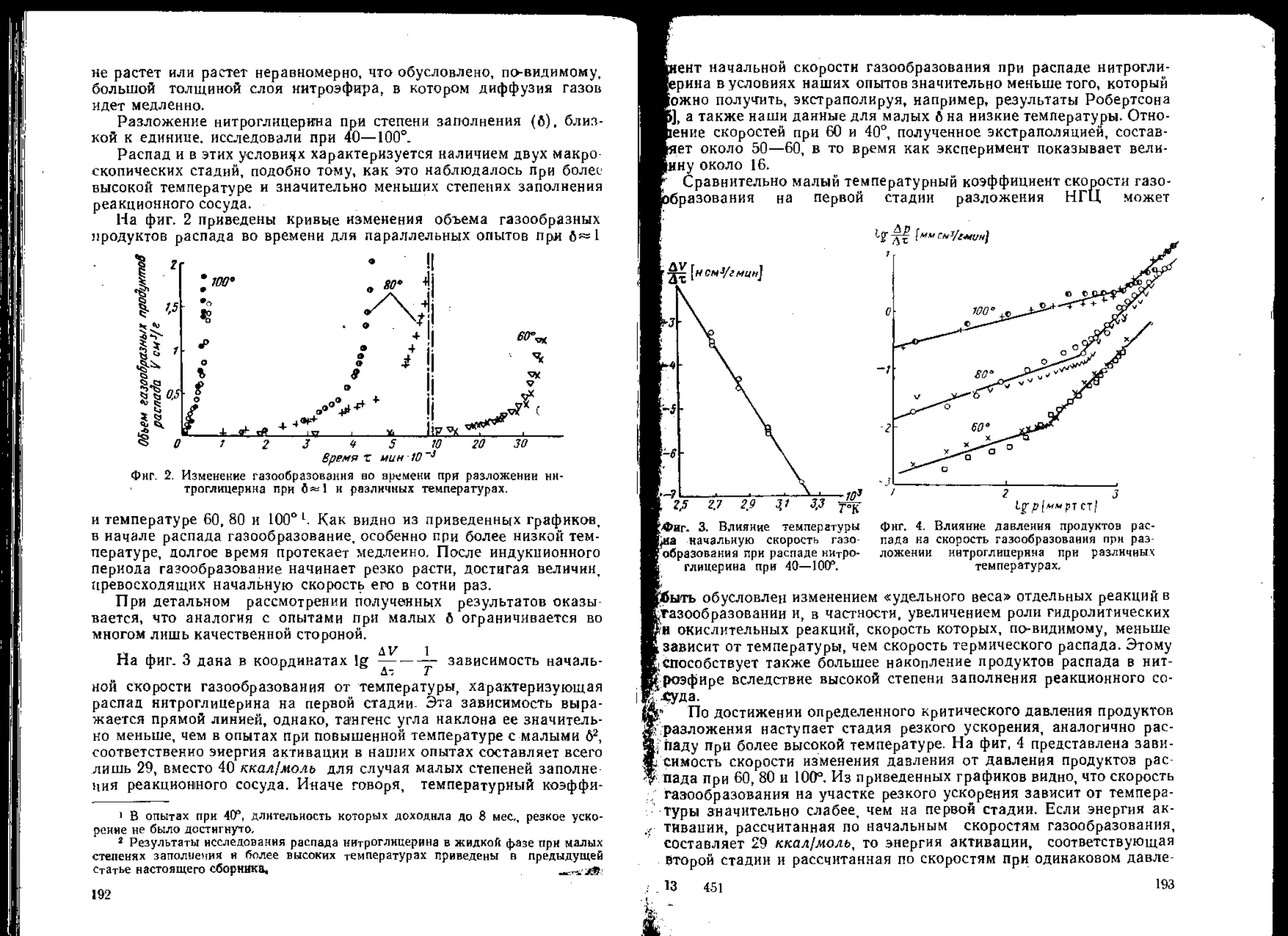
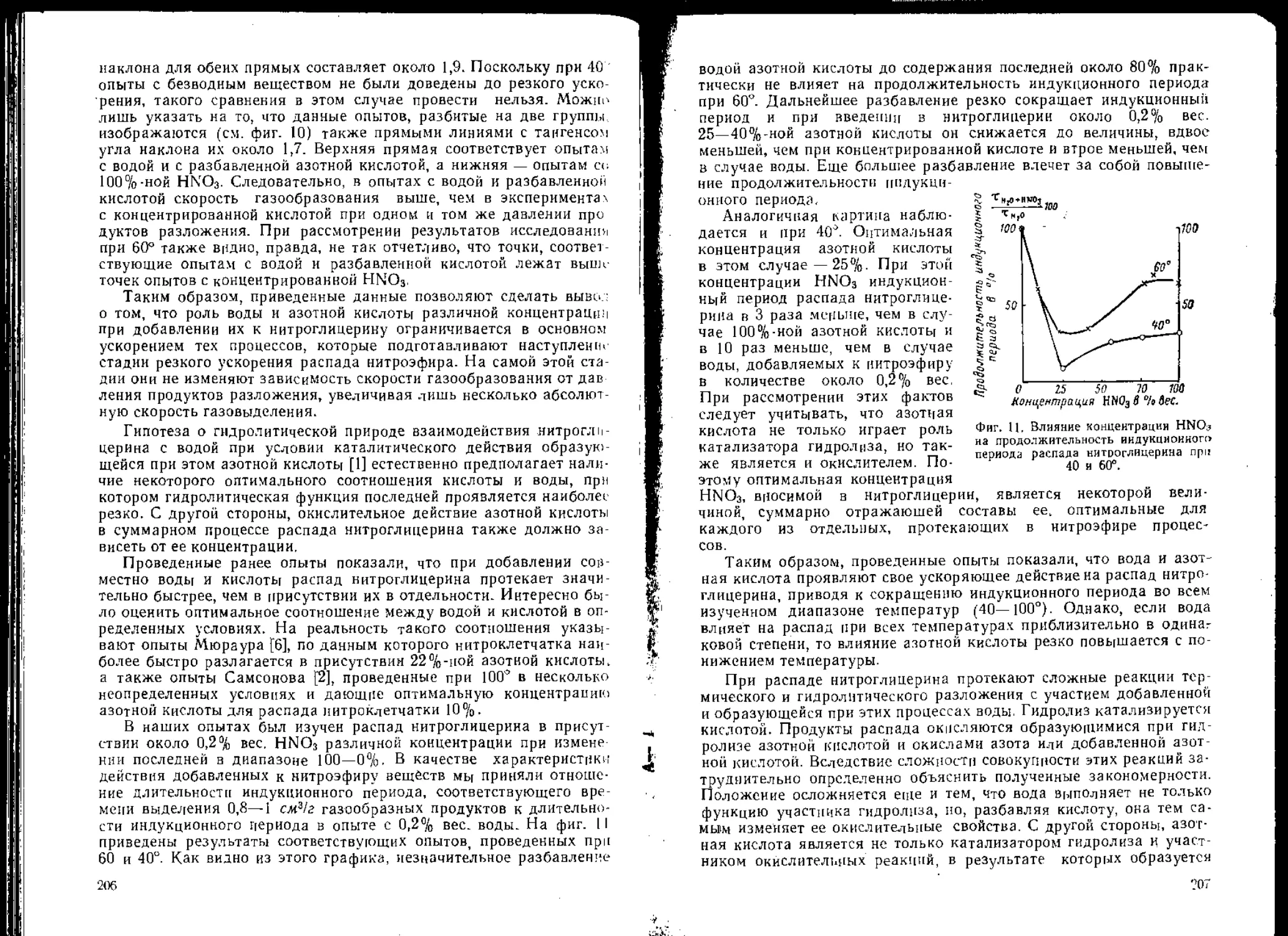
/























































































































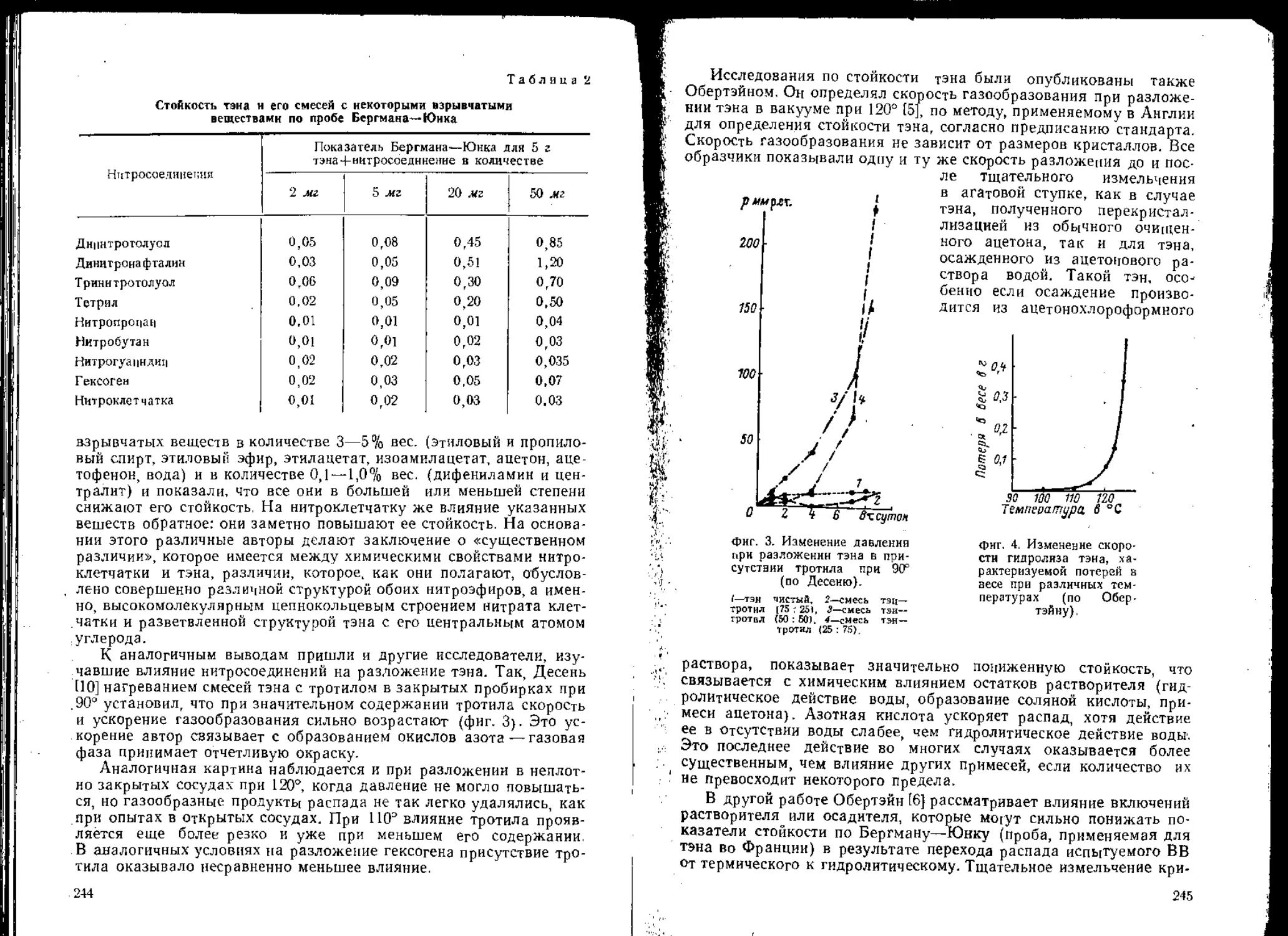


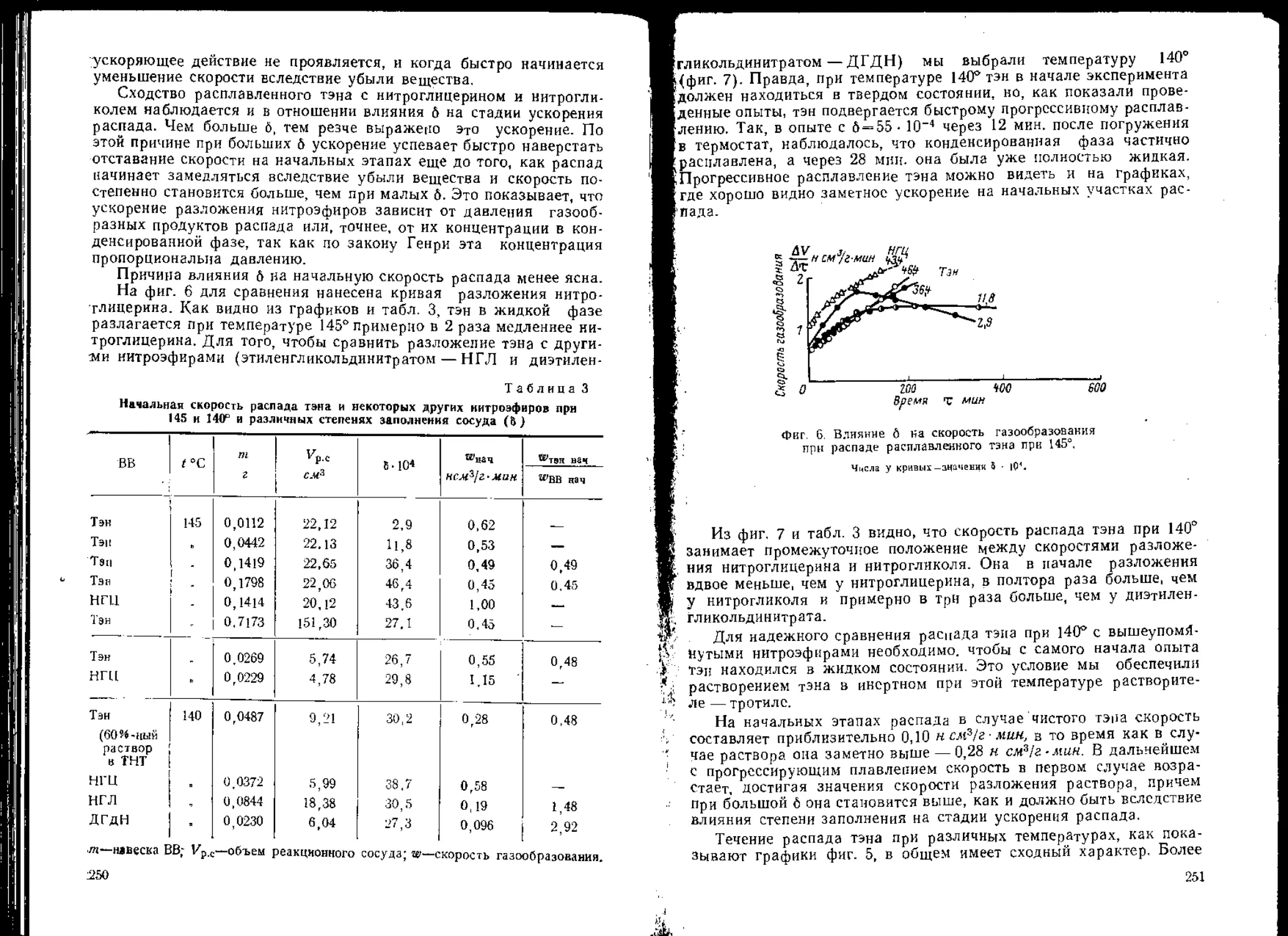






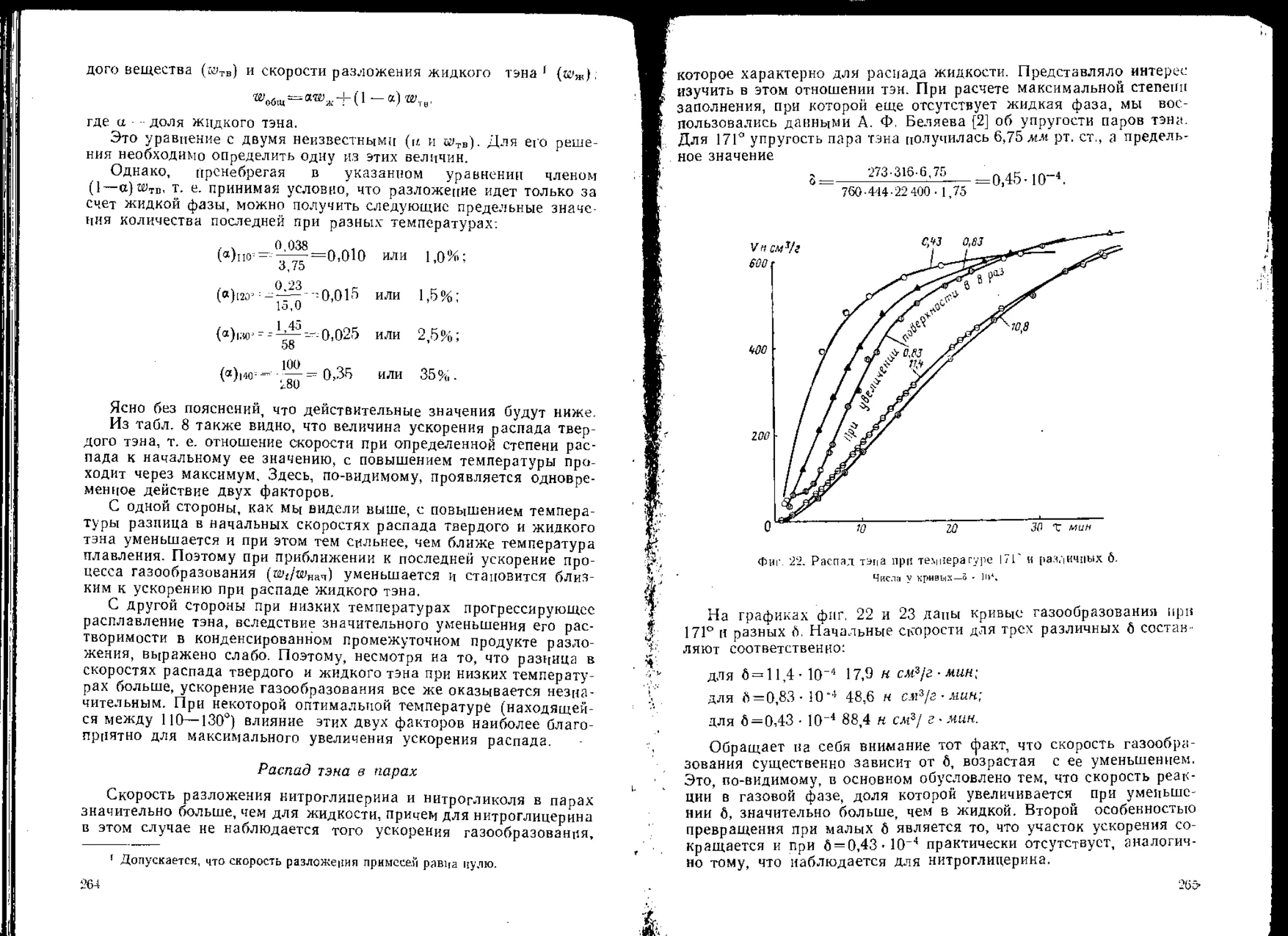
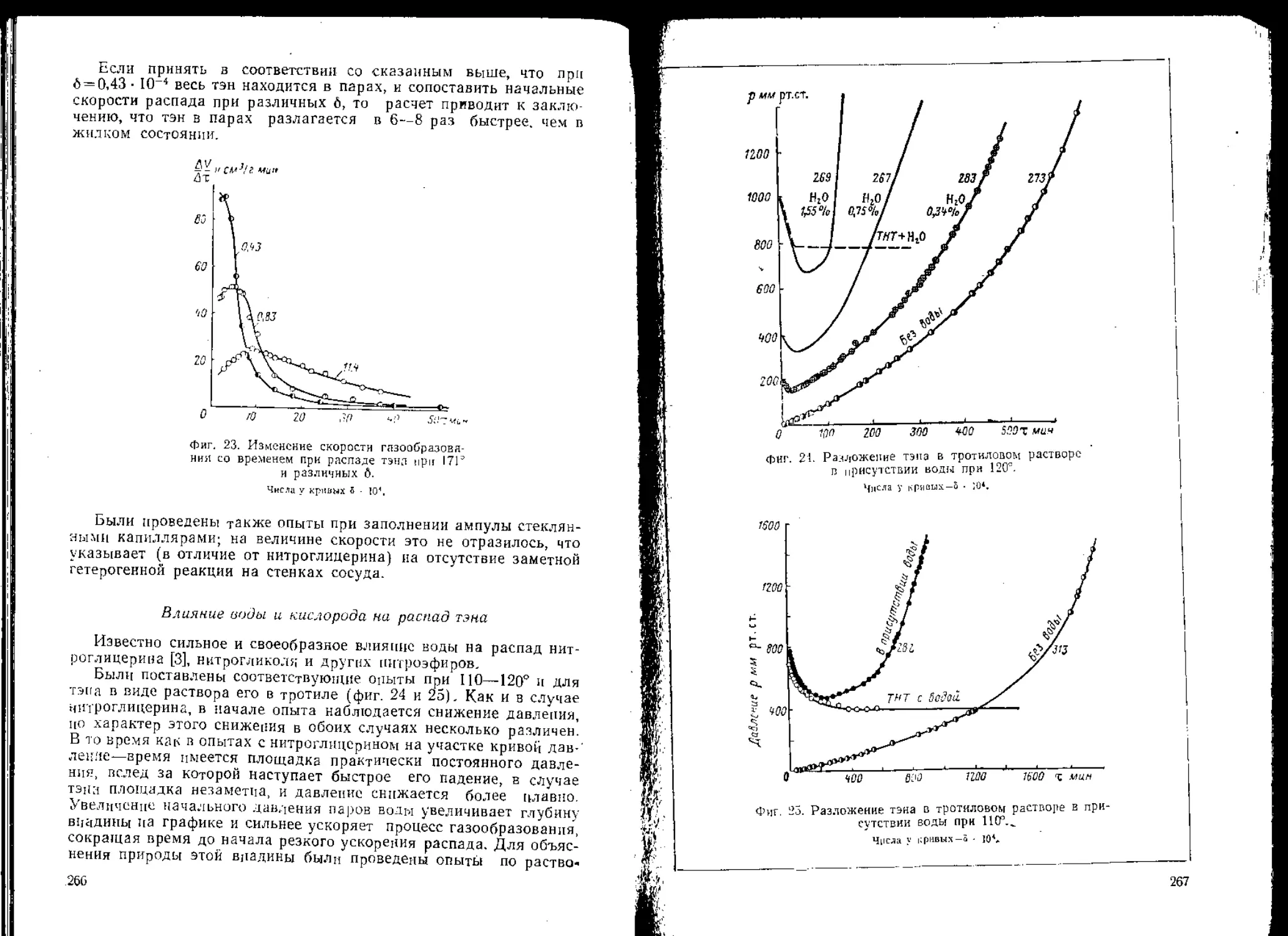























































































































































Автор: Горст А.Г. Гольбиндер А.И. Андреев К.К. Беляев А.Ф.
Теги: горное дело взрывчатые вещества
Год: 1963




Текст
И М I 1|МИ
ТЕОРИЯ
, ВС
ВЗРЫВЧАТЫХ S
ых
ВЕЩЕСТВ д7
' Д'- л
ва- |
t В !
СБОРНИК СТАТЕЙ ’л'
под редакцией /С. /<. Андреева, А, Ф, Беляева, А. И, Гольбиндера, А. Г. Горста по ем 1И- ИЮ
* ?и- ре Г. ( /ГО ,40- ; ми ту ае их эй :е- .0. к 0, ю
ГОСУДАРСТВЕННОЕ с-
НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО )-
ОБОРОНГИЗ о-
Москва 1963 гй гх
J 3
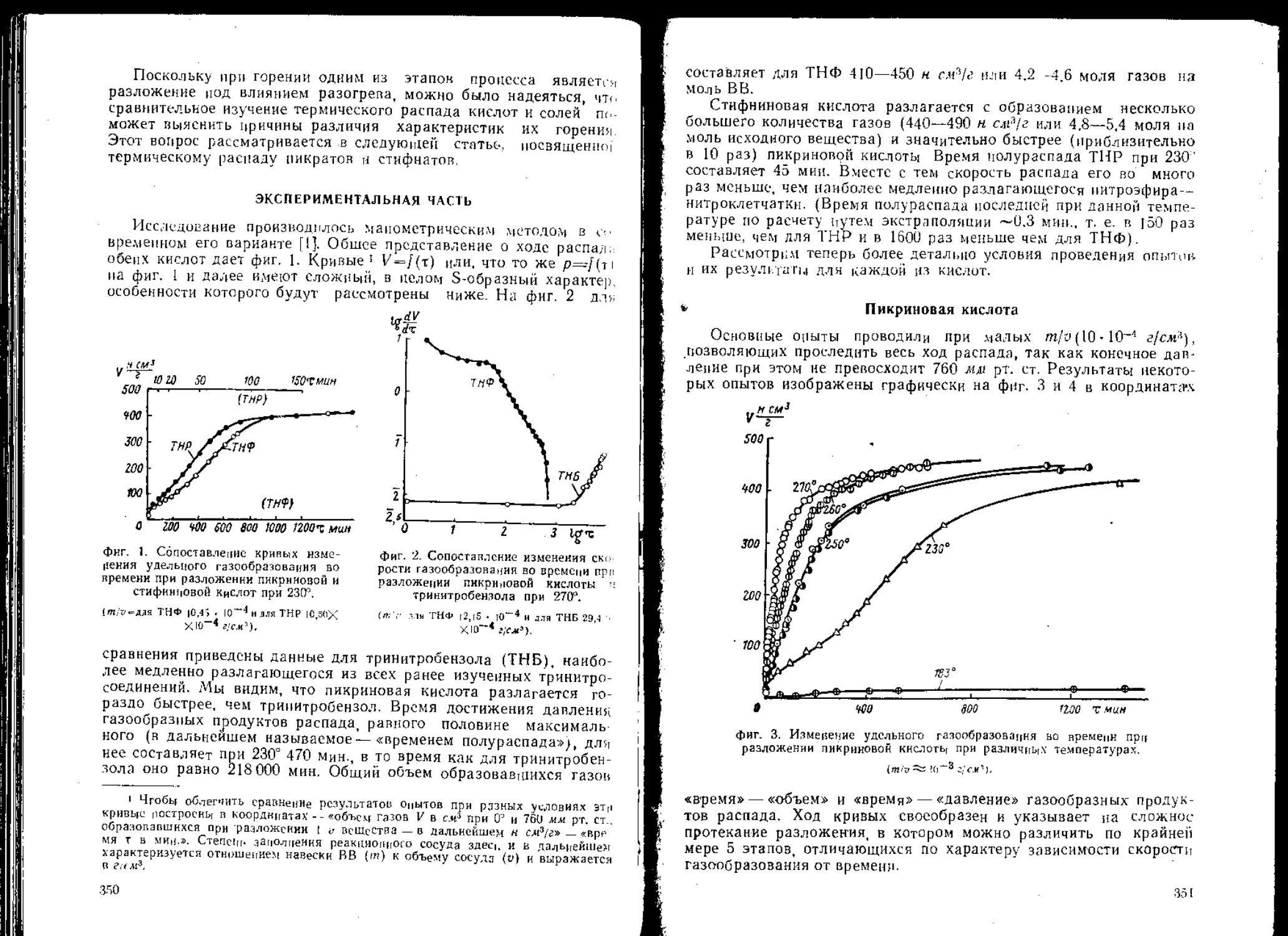
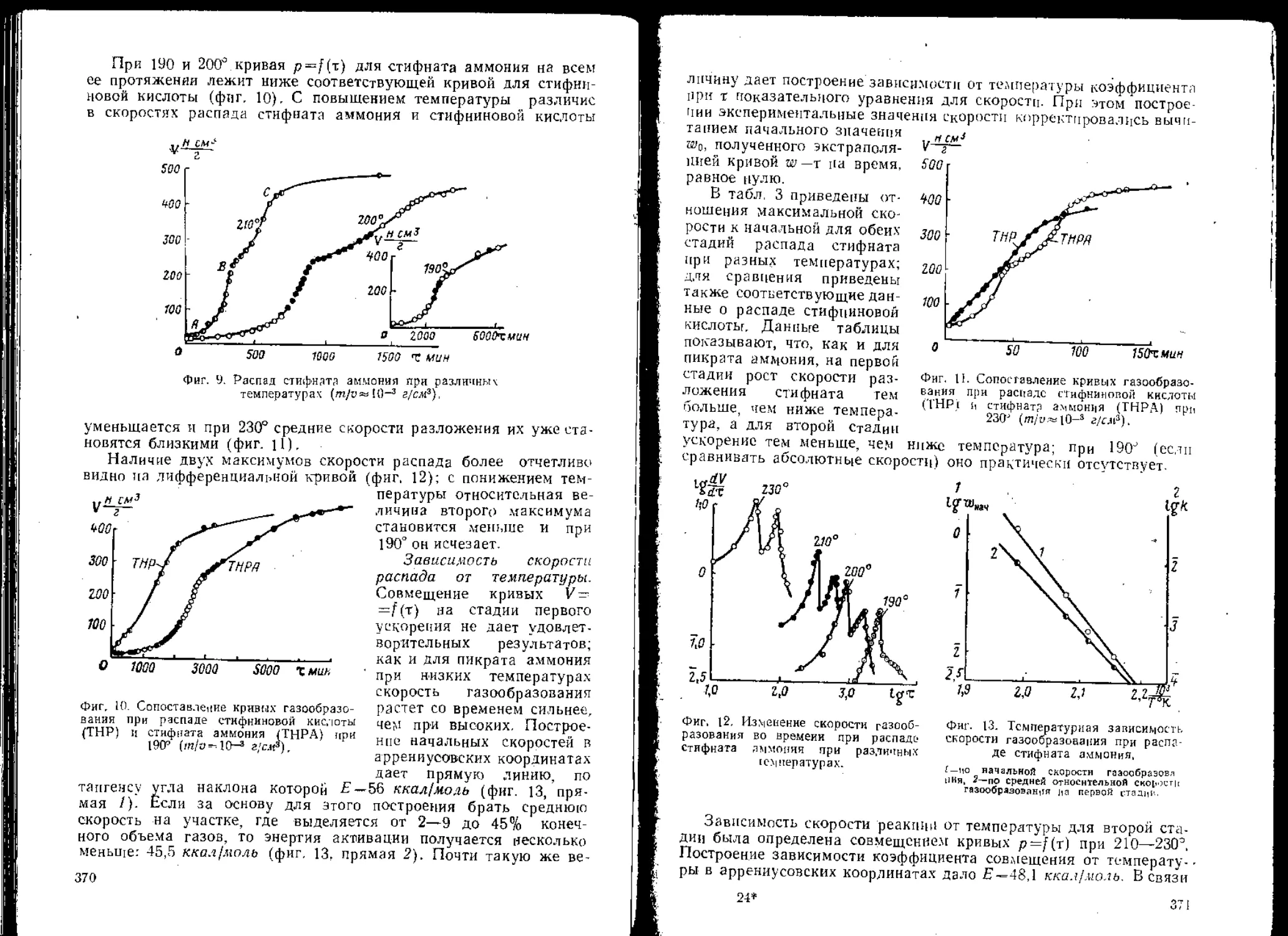
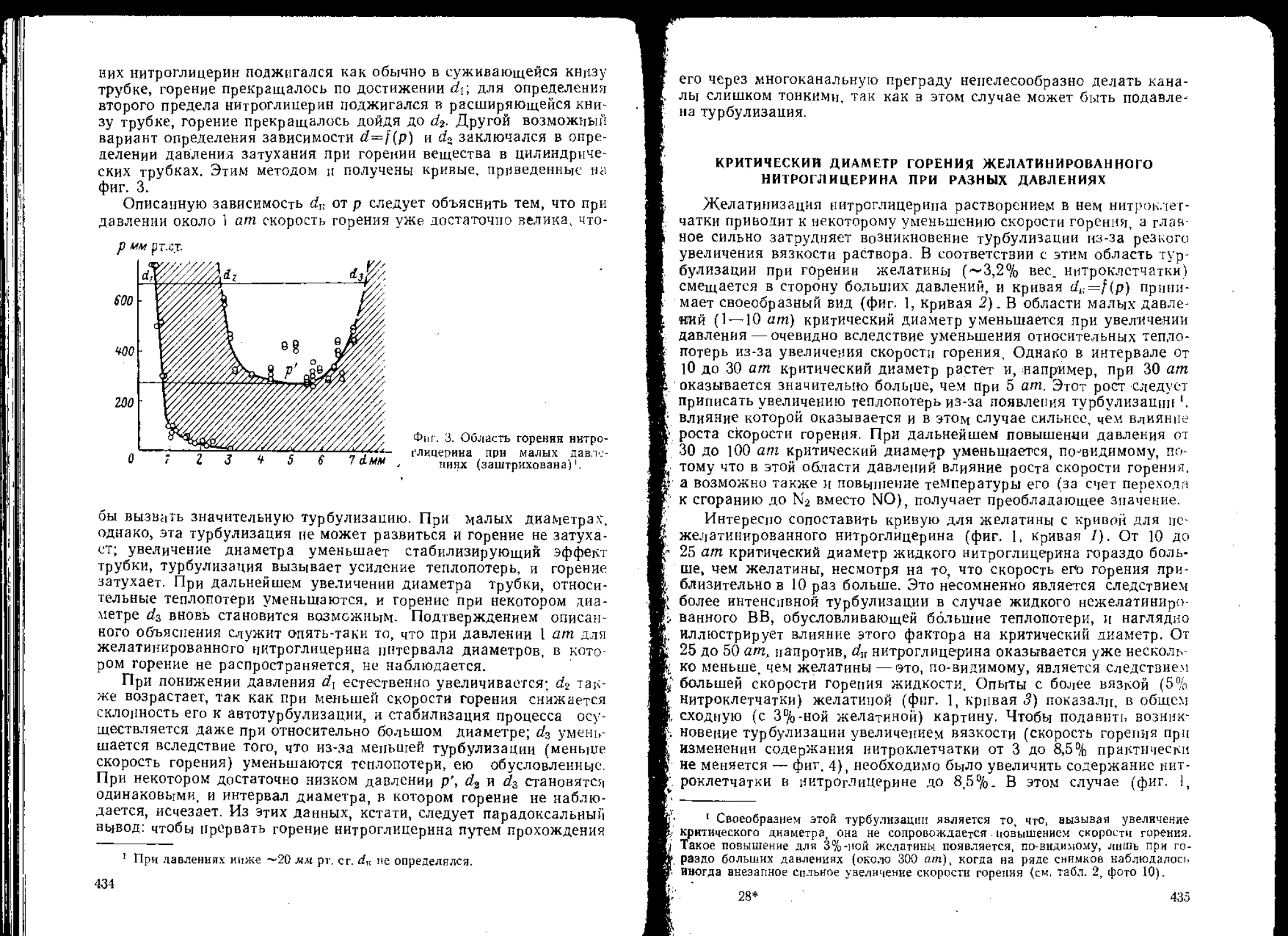
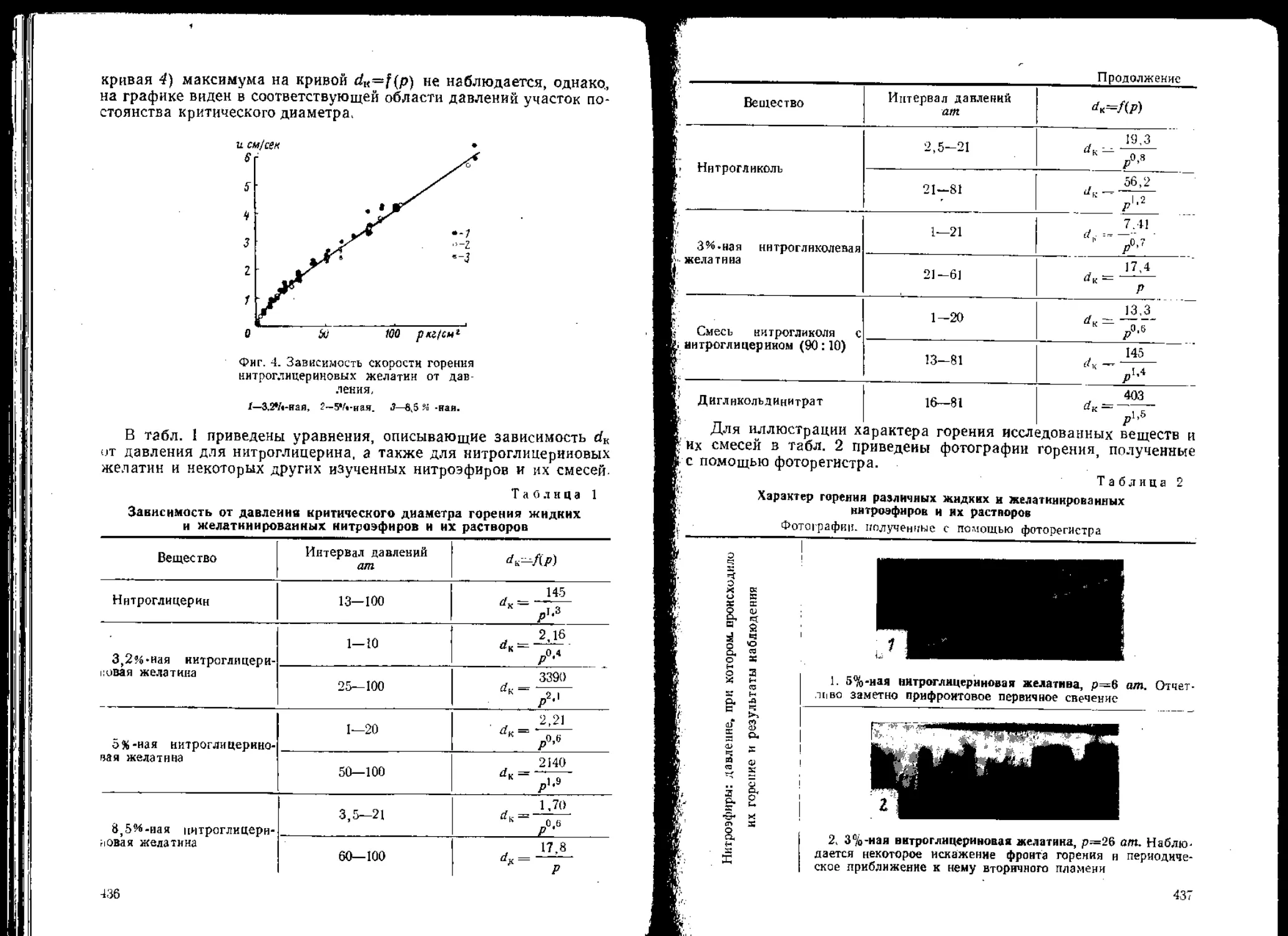
Сборник состоит нз четырех разделов. В первом разделе
освещаются вопросы чувствительности взрывчатых веществ
к механическим воздействиям, рассматриваемой на основе но-
вых представлений о возникновении взрыва при ударе. Статьи
второго раздела посвящены результатам исследовании тер-
мического распада *различных нитроэфиров и некоторых нитро-
соеднненнй. В третьем разделе содержатся исследования по
горению и вспышке взрывчатых веществ. В четвертом разделе
помещены статьи, относящиеся к различным вопросам теории
взрывчатых веществ.
Сборник предназначен для работников научно-исследова-
тельских институтов, конструкторских бюро н предприятий,
связанных с производством и применением взрывчатых ве-
ществ, а также для преподавателей, аспирантов и студентов
старших курсов втузов соответствующих специальностей.
|2Л
Зав. редакцией ииж. А. С, Займовская
>8.3 03 G,
ПРЕДИСЛОВИЕ
Отсутствие журнала, посвященного взрывчатым веществам, за-
трудняет и замедляет возможности ознакомления специалистов с
" результатами исследований по теоретическим проблемам горения,
_• детонации и медленного распада, В некоторой мере эта трудность
’• компенсируется изданием сборников статей по теории взрывчатых
веществ. Первый из них был издан Оборонгнзом в 1940 г,, второй —
(«Вопросы теории взрывчатых веществ») — Издательством Акаде-
мии наук СССР в 1947 г. Пять последующих сборников под назва-
нием «Физика взрыва» были выпущены ограниченным тиражом в
1952—55 гг.; они содержат много интересных исследований, выпол-
ненных главным образом в Институте химической физики АН СССР
и с 1959 г, широко цитируются в советской научной литературе по
теории взрывчатых веществ.
Настоящий сборник включает статьи, преимущественно по трем
разделам теории взрывчатых веществ — чувствительности к механи-
ческим воздействиям, медленному термическому распаду и горению
взрывчатых веществ.
Первый раздел начинается статьей основоположника современ-
ных представлений о механизме возбуждения взрыва при ударе
Н. А. Холево, впервые напечатанной в «Физике взрыва» в 1953 г,
Вторая статья представляет собой доклад известного английского
исследователя Ф. Ф. Боудена, сделанного им в Московском химико-
технологическом институте им. Д, И. Менделеева в 1959 г. Многими
остроумными опытами Ф. Ф. Боуден и его сотрудники показали ту
большую, иногда решающую роль, которую могут играть газовые
пузырьки при возбуждении взрыва, особенно в случае жидких
взрывчатых веществ. Работы К, К, Андреева и Ю. А, Теребнлиной
по возникновению взрыва при ударе на копре являются продолже-
нием и развитием представлений и исследований Н, А, Холево,
Последняя (8-я) статья первого раздела представляет собой обзор
зарубежных работ в этой области. Авторы этих работ приходят к
. выводам, аналогичным тем, которые впервые сделал Н. А, Холево.
Большая часть статей второго раздела относится к исследованию
термического распада нитроглицерина и в меньшей степени к рас-
паду других нитроэфиров н отчасти нитритов. Несмотря на неко-
торую односторонность использованной (манометрической) мето-
дики в этих работах удалось установить стадийность разложения
нитроглицерина и вскрыть физико-химическую сущность основных
4 51 3
стадий, а также влияния на распад различных примесей, в первую
очередь воды и кислот.
Три статьи второго раздела (22, 23 и 24) посвящены термичес-
кому распаду нитросоединений, в частности пикриновой и стнфни-
новой кислот и их солей. Установлен сложный многостадийный ха-
рактер разложения этих соединений; не наблюдается соответствия
кинетических характеристик распада и особенностей горения ука-
занных кислот и их солей; исходя из этого авторы приходят к суще-
ственному заключению, что процессы, протекающие в конденсиро-
ванной фазе, не являются ведущими при горении этих веществ.
Исследования, объединенные в третьем разделе, относятся к не-
скольким частным, но весьма существенным сторонам горения. Так
в работе Б, Н, Кондрикова (статья 29) показано парадоксальное
стабилизирующее влияние примеси азида свинца и повыщения дав-
ления на горение жидких нитроэфиров. В работах А. И, Гольбин-
дера описаны впервые установленное этим исследователем явление
дробного выгорания при горении многокомпонентных летучих сме-
сей, а также особенности самовоспламенения жидких смесей, их
горения и перехода последнего во взрыв. В работе Б. Н. Кондрикова
(статья 35) с помощью Нового простого метода количественно изу-
чалась интенсивность вспыщки взрывчатых веществ в зависимости
от температуры, навески и других факторов; установлен ряд инте-
ресных закономерностей, в частности уменьщение интенсивности
вспыщки некоторых веществ при повышении температуры и отсут-
ствие этого явления для инициирующих взрывчатых веществ, В ра-
ботах К- К- Андреева и В. В. Горбунова рассматриваются сущест-
венные факторы ускорения горения кристаллических взрывчатых
веществ — растрескивание их частиц под действием теплового удара
и проникновение горения в глубь порошкообразного взрывчатого
вещества; сделана попытка дать обобщенную схему нарушения
устойчивости горения порошкообразных взрывчатых веществ и уста-
новить основные условия, ведущие к этому нарушению.
Работы И. Я. Петровского и Л, В. Волкова, К. К- Андреева и
В. Г. Хотина посвящены технически актуальномувопросу детонации
и горения предохранительных взрывчатых веществ.
Наконец, в статьях, вошедших в четвертый раздел, освещаются
различные явления, связанные с возникновением детонации, и не-
которые другие вопросы.
Работы, рассмотренные в статьях 3, 7, 8—24, 26—29, 33, 35—37,
40—42 выполнены в Московском химико-технологическом институте
нм. Д. И. Менделеева, остальные—в других исследовательских
учреждениях.
Все статьи, включенные в сборник, поступили в издательство до
1 марта 1962 г.
Научное редактирование сборника выполнили профессора
К. К. Андреев (статьи 19—21, 25, 32, 38, 39), А. Ф. Беляев (статьи
26, 29—31, 34—36, 40—43), А. И. Гольбиндер (статьи 1—8, 11, 17,
22—24. 27, 28, 33, 37) и А. Г, Горст (статьи 9, 10, 12—16. 18).
I. ЧУВСТВИТЕЛЬНОСТЬ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
К МЕХАНИЧЕСКИМ ВОЗДЕЙСТВИЯМ
I Н. Л. ХОЛЕВО |
1. К ВОПРОСУ О ВОЗБУЖДЕНИИ взрыва
ПРИ ДЕФОРМАЦИИ ЗАРЯДА ВЗРЫВЧАТОГО ВЕЩЕСТВА 1
Как известно, при деформации заряда конденсированного ВВ
Может возникнуть химическое превращение, которое в зависимости
от скорости процесса называют: горением, вспышкой, взрывом или
детонацией; в дальнейшем мы будем рассматривать такое превра-
щение под общим названием «взрыв».
Вероятность возникновения взрыва при определенных условиях
деформации заряда является критерием чувствительности данно-
го ВВ к механическому воздействию, его способности взрываться.
Деформация заряда ВВ может происходить при ударе падаю-
щего на него груза, при действии ударной волны и при резком изме-
нении скорости движения заряда, например, в артиллерийских
снарядах в момент выстрела и при встрече с преградой.
Автором выдвинуты общие представления о механизме возбуж-
дения взрыва путем деформации небольшого заряда при ударе на
копре. Эти представления, после соответствующих уточнений, мож-
но распространить и на другие случаи действия внешних сил.
В настоящей статье рассматривается роль напряжений при воз-
буждении взрыва в случае деформации заряда ВВ.
РОЛЬ НАПРЯЖЕНИЙ ПРИ ВОЗБУЖДЕНИИ ВЗРЫВА
В ДЕФОРМИРУЕМОМ ЗАРЯДЕ ВВ
Общие замечания о деформации заряда при ударе на копре
Механизм явлений, происходящих при деформации заряда, очень
сложен. Вследствие этого автор ограничивается качественным тол-
кованием сущности отдельных явлений, хотя и не отрицает целесо-
образности попыток количественных расчетов, предпринятых, иа-
Пример, в свое время Ю. Б. Харитоном [8].
1 Статья подготовлена автором к печати в 1954 г. и была опубликована в
1955 г. ограниченным тиражом. В последуйщих сообщениях автор предполагал
иможить соображения о механизме возбуждения взрыва и относительной чувст-
вительности ВВ при ударе на копре, а также о возбуждении взрыва в других
условиях деформации заряда ВВ,
Принципиальная схема деформации заряда при ударе на копре
представлена на фиг. 1. Под действием падающего груза, облада-
ющего кинетической энергией А, верхний ударник, преодолевая со-
противление деформируемого заряда, оказывает давление на ВВ.
Первый период удара характеризуется увеличением удельного дав-
ления на ВВ до некоторой максимальной величины Р, а второй
период — его уменьшением.
На фиг. 1 показано состояние заряда в начале и конце первого
периода удара. Работа деформации заряда в этот период слагается
из работы сжатия и работы течения ВВ. Для качественного толко-
Заряд ВВ J^>V3apnu*u
Фиг. I. Принципиальная схема деформации заряда при ударе
на копре.
вания этого процесса можно написать в общем виде уравнение (1),
соответствующее концу первого периода удара.
Л=Л0+Ч.-|--^+^вв.
(1)
где А — энергия удара;
Ао — потенциальная энергия упругой деформации металла при-
бора и копра;
Ц7М— энергия, «поглощенная» металлом прибора и копра в ре-
зультате пластической деформации, внешнего трения и др.;
— работа сжатия заряда (V — объем заряда; Р — напряжение
2К
в заряде при ударе; Л — модуль всестороннего сжатия ВВ);
W'bb — работа течения заряда.
Во второй период удара потенциальная энергия упругих дефор-
pl у
маций Ло+"— трансформируется главным образом в энергию
отскока груза, которую можно определить по высоте отскока.
При рассмотрении уравнения (1) и фиг. 1 можно сделать следу-
ющие общие замечания.
I. Величина напряжений Р в заряде определяется его текуче-
стью и внешними условиями течения, причем текучесть заряда в
сильной степени зависит от температуры плавления ВВ, а внешние
условия течения от конструкции прибора, в котором деформируется
заряд.
2. По мере роста напряжений Р скорость течения заряда умень-
шается вследствие увеличения отрицательных ускорений движения
верхнего ударника.
6
3. Увеличение работы сжатия должно сопровождаться
» 2д
уменьшением работы течения И^вв заряда и наоборот.
В 1912 году на VIII международном конгрессе по прикладной
химии для испытания чувствительности ВВ при ударе на копре был
принят так называемый прибор Каста. Этот прибор, несколько изме-
ненный ГОСТ 2065—43, в дальнейшем называется «прибор № I».
Для создания условий деформации заряда, сопровождающихся
меньшими напряжениями, чем в приборе № 1, автор в 1946 г. пред-
ложил прибор № 2. Для возбуждения напряжений в заряде боль-
ших, чем при использовании прибора № 1, автор в 1950 г. разрабо-
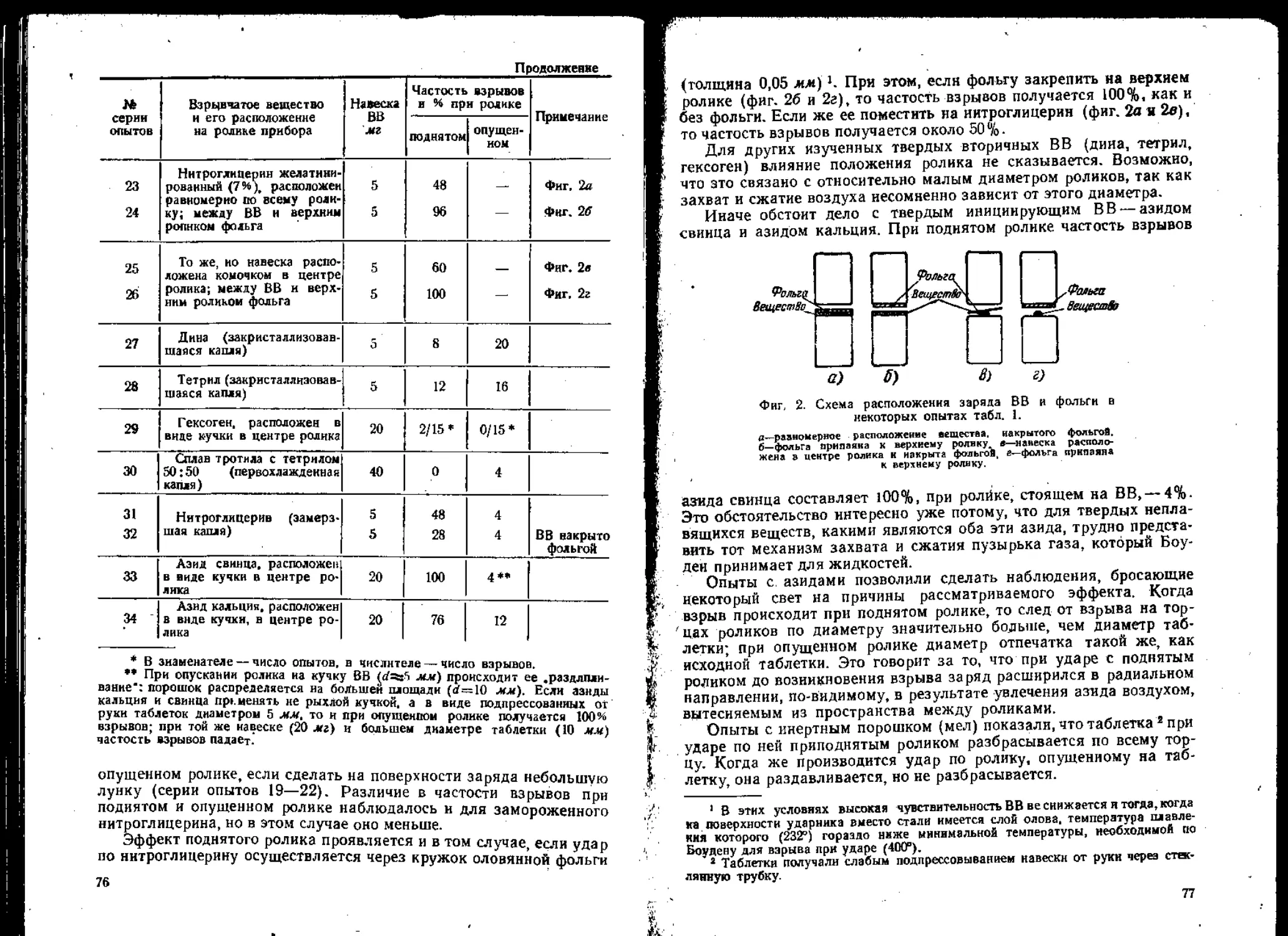
Фиг. 2. Схемы приборов для испытаний ВВ на копре.
тал прибор № 3 (схемы приборов № 1, 2 и 3 см. на фиг. 2). Приме-
няя в приборе № 3 для твердых ВВ кольцо и для жидких чашечку1
из различных цветных металлов (свинец, олово, алюминий, медь и
др.), можно возбуждать большие или меньшие напряжения в за-
ряде независимо от текучести ВВ. Получить желаемые напряже-
ния при деформации заряда в приборе № 3 можно также измене-
нием размера а на верхнем ударнике (см. фиг. 2).
Применение описанных выше приборов позволило осуществить
опыты, когда при постоянной энергии удара в заряде возбуждались
очень большие или очень малые напряжения, когда напряжения
эти определялись главным образом текучестью заряда (прибор № 2)
и когда текучесть заряда не оказывала определяющего влияния на
величину напряжений (прибор № 3), Результаты опытов позволили
сопоставить напряжения, возбуждаемые в заряде, и вероятность
возникновения взрыва, что дало основание для критики широко рас-
пространенных ошибочных представлений о роли напряжений
при деформации заряда ВВ.
1 В приборе с ударником диаметром 10 жж, кольцо имеет наружный диа-
метр 10, внутренний 6 и высоту 3 жж. После внесения 0,05 г ВВ и прессования вы-
сота кольца становится равной около 2,3 и внутренний диаметр около 4 жж. Ча-
шечка имеет внутренний диаметр 4, высоту 3 и толщину дна 0,3—0,5 жм.
7
Литературные сведения о роли напряжений
Каст [5], анализируя известные ему данные (до 1920 г.), сделал
вывод, что при ударе вероятность возбуждения взрыва тем больше,
чем больше удельное давление на ВВ. Подтверждением такого вы-
вода он считал известное из опыта увеличение чувствительности ВВ
в присутствии твердых частичек, на острых краях которых по мне-
нию Каста, «концентрируется» давление !. Роль давления Каст объ-
яснял возбуждением высокой температуры. Он допускал, однако,
и возможность непосредственного возбуждения взрыва, минуя теп-
ловую стадию.
Л. Веннен, Э- Бюрло и А. Лекорше [3] допускают, что в некото-
рых случаях удар вызывает колебания молекул другого характера,
чем колебания, вызванные повышением температуры.
Брунсвиг [2] считает, что помимо ряда теоретических соображе-
ний против предположения о промежуточном переходе энергии
удара в тепловую говорит отсутствие параллелизма между чувст-
вительностью к удару и температурой вспышки, что трудно объяс-
нить даже при учете различий в теплоемкости, теплопроводности,
коэффициенте упругости и т. п. Этот исследователь допускает, что
«давлению как фактору, преодолевающему сопротивления, принад-
лежит, вероятно, роль взаимного сближения атомов с большим хи-
мическим сродством».
В 1922 г. Юстров [14] высказал мысль, что чувствительность ВВ
при выстреле и при ударе на копре определяется критическими на-
пряжениями и что эти напряжения можно выявить эксперименталь-
ным путем.
Все эти общие представления о роли напряжений получили раз-
витие в исследованиях значительного числа специалистов. В ре-
зультате предложен ряд способов выявления критических напря-
жений при помощи прибора, принцип устройства которого такой же,
как и прибора № 1.
Понятие о напряжениях, как о факторе, определяющем чувстви-
тельность ВВ, и результаты исследований ученых, руководствовав-
шихся этим понятием, послужили основанием для ряда практиче-
ских и теоретических выводов.
Некоторые ученые делают вывод, что чувствительность, как не-
которое свойство ВВ, лучше всего характеризуется при ударе в
приборе № 1, в котором, по их мнению, отсутствует трение и чув-
ствительность зависит только от напряжений, возникающих в ре-
зультате сжатия заряда.
Такой вывод является обоснованием идей, которыми более 40 лет
назад руководствовался Каст, предлагая свой прибор для изучения
чувствительности ВВ. Однако представление о преимуществе при-
1 Представление об увеличении чувствительности в результате кконцентрацит|
давления на острых краях» широко распространено и вошло в учебники по тео-
рии ВВ. По-видимому, последнее обстоятельство возбуждало часто задаваемый
автору на заводах вопрос: «Острым или тупым инструментом безопасней вести
механическую обработку зарядов ВВ?».
8
бора № 1 ошибочно и, как это указано автором {10], может привести
к неправильным выводам об относительной опасности ВВ в слу-
жебных условиях.
Представление об определяющей роли напряжений привело не-
которых специалистов к отрицанию промежуточной тепловой ста-
дии возбуждения взрыва. Эти специалисты считают, что механи-
ческий удар может, непосредственно, без существенной тепловой
стадии вызывать взрывчатое разложение ВВ. Ими было выдвинуто
предположение, что механизм возбуждения взрыва при ударе сво-
дится к сильному и быстрому сближению атомов и молекул ВВ на
расстояния, при которых решающую роль начинают играть силы
отталкивания не теплового характера и, вследствие перемены зна-
ка возникающих при ударе инерционных ускорений, зти силы ока-
зываются в состоянии сообщить отдельным частям молекул и ато-
мов очень большие скорости, вследствие чего происходит разрыв
связей и наступает химическое разложение.
Известны работы ряда исследователей, сущность мнения кото-
рых о роли напряжений в возбуждении взрыва сводится к ука-
занному выше представлению [2], [4], [5], [14].
В связи с этим необходимо отметить интересные исследования
Ю. Н. Рябинина [6], [7], который изучал влияние статических напря-
жений до 50 тыс. ка/сл2 на термическое разложение ВВ. Он уста-
новил, что эти напряжения не только не увеличивают скорость раз-
ложения, но для ряда ВВ даже замедляют его. Бриджмен [11] изу-
чал влияние на ВВ статических напряжений до 100 тыс. кг/см2,
В результате испытания семнадцати ВВ он пришел к выводу, что
под действием таких напряжений взрыв не возбуждается.
Опыты Рябинина и Бриджмена отвергают утверждение некото-
рых исследователей [12 и др.] о возможности возбуждения взрыва
в результате непосредственного действия статического давления.
Однако эти опыты не дают ответа на вопрос о влиянии динамиче-
ских напряжений, подобных тем, которые возникают в ВВ, напри-
мер, при ударе на копре, когда время деформации заряда состав-
ляет 10’3—10ч сек.
Попытка экспериментальной проверки влияния динамических
напряжений была предпринята Тамманом и Крегером [13]. Однако
неудачное оформление опытов не дало возможности авторам сде-
лать определенный вывод.
На основании краткого изложения литературных сведений по
рассматриваемому вопросу можно сделать следующие выводы.
1. Выдвинутые в 20-х годах представления Каста, Брунсвига,
Эггерта, Юстрова и др. о напряжениях, как о факторе, определя-
ющем чувствительность ВВ, получили развитие в работах, выпол-
ненных на протяжении последующих 10—20 лет.
- 2. Представление об определяющей роли напряжений в возбуж-
дении взрыва послужило основанием для выдвижения гипотезы о
критических напряжениях, что и привело к выбору определенного
направления исследований устойчивости разрывного заряда артил-
лерийского снаряда при выстреле.
9
3. Представление об определяющей роли напряжений в возбуж-
дении взрыва служило теоретическим обоснованием мнения о пре-
имуществе способа выявления относительной чувствительности ВВ
при ударе в приборе № 1.
4, Широко распространенное представление о такой роли на-
пряжений в ВВ сопровождается у ряда авторов отрицанием проме-
жуточной тепловой стадии и выдвижением гипотез о возбуждении
взрыва в результате «уменьшения» или «увеличения» расстояний
между молекулами и атомами.
Экспериментальная проверка и обсуждение известных
в литературе сведений о роли напряжений
Автором впервые [10] установлено, что при ударе на копре не-
которые ВВ обладают меньшей чувствительностью в приборе № 1.
чем в приборе № 2, хотя в последнем возникают меньшие напря-
жения. чем в первом. Установление большей чувствительности ВВ
в приборе № 2 послужило основанием для критики широко распро-
страненного представления о роли напряжений в возбуждении
взрыва.
В табл. 1 приведены примеры, иллюстрирующие отмеченное яв-
ление. В ряде случаев (опыты 1, 2, 6, 10) частость взрывов в при-
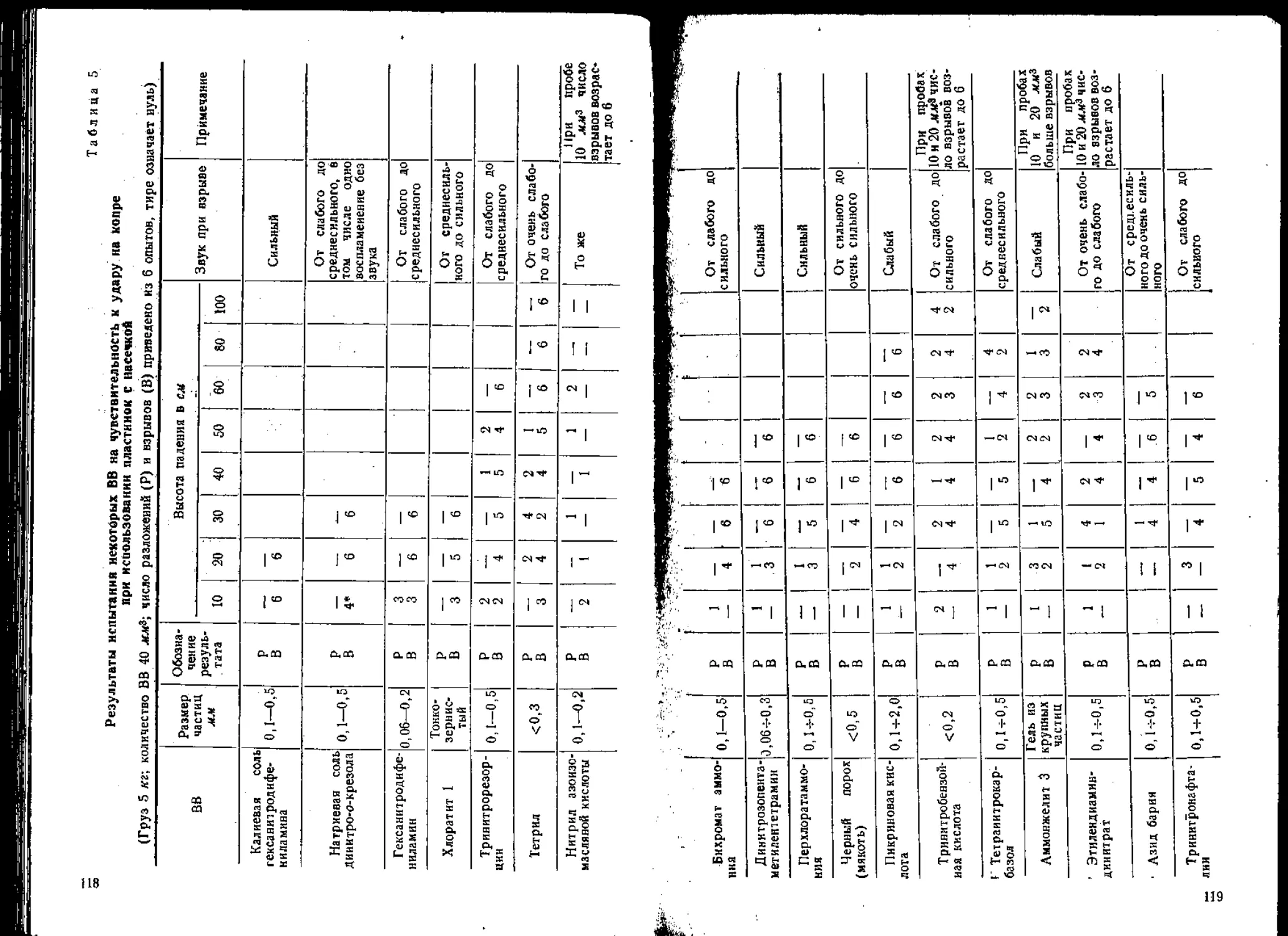
Таблица 1
Чувствительность ВВ в приборах №№ 1 и 2 (вес груза — 5 кг,
вес заряда ВВ — 0,05 г, число параллельных опытов — 20)
№ опыта Наименование ВВ Энергия Удара кГ м Частость взрывов И
прибор
№ 1 | № 2
1 Т ротил 4 20 0
2 Тетрил 4 100 0
3 Дымный порох 4 35 70
4 Гексоген 2,5 80 90
5 Гексоген-]-А1 (пудра) 50:50 4 0 —
Гексоген4-А1 (пудра) 50:50 2 — 100
б Гексоген-j-Al (пудра)-|-парафнн 45:45:10 2 40 0
7 КСЮз+угмь 87:13 4 0 —
КС1О3-|-уголь 87:13 2 — 100
8 NH4NO3-|-A1 (пудра) 80:20 4 0 —
NH4NOa-|-Al (пудра) 80 :20 2 — 100
9 Пироксилин 2.5 30 100
10 Пироксилий-7-па рафин (95:5) 2,5 100 0
11 | Стифнат свинца 2 0 100
10
боре № 1 больше, чем в приборе № 2. Вместе с тем, в таблице
приведены случаи, когда частость взрывов в приборе № 1 значи-
тельно меньше, чем в приборе № 2 (опыты 3. 5. 7, 8, 9, 11). что
совершенно не согласуется с представлением о напряжениях, как
об основном факторе, определяющем чувствительность ВВ, В табл. 1
приведены результаты опытов, когда в присутствии парафина чув-
. ствительность ВВ в приборе № 1 сильно увеличивается (сравните
опыты 5 и 6 или 9 и 10), что также нельзя объяснить, руководст-
вуясь старым представлением о роли напряжений в возбуждении
взрыва.
Таким образом, на основании данных табл. 1 можно сделать
вывод, что вероятность возбуждения взрыва не всегда находится в
. прямой зависимости от возникающих при ударе напряжений и, сле-
довательно. распространенные представления о роли напряжений
- в этом процессе не отвечают действительности.
Однако такой вывод оспаривают некоторые «защитники напря-
1 жений», утверждая, что ВВ обладают «избирательной» чувствитель-
>;. ностью к удару и к трению1. При этом чувствительность к удару,
по их мнению, является наиболее объективным критерием устойчи-
вости ВВ, обусловленной их химической природой. По мнению этих
ученых, прибор № 2 характеризует чувствительность только к тре-
. нию, результаты испытания в нем не отображают поэтому влияния
; напряжений и, вообще, не показательны. Соображения эти схола-
стичны и легко могут быть опровергнуты приводимыми ниже ре-
' зультатами опытов в приборе № 1.
Опыты в этом приборе провели с большим числом взрывчатых
и невзрывчатых веществ. При выборе веществ для испытаний при-
нимали во внимание главным образом их способность при ударе
выдавливаться в зазор прибора, которая обусловлена пределом
текучести н температурой плавления вещества.
В прибор № 1 помещали 0,05 г вещества и производили удар,
отмечая высоту отскока груза (только в случаях отказа) и степень
выдавливания вещества в зазор прибора. С каждым веществом про-
. водили до 50 испытаний, при этом для сравниваемых веществ соблю-
дали постоянными внешние условия удара.
Для характеристики метода исследования и выводов, которые
были при этом сделаны на фиг, 3 и в табл. 2, приведены результаты
изучения некоторых веществ.
На фиг. 3 вертикальные ряды точек соответствуют высотам от-
скока груза йо при высотах его падения h, равных 200, 400 и 600 мм.
Каждый вертикальный ряд (за исключением рядов V и VI, соответст-
вующих алюминиевой пудре и воде) получен в результате 20 парал-
лельных опытов.
Работа течения заряда И7ВВ в данном случае связана с высотами
падения й и отскока йо груза приближенным соотношением
0,005(ft — 1,70й„) кГ • м. (2)
। Под ударом они подразумевают деформацию заряда в результате динами-
ческого сжатня, а под трением — в результате течения.
И
В отдельных рядах на фиг. 3 отсутствуют точки, соответствую-
щие случаям взрыва. Число таких случаев отмечено цифрой в
кружке, расположенном над соответствующим рядом точек.
Фиг. 3. Высота отскока груза в сериях параллельных опытов с раз-
личными веществами на копре:
I—тротил. I Г—ди нитробензол, П1—тетрил, IV—ксилил, V—алюминиевая пудрз.
VI—вода.
(Цифры в кружке —число взрывов из 20 опытов).
Т э й л и ц а 2
Отношение средней высоты отскока к высоте падения груза
(см. фиг. 3)
Наименование вещества Температура затверде- вания °C Отношение A0:ft в и
Л мм
200 400 600
Тротил 80,7 38 19 33
Динитро бензол 90 36 25 21
Тетрил 128.8 46 45 48
Ксилил 181 52 33 47
Алюминиевая пудра >600 52 51 51
Вода 0 50 — 50
На основании данных фиг. 3, табл. 2 и некоторых наблюдений
можно сделать следующие выводы.
1. При ударе заряд в приборе № 1 сжимается и течет. Работа
течения заряда 117вв зависит от энергии удара и текучести ВВ.
В случаях, когда текучесть очень мала (алюминиевая пудра) или
12
когда текучесть ВВ и энергия удара невелики (ксилил при
Л = 200 мм), работа течения заряда, как это следует из уравне-
ния (2), небольшая, вследствие того, что заряд не выдавливается
в зазор прибора Ч В тех же случаях, когда происходит пластическое
и вязко-пластическое течение заряда в зазоре прибора, работа тече-
ния может достигать значительной величины, подчас превышающей
половину энергии удара.
Таким образом, широко распространенное представление о том,
что в приборе № 1 имеет место только сжатие заряда, не соответ-
ствует действительности, а утверждение, что работа течения веще-
ства в этом приборе мала, является следствием плохого изучения
, механизма деформации заряда.
Не имеет оснований также предложение некоторых исследова-
телей считать, что результаты испытаний в приборе № 1 характери-
зуют чувствительность к удару, а в приборе № 2 — к трению.
Оба прибора характеризуют чувствительность ВВ при кратко-
временной деформации сжатия и течения заряда; при этом, в при-
боре № 1 скорость течения меньше, а сжатие больше, чем в при-
боре № 2.
2. Наблюдаемое на фиг. 3 очень большое рассеяние результатов
при параллельных испытаниях объясняется не только различной
. величиной зазора а применявшихся приборах, но и рядом случай-
ных причин, влияющих на степень плавления заряда. Ввиду такого
большого рассеяния нельзя определить напряжения в зарядах в
случаях взрыва на основании измерения их в случаях отказа. Так,
например, нельзя думать, что напряжения, приведшие к взрыву
тетрила в 13 случаях (Л = 600 мм), равны напряжением в 7 случаях
отказа. Напряжения в случаях отказа могут очень сильно отли-
чаться от напряжений в случаях взрыва.
3. В вертикальных рядах на фиг. 3 отброшены точки, соответ-
ствующие случаям взрыва, Возникает вопрос: Какие высоты отскока
соответствуют отброшенным точкам? Если бы этим точкам соответ-
ствовали наибольшие значения высоты отскока, то представление
об определяющей роли напряжений в возбуждении взрыва в неко-
торой степени подтверждалось бы экспериментально. Ответ на по-
ставленный вопрос можно видеть в табл. 2, где дано изменение
отношения высоты отскока груза к высоте его падения hQ'.h при
увеличении высоты h. В случаях отсутствия взрывов отношение
Ло : й с увеличением h от 200 до 400 мм уменьшается (тротил, дини-
тробензол, ксилил). Если бы случаям взрыва соответствовали боль-
шие высоты отскока, то отбрасывание точек привело бы к дальней-
шему уменьшению отношения ha: h при увеличении высоты падения.
В этом случае, при Л = 600 мм отношение ho : h для тротила должно
было бы быть меньше 19, для ксилила — меньше 33 и для тетрила —
меньше 45%. В действительности же при переходе от высоты 400 к
высоте 600 мм отношение h^-.h не только не уменьшилось, но даже
1 Работа течения мала и в тех случаях, когда заряд обладает очень большой
текучестью (еода).
13
заметно увеличилось. Такой результат является следствием того,
что взрывам соответствуют преимущественно малые значения вы-
соты отскока, т. е. взрывы возбуждаются при меньших напряжен
ниях, но при большей работе течения — см. уравнение (2).
Таким образом, результаты опытов в приборе № 1, приведенные
на фиг. 3, а также в табл. 2 и 1 убеждают нас в том, что напряже-
ния не являются фактором, определяющим возбуждение взрыва н
что широко распространенные представления о такой роли напря-
жений не соответствуют действительности.
Действительная роль напряжений в возбуждении взрыва
При изучении роли напряжений в возбуждении взрыва прежде
всего необходимо было выяснить их влияние в случае когда течение
заряда отсутствует. Такая деформация заряда наиболее удачно
была получена в приборе № 3, в котором пластичность металла
кольца обеспечивала при ударе всестороннее давление на заряд,
а течение металла в зазоре прибора не возникало.
Опыты проводили с тротилом, который вносили в количестве
0,05 г в прибор № 3 с малым зазором и с алюминиевым кольцом.
После прессования (или после предварительных ударов при паде-
нии груза с небольшой высоты) производились испытания при па-
дении груза весом 5 кг с высоты 2000 лии. При этом взрывов не было.
При проведении второй серии таких же опытов с верхним удар-
ником, имеющим размер а, равный 9 лш (см. фиг. 2), или со свин-
цовым кольцом вместо алюминиевого, получал ась 100 % -ная
частость взрывов.
В первом случае, при ударе кольцо очень незначительно выдав-
ливалось в зазор прибора (после удара это проверяли, извлекая
кольцо из прибора) и, следовательно, происходило только всесто-
роннее сжатие заряда, с возникновением больших напряжений, но
при очень небольшом его течении. Во втором случае, в результате
частичного выдавливания алюминия в свободное пространство ме-
жду лысками ударника и муфтой или свинца в зазор прибора, имело
место большее течение заряда при меньших напряжениях 1, чем в
первом случае.
Так как в первом случае заряд подвергался всестороннему сжа-
тию при очень больших напряжениях, то возникло предположение,
что при деформации сжатия заряда, не сопровождающейся тече-
нием ВВ, взрыв не возбуждается.
Для проверки этого были проведены точно такие же опыты, но
со значительно более чувствительными ВВ, чем тротил — гексоге-
ном и тэном. Результаты опытов подтвердили наше предположение.
Особенно интересные результаты были получены с гремучей
ртутью, которая, как известно, характеризуется очень высокой чув-
ствительностью. Для проведения опытов применяли прибор, схема
1 О величине напряжений судили по высоте отскока груза при применении
нафталина вместо тротила.
•?>+’• которого приведена на фиг. 4. В медное кольцо с внутренним диа-
V метром 5 и высотой 2,5 jwjw помещали 0,05 г гремучей ртути и при
помощи пресса сдавливали ударники силой в 5000 кг. Прибор после
Ч' обжатия подвергали последовательным ударам на копре при паде-
\ нии груза весом 10 кг с высот 150, 250, 500, 750 и 1000 jhjh1. При
,i этом возбудить взрыв не удавалось. После удара с высоты 1000 лии
^’прибор (без поддона) переворачивали и муфту передвигали вниз;
$’В результате медное кольцо с гремучей ртутью оказывалось против
$ канавки муфты и при ударе могло выдавливаться в последнюю.
^ Подготовленный таким образом при-
I бор подвергали удару при падении
$ груза в 10 кг с высоты 150 мм. В этих
‘Т условиях взрыв гремучей ртути воз-
ж буждался. Явление неизменно повто-
•5й. рялось в значительном числе незави-
ж симых испытаний.
Таким путем установлено, что
ив случае всестороннего сжатия даже
такого чувствительного ВВ, как грему-
X чая ртуть, взрыв не возбуждается,
-Ж если течение заряда очень незначи-
тельно. Напротив, гремучая ртуть
при значительном течении заряда
•*; взрывается, хотя напряжения при
этом намного меньше, чем в первом
Фиг. 4. Прибор для опытов
с гремучей ртутью.
случае.
Описанные выше опыты с тротилом, гексогеном, тэном и грему-
; чей ртутью убедительно подтвердили предположение, что при все-
стороннем динамическом сжатии заряда, не сопровождающемся его
течением, взрыв не возбуждается 2.
Вместе с тем эти опыты, так же как и исследования, описанные
в предыдущем разделе, подтверждают высказанную автором [9]
мысль, что эффективность механического воздействия следует рас-
сматривать, принимая во внимание деформацию течения заряда,
а не деформацию его сжатия.
Дополнительным подтверждением этого могут служить опытные
данные С. М. Муратова, который, изучая относительную чувстви-
тельность нескольких ВВ в приборе № 1, получил парадоксальные
результаты, приведенные в табл. 3.
Неожиданные результаты опытов С. М. Муратова объясняются
тем, что он применял приборы № 1 с очень малым зазором между
1 Дальнейшее увеличение высоты падения груза не допускали из опасения
аиачительной деформации муфты прибора и возможного вследствие этого течения
заряда.
а По мнению автора этот вывод можно распространить и на случай возбуж-
дения больших напряжений, чем достижимые прн ударе на копре. Однако это
соображение не имеет практического значения, так как очень большие напряже-
ния неизбежно должны сопровождаться течением заряда, которое в обусловливает
возбуждение взрыва.
15
Таблица 3
Результаты испытания относительной чувствительности ВВ в приборе №1
Наименование ВВ Энергия удара кГ-м Частость взрывов %
Тротил 3 40
Гэп 3 100
Тетразен 3 60
Азид свинца 3 30
Гремучая ртуть 3 10
Стифнат свинца 4 10
ударником и каналом муфты, вследствие чего удар сопровождался
очень малым течением тех В В, которые обладают плохой текуче-
стью. В результате в зарядах таких, очень чувствительных, но обла-
дающих малой текучестью инициирующих ВВ, как гремучая ртуть,
стифнат свинца и азид свинца, взрыв возникал значительно труд-
нее, чем в зарядах тэна и тротила, которые обладают хорошей теку-
честью, Однако из этого нельзя сделать вывод, что названные ини-
циирующие ВВ обладают меньшей чувствительностью при дефор-
мации, чем тэн и тротил. Для выявления относительной чувстви-
тельности ВВ, обладающих различной текучестью, необходимо учи-
тывать условия течения вещества их зарядов.
Принимая во внимание результаты проведенных опытов, можно
сделать вывод, что возбуждение взрыва ВВ обусловлено течением
вещества заряда.
Возникает, однако, вопрос о характере влияния напряжений в
заряде, которые неизбежно сопровождают деформацию течения ВВ.
В табл. 1 приведены результаты опытов, в которых возбуждение
взрыва обусловлено течением заряда, сопровождающимся больши-
ми (опыты 1, 2, 6, 10) и меньшими (опыты 3, 5, 7, 8, 9, 11) напря-
жениями.
При очень больших напряжениях и малом течении тротилового
заряда в приборе № 3 с алюминиевым кольцом взрыв не возбуж-
дается, при меньших же напряжениях, но большем течении этого
заряда в приборе № 3 со свинцовым кольцом частота возбуждения
взрыва тротила 100%. Однако при еще меньших напряжениях, хотя
и большем течении заряда в приборе № 3 со свинцовым кольцом
н верхним ударником, имеющим фаски, взрыв не возбуждается.
Таким образом результаты этих опытов показывают сложный
характер зависимости вероятности возбуждения взрыва от напря-
жения.
Полагая, что возникновение взрыва обусловлено течением взрыв-
чатого вещества, определяющим параметром этого процесса можно
считать удельную скорость выделения энергии и в заряде при его
пластическом течении. Величина и тем больше, чем больше коэф-
16
(ициент внутреннего трения т] и градиент скорости течения ~,
егко показать, что
Можно предполагать, что роль напряжений сжатия связана с
IX влиянием на коэффициент внутреннего трения, особенно в слу-
1аях вязкого течения.
Практический интерес представляет зависимость скорости по-
глощения энергии и скорости течения ВВ от напряжений при посто-
1нной энергии удара.
Фиг. 5. Зависимость удельной скорости выделения энергии в
заояде (и) от возникающего в нем напряжения (Р) при
различных энергиях удара (Д) (Схематический график!.
При постоянной энергии удара с увеличением напряжений ско-
рость течения уменьшается (см. раздел 1). Это обстоятельство обус-
ловливает увеличение и при увеличении Р только до некоторой опти-
мальной величины. При дальнейшем же увеличении Р скорость
течения становится настолько мала, что и начинает уменьшаться.
Следовательно, при энергии удара Д=сопэ1, и=ф(Р) можно
представить кривыми, имеющими максимум, как это показано на
фиг. 5. Кривые на этой фигуре выражают зависимость и от Р при
энергиях удара
Ai<^2<SAa.
Допустим, что в приборах № 2, 1, Зс (свинцовое кольцо) и За
(алюминиевое кольцо) производят при энергии удара At деформа-
цию заряда ВВ-1, обладающего большой текучестью. Напряжения,
возникающие при деформации этого заряда в различных приборах,
показаны на фиг. 5 при помощи кружков, в которых написан индекс
2 451
17
Д12>О2>С?
соответствующего прибора1. В случае деформации заряда ВВ-2,
обладающего меньшей текучестью, чем ВВ-1, соответствующие
кружки располагаются несколько правее1 2; а в случае деформации
заряда ВВ-3, обладающего еще меньшей текучестью, кружки рас-
полагаются еще несколько правее.
Таким образом, Рим зависят от конструкции прибора и теку-
чести ВВ. При этом величина и в случае деформации заряда ВВ-1
очень мала в приборах № 2 и 1 вследствие малых напряжений, а в
приборах № Зс и За — вследствие очень малой скорости течения ВВ.
В случае деформации заряда ВВ-2 величина и наибольшая в при-
боре № 1, а в случае деформации заряда ВВ-3 величина и наиболь-
шая в приборе № 2.
С увеличением энергии удара (например, до А3), отмеченные
на фиг. 5 кружки также будут смещаться вправо. При этом ско-
рость выделения энергии и увеличивается до некоторого предела,
Однако смещение кружков вправо и увеличение и зависят от теку-
чести ВВ и конструкции прибора. Так, например, можно полагать,
что при увеличении энергии удара смещение кружков вправо для
ВВ-1 небольшое и сопровождается оно незначительным увеличе-
нием и в приборах № 2 и 1 и большим в приборе № Зс.
Фиг. 5 отображает только общую качественную зависимость и
от текучести заряда, конструкции прибора и энергии удара. В дей-
ствительности влияние отдельных факторов на величину и значи-
тельно сложнее. В частности, максимальная величина и сущест-
венно зависит от степени неоднообразия физических свойств заря-
да и от случайных отклонений (см. фиг. 3).
При помощи фиг. 5 можно, однако, объяснить роль напряже-
ний при течении заряда и ряд явлений, которые имеют место при
ударе на копре. Допустив, что частота взрыва находится в прямой
зависимости от и, легко объяснить результаты испытания на копре
(см. табл. 1 и 3),’которые нельзя понять, руководствуясь общепри-
нятыми пока представлениями о механизме удара. Например,
полагая, что текучесть тротила и тетрила (см. табл. 1) соответст-
вует текучести ВВ-2 (см. фиг. 5), легко объяснить нечувствитель-
ность этих ВВ в приборе № 2. Допущение, что текучесть стифната
свинца, пироксилина, смеси гексогена с алюминиевой пудрой и дру-
гими ВВ (см. табл. 1 и 3) соответствует текучести ВВ-3, объясняет
меньшую чувствительность этих ВВ в приборе № 1 по сравнению
с чувствительностью в приборе № 2.
Таким образом, разная чувствительность ВВ в приборах № 1
и 2 объясняется различной скоростью выделения энергии при де-
формации зарядов в этих приборах, а не схоластической «избира-
тельной чувствительностью ВВ к удару и к трению», как это пред-
ставляют некоторые исследователи.
1 Для определения величины напряжений нужно из центра кружка опустить
перпендикуляр на ось Р.
2 За исключением кружка, отвечающего напряжениям в приборе X® За.
В последнем величина напряжений существенно не зависит от текучести ВВ.
18
ОБЩИЕ ВЫВОДЫ
1. Экспериментальным путем установлено, что в случаях малой
скорости течения заряда взрыв не возбуждается даже и при очень
больших напряжениях. На основании этого можно считать, что в о л-
Суждение взрыва обусловлено течением заряда.
2, Эффективность действия механических сил при течении за-
ряда определяется удельной скоростью выделения механической
йергин, которая зависит от коэффициента внутреннего (и внеш-
его) трения и градиента скорости течения.
3. При постоянной энергии удара максимальная скорость выде-
ения энергии имеет место при некоторой оптимальной величине
1апряжений, которую можно достигнуть путем применения соответ-
твующей конструкции прибора.
4. Широко распространенные представления о критических на-
ряжениях ВВ противоречат опыту и, следовательно, являются
шибочными. Основанные на этих ошибочных представлениях ис-
ледования устойчивости заряда при выстреле оказались поэтому
есплодными.
5. Понятие об «избирательной» чувствительности ВВ «к удару»
: «к трению» — ошибочно, так как возбуждение взрыва опреде-
яется течением заряда, которое характеризуется коэффициентом
рения и градиентам скорости течениям
6. Ошибочным также является понятие о том, что прибор № I
характеризует чувствительность «к удару», а прибор № 2—-«к тре-
хию». Всевозможные способы механического воздействия (незави-
имо от конструкции прибора) могут характеризовать чувствитель-
юсть только при течении заряда. Однако коэффициент трения и
радиент скорости течения могут сильно изменяться в зависимости
>т способа механического воздействия.
7. Представление о том, что относительная чувствительность в
[риборе № 1 является наиболее надежным критерием относительной
«устойчивости» и безопасности заряда является ошибочным и пре-
пятствует развитию способов изучения чувствительности ВВ.
ЛИТЕРАТУРА
1. Баум Ф. А., Известия Артиллерийской академии‘РККА, XXVI, 1939.
2. Брунсвиг Г., Теория взрывчатых веществ, перевод с немецкого, М.—Л.,
Госхимтехиздат, 1932, стр. 27 и 35.
3. Вен нен Л,, Б ю р л о Э. и Лекорше А., Пороха и взрывчатые веще-
ства, перевод с французского, ОНТИ, 1936, стр. 98.
4. Дубинин С., Известия Артиллерийской академии РККА, XXIX, 1940.
5. Каст Г., Взрывчатые вещества и средства воспламенения, перевод с не-
мецкого, М.—Л., Госхимтехиздат, 1932, стр, 27,
6. Р я б и н и н Ю, И., Ж ФХ, 1946, 20, № 11.
7, Р я б и и и и Ю. Н., ДАН СССР, 1947, 58, № 2.
8, Харитон Ю, Б,, Сборник статей по теории ВВ, Оборонгиз, 1940,
стр, 177,
9. X о л е в о Н, А., Труды Казанского химико-технологического института
им. С. М. Кирова, 10, 1946.
2*
19
10. Холево Н. А., Труды Казанского хн мико-технологического института
им. С. М. Кирова, 11, 1947.
11. Bridgman Р., J. chetn. Phys., 1947,15. N 5.
12. Е g g е г t J., Chem. Zfg., 1920, 44.
13. TammanG., Kroger C.,Z. anorg. Chem., 1928, 189.
14. Ju s i ro w. 7 ges. Schiess- und Sprengstoll, 1922, 17. :
Ф. БОУДЕН
2. ПОСЛЕДНИЕ ИССЛЕДОВАНИЯ РАЗЛОЖЕНИЯ
ТВЕРДЫХ ВВ1
Взрыв твердых ВВ может быть вызван:
нагреванием до температуры воспламенения;
светом достаточной интенсивности;
ударом или толчком;
ультразвуковыми колебаниями;
действием электронов, а-частиц, нейтронов и осколков деления
ядра;
электрическим разрядом;
«самопроизвольным» мгновенным разложением растущего кри-
сталла.
В этом докладе рассмотрены вопросы, относящиеся к возбуж-
дению взрыва твердых ВВ тремя первыми видами воздействия.
ВОЗБУЖДЕНИЕ ВЗРЫВА НАГРЕВАНИЕМ
Исследователи лаборатории Кембриджского университета при-
держиваются, как и ученые СССР, того взгляда, что взрыв чаще
всего возникает от разогрева взрывчатого вещества. Условием его
возникновения является выделение тепла со скоростью, превыша-
ющей скорость его отвода, Теория этого сложного явления разра-
ботана в СССР Д. А. Франк-Каменецким. Согласно исследованиям,
проведенным в Кембриджском университете, при длительности ра-
зогрева порядка нескольких микросекунд размер нагретой области
находится в пределах 10-3—10~® см.
Можно показать на простом примере, что для возникновения
взрыва необходимо, чтобы определяющий размер кристалла был
не ниже некоторой критической величины. Если поместить кристал-
лы азида кадмия в термостат, имеющий температуру 320°, то только
кристаллы толщиной 10"3 см взрываются, кристаллы тоньше 10"® см
разлагаются без взрыва. Критическая толщина кристалла зависит
от температуры, как это и следует из теории Франк-Каменецкого.
Изучалось также термическое разложение кристаллов ВВ, про-
исходящее ниже температуры самовоспламенения. Для этой цели
г Настоящая статья является изложением доклада, прочитанного проф-
ессором Кембриджского университета Ф. Боуденом а МХТИ им. Менделеева
в марте 1959 г. Статья подготовлена к печати Б. Н. Кондриковым.
20
^применялся электронный микроскоп высокой разрешающей спо-
'собности, достигавшей для того микроскопа, которым располагала
лаборатория 5А. Это уже почти атомный размер. Проф. Ментером
была предпринята попытка обнаружить при помощи электронного
микроскопа молекулы в кристалле. Он исследовал под микроскопом
кристалл фталоцианина платины и получил картину, представлен-
ную на фиг. 1. Полоски —это ряды молекул, из которых построен
кристалл, расстояние между ними 12 А. Видно, что в одной области
Фиг. 1. Электронная микрофотография кристалла фталоцианина платины. (Уве-
личено приблизительно в 1 млн. раз; видны ряды молекул в кристалле; расстоя-
ние между атомными плоскостями 11,9А).
а—участок поверхности кристалла без дефектов, б—участок поверхности с дислокацией.
кристалл является совершенным. В других областях, однако, можно
обнаружить много различных дислокаций. Мы полагаем, что хими-
ческое превращение возникает предпочтительно на дислокациях.
Кристалл азида серебра при исследовании в электронном микро-
скопе (фиг. 2 и 3) разлагается в результате разогрева его потоком
электронов. На поверхности кристалла, на которой преимущест-
венно идет разложение, выделяются комочки серебра. На фиг. 2
зародыши заметны слабо. На фиг. За размер зародышей изменился
незначительно, а контрастность — очень заметно. На увеличенном
снимке (фиг. 36) видны зародыши и сетка серебра.
На фиг. 4 изображены лауэграммы кристалла азида серебра,
разлагающегося под действием потока электронов электронной ди-
фракционной камеры. На фиг. 4а показана электронограмма кри-
сталла азида в начале опыта. Уже на этой стадий можно видеть, что
азид начал разлагаться. На фиг. 46 относительные интенсивности
пятен несколько изменились. Изменение интенсивности еще отчет-
ливее заметно на фиг. 4в, некоторые пятна здесь очень ярки, но по-
ложение их пока не меняется. Интенсивность колец, характерных
для электронограммы металлического серебра, возрастает.
Следует отметить, что расстояние между атомами серебра в ази-
де примерно вдвое больше, чем в самом серебре, в связи с чем
по мере .разложения азида атомы серебра сближаются. После уда-
ления всего азота (фиг. 4д) они занимают нормальные, свойствен-
ные металлическому серебру положения.
Электронный микроскоп в сочетании с электронографией по на-
шему мнению является весьма важным инструментом для изучения
Фиг. 2. Электронная микрофотографий кристалла
а.т,»ла серебра в начале разложения, в момент, со-
ответствующий фиг. 4а,
разложения кристаллов. Электронный луч здесь одновременно при-
водит к разложению кристалла, сильно нагревая его, и позволяет
следить за ходом этого разложения (см. фиг. 5), В ряде случаев
такое совмещение может оказаться неудобным. Поэтому мы приме-
няли для исследования разложения кристаллов также растровый
электронный микроскоп, в котором электронный луч, отражающий-
ся от поверхности кристалла, обегает ее, подобно тому, как это про-
исходит в кинескопах, В этом случае мощность луча недостаточна
для того, чтобы разогреть кристалл и вызвать разложение. Такая
конструкция была разработана в Кембридже Смитом и Уэтли.
На фиг, 6 изображена иголочка азида серебра толшиной около 1 мк,
находящаяся на нагретой серебряной пластинке. Разложение на-
чалось в дефектах кристаллической решетки с того конца иголки,
который имеет хороший контакт с пластинкой. В результате кри-
сталл раздробился на мелкие кусочки, размер которых не превы-
шает 0,3 мк. В конце концов весь кристалл превращается во множе-
ство угловатых комочков или узелков серебра (фиг. 7),
Растрескивание и дробление кристаллов при нагревании наблю-
дается и в случае других ВВ. На фиг. 8 изображен кристалл азида
22
кадмия перед (а) ц после нагревания при 317" (б). Отчетливо вид-
но, что при нагревании кристалл растрескивается и разламывается
вАрль кристаллографических плоскостей. По нашему мнению, согла-
сующемуся с теми соображениями, которые были высказаны не-
Фиг. 3. Электронная микрофотография кристалла
азида серебра, изображенного на фиг. 2 в момент,
соответствующий фиг. 4г.
давно проф, К. К, Андреевым, дробление кристалла при нагревании
имеет большое значение для развития взрыва в нем. На фиг. 9й
изображен кристалл тэна, подвергнутый действию мощного источ-
ника света, а иа фиг. 96 тот же кристалл перед освещением. Взрыва
при этом не происходит, но весь кристалл покрывается множеством
трещин. Поэтому можно сделать вывод, что разложение кристаллов
ВВ начинается с поверхности в дислокациях и дефектах кристалли-
ческой решетки и приводит к раскалыванию кристалла на кусочки.
23
Фиг. 4. Разложение азида
серебра потоком электро-
нов. (Электронограмма
кристалла азида се-
ребра на различных ста-
диях разложения).
24
Фиг. 5. Электронная микрофотография кристалла нитрата ка-
лия, оплавляющегося под действием пучка электронов.
Фяг. 6, Частично разложившаяся фиг. 7. Иголочка азида серебра, раз-
И голочка азида серебра. рушившаяся в результате разложения
и кристаллографического фазового
превращения при ISO".
25
Фиг. 8. Кристалл азида кадмия перед (а) и после (б) нагревания
при 317° (увеличено в 440 раз).
Фиг. 9. Разложение и растрескивание монокристалла тэиа при действии
на него импульса света (энергия 900 дж. продолжительность 1,2 мсек.).
л—кристалл после действия светового импульса, б—первоначальный вид кристалла.
ВОЗБУЖДЕНИЕ ВЗРЫВА СВЕТОМ
Целый ряд работ, проведенных в Швейцарии и Кембридже, по-
казывает, что кристаллы ВВ могут разлагаться под действием света.
Если интенсивность источника света достаточно велика, то некото-
рые из особо чувствительных соединений взрываются. Кристалли-
ческие вторичные ВВ, в основном являющиеся ковалентными орга-
ническими соединениями, нс взрываются даже при энергии
2000 дж.
Эта форма возбуждения взрывчатого превращения интересна в
частности потому, что при малой продолжительности освещения
Фиг. 10. Схема установки для исследования воз-
буждения вэрь|ва светом.
J—нм пульса ;i я лампа, ложечка с ВВ. 3—держатель.
На схеме указаны размеры ячейки актинометра и ее
расположение (пунктир).
(а в наших опытах она составляла около 1 мсек) свет поглощается
тонким поверхностным слоем, в котором и начинается разложение,
более глубокие слои действием света не затрагиваются, в резуль-
тате чего удается избежать многих осложнений, сопровождающих
возбуждение взрыва теплом или ударом.
Некоторые результаты работ по действию света на кристал-
лы ВВ могут быть использованы при рассмотрении вопросов, свя-
занных с распространением взрыва и детонационными явлениями.
Более или менее подробно изучено лишь несколько веществ.
В их числе азид серебра, азид таллия, нитрид серебра, иодистый
азот и стифнат свинца. Наиболее детально исследовался азид
серебра.
Схема установки для исследования возбуждения взрыва светом
представлена на фиг. 10. Опыты показывают, что для возбуждения
взрыва требуется некоторое критическое количество лучистой энер-
гии. По-видимому, процесс возбуждения является фотохимическим:
нон азида под действием света превращается в радикал N3. Если
27
два радикала встречаются, то происходит сильно экзотермическая
реакция:
2N3 10 ккил.
Для того чтобы эта реакция прошла, нужно, во-первых, получить
два азидных радикала и, во-вторых, нужно, чтобы они встретились.
Если к ВВ примешать частички металла, то эти частички могут
действовать как ловушки электронов и будут отбирать электроны
у иона N", способствуя превращению его в радикал. Влияние этих
частиц на величину критической энергии светового импульса иллю-
Са&ерлгание золота 8 смеси с азиЗом$%вее.
Фиг. II. Увеличение чувствительности азида сереб-
ра к свету при добавлении частиц золота.
1—чувствительность азида с инертным разбавителем (рас-
чет), 2—чувствительность его по результатам опытов
с добавлением золота, Vc—критическое напряжение из
обкладках конденсатора в кв. (Световой импульс — искра
в воздухе от двух соединенных параллельно конденса-
торов емкостью по 8 лкф каждый).
стрирует фиг. 11. Действительно, добавление золота снижает крити-
ческую энергию, вместо того чтобы повышать ее за счет разбав-
ления вещества, так, как это следует из сопоставления кривых 1 и 2
фиг. 11.
Отметим также, что взрыв азидов под действием света часто со-
провождается расплавлением поверхностного слоя; естественно, что
реакции превращения иона в радикал и экзотермического взаимо-
действия двух радикалов в жидкой фазе проходят гораздо быст-
рее и легче, чем в твердой.
В случае ковалентных азидов и металлоорганических соедине-
ний, например, стифната свинца, первичный процесс, по-видимому,
не является фотохимической реакцией. Лучистая энергия в конеч-
ном счете превращается в тепловую, и взрыв развивается по нор-
мальному тепловому механизму. Разложение таких соединений ча-
сто происходит по свободно-радикальному механизму.
Если рассматривать ряд ионных неорганических азидов — KNs,
T1N3, AgNs, CuN3, мы обнаружим, что устойчивость по отношению
к действию тепла и света уменьшается от первого к последнему.
KNs—относительно устойчивое твердое вещество, в то время как
AgN3 легко разлагается и в некоторых условиях взрывается и дето-
28
нирует. Разность потенциала ионизации металла и сродства к элек-
трону азидного радикала в этом ряду возрастает. Было показано,
что параллельно изменяются и некоторые физические свойства. Из-
менения структуры, спектра поглощения, фотоэлектрической про-
водимости, показателя преломления, точки плавления указывают на
то, что по мере увеличения потенциала ионизации металла происхо-
дит отклонение от ионного типа решетки. Можно показать также,
что энергия, необходимая для того, чтобы возбудить электрон и
перевести его из полосы валентности в полосу проводимости, умень-
шается, что определяет скорость разложения азидов тяжелых ме-
таллов (T1N3, AgN3 и CuN3) и частично объясняет их неустойчи-
вость.
ВОЗБУЖДЕНИЕ ВЗРЫВА УДАРОМ
В основе возбуждения взрыва ударом как правило лежит про-
цесс разогрева вещества. Пути возникновения этого разогрева мо-
гут быть различными:
адиабатическое сжатие газовых включений;
разогрев при трении между поверхностями, ограничивающими
заряд, или между твердыми частицами в нем;
трение между кристалликами самого ВВ;
вязкостный разогрев ВВ при высоких скоростях сдвига;
' нагревание острого ударника при пластической деформации;
взаимодействие слабых ударных волн.
Мы рассмотрим лишь некоторые из этих путей.
Наиболее опасным в отношении возбуждения взрыва ударом яв-
, ляется присутствие в жидких, а также в некоторых твердых ВВ
небольшого количества воздуха в виде мелких пузырьков. Если для
' возбуждения взрыва нитроглицерина, свободного от пузырьков
, воздуха, необходима энергия порядка 10s—10е гсл, то при наличии
- - маленького пузырька диаметром 0,1 мм для возбуждения взры-
?’, ва достаточно энергии удара порядка 20—100 гем.
Воздух, находящийся в пузырьках, можно заменить другим
। каким-либо газом, при этом энергия удара, необходимая для воз-
буждения взрыва, будет изменяться, по-видимому, в меру измене-
ния отношения теплоемкостей этого газа при постоянном давлении
; и постоянном объеме ср/с„.
Наличие в жидком взрывчатом веществе пузырьков воздуха
, Способствует возбуждёнию вз рыва ударной " волной: - Интересные
^Ёдыты-были-проведены недавнб"Уиннингом. ОТГвзрывал заряд ни-
' .троглицерина (см. 'фиг. 12) рядом с трубкой, содержащей нитро-
глицерин с введенным в него пузырьком воздуха. Видно, что взрыв
в трубке возникает именно там, где находится этот пузырек.
При трении двух поверхностей с заключенным между ними ВВ
; причиной воспламенения также являются очаги разогрева. На
фиг. 13 представлена осциллограмма, полученная при возбуждении
взрыва трением. Взрыв возникает в тот момент, когда кривая резко
падает вниз. (На фиг. 13 этот момент отмечен вертикальной
29
Фиг. 12. Возбуждение взрыва нитроглицерина ударной волной в воде
(по Уинниигу).
Л—трубка с нитроглицерином, Б—пассивный заряд нитроглицерина с ма-
леньким пузырьком воздуха н нижней части, <а) н (б)—система перед взры-
вом и в момент его.
Фиг. 13. Запись катодным осциллографом термоэлектро-
движущей силы при скольжении ползуна из константана
по стальной поверхности, покрытой слоем нитроглицерина.
Слева масштаб температур в °C.
30
линией А). Если температура не достигает 400°, то взрыва
не наблюдается.
Интересны также опыты по возбуждению взрыва трением в при-
сутствии твердых частиц. Мы обнаружили, что важным свойством
такой частицы является ее температура плавления. Если она ниже
500°, то взрыва не происходит. Из приведенной ниже таблицы вид-
Таблица
Возбуждение взрыва тзна трением в присутствии примесей
|"' Примесь 11 Твердость по шкале Мооса * пл ас Частость взрывов
7 гл Без примесей 1,8 141 0
Азотнокислый аммоний 2-3 169 0
Бисульфат калия 3 210 0
Азотнокислое серебро 2—*3 212 0
Двухромовокислый натрий 2—3 320 0
Уксуснокислый натрий 1-5 324 0
Азотнокислый калий 2—3 334 0
Двухромозокислый калий 2—3 398 0
Бромистое серебро 2—3 434 50
.Хлористый свинец 2-3 501 60
. Йодистое серебро 2—3 550 100
Бура 3—4 560 100
Висмутит 2—2,5 685 100
. Стекло 7 800 100
. ; Каменная соль 2—2,5 804 50
. Медный блеск 3—3,5 1100 100
\ Свинцовый блеск 2,5—2,7 1114 100
кальцит 3 1339 100.
1ио, что вещества, обладающие различной твердостью, но близкими
Уймпературамн плавления оказывают в данных опытах приблизи-
тельно одинаковое действие.
fi Взрьгв циануртриазида может быть возбужден ударом металли-
ЯВЬскои иглой по наковальне, на которой он находится. Острый конец
Йглы при этом деформируется и нагревается, причем взрыв проис-
ХЪДйт при температуре около 300—350°. На фиг. 14 представлены
осциллограммы, полученные в опытах, в которых ударник с иглой
, из вольфрама бьет по стальной наковальне.
При энергии удара 1100 гем (фиг. 14а) максимальный подъем
температуры (250°) недостаточен для того, чтобы вызвать взрыв
,i
31
циануртриазида при начальной его температуре 95°. При дости-
жении температуры 300—350° (энергия удара 3300 гем - фиг. 146)
взрывы этого вещества происходят с частостью 100%.
В заключение рассмотрим вопрос о взаимном усилении слабых
ударных волн. На фиг. 15 представлены фотографии, полученные
в опытах Уиннинга по возбуждению взрыва нитроглицерина удар-
ной волной. Хорошо видно, что две или три ударные волны от актив-
Фнг. 14. Запись катодным осциллографом
подъема температуры при ударе вольфра-
мовой иглой по стальной наковальне.
о—разогрев до 250°, нет взрыва; б—разогрев до
300—350°, частость взрывов 100*/*.
ных зарядов совместно возбуждают взрыв пассивного заряда, в то
время как одна волна не может возбудить его.
В настоящее время в Кембриджском университете проводится
работа по изучению взрыва в отдельном кристалле. Те деликатные
методы, о которых было рассказано в начале доклада, могут быть
применены при изучении не только медленных, но и мгновенных
процессов. Это очень увлекательная область исследования, так как
в одном и том же кристалле можно наблюдать медленное разло-
жение и превращение, распространягогцееся со скоростью 7 км1сек.
Очень интересно установить, какая связь существует между медлен-
ным разложением и быстрыми процессами. Дальнейшее усовершен-
ствование аппаратуры и методов исследования могло бы внести
реальный вклад в развитие этой интересной области науки о взрыв-
чатых веществах. '
32
Фиг. 15. Возникновение взрыва при сложении ударных волн, распро-
страняющихся в воде (по Уиннингу).
Лг. Xj. Л], Л<—пассивные заряды нитроглицерина, Б—активные заряды, а)—систе-
ма перед взрывам, б)—системз в момент взрыва, в)—система в момент взрыва
при уменьшенных расстояниях между активными и пассивными зарядами.
3 451
33
, ВЫСТУПЛЕНИЯ ПО ДОКЛАДУ Ф. БОУДЕНА
Ледин Е. Е
Работы проф. Боудена являются новыми, они направлены на
изучение механизма возникновения взрыва, и с этой точки зрения
его доклад так же как и книга, переведенная на русский язык, пред-
ставляют безусловный интерес. Проф. Боуден рассказал нам о тон-
ких экспериментальных исследованиях с применением очень инте-
ресных методов, позволяющих изучать взрыв в его самой началь-
ной стадии. Те исследования, о которых рассказал проф. Боуден и
которые опубликованы в его монографии, являются новым вкладом
в теорию возникновения взрыва, и нам надо приветствовать проф.
Боудена, как принесшего это новое.
При положительном впечатлении от книги и доклада нельзя не
отметить некоторые особенности, которые кажутся мне очень су-
щественными. Проф. Боуден стоит на позициях теплового возбуж-
дения взрыва. Существо этого предположения заключается в том,
что возбуждение взрыва возникает только после того, как взрывча-
тое вещество приобретет достаточную температуру. Положение,
лежащее в основе тепловой теории, предполагает переход от меха-
нического воздействия на ВВ к химической реакции через разогрев,
который обязательно по этой теории должен предшествовать про-
цессу разложения. Это основное положение и является, на мой
взгляд, слабым местом теории. Окольные пути в природе не обяза-
тельны. Между возникновением взрыва и его продолжением не
должно быть разрыва, иначе трудно правильно о&ьяснить процесс
разложения; с другой стороны трудно представить себе универсаль-
ность температуры 500°, которая является условием взрыва, а не
его следствием. Получается как будто ВВ ждет, когда его нагреют
до 500°.
В условиях сжатия происходит взаимодействие между зарядом
и окружающей средой. Оно выражается в том, что вся система при-
ходит в напряженное состояние. Доказано однозначно, что это на-
пряженное состояние приводит к изменению не только внутрикри-
сталлического состояния, но и внутримолекулярного. Имеются вну-
тренние зависимости, которые выражают эту связь. Возьмем самый
простой пример: известно, что если есть простая связь ,
то межатомное расстояние г«1,7А; если же будет двойная связь
/С = С\ , то 1,4А; при тройной связи — С =С — имеем г=> 1,2А.
Когда атомы углерода сближаются между собой, то при этом воз-
никают внутренние связи между ними и теряются внешние. Надо
сказать, что энергия, расходуемая на изменение расстояния не есть
энергия активации—-это есть энергия связи, которую надо рассмат-
ривать отдельно и которая в конечном итоге приводит к разложе-
нию. Температура системы будет иметь значение, она определяет ее
чувствительность, но при механическом воздействии система изме-
34
. (Имтся в первую очередь вследствие изменения напряженности и ста-
бильности химической связи. Нельзя себе представлять, что система,
будучи нагружена, не изменяется химически, ее температура не
.Может поэтому играть определяющей роли в возникновении взрыва.
Уже постоянство критической температуры (5ОСГ) для большинства
веществ говорит о том, что эта температура или связана с методи-
кой, потому что она не является индивидуальной, или с процессом
.распространения, но отнюдь не с возникновением взрыва.
По тепловой теории схема возникновения взрыва такова: воз-
i Действие—температура—разложение (В—Т—Р); более вероятен
Другой механизм этого процесса: воздействие—разложение—темпе-
ратура (В—Р—Т). Температура является следствием, а не причн-
-Дой возникновения взрывчатого превращения.
V
. Андреев К. К-
Известно, что при ударе по взрывчатому веществу оно может
: ДЙюрваться. Вопрос о том, каким образом механическое воздействие
^Вызывает химическую реакцию, приводящую к взрыву, представ-
ит большой интерес. Исследования Бриджмэна, Рябинина, Боу-
и других исследователей показали, что даже очень высокие
'^.давления при статическом их приложении не вызывают взрыва и
^существенного ускорения химического превращения. Отсюда сле-
,;^ет, что те напряжения, которые при сжатии ударом создаются
УД0. взрывчатом веществе, сами по себе не вызывают взрыва. Есте-
ственно было поэтому предположить, что в динамических условиях
^действия оно может привести к разогревай ВВ, от которых и
'^извивается взрыв.
Какими путями могут возникать разогревы во взрывчатом веще-
ук-Йве при ударе? Боуденом и его сотрудниками многочисленными
. ЧЯ^роумными опытами показано, что, по крайней мере в случае жид-
'тГСтей, присутствие в ВВ газовых пузырьков может приводить к
. 'Шомному увеличению их чувствительности при ударе; взрывы воз-
'-/«ЙКают при несравненно меньшей энергии удара, чем в отсутствии
1И(Эырьков.
Л*;/Этому наблюдению исследователи дают естественное объясне-
У16, заключающееся в том, что пузырьки воздуха при ударе быстро
Ужимаются, сильно разогреваются и поджигают взрывчатое-веще-
Вопрос о том, как осуществляется это поджигание^ нельзя,
•$фнако, считать выясненным. По-видимому, здесь существенную
’WWb ИгРают условия теплообмена и массообмена между нагретым
.-^Уйырьком и окружающим его ВВ.
7^,. Известно, что если поджечь нитроглицерин при пониженном
‘Давлении в трубочке диаметром 3—4 мм и, ведя опыт в замкнутом
Объеме, дать давлению повышаться, то при 300—400 мм рт. ст. горе-
ИЧе ватухает вследствие возникшей турбулизации жидкости, кото-
.. так сильно охлаждает газы, что горение не может распростра*
уИЯТься. Усиленное поступление нитроглицерина в зону горения
^йЯОДШит его. При больших давлениях это же явление может приводить
не к затуханию, а к возникновению детонации, а при дальнейшем
повышении давления может вновь наблюдаться горение, но со зна-
чительно большей скоростью. Опыты шведского исследователя
Йоханссона показали, что аналогичные явления наблюдаются,
когда жидкость, содержащая пузырьки газа, подвергается удару.
Существенны далее условия течения процесса, которые отража-
ются на массообмене и передаче тепла от нагретого газа в ВВ и на
возможном его дополнительном разогреве в результате трения. По*
этому адиабатический разогрев газового пузырька не является
единственным фактором, который определяет исход опыта.
Из опытов, показывающих отсутствие заметного влияния давле-
ния на возникновение вспышки, нельзя делать заключение, что это
давление не может существенно влиять на возникновение и рас-
пространение взрыва при ударе. Происходит ли взрывная реакция
в форме горения, как это согласно исследованиям Боудена обычно
бывает вначале, или в форме взрыва — в обоих случаях давление
играет очень важную роль. Критический диаметр или размер, начи-
ная с которого распространение взрывчатого превращения стано-
вится возможным, сильно зависит от давления. Резкое понижение
давления при горении, как правило, приводит к его затуханию. По-
этому величина давления и длительность его существования явля-
ются важными факторами возникновения и развития взрыва при
ударе.
Разогрев пузырьков газа не является единственным путем, по
которому механическая энергия при ударе может переходить в тепло.
По-видимому, и в условиях удара на копре и во многих других слу-
чаях, решающее значение имеет течение ВВ и возникающие при
этом местные разогревы вследствие внутреннего трения. Эти пути
возникновения взрыва были изучены в исследованиях Н. А. Хо-
лево, показавшего, что во многих случаях при ударе взрыв возни-
кает, если есть возможность течения ВВ, и он не происходит, если
эта возможность отсутствует. Ряд парадоксальных явлений — влия-
ние инертных примесей, увеличивающих текучесть и чувствитель-
ность труднотекучих ВВ и уменьшающих чувствительность легко-
текучих, зависимость результатов удара от наличия канавки в муфте
прибора, повышение частости взрыва при понижении температуры
и другие явления естественно объясняются, исходя из основного
представления Н. А. Холево—нет течения вещества, нет и взрыва.
Опубликованные в 1958 г. работы немецких и шведских иссле-
дователей приводят их к тем же выводам о роли течения ВВ при
возбуждении взрыва ударом, которые десятью годами раньше были
сделаны Н. А. Холево. По-видимому, этот путь возникновения разо-
грева при ударе практически наиболее важен.
Большой интерес представляют исследования проф. Боудена по
горению и взрыву отдельных кристаллов взрывчатого вещества.
Макропроцесс горения, закономерности которого нас непосредст-
венно интересуют, представляет собой совокупность процессов в от-
дельных кристаллах в сочетании с взаимным их влиянием. Изучение
элементарных процессов несомненно поможет пониманию законо-
36
мерностей макропроцесса. Особенно интересным представляется
наблюдавшееся Боуденом самопроизвольное дробление кристаллов;
оно происходит даже при медленном разложении их и несомненно
играет существенную роль при горении и переходе его во взрыв.
Действительно это последнее явление несомненно связано с рез-
ким ускорением процесса. Исследования показали, что давление
само по себе относительно слабо увеличивает скорость горения.
Поэтому наиболее вероятным путем ускорения газообразования и
роста давления при горении является увеличение горящей поверх-
ности, которое и может возникать в результате интенсивной фраг-
ментации горящих частиц.
Изучение путей диспергирования является одной из очень инте-
ресных задач теории взрыва и мы надеемся, что исследования лабо-
ратории проф. Боудена внесут и в будущем много нового в этот
вопрос.
К. К, АНДРЕЕВ
3. к ВОПРОСУ О ФАКТОРАХ, ОБУСЛОВЛИВАЮЩИХ
ВОЗНИКНОВЕНИЕ ВЗРЫВА ПРИ УДАРЕ И ТРЕНИИ
( ИО МЕТОДАХ ОЦЕНКИ ЧУВСТВИТЕЛЬНОСТИ ВВ
К МЕХАНИЧЕСКИМ ВОЗДЕЙСТВИЯМ
О ФАКТОРАХ, ОБУСЛОВЛИВАЮЩИХ ВОЗНИКНОВЕНИЕ ВЗРЫВА
ПРИ УДАРЕ И ТРЕНИИ
Взрыв ВВ может быть вызван различными воздействиями, в
частности нагреванием, ударом, трением. Чувствительность взрыв-
чатого вещества к каждому из этих воздействий является важной
^Го характеристикой, ограничивающей зачастую область примене-
<1Шя и определяющей условия производства ВВ. Мерой чувствитель-
ности к данному воздействию являются очевидно определенные кри-
тические показатели последнего, переход через которые приводит
К взрыву. Однако ответ на вопрос о том, какие именно показатели
того или иного воздействия могут служить такой мерой, далеко не
-всегда прост и ясен, если неизвестен механизм возбуждения взрыва.
'Поясним это примером. Можно было предполагать, что чувстви-
тельность к тепловым воздействиям характеризуется некоторой
Критической для данного ВВ температурой [3], подобной темпера-
туре плавления. Теория теплового возникновения взрыва, в прин-
ципе приложимая и к конденсированным ВВ, показала, однако, что
не только температура, но и размеры нагретого заряда определяют
, возможность возникновения взрывчатого превращения. При более
же полном описании явления необходимо учитывать и длительность
нагрева, так как для многих ВВ реакция разложения даже в изотер-
мических условиях ее протекания является самоускоряющейся. При
экспериментальном определении для одного и того же ВВ могут.
вообще говоря, быть получены сильно различающиеся значения тем-
пературы вспышки. Причины этих различий однако, могут быть, как
правило, объяснены на основе изложенных выше соображений;
если учитывать мх при постановке опытов, то значения температуры
вспышки (а также и ее задержки) достаточно хорошо воспроизво-
димы и разброс экспериментальных данных относительно невелик.
Поэтому характеристика чувствительности ВВ к тепловым воздей-
ствиям в ее простейшем виде (температура вспышки) является до-
статочно определенной по величине и физическому смыслу.
Существенно иначе обстоит дело с чувствительностью к механи-
ческим воздействиям, в частности к удару, Результаты эксперимен-
тального определения в обычном их выражении (частость взрывов
при данной энергии удара) весьма сильно различаются у разных
исследователей по абсолютному значению; при этом изменение ча-
стости— от отсутствия взрывов до отсутствия отказов — может
происходить в весьма значительном интервале изменения энергии
удара, соизмеримом с ее абсолютным значением.
В связи с этим даже в лабораторных строго контролируемых ус-
ловиях определение чувствительности ВВ к удару построено на ста-
тистическом принципе, т, е. на определении частости взрывов, хотя
можно было бы представить себе подобную температуре вспышки
критическую высоту (энергию) или иной критический параметр
удара. При таком определении даже и у одного и того же исследо-
вателя колебания результатов параллельных серий опытов (обычно
каждая серия включает 25 опытов) нередко бывают весьма значи-
тельны; это наблюдается несмотря на то, что условия опыта, кото-
рые предположительно могут влиять на результат, поддерживаются
постоянными в пределах возможностей экспериментатора.
Такая относительно низкая определенность чувствительности ВВ
к удару обусловлена, по-видимому, рядом причин. Одной из этих
причин является сложность явления, во всяком случае гораздо боль-
шая, чем при тепловом возбуждении взрыва, которое в своеобраз-
ной форме является лишь одной из стадий возбуждения взрыва уда-
ром. Сложность явления выражается и в том, что на отдельные
стадии его развития одни и те же факторы могут иногда влиять по-
разному, благоприятствуя одним стадиям и подавляя другие.
В зависимости от условий действие отдельных факторов может при
этом меняться не только по величине, но и по направлению. Пояс-
ним сказанное, рассматривая развитие взрыва при ударе в соответ-
ствии с представлениями Н. А. Холево [4].
Как правило, действие удара, возбуждающего взрыв, приводит
к невсестороннему сжатию взрывчатого вещества, вследствие чего
возникает быстрое его течение. По причине неравномерности пос-
леднего возникает трение между слоями взрывчатого вещества 1
1 Возникновение разогревов возможно н в результате внешнего трения ме-
жду ВВ н поверхностью, вдоль которой оно перемешается, если трение между
ними достаточно велико. Трение инородных тугоплавких н твердых частиц, содер-
жащихся в ВВ, друг с другом млн о поверхности, между которыми оно заклю-
чено, тоже может привести к местным разогревая и взрыву.
38
и, в результате этого, местные разогревьи1, наиболее сильные в зо-
’I нах наибольшего градиента скорости течения. Если размеры и тем-
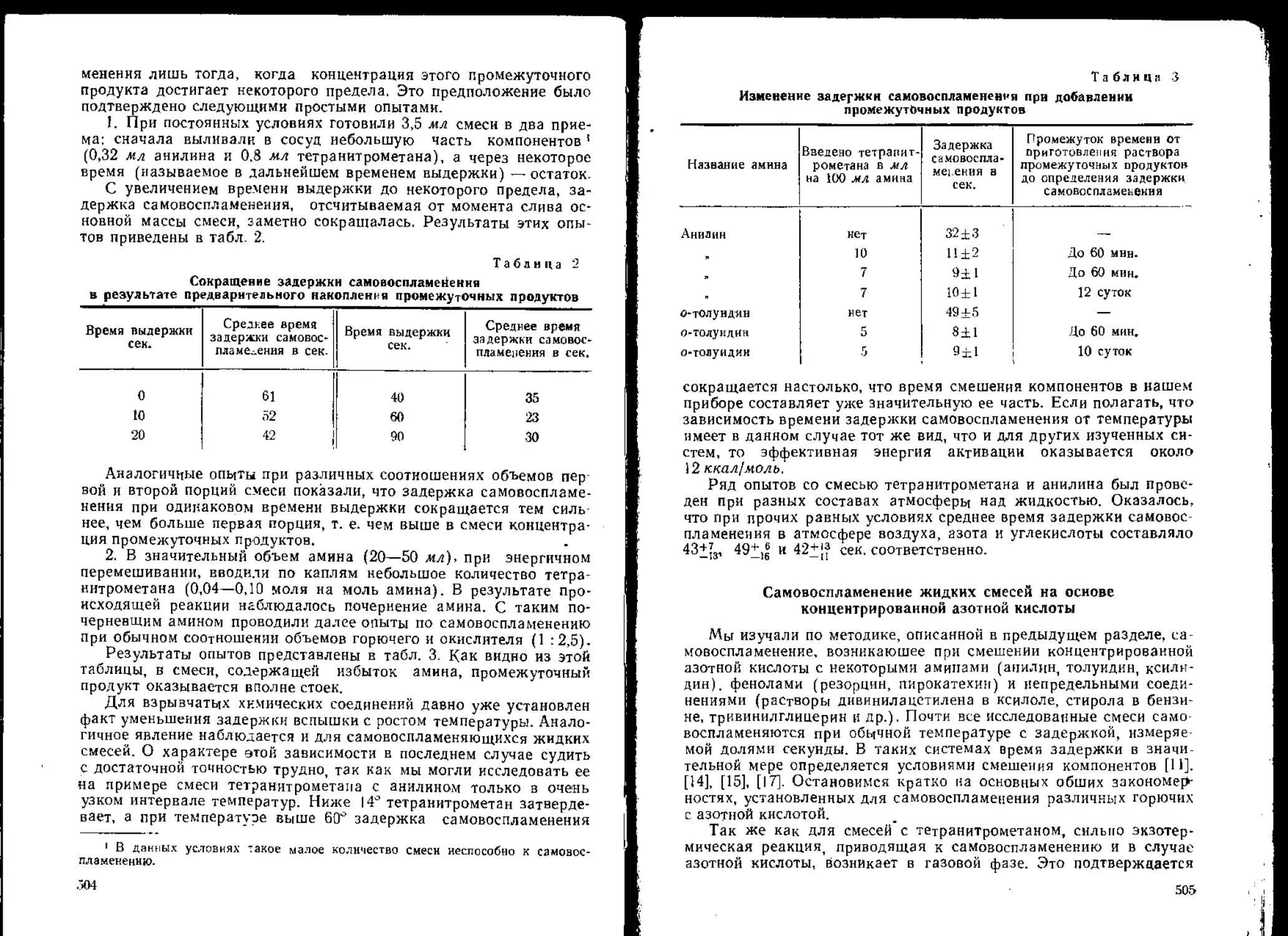
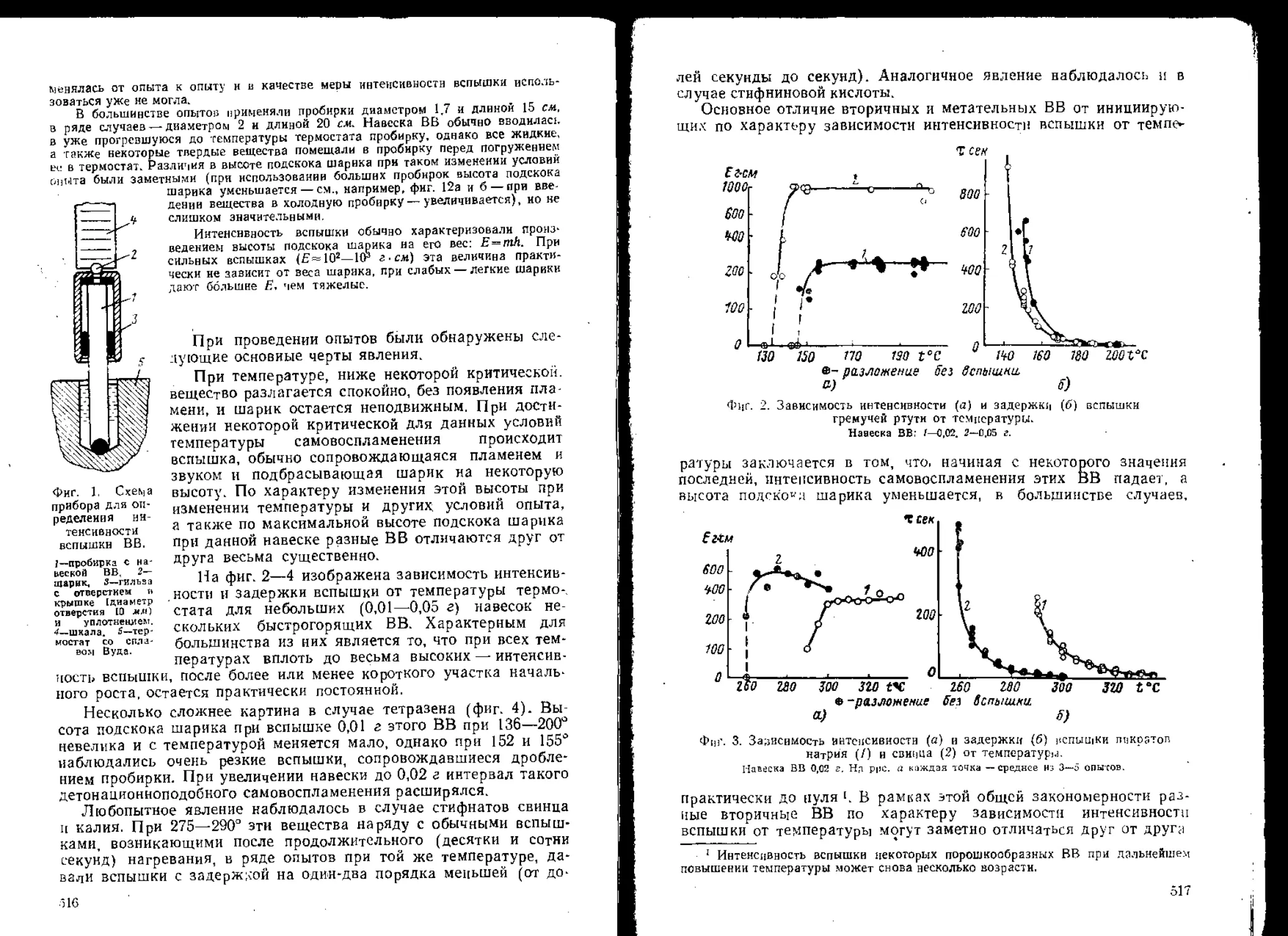
пература очага разогрева достаточно велики, то возникает воспла-
менение и горение, переходящее затем при благоприятных для этого
< условиях'во взрыв. Помимо размеров и температуры очага на воз-
можность возникновения взрыва может влиять также давление:
чем больше давление и чем дольше оно поддерживается, тем легче
воспламеняется ВВ; давление влияет также на переход горения
во взрыв.
Один из источников непостоянства условий и результатов опы-
; тов при ударе, так как он осуществляется на копрах, заключается
в возможной непараллельное™ соударяющихся поверхностей и в
некотором перекосе верхнего ролика, приводящих к тому, что на-
пряжения и скорость течения в разных частях заряда оказываются
2 различными. Помимо этого механически не вполне однородной мо-
жет быть и сама таблетка ВВ. Опыты с инертными пластмассами в
приборе № 2 [4] показали, что при ударе на копре по тонкой цилинд-
- рической таблетке форма ее обычно искажается в радиальном
< Направлении, т. е. деформация происходит неравномерно; далее пе-
С рсход от высот, при которых размеры таблетки не меняются, к высо-
; там, где деформация наблюдается при каждом опыте, требует
^Значительного, более чем вдвое, увеличения высоты падения груза.
цольшое влияние оказывает состояние поверхности роликов. Даже
'jibлабораторных условиях, где контролируется степень механической
.обработки и очистки поверхности, при испытаниях наблюдаются
‘ большие колебания частости взрывов от серии к серии опытов, ко-
Лррые явно выходят за возможные пределы случайных отклонений
'^указывают на систематический их характер.
• и1 Опыты с искусственным нанесением на поверхность роликов не-
Чбольших слоев смазки показали, что частость взрывов гексогена
‘ приборе № 2) при этом возрастает в два и более раза. Впрочем
V;1 Высокая частость взрывов наблюдалась и при некоторых способах
х«^*фомывки роликов, цель которых заключалась в удалении с них
-^Мазки. По-видимому, изменение состояния поверхности металла
Лайняет на адгезионную прочность контакта с ним ВВ. Это в свою
,'$Й®редь меняет профиль скоростей в текущем слое ВВ, притом в
Определенных условиях в сторону повышения возможности возник-
.йовения местных разогревов.
. 2,1 Очевидно, что в производственных условиях способы очистки
обработки поверхностей, соприкасающихся с ВВ, еще в мень-
степени обеспечивают постоянство ее состояния, в связи с чем
' возможны еще большие колебания частости взрывов.
Если испытание ведется в приборе № 1, то решающей, наиболее
Й*йряженной стадией деформации является течение ВВ в зазоре
.'Т?,!1—---
1 То, что решающую роль играют именно микро-, а не макроразогревы. пока-
зывает значительное влияние размеров кристалле^ на частость взрывов даже
. таких, низкоплавких ВВ как нитроглицерин. Если бьГ заряд, подвергающийся
Удару, расплавлялся полностью и затем уже в жидком виде взрывался, то размеры
4>исталлов не могли бы оказывать никакого влияния.
между роликом и муфтой. Величина же этого зазора может ме-
няться вдвое в зависимости от того, занимает ли ролик в муфте
центральное положение или прижат к одной из сторон.
Таким образом в приборе № 2, а еще в большей степени в № 1,
уже первая механическая стадия явления протекает очень непо-
стоянно. Это непостоянство может усиливаться двумя обстоятель-
ствами. Если вещество имеет относительно низкую температуру
плавления, то в тех местах, где начинается течение, происходит ме-
стное плавление, что ведет к резкому повышению текучести В В, так
как расплав служит своего рода смазкой Далее, мы обычно имеем
дело с веществом (порошок или застывший расплав), включающим
газовые (воздушные) пузырьки, которые при ударе сжимаются и
служат очагами повышения температуры, расплавления и местного
усиления течения ВВ z.
Колебания в температуре и размерах очагов разогрева, возника-
ющих при ударе, являются, по-видимому, одной из основных при-
чин непостоянства результатов испытания. Об этом можно судить по
тому, что при гомогенном разогреве значения характеристик воз-
никновения взрыва (температура или задержка вспышки) подвер-
жены гораздо меньшим колебаниям.
При возбуждении взрыва ударом непостоянство условий, в пер-
вую очередь давления, сказывается и на развитии очага воспламе-
нения, Может ли возникший разогрев при данной температуре вы-
звать взрывчатое превращение, во многом зависит от размеров
очага 1 * 3. В больших очагах, при прочих равных условиях, это пре-
вращение развивается легче, чем в маленьких по той общей при-
чине, что генерация энергии реакции, обеспечивающей распростра-
нение последней, происходит в объеме, пропорциональном кубу
линейного размера очага разогрева, а потери тепла — через поверх-
ность, пропорциональную квадрату его.
Число возникающих очагов разогрева зависит от однородности
как самого ВВ, так и условий его течения. В весьма однородных ВВ,
даже химически высоко реакционноспособных, возможен случай,
когда при относительно слабом воздействии возникает большое
число малых не развивающихся до конца очагов разогрева, тогда
как при значительной механической неоднородности взрывчатого
вещества большой, саморазвивающийся очаг при данной скорости
1 Оплавление может наступать и на стадии уменьшения давления при ударе.
Известно, что при повышении давления температура плавления повышается. По-
этому, если перемещение слоев ВВ начинается при очень больших давлениях, то
сначала, несмотря на повышение температуры, ЕВ будет в твердом состоянии.
При уменьшении давления удара температура плавления снижается, наступает
оплавление и соответственно усиливается течение,
г О роли газовых включений в ВВ и воздуха, захватываемого при ударе,
см. также настоящий сборник стр. 29, 72.
3 Известно, что в обычных условиях возбуждения вспышки ВВ нагревом она
происходит со значительной задержкой, обусловленной химическим развитием
самоускоряющегося экзотермического превращения. При тех высокотемператур-
ных и кратковременных разогрсвах, какие могут возникать при ударе, вряд ли
химическое развитие реакции успевает пройти в значительной степени и эта сто-
рона явления не получает существенного значения.
40
поглощения энергии возникает легче, то есть с большей вероят-
ностью.
Так как однородность вещества никогда не бывает полной или
одинаковой, то это также составляет источник непостоянства, отсут-
ствующий, по крайней мере в такой резкой степени, при возбуж-
дении взрывчатого превращения равномерным разогревом, когда
условие нарушения равновесия между теплоприходом и теплоотво-
дом относится ко всему объему В В.
Как вторую стадию следует рассматривать возникновение и раз-
витие горения хотя бы в одном из очагов разогрева. Часто эту
стадию трактуют с позиций теории теплового взрыва. Вряд ли это
наиболее удачный и исчерпывающий вариант подхода к данному
явлению.
В теории теплового взрыва рассматривается обычно некоторый
заряд, имеющий определенный размер и температуру, и устанавли-
вается при каких, взаимно связанных значениях этих параметров
теплоприход становится больше теплоотвода. Не говоря уже о том,
что при ударе совершенно не определенны и трудно определимы
форма и особенно размеры очага разогрева, а также распределе-
ние температуры в нем, их взаимосвязь может быть существенно
отличной от той, которая имеет место при обычном тепловом взрыве.
В теории теплового взрыва скорость тепловыделения выражается в
зависимости от температуры соотношением
Q0Be-^ri
где Qo тепловой эффект превращения единицы объема ВВ в те его
продукты, которые получаются при низкотемпературной реакции
распада, а остальные буквы имеют обычное свое значение. Как из-
вестно, при этой реакции органические ВВ обычно выделяют лишь
- часть своей потенциальной энергии. В условиях же удара Qo не яв-
•: Ляется постоянной величиной, но может заметно, а иногда и резко
' зависеть от давления и температуры, возрастая с их увеличением за
. . счет прохождения реакций превращения ВВ до конца.
Далее у тех взрывчатых веществ, у которых главную часть теп-
лового эффекта дает реакция в газовой фазе, скорость горения
сильно зависит от давления; в этом случае развитие очага разо-
»• грева (подразумевается разогрев, доведший ВВ до той стадии пре-
вращения, которая идет в газовой фазе со скоростью, зависящей
от давления) в значительной мере определяется давлением. В тео-
рии теплового взрыва конденсированных ВВ влияние давления
обычно не принимается во внимание, с известным на то основанием,
так как, например, температура вспышки от давления почти не
зависит [5]. Тепловой взрыв можно рассматривать поэтому лишь
как возможный начальный этап химического превращения при уда-
ре, особенно если разогревы слабы. Возникновение же горения при
ударе гораздо правильнее трактовать с позиций современных пред-
ставлений о воспламенении ВВ, согласно которым условием устой-
чивого горения является равенство тепловых потоков от продуктов
4t
горения к прогретому слою, перемещающемуся со скоростью горе-
ния, и от прогретого слоя к непрореагировавшему веществу.
Напомним, что не наблюдается параллелизма между чувстви-
тельностью к удару н температурой вспышки, которая по существу
своему является тепловым взрывом, лишь в известной мере ослож-
ненным химическим развитием реакции. В то же время такой парал-
лелизм существует по отношению к воспламеняемости, особенно
если последнюю брать при повышенных давлениях, соответственно
условиям возбуждения взрыва при ударе.
Есть еще один существенный аргумент против обычной трактов-
ки возникновения взрыва при ударе как теплового взрыва, по край-
ней мере для вторичных ВВ. Критический диаметр для горения этих
веществ при атмосферном давлении во много раз больше толщины
слоя ВВ, применяемого при испытании на чувствительность. По-
этому даже если бы возник тепловой взрыв с максимальным разме-
ром очага, равным толщине слоя ВВ, и с максимальной температу-
рой, равной температуре горения, то он все равно не привел бы к
воспламенению при атмосферном давлении. Необходимо в связи
с этим учитывать влияние давления, что и делает теория воспламе-
нения, j
Кстати рассмотрение возникновения взрыва при ударе с пози-
ций воспламеняемости дает естественное и простое объяснение ча-
сто наблюдающимся при опыте случаям так называемых неполных
взрывов, которые обычно представляют собой затухшее вследствие
падения давления или неразвившееся горение. Это объяснение при-
менительно ко второму случаю легко иллюстрировать следующим
простым опытом. Если коснуться шашки пороха накаленной прово-
лочкой и быстро ее отнять, то в месте прикосновения шашка вспы-
хивает, но не загорается. Очаг теплового взрыва, созданный прово-
лочкой, оказывается слишком малым, чтобы возник прогретый слой
толщины, необходимой для устойчивого распространения горения.
Таким образом условие теплового взрыва может быть выполнено,
а горение не возникнет, если объем, в котором произошел тепловой
взрыв, слишком мал.
Такая многогранность развития химических процессов при уда-
ре, включающая локальное химическое превращение (которое мо-
жет завершаться тепловым взрывом) и самораспространяющееся
превращение в форме горения и взрыва, развивающееся до конца
или прерывающееся на разных стадиях развития, чрезвычайно за-
трудняет экспериментальную характеристику чувствительности ВВ
при ударе. Мы оцениваем результат индивидуального испытания
лишь как взрыв или отсутствие взрыва, в то время как было бы
необходимо описать этот результат и при том количественно по
крайней мере .с трех сторон — возникновения разогревов и очагов
химической реакции, их роста в форме горения и перехода послед-
него во взрыв. При этом могут быть различными число очагов, раз-
меры и расположение каждого из них в заряде, от чего также зави-
сит итог процесса.
42
Эта сторона вопроса представляет и непосредственный практи-
ческий интерес. Наиболее опасно такое взрывчатое вещество, в ко-
тором легко возникают и легко распространяются очаги распада.
Если мы представим себе два ВВ, в одном из которых очаги рас-
лада легко возникают, но распространяются с трудом, а в другом
наоборот оин возникают с трудом, но легко распространяются, то
в зависимости от условий, в которых производится деформация,
. более опасным может быть либо первое, либо второе вещество. Прн
существующем методе испытания мы лишены однако возможности
расчленить возникновение и развитие взрыва.
Экспериментальная оценка возникновения очагов распада, осо-
бенно если они не развились в горение, очень субъективна; иногда
это небольшое изменение внешнего вида ВВ или его цвета (побу-
рение, почернение), причем по этим признакам не всегда можно
отличить оплавление и разложение; в другом случае это появление
' запаха продуктов разложения или местный разброс вещества.
/ Нужно добавить, что подчас очень трудно отличить разброс, воз-
/ .инкший в очаге химической реакции, от разброса, вызванного чисто
/ механическими причинами. Так, например, при снятии давления
1 < сильно сжатый гексоген обычно самопроизвольно разбрасывается,
’' ,иногда настолько интенсивно, что при этом возникает взрыв.
<'. / Вторая характеристика процесса — развитие горения при ударе
' .Тоже не оценивается количественно, хотя результаты испытания и в
„$том отношении могут быть существенно различными — горение
; может распространиться на весь заряд, может ограничиться частью
',7^йли частями его, может быть полным или неполным. Наконец, не-
' /у^егко установить, имел ли место при испытании переход горения во
.J взрыв, так как о наличии взрыва мы можем судить лишь по звуку,
; • то время как звук может возникнуть и в результате горения под
" .большим давлением.
' Единственное известное в настоящее время направление количе-
ствен но го подхода к оценке рассматриваемого явления, хотя также
.‘имеющее интегральный характер, состоит в определении степени
У/^Полноты- взрыва по количеству газов, образующихся при данном
Д'^дыте. Химические методы количественной оценки возникновения и
развития очагов реакции сложны. ,
; В связи с весьма низкой воспроизводимостью результатов испы-
' Г /гания некоторые исследователи высказывали мнение о непригодно-
’ /уСти копра как прибора для изучения чувствительности ВВ. Вряд
• ли можно с таким мнением согласиться, по крайней мере, если го-
ворить об определении чувствительности как практической характе-
ристики ВВ, ибо те влияния, которые сказываются на результатах
'Яри испытании на копре могут проявляться и при работе с ВВ на
/производстве.
Однако это заключение отнюдь не исключает целесообразности
использования для изучения ВВ таких более простых методов, как
определение чувствительности к трению или к передаче детонации
У на расстояние. Остановимся немного подробнее на последнем испы-
тании. На'основании опытов с рядом вторичных ВВ автор давно уже
ДУ 43
Ж
указывал на параллелизм в их чувствительности при ударе и в усло-
виях передачи детонации на расстояние [1].
Недавно этот параллелизм был вновь подтвержден и расширен
(А. Я. Апин), причем было показано, что наблюдается известное
сходство между результатами испытаний на копре и на передачу
детонации даже в отношении роли такой характеристики ВВ как
размеры кристаллов.
Однако чувствительность к передаче детонации характеризует
только одну, хотя быть может и наиболее важную сторону возбуж-
дения взрыва при ударе, а именно воспламеняемость ВВ при кратко-
временном сжатии ’ и действии газов высокой температуры и дав-
ления. Роль стадии генерации тепла при механической деформа-
ции ВВ, иначе говоря стадии образования очага высокой темпера-
туры, здесь уменьшается и стираются те особенности вещества,
в частности его текучесть г, которые в наибольшей мере сказыва-
ются именно на этой стадии. Разницы между чувствительностью
к удару и к трению в этих условиях испытания уловить нельзя.
В свою очередь проявляются некоторые особенности, специфичные
для данной формы возбуждения взрыва, например, резкое влияние
начальной плотности порошка, которое при ударе сказывается в
гораздо меньшей степени. Еще нагляднее односторонность испыта-
ния на передачу детонации выявляется в случае шашек бездымного
пороха, который, как известно, плохо передает детонацию на рас-
стояние, обладая в то же время высокой воспламеняемостью и чув-
ствительностью при ударе.
Поэтому испытание на передачу детонации является существен-
но полезным при оценке чувствительности к удару, особенно если
сравниваются вещества, близкие по физико-механическим свой-
ствам, когда различия в чувствительности к удару определяются не
ими, а термокинетическими свойствами ВВ. Однако, как ясно из
сказанного выше, не всегда восприимчивость к передаче детонации
в состоянии исчерпывающе характеризовать чувствительность ВВ
к удару.
Плохая воспроизводимость результатов испытания на копре
естественно сильно затрудняет точную оценку влияния тех или иных
характеристик ВВ на его чувствительность.
Однако в ряде случаев, в том числе и случаев, представляющих
непосредственный практический интерес, такая оценка возможна.
Напомним прежде всего результаты, полученные Н. А. Холево[4],
и толкование, которое он им дал. Этот исследователь показал, что
если ВВ обладает большой текучестью, то наибольшая частость
взрывов наблюдается в условиях затрудненного течения, которые
могут быть получены в приборе № 1. При испытании в этом при-
боре в ВВ возникают относительно большие напряжения, вслед-
1 Отметим кстати, что скорости деформации при передаче детонации и при
возбуждении взрыва ударом сильно отличаются.
* Вряд пи можно сомневаться, что флегматнзация азида свинца парафином
уменьшает расстояние передачи детонации; при испытании же на копре она, как
известно, увеличивает частость взрывов. . '
44
ствие чего возрастает коэффициент внутреннего трения и, соответ-
ственно усиливаются разогревы, образующиеся в веществе при его
течении. Напротив, при испытании ВВ с малой текучестью наиболь-
шая частость взрывов наблюдается в условиях облегченного тече-
ния, реализованных, например, в приборе №2.
Степень затрудненности течения в приборе № 1 зависит естест-
венно от величины зазоров, которая влияет на частость взрывов.
Как показывает опыт, для различных ВВ максимальная частость
взрывов получается при разных зазорах; при этом «оптимальную»
величину зазора пока не удается заранее предвидеть. Еще более
жесткие условия течения Н. А. Холево применил при испытании
жидких ВВ, помещая их в чашечку из текучего металла, например
свинца (см. стр. 7).
Можно облегчить течение вещества в приборе № 2, уменьшая
диаметр роликов (при той же удельной энергии удара) или диаметр
заряда. Кстати условия деформации, соответствующие тем, кото-
рые имеют место в приборе № 2, можно реализовать и в приборе
№ 1. Для этого надо расположить навеску ВВ в виде таблетки или
кучки, диаметр которой меньше диаметра роликов. Свободная коль-
цевая часть пространства между роликами и будет при ударе играть
роль канавки прибора № 2.
В. С. Козлов предлагал прибор со слегка закругленным перехо-*
дом от торцовой к цилиндрической поверхности, сочетающий усло-
вия приборов № 1 и 2. В этом приборе на начальной стадии осу-
ществляются условия прибора № 2, т. е. облегченного течения, а в
дальнейшем, когда свободное пространство, небольшое по объему,
заполнится ВВ и последнее начнет продавливаться в зазоры ме-
жду роликами и муфтой — условия прибора № 1. Таким образом
в приборе В. С. Козлова чувствительность ВВ может проявляться
в известной степени независимо от его текучести — этот прибор как
бы объединяет испытание в приборах № 1 и 2. В этом состоит
его преимущество, но одновременно и недостаток, заключающийся
в том, что остается неизвестным, в каких именно условиях — напря-
женного или облегченного течения — наиболее опасно В В. Добавим,
"что идея В. С. Козлова давно уже воплощена в действительность в
виде того прибора, который применяется в соответствии с ГОСТ
2065—43. В этом приборе благодаря наличию фасок на роликах,
имеется кольцевое пространство, играющее роль канавки прибора
№ 2; так как объем этой канавки, по сравнению с объемом ВВ, мал,
то в дальнейшем оно продавливается через зазоры между роликами
и каналом муфты, подобно тому, как это происходит в при-
боре Яа 1.
В прежнем, применявшемся и у нас штемпельном приборе Каста,
поршеньки не имели фасок; появление последних было связано с
переходом на подшипниковые ролики, которые эти фаски имеют.
Вряд ли авторы ГОСТа, допустившего применение роликов с фас-
. ками, учитывали возможность сильного влияния этих фасок на час-
тость взрыва некоторых (труднотекучих) ВВ, а тем более представ-
ляли себе физический смысл этого влияния. Возможно, что некото-
45
рые разногласия, возникавшие в свое время в оценке чувствитель-
ности отдельных ВВ вроде флегматизйрованного гексогена с алю-
минием, чувствительность которого оценивалась ниже чувствитель-
ности тротила, были обусловлены именно применением в одном слу-
чае приборов без фасок, а в другом с фасками при незнании роли
и сильного влияния последних на результаты испытания.
Как бы то ни было, испытание в приборе по ГОСТу дает обычно
приближенное представление о чувствительности ВВ; испытание в
приборах № 1 н 2 уточняет вопрос о том, в каких условиях — об-
легченного или затрудненного течения — взрывчатое вещество, в
соответствии с его текучестью, наиболее склонно к взрыву при
ударе.
Следует добавить, что хотя текучесть ВВ и может накладывать
сильнейший отпечаток на его поведение при ударе, кинетические
и термохимические характеристики ВВ ни в коей мере не утрачи-
вают своей роли, которая при резких их различиях может быть
определяющей.
Соотношение этих двух факторов чувствительности очень на-
глядно пояснил Н. А, Холево: если бы мы имели два ВВ, тождест-
венные по своим физико-механическим свойствам, то чувствитель-
ность определялась бы только их химико-кинетическими и термиче-
скими характеристиками. Если однако в действительности физико-
механические свойства различны, то чувствительность (частость
взрывов) ВВ зависит также и от них. Чем меньше различие физи-
ческих свойств сравниваемых ВВ, тем в большей мере их чувстви-
тельность определяется кинетическими и термическими характери-
стиками. <
В свою очередь роль физических факторов была бы выявлена в
чистом виде, если бы мы имели одинаковые по химико-кинетическим
и термохимическим характеристикам ВВ, различающиеся однако
по физическим свойствам. Кстати близкий к этому случай мы имеем,
если одно и то же ВВ может быть испытано при одинаковых внеш-
них условиях в различных агрегатных состояниях. Такое испыта-
ние реализовано, например, на способном к сильному переохлаж-
дению нитроглицерине; оно показало, как известно, существенное
различие частости взрывов жидкости и твердого вещества.
Из сказанного следует, что для полной и дифференцированной
характеристики чувствительности ВВ к механическим воздействиям
необходим комплекс испытаний, устанавливающих роль как хими-
ко-кинетических и термических, так и физических факторов в про-
цессе возбуждения взрыва. Первые в известной мере интегрально
могут быть характеризованы воспламеняемостью взрывчатого веще-
ства при повышенных давлениях, а также восприимчивостью его в
условиях передачи детонации на расстояние, вторые — испытанием
на копре в приборах № 1 и 2, а также определением чувствитель-
ности к трению. При последнем испытании условия трения в боль-
шей мере задаются экспериментатором, чем в приборах № 1 и 2,
где они определяются в первую очередь текучестью ВВ.
46
До настоящего времени комплексное испытание ВВ на чувстви-
тельность по ряду причин не проведено. Однако даже односторон-
ние испытания на копре позволили выявить ряд заслуживающих
внимания закономерностей.
Так при исследовании ВВ, однотипных по химической структуре,
йаблюдалось значительное влияние температуры плавления на ре-
зультаты испытания. Если температура плавления относительно
низка, то, как уже отмечалось, образующийся в начале течения на
поверхности частиц расплав играет роль своеобразной смазки, сни-
жая коэффициент трения и уменьшая частость взрывов в облегчен-
ных условиях течения, например, в приборе № 2. Напротив, в при-
боре № 1 частость взрывов может возрастать. Играет роль однако
Не только абсолютная величина температуры плавления, но и бли-
зость ее к температуре быстрого разложения, иными словами ско-
рость разложения образующегося расплава. Если эта скорость ве-
лика, то «эффект смазки» ослабляется и частость взрывов высока
ив условиях облегченного течения В условиях затрудненного те-
чения плавление обычно отсутствует и частость взрывов низка.
Интересно поведение систем, включающих легкоплавкую основу
j ’Й. относительно высокоплавкий труднотекучий порошок, чувстви-
тельный в условиях облегченного течения. Чувствительность таких
/ .систем особенно при большом содержании высокоплавкого компо-
нента может определяться в первую очередь именно его свойствами,
#. е. оказывается высокой. Свойства плавкой основы менее сущест-
венны. Особенно наглядно это проявляется для смесей азида
. ’ 'Ijtoema с парафином; в определенных условиях испытания,
:.,)югда частость взрывов азида близка к нулю как в приборе
М1, так и в приборе № 2, она резко увеличивается в присутствии
'1^>афина.
.Л'т Решающим фактором, определяющим увеличение частости взры-
в этих случаях, является, очевидно, увеличение текучести веще-
/Ч-ЙРрЙа и обусловленная этим возможность трения между частицами
Г./фердого высокоплавкого компонента. Возможность распростране-
/’ЙЙЯ взрывчатого превращения при добавлении легкоплавкого веще-
( «Тва может стать, а в условиях приведенного примера, несомненно
. Хгеййовится более низкой; но не она лимитирует в данном случае
'^Ювникновение взрыва.
' Если, однако, легкоплавкого компонента в смеси относительно
1Ййого и он химически относительно инертен, то флегматизирующее
действие на стадии развития взрыва может быть значительным,
ййк это и наблюдается, например, в 50%.ном «сплаве» гексогена с
тротилом.
С другой стороны и здесь может играть существенную роль из-
менение физических свойств (увеличение или уменьшение текуче-
... СТИ) системы.
1 Можно ожидать, что чувствительность эвтектических сплавов трудно теку-
чих ВВ будет в известных условиях ниже, чем их составных частей.
%' 47
А
ОБ ОЦЕНКЕ ЧУВСТВИТЕЛЬНОСТИ ВВ К МЕХАНИЧЕСКИМ
ВОЗДЕЙСТВИЯМ ПРИМЕНИТЕЛЬНО К ПРАКТИЧЕСКИМ УСЛОВИЯМ
При непостоянстве экспериментальных результатов для одного
и того же вещества, при большой и различной для разных ВВ зави-
симости частости взрывов от условий испытания (конструкция и
размеры прибора, толщина и диаметр заряда, величина зазоров
и т. д.) встает вопрос, как же оценивать чувствительность взрывча-
тых веществ применительно к запросам практики. Здесь уместно
процитировать те выводы, к которым пришли американские физико-
химики в итоге попыток определения чувствительности [6].
«В результате исследований однако было установлено, — и это
является открытием значительной практической важности — что
нельзя найти такой характеристики, как определенная механичес-
кая чувствительность взрывчатого вещества. Действительно, изме-
няя условия испытания, можно изменить даже порядок механиче-
ских чувствительностей взрывчатых веществ. Таким образом лабо-
раторные испытания не в состоянии установить абсолютную опас-
ность производственных операций, производимых над взрывчатыми
веществами. В лучшем случае применение различных испытаний
может дать лишь предостережение о том, что новое ВВ в общем
более опасно чем другое, уже применяющееся, быть может это
испытание сможет дать также некоторые указания относительно
особенно опасных условий».
Эти выводы по существу аналогичны тем, к которым незави-
симо и ранее пришел Н. А. Холево, вскрывший и физический смысл
непостоянства чувствительности.
Нужно однако добавить, что, если для какого-либо ВВ, меняя
соответствующие условия испытания, можно получить либо очень
высокую, либо очень низкую частость взрывов, то для веществ с
близкими физическими свойствами (например для многих тринитро-
соединений бензольного ряда) при обычных условиях испытания,
например в приборе по ГОСТ 2065—43, можно получить прибли-
женную сравнительную характеристику чувствительности, отража-
ющую в таком случае их кинетические и термохимические свойства.
Встает однако вопрос, в какой мере результаты такого или ком-
плексного испытания могут быть перенесены на условия обработки
и применения ВВ.
Ответ на этот вопрос весьма сложен в первую очередь вслед-
ствие недостаточного знания характера тех воздействий, которым
подвергается ВВ в практических условиях. Так, нет ясных, экспе-
риментально доказанных представлений о механизме возникнове-
ния преждевременных взрывов при выстреле, неожиданных взрывов
при прессовании, при шнековании и т. д. Мало может помочь и об-
ращение к практическим приемам уменьшения чувствительности
ВВ. Такое положение обусловлено прежде всего тем, что широкое
экспериментирование в этих вопросах не осуществлялось, так как
каждый эксперимент в натуральном масштабе, если он кончается
взрывом, стоит очень дорого. Обычно отрицательное заключение
48
о возможности применения для данного ВВ того или иного техно-
логического приема или способа использования основывается на
имевших место в практике единичных взрывах, которые могли быть
случайными.
Для уменьшения чувствительности ВВ к механическим воздей-
>. ствиям практика зиает два основных приема флегматизации; один
йз них заключается в применении сплавов, включающих ВВ с боль-
шой энергией взрыва и высокой чувствительностью и ВВ с меньшей
энергией взрыва и низкой чувствительностью, примерами могут слу-
жить сплавы пикриновой кислоты с динитронафталином, тэна с тро-
• тилом. В этом случае уменьшение чувствительности происходит
главным образом за счет снижения общей реакционной способности
ВВ и энергии его взрыва по сравнению с наиболее чувствительным
компонентом.
Однако широко применяется и другой способ флегматизации,
когда частицы высокочувствительного ВВ с высокой температурой
плавления обволакивают слоем нёвзрывчатого вязкого вещества;
типичным флегматизатором является парафин. В качестве флегма-
./уизатора может быть применено и малочувствительное легкоплав-
<ое взрывчатое вещество. Эту роль выполняет, например, тротил в
^/«сплавах» с гексогеном. Можно предполагать, что легкоплавкие
л/Ьязкие примеси играют роль смазки при течении ВВ, вызванном уда-
ллтюм или при треиии и уменьшают те разогревы, которые возникли
*7 <(ы в их отсутствии. Это их действие аналогично действию расплава,
образующегося на поверхности частиц легкоплавких ВВ при быст-
рой их деформации.
’ Цл Наряду с этим флегматизаторЫ несомненно играют и ту роль,
улучшают формуемость, в частности прессуемость взрывчатого
4 у^фещества. Это делает менее опасным и потому технически реальным
прессование, так как в случае применения флегматизатора бо-
'//Щве высокая плотность достигается при меньшем давлении' прессо-
вания, при этом не возникают те напряжения и тангенциальные
/ усилия, которые были бы неизбежны при достижении высокой
'.^Лртности заряда прессованием нефлегматизированного порошка,
/дарлее же плотный заряд при прочих равных условиях труднее де-
формируется.
'/о- Напомним в этой связи, что при испытании на копре нередко
/.присутствие флегматизатора увеличивает частость взрывов. Это
происходит в тех условиях, где отсутствие взрыва нефлегматизнро-
.й.анного ВВ обусловливается его недостаточной (для данных усло-
’•/ШЙ воздействия) текучестью. При медленных воздействиях (прес-
' Сование) это же увеличение текучести может оказывать обратное
Влияние. Повышенная текучесть ВВ, в частности способность его
Йлавиться без существенного разложения затрудняет, вообще го-
воря, возникновение взрыва при таких относительно медленных
воздействиях, каким ВВ подвергается в процессе шнекования и
Прессования. Благодаря текучести снимаются те локальные напря-
жения, которые могут возникать в ВВ при этих процессах. Вслед-
.4 451 49
ствие этого, во-первых, не возникают сильные местные разогревы;
во-вторых, реакция в очагах разогрева не развивается, даже, если
бы ови возникли.
Известно, что снаряжение боеприпасов шнекованием тротила
относительно безопасно. Объяснить это можно было бы относитель-
ной химической инертностью тротила. Однако вряд ли такое объяс-
нение достаточно, так как значительно более реакционноспособные
амматолы, частость взрыва которых при испытании на копре гораз-
до больше, чем тротила, также шнекуются. Очевидно поэтому
существенным фактором опасности шнекования является текучесть,
в частности текучесть, обусловленная возможностью плавления
без разложения. Если ВВ не обладает такой значительно выра-
женной текучестью, то большие напряжения сдвига, если они
возникнут, не смогут быть смягчены и вызовут критические разо-
гревы.
Однако слишком большая текучесть ВВ такая, например, какой
обладают маловязкие жидкости, может при известных предпосыл-
ках увеличить возможность взрыва. Представим себе, что осуще-
ствляется удар тугоплавкого металла о металл, покрытый слоем
взрывчатой жидкости, которая вследствие малой своей вязкости не
оказывает существенного сопротивления сближению соударяющих-
ся поверхностей. В этом случае особенно, если угол соударения
хотя бы немного отличается от прямого, сдвиг и деформация метал-
лических поверхностей, как известно, приводят к возникновению
сильных локальных разогревов. Если ВВ, как например, нитрогли-
церин, обладает большой реакционной способностью и способно
под большими давлениями гореть даже в тонком слое, то оно может
воспламениться н взорваться от трения металла о металл. Этот
взрыв затем передается остальной массе В В, не подвергнувшейся
непосредственному удару. Малая вязкость может, кроме того,
облегчить возникновение взрыва вследствие сжатия газовых
пузырьков.
Если вязкость нитроглицерина увеличена, например, растворе-
нием в нем нитроклетчатки, то обе эти возможности возникновения
взрыва реализуются труднее.
Таким образом и слишком малая и слишком большая текучесть
ВВ могут при известных условиях механических воздействий бла-
гоприятствовать возникновению взрыва.
В заключение коснемся того несколько схоластического аспекта
проблемы, в котором она ставилась некоторыми исследователями
в последнее время. Речь идет о попытках считать чувствительность
некоей абсолютной константой ВВ, обусловленной только его хи-
мической структурой. При этом указывалось, что и физические
свойства, роль которых подчеркивали Н. А. Холево и примыкающие
к нему исследователи, обусловлены химической структурой веще-
ства.
Сторонники этих взглядов забывают, что абсолютных свойств
вещества вообще нет, что все свойства в большей или меньшей мере
зависят от условий, в которых находится вещество. Поэтому только
50
силой привычки1, возникшей в те времена, когда чувствительность
была вещью в себе, когда ее физический смысл не был известен,
можно объяснить стремление привесить к каждому ВВ ярлычок
с числом его чувствительности и на этом считать задачу познания
последней законченной.
Далее, если бьг химик, зная состав и химическую структуру ВВ,
мог количественно характеризовать его физико-механические свой-
ства, то можно было рассматривать эти свойства, являющиеся важ-
ным фактором чувствительности, не непосредственно, а через функ-
циональную их зависимость от состава и структуры. Пока же этого
нет и мы не рассчитываем физико-механические свойства по составу
и структуре, а устанавливаем их путем непосредственного опреде-
ления. Помимо этого физико-механические свойства данного хими-
ческого соединения также не являются абсолютной его константой
и уже поэтому чувствительность ВВ может не быть постоянной
величиной. То, что она ею не является, можно иллюстрировать мно-
гими примерами. Так, естественно, чувствительность одного и того
же соединения в парообразном, жидком и твердом состоянии раз-
лична, чувствительность твердого вещества может сильно зависеть
от размеров кристаллов. Известно, что различные кристаллографи-
ческие модификации некоторых ВВ также различаются по чувстви-
тельности, Наконец и внешние условия, которые уже никак нельзя
включить в понятие химической структуры испытуемого вещества,
тоже влияют на чувствительность. Чувствительность к трению мо-
жет отличаться от чувствительности к удару, она зависит, в част-
ности, и эта зависимость объяснена Боуденом [2], от температуры
плавления материалов, между которыми осуществляется трение.
Таким образом чувствительность является переменной величи-
ной, зависящей от свойств ВВ и внешних условий, в том числе усло-
вий деформации. Возникает однако вопрос: нельзя ли эту зависи-
мость полностью или хотя бы частично выразить количественной
формулой? В принципе это возможно. Практически же, ввиду
большой сложности процесса, попытки в этом направлении (в том
числе и попытка Л. В. Дубнова, Эванса и др. исследователей) ус-
пеха не имели. Одновременно ясны отдельные определяющие
характеристики явления; это — из числа химико-кинетических и
термических свойств — тепловой эффект реакции, теплоемкость и
теплопроводность ВВ, объемная скорость тепловыделения в ее за-
висимости от температуры, скорость горения в зависимости от дав-
1 В самом деде, мы знаем, что чувствительность ВВ к детонации зависит от
величины кристаллов и от плотности порошка, но это нас уже не удивляет и мы
не ищем какой-то абсолютной чувствительности к детонации. Столь же мало осно-
вание искать ее применительно к чувствительности ВВ к удару. Зависимость от
условий воздействия отнюдь не является особенностью тех или иных характери-
стик ВВ. В сопротивлении материалов мы говорим о прочности на разрыв, на
сжатие, на сдвиг и это не вызывает удивления и не заставляет искать какую-то
единую абсолютную прочность, хотя такое понятие было бы несравненно проще
связано с составом и структурой. Как третий пример нужно напомнить понятие
хрупкости; его условность, зависимость хрупкости, например, от скорости дефор-
мации общеизвестна.
4*
51
ления (влияние этих факторов частично объединяется в воспламе-
няемости, а также в величине критического диаметра заряда); из
физико-механических свойств и условий деформации — коэффи-
циенты внутреннего и внешнего трения в их зависимости от давле-
ния и температуры, степень однородности ВВ (в частности наличие
и размеры газовых включений), давление и скорость его изменения
во времени, начальная температура, температура плавления, обо-
лочка, размеры отверстий в ней и другие характеристики условий
деформации.
ВЫВОДЫ
Возбуждение взрыва при механических воздействиях на взрыв-
чатое вещество обусловлено возникновением в нем местных разо-
гревов. Эти разогревы могут образовываться при неравномерном
течении взрывчатого вещества, при сжатии газовых пузырьков в
нем, при трении частиц тугоплавких примесей или твердых поверх-
ностей, между которыми находится взрывчатое вещество. Развитие
химической реакции, возникающей в очагах разогрева, до горения
и взрыва зависит от их температуры, размеров и от давления.
Ввиду того, что возбуждение взрыва определяется многими фак-
торами, главные из которых химико-кинетические и термохимиче-
ские характеристики взрывчатого вещества, его физические свой-
ства (текучесть) и условия деформации, чувствительность (частость
взрывов) при механических воздействиях может меняться в широ-
ких пределах.
Для интегральной оценки чувствительности необходимо опреде-
лять комплекс свойств взрывчатого вещества—частость взрывов
на копре в приборах № 1 и 2 и при испытании на трение, а также
способность к восприятию и передаче детонации на расстояние,
критические диаметры' заряда при горении и при детонации. При
этом нельзя забывать, что реальная опасность возникновения,
взрыва при механических воздействиях будет в сильной мере зави-
сеть также от условий этих воздействий, определяющих возмож-
ность течения взрывчатого вещества и возникновения при этом
местных разотревов, а также от поддержания давления, благоприят-
ствующего развитию начавшегося превращения до взрыва.
Эта статья была написана в 1956 г. Некоторые соображения,
развитые в ней, возникли под влиянием бесед с Н. А. Холево, кото-
рого автор в этой связи вспоминает с глубокой благодарностью.
ЛИТЕРАТУРА
[.Андреев К- К., Безопасность труда в горной промышленности, 1932,
12, 17-18.
,/2. Б о у д е н Ф. Ф. и И о ф ф е А. Д., Возбуждение и развитие взрыва,в твер-
дых и жидких веществах, перевод с английского, ИЛ, [955, стр. 23.
3. Сапожников А. В,( Теория взрывчатых веществ, Л,, ВТА РККА, 1926,
стр. [54.
52
4. X о л е в о Н. А., Труды Казанского хи мико-тех но логического института
км. С. М. Кирова, 1946, 10, 91; 1947, 11, 116.
5. В о w d е n F. Р.. Discussions of the Faraday Society, 1956, No 22,182.
6. Kistiakowsky G. B. and Connor R., Science in World War II vo].
Chemistry, 1946, p. 80.
А. А. АНДРЕЕВ, Ю. А. ТЕРЕБИЛИНА
4. О ВОЗБУЖДЕНИИ ВЗРЫВА ПРИ УДАРЕ И МЕТОДАХ
ХАРАКТЕРИСТИКИ ЧУВСТВИТЕЛЬНОСТИ ВЗРЫВЧАТЫХ
ВЕЩЕСТВ К МЕХАНИЧЕСКИМ ВОЗДЕЙСТВИЯМ
О МЕХАНИЗМЕ ВОЗБУЖДЕНИЯ ВЗРЫВА ПРИ УДАРЕ
• Под термином чувствительность взрывчатого вещества пони*
мается способность последнего реагировать на внешние воздействия
возникновением горения или взрыва. Способность эта может быть
выражена больше или меньше. Факторами, определяющими чувст-
вительность взрывчатого вещества к механическим воздействиям,
«|- являются свойства его и те условия воздействия, которые обеспечи-
. ; дают создание в заряде очага разогрева достаточной температуры
я размера, а также распространение и развитие до взрыва, начав-
шегося в нем превращения. В совокупности эти факторы опреде-
ляют опасность возникновения взрыва, т. е. чувствительность взрыв-
' /Чатого вещества, в определенных конкретных условиях. Они могут
быть разделены на две группы: химические в широком смысле этого
\. слова и механические.
у/ Поскольку первая во времени стадия изучаемого явления опре-
деляется преимущественно механическими факторами, мы начнем
‘ именно с них.
В настоящее время общепризнано, что взрыв при механических
: воздействиях возникает от местных разогревов, создаваемых этими
воздействиями. Пути возникновения разогревов могут быть раз-
ными Ь Один из них — неравномерное по скорости течение взрывча-
того вещества. Второй путь связан с наличием газовых пузырьков
й ВВ и третий —с местными разогревами от трения твердых ино-
родных частиц или металлических поверхностей наковальни и удар-
ника прибора при наличии между ними тонкого слоя взрывчатого
вещества.
Для возникновения местных разогревов существенны не одни
лишь физико-механические свойства ВВ, но и условия воздействия
на него. Так, чтобы взрывчатое вещество могло течь при сжатии,
.. создаваемом ударом, необходимо, чтобы оно не было замкнуто со
всех сторон, т. е. ему должна быть предоставлена возможность течь
в зазор. Далее очевидно, что от ширины и протяженности зазоров,
1 См. в настоящем сборнике статьи Н. А. Холево (стр. 5), Ф. Боудена (стр.
20) и К' К- Андреева (стр. 37).
через которые течет взрывчатое вещество при ударе, зависит воз-
никающее в нем давление; от величины же этого давления в свою
очередь зависит коэффициент внутреннего трения и скорость тече-
ния, а через них и интенсивность разогрева. Разогрев газового пу-
зырька также зависит от максимального давления и скорости
достижения последнего. Наконец, если мы имеем дело не с ударом
по нормали, при котором возникновение течения ВВ, а следова-
тельно, и трения определяется текучестью взрывчатого вещества,
а со скользящим ударом, то возможность возникновения разогревов
в этом случае может быть совершенно иной.
Таким образом возникновение локальных разогревов при меха-
нических воздействиях на взрывчатое вещество зависит от очень
многих факторов, совокупное влияние которых количественно не-
легко учесть и даже оценить. Это обстоятельство вносит большую
неопределенность в вопрос о том, приведет ли данное воздействие
к возникновению разогрева, заканчивающегося взрывом, или нет.
В этом отношении механическое воздействие как средство возбуж-
дения взрыва отличается от многих других, в особенности от теп-
лового. В последнем случае связь между причиной и следствием
гораздо более проста и поэтому легче может быть оценена.
Сказанное^ однако, относится не только к механической стороне
явления. Воспламенение и горение взрывчатого вещества при ударе
протекают в более сложных условиях, в сравнении с обычными ус-
ловиями соответствующих экспериментов, Главными факторами
здесь являются изменяющееся давление и малая толщина слоя
взрывчатого вещества. Известно, что согласно теории Михельсона-
Зельдовича количество тепла, которое нужно сообщить поверхност-
ному слою заряда ВВ, чтобы он воспламенился, зависит от скорости
горения, а через нее и от давления. Чем больше последнее, тем
меньше это количество. Оно зависит также от теплопроводности
взрывчатого вещества. Далее, если горение началось, но давление
быстро уменьшается, то оно может прекратиться, так как запаса
тепла в прогретом слое может не хватить для того, чтобы горение
продолжалось при низком давлении. Критический диаметр (точнее
критический размер) заряда при горении под атмосферным давле-
нием, как правило, гораздо больше, чем в обычных условиях воз-
буждения взрыва при ударе. Поэтому горение, возникшее при
ударе, может распространяться только в том случае, если при нем
будет поддерживаться достаточно высокое давление.
Наконец, как и в обычных условиях теплового взрыва, оказы-
вают свое влияние температура, достигнутая в очаге разогрева, п
размеры последнего. Поэтому, если рассматривать химические фак-
торы чувствительности, то нужно учитывать и те из них, которые
определяют возникновение теплового взрыва: скорость реакции,
вызываемой нагревом, и теплоту ее, определяющую температуру,
при ней достигаемую. Известно, что глубина превращения взрывча-
того вещества, как правило, зависит от давления и, по-видимому,
от температуры. Поэтому не всегда легко конкретизировать те зна-
чения константы скорости и теплоты реакции, которые фактически
54
являются определяющими при возбуждении взрыва ударом. Во вся-
ком случае их нельзя просто отождествлять с данными, получае-
мыми при медленном распаде или при детонации.
Таким образом и с химико-кинетической стороны возникновение
взрыва при ударе гораздо сложнее, чем при тепловом его возбуж-
дении, например, в обычных условиях определения температуры
вспышки взрывчатого вещества.
Наглядной иллюстрацией этого является несравненно большее
постоянство результатов определения температуры или задержки
вспышки в сравнении с определением характеристик чувствитель-
ности ВВ к удару. При этом, судя по наблюдениям характера дефор-
мации невзрывчатых веществ при ударе, непостоянство результатов
при возбуждении взрыва ударом в основном определяют механиче-
ские факторы, рассмотренные выше.
В связи со сложным характером явления и многочисленностью
факторов, от которых зависит возникновение взрыва при ударе,
практическое значение характеристик чувствительности, получен-
ных при определенных условиях испытания, — ограничено. Дейст-
вительно, если механические условия изменятся, а в практике они
могут меняться весьма широко, то и возможность возникновения
взрыва изменится. Поэтому принципиально правильнее для харак-
теристики конкретного ВВ в отношении чувствительности устанав-
ливать не частость взрыва, например, при ударе на копре, а некото-
рые более простые свойства взрывчатого вещества, ее определяю-
щие— термокинетические и физико-механические. Однако прибли-
женная характеристика этих свойств может быть получена и при
помощи копра. Недостатком метода определения чувствительности
ВВ на копре является по существу качественный характер резуль-
татов, а преимуществом то, что определение производится в усло-
виях, соответствующих тем, при которых возбуждается взрыв при
ударе (повышенное, быстро меняющееся давление, движение веще-
ства и др.).
Выше мы отмечали, что для возбуждения взрыва при ударе
имеют значение как физические, так и химические факторы. Сле-
дует, однако, подчеркнуть различие между ними. Химические фак-
торы неразрывно связаны с взрывчатым веществом и играют свою
роль при всех условиях, роль же физических факторов сказывается
лишь при определенных, более или менее конкретных для каждого
ВВ условиях. Поэтому, если мы имеем взрывчатое вещество, чув-
ствительность которого мала за счет химических факторов, т. е.
такое ВВ, для которого характерны малая воспламеняемость и
большой критический диаметр детонации, то для возбуждения взры-
ва его необходим сильный или большой по размерам местный разо-
грев. Это вещество будет взрываться только в тех условиях меха-
нических воздействий, какие обеспечивают возникновение такого
разогрева. Напротив, взрывчатое вещество, с теми же физическими
свойствами, но с высокой «химической» чувствительностью, будет
взрываться не только при этих механических воздействиях, но и при
55
многих иных, создающих более слабые разогреем, от которых пер-
вое взрывчатое вещество не взрывалось.
Сказанное отнюдь не означает, что взрывчатое вещество с боль-
шей «химической* чувствительностью всегда будет давать большую
частость взрывов. Напротив могут быть такие условия механичес-
ких воздействий, при которых частость взрывов «химически» менее
чувствительного вещества будет больше. Хорошим примером яв-
ляется азид свинца — вещество с высокой «химической» чувстви-
тельностью, которое при ударе на копре по нормали вследствие
низкой текучести взрывается с большим трудом, труднее, чем, на-
пример, тетрил, «химическая» чувствительность которого гораздо
меньше. Однако в среднем при тех разнообразных условиях меха-
нических воздействий, которые возможны в практике, взрывчатое
вещество с высокой «химической» чувствительностью будет более
опасным, чем ВВ, у которого чувствительность мала. Последнее же
будет опасным лишь при некоторых условиях воздействия, когда
конкретные механические свойства взрывчатого вещества создают
возможность возникновения достаточно интенсивных очагов высо-
кой температуры и развития начавшегося в них горения.
Так, например, по опытам Н. А. Холево тротил, в смеси с алю-
миниевой пудрой (80 : 20), при ударе между двумя стальными ро-
ликами диаметром 10 мм (прибор № 2) не дает взрывов даже при
большой энергии удара (4 кгм). Трудно заставить его взрываться и
от трения. Если, однако, поместить указанную смесь в прибор № 1,
где благодаря узким зазорам между роликами и муфтой течение
вещества затруднено, то частость взрывов при той же энергии
удара составляет 100%.
Приведем второй пример. Если на копре в приборе № 4 (см.
ниже) производить удар по нитроглицерину или азиду свинца,
слегка приподняв верхний ролик над ВВ, то может быть получена
частость взрывов 100%, предположительно вследствие захвата воз-
духа. При ролике, поставленном непосредственно на взрывчатое
вещество, она равняется нулю. Если же взять химически относи-
тельно малочувствительное взрывчатое вещество (например, тетрил
или гексоген), то наличие промежутка между верхним роликом
и ВВ не приводит к заметному повышению частости взрыва <
Вопрос о том, каким образом можно по результатам испытания
на копре оценить «химическую» и «механическую» стороны чувстви-
тельности ВВ, рассматривается ниже на конкретных эксперимен-
тальных данных.
Опыты проводили в приборе № 4, схема которого дана на фиг. 1.
Из схемы этого прибора видно, что он в основном соответствует при-
бору № 2 Н. А. Холево, главное отличие его состоит в том, что в
нем применены ролики большего диаметра (19 .м), а место стыка
их по всему периметру не соприкасается со стенками муфты, так
что ВВ при истечении, вызванном ударом, выбрасывается в неог-
раниченное ничем пространство. Особенности деформации навески
1 См. настоящий сборник, стр, 72.
56
Таблица I
Результаты опытов с некоторыми взрывчатыми веществами
иа иопре в приборе № 4
(Груз 10 кг, высота его падения 25 см, число опытов в каждой группе 25,
______отскок груза при холостом ударе 70% высоты его падения)_
Серия опытов Взрывчатое вещество На- веска мг Условия опыта Час- тость взрывов % Средний отскок груза при отказах % высоты падения
1 Гексоген 1 5 10 50 Навеска расположена кучкой в центре ролика 8 64 80 92 64 64 60 54
10 50 Навеска размещена равномер- но по всему торцу ролика 0 8 65 64
2 Тротил 5 Навеска расположена кучкой в центре ролика 0 61
5 Навеска размещена равно- мерно по торцу ролика 0 65
50 Таблетка диаметром 10 мм расположена в центре ролика 0 39
50 Навеска расположена кучкой в центре ролика 0 41
50 Навеска размещена равно- мерно по торцу ролика 4 35
3 Тетрил 5 Закристаллизовавшаяся капля 16 54
10 20 50 Навеска расположена кучкой в центре ролика 4 48 12 57 57 43
50 Навеска размещена равно- мерно по торцу ролика 24 50
4 Нитрогли- церин 2 5 1 Ю На нижний ролик наносится капля; под тяжестью веса верхнего ролика она расте- кается между торцами 4 8 0 64 60 61
5 Нитроглице- рин желати- нированный 2 5 10 На нижний ролнк наносится капля; под тяжестью веса верхнего ролика она расте- кается между торцами 4 4 0 66 67 65
6 Нитро- глицерин 10 В навеске находятся 2—< стальных крупинки толщиной 0,01—0,04 мм 28
57
-
Фиг, 1. Схема прибора
4 для испытания
взрывчатых веществ
на чувствительность при
ударе.
ВВ и возникновения взрыва в приборе № 4, так же как преиму-
щества и недостатки его конструкции, будут рассмотрены ниже,
а сейчас мы остановимся на результатах испытаний некоторых ВВ
в этом приборе на копре. Эти результаты приведены в табл. 1.
В опытах с каждым изучаемым ВВ меняли величину навески и
ее размещение на торце ролика. Помимо частости взрывов фикси-
ровали также высоту отскока груза после удара (в % от высоты
падения) в случаях отказов. Первая серия опытов выполнена с гек-
согеном, причем навеску ВВ в виде кучки
помещали в центре ролика. При маленькой
навеске (1 мг) частость взрывов была 8%,
при 5 мг она возросла до 64%, При даль-
нейшем увеличении навески в два и в 10 раз
частость взрывов продолжала расти. Если,
однако, большую навеску ВВ разместить
равномерно на торце ролика, то частость
взрывов резко падает. Это указывает на
то, что гексоген является труднотекучим
веществом, и то давление, которое дости-
гается при распределении энергии удара
на всю площадь ролика, к тому же
при малой толщине слоя, недостаточно для
того, чтобы вызвать течение вещества,
а следовательно, и взрыв. Это подтверж-
дается также сопоставлением высот от-
скока груза. При равномерно размещенном
слое ВВ высота отскока велика и прибли-
жается к наблюдаемой в холостых опытах,
что указывает на отсутствие течения ве-
щества; такая же высота отскока наблю-
дается при центральном расположении ма-
лой навески, что указывает в этом случае
на малую затрату энергии на течение ВВ.
При центральном расположении большой навески (50 мг) отскок
заметно уменьшается.
Возникает, однако, вопрос, чем же определяется низкая частость
взрывов при очень малых центрально расположенных навесках ВВ
(1 мг)? Можно было бы предположить, что значительная часть
энергии удара расходуется при деформации заряда, в процессе
которой диаметр его и сопротивление течению ВВ увеличиваются.
Когда же достигается такая толщина слоя ВВ, при которой продол-
жение течения вызвало бы взрыв, энергия груза настолько исчер-
пана, что течение ослабевает или прекращается. Однако против
этого объяснения говорит большая частость взрывов при навесках
10 и 50 мг по сравнению с 1 мг. Следует поэтому полагать, что при
малой навеске к моменту, когда сопротивление расплющенной таб-
летки становится значительным, мы уже попадаем в область таких
малых толщин, при которых тепловые потери не позволяют раз-
виться горению.
58
Таким образом по совокупности этих данных можно заключить,
что гексоген характеризуется большой «химической» чувствитель-
ностью, потому что он взрывается даже при очень малых навесках.
В связи с тем, что это ВВ обладает малой текучестью, высокая его
чувствительность может проявиться только в том случае, если усло-
вия деформации благоприятствуют возникновению течения заряда
(большое удельное давление и облегченные условия течения —
приборы № 2 или 4 с центральным расположением на-
вески) .
Рассмотрим теперь результаты опытов с тротилом. При навеске
5 н 50 мг мы не получаем взрывов при расположении ВВ как в се-
редине ролика, так и по всей площади его торца. Это следует объ-
яснить тем, что тротил обладает большой текучестью, что подтверж-
дается и величиной отскока, и одновременно он химически малочув-
ствителен.
При малых навесках он не взрывается, потому что толщина слоя
получается меньше критической. Тротил не взрывается и при боль-
ших навесках (большая толщина слоя), так как из-за значительной
текучести не возникает (при данных размерах ролика) напряже-
ния, достаточного для возбуждения взрыва при течении. Тетрил
занимает промежуточное положение. Текучесть его выше, чем гек-
согена: при навеске 50 мг. равномерно распределенной на ролике,
он взрывается с частостью во всяком случае не меньшей, чем при
центральном расположении, в то время как в случае гексогена
частость взрывов при равномерном размещении, как мы видели,
резко падала. Наряду с этим сопоставление величин отскоков пока-
зывает, что тетрил в отличие от гексогена достаточно сильно течет—
отскок (при равномерном размещении навески 50 мг) составляет
только 50% в отличие от 64% для гексогена. С другой стороны
тетрил «химически» менее чувствителен, чем гексоген: частость
взрывов тетрила становится мала (16%) уже при навеске 5 мг, при
которой гексоген дает еще 64% взрывов.
Поведение жидких ВВ в описанных условиях удара может быть
иллюстрировано опытами с нитроглицерином. Он дает в обычных
условиях испытания очень низкую, близкую к нулю частость взры-
вов при навесках от 2 до 10 мг. Текучесть нитроглицерина велика,
что подтверждается в частности большой величиной отскока. В нем
не возникают поэтому при ударе столь высокие напряжения, при
которых коэффициент внутреннего трения увеличился бы в степени,
достаточной для возникновения критических разогревов. Поэтому
хотя «химически» нитроглицерин и весьма чувствителен, эта чув-
ствительность в указанных условиях не может проявиться. Поведе-
ние нитроглицерина в этих условиях аналогично поведению тротила,
который хотя и менее текуч, но одновременно и гораздо менее
чувствителен химически.
В этой связи, однако, возникает вопрос: почему взрыв нитрогли-
церина, а также и тротила, гораздо легче возбудить в приборе № 1
и каким образом, не прибегая к этому прибору, можно характери-
зовать чувствительность таких взрывчатых веществ?
59
Как мы уже неоднократно отмечали, взрыв возникает при ударе
на копре, если возможно не только возникновение химического пре-
вращения вещества, но и распространение и развитие этого пре-*
вращения до взрыва.
Для возникновения превращения необходимо достаточно напря-
женное течение, а для распространения его — достаточная толщина
слоя вещества. В приборе № L реализуется сочетание обоих усло-
вий— напряженное течение в зазоре, точнее в том его месте, где
расширившийся при ударе ролик сужает просвет зазора, и более
толстый слой вещества в той части зазора, где ролик менее рас-
ширился, а также между торцами, где вещество не успело еще
выдавиться.
Чтобы воспроизвести такие условия, благоприятствующие воз-
никновению взрыва ВВ с большой текучестью, можно в принципе
идти двумя путями.
1. Увеличить размеры (высоту и диаметр) заряда, а соответст-
венно и роликов, равно как и вес груза; тогда большие напряжения
при течении будут возникать при толщине заряда еще превышаю-
щей критическую, и возникшие разогревы смогут развиться во
взрыв.
2. На торце ролика сделать лунку и поместить в нее несколько
больше ВВ, чем соответствует ее объему; в этом случае избыток
взрывчатого вещества при ударе будет выдавливаться между тор-
цами роликов в виде тонкого слоя аналогично тому, как это имеет
место в зазоре прибора № 1; возникшее на границе лунки воспла-
менение сможет распространяться в толстом слое взрывчатого ве-
щества, находящегося в лунке; при такой постановке опыта роль
зазоров прибора № 1 выполняет зазор между торцами роликов *.
Результаты опытов с нитроглицерином, выполненных по этой
схеме, приведены в табл.2.
Таблица 2
Частость взрывов нитроглицерина в приборе, имеющем ролик с лункой
(Объем лунки 0,01 см3, диаметр ее 5 мм, глубина 0,8 мм',
груз 10 кг, высота его падения 25 см)
Навеска в мг Число взрывон Число опытов
10 5/5
14 9/10
28 4/10
Другая возможность «проявления» потенциальной чувствитель-
ности текучего взрывчатого вещества состоит в том, чтобы искус-
ственно образовать в нем при ударе на копре очаги разогрева,
1 Если лунка заполнена лишь частично и нитроглицерин не попадает в зазор'
между торцами роликов, то взрывов не наблюдается; они возникают также труд-
нее, если в пространство между торцами выдавливается при ударе слишком мно-
го жидкости.
60
которые течение ВВ само по себе из-за малого коэффициента внут-
реннего трения создать не может. Это может быть достигнуто, на-
пример, введением во взрывчатое вещество немногих твердых ча-
стиц, трение которых о поверхность ударника или наковальни при-
водит к разогревам. Опыты подтверждают реальность этой
возможности (см. серия 6 в табл. 1).
Затруднить при опыте течение ВВ можно, поместив легкотеху-
чее вещество в чашечку из мягкого металла. В табл. 3 приведены
результаты соответствующих опытов с тротилом и нитроглице-
рином.
Таблица 3
Частость взрывов взрывчатых веществ,
помещенных в чашечку нз красной меди
(Объем чашечки 0,035 ем3, диаметр ее 5 мм, высота 1,8 мм;
груз 10 кг, высота его падения 25 ем)
Взрывчатое вещество Навеска в мг Число ВЭО’В"В
Число опытов
Тротил 50 7/25
Нитроглицерин 30 6/10
40 4/10
50 3/5
Таким образом приведенные результаты опытов хорошо согла-
суются с представлениями о механизме возбуждения взрыва при
ударе, выдвинутыми и развитыми Н. А. Холево. Эти представления
И должны служить для оценки чувствительности взрывчатых ве-
ществ к механическим воздействиям. Они позволяют применительно
К условиям удара приближенно дифференцировать роль химических
и физико-механических факторов чувствительности и соответствен-
но дают возможность получить более полное представление о свой-
ствах ВВ, чем при стандартном способе испытания.
О МЕТОДИКЕ ИСПЫТАНИЯ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
НА ЧУВСТВИТЕЛЬНОСТЬ ПРИ УДАРЕ
В настоящее время испытание ВВ на чувствительность при ударе
регламентировано ГОСТ 4545—48 н 2065—43. Эти стандарты пре-
дусматривают помещение взрывчатого вещества при испытании в
роликовый прибор, который является упрощенным вариантом штем-
пельного прибора Каста, предложенного последним в 1906 году.
В основу устройства штемпельного прибора были положены сле-
дующие, казавшиеся тогда бесспорными, соображения. Большие
колебания в результатах испытания, наблюдающиеся на копре,
вызываются непостоянством условий удара, в частности площади
заряда, на которую действует удар, н непараллельностью соударя-
ющихся поверхностей. При этом к ударному действию присоеди-
61
няется трение. Для устранения этих предположительных недостат-
ков и была принята кастовская, а затем и современная конструкция
прибора для испытания ВВ на копре.
В последнее десятилетие, однако, было показано, преимущест-
венно работами Н. А. Холево, что при ударе на копре взрыв во мно-
гих случаях возникает, по-видимому, не между торцами роликов,
а при продавливании ВВ под действием удара в зазор между роли-
ками и направляющей муфтой. В соответствии с этим даже если
при испытании было обеспечено постоянство площади удара и на-
правления движения соударяющихся поверхностей, но условия те-
чения ВВ после выхода его из пространства между торцами роликов
были различными, то частость взрывов может резко изменяться.
Роль этих условий наглядно демонстрируется результатами испы-
таний в предложенном Н. А. Холево приборе М21, Испытание в
приборах двух типов — с затрудненным (№1) и облегченным (№ 2)
течением заряда дает более полное представление о чувствитель-
ности ВВ, чем испытание в одном из них. Предусмотренный ГОСТ
2065—43 прибор, в котором ролики имеют фаски, занимает проме-
жуточное положение между приборами №№ 1 и 2.
Однако, если взрыв происходит в зазоре между роликами и
муфтой и возникновение его зависит от величины этого зазора, то
конструкцию такого прибора нельзя считать рациональной. В самом
деле величина зазора может незакономерно меняться от опыта к
опыту из-за смещения оси роликов относительно оси канала муфты.
При ударе выдавливание ВВ в зазор происходит обычно, как можно
было видеть в опытах, где В В вытекало за пределы канала, одно-
сторонне, очевидно там, где зазор оказался шире. Далее канал
муфты по идее должен выполнять только функцию направления
движения ролика и поэтому к соответствующим размерам (внут-
реннему диаметру) и обработке поверхностей его предъявляются
высокие требования. Поскольку же фактически он служит местом,
где возникает взрыв, то газы взрыва быстро эродируют канал и де-
лают его мало пригодным для точного направления движения ро-
лика. В известной мере это относится и к прибору № 2; хотя взрыв
в нем происходит не в зазоре, но газы могут выходить только через
канал муфты и поэтому они также вызывают его эрозию.
В свете этих соображений представлялось целесообразным так
изменить конструкцию прибора, чтобы горячие газы взрыва не по-
падали в канал направляющей муфты и не вызывали его быстрого
износа. Это и реализовано в приборе Ns 4, в котором муфта может
служить без замены длительное время. Помимо повышения посто-
янства условий испытаний это изменение конструкции прибора сни-
жает стоимость испытания. Муфта требуемого ГОСТом качества
(в отличие от роликов) обходится довольно дорого, а при испыта-
нии в приборах №№ I или 2 мощных ВВ деформация и эрозия ка-
нала наступают быстро и муфту приходится часто менять. Неко-
торые исследователи ограничивались даже лишь однократным
1 См. статью Н. А. Холево в настоящем сборнике (стр. 5).
62
использованием муфты, что очень удорожало испытание. Примене-
ние прибора № 4 существенно бы его удешевило и позволило бы
в одном приборе получать ту характеристику чувствительности,
которая до сих пор могла быть получена только при условии испы-
» тания ВВ в двух приборах — № 1 и 2.
Основное преимущество комплексного испытания в приборах
v № 1 и 2 заключается в том, что оно позволяет характеризовать,
не только термокинетическую, но и физико-механическую сторону
(в частности текучесть) чувствительности ВВ. Обе эти характери-
стики, как мы видели, могут быть установлены в предлагаемом,
приборе № 4 при следующей методике испытания твердых взрывча^
тых веществ: ВВ испытывают при двух расположениях навески на
торце нижнего ролика:
1) в виде слегка (от руки) подпрессованной таблетки диамет-
рам 5 мм, помещенной в центре ролика (такое расположение в
дальнейшем для краткости будем называть центральным);
2) в виде слоя равномерной толщины, распределенного по всему
торцу ролика (в дальнейшем будем называть «равномерное рас-
пределение»).
Испытания проводят с навесками нескольких величин1, причем
для каждой навески и каждого варианта ее распределения на ро-
лике выполняют определенное число (например, 25) параллельных
опытов, устанавливая частость взрывов. В опытах, где взрыва не
было, фиксируют высоту отскока груза после удара.
При центральном расположении создаются облегченные условия
течения ВВ, аналогичные реализуемым в приборе № 2, где соче-
тается малый диаметр роликов (большое давление при ударе) и на-
личие канавки. В случае центрального расположения навески (в
приборе № 4) течение облегчается концентрацией энергии удара на
малой площади заряда и относительно большим зазором между
роликами. Равномерное размещение заряда создает затрудненные
условия для течения ВВ, так как действие удара в этом случае
приходится на большую площадь и давление мало, а зазор между
роликами в начальной стадии удара меньше, чем при центральном
расположении навески.
Рассмотрим теперь толкование возможных результатов опытов-
с разными ВВ.
Допустим, что при равномерном размещении малой навески по-
лучена низкая частость взрывов. Это может произойти по двум:
причинам;
1) вещество термокинетически мало чувствительно и не взры-
вается в тонком слое;
2) его текучесть мала и оио не течет в данных условиях.
Повторяем опыт при центральном расположении навески, кото-
рое обеспечивает большее давление при ударе и облегчает тече-
ние ВВ. Если частость взрывов увеличивается, то отсутствие их или1
1 Судя по полученным опытным данным, достаточно полное представление
о чувствительности обычных ВВ можно получить при испытании двух навесок —
10 и 50 мг.
ОЗ-
меньшая частость при равномерном размещении были следствием
малой текучести. Примером таких веществ (табл. 4) могут слу-
жить гексоген, октоген, тэн, тринитрорезорцинат свинца и др.
Если, однако, для двух ВВ при маленькой навеске получается
малая частость взрывов как при равномерном, так и при централь-
ном расположении, то это еще не означает, что они имеют одинако-
вую низкую чувствительность. Малая частость взрывов может быть
обусловлена тем, что для обоих веществ при малой навеске слиш-
ком тонок слой ВВ, тоньше, чем необходимо для распространения
горения или взрыва. Различие между веществами тогда проявится
при больших навесках. Малочувствительное вещество по-прежнему
не будет давать взрывов; вещество же с большей чувствительностью
начнет их давать с большей частостью. В качестве примера можно
указать тринитроанилнн и стифнат калия. Оба они в малых навес-
ках не взрываются; однако тринитроанилин не взрывается и при
переходе к большим навескам, в то время как стифнат калия (при
центральном расположении) дает 100% взрывов.
Испытание при увеличенной навеске (50 мг) полезно и в том
отношении, что оно позволяет по величине отскока груза при отка-
зах судить о текучести вещества '.
Если текучесть мала, то ВВ не успевает значительно выдавиться,
затрата энергии на выдавливание его невелика и поэтому отскок
большой. Велик отскок и при центральном расположении заряда,
хотя как правило, он естественно меньше, чем при равномерном
размещении ВВ.
С другой стороны уменьшение отскока по сравнению с холостым
ударом дает возможность убедиться, что течение вещества при
ударе имеет место и что если при этом получаются отказы, то при-
чина их не в отсутствии течения.
Следует, однако, указать, что большой отскок может получиться
не только при малой текучести равномерно размещенного вещества,
но и в том случае, если текучесть относительно велика и затрата
энергии на выдавливание вещества соответственно мала. Это на-
блюдается, в частности, для некоторых легкоплавких ВВ, например,
для тротила. Характерно, что в этом случае при центральном рас-
положении навески получается больший отскок, по-видимому,
вследствие расплавления вещества при интенсивном течении и рез-
кого в связи с этим увеличения его текучести.
Сопоставление отскока и частости взрывов при разных навесках
и их расположениях позволяет в большинстве случаев более полно
характеризовать «химическую» и «механическую» сторону чувстви-
тельности испытуемого ВВ, чем при помощи каждого из этих пока-
зателей в отдельности.
Поясним это анализом данных табл. 4, где сведены результаты
испытания по предлагаемой методике 28 вторичных и инициирую-
щих ВВ.
1 При малых навесках различия в отскоках при испытании разных ВВ малы,
так как относительно мала затрата энергии иа выдавливание вещества.
64
Тринитробензол имеет значительную текучесть, о чем можно
судить по сильному уменьшению отскока не только при централь-
ном, но и при равномерном размещении заряда (навеска 50 мг).
Одновременно он химически мало чувствителен, и, чтобы возникали
взрывы, необходимы напряженные условия течения ВВ, которые
создаются прн навеске 10 мг и равномерном ее распределении. При
центральном расположении заряда течение облегчается ‘из-за боль-
шего удельного давления, а также из-за плавления ВВ при течении,
в результате напряженность течения становится меньше (при этом
следует учитывать, что температура плавления тринитробензола
относительно температуры вспышки низка).
Тротил в химическом отношении близок в триинтробензолу, но.
текучесть его значительно больше. При центральном расположении
; тротилового заряда отскок существенно больше, чем у трннитро-
бензола. Текучесть тротила столь значительна, что уменьшение На-
S', вески до 10 мг приводит к значительно меньшему (до 24% вме-
г' сто 56% для тринитробензола) росту частости взрывов даже при
равномерном размещении заряда. Не дает увеличения чаЬтости
взрывов и дальнейшее уменьшение навески тротила (до 2 мг)-, по-
; видимому толщина слоя ВВ оказывается прн этом меньше критиче-
ской для горения. Сочетание химической инертности со значитель-
ной текучестью и делает тротил одним нз наименее чувствительных
взрывчатых веществ, находящих практическое применение.
Пикриновая кислота ведет себя аналогично трннитробензолу.
/ Текучесть ее заметно меньше (отскок больше), но химическая Чуб-
S. ствительность больше. Максимум частости взрывов пикриновой
/ кислоты лежит как и у тринитробензола при навеске 10 мг и рав-
1. номерном ее размещении. Отличие от триннтробензола состоит в
том, что у пикриновой кислоты наблюдается значительная частость
. Взрывов и при центральном расположении заряда.
У тринитрорезорцина текучесть еше меньше, и при равномерном
распределении он показывает низкую частость взрывов при всех
навесках. Химически он чувствительнее пикриновой кислоты и дает
высокую частость при центральном расположении заряда даже при
наименьшей навеске.
Физически сходен с тринитрорезорцином тринитроксилол, но
кнмнчески он менее чувствителен и максимум частости взрывов
у него ниже.
Нитроамины (тринитроанилин и тринитроднамниобензол) ха-
рактеризуются низкой текучестью. При равномерном размещении
даже при 50 мг отскок соответствует холостому удару и частость
взрывов равна нулю. Однако частость взрывов остается низкой и
при центральном расположении навески, когда судя по отскоку,
: течение ВВ имеет место. Это свидетельствует, что и химическая
чувствительность нитроаминов мала, что впервые было отмечено
Ф. А. Баумом.
Тетрил по текучести близок к пикриновой кислоте, близок он
к ней и по частости взрывов в различных условиях.
1 5 451
Таблица 4
8
Результаты испытания взрывчатых веществ на копре в приборе ЛЬ 4
(Груз 10 кг, высота его падения 25 см, число опытов в каждой группе 25,
отскок груза при холостом ударе 72М высоты его падения)
Условные обозначения: Ц — центральное расположение; Р — равномерное распределение
№ по пор. Наименование вещества Темпе- ратура плавле- ния •С Располо- жение навески ВВ Величина навески ВВ
2 мг 10 мг 50 мг
% взры- вов Средний отскок груза при отказах в % высоты падения % взры- вов Средний отскок груза при отказах в % высоты падения % взры- вов Средний отскок груза при OTKiSix н откло- нения его от среднего значения з % высоты падения
1 Тринитробензол 122,5 ц 4 70 8 66 4 38^1
р 20 68 56 66 4 421м
2 Тринитротолуол 80,5 ц 0 68 0 66 0 47tf
р 4 68 24 67 4 42±!|
3 Пикриновая кислота 122,5 ц 12 69 32 68 36 47±?
р 0 69 64 69 24
4 Тринитрорезорцин 178 ц 32 70 32 62 84 40+«4 w-24
р 4 71 0 71 8 68±5
5 Тринитроксилол 182 ц 8 71 20 64 44 4g+ 12 ™-l3
р 4 70 12 66 36 62±b
6 Тринитроанилин 191,5 ц 0 72 4 62 12 46 ±30
р 0 72 0 71 0 70+2 fu-13
: Г - : -г - .
7 Тринитроднамино- 289 ц 4 70 32 66 ' 4 45+*
бензол р Р 70 О , 65 0 68 ±0
.8 : Тетрил 129 Ц : 16 72 20 66 16 45^14
Р , 8 72 0 . 66 44 52±*
9 ‘ Гексил 240 ц : 96 : 68 100 — 92 52 ±0
р 8 ; 69 8 63 : 8 65t;
10 ; Г ексоген 204 ц : 96 ; 72 92 65 92 54±1
р 0 72 . 0 68 0 66Ц
n . Октоген 280 И : 92 72 100 — —
P 0 . 72 0 71 4 68 ±3
12 Тэн 140 ц : 64 : 71 92 64 84 5211з
i р 0 70 0 67 8 681®
13 Дина 52 ц 16 72 44 65 60 61 ±3
р 4 72 68 68 88 6112
14 Гексанитродифенил 240 ц 28 70 68 62 44 481g
р 0 72 0 71 28 6611
.15. Гексанитродифе- 260 ц 92 72 100 60 50 ±4
нилоксид р 84 72 68 70 20 57^
16 Гекса нитрооксани- 331 ц 76 71 100 —г- 100 —
ЛИД р 0 72 4 70 8 671f
17 . Пикрат лития • — Ц ; 20 . 69 12 - 66 16 421^
р 0 71 0 67 12 7011
• • .— . . .. . ' .... 1
Продолжеяие
о Средний отскок грум при отказах и отклоне- ния его от среднего значения в & высоты падения 1 1 +1 1 "И + 1 + 1 1 [ 1 I 1 ° i +1 1 см о + 1 + 1 +1 1 ' Sig 1 8 га Й 1 1 1 1 1*" ! $ 1 ЙЙ
3 щ Ж ££ 3 “ | [§>«>£<= О | j | , ggg £> |<О£© JO
10 Mi I Средний отскок груза при отказах В 9-6 высоты падения 1^1 СО г О О О ] I I 1 1 Ср 1 f тмф » 0^01 | Ф 1 Ю | г- «5 г- 1 1 I | 1 © | о 1 ь- © £5 ю
И взры- вов 838° 8° 3*11) 18S8S83S2 3*
) г*~ Z Средний отскок груза при отказах в высоты падения 1 1 й $ Й й S й IS 13 IS 1 S 1 8S £ ° 1
к взры- вов । о-г о со <= g&g^ggg о « $ |
Располо- жение навески ВВ Си Си Си Си Си Си си CL
* Навеска 20 mi,
68
Гетероциклические N-нитрамины (гексоген и октоген) имеют
низкую текучесть, но химически чувствительны и с очень большой
частостью взрываются при центральном расположении навески,
Низкую текучесть и соответственно значительно более высокую
частость взрывов показывают гексил н тэн при центральном распо-
ложении заряда в сравнении с равномерным его размещением,
Гексанитродифенил имеет относительно низкую текучесть, по-
этому в отличие от тринитробензола в навесках 2 и 10 мг при рав-
номерном размещении взрывов не получается. С умеренной часто-
стью наблюдаются они лишь при навеске 50 мг. При центральном
расположении заряда частость взрывов гексанитродифенила в от-
личие от тринитробензола значительно больше. Аналогичную кар-
тину дает гексанитрооксанилид, но частость взрывов при централь-
ном расположении заряда у него больше, чем у гексанитроди-
феи ила.
Низкую текучесть и соответственно малую частость взрывов при
равномерном размещении заряда показывают пикраты и стифнаты
калия и лития; при центральном расположении частость взрывов у
них велика за исключением пикрата лития, что, возможно, связано
с большой его гигроскопичностью.
Азиды (аммония, свинца и калия) характеризуются низкой теку-
честью и соответственно малой частостью при равномерном распре-
делении навески, но вследствие высокой химической чувствитель-
ности частость взрывов при центральном расположении зарядов
этих ВВ становится велика.
К труднотекучим взрывчатым веществам относятся черный по-
рох и перхлорат аммония; при центральном расположении они дают
большую частость взрывов.
Поведение некоторых из изученных веществ весьма своеобразно.
Так, тетразен и гремучая ртуть текут плохо, но тем не менее взры-
ваются с большой частостью; гексанитродифенилоксид и дина не
различаются существенно по отскоку при центральном расположе-
нии и равномерном размещении заряда; они ие показывают осо-
бого различия и в частости взрывов.
В случае испытания инициирующих ВВ с их высокой химической
чувствительностью для возникновения горения и взрыва возможно
достаточно тех небольших локальных смещений, которые пред-
шествуют макротеченню. Для дины могут играть роль модифика-
ционные переходы при повышении давления.
В целом испытание по данному методу позволяет при помощи
одного прибора получить гораздо более,разностороннюю оценку
чувствительности твердых ВВ к удару, чем ее дает стандартный
способ. Изменяя при опыте условия течения ВВ путем изменения
расположения и величины навески и регистрируя частость взрывов
и величину отскока (при отказах), можно дать оценку взрывчатого
вещества в отношении опасности возникновения взрыва при раз-
личных условиях. Эта оценка не выражается в абсолютных едини-
цах. Такое выражение и не имеет смысла, поскольку, как мы ви-
дели, частость взрывов одного и того же ВВ при данной энергии
«9
воздействия 1*<ожет изменяться в Широких пределах. Основными
факторами этой частости нз свойств взрывчатого вещества являются
воспламеняемость'и текучесть. Решающее влияние могут иметь, как
ясно из вышесказанного, также условия механического воздействия.
Поэтому оценка чувствительности ВВ безотносительно к условиям
этого воздействия лишена практического смысла.
В описанных выше опытах мы пользовались грузом в 10 кг и
применяли только одну высоту падения 25 см лишь потому, что эти
условия наиболее употребительны. Естественно, что могут быть
применены разные высоты и грузы. Точно так же можно определять
не только частость прн данных условиях удара, но и нижний предел
условий, начиная с которого получаются взрывы.
Следует остановиться на вопросе об испытании жидких взрыв-
чатых веществ. Их текучесть слишком велика, чтобы при данном
диаметре роликов и скорости удара в результате течения образо-
вались разогревы, достаточные для возникновения взрыва. Напря-
женное течение возникает в условиях прибора № 4 лишь при столь
Тонком слое, который уже не способен взрываться. Поэтому даже
если способность к распространению взрыва у данного жидкого ВВ
относительно велика, как это имеет место в случае нитроглицерина,
частость взрывов у него получается близкой к нулю. Выявить чувст-
вительность нитроглицерина, а она высока, можно только, в том
случае, если обеспечить возникновение в нем разогрева при ударе
некоторыми особыми приемами. Можно, например, с этой целью
ввести в нитроглицерин твердые частицы, сделать лунку на
торце ролика или- поместить жидкость в чашечку нз относительно
мягкого металла. Наконец, можно обеспечить возникновение взры-
ва нитроглицерина при помощи пузырька газа, располагая навес-
ку ВВ на наковальне в виде кольца, как это делал Боуден, или
осуществляя удар несколько приподнятым верхним роликом1.
Реальная высокая опасность жидкого нитроглицерина в технике
обусловлена, по-внднмому, именно двумя последними условиями
удара.
Сказанное о жидких ВВ относится и к таким твердым, но очень
Текучим и одновременно химически мало чувствительным взрывча-
тым веществам, как тротил; Для него даже те наиболее напряжен-
ные условия течения, которые создаются при равномерном распре-
делении заряда, не приводят, как правило, к взрыву — при малых
навесках, потому что взрыв не может распространяться в столь
тонком слое, при больших — потому что он не возникает. Опять-
таки в этом случае, чтобы получить взрыв, надо сочетать условия
возникновения и распространения взрывчатого превращения. Это
наблюдалось в приборе № 1 е определенной величиной зазора, а в
приборах № 4 и 2 — в узком интервале условий опыта (см., на-
пример, табл. 4), при введении в тротил крупинок стали, а также
при ролике с лункой. Вероятно взрыв этого ВВ с большой час-
тостью можно получить и в обычных условиях испытания на копре,
1 См, настоящий сборник, стр. 72.
70
увеличив масштаб опыта, так'чтЬбы напряженное течение осущест-
влялось при большой толщине заряда. Такие условия в технике,
однако, редко встречаются, чем и обусловлена весьма малая чувст-
вительность тротила в практических условиях его использования.
В.заключение следует указать, что хотя описанный способ, ис-
пытания дает лучшее представление о чувствительности, чем стан-
дартный и он проще и дешевле в исполнении, все же принципиаль-
ная односторонность испытания на чувствительность при ударе, как
характеристики опасности взрывчатого вещества в обращении, со-
храняется и в нем. Эта односторонность заключается прежде всего
в том, что испытание на копре характеризует главным образом воз-
можность возникновения очагов взрыва, но не дает реального пред-
ставления о возможности его распространения. Кроме того, даже
возможность возникновения взрыва при обычном ударе по нормали
ограничивается текучестью вещества, почему, например, азид свин-
ца в этом случае может давать меньшую частость взрывов, чем
тротил. Этот последний недостаток может быть устранен приме-
нением при испытании скользящего удара. Для устранения же пер-
вого недостатка необходимо, испытание на копре дополнять опреде-
лением способности ВВ к распространению горения или взрыва в
тех условиях, для которых надо определить опасность в обращении
с данным взрывчатым веществом. Только такое дополнение позво-
лит установить, что например, перхлорат аммония, по частости
взрывов близкий к тэну и черному пороху, несравненно менее опа-
сен, чем тэн, поскольку критический диаметр для детонации тэна в
десятки раз меньше, чем у перхлората. Перхлорат аммония также
гораздо менее опасен, чем черный порох, потому что критический
диаметр для горения черного пороха при атмосферном давлении
около 1 леи, а у перхлората аммония около 33 мм. Очень низкие
критические диаметры для горения даже при атмосферном давле-
нии и для детонации, характеризующие инициирующие взрывчатые
вещества, являются главной причиной их высокой опасности в об-
ращении. В силу этих соображений следует полагать, что в буду-
щем определение способности к горению и детонации в сочетании
с испытанием на скользящий удар станет основным методом харак-
теристики опасности ВВ в обращении,
выводы
Предложен комплексный способ определения относительной чув-
ствительности ВВ к механическим воздействиям. Взрывчатое веще-
ство подвергают удару между торцами двух роликов значительного
диаметра, причем пространство, окружающее стык роликов — от-
крыто; изменяется величина навески и ее расположение на торце
ролика. Преимуществами этого способа по сравнению со стан-
дартным является большая возможность дифференцирования хими-
ческих и физических факторов чувствительности и рациональное
устройство прибора, существенно увеличивающее его долговечность
и постоянство условий испытаний и снижающее их стоимость.
71
К. К. АНДРЕЕВ. Ю. Л. ТЕРЕБИЛИ НА
5. К ВОПРОСУ о влиянии ВОЗДУШНЫХ ВКЛЮЧЕНИИ
НА ВОЗНИКНОВЕНИЕ ВЗРЫВА ПРИ УДАРЕ
Большинство исследователей обоснованно считает, что химиче-
ская реакция и взрыв прн ударе возникают не от механического
действия как такового, а в результате вызываемых последним ра-
зогревов. В вопросе о том, какими путями реально возникают разо-
гревы в различных ВВ и при разных условиях удара нет, однако,
полной ясности.
Две основные точки зрения получили наибольшее развитие в
последнее время. Н. А. Холево [2] как главный путь возбуждения
реакции при ударе рассматривает течение взрывчатого вещества и
местные его разогревы, возникающие вследствие внутреннего тре-
ния. Боуден [3] большую роль, особенно в случае жидких ВВ, при-
писывает включениям газов или паров, сжимаемых при ударе. Мно-
гочисленными опытами этого исследователя и его сотрудников [1]
было показано, что энергия удара, необходимая для возбуждения
взрыва жидких взрывчатых веществ, может очень сильно (на не-
сколько порядков) уменьшаться при введении в ВВ газовых пу-
зырьков.
Эти наблюдения были в работах Боудена естественно объяснены
тем, что в газах при адиабатическом их сжатии до некоторого опре-
деленного давления развивается гораздо большая температура, чем
в жидкости; нагретый пузырек газа разогревает н поджигает окру-
жающее его взрывчатое вещество.
Однако, будучи правильным, это объяснение, по-видимому, не
исчерпывает роли пузырьков. Существенны не только высокая
температура, возникающая при их сжатии, но и условия
взаимодействия, в частности теплообмена, разогретого газа с при-
легающим к нему взрывчатым веществом. Описываемые ниже опы-
ты поясняют сказанное.
Первоначальной целью этих опытов было сопоставление чувст-
вительности некоторых взрывчатых веществ в жидком и твердом
состоянии. Опыты проводились в приборе № 4 Чтобы обеспечить
одинаковые условия и в частности предотвратить выдавливание ча-
сти вещества при обычной постановке опыта с жидкостью в роли-
ковом приборе, когда верхний ролик своим весом давит на жид-
кость, испытуемое ВВ помещали на торце нижнего ролика в виде
капли, жидкой или замерзшей, а верхний ролик закрепляли при
помощи резинового колечка нли слабой пружинки иа небольшой
высоте (-^3 мм) над нижним роликом. Такие опыты, проведенные
с нитроглицерином, дали неожиданно высокую частость взрывов
при том же грузе и высоте падения, при которых в обычных усло-
виях испытания (верхний ролик перед ударом стоит на ВВ) ча-
стость взрывов была близка к нулю. Кроме того, было установлено,
что увеличение или значительное уменьшение количества жид-
1 См. настоящий сборник, стр. 56—58.
72
кого В В снижает частость взрывов. Таким образом оказалась, что
в данном случае результаты 'испытания особенно чувствительны к
условиям опыта.
Эффект поднятого ролика наблюдается не для всех жидкостей.
Так переохлажденный раствор тротила в тетриле (50 : 50) или жид-
кая дина не показывают его
по крайней мере прн данных
условиях опыта.
Аналогичные наблюде-
ния, по-видимому, были сде-
ланы и в работах Боудена с
сотрудниками, которые объ-
ясняли повышение чувстви-
тельности ВВ к удару захва-
том воздуха падающим гру-
зом. Поскольку действие воз-
духа более регулярно и силь-
но проявляется при наличии
углубления в ударнике или
прн неравномерном располо-
жении взрывчатой жидкости
на наковальне, Боуден уде-
лил этим последним усло-
виям опытов основное вни-
мание. Он отмечает, однако,
[1], что и при плоском удар-
нике, падающем на откры-
тый слой нитроглицерина,
большое значение имеет диа-
Фиг. 1. Влияние условий опыта на величи-
ну энергии удара, необходимую для воз-
буждения взрыва нитроглицерина (во
Боудену).
/—в ударвике имеется лунка с маленький пу-
зырьком rasa. 2—в слое ВВ на вакавальве
пузырьки воздуха, 3—ВВ расположено на иако-
вальве параллельными полосками, 4— ВВ ряска-
ложево на наковальне в виде отдельных капе-
лек, 5—удар производится плоским ударяикои
по пленке ВВ, не содержащей газовых пузырь-
ков. 6—то же по пленке ВВ, содержащей газо-
вые пузырьки.
метр ударника, с увеличе-
нием которого частость взрывов возрастает; при диаметре латун-
ного ударника 0,6 см взрыв вызвать очень трудно, а ударником диа-
метром 2,5 см гораздо легче, что естественно объяснить большей
вероятностью захвата воздуха. Боуден указывает также, что очень
толстые слон жидкости менее чувствительны, чем тонкие, предпо-
ложительно, потому что в первом случае пузырек более подвижен
н может ускользнуть прежде, чем давление повысится до значитель-
ной величины. Уменьшение частости взрывов наблюдается и при
слишком тонких слоях.
При соответствующих условиях вступление в действие «пузырь-
кового* механизма возникновения взрыва может приводить к огром-
ному повышению чувствительности ВВ, точнее к уменьшению ра-
боты удара, приводящей к возникновению взрыва. Представление
об этом влиянии дает взятая из работы Боудена фиг. 1, показыва-
ющая частость взрывов нитроглицерина в зависимости от энергии
удара при различных условиях опыта.
Боуден объясняет сделанные им наблюдения, особенно при опы-
тах с углублением в ударнике, адиабатическим разогревом воздуха,
ведущим к поджиганию взрывчатого вещества. Принципиальная
73.
возможность такого механизма воспламенения ВВ бесспорна. В не-
скольких работах, например [4], было показано, что если взрывча-
тое вещество поместить в цилиндр с газом и затем сжать этот газ
при помощи удара по поршню, то ВВ воспламеняется, В этой поста-
новке опыта механизм воспламенения, предлагаемый Боуденом,
реален. Однако при ударе по поверхности ВВ свободно падающим
ударником процесс воспламенения, по-видимому, протекает не-
сколько сложнее. Воздух, находящийся между ударником и нако-
вальней, по мере их сближения сжимается и приходит в движение
в радиальном направлении со: все возрастающей скоростью; при
сжатии он разогревается. Повышенная температура является одним
из основных факторов, определяющих возможность воспламенения
взрывчатого вещества. Второй фактор — повышение давления, кото-
рое, как известно, увеличивает воспламеняемость взрывчатого ве-
щества. Третий фактор —движение газа вдоль поверхности заряда;
это движение повышает коэффициент теплопередачи. Наконец, газ,
двигаясь вдоль поверхности заряда при достаточной скорости дви-
жения, может захватывать с собой капли или частицы ВВ. Если
этот захват не слишком мал (тогда его влияние мало) и не слиш-
ком велик (тогда поступление чрезмерно большого количества хо-
лодного вещества может снизить температуру разогретого возду-
ха), то он может сильно увеличить количество тепла, выделивше-
тося при сжатии, и лривести к воспламенению основной массы ВВ.
В табл. I приведены экспериментальные данные о влиянии при-
поднятости верхнего ролика прибора № 4 на частость взрывов не-
которых ВВ при испытании нх на копре. Для иллюстрации особен-
ностей расположения заряда ВВ в 23—26 сериях этих опытов на
фиг. 2 дана соответствующая схема.
Рассмотрим данные табл. I в свете высказанных выше сообра-
жений о механизме воспламенения ВВ воздухом, разогретым при
ударе.
Как для нитроглицерина, так и для ннтрогликоля частость
взрывов при поднятом ролике гораздо больше, чем при ролике,
поставленном на жидкость. Числа, приведенные в табл. 1, когда
они близки к нулю или к 100%, естественно неполностью характе-
ризуют явление, но резкая разница между результатами при раз-
ных положениях ролика несомненна. Для обоих ВВ наблюдается
некоторая оптимальная величина навески, увеличение или умень-
шение которой уменьшает частость взрывов. Для желатинирован-
ного нитроглицерина (серии опытов 14—18) эффект поднятого ро-
лика полностью сохраняется 1. Частость взрывов возрастает и при
1 Это обстоятельство было несколько неожиданным. Желатинированный ни-
троглицерин содержал, как обычно, много пузырьков воздуха и не обрабатывался
специально для их удаления. Тем не менее при поднятом ролике частость взрывов
резко возрастала. Это говорит за то, что пузырек, образующийся 'иа поверхности,
гораздо эффективнее в смысле возбуждения взрыва, чем находящийся в массе
вещества. В этой связи следует обратить внимание на следующее обстоятельство.
Если ВВ заранее накрывается роликом или фольгою, то устанавливается адге-
зионный контакт между ними. При ударе же поднятым роликом ВВ приходит
в сопри кос нов а ние с поверхностью металла так быстро, что адгезионный контакт
может не успеть установиться.
74
Таблица 1
Влияние при пп дня тост и верхнего ролика над нижним на частость взрывов
при испытании некоторых жидких и твердых взрывчатых веществ
на копре
(прибор № 4, вес груза 10 кг, высота падения 25 см,
число опытов в серии 25)
№ серии опытов Взрывчатое вещество и его расположение на ролике прибора Навеска ВВ мг Частость взрывов в 94 при ролике Примечание
ПОДНЯТОМ опущен- ном
1 0,25 76 —
2 0.5 84 0
3 5 100 8
4 Нитроглицерин в виде капли 10 92 83 0
5 20 —
6 50 20 —
7 * 10 — 40 В ВВ поло- жены 2—3
крупинки
стали
8 о.з 56 —
0 0,5 72 8
10 Нитрогликоль в виде кап- 1 92 —
11 ЛИ 2 72 ' 16
12 7,5 76 4
13 15 28 0
14 2 100 4
15 Нитроглицерин желатинн- 5 100 4
рованный (3^), расположен 80 п
10 комочком в центре ролика
17 25 56
Нитроглицерин желатини- 96
18 рованный (7И), расположен 5 4
комочком в центре ролика
19 20 — 12
20 Нитроглицерин желатини- 20 — 60 Лунка в навеске
21 рованный (1094), расположен комочком в центре ролика 50 .— 15
22 50 — 65 Лунка
* в навеске
75
Продолжение
м серин опытов Взрьщчатое вещество и его расположение на ролике прибора Навеска ВВ мг Частость взрывов в % при ролике Примечание
поднятом опущен- ном
23 24 Нитроглицерин желатини- рованный (7%), расположен равномерно по всему роли- ку; между ВВ и верхним роликом фольга 5 5 48 96 ' 1 1 Фиг, 1а Фиг, 16
25 26 То же, ио навеска распо- ложена комочком в центре ролика; между ВВ и верх- ним роликом фольга 5 5 60 100 1 1 Фиг. 2в Фиг, 2г
27 Дина (закристаллизовав- шаяся капля) 5 8 20
28 Тетрил (закристаллизовав- шаяся капля) 5 12 16
29 Гексоген, расположен в виде кучки в центре ролика 20 2/15* 0/15*
30 Сплав тротила с тетрилом 50:50 (переохлажденная капля) 40 0 4
31 32 Нитроглицерин (замерз- шая капля) 5 5 48 28 4 4 ВВ накрыто фольгой
33 Азид свинца, расположен в виде кучки в центре ро* лика 20 100 4**
34 - Азид кальцин, расположен в виде кучки, в центре ро- лика 20 76 12
* В знаменателе — число опытов, в числителе — число взрывов.
** При опускании ролика из кучку ВВ <а'=г5 Мм) происходит ее .раздлпли-
ванне": порошок распределяется на большей площади (</=10 мм). Если азнды
кальция и свинца пр», менять не рыхлой кучкой, а в виде подпрессоваиных от
руки таблеток диаметром 5 мм. то и при опущенпом ролике получается 100%
взрывов; при той же навеске (20 мг) и большем диаметре таблетки (10 мм)
частость взрывов падает.
опущенном ролике, если сделать на поверхности заряда небольшую
лунку (серии опытов 19—22), различие в частости взрывов при
поднятом и опущенном ролике наблюдалось и для замороженного
нитроглицерина, но в этом случае оно меньше.
Эффект поднятого ролика проявляется и в том случае, если удар
по нитроглицерину осуществляется через кружок оловянной фольги
76
(толщина 0,05 мм) К При этом, если фольгу закрепить на верхнем
ролике (фиг. 26 и 2г), то частость взрывов получается 100%, как и
без фольги. Если же ее поместить на нитроглицерин (фиг. 2а и 2в),
то частость взрывов получается около 50%.
Для других изученных твердых вторичных ВВ (дина, тетрил,
гексоген) влияние положения ролика не сказывается. Возможно,
что это связано с относительно малым диаметром роликов, так как
захват и сжатие воздуха несомненно зависит от этого диаметра.
Иначе обстоит дело с твердым инициирующим ВВ — азидом
свинца и азидом кальция. При поднятом ролике частость взрывов
Фиг, 2. Схема расположения заряда ВВ и фольги в
некоторых опытах табл. 1.
я—разном ерное расположение вещества, накрытого фольгой,
б—фольга припаяна к верхнему ролику. в—нанеска располо-
жена в центре ролика к накрыта фольгой, й—фольга припаяна
к верхнему ролику.
азида свинца составляет 100%, при ролйке, стоящем на ВВ, — 4%.
Это обстоятельство интересно уже потому, что для твердых непла-
вящихся веществ, какими являются оба эти азида, трудно предста-
вить тот механизм захвата и сжатия пузырька газа, который Боу-
ден принимает для жидкостей.
Опыты с азидами позволили сделать наблюдения, бросающие
некоторый свет на причины рассматриваемого эффекта. Когда
взрыв происходит при поднятом ролике, то след от взрыва на тор-
'цах роликов по диаметру значительно больше, чем диаметр таб-
летки; при опущенном ролике диаметр отпечатка такой же, как
исходной таблетки. Это говорит за то, что при ударе с поднятым
роликом до возникновения взрыва заряд расширился в радиальном
направлении, по-видимому, в результате увлечения азида воздухом,
вытесняемым из пространства между роликами.
Опыты с инертным порошком (мел) показали, что таблетка 2 при
ударе по ней приподнятым роликом разбрасывается по всему тор-
цу. Когда же производится удар по ролику, опущенному на таб-
летку, она раздавливается, но не разбрасывается.
1 В этих условиях высокая чувствительность ВВ ве снижается я тогда, когда
на поверхности ударника вместо стали имеется слой олова, температура плавле-
ния которого (232°) гораздо ниже минимальной температуры, необходимой по
Боудену для взрыва при ударе (40(f).
1 Таблетки получали слабым подлрессовыванием навескн от руки черта стек-
лянную трубку.
77
То, что эффект поднятого ролика наблюдается не только для
жидких нитроэфиров, но и для азидов свинца и кальция, естествен-
но, следует связать с относительно высокой воспламенямостью пос-
ледних, значительно большей при повышенных давлениях, чем дру-
гих изучавшихся ВВ. По-вндимому, это обусловлено тем, что коли-
чество энергии, заключающееся в сжатом воздухе, очень мало и
для воспламенения трудно воспламеняемых ВВ недостаточно.
Следует указать, что эффект поднятого ролика очень близок к
тому, который изучал Боуден [3] в опытах с ударником, имеющим
лунку с бортиком вокруг иее.
Фиг. 3. Схема расположения деталей прибора в опытах
по выявлению влияния лунки на возникновение взрыва
при ударе.
а—бортик лунки только касается капли вещества, б—еерхний
ролик без лунки только касается капли вещества, в—верхний
ролик опущен к бортик луикн стоит на нижнем ролике,
Г—пружинка для фиксации ролика оа определенной высоте,
верхний лату оный ролик. 3—лунка, 4— бортик, 5—капля ве-
щества, б—муфта с вырезом, 7—нижннй стальной ролик.
Мы провели ряд опытов с таким ударником. Полученные
результаты сведены в табл. 2, Схемы расположения деталей при-
бора и особенности его конструкции в этих опытах представлены
на фигурах 3 и 4.
Опыты подтвердили наблюдения Боудена о высокой частости
взрывов нитроглицерина в таких условиях (табл. 2, серия опытов
1—2). Для получения высокой частости необходимо, однако, чтобы
верхний ролик был поднят на некоторую высоту над нижним, так
чтобы края бортика были над каплей ВВ или хотя бы в пределах
капли, как на фиг. За. Если верхний ролик с лункой опущен на ниж-
ний (фиг. Зе), то высокой частости взрывов не наблюдается (табл. 2,
серии опытов 4—6).
Возможно, что действие воздуха в лунке и в этом случае, не ис-
черпывается его сжатием и разогревом. Мы пытались это проверить,
помещая нитроглицерин в чашечку из пластичного материала
(сплав пчелиного воска с пушечной смазкой) и подвергая его удару
роликом с лункой и без нее в приборе № 1 с малым зазором, запол-
ненным тем же сплавом (табл. 2, серии опытов 7—9, фиг. 4а и 46).
В этих условиях должно происходить только сжатие пузырьков; те-
чение же ВВ было затруднено большим сопротивлением вытеканию
вязкого материала в узких зазорах между роликом и муфтой;
однако в этих опытах такого влияния лунки, какое было заметно
при экспериментах с муфтой, устроенной по типу прибора № '4
(фиг. 4в и 4г), не было установлено и частость взрывов была, высока
78
Т а бли ца 2
Влияние устройства прибора н условий удара
на частость нарыва нитроглицерина
№ серий опытов Навеска ВВ мг Груз г Высота падения груза см Число взрывов Число опытов № фигуры, нллкстрирую- щей расположе ние деталей прибора
I. Повышенная частость взрывов при наличии на торце верхнего ролика лункИ-
с бортиком (верхний ролик приподнят над нижним и лишь касается
капли нитроглицерина)
1 4 260 50 9/10 За
2 4 260 50 0/10
3 4 2000 25 5/5 За
II. Низкая частость взрывов при ролике с лункой и бортиком,
но опущенном до касания с торцом нижнего ролька
4 4 260 115 0/5 Зв
5 4 2 000 25 , 2/9 Зе
6 4 10000 25 1/3 3<
III, Отсутствие влияния лунки с бортиком
при затрудненных условиях течения ВВ
7 40 260 50 2/2 4а
2/2 46
0/3 4л
3/3 4г
8 40 260 10 3/5 4а
2/5 46
0/3 4г
9 50 260 10 9/10 4а
6/10 46
0/10 4a
0/10 4л
как с лункой, так и без нее. Если исключить попадание нитроглице-
рина в зазор (опыты в приборе фиг. 4а и 4г), то частость взрывов в
отсутствии лунки резко уменьшается. Следовательно, сама по себе
лунка в этих условиях оказывается менее эффективной, чем узкий
зазор.
Вследствие того, что нитроглицерин очень чувствителен к удару,
если ему предоставлена возможность течь в узкий зазор, не удалось
79
проверить, будет ли проявляться эффект лунки в отсутствии тече-
ния этого ВВ при ударе.
В свете этих наблюдений вероятно, что при ролике с лункой, как
и в случае плоского ролика, существенную роль играет вытекание
сжатого и разогретого воздуха через узкий зазор между бортиком
лунки и нижним роликом. То, что такое течение имеет место, пока-
зывают опыты Боудена [5], который связывал его со взрывом в
лунке; в наших опытах вытекание с прорывом бортика лунки на-
блюдалось и при замене нитроглицерина на инертную жидкость.
Напряженное течение нитроглицерина при продавливании его через
Фиг. 4, Схема расположения деталей при-
бора в опытах, иллюстрирующих отсутствие
влияния лунки в затрудненных условиях
течения ВВ.
а—верхний ролик без луикк. прибор Л, !,
б—верхний ролик с лункой, прибор № I. в—верх-
ний ролик без лункн. муфта с вырезом, г—верх-
ний ролик с лункой, муфта с вырезом.
узкую щель, которому может способствовать сжатый в углублении
пузырек воздуха, по-видимому, является основной причиной легкого
возникновения взрыва в этих условиях.
В целом описанные наблюдения еще раз показывают многооб-
разие путей, которыми энергия механического воздействия может
концентрироваться во взрывчатом веществе в степени, достаточной
для возникновения взрыва. Величина энергии, необходимая для
возбуждения взрыва, очень мала и составляет ничтожную долю
общей энергии удара. Поэтому решающую роль для возникновения
взрыва может играть не столько энергия воздействия, сколько рас-
пределение ее во взрывчатом веществе. В тех условиях, которые
особенно благоприятствуют концентрации энергии, взрыв может
возникнуть от очень слабых воздействий.
Добавим, что условия удара, которые рассматриваются, в на-
стоящей статье, вполне реальны и в производстве. В связи с тем,
что эти условия одновременно довольно специфичны, возможно, что
именно их случайное воспроизведение и являлось причиной неко-
торых из неожиданных взрывов, происходивших при изготовлении
нитроэфиров и ВВ на их основе.
80
выводы
Частость взрывов некоторых высокочувствительных взрывчатых
веществ (нитроглицерин, азид свинца и др,) при ударе резко уве-
личивается, если ролик, передающий удар, не поставлен на веще-
ство, а находится на некоторой высоте над ним.
Этот эффект, по-видимому, связан с захватом, сжатием и тече-
нием воздуха, приводящим к воспламенению взрывчатого вещества.
ЛИТЕРАТУРА
1. Боуден Ф, Ф, и Иоффе А. Д„ Возбуждение и развитие взрыва в твер-
дых н жидких веществах, ИЛ, 1955.
2, Холево Н, А„ Сб. «Физика взрыва», АН СССР, 1955, вып, 3, стр. 16.
3. Bowden F. Р. et alia, Proc Roy. Soc., 1947, A188, 291.
4. См. напр. Bowden F. P. and Gurion 0. A., Proc. Roy, Soc., 1949,
Al 98, 347..
5. В о w d e n F. P. et alia, Proc. Roy. Soc., 1947, A188, 311.
К К. АНДРЕЕВ, Ю. А. ТЕРЕБИЛИНА
в. к ВОПРОСУ О МЕХАНИЗМЕ ВОЗНИКНОВЕНИЯ ВЗРЫВА
ПРИ ИСПЫТАНИИ НА ЧУВСТВИТЕЛЬНОСТЬ К УДАРУ
В РОЛИКОВОМ ПРИБОРЕ
Как показали исследования Н. А. Холево [1], [2], взрыв при ударе
на копре в условиях испытания в роликовом приборе происходит
вследствие течения сжатого ударом ВВ. Различие в скоростях дви-
жения разных слоев текущего ВВ обусловливает возникновение
внутреннего трения и местных разогревов, приводящих к. взрыву.
По Холево в роликовом приборе № 1 (фиг. 1) взрыв нередко
возникает не между торцами роликов, а в кольцевом зазоре между
роликом и муфтой. Это обстоятельство, если оно имело бы прямое
экспериментальное подтверждение, служило бы убедительным до-
казательством решающей роли течения ВВ в процессе возбуждения
взрыва ударом на копре. До сих пор, однако, о возникновении взры-
ва в кольцевом зазоре судили по косвенным данным, а именно по
более высокой частости взрывов текучих ВВ в приборе № 1 по
сравнению с прибором № 2. Результаты этих опытов допускали раз-
личное толкование. Так можно было бы допустить, что при наличии
канавки в приборе № 2 уменьшается сопротивление истечению ВВ
из пространства между торцами роликов, вследствие чего снижает-
ся давление, достигаемое при ударе, что и приводит к увеличению
числа отказов. Можно было допустить также, что в приборе № 1 на
возникновение взрыва влияет резкое (на 90s) изменение направле-
ния движения выдавливаемого.ВВ при переходе его из пространства
между торцами роликов в.кольцевой зазор, Дополнительным осно-
ванием в пользу последнего предположения представлялись резуль-
таты опытов с нитроглицерином и некоторыми другими ВВ в при-
6 451
81
боре № 41, где частость взрывов мала, хотя благодаря большому
диаметру роликов длина пути течения ВВ такая же, как и в при-
боре № L
Цель описанных ниже опытов состояла в том, чтобы непосред-
ственно доказать, что течение взрывчатого вещества в кольцевом
зазоре роликового прибора может приводить к взрыву. Наряду с
этим опыты должны были уточнить, почему такое течение приводит
к взрыву. Наконец, они позволили обнаружить, что в обычных
условиях испытания на копре разлагаются даже органические не-
взрывцатые вещества; это показывает, что течение при ударе при-
водит к значительным разогревам.
Фиг. 1. Схема роликового при-
бора Л° 1 для испытания чув-
ствительности взрывчатых ве-
ществ к удару.
Фиг. 2. Схема сборки прибора Ла 2 для
оценки влияния перемены направления те-
чения ВВ на частость взрывов;
а—канавка прибора заполнена воске и„ б—прибор
в перевернутом -виде.
ОПЫТЫ ПО ВЫЯСНЕНИЮ ВЛИЯНИЯ ИЗМЕНЕНИЯ В НАПРАВЛЕНИИ
ДВИЖЕНИЯ ВВ В ПРИБОРЕ № 1
В этих опытах было применено два варианта сборки прибора
№ 2, показанных на фиг. 2. В первом из них, канавка была заполнена
воском (фиг. 2а). Естественно было полагать, что наличие воска
будет смягчать предполагаемый удар и сделает менее резким изме-
нение направления течения от горизонтального к вертикальному.
Во втором варианте (фиг. 26) муфта была перевернута и созданы
условия течения заряда, близкие к тем, которые присущи прибо-
ру № 1. Результаты проведенных испытаний приведены в табл. 1.
При обоих вариантах постановки опыта частость взрывов практи-
чески оказалась одинаковой (серии 1 и 2). К этому следует доба-
вить, что частость взрывов жидкого и желатинированного нитро-
глицерина остается значительной, если вместо прибора № 1 приме-
нить прибор, предусмотренный ГОСТ 2065—43, с фасками у роли-
ков (32% Для жидкого нитроглицерина при навеске 0,03 г и 92%
для 10%-ного желатинированного при навеске 0,05 а). В этом при-
* См. настоящий сборник, стр. 56.
82
Таблица 1
Влияние условий течения ВВ на частость взрыва нитроглицерина
н тетргла при испытании на копре
(Груз 10 кг, высота падения 25 см, число опытов в серии 25)
№ серий На- веска мг За- зор * мк Условия и схема опыта % взры- вов Средний отскок груза при отказах см
Нитроглицерин. Влияние изменения направления течения ЕВ на частость взрывов
1 10 20 Фиг. 2а 24 16,0±0,5
2 10 20 Фиг. 26 28 16,3±g;®
Нитроглицерин. Влияние высоты верхнего зазора
и заполнения нижнего зазора восксм ]ia частость взрывов
3 10 27 Суммарная высота зазоров 18 мм: объем зазоров 8 мм$ Фиг. За 40 16,5Ц
4 10 2.7 Нижний зазор заполнен воском; верхний зазор высотою 7 мм, его объем 3 мм6. Фиг. 36 80 15,0±0
5 10 27 Нижний зазер заполнен воском; верхний зазер высотою 3 мм, его объем 1,3 жж3 Фиг. За 68 16,0ti;57
6 2 27 Суммарная высота зазоров 18 мм, их объем 8 .и.и3 Фиг. За 0/15** 17,0±g,6
7 2 27 Нижний зазер заполнен воском; Верхний зазор высотою 9 мм, его объем 4 .«.и3; 2 серии по 25 опытов Фиг. За 40 12 17,0±0,5 17,0±0,5
8 2 27 Нижний зазор заполнен воском; верхний зазор высотой 7 мм, его объем 3 жж3. 2 серии по 25 опытов Фиг. 36 80 80 17,0±0,5 17,0±0,5
9 2 27 Нижний зазор заполнен воском; верхний зазор высотою 3 мм, его объем 1,3 жж3 Фиг. Зе 40 17,0±0,5
10 2 27 Нижний зазор заполнен воском; верхний зазор высотою 2 мм, его объем 1 жж3 Фиг. Зе 12 17,О±О,5
6*
83
Продолжение
№ серий На- веска мг За- зор * мк Условии.и схема опыта % взры- вов Средний отскок груза прн отказах см
Тетрил. Влияние высоты верхнего зазора
и заполнения нижнего зазора воском на частость взрывов
11 20 40 Высота зазоров 18 мм Фиг. Зе 88 16,7^
Г2 20 Нижний зазор заполнен воском; верхний зазор высотою 7 мм, его объем 5 мм3 Фиг. 36" 72 i5,6t;:*
.13 20 40 Нижний зазор заполнен воском; верхний зазор высотою 2 мм, его объем 1,5 мм? Фиг. За 84 I4,8±®;|
14 20 40 Нижний зазор заполнен воском; верхний зазор высотою 9 мм,. его объем 6 мм? Фиг. Зг 80 14,0±2,5
15 50 40 Нижний зазор заполнен воском; верхний зазор высотою 7 мм, его объем 5 мм? Фиг. 36 40
16 50 40 Нижний зазор заполнен воском; верхний зазор высотою 2 мм, его объем 15 мм3 Фиг, За 28 8,0+Н * —0,0
17 50 27 Нижний зазор заполнен воском; верхний зазор высотою 7 мм, его объем 3 мм? Фиг. 26 16 16,5±0,5 _
18 50 27 Нижний зазор заполнен воском; верхний зазор высотою 3 мм, его объем 1,3 мм? Фиг. За 20
19 50 27 Часть нижнего зазора заполнена воском Фиг. 4 72 12,7
* Разность диаметров канала муфты и ролика.
»• В числителе число взрывов, в знаменателе число опытов.
боре перемена направления движения менее резка, чем при роли-
ках без фасок.
По совокупности этих данных следует заключить, что изменение
направления течения ВВ при испытании в приборе № 1 не оказы-
вает существенного влияния на результат опыта.
84
ОПЫТЫ С ОГРАНИЧЕНИЕМ ВОЗМОЖНОСТИ ИСТЕЧЕНИЯ
ЖИДКОГО ВВ
Для проведения этих опытов приборы собирались по схемам,
приведенным на фиг. 3 и 4. В приборе № 1 при обычной постановке
опытов (фиг. За) жидкость могла вытекать как через верхний, так
и через нижний зазор; частость взрывов при этом составляла 40%
(серия 3). Если нижний зазор заполнить воском (фиг. 36), то час-
тость взрывов возрастает вдвое (серия 4); если высота верхнего
зазора уменьшена с 7 до 3 мм (фиг. 3s), то даже в этом случае час-
тость взрывов, если и уменьшается, то незначительно (серия 5).
Фиг. 4. Схема сборки прибо-
ра № 2 для выяснения
влиянии выдавливания ВВ
в канавку на частость взры-
вов.
Фиг. 3. Схема сборки прибора № 1 для опре-
деления влияния ограничения истечения
жидкого ВВ на частость взрывов.
а—обычная сборка, б—ннжний зазор заполнен
воском, высота верхнего зазора 7 л.н, нижний за*
зор заполнен воском, высота верхнего зазора 3 **,«,
з—нижний зазор заполнен воском, высота верхнего
зазора 9 хж.
Систематическое изучение влияния высоты зазора было прове-
дено с навеской, уменьшенной до 2 мг. В этих условиях в приборе
№ 1, собранном по схеме За, взрывов не получалось (при 15 парал-
лельных опытах). Заполнение нижнего зазора воском при высоте
верхнего 9 мм (фиг. За) увеличило частость до 12—40%. После
уменьшения высоты верхнего зазора до 7 мм частость возросла до
80%, повторный опыт дал тот же результат. Дальнейшее уменьше-
ние высоты зазора до 3 мм (серия 9) понизило частость взрывов
вдвое, при зазоре высотой 2 мм она снизилась до 12%.
Интересно, однако, что увеличение высоты зазора до 9 мм при-
вело к уменьшению частости взрывов до 12—40%. Возможно, что
в последнем случае навеска недостаточна, чтобы быть продавлен-
ной через узкую часть зазора; не исключено также, что начавшееся
горение взрывчатого вещества гасится последующим снижением
давления из-за эжекционного эффекта.
Опыты с тетрилом показали, что он менее «чувствителен» к вы-
соте зазора и при навеске в 20 мг давал частость больше 80% как
в обычных условиях прибора № 1, так и при заполнении нижнего
зазора воском, а также и при уменьшении высоты верхнего зазора
до 2 мм. Увеличение навески при данном зазоре приводило к зна-
85
чительному уменьшению частости взрывов (серии 15, 16 и 171).
Возможно, что в случае тетрила при меньшей его текучести (по
сравнению с нитроглицерином) взрыв возникает между торцами
роликов, а роль зазоров сводится к тому, чтобы обеспечить некото-
рое сопротивление истечению. Если оно слишком велико, то ско-
рость истечения мала и взрыва не возникает (большая навеска или
узкий зазор, серии 18 и 19). Если оно меньше, то взрывы происхо-
дят с большей частостью, При большом зазоре (40 жк) и навеске
50 мг высота отскока груза почти вдвое меньше, чем при навеске
20 мг. Это показывает, что затрата энергии на выдавливание велика
и в первом из этих двух случаев значительно больше.
ОПЫТЫ ПРИ ОТСУТСТВИИ ВЗРЫВЧАТОГО ВЕЩЕСТВА МЕЖДУ
ТОРЦАМИ РОЛИКОВ
Схема сборки прибора для проведения этих опытов показана на
фиг. 5. Между торцами роликов помещали слой воска толщиною
0,5 лип, нижний зазор также заполняли воском. В верхний зазор
вводили навеску нитроглицерина. Для этого капельки его наносили
Фиг. 5. Схема сбор-
ки прибора № 1
для определения
чувствительности
ВВ при нахожде-
нии его только в
кольцевом зазоре
прибора.
в различных местах верхней части канала му-
фты, затем при медленном поворачивании
вводили верхний ролик; на торцы роликов ни-
троглицерин не попадал, Результаты испыта-
ний приведены в табл, 2. Частость взрывов
опытов серии 1 составляла 27%.
Во второй серии воск из пространства ме-
жду торцами был выдавлен последовательны-
ми ударами настолько, что отскок груза при
ударе был близок к получаемому при холо-
стом опыте (17,5 см). Частость взрывов при
этом была даже несколько больше (40%), чем
в предыдущей серии.
При четырехкратном повторении опытов в
несколько отличающихся вариантах (в отно-
шении толщины слоя воска, наличия или отсутствия фаски, вели-
чины навески) также были получены взрывы с большей или мень-
шей частостью, Эта частость была отчетливо больше в тех условиях,
в которых больше был отскок, т. е. тогда, когда достигались более
высокие давления,
В связи с тем, что при наличии воска между роликами взрыв
трудно отличить от отказа, были проведены аналогичные опыты
(серии 3, 4 и 5) в «газовом» приборе (описание его дано ниже),
позволяющем более надежно судить о наличии взрыва. Результаты
[ Отметим одно своеобразное наблюдение. В опытах серии 19, проводившихся
в условиях, близких к условиям серии |8, но отличавшихся тем,’ что взрывчатое
вещество выдавливалось не в свободное пространство, а в канавку (см. фиг. 4
и фиг. Зе), частость взрывов сильно увеличилась. Такое влияние канавки прояв-
лялось неоднократно и ранее; причина этого не вполне ясна.
86
Таблица 2
Чувствительность нитроглицерина
при Нальчик его только в зазоре прибора № 1
(Вес груза 10 кг, высота его падения 25 см, число опытов в серии 25)
№ серии опы- тов На- веска нитро- глице- рина мг Зазор Условия опыта (Схема сборки прибора фиг. 5) % взры- вов Средняя высота отскока груза при отказах' см
1 2 27 Таблетка воска высотою 0,5 мм\ нитроглицерин только в зазоре 27 5,0
2 2 27 Слой воска столь мал, что при уда- ре получается максимальный отскок, а воск при опыте в зазор не выдав- ливается 40 16,5
3 2 27 Опыты в .газовом" приборе На торце находится таблетка воска весом 0,05 г, Верхний ролик от руки придавлен к воску 32 1,1
4 2 27 На торце ролика слой воска такой малой толщины, что получается мак- симальный отскок, а воск при опыте в зазср не выдавливается Е6 16,0
5 4 27 Навеска нитроглицерина взята из расчета полного заполнения зазора 12 2,0
опытов в «газовом» приборе оказались примерно такими же, как
и в первых двух сериях.
Таким образом, проведенные испытания показали, что в роли-
ковом приборе взрыв при ударе может возникнуть и в том случае,
если ВВ между торцами роликов нет и оно находится только в за-
зоре между верхним роликом и муфтой.
Возникает естественно вопрос, каким же образом возбуждаются
эти взрывы вообще и особенно в тех случаях, когда между торцами
роликов нет вещества, которое выдавливалось бы при ударе и при-
водило в движение жидкое В В, находящееся в зазоре,
Наиболее вероятной причиной этого движения и взрыва являет-
ся деформация роликов, принимающих при ударе бочкообразную
форму, Раздаваясь, они вытесняют жидкость, заставляя ее продав-
ливаться через сузившуюся часть зазора, где и возникает взрыв.
Этот взрыв имеет возможность распространиться в прилегающую
более толстую часть кольцевого слоя сжатого взрывчатого веще-
ства. В подтверждение этого предположения о механизме возник-
новения взрыва в рассматриваемых условиях можно указать, что
при замене двух роликов на один удвоенной длины (он был изготов-
87
лен из несколько более мягкой стали) при 15 опытах с навеской
10 мг при зазоре 27 мк ни одного взрыва не было; не было их и в
четырех опытах при зазоре в 45 мк.
То, что при ударе в приборе № 1 текущее вещество подвергается
достаточно интенсивным тепловым воздействиям, показывают опы-
ты с воском, образующим при ударе облачко дыма. Чтобы коли-
чественно оценить протекающую при ударе реакцию, был применен
ранее разработанный «газовый» прибор, позволяющий измерить
объем газов, образующихся при ударе. Этот прибор (фиг. 6) пред-
0 7а
фиг. 6. Схема прибора для измерения
количества газов, выделившихся при
взрыве,
I—ударник» 2—крышка» 3—мембрана резино-
вая» 4— корпус, 5—ролики» б—муфта» 7—под-
дон.
ставляет собою камеру, имеющую подвижный ударник и отверстие
для отвода газов к U-образному манометру, заполненному дибутил-
фталатом; в камеру помещается обычный роликовый прибор (в дан-
ных опытах прибор № 1).
Опыты показали, что воск при ударе действительно разлагается,
образуя около 20 н см3 газов на а. Это подтверждает, что разогревы.
вызываемые механическим воздействием при ударе, охватывают
заметные объемы вещества и достаточно сильны, чтобы вызвать
разложение даже такого относительно термостойкого невзрывча-
того вещества как воск. Нитроглицерин, в котором разогрев, выз-
ванный ударом, является саморазвивающимся процессом, естест-
венно образует в случае полного взрыва гораздо больше
(около 700 н см3!г) газов.
Из совокупности рассмотренных данных следует заключить, что
взрыв жидкого взрывчатого вещества в роликовом приборе при
ударе происходит существенно иначе, чем эго ранее представляли
себе исследователи, Решающим фактором его возникновения яв-
ляется продавливание вещества через узкий зазор, которое приво-
дит к возникновению локального очага реакции. Эта реакция раз-
вивается до взрыва, если вблизи очага есть слой ВВ, толщина ко-
88
торого превышает критическую для распространения горения при
данном давлении. Именно невыполнение этих условий привело к
отсутствию взрывов в упомянутых в начале статьи опытах в при-
боре № 4.
Из сказанного ясно также, что те условия течения, которые при-
водят к возникновению взрыва в приборе № 1 или близком к нему
по условиям течения ВВ приборе по ГОСТу очень специфичны,
и поэтому результаты испытания в них дают лишь весьма услов-
ную характеристику чувствительности взрывчатого вещества.
ЛИТЕРАТУРА
1. Холево Н. А. Труды Казанского химико-технологического института
ям. С. М. Кирова, 1946,10. 91; 1947. 11, 116.
2. Хо лево Н. А., Сб, «Физика взрыва», АН СССР. 1955, вып. 3, стр. 16.
См. также настоящий сборник, стр. 5.
К. К. АНДРЕЕВ
7. ОБ ОСНОВНЫХ ФАКТОРАХ, ОПРЕДЕЛЯЮЩИХ
ОПАСНОСТЬ ВЗРЫВЧАТЫХ ВЕЩЕСТВ ПРИ
МЕХАНИЧЕСКИХ ВОЗДЕЙСТВИЯХ, И МЕТОДАХ
ЕЕ ОЦЕНКИ
За последнее десятилетие в представлениях о чувствительно-
сти ВВ к механическим воздействиям произошли радикальные сдви-
ги, которые привели к изменению экспериментальных методов ее
оценки. Главную роль в этом сыграли работы Н. А. Холево [4], [5],
вскрывшие физический смысл тех явлений, которые приводят к
взрыву при ударе на копре. На этой основе Н, А. Холево показал
и объяснил большую условность результатов стандартного и иных
методов испытания чувствительности ВВ к механическим воздейст-
виям. Эти результаты характеризуют чувствительность взрывчато-
го вещества (частость взрывов) лишь применительно к тем режимам
деформации заряда, которые реализуются в данном методе испы-
тания, и не могут быть перенесены на другие. С тех пор было пред-
ложено много вариантов метода испытания чувствительности ВВ
к механическим воздействиям — на копре н других приборах; в со-
вокупности своей они показывают, что частость взрывов для одного
и того же ВВ при данной энергии воздействия может изменяться в
оЧень широких пределах. Это обстоятельство очень затрудняет
практическую оценку возможности использования различных тех-
нологических приемов применительно к тому или иному ВВ. Одна
из главных причин этой трудности — отсутствие полных знаний о
характере воздействий, которым ВВ подвергается в технологиче-
ских процессах. Эту трудность можно в некоторой мере обойти, со-
поставляя опыт практики, характеризующий в известной степени
89
опасность различных операций применительно к отдельным взрыв-
чатым веществам, с опасностью (чувствительностью) последних в
разных условиях лабораторного испытания.
Такое сопоставление может быть использовано и для обратного
заключения — об опасности тех или иных новых технологических
приемов или процессов для известного или нового взрывчатого ве-
щества на основании поведения последнего при различных лабора-
торных испытаниях,
Известно, что для возбуждения взрыва при ударе необходимо,
чтобы, во-первых, в результате механического воздействия возник
очаг разогрева и, во-вторых, чтобы начавшаяся в нем реакция раз-
вилась в горение или даже взрыв. Возникновение очага разогрева
связано с механическими свойствами вещества, развитие реакции
в нем в большей степени зависит от кинетических и термохимиче-
ских характеристик ВВ.
Относительно легко ответить на вопрос о том, какие из этих
характеристик определяют опасность взрывчатого вещества. Они
могут быть сведены к двум интегральным показателям — воспла-
меняемости 1 и чувствительности к детонации, которые приближенно
могут быть характеризованы обоими критическими диаметрами —
для горения, с которого обычно начинается взрыв при ударе, и для
детонации.
Сложнее ответить на вопрос, какие показатели механических
свойств приводят к повышенной опасности, иначе говоря при про-
чих равных условиях опаснее ли более текучее или менее текучее
взрывчатое вещество. В общем виде ответ на этот вопрос не может
быть дан, так как есть конкретные условия, при которых опаснее
текучие вещества и другие, в которых опаснее менее текучие.
Все же в большинстве случаев можно считать более опасным
труднотекучее вещество. В самом деле для возникновения разо-
грева требуется достаточно большая скорость сдвига и давление.
Если вещество неподатливо, труднотекуче, то действие удара не
сможет смягчиться и возникнет большое давление, под которым
начнется смещение слоев вещества друг относительно друга (внут-
реннее трение) или относительно ударника (внешнее трение); есте-
ственно, что воспламеняемость при большом давлении будет велика.
1 Следует указать, что воспламеняемость зависит не только от термокинети-
ческих свойств вещества, jro и от давления. Поэтому чувствительность данного ВВ
также может меняться весьма сильно в зависимости от того, под каким давлением
в рем возник разогрев. Экспериментальные данные по воспламеняемости имеются
пока только для относительно низких давлений. В настоящее время проводится
ее изучение при высоких давлениях Методом определения критических диаметров
для горения. Добавим, что по крайней мере для низких давлений, как для инди-
видуальных взрывчатых веществ, так и для смес.-ii нс наблюдается параллелизма
между критическим диаметром для горения и для детонации. Так черный порох
(все компоненты которого в отдельности ре взрывчаты) горит легко при малом
диаметре заряда, но детонация его в литературе ire описана; тэн легко детонирует
(критический диамето детонации 2 .ш, но плохо горит (критический диаметр
горения 30 мм нри атмосферном давлении и около 4 мм при 25 ат), амматолы
(критический диаметр горения около 30 .ч.и) плохо горят и плохо детонируют,
динаммопы же горят гораздо лучше аммотолов, но детонируют хуже их.
90
Нужно при этом иметь в виду, что в практических условиях
удар, как правило, действует под некоторым углом (косой или
скользящий удар), В отличие от того, что происходит при ударе по
нормали при испытании на копре, например, в приборе № 1, в слу-
чае косого удара низкая текучесть вещества не может предохранить
его от сдвига. В этом случае смещение слоев ВВ друг относительно
друга или относительно ударника происходит принудительно и оп-
ределяется уже не столько текучестью, сколько тангенциальными
силами, действующими при косом ударе. Качественная грань между
ударом и трением здесь отсутствует, а количественное соотношение
определяется углом, под которым произведен удар; от него в соче-
тании с текучестью и коэффициентом внешнего трения зависит со-
отношение тангенциальных и нормальных усилий.
Испытание на чувствительность к трению отличается от испыта-
ния ударом по нормали только тем, что при трении нормальное и
тангенциальное усилия задаются независимо друг от друга.
Вследствие того, что при испытании на чувствительность к тре-
нию напряжения бывают обычно меньше, то наиболее опасными
оказываются такие вещества, у которых воспламеняемость велика,
т. е. критический диаметр даже при небольшом давлении — мал —
например, черный порох, гремучая ртуть, некоторые пиротехниче-
ские составы и т. п,
Вещества, легкотекущие из-за малого коэффициента внутрен-
него трения или трудногорящие (большой критический диаметр),
малочувствительны к трению; для взрыва таких веществ необхо-
димо возникновение большого давления. Именно по обеим этим
причинам тротил принадлежит к числу относительно малоопасных
в обработке взрывчатых веществ, тогда как азид свинца или гексо-
ген— к числу относительно наиболее опасных, Естественно также,
что опасность последних может быть уменьшена увеличением их
текучести, что и осуществляется в технике — нанесением на частицы
таких ВВ «смазки» (флегматизатора).
Следует добавить, что флегматизатор, по-видимому, играет роль
не только на первой стадии возбуждения взрыва ударом—стадии
возникновения разогрева при течении. Он препятствует также раз-
витию очага горения, смягчая тепловой удар горячих продуктов
превращения, который может вызвать растрескивание соседних ча-
стиц вещества и этим увеличить поверхность горения.
Еще раз подчеркнем, что эти соображения не универсально при-
ложимы— в ряде условий возбуждение взрыва может осуществ-
ляться иными путями — например поджиганием в результате сжа-
тия и разогрева газового пузырька (это сжатие может облегчаться,
если ВВ текучее), в результате трения твердых частиц посторонних
примесей во взрывчатом веществе друг о Друга, или трения их о на-
ковальню (ударник) и т. д. Кроме того, могут быть особые условия
течения, когда очень большая или напротив очень малая текучесть
может давать большую частость взрывов—тогда тротил может
быть чувствительнее азида свинца или аммиачная селитра чувст-
вительнее нитроглицерина.
91
Оценка опасности конкретных производственных процессов мо-
жет быть поэтому произведена только при полном знании характера
тех воздействий, которым подвергается ВВ; при отсутствии такого
знания необходимо проводить испытание с моделированием реаль-
ных воздействий.
О НАИБОЛЕЕ ЦЕЛЕСООБРАЗНЫХ ЛАБОРАТОРНЫХ МЕТОДАХ ОЦЕНКИ
ЧУВСТВИТЕЛЬНОСТИ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
К МЕХАНИЧЕСКИМ ВОЗДЕЙСТВИЯМ
Возникает вопрос, какие из существующих экспериментальных
методов определения чувствительности в наибольшей степени отра-
жают опасность вещества при механических воздействиях, если
исходить из рассмотренных выше общих соображений.
Испытание на копре в различных его вариантах [4], [5] (прибо-
ры № 1, по ГОСТу 2065—43 и № 2, а также модификации послед-
него) имеет тот принципиальный недостаток, что оно характеризует
преимущественно возможность возникновения очагов реакции, а не
распространения процесса. Принципиально слабой стороной этого
испытания является также то, что возможность течения н условия
его в значительной степени определяются текучестью вещества.
Если вещество очень малотекуче вроде азида свинца, то оно не
взрывается ни в одном из названных выше трех приборов; если оно
слишком текуче, то оно лишь с трудом, при очень узких зазорах,
взрывается в приборе № 1. В то же время в практике текучесть
играет, как было отмечено выше, значительно меньшую роль.
При испытании взрывчатых веществ на так называемую чувст-
вительность к трению, когда ударом перемещают пластинки, между
которыми заключено взрывчатое вещество, трение имеет место не-
зависимо от текучести вещества, хотя она и влияет на характер
трения, вызванного перемещением пластинок. Это обстоятельство
является существенным преимуществом данного метода по сравне-
нию с испытанием на копре,
Если текучесть вещества велика, то требуется, по-видимому,
весьма большое давление и некоторые дополнительные условия,
чтобы при испытании на чувствительность к трению возник взрыв.
Поэтому в опытах Боудена [3] в разработанном им приборе для ис-
пытания на чувствительность к трению многие ВВ, как например
тэн, взрыва не дают. В то же время эти вещества лэгко взрываются
в предложенном В. С. Козловым [I] варианте прибора Боудена, где
ВВ зажато между торцами двух роликов: закрепленного и подвиж-
ного. При горизонтальном ударе по боковой поверхности подвиж-
ного ролика он по идее В. С. Козлова должен перемещаться по
нормали к своей оси, В действительности же, так как прочности связи
верхнего торца ролика (сталь—сталь) и нижнего (сталь—ВВ или
ВВ—ВВ) не одинаковы, то ролик совершает движение по дуге. При
этом возникает не только треиие, но и скользящий удар. При теку-
чих ВВ происходит также треиие металла о металл, В этих усло-
виях взрываются даже относительно легкотекучие ВВ, включая
и нитроглицериновые пороха.
92
В сочетании с определением воспламеняемости (способности к
горению) результаты испытания на маятниковом приборе В. С. Коз-
лова (К-44-Ш), по-видимому, лучше, чем другие методы, могут
характеризовать практическую опасность ВВ при механических
воздействиях.
Этот вывод не означает ненужности испытаний ударом по нор-
мали на копре. Однако эти испытания приобретают дополнительный
характер, иллюстрируя поведение ВВ при некоторых особых усло-
виях механического воздействия. В этом отношении из новых ме-
тодов следует особо отметить испытание с приподнятым роликом [2],
которое позволяет очень наглядно выявить высокую воспламеняе-
мость, а значит и опасность взрывчатого вещества в тех условиях,
в которых она не выявляется обычным испытанием на копре.
Подчеркнем еще раз, что нельзя ограничиваться при определе-
нии чувствительности только испытанием на приборе Боудена—
Козлова без определения способности вещества к горению и к дето-
нации ‘. Хотя в некоторой мере способность к горению и к детона-
ции проявляют себя и при названном испытании, но это происходит
при неопределенных и больших '-давлениях. При обычных же в
практике условиях механических воздействий большое давление
создается только в очень малой части объема, занятого ВВ, боль-
шая же его часть находится при относительно малых давлениях.
Поэтому определение способности к горению сохраняет свое важ-
нейшее значение, причем это определение следует производить в той
области давлений, которая соответствует практическим условиям,
так как способность вещества к горению зависит от давления и
притом для разных ВВ в разной степени
выводы
Фундаментальными свойствами взрывчатого вещества, опреде-
ляющими его опасность при механических воздействиях, являются
термокинетические характеристики, проявляющиеся в критических
диаметрах для горения и для детонации.
Возможность возникновения при механических воздействиях
разогрева и давления, достаточных для возбуждения горения, за-
висит от конкретных условий деформации и текучести вещества.
Обычно она больше для труднотекучих веществ и может быть оце-
нена действием скользящего удара на приборе В. С. Козлова
(К-44-Ш).
1 Как иллюстрацию этого заключения укажем, что перхлорат аммония при
испытании на приборе Боудена—Козлова дает высокую частость взрывов; спо-
собность же его к горению очень низка (критический диаметр 33 мм), что и обус-
ловливает малую опасность перхлората в обращении. Добавление к перхлорату
текучих органических веществ уменьшает чувст вительчость при испытании на
приборе Боудена—Козлова, способность же к горению сильно возрастает; это
возрастание может получить преобладающее значение, что и приводит к увели-
чению опасности в обращении таких смесей по сравнению с чистым перхлоратом.
93
ЛИТЕРАТУРА
1, См, Андреев К- К, л Беляев А. Ф., Курс теории взрывчатых веществ,
Оборонгиз, 1960, стр. 323.
2, Андреев К. К. и Беляев А. Ф„ Курс теории взрывчатых веществ,
Оборонгиз, 1960, стр. 285. См. также настоящий сборник, стр 72.
3. Бо у Д е и Ф. Ф. и И о ф ф е А. Д., Возбуждение и развитие взрыва в твер-
дых и жидких веществах, ИЛ, 1955, стр. 25.
4. X о л е в о И. А. Труды Казанского химико-технологического института
нм, С, М. Кирова, 1946, *10, 91; 1947, 11, Ц6,
5, Холево И. А., Сб. «Физика взрыва», АН СССР, 1955, вып. 3, стр. 16.
См. также настоящий сборник, стр. 5,
К, К. АНДРЕЕВ
8, О НЕКОТОРЫХ ЗАРУБЕЖНЫХ ИССЛЕДОВАНИЯХ
ПО МЕТОДИКЕ ЭКСПЕРИМЕНТАЛЬНОГО ОПРЕДЕЛЕНИЯ
ЧУВСТВИТЕЛЬНОСТИ ВВ К УДАРУ
Из всех характеристик взрывчатых веществ чувствительность
к удару всегда принадлежала к числу наименее определенных, Эта
неопределенность относилась и к тому, какой из показателей уда-
ра— общую энергию, импульс, скорость или что-либо иное считать
фактором, определяющим возникновение взрыва.
Типичны для данных по чувствительности большие различия
между результатами, получавшимися разными исследователями для
одних и тех же взрывчатых веществ.
Наконец, испытание на чувствительность к удару является един-
ственным из испытаний взрывчатых веществ, при котором рекомен-
дуемое число параллельных опытов достигает 25—100; при этом
за характеристику чувствительности обычно принимается частость
взрывов при данной энергии удара или же та энергия, при которой
частость взрывов равна '/2 либо некоторой иной величине,
Распространенным было мнение, что непостоянство чувствитель-
ности связано только с побочными факторами—трудноустранимыми
дефектами методики определения на копре и что если бы их уда-
лось избегнуть, то было бы возможно установить чувствительность,
как некоторую абсолютную константу данного взрывчатого веще-
ства. Отголоски Этих настроений нашли в частности свое отражение
на состоявшейся в 1955 г. конференции по вопросам чувствительно-
сти ВВ. В ряде выступлений многократно подчеркивалось (и это
отразилось даже в решении конференции), что чувствительность
есть объективное свойство взрывчатого вещества, как будто факты,
указывающие на изменчивость чувствительности, на зависимость
частости взрывов от условий воздействия’, и выводы тех Исследо-
вателей, которые перестали считать эти факты случайными, побоч-
1 Кстати, сильно выраженный статистический характер результатов испытания
на чувствительность уже сам по себе указывает на большую зависимость резуль-
татов опыта от столь малых изменений его условий, что они оказываются вне
контроля экспериментатора.
94
ными и вскрыли их физический смысл, являются покушением на
материалистическое понимание этого физического явления.
Радикальное изменение представлений о чувствительности было
осуществлено Н, А. Холево, показавшим, что опасность возникно-
вения взрыва при механических воздействиях на взрывчатое веще-
ство зависит от его термокинетических характеристик, определяе-
мых химической структурой, от его механических свойств и от усло-
вий удара, которые отнюдь не исчерпываются энергией последнего.
В силу этого опасность взрыва при данной величине энергии удара
может для данного взрывчатого вещества меняться в весьма широ-
ких пределах и бессмысленно искать оторванно от условий удара
универсальную абсолютную характеристику опасности данного ве-
щества.
Мы не останавливаемся здесь более подробно на представлениях
и результатах опытов Н. А, Холево, поскольку они освещены в ряде
статей [2], [3].
Воззрения Н. А. Холево, однако, не сразу получили должное
признание среди исследователей, многим из которых трудно было
отрешиться от старых неправильных, но привычных взглядов на
чувствительность. В дальнейшем в ряде стран (США, Швеция,
Германия) были опубликованы работы, авторы которых приходят
к выводам, аналогичным сделанным Н. А. Холево. Рассмотрению
этих работ и посвящена настоящая статья.
ПРЕДСТАВЛЕНИЯ О ЧУВСТВИТЕЛЬНОСТИ Г. Б. КИСТЯКОВСКОГО
И Р. КОННОРА (США)
Во время Второй мировой войны крупные американские физи-
ки Г. Б, Кистяковский и Р, Коннор работали над исследованием
и разработкой ВВ. В отношении чувствительности взрывчатых
веществ к механическим воздействиям они естественно обра-
тились к тем методам ее определения, которыми пользовались в
гражданских (Горное бюро) и военных (Пикатинский арсенал)
учреждениях США. Однако Кистяковский и Коннор скоро убе-
дились в непригодности этих методов для абсолютной характе-
ристики чувствительности и, более того, в невозможности получения
такой характеристики, Не описывая экспериментальных данных,
которые привели их к этому выводу, они формулируют его в сле-
дующих положениях, которые приведем дословно,
«Было показано, что для возбуждения взрыва необходим быст-
рый удар, вызывающий сдвиг частиц и трение между ними. По-ви-
димому, поверхность частиц должна быть локально нагрета до
высоких температур. В равной степени важным фактором является
также замкнутость (confinement) вещества, Условия должны быть
таковы, чтобы газообразные продукты начальной реакции не могли
уходить, В противном случае для возбуждения взрыва должно быть
затрачено гораздо большее механическое усилие,.., Локальное вос-
пламенение, вызванное ударом на немногих маленьких участках
поверхности, должно быть поддержано путем задержания горячих
газообразных продуктов у этой поверхности. Тогда возрастающее
95
местное давление ускоряет реакцию и происходит взрыв. В против-
ном случае начальное воспламенение может легко затухнуть» [61.
И в другом месте:
«В результате исследований, однако, было установлено, — и это
является открытием значительной практической важности, — что
нельзя найти такой характеристики, как определенная механическая
чувствительность взрывчатого вещества. Действительно, изменяя
условия испытания, можно изменить даже порядок механических
чувствительностей ВВ».
Эти положения полностью согласуются с выводами, которые
были сделаны Н. А. Холево до опубликования американской ра-
боты. Следует добавить, что выводы американских исследователей
были опубликованы в их книге «Наука во II мировой войне», кото-
рая, по-видимому, мало известна европейским исследователям и в
частности тем из них, о работах которых речь будет идти ниже. Эта
неосведомленность относится вероятно и к работам Н. А. Холево,
впервые опубликованным в Известиях Казанского химико-техноло-
гического института нм. С. М. Кирова в 1946—1947 гг.
РАБОТА ЮНГБЕРГА (ШВЕЦИЯ)
Следующей страной, в которой были «открыты» и развиты пред-
ставления Н. А. Холево, была Швеция.
В работе С. Юнгберга [4] исследовано возбуждение взрыва при
ударе в приборах, аналогичных приборам №№ I и 2 Н. А. Холево,
Фиг. 1. Частость взрывов гремучего
студня в приборах № 1 и 2 по
Юнгбергу.
(Груз 2 кг, толщина слоя ВВ
0,4 мм, торцы роликов полирован-
ные, число опытов в серии 25).
/—прибор № 2 2—прибор № [р 5—прибор
№ 2с одним кружком фильтровальной
бумаги, 4—прибор № 2 с лаумя кружками
фильтровальной бумаги.
т. е. при отсутствии или наличии
в направляющей муфте кольцевой
канавки, куда может вытекать
взрывчатое вещество. Результаты
опытов с гремучим студнем пред-
ставлены на фиг. 1. В приборе
№ 2 при двухкилограммовом гру-
зе отдельные взрывы наблюда-
лись лишь начиная с высоты па-
дения 1,4 м. В приборе № 1 при
зазоре 0,05 мм 1 и высоте падения
груза 0,5 и I м частость взрывов
значительно больше. Если между
взрывчатым веществом и торцом
ролика с одной или с обеих сто-
рон положить кружок сухой филь-
тровальной бумаги или листик
асбеста, то уже при высоте паде-
ния 0,2 м наблюдается значитель-
ная частость взрывов. Если бума-
га смочена водой, .то взрывов не
наблюдается даже при увеличе-
нии высоты падения груза до
1 Уменьшение зазора в случае гремучего студня увеличивает частость
взрывов.
96
1,5 м. Все эти результаты естественно объясняются относительно
большой для прибора № 2 текучестью гремучего студня, вследствие
чего прн ударе не создается высокого давления; затруднение исте-
чения применением бумаги или помещением роликов в муфту без
канавки повышает давление при истечении, что приводит к взрыву.
Если применить ролики, торцы которых не полированы, а только
шлифованы, то частость взрывов сильно возрастает уже при малых
высотах падения груза, так как истечение ВВ затрудняется шеро-
Фиг. 3» Изменение давления во времени в
приборах № 1 и 2 при ударе по пласти-
лину.
(Груз 2 кг, высота его падения 0,5 тол-
щина слоя пластилина 0,4 мм, торцы ро-
ликов полированные).
/—без пластилина, 2—с пластилином и приборе
№ 2, 5—с пластилином в приборе № 1 при диа-
метрах канала муфты 8»05 о и ролика 8,00леи»
4—с пластилином в приборе № I при диаметрах
канала муфты и ролика 8,00 ля.
Фиг. 2. Устройство для
измерения давления
во времени при ударе
на копре.
/—накональая, 2—плекси-
гласовая изоляция, J—
защитное кольцо, 4—rpy3t
•7—ВВ, б—тензометриче-
ские датчики»
ховатостями на поверхности торцов. Если смочить шлифованные
торцы инертной жидкостью, то получается такая же низкая час-
тость взрывов, как на полированных роликах. Порошкообразные
труднотекучие ВВ, которые не выдавливаются даже при полиро-
ванных торцах, менее чувствительны к состоянию поверхности.
При помощи устройства, изображенного на фиг. 2, тензометри-
ческими датчиками было определено на катодном осциллографе
изменение во времени давления при ударе. Без ВВ или прн
механически гомогенном полностью замкнутом его слое удар имеет
упругий характер. Если слой состоит из пластичного вещества,
имеющего возможность выдавливаться, то во время выдавливания,
до тех пор пока оно не закончится, удар имеет неупругий характер.
Длительность выдавливания зависит от размеров отверстий, через
которые идет истечение, вязкости взрывчатого вещества и давления:
На фиг. 3 показаны кривые, полученные для слоя пластилина
толщиною 0,4 мм при полированных торцах и высоте падения груза
7 45 1 97
0,5 jh. При диаметрах ролика и канала муфты 8,00 лш возможность
вытекания вещества была практически устранена и кривые, без
пластилина и с ним, очень похожи друг на друга с тем лишь отли-
чием, что максимальное давление в последнем случае несколько
меньше, а длительность удара больше (кривые 1 и 4).
При муфте с каналом 8,05 мм наблюдается период выдавлива-
ния, при котором удар не упругий; за ним следует участок упругого
удара (кривая 3).
В приборе № 2 сначала наблюдается маленький подъем давле-
ния, заканчивающийся через 35 мксек; за это время пластилин
выдавливается и последующий ход кривой почти такой же, как и
Фиг. 4. Изменение давления во времени в приборе № I
при ударе по пластилину в зависимости от толщины его
слоя. (Груз 2 кг; высота его падения 1 м; диаметры канала
муфты 8,05 мм и ролика 8,00 мм; торцы роликов полиро-
ванные). Числа при кривых указывают толщину слоя
пластилина в
без пластилина (кривая 1). На фиг. 3 для опыта в приборе № 2
(кривая 2) не видно начала удара, так как длительность выдавли-
вания слоя 0,4 мм в другом опыте при высоте падения 1 м была
больше 125 мксек. Таким образом выдавливание длилось в течение
более чем 90 мксек, прежде чем повышение давления стало доста-
точным для регистрации его осциллографом.
На фиг. 4 изображены кривые изменения давления во времени
для пластилина при различной толщине его слоя. С увеличением
толщины слоя время выдавливания становится больше, а длитель-
ность упругой части удара меньше. Максимальное давление при
выдавливании приблизительно то же, что и при высоте падения
0,5 м, и является, по-видимому, при данном размере зазора харак-
теристическим для течения пластилина.
Для гремучего студня, по текучести существенно отличающегося
от пластилина, на аналогичных кривых период выдавливания выра-
98
жен менее отчетливо. На фиг. 5 и 6, где представлены результаты
опытов с гремучим студнем в приборе № 2 при толщине слоя ВВ
0,4 мм и высоте падения груза 1 м, видно лишь маленькое повы-
шение давления, которое заканчивается приблизительно через
40 мксек. Выдавливание должно было продолжаться по меньшей
мере 50 мксек, прежде чем осциллограф начал отмечать давление,
и при этом толщина слоя
должна была стать меньше
0,1 мм. Сорока микросекун-
дами позже почти все ВВ
было выдавлено, и последу-
ющее изменение давления во
времени имело тот же ха-
рактер, что и при упругом
ударе без ВВ.
Кривые для опытов со
слоем в 0,4 мм гремучего
студня при шлифованных
торцах без взрыва (фиг. 5)
и со взрывом (фцг. 6) очень
сходны друг с другом. На
восходящей части последней
кривой виден кратковремен-
ный минимум и несколько
небольших нарушений ее хо-
да. Сходное влияние взрыва
наблюдалось при помещении
слоя гремучего студня тол-
щиной 0,4 мм между двумя
бумажными кружками при
шлифованных торцах ролика
(кривая 3 фиг. 6) с тем лишь
отличием, что минимум дав-
ления был выражен отчет-
ливее. В обоих случаях
длительность взрыва была
мала по сравнению с време-
нем удара, что согласуется с
наблюдениями Ридила и Ро-
бертсона [7].
При шлифованных тор-
цах (в отличие от полиро-
ванных) ВВ остается в уг-
лублениях поверхности, ког-
да ее гребни приходят в кон-
такт и давление повышается.
При этом в точках контакта
возникает большое местное
давление и, возможно, что
Фиг. 5. Изменение давления во времени в
приборе № 2 при ударе по гремучему
студню. (Груз 2 кг, высота его падении
I м, толщина слоя ВВ 0,4 мм, торцы роли-
ков шлифованные).
В опыте с ВВ взрыва не было
Фиг. 6. Изменение давления во времени в
приборе № 2 при ударе по гремучему
студню. (Груз 2 кг, высота его падения
1 м, толщина слоя ВВ 0,4 ,«.и).
!—без ВВ, 2—с ВВ, торцы - роли кои шлифован*
ные, 3—с ВВ между диумя бумажными круж-
ками, торны роликов полированные.
В опытах с ВВ взрывы возникли.
7*
99
при этом в результате пластической деформации металла образу-
ются разогревы, поджигающие ВВ,
В опытах с гремучим студнем по кривым давление — время
очень трудно установить, имел ли место взрыв. Он проявляется в
виде кратковременных колебаний давления на восходящей ветви
кривой, но часто эти колебания не отличимы от случайных.
Чтобы установить момент взрыва, возникающее в результате его
свечение направляли через специальное отверстие в муфте на фото-
элемент. Такая система записи не безупречна, так как точка, в кото-
рой возникает взрыв, регистрируемый на кривой давление — время,
может не быть видима через отверстие, направленное на фотоэле-
мент. В табл. 1 приведено несколько значений времени от начала
Фиг. 7. Изменение давления во времени в приборе
№ 2 при ударе по гремучему студню н порошкооб-
разным тротилу н тэну. (Груз 2 кг; высота его
падения I л; толщина слоя гремучего студня
0,4 ля, тротила и тэна 0,2 лиг торцы роликов
полированные).
, ' /—без В В, 2—гремучий студень между двух бумажных
। кружков. 5—тротил, <—тзн.
Кривые сдвинуты qo осн времени.
В опытах с ВВ взрывы возникли
повышения давления до взрыва. Задержка взрыва убывает при
увеличении высоты падения. При шлифованных торцах роликов
момент воспламенения приблизительно совпадает с моментом за-
вершения выдавливания ВВ. При опытах на копре иной конструк-
ции наблюдались случаи возникновения взрыва после окончания
удара. Это, ио-видимому, было связано с тем, что для развития
взрыва требовалось некоторое время.
Были поставлены аналогичные опыты также с тэном и троти-
лом. Как показывает фиг. 7, кривые изменения давления для этих
ВВ сходны между собой и в свою очередь аналогичны кривым,
наблюдавшимся при гремучем студне, помещенном между бумаж-
ными кружками.
Таким образом опыты Юнгберга подтверждают вывод Н. А. Хо-
лево о том, что при ударе на копре происходит выдавливание ВВ
из пространства между торцами; от сопротивления этому выдавли-
100
Таблица 1
Время от начала повышения давления до взрыва
Высота падения см Скорость при ударе см[сек Время мксек Состояние торцов роликов
100 450 85-115 Полированные,
50 310 170—185 с двумя бумажны-
100 450 30—40 ми кружками Шлифованные
ванию, определяемого в частности величиной зазора между роли-
ком и муфтой, а также наличием канавки, и, естественно, свойст-
вами вещества, зависит, возникает взрыв или нет. Полученные кри-
вые давление — время дают конкретное и количественное представ-
ление об этой важной характеристике процесса.
РАБОТА КЕНЕНА, ИДЕ И ХАУПТА [5] (ЗАПАДНЫЙ БЕРЛИН)
Большое экспериментальное исследование для изыскания спо-
соба испытания взрывчатых веществ на опасность их в обращении
было проведено недавно в той лаборатории (бывший Химико-тех-
нический институт, ныне Управление испытания материалов), в ко-
торой Каст в начале столетия разработал современный способ опре-
деления чувствительности ВВ к удару.
Постановка вопроса
Немецкие исследователи подчеркивают необходимость разносто-
ронней характеристики восприимчивости ВВ к тому или иному виду
механического воздействия. Лишь по совокупности результатов этих
разносторонних испытаний можно делать заключение об опасно-
сти ВВ. Единой меры этой опасности в настоящее время не может
быть дано. Они справедливо отмечают также, что нужно различать
возможность возникновения взрыва при ударе от возможно-
сти его распространения по крайней мере для вторичных
взрывчатых веществ; для инициирующих последняя способность
велика, так что возбуждение взрыва, как правило, означает и его
распространение.
Основным методом определения чувствительности ВВ к удару
в Германии является известный метод испытания их на копре в
штемпельных приборах Каста, принятый до сего времени согласно
международному соглашению о железнодорожных перевозках для
характеристики взрывоопасности веществ. Показателем чувстви-
тельности по этому методу служит та минимальная высота падения
груза определенного веса, при которой наблюдается хотя бы один
отчетливый взрыв нз 6, а по позднейшим нормам из 4 опытов. Изу-
чению этого метода, а также его совершенствованию и была посвя-
щена рассматриваемая работа.
101
Проверка методики Каста
Устройство штемпельного прибора отвечало общим требованиям
Ленце и Каста, а именно: поверхность ударника была определен-
ной по площади и постоянной по состоянию, возможность бокового
смещения взрывчатого вещества и трения, к которому, как пред-
полагали эти авторы, чувствительност!,
взрывчатых веществ совершенно иная, чем
к удару, отсутствовала.
Детали Штемпельных приборов, исполь-
зованных в данной работе, имели размеры н
качество металла, приведенные на фиг, 8,
Исследователи в своих опытах ожидали
получить подтверждение тем результатам по
чувствительности, которые в свое время бы-
ли получены Кастом. В качестве одного из
наиболее чувствительных взрывчатых ве-
ществ была взята нитроклетчатка (13,3% N),
которая по Касту дает взрывы при падении
груза 5 кг с высоты 5 см. Кроме того, сле-
довало ожидать, в соответствии с общими
'взглядами Каста о росте числа взрывов с
высотой падения груза, что при 40 см все
6 опытов дадут взрывы.
Вопреки этим ожиданиям при 10 см
не было получено ни одного взрыва, при 20
н 30 только по одному, а при 40 см ни од-
ного, т, е. никакого роста частости взрывов
с увеличением высоты падения груза не на-
блюдалось. Так как по ориентировочным
опытам аналогично вели себя и другие изу-
чавшиеся Кастом ВВ (коллоксилин, тетрил,
гексил и пикриновая кислота), исследовате-
ли перешли к опытам с относительно более
чувствительными веществами — тэном и гек-
согеном, В приборах описанного типа и раз-
меров (зазор 0,03 мм) во всем интервале
высот от 10 до 40 см при грузе в 5 кг не
получилось нч одного взрыва. При повторе-
нии опытов с уменьшенным количеством
(20 .и3 вместо 50 лм13) отдельные взрывы
наблюдались при 20 и 30 см лишь с тэном, гексоген взрывов не
давал.
Таким образом, воспроизвести результаты Каста и соответству-
ющие данные других исследователей для тэна и гексогена не уда-
лось, Этот итог вполне естественен в свете исследований и сообра-
жений Н, А, Холево,
Чтобы понять все же, каким образом возникает взрыв, Иде и
сотрудники решили изучить поведение взрывчатого вещества при
102
o,i
Фиг, 8. Штемпельный
прибор Каста для опре-
деления чувствительности
ВВ к удару на копре.
I—верхний поршенек, 2—на-
пранляющая муфта, 5— ниж-
ний поршенек.
Материал—инструмент альмл я
сталь (Белер экстра МП.
термообработка — за кал на и
отпуск до твердости 60—
63 по Рокаеллу (шкала CS,
рабочая часть поршеньков
н канал муфты шлифован-
ные.
взрывчатого вещества
отказах и использовали для этой цели малочувствительные динит-
робензол и динитротолуол, а также тротил. В новых приборах —
при допустимых по Касту предельных энергиях удара (т. е. 2 кг до
высоты 60 см, 5 кг до 40 см, 10 кг до 24 см, 20 кг до 16 cjh), можно
было наблюдать, что вещество при ударе спрессовывается, в таб-
летку и лишь в небольшой части выдавливается в зазор; эта часть
несколько изменяется в зависимости от величины зазора, веса груза
и высоты падения. При более интенсивном воздействии, например
при грузе в 10 кг и падении его с высоты 50 см (количество веще-
ства 50 лш3), вещество было в основном выдавлено через зазор н
оказывалось на нижней стороне головок поршеньков. В опытах с
динитробензолом часть выдавленного вещества на поверхности бы-
ла слабо окрашена в сероватый цвет, т. е., по-видимому, вещество
было сильно нагрето и разложилось. При опытах с тротилом в од-
ном-двух местах на торцах поршеньков наблюдались воспламене-
ния, которые с током вещества распространялись в зазор и через
него наружу. Применение термочувствительных веществ, необра-
тимо меняющих окраску при достижении определенной темпера-
туры, показало, что между торцами поршеньков окраска не изме-
нилась. Выброшенное же из зазора вещество не только расплави-
лось (температура плавления 61°), но и переменило окраску.
Особенно трудно избегнуть выдавливания вещества в зазоры в
случае жидких и желатинообразных ВВ и, как правило, действие
взрыва проявляется здесь не на поверхности торцов поршеньков;
при этом взрыв возникает тем легче, чем меньше был в употреб-
лении прибор, т. е. чем меньше зазор между поршеньком и каналом
муфты.
Предотвращение возможности выдавливания вещества при
ударе сильно уменьшает вероятность взрыва, так что даже высоко
чувствительные вещества вроде хлоратита 1 или гремучей ртути не
взрываются при падении груза в 2 кг с высоты-60 см.
При менее точном изготовлении прибора или при износе послед-
него состояние его рабочих поверхностей и кромки поршеньков
ухудшается. Влияние этого ухудшения на поведение ВВ при ударе
было проверено следующим образом. В новых приборах с малым
зазором (0,03 леи), гладкой поверхностью и острыми кромками в
одном случае торцы порщеньков были сделаны шероховатыми пу-
тем удара по 20 Л1.мэ карбида кремния, а в другом — кромки их были
закруглены (г = 0,5 лоч), чтобы облегчить возможность вытека-
ния ВВ. Эти факторы были также изучены на старых штемпельных
приборах, зазор которых колебался в пределах 0,2—0,5 мм. Нако-
нец, при помощи специально устроенной муфты, которая позволяла
веществу течь также вниз, зазор был увеличен до 1 мм, В качестве
ВВ была применена тонковолокнистая нитроклетчатка (13,4% N),
которая, как указывалось выше, в нормированных приборах дает
лишь единичные взрывы.
Результаты испытаний представлены на фиг. 9. Из этих резуль-
татов видно, что наименее благоприятные для взрыва условия пред-
ставляет нормированный прибор, в котором наблюдались лишь
юз
два единичных взрыва. При других условиях (шероховатые поверх-
ности, закругленные кромки, увеличенный зазор) вероятность взры-
ва больше. Кастовское правило, что с увеличением высоты падения
грува увеличивается вероятность взрыва, во многих случаях выпол-
няется, однако, лишь при условии, если его требования к прибору—
малый зазор между поршеньком и каналом муфты, гладкая поверх-
Фиг, 9. Чувствительность нитроклетчатки (13,4% N,
длина волокна около 0,5 леи) к удару на копре в
кастовских приборах в зависимости от состояния
поверхности торцов поршеньков, формы их кромок и
величины зазора между ними и каналом муфты,
(Груз 5 кг. количество ВВ 50 ли3, число опытов в
каждой серии 6).
Условные обозначения:
клетки черные—взрыв, клетки заштриховамные—разложснис
без звука, клеткл белые—ни разложения, ни азрыаа.
ность торцов поршеньков и невозможность бокового выдавливания
ВВ—не соблюдаются.
Чтобы выяснить, имеют ли эти наблюдения общий характер, опы-
ты были повторены с кристаллическим веществом — тэном в виде
мелких частиц. Результаты этих опытов, изображенные на фиг. 10,
отличаются от представленных на фиг. 9 в том отношении, что пере-
ход от гладких торцов к шероховатым или же увеличение зазора
104
до 0,2—0,5 мм при гладких торцах не Приводит к повышению час-
тости взрывов, в то время как при всех остальных условиях наблю-
дается значительное ее увеличение.
Эти результаты подтверждают, что нормированный прибор не
пригоден для испытания порошкообразных ВВ, поскольку даже от-
носительно чувствительные ВВ, испытанные в нем, этой чувстви-
тельности не обнаруживают.
Фиг, 10. Чувствительность тэиа (размеры частиц
0,1—0,2 л,ч) к удару на копре в кастовских приборах
в зависимости от состояния поверхности торцов пор-
шеньков, формы их кромок и величины зазора между
ними и каналом муфты.
(Груз 5 кг, количество ВВ 20 мм3. число опытов в
каждой серии 6).
Условные обозначения те же, что на фиг, 9.
Опыты с тэном в виде крупных частиц (0,1—0,5 -им) показали
ббльшую его чувствительность (фиг. И); уже наличие шерохова-
тости дает небольшое увеличение частости взрывов, увеличение
зазора от 0,03 до 0,5 мм приводит к взрывам почти при каждом
опыте. Это опять-таки расходится с заключением Каста о том, что
при малых размерах частиц ВВ чувствительнее, чем при крупных
частицах.
105
Были поставлены также опыты с жидкими и желатинообразны-
ми взрывчатыми веществами. Одним из них был нитроглицерин,
который испытывали с грузом в 1 кг. Фиг. 12 показывает, что- по-
ведение жидкого ВВ совершенно иное. Для него подходит именно
нормированный прибор с гладкими торцами, малым зазором и ост-
Фиг, 11. Чувствительность тэна (размеры частиц
ОД—0,5 их) к удару на копре в кастовских приборах
в условиях, приведенных на фиг, 10.
рыми кромками поршеньков; в таком приборе чувствительность ВВ
соответствует указываемой Кастом.
Наличие фасок или шероховатости поверхности поршеньков при
малом зазоре приводит лишь к незначительному уменьшению час-
тости взрывов нитроглицерина; увеличение же зазора до 0,2— 1 мм
лишь при шероховатых поверхностях дает значительную частость
взрывов этого ВВ; в других изученных условиях взрывов не проис-
ходило.
Исследователи испытывали также желатинообразное взрывча-
тое вещество — аммонжелит 1, имеющий следующий состав: 37,7%
нитрогликоля, 4% тринитротолуола, 1,8% коллодионного хлопка,
52,3% аммиачной селитры, 4% древесной муки и 0,2% мумии. Как
106
Фиг. 12. Чувствительность нитроглицерина к удару на копре
в кастовских приборах в зависимости от состояния поверхности
торцов поршеньков, формы их кромок я величины зазора
между ними и каналом муфты.
(Груз 1 кг, количество ВВ 20 .ч.м3, число опытов в каждой
серии 6).
Условные обозначения те же, что на фиг. 9.
107
показали результаты испытаний, размер зазора в этом случае иг-
рает еще большую роль, его увеличение значительно уменьшает
частость взрывов при прочих равных условиях (см. фиг. 13),
Фиг. 13. Чувствительность аммовжелита 1 к удару на
копре в кастовских приборах в зависимости от состоя-
ния поверхности торцов поршеньков, формы их кро-
мок и величины зазора между ними и каналом муфты.
(Груз 5 кг, количество ВВ 60 лл3, число опытов в
каждой серии 6).
Условные обозначения те же, что на фнг. 9.
Объяснение полученных результатов и предположительный
механизм возбуждения взрыва при ударе
Своим результатам немецкие исследователи дают следующее
толкование, которое целесообразно привести частично даже тексту-
ально. «Рассмотрение полученных результатов приводит к заклю-
чению, что чувствительности, как свойства ВВ, вообще не сущест-
вует или во всяком случае, что ее нельзя установить испытанием
в штемпельных приборчиках, если выполнять требования Каста,
о предотвращении бокового выдавливания ВВ».
При хорошей пригонке поршеньков к муфте (зазор 0,03 jujm)
порошкообразные ВВ не проявляют своей чувствительности. Для
108
жидких же и желатинообразных ВВ эти условия соответствуют
максимуму чувствительности.
По-видимому, однако и в этом случае ВВ течет и взрывается
при продавливании с большой скоростью в узкий зазор, поскольку
взрывы происходят не между торцами поршеньков, а в зазоре. Если
же зазор сделать больше, то скорость выдавливания и интенсив-
ность воздействия на ВВ падают настолько, что взрывы наблюда-
ются лишь в единичных случаях или совсем прекращаются; это
может привести к ложному выводу, что такие опасные ВВ, как
нитроглицерин или динамиты, малочувствительны к механическим
воздействиям.
Порошкообразные ВВ при ударе сначала спрессовываются в
таблетку и лишь при большой энергии этого удара или при боль-
шом зазоре могут выдавливаться в последний. Это выдавливание
немецкие исследователи без достаточных к тому оснований считают
обусловленными присутствием между частицами взрывчатого веще-
ства воздуха, который при течении после сжатия, увлекает с со-
бой ВВ. На основании того, что при гладких торцах поршеньков и
наличии на них фасок увеличение зазора от 0,03 до 0,5 мж слабо
отражается на чувствительности как нитроклетчатки, так и тэна,
следует полагать, что в этих условиях опыта зазор не играет суще-
ственной роли, так как для выдавленного ВВ достаточно объема
кольца, образованного закругленной фаской. Следует добавить, что
за исключением очень чувствительных ВВ основное действие, о ко-
тором можно судить по следам взрыва на торцах поршеньков,
наблюдается на периферии заряда.
Целесообразность разработки новых методов
, На основе установленных закономерностей исследователи при-
шли к правильному выводу о непригодности стандартного кастов-
ского метода испытания ВВ на чувствительность к удару и о необ-
ходимости разработки нового, который отражал бы наиболее
опасные условия воздействия, могущие встретиться при производ-
стве и применении ВВ. Эти последние условия, кстати, не соответ-
ствуют тем, которые-реализуются в стандартном методе, основан-
ном на стремлении исключить возможность выдавливания и
сдвигов в ВВ.
Сделанное в ходе опытов наблюдение, что вероятность взрыва
возрастает в изношенных приборах, привело к мысли разработать
такую схему испытания, которая соответствовала бы условиям
деформации ВВ в таких приборах. В связи с этим возникла пока-
занная на фиг. 14 схема испытания при ударе по тонкому слою ВВ,
находящемуся между торцами двух стальных свободных роликов
без направляющей муфты, которая могла бы затруднить истече-
ние В В.
Поскольку некоторые ВВ проявили особенно высокую чувстви-
тельность при ударе между шероховатыми поверхностями, пред-
ставлялось целесообразным дополнительно провести испытание
109
также и в этих условиях. Наиболее подходящими для этой цели
оказались стальные пластинки с насечкой как у напильника.
Схема испытания ВВ на копре с применением стальных пласти-
нок с насечкой приведена на фиг. 15. Пластинки гладкие снизу
имеют двухрядную насечку, зубчики которой расположены под
углом около 68° друг к другу, расстояние между ними около 0,25зон;
I*
Фиг. 14. Схема испытания ВВ на
KOrtpe между двумя свободными
роликами.
/—вставка в груз [материал — иелегн-
розаниая сталь, содержащая [•/» С.
твердость 60 по Роквеллу — шкала С).
2—верхний ролик, нижний ролик,
4—центрирующее кольцо. 5— и а ко в а ль-
ни, d—стальной блок. Материал для
роликов — сталь \05 Сг 4, твердость 60—
63 по Роквеллу (шкала С), Торцы ро-
ликов тонко шлифованные и слегка
полированные.
Фиг. 15, Схема испытания ВВ на
копре между свободным роликом
и пдастицкой с насечкой,
/—вставка в груз [материал — легиро-
ванная Сталь, содержащая Р/» С, твер’
досуь 66 по Роквеллу — шкала С), 2—
ролик [материал — сталь 105 Сг 4,
твердость 60—63 по Роквеллу — шка-
ла С), 3—пластинка с иасецкой (раз-
меры 20X20X5 лмг материал — ыелегн'
роваиная сталь, содержащая 1,2—
L4fl/»C, твердость 60—63 по Роквел-
лу — шкала С) 4—наковальня (мате-
риал — сталь [05 Сг 4, твердость 60—
63 но Роквеллу--шкала С).
таким образом на 1 см2 имеется около 1500 пирамидальных ост-
риев. На пластинку помещается навеска испытуемого ВВ, на кото-
рую затем устанавливается ролик с фаской по краю. Фактический
диаметр торца ролика равен 9 мм. На поверхность с этим диамет-
ром приходится около 1000 остриев. Пластинка используется для
4 опытов, а затем заменяется новой.
Пригодность методов испытания ВВ по схемам, приведенным па
фиг. 14 и 15, была проверена исследователями на крупно-крнстал-
лическом тэне (0,1—0,5 jot), поведение которого в стандартном
приборе было ранее иллюстрировано фиг. 11. Результаты этих ис-
пытаний изображены на фиг, 16. Они кстати подтверждают вывод
о том, что взрыв возникает в результате течения ВВ, образование
же горячих точек в Боуденовском смысле само по себе еще недо-
110
/0 № 30 !0 ?ОЗОоО ю т зо ид
Высоко падения &ацза 8 cw
статочно для этого —в противном случае при ударе через стальной
шарик (см. 2 и 3 фиг. 16) чувствительность была бы больше, чем
через рол'ик, в полную противоположность тому, что имеет место
в действительности.
В соответствии с условиями проведенных испытаний исследова-
тели дают следующее определение чувствительности. Чувствитель-
ность при данных определенных условиях опыта есть та минималь-
ная энергия удара (произведение
высоты падения на вес груза),
при которой ВВ при непрерыв-
ном ряде в 6 опытов хотя бы од-
нажды дает взрыв, т. с. разла-
гается полностью или частично
со слышимым, хотя иногда м
очень слабым звуком.
При этом подразумевается,
что речь идет о веществе, способ-
ном к взрыву, что должно быть
предварительно установлено со-
ответствующими методами. Эта
оговорка существенна, поскольку
есть вещества, которые на копре
взрываются, но одновременно
характеризуются очень малой
способностью к распространению
взрыва. По-видимому, так об-
стоит дело с аммониевой солью
динитроортокрезола н бихрома-
том аммония.
Вторую оговорку приводим
текстуально: «Чувствительность к
Удару не является константой
вещества вроде плотности или
температуры плавления; более
того она согласно имеющемуся
опыту зависит от различных фак-
торов».
Во-первых, чувствительность обусловливается методом испыта-
ния и соответственно зависит от наличия оболочки, уплотнения,
расположения пробы и т. д. Во-вторых, играет роль состояние ис-
пытуемого ВВ, способ его получения, размеры частиц, возможно
также климатические условия при испытании. В-третьих, энергия
удара, определяемая как произведение веса груза и высоты паде-
ния, не является абсолютной характеристикой чувствительности,
Наблюдения показывают, что для некоторых ВВ указанное произ-
ведение, т. е. энергия, необходимая для получения взрыва, с умень-
шением веса груза уменьшается; по-видимому, играет роль резкость
удара, возрастающая с увеличением высоты падения груза. Влия-
ние этого фактора могло бы быть исключено проведением испыта-
Фиг. 16. Чувствительность тэнл (раз-
меры частиц 0,1—0г5 лип) к удару нз
копре в различных условиях опьпя.
(Груз 5 кг, количество ВВ 20 alii3.
число опытов в каждой серии 6).
/—прибор Каста нопъ|й, зазор 0.03 льи,
поршеньки без фасок, торцы их гладкие.
2- ВВ между стальными роликом I5X [5 м.«
к nJарнком диаметром 10 .о, 3—ВВ между
пластинкой 20 < 20 <.5 мл< с насечкой и
стальным Шариком диаметром [0 жж,
4--многократно применявшийся прибор
Каста с зазором от 0,2 до 0.5 мм и шеро
сонатой поверхностью торцов поршеньков,
т -ВВ между роликами ло схеме фиг. 14,
6—ВВ между пластинкой с наседкой
и стальным роликом по схеме фиг. [5
Условные обозначения те же. Что на фцг. 0,
111
ния при постоянной высоте падения, но этот прием имеет свои
недостатки.
Так как при ударе по ВВ наблюдается целая гамма переходных
явлений от отсутствия видимых изменений в нем до полного взры-
ва, то для некоторой определенности они условно разбиты на три
категории.
Отсутствие реакции — означает отсутствие разложения или из-
менения цвета, пригорелого запаха, искр или пламени.
Разложение (изменение окраски, частичное или полное исчез-
новение ВВ без пламени), иногда со слабым искреннем и без звука,
иногда с жестким металлическим звуком и со следами цветов по-
бежалости.
Взрыв (частичное или полное исчезновение ВВ), причем звук
может изменяться от очень слабого до очень сильного. Кроме того,
может наблюдаться пламя, вырывающееся из промежутка между
торцами роликов, и цвета побежалости на них размерами от була-
вочной головки до всей поверхности торца. В особых случаях не-
многие ВВ воспламенялись без звука. В этом случае явление обо-
значается не как разложение, а как взрыв, поскольку оно охватывает
всю пробу и чередуется со взрывом, такой взрыв обозначается в
приведенных ниже таблицах особо, например, звездочкой.
Исследователи по разработанным ими методам испытания чув-
ствительности ВВ к удару изучили ряд взрывчатых веществ. Ре-
зультаты этих опытов приводятся ниже.
Первый метод испытания, предназначенный для
порошкообразных ВВ
(удар по заряду, помещенному между двумя роликами без муфты)
Испытания проводили по схеме, приведенной на фиг. 14. На-
веска ВВ бралась по объему и изменялась в пределах 10—80 мм3.
Наиболее целесообразной оказалась проба в 40 лл3 при грузе
в 5 кг.
Применяли копры двух конструкций — один обычного кастов-
ского типа для грузов от 1 до 20 кг, другой — для грузов от 25 до
1000 г. Результаты опытов для разнообразных ВВ даны выборочно
в табл. 2.
Для некоторых взрывчатых веществ, в том числе и относительно
слабых (дннитрозопектаметилентетрамин, азид бария, бихромат
аммония и др.) можно, увеличивая высоту падения, добиться уве-
личения частости взрывов до получения их при всех 6 опытах; дру-
гие же ВВ, например, донарит, тетрил, пороховая мякоть этого
не дают; иногда наблюдается даже уменьшение частости взрывов
при увеличении высоты падения груза (тетранитрокарбазол).
Некоторые ВВ давали лишь единичные взрывы (например три-
нитронафталин) или взрывы лишь при больших высотах (например
азиды щелочноземельных металлов, пикриновая кислота). Для не-
которых веществ наблюдалось разложение без звука (аммиачная
112
Таблица 2
Результаты испытания некоторых ВВ на чувствительность
при ударе на копре между стальными роликами 10x10 и 15x15 мм
(Груз 5 /£г; количество ВВ 40 ммЗ; число разложений (Р) и взрывов (В)
приведено из 6 опытов; тире означает нуль)
ВВ Размер частиц мм Высо Ре- гр; зуль- тат 10 20 30 та падения ^за в см Звук взрыва
13 5С 60 80 1 00
Тэн 0,1—0,5 Р В 6 6 От слабого до сильного
Калиевая соль гек- санитродифениламина 0,1—0,5 Р В 4 5 6 Слабый
Нитроклетчатка 13,4% N Тонко- волок- нистая Р 111 В 3 4 5 6 Сильный
Натриевая соль ди- ки тро-о -крезола 0,1—0,5 Р В 2* 6* 6* 6* От очень слабого до слабого, частично воспламенение без звука
Коллоксилин 12.2М N Тонко- волок- нистый Р 1 В 15 6 5 6 От слабого до сильного
Гексоген 0,1—0,5 Р 111 В — 1 5 — 1 — 6 4 3 От слабого до средне-сильного
Г ексанитрод ифе ннл- амин 0,06—0,2 Р 2 11 В — 4 4 Qi 1 Cl 1 Cl 1 Слабый
Донарит 1 Крупно- зернис- тый Р - 2 2 В — 2 1 1 1 11 1 1 3 4 1 Слабый
Тринитрорезорци- ват свинца о,о5—о,; Р — В — 3 6 6 Очень сильный
Перхлорат аммония 0,1—0,5 I I _ I 5 4 6 1 Сильный
Пиротехнический состав 0,5— 1,( t I -2* 4 5 6 От слабого до с редне-сильного, в том числе 1 воспламенение без звука *
Тетрил <0,3 р — 1 в 12 13 3 -—112 2 Слабый
Пикриновая кислота 0,1—2, 1 г - 1 1 1 Д, CQ - 2 Слабый
Примечание, С пробами пикриновой кислоты 20 и 60 жж3 были
получены аналогичные результаты.
* Воспламенение без звука.
8 451
113
селитра), а другие вообще не реагировали на удар (гуанидиннит-
рат, нитрогуанидин, динитро-о-крезол, тринитротолуол),
Результаты, полученные при грузе в 1 кг для наиболее чувст-
вительных взрывчатых веществ из числа приведенных в табл, 2,
даны в табл, 3, Сопоставление этих таблиц позволяет сделать сле-
дующие выводы: при переходе от груза весом 5 кг к однокилограм-
мовому высота падения увеличивается меньше чем в 5 раз; крити-
ческая энергия удара при увеличении высоты падения уменьшается.
Груз в 1 кг не всегда оказывается достаточным для надежного воз-
буждения взрыва. Так в опытах с пироксилином несмотря на умень-
шение количества ВВ до 10 мм3 достигается только 50% взрывов,
для коллоксилина наблюдаются лишь единичные взрывы. Регу-
лярное увеличение частости взрывов с высотой падения груза пока-
зывает тэн, гексоген, калиевая соль гексанитродифениламина и тер-
мически нестойкий нитрил азоизомасляной кислоты.
Таблица 3
Результаты испытания некоторых ВВ на чувствительность при ударе
на копре между стальными роликами 10X10 и 15X15 мм
(Груз 1 кг\ количество ВВ 10 число разложений (Р) и взрывов (В)
приведено из 6 опытов; тире означает нуль)
ВВ Размер частиц мм Ре- зуль- тат Высота падении груза в см Звук взрыва
10 20 зс 40 50 60 80 100
Тэн 0,1—0,5 р в — 2 R 6 6 От слабого до сильного
Калиевая соль гек- санитродифеииламииа 0,1—0,5 р в — 2 1 2 4 1 2 6 6 3 3 Слабый
Гексоген 0,1—0,5 р в — 1 2 1 2 •4 4 5 От слабого до средне-сильного
Нитроклетчатка 13,4% N Тонко- волок- нистая р в — 1 1 1 2 1 3 1 2 1 1 2 3 От очень слабого до слабого, с пробой 40 мм3 сильный звук
Коллоксидин 12,2% N Тонко- волок- нистый р в — 1 1 1 3 — 2 2 1 Слабый
Натриевая соль ди- нитро-о-крезола 0,1—0,5 р в — — 1 [* 2 2* 2 1* 1 1* С пробой 20 мм3 реагирует чаще
Гексанитродифе- ннламиц 0,06—0,2 р в — 2 1 .3 2 Слабый
* Воспламенение без звука.
114
Результаты испытаний на копре различных ВВ исследователи
объясняют на основе следующих соображений.
Под действием удара падающего груза по достижении опреде-
ленного давления испытуемое порошкообразное ВВ начинает вы-
давливаться между торцами со значительной скоростью, что играет
существенную роль для возбуждения взрыва. Если возможность
выдавливания отсутствует, то чувствительность сильно умень-
шается.
Для порошкообразных ВВ при описанных условиях испытания
воздействие оказывается достаточным, чтобы трение при течении,
Фиг, [7, Венчик глубоких насечек на торцах стальных роликов
после взрыва тетрила. (Груз 5 кг, высота его падения 60 см,
количество ВВ 40 мм3, испытание в приборе, собранном по
схеме фиг. 14),
Слева торец нижнего ролнкэ, справа верхнего.
разламывание кристаллов и т. д. вызвало взрыв, жидкие же и
желатинообразные ВВ вследствие большей текучести требуют бо-
лее жестких условий течения.
В пользу такого объяснения говорят, в частности, наблюдения,
сделанные при опытах с тетрилом, который при Гладких торцах
обычно чаще разлагается без взрыва, чем со взрывом. При удале-
нии ацетоном остатков ВВ на периферии был обнаружен ряд ради-
альных глубоких насечек, которые были расположены как на ниж-
нем, так и на верхнем ролике и выглядели так, как будто были
выжжены микрогорелкой (фиг. 17). Так как других явлений, вклю-
чая слабые цвета побежалости, вблизи не обнаруживалось, следует
полагать, что эти насечки на торпах роликов появились в резуль-
тате локальных взрывов с высокой температурой их продуктов.
Тот факт, что насечки появляются только на периферии в узкой
кольцеобразной зоне шириною около 0,5 мм, указывает на то, что
лишь в ней возникают условия для взрыва. По-видимому, радиаль-
ное направление насечек связано с радиальным течением час-
тиц ВВ. За возбуждение взрыва на периферии торцов говорит
8* , 115
также расположение цветов побежалости, которые часто ограничи-
ваются периферийной зоной.
По сути дела, возбуждение взрыва при ударе имеет много об-
щих черт с возбуждением его при трении; последнее, возможно,
является необходимым условием возбуждения взрыва при ударе.
Второй метод испытания, предназначенный для
жидких и желатинообразных ВВ
(удар по заряду, помещенному между двумя роликами с муфтой)
Жидкие и желатинообразные ВВ не взрываются при испытании
по первому методу. Достаточно высокая частость взрывов, соответ-
ствующая их опасности в обращении, получается для жидких ВВ
в изношенных штемпельных приборах с шероховатыми торцами
поршеньков или в приборах с гладкими торцами (с фасками или
без фасок), но при наличии муфты с малым зазором. Эти условия
более определенны, поэтому при испытании жидких ВВ на них и
рекомендуется ориентироваться. Диаметр роликов был Принят рав-
ным 10 мм, толщина муфты — 3 л1л<, высота 13 jwjm и зазор (разность
диаметров) не больше 15 лк. Данные, полученные при испытании
нитроглицерина и нитрогликоля по этому методу, приведены
в табл. 4.
Таблица 4
Испытание жидких ВВ на чувствительность к удару на копре
между роликами 10x10 мм с закругленными фасками при наличии муфты
(Груз 0,1 кг; объем пробы ВВ Ю жж3, приведено число взрывов из 6 опытов)
Третий метод испытания чувствительности ВВ
(на пластинках с насечкой)
Испытания проводили по схеме, показанной на фиг. 15. Ре-
зультаты их приведены в таблицах 5 и 6. Данные таблиц 5 и 6
показывают большую чувствительность принятого метода испыта-
ния. Эта чувствительность проявляется в быстром увеличении час-
тости взрывов. Преимущества метода обнаруживаются не только
при испытании ВВ высокой чувствительности. Так пикриновая кис-
лота между роликами взрывается в отдельных случаях лишь при
высоте падения груза 100 см, на пластинке же с насечкой она ре-
гулярно дает слабые взрывы с тем же грузом (5 кг) начиная с вы-
116
соты 20 см. И при этом методе наблюдается увеличение частости
взрывов в случае уменьшения веса груза при данной энергии удара.
Преимуществом метода испытания ВВ на пластинках с васечкой
является возможность, не превосходя вес груза 20 кг, определять
чувствительность даже таких малочувствительных ВВ как динитро-
бензол, чего не удается сделать на роликовых приборах.
Большую легкость возникновения взрыва на пластинке с насеч-
кой по сравнению с роликами исследователи объясняют большей
затрудненностью в этом случае радиального течения ВВ. Признаки
этого течения при взрыве обнаруживаются по расположению и на-
правлению обычно обугленных остатков и по цветам побежалости
на поверхности торна верхнего ролика, которые часто начинаются
от отпечатков гребней насечки и направлены по каналам или ра-
диально.
Естественно предположить, что в точках контакта гребней насеч-
ки и торца ролика в результате перехода энергии деформации в
тепло возникают разогревы. Эти разогревы в сочетании с разру-
шением кристаллов при ударе и процессами течения ВВ в желоб-
ках вызывают взрыв.
Большая чувствительность рассматриваемого метода испыта-
ния ВВ обусловливается тем, что энергия удара распределяется
здесь неравномерно. Кроме того, в желобках пластинки остается
больше ВВ, чем на торце плоских роликов.
Общая характеристика чувствительности различных ВВ
по различным методам испытания
Сопоставление данных, полученных при испытании на пластин-
ках и в роликовых приборах, представлено в табл. 7.
По данным проведенных испытаний и в первую очередь испыта-
ния на пластинках с насечкой исследователи разделили изучен-
ные вещества на группы по чувствительности следующим образом:
1 группа: не чувствительны к удару (при грузе в 5 кг в данных
условиях испытания не наблюдается ни разложения, ни взрыва);
2 группа: мало чувствительны (при грузе в 5 или 1 кг наблю-
дается только разложение);
3 группа: чувствительны к удару (при грузе в 5 или 1 кг наблю-
даются взрывы, которых не происходит при весе груза 0,1 кг или
меньше).
При этом в соответствии с имеющимися опытными данными воз-
можно дальнейшее подразделение ВВ 3-й группы на следующие
подгруппы:
а) условно чувствительны к удару —- единичные взрывы;
б) чувствительны к удару, но частость не возрастает до 100%
или же взрывы происходят нерегулярно;
в) нормально чувствительны к удару в том смысле, что вероят-
ность взрыва равномерно и более или менее быстро возрастает
от 0 до взрывов при всех 6 опытах.
117
05
Таблица 5
Результаты испытания некоторых ВВ на чувствительность к удару на копре
при нс пользовании пластинок с насечкой
(Груз 5 кг-, количество ВВ 40 жз<з; число разложений (Р) и взрывов (В) приведено из 6 опытов, тире означает нуль)
ВВ Размер частиц мм Обозна- чение резуль- . тата Высота падения в см Звук при взрыве Примечание
10 20 30 40 50 ’60 80 100
Калиевая соль гекса н и 1 род и фе- нила мина 0,1—0,5 р в 6 6 Сильный
Натриевая соль д и и и тро-о- кр езо л а 0,1—0,5 р в 4* 6 6 От слабого до среднесильного, в том числе одно воспламенение без звука
Гексанитродифе- ниламин 0,06—0,2 р в 3 3 6 6 От слабого до среднесильного
Хлорат ит 1 Тонко- зернис- тый р в 3 5 6 От среднесиль- ного до сильного
Тринитрореэор- цни 0,1—0,5 р в 2 2 4 5 1 5 2 4 6 От слабого до среднесильного
Тетрил <0,3 р в 3 2 4 4 2 2 4 I 5 6 6 6 От очень слабо- го до слабого
Нитрил азоизо- мзсляной кислоты 0,1—0,2 р в 2 1 1 1 1 2 —* — То же При пробе 10 мм3 число взрывов возрас- тает до 6
Бнхромат аммо- ния 0,1—0,5 Р В 1 4 6 6 От слабого до сильного
Дииктрозопента- метилентетрамин 0,064-0,3 Р В 1 1 3 6 6 6 Сильный
Пер хлор а та мио- пия 0,14-0,5 р в — 1 3 5 6 6 Сильный
Черный порох (мякоть) <0,5 р в — 2 4 6 6 От сильного до очень сильного
Пикриновая кис- лота 0,14-2,0 р в 1 1 2 2 6 6 6 6 Слабый
Тринитробензой- иая кислота <0,2 р в 2 4 2 4 1 4 2 4 2 3 4 2 От слабого до сильного При пробах 10 н 20 мм3 чис- ло взрывов воз- растает до 6
К Тетраиитрокар- базол 0,14-0,5 р в 1 1 2 5 5 1 2 4 4 2 От слабого до среднееильного
Аммонжелит 3 Гель из крупных частиц р в 1 3 2 1 5 4 2 2 2 3 1 3 2 Слабый При пробах 10 и 20 мм3 больше взрывов
Этилендиамин- динитрат 0,14-0,5 р в 1 1 2 4 1 2 4 4 2 3 2 4 От очень слабо- го до слабого При пробах 10 и 20 мм3 чис- ло взрывов воз- растает до 6
Азид бария 0,14-0,5 р в 1 4 4 .6 5 От средьесиль- ного до очень силь- ного
Триннтронафта- ЛИИ 0,14-0,5 р в —• 3 4 5 4 6 От слабого до СИЛЬНОГО
Продолжение
ВВ Размер частиц мм Обозна- чение резуль- тата Высота падения в см Звук при взрыве Примечание
10 20 30 40 50 60 80 100
Аммониевая соль динитро-о-крезола <0,2 р в — 3 3 6 6 1 5 6 От слабого до среднесильного
Пиротехничес- кий состав 0,5—1,0 р в — 1 1 3 5* 4 4 От сильного до очень сильного; в том числе 1 вос- пламенение без звука
Оксицианид рту- ти 0,1—0,5 р в — 1 1 1 1 5 6 От сильного до очень сильного
Допарит 1 Зернис- тый р в — 2 1 2 2 1 2 1 Слабый При пробе 10 ст вы- соты 10 см чис- ло взрывов воз- растает до 6
Натриевая соль п-иитрсфенола (безводная) 0,06—0,2 р в — 1 2 3 4 1 3 5 5 От слабого до среднесильного
Азид стронции 0,1—0,5 р в — 1* 2* 2* 3 3* 1 5* От сильного до очень сильного; частично воспла- менение без звука При пробе 5 -ил3 от высо- ты 10 см число взрывов возрас- тает до 6
Азид ка льция 0,1—0,5 р в — — 1* 1* 1 2 2 Сильный При проб? б жж’ ст высоты 20 £.« число взрывов воз- растает до 6
!
Трииитролнилин 0,1—0,5 Р В — 1 — 2 3 Слабый Отдельные взрывы
Оксалат серебра <0,2 CuCQ — —- — — — 1 1 1 Слабый При пробе 60 мм3 от вы- соты 40 см чис- ло взрывов воз- растает до 6
Тринитротолуол <0,2 р в — 3 5 6 6 4 6
Гуанидиннитрат 0,1 -г-0,5 р в — — — — —
Динит робенэол Призмы 0,01—0,2 р в — — — — —
Динитро-о-кре- зол <0,2 р в — — — — —
* Воспламенение без звука.
и
Таблица б
8
Результаты испытания некоторых ВВ на чувствительность к удару на копре при использовании пластинок с насечкой (Груз 1 кг, количество ВВ 5 мм3, число опытов 6, условные обозначения те же, что в табл. 5)
ВВ Размеры частиц мм Обозна- чение резуль- тата Высота падения в см Звук при взрыве
10 | 20 30 40 50 60 80 100
Смесь хлората калия с красным фосфором 75/25 0,06—0,075 р в 6 6 Очень сильный
Ацетиленистое сереб- ро с примесью азотно- кислого серебра (из HNOn-раствора) <0,001 р в 6. 5. Сильный
Ацетиленистая ртуть Тонно- зернистая р в 4 6 6 От слабого до средне- сильного
Циануртриазид 0,02-0,55 р в 3 6 6 Сильный
Ацетиленнстое сереб- ро (из нейтрального раствора) р в 2 5 От слабого до средне- сильного
Ацетиленнстое сереб- ро (из NH3-paciBopa) 0,01 р в 2* 5* 6* Слабая вспышка
Азид свинца 0,01-0,1 р в 2 4 4 6 Очень сильный
i
. Ацетиленистая медь _ р в ’2* а* 6* 5*' 6* Слабый, вещество час- тично воспламеняется без звука, обильное выделе- ние сажи
- Перекись трициклоаце- тона <0,07 р в 1 3 5 б Сильный
Азид ртути (I) Зернистый р в 2 2 4 6 6 Очень сильный
Гёк са м ет ил ен т р нпер ок- сиддиамин 0,1 р в 1 3 2 3 6 6 Очень сильный
Тринитрорезорцина т свинца 0,05—0,2 р в 5 3 2 6 6 Очень сильный
Гремучая ртуть белая 0,05-0,8 р в — 3 5 5 5 4 6 Очень сильный
Тетразен 0,003-0,1 р в — 2 5 4 6 6 Сильный
Гремучая ртуть се- рая 0,05-0,8 р в — 2 4 4 4 2 6 Очень сильный
Щавелевокислая ртуть 0,06—0,5 р в — 4 4 6 6 Слабый
Эритриттетра нитрат 0,1-0,5 р в — —” 2 2 Очень сильный
Таи 0,1-0,5 р в — 2 1 Сильный
N * Воспламенение без звука.
4 группа: очень чувствительны к удару (взрывы происходят уже
при грузе в 0,1 кг или меньше при регулярном увеличении час-
тости) .
Деление это условно, но дает возможность ориентироваться при
практической оценке опасности ВВ.
Согласно данным испытания на пластинках с насечкой к пере
численным выше группам относятся:
к группе 7 — гуанидиннитрат, динитропроизводные бензола, ди-
нитрокрезол;
к группе 2 — тринитротолуол;
к группе 3 — а)триннтроанилин;
б) аммонжелит 3, тетранитрокарбазол, пиротехнический состав
на основе нитрата бария, железных опилок и алюминия, п-нитро-
фенолят натрия.
в) азотнокислые эфиры клетчатки—пироксилин и коллоксилин,
гексанитродифениламин и его калиевая соль, перхлорат и бихромат
аммония, азиды щелочно-земельных металлов, тринитросоединения
фенола, резорцина, бензойной кислоты и нафталина, натриевая и
Таблица 7
Чувствительность взрывчатых веществ, определенная между двумя
стальными роликами и на пластинках с насечкой,
выраженная как минимальная энергия удара,
при которой наблюдаются взрывы
(6 параллельных опытов; расположение ВВ в порядке уменьшения
чувствительности иа плэстинках с насечкой)
Взрывчатое вещество Размер частиц ВВ мм Минимальная энергия удара в кГ-м
между стальными роликами по схеме фиг. 14 па пластинках с насеч- кой по схеме фиг, 15
Вес груза а кг
5 1 0,1 5 1 0,1 0,025
Смесь хлората ка- лия с красным фос- фором 75/25 0,06— 0,075 0,003 0,003
Ацетиленистое се- ребро с примесью азотно-кислого сереб- ра (из НМОа-раство- ра) 0,001 1 0,5 0,003
Ацетиленистая ртуть Тонко- зернис- тая 0,06 0,003
Циануртриазид 0,02—0,55 0,04 0,003
Ацетиленистое се- ребро (иа нейтраль- ного раствора) 1 0,2 0,003
124
Продолжение
Взрывчатое вещество 1 Размер частиц ВВ » мм Минимальная энергия удара в кГ м
между стальными роликами по схеме фиг. 14 на пластинках с насеч- кой по схеме фиг. 15
Вес груза в кг
5 1 0,1 5 1 1 0,1 0,025
Ацетиленистое се- ребро (из NHa-pac- твора) Азид свинца Ацетиленистая медь Перекись трицик- лоацетона Азид ртути HgN3 0,01 0,01—0,1 0,07 Зернис- тый 0,1 0,08 0,01 0,03 0,003 0,003 0,003 0,003 0,003
Гексаметнлентри- пероксиддиаиин Трннитрорезорци- иат свинца Гремучая ртуть бе- лая Тетразен Гремучая ртуть се- рая Щавелевокислая ртуть Эритриттетранитрат Тэн Нитрил азоиаомас- ляной кислоты Хлоратит 1** Тетрил Нитроклетчатка 13,4И N Коллоксилин 12,2% N Калиевая соль гек- санитродифениламина Аммонжелит 1 *** 0,1 0,05—0,2 0,05—0,8 0,003—0,1 0,05—0,8 0,06—0,5 0,1—0,5 0,1—0,5 0,1—0,2 Тонко- зернис- тый 0,3 Тонко- волок- нистая Тонко- волок- нистый 0,1—0,5 Желати- нообраз- ный 1.5 1 3 0,5 0,5 0,5 2,5 °,5 0,5 0,5 0.6 0,2 0,2 0,2 0,5 0,3 0,4 0 2 0,1* 0,01 0,06 0,5 0,5 °.5 1 0,5 0,5 0,2 0,2 0,2 0,2 0,2 о,з 0,4 0,02 0,03 0,05 0,003 0,005 0,005 0,005 0,005 0,008 0,02 0,02
125
Продолжение
Взрывчатое вещество Размер частиц ВВ мм Минимальная анергия удара в кГ-м
между стальными роликами по схеме фиг. 14 на пластинках с насеч- кой по схеме фиг. 15
Вес груза в кг
5 I 0.1 5 1 0,1 0,025
Аммонжелит 3 **** Желатн- нообраз- 1 0,4
ный
Гексоген 0,1—0,5 1 о.з 0,4 од
Тринитробензойная 0,2 1 1 0,4
кислота
Донарит ***** Тонко- зернис- 1 0,6 1,5 0,4
тый
Дини тр оортокрезо- лят натрия 0,1—0,5 0,5 0.5 0,5 0,4
Нитроглицерин 0,03* 0,5 0,5
Тринитрорезорцин 0,1—0,5 1 0,5 0,5
Гекса ии тродифе- 0,06—0,2 1 0,8 0,5 0,5
ниламнн
Нитрогликоль 0,05* 1 0,5
Пикриновая кисло- 0,1—2,0 5 1 0,5
та
Азид кальция 0,1—0,5 5 2 0,6
Азид стронция 0,1—0,5 3 2 0,6
Азид бария 0,1—0,5 1 1,5 0,8
Бихромат аммония 0,1—0,5 1,5 1 0,8
Пороховая мякоть (75 И KNOa) 0,5 1,5 1 1
Динитрозопенгаме- тилентетрамин 0,06—0,3 1 1
Перхлорат аммония 0,1—0,5 1.5 1
Тетранитрокарба- 0,1—0,5 1 1
ЗОЛ
Этилендиамнпдини' трат 0,1—0,5 1.5 1
Тринитронафталин 0,1—0,5 1.5 1,5
Динитроортокрезо- 0,2 1,5 1,5
лят аммония
Пиротехнический состац ****** 0,5—1,0 2 1,5
ОксиЦианид ртути 0,1—0,5 2 1,5 1
126
* Значения получены между стальными роликами ЮхЮ мм, поме-
щенными в муфту.
, ** Хлоратит 1 состоит из КС1Оа (88.5и), древесной муки (8,5%)
Ж парафинового масла (3%).
'.. *** Аммокжелиг 1 состоит из аммиачной селитры (52,3%), нитрогли-
коля (37,7%), тринитротолуола (4%), коллодионного хлопка (1,8%), древесной
муки (4%) и мумии (0.2%).
**** Аммонжелит 3 содержит аммиачную селитру (55%), дигликоль-
дннитрат (22%), тринитротолуол (5%). динитротолуол (6%) и другие компо-
ненты.
***** Донарит 1 содержит аммиачную селитру (80%). ароматические
Аийтросоединения (14%). нитроглицерин (4%) и другие компоненты.
****** Пиротехнический состав содержит нитрат бария (52%), желез-
нце опилки (30%). декстрин (9%), А1 (7%) и другие компоненты.
’ аммониевая соли динитрокрезола, хлоратит I и донарит 1, газоге-
1 жерирующие вещества — динитрозопентаметилентетрамии и нитрил
; ваоизомасляной кислоты, тетрил, этилендиаминдинитрат, оксициа-
иид ртути, оксалат серебра, гексоген;
; к группе 4 — армстронгова смесь (КСЮз и красный фос-
: фор 75/25), органические перекиси — гексаметилентрипероксиддиа-
*-1Йин и перекись трициклоацетона, циануртриазид, соли тяжелых
. Металлов азотистоводородной кислоты (Pb, Hg), гремучей кислоты
. (Hg), стифниновой кислоты (РЬ), щавелевой кислоты (Hg) и аце-
- тилена (Ag, Hg, Си), тетразен и азотнокислые эфиры — эритрит-
.тетранитрат и пентаэритриттетранитрат.
В заключение исследователи подчеркивают значение, как фак-
тора возбуждения взрыва, узких щелей, через которые продавли-
вается ВВ особенно жидкое или желатинообрязное. Такие явления
могут иметь место при прессовании в прессформах, при смешении в
смесителях, если крылья мешалки чрезмерно приближаются к стен-
кам, в патронировочных машинках для желатинообразных ВВ
и т. д.
Другим существенным фактором возбуждения взрыва при ударе
является возможность выдавливания вещества, а также состояние
соударяющихся поверхностей (гладкие или шероховатые)..
127
ЗАКЛЮЧЕНИЕ
Рассмотренные экспериментальные данные и соображения зару-
бежных исследователей в основном согласуются с результатами
работ Н. А. Холево, проведенных ям и частично опубликованных
значительно ранее. Это относится к зависимости чувствительности
от возможности течения ВВ при ударе на копре (наличие и вели-
чина зазоров), от способности ВВ к течению (различие между поро-
шкообразными ВВ с одной стороны и жидкими и желатинообраз-
ными с другой), необязательность параллелизма между частостью
взрывов и высотой падения груза и др. Более того Н. А. Холево
в своих выводах продвинулся значительно дальше чем, например,
немецкие исследователи. Он не только констатировал непостоянство
чувствительности (частости взрывов) при различных условиях, но
и дал обоснованное объяснение 1 влиянию этих условий.
Новым в работе немецких исследователей является предложение
в качестве третьего метода (помимо приборов № 1 и 2 в немецкой
модификации) применять испытание ударом на пластинках с насеч-
кой, при котором чувствительность многих ВВ оказывается гораздо
выше, чем при других методах. По существу это не удивительно,
так как в методе на пластинках с насечкой удар вследствие затруд-
ненности истечения ВВ создает повышенное давление, и кроме того,
встреча гребней насечки с плоскостью ролика может приводить к
местным разогревай. Подобные разогревы Боуден [1] получал уда-
ром по игле, поставленной на наковальне, измерил возникающую
при этом температуру и показал, что легкогорящие ВВ, вроде циа-
иуртриазида, могут быть взорваны таким путем.
Для возбуждения горения трудногорящих ВВ нужно, кроме
разогрева, высокое давление, которое и создается при опытах на
пластинках с насечкой. Кроме того, в отличие от опытов с двумя
роликами, разогрев возникает, когда слой ВВ между пластинкой и
роликом еше имеет достаточную толщину для того, чтобы в нем
могло распространяться горение, что не всегда обеспечивается при
испытании легкотекучих ВВ между двумя роликами.
Положительная сторона испытания на пластинках с насечкой
состоит также в том, что местный разогрев создается в известной
мере независимо от свойств (текучести) ВВ и, если оно способно
гореть, то эта способность может проявляться. По существу удар
по ВВ на пластинках с насечкой является испытанием на воспла-
меняемость, хотя последняя устанавливается в данном случае при
несколько неопределенных условиях.
Когда мы ведем испытание в приборах № I и 2, то, чтобы
получить взрыв, мы должны обеспечить не только возможность его
распространения, но и создать условия для возникновения этого
взрыва. Отсутствие последних приводит к отказам. Например, азид
свинца в приборах № I и 2 не дает взрыва по этой причине, так
же как и нитроглицерин или тротил в приборе № 2.
1 См. настоящий сборник, стр. 5.
128
Учитывая эти соображения, следует признать испытание на пла-
стинках с насечкой полезным в том смысле, что если оно дает отри-
нательный результат, то это является доказательством низкой вос-
пламеняемости испытуемого ВВ.
Результаты опытов Юнгберга интересны, поскольку они показы-
вают, что то изменение напряжений во времени при ударе, которое
принимал Н. А. Холево, не имея прямых экспериментальных дока-
зательств, существует в действительности.
У читателя может возникндН'ь вопрос, если зарубежные авторы
в основном повторяют уже пройденный Н. А. Холево путь, то есть ли
необходимость для нас в рассмотрении этих работ.
Такое рассмотрение целесообразно, в частности, потому, что ра-
боты Н. А. Холево из-за большой оригинальности его представлений
не были своевременно в должной мере оценены многими нашими
исследователями и не получили того развития и использования,
которого они заслуживают. В этой связи то обстоятельство, что за-
рубежные исследователи, чтобы получить объективное представле-
ние о чувствительности ВВ, встали на тот же путь, который прошел
Н. А. Холево, и уже пришли к некоторым из его выводов, полезно
знать как широкому кругу лиц, ведущих работу с ВВ, так и осо-
бенно тем исследователям, которые до сих пор не смогли в должной
мере понять и оценить значение работ Н. А. Холево для науки о
взрыве и обеспечения безопасности при работе с ВВ.
Наряду с этим рассмотренные исследования содержат большой
.фактический материал по чувствительности большого числа ВВ при
’разнообразных условиях испытания, представляющий значитель-
ную ценность не только как подтверждение многих выводов Н. А. Хо-
лево, ио и сам по себе.
выводы
, Исследования американских, шведских и немецких исследовате-
лей по чувствительности взрывчатых веществ к удару привели их
'к заключению о сильной изменчивости чувствительности в зависи-
,'Мости от условий испытания и физико-механических свойств взрыв-
. чатого вешества. Обычное испытание на копре в штемпельных
приборах Каста, равно как и всякое другое подобное испытание, не
' может, взятое изолированно, дать полной характеристики опасно-
! сти ВВ в обращении. Поэтому при выборе методики испытания не-
- .Обходимо принимать во внимание физические свойства вещества
И характер тех воздействий, по отношению к которым требуется
. установить степень его опасности.
Рекомендованы новые варианты испытания ВВ на чувствитель-
. вость к удару — испытание между двумя роликами без муфты для
порошкообразных (точнее труднотекучих) ВВ (аналогично при-
; бору № 2 Н. А. Холево), между двумя роликами с муфтой при ма-
лом зазоре для жидких и желатннообразных ВВ (аналогично при-
бору № 1 Н. А. Холево) и между роликом и пластинкой с насечкой,
1 пригодное для ВВ различной физической структуры.
9 451 129
Результаты и выводы зарубежных исследователей в значитель-
ной их части повторяют и таким образом подтверждают те данные
и заключения, которые много раньше были П0ЛуЧены Н. А. Холево
в Казанском химико-технологическом институте.
16.
ЛИТЕРАТУрд
дых и SiX » ™°Р-
нн. С. М^КчюииЛММ^КоМТлИ'ив?’ хииико технологического института
3. Холево Н. А. Сб. «Физика взрыва» дН СССР, 1955 вып 3 стр
См. также настоящий сборник стр. 5. ’ к
4. LjungbergS., Nobel Helte, 1958, стр 4Q„48
оно О J ° е П е П Н” 1 d е к‘ М” Н а U р 1 W-, Explosivstoffe, 1958, 6,
6. Kistiakowski G, В, and Connor d „ и п
Chemislry, 1946, стр. 65.80. Г R” Sc,ence m WorId War П-
AI95^‘135ideal E' K a"d R°berlS0n A. J. B„ Proc. Roy. Soc.. 1949,
178,
vol.
II. ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ ВЗРЫВЧАТЫХ
ВЕЩЕСТВ
К. к, АНДРЕЕВ, Г. Н. БЕСПАЛОВ
9. О ВЛИЯНИИ ВОДЫ НА РАЗЛОЖЕНИЕ НИТРОГЛИЦЕРИНА
ПРИ ПОВЫШЕННЫХ ТЕМПЕРАТУРАХ
В ряде работ [1], [2], [5], [12], [13] установлены основные характе-
ристики термического распада нитроглицерина (НГЦ). В парах
(130—150°) разложение идет со скоростью, пропорциональной их
давлению, и убывает во времени по закону, близкому к закону реак-
ции первого порядка. При наличии жидкой фазы скорость газооб-
разования в том же интервале температур меньше, умеренно растет
со временем и слабо зависит от давления продуктов распада.
Если, однако, при разложении достигается некоторое критиче-
ское давление продуктов распада, то скорость газообразования на-
чинает быстро, приблизительно пропорционально квадрату давле-
ния, расти и может достигать очень больших значений, в сотни и
тысячи раз превосходящих начальное ее значение. «Критическое»
давление составляет около 200 льи рт. ст. при 100°. Оно не является
строго постоянной величиной: на ранних стадиях распада «крити-
ческое» давление больше; одной из возможных причин этого может
быть изменение состава продуктов разложения по мере его течения;
«критическое» давление увеличивается с ростом температуры, оче-
видно вследствие влияния последней на растворимость газов в жид-
кости.
t Наступление резкого ускорения газообразования по достижении
«критического» давления объясняется, в согласии с основной мы-
слью С. 3. Рогинского [5], [13], тем, что давление определяет кон-
центрацию газообразных продуктов распада в жидком НГЦ. По-
видимому, эти продукты — кислоты и вода — приводят к быстрому
развитию гидролитических и последующих окислительно-восстано-
вительных реакций, скорость которых гораздо больше, чем скорость
начального термического распада. Если разложение проводится при
больших степенях заполнения реакционного сосуда, когда газооб-
разные продукты распада почти целиком остаются в жидкости, то
резкое ускорение разложения наступает при 100° приблизительно
через 9 час, При 60° это время увеличивается до 550 час. Степень
распада, при которой наступает ускорение, имеет порядок десятой
доли процента.
9*
131
Все эти данные относятся к безводному НГЦ. В отдельных ра-
ботах имеются указания на ускоряющее влияние воды на разложе-
ние нитроглицерина или порохов на его основе [4], [10]. Эти указа-
ния, однако, имеют либо косвенный, либо качественный характер.
В то же время влияние воды иа разложение НГЦ представляет
значительный и разносторонний интерес. Технический нитроэфир и
различные смеси, в состав которых он входит, всегда содержат не-
которое количество воды. Как сложный эфир НГЦ способен к гид-
ролизу, Гидролиз образует продукты кислотного характера, которые
могут его ускорять. Наряду с этим продукты гидролиза, вступая в
окислительно-восстановительные реакции, образуют, в частности,
воду, которая также может способствовать ускорению разложения.
В связи с этим представлялось целесообразным изучить влияние
воды иа разложение НГЦ.
Результаты этого исследования, проведенного в основном ме-
тодом измерения давления образующихся газов при помощи мано-
метра типа Бурдона1, составляют содержание настоящей статьи.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Получение и очистка нитроглицерина
НГЦ был получен нитрованием динамитного глицерина безвод-
ной смесью химически чистых серной и азотной кислот (1:1) при
температуре 15°. После многократной промывки 3%-ным раствором
соды1 и водой НГЦ не содержал ни кислоты,
Фиг. 1, Прибор для моле-
кулярной перегонки нит-
роглицерина.
I—колбочка» 2—крышка>
3—отросток. 4—отвод.
ни щелочи и имел температуру затвердева-
ния 12,2°.
Для того чтобы избежать возможного
влияния примесей, содержащихся в НГЦ,
он подвергался очистке. Многократная кри-
сталлизация из расплава, как в стабильной,
так и в лабильной форме оказалась мало
эффективной: после троекратной перекри-
сталлизации температура затвердевания
повысилась всего лишь на 0,2°, Поэтому
была предпринята перекристаллизация из
эфира. После восьмикратной перекристал-
лизации НГЦ, освобожденный от эфира
длительной эвакуацией, имел температуру
затвердевания 13,0°. Дальнейшая пере-
кристаллизация не повышала эту вели-
чину.
Можно очищать НГЦ также молекулярной перепонкой. Преиму-
щество этого метода заключается, в частности, в том, что в НГЦ не
вводится никакого растворителя, полностью освободиться от кото-
1 Описание методики, ее преимуществ и слабых сторон приведено в [2].
132
t рого довольно трудно. Перегонка эта проводилась в приборе, изо-
| браженном на фиг. I.
F В колбочку 1 наливалось небольшое количество (30—40 г) НГЦ.
- Колбочка закрывалась пришлифованной крышкой 2 с пальцеобраз-
г ным отростком 3, куда заливался жидкий азот. Отвод 4 присоеди-
I нялся к высоковакуумной установке. Перегонка проводилась при
I остаточном давлении 10-4— 10-s лиг рт. ст. НГЦ нагревался до
! 40—50°. Оседавший на пальцеобразном отростке продукт собирал-
5 ся, причем для исследования применялась средняя фракция. После
; первой перегонки НГЦ становился совершенно бесцветным и имел
температуру затвердевания 12,9°, а после второй (и третьей) опе-
рации 13,Г. Нитроглицерин, перекристаллизованный 8 раз из эфира
| и затем подвергнутый молекулярной перегонке, имел такую же
г температуру затвердевания.
i В. В. Горбуновым была предпринята попытка очистить НГЦ
L путем перекристаллизации его из концентрированной азотной кис-
лоты (90%), не содержавшей окислов азота. После двукратной пе-
рекристаллизации и отмывки от кислоты был получен продукт с
L температурой затвердевания 13,0°.
S Из всех этих способов молекулярная перегонка является наибо-
t лее безопасным, надежным и сравнительно простым методом очист-
w ки нитроглицерина,
t Подготовка опыта
И . НГЦ тщательно освобождался от летучих примесей, которые мо-
и гут влиять на его распад. Для этого система собранных приборов
Г (фиг. 2) с нитроглицерином в реакционном сосуде 1 откачивалась-
£ сначала ротационным, а затем диффузионным насосом до остаточ-
ного давления 10"4—10-S мм рт, ст. Полностью удалить рее летучие
. вещества, растворенные в НГЦ трудно. Попеременное охлаждение
l .жидким азотом и нагревание реакционного сосуда теплой (30—4СГ)
ft: водой при непрерывной эвакуации облегчает их удаление. Вероят-
i У. ,но, в результате конвекции происходит перемешивание жидкости,
которое ускоряет выделение из нее растворенных летучих веществ.
Для перемешивания жидкости можно применить также магнитную
. мешалку. Чтобы обеспечить полноту удаления летучих веществ,
нитроглицерин особенно при больших навесках обычно в течение
суток находился под вакуумом. В этом случае не наблюдалось по-
вышения давления в реакционном сосуде после погружения его в
;термостат и кратковременного прогрева (2—3 мин,); в противном
случае давление возрастало на несколько миллиметров ртутного
столба.
Для введения воды после откачивания системы и освобождения
НГЦ от летучих растворимых примесей закрывались краны а и е
перед реакционным сосудом, Из предварительно заполненной
', водой 1 трубочки перед краном г, небольшое ее количество вводи;
1 Водя перед введением ее в трубочку кипятилась для удаления растворен-
ного в ней воздуха,
лось в пространство между кранами а и г, откуда пары воды при
открывании крана а заполняли всю систему, в том числе и ша-
рик 2. Давление паров воды (измерение его производилось мано-
метром 4), во избежание возможной конденсации, всегда было
меньше упругости пара при комнатной температуре и составляло
10—12 .ил* рт. ст, при 20° (давление водяного насыщенного пара
равно 17,5 мм рт. ст. при той же температуре). Затем кран б за-
крывался и пары воды, находящиеся в шарике 2, переводились в ре-
акционный сосуд 1. Для этого
открывался кран а при охла-
Фиг. 2, Схема установки дли подготовки
опытов^
/--реакционный сосуд с мембранным мано-
мегром, 2—шарик для дозировки водье и
определения объема реакционного сосуда,
3—ловушка, 4—ртутный манометр, 5—приспо-
собление для дозировки воды, б—манометр
МакЛеода, б, в, г, д, е—краны.
ждении реакционного сосуда
жидким азотом, Вода практи-
чески мгновенно конденсирова-
лась в сосуде, что было видно
по возвращению стрелки мано-
метра 1 в нулевое положение.
Зная объем шарика, давле-
ние и температуру паров воды
в нем, можно определить коли-
чество воды, вводимой в реак-
ционный сосуд, Изменяя дав-
ление паров НаО и число
операций, можно вводить в ре-
акционный сосуд любое необ-
ходимое количество воды.
Однако В. В. Горбуновым [3]
было установлено, что при та-
кой методике воды вводилось
в реакционный сосуд больше,
чем предполагалось. Это, по-
видимому, объясняется адсорбцией ее паров на стенке шарика
и конденсацией в виде тонкой пленки, Поэтому в шарике воды
находилось больше рассчитанного по закону идеальных газов,
Чтобы избежать этого, В. В. Горбунов подогревал шарик с парами
воды до 60 —100°; при этой температуре давление насыщенного
пара составляет 149—760 мм рт, ст,, что уменьшает адсорбцию
паров воды на стенках сосуда и, кроме того, позволяет работать
в интервале давлений (20 —50 мм рт. ст.), далеких от давлений
насыщенного пара при данной температуре, без снижения и даже
с увеличением точности измерения рн.о. По этой методике, вводя
определенное количество воды в сосуд с НГЦ и измеряя равно-
весное давление ее паров над нитроэфиром, исследователь опреде-
лил растворимость воды в НГЦ при 40, 60, 80 и 100°. Эти значения
растворимости использовались в настоящей работе для определе-
ния общего количества воды, находящейся в реакционном сосуде.
Когда требовались большие количества воды, она вводилась в
реакционный сосуд пипеткой. При этом во избежание улетучивания
воды откачивание системы производилось при непрерывном охлаж-
дении реакционного сосуда жидким азотом.
,134
После введения воды в реакционный сосуд перетяжка на тру-
бочке, соединяющей его с вакуумной системой, перепаивалась, и
мембранный приборчик был готов к эксперименту. Для проведения
опыта приборчик опускался в термостат так, чтобы весь реакцион-
ный объем находился при температуре опыта, Время прогрева
реакционного сосуда составляло 2—3 мин. Обычно опыт заканчи-
вали по достижении атмосферного давления, но в некоторых слу-
чаях его доводили до конечного давления, равного ~2000 мм рт. ст.
После окончания опыта мембранный приборчик вынимали из
термостата, и измеряли давление ркомн продуктов, неконденсирую-
щихся при комнатной температуре. Зная р1!Омн и давление при тем-
пературе опыта, можно подсчитать условное количество газов, кон-
денсирующихся при комнатной температуре, по уравнениям:
В = 100 —Л;
А = Ркомн^. 100>
рт комн
где А—% неконденсирующихся газов;
В—% газов, конденсирующихся при комнатной температуре;
р —давление в реакционном сосуде при температуре опыта
Т (°1<), мм рт. ст.;
рком„ — давление в реакционном сосуде при комнатной температуре
7'к0Мн(°К), мм рт. ст.
Этот расчет условен, так как при вычислении г В пренебре-
гается упругостью пара конденсирующихся газов, их раствори-
мостью в НГЦ, и предполагается, что реакция прекращается сразу
йосле извлечения реакционного сосуда из термостата.
Термостат представлял собой стеклянный сосуд емкостью 10 л,
изолированный снаружи асбестом и наполненный жидкостью (ди-
бутилфталат, глицерин или минеральное масло), нагревание кото-
рой осуществлялось погруженной в нее нихромовой спиралью.
Однородность температуры в термостате достигалась перемешива-
нием жидкости пропеллерной мешалкой, приводимой в движение
электромотором. Постоянство температуры поддерживалось при
помощи контактного термометра с магнитной головкой типа ТКМ,
соединенного с реле, выключающим или включающим нагреватель
при повышении или понижении температуры выше или ниже задан-
ной. Температура измерялась интервальным термометром. Колеба-
ния ее не превышали ±0,1° при 100”.
Результаты опытов
Вода плохо растворима в нитроглицерине и поэтому содержание
ее в нем можно изменять только в относительно узких пределах,
Это достигается изменением давления паров воды над нитроэфи-
ром. По закону Генри концентрация воды в НГЦ должна быть про-
порциональна этому давлению, Однако в ходе разложения, по-види-
135
мому, под влиянием его продуктов, растворимость воды в нитро*
глицерине может сильно возрастать. Растворенная вода может
связываться также химически. Поэтому играет роль не только вода,
первоначально растворенная в НГЦ, но и полное ее количество,
находящееся в реакционном сосуде.
Существенное влияние на ход процесса оказывает соотношение
между объемами НГЦ и реакционного сосуда, т. е, степень запол-
нения последнего -НГМ_ ) , так как оно определяет при данной
степени распада вещества давление, а следовательно, и концентра*
цию газообразных продуктов разложения в жидкости. Кроме того,
от величины б зависит распределение воды между жидкой и газо-
вой фазами. На растворимость как воды, так и продуктов распада
в НГЦ, а также на скорость реакции влияет температура. Таким
образом, течение распада нитроглицерина в присутствии воды за-
висит от ряда частично взаимно связанных факторов.
О количестве воды, введенной в реакционный сосуд, судили по
равновесному давлению ее паров (рн,о ). Зная растворимость НаО
в НГЦ, можно по этому давлению рассчитать количество воды,
растворенной в ннтроэфире и находящейся в газовой фазе, а сле-
довательно, и общее количество воды в реакционном сосуде.
Количество воды в жидкой фазе рассчитывается по формуле:
= kp^ofn- % вес. Н2ОЖ= А’рщо'ЮО,
где ^ — количество воды в НГЦ в г;
Рно—равновесное давление паров воды в реакционном сосуде
в мм рт. ст.;
& —количество воды в г, растворяющееся в 1 г НГЦ при
давлении ее паров в 1 мм рт. ст.;
т —навеска НГЦ в г.
Процентное содержание воды в НГЦ при данной температуре
определяется только давлением паров воды.
Количество воды в газовой фазе приближенно, но с достаточной
точностью определяется по закону идеальных газов:
Дн,о^ % вес> ы 0
_ Рню^100
mRT
где gr-количество воды в газовой фазе в г;
v— объем свободного пространства в см3;
М — молекулярный вес воды;
универсальная газовая постоянная
\ град. г-моль /
Т — температура опыта в ®К-
Количество воды в газовой фазе в процентах веса НГЦ зависит
не только от равновесного давления паров воды, но также от отно- 136
136
шения m/u, а следовательно, и от степени заполнения реакционного
сосуда Й, увеличиваясь с ее уменьшением.
Во всех последующих таблицах процентное содержание воды,
находящееся в реакционном сосуде, рассчитывалось по этим форч
мулам, причем использовались следующие значения k, найденные
В. В. Горбуновым [3].
<°С k в -------гНг°-------105
г НГЦ-j/j/ рт.ст.
80 1,93
100 Г01
120 0,66
Отношение количеств воды в жидкой фазе и газовой фазе зави-
сит только от mjc.
kmRT _r т kRT
---— ------—- ч-г —m , Г А" —---*
При максимально возможных Й вся вода, введенная в реакционный
сосуд, находится в жидкой фазе.
Влияние воды, находящейся в реакционном сосуде, на разложе-
ние НГЦ зависит от ее относительного количества, изменение кото-
рого существенно изменяет вид кривой р=Цт).
Небольшие количества воды не изменяют общего характера раз-
ложения безводного НГЦ, но увеличивают начальную скорость
газообразования и сокращают время до начала резкого его уско-
рения.
Дальнейшее увеличение количества воды ведет к изменению
вида кривой р=Цт): наблюдается индукционный период, в тече-
ние которого давление остается постоянным, затем следует паде-
ние давления, а потом, в зависимости от количества воды, более
или менее быстрый его рост.
Введение большого количества воды вновь изменяет характер
распада: после длительного и небольшого уменьшения давления
оно долго растет сначала с небольшой скоростью почти без уско-
рения, но затем через некоторое время, большее, чем даже для без-
водного НГЦ, наступает обычное резкое ускорение разложения.
Разложение нитроглицерина в присутствии малых
количеств воды
Влияние малых количеств воды на разложение НГЦ изучалось
при 100° (Й ^аО.ОЗ) и при 120° (Й» 0,006). Равновесное давление
паров ее составляло 8—14 мм рт. ст. при 100° и 10 мм при 120°.
Исходные и некоторые другие данные помещены в табл. 1 *.
Характер изменения давления, в общем, такой же, как и в слу-
чае разложения безводного НГЦ (фиг. 3 и 4). Давление растет сна-
чала медленно, но этот рост идет быстрее, чем для безводного НГЦ,
' Принятые в ней обозначения распространяются я на последующие таблицы1.
13Т
Таблица 1
Влияние малых количеств воды на разложение нмтрогл щеря на
t DC т г i • 10* ДНаО ММ рт. ст. % пес. НвО / Д У т X Д~ Лич н см3! г-мин туда мин.
0,2044 329 0 0 0,00129 2370
100° 0,2410 304 8 0,0204 0,00193 1665
0,2172 271 14 0,0383 0,00166 1420
120° 0,0643 57 0 0 0,034 1390
0,0556 63 10 0,079 0,042 1260
Условные обозначения:
т—навеска НГЦ; 5—степень заполнения; рц„о—равновесное давление па-
ров воды в реакционном сосуде; И вес. н,о—общее количество воды, находя-
i ! ДИ \
щееся в реакционном сосуде, по отношению к НГЦ; —-—) —началь-
\ Д1 'нач
над скорость газообразования, т700— время достижения давллыя 700 .«.«рт.
ст., которое соответствует стадии резкого ускорения газообразования.
и протекает с очень слабым ускорением, большим, однако, чем в
отсутствии воды. Спустя некоторое время, меньшее, чем для без-
водного НГЦ, наступает обычное для него резкое ускорение газо-
образования.
Фиг. 4. Разложение НГЦ в присутствии
малых количеств воды при 120° и
б = 0,006.
Числа у кривых—равновесное давление паров
воды и й 1В* Св скобках).
Фиг. 3. Разложение НГЦ в присут-
ствии малых количеств воды прн WO3
и Д =0.03.
Числа у кривых—равновесное давление паров
волы н ® 10* (в скобках).
Чем больше начальное давление паров воды, тем раньше насту-
пало резкое ускорение газообразования. Так, при 100’ (см. табл. I
и фиг, 3) увеличение рн,о от 0 (6=0,0329) до 8 (6=0,0304) и до
138
14 мм рт. ст. (6=0,0271) сократило время до начала резкого уско-
рения газообразования с 40 до 29 и 24 час. соответственно.
Как и в случае безводного НГЦ, в присутствии небольших коли-
честв воды газовая фаза, вначале бесцветная, становится в ходе
разложения бурой (NO2), причем интенсивность окраски постепен-
но усиливается.
Аналогично идет распад и при более низких температурах. Так,
по данным М. С. Плясунова при 60° и 6 = 0,80 (рн,о =29 мм рт. ст.)
через 360 час. после начала опыта наступает резкое ускорение газо-
образования; сухой НГЦ давал его через 550 час. (6 = 0,85).
Разложение нитроглицерина в присутствии умеренных
количеств воды
Своеобразная зависимость изменения давления во времени на-
блюдалась при умеренных количествах воды в реакционном сосуде.
Умеренные количества воды — относительное понятие; нижний пре-
дел их мало зависит от 6 и температуры и соответствует равновес-
Фнг. 5. Разложение НГЦ в присутствии умеренных
количеств воды прн 100° и 0.03.
Числа у кривых—порядковый номер кривой, равновесное давление
паров воды и 5 - jO4 (в скобках).
ному давлению паров Н2О примерно 30—50 мм рт. ст. Верхний
предел, как будет показано ниже, сильно зависит от этих факторов.
При 100°, 6 ~ 0,03 и различных давлениях паров воды
(50 мм рт. ст. и выше) процесс протекает следующим образом. Если
воды сравнительно немного, то после погружения реакционного со-
суда в термостат давление в нем быстро поднимается за счет испа-
рения воды, а затем некоторое время остается постоянным (фиг. 5,
кривые 2 и 3). Если же воды много, то она испаряется быстрее, чем
растворяется в НГЦ. Поэтому вначале давление выше равновес-
ного (кривые 4 и 5), а затем оно более или менее быстро (через
30—50 мин.) уменьшается до своего равновесного значения. Увели-
139
чение поверхности Н^Ц и, следовательно, уменьшение толщины
его слоя сокращает это время. Так при увеличении поверхности в
3 раза время достижения равновесного давления сократилось до
10 мин., т. е. было значительно меньше, чем в первом случае.
После достижения равновесного давления оно некоторое время
остается постоянным, затем быстро падает до некоторого минималь-
ного значения (рм), после чего начинает более или менее уско-
ренно расти. В конце падения давления газовая фаза приобретает
Фиг. 6. Разложение НГЦ в присутствии воды при 100°
и й=0,0060ч-0,0075.
Числа у кривых—порядковый номер кривой, равновесное давление па-
ров воды nJ Ю’ (в скобках J.
слабую, постепенно усиливающуюся в ходе дальнейшего разложе-
ния бурую окраску (NO2).
Такой же характер распада НГц в присутствии воды наблю-
дался при б ~ 0,006 н температурах 100 и 120° и при б ~ 0,03 и 80'
(фиг. 6, 7, 8).
Как видно из графиков, время до наступления падения давле-
ния— индукционный период — сильно колеблется, и при этом в об-
щем наблюдается тенденция к его росту при увеличении количества
воды. При 100° и 6^0,03 при повышении рн,о с 54 до 570 мм рт. ст.
(фиг. 5) индукционный период увеличивается с 35 до 70 мин. Сле-
дует отметить, что в этом случае из-за относительной медленности
растворения и небольшого по величине индукционного периода мо-
мент достижения равновесного давления и начала его падения ме-
нее отчетлив, чем при малых б и более низких температурах, прн
которых индукционный период больше.
Более сильные колебания индукционного периода наблюдались
при низких температурах и малых б. Так при 80° и б — 0,03 (фиг. 8)
период постоянства давления при изменении давления паров воды
от ~60 до ~240 мм рт. ст. колеблется от 600 до 2200 мин. При одной
и той же температуре более сильное изменение времени до начала
снижения давления при увеличении рн,о отмечалось при малых б-
140
Увеличение рнао со 136 мм рт. ст. при 6=0,0065 (фиг. 6, кривая 3)
до 330 жл (фиг. 6, кривая 6) ведет к. увеличению индукционного
периода со 180 до 1100 мин., тогда как при b ~ 0,03 приблизительно
Фиг. 7. Разложение НГЦ в присгтствни воды при 120’
и 0=0,0061^0,0068.
Числи у кривых—порядковый номер кривой* равновесное давление
ларов воды ив ГС* (в скобках).
Фиг. 8. Разложение НГЦ в присутствии воды прн 80°
J и 6 = 0,0258ч-0,035<
Числа у кривых—равновесное давление паров воды и & LQ*
{а скобках)*
в том же интервале давлений эта величина с 35 мин. (фиг. 5, кри-
вая 2) возрастает только до 50 мин. (фиг. 5 кривая 4) и даже при
/>н,о =569 мм рт. ст. (фиг. 5, кривая 5) она составляет лишь
70 мин.
141
Фяг. 9. Зависимость величины p^in от
начального давления паров воды при
разложении НГЦ в их присутствии.
Числа у кривых—температура опыта и S • (О1
(в скобках).
Повышение температуры, уменьшает не только величину индук-
ционного периода, но и диапазон его колебаний в зависимости от
содержания воды. Так, при 120°, рн,о =229 мм рт. ст. и 6 = 0,0063
(фиг. 7, кривая.?) величина его составляет 15 мин. и сравнительно
мало меняется с увеличением
равновесного давления паров во-
ды: даже при рн. о = 1409 мм
рт. ст. (фиг. 7, кривая 7) время
до начала снижения давления
увеличивается только в 2 раза.
Падение давления происходит
с возрастающей скоростью, кото-
рая затем уменьшается. Время
падения давления составляет:
при 120° и 6=^0,006 приблизитель-
но 7—10 мин., при 1003 и S~0,006
20—50 мин., при 100° и 6^0,03
35—45 мин., при 80° и б~0,03
160—240 мин.
При увеличении начального
давления паров воды при одной и
той же б рщш увеличивается
(фиг. 9). На фиг. 10 даны кривые
ц = /(/?н,о). Из графиков фиг. 9 и 10 видно, что ловы-
Т'н.о
шение температуры ведет к увеличению pmin, а следовательно, и
к уменьшению относительной величины падения давления.
О 200 ЧОО 600 800 р^ммртхп:.
Фиг. I0. Зависимость относительной величины па-
дения давления от начального давления паров
воды при разложении НГц в их присутствии.
Числа у кривых—температура опыта и ! 10* (в скобках).
После достижения минимума начинается более или менее уско-
ренный рост давления. Как видно из графиков фиг. 5—8, увеличе-
ние начального давления паров воды приводит к повышению ско-
рости роста давления и сокращению времени от момента прохож-
дения рпдпДО начала резкого ускорения газообразования.
14'2
Величина навески НГЦ очень слабо влияет на длительность ин-
дукционного периода. Из фиг. 11 видно, что увеличение навески
НГЦ примерно в 24 раза при одной и той же б, в присутствии
~1% вес. воды, практически не изменяет величины индукционного
периода, по на одну треть (на
чала резкого ускорения газооб-
разования после прохождения
давлением своего минимума.
Известную роль может иг-
рать величина поверхности
жидкости и толщина ее слоя,
так как эти факторы опреде-
40 мин.) сокращает время до на-
Фиг. 12. Изменение температуры НГЦ при
его разложении в присутствии воды прн
101Г.
Числа у кривых—давление паров воды и 5 . 10‘
(в скобках).
фиг. II. Влияние величины навес-
ки НГЦ иа разложение его в при-
сутствии воды.
J—/>pjQ-=408 мм рт. ст. (1.04% вес.),
4-0,0305. m=0,I84l г; 2-р,, -382 мл
п jQ
рт, ст. (1,04% вес.), 4-0,0276, т—4,4085 г.
ляют легкость диффузии, с одной стороны, паров воды в НГЦ и,
с другой, газов, образующихся при его разложении.
Несколько более быстрое ускорение реакции после рщщ при
большей навеске НГЦ может быть связано с саморазогревом за
счет тепла, выделяющегося при реакции. Опыты, проведенные сов-
местно с Б. С. Светловым, по определению температуры НГЦ, раз-
лагающегося в условиях, приближающихся к адиабатическим, по-
казали, что падение давления сопровождается небольшим, но
отчетливо заметным повышением температуры НГц (фиг. 12), при-
том тем большим, чем выше содержание воды.
Так, в присутствии 0,59 и 0,86% вес. воды температура НГЦ
превышает температуру термостата (100+0^5°) на 0,19 и 0,27"
соответственно. Затем температура падает до температуры термо-
143
стата. На стадии ускоренного роста давления температура НГЦ
повышается значительно сильнее и становится выше температуры
термостата более чем на 3° при достижении в реакционном сосуде
давления 1420 мм рт. ст.
Для того чтобы определить, как изменяется количество газов,
конденсирующихся при низкой температуре, в ходе разложения
НГЦ в присутствии воды реакционный сосуд время от времени
вынимался из термостата, и измерялось давление в нем при 20 и 0°.
Фиг. 13. Изменение давления и количества конденсирующихся газов (В)
при 20 и О’ в ходе разложения НГЦ при 80° в присутствии воды.
0-185 мм рт' CT/ 5=°i03^3J-
По давлению при температуре опыта 80° (эта температура была
выбрана, чтобы достаточно велик был индукционный период) и при
20 и 0° определялось содержание конденсирующихся газов. Резуль-
таты опыта показаны на фиг. 13. В индукционном периоде давление
как при высокой температуре (803), так и при низкой (20 и 0°) из-
меняется мало; следовательно, количество конденсирующихся газов
остается примерно постоянным. Падение давления сопровождается
уменьшением содержания конденсирующихся газов; при последую-
щем росте давления количество их увеличивается. Следует отме-
тить, что газы, не конденсирующиеся при температуре жидкого
азота, появляются только на стадии резкого ускорения газообра-
зования.
144
Влияние больших количеств воды на разложение НГЦ
Характерная картина разложения НГЦ в присутствии умерен-
ного количества воды, описанная в предыдущем разделе, при даль-
нейшем увеличении содержания НаО вновь изменяется. Так, в опыте
при рн,о =551 мм рт. ст. (6 = 0,0061), вначале наблюдалось не рез-
кое, а медленное и небольшое по величине падение давления:
(фиг. 6, кривая 8 и фиг. 14, кривая 7) давление за 1320 мни. упало
всего на 36 мм рт. ст. После достижения минимума оно начало,
опять-таки медленно, расти со скоростью, примерно равной началь-
Фиг. 14. Разложение НГЦ в присутствии воды при
100s и 6 = 0,006.
Числа у кривых—порядковый помер кривой, равновесное
давление паров воды • Ю4 (в скобках)»
ной скорости газообразования при разложении безводного НГЦ.
Резкое ускорение газообразования наступило значительно позже,
чем оно наолюдалось при разложении не только влажного, но н
безводного НГЦ. Так в том же опыте (фиг. 14, кривая 7) через
180 час. (~11 000 мин.) давление за 3 час. возросло до ат. Без-
водный же НГЦ в этих условиях начинает ускоренно разлагаться
уже через 135 час., а присутствие воды сокращает это время даже
’ до 17 час.
На протяжении всего опыта вплоть до начала резкого ускорения
роста давления, газовая фаза оставалась бесцветной; после чего
появилась бурая окраска, увеличивающаяся со временем по интен-
сивности. Такое же явление наблюдалось и при несколько меньшем
равновесном давлении паров воды (364 мм рт. ст., см. фиг. 14, кри-
вая 6). Различие заключается только во времени до начала резкого
' ускорения газообразования.
Если скорость газообразования в данном случае характеризует
скорость реакции, то приходится заключить, что при больших со-
держаниях воды ее влияние, в смысле ускорения, уменьшается.
Следует добавить, что в отдельных опытах как при 100°
(8~0,006), так и при 80° (6 ~ 0,03) не наблюдалось обычной кар-
10 451 145
I.;
Фиг. 15. Разложение НГЦ в
присутствии 11,04% вес. воды
при 100° и 6=0,034.
тины влияния воды (постоянство давления, быстрое падение и по-
следующий более или менее быстрый рост его) и в той области дав-
ления паров последней, для которой она характерна. Так, например,
при 100° и рн.о =344 мм рт. ст. (фнг. 14, кривая 5) в течение дли-
тельного времени давление медленно росло и лишь спустя ~ 180 час
рост его стал быстро ускоряться; при этом время до начала резкого
ускорения газообразования было значительно больше, чем оно
наблюдалось при разложении не только влажного, но и даже без-
водного НГЦ.
Такая же картина наблюдалась и при 80° (рн,о = 192 мм рт. ст.;
6=0,0258) (фиг. 8); ускорения газообразования не было в течение
690 час., причем за это время давление
возросло всего на 100 мм рт. ст. Это
указывает на то, что какие-то факторы,
находившиеся вне контроля экспери-
ментатора, могли оказывать сущест-
венное влияние на ход превращения
в присутствии воды и, в частности,
на сроки наступления резкого ускоре-
ния распада. По-видимому, роль этих
факторов возрастает с понижением
температуры и уменьшением б, так
как такого влияния не наблюдалось
при более высокой температуре (120°,
8^0,006), а также при большей сте-
пени заполнения (100° и 6^0,03). При
этих последних условиях описанного изменения характера кривой
p=f(r) не наблюдалось даже, при увеличении начального давле-
ния паров воды почти до 760 мм рт. ст. Добавление еще большего
количества воды (11%), при котором она находится в условиях
опыта не только в растворе, но и в капельно-жидком состоянии
(НГЦ мутный), значительно удлиняет индукционный период
(до ~6 час.); затем наступает очень небольшое падение давления,
переходящее в его рост с очень большой скоростью (фиг. 15).
Следует отметить, что перед началом падения давления вода,
по-видимому под влиянием образующейся кислоты полностью рас-
творяется в НГц, что можно заметить по исчезновению мути.
Влияние величины поверхности соприкосновения нитроглицерина
со стеклом на распад этого ВВ в присутствии воды 1
Сильные колебания величины индукционного периода и значи-
тельное его увеличение, наблюдавшиеся в некоторых опытах по раз-
ложению НГЦ в присутствии воды, можно было связать с влиянием
поверхности реакционного сосуда, состояние которой трудно под-
дается контролю. Чтобы выяснить возможность такого влияния,
были проделаны опыты с увеличенной поверхностью соприкоснове-
1 Эти опыты проводились студенткой А. Г. Казакевич.
146
ния НГЦ со стеклом. Это достигалось набивкой реакционного со-
суда стеклянными капиллярами или стеклянной ватой. Капилляры
(внешний диаметр 0,4—0,5 мм, длина 3— 4 мм) были полностью
покрыты НГЦ, при этом поверхность соприкосновения нитроэфира
со стеклом увеличивалась в 8—10 раз. Большее увеличение поверх-
ности достигалось применением стеклянной ваты, по которой рас-
пределялся нитроглицерин.
Опыты проводились при 100°
и 6^0,03, т. е. в таких усло-
виях, в которых не наблю-
далось аномального значи-
тельного увеличения вре-
мени до начала- резкого
ускорения газообразования,
и колебания величины ин-
дукционного периода были
незначительны.
Как видно на графике
(фиг. 16), увеличение по-
верхности н 8—10 раз не
изменяло общего вида кри-
вой p=f(T), но несколько
увеличивало время до на-
чала падения давления. Так
опыте, где равновесное
давление паров воды со-
ставляло 398 мм рт-' ст.
(кривая /), время достиже-
ния минимального давления
увеличилось на 20 мии. по
сравнению с обычным опы-
том (/4о = 408 лии рт- ст.,
кривая 2). Такое же увели-
чение этого времени наблюдалось и при меньшем ptIl0
вые 3 и 4). На последующее развитие процесса распада
ние поверхности значительного влияния не оказывало.
Сильное увеличение индукционного периода наблюдалось при
набивке реакционного сосуда стеклянной ватой, увеличивающей по-
верхность приблизительно в 30 раз. В этом случае при
/40 = 325 мм рт. ст. длительность индукционного периода с 76 мин.
возросла до 160 мин. (фиг. 17, кривая 2).
Интересно отметить, что такое же замедляющее действие ока-
зывает стеклянная вата, не находящаяся в непосредственном со-
прикосновении с НГц1. В этом случае при давлении паров воды,
равном 330 лци рт. ст, (кривая 3) величина индукционного периода
была приблизительно такой же, как и в случае распада НГЦ, рас-
't
в
> Фиг, 16. Влияние набивки реакционного со-
суда капиллярами на разложение НГЦ в
присутствии воды при 100°.
.«.к
мм
мм
мм
/—с набивкой,
2 -без набивки
5—с набивкой»
rf—без набивки,
ё «0.0330,
S-0.0305,
в-0,0292,
С-0.0311,
Vм
*40-°
'Чо-283
^н.о"271
рт. CT.i
рт. ст.;
рт, ст.;
рт. ст.
(ср. кри-
Увеличе-
1 Стеклянная вата находилась внутри мембранного манометра, НГЦ — на дне
реакционного сосуда.
10*
147
пределенного по стеклянной вате (train составляло 156 и 159 мин.
соответственно).
Еще более сильное влияние поверхности проявлялось при боль-
шом начальном давлении паров воды. Так в опыте, где
рн3о —460 мм рт. ст. (кривая 4), длительность индукционного пе-
риода составляла 900 мин., т. е. почти в 15 раз превышала обычную.
Такой характер разложения влажного НГЦ в реакционных сосу-
дах с набивкой показывает, что величина поверхности стекла, воз-
Фиг. 17. Влияние набивки реакционного сосуда стеклянной
ватой на разложение НГЦ в присутствии воды при lOCP.
1 —без набннкн, 5^0,0367, ^=293 мм рт, ст»; 2— с набивкой* 5=0,0350.
pu -=325 лсж рт. Ст/, J-c набивкой над НГЦ, 5-0,03, л -“330 мм
п2О г Н2О
рт. ст-» *-с набивкой. 5«=0,0291, р„ _-460 о рт» Ст.
HiU
можно из-за его слабощелочного характера, может оказывать
существенное влияние на распад НГЦ, значительно увеличивая
время до начала резкого ускорения газообразования, особенно при
больших количествах воды. Вероятно, этим отчасти можно объяс-
нить те колебания в величине индукционного периода, которые на-
блюдались при распаде НГЦ в присутствии воды.
Влияние степени заполнения сосуда (6) на разложение
нитроглицерина в присутствии воды
Увеличение степени заполнения реакционного сосуда означает
повышение давления продуктов разложения НГЦ при одной и той
же степени его распада и, следовательно, увеличение содержания
растворимых газообразных продуктов в НГЦ: Это приводит к бо-
148
лее раннему достижению критического давления газообразных про->
дуктов, после которого давление начинает резко расти.
В том случае, когда присутствует вода, увеличение степени за-
полнения при данном начальном давлении ее паров, кроме того,
означает уменьшение общего количества воды по отношению к НГЦ
при постоянном начальном содержании ее в жидкой фазе.
Влияние степени заполнения реакционного сосуда изучалось при
100° и начальном давлении паров воды —'100 мм рт. ст., причем б
Фиг. 18. Влияние степени заполнения реакционного сосуда
на разложение НГЦ при 100°
Г—8—0,0065, А, „-=136 мм рт, ст.; 2—5 =0.0126, р„ Л — 90 мм рт. ст.;
5—5=0,0284. „-*117 мм рт. ст.; 4—5=0,0547, р„ мм Рт» ст.;
Н
5—6-0,0927, „ — 101 мм рт. ст.; в-5=0.118; Р,_, „—117 мм рт. ст.;
ti^kJ C12U
Т—3=0,248. Рцд'ТМ мм рт. ст.
изменялась в интервале от 0,0065 до 0,25 (фиг. 18). Для возможно-
сти сравнения результатов опытов при различных б на графике по
оси ординат отложен объем (с учетом воды) газов, выделявшихся
при разложении, отнесенный к 1 г НГЦ, приведенный к нормаль-
ным условиям. Исходные и некоторые другие данные этих опытов
сведены в табл. 2.
Для всех этих б вид кривой р — J (т) один и тот же (см. фиг. 18):
период постоянства давления, падение его и последующий более или
менее быстрый рост. Увеличение б ведет к уменьшению относитель-
ного количества воды и соответственно к сокращению величины ин-
дукционного периода. По данным В. В. Горбунова при максимально
возможной б индукционный период составляет ~30 мин. (Tmtn при
6=0,857 — 43 мин.). После прохождения давлением минимума уско-
ренное развитие процесса идет тем быстрее, чем больше 6.
149
Таблица 2
Влияние степени заполнения реакционного сосуда на в присутствии воды прн 100° разложение НГЦ
тнгц г В 104 Рщо мм рт. ст. мг % вес. Н2О ^min мин. ^700 мин.
0,0678 65 136 0,774 1,14 180 945
0,1756 126 90 0,754 0,43 90 895
0,2883 284 117 0,894 0,31 67 605
0,5348 547 96 0,963 0,18 54 580
0,9189 927 101 1,38 0,15 46 415
1,2491 1180 117 1,99 0,16 60 295
2,1478 2480 104 2,58 0,12 52 305
Сказанное относится к сравнительно большим степеням запол-
нения (0,006—0,2). При малых б (порядка 0,001) значительная
часть воды (более 95%) находится в газовой фазе. В этих условиях
ие наблюдалось характерной картины разложения НГЦ в присут-
ствии воды (период постоянства давления, падение и ускоренный
его рост) в интервале начальных давлений ее паров от 10 до
300 мм рт. ст.
Данные этих опытов сведены в табл. 3.
Т а б л к'ц а 3
Разложение НГЦ в присутствии воды при 100° и малых В
^НГЦ г б 10* мм рт. ст. % вес. Н2О / ДУ \ \ 4т /на. f—) \ /щах ^тах мин.
0,0112 12 0 0 0,0039 0,027 33 500
0,0198 19 и 0,28 0,0026 0,100 22 000
0,0123 17 40 1,12 0,0021 0,010 35000
0,0141 20 318 8,02 0,0025 0,0035 1000
Условные обозначении’. 1 ДУ \
к 4 г / Д7 \ 4г f “НиЧаЛЬпал CKUpULTb i cJJUUUJ)aJUWdlUlH IS ft. jC'jUUM, нач 1 —скорость газообразования на максимуме в « см^г-мин: шах тшах— время достижения максимальной скорости в мип.
При разложении безводного НГЦ при 100° и 6=0,0012 давление
сначала ускоренно растет, а затем, после того, как скорость до-
стигла максимального значения, изменяется по кривой насыщаю-
щегося типа (фиг. 19, кривая /). Максимальная скорость газовы-
150
деления почти в 7 раз превышает начальную. Такая же зависимость
изменения давления от времени наблюдалась при разложении НГЦ
в присутствии небольших количеств воды (рц,о =10—40 лии рт. ст.,
03—1,1% вес.). Однако, как видно из фиг. 20, величина максималь-
ной скорости газообразования и время ее достижения отличны.
Вода в количестве 0,28% вес. (рн,о=11 мм рт. ст.) увеличивает
максимальную скорость в сравнении с безводным нитроэфиром бо-
лее чем в 3,5 раза, а в количестве 1,12% вес. (рн о=^0 мм рт. ст.)
уменьшает ее примерно в 3 раза (ср. на фиг. 20 кривые /, 2, 3).
в присутствии воды при 100° и малых б.
Числа у кривых—порядковый номер кривой» равновесное давление
парой воды и 5 * 10* (в скобках).
В первом случае максимальная скорость газообразования почти
в 40 раз превышает начальную, в то время, как во втором—всего
в 5 раз.
Увеличение количества воды до 8% вес. (рн,о=318 мм рт. ст.)
изменяет вид кривой р=/(т): вначале давление некоторое время
незначительно снижается, после чего возрастает сначала с неболь-
шим ускорением, а затем по насыщающейся кривой (см. фиг. 19,
кривая 4). На фиг. 20 (кривая 4) видно, что максимальная скорость
газообразования всего на 40% превышает начальную и почти в
8 раз меньше максимальной скорости распада безводного НГЦ.
Характерно, что в этом случае на протяжении всего опыта не на-
блюдалось бурой окраски газовой фазы, В предыдущих же опытах
газовая фаза, вначале бесцветная, постепенно бурела (NOS), а
затем, после достижения максимальной скорости газообразования,
окраска исчезала.
Из этой серии опытов видно, что существует наиболее эффектив-
ное количество воды, вызывающее более раннее и более сильное
ускорение газообразования; при большем ее содержании следует
Фиг. 20. Изменение скорости газообразования при разложе-
нии НГЦ в присутствии воды при 100’ и малых д.
Числа у кривых—порядковый номер кривой» равновесное* давление паров
воды в fi * 10* (в скобках)»
уже говорить о замедляющем влиянии, а достаточно большое коли-
чество Н2О замедляет распад настолько, что изменяет даже харак-
тер зависимости p—f (т).
Влияние предварительного нагревания нитроглицерина
на дальнейшее его разложение в присутствии воды
Можно было предполагать, что накопление кислоты в опреде-
ленной концентрации приводит к быстрому, вероятно гидролитиче-
скому, развитию реакции, которое проявляется в виде падения дав-
ления. При разложении безводного НГЦ на начальных стадиях
образуются кислые продукты, но гидролиза не наступает из-за не-
достатка воды. Если же в такой, предварительно разлагавшийся,
НГЦ добавить воды, то должна была бы сразу начаться быстрая
гидролитическая реакция, так как были бы выполнены оба усло-
вия, необходимые для ее быстрого протекания — наличие кислоты,
образованной термическим распадом, и воды. Если бы эта идея
была правильной, то, добавив к частично разложившемуся безвод-
ному НГЦ воды, можно было ожидать немедленного падения дав-
ления и последующего ускоренного газообразования.
Безводный НГц сначала нагревался при 80° 980 мин., т. е. в
течение времени, соответствующего длительности индукционного
периода, наблюдавшегося в присутствии воды. Затем в реакцион-
152
ный сосуд вводилась вода (ph.o^ISO мм рт. ст.), и НГЦ вновь
нагревался при той же температуре.
Данные этого и аналогичных опытов приведены в табл. 4.
Таблица 4
Влияние предварительного нагревания на разложение нитроглицерина
В присутствии воды при 80 и 100°
f°C /Пнгц г trl0< т МИИ. мм рт. ст, И вес. НаО Tmin мин. т700 МИИ,
80 0,3148 354 —**. 188 0,63 900 1735
0.2993 338 980 181 0,62 980 1660
0,3320 389 960* -200 -0,65 200 650
100 0,2850 336 157 0,38 66 390
0,3970 327 180 135 0,33 45 270
т — время предварительного нагревания НГЦ.
•Нагревание проводилось в присутствии небольшого ( — 4 мм рт. ст.) ко-
личества коды.
Чтобы наступило падение давления и дальнейшее развитие реак-
ции, потребовалось 850 мин. (фиг. 21, кривая 2), а для предвари-
тельно не разлагавшегося нитроэфира — 750 мин. (кривая 7). Та-
Фиг. 21. Влияние предвари-
тельного прогрева НГЦ на
его разложение в присутст-
вии воды при 80°.
7—без предварительного п согрева,
Pun-188 мм рт. ст., 5-0,0364;
м*.
2— прогрев прн 80' 9&0 мин.?
Л = 181 мм рт. ст.. В-*0,0338;
гцм >
5— Прогрев при 8GD 960 инн. в при-
сутствии иоды (рц мм
рт* ст*)ф q^3Q0 ММ рТ" СТ*"
“8-0,0389.
ким образом, предварительное разложение безводного НГЦ не
оказало существенного влияния на темпы' развития реакции в при-
сутствии воды.
Это. указывает на то, что либо при безводном распаде образо-
вание продуктов, стимулирующих гидролиз, идет гораздо медлен-
нее, чем в присутствии воды, либо, что при нем образуются иные
продукты, либо играют роль оба фактора. Чтобы выяснить это,
153
были проведены опыты более длительного предварительного нагре-
вания безводного НГЦ, а также опыты с порционным введением
воды. Безводный НГЦ нагревался при 100° в течение 180 мин. При
продолжении опыта с добавленной водой тт)в сократилось на
20 мин., т. е. приблизительно на 30%. Таким образом, кислые про-
дукты, стимулирующие гидролиз, образуются и при разложении
безводного НГЦ, но значительно медленнее, чем в присутствии
воды.
В другой серии опытов предварительное нагревание велось в
присутствии весьма малых (равновесное давление около
4 мм рт. ст.) количеств воды. При последующем введении больших
количеств последней сразу началось падение давления, через
200 мин. оно заканчивалось, и развивалось быстрое ускорение газо-
образования (фиг. 21, кривая 3). Это указывает на то, что доста-
точно очень малых количеств каких-то кислых промежуточных про-
дуктов, образуемых при разложении НГЦ в присутствии воды,
чтобы инициировать реакцию связывания воды и последующее рез-
кое ускорение разложения нитроэфира. Таким образом предвари-
тельное нагревание в присутствии воды особенно при порционном
ее введении гораздо сильнее стимулирует разложение НГЦ чем
«безводное» нагревание.
Изменение содержания кислоты в реакционном сосуде
при разложении нитроглицерина в присутствии воды 1
Если в присутствии воды происходит гидролитическое разложе-
ние НГЦ, то должны образовываться кислоты. Эти кислоты могут
оказывать каталитическое действие на процесс. Поэтому целесооб-
разно было определить изменение кислотности в ходе разложения
и кислоту, которой оно обусловлено.
Наиболее вероятно образование при разложении НГЦ в присут-
ствии воды азотной и азотистой кислот. Азотная кислота может об-
разоваться, во-первых, при гидролизе НГЦ по обычному пути, и,
во-вторых, в результате взаимодействия с водой двуокиси азота,
образующейся при термическом распаде НГЦ. В последнем случае
образуется также и азотистая кислота. Другим источником азоти-
стой кислоты может быть гидролитическая реакция иитроэфира,
аналогичная реакции Есо, наблюдавшейся Бейкером [6], [7] при ще-
лочном гидролизе некоторых алкилмононитратов, по которой обра-
зуется карбонильное соединение и ион NO^. Азотистая кислота мо-
жет образовываться также при восстановлении азотной кислоты.
Для того чтобы установить, какая реакция преобладает на на-
чальной (до рты) стадии разложения, были проведены опыты по
обнаружению и количественному определению ионов NO^~ и NO^.
Для определения в продуктах разложения иона NO” НГЦ раз-
лагался в присутствии воды при 100° (6 = 0,0898, рн,о = 105лии рт. ст.,
ртш = 34 мм рт. ст. через 50 мин.) в реакционном сосуде, прикреп-
1 Эта серия опытов была проведена студенткой Н. В. Гульцеаой.
154
ленном к мембранному манометру через трубочку с тонкой пере-
тяжкой, При достижении рпип мембранный приборчик вынимался
из термостата, реакционный сосуд охлаждался жидким азотом,
чтобы сохранить в нем все газы, конденсирующиеся при темпера-
туре кипения жидкого азота ’, после чего перетяжка перепаивалась.
Ампула с НГЦ и с конденсированными продуктами разложения
вскрывалась под раствором 0,1н. щелочи, причем весь объем ам-
пулы заполнялся этим раствором, что указывало на растворение в
нем всех газообразных продуктов. Раствор быстро отделялся от
НГЦ. Он обесцвечивал перманганат калия в кислой среде, давал
слабо окрашенное бурое кольцо с сульфатом железа в присутст-
вии разбавленной серной кислоты и выделял иод из раствора йоди-
стого калия в кислой среде. Сам нитроэфир таких реакций не
давал. Это служило доказательством присутствия ионов NO^ в по-
лученном щелочном растворе; судя по малой интенсивности ука-
занных реакций,можно заключить, что количество NO^-ионов было
невелико.
О наличии азотной кислоты судили по образованию осадка, ко-
торый появляется при приливании к подкисленному серной кисло-
той исследуемому раствору реактива нитрона (10%-ный раствор
дигидро-дифенил-фенилимино-триазола в 5%-ной уксусной кислоте)
[11], причем азотистая кислота перед определением разлагалась
твердым сульфатом гидразина.
Соотношение между азотной и азотистой кислотой определялось
как в газообразных продуктах разложения, так и в продуктах раз-
дюжения, растворяющихся в водном растворе щелочи. В обоих слу-
чаях проба для анализа отбиралась в момент, соответствующий
ршш. являющийся резкой границей между двумя макроскопически-
ми стадиями процесса разложения влажного НГЦ. В первом слу-
чае газообразные продукты разложения нитроглицерина собира-
лись в U-образной трубочке, охлаждаемой жидким азотом1 2, кото-
рая затем вскрывалась под определенным (15 или 25 мл) количе-
ством 0,1 н. раствора NaOH. Полученный таким образом раствор
анализировали на азотную и азотистую кислоты. Во втором случае
газообразные продукты собирали в реакционном сосуде при охлаж-
дении его жидким азотом. Сосуд этот затем отпаивали от мембран-
ного манометра и вскрывали под определенным количеством 0,1н.
раствора NaOH; затем производили отделение НГЦ. Дальнейшие
операции были одни и те же в обоих случаях.
Анализ проводился с помощью реактива нитрона по методу, опи-
санному Бушем [8]. По этому методу в отдельной пробе нитрит
окислялся перекисью водорода до нитрата, который совместно с уже
имевшимся в растворе нитратом осаждался в виде нитратиитрона;
в другой отдельной пробе нитрит разрушался сульфатом гидра-
зина, а оставшийся нитрат осаждался нитроном.
1 Оказалось, что все газы, образующиеся на этой стадии распада, полностью
конденсируются при температуре кипения жидкого азота,
2 Методика отбора газообразных продуктов описана ниже.
155
Следующий пример является типичным для определения соот-
ношения азотной и азотистой кислот, образующихся к моменту про-
хождения давления через минимум. Нитроглицерин в количестве
3,486 г разлагался в реакционном сосуде с мембранным маномет-
ром объемом 23,0 еж3 в присутствии паров воды (рн.о =367 мм рт. ст).
Через 55 мин. при pmin = 78 мм рт. ст. все газообразные продукты
переводились в нижнюю часть реакционного сосуда, охлаждаемую
жидким азотом, после чего сосуд отпаивался от мембранного мано-
метра и вскрывался под 25 мл 0,1 н. раствора NaOH. Из получен-
ного таким образом раствора отбиралось 5 мл так, чтобы не захва-
тить неразложившийся НГЦ. После подкисления серной кислотой
к нему приливалось 2 см3 3%-ного раствора перекиси водорода.
Раствор нагревали до 70°, пипеткой вводили в него 2 см3 3%-ной
H2SO4 и нагревали до 90°. В подготовленный таким образом раствор
добавляли 1,2 ел:3 реактива нитрона. Раствор с выпавшим осадком
охлаждали ледяной водой, осадок отфильтровывали и промывали
на фильтре небольшим количеством ледяной воды. Вес полученного
осадка 0,0605 г.
% (HNO3+ HNO )=5.°-°.0605*0,1679~ 100 =
* 31 3,4862
= 1,45 % вес. по отношению к НГЦ.
Вторые 5 мл раствора, нейтрализованные 2 н. серной кислотой, упа-
ривали до 1 мл и прикапывали к 0,05 г сухого сульфата гидразина,
после чего присутствующий в пробе нитрат осаждали так, как было
описано выше. Вес полученного осадка 0,058 г.
_. ziiNTZ-, s 5.0-0,058-0,1679* 100 , лл/ иг, г
% (HNO3)==---------- - ’ - —.=1,39% вес. по отношению к НГЦ.
3,4862
Исходные данные и результаты этого опыта и трех других сведены
в табл. 5.
Таблица 5
Определение азотной и азотистой кислот при разложении
нитроглицерина при 100° в момент, соответствующий у?т,п
тнгц г 5-104 Рнао мм рт. ст. ^min МИН. /*min мм рт. ст. И вес. (HNO3+HNO2) по отношению к НГЦ И вес. HNO3 по отношению к НГЦ
3,486 '865 367 55 78 1,45 1,39
2,129 784 285 58 70 2,16 2,12
2,149 815 480 47 ИЗ 1,79 1.76
2,084 809 430 55 95 1,87 1,82
156
В первых двух опытах азотная и азотистая кислоты определя-
лись по вышеописанной методике. В последних двух газообразные
продукты распада сначала собирались в U-образной трубочке, а
затем анализировались.
Несмотря на сравнительно малую точность определения (так
как в опытах использовалось небольшое количество НГЦ, не учиты-
валась растворимость продуктов распада в нитроэфире при опреде-
лении первым методом и не было гарантии полного извлечения
газообразных продуктов при аналитической работе по второму ме-
тоду), отчетливо видно, что количество азотистой кислоты состав-
ляет незначительную часть общей кислотности (не более 4%).
Таким образом, основным продуктом кислотного характера, об-
разующимся к моменту, соответствующему pmin, является азотная
кислота. Ее образование естественно можно объяснить гидролизом
НГЦ нормального типа, в результате которого образуются ионы NO3_
и гидроксильная группа у атома углерода. Это находится в согла-
сии с данными Фармера [9], обнаружившего при гидролизе НГЦ
разбавленной азотной кислотой динитрат глицерина. Обнаружен-
ные небольшие количества азотистой кислоты, как указывалось
выше, могут образоваться, во-первых, при разложении и восстанов-
лении выделяющейся азотиой кислоты, и, во-вторых, при растворе-
нии в воде двуокиси азота, отщепляющейся от НГЦ в результате
термического распада *, и, наконец, вследствие прохождения гидро-
литической реакции типа Есо, дающей ион NO^h карбонильное сое-
динение. Однако все эти реакции если и происходят, то в очень не-
большой степени.
Преобладающее присутствие в реакционном сосуде азотной кис-
лоты на стадии распада НГЦ до рты позволило впоследствии опре-
делять количество кислоты титрованием щелочью раствора, полу-
ченного обработкой газообразных продуктов распада водой,
принимая всю образующуюся кислоту за азотную. Для этого раз-
ложение НГЦ в присутствии воды проводилось в реакционном со-
суде, присоединенном к мембранному манометру через трубочку с
перетяжкой. На определенной стадии разложения мембранный
приборчик вынимался из термостата; после того, как газообразные
продукты охлаждением жидким азотом были переведены в нижнюю
часть реакционного сосуда, перетяжка перепаивалась. Ампулка
с НГЦ и продуктами разложения вскрывалась под водой3- Полу-
ченный раствор титровался 0,01 н- раствором NaOH (индикатор—
фенолфталеин). При расчете принималось, что рея кислотность
обусловлена азотной кислотой.
Определение кислотности проводилось на двух стадиях разло-
жения НГЦ в присутствии различных количеств воды при 100° и
1 По данным Н. В. Гульцевой при разложении безводного НГЦ при 100°
к 180 мин. в реакционном сосуде было найдено титрованием 0.13% (в пересчете
иа HNO3) кислоты, или ~0,04% к моменту времени, соответствующему рт<я. Это
находится в удовлетворительном согласии с данными по анализу азотистой кис-
лоты, образовавшейся при разложении НГц в присутствии воды (см. табл. 5).
1 Для удаления углекислоты вода предварительно кипятилась.
157
6=0,03—0,04: в начале падения давления (в конце индукционного
периода) и при ршщ. Исходные данные и результаты этих опытов
помещены в таблицах 6, 7 и 8.
Таблица 6
Кислотность в конце индукционного периода разложения нитроглицерина
в присутствии различных количеств воды при 100°
тнгц г 8-Ю4 Т’н.о мм рт. ст. % вес. Н?О р мм рт.ст. т мин. % вес. HNOa по отношению к НГЦ %HNO3 „
• 10“ ЛьсГ
0,3259 315 71 0,18 67 37 0,11 42
0,2716 319 88 0,22 78 55 0,17 35
0,3314 318 195 0,48 182 60 0,28 24
0,3666 324 222 0,54 214 43 0,22 23
0,2960 311 238 0,60 223 130 0,60 19,5
0,3041 313 267 0,64 254 90 0,48 20
0,3884 382 294 0,65 286 41,5 0,21 17,2
0,3276 314 363 0,91 360 48 0,20 11,5
0.3164 282 408 1,09 389,5 62 0,22 8,7
0,3091 333 406 0.98 387 61 0,22 8.9
т — время в мин. достижения давления р, при котором определялась кис-
лотность.
Таблица i
Кислотность в момент, соответствующий pmgn, при разложении
нитроглицерина в присутствии различных количеств воды прн 100“
^нгц г в. ки мм рт. ст. ^6 вес. НаО Anta мм рт. ст. Tm(ti МИИ. % вес. HNOa по отношению к НГЦ
0,3259 375 52 0,11 29 64 0,58
0,3684 404 146 0,31 49 60 1,41
0,3215 333 172 0,41 57 58 1,62
0,2837 330 222,5 0,54 62 72 2.21
0,3153 346 262,5 0,62 83 121 2,32
0,3110 370 316 0,72 82,5 78 2,74
0,4055 396 375 0,82 90 69 2,93
0,3346 364 375 0,86 89 87 2,94
0,2898 279 396 1,07 101,5 106 4,37
0,3070 333 510 1,23 136 103 4,74
0,3888 496 520 1,02 116 84 3,70
158
Таблица 8
Зявисимость от & кислотности в момент времени, соответствующий pmj„.
ври разложении нитроглицерина в присутствии приблизительно
постоянного количества воды при 100е
Лнгц г В-10« Т’н.о мм рт. ст. даНао MZ вес. нго Anin мм рт, ст. TmEn мни. % вес. HNOj по отношению к НГЦ
0,0780 82,2 266 1,42 1,82 106,5 169 5.98
0,1735 201 250 1.44 0,84 76 100 2,02
0,2679 202 226 2,04 0,76 76 72 2,57
0,8153 346 263 1,95 0,62 83 121 2,32
1,0034 870 298 4,52 0.45 82 55 1,20
2,0794 1714 252 6,46 0.31 59 53 1,06
таблиц 6, 7 и
8
сопоставления
что падение
давления
Из
сопровождается резким увеличением
начале падения давления мало зависит от начального количества
воды в реакционном сосуде и составляет ~0,2%. Только в тех опы-
тах, где индукционный период более обычного, эта величина не-
сколько увеличивается. Количество кислоты, определенное в мо-
мент, соответствующий зависит от начального количества воды
в реакционном сосуде: чем оно больше, тем больше и содержание
кислоты.
Как указывалось выше, увеличение 6 при постоянном начальном
У'у; давлении паров воды означает уменьшение количества Н2О, нахо-
видно,
кислотности. Кислотность В
'С*
У дященся в реакционном сосуде. Определение кислотности в момент
жЧЙим при разных S и рн,о=230—290 мм рт. ст. показало, что увели-
О 'Чение д уменьшает также и количество образующейся кислоты (см.
Табл. 8). На фиг. 22 представлены результаты опытов по определе-
у нию кислотности в момент, соответствующий ртш, при разложении
" , НГЦ в присутствии различных количеств воды и при различных 6.
^/'Зависимость изменения содержания кислоты от количества воды
изображается прямой линией с тангенсом угла наклона, приблизи-
У":.',«л®ио равным 3,6, что близко к отношению молекулярных весов
.азотной кислоты и воды—ЫЛ21-=з,5. Эквимолекулярное соотношение
У... Л*Н,0
. .’'i Воды, присутствующей вначале, и азотной кислоты, образующейся
"^..К моменту, соответствующему а также отсутствие каких-либо
т Других кислот в значительных количествах, дает довольно веское
• 'доказательство того, что при разложении НГЦ в присутствии воды
происходит его гидролиз обычного типа с образованием азотной
кислоты.
Если допустить, что в результате взаимодействия с водой одна
Нитратная группа в НГЦ превращается в спиртовую с образова-
159
нием азотной кислоты и динитрата глицерина 1, то оказывается, что
в присутствии 1% воды (рн,о=400 мм рт. ст.) при 6 0,03 обра-
зуется 3,5% вес. азотной кислоты. Это означает, что к моменту,
соответствующему pmln, разложилось приблизительно ~13% вес.
первоначального количества НГЦ. Разложение такого количества
нитроэфира происходит в очень короткий промежуток времени: вре-
мя падения давления составляет при 100° и 6 — 0,03 35—45 мин.
Фиг. 22. Кислотность в момент,
соответствующий pmto при раз-
ложении НГЦ в присутствии
различных начальных количеств
воды при 10СР.
НГЦ в присутствии
азотной кислоты и ди-
нитрата глицерина
при 100°.
7-НГЦ+8.6Ж вес. ДНГЦ+
+4,95! вес, HNOj. 3—0.0448;
2—НГЦ+1.04% вес. НаО,
J-0.0305.
Это падение, происходящее сначала с небольшой, но возрастающей
скоростью, после прохождения ее через максимум, замедляется;
при этом, чем больше начальное давление паров воды, тем больше
максимальная скорость падения давления.
Быстрое увеличение скорости падения давления свидетельствует
об автокаталитическом процессе: выделяющаяся азотная кислота
ускоряет реакцию взаимодействия НГЦ с водой. По мере израсхо-
дования воды падение давления замедляется. Возможно, автока-
талитическая реакция происходит и в течение индукционного пе-
риода. Однако для доказательства этого необходимо изучить кине-
тику изменения кислотности на этом участке разложения.
Азотная кислота, образовавшаяся в результате автокаталитиче-
ской реакции гидролиза НГЦ водой, по-видимому, вызывает даль-
нейшее развитие процесса превращения, проявляющееся в ускоря-
ющемся газообразовании.
1 Одновременно с этой реакцией может происходить гидролиз образующегося
ди нитрат а глицерина; однако вследствие меньшей его концентрации по сравнению
с концентрацией НГЦ первая реакция, вероятно является преобладающей.
160
Если этот вывод справедлив, то после добавления к НГЦ динит-
рата глицерина и азотной кислоты, должен сразу начаться ускорен-
ный рост давления, аналогичный тому, который наблюдается после
прохождения давления через минимум при разложении НГЦ в при-
сутствии воды. Действительно, НГЦ (6=0,0448) в присутствии
(сверх 100) 4,9% вес. азотной кислоты и 8,6% вес. симметричного
динитрата глицерина сразу же начинает ускоренно разлагаться
(фиг. 23, кривая 1).
На этой же фигуре для сравнения изображена кривая 2 измене-
ния давления во времени при разложении НГЦ в присутствии
1 % вес. воды. В этом случае при гидролизе НГЦ должно было об-
разоваться 3,5% вес. азотной кислоты и 10% вес. динитрата гли-
церина. Для того чтобы нагляднее сравнить оба опыта, за начало
отсчета времени был выбран момент, соответствующий ршш-
, Роль летучих и нелетучих продуктов
При разложении НГЦ в присутствии умеренных количеств воды
после индукционного периода и следующего за ним падения давле-
ния, наступает более или менее ускоренное газообразование. Паде-
ние давления сопровождается значительным увеличением содержа-
ния летучей кислоты в реакционном сосуде, образующейся в резуль-
тате взаимодействия НГЦ с водой. Оказалось, что это в основном
’ азотная кислота. Возможно, она и является причиной последующего
ускоренного разложения, сопровождающегося сильным газообразо-
ванием. Вероятно одновременно с выделением кислоты образуются
. и нелетучие продукты реакции, содержащие группу ОН (в частно-
сти, динитрат глицерина), которые могут принимать участие в даль-
нейшем развитии процесса. Для того, чтобы выяснить, какую роль
играют газообразные и конденсированные продукты при разложе-
нии влажного НГЦ после прохождения давления через минимум,
были поставлены следующие опыты
Во-первых, из НГЦ, разлагающегося в присутствии воды, в мо-
мент, соответствующий pmjn,‘ откачивали летучие продукты, после
чего его вновь нагревали.
Во-вторых, НГЦ, из которого после предварительного разложе-
ния были удалены летучие продукты распада, образовавшиеся к
моменту, соответствующему ртш, разлагали в присутствии вновь
введенных паров воды.
В-третьих, безводный НГЦ разлагали в присутствии летучих
продуктов распада.
Для проведения этих опытов НГЦ сначала разлагали в присут-
ствии воды в реакционном сосуде, соединенном через трубочку
(с перетяжкой для облегчения отпаивания) с вакуумным краном
особой конструкции* 2 (фиг. 24). Кран смазывался термостойкой
' Эти опыты были выполнены студенткой Н. В. Гульпевой.
2 Такой же кран применялся при введении воды и разлагавшийся НГЦ и при
откачивании из него летучих продуктов разложения (см. стр. 155),
11 451 161
малореакционноспособной смазкой, приготовленной из перфториро-
ванных углеводородов; для лучшей герметизации сверху пробка
крана заливалась ртутью (ртутное уплотнение). Благодаря этому
прибор мог погружаться в термостатирующую жидкость таким об-
разом, что весь реакционный объем находился при температуре
опыта. Во время опыта кран был закрыт. В тот момент, когда было
необходимо откачать газы, прибор вынимали из термостата и при-
соединяли к вакуумной установке. Для более быстрого и полного
Фиг, 24. Реакционный со-
суд с краном особой кон-
струкции для удаления
летучих продуктов разло-
жения.
Фнг. 25. Схема установки для удаления ле-
тучих продуктов разложения из реакцион-
ного сосуда с НГЦ.
I и 4—реакционные сосуды. У—вакуум-
ный крав, 3—и-обраэнкв трубочка.
удаления газов НГЦ периодически нагревался до ~40°, а ловушка
на вакуумной установке заливалась жидким азотом. Полностью
удалить летучие продукты разложения из НГЦ трудно: необходи-
мо, чтобы нитроэфир по крайней мере сутки находился под вакуу-
мом. Обычно под вакуумом НГЦ выдерживали двое суток. После
удаления летучих продуктов распада перетяжка, соединяющая ре-
акционный сосуд с кранами, перепаивалась. Если было необходи-
мо, перед этой операцией в реакционный сосуд обычным способом
вводили пары воды.
При подготовке опыта по разложению безводного НГЦ в при-
сутствии продуктов распада применялся следующий метод
(фиг. 25). Влажный НГЦ разлагали в реакционном сосуде /, со-
единенном с краном 2, описанным выше. После перерыва разложе-
ния эту часть конструкции прибора припаивали через U-образную
трубочку 3 к реакционному сосуду 4 с мембранным манометром и
навеской безводного НГЦ. Сосуд 4, в свою очередь, присоединялся
к вакуумной установке. После эвакуации этой части системы кран 2
162
открывали и летучие продукты разложения из сосуда 1 собирали
в U-образной трубочке 3, охлаждаемой жидким азотом. Собранные
в ней летучие продукты переводили в реакционный сосуд 4 путем
охлаждения его жидким азотом; при этом U-образную трубочку 3
нагревали до комнатной температуры.
Результаты опытов таковы. НГЦ сначала разлагался в присут-
ствии воды (рн,о = 198 мм рт. ст.) при 100° и 6=0,0277. После обыч-
ного в этих условиях падения давления и достижения через 68 мин.
минимального его значения (71 мм рт. ст.) все летучие продукты
тщательно откачивали, как описано
выше. Полученный таким образом
нитроэфир, возможно содержащий
малолетучие продукты распада,
образовавшиеся в результате пред-
варительного разложения его в при-
сутствии воды, подвергали дальней-
шему разложению при 100° и
6=0,0328. Оказалось, что в этом слу-
чае общий характер зависимости
такой же, как и при разло-
жении безводного НГЦ, не подвер-
гавшегося предварительному разло-
жению (фиг. 26). Начальные ско-
рости газообразования у них при-
близительно равны, однако, уско-
рение распада у НГЦ, предвари-
тельно подвергнутого разложению
в присутствии воды, началось на
7 час. раньше, что составляет около
15% времени до начала резкого
ускорения газообразования при разложении безводного НГЦ.
Если к НГЦ после удаления из него летучих продуктов, обра-
зовавшихся в нем в результате предварительного разложения в
присутствии воды, добавить пары Н2О и вторично подвергнуть его
нагреванию, то будет наблюдаться обычная в этих условиях кар-
тина: индукционный период, падение давления и ускоренный его
рост (фиг. 27, кривые 1 и 3), Продолжительность индукционного
периода остается приблизительно такой же (кривая I) или несколь-
ко увеличивается (кривая 5), однако последующий ускоренный рост
давления наступает в этих опытах несколько раньше в сравнении
с НГЦ, не подвергавшимся предварительному разложению (кри-
вая 2).
Из этих опытов следует, что образовавшиеся к моменту, соот-
ветствующему /7mfn> малолетучие продукты взаимодействия НГЦ
с водой не проявляют почти никакого действия на началь-
ной стадии последующего разложения как влажного, так .и.
безводного НГЦ, но, как указывалось выше, резкое ускорение газо-
образования у предварительно разлагавшегося НГЦ наступает не-
сколько раньше,
Фиг. 26. Распад НГЦ, предвари-
тельно разлагавшегося в присут-
ствии воды и освобожденного от
летучих продуктов разложения.
7 —НГЦ, предварительно разлагавшийся
в присутствии воды, 6=0,0328; 2—НГЦ,
не подвергавшийся предвзрнтелъвпму
разлвжевню, 6*0,0329,
163
' Сказанное справедливо только при условии полного удаления
летучих продуктов распада после предварительного разложения.
Если же они не полностью удалены (предварительно разлагавший-
ся НГЦ находился под вакуумом 12 час., вместо 48, как обычно),
то индукционный период у такого НГЦ практически отсутствовал:
давление, достигнув своего максимального значения (вследствие
испарения паров воды при разогреве), сразу начало падать и через
Фиг. 27, Влияние нелетучих продуктов распада
влажного НГЦ на повторное разложение его в
присутствии воды при 100°.
(—НГЦ, предварительно разлагавшийся, q—278 хх
рт, ст.. 1=0,0154; 2—НГЦ, предварительно не pas латавшийся,
р „-233 мх рт. ст. 1=0,0367; Л—НГЦ, предварительно
разлагавшийся. -«182 мм рт. ст., 6=0,0355; НГЦ.пре-
дварительно не разлагавшийся, Л“230 хм рт. ст., 1 =0,0323;
tt
5—НГЦ, предварительно разлагавшийся при неполной удале-
нки продуктов разложения перед повторным нагреванием,
Рт» мм Рт* ст.г 6*0,0459.
HfU
27 мин. перешло минимум (фиг. 27, кривая 5). Последующее газо-
образование продолжалось с большой скоростью и к 80-той мин.
резко ускорилось.
Если бы на развитие процесса, происходящего за падением дав-
ления, влияли только газообразные продукты разложения, а обра-
зовавшиеся нелетучие вещества не оказывали на него никакого
влияния, то ход распада чистого НГЦ в присутствии летучих про-
дуктов разложения, образовавшихся к моменту, соответствующему
pmin, и' ход разложения влажного нитроглицерина после прохож-
дений давлением своего минимума должны были бы быть одина-
ковы.
164
Фиг. 28. Влияние летучих продук-
тов распада влажного НГЦ на его
разложение при 100°.
т— нгц+н,О, о=40а мм ₽т- Ст->
8-О.ОЗОБ; 2-НГЦ+Н.О, А.л-=293 мм
рт* ст,. В—0.0367: НГЦ+летучие про-
дукты разложения, 0,0301; <#—НГЦ т ле-
тучие продукты разложения, S=0,0356*
Однако эксперименты показывают, что это не так. На фиг. 28
(кривые 3 и 4) представлено изменение давления во времени в опы-
тах, в которых к чистому, предварительно откачанному НГЦ, до-
бавлены летучие продукты разложения влажного нитроглицерина,
образовавшиеся к моменту, соответ-
ствующему pmtn. Для сравнения на
том же графике даны кривые раз-
ложения НГЦ в присутствии воды,
причем за начало отсчета времени
принят момент прохождения давле-
нием своего минимального значе-
ния (кривые I и 2).
Из опытов следует, что газооб-
разные продукты распада оказы-
вают более слабое влияние на раз-
ложение чистого НГЦ, чем на раз-
f ложение нитроэфира, содержащего
' нелетучие продукты разложения;
в последнем случае время до рез-
кого ускорения газообразования со-
кращается приблизительно вдвое.
Это находится в согласии с пре-
дыдущей серией опытов. Во всех
тех случаях, когда в НГЦ присут-
ствуют нелетучие продукты его раз-
s' ложения, образовавшиеся к мо-
менту, соответствующему pmln, уско-
рение газообразования наступает
' раньше, чем в их отсутствии, т. е. динитрат глицерина, образовав-
шийся в результате гидролиза, или более подвержен окислитель-
ному действию, или способствует распаду НГЦ.
г ’
' Разложение динитрата глицерина в присутствии воды
В связи с тем, что при разложении влажного НГЦ в результате
гидролиза возможно образование динитрата глицерина, который
затем может вступать в реакцию с водой и с кислотой, интересно
было изучить кинетику его разложения в присутствии воды. В этой
части работы мы не стремились детально и всесторонне исследо-
вать распад динитрата, а лишь пытались установить его основные
закономерности.
Опыты проводились при б » 0,03 и 100° в интервале начальных
(равновесных) давлений паров воды 60—400 мм рт. Ст., т. е. в таких
условиях 1, в которых при разложении влажного НГЦ наблюдалась
характерная манометрическая картина распада: индукционный
период, падение давления и его рост.
1 Растворимость воды в динитрате глицерина больше, чем в тринитрате; это
означает, что при данном равновесном давлении паров воды концентрация ее в
динитрате больше, чем в тринитрате.
165
Опыты по разложению несимметричного динитрата глицерина
показали такую же зависимость изменения давления во времени
(фиг. 29), как влажный НГЦ. Однако наблюдались и отличия. При
увеличении рн,о величина индукционного периода не возрастала,
как в случае НГЦ, а сокращалась. Так при рнйо = 57 мм рт. ст. вре-
мя достижения pmln составляло 150 млн., при рн,о=220 леи рт. ст.—
68 мин., а при рн,о = 412 мм рт. ст. всего 40 мин. В этом же интер-
вале давлений, как видно из графика, при разложении НГЦ в при-
Фиг. 29. Разложение несимметричного диннтрата
глицерина в присутствии воды при 100’.
/-ДНГЦ+Н,О, />н 0 =4(3 мм рт. ст., 3=0,0307: г-нгц+
+Н.о. />„„«408 мм рт. ст., 3=0,0305; f-ДНГЦ+Н.О,
*
/>Н ям Pt- ст., ®-0.0251; 4—НГЦ+Н2О. р^ О=230 мя
рт. ст. 4=0,0339; 5—ДНГЦ+НгО, р^ ц—57 jWJK рт. ст.,
5=0,03; 5-НГЦ +Н,о, />н Q=54 .мл ' рт. ст., 5=0,0259.
сутствни воды время достижения рты колеблется между 60 и
75 мин.
Другим существенным отличием распада динитрата глицерина
от распада тринитрата глицерина является то, что после достиже-
ния pffl!n давление растет с большей скоростью. Существенной раз-
ницы между распадом симметричного и несимметричного изомеров
дииитрата в присутствии воды не наблюдалось.
Одинаковый характер разложения тринитрата и динитрата гли-
церина указывает на то, что как в том, так и в другом случае в ос-
новном происходят одни и те же процессы. Однако различие в
структуре молекулы (наличие группы ОН) оказывает некоторое
влияние на ход разложения. В частности, вероятно наличием груп-
пы ОН, способной к относительно легкому окислению, следует объ-
яснить большую скорость газообразования на стадии ускоренного
роста давления.
166
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Основными причинами ускорения распада НГЦ, вызываемого
газообразными продуктами его разложения, является их окисляю-
щее и гидролизующее действие на промежуточные органические
продукты распада и иа сам нитроглицерин.
Аналогичное влияние могут иметь примеси — такие как вода,
обычно содержащаяся в нитроэфире, и кислоты, если НГЦ недоста-
точно отмыт от отработанной кислоты.
Вода вызывает гидролиз НГЦ, ведущий, как показал опыт, к
образованию азотной кислоты; скорость гидролиза естественно тем
больше, чем больше содержание воды. Однако эта скорость сильно
зависит также от содержания в НГЦ кислот, в частности азотной,
и это влияние может перекрывать влияние концентрации воды в том
смысле, что при малом содержании воды, но значительной кислот-
ности, гидролиз может идти гораздо быстрее, чем при большом со-
держания воды в НГЦ, но в отсутствии кислоты. При этом разбав-
ленная азотная кислота мало ускоряет гидролиз, концентрирован-
1< ная — также. Окисляющее действие азотной кислоты зависит от
' ее концентрации; при этом минимальная концентрация, начиная с
‘ которой сильно ускоряется развитие окисления, выше, чем та, при
которой наступает сильное ускорение гидролиза.
Это приводит к своеобразному и различному характеру влияния
; воды при разных ее содержаниях в сосуде и, в частности, в НГЦ,
, проявляющемуся в манометрических кривых при лабораторных
опытах.
При малых количествах воды ход разложения близок к тому,
который наблюдается для безводного НГЦ, но более сжат во вре-
мени. В координатах р=/(т) он может быть представлен кривыми
фиг. 3 и 4. Вначале рост скорости газообразования идет медленно;
по достижении некоторого «критического» давления скорость рас-
тет приближенно пропорционально квадрату суммарного давления
газообразных продуктов распада и воды.
При средних количествах воды стадийность процесса выра-
жена более резко. Отчетливо различаются три основные стадии
(фиг. 21, кривая /):
1) индукционный период, когда давление (водяных паров над
НГЦ) остается практически постоянным (АВ);
2) период быстрого падения давления (ВС);
3) период ускоренного роста давления (CD),
В индукционном периоде происходит накопление азотной кис-
лоты в НГЦ в результате «безводного» распада и взаимодействия
его продуктов с водой, а также в результате гидролиза, медленного
вначале и ускоряющегося по мере накопления кислоты.
Во втором периоде, начинающемся, когда содержание кислоты
достигает несколько десятых долей процента, пары воды переходят
в жидкий НГЦ и одновременно в результате гидролиза образуют
азотную кислоту. Ее растворимость в нитроэфире гораздо больше,
чем воды. Этим и определяется в основном соотношение давлений
167
до и после падения. Концентрация азотной кислоты в НГЦ в мо-
мент завершения падения давления в несколько раз (в зависимости
от количества воды, имевшейся в сосуде) больше, чем она была в
начале периода падения давления. Соответственно возрастает и
концентрация органических продуктов гидролиза.
В третьем периоде вследствие высокой концентрации азотной
кислоты, а также более легко, чем НГЦ, окисляющихся продуктов
гидролиза, основную роль получают окислительно-восстановитель-
ные процессы, сопровождающиеся обильным образованием газов
как бурых и конденсирующихся, так и неконденсирующихся (при
комнатной температуре). Процесс развивается тем быстрее, чем
больше было азотной кислоты на минимуме давления. Скорость
газообразования в этом периоде приближенно пропорциональна
квадрату давления.
При больших количествах воды (фиг. 6, кривая 8) давление
над НГЦ меняется относительно мало, и спустя большое время, ко-
торое может даже превосходить время до резкого ускорения при
распаде безводного нитроэфнра, наступает незначительное сниже-
ние давления, за которым следует обычно быстрый его рост. При
очень больших относительных количествах воды максимум скоро-
сти распада может быть даже меньше, чем прн «сухом» процессе
в тех же условиях. Основной причиной уменьшения ускорения в
последнем случае является, по-видимому, чрезмерное разбавление
водой азотной кислоты и других окислителей, тормозящее развитие
окисления.
Рассмотрим теперь подробнее три типа «водного» разложения.
Начнем со второго как наиболее типичного.
Если НГЦ содержит воду, то в нем идут два процесса — «без-
водное» разложение, не связанное с присутствием воды, и гидролиз.
Скорости обоих процессов вначале малы. Однако скорость первого
из них почти не растет, скорость же гидролиза увеличивается под
влиянием азотной кислоты, сначала образующейся преимуществен-
но в результате «безводного» распада. Вначале этот рост идет мед-
ленно, поэтому длительность индукционного периода в основном
определяется скоростью «безводной» реакции. В соответствии с
этим находится, согласно Б. С. Светлову, и температурная зави-
симость индукционного периода. Когда количество азотной кислоты
достигает 0,2—0,4% вес. количества НГЦ, гидролиз резко ускоряет-
ся. Причина резкости этого увеличения скорости не установлена —
возможно здесь играет роль, в частности, увеличение растворимости
воды в НГЦ, приводящее к переходу ее из газовой фазы в жид-
кость, что при опытах в замкнутом объеме проявляется в резком
падении давления. Во время этого падения практически вся вода
, путем гидролиза превращается в азотную кислоту, соответствую-
щее количество НГЦ образует, по-видимому, динитрат.
Сильное увеличение концентрации азотной кислоты в жидкости
вызывает окисление органических продуктов гидролиза, а также
и самого НГЦ; поскольку окисление дает воду, к нему прнсоеди-
168
няется гидролиз, поставляющий в свою очередь «пищу» для окис-
лительно-восстановительных процессов.
Продукты гидролиза окисляются легче, чем сам НГЦ, как это
подтвердил ряд опытов. Так при «водном» разложении дннитрата
глицерина рост давления в третьем периоде шел, при прочих равных
условиях, значительно быстрее, чем для НГЦ, хотя азотной кисло-
ты в этом случае может образоваться меньше, чем при «водном»
распаде тринитрата глицерина. Точно так же опыты с частично гид-
ролизовавшимся НГЦ, освобожденным тщательной эвакуацией от
летучих кислот, показали, что период ускорения газообразования
i
*
у него наступает кдк в присутствии воды, так и в ее отсутствии,
быстрее, чем для свежего нитроэфира. Наконец, летучие продукты
распада, будучи добавлены к свежему НГЦ, слабее ускоряли газо-
образование, чем это наблюдается в третьем периоде распада влаж-
. ного нитроглицерина.
Таким образом сначала последовательное, а затем и совместное
протекание гидролиза и окисления приводит к тому, что в присут-
ствии воды большая и растущая скорость распада НГЦ достигается
быстрее, чем в ее отсутствии. Так при б» 0,03 и 100° при «безвод-
I1 ном» распаде это произошло через 40 час., а в присутствии 1,5% вес.
воды — через 2 час.
, Если воды очень мало (например рн,о<30—50 мм рт. ст. прн
100° и б ~ 0,03), то скорость гидролиза вначале соответственно
f. мала, но так как кислотные продукты безводного распада (а также
>' .кислота, образуемая при развитии гидролиза) не разбавляются
существенно водой, быстро достигается такая концентрация кисло-
1 ты, при которой гидролиз идет с большой (относительно количества
воды) скоростью. Образование в этих условиях концентрированной
, (поскольку в сосуде мало воды, чтобы ее разбавить) азотной кис-
лоты, а также органических продуктов гидролиза приводит к раз-
витию окисления, более медленному, однако, чем при средних коли-
чествах воды, так как мало азотной кислоты.
Таким образом общий ход этого процесса получается сходным
с разложением безводного НГЦ с тем лишь отличием, что в послед-
нем случае отсутствие вначале воды, которая образуется лишь
позже в результате окисления, задерживает развитие гидролиза;
при наличии же даже небольшого количества воды он начинается
сразу и вскоре, благодаря значительной кислотности, идет
со сравнительно большой скоростью несмотря на то, что воды мало.
В итоге сохраняется ход кривой «давление-время», присущий
НГЦ, но процесс ускорения распада, связанный с накоплением
азотной кислоты и вызываемым ею окислением, развивается зна-
чительно быстрее. Так при рн,о = 14 мм рт. ст. давление, равное
700 мм, было достигнуто в присутствии малого количества воды
вдвое быстрее, чем для безводного НГЦ.
Если воды очень много, то она разбавляет кислоты, образую-
щиеся при «безводной» реакции, а затем и при гидролизе, и послед-
ний поэтому протекает вначале очень медленно. В результате этого
время до наступления третьего периода может возрастать. При
169
этом получается своеобразная картина: гидролиз задерживается,
а последующее ускорение распада, напротив, убыстряется, так как
образуется больше азотной кислоты и органических продуктов гид-
ролиза, В зависимости от условий, может преобладать как первое,
так и второе влияние, т. е. общее время от начала опыта до наступ-
ления сильного ускорения газообразования может как уменьшаться,
так и возрастать.
Хорошую иллюстрацию этих соотношений дает табл, 6; при оди-
наковой 6 увеличивалось давление паров воды, т, е, ее количество
и содержание в НГЦ; индукционный же период в общем менялся
мало и средняя скорость образования кислоты, отнесенная к кон-
г MHNOa 1
центрации воды-------—- , с увеличением последней уменьшалась,
L /'н.о'1 J
Причиной этого была очевидно меньшая средняя по времени кон-
центрация кислоты, образуемой «безводным» распадом, вследствие
чего снижалась скорость гидролиза. Добавим, что немногие опыты,
проведенные с динитратом глицерина, показали, что для него ин-
дукционный период сокращался при увеличении давления воды.
Возможно, что в этом случае концентрация воды играет большую
роль, а ускоряющее действие кислоты — меньшую, чем в случае
НГЦ,
При больших 6 обычно преобладающую роль играет сокраще-
ние длительности третьего периода и общее время развития про-
цесса сокращается при увеличении количества воды в НГЦ; при ма-
лых же б, например ~0,006, можно наблюдать и увеличение общего
времени при повышении содержания воды в НГЦ, причем оно мо-
жет становиться даже больше, чем для безводного нитроэфира.
Рассмотренные соображения объясняют и повышенную эффек-
тивность «порционного» введения воды. Если воды введено немного,
то концентрация кислоты скоро достигает за счет «безводного» рас-
пада таких значений, при которых гидролиз идет быстро и успевает
образовать много кислоты. При повторном добавлении воды ей не
приходится уже начинать процесс гидролиза с нейтрального начала
и суммарное время обоих нагреваний до падения давления сокра-
щается по сравнению с тем, которое наблюдается, если все коли-
чество воды добавить сразу.
Малое влияние предварительного «сухого» нагревания на пос-
ледующее разложение в присутствии воды требует дополнительного
объяснения. Следует допустить, что в этих условиях наряду с обра-
зованием высших окислов азота идет их восстановление ’, вероятно
до NO, и последующее добавление воды не приводит к образова-
нию кислот. Если же высшие окислы азота образуются в присутст-
1 В пользу этого допущения, как на это обратил наше внимание Б, С. Свет-
лов, говорит также сокращение времени до наступления резкого ускорения при
распаде безводного НГЦ в присутствии кислорода; по-видимому, кислород, быстро
окисляя NO, уменьшает роль восстановительных процессов, ведущих к уменьше-
нию концентрации высших окислов азота. Точно так же в случае термического
распада дягликольдинитрата, где высшие окислы азота быстро восстанавливаются
и не могут образовывать кислоты, присутствие воды не приводит к ускорению
распада.
170
вии воды, то она, взаимодействуя с ними, сразу дает кислоты и
разбавляет их настолько, что гидролиз (образующий азотную кис-
лоту) идет со значительной скоростью, а окисление (уничтожающее
высшие окислы азота) подавляется (напомним, что концентрация
азотной кислоты, сильно ускоряющая гидролиз, гораздо меньше,
чем соответствующая концентрация для окисления).
ВЫВОДЫ
1. Присутствие воды может оказывать сильное влияние на раз-
витие распада НГЦ при повышенных температурах,
2. При малых количествах воды сохраняется тип распада, при-
сущий безводному НГЦ, но протекает он быстрее,
3, При средних количествах воды наблюдается три периода рас-
пада: постоянство давления (индукционный период), падение дав-
ления и рост давления. Последний период протекает тем быстрее,
чем больше было воды, и это определяет уменьшение общего вре-
мени распада при увеличении количества воды,
4, При больших количествах воды распад может сильно замед-
ляться, и максимум скорости его может быть меньше, чем даже
: для безводного НГЦ.
5, Все эти закономерности объясняются исходя из предположе-
ния, что начальным этапом распада в присутствии воды является
„гидролиз, в нейтральной среде идущий медленно, но ускоряемый
кислотными продуктами «безводной» реакции и самого гидролиза.
Гидролиз ведет к накоплению концентрированной азотной кислоты
и органических продуктов, которые вызывают окисление, сопутст-
вуемое гидролизом и сопровождающееся выделением большого
количества газообразных продуктов.
ЛИТЕРАТУРА
1. Андреев К- К- н Глазкова А. П., ДАН СССР, 1955, 105, 286.
2, А и д р е е в К, К-, Г лаэкова А. П., М а у р и н а Н, Д. иСветлов Б. С.,
ЖФХ, 1958, 32, 1726,
3, Горбунов В, В., см, настоящий сборник стр, 219,
4. Л у к и н А, Я-, ЖФХ. 1932, 3, 406.
5, Рог янский С. 3. и С а по жн и ко в Л. М., ЖФХ, 1931, 2, 80.
6. В aker J. W., Еа st у D, М, J. chem. Soc., 1952, стр, 1193, 1208.
7, В а к е г J. W., Е a s t у D. М., Nature, 1950. 166, 156,
8. В Usch, Вег, 1906, 39, 1401,
9. F а г m е г R, С , J. chem. Soc., 1920, 117, 806,
10, М и г а о ur Н., Bull. Soc. chim. de France, Мёт., 1930, 47—48, 1259.
11, Po oth, Zs. anal, Chem,, 1909, 48, 375.
12, Robertson R., J. chem. Soc. 1909, 95. 1241.
13. Рогинский C. 3.. Phys. Zs. Sowjetunion, 1932, 1, 640.
К- к. АНДРЕЕВ, Г. Н. БЕСПАЛОВ
10. О ВЛИЯНИИ КИСЛОТ И СОДЫ НА РАСПАД
НИТРОГЛИЦЕРИНА В ПРИСУТСТВИИ ВОДЫ
Исследование разложения нитроглицерина (НГЦ) в присутст-
вии воды, результаты которого рассмотрены в предыдущей статье,
привело к заключению, что существенный этап распада представ-
ляет собой гидролиз НГЦ. Этот гидролиз в случае нейтрального
нитроэфира, идущий весьма медленно, может очень сильно уско-
ряться, если создается значительная концентрация азотной кислоты
в результате взаимодействия воды с окислами азота, образующи-
мися прн «безводном» распаде НГЦ. Быстрое протекание гидролиза
приводит к образованию столь больших количеств азотной кислоты,
а также динитрата глицерина, что развиваются более или менее
резко ускоряющиеся окислительно-восстановительные реакции ме-
жду ними, сопровождающиеся сильным газообразованием.
Из этого заключения вытекали два экспериментальных пути его
проверки, осуществленные в настоящей работе.
1. Добавив к НГЦ совместно с водой кислоту, мы должны сразу
или во всяком случае быстрее получить ту картину течения рас-
пада, которую дает нейтральный продукт по истечении индукцион-
ного периода.
2. Нейтрализация тем или иным способом образующейся кисло-
ты должна снять нли по крайней мере замедлить наступление того
изменения давления (постоянство, быстрое падение и ускоряющий-
ся рост), которое характерно для распада НГЦ и некоторых других
нитроэфнров в присутствии воды.
ВЛИЯНИЕ КИСЛОТ НА РАСПАД НИТРОГЛИЦЕРИНА
В ПРИСУТСТВИИ ВОДЫ
Было изучено влияние трех кислот: азотной, трнхлоруксусной
и щавелевой. Азотная кислота образуется при гидролизе и пред-
положительно является причиной его ускорения. Однако азогиая
кислота обладает не только кислотной, но и окислительной функ-
цией, и ее действие могло быть связано также с этой последней.
Поэтому кроме азотной, были изучены трихлоруксусная кислота,
являющаяся сильной кислотой, но не оказывающая окисляющего
действия, а также щавелевая кислота, сочетающая свойства кис-
лоты и восстановителя.
Методика эксперимента
Азотная кислота вводилась в НГЦ следующим образом. Нитро-
глицерин, освобожденный длительной эвакуацией в тонком слое от
летучих примесей, смешивался в определенном соотношении с
98,5%-ной азотной кислотой. В тех случаях, когда исследовалось
влияние больших количеств азотной кислоты (0,15—1% по отноше-
172
1нию к НГЦ), этот кислый НГЦ прямо вводился в реакционный со-
£суд. Если же изучалось разложение НГЦ при меньшем содержа-
Iнии HNO3, то в реакционный сосуд вводили приготовленный, как
['указано выше, кислый и предварительно откачанный нейтральный
^нитроглицерин в такой пропорции, чтобы получилось необходимое
[ соотношение между азотной кислотой и НГЦ. Во избежание улету-
.чнвания азотной кислоты откачивание для удаления воздуха из
' реакционного сосуда производилось при непрерывном охлаждении
последнего жидким азотом. Дозировка воды осуществлялась обыч-
ным способом.
Щавелевую и трихлоруксусную кислоты вводили в реакционный
. сосуд в виде водных растворов определенной концентрации, из ко-
торых вода удалялась откачиванием при комнатной температуре.
Остальные операции прн подготовке опыта оставались прежними.
Результаты опытов
Разложение нитроглицерина в присутствии малых количеств
азотной кислоты и малых количеств воды
’ При разложении НГЦ в присутствии небольшого количества
азотной кислоты (начальное давление ее паров в реакционном со-
суде составляло 13 мм рт. ст., что соответствует содержанию кис-
р мм рт.ст
000
7-
3 ь
t,z Г
600
Фиг. 1. Влияние малых количеств воды
« азотной кислоты иа разложение НГЦ
при 100°.
1-НГЦ+HNO,, PHN0 =13 мм рт. ст., 8=0.0346;
3-НГЦ+Н,0+НМО3, ₽н 0 нмо “14 JC« рт. ст..
, <-0.0381; 3—НГЦ+Н,О, ₽н q~’j4 мм рт. ст.,
8=0.0371; 4—НГЦ чистый, 8-0,0329.
J60-
1000
ZOOO Z мин
о
лоты приблизительно 0,03% вес. по отношению к НГЦ) без введе-
ния воды, газообразование шло (фиг. 1, кривая /) на начальном
этапе в три раза быстрее, чем при чистом нитроэфире, и время до
наступления резкого ускорения роста давления сокращалось при-
мерно вдвое,
Опыт, при котором в НГЦ были введены вода и кислота при-
близительно в равном количестве (кривая 2), также дал более
быстрое развитие процесса, чем оно наблюдалось в случае чистого
НГЦ, Интересно, однако, что при равном равновесном начальном
давлении вода (кривая 3) несколько сильнее ускоряла разложение
НГЦ, чем вода в сочетании с кислотой.
173
Разложение нитроглицерина в присутствии малых количеств
азотной кислоты и значительных количеств воды
Опыты при ~О,О2°/о азотной кислоты (по отношению к НГЦ)
(фиг. 2) и при различных равновесных начальных давлениях паров
воды и кислоты показали, что присутствие такого количества HN03
не меняет общей картины распада: газообразование качественно и
Фиг. 2. Влияние малых количеств азотной
кислоты на разложение НГЦ в присутствии
значительных количеств воды при 1ОСГ.
(—НГЦ+Н ,0+0,023% Bec.HNO,. л , ИК,Л =92 мм
•TlgU ”1“ П IN Uj
рт. ст., 4=0,0363; 2—НГЦ+Н.О+О 0|7% вес. HNO
РН O+HNO = 196 ММ рт’ ст“ 5 ’0'0287: 3-НГЦ +
+ Нг0+О,01э’к вес. HNOj. л _ =385 мм
HSO + HNO,
рт. ст., 4=0,03(70; 4-НгИ+ЩО, Рн о ям
Рт. ст.. 4=0,0290; 5-НГЦ+Н,О, д’ =408 мм
п и U
рт. ст,. 4=0,0305.
даже количественно близко к тому, которое дает одна вода; вели-
чина индукционного периода и время до резкого ускорения газо-
образования примерно одинаковы. Таким образом, при малом со-
держании азотная кислота относительно слабо влияет на те особен-
ности хода распада НГЦ, которые обусловлены присутствием воды.
Разложение нитроглицерина в присутствии больших количеств
(0,13—1,5% вес.) азотной кислоты и значительных количеств воды
Увеличение содержания азотной кислоты до 0,13% вес. значи-
тельно сокращает время до начала падения давления по сравнению
с опытом в присутствии воды, но без кислоты: время, соответствую-
щее ptain, сокращается с 65 до 23 мин, На последующем развитии
ускорения газообразования это количество кислоты однако почти
не отражается (фиг. 3). Лишь большое увеличение ёе содержания
174
(~1,5% вес.) сильно сокращает время до начала падения давления
при разложении влажного НГЦ, а также резко увеличивает уско-
рение газообразования. В этом случае уже нельзя говорить об ин-
Фиг. 3. Влияние 0,13% вес. азот-
ной кислоты на разложение НГЦ
в присутствии воды при 10(Л
Числа у кривых—равновесное давление
парой воды и кислоты н S * 10* (в скобках)-
дукционном периоде, как о периоде постоянства давления. Достиг-
нув максимума в результате разогрева, давление сразу же начинает
падать, после чего сначала сравнительно медленно, а затем быстро
р мм рт ст
в' То " ~2оЬ Тю sob too г ми»
Фиг. 4. Влияние 1,5% вес. азотной кислоты на разложе-
ние НГЦ в присутствии воды при 100°.
Числа у крирых—равновесное давление паров воды н кислоты
н 3 * 10* {я скобках)»
растет. При этом, чем больше добавлялось воды, тем позже насту-
пал минимум давления, но быстрее начиналось резкое ускорение
газообразования (фиг. 4).
175
Разложение нитроглицерина в присутствии трихлоруксусной
кислоты и воды
Опыты показали, что добавление трихлоруксусной кислоты н
количестве -—0,5% вес. по отношению к НГЦ сильно сокращает
индукционный период (Ттш=48 вместо 70 мин. в опыте с нейтраль-
ным НГЦ; фиг. 5, кривые / и 2).
Присутствие же 2,66% вес. трихлоруксусной кислоты совершен-
но уничтожает индукционный период — давление быстро падает и
через 19 мин. (кривая 5) проходит через свой минимум,
Фиг. 5. Влияние трихлоруксусной кис-
лоты на разложение НГЦ а присут-
ствии воды при КИР.
1— НГЦ нейтральный, „—230 мм рт.ст.,
5=0,0339; 5—Н Г U,4-0,48 % вес. ТХУ кислоты,
0^253 жж рт, ст., 5=0,0299; 4—НГЦ+
+2J56 X вес. ТХУ кислоты, _з«2Э0 жж
rigU
рт» ст., 5=0,0319,
Это показывает, что характер разложения, наблюдающийся на
начальном этапе распада влажного НГЦ, обусловлен кислотными,
а ие окислительными свойствами промежуточных продуктов, обра-
зующихся при взаимодействии НГЦ с водой.
Разложение нитроглицерина в присутствии щавелевой
кислоты и воды
Содержание в НГЦ сравнительно небольших количеств (0,08—
0,17% вес.) щавелевой кислоты (фиг. 6) ведет к некоторому увели-
чению индукционного периода (при 0,17% с 60 до 90 мин.). Кроме
того, в отличие от распада в отсутствии этой кислоты давление во
время индукционного периода не остается постоянным, а растет, и
при этом тем сильнее, чем больше добавлено кислоты. Минималь-
ное давление (после его падения) повышается, а относительная
величина падения давления соответственно уменьшается. Последу-
ющее ускорение газообразования развивается несколько быстрее.
Удлинение индукционного периода, по-видимому, связано с вос-
становлением двуокиси азота или азотной кислоты, предотвраща-
ющим рост кислотности НГЦ; о восстановлении, идущем с образо-
ванием газов, свидетельствует и некоторый рост давления в Индук-
ционном периоде; кислотность же, создаваемая 0,2% вес. щавеле-
вой кислоты, оказывается слишком малой, чтобы заменить азотную
176
кислоту, Более быстрое наступление ускорения распада предполо-
жительно также следует связать с восстановительным действием
щавелевой кислоты; окисли азота в сочетании с водой окисляют
сильнее, чем чистая азотная кислота,
При увеличении количества щавелевой кислоты до 0,71 —
0,77% вес. (фиг. 7) рост давления в индукционном периоде стано-
вится еще более выраженным и соответствует образованию
2,7—3,0 молей газа на моль щавелевой кислоты; падение давления
Фиг. 6. Влияние малых количеств щавелевой
кислоты на разложение НГЦ в присутствии
воды при IOOP.
/— НГЦ нейтральный, р ^-230 мм рт. ст., 6-0,03391
2— НГЦ+О,07йз$ вес, Н^О., РилА-256 лс.м рт. стЦ|
гЦО
5 = 0,0326; 3—НГЦ+0,16% вес. НЛО,, РНО = 246 *-*
рг, ст., S ,0.0313; <—НГЦ+0.17& вес. Н.с’о., Рн Q
= 262 мм рт, ст.. 6 0,0313.
Фиг. 7. Влияние значительных
количеств щавелевой кислоты
iia разложение НГЦ в присут-
ствии воды прн 10(Г.
1-игЦ+0,7!% нес. Н,С,О,.
ПсО
175 лслс рт, ст., 5 =0,0404; 2—НГИ+
-0т77?а вес. Н4Са0ъ ММ
рт.ст.. Б-0Д326; НГЦ+1.62% вес-
НаС.,О41 q 160 мм ртц ст
2 в = 0.0356.
наступает несколько раньше, но происходит гораздо менее резко н
меньше по величине. При еще большем содержании щавелевой кис-
лоты (1,6%) падения давления уже нет и наблюдается лишь неко-
торое кратковременное замедление его роста. Очевидно, при этой
концентрации проявляется уже ускоряющее действие щавелевой
кислоты на гидролиз, а ее восстанавливающее действие, увеличивая
концентрацию окислов азота, ускоряет и усиливает окисление и оп-
ределяемый им рост давления. В итоге оба этапа — гидролиз и окис-
лительно-восстановительные реакции—оказываются не разделен-
ными четко во времени, как это наблюдается в присутствии только
воды, а налагаются друг на друга и даже сливаются (фиг. 7, кри-
вая 5).
12 451
177
ВЛИЯНИЕ СОДЫ И МЕЛА НА РАСПАД НИТРОГЛИЦЕРИНА
В ПРИСУТСТВИИ ВОДЫ
Нейтрализация кислых продуктов распада НГЦ посредством
соды производилась двумя способами: введением небольших ко-
личеств ее в реакционный сосуд1 или припаиванием к последнему
ампулы, содержащей значительное количество NaaCO3. Преимуще-
ство первого способа заключалось в том, что вследствие тесного
контакта НГЦ с содой происходила быстрая нейтрализация обра-
зующихся кислот. К недостаткам этого способа следует отнести
Фиг. 8. Влияние небольших количеств соды на раз-
ложение безводного НГЦ при 100°.
Г—НгЦ+0.06И% вес. NaaCO3, i =0.0318; 2 - НГЦ нейтральный,
« =0,0329.
возможность омыляющего действия соды на нитроглицерин. По вто-
рому способу нейтрализоваться могут только летучие кислоты и.
кроме того, скорость их поступления в ампулу с содой ограничи-
вается диффузией через слой паров воды в соединительной трубке.
Прежде чем проводить разложение НГЦ при совместном при-
сутствии воды и соды был поставлен опыт с безводным НГЦ с не-
большим количеством введенной в него соды. Фиг. 8 показывает,
что сода, по крайней мере в малых количествах, не оказывает за-
метного влияния на распад безводного НГЦ.
Иначе обстоит дело в случае влажного НГЦ. Присутствие соды
значительно увеличивает время до начала падения давления. Так,
в присутствии 0,044% вес. Ыа2СОз (фиг. 9, кривая 4) продолжи-
тельность индукционного периода была примерно 120 мин., в то
время как в случае нейтрального НГЦ при таком же начальном
1 Сода вводилась в реакционный сосуд а виде водного раствора определенной
концентрации, из которого вода удалялась откачиванием при комнатной темпе-
ратуре.
178
давлении паров воды падение давления начиналось через 40 мин.
Кроме того, в присутствии соды давление в течение индукционного
периода не остается постоянным, а несколько возрастает (на
5—7 мм рт. ст.). Минимальное давление несколько увеличивается,
а относительное его снижение соответственно уменьшается (0,62
вместо 0,71).
Фиг. 9. Влияние небольших ко-
личеств соды на разложение
НГЦ в присутствии поды при
100°.
/—НГЦ нейтральный» jOTt _~293
рт. ст., 5=0,0367; 2-НГЦ+0,053X вес.
NajCOs, o“ail мм ₽*• ст"
5=0,0316; 3—НГЦ нейтральный,Ру ^=
-230 мм рт. ст., 1-0,0339; 4^НГЦ+
+0,044% вес. Na2COj, 236 мм
рт. ст., 5=0,0377’
Фиг. 10; Реакционный сосуд для
разложения НГЦ в присутст-
вии больших количеств соды
и мела,
По-видимому, образующаяся
кислота нейтрализуется содой, и,
когда последняя вся прореаги-
рует, наступает обычное в при-
сутствии воды падение давления
и последующий его ускоренный
рост. Меньшей растворимостью
в нитроглицерине СОз, образую-
щегося при нейтрализации соды,
но сравнению с азотной кислотой
следует объяснить некоторое по-
вышение давления в индукционном периоде и увеличение ртш-
Дальнейший рост происходит несколько медленнее, чем в отсут-
ствии соды.
Еще более длительное торможение распада НГЦ в присутствии
воды можно получить, если обеспечить нейтрализацию кислых лету-
чих продуктов распада. Для этого сравнительно большое количе-
ство соды или мела (в 5—9 раз больше НГЦ) вводили в боковую
ампулу, припаянную к реакционному сосуду (фиг. 10). Давление
паров воды в начале опыта составляло 135 мм рт. ст. В этом слу-
чае газообразование началось со значительной скоростью 1 (фиг. 11,
кривая 2), которая впоследствии стала уменьшаться и через
125 час. снизилась до своего постоянного значения, близкого по
величине к начальной скорости газообразования безводного НГЦ.
Рост давления с такой постоянной скоростью в дальнейшем про-
1 Предположительно в результате взаимодействия паров НГЦ с содой па
поверхности ее частиц.
12*
179
должался в течение 30 суток1. Аналогичный вид кривой р=/(т)
наблюдался при разложении безводного НГЦ в присутствии боль-
шого количества соды (кривая 3), но начальная скорость газообра-
зования в этом случае была несколько выше. На участке постоян-
ства ее значения в обоих случаях были близки по величине (ср. кри-
вые 2 и 3 фиг. И).
Как при распаде безводного НГЦ, так и в присутствии воды не
наблюдалось никакого ускорения газообразования несмотря на то,
что давление к концу опыта превышало две атмосферы. Безводный
Фиг. 11. Разложение сухого и влажного
НГЦ в присутствии больших количеств
соды и мела при 100°.
/—НГЦ без веды и соды, б “0.0057ц 2—НГЦ-(-
+ H,O + Nj2CO3, P^q“I35 рт. ст., 5=0(00-Э;
3-НГЦ+Ма?СОа, Е-0,0074; 4 - НГЦ+СаСОа(
0*^62 X* рт- ст.» Ч’-•0,0075.
НГЦ без соды (6 = 0,0067) (фиг. 11, кривая 1) в этих условиях на-
чинает ускоренно разлагаться уже через ~130 час., а в присутст-
вии воды намного раньше.
В обоих случаях газовая фаза на протяжении всего опыта оста-
валась бесцветной. Сода в ходе разложения приобретала коричне-
вую окраску, вероятно вследствие полимеризации и осмолеиия на
ней промежуточных продуктов распада; стенки реакционного со-
суда покрывались белым налетом.
Ускорение газообразования отсутствовало и в тех опытах, в ко-
торых сода была заменена мелом (фиг, 11, кривая 4). Характерно,
что в этом случае не наблюдалось участка с большой начальной
скоростью' газообразования: разложение в течение 20 суток шло. с
постоянной скоростью, приблизительно равной начальной скорости
распада безводного нейтрального НГЦ.
В Проведенных опытах при 100° ускорения газообразования не
происходило, лишь при условии, что разложение шло с не очень
1 В ходе опытов несколько раз делались перерывы (на 30 час.), отмеченные
на графике. После перерыва давление всегда на несколько миллиметров было
меньше, чем до него.
180
[ большой скоростью и продукты распада успевали диффундировать
t через слой жидкости и газа к соде. В противном случае наступает
F -падение давления и последующий ускоренный его рост, что наблю-
[ далось при разложении НГЦ, в присутствии воды при 12(Г и
| 6 = 0,0051 (фиг. 12, кривая 2), Если после перерыва на стадии рез-
[ кого ускорения роста давления снизить температуру опыта до 10СГ,
Фиг. 12. Разложение НГЦ в присутствии больших
количеств соды при 120 и 100s.
/—НГЦ без соды, Q=505 СТ1' И +
+ NaaCO,, рД =466 ** рт* ст., 6-0.0051.
п дО
то сильного ускорения газообразования не происходит в течение
последующих 130 час. нагревания (фиг. 12, кривая 2 после пере-
рыва).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Результаты опытов подтверждают и уточняют те предположен
ния, для проверки которых они были поставлены.
Введение азотной кислоты во влажный НГЦ может значительно
сократить и даже снять индукционный период. В случае безводного
нитроглицерина влияние азотной кислоты выражено гораздо сла-
бее и даже значительное ее содержание (0,3% вес. по отношению
к НГЦ) лишь вдвое сокращает время до наступления резкого уско-
рения распада нитроэфнра.
Следует отметить, что малые количества азотной кислоты, даже
в присутствии воды, слабо ускоряют распад. Это относится как к
малому, так и к значительному содержанию воды.
Если количество добавленной азотной кислоты превышает
0,1% вес. НГЦ, наблюдается значительное сокращение индукцион-
ного периода. На последующем ускорении газообразования это не
отражается, так как к моменту его наступления в результате гид-
't релиза образуется гораздо больше кислоты и дополнительное уве-
личение ее количества на 0,1 % относительно очень мало и не может
L заметно влиять на развитие процесса разложения нитроэфира.
Й
X
1Я1
Добавление 1,5% вес. азотной кислоты к влажному НГЦ не
только снимает индукционный период, но и приводит к значительно
более быстрому развитию последующего газообразования.
К сожалению точная количественная оценка влияния фактиче-
ской концентрации азотной кислоты и воды в НГЦ применительно
к этим опытам затруднительна по нескольким причинам. Во-пер-
вых, неизвестно, как при совместном присутствии в реакционном
сосуде воды и азотной кислоты с НГЦ они распределяются между
газовой и жидкой фазами; это распределение может быть тем бо-
лее существенным, что опыты проводились при относительно ма-
лых 6 и высокой (1ОСГ) температуре, когда относительное содер-
жание паров кислоты и воды в газовой фазе может быть значи-
тельным. Во-вторых, применявшийся метод дозировки воды, осно-
ванный на измерении объема ее паров при относительно низкой —
комнатной температуре, мот быть сопряжен с заметной неточностью
из-за адсорбции их на стенках измерительного шарика. В-третьих,
растворимость воды в НГЦ, содержащем те или иные дополни-
тельно введенные вещества (например, трихлоруксусную кислоту,
соду), может отличаться от ее растворимости в чистом нитроэфире.
Более надежны количества введенной в НГЦ азотной кислоты
и равновесное суммарное давление паров воды и кислоты, изме-
ренное при температуре опыта. Точно так жене вызывают сомнений
основные установленные закономерности разложения влажного
НГЦ в присутствии HNO3: сокращение продолжительности индук-
ционного периода, значительная величина необходимой для этого
концентрации HNO3 и меньшая чувствительность ц ней периода
ускорения.
Опыты с трихлоруксусной кислотой подтверждают, что первич-
ным фактором влияния кислот, в том числе и азотной, на разложе
ицс влажного НГЦ является их ускоряющее действие на гидролиз,
а не окислительная функция и что это последнее действие стано-
вится сильным лишь при значительных содержаниях азотной кис-
лоты в НГЦ.
Это подтверждается также опытами с щавелевой кислотой. Ма-
лое ее содержание в НГЦ (до —'0,2% вес.) удлиняет индукцион-
ный период, так как ускорение гидролиза при этих количествах
НЙС2О4 мало, а торможение образования азотной кислоты в резуль-
тате восстановления высших окислов азота велико. При большом
(0,7—1,6%) содержании щавелевой кислоты преобладает кислот-
ное действие, а относительно быстрое восстановление азотной кис-
лоты и частичный переход ее вследствие этого в окислы азота при-
водит к более раннему развитию газообразующих окислительно-
восстановительных реакций, которые не отделяются в этом случае
от гидролиза некоторым интервалом времени.
Еще более убедительным для подтверждения основного выво-
да— о решающей роли гидролиза как «спускового» механизма для
развития превращения НГЦ —являются результаты опытов с содой
и особенно с мелом. Они показывают, во-первых, что необходимым
условием для начала падения давления паров воды и последую-
18_>
I щего ускорения газообразования является накопление кислот, и,
I во-вторых, что эти кислоты являются летучими. Скорость гидролиза
I в отсутствии кислоты невелика. Поэтому гидролиз тогда начинает
| играть свою решающую роль, когда образуется достаточная кон-
I центрация кислоты. Количество последней должно соответствовать
; содержащемуся в НГЦ количеству воды, если воды мало, то до-
статочно малого количества кислоты, чтобы ее концентрация, а сле-
довательно, и величина константы скорости катализированного гид-
ролиза была велика. Так как скорость гидролиза пропорциональна
произведению константы и концентрации воды, то влияние кислоты
на константу скорости может преобладать над влиянием концент-
рации воды, и в целом процесс гидролиза (но не газообразования)
будет развиваться быстрее, чем в присутствии больших количеств
воды.
Если воды много, то требуется добавить больше кислоты и этим
создать условия для быстрого развития гидролиза. В этом случае
и последующее ускорение процесса распада НГЦ идет быстрее, так
как больше образуется кислоты в результате «превращения» в нее
воды, а кроме того, к ней добавляется кислота, введенная в нитро-
эфир в начале опыта.
Таким образом, наиболее опасным в условиях длительного пре-
бывания при повышенных (100°) температурах* является слабо
увлажненный НГЦ, если он недостаточно тщательно отмыт от кис-
лоты. Сильно увлажненный и особенно содержащий капельно-жид-
кую воду продукт менее опасен. Менее опасен был бы также и аб-
солютно безводный продукт, но в таком состоянии нитроглицерин
практически не реален, поскольку эффективными оказываются уже
очень малые концентрации воды в НГЦ, соответствующие ее пар-
циальному давлению в окружающей атмосфере всего 8—15 jhjh рт. ст.
Присутствие в нитроэфире примесей, не обладающих существенно
выраженными щелочными свойствами, но способными нейтрализо-
вать кислоты (например, мела), предотвращают развитие гидро-
лиза и последующее ускорение разложения нитроглицерина.
ВЫВОДЫ
1. Азотная кислота в малых концентрациях слабо ускоряет
разложение нитроглицерина в присутствии воды. Повышение кон-
центрации кислоты до десятых долей процента приводит к быст-
рому развитию гидролиза НГЦ; при еще большем увеличении
количества кислоты ускоряется также развитие после гидролиза
окислительно-восстановительных реакций.
2. Гидролиз НГЦ ускоряется также трихлоруксусной и щавеле-
вой кислотами. В последнем случае однако при малых концентра-
1 При более низких (6(Г) температурах, как показывают опыты Горбунова
и Светлова, этап ускорения распада НГЦ достигается тем быстрее, чем больше
воды растворено в нитроэфкре. По видимому, здесь образование кислот безводной
реакцией, скорость которой сильно уменьшается при понижении температуры, от-
ступает на втооой план н преобладающее влияние получает концентрация воды.
183
циях кислоты гидролиз задерживается из-за восстановительного
действия HjCgO* на продукты «безводного» распада.
3. Вещества, нейтрализующие кислоты (сода, мел), предупреж-
дают при распаде НГЦ в присутствии воды быстрое развитие гид-
ролиза и последующее ускорение газообразования в результате
окислительно-восстановительных реакций между продуктами гид-
ролиза.
Б. С. СВЕТЛОВ
11. О ТЕРМИЧЕСКОМ РАЗЛОЖЕНИИ НИТРОГЛИЦЕРИНА
В ЖИДКОИ ФАЗЕ
Изучение термического разложения полинитроэфиров Представ-
ляет большой теоретический и практический интерес. Однако выяс-
нение механизма распада их сильно затрудняется его сложностью.
Как показывают исследования [9], разложение даже алкилмононит-
ратов в газовой фазе является комплексом параллельно и последо-
вательно идущих реакций. Разложение полинитратов, изученное
менее детально, особенно в жидкой фазе, в условиях, приводящих
к резкому самоускорению, представляет собой несомненно еще бо-
лее сложный процесс [4], [5].
В настоящей работе описывается проведенное в интервале тем-
ператур 80—140° исследование термического разложения в жидкой
фазе одного из важнейших нитроэфиров — нитроглицерина (НГЦ)
и, в частности, самоускоряющегося его распада.
Изучение разложения НГЦ производилось манометрическим
методом с применением стеклянного манометра типа Бурдона 15].
Отношение объема нитроэфира к объему реакционного сосуда, обо-
значаемое в дальнейшем б, составляло 10-3—10“’.
Разложение нитроглицерина представляет собой, по крайней
мере, двухстадийный Процесс [4]. Особенно отчетливо это видно при
рассмотрении зависимости скорости газообразования от давления
в логарифмических координатах (фиг. 1), где течение распада опи-
сывается двумя прямыми линиями, идущими под разными углами.
Каждая из этих двух макроскопических стадий разложения обла-
дает своими характерными особенностями.
На первой стадии не наблюдается резкое самоускорение даже
при накоплении значительных количеств газообразных продуктов.
Отсюда был в свое время сделан, вопреки общепринятым ранее
представлениям, вывод о том, что газообразные продукты в этих
условиях не оказывают каталитического действия на термический
распад нитроглицерина [1].
Распад нитроглицерина на первой стадии можно характеризо-
вать средней величиной начальной скорости газообразования, рас-
считанной для интервала давления продуктов распада от его на-
чала до 40—80 мм рт. ст. Такой расчет не является вполне точным,
184
так как в 'Некоторых случаях иа начальном участке первой стадии
происходит небольшой рост скорости, отчетливо проявляющийся
при более высокой температуре и значительных степенях за-
полнения реакционного сосуда. Газообразование при этом может
ускоряться на указанном интервале отсчета скорости в 1,5—2 раза.
На начальную скорость газообразования оказывает значитель-
ное влияние величина б. Во всем изученном интервале температур
(140—80°) с увеличением б начальная скорость газообразования
падает.
Фиг. 1. Влияние давления газообраз-
ных продуктов разложения нитрогли-
церина на его распад прн различных
температурах и 6=0,03.
На фиг. 2 представлена зависи-
мость начальной скорости газообразо-
вания от величины б. Для рассмотре-
ния этой зависимости при различных
темпепатуоах ппиведены не абсолютнь
Фит. 2. Влияние степени запол-
нения реакционного сосуда на
начальную скорость газообразо-
вания при распаде нитроглице-
MS)
рииа. ~~ТГ —отношение
ж(Ъ = 10
начальной скорости при дай-
ной б к ее значению при
б=10-з.
Значения скоростей, а от-
ношения начальных скоростей при соответствующих значениях б
к начальной скорости при 6=10 . Как видно из графика, с ростом
температуры влияние б на начальную скорость газообразования
проявляется более резко. В области малых б начальная скорость
сильнее зависит от степени заполнения сосуда, чем в области высо-
ких и при б больше 3-10-2 она практически уже не меняется.
Следует отметить в связи с этим, что снижение начальной ско-
рости с ростом б является общей закономерностью для распада
многих изученных жидких нитроэфиров: для расплава пентаэрит-
риттетранитрата по данным Б. И. Кайдымова, а также для нитро-
гликоля, ди гликольди нитрат а и динитратов глицерина — по нашим
данным. Вместе с тем Б. С. Самсонов [2], [3] установил, что степень
заполнения сосуда не влияет на начальную скорость газообразова-
ния при распаде нитроцеллюлозы.
co
3. Влияние температуры на
на-
фиг.
чальную скорость газообразования
при распаде нитроглицерина.
2—5—l0-i-30 • Ю-4;г-г=300ч-450 Ю-4.
Объяснение этого явления затруднено отсутствием данных о де-
талях механизма разложения нитроэфиров. Однако можно пола-
гать, что причины, обусловливающие влияние б на начальную ско-
рость газообразования, заключаются не столько в растворимости
газов или в большей скорости распада нитроэфиров в газовой
фазе [5], сколько в протекании вторичных процессов окисления
органических веществ двуокисью азота, скорость которых связана
степенью распада, возможно в сочетании с обратимостью реак-
ции первичного отрыва NO2 от
молекулы нитроэфира.
Ввиду значительного и коли-
чественно различного при разных
температурах влияния б на вели-
чину начальной скорости, оценка
температурной зависимости ско-
рости газообразования была сде-
лана несколько формально; она •
определена отдельно для б = 10~3,
т. е. для степени заполнения ре-
акционного сосуда, в области ко-
торой она сильно влияет на на-
чальную скорость, и для б=10-1,
при которой начальная скорость
газообразования становится прак-
тически постоянной.
В обоих случаях зависимость
скорости газообразования от тем-
пературы подчиняется уравнению
Аррениуса. Энергия активации £\
наклона прямых в координатах
составляет 43,6 ккал/моль и для
подсчитанная по тангенсу угла
Др 1
1g —- нач. — —, для малой б
Дт Т
большой 39,3 ккал/моль (фиг. 3). Предэкспоненциальный множи-
тель В в уравнении Аррениуса соответствует для малых б 1018'4 и
для больших 1015-4 сек.-4.
Первые из полученных значений Е и В близки к тем,
которые подсчитаны по данным Робертсона из скоростей отщепле-
ния азота при распаде нитроглицерина в токе углекислого газа [10].
Расчет по результатам Робертсона дает Ё = 43,7 ккал!моль и
В — Ю10’64 сек.-1. Величина Е, полученная для больших б, близка
к энергии отрыва NO2 при распаде алкилмононитратов:
39,5 ккал/моль для метил нитрата [6] и 41,2 ккал/моль для этил нит-
рата [9].
В связи с этим следует указать, что разложение нитроглицерина
на первой стадии характеризуется наличием в газообразных про-
дуктах распада двуокиси азота, о чем судили как по изменению
окраски газовой фазы (побурение), заметному на глаз, так и по
специальным колориметрическим опытам, результаты которых по-
казали, что доля NO2 в газах достигает 40— 60% [7].
386 .
I
Вторая стадия разложения нитроглицерина, стадия резкого
ускорения газообразования, наступает лишь при том условии, если
давление образующихся газов достигает определенного критиче-
ского значения, которое в значительном интервале б(10~3—10~L),
является величиной приблизительно постоянной для данной темпе-
ратуры и, следовательно, не зависит от степени разложения.
Течение разложения при переходе во вторую стадию резко ме-
няется. Если на первой стадии (на участке индукционного периода)
скорость газообразования возрастала лишь в 2—3 раза и была про-
порциональна давлению в степени значительно меньшей единицы,
то на второй стадии она становится пропорциональной приблизи-
тельно квадрату давления газообразных продуктов разложения
(фиг. 1) и может превысить начальную скорость в сотни раз. Ана-
логичная картина распада получена для нитрогликоля, а также по
данным Б. И. Кайдымова для растворов пентаэритриттетранитрата
в тротиле при 120°.
Величина критического давления зависит от температуры, повы-
шаясь с ростом последней. Так при 80г (6 = 6—60-10-3) она состав-
ляет 60—80 мя рт. ст.; при 100° (6 = 1,5—60’10~3) 180—210 мм
рт. ст. и при 120° (6 = 3—60-Ю-3) 400—500 мм рт. ст. Опыты при
140° и 6=15- 10~3 и 6 = 29- 10-3 дали соответственно 1000 и 800 леи
рт. ст.; при меньших степенях заполнения реакционного сосуда раз-
ложение нитроглицерина при этой температуре не переходило
во вторую стадию несмотря на то, что давление газообразных про-
дуктов при распаде достигало 3500 мм. рт. ст.
Наличие критического давления, не зависящего непосредствен-1
но от степени распада, а также своеобразный характер разложе-
ния нитроглицерина на второй стадии связаны, по-видимому, с реак-
циями, в которых участвуют газообразные продукты, растворенные
в конденсированной фазе.
Можно допустить, что разложение нитроглицерина в жидкой
фазе протекает по двум путям. Первому из них соответствуют реак-
ции, которые не ускоряются газообразными продуктами распада.
Скорость этого процесса не растет с давлением. Второй путь — рас-
пад нитроглицерина при его взаимодействии с продуктами разло-
жения, причем скорость этого процесса пропорциональна квадрату
давления газообразных продуктов распада. Естественно полагать,
что начальная скорость газообразования характеризует протекание
распада по первому пути, а ускорение, наблюдаемое в опыте, яв-
ляется результатом развития реакций распада нитроглицерина по
второму пути. Отсюда для начального этапа, когда уменьшением
концентрации исходных веществ можно пренебречь, суммарная
скорость газообразования — = w + kp2, где w —скорость процес-
са, не ускоряемого давлением, т. е. начальная скорость газообра-
зования, р— давление газообразных продуктов и k — коэффициент
пропорциональности. Если справедливы указанные соображения,
то кривые =f(p) в логарифмических координатах дол-
187
жыы выражаться прямыми линиями с тангенсом угла наклона
около двух. Обработка результатов опытов, приведенных на фиг. 1,
действительно показывает, что в этих координатах получается пря-
мая линия, если за w принять величину, близкую к начальной
скорости газообразования (фиг. 4, прямая /). Для наглядного срав-
нения здесь же приведена кривая зависимости скорости суммарного
газовыделения от давления
для опыта при 100° (кри-
вая 2).
Исходя из изложенного,
следует полагать, что на-
чало второй стадии распада
является условным и соот-
ветствует тому моменту,
когда в жидкой фазе кон-
центрация ускоряющих ре-
акцию продуктов распада 1
становится значительной и
скорость их взаимодействия
с исходным веществом на-
чинает преобладать над ско-
ростью реакций, течение
которых не ускоряется про-
дуктами распада,
В пользу предположения
о ведущей роли газообраз-
ных продуктов распада при
самоускоряющемся разло-
Фиг. 4. Влияние давления газообразных
продуктов разложения нитроглицерина на
прирост скорости газообразования сверх на-
чальной ее величины при различных темпе-
ратурах.
/—при 80, 100 и |20°, 1—влияние давления на об-
щую скорость газообразовании при 100°.
жении нитроглицерина гово-
рят и опыты с чистым нитро-
глицерином в присутствии
газообразных продуктов, по-
лученных при разложении
этого нитроэфира на стадии
резкого ускорения в другом
опыте. С добавлением этих продуктов скорость с самого начал;)
опыта превышает в десятки и сотни раз начальную скорость газо-
образования при распаде чистого нитроглицерина. При этом, чем
больше добавлено продуктов, тем выше скорость распада.
Процесс разложения нитроглицерина на второй стадии представ-
ляет собой, по-видимому, преимущественно гидролиз нитроэфира с
образованием кислоты в сочетании с окислительными реакциями,
генерирующими воду.
Установленное влияние температуры на величину критического
давления может быть обусловлено с одной стороны изменением
1 Такими продуктами, по-видимому являются вода и образующаяся при
гидролизе нитроэфира азотиая кислота [[], а также и окислы азота, в частности
двуокись, которая представляет собой один из продуктов второй стадии рас-
пада [8].
188
растворимости газов с температурой и с другой тем, что скорость
। реакции, не ускоряемой продуктами распада (первичная реакция),
быстрее растет с температурой, чем скорость реакций, зависящих от
i давления. В результате этого для заметного Превышения скорости
реакций, зависящих от давления, над скоростью реакции, не зави-
I сящей от него, при более высокой температуре необходима большая
концентрация газообразных продуктов распада в жидкой
, фазе.
Вторая стадия разложения нитроглицерина характеризуется
крайне слабым влиянием температуры на скорость газообразова-
i ния. Как видно из графика (фиг. 1) при давлении, выше критическо-
, dp
> го, т. е. на участке резкого ускорения, точки зависимости Ig — =
= f(lgp) ПРИ 80, 100 и 120° ложатся практически на одну линию,
1 т. е. скорость газообразования зависит лишь от давления продуктов
( разложения, но не зависит от температуры. Зависимость, характери-
f зуюшая течение реакций распада нитроглицерина по второму пути
f, при тех же температурах (фиг, 4), составляет также одну линию.
У Отсутствие влияния температуры на скорость газообразования при
’ распаде нитроглицерина, обусловленном взаимодействием продук-
тов разложения с нитроэфиром, может быть связано с тем, что при
повышении температуры увеличение скорости реакции компенси-
! руется уменьшением концентрации реагирующих веществ вследст-
вне снижения растворимости газообразных продуктов в нитроглн-
; церине. Поэтому в данном случае температурную зависимость ско-
> рости процесса следует оценивать, учитывая влияние температуры
на растворимость продуктов распада в конденсированной фазе. Если
исходить из предположения о компенсации увеличения скорости
г. распада с температурой, уменьшением растворимости газов, то мо-
жно полагать, что энергия активации реакции разложения нитро-
глицерина с участием газообразных продуктов будет близка к теп-
лоте растворения их в этом ннтроэфире. Так как теплота раство-
рения газов в жидкостях, как правило, мала по сравнению с энер-
гией активации первичной реакции распада нитроглицерина
(40 ккал/молъ), то и энергия активации ведущих реакций на второй
стадии также мала.
Это обстоятельство, важное для оценки стойкости нитроэфиров,
следует учитывать при рассмотрении поведения этих веществ при
хранении и переработке. В реальных условиях, когда затруднено
удаление продуктов распада из массы вещества (большой объем и
низкая температура), разложение нитроэфира определяется в ос-
новном его взаимодействием с этими продуктами, обладающим рез-
ким самоускоряющимся характером и малым температурным коэф-
фициентом скорости. Поэтому характеристикой распада нитрогли-
церина, определяющей его стойкость при хранении и переработке,
является величина и температурная зависимость индукционного пе-
риода, при котором формируются условия развития последующих
реакций.
183
выводы
L Исследован термический распад нитроглицерина в жидкой
фазе при 80—140° в присутствии продуктов разложения.
2. Установлено наличие при распаде нитроглицерина двух-мак
роскопических стадий, протекание которых зависит лишь от содер
жания продуктов распада, но не от его глубины.
3. На первой стадии разложения газообразные продукты распа
да практически не оказывают ускоряющего влияния на скоростi
газообразования. Начальная скорость последнего зависит от степе-
ни заполнения реакционного сосуда и характеризуется нормальным
для нитроэфиров температурным коэффициентом этой скорости.
4. На второй стадии разложения скорость газообразования зави
сит от давления газообразных продуктов распада приблизительна
во второй степени и может превышать начальную скорость по край
ней мере в сотни раз.
5. Полученные результаты рассмотрены в свете предположения
об одновременном протекании двух процессов разложения нитро-
глицерина. Первый из них представляет собой самопроизвольное
Разложение, не ускоряемое газообразными продуктами распада,
[ри втором процессе происходит взаимодействие нитроглицерина
с продуктами его распада, как это предполагал С. 3. Рогинский [7].
со скоростью, зависящей от содержания этих продуктов.
Автор выражает большую признательность К. К- Андрееву за
постоянный интерес к работе.
ЛИТЕРАТУРА
I. А н д р е е в К. К., Г л а з к о в а А. П., ДАН СССР, 1955, 105, 286.
2. Андреев К.К, Самсонов Б. С., ДАН СССР, 1957, 114, 815.
3. Самсонов Б. С., Кандидатская диссертация, МХТИ, 1957.
4. Андреев К. К., ЖПХ. 1958.31.484—493.
5. Андреев К. К-, Глазкова А. П., М а у р и н а Н. Д., Светлов Б.
ЖФХ, 1958, 32, 1726.
6. Апин А. Я,Тодес О. М., Харитон Ю. Б., ЖФХ, 1936, 8, 866.
7. Рогинский С. 3. и Сапожников Л. М., ЖФХ, 1931, 2, 80.
8-Светлов Б. С., Научные доклады Высшей школы СССР, 1958,3, 422—425.
9. L е v у J. В., J, Amer. chem. Soc., 1954, 76, 3254, 3790.
10. R о b е г t s о n R.. J. chem. SocL, 1909, 95, 1241,
В. В. ГОРБУНОВ, Б. С. СВЕТЛОВ
12. О ВЛИЯНИИ ТЕМПЕРАТУРЫ ИА РАЗЛОЖЕНИЕ
НИТРОГЛИЦЕРИНА
Термический распад нитроглицерина и других нитроэфиров в
жидкой фазе исследован в основном при сравнительно высокой
температуре, а также относительно малой степени заполнения ам-
пулы в случае применения манометрического метода [1], [2], [3] и 14].
190
'Полученные прн этом результаты, интересные с точки зрения выяс-
нения механизма разложения, не могут быть, однако, непосредст-
венно использованы для описания поведения нитроэфиров в усло-
виях, наиболее интересных с точки зрения практики, т. е. при низ-
кой температуре и максимальной концентрации продуктов распада
в веществе.
' Чтобы воспроизвести указанные условия распада, мы применили
описанный ранее [1] метод, видоизменив его для максимального за-
полнения предварительно эвакуированной
ампулы и соединенного с ней стеклянного
манометра жидким нитроэфиром.
Для подготовки и проведения таких
опытов применили прибор, схема которого
изображена на фиг. 1. Стеклянный мано-
метр 1 с помощью тонкой перетяжки 2 со-
единен с сосудом 3, предназначенным для
предварительной эвакуации нитроэфира
в тонком слое. Опыт готовили следующим
образом. Сначала измеряли внутренний
объем реакционного сосуда 4 и стеклянного
Фиг. 1. Схема прибора
для изучения термическо-
го разложения жидких
нитроэфиров при макси-
малыюй степени заполне-
ния ампульь
/•—манометр со стрелкой*
2—перетяжка. 5—сосуд для
эвакуации ннтроэфира прн
подготовке опыта. 4—реак*
ционный сосуд, 5 в о—от-
воды для соединенна при-
бора с вакуумной установи
кой, 7—отросток для ваолэ>
нитроэфира.
манометра до места последующего пере-
паивания перетяжки 2, путем взвешивания
прибора с водой. После определения объ-
ема прибор присоединяли к вакуумной
установке при помощи соединительных ре-
зиновых трубок, надеваемых на гофриро-
ванные отростки 5 и 6 стеклянного мано-
метра и сосуда. Через отросток 7 в сосуд 3
помещали точную навеску ннтроэфира
(около 1 г), после чего этот отросток за-
паивали. Нитроэфир, находящийся в тон-
ком слое, откачивали не менее 6 час. при
давлении ]0“4—10-5 мм рт. ст. до полного
удаления летучих примесей, о чем судили
по отсутствию повышения давления в приборе после отключения
насосов. Затем нитроэфир переливали во внутреннюю полость
стеклянного манометра. Избыток нитроэфира при легком нагреве
струей водяного пара перегоняли из перетяжки обратно в сосуд 3.
Перетяжку после полной ее очистки от следов нитроглицерина бы-
стро перепаивали и прибор отсоединяли от вакуумной установки.
О количестве нитроэфира в приборе судили по разности первона-
чальной навески и навески, оставшейся после перепаивания пере-
тяжки 2 в сосуде 3. Подготовленный прибор соединяли отводом 5
с ртутным манометром, помещали в жидкостной термостат и изме-
ряли рост давления газообразных продуктов распада во времени.
Перед измерениями оказалось необходимым перемешивать содер-
жимое прибора многократной перегонкой пузырька газа из мано-
метра / в нижнюю часть реакционного сосуда и обратно, так как
в противном случае давление, особенно в начале опыта или почти
191
не растет или растет неравномерно, что обусловлено, по-видимому,
большой толщиной слоя нитроэфира, в котором диффузия газов
идет медленно.
Разложение нитроглицерина при степени заполнения (б), близ-
кой к единице, исследовали при 40—100°.
Распад и в этих условиях характеризуется наличием двух макро-
скопических стадий, подобно тому, как это наблюдалось при более
высокой температуре и значительно меньших степенях заполнения
реакционного сосуда.
На фиг. 2 приведены кривые изменения объема газообразных
продуктов распада во времени для параллельных опытов при б=1
время т мин tO
Фиг. 2. Изменение газообразования во времени при разложении ни-
троглицерина при б=И и различных температурах.
и температуре 60, 80 и 100° L. Как видно из приведенных графиков,
в начале распада газообразование, особенно при более низкой тем-
пературе, долгое время протекает медленно. После индукционного
периода газообразование начинает резко расти, достигая величин,
превосходящих начальную скорость его в сотни раз.
При детальном рассмотрении полученных результатов оказы-
вается, что аналогия с опытами при малых б ограничивается во
многом лишь качественной стороной.
На фиг. 3 дана в координатах 1g — —— зависимость началь-
ной скорости газообразования от температуры, характеризующая
распад нитроглицерина на первой стадии. Эта зависимость выра-
жается прямой линией, однако, тангенс угла наклона ее значитель-
но меньше, чем в опытах при повышенной температуре с малыми б2,
соответственно энергия активации в наших опытах составляет всего
лишь 29, вместо 40 ккал/моль для случая малых степеней заполне
ния реакционного сосуда. Иначе говоря, температурный коэффи-
1 В опытах при 40°, длительность которых доходила до 8 мес., резкое уско-
рение не было достигнуто.
г Результаты исследования распада нитроглицерина в жидкой фазе при малых
степенях заполнения и более высоких температурах приведены в предыдущей
статье настоящего сборника, .«^3*
192
может
о
-1
-<?г
v-6
образования на первой
Lg-p/wwpT ст/
фиг. 4. Влияние давления продуктов рас-
пада на скорость газообразования при раз-
ложении нитроглицерина при различных
температурах.
. .______ . in3
L 2,5 2.7 2.9 3,1 3.3
/Фиг. 3. Влияние температуры
|^иа начальную скорость газо-
образования при распаде нитро-
i; глицерина при 40—ЮСР.
ент начальной скорости газообразования при распаде нитрогли-
рина в условиях наших опытов значительно меньше того, который
ожно получить, экстраполируя, например, результаты Робертсона
], а также наши данные для малых б на низкие температуры. Отно-
ение скоростей при 60 и 40°, полученное экстраполяцией, состав-
ет около 50—60, в то время как эксперимент показывает вели-
ину около 16.
Сравнительно малый температурный коэффициент скорости газо-
стадии разложения
100°
1!
быть обусловлен изменением «удельного веса» отдельных реакций в
/газообразовании и, в частности, увеличением роли гидролитических
/и окислительных реакций, скорость которых, по-видимому, меньше
К зависит от температуры, чем скорость термического распада. Этому
И способствует также большее накопление продуктов распада в нит-
Ероэфире вследствие высокой степени заполнения реакционного со-
^суда.
fiS" По достижении определенного критического давления продуктов
^ разложения наступает стадия резкого ускорения, аналогично рас-
& паду при более высокой температуре. На фиг, 4 представлена зави-
Ж сим ость скорости изменения давления от давления продуктов рас-
Ж пада при 60, 80 и 100°. Из приведенных графиков видно, что скорость
газообразования на участке резкого ускорения зависит от темпера-
туры значительно слабее, чем на первой стадии. Если энергия ак-
./ тиваиии, рассчитанная по начальным скоростям газообразования,
составляет 29 ккал/люль, то энергия активации, соответствующая
Второй стадии и рассчитанная по скоростям при одинаковом давле-
; . 13 451 !93
к “
нии газообразных продуктов значительно меньше и составляет
15 ккал!моль.
Скорость газообразования на второй стадии пропорциональна
приблизительно второй степени давления продуктов разложения.
Наличие этой зависимости как при малых б и повышенных темпе-
ратурах, так и при больших б и относительно низких температурах
позволяет предположить о сходстве процессов, протекающих на
стадии резкого ускорения в широком диапазоне температур
(60—140*5-
Величина критического давления зависит от температуры, новы
шаясь с последней. Так при 60, 80 и 100° критическое давление со
ставляет соответственно около 250, 500 и 1000 мм рт. ст. Сопостав-
ление этих величин с критическими давлениями, полученными при
малых степенях заполнения показывает, что с увеличением б от 0,]
до 6 = 1 критическое давление увеличивается в 5—7 раз (при малых
б оно составляет для 80° около 70 и для ЮСГ около 200 мм рт. ст.).
Значительный рост критического давления при увеличении сте-
пени заполнения до максимальной ее величины может быть объяс-
нен тем, что на более ранних стадиях распада образуются преиму-
щественно менее активные продукты разложения, в результате чего
их давление над нитроэфиром, необходимое для перевода разложе-
ния во вторую стадию, должно быть выше. Однако можно показать,
что при некоторых допущениях степень заполнения реакционного
объема должна влиять на величину критического давления при не-
изменном составе продуктов разложения. Если допустить, что про-
дукты распада состоят из газа, ускоряющего распад и сравнительно
хорошо растворимого в нитроэфире, и газов, не участвующих в рас-
паде и к тому же плохо растворимых, то критическим давлением
окажется то общее давление продуктов распада, при котором дав-
ление ускоряющего газа достигает определенного н постоянного
значения для данной температуры (не зависящего от б).
Если принять указанные допущения, то зависимость критическо-
го давления от величины б будет выражаться уравнением
Ркпнт = Л 4" ---,
где А и В—постоянные коэффициенты, зависящие только от тем
пературы, причем
Рассмотрение этого выражения показывает, что при малых сте-
пенях заполнения (до 6 = 0,3) критическое давление Остается прак-
тически постоянным и лишь при б>0,3 с ростом последней оно за-
метно повышается, что в общем согласуется с экспериментом.
Полученные данные о распаде нитроглицерина позволили оцени-
нить влияние температуры на величину индукционного периода, за
продолжительность которого принимали время до выделения опре-
деленного количества газов, соответствующего давлению около
1000 мм рт. ст. При этом давлении распад НГЦ характеризуется,
независимо от температуры опыта, уже очень высокой скоростью
194
газообразования и большим содержанием в продуктах разложения
кислых веществ, количество которых по титрованию щелочью вод-
ной вытяжки соответствует концентрации их в нитроглицерине (в
пересчете на азотную кислоту) порядка процентов.
Фиг. 5 иллюстрирует влияние температуры на продолжитель-
ность индукционного периода для опытов с различными степенями
заполнения реакционного сосуда (как с максимальной, так и с мень-
шими значениями 6) в широком диапазоне температур (60—140°).
Фнг. 5. Влияние температуры на индукцион-
ный период распада нитроглицерина при
различных степенях заполнения реакцион-
ного сосуда,
Г--0-1, 2-8«0,03. .3-8 * 0,006,
Эта зависимость для изученных б описывается уравнением т — Ае ЙГ
и в координатах Igr-—выражается прямыми. Прямая 1 показы-
вает зависимость индукционного периода от температуры для рас-
пада нитроглицерина при максимальной степени заполнения реак-
ционного сосуда в интервале 100—60°. Величина Е, найденная пр
тангенсу угла наклона этой прямой, составляет 26 ккал/моль. Она
близка к величине энергии активации, подсчитанной по начальным
скоростям газообразования,
Величина Е, полученная из опытов с 6=^0,03 в диапазоне тем-
пературы 80—140° (прямая 2) составляет 28 ккал/моль и с 6=0,006
при 100—140° 32 ккал/моль (прямая 3).
Приведенные данные показывают, что на температурную зави-
симость индукционного периода при распаде нитроглицерина в при-
сутствии продуктов разложения влияет степень заполнения реакци,-
онного сосуда, а также, по-видимому, и температурный интервал,
носкольку с повышением б и понижением температуры Е умень;
шается с 32 до 26 ккал/моль.
Полученные результаты позволяют оценить поведение нитрогли-
церина в условиях его хранения. Экстраполяция показывает, что
13*
195
продолжительность индукционного периода распада нитроглицери-
на при 20“ в условиях, когда продукты разложения практически пол-
ностью находятся в нитроэфире, составляет всего 17 лет. Однако,
если продукты распада имеют возможность в той или иной степени
•удаляться из конденсированной фазы, то индукционный период ста-
новится соответственно больше. При 6^0,03 индукционный период
для 20° составляет уже около 100 лет. Дальнейшее уменьшение сте-
пени заполнения реакционного сосуда до 0,006 увеличивает индук-
ционный период до 2200 лет. При сравнении этих величин следует
учесть, что за время индукционного периода достигается различная
степень распада в зависимости от величины б. Если принять во всех
случаях количество газообразных продуктов при полном распаде
нитроглицерина цо = 6ОО см^г, то при б=«1 степень распада составит
около 0,2% при б— 0,03 около 3% и при 6=^0,006 около 17%. Не-
обходимо указать, что такая оценка степени распада по крайней
мере для опытов с максимальной б является весьма условной, вви-
ду значительного содержания кислых продуктов в нитроглице
рине на стадии резкого ускорения, о чем уже упоминалось выше.
С величинами индукционного периода, полученными для 20° в
присутствии продуктов распада, можно сопоставить результаты
экстраполяции данных Робертсона [5] для разложения нитроглице-
рина в условиях принудительного удаления газообразных продуктов
распада током COj. Оказывается, что в этих условиях степень рас-
пада 0,2%, соответствующая индукционному периоду при 6=0.
должна достигаться за 3000 лет; степень распада 3%, соответст-
вующая индукционному периоду при 6^0,03, —за 50 000 лет и, на-
конец, степень распада 17%, соответствующая индукционному пе-
риоду при 6^0,006,— за 300 000 лет. Указанные величины несрав-
ненно больше тех, которые вытекают из результатов опытов по
распаду нитроглицерина в присутствии продуктов разложения.
выводы
1. Изучено разложение нитроглицерина при низких температу-
рах (40—100°).
2. Показано, что разложение нитроглицерина при этих темпера-
турах протекает качественно сходно с распадом нитроглицерина
при более высоких температурах (80—140s). Это сходство заклю-
чается в наличии Двух макроскопических стадий, критического дав-
ления и приближенной пропорциональности скорости газообразо-
вания иа второй стадии квадрату давления продуктов распада.
3. Показано также, что наряду с известным сходством имеется
количественное различие в распаде нитроглицерина при низких и
при высоких температурах. Температурная зависимость начальной
скорости газообразования при низких температурах выражена сла-
бее. На второй стадии разложения критический инкремент скорости
1 Для расчета количества газообразных продуктов в этом случае за объем,
занимаемый ими, принимали весь объем реакционного сосуда.
193
газообразования составляет около 15 ккал [моль в отличие от высо-
ких температур, при которых скорость газообразования на второй
стадии от температуры практически не зависит.
4. Произведена оценка величины индукционного периода рас-
пада нитроглицерина при 20°. Полученные при этом результаты
- сопоставлены с влиянием степени заполнения реакционного сосуда
ва возможность удаления газообразных продуктов разложения из
нитроэфира.
5. Особенности разложения нитроглицерина при низких темпе-
ратурах рассмотрены в предположении о повышении на первой ста-
дии распада «удельного веса» реакций гидролитического и окисли-
тельного характера с относительно малым температурным коэффи-
циентом скорости, преобладающих на второй стадии.
6. Разработана методика изучения термического распада взрыв-
чатых веществ при максимальной степени заполнения реакцион-
ного сосуда, позволяющая расширить исследуемый диапазон тем-
пературы в область более низких температур.
Авторы выражают благодарность К. К. Андрееву за существен
ные указания при обсуждении работы и формулировании ее ре-
зультатов.
ЛИТЕРАТУРА
1. Андреев К- К., Глазкова А. П., М а у р и н а Н. Д., Светлов Б. С,.,
ЖФХ. 1958. 32, 1726.
2. А н д р е е в К. К.. Г л а з к о в а А. П.. ДАН СССР. 1955. 105, 286,
3. Андреев К- К.. Самсовов Б. С.. ДАН СССР, 1957, 114, 815.
4. А н Д р е е в К. К., ЖПХ, 1958, 31, 484—493.
5. Robertson R,,J. chem. Soc.. 1909, 95, 1241
В. В. ГОРБУНОВ, Б. С. СВЕТЛОВ
13. ВЛИЯНИЕ ВОДЫ И КИСЛОТЫ
НА САМОУСКОРЯЮЩИИСЯ РАСПАД НИТРОГЛИЦЕРИНА
Термическое разложение нитроглицерина в жидкой фазе в при-
сутствии продуктов распада представляет собой процесс, протека-
ющий двустадийно [3], [5]. Первая стадия — индукционный период —
характеризуется сравнительно малой и медленно растущей скоро-
стью разложения, вторая же стадия протекает с большим самоуско-
рением. На продолжительность индукционного периода влияют ус
ловия проведения опыта. Различные примеси, в частности вода,
кислота, двуокись азота, ускоряют разложение, сокращая этот
период. 1* -
Особенно сильно влияет вода и еще в большей степени вода с
кислотой или с двуокисью азота, образующей при взаимо-
197
действии с водой кислоту [I]. Это позволило сделать предположе-
ние о ведущей роли гидролитических процессов, протекающих при
разложении нитроглицерина как в присутствии воды и кислоты,
так и при самоускоряющемся распаде нитроэфира без добавок [5].
Опыты, проведенные в свое время для изучения влияния воды
и кислоты на разложение нитроглицерина, не дают количественных
представлений о роли концентрации примесей и температуры экс-
перимента, так как результаты этих опытов были получены по мано-
метрической методике или методом вспышки в ампулах (4] при ма-
лом отношении объема нитроэфира к объему ампулы. Поэтому зна-
чительная и иногда неопределенная часть примесей и продуктов
распада находилась в газовой фазе и количественная оценка их
состава и роли в процессе разложения нитроэфира могла быть про-
изведена лишь с учетом процессов диффузии и растворения, невы-
полнимым в условиях проведенных опытов, кроме того указанные
опыты проводились при относительно высокой температуре (в ос-
новном при 100° и выше) в узком ее интервале, что само по себе
затрудняло экстраполяцию результатов па более низкие темпера-
туры, интересующие практиков.
В связи с изложенным нами были поставлены опыты с приме-
нением манометрического метода при практически полном запол-
нении объема ампулы нитроэфиром, что позволило проследить раз-
ложение нитроглицерина в присутствии воды и азотной кислоты
(как раздельно, так и совместно) в интервале 40—100°.
Дозировку воды и кислоты осуществляли вымораживанием оп-
ределенных количеств их паров в реакционный сосуд с нитроэфи-
ром, предварительно освобожденным от летучих веществ путем
откачки. Точность дозировки, проверенная по «холостым» опытам
(без нитроглицерина), составляла ±3%.
Опыты по разложению нитроглицерина при 100° и максималь-
ной степени заполнения сосуда б показали (фиг. 1), что в присут-
ствии 0,2% вес. воды ускорение распада наступает значительно
быстрее (приблизительно в 1,5 раза), чем в присутствии того же
количества азотной кислоты, и в 2,5 раза быстрее, чем индивиду-
ального нитроэфира.
Аналогичный результат получен и при меныцей д (0,03) в при-
сутствии 0,5% вес. воды. В этом случае ускорение наступает и
3 раза быстрее, чем в опытах с таким же количеством азотной кис-
лоты. Следует отметить, что кривая давление—время в случае раз-
ложения нитроглицерина в присутствии воды прн д=г;1 имеет тот
же характер, что и при малых д, а именно: период постоянства дав-
ления, временное уменьшение его (правда небольшое) и последую-
щее резкое ускорение газообразования. При этом время до начала
уменьшения давления несколько меныпе того, которое наблюдается
при малых д и равном давлении паров добавленной воды.
Обращает на себя внимание насыщающийся в начале характер
кривой давление—время в опыте с азотной кислотой. По-виднмому,
на распад нитроглицерина в этот период накладывается разложе-
ние самой азотной кислоты.
193
На фиг. 2 и 3 приведены кривые разложения нитроглицерина в
присутствии воды и азотной кислоты при 60 и 40°. Как видно из
графиков, при 60° и определенном содержании воды распад нятро-
Фиг. 1. Влияние воды и азотной кислоты на разложение нитрогли-
церина при 100".
(Для НГЦ, без добавок приведены кривые двух, параллельных опытов).
глицерина сопровождается характерной впадиной на кривой p=f(r).
Распад в присутствии 100%-ной азотной кислоты протекает по
обычной для нитроглицерина кривой. Сначала давление растет
Фиг. 2. Влияние воды и
азотной кислоты на раз-
ложение нитроглицерина
при 60°.
1—0 14% вес. [00%-ной НМОз. 2—0,42% вес. 100%-ной
HNO3, 5—ОЛ8ЙН.О, 4—0,42%Н2О. 5—без добавок.
медленно, затем по истечении индукционного периода газообразо
вание резко ускоряется.
199
Опыты показали, что добавление к нитроглицерину води или
азотной кислоты значительно сокращает индукционный период пе,
сравнению с распадом сухого чистого нитроглицерина не тольк<
при повышенных, но и при низких температурах. Однако в послед-
нем случае азотная кислота по своему ускоряющему действию зна
чительно превосходит воду. При 60° индукционный период для чис-
того нитроглицерина составляет около 600 час., с добавкой 0,2 и
0,4% вес. воды — соответственно 230 и 160 час. В присутствии же
0,14% вес. азотной кислоты резкое ускорение наступает через 150.
Фиг. 3. Влияние воды и азотной кислоты на разложение нитроглицерина
при 40°.
J—0.16И вес. НЮ. 2- 0,34 ft. ЩО. 2-0, |7Я вес. 10044-ной HNOa.
а В опыте с 0,45% вес. кислоты^—через 120 час. Таким образом,
при 60° вода (0,2% вес.) сокращает время индукционного периода
распада нитроглицерина приблизительно в 2,5 раза, тогда как азот-
ная кислота даже в меньшем количестве (0,14% вес.) уменьшает
это время в 4 раза. Отношение продолжительности индукционного
периода при распаде данного нитроэфира в присутствии воды к
продолжительности этого периода для продукта, содержащего кис-
лоту, составляет около 1,5.
При 40° азотная кислота влияет еще сильнее, чем при 60°. В свя-
зи с тем, что опыты с безводным нитроглицерином при 40° не были
доведены до резкого ускорения распада вследствие их большой дли-
тельности, прямые данные по влиянию воды на индукционный пе-
риод в этих условиях отсутствуют. Косвенный метод, основанный
на экстраполяции температурной зависимости продолжительности
индукционного периода безводного продукта со 100, 80, 60” на 40 '
дает величину около 1 года. Отношение этой величины к времени
200
индукционного периода распада влажного нитроглицерина состав-
ляет около 2, что близко к отношению при 60°,
Опыты показывают, что индукционный период при 40° в при-
сутствии 0,17% вес, кислоты составляет 1300 час., а при 0,16% вес,
воды он равен 4600 час., т. е, кислота сокращает время до начала
резкого ускорения распада нитроглицерина по сравнению с водой
в 3,5 раза, тогда как при 60° этот период сокращался только в
1,5 раза.
Таким образом, с понижением температуры ускоряющее влияние
азотной кислоты на распад нитроглицерина растет, влияние же
воды остается, по-видимому, постоянным. Ускоряющее действие
кислоты проявляется с самого начала опыта. Начальные скорости
газообразования’ в случае добавления в нитроглицерин азотной
кислоты повышаются при 40 и 60° в 5—10 раз в зависимости от
количества добавленной кислоты, В опытах же с водой давление на
значительном участке распада практически не растет. В экспери-
ментах с умеренно разбавленной кислотой начальная скорость рас-
пада ниже, чем в случае концентрированной, но все же выше, чем
для индивидуального нитроглицерина. В присутствии разбавлен-
ной кислоты (до 20% HNO3) давление в начале долгое время или
совсем не растет (как в случае опытов с водой), или растет со ско-
ростью, меньшей, чем у одного нитроглицерина.
Увеличение начальной скорости газообразования в присутствии
крепкой азотной кислоты, сопровождаемое резким сокращением ин-
дукционного периода, обусловлено, по-видимому, развитием окис-
лительных процессов, удельное значение которых тем больше, чем
ниже температура и крепче кислота. Окислительные реакции при-
водят к образованию плохо конденсирующихся продуктов, а также
воды, участвующей в гидролитическом разложении нитроглицери-
на. В случае разбавленной кислоты, окисляющее действие которой
не так велико, протекает в основном гидролиз с образованием легко
растворимой в нитроэфире азотной кислоты. В этом случае давле-
ние растет слабо (когда удельный вес окислительных реакций еще
значителен) или даже совсем не растет,
В присутствии одной воды давление продолжительное время
остается постоянным в силу тех же причин, что и при высокой тем-
пературе и малых степенях заполнения реакционного сосуда. Гид-
ролиз нитроглицерина развивается очень медленно из-за нейтраль-
ности среды. Когда в результате распада и гидролиза нитроэфира
накапливается некоторое количество кислоты, гидролиз сильно
ускоряется вследствие резко выраженной автокаталитичностц
этого процесса, В известных условиях наблюдается даже снижение
давления, характерное для опытов с водой.
Таким образом, эксперименты показали, что общие закономер-
ности распада нитроглицерина при повышенных температурах в
присутствии воды и кислоты сохраняются и при низкой темпера -
1 Эту скорость рассчитывали как среднюю на участке распада от его начала
до выделения количества газов, соответствующих 50 мм рт. ст. сверх равновесного
давления добавки.
201
Фнг. 4. Влияние количества воды на разло-
жение нитроглицерина при 60=.
Числа у кривых — содержание воды в нитрогли-
церине в нес.
туре. Однако в этих условиях влияние указанных веществ прояв-
ляется количественно иначе. -
Описанные выше закономерности были установлены по резуль-
татам опытов, в которых к нитроглицерину добавлялась вода или
азотная кислота в основном в количестве около 0,2% вес.
С целью выявления влияния количества воды на распад нитро-
глицерина были проведены соответствующие опыты при 60° [.
На фиг. 4 даны кривые давление—время для распада нитрогли-
церина в присутствии различных количеств воды. Добавление к
нитроглицерину даже 0,05%
вес. воды заметно (прибли-
зительно на 30%) сокращает
индукционный период. Уве-
личение содержания воды
приводит к дальнейшему со-
кращению Этого периода.
Так, добавление 0,10% вес.
волы сокращает время до
начала резкого ускорения
распада нитроэфира на
40%, 0,18% вес, воды — на
60% и 0,42% вес. воды—
на 70%. Эти данные пока-
зывают, что с повышением
содержания воды, относи-
тельное ее влияние на вели-
чину индукционного перио-
да, по-видимому, уменьшает-
ся, Давление в отличие от
опытов с безводным нитро-
глицерином в начале или
растет очень медленно (ма-
лое содержание воды), или
остается постоянным на про-
тяжении всего индукционного периода. При добавлении 0,42%
воды кривая приобретает обычный для распада влажного нитро-
глицерина вид с характерной впадиной.
При 40° (см, фиг. 3) также наблюдается относительное умень-
шение влияния количества воды. Так, увеличение содержания воды
вдвое (с 0,16 до 0,34%) приводит к снижению индукционного пе-
риода с 4600 до 4000 час,, т, е. приблизительно на 10%. Это обус-
ловлено тем, что вода наряду с активной функцией (гидролиз) иг-
рает и роль разбавителя образующейся кислоты, что может приве-
сти к торможению как гидролитической, так и окислительной ре-
акций.
Влияние количества кислоты на распад нитроглицерина изу-
чали с применением HNOa различной концентрации. При 60° “уве-
Эти опыты были выполнены М. С. Плясуновым.
202
Фит. 5.
80%-ной
жение
Числа у
данной
Влияние количества 70 —
азотной кислоты на разло-
нитроглицерина при №.
кривых — содержание кислоты
концентрации в нитроглицерине
в вес.
Кличек не содержания 100%-ной азотной кислоты в нитроглицерине
В втрое (с 0,14 до 0,45% вес.) сократило индукционный период со
К 150 до 120 час., т, е. всего на 25%.
К Влияние количества 70—80%-ной азотной кислоты на распад
Е нитроглицерина показано на фиг. 5, Здесь увеличение содержания
Е кислоты указанной концентрации приблизительно в 4 раза сокра-
Е щает индукционный период на
Г 70%.
к Сходное влияние оказывает
| изменение содержания кислоты
Е. и при 40°. Повышение количества
& 70—80 %-ной азотной кислоты
I с 0,18 до 0,57% вес. сократило
г. время индукционного периода
к V 1200 до 400 час, (фиг. 6). Даль-
Ь нейшее увеличение количества
| кислоты до 2,5% вес, сократило
t указанное время лишь до 300 час.
г В этом можно видеть некоторое
Г сходство влияния количества до-
s.' бавленных к нитроглицерину ве-
в ществ, таких как вода или кис-
1} лота: относительное сокращение
н индукционного периода происхо-
| дит при малых количествах их
‘ резче, чем при больших.
1' Особо следует отметить тот факт, что в случае разбавленной
кислоты, ускоряющее влияние которой, как будет отмечено ниже,
проявляется наиболее сильно, можно наблюдать тормозящее
, влияние(
:т"ч наиболее сильно,
ее, если она взята в количестве, превышающем необходи-
мое для насыщения нитро-
эфира. На фиг. 7 представ-
лены результаты опытов по
разложению нитроглицери-
на в присутствии 22—
28%-ной азотной кислоты,
причем в одном из них этой
кислоты было 0,19% вес.
(полностью растворенной),
в другом же 1,9% вес., так
что часть ее не растворя-
лась в нитроэфире и нахо-
дилась при температуре
опыта в виде взвеси. В пер-
вом случае индукционный период составил 80 час., в то время как
во втором, в присутствии в десять раз большего количества кис-
лоты, время до резкого ускорения распада составило 280 час.
и было значительно больше индукционного периода в опыте
с 0,18% вес. воды. Резкое ускорение разложения наступило в этом
1000г
t fl 500
'U &_
'Ч
*э £
a 5
*
о,п
0,6?Чо
566 1000 -
время т час
Влияние количества 70—80%-ноii
кислоты на разложение нитрогли-
церина прн 4(Г.
кривых — содержание кислоты длиной
Фиг. 6.
азотной
Числа у
концентрации в нитроглицерине в вес.
203
случае лишь после того, как вся кислота растворилась в нитро-
эфире, о чем судили по исчезновению мути, При этом перед уско-
рением распада наблюдалась некоторое уменьшение давления
Установленное здесь тормозящее действие связано, по-видимому,
с тем, что нераствореиная вода, экстрагируя часть кислоты, сни-
жала тем самым содержание ее в нитроглицерине, пока она нс
вступала в реакцию гидролиза или не растворялась из-за повы-
шения общей кислотности.
Фиг, 7, Разложение нитроглицерина при 6Q'
в присутствии растворенной н частично не-
раствореннпй в нем 22—28%-ной азотной
кислоты и растворенной воды,
/—1,9*/» вес. частично нерасгвсреикой HNOa,
2—вес. растворенной водыч J—04[9V* вес,
растворенной НЫОэ, 4—чистый Ентроглацернн.
Фиг, 8, Влияние температуры на
продолжительность индукционного
периода при разложении нитрогли-
церина,
/—чистый нитроглицерин, 2—0,2*/* вес.
воды 5—0,2"/. вес. lW.-ной HNO3,
4—0,2*/. 20е/.-но Л НЬ'ОЭ1
Результаты опытов с добавлением азотной кислоты и воды, про-
веденные в широком температурном диапазоне (100—40°), позво-
лили экстраполяцией оценить влияние этих веществ на индукцион-
ный период при хранении нитроглицерина в нормальных условиях
(20°), когда практически все продукты, как добавленные в нитро-
эфир, так и образовавшиеся при распаде его, находятся в конден-
сированной фазе.
Влияние температуры на длительность индукционного периода
может быть изображено в координатах 1gт——-— прямыми ли-
ниями, На фиг. 8 эта зависимость представлена для опытов с 0,2%
100%-ной азотной кислоты (прямая <?), для опытов с 0,2% водь/
(прямая 2) для чистого нитроглицерина (прямая /) и для опытов
с 0,2% разбавленной HNQs (прямая 4). Экстраполяция показывает,
что при 20° индукционный период распада влажного нитроглице-
рина составляет 10 лет, кислого — 1,4 года, с добавлением разбав-
ленной азотной кислоты —3 месяца, безводного и нейтрального
нитроэфира — 17 лет.
204
Таким образом, эксперименты показывают, что вода и азотнан
кислота сильно влияют на течение распада нитроглицерина в ста-
дии индукционного периода, значительно сокращая его, В связи
с этим представлялось интересным выяснить, оказывают ли эти ве-
щества влияние на закономерности разложения нитроглицерина
в стадии резкого ускорения. Было показано [5], что скорость газооб-
разования при распаде безводного чистого нитроглицерина после
Фиг. 10, Влияние давления про-
дуктов распада на скорость газо-
образования при разложении ни-
троглицерина з присутствии воды
или азотной кислоты различной
концентрации при 40°,
0—0,17% вес. 100%-вой HNOS; к— 0,1814
вес, 75,4%-вой HNO3; д —40,28*4 вес,
55,6%-и с.й HNO,-,* -0,22% вес. 24,8%-мой
HNO3; ф —0,[б!4 вес, НаО.
Фиг. 9. Влияние давления продуктов рас-
пада на скорость газообразования при раз-
ложении нитроглицерина в присутствии во-
ды или азотной кислоты различной концен-
трации ври 60°,
0—0,14?! вес. 100%-вой HNO,; X—0,45% вес.
ЮО^-юй HNO3; 4—0,27% вес, S3,5%-ной HNOa;
д — 0,23% вес. 77%-вой HNO,; •- 0,20% вес,
41,54-вой HNO,' а-О.ЫЧ вес. 22,4%-ной НЫО3;
=0,1% вес, НгО;
* —чистый нитроглицерин.
начала резкого ускорения, независимо от степени заполнения со-
суда, пропорциональна приблизительно квадрату давления продук-
тов разложения. Эта закономерность сохраняется и в случае рас-
пада в присутствии воды и азотной кислоты различной концентра-
ции при 40 и 60°, На фиг, 9 и 10 приведены графики зависимости
скорости газообразования от давления продуктов распада в лога-
рифмических координатах для опытов с водой и азотной кислотой
различной концентрации. Как видно из фиг. 9, при 60° полученные
точки могут быть описаны в общем прямой линией, лежащей выше
прямой для индивидуального нитроглицерина. Это говорит о том,
что при одном и том же давлении продуктов распада в опытах в
присутствии воды или азотной кислоты скорость газообразования
выше, чем в опыте с индивидуальным нитроэфиром. Тангенс угла
205
наклона для обеих прямых составляет около 1,9. Поскольку при 40
опыты с безводным веществом не были доведены до резкого уско-
рения, такого сравнения в этом случае провести нельзя. Можно
лишь указать на то, что данные опытов, разбитые на две группы
изображаются (см. фиг. 10) также прямыми линиями с тангенсом
угла наклона их около 1,7. Верхняя прямая соответствует опытам
с водой и с разбавленной азотной кислотой, а нижняя — опытам со
100%-ной HNO3. Следовательно, в опытах с водой и разбавленной
кислотой скорость газообразования выше, чем в эксперимента\
с концентрированной кислотой при одном и том же давлении про
дуктов разложения. При рассмотрении результатов исследования
при 60° также видно, правда, не так отчетливо, что точки, соответ-
ствующие опытам с водой и разбавленной кислотой лежат выше
точек опытов с концентрированной HNO3,
Таким образом, приведенные данные позволяют сделать вывод
о том, что роль воды и азотной кислоты различной концентрацц’|
при добавлении их к нитроглицерину ограничивается в основном
ускорением тех процессов, которые подготавливают наступление
стадии резкого ускорения распада нитроэфира. На самой этой ста-
дии они не изменяют зависимость скорости газообразования от дав
ления продуктов разложения, увеличивая лишь несколько абсолют-
ную скорость газовыделения.
Гипотеза о гидролитической природе взаимодействия нитрогли-
церина с водой при условии каталитического действия образую-
щейся при этом азотной кислоты [1] естественно предполагает нали-
чие некоторого оптимального соотношения кислоты и воды, прн
котором гидролитическая функция последней проявляется наиболее
резко. С другой стороны, окислительное действие азотной кислоты
в суммарном процессе распада нитроглицерина также должно за-
висеть от ее концентрации.
Проведенные ранее опыты показали, что при добавлении соп-
местно воды и кислоты распад нитроглицерина протекает значи-
тельно быстрее, чем в присутствии их в отдельности. Интересно бы-
ло оценить оптимальное соотношение между водой и кислотой в оп-
ределенных условиях. На реальность такого соотношения указы-
вают опыты Мюраура [6], по данным которого нитроклетчатка наи-
более быстро разлагается в присутствии 22%-цой азотной кислоты,
а также опыты Самсонова [2], проведенные при 100’ в несколько
неопределенных условиях и дающие оптимальную концентрацию
азотной кислоты для распада нитроклетчатки 10%.
В наших опытах был изучен распад нитроглицерина в присут-
ствии около 0,2% вес. HNO3 различной концентрации при измене
нии последней в диапазоне 100—0%. В качестве характеристики
действия добавленных к нитроэфиру вещёств мы приняли отноше-
ние длительности индукционного периода, соответствующего вре-
мени выделения 0,8—1 cxt3/a газообразных продуктов к длительно-
сти индукционного периода в опыте с 0,2% вес. воды. На фиг. 11
приведены результаты соответствующих опытов, проведенных при
60 и 40“. Как видно из этого графика, незначительное разбавление
206
на продолжительность индукционного
периода распада нитцоглицернна при
40 и 60°.
водой азотной кислоты до содержания последней около 80% прак-
тически не влияет на продолжительность индукционного периода
при 60°. Дальнейшее разбавление резко сокращает индукционный
период и при введении в нитроглицерин около 0,2 % вес.
25—40%-ной азотной кислоты он снижается до величины, вдвое
меньшей, чем при концентрированной кислоте и втрое меньшей, чем
в случае воды. Еще большее разбавление влечет за собой повыше-
ние продолжительности индукци-
онного периода.
Аналогичная картина наблю-
дается и при 40°. Оптимальная
концентрация азотной кислоты
в этом случае — 25%. При этой
концентрации HNO3 индукцион-
ный период распада нитроглице-
рина в 3 раза мсцыпе, чем в слу-
чае 100%-ной азотной кислоты и
в 10 раз меньше, чем в случае
воды, добавляемых к нитроэфиру
в количестве около 0,2% вес,
При рассмотрении этих фактов
следует учитывать, что азотная
кислота не только играет роль
катализатора гидролиза, но так-
же является и окислителем. По-
этому оптимальная концентрация
HNO3, вносимой в нитроглицерин, является некоторой вели-
чиной, суммарно отражающей составы ее, оптимальные для
каждого из отдельных, протекающих в нитроэфире процес-
сов.
Таким образом, проведенные опыты показали, что вода и азот-
ная кислота проявляют свое ускоряющее действие на распад нитро-
глицерина, приводя к сокращению индукционного периода во всем
изученном диапазоне температур (40—100°). Однако, если вода
влияет на распад при всех температурах приблизительно в одинаг
ковой степени, то влияние азотной кислоты резко повышается с по-
нижением температуры.
При распаде нитроглицерина протекают сложные реакции тер-
мического и гидролитического разложения с участием добавленной
и образующейся при этих процессах воды, Гидролиз катализируется
кислотой. Продукты распада окисляются образующимися при гид-
ролизе азотной кислотой и окислами азота иди добавленной азот-
ной кислотой. Вследствие сложности совокупности этих реакций за-
труднительно определенно объяснить полученные закономерности.
Положение осложняется еще и тем, что вода выполняет не только
функцию участника гидролиза, но, разбавляя кислоту, она тем са-
мым изменяет ее окислительные свойства. С другой стороны, азот-
ная кислота является нс только катализатором гидролиза и участ-
ником окислительных реакций, в результате которых образуется
?07
вода, гидролизующая нитроэфир, ио она способна еамопроизволь-
но разлагаться с образованием воды и окислов азота,
К рассмотрению полученных в настоящей работе результатов
следует подходить, учитывая изменение «удельного веса» отдель-
ных реакций в общем протекании распада нитроглицерина, а так-
же температурную зависимость каждой реакции.
ВЫВОДЫ
I. Изучено термическое разложение нитроглицерина в присут-
ствии воды, а также азотной кислоты различной концентрации при
40—100° в условиях, когда добавленные к нитроэфиру вещества и
продукты распада практически полностью в нем растворены,
2, Показано, что вода сокращает индукционный период распа-
да нитроглицерина приблизительно в равной степени во всем изу-
ченном температурном диапазоне, тогда как азотная кислота сла-
бее, чем вода влияет на распад при высокой температуре и зна-
чительно сильнее, чем вода при низкой температуре.
3. С помощью экстраполяции оценено время индукционного пе-
риода при 20° для влажного и кислого нитроглицерина.
4. Показано, что при 40 и 60° наиболее сильно сокращает индук-
ционный период разложения нитроглицерина 25—40%-ная азотная
кислота.
5. Показано, что вода и азотная кислота различной концентра-
ции, введенные в нитроглицерин, влияют в основном на процессы,
сокращающие индукционный период. Стадия резкого ускорения
распада в опытах с добавлением воды и HNO3 показывает те же
закономерности, что и чистый нитроглицерин.
ЛИТЕРАТУРА
1. А н д р е е в К. К. и Г л а з к о в а А. П.. ДАН СССР. 1955, 105, 286.
2. Андреев К. К.. Самсонов Б. С., ДАН СССР, 1957 1 [4 815,
3. А н д р е е в К. К., ЖПХ, 1958, 31, 484—493.
4. Л у к и н А. Я.. ЖФХ, 1932, 3, 406.
5. Светлов Б, С., «Кинетика и катализ», АН СССР, 1961, 2, 38.
6, Muraour Н., Bull, Soc. chim. France, 1931, 49, 276; 1932, 51. 1094.
Б. С. СВЕТЛОВ
14. О РОЛИ ДВУОКИСИ АЗОТА
ПРИ САМОУСКОРЯЮЩЕМСЯ РАЗЛОЖЕНИИ
НИТРОГЛИЦЕРИНА
Двуокись азота является одним из первичных продуктов терми-
ческого распада нитроэфиров [6], [10]. В дальнейших стадиях этого
распада она может выполнять функцию окислителя органической
части молекулы, что было установлено на примере алкилмононитра-
тов [9], и, совместно с водой (образуя кислоты), функцию катализа-
тора гидролитического распада нитрата [1]. Особенно существенна
208
роль двуокиси азота в разложении при относительно низких темпе-
ратурах, когда ее растворимость в жидких иитроэфнрах велика.
Значение растворимости уже отмечалось ранее [5], [11], однако коли-
чественной ее оценки произведено ие было.
Настоящая работа 1 посвящена определению растворимости дву-
окиси азота в одном из практически наиболее важных нитроэфи-
ров — нитроглицерине.
Для исследования применяли очищенный двукратной перегонкой
в высоком вакууме нитроглицерин с 12,9“ Двуокись азота гото-
вили разложением нитрата свинца с последующей отгонкой кисло-
рода и сушкой РаО5.
Были использованы два метода определения растворимости: ве-
совой п манометрический, По первому методу 100—150 мг нитро-
глицерина помещали в чашечку кварцевых пружинных- весов, под-
вешенных и цилиндрическом сосуде, помещенном в термостат. По-
сле эвакуации в сосуд впускали двуокись азота, давление которой
поддерживали постоянным и контролировали по стеклянному ком-
пенсационному манометру типа Бурдона (см. [2]) с точностью
+0,5 мм рт. ст. Растворение заканчивалось примерно за 20 мни. Ко-
личество растворенных окислов азота устанавливали по привесу
нитроглицерина с точностью ±0,15 мг. Улетучивания нитроэфирз
ИЗ чашечки при опыте не происходило, что определяли в конце
опыта после откачки окислов азота.
По второму методу в ампулу, соединенную со стеклянным мано-
метром, помещали около 1 г нитроглицерина и после тщательной
эвакуации вводили в нее известное количество двуокиси азота при
охлаждении жидким азотом. Затем ампулу отпаивали от установ-
ки и помещали в термостат. Растворение двуокиси азота при энер-
гичном встряхивании ампулы происходило за 5 мин, По изменении!
давления с учетом равновесия МОгиДГ+О., по Боденштсйну [7] су-
дили о количестве двуокиси азота, растворившейся в нитроглице-
рине 2,
Весовым методом была определена зависимость растворимости
двуокиси азота в нитроглицерине от ее равновесного давления. Оп-
ределение производили при изменении давления окислов азота от
100 до 900 мм рт. ст- в интервале температур 20—80°.
Результаты экспериментов показывают, что растворимость дву-
окиси азота в нитроглицерине пропорциональна ее давлению в сте-
пени 1,5—2,3 (фцг. 1). При этом наблюдается рост этого показателя
с температурой.
Парциальное давление четырехокиси азота в равновесной систе-
ме NO2—NjO.t приближенно пропорционально общему' давлению
смеси в Степени 1,4—1,9. Это позволяет предположить, что в усло-
виях опыта в нитроглицерине растворяется в основном четырехокпсь
азота, В случае справедливости этого предположения количество
1 В проведении экспериментов принимал участие студент 10. В. Додонов.
г Обратимость растворения проверялась при последовательном нагревании и
охлаждении ампулы. Равновесные давления при этом воспроизводились с доста-
точной точность^ (±1—2 м.к рт. ст.).
14 45)
209
растворенных окислов азота, если растворимость их соответствует
закону Генри, должно было быть пропорционально парциальном'
давлению N2O4 в равновесной газовой смеси окислов азота. На
фиг. 2 приведена зависимость растворимости N, выраженной в мо-
лярных долях чстырехокиси азота в нитроглицерине, от рассчитан-
ного по Боденштейну [7] парциального давления N2O4.
Линейность связи растворимости с рассчитанным парциальным
давлением четырсхокисп подтверждает предположение о тохт, что н
Фиг. 1. Влияние общего давления окислоа
азота на растворимость их в нитроглице-
рине при различных температурах,
нитроглицерине преимущественно растворяется именно четырех
окись азота, В то же время, как видно из графика, влияние давления
N2O4 на его растворимость, соответствует закону Генри
N = К • Pn..o, ,
где N — концентрация N2O4 в нитроглицерине в молярных долях;
К — константа растворимости в д,и_[; ря.о, — парциальное давле-
ние чстырехокиси азота в м.и рт. ст.
Были также проведены опыты по определению влияния темпера-
туры на растворимость четырехокиси азота в нитроглицерине, для
чего при постоянном давлении окисдов азота температура опыта
менялась от 30 до 80°. Константы растворимости для каждого оти-
та в этом случае подсчитывали делением общей концентрация оки-
слов азота is растворе в молярных долях чстырехокиси па ее пар-
циальное давление в равновесной смеси.
Значения констант растворимости, полученные весовым и мано-
метрическим методами, приведены в таблице (см. стр. 211).
Как видно нз таблицы, результаты, полученные весовым и мано-
метрическим методами, количественно согласуются между собой,
310
T <i О ,'! II II
Зависимость константы растворимости четырсхокпсн азотл
в нитроглицерине (в .ц.и-i Ю4) от температуры
Метод Т е м 11 с р а т у р а Ли ii.ie:iHL- он
1 1 — —
определении 20 30 ; --Ю 70 I 60 70 3) _к.|< азг)!; .ил/ рт. С1
Весопой 17.3 11.6 j 7.72 7,6(1 I -1.7)3 1 3,62 ; 1 ,‘л9 ШИ—111)3 -
W.-3 7,37 7,88 4.73 3,2-1 ! 2,31
10,7 ' 7.3;) 7,23 ; 4,1() ' о н ' 4 .6(1
Манометри- ческий 16,3 17,3 10,8 1 7.3.- 11 7 8,од .3,127 ],{)! 1 7>,,37 ; 4,62 . 2,33 3,<;4 1.01‘ 2, Ю 61 । (при 36 ;;(' (при 86-
Среднее и наце- нке . 17.0 "т V) , 3,4 | ; | 2,0 2.1
I
* Растворимость оирсделятп при постоянной температуре и изменены:
давления. Коистангы подсчитаны ми кстом плнменыипх гсим.т ра гои.
** Растворимость определятд при постоянном лавлеппи и изменении тем-
пе ратурн,
Фиг. 2. Влияние нарцил.'п.ного дли,тения иа
се раствор iextocti, в iijit | к? гл tn icp hi i e при различных
температурах.
Зависимость константы растворимости четырехокиси азоту; в ни-
троглицерине от температуры им приведенным в таблице та а и’ем
выражается уравнением
. ,г зоба т ц)0 с, ,,
1g л :- ••-• - — - s,o >'.
RT
Ha oc-iiouairirii результатов проведенных экспериментов можно
сделать вывод о том, что в присутствии равновесной смеси NO2-
N2O4 нитроглицерин, при условии справедливости для данного слу-
чая закона Генри, растворяет преимущественно четьгрехокись азота
Иными словами, равновесие, определяющее соотношение NO2 н
N;jO., в растворе, смещено в сторону образования N2O4, что согла-
суется с определением констант указанного равновесия в большом
числе органических растворителей [8]. Таким образом, содержание
четырсхокпсп азота в нитроглицерине пропорционально парциаль-
ному ее давденшо дли общему давлению окислов азота в степени
I ,о—2.
Этот вывод можно сопоставит!, с некоторыми кинетическими за-
кономерностями, установленными памп при исследовании маномет-
рическим методом самоускоряющегося разложения нитроглицерин;:
при 80 — 110° и нитроглнколя при 100° в жидкой фазе. В указанных
условиях скорость газообразования, медленно растущая в начале
опыта, но достижении определенного условно критического давле-
ния газообразных продуктов распада начинает расти гораздо бы-
стрее-- -пропорционально приблизительно второй степени давления
газообразных продуктов разложения. Величина критического дав-
ления, как видно из приводимых ниже данных, значительно повы-
шается с температурой.
Изменение критического давления при распаде
нитроглицерина с температурой
Дкр рг- ст.
80 0,009—0,06 60—80
100 0,0015-0.0G 180—210
120 0,003—0.0Й -100—50(1
110 0’015 1000
140 0,029 800
Естественно допустить, исходя цз наличия критического давле-
нии, что резкое ускорение разложении питроЭфира наступает лишь
при достижении в жидкой фазе определенной концентрации раство-
ренных газообразных продуктов распада.
При сопоставлении указанных закономерностей самоускоряюще-
гося разложения нитроэфиров с влиянием давления окислов азота
на растворимость ХЕСЕ в нитроглицерине можно полагать, что рез-
кое ускорение распада обусловлено реакцией в жидкой фазе с уча-
стием четырехокисиазота. Скорость этой реакции пропорциональна
концентрации или парциальному давлению четЫрехокиси азота,
а следовательно давлению равновесной смеси окислов азота в сте-
пени, близкой к двум.
f Степень заполцентя реакционного госуда соответствует отношению объ-
ема иитрозфнра к объо.чу ямеетлы.
Посскольку /юля двуокиси дзота в газообразных продуктах раз-
ложения нитроглицерина при достижении критического давления
приближенно постоянна и составляет около 40—00% [6], то ста-
новится понятной Пропорциональность скорости распада квадрату
общего давления газообразных продуктов.
Если допустить, что установленная температур на я зависимость
растворимости справедлива в области более высокой температуры,
то экстраполяцией можно Оценить концентрацию четырехокисп азо-
та в нитроглицерине, соответствующую критическому давлению. Этт
концентрация в весовых процентах составляет:
при 80° 0,1-0,4-10“2%; прн 100° 0,3-0,810-!%;
при 120= 0,3- 0,8 10-2% „ при П0° 0,4-1,2.10^%.
При рассмотрении этих величин необходимо учесть относитель-
ную ограниченность данных о содержании двуокиси азота в газо-
образных продуктах распада прц достижении критического давле-
ния, а также возможное влияние на растворимость N2O4 других
продуктов разложения, которое определено не было.
Наличие критического давления, по-видимому, обусловлено тем.
что в начале распада концентрация чстырехокиси азота в нитрогли-
церине мала, следовательно, и скорость реакции с ее участием ii.i
сравнению с первичной реакцией также мала. По мерс разложения
нитроглицерина концентрация N2O4 в жидкой фазе растет и при
достижении критического давления опа достигает такого значения,
когда реакция с ее участием начинает преобладать над первичной.
Поэтому в известный момент скорость распада становится пропор-
циональной приблизительно второй степени давления продуктов
распада.
В данной работе не затронут детально вопрос о механизме само-
уокоряющегося разложения нитроглицерина, однако предположе-
ние о ведущей роли четырехокисп азота ие противоречит гипотезе
о гидролитическом характере распада с последующим взаимодей-
ствием промежуточных продуктов между собой [3]. Действительно,
поданным Г. Н. Беспалова [4], гидролитическое взаимодействие во-
ды с нитроглицерином в присутствии образующейся при этом ки-
слоты протекает очень быстро. Если исходить из этого, то можно
полагать, что при самоускоряющемся разложении нитроглицерина,
когда количество NO2 в продуктах распада велико и, следовательно,
велика скорость образования азс>тноп кислоты, скорость гидролити-
ческих реакций, катализируемых кислотой, также ведпка. Вероятно,
поэтому в последовательности двух процессов, один из которых вы-
зывает исчезновение воды за счет гидролиза, а другой — ее образо-
вание в результате окисления органических продуктов, реакцией,
управляющей скоростью разложения, является более медленная,
т, е, окисление. Полагая, что прц разложении нитроглицерина оки-
сление Протекает в основном с участием двуокиси азота, можно
Ожидать, что иа стадии резкого ускорения скорость разложения
должна быть пропорциональна концентрации N2O4, так как дву-
окись ассоциирует в четырехокнгь в растворе. В таком случае ско-
рость распада должна быть пропорциональна квадрату общего
давления газообразных-продуктов разложения, что и подтверждает
пПЫт-
Изучение термического разложения нитроглпколя показало, что
но достижении определенного критического давления продуктов рас-
пада скорость газообразования, как и в случае нитроглицерина.
Становится продй^гщ[опальной квадрату давления. Поэтому можно
полагать, чтбопг: санная схема самоускоряющегося разложения ни-
троглицерина приложима также и к распаду нптрог.чйколя.
ЛИТЕРАТУРА
I. А к ;i. р е е к К К., Г а л :> к о в а А. П., ДАН СССР, 1955, 105, 286.
2. Андреев К. К., I" л ;im;ob а А. П.. М а v р и и а Н. Д. Светлов Б. С'...
.ЖФХ, [958 32, 1726.
3. А ид реев К. К., ЖПХ, 1958, 31, 184 -493.
4. Бес п в .те в Г. II., Исследование термического разложенп!! нитроглицерина
в iHHK'yre.Tiiiiii воду.. Капдпдитсрал диссертация, МХТИ им. Д. И. Менделе-
ева, [96(1 г.
5. «'] у к и н А. Я., ЖФХ, 1932, 3, 406.
6. С во,- ,| о н 5. С., Намннле до,;,в и д, ,i Высшей ицсо.щм СССР, [958, 3,
122 425.
7, В о d е п s t <- i г. М., В о е s F., 7.. pliys. Cli., 1922, 100, 75, 121.
8. С н и с| a 1 1 1, P„ Traits. cfiern. Soc., [891, 59, 1076; [895, 794.
9. I. e v у ,1. В., Journal Airier. chem. So<-.. [954, 76, 3254, 3790,
|0. R о l:i <! r t s и г R., Journ, chem. Soc., [909,95, 124 [.
II. P о г n н с к i: .i C. 3., Pliys. Zs. SoVjetLinion, 1032, 1,640.
В. Я. ГОРБУНОВ, 5. С. СВЕТЛОВ
15. О РОЛИ КОНДЕНСИРОВАННЫХ продуктов
ПРН РАСПАДЕ НИТРОГЛИЦЕРИНА
Термическое Разложение нитроглицерина (НГЦ) в определен-
ных условиях протекает двустадийно: после относительно продол-
жительного индукционного периода наступает стадия резкого уско-
рения, характеризующаяся быстрым ростом скорости, которая при
этом становится пропорциональной приблизитедино квадрату дав-
ления газообразных продуктов [2] [6]. Сокращение индукционного
периода с. увеличением степени заполнения ампулы, необходимость
наличия некоторого «критического» давления для перехода первой
стадий во вторую, прямые опыты по действию газообразных про-
дуктов распада ;-:а разложение Г1ГЦ -- нее эти наблюдения под-
твердили ранее высказанное предположение [5] [7] о том, что в ука-
занном ускорении играют большую, если не решающую роль, про-
дукты распада, обладающие высокой летучестью. Этими продукта-
ми, по-видимому, являются вода, окислы азота и азотосодержашие
кислоты, Если содержание их в НГЦ достигает определенного уров-
ня, то как полагают [1][2] [3][6], Это и вызывает резко ускоренное
разложение, обусловленное гидролитическим и окислительно-вос-
214
стаиовительным взаимодействием указанных веществ с нитроэфи-
ром или с продуктами его распада. Однако наряду с летучими ве-
ществами в ходе разложения НГЦ, по-видимому, образуются также
и нелетучие промежуточные продукты [3], возможное влияние кото-
рых на расцад нитроглицерина еще не рассматривалось,
В настоящей работе была предпринята попытка показать нали-
чие и оценить роль конденсированных продуктов при распаде НГЦ.
Манометрическим методом [3] проводили разложение НГЦ, после
чего удаляли газообразные и легколетучие продукты и изучали раз-
ложение оставшегося вещества. Оказалось, что удаление летучих
веществ из частично разложенного нитроэфира происходит с очень
большим трудом и для его полною завершения необходимо приме-
нять вакуум (остаточное давление JO -4— |0‘5 мм рт. ст.) в сочетании
с. ловушкой, охлаждаемой жидким азотом в течение нескольких су-
ток. Полноту удаления летучих продуктов контролировали по изме-
нению давления при отключенных насосах и ловушке. Как пока-
зали опыты, газообразование при разложении полученного таким
образом вещества характеризуется, во-первых, очень большой на-
чальной скоростью, во-вторых, не ускорением, а замедлением во вре-
мени и. в-третьих, отсутствием бурой окраски газообразных про-
дуктов с малым содержанием в них газов, конденсирующихся при
комнатной температуре (около 20%). Лишь значительно позже,
когда наступает резкое ускорение, газовая фаза окрашивалась в
цвет двуокиси азота.
В цервой серин опытов НГЦ первоначально разлагался при степе-
ни заполнения сосуда 6 = 0,20 и температуре 100° до давления газо-
образных продуктов 2800 мм рт. ст., что соответствует около 10%
распада, После непрерывной эвакуации летучих веществ 1 в течение
трех суток, с этим продуктом опыты были продолжены прн 70—100"
и 6-0,03—0,04. Как видно из полученных результатов (фиг. 1), га-
зообразование во времени происходит по насыщающейся кривот
которая затем переходит в прямую со скоростью, во много раз пре-
вышающей скорость газообразования прн распаде свежего нитро-
глицерина на первой стадии- Если проводить опыт до больших дав-
лений (свыше 600—700 льи рт. ст- при ЮС"5), то наблюдается на-
ступление резкого ускорения распада.
На скорость газообразования частично разложившегося НГЦ
температура влияет значительно слабее, чем при распаде свежего
нитроглицерина, и температурный коэффициент приводит к величине
Е в уравнении Аррениуса около 20 ккал!моль.
Во второй серии опытов был использован НГЦ, полученный раз-
ложенцем при 6 — 0,24 и температуре 100° со степенью распада око-
ло 7%. Недостаточно эвакуированный продукт, имевший при 90
начальное давление около 30 .и.п. рт. ст,, разлагался по насыщаю-
щейся кривой лишь первые 40 мин-, после чего наступало резкое
ускорение распада. Хорошо откачанный продукт (начальное давле-
1 При подготовке каждого опыта ампула с продуктом предварительно зил-
KyiipoBa.iaci. не менее суток.
215
Нис около 8 мм рт. ст.) в аналогичных условиях (9(Г и iWO.04) дал
кривую газообразования, тождественную той, которая была полу-
чена в первой серии опытов.
Была предпринята попытка выделить конденсированные продук-
ты *, для чего частично разложенный 1П'Ц после тщательной два
куации был залит г. переохлажденном состоянии в пробирку-, ни
дне которой находились кристаллы питрогдицерцна в оставлен на
Фиг. I. Т-р^ическое разложение питрыт 1 лце] > и и а
со степенью распада [0% после удаления из iicro
газообразнык р дегколетучих продуктоп п сравне-
нии с<> епгжим НГЦ (температура опытен 7(| —10СТ.
б-11.03—0.04).
один месяц при температуре около 0°. В результате этого продукт
был разделен на две фракции: жидкую и твердую (в этих усло-
виях), которые использовались для дальнейшего изучения распада
МГЦ. Если термическое разложение продукта перед кристаллиза-
цией имело количественное сходство с разложением вещества пер-
вой серии (см. фНг- I), то выделенная жидкая фракция разлага-
лась значительно быстрее (примерно в 5 раз). Вещество твердой
фракции разлагалось при (10° со скоростью газообразования п ин-
дукционным периодом, приблизительно соответствующим свежему
нитроглицерину в этих же условиях (фиг. 2).
Здесь же приведены результаты опытов по распаду при 90—110'
жидкой фракции вещества, полученной после кристаллизации. Прн
’ Наличие нелетучих промежуточных продуктов при распаде НГЦ было под-
тверждено М. С. Плясу ноны.я по дегрессии температуры плавления, которая
н случае распада этого нзтроэфпра при 6=i),0J и [Э0а до 3%. сощанпла -Г.
216
100 и 110° кривые давление — время при разложении этого продук-
та на всем протяжении опыта имеют насыщающийся характер. Од-
нако эта зависимость не подчиняется‘уравнениям реакций первого
или второго порядка. Начальная скорость газообразования частич-
но разложенного нитроглицерина очень велика и приблизительно в
тысячу раз больше начальной скорости газообразования при рас-
паде свежего нитроглицерина. При 90° разложение вначале имеет
тот же характер, что и при более высокой температуре, по при до-
стижении некоторого давления (около 1000 лмг рт. ст.) ускоряется.
Ф0Г. 2. Турмнческое раз.'южение нитроглицерина со степеннк!
распада 7% после удаления из него газообразных и легко-
дстучих продуктов и кристаллизации, в сравнении со гвежлч
Ш'Ц (температура опытов 90—ИЩ 6=0,006).
1, 2, 3—жидкая фракция чос-ie криегялмтя.эацми, 7—то же, ко с
все. воды, 5—твердая фракция noejpj крггсгалдпэацнгг, <7--сипждГг НГЦ.
Добавление воды к жидкой фракции вещества в этом случае ке ме-
няет начальную стадию, но вызывает резкое ускорение распада зна-
чительно раньше. Кроме этих двух серин экспериментов, были про-
ведены отдельные опыты, которые показали, что картина распад;!
частично разложенного нитроглицерина, освобожденного на сзади;',
резкого ускорения от летучих продуктов, не зависит от того, какой
был первоначально применен нитроэфир: сухой, влажный нлП кис-
лый, Титрование щелочью с фенолфталеином водной вытяжки щ.1-
стично разложенного НГЦ после тщательной его эвакуации от ле-
тучих показано, что в продукте содержится около 1% кислоты в пе-
ресчете на щавелевую.
Совокупность полученных фактов можно объяснить, если исхо-
дить из следующих положений. При распаде НГЦ образуются про-
межуточные труднолетучие продукты, значительно менее стойкие
при нагревании, чем сам ннтроэфир. Содержание этих продуктов
мало и, по-видимому, не превышает 10%, в соответствии с малой
степенью распада НГЦ в условиях наших опытов. Разложение про-
межуточных продуктов приводит к образованию веществ, сравни-
тельно слабо ускоряющих распад НГЦ. Поэтому при нагревании
частично разложенного НГЦ газообразование вначале происходит
в основном в результате распада промежуточных продуктов. По ме-
ре уменьшения количества последних газообразование замедляется
и повышается удельный вес газообразования вследствие разложе-
Фиг. 3. Термический распад частично раадояадыого
j; итрог ли пер иi е а, после удаления из чего газооб-
разных и «-|ету"|ИХ продуктов в сражении со све-
жим НГЦ в Г|рисутствии 0,7% все. щавелевой кис-
лоты. (Температура опыта 100'; й =0,03),
/—’1ДСТИЧПО разложенный НГЦ (из ncpnciR CcpiiH oid.i-
тов), 2— I Н’Ц с 0,7% все. щавелспой кислогм.
пня НГЦ. Дальнейший распад может привести к самоускоряюшс-
муся процессу, определяемому в основном разложением Нитрогли-
церина.
Следует также отметить, что изменение газообразования во вре-
мени прн распаде частично разложенного НГЦ протекает качест-
венно сходно с распадом свежего НГЦ в присутствии щавелевой
к полоты (фиг. 3), который, по данным Г. И. Беспалова [4], на на-
чальной стадии протекает с большой, но уменьшающейся во времени
скоростью, в десятки раз превышающей скороств газообразования
при распаде чистого НГЦ 1. На последующей стадии наступает рез-
кое ускорение распада, причем газовая фаза на участке до начала
этой стадии остается бесцветной. Добавление к НГЦ водвг при боль-
’ Аналогичная кривая получается при разложении ди[ликольдииитрата в при-
сутствии щапелевой кислоты,
.'18
июм количестве щавелевой кислоты приводит к картине газообра-
зования, качественно сходной с той, которая получена для распада
частично разложенного нитроглицерина в присутствии воды. Так
как в условиях термического разложения НГЦ не исключено обра-
зование нелетучей кислоты (в результате окислительно-восстанови-
тельных реакции) 1, такое, сходство может не ограничиваться лишь
формальной стороной.
выводы
I. Освобождение разлагающегося на стадии резкого ускорения
нитроглицерина от газообразных ц легколетучих продуктов не вос-
станавливает первоначальную картину распада нитроэфира.
2. Газообразование при распаде частично разложенного нитро-
глицерина после освобождения его от летучих продуктов протекает
с очень большой, но убывающей со временем скоростью, темпера-
турный коэффициент которой меньше, чем при разложении чистого
нитроглицерина.
3. Распад частично разложенного нитроглицерина качественно
сходен с распадом этого нитроэфнра в присутствии щавелевой кис-
лоты.
4. Термический распад частично разложенного нитроглицерина
протекает так, как если бы в нем присутствовали трудиолетучне
продукты, константа скорости разложения которых значительно
превышает константу скорости распада самого нитроглицерина.
5. При самоускоряющсмся разложении нитроглицерина в при-
с\ ।ствнп продуктов распада скорость процесса определяется ис
только легколсту ним и продуктами, по также, по-видимому, и обра-
зованием нелетучих промежуточных продуктов разложения,
ЛИТЕРАТУРА
I. Андреев К. К-, Глазковз .A. 11., ДАН СССР, 195ь, 105, 28(.i.
А и д р ее в К. К-, ЖПХ, 1958, 31. 484- 493.
-С A ii ;1 реев К. К-, Г л а з к о в а A. 11.. М :r х р ,i ff ;r Н. Д., С в е т л о в Б. С.,
ЖФХ, [958. 32, 1726.
4. Б е с ее а л о в Г. Н.т Кл11липгг г'Си j.'iCcepTaansi, А1ХТ14 ни. .Че|:дс.тг-
«iia, 1960,
5. л у ,< (Е К А. Я., ЖФХ. 1932, 3, .-ЮЗ.
Светлов Б. С., «КиЕ<етпкз и катализ». АП СССР, 1961,2, 38.
7. Р о г и I, с в н if С. 3., Pliys. Zs. SciLvjelunion, 1932, 1. 640.
В. В. ГОРБУНОВ
16, О РАСТВОРИМОСТИ ВОДЫ В НИТРОГЛИЦЕРИНЕ
Вода оказывает сильное ускоряющее действие на термическое
разложение многих- ннтроэфиров. Это действие, как полагают, обу-
словлено гидролитическим взаимодействием ее с цитратом [11[3][5].
’ Анализ частично разложенного НГЦ после удаления летучих продуктов
подтверждает наличие в нем нелетучей кислоты,
919
Естественно, что оно количественно связано с содержанием воды
нитроэфире. Поэтому для детального его рассмотрения и, в частно-
сти, для уточнения механизма указанного взаимодействия пеобхо
Днмо знать растворимость воды в нитроэфирах. Настоящая работа
н основном посвящена определению растворимости воды в нитрогли-
церине (НГЦ), на примере которого наиболее подробно изучалась
роль последней в разложении нитроэфиров.
Для исследования применяли НГЦ, очищенный двукратной пере-
гонкой в высоком вакууме, с температурой плавления 12,9°. Опыты
проводили по манометрической методике {2] следующим образом:
в ампулу стеклянного манометра типа Бурдона объемом Ю—12 ел3
помещали около 0,5 а НГЦ; после эвакуации до полного удаления
летучих в ампулу вводили определенное количество паров воды.
Для этого стеклянный шар известного объема заполняли нарами
воды и измеряли их давление и температуру. Количество воды в ша-
ре рассчитывали по данным для перегретого пара [4]. Было уста-
новлено, что если в эвакуированную ампулу, не содержащую НГЦ.
вводить пары воды при отношении давления в измерительном шаре
к упругости насыщенного пара при той же температуре р/р6.=0,52 —
0,84, то после выдержки в термостате при 100° в ампуле устанав-
ливалось давление на 26—40% больше, чем это следовало по расче-
ту. Последнее, по-видимому, обусловлено адсорбцией воды на стен-
ках дозировочного шара, неучитывавшейся в условиях опыта. Вели-
чина этой адсорбции, по данным Мак-Бена [6], начинает резко уве-
личиваться при pips больше 0,6. Чтобы уменьшить роль адсорбции,
дозировочный шар с парами воды нагревали до температуры 60 —
100°. В связи с этКм одновременно увеличивается точность измере-
ния давления паров воды вследствие возрастания его абсолютно;!
величины. В результате давление паров воды в ампуле без НГЦ
при температуре опыта не преввцпало расчетное более чем ца
1,0—1,5%. После введения воды ампулу отпаивали от установки ц
помещали в жидкостной термостат. При энергичном встряхивании
ампулы растворение воды в НГЦ закапчивалось через 10—|5 мин..
О чем судили по установлению постоянного давления. Исходя цз
равновесного давления паров воды над НГЦ и общего количества
введенной воды, рассчитывали ее содержание в конденсированной
фазе. Определение растворимости производили при температурах
30 -90° в интервале давлений 16—120 мм рт. ст. Г
Поскольку молярная концентрация воды в растворе нс превыша-
ла 2%, а давление водяного пара было значительно меньше его зна-
чения для насыщенного состояния, предполагалось, что раствори-
мость воды в НГЦ, в условиях опытов, подчиняется закономерно-
стям растворимости газов в жидкостях. Константы растворимости
рассчитывали, соответственно закону Генри, делением концентра-
ции воды в НГЦ на равновесное давление се паров. Наибольшие
отклонения результатов параллельных опытов от средних значений
1 Обратимость растворения проверяли последовательным нагреванием и охла-
ждением ампулы. Разница между р ав е । овес е । Ь| м 11 давлениями при этом попу ч а л э о.
fie более 2 ,'-гл( рт. ст.
230
были при низкой п при высокой температурах и достигали 10%, что,
по-видимому, обусловливалось малыми абсолютными величинами
давлений в первом случае и протеканием распада НГЦ во втором Г
Это, в значительной степени, и определило выбранный для изучения
интервал температур. Зависимость константы растворимости от
температуры изображена на фиг. 1. Эта же зависимости может быть
1. Зависимость растворимости па-
от темпе-
Фиг. Э. Зависимость раствори;.]octi:
паров водь, в нитроглицер1П!е от
температурив координатах 1g К--1/Г.
Фиг.
ров воды в нитроглицерине
ратУрм.
выражена прямой линией в координатах 1gК— ЧТ (фиг. 2) н соот-
ветствующим ей уравнением 1g К = —9,114+ 1550ft *. В таблице
(см. стр. 222) приведены средние экспериментальные значения кон-
стант- растворимости и результаты экстраполяции их па более вы-
сокие и низкие температуры. Если предположить, что раствори-
мость воды в НГЦ подчиняется установленным закономерностям в
интервале давлений до упругости насыщенного пара, то можно
оценить и концентрацию насыщенного раствора.
В связи с тем, что нами также было изучено влияние воды на
термический распад дигликольдинитрата (ДГДН) и дииитроглицо-
рина (ДНГЦ), интересным оказалось сравнить растворимость Н2(>
в этих нитроэфирах с растворимостью ее в НГц. В интервале дав-
лении 23—240 мм рт. ст. содержание воды в ДГДН при 120° возра-
стает пропорционально увеличению давления ее паров (фиг. 3), что
указывает, в частности, на соответствие этой зависимости закону
Генри. Константа растворимости воды для этого иитроэфира не-
1 Отношение равновесного давления паров воды над НГЦ к упругости насы-
щенного пара при температуре опыта не превышало 0.5, вследствие чего влияние
адсорбции н условиях опытов было (незначительным.
* Константы этого уравнения рассчитаны по методу наименьших квадратов.
221
Т и б Л 1! it j
Влияние температуры на растворимость воды в НГЦ
Тернер,г,ури и ’С
Средние I'.'iTiiiii.ie
константы p^CiiiopKMCC i и К |<
.tf.if-1 I tv;
РИС ТСТ[ц,Ы KtiltC Гчсти рчС-
творимостд К н м.«—' I9G
Содержание вилм в насыщен-
ном растворе ее а 11ГЦ в
вес.
! 2(1 I 30 j 40 ! 50 ! 00
I 3 . 1 .. !_ Л.
70 1 80 ' 90 | J-’o
I ; J 1
сколько меньше чем для НГЦ и равна 5,2-10~е льц-1. Значителен-
большей растворявшей способностью обладает ДНГЦ, для которо-
го константа растворимости воды яри 120J равна 36-l0_fi л.чч.
ФПГ. 3. Р а ст вор и чп сю. воды в яитрогдидеричс.
дигликольдинитрате и Динитрогдииериие npi,
120° в зависимости от давления ее паров.
Последнее обстоятельство необходимо учитывать при рассмотрена''
опытов по изучению распада влажного НГЦ. так как одним из про-
дуктов реакции гидролиза, по-внлнмому, является ДНГЦ, наличие
которого может дополнительно увеличивать растворимость воды ’’
нитроглицерине.
Другим продуктом гидролиза НГЦ, как показал Г. Ц. Беспалое,
является азотная кислота, которая может оказывать влияние ;i;;
растворимость воды в пнтроэфире. Поэтому в настоящей работ
была оценена растворимость Н2О в кислом НГЦ. Добавление в си-
стему НГЦ-НоО еше одного летучего компонента привело к необ-
ходимости анализа газовой и жидкой фазы на содержание азотпРн
кислоты. Прибор для определения растворимости воды в кислом
НГЦ (фиг. 4) представлял собой комбинацию из двух стеклянных
манометров 1 типа Бурдона, соединенных торцовым краном 2.
В ампулу 3 вводили навеску НГЦ и эвакуировали прибор до уда-
ления летучих. После этого закрывали торцовой крап 2 и в сосуд 7
дозировали воду и азотную кислоту. Безводную азотную кислоту.
не содержащую окислов азота, получали непосредственно перед
опытом перегонкой из смеси азотнокислого калия и копчецтрцро-
Фиг. 5. Растворимость воды в кис-
лом и нейтральном нитроглицери-
не при 50 и 100° в зависимости
от давления ее паров. (Содержа-
ние HNO; в кислом НГЦ 0,3
. 0,6% вес.).
Фиг. 4. Схема прибора для [лучения
растворимости паров воль| в кислом
нитроглицерине.
I—манометры типа Бурдона. 2—торцоной
кран. 3—ампула для НГЦ, 4—сосуд дли
волы п I INOa, о—ампула.
/—КИСЛшЙ 14'11 прн .10°. 2—кислнй
НГ11 при 100=. 3—нейтральный ЦГ11
прн ЯР, 4—нейтральны!! НГЦ прн [00-
ванпой серной кислоты и дозировали ее в виде пара аналогично
воде. После введения добавок прибор помещали в термостат и от-
крывали торцовой крап. В результате увеличения объема системы
и растворения воды и азотной кислоты в НГЦ происходило умень-
шение давления, которое продолжалось обычно 15—20 мин. После
прекращения падения давления закрывали торцовой кран н извле-
кали прибор из термостата. Ампулу 5 охлаждали жидким азотом щ
после конденсации в ней паров воды и HNO3, отпаивали от прибо-
ра, Содержание HNO3 в ампуле 5 Определяли обратным титровани-
ем и рассчитывали по закону состояния идеальных газов давление
ее паров над раствором при температуре опыта. Давление паров
воды над раствором находили по разности между общим и расчет-
1 Точность дозировки проверяли титрованием, а также путем расчета ж
эакову состояния цдеаяьцЬ|Х газов. Обычно ошибка не превьнцала 5%.
пым давлениями паров HNO31. Таким образом устанавливали рав
повесный состав газовой фазы над НГЦ. Содержание воды и кис-
лоты в последнем определяли по разности между введенным коли-
чеством соответствующего вещества и количеством его в газовой
фазе. Эта методика применялась в том случае, когда после опреде
лендя растворимости проводили опыт по термическому раздоже
нню кислого НГЦ в присутствии воды, для чего служил стеклянный
манометр ампулы 3. Если термического разложения не проводили,
то отделяли жидкую фазу от газовой с помощью системы крапов.
Растворимость воды в Кислом нитроглицерине определяли прн
температурах 50 и 100° и содержании азотной кислоты в НГЦ
0,3— 0.6% вес. Зависимость растворимости воды в кислом НГЦ о?
давления ее паров дана па фиг. 5. Здесь же для сравнения приве-
чены прямые 3 и 4, характеризующие растворимость воды в ней-
тральном продукте при тех же температурах. Содержание воды- в
кислом НГЦ также возрастает пропорционально увеличению дав-
ления ее ларов. Сравнение показывает, что добавление HNOs К
НГЦ в условиях опыта увеличивает растворимость воды в нитро-
эфире в 5—7 раз. Таким образом, образующаяся при гидролизе
НГЦ азотная кислота не только выполняет функцию катализатора
и окислителя, но также способствует увеличению содержания воды
в жидкой фазе, а следовательно, и гидролизу.
ВЫВОДЫ
1. Определена растворимость воды в нитроглицерине при темпе-
ратурах 30—90° а в дигликольдинитрате и дицитроглнцерине — прн
I203.
2. Константа растворимости воды в нитроглицерине изменяется
в указанных пределах температуры от 95 10“е маГ1 до
13 10"6 мм~‘-т константа растворимости воды в днгликольдинитра-
[С несколько меньше (5,2- 1О'е мм~[ при 120°), а в дцнитроглипс
рине гораздо больше (36- 10“6 ялт’ при 120°), чем в нитроглицери-
не (6,6 Ю-6 мм~1 при 120°).
3. Растворимость воды в нитроглицерине, содержащем
0,3—0,6% вес. азотной кислоты, в 5—7 раз больше, чем раствори-
мость ее в нейтральном продукте при тех же температурах.
Автор выражает глубокую благодарность профессору К- К. Ан-
дрееву- и доценту Б. С. Светлову за постоянную помощь в работе.
ЛИТЕРАТУРА
i. Андреев К. К. и Глазкова А. П., ДАН СССР, 1955, 105,286.
2. А и д р ес в К. К.. Глазкова А. П„ Матрица Н. Д„ Светлов Б. С..
ЖФХ, 1958, 32, 1726.
1 Для правильности такого расчета необходимо, чтобь| общее давление смеси
паров волга и HNO3 равнялось сумме их давлений в отдельности в тех же усло-
виях. Как показали контрольные опыты, это условие соблюдается с точностью,
соответствующей точности дозировки.
22-1
3. Беспалов Г. Н., Исследование термического разложения нитроглицери-
на в присутствии воды, Кандидатская диссертация, МХТИ, i960 г.
4. Вукалович М. П., Термодинамические свойства воды и водяного пара,
Тосэиергоиздат, 1950.
5. Л у к и и А. Я- ЖФХ, 1932, 3, 406.
6. Мак-Бен Д., Сорбция газов и паров твердыми телами. Госхимтехиз-
дат, 1934.
К. К. АНДРЕЕВ
17. О ТЕРМИЧЕСКОМ РАСПАДЕ НИТРОГЛИЦЕРИНА
И ВОЗМОЖНОСТИ ПЕРЕХОДА ЕГО ВО ВЗРЫВ
Нитроглицерин (НГЦ) является старейшим и до сих пор наи-
более широко применяемым жидким нитроэфиром. Некоторые свой-
ства, отличающие нитроэфиры от веществ иного химического Строе-
ния, например и итр осоед имений и нитраминов ароматического
ряда, выражены у НГЦ особенно отчетливо. Эти особенности отно-
сятся к медленному химическому превращению, горению, вспышке
и чувствительности при ударе. В известной степени связанные друг
с другом они приводят к тому, что производство НГЦ является са-
мым опасным из всех производств вторичных взрывчатых веществ.
Медленный распад НГЦ в определенных условиях его протека-
ния способен чрезвычайно сильно самоускоряться. Это может при-
водить к тому, что даже при низких температурах, при которых
обычно ведется производственный процесс, достигаются такие
большие скорости распада и тепловыделения, что наступает силь-
ный локальный саморазогрев, приводящий к тепловому возникно-
вению горения— вспышке НГЦ. Горение же последнего неустойчи-
во и очень склонно к переходу во взрыв; в условиях вспышки эта
склонность выражена еще сильнее.
Мы рассмотрим в настоящей статье закономерности термическо-
го распада НГЦ и те условия, которые определяют возможность
возникновения вспышки в результате сильного самоускорения это-
го распада.
Скорость любой химической реакции зависит от темпера-
туры и, если реакция идет в газовой фазе или в растворе, также
от концентрации реагентов. В случае распада жидкого НГЦ,
если продукты его представляют собой газы, концентрация исход-
ного вещества не меняется и достаточно было бы рассматривать
только зависимость скорости реакции от температуры.
Однако в реальных условиях скорость медленного распада НГЦ
зависит также от времени. Эта зависимость обусловлена дву-
, мя причинами. Во-первых, медленный распад НГЦ самоускоряется
под влиянием продуктов реакции. Поэтому, если эти продукты не
удаляются тем или иным способом, то содержание их в НГЦ воз-
растает и соответственно растет скорость превращения. Во-вторых,
распад НГЦ идет с выделением тепла и, если это тепло не отво-
дить или отводить только частично, то температура вещества рас-
15 451
225
тет, соответственно растет и скорость реакции. Оба фактора само-
ускорения процесса взаимосвязаны: чем больше увеличилась ско-
рость распада из-за накопления продуктов последнего, тем болыш
будет н скорость тепловыделения, а следовательно, и тепловое ус
корение. Обратная зависимость химического самоускореиия от теп-
лового несколько сложнее, так как при повышенных температурах
продукты, ускоряющие разложение, представляют собой пары и
растворимость их с температурой уменьшается; поэтому, если эти
продукты имеют возможность и время, чтобы удалиться, то сум-
марное ускорение оказывается меньше, чем в том случае, когда
они полностью остаются в жидкости.
Следует иметь в виду также, что на развитие химического са-
моускорения превращения НГЦ значительное и сложное влияние
оказывают те примеси, которые или практически всегда (как
вода) в нем имеются или (как кислота) могут содержаться при
недостаточно тщательной отмывке его от отработанной кислоты.
Рассмотрим более детально обе возможности ускорения распа-
да НГц — тепловую и химическую.
ТЕПЛОВОЕ УСКОРЕНИЕ ЭКЗОТЕРМИЧЕСКОЙ ХИМИЧЕСКОЙ РЕАКЦИИ
ДО ВЗРЫВА
При течении экзотермической химической реакции в результат»
выделения тепла температура вещества становится выше темпера
туры окружающей среды. Поэтому происходит теплоотдача от ве-
щества к окружающей среде. На фиг. 1 представлена зависимость
Фиг. I. Зависимость теплоприхогы
(у() и теплоотвода (?г, <72. от
температуры Т.
теплоприхода и теплоотдачи q..
от температурь! Т.
Если теплоотдача равна теи-
лоприходу, то они находятся
в равновесии. Равновесие яв-
ляется устойчивым (точка Л),
если производная теплоотдачи по
температуре больше производной
теплоприхода по температуре.
Это условие, как видно из гра-
фика, выполняется, если темпера-
тура окружающей среды Т'о от-
носительно низка. Если она будет
выше (7^ или Eq) , то теплоотвод
iso вне станет меньше и равнове-
сия между теплоприходом и теп-
лоотводом не будет или оно будет
неустойчивым, как, например,
в точке касания В. Достаточно небольшого повышения темпера-
туры, чтобы теплоприход стал больше теплоотвода и наступил
самоускоряющийся рост температуры, доводящий превращение до
таких больших скоростей, при которых мы его называем взрывом.
226
Основными факторами этого процесса являются скорость хими-
ческого превращения, его тепловой эффект и размер заряда. Ско-
рость тепловыделения очевидно пропорциональна объему вещест-
ва, а теплоотвод — его поверхности. Поскольку объем растет про-
порционально кубу линейного размера, а поверхность его квадра-
ту, то ясно, что увеличение размера заряда будет благоприятство-
вать ускорению превращения ц переходу его в область неустойчи-
вости,
Д. А. Франк-Каменецкий [5] дал математический анализ явле-
ния для того случая, когда передача тепла осуществляется тепло-
проводностью, и показал, что нарушение устойчивости наступает,
если безразмерный параметр
В = (1)
становится больше некоторой определенной величины, зависящей
от формы заряда и равной для шара 3,32, для цилиндра 2 и для
плоского бесконечно протяженного слоя 0,88.
В формуле (|):
Q — тепловой эффект реакции в кал/см3;
\ — коэффициент теплопроводности в кал!см • сек град;
г--радиус цилиндра (шара) пли половина толщины плоского
слоя в см;
В — предэкспонецциальный множитель в уравнении Аррениуса
в сек~';
Е—энергия активации в кал/моль;
То — температура стенок сосуда в 3К-
Если выполняются условия, положенные в основу этого вывода,
т. е., если скорость реакции изменяется с температурой по закону
Аррениуса, а теплопередача происходит только путем теплопровод-
ности и отсутствуют другие осложнения, то, пользуясь формулой
(I), можно рассчитать для данного размера заряда ту температу-
ру, начиная с которой медленное превращение будет переходить
во взрывное, или решить обратную задачу — для данной темпера-
туры рассчитать тот минимальный размер заряда, начиная с кото-
рого происходит переход от медленного разложения к взрывному.
Сделаем оба расчета. Определим сначала, какова минимальная
температура вспышки для цилиндрического столбика Ь|ГЦ радиу-
сом | мм. Примем теплопроводность нитроглицерина равной
4-Ю“1 кал/СМ • сек • град. Тепловой эффект превращения примем
равным 800 кал/'г, т. е. приблизительно половине теплоты взрыва.
Известно, что при медленном термическом превращении основная
часть азота получается в виде эндотермической NO; соответственно
часть горючих элементов остается неполностью окисленной ц теп-
ловой эффект получается меньше, чем при реакции до азота, коды
и углекислоты. Впрочем величина теплового эффекта относительно
слабо влияет на результат расчета. Уменьшение Q.вдвое увеличи-
вает расчетную температуру вспышки прн Е — 40 000 кал/моль все-
15*
227
го на 5%. Кинетические характеристики принимаем по Р. Роберт-
сону [6] £ = 43 700 кал/моль, /?=1018-64 сек-Г
Подставляя в формулу (1) принятые значения параметров, по-
лучаем
800-1,6 q ]2inls>64e”hwy = 2
4-10—4 ’ 1,9867g
Откуда £— 1633.
Для заряда радиусом 0,5 леи аналогичным расчетом найдем
I критическую температуру равной I763 .
Мы видим, что полученные температуры' несколько ниже темпе-
ратуры вспышки НГЦ, определяемой обычными экспериментальны-
ми методами (около 200’). Расхождение невелико и физический
смысл его понятен. Вязкость НГЦ сильно уменьшается при повы-
шенных температурах, теплоотвод в нем происходит не только теп-
лопроводностью, но и конвекцией, в частности в результате разме-
шивания жидкости пузырьками выделяющихся газов. Это и ведет
к увеличению теплоотдачи, а следовательно, к повышению темпе-
ратуры вспышки.
Определим теперь, каков должен быть размер заряда НГЦ, что-
бы вспышка его произошла прп 2СР.
Используя формулу (1), записываем
800-1 .6 LS86 . 29а , 43700 <2
4-10-* 1,986-2932
Решая это уравнение, получаем г= 1,5 104 см. Следовательно
для того, чтобы при комнатной температуре разложение НГЦ мог-
ло путем саморазогрева ускориться до вспышки радиус заряда
должен быть более 150 м, т. е. во много раз больше чем размеры
тех аппаратов, в которых получают или хранят НГЦ в производст-
венных условиях.
Есть и другая сторона явления, учет которой также подчерки-
вает нереальность возникновения вспышки НГЦ при рассмотрен-
ных условиях и предпосылках.
Как видно из фиг. 1, взаимное расположение кривой теплопри-
хода и прямой теплоотдачи таково, что разность между теплопри-
ходом и теплоотводом, которая определяет скорость саморазогре-
ва, по мере повышения температуры до точки касания уменьшает-
ся и лишь при больших температурах начинает быстро расти.
Расчет показывает, что предвзрывиое повышение температуры для
цилиндрического заряда составляет 1,37
градусов или для
Е
1 986-
НГЦ 1,37 —'43'7^— =5,35°, Чтобы приближенно оценить мини-
мальное время, необходимое для саморазогрева НГЦ на 5,35°,
допустим, что процесс идет адиабатически, т. е. без отдачи тепла
во вне. Разогрев на 5,35° при теплоемкости НГЦ 0,35 кял/г. град
228
потребует 1,87 кал[г. При разложении 100% НГЦ выделяется по
нашему предположению 800 к.ал/г, то для выделения 1,87 кал1е
должно разложиться 0,234% НГЦ. Константа скорости распада
последнего при 20° равна 1,1 10“14 сек~~\ поэтому время, необхо-
димое для изотермического превращения 0,234% НГЦ составит
2,14 • 10й сек. или 7000 лет. Мы видим, что лаже если имеется
достаточно большой заряд НГЦ, в котором может возникнуть
вспышка путем теплового самоускорения, практически взрыва
не произойдет из-за крайне большого времени его задержки.
Означает ли одиако этот вывод, что в реальных условиях воз-
никновение вспышки НГЦ при низких температурах- невозможно?
На этот вопрос следует дать отрицательный ответ. Каким же
образом медленное разложение может развиться до вспышки?
Выше мы отмечали, что медленное химическое превращение
НГЦ, если газообразные продукты его не удаляются из жидкости,
развивается с самоускорещзем. Это химическое самоускорение в
случае НГЦ, а в известных условиях и при разложении других нит-
роэфиров, может стать очень большим. Оно и является той причи-
ной, которая непосредственно может довести скорость превраще-
ния НГЦ до величины, в миллионы раз превышающей начальную
скорость реакции (в отсутствии продуктов распада), и вызвать
этим быстрое тепловое самоускорение процесса.
В самом деле мы видели, что при 20° критический размер заря-
да НГЦ равен 150 л:. Если же скорость реакции станет в миллион
раз больше, то, повторив тот же расчет, мы получим критический
размер в 1000 раз меньше, т. е. 15 см. Больше того, нужно учиты-
вать, что химическое самоускорение также является экзотермич-
ным процессом и если оно даст даже относительно небольшое по-
вышение температуры, скажем, до 100°, то критический размер
снизится уже до 6 см.
Таким образом, решающим фактором возникновения взрыва в
результате медленного распада является развитие химического
(вследствие накопления продуктов реакции) самоускорения про-
цесса.
МЕДЛЕННЫЙ РАСПАД НГЦ ПРИ ПОСТОЯННОЙ ТЕМПЕРАТУРЕ
Рассмотрим более подробно основные экспериментальные зако-
номерности медленного изотермического распада НГЦ и особенно
самоускорения этого процесса.
Р. Робертсон [6], изучая разложение жидкого НГЦ в токе
инертного газа, уносящего летучие продукты, показал, что скорость
отшепления окислов азота постоянна, т. е., что разложение в этих
условиях идет без ускорения. По значениям скорости распада, по-
дученным Р. Робертсоном при разных температурах, и рассчитаны
те кинетические характеристики (энергия активации и предэкспо-
ненциальный множитель), которыми мы пользовались при расче-
тах по формуле (1).
2И1
Исследования лаборатории МХТИ (Г. Н. Беспалов, А. П. Глаз-
кова, В. В. Горбунов, I-I. Д. Маурнна, Al. С. Плясунов, Б. С. Свет-
лов. В. П. Шелапутипа) [I], [2], [3J, [4], проводившиеся манометри-
Фиг. 2. Изменение скорости газообразования во
времени на начальных этанах распада жидкого
КГЦ гт|! [4(Р и умеренных степенях заполнения
сосуда 6.
!—!: :6,< - |0~4; 2-Ь =п I0-4; S =29 И)—*. Скорости
гааообргз^лания шражола ’irEc,!tibi газов (При 0’и 7U0 -И-ц
рт. cii), выделяющихся в 1 к|и«. в пересчете на | г вещества.
иеским истодом, показали, что на начальном этапе разложение
НГЦ идет без существенного ускорения даже если не отводить
газообразные продукты рас-
пада (фиг. 2). Более того па
начальной стадии последние не
только не ускоряют, но даже
замедляют процесс; чем боль-
ше степень заполнения сосуда '
НГЦ, тем меньше начальная
скорость разложения (фиг. 3).
Однако так обстоит дело
лишь до тех пор, пока не до-
стигнуто некоторое критиче-
ское давление газообразных
продуктов распада. После это-
го начинается резкое ускоре-
ние превращения (фиг. 4). До
достижения критического дав-
ления скорость распада растет
пропорционально ^р'!1. Затем
Фиг. 3. ,3лbiichmocti- на’-алыюй ско|Н)сти
распада НГЦ (ш) от степени заполне-
ния сосуда б при температурах опыта
80—НО'.
По оси Бршнл fir.inxerio т ношение скорости ЭтИ СКОроСтЬ рЯСтСт Про-
распа.эт vyn лачиоЬ '• к скор-,-тн при 5 = |0~э в . цОрПИОналЪНО КВЭДрЭту ДЭВле-
нця (фиг. 5), что по предполо-
жению Б. С. Светлова связано с влиянием давления на концентра-
цию димера (КБОД в газах, а следовательно, и в жидкости.
1 Степенью заполнения Й здесь и в дальнейшем мы называем отношение объ-
ема ВВ к объему сосуда в котором проводится опыт,
_’30
Г Таким образом, одной цз особенностей самоускоряющеися реак
! ции является сильная ее зависимость от концентрации газообраз-
ных продуктов распада, растворенных в жидком НГЦ, что приво-
• дит к очень быстрому росту скорости процесса.
! Другая особенность этой реакции состоит в том, что когда раз-
। вилось самоускорение распада под действием газообразных
дуктов, влияние температуры» на скорость его оказывается
слабым (фиг. 6).
про-
очень
Фиг. 4, Ускорение распада НГЦ при [00° егод влия-
нием газообразных продуктов.
'1ие.и нал к гирь, ми—стелет заполнения cocy.-ia $ |(Й. f[,j
оси орди цат отложено количество выделившихся газон, ны ра-
женное в условных единицах (дзиленмел л рт, ст.,
женным ин объем сисУда н и /нигнипм ееч ВВ
в г и етз 1000).
Сочетание обеих этих особенностей приводит к тому, что мано-
метрическая методика при обычном ее выполнении (малые й) ока-
зывается малопригодной для исследования кинетики термического
распада НГЦ. Так как вещество в этом случае занимает только
очень малую часть объема сосуда, то большая часть газообразных
продуктов уходит в свободный объем последнего и не действует
на превращение в жидкой фазе. На практике же, когда мы имеем
Дело с большим объемом вещества, т. е. с большой толщиной его
слоя, продукты распада не успевают, диффундируя, покинуть ВВ
и прогрессивно накапливаются в нем.
: Обычная методика создает затруднения и при определении за-
висимости скорости реакции от температуры: повышение послед-
ней уменьшает растворимость газообразных-продуктов-распада в
жидкой фазе и это может снижать рост скорости и даже предотвра-
щать у-скоренне процесса, что н наблюдал В. С. Светлов в извест-
ных условиях опыта. Опять-таки в производственных объемах сни-
жение растворимости с температурой не обязательно успеет по
931
1
I
i 2 рт.с-rj
Условные обозначения;
i A'E=W'W-* ;D-5=W-«r’;
!. _промежуточные значения S >
I Фиг. 5. Зависимость удельной скорости газообра-
I зования (w) от давления газообразных продуктов
, распада НГЦ при 10(Г и разных степенях запол-
I нения сосуда б.
! Скорости выражена в условных единицах.
I
Фиг, 6. Зависимость удельной скорости га, \
зообразования (ш) от давления продуктов
распада НГЦ при разных температурах.
Скорость распада выражена в условных единицах.
232
упомянутым выше причинам уменьшить концентрацию газообраз-
ных продуктов в жидком НГЦ.
Чтобы приблизиться к реальным условиям, мы видоизменили
методику лабораторного опыта, применив заполнение сосуда нит-
роглицерином почти целиком; это обеспечивало практически пол-
ное сохранение в жидкой фазе газообразных продуктов превраще-
ния, В этих условиях резкое ускорение распада при 100° наступает
уже через 9—10 час., при 80° через 73—93 час. и при 60° через
550 час.
Эти данные позволяют экстраполяцией рассчитать соответству-
ющие значения времени наступления ускорения при более низких
температурах, Для 30° получим 3,2 года, для 20° — 17 лет. Полу-
ченные значения времени начала резкого ускорения распада НГЦ
хотя и велики, но они несравненно меньше, чем в случае разложе-
ния с удалением продуктов распада, когда время его даже для не-
которой небольшой доли ВВ, скажем 2%. составляет при 30 и 20°
соответственно 4800 и 56 500 лет.
ВЛИЯНИЕ НЕКОТОРЫХ ПРИМЕСЕИ НА МЕДЛЕННЫЙ РАСПАД НГЦ
Выше шла речь о распаде чистого НГЦ свободного от приме-
сей. Из примесей, которые обычно могут находиться в НГЦ, наи-
больший практический интерес представляют вода и Кислоты.
Вода растворима в НГЦ. По нашим определениям (В, В. Гор^
бунов, см. этот сборник стр, 219) в НГЦ растворяется при 20° 0,25,
при 30°-—0,31 и при 60° — 0,49% воды.
Однако содержание Н2О может быть и больше, так как она
может находиться в НГЦ не только в растворенном, но и во взве-
шенном виде. По этой причине технический НГЦ обычно бывает
мутным.
Вода оказывает значительное и своеобразное влияние на мед-
ленное превращение НГЦ. По опытам Г. Н. Беспалова (см. этот
сборник стр- 131) при повышенной температуре (80—100°) и малом
содержании Н2О кривая р —/(т) имеет тот же характер, что и в
отсутствии воды (фиг. 7), но время до начала ускорения распада
в первом случае меньше.
При большем содержании воды характер кривой существенно
меняется: длительное время давление остается постоянным и прак-
тически равным упругости паров воды над НГЦ, затем наступает
резкое падение его, обусловленное переходом воды в жидкую фа-
зу, После этого падения начинается более или менее быстрый рост
давления. Общее время до наступления резкого ускорения распа-
да в присутствии воды сильно сокращается по сравнению со вре-
менем для безводного продукта. Так, при Й —0,03 время до резкого
ускорения разложения безводного НГЦ составляло при 100s около
2400 мин., а при давлении паров воды, равном 400—500 мм, оно со-
кратилось до 150 мин. Пои максимальной б (около 1) при 100 и
60° в присутствии 0,2% HjO указанное время было в 2‘/2 раза
меньше, чем для безводного НГЦ.
233
Если добавить к НГЦ еще больше воды, то характер кривой
p = f(t) вновь меняется. Долгое время наблюдается очень медлен-
ное и слабое падение давления и лишь потом, по истечении време-
ни значительно большего,'чем даже для безводного НГЦ, наблю-
дается резкое ускорение процесса.
Таким образом, вода в малых количествах ускоряет превраще-
ние НГЦ, причем тем сильнее, чем больше ее введено. В больших
количествах она задерживает начало ускорения распада нитрогли-
церина.
Время т мин
Фиг. 7, Влияние еоды из распад НГЦ при Ю(Г и
различиях степенях заполнения сосуда б.
у кривых—лаьленяе варев шны над НГЦ г» рт- ст.
Н {в скобках) $ Iff-
Ускоряющее влияние на распад НГЦ оказывают и кислоты, и
частности азотная кислота. Начиная соответствующие исследова-
ния, мы, исходя из общераспространенных представлений, ожида-
ли, что очень малое содержание кислоты будет очень сильно уско-
рять разложение нитроглицерина, Опыты не подтвердили этого
ожидания. Так, в случае добавления к НГЦ HNO3 в количестве
0,3% по весу, время до наступления ускорения реакции сократилось
всего в два раза по сравнению с нейтральным продуктом (см, кри-
вую 4 фиг. 8),
Более того при сравнении равных (по давлению их паров над
НГЦ) количеств азотной кислоты п ;шды (кривые 2 и 3 фиг, 8),
оказалось, что при 100° кислота ускоряет распад НГЦ приблизи-
тельно так же как вода, совместное их присутствие при данном
давлении не сократило времени до начала ускорения процесса, На-
конец опыты в присутствии больших количеств воды (фиг, 9) по-
казали, что 0,02% азотной кислоты от веса НГЦ мало влияет па
течение распада. Так, например, при близких содержаниях воды
индукционный период в присутствии кислоты снизился только до
68 мин вместо 70 в ее отсутствии. Сильное влияние азотной кисло-
ты в присутствии воды проявляется лишь при относительно боль-
шом (~ 1,5%) содержании кислоты в НГЦ (фиг. Ю).
231
Все эти наблюдения объясняются допущением, что быстрое пре-
вращение, наступающее на стадии ускорения, обусловлено гидро-
лизом НГЦ, который сильно ускоряется кислотами в определенном
Фиг, 8- Влияние умеренных (0,3%) коли-
честв Н?<Оз на распад НГЦ при 100.
J — НГЦ без волы, 5 =0 0329; 2 -ПгЦ-i-
q-HnO+UNOa, р--14, $=0,0281; 3-НГЦ гН,О,
р=14, & 0,0271; 4—НГЦ+HNO,, р=ЕЗ, £ 0.0^46.
р- общее равновесное давление в начале опыта в
р-г. cfi., в—сг«1<?»ь заводнения сосу.чл нитроглице-
рином .
интервале их концентрации, и сопутствующими гидролизу окисли-
тельно-восстановительными реакциями между его продуктами с
возможным участием исходного нитроэфира. Если устранить влия-
Фиг. 9. Влияние малых (0,02%) количеств HNO3
на распад НГЦ при 10(7’ в присутствии воды.
Г—НГЦ+Н.0+0,023% вес. HNO,.
5-0,0363; 2-11ГЦ+НаО 4-0,01756 вес. HNO. р=.!Я1)
J 0.0287; 3-НГЦ+Н2О+0.0)9 sec. HNO,, р-385, « -0.Ю17.
р_обжее равновесное давление в начале опыти в лл рт. ст.
6— степень заполнения ссуда нитроглицерином.
ние кислот, например, при помощи соды, помещенной в сосуд, при-
паянный к ампуле с НГЦ. то в результате нейтрализации паров
235
кислот ускорения превращения не наступает как в присутстгши
воды? так и в ее отсутствии.
Вода, введенная в нейтральный НГЦ, действует относительна
медленно до тех пор, пока в результате «безводной» реакции или.
гидролиза в продукте не накопятся кислоты; если вода не введен;?,
то накопление кислот и воды (в результате окислительных реак-
ций) идет еще медленнее. Азотная кислота сама по себе действуй
медленно, в частности, потому, что требуется время для образова-
ния воды. Наиболее активной является вода в присутствии кисло-
ты, причем кислота эта не обязательно должна быть азотной.
Фиг. 10. Влияние значительных (1.5%) количеств
азотной квелоты на распад НГЦ при 1003 в присут-
ствии воды.
;_НГЦ нейтральный без воды, 8-0,0061: 2— НГЦ+ТКО, р-^205,
o=0f0060; J-HFU+HNOs. р=82,5г 8-0»00')2; 4-НГЦ+НаО + НЫО •<,
/7-^222. £ =0.0070; $-НГЦ+Н.Х)Ч-НЬГСЧ. р=5бО( 5-0,0068,
Р— общее равновесное давление & начало опыта н .м.м рт» ст»
е—степень заполнения сосуда нитроглицерином»
Следует добавить, что нами изучалось преимущественно влия-
ние смесей воды с азотной кислотой. Нельзя однако считать, что
это сочетание является наиболее эффективным стимулятором уско-
рения превращения НГЦ,
Опыты В. П. Шелапутиной показали, что весьма энергичным
ускорителем служит вода в сочетании с окисью и двуокисью азота
в определенных соотношениях. Так в одном из опытов при 803
(фиг. 11), в котором равновесное давление паров воды составляло
около 100 мм рт. ст., ускорение распада НГЦ наступило лишь через
2000 мин.,- при одновременном присутствии воды, NO и NOa резкий
рост скорости газообразования наблюдался уже через 50 мин. по-
сле начала опыта (см. кривую 2 фиг. 11).
По-видимому, в опытах с водой непосредственным продуктом
гидролиза является азотная кислота. Взаимодействуя с образовав-
шимися при гидролазе динитратом и тринитратом, она образует
двуокись азота я азотноватый ангидрид, окисляющее действие кото-
рых в присутствии воды значительно больше, чем окисляющее дей-
236
ствие азотной кислоты. Реакции взаимодействия азотной кислоты
и нитратов протекают относительно медленно, чем и определяется
Фиг. 11. Влияние окислов азота на распад НГЦ
при 80° в присутствии воды.
;_НГЦ+Н,о, О=284. *-0,0304; З-НГЦ+И.О+ЫО+ИО»,
РН о“246,,Р«о“3^ PNO," l00, s=O,03I2^-HrlJl+H>O+№+NO«
•Р«о = 13°’ PNO = u0’ 4~HTH+H1O+NO+
+ NO)h ^0=31. ^NO^30" PNO4-37’ i“°’0309: -e-расчетное
давление паров н газов» введевиы! а сосуд в начале
опыта (без учета их возможного взаимодействия и растворевня)
а хх рт, ст»; 6—стевень заполнения сосуда нитроглицерином»
Фиг. 12. Влияние окиси азота на распад НГЦ
при 80° в присутствии воды.
?-НГЦ+Н,О. Рн S-0.0304; 2-НГЦ+Н,О+Ы0, Рн Q=272,
PN0=ai4, 5=О,О&8; З-НГЦ+HjO+NO. дн О“238, ^0"Я'
SJ.0323-, 4-НГЦ+HjO+NO, ₽н Q-230, Л^о-50, t~°-0234-
/у—расчетное давление паров н газов, введенных в сосуд
в начале опыта (без учета нх возможного взаимодействия н растао-
рения) в жж рт, ст»; $—степень заполнения сосуда нитроглице-
рином.
значительный интервал времени между моментом падения давления
в опытах с водой и наступлением резкого его роста. Если сразу
ввести окислы азота, то окисление нитратов начинается бы-
стро.
Дополнительным свидетельством в пользу описанной последова-
тельности реакций являются результаты опытов по разложении
НГЦ в присутствии воды и окиси азота (фиг. 12). Индукционный
период в этих опытах сохранил свои характер (постоянство давле-
ния) и длительность. Однако с одной стороны отсутствует то паде-
ние давления по окончании индукционного периода, которое на-
блюдается при наличии воды, но в отсутствии окислов азота, н с
другой — ускорение газообразования после индукционного перио-
да развивается гораздо резче и быстрее. Отсутствие падения дав-
ления, может быть, связано с тем, что азотная кислота с окисью
азота образует азотистую кислоту, растворимость которой в НГЦ
предположительно меньше чем азотной, а окисляющее действие
больше. Соответственно на фоне быстрого роста давления в резуль-
тате окисления возможное небольшое падение давления за счет
растворения остается незамеченным.
МЕДЛЕННЫЙ РАСПАД НГЦ ПРИ НИЗКИХ ТЕМПЕРАТУРАХ
Описанные выше опыты были проведены прн относительно вы-
соких (80° и выше> температурах.
Опыты при более низких (40—80°) температурах (Б. С, Светлов
и В. В. Горбунов) длятся очень долго и поэтому были малочислен-
ны. Они показали, что закономерности, установленные при повы-
шенных температурах, качественно справедливы и для значитель-
но более низких температур, но имеют некоторые количественные
отличия.
Так при 100° и большой степени заполнения сосуда резкое ус-
корение в присутствии 0,2% вес. азотной кислоты наступает н
1,7 раз быстрее чем у безводного нейтрального НГЦ; вода дейст-
вует сильнее азотной кислоты и при том же содержании снижает
указанное время в 2,5 раза (фиг. 13).
При более низких температурах (60 и 40°) кислота и вода так-
же сокращают индукционный период, но кислота действует силь-
нее чем вода причем различие это возрастает при переходе от
60 к 40°. Кроме того, при низких температурах влияние добавок к
НГЦ в смысле относительного уменьшения индукционного периода
больше. Это неудивительно. При понижении температуры скорость
реакций разложения, образующих кислоты и воду, уменьшается;
соответственно снижается влияние этого источника ускоряющих
примесей и относительно возрастает роль ускорителей, введенных
извне:
Следует указать также, что во всех этих опытах речь шла о со-
держании добавок кислотного характера, хотя и небольшом, но
1 Нужно однако иметь в виду, что для поды в отличие от азотной кислоты
имеется некоторое оптимальное содержание, при котором индукционный период
наименьший. Оптимальное содержание установлено только для 100е. При низких
температурах соответстзующие опыты це проведены.
238
все же несравненно большем, чем те ничтожные их количества, ко-
торые регистрируются стандартным методом определения стойкости
НГЦ — иодокрахмальной пробой.
Фиг. 13. Влияние воды и азотной кислоты па распад НГЦ при 100'
и большой степени заполнения сосуда (д==1).
ОСОБЕННОСТИ РАЗВИТИЯ РАЗЛОЖЕНИЯ НГЦ ДО ВСПЫШКИ
Из рассмотренных экспериментальных данных следует, что
НГЦ способен к двум процессам превращения; один из них реали-
зуется при относительно высоких температурах и характеризуется
сильной зависимостью скорости превращения от'температуры. Ско-
рость второго процесса сильно зависит от концентрации продуктов
распада НГЦ (окислов азота и воды) и слабо от температуры. По-
этому при низких температурах, когда константа скорости первого
процесса ничтожна, фактическая скорость распада НГЦ может оп-
ределяться лишь вторым процессом. Эта скорость, однако, в начале
процесса очень мала, если только в НГЦ не введено достаточно
много кислоты и немного воды. При разложении первоначально
нейтрального и безводного НГЦ образуется двуокись азота и как
продукт окисления вода; если НГЦ содержит воду, то образование
азотной кислоты и затем окислов азота значительно ускоряется.
Казалось бы, что при низких начальных температурах именно
второй процесс и должен привести НГЦ к тепловому возникнове-
нию вспышки. Однако это, по-видимому, не так по той причине,
что скорость реакций второго процесса лишь очень слабо растет с
температурой. Соответственно скорость определяемого ими превра-
щения может достичь значений, при которых выражение Франк-
Каменецкого (1) становится равным критическому лишь при отно-
сительно высоких температурах. Расчет, выполненный по кинети-
ческим параметрам (Е и В) второго процесса, установленным для
этапа резкого ускорения, дает при г— 1 мм критическое значение
температуры —6Qp° (580е). Как мы видели, аналогичный расчет
239
для первого процесса—реакции, идущей в отсутствии продуктов
распада, дал значительно меньшую температуру (] 63°). Поэтому ।
при развитии ускорения распада продуктами реакции и при сопут-
ствующем ему разогреве раньше, чем будет достигнута критичес-
кая скорость для этого процесса, станет все быстрее и быстрее
идти и, наконец, достигнет критической скорости превращение, не
зависящее от продуктов разложения яитроэфира.
Здесь однако нужно сделать одну оговорку: скорость реакций
в присутствии газообразных продуктов распада сильно зависит от
их концентрации; вышеприведенные расчетные данные относятся
к тому случаю, когда давление этих продуктов над НГЦ невелико
(2 ат), так как при развитии процесса в обычных условиях трудно
представить себе возможность достижения более высоких давлений.
Однако, если такие условия имеются, то критические температуры
для обеих реакций сближаются. Если допустить, что установлен-
ные опытом для интервала температур 80—120° зависимость от
давления скорости самоускоряющегося процесса, а также ее неза-
висимость от температуры сохраняются и при более высоких зна-
чениях последней, то при 163° скорость процесса, идущего под вли-
янием продуктов распада, будет уже при 9 ат равна скорости его
в их отсутствии.
Если сильного повышения давления не может произойти, то раз-
витие реакции до зспышки принимает своеобразный трехстадий-
ный характер. На первой стадии, либо в результате первого про-
цесса (если температура не слишком низкая), либо вследствие гид-
ролиза нейтрального или кислого НГЦ образуются продукты
распада, что обусловливает развитие второго процесса. На следу-
ющей стадии этот процесс в результате накопления продуктов рас-
пада самоускоряется в такой степени, что происходит значительное
повышение температуры. На третьей стадии преобладающее значе-
ние получает первый процесс, тепловое ускорение которого и дово-
дит НГЦ до вспышки. Время до наступления вспышки определяет-
ся вторым процессом, а критические ее условия — первым. Разу-
меется, оба процесса идут не автономно, а совместно и они
взаимосвязаны, так что сказанное представляет собой естественно
известную схематизацию явления. Чем выше начальная темпера-
тура и более благоприятны условия для удаления газообразных
продуктов из жидкости или связывания их, тем больше выражена
роль первого процесса и наоборот.
Сказанное относится к безводному НГЦ. При наличии влаги
параллельно с первым идет гидролитический процесс; скорость по-
следнего больше, чем первого. Остальные кинетические характери-
стики этого процесса мало изучены. В остальном разложение до
вспышки н этом случае развивается аналогично безводному НГЦ.
ВЫВОДЫ
Распад НГЦ, идущий при удалении или связывании его продук-
тов очень медленно (при низких температурах) и без ускорения,
может сильно ускоряться, если продукты этого распада будут на-
240
капливаться. Наступление ускорения разложения происходит го-
раздо быстрее, если НГЦ содержит примеси — воду (в определен-
ных пределах) и кислоты или окислы азота. Особенностью реакций
самоускорения является сильная зависимость их скорости от кон-
центрации продуктов распада или примесей и слабая зависимость
от температуры. Вследствие этого, если достигается высокая кон-
центрация ускоряющих веществ, распад может привести к тепло-
вому возникновению вспышки даже при очень низких температу-
рах и относительно небольших размерах заряда,
ЛИТЕРАТУРА
!. А н д р с е в К. К. и Г л а з к о в а А. П., ДАН СССР, 1955, 105, 286.
2. А н д р е е в К- К., ЖПХ, 1958, 31, 484,
3. Андреев К. К-, Глазкова А, П,, М а у р и и а Н. Д. Светлов Б. С.,
ЖФХ, 1958,32, 1726.
4. М а у р и н а Н. Д., Термический распад нитроглицерина, Диссертация,
МХТИ им, Менделеева, 1951.
5. Фран к-К аменецкий Д. А,, Диффузия и теплопередача в химической
кинетике, АН, 1947, стр. 235.
6, Ro b er t so n R., J. chem, Soc„ 1909, 95, 1241.
К. К. АНДРЕЕВ, Б. И. КАНДЫМОВ
18. термический распад тэна
Введение в технику пентаэритриттетранитрата (тэна) потребо-
вало обстоятельного изучения его свойств, в частности химической
стойкости, тем более, что тэн принадлежит к числу эфиров азот-
ной кислоты, которые, как было известно, вообще говоря, сильно
уступают в этом отношении нитросоединениям ароматического
ряда и даже нитраминам.
Химическая стойкость тэна и смесей на его основе, методика
ее определения и влияние на нее некоторых примесей изучались
в ряде работ. Уже первые исследования показали, что химическая
стойкость тэна существенно больше, чем других нитроэфиров,
близких к нему по числу нитратных групп на углеродный атом, а
следовательно, и по энергии взрыва — нитроглицерина, нитрогли-
коля и даже значительно превосходящей их по стойкости нитро-
клетчатки. Так, по пробе Абеля при 75°, которую динамитный нит-
роглицерин выдерживает обычно в течение 15 мин., тэн не дает
реакции при 80° даже в течение 50 мин. Это обстоятельство обычно
связывали с более высокой симметрией молекулы тэна по сравне-
нию с нитроэфирами, имеющими линейную структуру [16], [17]. Так,
Штетбахер, а в дальнейшем и другие исследователи, полагают, что
расположение 4-х метоксинитро групп вокруг центрального углерод-
ного атома в тэне определяет его высокую химическую стойкость.
Такое же мнение высказывает Л. Авогадро, считающий, что тэн
16 451
241
благодаря своей симметричной структуре является наиболее стой-
ким из нитроэфиров.
Одно из обстоятельных исследований по этому вопросу было
опубликовано Пикардо [14], Он выдерживал в течение 450 дней
2 образчика тэна при 100 и 105° и через определенные интервалы
времени устанавливал изменение их свойств. Через каждые 15 дней
производилась проба при 120° с метилфиолетовой бумажкой и оп-
ределялась потеря в весе. С интервалом в 60 дней исследователь
выполнил несколько испытаний: пробы Талиани при 125°, Бергма-
Фиг. 1. Изменение pH при разложе-
нии тэна в присутствии различных ко-
личеств тротила (го Урбанскому и
др.) прн 120’.
Числа У кривых—содержание тротила
в смеси в вес.
Фиг. 2. Изменение давления при
разложении тэна в присутствии
различных количеств тротила (по
Урбанскому и др.).
Числа у кривых—содержание трот и.то
в смеси и вес.
на — Юнка при 132° и Анжели, а также определение содержания
азота по Лунге и температуры плавления. Последнее определение
проводилось также с кристаллами, конденсировавшимися в резуль-
тате возгонки на стенках сосуда.
Результаты испытаний показали, что стойкость не только не
падает со временем, но заметно повышается. При 105° этот процесс
идет быстрее, чем при 100°, Потеря в весе возрастает приблизитель-
но пропорционально времени. Содержание азота несколько возрас-
тает так же, как и температура плавления (140,3—141,2° в начале
опыта и 141,3—142,0° после 450-дневного нагревания при 105°). Су-
блимат имеет более высокую температуру плавления, чем исходный
тэн и более узкий интервал плавления (142,0—142,2°).
Из этих данных следует, что длительный нагрев приводит к суб-
лимации не только тэна, но и примесей, содержащихся в исходном
продукте, причем они либо возгоняются, не разлагаясь, либо раз-
лагаясь, образуют легко летучие и полностью удаляющиеся продук-
ты. В общем, эти опыты показывают, что даже очень длительное
нагревание тэна в открытом сосуде при 105° не приводит к замет-
242
ному разложению ВВ, а, напротив, повышает его чистоту и стой-
кость.
Урбанский, Квятковский и Миладовский [19] изучили влияние
нитропроизводных ароматического ряда на химическую стойкость
некоторых нитроэфиров, в частности, тэна. Исследование произво-
дилось путем определения pH смесей, нагревавшихся при 100- -
120° и по методу Талиани при 134,5°, модифицированному Гужо-
ном. Результаты опытов иллюстрируют графики фиг. 1 и 2. Нитро-
соединения снижают стойкость тэна, причем сильнее действуют при
данном процентном содержании (по весу) мононитросоединения,
затем ди- и Тринитросоединения. Это влияние сильнее проявляется
в первые часы нагрева. Производные бензола действуют несколько
сильнее, чем производные толуола. Интересно, что разложение
нитроманнита нитропроизводные не ускоряют.
Аналогичные результаты были получены Тонегутти [18], который
определил стойкость тэна и его смесей с тротилом по пробе Талиа-
ни при 120, 125 и 135°. Некоторые из его результатов приводятся
в табл. 1.
Таблица 1
Стойкость т>на и его смесей с некоторыми взрывчатыми веществами
по пробе Талнани
Температу- ра опытов °C Время достижения давления 300 мм рт. ст. в час
ТЭ1! Тэн с ЗОИ тнт Тэн с 50 И ТНТ Тэн с 10» пикриновой кислоты Тэн с 2054 нитрогли- церина Нитроклет- чатка
120 20,50 5,08 5,16 2,33 2,75 5,08
125 11,25 2,08 — 3,66
. 130 7,00 1,00 0.92 — 1,70
Из други? наблюдений Тонегутти следует отметить, что стой-
кость тетрил* при 120—125° в 3—4 раза больше, чем тэна, в то
время как пр* 13(р она вдвое меньше, чем у тэна. По мнению иссле-
дователя, это связано с тем, что в последнем случае температура
опыта слишком близка к критической температуре разложения.
Заключен^ Урбанского и Тонегутти подтверждаются и опыта-
ми Буржоля ’9] (габл. 2), который определял по пробе Бергмана--
Юнка влияние на стойкость тэна добавок различных ароматических
и алифатичегких нитросоединсний. Все ароматические нитросоеди-
нения, в наибольшей степени динитронафгалин, понижают стой-
кость тэна. Действие алифатических нитросоединений несравненно
меньше. Буржоль заключает: «Ароматические нитросоединения
действуют на тэн, разлагая его с образованием окислов азота. Ме-
ханизм этого действия, по-видимому, не известен».
Урбански! и Миладовский [20] в более поздней работе изучали
ио методу рЦ влияние на стойкость тэна при 1323 различных не-
16*
24'
Таблица 2
Стойкость тэна и его смесей с некоторыми взрывчатыми
веществами по пробе Бергмана—Юнка
Нптросоелянепня Показатель Бергмана—Юнка для 5 г тэна4-нитросоеднненне в количестве
2 мг 5 мг 20 мг 50 мг
Днпнтротолуол 0,05 0,08 0,45 0,85
Динитронафталин 0,03 0,05 0,51 1,20
Тринитротолуол 0,06 0,09 0,30 0,70
Тетрил 0,02 0,05 0,20 0,50
Нитропрог|аг| 0,01 0,01 0,01 0,04
Нитробутан 0,01 0,01 0,02 0,03
Нитрогуацндш| 0,02 0,02 0,03 0,035
Г ексоген 0,02 0,03 0,05 0,07
Нитроклетчатка 0,01 0,02 0,03 0.03
взрывчатых веществ в количестве 3—5% вес. (этиловый и пропило-
вый спирт, этиловый эфир, этилацетат, изоамилацетат, ацетон, аце-
тофенон, вода) и в количестве 0,1—-1,0% вес. (дифениламин и цен-
трали?) и показали, что все они в большей или меньшей степени
снижают его стойкость, На нитроклетчатку же влияние указанных
веществ обратное: они заметно повышают ее стойкость. На основа-
нии этого различные авторы делают заключение о «существенном
различии», которое имеется между химическими свойствами нитро-
клетчатки и тэна, различии, которое, как они полагают, обуслов-
лено совершенно различной структурой обоих нитроэфиров, а имен-
но, высокомолекулярным цепнокольцевым строением нитрата клет-
чатки и разветвленной структурой тэна с его центральным атомом
углерода.
К аналогичным выводам пришли и другие исследователи, изу-
чавшие влияние нитросоединений на разложение тэна. Так, Десень
ПО] нагреванием смесей тэна с тротилом в закрытых пробирках при
90° установил, что при значительном содержании тротила скорость
и ускорение газообразования сильно возрастают (фиг. 3). Это ус-
корение автор связывает с образованием окислов азота — газовая
фаза принимает отчетливую окраску.
Аналогичная картина наблюдается и при разложении в неплот-
но закрытых сосудах при 120°, когда давление не могло повышать-
ся, но газообразные продукты распада не так легко удалялись, как
при опытах в открытых сосудах. При 110° влияние тротила прояв-
ляется еще более резко и уже при меньшем его содержании,
В аналогичных условиях на разложение гексогена присутствие тро-
тила оказывало несравненно меньшее влияние.
244
Исследования по стойкости тэна были опубликованы также
Обертэйном. Он определял скорость газообразования при разложе-
нии тэна в вакууме при 120° [5], по методу, применяемому в Англии
для определения стойкости тэна, согласно предписанию стандарта.
Скорость газообразования не зависит от размеров кристаллов. Все
образчики показывали одну и ту же скорость разложения до и пос-
ле тщательного измельчения
в агатовой ступке, как в случае
тэна, полученного перекристал-
лизацией из обычного очищен-
ного ацетона, так и для тэна,
осажденного из ацетонового ра-
створа водой. Такой тэн, осо-
бенно если осаждение произво-
дится из ацетонохлороформного
фиг. 4, Изменение скоро-
сти гидролиза тэна, ха-
рактеризуемой потерей в
весе при различных тем-
пературах (по Обер-
тэйну),
Фиг. 3. Изменение давлении
при разложении тэна в при-
сутствии тротила при 90?
(□о Десеню).
<—тэн чистый. 2—смесь тэи—
тротил [75; 25), 3—смесь тэн—
тротил (50 : 50). 3—смесь тэн—
тротил (25 : 75),
раствора, показывает значительно пониженную стойкость, что
связывается с химическим влиянием остатков растворителя (гид-
ролитическое действие воды, образование соляной кислоты, при-
меси ацетона). Азотная кислота ускоряет распад, хотя действие
ее в отсутствии воды слабее, чем гидролитическое действие водьг.
Это последнее действие во многих случаях оказывается более
существенным, чем влияние других примесей, если количество их
! не превосходит некоторого предела.
В другой работе Обертэйн 16] рассматривает влияние включений
растворителя или осадителя, которые могут сильно понижать по-
казатели стойкости по Бергману—Юнку (проба, применяемая для
тэна во Франции) в результате перехода распада испытуемого ВВ
от термического к гидролитическому. Тщательное измельчение кри-
245
сталлов, обеспечивающее при последующем высушивании удаление
летучих примесей (вода, ацетон) приводит к резкому повышению
показателя стойкости. Отдельными опытами [7] было показано,
что скорость гидролиза тэна велика и особенно быстро она растет
с температурой выше 120° (фиг. 4). В одном опыте при 130° гидро-
лиз привел к взрыву.
Слабой стороной работ Обертэйна является проведение опытов
только ниже температуры плавления тэна. Влияние примесей мо-
жет быть двояким: химическим или физическим. Проводя сравни-
тельные опыты с жидким веществом, можно было бы исключить
влияние физического фактора. Поскольку этого не сделано, трудно
определить, какую часть ускоряющего влияния остатков ацетона,
воды и хлороформа следует отнести за счет перевода ими части
твердого тэна в жидкое состояние.
Одной из последних работ по выяснению влияния примесей на
стойкость тэна является исследование Л. Авогадро [8]. Он расши-
рил изучаемый интервал температуры вниз, определил влияние не
только тротила, но и некоторых веществ (диэтиленгликольдинит-
рат, пентаэритрнтацетат), которыми не занимались его предшест-
венники, а также пытался понять причину наблюдавшегося сниже-
ния стойкости тэна. По пробам Анжели при 120° и Талиани при
125° и несколько меньших температурах (до 90°) смеси тэна со
всеми тремя добавленными к нему веществами дают более или ме-
нее сильное снижение стойкости. Эти результаты подтверждаются
и потенциометрической пробой. Если, однако, провести эту пробу
при более низкой температуре (75°), лежащей ниже эвтектической
точки, то стойкость после 60-тидневного нагревания оказывается
для смеси с тротилом (50 : 50) даже несколько больше, чем для
тэна (изменение pH от 6,76 до 5,99 в первом случае и от 6,80 до
5,80 во втором). Полученные результаты Л, Авогадро сопоставля-
ет с растворимостью тэна в примешиваемых веществах; оиа состав-
ляет при 90 и 100° соответственно для тротила 25 и 66 частей на
100 частей растворителя, для пентаэритритацетата 30 и 100 частей
и для диэтиленгликольдинитрата 22 и 66 частей. Слабое влияние
примесей указанных веществ на стойкость гексогена Авогадро свя-
зывает с относительно низкой его растворимостью в них. Этим объ-
ясняется и уменьшение в случае тэна влияния примесей с пониже-
нием температуры и резкое повышение стойкости смесей ниже 75°
Отсюда Л, Авогадро справедливо заключает, что если смеси не
содержат жидкой фазы, то отсутствует и снижение стойкости.
Из приведенного обзора опубликованных по рассматриваемому
вопросу работ видно, что исследователи постепенно приближаются
к правильному пониманию механизма п значения влияния нитросо-
едплений и других примесей на стойкость тэна. Медленность этого
приближения имеет своеобразную причину. Все вышеупомянутые ис-
следователи забыли две работы 111], [13], опубликованные еще в
1920—21 гг., в которых на примере тетрила было показано, что
скорость разложения в расплаве или растворе гораздо больше, чем
в твердом состоянии. Это наблюдение полностью приложимо к слу-
246
чаю тэна, как было показано еще в 1939—1940 гг. в неопубликован-
ной работе К. К- Андреева и В. П. Маслова (МХТИ им. Менделе-
ева). В этой работе, поставленной для разъяснения результатов
исследования Урбанского, Квятковского и Миладовского, стойкость
тэна сопоставлялась по методу Талиани со стойкостью его смесей
с тротилом [. Было установлено, что при температурах ниже 140°
стойкость смесей значительно ниже стойкости тэна, в то время как
выше этой температуры (опыты проводились при 145°) разнима в
стойкости отсутствует. Было установлено также, что по крайней
мере на начальных стадиях скорость распада тэна в тротиловом
растворе не зависит от концентрации, т. е., что распад является ре-
акцией первого порядка. Из независимости скорости распада от
примеси нитросоединений при температуре выше температуры
плавления тэна (14Г) было сделано заключение, что «Скорость
разложения чистого тэна не меняется при добавлении к нему ни-
тросоединений, не образующих с ним химических соединений»; и
далее «Литературные указания по этому вопросу (подразумевают-
ся работы Урбанского, Квятковского, Миладовского) не соответст-
вуют действительности, вследствие упущения из виду различия
агрегатных состояний при проведении сравнительных опытов».
Рассматриваемая в настоящей статье работа имела целью в
плане обшего исследования термического разложения нитроэфиров
изучить термический распад тэна при различных условиях его те-
чения, а также влияние некоторых примесей к нему с тем, чтобы
попытаться дать объяснение ранее установленным особенностям
данного ВВ1 2.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методика работы
В настоящей работе был применен манометрический метод в
том его варианте [1], который имеет ряд существенных преимуществ
перед методами, применявшимися ранее: исключение перегонки
продуктов в холодные части прибора, а также влияние на процесс
распада воздуха, паров запорной и манометрической жидкостей.
Кроме того, вещество и продукты его разложения находятся в
контакте только со стеклом. Разложение испытуемого вещества на-
чиналось в вакууме или в атмосфере тех газов или паров, влияние
которых изучалось.
1 В работе были проведены также опыты по изучению установлен-
ного Урбанским и сотрудниками стабилизирующего влияния нитросоединений на
разложение нитроклетчатки. Исходной позицией было предположение, что распад
нитроклетчатки начинается поверхностной реакцией, которад. при покрывании
поверхности пироксилина жидкостью тормозится так же. как тормозится, напри-
мер, распад азида кальция парафином. Опыты показали, что парафин заметно
замедляет разложение нитроклетчатки; однако, учитывая обнаружившееся влия-
ние многих факторов на течение распада, авторы не считают свое предположение
окончательно подтвержденным этими опытами.
2 Более подробное описание методики и результатов работы см. [4].
247
Исследуемый тэн тщательно освобождался от летучих примесей,
которые могли влиять на его распад. Для этого система откачива-
лась до давления не выше 10-3 мм рт. ст. при одновременном на-
гревании реакционного сосуда водой с температурой 80—100°. При
подготовке опыта по разложению смесей тэн + ТНТ откачка от ле-
тучих примесей производилась в два этапа. Первоначально в реак-
ционный сосуд вводился только ТНТ, боковой отросток сосуда
закрывался резинкой с зажимом, и тротил откачивался в расплав-
ленном состоянии при температуре 80—90°. Периодическое охлаж-
дение и нагревание тротила в значительной степени облегчает его
освобождение от летучих примесей, так как последние очень бурно
выделяются в момент кристаллизации. После полного выделения
летучих примесей тротил охлаждался и вводилась навеска тэна,
после чего боковой отросток реакционного сосуда запаивался и
производилась откачка так же, как и одного ТНТ.
При исследовании применялся тэн заводского производства,
дважды перекристаллизованный из чистого ацетона охлаждением,
с температурой плавления 141,3° и с размерами кристаллов мень-
ше 1 мм.
Для опытов по разложению тэна в тротиловом растворе был
применен ТНТ с температурой плавления 80,6°.
Результаты
Распад тэна в расплаве
Для того, чтобы иметь возможность сопоставить тэн в отноше-
нии характера и скорости распада с другими изученными нитро-
эфирами, температура плавления которых гораздо ниже, было це-
лесообразно провести опыты, при температурах выше температуры
плавления тэна, т, е. с его расплавом. По обычной неавтоматизиро-
ванной методике это можно осуществить в интервале температур
от 141 до примерно 170°. При более высоких температурах разло-
жение с одной стороны идет слишком быстро, и с другой — период
полураспада становится соизмеримым со временем прогрева со-
суда.
Опыты были проведены при умеренных степенях заполнения ре-
акционного сосуда б, чтобы иметь возможность наблюдать течение
распада не только на начальных его этапах. Кривые газообразо-
вание— время и скорость газообразования — время в интервале
температур 145—171° (фиг. 5) показывают, что общий ход распада
сходен с тем, который наблюдается для других нитроэфиров (нит-
роглицерин, нитрогликоль, нитроклетчатка). Газообразование на-
чинается сразу после помещения реакционного сосуда в термостат
и идет с возрастающей скоростью, которая после прохождения
через максимум уменьшается. Газовая фаза приобретает желтую
окраску, интенсивность которой проходит через максимум и исче-
зает к концу распада.
218
Как это наблюдалось для нитроглицерина и нитрогликоля, с
увеличением 6 начальная скорость газообразования несколько
уменьшается. В области малых 6 это явление проявляется силь-
нее. Так, при 145° начальная скорость газообразования при
Ун см3/г
Фиг. 5. Изменение газообразования и eqp скорости со временем
при распаде тэна в расплаве в интервале температур 145—17Г
и 6 = 11-10-».
0=3- 10“Л составляла 0,62 н см3]г-мин, а при 6 = 45- 10-4 она рав-
нялась 0,45 нсмг/г-мин (см. табл. 3 и фиг. 6). Кривые скорость
газообразования — время при разных б, приведенные на фиг. 6,
показывают, что максимум скорости также уменьшается при уве-
личении 6. Следует оговориться, однако, что это справедливо толь-
ко для малых б, когда давление продуктов распада не велико и их
249
ускоряющее действие не проявляется, и когда быстро начинается
уменьшение скорости вследствие убыли вещества.
Сходство расплавленного тэна с нитроглицерином и нитрогли-
колем наблюдается и в отношении влияния б на стадии ускорения
распада. Чем больше б, тем резче выражено это ускорение. По
этой причине при больших 6 ускорение успевает быстро наверстать
отставание скорости на начальных этапах еще до того, как распад
начинает замедляться вследствие убыли вещества и скорость по-
степенно становится больше, чем при малых б. Это показывает, что
ускорение разложения нитроэфиров зависит от давления газооб-
разных продуктов распада или, точнее, от их концентрации в кон-
денсированной фазе, так как по закону Генри эта концентрация
пропорциональна давлению.
Причина влияния б на начальную скорость распада менее ясна.
На фиг. 6 для сравнения нанесена кривая разложения нитро-
глицерина. Как видно из графиков и табл. 3, тэн в жидкой фазе
разлагается при температуре 145° примерно в 2 раза медленнее ни-
троглицерина. Для того, чтобы сравнить разложение тэна с други-
ми нитроэфирами (этиленгликольдинитратом — НГЛ и диэтилен-
Таблица 3
Начальная скорость распада тэна и некоторых других нитроэфиров прн
145 н 140й и различных степенях заполнения сосуда (5 )
ВВ t °C m г С.И3 E-104 ЕРцач hcm^/Z'Muh ВЗЧ
“'ВВ иач
Тэн 145 0,0112 22,12 2,9 0,62 .—
Тэн ь 0,0442 22.13 H,8 0,53 ——
Тэп 0,1419 22,65 36,4 0,49 0,49
Тэн 0,1798 22,06 46,4 0,45 0.45
НГЦ 0,1414 20,12 43,6 1,00 —
Тэн 0,7173 151,30 27.1 0,45 —
Тэн 0,0269 5,74 26,7 0,55 0,48
НГЦ b 0,0229 4,78 29,8 1,15 —
Тэн 140 0,0487 9,21 30,2 0,28 0,48
(60 -ный раствор в ТНТ
НГН s 0,0372 5,99 38<7 0,58 —.
НГЛ 0,0844 18,38 30,5 0,19 1,48
ДГДН » 0,0230 6,04 27,3 0,096 i 2,92
ж—навеска ВВ; lzp.c—объем реакционного сосуда; w~скорость газообразования.
:250
tгликольдинитратом — ДГДН) мы выбрали температуру 140°
Цфиг. 7). Правда, при температуре 140° тэн в начале эксперимента
.должен находиться в твердом состоянии, но, как показали прове-
ренные опыты, тэн подвергается быстрому прогрессивному расплав-
лению. Так, в опыте с 6 = 55 10-4 через 12 мин. после погружения
!в термостат, наблюдалось, что конденсированная фаза частично
расплавлена, а через 28 мин. она была уже полностью жидкая.
’Прогрессивное расплавление тэна можно видеть и на графиках,
где хорошо видно заметное ускорение на начальных участках рас-
'пада.
Фиг. 6. Влияние б на скорость газообразования
при распаде расплавленного тэна при 145°,
Числа у кривых—значения 5 - |0\
Из фиг, 7 и табл. 3 видно, что скорость распада тэна при 140°
занимает промежуточное положение между скоростями разложе-
ния нитроглицерина и нитрогликоля. Она в начале разложения
вдвое меньше, чем у нитроглицерина, в полтора раза больше, чем
у нитрогликоля и примерно в трн раза больше, чем у диэтилен-
гликольдинитрата.
Для надежного сравнения распада тэиа при 140° с вышеупомя-
нутыми нитроэфирами необходимо, чтобы с самого начала опыта
тэн находился в жидком состоянии. Это условие мы обеспечили
растворением тэна в инертном при этой температуре растворите-
ле — тротиле.
На начальных этапах распада в случае чистого тэна скорость
составляет приблизительно 0,10 « см3/г мин, в то время как в слу-
чае раствора она заметно выше — 0,28 н см3/г-мин. В дальнейшем
с прогрессирующим плавлением скорость в первом случае возра-
стает, достигая значения скорости разложения раствора, причем
при большой б она становится выше, как и должно быть вследствие
влияния степени заполнения на стадии ускорения распада.
Течение распада тэна при различных температурах, как пока-
зывают графики фиг. 5, в общем имеет сходный характер. Более
J ад
252
J наглядно это видно из фиг. 8, где кривые У==/(т) представлены в
совмещенном виде. На значительной части распада изменение мас-
Г штаба времени приводит к хорошему совмещению кпивых во всем
----------------------------------------------------1 скорость
приблизительно 30% |/коа;
S
Фиг. 9.
коэффициента совмещения
вых V—f(r) от обратной
ратуры.
Зависимость логарифма
Л кри-
тсмпе-
Т температурном интервале от 145 до 171°. Абсолютная
'^газообразования растет до достижения
величина ее на максимуме в 2,5 ра-
за превышает начальное значение,
; после чего начинается уменьшение
| скорости.
!' По зависимости коэффициента
трансформации от температуры
можно рассчитать величину энергии
1 активации. Соответствующие дан-
{ ные, нанесенные на фиг. 9 и поме-
. щенные в табл. 4, хорошо уклады-
ваются на прямую линию и дают
<£=39 000 кал/моль. Если на на-
L чальном этапе считать распад моно-
му молекулярным, то константа скоро-
; сти распада, рассчитанная из на-
чальной скорости газообразования
Ж и 160° составляет 6,86 • 10-5 сек-1.
редэкспоненциальный множитель в уравнении Аррениуса полу-
чается равным 10[5.6 сек~[, J
$
Таблица 4
Зависимость скорости распада тэна от температуры
В 1С4 t°C 1 — -105 Т Д' * С08.М Iff ^совк
10,8 171 225,2 3,14 0,497
11,4 171 225,2 3,32 0,521
11,3 171 225,2 3,19 0,504
11,1 165 228,3 1,69 0 228
10,8 160 230,9 1,05 0,021
10,5 160 230,9 1,00 0,000
11,6 155 233,6 0,572 —0,243
10,8 150 236,5 0,373 —0,427
10,8 150 236,5 0,368 —0,434
11,8 145 239,2 0,203 —0 693
11,8 145 239,2 0,200 —0,699
* Ассви.— ^160°/^’.
253
Распад тэна в тротиловых растворах
Исследование распада тэна в растворах преследовало три ос
новные цели. Сопоставляя скорость распада расплава тэна и рас-
твора его в тротиле, можно было бы установить, влияет ли тро-
тил на распад тэна и каков характер этого влияния. Изменяя кон-
центрацию тэна в растворе, можно было бы выяснить, как она
влияет на скорость распада и, таким образом, установить порядок
ведущих реакций при распаде тэна. Наконец, изучение разложения
растворов позволило бы значительно расширить температурный
интервал исследования термического распада тэна в жидком со-
стоянии в область температур, лежащих ниже температур плавле-
ния тэна.
Сравнительные данные по распаду расплава тэна и его раство-
ров в тротиле при 145° приведены в табл. 5 и представлены на
фиг. 10 Ч Видно, что тротил не только не увеличивает скорость рас-
пада тэна, но напротив несколько снижает ее. Так, при переходе
от расплава к 40%-му раствору скорость уменьшается на 10—15%;
для 5%-ного раствора она составляет около 2/э скорости разложе-
ния расплава. Однако это уменьшение скорости с концентрацией
слишком мало, чтобы можно было сделать вывод о бимолекуляр-
ности ведущей реакции, и связано, по-видимому, с замедлением по-
следующих реакций или же с взаимодействием газообразных про-
дуктов распада тэна с тротилом, идущим с уменьшением объема.
Очень возможно также, что газообразные продукты распада рас-
творимы в тротиле, чем и обусловливается кажущееся уменьшение
их количества.
Таблица 5
Зависимость скорости распада растворов тэна в тротиле прн 145"
от концентрации
5-104 Концентрация тэна в рас- творе % вес. ^нач н см^/г мин wtsh-I-THT нач Wt-.100mhb. “'тэи+ТНТ
тэн.нач И1ТЭН
36,4 100 0,49 1,20 —
31,8 39 0,42 0,86 1,11 0,92
30,7 5 0,30 0,61 0,82 0,68
И,9 100 — — 1,26 —
16,4 13 — — 0,89 0,71
распаду
На фнг. 11 представлены результаты опытов по
50%-ных растворов тэна в тротиле при 120° и разных б.
1 Для опытов по распаду тэна в тротиловых растворах б рассчитывалась как
отношение объема навески тэна к сумме объемов свободного пространства и
навески.
254
О 100 200 300 т мин
AV/Лк нсмЗ/г-мин
Как и при 145° увеличение степени заполнения сосуда б приво-
дит к некоторому уменьшению скорости газообразования на на-
чальном этапе распада. Так,, например, начальная скорость газо-
образования при 6 = 70- 1СН со-
ставляет 0,0218 н смЦг мин, в то
время как при 6—273- 10-4 она
равняется 0,0150 н см3/гмин.
Общая картина развития га-
зовыделения сходна с той, кото-
рая наблюдалась при распа-
де нитроглицерина. На графике
отчетливо заметны два этапа
разложения: начальный этап с
медленно растущей скоростью га-
зообразования и этап быстрого ее
роста, наступающий тем раньше,
чем больше б, т. е. давление га-
зообразных продуктов распада.
Аналогичный характер имеет
течение распада 5%-ных ра-
створов тэна в тротиле при 100°,
как это видно из графиков
фиг. 12 и 13,
Переход от первого этапа ко
второму еще нагляднее виден
на фиг, 14 и 15, изображающих
в логарифмических координатах
зависимость скорости газообра-
зования от давления при 100 и 120°. Кривую каждого опыта можно
представить, как состоящую из двух прямых, идущих под разными,
углами наклона и соответственно отражающих два этапа про-
цесса разложения; начальный, когда скорость распада слабо зави-
сит от давления, и второй
— нсмуг-мин этап, когда скорость за-
fl, 02 г висит от него значительно
О 100 200 300 т мин
Фиг. 10. Влияние концентрации па
газообразование и его скорость при
распаде тэна в тротиловых растворах
при 145°.
Числа у кривых—тэна в тротиле и 5 »
Фиг. П, Изменение скорости газообразования
со временем при распаде тэна в 50%-ных тро-
тиловых растворах при 120° и различных д.
Числа у кривых—t . i0*.
более резко,
В табл. 6 приведены-
коэффициенты уравне-
ния, выражающего ли-
нейную зависимость меж-
ду логарифмами скорости!
и давления: lg w=lgА +
+BIgp на втором Этапе
распада. При 100° так же,
как в случае нитроглице-
рина, скорость газообра-
зования на втором этапе
растет пропорционально
давлению, приблизительно'
255.
Фиг. 12. Разложение тэна и нитроглицерина в 5%-лых троти-
ловых растворах при 100°.
Числа у кривых—Е • 10*.
Фиг. 13. Изменение скорости газообразования 5%-ных раство-
ров тэна и нитроглицерина в тротиле со временем при 100°
и различных 6.
Числа у кривых—S - 10*.
256
Фиг. 14. Зависимость lg w от Igp при распаде
5%-цых растворов тэна и нитроглицерина в тро-
тиле при 100’ и различных б.
Числя у кривых 10*.
Фиг, 15. Зависимость 1g ш от Igp при рас-
паде 50%-ных растворов тэна в тротиле при
120° н различных б.
Чисда у кривых-’S - 10*.
17 451
2
Таблица 6
Зависимость скорости распада тэна, нитроглицерина и их растворов в тротиле
от давления при 100 и 120°
(°C ВВ Содержание нитроэфира в растворе % вес. В .104 1g А в
Тэн 5 30,0 —5,39 1,40
Тэн 5 00,7 —6,84 1,94
100 Тэн 5 114,0 —7,27 1,90
НГЦ 5 51,0 —6,78 1,99
НГЦ чистый 29,0 — 2,4
НГЦ V 655,0 2,2
Тэн 50 70,0 —5,10 1,06
Тэн^ 50 273 0 —5,30 1,04
1'20 НГЦ чистый 75,5 — 2,0
НГЦ к ,216,0 — 2,1
При
однако, скорость распада тэна
во второй степени,
в тротиловом растворе на
»нач.’ Ю3)[нсм3/г-мин]-
Фиг. 16. Зависимость 1g от
ЦТ при разложении тэна в троти-
ловых растворах в интервале тем-
ператур 100—145°,
120°,
втором этапе растет пропорционально
первой степени давления, в то-
время как в случае чистого ни-
троглицерина она при той же
температуре растет пропорцио-
нально квадрату давления.
Экстраполяцией кривых w~ т
на время, равное пулю, были
определены начальные скорости
газообразования в интервале 100—
145° для распада 5%-ных
растворов тэна в тротиле (см.
табл. 7), Величина энергии ак-
тивации, определенная по углу
наклона прямой Ig'Wjaq— у- (фиг,
16), равна 40 100 кал'моль,- т, е,
практически та же, что и для
расплава при 145—171°, Если счи-
тать распад мономолекулярным на
начальном этапе, то константа ско-
рости при 110° составляет 8,57- 10-а сек-1, а предэкспоненциаль-
ный множитель в уравнении Аррениуса получается равным
[О15-8 сек-’.
258
Таблица 7
Зависимость скорости распада растворов тэна в тротиле от температуры
Г с 1 — -Ю5 т В 10-4 Содержание тэна в растворе вес. Х^нач' 1 н см^!г мин 1g (®нач'1№)
100 268,1 60,7 0,93 —0,032
110 261,1 303,0 20,0 3,75 0,574
120 254,4 46,7 4,5 15,50 1,193
130 248,1 59,0 5,8 58,00 1,764
145 239.2 30,7 5,0 300,00 2,477
Распад тэна в твердом состоянии
Если тэн в расплаве или в тротиловом растворе разлагается
со скоростью того же порядка, что нитроглицерин, то распад твер-
дого тэна протекает как в начале, так и по наступлении ускоре-
ния гораздо медленнее. Представление о различии в скоростях
твердого и жидкого тэна при 120° дает график фиг. 17 и табл. 8.
Т а б л и it а 8
Характеристики течения распада твердого тэна и его тротиловых
растворов при различных температурах
*°С Агрегат- ное со- стояние ВВ О'нач-ЮЗ « см^/г-мин нзч w$- Ю3 нем3*/г-мин v-io5 Т IgtoWt-lO3)
®^тя. нач
НО 875 твердый 0,038 99 0,99 26 261,1 —1,420
120 875 В 0,230 65 17,60 76 254,4 —0,638
130 279 м 1,45 10 34,50 24 248,1 0,161
135 52 « 4,50 90,00 20 245,1 0,653
140 55 п 100,00 2,8 318,00 3,2 242,2 2,000
НО 303 20%-ный раствор 3,75 - • 7,90 2,1 —
120 272 50%-ный раствор 15,00 —• • 24,50 1.6 —
130 59 6%-нын раствор 58,00 — 73,00 1,3 —
140 25 60%-ный раствор 280,00 342,00 1,2 -—
* —скорость в момент выделения 5 нсм^г газа.
Начальная скорость распада твердого тэна при 1202 э в 65 раз
меньше, чем для тэна в растворе и примерно в 130 раз меньше,
чем для чистого нитроглицерина. Как и в случае жидкого тэна на
кривых газообразования для твердого вещества можно различать
Фиг. 17. Изменение газообразования и его скорости
со временем при распаде Твердого тэна и тэна в
тротиловом растворе при 120?,
Числа у кривых—Б - Ю<.
2 этапа; начальный, со сравнительно медленным ростом скорости
газообразования, и этап быстрого роста ее со временем.
Характерным для разложения твердого тэна является сильное
ускорение газообразования в зависимости от степени распада. Так,
например, до выделения 5 н см?!г газа скорость газообразования
в случае распада твердого тэна возрастает приблизительно в
40 раз, в то время как при разложении тэна в тротиловом раство-
260
ре ока возрастает только в 1,6 раза. Подобный случай наблюдал
Фармер 111] при изучении распада тетрила. Для объяснения этого
факта он предположил, что происходит прогрессивное расплавле-
ние твердого вещества в ходе его распада.
Надо отметить, что па всем протяжении распада твердого тэна
при i20°, а также и в опытах при 110 и 130° не было обнаружено
появления расплава при быстром вынимании приборчика из термо-
стата и рассматривании кристаллов невооруженным глазом или
лупой, по-видимому, вследствие незначительного его количества,
хотя разложение было доведено до выделения 25 и 55 н см3/г газов.
Однако при температурах, близких к температуре плавления, на-
фиг. 18, Изменение газообразования во вре-
мени при распаде твердого тэна при ПО'
и б 104=875.
пример, 135°, нам удалось качественно наблюдать прогрессивное
плавление тэна. Так, при достижении 2,7% разложения исходного
вещества (^20 « см3/г газов) не было заметно наличия жидкой
фазы, но на стенках сосуда по мерс его охлаждения наблюдалось
образование новых, очень тонких игольчатых кристалликов, а ста-
рые крупные кристаллы уже не имели первоначальных резких очер-
таний. В дальнейшем при 3,7% разложения (~27 н см3/г газов) все
эти явления наблюдались еще резче, очертания старых кристаллов
были почти незаметны и вещество образовало сплошную массу.
Полное плавление наступило приблизительно при 6% разложения
вещества (выделение ~45 « см3/г газов). При 140° полное плавле-
ние наблюдалось гораздо раньше, а именно, при разложении толь-
ко 1% вещества (выделение 6—7 н слг3/г газов).
Результаты изучения распада твердого тэна в интервале
температур 110—140D (см. фиг. 18, 19, 20 и табл. 8), говорят о том,
что на рост скорости распада оказывает действие не только по-
степенный переход твердого вещества в жидкую фазу, но в фактор,
определяющий ускорение распада жидких нитроэфиров, т. с. рас-
творение газообразных продуктов распада в конденсированной
фазе.
Если бы распад ускорялся только за счет прогрессивного плав-
ления, то в момент полного расплавления вещества скорость газо-
образования при распаде тэна ниже его температуры плавления
361
Фпг. 19. Изменение газообразования во времени при распаде твердого
тэна прн 120—135е.
Числа у "кривых—температур* ' опыта в 'С и о - |(j*
(в скобках).
~~ -1Q3 .чск^/г-мин
Фрг. 20. Изменение скорости газообразовании во
времени при распаде твердого тэна в интервале
температур 120—135°.
Числа у кривых—тем перат ура опыта в 'С и / 10* (в скобках).
262
1е могла бы превышать начальной скорости распада его раствора.
До как показывает фиг. 17, скорость распада твердого тэна дости-
гает при 120° значений, в два с лишним раза превышающих на-
чальную скорость распада его раствора при той же температуре,
Несмотря на то, что не только не происходит полного плавления, но
я образование расплава столь незначительно, что он не был заме-
нен при рассмотрении кристаллов с помощью лупы.
Из графиков фиг. 17—20 и табл. 8 видно, что с повышением
температуры разница в начальных скоростях распада твердого
ТЭна и его раствора уменьшается
[и при приближении к темпера-
гуре плавления отношение ~
t г ^иач
стремится к единице.
(. Температурный коэффициент
’Скорости газообразования при
распаде твердого тэна быстро
(увеличивается с повышением
температуры и поэтому, если по-
строить график 1§-шнач— — (см.
фиг. 21), то получается линия
,Е отчетливой кривизной. Все это
i'ifoeopuT о том, что применение
обычного приема для расчета
л^нергии активации и В является
данном случае _________________
['Если рассчитать Е по двум точ-
^кам, соответствующим наибо-
4. лее низким температурам, то
8"она получается 51500 кал/моль,
§ температурах (130—135°) — 72 800
. Эти особенности распада твердого тэна наводят на мысль, что
Жидкая фаза, возможно, имеется уже в самом начале распада до
^образования заметных количеств конденсированного продукта, не-
F реводящего часть вещества в жидкое состояние. Можно предполо-
j'f-Жить, что в появлении жидкой фазы виноваты примеси, содержа-
сь'щиеся в малых количествах в тэне ’. В таком случае начальная
v скорость разложения тэна будет расти с температурой быстрее, чем
1 по закону Аррениуса; вследствие увеличения с ростом те.мперйту-
"ры растворимости вещества в примесях будет увеличиваться и ко-
т личество жидкой фазы. Если это так, то полученные нами началь-
Иые скорости распада тэпа в твердом состоянии являются сум-
мирными скоростями, слагающимися из скорости разложения тиср-
незаконным.
3
2
1
О
~1
-7-----.------Д---1---------7—
Z20 230 2^0 150 Z60
Фиг, 21, Зависимость 1g №чач от 1/^
при распаде тэна в интервале темпе-
ратур ПО—17Г.
-2.
а при двух более высоких
кал/моль.
1 Хившельвуд [13] при исследовании распада твердого тетрила также заклю-
чает, что даже после тщательной очистки тетрил, вероятно, еще содержит неболь-
шое количество примесей, которые, вызывая появление следов жидкой фазы,
' повышают начальную скорость его разложения.
263
до го вещества (к>тв) и скорости разложения жидкого тэна1 (&'»),
+ WTU'
где а - доля жидкого тэна.
Это уравнение с двумя неизвестными (it и штв). Для его реше-
ния необходимо определить одну из этих величин.
Однако, пренебрегая в указанном уравнении членом
(1—a)wTD. т. е. принимая условно, что разложение идет только за
счет жидкой фазы, можно получить следующие предельные значе-
ния количества последней при разных температурах:
(a)llO'J — -Л~~=0,010 или 1,0% 3,75
--0,015 или 1,5%; 15,0
= = 0,025 или 2,5%; 58
(«)|40 - -0,35 или 35%. L80
Ясно без пояснений, что действительные значения будут ниже.
Из табл. 8 также видно, что величина ускорения распада твер-
дого тэна, т. е. отношение скорости при определенной степени рас-
пада к начальному ее значению, с повышением температуры про-
ходит через максимум. Здесь, по-видимому, проявляется одновре-
менное действие двух факторов.
С одной стороны, как мы видели выше, с повышением темпера-
туры разница в начальных скоростях распада твердого и жидкого
тэна уменьшается и при этом тем сильнее, чем ближе температура
плавления. Поэтому при приближении к последней ускорение про-
цесса газообразования (wt/wKA4) уменьшается и становится близ-
ким к ускорению при распаде жидкого тэна.
С другой стороны при низких температурах прогрессирующее
расплавление тэна, вследствие значительного уменьшения его рас-
творимости в конденсированном промежуточном продукте разло-
жения, выражено слабо. Поэтому, несмотря на то, что разница в
скоростях распада твердого и жидкого тэна при низких температу-
рах больше, ускорение газообразования все же оказывается незна-
чительным. При некоторой оптимальной температуре (находящей-
ся между НО—130°) влияние этих двух факторов наиболее благо-
приятно для максимального увеличения ускорения распада.
Распад тэна в парах
Скорость разложения нитроглицерина и нитрогликоля в парах
значительно больше, чем для жидкости, причем для нитроглицерина
в этом случае не наблюдается того ускорения газообразования,
' Допускается, что скорость разложения примесей равна нулю.
264
которое характерно для распада жидкости. Представляло интерес
изучить в этом отношении тэн. При расчете максимальной степени
заполнения, при которой еще отсутствует жидкая фаза, мы вос-
пользовались данными А. Ф. Беляева [2] об упругости паров тэна.
Для 171° упругость пара тэна получилась 6,75 мм рт. ст,, а предель-
ное значение
760-444-22400 1,75
Фиг 22. Распад тэна при теянера гуре 171' и различных б.
Числа У кривых— о - 1и*.
На графиках фиг, 22 и 23 даны кривые газообразования при
171° и разных б, Начальные скорости для трех различных 6 состав
ляют соответственно:
для 6 = 11,4- IO-4 17,9 н см^г-мин;
для 6—0,83 • 10‘1 48,6 н см3/г • мин;
для 6 = 0,43 • 10-4 88,4 н cmz/ г • мин.
Обращает на себя внимание тот факт, что скорость газообра-
зования существенно зависит от 6, возрастая с ее уменьшением.
Это, по-видимому, в основном обусловлено тем, что скорость реак-
ции в газовой фазе, доля которой увеличивается при уменьше-
нии 6, значительно больше, чем в жидкой. Второй особенностью
превращения при малых 6 является то, что участок ускорения со-
кращается и при 6 = 0,43-10“4 практически отсутствует, аналогич-
но тому, что наблюдается для нитроглицерина.
265-
Если принять в соответствии со сказанным выше, что при
6 = 0,43 Ю-4 весь тэн находится в парах, и сопоставить начальные
скорости распада при различных 6, то расчет приводит к заклю-
чению, что тэн в парах разлагается в 6—8 раз быстрее, чем в
жидком состоянии.
Фиг. 23. Изменение скорости газообразова-
ния со временем при распаде тэна при 171а
и различных б.
Числа у кривых S 10\
Были проведены также опыты при заполнении ампулы стеклян-
ными капиллярами; на величине скорости это не отразилось, что
указывает (в отличие от нитроглицерина) на отсутствие заметной
гетерогенной реакции на стенках сосуда.
Влияние воды и кислорода на распад тэна
Известно сильное и своеобразное влияние воды на распад нит-
роглицерина [3], нитрогликоля и других ншроэфиров.
Были поставлены соответствующие опыты при 110—120° и для
тэиа в виде раствора его в тротиле (фиг. 24 и 25). Как и в случае
нитроглицерина, в начале опыта наблюдается снижение давления,
по характер этого снижения в обоих случаях несколько различен.
В то время как в опытах с нитроглицерином на участке кривой дав-'
ленде—время имеется площадка практически постоянного давле-
ния, вслед за которой наступает быстрое его падение, в случае
тэна площадка незаметна, и давление снижается более плавно.
Увеличение начального давления паров воды увеличивает глубину
впадины па графике и сильнее ускоряет процесс газообразования,
сокращая время до начала резкого ускорения распада. Для объяс-
нения природы этой впадины были проведены опыты по раство-
.266
р мм рт.ст.
фиг. 21. Разложение тэна в тротиловом растворе
в присутствии воды при 120е.
'(цела у кривых-5 ’,0*.
ДаЗлечие р ми рт.ст.
Фиг. 25. Разложение тэна в тротиловом растворе в при-
сутствии воды прн
Числа у кривых-3 ЮР
267
рению парообразной воды в расплавленном тротиле при НО—120 .
Как видно из фиг. 24 и 25, кривая снижения давления почти точно
следует ходу кривой для распада раствора тэна в тротиле в при-
сутствии воды. Это говорит в пользу ТОГО, Что основной Причиной
появления впадины является растворение водяного пара в жидкой
фазе.
Фиг. 26. Р.талоне пне твердого тэна в присутствии
иолы и кислорода при 12CF"1.
Г—»нгеты]| тэн; 2—в ирис уте твиц кислорода (pqis —402 -iuf
рт, ст») 3—й присутствии иолы (/V. 5М ж.к рт-’ст.); 4—
HgO
re npiicyicruHii воды it i сисл среда (р„ = 402 .«ret рт. ст,; р,, п ‘
Uj H-jU
154 гег.к рт. ст.). Чис-ia у кривых-S Ю‘,
Результаты исследования влияния воды на распад твердого
тэна при 120° показаны на фиг. 26. В течение около четырех часов
давление остается постоянным, в то время как при распаде веще-
ства без воды за это время давление возросло на 15 мм рт. ст. Из
графика видно, что вода в два с лишним раза сокращает индук-
ционный период до наступления резкого ускорения распада п
приводит к более сильному возрастанию скорости на этом
этапе.
Опыты по распаду жидкого тэна в присутствии кислорода
(фиг. 27) показали, что кислород не влияет на скорость газообра-
зования на начальном этапе распада, но сокращает время до на-
ступления резкого ускорения. Аналогичное влияние оказывает кис-
268
Ело род и при распаде твердого тэна (фиг. 26), Особенно сильно со-
р кращается время до наступления резкого ускорения при совмест-
| ном присутствии воды и кислорода (кривая 4),
f
io же при pn =324 рт» ст. Числа у кривых—5 * 10х-
О3
обсуждение результатов
Полученные данные по скорости распада тэна при различных
температурах, лежащих как выше, так и ниже температуры плавле-
ния, показывают необоснованность представлений о том, что повы-
шенная стойкость тэна определяется особенностями его химической
структуры. Если исключить влияние агрегатного состояния, т. е.
сравнивать жидкий тэп с другими жидкими же нитроэфирами, то
скорости их распада оказываются очень близкими так же, как и
основные кинетические характеристики. Это обстоятельство не яв-
ляется удивительным, так как первичной стадией при распаде нит-
роэфиров считают отрыв МО2-группы, а ожидать существенных раз-
личий в энергии активации этого процесса у таких веществ, как
например, метилнитрат, тэн и нитроглицерин, нет оснований.
Много общего у жидкого тэна с нитроглицерином и другими
нитроэфирами и в отношении развития распада во времени. Это
относится и к двухстадийности процесса, а также к влиянию степе-
ни заполнения реакционного сосуда взрывчатым веществом, увели-
чение которой уменьшает скорость газообразования на первой ста-
дии и увеличивает ее на второй. Если Это последнее влияние несом-
ненно связано с достижением определенного давления газообраз-
ных продуктов распада и соответственно некоторой критической
269
концентрации их в жидкости, то природа ускорения на начально!!
стадии не установлена. По-видимому, это ускорение является след
ствием протекания последовательных реакций, причем на первых
их стадиях газов выделяется меньше, чем на последующих.
Опыты по разложению раствора тэна в тротиле показывают,
во-первых, ошибочность вывода ряда предшествующих исследова-
телей о том, что тротил ускоряет распад тэна. В действительности
присутствие тротила ведет даже к некоторому замедлению газооб-
разования, но и это влияние невелико и тротил может рассматри.
ваться, как почти инертный растворитель. Отсутствие существенной
зависимости скорости распада от концентрации показывает, чти
основные реакции являются реакциями первого порядка. За это
говорит и то, что кинетические характеристики (Е и В) разложе-
ния тэна в тротиловых растворах практически те же, что и для его
расплава, причем В имеет нормальное значение. В этом отношении
наши результаты расходятся с данными А, Робертсона [15], кото-
рый получил для расплава значение энергии активации
47 000 кал/моль вместо 39 500 кал/моль для раствора. Это обстоя-
тельство, возможно, связано с тем, что Робертсон проводил опыты
при более высоких температурах (160—225°), когда заметная и
возрастающая с температурой часть вещества переходила в пары,
которые, как показано в настоящей работе, разлагаются Значи-
тельно быстрее.
Распад тэна при температурах ниже точки его плавления су.
щественно отличается от распада жидкости в первую очередь по
величине скорости начального этапа, которая в 40—100 раз (в за-
висимости от температуры) меньше, чем для расплава или раство-
ра. Это обстоятельство не является удивительным. Еще Хиншель-
вудом [13] и Фармером [11] на примере тетрила было установлено,
что расплав разлагается значительно быстрее твердого вещества.
Это явление наблюдалось позднее для ряда взрывчатых и невзрыв-
чатых веществ [12]. Причины его, однако, не установлены и то объ-
яснение, которое давал Хицшельвуд, трудно привести в количест-
венное согласие с экспериментальными данными. Таким образом,
различие в скоростях распада твердого и жидкбго тэна представ-
ляет собой частный случай общего явления, как это было показано
еще в 1939—1940 гг. К, К, Андреевым и В. П, Масловым, устано-
вившими, что на скорость распада жидкого тэна (при 145°) тротил
не влияет. Случай этот сложнее рассмотренного Хиншельвудом для
тетрила. Там разложение приводит к образованию устойчивого
промежуточного продукта (по Хиншельвуду — пикриновой кисло-
ты), накопление которого вызывает прогрессирующее ожижение
тетрила. Этот же продукт катализирует превращение. При распа-
де же тэна ожижение может происходить вследствие образования
малоустойчивого промежуточного продукта, концентрация которого
невелика и переход в жидкое состояние тзца может быть лннп-
частичным. Кроме того, в случае тэна сильное ускоряющее влияние
оказывают газообразные продукты распада и таким образом уско-
рение последнего имеет двойную природу. Во всяком случае уско-
270
i, ряющее влияние ожижения полностью объясняет наблюдение пред-
' шествующих исследователем относительно влияния примесей на
' стойкость тэпа. Так, естественно объясняется различием в молеку-
,лирных весах наблюдение Урбанского и сотрудников [19] о том, что
при равном весовом содержании тринитросоединения влияют сла-
. бее, чем динитро, а последние, в свою очередь, чем мононитро.
В принципе правильно объясняет Л, Авогадро [8] различие во вли-
янии тротила, пентаэритрнтацетата и питродиглнколя различной
растворимостью в них тэна. Обоснованными являются также его
. выводы, что при понижении температуры опыта снижение скорости
распада будет становиться из-за снижения растворимости меньше
и что ниже эвтектической точки оно отсутствует. Последний вывод
вполне соответствует заключению К. К' Андреева и В. П. Маслова
(1940 г,), сделанному ими исходя из отсутствия влияния тротила
1 на скорость распада жидкого тэна.
' Из различия скорости разложения в жидком и твердом агрегат-
. ном состояниях следует, в частности, что для обеспечения макси-
мальной стойкости тэна и аналогичных ему веществ следует доби-
ваться полной очистки продуктов от растворимых в них примесей.
Точно также нужно учитывать возможность ожижения и при ис-
' пользовании сплавов тэна. Правда, в этом случае следует иметь
; в виду, что снижение стойкости по указанной причине может па-
J блюдаться лишь, если часть продукта переходит в жидкое состоя-
‘ нпе, т, е. при относительно повышенных температурах. Нет оспова-
i1 ний ожидать, например, что тротил будет оказывать влияние на
скорость распада тэна при комнатной и близких к ней температу-
рах.
Значительное различие в скоростях распада твердого и жидкого
вещества может служить источником ошибки при определении ки-
нетических характеристик разложения при температурах ниже тем-
пературы плавления. Допустим, что продукт, который мы считаем
чистым, содержит небольшое количество растворимых примесей.
Если мы определим скорость распада при относительно низкой тем-
• пературе, то все вещество будет находиться в твердом состоянии
и разлагаться соответственно с малой скоростью. Определяя ско-
рость вблизи (но ниже) температуры плавления, мы будем иметь
часть продуктов в жидком состоянии и скорость распада будет
больше не только вследствие того, что температура выше, но и за
счет частичного ожижения вещества. Не учитывая этого последнего
обстоятельства и ведя расчет энергии активации по обычной фор-
муле E — R In мы получим завышенное значение Е а
J ktT2-T
как следствие, и В, Следует, однако, добавить, что если это объяс-
нение справедливо, то скорость распада твердого тэна, свободного
от примесей, при всех температурах во много раз меньше, чем
Жидкого, причем действительное соотношение скоростей меньше,
Чем давал эксперимент для температур, близких к температуре
плавления,
271
Независимо от объяснения сильной температурной зависимости
скорости, из самого этого факта следует, что скорость распада твер-
дого тэна при низких температурах действительно мала и много
меньше, чем для жидких нитроэфиров, включая и раствор тэна.
Все сказанное о твердом тэне относится к начальному этапу
распада; в дальнейшем, когда наступает частичное ожижение
тэна, а также проявляется ускоряющее действие газообразных про-
дуктов, скорость возрастает. Это возрастание больше, чем для
жидкого тэна, где действует один фактор ускорения, и различие в
скоростях становится меньше, но все же скорость для твердого тзна
при прочих равных условиях всегда остается меньшей.
Результаты изучения разложения тэна в парах интересны в том
отношении, что подобно другим нитроэфирам, тэн в этом агрегат-
ном состоянии характеризуется большей скоростью распада, хотя
разница между жидкостью и паром меньше, чем при переходе от
твердого состояния к жидкости; ускорение отсутствует; гетероген-
ной реакции на поверхности стекла, по-видимому, нет.
Аналогия с другими нитроэфирами наблюдается и в отношении
действия воды и кислорода, которые, не оказывая заметного влия-
ния на начальный этап разложения, сокращают время до наступле-
ния ускорения.
Все эти наблюдения при термическом разложении тэна уклады-
ваются в общую схему распада нитроэфирсв, согласно которой
начальной реакцией распада является отщепление группы NO2.
Двуокись азота затем окисляет органический остаток, восстанавли-
ваясь до NO. При этом накапливается вода и развивается гидро-
литическая реакция. Скорость этой реакции резко возрастает по
достижении определенной концентрации кислоты, образующейся
при гидролизе и путем взаимодействия NO2 с водой. Концентрация
кислоты и воды в жидкой фазе зависит также и от их давления над
жидкостью, которое в свою очередь определяется степенью запол-
нения реакционного сосуда веществом. В присутствии кислоты ско-
рость гидролитической реакции столь велика по сравнению со ско-
ростью реакции первого этапа, что наступление ее означает
сильный и резкий подъем скорости, который составляет одну из
основных особенностей распада нитроэфиров и при соответствую-
щих условиях может привести к тепловому возникновению
взрыва.
Добавление к тэну воды делает возможным с самого начала
протекание гидролитической реакции. Кроме того, при этой реак-
ции образуются не окислы азота, а, судя по данным для нитрогли-
церина, — непосредственно азотная кислота,
Ускоряющее действие кислорода следует приписать переводу
окиси азота в двуокись, что, с одной стороны, ускоряет окислитель-
ные реакции, а с другой, — способствует увеличению концентрации
кислоты. Особенно сильное влияние кислорода проявляется при
термическом разложении питроэфиров вроде нитроклетчатки и ди-
гликольдииитрата, продукты начальных этапов распада которых'
сильно восстанавливают NO2, В результате этого восстановления
272
[в отсутствие кислорода распад авто стабилизируется, то есть резкого
[его ускорения вообще не наступает, по крайней мерс при повы-
I шейных температурах (выше 100”).
ВЫВОДЫ
СОСТОЯНИИ
наиболь-
I Распад тэна в расплаве, растворе, парах и твердом
Г изучался манометрическим методом. Скорость распада
I шая в парах и наименьшая в твердом состоянии. Расплав и раствор
I (в тротиле) одинаковы по скорости разложения и занимают про-
I межуточное положение, близкое к другим нитроэфирам (нитрогли-
церин, нитрогликоль). Распад в парах идет со скоростью, убываю-
I щей во времени, В жидком и твердом состоянии наблюдаются две
Г макростадии распада: а) с малой слабо возрастающей скоростью,
Г не ускоряемая газообразными продуктами распада и б) характери-
I зующаяся сильным ростом скорости в связи с гидролитическим
[ действием образующейся воды, усиливаемым кислыми продуктами
[ распада,
г Скорость разложения твердого тэна возрастает также в резуль-
I тате его частичного ожижения продуктами распада. Сравнительно
[ высокая стойкость тэна при обычных температурах обусловлена
г, тем, что он находится в твердом агрегатном состоянии. Тротил
Е несколько снижает скорость разложения жидкого тэна и может
ускорять распад твердого тэна лишь постольку, поскольку часть
< последнего переходит в жидкое состояние.
ЛИТЕРАТУРА
1, А нд рее в К, К,, Глазкова А. П., М а у р и н а Н, Д иСветлов Б. С.,
ЖФХ, 1958, 32, 1726.
- 2. Беляев А. Ф., ЖФХ, 1948,22.91,
3, Б е с п а л о а Г. Н,, Исследование термического распада НГЦ в присутствии
ь воды. Диссертация, /АХТИ. 1959.
К 4, Кайдымов Б. И. Исследование термического распада пентаэритрит-тет-
I ранитрата. Диссертация, МХТИ, 1959.
ь' 5. А и b е г t е 1 п Р., Mem. des Poudres, 1951, 33, 175.
t 6. AuberteinP., Mem. des Poudres, 1953, 35, 103.
[’ 7. A u b e r t e i n P., Мёт. des Poudres, 1953, 35, 91,
8. A v о g a d г о L., di Cherr ione, Ann. Chi m., 1955, 43, 531.
9. В о u г j о 1 G.,, Mem, des Poudres, 1953, 35, 83.
10, DesseigneG., Мёт. des Poudres, 1950, 32, 137.
, 11. Farmer R. C_, Jourti. chem. Soc., 1920, 117, 1432.
12. В awn С. E. H., in Chemistry of the solid State; by Garner W, E,, But-
terworths scientific publ., 1955, 254.
13, Hinshelwood C, N., Journ. chein. Soc., 1921, 119—120, 72|.
14. P 1 с а г d о G., Annali dt Chimica applicata, 1942,32, 235.
15. Robertson A. J. B., Journ. Soc. chem. Ind., 1948, 67, 221.
16. Stettbacher A., Zs. g. Schjess- und Sprengstoff, 1916, 11, 112.
17. S t e 11 b a c h e г A., Zs. ang. Chem., 1928, 41, 716, 1226.
18. T о n e g u 11 i M„ La chimica e 1'lndustria, 1935, 17,517.
19, Urbanski T_, Kwiatkowski B. und Miladowski M-, Zs. g.
Schiess-und Sprengstoff, 1937, 32, 1, 29, 57, 85.
20. Urbanski T. und Miladowski M., Zs. g. Schiess-und Sprengstoff,
. 1938, 33, 247.
18 451
273
у,- .
Б. С. СВЕТЛОВ
19. ТЕРМИЧЕСКИЙ РАСПАД
ДИЭТИЛЕНГЛИКОЛЬДИНИТРАТА В ЖИДКОЙ ФАЗЕ
Из числа жидких нитроэфиров наиболее полно изучен в отноше-
нии термического распада нитроглицерин [2], [3], [4], [5]. Для него,
помимо основных кинетических характеристик, были установлены
следующие особенности распада; 1) наличие двух путей распада,
один из которых отличается очень сильной степенью самоускоре-
ния, в результате чего скорость разложения может увеличиваться
в сотни и более раз; 2) способность легко и с самоускорением вза-
имодействовать с добавленной водой.
Эти особенности обусловлены тем, что прн распаде нитроглице-
рина в нем наряду с другими продуктами накапливаются двуокись
азота [6], а также азотная и азотистая кислоты, образующиеся в
результате гидролиза нитроэфира и взаимодействия воды с окис-
лами азота. Указанные продукты и приводят к самоускоряющему-
ся разложению, развивающемуся вследствие гидролиза нитроэфи-
ра водой, образующейся в результате окислительных реакций. На-
копление двуокиси азота и кислот обусловлено, по-видимому, от-
носительно малыми скоростями их восстановления.
Нитрогликоль, а также по данным Б. И, Кайдымова и пента-
эритриттетранитрат в расплаве или в растворе в общем показыва-
ют те же закономерности распада, что и нитроглицерин.
Термический распад практически важного нитроэфира диэти-
ленгликольдинитрата до настоящего времени изучен мало. В отли-
чие от нитроэфиров типа нитроглицерина диэтиленгликольдинитрат
характеризуется наличием эфирной группировки, а также относи-
тельно малым числом нитратных групп, приходящихся на атом
углерода в его молекуле. Можно было ожидать, что указанное об-
стоятельство отразится на закономерностях разложения этого нит-
роэфира.
Для изучения термического разложения диэтиленгликольдинит-
рата была использована манометрическая методика со стеклянным
манометром типа Бурдона, позволяющая изменять степень запол-
нения реакционного объема веществом в широких пределах.
Диэтилетгликольдинитраг готовили обычным способом и после
тщательной отмывки от кислоты хранили в эксикаторе. Температура
затвердевания полученного нитрата в стабильной модификации
составляла 3,1,
Опыты проводились 5 в температурном интервале 60—150°. На
фигурах 1, 2, 3 приведены кривые газообразования во времени для
опытов при 100—150э с малыми и умеренными степенями запол-
нения ампул (6 = 0,0013—0,33)2. Все кривые изменения газообразо-
1 В проведении опытов принимали участие студенты О. И, Сергиенко и
М. П. Кошкин,
s Величина д соответствует отношению объема нитроэфира к объему ампулы,
в которой проводят опыт.
274
вания во времени имеют сравнительно простои вид. Рост давлении
газообразных продуктов разложения протекает монотонно с очень
малым и практически постоянным ускорением. Скорость газообра-
зования в процессе разложения возрастает лишь в 3—4 раза по
уравнению с начальной. В этом отношении диэтилепглнкольдини-
ррат существенно отличается от нитроглицерина, распад которого
в этих условиях сопровождается очень резким ускорением.
Начальная скорость газообразования при термическом разло-
жении диэтиленгликольдцнитрата зависит от б, уменьшаясь с твс-
i.
Фиг.
1. Термическое разложение дизгил^ц.щ-
кольдинитрата при 140 -150'.
Числа у крниых-степень :;а1к()лисни|Г ампулы ]<)-.
(Личением последней, подобно тому, как это наблюдается у нитро-
тлицерина и других жидких нитроэфиров. Зависимость начальной
[.скорости газообразования от температуры подчиняется уравнению
/Аррениуса и представлена на фиг. 4. Наклон прямых на этой фи-
гуре в координатах 1g — у приблизительно одинаковдля разных
i, серий опытов, отличающихся степенью заполнения реакционного
^.сосуда витроэфиром, в то время как абсолютные значения началь-
* вых скоростей газообразования изменяются с величиной б. Энер-
гия активации, подсчитанная для описанных опытов, составляет
|около 42 ккал/моль, а логарифм предэкспоненциальною множителя
16,5. Эти величины близки к тем, которые получены для распада
/нитроглицерина в тех же условиях. Абсолютные значения началь-
ных скоростей газообразования при распаде диэтилепгликольдииц-
Жирата приблизительно в 5 раз меньше, чем у нитроглицерина
й близки к скоростям газообразования при распаде питрогликодя
Я нитроцеллюлозы [1]. Для сравнения этих данных между собой
я таблице приведены значения констант скорости распада, подсчи-
танные по уравнению для моиомолекулярной реакции на начальном
Участке газообразования при б==0,01 (см. стр. 277).
''723
j r j W3 Л
'връиз'М дошхпроои ичмпароосяг tmpg
276
Константы скоростей газообразования при распаде некоторых нитро эф И ров в сек-1
Темпера-
тура в °C
Днэтиленгли-
кольдинитрат
120 2 10~7
100 1,2- 1(Г8
Нитроцсл- Ннтрогли-
люлоза церин
___ !
2-10“7 8-Ю-7
1,3 10 s 5-1(Г8
Нитрогль- Тэн а
коль растворе
Таким образом, диэтиленгликольдинитрат, несмотря на бли-
зость начальных скоростей его распада к скоростям распада дру-
гих жидких нитроэфиров, отличается от них отсутствием стадии
резкого ускорения распада.
Фиг. 5. Термическое разложение диэтилепгдиколь-
дицитрата при большой степени заполнения
ампулы.
Степень заполнения ампульг о: J—0,95; 2—0 т83; о —0,90- 4—0 60;
5-0,90; 6-0,63; 7-0,90; 5-0,77; 3-0Д5,
Известно, что увеличение степени заполнения ампулы способст-
вует развитию самоускоряющегося разложения такихнитроэфироз,
как нитроглицерин, нитрогликоль ц др., так как наступление ста-
дии резкого ускорения распада связано с активным участием в этом
процессе газообразных продуктов разложения [2], [3], [4]. Учитывая
это, можно было ожидать, что увеличение б позволит получить
более резкое ускорение и при распаде диэтиленгликольдинитрата.
Опыты проводились при 60—140° и б от 0,45 до 0,95. Результаты
их приведены на фиг. 5. При 140° газообразование было столь
быстрым, что кинетическую кривую снять не удалось.
Как видно из графиков, разложение в этих условиях протекает
приблизительно так же, как и при малых 6. В начале распада на-
блюдается некоторое ускорение в 2—3 раза, после чего рост дав-
ления происходит почти по прямотк Зависимость начальной скоро-
сти газообразования от температуры также подчиняется уравнению
Аррениуса (см. фиг, 4), причем энергия активации Е составляет
41 ккал!молъ, т. е, величину, практически равную наблюдаемой для
меньших 6. В этом отношении диэтпленгликольдинитрат сильно
отлпчается от нитроглицерина,
у которого величина Е при
низких температурах (40-
100'") и больших б сравнитель-
но мала (29 ккал/моль). В со-
ответствии с этим, если при
1001 разложение нитроглице-
рина протекает в 3 раза быст-
рее, чем днэтиленгликольднни-
трата, то при 60° разница ста-
новится значительно больше и
достигает 20—30 раз.
Таким образом, одной из от-
личительных особенностей рас-
пада диэтиленгликольдинитра
та является отсутствие в опи-
санных условиях опыта того
резкого самоускорения, которое
характеризует разложение дру-
гих изученных жидких нитро-
эфиров (нитроглицерин, иптро-
гликоль, расплав тэна).
Фиг. 6. Термическое разложение диэти-
лснггткольдинитрята в присутствии волы
;.ри 120" и степени заполнения axinv.Hit
6=0.030 >0,033,
(д),тср»кан1г<? золы & ?б вес,:
2-DJ3: >—0/24; 4-0.60.
Опыты, проведенные по разложению диЭтиленглякольдинитра-
та в присутствии воды (см. фиг. 6), показали другую отличитель-
ную особенность этого нитроэфира. В то время как распад нитро-
глицерина, нитро гликоля, а также в известных условиях пентаэри-
триттетрз цитрат?. в присутствии воды характеризуется участком по.
етоянства давлений, за которым следует отчетливая впадина на
кривой р=/(т) с последующим ускорением, наступающим значи-
тельно ращ.ще I' более резким, чем прц распаде сухого вещества,
газообразование при разложении влажного диэтиленгликольдинит-
рата протекаегточно так же, как сухого. Вода не влияет на разло-
женце этого ннтроэфира как в начале, так и на последующих ста-
диях распада. В этом отношении диэтиленгликольдинитрат сходен
с нитроклетчаткой -1].
Третья отличительная особенность, по-видимому, обусловливаю-
щая первые две. заключается в том, что в газообразных продуктах
разложения дпЭтдленгликольдинитрата не обнаруживается двуоки-
си азота. .Визуальное наблюдение показало отсутствие бурой ок-
раски газовой фазы в течение всего распада при различных его
условиях. Для белее надежной оценки были проведены специаль-
ные фотоэлектрокодориметрические опыты1, подтвердившие резуль-
тэты этих наблюдений. Диэтиленгликольдинитрат разлагали в
НЕ, сосуде, снабженном, кроме стеклянного манометра, кюветой
с плоскопараллельными стеклами. Через кювету проходил пучок
света, падающий на фотоэлемент, который фиксировал изменение
оптической плотности газообразных продуктов распада. При рас-
паде диэтиленгликольдннитрата не наблюдалось изменения опти-
ческой плотности газовой фазы, что указывает на отсутствие в ней
NO2 в рамках чувствительности метода, которая составляла по
предварительной градуировке I ,и,« рт. ст. двуокиси азота.
Учитывая отмеченные особенности, можно схематично предста-
ли вить разложение диэтиленгликольдннитрата следующим образом.
Первичным актом разложения, как это предполагают для нит-
роэфиров, является отрыв NO2, что согласуется с близостью на-
Ki. чальных скоростей газообразования и кинетических констант рас-
пада диэтиленгликольдипитрата и других нитроэфнров. Образую-
шаяся двуокись азота очень быстро восстанавливается, по-вцдимо-
му, до NO, не накапливаясь в сколько-нибудь значительных коли-
Щ чествах, в отличие от того, как это происходит при распаде других
нитроэфиров. Восстановление протекает не только при взаимодеи-
ствии двуокиси азота с другими продуктами распада диэтиленгли-
кольдинитрата, но, по-видимому, и при взаимодействии ее с самим
ннтроэфиром г.
Ведущей реакцией, определяющей кинетические константы рас-
пада, является реакция отрыва NO2, как более медленная.
К' Поскольку двуокись азота не накапливается, то не образуются
И;' азотная и азотистая кислоты, способные ускорять гидролитическое
Нг разложение нитроэфпра. Взаимодействие же пнтроэфира с водой,
К наличие больших количеств которой в продуктах распада обнару-
V, жено М. С. Плясуновым, в нейтральной среде, как показали опы-
, ты, идет с очень малой скоростью, Поэтому разложение дцэтилен-
Ж гликольдипитрата пе ускоряется в такой степени, как нитроглице-
К рнна, хотя окислительные реакции протекают со сравнительно
V большой скоростью,
К Сказанное не означает, что разложение диэтиленглнкольдпни-
Д/ трата при всех условиях сохранит описанный характер, Опыты
Ж. Б. А. Лурье по изучению распада этого нитроэфира в присутствии
Ж< ряда примесей показывают, чтс в определенных условиях можно
В;- наблюдать значительное ускорение разложения диэтиденгликольди-
Ж нитрата. Результаты этих опытов рассматриваются в следующей
ж- статье.
Ж Слабое ускорение газообразования, наблюдаемое при опыте,
Ж связано, по-впдимому, с прохождением последовательных реакций.
® включающих и себя как первичный отрыв NO2 с его восстаповле- 1 2
1 Эта методика была разработана В. П. Шелл пути noil, которой аптор ныря-
каст благодарность.
2 Опыты, проведенные Б. А. Лурье, показывают, что двуокжь азота, добавлен-
ная в количестве около J% к диэтилепгликольдинитрату, исчезает во много раз
быстрее, чем протекает разложение самого нитрата.
279
мнем, так и более глубокий распад образующихся при этом соеди-
нений. С этим согласуется, в частности, тот факт, что даже при
сравнительно больших 6 (до 0,03), где достигается по условиям
опыта лишь малая степень распада, состав газообразных продук-
тов успевает претерпевать определенное изменение. Так на началь-
ных этапах условная'доля конденсирующихся при комнатной тем-
пературе газов составляет 50%, в то время как по мере развития
разложения она заметно падает и при достаточно большой степени
распада снижается до 20—30%.
ВЫВОДЫ
1. Изучен распад диэтиленгликольдннитрата в жидкой фазе ts
присутствии продуктов распада при 60—150° и степени заполнения
ампулы нитроэфиром 0,001—0,9.
2. Зависимость начальной скорости газообразования подчиняет-
ся уравнению Аррениуса, где £ = 41— 42 ккал/моль и 1g В~ 16,5, что
очень близко к соответствующим константам разложения других
нитроэфиров в жидкой фазе.
3. В примененных условиях опыта распад диэтиленгликольдшш
трата протекает без резкого ускорения, а также не ускоряется до
бавлением воды.
4. Отсутствие в продуктах разложения диэтиленгликольдинитра-
та заметных количеств двуокиси азота может быть объяснено тем.
что окислительные процессы при распаде протекают значительно
быстрее, чем первичное отщепление двуокиси азота. Это обуслов-
ливает те особенности распада диэтиленгликольдннитрата, которые
отличают его от распада нитроглицерина, нитрогликоля и тэна.
5. При относительно низкой температуре (60° и ниже) разложе-
ние диэтиленгликольдннитрата протекает в десятки раз медленнее,
чем нитроглицерина, что в сочетании с малой склонностью к само-
ускореяию обусловливает сравнительно большую его стойкость.
ЛИТЕРАТУРА
I. Андреев К. К., Самсонов Б, С., ДАН СССР, 1957, 114, 815.
2. Андреев К. К., Глазкова А. П„ М а у р и и а Н. Д. и С в е т л о в Б. С.,
ЖФХ, 1958, 32, 1726.
3. А ч дреев К. К.. ЖПХ, 1958, 31, 484-493.
4. Р о г >i и с к и й С. 3., С а п о ж н и к о в Л, М., ЖФХ, 1931, 2, 80.
5. Светлов Б. С.г Научные доклады Высшей школы СССР, 1958, вып. 3
422—425.
6. R о b е f t. s о п R., J. chetn. Soc., 1909, 95, 1241.
Б. А. ЛУРЬЕ, Б. С. СВЕТЛОВ
20. О ВЛИЯНИИ НЕКОТОРЫХ ПРИМЕСЕЙ
НА ТЕРМИЧЕСКИЙ РАСПАД
ДИЭТИЛЕНГЛИКОЛЬДИНИТРАТА
Термическое разложение диэтиленгликольдинитрата (ДЭГДН)
без удаления продуктов распада протекает в известном отношении
сходно с разложением многих других полинитроэфиров. Близки как
начальные скорости газообразования, так и их зависимости от тем-
пературы, т. е. энергии активации. В связи с тем, что начальным
этапом распада нитроэфиров, вероятно, является разрыв связи
О—NO2, это сходство не удивительно.
Развитие распада ДЭГДН происходит, однако, без резкого уско-
рения газообразования, характерного для разложения таких нитро-
эфиров, как нитроглицерин (НГЦ), нитрогликоль (НГЛ), тэн и не-
которые другие [3]. Это ускорение, как полагают [I], [3], определяет-
ся гидролитическим разложением нитроэфира, самоускоряющимся
вследствие образования кислот в результате взаимодействия воды
с высшими окислами азота, накапливающимися при распаде этих
нитроэфиров [6], [8].
При термическом разложении ДЭГДН не наблюдается побуре-
ния газовой фазы, которое свидетельствовало бы об образовании
значительных количеств NO2. Эту особенность предположительно
определяет восстановительная природа нитроэфира и продуктов
его распада. Поэтому нами было изучено разложение ДЭГДН в
присутствии различных окислителей (О2, NO, NOa, HNO3), а также
восстановителя (НгСгО^).
Другой аспект исследования заключался в выяснении тех усло-
вий, при которых возможно развитие распада ДЭГДН е резким
самоускорением.
Разложение ДЭГДН в присутствии перечисленных примесей изучалось мано-
метрическим методом, описанным ранее [3]. Степень заполнения объема реакцион-
ного сосуда нитроэфиром (S) при 120“ в большинстве опытов составляла
0,014—0,04, а при 100 и 80’ 0,10—0.13.
Исследуемые примеси внодили в ампулу стеклянного манометра, содержа-
щего нитроэфир, предварительно освобожденный эвакуацией от летучих веществ,
Газообразные или легколетучие вещества вводили в прибор из сосуда, предназна-
ченного для измерения их количества, при охлаждении ампулы с нитроэфиром
жидким азотом.
Кислород применяли технический, осушенный в ловушке с жидким азотом;
окись азота готовили в нитрометре Луцге, а двуокись — смешением известного
количества окиси азота с избытком кислорода с откачкой последнего при охлаж-
дении жидким азотом после 1—1,5 час. взаимодействия; азотную кислоту вносили
в виде 98% HNO3 или 20% раствора се в ДЭГДН, после чего при охлаждении
ампулы жидким азотом эвакуировали из нее воздух; щавелевую кислоту добав-
ляли в виде порошка, причем в зависимости от того, изучался ли распад нитро-
эфира в присутствии разбавленной или концентрированной кислоты, применяли
ее кристаллогидрат или обезвоженные кристаллы.
Разбавление кислот водой, если это требовалось по условиям опыта, произ-
водили путем перевода ее паров в ампулу прн охлаждении последней.
Для определения в ходе опыта содержания двуокиси азота в газовой фазе
была применена колориметрическая методика, разработанная применительно к
281
распаду нитроэфиров в жидкой фазе В. П. Шелапутинай, которой авторы прино-
сят благодарность за любезно предоставленную возможность постановки этих
опытов, а также за помощь при.их проведении.
Схема соответствующею прибора приведена на фиг. 1. Кювета / с впвятшымн
,ч нее плоско-параллельными оптическими стеклами 2 соединена со стекля ц вы и
манометром обычного типа 3. применяемым для измерения общего давления. Ма-
нометр имеет ампулу 4 д.тя исследуемого нитроэфира.
После эвакуации воздуха и введения примеси кювета помещалась в термо-
стат 5, причем корны ее плотно закреплялись в гнездах Г>. Затем термостат запол-
нялся предварительно нагретой до определенной температуры жидкостью, после
чего включались перемешивание и регулируемый нагрев. Наличие кюветы нозво-
Фнг. i. Схема прибора для количественного
определения двуокиси азота в ходе распада
нитроэфиров:
1—кювета, 2—плоско-параллельные опгитескггс стик-
ла. 3—етекляинъгй манометр, 4—ампула для ннтро-
эфира, а—жидкостной термостат, 6—гнезда для кю-
веты, Г—источник света, 8—фотосопротипленгге,
3-контрольный светопровод, 10—зеркало, Ы—милли-
амперметр, 12— микроамперметр.
|яло измерять в процессе опыта не только давление, но и изменение оптической
плотности газовой фазы, которая в зависимости от содержания в рей двуокиси
азота окрашивалась в более или менее интенсивны Ге бурый цвет. Для этого луч
от источника света 7 проходил через кювету и фокусировался па фотосопротив-
лении 8. Постоянство силы света лампы, работавшей от аккумуляторов, контро-
лировали по миллиамперметру //. Оптическую плотность г азо ее ой ф.тзы определял ее
н каждый даЕШый леомсцт по силе тока в цепи фотосопротивленик, измеряемой
микроамперметром 12. Д.тя этого кювета нреДваритСлЬЕю градуировалась по дву-
окиси азота. С целью контроля за стабильпостью работы всей системы парал-
лельно со снятием результатов опыта проводили «холостые» измерения с помо-
ецью контрольного светопровода 9. При повороте зеркала 10 прекращается доступ
света к фотосопротивлс.|1шо 8 через кювету. В этом случае свет от своего источника
попадает нп фотосопротивленпе через контрольный светопровод 9, минуя кювету.
Дли затруднении возможного запотевииНН спЕгнческнх стекол 2 в результате
охлаждения их воздухом через торцовые отверстая кюветы концы последней цп
предложению В. 1[. Шелапутнной нагревались электроспиралыО-
О точности измерения количества двуокиси дзота но этому методу можно
судите,, если сопоставить давление Ь1О2 вещлснпой перед опытом, с количествам
ее, измеренным колориметрически. Разница между ними не превышала 10%.
ТЕРМИЧЕСКИЙ РАСПАД ДЭГДН В ПРИСУТСТВИИ КИСЛОРОДА
Кислород может существенно изменять картину газообразова-
ния при термическом распаде ДЭГДН. Разложение этого нитро-
эфира при 120° и достаточно большом давлении кислорода стано-
вится двустадийным процессом, что и следует из характера кривых,
изображенных на фцг. 2. Первая стадия характеризуется неболь-
шим уменьшением скорости газообразования, причем начальное ес
282
значение выше, чем при распаде чистого Нитроэфира. В начале вто-
рой стадии скорость газообразования резко возрастает и при боль-
шом содержании кислорода в десятки раз превосходит максималь-
ную скорость газообразования при распаде чистого ДЭГДН.
Еще резче проявляется влияние кислорода па разложение
ДЭГДН ирп более низких температурах. На фиг- 3 приведены ре-
зультаты опытов при ЮСЕ. При достаточно большим количестве
Фиг. 2. Влияние кислорода на термический распад ДЭГДН
при 120Л
•hrtJJt у кривых—степень ампулы нгг’грчэфпрп.м б 11?
— Ha'ia.'rbMoe давление кислорода в i/.v рь ст- при температуре
(Объс-Ч Ггродуктов :^десь и далео включает оГУы?\г Щ»ичсен).
о.
кислорода (р0 =300—400 -ил рт. ст.) разложение вначале сопро-
вождается быстрым падением давления со скоростью, в 7 раз пре-
вышающей скорость газообразования при распаде чистого ДЭГДН.
Давление падает на 20—30% от начального своего значения. Пос-
ле этого оно начинает расти сначала быстро, а затем более мед-
ленно, но в 7—8 раз быстрее, чем у чистого нитроэфира. Дальней-
ший распад нитроэфира в присутствии кислорода протекает также
очень быстро. При 80° влияние кислорода в общем то же, что и при
100% но падение Давления протекает относительно более резко. При
содержании кислорода, отвечающем начальному его давлению
До. = 200 мл рт. ст., давление за 50 час. упало приблизительно на-
половину.
Таким образом на определенном этапе кислород ускоряет раз-
ложенце ДЭГДН, причем тем больше, чем ниже температура. Са-
мое большое ускорение наблюдается или в начале второй стадии
28.3
(120°) или после падения давления (100—80°); в дальнейшем про-
исходит значительное падение скорости роста давления.
Следует отметить, что распад ДЭГДН в присутствии кислорода
сопровождается накоплением двуокиси азота в газовой фазе. Газо-
вая фаза через некоторое время после начала опыта окрашивается
в светло-бурый цвет, интенсивность которого увеличивается, прохо-
дит через максимум, а затем уменьшается. На несколько более позд-
них стадиях разложения газовая фаза становится снова бесцвет-
ной. Количественная оценка, проведенная с помощью колоримет-
Фиг. 3. Влияние кислорода на термический распад
ДЭГДН при 100?
Чнсда у кривых—степень заполнения ампулы нюроэфиром g.
Pq —начальное давление кислорода в ждг рт. ст, при темпе-
ратуре опыта,
рической методики, показала, что при начальном давлении кисло-
рода 200 мм рт. ст. (120° и 6 — 0,03) максимальное содержание
NO2 в газах (7% объемн.) достигается при возрастании давления
в 1,5 раза. В связи с этим можно было предположить, что влияние
кислорода на ускорение разложения нитроэфпра обусловлено обра-
зованием двуокиси азота при взаимодействии Ог с окисью азота.
Однако возможно, что образование NO2 не является единственной
причиной ускорения разложения ДЭГДН.
Опыты по нагреванию исходного спирта - диэтиленгликоля —
в присутствии кислорода показали, что при этом происходит по-
глощение кислорода со значительной скоростью (фиг. 4), причем
образуются продукты, обладающие свойствами перекисей и кислот.
Возможно, что аналогичные соединения образуются при взаимодей-
ствии кислорода с ДЭГДН и оказывают соответствующее влияние
на разложение последнего.
Кислород влияет на разложение не только ДЭГДН. Он ускоря-
ет по данным Самсонова [21 распад нитроцеллюлозы, которая в
присутствии больших количеств кислорода разлагается так же, как
и нитроглицерин. Разложение последнего может идти быстрее
2*1
в присутствии кислорода, особенно на стадии резкого ускорения
этого процесса. Так, например, при 100" и 6^=0,003 резкое ус-
корение разложения достигается быстрее (примерно в 2 раза) при
Фиг. 4. Взаимодействие дизтнлеигликоля с кис-
лородом при ТОСТ:
ci—зависимость скорости изменения давления от
величины последнего, б—изменение? давления во bjjsj
мени,
начальном давлении кислорода рОг=200—600 ММ рт. ст. При повы-
шенных температурах кислород способен даже качественно изме-
нять картину распада НГЦ. Так при 140° и 6-0,0023 НГЦ разла-
др v>.< рт.ст.
з Дт мин
5 lol
o' lob год зоо w
Время т мим
Фиг. 5. Влияние кислорода на термический
распад НГЦ при 140° и степени заполнения
ампулы нитроэфиром 6=s 0,0023.
—начальное давление к и с .пэр ода в мм рт. ст. при
температуре опыта.
гается с незначительной и практически постоянной скоростью, в то
время как в присутствии кислорода через некоторое время насту-
пает резкое ускорение разложения этого иитроэфира (см. фиг. 5).
28;
термический распад дэгдн в присутствии двуокиси азота
На фиг. 6 приведены кривые изменения давления во времени
для опытов при 120J. Эти опыты показывают, что газообразование
при термическом разложении ДЭГДН протекает тем быстрее, чем
больше введено двуокиси азота. При начальном Pno^ = 120 л<л< рт. ст.
газообразование происходит практически с той же скоростью, что
и в случае чистого нитроэфира. Повышение начального до
220—250 леи рт. ст. пли иначе говоря увеличение содержания дву-
Фиг. 6. Влияние двуокиси азота на термический распад
ДЭГДН при 120°,
Числа у кривых.-степень заполнения а^дулы нитроэфиром б.
-натальное видение дау окиси азота я мм рт- ст. прл темпе-
ратуре опыт,
окиси азота приводит к росту скорости газообразования в 1,5—2
раза, а при pN0. =410 мм рт. ст. — в 3 раза, по сравнению с опы-
том без добавления S'O^.
Прн 100° (фиг. ") влияние двуокиси азота на разложенце
ДЭГДН в общем сходно с тем, которое наблюдается при 120°.
Разница состоит лишь в том, что в начале опыта с большим содер-
жанием NO? появляется небольшой участок падения давления (на
фиг. 7 он не заметен; см. фиг. 9, где этот участок показан); даль-
нейший рост давления происходит в течение некоторого времени
почти по прямой, причем скорость газообразования при большом
содержании NOs выше скорости разложения чистого ДЭГДН в
3—10 раз. Относительно небольшое количество двуокиси азота
(начальное рыоа =60 мм рт. ст.) значительно (в 3 раза) снижа-
ет скорость газообразования на первых этапах разложения нитро-
эфира в сравнении с опытами при больших начальных рыо.; даль-
286
нейшее разложение идет с той же скоростью, что и у чистого про-
дукта.
Особенно сильно двуокись азота ускоряет распад ДЭГДН на
.начальном этапе при 80" (фиг. 8). При Pno.^75 мм рт. ст. ско-
рость газообразования, рассчитанная, как средняя в течение пер-
вых пяти часов опыта, превышает скорость в опыте с чистым
ДЭГДН в 20 раз, а при pNo, = 190 мм рт. ст. — в 200 раз. Кривые
давление—время па некотором этапе распада имеют насыщаю-
щийся характер.
Фиг. 7. Влияние двуокиси азота на термический
распад ДЭГДН при 100°.
4исла у кривых—степень заполнения ампулы ни’роэфиром 6.
—начальное давление двуокиси азота b .w.w рт. ст. При
температуре опыта.
Визуальное наблюдение за цветом газовой фазы, показывает,
что при разложении ДЭГДН в присутствии двуокиси азота, содер-
: жание последней не остается постоянным, а заметно уменьшается,
^.причем, если опыт проводить достаточно долго, то можно добиться
.полного обесцвечивания газовой фазы,
Учитывая, что NO^, являясь предположительно первичным про-
дуктом распада нитроэфиров, отсутствует в газообразных продук-
тах разложения ДЭГДН, оказалось интересным количественно
проследить за скоростью исчезновения двуокиси азота в опытах с
добавлением последней.
На фиг. 9 приведены кривые изменения давления двуокиси
азота во времени в сравнении с кривыми, характеризующими об-
щее давление. При рассмотрении этих кривых видно, что давление
двуокиси азота практически на протяжении всего опыта умень-
шается со скоростью, наибольшей вначале и монотонно изменяю-
щейся по сложному закону, который на отдельных участках может
быть апроксимирован уравнением — ^р",где п — больше едини-
d х
цы, но меньше двух,
Уменьшение давления двуокиси азота в течение некоторого
времени происходит намного быстрее
Фиг, 8. Влияние двуокиси азота на термиче-
ский распад ДЭГДН при 80'.
Числа у кривых- степень заполнения ампулы низро-
эфиром
л т -начальное давление двуокиси азота» хл рт» ст»
' N<)2
при температуре опыта»
увеличения общего давления.
На начальном участке при
140° скорость уменьшения
давления двуокиси азота
или иначе говоря исчезнове-
ния последней превышает
скорость увеличения общего
давления более чем в 2 раза,
а при 120° —в 5 раз. Прн
100° и особенно при 80°ско-
рость исчезновения NO;
уже настолько превышает
скорость общего газообра-
зования, что на кривой дав-
ление-время наблюдается
впадина, глубина и продол-
жительность которой при
80° больше, чем при 100°.
Если при этом сравнивать
участки после падения дав-
ления, то скорость уменьшения содержания NOs превышает ско-
рость общего газообразования в 2—5 раз,
Фиг. 9. Изменение содержания двуокиси азота при раз-
ложении ДЭГДН в ее присутствии.
О—общее давление*.
давление NO2»
Таким образом, разложение ДЭГДН в присутствии двуокиси
азота характеризуется ее исчезновением из газовой фазы со ско-
ростью, превышающей скорость общего газообразования.
ТЕРМИЧЕСКИЙ РАСПАД ДЭГДН В ПРИСУТСТВИИ КИСЛОТ
Основной особенностью разложения подкисленного ДЭГДН яв-
ляется резкий рост газообразования после небольшого индукцион-
ного периода. По прохождении максимума происходит быстрое
снижение скорости газообразования (фиг. 10, 11, 12 и 13). Газовая
фаза при этом, вначале бесцветная, окрашивается в бурый цвет
Фиг. И, Термический распад ДЭГДН
в присутствии концентрированной
азотной кислоты при 80° и степени
заполнения ампулы нитроэфиром
Й = 0,Н. (Содержание кислоты в нит-
роэфире 1,4% вес,).
/гаог
Фиг. 10. Термический распад ДЭГДН в
присутствии концентрированной азотной
кислоты при 120".
' Числа у кривых — степень заполнения ампулы
' нитроэфиром 6 и содержание кислоты в ни-
троз фи ре в V» вес. .
‘двуокиси азота, пропадающий затем по мере уменьшения скорости
газообразования. Доля NO2 в газообразных продуктах разложения
велика и достигает 30% прн добавлении |,2% щавелевой кислоты.
Наибольшее содержание двуокиси азота наблюдается вблизи мак-
симума скорости газообразования.
На фиг. 10 приведены кривые изменения общего давления во
'времени при разложении ДЭГДН в присутствии азотной кислоты
при 120°. Кислота оказывает свое влияние на ускорение разложе-
ния нитроэфира с самого начала опыта, увеличивая начальную
‘скорость газообразования по сравнению с чистым ДЭГДН в
50—100 раз в зависимости от содержания кислоты. К достижению
'максимума эта скорость увеличивается еще в 3 раза, после чего
.'резко падает.
С понижением температуры опыта относительная величина мак-
.симума скорости увеличивается; в то же время влияние кислоты
’Wa начальную скорость газообразования меняется мало. Так при
"'100° (6^0,1, содержание HNO3—1 — 1,5%) скорость в началеопы-
та в 30—40 раз выш§, чем у чистого нитроэфира; на максимуме
19 4S1 289
она возрастает еще в 15—30 раз. При 80° (фиг. 11) скорость по до-
стижении максимума уже в 60 раз выше, чем начальная. При
этом разложение протекает с явно выраженным индукционным пе-
риодом.
Разбавление добавленной к витроэфиру азотной кислоты во-
дой приводит к тому, что на начальном этапе разложения общее
давление (фиг. 12) после достижения равновесного состояния, со-
ответствующего количеству добавленной HNO3, не только не растет.
фиг. 12. Термический распад ДЭГДН в при-
сутствии азотной кислоты различной кон-
центрации при 12СГ.
Числа у кривых — степень заполнения ампулы
[гитроэфиром д и концентрация эзОтной кислоты
в вес. (Содержаще кислоты в нИтроэфнре
вес.).
но заметно уменьшается, достигает минимума и только потом
чинает увеличиваться со значительным ускорением. Величина
максимума скорости газообразования увеличивается с разбавле-
нием кислоты водой.
Чтобы исключить окисляющее действие азотной кислоты на ни-
троэфир или продукты его распада, была применена щавелевая
кислота, обладающая восстановительными свойствами. Однако это
не изменило общую картину разложения кислого ДЭГДН. На
фиг. 13 приведены кривые давление—время для опытов в присут-
ствии щавелевой кислоты различной концентрации. Эти опыты по-
казали, что безводная кислота обладает наибольшей способностью
ускорять газообразование, Она увеличивает начальную скорость
его более чем в сто раз. На максимуме, наступающем очень быст-
ро, скорость возрастает еще в два раза. 1
1 Наличие максимума скорости, возможно, связано с тем, что щавелевая
кислота к Е1ачалу опыта полностью не растворяется в иитроэфире. Растнорение ее
наступает лишь спустя некоторое небольшое время после погружения прибора
в термостат.
290
Прямейшие раствора щавелевой кислоты, содержащей около
20% воды, риволит к уменьшению начальной, а также максималь-
ной скорое^ газообразования по сравнению с опытом с безводной
добавкой в 1,5—2 раза. При использовании в опытах 36%-ной ща-
велевой кисдоты на начальном этапе наблюдается участок умень-
шения дав^ниЯ> подобно опытам с разбавленной азотной кисло-
той. Однакс в отличие от
этих опыто., в случае раз-
бавленной цавелевой кисло-
ты газооб[азовапие после
прохождения минимума дав-
ления происходит значитель-
но медленье, цсм с менее
разбавлений’или с сухой
кислотой.
Таким образом, разложе-
ние ДЭГД1) в присутствии
изученных :ислот протекает
значительш быстрее и с
большим ускорением, чем в
их отСутствщ. Основным от-
фиг. 13; Термический распад ДЭГДН в при-
сутствии щавелевой кисдотц различной кон-
центрации при 120°.
Числа у кривых — степень заполнения ампулы
ннтрозфяром 6 и концентрация щавелевой кисло-
ты в ’/» вес. (Содержание в нитрозфире
:и 34. .
личием в действии азотной
и щавелевое кислот являет-
ся количественно различное
влияние во1ы на разложе-
ние кисло-q иитроэфира.
В случае Потной кислоты
разбавление водой приво-
дит к более резкому ускорению разложения ДЭГД в сРа
с действием крепкой кислоты; разбавленная щавеле^я
ускоряет эт-)Т процесс слабее, чем концентрированная.^ УТКгпД
Следует отметить, что азотная кислота ускоряет, расц^д МП
значительнс сильнее, чем разложение НГЦ.
ТЕРМИЧЕСКИЙ РАСПАД ДЭГДН В ПРИСУТСТВИИ ОКИСИ АЗОТА
Как показали опыты, окись азота может существенно изменять,
особенно в начале разложения нитроэфира, характер газообразо-
вания. Так При 100° (фиг. 14) добавление окиси азота в количест-
ве, соответствующем начальному его давлению pNo=10S мм рт. ст.
при температуре опыта приводит к тому, что общее давление р
в течение примерно Ю час. снижается, причем величина этото сни-
жения составляет 20% начального давления. Затем общее давле-
ние начинает увеличиваться приблизительно с той же скоростью,
что и в опь1те с чистым ДЭГДН. Повышение количества окиси
азота увеличивает абсолютную величину снижения общего давле-
ния, нс изменяя относительного значения ее. Скорость газообразо-
вания после прохождения минимума давления растет несколько
быстрее прц большем содержании окиси азота, Аналогичная в об-
тем картина наблюдается при 120 и 80э. При 120° снижение дав-
ления происходит лишь при содержаниях окиси азота, отвечающих
начальному ее давлению 575 мм рт. ст. и более при температуре
опыта. В этих условиях снижение давления невелико и составляет
лишь 3% начального. При меньшем количестве NO давление не
падает; рост его в начале разложения происходит медленнее, чем
в опыте с чистым нитроэфиром.
Время х ми//
Фиг. (4. Влияние окиси азота на термический распад
ДЭГДН при 10(Г,
Числа у кривых—степень заполнении ампулы нитроэфиоом 8 и
начальное давление окиси азота и мм ;>т. ст. при температуре опыта.
(Ось ординат р ят рт. ст,)
При 80э снижение давления наблюдается на протяжении
100 час., происходит оно более резко и по величине составляет око-
ло 30% начального давления окиси азота.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Полученные результаты опытов показывают, что вещества, при-
мененные в качестве примесей к ДЭГДН, в той или иной степени
изменяют картину газообразования при разложении этого нитро-
эфира, причем не только количественно, но и качественно.
Распад ДЭГДН в присутствии кислорода, двуокиси и окиси
азота, а также сильно разбавленных кислот в определенных усло-
виях характеризуется прежде всего взаимодействием газовой фазы
с жидкостью, приводящим к падению давления в результате
связывания газообразных продуктов. Если уменьшение давления
при разложении ДЭГДН в присутствии разбавленной кислоты с
последующим резким ускорением может быть объяснено прохож-
дением гидролитической реакции с образованием хорошо раство-
римой в нитроэфире азотной кислоты, как это предполагается для
аналогичного случая в НГЦ Ц], [4], тр взаимодействия кислорода,
292
двуокиси и даже окиси азота с нитрозфиром, протекающего с
уменьшением объема, ранее не наблюдалось и оно является, по-
’ видимому, следствием других процессов.
t Рассмотрим возможные пути протекания реакции ДЭГДН с
указанными веществами.
Сопоставляя результаты опытов по распаду ДЭГДН в присут-
г ствии кислорода с опытами по взаимодействию последнего с дизти-
; ленгликолем, а также, учитывая способность простых эфиров об-
, разовывать малолетучие перекисные соединения, можно предполо-
; жить, что аналогичные реакции происходят и в случае ДЭГДН.
Образующиеся продукты, по-видимому, ограниченно стойки при
нагревании, поскольку величина снижения давления, характеризу-
i ющая образование указанных соединений, сильно зависит от тем-
;! пературы. С этим согласуется также и тот факт, что при анализе
продуктов взаимодействия диэтиленгликоля с кислородом, коли-
чество последнего, способного вытеснять йод из йодистого калия,
j составляет меньше 10% вступившего в реакцию газа, остальная
Г часть которого находится, по-видимому, в виде продуктов распада
£ перекисей и, в частности, в виде кислоты.
f Большая скорость газообразования при дальнейшем разложе-
нии ДЭГДН, наблюдающаяся после прохождения минимума дав-
1; ления, вероятно, обусловлена совокупностью процессов, включаю-
!'• щей в себя распад образовавшихся продуктов, а также участие их
у. и, в частности, кислот, в разложении самого нитпоэфира. Кроме
ji этого, кислород может способствовать разложению ДЭГДН, окис-
Л ляя окись азота до двуокиси, которая может, образуя с водой кис-
лоты, ускорять развитие гидролиза основного продукта.
Уменьшение давления в начальный период нагревания ДЭГДН
I. в присутствии двуокиси азота может быть следствием увеличения
; растворимости последней в разлагающемся нитроэфире или обра-
зования сравнительно малолетучего соединения, аналогичного тому,
у которое получается при взаимодействии двуокиси азота с ди-
". этиловым эфиром [5]. Последнее предположение косвенно подтвер-
ждается большой скоростью взаимодействия диэтиленгликоля с
двуокисью азота с образованием малолетучих продуктов.
Поскольку окись азота растворяется в ДЭГДН намного хуже
двуокиси, то уменьшение давления при распаде этого нитроэфира
g в присутствии NO, по-видимому, происходит в результате образо-
вания конденсированных соединений. Однако окись азота, в отли-
т чие от кислорода и двуокиси, пе взаимодействует с диэтиленглико-
Г лем. Поэтому можно предположить, что на место отрывающейся
> при распаде молекулы нитроэфира двуокиси азота вступает окись
f последнего, образуя нитрит, как это предположено для Зтилпитра-
та {7], a NO2 связывается в малолетучее соединение.
? < Отсутствие случаев уменьшения давления в начальный период
разложения ДЭГДН при добавлении двуокиси азота и высокой
у температуре, а также значительное увеличение при этом скорости
!' газообразования в сравнении с распадом чистого нитроэфира пока-
I зывают, что наряду с предполагаемыми выше процессами проте*
293
кают реакции, приводящие к образованию продуктов, труднокон-
деясирующихся при температуре опыта. Все это, а также относи-
тельно быстрое исчезновение двуокиси азота позволяет предпола-
гать, что NOa способна реагировать непосредственно с ДЭГДН.
Поэтому при разложении чистого нитроэфира не накапливается
первичный продукт реакции—двуокись азота; образуясь—она
быстро вступает в дальнейшие реакции в отличие от НГЦ, рас-
пад которого характеризуется накоплением значительных коли-
честв NO2..
Азотная кислота, по-видимому, также способна к непосредствен-
ному окислению ДЭГДН, так как газообразование при распаде по-
следнего в присутствии этой кислоты протекает очень быстро.
Ускорение, которое наблюдается при этом, обусловлено, вероятно,
совокупностью нескольких реакций, которые включают в себя
гидролиз, ускоряющийся по мере разбавления кислоты водой, об-
разующейся в результате происходящего окисления нитроэфира.
Кривые газообразования при распаде ДЭГДН в присутствии
азотной и щавелевой кислот качественно схожи, однако причины
наблюдаемого ускорения этого распада различны, поскольку щаве-
левая кислота не обладает окисляющей способностью. Ускоряю-
щее действие щавелевой кислоты па разложение ДЭГДН можно
рассмотреть в свете данных по распаду других нитроэфиров в при-
сутствии кислот.
По данным Б. С. Самсонова безводная серная кислота сильно
ускоряет распад нитроцеллюлозы с самого начала опыта. По
данным В. В. Горбунова то же происходит и с НГЦ. Это можно
было бы объяснить непосредственным взаимодействием кислоты
с питроэфиром с отщеплением от последнего азотной кислоты по
реакции переэтерификации. Вместе с тем, Г. Н. Беспаловым было
показано, что сухая щавелевая кислота также очень сильно (в де-
сятки раз) ускоряет начальный этап разложения НГЦ. Возможно,
что влияние безводной щавелевой кислоты на распад нитроэфиров
аналогично по своей природе действию на них H2SO4. Если при
разложении ДЭГДН в присутствии щавелевой кислоты происхо-
дит переэтерификация, то образующаяся азотная кислота удаляет-
ся из сферы реакции, вследствие своей летучести и в результате
окисления ДЭГДН, щавелевой кислоты п продуктов реакции. По
этой причине происходит быстрое газовыделение.
Разбавление азотной кислоты водой приводит в начале опыта
к уменьшению давления, после чего наступает более резкое, чем
н случае безводной кислоты повышение его в результате ускорен-
ного газообразования. Разбавление же шавелевой кислоты водой
напротив влечет за собой замедление распада нитроэфира. Такое
различие может быть объяснено тем, что вода, сильно подавляя
реакцию непосредственного взаимодействия нитроэфира со щаве-
левой кислотой, относительно медленно гидролизует ДЭГДН
в присутствии этой кислоты.
Все исследованные вещества, за исключением окиси азота, спо-
собны в определенных условиях сильно (в десятки и даже сотни
294
раз) ускорять газообразование при распаде ДЭГДН. Однако, в от-
личие от распада НГЦ, это ускорение ограничивается некоторым
не слишком большим максимумом скорости, наступающим уже на
сравнительно ранних стадиях разложения и определяемым поэто-
му не израсходованием нитроэфира, а какими-то другими причи-
нами. Наличие максимума можно сопоставить с изменением
окраски .газовой фазы, указывающей на присутствие в ней дву-
окиси азота в случае больших скоростей разложения. Содержание
NO2 в газовой фазе проходит также через максимум, который во
времени располагается вблизи максимума скорости газообразова-
ния. Затем, по мере падения этой скорости уменьшается и содер-
жание в газовой фазе двуокиси азота. Все это позволяет предпо-
ложить, что последующее торможение действия добавленных
к нитроэфиру веществ происходит вследствие исчезновения послед-
них. Исчезновение этих веществ обусловлено взаимодействием их
с ДЭГДН и продуктами распада последнего. Разложение же
самого ДЭГДН, если и генерирует ускоряющие вещества, то со ско-
ростью, меньшей, чем скорость их взаимодействия с нитроэфиром,
Таким образом, термическое разложение ДЭГДН в присутствии
различных веществ, хотя и может сильно ускоряться на определен-
ном этапе, но в конечном итоге наступает замедление распада, обус-
ловленное способностью его к автоторможению, проявляющемуся,
в частности, в случае чистого нитроэфира в отсутствии стадии рез-
кого ускорения разложения. Это свойство существенно отличает
ДЭГДН от НГЦ, НГЛ и некоторых других нитроэфиров, харак-
терной особенностью распада которых является самоускоряющееся
разложение, развивающееся в результате действия продуктов раз-
ложения.
ВЫВОДЫ
I. Изучен термический распад диэтиленгликольдинитрата
в присутствии кислорода, двуокиси и окиси азота, азотной и щаве-
левой кислот при 80—120°.
2. Обнаружено, что жидкий диэтиленгликольдинитрат способен
при повышенных температурах к взаимодействию с кислородом,
двуокисью и окисью азота, идущему с уменьшением объема этих
газов.
Связывание кислорода и двуокиси азота установлено также для
самого диэтиленгликоля.
3. Показано, что концентрированные азотная и щавелевая кис-
лоты ускоряют разложение диэтиленгликольдинитрата в десятки и
даже сотни раз. Разбавление азотной кислоты усиливает ускоре-
ние газовыделепня, в то время как в случае щавелевой кислоты
разбавление тормозит разложенце.
4, Резкое ускорение, наблюдающееся при распаде диэтиленгли-
кольдинитрата в присутствии некоторых веществ, сопровождаю-
щееся накоплением NO2, сменяется по прохождении скорости рас-
пада через максимум замедлением газообразования и постепенным
исчезновением Двуокиси азота,
295
5. Полученные данные рассмотрены в свете предположения
о способности диэтиленгликольдннитрата к автоторможению раз-
ложения,
G. Сделано предположение о непосредственном взаимодействии
диэтиленгликольдннитрата со щавелевой кислотой.
В заключение авторы считают своим приятным долгом выра
зить глубокую благодарность К. К, Андрееву за ценные критиче-
ские замечания, сделанные при просмотре рукописи статьи.
ЛИТЕРАТУРА
I. Андреев К. К., Глазкова А. П., ДАН СССР, 1955, 105, 286.
2. Андреев К. К., Самсонов Б. С, ДАН СССР. 1957, 114, 815,
3. Андреев К. К., Глазкова А. П..М аурина Н, Д., Светлов Б. С ,
ЖФХ, 1958, 32, 1726,
4. А н д реев К. К-, ЖПХ, 1958, 31, 484—493.
5, Блюмберг Э. А, Пнкаева В. Л., Эмануэль И М., ЖФХ, 1955,
29, 1569—81.
6. Светлов Б, С_, Научные доклады Высшей школы, 1958, 3, 422.
7. Levy J. В., J, Am, chem. Soc., 1954, 75. 3254, 3790
8. R о b е г t s о и R., J. chem. Soc., 1909, 95, 1241.
Б. H. КОНДРИКОВ
21. ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ ДИНИТРИТА
ЭТИЛЕНГЛИКОЛЯ И ТРИНИТРИТА ГЛИЦЕРИНА
Выяснение механизма термического разложения нитроэфиров
существенно затрудняется сложностью этого процесса, обуслов-
ленной, в частности, тем, что один из первичных продуктов распа-
да— двуокись азота — является энергичным окислителем, легко
взаимодействующим с другими продуктами разложения, а часто и
с самим нитроэфиром. Роль процессов окисления можно значи-
тельно уменьшить, если при изучении высокотемпературных пре-
вращений перейти от эфиров азотной к эфирам азотистой кислоты,
механизм распада которых, ввиду подобия химического строения
этих соединений, сходен с механизмом распада нитроэфиров, но
как правило, включает отщепление не двуокиси, а окиси азота —
окислителя несравненно более слабого. Поэтому в течение вот уже
более двух десятилетий исследование механизма термического рас-
пада нитроэфиров неразрывно связано с изучением распада орга-
нических нитритов. Однако до самого последнего времени исследо-
вания термического распада эфиров азотистой кислоты ограничи-
вались в основном мононитритами одноатомных спиртов; полини-
триты— соединения наиболее интересные, ввиду большего родства
их с широко применяющимися полинитратами—почти не исследова-
лись. Настоящая работа представляет собой одну из первых попы-
ток изучения термического разложения полинитритов многоатом-
ных спиртов,
296
ТЕРМИЧЕСКИЙ РАСПАД ЭФИРОВ АЗОТИСТОЙ КИСЛОТЫ
(обзор)
Первые систематические исследования термического разложе-
ния эфиров азотистой кислоты были предприняты Стиси с сотруд-
никами в Канаде [46]—[55]. Кинетика разложения метил- (46], [50],
[52], этил- [47], [54], н-пропил- [48], [54], изопропил- [49] и н-бутилнит-
ритов [51] изучалась манометрическим методом при 170—240° и
давлении 0,005—50 см рт. ст. (для метилнитрита — до 3000 см
рт. ст,), Оказалось, что распад этих соединений в основном гомо-
генен, и рост давления подчиняется закону реакции первого поряд-
ка. Отношение давления конечных газообразных продуктов распа-
да к начальному давлению паров нитрита мало зависит от
концентрации п температуры и изменяется от 1,6 для к-бутилнит-
рита до 2,0 для н-пропил- и изопропилнитритов,
Газообразные при комнатной температуре продукты содержат
80—98%NO, 1 — 10% СО и небольшие количества других газов,
природа которых не выяснялась. В продуктах, конденсирующихся
при комнатной температуре, были обнаружены альдегиды и
спирты. Энергия активации распада для всех членов ряда оказа-
лась почти одинаковой и близкой к 37 ккал моль.
На основании результатов исследования Стиси заключил, что
термический распад алкилмононитритов представляет собой про-
стую мономолекулярную реакцию
RCH.ONO -Ь RCH2OH-]--~ RCHO-RNO (1)
скорость которой определяется скоростью разрыва связи RCH2O—
NO
RCH2O—NO^RCH2O-+NQ (2)
а энергия активации отвечает энергии разрыва этой связи. Меха-
низм реакций, следующих за разрывом связи О—NO, был предло-
жен Райсом [44] на основании некоторых косвенных данных, полу-
ченных им при изучении термического распада этилнитрита при
400—500°. Этот механизм включал следующие основные реакции:1
СН3СН2О- 4-CH3CH2ONO^CH3CH2OH + CH3CHONO (3)
CH3CHONO^CHSCHO+NO ' (4)
1 Предполагалось, что при малых давлениях возможны также реакции:
СН3СН2О - - СНз +СН2О
СН3 +CH3CH,ONO~ CH4+CH3CHONO
Первая из них — основной путь распада этоксила —в настоящее время довольно
надежно обоснована экспериментально. Вторая недавно была отвергнута в ре-
зультате получения взаимодействия мс гилцитрита с продуктами разложения трет-
бутнллерекиси [30], В работе [30] была предложена иная реакция взаимодействия
метила с нитритом
СНз 4-CH3ONO- CH3NO +СН3О.
использовавшаяся также П. Греем для интерпретации данных по распаду трет-
бутилнитрита [29].
297
Несмотря на то, что никаких прямых доказательств существования
реакций (3) и (4) Райсу получить не удалось, этот механизм около
двух десятилетий считался общепринятым и был распространен не
только на распад всех остальных эфиров азотистой кислоты как
в газовой, так и в жидкой фазе [31], но и на распад алкилнитра-
тов [41].
Существенно отметить, что поскольку распад нитритов — это
реакция первого порядка, скорость обратной ступени реакции (2)
RCH2O> +NO^RCH2ONO (5)
(т. е. обычного взаимодействия окиси азота с радикалом) при та-
ком механизме должна быть значительно меньше, чем скорость
реакции (3) !.
Прямая экспериментальная проверка справедливости этого за-
ключения была предпринята Леви [34], изучавшим с помощью ин-
фракрасного спектрометра реакцию перекиси этила в присутствии
окиси азота и этилнитрита при 181° и давлении порядка нескольких
см рт. ст. При этой температуре скорость распада перекиси очень
велика, и этилнитрит в течение нескольких минут должен был бы
разложиться по реакции (3). Однако ни малейших следов разло-
жения обнаружено не было. В то же время скорость реакции (5)
была весьма значительной: за 1—2 мин. около 20%, а за 4—
6 мин. — около 60% перекиси в присутствии NO переходило в этил-
иитрит г.
Механизм Райса, таким образом, оказался несостоятельным по
крайней мере для той области условий, для которой получена
основная масса экспериментальных данных по распаду нитритов
и нитратов.
Иной механизм был предложен Леви. Экспериментальной осно-
вой этого механизма являлось исследование термического распада
этилнитрита, проведенное им с помощью инфракрасного спектро-
метра [37]. В продуктах распада при 160—200° были обнаружены
ацетальдегид (с выходом 60—90% по отношению к этилнитриту),
закись азота (20—25%), а также небольшие количества этилового
спирта и синильной кислоты. Скорость исчезновения этилнитрита
подчинялась закону реакции первого порядка. При добавлении NO
скорость распада почти не изменялась, выход ацетальдегида уве-
1 Действительно, обозначив константы скоростей реакции буквой k с индек-
сом. соответствующим номеру уравнения реакции, получим (в обычном предпо-
ложении о стационарности концентрации этоксила)
£[n [C2H5ONO] t , k2k [СДДСЖО]
V1NO1 -I К I32hTono]“ ’
при ks [NO] « I.^ONO]
это — уравнение реакции первого порядка.
2 Аналогичное явление наблюдал впоследствии Хиншельвуд с сотрудника-
ми [22], обнаруживший, что перекись трет-бутила при 160" в присутствии NO
почти количественно переходит в трет-бутилцитрит.
298
1личивался до 100%, a N..0— до 50%, Добавление ацетальдегида
^приводило к увеличению скорости распада, уменьшению выхода
[. NsO и некоторому возрастанию количества HCN в продуктах раз-
ложения. Механизм, предложенный для объяснения этих результа-
нтов, включал следующие основные реакции:
' CHjCH,,ONO ф СН3СН2О--[-NO
CH3CH;O-+NO Т> CH3CHO + NOH
СН3СН2О- СНз'+СН.О1
2NOH -+ NjO+H2O
СН3СНгО- 4-СН3СНО сн3сн2он + сн3со-
CiV+CO
(6)
(7)
(8)
(9)
(10)
Рассмотрение реакций (6) и (7), определяющих скорость распа-
да в обычных условиях и особенно при добавлении NO, приводит
к кинетическому уравнению реакции первого порядка
41п[С2Н5ОМО] Ж д,
/Е[ -i-
dx
&
Л. из которого, в частности, следует, что энергия активации разрыва
Ж связи О—NO в этилнитрите меньше экспериментально опреде-
Ж' ляемой величины (см. табл. 3 на стр. 326) на энергию активации
реакции (7), по-видимому, несколько ккал/моль.
Впоследствии этот механизм был распространен на другие
алкилмононитриты [38], а также с успехом использован для интер-
претации полученных Поллардом и сотрудниками [42], [43] резуль-
татов исследования термического разложения этилнитрата [10],
В отличие от разложения алкилмоноиитритов термический рас-
пад полинитритов — это область почти совершенно не исследован-
ная, Лишь совсем недавно в литературе появились данные о тер-
мическом разложении дйнитритов нескольких гликолей при темпе-
ратурах около 300° [32], [33].
Интересно, что и для всех исследовавшихся динитритов первой
ступенью распада, по-видимому, является разрыв связи О—NO с
образованием оксирадикала и NO. Дальнейшая судьба оксиради-
кала зависит от его структуры. При разложении одних динитритов
(пропан- [,2-; бутан- 2,3- и 1,4-, гексан- 2,5- и некоторых других)
радикал распадается или перегруппировывается, отшепляя NO.
Радикалы других динитритов (пропан- 1,3-, бутан- 1,3-, пентан- 2,4-
и |,5- и 2,5- диметилгексан- 2,5-) дают при распаде не окись, а дву-
u'r
1 Прн 160—200° для констант скорости реакций (7), (8) и обратной ступени
т •’ реакции (6) выполняется следующее соотношение: Л| ;> Как показывают
т* результаты некоторых работ [33] при повышении температуры до 300’ скорость
У реакции (8) существенно увеличивается и приближается к скорости реакции (7),
99
окись азота, значительные количества которой (вместе с продук-
тами окисления двуокисью азота органических веществ) обнару-
живались в продуктах реакции.
Было установлено, что при разложении динитритов вблизи
300° не играют никакой роли реакции типа (7), (10) и др. столь от-
четливо проявляющиеся при распаде мононитритов вблизи 200°.
Поскольку в данном случае были иными и условия проведения
опытов и химическое строение, исследуемых веществ, ответить на
вопрос о причинах различий в механизме распада авторы, естест-
венно, не могли.
Результаты этих работ использовались для интерпретации дан-
ных, полученных при изучении распада нитроцеллюлозы при горе-
нии ее в вакууме [57].
Несмотря на то, что в этих работах, проводившихся почти од-
новременно с нашими, был выполнен значительный объем исследо-
ваний по распаду динитритов многоатомных спиртов, целый ряд
вопросов остался незатронутым. Прежде всего совершенно не изу-
чался распад полинитритов при более низких (100—200°) темпе-
ратурах, при которых было получено в свое время особенно много
требующих интерпретации данных по разложению полинитратов.
Отсутствовали какие бы тони было кинетические исследования
распада полинитритов. Не имелось никаких сведений о разложе-
нии наиболее интересных из них-—динитрита простейшего много-
атомного спирта — этиленгликоля (как это ни странно, американ-
ские исследователи, получившие и изучавшие распад более десятка
динитритов, ни словом о нем не упоминают) и аналога нитрогли-
церина — глицеринтринитрита.
В настоящей статье изложены основные результаты выполнен-
ных нами исследований кинетики и механизма термического рас-
пада динитрита этиленгликоля и тринитрита глицерина [9], [10], [5].
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ ДИНИТРИТА ЭТИЛЕНГЛИКОЛЯ
Получение динитрита этиленгликоля
Динитрит этиленгликоля получали двумя методами. По пер-
вому из них [21] раствор глицеринтринитрита в этиленгликоле на-
гревали в перегонной колбе до 100—105° и отгоняли образующий-
ся по обычной обменной реакции гликольдинитрит. Тринитрит гли-
церина предварительно готовили [39] продолжительным пропуска-
нием N2Oj через глицерин, сушили и перегоняли в токе воздуха
при 60—100°.
Этот громоздкий, неудобный метод впоследствии был .заменен
получением динитрита этиленгликоля, аналогично нитритам дру-
гих одноатомных и двухатомных спиртов [6], [32] этерификацией
гликоля водным раствором азотистой кислоты. К охлаждаемому
смесью льда и соли водному раствору двух эквивалентов этилен-
гликоля и около трех — нитрита натрия при интенсивном переме-
шивании добавляли по каплям два эквивалента разбавленной со-
300
ляной кислоты. Температура массы не поднималась выше ми-
нус 2°. Сразу же после окончания слива динитрит сепарировали и
обрабатывали безводной содой. Затем его сушили сульфатом нат-
рия и перегоняли под атмосферным (при температуре около 96°)
и под пониженным давлением. Полученную таким образом подвиж-
ную желтую жидкость с характерным запахом освобождали от
тазов и в вакууме переводили в запаиваемые ампулы. Элементар-
ный анализ ее дал следующие результаты.
Найдено %; С 19,78; 19,97; Н 3,60; 3,50; N 23,15; 23,20.
Вычислено для C2H4O4N2 %: С 20,00; Н 3,36; N 23,33.
Показатель преломления — 1,3884,
Молекулярный вес, определенный манометрическим методом,
120±0,6, Теплота испарения, рассчитанная из зависимости упру-
гости пара от температуры в интервале 16—100° по наклону пря-
мой в координатах lg р~ 1/Т (см. фиг. 1), равна 8,8 ккал/моль.
Методика эксперимента
Кинетику распада в газовой фазе изучали измерением давле-
ния в статической системе стеклянным манометром типа Бурдона
.при 120—190° и начальном давлении 50—1000 мм рт. ст.
от темпе-
Фиг. 1.
Зависимость упругости паров дияитрпта этиленгликоля
ратуры.
Л —в координатах р—1°, б —г» координатах if р —4
Г
сосуд ис-
Для введения динитрита и добавок в реакционный
пользовалась вакуумная установка обычного типа. Ампулу с дини-
‘ тритом помещали в запаиваемый ампуловскрыватель (фиг. 2),
Присоединяли последний к установке и, откачав систему до оста-
точного давления 10~2—10~4 мм рт. ст., обламывали кончик ампу-
;,лы поворотом крана вскрывателя.
При хранении даже в запаянных ампулах и на холоду дипитрит
заметно разлагается, поэтому после вскрытия ампулы легколету-
чие продукты распада удалялись продолжительным откачиванием,
301
затем часть нитрита переводили в вакууме в реакционный сосуд,
охлаждая последний жидким азотом, отогревали сосуд до комнат
Фиг. 2. Ампуло-
вскрыватель.
ной температуры и снова откачивали динитрит
для удаления остатков легколетучих примесей.
Готовый к проведению опыта реакционный
сосуд отпаивали и, определив по давлению па-
ров при 100э количество нитрита в сосуде, поме-
щали его в жидкостной термостат, температура
которого поддерживалась постоянной в пределах
±0,2:’. Время прогрева вещества до температуры
термостата обычно не превышало 0,5 мин.
Газообразные конечные продукты распада
Вначале анализировали методом, включающим
выделение из смеси с помошью глубокого охлаж-
дения двух фракций: NO, СО и N2— в первой
(охлаждение до минус [65°) и N2O и СОг — во
второй (минус 120°), NO поглощали подкислен-
ным раствором FeSO4, СОг—60%-ным раство
ром КОН, а содержание Ns и N2O в отдельных
фракциях определяли по разности.
Был использован также метод, по которому
углекислоту, а вслед за ней окись азота с избыт
ком кислорода поглощали 60% -ним раствором
КОН, закись азота сжигали с водородом (при-
чем немного СО окислялось закисью до СО2), а
окись углерода — с кислородом на раскаленной
платиновой спирали. Образующуюся двуокись
углерода поглощали щелочью.
Оба метода были проверены на искусственных смесях, погреш-
ность не превышала I —1,5% абс.
Результаты экспериментов
Влияние начальной концентрации
Манометрические кривые, полученные при |70° и различных на-
чальных давлениях паров гликольдинитрита, приведены на
фиг. 3—5. Основная особенность разложения гликольдинитрита,
характеризуемая этими кривыми, заключается в том, что при до-
статочно высоком (выше ~150 мм !) начальном давлении оно име-
ет ярко выраженный двустадийный характер, причем на первой
стадии скорость распада снижается сначала резко, а затем более
медленно, на второй — растет, проходит через максимум и быстро
уменьшается до нуля.
При малых начальных давлениях скорость газообразования не-
прерывно уменьшается со временем, кинетические кривые имеют
' Все концентрации газообразных продуктов здесь и далее выражены в
мм рт. ст. при температуре опыта,
302
обычный вид (при 180° и начальном давлении около 70 мм умень-
шение скорости распада со временем происходит по закону, близ-
кому к мономолекулярному — см. фиг: 8,6). Начальная скорость
Фиг. 3. Кинетика газообразования при
распаде гликольдинитрита при 170’.
д; 0—248, А—249, 4-—251 « к рт» Ст, /—н коор-
динатах р/ри — т; 2—н ииирдииахзх w—т (да—ско-
рое pocifl давления).
газообразования ’ от концентрации динитрита не зависит. Однако
на последующих ступенях первой стадии распада такая вависи-
Фиг. 4. Влияние начальной концентрации на разложение
гликольдипигрита при 170э.
рг;. /—76; 2—22,; 3—320; 4—405; 5—659 л At рт. ст.
мость обнаруживается, а именно: в один н тот же момент времени
(или при одном и том же относительном повышении давления)
1 Здесь и в дальнейшем речь идет о скорости роста давления, отнесенной к
dp 1
начальной концентрации дипитрита: — ' , или w мин,"’,
303
в разных опытах скорость тем ниже, чем выше начальная концент-
рация динитрита (фиг. 5). Это влияние концентрации вполне опре-
деленно, но невелико: при повышении 1начального давления от 100
Фиг. 5. Влияние начальной концентрации на
скорость газообразования при разложении
‘ гликольдинитрита при 170’.
рр. 1-76; 2—115; 3—221; <-270; 5-320; 6-659;
/—920 JfM рт* ст.
Фиг. 6. Зависимость скорости на максимуме (uimai).
времени ее достижения (т) и давления, при котором она
достигается (р), от начальной концентрации паров гли-
кольдинитрита (ро).
Р координатах; /_ «.'№аг—р.; 2—г-р,,; ——р.,.
до 400 мм скорость газообразования на средних ступенях первой
стадии распада уменьшается в 1,2—1,4 раза, увеличение ро сверх
400 мм не вызывает дальнейшего снижения скорости.
304
> Максимальная скорость газообразования при повышении на-
! чального давления до 600—800 мм растет, а затем несколько сни-
жается. Время достижения максимума и давление, при котором
i он достигается, при этом уменьшаются (см. фиг. 6).
Концентрация динитрита определенным образом влияет также
на состав конечных продуктов распада, что проявляется, в част-
ности, в изменении при повышении начального давления количе-
ства продуктов, не конденсирующихся при разных температурах
; (см. фиг. 7).
Фнг. 7. Зависимость количества конечных продук-
тов распада, не конденсирующихся при различных
температурах, от начального давления.
Время разложения при повышении начального давления суще-
^ствеино сокращается. При 70—100 мм оно составляет, по крайней
.А: мере, 450—500, при 140—150 мм — 250—300, при 200 мм и более
высоком давлении — 150—200 мин.
Влияние температуры
При повышении температуры влияние концентрации на ско-
Ж... рость как первой, так и второй стадий распада существенно
уменьшается. Из фиг. 9 видно, что при 190° увеличение начального
^Давления со 140 до 300 мм не изменяет скорости и не позволяет
/^.обнаружить двустадийности разложения. Роль второй стадии рас-
Ж/ пада при повышении температуры значительно снижается, и отно-
J».’ шение начальной скорости к скорости на максимуме возрастает:
№ При 170° и начальном давлении 276 мм (фиг. 5) оно равняется 1.8,
Ж-: а при 180° и 299 лл (фиг. 8)—3,1.
Ж' Начальная скорость газообразования при повышении темпера-
Ц; туры растет.- При 170° она составляет 5,8* 10-4, при 180° —
15-10-4, при 190°—33,3 I О-4 сек-1. Энергия активации распада
динитрита, рассчитанная по этим значениям скоростей, получается
равной 35,6 ккал/моль.
20 451
305
Заметно увеличивается при повышении температуры объем га-
зообразных конечных продуктов распада (при Ро = 300—400 мм и
170° он равен 2,9 моля на моль нитрита, при 190° — 3,2 моля на
моль).
Фиг. 8, Кинетика распада гликольдннитрита при 180°.
299i 2 и 4—65,6 ллс рт* ст.» а—в координатах р/р0—“• в "Ч б—н координатах
{Ркои^’-’-
Фиг. 9. Кинетика распада гликольдипитрита
при ISO3.
₽): Г II 2—143; Л-305 -UJK рт, ст.
Влияние окиси азота т
Добавление окиси азота (см. фиг. [0, И и табл. )) существенно
уменьшает начальную скорость роста давления1 2, причем эффект
добавления каждой новой порции NO меньше, чем предыдущей, а
1 В проведении опытов с добавлением NO принимал участие студент
В. И. Комков.
2 Уменьшается начальная скорость газообразования н при добаплспии к ди*
нитриту NOz
при достижении некоторой предельной концентрации окиси азота
исчезает совсем. При повышении температуры эта предельная
концентрация несколько увеличивается, а эффект добавления NO—
уменьшается (фиг. II).
1 dp Др
Фиг. 10. Вл и нние окиси азота на распад гликольдини*
трита при 170е и ро=250—-260 леи рт. ст.
₽NO: >~0; 2~2Ь‘ мм ст' (4;i!
Следует отметить, что снижение начальной скорости зависит
не от соотношения между концентрациями динитрита и окиси
азота, а только от концентрации NO (см. табл. I).
Фиг. II. Зависимость на-
тальной скорости газооб-
разования при распаде
гликольдннитрита при
170 и 190° от давления
окиси азота. По оси ор-
динат — отношение на-
чальной скорости газооб-
разования в присутствии
NO к начальной скорости
при тон же температуре
без добавления NO.
Существенно также, что, значительно уменьшая начальную
скорость, окись азота, добавленная в умеренных (до —200 мм)
количествах, практически не влияет на скорость последующих
ступеней распада (см. фиг. 10).
с
Влияние окиси азота на начальную скорость газообразования при термическом распаде гликольдинитрита при 17(Г
586 1 313 i 1г87 1,03
СО
60; 01. о
О
04
28 00 ОО ©
СМ 04
О
ь- СО 187 23
»—< о" »—I
Cl см СО 38 1
о 23 —
ю LZZ 57
о о
04 о
03 24 04 S 52
со ©
»—I О —<
СО
э 60 со 04
см О —•
04 со
Г*-1 о О <30
МО о
—
о 81
со «о
со о t—<
ю
о СО S
со о>
СО о t—<
ю 197 5?
04 м
СМ © 04
СМ □> 130 1 00
04 о *
о см
О ю
со
£ те
S сх и
h О
о а,
S о*
4? ° • ч
S 0J (- «
£ е. Ш О '
ч ~ Ч о. I
S ч «_> Q =
я м со И S
ч о О
CQ 4 а. о
8д= 4) Q — пЗ
20 -а ПЭ Г я 3? Ч ед ВЗ w ИЛЬИ НИЯ 1
о S’ QJ те
х S X 1 Г О
«о
1 * t=£
Состав газообразных продуктов распада
Результаты анализа газообразных конечных продуктов распада
динитрита этиленгликоля при 170° приведены в табл. 2. Весьма су-
щественным фактом является наличие в газах значительных коли-
честв двуокиси углерода. Прн очень большой начальной концент-
рации паров динитрнта на второй стадии распада наблюдается
преходящее пожелтение газовой фазы, указывающее на образова-
ние двуокиси азота. Если температуру на второй стадии быстро
понизить со 170 до Ю03, то образование все возрастающих коли-
честв NO2 можно отметить и при обычных концентрациях гликоль-
динитрита. NO2 обнаруживается в промежуточных продуктах при
умеренных концентрациях динитрита также и в том случае, если
распад е самого начала проводить при низкой (1203) температуре.
Таблица 2
Состав газообразных конечных продуктов распада днннтрита этиленгликоля
прн 17(Г
Начальное давление динитрнта, мм рт. ст. Вещество, Надальнее давление добавленного вещества мм рт, ст. Рксн/Ро ПрИ температурах на Молей газа на моль динитрнта
добавленное к динитриту 170° 106° комнат- ной NO n2o СО со2 N»
184 — 3,00 2,54 2,08 1,54 0,10 0,16 0,22 0,06
276 — -- 2,89 2,44 2,10 1,55 0,13 0,15 0,19 0.08
390 34 2,92 2,46 2,18 1,46 0,16 0,25 0,25 0,06
260 NO 62 2,92 2,43 2,16 1,52 0,12 0,18 0,27 0,07
295 67 2,91 2,44 2,16 1,46 0,17 0,20 0,28 0,05
281 228 2,88 2,40 2,15 1,50 0,16 0,22 0,21 0,06
291 Н20 67 - — — 1,57 0,12 0,25 0,21 0,03
Следует отметить, что изменение начального давления, добав-
ление окиси азота и воды слабо влияют на состав газообразных
конечных продуктов распада.
Конденсирующиеся продукты распада
Конденсирующаяся при комнатной температуре после оконча-
ния опытов с достаточно большим (более ~200 мм при 170°) на-
чальным давлением бесцветная вязкая жидкость, буреющая при
продолжительном нагревании, хорошо растворяется в воде и
ЗОУ
ЗОЯ
имеет кислую реакцию на лакмус. Она содержит гликолевую кис-
лоту (проба с раствором FeCl3 и Фенола в воде [8] и проба
с 2,7-диоксинафталином) и, по-видимому, гликолевый альдегид
(пожелтение водного раствора продуктов при добавлении несколь-
ких капель концентрированной щелочи [23]). Щавелевая кислота
(проба с CaSCU и проба с дифениламином [26]) и этиленгликоль
(реакция с бурой и проба с CuSO4 в щелочной среде [4]) в ней не
Фиг. 12. Кинетика образования при распаде гликольдинитрита
продуктов, не конденсирующихся при 170’ и конденсирующихся
при 100’. Температура опыта 170Т
Продукты, не конденсирующиеся прн J70 и 100” — верхняя н средняя
кривые, конденсируют неся при 100° —нижняя кривая (ось ординат
справа).
ро * I—332, 2— 302, 5—332 а« рт. ст. (опыт без охлаждения), ip—разность
между давлением в сосуде дрн 170° и давлением при 100°. Все давлении
нырзжены Б АЛ рт. СТ. Прн ]70°.
обнаружены. В продуктах присутствуют также вода и формальде-
гид, причем количество последнего, по-влднмому, тем больше, чем
выше температура опыта.
Были проведены также эксперименты по определению кинетики
образования при распаде динитрита этиленгликоля продуктов,
конденсирующихся при понижении температуры. В этих экспери-
ментах реакционный сосуд, нагревавшийся при 170°, время от вре-
мени быстро охлаждали до 100°, замеряли давление при этой тем-
пературе и снова быстро нагревали до температуры опыта. Кри-
вые, полученные в этих опытах, приведены на фиг, 12—13. Отчет-
ливо видно, что труднолетучие продукты, образуются исключитель-
но на второй стадии разложения, и повышение давления влияет
310
L на их образование лишь в той мере, в какой оно влияет на ход
второй стадии (приближая момент ее наступления и увеличивая
скорость реакции).
Фиг, 13. Кинетика образования продуктов распада гли-
кольдинитрита, конденсирующихся при 10(Г, в опытах
при 170°,
ро: 1—184, 2—332, 3—535 мм рт. ст,, Лр—концентрация продук-
тов, конденсирующихся при |(ХР, выраженная в мм рт. ст.
при J7(P.
Добавление к гликольдинитриту конденсирующихся продуктов
распада и увеличение поверхности
Своеобразно влияет на газообразование добавление к гликоль-
динитриту конденсирующихся при комнатной температуре продук-
тов разложения и увеличение отношения поверхности реакцион-
ного сосуда к его объему. Добавление одного из конденсирующих-
ся продуктов — воды (фиг, 14) и увеличение поверхности набивкой
сосуда стеклянными трубочками (фиг. 15) не оказывают влияния
на скорость начальных этапов распада, однако на средних этапах
кривые в этих опытах идут заметно выше, чем в обычных, что ука-
зывает на увеличение «удельного веса» реакций второй стадии. До-
бавление конечных конденсирующихся при комнатной темпера-
туре продуктов распада, очень гигроскопичных и, несмотря на про-
должительную эвакуацию, по-видимому, содержащих немного
воды, еще сильнее действует на вторую стадию и несколько уско-
ряет первую (фиг. 16), Эффективнее всего на распад влияет совме-
стное присутствие значительных количеств воды и конденсирую-
311
%
3
Фнг. 14. Влияние воды на распад глнкольдинитрнта
при 170’ и po&260-290 мм рт. ст.
Р1ЬО: 1-7», 2-67. 3-0 мм рт. Ст.. ( Зр-р—Pj^q)-
Фиг. 15. Влияние величины поверхности
на разложение гликсльдниитрита при ]70°
и /?о=--500 лги рт. ст.
S/P : /—10. 2—? ел"-1.
Фиг. 16, Влияние конечных конденси-
рующихся при комнатной температуре
продуктов распада на разложение
гликольдинитрита при 17(Г.
I— Рл=220 мм рт. ст., с добавлением про-
дуктов распада. 2—рв-248 мм рт. ст„ Чи-
стый гликольдкиитрит.
312
щихся продуктов: начальная скорость возрастает в несколько раз
и реакция заканчивается примерно вдвое быстрее, чем в опытах
с чистым динитритом (фиг. 17). На стенках сосуда в этих опытах
Фиг. 17. Влияние конечных конденси-
рующихся при комнатной температу-
ре продуктов распада и ноды на раз-
ложение гликольдннитрита при 170°.
/—с добавлением продуктов распада и воды
(рй=*2В0; о" 100 яа pu ст J; 2— чистый
гликольдквнтрнт (рс«27б млс рт.съ)
появляется белый налет, заменяющийся при охлаждении тонким
слоем бесцветной прозрачной жидкости. Газовая фаза в начале
опытов желтеет или буреет, а затем медленно обесцвечивается.
Механизм термического разложения динитрита этиленгликоля
Двустадийность распада
Полученные при изучении термического разложения динитрита
этиленгликоля данные показывают, что распад даже этого про-
стейшего полинитрита весьма сложен.
Как уже отмечалось выше, характер кинетических кривых рас-
пада при не слишком высокой температуре и достаточно большой
начальной концентрации указывает на то, что разложение вклю-
чает по крайней мере две разделенные во времени макроскопиче-
ские стадии [16], каждая из которых, очевидно, состоит из несколь-
ких элементарных реакций. Это предположение подтверждается
рядом других экспериментальных данных: значительным влиянием
начальной концентрации, а также набивки реакционного сосуда
стеклянными трубочками на вторую стадию распада и весьма сла-
бым— на первую; существенным влиянием окиси азота на пер-
вую стадию и практическим отсутствием такого влияния — на вто-
рую; появлением на второй стадии труднолетучих продуктов рас-
пада и двуокиси азота и отсутствием их на первой и т. д.
Первая стадия распада. Близость энергии активации реакции,
определяющей начальную скорость распада гликольдинитрита
(36 ккал/моль) к величинам, полученным для мононитритов (34 —
38 ккал/моль—см. табл. 3 на стр. 326), образование NO и N2O,
сходство характера влияния NO на начальную скорость распада
гликольдинитрита с предсказанным Леви для изопропилнитрита
[38] и ряд других фактов позволяют допустить, что механизм на-
31»
чальной стадии разложения динитрита сходен с механизмом рас-
пада мононитритов, принятым Леви [37], [38] и включает следую-
щие основные реакции:
CH,ONO л CHaONO
I I +NO (И)
ch2ono сн,о-
CHjONO к CH,ONO
Г -4-NO^I + NOH (12)
CH2O- CH о
ch2ono k
] 2CH2O-|-NO (13)
CH2O-
2NOH-> N3O + H2O (14)
В присутствии NOj возможна также реакция
ch2ono ch2ono
I +NO2 I . . (15)
CHjO- CHjONO,
Кинетическая обработка уравнений (И) — (13) в предположе-
нии, что концентрация радикала СНЙ • ONO • СН2О * стационарна,
дает следующее выражение для скорости изменения давления на
первой стадии
MWAixu fCHiONO’
up fejJNOj-T^efe^ । *
(A; 4“ Aj) [NO)-]-A3 CHaONO
Для начальной скорости газообразования при распаде имеем
w0 = —I—]=3fc, или fe=--w0.
Ра \ dt /а 3
Это согласуется с независимостью ее от концентрации и позволяет
определить константы скорости по значениям начальных скоростей
газообразования, а также постоянные В и Е уравнения Аррениуса
для k. Используя данные, приведенные на стр. 305, получаем
й=10г3,8ехр (-35,6- 103Ж) сек-'.
Величина 35,6 ккал/моль, по-видимому, равняется энергии разрыва
связи О—NO в динитрите этиленгликоля. Предэкспонент в этом
случае так же, как алкилмононитритов (см. табл. 3), и в отли-
чие от многих нитратов [1] имеет нормальное значение.
Для опытов с добавлением окиси азота начальная скорость га-
зообразования определяется выражением
щ,ко = 1 ( \____,k (NOlo +
Ро \ d~ /о (&j + [МО)а4~^з
(16)
314
итсюда, в частности, следует, что в согласии с опытом начальная
скорость зависит не от соотношения между концентрациями NO и
динитрита, а только от концентрации NO и что с увеличением этой
концентрации она уменьшается, приближаясь к предельному зна-
1 k Ао
учению, равному ---
Вычитая из уравнения (16) соотношение ЗА, после преобра-
зований получим
(№_1° + + [NO]0
®о~ fe(3fej+2A2) ' ft(3fe(4-2ft2)1 u
(17)
^Рассматривая выражение (17), видим, что в случае приложи-
мости постулируемого механизма к первой стадии
йнольдинитрита мы должны для опытов
sc добавлением различного количества
ркиси азота получить прямую
’л=Л4-В[МО]о в координатах:
[NO]0
[NO]0;
распада гли-
Фиг. 18. Влияние окиси азота
на начальную скорость газооб-
разования прн распаде гли-
кольдиннтрита при 170°,
I—внутренний масштаб, 3—внешний
масштаб.
-------Й77 ММ‘МИН.
wo — wa
i На фиг. 18 зависимость между кон-
центрациями NO и начальной ско-
ростью представлена в координатах
'(NO]o, х. Как видно, во всем интервале
концентраций NO точки хорошо укла-
дываются на прямую. Коэффициенты
.Уравнения этой прямой, рассчитанные
irto методу наименьших квадратов, рав-
Jhh: 4 = 6,6 Ю2 мм • мин, В = 39,3 мин.
у Зная их, а также учитывая, что
|&=1,9-1СН сек-1, а предельное зна-
пение =17.]0“4 сек-’,
L ° A|+A2
?МОжно рассчитать отношения констант
(скоростей вторичных реакций первой
!,.Стадии. Получим: Аа/А^б, A3/Ai^l,2- 102, т. е. Аз]>А2>А[ (концент-
рации в мм рт. ст.).
Напомним, что при распаде этилнитрита вблизи 200° [37] на-
"т.|блюдалось обратное соотношение между скоростями соответствую-
у щих реакций (см. сноску на стр. 299).
Принятый механизм объясняет также образование формальде-
гида, окиси и закиси азота при распаде гликольдинитрита, влияние
NO2 на начальную скорость и ряд других фактов. Если учесть от-
щепление окиси азота, угнетающее действие ее и существование
предельной концентрации NO, то легко объяснить наблюдаемое
315
самоторможение реакции иа первой стадии и зависимость степени
его от начальной концентрации динитрита
Вторая стадия распада. Однако основной особенности распада
гликольдинитрита — его ярко выраженной двустадийности со зна-
чительным увеличением скорости второй стадии при повышении
концентрации—этот механизм, предложенный для интерпретации
одной нз классических мономолекулярных реакций, естественно,
объяснить не может. Путь к объяснению особенностей второй ста-
дии распада указывают опыты по определению кинетики образо-
вания труднолетучих продуктов и по добавлению этих продуктов
к свежему динитриту. Действительно, если сопоставить ускоряю-
щее действие труднолетучих продуктов с тем фактом, что обра
зуются они на стадии самоускорения распада и тем быстрее, чем
скорость газообразования на этой стадии больше, то естественно
будет предположить, что самоускорение разложения гликольди-
нитрита объясняется каталитическим действием этих продуктов.
Сопоставив эти наблюдения с образованием двуокиси азота
в промежуточных, а углекислоты и органических кислот — в ко-
нечных продуктах распада, а также с влиянием на распад воды
(самой по себе п в сочетании с другими продуктами) и легкой гид-
ролизуемостью динитрита в жидком состоянии, мы предположили,
что реакцией катализируемой труднолетучими продуктами, яв
ляется взаимодействие гликольдинитрита и быть может промежу-
точных продуктов его распада с водой, первоначально образую-
щейся, например, по реакции (14).
Это взаимодействие, подобно низкотемпературному гидролизу
нитритов, приводит к замене нитритной группы на оксигруппу® и
образованию азотистой кислоты. Последняя разлагается па окись,
и двуокись азота и воду. Двуокись азота окисляет находящиеся
в большом избытке органические продукты и дает СО, СОг и неко-
торое дополнительное количество воды, а также другие вещества,
в том числе органические кислоты.
Трудно предположить, чтобы реакции гидролитического харак-
тера совершались в объеме. Реакции взаимодействия нитритов
с водой — это ионные процессы, скорость прохождения которых
в газовой фазе в обычных условиях ничтожно мала. Влияние по-
г По аналогии с распадом алкил мононитритов можно заключить, что после-
довательность реакций (11)—(14) далеко не исчерпывает всего многообразия
превращений, происходящих на первой стадии распада гликольдинитрнта и аклю
чающих, возможно, распад мононитритов, взаимодействие оксирадикала с карбо
нильными соединениями, ннтроксилом и т. д. Однако опытных данных, требующих
постулирования этих ступеней, имеется пока слишком мало,
i Отсутствие в продуктах распада значительных количеств этиленгликоля
можно объяснить тем, что при высокой температуре он весьма быстро окисляется
двуокисью азота. Опыты по окислению гликоля NOa при 15(Г подтвердили это
предположение и показали, кроме того, что в газообразных продуктах окисления
концентрация СО2 значительно больше, чем СО (ср. табл. 2). Напомним, что при
окислении двуокисью азота формальдегида и глиоксаля (основных органических
продуктов распада динитрита в том случае, если бы он целиком следовал меха-
низму Леви) наблюдается обратное соотношение между концентрациями
СО и СО2.
316
ерхности на самоускорение н катализ разложения именно трудно-
етучими продуктами указывают на то, что эти реакции идут на
тенке сосуда в тонкой пленке покрывающей ее жидкости.
Труднолетучие продукты, оседая на поверхности стекла и аб-
орбируя молекулы динитрита и воды, облегчают и ускоряют их
заимодействие. Вероятно, играет роль также кислотный характер
;екоторых продуктов '.
Интересно отметить, что появление в продуктах распада окиси
дота, существенно уменьшая скорость газообразования, очень сла-
<5о влияет на скорость распада нитрита (при 170° 1,9.10”4 сек.-’,
д предельное Значение константы скорости в присутствии
ЫО: k — 1,7 • 10~4 сек.-1). В связи с этим влияние начальной
концентрации динитрита на скорость первой стадии распада весь-
'ма невелико. В основе второй стадии лежат полимолекулярные
реакции, поэтому при увеличении концентрации динитрита ско-
рость и роль реакций этой стадии в процессе разложения сущест-
венно возрастают.
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ ТРИНИТРИТА ГЛИЦЕРИНА
(Работа, результаты' которой приведены в данном разделе,
выполнена совместно с Е. В. Васильевым)
Получение и некоторые свойства глицеринтринитрита
Тринитрит глицерина получали по О. Массону [39], пропуская
через динамитный глицерин, налитый в охлаждаемый водой стек-
лянный пллиндр, сильный ток смеси окиси и двуокиси азота.
Смесь окислов в свою очередь получали, добавляя воздух к газу,
'Выделяющемуся при действии серной кислоты на нитрит натрия.
Продукт сушили сульфатом натрия и трижды перегоняли в токе
азота при 60—100°, отбирая каждый раз средние 0,6—0,8 дестил-
лата. Затем нитрит откачивали для удаления растворенных в нем
газов и в вакууме переводили в ампулы, которые хранили затем
в темном месте на холоде. Чистый нитрит — светло-желтая по-
движная жидкость с характерным запахом. При хранении на воз-
духе в течение 2—3 дней разлагается, окрашиваясь в темно-зеле-
ный цвет, в запаянных ампулах его удается сохранять в течение
2—-3 месяцев.
Результаты элементарного анализа свежеперегнанного про-
дукта:
Найдено %: С—20,20; 20,29; Н—2,90; 3,00г. Вычислено % для
C3H5O6N3 : С—20,11; Н—2,79.
1 На взаимодействие динитрита с продуктами распада, помимо прямых опы-
тов добавления продуктов, указывает также тот факт, что при достаточно боль-
шой концентрации динитрита аремя разложения значительно (в 2—3 раза) меньше
того, которое требуется для завершения мономолекулярной реакции распада.
2 . Надежных определений азота в тринитрите провести не удалось.
317
Определение молекулярного веса, проведенное манометричес-
ким методом, дало в двух опытах значения 181,5 и 178 (расчетный
молекулярный вес— 179).
Зависимость упругости пара от температуры в интервале
О—130° представлена на фиг. 19. Теплота испарения, определен-
ная по наклону прямой в координатах Igp— у~, составляет
10,1 ккал/моль, температура кипения, полученная экстраполяцией
Фиг. 19. Зависимость упругости пара глицеринтринитрита от темпе-
ратуры.
с—s координатах p—i, б—в координатах ig р—
кривой давление—температура на 760 мм рт. ст., равна 154° (ги
Массону [39] приблизительно 150°). Температура затвердевания
от —60 до —58° С.
Методика подготовки и проведения опытов по термическому
разложению тринитрита глицерина в основном аналогична опи-
санной выше для гликольдинитрита.
Результаты экспериментов
Влияние начальной концентрации
Опыты по термическому разложению глицеринтринитрита
проводились при 100—160°. Влияние начальной концентрации на
распад особенно подробно исследовалось при 150°. Кинетические
кривые, полученные при этой температуре, приведены на фиг. 20.
318
Сходство их с кривыми, получавшимися для динитрита этилен-
гликоля, например, прн 170° совершенно очевидно: отчетливая
двустадийность разложения с падением скорости на первой ста-
дии и ростом — на второй, независимость начальной скорости от
Фиг. 20. Кинетика газообразования при распаде
глицеринтринитрита при 150°.
Z—545; 2—ЗЮ; 5—2i5; 4—1421. 5—59 Мм рт» ст,.хт—и коор-
динатах б—в координатах и,—т.
Концентрации, увеличение при повышении концентрации максималь-
ной скорости и уменьшение времени ее достижения позволяют го-
ворнть об этом с достаточной определенностью. Ясно видны п
некоторые различия. При близких временах полного разложения
роль реакций второй стадии при распаде тринитрита значительно
319
больше, чем при распаде динитрита: отношение максимальной ско-
рости к начальной при ро^2ОО мм, например, в первом случае
около 3 (150°), во втором —около 0,4 (170°). Скорость на первой
стадии разложения падает менее резко, чем в случае гликольдини-
трита, и угнетающее влияние начальной концентрации на скорость
первых этапов распада не проявляется.
Газовая фаза на второй стадии разложения при обычных кон-
центрациях тринитрита окрашена в светло-желтый цвет (в случае
динитрита при этих концентрациях она не окрашивается даже при
140°). Количество конечных продуктов, не конденсирующихся при
температуре опыта, несколько (около одного моля на моль исход-
ного вещества) больше, чем при распаде динитрита. Количество
конечных продуктов, конденсирующихся при снижении температу-
ры до комнатной, составляет ~0,4 моля на моль.
Влияние температуры
Изменение температуры влияет на распад тринитрита глицери-
на так же, как на распад динитрита этиленгликоля: при понижении
температуры роль реакций второй стадии увеличивается, соотноше-
ние между скоростью на максимуме и начальной возрастает (при
ро'-'ЗОО мм (®тах/^о) мз=5,4; (К'тах/и’о) 160’ =2,2). Увеличивается
при понижении температуры и содержание двуокиси азота в про-
дуктах второй стадии распада. Лишь слегка желтая при 150—160°
газовая фаза при 100° вблизи максимума скорости становится кир-
пично-красной.
При значительном (до 100—120°) понижении температуры ха-
рактер разложения несколько меняется: скорость на начальной
стадии не уменьшается, а медленно растет, и кривая превращается
в кинетическую кривую обычной автокаталитической реакции
(фиг. 21), ! *'
Учитывая сходство между распадами динитрита этиленгликоля
и тринитрита глицерина, мы провели опыт по разложению дини-
трита при низкой температуре в надежде получить и для него обыч-
ную автокаталитическую кривую с одним экстремальным значе-
нием скорости. Эти ожидания оправдались: при 120° (фиг. 22) динит-
рит разлагался с постоянно возраставшей скоростью, на максимуме
превысившей начальную в 17 раз. Газовая фаза вблизи максимума
была окрашена в ярко-желтый цвет.
Начальная скорость газообразования при 143—160° растет с
температурой несколько быстрее, чем в случае гликольдинитрита
(при 170—190°), энергия активации при расчете по начальным ско-
ростям в этом интервале температур составляет 41,4 ккал/моль'
(фиг. 23). Предэкспонент также высок: 10’7’8 сек.-1.
1 Следует отметить, что в этом отношении исследовавшиеся полииитриты со-
вершенно аналогичны соответствующим полинитратам: динитрит и динитрат [2]
гликоля имеют энергию активации распада около 36 ккал/моль, тринитрит и
трннитрат [2] глицерина —41—43 ккал/моль. Причины этого различия между ди- и
тринитратамн и нитритами все еще недостаточно ясны.
320
31 451
321
При 120° начальная скорость существенно выше, чем это тре-
буется по уравнению Аррениуса; по-видимому, при этой темпера-
туре на первую стадию начинает накладываться вторая.
Фиг. 22. Распад динитрнта этиленгли-
коля при 120’ и ро=78О мм рт. ст.
Фиг. 23. Зависимость на-
чальной скорости газообра-
зования при распаде глице-
риитринитрита от температу-
ры в интервале 120—iSO*3.
Влияние окиси азота
Добавление окиси азота к тринитриту глицерина так же, как
к динитрнту гликоля, уменьшает скорость газообразования на пер-
вой стадии и почти не влияет на вторую. Характер уменьшения на-
чальной скорости при добавлении возрастающих количеств NO
Фиг. 24, Влияние NO на на-
чальную скорость газообразо-
вания при распаде глицерин-
1ринитрита при 150’.
(фиг. 24) сходен с тем, который наблюдается для динитрита. До-
бавление к тринитриту больших количеств NO уменьшает началь-
ную скорость распада в пределе приблизительно в 4 раза против
3 в случае гликольдинитрита при 170—1902 Правда, это предель-
ное подавление газообразования достигается при добавлении не
менее чем 500 мм NO, тогда как для уменьшения скорости до пре-
дела в случае дппитрита при 170° достаточно всего 200 мм этого
газа (см. фиг. 11).
322
Конечные продукты распада и их влияние на ход разложения
Конденсирующиеся при комнатной температуре конечные про-
дукты распада1 — темно-бурая жидкость, несколько менее вязкая,
чем в случае динитрита, хорошо растворяются в воде и имеют кис-
лую реакцию на конго-рот. Титрование водного раствора этих
продуктов щелочью показало, что на карбоксильные группы заме-
няется около 0,1 всех — ONO групп, содержащихся в тринитрите.
Добавление конечных конденсирующихся продуктов к свежему
нитриту, как показывает фиг, 25, существенно увеличивает ско-
Фиг, 25. Влияние конденсирующихся
продуктов распада па разложение
глицеринтринитрита при 150°,
f и 2—тринитрит с добавлением конден-
сирующихся продуктов, свежий нитрит-
фиг. 26, Влияние воды на распад
тринитрита глицерина при ISCT.
I—мм Рт, ст,, 2—без добавле-
ния поды.
рость и изменяет характер распада, превращая самоускоряющую-
ся реакцию в быстро замедляющуюся.
Эти опыты проводились следующим образом. Конденсирующие-
ся продукты от предыдущего опыта, растворенные в небольшом
количестве воды, помещали в чистый реакционный сосуд и продол-
жительное время откачивали, нагревая его до 80—100° для уда-
ления воды. После этого вводили тринитрит в специальный отрос-
ток, припаянный к сосуду, затем последний отпаивали и помещали
в термостат.
Следует отметить, что несмотря на продолжительную предвари-
тельную эвакуацию, конденсирующиеся очень гидрофильные про-
дукты, по-видимому, все же удерживали немного воды, В одном из
двух опытов (кривая 2), приведенных на фиг. 25, вола удалялась
более тщательно, чем в Другом. Скорость разложения в этом о^ыте
соответственно была ниже.
1 Газообразные продукты распада не анализировались. Известно лить, что
значительную долю их составляет окись азота, дающая сильное побурение газовой
фазы при впускании в сосуд воздуха.
21*
323
Добавление воды действует на распад тринитрита в сравнении
с динитритом гораздо сильнее (ср. фиг. 26 и 14), и не только при-
ближает наступление второй стадии, но и значительно увеличивает
скорость на первой.
Нужно сказать, что несмотря на специально принимавшиеся
меры предотвращения взаимодействия добавляемых веществ с
у жидким (не успевшим еще пре-
вратиться в пар) нитритом в про-
цессе прогрева реакционного сосу-
Фиг. 27. Влияние отношения поверх-
ности сосуда к его объему на распад
глицеринтрипитрита прн 15(Г.
s/-j: 1—10; 2-2 сл~1.
да до температуры термостата,
реакция в жидкой фазе все же,
по-видимому, протекала н могла
повлиять на ход опыта.
Увеличение отношения поверх-
ности реакционного сосуда к его
объему (фиг. 27) так же как при
распаде дицитрита заметно при-
ближало наступление второй ста-
дии и почти не влияло на
начальную скорость газообразо-
вания.
Обсуждение результатов
Вряд ли можно сомневаться,
сравнивая результаты опытов,
проведенных с тринитритом глице-
рина и динитритом гликоля, в
глубоком сходстве механизмов их
распада. Первое, о чем можно го-
ворить с достаточной определен-
ностью — это двустадийный ха-
рактер распада этих соединений.
Реакции первой стадии разложения тринитрита, по-видимому,
аналогичны реакциям распада алкилмононитритов и гликольдинит-
рита на этой стадии, и основные из них включают отщепление сред-
ней или крайней группы NO, распад оксирадикалов и реакции их
с окисью азота, распад образовавшихся при этом нитритов окси-
альдегидов и т. д.
Основываясь на результатах, полученных недавно (32] при изу-
чении распада I, З-буУиленгликольднпитрита, средняя NO-rpynna
которого отшепляется в 14 раз быстрее, чем крайняя, а также на
старых опытах Фармера [25] и Делпи [20], обнаруживших, что при
омылении нитроглицерина образуется в основном симметричный
динитрат глицерина, можно ожидать, что скорость отшепления
средней NO-группы в тринитрите глицерина больше, чем край-
них прямых указаний на это однако не имеется.
। Этому предположению противоречат опыты с динитратами глицерина, про-
веденные Елинеком (7). В этих опытах было показано, что при 140—16(Р скорости
распада паров симметричного и несимметричного динитратов разлнчЭ|0тся мало.
324
Опыты с добавлением NO при 150° показывают, что скорость
распада оксирадикалов в случае глииеринтринитрита, по-видимо-
му, выше, чем у гликольдинитрита (даже при 190э). Для предель-
ного угнетения газообразования при разложении тринитрита необ-
ходимо добавить значительно больше NO. Об этом свидетельствуют
менее резкое падение скорости на первом этапе распада трини-
трита и отсутствие угнетения газообразования при повышении на-
чального давления. «Удельный вес» реакций радикалов с NO, даю-
щих нитроксил, соответственно меньше, чем в случае распада ди-
нитрита, по-видимому, еще меньше роль обратной ступени реакции
распада молекулы нитрита. Экспериментальных данных для более
детального обсуждения механизма первой стадии не имеется.
Вторая стадия разложения тринитрита, по-видимому, так же,
как вторая стадия распада дицитрита, включает гидролиз нитрита
на стенке в тонком слое покрывающих ее конденсированных про-
дуктов и окисление органических веществ двуокисью азота, обра-
зующейся при распаде азотистой кислоты.
Роль реакций гидролиза и окисления при распаде глицеринтри-
ннтрита очевидно значительно больше, чем в случае разложения
гликольдинитрита. На это указывает более сильный, чем для гли-
кольдинитрита, рост скорости в ходе разложения, образование зна-
чительных количеств двуокиси азота при тех условиях, при кото-
рых ни малейших следов ее не заметно в случае гликольдинитрита,
значительно более сильное влияние воды и конденсирующихся про-
дуктов распада и г. д.
О МЕХАНИЗМЕ ТЕРМИЧЕСКОГО РАЗЛОЖЕНИЯ ЭФИРОВ АЗОТИСТОЙ
И АЗОТНОЙ КИСЛОТ
Эфиры азотистой кислоты
Газофазный термический распад алкилмононитритов вблизи
200°—одна цз простых и детально изученных пиролитических ре-
акций. Наиболее вероятные основные ступени ее—разрыв связи
О—NO, взаимодействие образовавшейся окиси азота с оксирадика-
лом и распад последнего;
RCH2ONO^> RCHaO- 4-NO RCHO4-NOH
RCH2O~ R-+CH.0
Суммарная скорость Процесса определяется скоростью первой
его ступени и изменяется по закону мономолекулярной реакции.
Энергия активации этой рсакции находится в пределах
34—38 ккал/моль, предэкспоненпиальный фактор в уравнении Ар-
рениуса имеет нормальное значение (10,34-10|4 сек.-1).
Увеличение и усложнение молекулы в ряду алкилмононитритов
приводит к увеличению скорости распада. В табл. 3 приведены
константы уравнения Аррениуса и константы скорости распада
ряда мононитритов при 190э. Видно, что при переходе от одного го-
молога к следующему скорость реакции увеличивается приблизи-
тельно вдвое.
Таблица 3
Скорость термического распада алкилнитритов
Вещество Интервал температур °C Энергия активации £. МОЛЬ 1g В, (сек.-1) Начальная сксрэсть распада и k при 190° сек.“1. Ю4 Ссылки на лите- ратуру
Метил нитрит 193—241 36,4 13,26 0,97; 0,48 [46]
Этнлнитрит 190—241 37,7 14.14 1,89; 0,94 ]47]
161—201 37,5 13,79 — [37]
• 430—530 34,3±3 — — 144]
«- Пропили итрит 170-210 37,65 14,44 3,95; 1,98 ]48]
200—233 34,7 13,20 — [13]
Изопропилннтрит 170-210 37,0 14,10 3,70; 1.85 [49]
я- Бутилнитрит 170-212 37 14,48 8,88; 4,44 [55]
202—239 36,2 13,66 — ]13)
Диннтрнт этилен- гликоля 170—190 35,6 13,8 33; И
ТрииитрИт глице- рила 143—160 41.6 17,8 140*; 47
* По расчету.
Увеличивается при усложнении молекулы и скорость распада
оксирадикала. При распаде этилнитрита эта реакция не играет
почти никакой роли и во всяком случае скорость ее неизмеримо
меньше, чем скорость образования нитрита из этоксила и NO [37],
при распаде н-пропилнитрита она уже весьма существенна [38], а
в случае трет-бутилнитрита при 201°, как показывает простой рас-
чет по данным Леви [38]. константа скорости распада трет-бутокси-
ла (&з) в несколько сотен раз больше, чем константа скорости об-
разования нитрита из грег-бутоксила и NO (&,) (концентрации в
мм рт. ст.).
Если молекулу этилнитрита усложнит!,, заменив водород р-уг-
леродного атома не на СН3, а на ONO, г. е. превратить этилнит-
рит в динитрит этиленгликоля, то на начальной стадии распада это
приведет к увеличению скорости разрыва связи О—NO раз в 5—6
большему, чем при введении СН3-группы (см. табл- 3), а также
к некоторому увеличению скорости распада оксирадикала, без
принципиального изменения механизма этой стадии.
То же самое произойдет при переходе от пропилпитрита к три-
нитриту глицерина: основные реакции начальной стадии распада
326
сохранятся, но скорость разрыва связи О—NO и скорость распада
оксирадикалов существенно увеличатся.
Если для этилнитрита соотношения между скоростями основ-
ных реакций можно представить в виде
> k-» Аз<
то для гликольдинитрита
^3,
а для глицеринтринитрита, по-видимому:
При повышении температуры соотношения между отдельными
скоростями процесса очевидно изменяются. Скорость распада ок-
сирадикала вероятно растет быстрее, чем скорость реакций взаимо-
действия его с окисью азота. На это, помимо некоторых косвен-
ных данных, указывают результаты работ американских исследова-
телей [33], не обнаруживших в продуктах распада вицинальных
динитритов прн 260—280° иных органических соединений, кроме
альдегидов, образовавшихся в результате разрыва связи между
а—а' атомами углерода, а также результаты исследований Пол-
ларда с сотрудниками [43], установивших присутствие в продук-
тах распада этилнитрата значительных количеств формальдегида
при ~3003 и не нашедших его при ~200°.
Это предположение подтверждается и опытом с динитритом
этиленгликоля, проведенным нами при 290°. Давление в этом слу-
чае возросло не в 2,8—3,1 раза, как в опытах при 170°, а в ~3,8
раза, вплотную приблизившись к тому давлению, которое должно
было бы создаться, если бы динитрит разлагался по уравнению
CH.ONO
| -> 2CH.O + 2NO
CH,ONO
Скорость реакции распада нитроксила при повышении темпе-
ратуры, по-видимому, также растет несколько быстрее, чем ско-
рость рекомбинации оксирадикала и NO.
Таким образом, если рассматривать только начальные этапы
распада, переход от мононитритов к полинитритам принципиаль-
ных изменений в механизме разложения не вызывает. Несмотря на
то, что процесс этот очевидно становится более сложным, основ-
ные закономерности его остаются теми же. Естественно меняются
лишь скорости отдельных основных реакций и соотношения меж-
ду константами этих скоростей, прежде всего при распаде оксира-
дикала и в реакциях его с окисью азота (не в пользу последних).
Однако, если мы от начальных этапов распада перейдем к рас-
смотрению всего процесса в целом, мы сразу же натолкнемся на
существенное, принципиальное отличие механизма газофазного
пиролиза полинитрнтоз от мононитритов. Заключается оно в нали-
чии у полинитритов в определенных условиях второй самоускоряю-
327
щейся и существенно полимолекулярной стадии распада, а также
в появлении в промежуточных продуктах двуокиси азота, как по-
казано выше, тесно связанное со степенью развития самоускоряю-
щейся реакции.
По аналогии с предположением американских исследователей
[32], обнаруживших NO2 в продуктах распада некоторых невици-
нальных динитритов вблизи 300° можно было бы представить себе,
что двуокись азота возникает при распаде оксирадикзла, образо-
вавшегося в результате отрыва одной NO-группы от молекулы
нитрита. Однако, по-видимому, в условиях наших опытов эта реак-
ция роли не играет. Основное экспериментальное противоречие
предположению об образовании NO3 при распаде оксирадикалов
заключается в результатах опытов со снижением температуры на
второй стадии разложения динитрита этиленгликоля со 170 до 100°:
несмотря на то, что при 1003 скорость термического распада динит-
рита (k) практически равна нулю, концентрация NOa продолжи-
тельное время (около получаса и более) увеличивается, причем
газовая фаза в реакционном сосуде из совершенно бесцветной пре-
вращается в темно-красную. Разумеется никакие оксирадикалы,
распадающиеся с образованием NOa, такой картины процесса дать
бы не смогли. По-видимому, в настоящее время единственно обо-
снованным предположением, логично объясняющим как этот факт,
так и совокупность остальных экспериментальных данных, относя-
щихся ко второй стадии распада полинитритов, является предпо-
ложение о том, что последние гидролизуются на стенке сосуда в
пленке покрывающих ее труднолетучих продуктов разложения. Об-
разующаяся при этом азотистая кислота быстро распадается с об-
разованием двуокиси азота, которая и обеспечивает прохождение
реакций окисления, дающих обнаруженные в конечных продуктах
Органические кислоты и углекислоту.
Предположение о гидролитической реакции на второй стадии
разложения полинитритов хорошо согласуется с легкой гидроли-
зуемостщо эфиров азотистой кислоты — явлением общеизвестным
и давно описанным в литературе [24]. Реакция гидролиза нитрита
может быть представлена в виде
RONO-I-H.O ROH + HONO
причем молекула, так же как при термическом распаде, рвется по
связи О—NO [18]. [19]. Эта реакция катализируется кислотой, и ско-
рость кислого гидролиза, равно как и начальная скорость терми-
ческого распада, при переходе от низших членов ряда к высшим
увеличивается [18], [45].
Можцо полагать, что в еще большей степени она увеличивает-
ся прн переходе от мононитритов к полинитритам. На это указыва-
ет, например, крайне низкая стойкость последних при обычных
температурах, несовместимая со сравнительно малой скоростью их
термического распада при повышенных температурах. Действи-
тельно, гликольдинитрнт почти полностью разлагается при хране-
нии на воздухе, окрашиваясь в темно-зеленый цвет (HNO3), в те-
328
|^ение трех-шести дней, глицеринтринитрит — в течение двух-трех
йдней, тогда как этилнитрит и другие мононитриты, насколько мож-
№0 судить по литературным данным, могут храниться в темноте
[весьма продолжительное время1.
& Аналогичная картина наблюдается при действии воды, которая
[на глазах разлагает динитрит этиленгликоля и особенно тринитрит
[ В)
№
К1, Фиг. 28. Распад динитрнта 1,4-бутиленгликоля.
В:. а—при |70’ и р„-370 мм рт, с г., б—при ISO’ и р0«-4<М мм
к ’ рт. ст. 1—в координатах ——т; 2—s координатах is— т.
К р“
^глицерина, но относительно медленно реагирует с этил- или пропил-
(нитритами.
ft Очевидно, переход от алкилмононитритов к динитриту этилен-
Мликоля и тринитриту глицерина увеличивает не только и не столь-
Кко скорость термического распада, сколько скорость гидролиза
^нитрита г. Можно полагать, что существенную роль при этом игра-
г<ет не только увеличение количества нитритных групп в молекуле,
К ’ Гидролитические реакции очевидно играют Значительную роль и при высоко-
Ктемпературьом жидкофазном распаде эфиров азотистой кислоты. Результаты ре-
Вфкоторых, к сожалению правда немногочисленных, работ по разложению нитритов
Я/В жидкой фазе [31], [33] могут быть легко объяснены с привлечением этого пред-
К''лоложения.
Ж“- J При этом введение второй нитритной группы в молекулу, в которой одна
Чфтакзя группа уже имеется, по-видимому, является более эффективным, чем вве-
А;] Хенне Третьей — 0N0 группы при уже имеющихся двух.
329
но и то, что эти группы расположены рядом друг с другом. Если
это последнее предположение справедливо, то разъединение нит-
ритных групп в молекуле полинитрита, например, СН2-группами,
должно уменьшить роль реакций второй стадии при термическом
разложении эфира.
Для того чтобы проверить справедливость этого заключения,
мы провели опыты по распаду динитрита 1,4-бутиленгликоля
Оказалось, что разложение этого соединения при 150 — 190° в ос-
новном протекает аналогично распаду динитрита этиленгликоля и
тринитрита глицерина с той только разницей, что вторая стадия
у него при обычных начальных концентрациях испытуемого ве-
щества в реакционном сосуде становится отчетливо выраженной не
при 180°, как в случае этиленгликольдинитрита, а лишь при 150°
(см. фиг, 28), т. е. «удельный вес» ее в процессе разложения сущест-
венно меньше, чем при распаде этиленгликольдинитрита1 2 3. Следует
особо подчеркнуть при этом, что скорость собственно термического
распада молекулы при переходе от 1,2-этиленгликольдинитрита
к 1,4-бутиленгликольдннитриту почти не меняется. Отметим также,
что при 280—300° 1,4-бутиленгликольдинитрит (в противополож-
ность, например, 1,3-бутиленгликольдинитриту) разлагается без
образования NO2 [32], и, следовательно, возможность появления вто-
рой стадии вследствие накопления двуокиси азота, образовавшейся
в результате распада оксирадикала, у него отсутствует-
Эфиры азотной кислоты
Результаты описанных выше исследований позволяют сделать
некоторые предположения относительно механизма термического
распада нитроэфиров.
Механизм термического распада алкилмононитратов вблизи
200°, основанный на многочисленных исследованиях пиролиза нит-
роэфиров (основные из них [3], [17], [27], [35], [36], [41], [421, [431),
алкилмононитритов и окисления альдегидов двуокисью азота [28],
[40] в настоящее время может быть представлен в виде следующих
основных реакций [10]:
RCH2ONO2 RCH2O- -[-NO.,
RCHjO- -> R-+CH.0
RC1EO- -[-NO RCHvONO
RCH,O-+N0 -> RCHO^NOH
RCHO, CH,OfNO2^> NO, CO, CO2, H2O
И Др. Продукты.
1 Эти опыты были проведены В. Г, Хотиным,
2 Возможно, что метиленовые группы уменьшают удельный вес реакций вто-
рой стадии распада не только в том случае, когда они наедены между группа-
ми — ОХО. Ясность в этот вопрос могло бы внести изучение термического распада,
например, 2, 3-бутиденгликодьдинитрита.
330
2N0H N..0 J-H.O.
,Можно предположить, что, поскольку переход от мононитритов к
полинитрптам не сопровождается существенным изменением меха-
низма первой стадии распада, основные реакции начальных этапов
разложения полинитратов также будут аналогичны приведенным
выше для мононитратов. В случае нитрогликоля это могли бы
быть следующие реакции:
CH2ONOa СН2О-
I | +NO,
CH2ONO2 CHaONO2
СН2О-
| 2CH..O-]-NOa
ch,ono2
CH2O-]-N02 NO, CO, CO2, H2O
CH2O- ch2ono
[ +NOi|
ch2ono2 ch2ono,
CH2O- CHO
| H-NO 4 | +NOH
ch2ono2 ch2ono2
2NOH -> N2OH-H2o
• и T. Д.
Основываясь на результатах, полученных при изучении распа-
да диннтрита этиленгликоля, можно заключить, что скорость рас-
пада оксирадикала (А3) в случае нитрогликоля очевидно будет
больше, чем в случае этилцитрата, а соотношение констант скорос-
тей реакций и k2, по-видимому, изменится не в пользу образо-
вания нитрнт-нитрата,
Исследования термического распада паров нитрогликоля, про-
веденные в лаборатории МХТИ им. Д. И. Менделеева [2], [10], [12],
[14] и др. подтверждают этот механизм. Скорость распада, так же
как в случае мононитратов, не зависит от начальной концентрации
нитрата, энергия активации распада (35—36 ккал/моль) близка
к значению ее для мононитратов, газообразные продукты распада
содержат NO?, NO, СОг, СО, N»; при добавлении окиси азота в про-
дуктах обнаруживается N2O [10], При разложении образуются так-
же вода, формальдегид и щавелевая кислота, При добавлении оки-
си азота начальная скорость газообразования при 170° уменьшает-
ся (фиг. 29), причем характер зависимости ее от концентрации
NO такой же, как и при распаде гликольдинитрита. На последую-
щие стадии распада добавление NO не влияет. При более низкой
температуре (150°) £14] добавление NO, так же как в случае раз-
ложения этилнитрата в определенной области условий [14], [42] не
331
влияет на начальную скорость газообразования, но значительно
уменьшает скорость распада на средних этапах (фиг. 30).
Интересно отметить, что при температурах ниже 1703 зависи-
мость скорости газообразования при распаде нитрогликоля от вре-
мени имеет максимум, положение и величина которого не зависят
от концентрации нитрогликоля [2]. Это явление можно было бы
объяснить развитием окислительных реакций, роль которых дол-
жна возрастать по мере накопления в продуктах распада двуоки-
Фиг, 29, Влияние NO на распад нитрогликоля
при 170’.
д—изменение скорле’и гзэпабраэопаннн ко эр^мени при
(начальная концентрация ниiрогликсля) и О — Ь>7
NO
я 0;+—196 и 14; • и г.0 мл рт, сгг; б—нзиенение
начальной скорости распали при добавлении NO.
си азота и органических веществ. Однагэ в этом случае величина
и положение максимума очевидно доля ны были бы зависеть от
начальной концентрации нитрата. Поскольку такой зависимости не
наблюдается, можно предположить, что ускорение газообразова-
ния связано с накоплением и последующим разложением проме-
жуточных продуктов распада нитрогликоля, например нитрата гли-
колевого альдегида или нитрит-цитрата гликоля. В этом случае
независимость скорости реакции от концентрации нитрата более
объяснима. Уменьшение этого эффекта при повышении температу-
ры и полное исчезновение его при 17СГ можно было бы связать
с тем, что роль реакций образования насыщенных мононитратов
при возрастании температуры уменьшается и все большая часть
оксирадикалов распадается на формальдегид и NOj,
Может быть именно поэтому в случае нитроглицерина, скорость
распада окенрадикадов которого несомненно значительно выше,
чем нитрата глиоксила (CH2ONO3CH2O •), максимума на кривой
при распаде НГЦ в парах не наблюдается, и зависимость
скорости от времени для него при всех температурах в интервале
332
140—160 описывается кривыми вида, изображенного на
фиг. 29, а
По-видимому, в случае нитроглицерина реакции, ведущие к об-
разованию нитритов, так же как иные реакции оксирадикалов с
NO, играют еще меньшую роль, чем при распаде нитрогликоля Об-
разованию значительных ко-
личеств нитритов при распа-
де нитроглицерина и нитро-
гликоля препятствует так-
же то обстоятельство, что
эти нитриты легко разлага-
ются водой, которая всегда
образуется при распаде по*
линитратов.
Изложенные выше сооб-
ражения согласуются с ре-
зультатами опубликованной
недавно работы Уэринга, ис-
следовавшего распад нитро-
глицерина при 140—160s с
помощью инфракрасного
спектрометра [56]. Было об-
наружено, что скорость рас-
пада паров нитроглицерина
подчиняется уравнению ре-
акции первого порядка и
уменьшается в присутствии
двуокиси азота. Никаких по-
лос поглощения нитритов в
спектре продуктов распада
не обнаруживается. Сдвига
частот ONO2-rpyniibi в ходе
распада также не наблю-
дается; по мнению Уэрицга,
это свидетельствует о боль-
шой скорости распада со-
держащих нитратную груп-
Фиг. 30, Влияние окиси азота на распад
этилнитрата при |81° и ннтрогликоля при
150= (по данным М. С. Плясунова [|4]>,
а—э11!Лии:ра: при д, (кончен Гранин ни ра а) и
Z—333 н 0; 2—342 и 4В0; 247 и 43й ‘мм р*. с г,
б—rttcrpc,гликоль прн р„ rr J-i2O и Q; 2— JI& н
и £31; 4—'/5 и 443 лед рт. ст,
^р-рио.
2|4; 3—123
пу промежуточных Продук-
тов, Механизм распада, предложенный этим исследователем,
аналогичен написанному выше для питрогликоля с учетом уже упо-
минавшихся изменений в скоростях отдельных реакций.
Следует отметить, что Уэринг считает реакцию распада паров
нитроглицерина гомогенной. Это противоречит результатам, полу-
ченным другими исследователями [2], [11], [12]. Некоторые из этих
результатов изображены на фиг. 31 (см. [12]), Несмотря на то, что
1 Отсутствие максимума скорости в случае моионидратов можно также объ-
яснить чрезмерно малой скоростью распада алкнлмононнтрнтов — основных про-
межуточных продуктов разложения мононитроэфиров.
333
кривые, приведенные на фиг. 31, получены манометрическим мето-
дом, различие в скоростях реакции в сосудах без капилляров и
с ними настолько велико, что сомневаться в значительной гетеро-
Фиг. 31. Влияние набивки реакционного сосуда стеклян-
ными капиллярами на распад паров нитроглицерина при
140° (по Н. А. Межевых [12]).
(m/и г/сл’) 1о* и S см1: 1-0.4 и 2000 : 2—0.5 и 1000; 3—0,5 и 1000:
4—о,7 и 1000; 5-0,5 и 100; 0-0,8 и [00.
<m!v г!см3, 10* и г скЧУ-В.З „ |00 (без ипВннки); 2—5,5 и 700 (сгск-
лйи1гы& капилляры); 3—5,6 и *-000 (с <екля|fпая ваш); 9,2 и 10 000
(кварцевый песак)^
гениости распада нитроглицерина вряд ли возможно. Кстати, рас-
пад нитрогликоля, как это следует например из фиг. 32, взятой из
той же работы [12], — реакция существенно гомогенная.
334
Известно, что при распаде полинитратов в жидкой фазе боль-
шую и при понижении температуры все увеличивающуюся роль
«грают гидролитические реакции. Гидролиз полинитратов, так же
как гидролиз нитритов, в присутствии кислоты существенно уско-
ряется. Можно предположить, что скорость гидролитических реак-
ций и возможно каталитическое действие на них кислот увеличи-
вается прн накоплении в молекуле не только ONO, но и
ONCh-групп. Быть может, особенно эффективным также является
их соседнее расположение Г
Вероятно, в значительной степени именно этим обстоятельством,
а не увеличением, как это еще иногда полагают, «лабильности»
молекулы или уменьшением энергии активации ее распада объяс-
няется существенное снижение химической стойкости при хранении
по ряду ннтроэфиров с увеличивающимся числом нитратных групп
(при переходе от алкилмононитратов к нитрогликолю, нитроглице-
рину, эритриттетранитрату и т. Д-) 1 2.
ВЫВОДЫ
Термический распад алкилмоноиитритов в газовой фазе вблизи
200° изучался многими исследователями и представляет собой от-
носительно простую реакцию, основные ступени которой — обрати-
мый разрыв связи О—NO, взаимодействие окиси азота с окси-
радикалом с образованием нитроксила и карбонильного производ-
ного и распад оксирадикала.
Газофазный термический распад динитрнта этиленгликоля, изу-
чавшийся нами при 120—190° и начальной концентрации
'50—1000 л.и рт. ст., более сложен и включает по крайней мере две
макроскопические стадии. Близость энергии активации распада
этого соединения к величинам, получаемым для мононитритов,
' влияние окиси азота ца скорость, гомогенность, образование
NO, NaO и СНгО и ряд других фактов позволяют предположить,
что механизм первой стадии распада этого динитрита аналогичен
механизму распада мопонитритов.
Реакции второй стадии имеют самоускоряющцйся в значитель-
ной степени гетерогенный характер и «удельный вес» их существен-
но возрастает при увеличении концентрации динитрнта и снижении
' температуры опыта. Влияние воды, содержащих кислоту трудно-
летучих продуктов, образование двуокиси азота и углекислоты и
ряд других фактов можно объяснить, предположив, что в основе
Второй стадия распада лежат реакции гидролитического характера.
1 Следует отметить, что скорость гидролитических реакций в ряде случаев,
по-видимому, может существенно увеличиваться при накоплении в молекуле не
ТОЛЬКО НИтрИтЦЫХ или нитратных, но и нитрогрупп.
2 Другим важным фактором, приводящим к уменыпенгпо химической стойко-
сти при увеличении количества ОКОг-групп в молекуле нитрата, является накоп-
ление прн распаде богатых кислородом нитроэфиров значительных количеств выс-
ших окислов и кислот азота, приводящее к значительному увеличению скорости
реакций окисления, продукты которых в свою очередь участвуют в гидролитиче-
ских реакциях [15].
335
протекающие на стенке реакционного сосуда в тонкий аденке кон-
денсирующихся на ней продуктов.
Таким образом, увеличение числа ONO-групп в молекуле нитри-
та от одной до двух, сравнительно слабо увеличивая скорость тер-
мического распада молекулы, существенно повышает скорость гид-
ролитических реакций, делая их возможными даже при распаде
нитрита в парах при весьма высокой температуре.
Тринитрит глицерина в газовой фазе при 100—-160° разлагается
аналогично гликольдинитриту. Распад его также двустадиен, и
влияние температуры, концентрации окиси азота, воды и конденси-
рующихся продуктов распада, увеличения поверхности и других
факторов на течение этих стадий удовлетворительно описывается
схемой, аналогичной той, которая предложена для гликольдинит-
рита.
Скорость разрыва связи О—NO в тринитрите глицерина в не-
сколько раз больше, чем в динитрите этиленгликоля, различие в
скоростях реакций второй стадии еще больше.
Некоторые результаты опытов с динитритом 1,4-бутиленглико-
ля показали, что разделение ONO-групп в полинитритах СН2-груп-
пами слабо влияет на скорость распада, по заметно уменьшает
склонность процесса к самоускорению, по-види.мому, снижая ско-
рость реакций полинитрита с водой.
На основании результатов, полученных при изучении распада
поликитритов, сделаны некоторые выводы относительно механизма
термического распада нитроэфиров.
ЛИТЕРАТУРА
I. Андреев К. К-, Термическое разложение и горение взрывчатых веществ.
М.г Госэцергоиздат, 1957,
2. А и д р е е в К. К. Глазкова А. П.,Маурика Н, Д и Светлов Б. С..
ЖФХ. 1958, 32, 1726.
3. А и и н А. Я , Т о д е с О. М.г Ха р и тон Ю. Б., ЖФХ, 1936, 8, 866,
4. Вайбель, Идентификация органических соединений, ИЛ, 1957.
5. Васильев Е. В., Исследование термического разложения глицерянтри-
нитрита, дипломная работа, МХТИ нм. Д. И. Менделеева, [959.
6. Гаттерма н-В и л а и Д, Практические работы по органической химии.
Госхимиздат, 1948. стр. 187.
7. Елннек. Изучение термического разложения динитратов глицерина, дип-
ломная работа, МХТИ нм. Д. И. Менделеева, 19,59.
8. Кларк Г., Руководство по качественному и количественному органиче-
скому анализу, Госхимиздат, 1934.
9. К о и д р и к о в Б. Я.. ЖФХ, 1958, 32, 1 [75. Научные доклады высшей шко-
лы, химия и химическая технология. 1959, 19.
10. Кендриков Б. Н., Изучение термического разложения гликольдииит •
рита. диссертация, МХТИ им. Д. И. Менделеева. 1953.
И. Маури на Н. Д., Термический распад нитроглицерина, диссертация.
МХТИ нм. Д. И. Менделеева. 19,5|.
12. Межевых Н. А„ Влияние поверхности стекла на термическое разложе-
ние нитрозфиров, дипломная работа, МХТИ им. Д. И. Менделеева, 1957.
|3. Нагиев М. ф., Петрова 3. Г., Султанова А. И., ДАН, 1956.
106, 573.
14. Плясунов М. С,, Влияние окиси и двуокиси азота и ацетальдегида на
термический распад ннтроглнколя в парах, дипломная работа, МХуИ им.
Д. И. Менделеева, 1956.
.336
31). Jest P„ P h
31. К о г п Ы u hi
32. К u h n L. P..
33. Kuhn
34. Levy
35. Levy
36. Levy
37. Lev у
38. Levy
R,; 15. Светлов Б. С., См. настоящий сборник, стр. 184, 208.
16. Э м м а н у э л ь Н. М., Сб. «Вопросы химической кинетики, катализа и ре-
[ акционной способности», АН СССР, 1955, стр. 117.
Г' 17. Adams G. К-, Bawn С. Е. И,, Trans. Faraday Soc., 1919, 45, 494.
11 18. Allen A. D., J. chem. Soc., 1954, 1968.
19. Anbar M., Dostrovski Samuel D. Yoffe A., J. chcm.
t-Soc, 1954, 3603.
Г 20. Berl E„ Delpy M„ Bcrichte, 1910.43. 1424.
t.: 21. Bert Oni C., Gazz. chi,n. ita|., 1885, 15, 353.
f 22. Birss F. W., Dauby C. J., Hinshelwood C. N., Proc. Roy.
|;Soc„ 1957. A239, 154.
В 23. Fischer E., Landsteiner K., Bcrichte. 1892, 25, 2,552.
с 24. Fischer W. H,, Z. phys. Chem., ]9< 9, 65, 61.
ft' 25. Farmer R. C., J. them. Soc., 1920. 117, 806.
!: 26. Feigl F., <Spot Tests», New York, 19.54, vol. 2.
1 27. Cm. nanp. Gray P„ Proc. Roy. Stic.. 1954, A221, 462.
\ 28. Gray P.r Yoffe A. D., Chem. Revs., 1955. 55. 1069
j, 29. Gray P., Chem. and Ind.. 193|), 120
i I 1 i p s "L., Proc, c he in. Soc., I960, 73.
N., Oil veto E. P.. J. Am. chem. Soc., 1919, 71.226,
Wright R., De Angelis L„ J. Am. chem. Soc.,
Ji'1954, 76. 328; 1956, 78, 2719.
L. P._ De Angelis L., J. Am. chem. Soc., 1956, 52, 59.
J. B., J. Am. chem. Soc., 1953. 75, 1801.
J. B„ J. Am. chem. Soc-. 1954. 76. 32-54, 379'1.
J. B., Adrian F. J., J. Ain. them. Soc., 1955, 77, 2015.
J. B„ J. Am. chem. Soc., 1956. 78, 762.
J. B.. hid. Eng. Cherti. 1956, 48, 762.
!• 39. Masson O.. Berichie. 1883, 16. 1697.
K, • 40. Pedler Л. E., Pollard F. H.. Trans. Faraday Soc., 1957, 53, 44.
R -41. Phillips L._ Nature, 1950, 165, 564
У 42. Pollard F. H„ Wyatt R. M. H.. Marshall H. S. B., Nature.
[ 1950, 165,564;
S Pollard F. H., Marshall F|. S. B., Pedler Л. E., Nature.
К1953, 171, 1154;
Po 11 a r d F. H., P e d 1 e r A. E., f l a r d у C. G., Nature, 1954, 174, 979,
I. 4,‘J. Pa I |ard F. H., Marshall H. S. B.. Pedler A. E., Trane. Fa-
B'raday Soc., 1956, 52, 959.
E" 44. Rice F. O., R a d 0 w s k a s E. 1.., J. Am. chem. Soc.. 1935, 57, 350.
b- 45. S c r a b a 1 A., Zahorka A., Welmanii K., Z. phys. Chem., 1939,
I..A183. 345.
46. S t e a c i с E. W. R.r Shaw G., Proc. Roy. Soc-, 1931, A146, 388.
в.' 47. S t e a c i e E. W. R., Shaw G., J. chem. Phys., 1934,' 2. 345.
fe. 48. Steacie E. W. R., Shaw G.. J. chem- Phys., 19-35, 3. 314.
И, 49. Steacie E. W. R., Shaw G.. Proc. Roy. Sue.. 1935, A151, 685..
K, 50. Steacie E. W. R., Calder D.. J. chem. Soc., 1936, 4, 95.
B', 5). Steacie E. W. R., Smith W.. J. chem. Phys., E'3J, 4, 504.
I; 52. Steacie E. W. R., Rosenberg S.. J. chem. Phys., 1936.4,223.
J1.. 53. S tc a c i e E. W. R., Katz S. and oth.. Саги J. Res. 1936, Bl4, 268.
L 54. S I e a c i e E. W. R., Katz S.. J- chem. Phys., 1937, 5. 125.
B.' 55. Steacie E. W. R., ’Atomic and Free Radical Reactions», 2 nd Ed.,
Й New York- 1951, vol. 1.
F - 56. W a r i n g С. E., Armed Forces chem. J., 1959. 13, № 6. 28.
f 57. W о I f г о 111 M. L„ F г a z e r J- 11- К u h n L. P. and oth., J. Am- chem.>
7 Soc., 1955, 77, 6573; 1956. 78, 4695;
Wolfrom M. L.. Chanev A.. Wain P. M., J. Am. chem. Sos.,
I 1958, 80. 946.
t; 22 4.51 337
Ю. Я. МАКСИМОВ
22, ТЕРМИЧЕСКИЙ РАСПАД ПАРОВ
НИТРОПРОИЗВОДНЫХ БЕНЗОЛА
Стойкость полинитросоединений ароматического ряда по срав-
нению с ВВ других классов, например, нитроэфирами очень вели-
ка, В связи с этим изучению термического распада нитросоедине-
ний до последнего времени уделялось мало внимания; сколько-ни-
будь подробные исследования термического распада, впрочем весь-
ма немногочисленные, 'имеются в литературе только для тринитро-
толуола,
В последнее время в связи с возникшей потребностью нефтя-
ной промышленности во взрывчатых веществах, способных длн
тельное время выдерживать относительно высокую температуру,
вопросы термического распада нитросоединений приобрели прак-
тический интерес.
Для того, чтобы поиски таких ВВ вести более обоснованно, чем
это делалось до сего времени, целесообразно установить некоторые
закономерности, связывающие особенности термического распада
нитросоединений с их химической структурой н агрегатным состоя
нием вещества. Как одно из первых исследований в этом направ-
лении и было проведено изучение термического распада паров нит
ропроизводных простейшего ароматического соединения бензола
Результаты этого исследования изложены в данной статье,
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Вещества и методика
Мононитробензол. Фракция, выделенная из технического про-
дукта путем двукратной перегонки в вакууме. Температура затвер-
девания 5,4—5,5°, температура кипения при 760 мм рт, ст. 210,5°
1,3~динитробензол. Технический продукт подвергали двукрат
ной перекристаллизации из ацетона и однократной из метанола.
Температура плавления 89,5—90,0°, температура кипения при
760 мм рт. ст. ~ 300э.
1.3,5-тринитробензол. Прозрачные пластинчатые кристаллы
зеленоватого оттенка, получен при двукратной перекристаллиза-
ции лабораторного продукта из этанола. Температура плавления
122,0—122,5°.
Термическое разложение этих нитросоединений изучали мано-
метрическим методом в стеклянном сосуде цилиндрической формы
объемом 20—25 см3 со стеклянным манометром типа Бурдона [1].
Остаточное давление воздуха в сосуде перед опытом составляло
10’3-=-10 ’4 мм рт, ст. Разложение вели в термостате, наполненном
жидким оловом. Температуру в термостате поддерживали постоян-
ной с колебаниями в пределах ±0,5°. В ходе распада измеряли
ззя
давление в сосуде. По этим данным строили затем кривые р —
графическим дифференцированием которых получали изменение
скорости распада со временем.
Результаты экспериментов
Мононитробензол
- Скорость роста давления при термическом разложении паров
нитробензола уменьшается во времени {фиг. !) Общий прирост
давления в конце распада в интервале температур 395—445°, в ко-
тором было наиболее удобно изучать разложение, составляет
Фиг. 1. Термический распад мононитробензола в парах при 415=.
л) Изменение относительной скорости газообразования со временем при раз-
личном начальном давлении лзрев нитробензола
N кривой 12 3 4 5
pt мм 80 204 575 Т ТЭД |000ч
J;'zr 2 2 2 2 20
Здесь н далее па фиг 2 и 3 от irninenne суммарной поверхности стекла к
свободному объему сосуда; А—начальное давление паров внтросоедннсвия
в мм рт, ст,
б) Зависимость константы скорости от начального длвленнэ парив.
170—200% по отношению к начальному давлению паров. Почти с
самого начала опытов на стенках реакционного сосуда образуется
бурый налет, интенсивность окраски которого увеличивается
по мере протекания распада. Кроме этого, на более поздних эта-
пах разложения наблюдается также осаждение на дне сосуда тон-
кодисперсного порошка темно-серого цвета.
Из данных, приведенных на фиг. 1, видно, что начальная ско-
рость газообразования зависит от концентрации вещества (началь-
ного давления). Скорость реакции при малых начальных давле-
ниях быстро растет с начальной концентрацией вещества, а затем
22;
332
этот рост замедляется. Своеобразное влияние оказывает начальное
давление паров на характер кривых скорость — время. При малых
начальных давлениях темп падения скорости газообразования убы-
вает по мере течения распада. С начала и до конца разложения
скорость в этом случае уменьшается по закону реакции первого по-
рядка. При начальном давлении паров более 500 мм рт. ст. ско-
рость газообразования па начальном этапе падает медленнее, чем
на последующем, н на кривой наблюдается точка перегиба (кри-
вые 3 и 4 фиг. 1, а).
Исходя из предположения, что особенности распада нитробен-
зола могут быть связаны с течением гетерогенной реакции на стек-
ле, мы провели опыты, в которых поверхность стекла была увели-
чена наполнением сосуда капиллярными трубками (кривая -5
фиг. 1, а). Характерным для разложения в этих условиях является
более быстрое падение скорости газообразования и заметное (на
10—20%) уменьшение конечного давления газообразных продуктов
распада.
Если по температурной зависимости начальной скорости распа-
да (углу наклона начального участка кривых р=?(т)) при давле-
нии паров мононитробензола 400—500 мм определить Е (фиг. 4),
то константа скорости, рассчитанная для реакции первого порядка,
выразится уравнением
53 400
k = 101г'65-е АТ сек~’,. (1)
Динитробензо а
Термическое разложение 1,3-динитробензола, изученное в ин-
тервале температур 345—410° и при давлении паров 200—800 мм
рт. ст., как видно из кривых фиг. 2, протекает с ускорением газо-
образования.
Появление па поверхности стекла бурого налета и темно-серого
порошка на дне сосуда в результате расцада диннтробензола, ка-
чественно схоже с наблюдениями при термическом разложении
мононитробензола. Кроме того при распаде динитробензола, как
и двух других нитросоединений бензола, образуется некоторое коли-
чество продуктов, конденсирующихся при комнатной температуре.
Во всех опытах с динитробензолом при вскрытий реакционного со-
суда обнаруживался сильный запах горького миндаля. Общее коли-
чество газов, образующихся при распаде, составляет около 3 молей
на моль вещества.
Кривые р — f(t), приведенные на фиг. 2, показывают, что уве-
личение исходной концентрации паров двояко влияет на распад,
С одной стороны, рост начального давления паров до некоторого
предела (при 395° около 300 мм рт. ст,) увеличивает начальную
скорость газообразования, аналогично наблюдавшемуся при раз-
ложении мононитропроизводного. Однако величина предельного
давления в случае дицитробензола значительно меньше. С другой
310
стороны-, увеличение концентрации во всем изученном диапазоне
давлений приводит к возрастанию максимального значения скоро-
сти. Зависимость максимальной скорости газообразования от на-
чального давления при 395э хорошо описывается уравнением
=8,83-10“4-^зй. (21
\ dt /гаах
где скорость роста давления выражена в мм/мин, а начальное дав-
ление — в мм рт. ст.
Фиг. 2, Изменение относительной скорости газообразо-
вания со временем в процессе разложения 1,3-ди1|итри-
бензола в ларах при 395° и различном начальном дав-
лении.
N кривой ] 2
р„ мм 135 307
У» см~1 2 2
.7 •/ j
450 "40 1565
2 2 20
Увеличение поверхности стекла в 10 раз ведет в отличие от
мононитробензола к возрастанию скорости в первой половине рас
пада (кривая 5 фиг. 2). Максимум скорости в этом случае боль-
ше, а этап падения ее наступает раньше, чем в опытах без запол-
341
нения сосуда стеклянными трубками. Давление газообразных про-
дуктов в конце разложения так же, как и в случае монопитробен-
зола, при увеличении поверхности стекла становится заметно
меньше.
Константа скорости реакции первого порядка рассчитана на
начальном этапе распада динитробензола в изученной области тем
ncp.iTyp при давлении паров 550— 750 мм рт. ст. по тому же спо-
собу. что п в случае мононитробеизола (см, фиг. 4). Зависимость
константы скорости от температуры выражается уравнением.
л:
k сек.”1 (3)
Уменьшение отношения максимальной скорости роста давления
к начальной (—’Н с увеличением температуры (см. таблицу) мо-
\ М> •
жет быть объяснено тем, что скорость вторичных реакций зависит
от температуры в меньшей степени, чем скорость реакции в нача-
ле процесса. Максимальная скорость газообразования достигается
независимо от температуры опыта при давлении, составляющем
55- 60% конечного его значении.
Таблиц ;г
Ускорение газообразования прн разложении паров 1,3-дннитробензола
и 1,3,5-тринитробензола
[.-Ч-лииитробензол ! ,3,5-триг:итробелзол
Темггора- тура Начальное давление парой мм ртц ст_ Величина ускорения gMiax. wQ Темпера- тура °C У [зч. льное давление на ров ММ pTi ст. Величвна ускорения, «'так w0
;.;70 548 4,67 312 92 2,24
380 568 3.62 312 137 3,74
395 307 1,62 312 233 6,22
395 450 2,12 312 247 11,4
395 743 2,64 329 [96 2,64
410 700 2,33 353 141 1,42
Тринитробензол
Разложение ],3,5-тринитробепзола в парах изучали в области
температур 270—355" Оно характеризуется резко выраженным ус-
корением газообразования со временем (фиг, 3). Судя по интен-
сивности окрашивания стенок сосуда, общее количество вещества
бурого цвета, выделяющегося в виде налета на стекле, мало, а тем-
но-серый порошок, всегда имеющийся в продуктах распада моно-
и динитронроизводных бензола, в этом случае не был обнаружен.
342
'Количество газов в конце разложения составляет 4—4,5 моля ‘на
моль тринитробензола.
В широком диапазоне давлении не обнаруживается влияния
исходного давления паров тринптробеизола на начальную ско-
рость газообразования. Однако на этапе ускорения последнего уве-
личение исходной концентрации трннитробсцзола приводит к зна-
чительному возрастанию скорости распада. При давлении, не
слишком близком к упругости насыщенного пара трипитробедзола,
Фиг. 3. Изменение относительно!'! скорости газооб-
разования со временем для разложения тринитро-
берзола в парах при 312э и различном начальном
давлении.
N кривой 1 2 а 4 >
р0 мм Э2 [37 ЙГ) ЫГ [33
при разных температурах наблюдается прямая пропорциональности
между максимальным значением относительной скорости газообра-
зования и начальным давлением паров изучаемого вещества. Одна-
ко при приближении к давлению насыщенного пара максимальная
скорость распада тринитробензола резко увеличивается *.
В обшем случае максимальная скорость газообразования дости-
гается в изученном интервале температур при давлениях, составля-
ющих 49—55% конечного давления продуктов распада.
1 Опыты по разложению жидкого трннитробеизола, результаты которых буду,
опубликованы отдельно, показывают, что скорость распада этого вещества в жид-
ком состоянии в несколько раз больше, чем в парах.
При увеличении поверхности стекла происходит резкое увели
чение скорости распада в начальной стадии и существенно изме-
няется характер кривой р=/(т). Так при 312° (фиг. 3) увеличение
поверхности стекла в десять раз ведет к пропорциональному воз
растанпю начальной скорости распада. Дальнейшее течение по-
следнего характеризуется быстрым монотонным падением скорости
вплоть до конца разложения. Общее количество газообразных
продуктов в этом случае на 10—15% меньше по сравнению с опы-
фиг. 4. Зависимость начальной скорости распада от
температур^ для разложения паров питропроизвод-
ных бензола.
MoH0HHTpci6e!t3cia, 2—днннтробензгнт. 3~трншпробензол.
тами без наполнения сосуда стеклом. Применение кварцевых ка
пилляров и битого стекла вместо капилляров не изменяло общей
картины явления. Проведение разложения в сосуде, стенки кото-
рого покрыты налетом конденсированных продуктов распада три
нитробензола, не приводило к изменению начальной скорости га-
зообразования.
Определение температурной зависимости начальной скорости
разложения тринитробензола при s/u = I—2 сдс i (фиг. 4) дало зна-
чение константы скорости, выражаемое уравнением
5 (S»o
А=1о1ад0-/ сек-' (4)
Уменьшение с ростом температуры величины а'О|ах/ш0, значения
которой приведены в таблице (см. стр. 342), указывает, как и в
случае динитробецзола, йа более слабую, чем для начальной реак-
ции, температурную зависимость скорости вторичных реакций, опре-
деляющих распад на Этапе ускорения.
3 (4
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Термическое разложение паров моно-, ди- и тринитробензол а
наряду с различиями, обусловленными особенностями химической
структуры (числом NO2-rpynn в молекуле), имеет ряд общих черт,
присущих распаду соединений, которые содержат в молекуле связи
С—N. Для всех трех веществ энергия активации, определенная
на начальном этапе разложе-
ния, практически одинакова
(разница не превышает точно-
сти определения) и лежит в
пределах 51,9—53,5 ккал/моль
(фиг. 4). Близкое соответствие
полученной величины и изме-
ренной ранее энергии актива-
ции распада нитрометана
(53,6 ккал/моль) [2], [4], указы-
вает на идентичность первич-
ных реакций разложения этих
веществ В свою очередь энер-
гия активации распада нитро-
метана близка по величине k
энергии связи С—N, которая
по Котреллу [2], [4] составляет
52—57 ккал. Это дает основа-
ние считать, что. распад нитро-
соединений происходит именно
по связи С—N 1
Другой характерной особен-
ностью разложения нитропро-
изводных бензола является за-
висимость константы скорости
начальной реакции разложе-
ния от концентрации вещества
в объеме. Значительная при
Фнг. 5, Проверка подчинения распаду
ароматических нитросоединсний автока-
талитическому закону.
Условия опытов
Л1оионитрсбея-
зол (МНБ) ,
Диннтр< )бец-
эол (ДНБ) .
Тринитрибеп-
зил (ТцБ)
Начальное
Температура ; дапленпс
Г'С паров
•жл ртч ст,
4тЗ Ц50
395 307
3]2 230
малом давлении паров, эта зависимость постепенно уменьшается
с увеличением давления. Величина предельного давления (при ко-
тором влияние исходной концентрации на начальную скорость рас-
пада уже отсутствует), резко уменьшается с увеличением чнслг,
NO2-rpynn в молекуле. Так предельное давление для распада мо-
нонитробензола при 415° составляет около 1000, а в случае динитро-
бензола при 395° — около 300 л.и рт. ст. При разложении паров
трннитробензола минимальное начальное давление паров в опытах-
(92 мм рт. ст.) оказалось уже выше предельного. Объяснение этого
’Однако более позднее исследование распада цитроэтана и нитропропана
показало, что пиролиз этих нитросоединений идет не только через разрыв связи
С—N. Американские исследователи Гилленбранд и Килпатрик считают, что при
распаде нитрометана, по крайней мере частично, происходит также внутримоле-
кулярная перегруппировка [5].
345
явления изменением механизма мономолекулярной реакции, как
это происходит в области малых концентраций, когда скорость
процесса определяется числом соударений между молекулами, и
превращение формально подчиняется бимолекулярному закону,
кряд ли отвечает действительности. Например, для алифатических
уч'левородоров с числом углеродных атомов в молекуле !, 2 и Заяв-
ление паров вещества, при котором происходит такое изменение
механизма, значительно меньше измеренного для нитронроизвод-
нь[Х бензола ц соответственно равно 100, 10 и 1 дпч рт, ст, Против
этого объяснения говорит также тот факт, что разложение моцо-
нНтробензола при малой концентрации подчиняется уравнению
первого, а не второго порядка, как следовало бы ожидать, если бы
рассматриваемое объяснение было справедливо.
В целом термическое разложение нитросоедииений несомненно
весьма сложный процесс, включающий, вероятно, большое число
элементарных реакций 1. Поэтому па основании результатов лишь
одних кинетических исследований, проведенных манометрическим
методом, невозможно судить о конкретном механизме распада, хотя
они в известной степени и позволяют определить тип сложной
реакции. Так, распад всех трех изученных веществ на значительном
участке (до выделения газов в количестве 50%) следует автоката-
литическому закону
(« — x)-\-k?x{a — х), (5)
где а -начальное количество вещества, х — количество разложив-
шегося вещества, ~ константа скорости мономолекулярной ре-
акции и — константа скорости автокаталитической реакции.
Фиг. 5, на которой результаты опытов представлены в координа-
тор
d~. р
тах :——— , показывает, что Экспериментальные точки хорошо
Ркон^Р Рп
ложатся на прямые, как и должно быть по уравнению (5). Полу-
ченные результать! согласуются с имеющимися в литературе указа-
ниями об автокаталитической природе разложения тринитротолуола
в жидком состоянии [3], [6]. Кинетический закон автокатализа со-
блюдается, как следует из наших данных, в течение значительной
доли периода разложения и при распаде нитросоединений бензола
в газообразном состоянии.
Для разложения нитропроизводных бензола в парах характер-
но влияние поверхности. Увеличение поверхности, как показывают
полученные результаты, может оказывать двоякое влияние на
распад. С одной стороны, оно, в случае разложения ди- и тринит-
г Достаточно сказать, что на основании кинетических данных и результатов
анализа продуктов распада простейшего нитросоединения — нитрометана — Кот-
релл с сотрудниками [4], [5] допускают при разложении этого вещества по крайней
мере 5 реакций,
.346
□производных, ведет к увеличению скорости газообразования на
ачальном этапе и, с другой стороны, к уменьшению ее почти на
сем протяжении распада.
Ускоряющее действие развитой поверхности стекла на разло-
жение проявляется для различных иитросоединеиий по-разному.
!сли в случае монопитробензола такое влияние практически отсут-
твует, то для динитробецзола оно уже хорощо заметно, а в случае
ринитробензола начальная скорость увеличивается пропорццо-
ально увеличению поверхности стекла. Одной из возможных при-
ми различного влияния Стеклянной поверхности на распад трех
птросоединеннй может быть то, что, вследствие различия в их
емпературах кипения относительное давление паров po/ps 1 (отпо-
тение давления пара в опыте к давлению насыщенного пара) для
азных веществ было различным. В опытах с мононитробензолом
тносительное давление не превышало 0,05, для диивтробензола
но было около 0,1, а в случае трннптробензола 0,5 и более.
I этой связи следует отметить отсутствие влияния химической прц-
оды Стенки на картину распада тринитробепзола. Эти факты, а
акже то, что в жидком состоянии скорость разложения тринитро-
ензола больще, чем в парах2, указывают, что причиной ускоряю-
щего действия развитей поверхности стекла на разложение нитро-
осдинений бензола в парах может быть образование на стекле
жидкой пленки (состоящей из исходного вещества и продуктов его
аспада), в которой разложение протекает значительно быстрее,
ем в парах.
Уменьшение скорости газообразования- на последующих этапах
аспада под влиянием увеличения поверхности обусловлено, по-ви-
.имому, уменьшением общего количества газообразных продуктов
связано с образованием на стенке богатого углеродом налета,
'акое влияние стенки резче выражено в случае мононитробензола
несколько слабее при разложении тринитробензола. Аналогичное
лияние поверхности стекла наблюдалось также при разложении
штрометана [5], Объяснение этого явления полимеризацией на
тенке образующихся при распаде ненасыщенных углеводородов,
(энное Котреллом, правдоподобно и тем более реально для соеди-
нений бензольной структуры, в которой уже имеются ненасыщен-
ные связи.
В течение термического разложения изученных трех нитропро-
13волных бензола имеется ряд различий, обусловленных особен-
1 Отношение ра,'р. может служить характеристикой количества вещества, ад-
сорбированного на поверхности твердого тела.
а Аналогичный вывод вытекает также из результатов работы А. Робертсоны,
Научавшего разложение тротила [61. Это явление, вероятно, присуще разложению
лишь ннтросоединений, поскольку, например, в случае распада алкилннтратов на-
блюдается обратное влияние агрегатного состояния [1]. Одной из причин такого
влияния агрегатного состояния па скорость разложения ннтросоединений можно
считать образование нитрита при ассоциации органического радикала с NOS. Об-
разование нитрита, отличающегося .от 1гитросоедииения значительно меньшей
"Стойкостью, идет наиболее успешно в жидком состоянии, причем понижение тем-
пературы й повышение давления способствуют протеканию реакции ассоциации
по этому пути [7].
347
ностями их химической структуры. К ним относятся различия в на
чальной скорости распада, которая увеличивается при переходе oi
моно- к тринитропроизводному. Константа скорости реакции равн.
3,2-10-5 сек.-1 для мононитробензола при температуре 407”, дина
тробензола — при 397° и тринитробензола — при 352°. При переход,
от динитро- к тринитробензолу скорость распада увеличивается
больше, чем при переходе от моно- к динитропроизводном\
В связи с тем, что энергия активации начальной реакции распада
для всех трех веществ одинакова, изменение скорости при переходи
от моцо- к тринитросоединению происходит лишь вследствие уве
личения предэкспоненпиального множителя.
Кроме того, количество нитрогрупп в молекуле оказывает суше
ственное влияние на характер распада. Если разложение монони
тробензола протекает с уменьшением скорости во времени, п
в случае ди- и тринитропроизцодных наблюдается ускорение, коте
рое в случае трпнитробензола выражено больше. Величина уско
рения распада, характеризуемая отношением при разле
женин обоих вешеств тем больше, чем ниже температура и больше
начальная концентрация паров вещества. Влияние этих факторов
на величину ускорения выражено сильнее у тринитробензола, чем
у динитробензола. Для первого понижение температуры на 40° (oi
350° до 310°) ведет к увеличению wmAX/wo от 1,5 до 4,5, а увеличь
ние начального давления приводит к пропорциональному росте
максимальной скорости; для второго такой же перепад темпера
туры (от 410° до 37(Г) вызывает изменение от 2,3 до 4,7.
а максимальная скорость возрастает пропорционально давление
паров в степени !/з.
Таким образом начальные стадии распада трех изученные
вешеств обладают одинаковыми кинетическими характеристиками
что указывает, вероятно, на общность механизма реакций на этом
этапе. В то же время, при переходе от моно- к тринитробензолу
наблюдается существенное изменение характера последующи1'
стадий разложения. В зависимости от числа нитрогрупп в моле
куле может не только существенно изменяться соотношение скорм
стен отдельных этапов распада, но и сам механизм последнем
При увеличении числа нитрогрупп в молекуле реакционная способ
ность исходного вещества и промежуточных продуктов первой стг
дии распада (такими продуктами могут быть радикалы фенил
нитрофенил и динитрофенил) может, по-видимому, изменяться hi
одинаково. Соответственно введение очередной нитрогруппы по
разному изменяет скорость первичной н последующих реакции
Кроме того, при разложении нитропроизводных бензола определен
ную и различную для моно-, ди- и тринитробензола роль, вероятна
играют также окислительные реакции, являющиеся, по-видимомс.
одним из основных путей газообразования при распаде. В польза
этого говорит и тот факт, что при переходе от моцо- к тринитро
бензолу общее количество газов увеличивается от 1,7—2 до 4
5 молей на моль вещества.
348
ЛИТЕРАТУРА
1. Андреев К. К., Глазкова А. П., Маурина Н. Д Светлов
1ФХ, 1958, 32, 1726.
2. К о т р е л л Т. Л., Прочность химических связей. ДА., ИЛ, 1956.
3. Р о г н н с к и н С. 3. М а г и д А. М., ЖФХ, 1931 2, 262.
4. С о 11 г е 11 Т L., " ‘
, 584.
5. Cottrell Т. L,
1089.
Hillenbrand L.
6. R о b е г t s о л А,
7 G г а у Р., Y о f f е A. D., Chem. Rev.. 1955, 55, 1069.
Graham Т, Е., R е i d Т. J., Trans.
Faraday
Soc.
17,
Graham T. E., R e 1 d T. J., Trans.
I., Kilpatrick M. L., J. ch. Phys.,
Inns. E'er: d у Soc.. 1948,44,977.
Б. С„
1951.
Faraday
1953. 21,
Soc.,
1951,
525.
К. К. АНДРЕЕВ, ЛЮ-БАО-ФЕН
23, ТЕРМИЧЕСКИЙ РАСПАД ПИКРИНОВОЙ
И СТИФНИНОВОЙ КИСЛОТ
Термический распад пикриновой ц стифниновой кислот пред-
тавляет интерес в двух аспектах.
В определенных условия^ промышленного применения (взрывы
1 глубоких скважинах) возникла потребность во взрывчатых ве-
цествах, способных в течение длительного времени выдерживать
'действие относительно высоких температур. В связи с тем, что
Йитросоедцнения ароматического ряда характеризуются вообще го-
воря гораздо меньшей скоростью распада, чем ВВ многих других
классов, то естественно поиски термостойких веществ были в пер-
вую очередь начаты среди них. Для того чтобы направленно^и
обоснованно вести такие поиски, целесообразно установить влия-
ние тех или иных заместителей в ароматических полинитросоеди-
^нениях на характер и скорость термического распада. Изучение
<распада оксипроизводных тринптробензола — тринитрофенола
,(ТНФ) и тринитрорезорцина (ТНР) явилось частью исследований
(р этой области.
j Помимо этого термический распад пикриновой и стифниновой
’ для пони-
в отно-
«ислот интересен в сопоставлении с распадом их солеи
.мания резкого различия между этими солями и кислотами
•шениц скорости и способности к горению.
У солей ряда металлов способность к горению гораздо
:и скорость горения существенно выше, чем у кислот, хотя
образования солей больше, чем кислот и соответственно
-тепловой эффект их полного превращения при горении пли взрыве.
Кроме того, как показали недавние исследования [2], [4], завн-
'симость скорости горения от давления для некоторых пикратов и
‘стифнатов очень своеобразна и отличается от зависимости, уста-
'новленной для кислот.
больше
теплота
меньше
1 О распаде солей пикриновой и стифниновой кислот см. па стоящий сборник
4стр. 363.
319
Поскольку при горении одним из этапом процесса является
разложение иод влиянием разогрева, можно было надеяться, чъ
сравнительное изучение термического распада кислот и солей по-
может выяснить причины различия характеристик их горения
Этот вопрос рассматривается в следующей статье-, носвященжн
термическому распаду пикратов и стифнатов,
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследование производилось манометрическим методом в с
временном его варианте [1], Обшее представление о ходе распад;:
обеих кислот дает фиг, 1, Кривые 1 К=/(т) или, что то же p=^jh\
иа фиг, 1 и далее имеют сложный, в целом S-образный характер,
особенности которого будут рассмотрены ниже. На фиг. 2 для
фиг. 1. Сопоставление кривых изме-
рения удельного газообразования во
нремени при разложении пикриновой и
стифкниповой кислот при 2303.
(т.-Т^лдя ТНФ [0.4S . I0""1 и для THP |С,о(1Х
Х1Ь—4
фиг. 2. Сопоставление изменения ско-
рости газообразовании во времени при
разложении пикриновой кислоты и
тринитробензола при 270”.
(л г -ли ТНФ г2,г5 • 10“4 и для THE 29,4
ХЮ~* г;смг).
сравнения приведены данные для тринитробензола (ТНБ), наибо-
лее медленно разлагающегося из всех ранее изученных тринитро-
соединений. Мы видим, что пикриновая кислота разлагается го-
раздо быстрее, чем трииитробензол. Время достижения давления
газообразных продуктов распада, равного половине максималь
ного (в дальнейшем называемое — «временем полураспада»), для
нее составляет при 230° 470 мин., в то время как для тринитробен-
зола оно равно 218 000 мин. Общий объем образовавшихся газов
। Чтобы облегчить сравнение результатов Опытов при разных условиях этГ1
кривые построены и координатах -- «объем газов V в сл:3 при О” и 76U мм рт, ст.,
образовавшихся при разложении I а вещества — в дальнейшем н см3/г» — «ври
мя т в мин.». Степень заполнения реакционного сосуда здесы и в дальнейшем
характеризуется отношением навески ВВ (т) к объему сосуда (v) и выражается
350
составляет для ТНФ 410—450 н. см^/г или 4.2 -4,6 моля газов на
моль ВВ.
Стифниновая кислота разлагается с образованием несколько
большего количества газов (440—490 н см^г или 4,8—5,4 моля па
моль исходного вещества) и значительно быстрее (приблизительно
в 10 раз) пикриновой кислоты Время полураспада ТНР при 230’
составляет 45 мин. Вместе с тем скорость распада его во много
раз меньше, чем наиболее медленно разлагающегося нитроэфира—
нитроклетчатки. (Время полураспада последней при данной темпе-
ратуре по расчету путем экстраполяции ~0.3 мни., т. е. в [50 раз
меньше, чем для ТНР и в 1600 раз меньше чем для ТНФ).
Рассмотрим теперь более детально условия проведения опытов
н их результаты для каждой из кислот.
Пикриновая кислота
Основные опыты проводили при малых т/а(10-10~4 г/смл),
.позволяющих проследить весь ход распада, так как конечное дав-
ление при этом не превосходит 760 лл рт. ст. Результаты некото-
рых опытов изображены графически на фиг. 3 и 4 в координата**
фиг. 3. Изменение удельного газообразования но времени при
разложении пикриновой кислоту при различиях температурах.
(«п/г=к 2/с.и’).
«время» — «объем» и «время» — «давление» газообразных- продук-
тов распада. Ход кривых своеобразен и указывает на сложное
протекание разложения, в котором можно различить по крайней
мере 5 этапов, отличающихся по характеру зависимости скорости
газообразования от времени.
35!
Первым этапом, отчетливо наблюдающимся при низких темпе-
ратурах и больших m/v, является индукционный период, на протя-
жении которого газоваделения практически не наблюдается. В ка-
честве примера на фиг. 4 приведены начальные участки нескольких
кривых р— /(т), полученных при 183° и различных m/v. В этих ус-
ловиях индукционный период может составлять десятки минут
(участок ОВ на фиг, 4), Если поднять температуру опыта на 50°
или более, то индукционного периода це обнаруживается (фиг, 3).
«Риг. 4. Начальный участок кривых
р=^/(т) при разложении пикриновой
кислоты при 183°.
(Числа у криных -miv в гея’ - 10*).
Фиг* 5. Проверка подчинения
распада пикриновой кислоты на
третьем этапе закону реакции
первого порядка.
Услоиия опыта: га/Т1=1т |0—г
г' = |£3\ И 0 --конечный объем газо-
образных продуктов разложения; У^
нх объем в момент времени т* (Иехо.т,-
н?гн крист приведена на фиг* 6),
По-видимому, он уменьшается настолько, что становится близок
к времени прогрева прибора.
Следующий этап — ускорение газообразования (участок ВС на
фиг. 4), которое при 183° длится около 50 мин. На втором этапе,
по-видимому, происходит развитие той реакции, которая идет
в индукционном периоде, отличаясь от него тем, что величина
давления и скорость нарастания последнего возрастают настолько,
что становятся доступными измерению применявшимся мето-
дом.
Третьим этапом (участок CD на фиг. 4) является быстрая реак-
ция (средняя скорость газовыделення, например, при 183° состав-
ляет 0,03 н сл!3/а в мин,). Скорость этой реакции непрерывно па-
дает во времени по закону, близкому к закону протекания реакций
первого порядка (фиг, 5).
Третий этап отчетливо замете!! и при более высоких температу-
рах — вплоть до 250э. По-видимому, температурная зависимость
скорости распада на этом этапе относительно слаба. Образующие-
ся газы в основном (около 80%) не конденсируются при комнат-
ной температуре; температура плавления пикриновои кислоты
352
к концу третьего этапа лишь немного понижается (по данным
опыта при 183° на 0,2°).
При 183° и малых m/v скорость газообразования и количество
образующихся газов на третьем этапе больше, чем при больших
m/v (фиг. 6).
Фиг. 6. Влияние m/v на удельное газообразование при
распаде пикриновой кислоты прн 183°. (Числа у кри-
вых— m/v в г/см3 104).
Такая зависимость от m/v позволяла предположить, что газо-
(или давление) -тормозят развитие
предположению противоречат од-
образные продукты распада
данного этапа процесса. Этому
нако результаты опыта с пе-
рерывом, когда после прохож-
дения третьего этапа газы бы-
ли откачаны и опыт продол-
жен, Повторения процесса не
последовало, а сразу начался
следующий этап, но шел он с
большей, чем обычно, ско-
ростью (фиг. 7).
Другим возможным предпо-
ложением было рассмотрение
первых трех этапов процесса,
как последовательных стадий
развития качественно одних и
тех же реакций при количест-
венном различии их скоростей
на разных стадиях. Развитие
фиг. 7. Влияние эвакуации продуктов
распада после начального подъема дав-
ления на последующее газообразование
при т/ояаЮ- 10-’ г/см? и 250°.
эаакуацни, 2— после эвакуации и
о—параллельный опыт без эвакуации.
реакции начинается сразу, как
только вещество прогрелось, но идет вначале столь медленно, что
не улавливается ртутным манометром (первый этап), затем сле-
дуют периоды регистрируемого ускорения газообразования (второй
этап) и падения скорости (третий этап).
23 451
353
Такая картина была бы естественной в случае развития авто-
каталитической или коисекутивных реакций. Можно было бы
предположить также, что на первых трех этапах мы имеем дело
с разложением малостойкой примеси, содержащейся в пикриновой
кислоте; однако это предположение пришлось бы дополнить допу-
щением, что примесь разлагается с самоускорением. Далее в этом
случае удельное количество образующихся газов не должно было
бы зависеть от т/и, а фактически газообразование, как мы видели,
существенно уменьшается при увеличении т/и. Наконец, дополни-
Фиг. 8, Влияние m/v на удель-
ное газообразование при распа-
де пикриновой кислоты при 25(У\
(Числа у кривых — m/и п
г/см3 104),
Фиг, 9, Влияние эвакуации продук-
тов распада на четвертом Этапе
ira удельное газообразование прн
разложении ТНФ 10-3г/см3.
Г=230’).
J —до эвакуации, 2— после эвакуации
(для удобства сравнения кривая по-
строена так же, как продолжение кри-
вой /)„ 3—параллельный опыт без эва-
куации.
тельная очистка ТНФ не изменила характера кривых, на которых
по-прежнему наблюдались начальные этапы распада.
Четвертый этап характеризуется значительно (иЗ—9 раз) мень-
шей скоростью газообразования, чем максимальная скорость на
третьем этапе, причем на четвертом этапе скорость не падает как
на третьем, но слабо растет во времени, 'Рост этот выражен тем
сильнее, чем ниже температура. Так при 230° максимальная ско-
рость газообразования превосходит начальную для четвертого
этапа в 1,9 раза, а при 260° только на 6% (в обоих случаях
mjv — 10 • 10-4 г/сл13) 1.
Скорость четвертого этапа существенно зависит также от m/v,
возрастая С его увеличением или, что то же, с ростом давления га-
зообразных продуктов распада, как при 183°, так и при 250° (фиг. 6
г Сравнение производится для скоростей удельного газообразования, отнесен-
ных к исходному количеству вещества. Если отнести их к количеству неразло-
жившегося в данный момент вещества, то они будут меняться, естественно,
сильнее.
35+
и 8), При 183° увеличение щ/о в 10 раз привело к увеличению ско-
рости газообразования на 4-м этапе вдвое.
Чтобы проверить естественное предположение, что большая
скорость и большее ускорение на четвертом этапе прн увеличенных
m/v обусловлены большим давлением (концентрацией) газообраз-
ных продуктов распада были поставлены (при 230°) опыты по от-
качиванию этих продуктов на четвертом этапе вблизи максимума
скорости. Это однако почти не отразилось на величине скорости
при продолжении опыта (фиг, 9), но дальнейшего роста ее, наблю-
давшегося в обычных условиях, почти не происходило и в целом на
протяжении 400 мин. средняя скорость газообразования после от-
качки газов была в I1/» раза меньше, чем без нее. Такое же влия'
ние оказывает замена газообразных продуктов распада азотом.
Это говорит о том, что па процесс распада влияет не давление как
таковое, а химическая природа газа, производящего это давление
Чтобы выяснить, как влияют конденсированные продукты1 рас-
пада на его течение к пикриновой кислоте, разложившейся на 25%
(по объему газообразных продуктов), была добавлена свежая
в том же количестве, какое было взято для первого опыта и разло-
жение продолжено.
Начальный подъем давления (третий этап) был выражен го-
раздо слабее, в остальном же разложение шло очень сходно с рас-
падом свежей кислоты при том же m/v, Это говорит не в пользу
гипотезы о катализе процесса конденсированными продуктами рас-
пада как причины ускорения последнего; следует однако отметить,
что ускорение в целом выражено относительно слабо и поэтому
количественный учет влияния различных факторов и в частности
роли конденсированных продуктов на процесс распада — затруд-
нен.
Добавим, что большее ускорение реакции при низких темпера-
турах, обычное для автокаталитических процессов, говорит за то,
что на четвертом этапе мы имеем дело с реакциями, имеющими
различную температурную зависимость — при этом скорость уско-
ряющейся реакции в меньшей степени зависит от температуры,
чем реакции, протекающей без ускорения.
Так как характер кривой «давление—время» меняется с тем-
пературой, то полного совмещения кривых р=/(т) путем измене-
ния масштаба времени получить не удается. Поэтому определение
температурного коэффициента скорости было произведено сопо-
ставлением средних скоростей при разных температурах на участ-
ках сходного типа (четвертый этап). Эти скорости в аррениусов-
ских координатах дают прямую (фиг. 10).
Наконец как пятый этап можно рассматривать период падения
абсолютной скорости газообразования, подобный тому, какой на-
блюдается для любой реакции в результате уменьшения концент-
рации реагентов. В пользу такого толкования говорит и попытка
представить последний участок кривой в обычных координатах
реакции первого порядка (фиг. 11). Мы видим, что на некотором,
вообще говоря небольшом, участке уравнение реакции первого
23*
355
порядка выполняется. При меньших временах константа скорости
падает, как этого и следовало ожидать. Таким образом, по суще-
ству может быть следовало бы четвертый и пятый этапы объеди-
нить и рассматривать их как два периода одного и того же про-
цесса, включающего реакцию, скорость газообразования которой
пропорциональна наличному количеству исходного вещества, и ав-
токаталитическую реакцию, скорость которой пропорциональна
концентрации продуктов распада в простейшем случае в первой
степени, Фиг. 12 показывает, что при построении графика в коорди-
натах, соответствующих закону автокаталитической реакции, па
Фиг. Ю. Температурная зависи-
мость средней скорости газооб-
разования при распаде пикри-
новой кислоты (четвертый
этап).
Фиг. 11. Проверка подчинения распада
пикриновой кислоты на конечном этапе
закону реакции первого порядка
(m/u-slO-3 г/с.м3, Г=230“) а—степень
распада = Ут/УКОп.
участке, где а меняется от 17 до 46%, линия скорости представ-
ляет собой прямую.
Выше течение распада ТНФ было иллюстрировано главным
образом графиками изменения удельного объема образовавшихся
газов во времени. Сложный характер распада еще более наглядно
виден на графиках в координатах; «скорость газообразования» —
«время» (фиг. 13). В частности на кривой видно наличие двух
максимумов скорости — одного соответствующего концу второго
этапа, и другого — наблюдающегося на четвертом этапе; при
этом первый максимум значительно больше второго; при
mjv — 10.45 • 10~4 г/см3 и 230° отношение скоростей на максимумах
равно 6,86.
Помимо описанных выше опытов по распаду жидкой пикрино-
вой кислоты были проведены эксперименты по разложению ее в па-
рах. Скорость распада ТНФ в парах при 290° во много раз меньше,
чем в жидком состоянии. Она зависит от начального давления па-
ров, возрастая с его увеличением сначала слабо, а затем гораздо
сильнее (фиг. 14). Одновременно меняется и характер кривой
— — = /(-),описывающей изменение скорости газообразования со
Лт />0
временем (фиг, 15). При малом давлении паров (30 мм рт. ст.)
кривая имеет слабо выраженный максимум и наступает он поздно.
С повышением давления максимум становится больше и наступает
Фиг. 12. Про перка г год1 ги цен ил распада
пикриновой кислоты автокаталитическо-
му закону (т/у«!0 3 г/с.чр).
и—степень рлгл.иа -!•'
газообразования во времени при
распаде пикриновой кислоты
(m/osilCH г/см3, Г—230°, римские
цифры у кривой - этапы распада).
раньше. При начальном давлении ТНФ 110 мм рт. ст. скорость
наибольшая в начальный момент и быстро надает со временем по
закону, близкому к закону реакции первого порядка.
Существенным осложняющим фактором является сильное влия-
ние поверхности стекла на реак-
цию распада в парах Увеличе-
ние поверхности в 10 раз путем
введения в реакционный сосуд
стеклянных капилляров увеличи-
вает начальную скорость реакции
больше, чем в 100 раз (фиг. 16).
Очевидно, что влияние гетеро-
генной реакции должно сущест-
венно сказываться и в опытах
разложения паров без искус-
ственного увеличения поверхно-
сти, особенно, если давление их
находится не далеко от насыще-
Фиг. 14, Рост давления во времени
при распаде паров пикриновой кисло-
ты при 29CF,
(Числа у кривых — начальной да я?) е пне
паров ТНФ о лм рт. ст-)э
ния; это в частности может быть
причиной сильного ускорения
реакции при р=110 по сравне-
нию с р = 87 мм рт. ст. (фиг. 15);
ускорение по этой причине воз-
можно и в опытах по распаду жидкости при малых т/и, Действи-
тельно опыты при 230° и т/с =10- 10~4 г/см3 показали, что в сфе-
357
рическом реакционном сосуде диаметром 6 мм (s/o = 6,l дм'1) ско-
рость на максимуме значительно (в 1,3 раза) больше, чем прц
диаметре сосуда 40 ,п,и (s/v — 1,5 сл1_[).
Фиг. 16. Влияние заполнения сосуда сте-
клянными капиллярами на распад пик-
риновой кислоты в парах при 290'5.
(miv 2 10-4 г/с.н3, ро=30 Л1Л1 рт. ст., чис-
ла у кривых—величина отношения в си-1
суммарной поверхности стекла з к объ-
ему сосуда о).
Фиг. 15. Изменение относительной ско-
рости газообразовании во времени
при распаде паров пикриновой кисло-
ты прн 290’.
(Числа у кривых — начальное давление
паров ТНФ в мм рт. ст.).
Интересно,, что при разложении в парах в сосуде с увеличенной
поверхностью образуется несколько больше газов, чем без добавки
стеклянных капилляров, но затем давление их медленно падает,
очевидно в результате последующей реакции, проходящей с обра-
зованием конденсированных продуктов.
Стифнмновая кислота
Известной трудностью при изучении распада стнфниновой кис-
лоты = 177,5—178°) является то, что она относительно прочно
удерживает воду. Откачивание при комнатной температуре и при
100’ в течение двух часов, по-видимому, оказывается недостаточ-
ным, чтобы се удалить. Лишь эвакуация при 180°, когда стифнино-
вая кислота уже находится в жидком состоянии, в течение 5 мин.
исключала быстрый подъем давления, наблюдавшийся при исполь-
зовании приведенных выше режимов откачивания, Эта эвакуация
меняет н характер начального подъема давления — из насыщаю-
щегося он становится ускоряющимся. По-видимому, присутствие
воды ускоряет первичные реакции, которые без нее развиваются
1 Аналогичное явление наблюдается и для других изученных иитросоедннений
(см. настоящий сборник стр. 316).
358
медленнее. Речь при этом идет о весьма малых количествах воды,
потому что уже через 10 мин. общее количество газов в опытах
с откачкой при 100 и при 180° становится почти одинаковым. Ил-
люстрирует это фиг. 17. Некоторое влияние на ход кривых мог
иметь прогрев (время быстрого роста давления составляет всего
около 8 мин).
Фиг. 17. Влияние условий эвакуации на распад
стифниновой кислоты при различных темпера-
турах (т/о«10-10-4 г/см?}. Температура, при
которой производилась эвакуация реакционного
сосуда:
J —[80°^ 2--100° с жидким азотом, 3 — }80°ц 4—WCP,
5—ЮСР с ЖИДКИМ азотом, 6—18СП.
Общая картина распада стифниновой кислоты, изображенная
на фиг. 18, довольно отчетлива и аналогична наблюдавшейся для
пикриновой кислоты. Она включает небольшое начальное ускоре-
ние газообразования (первый этап), разложение со значительной,
но быстро уменьшающейся скоростью (второй этап), участок
почти постоянной слабо растущей скорости (третий этап) и, нако-
нец. ее падение (четвертый этап).
Первый этап обнаруживается лишь в опытах, проведенных при
наиболее низких из изученных температур: 180, 190 н 200°, В неко-
торой мере это относится и ко второму этапу, длительность кото-
рого составляла при этих температурах около 10 мин,, а количество
образовавшихся газов 20—35 « с.п1 * 3/а или 0,22—0,35 молей на моль
стифниновой кислоты *. При этом образуются в основном газы, нс
1 Максимальное количество газов, которое может образовать стифпиновая
кислота, составляет по гипотетическому уравнению
CsH(OH})(NOs)3-6CO . I'/sNsH-l'/aibO-r’/A'
S’/i молей на моль или fi46 см3/г.
конденсирующиеся при О3. Был проведен также опыт по разложе-
нию твердой стифниновой кислоты при 150°; давление быстро под-
нялось до 46 мм рт. ст., после чего в течение 72 час. оно оставалось
постоянным. Это обстоятельство указывает на то, что скорость
н см3
г
500
Фиг. 18. Распад стифниновой кислоты при различ-
ных температурах (т/и^10—3 а/си3).
разложения твердой стифниновой кислоты значительно меньше,
чем ее расплава.
Второй этап, т. е. участок быстро падающей скорости заметен
на графиках опытов при 220 и 230° (фиг. 18). При 240 и 250"
давление поднимается слишком быстро, чтобы участок падения
скорости можно было обнаружить и отделить от разогрева.
Фиг. 19. Влияние m/у на распад стифниновой
кислоты при 226°.
(Числа у кривых —вел1||;11Нл т-г' и . (0*).
Два опыта при 2203 были проведены при m/v. различающихся
в 4 раза (фиг. 19). При большем m/v газообразование на втором
этапе было немного больше. По-видимому1 оно несколько растет
также с увеличением температуры, как это показывает сопостав-
ление опытов при 190 и 200° (фиг. 17).
360
Фиг. 20. Температурная зави-
симость средней скорости газо-
образования при распаде стиф-
нииовой кислоты (третий этап).
Третий этап — слабо ускоряющееся газообразование—охваты-
вает основную часть распада. Скорость газообразования здесь го-
раздо меньше (в 6—8 раз), чем па предыдущем этапе. Мал и рост
скорости; при 200° отношение максимальной и минимальной ско-
ростей составляет 1,56, при 230°—1,48,
при 240^—1,42 и при 250°—1,20. Ско-
рость газообразования на этом этапе
не зависит от m/v (фиг. 19); опыты
довольно хорошо воспроизводятся.
Если ход реакции на третьем этапе
выразить автокаталитическим уравне-
нием, то соотношение констант авто-
каталитической
реакций для стифниновой
оказывается вдвое больше,
пикриновой,
мость скорости реакции при
ТНР была определена сопоставлением
средних скоростей на участке, где
степень превращения меняется от 22
до 45% (фиг. 20). В координатах
Аррениуса получается прямая. Ско-
рость распада стифниновой кислоты при
и моиомолекулярнои
кислоты
чем для
Температурная зависи-
распаде
230 и 250э приблизительно
в 10 раз больше, чем пикриновЪй и различие это мало меняется
с температурой,
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Пикриновая кислота разлагается гораздо быстрее тринитробен-
зола, при 270°, например, почти в 200 раз. Однако это обстоятель-
ство не является ее исключительной особенностью. Все заместите-
ли увеличивают скорость распада трицитробензола. В частности
скорость распада метилтринитробензола — тротила того же по-
рядка, что и окситринитробензола — пикриновой кислоты. Глав-
ным отличием распада пикриновой кислоты от тринитробензола к
других его замещенных и большинства ВВ вообще является очень
слабое по сравнению с ними ускорение распада на основном уча-
стке, а в начале распада даже замедляющееся его течение. Та же
особенность присуща стифниновой кислоте, у которой она еще бо-
лее выражена. Согласно исследованиям Ю. Я- Максимова она ха
рактерна также для гексанитродифениламина и гексанитрооксани-
лида, т. е. для всех изученных нитросоединений ароматического
ряда, содержащих подвижный атом водорода, придающий соеди-
нению кислотные свойства.
Другой особенностью распада обеих кислот является отчетливо
выраженный его стадийный характер. При этом первая стадия,
включающая индукционный период, рост скорости и ее падение
протекает относительно быстро и поэтому очень заметна.
361
Влияние степени заполнения сосуда, или иначе, давления газо-
образных продуктов распада, проявляется относительно слабо,
особенно для стифниновой кислоты. Этим подтверждается в част-
ности ранее высказывавшееся мнение о характерном отличии нит-
росоединений ароматического ряда от нитроэфиров, заключающем-
ся в том, что для них ускорение разложения относительно невели-
ко, а также что оно наступает сравнительно поздно. В парах пи-
криновая кислота разлагается значительно медленнее, чем в жид-
ком состоянии. Это, кстати, способствует более резкому проявле-
нию верхнего температурного предела вспышки, который был
установлен для этого вещества.
Реакция в парах, подобно тому как это наблюдается для мно-
гих других нитросоединений, сильно ускоряется поверхностью
стекла. Это обстоятельство существенно осложняет количествен-
ную интерпретацию результатов, получаемых в стеклянных сосу-
дах, а также перенос сделанных выводов па разложение
в сосудах из других материалов.
Зависимость скорости распада пикриновой кислоты от темпера-
туры невелика; средний температурный коэффициент в интервале
183—250* 2 э составляет (для основного—третьего этапа распада)
1,12; это соответствует энергии активации £ = 38,6 ккал/моль и
1g В = 11,6. Для стифниновой кислоты соответственно £ —
= 34,6 ккал/моль и 1g 5= 11,2. Найденные нами значения Е и В для
пикриновой кислоты сильно расходятся с приводимыми в литера-
туре [3] (£ = 58 000 кал/моль и lg 5=22,5). Возможной причиной
этого является то, что при расчете констант ранее не была учтена
сложность кинетических кривых, вследствие чего сравнивали ско-
рости реакции иа участках, соответствующих разным этапам рас-
пада, например, индукционный период при низкой температуре в
период быстрого роста давления — при повышенной. В пользу
этого предположения говорит и то, что константы скорости, рас-
считанные по литературным данным [5] и полученные нами раз-
личаются гораздо меньше, чем их зависимость от температуры. !
Видно также, что предэкспоненциальный множитель не только не
превосходит обычной для мономолекулярных реакций2 величины,
но даже несколько ниже ее,
Сопоставление с данными, полученными Ю. Я. Максимовым
для тринитробензола 3, показывает, что отмеченное выше различие
скоростей распада этого ВВ и изученных нами кислот в основном
определяется значительной разницей ц энергиях активации. Для
! Для четвертого этапа по фарфору скорость газообразования составляет
(1,0070 С/.Д/е мин, по нашим данным 0,0048 c.irye мин. Аналогично для третьего
этапа Фармер дает 0,033 см^/г-мин, паши опыты —0,041 мин. Если учесть
к тому же, что m'u могли быть различны (Фармер нс указывает значения этой
характеристики), то согласие следует признать удовлетворительным,
2 Прямых опытов для подтверждения того, что начальный этап жидкофаз-
ного распада явлнетея реакцией первого порядка, мы tie проводили; пет однако
оснований ожидать, что пикриновая и стнфниновая кислоты являются в этом от-
ношении исключением.
3 См. настоящий сборник, стр, 338,
362
тринитробензола по Максимову £ = 44,0 ккал!моль, 1g В=11,2.
Если, что вероятно, начальным этапом распада тринитросоедине-
ния является отрыв от молекулы одной нитрогруппы, то наличие
заместителя в бензольном кольце существенно облегчает этот от-
рыв подобно тому, как оно облегчает и вступление витротруппы
в это кольцо при нитровании.
выводы
Изучен при помощи стеклянного манометра распад пикриновой
и стифниновой кислот в интервале температур 183—270°.
Распад этот протекает сложным образом и включает ряд ста-
дий, различающихся характером зависимости скорости от време-
ни; в целом он протекает без значительного ускорения и в этом
отношении отличается от распада тринитробензола, тринитротолу-
ола и многих других нитросоединений ароматических углеводоро-
дов.
Определены кинетические характеристики для основной стадии
распада; для пикриновой кислоты Е = 38,6 ккал/моль, В = 10!1-6, пе-
риод полураспада при 230°—470 мин.; для стифниновой кислоты
Е = 34,6 ккал/моль, В=10[1'2, период полураспада при 230° —
45 мин. В парах пикриновая кислота разлагается значительно мед-
леннее, чем в жидкой фазе, причем наряду с гомогенной происхо-
дит со значительной скоростью гетерогенная реакция на поверх-
ности стекла.
ЛИТЕРАТУРА
i. А и д р е е в К. К., Глазкова А, П., Мау рина Н. Д. и Светлов Б. С.,
ЖФХ, 1958, 32, 1726,
2. Беляев А, ф. и Беляева А. Е., ДАН СССР, 1946, 52, 507; Беля-
ев А. ф. иКовдрашков Ю. А., ДАН СССР, 1960, 131, 364,
3. РогинскийС. 3., ЖФХ, 1932, 3,419.
4. Светлов Б, С. и Фогельзанг А, Е., ДАН СССР, 1961, 137, 654,
5, F а г m е г R. С., J chem, Soc., 1920, 117, 1432.
К. К. АНДРЕЕВ. ЛЮ-БАО-ФЕН
24. ТЕРМИЧЕСКИЙ РАСПАД АММОНИЕВЫХ,
КАЛИЕВЫХ И СВИНЦОВЫХ СОЛЕИ
ПИКРИНОВОЙ И СТИФНИНОВОЙ КИСЛОТ
В отличие от кислот аммониевые, калиевые и свинцовые соли
тринитрофепола и тринитрорезорцина не плавятся и поэтому раз-
лагаются при повышенной температуре, оставаясь в твердом со-
стоянии. Кроме того, они нелетучи и поэтому химическая реакция
разложения, по крайней мере на первых ее стадиях, протекает
в твердой фазе. Известное исключение представляют пикрат и
363
стифнат аммония, которые подобно другим аммониевым солям за-
метно диссоциируют при повышенных температурах, в результате
чего становится возможным разложение паров кислот и взаимо
действие их с аммиаком в газовой фазе.
Как известно, соли пикриновой и стифниновой кислот (опять-
таки за исключением аммониевых солей) резко отличаются по ско-
рости и закономерностям горения от самих кислот. Одной из задач
исследования термического распада солей было попытаться обна-
ружить такие его особенности, которые позволили бы объяснить
указанные отличия в отношении горения.
Исследование термического распада вели манометрическим ме-
тодом с применением стеклянного манометра типа Бурдона [1].
В статье приняты следующие условные обозначения:
т —время в мин,;
V— объем газов, образовавшихся при разложении 1 г вещества,
приведенный к нормальным условиям ^*см j;
„ н сл3
те — скорость газообразования в -——- ;
г-мин
mjv — степень заполнения реакционного сосуда (т —навеска веще-
ства в г, v —объем сосуда в см3) в г]см3;
р~давление газообразных продуктов распада вещества в мм
рт. ст.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
1. Разложение аммониевых солей пикриновой
и стифниновой кислот
Пикрат аммония
Пикрат аммония очищали однократной перекристаллизацией из горячей воды.
Он не плавится при нагревании, по крайней мере, в интервале температур
200—270°. в котором вели опыты.
Стенки реакционного сосуда в конце опыта оказываются покрытыми корич-
невым налетом, интенсивность которого увеличивается но направлению ко дну
сосуда. Помимо зтого, на дне сосуда остается черный рыхлый порошок.
Конечный объем газообразных продуктов составляет 5,6—5,8 молей на моль
пикрата (510—530 си3/г), т, е. несколько болыпе, чей для пикриновой кислоты (см.
стр, 351 настоящего сборника).
Общий характер кривых V=f(r) или, что то же, р=/(т) при
разных температурах (200—270°) представлен на фиг. 1. Мы ви-
дим, что как ц для пикриновой кислоты, эти кривые имеют S-об-
разный характер; в случае пикрата однако эта 3-образность выра-
жена сильнее: ускорение газообразования на первом участке кри-
вой—до точки перегиба — значительно больше. Об этом можно
судить по данным табл. 1, где для обоих веществ приведены отно-
шения максимальной скорости газообразования к начальному ее
значению.
364
Таблица 1
Отношение максимальной скорости газообразования к начальной при разложении
пикрата аммония и пикриновой кислоты при разных температурах
(т/и= (Ю-т-12)' 1(Н г/сч3)
Температура °C Пикрат аммония I ;икрнновая кислота
Я’т ах/к'нач а,иаах/^‘нач
230 17,5 1,9
250 11,7 1,2
260 10,4 1,1
270 5,5 1.0
Еше нагляднее указанное различие видно на фигурах 2 и 3, где
для двух температур (230 и 270°) сопоставлены кривые ^=f(r)
для соли и кислоты.
Фиг. I. Распад пикрата аммония при различных температурах
(тД'иЮ-3 е/сл3).
На начальных этапах распада скорости разложения пикрата
аммония меньше, чем пикриновой кислоты, но растет она во вре-
мени значительно быстрее. При низких температурах 230° кривая
V—f(r) пикрата почти на всем своем протяжении идет ниже кри-
вой для кислоты и пересекает последнюю лишь при V, близком
к 400 н см3!г. При повышении температуры начальная скорость
распада пикрата растет сильнее, чем для кислоты и, хотя ускоре-
ние распада во времени для него с ростом температуры умень-
шается, пересечение кривых V=f(r) происходит раньше: при 250°,
когда и при 270°, когда р=[/4Ркон-
Другой особенностью манометрических кривых разложения пи-
крата аммония является отчетливо выраженный двустаднйный его
характер, выражающийся в данном случае в том, что после дости-
365
жения степени разложения около 50% скорость газообразования
сразу сильно (в 2—5 раз) уменьшается и рост ее резко переходит
в медленное падение. Эта перемена закона изменения скорости га-
н см3
Фиг. 2. Сопоставление кривых газообразования при распаде
пикриновой кислоты (ТНФ) и пикрата аммония при 230°
(т/ия=10-э г/с№).
зообразования отчетливо видна даже на интегральных графиках
особенно при 230—260°; еще нагляднее она выявляется на диффе-
Фиг 3. Сопоставление кривых газо-
образования при распаде пикриновой
кислоты (ТНФ) и пикрата аммония
при 27СР (m/v-lQ-3 a/c.ti3).
Фиг, 4. Изменение относительной ско-
рости газообразования во времени
при распаде пикрата аммония
(т/ояаЮ-3 г/сл«э).
ренциальных кривых, если по оси ординат откладывать не абсо-
лютную, а относительную скорость, т- е, отношение скорости реак-
ции к количеству непрореагировавщего вещества (фиг. 4); в этом
366
случае видно, что происходит не только падение скорости, но и по-
следующий рост ее относительного значения, т. е. что кривая
т’отн = Лт) имеет два максимума, причем первый из них значитель-
но больше.
Зависимость скорости распада от температуры была определе-
на построением аррениусовского графика для начальной скорости
в интервале 200—270°, Точки удовлетворительно укладываются на
прямую (фиг. 5, прямая /), по тангенсу угла наклона которой
энергия активации Е получается равной 57,2 ккал/моль; так как
абсолютная величина скорости газообразования относительно ве-
Фиг. 5. Температурная зависимость скоро-
сти газообразования при распаде пикрата
аммония.
/—но начальным скоростям газообразования,
2—по средней относительной скорости гаэообра*
эоп;п1ия, 3—но скорости газообразования а из'
Чале второй стадии, 4—по значениям константы
скорости разложения,
лика (&2ооз =2,9 • 10 асек._|), то соответственно предэкспоненцналь-
ный множитель В получается большим (~10!Э).
Сильная зависимость начальной скорости распада пикрата
аммония от температуры, значительно большая, чем для пикрино-
вой кислоты, и приводит к тому, что при высоких температурах он
разлагается в среднем быстрее, чем пикриновая кислота, а при
низких медленнее.
Рост начальной скорости в данном случае оказывается более
: существенным, чем уменьшение ускорения при повышении темпе-
ратуры. В соответствии с этим находятся и результаты определе-
I ния температурной зависимости средней скорости газообразования
(на участке изменения степени распада от 3-411 до 45%), давше-
го £ = 43 ккал/моль ^фиг. 5, прямая 2). То, что температурная за-
висимость средней скорости оказывается меньше, чем для на-
чальной, естественно объясняется большим ускорением распада
1 при низких температурах.
Как указывалось, разложение пикрата аммония протекает
двухстадийно. Скорость на второй стадии естественно также воз-
растает с температурой, однако слабее, чем на первой. Поэтому от-
367
ношение максимальной скорости распада на первой стадии к на-
чальной скорости на второй при повышении температуры увеличи-
вается (см. табл. 2).
Таблица 2
Скорости газообразования на различных стадиях распада пикрата аммония
(т/о=(10-Н2) 1(Н г/с.мэ)
Температура °C (^Ггпат) 1 н см^г-мин н см^/гмин (^гпах) [ (»нач>!
200 0,069 — —
230 0,98 0,54 I.8
250 4,2 1,7 2.5
260 9,1 3,1 2,9
270 21,7 4,8 4,5
Если начальные скорости второй стадии при разных темпе-
ратурах нанести на график в аррениусовских координатах, то
также получается прямая, которая дает Е = 32,2 ккал/моль (фиг, 5
прямая <?).
Изменение скорости распада со временем. Рост скорости газо-
образования во времени значительно сильнее при низких темпера-
Фиг. 6. Проверка подчинения первой
турах, чем при высоких, поэтому
изменением .масштаба времени
не удается совместить кривые
V=f(T).
При построении зависимости
а»=/(т) в логарифмических коор-
динатах прямой не получается.
Построение графика на основе
уравнения автокаталитической
реакции
— = (а. — х)~ ^хп (-Z -х)
d~.
стадии распада пикрата аммония при
20СР автокаталитическому закону. позволяет ОПИСЭТЬ значительную
часть первой стадии распада при
200—250°. но при п> 1 и различном для разных температур (л = 5
для 200, 230, 250° и zi = 3 для 270°). На фиг. 6 представлен соот-
ветствующий график, характеризующий первую стадию распада
пикрата аммония при 200°.
Хорошее приближение к прямой лает построение эксперимен-
тальных данных в координатах Ig(V— 1%) =/(т), где Ео близко
к тому объему, который соответствует начальному давлению, воз-
никающему при разогреве пикрата аммония до температуры опы-
та. В интервале, где образуется от 34-13 до 40% газообразных про-
368
дуктов (т. е. от 14<-70 до 210 нсмг/г) получается прямая (фиг. 7).
Расчет зависимости тангенса угла наклона прямых от температуры
приводит к £' = 40,5 ккал/моль (фиг- 5, прямая 4), не далекому от
значения, полученного для средних скоростей.
Влияние степени заполнения сосуда (wjv) на течение распада.
При 250 и 270° величина m/v относительно слабо влияет на ход
распада; увеличение m/v уменьшает начальную скорость газообра-
Фиг, 7, Спрямление кривой
I7 = f(r) разложения пикрата
аммония при 25(Т (M/W
- 10~э е/см\ 1-'о принято ран-
ным 21 н см3/г).
Фиг. 8. Влияние m/v на газообразова-
ние прн распаде пикрата аммония при
27СР. (Числа у кривых — m/v в
е/см3 10*).
зования, но рост скорости идет быстрее и максимум ее больше;
увеличение m/v повышает также скорость газообразования на вто-
рой стадии распада (см. фиг. 8).
Стифнат аммония
Стифнат аммония был получен из горячего (80°) насыщенного раствора стиф-
ниновой кислоты ((пя 178?) нейтрализацией избытком 25%-ного раствора аммиака.
При охлаждении выпадали оранжевые кристаллы стифната аммония. Продукт очи-
щали перекристаллизацией из горячей воды.
Конечный объем газообразных продуктов распада составляет 6,2—6,7 мо-
лей на моль стифната (500—540 нсл^/г).
По внешней картине и общему характеру разложения стифнат
аммония сходен с пикратом (фиг. 9).
На первой стадии распада наблюдается ускорение газообразо-
вания (участок АВ на фиг. 9); по достижении давления, состав-
ляющего около 40% от конечного, скорость газообразования резко
(в 5—6 раз) падает, но затем начинает вновь расти (участок ВС).
Таким образом, в отличие от пикрата аммония рост скорости рас-
пада на второй стадии выражен сильнее — увеличивается не толь-
ко относительная, но и абсолютная скорость. Этот общий характер
кривой V=f(r) или, что то же р=/(т) наблюдается во всем изучен-
ном интервале температур, но при самой низкой из них (190°)
роста абсолютной скорости на второй стадии уже нет — опа дли-
тельное время остается постоянной.
24 451
369
При 190 и 200° кривая p=f(x) для стифната аммония на всем
ее протяжении лежит ниже соответствующей кривой для стифни-
новой кислоты (фиг, 10), С повышением температуры различие
в скоростях распада стифната аммония и стифниновой кислоты
уменьшается и при 23СГ средние скорости разложения их уже ста-
новятся близкими (фиг. 11),
Наличие двух максимумов скорости распада более отчетливо
видно па дифференциальной кривой (фиг. 12); с понижением тем-
Фиг, 10. Сопоставление кривнх газообразо-
вании при распаде стифниновой кис,юты
(ТНР) и стифната аммония ITHPA) при
19(Р (mjv —10—3 г'с.«®).
пературы относительная ве-
личина второго .максимума
становится меньше и при
190° он исчезает.
Зависимость скорости
распада от температуры.
Совмещение кривых V -
=f (т) на стадии первого
ускорения не дает удовлет-
ворительных результатов;
как и для пикрата аммония
при низких температурах
скорость газообразования
растет со временем сильнее,
чем при высоких. Построе-
ние начальных скоростей в
аррениусовских координатах
дает прямую линию, по
тангенсу угла наклона которой £—56 ккал1моль (фиг. 13, пря-
мая /). Если за основу для этого построения брать среднюю
скорость на участке, где выделяется от 2—9 до 45% конеч-
ного объема газов, то энергия активации получается несколько
меньше: 45,5 ккал(моль (фиг, 13, прямая 2). Почти такую же ве-
370
личину дает построение зависимости от температуры коэффициента
при т показательного уравнения для скорости. При этом построе-
нии экспериментальные значения скорости корректировались вычи-
танием начального значения
кг/о, полученного экстраполя-
цией кривой и/—т па время,
равное нулю.
В табл, 3 приведены от-
ношения максимальной ско-
рости к начальной для обеих
стадий распада стифната
при разных температурах;
для сравнения приведены
также соответствующие дан-
ные о распаде стифниновой
кислоты. Данные таблицы
показывают, что, как и для
пикрата аммония, на первой
стадии рост скорости раз-
ложения стифната тем
больше, чем ниже темпера-
тура, а для второй Стадии
Фиг. II. Сопоставление кривых газообразо-
вания при распаде стифнинопой кислоты
(ТНР г и стифнатр аммония (ТНРА) при
230J (m/'u.-elO-s г/с.и3).
ускорение тем меньше, чем ниже температура; при 190J (если
сравнивать абсолютные скорости) оно практически отсутствует.
Фиг. 12, Изменение скорости газооб-
разования во времени при распаде
стифната аммония при различных
tcii пературах.
Фиг. 13. Температурная зависимость
скорости газообразования при распа-
де стифната аммония,
£—мо начальной скорости гаэообраэовя
ния. 2—по средней относительной скорости
газообразования на первой стадии-
Зависимость скорости реакшы от температуры для второй ста-
дии была определена совмещением кривых p=f(r) при 210—230°,
Построение зависимости коэффициента совмещения от температу-
ры в аррениусовских координатах дало £—48,1 ккад/молб. В связи
24*
Л
Таблица 3
Отношение максимальной скорости газообразования к начальной для двух
стадий распада стнфната аммония (т/о=(9,4—10,7) 10-4 г/сл3)
Стифнат аммония Стифннноваи кислота
Температура / о-шах \ / «’max \ ®’тах
°C X И'нач f 1 \ ^нач / г Weaq
190 38 1,0 1,6
200 34 2,0 1,6
210 21 2,3 —
230 8 7 3,7 1,5
с тем, что кривая p=f(x) для 190° уже не может быть совмещена
с кривыми газообразования при более высоких температурах, тем-
пературная зависимость скорости распада стифната аммония была
рассчитана также исходя из начальных скоростей второй стадии;
в этом случае Е получается рав-
ной 40,2 ккал/моль. Таким обра-
зом, как и у пикрата аммония,
скорость второй стадии распада
стифната значительно слабее за-
висит от температуры, чем ско-
рость первой стадии.
Изменение скорости распада
со временем. Построение этой за-
Фиг. 14. Проверка подчинения пока-
зательному закону распада стифната
аммония на стадии первичного уско-
рения при различных температурах
г/слг3).
и 230° прямые получаются
Ig им)—т, где ffi'o—некоторые
висимости в логарифмических ко-
ординатах, так же как и для пик-
рата аммония, не дает прямой
линии. Такая прямая получается
в полулогарифмических коорди-
натах 1g—----т для температур
190 -и 200° (фиг. 14). При 210
при построении в координатах
начальные значения скорости
распада, различные для разных температур. -
Значительная часть первой стадии распада стифната аммония
удовлетворительно описывается также уравнением автокаталити-
ческой реакции
— =• A, (а — х)--k,,x " (а — х),
причем п заключается в пределах 3,5—4 для интервала темпера-
тур 190—210° и равно 5 при 230°.
372
Влияние степени заполнения сосуда (m/v) на течение распада.
Как и для пикрата аммония увеличение tn!v уменьшает (хотя и
относительно слабо) начальную скорость газообразования при
210’, но ускоряет ее рост во времени; точно так же увеличивается
Фиг. 15. Влияние m/v на скорость распада стнфпата ам-
мония прн 210Р.
(Числа у кривых—Л;'?' и г.'см* - |04).
скорость на второй стадии (фиг. 15). Численные значения, харак-
теризующие эту зависимость, приведены в табл. 4.
Таблица 4
Влияние m/v на распад сти фиата аммонии при 210°
m/v-lW г /см3 1 н слР/г-лшн (ffi,max)[ « см3/г-мин (й'начЪ j « см3/г-мин\ / ®’тах\ | (^'тах)[ | (®яач)2
1 те’иач Л
9.8 1 о,ц 2,3 0,3-5 ; 21 1 0,0
43,4 ! 0,0.5 5.0 1.4 100 3,6
Основные отличия закономерностей распада пикрата и стифната
аммония от закономерностей разложения пикриновой и стифнино-
вой кислот.
Сложная картина многостадийного развития реакции разло-
жения пикриновой кислоты 1 на начальном этапе, включающая ин-
дукционный период, затем ускорение газообразования, быстрый его
рост, падение скорости и последующее медленное ускорение, за-
меняется в случае пикрата и стифната аммония двухстадийпым
протеканием разложения. Первой стадией является сильно уско-
ряющееся газообразование, проходящее до достижения приблизи-
тельно половины конечного давления, после чего скорость резко
1 Данные о распаде пикриновой и стифниноной кислот cvl. настоящий сбор-
ник стр. 349.
падает и начинается вторая, также самоускоряюгцаяся, стадия
процесса.
Начальная скорость в случае солей меньше, чем при распаде
кислот, но растет опа со временем значительно быстрее, особенно
прн низких температурах. Соответственно, давление газообразных
продуктов распада солей прн повышенных температурах сравни-
тельно рано становится больше, чем при распаде кислот (фиг. 3).
При меньших температурах скорости распада солен и кислот вы-
равниваются позже (фиг. 2).
Иначе говоря, в изученном интервале температур средняя ско-
рость распада солен несколько ниже, чем кислот при умеренных
температурах; при высоких температурах опа приближается к ско-
рости распада кислот.
Сильнее зависит скорость распада солей и от температуры.
Это различие проявляется особенно отчетливо, если сравнивать
начальные скорости н слабее—при сравнении средних скоростей.
Увеличение m!v в случае солей несколько уменьшает началь-
ные скорости газообразования, но увеличивает аналогично тому,
что наблюдалось для пикриновой кислоты, рост скорости во вре-
мени. а также величину и рост скорости па второй стадии,
2, Разложение калиевых солей пикриновой
и стифниновой кислот
Пикрат кал 11 я
Пикрат калия был получен прилипанием раствора углекислого калия (7%)
к горячему насыщенному водному раствору пикриновой кислоты (%., 122.4;).
Полученный мелкокристаллический порошок просеивали через сито с размером
японки 0,155 ,п.ч, причем Для опытов брали фракцию, прошедшую через это сито.
Чистоту пикрата контролировали определением содержания калия (получено
14,45%, теоретически 14,60%).
Конечный объем газообразных продуктов распада составляет 3,8 молей на
моль пикрата калия (320 яс,и3/г), т. е. меньше чем для пикриновой кислоты
(410 -460 нсл3/а) в пикрата аммония (510—530 « сл3/г). В конце опыта на дне
реакционного сосуда остается черный порошок.
Сводные результаты исследования распада пикрата кадия
и интервале температур 250—300°, в котором пикрат находится
и твердом состоянии, приведены на фиг. 16.
У пикрата калия кривая К=/(т) или, что то же p=f(x) имеет
S-пбразную форму как для кислоты или пикрата аммония. Однако
индукционный период в случае пикрата калия гораздо больше
1 прн 250°, например, около 1000 мин,), а последующий рост ско-
рости газообразования и ее падение протекают значительно резче.
Этот характер изменения скорости со временем при разных тем-
пературах особенно отчетливо виден на дифференциальных кри-
вых (фиг, 17). Кривые V=f(-r) хорошо воспроизводятся и совме-
щаются (для разных температур) изменением масштаба времени
(фцг, 18). Скорость распада пикрата калия в несколько раз мень-
ше, чем пикриловой кислоты или пикрата аммония (фиг. 19);
374
Фиг.
ПИЯ
при
17. Изменение скорости газообразова-
во времени при распаде пикрата калия
различных температурах =
= 10-3 е1см>).
375
600ъмин(1,1)
176
разница между ними особенно велика вначале, когда разложение
пикрата калия находится еще в стадии индукционного периода.
Зависимость скорости распада от температуры. По зависимо-
сти коэффициента совмещения (&сов) от температуры в интерва-
ле 250—300°, изображающейся в аррениусовских координатах пря-
мой линией (фиг. 20), Е равна 41,2 ккал/моль. Энергия активации
была рассчитана также по температурной зависимости индукци-
онного периода тШ1Ц, за который принималось время выделения
Фиг. 20. Температурная зависимость ско-
рости газообразования при распаде пик-
рата калия.
/—по оеличине коэффициента трансферта
цин, ло величинам индукционного периода.
Фиг. 21. Зависимость скорости рас-
пада пикрата калия от времени.
10 « см3/г газов, что соответствовало приблизительно 3% распада
(фиг. 20). В этом случае Е — 42 ккал/моль.
Изменение скорости распада со временем. Эту зависимость оп-
. dV . dV
ределяли построением кривых в координатах 1g-------т и 1g— —
— IgT (фиг. 21). В обоих случаях некоторый участок кривой пря-
молинеен. При этом в логарифмических координатах этот участок
больше, чем в полулогарифмических. Однако при повь|шении
температуры показатель степени изменяется от 5,5 (при 250°) до
4,5 (при 290°).
Определяли также влияние tn/v на течение распада пикрата ка-
лия. Изменение m/v от 10-!0'J г/см2 до 100. 10'4 а/с.и3 при
280—300° не отражалось на индукционном периоде, но несколько
ускоряло последующее развитие реакции. Это последнее влияние
m/v сходно с тем, которое наблюдалось при разложении большин-
ства изученных в настоящей работе веществ.
О д поз а меще н ны й стифнат калия
Однозамещенный стифнат калия получали добавлением рассчитанного коли-
чества KsCQa к взнеси стифниновой кислоты ((пп 178,4') в кипящей воде при пере-
мешивании. Стифнат кадия после охлаждения выпадает в виде мелких желтых
377
кристаллов. Содержание калия в продукте было найдено равным 13,7%, по теории
в однозамещенном стифнате оно равно 13,8%. Для исследования были взяты кри-
сталлы, которые проходили через сито с размером отверстия 0,22 мм и задержи-
вались на ситс с отверстием 0,19 мм.
По общему характеру кривых V=/(t) (см. например фиг. 22)
распад однозамещенного стифната калия в изученном интервале
относительно низких температур 170—200°1 очень сходен с рас-
падом кислот, особенно пикриновой. Первым этапом распада
стифната является индукционный период (фиг. 23), очень корот-
кий при температурах выше 190°, но отчетливо заметный при
Фиг. 22. Распад одпозавдщен-
него стифната калия при 190е
10-9 г/см3).
Фиг. 23. Начальный участок кривых У=)(т)
при распаде однозамещенного стифната
кадия прн различных температурах
г/см3)'.
170°, когда его длительность составляет около 20 мин. Второй
этап представляет собой кратковременное ускорение газообразо-
вания. На третьем этапе газообразование протекает быстро, по
со скоростью, убывающей во времени. Максимальное давление,
достигаемое на третьем этапе, возрастает с температурой; при
170° оно соответствует 10 н смИг, при 200° около 75 н см?[г. На
четвертом этапе скорость (абсолютная) почти постоянна, а точ-
нее очень слабо растет со временем: так при 190° максимум ско-
рости всего на 20% выше ее значения в начале четвертого этапа:
по достижении давления, составляющего несколько больше поло-
вины конечного, скорость газообразования отчетливо уменьшает-
ся и начинается пятый этап, идущий вновь с некоторым, более
значительным ростом скорости (фиг. 24). На последнем, шестом
этапе, наблюдается естественное падение скорости.
Все эти особенности распада еще более, отчетливо видны на
дифференциальных кривых для абсолютной и особенно для отно-
сительной скорости (фиг. 24).
При сходстве общего характера кривых распада однозаме-
щенного стифната и стифниновой кислоты количественно они раз-
1 Выше 2Q0” происходила вспышка.
378
.тачаются весьма значительно. Это различие, мало заметное на
начальных этапах, становится больше на четвертом этапе, на ко-
тором, например при 200°, скорость распада однозамещенного
стифната в 6 раз меньше, чем стифниновой кислоты (фиг. 25).
Фиг, 24. Изменение относительной скоро-
сти газообразования во времени при рас-
паде однозамещенного стифната на дня
(т/и^Ю*3 г/с.и3).
Температурная зависимость скорости распада стифната была
определена для начальных скоростей четвертого этапа по прямой
Аррениуса; энергия активации получается равной 47,6 ккал/моль.
Фиг. 25. Сопоставление кривых разло-
жения стифниновой кислоты (ТНР),
ее однозамещенной (THPKi) и двух-
замешенной (ТНРКг) калиевых солей
при 200° (т/ои1(Н г/елг3).
Фиг. 26. Влияние m/v на разложение
однозамещешюго Стифната калия при
195°. (Числа у кривых — m/v в
г/с.м3 КУ).
Изменение tn/v слабо влияет на начальные этапы распада, но
заметно увеличивает скорость его на четвертом этапе (фиг. 26).
379
Д в у замещенный стифнат калия
Двузамещенный стифнат калия получали взаимодействием углекислого калия
с насыщенным водным раствором стифниновой кислоты в виде мелких оранжевых
кристаллов. Полученный продукт просеивали через сито с размером ячейки
0.11 .им. Содержание калия в двузамещенном стифнате составило 24.24/п (теоре-
тически оно равно 2-1,3%).
Изучение термического распада двузамещенного стифната кали!| проводили
в интервале 200—240°: при 250° в условиях опыта уже происходила вспышка.
В связи с тем, что двузамещенный стифнат Кристаллизуется с одной молекулой
воды, для ее удаления реакционный сосуд откачивали в течение часа при ком-
натной температуре и затем в течение часа при 1203. Впрочем присутствие парой
воды слабо отражается на ходе распада, как это показали сравнительные опыте:
но разложению двузамещенного стифната при удалении воды и без него.
На фиг. 27 приведены результаты опытов при разных темпера-
турах, позволяющие видеть общий характер зависимости У=Дт)
или, что то же p=f(x). По графикам фиг, 27 и особенно по диф-
ференциальным кривым (фиг. 28) можно различить три этапа: ин-
дукционный период, период ускорения и период падения скорости,
начинающийся после достижения V’^80 н см^}г, что соответствует
— 35% Укон- При повышении температуры опыта увеличивается,
как видно из фиг. 28, максимум абсолютной скорости; однако в от-
лпчце, например, от стифната аммония возрастает также и относи-
тельное увеличение скорости (отношение максимальной скорости
к ее начальному значению). Так при переходе от 2)0 к 230° отно-
шение зута1/а.'нач возрастает с 2,3 до 5 (фиг. 29).
По характеру распада, его ускорению и величине скорости дву-
замещенный стифнат калия резко отличается от однозамещенчого.
а следовательно, и от стифниновой кислоты (фиг. 25). При 200э
двузамещенный стифнат разлагается в 6 раз медленнее, чем одно-
замещенный и в 23 раза медленнее чем кислота (сравнение произ-
водилось по средней скорости газовыделения на участке от начала
опыта до выделения 150 н см3/г газов). При 220° разница в скоро-
стях разложения становится меньше,
380
Зависимость скорости распада от температуры определили, во-
первых, для индукционного периода, за который было принято вре-
мя выделения 10 н см?/г газов. В аррениусовских координатах эта
зависимость выражается прямой и по тангенсу угла наклона пос-
Фиу. 28. Изменение скорости газообразования во
времени при распаде двузамещенного стифната
калия прн различных температура^
pn/u^lO-3 г/сл3).
Фиг, 29. Изменение относительней) скорости
газообразования во времени при распаде
двузамещенного стифната калия при раз-
личных температурах г/см1).
.тедней энергия активации получается равной 44,8 ккал/моль
(фиг. 30, прямая /).
Во-вторых, ее определяли но времени достижения макси-
мума скорости, иначе говоря по средней скорости на этапе
ускорения. Этот .метод (фиг.
30, прямая 2) дает Е=^
= 52,9 ккал/моль.
В-третьих, температур-
ный коэффициент скорости
определялся на участке за-
медления распада по выра-
жению для констант скоро-
сти реакции первого поряд-
ка (фиг. 30, прямая 5),
в этом случае энергия акти-
вации оказалась равной
50,7 ккал/моль.
В-четвертых, температур-
ную зависимость определя-
ли по изменению тангенса
угла наклона прямолиней-
ного участка кривой lg V—т,
хотя к сожалению этот участок был невелик (степень распада
от 5 до 15% при 200—210° и от 5 до 27% при 220—240°); данный
метод также дает прямую в аррениусовских координатах и
Е=51,7 ккал/моль.
381
Изменение скорости распада со временем. Попытка совмеще-
ния кривых V--=/(t) для опытов при разных температурах путем
изменения масштаба времени не дала положительного результата.
Ускорение при высоких температурах больше, чем при низких в
противоположность тому, что можно было бы ожидать, например.
совместном протекании мономолскулярной и автокаталитичсс-
реакций. Построение в
при
кой
Фиг. 30. Температурная зависимость скоро-
сти газообразования при распаде двузаме-
шенного стифната калия (т/о«10—3 е/с.п3).
/—по величинам индукционного периода, 2~пи
временам достижения максимума скорости.
J—по значениям константы скорости газообразен
ванин на этапе замедление распада.
координатах степенной зависимости
(lg V—ig с) дает прямые
в значительном интервале
разложения, но показатель
степени меняется, возрастая
с увеличением температуры
от 2,4 1[ри 200rj до 5,9 при
240°.
Прн построении кривой
lg V—т прямолинейным по-
лучается лишь небольшой се
участок. Таким образом за-
висимость скорости газооб-
разования от времени не
укладывается для двузамс-
щецного стифната калия
в простые соотношения то-
похимического н ценного
распада.
Влияние степени заполне-
ния сосуда rn/t' на течение
распада. Как это наблюда-
лось п при распаде пикрата кялия. в случае двузамещенного стиф-
ната, изменение m/а от 10- КГ4 г/см3 до (15 • 10"4 г/слР при 230—240'
нс отражалось на индукционном периоде, но несколько ускоряло
последующее развитие реакции.
Общие закономерности, отличающие распад калиевых солей
пикриновой и стифниновой кислот
Одна из трех солей — однозамещенный стифнат калия — резко
отличается в отношении характера своего распада от двух других.
Расцад одпозамещенпого стифната качественно очень сходен с рас-
падом пикриновой и стифниновой кислот, но в сравнении с послед-
ней протекает, особенно при низких температурах, значительно
медленнее.
Пикрат н двузамещенный стифнат калия близки ио типу распа-
да; наблюдается индукционный период, этап ускорения и падение
скорости. Стифнат разлагается значительно (в 40 раз) быстрее,
чем пикрат. Обе соли распадаются гораздо медленнее, чем соответ-
ствующие им кислоты. Кривые V=f{r) для опытов по распаду пи-
крата калия при разных температурах на участке ускорения хоро-
шо совмещаются друг с другом путем изменения масштаба оси
382
времени. В случае двузамещенного стифната такого совмещения
получить не удается, так как при высоких температурах ускорение
выражено сильнее, чем при низких.
Участок ускорения для двузамещенного стифната не уклады-
вается полностью в простейшие выражения топохимической и цеп-
ной реакций; на участке падения скорость распада этого стифната
подчиняется закону реакции первого порядка. Известное сходство
кривые 1'=) (т) среднего стифната и пикрата обнаруживают с кри-
выми аммониевых солен, однако без столь отчетливо выраженной
стадийности как для этих последних.
3. Разложение свинцовых солей пикриновой
и стифниновой кислот
Пикрат свинца
Для получения пикрата свинца к насыщенному раствору пикриновой кислоты
в воде небольшими порциями добавляли намельченный углекислый свинец, Полу-
ченный раствор фильтровали в горячем состоянии и фильтрат охлаждали. Пикрат
свинца высаживался в виде мелких желтых кристаллов, отфильтровывался, про-
мывался бензолом и сушился в течение нескольких часов при 80°.
Содержание свинца в полученном продукте 30,3%, теоретически оно в моно-
гидрате пикрата свинца составляет 30,-1%. Пикрат свинца просеивали через сито
с размером ячейки 0,15 мм.
После разложения пикрата свиниа на дне сосуда оставался черный порошок
Налета на стенках сосуда, подобного тому, который образуется при распаде пик-
риновой кислоты и пикрата аммония, не наблюдалось.
Распад пикрата свинца изучали в интервале температур 230-
260°. При 260° и навеске 20 мг разложение в двух проведенных
опытах через 20 мин закончилось вспышкой. При уменьшении на-
вески до 5 мг распад проходил до конца без вспышки.
Распад начинается с быстрого роста давления (в процессе про-
грева), соответствующего выделению 35 « елр/а газов при
240—260°. Эти газы представляют собой, очевидно воду !, связан-
ную пикратом и ие удалившуюся при обычной подготовке опыта
(откачивание диффузионным насосом в течение 2 часов при ком-
натной температуре).
Кривые V=f(%) при низких температурах показывают плавный
подъем без отчетливо выраженного индукционного периода; ско-
рость газообразования возрастает в 9-—18 раз по сравнению с на-
чальным ее значением (фиг. 31). Наибольший рост (табл. 5)
наблюдается при самой низкой температуре опыта (230°); при по-
вышении температуры ускорение уменьшается, но при 260° вновь
несколько возрастает, что впрочем может быть уже связано с само-
разогревом.
По достижении давления, составляющего около половины ко-
нечного, скорость проходит через максимум и начинает падать.
Однако так обстоит дело лишь вблизи нижней границы изученного
интервала температур (230—240°). При повышенных температурах
1 Выделение 1 моля воды соответствует 33,0 н см3/г моногидрата.
383
Таблица 5
Влияние температуры иа течение распада пикрата свинца
[m/v= (8,9-5-10,5). 10-4 г/см3]________________________________________
ГС (^нач); (a’max)l « см31г-мин\ (№Яач)2 н сл^г-мин (T'maih н см^/г-мин / \ \ wHa4 /1 («mail {юнэч)2
230 0,023 0,41 — — 17 —
240 0,094 1,2 — — 13 —
250 0,23 2,2 2,2 3,1 10 1,0
250 0,28 2,4 2,4 3,0 8,6 1.0
_'6О (1,89 11,0 3,6 7,5 12,-, з, I
(250 и 260°) кривая уже показывает отчетливую двустадийность
процесса, подобную наблюдавшейся у стифната аммония. Дву-
стадийность особенно ясно видна на дифференциальных кривых
(фиг, 32), показывающих наличие двух максимумов скорости.
Сопоставление кривых V=/(t) при 230° (фиг, 33) показывает,
что средняя скорость распада пикрата свинца значительно меньше,
чем пикриновом кислоты; однако, это различие связано главным
образом с тем, что разложение пикрата не сразу достигает макси-
мальной скорости; последняя же близка к максимальной скорости
распада кислоты (фиг. 34).
При 250 и 260° (фиг. 35) кривая V — f(r) для пикрата свинца,
уже начиная с половины распада, пересекает кривую для кисло-
ты. Иначе говоря, скорость распада этого пикрата быстрее растет
с температурой, чем скорость распада кислоты аналогично тому,
как это наблюдается и для пикрата аммония.
Зависимость скорости распада от температуры была определена
по коэффициентам прямых в координатах 1g w—т. Полученные
384
Фиг, 32, Влияние температуры на характер кривых
dV
= ИТ> при распаде пикрита евипця
г/см3).
Фиг. 33, Сопоставление кривых V=f(x) при
разложении пикриновой кислоты (ТНФ),
пикрата аммония и пикрата саинца при 230°
jmiiiai"-’ г/см*).
25 451
385
dV
Фиг, 34. Сопоставление изменения скорости
газообразования во времени при распаде
пикриновой кислоты (!) и пикрата свин-
ца (2) при 230’ (m/t>=sl(W г/м3).
Фиг. 35. Сопоставление кривых газообразо-
вания при распаде пикриновой кислоты
(ТНФ), ее аммониевой и свинцовой солен
при 2509 (m/v^lO—3 г/ск3).
dV Н См3
dT г-мин
Фиг. 36. Влияние m/v на харак-
тер изменения скорости газо-
образования во времени при
распаде пикрата свинца при
250’.
I Чис ла у кривых—m в - Ю1)
38С
значения укладываются на аррениусовскую прямую, по тангенсу
угла наклона которой £ = 60,2 ккал/моль. Энергия активации была
рассчитана также по значениям средних скоростей в интервале,
где образуется от 30—50 до 140 н сл3/г газов; в этом случае
Е = 58,7 ккал/моль. Расчет по начальным скоростям распада дает
энергию активации 65,1 ккал/моль. Таким образом, по всем трем
методам оценки скорость реакции распада пикрата свинца сильнее
зависит от температуры, чем скорость разложения рассмотренных
ранее солей.
Изменение скорости распада со временем. При 230—260° в полу-
логарифмических координатах (1g —г) на значительном участ-
ке получается прямая,
При построении экспериментальных данных в логарифмиче-
ских координатах получаются кривые, обращенные выпуклостью к
оси абсцисс. Если, однако, по оси ординат откладывать lg(oi—Wq),
то в некотором среднем интервале распада, соответствующем сте-
пени превращения от 12 до 43%, получаются прямые, но показа-
тель степени п в уравнении w—w0 — kxn колеблется от 2,0 до 2,5.
Влияние степени заполнения сосуда m/v на течение распада.
Определения были сделаны при относительно высокой температуре
(250°—см. фиг. 36). В отличие от других солей m/v сильно влияет
на течение распада пикрата свинца на всем протяжении процесса:
при увеличении m/v повышается скорость нарастания давления на
участке ускорения, сильно возрастают максимумы скорости, осо-
бенно первый (табл. 6). Начальная скорость при увеличении m/v
возрастает относительно слабо, но сильно увеличивается ускорение
и соответственно максимальное значение скорости.
Таблица б
Влияние m/v на распад пикрата свинца при 250°
m/v-10< г/см3 (®нвч)[ и сма/г-мин н см3/г-мин (а'нач)г н см3/г -мин н. (^max)s смЗ/г^мин / »тах \ \ к'нач /1 £ |
9,0 0,23 2,2 2 2 3,1 9,6 1,4
47,1 0,30 7.3 3,1 4,5 24 1,а
Из табл. 6 видно, что отношение максимальной скорости распа-
да к начальной возрастает на первой стадии в 2’/г раза при увели-
чении m/v в 5 раз; при повышенных m/v первый максимум больше
второго.
Стифнат свинца
Стифнат свинца был получен осаждением его из водного раствора стифната
натрия водным раствором нитрата свинца. К насыщенному раствору стифниновой
кислоты (<пл 178,4°) небольшими порциями присыпали бикарбонат натрия. При
этом происходит бурное выделение углекислого газа и образование стифната нат-
рия. Полученный раствор фильтровали от механических примесей, к нему добав-
ляли 0,5 мл 98—99%-ной уксусной кислоты. Затем раствор нагревали до 70—80“
25* 387
и ;быстро приливали в него Wo-ный фильтрованный раствор нитрата свинца. При
последующем охлаждении стифнат свинца выпадал в виде мелких кристаллов
оранжевого цвета. Качество продукта проверяли определением содержания в нем
свинца. Содержание последнего было найдено равным 44,2% (теоретически и
моногидрате стифната содержится свинца 44.3%), Стифнат свинца был отсеян
между ситами с размерами ячеек 0,19 н 0,16 леи.
В первом опыте кристаллизационную воду удаляли путем эвакуации прн
нагревании вещества до 120—125° в течение двух часов. При этом на стенке
реакционного сосуда образовался желтоватый налет. Для удаления этой примесн
стифнат свинца подвергали предварительной эвакуации прн 12(Р в течение трех
часов, после чего пересыпали в другой сосуд и на воздухе вновь превращали в
цоногндрат. Дальнейшие опыты вели именно с этим регидратированным стиф-
и атом.
Фиг. 37. Распад стифната свин-
ца при различных температурах
(>п/и=:10-3 г/см3).
Фиг, 38. Сопоставление кривых газо-
выделения прн распаде стифниновой
кислоты (ТНР) и ее солей: ам-
мониевой (ТНРА), однозамещенной
(THPKi) и двузамещенной (ТНРКД
калиевых н свинцовой (ТНРС) при
20СР (ra/ii.aalO-3 г/см3).
Термический распад стифната свинца (фиг. 37) изучали в ин-
тервале температур 200—230°; при 240э (навеска 20 мг) уже через
25 сек. произошел взрыв. Индукционного периода не наблюдается.
Быстрое, но небольшое начальное повышение давления, по-видимо-
му, связано с выделением остатков кристаллизационной воды. От-
четлив двухстадийный характер распада; обе стадии идут со сла-
бым ускорением (табл. 7), заметным даже на интегральной кри-
вой, но еще более отчетливо на дифференциальной. Переход от
первой стадии ко второй происходит после выделения примерно
40 н см?[г газов, т. е. около полного количества газов, выделяю-
щихся при распаде.
Сопоставление скоростей распада стифната свинца, кислоты и
других ее солей (фиг. 38) показывает, что при 200° стифнат свин-
ца разлагается значительно медленнее (в 3—4 раза на первой и в
8—10 раз иа второй стадии), чем кислота, а также стифнат аммо-
ния и однозамещеняый стифнат калия, но быстрее, чем дв-узаме-
388
Таблица 7
Влияние температуры на термический распад стифната свинца
[m/v=-(9,8-bl 1,9)-10-4 г/сл<3]
f °C н ему г-мин к смЗ/г мпн (^нач)а и см^г - un )? н сжЗ/г-лия . -- g s- Е 31 § 3 ! 8 £ ь з л to
(Ш,1ач)2
200 0,063 0,11 0,034 0,055 1,8 1,6 3,3
210 0,10 0,21 0,078 0.11 2,1 1,5 2.8
220 0.21 0,52 0,16 0.28 2,4 1.8 3,2
230 0,48 1,25 0,31 0,56 2,6 1,8 4,0
щенный стифнат калия. При 230° (фиг. 39) наблюдается аналогич-
ная картина с тем лишь отличием, что кривая У=/(т) двузамешен-
ного стифната калия, скорость
распада которого быстрее растет
с температурой, пересекает кри-
вую стифната свинца.
Зависимость скорости распада
от температуры была определена
различными путями. По сред-
ней скорости на участке, где
выделяется от 10 до 35 н смл/г
газов, т. е, на первой стадии
распада была построена аррениу-
совская прямая, которая дала
энергию активации, равную
37,8 ккал/моль.
Близкая величина энергии ак-
тивации (36,4 ккал/моль} полу-
чается построением прямой Арре-
ниуса по значениям констант по-
казательного уравнения для вто-
рой стадии, а также по на-
чальным скоростям последней
(34,8 ккал/моль}.
Изменение скорости распада
Фиг. 39. Сопоставление кривых газо-
выделения при распаде стифниновой
кислоты (ТНР) и ее солей: аммоние-
вой (ТНРА), двузамещенной калиевой
(ТНРКз) и свинцовой (ТНРС) при 230s
(m/p«10-3 г/с .и3),
временем. Для первой стадии
распада во всем изученном интервале температур изменение газо-
образования может быть выражено соотношением
lg (V— Vo) = п 1g r-J-A,
где Vo = 2 смЧг (фиг. 40). Однако величина п не постоянна и воз-
растает с температурой от 1,14 при 200° до 1,42 при 230°.
Для второй стадии при всех четырех температурах (200—230°)
получаются удовлетворительные прямые в полулогарифмических
координатах lg V—т (фиг. 41).
Как уже отмечалось, рост скорости по мере течения распада
на обеих стадиях относительно мал; ускорение увеличивается с
389
0,0 ? г з ig"c
Фиг. 40. Спрямление кривых зависимости
количества образовавшихся газов от време-
ни на первой стадии распада стифната
евнниа (m>v~ 1Q-9 г/сл9).
(ZZ0°)
Фиг. 41. Спрямление кривых зависимости коли-
чества выделившихся газов от времени на
второй Стадии распада стифната свинца
(т;и == iQ-9 г/сл5).
390
температурой на первой стадии в пределах от 1,8 (при 200°) до
2,6 раза (при 230°); на второй стадии увеличение ускорения еще
меньше (1,6—1,8 раза).
Влияние степени заполнения сосуда на течение распада. При
увеличении т/о скорость распада и максимум ее растут (фиг. 42),
Фиг. 42. Влияние m/v на зависимость ско-
рости распада стифната свинца от времени
на второй стадии при 20СР.
(Числа у кривых—‘m/v и |04)*
хотя и не слишком сильно. Так увеличение m/v в 10 раз (от 12 до
120 •10-4 г/см3) привело и увеличению максимальной скорости
только в два раза.
Общие закономерности, отличающие распад свинцовых солей
пикриновой и стифниновой кислот
Характерными особенностями распада обеих свинцовых солей
является двухстадийность (для стифната свинца отчетливо выра-
женная при всех температурах опыта, для пикрата свинца более
заметная при высоких температурах) и отсутствие индукционного
периода.
Основные различия в распаде свинцовых солей заключаются в
сильной зависимости скорости его от температуры для пикрата
свинца (Ел^бО ккал/моль), значительно большей, чем для кислоты
(Е^37 ккал/моль); в случае стифната свинца, напротив, энергия
активации относительно мала (~35 ккал/моль) и близка к таковой
для кислоты (Е = 34,6 ккал/моль). Скорость распада пикрата свин-
ца относительно сильно растет во времени и при увеличении m/v.
Для стифната эти зависимости выражены значительно слабее.
При низких температурах скорость газообразования на протя-
жении большей части распада пикрата свинца меньше, чем в слу-
чае пикриновой кислоты; однако, температурная зависимость
391
скорости распада соли больше, чем кислоты, также больше и ус-
корение распада во времени; поэтому при повышении температу-
ры пикрат разлагается медленнее кислоты лишь в первой половине
распада; во второй — соотношение скоростей обратное,
В случае стифната скорость распада как при низких, так и при
повышенных температурах на всем протяжении остается значи-
тельно меньше скорости распада кислоты.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Общие закономерности термического распада пикриновой
и стифниновой кислот и их аммониевых, калиевых
и свинцовых солей
В разложении перечисленных кислот и их солей наблюдаются
как некоторые общие черты, так и значительные различия. Для
облегчения рассмотрения основные особенности распада схемати-
чески представлены в табл. 8; значения энергии активации для изу-
чавшихся веществ приведены в табл. 9.
392
Jtj
Время И,1ТеРвал полура- температу- спада Вещество ры и игре- Характер раснала при />30° ^2 Гатосе со- т стояние 1 - в мин,
чоо Стифнат аммс- 190—230 ^00 пия твердый 300 ( Пикрат калия , 250—300 твердый t00 300 Олпозаметеп- 170—200 иый стифнат твердой кадия t00 230" 1 65 — 1,44
50 ТОО 250" Г / 8600 18,3 —
4000 1000 . —/ 262*** -- 5,83 / вспышка —200°
ТОО ЮООО 20000
Ov(J 200 Дву заменен- >00—240 ный стифнат твердый too калия । 300 Пикрат свинца 230—260 : твердый -рр 230" 210 4,67 / вспышка У —250°
200 $00 f^o" /Тж / / 920 1,96 — . / у' вспышка ^260°
i ISO 100 Стифнат свинца 200—230 cQ твердый ТОО -200 230 е ’ 143 — 3,18 /взрыв t < 94(1° 1
_______ • КО 200
* /([—Отношение периодов
* К%—отношение периодов
* —определено расчетом.
полураспада пикратов п
полураспада стифнатов и
пикриновой кислоты,
стифниновой кислоты.
393
Таблица 9
Энергия активации Е термического распада пикриновой
и стифниновой кислот и их солей
Вещество Интервал темпе- ратуры °C Е ккал/моль Примечание
Пикригюная кис- лота 183—270 38,6 Е определена но средним значе- ниям скорости при 5—50% распада
Пикрат кали а 250—300 41,2 Е определена по совмещению кри- вых I/—/(д)
Пикрат аммониа 200—270 43 40,5 Е определена по средней скорости при распаде от 3—11 до 45% (пер- вое значение) и по тангенсам угла наклона прямых 1g (V—1/0)=Л-|-At
Пикрат свинца 230—260 60,2 58,7 Е определена по тангенсам угла наклона прямых 1g w=j44At (первое Значение) и по средней скорости при 10—50% распада
Стифннновая кис- лота 180—250 34,6 Е определена по значениям Сред- них скоростей при 22—45% распада
Стифнат калия однозамещенный 170—200 47,6 Е определена по значениям скорос- ти при <5—35% распада
Стифнат калия двузамещецный 200—240 1 51.7 52,9 Е определена по тангенсам угла наклона прямых 1g прн 5— 27% распада (первое значение) и по средней скорости на этапе ускоре- ния
Стифнат аммония 190—230 45,5 45,5 Е определена по средней скорости при распаде от 2—9 до 45% (первое значенье) п по углу наклона прямых 1g w_--.4-|-£t
Стифнат свинца 200—300 34,8 30,4 Е определена по значениям скорос- ти при —30% распада (первое зна- чение) и по углу наклона прямых 1g V_A-kz
Одной из особенностей распада, присущей всем изученным со-
единениям, является рост скорости, притом даже абсолютной, во
времени на значительном участке распада. Падение этой скорости,
обусловленное уменьшением концентрации (а в данном случае коли-
394
" чества) реагентов, которое для реакций обычного типа наблюдает-
ся с самого начала распада, в рассматриваемом случае наступало
относительно поздно. Те кратковременные участки падения скоро-
сти, которые наблюдались для некоторых из изученных веществ,
связаны не с уменьшением количества реагентов, а с временным
уменьшением роли фактора ускорения.
Другой особенностью является уменьшение скорости распада
при переходе от кислоты к солям. Это обстоятельство представляет
определенный интерес и в плане возможности использования солей
кислот в качестве термостойких веществ; если учесть большой ин-
дукционный период распада пикрата калия, то начальная скорость
; его разложения оказывается намного меньше, чем у других ве-
! шести; возможно также сочетание пикрата калия с кислородосо-
держащими солями для увеличения кислородного баланса и тепло-
' ты взрыва полученной смеси.
Если характеризовать уменьшение скорости газообразования
отношением «периодов полураспада» солей и соответствующих
кислот, то оно составляет при 230э для пикратов от 1,5 (для пикра-
та аммония) и 2 (для пикрата свинца) до 18 (для пикрата калия).
г Для стифнатов соответствующие числа составляют 1,4 (стифнат
J аммония), 5 и 6 (дву- и однозамещенные соли калия) и 3 (стнф-
; нат свинца). Следует, однако, оговориться, что указанные отноше-
f ния условны, так как закон изменения скорости существенно раз-
’ личен — в частности, для пикрата калия большой период полурас-
пада в значительной мере определяется большим индукционным
: периодом, предшествующим росту скорости. Кроме того, в виду
' различий в температурной зависимости скорости, отношения «пе-
риодов полураспада» различны при разных температурах.
Из закономерностей, общих не всем, а лишь части изученных
соединений, можно указать на стадийность распада.
Собственно говоря, всякая реакция, протекающая с ускорени-
, ем, может рассматриваться как стадийная, поскольку она вклю-
чает стадию ускорения и стадию падения скорости. Однако такого
рода стадийность неизбежна и в этом смысле тривиальна.
' Стадийность распада, наблюдавшаяся для изучавшихся кислот
I и некоторых их солей, имеет иной и неодинаковый характер.
В общем основным типом этой стадийности можно считать со-
четание двух этапов ускорения, подобных наблюдающимся в слу-
чае стифната аммония (см. фиг. 12), но с большими вариациями
; как в величине ускорения и в доле газообразования на каждом из
; этих этапов, так и в относительном времени их протекания. В неко-
j торых случаях в начале разложения наблюдается индукционный
L период более (пикрат и двузамешепный стифнат калия) или менее
(однозамещеппый стифнат калия, пикриновая кислота) длитель-
. ный.
Ускорение обычно сильнее выражено на первом этапе, чем на
втором. Обычно также с повышением температуры относительное
ускорение уменьшается, хотя максимум скорости по абсолютной
395
своей величине естественно растет. Однако в случае двузамещеп-
ного стифната калия наблюдается обратная картина.
По абсолютной величине начальная скорость распада солей,
как уже отмечалось, обычно меньше, хотя и в разной степени, чем
соответствующих кислот. Так как, однако, ускорение у солей боль-
ше, то на определенной ступени распада соль и кислота могут
меняться местами, особенно если рассматривать не одну какую-то
температуру, а весь изученный интервал.
Температурная зависимость Для кислот на основном этапе рас-
пада невелика ц характеризуется энергией активации в пределах
35—39 ккал/моль. Сравнительно малая константа скорости распа-
да (для пикриновой кислоты, например, при 200° & = 6,3- 10-7 сек-1,
если константу эту рассчитывать как для мономолекулярной реак-
ции) обусловлена малыми значениями предэкспоненциального
множителя.
Для солей зависимость скорости от температуры больше. Раз-
ница энергий активации распада солей и соответствующих кислот
невелика для стифнатов аммония и свинца и пикратов аммония и
калия, но значительнее для одно- и двузамещенного стифнатов ка-
лия и особенно для пикрата свинца.
Рост скорости во времени во многих случаях и на более или
менее значительном участке может быть описан степенной или по-
казательной формулой. Иногда, чтобы получить прямую в этих
координатах, приходится вычитать начальные значения скорости
или объема газообразных продуктов.
Увеличение m/о, как правило, увеличивает1 скорость распада,
обычно сильнее на второй, чем на первой стадии, очевидно в ре-
зультате увеличения давления газообразных продуктов распада.
Это ускорение, однако, различно для разных веществ.
Зависимость между кинетическими особенностями распада
пикриновой и стифниновой кислот и их солей
и характеристиками вспышки и горения
Одной из перспективных задач исследования было выяснение
наличия возможного соответствия между свойствами исследован-
ных веществ в отношении их вспышки, воспламеняемости и горе-
ния, с одной стороны, и медленного термического распада,
с другой.
, Рассмотрение этого вопроса, однако, затрудняется тем обстоя-
тельством, что до настоящего времени вспышка пикриновой и стиф-
ниновой кислот и особенно их солей изучена недостаточно; в из-
вестной мере сказанное приложимо и к горению—в этом отноше-
нии нет систематических данных для пикрата и стифната аммония,
а также и самой стифниновой кислоты. Вследствие этого сопостав-
1 Для пикриновой кислоты, пикрата и стифната аммония увеличение wi/c-'
приводит к некоторому уменьшению скорости газообразования па начальном его
этапе.
396
1 ление характеристик различных форм химического превращения
изученных веществ имеет пока до получения более полных данных
по вспышке и горению предварительный характер.
Соответствия между кинетическими характеристиками и
вспышкой можно было ожидать в двух аспектах — температура
вспышки должна быть тем ниже, чем больше константа скорости
распада *; время задержки должно быть больше у веществ с силь-
но выраженным индукционным периодом или периодом ускорения
разложения, причем у таких веществ интервал между температу-
рой, при которой вспышка не происходит, и температурой вспыш-
ки с некоторой малой задержкой должен быть относительно велик.
Далее можно было бы сопоставлять кинетические характеристи-
ки и интенсивность вспышки, хотя эта последняя в большей мере
зависит от закономерностей горения (скорости горения при повы-
шенных температурах ц ее зависимости от давления), чем непо-
средственно от кинетики медленного распада.
Некоторое соответствие между скоростью распада и минималь-
ной температурой вспышки действительно намечается. Так стифни-
новая кислота даже при меньшей навеске (40 мг) начинает давать
вспышки при более низкой температуре, чем пикриновая кислота
(100 мг). Минимальная температура вспышки пикрата калия
(20 мг) лежит выше, чем для стифната калия или пикрата свинца.
Еще меньше дачных имеется о воспламеняемости изученных
веществ. Можно лишь обратить внимание на высокую воспламе-
няемость стифната свинца, возможно связанную с большой ско-
ростью его горения и соответственно малой толщиной прогретого
слоя. Есть сомнения, можно ли ее в данном случае характеризо-
вать, как для вторичных ВВ, критическим диаметром для горения.
Минимальная скорость, начиная с которой возможно горение
многих быстрогорящих ВВ, очень велика по сравнению с тем, что
наблюдается для вторичных ВВ. Кроме того, опыты Фогельзанга
показали, что увеличение диаметра заряда для гидрата пикрата
свинца с 4 до 7 мм не снижает критического давления для горения.
Наконец, непосредственное определение воспламеняемости было бы
затруднено тем, что некоторые из изучавшихся ВВ не горят при
атмосферном давлении.
При сопоставлении кинетики и характеристик горения можно
было бы в первую очередь выяснить, есть ли соответствие между
скоростью горения и скоростью распада. При одинаковом типе го-
рения чем больше скорость распада, тем больше должна бы быть
скорость горения. При этом следовало бы учитывать также темпе-
ратурную зависимость скорости распада: если для двух веществ
скорости их разложения при некоторой температуре протекания
медленного процесса одинаковы, но по-разному зависят от темпе-
1 Точнее скорость тепловыделения. Предполагается, однако, что скорость теп-
ловыделения пропорциональна скорости распада или скорости газообразования,
которой непосредственно характеризовали скорость распада в настоящем иссле-
довании.
397
ратуры, то при более сильной зависимости скорость распада при
высокой температуре, а следовательно, и скорость горения, должна
быть больше.
Далее можно было сопоставлять кинетику и способность ве-
ществ к горению, характеризуемую минимальным давлением или
другим критическим параметром горения. Наконец, можно было бы
искать соответствия между кинетикой и зависимостями скорости
горения от давления и от начальной температуры.
Начнем с величины скорости горения. Известно, что в этом от-
ношении существует, по крайней мере при низких давлениях, рез-
кое различие между медленно горящими пикриновой кислотой и ее
аммониевой солью и быстро горящими пикратом калия и особенно
пикратом свинца. Никакого соответствия этому различию не обна-
руживается при сопоставлении скоростей распада этих соединений.
Пикрат калия разлагается значительно медленнее пикриновой кис-
лоты, причем близки и температурные зависимости их скоростей.
Медленнее, чем пикриновая кислота, разлагается и пикрат свинца;
правда температурный коэффициент скорости его разложения ве-
лик и соответственно можно было бы ожидать более быстрого рас-
пада пикрата при высоких температурах. Однако, как видно на
примере стифпатов, этот путь подхода не оправдывается.
Сравнение со стифниновой кислотой в отношении кинетики и-
скорости горения провести не удается, так как характеристики го-
рения кислоты не определены с достаточной достоверностью.
Однако стифнат аммония горит очень медленно, а стифнат свинца
очень быстро. В то же время как скорость распада стифната свин-
ца, так и ее температурная зависимость заметно меньше, чем у
стифната аммония.
Отсутствие соответствия между скоростью распада и скоростью
горения кислот и их солей могло бы быть связано с различиями
в летучести, точнее в относительной способности к экзотермической
реакции в конденсированной фазе. Однако такого различия не об-
наруживается и при сравнении нелетучих солей друг с другом. Так.
стифнат свинна разлагается медленнее, чем оба стифната калия, а
горит значительно быстрее их. Пикрат калия горит (при 20 от)
даже несколько быстрее, чем пикрат свинца, а разлагается он
(при атмосферном давлении) заметно медленнее,
Не обнаруживается соответствия между характеристиками го-
рения веществ и медленного их распада и в том случае, если по-
следний оценивать по температурной зависимости скорости или по
характеру кинетической кривой. Так, если скорость распада пикра-
та свинпа значительно сильнее зависит от температуры (в сравне-
нии с пикриновой кислотой), то о стифнатах свинца и аммония
этого сказать нельзя. Если быстро горящий пикрат калия разла-
гается со значительным индукционным периодом, так же как и
двузамещенный стифнат калия, то у еще быстрее горящих стифна-
та и пикрата свинца индукционного периода не наблюдается. То Же
можно сказать, сопоставляя степень самоускорения распада и вли-
яние m/v на его течение.
398
Если в качестве характеристики горения использовать не ско-
рость, а способность вещества к горению, то также не обнаружи-
вается отчетливой зависимости между этой способностью и харак-
теристиками медленного распада, Из изученных веществ (в отно-
шении способности их к горению) легче всего при наименьшем
давлении (0,02 ат) горит стифнат свинца и труднее всего—пикрат
свинца (18 ат). Пикрат свинца разлагается (при 230°) в 6 раз мед-
леннее стифната 1 и можно .было бы с этим связать его меньшую
способность к горению. Однако двузамещенный стифнат калия го-
рит труднее (с 3 ат), чем однозамещенный, хотя скорость распада
первого из них больше, чем второго,
Не выявляется соответствия и между скоростью горения и спо-
собностью к нему. При 20 ат, правда, быстрее других горят стиф-
нат свинца и однозамещенный стифнат калия, для них минималь-
ное давление низко; однако пикрат калия и пикрат свинца горят
с почти одинаковой скоростью, но сильно различаются по способ-
ности к горению. Добавим еще, что моногидрат пикрата свинца
горит не хуже обезвоженного, причем скорость горения его даже
больше.
До сих пор мы пытались сопоставить характеристики различ-
ных форм химического превращения изучавшихся веществ несколь-
ко формально с тем, чтобы, если удастся обнаружить такое соот-
ветствие, перейти к рассмотрению его физического смысла.
Если подойти к делу несколько иначе и представить себе, какие
связи можно было ожидать, то помимо естественного параллелиз-
ма между скоростью распада и скоростью горения можно было
предположить следующие закономерности:
а) чем больше температурная зависимость скорости распада,
тем легче и быстрее должно гореть ВВ, так как при горении нагрев
идет несомненно до более высоких температур, чем при проводив-
шихся опытах по разложению. Этого, однако, не наблюдается
(пикрат свинца, двузамещенный стифнат калия); напротив, легче
всего горит стифнат свинца с малой температурной зависимостью
скорости термического распада;
0) если начальная стадия распада, ведущая весьма вероятно
к диспергированию вещества, является лишь слабо экзотермичной,
то чрезмерная скорость ее, не подкрепляемая протекающими по-
близости экзотермичными процессами, может затруднять распро-
странение процесса; однако, если меньшую способность к горению
пикрата свинца и двузамещенного стифната калия можно было бы
объяснить таким образом, то это объяснение трудно было бы рас-
пространить па остальные изученные соли;
в) при большом индукционном периоде вещество успевает про-
греться и реагирует, будучи нагрето, быстрее; в этом смысле боль-
шой индукционный период должен облегчить распространение го-
рения; это, однако, не 'наблюдается, и стифнат свинца, разлагаю-
’ Следует, однако, оговориться, что данные по горению относятся к моногид-
рату стифната свинца, а по разложению — к обезвоженному веществу.
399
щийся без заметного индукционного периода, горит легче, чем
пикрат калия, имеющий наибольший индукционный период.
В общем не удается для всех изученных соединений найти та-
кие черты термического распада, которые объяснили бы особенно-
сти их горения.
Может быть это связано с тем, что при горении существенна
скорость тепловыделения, а она не обязательно должна быть тож-
дественной скорости газовыделения, . которой характеризовался
термический распад.
Возможно, однако, что соответствие отсутствует по более глу-
бокой причине, заключающейся в том, что реакции, определяющие
распространение горения, не те или не только те, от которых зави-
сит течение медленного распада.
В частности, применительно к горению с диспергированием мож-
но себе представить, что частицы образуются в результате мало-
экзотермичной реакции и затем продолжают реагировать в фор-
ме горения с поверхности в окружающих их газах. Основной тепло-
приход, вызывающий диспергирование нового слоя, определяется
именно реакциями горения частиц; диспергирование является не
ведущим, а ведомым этапом процесса и в этом смысле относитель-
но мало влияет на возможность горения и скорость его распростра-
нения. Впрочем, диспергирование также может оказывать свое
влияние на скорость процесса: если частины получаются крупные,
то время их сгорания становится больше и это эквивалентно
уменьшению скорости тепловыделения, а следовательно, и горения.
Должно быть известное соответствие между скоростью горения
взвеси и скоростью диспергирования; если последняя слишком ве-
лика, то частицы не успевают сгорать или сгорают слишком дале-
ко от поверхности ВВ, передают, ему слишком мало тепла и поэто-
му горение не распространяется. Повышение давления, ускоряя
горение частиц, сильно увеличивает скорость перемещения фронта
горения и переводит зону тепловыделения все больше и больше в
конденсированну[о фазу. Когда этот переход совершится, дальней-
шее увеличение давления не ускоряет горения; если впрочем газо-
образные продукты способны к экзотермическим реакциям, то дав-
ление оказывает на эти реакции и на скорость горения тот же эф-
фект, который оно производит в случае вторичных ВВ.
ВЫВОДЫ
1. Изучена кинетика медленного термического распада аммо-
ниевых, калиевых и свинцовых солей пикриновой и стифниновой
кислот.
2. Распад всех изученных веществ имеет самоускоряющийся
многостадийный характер, различный для разных веществ, схема-
тически изображенный в графе 3 табл. 8.
3. Скорость распада всех солей данной кислоты в изученной об-
ласти температур в целом ниже, чем скорость распада самой кис-
лоты-
400
4. Калиевые соли распадаются медленнее, чем свинцовые, ко-
торые в свою очередь разлагаются медленнее аммониевых.
5. Температурные коэффициенты скорости распада для солей в
общем больше, чем для соответствующих кислот; особенно велик
температурный коэффициент скорости распада пикрата свинца.
6. Скорость распада стифниновой кислоты и ее солей значи-
тельно больше, чем пикриновой кислоты и соответствующих ее со-
лей; «период полураспада» при 230° для стифниновой кислоты,
стифната аммония, однозамешенного стифната калия, двузамещен-
ного стифната калия и стифната свинца составляет соответственно
45, 65, 260, 210 и 143 мин.; для пикриновой кислоты и ее аммоние-
'вой, калиевой и свинцовой соли — 470, 720, 8600 и 920 мин. Силь-
но повышенный «период полураспада» пикрата калия частично
связан с большим индукционным периодом, наблюдающимся при
его разложении.
7. Не обнаруживается отчетливого соответствия между кине-
тическими характеристиками распада изученных взрывчатых ве-
ществ и их способностью к горению, а также скоростью послед-
него.
8. Высказаны гипотезы о механизме горения быстрогорящих
веществ, объясняющие возможность отсутствия решающего влия-
ния скорости термического распада на скорость горения и способ-
ность к нему.
ЛИТЕРАТУРА
1. Андреев К- К., Глазкова А. П., Маурина Н. Д. и С в е т л о н Б. С.,
ЖФХ, 1958, 32, 1726.
А. А. ШИДЛОВСК.ИЯ
25. ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ НИТРАТА АММОНИЯ
Известен ряд веществ, ускоряющих [3], [4], [5] или замедляющих
[1], [2] термическое разложение нитрата аммония. Известно также,
что скорость термического разложения нитрата аммония значи-
тельно возрастает после разрушения его кристаллической решетки
(Гпл МН4\О3169э) и, следовательно, наиболее энергично процесс
разложения протекает в жидкости — расплаве нитрата аммония.
Ранее нами указывалось [4], что наиболее активными катали-
заторами для ускорения термического разложения расплава нитра-
та аммония являются соединения шестивалентного хрома: трех-
окись хрома (СгОз) и бихроматы или хроматы калия "или натрия.
Однако проведенные нами опыты показали, что при /°=200° для
полного разложения в течение 3—6 мин. небольшого количества
(1 г) нитрата аммония требуется значительное содержание ката-
лизатора (2—3% от веса разлагаемого вещества).
При меньшем количестве катализатора (1%) интенсивного раз-
ложения нитрата аммония не наступало, и потеря в весе за то же
время при той же температуре не превышала 2—3%.
26 451 401
Нам представилось интересным установить, как будут вести
себя катализаторы при значительно увеличенной навеске распла-
ва нитрата аммония, а именно 10 и 50 г. Во всех опытах нитрат ам-
мония нагревался на воздушной бане до 200±3° (внешняя обо-
лочка воздушной бани нагревалась на песчаной бане).
В качестве резервуара для расплава NH4NO3 при опытах с 1 г
служила пробирка диаметром 15 мм, с 10 г — пробирка диаметром
35 мм и с 50 г — стакан диаметром 60 мм.
После доведения температуры расплава NH4NO3 до 200° на его
поверхность всыпалась навеска порошка СгО3 или КаСг2О7 в коли-
честве от 0,01 до 0,3 г. Спустя 1—2 мин. происходило растворение
катализатора, а затем при достаточном количестве его начиналось
бурное разложение нитрата, сопровождающееся сильным газооб-
разованием и значительным вспениванием жидкости. Температура
расплава при этом повышалась (в опытах с 50 а—-до 290—320°) и
в связи с этим изменялся состав продуктов разложения: вместо
выделявшейся в начале бесцветной закиси азота, появлялась бурая
двуокись азота; весь процесс разложения заканчивался в течение
6—8 мин. и на дне и стенках сосуда оставался только тонкий оран-
жево-бурый налет соединений хрома.
При недостаточном количестве катализатора он успевал рас-
пределиться равномерно по всей массе расплава, расплав окраши-
вался в равномерный светло-оранжевый цвет, и больше никаких
явлений не наблюдалось.
Кроме визуального контроля, определялась потеря в весе нитра-
та аммония, произошедшая после его разложения. Результаты
опытов приведены в таблице.
Термическое разложение нитрата аммония с каталитическими добавками—
соединениями Ст (VI)—при температуре 20О±3°
Навеска катализатора г Потеря в весе NH4NO3 в % за 6 мин. при весе расплава (в скобках указан % катализатора)
1 г 10 г 50 г
СгО3 0,01 3(1.0) 5(0.1) —
CrOj 0,02 100(2,0) 100 (0,2) —
СгО3 0,03 — — 3(0,06)
СгО3 0,05 — — 100(0,10)
К2СГ2О7 0,0? 5(1,0) — —
К2Сг2О7 0,02 98(2,0) 2(0,2) —
KjCr2O7 0,03 — 95(0,3) —
KjCr2O7 0,20 — — 3(0,4)
KjCr^O? 0,30 — — 100(0,6)
402
Как видно из таблицы, количество катализатора, необходимое
для полного разложения расплава NH4NO3, растет не пропорцио-
нально весу последнего, а значительно медленнее. Так для разло-
жения 1 г NH4NO3 необходимо ввести 0,02 г СгОя, т. е. 2%, а для
разложения 50 г всего 0,05 г или 0,1% веса нитрата. В несколько
менее резкой форме то же наблюдается и для бихромата калия.
В опытах с 50 г расплава интенсивное разложение нитрата на-
чиналось еще до равномерного распределения катализатора по
всей массе вещества. Следовательно, при большой массе расплава
действие катализатора (% которого меньше, но абсолютное коли-
чество больше) в значительной мере является местным; очевидно,
главным моментом для возбуждения бурного разложения NH4NO3
является создание в расплаве зоны, в которой бы имелась доста-
точно большая концентрация катализатора.
Можно предполагать, хотя по техническим причинам мы и не
смогли провести соответствующих опытов, что для полного разло-
жения десятков и даже сотен кг нагретого до 200° расплава
МН4МОз достаточно будет внести в него концентрированно в одном
месте 2—3 г вышеупомянутых катализаторов для возбуждения в
расплаве очага интенсивного разложения, которое затем распро-
странится по всей массе нитрата. Укажем также, что при введении
в нитрат аммония некоторых'веществ (например, NaNO3 или др.),
понижающих температуру его плавления, полученный расплав,
вероятно, может стать чувствительным к воздействию катализато-
ров разложения и в интервале более низких температур, а именно
ЛИТЕРАТУРА
I. Розмаи Б. Ю,, О термической стойкости аммиачной селитры. Изд. ин-
ститута инженеров иодного транспорта, Л,, 1957.
2. Р о з м а и Б. Ю., Б о р о д к и и а Л. И-, ЖПХ, J959, 32, 280, Р о з м а и Б. Ю„
ЖПХ, 1960,33, 1253.
3. Ш и д л о в с к и й А. А., Труды МХТИ, 1956, 22, 84.
4, Шндловский А, А,, Известия Высшей школы, <Химия и химическая
технология», 1958, 1, Ns 3, Ю5,
5. Hainer R, М., Fifth Symposium on Combustion, N. ¥., [955, 224.
III. ГОРЕНИЕ И ВСПЫШКА ВЗРЫВЧАТЫХ ВЕЩЕСТВ
К. К. АНДРЕЕВ
26. К ВОПРОСУ О ФАКТОРАХ, ОПРЕДЕЛЯЮЩИХ
ЗАВИСИМОСТЬ СКОРОСТИ ГОРЕНИЯ ВЗРЫВЧАТЫХ
ВЕЩЕСТВ ОТ НАЧАЛЬНОЙ ТЕМПЕРАТУРЫ И ДАВЛЕНИЯ
ОБЩИЕ СООБРАЖЕНИЯ
Известно, что для твердого горючего, применяемого в полузамк-
нутом объеме, желательна возможно малая зависимость скорости
горения от начальной температуры и давления. Если скорость го-
рения растет пропорционально давлению или быстрее, то такое
горючее не может быть использовано в указанных условиях. Зна-
чение зависимостей скорости горения от давления и от темпера-
туры наглядно видно из уравнения баланса прихода газа при го-
рении и его расхода
(П
где иы — массовая скорость горения;-
S—поверхность горящего заряда;
а — площадь критического сечения сопла;
А —коэффициент расхода;
р— давление в камере.
Если выразить, как это часто делают, зависимость скорости
горения от начальной температуры и давления эмпирическим со-
отношением
Вр*
——,
где В, А[ и Bi — постоянные, то подставляя (2) в (1), получаем
B‘P'S =Аар.
Ai — ВуТ0
Определяя из (2а) равновесное давление, имеем
(2)
(2а)
Р =
(3)
Из уравнения (3) видно, что зависимость равновесного давления от
параметров заряда и камеры определяется зависимостью скорости
горения от давления (через величину v).
404
Зависимость равновесного давления от начальной температуры
обусловливается не только зависимостью скорости горения от тем-
пературы, но и одновременно ее зависимостью,от давления. Дей-
ствительно из (3)
l _ I А. .
аТй р 1—у —BjTq
Иными словами, относительное изменение равновесного давления
при изменении начальной температуры горючего определяется как
ее влиянием на скорость горения (через второй множитель), таки
зависимостью скорости горения от давления (через первый множи-
тель) .
Представим себе два горючих, скорость горения которых оди-
наково зависит от температуры, возрастая при ее повышении на
100° в два раза; если v их различны, давление при горении в по-
лузамкнутом объеме при этом изменении температуры возрастет
в разное число раз. Если, например, vt = 0,5, a v2=0,75, то давление
увеличится соответственно в 4 и в 16 раз. Поэтому изыскание го-
рючего с малой зависимостью скорости горения от температуры
лишь в том случае представит практический интерес, если это не
приведет к повышенной зависимости" скорбстй’ГОрёния от давле-
ния. В дополнение к сказанному следует уточнить тот смысл, кото-
рый вкладывается в понятие «зависимость скорости горения от дав-
ления». Если рассматривать эту зависимость, как это сделано
выше, в обычной форме
им = ?р\ (5)
то ив двух величин, определяющих величину скорости при данном
давлении, для равновесия при горении в полузамкнутом объеме
существенна только одна. В самом деле барический коэффициент
скорости горения
. 1 1
dp ’ ик ₽/? р
при данном давлении зависит только от величины v, но не от р.
Определим теперь температурный коэффициент скорости горе-
ния. Это можно сделать двумя путями. Если принять по Беляеву и
Зельдовичу [1], что экзотермические реакции идут только или пре-
имущественно в газовой фазе, и что скорость гсфения соответствен-
но' определяется "реакцией, идущей при температуре, практически
равной температуре горения, то зависимость скорости от темпера-
туры горения, а через нее и от начальной температуры, может быть
выражена соотношением
(7)
где Тт — температура горения, связанная с начальной температу-
рой Го соотношением
(8)
ср
405
в котором Q — тепловой эффект горения, а ср—средняя теплоем-
кость продуктов горения от То до Тг> и k — постоянные, причем
значение k зависит от порядка реакции и для мономолекулярной
реакции равно 3/г. Из (7) имеем
1 1 /. , 1 Е \ ,,,,
—=-----=----- -----------). (9)
ДГг вм ГД 1 2/? Гг / ' '
Из (9) видно, что температурный коэффициент скорости горения
с ростом начальной температуры должен уменьшаться, хотя и не-
значительно При этом он тем меньше, чем выше температура го-
рения и чем меньше энергия активации ведущей реакции. Это же
означает, что для веществ с большой скоростью горения указан-
ный коэффициент должен быть меньше, чем для веществ с малой
скоростью, так как большая скорость горения означает высокую
температуру протекания ведущей реакции или малую энергию ее
активации, или то и другое вместе.
Второй путь оценки зависимости скорости горения от темпера-
туры1 основан на том, чтобы, также следуя Зельдовичу [5], принять,
что скорость горения определяется скоростью распространения теп-
ловой волны, создаваемой реакцией в конденсированной фазе;
тогда ее зависимость от температуры выразится соотношением
и
(10)
где и —скорость горения, равная скорости тепловой волны;
р'—плотность конденсированной фазы;
с'— ее теплоемкость;
7К — максимальная температура, до которой может быть нагрета
конденсированная фаза, в случае летучих веществ — тем-
пература кипения;
т/—коэффициент теплопроводности конденсированной фазы;
Q'— тепловой эффект реакции;
А'— предэкспоненциальный множитель н выражении для скорости
реакции в конденсированной фазе;
Е’— энергия активации реакции в конденсированной фазе,
Обозначая
2ri'Q' А'е~£г^Гк через <р,
получим зависимость скорости горения от начальной температуры
в виде
(Юа)
' к — 2 О
Это выражение сходно с тем, которое вытекает из гипотезы Ми-
1 Изменение начальной температуры не может быть большим по сравнению
с температурой горения,
406
хельсона и Малляра—Ле-Шателье [7]; они, как известно, рассматри-
вали распространение горения как следствие разогрева газообраз-
ными продуктами горения непрореагировавшего вещества до неко-
торой критической температуры, аналогичной температуре вспыш-
ки. Обоснованная и конструктивная критика этой гипотезы как
универсального механизма горения дана в работах Я- Б. Зельдо-
вича и Д. А. Франк-Кдмеиенкого Е4],
Реальность первого пли второго механизма Зельдовича в кон-
кретном случае определяется свойствами вещества и условиями, в
которых идет горение. Если вещество мало летуче, то это благо-
приятствует протеканию реакции в конденсированной фазе. Ско-
рость реакции в газовой фазе зависит от давления, под которым
идет горение, так как она пропорциональна давлению в первой
(мономолекулярные реакции) или более высокой степени. По этой
причине можно представить себе такой случай, когда при низких
давлениях преобладающее значение имеют процессы в конденсиро-
ванной фазе, а при более высоких — в газовой,
По-видимому именно такой случай представляет собой горе-
ние унитарных горючих на основе нитроклетчатки и жид^щс нитро-
эфиров. При очень низких давлениях скбростьторения не зависит
от давления 16]. Это показывает, что газофазная реакция не являет-
ся ведущей в этих условиях.
В области умеренных давлений (атмосферы) скорость горения
столь сильно зависит от температуры, что при расчете по формуле
(7) это приводит к нереально большим значениям энергии актива-
ции. Наряду с этим температурный коэффициент скорости горения
возрастает с увеличением начальной температуры, что находится
в противоречии с зависимостью (7) и согласуется с выражением
(10а). Наконец, температурный коэффициент скорости зависит от
давления, уменьшаясь с его увеличением, что опять-таки нельзя
согласовать с зависимостью (7). Все это заставляет предполагать,
что при низких давлениях, особенно, если начальная температура
горючего высока, скорость горения определяется реакцией в кон-
денсированной фаз?, а при высоких — в основном в газовой,
В промежуточной области распространение горения определяет-
ся совокупностью процессов, происходящих в обеих фазах; чем
ниже давление, тем большую роль играет превращение в конденси-
рованной фазе и наоборот. Для тех условий, когда справедливо
соотношение (10а), температурный коэффициент скорости горения
определяется выражением
=----1---. (11)
dTa ч 7\ — То
АНАЛИЗ ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ
Попытаемся сопоставить экспериментальные данные с рассмот-
ренными в предыдущем разделе общими соображениями.
В связи с тем, что существующая теория Зельдовича—Беляева
рассматривает горение индивидуальных химических соединений, за
407
основу прн этом сопоставлении мы взяли данные, полученные для
ряда ВВ, принадлежащих к тем же классам химических веществ
(нитроэфиры и нитросоединения), к которым относятся основные
компоненты современных унитарных горючих. Далее при атмо-
сферном и умеренно повышенных давлениях процесс горения не-
редко может осложняться побочными не учитываемыми влияниями
(теплопотери во вне, переменная неполнота реакции, удаленность
зоны максимальной температуры от горящей поверхности и др.).
Поэтому основные определения, относящиеся к установлению зави-
симости скорости горения от давления, были проведены при давле-
ниях, лежащих выше 50 и во многих случаях до 1000 ат.
Основной задачей сопоставления экспериментальных результа-
тов с выводами теории была проверка справедливости последней-
Если бы удалось подтвердить эту справедливость хотя в некоторых
простейших условиях, например, при высоких давлениях и для ин-
дивидуальных соеяппший. то тем саЯым теория получила бы более
прочную опору, чем она сегодня ее имеет, и это позволило бы сде-
лать следующий шаг к описанию горения многокомпонентных си-
стем и к учету тех нарушений, которые обусловливаются пониже-
нием давления.
Результаты исследования зависимости скорости горения от дав-
ления для ряда изученных веществ приведены в табл. 1 и графи-
чески представлены на фиг. 1.
Таблица 1
Зависимость скорости горения некоторых взрывчатых веществ
от давления
Взрывчатое вещество Интервал давление ат Значения коэффициентов в формуле Им=Л ьВр’ * Скорость горения им при 100 ат г!сл^сек
А В V
Тротил 20—950 0,04 0,00716 1 0,76
Пикриновая кислота 25—950 0,14 0,00805 1 0,95
Тетрил 10—250 0,0 0,0630 0,695 1,55
Г ексоген 100— 100С 0,90 0,0216 1 3,06
Дина 20—100 0,12 0,00912 1 1,02
Тэн 15—750 0,0 0,0193 1 1,93
Нитроглнколь желатини- рованный (97:3) 1—150 0,075 0,0315 1 3,22
Нитроглицерин желатини- рованный (95 • 5) 10—320 0.8 0,0592 1 6,72
* ии выражено а г.см^сек, а р в кг!см2.
Мы видим, что для почти всех веществ зависимость скорости
горения от давления в изученном интервале последнего близка к
408
линейной; при этом во многих случаях особенно для летучих и мед-
ленно разлагающихся веществ член А в выражении им = Д + Вр’
близок к нулю, a v близко к 1, так что указанная зависимость пре-
вращается в прямую пропорциональность. С точки зрения теории
такой результат означает, что ведущ&йпртгторении является бимо-
лекулярная реакция. Он же показывает, что по крайней мере в
изученном интервале давлений в большинстве своем эти взрывча-
тые вещества сами по себе мало пригодны для устойчивого горе-
ния в полузамкнутом объеме.
При давлении 100 ат скорости горения приведенных в табл. I
веществ различаются максимально в 9 раз. Наименьшую скорость
имеет тротил (0,76 г/см2сек), наибольшую (6,7 г/см2сек) желатини-
рованный нитроглицерин, характеризующийся высокой теплотой
горения. .
В связи с этим возникает вопрос, нельзя ли как основной пара-1
метр, определяющий величину скорости горения ВВ, рассматривать
теплоту (или температуру) его горения. Известно, что в порохове-
Фиг. 1. Зависимость скорости горения некоторых
взрывчатых веществ от давления.
/—тротил, 2—пикриновая кислота, 3—тетрил, 4—гэн,
5—гексоген, 6—желатинированный нитроглицерин-
дении такая связь для баллиститных порохов принимается как
у нас, так и за рубежом. У нас она обычно выражается формулой
Особого технического бюро (ОТБ) [2]
«1= (0,16<2ж—32) • Ю’4,
где иг—расчетная скорость горения при 1 ат в г/см2сек, а
Q®—теплота горения при воде жидкой в больших калориях
на 1 кг пороха.
409
Если расчетную скорость отнести к давлению, равному
100 кг! см2, то формула примет вид
и100«(0,16рж-32) • 10-2.
Мюраур [9] на основании своих многочисленных исследований,
которые привели его к формуле, выражающей зависимость скоро-
сти горения от давления
и = а/2 + 7>/2р,
установил, что коэффициент b (скорость горения выражена в
мм/сек) связан с температурой горения соотношением
lg(1000fr) =1,214 + 0,30877/1000 или
lg fr/2- —2,087+0,30877/1000.
Таким образом в обоих случаях предполагается, что скорость
горения зависит только от теплоты, выделяющейся при горении,
или температуры, при нем достигаемой, но не от кинетики химиче-
ского превращения.
Это допущение может быть истолковано по-разному. Так, мож-
но себе представить, что промежуточные реакции различны, но ве-
Фиг. 2. Зависимость скорости горения
некоторых взрывчатых веществ от его
расчетной теплоты.
Фиг. 3. Зависимость скорости горения
некоторых взрывчатых вешеств от его
расчетной температуры-
дущая реакция одна и та же, например, взаимодействие между
окисью азота и окисью углерода. В таком случае скорость распро-
странения превращения действительно зависела бы лишь от темпе-
ратуры, которая при нем достигается, Можно было бы допустить
также, что ведущим является превращение какого-либо одного из
основных компонентов, другие же играют роль разбавителей и на
скорости процесса отражаются лишь в той мере, в которой их при-
сутствие изменяет температуру горения, Не исключено было, нако-
нец, что формулы ОТБ и Мюраура имеют приближенный характер
и отражают лишь тот факт, что скорость реакций при горении уве-
личивается с температурой. Сопоставлением теплот, температур и
скоростей горения взрывчатых веществ, относящихся к различным
классам химических соединений, можно было выяснить, какое из
НО
приведенных допущений справедливо. В табл, 2 и на графике
фиг, 2 приведены для некоторых ВВ значения теплот горения н
скоростей последнего при 100 ат.
Мы видим, что для пяти веществ — питрогликоля, гексогена,
тетрила, пикриновой кислоты и тротила зависимость между ско-
ростью горения и теплотой близка к линейной и может быть вы-
ражена соотношением «юо= (0,285Q—124) • 10“2, Значительно ниже
прямой лежат точки для тэна и дины. На фиг. 3 и 4 показаны за-
висимости и lguM=/(7r). Первая из них естественно ана-
логична зависимости от Qp.w- Точки, выражающие вторую зави-
симость, могут быть уложены на две параллельных прямых;
1 :lguioo=—1,115+0,470^/1000 и 2: lg и100—1,495+0.470fr/] 000.
Таким образом из сопоставления экспериментальных данных с
обеими формами эмпирической зависимости скорости горения от
теплового его эффекта вытекает, что эти зависимости не универ-
сальны, имеют приближенный характер и отражают лишь тот
Таблица 2
Теплота, температура и скорость горения некоторых взрывчатых веществ
Взрывчатое вещество Теплота горения Qp.« при 100 ат по расчету при воде жидкой ккал!кг Температура горения ТГ при посто- янном давлении по расчету °К Массовая скорость горения и„ при 100 ат г/сМ'Сек Температура горения 7* г.л с учетом диссоциации
Тротил 638 1980 0,76 1980
Пикриновая кислота 8С6 2475 0,95 2475
Тетрил 9.9 2775 1,55 2775
v Гексоген 1352 3510 3,06 3325
Лица 1270 3090 1,02 3080
' Тэн 1525 3880 1,93 3355
Нитрогликоль жедатинп- рованньй (97; 3) 1750 4250 3,2 3360
Нитроглицерин желатини- рованный (95; 5) 1610 4115 6,7 3330
факт, что скорость горения, вообще говоря, растет с увеличением
температуры последнего.
Зависимость скорости горения от температуры описывается и
теорией горения Беляева—Зельдовича, Представляло интерес вы-
яснить, не будет ли эта зависимость лучше согласовываться с фак-
тами, График фиг, 5 дает ее в координатах 1g
I
т '
J г
Мы видим, что общий характер этой зависимости аналогичен
рассмотренным раньше. Изученные вещества могут быть уложены
на две прямые, причем для нитроэфнров тангенс .угла наклона пря-
мой больше, чем для нитросоединений. Следовательно, и это сопо-
ставление подтверждает сделанный выше вывод о приближенном
характере связи между теплотой и скоростью горения даже для
однотипных по характеру горения ВВ, а также иллюстрирует
сходство всех рассмотренных зависимостей.
Таким образом, не только температура горения, но и кинетика
химического превращения является существенным фактором, оп-
ределяющим величину скорости горения. Это заключение стано-
вится еще более убедительным, если для ряда веществ температу-
ру горения рассчитать с учетом диссоциации. Во внутренней бал-
листике ствольной артиллерии этого обычно не делается, так как
мы имеем дело с большими давлениями, где диссоциацией можно
пренебречь. Для условий баллистики полузамкнутого объема, в
частности, для тех давлений, к которым относятся рассматривае-
мые нами результаты, учет диссоциации приводит к существенному
изменению температуры горения; это изменение тем больше, чем
выше температура горения и чем больше кислородный баланс ВВ.
В табл. 2 приведены значения температуры горения для восьми
взрывчатых веществ без учета и с учетом диссоциации. Мы видим,
что в последнем случае температуры
гликоля и нитроглицерина очень
3340 + 20° К, тогда как скорости
горения некоторых из них раз-
личаются в 2—3 раза.
для гексогена, тэна, нитро-
близки и находятся в пределах
фиг. 5. Зависимость скорости горения
некоторых взрывчатых веществ от его
расчетной температуры в координатах
Фиг. 4. Зависимость скорости горения
некоторых взрывчатых веществ от его
расчетной температуры в координатах
lg U«—Tt.
Вывод о том, что скорость горения летучего взрывчатого ве- ’
щества зависит не только от температуры горения, не противоречит
теории Беляева—Зельдовича, которая как второй существенный
412
фактор данной зависимости рассматривает энергию активации хи-
мического превращения при горении. Однако, если различие в ско-(
рости этого процесса для близких по температуре горения взрыв-
чатых веществ определяется различиями в энергии активации, то
это должно проявиться и в различии зависимости скорости горе-
ния от температуры. Чтобы проверить этот вывод, были проведены
опыты по определению зависимости скорости горения двух взрыв-
чатых веществ (тетрила и тэна) от температуры горения. Измене-
ние последней может осуществляться двумя путями— изменением
начальной температуры взрывчатого вещества ^ТГ = ТО+ или
разбавлением его инертной примесью. Последний путь допускает
изменение температуры горения в более широких пределах, но вы-
бор примеси, которая была бы действительно инертной, затрудни-
телен и может быть подтвержден лишь сопоставлением данных,
полученных обоими путями. В рамках настоящей работы был реа-
лизован лишь второй путь с-применением для теплового разбавле-
ния взрывчатого вещества воды, являющейся одним из продуктов
горения и химически достаточно инертным веществом.1
Результаты' опытов даны в табл. 3, а результаты расчетов, про-
веденных на основе этих данных, представлены на фиг. 6.
Таблица 3
Влияние воды на скорость горения тетрила и тэна
при давлении 10) ат
Содержание воды
в % по весу
г!см2сёк
Тг °К
по расчету
Примечание
Тетрил (по Поясову)
0 0 1,55 1,56 2795
2.38 1,32 2675 Плотность образчиков
2.68 1,35 2640 была в пределах 1,21— 1,35 г!емл
4.38 1,20 253.5
5,98 1.12 2485
Тэн (по Поповой)
0 1,93 3880
1.63 1,75 3775 Плотность образчиков заключалась в пределах
2,49 1,60 3735
3,62 1,39 3645 1,2—1,3 г!см^
5,40 1,18 353.5
Мы видим, что, во-первых, изменение скорости горения с рас-
четной его температурой в координатах 1g и2]Т^ — 1 /Тт изображает -
' Эти опыты были проведены студентом В. А. Попсовым (тетрил) и инжене-
ром П. П. Поповой (тэн),
413
18,0
11,3
ИЛ
/Д7
11,5
''ЦЧ
2,5 2,7 W 3# 3,8 ‘i.Of-IO*
Фиг. 6. Зависимость скорости
горения тетрила и тэна от рас
четной его температуры, регу-
лируемой содержанием воды.
I—тетрил, 2—тэн.
ся прямой линией. Таким образом это требование теории выпол-
няется. Во-вторых, по наклону этой прямой можно вычислить
энергию активизации для данного процесса. Для тетрила Она
составляет около 7 ккал/моль, если
считать реакцию бимолекулярной и
принять, что скорость ее пропорцио-
нальна квадрату концентрации. Для
тэна аналогично получается около
47 ккал/моль. Мы видели выше, что
если судить по температурной шкале,
тэн относится к медленногорящим
веществам. Результаты определения
„ зависимости скорости горения от его
. температуры согласуются с этим выво-
дом. При прочих равных условиях
большая энергия активации означает
меньшую скорость горения. Добавим
также, что относительно низкая ско-
рость горения тэна (и дины) показы-
вает, что скорости горения не изменя-
ются параллельно скоростям медленного термического распада,
так как тэн, равно как и дийа, разлагается значительно быстрее,
чем нитросоединения или нитрамины ароматического ряда; при
этом у нитроэфиров выше и расчетные температуры их горения.
О теоретической связи между температурой
(ТЕПЛОТОЙ ГОРЕНИЯ) И ЗАВИСИМОСТЬЮ СКОРОСТИ ГОРЕНИЯ
ОТ НАЧАЛЬНОЙ ТЕМПЕРАТУРЫ И ОТ ДАВЛЕНИЯ
В связи с тем, что в рамках проведенных хотя и ограниченных
исследований наблюдавшиеся закономерности горения не проти-
воречат теоретическим, мы имеем основание рассматривать и дру-
гие интересующие технику зависимости, исходя из существующей
теории. С этих позиций и рассмотрим вопрос о температурном ко-
эффициенте скорости горения и его зависимости от теплоты горе-
ния. Обычно считают, что высококалорийным твердым унитарным
горючим свойственна большая зависимость скорости горения от
температуры, чем горючим меньшей калорийности. Этот вывод сде-
лан на основе практического опыта, полученною с горючими, кало-
рийность которых менялась путем изменения содержания нитро-
глицерина. Выше мы видели, что температурный" коэффициент»
связан с температурой зависимостью (9)
. -L=J- (k _l _L. 2L \
Tr \ 2R TT /
из которой следует, что он тем меньше, чем выше температура го-
рения.
В свете этих данных естественно предположить, что увеличе-
ние температурной зависимости скорости горения горючих на ос-
нове нитроглицерина при увеличении содержания последнего обу-
414
словливается не увеличением собственно калорийности смеси, а ки-
нетическими особенностями (большая энергия активации Е) нит-
роглицерина. Если это единственное противоречие с теорией может
быть таким образом устранено, то сохраняется, как вывод из тео-
рии, положение, что «горячие» горючие должны проявлять мень-
шую зависимость скорости горения Ът начальной температуры, чём'
«холодные»! Экспериментальная'проверка этого заключения предо-
ставляет несомненный интерес. Если оно подтвердится, можно бу-
дет ориентироваться на поиски таких веществ, которые сочетали
бы большую теплоту горения с малой его энергией активации. До-
бавим, что такие вещества одновременно были бы относительно
быстрогоращтгит-так как согласно теории сочетание высокой тем-
пературы горения и малой энергии активаций должно (при прочих
равных .условиях) привбдить“к'больщбй~сК<3'рстсти горения.
Коснемся теперь вопроса о зависимости скорости—горения от
давления. Теория связывает его с порядком п ведущей реакции,
устанавливая пропорциональность скорости горения давлению в
степени «/2. Определенная опытом при повышенных давлениях
пропорциональность скорости горения первой степени давления мо.-
жет быть истолкована как следствие бимолекулярноСти ведущей"
реакции. Однако тот же опыт показывает, что при данлщшяхшиже
100 ат скорость горения иногда сильнее или слабее зависит от дав-
ления. Первое может быть связано с изменением полноты реакции
при низких давлениях вблизи того, при котором становится воз-
можным горение; второе — вблизи начала линейного участка из-
менения скорости горения—с возможностью частичного протека-
ния реакции в конденсированной фазе.
Когда экзотермическая реакция в значительном интервале дав-
ления идет в заметной степени в конденсированной фазе, как это,
например, происходит при горении нитроклетчатки, скорость горе-
ния растет с давлением медленнее, чем по закону прямой про-
порциональности. Наконец, можно представить себе в качестве ве-
дущей и реакцию первого порядка, как это, судя по данным англий-
ских исследований [8], имеет место при горении гидразина. Ради-
кальным путем для снижения зависимости скорости горения от
давления является использование веществ вроде пикрата калия, £
скорость горения которых от давления не зависит, по-видимому, з
потому, что ведущая реакция в этом случае идет в конденсирован-т
ной фазе. При известных предпосылках при таком характере горе-И
ния мала зависимость скорости горения и от начальной температуры.
Опыты, результаты которых использованы в настоящей статье,
были проведены А. П. Глазковой, И. А. Терешкиным 13] и П. П. По-
повой, большая часть расчетов—И. А. Терешкиным. Автор выра-
жает им свою признательность.
ВЫВОДЫ
1. Зависимость скорости горения от давления для ряда вторич-
ных взрывчатых веществ, принадлежащих к классу нитроэфиров и
нитросоединений, близка к прямой пропорциональности. Отсюда
следует, согласно теории Зельдовича—Беляева, что ведущая реак-
ция при горении веществ этого класса является бимолекулярной.
2. Скорость горения ряда взрывчатых веществ может быть при-
ближенно связана с теплотой и температурой горения линейными
зависимостями. Подобная же зависимость получается в координа-
тах lg«s/Tr3--
4 г
3. Количественный анализ последней зависимости показывает,
что она имеет, как и первые две зависимости, эмпирический и при-
ближенный характер.
4. Показано методом разбавления инертной примесью на двух
веществах (тэн и тетрил), что зависимость скорости горения отего
расчетной температуры одинакова по характеру и согласуется с
теоретической, но количественно является различной: более силь-
ной для тэна, чем для тетрила.
ЛИТЕРАТУРА
L Беляев А,Ф„ ЖФХ, 1938, 12, 93; 1940, 14, 1009.
2. См. напр. Будников М. А., Левкович Н. А., Быстров И. В., С и-
ротинский В. Ф. и Шехтер Б. И., Взрывчатые вещества и пороха. М...
Оборонгнз, 1955, стр. 330.
3. Глазкова А. П. и Терешкин И. А., ЖФХ, 1961, 35, 1623.
4. Зельдович Я. Б. и Франк-Каменецкий Д. А., ЖФХ, 1938,
12, 100.
5. Зельдович Я. Б., ЖЭТ Ф, [942. 12, 498.
6. Похил П. Ф., Сб. «Физика взрыва», АН СССР, 1953, № 2, стр. 192.
7. См. напр. Харитон Ю. Б., Дополнительная статьи в книге Г. Бруисвига,
Теория взрывчатых веществ, М-, Госхнмиздат, 1932, стр. 245.
8. Adams G. К. and Stocks G. W_, Fourth Symposium on Combustion,
Baltimore, The Williams and Williams Co., 1953, p. 239.
9. Muraour H.f Cahiers de physique, premiere serie, 1942,p.29.
X. К. АНДРЕЕВ
27. О ГОРЕНИИ НИТРАТОВ НЕКОТОРЫХ СПИРТОВ
Горение взрывчатых веществ обычно вызывается их поджигани-
ем. Оно может возникнуть также при местном разложении, если
последнее приводит к разогреву, достаточно сильному для того,
чтобы он закончился локальным самовоспламенением. Характер
возникшего горения и степень его устойчивости зависят от свойств
вещества и условий горения.
Для нитратов спиртов типичные особенности процесса горения
были установлены в основном при исследовании нитрогликоля
(НГЛ) и нитроглицерина (НГЦ). Результаты этого исследования и
их объяснение рассматриваются в настоящей статье.
ПРЕДЕЛЬНЫЕ УСЛОВИЯ НОРМАЛЬНОГО (НЕВОЗМУЩЕННОГО)
ГОРЕНИЯ
Опыты, поставленные по обычной методике (горение жидкости,
налитой в узкую стеклянную пробирку в приборе постоянного дав-
ления с окнами, фотографическая регистрация характера и скоро-
416
сти распространения процесса), показали, что горение возможно
лишь если диаметр заряда, давление и температура не слишком
малы [3], Влияние этих параметров определяется тем, что при слиш-
ком низком давлении и температуре скорость химических реакций,
а следовательно, и тепловыделения оказывается недостаточной,
чтобы компенсировать при малом диаметре теплопотери продуктов
горения и прогретого слоя вещества во вне. Повышение скорости
реакций посредством увеличения давления или температуры, а так-
же уменьшение относительных теплопотерь путем увеличения диа-
метра н повышения начальной температуры вещества устраняют
преобладание теплопотерь над теплоприходом и горение становит-
ся возможным. Так, если ограничиться давлением, то НГЛ в про-
бирках диаметром 4 лии при комнатной температуре горит, начи-
ная с давления 300 мм рт. ст., а НГЦ в аналогичных условиях —
при давлении 24 мм и выше.
ПЕРЕХОД НОРМАЛЬНОГО ГОРЕНИЯ НИТРОГЛИКОЛЯ
НА турбулентный режим
При увеличении давления скорость горения НГЛ растет прибли-
женно пропорционально давлению подобно тому как это наблю-
дается для многих твердых веществ, например, для тринитротолу-
ола, тринитрофенола и др. Однако в то время как для тринитрото-
луола и пикриновой кислоты эта зависимость выполняется во всем
большом изученном интервале дав-
ления (до 1000 ат), для НГЛ ско-
рость горения растет пропорцио-
нально давлению лишь приблизи-
тельно до 20 ат [4]. При этом дав-
лении характер горения .меняется;
поверхность жидкости перестает
быть ровной и горизонтальной и пе-
риодически искажается; процесс
принимает пульсирующий характер
и представляет собой как бы после-
довательность чередующихся вспы-
шек, разделенных периодами замед-
фнг. 1. Зависимость скорости горе-
ния нитрогликоля от давления.
/—жидкость^ 2—цйтроглнколь, жела-
тинированный коллоксилином (97 ; 3).
ленного горения,
Переход на пульсирующий ре-
жим сопровождается сильным (при-
. близительно в десять раз) увеличе-
нием средней скорости горения; в
переходной области значения скорости сильно колеблются. При
дальнейшем увеличении давления скорость растет линейно и го-
раздо быстрее, чем на нормальном режиме (фиг. 1), и неравномер-
ность горения не улавливается.
Следует указать еще на одну особенность горения, проявляю-
щуюся с ростом давления. При низких давлениях горение идет со
слабым свечением, прилегающим к поверхности жидкости, причем
пламя имеет обычную форму — конуса, обращенного вершиной
27 451
417
3. Зависимость скорости горения
^гликоля от давления при разных
температурах.
вверх. При давлении около 10 ат (величина этого давления умень-
шается при увеличении диаметра трубки) на значительном (около
2 см) расстоянии от поверхности жидкости появляется яркое голу-
боватое пламя в виде конуса, обращенного вершиной вниз. Возник-
новение вторичного пламени сопровождается резким изменением
состава газов — окись азота полностью восстанавливается до азо-
та. Это ведет к сильному (приблизительно вдвое) увеличению теп-
лоты и, соответственно, температуры горения. Появление горячего
вторичного пламени не отражается, однако, ни на скорости, ни на
характере горения. При дальнейшем повышении давления расстоя-
ние между первичным и вторичным пламенем уменьшается и при
60 и более атмосферах становится неразличимым.
Описанные особенности иллюстрируются фотоснимками фиг. 2.
Особенно отчетливо пульсация видна на них благодаря наличию
яркого вторичного пламени
(фиг. 2, фото 3, а). В тот момент, vc
когда происходит искажение по-
верхности, прогретый слой веще-
ства охлаждается и испарение
замедляется. В результате этого
вторичное пламя приближается к
поверхности жидкости, а затем
отбрасывается от нее, когда про-
исходит бурное разложение в об-
разовавшемся, утолщенном про-
тив нормального, прогретом слое.
Повышение начальной темпе- j
ратуры, увеличивающее скорость
нормального горения, уменьшает
давление, при котором появляет-
ся турбулентный режим и усили-
вает зависимость скорости горе- ?
ния от давления на этом режиме
(фиг. 3).
Метилнитрат и диэтиленглп- О
кольдинитрат ведут себя анало-
гично НГЛ. Для первого из них, и
горящего с относительно большой
скоростью (0,14 г/см2сек при
I ат) переход на турбулентный режим происходит при давлении око-
ло 2 ат, для медленно горящего дигликольди нитрата это наблю-
дается примерно при 55 ат. При этом же давлении переходит на
пульсирующий режим и горение жидкой переохлажденной до ком-
натной температуры дины., скорость горения которой также отно-
сительно мала.
НАРУШЕНИЕ НОРМАЛЬНОГО РЕЖИМА ГОРЕНИЯ НИТРОГЛИЦЕРИНА
При горении НГЦ наблюдаются явления, сходные с описанны-
ми выше, но с некоторыми количественными и качественными от-
личиями [2]. При давлении выше 24 мм рт. ст. скорость горения
27* 419
418
НГЦ, значительно превышающая скорость горения НГЛ, медленно
растет с давлением; при давлении около 1 ат при поджигании на-
блюдается нечто вроде локальной вспышки, сопровождающейся
расплескиванием жидкости и горения в этом случае не возникает.
Более того, если НГЦ зажжен в трубке диаметром около 4 мм при
низком давлении и горение идет в замкнутом объеме так, что дав-
ление растет, то по достижении около 300 мм рт. ст. горение само-
произвольно затухает. Давление затухания сильно зависит, как
показали опыты Г. Н. Беспалова от диаметра трубки. При диамет-
ре 2,6 мм горение затухало уже при 150 мм рт. ст. При диаметре
трубки 10 мм горение происходило до давления 1050 мм рт. ст.
Таким образом можно наблюдать горение НГЦ и при атмосферном
давлении. Однако этого удалось достигнуть только при одном из
разнообразных изучавшихся способов поджигания — поджигании
Фиг. 4. Зависимость скорости горения
нитроглицерина от давления.
при пониженном давлении с последующим медленным ростом дав-
ления до атмосферного. Примене-
ние других способов вызывало
турбулизацию и как . следствие
этого затухание горения. Ско-
рость горения НГЦ при атмо-
сферном давлении составляет
0,146 см/сек. или 0,23 г/см? сек.
Из сказанного следует, что в
случае НГЦ повышение давле-
ния, увеличивающее скорость го-
рения, не только не способствует
последнему, но оказывает обрат-
ное действие. Это действие одна-
ко ограничивается некоторым ин-
тервалом давления: выше 11 ат
вновь наблюдается горение [6],
но идет оно уже, подобно горе-
нию НГЛ, на турбулентном режиме со скоростью постоянной, но
гораздо большей, чем это соответствовало бы нормальному горе-
нию (фиг. 4). Своеобразное явление наблюдалось яри некоторых
опытах с НГЦ при .повышенной температуре (около 100°): пламя
как бы проникало вдоль одной стороны стенки трубки на большую
глубину и поверхность горящей жидкости становилась почти верти-
кальной. Возможно, что это явление представляет собой пульсацию
очень большого периода.
Таким образом, основные отличия НГЦ от НГЛ заключаются,
во-первых, в большей скорости горения, во-вторых, в том, что тур-
булентность последнего возникает у НГЦ при меньшем давлении
(1/2 ат), при котором еще нет вторичного пламени,'и, в-третьих,
в том, что эта турбулентность приводит не к ускоренному пульси-
рующему горению, а к затуханию. Непосредственной причиной за-
тухания является, по-вндимому, резкое усиление теплопотерь при
переходе горения на турбулентный режим, что приводит к силь-
1 См. настоящий сборник стр. 432.
420
ному увеличению критического диаметра. Опыты Г. Н. Беспалова
показали, что при турбулентном режиме, несмотря на большую
скорость горения критический диаметр гораздо больше, чем при
нормальном горении и быстро растет при понижении давления.
Если экстраполировать полученную зависимость dtI=/(p) на одну
атмосферу, то значение критического диаметра получается равным
-—15 см. Если уменьшить турбулизацию увеличением вязкости нит->
роглицерина путем растворения в нем нитроклетчатки, то критиче-
ский диаметр сильно снижается, например, при 15 ат с 4 до 0,5 мм.
Турбулизацию можно предотвратить также уменьшением диа-
метра трубки. Оказалось, что при атмосферном давлении возмож-
ность горения нитроглицерина можно обеспечить не только увели-
чивая диаметр трубки, но и применяя малые диаметры ее (от 0,5
до 1,5 лтл1), несмотря на то, что при малых диаметрах удельная
поверхность теплоотвода увеличивается. Это опять-таки показы-
вает, что основным фактором увеличения теплопотерь при затуха-
нии горения НГЦ в области низких давлений является возникнове»
ние турбулизации.
Применив трубки, достаточно большого диаметра, например,
20 см, можно было бы, по-видимому, и для НГЦ получить, подобно
тому как это наблюдалось для НГЛ, горение при всех давлениях,
начиная от нижнего предела, равного нескольким миллиметрам
ртутного столба, сначала нормальное, а затем турбулентное. Про-
верка этого заключения не осуществлена, так как она потребова-
ла бы применения слишком больших, опасных в условиях лабора-
тории, количеств НГЦ.
НАРУШЕНИЕ НОРМАЛЬНОГО РЕЖИМА ГОРЕНИЯ
ПЕНТАЭРИТРИТТЕТРАНИТРАТА (ТЭНА)
Возникновение турбулентности во фронте горения естественно
представить себе в случае горения жидкостей. Однако, если горит
твердое плавящееся вещество, то часть прифронтового прогретого
слоя находится в жидком состоянии. Опыты, проведенные с тэном,
показали, что при низких давлениях, когда толщина прогретого
слоя и расплавленной его части относительно велика, влияние тур-
булизации может проявиться. В определенных условиях опыта
(а! = 7 мм, f°= 100°) тэн способен гореть при атмосферном давлении.
Однако при повышении давления до 1,5 ат горение прекращается.
Способность к горению у этого ВВ появляется вновь лишь при
значительно больших давлениях — с 16 ат.
Затухание горения в интервале давления 1,5—16 ат можно свя-
зать с наступлением турбулизации в расплавленной части прогре-
того слоя. При относительно малой скорости горения твердого ве-
щества (0,031 г1см?сек при атмосферном давлении) толщина про-
гретого слоя сравнительно велика. В этих условиях тэн ведет себя
аналогично жидкому НГЦ. Отличие состоит в том, что при повы-
шенных^ давлениях, когда НГЦ горит на турбулентном режиме с
большой скоростью, для твердого тэна повышенной скорости, соот-
ветствующей турбулентному режиму, не наблюдается. Это неуди-
421
вительно, так как при больших давлениях, точнее прн соответству-
ющих им больших скоростях горения, толщина прогретого расплав-
ленного слоя у тэна становится столь малой, что турбулентность
не может в нем развиться.
Опыты с жидким тэном при 140° показали, что при давлении
около 8 ат горение его переходит на турбулентный режим с соот-
ветствующим сильным увеличением скорости.
Следует добавить, что как в случае НГЦ, так и в случае тэна
затухание наблюдается при относительно малых скоростях горе-
ния. Возможно, что турбулизация в пределах прогретого слоя на-
чинается раньше, чем она распространяется на холодные слои
жидкости и проявляется в макроперавномерности процесса горе-
ния.
ВЛИЯНИЕ ПОДВИЖНОСТИ ЖИДКОСТИ НА НАРУШЕНИЕ
НОРМАЛЬНОГО РЕЖИМА ГОРЕНИЯ
Уменьшение подвижности жидкости, в частности увеличение ее
вязкости путем растворения в ней небольшого количества высоко-
полимерного вещества (например нитроклетчатки или полиметил-
метакрилата) затрудняет появление турбулентности и соответст-
венно более или менее значительно увеличивает интервал давления,
в котором наблюдается нормальное горение. Так, для НГЛ, жела-
тинированного коллоксилином (97:3), — см. фиг. 1—нормальное
горение наблюдалось во всем изучавшемся интервале давления (до
150 ат), в то время как горение жидкого НГЛ переходило на тур-
булентный режим уже при 20 ат.
Так же влияет желатинизация НГЦ на способность его к горе-
нию. Чем сильнее повышается вязкость, тем выше то давление, при
котором это ВВ утрачивает способность к горению; при значитель-
ном ее повышении, интервала давления, в котором вещество не
способно к горению, не наблюдается.
Интересно, что увеличение вязкости жидкости приводит к неко-
торому уменьшению скорости горения, тем более значительному,
чем сильнее повышена вязкость и, по-видимому, чем больше ско-
рость горения жидкости. Это снижение скорости наблюдается и в
том случае, если полимер (нитроклетчатка) имеет большую ско-
рость горения, чем жидкость, в которой его растворяют,
Не исключено, что в случае маловязкой жидкости вследствие
кипения поверхность фронта горения приобретает некоторый мик-
рорельеф, выраженный в большей степени, чем у вязкой желатины.
Возможно, однако, также, что в изучавшихся условиях горения су-
щественную роль играет диффузия в конденсированной фазе1, ско-
рость которой уменьшается с увеличением вязкости.
Нарушение нормального режима горения и стабилизация его
повышением вязкости наблюдались не только для индивидуальных
1 Вследствие преимущественного отшеплеиия нитроэфиром окислов азота и rte
рехода нк в газовую фазу горящая поверхность жидкости обедняется кислородом,
который поступает к ней путем диффузии окислов азота, выделяемых и глубже
расположенных слоях вещества.
422
жидких веществ, в частности нитратов спиртов. Уиттакер НО] и др.
описывают это явление для многих двух- и поликомпонентных рас-
творов горючих (2-нитропропан, нитрил себациновой кислоты) в
окислителях (азотная кислота, четырехокись азота) и связывают
его с увлечением при испарении горящей жидкости ее капелек от-
текающим паром. Этими исследователями изучалось влияние вяз-
кости, упругости паров горючего, диаметра и формы трубки. Уве-
личение вязкости, достигавшееся растворением полимстилметакри-
лата, уменьшало скорость горения, которая стремилась к некоторо-
му пределу, и увеличивало давление перехода на турбулентный
режим. Это давление несколько уменьшалось при увеличении диа-
метра трубки. При 14,6 ат смесь 2-нитропропана с 97%-ной азот-
ной кислотой горела со скоростью 0,114 см[сек; при содержании
в этой смеси 0,1, 0,5 и 0,75% полимегилметакрилата скорость горе-
ния оказалась равной 0,079, 0,076 и 0,074 сл</сек> а критическое
давление перехода горения на турбулентный режим жидкой и же-
латинированных смесей нитропропана и азотной кислоты составило
76, 83, 117 и 130 ат соответственно.
Опыты с желатинированными жидкостями позволили выяснить
также вопрос о том, не обусловлено ли появление пульсации горе-
ния вторичным пламенем. В случае желатины это пламя появляет-
ся при том же давлении и расположено даже несколько ближе к
поверхности, чем при горении жидкости; в то же время пульсирую-
щее горение наблюдается в первом и отсутствует во втором случае
даже и тогда, когда вторичное пламя располагается совсем близко
у поверхности горящего вещества. Отсутствие прямой связи между
появлением вторичного пламени и переходом горения на турбулент-
ный режим подтверждается также тем фактом, что для некоторых
жидкостей, например для НГЦ, этот переход происходит при столь
низких давлениях, когда вторичного пламени еще нет.
ТЕОРИЯ ЯВЛЕНИЯ
Независимо от экспериментальных исследований, Л. Д. Ландау
(7] рассмотрел теоретически горение жидкости, происходящее пу-
тем перехода ее в пары и химической реакции в парах. Он показал,
что фронт горения является устойчивым лишь в том случае, если
скорость горения не превосходит некоторого критического значе-
ния, которое определяется соотношением
«кр=(4<хк£р2грх)1'4, (1)
где «вр—критическое значение массовой скорости горения;
хк— поверхностное натяжение на границе между жидкостью
и ее насыщенным паром;
ускорение силы тяжести;
Рг —плотность газообразных продуктов горения;
— плотность жидкости.
Смысл вывода Л. Д. Ландау заключается в том, что при горе-
нии горизонтальная поверхность жидкости становится неустойчи-
423
вой, возникшее искажение ее стремится расти по амплитуде. Этому
росту однако препятствует сила тяжести и сила поверхностного на-
тяжения. До значения скорости, соответствующего выражению (1),
эти силы в состоянии предотвратить развитие турбулентности, при
больших значениях скорости оно наступает.
Формулу (1) можно представить 15] в виде
Р -1/2 5/4
«кр = Т^т-Рг Рж . (2)
Л1
где Р — парахор; М — молекулярный вес жидкости; у — постоян-
ная, равная в системе CGS 7,91.
Величина Р/М для органических жидкостей меняется в узких
пределах, так же как и плотность жидкости; относительно слабо
изменяется и плотность газов при постоянном давлении. Поэтому
формула (2) приближенно может быть представлена в виде
(3)
Турбулизация фронта горения может быть предотвращена так-
же путем повышения вязкости жидкости. В. Г. Левич [8] показал,
что критическое значение скорости горения связано с вязкостью со-
отношением
«кр=(з/WrW'3. (4)
где -л — вязкость жидкости.
Таким образом, если поверхностное натяжение и сила тяжести
не в состоянии предотвратить возмущение фронта горения, турбу-
лизация все же может не возникнуть, если вязкость жидкости до-
статочно велика. Однако необходимая величина вязкости довольно
высока и, например, для скорости горения, свойственной НГЦ при
атмосферном давлении, составляет около 1 пуаза, т. е. в три раза
рольше его вязкости при комнатной температуре. Поэтому, чтобы
стабилизирующее влияние вязкости проявилось при горении нитра-
тов спиртов ее требуется повышать искусственно, например, раст-
ворением высокополимерных веществ.
Зависимости Л. Д. Ландау и В. Г. Левина объясняют законо-
мерности, установленные опытом. Так как основным фактором, оп-
ределяющим наступление турбулентного режима, является величи-
на скорости горения при данном давлении, то естественно, что
раньше всего (т. е. при наименьшем давлении) турбулизация про-
является при горении наиболее быстрогорящего НГЦ за ним сле-
дуют метилнитрат, НГЛ и ди гликольди нитрат. Понятно и способ-
ствующее появлению турбулентности влияние давления и началь-
ной температуры. При повышении давления критическое значение
скорости горения растет (фиг. 5), но относительно медленно, про-
порционально корню квадратному из давления, скорость же горе-
ния, как показывает опыт, увеличивается приближенно пропорцио-
нально давлению и естественно при некотором давлении становит-
ся больше критической.
424
Следует указать, что переход горения на турбулентный режим
должен произойти также и потому, что по мере приближения дав-
ления, под которым идет горение, к критическому значению (в-
обычном физико-химическом значении этого термина) поверхност-
ное натяжение уменьшается до нуля и в соответствии с формулой
(1) падает до нуля и критическое значение скорости. Критическое
фиг. 5. Зависимость скорости горении
и критического (по Ландау) ее зна-
чения от давления и начальной темпе-
ратуры.
же давление, как показывает опыт, для многих органических
жидкостей лежит вблизи 50 ат.
Из приведенных формул и из графика фиг. 5 ясно, что переходу
горения на турбулентный режим способствует также, как это мы
видели на примере НГЛ, повышение начальной температуры ве-
щества, так как оно увеличивает скорость горения; критическое же
значение скорости от начальной
температуры практически не за-
висит.
Согласуются с теоретическими
заключениями и наблюдения от-
носительно влияния вязкости на
рассматриваемое явление в том
смысле, что легко стабилизуется
горение медленно горящего НГЛ,
а быстрогорящий НГЦ требует
для поддержания нормального
режима горения большего содер-
жания растворенной нитроклет-
чатки.
Поразительное влияние умень-
шения подвижности жидкости на
устойчивость ее горения иллюстрируют опыты Б. Н. Кондрикова
t(cM. стр- 443) с НГЦ, в которых подвижность жидкости уменьша-
лась добавлением к ней азида свинца. Смесь равных (по весу)
количеств НГЦ и азида свинца при давлениях от 6 до 40 ат горит,
^как правило, без перехода во взрыв, в то время как сам НГЦ при
фоджигании в интервале от 6 до 11 ат затухает или дает взрыв,
а азид свинца детонирует при всех давлениях. Более того, при тех
повышенных давлениях, при которых НГЦ вновь начинает гореть,
но уже на турбулентном режиме, т. е. с большой скоростью, добав-
ление к нему азида свинца, очевидно затрудняя турбулизацию,,
резко (примерно вдвое) снижает линейную скорость горения. Пос-
леднее наблюдается при горении жидкого НГЛ с азидом свинца &
(области турбулентного режима, т. е. при давлениях выше 20 ат.
' О СВЯЗИ МЕЖДУ ТУРБУЛЕНТНЫМ ГОРЕНИЕМ И ПЕРЕХОДОМ
! ГОРЕНИЯ ВО ВЗРЫВ
Уменьшение подвижности жидкости, в частности, путем увеличен
иия ее вязкости является эффективным способом предупреждения-
(перехода горения с нормального на турбулентный режим. Этот
гслособ используется в современном пороходелии, обеспечивая ус-
425*
Температура, термостата t °C
Фиг. 6. Зависимость интенсивности
вспышки 50%-ной нитроглицериновой
желатины от температуры термостата.
скорость газообразования столь
тойчивость горения бездымных порохов на труднолетучем раство-
рителе. Предупреждение возникновения турбулентного горения яв-
ляется так же важным фактором предотвращения перехода его во
взрыв. Опыты, поставленные в свое время [I] по горению жидких
нитратов (НГЦ и НГЛ) в железной трубке, закрытой диском, вы-
рывающимся при определенном давлении, показали, что горение
указанных жидкостей легко, при малой прочности диска, переходит
во взрыв. В то же время желатинированный НГЦ с содержанием
всего 7% нитроклетчатки устойчиво горел во всем изучавшемся
интервале прочностей диска (до 1200 кг/см'2, заряд 200 а).
Известно, однако, что при поджигании сильно нагретого грему-
чего студня [9] он взрывается; подобный же взрыв происходит и в
условиях определения его температуры вспыщки. Мало того, опыты
показали, что можно очень сильно увеличить интенсивность вспыш-
ки даже в случае желатин, со-
держащих гораздо больше (до
50%) нитроклетчатки. Эти опыты
Б. Н. Кондриков и И. В. Бабай-
цев ставили следующим образом
(см. стр. 515). Маленькую навеску
желатины (0,02 а) вводили в про-
бирку, помещенную в термостат.
Определялась интенсивность воз-
никающей вспышки в зависимо-
сти от температуры термостата.
Мерой этой интенсивности слу-
жила высота подброса шарика,
свободно закрывающего устье
пробирки. Характер установлен-
ной зависимости своеобразен
(фиг. 6) и объясняется следую-
щим образом. При низких темпе-
ратурах происходит лишь мед-
ленное разложение, при котором
мала, что шарик не подбрасы-
вается; при высоких температурах быстро, прежде чем желатица
успеет прогреться, происходит воспламенение; горение при этом
идет по макроповерхности кусочка и скорость газообразования
опять-таки недостаточна, чтобы подбросить шарик. При промежу-
точных температурах желатина успевает прогреться до воспламе-
нения. Однако не только этот прогрев сам по себе увеличивает ско-
рость горения, существенно и другое, В результате химических про-
цессов, в частности деполимеризации нитроклетчатки, вязкость же-
латины снижается настолько, что горение ее происходит с интен-
сивной турбулизацией и диспергированием вещества выделяющи-
мися газами. Все это приводит к столь сильному развитию поверх-
ности горения, что вспышка принимает характер взрыва, дробящего
пробирку и сильно подбрасывающего шарик, подобно тому, что
дает в аналогичных условиях нежелатинированный НГЦ.
426
Таким образом, переход горения жидкости на турбулентный ре-
ким существенно благоприятствует возможности возникновения
(Зрыва. Это неудивительно. Основным отличием детонации от нор-
иального горения является то, что при ней реакция идет под гораз-
[O большим давлением и в форме горения взвеси частиц вещества,
[то и определяет гораздо большую скорость процесса. Таким об-
)азом для осуществления перехода горения во взрыв должно воз-
>асти давление и образоваться взвесь ВВ. Впрочем, достаточно
1аже только образования взвеси: если суммарная поверхность
te горящих частиц, т. е. поверхность горения, достаточно велика,
о давление повысится автоматически.
Сказанное относится, в частности, и к горению НГЦ, Даже в
}бычных условиях лабораторной пробы на температуру вспыщки
)н дает, как известно, интенсивный взрыв. Тем больше оснований
)жидать взрыва при возникновении вспышки НГЦ в производст-
венных условиях, где размер зоны разогрева может быть гораздо
Дольше. Поэтому из-за легкости возникновения турбулентного го-
)ения и низких давлений, при которых оно начинается, самовоспла-
менение НГЦ в производственных условиях практически всегда
зриводит к взрыву.
О ДВОЙСТВЕННОМ ВЛИЯНИИ ДАВЛЕНИЯ НА ПЕРЕХОД
ГОРЕНИЯ ВО ВЗРЫВ
Существенным фактором перехода горения во взрыв считают,
i небезосновательно, повышение давления. Если горит порошкооб-
разное вещество, то повышение давления до определенной величи-
ны приводит к проникновению горячих продуктов горения в глубь
|аряда и, если при этом относительно быстро образуется достаточ-
но толстый слой горящей взвеси, то это может привести к перехо-
ду горения во взрыв. В случае жидкостей повышение давления, как
Вы видели, приводит к возникновению турбулентности, которая в
(вою очередь способствует переходу горения во взрыв. По-видимо-
Йу, для такого перехода более благоприятно наступление турбули-
зации при низких давлениях, когда скорость образования взвеси
келика, а линейная скорость ее сгорания мала и происходит поэто-
ву накопление частиц или капелек вещества.
L Возможно и другое явление. При чрезмерно интенсивной турбу-
пизации охлаждение пламени поступающей в него жидкостью ста-
новится слишком большим и горение прекращается. Однако, если
ври затухании прогрелось большое количество жидкости и до до-
статочно высокой температуры, то в ней мож*ет развиться самоус-
Воряющееся разложение, которое может привести к превышению
геплоприхода над теплопотерями и к вспышке, а следовательно, в
Мучае НГЦ, и к взрыву. Затухание и взрыв в этих условиях сосед-
ствуют и турбулизация может приводить к взрыву не сама по себе,
ве из-за увеличения скорости горения, а через образование доста-
точно большого объема нагретого вещества, в котором может раз-
виться тепловой взрыв. Локальный взрыв прогревшегося при зату-
хаиии НГЦ приводит к взрыву всей остальной «холодной» части
вещества. Взрыв такого типа легко осуществить, создавая искус-
ственно локальный разогрев поверхностного слоя жидкости, с по-
мощью раскаленной спирали, введенной в НГЦ на некоторую глу-
бину (1,5—2 Л(ж). Когда спираль находится на поверхности, то
поджигание вызывает лищь слабую вспышку, сопровождающуюся
разбрызгиванием жидкости и не приводящую ни к горению, ни
к взрыву.
В условиях описанных опытов по поджиганию сйиралью повы-
шение давления, предотвращая затухание, может оказывать на
горение стабилизирующее действие. Это действие наглядно иллю-
стрируют опыты Б. Н. КонДрикова со смесями жидких нитратов с
азидом свинца при горении их в области умеренно повышенных
Фиг. 7. Зависимость скорости
горения смеси нитроглицерина
и азида свинца (50 : 50) от дав-
ления.
/—смесь с нежелэтмннровамным
НгН. 2—смесь с желатинирован-
ным НГЦ.
давлений. Так, смесь равных (по весу) количеств НГЦ и азида
свинца при 3,7 ат дала неполный взрыв, при 4,9 ат происходило за-
тухание, а при 5,4 и более высоких давлениях (до 40 ат) наблюда-
лось горение, как правило, без перехода во взрыв (фиг. 7). Анало-
гичная картина наблюдается также для слабожелатинированного
НГЦ; он сам по себе благодаря повышенной вязкости способен к
устойчивому горению; добавление азида свинца (50%) лишает его
этой способности при низких давлениях — поджигание приводит к
затуханию; повышение давления до 3 и более атмосфер возвращает
способность к горению.
Сказанное относится и к смесям на основе НГЛ (фиг. 8) и ди-
гликольдинитрата; при этом чем меньше скорость горения нитрата,
тем больше то давление, начиная с которого восстанавливается
способность его к горению.
Эти наблюдения естественно объясняются исходя из представ-
ления о том, что переход горения во взрыв связан с охватом хими-
ческой реакцией при поджигании слоя вещества некоторой критиче-
ской толщины. Взрыв этого слоя, точнее взвеси частиц вещества,
образовавшихся при развитии реакции в нем, если он приводит к
возникновению скачка давления достаточной величины, вызывает
428
зрыв остальной части заряда. Если скачок этот недостаточен, то
происходит затухание или при малой амплитуде возникает пульси-
ующее горение. Повышение давления в условиях описанных опы-
ов затрудняло утолщение реагирующего слоя до необходимой для
озникновения взрыва толщины.
Условные обозначения:
9 ~ горение , О — затухание ; 3~-бзрыб
Фиг. 8. Влияние состава, давления и способа поджигания
на поведение смесей слабожелатннированного ннтрогли-
коля и азида свинца.
а—поджигание непосредственно спиралью, б—поджигание по-
средством слоя слабожелатинироэаниого НГЛ. Каждый квадро
Тик на графике соответствует одному опыту, Числа, располо-
женные вдоль осн абсцисс, означают давление (или интервал
давления), при котором производились опыты. Числа у фигур-
ных скобок — содержание азида свинца в его смеси с желати-
нированным нитроглкколем.
ВЫВОДЫ
Зависимости скорости горения от давления и начальной тем-
пературы для жидких вешеств, в частности нитратов спиртов, отли-
аются вследствие турбулизации фронта, наступающей при опре-
;еленных условиях горения, от тех, которые наблюдаются для твер-
,ых вешеств, Основными факторами, определяющими переход го-
>ения на турбулентный режим, являются величина скорости горе-
ия и давление. Турбулизация приводит к резкому увеличению
юличества жидкости, поступающей в зону горения, что Уюжет при
низких давлениях вызвать затухание его, а при высоких — сильно
увеличить количество сгорающего вещества. Возникновение при
Повышении давления турбулентного горения может приводить к пе-
реходу последнего во взрыв, особенно при низких давлениях.
В известных условиях повышение давления может стабилизиро-
вать горение.
429
ЛИТЕРАТУРА
1. А н д р е е э К. К., ДАН СССР, 1940, 29, 469.
2. А н д р е е в К. К-, ДАН СССР, 1945, 50, 277.
3. А н д р е е в К. К., ЖФХ, 1946, 20, 467.
4. Ан д р еев К. К-, ДАН СССР, 1946, 51, 123.
5. А н д р ее в К. К., ДАН СССР, 1946, 54, 39.
6. Беляев А. Ф. и Комкова Л. Д., Сб. «Физика взрыва» АН СССР, 1953,
№ 2, стр. 175.
7. Л а нда v Л. Д„ ЖЭТФ, 1944, 14, 240.
8. Л е в и ч В, Г„ ДАН СССР, 1956, 109, 975.
9. MuraourH., Wohlgemuth 1., Chimie et Industrie, 1936, 36, 472-
10. Whittaker A. G., Donovan T. M. and Williams H., Journ. phys.
Chem , 1958, 62, 908.
, К. К. АНДРЕЕВ. Г. H. БЕСПАЛОВ
28. О ГОРЕНИИ НИТРОГЛИЦЕРИНА
В отношении способности к горению нитроглицерин отличается
от других изученных с этой точки зрения нитроэфиров некоторыми
особенностями [I]. В обычных условиях лабораторного опыта (под-
жигание накаленной спиралью при комнатной температуре и ат-
мосферном давлении в стеклянной трубке диаметром несколько
миллиметров) нитроглицерин не горит. Чтобы получить горение,
необходимо вести опыт прн пониженном давлении (1/2 ат). Горе-
ние, но уже на турбулентном режиме, наблюдается также при дав-
лениях (р) больше ~11 ат [2], причем скорость его (и) оказывает-
ся на порядок больше, чем можно было ожидать, экстраполируя
зависимость и от р, полученную в области пониженных давлений.
Утрата нитроглицерином способности к горению при давлении
выше 1/2 ат была объяснена переходом горения на турбулентный
режим. В области низких давлений, когда химическое превращение
при горении идет с неполным тепловыделением, составляющим
только около половины максимально возможного, поступление в
результате турбулизации большого количества жидкости в относи-
тельно холодную и малоплотную зону горения прекращает хими-
ческую реакцию, так как теплопотери становятся слишком боль-
шими. Из этого объяснения следует, что уменьшив относительные
теплопотери, например увеличением диаметра трубки, можно было
бы затруднить затухание, а в пределе при достаточно большом диа-
метре, вообще предотвратить его. Горение нитроглицерина было бы
тогда сходно с горением, например, нитрогликоля. Как известно
(1], в последнем случае наблюдаются две области по давлению —
нормального (до ^-20 ат) и турбулентного (выше 20 ат) горения —
без интервала давления, в котором горение не распространяется.
Если это так, то затухание горения нитроглицерина при повы-
шении давления можно рассматривать как следствие значительно'
го увеличения критического диаметра горения при переходе на
турбулентный режим. Увеличивая диаметр трубки в этом случае,
можно было бы повышать давление затухания и, в конце концов,
430
поднять его до 11 —15 ат, когда горение на турбулентном режиме
распространяется устойчиво при диаметре трубки всего 5 мм. Веро-
ятно, было бы даже достаточно повысить давление затухания нор-
мального горения до меньшей, чем указано, величины, так как оно
при горении на турбулентном режиме понижается при увеличении
диаметра. Таким образом, увеличение диаметра трубки сближало
бы обе — нижнюю и верхнюю границы интервала отсутствия го-
рения по давлению и должно было бы привести к их слиянию, т. с.
к исчезновению этого интервала.
Наряду с указанным вариантом, который можно было условно
обозначить как превращение нитроглицерина в нитрогликоль, воз-
можен и другой — превращение нитрогликоля в нитроглицерин.
Создав условия усиленного теплоотвода, можно ожидать и для
нитрогликоля прекращения горения при переходе последнего на
турбулентный режим. Этими условиями может быть малый диа-
метр трубки или, лучше, нахождение ее вместо воздуха в воде или
иной жидкости.
Проверка этих соображений и явилась одной из задач настоя-
г щей работы. Решение ее оказалось, однако, затруднено тем обстоя-
тельством, что диаметр, начиная с которого нитроглицерин спосо-
бен к горению во всем интервале давлений, по-видимому, настоль-
ко велик, что делает опыты в лабораторных условиях цебезопас-
: иыми.
' Чтобы избежать применения больших зарядов, мы пошли в па-
,. правлении увеличения вязкости нитроглицерина путем растворения
в нем нитроклетчатки (желатинизации). Повышение вязкости за-
трудняет турбулизацию и смягчает ее последствия 11].
КРИТИЧЕСКИЙ ДИАМЕТР ГОРЕНИЯ ЖИДКОГО НИТРОГЛИЦЕРИНА
ПРИ РАЗНЫХ ДАВЛЕНИЯХ
Для определения критического диаметра горения нитроглице-
рин наливали в коническую стеклянную трубку с углом при вер-
шине не более 10°. Чтобы обеспечить постоянные условия теплоот-
вода, трубку помещали в стакан с водой. Опыты проводили в бом-
бе постоянного давления в атмосфере азота. Поджигание осущест-
вляли с широкой стороны конической трубки накаленной нихромо-
вой проволочкой. Критическим диаметром горения (rfK) считали
1 тот диаметр, ниже которого горение не распространялось.
Как показали опыты, критический диаметр горения нитроглице-
, рина в области повышенных давлений не зависит от того, находит-
ся ли трубка с нитроглицерином в воде или в газе, что по-видимо-
- му, следует объяснить большой скоростью горения в этих условиях,
вследствие чего относительная величина теплопотерь из быстро пе-
ремещающейся зоны реакции не успевает измениться за счет
изменения коэффициента теплоотдачи наружной поверхности сте-
клянной трубки.
Результаты опытов при повышенных давлениях (100—13 ат),
когда нитроглицерин горит на турбулентном режиме, показывают, '
Что критический диаметр затухания сильно увеличивается при
431
понижении давления. Кривая, описывающая этот рост (фиг. 1, кри-
d
K^pl,3l
вая 1), может быть представлена
уравнением
где dK —
критический диаметр в мм, а р— давление в кг[см?. При переходе
-от 100 к 13 am dK возрастает с 0,35 до 3,5—5 мм. Если полученная
зависимость будет выполняться и при дальнейшем уменьшении р,
du мм
Фиг. 1. Зависимость критического диа-
метра горения (dH) нитроглицерина н
^го желатин от давления (р) в интер-
вале выше атмосферного.
1—нитроглицерин 14—в окружении воз-
духа; . —в окружении воды), 2—3.2*/*-нал
желатина, <3—5*/*-иая желатина^ 4—8,5’/4-ная
желатина.
то критический диаметр при
атмосферном давлении должен
составлять 14,5 см.
В интервале давлений ниже
атмосферного, где горение идет
без видимой пульсации, при
Фиг. 2. Зависимость кри-
тического диаметра горе-
ния нитроглицерина от
давления в интервале
150—1000 мм рт. ст.
повышении р критический диаметр возрастает1 (фиг. 2). При
р=150 мм рт. ст. критический диаметр составляет 2,5 мм. при
390 мм — ~7 мм и при Ю50—10 мм.
1 Методически эти опыты ставили не как определение диаметра затухания прн
горении в конических трубках, а как определение давления затухания при горении
в цилиндрических трубках различного диаметра под медленно растущим давле-
нием. В этих опытах по определению критического диаметра при давлениях ниже
атмосферного трубку с нитроглицерином в воду не помещали, так как это при-
водило к слишком большому dK (более 12 мм). Во всех остальных случаях, опи-
санных ниже, трубка помещалась в воду.
432
Это обстоятельство несколько неожиданно, так как обычно при
(увеличении давления, т. е. при увеличении скорости горения, теп-
лопотери, а с ними и критический диаметр, уменьшаются. По-види-
мому, установленная данными опытами своеобразная зависимость
обусловлена тем, что с вызванным ростом давления увеличением
скорости горения усиливается турбулизация его фронта, что и уве-
личивает теплопотери. Это усиление теплопотерь вследствие появ-
ления турбулизации оказывается более сильным, чем их уменьше-
ние из-за увеличения скорости горения. Таким образом увеличение
критического диаметра является более чувствительным показате-
лем начала турбулизации горения, чем увеличение его скорости
и появление пульсации. Возможность наблюдения последней, на-
пример, при фотографической регистрации процесса зависит от раз-
решающей способности аппаратуры. Обычно пульсацию удается
зафиксировать лишь при развившейся турбулентности. В пользу
этих соображений говорит то обстоятельство, что критический диа-
метр желатинированного нитроглицерина, прн горении которого
турбулизация в основном подавлена, в той же области давлений
ф сравнении с нежелатинированным веществом гораздо меньше п
(уменьшается при повышении давления: критический диаметр
3%-ной нитроглицериновой желатины составляет при атмосфер-
ном давлении приблизительно 2 мм, а нитроглицерин в этих усло-
виях не горит даже при диаметре трубки равном 12 мм.
( Следует указать, что в опытах по определению критических ус-
ловий горения необходимо Очень «мягкое» поджигание. Так уда-
лось получить горение нитроглицерина даже при атмосферном
(давлении, однако, лишь при единственном из различных испытанных
способов зажигания, а именно поджигании при пониженном дав-
лении с последующим его ростом до I ат. По-видимому, обычные
способы поджигания способствуют возникновению развитого турбу-
лентного режима горения, при котором критический диаметр го-
раздо больше, чем при нормальном горении.
Последующие опыты 1 показали, что зависимость возможности
; горения жидкого нитроглицерина от диаметра трубки (rf) является
; более сложной (фиг. 3). Если при низких давлениях (140—150 мм
; рт. ст.) наблюдается только обычный нижний предел диаметра,
( очевидно обусловливаемый теплопотерями нормального горения, то
^'при больших давлениях, например, при I ат, горение, устойчиво
j идущее при малых диаметрах, перестает распространяться при не-
сколько ббльших его значениях и вновь приобретает эту способ-
‘ ность при дальнейшем увеличении диаметра. При этом скорости
! горения в трубках малого и большого диаметров приблизительно
одинаковы и равны 0,14—0,18 см!сек. Соответственно и экспери-
ментальная методика установления обоих критических значений
диаметра (rfj и была различной. Для определения первого из
। В этой серии опытов применяли нитроглицерин с температурой затвердева-
ния 11,8?. Результаты, полученные с этим нитроглицерином, количественно не-
сколько отличались от описанных выше, но общая картина сохранялась.
28 451 . 433
них нитроглицерин поджигался как обычно в суживающейся книзу
трубке, горение прекращалось по достижении d\\ для определения
второго предела нитроглицерин поджигался в расширяющейся кни-
зу трубке, горение прекращалось дойдя до d?. Другой возможный
вариант определения зависимости d~f(p) и d% заключался в опре-
делении давления затухания при горении вещества в цилиндриче-
ских трубках. Этим методом и получены кривые, приведенные на
фиг. 3.
Описанную зависимость dK от р следует объяснить тем, что при
давлении около 1 ат скорость горения уже достаточно велика, что-
Фиг. 3. Область горевин нитро-
глицерина при малых давле-
ниях (заштрихована)
бы вызвать значительную турбулизацию. При малых диаметрах,
однако, эта турбулизация не может развиться и горение не затуха-
ет; увеличение диаметра уменьшает стабилизирующий эффект
трубки, турбулизация вызывает усиление теплопотерь, и горение
затухает. При дальнейшем увеличении диаметра трубки, относи-
тельные теплопотери уменьшаются, и горение при некотором диа-
метре вновь становится возможным. Подтверждением описан-
ного о&ьяснения служит опять-таки то, что при давлении I ат для
желатинированного нитроглицерина интервала диаметров, в кото-
ром горение не распространяется, не наблюдается.
При понижении давления d> естественно увеличивается; d? так-
же возрастает, так как при меньшей скорости горения снижается
склонность его к автотурбулизации, и стабилизация процесса осу-
ществляется даже при относительно большом диаметре; d3 умень-
шается вследствие того, что из-за меньшей турбулизации (меньше
скорость горения) уменьшаются теплопотери, ею обусловленные.
При некотором достаточно низком давлении р’, d2 и d& становятся
одинаковыми, и интервал диаметра, в котором горение не наблю-
дается, исчезает. Из этих данных, кстати, следует парадоксальный
вывод: чтобы прервать горение нитроглицерина путем прохождения
1 При давлениях ниже -"20 мм рт. cr. d,, не определялся.
434
его через многоканальную преграду нецелесообразно делать кана-
лы слишком тонкими, так как в этом случае может быть подавле-
на турбулизация.
КРИТИЧЕСКИЙ ДИАМЕТР ГОРЕНИЯ желатинированного
НИТРОГЛИЦЕРИНА ПРИ РАЗНЫХ ДАВЛЕНИЯХ
л
Желатинизация нитроглицерина растворением в нем нитроклет-
чатки приводит к некоторому уменьшению скорости горения, а глав-
ное сильно затрудняет возникновение турбулизации из-за резкого
увеличения вязкости раствора. В соответствии с этим область тур-
булизации при горении желатины (~3,2°/о вес. нитроклетчатки)
смещается в сторону больших давлений, и кривая di: = fi,p) прини-
мает своеобразный вид (фиг. 1, кривая 2). В области малых давле-
ний (1 — 10 ат) критический диаметр уменьшается при увеличении
давления — очевидно вследствие уменьшения относительных тепло-
потерь из-за увеличения скорости горения, Однако в интервале от
10 до 30 ат критический диаметр растет и, например, при 30 ат
оказывается значительно больше, чем при 5 ат. Этот рост следует
приписать увеличению теплопотерь из-за появления турбулизации 1.
влияние которой оказывается и в этом случае сильнее, чем влияние
роста скорости горения, При дальнейшем повышении давления от
30 до 100 ат критический диаметр уменьшается, по-видимому, по-
тому что в этой области давлений влияние роста скорости горения,
а возможно также и повышение температуры его (за счет перехода
к сгоранию до N2 вместо NO), получает преобладающее значение.
Интересно сопоставитв кривую для желатины с кривой для цс-
жедатинированного нитроглицерина (фиг. 1, кривая 1). От 10 до
25 ат критический диаметр жидкого нитроглицерина гораздо боль-
ше, чем желатины, несмотря на то, что скорость его горения при-
близительно в 10 раз больше. Это несомненно является следствием
более интенсивной турбулизации в случае жидкого нежелатиниро-
ч ванного ВВ, обусловливающей большие теплопотери, и наглядно
иллюстрирует влияние этого фактора на критический диаметр. От
25 до 50 ат, напротив, с?1Г нитроглицерина оказывается уже несколь-
i ко меньше, чем желатины — это, по-видимому, является следствием
) большей скорости горения жидкости. Опыты с более вязкой (5%
нитроклетчатки) желатиной (фиг. 1, кривая 3) показали, в общем
I, сходную (с 3%-ной желатиной) картину. Чтобы подавить возннк-
новение турбулизации увеличением вязкости (скорость горения прн
изменении содержания нитроклетчатки от 3 до 8,5% практически
не меняется — фиг. 4), необходимо было увеличить содержание лит-
роклетчатки в нитроглицерине до 8,5%. В этом случае (фиг. ],
i- 1 Своеобразием этой турбулизации является то, что, вызывая увеличение
^критического диаметра, она не сопровождается . повышением скорости горения,
j Такое повышение для 3%-ной желатины появляется, по-видимому, лишь при го-
p. раэдо больших давлениях (около 300 ат), когда на ряде снимков наблюдалось
Г. иногда внезапное сильное увеличение скорости горения (см, табл. 2, фото 10).
ь 28* 435
кривая 4) максимума на кривой dK=f{p) не наблюдается, однако,
на графике виден в соответствующей области давлений участок по-
стоянства критического диаметра.
Фиг. 4. Зависимость скорости горения
нитроглицериновых желатин от дав-
ления,
/—3,2*А-ная, 2-5,^-ная. ?—вц5 И -ная.
В табл. 1 приведены уравнения, описывающие зависимость dK
от давления для нитроглицерина, а также для нитроглицериновых
желатин и некоторых других изученных нитроэфиров и их смесей.
Таблица I
Зависимость от давлений критического диаметра горения жидких
и желатинированных нитроэфиров и их растворов
Вещество Интервал давлений ат Л»
Нитроглицерин 13—100 145 = —— р’<3
3,2%*ная нитроглицери- новая желатина 1-10 . V6 U к —— к
25—100 3390 аК~
5%-ная нитроглицерино- вая желатина 1—20 2,21 ' Дк = —1 />,е
50—100 2140 ак = ——- Р1.э
8,5%-ная нитроглицери- новая желатина 3,5—21 1,70 0,6 Р
60—100 17,8 Р
436
Продолжение
Вещество Интервал давлений ат dK=f(p)
Ннтрогликолъ 2,5-21 19,3 ~ />3
21—81 56,2 _ —!— Z2
3^*ная нитрогликоленая 1—21 rf,, - 7.41
желатина 21-61 17,4 Р
Смесь нитрогликоля с 1-20 13,3 dK — /е
нитроглицерином (90:10) 13-81 145 rf,( ~ I 4 /> ’
Дигликольдинитрат 16—81 403
Для иллюстрации характера горения исследованных веществ и
Их смесей в табл. 2 приведены фотографии горения, полученные
с помощью фоторегистра.
Таблица 2
Характер горения различных жидких и желатинированных
нитроэфиров и их растворов
Фотографии, подученные с помощью фоторегистра
i’
g.
в
X
и
П
£
сЧ
в
о
3
1. 5%-иая нитроглицериновая желатина, ₽=6 ат. Отчет-
ливо заметно прифронтовое первичное свечение
В
Ф
&
5
а
ж
2к 3%-ная нитроглицериновая желатина, р=26 ат. Наблю-
дается некоторое искажение фронта горения н периодиче-
ское приближение к нему вторичного пламени
437
Продолжение
Пнтрбэфиры; давление, при котором происходило их горение и результаты наблюдения
3. 5%-ная нитроглицериновая желатина, р=42 ат. При-
фронтовое первичное снечение н гораздо более яркое вто-
ричное пламя, отделенное от перного темной зоной
4. 10%-ный раствор нитроглицерина в ннтрогликоле,
р=24 ат, Начавшееся на нормальном режиме горение пере-
ходит на турбулентный режим
5. 10%-ный раствор нитроглицерина в ннтрогликоле,
р=16 ат. Нерегулярное искажение фронта горения — пуль-
сирующее горение
6. 10%-ный раствор нитроглицерина в ннтрогликоле,
р=26 ат, Периодическая турбулизация с частотой 80 сен-1
438
Продолжение
Ншроэфиры: давление, при котором происходили их горение и результаты наблюдения
7. Ди гликольдинитрат, р=81 ат. Горение с большой ско-
ростью; при переходе горения из трубки большого диамет-
ра (4.8 .и.н) в толстостенный капилляр (1 леи) наблюдается
уменьшение скорости горения и исчезновение вторичного
I пламени
8. 10%-ный раствор нитроглицерина в нитрогликоле,
р=9 ат. Многозональная структура первичного пламени
9. 5%-ная нитроглицериновая желатина, р~213 ат. По-
степенное увеличение скорости по мере горения заряда
10. 3%-ная нитроглицериновая желатина, р—361 ат. Дву-
кратное резкое увеличение скорости горения
439
критический диаметр ГОРЕНИЯ НИТРОГЛИКОЛЯ, смеси его
С НИТРОГЛИЦЕРИНОМ И ДИГЛИКОЛЬДИНИТРАТА ПРИ РАЗНЫХ
ДАВЛЕНИЯХ
На фиг. 5 (кривая /) показано изменение dK с давлением для
нитрогликоля. Во всей области давления от 1 до 80 ат критический
диаметр уменьшается от 13 мм при 1 ат 1 до 0,4 мм при 80 ат. Од-
нако в интервале давлений 10—20 ат в тех случаях, когда горение
шло с пульсацией, затухание происходило при заметно большем
Фиг. 5. Зависимость критического диа-
метра горения нитрогликоля, смеси его
с нитроглицерином (90:10) и 3%-ной
нитрогликолевой желатины от давления,
диаметре, несмотря на то, что
скорость горения в этих услови-
ях была гораздо больше.
Чтобы более отчетливо вы-
явить изменение d# при переходе
горения на пульсирующий ре-
жим, опыты были продолжены
с применением растворов нитро-
глицерина в нитрогликоле (фиг. 5.
кривая 2). 10%-ный раствор ни-
троглицерина в нитрогликоле го-
рит несколько быстрее, чем чи-
стый нитрогликоль, и соответст-
венно переход на турбулентный
режим наблюдается при несколь-
ко меньшем давлении-------15 ат-
Кривая d„ = f(p) имеет максимум
вблизи этого давления; правда
из-аа разброса точек он выражег
не очень отчетливо.
1--яитроглнколь. д—itWt-ный раствор нитро- Была сделана также попытка
глицерина в нитрогликоле. ннтро-
глнколеная желатина. ПОЛучИТЬ ЗЗТухаНИС ГОрСНИЯ НН
трогликоля при переходе на тур^
булентный режим. В обычных условиях лабораторного эксперимен-
та для нитрогликоля не наблюдается интервала давления, разделя-
ющего область нормального и турбулентного горения, в котором
горение не распространялось бы. Вероятной причиной этого отли-
чия нитрогликоля от нитроглицерина является то обстоятельство,
что для первого переход на турбулентный режим наблюдается при
большем давлении (-—20 ат), когда горение идет уже с полным
тепловыделением и со значительной скоростью, и преобладание
теплопотерь над теплоприходом не осуществляется.
Теплопотери можно увеличить, уменьшая диаметр трубки. Это.
однако, не привело к затуханию горения, по-видимому, потому что
уменьшение диаметра затрудняло возникновение турбулизации по-
добно тому, как это наблюдалось для нитроглицерина. Другая воз-
можность заключалась в повышении температуры опыта, снижаю-
щем, как известно, давление перехода горения на турбулентный
1 Если трубочка окружена не водой, а воздухом, то критический диаметр
значительно меньше (— 1 ле.ч).
440
> режим. Кроме того, трубку с горящей жидкостью помещали для
усиления теплоотвода в воду при соответствующей температуре.
В этих условиях при 40° переход горения на турбулентный режим
происходил при давлении около 8 ат. В значительной части опытов,
проведенных как в конических, так и в цилиндрических трубках,
наблюдалось затухание горения при диаметрах 4—10 мм (фиг, 6),
, причем оно наблюдалось в тех случаях, когда горение начиналось
сразу на турбулентном режиме.
Фиг. 6. Зависимость критического диа-
метра горения нитрогликоля от дав-
Фиг. 7. Зависимость критического
диаметра горения дигликольдиннт-
леиия при 40 и 20°. рата от давления,
Г. Таким образом, затухание горения при переходе на турбулент-
( ный режим можно наблюдать и для нитрогликоля, хотя оно для
1 этого взрывчатого вещества и не столь четко выражено, как для
’ нитроглицерина.
Критический диаметр желатинированного нитрогликоля (фиг. 5.
' кривая 3} значительно меньше, чем для жидкого при всех давле-
i, ниях, возможно из-за уменьшения роли конвективной теплоотдачи;
различие особенно велико в области тех давлений, где горение
* жидкости переходит на турбулентный режим.
Диаметр затухания определялся также Для горения дигликоль-
динитрата. При атмосферном давлении критический диаметр велик
(~14 мм) очевидно в связи с малой скоростью горения При по-
вышении давления он уменьшается (фиг. 7). При давлении выше
55 ат, горение ди гликольди нитрата переходит на турбулентный ре-
жим, о чем, в частности, можно судить по большой скорости горе-
ния в трубках значительного диаметра (фиг. 8), Однако эта ско-
рбеть сильно уменьшается при уменьшении диаметра трубки, оче-
видно, вследствие его стабилизирующего влияния. В связи с тем,
что определение диаметра затухания производилось без одновре-
1 В трубочке, окруженной воздухом, критический диаметр при атмосфер-
ном давлении составляет ~5 мм.
441
менного определения скорости горения, остается неясным, отно-
сятся ли значения при давлениях выше 55 ат к турбулентному '
или нормальному горению.
Фиг. 8. Зависимость скорости горения дигли-
кольдинитрата (при повышенных давлени-
ях) от давления в трубках различных диа-
метров.
.'--5 .ик. ?—I .н.п.
ВЫВОДЫ
1. Исследовано горение нитроглицерина в жидком и желатини-
рованном нитроклетчаткой виде и некоторых других нитроэфиров
в интервале давлении 0,01 —100 ат.
2. Критический диаметр и скорость горения при атмосферном
давлении для исследованных нитроэфпров составляют:
Нитроглицерин...............
Нитрогликоль ...............
Диг-тикольдини гр;п ........
н „ч.ч
0,5
1,0
и ц см)сек
' 0,14
0.030
0,012
3. В интервале давлений 20—100 ат критический диаметр горе-
ния указанных китроэфиров уменьшается при увеличении давления
X
по закону —z-, где/г заключается между 1,15 и 1,48.
р
4. Переход горения жидкости (нитроглицерин, нитрогликоль)
на турбулентный режим приводит к сильному увеличению критиче-
ского диаметра.
5. Малые диаметры трубки стабилизируют горение нитроглице-
рина, затрудняя переход с нормального на турбулентный режим;
поэтому н известных условиях можно наблюдать три критических
значения диаметра: d{— наименьший, при котором возможно нор-
мальное горение, d2 — наибольший, выше которого появление тур-
булизации приводит к затуханию из-за увеличения теплопотерь во
вне, и — выше которого горение вновь становится возможным
142
вследствие уменьшения относительной поверхности теплоотвода.
При атмосферном давлении критические значения диаметра со-
ставляют соответственно 0,5; 2,6 и 7,5 мм.
6. Увеличение вязкости нитроглицерина путем растворения в
нем высокополимера (нитроклетчатки), затрудняя турбулизацию,
уменьшает возрастание критического диаметра (при переходе горе-
ния на турбулентный режим), а при достаточной вязкости — сни-
мает его.
ЛИТЕРАТУРА
1. Андреев К, К., Сб. статей по теории взрывчатых веществ, под ред.
К. К. Андреева и Ю. Б. Харитона. Оборонгиз, 1940, стр. 39;
А и д р е е в К. К., ЖФХ, 1946, 20, 467;
А и д р е е в К. К., ДАН СССР, 1945, 50, 277;
Андреев К. К., Термическое разложение и горение взрывчатых веществ,
Госэиергоиздат, 1957.
2, Б е л и е в А. Ф.. Комкова Л. Д., Сб. «Физика взрыва», АН СССР, 1953,
.V» 2, стр. 175-
£. Я. КОН Д РИКО В
29, О ГОРЕНИИ СМЕСЕЙ ИНИЦИИРУЮЩИХ ВВ
И ЖИДКИХ НИТРОЭФИРОВ
Основная особенность инициирующих ВВ заключается в том.
что горение их переходит в детонацию при таких условиях (неболь-
шой открыто расположенный заряд, обычные давление и темпера,
тура), при которых ВВ других классов горят устойчиво и спокой-
но. Причины этого различия не вполне ясны. Одной из них,
очевидно, является большая скорость горения инициирующих ВВ,
достигающая при атмосферном давлении десятков сантиметров в се-
кунду. Однако эта причина, по-видимому, не единственная. Извест-
но, что некоторые взрывчатые вещества, например пикраты метал-
лов, также обладают весьма высокой скоростью горения и тем не
менее не относятся к классу инициирующих ВВ, С другой стороны,
такие типичные инициирующие ВВ, как азид свинца, вообще не го-
рят и при поджигании сразу же, без какого бы то ни было периода
горения дают детонацию.
Вследствие значительной неустойчивости горения инициирую-
щих ВВ, изучать этот процесс и причины перехода его во взрыв до-
вольно трудно. Между тем, известно, что некоторые смеси иници-
ирующих ВВ с инертными веществами, а также вторичными ВВ
способны гореть вполне устойчиво. Так, смеси азида свинца с жела-
тинированными нитроглицерином и нитроклетчаткой [2], [3], [5]
устойчиво горят при атмосферном давлении даже при содержании
в них до 30—40% азида.
Представляло интерес выяснить влияние давления на характер
и скорость горения подобных смесей при различном содержании
ааида и вблизи области условий, приводящих к переходу горения во
44з
взрыв, В качестве вторичных ВВ применялись жидкие нитрозфиры
(нитрогликоль, нитроглицерин и дигликольдинитрат), обычно жела-
тинированные небольшим количеством коллоксилина. Из иници-
ирующих ВВ, помимо азида свинца, частично изучались также
стифнат свинца и пикрат калия,
МЕТОДИКА ПРОВЕДЕНИЯ ЭКСПЕРИМЕНТОВ
Жидкие нитроэфиры желатинировали небольшим количеством
(3%) коллоксилина (12% N) и осторожно смешивали с иницииру-
ющим ВВ, просеянным через шелковое сито с 39 отверстиями на
сантиметр, Смешение производилось деревянной палочкой в фар-
форовой хорошо глазурованной чашке. Полученную смесь засасы-
вали или набивали в трубки молибденового стекла с оплавленными
концами. Диаметр трубки обычно составлял ~5 мм, высота заряда
1—2 см. Определение плотности зарядов одной из приготовленных
таким образом смесей показало, что эта плотность близка к теоре-
тической,
При повышенных (до 70 ат) давлениях опыты проводили в бом-
бе для изучения горения ВВ при постоянном давлении, Опыты в этой
бомбе проводили в атмосфере азота, реже — углекислоты, Поджи-
гание производилось накаливаемой током нихромовой проволоч-
кой через слой желатинированного нитрогликоля или непосредст-
венно. Скорость и характер горения фиксировали при помоши фо-
торегистра, а в ряде случаев — кинокамеры АК-16-
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТОВ
Смеси азида свинца с нитрогликолем
При поджигании смеси слабо желатинированного нитрогликоля
с 10% азида свинца на воздухе в трубочках диаметром 5 мм после
сгорания слоя толщиной около 2 мм происходит затухание.
В трубках диаметром 7 мм выгорает слой толщиной 3—4 мм, а
при диаметре 10,5 мм—слой 4—5 мм, Горение сопровождается силь-
ным частым треском, иногда из трубки вылетают кусочки смеси
размером около миллиметра.
Повышение давления стабилизирует горение и тем легче, чем
больше диаметр трубки. При диаметре 5 мм смесь затухала при 4
и 5 ат ц начинала гореть лишь с 6 ат. При диаметре трубки 7
горение .происходило и прн 4 ат. При более высоких давлениях
смесь горит устойчиво со скоростью практически равной скорости
горения чистой желатины (фиг, 1). Вторичное пламя смеси, отчет-
ливо видное на фотографиях, начиная с давления 8 ат, не тусклое
красноватое, как при горении одного желатинированного нитрогли-
коля, а яркое белое. Ширина темной зоны между ним и поверхно-
стью конденсированной фазы близка к ширине этой же зоны в слу-
чае горения желатинированного нитрогликоля (фиг. 2), При дав-
лении около 20 ат на некоторых фотографиях (фиг. 3) видны 4 зо-
ны волны горения,
444
При больших концентрациях азида в смеси поджигание ее при-
водит не только к затуханию, но и к взрыву заряда, Из фиг. 8, по-
мещенной в статье 27 (см, стр, 429 сборника) и отображающей
влияние состава, давления и способа воспламенения на поведение
смесей азида свинца с желатинированным нитрогликолем, видно,
что, повышая давление, можно предотвратить затухание или взрыв
Точки, е на прямой. 3 -опыты в
атмосфере CQZ ; See остальные -
- S атмосфере азота.
Фиг. 1. Влияние давления на скорость горения
смесей азида свинца с желатинированным нитро-
гликолем.
процентное содержание PbNe в смеси: 0—0; [0;
^—30: jf—50. 4—60; штрих-пунктирная линия А отделяет
область устойчивого горения от области, в которой смеси
затухают или взрываются.
и получить устойчивое горение смеси. Возникновение взрыва об-
легчается поджиганием смеси непосредственно раскаленной про-
волочкой, особенно если она введена в вещество на некоторую глу-
бину. Вполне естественно, что при большом содержании азида
взрыв происходит легче, чем при малом.. При содержании в смеси
75% PbNe возбудить устойчивое горение ее не удалось даже при
повышении давления до 54 ат.
Давление, начиная с которого смесь становится способной
к устойчивому горению, при повышении содержания азида в ней
: существенно растет. Этот рост выражается соотношением
ркр = 4,10е[дап, кг]см2 (см. фиг. 4),
/ где m — содержание азида свинца в смеси с желатинированным
нитрогликолем в г/смл.
445
Скорость горения подобных смесей (см. фиг. 1) приближенно
пропорциональна давлению. Зависимость коэффициента пропор-
циональности (В) от состава смеси может быть представлена
в виде
В = 0,035 е1’31тг/смгсек : кг] см2 (см. фиг. 4).
W
Давление р кг / гмг
Условные обозначения :
на кривой 1 е и х - содержание
хлорида свинца, в смеси 10 и 30
на кривой. 2: о и + -содержа-
ние азида, свинца Си 'О °]о
соответственно;
на кривой 3: 9 и 9 - то же
50 и 60 % ;
на кривой У : Д- то же 30 °]а
Фиг. 2, Влияние давления на ширину
темной зоны прн горении смесей азида
и хлорида свинца с желатинированным
нитрогликолем.
I—смеси с РЬСЬ. 2. 3. 4—смеси с РЬХ6.
(30%-ная смесь азида свинца недостаточно
однородна н горит неравномерно).
На фиг. 5 приведены фотографии горения смесей азида свинца
и желатинированного нитрогликоля при содержании 30, 50 и 60%
PbNs. Пламя при горении этих смесей обычно состоит из трех
Фиг. 3. Горение смеси азида свинца с желатинированным нитро-
гликолем ]0:90 при давлении 22 ит (негатив).
зон, ширина темной зоны меньше, чем при гореции желатинирован-
ного нитрогликоля (фиг. 2).
Для изготовления смеси с 30% PbNs, в отличие от других сме-
сей применялся азид, не протертый через сито и содержавший на-
ряду с тонкодисперсным веществом небольшие комочки слипшего-
446
ся порошка. Эта смесь особенно легко взрывалась при низких дав-
лениях, а при повышенных — горение ее, хотя и было вполне устой-
чивым, но происходило крайне неравномерно (фиг. 5,а).
Фиг. 4. Зависимость скорости горения и
критического давления от состава для сме-
сей азида свинца с желатинированным нит-
рогл иколеи.
/—критическое давление. 2 —коэффициент про-
порциональности В н уравнении «м=Вр.
Смеси азида свинца с нитроглицерином и диглнкольдинитратом
Смеси азида свинца с другими жидкими ннтроэфирами вели
себя аналогично смесям его с желатинированным яитрогликолем,
Так, смесь равных количеств азида и слабо желатинированного
нитроглицерина при 2 ат затухала, ио уже начиная с 3 ат и выше
устойчиво горела со скоростью, приближенно линейно растущей и
давлением (фиг. 6).
Аналогично вела себя и смесь 50:50 азида свинца с нежелати-
нированным нитроглицерином. При поджигании ее при 3,7 ат про-
изошел неполный взрыв, при 4,9 ат смесь затухла, при 5,4 ат — она
^загорелась и спокойно сгорела. На расстоянии около сантиметра
над поверхностью конденсированной фазы наблюдалось желтова-
; тое вторичное пламя. При более высоких давлениях смесь хорошо
воспламеняется и устойчиво горит со скоростью, несколько боль-
.шей (фиг. 6), чем скорость горения желатинированной смеси, но
> значительно (приблизительно вдвое) меньшей, чем скорость горения
^ нитроглицерина без азида свинца [6]. Лишь в одном опыте (при
t 38 ат) в конце горения наблюдался слабый взрыв.
* Смесь на основе желатинированного дигликольдииитрата 50 : 50
при умеренно повышенных давлениях (38 и 47 ат) не горела или,
прогорев немного, затухала (51 ат) г при 53; 55 и 60 am наблюдалось
447
Фиг, 5. Горение смесей азида свинца с желатини-
рованным иитрогликолем.
Процентное содержание PbVt и смеси к давление.
a—50V. в 2[ ат (позитив), б—60’/. и 32 ат (негатив, и
конце горения взрыи, хорошо виден переход от горения
слоя желатин кров а иного ннтрогликоля к горению смеси),
a—50V. и 65 ат (негатив), г—-301/. и 25 ат (негатнн, горе-
ние устойчивое, во не равномерное).
I
448
устойчивое горение со скоростью 1 —1,5 см]сек. На фотографиях
видна яркая узкая полоска пламени вблизи поверхности конденси-
рованной фазы (фиг. 7), горение сопровождается множеством сла-
бых вспышек, придающих фотографии характерный полосатый вид.
Фиг. 6. Зависимость скоро-
сти горения смесей азида
свинца с нитроглицерином
50:50 от давления.
/—нитроглицерин иежелатнин-
роввнный, 2—нитроглицерин же-
лэтилированный.
Фиг. 7. Горение смеси азида свинца с желатинирован-
ным дигликольдинитратом 50 ; 50 при давлении 60 ат
(негатив).
Смеси стифната свинца с нитрогликолем
Смесь 30% тринитрорезорцината свинца (ТНРС) с желатини-
рованным нитрогликолем при атмосферном давлении в трубках
диаметром 5 и 7,5 мм затухает. При диаметре 10,5 мм смесь мед-
ленно горела с частым сильным треском, При повышенном давлении
смесь горит равномерно и устойчиво со скоростью, близкой к скоро-
сти горения смеси нитрогликоля с 30% азида свинца (фиг. 8 и 9).
При увеличении содержания ТНРС до 50% скорость горения и
давление, начиная с которого смесь горит устойчиво, повышаются,
и распространение пламени становится неравномерным (см.
фиг. 10 и 11).
Смесь, содержащая 60% ТНРС, при атмосферном давлении дает
взрыв (при поджигании как непосредственно раскаленной прово-
лочкой, так и через слой желатинированного нитрогликоля). При
29 451
449
Фиг, 8, Горение смеси 30% ТНРС с желатиниро-
ванным иитроглнколем при давлении 20 ат
(негатив).
Фиг. 10. Горение смеси 50% ТНРС с
желатинированным нитрагликолем прн
давлении;
б—5[. в—37 ат (позитив).
Фиг. 9, Влияние давления на ско-
рость горения снесен стифната н
азида свинца с желатинированным
ннтрогликоле м<
/ и 4—смсси с ТНРС 5£) и 30%. 2 и
3—смеси с PbN* — 50 и 30*/» (соответ-
ственно) ,,
450
овышенных давлениях смесь горит, хотя и крайне неравномерно.
V без перехода во взрыв, причем время сгорания заряда по мере
повышения
вается, т,
горения
фиг, 11).
Фиг. 11. Горение смеси 60%
ТНРС с желатинированным
нитрогликолем при давлении;
□—37, 6—51, в—62 ат (цегаткв).
давления увеличи-
е. средняя скорость
уменьшается (см.
Смеси пикрата калия
с дигликольдинитратом
и нитрогликолем
Влияние состава на горение
смеси пикрата калия с желати-
нированным днглпкольдинитра-
том при атмосферном давлении
в трубках диаметром 11 мм ил-
люстрируется фиг. 12 [. Смесь,
содержащая около 5% пикрата,
горит со звучным потрескива-
нием и со скоростью, близкой
Фиг. 12. Зависимость скоро-
сти горения смесей пикрата
калия с желатинированным
дигликольдинитратом при
атмосферном давлении от
состава.
4 скорости горения чистой желатины. Смеси с 8,9 и 19% пикрата
ie горят, а начиная с 24% вновь становятся способными к горе-
нию, причем скорость последнего в несколько раз выше, чем
(истого дигликольдинитрата. При увеличении содержания пикрата
) смеси потрескивание, сопровождающее горение, учащается и при
И)—70% переходит в ровное сильное шипение. Смесп пикрата
Эти опыты были проведены студентом В. И. Козловым.
29*
451
калия с желатинированным нитрогликолем вели себя аналогично,
но горели быстрее и при увеличении содержания пикрата горение
их прекращалось позже, чем дигликольдинитратных смесей.
Фиг, 13, Влияние давления иа ско-
рость говения (и) и ширину темной
аоцы (Л) прн горении смеси равных
количеств пикрата калия н желатини-
рованного нитрогликоля,
Ч— ширина темной зоны прн горении
смеси, о—ширина темной зоны прн го-
рении желатинированного ннтрогликоля
без добавления пикрата.
Зависимость скорости горения смеси равных количеств пикрата
калия и желатинированного питрогликоля от давления представле-
на на фиг, 13. При поджигании слоем желатинированного нитрогли-
Фиг. 14. Горение смеси пикрата калия с желатини-
рованным иитрогликолем прн поджигании через слой
желатинированного нитрогликоля, давление 31 ат
(позитив).
Фиг, 15, Неравномерное горение смеси пикрата калия с желати-
нированным нитро гликолем при поджигании непосредственно
раскаленной проволочкой, давление 20 ат (позитив),
коля эта смесь в интервале 1—75 ат горит устойчиво и равномерно,
В волне горения ее видны три зоны (фиг. 14), причем и первич-
ное и вторичное пламена более тусклы, чем в случае смесей, со-
452
держащих соли свинца. Зависимость ширины темной зоны от дав-
ления довольно своеобразна (фиг, 13); при умеренно повышенных
давлениях эта зона значительно ^же, чем при горении чистой же-
латины, при более высоких давлениях это различие исчезает.
В том случае, когда смесь воспламенялась непосредственно про-
волочкой, горение было неравномерным, неустойчивым и скорость
;его по фотографиям полученным на фоторегистре (фиг, 15) опре-
делить не удавалось.
1,0
0,5
и см/сек
1,5 г
Смеси, содержащие хлорид свинца
Увеличение скорости горения, изменение критического диаметра,
уменьшение толщины темной зоны и другие эффекты, наблюдав-
шиеся при добавлении к нитроэфирам азида свинца и ТНРС, можно
;было бы приписать каталитическому
.'влиянию содержащегося в них свинца.
,Для того чтобы установить возможную
роль этого фактора, мы провели опыты
по горению смесей желатинированного
[нитрогликоля с хлоридом двухвалентного
свинца. Оказалось, что добавление Ю и
30% РЬСЬ почти не влияет на линейную
скорость горения нитрогликоля (фиг. 16)
и, следовательно, несколько увеличивает
массовую скорость. Очевидно, однако,
что этот эффект не сравним с действием
^зида свинца и ТНРС на скорость горе-
ния и, следовательно, роль катализа
в увеличении скорости горения нитро-
эфйров при добавлении к пим быстро-
ГОрящих соединений свинца невелика.
Пламя при горении смесей, содержащих
:лорид свинца, имеет сложную полизо-
’альную структуру, причем на некоторых
ютографиях можно различить до пяти
ервой темной зоны несколько превышает ширину ее при горении
0 2Q W р яг/см1
УслцЗте обозначения;
Содержание злорида, сваяца,
в смеси:
Фиг, 16. Влияние давления
на скорость горения смесей
желатинировав кого нитро-
гликоля с хлоридом свинца.
зон (фиг. 17). Ширина
Фиг. 17. Горение смеси 30% PbCh с желатинированным нитроглико-
лем при давлении 50 ат (негатив).
селатцнированного нитрогликоля и тем более — высокопроцент-
[ых смесей его с азидом свинца (см. фиг. 2), что также указывает
а различие в механизмах действия этих веществ.
453
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Основной результат описанных выше опытов заключается в цы-
явлении своеобразного влияния давления на возможность горения
смесей инициирующих ВВ с жидкими нитроэфирами. Неустойчивое
при атмосферном или умеренно повышенном давлении горение этих
смесей, как правило, вполне устойчиво при высоких давлениях. Этот
эффект проявляется для смесей нитроэфиров как с азидом, так и
со стифнатом свинца. Парадоксальность его особенно наглядна в
тех случаях, когда повышение давления предотвращает не только
затухание, ио и взрыв смесей (см. например данные для смеси
желатинированного нитрогликоля с 30% PbNe на фиг. 8 статьи 27,
стр. 429). Своеобразие такого влияния давления проявляется еще
и в том, что смесь 50:50 азида свинца с нитроглицерином, способ-
ность которого к нормальному (невозмущенному) горению гораздо
меньше, чем нитрогликоля, горит, начиная с меньших давлений, чем
нитрогликолеаая смесь. В первом случае горение начинается с 3 ат,
во втором — лишь с 20 ат.
Совершенно очевидно, что объяснить эти явления, исходя из
обычных представлений о механизме возмущения горения и пере-
хода его во взрыв [1] было бы затруднительно. Действительно,
обычные представления основаны на многочисленных фактах пря-
мо противоположного влияния давления на устойчивость горения
ВВ и логично объясняют их тем, что при возрастании давления уве
личиваются скорость горения и динамическое повышение давления.
При достижении некоторого критического давления нагретые газо-
образные продукты начинают проникать в глубь заряда, поверх-
ность горения увеличивается, скорость его и динамическое повыше-
ние давления прогрессивно возрастают, горение становится нерав-
номерным, пульсирующим или переходит во взрыв.
Наблюдавшееся в наших опытах своеобразное влияние давле-
ния на устойчивость горения ВВ может быть, однако, естественно
объяснено, если исходить из представления [2], [3] о том, что пере-
ход горения во взрыв связан с охватом химической реакцией слоя
взрывчатого вещества некоторой критической толщины. Взрыв это-
го слоя, точнее взвеси частиц ВВ, образовавшихся при реакции в
нем, если он приводит к образованию достаточно большого скачка
давления, вызывает взрыв остальной части заряда. Если скачок
давления недостаточен, то происходит затухание, вследствие нару-
шения прогретого слоя, дли при малой амплитуде этого скачка го-
рение приобретает пульсирующий характер. Возникновение взрыва
взвеси зависит также от времени пребывания инициирующего ВВ
в прогретом состоянии. Если это время меньше периода индукции,
то слой смеси сгорает прежде, чем инициирующее ВВ успевает
дать вспышку.
На то, что важно не непосредственно давление, а именно тол-
щина прогретого и реагирующего слоя и длительность его сущест-
вования, указывает зависимость явления от скорости горения ни-
троэфира, Смеси на основе нитроглицерина, горящего быстрее, чем
454
ннтрогликоль, приобретают способность гореть устойчиво, начиная
с менылнх давлений; медленно горящие смеси на основе дигликоль-
динитрата, напротив, дают устойчивое горение лишь при значитель-
но больших давлениях, чем нитрогликолевые. Скорости горения
при давлениях, начиная с которых смеси 50 : 50 на основе нитрогли-
коля и дигликольдинитрзта горят устойчиво (20 в первом и —'50 ат
во втором случае) приближенно равны и составляют около 1 см1сек.
Легко объяснить также с этой точки зрения совершенно не-
обычное явление, наблюдающееся при добавлении наименее спо-
собного к горению инициирующего ВВ — азида свинца к одному из
наиболее легко взрывающихся и трудно горящих вторичных ВВ —
нежелатинированному нитроглицерину, В той области давлений,
в которой нитроглицерин из-за турбулизации поверхности жидко-
сти не горит, а азид, как ему свойственно, детонирует, смесь рав-
ных количеств этих веществ оказывается способной к устойчивому
горению. Выше уже отмечалось, что при тех повышенных давле-
ниях, при которых нитроглицерин вновь начинает гореть, но уже на
турбулентном режиме, т. е, с большой скоростью, добавление к не-
му азида свинца, очевидно затрудняя турбулизацию, резко (при-
мерно вдвое) снижает линейную скорость горения. Последнее на-
блюдается и при горении жидкого нитрогликоля с азидом свинца
. при давлениях выше 20 ат.
Если при поджигании прогревается более толстый слой смеси,
вероятность возникновения взрыва в ней, очевидно, возрастает.
Этот случай иллюстрируют опыты с 30 %-ной смесью азида свинца
,е нитрогликолем при поджигании накаливаемой током проволочкой
. (фиг. 8 статьи 27, стр. 429). В том случае, если способность си-
стемы к взрыву менее значительна, чем смесей с азидом свинца,
поджигание проволочкой может привести не к взрыву, а к неравно-
,мерному возмущённому горению в тех условиях, в которых при бо.
бее «мягком» поджигании система горит равномерно и спокойно.
Так ведет себя, например, смесь с пикратом калия (ср. фиг. 14
, и 15).
Тепловой взрыв, приводящий к затуханию смеси, не обязатель-
но должен охватывать весь прогретый слой. Выше отмечалось, что
(торение исследовавшихся смесей при атмосферном давлении со-
провождается частым потрескиванием — очевидно микровспышка-
^ми частиц быстрогорящего ВВ или их агрегатов. Эти микровспыш-
:,Ки, перемешивая прогретый слой и усиливая теплоотвод, мешают
распространению горения и при достаточном содержании п смеси
быстрогорящего вещества приводят к затуханию, В том случае,
1,если быстрогорящее ВВ способно к самостоятельному горению, при
(значительном содержании его в смеси, последняя может нанять го-
(реть вновь, но уже на режиме,' свойственном чистому быстрогоря-
Ицему веществу (при малых содержаниях быстрогорящего ВВ это-
ГМу, очевидно, мешает нитроэфир, в частности тем, что поглощает
ьтепло на свое испарение). Именно таким образом ведут себя смеси
»с пикратом калия, причем смесь с нитрогликолем затухает при
большем содержании пикрата, чем с медленно горящим дигликоль-
455
динитратом. Картина, по-видимому, сходная с описанной выше, на-
блюдалась А. Ф. Беляевым [5] для смесей стифната свинца с три-
нитротриазидобензолом.
Полученные данные позволяют также оценить скорость горения
азида свинца с большим приближением, чем это можно было сде-
лать на основании предшествующих работ. Подставляя в уравнение
uM = 0,035 (а/см2сек) значение дт = 4,71 а/см3 (удельный вес
а-азида свинца), мы получим линейную скорость горения чистого
азида свинца при атмосферном давлении равной 3,5 см/сек.
Напомним, что по Беляеву [5] скорость горения азида свинца
по крайней мере на порядок больше и составляет 50—100 см/сек.
Следует отметить, однако, что эта величина получена экстраполя-
цией на чистый азид скоростей горения смесей его с гремучей
ртутью прн температуре жидкого азота. Содержание азида в сме-
си не превышало 15% (при этой концентрации горение переходи-
ло в детонацию). Очевидно, что надежность величины, полученной
столь далекой экстраполяцией, при таком экзотическом горючем
как гремучая ртуть, да к тому же в совершенно неизведанной обла-
сти низких температур весьма невелика.
По-видимому, скорость, с которой горел бы азид свинца, если
бы он мог гореть при атмосферном давлении, значительно ниже.
На это указывают, кроме всего прочего, результаты опытов по го-
рению смесей азида свинца с гремучей ртутью и со стифнатом свин-
ца, проведенных Б. С. Светловым еще в 1952 г. При изучении горе-
ния этих смесей (с .добавлением небольших количеств битума)
в манометрической бомбе им было показано, что введение в смесь
азида очень сильно уменьшает скорость горения стифната и прак-
тически не изменяет скорости горения гремучей ртути. На этом ос-
новании естественно предположить, что скорость горения азцда
свинца мало отличается от скорости горения гремучей ртути (при
атмосферном давлении ~1,5 см]сек. [7]) и значительно меньше, чем
стифната свинца (~30 см/сек) [8].
Если это предположение справедливо, то объяснение отсутствия
у азида свинца способности к устойчивому горению феноменально
большой скоростью горения этого вещества, очевидно, ошибочно.
По-видимому, соображения, заключающиеся в том, что взрыв азида
свинца при поджигании обусловлен интенсивным диспергирова-
нием и взрывоподобным сгоранием слоя образовавшейся взвеси [4].
являются более обоснованными и лучше отражают сущность яв-
ления.
Автор глубоко благодарен Профессору К. К- Андрееву за по-
мощь в проведении настоящей работы и объяснении ее результатов.
ЛИТЕРАТУРА
1. Андреев К. К., Термическое разложение и горение взрывчатых веществ.
Госэнергоиздат, 1957.
2, А н д р е е в К. К., Proc. Roy. Soc., 1958, А 240, 257.
3. А н д р е е в К. К-, Изв. АН СССР ОТН, Энергетика и автоматика, 1959.
№ 4, стр. 188.
4. Андреев К. К, и Кондриков Б. Н., ДАН, 1961, 137, вып. 1.
456
5. Беляев Л. ф,, Сб. «Физика взрыва», АН СССР, 1952, № 1, стр. 185.
6. Беляев А. ф., Сб. «Физика взрыва», АН СССР, 1952, № 2, стр, '67
7. Беляев А. Ф. и Коротков А. И., Сб. «Физика взрыва» АН СССР
1952, № 1, стр. 177.
8. Светлов Б. С. и Фогельзанг А. Е., ДАН СССР, 1961, 137, вып. 3.
А. И. ГОЛЬБИИДЕР
30. НЕКОТОРЫЕ ЗАКОНОМЕРНОСТИ ГОРЕНИЯ ЛЕТУЧИХ
МНОГОКОМПОНЕНТНЫХ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
При горении некоторых жидких взрывчатых смесей (в дальней-
шем — ЖВС) были обнаружены своеобразные изменения скорости
и характера горения по мере его распространения. В случае летучих
взрывчатых веществ, к которым относятся изученные Ж^С, меха-
низм явления, названного дробным выгоранием, кратко заключает-
ся в следующем.
В процессе горения, как известно [2], начальной стадией может
являться испарение В В, а химические реакции, и, соответственно,
выделение тепла, идут, главным образом, если не исключительно
, в газовой фазе, в парах. Для тайих веществ термокинетнческне ха-
рактеркстики ведущей реакции горения, а следовательно, также
скорость и характер последнего, определяются не составом конден-
сированного ВВ, а составом паров над ним. При горении индиви-
. дуальных ВВ состав пара совпадает с составом конденсированной
. фазы и во всяком случае, остается постоянным**при горении в не-
г изменных условиях. Напротив, когда ВВ является смесью несколь-
; ких компонентов, упругость паров которых при одинаковой темпе-
ратуре существенно различна, состав пара может более или менее
значительно отличаться от состава жидкости и изменяться по мере
ее испарения (выгорания). Изменение состава пара ведет к изме-
нению скорости и характера горения. В определенных условиях
составы прогретого поверхностного слоя и остальной жидкости мо-
гут достаточно быстро выравниваться. В таких случаях наблюдает-
. ся более или менее непрерывное изменение состава смеси и, соот-
ветственно, характеристик ее горения по мере распространения по-
следнего. В других случаях, когда условия для выравнивания кон-
центраций неблагоприятны, соответствующие изменения составов
жидкости и пара, а следовательно, и скорости горения, будут про-
исходить лишь в процессе выгорания сравнительно тонкого прогре-
того слоя. В таких условиях дробное выгорание может приводить
к периодическим изменениям характеристик горения — пульсациям,
различной частоты и амплитуды.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В большей части описываемых ниже опытов, ЖВС сжигали на
воздухе при атмосферном давлении и комнатной температуре
в стеклянных трубках с внутренним диаметром около 0,5 см. Зажи-
гание производили проволочной спиралью, накаливаемой током.
457
Смеси с тетранитрометаном
В этих условиях стехиометрическая смесь бензола и тетранитро-
метана (содержание бензола 13,7% вес.) сначала горит с весьма
малой скоростью — около 0,01 см1сек. Поверхность горящей жидко*,
сти остается в это время совершенно ровной, примыкающий к ней'
фронт пламени светится чрезвычайно слабо, а над срезом трубки
наблюдается вторичное пламя. В дальнейшем такой режим мы на*
зываем горением с малой скоростью.
По мере сгорания скорость горения растет сначала медленно, а
затем быстрее. Исходная высота столба ЖВС в трубке составляла
в наших опытах 5—10 см. После того как сгорит 40—45% исход-
ного количества смеси, скорость горения сравнительно быстро (на
участке 3—5 мм) увеличивается в 2—3 раза и достигает 0,05—
0,06 см!сек,. При этой скорости явно изменяется и характер горения:
появляется яркое пламя над мениском ЖВС, в поверхностном слое
горящей жидкости наблюдается выделение многочисленных мелких
пузырьков, вторичное пламя исчезает. В таком режиме, называемом
в дальнейшем горением с большой скоростью, процесс идет до
конца.
По мере увеличения содержания окислителя в смесях тетранитро*
метана и бензола среднее значение малой скорости 1 увеличивается,
а длина участка, на котором существует этот режим горения — со-
кращается. При содержании тетранитрометана в смеси 90% вес. и
более горение с момента поджигания идет с большой скоростью.
По мере увеличения содержания бензола в исходной смеси, в об-
ласти отрицательного кислородного баланса, наблюдается умень*
шенне среднего значения малой скорости и увеличение длины уча-
стка, на котором горение идет в этом режиме. Наконец, при содер*
жании бензола более 20% вес., горение в принятых условиях опыта
(диаметр трубки, температура) становится невозможным. Резуль*
таты опытов со смесями разного исходного состава представлены
на фиг. 1, где помимо значений малой и большой скоростей горения
нанесены также равновесные кривые начала кипения и конденсации
ЖВС, рассчитанные по упругостям паров чистых компонентов в
предположении, что смесь является идеальной. Кроме того, на гра*
фике нанесены и экспериментальные точки «начала кипения»1 2 3.
Мы отмечали выше, что после того как установится режим боль*
шой скорости, горение при этом режиме идет до конца. Скорость
горения, однако, не остается постоянной, а весьма медленна умень*
шается. Постепенное уменьшение большой скорости наблюдается
1 Здесь и в большинстве дальнейших опытов скорость горения определяли
как отношение длины некоторого участка столба жидкости (обычно не менее
1 е.ч) к времени его сгорания, измеренному секундомером.
3 В связи с очевидными трудностями исследования равновесна «жидкость-
пар» для высокочувствительных ЖВС, мы ограничились определением температур
начала кипения по методу М. Н. Сиволобона [7]. Контрольные опыты с хорошо
изученными невзрывчатым и бинарными системами показали, что определенные
таким методом температуры начала кипения оказываются на 3—7° выше равно*
весных.
458
при горении как смесей такого исходного состава, при котором на
начальном участке процесс идет с малой скоростью, так и смесей
с положительным кислородным балансом, где такого участка нет,
При горении слоя ЖВС начальной высотой 6,5 см, измеряли время
сгорания трех участков длиной по 2 см. Для смеси 10% бензола и
90% тетранитрометана сре,
стке измерения составила
0,06 см!сек, а на ниж-
нем— 0,05 см! сек. Для
смеси, содержавшей 7%
вес. бензола, в аналогич-
ных условиях соответст-
вующие скорости были
0,050 и 0,035 см!сек.
С ростом начального
давления скорость горе-
ния рассматриваемой
ЖВС, также как других
летучих ВВ, возрастает.
При этом изменения ско-
рости и характера горе-
ния по мере выгорания —
ослабевают и начиная
с давления около
1,5 кг1смг становятся не-
уловимыми; фронт горе-
ния распространяется с
видимой равномерностью
по всей длине трубки *.
Изменения скорости и
характера горения по
мере выгорания более или
блюдались и при горении некоторых других бинарных ЖВС тетра-
нитрометана с жидкими горючими, например, с метанолом, этано-
лом, этилннтратом, диэтиловым эфиром, толуолом и др. Из числа
изученных ЖВС, мы приведем данные для смеси с этилннтратом,
где эти изменения наиболее резки и своеобразны. Если определять
зависимость «средняя скорость горения — состав ЖВС», то для
смесей тетранитрометана с этилнитратом можно отметить в опре-
деленном интервале концентраций—резкий максимум скорости.
Средние скорости, рассчитанные по времени сгорания слоя смеси
высотой около 5 см, представлены на фиг. 2. В интервале концен-
траций этилнитрата от 28 до 35% скорость горения резко воз-
растает; почти сразу после поджигания горение переходит на тур-
булентный режим, а в двух случаях (отмечены на фиг. 2 стрел-
скорость горения на
верхнем уча-
725
ПЛ
Р,025-_
« 775
I W
^100
B-S5F
90 -
85
30 20 Ю
10 80 90
Состав ЖВС в % вес.
л-зксперииентшпные точки начала,
кипения
Фиг. 1. Зависимость средней-" скорости горения
смеси тетранитрометана и бензола от состава.
/—средняя скорость рас пространен ян пламени на
участке горения с малой скоростью, Передняя ско-
рость распространения пламени на участке горения
с большой скоростью, 1? и 4—равновесные кривые
температур конденсации и кипения.
Пунктиром отмечен стехиометрический состав.
с
%
0 бензол
lOOTem/MHum*
рометан
менее подобные описанным выше, на-
1 При повышении начального давления выше 2 кг/см2 горение переходит на
турбулентный режим, а при еще более высоком давлении наблюдается переход
горения в детонацию. Эти явления мы здесь не рассматриваем,
459
ками) возникал взрыв. При быстром горении явления дробного
выгорания—отсутствуют. Горение смесей, начальный состав кото-
рых лежит вне указанного интервала концентраций, идет со ско-
ростью, сильно изменяющейся по мере распространения. Ряд сме-
сей тетранитрометана и этилнитрата различного начального со-
става мы сжигали в трубках длиной 6—8 см и измеряли время сго-
рания на различных участках слоя жидкости.
Результаты опытов представлены на фиг. 3. Скорости горения,
рассчитанные по времени сгорания определенных отрезков по дли-
Средняя скорость горения 5 см/ сек
фиг. 2. Зависимость средней скорости
горения смеси этилнитрата и тетрапи-
трометапа от состава.
фиг. 3. Изменение скорости горения по
мере выгорания смесей этилнитрата и
тетранитрометака разного начального
состава.
(Числа у кривых — содержание эгилвитрага
в исходной смеси а •/« вес.).
не заряда, отнесены при построении графика к серединам соответ-
ствующих отрезков. Каждая из кривых фиг. 3 построена по 2—4 па-
раллельным опытам. Из графика фиг. 3 видно, что горение смесей,
содержащих значительный против стехиометрии избыток горючего,
ускоряется по мере распространения процесса. Напротив того, ско-
рость горения смесей с большим избытком окислителя по мере сго-
рания уменьшается, подчас так значительно, что горение затухает.
При горении многих смесей тетранитрометана с жидкими орга-
ническими горючими систематических изменений скорости распро-
странения пламени мы не наблюдали и горение шло с видимой (при
визуальном наблюдении) равномерностью. К числу таких ЖВС от-
носятся, например, растворы в тетранитрометане октанола, нитро-
бензола, дигликольдинитрата, мононитробензола, моно- и динитро-
толуола и др.
460
He приводя всех экспериментальных данных по каждой из изу-
ченных ЖВС, мы перечислим ниже лишь основные общие законо-
мерности, обнаруженные в ходе исследования;
1. В смесях, которые по упругостям и составу паров существен-
но отклоняются ’ от соответствующих параметров идеальных си-
стем— явлений дробного выгорания, мы, как правило, не наблю-
дали. Равновесие жидкость—пар для ЖВС тетранитрометана с ор-
ганическими горючими, насколько нам известно, никем не изуча-
лось. В связи с этим нет достаточно достоверных данных о природе
и характере отклонений от идеальности для разных смесей. В не*
которых случаях, примером которых может служить смесь тетрани-
трометана с фурфуролом, образуется химическое соединение; в дру-
гих —возможно образование азеотропов.
2. Явления дробного выгорания такого характера, как описан-
ные выше, для смесей бензола или этилнитрата с тетранитромета-
ном мы наблюдали только в системах, где компонент, обладающий
большей упругостью пара, имел одновременно и плотность, суще-
ственно меньшую, чем второй компонент ЖВС. Так, Скорость горе-
ния изменялась по мере распространения процесса для смесей с ме-
танолом и этанолом, которые имеют большую упругость пара и
меньшую плотность, чем тетранитрометан, но для системы окта-
нол—тетранитрометан, где окислитель, обладающий большей плот-
ностью, является более летучим компонентом — таких изменений
мы не наблюдали.
3. Если сопоставлять свойства бинарных ЖВС тетранитромета-
на с несколькими горючими, близкими Друг другу по химической
природе, например, с гомологами, то можно видеть, что дробное
выгорание наблюдается тем отчетливее, чем больше разность упру-
гостей пара и плотностей компонентов смеси. Так, изменение ско-
рости горения по мере распространения его весьма значительно для
ЖВС с бензолом и почти неуловимо для смеси с толуолом; анало-
гичные соотношения можно отметить для ряда метанол—этанол—
пропанол—бутанол и других.
Смеси с азотной кислотой и четырехокисью азота
Дробное выгорание характерно для многих ЖВС не только с
тетранитрометаном, но и с другими летучими окислителями. Рас-
смотрим, например, данные, полученные для смесей четырехокиси
' азота и нитробензола. При атмосферном давлении и комнатной тем-
пературе, в трубках диаметром около 1 см, смеси указанных ком-
понентов нулевого (содержание четырехокиси азота 71,8% вес.) и,
тем более, положительного кислородного баланса—к горению не-
способны. Смеси с значительным против стехиометрии избытком
горючего (содержание нитробензола 36% вес. и более) горят в
1 Показателем и мерилом такого отклонения мы считали разность между тем-
пературой начала кипения определенной экспериментально (см. сноску1 на
стр. 458) и найденной расчетом, выполненным по законам идеальных смесей.
461
J;
принятых условиях, Горение распространяется с небольшой, по-
степенно снижающейся, скоростью (на начальном участке около
0,01 см[сек.) и затухает после выгорания некоторой доли смеси; эта
доля несколько увеличивается с ростом содержания нитробензола
в исходной смеси.
Сходные явления мы наблюдали и при горении ЖВС на основе
концентрированной (93—98%) азотной кислоты. Смесь этого оки-
слителя с мононитробензолом при атмосферном давлении способ-
на к горению лишь в определенном интервале составов. При этом
после выгорания некоторой части смеси скорость горения умень-
шается настолько, что дальнейшее распространение процесса
в трубках малого диаметра становится невозможным. Средняя ско-
рость горения и длина сгорающего слоя ЖВС растут по мере уве-
личения содержания горючего. Результаты некоторых опытов, про-
веденных в стеклянных трубках диаметром 9 мм, при температуре
50э представлены в таблице. При повышенной температуре скорость
горения данной ЖВС также, как и других ВВ —увеличивается, од-
новременно, с ростом температуры растет и относительная длина
слоя ЖВС, сгорающего до затухания.
Таблица
Длина слоя ЖВС концентрированной азотной кислоты и моноиитробеизола,
сгорающего при атмосферном давлении
(Начальная высота слоя 6.) мм. нт "ильная температура 50\
внутренний диаметр трубки 9 мм)
Содержанье горючего в смеси % сбъемн. Длина сгоревшего слоя в мм
максимальная средняя из 3—5 опытов Примечание
27 0.5 .. _ - —
29 4 2 —
31 10 7 Состав нулевого кислородного ба- ланса
33 37 15
35 60 30 *
Значительное количество опытов было выполнено со смесями
азотной кислоты и дихлорэтана. Здесь также во многих случаях на-
блюдалось затухание после выгорания части смеси, причем с изме-
нением соотношения между компонентами можно отметить следую-
щее: смеси с отрицательным кислородным балансом горят с очень
малой скоростью и быстро затухают. По мере увеличения содержа-
ния окислителя начальная скорость горения и длина сгорающего
слоя ЖВС — возрастают. Максимумы скорости горения и длины
сгорающего слоя наблюдаются для смесей с примерно двойным
избытком кислоты против стехиометрии; дальнейшее увеличение ее
содержания приводит к снижению начальной скорости горения и
сокращению длины сгорающего слоя. На фиг. 4 на участок тре-
462
.угольной диаграммы нанесены результаты опытов по сжиганию
[тройных смесей дихлорэтан—азотная кислота—вода в стеклянных
- трубках диаметром 5—7 мм. при атмосферном давлении и темпера-
туре 40°. В этих опытах мы условно считали, что смес[> неспособна
к горению, если последнее затухает раньше, чем выгорит слой
толщиной 5 мм. Из графика видно, что определенная таким обра-
зом область составов, способных к горению, расположена несим-
: метрично относительно линии, соединяющей составы нулевого кис-
Фиг. 4. Участок треугольной диаграммы для смесей
дихлорэтан—азотная кислот а—вода.
О —горение распространилось по трубке более чем на
~" '' _ ______ олеума,
затухло* на участке длиной менее 0,5 ок,
.г_____г______ т-:. ___", 2—граница
области расслоения смесей с добавками олеума, 3—линия
0,5 ел, X—то же, для смесей с добавками
• —горение , .....’
/—граница горючести для тройных смесей,
стехиометрических составов.
к горению.
'дородного баланса. Смеси, содержащие большой избыток окисли-
теля, еше способны к горению при значительном разбавлении
инертным компонентом (водой). В дальнейших опытах к ЖВС, ко-
торые по составу находились на пределе горючести, для данных
условий, мы добавляли различные количества олеума (около 20%
свободного SO3). Оказалось, что дальнейшее разбавление жидко-
сти инертным веществом делает ее вновь способной
3 опытах со смесями, содержавшими олеум, сгорал слой более
Б мм для всех изученных составов вплоть до такого содержания
рлеума, при котором начинается расслоение смеси. Ориентировоч-
ная граница области, где дихлорэтан, азотная кислота, вода и
[верная кислота еще образуют однофазные системы, также показана
На фиг. 4; все испытанные смеси, находящиеся внутри этой обла-
сти, оказались способными к горению. В опытах со смесями, содер-
кавшими добавку олеума, горение никогда не распространялось
463
41
отдельных участков высотой 0,5
5 0,08
J_____I____I____1
* о 20 W 60 80 100
Относительная Зли на сгорев-
шего слоя Ж8С 8 %
Фиг. 5. Изменение скорости горения
по мере выгорания смеси дихлорэтана
и азогной кислоты при разных темпе-
ратурах.
до конца трубки. Перед затуханием наблюдалось помутнение
остатка жидкости, обусловленное началом расслоения на две жид*
кие фазы.
Изменение скорости горения по мере выгорания в стехиометри-
ческой смеси дихлорэтан — азотная кислота (97,3% HNO3) можно
видеть по результатам опытов, приведенным на фиг. 5. В этих опы-
тах так же, как в показанных на фиг. 3, измеряли время сгорания
см по длине трубки, рассчитывали
среднюю скорость горения на
данном участке и для построе-
ния графика относили эту ско-
рость к середине соответствую-
щего участка.
Следует особо подчеркнуть,
что дробное выгорание, наблю-
давшееся у некоторых из изу-
ченных ЖВС, происходило лишь
при сравнительно медленном го-
рении и при низком давлении.
С ростом давления, под которым
идет горение, изменения ско-
рости последнего по мере выго-
рания ЖВС становятся все сла-
бее, и, начиная с некоторого дав-
ления, — больше не наблюдаются.
Для разных ЖВС это давле-
ние различно; в опытах с составами, близкими к стехиометриче-
ским, оно составляло около 1,5 к^см2 для смеси тетранитрометана
и бензола, около 5 кг]см? — для смеси азотной кислоты и дихлор-
этана, и более 10 кг]см?— для смеси азотной кислоты и нитро-
бензола.
Растворы полимера в жидком ВВ
Явления дробного выгорания мы наблюдали не только для ЖВС
типа горючее—окислитель. В работе, выполненной совместно
с В. В, Горячевым [4], описаны изменения скорости и характера
горения по мере распространения его для жидких ВВ, слегка загу-
щенных растворением высокомолекулярных соединений (полиме-
тилметакрилат).
В случае медлецногорящего этилнитрата эти изменения того
же характера, что и для некоторых из описанных выше ЖВС: ско-
рость горения загущенного нитроэфира уменьшается по мере рас-
пространения процесса вплоть до предела, при котором горение в
трубках данного диаметра становится невозможным из-за тепловых
потерь.
В случае быстрогорящего метилнитрата растворение хотя бы
небольших количеств полимера резко меняет характер горения —
оно становится пульсирующим, короткие периоды относительно
медленного распространения чередуются с более или менее интен-
464
сивными вспышками, Заметная доля ВВ быстро сгорает именно в
результате таких вспышек, что приводит к уменьшению общего
времени сгорания столбика ВВ данной высоты, т, е, как бы к росту
скорости горения. По мере увеличения содержания полимера в рас-
творе — частота и интенсивность пульсаций — возрастают. Как
показала скоростная киносъемка, горение слегка загущенного ме-
тилнитрата состоит из ряда, примерно одинаковых циклов. В нача-
ле каждого цикла поверхность горящей жидкости опускается мед-
ленно, оставаясь все время ровной; скорость горения на этом уча-
стке меньше, чем для незагущенного метилнитрата (около
0,08 см!сек при содержании 0,1 % вес, полимера и около 0,05 см]сек.
при добавлении его в количестве 0,25%, незагущенвая жидкость —
0,12 см]сек). В дальнейшем в поверхностном слое жидкости появ-
ляются пузырьки, образуется слой пены, растущей по толщине и,
наконец, происходит резкий выброс поверхностного слоя в зону пла-
мени и быстрое его сгорание в виде взвеси отдельных капель. После
сгорания взвеси цикл повторяется. Описанные пульсации, также,
как другие проявления дробного выгорания подавляются с ростом
начального давления.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Изучение горения смесей особенно наглядно показало, что
именно состав пара, а не самой жидкости определяет возможность
горения в данных условиях, скорость и другие характеристики про-
цесса. Весьма характерны в этом отношении результаты опытов со
смесями дихлорэтана и азотной кислоты, приведенные в экспери-
ментальной части. Разбавление смеси водой делает горение невоз-
можным из-за неблагоприятного состава пара. При дальнейшем
разбавлении жидкости балластным веществом (олеум), горение
становится возможным, так как вода прочно связывается в моно-
гидрат серной кислоты и в паровую фазу вначале не поступает. В
^случае смеси нитробензола и четырехокиси азота, окислитель име-
ет при одинаковой температуре значительно большую упругость
papa, чем горючее. Поэтому, если состав жидкости соответствует,
Цапример, нулевому кислородному балансу, состав пара над нею
содержит такой большой избыток окислителя, при котором горение
Ьод атмосферным давлением в трубке малого диаметра — невоз-
[ожно. Аналогичным образом могут быть объяснены и результаты
ПЫтов с другими смесями.
Решающая роль состава пара позволяет объяснить и наблю-
;авшиеся явления дробного выгорания ЖВС. Рассмотрим несколь-
;о Подробнее данные по горению смеси тетранитрометана и бензо-
ta. Ввиду того, что упругость паров бензола заметно выше, чем
етранитрометана, концентрация горючего в парах должна быть
ущественно больше, чем в жидкости. По расчету, выполненному
<Ля идеальной смеси, при содержании бензола в жидкости около
5% вес., содержание его в парах составит более 40%. В результа-
е, над жидкостью стехиометрического состава пар будет содержать
D 451 465
значительный избыток горючего, что и обусловливает очень малую
начальную скорость горения. В процессе горения жидкость прогре-
вается с поверхности, соответственно с поверхности происходит и
испарение; поверхностный слой ЖВС обедняется бензолом и обо*
гащается тетраннтрометаном. Одновременно происходит выравни-
вание состава поверхностного слоя и всей остальной жидкости,
в первую очередь, при помощи гравитационных токов, в связи с тем.
что плотность тетранитрометана почти в два раза выше плотности
бензола. По мере обогащения окислителем поверхностного слоя
плотность его становится больше плотности нижележащих слоев
жидкости, что благоприятствует перемешиванию и выравниванию
состава жидкости на значительную глубину от поверхности. Вырав-
ниванию концентраций различных слоев жидкости способствует
также и диффузия, но скорость последней в жидкостях существен-
но меньше, чем скорость конвективного перемешивания по описан-
ному выше механизму. Когда скорость горения очень мала, как это
наблюдается в случае смесей бензола с тетранитрометаном нуле-
вого или, тем более, отрицательного кислородного баланса, вырав-
нивание состава поверхностного слоя и остальной жидкости успе-
вает происходить на значительную глубину. Если слой жидкости
не велик, как это было в наших опытах (10—20 диаметров), то
можно ожидать, что состав всей жидкости в процессе горения ус-
певает выравниваться. Тогда, по мере распространения горения бу-
дет происходить изменение состава всей жидкости подобное тому,
которое наблюдается при простой перегонке бинарных смесей из
компонентов с различной летучестью. Обогащение жидкости
труднолетучнм компонентом (в данном случае тетранитро-
метаном), вызывает соответствующее изменение состава пара; со-
отношение между количествами горючего и окислителя в ларах
приближается к стехиометрическому для ведущей реакции горения,
соответственно возрастают температура в пламе'ни и скорость реак-
ции, фронт пламени приближается к поверхности жидкости и ско-
рость горения увеличивается.
Рассмотренная выше картина дробного испарения компонентов
смеси с поверхности и выравнивания концентраций поверхност-
ного слоя и остальной жидкости может наблюдаться только при
достаточно малой скорости горения. Если эта скорость по каким-
либо причинам (повышение давления или температуры, изменение
состава смеси) возрастет, то глубина, на которую успевает про-
изойти перемешивание и выравнивание состава за время сгорания
прогретого поверхностного слоя — уменьшается. При достаточно
большой скорости горения процессы дробного испарения и обу-
словленного им дробного выгорания проявляются очень слабо и
лишь при сгорании прогретого слоя жидкости. В результате могут
возникнуть периодические изменения скорости и характера горе-
ния. При малой толщине прогретого слоя, соответствующей боль-
шой скорости горения, частота таких изменений — велика, а ам-
плитуда — очень мала, так что они существенной роли не играют
и даже не улавливаются при обычной постановке опытов.
46S
Г Дробное выгорание не наблюдается в ряде случаев и прн ма-
|лой скорости горения. Тривиальными причинами такого явления
Смогут быть образование азеотропов или сравнительно устойчивых
L химических соединений компонентов смеси в жидкой фазе. В слу*
[чаях, когда плотность более летучего компонента смеси выше, чем
[ плотность менее летучего, дробное испарение приводит к умень-
в- шению плотности верхнего слоя жидкости. В таких случаях переме*
L шнвание может происходить только за счет диффузии, т. е. крайне
г медленно. Именно поэтому мы не наблюдали явлений дробного
г. выгорания, например, для бинарных ЖВС тетранитрометана с ок-
Е'-танолом и ксилолом, в то время как они отчетливо наблюдаются
К для смесей того же окислителя с метанолом, этанолом и бензолом,
t Можно ожидать далее, что дробное испарение перестанет играть
* роль и в случае горения ВВ в таких условиях, когда существенную
J роль начинают играть реакции, идущие в конденсированной фазе.
Г-- Следует отметить, что явления дробного испарения при rope-
s'. нии невзрывчатых жидкостей наблюдались неоднократно, Напри-
S. мер, при горении таких многокомпонентных смесей, как нефть и не-
L которые нефтепродукты [3], [6] или водные растворы спиртов [8].
» Возникновение пульсаций в результате дробного выгорания
л мы наиболее отчетливо наблюдали в случае растворов полиметил*
t метакрилата в летучем нитроэфире — метилнитрате. В свете пред*
t шествующих работ (см. напр. [1], [9]) возникновение пульсации при
[’увеличении вязкости представлялось неожиданным. В цитирован-
I ных работах было показано, что загущение стабилизирует горение
№ и желатинированные ВВ горят равномерно в тех условиях, в ко-
и торых те же вещества, но при малой вязкости горят с пульсация-
& ми. Известны и теоретические обоснования стабилизирующего
к влияния вязкости [5].
|- В изученных нами случаях возникновение пульсаций при загу-
I щении определяется тем, что применявшийся полимер гораздо ме*
в нее летуч, чем жидкое ВВ, которое им загущали и, в то же время
кон сам по себе неспособен к экзотермическому превращению в кон-
£денсированной фазе. При горении происходит дробное испарение
L прогретого слоя и содержание летучего компонента в нем умень-
мшается. Соответственно возрастает вязкость этого слоя, поверх*
Юность которого становится сравнительно прочной пленкой, препят-
[ ствующей дальнейшему удалению пузырьков пара. Повышение
К вязкости прогретого слоя затрудняет одновременно выравнивание
Е его состава со всей остальной жидкостью. В результате роста вяз*
|кости скорость горения ЖВС снижается, толщина прогретого слоя
Еувеличивается и, он превращается в пену из пузырьков пара, зам*
К инутых вязкой пленкой. Когда давление пара в пузырьках ставо-
рвнтся достаточно велико, пленка прорывается. При этом в зону
| горения сразу выбрасывается значительное количество пара и ув*
[леченных им капель жидкости, которые быстро сгорают в форме
^.вспышки. после чего цикл явлений повторяется. Средняя скорость
& распространения такого процесса может быть больше, чем для
^равномерного горения незагущенного вещества. При повышении
». 30* 467
давления, под которым идет горение, режим пульсации, обуслов-
ленный испарением летучего компонента — подавляется. Возмож-
но также, что пульсации перестают быть заметны из-за уменьше-
ния их амплитуды и увеличения частоты, вследствие роста скоро-
сти горения и сокращения толщины прогретого слоя.
Рассмотренная картина наблюдается при сравнительно боль-
шой скорости горения, характерной для метилнитрата. В случае
несколько менее летучего и медленногорящего ВВ —этилнитрата,
то же явление выражено не так отчетливо. При медленном горе-
нии испарение более летучего компонента может привести не к
локальному изменению состава тонкого прогретого слоя, а к по-
степенному изменению состава в слое значительной толщины.
Амплитуда пульсаций при этом уменьшается. В случае этилнит-
'рата, это явление приводит к постепенному обогащению полиме-
ром слоя возражающей толщины и падению скорости горения,
вплоть до затухания его при малом диаметре трубки из-за тепло-
вых потерь.
В заключение отметим, что явления дробного испарения воз-
можно не являются единственным механизмом, приводящим к
различию в скоростях выгорания отдельных компонентов смеси.
Для труднолетучих многокомпонентных систем существенные раз-
личия в скоростях реакций компонентов могут, при определенных
условиях играть ту же роль, которую в случае летучих ВВ играют
различия в упругости пара.
ЛИТЕРАТУРА
I, Андреев К. К.. Термическое разложение и горение ВВ. М.—Л., Госэнер-
Тоиздат, 1957. стр. 130—133; ЖФХ, 1946, 20, 467.
2, Беляев А. Ф., Сборник статей по теории взрывчатых веществ под рел.
К, К, Андреева и Ю. Б. Харитона, Оборонена, 1940, стр. 21—38; докторская диссер-
тация ИХФ АН СССР, 1946 г.
3. Б л и и о в В. И, ДАН СССР, 1953, 89, 101.
4. ГольбиндерА. И. и Горячев В. В., ЖФХ, 1961, 35, 1808.
5. Л е в и ч В. Г., ДАН СССР, 1956, 109, 975.
6. Худяков Н. Г. Изв. АН СССР, ОТН, 1945. 10—И, 1185; 1948, 4, 579,
1951, 7, 1015.
7. Si voloboU М. N.,Berichte, 1891,24,944.
8. Weise Н., Lorell 1., Wood В. I., Fifth Symposium on Combustion.
1955, p. 132.
9, Whittaker A. I., Donovan T, M. Williams H., J. phys. Chem., 1958
62, № 8, 908.
А. И. ГОЛЬБИНДЕР
31. ГОРЕНИЕ САМОВОСПЛАМЕНЯЮЩЕЙСЯ ВЗРЫВЧАТОЙ
СМЕСИ И ПЕРЕХОД ЕГО В ДЕТОНАЦИЮ
Изучение явлений перехода горения взрывчатых веществ в де-
тонацию представляет существенный интерес во многих отноше-
ниях. В случае самовоспламеняющихся жидких взрывчатых смесей
468
наблюдается.ряд особенностей в условиях перехода горения в дето-
нацию, которые были исследованы на примере смесей тетранитро-
метана и органических аминов, в первую очередь анилина.
Смесь тетранитрометана и анилина, состава близкого к усло-
вию нулевого кислородного баланса, самовоспламеняется после
некоторого индукционного периода, который в обычных условиях
(атмосферное давление, комнатная температура) составляет око-
ло минуты. Исследование процесса самовоспламенения, в частно-
сти реакций, протекающих в период задержки, позволило уста-
новить следующие характерные черты этого явления.
1. В течение индукционного периода температура жидкости
практически не отличается от начальной. Очень небольшой разо-
грев (2—9’) наблюдается лить к самому концу этого периода.
2. В конце индукционного периода начинается ускоряющееся
образование газов. Пузырьки последних прорываются через по-
верхность жидкости, увлекая за собой мельчайшие ее капли.
3. Сильно экзотермическая реакция, приводящая к воспламе-
нению, возникает в газовой фазе, или точнее, в газопарокапельной
взвеси над поверхностью жидкости. Воспламенение газов или па-
ров приводит к горению жидкой смеси.
Горение самовоспламенившейся смеси распространяется с зна-
чительной скоростью, приближающейся, или даже превышающей
критическую скорость, при которой возникает автотурбулизация
поверхности горящей жидкости вследствие эффекта Ландау [6]’.
Так, в трубках диаметром 13 и длиной 50 мм при 18—21°, средняя
скорость горения такой смеси составляла от 0,44 до 0,80 г/см2сек.
Характерна при этом неравномерность распространения горения,
сильные пульсации пламени и значительный (в 1,5—2 раза) раз-
брос времени сгорания участков постоянной длины и параллель-
ных опытах. Горение в трубках сопровождается резким свистя-
щим звуком. Примыкающая к поверхности жидкости зона первич-
ного пламени имеет значительную ширину и ярко светится. Вто-
ричное пламя над срезом трубки едва заметно из-за малой ярко-
сти. Трубки даже из тугоплавкого стекла заметно оплавляются
при горении смеси, что не наблюдается в случае других жидких ВВ,
Так же как и для других взрывчатых жидкостей, при переходе
На пульсирующий режим в определенных условиях можно наблю-
,ать затухание горения. В случае самовоспламеняющихся смесей
ассматриваемого типа (и это отличает их, например, от нитро-
фиров) такое затухание является лишь временным. Реакции, црн-
едшие к самовоспламенению, не прекращаются после затухания,
бразование активных газообразных веществ продолжается и они
1Новь вспыхивают над поверхностью жидкости. Таким образом,
орение смеси тетранитрометана и анилина (а, вероятно, и других
минов) может протекать в форме чередующих’ся вспышек и за-
1 Значение критической скорости для данной смеси Определить затрудни
ельно Мы полагаем, что она того же порядка, как установленная К. К. Андрс-
вым [I] для нитроэфиров и найденная нами Для некоторых жидких несановос-
ламеняющихся смесей, т. е. 0,25—0,3 г/см2 сек.
460
туханий. В отдельных случаях перерывы горения достигают
0,5—1,5 сек., в других—они так малы, что при визуальном наблю-
дении заметить их нельзя.
Рассматриваемая смесь способна к самовоспламенению и горе-
нию в довольно широком интервале концентраций. Наибольшая
скорость горения наблюдается в случае смеси, содержащей около
20% вес. анилина (нулевому кислородному балансу соответствует
содержание 15,5% вес. анилина). Дальнейшие опыты проводились
со смесью, состав которой отвечает максимуму скорости горения.
Во многих случаях горение самовоспламенившейся смеси тет-
ранитрометана и анилина, переходило в детонацию. Для выявле-
ния условий этого перехода была поставлена серия опытов, в ко-
торых меняли общую массу приготовляемой смеси и высоту слоя
ее в трубке.
В этих опытах тетранитрометан и анилин, взятые в таких ко-
личествах, чтобы содержание последнего в смеси составляло 19,5%
вес., одновременно через общую воронку сливали в стеклянную
трубку. Воспламенение возникало через 35—55 сек. В части опы-
тов смесь сгорала до конца, в других—возникала детонация. Ре-
зультаты этих опытов сведены в табл. 1. Высоту детонировавшего
слоя смеси определяли по длине отпечатка на металлической пла-
стинке, в контакте с которой находилась трубка.
Из данных таблицы видно, что горение самовоспламенившейся
смеси переходит в детонацию в том случае, если высота слоя жид-
кости превышает некоторую критическую величину. Сопоставляя
результаты опытов в трубках разного диаметра, нетрудно убе-
диться, что именно высота слоя жидкости, а не общая ее масса,
является критическим условием перехода горения в детонацию.
В опытах фиксировали также время от момента самовоспламе-
нения до возникновения детонации и длину сгоревшего слоя смеси,
как разность между исходной высотой столба жидкости в трубке
и длиной отпечатка на металлической пластинке. В тех случаях,
когда горение переходило во взрыв, нам не удалось обнаружить
какую-либо закономерность в изменении длины участка жидкости,
сгоравшей до возникновения детонации. В части параллельных
опытов можно было наблюдать детонацию практически в момент
воспламенения, а в других опытах детонация возникала после го-
рения в течение нескольких секунд.
С ростом начального давления время сгорания слоя смеси оп-
ределенной высоты существенно сокращается (скорость горения
возрастает), а переход горения в детонацию резко облегчается.
В серии опытов, проведенной в бумажных трубках диаметром око-
ло 7 мм, проклеенных жидким стеклом, при высоте слоя смеси
всего 4 см, были получены результаты, приведенные в табл. 2-
Другие изученные амины ведут себя в смеси с тетранитромета-
ном аналогично анилину. Смеси тетранитрометана с о-толуидином,
по времени задержки самовоспламенения и скорости горения близ-
ки к соответствующим смесям анилина. Критическая высота слоя
смеси в стеклянной трубке, при которой горение переходит в дето-
470
Таблица 1
Горение смеси тетранитрометаиа и анилина (80,5:19,5)
прн различной высоте слоя
Внутренний диаметр трубки мм Объем смеси мл Высота слоя смеси см Частость детонации *
10 3,5 5 0/5
13,5 7,0 5 0/5
18 12,7 5 0/3
10 4,9 6 0/3
21 49 14 0/5
5,6 3,5 14 1/4
13,5 21,0 15 0/3
21 50 14 1/4
8 8,8 18 2/4
8 10,9 22 3/4
10 21 27 1/1
5 5,9 30 8/8
8 14 28 3/3
10 22,1 28 2/4
5 7,9 40 2/2
10 35 45 3/3
8 26 52 3/3
10 42 53 4/4
* В числителе число случаев детонации, а в знаменателе число параллель*
[X опытов.
Таблица 2
^Начальное давление ,> кГ[сл&. Время сгорания слоя высотой 4 см в сек. Частость детонации
минимум максимум
L 1 .о 16,0 20,2 0/8
1,8 0,18 7,0 2/8
J 2>2 0,24 0,84 4/8
471
нацию в случае о-толуидина оказалась также около 10 см. Смеси
ксилидина 1 с тетранитрометаном самовоспламеняются с задерж-
кой около 1,5 сек.; горение идет еще быстрее, чем для смеси с ани-
лином, с очень сильными колебаниями скорости, отдельными вспыш-
ками и пульсациями пламени. При таком режиме горения говорить
о какой-либо характерной скорости распространения последнего—
весьма трудно.. Многочисленные опыты со смесями, содержавшими
от 11,5 до 37% вес. ксилидина (нулевому кислородному балансу
соответствует содержание ксилидина 14,7% вес.), при общем объ-
еме жидкого ВВ от 1,0 до 50 мл показали, что независимо от коли-
чества смеси она сгорает до конца, если высота слоя ВВ не более
3 см. При большей высоте слоя — воспламенение регулярно приво-
дит к детонации.
В случае смеси триэтиламина и тетранитрометана самовоспла-
менение возникает с весьма малой задержкой. Нельзя поэтому без
специальных устройств успеть за время задержки смешать сколь-
ко-нибудь значительные объемы горючего и окислителя. При про-
стом одновременном сливе компоненты не успевают смешаться и
просто разбрасываются при воспламенении небольшой доли их.
успевшей образовать смесь. Воспламенение происходит в форме
очень резкой вспышки. В тех опытах, где путем охлаждения ком-
понентов или разбавлением триэтиламина несколько увеличивали
время задержки, даже при высоте слоя в 1,5—3 см, получали прн
самовоспламенении сильный взрыв.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Существенная роль высоты столба жидкости в процессе пере-
хода горения в детонацию может, в данном случае, быть объяс-
нена следующим образом.
В самовоспламенившейся и горящей смеси продолжаются реак-
ции между тетранитрометаном и амином, в результате которых
образуются газы. Пузырьки газов подымаются к поверхности
жидкости и вызывают ее возмущение (отрыв капель, возникновение
рельефа). Если полагать, что реакции, приводящие к газообразова-
нию, идут более или менее равномерно во всем объеме жидкости,
то очевидно, что количество пузырьков, проходящих в единицу вре-
мени через единицу площади поверхности жидкости будет тем
больше, чем больше высота слоя жидкости. Начиная с некоторой
критической высоты слоя, поток газа через поверхность жидкости
вызывает интенсивное диспергирование ее и образование доста-
точно толстого слоя газо-капельной взвеси, последующий взрыв
которой и является по Андрееву [2] причиной перехода горения в де-
тонацию.
Для подтверждения предлагаемого механизма были поставле-
ны специальные опыты, в которых различную интенсивность потока
1 Техническая смесь изомеров.
472
газов можно было получить при одинаковой и достаточно малой
высоте столба жидкости. Смесь тетранитрометана и анилина, та-
кого же состава, как в основной серии опытов, приготовляли в ко-
личестве 2 мл в трубке с внутренним диаметром о мм и в количе-
стве 25 мл в трубке диаметром 18 мм. В обоих случаях высота
столба жидкости была 10 см и во всех опытах (по 6 с трубками
каждого диаметра) смесь сгорала до конца. Затем опыты были
повторены в комбинированной трубке, нижняя часть которой дли-
ной около 4 см имела диаметр 18 мм, а верхняя — 5 лш. Количе-
ство смеси (11,5 мл) выбрали таким образом, что общая высота
слоя ее в комбинированной трубке составила также 10 см. При
выбранных размерах интенсивность потока пузырьков газов через
поверхность жидкости должна быть такой же, как в трубке по-
стоянного диаметра с высотой слоя смеси 50 см. Во всех 5 опытах
в комбинированных трубках горение самовоспламенившейся смеси
перешло в детонацию, которая всегда начиналась в узкой части
трубки.
В случае самовоспламеняющейся жидкой взрывчатой смеси,
образующиеся и прорывающиеся через поверхность жидкости газы
представляют собой, как было указано, активные продукты, спо-
собные к быстрой экзотермической реакции. Это обстоятельство
определяет механизм вспышки газо-капельной взвеси и обуслав-
ливает возможность дополнительного усиления динамического дей-
ствия такой вспышки вследствие одновременного воспламенения
над поверхностью жидкости и внутри пузырька, подходящего к ней
или одновременной вспышки в ряде пузырьков, разделенных тон-
кими прослойками жидкости.
Как показывают описанные здесь опыты, жидкие самовоспла-
меняющиеся смеси ведут себя подобно инициирующим ВВ— регу-
лярный переход горения в детонацию наблюдается в определенных
условиях при атмосферном давлении.
Помимо отмеченных выше явлений, облегчающих образова-
ние и вспышку слоя газо-капельной взвеси и увеличивающих ее
интенсивность, наличие пузырьков газов в жидкости должно су-
щественно повышать чувствительность последней к воздействию
быстрого подъема давления. Хорошо известны данные, например,
Ф. Боудена [4] о том, насколько наличие пузырьков газа повышает
чувствительность жидкого ВВ к механическому удару. Аналогич-
ную роль играют пузырьки и при действии на жидкое ВВ вспышки
взвеси. Очевидно, что в данном случае, когда пузырьки заполнены
не воздухом, содержащим незначительное количество паров ВВ,
как это имеет место, например, в случае нитроглицерина, а реак-
ционноспособной смесью газов (паров) в состоянии, близком к са-
мовоспламенению, достаточен относительно слабый подъем дав-
ления для того, чтобы вызвать воспламенение в пузырьке. Одно-
временное воспламенение в ряде пузырьков (в результате вспышки
взвеси над слоем жидкости) приводит к детонации. 1
Своеобразным вариантом газо-жидкостной взвеси в рассматри-
ваемых системах может быть образование слоя пены из пузырьков
473
'реакционно-способных газов, разделенных пленками жидкого ВВ.
Если высота слоя пены превысит некоторый минимум, то горение
его может перейти в детонацию в результате сложения слабых
ударных волн, по механизму, аналогичному механизму возникнове-
ния детонации в газах [5]. Детонация слоя пены вызывает взрыв
жидкой смеси. Источниками слабых ударных волн могут быть
либо газодинамическое ускорение горения пены за счет прогрессив-
ного роста поверхности горящей жидкости, либо несколько после-
довательных вспышек газа в отдельных пузырьках пены.
В подтверждение того, что взрыв возникает в слое пены, можно
привести следующие косвенные данные: во многих случаях, когда
происходила детонация смеси, на пластинках, к которым прикреп-
лялись трубки, можно было отметить резкое изменение интенсив-
ности «отпечатка». На участке, к которому прилегала верхняя часть
трубки, наблюдалось относительно слабое механическое действие,
а в нижней части—значительно более сильное. Можно предполо-
жить, что слабый отпечаток получается на участке взрыва пены,
которая по сравнению с жидкостью является зарядом малой плот-
ности *.
С представлением о возникновении взрыва в газе или двухфаз-
ном слое хорошо согласуются и результаты опытов при разных
давлениях (табл. 2). С ростом начального давления облегчается
возникновение взрыва жидкости, подобно тому, как это наблюдали
в свое время К- К- Андреев и В. П. Маслов [3] или Е. В. Рдултов-
ская и Ю. Б. Харитон [7] для взрывов гремучего газа.
ЛИТЕРАТУРА
1. Андреев К. К., Термическое разложение и горение взрывчатых веществ,
М.—Л., Госэнергоиздат, 1957, стр. 251.
2. Андреев К. К., Термическое разложение и горение взрывчатых веществ,
М.—Л.. Госэнергоиздат, 1957, стр, 272, Proc. Roy. Soc., 1958, А246, 257.
3. Андреев К. К. и Мас лов В. П., Сборник статей по теории взрывчатых
веществ. Оборонно, 1940, стр. 150.
4. Боуден Ф. и Иоффе А., Возбуждение и развитие взрыва в твердых и
жидких веществах. ИЛ, 1955, стр. 33.
5. Зельдович Я. Б, и Компаиеец А, Сч Теория детонации, Гостех-
теоретиздат, 1955, стр. 183
6. Л а н д а у Л. Д., ЖЭТФ, 1944, 14, 240.
7. Рд ултовская Е. В, иХарнтонЮ. Б., ДАН, 1939, 23, 530.
Я. Я. ПЕТРОВСКИЕ. л. в. волков
32. О ПРИЧИНАХ НЕПОЛНОЙ ДЕТОНАЦИИ ВВ В ШПУРАХ
При ведении взрывных работ в шахтах довольно часто наблю-
даются случаи неполной детонации зарядов ВВ в шпурах. Это ме-
шает нормальной работе. Неполная детонация даже немногих за-
* По независящим от автора причинам не было возможности измерить ско-
рость детонации на различных участках зарядов.
474
рядов снижает эффективность буровзрывных работ, так как нару-
шает рассчитанную схему взаимодействия зарядов в забое. Кроме
того, и это главное — она имеет прямое отношение к двум основ-
ным источникам травматизма, происходящего от взрывных работ-
Известно [2], что около 40% случаев этого вида травматизма
связано с ликвидацией отказавших зарядов, со взрывами их при
разбуривании забоя, с попаданием патронов ВВ в погрузочные
машины и т. д. Кроме того, неполная детонация является причиной
выгорания зарядов ВВ в шпурах — крайне опасного явления в
шахтах с газопылевым режимом. Взрыв части заряда развивает
в замкнутой полости шпура большое давление газов, имеющих вы-
сокую температуру. В ©тих условиях, как доказано исследования-
ми МакНИИ [1] и рядом зарубежных работ, все ВВ способны
гореть. Если по истечении некоторого времени полость шпура
вскрывается, то горящая масса выбрасывается в призабойное про-
странство, атмосфера которого к этому времени часто становится
взрывоопасной, даже если она и не была такой перед началом
взрывания. Конечно, далеко не каждый случай неполной детона-
ции приводит к возникновению выгорания. Для этого необходимо,
чтобы полость шпура не была вскрыта взрывом части заряда и
чтобы данное ВВ обладало значительной склонностью к горению.
Практика буровзрывных работ позволяет сделать следующие
! обшие выводы, на которые следует опираться при постановке
; исследований конкретных причин возникновения неполной дето-
нации.
1. Неполная детонация характерна главным образом для про-
ходческих работ, где применяются длинные заряды.
2. Неполная детонация чаще наблюдается при использовании
ВВ с относительно низкой детонационной способностью.
3. Неполная детонация наиболее характерна для угольных
шахт, в то время как в горнорудной промышленности, а за рубе-
жом также и в угольной промышленности это явление редкое.
По-видимому, последнее обстоятельство является следствием
существенных отличий в технике буровзрывных работ в угольной
промышленности СССР, а именно:
здесь используются ВВ, имеющие сравнительно низкую дето-
национную способность;
в горнорудной промышленности, а за границей также и на
проходческих работах в шахтах, применяется перфораторное бу-
!: рение, что обеспечивает получение шпуров, хорошо очищенных
от буровой муки, тогда как в угольных шахтах в проходческих за-
' боях шпуры бурят электросверлом, в связи с чем они плохо очи-
; щаются от буровой муки;
в горнорудной промышленности
>ние и раздавливание патронов при
1 прещено в отечественной угольной промышленности.
Исследованию явления неполной детонации и выгорания за-
1 рядов в шпурах посвящен ряд работ за рубежом (Тафанель и Дот-
риш, Фрипиа, Гримшоу, Одибер, Дуэйт, Сарториус) и в Совет-
475
широко применяется разреза-
досылке их в шпуры, что за-
ском союзе (Рудаковский, Дубнов, Галаджий). Установлено, что
в условиях производства действует ряд факторов (таких как не-
досылка патронов, образование между ними пробок или пересы-
пок из буровой муки, боковой или радиальный зазор между по-
верхностью заряда и стенками шпура и пр.), которые могут при-
вести к неполной детонации даже при нормальном качестве ВВ.
Для предотвращения последней в угольных шахтах проведено
два мероприятия, которым в свое время придавалось большое
значение. Во-первых, было введено правило, запрещающее раз-
давливать патроны в шпурах и обязывающее досылать их в шпур
сплошной колонкой. Это преследовало цель избежать образования
между патронами пересыпок буровой муки, которые, если судить
по данным МакНИИ [1], являются основной причиной неполной
детонации. Во-вторых, в целях повышения детонационной способ-
ности зарядов диаметр патронов был увеличен с 32 до 36 чьи. По-
следующая практика буровзрывных работ не показала, однако,
ожидаемого резкого снижения числа случаев неполной детонации
и выгорания зарядов. Это привело к выводу, что причины непол-
ной детонации шпуровых зарядов более сложны и требуют более
детального изучения. В связи с этим в 1957—58 гг. авторами в
бывшем Всесоюзном научно-исследовательском угольном институ-
те (ВУГИ) была проведена работа [5], результаты которой кратко
излагаются в настоящей статье.
ВЛИЯНИЕ ВЕЛИЧИНЫ ВОЗДУШНОГО ПРОМЕЖУТКА между торцами
ПАТРОНОВ НА ПЕРЕДАЧУ ДЕТОНАЦИИ
Исследовались аммонит .V® 8 и победит ПУ-2 в патронах диа-
метром 32 и 36 Л1Л, весом соответственно 200 и 250 г и плотностью
ВВ в патронах 0,98—1,02 а/с.и3. Критический диаметр, по нашим
определениям, оказался для аммонита № 8 равным 12, а для по-
бедита ПУ-2 — 8 льн. Расстояния передачи детонации по стандарт-
ной методике 1 приведены в табл. 1.
Таблица I
Расстояние передачи детонации через воздушный промежуток
для аммонита № 8 и победита ПУ-2
Наименование ВВ Диаметр патронов ММ Расстояние передачи детонации черен воздушный промежуток в см
На открытом воздухе В стальной трубе без зазора
Победит ПУ-2 32 5 48
Победит ПУ-2 .36 ю >100
Аммонит № 8 36 3 14
1 Количество подтверждений было увеличено с 3 до |0 опытов на точку.
476
В качестве модели шпура были взяты стальные трубы с тол-
щиной стенок 3 Л1Л1. Внутренний диаметр труб при испытании пат-
ронов диаметром 32 мм был равен 36 мм, а для патронов диамет-
ром 36—39 лш. Для устранения зазора между боковой поверхно-
стью патронов и стенками трубы патроны обертывали картоном.
Забойку не применяли. О полноте детонации пассивного патрона
судили по характеру разрушения трубы и отсутствию остат-
ков ВВ. Достаточность такого критерия была доказала путем со-
поставления результатов опытов с обжатием стандартных свинцо-
вых столбиков, приставлявшихся к заднему торцу пассивного
патрона. Участки труб между активным и пассивным зарядами
не разрушались и не раздувались при взрыве.
Максимальные расстояния, на которых наблюдалась безотказ-
ная передача детонации (подтвержденные 10 повторными опыта-
ми), приведены в табл. 1.
На больших расстояниях наблюдается нарастание вероятно-
сти отказа, но очень медленное. Так, аммонит № 8 в патронах
диаметром 36 мм дает частость передачи детонации: при 14 см —
100%, 15 — 90%, 20 — 64%, 30 — 55%, 40 — 33%.
Резкое повышение расстояния передачи детонации при поме-
щении зарядов в трубу объясняется тем, что ударная волна и про-
дукты взрыва, распространяясь в трубе, гораздо дольше сохра-
няют скорость и давление, чем на открытом воздухе, и, следова-
тельно, могут инициировать патроны ВВ на значительно больших
расстояниях. Это иллюстрируют фотографии, полученные с помо-
щью прибора зеркальной развертки СФР. На фиг. 1 показано рас-
пространение продуктов детонации с торца активного патропа на
открытом воздухе, а на фиг. 2 —в стальной трубе. Отчетливо вид-
но, что в трубе скорость продуктов взрыва снижается медленно,
а на открытом воздухе — быстро. Чем больше продукты взрыва
имеют возможность расширяться в стороны (т. е. чем больше от-
ношение диаметра трубы к диаметру патрона), тем больше картина
явлений будет приближаться к передаче детонации на от-
крытом воздухе. Небольшие расстояния передачи детонации1, по-
лученные МакНИИ [1] в стальной мортире, объясняются, цо-види-
мому, тем, что канал мортиры был очень большого диаметра.
Результаты проведенных опытов позволяют отметить следую-
щее.
: 1. Расстояние безотказной передачи детонации в трубах зна-
чительно больше, чем на открытом воздухе (для аммонита № 8 —
; почти в 5 раз, для победита ПУ-2 — в 10 раз).
2. Замена безнитроглицериновых аммонитов низкопроцентны-
ми нитроглицериновыми ВВ практически ликвидирует опасность
;возникновения неполной детонации заряда вследствие недосылок
Отдельных патронов (при отсутствии других причин), так как не-
’ досылки на расстояния 50 и 100 см в практике маловероятны.
1 Близкие к расстояниям передачи детонации ia открытом воздухе.
477
Фиг. 1. Движение ударной полны и продуктов детонации
ОТ конца активного заряда на открытом воздухе.
(Аммонит Ns 8, диаметр заряда 36 мм, вес 200 г, плотность
1,0 г/см3).
I—заряд. 2—изоляционная лента. 3—деревянная дощечка.
Фиг. 2. Движение ударной волны и продуктов детонации от конца
активного заряда в стальной трубе.
(Аммонит № 8, диаметр заряда 36 мм, вес 200 г, плотность 1,0 г/см3),
/—заряд, 2—стальная труба диаметром 39 -к-м с рядом отверстий для
съемки, я?—-стальная пластинка,
478
3, Стандартное испытание на передачу детонации на открытом
воздухе не только не дает абсолютных данных о расстояниях пере-
дачи детонации в шпурах, но н не показывает правильного соот-
ношения между этими расстояниями для различных ВВ, Поэтому
стандартное испытание следует рассматривать лишь как средство
контроля качества каждого данного ВВ.
ВЛИЯНИЕ ВЕЛИЧИНЫ ПЕРЕСЫПОК БУРОВОЙ МУКИ МЕЖДУ
ПАТРОНАМИ НА ПЕРЕДАЧУ ДЕТОНАЦИИ
Предварительно было проведено определение длины пересы-
пок, которые могут образоваться при досылании патронов через
плохо очищенные шпуры. Для этого в блоке породы были пробу*
рены колонковым электросверлом сквозные шпуры диаметром
45 мм. Шпуры практически не очищались от буровой муки, если
не считать удаления некоторого количества ее при извлечении бу-
ровой штанги. Буровая мука была сухая. Сквозь эти шпуры про-
талкивались патроны диаметром 36 мм. Длина пересыпок, соби-
равшихся при этом перед патроном, обычно не превышала 2 и
в очень редких случаях 2,5 см (измерение производилось после
поджатия пересыпки патроном).
Аналогичные опыты были также проведены в ВУГИ под ру-
ководством Н, Г, Петрова [3], Из ряда алебастровых слепков шпу-
ров, пробуренных в шахте, был выбран образец с наибольшими
искривлениями. По нему была изготовлена из цемента разъемная
модель шпура, В канал этой модели насыпалась буровая мука и
проталкивались патроны ВВ. Так же как и в наших опытах, пере-
сыпок длиной более 2—2,5 см не наблюдалось.
В работах МакНИИ [1] патроны аммонита № 8 диаметром
32 мм в мортире давали 8% отказов при пересыпке угольной муки
длиной всего в 1 см, а при такой же пересыпке инертной муки —
56%, Угольная пересыпка в 3 щи длиной давала 100% отказов.
Победит № 6 при угольной пересыпке длиной 1 см дал 12% отка-
зов, а при 3 см —88%,
Чтобы выяснить, не связаны ли малые расстояния передачи де-
тонации, полученные МакНИИ, с наличием радиального зазора
между патронами и стенками шпура, мы провели опыты в отсутст-
вии этого зазора.
Испытания вели в стальных трубах. В качестве материала пе-
ресыпки применялись песок, глинистый сланец и уголь. Пересып-
; ка уплотнялась усилием до 5 кг и занимала все пространство
между патронами. Радиальный зазор заполнялся картоном, за-
бойка не применялась.
С увеличением длины пересыпки вероятность передачи дето-
, нации очень быстро снижается. Так, например, для победита
; ПУ-2 при диаметре патрона 32 мм это подтверждается следующи-
г ми данными:
47?
Длина песчаной пересыпки в см 5,2 5,5 6,0
Частость передачи детонации в % 100 80 66
Результаты испытаний, подтвержденные 10 повторными опытами,
приведены в табл. 2.
Таблица 2
Передача детонации через различные пересыпки в случае отсутствия
радиального зазора между зарядом и стенками шпура
Наименование ВВ Диаметр патроуов .и.и Предельные расстояния безотказной передачи детонации через пересыпки в см
Песок Сланец Уголь
Победит Пу-2 32 5,2 6.0 3,5
36 5,2 — —
Аммонит № 8 32 3.2 — —
36 3,5 — 4,5
Из табл. 2 следует, что предельное расстояние передачи дето-
нации через пересыпки очень мало изменяется с изменением диа-
метра патронов. Отсюда ясно, что переход на патроны большего
диаметра, без повышения требований технических условий по по-
казателю передачи детонации на воздухе [, ухудшит передачу ее
через пересыпки в шпурах, Перевод буровзрывных работ в шах-
тах на патроны увеличенного диаметра был произведен без ка-
ких-либо изменений технических условий на ВВ и поэтому не при-
вел на практике к снижению числа случаев неполной детонации
и выгорания зарядов в шпурах.
Возникает вопрос о механизме передачи детонации при разме-
рах пересыпок, близких к предельным. Инициируется ли пассив-
ный заряд ударной войной, проходящей по пересыпке, или же пе-
ресыпка инициирует ВВ ударом всей своей массы? Для выяснения
этого были проведены следующие опыты.
Патроны аммонита № 8 диаметром 36 мм вставлялись в сталь-
ную трубу без радиального зазора, К активному патрону вплот-
ную примыкала песчаная пересыпка длиною 2—2,5 см, т. е. мень-
ше предельной. Между пересыпкой и пассивным патроном остав-
лялся воздушный промежуток в 10 ext, т. е. также немного меньше
предельного. В стенке трубы на участке воздушного промежутка
просверливали 20 отверстий диаметром 10 мм для уменьшения
степени сжатия воздуха при движении пересыпки. Во всех опытах
детонация передавалась полностью, Это говорит в пользу механиз-
* Такое повышение но существу явилось бы требованием сохранения качества
ВВ на прежнем уровне, так как расстояние передачи детонации в стандартной
пробе сильно зависит от диаметра патронов ВВ,
480
ia передачи детонации ударом всей массы материала, так как
.оздушный промежуток в сочетании с пересыпкой резко ослабляет
дарную волну, но мало влияет на скорость движения центра тя-
сести пересыпки. Увеличение воздушного промежутка до 30 см
ривело к отказам. Аналогичные результаты были получены и без
тверстнй в трубе.
Имеются и другие доводы, говорящие в пользу высказанного
:ами предположения о механизме передачи детонации через пере-
:ыпки между патронами заряда в шпуре. Если передача детона-
дии через пересыпки происходит так, как мы предполагаем, то
касса предельных пересыпок, приходящаяся на единицу площади
поперечного сечения зарядов, должна быть близкой для разных
материалов. Данные соответствующего подсчета приведены в
габл. 3,
Таблица 3
Наименование ВВ Диаметр патронов мм Удельная масса предельных пересыпок в г на сл(2 поперечного сечения заряда
Песок Сланец Уголь
Зобедит ПУ-2 32 8,2 9,0 6,8
Кммонит № 8 36 5,7 — 3,6
Таким образом, наше предположение подтверждается с точно-
стью + 15% для победита ПУ-2 и +22% для аммонита № 8.
По-видимому, решающее значение в таком механизме пере-
дачи детонации имеет скорость движения пересыпки. Количество
Движения, сообщаемое пересыпке взрывом данного ВВ, в наших
Условиях сохраняется примерно постоянным для всех величин
гересыпок, так как оно определяется главным образом временем
'азрыва стенок трубы. Поэтому с увеличением пересыпки ско-
ость движения ее,изменяется обратно пропорционально массе и,
,ойдя до известного предела, перестает возбуждать детонацию
; пассивном заряде.
На фиг. 3 приведен полученный с помощью СФР снимок пере-
дачи детонации в стальных трубах через предельную пересыпку
геска между патронами аммонита № 8, имеющими диаметр 36 мм.
1ремя задержки передачи детонации через пересыпку достигает
'0 мксек.
, Для открытых зарядов, где свободный разлет продуктов взрыва
резко снижает импульс, получаемый пересыпкой, получено 100%
отказов передачи детонации через пересыпку величиною всего
г1 см, как для аммонита № 8, так н для победита ПУ-2.
В свете изложенного можно легко объяснить резко занижен-
ные величины предельных пересыпок, полученные в МакНИИ: ос-
новные испытания там проводились при очень больших радиаль-
ных зазорах, приближающих характер явлений к передаче через
31 451 481
пересыпку на открытом воздухе. Неудивительно, что в МакНИИ
получили отказы даже при пересыпке в 1 см. Влияние таких ма-
лых пересыпок было расценено как решающее, поскольку они
очень вероятны в производственных условиях.
По нашим данным, величины предельных пересыпок, при кото-
рых не наблюдаются отказы в передаче детонации, в случае от-
сутствия радиального зазора, значительно превышают величины
вероятных пересыпок, встречающихся в производственных усло-
виях. Из этого следует, что практически возможные пересыпю
Фиг. 3. Передача детонации в стальной трубе при отсут-
ствии радиального зазора, через пересыпку из песка
длиной 3,5 с.и.
(Аммонит № 8, диаметр патронов 36 .ил, вес 250 г,
плотность 1.0 г/с,и3').
/—активный патрон, 2—пересылка, 3—пассивный патрон,
4—Стальная труба.
сами по себе, при ликвидации радиального зазора (например пу-
тем разрезания и раздавливания патронов при досылке в шпуры),
не могут дать той частости неполных детонаций шпуровых заря-
дов, которая наблюдается в шахтах в настоящее время.
Несмотря на это вопрос борьбы с возможностью образования
пересыпок между патронами шпуровых зарядов остается весьма
актуальным. Радикальным решением этого вопросы было бы вне,т
реиие промывки при вращательном бурении шпуров, которую на-
чали применять на шахтах Кузбасса, Целесообразны и другие
способы, как например:
а) Придание торцам патронов В В выпуклой (обтекаемой)
формы. Такой патрон не будет собирать перед собой большую пе-
ресыпку.
б) Разработка простейших приспособлений для улучшения очи-
стки шпуров.
482
в) Увеличение диаметра штанг, с максимальным приближе-
нием его к диаметру резца, и запрещение применении штанг после
определенного износа.
г) Применение цилиндрических резцов, уменьшающих искрив-
ление шпуров при бурении.
д) Применение ВВ, имеющих большие расстояния передачи
детонации через пересыпку.
О НЕПОЛНОЙ детонации зарядов вследствие попадания
в них патронов с утраченной детонационной способностью
Патрон ВВ с утраченной детонационной способностью, попа-
дая в заряд, действует как пересыпка длиной 20—23 см и обяза-
тельно приводит к неполной детонации.
Патрон ВВ может потерять детонационную способность в мок-
ром шпуре в результате замокания. Для предупреждения послед-
него следует применять водоустойчивые ВВ. В СССР разработано
несколько способов придания ВВ водоустойчивости, вполне доста-
точной для большинства условий работ, встречающихся в уголь-
ной промышленности. Назрело время выпуска подавляющего ко-
личества ВВ в водоустойчивом варианте.
Патроны могут утратить детонационную способность также за
время перевозки и хранения на складе. Аммониты не только имеют
относительно низкую детонационную способность, но и быстро те-
ряют ее вследствие увлажнения и слеживания при хранении, осо-
бенно при повышенной влажности и колебаниях температуры, Па-
рафинированная оболочка частично помогает бороться с этими
вредными явлениями. Однако при длительной транспортировке ВВ
от заводов к потребителям в отдельных патронах возникают не-
большие, труднообнаруживаемые нарушения герметичности обо-
лочки. При последующем хранении на складах такие патроны те-
ряют детонационную способность гораздо быстрее, чем патроны с
неповрежденной оболочкой. Если в хранящейся партии ВВ таких
патронов несколько процентов, то они почти не обнаруживаются
при испытании ид передачу детонации, т. е. трудно поддаются кон-
тролю. Но даже 2% таких патронов будут давать при взрывании
в каждом штреке в среднем два отказа. В связи с этим явление по-
тери детонационной способности отдельными патронами приносит
: промышленности больший вред, чем общее равномерное снижение
I детонационной способности всей партии, которое легко обнаружн-
вается,
t Для снижения возможности подобных явлений необходимо
. улучшить укупорку ВВ и снизить фактические сроки от момента
J. изготовления взрывчатых веществ до их использования.
i В настоящее время патроны при выдаче взрывнику прокалы-
1 вают игольчатыми штампами, что нарушает их герметичность. Не-
!» израсходованные патроны возвращаются на подземные шахтные
j склады и быстро теряют там детонационную способность, Необхо-
j димо отказаться от прокалывания патронов ВВ на шахтах и ввести
31*
483
печатание номерных знаков на оболочках патронов при изготов-
лении ВВ на заводе.
Следует расширить применение взрывчатых веществ, содержа-
щих до 10—15% нитро*афиров и обладающих поэтому повышен-
ной детонационной способностью, которая дольше сохраняется
при хранении ВВ.
В связи с тем, что для Советского Союза характерны продол-
жительные сроки храпения взрывчатых веществ, очень важно сни-
жение их слежцваемости. В этом отношении целесообразно ис-
пользовать результаты работ, проведецых в Англии [7] и Польше,
где положительный эффект был достигнут введением в состав ВВ
небольшого количества некоторых веществ (например, фуксина).
ВЛИЯНИЕ РАДИАЛЬНОГО ЗАЗОРА МЕЖДУ БОКОВОЙ ПОВЕРХНОСТЬЮ
ПАТРОНОВ И СТЕНКАМИ ШПУРА
Зазор между боковой поверхностью патронов ВВ и стенками
шпура, называемый сокращенно радиальным, может приводить к
затуханию детонации заряда даже тогда, когда на воздухе патро-
ны имеют нормальную детонационную способность и досланы в
шпуре одни к другому впритык без каких-либо пересыпок. Это
явление исследовалось С. И, Рудаковским (1938 г.). А, Е, Пере-
верзевым и А. А, Ильющиным (1947 г.) и Л. В, Дубновым
(1954 г,),
Для объяснения механизма этого явления предложено не»
сколько гипотез. Наиболее вероятная причина затухания детона-
ции заключается в том, что продукты взрыва, распространяясь
в зазоре со скоростью, превышающей скорость детонации, уплот-
няют ВВ впереди фронта детонационной волны.
Для описываемых ниже исследований был применен победит
ПУ-2 из партии, использованной в предыдущих опытах, и аммонит
№ 8 новой партии, оказавшейся несколько лучше прежней (рас-
стояние передачи детонации по стандартной методике, но при 10
повторных испытаниях на точку, оказалось равным 4 см для пат-
ронов диаметром 32 мм и 5 см— для 36 AtAt патронов, тогда как
для прежней партии при диаметре 36 мм оно было равно 3 сл().
Опыты проводили в стальных трубах с внутренним диаметром
39 мм для 32 мм патронов и 44,5 мм для патронов диаметром
36 ,и,и, т. е, радиальные зазоры были выбраны возможно ближе
к существующим на производстве. Толщина стенок трубы 3 мм.
Заряд аммонита составляли из трех патронов, заряд победита—
из четырех. Патроны в трубе располагались впритык один к дру-
гому. Со стороны боевика забойку пе применяли; противополож-
ный конец трубы в большинстве случаев забивали на 5—7 см сме-
сью песка и глины или приставляли к массивной стальной пре-
граде Результаты испытаний приведены в табл. 4.
1 Как показали опыты, эта забойка не влияет на частоту отказов.
484
Таблица 4
Неполная детонация зарядов ВВ в стальных трубах
при наличии радиального зазора
ВВ Диаметр патронов мм Количестно опытов Количество полных детонаций Количество неполных детонаций * % неполных детонаций
Аммонит № 8 32 18 3 15 83
(3 патрона) 36 5 I 1 20
Победит ПУ-2 32 4 2 2 50
(4 патрона) 36 9 9 0 0
* Обрывы детонации в зарядах аммонита № 8 происходили чаще всего на
третьем патроне (иногда даже в копие второго), а в зарядах победита ПУ-2 —
только на четвергом патроне.
Данные табл, 4 показывают, что радиальный зазор даже сам
по себе, без осложняющих обстоятельств, может приводить к не-
полной детонации зарядов из аммонита и победита нормального
качества.
Из табл. 4 также следует, что возникновение неполной дето-
нации зарядов зависит от свойств ВВ, диаметра патронов и длины
заряда. Аммонит № 8 дает неполные детонации значительно чаще,
чем победит ПУ-2. Для обоих ВВ уменьшение диаметра патронов
приводит к повышению вероятности обрыва детонации.
Особенно важно отметить, что вероятность обрыва детонации
возрастает с увеличением длины заряда, Если бы заряды аммо-
нита № 8 мы составляли из двух патронов, то случаи неполной
детонации были бы значительно реже, потому что большинство
обрывов детонации для этого ВВ происходило на третьем патроне.
А если бы заряды победита ПУ-2 составляли не из четырех, а из
трех патронов, то случаи неполной детонации для него вообще пе
были бы зафиксированы. Это соответствует данным практики, где
подавляющее большинство случаев неполной детонации происхо-
дит при применении длинных зарядов (например, на проходческих
работах),
В наших экспериментах неполные детонации происходили зна-
чительно чаще, чем в условиях производства. Это означает, что
па рассматриваемое явление сильно влияют условия опыта.
Для более детального выяснения механизма обрыва детонации
мы провели несколько серий экспериментов,
С помощью прибора СФР в режиме лупы времени (покадро-
вая съемка) был снят процесс детонации зарядов ВВ в стальных
трубах при наличии радиального зазора (фиг, 4), Труба с внут-
ренним диаметром 39 в этом опыте располагалась горизон-
тально, Вдоль верхней и нижней образующих трубы были про-
сверлены отверстия диаметром 4 мм, Заряд аммонита № 8 диа-
485
метром 32 мм располагался внутри трубы с радиальным зазором,
прилегая к нижнему ряду отверстий. Поэтому в нижние отверстия
газы начинают вырываться только после прохождения детонацион-
ной волны. Опережающий поток газов, двигаясь по зазору, обна-
руживается с помощью верхнего ряда отверстий.
Труба, оклеенная бумагой с черными полосками против отвер-
стий, была заснята до взрыва на всех кадрах и служила таким
Фиг. 4. Детонация заряда в трубе с зазором.
(Аммонит № 8, диаметр патронов 32 мм, заряд из трех патронов).
Z—заряд, 2—стальная труба, J—лесок, 4—пластилин, 5—эона подпрес-
совки, 6—фронт струй газа, 7—продукты детонации внутри трубы.
Первый кадр снят через 50 —60 лксек после инициирования заряда.
образом в качестве масштаба и репера. При взрыве частота съем-
ки была 150 000 кадров в секунду. На фиг, 4 показаны только
4 кадра, выбранные с интервалами 40 мксек. Расстояние между
соседними полосками на трубе равно 2 см. Скорость фронта опе-
режающего потока продуктов взрыва в зазоре в среднем равна
3500 м!сек, а скорость детонации — 2700 м}сек. На фиг, 4 видно,
как опережение возрастает от 8 см на первом кадре до 18 на чет-
вертом, Этот быстрый 1[Оток газов обладает высоким давлением
и может уплотнять ВВ впереди детонационной волны. При таком
486
длины зарядов, так как при этом увеличивается опе-
Шлотнении не только возрастает плотность ВВ, но и уменьшается
[иаметр патронов,
Известно, что для смесевых ВВ с увеличением плотности заряда
нижается детонационная способность (увеличивается критиче-
ский диаметр). Поэтому при подпрессовке патронов опережаю-
дим потоком сазов диаметр их может оказаться меньше критиче-
ского и детонация затухнет. Это и наблюдается в наших опытах и
В реальных условиях для промышленных ВВ, причем гораздо
чаще для тех ВВ, у которых критический диаметр больше и более
(быстро возрастает с плотностью. Именно этими свойствами отли-
чается аммонит № 8 по сравнению с победитом ПУ-2. По той же
'Причине чаще бывают отказы при употреблении ВВ пониженного
:ачества (по изготовлению или из-за увлажнения, замокания, сле-
живания), так как понижение качества ВВ увеличивает его кри-
ическиц диаметр-
Уплотнение заряда давлением газов, движущихся в зазоре,
:осит динамический характер, так как совершается за малые про-
[ежутки времени. Поэтому степень уплотнения заряда, достигае-
ма к моменту подхода детонационной волны, зависит не только
it давления газов, но и от времени их действия, т. е. от величины
переженил. Этим объясняется увеличение вероятности отказов с
величением
ежение.
Большую роль играет величина зазора. При очень большом за-
оре (в пределе — на открытом воздухе) опережающий поток не
южет создаться, так как газы имеют возможность свободного
асширения во все стороны. При очень малом зазоре (в преде-
е — без зазора) опережающий поток встречает слишком большое
□противление и не может намного опередить детонационную
олну. Очевидно, существует некоторая наиболее опасная величи-
а зазора, при которой явление обгона наиболее резко выражено;
а ряды, которые безотказно детонируют на открытом воздухе н
ем более в трубе без зазора, дают при этой «опасной» величине
азора большой процент отказов.
В настоящей работе мы не задавались целью установить нам-
олее опасные величины зазоров, а исследовали только величины,
лизкие к наблюдаемым на производстве. Однако тот факт, что
наших опытах частота отказов была значительно выше, чем на
роизводстве, говорит о большей жесткости условий лаборатор-
ых экспериментов. Это объясняется тем, что трубы, в которых
роводились опыты, дают прямолинейный зазор с ровными степ-
ами, а в реальных шпурах стенки извилисты. Последнее резко
величивает сопротивление движению газов по зазору и умень-
шает возможность обгона. Таким образом, шпур с неровными стен-
ами эквивалентен в этом отношении трубе с меньшим радналь-
ым зазором.
Мы поставили ряд опытов для проверки изложенных сообра-
жений о механизме возникновения отказов при радиальном зазоре,
тли эти соображения правильны, то наличие зазора должно не
487
только не мешать, но даже помогать прохождению детонации в за-
рядах из таких ВВ, у которых критический диаметр при увеличе-
нии плотности не растет, а уменьшается, К ВВ такого типа отно-
сятся тротил, гексоген, пикриновая кислота, тэн и т. п. Мы провел:,
опыты с тротилом плотности 1 г[см?. Схема этих опытов пред
ставлена на фиг. 5, На открытом воздухе заряды тротила диамет-
ром 8 мм безотказно детонировали, а при диаметре 6 мм детона
ция затухала, т. е. критический диаметр при этой плотности бы,:
равен примерно 7- -8 мм. В дюралевой трубе с внутренним лиа
Фиг. 5. Схема опытов по детонации заряда поро-
шкообразного тротила при диаметре меньше кри-
тического:
а) на открытом воздухе, б) в трубе.
/—электродетонатор, Ч—дополнительный детонатор из
тротила дна метро и 9 мм, длиной 40 леи, плотностью
1,0 г/сж’. 3—патроны тротила диаметром S мм, длиной
290 мм каждый, плотностью 1,0 г)см\ 4— картонная по-
лоска, 5—стальная плита, 6—дюралюминиевая труба
с внутренним диаметром 11,7 мм и толщиной стенки
2,2 мм
метром 11,7 и наружным 16 мм безотказно детонировали заряде,
диаметром 8 мм, длиной 50 см. Тогда мы поместили в такую же
трубу заряд диаметром 6 мм, т. е. меньше критического. Чтобы
создать в зазоре мощный поток газов высокого давления, заряа
инициировался промежуточным детонатором из того же тротила,
но диаметром 9 мм (в опытах на открытом воздухе применяло^
такое же инициирование). Детонация прошла по всему заряду дли
ной 58 см. Опыт повторен 7 раз с тем же результатом. Это гово-
рит о наличии подпрессовки ВВ опережающими газами.
К аналогичным выводам независимо от нас пришел Иогансог
[9], который в 1958 году в Швеции исследовал влияние радиан и
ного зазора на детонацию аммон-желатиндинамитов.
Рассмотрим теперь сочетание радиального зазора с пересыпка
ми между патронами. На производстве такое сочетание возмож-
но, так как правило о досылании патронов в шпур сплошной ко
лонкой не всегда выполняется, да и не всегда оно осуществимо
Кроме того, небольшие пересыпки между патронами могут обра
зоваться и при соблюдении этого правила.
188
Мы видели (см. фиг. 3), что даже небольшие пересыпки сильно
задерживают распространение детонационной волны и увеличи-
вают отставание ее от опережающего потока газов, усугубляя
вредное влияние зазора, Поэтому были проведены опыты по изу-
чению совместного действия пересыпки и радиального зазора.
В качестве материала для пересыпки использовали песок. На
фиг- 6 представлена схема этих опытов. Победит ПУ-2 в патронах
диаметром 36 л.и при пересыпке в 2 ext дал 10% отказов, а при
пересыпке в 3 см отказы были массовыми. Аммонит № 8 в патро-
в)
Фиг. 6. Схема оцытои по изучению совместного
действия пересыпки между патронами и радиаль-
ного зазора на прохождение детонации:
а) с отражающей поверхностью; б) без отражаю*
щей поверхности; в) с перегородками в радиаль-
ном зазоре.
/—пересылка, 2— радиальный зазор» 5—отражающая по*
верхность, 4—перегородки из изоляционной ленты.
нах диаметром 36 мм уже при пересыпках 0,5—1 см дал 11 от-
казов из 15 опытов.
Легкие перемычки (из свернутой жгутиком изоляционной лен-
ты), преграждавшие сечение радиального зазора, уничтожили
вредное влияние последнего: победит ПУ-2 диаметром 32 и 36 мм
безотказно детонировал при песчаной пересыпке толщиной 4 см,
тогда как без этих перемычек такая пересыпка приводила к 100%
отказам. Это говорит о том, что достаточно раздавить в шпуре
хотя бы только конец каждого патрона, чтобы ликвидировать вред-
ное влияние радиального зазора на полноту детонации.
Сочетание радиального зазора (если этот зазор не слишком
велик) с недосылкой, очевидно, не должно сильно ухудшать пере-
дачу детонации по сравнению с недосылками без зазора, так как
опережающий поток газов, резко расширяясь в воздушном про-
межутке между патронами, ослабевает и сливается с основной
массой продуктов детонации, движущихся с торца активного па-
трона. Мы убедились на опыте, что при наличии радиального за-
зора расстояние передачи детонации через воздушный промежуток
489
- Д'»* SX
а ай?1.и № 8 диамет-
‘Z 36P.i!.u в трубе с внутренним диаметром 44,0 мм - И (ср.
ТабИтак мь, разобрали влияние радиального зазора на детона-
ционную способность шнурового заряда. Мы доказали, что ради-
ционнум и „„„„«-тппцет основную опасность в отношении воз-
?,Х'венп приводящей нередко к еыеор,-
ию заряда, Особенно усиливают вредное деиствие зазора пере-
сыпки которые очень вероятны в условиях паш^ угольных шахт.
Простейшей в то же вр^« наиболее действенной мерой борьбы
с вредным влиянием радиального зазора является его ликвидация
путем надрезания оболочки патронов и раздавливания их в шпу-
ре забойником. Внедрение такого способа заряжания следует про-
вести на проходческих рвотах, потому что там чаще всего наблю-
дается неполная детона^я, а также потому Что раздавливание
патронов повышает эффективность буровзрывных работ при про-
ходке [41 Действующие правила, требующие посылать патроны
в шпур сплошной колоний и не нарушать целостИ их, препятст-
вуют внедрению разрезав и раздавливания патронов и поэтому
‘ЮЛПпи впедрениТраздавливаиия необходимо одновременно при-
а Грмег> Тппиданию геем ВВ водоустойчивости и неслеживае-
Х^ ^о^Хно^дост-нуть путем изменения состава ВВ и
улу|шлХ77яеу^ к°р°тК11е заРяды- ргаль’
ного зазора невелико и раздавливание патронов нецелесообразно,
тем более что оно, повышая бризантное действие взрыва, ведет
к нежелательному переиз^льчению угля-
О ДЕТОНАЦИОННОЙ способности шпуровых ЗАРЯДОВ
д Speccobahh СКАЛЬНОГО аммонита № 1
Пои внедрении в горнорудной промышленности мощного непре-
ири и ДР прессованного скального аммонита № 1
° S Междуведомственная комиссия по взрывному делу при
Ии9ХутХТго дела ДН СССР в 1958 году обратилась в БУГИ
с просьбой исследовать причины этого явления^
Для проведения испытании был доставлен скальный аммонит
№ |в патронах диаметром 36 мм, весом 250 г. Каждый патрон
состоял из двух шашек плотностью 1,45 г/см , помещенных в бу-
мажиую гильзу с последующей парафинировав. Бризантность
этого ВВ при ^плотности 1,0 г/см* равна 20 мм, а работоспособ-
Н°СКаГ Уже Сказывалось Для горнорудной промышленности, где
применяется перфоратор^ бурение, возможность образования
пересыпок в шпурах не характерна. В связи с вь1СОкОй йлотностью
490
ВВ трудно было также ожидать переуплотнения его продуктами
К; взрыва, обгоняющими фронт детонации в радиальном зазоре. Это
подтвердилось в лабораторных условиях: в стальных трубах с вну-
В треиним диаметром 44,5 мм случаев затухания детонации в заря-
В- дах скального аммонита № 1, составленного из четырех патронов,
В не было.
В Поэтому причину неполных взрывов следовало связывать с ноз-
В можностью недосылки патронов в сочетании с обратным иниции-
В рованием.
В В горнорудной промышленности широко применяют огневое
В взрывание. Чтобы избежать отсечки огнепроводного шнура взры-
В вами шпуров предыдущей серии, боевик досылается в шпур в чи-
|В еле первых патронов. При этом могут создаться условия, при ко-
В торых часть заряда инициируется через воздушный промежуток
той стороной боевика, с которой в него вставлен капсюль (обрат-
В ное инициирование). Для проверки влияния воздушного проме-
В жутка в сочетании с обратным инициированием были проведены
В опыты в стальных трубах диаметром 39 мм без устранения ради-
В ального зазора. Результаты этих опытов даны в табл. 5. Кроме
В того, в ней приведены данные о передаче детонации через воздуш-
В ный промежуток на открытом воздухе, а также через пересыпки
В на открытом воздухе и в трубах. Исследования передачи детонации
В через пересыпки были предприняты с целью накопления общих
В: сравнительных данных.
Таблица 5
Передача детонации между патронами прессованного
скального аммонита № 1 в различных условиях
Условия инициирования Максимальное расстояние безотказной передачи детонации см
Через воздух Через песок
на открытом воздухе в трубе иа открытом воздухе в трубе
Прямое 7 20 1,5 4
Обратное 1,5 10 — 2
г Из табл. 5 следует, что в случае обратного инициирования в
I трубе при воздушном .промежутке, превышающем 10 см, уже по-
f-лучаются отказы. Такая величина недосылки вполне реальна для
f производственных условий и случаи неполной детонации, по-видп-
| мому, следует объяснить именно этим.
( Обращает на себя внимание, что во всех видах проведенных
[ испытаний обратное инициирование резко ухудшает передачу де-
I тонации.
L Интересно отметить также то, что прессованный скальный ам-
| монит № 1, имея высокий показатель передачи детонации по
I стандартной методике (на уровне Предохранительных питроглп-
1‘Ч
цериновых ВВ), через пересыпку в трубе передает детонацию ху-
же, чем победит ПУ-2, а через воздушный промежуток в трубе —
в несколько раз хуже. Таким образом, даже, мощные аммониты с.
высокими детонационными свойствами в условиях шпура уступа-
ют по ряду важных показателей низкопроцентным нитроглицери-
новым ВВ, имеющим значительно меньшую мощность. Это дает
также еще один пример того, что стандартная проба на передачу
детонации на открытом воздухе не отражает поведения ВВ в
шпуре.
ПРЕДВАРИТЕЛЬНАЯ ПРОВЕРКА ЛАБОРАТОРНЫХ ИССЛЕДОВАНИЙ
ШАХТНЫМИ ИСПЫТАНИЯМИ
В ходе выполнения программы промышленных испытаний но-
вых ВВ [6] попутно и в ограниченном масштабе была проведена
проверка влияния раздавливания патронов на частоту случаев
неполной детонации.
В шахте «Южная» треста «Рутченковуголь» (Донбасс!
в июле—августе 1958 года при проходке квершлага проводились
сравнительные испытания нового ВВ, которому был присвоен ин-
декс Э-3, и штатного аммонита ПЖВ-20.
Э-3 является высокопредохранительным ВВ, изготовление и
испытание которого предложил ВУГИ. По своему составу оно было
подобно английскому ВВ «Юниджекс» [8]. Не имея предохранитель-
ной оболочки, Э-3 не воспламеняет метан открытыми зарядами ве
сом до 300 г. Такие высокие предохранительные свойства достиг-
нуты путем замены аммиачной селитры (NH4NO3) и хлористого на-
трия, которые обычно применяются в предохранительных отечест-
венных ВВ, обменными солями: натровой селитрой (NaNOs) и хло-
ристым аммонием (NH4C1). При взрывной реакции происходит об-
мен катионами и выделяется хлористый натрий в высокодисперсном
состоянии с большой удельной поверхностью, что резко повышает
эффект пламегашения. Таким путем можно при той же антигризут-
ности добиться большей энергии взрывчатого вещества с обмен-
ными солями, чем у ВВ с пламегасителем (NaCl), введенным в со-
став в готовом виде.
Замена обычных компонентов обменными солями одновременна
практически решила в данном случае проблемы слеживаемости и
гигроскопичности, потому что новые компоненты являются малого-
гроскопичными и не слеживаются. После складского хранения в
течение года такое ВВ не слежалось п сохранило нормальное ка-
чество.
Кроме того, повое ВВ имеет очень высокую водоустойчивость,
в несколько раз большую, чем у ПЖВ-20, аммонита В-3 или аммо-
нита № 6 ЖВ. Это достигнуто введением в его состав не только сте-
арата кальция, но также натрий-карбоксиметилцеллюлозы и као-
лина. Нятрий-карбоксиметилцеллюлоза в воде очень быстро набу-
хает, и патрон покрывается слизистой массой, прекращающей до-
ступ воды внутрь патрона. Каолин набухает медленнее, но зато оц
делает гель более консистентным; с этой целью он и вводится.
-192
Для обеспечения хорошей детонационной способности в состав
Э-3 введено около 15% слегка желатинированного нитроглицерина
(труднозамерзающая смесь). В результате ВВ получило структуру
жирного тяжелого порошка с плотностью 1,25 г/см3. Патроны легко
раздавливались в шпурах, заполняя все сечение, В отношении
транспортировки и хранения новое ВВ относится, как [1 победиты,
к классу аммонитов.
Э-3 дает обжатие свинцовых столбиков не более 7—7,5 мм и
имеет работоспособность примерно 185 см3. Однако за счет повы-
шенной плотности ВВ и раздавливания патронов достигалась высо-
кая концентрация энергии в единице объема шпура, что позволило
сохранить удельный расход бурения на том же уровне, что и для
ПЖВ-20, обладающего более высокой в сравнении с Э-3 работо-
способностью (ПЖВ-20 применялся без раздавливания патронов).
При лабораторных испытаниях Э-3 в патронах диаметром 36 ж-и
передавал детонацию на открытом воздухе только на 3 см, т. е.
близко к наименьшим показателям аммонита № 8. При наличии ра-
диального зазора заряды Э-3 в стальных трубах давали затухание
детонации на 3 и 4 патронах. Однако при отсутствии радиального
зазора это ВВ безотказно передавало детонацию в стальных тру-
бах через песчаные пересыпки в 4,5 см, т. е. значительно лучше ам-
монита № 8 и близко к победиту ПУ-2. Мы полагали, что Э-3 будет
хорошо детонировать в шпурах при раздавливании патронов.
В квершлаге шахты Э-3 испытывалось только в виде разрезан-
ных и раздавленных патронов, досланных в шпуры по одному. Ам-
монит ПЖВ-20 испытывался как целыми патронами, посылаемыми
в шпур согласно действующим правилам сплошной колонкой, так и
в двух циклах с применением разрезания и раздавливания патро-
нов. Бурение шпуров производилось электросверлом, и каких-либо
особых мер по их очистке не предпринималось, поэтому они были
засорены буровой мукой. Данные этих испытаний приведены в
табл.6.
Таблица 6
Результаты шахтных испытаний аммонита ПЖВ-20
и ВВ типа Э-3 в различных условиях заряжания
ВВ и условия заряжания Количество взорванных шлу ров Количество случаев неполной детонации
Аммонит « ПЖВ-20
Патроны Аммонит целые, досылка колонкой ПЖВ-20 123 5
Патроны по одному ВВ типа раздавливались и досылались Э-3 48 0
Патроны по одному раздавливались и досылались 112 0
493
Таким образом, аммонит ПЖВ-20 при принятой на шахтах тех-
нологии взрывных работ и тщательном выполнении правил давал
одну неполную детонацию на каждых 25 взорванных шпуров, Прн
введении раздавливания того же аммонита 48 взорванных шпуров
не дали ни одного случая неполной детонации. Э-3 в 112 взорванных
шпурах также не дал ни одного случая неполной детонации, хотя
детонационная способность его, определяемая по стандартной мето-
дике, невысока. Шахтные испытания подтвердили выводы лабора-
торных исследований; однако они проведены в ограниченном мас-
штабе, и делать по ним окончательные выводы преждевременно.
ОСНОВНЫЕ ВЫВОДЫ
1. Одной из основных причин неполной детонации шпуровых за-
рядов в угольных шахтах является наличие радиального или боко
вого зазора между патронами и стенками шпура. Продукты взры-
ва, двигаясь по этому зазору, опережают фронт детонации и уплот-
няют ВВ впереди него, что может в процессе взрыва приводить к
потере детонационной способности заряда.
2. Вредное влияние радиального зазора особенно усиливается
цри сочетании его даже с небольшой пересыпкой буровой муки, ко
торая резко замедляет передачу детонации между патронами и тем
самым облегчает переуплотнение ВВ опережающим потоком газов.
Сочетание радиального зазора с небольшими пересыпками (]--
2 сл() следует считать основной причиной высокой частости непол-
ных детонаций в угольной промышленности не только предохрани-
тельных аммонитов, но и победитов.
3. Основными направлениями борьбы с неполной детонацией
зарядов в шпурах должны быть следующие;
а) устранение радиального зазора между боковой поверхностью
патронов и стенками шпура, что может быть вначале осуществлено
внедрением известного метода разрезания и раздавливания патро-
нов при досылке их в шпур, а в дальнейшем — разработкой и вцел-
рением механических методов заряжания шпуров;
б) улучшение очистки шпуров от буровой муки, так как устра-
нение радиального зазора, хотя и снижает вредное влияние пере-
сыпок, но не исключает его;
в) улучшение детонационных свойств предохранительных
взрывчатых веществ, особенно в отношении склонности к перепрес-
совке, и обеспечение сохранения качества ВВ в процессе перевозки,
хранения и использования.
4. Проведенные исследования доказали несостоятельность оцеш
ки детонационных свойств ВВ в шпурах на основании результатов
испытания на передачу детонации на открытом воздухе. Для пра-
вильной оценки необходимо пользоваться испытаниями по типу
описанных в статье.
494
ЛИТЕРАТУРА
I. Галаджин Ф. М. О причинах выгорания взрывчатых веществ в шпу-
рах при взрывных работах, Бюллетень № 8 МакНИИ. 1958, стр. 29.
2. Зайцев А. П., Совещание по вопросам безопасности буровзрывных ра-
бот в угольной промышленности, Безопасность труда в промышленности, 1961.
М 1, стр. 34-
3. Петров Н. Г., Исследование процесса заряжании п забойки шпуров (ис-
следование коэффициента заряжания), Отчет ВУГИ, 1958.
4, Петровский И. Я-, Разработка технических требований Na более эф-
фективные ВИ для конкретных гор но-геологических условий работ в угольной
промышленности, Отчет ВУГИ, № 1299, 1958.
5, Петровский И. Я-> Исследование причин, вызывающих отказы ц выго-
рания зарядов ВВ в шпурах, и разработка практических путей по их устранению,
Отчет ВУГИ № 1339, 1958.
6, Петровский И. Я.. Крутов И. В., Промышленные, испытания новых
предохранительных ВВ, Отчет ВУГИ, 1958.
7. Shepherd W. С. F., Grimshaw Н. С., The development of safer
mining explosives, Safety in Mines Research and Testing Branch, Ministry of Fiiel
and Power, July 1950-
8. Та у 1 о г G., The Colliery Guardian, 181, .M 4687, 1950.
9, Johansson С. H., S с 1 b e г g H. L., Per so n A., S j о e 1 f п T., Channel
effect in detonation in lubes with an open space between the charge and the tube
wall, Communications du XXXI Congres International de Chimie Industrielle,
Liege. 1958, pp. 57—66.
К. К. АНДРЕЕВ, В. Г, ХОТИН
33. К ВОПРОСУ О ФАКТОРАХ, ОПРЕДЕЛЯЮЩИХ
ВОЗМОЖНОСТЬ ВЫГОРАНИЯ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
В ШПУРАХ
Вопрос о причинах и возможностях предотвращения выгорания
зарядов ВВ в шпурах стал в настоящее время одной из наиболее
острых проблем обеспечения безопасности взрывных работ в уголь-
ных шахтах.
Возникновение горения заряда ВВ нежелательно по многим
причинам.
Во-первых, оно создает опасность запаздывающего взрыва, так
как горение по истечении некоторого времени может перейти во
взрыв. Этим взрывом может быть поражен взрывник, вернувшийся
в забой. Во-вторых, при горении образуется гораздо больше ядови-
тых газов (NO и СО)', чем при детонации, что может вызвать от-
равление работающих в, шахте, В третьих, если вместо взрыва про-
исходит выгорание, то ВВ не совершает той работы, которую оно
должно выполнить. В-четвертых, и для угольных шахт это самое
главное, горение ВВ гораздо легче, чем его взрыв, воспламеняет
метановоздушную смесь или угольную пыль.
При взрывных работах детонацию зарядов возбуждают взры-
вом капсюля-детонатора. Выгорание при этом может возникнуть
лишь в том случае, если хотя бы часть заряда не взорвалась. Ос-
495
новным же свойством ВВ, определяющим возможность отказа
взрыва, является его детонационная способность, характеристиками
которой могут служить критический диаметр детонации и способ-
ность к передаче последней на расстояние.
Возможность выгорания отказавшего заряда, естественно, зави-
сит от способности взрывчатого вещества к горению, которая может
быть характеризована, например, критическим диаметром горения.
Обе указанные характеристики могут существенно зависеть от
плотности ВВ. При этом для аммонитов, в том числе и аммонитов
с небольшим содержанием нитроэфиров, детонационная способ-
ность с увеличением плотности уменьшается, способность же к го-
рению, как правило, растет [1]. Следовательно, с точки зрения воз-
можности выгорания повышение плотности вдвойне нежелательно.
Уплотнение ВВ в условиях взрывных работ может произойти
разными путями; вещество может быть сжато при заряжании щпу-
ра; оно может быть уплотнено также газообразными продуктами де-
тонации, опережающими детонационную волну при наличии некото-
рого зазора между стенками шпура и зарядом (канальный
эффект1); уплотнение может произойти в результате действия
ударной волны от взрыва соседнего шпура, что возможно при ко-
роткозамедленном взрывании, и даже при одновременном подрыве
ряда шпуров вследствие разброса во времени срабатывания элек-
тродетонаторов.
Очевидно, что влияние сжатия при данном воздействии должно
существенно зависеть от уплотняемости ВВ, которая может быть
характеризована зависимостью плотности от давления прессования.
Итак, три основных фактора определяют возможность выгора-
ния заряда в нормальных условиях ведения взрывных работ—дето-
национная способность ВВ, его способность к горению и зависи-
мость этих способностей от возможных при взрывных работах изме-
нений вещества.
Настоящее исследование и было посвящено определению ука-
занных зависимостей для двух промышленных взрывчатых веществ
на основе аммиачной селитры — предохранительного аммонита
ПЖВ-20, состоящего из аммиачной селитры (64%), тротила (16%)
и хлористого натрия (20%), и победита ВП-1, содержащего 9%
жидких нитроэфиров (дигликольдинитрат и нитроглицерин).
Определение уплотняемости производили на механическом прес-
се. Навеску ВВ в 10 г сжимали в стеклянной трубке диаметром
21,2 мм металлическим пуансоном диаметром 21,1 мм, перемещаю-
щимся с постоянной малой скоростью (25 мм/мин). Плотность рас-
считывали по кривой, записанной самописцем и показывающей из-
менение высоты слоя прессуемого ВВ с давлением.
На фиг. 1 показана зависимость плотности заряда (6) от давле-
ния прессования (р) для обоих взрывчатых веществ. Мы видим, что
1 В свете новейших исследований следует заметить, что уплотнение заряда
не является единственным фактором, определяющим затухание детонации в ре-
зультате канального эффекта,
196
уплотняемость победита, содержащего жидкие 'нитроэфиры, смачи-
вающие порошок, гораздо выше, чем аммонита, не содержащего
жидких компонентов, даже при отсутствии в нем волокнистых до-
бавок, препятствующих уплотнению.
Определение детонационной способности производили в двух
сериях опытов. В первой из них заряд ВВ вводили в бумажную ко-
ническую гильзу с толщиной стенки 0,17 мм, длиной около 25—
30 см, в одном случае без уплотнения (средняя плотность ПЖВ-20
1,02—1,08 г/см3, ВП-1 0,73—0,84 г/см3) и в другом с уплотнением
текстолитовым пуансоном от руки
до максимально достижимой при
этом плотности (для ПЖВ-20
1,23—1,26 г/см3, для ВП-1 1,48—
1,54 г/см3). Подрыв полученных
зарядов 1 на стальных пластинах
показал, что в зарядах малой
плотности детонация прекраща-
лась при близких диаметрах для
обоих взрывчатых веществ (6,5—
6,7 лш для ПЖВ-20 и 5,8—5,9 мм
для ВП-1); при плотной набивке
бумажной гильзы детонация пре-
кращалась для ПЖВ-20 при диа-
метре 8,3—8,5 лш, для победита
же — при гораздо большем диа-
, метре (20—26 .илг}2,
Фиг. 1. Зависимость плотности от дав-
ления прессования для предохрани-
тельного аммонита ПЖВ-20 и побе-
дита ВП-1.
Чтобы точнее определить зависимость критического диаметра де-
тонации (i/кд) от плотности, опыты были повторены, причем при-
. меняли цилиндрические телескопические заряды в стеклянных
Трубках, уширенных на инициируемом конце. Заряды подрывали
i на латунных пластинах и по отпечатку судили о прохождении дето-
нации.
। На фиг. 2 представлены результаты этих опытов. На фиг. 3 они
! приведены в сопоставлении с данными по критическому диаметру
;,горения (t/кг) [1]. В качестве аргумента на фиг. 3 по оси абсцисс
отложено давление прессования.
Мы видим, что основное преимущество победита, которое до-
стигается введением в его состав сенсибилизирующих нитроэфиров
;:и заключается в повышенной детонационной способности, реально
'Лишь в том случае, если исключена возможность его уплотнения.
' > В качестве инициатора, как и в практических условиях, применяли электро-
''детонатор ЭД-8-56 (гремучертутно-тетрнловый).
8 Образчик ВП-1, с которым производились опыты, был приготовлен раньше
образчика ПЖВ-20 хотя и находился в пределах гарантийного соока хранения.
Чтобы выяснить, не связана ли сильная зависимость детонационной способности
.победита от плотности с повышенной способностью его к старению, опыты были
Повторены со свежеприготовленным в лаборатории образчиком. Критический диа-
метр детонации был несколько меньше, но по-прежнему гораздо сильнее, чем для
"7ЖВ-20, зависел от плотности и особенно От давления прессования.
12 451
497
Если же, как это обычно бывает в условиях практического примене-
ния, такое уплотнение происходит, то повышенная уплотняемость
может легко свести на нет преимущества победита и он будет вести
себя в отношении возможности отказов и особенно выгораний даже
хуже, чем обычные предохранительные аммониты без сенсибили-
заторов, например аммонит ПЖВ-20, состав которого, кстати, от-
Фиг. 2. Зависимость критического
диаметра детонации от плотности
для предохранительного аммонита
ПЖВ-20 и победита ВП-1.
Фиг, 3. Зависимость критических диамет-
ров детонации и горения от давления
прессования для предохранительного ам-
монита ПЖВ-20 и победита ВП-1.
нюдь нельзя считать рациональным. Кривые фиг. 2 иллюстрируют
эти соотношения. Действительно, лишь при насыпной плотности
(меньше I г/слг4) критический диаметр детонации победита несколь-
ко меньше, чем для ПЖВ-20. При уплотнении он растет, однако,
быстрее, чем для ПЖВ-20, и уже при плотности 1,2 г/см3 стано-
вится больше, чем критический диаметр детонации ПЖВ-20.
Из сказанного ясны также пути устранения установленного не-
достатка, из которых отметим здесь два: введение в победит (в до-
статочном, соответственно содержанию жидкости, количестве) ве-
ществ, препятствующих уплотнению, и замена жидких сенсибили-
заторов твердыми [2].
ЛИТЕРАТУРА
1. Андреев К. К- и Попова П. П., ДАН, 1960, 134, 1142.
2. Андреев К. К., Смоляницкий В. 3. и Бахаревич Н. С. Автор-
ское свидетельство № 1894/351529 с приоритетом от 6 февраля 1947. L. Medard,
Mem. des poudres, 1950, 32, 219.
498
.1 И. ГОЛЬБИНДЕР
34. САМОВОСПЛАМЕНЕНИЕ ЖИДКИХ ВЗРЫВЧАТЫХ
СМЕСЕЙ
Исследования самовоспламенения (вспышки) взрывчатых ве-
ществ выполняются, как правило, с индивидуальными взрывчатыми
соединениями [4]. В данной работе приведены некоторые результа-
ты исследования вспышки жидких взрывчатых смесей.
Воспламенение систем такого рода обычно изучается примени-
тельно к условиям горения в жидкостных реактивных двигателях,
где соответствующие жидкости смешиваются и воспламеняются в
виде мельчайших капель [1], [11], [13]. В нашей работе исследовано
самовоспламенение смесей в виде компактных жидких зарядов1.
Помимо интереса, который может представлять такое исследование
для решения некоторых прикладных задач, оно позволяет также
расширить представления о механизме самовоспламенения взрыв-
чатых веществ вообще.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Самовоспламенение жидких смесей тетранитрометана
с органическими аминами
Как известно [11] при смешении в обычных условиях тетранитро-
метана со многими жидкими органическими аминами происходит
самовоспламенение. Мы изучали это явление для пяти аминов: ани-
лина, о-толуидипа, экстралина (смесь анилина, моно- и диметил-
анилипа), ксилидина (техническая смесь изомеров) и триэтилами-
на. Наиболее детально исследованы смеси с анилином, для которых
задержка самовоспламенения при комнатной температуре состав-
ляет несколько десятков секунд. Как было показано специальными
опытами, описание которых мы для краткости опускаем, время вза-
имного растворения компонентов данной смеси в обычных для
наших опытов условиях (выливание тетранитрометана в сосуд с
анилином) составляет лишь незначительную долю индукционного
периода.
В части опытов задержку самовоспламенения определяли с по-
мощью индуктометра с фотоэлементом, схема которого показана
на фиг. 1. Определенное количество амина помещали в тигель, уста-
новленный в приборе так, чтобы луч света от осветителя проходил
над уровнем жидкости и попадал на фотоэлемент. При выливании
в тигель тетранитрометана, струя последнего пересекала луч све-
та и уменьшая освещенность фотоэлемента, вызывала изменение
фототока, регистрируемое на осциллограмме. Момент воспламене-'
ния фиксировали на той же осциллограмме по скачку фототока,
1 Значительная часть экспериментов выполнена во Всесоюзном научно-иссле-
довательском институте буровой техники при участии С. А. Ловля, Н. И. Коз л о
вой, Г. В. Димза, которым автор выражает искреннюю благодарность.
32* 499
вызванному свечением пламени. В другой части опытов, когда ин-
дукционный период был сравнительно велик, время задержки опре-
деляли визуальным наблюдением с секундомером.
г
Фиг. I. Схема индуктометра.
/—тигель с горючий, 2—пробирка с окислителем, J—осветитель, А— фотоэлемент.
Задержка самовоспламенения зависит от концентрации компо-
нентов в составе смеси. Характер этой зависимости виден на фиг. 2,
где представлены результаты
Концентрация анилина в смеси в
а/о объемн.
Фиг. 2, Зависимость задержки само-
воспламенения смеси тстранитромета-
Н’ й анилина от состава.
/—нижний и 3—верхний концентрацион-
ные .пределы самовоспламенения, £—сте-
хиометрический состав, + йоспламенеиис,
—’ беспламенное разложение.
sItob со смесью тетранитрометана
и анирина при 15—17°; общий
объем смеси оставался постоян-
ным, равным 3,5 мл. Как видно
из графика, в области смесей
с отрицательным кислородным
балансом, индукционный период
сравнительно слабо меняется
с составом. Аналогичные резуль-
таты были получены и в опытах
с ксилидином и о-толуидином.
В связи с этим в большинстве
дальнейших экспериментов мы
работали со смесями, имеющими
некоторый избыток горючего про-
тив стехиометрии, и применяли
постоянные для различных аминов
объемы горючего (I мл) и окис-
лителя (2,5 мл). Таким путем
обеспечивалось постоянство усло-
вий смешения, которое в данном
интервале концентраций сильнее
влияет на время задержки, чем
небольшие колебания молекуляр-
500
ного состава. Результаты опытов, проведенных в описанных усло-
виях с 4 аминами, представлены в табл. !.
Таблица 1
Задержка самовоспламенения бинарных смесей тетраннтрометана
(2,5 мл) с аминами (во 1 .«л)
Амин Количество опытов Время задержки сек. Температура опыта °C
Анилин 10 38±9 16
о-Тслуидиг! 12 7,8±3,5 . 24
Ксилидин 22 1,1 ±0,4 22
Трнэтиламнн 15 0,42±0,04 20
Из исследования вспышки обычных В В известно влияние массы
нагреваемого вещества на минимальную температуру самовоспла-
менения. Малые навески разлагаются без воспламенения при той
температуре, при которой большие воспламеняются. Обычно явле-
ния такого рода объясняют, исходя только из тепловых представ-
лений, тем что с увеличением навески скорость тепловыделения,
пропорциональная объему вещества, растет быстрее, чем скорость
теплоотвода, пропорциональная его поверхности.
Влияние массы мы наблюдали и для рассматриваемых здесь
самовоспламеняющихся смесей. В определенных условиях воспла-
менение смеси анилина и тетранитрометана постоянного состава
происходило лишь в том случае, когда общее количество ее превы-
шало 1,2 мл. В других опытах, также при постоянном составе сме-
си, отмечено небольшое сокращение времени задержки с увеличе-
нием общей массы жидкого ВВ.
Можно было бы думать, что и в данном случае влияние массы
обусловлено только относительным изменением скорости тепловьь
деления и теплоотвода с изменением количества смеси. Опыты со
смесью анилина и тетранитрометана показали, однако, что одни
только тепловые процессы не могут объяснить механизм рассмат-
риваемого явления. Приведем из этой серии экспериментов следу-
ющие характерные данные.
Равные количества смеси при постоянном соотношении объе-
мов горючего и окислителя (I :2,5) получали в двух сосудах оди-
наковых размеров. Один из сосудов был стеклянным и находился
при опытах на воздухе, другой — стальным и был помещен в энер-
гично перемешиваемую воду той же температуры. Несмотря на
существенные различия в условиях теплоотвода, минимальное коли-
чество смеси, при котором хотя бы в одном из пяди параллельных
опытов возникает самовоспламенение, и продолжительность ицдук:
ционного периода оказались одинаковыми в обоих сосудах.
Минимальная высота слоя смеси, при которой еще возможно
воспламенение, оказывается практически постоянной. Так, в алю-
миниевых стаканчиках диаметром от 8 до 28 мм, при комнатной
501
температуре, минимальная высота слоя смеси, при которой еще
возникает воспламенение, оказалось одинаковой и равной 2 AtAt1.
Таким образом минимальный объем жидкой смеси, с которым еще
наблюдается самовоспламенение при постоянной начальной темпе-
ратуре, увеличивается пропорционально квадрату диаметра.
Далее, готовили по 1,2 мл смеси в стеклянных сосудах, форма и
размеры которых показаны на фиг. 3. При этом в открытом сосуде
2 воспламенений не было, а в полузамкнутом 1 они возникали ре-
гулярно. При изменении в последующих опытах величины заряда
оказывалось, что в полузамкнутых сосудах минимальное количест-
Фиг. 3, Стеклянные сосу-
ды для опытов по само-
воспламенению смеси те’
транитрометана н ани-
лина.
/—полузамкнутый, 2— откры-
тый.
от дна тигля, так что
во смеси, при котором еще возникает вос-
пламенение, при прочих равных условиях,
в 2—4 раза меньше, чем в открытых сосу-
дах. Таким образом, здесь наблюдалось
существенное различие в условиях возник-
новения вспышки при весьма близких усло-
виях теплоотвода.
Для изучения тепловых явлений в тече-
ние индукционного периода в дальнейших
опытах на индуктометре, показанном на
фиг. 1, в тигель вводили спай малоинерци-
онной термопары. В части параллельных
опытов спай термопары помещали недалеко
после слива окислителя он оказывался в жид-
кости. В других опытах спай находился на высоте 3—7 мм над
уровнем жидкой смеси в тигле. Изменение э. д. с. в данном случае
записывали через усилитель на той же осциллограмме, что и изме-
нение фототока. Одна из таких осциллограм показана на фиг. 4.
В процессе исследования желательно было зарегистрировать из-
менение температуры в жидкости и в парах над нею в одном и том
же опыте. В связи с тем, что в нашем распоряжении был только
один усилитель термо-э.д.с., часть параллельных опытов провели
с дифференциальными термопарами, один спай которых помещали
в жидкую смесь, а другой — над нею.
В результате большого числа опытов было установлено, что в
течение всего индукционного периода температура жидкости ос-
тается практически постоянной и равной начальной температуре
компонентов смеси. Воспламеняется и затем горит практически
холодная смесь. Лишь перед самым воспламенением, а иногда да-
же после него, термопара, помещенная в жидкость, начинает отме
чать слабый и относительно медленный разогрев. С изученными
аминами, к моменту появления пламени, максимальный разогрев,
отмеченный термопарой, спай которой помещен в жидкость, не пре-
вышал 15, а в большинстве опытов был меньше 10°.
1 При отсутствии самовоспламенения, через короткое время после смешения
компонентов возникает беспламенное разложение: смесь как бы вскипает, бурно
выделяется газ, часть жидкости разбрызгивается, а остаток приобретает смоло-
образную консистенцию. _____________________________________________________
502
Быстрый разогрев газов или паров над жидкостью во всех слу-
чаях предшествует воспламенению. В газовой фазе подъем темпе-
ратуры происходил со скоростью порядка многих сотен градусов в
секунду. Наша система записи были слишком инерционной и по-
этому максимальная температура, зарегистрированная ею к мо-
менту появления пламени, не превышала нескольких десятков гра-
дусов. Во всех опытах с дифференциальными термопарами перед
воспламенением отмечен быстрый относительный разогрев спая,
помещенного над жидкостью.
Фиг. 4. Образец осциллограммы при исследовании самовоспламенения жидких
взрывчатых смесей.
1—масштаб времени (кривая эталонной частоты!, 2—кривая изменения фототока, 3—кривая
изменения терио-э. д, с., 4—нулевая линяя, 5—момент слива окислителя, б—момент появле-
ния пламени.
Отметим еще одно важное наблюдение: для всех исследованных
смесей самовоспламенению предшествует «кипение», при котором
выделяющиеся газы или пары увлекают мельчайшие капельки
жидкости. Над жидкостью появляется своего рода «дым». Это яв-
ление отчетливо фиксируется на осциллограммах по уменьшению
фототока перед воспламенением и легко наблюдается в случаях
смесей с большим временем задержки—визуально, а с малым —
при скоростной киносъемке. Интенсивное газовыделение начинает-
ся, как Правило, в самом конце индукционного периода.
Было сделано предположение, что основным процессом, Проте-
кающим в течение значительной части индукционного периода, яв-
ляется термонейтральная или слабоэкзотермическая реакция ами-
на с окислителем, ведущая к образованию конденсированного,
сравнительно стойкого, промежуточного продукта. Дальнейшие
стадии Процесса идут со скоростью, достаточной для самовоспла-
502
менения лишь тогда, когда концентрация этого промежуточного
продукта достигает некоторого предела. Это предположение было
подтверждено следующими простыми опытами.
1. При постоянных условиях готовили 3,5 мл смеси в два прие-
ма: сначала выливали в сосуд небольшую часть компонентов ’
(0,32 мл анилина и 0.8 мл тетранитрометана), а через некоторое
время (называемое в дальнейшем временем выдержки) — остаток.
С увеличением времени выдержки до некоторого предела, за-
держка самовоспламенения, отсчитываемая от момента слива ос-
новной массы смеси, заметно сокращалась. Результаты этих опы-
тов приведены в табл. 2.
Таблица 2
Сокращение задержки самовоспламенения
в результате предварительного накопления промежуточных продуктов
Время выдержки сек* Среднее время задержки самовос- пламенения в сек. Время выдержки сек* Среднее время задержки самовос- пламенения в сек*
0 61 40 35
10 52 60 23
20 42 90 30
Аналогичные опыты при различных соотношениях объемов пер-
вой и второй порций смеси показали, что задержка самовоспламе-
нения при одинаковом времени выдержки сокращается тем силь-
нее, чем больше первая порция, т. е. чем выше в смеси концентра-
ция промежуточных продуктов,
2. В значительный объем амина (20—50 мл), при энергичном
перемешивании, вводили по каплям небольшое количество тетра-
нитрометана (0,04—0,10 моля на моль амина). В результате про-
исходящей реакции наблюдалось почернение амина. С таким по-
черневшим амином проводили далее опыты по самовоспламенению
при обычном соотношении объемов горючего и окислителя (1 :2,5).
Результаты опытов представлены в табл. 3. Как видно из этой
таблицы, в смеси, содержащей избыток амина, промежуточный
продукт оказывается вполне стоек.
Для взрывчатых химических соединений давно уже установлен
факт уменьшения задержки вспышки с ростом температуры. Анало-
гичное явление наблюдается и для самовоспламеняющихся жидких
смесей. О характере этой зависимости в последнем случае судить
с достаточной точностью трудно, так как мы могли исследовать ее
на примере смеси тетранитрометаиа с анилином только в очень
узком интервале температур. Ниже И13 тетранитрометан затверде-
вает, а при температуре выше 60° задержка самовоспламенения
1 В данных условиях -экое малое количество смеси неспособно к самовос-
пламенению.
.504
Таблица 3
Изменение задержки самовоспламенения при добавлении
промежуточных продуктов
Название амина Введено тетранит- рометана в мл на 100 мл амина Задержка самовоспла- метения а сек. Промежуток времени от приготовления раствора промежуточных продуктов до определения задержки самовоспламенения
Анилин нет 32±3 —
10 11 ±2 До 60 мни.
7 9±1 До 60 мин.
в 7 10±1 12 суток
о-толуидин нет 49+5 —
О-ТОлуидНн 5 8±1 До 60 мин.
о-толуидин 5 10 суток
сокращается настолько, что время смешения компонентов в нашем
приборе составляет уже значительную ее часть. Если полагать, что
зависимость времени задержки самовоспламенения от температуры
имеет в данном случае тот же вид, что и для других изученных си-
стем, то эффективная энергия активации оказывается около
12 ккал!моль.
Ряд опытов со смесью тетранитрометана и анилина был прове-
ден при разных составах атмосферы над жидкостью. Оказалось,
что прн прочих равных условиях среднее время задержки самовос-
пламенения в атмосфере воздуха, азота и углекислоты составляло
43+7,, 494',е и 42+1? сек. соответственно.
— Ю —10 — 11
Самовоспламенение жидких смесей на основе
концентрированной азотной кислоты
Мы изучали по методике, описанной в предыдущем разделе, са-
мовоспламенение, возникающее при смешении концентрированной
азотной кислоты с некоторыми аминами (анилин, толуидин, ксили-
дин). фенолами (резорцин, пирокатехин) и непредельными соеди-
нениями (растворы дивинилацетилена в ксилоле, стирола в бензи-
не, тривинилглицерин и Др.). Почти все исследованные смеси само-
воспламеняются при обычной температуре с задержкой, измеряе-
мой долями секунды. В таких системах время задержки в значи-
тельной мере определяется условиями смешения компонентов [И].
[14], [15], [17]. Остановимся кратко на основных обших закономер-
ностях, установленных для самовоспламенения различных горючих
с азотной кислотой.
Так же как для смесей с тетранитрометаном, сильно экзотер-
мическая реакция, приводящая к самовоспламенению и в случае
азотной кислоты, возникает в газовой фазе. Это подтверждается
505
как нашими записями термо-э.д.с. при различном расположении
спаев термопар, так и данными скоростной киносъемки и другими
исследованиями [11], [14].
В отличие от того, что наблюдалось для смесей на основе тет-
ранитрометана, с момента смешения концентрированной азотной
кислоты и реакционноспособного горючего, в конденсированной
фазе возникает экзотермическая реакция и температура жидкости
быстро растет.
В течение короткого индукционного периода термопара, поме-
щенная в жидкость, показывает приблизительно постоянную темпе-
ратуру от 80 до 1303, в зависимости от рода горючего и состава
смеси. При этом идет не только газообразование вследствие проте-
кания химической реакции между горючим и окислителем, но и ки-
пение. Именно кипение и ограничивает, по-видимому, максимальную
температуру жидкости, которая остается примерно той же после
воспламенения, вплоть до момента, когда фронт пламени прибли-
зится к спаю термопары. Таким образом, в случае смесей с азот-
ной кислотой воспламеняется и горит взрывчатое вещество, нагре-
тое теплом первичных стадий реакции до температуры кипения.
Вспышка жидких взрывчатых смесей тетранитрометана
с органическими горючими при нагревании
Тетранитрометан хорошо растворяет различные органические
жидкости и со многими из них образует растворы, устойчивые, по
крайней мере в небольших количествах, при обычной температуре.
Большая часть опытов по изучению вспышки жидких взрывча-
тых смесей при нагревании была проведена в следующих условиях:
массивный латунный блок нагревали с помощью электропечи.
В блоке было просверлено 4 гнезда диаметром 20 мм, заполненных
сплавом Вуда. В одно из гнезд погружали термометр, а в осталь-
ные помещали стеклянные пробирки диаметром 15—17 мм, при
погружении их в сплав на половину своей длины. После того как
устанавливалась заданная для каждого опыта температура, в про-
бирку вводили пипеткой около 0,1 г (0,06—0,08 мл) исследуемой
смеси. В опытах фиксировали наличие или отсутствие вспышки,
характер последней, интервал времени от введения жидкости в про-
бирку до вспышки.
Испытанные нами смеси являются легколетучими веществами,
поэтому в малых количествах оцп давали вспышку лишь при от-
носительно высокой температуре. При меньших температурах про-
исходит лишь быстрое испарение смеси, иногда с заметным раз-
ложением. Ни одна из изученных смесей це вспыхивала в этих
условиях при температуре ниже 160°.
По той же причине (быстрое испарение малой навески) мы не
наблюдали вспышки с задержкой больше 10 сек. Если за это время
вспышка не произошла, то она не могла возникнуть и в дальней-
шем, так как малое количество смеси успевало улетучиться.
506
Таблица 4
Температура и характер вспышки бинарных смесей на основе
тетра н нтрометана
(Условные обозначения в графе „Характер вспышки"; 1 — беззвучное
воспламенение; 2—вспышка со свистящим звуком; 3 ~ вспышка с „хлопком";
4—вспышка с сильным „хлопком"; 5 — взрыв с дроблением пробирки)
Наименование горючего Температу- ра вспышки °C Температу- ра начала кипения смеси °C Массовая ско- рость горения при темпера- туре начала кипения г/см2сех Характер вспышки
Бензольная головка 180 11 0,3 4
Стирол 230—270 112 0,3 4 и 5
Фурфурол 240 122 0,25 5
Тривинилглицерин 278 — — 3
Керосин тракторный 285 123 — 3
Бензальдегид 280 113 — 4
Бензин Б-70 290 — 3
Соляровое масло 300 117 — 2 и 3
Этанол 300 90 0,14 3
Автол 310 125 — 3
Фракция бензина Б-70, 325 90 — 5
выкипающая до 100°
Ксилол (смесь изомеров) 340 — — 3 и 4
Грозненский бензин лря- 370 114 0.15 4
мой гонки
Монон и т роме тан 435 — — ]
Толуол 500 140 0,08 о
Нитробензол 510 129 0.16 3
Дихлорэтан 510 120 0,07 1
Бензол 520 104 0,15 1 2
В табл. 4 приведены результаты исследования 18 бинарных
смесей тетраннтрометана с различными жидкими горючими. «Тем-
пературой вспышки» мы условно назвали минимальную температу-
ру металлического блока, при которой воспламенение происходило
с задержкой не более 5 сек. В этой же таблице приведены для
некоторых смесей температуры «начала кипения», определенные
по способу М. Н. Сиволобова [18], и значения массовой скорости
горения при этой температуре, рассчитанные экстраполяцией дан-
ных, полученных в интервале 20—70°. Для всех смесей испытыва-
лись составы, близкие к нулевому кислородному балансу.
С увеличением количества нагреваемого вещества температура
вспышки заметно снижается. Так, в количестве около 0,1 мл смесь
507
тетранитрометана и бензола вспыхивает только при температуре
печи выше 500°. При кипячении 25 мл смеси того же состава (тем-
пература жидкости не выше 130°) через 3—5 мин наблюдалась
вспышка, в большинстве опытов заканчивавшаяся взрывом. Связь
температуры вспышки и массы нагреваемого вещества наглядно
0 20 1/0 60 80 Ю0 Стирал
Состав горючего в°/о объем».
Фиг. 5. Зависимость температуры вспыш-
ки и скорости горения тройной смеси те-
тр анитрометан—бензол—стирол от соста-
ва горючего.
/—температура вспышки, 2—скорость горения,
(точками показаны опыты, ь которых вспышка
имела характер взрыва).
чивая содержание стирола.
Результаты опытов Представлены на фиг. 5. Из графика видно, что
введение в состав горючего весьма малого количества стирола
(0,7—1,0% объемн.) вызывает снижение температуры вспышки
примерно на 200°; дальнейшее увеличение содержания стирола
вызывает лишь относительно медленное ее падение. С увеличе-
нием содержания Стирола в горючем растет и скорость горения,
вначале медленно, а затем быстрее; одновременно усиливается
интенсивность вспышки. Аналогичные результаты были получены
в опытах с тройной смесью тетранитрометан—бензол—фракция
бензина пиролиза (бензольная головка).
Для бинарных смесей тетранитрометана с фурфуролом, моно-
нитробензолом, мононитрометаном и ксилолом была исследована
зависимость температуры вспышки от соотношения количеств го-*
видна также на примере сме-
си тетранитрометана с легкой
фракцией пиролиза нефти,
так называемой бензольной го-
ловкой, содержащей значи-
тельные количества непредель-
ных углеводородов. Такая
смесь состава, близкого к нуле-
вому кислородному балансу,
взятая для опыта в количестве
0,05 мл, воспламеняется при
190°; та же смесь в количестве,
немного превышающем 1 мл,
самовоспламеняется при 40—
45°; если приготовить одновре-
менно более 20 мл этой смеси,
то воспламенение ее наступает
уже при 15—18°.
Далее была проведена се-
рия опытов с тройными сме-
сями. В качестве окислителя
в них применяли, как и рань-
ше, тетранитрометан, а в ка-
честве горючего — смесь бен-
зола и стирола. Соотношение
горючего и окислителя во всех
опытах поддерживали близким
к нулевому кислородному ба-
лансу, а состав горючего ме-
няли от опыта к опыту, увели-
508
рючего и окислителя в составе смеси. Наибольшее различие в
температуре вспышки наблюдалось в системе тетранитроме-
тан—мононитрометаи, где при нулевом кислородном балансе
вспышка возникает при 435Q, а в смеси с большим избытком окис-
лителя— при 330°. Для других смесей изменение температуры
вспышки с составом не превышало 30° для всего изученного ин-
тервала концентраций (кислородный баланс от минус 40 до
плюс 30). Во всех изученных смесях минимум кривой «темпера-
тура вспышки—состав» оказывался в той же области концентра-
ций, что максимум кривой «скорость горения—состав».
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Рассмотрим сначала результаты, полученные при изучении са-
мовоспламенения смесей тетранитрометана и аминов. Экспери-
ментально установлено, что к моменту воспламенения разогрев
в конденсированной фазе очень мал. Даже после того как про-
изошло воспламенение, температура жидкости, вплоть до момен-
та, когда фронт горения подходит к спаю термопары, остается
близкой к начальной температуре компонентов смеси. Измерен-
ный в опытах «предвзрывной разогрев» жидкости меньше, чем
рассчитанный по теории теплового воспламенения [12] для случая
кондуктивной теплопередачи и заведомо меньше, чем при конвек-
тивном теплообмене в жидкости, размешиваемой пузырьками
образующихся газов. Это обстоятельство уже само по себе гово-
рит о том, что в данном случае нельзя считать, что самовоспла-
менение возникает вследствие теплового самоускорения процесса,
протекающего в конденсированной фазе.
Другие данные подтверждают эту точку Зрения. Прежде всего
здесь необходимо отметить результаты прямых измерений темпе-
ратуры в жидкости и в газовой фазе над нею. показывающие, что
быстрый разогрев, предшествующий самовоспламенению, возни-
кает в газовой фазе. Нарастание температуры газопаровой смеси
непосредственно перед вспышкой идет с очень большой скоростью,
т. е. так, как этого следует ожидать по теории [12] для теплового
самовоспламенения в газовой фазе. Кривые, полученные при из-
мерении температуры жидкости, существенно отличаются От по-
лученных для газа не только по величине максимального разогре-
ва перед воспламенением или по положению начала подъема тем-
пературы относительно момента появления пламени, но и по ха-
рактеру кривой «температура—время». В жидкости подъем темпе-
ратуры, в течение всего времени предшествующего воспламене-
нию и даже некоторое время после него, происходит значительно
медленнее, чем в газе.
Весьма показательны также результаты опытов в двух типах
сосудов, показанных на фиг. 3.,Условия теплообмена жидкости
с окружающей средой в обоих сосудах одинаковы, а критические
условия самовоспламенения в них существенно различаются. Эти
509
различия обусловлены особенностями протекания реакций в га-
зовой фазе, завершающихся вспышкой.
При тождественности условий отвода тепла от реагирующего
газа скорость реакции, приводящей к воспламенению, а следова-
тельно, и критические условия его возникновения, должны опре-
деляться концентрацией реакционноспособных газов. В рассмат-
риваемых опытах эта концентрация зависела от соотношения
между скоростью поступления соответствующих газов (или па-
ров) из жидкости и скоростью разбавления их окружающим воз-
духом. В простейших предположениях количество газов, выде-
ляющихся в единицу времени на единицу поверхности жидкости
(удельная скорость газообразования), должно быть пропорцио-
нально высоте слоя последней. Вблизи от Критических условий эта
концентрация оказывалась недостаточной для воспламенения в
открытом сосуде малой высоты, где происходит быстрое разбав-
ление реакционноспособных газов (или паров) в результате пере-
мешивания с окружающим воздухом. В полузамкнутом сосуде
условия для образования газовой (или паро- газо-капельной)
смеси, необходимой концентрации, оказываются благоприятными.
В открытом цилиндрическом сосуде постоянного диаметра, по
мере увеличения объема приготовляемой смеси, и, соответственно,
высоты ее слоя, удельная скорость газообразования возрастает.
Начиная с некоторой критической высоты слоя жидкости, эта
скорость становится достаточно большой для того, чтобы, несмот-
ря на разбавление выделяющихся газов или паров окружающим
воздухом, могла создаться необходимая для самовоспламенения
концентрация их.
На первый взгляд изложенным представлениям о возникнове-
нии вспышки в газовой фазе противоречат результаты опытов,
проведенных в атмосфере воздуха, кислорода и углекислоты, где
наблюдалась практически одинаковая задержка. На самом деле
противоречия здесь нет. Задержка самовоспламенения, состав-
ляющая для смеси тетранитрометана и анилина десятки секунд,
является суммой времен протекания по крайней мере двух раз-
личных макроскопических стадий процесса. Первая из них проте-
кает в конденсированной фазе и продолжительность ее зависит
от времени накопления активных Промежуточных продуктов и
развития химического самоускорения процесса, приводящего к
образованию газов. Вторая стадия определяется временем созда-
ния необходимой концентрации реакционно-способных веществ н
газовой фазе и тепловым самоускорением идущей здесь реакции
до вспышки. Состав газовой Среды, очевидно, может повлиять
только на вторую стадию процесса. Интервал времени от начала
газовыделения до появления пламени составляет, как показывает
опыт, лищь незначительную долю суммарного времени задержки.
Понятно, что даже существенное изменение этой доли, в связи е
изменением состава атмосферы, не отражается заметным образом
на экспериментально определяемой задержке. Дополнительно сле-
дует отметить, что опыты в атмосфере различных, газов мы про-
510
водили в условиях, далеких от критических параметров самовос-
пламенения. Можно полагать, что например, при меньшей высоте
слоя жидкой смеси, влияние состава атмосферы проявилось бы
сильнее.
Интересные выводы позволяют сделать и результаты опытов,
выполненных в разных условиях теплоотвода. То, что с измене-
нием последних практически не меняется время задержки воспла-
менения, можно было бы объяснить, аналогично предыдущему,
значительным преобладанием времени изотермического химиче-
ского самоускорения над продолжительностью теплового само-
ускорения.
Опытами установлено, однако, что не только задержка, но и
критические условия воспламенения (в данном случае минималь-
ная масса жидкого ВВ) остаются постоянными При изменении
условий теплоотвода. Последнее обстоятельство подтверждает
предлагаемый механизм, согласно которому тепловое самоускоре-
ние (тепловой взрыв) происходит в газах, парах или газокапель-
ной взвеси над жидкостью. Напомним, что в части опытов этой
серии применяли стеклянные сосуды, окруженные воздухом, а в
других — стальные такого же размера, помещенные в воду. Как
показывает расчет, коэффициент теплопередачи от находящейся
внутри сосуда жидкости к окружающей среде меняется по. край-
ней мере в несколько десятков раз, в то время как для газа, на-
ходящегося внутри сосуда, этот коэффициент увеличивается, при
таком же изменении условий, не более чем в полтора-два раза.
Если бы решающую роль играл тепловой взрыв в конденсирован-
ной фазе, то, как показывает расчет, критический размер заряда
должен был бы существенно измениться при переходе от стеклян-
ного сосуда к стальному. Этого в опытах не наблюдалось.
В случае, когда в смесь тем или иным способом вводят зара-
нее приготовленные промежуточные продукты взаимодействия тет-
ранитрометана и анилина — время задержки самовоспламенения
резко сокращается (см. опыты в табл. 2 и 3). Это показывает,
что основную часть индукционного периода занимают реакции в
конденсированной фазе, приводящие к образованию активного
промежуточного продукта. Выяснение механизма и кинетики этих
реакций было предметом специального исследования. Здесь мы
ограничимся лишь указанием на то, что к концу индукционного
периода свободный амин в смеси практически отсутствует. Само-
ускоряющееся взаимодействие продукта первичной реакции с тет-
ранитрометаном приводит в дальнейшем к образованию газов
(паров), воспламеняющихся, когда их концентрация достигнет не-
которого предела.
Самовоспламенение жидких взрывчатых смесей, содержащих
в качестве окислителя азотную кислоту, также возникает в газо-
вой фазе. Однако в ходе предпламенных процессов здесь наблю-
дается отчетливое различие. В случае смесей с азотной кислотой
процессы, протекающие в конденсированной фазе, сильно экзо-
термичны, вследствие чего жидкость интенсивно нагревается. Со-
611
держание в смеси сравнительно легколетучих компонентов есте-
ственно ограничивает максимальный разогрев жидкости темпера-
турой кипения. В связи с этим в газофазных реакциях, поивопя-
щих к самовоспламенению, наряду с промежуточными продуктами,
образовавшимися в жидкой фазе, могут принимать участие так-
же пары исходных веществ или продукты их термического разло-
жения. Если в случае смесей тетранитрометана с аминами вос-
пламенялась и в дальнейшем горела холодная жидкость, то вос-
пламенение и горение смесей с азотной кислотой происходит, ког-
да жидкость нагревается до кипения *.
Самовоспламенение жидких взрывчатых смесей при повышен-
ной температуре, достигаемой путем нагрева их от внешнего ис-
точника (тепловая вспышка), имеет много обшего с вспышкой
обычных взрывчатых веществ. Отметим с этой точки зрения от-
четливую связь между интенсивностью вспышки, скоростью и ха-
рактером горения, которые для индивидуальных взрывчатых ве-
ществ установил К. К. Андреев [3]. Как мы видели, наиболее рез-
кий, взрывной характер имеет вспышка смесей тетранитрометана
с бензольной головкой, стиролом и фурфуролом. Эти смеси горят
со сравнительно большой скоростью, горение их неустойчиво и
даже при обычной температуре, в стеклянных трубках диаметром
5—7 мм наблюдались случаи перехода горения во взрыв. При по-
вышенной температуре, достигаемой в жидкости в момент тепло-
вой вспышки, устойчивость горения еще меньше и даже в иссле-
дованных малых количествах смеси вспыхивают с большой интен-
сивностью.
Для некоторых индивидуальных ВВ было обнаружено [2] яв-
ление «верхнего предела» температуры вспышки. Это явление на-
блюдается для веществ, имеющих сравнительно низкую темпера-
туру кипения, в условиях, когда время химического самоускоре-
ния реакций распада оказывается больше времени испарения.
Исследованные жидкие смеси летучи, поэтому для многих из них,
например, для смесей с бензолом, толуолом, дихлорэтаном, в ус-
ловиях опытов с малыми количествами жидкости, явление «верх-
него предела» наблюдается во всем интервале температур: от ком-
натной до температуры около пятисот градусов, когда уже пра-
вильнее говорить не о вспышке ВВ в обычном смысле, а о под-
1 Явления, протекающие в процессе горения, мы здесь не рассматриваем. От-
метим ли[иь одно обстоятельство, существенное как для воспламенения, так и для
последующего горения взрывчатой жидкости. Тетранитрометаи хорошо растворяет
жидкие органические горючие. В связи с этим при значительных задержках само-
воспламенения всегда успевает образоваться гомогенный раствор. Азотная кислота
и большинство жидких горючих пе обладают взаимной растворимостью. Кроме
того, задержка самовоспламенения для смесей с азотной кислотой очень мала,
поэтому к началу его. как правило, однородного раствора ие получается; воспла-
меняется и горит в этом случае взвесь капель одного компонента в другом. Раз-
меры капель, а следовательно, и поверхность контакта между реагирующими
веществами, зависят как от физико-химических свойств обеих смешиваемых жид-
костей, так и от условий их смешения. Во многих случаях именно влияние послед-
него фактора определяет время задержки самовоспламенения и состояние горя-
щей смеси.
513
жигании его паров нагретой, поверхностью. Если увеличивать мас-
су В В, то при тех же условиях нагрева время испарения растет
и самоускорение распада успевает произойти раньше, чем испа-
рится все вещество. Верхний предел температуры вспышки таким
образом может быть снижен, что и наблюдалось в опытах со сме-
сью тетранитрометана с бензолом и др.
Наглядное представление об относительной роли в процессе
вспышки химического самоускорения и испарения вещества при
нагревании ^ают результаты опытов с тройной смесью тетранитро-
метан—бензол—стирол. Смесь тетранитрометана и бензола легко
испаряется, а в химическом отношении сравнительно инертна. Со-
ответственно, самовоспламенение этой смеси возникает только при
такой температуре, печи, при которой вспыхивает смесь паров.
Температура воспламенения смеси чистых паров сравнительно вы-
сока, в данном случае около 500°. После добавления к бинарной
смеси небольшого количества (около 0,5% объемн.) стирола, ус-
ловия испарения капли смеси существенно измениться не могли.
Резкое снижение температуры вспышки тройной смеси объясняет-
ся тем, что наличие в системе реакционноспособного стирола обус-
лавливает при более низкой температуре печи протекание с доста-
точной скоростью процессов образования активных продуктов, так
что концентрация их, необходимая для самовоспламенения, соз-
дается за время меньшее, чем время полного испарения.
Если компоненты смеси имеют различную упругость пара, то,
как известно, состав паровой фазы может отличаться от состава
жидкости. Как известно [7], [8], при горении жидких смесей легко-
летучих веществ это обстоятельство вызывает своеобразные явле-
ния дробного выгорания, обусловленные различиями составов
жидкости и паров над нею, а также их изменением по мере рас-
пространения горения. Дробное испарение играет определенную
роль и при тепловой вспышке смесей, что видно, например, по ре-
зультатам опытов со смесями одних и тех же компонентов при раз-
ных соотношениях между ними. В этим опытах было установлено
совпадение областей концентраций, в которых наблюдается мак-
симум скорости горения и минимум температуры вспышки. При
опытах по тепловому самовоспламенению, выполненных с очень
малыми количествами жидкой смеси, влияние дробного испаре-
ния относительно невелико. Капля смеси, в большей своей части,
успевает испариться за время меньшее, чем задержка самовос-
пламенения. Различия в составах жидкости и пара, обусловлен-
ные разницей в упругостях паров компонентов, оказываются при
этом сглаженными.
Сравнительно давно А. Ф. Беляев [5] указывал на то, что
вспышка летучих ВВ возникает в паровой фазе. Это положение
по существу никем не подвергается теперь сомнению, однако до
сего времени не было Прямых его экспериментальных подтверж-
дений. В работах по теории теплового взрыва ВВ, в том числе и
выполненных в недавнее время'[9], [10], [16], это существенное об-
стоятельство также фактически нс учитывается. При исследовании
33 451 513
жидких смесей получены прямые экспериментальные подтверж-
дения того, что самовоспламенение возникает в газовой фазе.
Для изученных в данной работе смесей наблюдалось два раз-
личных режима самовоспламенения.
1. В конденсированной фазе идет, без существенного разогре-
ва вещества, самоускоряющийся процесс его превращения в реак-
ционноспособные газы. Когда концентрация последних превысит
некоторый предел, реакции между ними завершаются тепловым
с ан оу ско рением (тепловым взрывом), приводящим к вспышке. Тем-
пература конденсированного вещества, воспламеняющегося по
этому режиму, может быть значительно ниже температуры его
кипения. В связи с этим пары исходного вещества могут не играть
существенной роли в газофазных реакциях, завершающихся
вспышкой.
2. В конденсированной фазе идут экзотермические реакции.
В результате химического и теплового самоускорения этих реак-
ции возникает прогрессивный саморазогрев. Максимальная тем-
пература, однако, ограничивается температурой кипения самого
взрывчатого вещества или продуктов первичных стадий его пре-
вращения [. Тепловой взрыв и воспламенение происходит, как и
в предыдущем случае, в газовой фазе, но теперь в них, наряду
с продуктами первичных реакций, могут принимать участие и пары
взрывчатого вещества.
Механизмы самовоспламенения смесей и индивидуальных
взрывчатых веществ должны иметь много общего. Можно, поэтому,
ожидать, что описанные два режима самовоспламенения будут
наблюдаться и для некоторых взрывчатых соединений.
ЛИТЕРАТУРА
1. Альтман Д. и Пеннер С., Горение жидких топлив, CC5. «Процессы го-
рения» (перевод с английского), Физматгиз. 1961, стр. 392—430.
2. Андреев К, К, Сборник статей по теории взрывчатых веществ, Оборон-
гиз, 1940, стр. 137.
3, Андреев К. К., ЖПХ, 1948, 21, 462.
4. Андреев К- К.. Термическое разложение и горение взрывчатых веществ.
Госэнергоиздат, 1957, стр. 277.
5. Беляев А, Ф„ ЖФХ, 1946, 20, 613.
6. Беляев А. Ф„ ЖПХ, 1950, 23, 432.
7. Гольбин дер А, И., Горячев В. В.. ЖФХ, 1961, 35, 1808.
8. Г о.л ь б и н д е р А. И., Настоящий сборник, с;р. 457.
9. Мержанов А, Г., Дубовицкий Ф. 14., ДАН. 1958, 121, 618; 1959;
124, 362.
10. Мержанов А. Г.,Абрамов В. Г., Д v б о в и цк и й Ф. И., ДАН, 1959,
128, 1238.
11. Паушкия Я. М,, Химический состав и свойства реактивных топлив,
АН СССР. 1958, стр. 327,
12. Семенов Н. Н., О некоторых проблемах химической кинетики и реак-
ционной способности, изд. 2-е, АН СССР, 1958, стр. 421,
1 Такую же роль, как кипение, может в некоторых случаях играть эндотер-
мическое разложение исходного вещества или промежуточного продукта,
13, Ш аулов Ю. X,, Лернер М. О., Горение в ЖРД, Оборонгиз, 1901,
стр. 122—127.
14. Bernard М,, Fifth Symposium on Combustion, 1955, p. 217.
!5. Barrere A., Moutet A., Fifth Symposium on Combustion, 1955.
p. 177.
16. Gray, Harper, Trans. Faraday Soc,, 1959, 55, 581.
17. Kilpatrick et al.. Fifth Symposium on Combustion, p. 196; J. phvs.
Chem., 1953. 57, 385.
18. S i v о 1 о b о f f M. N., Berichte, 1891, 24, 944.
Б. H. КОНДРИК.ОВ
35. ОБ ИНТЕНСИВНОСТИ ВСПЫШКИ ВЗРЫВЧАТЫХ
ВЕЩЕСТВ
Способность к самовоспламенению при повышенной темпера-
туре—одно из самых общих свойств всех взрывчатых систем и,
в частности, конденсированных взрывчатых веществ. Основные
характеристики самовоспламенения — условия, при которых оно
становится возможным (так называемые критические условия) и
зависимость индукционного периода (задержки вспышки) от тем-
пературы — изучались многими исследователями и для ряда ВВ
известны довольно хорошо. Однако эти характеристики по суще-
ству описывают лишь развитие подготовительных, предпламен-
ных реакций, совершенно не касаясь того, как развивается Процесс
после возникновения пламени. Известной характеристикой распро-
странения пламени при вспышке может служить интенсивность
последней, определяемая скоростью газообразования п повышением
давления при самовоспламенении. К сожалению, у. большинстве
работ эта сторона вопроса почти не затрагивается. В лучшем слу-
чае отмечается лишь характер вспышки, оцениваемый по силе звука
да по тому, дробится или остается целым при вспышке стеклянный
сосуд,в котором она происходит.
Задача настоящей работы заключалась в том, чтобы с помо-
щью простейшей методики сравнить между собой различные ВВ
по интенсивности их теплового самовоспламенения. В качестве
меры интенсивности использовалась высота или энергия подскока
шарика, лежащего на отверстии пробирки, в которой вспыхивала
небольшая навеска ВВ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методика проведения опытов заключалась в следующем. Навеску ВВ вво-
дили в стеклянную пробирку, нижняя часть которой на глубину 5—7 см погружа-
лась в термостат со сплавом Вуда, а верхняя была вставлена в массивную метал-
лическую гильзу с отверстием, закрыты^ стальным или стеклянным шариком
(фиг. 1). Высота подскока шарика при вспышке определялась визуально с помо-
щью шкаль;. Задержка вспышки определялась ио секундомеру. Температура тер-
мостата поддерживалась постоянной с точностью до одного градуса.
Высота подскока шарика отмечалась только в тех опытах, в которых пробирка
оставалась целой. Если при вспышке пробирка дробилась, высота подскока сильно
33* 515
менялась от опыта к опыту и и качестве меры интенсивности вспышки исполь-
зоваться уже не могла.
В большинстве опытов применяли пробирки диаметром 1,7 и длиной 15 см,
в ряде случаев —диаметром 2 и длиной 20 си. Навеска ВВ обычно вводилась
в уже прогревшуюся до температуры термостата пробирку, однако все жидкие,
а также некоторые твердые вещества помещали в пробирку перед погружением
ее в термостат. Различия в высоте подскока шарика прн таком изменении условий
опыта были заметными (прн использовании больших пробирок высота подскока
шарика уменьшается — см., например, фиг. 12а и б — при вве-
дении вещества в холодную пробирку — увеличивается), ио не
слишком значительными.
Интенсивность вспышки обычно характеризовали пронз-
ведением высоты подскока шарика на его вес: E^mh. При
сильных вспышках (Е=102—1(г г см) эта величина практи-
чески не зависит от веса шарика, при слабых — легкие шарики
дают большие Е, чем тяжелые.
Фиг. 1, Схема
прибора для оп-
ределения ин-
тенсивности
вспышки ВВ.
J—пробирка с на-
ьеской ВВ, 2—
т«апнк. 3—гилъзэ
и уплотнением.
4—шкала. 5—тер-
мостат со спла-
вом Вуда.
При проведении опытов были обнаружены сле-
дующие основные черты явления.
При температуре, ниже некоторой критической,
вещество разлагается спокойно, без появления пла-
мени, и шарик остается неподвижным. При дости-
жении некоторой критической для данных условий
температуры самовоспламенения происходит
вспышка, обычно сопровождающаяся пламенем и
звуком и подбрасывающая шарик на некоторую
высоту. По характеру изменения этой высоты при
изменении температуры и других условий опыта,
а также по максимальной высоте подскока шарика
при данной навеске разные ВВ отличаются друг от
друга весьма существенно.
На фиг. 2—4 изображена зависимость интенсив-
с ышке₽7диаметр -Н0СТ11 11 задержки вспышки от температуры термо-,
отверстия ю жл) стата для небольших (0,01—-0,05 г) навесок не-
скольких быстрогорящих ВВ. Характерным для
большинства из них является то, что при всех тем-
пературах вплоть до весьма высоких — интенсив-
ность вспышки, после более или менее короткого участка началь-
ного роста, остается практически постоянной.
Несколько сложнее картина в случае тетразена (фиг. 4). Вы-
сота подскока шарика при вспышке 0,01 г этого ВВ при 136—200°
невелика и с температурой меняется мало, однако при 152 и 155°
наблюдались очень резкие вспышки, сопровождавшиеся дробле-
нием пробирки. При увеличении навески до 0,02 г интервал такого
детонационноподобного самовоспламенения расширялся.
Любопытное явление наблюдалось в случае стифнатов свинца
и калия. При 275—290° эти вещества наряду с обычными вспыш-
ками, возникающими после продолжительного (десятки и сотни
секунд) нагревания, в ряде опытов при той же температуре, да-
вали вспышки с задержкой на один-два порядка меньшей (от до-
516
лей секунды до секунд). Аналогичное явление наблюдалось и в
случае стифниновой кислоты.
Основное отличие вторичных и метательных ВВ от инициирую-
щих по характеру зависимости интенсивности вспышки от темпе-
Фиг. 2. Зависимость интенсивности (о) и задержки (б) вспышки
гремучей ртути от температуры.
Навеска ВВ: I—0,02. 2—0,05 г.
ратуры заключается в том, что, начиная с некоторого значения
последней, интенсивность самовоспламенения этих ВВ падает, а
высота подскока шарика уменьшается, в большинстве случаев.
a) g)
Фиг. 3. Зависимость интенсивности (а) и задержки (б) вспышки пикратов
натрия (!) н CBHijua (2) от температур,,!.
Навеска ВВ 0,02 г. Нз ррс. а каждая точка — среднее из 3—5 опытов.
практически до нуля L В рамках этой общей закономерности раз-
ные вторичные ВВ по характеру зависимости интенсивности
вспышки от температуры могут заметно отличаться друг от друга
1 Интенсивность вспышки некоторых порошкообразных ВВ при дальнейшем
повышении температуры может снова несколько возрасти.
517
Так как динитронафталин детонирует только от промежуточного
детонатора, были проведены контрольные опыты с инертным веще-
ством (уротропином); обжатие свинцовых цилиндров в этих опы-
тах происходило в результате детонации тетриловой шашки. При
подрыве инертного вещества Ю-граммовой тетриловой шашкой
в трубке диаметром 40 мм и высотой 100 мм получается обжатие
свинцового цилиндра 9,0 мм, а в трубке того же диаметра, но дли-
ной 200 мм — 7—8 мм. Тетриловая шашка весом 5 г в этих же ус-
ловиях дает обжатие свинцовых столбиков соответственно 4 и
1 -2 мм.
В табл. 5 приведены результаты опытов с динитронафталином,
при плотности 0,45—0,60 г'.см?.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
На примере изученных динитросоединений еще раз потвержде-
но, что наличие оболочки, оказывающей сопротивление разбросу
взрывчатого вещества, делает возможным детонацию зарядов та-
кого диаметра, при котором без оболочки детонация затухает.
Наиболее характерны в этом отношении опыты с динитронафтали-
ном, В то время как в бумажной гильзе это вещество не детонирует
даже при диаметре заряда 200 мм, в стальной трубке детонация его
происходит при диаметре 30 мм.
Анализ полученных результатов позволяет оценить влияние, ко-
юрое оказывают различные характеристики оболочки на детона-
ционную способность заряда. Помимо инерционного сопротивле-
ния оболочки, отчетливо проявляется роль ее прочности. Так в опы-
тах с динитрофенолом (табл. I) легкая, но обладающая существен-
ной прочностью на разрыв оболочка из жести обеспечивает дето-
нацию заряда такого диаметра, при котором в случае несвязанной,
но более массивной оболочки из песка взрыв затухает. Аналогич-
ные результаты наблюдались и в случае динитротолуола (табл. 2),
где заряд плотностью 0,6 г!см? детонировал в прочной оболочке
нз жести и не взрывался в оболочке равной массы из стекла.
В случае зарядов динитронафталииа диаметром 30 мм наблюда-
лась полная детонация в стальной трубке и затухание ее при
взрыве в оболочке из того же материала, но составленной из от-
дельных полос и поэтому лишенной прочности (см, табл. 5).
Можно отметить, что наряду с инерционным сопротивлением
и прочностью оболочки как одного целого, существенное влияние
на возможность распространения детонации оказывает также сжи-
маемость материала, его способность уплотняться под действием
взрыва. Так в случае динитрофенола (табл. 1) малосжимаемая
оболочка из воды или парафина обеспечивает детонацию заряда
диаметром 29 мм, в то время как в легко уплотняемой оболочке
из песка взрыв заряда такого диаметра затухает. При сопоставле-
нии этих опытов следует учесть, что средняя плотность и, следова-
тельно, инерционное сопротивление оболочки как целого в случае
песка больше, чем в случае воды, но при быстрых воздействиях
55(5
вода, по-видимому, обладает, в отличие от песка, некоторым сопро-
тивлением на разрыв. Такая же картина получена в опытах с заря-
дами из динитротолуола плотностью 0,6 г/слР в оболочках из воды
или глины, с одной стороны, и из песка — с другой. С рассматри-
ваемой точки зрения интересны результаты, полученные с динитро-
хлорбензолом. Заряд этого вещества, окруженный легкой и сравни-
тельно непрочной, но малосжимаемой оболочкой из стекла, детони-
рует при диаметре 40 мм. Если окружить такой заряд легко уплот-
няемой оболочкой из того же динитрохлорбензола толщиной 15-илг.
имеющей общую массу в 6—7 раз больше, чем масса стеклянной
оболочки, то детонация в заряде, диаметр которого увеличен до
70 мм, затухает.
Опыты с зарядами динитротолуола разной плотности еще раз
(ср. табл. 2) подтвердили, что критический диаметр распростра-
нения детонации в нем растет с увеличением плотности, как это
наблюдалось ранее для смесевых взрывчатых веществ, например
аммонитов.
Полученные результаты показывают далее, что все исследован-
ные динитросоединепия в виде порошкообразных зарядов малой
плотности в относительно прочных оболочках и при малом диа-
метре способны детонировать. Наибольшую опасность с этой точ-
ки зрения представляют динитрофенол, динитротолуол и динитро-
х.Юрбензол. Эти результаты должны учитываться при рассмотре-
нии вопросов опасности взрыва в производстве и применении со-
единений такого типа.
выводы
1. Изучено влияние оболочек различного характера на бри-
зантное действие взрыва некоторых динитросоединений бензоль-
ного ряда.
2. Установлено, что чем меньше детонационная способность ве-
щества, тем большее влияние иа устойчивость детонации оказы-
вает оболочка,
3. Показано, что па детонационную способность исследуемых
веществ, помимо инерционного сопротивления оболочки, большое
влияние оказывает ее прочность.
Можно отметить также, что наряду с инерционным сопротив-
лением и прочностью оболочки существенное влияние на возмож-
ность распространения детонации оказывает ее сжимаемость, спо-
собность уплотняться под действием взрыва.
ЛИТЕРАТУРА
I. БеляевА. Ф., ДАН СССР, 1945. 50, 299.
2. Блинов И. Ф., Научные доклады Высшей школы. Химическая технология,
ЛЬ 4. 1958, стр, 640.
3. Блинов И. Ф., «Химическая промышленность» 1959, .№ 5, стр. 419.
4. Харитон Ю. Б., О детонационной способности ВВ, Сб. «Вопросы тео-
рии ВВ», АН СССР, 1947.
557
Одни из них (пироксилин, тетрил, дина, вьтсок о процентные
растворы нитроклетчатки в нитроглицерине, тротил *) характери-
зуются тем, что при повышении температуры увеличение ицтен-
т сен
130 150 170 130 t°c
а)
&~раз<лвжение 5ез вспь
130 150 ПО 190 VC
.; * взрыв с дровыекаем
проварки.
Фиг. 4. Зависимость интенсивности (а) и задержки (б)
вспышки тетразена от температуры.
Навеска ВВ: ! 0,(11. 2—0.01’ г.
Фиг. 5. Зависимость иг|тс;:снвггости (l't
и задержки (2) вспышки 0,05 г пиро
ксилина от температуры.
Фиг. 6. Зависимость интенсивно-
сти (/) н задержки (2) вспышки
раствора коллоксилина в Нитро-
глицерине (50 : 50) от температуры.
Навеска ВВ 0,02 е. Каждня точка на
кривой т=/(Н—среднее на 5—Ю опытов-
сивности их вспышки до максимальной происходит постепенно, в
значительном (иногда десятки градусов) интервале температур
(фиг. 5—8).
1 Тротил (фиг. 8) отличается От других ВВ тем, что прн достижении некоторой
достаточно высокой температуры (в условиях наших опытов ~365°) самовоспла-
менение его внезапно прекращается и шарик более уже не подбрасывается. До-
стигается так называемый верхний предел самовоспламенения [1].
518
Другие ВВ (дигликольдинитрат, гремучий студень, отчасти
тэн) при переходе через критическую температуру самовоспламе-
нения дают вспышку максимальной интенсивности либо сразу же,
Фиг. 7. Зависимость интенсивно-
сти (/) и задержки (2) вспышки
тетрила от температуры.
Фиг. 8. Зависимость интенсивности (/) и
задержки (2) вспышки тротила от тем-
пературы.
Навеска ВВ Од г,
Навеска ВВ Пд г.
либо в очень узком (один-даа градуса) интервале температур, пос-
ле чего высота подскока шарика постепенно уменьшается
(фиг. 9—11).
Фиг. 9. Зависимость интенсивности (/)
и задержки (2) вспышки диг.тико.тр-
динитрата от температуры.
Навеска В В 0,05 г.
10. Зависимость интенсивности (/)
и задержки (2) вспышки гремучего студ-
ня (10% кодлоксн-тина) от температуры.
Навеска ВВ 0,02 г. Каждаи точка на кривой
-,=/(<}—среднее из 3—5 опытов.
Своеобразное поведение обнаруживает нитроглицерин (фиг. 12,
опыты проводились в пробирках диаметром 1,7 и 2 см). При до-
стижении критической температуры самовоспламенения одна кап-
ля ("*0,02 г) нитроглицерина сразу же подбрасывает шарик на
значительную высоту, большую, чем максимальная высота при
вспышке тэна и дикликольдинитрата и близкую к максимальной
Фиг. 11. Зависимость интенсивности (!) я
задержки (2) вспышки тэна от температуры.
Навеска ВВ 0,05 г вводится в вагретую пробврку
диаметром 1,7 и высотой 15 см (ср. фиг. [6).
Фиг 12. Зависимость интенсивности (/) и задержки (2) вспышки нитро-
глицерина от температуры.
а—вробнрка высотой 20 и Диаметром 2 см, б—пробирка высотой [5 и диаметров
1,7 см.
Навеска ВВ~ 0.02 г (одна капля).
высоте при вспышке гремучего студня. Однако при дальнейшем
повышении температуры, в противоположность тому, что наблю-
дается в случае гремучего студня, дигликольдинитрата или тэна,
интенсивность вспышки НГЦ не падает, а быстро растет, достигая
520
—'1000 г-см, причем в некоторых из этих опытов вспышка дробит
пробирку на куски. При температуре около 290° интенсивность
вспышки резко уменьшается, а при 300—310° становится равной
нулю. Обращает на себя внимание значительный разброс резуль-
татов, особенно при опытах в больших пробирках в области
230-280°
То же самое наблюдается в случае нитрогликоля (фиг. 13) с
той лишь разницей, что дробления пробирки не происходит, а раз-
брос результатов так велик, что в интервале 210—300° зависимость
Ф -разложение без вспышки. % коллоксилина.
Фиг. 14. Зависимость максимальной
интенсивности вспышки растворов
коллоксилинз в нитроглицерине от их
состава.
Навеска ВВ 0.02 г.
Фиг. 13. Зависимость интенсивно-
сти (?) и задержки (2) вспышки нит-
рогликоля от температуры.
Навеска ВВ ^0.05 г (две капли).
интенсивности вспышки нитрогликоля от температуры в коордИ'
патах Е—Т выражается уже не кривой, а некоторой довольно об-
ширной областью.
Максимальные интенсивности вспышки исследовавшихся ве-
ществ и некоторые другие характеристики опытов приведены в
.табл. 1.
Наибольшей из вторичных ВВ интенсивностью вспышки обла-
дают иитроэфиры —- нитроглицерин, метилнитрат, пироксилин, нит-
рогликоль, наименьшей— нитроамины и ароматические нитросо-
единения, особенно термостойкие — гексил, гексанитродифенил-
сульфид, гексоген, тротил. Из ароматических нитросоединений вы-
деляется стифниновая кислота, в навесках —0,04 г подбрасываю-
щая, хотя и весьма нерегулярно, шарик почти так же высоко, как
и нитрогликоль.
Быстрогорящие ВВ по интенсивности вспышки отличаются друг
от друга мейьше, чем вторичные, причем' ни для одного из них она
не достигает максимального значения, наблюдаемого для нитро-
глицерина.
521
Таблица 1
Максимальные интенсивности вспышки некоторых инициирующих
и вторичных ВВ *
Интервал тем- Максималь- Температура,
ператур, в ко- при которой
Взрывчатое вещество тором иауча - наблюдается
г лась и была вспышки вспышка макси-
получена вспышка ** °C (^та1г * ХЮ-2 мальной интен- сивности °C
Гремучая ртуть ! 0,02 | 0,05 /47—200 J3G—2W 2,5 10 155—200 145—200
Тетразен 1 0,0] /36—900- 0,8 140
1 0,02 142—180 2,4 165
Пикрат натрия 0,02 290—33.5 3,1 305—335
Пикрат калия 0,02 310—370 2,9 320
Пикрат свинца 0,02 266—300 ,5,6 270
Гексилат калия 0,02 325- -375 8 345
Нитроглицерин —0,02 /35,5—320 11 225—290
Нитрогликоль * -0.05 202.5 -308 8 21.5—270
Метилиитрат * -0,02 267—270 8,1
Пигликольдннитрат * -0,05 200,5—242 0,7 200,5
Тэн 1 0 05 264—976 1 206
1. 0.3 /53—218 1.6 202
Ли 11а * 1 0,05 209—243 1 250
1 0,2 262—320 0,5 230
Гремучий студень (10% коллоксилина) 0,02 /35—235 2,8 195
Пироксилин порошке- 0,05 176—2О5 8 195
образный Пироксилиновый порох 0,1 170—205 1
Тетрил 0,1 202—265 1 225
Тротил 0,1 ' 309- -36.5 1 365
Гексоген 0.1 230—305 1
Окто гс [I 0,1 26.5—330 0—0,2
1сксил- 0,1 2о0—36-j
Гексанитроднфенилсуль- 0,1 280 -410 1
фин t
Пикриновая кислота*** 0,1 300—325
Пикрат аммония **** 0,02 260—325
Стифиицовая кислота -0,04 280—320 । до 4 1
* Опыты с пешее свами, отмеченными звездочкой, пронодились в про-
бирках диаметром 2 и высотой 20 см, остальные —в пробирках диаметром 1,7
II высотой 15 см.
•=* Критические темлераууры самовоспламенения набраны курсивом.
*** Из 26 опытов в трех очень слабые вспышки, в остальных — термиче-
ское разложение,
*•** Иа 15 опытов в очень слабые вспышки, в остальных — термическое
разложение.
Интересно проследить, как зависит максимальная интенсивность
вспышки растворов коллоксилина в нитроглицерине от их состава
(фиг. 14)’. При содержании 10% коллоксилина максимальная
интенсивность вспышки снижается очень сильно. При увеличении
содержания коллоксилина до 60% эта величина остается прибли-
женно постоянной, а затем снова резко падает, уменьшаясь при
70% коллоксилина более чем в два раза, а при 75% практически
до нуля. Эту зависимость можно сопоставить с тем, что при уве-
личении навески вызвать взрыв растворов, содержащих 60 и мень-
ше процентов коллоксилина в нитроглицерине, очень легко. При
Фиг. 15. Влияние величины навески и природы газа, запол-
няющего пробирку, па интенсивность (а) и задержку (б)
вспышки дины.
Навеска ВВ: J и 2—0г05 г, 3 и 4—0г2 е.
Газ. заполняющий пробирку! / и «У—воздух, 2 и 4—COj.
определенных условиях 60%-ная желатина дает взрыв с дробле-
нием пробирки уже при навеске 0,1 г. В то же время 70%-ный
раствор не дробит пробирку даже прн навеске 1 г.
Вообще говоря, при увеличении навески ВВ естественно ожи-
дать повышение интенсивности самовоспламенения. Действитель-
но, для многих ВВ (инициирующие и быстрогорЯщие, нитрогли-
церин, пироксилин, гремучий студень) при увеличении навески
интенсивность вспышки существенно возрастает. В случае тэна
при навесках от 0,05 до 0,3 г она растет слабо, однако 1—2 г тэна
взрываются, дробя пробирку на мелкие куски. Такой же взрыв,
по после продолжительного периода горения, дают н 2 г дигли-
кольдинитрата, Порошкообразный пироксилин дает сильный взрыц
уже при навеске 0,5 а. Значительно повышает интенсивность
вспышки увеличение навески в случае пикриновой кислоты.
Однако некоторые вторичные ВВ обнаруживают иное поведе-
ние: при увеличении навески интенсивность их вспышки не возра-
стает, а уменьшается. Типичным примером такого вещества являет-
ся дина (фиг. 15), в случае которой увеличение навески в 4 раза
(с 0,05 до 0,2 а) вдвое уменьшает максимальную интенсивность
вспышки.
1 Эти опыты проведены И. В. Бабайиеным.
Таблица 2
Влияние С02 на характеристики вспышки некоторых ВВ"'
Взрывчатое вещество На- веска г Темпера- тура °C В воздухе В СО2
хю-= т сек (jEmaxZ-<ur)X Х10-2 Ct'К
Дигликольди нитрат 0,05 206—207 0,7 8,6 0 18,7
Нитрогликоль 0,05 211—212 3.4 9,8 2,2 8,8
Метнлнитрат 0,02. 267—270 8,1 2.9 0,9 3,2
Пироксилин 0,05 189—191 2,9 66 1,5 ©С.
* Значения Е и т—средние из нескольких опытов.
Для того чтобы выяснить влияние на интенсивность вспышки
процессов, происходящих в газовой фазе, были проведены опыты,
при которых воздух в пробирках был заменен на углекислый газ
Фиг. 16. Влияние природы газа, заполняющего пробирку, на интенсивность (и)
и задержку (б) вспыщки тэна.
Навеска ВВ 0,05 a. f—а воздухе, 2—в СО-».
Точки и» оси абсцисс при низкой температуре — разложение без вспышки. Навеска вво-
дился в пробирку диаметром 2 и высотой 20 см перед погружением последней в термо-
стат (ср. фиг. И).
(табл. 2, фиг. 15, 16 и 17). Результаты этих опытов показывают,
что состав газовой фазы играет существенную роль при самовос-
пламенении не только легколетучего метилнитрата, который иначе
как в газовой фазе вспыхнуть и не может, но и при вспышке таких
труднолетучих веществ, как дигликольдпнцтрат и тэн, и даже сов-
сем нелетучего пироксилина.
Интересное поведение обнаруживает дина (фиг. 15), При ма-
лой (0,05 а) навеске максимальная интенсивность вспышки ее в уг-
лекислоте значительно меньше, чем в воздухе, при большой
(0,2 г) — обе интенсивности практически совпадают друг с другом,
а также с интенсивностью вспышки 0,05 г в атмосфере СО2.
.21
В случае нитроглицерина замена воздуха на углекислоту су-
щественно уменьшает интенсивность вспышки при низких темпера-
турах и практически не влияет на нее при высоких (фиг. 17).
Следует отметить, что замена воздуха на СОа в большинстве
случаев угнетающе действует не только на интенсивность вспышки,
но и на способность веществ к самовоспламенению, заметно по-
вышая критическую температуру вспышки.
Фиг. J7. Влияние состава газовой фазы на интенсивность (а) и за-
держку (б) вспышки нитроглицерина.
Навеска В В —0,02 г. /—в воздухе, 2—в СОа,
Характерной особенностью 'большинства жидких и плавящихся
ВВ, отмечавшейся еще А, Ф. Беляевым [3], является образование
перед вспышкой иногда весьма значительных количеств пены. Как
показывает визуальное наблюдение, количество и консистенция
пены изменяются от вещества к веществу и существенно зависят
от условий проведения опыта. В частности, при повышении темпе-
ратуры количество пены значительно уменьшается, а при достаточ-
но высокой температуре она не образуется вовсе. Существенно от-
метить, что образование пены способствует увеличению интенсив-
ности вспышки, а в случае больших (1—2 г) навесок таких ВВ, как
дигликольдинитрат и тэн, обусловливает возможость их взрыва.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Вспышка ВВ, как известно, представляет собой горение нагре-
того вещества, возникшее путем его самовоспламенения. Интенсив-
ность вспышки определяется скоростью газообразования при горе-
нии, а последняя для большинства ВВ в основном — скоростью и
поверхностью горения 1.
1 Скорость газообразования, естественно, зависит также от удельного объема
газообразных при температуре горения продуктов. Здесь эту сторону вопроса мы
рассматривать не будем.
Значение нормальной скорости горения при вспышке вещества
совершенно очевидно. Отчетливо проявляется оно, например, при
сравнении большинства вторичных ВВ с быстрогорящими, особен-
но пикриновой кислоты и гексила с пикратами и гексилатами ме-
таллов (см, табл. 1).
Не менее очевидно, однако, что нормальная скорость горения
не является единственно определяющим процесс фактором. Гексо-
ген и тетрил горят быстрее, чем днгликольдинитрат и нитрогликоль,
между тем вспышка 0,1 г гексогена даже не Приподнимает шарик,
тетрил дает Е равную всего 15 гем, тогда как дигликольдинитрат
(при навеске 0,05 г) дает £=70 гем, а нитрогликоль по интенсив-
ности вспышки приближается к гремучей ртути. Наиболее вероят-
ной причиной такого различия является разница в величине поверх-
ности горения при вспышке этих веществ.
Поверхность горения при самовоспламенении ВВ тем больше,
чем больше возможность проникновения пламени в глубь заряда,
т. е. с одной стороны, чем больше температура газов и воспламе-
няемость вещества, а с другой —чем вещество рыхлее и пористее,
чем меньше его прочность, плотность, вязкость и т. п. Наглядно
иллюстрируют это утверждение опыты с порошкообразным пирок-
силином и пироксилиновым порохом (см. табл. 1), Первый из них
при вспышке подбрасывает шарик почти так же высоко, как гре-
мучая ртуть, а второй вообще не подбрасывает его. Очевидно, рых.
лость и пористость порошкообразного пироксилина при достаточке
высокой воспламеняемости нагретого вещества обеспечивают про-
никновение пламени в глубь навески и бурное интенсивное ее сго-
рание.
Аналогичным образом ведут себя и те жидкие В В (типичный
пример их нитроглицерин), скорость горения которых при повы-
шенной температуре Превышает критическую скорость горения
жидкости по Ландау—Андрееву [2]. В атом случае горение жид-
кости идет на турбулентном режиме, газообразные продукты про-
никают в массу ВВ и вспышка принимает характер взрыва.
Однако скорость горения многих вторичных ВВ (нитрогликоля,
дигликольдинитрата, тэна и др.) даже при температуре самовос-
пламенения все еще очень далека от критической, между тем
вспыхивают они весьма интенсивно, в определенных условиях —
со взрывом. Очевидно, при нагревании эти вещества претерпевают
такие превращения, которые позволяют пламени проникать в
глубь заряда даже при сравнительно малой нормальной скорости
чго распространения. Можно полагать, что основным из этих прев-
ращений является вспенивание ВВ. По-видимому, легкая, мало-
вязкая пена есть именно то .состояние вещества, при котором го-
рение особенно легко проникает в глубь его, и, охватив большую
поверхность, создает повышение давления, характерное для
взрыва.
Распространение горения в слое пены, 'помимо всего прочего,
зависит от ее консистенции. Легкая подвижная пена, образующая-
ся при нагревании таких ВВ, как ди гликольдинитрат и тэн, сго-
рает быстро и, если количество ее достаточно велико, дает взрыв.
Вещества, плавящиеся при высокой температуре, особенно термо-
стойкие ВВ обычно дают пену более густую, вязкую, сгорающую
сравнительно медленно и спокойно.
Другим важным фактором, определяющим интенсивность са-
мовоспламенения ВВ в условиях наших экспериментов, является,
как показывают опыты по вспышке в атмосфере углекислоты,
превращение вещества в реакционноспособные газы и пары и об-
разование смеси их с воздухом. Склонность газовых систем к тур-
булизации фронта и увеличению поверхности горения общеизвест-
на, естественно ожидать поэтому, что превращение конденсирован-
ного вещества в способный к горению газ уже само по себе зна-
чительно увеличит скорость его сгорания, С другой стороны, горе-
ние газовой смеси в результате повышения давления, разбрасы-
вания вешества струями горящих газов, проникновения их в глубь
рыхлого ВВ и т. п, может привести к увеличению скорости сгора-
ния или даже взрыву самого конденсированного вещества. Нако-
нец, при взрыве газовой смеси может возникнуть и ударная волна,
достаточно интенсивная, чтобы возбудить взрыв или детонацию
в конденсированном веществе, тем более нагретом и вспенившемся.
Можно заключить, следовательно, что в случае большинства
вторичных и метательных ВВ для возникновения вспышки значи-
тельной интенсивности необходимо, чтобы при нагревании веще-
ства образовались большие количества либо маловязкой и легкой
пены, либо горючей газовой смеси [.
Рассмотрим теперь в свете высказанных выше предположений
основную из полученных в настоящей работе закономерностей —
зависимость интенсивности вспышки ВВ от температуры. Для
большинства ВВ переход через критическую температуру самовос-
пламенения сопровождается более или менее плавным увеличени-
ем интенсивности вспышки. Основная причина этого заключается,
вероятно, в том, что при повышении температуры уменьшается
степень разложений вещества перед вспышкой, т. с. увеличивает-
ся количество сгорающего ВВ и уменьшается разбавление его
продуктами распада.
В случае самовоспламенения тех веществ, у которых при ма-
лой навеске вспышка значительной цли даже максимальной ин-
тенсивности возникает сразу же по достижении критической тем-
пературы самовоспламенения, основную роль в процессе горения
в этих условиях играют, по-виднмому, газообразные Продукты в
смеси с воздухом. Легко видеть, что наибольшее количество по-
следних перед вспышкой образуется именно вблизи критических
условий. Вспышка малых количеств дигликольдипитрата. нитро-
гликоля, нитроглицерина, гремучего студня, а может быть и тэна
при температурах, близких к критической, по-видимому, происхо-
дит именно таким образом.
1 Образование слоя йены или газовой смеси несомненно может влиять не
только на интенсивность, но и на другие характеристики теплового самовоспла-
менения — критические условия и задержку вспышки.
527
Интенсивность вспышки вторичных и метательных ВВ, начи-
ная с некоторой температуры, снижается. Объяснить это явление
можно, предположив, что при высоких температурах вещество
вспыхивает раньше, чем основная масса его успеет подготовиться
к вспышке — превратиться в пену или газы и пары, а при доста-
точно высокой температуре даже просто прогреться. Скорость сго-
рания вещества при этом уменьшается, интенсивность вспышки
падает.
Инициирующие ВВ, быстро горящие при любой температуре, в
такой подготовке не нуждаются и дают вспышку значительной и
постоянной интенсивности при всех температурах выше некоторой
предельной (ниже ее перед вспышкой разлагается слишком боль-
шая доля вещества). Впрочем, зависимость интенсивности вспыш-
ки от температуры, полученная для тетразена, позволяет пола-
гать, что и в случае некоторых инициирующих ВВ подготовка ве-
щества в индукционном периоде может существенно влиять на
интенсивность самовоспламенения.
Автор глубоко благодарен профессору К. К- Андрееву за по-
мощь при Проведении работы и обсуждении ее результатов.
ЛИТЕРАТУРА
1. Андреев К. К-, Сборник статей по теории ВВ под ред. К. К. Андреева
и Ю. Б. Харитона, Оборонгиз, 1940, стр. 137.
2. Андреев К, К., Термическое разложение и горение ВВ, Госэнергоиздат,
1957. стр. 280 и 248.
3, Бел яев А, Ф., ЖПХ, 1950, 23, 432.
к. к.'ан-дреев, В. В. ГОРБУНОВ
36. О ТЕРМОСТАБИЛЬНОСТИ КРИСТАЛЛОВ
ВЗРЫВЧАТЫХ ВЕЩЕСТВ
В ряде исследований по горению и термическому разложению
монокристаллов взрывчатых веществ отмечается растрескивание
и разрушение их при этих процессах [3]. К- К- Андреевым было
высказано предположение, что одной из причин растрескивания
может быть тепловой удар, т. е. быстрое нагревание кристаллов
с поверхности [1]. С различиями в склонности к растрескиванию
он связывал различное поведение порошкообразных ВВ при под-
жигании их в замкнутом объеме. Предполагалось, что растрески-
вание кристаллов происходит при нагревании их горячими газо-
образными продуктами горения, проникающими в поры между
частицами, составляющими заряд порошкообразного В В. Увели-
чение поверхности при растрескивании кристаллов может привести
к возрастанию газообразования и переходу горения во взрыв.
528
В связи с этими соображениями мы поставили опыты по опре-
делению термостабильности кристаллов ВВ и ее роли в процессе
горения. Термостабильность или чувствительность кристаллов к
тепловому удару в предварительных экспериментах определялась
бросанием больших кристаллов в нагретую воду. Устанавливали
максимальный перепад температур (разность между начальной
температурой кристалла — обычно 18—20° — и температурой
воды), при котором в кристалле отсутствовали какие-либо види-
мые изменения (трещины,
раскалывание).
Оказалось, что кристал-
лы многих ВВ весьма чув-
ствительны к тепловому
удару. Наибольший перепад
температур выдерживают
кристаллы тротила и пикри-
новой кислоты (около 45°),
кристаллы тэна и гексогена
растрескивались уже при
перепаде 15—20°.
К недостаткам этой ме-
тодики следует отнести не-
определенность времени теп«
левого воздействия на кри-
сталлы и существенные раз-
личия в условиях опытов от
Фиг. I. Схема устанонки для определения
термостабильностн кристаллов ВВ.
/—кварцевая трубка с платиновой спиралью.
2—термопара. J—затаор. 4—телефонный элемент,
S—кристалл ВВ, fi—ЭНО-[, 7—ЭПВ-01.
тех, которые имеют место
при экспериментах по изучению перехода горения ВВ во взрыв.
Поэтому дальнейшие опыты мы проводили по другой методике,
которая позволила оценить термостабильность кристаллов ВВ по
характеру изменений, возникающих в них при свободном падении
через печь. Эта печь представляла собой кварцевую трубку (диа-
метр 25 мм, длина 300 ли*), обогреваемую платиновой спиралью.
Схема установки для определения термостабильностн кристаллов
ВВ дана на фиг. 1. Для измерения и регулирования температуры
печи применяли термопару 2 и электронный потенциометр
ЭПВ-01 7. Градиент температур по длине печи не превышал 1°/сл1
при температуре печи 500°. Кристалл 5 помещался на лепестки за-
твора 3. установленного на верхнем обрезе печи, и при открывании
затвора падал через печь. В качестве приемного устройства
применяли телефонный элемент 4, на мембрану которого падал
кристалл или его осколки. Время от момента открывания затвора
до падения кристалла или его осколков на мембрану регистриро-
вали осциллографом ЭНО-1 6. Для кристаллов весом от 2 до
200 мг это время, за вычетом времени пролета через холодные
части прибора, составляло 0,24—0,28 сек. По аналогии с предыду-
щими опытами вначале термостабильность оценивали по мини-
мальной температуре печи, при которой на кристалле появлялись
трещины. Для большинства изученных ВВ появление трещин
34 451
529
наблюдалось при температурах печи 200—300°. Однако, несмотря
на близкие значения температуры возникновения трещин, прн
дальнейшем повышении температуры печи для одних веществ на-
блюдалось интенсивное дробление кристаллов, для других (тет-
рил, дина, перхлорат аммония) только увеличение числа трещин
Поэтому в последующих опытах мерой чувствительности к дей-
ствию теплового удара служило число осколков, образовавшихся
при данной температуре печи2, и изменение этого числа при уси-
лении нагрева ее.
Фиг. 2. Зависимость интенсивности дробления крис-
таллов ВВ от температура печи.
/—тротил, 2—пнкрмнояая кислота. 5—стифнинояая кислота,
4—стифнат калии, о—тэн. 6—гексоген. 7— пикрат калия.
Зависимость интенсивности дробления кристаллов некоторых ВВ
от температуры печи представлена на фиг. 2. Каждая точка на
графике этой фигуры усредняет результаты не менее 5 опытов.
Видно, что для всех изученных ВВ количество осколков линей-
но растет при увеличении температуры печи. Наиболее склонны
к растрескиванию, особенно при высоких температурах, пикрат
калия и гексоген. Умеренное дробление наблюдалось для кристал-
лов стифниновой кислоты и стифната кадия. Прямые в координатах
число осколков — температура печи па фиг. 2 имеют различный
наклон, поэтому положение вещества в ряду термостабильностн
при изменении температуры может меняться. Так при температуре
1 В этих и последующих опытах применяли кристаллы весом от 20 до 60 лл
Изменение веса кристаллов в таких пределах не меняло качественной картины
растрескивавип-
® При падении через холодную печь никакого изменения в кристаллах не
наблюдал ос и.
530
печи 500° Кристаллы тэна образуют в 5 раз больше осколков, чем
стифниновая кислота; при температуре 700° число осколков
обоих В В почти одинаково. Возможно, что резкое увеличение
числа осколков кристаллов стифниновой кислоты при температу-
рах выше 500° связано с достижением условий, при которых
происходят качественные изменения в их структуре, в результате
чего чувствительность кристаллов этого ВВ к тепловому удару
резко увеличивается. Высокой термостабильностью отличаются
кристаллы тротила, пикриновой кислоты, тетрила, дины и перхло-
рата аммония. Даже при температурах печи 1000— П 00° кристал-
лы этих веществ дробились не более чем на 5—7 осколков. На
кристаллах дины появлялись трещины при 300—600°, при более
высоких температурах происходило расплавление кристаллов это-
го вещества.
Интересно было оценить термостабильность кристаллов ВВ
при высоких температурах, близких к тем, которые приводят
к воспламенению кристалла при падении его через печь. Вспышка
наблюдалась для гексогена, октогена, пикрата калия, стифниновой
кислоты и стифната свинца. Вследствие большого числа осколков
и капель расплава вблизи вспышки, оценка термостабильности по
числу осколков в этом случае менее надежна; однако, по крайней
мере, для кристаллов гексогена (фиг. 2) число осколков при повы-
шении температуры печи вплоть до вспышки (900°) продолжало
расти приблизительно так же, как и при более низких температу-
рах. Мелкие (4—5 мг) кристаллы азида свинца не растрескивались
при температуре печи 550° и взрывались при 600°. Вплоть до
вспышки при 700э не растрескивались также кристаллы стифната
свинца. Следует указать, что близкие к ним по весу кристаллы
гексогена в этих условиях дробились на 15—20 осколков.
В предыдущих опытах была изучена термостабильность боль-
ших кристаллов ВВ. В реальных условиях мы имеем дело с мелки-
ми кристалликами и их обломками. Интересно было изучить влия-
ние размеров кристаллов на интенсивность их дробления под дей-
ствием теплового удара. При температуре печи 500 и 700° была
изучена термостабильность кристаллов тэна и гексогена различ-
ных размеров. Согласно принятой методике мерой чувствитель-
ности к тепловому удару служило число осколков, образующих-
ся при дроблении кристалла. Вначале рассчитывали удельную по-
верхность кристалла (см$/г), предполагая, что он имеет форму ша-
ра и соответствующий вес. Так же определяли расчетом удельную
поверхность осколков после опыта. Зависимость интенсивности
дробления от размеров кристаллов (расчетного диаметра шара)
тэна и гексогена при температуре печи 500 и 700° изображена на
фиг. 3 (график о)- Здесь же показано увеличение удельной по-
верхности в результате дробления кристаллов при температуре
печи 500Р (график б). При всех изученных размерах кристаллов,
гексогец дал больше осколков, чем тэн. Однако это различие
уменьшается с увеличением размеров кристаллов. Так при дроб-
лении кристаллов, имеющих диаметр около I мм, гексоген даст
34* 5Д[
в 5,5 раза больше осколков, чем тэн; при увеличении диаметра до
4 мм это отношение уменьшается до 2,5 раз. Абсолютное увели-
чение поверхности для мелких кристаллов больше, чем для круп-
ных. В то же время относительное увеличение удельной поверхно-
сти возрастает с увеличением размеров исходных кристаллов.
Расчетный диаметр кристалла, в мм
40 20 10
Удельная поверхность кристалла, .
до опыта, $кр см z/z
Фиг. 3. Влияние размеров кристаллов ВВ на
интенсивность их дробления под действием
теплового удара.
Данные, полученные при изучении термостабильности кристал-
лов различных В В, показывают, что последние сильно различают-
ся в этом отношении.
Интересно сопоставить чувствительность вещества к теплово-
му удару с устойчивостью горения его под возрастающим давле-
нием. Для этого было изучено горение ВВ в стальном стакане,
закрытом свинцовым диском, срезающимся по достижении опре-
деленного давления [2]. Определяли критическую толщину диска,
532
прн которой происходило дробление трубки, близкое к взрывному.
Если толщина диска меньше критической, то наблюдается только
вырывание его, причем иногда горение оставшегося заряда прекра-
щается. В одном из опытов после вырывания диска большая
часть заряда (около 70%), состоявшего из кристаллов гексогена
размером 5—7 мм, осталась в трубке. Наибольшему дроблению
подверглись кристаллы верхних слоев заряда, непосредственно
соприкасавшиеся с продуктами горения воспламенителя \ Кри-
сталлы, лежащие в глубине заряда, остались без изменения. Ни-
же приведены результаты ситового анализа той части заряда гек-
согена, которая осталась в трубке после прекращения горения.
Размеры кристаллов в мм Вес фракции в г Содержание в % вес.
До опыта
от 5 до 7 ............................ 50
После опыта
от 5 до 7 ................ 15 7 45,2
от 3 до 5 , .......................... 9,0 25,9
от 2 до 3 .......................... 3,0 8,6
от 1 до 2 .......................... 1,2 3^5
от 0,5 до 1 .......................... 2,9 8.3
менее 0,5............................. 3,0 8,5
34,8 100,0
Горение тэна и гексогена (размеры кристаллов около 0,25 мм)
переходит во взрыв при толщине свинцового диска 1,1 мм. Можно
предполагать [1], что нанесение на поверхность кристалла пленки
инертного вещества уменьшит резкость нагрева, растрескивание
и, следовательно, склонность к переходу горения во взрыв. Дей-
ствительно, наши опыты показали, что горение тэна, содержащего
4,3% флегматизатора2, распределенного на поверхности кристал-
лов, переходит во взрыв лишь при толщине свинцового диска не
менее 5 мм, а гексоген с таким же количеством флегматизатора
не дает взрыва, даже если трубка закрыта стальным дискам тол-
щиной 6 мм. Склонность к переходу горения во взрыв сильно по-
нижается только в том случае, если флегматизатор в виде плен-
ки покрывает поверхность кристаллов. Простое добавление к гек-
согену инертного вещества, например, крахмала в количестве 9%
приводит к увеличению критической толщины диска всего лишь
до 1,5 мм (вместо 1,1 мм для гексогена без добавок). Все эти дан-
ные говорят за то, что увеличение поверхности кристаллов в ре-
зультате растрескивания играет существенную роль при переходе
горения порошкообразных ВВ под возрастающим давлением во
взрыв.
1 В данном случае дробление кристаллов происходило не под влиянием скач-
ка давления (около 10 ат), так как опыты показали, что в кристаллах гексогена
не наблюдается каких-либо изменений при всестороннем резком сжатии их до
давления 70 ат и последующем резком падении давления.
2 В качестве флегматизатора применяли смесь стеарина и церезина.
3 Смесь гексогена с 9% крахмала имеет такую же скорость и расчетную
температуру горения, как и гексоген с 4,3% флегматизатора.
533
выводы
' 1. Разработана методика оценки чувствительности кристаллов
ВВ к тепловому удару, осуществляемому нагретым газом.
2. Определена чувствительность к тепловому удару кристаллов
ряда ВВ при температурах печи 300—1100°. Наиболее склонны
к растрескиванию кристаллы гексогена и пикрата калия. Кристал-
лы тротила, пикриновой кислоты и тетрила мало чувствительны
к тепловому удару.
3. Изучено влияние размеров кристаллов тэна и гексогена на
чувствительность их к тепловому удару прн температуре печи 500
и 700°. С увеличением размеров исходных кристаллов чувствитель-
ность эта возрастает.
4. Полученные данные сопоставлены с некоторыми закономер-
ностями, наблюдающимися при переходе горения ВВ в замкнутом
объеме во взрыв.
ЛИТЕРАТУРА
I. Андреев К. К-, Научные доклады Вмешен школы, Химия и химическая
технология, № 2, 1959, стр, 244—247.
2. Андреев К- К., Термическое разложение и горение взрывчатых веществ,
А!.. Госэнергоиздат, 1957. стр. 259.
3. См, работы, цитируемые в книге: Боуден Ф. ф. н Иоффе А. Д., Быст-
рые реакции в твердых телах. ИЛ, 1962
К. К. АНДРЕЕВ
37. к ВОПРОСУ О ПЕРЕХОДЕ ГОРЕНИЯ ВЗРЫВЧАТЫХ
ВЕЩЕСТВ ВО ВЗРЫВ
Существенной особенностью ВВ является то, что при их Хими-
ческом превращении выделяется тепло и образуются газы. В то же
время это превращение может ускоряться теплом (если его выде-
ление приводит к повышению температуры) и газами (если их об-
разование приводит к повышению давления, а превращение идет
в форме горения). Однако наряду с этими факторами, ускоряющи-
ми процесс, имеются факторы, противодействующие ускорению. Это
в первом случае теплоотвод во вне, уменьшающий повышение тем-
пературы и во втором случае — газоотвод, приводящий к снижению
давления.
Если влияние тех и других факторов уравновешивается, то ско-
рость процесса возрастает лишь до некоторого равновесного зна-
чения. Если факторы ускорения преобладают, то скорость химиче-
ского превращения не останавливается на этом пределе, а продол-
жает расти, стремясь к своему максимальному значению.
При гомогенном протекании химического превращения во всем
объеме заряда ВВ это происходит, если скорость выделения тепла
становится больше и растет с температурой быстрее, чем скорость
534
его отдачи окружающей среде. Тогда температура ВВ растет, со-
ответственно увеличивается скорость превращения, и оно может
завершиться взрывом.
Представим себе горящий заряд порошкообразного ВВ с неко-
торой удельной внутренней поверхностью, причем горение происхо-
дит по всей Этой поверхности. Если скорость образования газов
больше, чем скорость их отвода за пределы заряда, то такое горе-
ние становится самоускоряющиМся, Давление в заряде растет и со-
ответственно увеличивается скорость горения.
При тепловом самоускорении одним из основных параметров
является размер заряда. Теплоприход пропорционален кубу разме-
ра, теплоотвод (при конвективном теплообмене) —его квадрату.
Поэтому, увеличивая размер заряда, мы можем всегда прийти
к критическому его значению, выше которого процесс станет само-
ускоряющимся. Точно так же и при горении в рассматриваемых
условиях, когда количество генерируемых газов пропорционально
объему заряда (при данной удельной внутренней поверхности ВВ),
а отводимое их количество — его поверхности, должен быть неко-
торый критический размер, при превышении которого скорость га-
зовыделения становится больше скорости газоотвода.
Другим основным параметром при гомогенном протекании хи-
мического превращения и тепловом его самоускорении является на-
чальная температура, которая определяет исходную скорость реак-
ции, а следовательно, и скорость выделения тепла. При тепловом
возникновении взрыва критические значения обоих указанных па-
раметров (размера заряда и начальной температуры), как извест-
но, связаны между собой.
Когда реакция идет в форме горения таким вторым параметром,
от которого также зависит и критический размер, является на-
чальное давление. Это давление определяет скорость горения,
а следовательно, и начальную скорость газообразования. Ту же
роль, что давление, играет удельная поверхность вещества, так как
скорость газообразования равна произведению скорости горения на
его фактическую поверхность.
ОСНОВНЫЕ УСЛОВИЯ. НЕОБХОДИМЫЕ ДЛЯ ВОЗНИКНОВЕНИЯ
САМОУСКОРЯЮЩЕГОСЯ ГОРЕНИЯ ПОРОШКООБРАЗНЫХ
ВЗРЫВЧАТЫХ ВЕЩЕСТВ
Есть однако, существенное различие между обоими механизмами
развития самоускоряющегося превращения в отношении их воз-
буждения.
Чтобы возникло тепловое самоускорение процесса, достаточно
заряд ВВ некоторого размера нагреть до некоторой температуры.
Чтобы взрыв возник путем горения, надо, чтобы достаточно
большой заряд, состоящий из множества частиц, загорелся по всей
своей внутренней поверхности. Это не так просто обеспечить. Если
удельная поверхность велика, т. е. размеры частиц и зазоры меж-
ду ними малы, задача усложняется еще больше. Когда мы зажига-
535
ем заряд извне, то горение, особенно при малой газопроницаемости,
либо ограничивается макроповерхностью заряда, либо проникает
в него лишь на небольшую глубину. При локальном поджигании
изнутри порошкообразного заряда большая часть ВВ, окружающего
очаг поджигания, разбрасывается незагоревшейся от повышения
давления, вызванного горением, начавшимся в очаге.
Горение может проникнуть в глубь порошкообразного заряда
в двух случаях.
1. Горение ВВ идет с большой скоростью, благодаря чему у по-
верхности заряда образуется значительное динамическое по-
вышение давления. Наряду с этим химические реакции протекают
быстро и уже при низких давлениях образуются продукты полного
превращения, имеющие высокую температуру, т. е. большую вос-
пламеняющую способность. Третьим условием, в известной мере
связанным с двумя первыми, является высокая воспламеняе-
мость В В.
Если эти условия выполняются в достаточной мере, то даже
при постоянных и низких давлениях горение проникает в глубь по-
ристого заряда и может переходить во взрыв. Именно так обстоит
дело в случае инициирующих взрывчатых веществ.
Вторичные взрывчатые вещества не удовлетворяют условиям,
обеспечивающим переход горения при низких постоянных давлениях
во взрыв. Во-первых, превращение этих ВВ при горении идет срав-
нительно медленно и при атмосферном давлении не завершается,
вследствие чего температура продуктов их горения гораздо ниже1,
чем при полном превращении, достигающемся при высоких давле-
ниях. Во-вторых, скорость горения вторичных взрывчатых веществ,
а следовательно, и динамическое повышение давления во фронте
горения значительно меньше, чем у инициирующих ВВ. В-третьих,
у них меньше и воспламеняемость.
При горении под повышенными постоянными давлениями, соз-
даваемыми впуском в бомбу инертного газа перед поджиганием ВВ,
реакции идут быстрее и полнее, скорость горения также больше,
но проникновению газов в глубь заряда и воспламенению ими
частиц взрывчатого вещества препятствует охлаждение инертным
газом, разбавляющим ВВ.
2. Наиболее реальный способ, обеспечивающий горение порош-
кообразного заряда по всей внутренней поверхности, состоит в под-
жигании ВВ в замкнутом сосуде, в котором горение идет под воз-
растающим давлением, заставляющим газообразные продукты про-
никать в глубь порошка.
Чем больше сопротивление стенок сосуда, определяющее макси-
мальное давление, которое в нем может быть достигнуто, тем толще
слой вещества, в который может проникнуть горение. Если этот
1 Температуры горения при атмосферном давлении по расчету, основанному на
конечном составе газообразных продуктов горения и для некоторых ВВ прибли-
женному (из-за незнания теплоты образования конденсированных продуктов),
составляют для тротила около 2000°, тетрила — 240(Р, гексогена — 3200°. гре-
мучей ртути — 3900^, диазодинитрофенола — 3300' и пикрата калия — 2800° К.
536
слой толще критического, то дальнейший рост давления в нем уже
не зависит от наличия оболочки (представим себе, что она
исчезла в тот момент, когда горение охватило поверхность заряда),
а идет имманентно, определяясь теми параметрами, которые были
упомянуты выше. Таким образом, роль сопротивления оболочки при
поджигании в замкнутом объеме состоит не столько в том, что
это сопротивление влияет (через давление) на полноту превраще-
ния, на расстояние от поверхности заряда, на котором эта полнота
достигается, и на скорость горения, сколько в том, что оно опреде-
ляет глубину проникновения газов, а вместе с ними и горения,
в толщу заряда.
Известно, что горение некоторых ВВ, например, тона и гексоге-
на, при поджигании их в замкнутом объеме переходит во взрыв го-
раздо легче при меньшей прочности оболочки, чем многих других
ВВ, например, тетрила или тротила. В то же время разница в ско-
ростях их горения (особенно при умеренно повышенных давлениях)
не так велика, чтобы объяснить это различие. Различие же в тем-
пературах горения этих веществ велико и, по-видимому, оно играет
решающую роль.
Добавление небольших количеств алюминия к аммонийным со-
лям кислородных кислот, способным к внутримолекулярному оки-
слению, очень резко увеличивает склонность горения к переходу все
взрыв *, несмотря на то, что скорость нормального горения этих со”
лей от добавки алюминия не повышается. Влияние алюминия
в данном случае заключается очевидно в повышении температуры
продуктов горения на начальном его этапе — более горячие газы,
проникая в порошок, легче воспламеняют его частицы.
Интересно, что наблюдается отчетливый параллелизм между
склонностью горения к переходу в детонацию и детонационной спо-
собностью порошкообразных В В. По-видимому, проникновение
газообразных продуктов горения в глубь заряда играет существен-
ную роль не только при возникновении детонации, но и при ста-
ционарном ее распространении.
ЗНАЧЕНИЕ РАЗМЕРОВ ЧАСТИЦ ПОРОШКООБРАЗНОГО ВВ
И НЕКОТОРЫХ ДРУГИХ ФАКТОРОВ ПРИ ПРОНИКНОВЕНИИ
г ГОРЕНИЯ В ГЛУБЬ ЗАРЯДА
Роль величины горящей поверхности как фактора, определяю-
щего скорость газообразования, не требует пояснений. Чтобы обес-
печить наибольшую скорость газообразования с единицы объема
заряда желательно, чтобы частицы ВВ были очень мелкими. Одна-
ко тогда проникновение газов в глубь заряда затруднено и, кроме
того, они охлаждаются сильнее, Все это мешает охвату горением
внутренней поверхности заряда особенно, если она велика. Наибо-
лее благоприятен для перехода горения во взрыв тот случай, когда
1 Повышается также детонационная способность — критический диаметр дето-
нации сильно уменьшается.
537
частицы ВВ не слишком мелкие, газы проникают глубоко и их
тепла хватает, чтобы поджечь всю первоначальную поверхность.
После того, как эта поверхность загорелась, частицы самопроиз-
вольно измельчаются путем растрескивания, например, в резуль-
тате теплового удара. Тогда оказываются обеспеченными оба усло-
вия, благоприятствующие ускорению: поджигание во всем объеме
и большая поверхность горения [.
Если превращение быстро доходит до конечных продуктов, то
поверхность твердого ВВ оказывается в контакте с газами чрез-
вычайно высокой температуры. Это создает особенно благоприят-
ные условия для увеличения поверхности горения в результате рас-
трескивания частиц. Возможно, что именно такие условия реализу-
ются в случае азида свинца и подобных ему ВВ,
Можно представить себе и отрицательное влияние растрескива-
ния на развитие процесса. Если оно происходит слишком легко
и быстро при проникновении в заряд первых порций слабо нагре-
тых газов, то уменьшаются размеры частиц и пор, что может за-
труднить дальнейшее проникновение газов. Наряду с этим растре-
скивание, ведущее к обнажению большой поверхности, может при-
вести к слишком сильному охлаждению газов с соответствующими
последствиями.
Следует отметить одно обстоятельство, осложняющее в случае
мелких частиц «внутреннее» поджигание. Газопроницаемость по-
рошка в этом случае мала, давление газов будет выравниваться
в толще вещества медленно, возникнет градиент давления, под
влиянием которого порошок может уплотняться. Уплотнение это
будет выражено тем сильнее, чем (при данной газопроницаемости)
меньше плотность вещества, чем легче оно уплотняется, чем боль-
ше и чем быстрее растет давление продуктов горения воспламе-
нителя.
Рассмотрим еще некоторые явления, возникающие при распро-
страненном способе поджигания порошкообразного заряда 1 2 в замк-
нутой или полузамкнутой емкости, значительно осложняющие
картину.
Горячие газы воспламенителя3 приходят в контакт с открытым
торцом заряда и нагревают ВВ с поверхности. На дальнейшее раз-
витие процесса существенное влияние оказывает относительная
плотность поджигаемого вещества. Если ВВ спрессовано до боль-
шой плотности, то действие газов ограничивается воспламенением
1 Увеличение поверхности н результате растрескивания наблюдалось в опытах
при низких и умеренно повышенных давлениях. Большие давления могут, сжи-
мая частииы, препятствовать расхождению образовавшихся при растрескивании
осколков и таким образом затруднить рост эффективной поверхности горения.
5 ВВ помещено в открытую с одного конца гильзу дз негорючего в данных
условиях материала, установленную в емкость.
3 Очевидно, что такую же роль могут играть продукты горения самого ВВ,
Если воспламенитель газов не образует (например, накалеииая проволока), то
с самого начала горения, а если воспламенитель газовый, то через некоторое вре-
мя, проникновение тепла в глубь заряда осуществляется фильтрацией газообраз-
ных продуктов горения самого ВВ.
-538
заряда с поверхности. Если относительная плотность его не так ве-
лика, т. е., если между частицами есть значительные зазоры, то
газы проникают в глубь заряда, разогревая при Этом частицы ВВ.
Сами газы по мере их углубления в заряд естественно охлаждаются.
Соответственно различной температуре газов по глубине проникно-
вения будет различаться и действие их на частицы ВВ. Схематиче-
ски можно выделить три зоны от внешней поверхности заряда
в глубину.
В первой из них, ближайшей к торцу заряда, газы достаточно
горячи, чтобы быстро воспламенить частццы с поверхности. Части-
цы загораются в этом случае после прогрева тонкого поверхностно-
го слоя вещества и горят с нормальной скоростью подобно тому,
как горят зерна пороха при выстреле или при обычном опыте в ма-
нометрической бомбе.
Во второй зоне газы холоднее и разогрев частиц ВВ происходит
медленнее. Воспламенение возникает с некоторой задержкой и
к моменту возникновения горения частицы успевают прогреться
полностью или по крайней мере на значительную глубину. В связи
с этим горение протекает быстрее, чем если бы горели непрогрев-
шиеся частицы, и скорость газообразования выше, чем при нор-
мальной для данной начальной температуры и давления скорости
горения по той же поверхности. Здесь возможен и случай, когда
частица к моменту воспламенения ее с поверхности нагрелась до
столь высокой температуры, что в ней идет, наряду с горением с по-
верхности быстрая объемная реакция. Общая скорость превраще-
ния и газообразования в этом случае будет наибольшей.
Наконец в третьей зоне газы охладились настолько, что они уже
не в состоянии поджечь частицы с поверхности. Однако эти газы
могут разогреть ВВ настолько, что при достаточной толщине про-
гретого слоя в нем с соответствующей задержкой возникнет тепло-
вой взрыв. Если ВВ плавящееся, то разогрев может привести
к плавлению и агломерации частиц, ведущим к уменьшению внут-
ренней поверхности. Эти явления особенно существенны в случае
ВВ с низкой температурой плавления. Если, однако, заряд расплав-
ляется полностью или вообще образуется достаточно толстый слой
жидкости, то переход от твердого к жидкому состоянию может сде-
лать возможными новые пути ускорения горения. Известно, что
для некоторых жидких ВВ (например, нитроглицерина, нитрогли-
коля) поджигание в замкнутом объеме легко приводит к взрыву.
ВЫВОДЫ
Рассмотрена принципиальная аналогия между возможностью
ускорения гомогенного экзотермического превращения в заряде ВВ
и горения этого заряда по внутренней поверхности. На этой основе
установлены важнейшие условия, определяющие возможность пе-
рехода во взрыв горения твердых взрывчатых веществ, в первую
очередь порошкообразных.
.539
IV. РАЗНЫЕ ИССЛЕДОВАНИЯ
А. А. ШИДЛОВСКИЙ
38. ВОДА КАК ОКИСЛИТЕЛЬ В РЕАКЦИЯХ
С НЕОРГАНИЧЕСКИМИ ВЕЩЕСТВАМИ
Общеизвестно, что вода представляет собой весьма прочное хи-
мическое соединение: теплота образования воды жидкой равна
68,32 ккал/г-моль, воды парообразной 57,80 ккал/г-моль. Извест-
но также, что экзотермические реакции воды с активными (щелоч-
ными или щелочно-земельными) металлами легко протекают уже
при комнатной температуре. Ранее нами было установлено [3], [4].
что в условиях высокой температуры и давления реакции взаимо-
действия воды с такими металлами, как магний или алюминий, мо-
гут протекать также в форме горения или взрыва.
2те
/iMe+mH2O=7We„Om-j-»iHt (1)
Вода при этом выполняет роль окислителя — приемщика электро-
нов. Но окислительные свойства воды могут проявляться и в дру-
гих реакциях: при взаимодействии ее с неметаллами, обладающими
заметно выраженными восстановительными свойствами, а также
с низшими окислами некоторых металлов.
Высоким экзотермическим эффектом сопровождаются реакции
взаимодействия воды с бором и кремнием [
ЗН20жидк- + 2В«р = ВгОзвр.'4- ЗН2—|—97 ккал (2)
2Н20жндк. —|— Sixp. = SiO2„p. 2Н2 4- 69 ккал (3)
Реакция (2) протекает лишь при температуре красного каления,
реакция (3) и при комнатной температуре, если в воде присутст-
вует некоторое количество щелочи [2].
Взаимодействие воды с фосфором, выражаемое уравнением
5Н2ОЖидк. + 2Рбел. = РгОзкр. + 5Н2 + 18 ккал, (4)
сопровождалось бы выделением сравнительно небольшого количе-
ства тепла (<7 = 118 ккал на кг смеси реагирующих веществ).
г Теплота реакций во всех случаях вычислялась нами на основании данных
справочника [6]. При протекании реакций (2) и (3) наряду с водородом
могут образоваться также гидриды бора илн кремния.
540
Известно, что при обычных условиях система (Н2О + Р) доста-
точно химически устойчива. Однако протекание вторичной экзо-
термической реакции
РгОйкр, + ЗН20жидк. = 2НзРО4нр. -р 47 ккал (5)
дает дополнительное тепло и в связи с этим реакционная способ-
ность системы (Н2О+Р) повышается.
Как известно, реакция
4Н2Ожидк. 4- Рбел. — HgPOinp. -р 2,5Н2'4- 33 ккал (6)
(<7 = 320 ккал/кг) нашла себе, правда, сравнительно недавно, про-
мышленное применение [1].
Взаимодействие воды с углеродом является процессом эндотер-
мическим
H2Onapi+ Сграфит ~ СО -j- Н2 — 31 ккал (7)
и потому практически реализуется только при высоких температу-
рах. Также эндотермическими и потому более трудно осуществи-
мыми явились бы процессы взаимодействия воды с неметаллами:
As, S, Se и Те, например
ЗН20цар—|—2Азкр,=Аз20зкр, -f- ЗН2— 16 ккал (8)
Наши расчеты показывают, что реакции окисления водой
(жидкой) низших окислов некоторых металлов могут протекать
с выделением тепла, например
ЗпОкр.+^гОжидн. — ЗпОгкр,4- Н24*2 ккал (9)
Nb2O4Kp. + H2OffiHAH. = Nb2O5Kp1 + H2-p7 ккал (10)
Возможно, что экзотермическим будет также и процесс окисле-
ния водой окиси германия (GeO) в двуокись; расчет здесь не мо-
жет быть произведен, так как в термохимических справочниках от-
сутствует значение ДН для GeOKp.
Переходя к рассмотрению взаимодействия воды с низшими оки-
сла ми неметаллов, укажем прежде всего на общеизвестную
реакцию
СОгаэ 4* Н2ОПар= СО2газ*рН2 4* 9 ккал (11)
Особо должен быть поставлен вопрос об окислении сернистого газа.
Реакция
Н20жиди.'4* SO2raa = ^Оз^р, 4* Н2 — 29 ккал (12)
является эндотермической, так же как и реакция
Н2Опар 4- SO*™ ~ SO3Kp. -|- Н2— 18 ккал (13)
Однако вторичная экзотермическая реакция
SOaKp.+H2Onap= р26 ккал (14)
541
приводит к тому, что взаимодействие SO2 с избытком водяного пара
является уже экзотермическим и потому более легко осуществи-
мым процессом
2Н2ОПар 4- SO2ra3 = 4* 8 ккал (15)
Тепло, выделяющееся при гидратации серной кислоты избытком во-
дяных ларов, должно еще повысить экзотермичность этой реакции.
Вместе с тем едва ли можно надеяться получить водород в ре-
зультате взаимодействия сернистого газа с водяным паром, так как
Н2 будет восстанавливать избыток SO2, превращая его в серу
2Н2 -|- SO2 = 2Н2Опар Sup. 45 ккал (16)
и, следовательно, суммарно реакция между водяным паром и серни-
стым газом выразится уравнением
2Н2Опар-|-ЗЗО2гаэ—2Н25О4ЖПд1( |-5)ф.--}-59 ккал (17)
По указанию Барнета и Вильсона [5], эта, по их выражению,
сложная и мало понятная реакция диспропорционирования проте-
кает при 150°. Нам же представляется, что механизм этой реакции
может быть объяснен последовательным протеканием реакций (15)
и (16).
Следует заметить также, что водяной пар может играть роль оки-
слителя и по отношению к другим классам химических соединений:
например гидридов многих металлов и некоторых неметаллов
(SiH4; РН3). боридов и силицидов многих металлов и др.
Из изложенного явствует, что вода может, в особенности при по-
вышенной температуре, принимать весьма активное участие в са-
мых разнообразных химических реакциях между неорганическими
веществами. Возможно, что наиболее экзотермические из вышеопи-
санных реакций, например реакции (2) и (3) при подборе соответ-
ствующих условий температуры и давления удастся осуществить
в форме процессов горения или взрыва.
ЛИТЕРАТУРА
1, Вольфкович С. И., Роговин 3. А., Руденко Ю. П„ Шмаиен-
к о в И. В., Общая химическая технология, Госхимнздат, 1960 г., т. 11, стр. 55.
2, Некрасов Б. В., Курс обшей химии, Госхимиздат, 1952, стр. 539 н 490.
3. ШидловскнйА. А,, ДАН СССР, 1946, 51, вып. 2, 127.
4. Ш и д л о в с к и й А, А., ЖПХ, 1946, 19, 371.
5. В а г и е 1 t Е., W i ] s о п С. L., Inorganic Chemislry, London, 1959, р. 487.
6. Rossini F., Wagman D., Evans W., Levine S., Jaffe I., Selected
Values of chemical thermodynamic Properties, Washington, 1952,
542
А. А. ШИДЛОВСКИЯ
39. РАСШИРЕНИЕ ОБЛАСТИ ПРИМЕНЕНИЯ
ТЕРМОХИМИЧЕСКОГО ЗАКОНА ПОСТОЯННЫХ РАЗНОСТЕЙ.
ПРЕДВЫЧИСЛЕИИЕ НЕИЗВЕСТНЫХ ТЕПЛОТ
ОБРАЗОВАНИЯ СОЛЕЙ АММОНИЯ
В работе А, Ф. Капустинского и нашей [4] показана возможность
применения термохимического закона постоянных разностей ддя
приближенного вычисления теплот образования твердых неоргани-
ческих соединений,
Продолжив эту работу, мы сопоставили между собой попарно
теплоты образования 27 пар кристаллических солей калия и аммо-
ния. Соответствующие данные этого сопоставления приведены
в табл. 1. Теплоты образования большинства солей взяты нами из
справочника [61; для солей с анионами S2O^“,SO^~* MgSO4 • 6Н2О
и [Fe(CNsP“• ЗНаО — из справочника [1]; для иодата аммония зна-
чение теплоты образования найдено экспериментально [4], Данные
по теплотам образования водных растворов взяты для разведе-
ния со, или если таких данных нет, то для максимального разведе-
ния. Как видно из табл, 1, среднее отклонение от разности 28,4 ккал
составляет для кристаллических солей калия и аммония
+2 ккал[г-экв. Эти отклонения по своей величине распределяются
следующим образом.
Отклонение в ккал Чиело сопоставлений
от 0 до 1 11)
от 1 до 2 4
от 2 до 3 8
от 3 до 4 3
от 4 до 6 2
Наиболее значительные отклонения дают анионы;
F“ — 5,5 ккал; С10“ -j- 5,8 ккал.
Так как для водных растворов солей калия и аммония средняя
разность в теплотах образования составляет 28,4+0,3 ккал (см.
табл, 1) и наибольшие отклонения не превосходят + 0,9 ккал, то,
очевидно, что значительные отклонения для понов F~ и СЮ^- свя-
заны с различием в теплотах растворения солей калия и аммония.
Действительно, как видно из приводимых ниже данных, теплоты
растворения этих солей не равны (—АН растворения ккал/а-экк).
К'? NHi' Разность (А)
F~ —4,2 —1,2 -[-5,4
С10Г -12,2 -6,3 -5,9.
Отклонения от разности (28,4) и разности в теплотах растворе-
ния (А) полностью уравновешивают друг друга.
543
Таблица 1
разность в теплотах образования (—ДН2де) кристаллических солей калия
н аммония и их водных растворов
Кристаллические соли Водные растворы солей
м по пор. Анионы 4* + т X Z +•» § X ? z « ! х+ § < и: к Отклонение от разности! 28,4 зГ Z +„ § Г ? 1 + Г + § < 'х а Отклонение от разности 28,4
1 Р- 134,5 [[[,6 22,9 —5,5 138,7 110,4 28,3 —0,1
2 ci- [04,2 75,4 28,8 +0,4 101,1 71,8 29,3 +0,9
3 Вт- 93,7 64,6 29,1 +0,7 88,9 60,7 28,2 —0,2
4 J- 78,3 48,3 30,0 + 1.6 73,4 45, [ 28,3 —0,1
5 сю; 103,6 69,4 34,2 4-5,8 9[ ,4 63,1 28,3 —0,1
6 JOf 121,5 94,0 27,5 -0,9 115,0 86,4 28,6 +0,2
7 NOf 88,5 63,1 25,4 —3,0 85,2 57, [ 28,[ —0,3
8 NO3- 117,8 87,3 30,5 +2,1 109,4 81,1 28,3 —0,1
9 CN- 26,9 0,0 26,9 — 1,5 24, [ —4.6 28,7 +0,3
10 CNS- 48,6 20 28,6 -Ь0.2 42,8 14,5 28,3 —о,1
11 HSO^ 276,8 244,8 32,0 +3,6 274.3 245,9 28,4 0,0
12 HS- 63,2 38,1 25,1 —3,3 64, [ 35,1 29,0 4-0,6
13 h2po7 374,9 346,7 28,2 —0,2 370,4 342,9 27,5 —0,9
14 HCO^ 229,3 203,7 25,6 —2,8 224,5 196,9 27,6 —0,8
15 нсог 158 132,8 25,2 —3,2 157,7 129,8 27,9 —0,5
[6 CH3CO2 [73,2 [47,8 25,4 -3,0 176,9 148,1 28,8 +0,4
[7 SO^- 342,7 281,9 30,4 +2,0 337,0 289,4 28,3 —0,1
18 sop 266,9 212,0 27,4 —1,0 269,1 211,3 28,9 +0,5
[9 SjOl 361,1 299,6 30,7 +2,3 350, [ 293,3 28,4 —0,0
20 S20g 458,3 396,4 3[ ,0 +2,6 445.3 387,8 28,7 +0,3
21 CrO*~ 330,5 275,2 27,6 —0,8 326,0 269,4 28,3 -0,1
22 Cr2O|“ 485,9 425,5 30,2 +48 468,7 412,6 28,0 —0,4
23 P1C1J- 254,2 195,5 29,4 +1,0 242,8 186,9 27,9 —0,5
24 c20a4- 320,8 268,7 26,0 —2,4 317,1 260,6 28,2 —0,2
25 SO2“ZnSO4-6H2O [0[ 1,8 955,6 28,1 —0,3 — — — —
26 SO4-MgSO4‘6H2O [08[,8 1024,4 28,7 +0,3 — — — —
27 [Fe(CN)J4-.3H2O 340,3 2 [6, 31,1 +2,7 [[9,5 4,2 28,8 +0,4
Средняя разность 28,4 ккал Средняя разность 28,4 / СКОЛ
Среднее отклонение £2 ккал Среднее отклонение £0,3 ккал
544
Выяснение причин различия н теплотах растворения солей калия
и аммония требует дальнейшей теоретической и экспериментальной
работы.
Используя разность K+—NH + —28,4 ххал/г-эхо, мы прибли-
женно вычислили неизвестные до сего времени теплоты образования
для некоторых хорошо изученных солей аммония, свойства и спосо-
бы получения которых описаны в литературе [5J.
Результаты этих вычислений приведены в табл. 2.
Таблица 2
Теплоты образования кристаллических солей аммония (в ккал/г-моль)
А. Постоянная разность К+—NH^=28,4 ккал/г-эк в
Аннон —AHjjj солей калия ккал/г-моль [6] Вычисленная —ДН!Эй солей аммония, в ккал/г-моль
ReO; 264,0 235,6 ±2
SeO*~ 266,9[2] 210,1±4
SnClg- 362,9 306,1 ±4
PdCl4~ 26[,6 2С4.8±4
PiClt 30[,0 244,2±4
AHI" 777,6 692,4± 6
Б. Постоянная разность Na+—NH*=22,6 ккал/г-экв
Анион —AHjga солей натрия ккал/г-моль {6 J Вычисленная —AHj^g солей аммония в ккал/г-моль
SeO*- 258 212,8±4
Aiii- 758.5 691,7±6
Указанные в последней графе справа табл. 2 пределы отклоне-
ния не превышают 1,5—2,0% от величины теплоты образования
рассматриваемых солей.
Используя постоянную разность Na+ —NHi, равную
‘22,6 ккал 1г-экв [4], мы получили данные, помещенные в табл. 2Б.
Предвычисления для солей аммония, сделанные по разностям
К+ — NHt" «Na+ — NH4, близки друг к другу.
В заключение укажем на имеющееся в справочнике 161 явно оши-
бочное значение ДН = —598 для КаСгзО?. СгОз (КаСг^Ою).
Для трихромата аммония (МЩгСгзОю нами эксперименталь-
ным путем получено значение — ДН298 = 580±6 ккал/г-моль [3].
35 451
546
Используя "разность К+—NH + — 28,4 ккал, мы получаем для
КаСг3О]0 значение:
ДН298 = — (580±6) —28,4 -2— —637±6 ккал/г-моль.
выводы
1. На примерах солей калия и аммония с одинаковыми аниона-
ми показано, что термохимический закон постоянных разностей
может быть с успехом использован для приближенного вычисления
теплот образования кристаллических солей, которые имеют общий
катион или анион, равные или близкие значения теплот растворе-
ния а также являются сильными электролитами
2. С точностью ±2% (±2 ккал/г-экв) вычислены'неизвестные
теплоты образования для перрениата, селеиата, гексахлоростанна-
та, тетрахлорпалладиата, гексахлороплатината и гексафторалю-
мината аммония.
ЛИТЕРАТУРА
1. Брнцке Э. В., Капустииекий А. Ф., Веселовский Б. К., Ша-
мовскяй Л. М., Ченцова Л. Г., Анваер Б. И., Термические константы
неорганических веществ, АН СССР. 1949.
2. Зубова Г. А., Термодинамические свойства труднорастворимых селена-
тов. Кандидатская диссертация, МХТИ им. Д. И. Менделеева, 1958.
3, Капустииекий А.Ф., Ш ид л о век и й А. А., Изв. АН СССР (сектор
платины), 1955, вып, 30, стр. 31.
4, К а п у с т и н с к и й А. Ф„ Ш и д л о в с к и й А. А., Ш и д л о в с к а я Ю. С.,
Изв. АН СССР, ОХН, 1958, № 4, стр. 385. .
5. Руководство по препаративной неорганической химии, под ред. Г. Брауэра,
ИЛ, 1956 (перевод с немецкого).
6, Rossini F_. W a g m а п D„ Evans W.. L e v [ n e $., J a f f e 1., Selected
Values of chemical thermodynamic Properties, Washington, 1952.
Ю. Д. МАКСИМОВ
40. УПРУГОСТЬ ПАРОВ НИТРОПРОИЗВОДНЫХ БЕНЗОЛА
Имеющиеся в литературе данные об упругости паров моно- и
динитробензола вплоть до температуры кипения [2], [3] не вызывают
сомнений’, так как соответствующие измерения производились в об-
ласти температур, при которых стойкость этих соединений доста-
точно высока. Иначе обстоит дело с измерением упругости паров
тринитробензола, который, в отличие от первых двух веществ, раз-
лагается с заметной скоростью уже при температуре ниже темпе-
ратуры кипения. Это обстоятельство затрудняет непосредственное
определение температуры его кипения. А. Ф. Беляев [1], определяв-
ший ее по улетучиванию навески ВВ в 2 мг с поверхности нагрето-
го металлического блока, дает приближенное значение 315°. Вели-
чина скрытой теплоты испарения, найденная им по времени уле-
тучивания навески этого ВВ при различных температурах, состав-
ляет около 18,5 ккал/моль. Однако экспериментальная' проверка
546
Фиг. I. Температурная зависимость
давления насыщенного пара для ве-
ществ ряда бензол—тринитробензол.
I—бензол, 5—мо но нитробензол, .?—диннтро-
бензол, 4—три нитробензол (по данным
А. Ф, Беляева! я 5—трннитробензол (по
нашнм данным).
значений давления паров, вычисленных по температуре кипения н
теплоте испарения, найденным А- Ф. Беляевым, показала расхож-
дение между результатами опытов и его данными. Это послужило
поводом для более детального изучения упругости паров тринитро-
бепзола.
Для определения давления паров этого вещества при различ-
ной температуре навеска его в 0,05 г нагревалась в тщательно эва-
куированном (до 10~3 —10 4 мм рт. ст.) и запаянном стеклянном со-
суде, снабженном манометром
Тина Бурдона. Температура бани
со сплавом олова и евицца, в ко-
торой находился сосуд с испы-
туемым ВВ, поддерживалась по-
стоянной с. точностью ±0,5 \
Были измерены также упруго-
сти паров для моно- п дицитро-
производных бензола, Измерения
проводились с одним образчиком
вещества При последовательном
повышении температуры. Спе-
циально поставленный опыт с ди-
нитробензолом показал, что для
прогрева сосуда с веществом от
комнатной температуры до 250°
требуется 2—3 мин. Перед изме-
рением при каждой температуре
производилась выдержка в тече-
ние 5—10 мин. Результаты опы-
тов приведены в табл. 1 и на фиг. 1. Данные, полученные по при-
мененной методике, хорошо согласуются с имеющимися в литера-
туре.
Измерение упругости паров тринитробензола при температуре
200—25(У5 проводилось с одним образчиком, при этом степень за-
полнения сосуда веществом была 10-3 г]смя Надежность сделан-
ных в этом опыте измерений подтверждается тем, что давление
в сосуде, измеренное при комнатной температуре после опыта, было
менее 1 мм рт. ст. При повышении температуры до 270 и более
градусов скорость разложения тринитробензола увеличивается на-
столько, что за время, необходимое для одного измерения, образует-
ся заметное количество газов, и поэтому определение упругости па-
ров ВВ становится малонадежным. С целью исключить влияние
разложения для каждого определения бралась новая порция веще-
ства, и упругость паров определялась путем экстраполяции давле-
ния, растущего в сосуде при разложении, на время, равное времени
' При изучении термического распада трннитробепзола было установлено, что
скорость разложения его в жидком состоянии больше, чем в парах. Поэтому жела-
тельно было брать малую степень заполнения сосуда с тем, чтобы количество
вещества в жидкой фазе было минимальным.
36 451
547
прогрева. Из фиг. 2, на которой изображены кривые роста давления
со временем в случае нагревания тринитробензола при 312°, можно
видеть, что воспроизводимость данных при определении упругости
паров в этих условиях является вполне удовлетворительной.
Полученные результаты измерения давления насыщенного пара
тринитробензола приведены в табл. 1, а соответствующие точки
нанесены на график Igp—1/Т (фиг. 1). Для сравнения на этом гра-
фике изображена линия 4, построенная по данным А. Ф, Беляева.
Фиг. 2. Рост даилепия во времени в слу-
чае разложения тринитробе||.чола при 312“
и различных степенях заполнения реак-
ционного сосуда (в г/см3 104):
/—18.[. 2—37.[, 3—81,8.
Из фиг. 1 видно, что во всем изученном интервале температур зна-
чения упругости паров тринитробензола, полученные нами, лежат
ниже вычисленных по Беляеву. Температура кипения, определенная
экстраполяцией полученных данных на давление 760 мл рт. ст., со-
ставляет 350°, что на 35° выше значения, найденного А. Ф. Беляе-
вым. Это различие обусловлено, вероятно, тем, что в наших опытах
при измерении давления паров роль фактора разложения была све-
дена до минимума, в то время как в работе А. Ф, Беляева учет это-
го фактора был весьма затруднителен.
Совокупное рассмотрение зависимости p=f (т) для веществ
ряда бензол-тринитробензол (фиг. 1) также говорит в пользу на-
дежности полученных результатов.
Температурная зависимость упругости паров рассматриваемых
нитросоединений вблизи температуры кипения может быть выра-
жена уравнением
_________у
р =А-е .
Коэффициенты уравнения, температура кипения и отношение
Трутона для интересующих нас четырех веществ сведены в табл. 2.
Из данных этой таблицы видно, что величина отношения Трутона.
равная для бензола 20,8, при последовательном введении в его
548
молекулу групп NO2 закономерно возрастает, достигая для три-
питробензола значения 28—30, которое в полтора раза превышает
нормальное значение этого отношения (21,0). Подобная закономер-
ность впервые наблюдалась А. Ф. Беляевым [1] на примере азотно-
кислых эфиров алифатических спиртов и имеет, вероятно, общин
характер для органических соединений, содержащих в молекуле
Таблица 1
Упругость паров нитропроизводных бензола при различной температуре
Вещество Темпера- тура °C Давление насы- щенного пара ММ рт, СТь 1 -103 7° К lg Р
239 1430 1,95 3,156
250 1775 1,91 3,250
Нитробензол 259 2167 1,88 3,360
269 2512 1,84 ‘3,400
280 3022 1.81 3,481
291 3582 1,78 3,551 :
252 220 1,91 2,343
1. З-динитробепзол 253 230 1,90 2,362
290 687 1,78 2.837
291.5 716 1,77 2.8Г6
202,5 11 2,10 1,012
210,5 14 2,97 1,146
223,5 22 2,01 1,342
234,5 31 1,98 1,492
245,5 46 1,91 1,663
250 56 1.91 1,748
250 52 1,91 1,716
256 64,5 1,89 1,810
1, 3, 5*трщ|итробензол 267 89 1,8,5 1,949
271 100 1,84 2,000
271 101 1,84 2,004
271 102 1,84 2,069-
288 165 1,79 2,218 '
288 166 1,79 2,220
292 184 1,77 2,26,5
312 287 1,71 2,458
312 28л 1,71 2,455
. 312 293 1,71 ' 2,467
36*
549
Таблица 2
Величины, характеризующие кипение нитропроизводных бензола
Вещее тво 1 1g А Скрытая тепло- та исп: pci пи» нргг ? k к ал/Af оль Температура ьипепия >гри 760 мм рт. ст. 5 С Отношение Трутона к т J 1ГИ11
—
Бензол 7,44 7 З-Г’О 80,1 20,8
Моиопитробеяэол , 8,03 11 600 210,5 23,8
1, З-динитробсггзол ; 8,55 14 500 300 25,3
1. 3, 5-тринцтробщшол по нашим данным ; 9, Об 17 504 350 28,1
по дяштым Беляева 11 р ri м с ч а и и с. Значении I8 5CU j 315 IgA даны Д-'|Я р н мм. рт. ст. 31,5
NOa-группы. В связи с этим следует сказать, что распространенное
ранее среди некоторых исследователей мнение о неизменности нор-
мального значения отношения Трутона для соединений этого типа,
по-видимому, является ошибочным.
ЛИТЕРАТУРА
I. БеляевА, Ф., ЖФХ, 1948, 22, 91.
2, Ihrjg, USP, [838311.
3. W у [ е г R,, Helv. chim. acta, 1932, 15, 23.
| И. Ф. БЛИНОВ Л. М. СВЕТЛОВА
41. ВЛИЯНИЕ ОБОЛОЧКИ НА ДЕТОНАЦИОННУЮ
СПОСОБНОСТЬ НЕКОТОРЫХ ДИНИТРОСОЕДИНЕНИИ
БЕНЗОЛЬНОГО РЯДА1
На детонационную способность взрывчатых веществ большое
влияние оказывает наличие оболочки. Она замедляет радиальный
разброс взрываемого вещества, а следовательно, согласно прин-
ципу Ю. Б, Харитона [4], уменьшает критический диаметр заряда.
Именно поэтому вещество, не способное устойчиво детонировать
при данном диаметре заряда в отсутствии оболочки, становится
детонаццонноспособцым при том же диаметре заряда с оболочкой.
г Настоящая работа была выполнена под руководством И, Ф. Блинова, ко-
торому преждевременная смерть помещала завершить ее.
Автор статьи выражает искреннюю благодарность А. И. Годьбиндеру за по-
мощь в подготовке рукописи п ценные советы ври обсуждении результатов.
550
Особенно сильно снижает критический диаметр заряда прочная, не-
разрывающаяся при взрыве оболочка.
Исследуя детонацию аммиачно-селитренных смесей, А. Ф. Беля-
ев пришел к выводу [1], что вследствие очень малого времени про-
текания взрывных процессов, действие оболочки определяется и ос-
новном ее инерционным сопротивлением (массой). Однако, оболоч-
ки из сыпучих материалов оказывают меньшее влияние на устой-
чивость детонации, чем оболочки равной массы из прочных мате-
риалов, создающих дополнительное сопротивление разлету цепро-
реагировавшего взрывчатого вещества.
А. Ф. Беляев указывает также, что увеличение диаметрального
размера заряда эквивалентно в общем окружению заряда мень-
шего диаметра сыпучей инертной оболочкой равной массы.
Влияние свойств оболочки на возможность и скорость распро-
странения детонации проявляется в том случае, когда заряд ВВ
имеет диаметр, далекий от предельного, соответствующего наиболь-
шей скорости детонации. Изучение влияния оболочки иа детона-
ционную способность имеет значение поэтому для тех взрывчатых
веществ, у которых достаточно велик критический диаметр заряда
и наблюдается значительная разница между критическим и пре-
дельным диаметрами, К числу подобных систем относятся, наряду
со смесями, которые главным образом и изучались с этой точки
зрения, также вещества, обладающие малой взрывчатостью, в ча-
стности ароматические динитросоединения. Можно было ожидать,
что исследование таких соединений даст наиболее полное и нагляд-
ное представление о влиянии различных характеристик оболочки
(плотность, прочность, сжимаемость) па детонационную способ-
ность зарядов. Вместе е тем, изучение влияния оболочки иа детона-
ционную способность таких соединений представляет значительный
практический интерес и для оценки реальной опасности взрыва в
различных условиях их производства, хранения и применения.
В связи с этим, в числе других работ по изучению взрывчатых
свойств динитросоедннений [2], [3], было проведено исследование по
выяснению влияния оболочки на детонационную способность заря-
дов из этих веществ. Результаты этого исследования изложены
ниже.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Опыты проводили с техническими продуктами заводского изго-
товления. В качестве объектов исследования применяли:
динитрофепол с температурой плавления . . . . . , 114,5е
динитротолуол . , ... . . . 63,5°
дигштрох лор бензол . . ... . . . 50°
диннтроаннлин . . . 178'
динитропафталнн . • „ . . . . . 155s
Ранее было установлено [2], что критический диаметр детона-
ции зарядов из подобных ВВ увеличивается с ростом их плотно-
551
сти. В связи с этим, в настоящей работе применяли порошкообраз-
ные заряды с плотностью, близкой к насыпной плотности соответ-
ствующих веществ. Все опыты проводили с кристаллическими про-
дуктами, на 100% проходившими через сито с И отверстиями на см.
Были испытаны оболочки различной прочности, плотности п
сжимаемости: стальные трубы, трубки, свернутые из листового
свинца и железа, стеклянные и керамиковые трубки, оболочки, со-
ставленные из Отдельных стальных полос, оболочки из жидкостей,
сыпучих (песок) и пластичных материалов.
В опытах с оболочками из жидкости, глины или песка заряд ис-
пытуемого вещества, находящийся в гильзе из парафинированной
бумаги, помещали в гильзу большего диаметра; свободное прост-
ранство между стенками заполняли жидкостью, пластичной глиной
пли сухим песком.
В период выполнения данной работы мы пе имели возможно-
сти измерять скорость детонации. Результат опыта оценивали по
деформации свинцового цилиндра, на котором взрывали исследуе-
мый заряд в соответствующей оболочке. Опыт проводили анало-
гично стандартному испытанию на бризантность.
Для более полного суждения об устойчивости взрыва применяли
удлиненные заряды; в большей части опытов длина заряда была
100 мм (2,5—3,5 диаметра), в отдельных опытах применяли заряды
длиной 200 мм. Детонацию исследуемого заряда возбуждали кап-
сюлем-детонатором № 8. Исключение составили опыты е динитро-
нафтадином, где применяли промежуточные детонаторы из прес-
сованного тетрила весом 5 и 10 г.
При оценке результатов опытов следует учитывать, что нали-
чие оболочки влияет на них двояким образом. С одной стороны,
затрудняя разлет продуктов детонации, оболочка уменьшает «хи-
мические» потери, в связи с чем возрастает полнота превращения
исходного ВВ в продукты взрыва и скорость детонации увеличи-
вается. С другой стороны, замедляя радиальный разлет, оболочка
усиливает механическое действие продуктов взрыва на свинцовый
цилиндр. Это последнее влияние оболочки будет сказываться даже
в том случае, если испытывать заряды такого ВВ, для которого
применяемый диаметр будет больше предельного, ц полнота пре-
вращения, а следовательно и скорость детонации, не будут изме-
няться из-за наличия оболочки.
Принятая нами методика выполнения опыта не позволяет раз-
делить влияние указанных двух факторов. Кроме того, мы испы-
тывали заряды различных диаметров. Изменение диаметра заряда,
так же как и наличие и качество оболочки, могут влиять на обжа-
тие свинцового цилиндра обоюдно. В связи с этим получаемые
результаты позволяют надежно различать только случаи наличия
или отсутствия взрыва исследуемого заряда, но не дают надеж-
ных данных для суждения о полноте превращения и скорости де-
тонации в каждом случае.
В табл. 1 приведены результаты опытов с динитрофенолом, при
плотности зарядов 0,62—0,65 г[см\ Заряды такой плотности в тон-
552
кой бумажной гильзе детонируют при диаметре 40—45 мм; в от-
дельных опытах при диаметре заряда 40 мм наблюдается затуха-
ние детонации
Таблица 3
Влияние оболочки на детонационную способность дннитрофенола
Md терна л сболочкн Диаметр обо- лочки впутрен- ницбгаруяшый ям Вес оболочки па I см дайры заряда г/см Обжатие свин- цового цилинд- ра мм
•Стальная цельнотянутая трубка 29/39,4 45 20,8
Свинцовая трубкт 29/35 47 15,5
Вода 29/77 40 9,5
29/101 63 7,5
Парафин 29/73 40 9,3
Песок 29/63 40 1,2
29/59 41 1,3
Жестяная трубк, 30/31 5,2 7,8
30/32 8,4 10,4
Стальные полосы ннфимой 4— 35/43 38 20,3
5 мм 20,0
Свинцовая труби* 35/41 47 18,1
Керамиковая тр;бка 35/47 14 16,3
17,5
Вода 35/76 10 14,7
Песок I 35/56 40 15,5
14,2
Жестяная трубк» 35/36 3 12,6
То же 35/37 6 12,6
В табл. 2 [риведены результаты опытов с динитротолуолом, при
плотности зарядов 0,6 и 0,8 г/см3. Дипитротолуол при плотности
0,6 а/сл3 испытывался в бумажной или картонной гильзе. Крити-
ческий дна метр в этих условиях около 45 мм.
В табл. 3 1риведены результаты опытов с динитрохлорбензолом
при плотностг 0-8—0,85 г/см2.
В табл. 4 приведены результаты опытов с динитроанилицом при
плотности за)ядов 0,7—0,8 г/см3. Без оболочки динитроанилин де-
тонирует в заряде диаметром 60 мм.
Динитроюфталин обладает очень низкой детонационной спо-
собностью. Заряд динитронафталина диаметром 200 мм (высота
200 мм) в бу»ажной оболочке с промежуточным детонатором весом
100 г из прессованного тетрила, полностью не детонирует.
553
Таблица 2
Влияние оболочки на детонационную способность динитротолуола
Материал оболочки Диаметр обо- лочки BliyipOH- иий/на р у жь । а й мм Вес оболочки на 1 cif ДЛН[1Г1| заряда г /с.и Обжатие свин- цового цилинд- ра мм
Плотность зарнлов 0,6 a/c_4f:3
Ста.тьияя цельнотянутая трубка 40/50 57 26,0
Составная оболочка из сталь- 40/48 42 . 20,0
ных полос шириной 4--5 мм
Стальная трубка со нгвом 40/48 88 16,1
Стальная трубка со швом, раз- 40/48 38 12.8
резанная вдоль
Свинцовая трубка 40/44 30 21,3
Вола 40/80 40 15,0; 13,7
Глина 40/(U 40 17,0; 14,6
Песок 40/70 40 2,4; 2,6
Жестяная трубка 40/41,2 3 15,2; 15,6
Стеклянная трубка 40/42,5 3,5 0,8
Плотность зарядов 0,8 г! см3
Стальная трубка со швом 40/48 40 26,4
Стальная трубка со шиом. раз- 40/48 40 25,0
резанная вдоль
Свинцовая трубка 40/46 40 26,0
Вода 40/80 40 1,2; 1,2
Глина 4C/G4 40 1,5
Жестяная трубкл 40/41,2 7 1,0; 0,8
Таблица 3
Влияние оболочки на детонационную способность динитрохлорбензола
Материал оболочки Диаметр обо- лочки вцутрсп- ний/каружный мм Вес оболочки из 1 см длины парада г/сж Обжатие свин- цового цилинд- ра _ил/
Жесть 40/41 3 17,5; 17,7
Стекло 40/42 3,2 11,0; 10,7
Бумага 40/48,8 — 0,4
То же 70/70,8 •— о.з
В 80/80,8 — 10,2
554
Таблица 4
Влияние оболочки на детонационную способность диннтроанилииа
Материал оболочки Диаметр обо- лов г и внутреи- 1гий/|1ар}жнь|й мм Вес оболочки на 1 ем длины заряда г/см Обжатие свпп- НОВОГО ЦИЛИН Л“ ра
Стальная трубка 30/40,8 45 ГО \п
СтзЛ| пая трубка, разреза и пан 30/40,6 45 12,4; 12,7
вдоль и а 2 части СтаЛ1 ныс элементы я колике- ,30/38 30 0,6; 1,3
сгве 16 шт. Свиньи Э^/37,6 4а 2,8; 2,5
Медь 14/23 г> Полная летон.т-
* О результатах опь|та суди ЛИ ПО Нроб|1Т1цО сгщицоных п л а ция в 2 опы- тах * гтинок.
Таблица -5
Влияние оболочки на детонационную способность динитгонафталина
Материал оболочки Диаметр оболочки вкутре । - ьий/наруж- иый мм Вес оболоч- ки па 1 ем длины заря- да г/см Вес тетри- лового де- тонатора г Обжатие СВ[1| ЦОВОг'О цилипдр.г мм
Стали ая цельнотянутая трубка 40/50,8 45 5 18,7; 20,7
Сталигая трубка со шном 40/48 40 Fj 8,3; 8,3
Состав! ая «..белочка из сталтпых полос, шириной 4—f мм 40/50 40 4,7; 5,6
Свинцовая трубка 40/46 43 0 8,4; 8,4
Сталг н я цельнотянутая трубь а 40/50 .11) 10 22,0; 24,2
Стаж и я трубка со швом 40/48 40 10 21,0- 19.5
Составг ая «.болонка из стальных полос шириной 4 —г мм 46/48 42 10 8,0; 7,8
Свит новая трубка 40/48 Д7 10 19,3' 20,6
Сталг!ая цельнотянутая труби а 30/40,8 40 16,1; 16.0
Стал, ная цельнотянутая трубьа, разрезанная вдоль 30/40 37 о 5,6; 3,о
Cocti Bi ая «болочка из сталь(.ьх полос шириной 4—5 мм 30/43 48 5 0,8; 2,0
Стальигя цельнотянутая 30/40 3,5 10 15,2
трубка * XM0 37 11 16,1
Сталь пая цель и о тянутая 30/40 34 10 7,2
трубь;, разрезанная вдоль ,30/40 37 10 12 4
Стальг ая цельнотянутая труб, а из элементов* 30/40 30 10 0,8
* Применялись трубки длиной 200 мм.
555
Я. Ф. БЛИНОВ , Л. М. СВЕТЛОВА
42. О ВЛИЯНИИ РАЗМЕРОВ ОЧАГА ИНИЦИИРОВАНИЯ
НА ДЕТОНАЦИОННУЮ СПОСОБНОСТЬ
МАЛОЧУВСТВИТЕЛЬНЫХ ВВ
При изучении взрывчатости динитросоедийений было обнару-
жено своеобразное и интересное явление, противоречащее, на пер-
вый взгляд, известным данным о влиянии диаметра заряда на воз-
можность распространения детонации. Порошкообразные заряды
малой плотности прн возбуждении детонации одним и тем же ини-
циатором устойчиво детонировали при малом диаметре заряда,
в то время как в аналогичных зарядах большего диаметра детона-
ция затухала,
Результаты экспериментов, проведенных для изучения этого
явления, изложены ниже.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методика экспериментов была очень проста: из исследуемых
веществ готовили в различных оболочках заряды, плотностью
близкой к насыпной. Как правило, применяли заряды, длина ко-
торых равнялась 3—4 диаметрам. Взрыв инициировали капсюлем-
детонатором № 8. В отдельных опытах применяли также проме-
жуточные детонаторы в виде насыпных или прессованных заря-
дов из мощных ВВ (тэн, гексоген). О результатах опыта судили
по деформации оболочки заряда, по отпечатку на металлической
пластинке, находившейся в контакте с зарядом, или по обжатию
свинцового цилиндра, на котором устанавливали испытуемый за-
ряд. При выполнении данной работы мы не имели возможности
определять скорость детонации, В качестве обьекта исследования
применяли технические продукты в виде кристаллического порош-
ка, просеянного через сито с 11 отверстиями на см.
В описанных условиях 2,4-динитротолуол с плотностью заряда
0,5—0,65 г/см3 при инициировании капсюлем-детонатором устой-
чиво детонирует в стальной трубе с внутренним диаметром 31 мм
(длина заряда 100 мм). В тех же условиях, в трубе диаметром
104 мм наблюдалось затухание детонации с разбросом большей
части вещества.
Такие же результаты были получены при испытании динитро-
анилина и пикраминовой кислоты. Эти вещества в зарядах с плот-
ностью 0,41—0,70 г/см3 устойчиво детонируют в медной трубке
с внутренним диаметром около 14 мм (толщина стенки I—2 мм)
и в свинцовой диаметром 10 мм с толщиной стенки 2 мм при ини-
циировании капсюлем-детонатором, В стальных трубах диаметром
40 мм в аналогичных условиях устойчивой детонации не наблю-
дается.
Опыты с этими веществами были поставлены также по схеме,
показанной на фиг. 1. Заряды в узкой медной (</=10—15 мм)
558
J
5
6
трубке, частично разбрасывалось,
его оставалась в виде сильно уплот-
слоя на стенках трубки, В отдельных
где было произведено определение
остатка, она оказалась равной
и широкой стальной (с/ = 40 мм) трубках имели одинаковую плот-
ность, которая в разных опытах с динитроапилином изменялась
от 0,64 до 0,82 п с пикраминовой кислотой от 0,41 до 0,70 г/см3.
В отдельных опытах узкая трубка только соприкасалась с зарядом
и широкой трубке, в других — была погружена в него па глуби-
ну от 1 до 5 см. Во всех опытах детонация распространялась в
трубке меньшего диаметра и затухала прн переходе в более ши-
рокую трубку. Вещество, находившееся в ши-
рокой
а часть
венного
опытах,
плотности
1,33—1,43 г/см3. Результаты опытов не изме-
нились в случае, когда широкая часть трубки
была снабжена навинчивающейся крышкой.
Существенно отметить, что наблюдавшееся
затухание взрыва в зарядах большого диа-
метра связано не с невозможностью распро-
странения детонации в данных опытах, а
с. условиями инициирования. Так, в случае
когда применяли промежуточные детонаторы
из тэна или гексогена, хотя бы в виде насып-
ных зарядов, детонация динитросоединений
в трубках большого диаметра распространя- -
ласъ устойчиво, если только диаметр этих де-
тонаторов превышал некоторый минимум.
Более того, устойчивую детонацию можно
было наблюдать в зарядах большого диа-
метра и в том случае, когда она возбуждалась
зарядами того же динитросоединения мень-
шего диаметра, если переход от малого
к большому диаметру не был резким. Соот-
ветствующие опыты были проведены с динитроанилином и пикра-
миновой кислотой в конических зарядах. Исследуемое вещество
насыпали в оболочки из листового свинца толщиной 2 мм, имев-
шие форму усеченного конуса с диаметром меньшего основания 10
и большего — 45 мм (высота заряда 135 мм). Взрыв возбуждали
Капсюлем-детонатором. При инициировании со стороны меньшего
основания происходила полная детонация, а со стороны боль-
шего — отказ.
Своеобразное влияние на взрывчатость исследуемых зарядов
оказывает плотность. Ранее было установлено [2], что с ее увели-
чением детонационная способность динитросоединений ухудшает-
ся (растет критический диаметр). В то же время, При понижении
плотности наблюдаются отказы при тех способах инициирова-
ния, которые обеспечивают взрыв зарядов большей плотности.
Так, в описанных опытах с пикраминовой кислотой в конусах с
оболочкой из свинца наблюдался полный взрыв при инициирова-
Фиг. I. Схема опытов
с динитроанилином и
пикраминовой кисло-
той.
Г — капсюль * детонатор,
2—медная трубка с ВВ,
5—стальная трубка с ВВ,
4—стальная пластинка,
5—свинцовый цилиндр,
б—стальная плита.
559
нии с большего основания капсюлем-детонатором заряда плот-
ностью 0,65 а/сти3 и отказ при плотности 0,60 г/см3. Подобные же
явления наблюдаются и в случае очень мощного промежуточного
детонатора. Так, при использовании в качестве такого детонатора
прессованной тетриловой шашки весом 20 г, происходила полная
детонация заряда из динитроанилина с плотностью 0,71—0,72 г] см3
и диаметром 70 ж.ч; аналогичный заряд с плотностью 0,56 a/cxt3
в этих условиях инициирования давал отказ. Для пикраминовой
кислоты такие же результаты были получены в трубках диаметром
40 мм, при плотности зарядов 0,8 и 0,5 г/см3, соответственно.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Исходя из общих представлений об условиях устойчивости де-
тонации, развитых Ю. Б. Харитоном [3], полученные результаты
могут быть объяснены следующим образом.
Исследованные динитросоединения являются системами с ма.
лой взрывчатостью и критический диаметр детонации для зарядов
из них без оболочки — велик. В прочной и массивной оболочке
(металлическая трубка), замедляющей разлет вещества, становит-
ся возможной детонация таких соединений в заряде малого диа-
метра. В описанных выше опытах взрыв инициировали в заряде
малой плотности и большого диаметра таким способом, что в пер-
вый момент реакция возникала только в ограниченном по диаметру
участке заряда (например, капсюлем-детонатором или по схеме
фиг. 1), Окружающее очаг инициирования вещество, реакционная
способность которого в условиях взрыва мала, может вести себя
как инертная оболочка. Малая плотность вещества делает инер-
ционное его сопротивление незначительным. Оболочка из вещества
малой плотности легко уплотняется. В результате этого уплотне-
ния происходит быстрый разлет непрореагировавшего вещества и
продуктов неполной реакции из очага инициирования, что и ведет
к затуханию взрыва в этом очаге. Если слой уплотняющегося ве-
щества между границей очага инициирования н металлической
оболочкой мал, как это имеет место в коническом заряде при ини-
циировании с меньшего основания конуса, то разлет вещества
вследствие уплотнения ограничивается и не может привести к за-
туханию детонации.
Понятно, что при одинаковых размерах очага инициирования
разлет ВВ в результате уплотнения окружающих слоев будет тем
меньше, чем больше их плотность. Именно поэтому наблюдается,
при определенных условиях инициирования, детонация зарядов
большой плотности и отказ их при меньшей плотности.
В том случае, когда инициатор создает очаг инициирования
достаточно большого размера —устойчивая детонация возникает
и в заряде, заключенном в трубку большого диаметра. Если, од-
нако, применить еще более мощный инициатор (прессованный
заряд мощного ВВ), то в зарядах малой плотности сжатие, при-
водящее к уплотнению вещества, распространяется быстрее, чем
560
взрывчатое превращение. Впереди фронта реакции, возникшей л
определенном слое динитросоединения, оказывается уплотненное
вещество, восприимчивость которого к детонации мала. Это явле-
ние аналогично описанному А. Ф. Беляевым [1] затуханию дето-
нации зарядов аммиачной селитры и смесей на ее основе при
слишком сильном инициировании.
ЛИТЕРАТУРА
I, Б е л я е в А, Ф-, ДАН СССР, J943, 26, 197.
2. Блинов И, Ф., Научные доклады Высшей школы, Химия и химическая
технология, № 4, 1958, стр. 640,
3. Харитон Ю. Б. О детонационной способности ВВ, Сб. ^Вопросы тео-
рии ВВ», АН СССР, 1947.’
Г. Г. РЕ МП ЕЛЬ
43. ОПРЕДЕЛЕНИЕ СКОРОСТЕЙ УДАРНЫХ ВОЛН,
НЕОБХОДИМЫХ ДЛЯ ВОЗБУЖДЕНИЯ Д ЕТОНАЦИИ ВВ
Исследование явлений, происходящих при возбуждении дето-
нации, представляет не только теоретический, но и практический
интерес. Эти исследования имеют особое значение для работ в
шахтах, опасных по газу и пыли.
Элементы детонационной цепи должны быть подобраны таким
образом, чтобы обеспечить устойчивую детонацию всего заряда
с «нормальной» для него скоростью. Разумеется, это требование
может быть выполнено только для зарядов, способных к устойчи-
вой детонации.
Процессы, протекающие при возбуждении детонации, более
сложны, чем явления, возникающие при распространении сфор-
мировавшейся детонационной волны. Однако возбуждение дето-
нации должно в значительной мере регулироваться теми же об-
стоятельствами, которыми регулируется устойчивость ее. Следо-
вательно, необходимой предпосылкой для изучения процесса воз-
буждения взрыва явилось изучение явлений, определяющих устой-
чивость детонации.
Только после того, как в результате работ Ю. Б. Харитона [5J,
А. Ф. Беляева [3], А, Я. Апина [1]> В. К- Боболева [4] и др. были
установлены основные закономерности, определяющие устойчи-
вость детонации и зависимость ее скорости от диаметра заряда,
размера частиц и других факторов, стало возможным плодотвор-
ное изучение процесса возбуждения детонации. Большинство бо-
лее ранних работ, посвященных этому вопросу, носило характер
эмпирического подбора необходимых размеров детонаторов (бое-
виков) или допустимой толшины прослоек между детонатором и
зарядом в тех или иных частных случаях без физической интер-
претации полученных данных.
561
По мнению большинства исследователей, возникновение дето-
нации связано с образованием в заряде ударной волны. Если эта
волна обладает достаточной интенсивностью, то за ее фронтом на-
чинается бурная химическая реакция. Возникает детонация. Оче-
видно, что для возбуждения ее ударная волна должна иметь па-
раметры (в первую очередь скорость), которые пе меньше крити-
ческих величин, необходимых для развития реакции за фронтом
волны. Можно было ожидать, что эти критические величины дол-
жны зависеть ие только от таких характеристик заряда, как его
химически)! состав и плотность, но и От условий разлета продук-
тов взрыва, т- е. от тех условии, которые определяют возможность
устойчивой детонации.
Очевидно, что для выяснения закономерностей, определяющих
возбуждение детонации, в первую очередь надо знать величину
скорости инициирующей ударной волны, необходимой для возник-
новения детонации, и зависимость этой скорости от характеристик
заряда.
Единственные данные о необходимых скоростях инициирую-
щих ударных волн были получены Б. И. Шехтером [2], который
установил, что в зарядах из прессованного тротила и флегматизи-
рованного гексогена диаметром 23 мм (т, е. при диаметрах при-
мерно в 10 раз больше критических для данных ВВ) детонация
возникает в том случае, если ударная волна имеет скорость
2400—3000 м}сек. Глубина и площадь фронта волны в опытах
Б, И, Шехтера оставались постоянными.
Ударные волны в инициируемом заряде создавались этим ис-
следователем при помощи волн, распространяющихся по инертным
прослойкам, (вода, сталь и Др,), что устраняло возможность непо-
средственного воздействия продуктов взрыва активного заряда (де-
тонатора) на пассивный заряд.
Однако после опытов Б, И, Шехтера по-прежнему оставалось
неизвестным, как будут меняться минимальные скорости ударных
волн, необходимые для возбуждения детонации, в зависимости от
диаметра пассивного заряда, величины его частиц и других фак-
торов.
Неясным оставался также вопрос, каковы должны быть скоро-
сти инициирующей волны в условиях, чаще всего встречающихся
на практике при непосредственном контакте активного и пассивно-
го зарядов.
Для выяснения этих вопросов была проведена работа, резуль-
таты которой излагаются в настоящей статье.
Непосредственное определение скорости ударных волн, возни-
кающих в начале пассивного заряда, представляет значительные
трудности. Однако о скорости ударной волны можно судить по
скорости детонации в том случае, если в пассивном заряде сразу
возникает детонация п если возможна запись ее истинной скорости.
При оптических методах регистрации скорости детонации не-
прозрачных зарядов, последнее условие выполняется только в том
562
случае, если волна с самого начала распространяется с постоянным
радиусом кривизны. Если происходит увеличение этого радиуса,
то выход волны на поверхность в начале заряда происходит с боль-
шей скоростью, чем движение по оси, в результате чего на началь-
ном участке записывается завышенное значение скорости,
О скорости ударной волны, возникающей в начале инициируе-
мого заряда, можно также судить по скорости детонации активного
заряда или скорости ударной волны, распространяющейся по инерт-
ной прослойке, через которую передается детонация.
Хотя скорость ударной волны, возникающей в начале заряда, не
равна скорости детонации активного заряда или скорости волны
в инертной прослойке, она должна быть однозначно связана с ними.
Чем больше скорость детонации активного заряда, тем при про-
чих равных условиях скорость ударной волны, возникающей в
иницийруемом (пассивном) заряде, будет больше.
В частном случае, когда плотность и сжимаемость вещества
активного заряда будут равны плотности и сжимаемости вещества
пассивного заряда, скорость ударной волны в начале пассивного
заряда будет равна скорости детонации активного заряда (при
наличии контакта активного и пассивного зарядов).
Таким образом, о скорости ударной волны, необходимой для
возникновения детонации в пассивном заряде, можно судить по
скорости детонации, возникающей в начале заряда, если есть уве-
ренность в отсутствии искажений прн ее записи, и по скорости де-
тонации активного заряда или по скорости ударной волны в инерт-
ной среде, примыкающей к заряду.
Способность ударной волны возбуждать за своим фронтом ин-
тенсивную химическую реакцию или иначе говоря способность
ударной волны, созданной в заряде, к переходу в детонационную
зависит не только от скорости, но и от ее глубины (точнее, от ха-
рактера изменения давления за фронтом волны), площади и фор-
мы фронта волны.
Наиболее благоприятные условия для возникновения детонации
создаются при действии плоской волны или волны, имеющей ма-
лую кривизну и большую глубину. При малой кривизне фронта
инициирующей ударной волны, создающейся в начале пассивного
заряда, устраняется также возможность значительных искажений
записи скорости детонации на начальном участке заряда.
Поэтому при проведении опытов было желательно производить
инициирование волнами с возможно большей глубиной зоны сжа-
тия и большим радиусом кривизны фронта волны. Этого можно
было достичь, используя в качестве детонаторов заряды большой
высоты, заключенные в прочную массивную оболочку или имею-
щие большой диаметр.
В качестве пассивных зарядов было решено в первую очередь
использовать заряды из литого и прессованного тротила, В даль-
нейшем предусматривается работа с зарядами из практически ис-
пользуемых аммонитов.
36*
563
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Было поставлено три группы опытов.
1. Заряды из литого тротила инициировались взрывами зарядов
из прессованной смеси тротила с тальком.
2. Такие же заряды инициировались взрывами зарядов из
насыпного тротила или его смеси с алебастром.
3. Детонация в зарядах из прессованного тротила возбужда-
• лась ударной волной, распространявшейся по воде.
Возбуждение детонации зарядов из литого тротила взрывами
прессованных зарядов из смеси тротила с тальком
В качестве пассивных зарядов применялись заряды из литого
мелкокристаллического тротила, Заряды изготовлялись путем от-
ливки в разъемные металлические формы различных диаметров.
В качестве активных зарядов использовались прессованные за-
ряды диаметром 32 мм из смеси тротила с тальком, помещенные
в стальную трубу с внутренним диаметром 32 л.н и толщиной сто
нок 4 мм. В трубу помещали 10 шашек, Вследствие большой
длины заряда и наличия массивной оболочки волна, возникаю-
щая на торце такого заряда, должна была иметь большую глубиш
и большой радиус кривизны.
Тротил и тальк перед приготовлением смеси просеивались че
рез сито № 24. -
Во всех опытах определялась скорость детонации активного
заряда и фиксировалось либо возникновение детонации, либо от
каз в пассивном заряде. В части опытов скорость детонации изме-
рялась при помощи фоторегистра с зеркальной разверткой
(СФР-2). В этом случае определялась и скорость детонации в на-
чале пассивного заряда. В остальных опытах скорость детонации
активного заряда измерялась по методу Дотриша.
В опытах с фоторегистром активный и пассивный заряды при-
креплялись к деревянной рейке или латунной пластинке так, что
бы их торцы соприкасались, а оси совпадали,
В стенке трубы, в которую был заключен активный заряд, име-
лась щель, обращенная к фоторегистру. О полноте детонации ини-
циируемого (пассивного) заряда судили по фотоснимку и отпе-
чатку на латунной пластинке.
В тех случаях, когда скорость детонации активного заряда оп-
ределялась по методу Дотриша, активный и пассивный заряды
укладывались на тонкий стальной лист. О полноте детонации пас-
сивного заряда судили по отпечатку на листе.
Йзмеиение скорости детонации активного заряда достигалось
путем изменения плотности и содержания компонентов в шашках
из смеси тротила с тальком.
Схема опыта с фоторегистром показана на фиг. 1; образны по-
лученных снимков детонации активного и пассивного зарядов —
на фиг. 2 и 3
5S4
Фиг, 1. Схема инициирования заряда из литого тротила
зарядом из прессованной смеси тротил—тальк при изме-
рении скорости детонации фоторегнстром.
/—фоторЕгистр, 2—капсюль-детонатору J—промежуточный дето-
натор, *?—активный зарад (смесь тротила с тальком}. 5—сталь-
ная труба, б—пассивный зарад (литой тротил}. 7—латунная
пластинка, <?—;деревянная рейка, щель а трубе.
Фиг. 2, Снимок, полученный во время опыта по инициированию
заряда диаметром 22 мм из литого тротила.
I—коней активного заряда, ?—след детонадни пассивного наряда.
37 451
565
На фиг. 2 показан снимок, полученный во время опыта по ини-
циированию заряда диаметром 22 мм из литого мелкокристалли-
ческого тротила взрывом заряда из прессованной смеси 70% тро-
тила и 30% талька диаметром 32 мм без оболочки. Скорость де-
тонации смеси была 5400 м/сек. В литом тротиле детонация воз-
никла и распространялась до конца заряда также со скоростью
5400 м/сек.
Фиг. 3. Снимок, полученный во время опыта по
инициированию заряда диаметром 32 лш из литого
тротила.
1—след детонации актнаного заряда, видамый в щель.
2—граница активного и пассивного зарядов. 5—участок
разгона детонации пассивного заряда. 4—конец участка
раагона, 5—участок устойчивой детонации пассяняого
заряда.
На фиг. 3 показан снимок, полученный во время опыта по ини-
циированию заряда диаметром 32 мм из литого мелкокристалли-
ческого тротила взрывом заряда из прессованной смеси 45% тро-
тила и 55% талька диаметром 32 жж в стальной трубе с толщиной
стенок 4 жж. В стенке нижней части трубы имелась щель шири-
ной 4 мм. В данном опыте скорость детонации активного заряда
в момент подхода к литому тротилу была 3300 м/сек (свечение
слабое). Скорость детонации, возникшей в литом тротиле, внача-
ле была примерно равна скорости детонации активного заряда.
Однако на расстоянии 30—35 мм от начала пассивного заряда
скорость его детонации увеличилась до 6500 м/сек, после чего она
распространялась с этой же скоростью.
566
Таблица 1 Возбуждение детонации литого тротила взрывами прессованных зарядов из смеси тротила с тальком I Определено на фоторегистре Скорости, при которых детонация пассивного заря- да не возникает в м/сек ; 2300 3100 I 3200 2800 2600 3400 3600 3700
Скорости, при которых воз- никает детонация пассив- ного заряда в м/сек 3500 3400 3400 3300 3000 2600 - - 1
3900 3200 3300 — 5500 5400 3500 - 21 ! — . — — | — - I 4400 ' - (
I Определено по Дотришу Скорости, при которых детонация пассивного заря- да не возникает в м/сек Отказов не было То же 2000 2050 2400 - -- 2000 2450 3000 3300 3300 — 3000 3150 3400 3600 3750 3900 4100 —
Скорости, при которых воз- никает детонация пассив- ного заряда в м/сек 3400 3350 3000 5650 4700 4200 3750 3600 3050 S S 8 8 § ® § । 8 1 8 8 8 3 8 8 8 , СЧ Ь- CO LO об = ip | ''г со со со го сч XOCOCOCOCOCOCJOJ 5400 4750 4600 4200 3900 3300 6600 5100 4200
Диаметр пассивного заряда из литого тра- тила в мм 80 40 04 со <D 8
37*
567
В большинстве опытов, при которых наблюдались взрывы пас-
сивных зарядов, детонация литого тротила возникала без задер-
жки и скорость ее вначале была равна скорости детонации актив-
ного заряда из смеси тротила с тальком, а затем увеличивалась
или оставалась постоянной.
Однако в некоторых опытах происходила задержка или умень-
шение скорости детонации при переходе волны из прессованной
смеси в литой тротил. К сожалению, в таких опытах четкой кар-
тины явлений, происходящих в месте контакта пассивного и ак-
тивного зарядов, получить не удалось. Зависимость этого явления
от скорости детонации активного заряда также не была обнару-
жена. Например, задержка наблюдалась прн скорости детонации
активного заряда 3500 м/сек, тогда как при скорости детонации
активного заряда 3300 м/сек пассивный заряд из литого тротила
начинал детонировать без задержки (фиг. 3).
Из анализа полученных снимков могут быть сделаны следую-
щие выводы:
При инициировании литого тротила взрывами зарядов из прес-
сованной смеси тротил—тальк в пассивном заряде в большинстве
случаев возникает детонационная волна, скорость которой равна
скорости детонации активного заряда. Эта скорость быстро уве-
личивается. На расстоянии 30—60 мм от начала пассивного за-
ряда скорость детонации достигает своего нормального значения
(для данного диаметра заряда из литого тротила). На основании
этого можно считать, что скорость ударной волны, возникающей
в литом тротиле, примерно равна скорости детонационной волны
в прессованной смеси тротила с тальком.
При возбуждении детонации в зарядах из литого тротила ма-
лых диаметров, близких к критическому, заряды могут детониро-
вать с постоянной скоростью, заданной им активным зарядом (во
всяком случае при длине заряда до 200 мм).
Искажение, обычно возникающее в начале пассивного заряда
при измерении скорости детонации на фоторегистре, в проведен-
ных опытах было устранено, или во всяком случае, сведено к ми-
нимуму.
В табл. 1 приведены результаты опытов, в процессе которых
рпределялась минимальная скорость детонации активных зарядов
из смеси тротила с тальком, необходимая для возбуждения дето-
нации в литом мелкокристаллическом тротиле,
Возбуждение детонации зарядов из литого тротила
взрывами зарядов малой плотности из измельченного тротила
или из его смесей с алебастром
Активные заряды представляли собой бумажные цилиндры
длиной 0,6 xt, заполненные измельченным тротилом, просеянным
через сито № 24, или смесями этого тротила с алебастром.
Пассивные заряды --тротиловые, литые.
568
Активные и пассивные заряды укладывались на стальные лис-
ты так, чтобы их торцы соприкасались, а оси совпадали. У иниции-
рующих зарядов измерялась скорость детонации по Дотришу.
О полноте детонации инициируемого заряда судили по отпечатку
на плите.
Характеристики инициируемых и инициирующих зарядов и ре-
зультаты опытов приведены в табл, 2,
Таблица 2
Возбуждение детонации литого туотила взрывами зарядов малой
плотности из измельченного тротила или смесей с алебастром
Пассивный заряд Активный заряд Результаты опыта
Вещество Дна* метр мм Вещество Диа- метр мм Плот- ность г[см? Скорость детона- ции м^сек
Тротил ли- 100 Тротил насыпной, из- 44 0,9 4600 Детонация
той, отливка мельченный
мелкокри- сталличес- 80 То же 88 1,0 5000 я
кая 80 80 1,0 5000 я
44 я 44 1,0 5000 я
40 32 0,95 46ft) я
34 я 32 10 4800 я
32 . 32 1,0 4700 т»
32 ы 32 1,0 5000
32 • 32 10 4600 р
21 п 80 0,95 4600 т»
26 ы 80 0,96 4700 V
80 Смесь тротила с
алебастром 80/20 80 1,0 4450 >1
80 То же 70/30 80 1,0 4100 ц
80 50/50 80 1,° 2450
80 40/60 80 1,0 — р
60 60/40 60 1,1 3000 »|
52 50/50 80 1,1 2450 я
40 50/50 80 1,1 2700 Затухание
32 50/50 80 1,1 2700 »
32 50/50 150 1,2 2850 Детонация
’ Возбуждение детонации зарядов из прессованного тротила
ударными волнами, распространявшимися в воде
Сборка активного и пассивного зарядов в-этих опытах произ-
водилась по схеме, приведенной на фиг. 4.
569
На дно пробирки 2 из ацетилцеллюлозы с внутренним диамет-
ром 23,5 лш устанавливались 2 шашки из флегматизнрованного
гексогена (плотность 1,58—1,60 a/cxt3), диаметром 23 леи и об-
щей высотой 57—-60 мм, служившие во всех опытах активным за-
рядом 3. Затем в пробирку наливалась вода до тех пор, пока вы-
сота ее слоя над шашками не достигала нужной величины.
Фиг. 5. Снимок, полученный при передаче де*
тонации через слой воды.
!—след детонации активного заряда. 2—конец ак*
тявного ааряда. 3—начало детонации пассивного аа-
ряда, след продуктов детонации активного заряде,
двигающихся за выбрасываемой водой. 5—след де*
тонации пассивного заряда.
Фиг. 4. Схема
опыта возбуж-
дения детона-
ции через слой
воды.
/—пассйяный за*
ряд. 2—пробирка,
j—активный эй'
ряд. капсюль-
детонатор. 5—во-
да. tf—проволо ка,
При опытах с пассивными зарядами из прессованного троти-
ла, просеянного через сито № 52, с диаметром от 3,85 до 23 Л1Л<
я пробирку опускали медную проволоку 6 с привязанным к ней
столбиком шашек, представлявшим собой пассивный заряд 1.
Торец нижней шашки приводили в соприкосновение с поверхно-
стью воды так, чтобы между шашкой и водой не было воздушной
прослойки и чтобы заглубление заряда в воду не превышало
0,5 лш. При опытах с пассивными зарядами диаметром 23 мм, за-
ряд, состоявший из двух шашек, опускали на нитках в пробирку,
привязанную к латунной пластинке.
О полноте детонации пассивного заряда судили по деформа-
ции медной проволоки или латунной пластинки.
В части опытов скорость детонации определялась с помощью фо-
торегистра.
На фиг. 5 показан снимок, полученный при передаче детона-
ции от активного заряда пассивному через слой воды.
570
Активным зарядом в этом опытё служили две шашки из прес-
сованного флегматизированного гексогена общей высотой 60 мм,
а в качестве пассивного заряда— шашки диаметром 8 мм из из-
мельченного и просеянного через сито № 52 тротила с плотностью
1,59 г/с-и3. Толщина слоя воды составляла 18 мм.
На фиг. 5 виден участок разгона детонации в пассивном заря-
де. Свечение на участке разгона было слабым. Средняя скорость
на участке разгона была 4500—5000 м!сек. Скорость ударной вол-
ны в воде на расстоянии 18 мм от пассивного заряда диаметром
23 мм равнялось, приблизительно, 3900 м[сек. Переход к нормаль-
ной скорости детонации 6700 м[сек. сопровождался резким возра-
станием интенсивности свечения.
На фиг. 5 виден также след продуктов детонации активного
заряда, двигающихся за выбрасываемой водой. Этот след под-
ходит к пассивному заряду позже, чем в последнем возникает де-
тонация.
Результаты опытов по передаче детонации через слой воды
приведены в табл. 3.
•Таблица 3
Возбуждение детонации прессованного тротила через слой воды
Пассивный заряд Толщина слоя воды между активным и пассивным за- рядами в мм Результаты опыта Толщина слоя воды, прн которой обеспе- чивается детонация зарядов данного диаметра в мм
Диаметр мм Плотность ZjCM^
23 1,58 30 Отказ
23 1,57 30
23 1,57 28
23 1,57 25 Детонация 25
23 1,57 25 -
10 1,58 28 Отказ
10 1,58 25 Детонация 25
8 1,59 20 Отказ
8 1 59 18 Детонация 18
8 1’59 18
5,85 1,58—1,61 28 Отказ
5,85 1,58—1,61 25 *
5,85 1 58—1,61 20 Детонация
5,85 1,58—1,61 18 к
5,85 1,58—1 61 15
5,85 1,56—1,58 20 Отказ
5,85 1 56—1,58 20
5,85 1,56—1,58 18 Детонация 18
5.85 1,51—1,55 25 Отказ
3,85 1,56—1,61 15 В
3,85 1,56—1,61 15
3,85 1,5 —1,56 15
3,85 1,56—1,61 12 . Детонация 12
3,85 1,56—1,61 12
3,85 1,5 — 1,56 12 ' •
571
В’табл. 4 приведены минимальные скорости, которые должна
иметь ударная волна в воде для возбуждения детонации в заря-
дах разных диаметров из прессованного измельченного тротила,
и расчетные значения скоростей ударных волн, возникающих в
прессованном тротиле.
Приближенное вычисление последних было проведено в пред-
положении, что уравнение состояния прессованного тротила при
больших давлениях имеет вид
Р^?к,
где р—плотность ВВ, й=8. Пассивные заряды имели плотность
1,58s/cjh3,
Таблица 4
Минимальные скорости ударной волны в воде для возбуждения
детонации прессованных тротиловых зарядов различного диаметра
Диаметр пассивного заряда мм Прослойка воды, через которую пере- дается детонация, мм Скорость ударной волны в воде м/сек Скорость ударной волны в прессован- ном тротиле м/сек
10 25 3160 2700
5,85 18 3860 3600
5,85 12 4760 4500
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Минимальные скорости ударных волн, необходимые для воз-
никновения детонации в зарядах различных диаметров из литого
и прессованного тротила, приведены в табл. 5.
В табл. 5 показана весьма удобная для анализа результатов
испытаний зависимость минимальной скорости инициирующей
ударной волны от относительного диаметра, т. е. от величины от-
ношения d/dKp, где d — фактический диаметр заряда и —кри-
тический диаметр. Использование такой зависимости было пред-
ложено А. Ф. Беляевым [3].
Критический диаметр для мелкокристаллического литого тро-
тила принимался равным 20 мм, для прессованного — 3,5 мм.
Зависимость минимально необходимых скоростей инициирую-
щей волны (Диниц) от абсолютного и относительного диаметров
заряда изображена на фиг. 6 и 7, На этих же фигурах показана
зависимость скорости установившейся детонации Дзар от диамет-
ра заряда для мелкокристаллического литого тротила (по лите-
ратурным данным),
Скорость инициирующей ударной волны, которую надо создать
в начале заряда для возникновения в нем детонации, зависит от
его диаметра. Чем меньше диаметр заряда, тем больше должна
быть скорос’гь волны. При диаметрах заряда, которые значительно
больше критических, скорость ударной волны, обеспечивающей
572
Таблица 5
Зависимость минимальных скоростей ударных волн для возбуждения
детонации литых и прессованных тротиловых зарядов от относительного
диаметра последних
Характеристика активных зарядов Характеристика пассивных зарядов Диаметр пассив- ного за- ряда мм d/d^ Минимальная скорость ини- циирующей волны м/сек
Шашки диаметром МелкокристаллН' 22 1,10 4200
32 мм нз прессованной смеси тротила с таль- ческий литой тротил (р= 1,58-г1,59 г/сжЗ) 23 1,14 3800
ком в стальной трубе 26 1,30 3300
32 1,60 2600
Насыпной измельчен- Литой мелкокри- 32 1,60 2850—2700
ный тротил нли его сме- си с алебастром сталлический тротил 52 2,40 2450
Шашки диаметром Прессованный тро- 3,85 1,10 4500
23 мм из флегма тизи- тил, просеянный пе- 5,85 1,65 3600
роваиного гексогена. ред прессованием
Возбуждение детонации через сито № 52 10,00 2,85 2700
производилось ударной (р=1,5-е-1,61 г/сл3)
волной, распространяв- шейся в воде
возбуждение детонации, приближается к скорости звука в ве-
ществе.
Значения последней определялись в отдельной работе. В ли-
том тротиле при плотности 1,58 г/см? скорость звука 2100 ж/сйк,
в прессованном при той же плотности— 1900 м/сек, При диамет-
рах, близких к критическому, минимальная скорость инициирую-
щей ударной волны приближается к так называемой критической
скорости детонации, т. е. к скорости, с которой обычно детонирует
заряд критического диаметра. Для литого и прессованного тротила
эти скорости примерно равны 4200—4500 м/сек.
Следует отметить, что при использованной в настоящей работе
методике, когда инициирующие волны имели большую глубину и
малую кривизну, создавались особо благоприятные условия для
возбуждения детонации. При меньших глубинах инициирующей вол-
ны, что обычно бывает на практике, для возбуждения детонации
потребуются несколько большие скорости инициирующей ударной
волны.
Как мы видели с уменьшением диаметра заряда растет мини-
мальная скорость инициирующей волны, необходимая для воз-
буждения в нем детонации. Это явление указывает на то, что при
малой скорости волны увеличиваются относительные потери из
зоны реакции, создающейся в начале заряда. Увеличение потерь
может иметь место только в том случае, если с уменьшением ско-
рости одновременно растет длина зоны этой реакции. Если увели-
573
574
чение времени реакции с уменьшением скорости волны и темпера-
туры на ее фронте очевидно и никаких проверок не требует, то
возрастание длины зоны реакции с уменьшением скорости ини-
циирующей волны само по себе не обязательно.
В самом деле длина зоны реакции /~Дт,
где Д—скорость детонации в м!сек\
t — время реакции в сек.
С уменьшением скорости инициирующей волны I может остать-
ся постоянной, так как с уменьшением Д увеличивается т, и про-
изведение их может не меняться.
Полученные данные показывают, что в действительности (во
всяком случае для тротиловых зарядов) длина зоны реакции с
уменьшением скорости волны возрастает, а это может случиться
только тогда, когда т возрастает быстрее, чем уменьшается Д.
Скорость детонации, возникающей в начале пассивного заряда,
зависит от скорости детонации активного заряда или от скорости
ударной волны в среде, через которую детонация передается.
Как видно из графиков фнг. 6 и 7, кривые зависимости скоро-
сти инициирующей волны и скорости детонации от диаметра заря-
да пересекаются при d—d^p.
Скорость, соответствующая точке пересечения, является, оче-
видно, критической скоростью детонации. Следует -отметить, что
до сего времени определение критической скорости детонации
встречало значительные трудности так как вблизи критического
диаметра скорость детонации очень быстро падает и достаточно
небольшого ухудшения качества заряда для того, чтобы распро-
странение детонации стало невозможным.
Определение критических скоростей по точкам пересечения
кривых Диниц=/(^) и Дзар=/(^) позволяет уточнить их значения
для различных ВВ.
Как видно из графика фиг. 6 для литого тротила Дкр=
= 4500—4600 м[сек. В соответствии с данными А. Я. Апина [1] и
В. К- Боболева [4] критическая скорость детонации от величины
зерна ВВ не зависит.
Таким образом, для возникновения детонации в зарядах, диа-
метр которых равен критическому, надо создать ударную волиу,
скорость которой должна быть не меньше нормальной скорости
детонации таких зарядов.
По мере увеличения диаметра заряда необходимая скорость
инициирующей ударной волны уменьшается, асимптотически при-
ближаясь к скорости звука в данном ВВ.
Как видно из кривых фиг. 7, скорости ударных волн, которые
надо создать для возбуждения зарядов из литого и прессованного
тротила, при одинаковых dfdxp довольно близки между собой.
Некоторое увеличение скорости ударной волны, необходимой
для возникновения детонации зарядов из прессованного тротила,
вероятнее всего объясняется тем, что возникновение детонации
при инициировании литого тротила несколько облегчалось теп-
575
левым воздействием продуктов взрыва инициирующего заряда.
При возбуждении прессованного тротила детонация передавалась
через слой воды и тепловое воздействие поэтому было исключено.
•
ОБЩИЕ ВЫВОДЫ
1. Скорость инициирующей ударной волны, которую надо соз-
дать в заряде для того чтобы возникла детонация, зависит от диа-
метра заряда. Для зарядов критического диаметра Диниц=Дкр-
По мере увеличения диаметра Диниц уменьшается асимптотиче-
ски, приближаясь к скорости звука в веществе.
2. С уменьшением скорости детонационной волны в тротиловых
зарядах увеличивается длина зоны химической реакции за фронтом
волны.
3. Точное значение критической скорости детонации (скорости
с которой детонирует заряд при диаметре, равном критическому)
может быть определено по точке пересечения кривых Диниц=Н^)
иДзар = /(^)«
ЛИТЕРАТУРА
1. Апин А. Я-, О детонации и взрывном горении ВВ, ДАН СССР, 1945,
50, 285.
2. Баум Ф. А., С т а н ю к о в и ч К. П., Ш е х т е р Б. И., Сб. «Физика взрыва».
Госфизматгиз, 1959, стр. 272—291.
3. Беляев А. Ф., Условия устойчивости детонации амселитренных ВВ,
Сб. «Вопросы теории ВВ», АН СССР, 1947, стр. 29.
4. Б о б о л е в В. К-, О предельных диаметрах зарядов химически однородных
ВВ, Диссертация, Институт химической физики, 1947.
5. Харитон Ю. Б., О детонационной способности ВВ, Сб. «Вопросы тео-
рии ВВ», АН СССР, 1947, стр. 7.
СОДЕРЖАНИЕ
Стр,
Предисловие.............................................................. 3
I. Чувствительность взрывчатых веществ к механическим воздействиям
1.| И. А. Холево~| . К вопросу о возбуждении взрыва при деформации
заряда взрывчатого вещества........................................ 5
2. Ф. Боуден. Последние исследования разложения твердых ВВ............. 20
Выступление Е. Г. Ледина по докладу Ф. Боудена.................... 34
Выступление; К. К. Андреева по докладу Ф. Боудена ....... 35
3. К. К. Андреев. К вопросу о факторах, обусловливающих возникновение
взрыва при ударе и треиии и о методах оценки чувствительности ВВ
к механическим воздействиям........................................... 37
4. К. К, Андреев, Ю. А. Теребилина. О возбуждении взрыва при ударе и
методах характеристики чувствительности взрывчатых веществ к
механическим воздействиям.............................................. 53
5. К. X. Андреев, Ю. А. Теребилина. К вопросу о влиянии воздушных
включений на возникновение взрыва при ударе........................... 72
6. X. К. Андреев, Ю. А. Теребилина. К вопросу о механизме возникновения
взрыва при испытании на чувствительность к удару в роликовом
приборе ............................................................... W
7. К. К. Андреев. Об основных факторах, определяющих опасность взрыв-
чатых веществ при механических воздействиях и методах ее оценки 39
3. X. X. Андреёв. О некоторых зарубежных исследованиях по методике
экспериментального определения чувствительности ВВ к удару ... 94
П. Термическое разложение взрывчатых веществ
9. X. X. Андреев, Г. Н. Беспалов. О влиянии воды иа разложение нитро-
глицерина при повышенных температурах................................ 131
10. X. X. Андреев, Г. Н. Беспалов. О влиянии кислот и соды на распад
нитроглицерина в присутствии воды ................................... 172
II. Б. С. Светлов. О термическом разложении нитроглицерина в жидкой
фазе............................................. ................... 194
12. В. В. Горбунов, Б. С. Светлов. О влиянии температуры на разложенце
нитроглицерина......................: ............................... 190
577
Стр^
13. В. В. Горбунов, Б, С. Светлов. Влияние воды и кислоты на самоускоря-
ющийся распад нитроглицерина........................................ 197
14. Б. С. Светлов. О роли двуокиси азота при самоускоряющемся разложе-
нии нитроглицерина.................................................. 203
15. В. В. Горбунов, Б. С. Светлов. О роли конденсированных продуктов при
распаде нитроглицерина.............................................. 214
16. В. В. Горбунов. О растворимости воды в нитроглицерине............ 219
17. К. К. Андреев. О термическом распаде нитроглицерина и возможности
перехода его во взрыв............................................... 225
18. К. К. Андреев, Б. И. Кайдымов. Термический распад тэна......... 241
19. Б. С. Светлов. Термический распад диэтиленгликольдннитрата в жид-
кои фазе............................................................ 274
*
20. Б. А. Лурье, Б. С. Светлов. О влиянии некоторых примесей на терми-
ческий распад диэтиленгликольдинитрата........................... . 281
21. Б. И. Кондриков. Термическое разложение динитрита этиленгликоля и
тринитрита глицерина ............................................... 296
22. Ю. Я. Максимов. Термический распад паров нитропроизводных бензола 338
23. К, К. Андреев, Лю-Бао-фен. Термический распад пикриновой и стифни-
новой кислот........................................................ 349
24. К. К. Андреев, Лю-Бао-фен. Термический распад аммониевых, калиевых
и свинцовых солей пикриновой и стифниновой кислот................... 363
25. А. А. Шидловский. Термическое разложение нитрата аммония .... 401
1Й. Горение и аспышка взрывчатых вещеста
*
26. /С /С Андреев. К вопросу о факторах, определяющих зависимость ско-
рости горения взрывчатых веществ от начальной температуры и
давления............................................................ 404
27. К. К. Андреев. О горении нитратов некоторых спиртов...............416
28. К. К. Андреев, Г. И. Беспалов. О горении нитроглицерина.......... 430
29. Б. И. Кондриков. О горении смесей инициирующих ВВ и жидких
. нитроэфиров....................................................... 443
30. А. И. Гольбиндер. Некоторые закономерности горения летучих много-
компонентных взрывчатых веществ..................................... 457
31. А. И. Гольбиндер. Горение самовоспламеняющейся взрывчатой смеси
и переход его в детонацию........................................... 468
32. И. Я. Петровский, Л. В. Волков. О причинах неполной детонации ВВ
в шпурах........................................................... 474
33. К. К. Андреев, В. Г. Хотин. К вопросу о факторах, определяющих воз-
можность выгорания взрывчатых веществ в шпурах...................... 495
34. А. И. Гольбиндер. Самовоспламенение жидких взрывчатых смесей . . . 499
35. Б. Н. Кондриков. Об интенсивности вспышки взрывчатых веществ ... 515
36. К. К. Андреев, В. В. Горбунов. О термостабильности кристаллов взрыв-
чатых веществ....................................................... 528
578
Стр-
37. К. К. Андреев. К вопросу о переходе горения взрывчатых веществ во
взрыв................................................................. 534
IV. Разные исследования
38. А. А. Шидловский. Вода как окислитель в реакциях с неорганическими
веществами............................................................ 540
39. А. А. Шидловский. Расширение области применения термохимического
закона постоянных разностей. Предвычисление неизвестных теплот
образования солей аммония............................................. 543
40. Ю. Я. Максимов. Упругость паров иитропроизводных бензола .... 540
41. \И. Ф. Блинов ,Л. М. Светлова. Влияние оболочки на детонационную
способность некоторых динитросоединений бензольного ряда .... 550
42. И Ф. Блинов , Л. М. Светлова. О влиянии размеров очага инициирова-
ния на детонационную способность малочувствительных ВВ .... 553
43. Г. Г. Ремпель. Определение скоростей ударных волн, необходимых для
561
возбуждения детонации ВВ
,3-i
i
14.;
15.!
.s’
17.
!8.|
19. j
20.
21.
22.;
23. i
24.
25.
26.
27.
28.
29.
30.
31
32
33
34
35
36
57
ТЕОРИЯ ВЗРЫВЧАТЫХ ВЕЩЕСТВ
ИадательскиА редактор А. А. Степанова Техн. ред. А. Я. Новик
Т-13666 Подписано в печать 24/ХП 1962 г. Учетио-иэд. а. 37,2
Формат бумаги 60х921/1б=18,13 бум. л.—36,25 печ. л.
Цена 2 р. 80 к. Тираж 2640 экз. Заказ 451/5075
Типография Оборонгиза
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Стр. Строка Напечатано Должно быть
3 19 снизу 1953 1955
51 8 сверху было было бы
130 6 снизу Ide К. М. Ide К, Н
131 7 сверху давлению, п убывает давлению и убывающей
144 ФИГ. 13, ось ординат в % вес в %
166 Фнг. 29, ось ординат 500 600
167 18 сверху кислоты зависит кислоты тоже зависит
172 7, 8 сверху гидролиз в случае нейтрального нитроэфира. гидролиз, в случае нейтрального ннтроэфира
173 11 снизу 0,03 0,3
186 22 сверху 3 = 10“1 а = ю-2
186 1 снизу (7J [8]
234 Подпись к фиг. 7 10-4 10*
236 4 снизу образовавшимися образовавшимся
237 Подпись к фиг. 12 РнгО’ Т’НгО = 284
247 16 Снизу влияние ВЛИЯНИЯ
271 7 снизу TTj-r 7-2-7!
297 Сноска, 6 сверху получения изучения
323 13 снизу 80—100° для 80—100°, для
329 Фиг. 286, ось ординат 1№ 10^
341 Формула (2) 1,36 0,36
342 Формула (3) 52 900 52600
358 Сноска относится к стр. 357
394 Табл. 9, послед- няя строка 200—300 200 -230
424 Формула (2) -1/2 Рг 1/2 рг
425 8 снизу наблюдается прн наблюдается и при
451 Фиг. 11 в 40 мсек мсек
495 21 сверху Sc 1berg Selberg
517 Ось ординат рис. 2а и За должна быть разорвана между нулем и 100,
548 11 снизу АО
Зак. 451^5075
W03C