н~ Нь+ и Н + Нь"\ При этом каждый ионный член входит в
полную волновую функцию наравне с «ковалентным» (гайт-
лер-лондоновским). Метод МО преувеличивает вклад ион¬
ных состояний, в силу чего энергия Е = E(R) стремится при
R-* оо к более высокому значению, чем 2£н и диссоциа¬
тивный предел получается неправильным.
МОЛЕКУЛЯРНЫЕ ОРБИТАЛИ ГОМОЯДЕРНЫХ
ДВУХАТОМНЫХ МОЛЕКУЛ И ИХ ИОНОВ
Квантовые числа молекулы
Свойственная изолированным атомам сферическая сим¬
метрия при образовании между ними химических связей
утрачивается. Поэтому N- и одноэлектронные состояния
жесткой молекулы классифицируют с учетом симметрии ее
ядерного полиэдра, которая может быть самой разнообраз¬
ной.
Ниже в качестве примера мы рассмотрим молекулы,
принадлежащие точечной группе симметрии К их чис¬
лу относятся все двухатомные гомоядерные и много¬
атомные гетероядерные линейные структуры с центром ин¬
версии: НС=СН, 0=С=0 и т.д.
Из классической механики известно, что при движении
частицы в симметричном относительно оси z поле, проекция
ее момента импульса на эту ось (Lz) сохраняется. Аналогич¬
но в квантовой механике для линейных молекулярных си¬
стем (как гомо-, так и гетероядерных), в которых поле обла¬
* Только в методе ВС буква ср обозначала АО, а здесь-МО,
тогда как для атомных функций введено обозначение X.
202
дает аксиальной симметрией, имеет место сохранение
i-компоненты полного момента*, что математически выра¬
жается следующим коммутационным соотношением:
[Я, LJ = О
где Lz-оператор проекции полного электронного орбитального
момента импульса на ось молекулы г.
В то же время операторы Lr и L„ не коммутируют с Н:
[Я, Ц # 0; [Я, Ly] Ф 0.
Физический смысл приведенных коммутационных соот¬
ношений понять нетрудно, если вспомнить, что оператор L.
связан с поворотом вокруг оси z. В силу аксиальной симме¬
трии линейной молекулы ее гамильтониан остается неиз¬
менным относительно такого поворота. Вместе с тем, вра¬
щение электронной оболочки** вокруг осей хну приводит
к разрушению молекулы, так как электронная плотность
при этом «уходит» от ядер. Так как
L2 = L2X+ L) + L\
то: [Я, L2\ Ф 0
Последнее означает, что мы не можем характеризовать
злектронную оболочку молекулы ни квантовым числом ква¬
драта ее полного орбитального момента, ни совокупностью
квантовых чисел /, характеризующих одноэлектронные ор¬
битальные моменты.
Таким образом, iV-электронная молекулярная волновая
функция *Р(1, 2,..., N) будет собственной функцией операто¬
ров й и L.:
ЯЧ> = £Т; L2¥ = А/¥
где М -собственное значение оператора L,.
* Это утверждение справедливо в пренебрежении спин-орби-
гальным взаимодействием.
** Ядра в нашем рассмотрении полагаются фиксированными
(адиабатическое приближение).
14*
203
Кроме того, в одноэлектронном приближении Lz можно
представить в виде суммы соответствующих одноэлек¬
тронных операторов lz(i):
Учитывая аксиальную симметрию рассматриваемых
здесь систем, удобно перейти к цилиндрическим координа¬
там, т.е. задавать положение точки в пространстве пере¬
менными z, р (расстоянием по нормали от оси z) и углом
поворота ф° в плоскости ху вокруг оси z (рис. 36). В этой си¬
стеме координат
Полную волновую функцию линейной молекулы пред¬
ставим, как и ранее, в виде детерминанта Слэтера, соста¬
вленного из МО, являющихся собственными функциями
оператора / (для простоты воспользуемся однодетерми-
нантным приближением). В цилиндрической системе коорди¬
нат эти МО примут вид:
где т- квантовое число, определяющее величину z-компоненты
одноэлектронного момента импульса:
N
4( 1,2 JV)= £(г0)
(78)
i = 1
/2срт = тАср,
При этом т принимает цело¬
численные значения, которые час¬
то обозначают буквами:
т = О, +1, —1, + 2, —2,...
Рис. 36. Цилиндрическая система координат
(р, ф°, г):
х = pcoscp0; у = psincp0; z = z.
204
или, иным способом:
|т| = 0, 1, 2,...
on 5
МО и симметрия молекулы
Посмотрим теперь, как будут преобразовываться МО tpm
при операциях симметрии, характерных для любых ли¬
нейных молекул. Начнем с операции поворота молекулы во¬
круг ее оси z на произвольный угол ф£(Сф=(г)):
о
<V" Ф" = C.p-'-ytp, г)в'“ф' =
О о
г, Ч Йи(ф° + ф°) ,
= /(р, z)e у
Мы видим, что квантовое число т при таком преобразо¬
вании не изменяется. Теперь рассмотрим два важных
частных случая.
1) Пусть т = 0 (ст-МО). Тогда
£<?:‘х,Фт=о~ с.р^Лр,-) ™ Ар.-)
г. ,. ст-МО инвариантны относительно вращений вокруг оси
. г;\> является их характерной особенностью.
2) Если тФ 0 и поворот совершается на угол кп (к = О,
± 1, ±2, ...), то, используя формулу Эйлера (eni = — 1):
&Ъп + о = /(Р. ^ + кп) - /(p. z)e^°eimkn = (- 1ГЧ
т. е. в зависимости от величины и знака т (т Ф 0) и угла по¬
ворота (кратного тс) орбиталь меняет или не меняет знак.
Более детально этот вопрос читатель может разобрать самостоя¬
тельно.
205
Двукратное вырождение
Группы симметрии линейных молекул включают в себя
бесконечное число плоскостей gv, проходящих через ось г.
Отражение в любой из этих плоскостей преобразует МО
следующим образом:
6,Фт = ovf(p,z)eMf = Др, г) е ~ ‘тф = ф_т (79а)
так как при данной операции симметрии происходит замена
переменной ф°: ф° -*■ — ф°.
Если т = О, нетрудно убедиться, что
6рФ—0 = Фт = 0 (796)
Таким образом, сг-МО не изменяются и при отражениях
в плоскостях ст„. Если же т ф 0, то преобразования bv сме¬
шивают МО с одинаковыми по абсолютной величине, но
противоположными по знаку квантовыми числами т, что
позволяет отнести функции фт и ф_т к одному и тому же
НП. Иными словами, все орбитали линейной молекулы,
кроме орбиталей a-типа, двукратно вырождены.
Симметрия термов
До сих пор мы рассматривали симметрию одноэлек¬
тронных состояний. Теперь коснемся симметрии элек¬
тронных термов. Можно доказать (мы здесь этого делать не
будем), что детерминант Слэтера, построенный из орбита-
лей фш (которые, напоминаем, являются собственными
функциями оператора /г) будет собственной функцией опера¬
тора Lz. Естественно, при этом возникает следующий во¬
прос: как связаны собственные значения Lz (мы обозначили
их выше буквой М) с собственными значениями /2? Иными
словами, как связаны числа М и mk?
Прежде чем написать нужную формулу, заметим, что ка¬
ждая МО фт может входить в каждый столбец детерминан-
206
!а Слэтера либо один раз, например, в случае конфигура¬
ции:
% %i
(л-МО) (я'—МО)
ф1(1)а(1)ф1(2)а(2)
ф_1(1)а(1)ф_1(2)а(2)
1/2
-7— [ф1(1)ф-1(2) - ф1(2)ф_1(1)]а(1)а(2)
1/2
(80)
либо два раза (с противоположными спиновыми сомножите¬
лями), скажем, при конфигурации
<Pi(l)a(l)<Pi(2)a(2)
ф1(1)(3(1)ф1(2)Р(2)
AL 4'(l,2) = 4=-det
г-MO)
= —^[а(1)Р(2) - а(2)Р(1)]ф1(1)ф2(2)
]/*
Поэтому в общем случае можно записать:
м = Z тк' + 2Итк- (81)
г.хе тк-квантовые числа данного набора МО; индекс к' нумерует
орбитали, встречающиеся в слэтеровском детерминанте один раз,
а к"-орбитали, входящие в него дважды.
Отсюда нетрудно видеть, что для первого из указанных
выше случаев М — 1 + ( — 1) = 0, а для второго: М = 2 х
х 1 = 2.
Обозначения
В дальнейшем, однако, мы будем использовать для ха¬
рактеристики электронных термов не само число М, а его
абсолютное значение А = | М |. Это связано со следующим
обстоятельством. Если ¥ (1, ..., N)~собственная функция
оператора Lz, то и ст^ также будет его собственной функ-
207
цией, но с противоположным по знаку собственным значе¬
нием* :
1г№)=-М№)=^-м
Следовательно, функции Ч/м и 'Р.д* отвечают одному
и тому же энергетическому терму Ек, т. е. (при Л ф 0) со¬
ответствующие состояния, отличающиеся направлением
проекции орбитального момента на ось молекулы, дважды
вырождены.
Термы с различными значениями Л обозначаются за¬
главными греческими буквами:
А = 0, 1, 2, ...
ъ п д
Особенность S-термов
S-термы, как следует из сказанного, невырождены. Но
у них есть другая особенность, связанная не с изменением
знака М (ибо в этом случае М = 0), а с изменением (или
с неизменностью) знака самой волновой функцией ¥ (1, 2,
..., N). Рассмотрим два примера.
Пример 1. Допустим, что детерминант Слэтера составлен толь¬
ко из ст-МО, которые, повторяем, не меняются при операциях
(см. (79)). Тогда и сам детерминант (т. е. волновая функция 4* (1, 2,
..., N)) не изменится при действии оператора av (1, 2, ..., N).
Пример 2. Если же в слэтеровский детерминант входят, ска¬
жем, функции ф + 1 и ф _ j [см. (80)], то тогда преобразование аи из¬
менит знак волновой функции
* Это равенство вытекает из формул (78), (79а), (81) и из соотно¬
шения ст„(1, 2, ..., N) = а„(1) х ... х a„(jV), показывающего, что опе¬
рация отражения координат всех электронов в плоскости, проходя¬
щей через ось молекулы, тождественна произведению таких
операций отражения для каждого из них.
208
Отсюда вывод: необходимо различать S-термы, волновая
функция которых не меняется вовсе при отражении в плоскости
aw, и 2-термы, волновая функция которых при этой операции
симметрии меняет знак. Первые обозначают символом 2+, а вто¬
рые —
Все, что говорилось до сих пор, справедливо для любых
линейных молекул. Если же система имеет симметрию Z)oofc,
то появляются дополнительные особенности. Они связаны
с наличием в таких молекулах центра инверсии и плоскости
симметрии стЛ. Посмотрим, как будут преобразовываться
МО фт при отвечающих этим элементам симметрии опера¬
циях I и сгЛ:
Как видим, обе операции значения квантового числа
т не изменяют. Сама же МО может менять или не менять
знак в зависимости от вида сомножителя /(р, z) и четности
т. Принято различать МО четные (д) и нечетные (и) относи¬
тельно операции инверсии I, При этом свойство четности
МО однозначно определяет и их симметрию относительно
операции аЛ, так как
Четность МО
= (- 1Г/(Р. - г)*”''"
<4>т = (- 1Г/(Р, - г)е“”9’ = (- 1Га»Ч>,
209
Четность многоэлектронных состояний
Построенный из орбиталей срт д и фж и детерминант Слэ-
тера также будет обладать определенной четностью, кото¬
рая зависит только от числа МО фт и в нем. Если таких МО
четное число в детерминанте, то и сам он будет обладать
признаком четности (Ч^); если число функций фш „ нечетно,
то iV-электронное состояние молекулы будет нечетным (*?„).
A-У двоение
Волновая функция ¥ (1, 2, ..., N) и соответствующий ей
электронный терм линейной молекулы характеризуются
значением квантового числа А z-компоненты полного орби¬
тального момента импульса. Если в такой молекуле есть
еще центр инверсии (группа симметрии Dxh), то функция
(1, 2, ..., N) и электронный терм характеризуются также
определенной четностью. При А Ф 0 терм двукратно выро¬
жден, однако это вырождение приближенное и связано с не-
учетом влияния вращения молекулы на ее электронные со¬
стояния. Как только это влияние учитывается, термы с А ^
ф 0 расщепляются на два близких уровня. Это явление
называют А-удвоением. *
Полный спин молекулы
Кроме названных характеристик каждому терму (и ка¬
ждому состоянию *Р) отвечает полный спин S всех электро¬
нов молекулы. Если S ф 0, то имеет место вырождение
кратности (2S + 1) по направлениям полного спина. Число
(2S + 1) называется мулътиплетностъю (или спиновой мулъ-
типлетностъю) терма и пишется в виде верхнего левого
индекса у буквенного символа терма, например: 3Е*, ХП.
* Эффект А-удвоения существенен только для П-термов. Заме¬
тим также,,что симметрия линейных молекул по отношению к пло¬
скости <jv приводит к различиям между термами Z+ и £ ~.
210
В подавляющем большинстве случаев основной терм
двухатомной молекулы-это *Е + , а для гомоядерных моле¬
кул 11>д . Есть и исключения, например 3Е~ для 02, 2П для
NO.
Четверка простейших и...
Теперь, ознакомившись с основными характеристиками
электронной структуры двухатомных молекул, мы можем
перейти к качественному рассмотрению их МО. Начнем
с простейших гомоядерных систем: Н2, Н2, Не2 и Не2.*
Характеристика
молекулы
+
н2
н2
+
Не2
(Не2)
Молекулярно-орбиталь¬
ная конфигурация
(од1 S)1
(ct31s)2
(Vs)2
(cr/ls)1
(a9ls)2
(au*ls)2
Основной терм
2Т +
+
‘-‘в
%+
—
Ое, кДж/моль
273
458,5
231
—
Re, нм
0,106
0,074
0,108
—
К
1/2
1
1/2
0
Как видно из таблицы, их молекулярные орбитали образо¬
ваны только 1 s-атомными орбиталями. Заселение этих МО
электронами происходит в порядке возрастания орби¬
тальных энергий (хотя, вообще говоря, это не обязательно,
ибо, как и в случае атомных систем, электронная конфигура¬
ция определяется минимумом полной энергии системы, ко¬
торая не равна сумме орбитальных энергий).
* В обозначении МО будем указывать ее тип (а, я, ....), чет¬
ность {д или и) и АО, из которых она построена. Звездочка у симво¬
ла МО означает, что последняя имеет антисвязывающий характер
(см., например, рис. 35).
211
В 1928 г. Г. Герцбергом было предложено молекулярно¬
орбитальное определение понятия кратности химической
связи для двухатомных гомоядерных молекул:
2
св антисв
где п и и -заселенности связывающих и антисвязывающих
МО.
Впоследствии это определение было распространено на
более сложные системы.*
Особого внимания заслуживает молекула Li2 и ион Li2.
Электронная конфигурация Li2: (<rels)2 (o^ls)2 (og2s)2 и К =
= Ц* = 1. Казалось бы, эха молекула по оаоей элек-
тронной структуре подобна молекуле водорода. Но почему
же тогда так резко отличаются величины De и Re для Li2 и
Н2:
£>е(Н2) = 458,5 кДж/моль De(Li2) = 109,2 кДж/моль
Re (Н2) = 0,074 нм Re (Li2) = 0,267 нм
Но главное-из качественной схемы МО не ясно, почему
при переходе от Li2 к Li2, т.е. при удалении электрона со
связывающей МО ct92s, система стабилизируется-
De(Li2) «150 кДж/моль.**
Следует заметить, что в межъядерной области молекулы
Н2 разностная электронная плотность Др (определяемая как
разность электронных плотностей молекулы и составляю¬
щих ее свободных атомов) всюду положительна, тогда как
* См. Дмитриев И. С. и Семенов С. Г. Квантовая химия, ее
прошлое и настоящее. М.: Атомиздат, 1980, с. 143-147.
** Кстати, и для остальных молекул Х2 (где Х-атом щелочно¬
го металла) переход Х2 -> Х2 сопровождается увеличением De на
20-30 кДж/моль.
212
в молекуле Li2 Ар отрицательна у ядер и достигает положи¬
тельного максимума в центре связи. Таким образом, элек¬
тронный заряд молекулы Н2 сильно «сжат» между ядрами;
а у Li2, хотя и есть перекрывание 2s-AO, подобное сжатие
заряда отсутствует в силу наличия атомных Ь2-оболочек
и в межъядерной области появляются узловые плоскости.
Замечание о характере МО
Приведенное выше определение связывающей и антисвя¬
зывающей МО следует уточнить. Эта характеристика мо¬
лекулярной орбитали не имеет прямого отношения к вели¬
чинам орбитальных энергий. Поэтому лучше было бы
считать МО связывающей, если удаление находящегося на
ней электрона приводит к уменьшению энергии связи, а доба¬
вление-к ее увеличению (для антисвязывающих МО-наобо¬
рот). Но и это определение не совсем удачно, так как в рас¬
смотренном выше случае молекулы Li2 удаление одного
электрона с верхнего дважды занятого уровня приводит
к упрочнению химической связи и потому <т 2s-MO должна,
в соответствии с приведенным определением, рассматри¬
ваться как антисвязывающая. Но тогда становится непо¬
нятным сам факт существования молекулы Li2 (и иона Li2),
ибо оказывается, что в нем исв < пантисв (К < 0).
Поэтому более строгое определение будет такое; связы¬
вающая-это МО, удаление с которой всех электронов
приводит к ослаблению (и даже исчезновению) химической
связи.
Разумеется, и это определение не универсально, так как
оно ограничено только случаем двухатомных молекул.
Заметим, что удаление любого электрона из атома или
молекулы требует затрать? энергии и всегда увеличивает
полную (отрицательную) энергию системы. Энергия же хи¬
мической связи, представляющая собой малую разность двух
больших величин (полной энергии молекулы и суммы пол¬
213
ных энергий составляющих ее изолированных атомов), мо¬
жет изменяться при этом различным образом.
В случае многоатомных молекул уже нельзя говорить
о связывающих и антисвязывающих МО вообще, но лишь
о связывающем и разрыхляющем характере данной МО
в области той или иной связи.
Завершая наш рассказ о МО двухатомных молекул, мы
советуем читателю внимательно изучить таблицу, в которой
даны их основные характеристики. Особое внимание обраг
тите на следующие обстоятельства:
а) инверсию ад2pz~ (z-ось молекулы) и двух выро¬
жденных я„2р-МО при переходе от N2 к 02, которую обыч¬
но связывают с различием орбитальных энергий 2s- и 2р-АО
атомов N и О (для N Де = e2s - е2р ж 579 кДж/моль, тогда
как для О As % 1447,5 кДж/моль);
б) наличие двух неспаренных электронов в молекулах В2
и 02, что обусловливает их парамагнетизм;
в) особую прочность молекулы N2;
г) взаимосвязь в изменении величин К, De и R*.
Заметим, что во всех приведенных в таблице молекулах
МО типа п„ являются сильносвязывающими, а уровни типа
7г9-сильноантисвязывающими. Характер же cr92pz-MO не¬
сколько изменяется: в N2 этот уровень оказывается почти
не связывающим, тогда как в 02 и F2 он имеет связываю¬
щий характер. Уровень au2s в Т^-слабоантисвязывающий,
в 02-несвязывающий. Упрочение уровня a-связи (ст92рг)
свидетельствует о постепенной потере 2s-AO валентного
характера в атомах элементов конца II периода и об уве¬
личении роли 2р-АО в образовании химических связей.
* Подробное обсуждение этих вопросов читатель сможет най¬
ти почти в любой книге по теории строения молекул, см., напри¬
мер: В. И. Минкин, Б.Я.Симкин, Г. М. Миняев. Теория строения
молекул. М.: Высшая школа, 1979, с. 122-129.
214
Вообще следует заметить, что приведенными в таблице
молекулярно-орбитальными диаграммами следует пользо¬
ваться осторожно, так как многие выводы, сделанные без
учета конкретных величин орбитальных энергий, карт рас¬
пределения электронной плотности и других количест¬
венных характеристик молекул, могут быть ошибочными.
Изложенный выше качественный подход можно распро¬
странить и на гетероядерные молекулы. При этом необходи¬
мо иметь в виду, что молекулярные орбитали будут состав¬
лены из АО различных атомов и вклад этих АО будет,
вообще говоря, различен. В результате МО оказываются по¬
ляризованными, а соответствующие молекулярно-орби¬
тальные диаграммы уже не будут иметь того симметрично¬
го вида, который имел место для гомоядерных систем.
В качестве примера на рис. 37 приведена такая диаграмма
для молекулы воды (указаны только занятые в основном со¬
стоянии валентные МО).
Молекула Н20 относится к точечной группе симметрии
C2v, которая имеет четыре неприводимых представления
(НП): А1, А2, В1 и В2. Ниже дана классификация валентных
АО атомов кислорода и водорода по этим НП (направление
координатных осей указано на рис. 37):
НП группы С2,. АО, преобразующиеся по данному НП
А\ IsO, 2sO, 2pzO\ 1 sHa + lsHb
Согласно данным квантовохимического расчета, МО мо¬
лекулы Н20 имеют вид*:
* Мы использовали данные неэмпирического расчета Шала
и Эллисона (1955 г.). Орбиталь (pl0i представляет собой почти чи¬
стую ls-AO кислорода.
Если атомы разные...
А 2
Bi
В2
2 ру0
2рх0 ', ЬЯД - UHb
216
Фа», = - 0,029IsO + 0,8452s0 + 0,1332pzO + 0,178 (lsHa + 1 sHb)
cplb2 = 0,5432pxO + 0,776 {lsHa - 1 sHb)
Фзв1 = — 0,026IsO - 0,4602sO + 0,8282pzO + 0,334 (lsHa + 1 sHb)
Ф1Ь, = 2 Py°
МО молекулы H20
Рис. 37. Молекулярно-орбитальная диаграмма молекулы воды.
На графике справа показано направление координатных осей.
15-918 217
Таким образом, орбиталь <р1Ьз имеет связывающий ха¬
рактер для О—Н- и антисвязывающий для Н—Н-взаимо-
действий, в отличие от ср3а1, которая является связывающей
для взаимодействий обоих типов. Обе МО (<р3в1 и фц,2) от¬
вечают ст-связи О—Н. Уровень 1 Ъх соответствует неподелен-
ной паре 2р„-электронов атома кислорода, его можно рас¬
сматривать как слабосвязывающий. Орбиталь ф2а1 сущест¬
венного участия в химической связи не принимает, она
образована в основном 2s-AO кислорода.
Молекулярная орбиталь ф; определяется обычно как соб¬
ственная функция некоторого одноэлектронного гамильто¬
ниана, в качестве которого в принципе должен использо¬
ваться оператор Хартри-Фока (фокиан), так как именно он
оптимальным образом учитывает согласованное взаимодей¬
ствие электронов в молекуле. Практически же этот оператор
часто аппроксимируется полуэмпирическим модельным
одноэлектронным оператором.
Но каким бы оператором ни пользовались при расчете
молекулярных систем, всегда предполагается, что симмет-
КОНЦЕПЦИЯ ГИБРИДИЗАЦИИ
Канонические и локализованные МО
V
рия гамильтониана отвечает
симметрии молекулы, а его
собственные функции преоб¬
разуются по неприводимым
представлениям точечной
группы симметрии молеку¬
лы. Такие МО называются
каноническими.
Рассмотрим в качестве
примера канонические МО
Рис. 38. Направление химических связей
в молекуле метана.
218
молекулы метана СН4 (симметрия Td) в приближении МО
ЛКАО (рис. 38):
НП е
группы Td
Канонические МО метана
1
А\ ед Ф0 = я2 sC + —bilsHi + lsH2 + 1 sH3 + ЬЯ4)
Ф! = с2рхС + —d(isHt + 1 sH2 - 1 sH3 - IsffJ
T2 et2 ф2 = c2pyC + —d(lsHl - UH2 + 1 sH3 - 1 sHJ
Ф3 = c2pzC + —d{lsHt - 1 sH2 - 1 sH3 + lsH4)
Одним из наиболее характерных свойств канонических
МО является их существенно делокализованный характер.
Так, приведенные выше МО ф0-фз метана охватывают все
пять ядер этой молекулы.
Поэтому канонические МО не отражают эксперименталь¬
но наблюдаемые аддитивность и трансферабельность.*
Описание структуры молекул (особенно насыщенных)
в терминах канонических МО не отвечает представлениям
классической структурной химии. Однако канонические МО
можно преобразовать так, чтобы получить новые МО, лока¬
лизованные на отдельных атомах и связях. Для молекулы
метана локализованные МО (ЛМО) ф,- будут иметь вид:
* Трансферабельность-переносимость из одной молекулы
в другую, структурно родственную, ряда молекулярных свойств.
К примеру, длины связей С—С и С—Н во всех насыщенных угле¬
водородах с точностью до 0,5% постоянны, а энергии атомизации
алканов с точностью до 2% равны сумме средних энергий разрыва
всех связей С—С и С—Н.
15*
219
1
Ф! = — с (2sC + 2рхС + 2руС + 2pzC) + dlsHj + (а - c)2sC +
+ 1 (b-d)G(H)
1
ф2 = — с (2sC + 2рхС - 2руС - 2р2С) + <ШЯ2 + (а - c)2sC +
+ j(b-d)o(H)
1
Фз = — c(2sC - 2рхС + 2р„С - 2pzC) + dlsH3 + (а - c)2sC +
+ у(Ь - Л)ст(Я)
1
Ф4 = — с (2sC - 2рхС - 2рС + 2рС) + dlsH4 + (а - с) 2sC +
+ у (Ь - d) ст (Я)
где ст(Я) = lsffj + ЬЯ2 + ЬЯ3 + ЬЯ4.
Если положить а = с и b — d, что эквивалентно допуще¬
нию абсолютной локализации МО на связях С—Н„ то вид
JIMO упрощается:
ф! = у (2sC + 2 рхС + 2 руС + 2ргС) + blsHx
ф2 = ^(2sC + 2рхС - 2руС - 2pzC) + b\sH2
Фз = ~(2sC - 2РхС + 2руС - 2р2С) + blsH3
ф4 = ^(2sC — 2 рхС — 2 руС + 2 pzC) + blsHi
Линейные комбинации валентных 2s- и 2р-АО атома
В дальнейшем для краткости мы будем обозначать ги¬
бридные орбитали символом hb а чистые AO-s, рх, ру и pz.
Эти гибридные АО, как и JIMO фх _4, эквивалентны, т.е.
при операциях группы симметрии Td либо не изменяются,
либо переходят друг в друга. Кроме того, если каждой кано¬
нической МО возможно сопоставить некую орбитальную
энергию е;, то всем эквивалентным Л МО отвечает одно об¬
щее значение 8 одноэлектронной энергии, в данном случае:
Концепция гибридизации получила широкое распростра¬
нение главным образом при обсуждении стереохимических
проблем. Однако не следует думать, что именно характер
гибридизации электронных облаков определяет геометрию
молекулы. В действительности дело обстоит как раз наобо¬
рот -исходным моментом при определении типа гибридиза¬
ции является известная пространственная симметрия моле¬
кулы. Когда же от данной молекулы (например, СН4)
переходят к другим, гомологичным соединениям (скажем,
насыщенным углеводородам) и утверждают, что вследствие
лр3-гибридизации электронных облаков атомов углерода его
углерода с коэффициентом представляют собой ги¬
бридные АО htC:
hxC = —(2 sC + 2 рхС + 2 руС + 2 pzC)
h2C = у (2sC + 2рхС - 2руС - 2pzC)
h3C = y(2sC - 2pxC + 2pyC - 2pzC)
h4C = —(2 sC — 2 pxC — 2 pyC + 2pzC)
h2C =
h3C =
6 = = e2 = E3 = e4 = (8ei +-3e(2)/4
221
соседи должны находиться в тетраэдрических или близких
к ним углах, то создается иллюзия, будто причиной такой
геометрической структуры углеводородов является ^-ги¬
бридизация. На самом же деле в основе подобных рассужде¬
ний лежит предположение (очень часто оправдывающееся
экспериментально) о сходстве геометрической структуры
рассматриваемых молекул.
Более того, квантовомеханические расчеты электронной
структуры молекулы метана показали, что тетраэдрическая
конфигурация этой молекулы отвечает наибольшей, по срав¬
нению со всеми другими возможными для нее конфигура¬
циями, электронной энергии. И только благодаря тому, что
этой конфигурации соответствует минимум энергии оттал¬
кивания ядер, в результате чего полная энергия молекулы
(равная сумме ее электронной и ядерной энергий) оказывает¬
ся все же минимальной, связи С—Н в метане направлены
в углы тетраэдра. Таким образом, геометрия молекулы не
обусловлена данным типом гибридизации. Последняя лишь
устанавливает соответствие между взаимным расположе¬
нием ядер и пространственным распределением электронной
плотности. Но это не единственная, и даже не главная в со¬
временной теории строения молекул, функция концепции
гибридизации.
Дело в том, что свойства молекулярных систем можно
разбить на два класса: одноэлектронные и коллективные.
Одноэлектронными называют те свойства, которые в первом
приближении связаны с поведением отдельных электронов
(например, потенциалы ионизации, электронные спектры).
Коллективные же свойства уже в первом приближении свя¬
заны с поведением всех электронов молекулы. Примерами
коллективных свойств могут служить полная энергия моле¬
кулы, суммарная энергия ее связей, дипольный момент, рав¬
новесные межъядерные расстояния.
Иными словами, коллективные свойства (а как раз с ни¬
ми чаще всего имеют дело химики-экспериментаторы) опре¬
деляются суммарным, общим распределением электронной
222
плотности, а не одним каким-либо электронным облаком
(орбиталью) или группой; облаков. В этом случае не столь
существенно, какую форму будут иметь эти отдельные элек¬
тронные облака, главное-правильно определить распреде¬
ление суммарной электронной плотности в пространстве
молекулы. «Разложить» же эту суммарную плотность по
атомам и связям можно очень многими способами, подобно
тому как при решении физических задач можно многими
способами разложить какой-либо вектор на компоненты.
Конечно, тут открывается большой простор для фанта¬
зии теоретика (деформируй отдельные электронные облака
атомов молекулы так, или почти так, как хочешь, благо ма¬
тематика это позволяет!). Можно сосредоточить (локализо¬
вать) электронную плотность частично на атомах (в виде
электронных пар внутренних оболочек атомов или непо-
деленных электронных пар валентной оболочки), а частично
на химических связях (локализация электронов в поле двух
ядер отвечает двухцентровому взаимодействию атом-атом,
которое описывается классической символикой валентного
штриха), а можно пользоваться и делокализованными орби¬
талями, охватывающими в принципе все атомные ядра мо¬
лекулы. Разумный теоретик стремится воспользоваться этой
свободой для того, чтобы построить модель, приемлемую
для химика и пригодную для описания данного класса
свойств.
Как показывает квантовомеханический анализ, локализо¬
ванные на определенных связях и определенным образом
направленные в пространстве гибридные орбитали оказы¬
ваются оптимальными для рассмотрения коллективных
свойств молекулы. Кроме того, в терминах локализованных
гибридных орбиталей могут быть наилучшим образом
объяснены такие особенности молекулярных систем, как
трансферабельность и аддитивность ряда свойств связей.
Но как только переходят к описанию одноэлектронных
свойств молекул, требуется введение более общих и физиче¬
223
ски более адекватных представлений о делокализации элек¬
тронов, т. е. учета того обстоятельства, что в действительно¬
сти движение электрона не ограничивается областью двух
отдельных ядер многоатомной молекулы.
В ряде случаев гибридные атомные орбитали (ГАО) могут
быть определены из соображений симметрии и дополни¬
тельных условий, например условия их орто-нормированно-
сти
и предположения, согласно которому каждая ГАО ориенти¬
рована по определенной химической связи или, как еще го¬
ворят, по линии связи атомов, т. е. по линии, проходящей че¬
рез атомные ядра.
В качестве примера обратимся к случаю гибридных АО,
построенных из четырех орбиталей: одной 2s и трех 2р. Эти
ГАО реализуют четыре ст-связи центрального атома А
с четырьмя, вообще говоря, различными лигандами: L1} L2,
L3, и L4, так что связи А - L. (i = 1, 2, 3, 4) могут быть не¬
эквивалентными. Тогда i-ю ГАО можно представить в виде
причем вклады образующих ее 2s- и 2р-орбиталей опреде¬
ляются следующими выражениями:
Как построить гибридную АО?
(83)
^ = N (а^ + btpx + ctPy + dipz)
(84)
s-вклад:
**i
af + bf + cf + df
(85a)
bf + cf + df
(85c)
p- вклад:
af + bf + cf + df
224
Обозначая отношение p-вклада к s-вкладу через Xf,
, bf cf df
xf = 4- + 4- + 4- <86)
af af at
можно записать формулы (85a) и (856) иначе:
1
s-вклад в ГАО /i,:
p-вклад в ГАО ht:
1 + Xf
1 4- Xf
Кроме того, суммы s- и p-вкладов по всем четырем Г АО
равны, соответственно, 1 и 3:
1-г
i=i 1
+ Xf
f X?
i-i 1 +
= 1 (87а)
= 3 (876)
В состав гибридных орбиталей могут входить не все три
2р-АО, а только две или одна из них, тогда как остальные
остаются чистыми (негибридизованными) р-орбиталями
и могут принимать участие в образовании я-связей. При
этом из к исходных АО образуются к орто-нормированных
гибридных АО. Разумеется, если в гибридизации участвуют
не три, а, скажем, две р-АО (что приводит к образованию
трех ГАО), то условие (876) надо записать так:
= 2
у —
А 1 + X?
На параметр гибридизации Xt накладывается условие:
Я^ 0. Значению = 0 отвечает случай ht = s, a Xt = оо,
-hi = р.
225
Соотношение (86) позволяет формально рассматривать
величину Xi как вектор с компонентами bjah cjcii и dja^
Нормировочный коэффициент в формуле (84) нетрудно
найти из условия орто-нормированности ГАО и образую¬
щих их АО:
Можно показать, что fapjdV = cos ау, где а^-угол ме¬
жду направлениями i-й и j-й ГАО. Тогда условие ортого¬
нальности гибридных орбиталей приобретает простой вид
Из формулы (88) следует, что, во-первых, ^ 1, а во-
вторых, (Х.( ^ 0 при любом i), cos a tj <0 и а tj ^ 90°. Послед¬
нее означает, что угол между двумя spk-TАО не может быть
меньше 90°.
Косинусы углов oty и параметры гибридизации связаны
так называемой формулой Ван-Флека (1933 г.)
cosa12cosa34 = cosa13cosa24 = cosa14cosa23 = (A.1X,2^3?i4)''1
Последние три слагаемые можно записать как скалярное
произведение векторов А,,- и р= (рх, рт р2), тогда:
1 + XiXj cos а у = 0 (i ф j)
откуда
cos ai;. = — 1
(88)
откуда
i/mr
(s + Xtp)
(89)
226
Если теперь повернуть систему координат так, чтобы ось
z совпала с направлением вектора Хг, которое можно задать
единичным вектором щ, то формула (89) примет вид:
^ = 1 (s + lipnd = ~т=== (s + XiPi)
i/mr /r+xf
Условие ортогональности ГАО (83) тогда следует запи¬
сать так:
1 + kiXjlpipjdV= О
Дигональные гибриды
Посмотрим теперь, как изложенная теория «работает»
в конкретных случаях, например при описании линейных
молекул или линейных фрагментов симметрии С^:
где А-центральный атом, образующий две неэквивалентные ст-свя¬
зи с двумя различными лигандами L и L'.
В этом случае можно записать следующие выражения
для орбиталей атома А:
Й! = (s + x1p1)//r+~xf", где рх = р2
h2 = (s + Х2Р2)/]/тГ, гДе Pi= ~Pi = ~Px
hi = Px (^з = 00) не участвуют в обра-
= Ру (Х4 = оо) зовании а-связей
Из условия (88) получаем
cosa12 = cos 180° = — 1 = - 1ДД2
откуда: Х2 = 1/^ (90)
Тогда:
*!=(« + KPzW1 + Ч
h2 = (s + ~Р2)/l/T+l/xf - (1,5 - p,)/|/l + Xf
л,х
В случае эквивалентных a-связей реализуемых ГАО и
h2 атома А во фрагменте L—А—L симметрии имеет
место равенство Хг = Х2 = X, так что А, = 1-см. (90). Полу¬
чающиеся две sp-ГАО, называемые дигоналъными, имеют
вид:
hl = dil = ~U(S + Pz)
1/2
h2 = di2 = — (s - pz)
1/2
Тригональные ГАО
В тех плоских молекулах и молекулярных фрагментах,
где центральный атом образует три эквивалентные а-связи,
углы между которыми равны 120° (AL3, симметрия D3h),
ГАО могут быть представлены так:
h1 = (s + Xpy)f\f\ + к2 здесь учтено, что в силу эк¬
вивалентности связей:
А.1 = Х2 = А<з — X
h2 — (s + Хр2)/|/1 + А2
h3 — (s + Хр3)/\/ 1 + X2
hA = Pz (К = 00)
ось z перпендикулярна пло¬
скости молекулы
Для определения значения X можно воспользоваться ли¬
бо условием (88), либо (87а или б). Во всех случаях получаем
X — у 2, например:
cos 120° = -1/2 = -1/Х2
X = |/2
228
Тогда:
= + (1=1, 2,3)
^4 = Pz
Эти ГАО нетрудно выразить непосредственно через рх- и
ру-АО, надо только помнить, что p-функции преобразуются
как векторы:
Pi = Рх
1
р2 = —рх cos 60° + ру cos 30° = -—Рх +
+
+ —Ру
1
Рг = ~РХ cos 60° - ру cos 30° = -урх -
_VLv
2 Ру
Таким образом, учитывая приведенные выше формулы,
окончательно получаем:
1 п
tri\ = (s + l/2Pi)/l/3 = —+ j—px
tri2 = (s + j/2p2)/|/3 = -^r-s i-rpx + —7=-Pj,
j/3 j/б |/2
tri3 = (s + |/2р3)Д/з = j=r- s i
У
htfjf
во°Л
а=120°
/
'Н
ь/
п3
fi ' 1/6 Px I/2 P’
И снова CHJ
И, наконец, для четырех эквивалентных тетраэдрических
ГАО, рассуждая аналогичным образом, получаем
= Х2 — Х3 = Я.4 = X,
cosa = cosl09°28' = — 1/3 = — 1/Х2
229
откуда
Х = |/3; А, = у(5 + ]/Зр;) (i = I, 2, 3, 4)
или, в соответствии с рис. 38:
а а а /-
Pi = Рх COSy + ру cosy + pz COS у = (р, + ру + Р2)/|/3
Pi = (Рх - Ру - Р,)/|/3
Рз = ( - Рх + Ру - Pz)/V*
Рл = ( ~ Рх ~ Ру + Pz)ll/З
то 5р3-гибридные орбитали можно записать в уже знакомом
нам виде:*
h! = trj = (s + рх + ру + рг)/2
/?2 = tr2 = (s + px - py - pz)/2
A3 = tr3 = (s - px + py - pz)/2
/i4 = tr4 = (s - px - py + pz)/2
Пять ГАО углерода?!
Выше мы изложили традиционные квантовохимические
представления о гибридизации атомных орбиталей на тра¬
диционных примерах (С02, НС = СН, Н2С = СН2, СН4,
BF3 и т.д.). Однако эти представления, которые по праву
можно назвать классическими, в ряде случаев оказываются
неприменимыми. Одним из таких случаев является молеку¬
* Детально вопрос об spk-VАО рассмотрен в статье: Bin-
gel W. A., Liittke W. Hybrid Orbitals and their Applications in Structural
Chemistry-Angew. Chem. Intern. Edition in English, 1981, vol. 20,
№ u, p. 899-990.
230
Рис. 39. Молекула 1,6-С2В4Н6.
ла 1,6-дикарба-клозо-гексабо-
рана (рис. 39), где четырех
валентных АО углерода не¬
достаточно для построения
пяти ортогональных ГАО.
Однако при отказе от тре¬
бования ортогональности,
как было показано С. Г. Се¬
меновым,* удается постро¬
ить линейно-зависимый на¬
бор неортогональных JIMO,
преобразующихся друг в дру¬
га при операциях симме¬
трии DAh. Эти 15 J1MO (6 двухцентровых, локализованных
на связях СН и ВН; 8 трехцентровых, локализованных на
связях СВ2 и одна четырехцентровая, тождественная канони¬
ческой 1Ь2з-МО, «охватывающей» атомы бора) с элек¬
тронными заселенностями ж 2, не могут быть переведены
унитарным преобразованием в исходные 13 канонических
МО (сравни с рассмотренным выше случаем молекулы
метана).
Из пяти ГАО углерода одна ориентирована к соседнему
атому водорода, а четыре других, эквивалентных ГАО,-в
направлении пары соседних атомов бора.
Вообще следует отметить, что в последние 10-15 лет пер¬
воначальная концепция гибридизации заметно видоизмени¬
лась **, однако неизменными остались ее взаимосвязь с лока¬
лизацией молекулярных орбиталей, а тем самым и ее статус
* Семенов С. Г. Квантовохимическое исследование электрон¬
ной структуры 1,6-дикарба-клозо-гексаборана в приближении
ППДП.- Журнал структурной химии, 1981, т. 22, № 5, с. 164-166.
** Подробнее см.: Дмитриев И. С., Семенов С. Г. Квантовая хи¬
мия-ее прошлое и настоящее. М.: Атомиздат, 1980. 160 с.
231
теории, связывающей классические и квантовохимические
структурные представления.
* * *
Наш рассказ подошел к концу. Разумеется, в него не во¬
шло многое из того, что составляет предмет размышлений
и поисков современного химика, специализирующегося в ин¬
тересной и трудной области теории строения молекул. Тем,
кого увлекло знакомство с основами квантовой химии, мы
рекомендуем для дальнейшего чтения книги Р. Мак-Вини
и Б. Сатклифа (Квантовая механика молекул. М.: Мир, 1972.
380 с.) и Л. Цюлике (Квантовая химия. Основы и общие ме¬
тоды. М.: Мир, 1976. 512 с.), а также систематическое зна¬
комство с оригинальной литературой.
Игорь Сергеевич Дмитриев
ЭЛЕКТРОН
ГЛАЗАМИ ХИМИКА
Редактор С.Л. ТОМАРЧЕНКО
Художник Б.Н. ОСЕНЧАКОВ
Художественный редактор Д. Д. НЕКРАСОВА
Техн. редактор 3. Е. МАРКОВА
Корректор М.З. БАСИНА
ИБ № 1459
Сдано в набор 30.07.82. Подписано в печать 03.02.83. М-23816. Формат бумаги 70 х 100 1/32.
Бумага офсетная № 2. Гарнитура тайме. Офсетная печать. Уел. печ. л. 9,35.
Уел. кр.-отт. 19,03. Уч.-изд. л. 10,06. Тираж 75 000 экз. Зак. 918. Цена 30 коп.
Изд. № 2244.
Ордена «Знак Почета» издательство «Химия». Ленинградское отделение.
191186, г. Ленинград, Д-186, Невский пр., 28.
Можайский полиграфкомбинат Союзполиграфпрома при Государственном комитете
СССР по делам издательств, полиграфии и книжной торговли
г. Можайск, ул. Мира, 93.
30 коп.
Welcome to site
www.twirpx.com