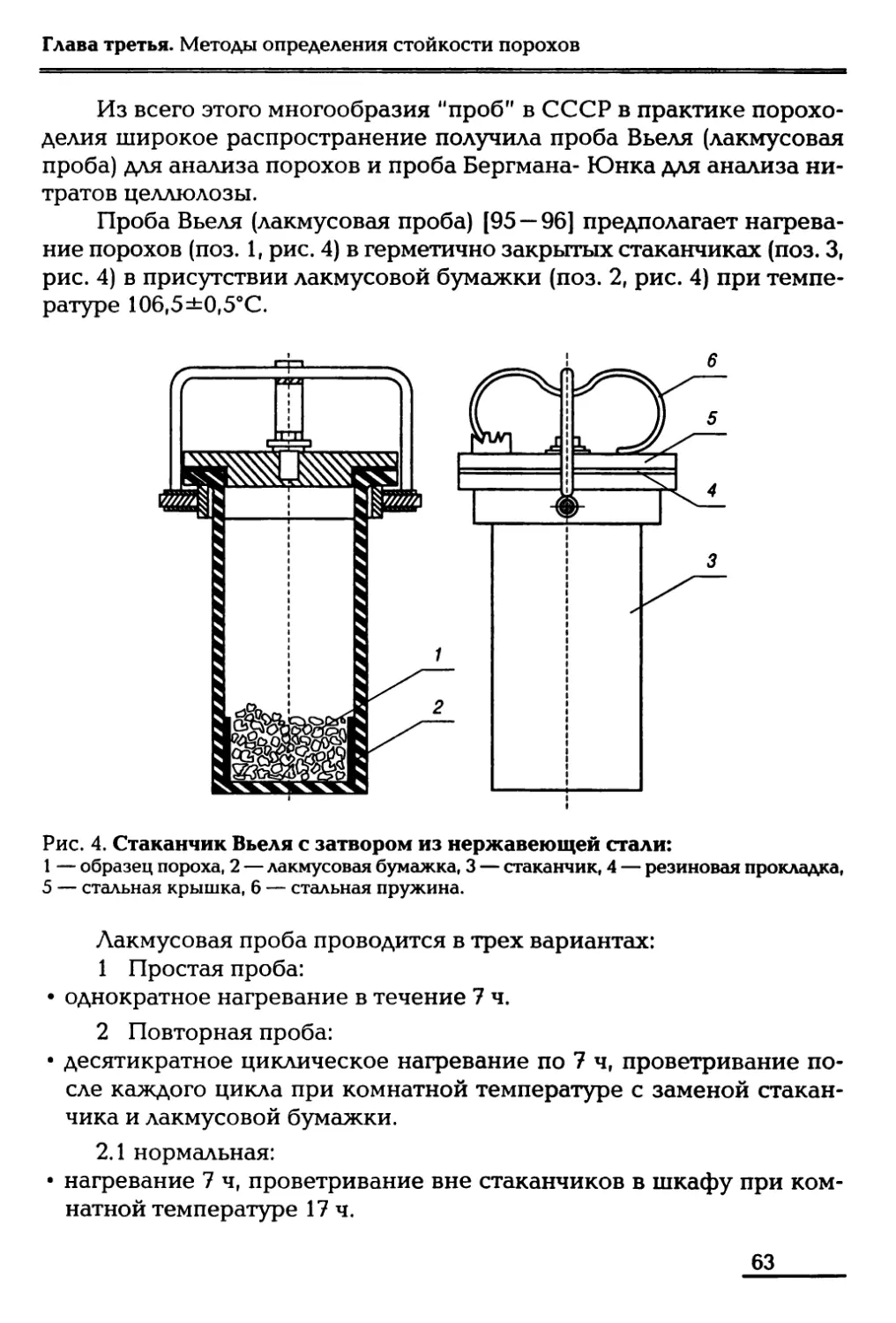
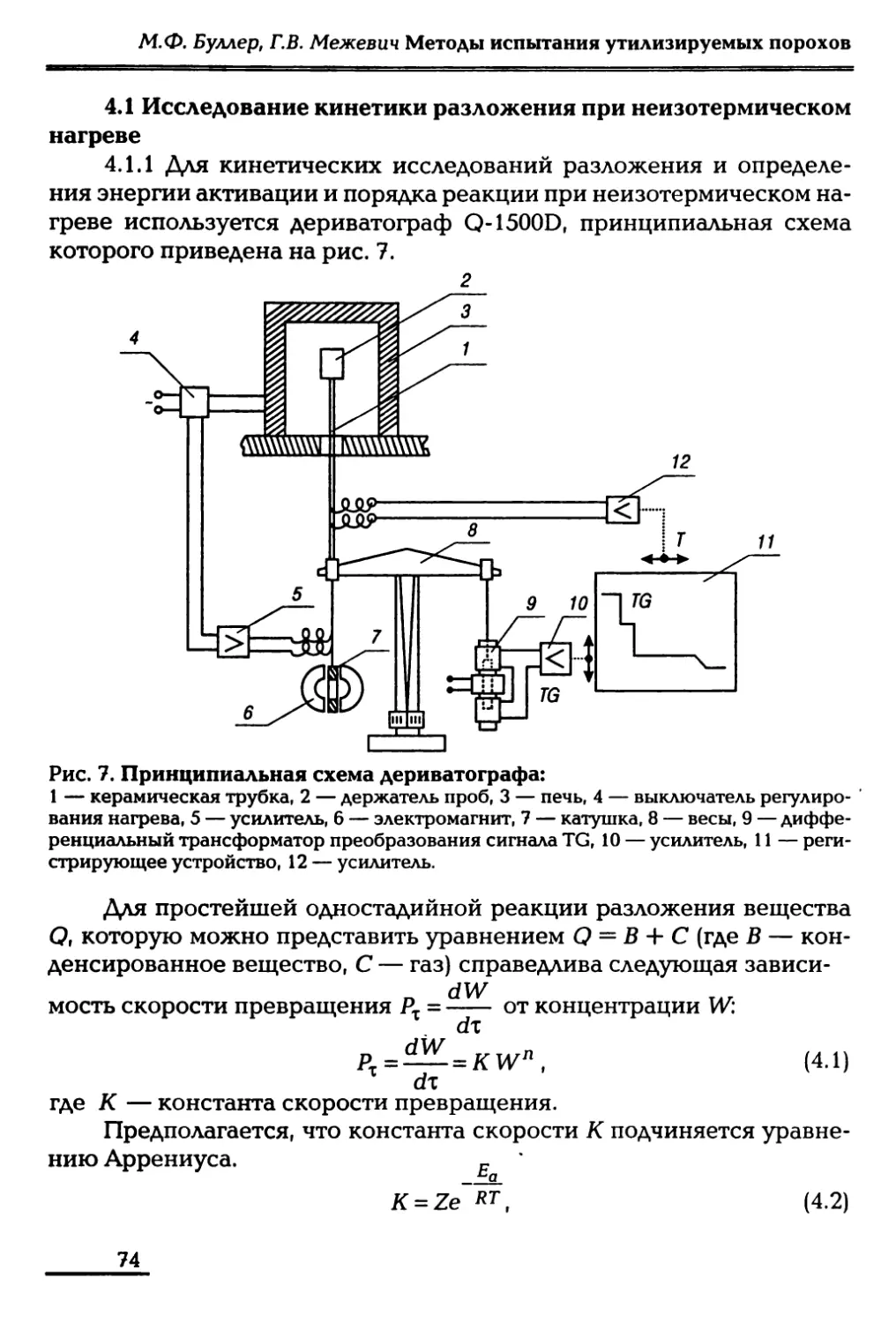
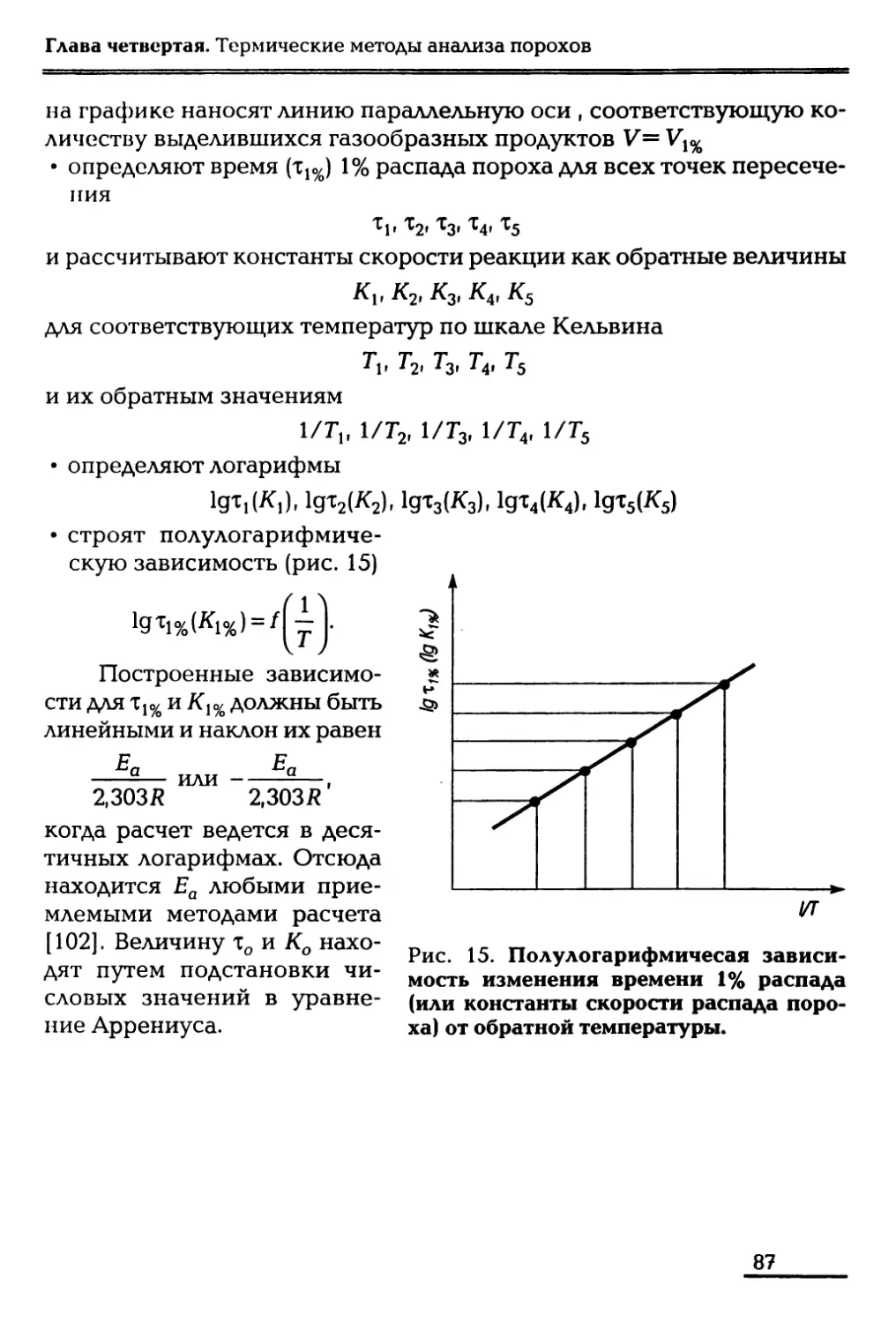
/


























































































Автор: Буллер М.Ф. Межевич Г.В.
Теги: фейерверки пиротехника химическая технология химические производства взрывчатые вещества порох
ISBN: 966-8311-08-6
Год: 2005




Текст
Министерство промышленной политики Украины
Государственный научно-исследовательский
институт химических продуктов
М.Ф. Буллер
Г.В. Межевич
МЕТОДЫ ИСПЫТАНИЯ
УТИЛИЗИРУЕМЫХ
ПОРОХОВ
Шостка • 2005
ББК 35.63
Б90
УДК 662.1 + 662.2 + 662.3 + 543.5
В книге излагаются современные методы исследования состава
и стойкости утилизируемых порохов (дымных и пироксилиновых).
Книга рассчитана на работников научно-исследовательских
институтов, учебных заведений, заводских лабораторий.
alverworld.com
Рецензенты:
член-кор. НАН Украины Г.А. Ковтун
д-р хим. наук Ф.А. Чмиленко
канд. техн. наук А.М. Мальцев
Научный редактор
д-р техн. наук С.П. Фомин
Печатается по решению Пленума научно-технического совета
Государственного научно-исследовательского института химических
продуктов, протокол № 12 от 22.07.2005 года.
Буллер М.Ф., Межевич Г.В.
Б90 Методы испытания утилизируемых порохов/М.Ф. Буллер, Г.В. Межевич. —
К.: Изд-во ООО «ДНА», 2005. — 94 с.: ил. — Библиогр.: с. 88 — 94.
ISBN 966-8311-08-6
© М.Ф. Буллер, Г.В. Межевич, 200Л
© Оригинал-макет, художественное оформление.
Издательство ООО «ДИА», 2()0.‘>
ОГЛАВЛЕНИЕ
Предисловие 5
История начала пороходелия 7
Глава первая Физико-химический анализ компонентного
состава дымных порохов 13
1.1 Определение массовой доли нитрата калия 18
1.2 Определение массовой доли серы 21
1.3 Определение массовой доли влаги 22
1.4 Определение массовой доли углерода
в древесных углях 23
Глава вторая Хроматографический анализ
пироксилиновых порохов 27
2.1 Определение массовой доли дифениламина 36
2.2 Определение компонентного состава нитрозо-
и нитропроизводных дифениламина 39
2.3 Определение массовых долей дифениламина
и его N-нитрозо - и нитропроизводных 43
2.4 Определение массовой доли дифениламина
и симметричной диметилдифенилмочевины
(централит II) 47
2.5 Определение массовых долей этилового
спирта и этилового эфира 53
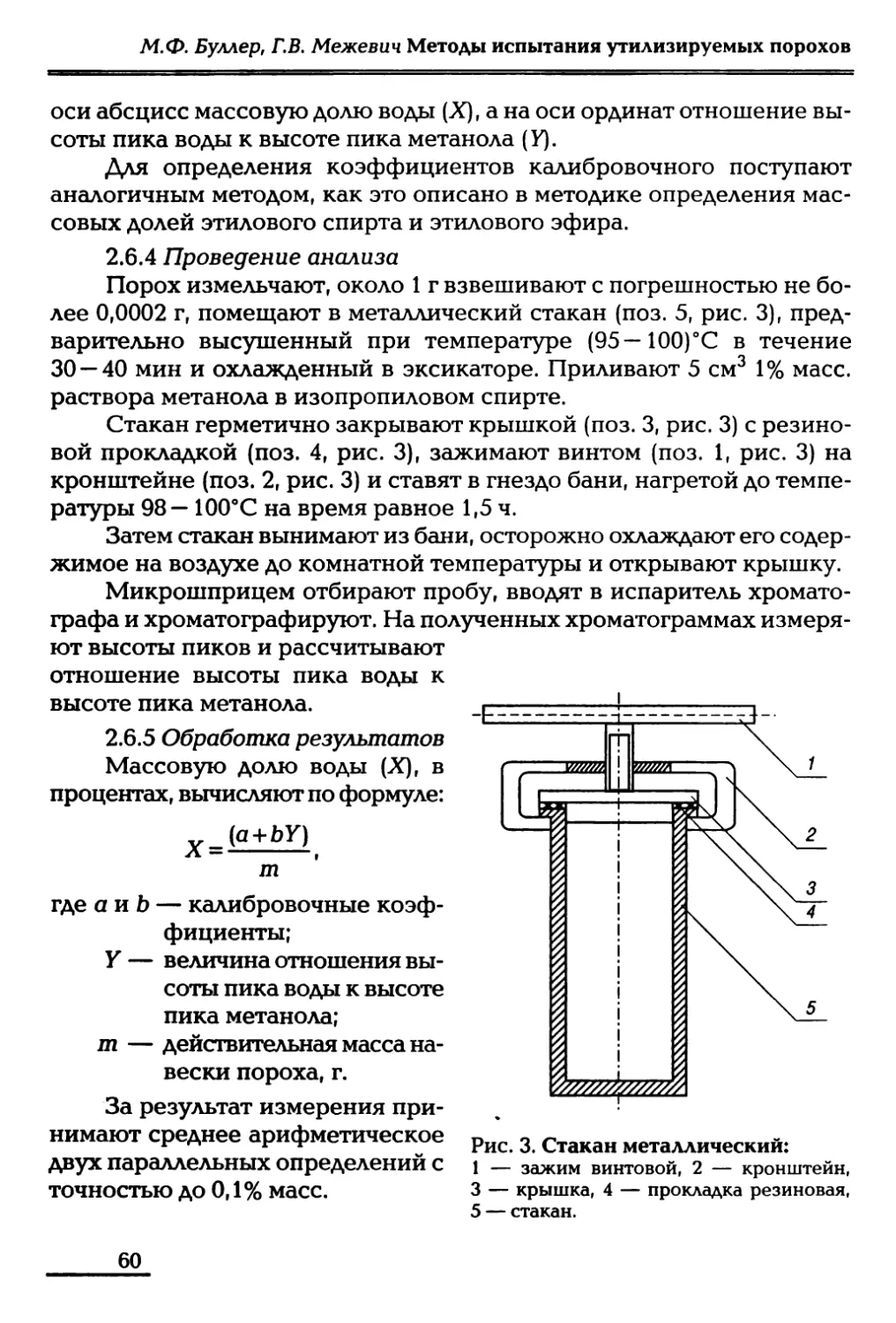
2.6 Определение массовой доли воды 58
Глава третья Методы определения стойкости порохов 61
3.1 Определение гигроскопичности дымных порохов 65
3.2 Определение химической стойкости нитратов
целлюлозы 66
3.3 Определение химической стойкости
пироксилиновых порохов 69
Глава четвертая Термические методы анализа порохов 73
4.1 Исследование кинетики разложения
при неизотермическом нагреве 74
4.2 Исследование кинетики разложения
при изотермическом нагреве 79
Литература 88
КОШЕЛЕВ
Вячеслав Владимирович
15 августа исполнилось бы 90 лет со дня рождения
прекрасного педагога, доктора химических наук, про¬
фессора кафедры "Химия и технология высокомолеку¬
лярных соединений” Ленинградского технологического
института им. Ленсовета Кошелева Вячеслава Владими¬
ровича.
Вячеслав Владимирович родился в г. Рошаль, Москов-
(1915-2000 ГГ.) ской обл. Руководством завода был направлен на учебу в
Ленинград. По окончанию ВУЗа был оставлен на даль¬
нейшую учебу в аспирантуре как талантливый инженер-
химик.
Прошел Вячеслав Владимирович и тяжелые дороги
Великой Отечественной Войны. Родина высоко оценила
его солдатский подвиг, он награжден многими орденами
и медалями.
После войны Вячеслав Владимирович продолжил ра¬
боту и учебу на кафедре, посвящая основное время под¬
готовке научных и инженерных кадров для оборонной
промышленности.
Многие наши сотрудники помнят его со студенческой
скамьи. Вячеслав Владимирович тесно сотрудничал с ру¬
ководством нашего института в сфере подготовки науч¬
ных кадров высшей квалификации, у многих он был на¬
учным руководителем при подготовке диссертации.
Под его руководством кандидатами наук стали: Ме-
зин Рудольф Михайлович, Кочергин Вячеслав Василье¬
вич, Жангужина Альфия Наильевна, Буллер Михаил
Фридрихович, Петлякова Лидия Дмитриевна, Бабкина
Татьяна Николаевна, Мараховская Александра Юрьевна.
Все, кто сталкивался с Вячеславом Владимировичем, с
теплотой вспоминают его кабинет на кафедре и квартиру
на "Червоного Казачества", где всегда царила атмосфера
душевной теплоты, моральной и материальной поддержки.
Для многих сотрудников НИИХП он по праву и приз¬
нанию стал Учителем.
ПРЕДИСЛОВИЕ
Посвящается памяти профессора
Вячеслава Владимировича Кошелева
В настоящее время при взрывных работах в карьерах
достаточно широко применяются утилизируемые
пороха как компоненты смесевых промышленных взрывчатых ве¬
ществ. Большую долю применяемых порохов составляют дымные и
пироксилиновые пороха.
Повторное применение порохов возможно в случае, если они
обладают соответствующими физико-химическими свойствами,
дающими возможность безопасно проводить взрывные работы в
карьерах.
Уровень соответствия утилизируемых порохов требуемым фи¬
зико-химическим свойствам определяется:
• химическим составом;
• химической стойкостью;
• чувствительностью к механическим воздействиям.
Для оценки физико-химических свойств разработаны различ¬
ные методики. Методики разрабатывались с момента появления по¬
рохов и постоянно совершенствуются.
Методики по определению химического состава дымных поро¬
хов описаны в монографии Н.Г. Шиллинга "Курс дымных порохов"
год издания 1940, М.: Оборониздат, 275 с, а методики по определению
состава и стойкости пироксилиновых порохов — в монографии
Г.К. Клименко "Методы испытания порохов", год издания 1941, М.:
Оборониздат, 299 с. С тех пор прошло достаточно много времени и
методики, описанные в данных монографиях, устарели и не соответ¬
ствуют современному уровню развитию аналитической химии и при¬
боростроения. Многие вышеописанные методики в настоящее время
не применяются в практике лабораторного анализа порохов.
5
Предисловие
Теория и методы испытания порохов на чувствительность к ме¬
ханическим нагрузкам хорошо освещены. Достаточно назвать моно¬
графии авторов Н.А. Холево "Чувствительность взрывчатых веществ
к удару", год издания 1974, М.: Машиностроение, 136 с. А.В. Дубовик
и В.К. Боболева "Чувствительность жидких взрывчатых систем к уда¬
ру", год издания 1978, М.: Наука, 232 с.
Указанное выше побудило нас осветить современные методы
анализа химического состава и стойкости артиллерийских порохов.
Кроме того, мы посчитали нужным изложить историю начала поро-
ходелия с кратким описанием технологии получения дымных и пи¬
роксилиновых порохов.
Книга обобщает практически весь экспериментальный материал
в области аналитической химии и стабильности дымных и пирокси¬
линовых порохов, наработанный за 30 лет в аналитическом отделе
ГосНИИ химических продуктов (г. Шостка). Главы 1,3,4 и история на¬
чала пороходелия написаны М.Ф.Буллером, глава 2 — Г.В.Межеви-
чем и М.Ф. Буллером.
Авторы весьма признательны член-корр. НАН Украины, доктору хи¬
мических наук, профессору Г.А. Ковтуну, доктору химических наук,
профессору Ф.А.Чмиленко, кандидату технических наук А.М. Маль-
чеву за рецензирование и высказанные при этом ценные советы и за¬
мечания. Авторы признательны также своей сотруднице О.А. Ново¬
бранец за компьютерный набор рукописи.
6
ИСТОРИЯ НАЧАЛА
ПОРОХОДЕЛИЯ
Т/Т3°бРетение пороха (дымного) вообще приписыва-
х Хется английскому химику Рожеру Бакону, жившему
в начале 13 века. Предполагается, что порох был известен азиатским
народам (китайцам) еще ранее. Достоверно известно, что дымный по¬
рох стал употребляться в военном деле в течение 14 века. В 1308 году
испанцы применили его при осаде Гибралтара, во Франции в 1338 году
появилось огнестрельное оружие, в 1378 году было употреблено боль¬
шее количество дымного пороха в войне венецианцев с генуэзцами [1].
На Руси порох стал известен позже. В Голицинской летописи
времен царствования Алексея Михайловича написано: “В лето 1389
год вывезли из Немец армата на Русь и огненную стрелу и оттого ча¬
су уразумели из них стреляти. В 1395 году в княжение Василия Дми¬
триевича сгорело в Москве несколько дворов от делания пороха".
При Петре 1 в 1715 году вокруг Петербурга были построены три
государственных (казенных) завода (Охтенский, Петербургский, Се-
строрецкий). По мнению Петра 1, казенные заводы должны были по¬
высить и усреднить качество изготовляемого пороха [2]. Позднее бы¬
ли построены Шосткинский (1765 год) и Казанский (1788) заводы.
До середины XIX века дымный порох оставался единственным
метательным и взрывчатым веществом. Дымный порох представляет
собой механическую смесь нитрата калия (селитры), древесного угля
и серы. Чаще всего для изготовления пороха (начиная с конца XIX ве¬
ка) брали 75% селитры, 10% серы и 15% угля.
Быстрое развитие горного дела в 17 веке потребовало усовершен¬
ствования ручного труда. Тирольский горняк Гаспар Бейндль в 1627 го¬
ду провел первые опыты взрыва в руднике [3]. Наступила эра примене¬
ния дымного пороха в качестве промышленного взрывчатого вещества.
К составу современных дымных порохов в большей степени под¬
ходит уравнение реакции горения, данное Г. Кастом.
74KN03 + 30S + 16С6Н20 = 56С02 + 14СО + ЗСН4 + 2H2S + 4Н2 + 35N2 +
+ 1 9К2СОэ + 7K2S04 + 8K2S203 + 2KCNS + (NH4)2C03 + C + S
7
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
Один кг такого пороха дает при взрыве (горении) 0,57 кг конденси¬
рованных веществ и 0,43 кг газообразных веществ. Коэффициент полез¬
ного действия, совершаемый дымным порохом при стрельбе или при
взрывах, достаточно мал—порядка 50% из-за малого количества образо¬
вавшихся газов. В воздух разлетается большое количество горячих твер¬
дых продуктов (копоть), образовавшийся дым негативно действовал на
воинов, мешал наблюдать за действием противника. Это подталкивало
ученых искать новые виды порохов и взрывчатых веществ, которые не
имели бы отрицательных свойств, присущих дымному пороху.
В 1845 году Хр. Фр. Шенбайн открыл пироксилин [4 — 6] (нитрат
целлюлозы), в 1847 году А. Собреро в Турине открыл нитроглицерин
[7]. Г. Каст дает следующие уравнения реакции горения соответ¬
ственно для
пироксилина 2С24Н29О9 (0N02) 11 = 24С02 + 24СО + 12Н20 + 17Н2 +11N2
и нитроглицерина 32C3H5(0N02)3 = 96С02 + 80Н2О + 6NO + 45N2 + 502
Из уравнений реакций видно, что продуктами горения пирокси-
линов и нитроглицеринов являются только газообразные вещества.
Эти открытия стали отправными точками по созданию следую¬
щего поколения метательных взрывчатых веществ — бездымных по¬
рохов. Понадобились десятки лет, чтобы эти открытия стали востре¬
бованы военной промышленностью. Первый шаг к этому сделал Аль¬
фред Нобель. В результате многочисленных опытов Нобель (1866 год)
создал практически применимое взрывчатое вещество на основе ни¬
троглицерина — гурдинамит [8]. В 1875 году Нобель открыл, что ни¬
траты целлюлозы могут набухать в нитроглицерине, образуя доста¬
точно устойчивую желатинообразную массу, обладающую мощными
взрывчатыми свойствами.
Нитрат целлюлозы состоит главным образом из тринитрата цел¬
люлозы — полимерного вещества, отвечающего общей формуле
[C6H702(0N02)3]n. Это сложный эфир азотной кислоты, который по
своему строению подобен тринитрату глицерина С3Н5(0Ы02)з, и э го
сходство является причиной растворимости твердого высокомолеку¬
лярного нитрата целлюлозы в жидком сложном эфире глицерин.! п
азотной кислоты. Приготовленное взрывчатое вещество этого tiiii.i,
названное Нобелем "гремучий желатин", содержало 92% тринигр.гт
глицерина и 8% нитрата целлюлозы. Это одно из наиболее мощных
бризантных веществ из известных взрывчатых веществ примети те л
для взрывания скал. При более низком соотношении тринитратп i ли
церина и нитрата целлюлозы получаются медленно сгорающие смгси,
пригодные для заполнения снарядов.
8
История начала пороходелия
Для оружия с нарезными стволами необходимо очень медленное
горение пороха. Первоначальный толчок снаряда не должен сопро¬
вождаться возникновением чрезмерного давления в почти гермети¬
чески замкнутом пространстве, где заключена свинцовая пуля или
снаряд, плотно пригнанные в винтообразном канале ствола оружия.
Все попытки применять энергию нитратов целлюлозы и глицерина
для стрельбы не имели успеха ввиду их разрушительного действия.
Все усилия Ленка (1847 г.), Отто (1855 г.), и многих других исследователей,
пытавшихся уплотнить рыхлый пироксилин прессованием, заполнени¬
ем пустот склеивающими веществами, не дали желаемых результатов.
Получалось так, что нитраты целлюлозы и глицерина к данному
моменту не имели перспектив в качестве метательных средств, поэто¬
му исследования по совершенствованию составов на основе дымного
пороха не прекращались.
Несомненным успехом считалась разработка Г. Шульцем (1865 г.)
охотничего пороха [9] , изготовляемого путем нитрования древесины
(или слабообожженной древесины) и пропитывания последней кали¬
евой или бариевой селитрами. Этот порох, до появления первого военно¬
го пироксилинового пороха (1884 г.), был единственным применением
нитратов целлюлозы в качестве метательного средства (пороха). Важен
факт, что в этом порохе нитрат целлюлозы был частично желатинизи-
рован (пластифицирован) с поверхности спиртоэфирной смесью.
С этого момента оставалось сделать всего один шаг к созданию
военного пироксилинового пороха на основе полностью желатинизи-
рованного нитрата целлюлозы. Хотя Фолькману в Вене (1871 г.) были
известны основные принципы производства пироксилинового поро¬
ха (полная желатинизация нитрата целлюлозы), как это мы понимаем
на современном этапе, однако с применением нитратов целлюлозы в
желатинизированном виде для военных целей произошла задержка.
Опыт военных кампаний до 1871 г. потребовал повышения мощ¬
ности орудий, эту задачу надо было решить, по логике, за счет созда¬
ния технологии пироксилинового пороха. Но, увы, пошли по пути со¬
вершенствования состава дымного пороха. Эта задача на недолгое
время была решена путем прибавки к калиевой селитре невзрывча¬
тых горючих веществ. На заводах Круппа с 1882 года выпускался
призматический бурый порох, который успешно использовался как в
орудиях крупного, так и мелкого калибра. Для ручных ружей все-та-
ки был необходим порох, который полностью переходил при горении
в газообразные продукты.
Перед тем как перейти к следующему этапу описания эволюции
пороха, необходимо отметить, что в этот период назрела объективная
9
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
необходимость изучения процессов горения порохов во взаимосвязи
с движением снаряда на траектории [10]. И эта задача начала успеш¬
но решаться совместными усилиями ученых: химиков, металлургов,
математиков. Становится ясно, что альтернативы порохам на основе
нитратов целлюлозы и глицерина уже не может быть.
Дуттенгофером (1883 г.) по прототипу пороха Шульце был разра¬
ботан порох на основе нитрованной слабообожженной древесины,
пластифицированной уксусным эфиром. Полученная желатинооб¬
разная масса подсушивалась, пропускалась через вальцы и зерни¬
лась, как дымный порох. Такой порох с 1884 г испытывался в войсках
и показал неплохие результаты.
Вьелем (1884— 1885 г.) при проведении лабораторных работ [11]
было показано, что путем желатинизации можно регулировать ско¬
рость горения пороха. Результатом работы Вьеля было создание лен¬
точного пороха для полевых орудий. В качестве пластификатора
Вьель использовал смесь спирто- эфирного растворителя с добавкой
амилового спирта. Использование амилового спирта было недоста¬
точно оправдано, так как в готовом порохе его содержание доходило
до 5% масс. Это было причиной нестабильности свойств пороха из-за
нежелательной летучести пластификатора (амиловый спирт). Более
ранние образцы Вьель готовил с помощью уксусного эфира или ами¬
лацетата. Оставался один шаг для создания пироксилиновых (одно¬
основных) и баллиститных (двухосновных) порохов, которые являют¬
ся прообразами современных порохов.
Этот шаг сделали великий русский ученый Д.И. Менделеев (1890 г),
создавший пироколлодий [12], и великий изобретатель Альфред Но¬
бель (1889 г), создавший "кордит" [8]. Великий ученый предложилже-
латинизировать нитраты целлюлозы с содержанием азота 12,2 12,5%
смесью этилового спирта и этилового эфира, великий изобретатель
использовал летучий растворитель для получения кордита. Основная
заслуга Д.И. Менделеева в том, что он создал русскую научную шко¬
лу пороходелия, при нем начались фундаментальные исследования в
этой отрасли. Менделеевым и его учениками предложены технологии
обезвоживания пироксилина этиловым спиртом [13], флегматизации
пороха, правило составления нитрующих смесей для целлюлозы.
Первый русский пироксилиновый порох был получен в 1890 году
на Охтенском пороховом заводе для трехлинейной винтовки Мосина.
К началу XX века в России уже делались пироксилиновые пороха и
для пушек. Первая мировая война показала, что Россия еще не смогла
полностью обеспечить свою армию собственными порохами, более
двух третьих потребности в порохах Россия покрывала за счет ино¬
земных поставок (в основном из США).
10
История начала пороходелия
В 1889 г. изобретение А. Нобеля было взято на вооружение в Ан¬
глии, изготавливаемый бездымный порох — кордит, содержал 30%
тринитрата глицерина, 65% нитрата целлюлозы и 5% минерального
геля (неочищенный вазелин). При таком высоком содержании нитра¬
та целлюлозы желатинизирование не может быть достигнуто под
влиянием одного тринитрата глицерина и для образования однород¬
ного геля добавляли ацетон, который является растворителем для
обоих компонентов. Паста, смоченная растворителем, замешивалась
в смесителе в густую массу, которая затем выдавливалась в виде шнура
(шнур по-английски "cord" — отсюда название “кордит") различных
размеров и с различным количеством “каналов". Шнур разрезали на
куски и тщательно сушили для удаления ацетона, большую часть ко¬
торого регенерировали.
И в заключении вкратце опишем технологические схемы изгото¬
вления дымных и пироксилиновых порохов.
Основными стадиями технологического процесса изготовления
дымных порохов [14] являются:
1 Подготовка (изготовление) компонентов
1.1 очистка и чашуирование серы
1.2 изготовление древесных углей, путем обжига древесины
требуемой породы
1.3 нитрат калия (калиевая селитра) специальной подготовки на
заводах не проходит
2 Измельчение компонентов
Измельчение компонентов возможно по различным вариантам:
— измельчение компонентов по отдельности
— измельчение древесного угля и серы совместно
Последний вариант наиболее распространен.
3 Приготовление тройной смеси
Смешение компонентов нужного соотношения с дополнительным
измельчением до необходимых размеров частиц с отбраковкой ча¬
стиц больших по размеру.
4 Уплотнение состава (получение пороховой лепешки)
Уплотнение состава проводят на прессах при температуре 95—110°С.
5 Получение порохового зерна
Пороховое зерно получают дроблением уплотненного тройного
состава, пороховую пыль отделяют от зерна.
6 Придание пороховому зерну потребительских свойств
Требуемые потребительские свойства пороха достигаются опера¬
циями полировки, чистки и сортировки.
И
МФ. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
7 Усреднение и укупорка
Придание однообразия свойствам пороха проводится смешением
малых партий в большую общую партию. Укупорка способствует
сохранности свойств пороха.
Основными стадиями технологического процесса изготовления
пироксилиновых порохов являются [15]:
1 Подготовка (изготовление) компонентов
1.1 Изготовление стабилизированного пироксилина нитрацией
целлюлозы
1.2 Изготовление этилового эфира
Остальные компоненты поступают на завод в готовом виде.
2 Обезвоживание пироксилина
Обезвоживание пироксилина проводят вытеснением воды этило¬
вым спиртом.
3 Приготовление пороховой массы
Приготовление пластичной пороховой массы проводят обработкой
пироксилина спирто-эфирным растворителем. На этой стадии в
пороховую массу добавляют стабилизатор химической стойкости.
4 Прессование пороховой массы и удаление растворителей
Прессование пороховой массы проводят для окончательной ее
пластификации и создания "влажных" пороховых заготовок —
шнуров, лент. Пороховые заготовки подвергают провяливанию
для удаления летучих растворителей. Спирт и эфир улавливают и
используют повторно.
5 Резка заготовок на пороховые элементы и их разымка
Резка предусматривает получение из провяленных заготовок порохо¬
вых элементов (зерна, трубки, пластины). Разымка проводится с це¬
лью отбраковки пороховых элементов по геометрическим размерам.
6 Предварительная сушка, вымочка, сушка, увлажнение и мешка пороха.
Предсушка проводится для того, чтобы понизить содержание раство¬
рителя в порохе с 15 — 20% после предыдущей операции до 9 — 10%.
В процессе вымочки (выдержки пороха в водной среде) содержа¬
ние растворителей достигает нормы, а содержание влаги увеличи¬
вается. Избыток влаги удаляется из пороха сушкой, окончательная
корректировка содержания спирта, эфира и воды в порохе прово¬
дится увлажнением.
7 Усреднение и укупорка
Операция проводится для придания и сохранения однообразия
свойствам общей партии пороха.
При получении специальных марок пироксилиновых порохов
могут быть предусмотрены дополнительные операции — флегма гпза-
ция, графитовка и др.
12
ГЛАВА
ФИЗИКО-ХИМИЧЕСКИЙ
АНАЛИЗ КОМПОНЕНТНОГО
СОСТАВА ДЫМНЫХ
ПОРОХОВ
Еще в 1844 году в сочинениях "О порохе " полковни¬
ка артиллерии Тиммерганса [16] говорится о
необходимости физико-химического анализа дымных порохов. В
книге подробно описаны известные в то время методы анализа хими¬
ческого состава дымных порохов.
Согласно действующей в настоящее время нормативной докумен¬
тации определение массовой доли нитрата калия проводится следую¬
щими методами: химическим, весовым, объемным и электрохимиче¬
ским (кондуктометрическим). Весовой метод, получивший благодаря
своей простоте наибольшее распространение, обладает существен¬
ными недостатками. При обработке образца пороха горячей водой с
последующим отделением на фильтре твердого остатка, в раствор пе¬
реходит не только нитрат калия, но и водорастворимые вещества дре¬
весного угля, а также мельчайшие твердые частицы серы и угля. Эти
дополнительные вещества становятся источником ошибки, завышая
результаты определения массовой доли нитрата калия весовым мето¬
дом. Повысить точность весового метода можно путем учета количе¬
ства водорастворимых веществ древесных углей, для этого при анализе
вводится холостой опыт — определение количества водораствори¬
мых веществ древесного угля. Методы, предусмотренные норматив¬
ной документацией, довольно продолжительны во времени и трудо¬
емки. Много времени затрачивается на пропускание раствора через
катионит при применении объемного метода, на выпаривание и суш¬
ку фильтрата при применении весового метода и на построение кали¬
бровочного графика при применении кондуктометрического метода.
13
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохом
Ускорить определение массовой доли нитрата калия в дымных
порохах весовым методом можно, если выпаривать и сушить не весь
раствор, как это предусмотрено нормативной документацией, а его
аликвотную часть, с последующим пересчетом на весь объем ра¬
створа.
V. Ohman и G. Laurem [17] отработали методику интерферомет¬
рического определения массовой доли нитрата калия в дымном по¬
рохе, которая позволяет сократить время анализа до 30 минут. По
этому методу навеску пороха растворяют в дистиллированной воде,
затем кипятят раствор в течение нескольких минут. Твердую фазу
(сера + древесный уголь) отделяют от раствора путем фильтрова¬
ния, промывают фильтр горячей водой. Собранный фильтрат охлаж¬
дают до + 20°С и производят измерения на интерферометре. По ка¬
либровочному графику определяют содержание нитрата калия. Для
построения калибровочного графика используют чистые растворы
нитрата калия.
Авторами [18, 19] разработана кондуктометрическая методика,
которая позволяет определять содержание нитрата калия по величи¬
не электропроводности. Отличие данного метода [18] от интефероме-
трического заключается в том, что для построения калибровочного
графика используются не растворы нитрата калия, а водные раст¬
воры эталонных образцов дымного пороха [20]. Предложенный спо¬
соб обладает достаточной точностью и хорошей воспроизводи¬
мостью.
Авторами [21] предложен способ определения массовой доли ни¬
трата калия методом УФ-спектроскопии. Метод основан на экстрак¬
ции нитрата калия горячей дистиллированной водой из дымного по¬
роха и измерении оптической плотности полученного экстракта в
УФ-области спектра. В предложенном способе практически устраня¬
ется влияние твердых взвешенных частиц в результате значительно¬
го разбавления аликвотной части фильтрата при фотометрировании
в УФ области спектра. Влияние же водорастворимых компонентов
древесного угля исключается выбором интервала спектрофотометри-
рования в диапазоне длин волн 195 — 210 нм. Данный способ отлича¬
ется высокой точностью и непродолжительностью анализа, что по¬
зволяет рекомендовать его для широкого использования.
Среди методов определения содержания массовой доли нитрата
калия, которые применяются при анализе руд и катализаторов, име¬
ются и такие, которые с успехом могли бы применяться и при анали¬
зе дымных порохов.
14
Глава первая. Физико-химический анализ компонентного состава дымных порохов
Нитрат калия в воде диссоциирует на ионы калия и нитрат-ионы,
поэтому концентрацию нитрата калия можно определить как по ко¬
личеству ионов калия, так и по количеству нитрат-ионов.
К наиболее общим методам определения концентрации ионов
калия относятся методы фотометрии пламени [22 — 24], радиометриче¬
ский [25]. Часто применяются и ионоселективные электроды [26 — 28].
В.Н. Басов [29] использовал тетрофенилборат натрия при ампероме¬
трическом определении ионов калия.
При определении нитрат-ионов используют, в основном, коло¬
риметрические [30], полярографические [31] и потенциометрические
[32 — 33] методы.
Полярографические и колориметрические методы дают значи¬
тельные относительные ошибки (15—18%). Применение колориме¬
трических методов при анализе дымного пороха затруднено в связи с
тем, что фильтрат дымных порохов некоторых марок имеет желтова¬
тую или коричневую окраску. Потенциометрическое определение
нитрат-ионов основано на титровании в среде 80%-ной серной кислоты
раствором хлорида титана или раствором ацетанилида [34]. В качестве
индикаторного электрода используется платиновый электрод. Если
нитраты определяют в присутствии окислителей и окрашенных ве¬
ществ, то средой при потенциометрическом титровании является
концентрированная фосфорная кислота с удельной плотностью
1,85— 1,94 г/см3 [35]. Потенциометрические методы непродолжитель¬
ны, относительная ошибка составляет до 0,9%.
R. Сох [36] рекомендовал хемилюминисцентный метод опреде¬
ления нитрат-ионов, основанный на восстановлении N03 до NO и ре¬
акции NO с Оэ, сопровождающейся свечением [37]. Для определения
нитрат-ионов применяют и ионселективные электроды [22].
Ю.И. Беляев и др. [38] предложили кинетический способ, в кото¬
ром о концентрации электролита в определенном объеме судят по
времени нагревания анализируемой пробы до момента кипения.
Определение массовой доли серы в дымных порохах в настоя¬
щее время по действующей нормативной документации проводят
сульфитным методом.
Данный метод широко распространен в лабораторной практике
и используется при определении свободной серы в различных при¬
родных минералах [39].
Для сокращения времени анализа и его упрощения предложено
[40] проводить обработку дымного пороха водным раствором серни¬
стокислого натрия в присутствии неионогенного поверхностно-ак-
15
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохом
тивного вещества на основе оксиэтилированных высших жирных
спиртов или оксиэтилированных алкилфенолов в количестве 0,2 — 0,5%
масс, в водном растворе. Это позволяет на 10 —15% уменьшить время
проведения анализа.
Известен так же весовой способ [41, 42] определения массовой
доли серы экстракцией ее из дымного пороха сероуглеродом или бен¬
золом. Содержание серы определяют по разности массы навески дым¬
ного пороха до и после экстракции. Данный метод продолжителен и при¬
менение его усложнено таким токсичным растворителем как бензол.
Е. Camera и D. Pravisani [43] определяли содержание серы в дымных
порохах газохроматографически после извлечения серы экстракцией
сероуглеродом. Полученный экстракт хроматографировали на колон¬
ке, заполненной цеолитом, предварительно обработанным силико¬
ном Е301. Газохроматографический анализ является несколько менее
точным, чем весовой анализ, выполненный только после экстракции,
но обладает преимуществом по времени.
Предлагались и методы, основанные на окислении серы хлором,
перманганатом калия или перекисью водорода, и титровании хлоридом
бария образующейся серной кислоты. Менее сложным и непродол¬
жительным является метод V. Ohmana. Метод основан на сжигании
остатка (после удаления нитрата калия), дымного пороха в калориме¬
трической бомбе, заполненной кислородом. Образовавшуюся серную
кислоту оттитровывают щелочью в присутствии фенолфталеина.
И. Кубант [44] предложил титрометрический способ определе¬
ния массовой доли серы в углях и других материалах, основанный на
предварительном извлечении серы бензолом, взаимодействии серы с
сульфитом натрия в горячей диоксановой среде и иодометрическом
определении образовавшегося иона S203“2.
В настоящее время общепринято, что массовые доли нитрата калия
и серы в дымных порохах определяют прямыми способами, а массовую
долю древесного угля рассчитывают по разности с учетом влажности.
Необходимо остановиться и на анализе древесных углей, ис¬
пользуемых в производстве дымного пороха. Данный компонент в
большой степени определяет качество дымных порохов, поэтому дре¬
весный уголь всегда изготавливался на заводах, производящих дым¬
ные пороха. Одним из основных физико-химических показателей
древесных углей является содержание углерода или степень обжига.
Длительное время массовую долю углерода в древесных углях
определяли методом сжигания по ОСТ 84-2281-86 с абсорбционно-ве¬
совым окончанием. Метод трудоемок, продолжителен и не отвечает
16
Глава первая. Физико-химический анализ компонентного состава дымных порохов
потребностям производства и современному уровню развития анали¬
тической химии.
Этот способ анализа содержания углерода в древесных углях
был усовершенствован по принципу автоматического кулонометри¬
ческого титрования в растворе продуктов сжигания [45]. Анализ про¬
изводится при помощи экспресс-анализатора на углерод, который
выпускается серийно. Величина относительной погрешности при
определении массовой доли углерода в древесных углях экспресс-ме¬
тодом изменяется для различных образцов в пределах 0,2-1,3%.
Предложенный метод существенно сокращает время проведения
анализа, в среднем на один анализ с 6,5 до 0,6 часа.
В некоторых случаях при экспресс-анализе древесного угля
можно применить и методику, описанную в [46]. Метод основан на
фотометрировании водно-щелочного экстракта древесного угля в УФ
области спектра. Интенсивность окраски водно-щелочных экстрак¬
тов зависит от содержания углерода.
И, наконец, остановимся на схеме анализа компонентного соста¬
ва дымных порохов.
Исходя из того, что технологический процесс производства дым¬
ного пороха построен таким образом, что сначала готовится серо¬
угольная смесь (двойная смесь), а затем тройная смесь, путем добав¬
ления определенного количества нитрата калия, то в производстве
дымного пороха предусмотрено проведение анализов как двойной
смеси (пофазный контроль), так и готового пороха.
Целесообразно упростить аналитический контроль в производ¬
стве дымного пороха следующим образом. При анализе дымного по¬
роха производить только определение массовой доли нитрата калия,
а содержание серы и угля выполнять расчетным путем, учитывая ре¬
зультаты анализа компонентного состава двойной смеси. Содержа¬
ние серы (W) в дымных порохах в% масс, определять по формуле:
W = (100-Х) У/100,
где X — содержание нитрата калия, % масс.;
Y — содержание серы в двойной смеси, % масс.
Содержание древесного угля (G) в % масс, определять по формуле:
G = 100 —X— W.
Введение такой схемы анализа позволит сократить затраты на
проведение анализа дымного пороха и снизить себестоимость самого
пороха.
17
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
1.1 Определение массовой доли нитрата калия
1.1.1 Метод основан на использовании катионита, способного
обменивать свои подвижные положительные ионы на катионы, нахо¬
дящиеся в растворе
R—Na+K+ = R—K+Na+,
где R — макромолекула, к которой присоединена активная группа.
1.1.2 Аппаратура, посуда, материалы и реактивы
• весы аналитические с погрешностью взвешивания не более 0,0002 г;
• стаканы стеклянные вместимостью 50 мл;
• воронки стеклянные лабораторные;
• воронки делительные цилиндрические типа VIII вместимостью 250
и 1000 мл;
• колбы мерные вместимостью 200 мл;
• колбы конические вместимостью 500 мл;
• бюретки вместимостью 50 мл;
• пипетки вместимостью 50 мл;
• палочки стеклянные;
• фильтры бумажные диаметром 110 мм или бумага фильтровальная;
• штатив;
• вата стеклянная;
• натр едкий, 5%-ный и 0,1 М водные растворы;
• натрий хлористый, насыщенный водный раствор;
• кислота соляная 5, 10, 15%-ные водные растворы;
• кислота серная х.ч.;
• катионит марки КУ-2-8;
• дифениламин, 1%-ный раствор в концентрированной серной ки¬
слоте;
• аммоний роданистый, 8%-ный водный раствор;
• метиловый оранжевый, 0,1%-ный водный раствор;
• индикатор смешанный (две части 0,2%-ного спиртового раствора
метиленового красного и часть 0,05%-ного водного раствора мети¬
ленового синего);
• метиловый красный, 0,2%-ный спиртовой раствор;
• нитрат калия, 0,1 М водный раствор;
• вода дистиллированная.
1.1.3 Подготовка к анализу
1.1.3.1 Подготовка катионита
Катионит заливают в стакане насыщенным водным раствором
натрия хлористого так, чтобы над поверхностью катионита был слой
18
Глава первая. Физико-химический анализ компонентного состава дымных порохов
раствора не менее 30 мм, и оставляют набухать на 6 —12 ч. Набухший
катионит помещают в делительную воронку, заливают его полностью
5%-ным раствором натра едкого и выдерживают в течение 3 —4 ч.
После чего щелочь из воронки удаляют со скоростью 10 — 15 мл/мин и
к катиониту приливают свежую порцию щелочи. Такую обработку
катионита проводят до тех пор, пока не исчезнет окраска выходящего
раствора. Расход щелочи при этом должен составлять 7—10 объемов
на один объем катионита.
После щелочной обработки катионит промывают 10 объемами
дистиллированной воды (кран воронки полностью открыт). Далее че¬
рез катионит пропускают со скоростью 10—15 мл/мин кислоту соля¬
ную : вначале 5 объемов 5%-ного раствора, затем 5 объемов 10%-ного
раствора и затем 15%-ный раствор до исчезновения в вытекающем
растворе ионов железа (проба с аммонием роданистым не должна да¬
вать розового окрашивания).
После кислотной обработки катионит промывают дистиллиро¬
ванной водой до нейтральной реакции по индикатору метиловому
оранжевому, при этом скорость пропускания воды максимально воз¬
можная. Катионит хранится под слоем дистиллированной воды высо¬
той не менее 10 мм.
1.1.3.2 Заполнение ионообменной колонки
В качестве ионообменной колонки используют цилиндрическую
делительную воронку вместимостью 250 мл, на дно которой помещают
вату для фиксации нижнего слоя катионита. Ионообменную колонку
устанавливают с помощью штатива в вертикальном положении.
Кран закрывают, колонку наполняют на три четверти ее объема
дистиллированной водой, и набухший катионит переносят в колонку,
одновременно открывая кран. Когда слой катионита достигнет высоты
10—12 см, кран закрывают.
Перед пропусканием анализируемого раствора через колонку
зерна катионита должны быть отмыты дистиллированной водой до
нейтральной реакции по индикатору метиловому оранжевому, при
этом скорость вытекающего раствора из колонки должна быть мак¬
симально возможной.
После анализа 50 — 55 проб катионит регенерируют.
1.1.3.3 Регенерация зерен катионита в колонке
Отработанный катионит можно многократно регенерировать.
Для этого через колонку пропускают 600 — 800 мл 10 — 15%-ного водного
раствора кислоты соляной со скоростью 10—12 мл/мин. После чего
19
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
катионит с максимальной скоростью промывают дистиллированной
водой до нейтральной реакции по метиловому оранжевому.
Для проверки полноты регенерации катионита через колонку про¬
пускают 20 мл 0,1 М водного раствора нитрата калия и в собранном ра¬
створе (элюате) количественно определяют содержание азотной кисло¬
ты. Если катионит полностью регенерирован, то в полученном элюате
будет оттитровано 20 ±0,05 мл точно 0,1 М водного раствора азотной
кислоты. При получении заниженного результата операцию регене¬
рации повторяют вновь, после чего проверяют полноту регенерации.
1.1.4 Проведение анализа
Около 1 г пороха, измельченного до порошкообразного состояния,
взвешивают с погрешностью не более 0,0002 г, помещают в стакан и
приливают 30 — 40 мл воды, нагретой до 60 — 70°С.
Раствор в стакане перемешивают стеклянной палочкой и филь¬
труют через бумажный фильтр в мерную колбу; при этом осадок из
стакана переносят на фильтр. Осадок на фильтре промывают горячей
водой до полного удаления калия нитрата. Последняя порция фильт¬
рата должна давать отрицательную реакцию на нитрат-ион с раство¬
ром дифениламина.
Содержимое колбы охлаждают, доводят дистиллированной водой
до метки и взбалтывают. Из мерной колбы пипеткой отбирают 50 мл ра¬
створа и пропускают его со скоростью 10—15 мл/мин через колонку с
катионитом, собирая раствор в коническую колбу. Для полного перево¬
да в элюат азотной кислоты, выделившейся в процессе обмена, колонку
промывают в ту же колбу 150 — 200 мл дистиллированной воды до ней¬
тральной реакции по метиловому оранжевому. Первую порцию дистил¬
лированной воды (примерно 30 мл) пропускают через колонку со ско¬
ростью 10—15 мл/мин, последующие — с максимальной скоростью.
Элюат титруют раствором едкого натра в присутствии 5 — 6 ка¬
пель смешанного индикатора или 3 — 4 капель метилового красного до
перехода окраски раствора соответственно из малиновой в светло-зе¬
леную или розовой в желтую.
1.1.5 Обработка результатов
Содержание нитрата калия (X) в % масс, вычисляют по формуле:
Y_yo,oioiiyriooioo
V2 m(100-W)
где V — объем точно 0,1 М раствора едкого натра, израсходованный
на титрование, в мл;
20
Глава первая. Физико-химический анализ компонентного состава дымных порохов
V{ — объем мерной колбы в мл;
V2 — объем пипетки в мл;
0,01011 — количество калия нитрата, соответствующее 1 мл точно 0,1 М
раствора натра едкого, в г;
т — навеска пороха в г;
W — влажность пороха в % масс.
Производят два параллельных определения, по результатам ко¬
торых вычисляют среднее арифметическое с погрешностью не более
0,1%. Расхождение между результатами параллельных определений
не должно превышать 0,5% масс.
1.2 Определение массовой доли серы
1.2.1 Метод основан на взаимодействии серы с сернистокислым
натрием при кипячении в водной среде (S + Na2 S03 —> Na2 S203) и от-
титровывании образовавшегося гипосульфита водным раствором
йода (2Na2S203 12 —^ ^ 2Na IЧ- Na2S^O0).
1.2.2 Аппаратура, посуда, материалы и реактивы:
• весы аналитические с погрешностью взвешивания не более 0,0002 г;
• воронки стеклянные лабораторные диаметром 56±5 мм;
• колбы конические вместимостью 100 и 250 мл;
• холодильник стеклянный, или стеклянные трубки;
• цилиндр измерительный вместимостью 50 мл;
• бюретки вместимостью 50 мл;
• баня песчаная;
• фильтр бумажный диаметром 110 мм или бумага фильтровальная;
• натрий сернокислый безводный;
• формалин технический;
• кислота уксусная, 10%-ный водный раствор;
• иод, 0,05 М водный раствор (коэффициент устанавливают по 0,05 М
водному раствору тиосульфата натрия в присутствии 2 мл 5%-ного
водного раствора крахмала);
• фенолфталеин, 1%-ный спиртовой раствор;
• спирт этиловый;
• крахмал водорастворимый;
• вода дистиллированная.
1.2.3 Проведение анализа
Около 0,5 г пороха, измельченного до порошкообразного состоя¬
ния, взвешивают с погрешностью не более 0,0002 г, помещают в ко¬
ническую колбу вместимостью 100 мл, добавляют 1 — 2 мл этилового
спирта, 1,0 —1,5 г безводного сернистокислого натрия и 50 мл дистил-
21
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых иорохов
лированной воды. Колбу соединяют с обратным холодильником и ки¬
пятят раствор 10 мин на песчаной бане. При кипячении рекомендует¬
ся несколько раз взбалтывать содержимое колбы.
По окончании кипячения раствор фильтруют через бумажный
фильтр в коническую колбу вместимостью 250 мл. Нерастворимый
остаток на фильтре промывают 150 —200 мл дистиллированной воды,
нагретой до 60 — 70°С, до полного удаления тиосульфат-иона. Послед¬
няя порция фильтрата должна давать посинение от одной капли иода
в присутствии 0,5 мл водного раствора крахмала. После этого к
фильтрату добавляют 5 мл формалина для связывания непрореагиро¬
вавшего натрия сернистокислого. Выделившийся при этом натр ед¬
кий нейтрализуют в присутствии фенолфталеина раствором уксус¬
ной кислоты до кислой реакции.
После нейтрализации серноватистокислый натрий оттитровыва-
ют раствором иода в присутствии 2 мл раствора крахмала до синего
окрашивания.
1.2.4 Обработка результатов
Содержание серы (X) в % масс, вычисляют по формуле:
у-доозг-юо-юо
jn(lOO-W)
где V — объем точно 0,05 М водного раствора иода, израсходован¬
ный на титрование, в мл;
0,0032 — количество серы, соответствующее 1 мл точно 0,05 М ра¬
створа иода, в г;
т — навеска пороха в г;
W — влажность пороха в% масс.
Производят два параллельных определения, по результатам ко¬
торых вычисляют среднее арифметическое с погрешностью не более
0,1%. Расхождение между результатами параллельных определений
не должно превышать 0,2%.
1.3 Метод определения массовой доли влаги
1.3.1 Метод основан на определении потери массы пороха при сушке
1.3.2 Аппаратура и посуда
• весы аналитические с погрешностью взвешивания не более 0,0002 г;
• шкаф сушильный;
• стаканчики для взвешивания (бюксы) типа СН, с номинальным ди¬
аметром 58 мм;
• эксикатор с хлористым кальцием.
22
Глава первая. Физико-химический анализ компонентного состава дымных порохов
1.3..3 Проведение анализа
Около 10 г пороха помещают в сухой, предварительно взвешен¬
ный бюкс, и взвешивают с погрешностью не более 0,0002 г.
Бюкс с порохом помещают в сушильный шкаф, снимают с него
крышку и сушат при температуре 100±2°С в течение 30 мин. Крупно¬
зернистый порох сушат при той же температуре в течение 60 мин.
Затем (не вынимая из сушильного шкафа) бюкс закрывают крыш¬
кой, охлаждают в эксикаторе и взвешивают.
1.3.4 Обработка результатов
Содержание влаги (X) в % масс, вычисляют по формуле:
х = т\ ~ т2
т
•100,
где т{ — масса бюкса с порохом до сушки в г;
Л72 — масса бюкса с порохом после сушки в г;
т — навеска пороха в г.
Производят два параллельных определения, по результатам ко¬
торых вычисляют среднее арифметическое с погрешностью не более
0,1%. Расхождение между результатами параллельных определений
не должно превышать 0,1%.
1.4 Определение массовой доли углерода в древесных углях
1.4.1 Метод основан на сжигании пробы древесного угля в токе
кислорода. Образовавшийся при этом диоксид углерода поглощается
в электролитической ячейке раствором, содержащим хлористый
стронций и хлористый калий, создавая кислую среду:
со2+н2о = н2со3
H2C03+SrCl2 = SrC03l + 2НС1
НС1 = Н+ + СГ
При этом изменяется ЭДС электродной системы и начинается
прохождение тока через ячейку, сопровождающееся электрохимиче¬
ской реакцией. Во время протекания генераторного тока катионы во¬
дорода восстанавливаются:
2Н+ +2е~ = Н2Т
На цинковом аноде образуются ионы цинка, взаимодействую¬
щие со вспомогательным электролитом, состоящим из хлористого ка¬
лия и железистосинеродистого калия. Выделившаяся комплексная
соль цинка выпадает в осадок:
23
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
Zn — 2е~ = Zn2+
3Zn С12 + 2К4 Fe(CN)6 = K2Zn3 [Fe(CN)6]2l + 6KC1
В ходе электрохимической реакции восстанавливается номи¬
нальное значение pH раствора в электролитической ячейке.
1.4.2 Аппаратура, посуда, материалы и реактивы
• экспресс-анализатор на углерод типа АН-7529;
• весы лабораторные общего назначения 2 класса точности с наи¬
большим пределом взвешивания 200 г;
• меры массы общего назначения Г-2-210;
• шкаф сушильный, обеспечивающий температуру нагрева (100±5)°С;
• кислород газообразный;
• печь муфельная, обеспечивающая температуру нагрева 900-ь1000 °С;
• лодочки фарфоровые типа АС-2;
• эксикатор без крана;
• стаканчики для взвешивания типа СН;
• кальций хлористый обезвоженный;
• алюминия оксид;
• углерод технический;
• калий хлористый;
• калий железистосинеродистый 3-водный;
• кислота борная;
• стронций хлористый 6-водный;
• вода дистиллированная.
• шпатель номер 1;
1.4.3 Подготовка к выполнению измерений
1.4.3.1 Пробы древесного угля, отобранные для анализа, сушат в
сушильном шкафу в течение 1 ч при температуре (100±5)°С.
1.4.3.2 Лодочки для сжигания помещают в муфельную печь и
прокаливают в течение б ч при температуре 900 — 950°С.
1.4.3.3 Оксид алюминия прокаливают в тех же условиях, что и ло¬
дочки. Прокаленные лодочки и оксид алюминия хранят в эксикаторе.
1.4.3.4 Подготовка контрольных проб: помещают в фарфоровую
лодочку 0,01 г технического углерода с известной массовой долей
углерода и сверху насыпают равномерный слой 0,1 г инертного веще¬
ства (оксида алюминия).
1.4.3.5 Пробы древесного угля помещают в фарфоровые лодоч¬
ки. Величину навески древесного угля назначают таким образом, что¬
бы масса углерода в ней была не более 0,01 г. Навеску древесного угля
засыпают сверху равномерным слоем 0,1 г оксида алюминия.
24
Глава первая. Физико-химический анализ компонентного состава дымных порохов
Пример 1: Если ожидаемая массовая доля углерода в древесном
угле 50%, то требуемая навеска 0,02 г.
1.4.3.6 В соответствии с руководством на экспресс-анализатор
АН-7529 выполняют пуск прибора, подготовку датчика АН-7529, регу¬
лировку газового тракта, подготовку к работе устройства сжигания, про¬
верку и компенсацию “холостого счета", проверку рабочей точки (при
температуре в печи 120(М250°С). Проверку "холостого счета" произво¬
дят один раз в смену и после установки нового баллона с кислородом.
1.4.4 Выполнение измерений
1.4.4.1 Готовят:
• три навески, содержащие по 0,1 г оксида алюминия для "холостых
проб";
• три навески технического углерода — по 0,01 г в смеси с 0,1 г оксида
алюминия — для насыщения газового тракта;
• три контрольные пробы для градуировки прибора;
• требуемое количество проб древесного угля для проведения анализа.
1.4.4.2 С помощью десяти кнопок переключателя "установка
навески" устанавливают на индикаторе "навеска" значение массы
проб, умноженное на коэффициент пересчета К = 10 (на порядок выше).
Пример 2: масса навески анализируемой пробы составляет 0,02 г.
На индикаторе "навеска, г" устанавливают 0,20 г.
1.4.4.3 Включают таймер "время, мин" и устанавливают на нем
5 минут.
1.4.4.4 Открывают затвор трубки печи и медным крючком уста¬
навливают лодочку с навеской в рабочую зону трубки печи устрой¬
ства сжигания. Температура в рабочей зоне 120СМ-1250вС.
1.4.4.5 Закрывают затвор трубки печи и нажимают кнопку
"сброс", расположенную на передней панели экспресс-анализатора.
1.4.4.6 По окончании горения навески, о чем свидетельствует
стабильность показаний на табло индикатора "%С", снимают показа¬
ния на табло индикатора "%С".
1.4.4.7 Открывают затвор и извлекают лодочку сжигания.
1.4.4.8 После сжигания "контрольной пробы", результат на табло
индикатора "%С" должен соответствовать значению массовой доли
углерода в анализируемой контрольной пробе. Если нет соответ¬
ствия, производят корректировку градуировки анализатора.
1.4.5 Вычисление результатов
1.4.5.1 Массовую долю углерода в контрольной пробе, пропор¬
циональное значение которой ()* высвечивается на табло индикато¬
ра, вычисляют по формуле:
25
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
СК =
0.01'Ст
М
К,
где Сэт — массовая доля углерода в техническом углероде по паспор¬
ту, %;
М — масса навески, г, выставленная на табло индикатора "наве¬
ска" согласно планируемой для анализируемых проб дре¬
весного угля (пример, 2);
X — значение "холостого счета", %;
К — коэффициент пересчета равный 10;
0,01 — масса контрольной пробы, г.
1.4.5.2 Массовую долю углерода в древесном угле вычисляют по
формуле:
С= (А —X) К,
где А — показания индикатора, "%" экспресс-анализатора, получен¬
ные при анализе пробы, %;
X — значение "холостого счета", %;
К — коэффициент пересчета равный 10.
По полученным результатам вычисляют среднее арифметиче¬
ское значение, округляемое до 0,1%. Расхождение между результатами
двух параллельных определений не должно превышать 1,0%.
26
ГЛАВА
ХРОМАТОГРАФИЧЕСКИЙ
АНАЛИЗ
ПИРОКСИЛИНОВЫХ
ПОРОХОВ
Хроматография — одно из наиболее впечатляющих
достижений физической и аналитической химии —
была открыта в начале XX века русским ученым М.С. Цветом в ходе
исследований состава листьев растений и механизма поглощения
ими солнечной энергии.
Хроматография — физико-химический метод разделения, осно¬
ванный на распределении разделяемых компонентов между двумя
фазами — одна из них неподвижная, другая — подвижная, непрерыв¬
но протекающая через неподвижную фазу. В отличие от других мето¬
дов разделения, также основанных на распределении веществ между
фазами, хроматография — метод динамический, так как разделение
происходит в потоке подвижной фазы.
Хроматографические методы, в основном, применяются в ана¬
литической химии, где на основе разделения смесей проводится их
качественный и количественный анализ и, как следствие этого, мож¬
но дать следующее определение хроматографии как аналитического
метода. Хроматография — это физико-химический метод анализа
сложных смесей (газов или жидкостей) путем предварительного раз¬
деления их при движении по слою сорбента за счет различий молеку¬
лярного взаимодействия (в общем случае за счет различной сорбиру-
емости) и последующего определения разделенных компонентов на
выходе из хроматографической колонки с помощью специальных
датчиков — детекторов.
Хроматографические методы применяют также для определе¬
ния физико-химических свойств веществ, при этом чаще всего полу-
27
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
чают хроматограмму лишь одного вещества, в связи с чем признак
разделения становится несущественным.
При классификации хроматографических методов учитывают
природу атомно-молекулярного взаимодействия компонентов смеси
с неподвижной фазой, агрегатное состояние фаз, технику хромато¬
графического анализа.
В зависимости от природы взаимодействия, обусловливающего
распределение компонентов между подвижной и неподвижной фаза¬
ми, выделяют следующие основные методы хроматографии: адсорб¬
ционный; распределительный; ионообменный; молекулярно-сито¬
вый и др.
В адсорбционной хроматографии разделение компонентов смеси
основано на их различной адсорбционной способности по отношению
к высокоразвитой активной поверхности адсорбента, В распредели¬
тельной хроматографии разделяются соединения, отличающиеся по
значениям коэффициентов распределения между подвижной газообраз¬
ной или жидкой фазой и неподвижной жидкой фазой, нанесенной на
поверхность твердого инертного носителя. На практике разделение
компонентов смеси, как правило, происходит при одновременном
действии различных сил.
В зависимости от агрегатного состояния подвижной фазы разли¬
чают газовую [47] и жидкостную хроматографию [48].
Газовая хроматография (ГХ), в свою очередь, подразделяется на
газоадсорбционную (неподвижная фаза — твердое тело) и газо-жид¬
костную (неподвижная фаза — жидкость).
В зависимости от агрегатного состояния неподвижной фазы
жидкостная хроматография делится на жидко-жидкостную распре¬
делительную (неподвижная фаза — жидкость) и твердо-жидкостную ад¬
сорбционную или ионообменную (неподвижная фаза — твердое тело).
В жидкостной молекулярно-ситовой хроматографии неподвиж¬
ная фаза может быть твердым телом (например, сита типа цеолитов)
или гелем (эксклюзионная хроматография).
Высокоэффективный вариант жидкостной хроматографии (ВЭЖХ)
отличается использованием сорбентов с размером зерен 3—10 мкм,
что обеспечивает быстрый массоперенос при очень высокой эффек¬
тивности разделения [49, 50]. В зависимости от техники выполнения
анализа различают хроматографию на колонках, в тонком слое сор¬
бента (ТСХ) [51] и на бумаге.
По цели и масштабам процесса разделения веществ хроматогра¬
фия делится на аналитическую, препаративную и промышленную.
28
Глава вторая. Хроматографический анализ пироксилиновых порохов
Основная цель аналитической хроматографии — качественный
и количественный анализ компонентов смеси (масса исследуемой
пробы обычно незначительна); цель препаративной хроматографии —
получить в лабораторных масштабах образцы чистых веществ (обыч¬
но не более 1 кг), а цель промышленной хроматографии — очистка и
выделение больших партий товарной продукции на производствен¬
ных установках.
В практике аналитического контроля состава и при исследованиях
пироксилиновых порохов чаще всего используют ГХ, ВЭЖХ и ТСХ.
Для решения задач аналитического определения компонентного
состава пироксилиновых порохов существенное значение имеет га¬
зовая хроматография. Раздельное определение малых количеств уда¬
ляемых и неудаляемых сушкой летучих веществ (вода, этиловый
спирт, диэтиловый эфир, этилацетат, ацетон и другие легколетучие
растворители), определение стабилизатора химической стойкости —
дифениламина и, применяемых в некоторых случаях, флегматизато-
ров, пламегасителей и других дополнительных компонентов — вот
далеко незаконченный перечень вопросов, которые может разре¬
шить газовая хроматография в интересах производства и исследова¬
ния пироксилиновых порохов. Нормативные документы и лаборатор¬
ные методики, разработанные на основе метода газовой хроматогра¬
фии позволяют осуществлять операционный контроль процесса
изготовления пироксилиновых порохов и полуфабрикатов для них,
проводить оценку качества готового пороха, служить инструментом
при исследованиях и разработке новых видов пироксилиновых поро¬
хов. При исследовании и анализе пироксилиновых порохов методом га¬
зовой хроматографии обычно используют детектор по теплопроводно¬
сти (катарометр), реже применяют детектор ионизации пламени
(ДНИ). В качестве подвижной фазы, в подавляющем большинстве слу¬
чаев, используют гелий (благодаря инертности и выской теплопровод¬
ности). Более теплопроводный водород опасен и применение его огра¬
ничено. В качестве неподвижной фазы для вышеуказанных целей ши¬
рокое применение нашли полиэтиленгликоли различной молекулярной
массы (1000 — 20000) при определении спирта, эфира и других легколе¬
тучих растворителей, различные высококипящие кремнийорганиче-
ские соединения (например, силиконовый эластомер СКТФТ-50Х) —
при определениии массовой доли дифениламина и пористые полимер¬
ные сорбенты типа "Полисорб" — при определении массовой доли воды.
Одна из первых работ [52], связанных с применением газовой
хроматографии к анализу продуктов производства пироксилиновых
29
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
порохов, была посвящена определению воды, этилового спирта, аце¬
тона, изо-пропилового спирта и гептана в нитроцеллюлозе. К этому
моменту авторы [53] уже осуществляли газохроматографическое
определение остатков растворителей в производстве бездымных но-
рохов. Раздельное определение "летучих" (спирт, эфир, этилацетат,
ацетон) проводили, так называемым, кинетическим методом с газо¬
хроматографическим окончанием (контролем дистиллята). Из 5— 15 г
измельченного пироксилинового зерна экстрагировали в течение
10 —16 ч "летучие” компоненты. Экстракт разделяли на колонне вме¬
сте с "жидким носителем". В качестве носителя авторы выбрали н-
пропанол — для спирта и толуола; метанол — для эфира и 50%-ный
водный этанол — для ацетона. Через каждые 20 — 30 мин дистиллят
подвергали газохроматографическому анализу. Определение состава
дистиллята происходило в течение 5 —7 ч.
Эта работа [53] явилась основополагающей, для совершенство¬
вания и разработки новых методик определения массовой доли лету¬
чих веществ. В первую очередь было уделено внимание сокращению
продолжительности подготовки пробы к газохроматографическому
анализу и самого анализа. С целью извлечения летучих веществ ис¬
пользовали продувку образца пороха нагретым воздухом или газом-
носителем — это был продолжительный процесс и получалась сильно
разбавленная паро-воздушная смесь. Растворение навески пороха в
подходящем органическом растворителе приводило к получению
очень вязкого раствора, который было затруднительно ввести в испа¬
ритель хроматографа. Омыление навески образца приводило к обра¬
зованию разбавленных водных растворов анализируемых компонен¬
тов, что сказывалось на точности определения. Выход был найден в
использовании омыления с одновременной отгонкой выделяющихся
летучих компонентов, при этом основное внимание было направлено
на аппаратурное оформление совмещенного процесса. Результатом
этих работ стали методики определения массовой доли "летучих" ве¬
ществ как для пофазного контроля производства пироксилиновых
порохов, так и для оценки их содержания в готовом порохе [54 — 56].
Разработанные методики легли в основу многочисленных норматив¬
ных документов.
Применение полимерных сорбентов типа Porapak Q (отечест¬
венный аналог Полисорб I) для определения воды в гидрофильных
макромолекулярных образцах типа декстранов [57] подтолкнуло к
внедрению их при разработке методик определения массовой доли
воды в сырье, полупродуктах и готовом порохе на всех технологиче-
30
Глава вторая. Хроматографический анализ пироксилиновых порохов
ских стадиях производства пироксилинового пороха (исходная целлю¬
лоза, нитроцеллюлоза после водоотжима и обезвоженная нитроцеллю¬
лоза, пороховая масса, пороховой шнур и порох на стадиях изготовле¬
ния) [58, 59]. Методика определения массовой доли воды в порохе была
защищена патентом [60] и легла в основу соответствующего ГОСТа [61].
Разработка методик определения стабилизатора химической
стойкости пироксилиновых порохов — дифениламина [62 — 64] при¬
вела к оформлению двух способов подготовки образца к хроматогра¬
фированию. Это экстрагирование дифениламина подходящим орга¬
ническим растворителем, в состав которого предварительно введено
вещество, служащее внутренним стандартом при количественной
интерпретации результатов анализа, — при определении массовой
доли дифениламина в полупродуктах производства пироксилинового
пороха (пороховая масса, шнур после прессования и зерно после рез¬
ки). Для определения массовой доли дифениламина в мягком зерне
на стадиях провяливания, сушки, вымачивания и в готовом порохе
[62] анализируемая проба подвергалась омылению водным раствором
гидроксида натрия или калия с одновременным экстрагированием ди¬
фениламина раствором внутреннего стандарта в подходящем экстра¬
генте. Разработаны методики, позволяющие определять массовую до¬
лю дифениламина [63] и при наличии симметричной диметилдифе-
нилмочевины (централита И) [64] в утилизируемых порохах.
Для обнаружения химических изменений, т.е. в качестве метода
для контроля содержания дифениламина и его производных в про¬
цессе хранения порохов, наиболее часто используется ТСХ [65] и, в
последнее время, ВЭЖХ [66, 67].
Для разделения компонентов порохов, нитропроизводных ста¬
билизаторов химической стойкости методом ТСХ применяли опреде¬
ленный крут растворителей. Производные дифениламина разделяли
с помощью бензола; хлороформа; смеси бензола и петролейного эфи¬
ра, содержащего 10% метанола, в соотношении 1:1; бензола и петро¬
лейного эфира в соотношении 1:1; бензола, четыреххлористого угле¬
рода и дихлорэтана в соотношении 5:3:5; бензола и хлороформа в
соотношении 1:1; бензола и метанола в соотношении 4:1; диизопро-
пилового эфира и петролейного эфира в соотношении 3 : 7.
Идентификацию N-нитрозо- и нитродифениламинов, разделен¬
ных методом ТСХ, проводили аэрозолем спиртового раствора гидрок¬
сида калия (реагент готовили растворением максимального количе¬
ства гидроксида калия в водном растворе этилового спирта 50/50);
опрыскиванием раствором 5 г бихромата калия в смеси 100 мл уксус-
31
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
ной кислоты, 50 мл концентрированной серной кислоты и 100 мл воды.
В качестве идентифицирующих реагентов исследованы также раство¬
ры ванилина и сульфаниловой кислоты. Пятна динитропроизводиых
дифениламина окрашивали спиртовым раствором гидроксида натрия.
Авторы [65] считают, что наилучшие результаты при идентифика¬
ции дифениламина и его N-нитрозо- и нитропроизводных получают с
применением спиртового раствора фосфорномолибденовой кисло¬
ты. Окраска пятен при этом очень насыщенная и долго не исчезает.
Для обнаружения химических изменений 1 г измельченного по¬
роха экстрагировали 10 мл метиленхлорида в течении 17 ч. Процесс
экстрагирования можно резко ускорить применением устройств для
взбалтывания. Производные дифениламина после ТСХ-разделения
идентифицировали после опрыскивания хроматографической пла¬
стинки раствором 6,6 г фосфорномолибденовой кислоты в 100 мл эти¬
лового спирта с использованием значений отношений длины пробега
пятна вещества к длине пробега фронта растворителя (Rf) для индиви-v
дуальных компонентов (при отсутствии чистых веществ использова¬
ли литературные данные).
В последние годы появилось много работ [68 — 93] с использовани¬
ем метода ВЭЖХ, имеющей значительные основные преимущества
перед ТСХ (непосредственное количественное определение) и перед
ГХ — разделение и идентификация компонентов при температуре,
близкой к комнатной.
Метод ВЭЖХ позволяет быстро и с высокой точностью опреде¬
лять взрывчатые вещества. Например, при введении в хроматограф,
сопряженный с УФ-детектором, 0,1 см3 раствора нитрофенола, со¬
держащего по 1 • 10“5 г/см3 гексогена, октогена и тетрила, разделение
и идентификация взрывчатых веществ происходит за 10 мин [68].
Минимальная пороговая концентрация составляет 8 -10“10 г/см3 для
нитробензола и 2,6-динитротолуола и 3 -10“9 г/см3 — для 2-нитрото¬
луола и 3-нитротолуола (детектор — электронная ловушка).
Для исследования и анализа пироксилиновых порохов применяют
как нормально-фазовый вариант ВЭЖХ (НФ ВЭЖХ), так и обращенно-
фазовый вариант (ОФ ВЭЖХ). В НФ ВЭЖХ используют полярный
адсорбент и неполярный (или слабополярный) элюент. В настоящее
время наиболее распространенным полярным адсорбентом является
силикагель. Кроме силикагеля к полярным адсорбентам относятся
CN —, NH2 —, диол — и ряд других химически модифицированных си¬
ликагелей с различными функциональными полярными группами на
поверхности адсорбента. В качестве элюента в НФ ВЭЖХ использу-
32
Глава вторая. Хроматографический анализ пироксилиновых порохов
ют гексан, н-пентан и другие неполярные растворители. Для регули¬
рования элюирующей силы неполярного элюента пользуются поляр¬
ной добавкой (с увеличением количества полярной добавки увеличи¬
вается элюирующая сила элюента). В тех случаях, когда необходимо уме¬
ньшить время удерживания адсорбата без существенного ухудшения
разрешения пиков, в элюент добавляют вещество, способное образо¬
вывать цепочные ассоциаты в растворе. К таким веществам, в первую
очередь, относятся спирты — метиловый и этиловый (от 0,5% до 20%
по объему).
В ОФ ВЭЖХ используют неполярный адсорбент и полярный
элюент. В качестве адсорбента в ОФ ВЭЖХ обычно применяют сили¬
кагели, на поверхности которых привиты нормальные углеводород¬
ные цепочки с 8, 16 или 18 углеродными атомами. В качестве элюента
в ОФ ВЭЖХ применяют, в основном, водно-метанольные, водно-аце¬
тонитрильные или водно-метанольно-ацетонитрильные смеси. Орга¬
нические растворители, используемые в смеси с водой в элюенте для
ОФ ВЭЖХ, называют модификаторами. Для увеличения селективности
и улучшения разрешения можно использовать смешанный модифика¬
тор. Часто в качестве модификаторов используют буферные растворы
[50]. Наиболее универсальным элюентом в ОФ ВЭЖХ является смесь:
ацетонитрил — 0,03 М раствор метафосфата калия, диэтиламинфос-
форная кислота. При анализе ряда близких по составу, строению и
химическим свойствам взрывчатых веществ наиболее успешное раз¬
деление компонентов было достигнуто при применении в качестве
противоиона однозамещенного фосфата тетрабутиламмония [50].
Основным недостатком НФ ВЭЖХ, являются трудности, связан¬
ные с регенерациеей колонок, заполненных силикагелем. Ухудшение
работы таких колонок связано с накоплением воды. Регенерация в та¬
ких случаях продолжается сутками и не всегда приводит к нужному ре¬
зультату, однако такие колонки еще можно использовать с элюентом,
содержащим амин или органическую кислоту, т.е. для других анализов.
В то же время, следует отметить, что порядок элюирования произ¬
водных дифениламина в НФ ВЭЖХ [69] (дифениламин; 2-нитродифе-
ниламин; N-нитрозодифениламин; 2,2'-динитродифениламин; 4-нитро¬
дифениламин; 4,4'-динитродифениламин), соответствует последова¬
тельности расположения пятен тех же производных на тонкослойных
хроматограммах при использовании бензола в качестве подвижной
фазы [65].
Для анализа порохов и ВВ жидкостную хроматографию активно
начали применять в начале 70-х годов прошлого столетия. Одной из
33
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
первых, в этом плане, является работа [70], в которой на исскустненной
смеси компонентов ВВ рассмотрены методы разделения нитроэфи¬
ров, нитроароматических соединений и нитраминов и дано описание
применения метода для анализа твердых ракетных топлив. Анализ
ранних работ, опубликованных в зарубежной печати по возможно¬
сти применения ЖХ для определения ароматических соединений,
аминов, нитросоединений и нитраминов, приведен в обзоре [71].
Известны работы по количественному определению тетрила и
продуктов его разложения [72], взрывчатых веществ в почве и воде
[73], следовых количеств тротила [74].
Для исследования и анализа порохов и взрывчатых веществ ис¬
пользовали как НФ ВЭЖХ, так и ОФ ВЭЖХ.
Методом НФ ВЭЖХ проводили определение дифениламина,
симметричной диметилдифенилмочевины (централит II) и их произ¬
водных в стабилизированных порохах [69, 75, 76] и анализировали
смеси известного состава, основными компонентами которых были
нитроглицерин, диэтиленгликольдинитрат, 2,4-динитротолуол, ни¬
тродифениламин, симметричная диметилдифенилмочевина, N-дифе-
нилмочевина, дифенилуретан (акардит) и дифениламин [77].
Детектирование разделенных компонентов проводили с помо¬
щью ультрафиолетового (СФД-УФ), электрохимического (ЭХД) и
других типов детекторов. Трудности применения метода ЖХ связа¬
ны, в основном, с тем, что ни один из детекторов (спектрофотометри¬
ческий на ультрафиолетовую область спектра — УФД, электрохими¬
ческий, спектрометрический, кулонометрический, термический и др.)
не обеспечивает высокой точности определения содержания широ¬
кого класса взрывчатых веществ (с С — N02 —, О — N02 —, N — N02 —
связями, галоидсодержащих взрывчатых веществ и т.д.). Действуют
детекторы, преимущественно, избирательно.
Большинство работ по применению ОФ ВЭЖХ для анализа и ис¬
следования порохов и взрывчатых веществ выполнены с применени¬
ем в качестве детектора СФД-УФ, однако, имеется ряд работ с приме¬
нением ЭХД [78 — 84]. Это работы по определению остатков ружей¬
ного и артиллерийского выстрелов на руках, одежде и в почве [76 — 82],
определение следов ароматических аминов в морской воде [83] и
определению нитрозаминов [84]. Авторы [85, 86] изучали разложение
дифениламина в ракетных топливах на основе нитроцеллюлозы с ис¬
пользованием двойного амперометрического детектирования.
Из последних работ, посвященных исследованию порохов и
взрывчатых веществ методом ВЭЖХ, следует отметить [87 — 93]. В ра-
34
Глава вторая. Хроматографический анализ пироксилиновых порохов
боте [87] проводили определение нитроглицерина, нитрогликоля,
тринитротолуола и 2,4-динитротолуола в хлороформовых экстрактах
динамитов на колонке 150 х 4 мм, заполненной Lichrosorb RP (5 мкм),
с использованием в качестве элюента смеси метанола с водой в соот¬
ношении 60 : 40 и СФД-УФ при длине волны 230 нм.
Авторы патента [88] описывают способ разделения симметричной
диэтилдифенилмочевины (централита-1) и продуктов ее превращений
в метательных зарядах на основе нитроцеллюлозы. Около 1 г измельчен¬
ного пороха экстрагировали метанолом с добавлением внутреннего
стандарта (ацетанилид) при озвучивании ультразвуком в течение 1,5 ч.
Применялась колонка, заполненная силикагелем С18 (10 мкм); по¬
движная фаза: смесь метанола и воды при соотношении 2 : 1 (расход
составлял 1,5 мл/мин), использовали СФД-УФ при длине волны 230
нм (варианты: 230 — 254 нм). Наблюдался следующий порядок элюи¬
рования компонентов: ацетанилид — нитроглицерин — изомеры ди-
нитроцентралита и тринитроцентралита — мононитроцентралит —
централит — дибутилфталат.
Авторы [89] анализировали взрывчатые вещества и продукты их
разложения в долго хранившихся снарядах методами газовой и жид¬
костной хроматографии. В случае жидкостной хроматографии ис¬
пользовали колонку (25 см х 4,6 мм) с Supelcosil LC-18 (5мкм), подвиж¬
ной фазой служила смесь (55:45) метанола с 0,025 М фосфатным бу¬
ферным раствором с pH = 3 при расходе 1,5 мл/мин, проба составляла
30 мкл; СФД-УФ (254 и 215 нм), или применяли колонку с Supelcosil
LC-CN (5 мкм) при тех же самых условиях.
В [90,91] определяли производные дифениламина в австралийских
одноосновных артиллерийских порохах (дифениламин и его N-ни-
трозо- и С-нитропроизводные) и исследовали механизм превращения
производных дифениламина при 80°С. Относительное содержание 2-ни-
тродифениламина и 4-нитродифениламина изменялось в диапазоне
от 32 : 68 до 63 : 37. Авторами установлено, что 4-нитродифениламин
более стабилен, чем 2-нитродифениламин, а также обсуждается ме¬
ханизм перехода N-нитрозаминов в изомерные нитропроизводные.
Разработаны методики [92, 93] разделения стабилизатора хими¬
ческой стойкости — 2 нитродифениламина и его основных производ¬
ных в ракетных топливах и артиллерийских порохах. В работе [93] ко¬
лонку (250 х 4 мм) с обращенной фазой Nucleosil С18 термостатиро-
вали при температуре 35°С (в основном, для исключения влияния
дрейфа при колебании комнатной температуры). В качестве подвиж¬
ной фазы использовали трехкомпонентную смесь изо-пропанола,
35
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
ацетонитрила и воды в соотношении (по объему) 16 : 38 : 46, соответ¬
ственно при расходе 0,55 мл/мин. Детектирование осуществляли с
помощью многоволнового СФД-УФ.
Авторами [66] разработан способ определения массовой доли ди¬
фениламина и его производных в старых (утилизируемых) порохах,
благодаря чему появилась возможность оценки состояния стабилиза¬
тора в порохах во время хранения.
2.1 Определение массовой доли дифениламина
2.1.1 Определение основано на разложении образца пироксилино¬
вого пороха 30% масс, водным раствором щелочи с одновременным
экстрагированием дифениламина раствором гексадекана в гептане
(или в н-бутиловом спирте) с последующим газохроматографическим
анализом экстракта.
2.1.2 Аппаратура, материалы и реактивы
• хроматограф газовый аналитический с детектором по теплопровод¬
ности;
• баня водяная;
• весы лабораторные с наибольшим пределом взвешивания 200 г;
• набор гирь;
• линейка измерительная металлическая длиной 300 мм и ценой деле¬
ния 1 мм;
• термометр стеклянный, диапазон измерения температуры от 0 до
плюс 100°С, с ценой деления шкалы 1°С;
• часы механические или электронные;
• микрошприц вместимостью 10 мкл;
• насос вакуумный;
• электроплитка бытовая с закрытым обогревом;
• колбы конические вместимостью 50; 100 или 250 см3;
• колбы мерные вместимостью 100 см3;
• пипетка вместимостью 10 см3;
• холодильник лабораторный шариковый;
• воронка диаметром 50 мм;
• чаша выпарительная фарфоровая вместимостью 100—150 см3;
• гелий газообразный сжатый;
• гексадекан;
• Н-гептан;
• раствор гексадекана в н-гептане с в соотношении 1 : 200;
• раствор гексадекана в н-бутиловом спирте в соотношении 1 : 200;
• спирт н-бутиловый;
36
Глава вторая. Хроматографический анализ пироксилиновых порохов
• дифениламин;
• метилен хлористый;
• жидкая фаза: трифторпропилметилсиликон СКТФТ-50Х;
• калия гидроксид (или натрия гидроксид), 30% масс, водный раствор
• твердый носитель — хромосорб 11W", хезосорб силанизированный
или порохром;
• вата медицинская гигроскопичная;
• стеклоткань прокаленная;
• вода дистиллированная.
2.1.3 Подготовка к анализу
2.1.3.1 Подготовка сорбента и хроматографической колонки
Твердый носитель, количество которого определяют в зависимо¬
сти от объема заполняемой колонки, помещают в выпарительную ча¬
шку и заливают раствором жидкой фазы СКТФТ-50Х (15% масс.
СКТФТ-50Х по отношению к твердому носителю) в хлористом мети¬
лене. Количество раствора хлористого метилена берут из расчета 2 : 1
к объему твердого носителя. Полученную массу выдерживают не ме¬
нее 30 минут, позволяя хлористому метилену испариться. Оставший¬
ся растворитель удаляют на водяной бане при температуре (50±2)°С
при осторожном перемешивании сорбента. Работы проводят в вы¬
тяжном шкафу.
При заполнении U-образных колонок подготовленный сорбент
засыпают в чистую сухую колонку небольшими порциями, слегка по¬
стукивая по ней деревянной палочкой, обеспечивая равномерное
уплотнение сорбента.
При заполнении спиральных колонок один из концов колонки
закрывают тампоном из стеклоткани и подключают к водоструйному
насосу. Колонку закрепляют в штативе таким образом, чтобы спи¬
ральные витки располагались горизонтально. К открытому концу ко¬
лонки прикрепляют воронку для удобства засыпки сорбента. Создав
разрежение, начинают осторожно засыпать подготовленный сорбент
с одновременным постукиванием по колонке деревянной палочкой
для более равномерного ее заполнения. О заполнении колонки судят
по прекращению убыли сорбента в воронке. Оставшийся в воронке
сорбент и небольшое его количество из конца колонки осторожно
ссыпают. Сняв воронку, закрывают открытый конец колонки тампо¬
ном из стеклоткани.
Вновь заполненную колонку помещают в термостат хроматогра¬
фа, подсоединяют одним концом к линии газа-носителя, и, не присо¬
единяя ее другим концом к детектору, продувают газом-носителем с
37
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
расходом 30 — 50 см3 /мин в течение от 5 до 8 ч, постепенно повышая
температуру термостата колонок от комнатной до 180°С. После этого
свободный конец колонки подсоединяют к детектору.
Монтаж, наладку и вывод хроматографа на рабочий режим про¬
изводят в соответствии с руководством по эксплуатации соответ¬
ствующего типа хроматографа.
2.1.3.2 Условия проведения анализа:
• длина колонки, м
• внутренний диаметр колонки, мм
• температура термостата колонок, °С
• температура термостата детектора, °С
• температура испарителя, °С
• ток детектора, мА
• расход гелия на выходе из колонки, см3/мин
• скорость протяжки диаграммной ленты, мм/ч
• объем анализируемой пробы, мкл
• внутренний стандарт
1.0 -3,0
3.0 -4,0
130 -200
130 -200
300 -325
90-140
30-120
60 -720
1 -8
гексадекан
Допуски в условиях проведения анализа уточняются при подбо¬
ре оптимальных условий разделения для данной колонки.
2.1.3.3 Приготовление стандартного раствора
В колбу вместимостью 100 см3 помещают навеску равную 0,15 г
дифениламина, взвешенную с точностью до четвертого десятичного
знака. Добавляют 30 см3 раствора н-гептана с гексадеканом (или ра¬
створа н-бутилового спирта с гексадеканом) и 30 см3 30% масс, водно¬
го раствора гидроксида натрия (или гидроксида калия), перемешива¬
ют вручную и дают отстояться до разделения слоев.
Стандартный раствор необходимо проверять один раз в сутки и
хранить в прохладном месте (холодильнике).
2.1.4 Проведение анализа
Порох (10,0000±0,5000) г, предварительно измельченный и высу¬
шенный до постоянной массы, взвешивают с точностью до четвертого
десятичного знака. Помещают навеску в реакционную колбу, прилива¬
ют 30 см3 30% масс, водного раствора гидроксида натрия (или гидрокси¬
да калия), 30 см3 раствора гексадекана в гептане (гексадекана в н-бу-
тиловом спирте). Колбу присоединяют к обратному холодильнику.
Содержимое колбы кипятят на электроплитке до полного разложе¬
ния пороха (10 — 30 минут). После этого кипятят еще 5 минут. Затем
электроплитку отключают. Не отсоединяя от холодильника, колбу с
содержимым охлаждают на воздухе, отсоединяют от холодильника и
38
Глава вторая. Хроматографический анализ пироксилиновых порохов
закрывают пробкой. Перемешивают содержимое энергичным встря¬
хиванием и дают отстояться до разделения слоев.
Из верхнего слоя микрошприцем отбирают пробу и вводят в ис¬
паритель хроматографа. Хроматографируют пробу по уточненным
условиям проведения анализа. Предварительно проводят анализ и
стандартного раствора.
2.1.5 Обработка результатов
На хроматограммах измеряют высоты (площади) пиков дифени¬
ламина и гексадекана для анализируемого и стандартного растворов.
По полученным данным рассчитывают отношения высот пиков дифе¬
ниламина к высоте пика гексадекана для стандартного (Ct) и анализи¬
руемого (С2) растворов.
Массовую долю дифениламина X, в процентах, вычисляют по
формуле: v 1,5С210
Сгт '
где 1,5— массовая доля дифениламина в стандартном растворе в рас¬
чете на 10 г пороха, % масс.;
Cj — отношение высоты пика дифениламина к высоте пика гек¬
садекана в стандартном растворе;
С2 — отношение высоты пика дифениламина к высоте пика гек¬
садекана в анализируемом растворе;
т — масса навески пороха, г;
10 — рассчетная масса навески пороха, г.
Проводят два параллельных определения, по результатам кото¬
рых рассчитывают среднее арифметическое значение, округляемое
до десятых долей процента.
Расхождение между результатами параллельных определений
не должно превышать 0,1%.
2.2 Определение компонентного состава нитрозо- и нитропро¬
изводных дифениламина
2.2.1 Определение основано на экстрагировании дифениламина и
его производных из навески измельченного пороха метиленхлори-
дом с последующим анализом полученного экстракта методом тонко¬
слойной хроматографии.
2.2.2 Аппаратура, материалы и реактивы
• набор оборудования для тонкослойной хроматографии (прибор для
нанесения тонких слоев регулируемой толщины; рабочий шаблон
для установки стеклянных пластинок; стойка для сушки и хранения
39
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
стеклянных пластинок после нанесения слоя; разделительная каме¬
ра с пришлифованной крышкой; прибор для опрыскивания)
• весы лабораторные общего назначения 2 класса точности с наи¬
большим пределом взвешивания 200 г;
• весы лабораторные квадрантные с наибольшим пределом взвеши¬
вания 160 г или 500 г;
• набор гирь общего назначения 2 класса точности общей массой 210 г;
• ножницы рычажные;
• прибор для встряхивания пробирок и колб;
• шкаф сушильный воздушный, обеспечивающий температуру 110°С;
• термометр стеклянный ртутный, диапазон измерения температуры
от 0 до плюс 150°С, с ценой деления шкалы 1°С;
• ножницы рычажные;
• часы механические или электронные;
• микрошприц вместимостью 10 мкл;
• пипетки вместимостью 10 см3;
• пластинки стеклянные;
• склянки пенициллиновые с пробками из самозатягивающейся ре¬
зины;
• цилиндр мерный лабораторный вместимостью 50 см3;
• чаша выпарительная фарфоровая вместимостью 100 см3;
• эксикатор;
• бензол;
• метилен хлористый;
• дифениламин;
• дифениламина нитрозо- и нитропроизводные;
• кислота фосфорномолибденовая;
• спирт этиловый;
• силикагель для тонкослойной хроматографии;
• гипс для тонкослойной хроматографии;
• вода дистиллированная;
• вата медицинская гигроскопическая.
2.2.3 Подготовка к анализу
2.2.3.1 Подготовка хроматографических пластинок
2.2.3.1.1 На рабочем шаблоне укладывают впритык друг к другу
5 чистых сухих стеклянных пластинок размером 160 х 235 мм или
10 пластинок размером 80 х 235 мм. В начале и в конце ряда укладыва¬
ют менее качественные пластинки. Упор шаблона должен находиться
справа от работающего.
2.2.3.1.2 На крайнюю левую пластинку помещают открытый при¬
бор для нанесения слоев. Устанавливают щель величиной 0,25 мм.
40
Глава вторая. Хроматографический анализ пироксилиновых порохов
2.2.3.1.3 В сухую фарфоровую чашку помещают 25,0 г силикагеля
для тонкослойной хроматографии, зернением 5 — 40 мкм, содержаще¬
го 13% масс, гипса. При медленном перемешивании добавляют 35 см3
дистиллированной воды и растирают до образования однородной
массы, не содержащей комков и пузырьков воздуха. После этого до¬
бавляют при перемешивании еще 15 см3 воды. Суммарное время пе¬
ремешивания не должно превышать 100 с.
2.2.3.1.4 Подготовленной суспензией заполняют прибор для на¬
несения слоев, проводят вдоль его фронта пальцем, смоченным водой
(для снижения трения в момент старта).
Перемещают рукоятку прибора на 180°, позволяя суспензии вытечь
на стартовую пластинку, и равномерно передвигают прибор до упора
на рабочем шаблоне. Затем прибор снимают, разбирают, моют и сушат.
2.2.3.1.5 Пластинки с нанесенным слоем слегка раздвигают и
подсушивают 20 мин на воздухе, не снимая с шаблона. После чего
проводят активацию их нагреванием в течение 30 мин в сушильном
шкафу при температуре 110°С.
2.2.3.1.6 Активированные пластинки извлекают из шкафа, ох¬
лаждают на воздухе и помещают в эксикатор. Допускается проводить
активирование непосредственно перед применением хроматографи¬
ческих пластинок для анализа.
2.2.3.2 Подготовка образца к анализу
2.2.3.2.1 Около 1 г измельченного образца пороха помещают в пе¬
нициллиновую склянку, приливают 10 см3 метиленхлорида, закрыва¬
ют пробкой с зажимом, закрепляют на аппарате для встряхивания
пробирок и колб и встряхивают в течение 7 — 8 ч.
2.2.3.3 Подготовка разделительной камеры
2.2.3.3.1 Внутренние боковые поверхности камеры обкладывают
фильтровальной бумагой. В камеру вносят 150—170 см3 бензола, сма¬
чивая при этом фильтровальную бумагу, и закрывают камеру при¬
шлифованной крышкой для насыщения.
2.2.4 Проведение анализа
2.2.4.1 Хроматографическую пластинку извлекают из эксикатора
и снимают лезвием узкий слой адсорбента у боковых граней, чтобы
предотвратить уход растворителя в процессе анализа.
2.2.4.2 В 1,5-2,0 см от нижнего обреза пластинки отмечают ли¬
нию старта и в, приблизительно, 10 см от линии старта обозначают
ожидаемую линию фронта растворителя.
2.2.4.3 Пробы экстрактов, полученных по п. 2.2.3.2.1, наносят ми¬
крошприцем в количестве 5—10 мкл на линию старта в 1,5-2,0 см
41
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
друг от друга. После нанесения каждой пробы несколько раз промы¬
вают микрошприц метиленхлоридом и протирают иглу ватой.
При необходимости, на эту же (или на отдельную) пластинку нано¬
сят пробы индивидуальных веществ (свидетелей), наличие которых
предполагается в анализируемой смеси.
2.2.4.4 Пластинку по п 2.2.4.3 помещают в разделительную камеру,
подготовленную по п. 2.2.3.3.1, закрывают крышкой и выдерживают
ее там до тех пор, пока растворитель не достигнет линии фронта,
обозначенной на пластинке.
2.2.4.5 Пластинку извлекают из камеры, позволяют стечь остаткам
растворителя и подсушивают на воздухе. Подсушенную пластинку
устанавливают у задней стенки вытяжного шкафа и опрыскивают
спиртовым раствором фосфорномолибденовой кислоты (6,6 г кислоты
в 100 мл этилового спирта), вызывающей окрашивание пятен разде¬
ленных компонентов. Для усиления окраски пятен пластинку выдер¬
живают в течение 30 мин в сушильном шкафу при 110°С. Затем пла¬
стинку извлекают и охлаждают.
2.2.5 Идентификация разделенных компонентов
2.2.5.1 На полученных тонкослойных хроматограммах (см. рис. 1)
отмечают окраску каждого пятна
старта до центра пятна.
2.2.5.2 По полученным дан¬
ным определяют цвет пятен и
рассчитывают значения Rf как
для комонентов анализируемого
экстракта пороха, так и для инди¬
видуальных соединений (свиде¬
телей).
Rf — Li/L2 i
где Lj — расстояние от линии
старта до центра пятна
компонента, мм;
12 — расстояние от линии
старта до линии фронта
растворителя, мм.
2.2.5.3 Полученные результа¬
ты (Rf и цвет соответствующего
пятна) сравнивают с аналогичны¬
ми для индивидуальных компо-
и замеряют расстояние от линии
1 2 3 4 5
Рис. 1. Тонкослойная хроматограмма
экстракта пороха:
1 — экстракт, 2 — 5 — "свидетели": 2 — ди¬
фениламин, 3 — 2-мононитро-дифенила-
мин, 4 — N-нитрозодифениламин, 5 —
4 -мононитродифениламин.
42
Глава вторая. Хроматографический анализ пироксилиновых порохов
нентов, входящих в состав разделяемой смеси. В результате делают за¬
ключение о составе производных дифениламина в анализируемом об¬
разце пороха.
2.2.5.4 При отсутствии (частичном или полном) индивидуальных
компонентов (производных дифениламина — свидетелей) пользуют¬
ся данными, приведенными в табл. 1.
Таблица 1. Значения Rf и окраска пятен дифениламина и его производных
после разделения бензолом на силикагеле и проявления раствором фосфор¬
номолибденовой кислоты
Вещество
Значение Rf
Цвет пятна
Дифениламин
0,51
синий
2-нитродифениламин
0,47
красно-коричневый
N-нитрозодифениламин
0,32
синий
2,4 -динитродифениламин
0,30
оранжевый
2,2 ,4-тринитродифениламин
0,23
желтый
4-нитродифениламин
0,19
желто-зеленый
2,4,4 -тринитродифениламин
0,15
желтый
4,4 -динитродифениламин
0,06
желтый
Порядок элюирования и окраска пятен соответствующих компо¬
нентов сохраняются. В зависимости от условий, экспериментальные
значения Rf могут несколько отличаться от приведенных в табл. 1.
2.3 Определение массовых долей дифениламина и его N-нитрозо-
и нитропроизводных
2.3.1 Определение массовых долей дифениламина и его N-ни-
трозо- и нитропроизводных в пироксилиновых порохах основано на
экстрагировании из пороха дифениламина и его производных и хро¬
матографировании полученного экстракта на микроколоночном
жидкостном хроматографе 1,Милихром-4ВУФЭ" со спектрофотоме¬
трическим детектором при изократическом элюировании.
2.3.2 Средства измерений, материалы и реактивы
• хроматограф жидкостной микроколоночный "Милихром-4ВУФЭ"
или другой ВЭЖХ хроматограф со спектрофотометрическим детек¬
тором в УФ-области, персональным компьютером IBM PC и програм¬
мным обеспечением для обработки хроматографических пиков;
• колонка КАХ4-64 (80);
43
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
• весы лабораторные с наибольшим пределом взвешивания 200 г;
• набор гирь;
• аппарат для встряхивания пробирок и колб любого типа;
• ножницы рычажные;
• часы механические или электронные;
• воронка фильтрующая диаметром 20 или 30 мм пористостью 40 или
100 мкм;
• колбы мерные;
• пипетки вместимостью 5 или 10 см3;
• склянки пенициллиновые с пробками из самозатягивающейся резины;
• цилиндр мерный вместимостью 50 см3;
• шприцы медицинские вместимостью 1 см3, 2 см3 или 5 см3;
• ацетанилид — 1% раствор в метаноле (раствор внутреннего стан¬
дарта);
• вата медицинская гигроскопическая;
• вода дистиллированная;
• вода бидистиллированная;
• гелий газообразный сжатый;
• метанол для ВЭЖХ;
• дифениламин;
• N-нитрозодифениламин;
• 4-Нитродифениламин;
• 2-Нитродифениламин;
• динитропроизводные дифениламина;
• тринитропроизводные дифениламина.
2.3.3 Подготовка к анализу
2.3.3.1 Подготовка хроматографа
Вывод хроматографа на рабочий режим проводят в соответствии
с инструкцией по эксплуатации жидкостного микроколоночного хро¬
матографа "Милихром-4ВУФЭ". Большие положительные значения
фона свидетельствуют о попадании воздуха в рабочую кювету детек¬
тора, а большие отрицательные значения фона — о попадании возду¬
ха в кювету сравнения. В обоих случаях необходимо удалить воздух
элюентом при помощи медицинского шприца.
2.3.3.2 Приготовление элюента (подвижной фазы)
Для определение массовой доли дифениламина и его N-нитрозо —
и нитропроизводных в пироксилиновых порохах применяют элюент,
состоящий из метилового спирта и бидистиллированной воды при со¬
отношении компонентов 70 : 30 (по объему). После смешения компо¬
нентов элюент перед применением должен быть профильтрован че-
44
Глава вторая. Хроматографический анализ пироксилиновых порохов
рез фильтрующую воронку и дегазирован от растворенного в нем
воздуха. Дегазацию проводят барботажем через элюент потока газо¬
образного гелия в течение 2 — 5 мин.
2.3.3.3 Приготовление эталонного раствора
В пенициллиновую склянку помещают 0,01 г дифениламина,
0,002 г N-нитрозодифениламина, 0,001 г 4-нитродифениламина и
0,001 г 2-нитро-дифениламина, взвешенных с точностью до четверто¬
го десятичного знака, что соответствует массовой доле дифенилами¬
на 1,0% масс., массовой доле N-нитрозодифениламина — 0,2% масс.,
массовым долям 4-нитродифениламина и 2-нитродифениламина —
0,1% масс, на навеску пороха 1 г. В склянку приливают 5 см3 раствора
ацетанилида в метаноле (1% масс.), растворяют введенные компонен¬
ты при взбалтывании вручную и добавляют 5 см3 элюента.
Действительную массовую долю дифениламина или его производ¬
ного (X,) в эталонном растворе, в процентах, вычисляют по формуле
х,-™.
Mt
где Y — расчетная массовая доля дифениламина или его производ¬
ного в эталонном растворе, % ;
М — расчетная масса навески дифениламина или его производ¬
ного, г;
М( — действительная навеска дифениламина или его производ¬
ного, г.
Перечень нитропроизводных и их массовая доля при пригото¬
влении эталонного раствора определяется возрастом пороха. Эталон¬
ный раствор необходимо проверять один раз в сутки и хранить в
прохладном месте (холодильнике).
2.3.3.4 Условия проведения анализа
Колонка КАХ4-64 (80):
• длина колонки, мм 64 (80)
• внутренний диаметр колонки, мм 2
• обращенно-фазовый адсорбент Силасорб С18
с размером частиц, микрон 5 — 8
• скорость набора элюента, мкл/мин 999
• расход элюента, мкл/мин 70
• объем ступени элюента, мкл до 2500
• объем на регенерацию колонки, мкл 70
• объем буфера, мкл 6
• объем анализируемой пробы, мкл 1—2
45
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
• длина волны детектора, нм 220
• масштаб регистрации, единиц оптической плотности .... 1,0
• время измерения, с 0,6
• чувствительность регистратора, В 0,1
• скорость передвижения диаграммной ленты, мм/с 0,1
2.3.3.5 Проведение испытаний
Около 1 г высушенного и измельченного пороха, взвешенного с
точностью до четвертого десятичного знака помещают в пенициллино¬
вую склянку. Заливают в склянку 5 мл раствора ацетанилида в мета¬
ноле (1% масс.) закрывают ее пробкой и периодически встряхивают в
течение 30 мин. Затем оплывший образец перемешивают микрошпате¬
лем, добавляют 5 мл водного элюента, снова тщательно перемешивают,
отжимают осадок в склянке стеклянной палочкой с приплюснутым кон¬
цом, экстракт отфильтровывают через фильтровальную бумагу или
стеклянный пористый фильтр, отбирают пробу и хроматографируют.
Допускается применять аппарат для встряхивания пробирок и
колб. Экстрагирование продолжают не менее двух часов. Пробу на
анализ отбирают непосредственно из пенициллиновой склянки.
Выполняют по два параллельных хроматографирования как для
эталонного, так и для анализируемого раствора.
При увеличении сопротивления колонки проводят в конце рабо¬
чего дня регенерацию метанолом и на следующее утро регенерацию
метанолом повторяют.
2.3.3.6 Обработка результатов испытаний
После завершения хроматографирования с помощью компью¬
терной программы выполняют первичную математическую обработ¬
ку данных — рассчитывают площади пиков анализируемых компо¬
нентов и внутреннего стандарта.
Массовую долю анализируемого компонента (дифениламин или
его производного) в порохе, X, в процентах, вычисляют по формуле:
^ _ Xj • С2/ • т
Си
где X,- — массовая доля компонента в эталонном растворе, в процентах;
Си — отношение площади пика дифениламина (или его произ¬
водного) к площади пика внутреннего стандарта в эталон¬
ном растворе;
Съ — отношение площади пика дифениламина (или его произ¬
водного) к площади пика внутреннего стандарта в анализи¬
руемом растворе;
46
Глава вторая. Хроматографический анализ пироксилиновых порохов
т — расчетная масса навески пороха, г.
т{ — фактическая масса навески пороха, г;
По результатам параллельных определений рассчитывают среднее
арифметическое значение, округляемое до второго десятичного знака.
2.4 Определение массовой доли дифениламина и симметрич¬
ной диметилдифенилмочевины (централит II)
2.4.1 Определение по методу А основано на разложении образца
пороха водным раствором гидроксида натрия или гидроксида калия с
одновременным экстрагированием стабилизатора соответствующим
экстрагентом с последующим газохроматографическим анализом по¬
лученного экстракта.
Определение по методу Б основано на экстрагировании стабили¬
затора из образца пороха с последующим газохроматографическим
анализом экстракта.
Определение дифениламина в порохах, содержащих в своем со¬
ставе динитротолуол, необходимо проводить по методу Б.
2.4.2 Метод А
2.4.2.1 Средства измерений, посуда, материалы и реактивы
• хроматограф газовый с детектором по теплопроводности;
• баня водяная;
• весы лабораторные общего назначения 2 класса точности с наи¬
большим пределом взвешивания 200 г;
• весы лабораторные квадрантные с наибольшим пределом взвеши¬
вания 160 или 500 г;
• набор гирь общего назначения 2 класса точности общей массой 210 г;
• ножницы рычажные;
• термометр жидкостный стеклянный, диапазон измерения темпера¬
туры от 0 до плюс 100°С, с ценой деления шкалы 1°С;
• часы механические или электронные;
• микрошприц вместимостью 10 мкл;
• насос вакуумный;
• электроплитка бытовая с закрытым обогревом;
• колбы конические вместимостью 50, 100 или 250 см3;
• колбы мерные вместимостью 100 или 1000 см3;
• пипетки вместимостью 1, 5 или 10 см3;
• холодильник лабораторный типа ХШ;
• цилиндр мерный лабораторный вместимостью 50 см3;
• чаша выпарительная фарфоровая № 3 или № 4;
• гелий газообразный сжатый;
• н-гептан;
47
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
• метилен хлористый;
• спирт н-бутиловый;
• дибутилсебацинат;
• раствор внутреннего стандарта: 12—15 см3 дибутилсебацината в
1000 см3 н-бутилового спирта;
• дифениламин;
• централит-И;
• жидкая фаза — трифторпропил(25%)метилсиликон СКТФТ-50Х
для хроматографии или силиконовый эластомер SE-30, или цианоэ-
тил (25%) метилсиликоновый эластомер ХЕ-60;
• натрия гидроксид или калия гидроксид, водный раствор с массовой
долей 30%;
• натрия хлорид или калия хлорид;
• твердый носитель для газовой хроматографии — полихром-1 или
другой инертный носитель;
• вата медицинская гигроскопическая;
• стеклоткань прокаленная.
2.4.2.2 Подготовка к испытанию
2.4.2.2.1 Подготовка сорбента и хроматографической колонки
Твердый носитель, количество которого рассчитывают в зависи¬
мости от объема заполняемой колонки, заливают в выпарительной
чашке раствором жидкой фазы СКТФТ-50Х (15% СКТФТ-50Х по от¬
ношению к массе твердого носителя) в хлористом метилене. Для при¬
готовления раствора хлористый метилен берут в соотношении 2 : 1 к
объему твердого носителя. Полученную массу выдерживают на воз¬
духе не менее 30 минут, позволяя испариться свободному хлористому
метилену. Затем оставшийся растворитель удаляют на водяной бане
при температуре (50±2)°С при осторожном перемешивании сорбента.
Заполнение колонки сорбентом, монтаж, наладку и вывод хро¬
матографа на рабочий режим производят в соответствии с руковод¬
ством по эксплуатации соответствующего типа хроматографа.
Свежеприготовленную колонку помещают в термостат хромато¬
графа и, не присоединяя ее к детектору, продувают гелием с расхо¬
дом 30 — 50 см3 /мин в течение времени от 5 до 8 ч, постелено повы¬
шая температуру термостата колонок от комнатной до 190°С.
2.4.2.2.2 Монтаж, наладку и вывод хроматографа на рабочий ре¬
жим производят в соответствии с руководством по эксплуатации.
2.4.2.2.3 Условия проведения испытаний:
• длина колонки, м 0,5 —3,0
• внутренний диаметр колонки, мм 3,0-4,0
48
Глава вторая. Хроматографический анализ пироксилиновых порохов
• температура термостата колонок, °С 130 —200
• температура термостата детектора, °С 200 — 300
• температура испарителя, °С 300
• ток детектора, мА 90 — 160
• расход гелия на выходе из колонки, см3/минуту 30—120
• скорость протяжки диаграммной ленты, мм/ч 600 — 720
• объем анализируемой пробы, мкл 1 — 8
Допуски в условиях испытания даны для возможности подбора
оптимальных условий для конкретного прибора.
2.4.2.2.4 Приготовление эталонного раствора для определения
массовых долей дифениламина и централита II
В колбу вместимостью 100 см3 помещают 0,05 г дифениламина и
0,1 г централита II, взвешенных с точностью до четвертого десятично¬
го знака, что соответствует массовой доле дифениламина — 1,0% и
централита II — 2,0% в пересчете на навеску пороха 5 г, добавляют 10 см3
н-гептана (н-бутилового спирта), 20 см3 раствора внутреннего стан¬
дарта и 30 см3 раствора гидроксида натрия, перемешивают вручную и
дают отстояться до разделения слоев.
Действительную массовую долю дифениламина (централита И) в
эталонном растворе (X), в процентах, вычисляют по формуле:
где Y — расчетная массовая доля дифениламина (централита II) в
эталонном растворе,%;
М — расчетная масса навески дифениламина (централита И), г;
т — действительная навеска дифениламина (централита И), г.
Для дифениламина: Y= 1,0; М = 5,0, а для централита: Y = 2,0; М = 5,0.
Эталонный раствор необходимо проверять один раз в сутки и
хранить в прохладном месте (холодильнике).
2.4.2.2.5 Подготовка пробы к анализу
Порох измельчают до частиц размером не более 5 мм и взвеши¬
вают (5,0 ±0,1) г с точностью до четвертого десятичного знака, поме¬
щают в реакционную колбу, приливают 30 см3 раствора гидроксида
натрия или гидроксида калия, 10 см3 н-гептана (н-бутанола) и присое¬
диняют обратный холодильник.
Содержимое колбы кипятят на электроплитке до полного разло¬
жения испытуемого образца (10 — 30 минут). Дополнительно кипятят
еще 5 минут. Затем электроплитку отключают. Не отсоединяя от хо¬
лодильника, колбу с содержимым охлаждают на водяной бане с тем-
49
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
пературой (20±5)°С или на воздухе. При применении н-бутилового
спирта вносят в колбу высаливающее вещество — около 10,0 г хлорида
натрия или хлорида калия. Вносят в колбу 20 см3 раствора внутренне¬
го стандарта. Закрывают колбу пробкой, перемешивают содержимое
энергичным встряхиванием и дают отстояться до разделения слоев.
2.4.2.3 Проведение испытания
2.4.2.3.1 Из верхнего слоя подготовленной пробы по п. 2.4.2.2.5.2
микрошприцем отбирают аликвоту и вводят в испаритель хромато¬
графа. Хроматографируют при условиях, указанных в п. 2.4.2.2.3.
Сначала проводят анализ эталонного раствора, а затем анализ иссле¬
дуемого раствора. После каждого ввода пробы микрошприц промы¬
вают н-гептаном или н-бутиловоым спиртом. При анализе порохов,
содержащих вазелиновое масло или церезин, пробу отбирают через
фильтр из ваты, намотанной на кончик иглы микрошприца.
2.4.2.4 Обработка результатов
Обработку результатов анализа выполняют на компьютере с по¬
мощью программы 11 Ассистент химика" или другой аналогичной про¬
граммы.
При отсутствии компьютера и выводе хроматографической ин¬
формации на соответствующий регистратор, поступают следующим
образом.
На хроматограммах измеряют площади (высоты) пиков дифени¬
ламина и централита II и внутреннего стандарта для анализируемого
и стандартного растворов. По полученным данным рассчитывают от¬
ношения площадей (высот) пиков дифениламина и централита II к
площади (высоте) пика внутреннего стандарта для анализируемого и
эталонного растворов.
Массовую долю дифениламина (централита И) в порохе (Z), в
процентах, вычисляют по формуле:
,-ХС2Р
^ “ _ г
С\-Щ
где X — массовая доля дифениламина (централита II) в эталонном
растворе, в процентах;
Сх — отношение площади (высоты) пика дифениламина (центра¬
лита II) к площади (высоте) пика внутреннего стандарта в
эталонном растворе;
С2 — отношение площади (высоты) пика дифениламина (центра¬
лита И) к площади (высоте) пика внутреннего стандарта в
анализируемом растворе;
50
Глава вторая. Хроматографический анализ пироксилиновых порохов
тх — фактическая масса навески пороха, г;
Р — рассчетная масса навески пороха, 5 г.
Расхождение между результатами параллельных определений не
должно превышать 0,1% для дифениламина и 0,2% для централита И.
2.4.3 Метод Б
2.4.3.1 Средства измерений, посуда, материалы и реактивы
• хроматограф газовый аналитический с детектором по теплопровод¬
ности;
• баня водяная;
• весы лабораторные общего назначения 2 класса точности с наи¬
большим пределом взвешивания 200 г;
• весы лабораторные квадрантные с наибольшим пределом взвеши¬
вания 160 или 500 г;
• набор гирь общего назначения 2 класса точности общей массой 210 г;
• ножницы рычажные;
• термометр жидкостный стеклянный, диапазон измерения темпера¬
туры от 0 до плюс 100°С, с ценой деления шкалы 1°С;
• часы механические или электронные;
• микрошприц вместимостью 10 мкл;
• насос вакуумный;
• сито из сетки или перфорированное сито из нержавеющей стали
или латуни с диаметром отверстия (стороной квадрата) 2 мм;
• шкаф сушильный, обеспечивающий температуру 70 —80°С;
• электроплитка бытовая с закрытым обогревом;
• колбы конические вместимостью 50, 100 или 250 см3;
• колбы мерные вместимостью 100 или 1000 см3;
• пипетки вместимостью 1, 5 или 10 см3;
• холодильник лабораторный типа ХШ;
• цилиндр мерный лабораторный вместимостью 50 см3;
• чаша выпарительная фарфоровая № 3 или № 4;
• гелий газообразный сжатый;
• ацетон;
• метилен хлористый;
• сквалан (2, 6, 10, 15, 19, 23-гексаметилтетракозан);
• экстрагент — раствор ацетона в метилене хлористом в соотноше¬
нии 15:85;
• раствор внутреннего стандарта — 5%-ный раствор сквалана в эк¬
страгенте;
• дифениламин;
• централит-Н;
51
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
• сорбент для газовой хроматографии — инертон AW, зернением
0,25 — 0,36 мм, пропитанный 5% силиконового эластомера SE-30
("СйетароГ, Praha);
• вата медицинская гигроскопическая;
• стеклоткань прокаленная.
2.4.3.2 Подготовка к испытанию
2.4.3.2.1 Подготовка хроматографической колонки по п. 2.4.2.2.1
2.4.3.2.2 Монтаж, наладку и вывод хроматографа на рабочий ре¬
жим проводят в соответствии с руководством по эксплуатации.
2.4.3.2.3 Условия проведения испытаний по п. 2.4.2.2.3
2.4.3.2.4 Приготовление эталонного раствора
Эталонный раствор для определения массовой доли дифенила¬
мина и централита II как при раздельном, так и при совместном при¬
сутствии в порохе, методом Б готовят следующим образом.
В колбу вместимостью 100 см3 помещают 0,075 г дифениламина
и 0,1 г централита И, взвешенных с точностью до четвертого десятич¬
ного знака, что соответствует массовой доле дифениламина — 1,5%
и централита II — 2,0% в пересчете на навеску пороха 5 г, добавляют
30 см3 экстрагента и перемешивают.
Действительную массовую долю дифениламина и централита II в
эталонном растворе, в процентах, вычисляют по формуле п. 2.4.2.2.4, где
для дифениламина: Y = 1,5; М = 5,0, а для централита II: Y = 2,0; М = 5,0.
Эталонный раствор необходимо проверять один раз в сутки и
хранить в прохладном месте (холодильнике).
2.4.3.2.5 Подготовка пробы к анализу
Пробу пороха измельчают рычажными ножницами с отсевом
крупных частиц на сите с отверстием 2 мм или другим способом, по¬
зволяющим получать частицы не крупнее указанных.
Около (5 =*=0,1) г измельченного пороха взвешивают с точностью
до четвертого десятичного знака, помещают в коническую колбу вме¬
стимостью 100 см3 и заливают 30 см3 экстрагента.
Колбу присоединяют к обратному холодильнику и помещают на
водяную баню. Температура бани при экстрагировании должна быть
40 —45°С. По окончании экстрагирования колбу отсоединяют от хо¬
лодильника и экстракт декантируют нерез воронку в мерную колбу
вместимостью 100 см3. После слива остаток в колбе заливают новой
порцией экстрагента в количестве 30 см3 и снова экстрагируют. Та¬
ким образом экстрагирование проводят три раза по 20 мин свежими
порциями экстрагента.
По окончании экстрагирования остаток в колбе промывают 5 см3
экстрагента, который сливают в ту же мерную колбу. Туда же вводят
52
Глава вторая. Хроматографический анализ пироксилиновых порохов
5 см3 раствора внутреннего стандарта. Раствор в колбе доводят до
метки экстрагентом и тщательно перемешивают.
2.4.3.3 Проведение испытания
2.4.3.3.1 Из раствора, полученного по п. 2.4.3.2.5, микрошприцем
отбирают пробу и вводят ее в испаритель хроматографа.
2.4.3.4 Обработка результатов
Обработку результатов анализа выполняют в соответствии с п. 2.4.2.4.
2.5 Определение массовых долей этилового спирта и этилового
эфира
2.5.1 Определение массовой доли летучих веществ, неудаляемых
сушкой, — этилового эфира и этилового спирта основано на разложе¬
нии образца пороха 30% водным раствором щелочи с одновременным
экстрагированием определяемых компонентов н-пропиловым спир¬
том и последующим газохроматографическим анализом экстракта.
2.5.2 Аппаратура, материалы и реактивы
• хроматограф газовый аналитический с детектором по теплопровод¬
ности;
• баня водяная;
• весы лабораторные с наибольшим пределом взвешивания 200 г и
допускаемой погрешностью взвешивания 0,00075 г;
• набор гирь для химических анализов и взвешиваний с высокой точ¬
ностью общей массой 210 г;
• линейка измерительная металлическая длиной 300 мм, с ценой де¬
ления 1 мм;
• термометр стеклянный, диапазон измерения температуры от 0 до
плюс 100°С, с ценой деления шкалы 1°С;
• микрошприц вместимостью 10 мкл;
• секундомер;
• насос вакуумный;
• шкаф сушильный, обеспечивающий температуру нагрева 100°С;
• электроплитка закрытого типа;
• алонж изогнутый;
• воронка диаметром 50 мм;
• каплеуловитель;
• колба коническая вместимостью 100 см3 с узким горлом и градуировкой;
• пипетка вместимостью 1 см3 с ценой деления 0,01 см3;
• пипетка вместимостью 5 или 10 см3 с ценой деления 0,1 см3;
• холодильник лабораторный с прямой трубкой длиной 300 — 400 мм;
• цилиндр мерный с носиком вместимостью 50 или 100 см3;
53
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
• чашка выпарительная фарфоровая вместимостью от 100 до 150 см3;
• вакуумная смазка;
• стеклоткань прокаленная;
• калия гидроксид (или натрия гидроксид), 30% масс, водный раствор;
• твердый носитель — инертный носитель для газовой хроматографии;
• ацетон;
• гелий газообразный сжатый;
• метилен хлористый;
• полиэтиленгликоль с молекулярной массой 15000 — 20000;
• спирт н-пропиловый;
• спирт этиловый;
• эфир этиловый;
• вода дистиллированная.
2.5.3 Подготовка к анализу
2.5.3.1 Подготовка сорбента и хроматографической колонки
Необходимое количество твердого носителя, рассчитанное в зави¬
симости от объема заполняемой колонки, обрабатывают в выпаритель¬
ной чашке раствором полиэтиленгликоля в хлористом метилене (25%
полиэтиленгликоля по отношению к массе твердого носителя) в объеме,
достаточном для смачивания всей поверхности твердого носителя.
После тщательного смешивания раствора полиэтиленгликоля с
твердым носителем и выдержкой при комнатной температуре в течение
30 минут, растворитель отгоняют на водяной бане при температуре
(50±2)°С при постоянном осторожном перемешивании сорбента.
Работы проводятся в вытяжном шкафу.
Высушенный сорбент используют для наполнения колонки.
Присоединение колонки к газовой линии, монтаж, наладку и вы¬
вод хроматографа на рабочий режим производят в соответствии с ру¬
ководством по эксплуатации соответствующего типа хроматографа.
2.5.3.2 Условия проведения анализа:
• длина колонки, м 1,0 —3,0
• внутренний диаметр колонки, мм 3,0-4,0
• температура термостата колонок, °С 80 — 110
• температура термостата детектора, °С 130 — 160
• температура испарителя, °С 200 — 210
• ток детектора, мА 90 — 130
• расход гелия, см3/мин л 30 — 120
• скорость протяжки диаграммной ленты, мм/ч 600 — 720
• объем анализируемой пробы, мкл 1 —8
• внутренний стандарт ацетон
54
Глава вторая. Хроматографический анализ пироксилиновых порохов
Допуски в условиях проведения анализа уточняются при подборе
оптимальных условий разделения для данной колонки.
2.5.3.3 Построение калибровочного графика и определение ка¬
либрованных коэффициентов
В 7 колб вместимостью 100 см3 поочередно приливают 30 см3 ди¬
стиллированной воды, 20 см3 н-пропилового спирта и 0,2 см3 ацетона
(внутренний стандарт). Далее к содержимому колб добавляют соответ¬
ственно 0,03; 0,07; 0,09; 0,11; 0,13; 0,15; 0,19 г этилового спирта и этило¬
вого эфира, что будет соответствовать массовой доле этилового спирта
и этилового эфира 0,3; 0,7; 0,9; 1,1; 1,3; 1,5; 1,9%, в 10 г пороха. Навеска
этилового спирта берется в пересчете на абсолютное его содержание.
Содержимое колб перемешивают в течение 10 — 20 секунд, отби¬
рают микрошприцем пробу и хроматографируют. Анализ раствора
каждой колбы проводят три раза. На полученных хроматограммах за¬
меряют высоту пиков этилового спирта, этилового эфира и ацетона.
По трем измерениям выводят среднюю арифметическую величину.
Рассчитывают отношение высоты хроматографического пика
этилового спирта к высоте хроматографического пика ацетона и от¬
ношение высоты хроматографического пика этилового эфира к вы¬
соте хроматографического пика ацетона для каждой концентрации.
Строят калибровочный график в координатах: X — массовая доля
анализируемого вещества (этиловый спирт или этиловый эфир), Y—
отношение высоты пика анализируемого вещества этилового спирта
или этилового эфира к высоте пика ацетона.
Для определения коэффициентов калибровочного графика ис¬
пользуют метод наименьших квадратов, принимая во внимание тот
факт, что в узком диапазоне значений калибровочный график аппрок¬
симируется прямой линией вида Х=а + bY, где айв коэффициенты.
Заполняют таблицу результатами анализа калибровочных сме¬
сей отдельно для этилового спирта и отдельно для этилового эфира.
г,
X?
х.к,
11
ШШЙШ
*7
г.
*7
XiYj
£2 ^
“о"
II
ZY=
(ЕУ)2=
zx2=
1XY=
55
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
По результатам вычисляют коэффициенты а и Ъ по формулам:
b_nS*y-£*Zy
при л = 7 — число точек градуировочного графика (количество кали¬
бровочных смесей).
Затем проверяют линейную зависимость между X и Y. Для этого
вычисляют коэффициент корреляции т2 по формуле:
[»I,Y2-СгЭДлХХ2--Gx/]'
При значениях т2 в пределах 0,98—1,04 коэффициенты аиЪ ис¬
пользуют для расчета массовой доли этилового спирта и эфира в по-
рохах.
При значениях т2 , не укладывающихся в пределы 0,98— 1,04, ка¬
либровочную процедуру повторяют вновь.
Расчет калибровочных коэффициентов можно проводить, ис¬
пользуя известную программу Microsoft Excel.
2.5.4 Проведение анализа
Порох массой (10,0 0,5) г, предварительно измельченный и высу¬
шенный до постоянной массы взвешивают с точностью до четвертого
десятичного знака. Навеску помещают в реакционную колбу (поз. 1,
рис. 2), приливают 30 см3 раствора гидроксида натрия (или гидроксида
калия), 10 см3 н-пропилового спирта и 0,2 см3 ацетона.
В приемную колбу (поз. 6, рис. 2), вместимостью 100 см3, прили¬
вают 10 см3 н-пропилового спирта, 10 см3 дистиллированной воды и
присоединяют через алонж (поз.4, рис. 2), к холодильнику (поз. 3, рис. 2).
Длина трубки алонжа должна быть такой, чтобы конец ее был погру¬
жен в раствор на 0,5— 1,0 см.
Для исключения переброса раствора из приемной колбы в холо¬
дильник и реакционную колбу в стенке трубки алонжа делают отвер¬
стие диаметром 1,0-1,5 мм.
Для выравнивания давления внутри перегонной установки с ат¬
мосферным давлением в пробку (поз. 5, рис. 2), соединяющую алонж
и приемную колбу, вставляют трубку (поз. 7, рис. 2), внутренним диа¬
метром около 2 мм.
56
Глава вторая. Хроматографический анализ пироксилиновых порохов
Рис. 2. Установка для разложения:
1 — колба реакционная; 2 — плитка электрическая, 3 — холодильник, 4 — алонж, 5 —
пробка с отверстием, 6 — колба приемная, 7 — трубка.
Реакционную колбу присоединяют к холодильнику и кипятят ее
содержимое на электроплитке (поз. 2, рис. 2), до полного разложения
пороха. Объем отгона в приемной колбе должен составлять 50 см3 (по
метке на приемной колбе). Содержимое премной колбы охлаждают,
микрошприцем отбирают пробу, вводят ее в испаритель хроматографа.
Проводят два параллельных определения.
На хроматограмме пики компонентов выходят в следующем по¬
рядке: этиловый эфир, ацетон (внутренний стандарт), этиловый
спирт, н-пропиловый спирт. На полученных хроматограммах измеря¬
ют высоты пиков компонентов и рассчитывают отношение высоты
пика этилового эфира к высоте пика ацетона и высоты пика этилово¬
го спирта к высоте пика ацетона.
2.5.5 Обработка результатов
Массовую долю анализируемого вещества X, в процентах, вычи¬
сляют по формуле:
X =
(а + ЬУ)10
т
где а и Ь — калибровочные коэффициенты;
Y— величина отношения высоты пика определяемого компо¬
нента к высоте пика ацетона.
т — действительная масса навески пороха, г;
10 — расчетная масса навески пороха, г.
За результат анализа принимается среднее арифметическое зна¬
чение, округляемое до десятых долей процента.
57
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
2.6 Определение массовой доли воды
2.6.1 Определение основано на экстрагировании воды из пороха
изопропиловым спиртом с последующим газохроматографическим
анализом экстракта.
2.6.2 Аппаратура, материалы и реактивы
• хроматограф газовый аналитический с детектором по теплопровод¬
ности;
• весы лабораторные с наибольшим пределом взвешивания 200 г;
• шкаф сушильный;
• баня обеспечивающая термостатирование при температуре 100°С,
с крышкой и отверстиями для металлических стаканов;
• часы;
• термометр ртутный стеклянны лабораторный с пределом измере¬
ния температуры от 0°С до 150°С и ценой деления 0,5°С или 1°С
• эксикатор;
• микрошприц вместимостью 10 мкл;
• стакан металлический с крышкой, резиновой прокладкой из вулкани¬
зированного каучука или силиконовой резины и винтовым зажимом;
• колбы мерные вместимостью 25 см3, 100 см3 и 1000 см3;
• пипетки вместимостью 1 см3 с ценой деления 0,02 см3, 5 см3 с ценой
деления 0,05 см3 и 10 см3 с ценой деления 0,01см3;
• бумага фильтровальная или ткань мягкая;
• твердый носитель — полисорб 1 (фракции 0,3-0,5 мм);
• гелий газообразный сжатый;
• кальций хлористый, прокаленный;
• цеолит типа А, прокаленный при температуре 250 — 300°С в течение 5 ч;
• спирт изопропиловый;
• метанол;
• раствор метанола в изопропиловом спирте с массовой долей 1%.
2.6.3 Подготовка к испытанию
2.6.3.1 Подготовка прибора
Подготовку и заполнение колонки сорбентом проводят извест¬
ным методом. Заполненную колонку не присоединяя к детектору
продувают потоком сухого газа-носителя в течение 3 —5 ч, повышая
температуру термостата колонок в течение указанного времени от 30
до 170°С (для удаления сорбированных веществ с поверхности и из
пор полисорба 1). После чего температуру термостата колонок сни¬
жают до комнатной, колонку присоединяют к детектору.
2.6.3.2 Монтаж, наладку и вывод хроматографа на рабочий режим
проводят в соответствии с требованиями инструкции на хроматограф.
58
Глава вторая. Хроматографический анализ пироксилиновых порохов
2.6.3.3 Условия проведения анализа:
• длина колонки, мм 2000
• диаметр колонки, мм 3,0-4,0
• температура термостата колонки, °С 100 — 130
• температура испарителя, °С 200 —250
• температура детектора, °С 100 — 130
• расход газа-носителя, мл/мин 40 — 120
• объем вводимой пробы, мкл 4—8
Допуски в условиях проведения анализа уточняются при подбо¬
ре оптимальных условий разделения для данной колонки.
2.6.3.4 Подготовка растворителя
В колбу заливают 1000 см3 изопропилового спирта и засыпают
250 — 300 г свежепрокаленного цеолита и выдерживают не менее 48 ч.
Изопропиловый спирт в процессе работы постоянно держат над
цеолитом. Периодически, не менее одного раза в сутки определяют
массовую долю воды в 1% масс, растворе метанола в изопропиловом
спирте.
2.6.3.5 Посуда, применяемая при проведении анализа и построе¬
нии градуировочного графика, должна быть высушена при темпера¬
туре (95 — 100)°С и храниться в эксикаторе над хлористым кальцием.
2.6.3.6 Построение градуировочного графика
В мерную колбу вместимостью 100 см3 приливают 1 см3 дистилли¬
рованной воды. Содержимое колбы доводят до метки изопропиловым
спиртом.
В мерные колбы (7 штук) вместимостью 25 см3 переносят полу¬
ченный раствор в количестве 2,50; 3,75; 5,00; 6,25; 7,50; 8,75; 10,00 см3.
Это соответствует 0,50; 0,75; 1,00; 1,25; 1,50; 1,75; 2,00% воды на массу
пороха равную 1 г.
В каждую колбу добавляют по 5 см3 раствора метанола в изопро¬
пиловом спирте. Его готовят следующим образом: в мерную колбу,
вместимостью 100 см3, приливают 5 см3 метанола и доводят объем до
метки изопропиловом спиртом.
Объем каждой колбы после добавления раствора метанола в изо¬
пропиловом спирте доводят до метки изопропиловым спиртом.
Из полученных растворов микрошприцем отбирают пробы и
хроматографируют.
Для построения каждой точки градуировочного графика приго¬
товление растворов и измерение их концентрации проводят три раза.
По результатам трех измерений вычисляют среднее арифметическое
с точностью до 0,05 и строят градуировочный график, откладывая на
59
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
оси абсцисс массовую долю воды (X), а на оси ординат отношение вы¬
соты пика воды к высоте пика метанола (У).
Для определения коэффициентов калибровочного поступают
аналогичным методом, как это описано в методике определения мас¬
совых долей этилового спирта и этилового эфира.
2.6.4 Проведение анализа
Порох измельчают, около 1 г взвешивают с погрешностью не бо¬
лее 0,0002 г, помещают в металлический стакан (поз. 5, рис. 3), пред¬
варительно высушенный при температуре (95—100)°С в течение
30 — 40 мин и охлажденный в эксикаторе. Приливают 5 см3 1% масс,
раствора метанола в изопропиловом спирте.
Стакан герметично закрывают крышкой (поз. 3, рис. 3) с резино¬
вой прокладкой (поз. 4, рис. 3), зажимают винтом (поз. 1, рис. 3) на
кронштейне (поз. 2, рис. 3) и ставят в гнездо бани, нагретой до темпе¬
ратуры 98 — 100°С на время равное 1,5 ч.
Затем стакан вынимают из бани, осторожно охлаждают его содер¬
жимое на воздухе до комнатной температуры и открывают крышку.
Микрошприцем отбирают пробу, вводят в испаритель хромато¬
графа и хроматографируют. На полученных хроматограммах измеря¬
ют высоты пиков и рассчитывают
отношение высоты пика воды к
высоте пика метанола.
2.6.5 Обработка результатов
Массовую долю воды (X), в
процентах, вычисляют по формуле:
х = (о + ЬУ)1
т
где а и Ь — калибровочные коэф¬
фициенты;
Y— величина отношения вы¬
соты пика воды к высоте
пика метанола;
т — действительная масса на¬
вески пороха, г.
За результат измерения при¬
нимают среднее арифметическое
двух параллельных определений с
точностью до 0,1% масс.
Рис. 3. Стакан металлический:
1 — зажим винтовой, 2 — кронштейн,
3 — крышка, 4 — прокладка резиновая,
5 — стакан.
60
ГЛАВА
МЕТОДЫ ОПРЕДЕЛЕНИЯ
СТОЙКОСТИ ПОРОХОВ
Стабильность физико-химических свойств является
основным требованием к порохам при практиче¬
ском их использовании. Задача производителя — производство поро-
хов с высокими и постоянными качествами, задача потребителя —
сохранение первоначальных свойств до момента использования. Вре¬
мя этого цикла и представляется как срок хранения. Чем выше срок
хранения, тем и выше эффективность использования порохов.
Срок хранения порохов -определяется совокупностью ряда фак¬
торов, в число которых входят, в основном, условия хранения (темпе¬
ратура, влажность) и определенные, присущие каждому виду пороха,
исходные качества, определяющие его способность более или менее
длительно сопротивляться процессу разложения.
Первые опыты производства и эксплуатации дымных порохов,
показали, что "способность же пороха хорошо сохраняться какъ въ
магазинахъ, такъ и во время перевозки, зависитъ отъ фигуры, поли¬
ровки и твердости зерна, отъ количества употребленной серы, а въ
особенности отъ качества селитры, отъ которой зависитъ поглощеше
порохомъ воды, главной причины порчи пороха" [1]. Из этого четко
видны рекомендации изготовителю и потребителю. Изготовитель
должен применять чистые компоненты, хорошо их измельчать, плот¬
но их спрессовывать, поверхность хорошо сглаживать, а потребитель
должен "держать порох сухим". Следуя этим рекомендациям, можно
было бы дымные пороха хранить сколько угодно долго.
Методов определения стойкости дымных порохов при нагревании
не существует. В случае с дымными порохами правильнее говорить о
физической стойкости. В данной ситуации методом, который косвенно
характеризовал бы стойкость дымных порохов, является метод оценки
гигроскопичности. Метод по гигроскопичности [94] характеризует спо¬
собность пороха поглощать влагу из паровой фазы. Эта способность и
является обобщающим фактом качества и стабильности дымных порохов.
61
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
Появление взрывчатых веществ, которые в основном предста¬
вляют собой производные органических веществ, и пироксилиновых
порохов, основу которых составляют производные природного поли¬
мера (целлюлозы), поставило задачу более глубокого исследования
процесса химической стойкости данных соединений. Возникла
необходимость исследования механизма и кинетики разложения
взрывчатых веществ и пироксилиновых порохов и создание норма¬
тивных методов оценки их стойкости.
Первые обобщенные результаты исследования химической
стойкости находим у А. Сапожникова [1]. Он пишет, что “причины,
который могуть иметь особенно значительное вл1яше на химическую
стойкость взрывчатыхъ веществъ бываютъ двухъ родовъ. Первая и
самая важная изъ нихъ — степень чистоты получаемого при фабри-
кащи продукта въ смыслъ отсутств1я въ немъ постороннихъ прими-
сей, въ особенности применяемыхъ при нитрацш кислотъ азотной и
серной хотя бы въ самыхъ незначительныхъ количествахъ. Вторая
причина — это услов1я хранешя, при чемь приходится считаться глав-
нымъ образомъ съ температурой хранилища и съ состояшем воздуха
въ отношенш содержашя въ немъ влажности".
Сапожников дает и описание известных на тот момент методов
(проб) оценки химической стойкости — проба Абеля, проба Гесса,
проба по температуре вспышки, проба Вьеля, проба Бергмана-Юнка,
проба по потере массы. Обилие методов на тот период (1912 год) ука¬
зывает на важность оценки химической стойкости и широкий круг
исследователей в данной области. Испытания порохов по описанным
пробам проводятся при повышенной температуре, как сейчас гово¬
рят — проводят форсированное старение.
Более полное описание методов испытания химической стойко¬
сти порохов представлено Г.К. Клименко [41].
Им дана следующая классификация методов испытания стойкости:
• количественные методы;
• методы, основанные на термическом разложении в отсутствии про¬
дуктов разложения;
• методы, основанные на термическом разложении в присутствии
продуктов разложения;
• методы, основанные на определении температуры вспышки и вре¬
мени, протекающего до вспышки;
• методы, основанные на изучении разложения с явно выраженным
процессом автокатализа;
• методы без нагревания испытываемого пороха.
62
Глава третья. Методы определения стойкости порохов
Из всего этого многообразия "проб" в СССР в практике порохо-
делия широкое распространение получила проба Вьеля (лакмусовая
проба) для анализа порохов и проба Бергмана- Юнка для анализа ни¬
тратов целлюлозы.
Проба Вьеля (лакмусовая проба) [95 — 96] предполагает нагрева¬
ние порохов (поз. 1, рис. 4) в герметично закрытых стаканчиках (поз. 3,
рис. 4) в присутствии лакмусовой бумажки (поз. 2, рис. 4) при темпе¬
ратуре 106,5±0,5°С.
Рис. 4. Стаканчик Вьеля с затвором из нержавеющей стали:
1 — образец пороха, 2 — лакмусовая бумажка, 3 — стаканчик, 4 — резиновая прокладка,
5 — стальная крышка, 6 — стальная пружина.
Лакмусовая проба проводится в трех вариантах:
1 Простая проба:
• однократное нагревание в течение 7 ч.
2 Повторная проба:
• десятикратное циклическое нагревание по 7 ч, проветривание по¬
сле каждого цикла при комнатной температуре с заменой стакан¬
чика и лакмусовой бумажки.
2.1 нормальная:
• нагревание 7 ч, проветривание вне стаканчиков в шкафу при ком¬
натной температуре 17 ч.
63
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
2.2 ускоренная:
• нагревание 7 ч, проветривание вне стаканчиков в шкафу при ком¬
натной температуре 2 ч.
3 Повторная (нормальная и ускоренная проба) до одного часа:
• испытание является продолжением испытания по повторной пробе
и проводится до тех пор, пока в последнем цикле красное окраши¬
вание лакмусовой бумажки или бурые пары появятся в течение
первого часа от начала одного из последних циклов.
Показателем химической стойкости по пробе Вьеля служит время,
от момента начала и до окончания испытания:
по простой пробе:
• не менее 7 ч;
по повторным пробам (нормальной и ускоренной):
• не менее 70 ч (10 циклов по 7 ч);
по повторным пробам (нормальной и ускоренной) до одного часа:
• сумма времени всех циклов.
В конце 80 годов прошлого столетия пробу Вьеля заменили на
пробу по газовыделению.
Проба Бергмана-Юнка [97] применяется до настоящего времени.
Во многих странах, производящих пироксилиновый порох, для
оценки химической стойкости до настоящего времени применяется
так называемая проба "нагреванием при 65°С " [98]. Результатом ис¬
пытания принимается время до появления паров красного (бурого)
цвета при нагреве навески пороха при 65°С. Навеска, которая берет¬
ся для испытания в США и Франции, составляет 45 г, Дании — 5 г, в
Швеции — 250 г. При этом в Швеции считается, что если порох выдер¬
жал нагревание равное или свыше 6 месяцев, то порох может хранить¬
ся без ограничений, если порох выдержал нагревание равное 4 — 6 ме¬
сяцев, то его нельзя хранить.
Существует ускоренный вариант данной пробы. Он основан на
нагревании навески пороха в стандартной пробирке при 134,5°С и
определении времени до начала изменения цвета метилвиолетовой
бумажки, появления бурых паров и вспышки. Для стойких пирокси¬
линовых порохов параметры данной пробы следующие:
• изменение цвета бумажки — 1 ч;
• образование бурых паров — 2 ч;
• вспышка — 5 ч.
Данная проба нашла широкое распространение в США ( так
называемая "американская проба”).
64
Глава третья. Методы определения стойкости порохов
3.1 Определение гигроскопичности дымных порохов
3.1.1 Определение основано на выдержке первоначально сухого
дымного пороха в атмосфере воздуха с влажностью 92%, в течение 12 ч
и расчета количества поглощенной влаги.
3.1.2 Аппаратура и реактивы
• весы аналитические с погрешностью взвешивания не более 0,0002 г;
• набор гирь общей массой 210 г;
• баня водяная;
• эксикатор;
• калия нитрат;
• вода дистиллированная.
3.1.3 Подготовка к анализу
3.1.3.1 Приготовление насыщенного водного раствора нитрата
калия
Около 1200 г нитрата калия растворяют в 1 л дистиллированной
воды при температуре 60±5°С.
3.1.3.2 Подготовка эксикатора
Приготовленный горячий раствор нитрата калия наливают в эк¬
сикатор в таком количестве, чтобы высота слоя составила примерно 3 см.
Раствор в эксикаторе охлаждают, после чего эксикатор плотно зак¬
рывают крышкой.
3.1.4 Проведение анализа
Образец дымного пороха около 10 г, высушенный до постоянной
массы, помещают в бюксе в эксикатор. В эксикатор устанавливают
одновременно не более 6 бюксов. С бюксов снимают крышки и кладут
их рядом с бюксами. Эксикатор закрывают крышкой. Бюксы выдер¬
живают в эксикаторе 12 ч при температуре 20=*=2вС. Снимают крыш¬
ку с эксикатора, бюксы закрывают, вынимают их из эксикатора и
взвешивают.
3.1.5 Обработка результатов
Гигроскопичность пороха (X) в процентах вычисляют по формуле
х=т\ т2..100|
т
где тх — масса бюкса с навеской после увлажнения, г;
т2 — масса бюкса с навеской до увлажнения, г;
т — навеска пороха, г.
Производят два параллельных определения, по результатам кото¬
рых вычисляют среднее арифметическое с погрешностью не более 0,1%.
65
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
3.2 Определение химической стойкости нитратов целлюлозы
3.2.1 Определение химической стойкости основано на определе¬
нии количества окислов азота, выделенных 1 г нитроцеллюлозы при
нагревании в течение 2 ч при температуре (132,0±0,4) °С. Количествен¬
ное определение окислов азота основано на химических реакциях:
KI03 + 5KI + 6HN03 = 3I2 + 6KN03 4- ЗН20
I2 + 2Na2S203 = 2NaI + Na2S4Oe
3.2.2 Аппаратура, материалы и реактивы
• аппарат для определения стойкости;
• весы лабораторные с наибольшим пределом взвешивания 200 г;
• набор гирь для технических анализов и взвешиваний с повышен¬
ной точностью общей массой 210 г;
• термометр стеклянный, диапазон измерений температуры от 0 до
плюс 150°С, с ценой деления шкалы 0,2°С;
• часы;
• бюретка с автоматическим нулем и склянкой, вместимостью 25 см3
с ценой деления 0,1 см3;
• воронка;
• колба коническая вместимостью 100 см3 или 250 см3;
• пипетка вместимостью 25 см3 с ценой деления 0,2 см3 или 0,1 см3;
• пипетка вместимостью 1 см3 с ценой деления 0,01 см3;
• пробирка для определения стойкости с двумя метками и поглоти¬
тельной насадкой;
• пробка резиновая;
• стаканчик для взвешивания или лоток из алюминия размером
(100±5) мм х (60±5) мм;
• цилиндр мерный вместимостью 100 см3 с ценой деления 1,0 см3;
• вазелин для технических целей;
• калий иодноватокислый, водный раствор с массовой долей калия
иодноватокислого 3,0%;
• калий йодистый, водный раствор с массовой долей калия йодистого
5,0%;
• натрий серноватистокислый, водный раствор с концентрацией нат¬
рия серноватистокислого 0,05 моль/дм3;
• кислота азотная, водный раствор с концентрацией 0,02 моль/дм3;
• крахмал растворимый, выдерживающий испытания на чувстви¬
тельность к иоду по 3.2. 3.3;
• вода дистиллированная.
3.2.3 Подготовка к анализу
3.2.3.1 Подготовка пробы нитроцеллюлозы
66
Глава третья. Методы определения стойкости порохов
Образцы нитроцеллюлозы перед испытанием сушат при темпе¬
ратуре 110°С в течение 1 ч.
3.2.3.2 Приготовление раствора крахмала
Раствор крахмала с массовой долей 0,4% приготавливают следу¬
ющим образом: 1 г крахмала смешивают с 5 —10 см3 воды, превращая
в жидкую кашицу, которую медленно вливают в 250 см3 кипящей воды.
Раствор кипятят в течение 2 минут и охлаждают.
3.2.3.3 Определение чувствительности крахмала к иоду
3.2.3.3.1 Применяемые реактивы и растворы
• вода дистиллированная;
• иод металлический, водный раствор с концентрацией йода 0,1 моль/дм3;
• натрий серноватистокислый, водный раствор с массовой долей нат¬
рия серноватистокислого 0,05 моль/дм3.
3.2.3.3.2 Проведение анализа
0,5 см3 раствора крахмала, приготовленного по 3.2.3.2, помещают
в коническую колбу вместимостью 250 см3, прибавляют 100 см3 воды
и 0,05 см3 раствора иода.
Крахмал считают соответствую¬
щим требованиям по качеству если по¬
сле прибавления раствора иода наблю¬
дается синее окрашивание раствора,
исчезающее при прибавлении 0,05 см3
раствора серноватистокислого натрия.
3.2.4 Проведение анализа
2,00 г нитроцеллюлозы, подгото¬
вленной по 3.1, взвешивают в стаканчи¬
ке. Образец нитроцеллюзы при помощи
воронки помещают в пробирку (поз. 1,
рис. 5) для определения химической
стойкости. Для равномерного распреде¬
ления, уплотнения и меньшего оседа¬
ния нитроцеллюлозы на стенках про¬
бирки ее слегка постукивают дном о по¬
верхность стола. При этом навеска
должна уплотниться до метки 1.
Пробирку, наполненную нитро¬
целлюлозой, соединяют с поглотитель¬
ной насадкой (поз. 2, рис. 5). Шлиф нас¬
адки перед соединением должен быть
смазан вазелином, чтобы в пробирку не
Рис. 5. Пробирка для опреде¬
ления стойкости:
1 — пробирка, 2 — поглотительная
насадка, 3 — колпачок
67
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
попал воздух. В насадку наливают 20 — 25 см3 бидистиллированной
воды и на трубку насадки надевают колпачок (поз. 3, рис. 5).
Подготовленную пробирку вставляют в гильзу термостата, на¬
гретого до температуры (132,0±0,4)°С, и захватывают за шейку про¬
бирки металлическим держателем, соединенным со шнуром.
Дно пробирки должно отстоять от дна гильзы термостата на 20 мм,
что обеспечивается введением в гильзу деревянного столбика соот¬
ветствующей высоты, изготовленного из древесины лиственной по¬
роды. Гильзы с пробиркой должны входить в термостат на 0,75 своей
высоты. Одновременно в центральную гильзу термостата помещают
пробирку без насадки с термометром, укрепленным с помощью пробки.
Середина ртутного резервуара термометра должна находиться
на расстоянии около 25 мм от дна пробирки.
После 2 ч нагревания при температуре (132,0=Ь0,4)°С пробирку с
нитроцеллюлозой вынимают из гильзы термостата при помощи шну¬
ра и охлаждают над гильзой в течение 30 мин (при охлаждении вода
из насадки засасывается внутрь пробирки). Перелив остатка воды из
поглотительной насадки в пробирку проводят на лабораторном столе.
Насадку тщательно несколько раз промывают водой, и промывные
воды собирают в пробирку. Содержимое пробирки хорошо встряхи¬
вают и доводят дистиллированной водой до отметки 50 мл.
В коническую колбу наливают 5 см3 раствора йодистого калия, 3 см3
раствора иодноватокислого калия и перемешивают. Затем из пробирки
отбирают пипеткой 25 см3 водной вытяжки (в случае необходимости
содержимое пробирки фильтруют) и приливают ее к содержимому
конической колбы. Выделившийся иод оттитровывают раствором
серноватистокислого натрия.
В конце титрования в качестве индикатора добавляют 4 — 5 ка¬
пель раствора крахмала и титруют до обесцвечивания раствора.
3.2.5 Обработка результатов
Объем окислов азота X, см3, выделенных 1 г нитроцеллюлозы,
вычисляют по формуле
Х = [У + (У2-^)]1,12лХ,
где V — объем 0,05 моль/дм3 раствора серноватистокислого натрия, из¬
расходованного на титрование испытуемого раствора, см3;
Vj — объем 0,05 моль/дм3 раствора серноватистокислого натрия,
израсходованного на титрование испытуемого раствора
при определении щелочности, см3;
68
Глава третья. Методы определения стойкости порохов
V2 — объем 0,05 моль/дм3 раствора серноватистокислого натрия,
израсходованного на титрование при контрольном опреде¬
лении, см3;
п = 1 при титровании 25 см3 водной вытяжки или п = 0,5 при титро¬
вании всего объема вытяжки;
1,12 — количество окислов азота, соответствующее 1 см3 точно
0,05 моль/дм3 раствора серноватистокислого натрия, см3;
К — поправочный коэффициент для приведения концентрации
раствора серноватистокислого натрия к точно 0,05 моль/дм3.
Производят два параллельных определения, по результатам кото¬
рых вычисляют среднее арифметическое и округляют его до 0,01 см3.
Каждое из параллельных определений должно соответствовать
норме, установленной для нитроцелюлозы.
3.3 Определение химической стойкости пироксилиновых порохов
3.3.1 Метод основан на измерении давления газообразных про¬
дуктов термического разложения пороха в герметично закрытом ре¬
акционном стакане (первичный преобразователь) при температуре
125,0±0,5°С в течение 4,5 ч.
3.3.2 Аппаратура, материалы и реактивы
• измерительно-вычислительный комплекс "Вулкан-В", состоящий
из 4 термостатов (в термостате имеется 8 гнезд для помещения стакан¬
чиков (первичных преобразователей), первичных преобразователей,
индивидуальных терморегуляторов, блока пневмоэлектрологики,
измерительных преобразователей давления, электробарометра, пу¬
скателя, термометра цифрового, информационно- измерительной
системы и цифропечати;
• весы лабораторные с наибольшим пределом взвешивания 200 г;
• набор гирь для химических анализов и взвешиваний с высокой точ¬
ностью общей массой 210 г;
• термометр стеклянный, диапазон измерения температуры от 0 до
плюс 150°С, с ценой деления шкалы 2°С;
• шкаф сушильный, обеспечивающий температуру нагрева до 100°С;
• эксикатор диаметром от 136 до 290 мм;
• коробочки из алюминия штампованные размером 85 х 60 х 30 мм;
• марля;
• прокладки уплотнительные из резины маслокислотостойкой, выдер¬
живающие температуру (125±5)°С;
• ацетон;
• спирт этиловый;
69
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
• кальций хлористый
• воздух сжатый, давлением от 0,25 до 0,30 МПа, очищенный от мас¬
ла и влаги
• вода дистиллированная.
3.3.3 Подготовка к анализу
В основу работы измерительно-вычислительного комплекса
"Вулкан-В" заложен компенсационный принцип измерения, по кото¬
рому давление газообразных продуктов разложения (Рр) при нагре¬
вании пороха в реакционном объеме (первичного преобразователя,
поз. 6, рис. 6) вызывает прогиб мембраны (поз. 5, рис. 6) и замыкание
ее контакта с иглой (поз. 6, рис. 6). Сжатый воздух (Рк), подаваемый в
компенсационный объем первичного преобразователя, в этот момент
возвращает мембрану в исходное состояние до размыкания контакта.
При этом регистрируется давление компенсации.
I 3.3.3.1 Подготовка измери-
Рис. 6. Первичный преобразователь:
1 — вкладыш с образцом, 2 — реакционный
стакан, 3 — накидная гайка, 4 — прокладка
резиновая, 5 — мембрана, 6 — контактная
игла, 7 — мембранный узел.
тельно-вычислительного ком¬
плекса "Вулкан-В"
Монтаж, наладку и вывод
измерительно-вычислительно¬
го комплекса "Вулкан-В" на ра¬
бочий и температурный режи¬
мы производят в соответствии с
руководством по эксплуатации.
Прокладки резиновые
(уплотнительные, поз. 4, рис. 6)
проверяют визуально на отсут¬
ствие трещин при изгибе.
3.3.4 Проведение анализа
3.3.4.1 Образцы пороха
массой 1 г, предварительно из¬
мельченные и высушенные до
постоянной массы, взвешивают
с точностью до третьего деся¬
тичного знака. Помещают их во
вкладыши (поз. 1, рис. 6), кото¬
рые вставляют в реакционные
стаканы (поз. 2, рис. 6) первич¬
ного преобразователя. При по¬
мощи накидной гайки (поз. 3,
рис. 6) реакционные стаканы
70
Глаш! третьи. Методы определения стойкости порохов
подсоединяют к мембранным узлам (поз. 7, рис. 6) первичного преоб¬
разователя.
3.3.4.2 Собранные таким образом первичные преобразователи,
не допуская их переворачивания, поочередно, в соответствии с их но¬
мерами, помещают в гнезда термостата, предварительно нагретого до
температуры (125,0±0,5)°С. На первичный преобразователь надева¬
ют термоизоляционные заглушки. Подсоединяют пневморазъемы
термостата к первичным преобразователям и пневмотумблером подают
сжатый воздух в термостаты.
3.3.4.3 Проверяют герметичность линии сжатого воздуха, для
этого нажимают кнопку "ИЗМ" в ручном режиме и следят за повы¬
шением давления в линии. Если давление в линии установилось ниже
266,62 кПа (2000 мм рт. ст.), то находят течь и устраняют ее.
3.3.4.4 Устанавливают интервал опроса первичных преобразова¬
телей "10 МИН.", время — "00 Ч 00 МИН" и кнопкой "АВТ." на лице¬
вой панели блока управления измерительно-вычислительного ком¬
плекса "Вулкан-В" включают автоматический режим работы.
3.3.4.5 После первого часа термостатирования образцов, интер¬
вал опроса первичных преобразователей устанавливают "30 МИН".
3.3.4.6 Испытание образцов проводят в течение 4,5 часов, после
чего измерительно-вычислительный комплекс "Вулкан-В" отключают
от электросети и от линии сжатого воздуха. Первичные преобразовате¬
ли извлекают из термостата, охлаждают до комнатной температуры и
отсоединяют от воздушной линии.
3.3.4.7 Реакционные стаканы отсоединяют от мембранных узлов
и из стаканов извлекают вкладыши с порохом. Реакционные стаканы,
вкладыши и мембранные узлы протирают марлей, смоченной ацето¬
ном, затем промывают дистиллированной водой и протирают марлей,
смоченной этиловым спиртом, и сушат при температуре 95— 100°С в
течение 1 ч.
Резиновые прокладки промывают спиртом этиловым и сушат
при температуре 95— 100°С не менее 1 ч.
3.3.5 Обработка результатов
За результат испытания принимают давление газовыделения,
которое вычисляют по формуле
Р3,5 = Р4,5 *“ Р\,0*
гдеР35— давление газовыделения от собственного термораспада за
3,5 ч, кПа (мм рт. ст.);
71
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
Р4 5 — давление газовыделения за 4,5 ч, кПа, (мм рт. ст.);
Pj о — давление в ПП за первый час термостатирования (давление
за счет расширения воздуха и испарения летучих веществ),
кПа, (мм рт. ст.).
Проводят два параллельных определения. Расхождение между
результатами параллельных определений не должно превышать 2,66 кПа
(20 мм рт. ст.). За результат анализа принимают наибольшее значение
давления газовыделения.
72
ГЛАВА
ТЕРМИЧЕСКИЕ МЕТОДЫ
АНАЛИЗА ПОРОХОВ
Термический анализ является одним из методов фи¬
зико-химического анализа и служит для исследо¬
вания процессов, происходящих в веществе при нагревании или ох¬
лаждении. Термин "термический анализ" объединяет целую группу
методов [99], которые отличаются между собой измеряемой характери¬
стикой и аппаратурным оформлением. Если измеряется температура
образца, метод называют термографией, масса — термогравиметри¬
ей, размер — дилатометрией, количество выделившегося тепла — ка¬
лориметрией и т.д. Все методы термического анализа объединяет
то, что образец исследуемого вещества непрерывно нагревается или
охлаждается по какому-то определенному заданному закону, напри¬
мер линейному, когда скорость нагрева (охлаждения) постоянна, или
ступенчатому, когда скорость нагрева меняется дискретно. Во всех
методах термического анализа эксперименты проводят по абсолют¬
ной или дифференциальной схемах. При абсолютной схеме реги¬
стрируемую характеристику измеряют непосредственно в исследуе¬
мом веществе, а при дифференциальной — разность величины этой
характеристики, относящейся к исследуемому веществу и специаль¬
ному эталону.
В любом веществе под действием тепла могут развиваться раз¬
личные физические превращения (испарение поверхностной влаги,
плавление, кипение, рекристаллизация) и химические реакции (де¬
струкция, деструктивное структурирование, взаимодействие между
компонентами смеси). Изучать последовательность и кинетические
закономерности таких превращений возможно лишь по комплексу
методов термического анализа, в частности- термографии (ДТА) и
термогравиметрии (TG), в которых исследования проводятся в неизо¬
термическом режиме, т.е. в условиях непрерывно и монотонно ме¬
няющейся температуры.
73
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
4.1 Исследование кинетики разложения при неизотермическом
нагреве
4.1.1 Для кинетических исследований разложения и определе¬
ния энергии активации и порядка реакции при неизотермическом на¬
греве используется дериватограф Q-1500D, принципиальная схема
которого приведена на рис. 7.
2
Рис. 7. Принципиальная схема дериватографа:
1 — керамическая трубка, 2 — держатель проб, 3 — печь, 4 — выключатель регулиро¬
вания нагрева, 5 — усилитель, 6 — электромагнит, 7 — катушка, 8 — весы, 9 — диффе¬
ренциальный трансформатор преобразования сигнала TG, 10 — усилитель, 11 — реги¬
стрирующее устройство, 12 — усилитель.
Для простейшей одностадийной реакции разложения вещества
О, которую можно представить уравнением О = В + С (где В — кон¬
денсированное вещество, С — газ) справедлива следующая зависи-
n dW
мость скорости превращения Рт = от концентрации W:
dx
P- = ™-=KWn, (4.1)
dx
где К — константа скорости превращения.
Предполагается, что константа скорости К подчиняется уравне¬
нию Аррениуса. Р
__£о_
K = Ze RT, (4.2)
74
Глава четвертая. Термические методы анализа порохов
где Z — предэкспоненциальный множитель;
Еа — энергия активации;
R — универсальная газовая постоянная;
Т — абсолютная температура.
Уравнение Аррениуса (4.2) только в некоторых случаях точно
применимо к деструкции полимеров (порохов) из-за сложности этого
процесса, представляющего собой ряд последовательных и парал¬
лельных реакций, разграничить которые зачастую не представляется
возможным.
Предложено несколько методов определения кинетических пара¬
метров термодеструкции высокомолекулярных соединений, основан¬
ных на математической обработке кривых TG, из которых наиболь¬
шее применение нашел метод Фримена и Кэррола [100J. Согласно
этому методу для текущей скорости разложения Рт конденсирован¬
ного вещества Q, соответствующую температуре Т в данный момент
времени, справедливо выражение
Pr=^ = -e'*TWn, (4.3)
dT q
где Рт— скорость разложения, мг/К;
q — скорость нагрева, К/мин;
W — масса образца, расходуемого в реакции, мг;
Т — температура, К;
Еа — энергия активации, кДж/моль;
п — порядок реакции;
R — универсальная газовая постоянная, кДж/моль К.
Если уравнение 4.3 применять для двух температур при q = const,
то после логарифмирования и вычитания одного из другого получа¬
ются следующие выражения:
Таким образом, из одной кривой TG могут быть найдены величи¬
ны Еа и п. Для этого необходимо построить зависимости lg Р от lg W и
lg Р от (1/Г). Наклоны прямых равны в первом случае порядку реак¬
ции (п) и во втором — энергии активации (Еа).
(4.4)
(4.5)
(4.6)
75
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
Преимущество этого метода в том, что для непрерывного изучения
кинетики в широком интервале температур требуется сравнительно
немного данных. Основной недостаток заключается в нахождении
скорости разложения путем проведения касательных к крутым
участкам термограммы.
4.1.2 Аппаратура, посуда, реактивы
• дериватограф типа Q-1500D фирма МОМ ВНР;
• весы лабораторные типа ВАР-200;
• печь муфельная типа СНОЛ-1;
• секундомер механический;
• расходомер пенный;
• измельчитель;
• оксид алюминия, прокаленный при 1000°С;
• спирт этиловый;
• вода дистиллированная;
• гелий газообразный.
4.1.3 Подготовка к испытанию
Образец полимера (пороха) измельчают до размеров не более
1x1 мм.
Вывод дериватографа на рабочий режим проводят согласно ин¬
струкции по эксплуатации.
Условия проведения испытания:
• инертное вещество оксид алюминия
• навеска образца, мг 50 — 200
• навеска инертного вещества, мг 50 —200
• интервал температур, °С 20 — 500
• скорость нагрева печи, °С/мин 2,5 — 10,0
• чувствительность термовесов ГС, мг 50 — 100
• чувствительность канала TG самописца 250 — 500
• чувствительность канала Д ТА самописца 100 — 1000
• чувствительность канала Д ГС самописца 250—2500
• чувствительность Т, °С 500 — 1000
Оптимальные условия дифференциально-термического анализа
подбираются для конкретного вида полимера (пороха).
4.1.3 Проведение испытания
4.1.3.1 Проведение испытания в атмосфере воздуха
Один тигель с помещенной в него навеской (50 — 200 мг) инерт¬
ного вещества устанавливают на дальний спай термопары, а другой
(пустой) тигель без навески — на ближний спай термопары. При вы-
76
Глава четвертая. Термические методы анализа порохов
бранной чувствительности TG устанавливают шкалу весов на ноль и
арретируют весоизмерительную систему.
Навеску пороха, равную навеске инертного вещества, помещают
в предварительно снятый с термопары пустой тигель. Постукиванием
тигля по твердой поверхности добиваются равномерного уплотнения
навески по объему тигля. На термовесовом блоке переключателем
устанавливают величину навески образца. Затем вновь помещают ти¬
гель с навеской пороха на ближний спай термопары и опускают печь
(поз. 3, рис. 7).
Задают выбранный режим работы дериватографа, оптимальный
для конкретного исследуемого образца. Проводят термический анализ
образца согласно инструкции по эксплуатации прибора.
По достижении заданной температуры нагрева отключаются
печь и блоки регистрации. Печь поднимают, после охлаждения осно¬
вания печи снимают тигли с термопары.
Охлажденные тигли промывают дистиллированной водой, проти¬
рают ватой или бязью, смоченной в этиловом спирте, и прокаливают
в муфельной печи при 800— 1000°С в течение 10 — 20 мин.
Проводят два параллельных испытания.
4.1.3.2 Проведение испытания в инертной среде
Испытания образцов полимера в инертной среде проводят анало¬
гично испытаниям по пункту 4.1.3.1, предварительно установив подачу
гелия в печь со скоростью 20 — 40 мл/мин (по пенному расходомеру).
По окончании испытания отключают подачу гелия.
4.1.4 Обработка резуль¬
татов [101]
4.1.4.1 По результатам
экспериментов получают де-
риватограмму, на которой на¬
несены 4 кривые: 1 — кривая
Т — изменение температуры
в образце, 2 — кривая ДТА —
изменение скорости темпера¬
туры образца, 3 — кривая TG—
изменение массы образца, 4 —
кривая ATG — изменение
скорости массы образца. Ти¬
пичная дериватограмма (без
кривых ДТА и ДГС) представ¬
лена на рисунке 8.
Рис. 8. Дериватограмма образца.
77
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
4.1.4.2 Энергию активации и порядок реакции процесса де¬
струкции пороха рассчитывают для участка изменения массы вещест¬
ва (кривая TG) до момента вспышки по экспериментальной деривато-
грамме. На этой части кривой TG отмечают четыре точки и определяют
в каждой точке:
• убыль массы (мг) — Wv W2, W3, W4;
• соответствующие ей температуры (°С) — tv t2, t3, t4;
• соответствующие ей температуры по шкале Кельвина — Tv Т2, Т3, Т4;
• обратные величины— 1 /Т{, 1 /Г2, 1 /Т3, 1 /Т4;
• температурные скорости разложения — РГ1, Рп, Р73, РТ4.
Скорость разложения в каждой точке кривой TG равна тангенсу
угла наклона касательной к кривой в заданной точке, т.е для точки 2
температурная скорость Рт разложения полимера равна:
Д G ВС
Рт = ——- = tgBAC = ——
1 АТ * АВ
Далее с учетом уравнения (4.6)
• рассчитывают — Рх1, Рх2, Рх3, Рх4;
• определяют логарифмы — lgPTlr 1дРТ2* 1д^тз»
• определяют логарифмы — lgVV^, lgVV^, lgVV^, lgVl^;
• строят графические зависимости:
• логарифмическую, Р= f(W) (рис. 9)
• полулогарифмическую lgPx=/^j (рис. 10)
Построенные зависимости в обоих случаях должны быть линей¬
ными. Из логарифмической зависимости Р =/ (W) определяют поря-
Рис. 9. Логарифмическая зависи- Рис. 10. Полулогарифмическая зависи¬
мость изменения скорости превра- мость изменения скорости разложения
щения от концентрации вещества, вещества от обратной температуры.
78
Глава четвертая. Термические методы анализа порохов
Из полулогарифмической зависимости скорости превращения
от обратной температуры определяют константу скорости К:
tf=tgrp =
AigPt
#)
(4.8.)
Поскольку из уравнений (4.1 и 4.2) следует, что
Еп
К = -
2,303 *8,31 • /С
2,303R
то Еа = —г- ■ = 19,139-10 3 К (кДж/моль);
а 1000
при Д = 8,31 кДж/моль • К.
4.2 Исследование кинетики разложения при изотермическом
нагреве
4.2.1 Исследование кинетики термического разложения порохов
при изотермическом нагреве обычно проводят на установке с исполь¬
зованием стеклянного реакционного сосуда с мембранным маноме¬
тром Бурдона (поз. 2, 3, 4, 5, б, 8, рис. 12) ложечного типа.
Установка для исследования термического разложения порохов
в вакууме состоит из стеклянных манометров Бурдона, вакуумной
установки для вакуумирования манометров Бурдона (рис. 11) вакуум¬
ной установки для проведения кинетических исследований (рис. 12)
и жидкостного термостата (поз. 7, рис. 12).
Рис. 11. Схема высоковакуумной установки:
1 — стеклянный манометр Бурдона, 2 — вакуумный шланг, 3 — калиброванный шарик,
4 — сосуд Дьюара, 5,6,7,8,9,10—двуходовые вакуумные краны, 11 — ртутный манометр,
12 — вакуумметр, 13 — диффузионный масляный насос, 14 — форвакуумная емкость,
15, 16 — трехходовые вакуумные краны.
79
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
Рис. 12. Установка по исследованию кинетики разложения:
1 — навеска, 2 — реакционный сосуд манометра Бурдона, 3 — отросток для ввода наве¬
ски, 4 — отросток для припайки манометра к высоковакуумной установке, 5 — компен¬
сационный сосуд манометра Бурдона, б — серповидная мембрана, 7 — жидкостной
термостат, 8 — стрелка манометра Бурдона, 9 — бумажные указатели, 10 — вакуумный
шланг, 11 — ртутный манометр, 12 — вакуумные краны, 13 — расширитель.
Вакуумная установка (рис. 11) для откачивания манометров Бур¬
дона (поз.1, рис. 11) состоит из ртутного манометра (поз.11, рис. 11)
для контроля перепада давления, рессивера и буферных емкостей
(поз. 14, рис. 11), которые служат для уменьшения толчков при сбросе
давления из системы вакуумных кранов (поз. 5, 6, 7, 8, 9, 10, рис. 11),
которые хорошо притерты, термопарного вакуумметра (поз. 12, рис. 11),
форвакуумного и диффузионного (поз. 13, рис. И) насосов.
Вакуумная установка для проведения кинетических исследова¬
ний (рис. 12) состоит из манометра Бурдона, ртутного манометра
(поз. 11, рис. 12) для измерения давления воздуха, подаваемого для
компенсации давления газообразных продуктов разложения, систе¬
мы вакуумирования и системы краников (поз. 12, рис. 12) для регули¬
рования давления в системе.
Манометр Бурдона представляет собой стеклянный прибор, со¬
стоящий из реакционного сосуда (поз. 2, рис. 12), тонкостенной сте¬
клянной мембраны серповидного сечения (поз. 6, рис. 12), заканчи¬
вающегося стрелкой (поз. 8, рис. 12), и трубки окружающей мембра¬
ну (поз. 5, рис. 12). Реакционный сосуд, в который помещается
навеска образца (поз. 1, рис. 12), сообщается с внутренним простран¬
ством мембраны. Образующиеся при разложении пороха газы давят
на мембрану и отклоняют стрелку от первоначального положения.
При подаче воздуха в трубку, окружающую мембрану (поз. 5, рис. 12),
80
Глава четвертая. Термические методы анализа порохов
стрелка возвращается в нулевое положение. Давление подаваемого
воздуха отсчитывается по ртутному манометру (поз. 11, рис. 12).
Манометр Бурдона вышеописанной конструкции является при¬
бором одноразового использования. Часто при исследованиях ис¬
пользуется и манометр Бурдона, конструкция которого позволяет его
использовать неоднократно (рис. 13).
Реакционная часть данного манометра (поз. 8, рис. 13) является
разъемной и состоит из двух частей: верхней части (I, рис. 13) имею¬
щей связь с серповидной мембраной (поз. 5, рис. 13) и связь с измери¬
тельным объемом посредством соединительной трубки (поз. 3, рис. 13)
через отверстие (поз. 1, рис. 13) и
нижней части (II, рис. 13) в виде
пробирки, оканчивающейся кер¬
ном с отверстием (поз. 10, рис. 13).
Отверстия в верхней (поз. 1, рис. 13)
и нижней частях (поз. 10, рис. 13)
должны совпадать по горизон¬
тальной плоскости.
На вакуумных установках,
можно проводить следующие ви¬
ды исследований.
I. Определение температу¬
ры начала термического разло¬
жения, протекающего с измери¬
мой скоростью.
Опыт проводят с навеской
пороха 0,1 г в реакционном сосуде
объемом — 28 см3. Температура
термостата, начинается с 150 °С,
повышается на 10°С через каж¬
дый час. Нагревание ведут до мо¬
мента, когда нарастание давления
газообразных продуктов в реак¬
ционном сосуде будет проходить
со скоростью 30 — 50 мм рт. ст. в час.
Найденная температура и будет
температурой разложения, проте¬
кающего с заметной скоростью.
II. Определение температуры
интенсивного разложения пороха.
Рис. 13. Стеклянный манометр Бур¬
дона разборный:
/ — измерительная часть:
I — отверстие, 2 — крючки, 3 — соедини¬
тельная трубка, 4 — верхняя часть реак¬
ционного сосуда, 5 — серповидная мембра¬
на, 6 — измерительный (компенсационный)
объем, 7 — стрелка.
II — реакционная часть
8 — реакционный съемный сосуд, 9 —
крючки, 10 — отверстие.
81
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
Опыт проводится так же, как и по пункту 1, но нарастание давле¬
ния газообразных продуктов при найденной температуре должно
быть 50 — 60 мм рт. ст. за час.
III. Кинетические исследования термического разложения пороха
Опыт проводят с навеской 1 г в реакционных сосудах объемом
28 см3 при отношении массы навески к объему — 0,0357 г/см3. Для до¬
стоверного расчета кинетических параметров необходимо проводить
испытания при четырех-пяти температурах с интервалом 10°С. Темпе¬
ратура первого кинетического опыта выбирается на основании резуль¬
татов определения температуры начала заметного разложения пороха.
IV. Определение полного разложения пороха до завершения
Опыт проводят в реакционных сосудах объемом 100 см3 при тем¬
пературе на 20 — 40°С выше температуры кинетического опыта, соот¬
ветствующего термораспаду, протекающему с максимальной скоро¬
стью, при отношении массы навески к объему — 0,0357 г/см3. Наве¬
ска пороха для данного опыта 0,1 г. Опыт полного разложения
считается законченным, если давление газообразных продуктов раз¬
ложения не повышается в течение 5 — 6 часов.
4.2.2 Аппаратура, посуда, реактивы
• стеклянная высоковакуумная установка;
• стеклянная установка по исследованию кинетики разложения;
• стеклянный манометр Бурдона (неразборный или разборный);
• термостат жидкостной;
• компрессор;
• горелка ручная для стеклодувных работ;
• зажигалка газовая электрическая;
• полиэтилсилоксановая жидкость ПЭС-5;
• шланг вакуумный резиновый;
• эфир этиловый;
• пропан-бутан в баллонах;
• сосуд Дьюара;
• азот жидкий;
• течеискатель;
• ножницы;
• пружинки металлические;
• зажим металлический.
4.2.3 Подготовка к испытанию
4.2.3.1 Подготовка и заполнение манометров Бурдона
4.2.3.1.1 Подготовка и заполнение неразборного манометра Бурдона
82
Глава четвертая. Термические методы анализа порохов
Неразборный манометр Бурдона подсоединяют к вакуумной
установке для откачивания воздуха, как показано на рис. 11. Компен¬
сационный сосуд (поз. 5, рис. 12) манометра Бурдона посредством
отрезка вакуумного шланга (поз. 10, рис. 12) длиной около 25 см сое¬
диняется с высоковакуумной установкой через двухходовой кран
(поз.5, рис. 12). Места надевания вакуумного шланга на стеклянные
части установки протирают вакуумной смазкой. Реакционный сосуд
манометра Бурдона (поз. 2, рис. 12) соединяют с вакуумной установкой
путем приварки стеклянного отростка (поз. 4, рис. 12) к стеклянной
трубке в виде колена в месте "а" со стороны двухходового вакуумного
крана (поз. 7, рис. 11). Свободный стеклянный отросток (поз. 3, рис. 12)
закрывают резиновой пробкой и проверяют герметичность маноме¬
тра Бурдона течеискателем. При обнаружении течи в манометриче¬
ском приборе в систему впускают воздух и пропаивают место течи.
После пропайки манометрический прибор повторно вакуумируют и
проверяют на герметичность.
В реакционный сосуд (поз. 2, рис. 12) через специальный отро¬
сток (поз. 3, рис. 12) с бумажного лоточка ссыпают навеску пороха.
Остатки навески протирают ваткой, и отросток запаивается. Двуххо¬
довые краны (поз. 5, б, 7, рис. 11) открывают, а краны (поз. 8,9,10, рис.
11) закрывают. Включают форвакуумный насос и осторожно, непре¬
рывно наблюдая за стрелкой манометрического прибора, поворачи¬
вают трехходовой кран (поз. 15, 16, рис. 11) до полного соединения
форвакуумного насоса с вакуумной установкой. Кран (поз. 9, рис. 11)
на короткое время открывают и вновь закрывают. После достижения
остаточного давления порядка 10”1 мм рт.ст. систему вакуумируют
форвакуумным насосом еще в течение 15 минут.
Контроль за вакуумом осуществляют по вакууметру следующим
образом: открывают кран (поз. 10, рис. И) на ионизационную лампу
(поз. 12, рис. 11), включают вилку измерительного прибора в сеть на
220 В, тумблер "ток накала измерения" ставят в положение "измере¬
ние" и производят отсчет по шкале прибора. Кран (поз. 10, рис. 11)
вновь закрывают.
Для получения более глубокого вакуума порядка 4 • 10 “3 мм рт. ст.
запускают в работу диффузионный масляный насос (поз. 13, рис. 11).
Сосуд Дьюара (поз. 4, рис. 11) вакуумной установки заполняют с жид¬
ким азотом (или с сухим льдом).
По достижению заданного вакуума в реакционном сосуде мано¬
метра Бурдона вакуумный шланг пережимают зажимом по месту "в",
отпаивают реакционный сосуд от вакуумной установки (одновремен¬
но запаивают и стеклянный отросток реакционного сосуда) по месту
83
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
“а". И, наконец, краны (поз. 5,7, рис. 11) закрывают, а ножницами пе¬
ререзают шланг выше зажима на 7 — 10 см.
4.2.3.1.2 Подготовка и заполнение разборного манометра Бурдона
Измерительную часть (I) стеклянного манометра Бурдона раз¬
борного (рис. 13) посредством вакуумного шланга длиной около 25 см
соединяют с высоковакуумной установкой через вакуумный двуххо¬
довой кран (поз. 5, рис. 11).
Поверхности соединения измерительной части (муфта) и реак¬
ционной части (керн) промазывают тонким слоем вакуумной смазки
и соединяют их так, чтобы отверстия (поз. 1, 10, рис. 13) совпали. Со¬
единение проводят путем вращения обеих частей в противополож¬
ном направлении с небольшим поддавливанием до момента образова¬
ния прозрачного слоя вакуумной смазки. Проверяют герметичность
собранного манометра Бурдона течеискателем. В случае обнаруже¬
ния течи на манометре Бурдона ее пропаивают, если этого сделать не¬
возможно, то манометр Бурдона заменяют.
После проверки манометра Бурдона на герметичность его разби¬
рают и в реакционную часть вносят навеску пороха. Обе части мано¬
метра Бурдона вновь качественно соединяют при этом отверстия
(поз. 1, 10, рис. 13) должны совпадать. На крючки (поз. 9, рис. 13) на¬
девают металлические пружинки для фиксации.
Проводят вакуумирование на высоковакуумной установке. По¬
сле достижения необходимого вакуума (~10“2 мм рт. ст.) пружинки с
крючков снимают. Реакционную часть манометра Бурдона поворачива¬
ют относительно измерительной части на 180 градусов и вновь наде¬
вают на крючки пружинки. Зажимом пережимают вакуумный шланг,
отключают вакуумирование. Ножницами отрезают манометр Бурдо¬
на с пережатым резиновым шлангом от высоковакуумной установки.
По окончании вакуумирования перекрывают краны (поз. 5, 6, 7,
9, 10, рис. 4.5), закрывают трехходовые краны (поз. 15, 16, рис. 11), вы¬
ключают диффузионный и форвакуумный насосы, в установку впу¬
скают воздух путем открывания крана (поз. 8, рис. 4.5).
4.2.3.1.3 Проведение кинетических исследований
Подготовленный по пункту 4.2.3.1 или пункту 4.2.3.1.2 манометр
Бурдона посредством вакуумного шланга (поз. 10, рис. 12) подсоеди¬
няют к установке по исследованию кинетики разложения.
На внешнюю поверхность компенсационного сосуда в месте
окончания стрелки наклеивают по окружности две полоски бумаги
(поз. 9, рис. 12) на расстоянии 5 — 6 мм друг от друга. На каждую поло¬
ску по вертикали наносят по линии, которые должны совпадать в вер-
84
Глава четвертая. Термические методы анализа порохов
тикальном направлении между собой и "нулевым" положением
стрелки серповидной мембраны.
Ус тановку вакуумируют и сообщают с манометром Бурдона (зажим
убирают). Термостат нагревают до нужной температуры, манометр
Бурдона погружают в нагретую жидкость до уровня ниже на 80 —100 мм
от верхнего края стрелки. С этого момента отмечают время начала
опыта. В течение опыта отклонение стрелки (вправо) мембраны компен¬
сируют подачей порции воздуха во внутреннюю полость компенсаци¬
онного сосуда манометра Бурдона через кран "напуска" (поз. 12, рис. 12).
Перед началом опыта в рабочий журнал записывают природу об¬
разца, массу навески, температуру опыта, объем реакционной части,
атмосферное давление. При проведении опыта записывают время заме¬
ра, температуру и давление по ртутному манометру (поз. 11, рис. 12).
По окончании опыта манометр Бурдона вынимают из нагретой
жидкости. При охлаждении продуктов разложения стрелка мембра¬
ны отклоняется влево. Для возвращения стрелки в нулевое положе¬
ние из внутренней полости компенсационного сосуда откачивают
воздух порциями. После полного охлаждения манометра Бурдона
фиксируют показание давления по ртутному манометру.
Металлическим зажимом пережимают вакуумный шланг и ма¬
нометр Бурдона отсоединяют от установки.
Разборные манометры Бурдона готовят для повторного приме¬
нения, неразборные — утилизируют.
4.2.4 Обработка результатов
4.2.4.1 Результаты кинетических исследований представляют в
виде графика в координатах: количество выделившихся газообраз¬
ных продуктов (V) в см3 приведенных к нормальным условиям (давле¬
ние 760 мм рт.ст., температура 273°С) и навеске равной 1,0 г — время
(х) термостатирования при заданной температуре.
4.2.4.2 Количество выделившихся газообразных продуктов (V) в
н см3/г рассчитывают по формуле:
У=Р{К,
где Р, — давление газообразных продуктов при температуре испы¬
тания,
К — коэффициент, равный
Ро-Тх-т'
где Т0 = 273°С, Vp — объем реакционного сосуда, см3, Р0 — 760 мм рт.ст.,
Т — температура испытания, °С, m — навеска пороха, г.
85
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
4.2.4.3 Объем реакционного сосуда (Ур) определяют по массе
(объему) дистиллированной воды (V^) за вычетом объема занимаемо¬
го самим образцом пороха (VJ
V =V — V
vp УВ ¥П‘
Навеску пороха (ш) рассчитывают исходя из заданной степени
заполнения реакционного сосуда (А)
M=A*Vp.
4.2.4.4 Для определения энергии активации (Еа) и предэкспонен-
ты ( К0 или т0) уравнения Аррениуса
Ki%=K0e XT;
Eg
T\%=T0eRT ,
где Kx%—константа скорости реакции 1% распада пороха при темпе¬
ратуре равной Г,
т1%—время реакции 1% распада пороха при температуре, равной Г.
проводят серию опытов:
• кинетические исследования при четырех-пяти температурах с ин¬
тервалом 5— 10°С и строят для них кинетические графики (рис. 14)
• опыт по полному разложе¬
нию пороха до завершения
и определяют максималь¬
ный объем газообразных
продуктов в пересчете на 1 г
пороха (Vmax)
4.2.4.5 Расчет кинетиче¬
ских параметров проводят в
следующей последовательно¬
сти:
• на график (рис. 14) наносят
кинетические кривые газо-
выделения
• рассчитывают объем газо¬
образных продуктов, соот¬
ветствующих 1% разложе¬
нию пороха
У1% = 0,01 vmax
Рис. 14. График зависимости количества
выделившихся газообразных продуктов
(V) нсм3/г во времени т при температу¬
рах Гр Г2, 7*з, 7*4, 7*5.
86
Глава четвертая. Термические методы анализа порохов
на графике наносят линию параллельную оси г соответствующую ко¬
личеству выделившихся газообразных продуктов ^1%
• определяют время (т1%) 1% распада пороха для всех точек пересече¬
ния
Ti, т2, т3( т4, т5
и рассчитывают константы скорости реакции как обратные величины
К{1 К2, К3, К4, К5
для соответствующих температур по шкале Кельвина
Т\, Т2, Т3, Т4, Т5
и их обратным значениям
1 /Г1г 1 /Т2, 1/Т3, 1 /Г4, 1 /Т5
• определяют логарифмы
19тз(^з)' l9T4(^Q)» ^9^5(^5)
• строят полулогарифмиче¬
скую зависимость (рис. 15)
Построенные зависимо¬
сти для т10/о и Кх% должны быть
линейными и наклон их равен
или --
2,303# 2,303#
когда расчет ведется в деся¬
тичных логарифмах. Отсюда
находится Еа любыми прие¬
млемыми методами расчета
[102]. Величину т0 и К0 нахо¬
дят путем подстановки чи¬
словых значений в уравне¬
ние Аррениуса.
Рис. 15. Полулогарифмичесая зависи¬
мость изменения времени 1% распада
(или константы скорости распада поро¬
ха) от обратной температуры.
87
ЛИТЕРАТУРА
1. Сапожников A. Teopin взрывчатыхъ веществъ. Курсъ Михайловской Ар-
тиллершской Академш. С.-Петербурга, 1912. — 369 с.
2. Лукьянов П.М. История химических промыслов и химической промы¬
шленности России до конца XIX века / Под ред. С.И. Вольфковича. — М.:
Изд-во Академии наук СССР, 1961. — Т. 3. — 659 с.
3. Бейлинг К., Дрекопф К. Взрывчатые вещества и средства взрывания. —
М.: Оборонгиз, 1941. — 304 с.
4. Брунсвиг Г. Бездымный порох. — М., А.: Государственное химико-техно¬
логическое издательство, 1933. — 368 с.
5. Сарыбаева Р.И., Щелохова Л.С. Химия азотнокислых эфиров целлюлозы. —
Фрунзе “Илим", 1985, — 164 с.
6. Найман ИМ., Терлецкий НА. Непрерывное измельчение нитроклетчат¬
ки. — М.: Оборонгиз, 1945. — 112 с.
7. Тарасов А.П. Производство бездымных порохов. — М.: Дом техники,
1963. —300 с.
8. Физер Л., Физер М. Органическая химия, углубленный курс: Пер. с анг. —
М.: Химия, 1970. — Т. 1. — 688 с.
9. Броуне С А. Технология пороха (практическое пороходелие), Часть И. Пи¬
роксилиновый бездымный порох. — Казань: Издание спецфака К.Х.Т.И.,
1932. —225 с.
10. Нобль, Эйбль. Изследоваше взрывчатыхъ составовъ. Действ1е воспламе-
неннаго пороха. — С.-Петербургъ, 1878. — 132 с.
И. Забудскш ГА. Взрывчатыя вещества. Теоретическая часть. — С. — Пе¬
тербурга, 1907. — 295 с.
12. Закощиков А.П. Нитроцеллюлоза. — М.: Издательство оборонной промы¬
шленности, 1950. — 372 с.
13. Орлова Е.Ю. Химия и технология бризантных взрывчатых веществ. — Л.:
Химия, 1973. — 688 с.
14. Горст А.Г. Пороха и взрывчатые вещества. — М.: Машиностроение, 1972. —
208 с.
15. Егоров Т.С. Производство бездымного пироксилинового пороха. — М.:
Объединенное научно-техническое издательство, 1935. — 154 с.
16. Тиммерганс. Начальные основашя артиллерш. 4.1. О порохе. С.-Петер¬
бурга, 1844, — С. 272-280, 379 с.
88
Литература
17. Ohman V., Laurem G.// Anal. Chemi. — 1936. — Vol. 107 — P. 409.
18. Способ определения содержания калиевой селитры в дымном порохе:
А.с. № 146336 СССР, МКИ G 01 N 33/22, С 06 В 31/04/ А.А.Желтоножко,
М.Ф. Буллер, З.М.Егорчева, Т.А. Тосенко (СССР). — 2264479/23; Заявле¬
но 24.09.79; не публикуется.
19. Способ автоматического определения содержания калия в солях: А.с.
№ 647593 СССР, МКИ G 01 N 27/06 / А.Г. Злобинский, Г.Г. Колпиков, В.А.
Миронов, Л.А. Рейбман и В.Н. Станишевский (СССР). — 2161515/18-25;
Заявлено 11.08.75; Опубликовано — 1979. № 6 — С. 143
20. Способ определения калиевой селитры в дымном порохе: А.с. № 162359
СССР, МКИ G 01 N 33/22, С 06 В 31/04, G 01 N 5/04 / М.Ф. Буллер
(СССР). — 2281271; Заявлено 16.06.80; не публикуется.
21. Способ определения калиевой селитры в дымных порохах: А.с. № 324264
СССР, МКИ G 01 N 21/00, С 06 В 21/00 / А.В. Макаревич, М.Ф. Буллер,
С.П. Ярманова (СССР). — 4526769; Заявлено 31.01.90; не публикуется.
22. Дж.Фритц, Г. Шенк. Количественный анализ. Пер. с анг. — М.: Мир,
1978. — 557 с.
23. Капоп Takahiro, Sugita Shizuo. Определение ионов калия и натрия с помо¬
щью автоанализатора // Bull. Sol. Sea. Water Sei Jap. — 1979. — Vol. 33,
№ 2. — P. 116-118.
24. Потапова В.Г., Золотовицкая Э.С. Определение калия в окиси алюминия
для синтетического корунда: Сб. Научн. тр. ВНИИ монокристаллов,
сцинтилляционных материалов и особо чистых химических веществ. -
М.: Наука, 1979, № 4. — С. 136-138.
25. Вельды М.П. Бета-радиометрия калия в пробах сложного состава // Ме¬
тоды и средства аналитического контроля в калийной промышленности. —
Л.: Химия, 1979. — С. 30 — 39.
26. Вещество для индикаторной мембраны ионоселективных электродов для
определения ионов К+, NH4+, Cs + : А.с. 647594 СССР, МКИ G 01N 27/30,
G 01 N 27/40 / А.В. Гордиевский, Ю.И. Урусов, А.Я. Сырченков, А.Е. Ко¬
сов, А. Ф. Жуков (СССР). — № 1974151/18-25; Заявлено 27.11.73; Опубл.
Бюл. №6, 1979.— С. 143.
27. Гусев Ю.Н. и др.// Методы анализа и контроля качества продукции в хи¬
мических производствах. — 1979. — № 3. — С. 20 — 23.
28. Одновременное определение нитрата и нитрита с помощью ионселек-
тивных электродов. Фудзинага Таитиро, Окадзаки Сатоси, Хара Хиро-
кадзу/ Полярографии, Rev. Polarogr. — 1979. — Vol. 25. № 1-6. — С. 52.
29. Басов В.Н. и др.// Методы анализа и контроля качества продукции в хи¬
мических производствах. — 1979. — № 2. — С. 45 — 48.
30. Saltzman В.Е., Wartbung А.Е. Presion Flow Dilution System for Standart Low
Concentration of Nitrogen Dioxide// Anal. Chemi. — 1965. — Vol. 37, — P. 1261.
31. Montgomery A.C., Dymock I.E. The determination of nitrate in water // Ana¬
lyst. — 1961. — Vol. 86, — P. 1623.
32. Агасян П.К., Николаева E.P. Основы электрохимических методов анализа
(потенциометрические методы). М.: Изд-во МГУ, 1986. — 166 с.
89
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
33. West Р. W., Lules G.Z. Determination of nitrate// Analyt. Chem. Acta. — 1966. —
Vol. 23. — P. 227.
34. Грибова E.A., Черкасский АЛ., Баврина Ю.П., Стерлина Л.И. Прямое по¬
тенциометрическое определение азотной кислоты и нитратов в смесях с
другими кислотами и солями // Заводская лаборатория. — 1980. №7. —
С. 595-596.
35. Способ определения азотной кислоты и нитратов: А.с. 658083 СССР,
МКИ G 01 В 21/00, G 01 N 27/46/ С.Г.Дятел (СССР). № 2313289/23-26; За¬
явлено 06.01.76; Опубл. 1979, Бюл. №15. -75с.
36. Сох R. Determination of nitrate and nitride at the parts per billion level by che¬
miluminescence // Anal. Chem. — 1980. — Vol. 52, №2. — P. 332 — 335.
37. Артищев H.B., Крапивина С.Ф., Ершов В.А. Хемилюминесцентная реак¬
ция окиси азота и озона и ее аналитическое применение // Журн. Всес.
Хим. о-ва им. Д.И. Менделеева. — 1980. 26. №1. — С. 116—117.
38. Способ определения концентрации электролита: А.с. 693210 СССР, МКИ
G 01 N 27/06, G 01 N 25/20 / Ю.И. Беляев, В.С.Прохоров, П.И.Стальнов,
М.В.Кулаков (СССР). -№ 2515412/18-25; Заявлено 02.08.77; Опубл. 1979,
Бюл. № 39. — 141с.
39. Менковский М.А. Природная сера. — М.: Химия, 1972. — 240 с.
40. Способ определения содержания серы в дымном порохе: А.с. 284508
СССР, МКИ G 01 N 31/16, С 06 В 21/00 / М.Ф. Буллер, А.В. Макаревич,
А.М. Лыч (СССР). — № 3190808; Заявлено 28.01.88; не публикуется.
41. Клименко Г.К. Методы испытания порохов. — М.: Гос изд-во оборон,
пром-ти. 1941. — 299 с.
42. Шиллинг Н.А. Курс дымных порохов. — М.: Госизд-во оборон, пром-ти.
1940. — С. 276.
43. Camera Е., Pravisani D. Analisi gascromatografica dello zolfo in una polvere
nera // La Chimica e L' industria. — 1965. — Vol. 47. №11. — P. 1210- 1212.
44. Пат. 175286ЧССР, МКИG01N31/16. Zpusobstanoveniobsahuelementar-
ne siry: Пат. 175286 ЧССР, МКИ G01N31/16/J. Kubant (ЧССР), заявл.
6.11.75 № 7471 -75, опубл. 15.11.78.
45. Макаревич А.В., Черникова Н.И., Буллер М.Ф. Метод определения массо¬
вой доли углерода в древесных углях для дымных порохов// Передовой
производственный опыт. — 1990. — № 11, — С. 34 —35.
46. Способ определения массовой доли углерода в древесных углях: А.с.
147144 СССР, МКИ С 01В 31/00, G 01 N 21/24/ М.Ф. Буллер (СССР). —
2259682/26; Заявлено 27.06.79; не публикуется.
47. ДЛ.Вяхирев, А.Ф.Шушунова. -Руководство по газовой хроматографии.
Учеб, пособие для ун-тов. — М.: "Высшая школа", 1975. — 302 с.
48. Перри С., Амос Р., Брюер П. Практическое руководство по жидкостной
хроматографии — М.: Мир. — 1974. — 260 с.
49. Стыскин Е.Л., Ициксон Л.Б.. Брауде Е.В. Практическая высокоэффектив¬
ная жидкостная хроматография. — М.:Химия. — 1986. — 288 с.
50. Сычев С.Н. Методы совершенствования хроматографических систем и
механизмы удерживания в ВЭЖХ. — Орел: ОрелГТУ. — 2000. — 212 с.
90
Литература
51. К). Кихнер. Тонкослойная хроматография: Пер. с анг. — М.: Мир, 1981. —
Т.1. — 616с.
52. Rice D.D., Trowell J.M. Газохроматографическое определение летучих в
твердых телах и высококипящих жидкостях. //Analyt.Chem. — 1967. —
Vol. 39, № 2. — Р. 157— 162.
53. Me hi hose K.t Rudo W. Газохроматографическое определение остатков ра¬
створителей в производстве бездымных порохов. //Explosivstoffe. —
1968. — Vol. 16, № 6. — Р. 121 -135.
54. Межевич Г.В., Егорчева З.М. Применение газовой хроматографии для по-
фазного контроля производства пироксилиновых порохов. /НИИ хими¬
ческих продуктов. — Шостка, 1975. — 11 с. — Рус. — Деп. в ЦНИИНТИ
СССР № 6842 // Анот. в ж. Информация о новых поступлениях литера¬
туры, № 7, 1976.
55. Межевич Г.В. Применение газовой хроматографии в анализе и производ¬
стве пироксилиновых порохов. Обзор. /НИИ химических продуктов. —
Шостка, 1975. — 31 с. — Рус. — Деп. в ЦНИИНТИ СССР № 1149 // Анот.
в ж. Информация о новых поступлениях литературы, № 7, 1976.
56. Способ определения содержания этйлацетата в высокоэнергетических
порохах: — А.с. № 330859 СССР МКИ GOln 31/08/ Г.В. Межевич, Н.В. Крав¬
ченко (СССР). — № ; Заявлено 01.10.91; не публикуется.
57. Pasika W.M., West III А.С. Количественное определение воды в гидро¬
фильных макромолекулярных образцах методом газовой хроматогра¬
фии. // Analyt. Chem. — 1971. — Vol. 43, № 2. — Р. 275-276.
58. Межевич Г.В., Мезина Ю.М., Егорчева З.М. Газохроматографическое
определение содержания воды в пироксилиновом порохе. /НИИ хими¬
ческих продуктов.- Шостка, 1975. — 14 с. — Рус. — Деп. в ЦНИИНТИ
СССР № 6843 // Анот. в ж. Информация о новых поступлениях литера¬
туры, № 7, 1976.
59. Межевич Г.В., Исаев Е.Н. Определение крепости остаточного спирта в обез¬
воженном пироксилине. // ВСМ, серия II. — 1975, вып. 2(25). — С. 60 — 66.
60. Способ определения содержания воды в пироксилиновых порохах: А.с.
№ 89195 СССР МКИ G 01п 31/08 / G 01п 33/22 / Г.В. Межевич, Ю.М. Ме¬
зина (СССР). № ; Заявлено 06.08.75; не публикуется.
61. ГОСТ 8660-80 Пороха пироксилиновые. Методы определения массовой
доли летучих веществ. — Изд. стандартов, 1980.
62. Межевич Г.В., Мезина Ю.М., Егорчева З.М. Экстракционно-газохромато¬
графическое определение дифениламина в пороховой массе. // ВСМ, се¬
рия II. — 1975, вып. 12(35) — 13(36). — С. 42-45.
63. Межевич Г.В., Буллер М.Ф. Определение стабилизаторов химической
стойкости в утилизированных порохах и топливах. // XiM. пром. Укра!-
ни. — 2002, № 6(53). — С. 11 - 13.
64. Пат. 94 А Украша, МКИ С 06 В 21/00. Cnoci6 визначення cra6i\i3aTopiB
xiMi4Hoi стшкостп Пат. 94 А Украша, МКИ С 06 В 21/00 Г.В. Межевич,
Т.Ю. Козинець, H.I. Чернжова, М.Ф. Буллер, О.Ю. Мараховська, С.П. Яр-
манова (Украша); ДержНД1ХП — 200204333396; Заявл. 23.04.02.
91
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
65. Межевич Г.В., Козинец Т.Ю. Исследование превращений стабилизатора
ДФА в процессе производства и хранения пироксилиновых порохов.
Применение метода тонкослойной хроматографии. Сообщение 1. Выбор
условий разделения. //ВСМ, серия II. — 1977, вып. 13(77). — С. 50 — 55.
66. Cnoci6 визначення дифен1лам1ну та його ттрозо- та нпрозамщених в ni-
оксилшових порохах: Деклар. пат. на корисну модель № 2991/ Межевич Г.В.,
Маренець М.О., Закотей В.Г., Буллер М.Ф.(Украша); ДержНД1ХП. —
№2004021404; Заявлено 15.09.2004
67. Буллер М.Ф., Межевич Г.В., Маренец МЛ., Закотей В.Г. Утилизированные
пороха. Определение массовой доли дифениламина. // XiM. пром. Укрш-
ни. — 2004, № 6(65). — С. 43-46.
68. Krul /.5., Катр M.J. Анализ взрывчатых веществ//СЬеш. Ztg. -1980. -Vol.
104, № 10. -Р. 293 — 294.
69. Gorin Р., Lebert At, Stephan M., Zeller В. ЖХВД для определения ДФА, цен-
тралита и их производных в стабилизированных порохах // Informs chim. —
1976, № 158. — Р. 209-212.
70. Doali J. О., Juhasz АЛ. Применение ВЭЖХ к количественному анализу со¬
единений, используемых в производстве ТРТ и ВВ. //J. Chromat. Sci. —
1974.—Vol. 12, № 1. —P.51-56.
71. Жидкостная хроматография н ее применение для анализа взрывчатых
веществ: Реф. обзор. Депонир. рукопись. / Н.Н. Чеблакова, Н.И. Куроч¬
кин, Е.Е. Грязин, А.А. Спутницкий; ОНТИ-0512. — Б.м. — 1979. — 24 с.
72. Farey M.G., Wilson S.E. Количественное определение тетрила и продуктов
его разложения методом ВЭЖХ//J.Chromatogr. — 1975. —Vol. 114, № 1. —
Р. 261-265.
73. Burkitt D., В irk 5., Delaney J. Определение ВВ в почве и воде методом высо¬
коэффективной жидкостной хроматографии //Abstr.Pap. Pittsburg Conf.
And Exsplos. Anal.Chem. and AppLSpektroscop. New Orleans. La. 25 Febr. —
1 March, 1985. — Vol.l, — P. 896.
74. Voyksner R.D. Определение следовых количеств ВВ методом сочетания
ВЭЖХ и масс-спектрометрии с термораспылением // J.Chromatogr. —
1986. — Vol. 354, № 2. — Р. 393-405.
75. Poyet J-М., Prigent Н., Vignand М. Применение жидкостной хроматогра¬
фии высокого давления к качественному и количественному анализу ВВ
// Analysis. — 1976. — Vol. 4, № 2. — Р. 53 - 57.
76. Lebert М., Stephan М., Zeller В. Определение ДФА, централита и их произ¬
водных в порохах и РТ методом ВЭЖХ //Centre de Recherches du Bouchet
Societe Nationale des Poudres et Explosifs. 91710 Vert-le-Petit, France.
77. Fariwar-Mohsenie M., Ripper £, Haberman K.H. Анализ взрывчатых ве¬
ществ методом жидкостной хроматографии высокого давления. // Fres-
enius Z. Anal. Chem. — 1979. — Vol. 296, № 2-3. — P. 152- 155.
78. Lloyd J.B.F. Анализ взрывчатых микропримесей методом ВЭЖХ с элек-
тро-химическим детектированием //J. Chromatogr. — 1983. — Vol. 261,
№3. —P.391-406.
92
Литература
79. Dahl 1).11, Slahck S.C., Lott P.F. Определение следов ружейного выстрела
ме тодом ВЭЖХ с электрохимическим детектированием //Microchem. J. —
1985. — Vol. 31, N° 2. — Р. 145- 160.
80. Lloyd J. В. F. Следы ДФА на тампонах и обрезках одежды: выделение и раз¬
деление методом ВЭЖХ с электрохимическим детектированием //Апа-
lyl. Chem. — 1987. — Vol. 59, № 10. — Р. 1401 - 1404.
81. Dahl D.B., Lott P.F. Анализ остатков выстрела по стабилизатору методом
ВЭЖХ с электрохимическим детектором и анализ металлических остат¬
ков методом атомно-абсорбционной спектроскопии //Microchem. J. —
1987. — Vol. 35, № 3. — Р. 347-359.
82. Dahl D.B., Cayton J.C., Lott P.F. Анализ остатков артиллерийского выстре-
ла//М1сгос!1ет. J. — 1987. — Vol. 35, № 3. — Р. 360-364.
83. Varney M.S., Preston M.R. Определение следов ароматических аминов в
морской воде методом ВЭЖХ с электрохимическим детектированием
//J. Chromatogr. — 1985. — Vol. 348, № 1. — Р. 265-274.
84. Thomas М.В., Msimanga Н., Sturrock Р.Е. Применение электрохимическо¬
го детектирования с разверткой потенциала при определении нитроза-
минов методом ЖХ // Anal. Chim. Acta. — 1985, N° 174. — Р. 287 — 291.
85. Bergens A., Danielsson R. Разложение дифениламина в ракетных топливах
на основе нитроцеллюлозы . I. Оптимизация численной модели для дан¬
ных концентрация — время для дифениламина и основных продуктов
его разложения, определяемых методом жидкостной хроматографии с
двойным амерометрическим детектированием//Та1ап1а. — 1995. —Vol. 42,
№2. — Р. 171-183.
86. Bergens А. Разложение дифениламина в ракетных топливах на основе ни¬
троцеллюлозы. II. Применение численной модели для данных концентра¬
ция — время, определенных методом жидкостной хроматографии с де¬
тектированием при двух длинах волн //Talanta. — 1995. — Vol. 42, N° 2. —
Р. 32-35.
87. Boj S., Dawid M., Ambrozek M. Анализ динамитов методом ВЭЖХ. //Chem.
Anal. — 1993. — Vol. 38, № 3. — Р. 323-329.
88. Пат. 156256 Польша, МКИ С 06 В 25/00, G 01 N 30/02. Способ разделения
централита-1 и продуктов его превращений в метательных зарядах на ос¬
нове нитроцеллюлозы и нитроглицерина: Пат. 156256, Польша МКИ С 06
В 25/00, G 01 N 30/02.
89. Hastenteufel S.t Schmidt М., Wagner R. Старые запасы оружия. Актуаль¬
ный вызов аналитической химии окружающей средыУ/Lab. Ргах. —
1992. Vol. 16, № 9. — Р. 888-892, 894.
90. Curtis N.J., Rogasch Р.Е. Определение производных ДФА в австралийских
артиллерийских порохах методом ВЭЖХ //Propellants, Explos., Руго-
techn. — 1987. — Vol. 12, № 5. — Р. 158- 163.
91. Curtis N. Y. Изомерное распределение нитропроизводных дифениламина
в артиллерийских порохах: химия нитрозамина //Propellants, Explos., Ру-
rotechn. — 1990. — Vol. 15, № 5. — Р. 222-230.
93
М.Ф. Буллер, Г.В. Межевич Методы испытания утилизируемых порохов
92. Doali J.O., Juhasz АЛ. Определение 2-нитродифениламина в двухоснов¬
ных порохах методом высокоэффективной жидкостной хроматографии
//Analyt. Chem. — 1976. — Vol. 48, № 13. — Р. 1859- 1860.
93. Kansas L., Robertson D. Определение 2-нитродифениламина и его основ¬
ных производных в двух- и трехосновных порохах // Propellants, Explo¬
sives, Pyrotechnics. — 1994. — Vol. 19, № 4. — P. 171 — 173.
94. ГОСТ 8065-72 Пороха дымные. Метод определения гигроскопичности. —
Взамен ГОСТ 8065-56; Введ 01.07.73. — М.: Изд-во стандартов, 1976. —
С. 19-21, 32с.
95. ГОСТ 1736-42 Методы испытаний пироксилиновых порохов. Проба Вве¬
дя при 106,6±0,5°С — Взамен СГ 1047Т разд. 7., и СГ 1048Т, разд. 104
Введ. 01.01.43 — М.: Стандартгиз, 1943. — 47 с.
96. ГОСТ 11857-77. Методы определения стойкости по лакмусовой пробе. —
Взамен ГОСТ 11857-66; Введ. 01.07.78 — М.: Изд-во стандартов, 1977, 12 с.
97. ГОСТ В 10836-75 Нитроцеллюлоза. Метод определения химической
стойкости. — Взамен ГОСТ 10836-64; Введ 01.07.76. — М.: Изд-во стан¬
дартов, 1975. — С. 44 — 49, 61 с.
98. Хансон Я. О контроле за состоянием ракетных топлив при хранении //
Труды Международной конференции "Химическая проблема стабиль¬
ности взрывчатых веществ". Тюрингия: май 25 — 27, 1970. — Р. 1 — 13.
99. Уэнгландт У. Термические методы анализа. — М.: Мир, 1978, 528 с.
100. Freeman E.S., Carroll В. The application of thermoanalytical techniques to re¬
action kinetics. The thermogravimetric evaluation of the kinetics of the de¬
composition of calcium oxalate monohydrate//J. Phys. Chem. — 1958. —
Vol. 62. — P. 394
101. Методические указания. Tермический анализ полимеров. Составители —
М.В. Виноградов, Э.С. Шульгина. — Л.: АТИ им. Ленсовета. — 1981.— 30 с.
102. Эмануэль Н.М. Кнорре Д.Г. Курс химической кинетики. Изд. 3-е, пере-
раб. и доп. — М.: Высшая школа, 1974, — С. 46 — 49, 400 с.
94