/
























































































































































































































































































































































































































Автор: Ригден Д.Д.
Теги: молекулярная биология общая биофизика, общая биохимия и общая физиология биология биоинформатика
ISBN: 978-5-9710-0842-2
Год: 2013




Текст
ББК 28.07 30.16
Структура и функционирование белков: Применение мсю юн бнпмнформиики.
(Под руководством Даниэля Джона Ригдена.) Пер. с англ. / I Io i рсД
В. Н. Новоселецкого. М.: УРСС: ЛЕНАНД, 2014. — 424 с., пн нк i
Молекулы белков лежат в основе почти всех биологических проц - сон Ученым всегда были любопытны как белки, участвующие в метаболических nyiax. in и и молекулярные основы их функционирования. Однако в эру системной биол <м и и еще । нищие внимание уделяется полному пониманию работы всей совокупности белков opi дин iMa, ст прогеома. Все более важно, что мы не только понимаем все стороны данной функции, и «и функций, какого-либо белка, но и то, что наше знание распространяется на все компоненты и мучаемой системы или организма и так далеко, насколько это возможно. 1кч всестороннего анализа информации попытки синтеза и расчетов не смогут выйти за рамки приблищ тиной реальности.
Книга «Структура и функционирование белков: Применение мет юн ( ноинформа гики» представляет собой уникальный обзор современного состояния вопросов моделирования структуры белков и предсказания их функции. Книга написана ведущими специалистами в своей области, прекрасно иллюстрирована и содержит ссылки на доступные ерверы и другие ресурсы, которые читатель, возможно, захочет использовать в своей научной работе. В конце каждой главы описываются перспективы развития и наиболее актуальные проблемы соответствующих областей науки.
На сегодняшний день научное сообщество довольно близко подошло к объяснению явлений, природа которых до недавних пор была не ясна, — таких как обмен доменов, круговая перестановка, образование фибрилл, белки с присущей неупорядоченностью и многими другими. В 2008 году мы сталкиваемся с метаморфными белками, исследование которых может значительно поспособствовать нашему пониманию пространства типов укладки белков. И несмотря на то, что структуры белков непрерывно готовят нам новые трудности, совершенно ясно, что биоинформатика структуры и функции белков на протяжении многих лет будет оставаться одной из самых востребованных и волнующих областей исследований.
Книга рассчитана на студентов, аспирантов и специалистов, ингресующихся вопросами молекулярного моделирования и биоинформатики.
Translation from English language edition: From Protein Structure to Function with Bioinformatics by Daniel John Rigden (Ed.)
Формат 60x90/16. Печ. л. 26,5. Зак. № ЗМ-28.
Отпечатано в ООО «ЛЕНАНД». 117312, Москва, пр-т Шестидесятилетия Октября, 11 А, стр. 11.
ISBN 978-5-9710-0842-2
(ЛЕНАНД)
ISBN 978-5-453-00057-9
(УРСС)
11810 ID 180705
9 785971 008422
© Springer Netherlands is a part of Springer Science + Business Media, 2009.
All rights reserved
© УРСС, 2013
ИЗДАТЕЛЬСКАЯ I 1ПСС Г-Д—] ГРУППА UnOD
Jk E-mail: URSS@URSS.ru
ГСК/ Тел./факс (многоканальный):
Д +7(499)724 25 45
v Каталог изданий в Интернете:
urss http://URSS.ru
Оглавление
Введение.........................................................10
Литература...................................................13
Глава 1
Предсказание структуры белков ab initio....................14
1.1. Введение....................................................15
1.2. Энергетические функции......................................17
1.2.1. Рациональные энергетические функции...................17
1.2.2. Сочетание эмпирических энергетических функций и сборки из фрагментов.......................................23
1.3. Методы конформационного поиска..............................28
1.3.1. Моделирование методом Монте-Карло.....................29
1.3.2. Молекулярная динамика.................................31
1.3.3. Генетические алгоритмы................................31
1.3.4. Математическая оптимизация............................32
1.4. Отбор моделей...............................................32
1.4.1. Рациональная энергетическая функция...................33
1.4.2. Эмпирическая энергетическая функция...................34
1.4.3. Функция совместимости структуры и последовательности..35
1.4.4. Кластеризация макетов структур........................36
1.5. Замечания и обсуждение......................................37
Литература...................................................39
Глава 2
Распознавание фолда..............................................44
2.1. Введение....................................................44
2.1.1. Важность «слепых» испытаний: соревнование С ASP.......45
2.1.2. Предсказание структуры ab initio и моделирование по гомологии.................................46
2.1.3. Пределы пространства типов укладки....................48
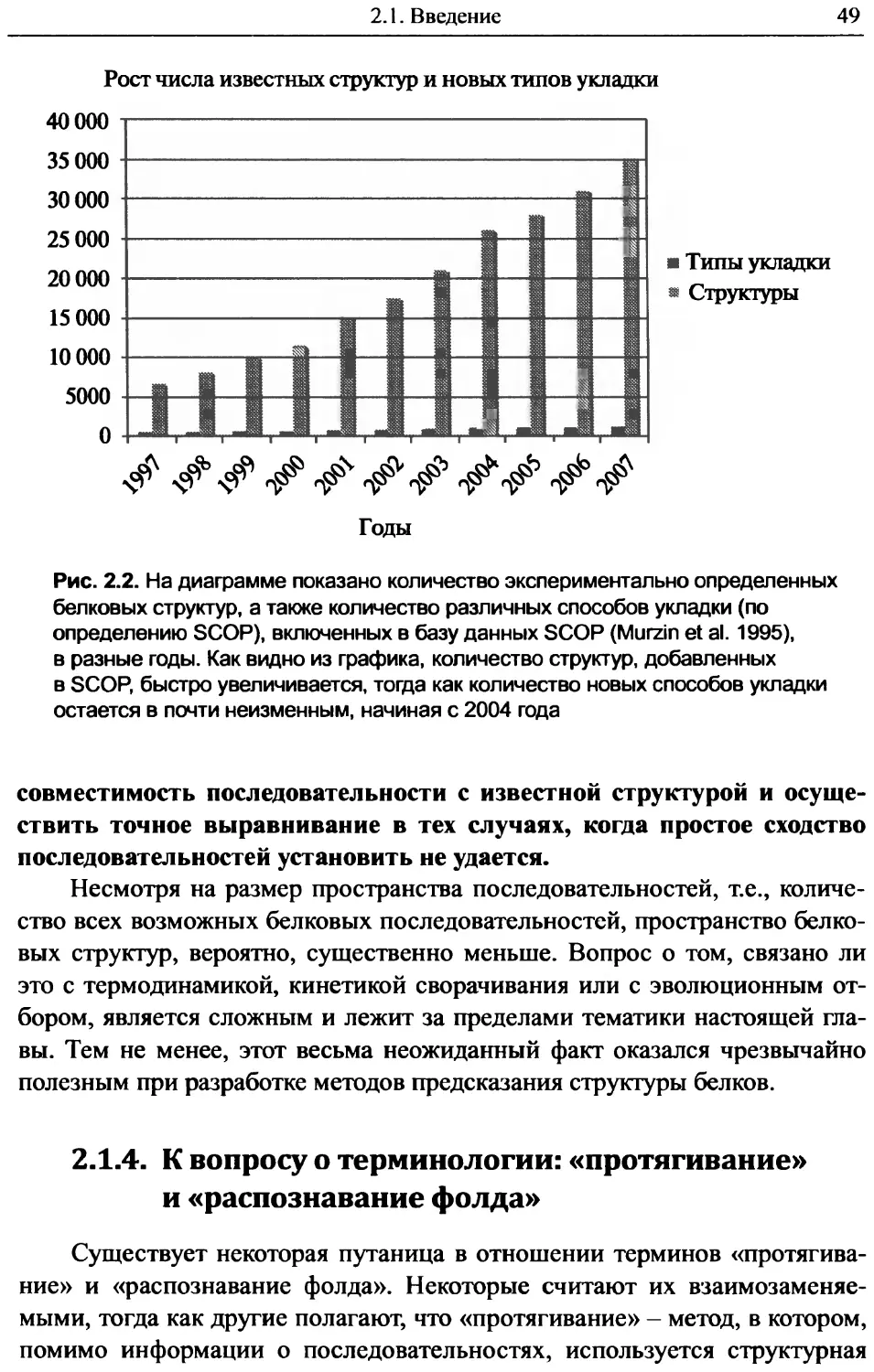
2.1.4. К вопросу о терминологии: «протягивание» и «распознавание фолда».......................49
2.2. «Протягивание»..............................................50
2.2.1. Эмпирические потенциалы...............................51
2.2.2. Поиск выравнивания....................................54
2.2.3. Эвристические правила выравнивания....................56
4
Оглавление
2.3. Определение отдаленной гомологии без протягивания............59
2.3.1. Использование предсказанных структурных свойств........60
2.3.2. Профили последовательностей и скрытые марковские модели.63
2.3.3. Классификация типов укладки и метод опорных векторов...67
2.3.4. Согласованные подходы...................................68
2.3.5. Проход по сети гомологов................................70
2.4. Точность выравнивания, качество моделей и статистическая значимость.......................................71
2.4.1. Алгоритмы создания выравниваний и оценка...............72
2.4.2. Оценка статистической значимости.......................74
2.5. Веб-инструменты для распознавания элементов укладки..........75
2.6. Перспективы..................................................77
Литература....................................................80
Глава 3
Сравнительное моделирование структуры белков......................83
3.1. Введение.....................................................83
3.1.1. Структура определяет функцию...........................83
3.1.2. Последовательности, структуры и структурная геномика...84
3.1.3. Методы предсказания структуры белков...................85
3.2. Этапы сравнительного моделирования структуры белков..........88
3.2.1. Поиск структур, потенциально родственных с мишенью.....90
3.2.2. Отбор шаблонов.........................................92
3.2.3. Выравнивание последовательности со структурой..........94
3.2.4. Построение модели......................................97
3.2.5. Оценка моделей........................................109
3.3. Эффективность методов сравнительного моделирования..........110
3.3.1. Точность методов......................................110
3.3.2. Ошибки в сравнительных моделях........................112
3.4. Применение сравнительного моделирования.....................114
3.4.1. Моделирование одиночных белков........................114
3.4.2. Сравнительное моделирование и проект исследования структуры белков......................115
3.5. Заключение..................................................116
Литература...................................................117
Глава 4 Предсказание структуры мембранных белков.........................126
4.1. Введение....................................................126
4.2. Структурные классы..........................................127
4.2.1. Пучки альфа-спиралей..................................127
4.2.2. Бета-бочонки..........................................129
Оглавление
5
4.3. Особенности кристаллизации мембранных белков.............130
4.4. Базы данных..............................................132
4.5. Множественные выравнивания последовательностей...........133
4.6. Предсказание топологии трансмембранных белков............135
4.6.1. Альфа-спиральные белки.............................135
4.6.2. Белки, имеющие структуру р-бочонка.................140
4.6.3. Полногеномный анализ...............................142
4.6.4. Наборы данных, гомологичность, точность и перекрестная проверка..........................142
4.7. Предсказание пространственной структуры..................145
4.8. Перспективы развития методов предсказания структуры мембранных белков......................148
Литература................................................149
Глава 5
Методы биоинформатики для изучения структуры
и функций неупорядоченных белков..................................153
5.1. Идея неупорядоченности белков................................154
5.2. Свойства последовательностей БПН.............................155
5.2.1. Необычный аминокислотный состав БПН....................156
5.2.2. Паттерны последовательностей БПН.......................156
5.2.3. Низкая сложность последовательностей и неупорядоченность.157
5.3. Предсказание неупорядоченности...............................158
5.3.1. Предсказание областей с низкой сложностью..............159
5.3.2. Графики «заряд-гидрофобность»..........................159
5.3.3. Методы предсказания на основе предрасположенности......159
5.3.4. Методы предсказания на основе отсутствия выраженной вторичной структуры...........................................161
5.3.5. Алгоритмы машинного обучения...........................162
5.3.6. Предсказание на основе потенциалов контакта............163
5.3.7. Для предсказания неупорядоченности достаточно сокращенного алфавита..............................165
5.3.8. Сравнение методов предсказания неупорядоченности.......165
5.4. Функциональная классификация БПН.............................166
5.4.1. Функциональная классификация БПН на основе генной онтологии.....................................................166
5.4.2. Классификация БПН на основе механизма действия.........167
5.4.3. Структурные элементы БПН, связанные с функционированием..171
5.5. Предсказание функций БПН.....................................173
5.5.1. Корреляция модели неупорядоченности и функции..........174
5.5.2. Предсказание коротких мотивов распознавания в БПН......175
5.5.3. Прогнозирование СМОР...................................176
6
Оглавление
5.5.4. Сочетание информации о последовательности и неупорядоченности: участки фосфорилирования и мотивы связывания СаМ......................................177
5.5.5. Поддержание неупорядоченности..........................178
5.6. Ограничения методов предсказания функций БПН.................179
5.6.1. Быстрая эволюция БПН...................................179
5.6.2. Независимость последовательности и функции и неопределенность...........................................180
5.6.3. Консервативность и неупорядоченность...................182
5.7. Заключение...................................................182
Литература....................................................183
Глава 6
Функциональное разнообразие в элементах упаковки и надсемействах..............................188
6.1. Определение функций..........................................189
6.2. От способа укладки к функции.................................192
6.2.1. Определение способа укладки............................192
6.2.2. Связь между способами укладки и предсказание функций...195
6.3. Разнообразие функций гомологичных белков.....................199
6.3.1. Определения............................................199
6.3.2. Эволюция белковых надсемейств..........................201
6.3.3. Дивергенция функций в ходе эволюции белков.............203
6.4. Заключение...................................................213
Литература....................................................214
Глава 7
Предсказание функции белка по свойствам его поверхности......................................218
7.1. Способы представления поверхности............................218
7.1.1. Поверхность ван-дер-Ваальса............................218
7.1.2. Молекулярная поверхность (поверхность без растворителя).219
7.1.3. Поверхность, доступная растворителю....................220
7.2. Свойства поверхности.........................................220
7.2.1. Гидрофобность..........................................220
7.2.2. Электростатические свойства............................222
7.2.3. Консервативность поверхности...........................222
7.3. Предсказание функций по свойствам поверхности................224
7.3.1. Гидрофобная поверхность................................224
7.3.2. Электростатическая поверхность.........................225
7.3.3. Консервативность поверхности...........................226
7.3.4. Сочетание свойств поверхности для предсказания функций.226
Оглавление
7
7.4. Взаимодействие лиганда с белком..........................227
7.4.1. Свойства взаимодействий лиганда с белком...........227
7.4.2. Предсказание расположения активного центра.........227
7.4.3. Предсказание чувствительности к лекарствам.........231
7.4.4. Аннотация сайтов связывания лигандов...............232
7.5. Белок-белковый интерфейс.................................233
7.5.1. Свойства белок-белкового интерфейса................233
7.5.2. Активные точки белковых интерфейсов................234
7.5.3. Предсказание расположения интерфейса...............236
7.6. Заключение...............................................238
Литература................................................238
Глава 8
Пространственные мотивы.......................................241
8.1. Предыстория и значение...................................242
8.1.1. Что такое функция?.................................244
8.1.2. Структурные мотивы: определение и область действия.245
8.2. Обзор методов............................................246
8.2.1. Поиск мотивов......................................246
8.2.2. Определение и подбор мотивов.......................247
8.2.3. Интерпретация результатов..........................250
8.3. Специфичные методы.......................................255
8.3.1. Мотивы, заданные пользователем.....................255
8.3.2. Обнаружение мотива.................................260
8.4. Аналогичные методы.......................................271
8.4.1. Гибридные описания «точка-поверхность».............271
8.4.2. Одноточечные описания..............................272
8.5. Использование молекулярного докинга при аннотировании функции.....................................273
8.6. Обсуждение...............................................276
8.7. Заключение...............................................278
Литература................................................278
Глава 9
Динамика белков: от структуры к функционированию..............282
9.1. Молекулярно-динамические расчеты.........................282
9.1.1. Принципы и приближения.............................283
9.1.2. Приложения.........................................286
9.1.3. Ограничения и улучшенные алгоритмы сэмплирования...292
9.2. Анализ главных компонент.................................297
8
Оглавление
9.3. Алгоритмы сэмплирования коллективных координат..............301
9.3.1. Коллективная динамика.................................301
9.3.2. TEE-REX...............................................302
9.4. Методы предсказания функциональных мод......................307
9.4.1. Анализ нормальных мод.................................307
9.4.2. Модели эластичных сетей...............................308
9.4.3. CONCOORD..............................................308
9.5. Итоги и перспективы.........................................313
Литература...................................................315
Глава 10
Интегральные серверы для предсказания функции по структуре.............................................320
10.1. Введение...................................................320
10.1.1. Задача предсказания функции по структуре.............321
10.1.2. Методы предсказания структура-функция................323
10.2. ProKnow....................................................325
10.2.1. Подбор типа укладки..................................325
10.2.2. Структурные мотивы...................................328
10.2.3. Гомология последовательностей........................328
10.2.4. Мотивы в последовательности..........................328
10.2.5. Взаимодействия белков................................328
10.2.6. Объединение предсказаний.............................329
10.2.7. Успешность предсказания..............................329
10.3. ProFunc....................................................330
10.3.1. Основанные на структуре методы, используемые ProFunc.332
10.3.2. Оценка структурных методов...........................339
10.4. Заключение.................................................342
Литература...................................................343
Глава 11 Примеры: предсказание функции структур, полученных в проектах по структурной геномике....................345
11.1. Введение...................................................345
11.2. Примеры масштабного предсказания функции белков............347
11.3. Несколько особых примеров..................................355
11.4. Коллективное аннотирование.................................362
11.5. Заключение.................................................363
Литература.................................................365
Оглавление
9
Глава 12
Предсказание функции белков на основе их теоретических моделей............................367
12.1. Введение................................................368
12.2. Модели белков как общедоступный ресурс..................370
12.2.1. Качество моделей.................................371
12.2.2. Базы данных моделей..............................372
12.3. Точность и добавленная ценность основанных на моделях предсказаний...........................374
12.3.1. Реализация.......................................377
12.4. Практическое применение.................................379
12.4.1. Пластичность остатков каталитического центра.....380
12.4.2. Картирование мутаций.............................382
12.4.3. Комплексы белков.................................383
12.4.4. Предсказания функции на основе моделей ab initio.385
12.4.5. Предсказание специфичности к лигандам............388
12.4.6. Моделирование структуры изоформ, полученных альтернативным сплайсингом.....................390
12.4.7. От общей функции к молекулярным деталям..........391
12.5. Что дальше?.............................................392
Литература................................................393
Указатель основных сокращений и наименований................................................397
Приложение Цветная версия иллюстраций....................................399
Введение
Молекулы белков лежат в основе почти всех биологических процессов. Ученым всегда были любопытны как белки, участвующие в метаболических путях, так и молекулярные основы их функционирования. Однако в эру системной биологии еще больше внимание уделяется полному пониманию работы всей совокупности белков организма, его протеома. Все более важно, что мы не только понимаем все стороны данной функции, или функций, какого-либо белка, но и то, что наше знание распространяется на все компоненты изучаемой системы или организма и так далеко, насколько это возможно. Без всесторонней информации попытки синтеза и расчета не выйдут за рамки приближения реальности.
Для полномасштабного анализа функий белков был создан ряд постгеномных технологий, но зачастую этот анализ ограничивается ценными, но не полными результатами вроде «белок А участвует в делении клетки» или «белки В и С взаимодействуют». Выяснение деталей молекулярного функционирования оказывается гораздо более дорогим, и проводится в лабораториях, воодушевленных специалистами по биоинформатике на заполнение пробелов в наших знаниях. Сравнение аминокислотных последовательностей белков разных видов является основой для компьютерного аннотирования функций белков, хотя запутанные механизмы, которыми эволюция связывает структуры и функцию, часто ограничивают точность и применимость предсказаний. Более того, маловероятно, что истинно новая функция будет предсказана исключительно на основе анализа последовательностей, хотя «сиротская» (orphan) активность - известный биохимический процесс, для которого еще не определены ответственные за него белки, - несомненно существует. Например, некоторые проблемы такого рода могут возникнуть потому, что хотя структура белка и определяется его последовательностью, функция белка определяется в первую очередь его структурой, поэтому несколько незначительных отличий между последовательностями белков могут оказаться значимыми при рассмотрении пространственной структуры белка в целом.
Аксиома, что структура определяет функцию, и поэтому может быть использована для предсказания этой функции, является краеугольным камнем таких областей, как предсказание структуры и структурное аннотирование функций, которые охватываются в этой книге. И хотя структур-
Введение
11
ная геномика обрушила на исследователей вал результатов, все еще остаются неизвестные функции, которые стимулируют разработку структурных методов предсказания функции, и эти методы могут быть применены к модельным структурам хотя бы в некоторой степени. Таким образом, первые главы этой книги охватывают построение структур белков исходя из их последовательностей или хотя бы получение какой-то информации об этих структурах. Затем в книге обсуждаются различные пути, по которым знание структуры приводит к предсказанию функции, и, наконец, в последних двух главах речь идет о реальном применении результатов структурной геномики или моделей белков.
Глава 1 посвящена стремительному развитию методов моделирования ab initio. Этот подход все лучше подходит для точного предсказания укладки белковой цепи или в некоторых случаях даже деталей на уровне расположения атомов, например, для маленьких белков, для которых не удается выявить сходства с уже известными структурами. Глядя на недавние результаты, удивительно вспоминать сейчас, что не далее, как в 1997 году, А.Леск, оценивая результаты CASP2 (конкурса по предсказанию структуры белков) заявлял: «Я считаю результаты... разочаровывающими, или даже отрезвляющими, и многие коллеги разделяют это мнение. За исключением одной мишени, все предсказания увенчались не более чем частичным успехом.» (Lesk, 1997). Главы 2 и 3 посвящены обсуждению структур и моделированию новых структур на основе уже известных. Сравнительное моделирование, рассматриваемое в Главе 3, - это сложившаяся и важная методика, позволяющая во многих случаях последовательно создавать надежные модели. Также важно, что про полученные модели сразу известно, в какой части они более надежны, в какой — менее. Глава 2 касается распознавания укладки белка по его последовательности, которое часто является информативным само по себе (Глава 6), в то время как просто сравнение последовательностей оказывается недостаточным. Однако не менее важно, что распознавание фолда расширяет границы применимости сравнительного моделирования, и это приводит к возрастанию числа моделей, которые могут быть построены по одной экспериментальной структуре. Для мембранных белков, о которых пойдет речь в Главе 4, подходы структурной биоинформатики ограничены по фундаментальным соображениям - число известных пространственных структур все еще мало. По этой причине в Главе 4 детально рассматриваются и вопросы предсказания топологии различных классов белков, что, по сути, является предсказанием структуры низкого разрешения. В Главе 5 речь идет о завораживающем классе белков, которые, будучи изолированными, демонстрируют отсутствие внутренней упорядоченности, но приобретают
12
Введение
её при взаимодействии с другими молекулами. Такие белки, изучение которых расцвело в последнее десятилетие, имеют свои собственные идиосинкратические правила соответствия между структурой и функцией. В Главе 5 эти вопросы также обсуждаются.
Вторая часть этой книги, озаглавленная «От структур к функциям» и начинающаяся с Главы 6, открывает обсуждение вопроса, как изменяются и эволюционируют функции белков в контексте типов их укладки, или фолдов, или типов укладки в надсемействах. Некоторые типы, будучи реально обнаруженными или только предполагаемыми, являются надежными признаками конкретных функций, что важно при предсказании функции на основе структуры; другие типы укладки - суперфолды - обеспечивают реализацию разнообразных функций. Взаимодействия белков с лигандами происходит непременно на поверхности белка, поэтому не удивительно, что многие аспекты геометрии этой поверхности и её свойства могут быть успешно использованы для предсказания функции. Такие методы рассматриваются в Главе 7. В Главе 8 обсуждаются паттерны локальной структуры, которые могут иметь тесное отношение к связыванию лигандов или катализу. Такие паттерны возникают из-за консервативности или конвергентной эволюции эффективных каталитических центров, а также ограничений на связывание, наложенных физико-химическими свойствами конкретного низкомолекулярного соединения. Кроме того, в Главе 8 рассматриваются последние успехи в применении докинга низкомолекулярных соединений для предсказания специфичности ферментов. Зачастую незамеченной остается связь между функционированием белка и его динамикой. Структуры белков не статичны, и их движения, большие или малые, часто являются ключевыми для функционирования. Молекулярная динамика и сходные с ней методы конформационного сэмплирования и анализа рассматриваются в Главе 9, в которой также представлены примеры того, как рассмотрение динамики проясняет наше понимание функционирования белков. Вместе со все нарастающим числом и спектром методов предсказания функции белка по его структуре, целесообразным становится одновременное применение нескольких методов в рамках интегральных веб-серверов. Такой подход удобен для пользователя, а также позволяет делать консенсусные предсказания. В Главе 10 описаны возможности и функционирование веб-серверов ProFunc и ProKnow, реализующих этот подход. В Главе 11 обсуждается опубликованная работа, в которой основанные на структуре методы были применены к предсказанию функции белков, полученных в рамках проектов по структурной геномике. Это позволило получить ценную картину того, какой из методов обычно оказывается наиболее информативным. Глава завершается обсуждением
Литература
13
последних тенденций в направлении коллективного аннотирования как способа преодоления узких мест в аннотировании таких белков. Глава 12 охватывает приложения структурных методов к структурам моделей, полученным как с помощью сравнительного моделирования, так и с помощью методов ab initio. Наряду с большим количеством примеров обсуждается опубликованная работа, в которой оценивается точность моделей с функционально-значимой точки зрения, а также применимость различных методов моделирования.
Цель этой книги состоит в предоставлении современного взгляда на состояние дел в предсказании структуры белков и основанном на структуре предсказании функции белков. Каждая глава содержит ссылки на доступные веб-серверы и другие ресурсы, которые читатель может пожелать использовать в своей работе. В конце каждой главы авторы намечают направления дальнейшего развития и ожидаемые затруднения в соответствующих областях. Когда написание книги уже подходило к концу, появилось сообщение о значительном успехе в давнишней проблеме - улучшении сравнительных моделей (Jagielska et al. 2008). Тем не менее, создается впечатление, что структуры белков непрерывно готовят нам новые трудности. Стоило нам почувствовать, что научное сообщество приблизилось к объяснению явлений, природа которых до сих пор была не ясна, таких как обмен доменов, круговая перестановка, образование фибрилл, белки с присущей неупорядоченностью и многими другими, как мы сталкиваемся с метаморфными белками (Murzin 2008), исследование которых может значительно поспособствовать нашему пониманию пространства типов укладки белков. Смогут ли методы биоинформатики хотя был предсказать, какие белки могут видоизменяться между двумя типами укладки? Трудно сказать, но совершенно ясно, что биоинформатика структуры и функции белков на протяжении многих лет будет оставаться волнующей областью исследований.
Литература
Jagielska A, Wroblewska L, Skolnick J (2008) Protein model refinement using an optimized physics-based all-atom force field. Proc Natl Acad Sci USA 105:8268-8273
Lesk AM (1997) CASP2: report on ab initio predictions. Proteins Suppl 1:151-166 Murzin AG (2008) Metamorphic proteins. Science 320:1725-1726
Глава 1
Предсказание структуры белков ab initio
Жу-ён Ли, Ситао By, Ян Жанг
Несмотря на пятьдесят лет усилий, проблема предсказания пространственной структуры белков по аминокислотной последовательности по-прежнему не решена. Если у исследуемого белка существует гомолог с известной структурой, задача сравнительно проста: копируя каркас известной структуры, можно построить модель высокого разрешения. Однако такая процедура моделирования не помогает ответить на вопросы о том, как и почему белок приобретает характерную для него структуру. В тех случаях, когда структурных гомологов (иногда аналогов1) исследуемого белка не существует или они существуют, но гомологию не удается установить, модели приходится строить «с нуля». Эта процедура, называемая моделированием ab initio, важна для решения проблемы предсказания структуры белков; также она может оказаться полезной для понимания физико-химических принципов сворачивания белков в природе. В настоящее время точность методов моделирования ab initio невысока, а успех ограничивается белками небольшого размера (менее 100 остатков). В настоящей главе приведен обзор методов моделирования ab initio. Особое внимание *
Jooyoung Lee
Center for Bioinformatics and Department of Molecular Bioscience,
University of Kansas, Lawrence, KS, 66047, USA
School of Computational Sciences, Korea Institute for Advanced Study,
Seoul, 130-722, Korea
Sitao Wu and Yang Zhang
Centre for Bioinformatics and Department of Molecular Bioscience,
University of Kansas, Lawreance, KS, 66047, USA
e-mail: yzhang@ku.edu
1 Напомним, гомологами называются белки, имеющие общего предка, аналогами - выполняющие сходную функцию. Прим, перев.
1.1. Введение
15
уделено трем основным факторам алгоритмов моделирования: энергетической функции, конформационному поиску и отбору моделей. Обсуждаются достижения и успехи различных алгоритмов.
1.1. Введение
В связи с выдающимися успехами проектов секвенирования генома количество доступных последовательностей белков растет экспоненциально. Однако из-за технических сложностей, существенных временных и трудовых затрат при экспериментальном определении структуры белков количество доступных белковых структур сильно запаздывает. К концу 2007 года в базе данных UniProtKB (Bairoch etal. 2005) (http://www.ebi.ac.uk/swissprot) насчитывалось 5,3 миллиона белковых последовательностей. Однако соответствующее количество белковых структур в базе данных the Protein Data Bank (PDB) (Berman et al. 2000) (http://www.rcsb.org/pdb) составляло лишь около 44000 - менее 1 % от числа белковых последовательностей2. Как видно из рис. 1.1, разрыв стремительно увеличивается. Таким образом, развитие эффективных компьютерных алгоритмов предсказания пространственной структуры белков по последовательности является, возможно, единственным способом сократить этот разрыв.
В зависимости от того, насколько белки, использующиеся для построения модели, похожи на исследуемый белок, методы предсказания структуры белков делятся на две категории. Если белки, близкие по структуре к исследуемому, удалось обнаружить в библиотеке PDB, можно построить модель исследуемого белка, копируя каркас белков известной структуры (шаблонов). Такая процедура называется сравнительным моделированием или моделированием на основании гомологии (англ, template-based modeling, ТВМ) (Karplus etal. 1998; Jones 1999; Shi etal. 2001; Ginalski etal. 2003b; Skolnick etal. 2004; Jaroszewski etal. 2005; Soding 2005; Zhou and Zhou 2005; Cheng and Baldi 2006; Pieper et al. 2006; Wu and Zhang 2008) и будет обсуждаться в последующих главах. Методы сравнительного моделирования, как правило, позволяют создавать модели высокого разрешения, однако эта процедура не может помочь в понимании физико-химических принципов, которые лежат в основе процесса укладки белков.
Если белковых шаблонов нет в наличии, пространственную модель белка приходится строить «с нуля». Эта процедура имеет несколько названий, например, моделирование ab initio (Klepeis etal. 2005; Liwo et al. 2005;
2 На начало 2013 года в базе данных UniProt/TrEMBL насчитывается около 29 миллионов последовательностей, а в базе данных PDB 87 тысяч структур. Прим, перев.
16
Глава 1. Предсказание структуры белков ab initio
Рис.1.1. Рост числа доступных последовательностей белков (левая ось ординат) и числа расшифрованных структур белков (правая ось ординат) за последние 12 лет. Отношение числа последовательностей к числу структур быстро возрастает. Данные получены из баз данных UniProtKB (Bairoch et al. 2005) и PDB (Berman et al. 2000).
Wu et al. 2007), моделирование de novo (Bradley et al. 2005), физическое моделирование (Oldziej et al. 2005) или свободное моделирование (Jauch et al. 2007). В настоящей главе, чтобы избежать путаницы, будет использоваться термин «моделирование ab initio». В отличие от сравнительного моделирования, успешное моделирование ab initio может помочь ответить на основные вопросы о том, как и почему белок приобретает характерную для него структуру из множества возможных.
Как правило, моделирование ab initio сопровождается конформационным поиском, условия которого определяются специально заданной энергетической функцией. Результатом этой процедуры обычно является ряд конформаций (структурных макетов), среди которых осуществляется отбор окончательных моделей. Таким образом, успешное моделирование ab initio определяется тремя факторами: 1) точная энергетическая функция, согласно которой среди множества структурных макетов самому стабильному с точки зрения термодинамики состоянию отвечает природная структура белка; 2) эффективный метод поиска, с помощью которого можно быстро определить низкоэнергитические состояния посредством конформационного поиска; 3) отбор из множества структурных макетов моделей, наиболее близких к природной структуре.
1.2. Энергетические функции
17
Настоящая глава содержит обзор современного состояния области исследований, которая связана с прогнозированием структуры белков ab initio. Приведенный обзор не является ни полным (не включает информацию обо всех существующих методах ab initio), ни глубоким (не содержит исчерпывающей информации об основах и назначении методов). Для сравнительного изучения различных методов моделирования ab initio читателям рекомендуется обратиться к одному из последних обзоров по теме (Helles 2008). Оставшаяся часть главы организована следующим образом. Подробно описаны три основные проблемы моделирования ab initio -энергетическая функция, алгоритм конформационного поиска и схема отбора моделей. Обсуждаются новые и перспективные идеи по улучшению эффективности и результативности предсказания. Наконец, обсуждаются современные успехи и проблемы моделирования ab initio.
1.2. Энергетические функции
В этом разделе будут рассмотрены энергетические функции, используемые для моделирования ab initio. Следует отметить, что часто энергетические функции тесно взаимосвязаны с процедурами поиска, и при разделении этих компонентов процедура моделирования утрачивает свою производительность/достоверность. Используемая классификация функций включает две группы: а) рациональные энергетические функции; б) эмпирические энергетические функции. Принадлежность к определенной группе зависит от того, используются ли статистические данные известных пространственных структур белков. Несколько наиболее перспективных методов из каждой группы, отличающихся точностью моделирования и уникальностью, обсуждаются более подробно. В таблице 1.1 приводится список методов моделирования ab initio, а также информация о таких свойствах различных методов, как энергетические функции, алгоритмы конформационного поиска, методы отбора моделей и характерные времена вычислений.
1.2.1. Рациональные энергетические функции
В случае строго рационального ab initio метода описание взаимодействия между атомами опирается на законы квантовой механики и кулоновский потенциал, при этом используются лишь некоторые фундаментальные постоянные, такие как заряд электрона и постоянная Планка. Описания атомов представлены типами атомов, в которых значимо лишь
18
Глава 1. Предсказание структуры белков ab initio
Таблица 1.1. Список алгоритмов моделирования ab initio, рассмотренных в этой главе, а также их энергетические функции, методы конформационного поиска, схемы отбора моделей и типичное процессорное время в расчете на одну мишень
Алгоритм и адрес сервера Тип силового ПОЛЯ Метод поиска Отбор моделей Затраты процессорного времени
AMBER/ CHARMM/ OPLS (Brooks et al. 1983; Weiner et al. 1984; Jorgensen and Tirado-Rives 1988; Duan and Kollman 1998; Zagrovic et al. 2002) Рациональное Молекулярная динамика (МД) Наименьшая энергия Годы
UNRES (Liwo et al. 1999, 2005; Oldziej et al. 2005) Рациональное Отжиг в конформационном пространстве (CSA) Кластеризация/ свободная энергия Часы
ASTRO-FOLD (Klepeis and Floudas 2003; Klepeis et al. 2005) Рациональное aBB/CSA/ мд Наименьшая энергия Месяцы
ROSETTA (Simons et al. 1997; Das et al. 2007) http://www.robetta.org Рацио-нально-эмпири-ческое Монте Карло (МК) Кластеризация/ свободная энергия Месяцы
TASSER/Chunk-TASSER (Zhang and Skolnick 2004a; Zhou and Skolnick 2007) http://cssb.biology.gatech.edu/ skolnick/webservice/MetaTASSER Эмпирическое МК Кластеризация/ свободная энергия Часы
I-TASSER (Wu et al. 2007; Zhang 2007) http://zhang. bioinformatics.ku.edu/ITASSER Эмпирическое МК Кластеризация/ свободная энергия Часы
количество электронов для каждого типа (Hagler etal. 1974; Weiner etal. 1984). Однако до настоящего времени серьезных попыток использования методов, в основу которых была бы положена квантовая механика, не предпринималось просто потому, что вычислительные ресурсы, необходимые для таких расчетов, значительно превосходят доступные на сегодняшний день. Без квантово-механического рассмотрения взаимодействий отправной точкой при моделировании белков ab initio по сути становится
1.2. Энергетические функции
19
использование силовых полей, оперирующих с большим числом типов атомов; химические и физические свойства атомов для каждого типа близки к параметрам, рассчитанным на основе структур кристаллов или квантово-механической теории (Hagler etal. 1974; Weiner etal. 1984). Известными примерами таких полноатомных рациональных силовых полей являются AMBER (Weiner et al. 1984; Cornell et al. 1995; Duan and Kollman 1998), CHARMM (Brooks etal. 1983; Nena etal. 1996; MacKerell Jr. etal. 1998), OPLS (Jorgensen and Tirado-Rives 1988; Jorgensen etal. 1996) и GROMOS96 (van Gunsteren etal. 1996). Потенциалы этих силовых полей содержат члены, связанные с длиной связей, величиной валентных и торсионных углов, взаимодействиями Ван-дер-Ваальса и электростатическими взаимодействиями. Основные различия между ними заключаются в выборе типов атомов и параметров взаимодействий.
Для изучения процесса укладки белков классические силовые поля часто использовались в сочетании с моделированием методом молекулярной динамики (МД). Однако с точки зрения предсказания структуры белков результаты были не совсем успешными. (Об использовании МД для выявления функции белков на основании данных об известных белковых структурах см. в Главе 10). Первым значительным успехом использования МД для изучения процесса ab initio сворачивания белка стала, вероятно, работа Дуана и Кольмана 1997 года. Они моделировали головку виллина (36-членный фрагмент) в явно заданном растворителе в течение 6 месяцев на параллельных суперкомпьютерах. Структуру высокого разрешения окончательной упаковки белка получить не удалось, однако лучшая из полученных моделей имела отклонение от нативной структуры в пределах 4,5 А (Duan and Kollman 1998). Панде и его коллеги недавно провели моделирование сворачивания этого небольшого белка с использованием Folding@Home -компьютерной системы, рассредоточенной по всему миру (Zagrovic etal. 2002). Отклонение от нативной структуры составило 1,7 А, а общее время моделирования - 300 мс, или около 1000 лет процессорного времени. Несмотря на эти весьма значительные усилия, моделирование методом МД с использованием полноатомных силовых полей отнюдь не является стандартным методом предсказания структуры белков среднего размера (около 100-300 остатков). Более того, систематическая оценка достоверно-сти/точности полученных результатов не проводилась даже для белков небольшого размера.
Еще одна возможная область применения рациональных силовых полей в моделировании методом МД - улучшение «качества» белковых структур. Целью в данном случае является приближение структур белковых моделей, начиная со структур с низким разрешением, к нативной
20
Глава 1. Предсказание структуры белков ab initio
структуре белка за счет улучшения локальной упаковки боковых цепей и основной пептидной цепи. Когда исходная модель близка к нативной структуре, направленные конформационные изменения относительно малы, а значит, время моделирования будет значительно меньше того, которое потребовалось бы для моделирования сворачивания белка ab initio. Одним из первых успешных примеров уточнения структуры белка с использованием МД была «лейциновая застежка» GCN4 (димер размером 33 остатка) (Nilges and Brunger 1991; Vieth etal. 1994). Неупорядоченная структура димера низкого разрешения (2-3 А) была сначала собрана методом моделирования Монте-Карло (МК), а затем улучшена методом МД. С помощью ограничений, характерных для спиральной конформации, наложенных на двугранные углы, Сколник и его коллеги (Vieth et al. 1994) смогли получить уточненную структуру белка GCN4, для которой среднеквадратичное отклонение (СКО, англ, root mean square deviation, RMSD) основной цепи составило менее 1 А. Использовались силовое поле CHARMM (Brooks et al. 1983) и модель воды TIP3P (Jorgensen et al. 1983).
Позже Ли и соавт. (Lee etal. 2001), используя AMBER 5.0 (Case etal. 1997) и модель воды TIP3P (Jorgensen etal. 1983), предприняли попытку улучшить качество 360 структурных моделей низкого разрешения, созданных программой ROSETTA (Simons etal. 1997) для 12 белков небольшого размера (менее 75 остатков). Однако они пришли к выводу, что систематического улучшения качества структур достичь не удалось (Lee et al. 2001). Фэн и Марк (Fan and Mark 2004) попытались улучшить структуру 60 моделей, созданных ROSETTA для 11 небольших белков (менее 85 остатков), используя GROM ACS 3.0 (Lindahl etal. 2001) и явно заданную модель воды (Berendsen etal. 1981). Сообщается, что для 11 из 60 моделей удалось улучшить значения СКО на 10%, однако для 18 из 60 моделей значения СКО ухудшились после процедуры уточнения структуры. Чен и Брукс (Chen and Brooks 2007) использовали CHARMM22 (MacKerell Jr. etal. 1998) для уточнения структуры пяти мишеней CASP63 (размером 70-144 остатка), полученных при помощи сравнительного моделирования. В четырех случаях было достигнуто уменьшение СКО на величину до 1 А. В работе была использована неявно заданная модель растворителя, основанная на обобщенном приближении Борна (generalized Bom (GB) approximation) (Im et al. 2003), что значительно ускорило вычисления. Кроме того, в ходе процедуры уточнения структуры налагались пространственные ограничения, имевшиеся в начальных моделях (Chen and Brooks 2007).
3 CASP - конкурс критической оценки методов предсказания структуры белков (Critical Assessment of Structure Prediction). Прим, перев.
1.2. Энергетические функции
21
Заслуживают внимания результаты, которые получили Сумма и Левитт (Summa and Levitt 2007). Они использовали различные потенциалы молекулярной механики (ММ), а именно AMBER99 (Wang et al. 2000; Sorin and Pande 2005), OPLS-AA (Kaminski etal. 2001), GROMOS96 (van Gunsteren et al. 1996) и ENCAD (Levitt et al. 1995), для улучшения структуры 75 белков с помощью процедуры минимизации энергии in vacuo. Было установлено превосходство эмпирических атомных контактных потенциалов над потенциалами ММ: в случае применения первых структурные макеты почти всех тестируемых белков приближались к нативным состояниям, тогда как при применении последних, за исключением AMBER99, макеты структур по сути отдалялись от нативных состояний. Возможно, неудовлетворительные результаты при использовании потенциалов ММ были частично обусловлены выполнением моделирования в вакууме, без сольватации. Полученные данные демонстрируют возможности сочетания эмпирических потенциалов и физических силовых полей для уточнения структуры белков.
Применение рациональных потенциалов и связанного с ними МД-мо-делирования не дало ожидаемых результатов в области предсказания структуры белков. В то же время, методы быстрого поиска (такие как моделирование методом Монте-Карло и генетические алгоритмы), основанные на рациональных потенциалах, зарекомендовали себя перспективными как при предсказании белковых структур, так и при повышении их качества. Один из примеров использования этих методов - продолжающийся проект Шераги и его коллег (Liwo et al. 1999, 2005; Oldziej et al. 2005), которые разрабатывают рациональный метод предсказания структуры белков исключительно на основе термодинамической гипотезы. Метод сочетает использование крупнозернистого потенциала UN RES с алгоритмом глобальной оптимизации, который называется отжигом в конформационном пространстве (Oldziej et al. 2005). В потенциале UNRES каждый аминокислотный остаток описывается двумя взаимодействующими соединенными частицами: атомом Са и центром боковой цепи остатка. Это по сути уменьшает число атомов в десять раз, что дает возможность исследовать полипептидные цепи размером более 100 остатков. Время предсказания в таком случае можно снизить до 2-10 часов. Энергетическая функция UNRES (Liwo et al. 1993) содержит член, отвечающий за вклад всех парных взаимодействий между частицами системы, а также дополнительные члены, такие как локальная энергия и энергия корреляции. Модели UNRES с низкой энергией затем преобразуются в полноатомные с помощью силового поля ЕСЕРР/3 (Nemethy et al. 1992). Хотя многие параметры энергетической функции рассчитываются с использованием методов квантовой
22
Глава 1. Предсказание структуры белков ab initio
механики, некоторые из них все же получают, используя функции распределения и корреляционные функции для данных, из базы PDB. В связи с этим может возникнуть вопрос, насколько природа описанного подхода является неэмпирической, или ab initio. Тем не менее, среди доступных методов моделирования ab initio этот метод, пожалуй, является одним из наиболее достоверных (в терминах применения полной глобальной оптимизации к рациональной энергетической функции). С 1998 года он систематически применялся для исследования множества мишеней CASP. Наиболее заметные успехи при прогнозировании этим методом были достигнуты для Т061 из CASP3. Для созданной модели а-спирального белка размером 95 остатков значение СКО от нативной структуры составило 4,2 А. Точность моделей, полученных для белка другими методами, была значительно ниже. Впервые четко показано, что качество моделей мишеней, полученных с помощью метода ab initio, может быть лучше, чем моделей, полученных с помощью методов, в основе которых лежит использование шаблона. В CASP6 упаковка мишени структурной геномики ТМ0487 (Т0230, 102 остатка) была осуществлена с помощью этого метода с точностью 7,3 А. Тем не менее, крайне малое количество моделей, полученных исключительно с использованием методов моделирования ab initio, а также лучшая, но все еще низкая, точность таких моделей стали причиной отсутствия должного интереса у научного сообщества, где пользуются большим спросом точные модели белка.
Еще один пример метода рационального подхода к моделированию -это многостадийный иерархический алгоритм ASTRO-FOLD, который был предложен Флудасом и его коллегами (Klepeis and Floudas 2003; Klepeis et al. 2005). Сначала на основании вычисления функции свободной энергии перекрывающихся олигопептидов (как правило, пентапептидов) и всех возможных контактов между парами гидрофобных остатков осуществляется предсказание элементов вторичной структуры (а-спиралей и р-тяжей). Используются члены свободной энергии, которые отражают вклад энтропии, образования полостей, а также поляризационный и ионизационный вклад каждого олигопептида. Затем рассчитанная предрасположенность к образованию той или иной вторичной структуры преобразуется в верхнюю и нижнюю границы для значений двугранных углов основной цепи белка, а также в ограничения, налагаемые на расстояния между атомами Са. После этого в ходе глобальной минимизации в полноатомном силовом поле ЕСЕРР/3 создается окончательная модель третичной структуры полноразмерного белка. Описанный подход успешно применялся для предсказания структуры а-спирального белка размером 102 остатка двойным слепым методом (однако открытая проверка сообщест
1.2. Энергетические функции
23
вом для сравнения относительной производительности этого и других методов не проводилась). СКО атомов Са предсказанной модели от экспериментальной структуры составило 4,94 А. Метод глобальной оптимизации, используемый при таком подходе, сочетает в себе метод а-ветвей и границ (аВВ), отжиг в конформационном пространстве (CSA) и МД-моделиро-вание (Klepeis and Floudas 2003; Klepeis et al. 2005). Относительную производительность этого метода при определении белковых структур еще предстоит оценить в будущем.
Тейлор и его коллеги (2008) недавно предложили новый подход. Построение структурных моделей белка осуществляется с помощью перебора возможных топологий в крупнозернистом представлении с учетом заданных определений вторичной структуры и ограничений физических контактов между элементами вторичной структуры. В основе оценки конформаций лежат компактность структуры и экспонированность элементов. Конформации, получившие наиболее высокие оценки, затем отбирают для дальнейшего уточнения (Jonassen et al. 2006). Авторы успешно осуществили упаковку набора из пяти белков с укладкой типа «ар-сэндвич» размером до 160 остатков, при этом для первой модели значение СКО от природной структуры составило 4-6 А. Но опять-таки, несмотря на то, что метод вызывает интерес с точки зрения методологии, его производительность в открытых слепых экспериментах на белках с различными типами укладки еще предстоит выяснить.
В последней разработке ROSETTA (Bradley et al. 2005; Das et al. 2007) рациональный атомный потенциал используется на второй стадии усовершенствования структуры методом Монте-Карло, которой предшествует сборка фрагментов с низким разрешением (Simons etal. 1997). Особенности этого метода обсуждаются в следующем разделе.
1.2.2. Сочетание эмпирических энергетических функций и сборки из фрагментов
В основу эмпирического потенциала положены эмпирические энергетические термы4, которые установлены на основе статистических данных об известных белковых структурах, размещенных в базе данных PDB. Эти энергетические термы, согласно Сколнику (2006), можно разделить на две группы. К первой группе принадлежат общие энергетические члены и энергетические члены, не зависящие от аминокислотной последователь
4 По устоявшейся терминологии, терм - это одно из слагаемых энергетической функции. Прим, перев.
24
Глава 1. Предсказание структуры белков ab initio
ности, например, вклад водородных связей или жесткость основной цепи пептида (Zhang et al. 2003). Вторая группа содержит энергетические термы, зависящие от аминокислотного состава или последовательности белка, например, потенциал парных взаимодействий остатков (Skolnick et al. 1997), потенциал атомных взаимодействий, зависящих от расстояния (Samudrala and Moult 1998; Lu and Skolnick 2001; Zhou and Zhou 2002; Shen and Sali 2006), член, отражающий предрасположенность к формированию той или иной вторичной структуры (Zhang et al. 2003, 2006; Zhang and Skolnick 2005a).
В большинстве эмпирических силовых полей учитывается предрасположенность вторичной структуры, однако локальную структуру белка, вероятно, довольно сложно воспроизвести при упрощенном моделировании. Иными словами, в природе для разнообразных белковых последовательностей характерны, как правило, либо спиральные, либо распрямленные элементы структуры в зависимости от едва уловимых различий в локальном и глобальном окружении последовательностей, однако пока еще не созданы силовые поля, которые могут воспроизводить такие тонкие различия должным образом. Один из способов обойти эту проблему состоит в непосредственном использовании для сборки пространственных моделей фрагментов вторичной структуры, полученных на основе анализа последовательностей или в ходе выравнивания профилей. Дополнительное преимущество такого подхода заключается в том, что использование вырезанных фрагментов вторичной структуры может значительно снизить энтропию при конформационном поиске.
В настоящем разделе представлены два метода предсказания структуры белков, в основу которых положены эмпирические энергетические функции. Показано, что эти методы принадлежат к числу наиболее успешных методов предсказания структуры белков ab initio (Simons etal. 1997; Zhang and Skolnick 2004a).
Одна из наиболее широко известных идей в моделировании ab initio была впервые предложена Боуи и Эйзенбергом. Они создавали белковые модели, собирая небольшие фрагменты (преимущественно нонамеры), взятые из базы данных PDB (Bowie and Eisenberg 1994). Используя аналогичную идею, Бейкер и его коллеги разработали метод ROSETTA (Simons et al. 1997), который оказался весьма успешным при свободном моделировании мишеней в экспериментах CASP. Это привело к тому, что подход, основанный на сборке фрагментов, стал очень популярным в научном сообществе. В последних версиях ROSETTA (Bradley etal. 2005; Das etal. 2007) авторы сначала создавали упрощенные модели, конформации которых были представлены тяжелой основной цепью белка и атомами Ср. На
1.2. Энергетические функции
25
второй стадии ряд отобранных моделей низкого разрешения проходил процедуру уточнения структуры с использованием полноатомной рациональной энергетической функции, которая включала взаимодействия Ван-дер-Ваальса, свободную энергию парных взаимодействий с растворителем и зависящий от ориентации потенциал водородных связей. Блок-схема двухстадийного моделирования представлена на рис. 1.2; подробности описания энергетических функций можно найти в ссылках (Bradley et al. 2005; Das et al. 2007). В ходе конформационного поиска осуществляется большое количество циклов минимизации энергии методом Монте-Карло (Li and Scheraga 1987). Наиболее ярким примером применения этого двухстадийного протокола является слепое предсказание структуры мишени ab initio (Т0281 из CASP6, 70 остатков), для которой СКО атомов Са от кристаллографической структуры составило 1,6 A (Bradley et al. 2005). В CASP7 широкое сэмплирование осуществлялась с помощью распределенных сетевых вычислений Rosetta@home, что давало возможность использовать около 500000 часов процессорного времени для каждого домена мишени. Одна из мишеней, Т0283, была создана в ходе моделирования по шаблону, однако моделирование осуществлялось ROSETTA с использованием протокола ab initio. Полученная таким образом модель имела CKO = 1,8 А для 92 остатков из 112 (рис. 1.3, слева). Несмотря на значительные успехи, описанная процедура является довольно затратной в отношении вычислительных ресурсов, что препятствует ее повседневному использованию.
Заметные успехи алгоритма ROSETTA, а также ограниченная доступность энергетических функций этого метода привели к тому, что некоторые исследовательские группы приступили к самостоятельной разработке энергетических функций, основанных на идее ROSETTA. К числу программ-производных ROSETTA принадлежат Simfold (Fujitsuka etal. 2006) и Profesy (Lee et al. 2004); их энергетические функции содержат следующие термы: потенциал взаимодействий Ван-дер-Ваальса, потенциалы двугранных углов основной цепи белка, потенциал гидрофобных взаимодействий, потенциал водородных связей для основной цепи белка, ротамерный потенциал, терм энергии парных взаимодействий, потенциал парных взаимодействий р-тяжей и терм, контролирующий радиус компакгизации белка. Однако результаты, полученные при предсказании этими методами, были лишь отчасти успешными по сравнению с ROSETTA.
Еще один успешный подход свободного моделирования - это программа TASSER Чжана и Сколника (2004а), которая строит пространственные модели белков исключительно с использованием эмпирических методов. Последовательность мишени сначала «протягивается» через набор репрезентативных белковых структур в процессе поиска возможных
26
Глава 1. Предсказание структуры белков ab initio
Модели низкого разрешения
Фаза И: Оптимизация на атомарном уровне с помощью рациональных потенциалов
Конечная модель с атомарным разрешением
Рис. 1.2. Блок-схема протокола программы ROSETTA
способов укладки. Затем близкие фрагменты (более 5 остатков) извлекают из областей, выровненных в ходе протягивания, и используют при повторной сборке полноразмерных моделей. Области, которые не удалось выровнять, строят с помощью методов моделирования ab initio (Zhang et al. 2003). Конформация белка в TASSER представлена набором Са-атомов и центрами масс боковых цепей. Процесс повторной сборки осуществляется с помощью параллельного моделирования методом Монте-Карло. Энергетические потенциалы TASSER содержат информацию о предсказанных
1.2. Энергетические функции
Рис. 1.3. (Цветную версию рисунка см. на вклейке.) Два примера успешного свободного моделирования из CASP7. Т0283 (слева) - мишень сравнительного моделирования (из Bacillus halodurans) размером 112 остатков. Модель построена с помощью полноатомного метода ROSETTA (гибридный подход, сочетающий в себе физические и эмпирические методы) (Das et al. 2007) на основе свободного моделирования. Оценка (TM-score) составляет 0,74 (Zhang and Skolnick 2004b); значение CKO -1,8 A для 92 остатков (общее значение CKO - 13,8 А из-за неправильной ориентации С-концевой спирали). Т0382 (справа) - мишень сравнительного моделирования (из Rhodopseudomonas palustris CGA009) размером 123 остатка. Модель построена с помощью метода I-TASSER (исключительно эмпирический подход) (Zhan 2007). Оценка составляет 0,66; СКО - 3,6 А. Синим и красным цветами показаны модельная и кристаллографическая структуры, соответственно
предрасположенностях вторичной структуры, водородных связях основной цепи, различных коротко- и дальнодействующих корреляциях и энергии гидрофобных взаимодействий, которая основана на статистических данных о структурах из библиотеки PDB. Вклады эмпирических энергетических потенциалов оптимизированы с использованием большого набора структурных макетов (Zhang et aL 2003), что приводит к согласованию сложных взаимосвязей между различными потенциалами взаимодействия.
Существует несколько новых версий TASSER. Одна из них - Chunk-TASSER (Zhou and Skolnick 2007), принадлежащая группе Сколника. Здесь последовательности мишени сначала разделяют на подпоследовательности («куски», англ, «chunks»), каждая из которых содержит три последовательных стандартных элемента вторичной структуры (спирали и/или тяжа). Такие подпоследовательности затем сворачиваются независимо. Наконец, на основе моделей подпоследовательностей устанавливаются пространственные ограничения, которые используются для последующего моделирования TASSER.
Еще одна версия - I-TASSER (Wu et al. 2007) - уточняет положение центров масс кластеров TASSER в ходе многократных этапов моделирования методом Монте-Карло. На основании моделей, полученных в первом цикле моделирования TASSER, и структурных шаблонов, определенных с помощью выравнивания в ходе моделирования по шаблону с использованием данных библиотеки PDB, устанавливаются пространствен-
28
Глава 1. Предсказание структуры белков ab initio
Рис. 1.4. (Цветную версию рисунка см. на вклейке.) Блок-схема программы моделирования структуры белков I-TASSER
ные ограничения, которые затем используются во втором цикле моделирования. Целью моделирования является устранение стерических наталкиваний и уточнение топологии. Блок-схема алгоритма I-TASSER приведена на рис. 1.4. Несмотря на то, что в ходе процедуры используются структурные фрагменты и пространственные ограничения шаблонов, полученных в ходе протягивания, с помощью метода часто удается построить модели, обладающие корректной топологией, даже в тех случаях, когда топологии шаблонов, составляющих модель, некорректны. В CASP7 из 19 мишеней для свободного моделирования и моделирования по шаблону с помощью 1-TASSER удалось построить модели с корректной топологией (3-5 А) для 7 последовательностей размером до 155 остатков. На рис. 1.3 (справа) приведен пример Т0382 (123 остатка), для которого начальные шаблоны имели неверную топологию (более 9 А), однако окончательная модель на 3,6 А отличалась от структуры, полученной методом рентгеноструктурного анализа. Недавно Хеллесом было проведено сравнительное исследование 18 алгоритмов прогнозирования ab initio. Он пришел к заключению, что I-TASSER - один из лучших методов по таким показателям, как точность моделирования и затраты процессорного времени, приходящиеся на мишень (Helles 2008).
1.3. Методы конформационного поиска
Успех моделирования белковых структур методами ab initio зависит от наличия действенного метода конфомационного поиска, с помощью ко-горого можно эффективно находить глобальный энергетический минимум
1.3. Методы конформационного поиска
29
структуры при заданной функции энергии со сложной энергетической поверхностью. Исторически наиболее популярными методами исследования конфомационного пространства макромолекул, таких как белки, являются моделирование методом Монте-Карло и молекулярная динамика. Моделирование сложных систем, таких как белки, каноническими методами МД/МК с условием полного исследования конформационного пространства обычно требует огромных вычислительных ресурсов. Опыт прямого применения МД для получения природной структуры белков не содержит по-настоящему успешных примеров. Одна из возможных причин таких неудач, возможно, заключается в том, что время моделирования, необходимое для упаковки небольшого белка, составляет миллисекунды, что в 1012 раз больше типичного шага интегрирования, значение которого составляет фемтосекунды (1015). Основная техническая сложность, возникающая при применении метода МК, является следствием того, что энергетическая поверхность конформационного пространства белка обычно довольно «пересеченная» и содержит множество энергетических барьеров, которые могут легко блокировать процедуру МК-моделирования.
В этом разделе обсуждаются последние разработки в области методов конформационного поиска, направленные на решение обозначенных выше проблем. Проиллюстрированы ключевые идеи методов конформационного поиска, которые используются в различных процедурах ab initio и связанных с ними методах моделирования белков. За подробностями читателю рекомендуется обратиться к соответствующим ссылкам. В отличие от различных энергетических функций, которые используются в моделировании методами ab initio, в поисковых методах в принципе должна существовать возможность переноса элементов из одного метода моделирования белков в другой, как и в случае науки и технологии в целом. В настоящее время не существует единого метода, который бы отличался исключительной производительностью при решении любых исследовательских задач. Исследование и систематическое увеличение производительности различных поисковых методов - проблемы, которые еще только предстоит решить.
1.3.1. Моделирование методом Монте-Карло
Алгоритм имитации отжига (ИО) (Kirkpatrick et al. 1983) - возможно, наиболее популярный метод конформационного поиска. Принципы ИО просты и понятны, метод можно с легкостью применять для решения любых задач по оптимизации структуры. Обычно в ИО для создания набора конформационных состояний, которые подчиняются классическому больцмановскому распределению энергии при заданной температуре, ис
30
Глава 1. Предсказание структуры белков ab initio
пользуется алгоритм МК, предложенный Н. Метрополисом. На начальном этапе ИО выполняется расчет при высокой температуре, за которым следует серия расчетов с постепенным снижением температуры. (Отсюда название метода - имитация отжига.) Именно в силу простоты ИО эффективность осуществляемого конформационного поиска невысока по сравнению с другими, более изощренными методами, которые обсуждаются ниже.
В тех случаях, когда энергетическая поверхность исследуемой системы неровная (из-за множества энергетических барьеров), расчеты методом МК имеют свойство «застревать» в метастабильных состояниях, которые в дальнейшем искажают распределение отобранных‘Состояний, нарушая эргодичность выборки. Чтобы обойти это нарушение, было разработано множество методик. Одна из них основана на использовании обобщенного ансамбля вместо канонического, который обычно используется при моделировании. Первоначально метод имел разные названия, в том числе муль-тиканонического ансамбля (Berg and Neuhaus 1992) и энтропийного ансамбля (Lee 1993). Идея, лежащая в основе метода, состоит в том, чтобы ускорить переход между состояниями, разделенными энергетическими барьерами. Осуществляется это посредством изменения вероятности перехода таким образом, что вид окончательного распределения энергии выборки сменяется с колоколообразного на более плоский.
Еще один популярный метод, близкий к описанному, - МК-метод обмена реплик (replica exchange МС method, REM) (Kihara etal. 2001), при котором одновременно выполняется ряд расчетов методом МК в выбранном диапазоне температур. Время от времени делаются попытки обменять структуры (или, равнозначно, температуры) соседних запусков для сэмплирования состояний в широком диапазоне значений энергии, что дает возможность преодолеть энергетические барьеры. Параллельное гиперболическое сэмплирование (parallel hyperbolic sampling, PHS) (Zhang etal. 2002) является расширением метода REM для понижения энергетического барьера путем введения динамически деформирующейся энергии с помощью обратного гиперболического синуса.
Метод Монте-Карло с минимизацией (Monte Carlo with minimization, MCM), первоначально разработанный Ли и Шерагой (Li and Scheraga 1987), успешно применялся для конформационного поиска в энергетической функции высокого разрешения программы ROSETTA. В этом методе возбужденные белковые структуры после локальной минимизации энергии перераспределяются между локальными энергетическими минимумами. Для данной структуры А, находящейся в локальном энергетическом минимуме, в ходе случайного возмущения и последующей локальной минимизации энергии создается тестовая структура В. Чтобы определить, насколько структура В
1.3. Методы конформационного поиска
31
приемлема по сравнению со структурой А, используется стандартный алгоритм Метрополиса - рассчитывается различие в энергии двух состояний.
1.3.2. Молекулярная динамика
При расчете МД (которая подробно обсуждается в главе 10) на каждом шаге движения атома осуществляется решение уравнений движения Ньютона. Это, возможно, самый надежный метод, в котором процессы, происходящие в белках, описываются на атомном уровне. Метод, таким образом, чаще других используется для изучения способов укладки белка (Duan and Kollman 1998). Большое время расчета является одной из основных проблем метода, поскольку шаг по времени обычно имеет порядок фемтосекунд (10'15 с), тогда как время самой быстрой в природе упаковки небольшого белка (менее 100 остатков) лежит в миллисекундном диапазоне. К настоящему времени не предпринималось серьезных попыток выполнения полноатомных расчетов МД для предсказания структуры белка, исходя из распрямленной или неупорядоченной структуры5. В случаях, когда доступна модель низкого разрешения, расчеты МД часто выполняются для уточнения структуры, поскольку считается, что конформационные изменения будут незначительными. Заслуживает внимание подход, использованный в недавней работе Шераги с коллегами, которые реализовали расчет МД в пространстве торсионных углов с использованием крупнозернистого энергетического потенциала UNRES (см. обсуждение выше).
1.3.3. Генетические алгоритмы
Отжиг в конфомационном пространстве (conformational space annealing, CSA) (Lee etal. 1998) является одним из самых успешных генетических алгоритмов. Используя алгоритм локальной минимизации энергии, как в методе Монте-Карло с минимизацией, и концепцию отжига в конформационном пространстве, сначала проводится поиск локальных минимумов во всем конформационном пространстве, а затем, по мере уменьшения радиуса отсечки, поиск сужается до низкоэнергетических областей меньшего размера. Радиус отсечки означает здесь степень сходства между двумя конформациями и определяет разнообразие конформационной по
5 Нужно заметить, что такие попытки были предприняты чуть позже. Так, рассмотрение МД виллина и его переход из неупорядоченной структуры в упорядоченную, было выполнено в недавней работе Shaw D.E. etal. (2010). Atomic-level characterization of the Structural Dynamics of Proteins. Science 330, 341-346.
32
Глава 1. Предсказание структуры белков ab initio
пуляции. Радиус отсечки играет роль температуры в стандартном методе ИО, и первоначально ему присваивается большое значение для увеличения конформационного разнообразия. В процессе поиска это значение постепенно уменьшается. Отжиг в конформационном пространстве успешно применялся для решения различных задач глобальной оптимизации, в том числе для предсказания структуры белков, которое комбинировалось с моделированием методами ab initio в UNRES (Oldziej et al. 2005) и ASTROFOLD (Klepeis and Floudas 2003; Klepeis et al. 2005), а также, независимо, co сборкой фрагментов в Profesy (Lee et al. 2004).
1.3.4. Математическая оптимизация
Поисковый алгоритм а-ветвей и границ (a branch and bound, аВВ), предложенный Флудасом с коллегами (Klepeis and Floudas 2003; Klepeis et al. 2005), является уникальным в том смысле, что он математически точен, тогда как все остальные методы, обсуждавшиеся в этом разделе, являются стохастическими и эвристическими. Пространство поиска последовательно делится на две половины, в то же время определяются нижняя и верхняя границы (НГ и ВГ) глобального энергетического минимума для каждой области фазового пространства. Верхняя граница - это просто лучшее из недавно определенных значений локального энергетического минимума, а оценка для нижней границы делается на основе модифицированной энергетической функции, увеличенной на квадратичный член рассекающих переменных с коэффициентом а (отсюда название аВВ). При высоком значении а модифицированная функция энергии имеет лишь один энергетический минимум, значение которого служит нижней границей. Рассечение фазового пространства сопровождается оценкой НГ и ВГ для каждого рассеченного фазового подпространства. Фазовые подпространства, НГ которых выше глобальной ВГ, исключаются из поиска. Процедура продолжается до момента определения глобального минимума посредством определения фазового подпространства, для которого НГ совпадает с ВГ. В случаях, когда удается найти решение, полученный результат является математически точным, однако метод все еще не используется для больших белков с большим числом степеней свободы.
1.4. Отбор моделей
В ходе моделирования методами ab initio обычно создается множество структурных макетов. Важной проблемой моделирования является вы
1.4. Отбор моделей
33
бор соответствующих моделей, которые по структуре близки к природному состоянию белка, в связи с чем возникла новая область исследования, названная «методы оценки «качества» упаковки моделей» (Model Quality Assessment Programs, MQAP) (Fischer 2006). В целом, подходы к отбору в моделировании можно разделить на два типа, а именно, основанные на энергетической функции и основанные на функции свободной энергии. В случае энергетических подходов используются разнообразные специфические потенциалы, а окончательным предсказанием структуры является состояние с наименьшей энергией. В подходах, основанных на свободной энергии, свободная энергия заданной конформации R может быть записана как
F(R) = -кВТ in Z(7?) = -JlBTlnje к‘т dQ. (1)
где Z(R) - ограниченная функция распределения, которая пропорциональна частоте встречаемости структур вблизи R в ходе моделирования. Ее можно оценить с помощью процедуры кластеризации при заданном значении отсечки по СКО (Zhang and Skolnick 2004с).
Из множества методов отбора моделей, основанных на свободной энергии, в настоящем разделе обсуждаются три энергетические/оценоч-ные функции: 1) рациональная энергетическая функция; 2) эмпирическая энергетическая функция; 3) оценочная функция, которая описывает соответствие между последовательностью мишени и структурами моделей. К числу программ оценки качества моделей принадлежит еще один популярный метод, в котором используется согласованная конформация, определенная на основе предсказаний, полученных при применении различных алгоритмов (Wallner and Elofsson 2007). Эта группа методов также известна как метасерверы (Ginalski et al. 2003а; Wu and Zhang 2007). Суть этих методов близка к методу кластеризации, поскольку в обоих случаях принимается допущение, согласно которому наиболее часто встречающееся состояние ближе всего к природному. Подход используется главным образом для отбора моделей, созданных веб-серверами с использованием методики протягивания (Ginalski et al. 2003; Wallner and Elofsson 2007; Wu and Zhang 2007).
1.4.1. Рациональная энергетическая функция
Для разработки полноатомных рациональных энергетических функций Лазаридис и Карплус (1999а) использовали потенциалы сольватации силовых полей CHARMM19 (Neria etal. 1996) и EEF1 (Lazaridis and Karplus 1999b), на основе значений которых осуществлялась дифферен-
34
Глава 1. Предсказание структуры белков ab initio
циация природной структуры белка и макетов, созданных при протягивании через другие белковые структуры. Было установлено, что энергия нативного состояния в большинстве случаев ниже энергии макетов. Позже при разработке рациональных энергетических функций использовалось поле CHARMM и концепция сплошной среды для растворителя (Petrey and Honig 2000), поле CHARMM и метод GB (Dominy and Brooks 2002; Feig and Brooks 2002), OPLS и GB (Felts et al. 2002), поле AMBER и метод GB (Lee and Duan 2004), поле AMBER и потенциал сольватации Пуассона-Больцмана для нескольких наборов структурных макетов (в том числе наборы макетов Парка-Левитта (Park and Levitt 1996), Бейкера (Tsai etal. 2003), Сколника (Kihara etal. 2001; Skolnick etal. 2003) и CASP (Moult etal. 2001)) (Hsieh and Luo 2004). Все эти авторы получили близкие результаты, т.е., значения энергии, получаемые при использовании этих потенциалов, для природных белковых структур ниже, чем для макетов. Может показаться, что успешное применение модели дифференциации на основе физических потенциалов противоречит данным, полученным в результате применения других, менее успешных физических методов. Недавно было показано, что сочетание потенциалов поля AMBER и метода GB позволяет отличать природную структуру лишь от грубо минимизированных макетов, полученных в TASSER (Zhang and Skolnick 2004a; Wroblewska and Skolnick 2007). После расчета МД макетов в течение 2 нс ни одна из нативных структур не обладала значением энергии, которое было бы ниже, чем минимальное значение энергии макета, а корреляция между значением энергии и СКО была близка к нулю. Эти результаты частично объясняют несоответствие между широко освещаемыми успехами метода дифференциации макетов с использованием физических потенциалов, с одной стороны, и менее успешными результатами методов упаков-ки/уточнения структуры белков, с другой стороны.
1.4.2. Эмпирическая энергетическая функция
В 1990 году Сиппл, используя статистические данные о белках известной структуры, размещенные в базе данных PDB, разработал потенциал парных взаимодействий остатков (Sippl 1990); последняя версия метода - PROSA II (Sippl 1993; Wiederstein and Sippl 2007). С тех пор появилось множество различных эмпирических потенциалов, в том числе потенциал атомных взаимодействий, потенциал сольватации, потенциал водородных связей, потенциал торсионных углов и др. В крупнозернистых потенциалах каждый остаток представлен либо одиночным атомом, либо несколькими атомами. Так, существуют потенциалы на основе атомов Са
1.4. Отбор моделей
35
(Melo etal. 2002), атомов Ср (Hendlich etal. 1990), центров масс боковых цепей (Bryant and Lawrence 1993; Kocher et al. 1994; Thomas and Dill 1996; Skolnick et al. 1997; Zhang and Kim 2000; Zhang et al. 2004), центров масс боковых цепей и атомов CQ (Berrera et al. 2003). Один из наиболее широко используемых потенциалов - RAPDF - является полноатомным, учитывает характерные особенности остатков и зависит от расстояния (Samudrala and Moult 1998). При расчете потенциала учитывается расстояние между 167 специфическими псевдоатомами аминокислот. Позже появились другие атомные потенциалы с различными исходными состояниями, в том числе KBP (Lu and Skolnick 2001), DFIRE (Zhou and Zhou 2002), self-RAPDF (Wang et al. 2004), victor/FRST (Tosatto 2005), DOPE (Shen and Sali 2006). По утверждениям создателей использование этих потенциалов позволяет выявлять нативную структуру белка среди структурных макетов. Однако проблема отбора моделей, близких к нативной структуре, среди множества макетов в этих методах по-прежнему не решена (Skolnick 2006). Эта проблема, по сути, является более важной, чем распознавание нативной структуры, поскольку фактически на сегодняшний день не существует белковых молекул, структура которых была бы определена методами компьютерного моделирования. Согласно результатам экспериментов CAFASP4-MQAP 2004 года (Fischer 2006), наиболее высокой производительностью отличались энергетические функции Victor/FRST (Tosatto 2005) и MODCHECK (Pettitt et al. 2005). Функция Victor/FRST содержит полноатомный потенциал парных взаимодейстий, потенциал сольватации и потенциал водородных связей. В состав MODCHECK входит потенциал взаимодействий атомов Ср. На соревнованиях CASP7-MQAP в 2006 году лучшую производительность показал метод Peons, разработанный группой Элофссона (Wallner and Elofsson 2007).
1.4.3. Функция совместимости структуры и последовательности
Третью группу программ для оценки качества моделей составляют методы, в которых лучшие модели отбираются не только на основе энергетических функций, а используется критерий «совместимости» последовательностей мишеней и модельных структур. Самым ранним и по-прежнему успешным является метод Люти с соавт. (Luthy et al. 1992), которые для определения качества структур использовали метод протягивания и соответствующую оценочную функцию. Позже в другом методе (Colovos and Yeates 1993) использовалась квадратичная функция ошибок для описания невалентных взаимодействий между атомами СС, CN, СО, NN, NO
36
Глава 1. Предсказание структуры белков ah initio
и 00, и в макетах, близких к нативным, было обнаружено меньшее количество ошибок, чем в остальных. Метод Verify3D (Eisenberg etal. 1997) является улучшением метода Люти с соавт. за счет усреднения оценок индивидуальных остатков по окну шириной в 21 остаток6. В методе GenThreader (Jones 1999) для разделения природных и неприродных структур используются нейронные сети. Входные данные GenThreader включают энергию парных взаимодействий, энергию сольватации, оценку выравнивания, размер выравнивания, а также размеры последовательности и структуры. Аналогичным образом, нейронные сети составляют основу метода ProQ (Wallner and Elofsson 2003), который используется для предсказания качества структуры макетов. Входными данными для ProQ являются контакты, площадь, доступная растворителю, форма белка, вторичная структура, оценка структурных выравниваний между макетами и шаблоном, доля областей белка, смоделированных на основе шаблонов. Недавно была разработана консенсусная программа ModFold (McGuffin 2007), в состав которой вошли ProQ (Wallner and Elofsson 2003), MODCHECK (Pettitt etal. 2005) и ModSSEA. Авторами было показано, что производительность метода выше, чем производительность входящих в его состав отдельных программ.
1.4.4. Кластеризация макетов структур
С целью определения состояния с наименьшим значением свободной энергии во многих методах моделирования ab initio используются адаптированные методики кластеризации структур. В работе (Shortle et al. 1998) конформация в центре самого большого кластера для всех 12 рассмотренных случаев была ближе к нативной структуре, чем большая часть шаблонов. Структуры центра кластера принадлежали к 1-5% структур, наиболее близких к нативным.
Чжан и Сколник разработали метод итеративной кластеризации структур SPICKER (Zhang and Skolnick 2004с). Использовались 1489 репрезентативных тестовых белков, для каждого из которых было построено до 280 000 структурных макетов. Лучшие модели среди пяти наиболее высоко оцененных моделей каждого белка оказались в числе лучших 1,4% среди всех использованных макетов. Для 78% из этих 1489 белков СКО между лучшими из пяти наиболее высоко оцененных моделей и структурой макета, наиболее близкой к природной, составило менее 1 А.
6 Так в оригинале. На самом деле Verify3D является лишь программным воплощением метода Люти с соавт. Прим, перев.
1.5. Замечания и обсуждение
37
В методе моделирования ab initio ROSETTA (Bradley et al. 2005) кластеризация структурных макетов используется для отбора моделей низкого разрешения. Затем структура моделей уточняется в ходе полноатомного моделирования, результатом которого являются окончательные модели. В случае методов TASSER/I-TASSER (Zhang and Skolnick 2004а; Wu etal. 2007) используются тысячи макетов моделей, полученные в результате расчета МК; их кластеризация осуществляется с помощью метода SPICKER (Zhang and Skolnick 2004с). Окончательными моделями выступают центры кластеров. В методе, разработанном Шерагой с коллегами, осуществляется кластеризация структур, а затем - отбор структур с наименьшими значениями энергии (Oldziej et al. 2005).
1.5. Замечания и обсуждение
Моделирование структуры белка методами ab initio на основе одной лишь его аминокислотной последовательности принято считать «Святым Граалем» предсказания структуры белков (Zhang 2008), поскольку успех в разработке таких методов означал бы полное и окончательное решение проблемы. Помимо создания пространственных структур, моделирование ab initio может помочь нам понять принципы, лежащие в основе сворачивания белков в природе. Решить эту задачу можно не только на основе методов сравнительного моделирования, в которых пространственная структура строится в процессе копирования каркаса других, уже известных структур.
Идеальной схемой моделирования методами ab initio представляется эксперимент, в котором в качестве взаимодействующих частиц рассматриваются атомы белка, взаимодействие определяется точным физическим потенциалом, а исследование процесса сворачивания белка осуществляется в ходе решения уравнений движения Ньютона на каждом шаге. Был выполнен ряд таких молекулярно-динамических расчетов с применением классических силовых полей CHARMM и AMBER. Хотя МД моделирование является крайне важным методом изучения процесса упаковки белков, однако успехи, достигнутые при применении этого метода для предсказания структуры, весьма ограничены. Одной из причин является чрезвычайно большие вычислительные ресурсы, необходимые для моделирования белков средних размеров. С другой стороны, в области развития эмпирических (или гибридных, рациональных и эмпирических) методов наблюдается быстрый прогресс; появляется множество примеров успешных моделей белков размером до 100 остатков, как низкого, так и среднего каче
38
Глава 1. Предсказание структуры белков ab initio
ства, часто с правильной топологией. Также, хотя и крайне редко, появляются сообщения об успешном получении моделей высокого разрешения (менее 2 А для атомов Са) (Bradley et al. 2005).
В современных методах ab initio при предсказании структуры белков обычно используется максимальное количество информации об известных структурах. На то есть несколько причин. Во-первых, использование локальных фрагментов структуры, полученных непосредственно из структур, размещенных в базе данных PDB, способствует уменьшению степеней свободы и энтропии конформационного поиска, а также сохранению точности нативных структур белков. Во-вторых, применение эмпирического потенциала, разработанного с использованием большого количества статистических данных об известных структурах, способствует поддержанию хрупкого баланса сложных взаимосвязей между различными источниками энергетических термов (Summa and Levitt 2007). Термы эмпирического потенциала тщательно параметризованы. Благодаря успехам в области методов конформационного поиска часть вычислений и процедур носит полуавтоматический характер. В силу этих причин точность методов ab initio при применении к белкам размером 100-120 остатков заметно увеличилась в последнее десятилетие.
Для дальнейших улучшений необходимы как параллельные разработки точных функций потенциальной энергии, так и методы эффективной оптимизации. Это значит, что большое значение имеют независимое иссле-дование/развитие функций потенциальной энергии; в то же время, необходима регулярная оценка эффективности различных методов конфомацион-ного поиска, которая позволит независимо исследовать как преимущества, так и ограничения доступных поисковых методов.
Важно отметить, что методы предсказания ab initio, основанные исключительно на физико-химических принципах взаимодействия, в настоящее время заметно отстают от методов биоинформатики и методов моделирования на основе информации о структуре с точки зрения точности и скорости моделирования. Тем не менее, доказано, что физические атомные потенциалы полезны при уточнении деталей упаковки атомов боковых цепей и основной пептидной цепи. Таким образом, развитие сложных методов, сочетающих как эмпирические, так и физические энергетические потенциалы, возможно, представляет собой перспективный подход к решению проблемы моделирования ab initio.
Благодарности. Проект частично поддержан Фондом KU Start-up Fund 06194, Фондом Альфреда П.Слоана и Грантом №R01GM083107 Национального института общих медицинских наук.
Литература
39
Литература
Bairoch A, Apweiler R, Wu CH, et al. (2005) The Universal Protein Resource (UniProt). Nucleic Acids Res 33(Database issue):D154-159
Berendsen HJC, Postma JPM, van Gunsteren WF, et al. (1981) Interaction models for water in relation to protein hydration. Intermolecular forces. Reidel, Dordrecht, The Netherlands
Berg BA, Neuhaus T (1992) Multicanonical ensemble: a new approach to simulate first-order phase transitions. Phys Rev Lett 68(1 ):9—12
Berman HM, Westbrook J, Feng Z, etal. (2000) The protein data bank. Nucleic Acids Res 28(l):235-242
Berrera M, Molinari H, Fogolari F (2003) Amino acid empirical contact energy definitions for fold recognition in the space of contact maps. BMC Bioinformatics 4:8
Bowie JU, Eisenberg D (1994) An evolutionary approach to folding small alpha-helical proteins that uses sequence information and an empirical guiding fitness function. Proc Natl Acad Sci USA 91(10):4436-4440
Bradley P, Misura KM, Baker D (2005) Toward high-resolution de novo structure prediction for small proteins. Science 309(5742): 1868-1871
Brooks BR, Bruccoleri RE, Olafson BD, et al. (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4(2): 187-217
Bryant SH, Lawrence CE (1993) An empirical energy function for threading protein sequence through the folding motif. Proteins 16(1 ):92—112
Case DA, Pearlman DA, Caldwell JA, et al. (1997) AMBER 5.0, University of California, San Francisco, CA.
Chen J, Brooks CL (2007) Can molecular dynamics simulations provide high-resolution refinement of protein structure? Proteins 67(4):922-930
Cheng J, Baldi P (2006) A machine learning information retrieval approach to protein fold recognition. Bioinformatics 22(12): 1456-1463
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2(9): 1511-1519
Cornell WD, Cieplak P, Bayly Cl, et al. (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117:5179-5197
Das R, Qian B, Raman S, etal. (2007) Structure prediction for CASP7 targets using extensive allatom refinement with Rosetta@home. Proteins 69(S8):118-128
Dominy BN, Brooks CL (2002) Identifying native-like protein structures using physics-based potentials. J Comput Chem 23(1): 147-160
Duan Y, Kollman PA (1998) Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science 282(5389):740-744
Eisenberg D, Luthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with threedimensional profiles. Method Enzymol 277:396-404
Fan H, Mark AE (2004) Refinement of homology-based protein structures by molecular dynamics simulation techniques. Protein Sci 13(1 ):211-220
Feig M, Brooks CL (2002) Evaluating CASP4 predictions with physical energy functions. Proteins 49(2):232-245
Felts AK, Gallicchio E, Wallqvist A, et al. (2002) Distinguishing native conformations of proteins from decoys with an effective free energy estimator based on the OPLS all-atom force field and the Surface Generalized Bom solvent model. Proteins 48(2)404—422
Fischer D (2006) Servers for protein structure prediction. Curr Opin Struct Biol 16(2): 178-182
Fujitsuka Y, Chikenji G, Takada S (2006) SimFold energy function for de novo protein structure prediction: consensus with Rosetta. Proteins 62(2):381—398
Ginalski K, Elofsson A, Fischer D, et al. (2003a) 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics 19(8): 1015-1018
Ginalski K, Pas J, Wyrwicz LS, et al. (2003b) ORFeus: detection of distant homology using sequence profiles and predicted secondary structure. Nucleic Acids Res 31 (13):3804—3807
Hagler A, Euler E, Lifson S (1974) Energy functions for peptides and proteins L Derivation of a consistent force field including the hydrogen bond from amide crystals. J Am Chem Soc 96:5319-5327
40
Глава 1. Предсказание структуры белков ab initio
Helles G (2008) A comparative study of the reported performance of ab initio protein structure prediction algorithms. J R Soc Interface 5(21)387-396
Hendlich M, Lackner P, Weitckus S, etal. (1990) Identification of native protein folds amongst a large number of incorrect models. The calculation of low energy conformations from potentials of mean force. J Mol Biol 216(1): 167-180
Hsieh MJ, Luo R (2004) Physical scoring function based on AMBER force field and Poisson-Boltzmann implicit solvent for protein structure prediction. Proteins 56(3):475-486
Im W, Lee MS, Brooks CL (2003) Generalized bom model with a simple smoothing function. J Comput Chem 24( 14): 1691-1702
Jaroszewski L, Rychlewski L, Li Z, etal. (2005) FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res 33(Web Server issue): W284-288
Jauch R, Yeo HC, Kolatkar PR, et al. (2007) Assessment of CASP7 structure predictions for template free targets. Proteins 69(Suppl 8):57-67
Jonassen I, Klose D, Taylor WR (2006) Protein model refinement using structural fragment tessellation. Comput Biol Chem 30(5):360-366
Jones DT (1999) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol 287(4):797-815
Jorgensen WL, Tirado-Rives J (1988) The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc (110): 1657-1666
Jorgensen WL, Chandrasekhar J, Madura JD, etal. (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926-935
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS AllAtom Force Field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225-11236
Kaminski GA, Friesner RA, Tirado-Rives J, et al. (2001) Evaluation and Reparametrization of the OPLS-AA Force Field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem В 105:6474-6487
Karplus K, Barrett C, Hughey R (1998) Hidden Markov models for detecting remote protein homologies. Bioinformatics 14:846-856
Kihara D, Lu H, Kolinski A, et al. (2001) TOUCHSTONE: an ab initio protein structure prediction method that uses threading-based tertiary restraints. Proc Natl Acad Sci USA 98(18): 10125— 10130
Kirkpatrick S, Gelatt CD, Vecchi MP (1983) Optimization by simulated annealing. Science 220(4598):671-680
Klepeis JL, Floudas CA (2003) ASTRO-FOLD: a combinatorial and global optimization framework for Ab initio prediction of three-dimensional structures of proteins from the amino acid sequence. Biophys J 85(4):2119-2146
Klepeis JL, Wei Y, Hecht MH, et al. (2005) Ab initio prediction of the three-dimensional structure of a de novo designed protein: a double-blind case study. Proteins 58(3):560-570
Kocher JP, Rooman MJ, Wodak SJ (1994) Factors influencing the ability of knowledge-based potentials to identify native sequence-structure matches. J Mol Biol 235(5): 1598-1613
Lazaridis T, Karplus M (1999a) Discrimination of the native from misfolded protein models with an energy function including implicit solvation. J Mol Biol 288(3):477—487
Lazaridis T, Karplus M (1999b) Effective energy function for proteins in solution. Proteins 35(2): 133-152
Lee J (1993) New Monte Carlo algorithm: entropic sampling. Phys Rev Lett 71 (2):211-214
Lee J, Scheraga HA, Rackovsky S (1998) Conformational analysis of the 20-residue membranebound portion of melittin by conformational space annealing. Biopolymers 46(2): 103-116
Lee J, Kim SY, Joo K, et al. (2004) Prediction of protein tertiary structure using PROFES Y, a novel method based on fragment assembly and conformational space annealing. Proteins 56(4):704-714
Lee MC, Duan Y (2004) Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized bom solvent model. Proteins 55(3):620-634
Литература
41
Lee MR, Tsai J, Baker D, et al. (2001) Molecular dynamics in the endgame of protein structure prediction. J Mol Biol 313(2):417-430
Levitt M, Hirshberg M, Sharon R, et al. (1995) Potential-energy function and parameters for simulations of the molecular-dynamics of proteins and nucleic-acids in solution. Comput Phys Commun 91 (l-3):215-231
Li Z, Scheraga HA (1987) Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc Natl Acad Sci USA 84(19):6611-6615
Lindahl E, Hess B, van der Spoel D (2001) GROM ACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 7:306-317
Li wo A, Pincus MR, Wawak RJ, et al. (1993) Calculation of protein backbone geometry from alpha-carbon coordinates based on peptide-group dipole alignment. Protein Sci 2( 10): 1697— 1714
Li wo A, Lee J, Ripoil DR, et al. (1999) Protein structure prediction by global optimization of a potential energy function. Proc Natl Acad Sci USA 96(10):5482-5485
Liwo A, Khalili M, Scheraga HA (2005) Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc Natl Acad Sci USA 102(7):2362-2367
Lu H, Skolnick J (2001) A distance-dependent atomic knowledge-based potential for improved protein structure selection. Proteins 44(3):223-232
Luthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356(6364):83-85
MacKerell Jr. AD, Bashford D, Bellott M, et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem В 102 (18):3586—3616
McGuffin LJ (2007) Benchmarking consensus model quality assessment for protein fold recognition. BMC Bioinformatics 8:345
Melo F, Sanchez R, Sali A (2002) Statistical potentials for fold assessment. Protein Sci 11 (2):430— 448
Moult J, Fidelis K, Zemla A, et al. (2001) Critical assessment of methods of protein structure prediction (CASP): round IV. Proteins(Suppl 5):2-7
Nemethy G, Gibson KD, Palmer KA, et al. (1992) Energy parameters in polypeptides. 10. Improved geometric parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J Phys Chem В 96: 6472-6484
Neria E, Fischer S, Karplus M (1996) Simulation of activation free energies in molecular systems. J Chem Phys 105(5): 1902 -1921
Nilges M, Brunger AT (1991) Automated modeling of coiled coils: application to the GCN4 dimerization region. Protein Eng 4(6):649-659
Oldziej S, Czaplewski C, Liwo A, et al. (2005) Physics-based protein-structure prediction using a hierarchical protocol based on the UNRES force field: assessment in two blind tests. Proc Natl Acad Sci USA 102(21 ):7547-7552
Park B, Levitt M (1996) Energy functions that discriminate X-ray and near native folds from well constructed decoys. J Mol Biol 258(2):367-392
Petrey D, Honig В (2000) Free energy determinants of tertiary structure and the evaluation of protein models. Protein Sci 9(11 ):2181-2191
Pettitt CS, McGuffin LJ, Jones DT (2005) Improving sequence-based fold recognition by using 3D model quality assessment. Bioinformatics 21(17)3509-3515
Pieper U, Eswar N, Davis FP, et al. (2006) MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 34(Database issue):D291-295
Samudrala R, Moult J (1998) An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. J Mol Biol 275(5):895-916
Shen MY, Sali A (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci 15(11):2507-2524
Shi J, Blundell TL, Mizuguchi К (2001) FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol 310(1 ):243—257
42
Глава 1. Предсказание структуры белков ab initio
Shortle D, Simons KT, Baker D (1998) Clustering of low-energy conformations near the native structures of small proteins. Proc Natl Acad Sci USA 95( 19): 11158-11162
Simons KT, Kooperberg C, Huang E, etal. (1997) Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol 268(1 ):209-225
Sippl MJ (1990) Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol 213(4):859-883
Sippl MJ (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17(4):355-362
Skolnick J (2006) In quest of an empirical potential for protein structure prediction. Curr Opin Struct Biol 16(2): 166-171
Skolnick J, Jaroszewski L, Kolinski A, et al. (1997) Derivation and testing of pair potentials for protein folding. When is the quasichemical approximation correct? Protein Science 6:676-688
Skolnick J, Zhang Y, Arakaki AK, et al. (2003) TOUCHSTONE: a unified approach to protein structure prediction. Proteins 53(Suppl 6):469-479
Skolnick J, Kihara D, Zhang Y (2004) Development and large scale benchmark testing of the PROSPECTOR 3.0 threading algorithm. Protein 56:502-518
Soding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21(7):951-960
Sorin EJ, Pande VS (2005) Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys J 88(4):2472-2493
Summa CM, Levitt M (2007) Near-native structure refinement using in vacuo energy minimization. Proc Natl Acad Sci USA 104(9):3177-3182
Taylor WR, Bartlett GJ, Chelliah V, et al. (2008) Prediction of protein structure from ideal forms. Proteins 70(4): 1610-1619
Thomas PD, Dill KA (1996) Statistical potentials extracted from protein structures: how accurate are they? J Mol Biol 257(2):457-469
Tosatto SC (2005) The victor/FRST function for model quality estimation. J Comput Biol 12(10): 1316—1327
Tsai J, Bonneau R, Morozov AV, et al. (2003) An improved protein decoy set for testing energy functions for protein structure prediction. Proteins 53(l):76-87
van Gunsteren WF, Billeter SR, Eising AA, et al. (1996) Biomolecular simulation: the GROMOS96 manual and user guide. VDF Hochschulverlag AG an der ETH, Zurich. Vieth M, Kolinski A, Brooks CL, etal. (1994) Prediction of the folding pathways and structure of the GCN4 leucine zipper. J Mol Biol 237(4):361-367
Wallner B, Elofsson A (2003) Can correct protein models be identified? Protein Sci 12(5): 1073— 1086
Wallner B, Elofsson A (2007) Prediction of global and local model quality in CASP7 using Peons and ProQ. Proteins 69(S8): 184-193
Wang JM, Cieplak P, Kollman PA (2000) How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? JComput Chem 21 (12): 1049-1074
Wang K, Fain B, Levit M, et al. (2004) Improved protein structure selection using decoy-dependent discriminatory functions. BMC Struct Biol 4(8)
Weiner SJ, Kollman PA, Case DA, et al. (1984) A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc 106: 765-784
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(Web Server issue): W407-410
Wroblewska L, Skolnick J (2007) Can a physics-based, all-atom potential find a protein’s native structure among misfolded structures? I. Large scale AMBER benchmarking. J Comput Chem 28(12):2059-2066
Wu S, Zhang Y (2007) LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res 35(10):3375-3382
Литература
43
Wu S, Zhang Y (2008) MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins 72(2):547-556
Wu S, Skolnick J, Zhang Y (2007) Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol 5:17
Zagrovic B, Snow CD, Shirts MR, et al. (2002) Simulation of folding of a small alpha-helical protein in atomistic detail using worldwide-distributed computing. J Mol Biol 323(5):927-937
Zhang C, Kim SH (2000) Environment-dependent residue contact energies for proteins. Proc Natl Acad Sci USA 97(6):2550-2555
Zhang C, Liu S, Zhou H, et al. (2004) An accurate, residue-level, pair potential of mean force for folding and binding based on the distance-scaled, ideal-gas reference state. Protein Sci 13(2):400-411
Zhang Y (2007) Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 69(Suppl 8):108-l 17
Zhang Y (2008) Progress and challenges in protein structure prediction. Curr Opin Struct Biol 18(3):342-348
Zhang Y, Skolnick J (2004a) Automated structure prediction of weakly homologous proteins on a genomic scale. Proc Natl Acad Sci U S A 101:7594-7599
Zhang Y, Skolnick J (2004b) Scoring function for automated assessment of protein structure template quality. Proteins 57:702-710
Zhang Y, Skolnick J (2004c) SPICKER: a clustering approach to identify near-native protein folds. J Comput Chem 25(6):865-871
Zhang Y, Skolnick J (2005a) The protein structure prediction problem could be solved using the current PDB library. Proc Natl Acad Sci USA 102:1029-1034
Zhang Y, Skolnick J (2005b) TM-align: a protein structure alignment algorithm based on the TMscore. Nucleic Acids Res 33(7):2302-2309
Zhang Y, Kihara D, Skolnick J (2002) Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins 48(2): 192-201
Zhang Y, Kolinski A, Skolnick J (2003) TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys J 85(2): 1145-1164
Zhang Y, Hubner I, Arakaki A, et al. (2006) On the origin and completeness of highly likely single domain protein structures. Proc Natl Acad Sci USA 103:2605-2610
Zhou H, Skolnick J (2007) Ab initio protein structure prediction using chunk-TASSER. Biophys J 93(5): 1510—1518
Zhou H, Zhou Y (2002) Distance-scaled, finite ideal-gas reference state improves structure derived potentials of mean force for structure selection and stability prediction. Protein Sci 11(11):2714—2726
Zhou H, Zhou Y (2005) Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins 58(2):321-328
Глава 2
Распознавание фолда
Лоуренс А. Келли
Распознавание типов укладки (фолдов) связано с предсказанием пространственной структуры белка по аминокислотной последовательности, в основу которого положено определение весьма отдаленных гомологов или аналогов с известной структурой. Таким образом, описанный подход занимает промежуточное положение между изучением сворачивания белка методами ab initio и моделированием на основе близкой гомологии. В настоящей главе представлен исторических обзор этой области исследований, а также приводится информация о последних успехах - от протягивания и сравнения профилей последовательностей до современных мета-серверных консенсусных подходов и анализа сетей гомологов.
2.1. Введение
Аминокислотная последовательность белка определяет его структуру, которая, в свою очередь, определяет его биологические функции и механизм действия. Сворачивание белка представляет собой связующее звено между набором инструкций для живой материи и самой живой материей. С этой ключевой парадигмой биохимии связана почти каждая четвертая Нобелевская премия в области химии начиная с 1956 года (Seringhaus and Gerstein 2007). В 2005 году журнал «Science» назвал проблему сворачивания белка одной из 125 крупнейших нерешенных проблем науки (Science Editorial 2005).
К моменту написания настоящей главы* 1 в сотнях секвенированных геномов было обнаружено более 5,8 миллиона уникальных белковых по-
Lawrence A. Kelley
Structural Bioinformatics Group, Department of Biological Sciences,
Imperial College London, SW7 2AY, UK
e-mail: l.a.kelley@ic.ac.uk
1 2009 год. Прим, перев.
2.1. Введение
45
следовательностей. Это число экспоненциально растет в течение последних двух десятилетий и, вероятнее всего, будет расти еще быстрее. В ходе новых метагеномных проектов, к числу которых принадлежит проект секвенирования методом дробовика случайных образцов морской воды по всему миру каждые 200 миль, в каждом барреле морской воды обнаруживают 1,3 миллиона генов и 50,000 новых видов организмов. С помощью приборов одиночного секвенирования сегодня можно в течение 24 часов секвенировать 100 миллионов пар оснований, и, вероятнее всего, эта скорость будет расти, а стоимость падать.
Между тем, несмотря на прогресс методов высокопроизводительной структурной геномики и большое количество ЯМР- и кристаллографических роботов, ведущих работу по определению структуры белков 24 часа в сутки, к настоящему времени установлено лишь 50,000 белковых структур2.
2.1.1. Важность «слепых» испытаний: соревнование CASP
За последние 30 лет было разработано огромное разнообразие методов, нацеленных на решение проблемы предсказания структуры белков в целом и распознавание способов укладки в частности. Как и в случае других научных начинаний, в данном случае крайне важно, чтобы любая новая методика была полностью протестирована «экспериментально». Именно поэтому возник конкурс критической оценки методов предсказания структуры белков CASP (Critical Assessment of Structure Prediction) (http:// predictioncenter.Ilnl.gov/; Moult etal. 2007). Целью конкурса, или рабочего совещания, CASP (который проводится каждые два года) является моделирование природного состояния аминокислотной последовательности, структура которой неизвестна. Однако есть важная особенность - организаторам конкурса структура известна. Структуры белков, предлагаемых к рассмотрению в конкурсе, определяется экспериментальными методами незадолго до конкурса, однако эти данные становятся доступны научному сообществу лишь после его завершения. В результате, эксперты конкурса находятся в довольно необычной ситуации: им известна пространственная структура ряда белков, о которой ничего не известно специалистам по предсказанию.
CASP функционирует как система «слепой» экспериментальной оценки различных методов предсказания структуры в реальных условиях,
2 Напомним, что на начало 2013 года число структур в базе данных PDB составляет около 87 тысяч. Прим, перев.
46
Глава 2. Распознавание фолда
поэтому именно идея соревнований CASP использовалась для определения набора методов, которые будут описаны в этой главе. Это вовсе не означает, что методы, не описанные здесь, не дают достоверных результатов, которые по каким-то причинам не были продемонстрированы в CASP. В последние годы были разработаны буквально сотни методов предсказания структуры, и чтобы не перегружать читателя, результаты соревнований CASP используются как фильтр. Обзор последних результатов CASP7 представлен в дополнительных материалах CASP7 (Moult et al. 2007).
2.1.2. Предсказание структуры ab initio и моделирование по гомологии
Если мы надеемся однажды описать структуру сколь-нибудь заметной доли белков в природе, не прибегая к открытию каких-либо революционных экспериментальных методов, то нам понадобится подход, который позволит предсказывать структуру на основе последовательности с использованием вычислительных методов. После того, как в 1961 г. Анфинсен показал, что после денатурации рибонуклеаза может проходить процесс рефолдинга, сохраняя при этом ферментативную активность, стала популярной идея о том, что информация, необходимая белку для обретения окончательной конформации, закодирована в его последовательности. В результате для прогнозирования структуры белков течение последних десятилетий использовалось сочетание «чистых» методов, в которых в качестве входных данных используется лишь аминокислотная последовательность, и законов физики (или их приближений). В этом направлении были достигнуты определенные успехи, которые описаны в главе 1 настоящей книги. Однако в целом эти методы отличаются либо громоздкими вычислениями, что делает их применение на практике затруднительным, либо низкой производительностью и неточными результатами при исследовании любых систем, кроме белков небольшого размера (менее 100 аминокислотных остатков). Физический подход может показаться единственно верным решением проблемы сворачивания, однако предсказание структуры белков имеет большое практическое значение, а значит, необходимо принять имеющиеся ограничения и двигаться, хотя бы и временно, в направлении поиска более прагматичного решения. Такой подход привел к тому, в области методов предсказания белковой структуры акцент сместился от физики к глубинному анализу, или «добыче», данных (data mining).
Давно известно, что похожие белковые последовательности сворачиваются в похожие структуры. Потому, имея новую белковую последовательность, структуру которой предстоит установить, (называемую в даль-
2.1. Введение
47
Рис. 2.1. (Цветную версию рисунка см. на вклейке.) Схематичное представление упрощенного алгоритма построения модели с помощью выравнивания последовательностей исследуемого белка и шаблона. Показано выравнивание последовательности известной структуры («известная последовательность») и исследуемой последовательности. Размытыми линиями показаны вставки и делеции; красными буквами - аминокислотные замены. Остатки окрашены согласно биофизическим свойствам. Тонкие волнистые линии соединяют соответствующие положения в исследуемой и известной последовательности.
нейшем «исследуемая последовательность») достаточно просто проверить, существуют ли похожие последовательности, структура которых уже известна. Если существуют последовательности с высокой степенью подобия, процесс определения структуры легко осуществить, применяя методы выравнивания аминокислотных последовательностей. Используя простой способ оценки подобия типов аминокислот, такой как оценочная матрица BLOSUM, в сочетании с алгоритмом динамического программирования, таким как алгоритм Смита-Уотермана, можно быстро и оптимально (согласно оценочной функции) выровнять две последовательности.
Имея выравнивание последовательности по известной структуре (далее называемой «шаблон»), можно затем построить грубую модель простым копированием соответствующих пространственных координат шаблона и переименованием аминокислотных остатков в соответствии с эквивалентными остатками из выравнивания (рис. 2.1).
Далее модель можно улучшить, используя множество методов моделирования по гомологии, описанных в соответствующей главе настоящей книги. Преимущества этого подхода очевидны: он является быстрым в вычислительном отношении, а точность итоговой модели очень высока при условии высокой степени сходства исследуемой последовательности и последовательности шаблона. Это обстоятельство немедленно указывает на ограничение метода. Если нет похожей последовательности с уже известной структурой, то никакого результата не удастся получить вовсе.
Таким образом, поиск решения для проблемы предсказания белковых структур осуществляется по двум направлениям. Первое направление, основанное на общих физических принципах, нацелено на создание понятного и универсального метода, который позволял бы предсказывать струк
48
Глава 2. Распознавание фолда
туру по последовательности, а также предоставлял бы возможности для дизайна белков, изучения динамики и решения множества других важных задач. Однако создание такого метода сопряжено с экспериментальными сложностями и, вероятно, в ближайшие годы останется нереализованным с вычислительной точки зрения. Другое направление представляет собой простой и понятный, но существенно ограниченный эвристический метод моделирования по гомологии, с помощью которого можно получать модели высокого качества, но лишь в очень ограниченном числе случаев. Именно для решения этой проблемы существует метод, известный как «распознавание фолда», который был создан, чтобы соединить два противоположных направления исследований.
2.1.3. Пределы пространства типов укладки
Напомним некоторые ключевые сведения о природе белков. В базе данных белковых структур размещено около 50000 экспериментально определенных белковых структур (Berman et al. 2000). В базе данных SCOP (Structural Classification of Proteins) (Murzin et al. 1995) эти структуры объединены лишь в 1100 групп с уникальным типом укладки (уникальной топологией) и около 1800 надсемейств (эволюционно связанных белковых семейств). Количество белков, структура которых известна из экспериментов, увеличивается с каждым днем, тогда как количество новых типов укладки растет крайне медленно. Более того, скорость обнаружения новых фолдов, похоже, начинает снижаться (рис. 2.2). Эти результаты привели к широкому признанию мнения, согласно которому число способов укладки, встречающихся в природе, конечно и невелико (Marsden etal. 2006). В структурных базах данных есть сотни, если не тысячи примеров, которые свидетельствуют, что структуры с высокой степенью сходства могут иметь совершенно разные последовательности. Таким образом, хотя справедливо, что последовательности с высокой степенью сходства имеют весьма похожие структуры, справедливо также и то, что похожими структурами могут обладать существенно отличающиеся друг от друга последовательности.
Таким образом, получается, что для любой последовательности, выбранной из базы данных секвенированных геномов, высока вероятность существования структуры, которая уже встречалась исследователям ранее. Главный вопрос состоит в том, как из 50000 структур выбрать правильный шаблон и каким образом осуществить выравнивание интересующей последовательности по этой структуре. Распознавание фолда связано с поиском оценочных функций, которые могут надежно определить
2.1. Введение
49
Рис. 2.2. На диаграмме показано количество экспериментально определенных белковых структур, а также количество различных способов укладки (по определению SCOP), включенных в базу данных SCOP (Murzin et al. 1995), в разные годы. Как видно из графика, количество структур, добавленных в SCOP, быстро увеличивается, тогда как количество новых способов укладки остается в почти неизменным, начиная с 2004 года
совместимость последовательности с известной структурой и осуществить точное выравнивание в тех случаях, когда простое сходство последовательностей установить не удается.
Несмотря на размер пространства последовательностей, т.е., количество всех возможных белковых последовательностей, пространство белковых структур, вероятно, существенно меньше. Вопрос о том, связано ли это с термодинамикой, кинетикой сворачивания или с эволюционным отбором, является сложным и лежит за пределами тематики настоящей главы. Тем не менее, этот весьма неожиданный факт оказался чрезвычайно полезным при разработке методов предсказания структуры белков.
2.1.4. К вопросу о терминологии: «протягивание» и «распознавание фолда»
Существует некоторая путаница в отношении терминов «протягивание» и «распознавание фолда». Некоторые считают их взаимозаменяемыми, тогда как другие полагают, что «протягивание» - метод, в котором, помимо информации о последовательностях, используется структурная
50
Глава 2. Распознавание фолда
информация. В настоящей главе будет использоваться третий подход! Термин «распознавание фолда» будет использоваться в качестве общего термина для обозначения любого метода, с помощью которого можно установить отдаленные или тонкие связи между последовательностью и известной структурой. Термин «протягивание» (описанный в следующем разделе) используется просто для обозначения методов, в которых предпринимается попытка явно моделировать парные взаимодействия между аминокислотами в пространственной структуре. Сюда не относятся гораздо более простые алгоритмы, в которых используется комбинация одномерных строк, содержащих данные о предсказанных структурных свойствах, и информации о последовательности (эти методы описаны в разделе 3.2).
2.2. «Протягивание»
Какую информацию можно извлечь из того факта, что многие последовательности, значительно отличающиеся друг от друга, обладают похожей пространственной структурой? Существует большое количество данных, свидетельствующих, что встречающееся в природе (нативное) состояние белка находится в широкой и глубокой энергетической «яме». Сворачивание белка в характерную для него (зачастую, но не всегда, уникальную) структуру является результатом энергетически выгодных взаимодействий между аминокислотами внутри структуры, а также между аминокислотами и окружающим растворителем.
Если бы нам удалось понять, какие пространственные взаимодействия и взаимодействия с растворителем стабилизируют данную структуру, это дало бы возможность предсказывать последовательности, совместимые с этой структурой, а также создавать последовательности, соответствующие этой структуре. В этом и состоит концепция протягивания. Имея последовательность, структуру которой необходимо предсказать, исследователь выравнивает, или «драпирует», исследуемую последовательность относительно всех известных структур баз данных. Для каждого случая рассчитывается оценка, которая дает представление о том, насколько данная структура выгодна для рассматриваемой последовательности. Структура с наиболее выгодной оценкой и становится предсказанием. Но каковы эти выгодные взаимодействия, и как вычислить их величину? К счастью, благодаря упорной работе множества экспериментаторов по всему миру, существует база данных нативных белковых структур - база данных энергетически выгодных взаимодействий.
2.2. «Протягивание»
51
Путем тщательного статистического анализа распределения различных типов аминокислот в известных белковых структурах можно устанавливать четкие связи «последовательность-структура» и использовать их для решения проблем предсказания. Эти эмпирические, или «основанные на знаниях», силовые поля широко используются во всех методах предсказания структуры белка, а их ключевая роль в моделировании ab initio означает, что множество деталей, касающихся данной проблемы, можно найти в соответствующей главе. Тем не менее, краткий обзор будет не лишним.
2.2.1. Эмпирические потенциалы
Чтобы эмпирически установить правила, связывающие белковую последовательность с пространственной структурой, необходимо: 1) располагать большим количеством примеров последовательностей и соответствующих им структур; 2) выбрать ряд структурных свойств белка для анализа. Простой иллюстрацией метода является разработка потенциала сольватации. Любой глобулярный белок в нативном свернутом состоянии имеет ряд остатков, погруженных в (сильно гидрофобную) внутреннюю область и ряд (сильно гидрофильных) остатков на поверхности, обращенных к окружающему растворителю. Такие остатки называют заглубленными и экспонированными, соответственно. Несложно рассчитать, в какой мере данный остаток R белка с известной структурой экспонирован или заглублен. Один из методов, хотя и грубый, состоит в простой оценке количества остатков, находящихся в пределах определенного расстояния от R (как правило, используются более сложные методы, Richmond 1984; Kabsch 1983). Таким образом, можно составить список всех остатков во всех известных белковых структурах с соответствующей информацией о степени доступности для растворителя (относительно соседей). Имея в распоряжении такие данные, можно использовать разнообразные статистические методы для установления имеющихся связей между типом аминокислоты и ее предрасположенностью к нахождению на поверхности или внутри белка. К числу распространенных относятся методы, основанные на статистической механике или баесовской статистике (для сравнения с другими методами см. (Xia and Levitt 2000)). Впервые предложенные Танака и Шерага (1976), а позже усовершенствованные Сипплом (1990), Мязава и Джерниган (1996), эти методы основаны на статистике Больцмана.
Сначала принимается допущение, что белковые структуры в базе данных представляют собой своего рода ансамбль и что уровни экспони-рованности остатков каждого типа в белках распределены согласно распределению Больцмана. Затем рассчитывается потенциал средней силы,
52
Глава 2. Распознавание фолда
который обусловливает наблюдаемое статистическое распределение по уравнению Больцмана. «Энергия», связанная с данным свойством р, определяется уравнением:
£(p) = -log
ПрьЛР)
П'цХр)
где nobs(p) - наблюдаемое значение р, а пехр(р) - «ожидаемое» значение р в эталонной структуре, для которого не предполагается наличие специфических взаимодействий или предпочтений.
Применение этого подхода обычно предполагает дискретизацию расстояний и создание справочной таблицы со значениями силового поля, при этом непрерывно дифференцируемые функции молекулярной механики не используются (однако бывают исключения). При протягивании эта справочная таблица позволяет определить значение «энергии» для данной комбинации структуры и последовательности. Каждый аминокислотный остаток в модели будет иметь некоторую степень экспонированно-сти/заглубленности. В зависимости от типа остатка в рассматриваемой последовательности, можно вносить в таблицу значения вероятности обнаружения, скажем, валина, экспонированного на 30%. Энергию всей модели можно определить простым суммированием значений энергии по всем остаткам в модели. (Обратите внимание, что суммирование можно использовать благодаря логарифмическому члену в уравнении.)
Более сложная, но и более продуктивная энергетическая функция используется при рассмотрении взаимодействующих пар аминокислот. В этом случае можно рассчитать частоту, с которой аминокислоты определенного типа встречаются вблизи других аминокислот, например, как часто остаток лейцина можно наблюдать на расстоянии 4 А от остатка валина. Как и ранее, получаемые статистические данные аккумулируются для всех возможных пар остатков 20 типов. Затем рассчитываются ожидаемые частоты, которые отражают, как часто аминокислота данного типа наблюдается в пределах заданного расстояния.
В действительности типичные потенциалы парных взаимодействий, широко использующиеся на практике, разработаны на значительно более высоком уровне детализации. Для читателей, хорошо знакомых с математикой, ниже мы приводим подробное рассмотрение широко распространенного потенциала парных взаимодействий. Остальные могут без стеснения пропустить эту часть раздела.
Можно ввести классификацию контактов по расстоянию до определенного порога (скажем, 30 А) и отнести их к соответствующим интервалам. Интервалы расстояний можно затем разбить далее по удаленности
2.2. «Протягивание»
53
контактирующих остатков в последовательности на ближний диапазон (скажем, от 3 до 9 разделяющих остатков) и дальний диапазон (более 9 разделяющих остатков). Кроме того, даже при наличии 50000 структур в базе данных можно столкнуться с проблемой недостатка данных, если используется такое большое количество подразделов. Потому вводится схема присвоения весовых коэффициентов наблюдениям (член Л/^т), которая по сути лишь рассчитывает встречаемость результата, если он наблюдался 1 /о раз. Теперь энергия Е/ для пары остатков ij9 разделенных к остатками и лежащих в диапазоне расстояний I, рассчитывается по формуле:
El = RT\n[l + Mijka]-RT\n
l + MiJka
где Mijk - частота встречаемости для пары остатков ij\ разделенных к остатками, о - наблюдаемый вклад (часто задается \/50),fkj(l) - относительная частота встречаемости пары ij, разделенной к остатками в интервале расстояний /:
луло Мик '
где fk*(l) - относительная частота всех пар, разделенных к остатками в интервале расстояний /:
...
f(l)=
Здесь R - количество типов остатков, a N - количество классов разделения последовательностей. Парный потенциал для данного белка определяется как сумма энергий для контактов «остаток-остаток» в рамках данных параметров разделения.
На детали методики расчета потенциалов может оказывать влияние множество различных обстоятельств. Так, в основу силового поля может быть положено лишь расстояние между атомами Са основной цепи, и этого будет достаточно для предварительного распознавания грубой топологии структуры. Некоторые исследователи увеличивают количество участков атомных взаимодействий, что, вероятно, положительно влияет на учет водородных связей. Область применения уравнения Больцмана не ограничивается расстояниями. Некоторые исследователи учитывают зависимости, связанные с различными углами, в том числе, углами упаковки между бета-тяжами. В силовом поле могут по-разному учитываться вклады остатков, разделенных различными расстояниями, то есть, исследователь
54
Глава 2. Распознавание фолда
может использовать различные функции для остатков, расположенных близко в последовательности (i, /+3), и остатков, расположенных дальше друг от друга (/, i+n\ п > 10), как это уже упоминалось выше.
Очевидно, что вычислительная мощность метода протягивания в значительной степени ограничивается производительностью энергетической функции. В результате, множество как уже завершенных, так и современных исследований ставят своей задачей разработку более точных и, следует надеяться, более производительных эмпирических потенциалов.
2.2.2. Поиск выравнивания
В случае наличия функции потенциальной энергии, с помощью которой можно получить оценку для данной модельной структуры белка, исследователь сталкивается с непростой задачей: как найти выравнивание последовательности по структуре, для которого значение потенциальной функции будет минимальным (максимальным). Если не принимать во внимание тот факт, что в процессе эволюции в последовательности белка появляются вставки и делеции, можно использовать методику «протягивания без разрывов». Этот подход подразумевает простое «проскальзывание» последовательности через структуру, при этом учитывается и оценивается каждое выравнивание без разрывов. Преимуществом подхода является высокая скорость вычислений, а существенным недостатком - отсутствие возможности учитывать разрывы. Вставка или делеция лишь одного остатка вызывает смещение считывающей рамки, в результате чего выравнивание, которое в других условиях было бы определено как высококачественное, не учитывается. Таким образом, принимать во внимание разрывы крайне важно для учета природы эволюционных вариаций.
Однако именно учет таких разрывов превращает простую задачу в NP-сложную, для которой не существует быстрого (за полиномиальное время) решения. Полный перебор всех возможных выравниваний с разрывами для исследуемых последовательности и структуры невыполним по очевидным причинам, в особенности, если поиск осуществляется в базе данных, которая содержит тысячи структур. В случае обычного выравнивания последовательностей, при котором не учитывается вклад парных взаимодействий между любыми двумя остатками одного и того же белка, такую проблему выравнивания можно решить с помощью рекурсивного процесса динамического программирования. Но когда различные вклады парных взаимодействий, такие как физические потенциалы, учитываются, динамическое программирование использовать нельзя. В классическом динамическом программировании выравнивание остатка исследуемой
2.2. «Протягивание»
55
Таблица 2.1. (а) Оценочная матрица BLOSSUM (Seq)
А R N D С Q F Р S Т W У V
А 4 -1 -2 -2 0 -1 -2 -1 1 0 -3 -2 0
R -1 5 0 -2 -3 1 -3 -2 -1 -1 -3 -2 -3
N -2 0 6 1 -3 0 -3 -2 1 0 -4 -2 -3
D -2 -2 1 6 -3 0 -3 -1 0 -1 -4 -3 -3
С 0 -3 -3 -3 9 -3 -2 -3 -1 -1 -2 -2 -2
Q -1 1 0 0 -3 5 -3 -1 0 -1 -2 -1 -2
F -2 -3 -3 -3 -2 -3 6 -4 -2 -2 1 3 -1
Р -1 -2 -2 -1 -3 -1 -4 7 -1 -1 -4 -3 -2
S 1 -1 1 0 -1 0 -2 -1 4 1 -3 -2 -2
Т 0 -1 0 -1 -1 -1 -2 -1 1 5 -2 -2 0
W -3 -3 -4 -4 -2 -2 1 -4 -3 -2 11 2 -3
У -2 -2 -2 -3 -2 -1 3 -3 -2 -2 2 7 -1
V 0 -3 -3 -3 -1 -2 -1 -2 -2 0 -3 -1 4
(б) Простая матрица оценки вторичной структуры (SS)
Предсказанная / известная а-спираль р-ТЯЖ петля
а-спираль +1 -1 -1
Р-тяж -1 +1 -1
петля -1 -1 +1
(в) Простая матрица оценки экспонированности (Solv)
Предсказанный / известный Заглубленный Экспонированный
Заглубленный +1 -1
Экспонированный -1 +1
последовательности относительно структуры предполагаемого шаблона оценивается с помощью простой справочной таблицы (например, BLOSUM или особой оценочной матрицы профиля/положения (PSSM, от «profile/position specific scoring matrix»); см. ниже, а также в табл. 2.1). В то же время, при использовании метода протягивания оценка выравнивания
56
Глава 2. Распознавание фолда
остатка последовательности относительно остатка структуры определяется на основании того, каким образом были выровнены все остальные остатки, с которыми исследуемый участок может взаимодействовать.
2.2.3. Эвристические правила выравнивания
Поскольку проблема протягивания формально не решаема (является NP-сложной), было разработано множество эвристических правил, предназначенных для высокочувствительного отбора возможных выравниваний в процессе поиска псевдооптимального решения за разумное вычислительное время. Один из подходов состоит в использовании ограничений для положения и размера разрывов. Суть состоит в том, что разрывы допускаются только в наиболее вероятных областях структуры шаблона, например, между консервативными элементами вторичной структуры (Madej etal. 1995). Еще один подход известен как «замороженное приближение» (рис. 2.3, слева) (Westhead etal. 1995). Как уже было отмечено выше, основная проблема при расчете оценки выравнивания определенного остатка относительно положения в структуре шаблона состоит в том, что выравнивание всех остальных остатков, то есть, окружение исследуемого остатка, должно быть известно. А поскольку оно неизвестно, при использовании фиксированного приближения за него принято принимать окружение последовательности шаблона. Это изящное и простое решение будет, несомненно, удачной аппроксимацией в тех случаях, когда исследуемая последовательность близка к последовательности шаблона. Однако этот способ неприемлем, если между последовательностями имеются существенные различия, то есть, именно в том случае, который нас интересует.
Более утонченный вариант фиксированного приближения разработан Сколником и Кихара (Skolnick and Kihara 2000) и носит название «размороженное приближение» (рис. 2.3, справа). В этом случае источником окружения остатка служит не структура шаблона, а первичное пробное выравнивание исследуемой последовательности относительно структуры шаблона, полученное с использованием классических методов выравнивания на основе профилей. Это значит, что по крайней мере окружение исследуемого остатка основано на верной последовательности. Тем не менее, достоверность получаемых в итоге значений энергии существенно зависит от качества первичного выравнивания, которое, как мы помним, являлось проблемой изначально. Чтобы уменьшить зависимость метода от первичного выравнивания, процесс многократно повторяют, каждый раз выравнивая последовательность с учетом вклада, который вносят оценки протягивания, полученные в ходе предшествующих повторений.
2.2. «Протягивание»
57
Последовательность
шаблона L.FDLCDLIPV - - CGFA
Исследуемая huminIfrrt'1 itui i-^
последовательность
Рис. 2.3. (Цветную версию рисунка см. на вклейке.) Схематичное представление протягивания. Слева: замороженное приближение: здесь остаток М исследуемой последовательности предварительно выровнен относительно остатка V структуры шаблона. Затем производится оценка эмпирического потенциала для остатка М в окружении остатков G, L и F, взятых непосредственно из нативной структуры шаблона. Справа: размороженное приближение. Сначала выполняется пробное выравнивание. Можно использовать, например, методики выравнивания профилей. Теперь за окружение остатка М теперь принимается окружение из пробного выравнивания. Тенями показаны остатки исходного шаблона
Довольно сложный подход, разработанный Джонсом с сотр. (Jones etal. 1992) и названный «двойным динамическим программированием», используется в программе THREADER. Эффективность подхода была продемонстрирована в ранних испытаниях CASP. Полное описание методики лежит за пределами настоящей главы, однако общая идея состоит в выравнивании одиночного положения в исследуемой последовательности относительно одиночного положения в структуре шаблона. Затем для выравнивания оставшейся части последовательности используется традиционный алгоритм выравнивания, который позволяет оптимизировать потенциал относительно этого фиксированного положения. Оптимальное выравнивание, обнаруженное таким образом, позже добавляют в оценочную матрицу. Процесс повторяют для каждой возможной пары остатков последовательности и структуры (или по крайней мере для большого количества значимых пар), при этом информация об оптимальном выравнивании всякий раз заносится во вторичную оценочную матрицу. Наконец, вторичная оценочная матрица используется для создания окончательного выравнивания, в котором учитывается максимально возможное количество накопленных оптимальных выравниваний. Именно из-за такого двухуровневого выравнивания метод называется двойным динамическим программировани
58
Глава 2. Распознавание фолда
ем. Первоначально метод предназначался для разделения проблемы протягивания на множество небольших задач, сочетание решений которых давало однозначный ответ. Эта идея позже неоднократно использовалась во многих методах, описываемых ниже.
Алгоритм сэмплирования Гиббса применялся для решения проблемы протягивания Бриантом (Bryant 1996). В случае применения этой методики на первом этапе выполняется случайное выравнивание. На каждом шаге алгоритма случайным образом выбирается элемент вторичной структуры ядра С, для него создаются все возможные альтернативные выравнивания, для каждого нового выравнивания рассчитывается оценка S, а затем осуществляется выбор нового выравнивания с вероятностью, пропорциональной exp(-S/kT), где к - постоянная Больцмана, а Т - воображаемая «температура» системы. Каждая новая итерация предполагает использование нового случайным образом выбранного элемента ядра в качестве мишени для выравнивания. Используется протокол имитации отжига, с помощью которого «температура» системы со временем медленно снижается. Использование высокой температуры на начальном этапе приводит к тому, что выравнивания с низкими оценками учитываются так же часто, как и выравнивания, оцениваемые высоко. Это приемлемо для начального этапа моделирования, поскольку вероятность получить полное выравнивание высокого качества случайным образом очень мала. Тем не менее, по мере того, как температура падает, постепенно снижается вероятность учета выравниваний с низкими оценками, и система «оседает» на выравнивание с глобально низкой энергией. Имитация отжига широко используется для решения проблем оптимизации в биоинформатике и других областях. Метод не гарантирует нахождение выравнивания, соответствующего глобальном оптимуму, однако характеризуется быстротой и высокой производительностью.
При использовании алгоритма протягивания «разделяй и властвуй» (Xu etal. 1998) осуществляется многократное разделение структуры на подструктуры, для которых решается проблема выравнивания, и полученные для них решения объединяются для нахождения глобального оптимального выравнивания. Схожим образом в алгоритме поиска ветвей и границ (Lathrop and Smith 1996) пространство поиска протягивания многократно делится на подпространства меньшего размера, среди которых осуществляется отбор наиболее удачного подпространства, которое затем снова делится. На завершающем этапе наиболее удачное из отобранных подпространств содержит лишь одно выравнивание, которое и является глобальным оптимумом. Нахождение глобального оптимума требует очень больших временных затрат, в связи с чем была разработана версия про
2.3. Определение отдаленной гомологии без протягивания
59
граммы, условно называемая «Всегда готов!», (Lathrop 1999), с помощью которой быстро удается получить довольно точное приближение. Чем дольше процесс выполнения программы, тем точнее получаемые результаты. В конечном счете, программа возвращает выравнивание, соответствующее глобальному оптимуму.
Еще один подход, тесно связанный с описанным, - протягивание белка с помощью линейного программирования (Xu et al. 2003). Линейное программирование - общий метод, который используется для решения сложных задач при наличии ряда ограничений. В случае протягивания к числу таких ограничений часто относится представление о том, что выравнивание определенной области исследуемой последовательности относительно структуры подразумевает аналогичное выравнивание последующих (предыдущих) частей последовательности относительно соответствующих последующих (предыдущих) частей структуры. Ограничения такого типа, будучи скорее логическими переменными, чем непрерывными, могут все вместе рассматриваться как задача целочисленного программирования. Такие задачи часто решают, представляя их менее строго - как непрерывные задачи линейного программирования, после чего следует применение модели ветвей и границ.
Приведенный обзор некоторых методов протягивания демонстрирует, насколько широкий набор инструментов из области физики, математики и компьютерных наук использовался для решения этой сложной задачи в последние 15-20 лет. Тем не менее, до сих пор не существует единого метода, который обладал бы выраженным превосходством над другими методами в этой области. Несмотря на наличие высокопроизводительных и точных методов выравнивания последовательности относительно структуры, основанных на использовании энергетической функции, именно энергетическая функция представляется тем «слабым звеном», которое является основной причиной низкой производительности.
2.3. Определение отдаленной гомологии без протягивания
Подход с использованием протягивания первоначально был разработан для решения проблемы определения совместимости последовательности с известной структурой. Число способов белковой укладки в природе конечно. Это указывает на то, что при наличии соответствующих энергетической функции и алгоритма выравнивания использование методов на основе протягивания может оказаться успешным, тогда как методы на ос
60
Глава 2. Распознавание фолда
нове последовательностей не приносят желаемых результатов. Применение методов на основе последовательностей требует наличия некоторой ощутимой гомологии между исследуемой последовательностью и известной структурой; для использования методик на основе протягивания наличие определяемой гомологии не требуется.
В раннем периоде развития методов поиска потенциальных гомологий по базам данных последовательностей преобладали BLAST и другие подобные подходы. В их основе лежала идея использования общей оценочной функции, такой как матрицы BLOSUM или РАМ, которые показывали вероятность мутационного перехода одного типа аминокислоты в другой на основе ряда надежно выровненных блоков схожих белковых последовательностей. Это были простые справочные таблицы размером 20x20, в которых содержались оценки для соответствия между любыми парами аминокислот в выравнивании. Таким образом, выравнивание гидрофобного остатка относительно другого гидрофобного остатка (например, лейцина относительно валина) получало хорошую оценку, а выравнивание непохожих остатков (например, глутамата относительно триптофана) - плохую оценку. Сочетание такой оценочной функции с алгоритмом стандартного динамического программирования позволяло получить относительно невысокую производительность при определении гомологических связей. Если задаться целью поиска по базам данных последовательностей с известной структурой, а затем построить модель на основе полученного выравнивания, то можно получить одну из самых простых методик предсказания структуры белка (рис. 2.4а).
Очевидный недостаток этого подхода состоит в том, что с помощью простых оценочных функций размером 20x20 можно успешно определять лишь близкую гомологию (более 30% идентичности последовательностей). Известно, что в ниже этой границы идентичности последовательности могут существенно дивергировать, тогда как структуры обладают высокой степенью сходства. Таким образом, при использовании обсуждаемого подхода не учитывается множество гомологичных связей, определение которых позволит значительно улучшить качество предсказаний структуры белков.
2.3.1. Использование предсказанных структурных свойств
Одна из самых ранних попыток превратить распознавание гомологии в нечто большее, чем простое сопоставление последовательностей, была предпринята Боуи и сотр. (Bowie etal. 1991). В основе метода лежит тот
2.3. Определение отдаленной гомологии без протягивания
61
Рис. 2.4. (Цветную версию рисунка см. на вклейке.) Схематическое представление развития методов распознавания фолда на основе последовательностей. В левой части каждого рисунка представлена исследуемая последовательность. Серой ячейкой справа на рисунках показана база данных шаблонов известной структуры. Стрелки указывают на процедуру сравнения исследуемой последовательности с определенным шаблоном, а) Простое сравнение аминокислотной последовательности с последовательностями из базы данных, б) Сопоставление с учетом информации о предсказанной (исследуемый белок) и известной (шаблон) вторичной структуре. Волнистыми линиями показаны альфа-спирали, ромбами - бета-тяжи.
в) Исследуемая последовательность представлена профилем из множества последовательностей, PSSM или HMM (СММ) (цветная решетка). Каждому ряду решетки соответствует гомологичная последовательность, каждой колонке -положение в последовательности, г) Ситуация, противоположная (в). Теперь осуществляется поиск исследуемой последовательности в библиотеке профилей. д) Сравнение «профиль-профиль» (вторичная структура по-прежнему представлена простой трехбуквенной строкой), е) То же, что и (д), однако вторичная структура представлена профилем. Для каждого положения последовательности характерны определенные значения вероятности каждого из трех типов вторичной структуры. Обратите внимание, что в данном случае исследователь, вероятно, использует предсказанную вторичную структуру шаблонов, несмотря на то, что фактическая вторичная структура известна. Показано, что метод отличается высокой производительностью, (e.g. Bennett-Lovsey et al. 2008)
62
Глава 2. Распознавание фолда
факт, что определенные структурные свойства белковых последовательностей можно предсказывать в отсутствие точного шаблона. Особенно примечательно, что вторичную структуру, то есть, положение альфа-спиралей и бета-тяжей, теперь можно предсказывать с точностью до 80%, используя такие программы как PSIPRED (Jones 1999а). Учитывая, что структура более консервативна, чем последовательность, пара белков, обладающих отдаленной гомологией, будет содержать схожие паттерны элементов вторичной структуры даже в отсутствие любого очевидного сходства последовательностей. Кроме того, с относительно высокой точностью можно предсказывать степень доступности остатка растворителю (см., например, Kim and Park 2003), а также наличие в структуре крутых поворотов типа бета-шпилька (например, Kumar et al. 2005).
Такие предсказанные структурные свойства обеспечивают наличие более подробной информации о структуре белков, которую затем можно использовать наряду с сопоставлением последовательностей. При выравнивании двух аминокислот исследуемой последовательности и последовательности шаблона можно рассчитать совместимость на основе матрицы мутаций, такой как BLOSUM, а также члены, связанные с сопоставлением вторичных структур и доступностью для растворителя:
Sy =Seqij+SSij+Solvy,
где Sy - общая оценка сопоставления остатка i в исследуемой последовательности с остатком j в последовательности шаблона, Seqy - оценка, полученная для сопоставления i nJ в матрице BLOSUM, SSy - оценка для сопоставления предсказанного типа вторичной структуры остатка / с известным типом вторичной структуры остатка у, и Solvy — оценка для сопоставления степени заглубленности остатка i с известной степенью заглубленно-сти остатка j. Простые версии таких оценочных функций представлены в таблицах 2.16 и 2.1 в, где одинаковые степени (например, сопоставление спирали со спиралью) получают оценку +1, а все остальные сочетания -оценку -1. Часто функции детально разработаны и основаны на эмпирических наблюдениях частот, с которыми отличающиеся степени оказываются выравнены в случаях известных гомологов. Этот процесс аналогичен продвижению от простой матрицы сопоставления последовательностей на основе идентичности к более чувствительной матрице BLOSUM-типа.
Идея сочетания информации о последовательности и вторичной структуре в ходе поиска по базам данных схематично представлена на рис. 2.46. Методы, в основу которых положена эта идея, значительно превосходят методы стандартного поиска последовательностей по производительности и оказываются на порядки быстрее в вычислительном отношении, чем
2.3. Определение отдаленной гомологии без протягивания
63
большинство алгоритмов протягивания. Это свойство является чрезвычайно важным при поиске по большим базам данных шаблонов.
На начальном этапе развития международного соревнования С ASP подходы на основе протягивания в целом отличались наиболее высокой производительностью, а описанные выше гибридные подходы «последовательность-структура» вплотную следовали за ними. Однако вскоре последовало смещение методов протягивания с лидирующих позиций, чему способствовали два обстоятельства: 1) стремительное увеличение размеров баз данных последовательностей и 2) развитие метода PSI-BLAST.
2.3.2. Профили последовательностей и скрытые марковские модели
В то время как базы данных последовательностей быстро увеличивались в размерах в соответствии с распространенными по всему миру попытками секвенирования генома, развитие технологий, направленных на эффективное использование получаемой информации, находилось лишь в начале своего пути. Простой подход, использованный Парком и сотр. (Park etal. 1997), демонстрирует, каким образом две гомологичные последовательности, дивергировавшие далеко за пределы точки, в которой их гомология определяется простым прямым сравнением, можно связать с помощью третьей последовательности, являющейся подходящим промежуточным звеном для двух исследуемых. Такой «перескок» по пространству последовательностей, известный как поиск промежуточной последовательности, обладал очевидным потенциалом, а в программе PSI-BLAST (Altschul et al. 1997) был разработан усовершенствованный подход. Вместо использования оценочной матрицы фиксированного размера 20x20 для каждого белка, а также для каждого положения остатка в белке, можно было создать оценочную матрицу размером лх20, или профиль, который содержал бы информацию о специфических мутационных предрасположенностях каждого положения конкретной белковой последовательности. По этой причине такой профиль часто называют позиционно специфической оценочной матрицей (PSSM, от «position specific scoring matrix»).
После того, как проведено первичное стандартное сканирование BLAST с целью обнаружения относительно близких гомологов, выполняется (псевдо-)множественное выравнивание последовательностей этих гомологов относительно исследуемой последовательности. Выравнивание позволяет получить статистику наблюдаемых мутаций для каждого положения исследуемой последовательности. Эти статистические данные являются основой новой оценочной матрицы, которую затем можно использовать на
64
Глава 2. Распознавание фолда
последующих этапах поиска. Такой процесс поиска гомологов, создания новой оценочной функции и повторного поиска с использованием этой новой оценочной функции может повторяться множество раз (обычно от 5 до 10) и носит название PSI-BLAST (от «Position Specific Iterated BLAST»). Сочетание такого эффективного итеративного подхода с информацией из постоянно растущей базы данных последовательностей позволило значительно усовершенствовать процесс определения крайне отдаленной гомологии. На соревновании CASP4 исследовательские группы, использовавшие указанный подход (PSI-BLAST или его вариацию), продемонстрировали более высокую производительность по сравнению с успешными ранее исследовательскими группами, основу работы которых составлял метод протягивания.
Причина успешности подхода PSI-BLAST заключается в учете того обстоятельства, что для каждого положения белковой последовательности характерно собственное значение эволюционно давления. Так, глицин в определенном положении может быть высоко консервативен, если его присутствие обеспечивает наличие в белковой цепи довольно крутого поворота, необходимого для поддержания топологии. Любая мутация в таком положении может оказаться летальной из-за возможного нарушения правильного сворачивания белка. В другом положении остаток глицина может находиться под минимальным давлением отбора, располагаясь в высоко изменчивой области петли. Соответственно, при выравнивании исследуемой последовательности по структуре присутствие первого остатка глицина обязательно, тогда как природа второго остатка может изменяться. Именно учет мутационной предрасположенности, которая определяется, среди прочего, положением остатка, делает подход гораздо более чувствительным при определении отдаленной гомологии.
Одним из наиболее типичных способов применения созданных в PSI-BLAST профилей является поиск профиля исследуемой последовательности среди последовательностей базы данных PDB или, наоборот, поиск исследуемой последовательности в базе профилей шаблонов. Профили не всегда создаются с помощью PSI-BLAST. Так, профили на основе скрытых марковских моделей (СММ) (HMMs, от «Hidden Markov Models») создаются с использованием множественных выравниваний последовательностей, однако содержат больше информации, чем стандартные профили. Например, в них содержится информация о положениях наиболее типичных вставок и делеций, а также о вероятностях замен при переходах от сопоставляемых состояний и к сопоставляемым состояниям для каждого положения цепи. Опять-таки, это часто связано с использованием предсказанных структурных свойств, таких как вторичная структура. Альтер
2.3. Определение отдаленной гомологии без протягивания
65
нативные подходы, в основе которых лежат принципы «последовательность-профиль» и «профиль-последовательность», схематично представлены на рис. 2.4в и 2.4г.
Улучшенные профили и СММ можно создавать, используя структурные выравнивания отдаленных гомологов, а также добавляя последовательности неизвестной структуры, которые можно легко выровнять относительно каждой из имеющихся структур (Kelley et al. 2000; Tang et al. 2003). Однако использование структурных выравниваний для создания профилей более высокого качества часто приводит к незначительным улучшениям в определении отдаленных гомологов или точности выравнивания. Это, вероятно, связано с тем, что выравнивания последовательностей на основе структурных выравниваний не обладают свойством однозначности, особенно в случае наличия больших вставок или делеций, а также в случае значительных изменений структуры. Эти обстоятельства могут приводить к некорректным выравниваниям между рядами последовательностей, связанных с каждой из структур. Решение (Zhou and Zhou 2005), использованное ими в методе SP3, который оказался успешным, состоит в том, чтобы создавать белковые фрагменты и использовать их для построения профилей.
В последние годы скрытые марковские модели широко используются различными исследовательскими группами - с их помощью удается получать хорошие результаты. Как уже упоминалось выше, одно из ключевых преимуществ СММ по сравнению с относительно более простыми профилями, создаваемыми PSI-BLAST, состоит в наличии дополнительной информации, которая касается разрывов и соседних остатков. Однако как для профилей, так и для СММ определяющее значение имеет множественное выравнивание последовательностей, на основе которого они получены. Последовательности и качество выравнивания представляются более важными для качества профиля характеристиками, чем точность статистических методов, использующихся при выравнивании в процессе создания профиля. В результате многие исследовательские группы сочли полезным осуществлять сбор гомологичных последовательностей с помощью PSI-BLAST, а для создания более точного множественного выравнивания использовать отдельную более производительную программу.
Как было показано недавно, выравнивания типа «профиль-профиль» и «СММ-СММ» как обобщения методов выравнивания «последовательность-профиль» или сравнения «последовательность-СММ» характеризуются значительно более высокой производительностью. Таким образом, вместо использования профилей (или СММ) лишь для исследуемой последовательности или последовательности шаблона, их используют для
66
Глава 2. Распознавание фол/ш
обеих этих последовательностей и сравниваю! дру! с jipyioM (рис. 2.4д). Каждое положение в последовательности можно р1нч м4нрннагь как вектор вероятностей. В случае простых профилей испольiycicN 20-тимерный вектор вероятностей (по одному измерению на каждый imii аминокислотного остатка). Положение в исследуемой послсдо1инс;1ыюсги схоже с положением в структуре шаблона в том случае, если оба ни положения находятся под одинаковым эволюционным давлением, ко герое дало бы сходные векторы вероятностей для этих положений. Дли сраннсния таких векторов в последнее время было разработано множсспю различных методик (самая простая из которых - скалярное произведение); почти все они превосходят более простые методы оценки форма!а «последовательность-профиль» (см., например, Rychlewski etal. 2000; Ohlsen etal. 2004; Soeding 2005; Bennett-Lovsey et al. 2008).
В свете успехов методов «профиль-профиль» мhoi не исследовательские группы изменили процесс предсказания таким образом, что теперь в нем учитываются профили вторичной структуры, где вмеею простого предсказания одного из трех состояний (альфа-спирали, бета-лижа или петли) рассчитывается вероятность каждого состояния, которая затем рассматривается как вектор. Были получены результаты, подтверждающие более высокую производительность такого подхода (Tang et al. 2003; Bennett-ixwsey et al. 2008). Схематическое изображение подхода представлено на рис. 2.4д.
Производительность методов предсказания с использованием профилей росла благодаря улучшению и увеличению баз данных последовательностей, усовершенствованию процедур создания профилей и улучшению алгоритмов сопоставления профилей. По мере такого роста производительности важность дополнительной информации о предсказанных структурных свойствах, казалось, уменьшалась в сравнении с се первоначальной определяющей ролью в ранних методиках Боуи и сотр. (Bowie et al. 1991). В основе наиболее успешных методик предсказания вторичной структуры обычно лежат алгоритмы машинного обучения, такие как искусственные нейронные сети или метод опорных векторов, обучение которых осуществляется на окнах профилей последовательностей, созданных в PSI-BLAST. Причина, по которой использование этой информации дает лишь ограниченные результаты, вероятно, кроется в недостатке новых, или «независимых», данных. Исходными данными для предсказания вторичной структуры, как правило, являются те же профили, которые используются для сопоставления последовательностей. Таким образом, можно утверждать, что большая часть информации при предсказании вторичной структуры, вероятно, уже закодирована в профиле, из которого она была получена.
2.3. Определение отдаленной гомологии без протягивания
67
2.3.3. Классификация типов укладки и метод опорных векторов
Распознавание фолдов является проблемой классификации. Ее можно рассматривать как ряд вопросов о том, сворачивается ли рассматриваемая последовательность в тот или иной тип укладки из всего их многообразия. Найти решение в таком случае можно с помощью методов машинного обучения. Если известны свойства исследуемой последовательности, S, такие как ее аминокислотный состав, родственные последовательности, предсказанная вторичная структура и т.п., можно определить наиболее вероятный тип укладки, обладающий свойствами s, из некоторого набора типов укладки F. Такие классификаторы грубо можно разделить на генеративные и дискриминативные. Типичным генеративным классификатором является наивный байесовский классификатор. Суть в том, чтобы определить относительную важность каждого свойства (параметры модели) для предсказания типа укладки путем анализа частот, с которыми данные свойства встречаются у элементов данного класса в определенном обучающем наборе.
В качестве примера использования наивного байесовского классификатора приведем операцию определения наиболее вероятного значения Fnh при заданных значениях sb s2, ... sn. В результате имеем:
В общем случае P(st \fj) можно оценить по формуле:
_пс +тр п + т где:
п - количество обучающих примеров, для которых f-ff, пс - количество примеров, для которых f=fjns = sy, р - априорная оценка для Pfa | fj)\
т - эквивалентный объем выборки (весовой терм для априорной оценки).
Существует выраженное сходство между этим подходом и методами, которые описаны выше и используются для задания энергетических функций.
В отличие от генеративных классификаторов, где вероятности определяются с использованием обучающих примеров, в случае дискримина-тивных классификаторов предпринимается попытка достичь максимальной предсказательной точности непосредственно на обучающем наборе.
68
Глава 2. Распознавание фолда
Нейронные сети и метод опорных векторов - это дискриминативные классификаторы, которые широко используются в вычислительной биологии (см., например, Busuttil et al. 2004; Garg et al. 2005; Nguyen and Rajapakse 2003; Bradford and Westhead 2005).
Использование метода опорных векторов (англ, support vector machines, SVM) позволяет определить границу решений, или гиперплоскость, которая разделяет входные данные на два класса (например, тип укладки А и тип укладки не-А) на основе значения вектора свойств s. В наиболее сложных случаях данные невозможно разделить, используя линейную функцию входных свойств. В методе опорных векторов проблема нелинейности решается с помощью кернфункции k(sb Sj), которая оценивает степень подобия пар входных примеров sb Sj. В процессе обучение осуществляется сравнение каждого из примеров, как положительного, так и отрицательного с другими примерами рассматриваемого ряда с помощью кернфункции, создающей матрицу значений подобия размера п *п, где п -количество обучающих примеров. Хитрость состоит в том, что с помощью кернфункции - как правило, простой и быстрой в вычислительном отношении - можно представить данные в пространстве свойств более высокой размерности, где затем их можно разделить линейно. Определенная таким образом граница решений включает обычно лишь небольшое количество обучающих примеров, которые располагаются на самой границе решений и известны опорные векторы из-за своей способности «поддерживать» границу, подобно тому, как распорки служат опорой строения.
Метод опорных векторов используется для определения отдаленной гомологии, в том числе в методах SVM-Fisher (Jaakkola et al. 2000), SVM-k-spectrum (Leslie et al. 2002), SVM-pairwise (Liao and Noble 2003), SVM I-sites (Hou et al. 2003) и SVM-mismatch (Leslie et al. 2004).
Все эти методики являются в каком-то смысле методиками «чистого» распознавания, поскольку при осуществлении моделирования не создается окончательное выравнивание. Вместо этого с некоторой вероятностью определяется принадлежность исследуемой последовательности к тому или иному классу. В некоторых случаях это может быть полезно, однако зачастую исследователь стремится получить пространственную модель исследуемой последовательности, и потому для выполнения (нетривиальной) стадии выравнивания необходима дополнительная система.
2.3.4. Согласованные подходы
В последних экспериментах CASP было показано, что консенсусные методы, в которых данные нескольких серверов для распознавания фолдов
2.3. Определение отдаленной гомологии без протягивания
69
объединяются в общее предсказание, обладают значительным преимуществом. Эти «метасерверы» заметно превосходят многие индивидуальные методы, на основе которых они разработаны. К числу таких индивидуальных методов принадлежат выравнивание последовательности относительно профиля, скрытые марковские модели, выравнивание профиля относительно профиля и протягивание.
К числу наиболее популярных методик, в которых сочетание предсказаний объединяется в метасерверах, принадлежат Peons (Wallner and Elofsson 2005), 3D-Shotgun (Fischer 2003) и 3D-Jury (Ginalski et al. 2003). Самым простым, но от этого не менее производительным является метод 3D-Jury. В методе осуществляется сравнение пространственных моделей, созданных с помощью различных серверов, путем выравнивания их структур. Затем проводится переоценка моделей на основе их структурного сходства с остальными моделями группы. Таким образом, если в нескольких относительно независимых системах предсказания типов укладки были выбраны близкие шаблоны, а затем созданы близкие выравнивания, такие модели впоследствии получат более высокую оценку по сравнению с остальными, менее типичными моделями. В методе Peons этот подход 3D-Jury сочетается с использованием нейронной сети, которая обучена отличать модели, обладающие свойствами, общими для всех белковых структур, от моделей, у которых такие свойства отсутствуют (подобно тому, как это реализовано в эмпирической функции энергии в методе протягивания). Наконец, в методе 3D-Shotgun для каждого остатка каждой модели рассчитывается оценка по методу 3D-Jury, после чего из наиболее общих, или «популярных», частей создается новая модель. Это может привести к сильной фрагментации модели, и, несмотря на то, что на устранение этого недостатка был нацелен целый ряд экспериментов, проблема по-прежнему не решена.
Обширное исследование причин высокой производительности метасерверов было проведено в работе (Bennett-Lovsey et al. 2008). Авторы пришли к выводу, что улучшения по большей части заключаются не в исключении отдаленных гомологов per se, а в повышении точности, т.е., исключении ложноположительных результатов. Это явление связано с тем, что при сочетании множества различных систем предсказания структуры вероятность того, что все они совершают одну и ту же ошибку, значительно меньше вероятности согласованного результата. Любая особенность последовательности, которая может вызвать отказ в работе одного или двух методов предсказания, вряд ли будет иметь такое же влияние на большинство методов. Объединение классификаторов и алгоритмов предсказания в ансамбли с целью повышения производительности - устояв
70
Глава 2. Распознавание фолда
шаяся область исследований, которая занимает свое место между статистическим распознаванием образов и машинным обучением (Jain et al. 2000; Kuncheva and Whitaker 2003). К сожалению, даже спустя несколько десятилетий исследований мы не научились, используя основы теории, создавать оптимальные ансамбли. В результате основным принципом работы метасерверов, как правило, является метод проб и ошибок.
2.3.5. Проход по сети гомологов
Мы уже видели на примере PSI-BLAST и поиске промежуточных последовательностей, как сочетание ряда взаимосвязей между гомологами может привести к появлению продуктивных поисковых методов. В последних работах положено начало изучению этой сети взаимосвязей на еще более высоком уровне детализации. В подходах на основе профилей делается попытка создания одинарного статистического представления для ряда родственных белков - своего рода «усредненное» представление. Такие подходы, однако, исключают из рассмотрения большую часть информации, имеющуюся в этой сети взаимосвязей. Бейтман и Финн (2007) использовали простой подход по восстановлению части этой информации. В их методе выполняется сравнение результатов двух независимых процедур поиска по профилю и ставится вопрос, является ли число найденных последовательностей, общих для обеих процедур, большим, чем это могло бы оказаться случайно. Если рассматриваемые последовательности близкородственны, то для их профилей будет найдено большое количество общих последовательностей. В противном случае найденные по их профилям последовательности будут иметь только случайное сходство. Такой подход аналогичен исследованию структуры первого порядка сети гомологов, т.е. сравнению соседей одной последовательности с соседями другой. Этот простой подход оказался весьма эффективным при выявлении гомологии (выравниваний, созданных этим методом, нет) и значительно превосходит современные методы сравнения профилей.
Уэстон с сотр. (2004) в своем алгоритме Rankprop более глубоко использовали общую структуру сети гомологов. Ключевым нововведением, которое привело к успеху поисковой машины Google, является её способность использовать общую структуру, делая предположение о ней исходя из структуры локальных гиперссылок сети. Алгоритм Pagerank поисковой системы Google моделирует поведение случайного пользователя сети, который случайно нажимает на последующую ссылку, а также периодически перескакивает на случайную страницу. Веб-страницы ранжируются в соответствии с распределением вероятностей итоговых случайных маршру
2.4. Точность выравнивания и качество моделей
71
тов движения. На начальном этапе в алгоритме Rankprop используется сеть подобия последовательностей белков, предварительно рассчитанная при использовании всей базы данных последовательностей. По аналогии с процессом диффузии, интересующий исследователя белок попадает в сеть, после чего информация о ссылках на последовательность этого белка (о связях между последовательностью этого белка и близкими последовательностями других белков) распространяется по сети к соседям, соседям соседей и т.д. Затем белки базы данных ранжируются в соответствии с количеством ссылок, которое они получили на рассматриваемый белок. Показано, что такой подход превосходит стандартные методы поиска профилей по последовательности и сопоставим с методами поиска профилей по профилю, несмотря на то, что для создания первичной сети подобия используется PSI-BLAST.
Наконец, Хегер и его коллеги (2008) разработали алгоритм Maxflow, способный осуществлять проход по крупным сетям гомологов на уровне индивидуальных остатков. Алгоритм выполняет поиск согласованно выровненных пар остатков в сети парных выравниваний. Отличие этого метода от других состоит в том, что она нацелен на создание выравниваний, что имеет ключевое значение при моделировании белков.
Все эти новые подходы, основанные на использовании сетей, являются весьма полезными разработками для выявления гомологии. Серьезным недостатком этой группы методов являются огромные вычислительные ресурсы, необходимые для создания сетей подобия белков масштаба «каждый с каждым». Представляется очевидным, что производительность этих методов возрастет, если создать по-настоящему полную сеть на основе современных баз данных, которые содержат около 6 миллионов последовательностей. Однако сокращенные базы данных, которые содержат только последовательности с идентичностью менее 50% и имеют гораздо меньший размер, согласно результатам исследований, обладают той же, если не более высокой, производительностью, что и полные базы данных. Также интересно отметить, что область исследований, связанная с распознаванием гомологии, в скором времени, вероятно, получит толчок в развитии, основой которого послужат новые методики на основе теории графов.
2.4. Точность выравнивания, качество моделей и статистическая значимость
В распознавании фолда можно выделить две проблемы: 1) выявление подходящего шаблона и 2) выравнивание с этим шаблоном. Очевид
72
Глава 2. Распознавание фолда
но, что любой полезный метод предсказания структуры белков на основе моделирования по шаблону должен как минимум обладать способностью выявить подходящие шаблоны. Тем не менее, качество получаемой модели не зависит от качества определения шаблона и целиком определяется качеством выравнивания. Ошибки в выравнивании, несмотря на использование шаблона высокого качества, будут в любом случае приводить к построению модели низкого качества.
До настоящего момента мы полагали, что система, способная точно определить шаблоны, будет также создавать точные выравнивания. Исключения составляют некоторые классификаторы на основе метода опорных векторов, которые обсуждаются в разделе 3.3. Несмотря на то, что упомянутое допущение, как правило, справедливо, существует множество случаев, когда оно не выполняется. Во-первых, в большинстве описанных систем шаблоны по сути ранжируются с использованием некоторой оценки выравнивания. Иными словами, в таких методах всегда существует модель с самой высокой оценкой. Однако тот факт, что данное выравнивание оценивается выше других, не означает, что оно не содержит ошибок. Во-вторых, в большинстве из этих методов предпринимается попытка определить признаки чрезвычайно отдаленной гомологии. В этом качестве методы могут просто определять некоторые консервативные мотивы, или «пятна» сходства, прерываемые длинные участками последовательности, для которых сходство установить нельзя. В итоге создается выравнивание, которое по существу представляет собой «шум». Это, в свою очередь, приводит к большим погрешностям в создаваемой пространственной модели.
По этим причинам многие группы исследуют методы повышения точности выравнивания и предсказания качества получаемых моделей. Существует три способа, с помощью которых можно решить проблему точности выравнивания: 1) непосредственное усовершенствование алгоритмов создания выравниваний; 2) создание большого количества выравниваний и разработка системы отбора лучшего варианта; 3) построение пространственных моделей на основе множества большого количества выравниваний и оценка полученных моделей.
2.4.1. Алгоритмы создания выравниваний и оценка
Как уже было показано, использование информации о белковой эволюции в форме профилей или скрытых марковских моделей, а также информации о предсказанной вторичной структуре повышает степень определения гомологии, что, как правило, сопровождается соответствующим повышением точности выравнивания (Elofsson 2002). Захария и соавт.
2.4. Точность выравнивания и качество моделей
73
(Zachariah et al. 2005) показали, что при построении выравнивания с помощью динамического программирования использование более точной модели инициации и удлинения разрывов не улучшает процедуру установления гомологии, однако значительно повышает точность выравнивания.
Успешный подход был предложен на последнем совещании CASP (Venclovas and Margelevicius 2005). В этой процедуре ряд последовательностей, которые перекрывают пространство последовательностей между искомой последовательностью и шаблоном (шаблонами), используются для инициирования дополнительных процедур поиска в PSI-BLAST по неизбыточной базе данных последовательностей. Затем выравнивания искомой последовательности относительно шаблона (шаблонов) извлекаются из результатов поиска и проходят процедуру анализа согласованности. Для областей, в которых создается один преобладающий вариант выравнивания, это выравнивание считается достоверным, в то время как области, в которых согласованность выравнивания исследуемой последовательности относительно шаблона отсутствует, определяются как недостоверные. Таким образом, точность выравнивания можно увеличить, осуществив поиск согласованных выравниваний. Эта концепция близка к идее, используемой в 3D-Jury, где осуществляется поиск согласованного решения для пространства структур. Прасад и соавт. (2004) применяют близкий подход, используя пять различных методов для создания выравниваний и поиска среди них согласованного выравнивания.
Тресс и его коллеги (2003) исследовали распределение оценок профилей «остаток-остаток» по длине выравнивания. Было установлено, что области точного выравнивания можно достоверно определять на основе присутствия смежных участков с высокими значениями оценочной функции для остатков.
Как уже упоминалось ранее, подход на основе динамического программирования или СММ гарантирует построение «оптимального» выравнивание при заданной оценочной функции. Однако оценочные функции не совершенны. В связи с этим возможно существование большого количества выравниваний, близких к «оптимальному» и характеризующихся достаточно высокой оценкой, которые в действительности могут оказаться более точным с точки зрения структуры. Аналогичным образом, алгоритмы выравнивания нуждаются в определенной параметризации вероятности вставок и делеций, а параметры не могут быть универсальными для всех белков. В связи с этим Ярошевский и его сотрудники (2002) провели систематическое исследование выравниваний, близких к «оптимальному», варьируя параметры выравнивания и ослабляя наиболее выраженный ход процедуры выравнивания с помощью матрицы динамического программирования. При
74
Глава 2. Распознавание фолда
этом они установили, что в ходе ограниченного поиска «вблизи» оптимального выравнивания удается обнаружить выравнивания гораздо более точные, чем «оптимальное» с точки зрения значения оценочной функции. В результате остался открытым вопрос о том, как достоверно отобрать такие улучшения выравнивания из большого множества вариантов.
Чивиан и Бейкер (2006) попытались решить эту проблему путем создания моделей на основе выравниваний и оценки каждой модели с использованием сочетания кластеризации структур (например, методом 3D-Jury) и тонко настроенной энергетической функции пространственной структуры белка. Кроме того, Уолнер и Элофсон (2006) обучали нейронную сеть на окружениях остатков и оценках «профиль-профиль» набора белковых моделей для создания алгоритма предсказания качества моделей. Наконец, МакГаффин (McGuffin, 2008) использовал несколько программ для оценки качества моделей наряду с методами кластеризации структур, такими как 3D-Jury, в качестве входных данных для предиктора на основе нейронной сети.
2.4.2. Оценка статистической значимости
Для того чтобы методы, описанные в этой главе, имели практическое применение в широком биологическом сообществе, необходимы надежные способы оценки ошибок. Если молекулярный биолог сталкивается с предсказанием без указания вероятности точности прогноза, такой прогноз является практически бесполезным. При поиске последовательностей, поиске по библиотеке способов укладки или поиске по набору моделей, полученных на основе протягивания, результаты имеют общий вид -списка оценок. Известно, что при сравнении последовательности с библиотекой потенциальных моделей большинство из этих моделей оказываются неправильными. Таким образом, большинство оценок «последовательность-структура» можно рассматривать как фоновый шум. Затем можно использовать статистические показатели, чтобы рассчитать, превосходит ли данная ошибка фоновый шум и если да, то насколько.
В настоящее время не существует общего аналитического описания формы распределения оценок протягивания или распознавания укладки по различным моделям и последовательностям, хотя хорошо известно, что распределение оптимальных оценок не является нормальным. Для содержащего пропуски локального выравнивания двух последовательностей или последовательности относительно профиля распределение оптимальных оценок выравниваний может быть аппроксимировано распределением экстремального значения. Такие системы, как BLAST, PSI-BLAST, скрытые марковские мо
2.5. Веб-инструменты для распознавания элементов укладки
75
дели и многие методы «последовательность-профиль» и «профиль-профиль», подстраивают распределения оценок своих выходных данных под распределение экстремальных значений, из которого затем можно рассчитать вероятность ошибки первого рода и математическое ожидание.
В некоторых методах на основе профилей для приближения распределения оценок используется нормальное распределение и стандартизованные значения. Эти значения вычисляются с помощью среднего значения и стандартного отклонения оценки выравнивания рассматриваемой последовательности с библиотекой всех структурных моделей. Аналогичным образом, во многих методах протягивания оптимальная грубая оценка используется в качестве первичного показателя совместимости структуры и последовательности и определяется статистическая значимость оценки при учете предположения о нормальном распределении оценок последовательностей, протянутых через библиотеку доступных моделей. В методе протягивания с сэмплированием по Гиббсу (Bryant 1996) значимость оптимальной оценки определяется сравнением с распределением оценок, полученных протягиванием перемешанной рассматриваемой последовательности через ту же самую структурную модель. Распределение перемешанных оценок предполагается нормальным. В последнее время многие системы распознавания фолда отказываются от любых явных статистических расчетов и вместо этого полагаются на методы машинного обучения, такие как нейронные сети и методы опорных векторов, для прогнозирования оценки точности.
Однако зачастую наиболее передовые системы предсказания структуры, пытающиеся использовать крайне далекие гомологические отношения, являются высокоэмпирическими и в общем случае не имеют надежных статистических показателей вероятных ошибок. Читателю важно понимать, что предсказание структуры белков - это очень неточная наука, и поэтому нужно быть осторожным при толковании полученных результатов. Наиболее ценным инструментом при таком толковании является неизменно биологическое понимание изучаемого гена или системы.
2.5. Веб-инструменты для распознавания элементов укладки
Большое количество систем распознавания фолда размещено в сети с возможностью бесплатного доступа для использования в академических целях. Некоторые примеры таких систем приведены в таблице 2.2. В последнем соревновании CASP7 методы I-TASSER, HHpred, Roberta и Peons
76
Глава 2. Распознавание фолда
Таблица 2.2. Популярные веб-серверы для распознавания удаленной гомологии/фолда. «Согласованный» означает, что при работе сервера результаты различных независимых серверов используются для создания общего прогноза; «Одиночный» означает, что при работе сервера используются лишь собственные локальные методы. В колонке «Построение модели/оценка достоверности» указано, осуществляется ли сервером создание выходного файла с пространственными координатами потенциальной модели («Модель»), а также выставление оценки, которая позволяет судить о достоверности модели (значения Z, Р, Е и др.). В колонке «РФ/аЬ initio» указан метод: «РФ» - если полученные результаты основаны на методах отдаленной гомологии/распознавания фолда; «аЬ initio» - если дополнительно осуществляется построение модели в отсутствие шаблона
Название сервера Веб-адрес Согласованный / одиночный Построение модели / оценка достоверности РФ/ ab initio
I-TASSER http://zhang.bioinfor- matics.ku.edu/I-TASSER/ Одиночный Модель + достоверность РФ + ab initio
Phyre http ://www. imperial .ac.uk/ phyre/ Одиночный Модель + достоверность РФ
SAM-T06 http ://www. soe. uese. edu/ compbio/SAM_T06/T06-query.html Одиночный Модель + достоверность РФ
HHpred http://toolkit.tuebingen. mpg.de/hhpred Одиночный Достоверность РФ
GenThreader http://bioinf.cs.ucl.ac.uk/ psipred/psi form, htm Одиночный Значение Р РФ
PCONS http://pcons.net/ Согласованный Модель + оценка Peons РФ
Bioinfo http://meta.bioinfo.pl Согласованный Модель + значение Z РФ
FFAS http ://ffas. Ij erf. edu Одиночный Оценка FFAS РФ
Roberta http ://robetta. bakerl ab. org/ Одиночный Модель + достоверность РУ + ab initio
SP4 http ://sparks. informatics. iupui.edu/SP4/ Одиночный Модель + значение Z РУ
продемонстрировали высокую производительность. Peons, Bioinfo, и Genesilico являются метасерверами, или консенсусными серверами, которые осуществляют сбор результатов моделирования независимых серверов и работают с полученными моделями, используя структурную класте
2.6. Перспективы
77
ризацию или методы машинного обучения. Метасерверы, как правило, превосходят по производительности любые независимые серверы. Сервер Roberta, созданный в лаборатории Давида Бейкера, не ограничен распознаванием укладки и может создавать целый спектр предсказаний структуры белков, от сравнительных моделей до моделей ab initio. Разработанный в последнее время сервер 1-TASSER был создан с целью распознавания укладки, однако на совещании CASP7 были продемонстрированы некоторые обнадеживающие результаты использования I-TASSER в моделировании ab initio.
Время выполнения полного цикла моделирования для большинства этих серверов составляет, как правило, меньше часа, с важной оговоркой, что время эксперимента в значительной степени зависит от количества задач в очереди в данный момент времени. Простота использования и интерпретации результатов этих систем варьируется в широких пределах, а пригодность результатов для данного пользователя в значительной степени зависит от его исследовательского опыта. Кроме того, при решении задачи предсказания всегда полезно использовать несколько серверов для исключения из рассмотрения ложных результатов.
2.6. Перспективы
Ни в одном из наиболее успешных методов в последних соревнованиях CASP не использовалось одно лишь протягивание. Во многих методах протягивание не используется вообще. В некоторых методах для оценки потенциальных моделей по завершении построения или в сочетании с методами на основе профилей используются эмпирические потенциалы (см., например, Jones 1999b; Zhang 2007). Первичное преобладание подходов на основе протягивания и последующее снижение их популярности поднимают ряд интересных вопросов. Продолжительные дебаты в области структурной биологии касаются понятий гомологии и аналогии. Очевидно, множество различных последовательностей может иметь схожую укладку. Многие исследователи объясняют это явление процессом дивергентной эволюции общей предковой последовательности под давление отбора в пользу конкретной структуры. Однако некоторые исследователи полагают, что в случаях, когда наблюдаются существенные различия в последовательностях, имеющих одну и ту же укладку, могут иметь место прецеденты конвергентной эволюции, т.е., независимой эволюции в сторону одного и того же способа упаковки при отсутствии общего предка. Это явление сродни конвергентной эволюции крыла летучей мыши и птицы.
78
Глава 2. Распознавание фолда
Существуют яркие примеры конвергентной эволюции в белках, когда близкие локальные элементы структуры независимо развивались в нескольких случаях. Вероятно, наиболее известным примером является каталитическая триада Ser/His/Asp (Dodson and Wlodawer 1998), обнаруженная по крайней мере в пяти различных белковых укладках, которые сложно считать гомологичными. Такие доказательства в пользу конвергентной эволюции предполагают, что методы протягивания могут оказаться полезны там, где подходы «последовательность-профиль» терпят поражение. И все же, использование протягивания, как представляется, постепенно сходит на нет: происходит его замещение методами на основе последова-тельностей/профилей.
У этого процесса может быть несколько причин. Во-первых, остается открытым вопрос о том, как проходит сворачивание белка: для целой молекулы или локальных структур, возникших в ходе эволюции множество раз. Природа, возможно, несколько раз «натолкнулась» на простые способы укладки, такие как четырехспиральные пучки, но для более сложных структур такое явление сложно явно доказать. Для некоторых типов укладки, таких как бочонки Т1М и Р-трилистники, ранее считавшихся примерами конвергенции, появляется все больше данных о гомологии, установленных благодаря повышению чувствительности методов сравнения последовательностей (Copley and Bork 2000; Ponting and Russell 2000).
Во-вторых, даже если истинные аналоги существуют, методы на основе последовательностей могут определять их благодаря общим биофизическим предрасположенностям, необходимым для данной укладки, что, в свою очередь, будет отражено в высококачественном профиле, построенном с использованием множества отдаленных гомологичных последовательностей. В-третьих, широко признанным методом оценки качества предсказаний белковых структур является соревнование CASP. Неприятным побочным эффектом популярности CASP является то, что методы, которые способны установить связи для способов укладки аналогичных белков, как правило, остаются незамеченными из-за наличия методов, способных точно выровнять белковые последовательности относительно близких гомологов из постоянно растущих структурных баз данных белков. Если в базах данных структур можно установить гомологичные связи, это неизменно будет обеспечивать создание лучшей модели по сравнению с аналогичными связями. Т.е., по мере роста баз данных последовательностей и структур необходимость установления аналогии уменьшается, поскольку: а) расширяются возможности более глубокого поиска в пространстве последовательностей, б) появляется все больше близких структурных шаблонов на выбор.
2.6. Перспективы
79
Это приводит нас к выводу о том, что простое определение структур небольшого числа тщательно отобранных белков (Marsden et al. 2007) позволило бы относительно точно моделировать большую часть последовательностей генома. С точки зрения перспектив дизайна новых фолдов или предсказания способов укладки ab initio такие результаты неудовлетворительны. Но для эффективной технологии определения принадлежности структуры к тому или иному геному этого вполне достаточно при условии, что в наличии имеется достаточное количество тщательно подобранных структур.
Неясно, в какой степени улучшение предсказаний структуры обусловлено увеличением размера баз данных и в какой степени - усовершенствованием алгоритмов. База данных последовательностей увеличивается в размерах в геометрической прогрессии, однако количество значимой информации растет отнюдь не так быстро. Подавляющее большинство последовательностей, добавляемых в базу данных последовательностей ежегодно, очень похоже на уже имеющиеся в базе. В последних работах (Chubb, Kelley and Sternberg, рукопись в стадии подготовки) показано, что, несмотря на увеличение размеров баз данных, определение гомологии с помощью стандартных инструментов (таких, как PSI-BLAST) вышло на плато. Таким образом, сложно представить себе дальнейшее значительное увеличение обнаружения гомологии лишь на основе информации из баз данных последовательностей. Неясно также, насколько последние улучшения методов предсказания структуры ab initio могут быть связаны с ростом структурных баз данных, которые содержат фрагменты структуры, оптимальные для использования в методах сборки фрагментов (Zhang and Skolnick 2005).
Базы данных последовательностей и структур будут продолжать расти. Даже если бы развитие алгоритмов предсказания структуры сегодня прекратилось, точность предсказаний структуры продолжала бы расти. Опуская вопросы белкового дизайна, предсказание структуры - практический опыт, полезный для сокращения времени и затрат на определение структуры белка.
Стремление «решить» проблему сворачивания белков по сей день считается одним из «священных Граалей» молекулярной биологии. Но даже в отсутствие такого «решения» представляется вероятным, что в течение разумного периода времени нам удастся получить точные и полезные модели если не для всех, то для большинства белков, встречающихся в природе. Независимо от того, сколько лет (5, 10 или 50) изобретательности и кропотливой работы потребуется от экспериментаторов, определяющих структуры и геномы, и специалистов по моделированию, извлекающих полезную информацию из результатов экспериментов, оно того стоит. И теперь это уже лишь вопрос времени.
80
Глава 2. Распознавание фолда
Белковый дизайн, однако, остается очень разносторонней и сложной задачей, основанной на глубоком понимании процесса сворачивания белков. Понять процесс белковой укладки - значит понять, как «программное обеспечение» ДНК становится «аппаратным обеспечением», или «оборудованием» функциональных белков. Это значит на фундаментальном уровне понять природу живых существ. Тем не менее, возможно, для проблемы сворачивания белка не существует элегантного решения. Природе не обязательно использовать элегантное решение - достаточно того, которое работает. С неохотой мы вынуждены признать, что, возможно, нам придется довольствоваться лишь сложным предсказательным аппаратом. Тем не менее, остаются сильны надежды на то, что отыщется простое, вычислительно доступное и до сих пор не открытое объяснение процесса сворачивания белков.
Благодарности. Я бы хотел поблагодарить доктора Бенджамина Джефферис за его активную помощь при подготовке иллюстраций к этой главе.
Литература
Altschul SF, Madden TL, Schaffer AA, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402
Bateman A and Finn RD (2007) SCOOP: a simple method for identification of novel protein super-family relationships. Bioinformatics 23:809-814
Bennett-Lovsey RM, Herbert AD, Sternberg MJ, et al. (2008) Exploring the extremes of se-quence/structure space with ensemble fold recognition in the program Phyre. Proteins. 70:611-625
Berman HM, Westbrook J, Feng Z, et al. (2000) The protein data bank. Nucleic Acids Res 28:235-242
Bowie JU, Liithy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253:164-170
Bradford JR, Westhead DR (2005) Improved prediction of protein-protein binding sites using a support vector machines approach. Bioinformatics 21:1487-1494
Bryant SH (1996) Evaluation of threading specificity and accuracy. Proteins 26(2): 172-185 Busuttil S, Abela J, and Pace GJ (2004) Support vector machines with profile-based kernels for remote protein homology detection. Genome Inform Ser Workshop Genome Inform 15:191— 200
Chivian D, Baker D (2006) Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Res 34:el 12
Copley RR, Bork P (2000) Homology among (beta/alpha)(8) barrels: implications for the evolution of metabolic pathways. J Mol Biol 303:627-641
Dodson G, Wlodawer A (1998) Catalytic triads and their relatives. Trends Biochem Sci 23:347-352 Elofsson A (2002) A study on protein sequence alignment quality. Proteins 46:330-339
Fisher D (2003) 3D-SHOTGUN: a novel, cooperative, fold-recognition meta-predictor. Proteins 51:434^41
Garg A, Bhasin M, Raghava GP (2005) Support vector machine-based method for subcellular localization of human proteins using amino acid compositions, their order and similarity search. J Biol Chem 280:14427-14432
Литература
81
Ginalski К, Elofsson A, Fischer D, et al. (2003) 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics 19:1015-1018
Heger A, Mallick S, Wilton C, et al. (2008) The global trace graph, a novel paradigm for searching protein sequence databases. Bioinformatics 23:2361-2367
Hou Y, Hsu W, Lee ML, et al. (2003) Efficient remote homology detection using local structure. Bioinformatics 19:2294-2301.
Jaakkola T, Diekhans M, Haussler D (2000) A discriminative framework for detecting remote protein homologies. J Comput Biol 7:95-114
Jain AK, Duin RPW, Mao JC (2000) Statistical pattern recognition: A review. IEEE Trans Pattern Anal 22:4-37
Jaroszewski L, Li W, Godzik A (2002) In search for more accurate alignments in the twilight zone. Prot Sci 11:1702-1713
Jones DT (1999a) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195-202.
Jones DT (1999b) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol 287:797-815
Jones DT, Taylor WR, Thornton JM (1992) A new approach to protein fold recognition. Nature 358:86-89
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577-2637
Kelley LA, MacCallum RM, Sternberg MJ (2000) Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol 299:499-520
Kim H, Park H (2003) Prediction of protein relative solvent accessibility with support vector machines and long-range interaction 3D local descriptor. Proteins 54:557-562
Kumar M, Bhasin M, Natt NK, et al. (2005) BhairPred: prediction of beta-hairpins in a protein from multiple alignment information using ANN and SVM techniques. Nucleic Acids Res 33 (Web Server issue): 154-159
Kuncheva LI, Whitaker CJ (2003) Measures of diversity in classifier ensembles and their relationship with the ensemble accuracy. Mach Learn 51:181-207
Lathrop RH (1999) An anytime local-to-global optimization algorithm for protein threading in theta (m2n2) space. J Comput Biol 6(3-4):405-418
Lathrop RH, Smith TF (1996) Global optimum protein threading with gapped alignment and empirical pair potentials. J Mol Biol 255:641-665
Leslie C, Eskin E, Noble WS (2002) The spectrum kernel: a string kernel for SVM protein classification. Рас Symp Biocomput 564-575
Leslie CS, Eskin E, Cohen A, et al. (2004) Mismatch string kernels for discriminative protein classification. Bioinformatics 20:467-476
Liao L, Noble WS (2003) Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J Comput Biol 10:857-868 Madej T, Gilbrat J-F, Bryant SH (1995) Threading a database of protein cores. Proteins 23:356-369 Marsden RL, Lee D, Maibaum M, et al. (2006) Comprehensive genome analysis of 203 genomes provides structural genomics with new insights into protein family space. Nucleic Acids Res 34:1066-1080
McGuffin U (2008) The ModFOLD server for the quality assessment of protein structural models. Bioinformatics 24:586-587
Miyazawa S, Jemigan RL (1996) Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol 256(3):623-644
Moult J, Fidelis K, Kryshtafovych A, et al. (2007) Critical assessment of methods of protein structure prediction - Round VII. Proteins 69 S8:3-9
Murzin AG, Brenner SE, Hubbard T, et al. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536-540
Nguyen MN, Rajapakse JC (2003) Multi-class support vector machines for protein secondary structure prediction. Genome Inform Ser Workshop Genome Inform 14:218-227
Ohlson T, Wallner B, Elofsson A (2004) Profile-profile methods provide improved fold-recognition: a study of different profile-profile alignment methods. Proteins 57:188-197
82
Глава 2. Распознавание фолда
Park J, Teichmann SA, Hubbard T, et al. (1997) Intermediate sequences increase the detection of homology between sequences. J Mol Biol 273:349-354
Pearson WR (1998) Empirical statistical estimates for sequence similarity searches. J Mol Biol 276:71-84
Ponting CP, Russell RB (2000) Identification of distant homologues of fibroblast growth factors suggests a common ancestor for all beta-trefoil proteins. J Mol Biol 302:1041-1047
Prasad JC, Vajda S, Camacho CJ (2004) Consensus alignment server for reliable comparative modeling with distant templates. Nucleic Acids Res 32:W50-W54
Richmond TJ (1984) Solvent accessible surface area and excluded volume in proteins. Analytical equations for overlapping spheres and implications for the hydrophobic effect. J Mol Biol 178:63-89
Rychlewski L, Jaroszewski L, Li W, Godzik A (2000) Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci 9:232-241
Science Editorial (2005) So much more to know. Science 309:78-102
Seringhaus M, Gerstein M (2007) Chemistry Nobel rich in structure. Science 315:40-41
Sippl MJ (1990) Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol 213:859-883
Skolnick J, Kihara D (2000) Defrosting the frozen approximation: PROSPECTOR - a new approach to threading. Proteins 42:319-331
Seeding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951— 960
Tanaka S, Scheraga HA (1976) Medium- and long-range interaction parameters between amino acids for predicting three-dimensional structures of proteins. Macromolecules 9:945-950
Tang CL, Xie L, Koh IY, et al. (2003) On the role of structural information in remote homology detection and sequence alignment: new methods using hybrid sequence profiles. J Mol Biol 334:1043-1062
Tress ML, Jones D, Valencia A (2003) Predicting reliable regions in protein alignments from sequence profiles. J Mol Biol 330:705-718
Venclovas C, Margelevicius M (2005) Comparative modeling in CASP6 using consensus approach to template selection, sequence-structure alignment, and structure assessment. Proteins(Suppl 7):99-105
Wallner B, Elofsson A (2005) Pcons5: combining consensus, structural evaluation and fold recognition scores. Bioinformatics 21:4248-4254
Wallner B, Elofsson A (2006) Dentification of correct regions in protein models using structural, alignment, and consensus information. Prot Sci 15:900-913
Westhead DR, Collura VP, Eldridge MD, et al. (1995) Protein fold recognition by threading: comparison of algorithms and analysis of results. Protein Eng 8:1197-1204
Weston J, Elisseeff A, Zhou D, et al. (2004) Protein ranking: from local to global structure in the protein similarity network. PNAS 101:6559-6563
Xia Y, Levitt M (2000) Extracting knowledge-based energy functions from protein structures by error rate minimization. Comparison of methods using lattice model. J Chem Phys 113:9318-9330
Xu J, Li M, Kim D, et al. (2003) RAPTOR: optimal protein threading by linear programming. J Bioinform Comput Biol 1:95—117
Xu Y, Xu D, Uberbacher EC (1998) An efficient computational method for globally optimal threading. J Comput Biol 5:597-614
Zachariah MA, Crooks GE, Holbrook SR, Brenner SE (2005) A generalized affine gap model significantly improves protein sequence alignment accuracy. Proteins 58:329-338
Zhang Y (2007) Template-based modeling and free modeling by I-TASSER in CASP7. Pro-teins(Suppl 8): 108-117
Zhang Y, Skolnick J (2005) The protein structure prediction problem could be solved using the current PDB library. Proc Natl Acad Sci USA 102:1029-1034
Zhou H, Zhou Y (2005) Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins 58:321-328
Глава 3
Сравнительное моделирование структуры белков
Андраш Физер
Предпосылкой к пониманию функционирования клетки на системном уровне является представление о пространственной структуре белков, которые служат посредниками в биохимических взаимодействиях. Стремительное увеличение числа доступных белковых последовательностей заложило основу для реализации следующего шага проектов геномного масштаба, который заключается в определении пространственной структуры для каждого известного белка. Для достижения этой амбициозной цели дорогостоящие и медленные эксперименты по определению структуры подкрепляются теоретическими подходами в исследованиях. В настоящей главе представлен обзор текущего состояния дел в области структурного моделирования и ее последние достижения. Особое внимание уделено методикам сравнительного структурного моделирования.
3.1. Введение
3.1.1. Структура определяет функцию
Функциональная характеристика белков - одна из наиболее актуальных проблем современной биологии. Несмотря на то, что белковые последовательности содержат ценную информацию о функциях, часто функционально значимые остатки невозможно определить в силу высокой пластичности последовательностей (Todd et al. 2002). Так, в случае ферментов одну и ту же функцию могут выполнять белки, для которых идентичность
Andras Fiser
Department of Biochemistry, Albert Einstein College of Medicine, 1300 Morris
Park Ave, Bronx 10461, NY, USA
e-mail: andras@fiserlab.org, http://wwyv.fiserlab.org
84
Глава 3. Сравнительное моделирование структуры белков
последовательностей составляет более 40%. Однако если идентичность последовательностей снижается до 30-40%, достоверно можно предсказать лишь первые три индекса номенклатуры Комиссии по ферментам (КФ, от «Enzyme Commission», ЕС), и точность такого прогноза составит лишь 90%. При идентичности последовательностей менее 30% для описания функций требуется информация о структуре белка. В tq же время, согласно оценкам, для 75% гомологичных ферментов идентичность положений остатков составляет менее 30% (Todd et al. 2001). Еще одно количественное исследование дивергенции последовательностей и функций основано на классификации функций 6828 белковых семейств в системе генной онтологии (Sangar et al. 2007). Показано, что среди гомологичных белков доля дивергентных функций резко снижается, если порог идентичности последовательностей составляет 50% и выше. Однако даже если идентичность последовательностей составляет 50% и выше, использование аннотаций одних гомологов для описания других ведет к ошибочному соотнесению совершенно разных функций в 6% случаев.
Функциональному описанию белка часто способствует наличие пространственной структуры. Данные, которые можно извлечь из пространственной структуры, варьируют от таких низкоуровневых описаний функции, как подтверждение типа укладки (G Wu et al. 2000) и предположения об общей функциональной роли белка, до описаний высокого уровня, таких как специфичность лигандов, а также создание ингибиторов в рамках разработки лекарств на основе структурных данных (Becker et al. 2006; Evers et al. 2003).
3.1.2. Последовательности, структуры и структурная геномика
Реализация крупномасштабных проектов секвенирования генома привела к тому, что к настоящему времени известно около шести миллионов уникальных последовательностей (Apweiler etal. 2004; С. Н. Wu etal. 2006). Если к этим сведениям из общедоступных баз данных добавить ме-тагеномные данные проекта «Craig Venter’s Global Ocean Survey», число известных последовательностей удвоится (Rusch etal. 2007; Venter etal. 2004; Yooseph et al. 2007). В то же время, количество белков, пространственная структура которых определена в экспериментах с использованием рентгеновской кристаллографии или спектроскопии ядерного магнитного резонанса (ЯМР), насчитывает лишь около 50000. Поскольку методики определения пространственной структуры являются сложными и длительными, доля белков, для которых экспериментально определены пространственные модели, будет и дальше сокращаться, составляя величину менее современного значения - 1%. Чтобы сократить разрыв между количеством
3.1. Введение
85
известных последовательностей и пространственных моделей, необходимо применение вычислительных методов.
В 2000 году по всему миру были запущены проекты в области структурной геномики. Одной из ключевых целей являлось экспериментальное определение пространственной структуры нескольких тысяч тщательно отобранных последовательностей белков с неизвестной структурой. Определенные таким образом структуры можно было бы использовать в качестве шаблонов при компьютерном моделировании структуры белков с близкими последовательностями. Количество таких родственных белков в 100 раз превышает число структур, определенных экспериментально (Burley et al. 1999). Эти повсеместные усилия становятся главным источником экспериментально определенных белковых структур: 75% новых элементов укладки, размещенные в PDB в последние годы, появились в результате проектов структурной геномики (Burley et al. 2008). В то же время, такие экспериментальные исследования подчеркивают значение теоретических методов моделирования структуры, поскольку более 99% пространственных моделей, которые еще только предстоит построить, будут получены с помощью вычислительных методов (Manjasetty et al. 2007).
3.1.3. Методы предсказания структуры белков
Исследование принципов, определяющих пространственную структуру природных белков, можно осуществлять либо на основе физических законов, либо на основе теории эволюции. В зависимости от того, какие данные положены в основу методов предсказания структуры белков, сами методы делятся на две группы (Fiser et al. 2002).
Первую группу составляют методы ab initio, или методы моделирования без использования шаблонов, которые обсуждались в главе 1. Предсказание структуры здесь осуществляется лишь на основании данных о последовательности (Bonneau and Baker 2001; Pillardy et al. 2001). Предполагается, что природной структуре белка соответствует глобальный минимум свободной энергии, который достигается в течение времени существования молекулы. Методы нацелены на определение этого минимума с помощью исследования множества возможных белковых конформаций (Dill and Chan 1997; Sali et al. 1994).
Вторую группу методов называют моделированием по шаблону. Она включает методики «протягивания», в результате которых исследователь получает полное описание пространственной структуры для молекулы-мишени (J. Xu et al. 2007) (см. также главу 2), и сравнительное моделирование (Fiser 2004). В основе этой группы методов лежит сходство, которым объединены большинство моделируемых последовательностей и по край
86
Глава 3. Сравнительное моделирование структуры белков
ней мере одна известная структура. Сравнительное моделирование касается тех случаев моделирования по шаблону, когда определен не только способ укладки из доступного набора шаблонов, но также построена полноатомная модель (Marti-Renom etal. 2000). Если структура по крайней мере одного белка семейства определена экспериментально, структуру других членов семейство можно смоделировать с помощью выравнивания относительно известной структуры. Предсказание структуры белков с помощью методов сравнительного моделирования возможно благодаря тому, что незначительные изменения в белковой последовательности, как правило, приводят к незначительным изменениям пространственной структуры (Chothia and Lesk 1986). Предсказанию также способствует тот факт, что пространственная структура белков, принадлежащих к одному семейству, более консервативна, чем их аминокислотная последовательность (Lesk and Chothia 1980). Таким образом, если сходство между двумя белками можно установить на уровне последовательностей, обычно можно предполагать также и сходство структуры. Методы сравнительного моделирования, или моделирования по шаблону, применяются все более широко в связи с тем, что количество различных способов укладки, которые встречаются в белках, довольно ограничено (Andreeva et al. 2008; Chothia et al. 2003; Greene et al. 2007).
Оба подхода к предсказанию структуры белков имеют свои преимущества и ограничения. В принципе, методы ab initio можно применять для моделирования любых последовательностей. Тем не менее, в связи с тем, что укладка белка является сложным процессом, а наше понимание этой проблемы все еще ограничено, в результате применения методов ab initio обычно удается получить модели низкого разрешения. Несмотря на значительный прогресс в области предсказания структуры белков методами ab initio (R. Das et al. 2007), их по-прежнему можно применять лишь к ограниченному числу последовательностей размером около 100 остатков. Сопоставление результатов моделирования с эталонными структурами показывает, что полное и верное представление о способе укладки большинства мишеней с помощью методик моделирования ab initio получить все еще нельзя (Jauch etal. 2007). Прогресс наших представлений о точности и производительности доступных к настоящему времени силовых полей и методик отбора в значительной степени обусловлен выдающимися успехами в области вычислительных возможностей. Для более полного их использования в последнее время было запущено несколько крупнейших в своем роде исследовательских проектов, которые, по ожиданиям, значительно поспособствуют углублению наших представлений о процессе белковой укладки. К числу таких проектов, среди прочих, относятся Rosetta@home (http://boinc.bakerlab.org/rosetta/), Folding@home (http://folding. stanford.edu/) и проекты Blue Gene, поддерживаемые IBM.
3.1. Введение
87
В проектах Rosetta@home и Folding@home изучение процесса белковой укладки или моделирование осуществляется посредством запуска расчетов на персональных компьютерах пользователей-волонтеров, которые объединены в сеть из миллиона процессоров по всему миру. В IBM для решения тех же исследовательских задач создан Blue Gene - вычислительный кластер, пиковая производительность которого оценивается в 596 терафлоп. В настоящее время различные вариации компьютеров Blue Gene занимают четыре из десяти первых мест в списки 500 наиболее мощных суперкомпьютеров ТОР500 по состоянию на ноябрь 2007 года (http://www. research.ibm.com/bluegene/).
В отличие от методов ab initio, сравнительное моделирование белковых структур позволяет получать модели, которые по качеству сопоставимы со структурами низкого разрешения, полученными методом рентгеновской кристаллографии, или со структурами среднего разрешения, полученными методом ЯМР. Однако применение методов сравнительного моделирования ограничено теми последовательностями, которые можно с уверенностью использовать при сопоставлении с известными структурами. В настоящее время вероятность обнаружить близкие белки с известной структурой для последовательности, случайным образом выбранной из генома, варьирует в пределах от 30 до 80% в зависимости от генома. Около 70% всех известных последовательностей содержат по крайней мере один домен, для которого можно определить связь по крайней мере с одним белком известной структуры (Pieper et al. 2006). Это количество более чем на порядок превышает число белковых структур, определенных экспериментально и размещенных в PDB (Berman et al. 2007). Методы сравнительного моделирования применяются для определения структуры белков все шире, поскольку растет количество белковых структур, определенных экспериментально. Эта тенденция становится еще более выраженной благодаря проекту Исследования структуры белков (PSI, от «Protein Structure Initiative»), целью которого является определение по крайней мере одной структуры для каждого белкового семейства (Burley et al. 2008; Vitkup et al. 2001). Пятилетний период исследования возможностей осуществления этого проекта структурной геномики и технологии накопления данных (PSI-1, 2000-2005 годы) сменился «стадией образования продукта» (PSI-2, 2005-2010 годы). Вполне возможно, цели проекта по существу будут достигнуты менее чем за 10 лет, что сделает возможным применение методов сравнительного моделирования для исследования большинства белковых последовательностей.
Как мы увидим, на практике при моделировании по шаблону всегда используется информация, которая с самим шаблоном не связана - это
88
Глава 3. Сравнительное моделирование структуры белков
различные силовые ограничения от общих статистических данных до молекулярно-механических силовых полей. В результате повышения качества силовых полей и алгоритмов поиска в большинстве успешных методов все чаще исследуется независимое от шаблона конформационное пространство (R. Das etal. 2007; Y. Zhang 2007). Аналогично, в большинстве удачных методов моделирования ab initio для построения моделей по сути используются фрагменты известной структуры (Bystroff and Baker 1998; Zhou et al. 2007). Разумно было бы по-отдельности обсуждать два фундаментальных принципа, лежащих в основе методов структурного моделирования, однако, согласно последним тенденциям, наибольший интерес вызывают методы, сочетающие оба принципа. Методы моделирования ab initio могут пролить свет на динамику процесса упаковки белка, тогда как на практике эффективное моделирование структуры почти всегда включает определенные вариации моделирования по шаблону.
3.2. Этапы сравнительного моделирования структуры белков
При сравнительном моделировании структуры белков, или моделировании по шаблону (по гомологии), пространственная модель белка неизвестной структуры (мишени) строится на основе известной структуры одного или более близких к исследуемому белков (шаблонов) (Blundell etal. 1987; Fiser 2004; Ginalski 2006; Greer 1981; Marti-Renom etal. 2000; Petrey and Honig 2005). Необходимыми условиями для получения модели удовлетворительного качества являются: а) заметное сходство между последовательностями мишени и шаблона; б) правильное выравнивание этих последовательностей.
Все современные методы сравнительного моделирования включают пять последовательных этапов. Первых этап - поиск белков с известной пространственной структурой, которые близки к последовательности мишени. Второй этап - выбор структур, которые будут использоваться в качестве шаблонов. Третий этап - выравнивание последовательностей относительно последовательности мишени. Четвертый этап - построение модели мишени исходя из выравнивания ее последовательности относительно структур шаблонов. Последний этап - оценка модели с использованием разнообразных критериев.
Существует несколько компьютерных программ и веб-серверов, в которых процесс сравнительного моделирования автоматизирован (таблица 3.1). Веб-серверы полезны и удобны в использовании (Battey et al. 2007; Fernandez-Fuentes et al. 2007a; Rai et al. 2006; Y. Zhang 2007), однако наилучших
3.2. Этапы сравнительного моделирования структуры белков
89
Таблица 3.1. Названия и URL некоторых онлайн-инструментов, полезных при решении различных задач сравнительного моделирования
Распознавание укладки с помощью поиска по базам данных
BLAST/PSI-BLAST FastA/ SSEARCH FFAS03 www.ncbi.nlm.nih.gov/BLAST/ www. ebi .ac.uk/fasta3 3 ffas.ljcrf.edu/ffas-cgi/cgi/ffas.pl
Распознавание укладки с помощью протягивания
PHYRE/3D-PSSM FUGUE LOOPP MUSTER SAM-T06 Prospect PSIPRED UCLA-DOE 123D www.sbg.bio.ic.ac.uk/~3dpssm www-cryst.bioc.cam.ac.uk/~fugue cbsuapps.tc.comell.edu/ zhang.bioinformatics.ku.edu/MUSTER www.soe.ucsc.edu/research/compbio/SAM_T06/T06-query.htmlcompbio.oml.gov/structure/prospect bioinf.cs.ucl.ac.uk/psipred/psiform.html www.doe-mbi.ucla.edu/Services/FOLD123d.ncifcrf.gov
Инструменты выравнивания последовательностей
Smith-Waterman ClustalW MUSCLE T-COFFEE PROMALS PROBCONS jaligner.sourceforge.net/ www.ebi.ac.uk/clustalw/ www.drive5.com/lobster/ tcoffee.vital-it.ch prodata.swmed.edu/promals/promals.php probcons.stanford.edu
Сравнительное моделирование, моделирование петель и боковых цепей
МММ М4Т MODELLER MODWEB I-TASSER HHPRED 3D-JIGSAW CPH-MODELS COMPOSER SWISS-MODEL FAMS WHATIF PUDGE 3D-JURY RAPPER ESYPRED3D CONSENSUS PCONS www.fiserlab.org/servers/MMM www.fiserlab.org/servers/M4T www.salilab.org/modeller/modeller.html modbase. compbio .ucsf. edu/Mod Web20-html/modweb. html zhang.bioinformatics.ku.edu/I-TASSER/ toolkit.tuebingen.mpg.de/hhpred www.bmm.icnet.uk/servers/3djigsaw/ www.cbs.dtu.dyk/services/CPHmodels/www.cryst.bioc.cam.ac.uk swissmodel.expasy.org/workspace www.pharm. kitasato-u .ac.jp/fams www.cmbi.kun.nl/whatif/ wiki.c2b2.columbia.edu/honiglab_public/index.php/Software meta.bioinfo.pl mordred.bioc.cam.ac.uk/~rapper www.fundp.ac.be/sciences/biologie/urbm/bioinfo/esypred/ structure.bu.edu/cgi-bin/consensus/consensus.cgi pcons.net
90
Глава 3. Сравнительное моделирование структуры белков
Окончание таблицы 3.1
Моделирование петель
ARCHPRED MODLOOP WLOOP fiserlab.org/servers/archpred salilab.org/modloop bioserv.rpbs.jussieu.fr/cgi-bin/
Моделирование боковых цепей
SCWRL IRECS dunbrack.fccc.edu/SCWRL3 .php irecs.bioinf.mpi-inf.mpg.de/index.php
Оценка модели
PROCHECK Prosa-web WHATCHECK VERIFY3D ANOLEA AQUA PROQ www.biochem.ucl.ac.uk/~roman/procheck/procheck.html prosa.services.came.sbg.ac.at/prosa.php swift.cmbi.ru.nl/gv/whatcheck nihserver.mbi.ucla.edu/Verify_3D protein.bio.puc.cl/cardex/servers/anolea/ urchin.bmrb.wisc.edu/~jurgen/Aqua/server/ www.sbc.su. se/~bjom w/ProQ/ProQ. cgi
результатов на сегодняшний день удается достичь при неавтоматизированном использовании различных инструментов моделирования экспертами (Корр et al. 2007). Принятие сложных решений по отбору наиболее подходящих в структурном и общебиологическом отношении шаблонов, оптимальному сочетанию различной информации о шаблонах, уточнению выравниваний в нетривиальных случаях, отбору сегментов для моделирования петель, включению в модели кофакторов и лигандов и определению внешних ограничений требует экспертного подхода, который сложно полностью автоматизировать (Fiser and Sali 2003а), хотя в этом направлении предпринимается все больше попыток (Contreras-Moreira et al. 2003; Fernandez-Fuentes et al. 2007b).
3.2.1. Поиск структур, потенциально родственных с мишенью
Сравнительное моделирование обычно начинается с поиска в базе данных Protein Data Bank (PDB) (Berman etal. 2007) белков известной структуры, при этом последовательность мишени используется в качестве запроса. Этот поиск, как правило, осуществляется посредством сравнения последовательности мишени с последовательностью каждой из структур в базе данных.
Существует два основных класса методов сразнения белков, которые используются при определении способов укладки. Методы одного класса
3.2. Этапы сравнительного моделирования структуры белков
91
выполняют сравнение последовательности мишени с каждым из шаблонов в базе данных независимо и реализуются с помощью парного сравнения последовательностей (Apostolico and Giancarlo 1998). К настоящему времени проведена полная оценка этих методов поиска последовательностей (Pearson 2000; Sauder et al. 2000) и определения способов укладки (Brenner etal. 1998). Наиболее популярными программами этой группы являются FASTA (Pearson 2000) и BLAST (Schaffer et al. 2001). Для повышения чувствительности при поиске последовательностей можно использовать информацию об эволюции белков в форме множественного выравнивания последовательностей (Altschul etal. 1997; Henikoff etal. 2000; Krogh etal. 1994; Marti-Renom etal. 2004; Rychlewski etal. 2000). В таких методах сначала осуществляется поиск всех имеющихся в базе данных последовательностей, для которых можно установить четкую связь с мишенью и легко осуществить выравнивание. Множественное выравнивание этих последовательностей является профилем последовательности мишени, в котором в неявном виде содержится дополнительная информация о расположении и паттерне эволюционно консервативных остатков белка. Наиболее широко известная программа из этого класса - PSI-BLAST (Altschul etal. 1997), в которой применяется алгоритм эвристического поиска коротких мотивов. Следующий шаг в направлении повышения чувствительности метода - предварительный расчет профилей последовательностей для всех известных структур и дальнейшее использование алгоритма парного динамического программирования для сравнения двух профилей. Этот прием применялся, среди прочих программ, в COACH (Edgar and Sjolander 2004) и FFAS03 (Jaroszewski etal. 1998, 2005). Построение скрытых марковских моделей (СММ) на основе профилей - еще один чувствительный метод определения универсальных консервативных мотивов в последовательностях (Karplus et al. 1998). Значительного улучшения методов, основанных на СММ, удалось достичь за счет включения информации о предсказанных элементах вторичной структуры (Karchin et al. 2003; Karplus et al. 2005). Еще одна разработка, используемая в этой группе методов, состоит в применении СММ, основанных на филогенетических деревьях, когда отбор различных подмножеств последовательностей для анализа профиля СММ осуществляется в каждом узле эволюционного дерева (Edgar and Sjolander 2003). Способствовать поиску шаблона может также определение промежуточных последовательностей, которые гомологичны обеим рассматриваемым последовательностям (John and Sali 2004; Sauder et al. 2000). Эти более чувствительные методы определения способа укладки особенно полезны при поиске выраженных структурных связей, когда идентичность последовательностей мишени и образца со
92
Глава 3. Сравнительное моделирование структуры белков
ставляет менее 25%. Более точные профили последовательностей и структурные выравнивания можно построить с помощью методов, основанных на согласовании, таких как Т-Coffee (Moretti et al. 2007), PROMAL (и PROMAL3D для структур) (Pei and Grishin 2007; Pei et al. 2008), ProbCons (Do et al. 2005) и др. Более подробную информацию о методах множественного выравнивания последовательностей можно найти в последних обзорах (Edgar and Batzoglou 2006; Notredame 2007).
В основу второго класса методов положено парное выравнивание последовательности белка и структуры белка; при этом осуществляется поиск соответствия между последовательностью мишени и пространственными профилями из базы данных или «протягивание» через библиотеку типов пространственной укладки. Этот класс методов также называют определением типа укладки, протягиванием или сопоставлением пространственных шаблонов (Bowie et al. 1991; Finkelstein and Reva 1991; Jaroszewski et al. 1998; Jones 1999; Shi et al. 2001; Sippl 1995). Они подробно обсуждались в главе 2 и особенно полезны, когда составление профилей последовательностей невозможно в силу недостатка известных последовательностей, для которых можно установить четкую связь с мишенью или потенциальными шаблонами.
Методы поиска шаблонов «превосходят» потребности сравнительного моделирования в том смысле, что они позволяют обнаруживать последовательности настолько отдаленные, что построение надежных сравнительных моделей для них невозможно. Причина этого в том, что установление взаимосвязей между последовательностями часто основано на коротких консервативных сегментах, в то время как для успешного выполнения сравнительного моделирования требуется общее корректное выравнивание всей моделируемой части белка. В этом заключается важное различие между распознаванием укладки и сравнительным моделированием: оба метода основаны на использовании шаблонов и нацелены на создание описания пространственной структуры мишени, однако целью методов распознавания укладки является определение общей пространственной формы последовательности мишени или по крайней мере класса форм, к которому мишень принадлежит, тогда как сравнительное моделирование нацелено на создание полноатомной модели последовательности мишени.
3.2.2. Отбор шаблонов
После того, как с помощью алгоритмов поиска сформирован список шаблонов, необходимо отобрать один или несколько шаблонов, подходящих для решения конкретной задачи моделирования. При отборе шаблонов необходимо учитывать ряд факторов.
3.2. Этапы сравнительного моделирования структуры белков
93
3.2.2.1. Общие соображения при отборе шаблона
Наиболее простое правило отбора шаблона - выбрать структуру, сходство последовательности которой с исследуемой последовательностью максимально. Семейство белков, к которому принадлежат мишень и шаблоны, может включать несколько подсемейств. Построение множественного выравнивания и филогенетического дерева (Felsenstein 1981) может помочь при отборе шаблона из подсемейства, наиболее близкого к последовательности мишени. Также следует принять во внимание наличие сходства между «окружением» шаблона и окружением, в котором будет осуществляться моделирование мишени. Термин «окружение» в данном случае используется в широком смысле и подразумевает все, что не является собственно белком (например, растворитель, pH, лиганды, взаимодействия на уровне четвертичной структуры). Если это возможно, в общем случае следует использовать шаблон, связанный с теми же или близкими лигандами, что и исследуемая последовательность. Качество структур, определенных экспериментально, - еще один важный фактор при отборе шаблонов. Разрешение и фактор достоверности в случае кристаллической структуры и количество ограничений на остаток в случае ЯМР-структуры являются показателями их точности. Так, если два шаблона характеризуются близким уровнем сходства последовательности с мишенью, в общем случае следует использовать шаблон, определенный с более высоким разрешением. Критерии отбора шаблонов зависят также от цели, с которой создается сравнительная модель. Например, в случае построения модели «белок-лиганд» при отборе наличие в шаблоне близкого лиганда, вероятно, важнее, чем разрешение.
3.2.2.2. Преимущества использования нескольких шаблонов
Необязательно выбирать для моделирования единственный шаблон. На самом деле, оптимальное использование нескольких шаблонов увеличивает точность модели (Fernandez-Fuentes et al. 2007а, b; Sanchez and Sali 1997; Venclovas and Margelevicius 2005); однако не все программы моделирования поддерживают возможность использования нескольких шаблонов. Преимущество сочетания нескольких шаблонных структур может быть двойным. Во-первых, структуры шаблонов, имея небольшое перекрывание между собой, могут быть выровнены с различными доменами мишени, и в таком случае с помощью процедуры моделирования можно построить гомологичную модель всей последовательности мишени. Во-вторых, структуры шаблонов можно выравнивать с одной и той же частью мишени, а модель строить с использованием того шаблона, который является лучшим для конкретной части исследуемого белка.
94
Глава 3. Сравнительное моделирование структуры белков
Более совершенный способ отбора подходящих шаблонов предполагает создание и оценку моделей для каждого потенциального шаблона и/или их сочетаний. Оптимизированные полноатомные модели можно затем оценить с помощью энергетической или оценочной функций, таких как стандартизованная оценка PROSA (Sippl 1995) или VER1FY3D (Eisenberg et al. 1997). Эти способы оценки обычно довольно точны и позволяют отобрать из числа созданных моделей наиболее адекватные (Wu etal. 2000). Такой метод проб и ошибок можно рассматривать как ограниченное протягивание (т.е. последовательность мишени продевается через близкие структуры шаблонов). Однако эти подходы хороши только при отборе различных шаблонов на глобальном уровне.
В недавно разработанном методе множественного картирования с различными шаблонами М4Т (от «Multiple Mapping Method with Multiple Templates») отбор и сочетание многочисленных структур шаблонов осуществляется посредством многократной кластеризации, при которой учитываются «уникальный» вклад каждого шаблона, сходство их последовательностей между собой и с последовательностью мишени, а также экспериментальное разрешение (Fernandez-Fuentes etal. 2007а, b). Полученные в итоге модели по качеству систематически превосходят модели, при создании которых используется единственный лучший шаблон.
Еще одно важное наблюдение, полученное в ходе обсуждаемых исследований, заключается в том, что если идентичность последовательностей составляет менее 40%, то модели, построенные с использованием нескольких шаблонов, точнее, чем построенные с использованием единственного шаблона. Эта закономерность становится все более выраженной по мере отдаления рассматриваемых пар «мишень-шаблон». В то же время, преимущество использования нескольких шаблонов постепенно исчезает при идентичности последовательностей мишени и шаблона 40% и выше (рис. 3.1). Это свидетельствует о том, что в указанном диапазоне средние различия между структурами шаблона и мишени меньше, чем средние различия между структурами разных шаблонов, характеризующихся высокой степенью сходства с мишенью (Fernandez-Fuentes et al. 2007b).
3.2.3. Выравнивание последовательности со структурой
При построении модели во всех программах сравнительного моделирования используется принятый в качестве допущения список структурных соответствий между остатками мишени и шаблона. Этот список составляется при выравнивании последовательностей мишени и шаблонов.
3.2. Этапы сравнительного моделирования структуры белков
95
Идентичность последовательностей мишени и шаблона (%)
Рис. 3.1. Сравнение точности (ось у) моделей, построенных для одного и того же набора из 765 последовательностей белков-мишеней.
Построение осуществлялось с использованием одного шаблона (показан только результат с лучшим значением математического ожидания, столбцы темно-серого цвета) или нескольких шаблонов (столбцы светло-серого цвета). Степень идентичности последовательностей (ось х) рассчитывалась для результата с наибольшим значением математического ожидания и последовательностью исследуемого белка. Планками погрешности показана стандартная средняя ошибка
Такое выравнивание осуществляется во многих методах поиска шаблонов, иногда оно может непосредственно использоваться при моделировании в качестве входных данных. Тем не менее, часто, в особенности в сложных случаях, это начальное выравнивание не является лучшим, например, при идентичности последовательностей менее 30% (где идентичность последовательностей определяется как число идентичных положений в выравнивании, нормированном на длину последовательности мишени). Методы поиска обычно настроены на определение отдаленных связей, которое на практике часто осуществляется посредством сравнения локальных мотивов, а не полноразмерного оптимального выравнивания. Таким образом, после того, как шаблоны отобраны, метод следует использовать для выравнивания шаблонов относительно последовательности мишени. Выравнивание относительно легко получить в тех случаях, когда идентичность последовательностей мишени и шаблона составляет 40% и выше. Если
96
Глава 3. Сравнительное моделирование структуры белков
идентичность последовательностей менее 40%, точность выравнивания становится главным фактором, определяющим качество полученной модели. Ошибочное выравнивание лишь одного остатка приводит к ошибке в модели, величина которой составляет около 4 А.
3.2.3.1. Использование структурной информации при выравнивании
Выравнивания в сравнительном моделировании составляют уникальную группу, поскольку с одной стороны в них всегда находится пространственная структура - шаблон. Следовательно, качество выравнивания можно улучшить, если учесть информацию о структуре шаблона. Так, следует избегать вставок в элементах вторичной структуры, в заглубленных областях или между остатками, которые располагаются далеко друг от друга в пространстве. Такие критерии учитываются в некоторых методах выравнивания (Blake and Cohen 2001; Jennings et al. 2001; Shi et al. 2001).
В тех случаях, когда в наличии имеется несколько структурных шаблонов, можно сначала наложить их друг на друга для получения множественного структурного выравнивания, которое позволит выявить остатки, консервативные в структурном отношении (Al Lazikani etal. 2001; Petrey etal. 2003; Reddy etal. 2001). На следующем этапе осуществляется выравнивание последовательности мишени относительно имеющегося множественного структурного выравнивания. Преимущества использования нескольких структур и нескольких последовательностей заключаются в получении структурной и эволюционной информации о шаблонах, а также эволюционной информации о последовательности мишени. Использование такой информации часто приводит к том, что для моделирования удается получить выравнивание, качество которого выше по сравнению с аналогичными результатами методов парного выравнивания последовательностей (Jaroszewski et al. 2000; Sauder et al. 2000).
В методе множественного картирования МММ (от Multiple Mapping Method) прямо используется информация о пространственной структуре (Rai and Fiser 2006; Rai et al. 2006). Минимизация ошибок выравнивания в МММ осуществляется за счет отбора и оптимального склеивания фрагментов, для выравнивания которых применялись различные методы. Фрагменты составляют ряд различных выравниваний, использующихся в качестве входных данных. Критерием отбора фрагментов служит оценочная функция, на основании которой определяется предпочтительное положение фрагмента последовательности мишени в структурном окружении шаблона. В оценочную функцию входит четыре члена, которые используются для оценки совместимости альтернативных переменных сегмен
3.2. Этапы сравнительного моделирования структуры белков
97
тов белкового окружения: а) специфичные к окружению матрицы замены FUGUE (Shi etal. 2001); б) матрица аминокислотных замен BLOSSUM (S. Henikoff and Henikoff 1992); в) специальная матрица замен НЗР2, которая предназначена для оценки степени совпадения предсказанной вторичной структуры последовательности мишени с наблюдаемой вторичной структурой и типа доступности остатков шаблона (Luthy etal. 1991); г) полученный на основе статистических данных потенциал парных взаимодействий остатков (Rykunov and Fiser 2007). По сути, в МММ выполняется ограниченное обратное протягивание коротких фрагментов: задачей при осуществлении протягивания является не определение правильного способа укладки, а выбор из множества доступных картированных альтернатив выравнивания для сегментов последовательности, которые протянуты через одну и ту же укладку. Такие локальные картирования определяются для остальной части модели, при этом выравнивания являются основой последовательного решения и задают границы оценки.
3.2.4. Построение модели
При обсуждении этапа построения модели в сравнительном структурном моделировании полезно выделить две части: моделирование с использование шаблона и моделирование без использования шаблона. Такое разделение необходимо, поскольку определенные участки мишени должны строиться без использования шаблонов. К таким участкам относятся разрывы в последовательности шаблона при выравнивании мишени по шаблону. Моделирование этих областей обычно называют проблемой моделирования петель. Очевидно, что эти петли отвечают за большинство характерных отличий мишени от шаблона, и поэтому именно они определяют структурные, а следовательно, и функциональные различия. В отличие от петель, оставшаяся часть мишени, в частности ядро с консервативной упаковкой, строится с использованием структурной информации о шаблоне. Сначала мы рассмотрим некоторые основные подходы из этой последней части - моделирования с использованием шаблона. Этот способ также является первым логичным шагом при построении модели, поскольку моделирование с использованием шаблона обеспечивает структурный каркас для любого последующего моделирования петель.
З.2.4.1. Моделирование с использованием шаблона
Моделирование посредством сборки твердых тел
Первый в сравнительном моделировании и все еще широко использующийся подход состоит в том, что модель создается на основе неболь-
98
Глава 3. Сравнительное моделирование структуры белков
шого числа твердых тел, которые получают при выравнивании с шаблонами белковых структур (Blundell et al. 1987; Browne et al. 1969; Greer 1990). Такой подход основан на естественном разбиении белковой структуры на консервативные области ядра, соединяющие их петли с высокой изменчивостью и боковые цепи, обрамляющие основную цепь белка (Topham et al. 1993). Подход используется в широко распространенной программе COMPOSER (Sutcliffe etal. 1987). Точность модели можно несколько повысить, если использовать более одного структурного шаблона для построения каркаса, а также если усреднить шаблоны с учетом их весов, определенных на основе сходства их последовательностей с последовательностью мишени (Srinivasan and Blundell 1993).
Моделирование посредством сопоставления сегментов или реконструирования координат
Большинство существующих гексапептидных сегментов белковых структур с помощью процедуры кластеризации можно объединить лишь в 100 структурно различных классов (Unger et al. 1989). Это обстоятельство составляет теоретическую основу метода преобразования координат. Используя информацию о положении атомов в структурах шаблонов, определяя и собирая короткие полноатомные сегменты, можно построить структурные модели исследуемых белков. Положения атомов, которые обычно используются в этом методе, соответствуют положениям Са-атомов сегментов, которые при выравнивании структуры шаблона и последовательности мишени являются консервативными. Такие полноатомные сегменты можно получить либо в ходе сканирования всех известных белковых структур, в том числе, не связанных с моделируемой последовательностью (Claessens et al. 1989; Holm and Sander 1991), либо в ходе конформационного поиска, который ограничен энергетической функцией (Bruccoleri and Karplus 1990; van Gelder et al. 1994). Так, в общем методе моделирования на основе сопоставления сегментов (SEGMOD) (Levitt 1992) положения некоторых атомов (обычно Са-атомов) используются для поиска по репрезентативной базе данных всех известных белковых структур. С помощью этого метода можно моделировать главную цепь, боковые цепи и разрывы. Некоторые методы моделирования боковых цепей (Chinea et al. 1995), а также группу методов моделирования петель, основу которых составляет обнаружение подходящих фрагментов в базах данных известных структур, (T.A.Jones and Thirup 1986) можно рассматривать как методы сопоставления сегментов и реконструирования координат.
3.2. Этапы сравнительного моделирования структуры белков
99
Моделирование посредством удовлетворения пространственным ограничениям
В методах этой группы на начальном этапе для последовательности мишени устанавливается множество структурных ограничений. Для этого используется выравнивание мишени с родственными белками. По концепции процедура близка методу определения белковых структур на основе ограничений, установленных в экспериментах ЯМР. Ограничения обычно устанавливают, используя допущение, согласно которому соответствующие расстояния между остатками структур мишени и шаблона в процессе выравнивания одинаковы. Эти основанные на гомологии ограничения обычно дополняются стереохимическими ограничениями - длин связей, углов связей, двугранных углов, а также невалентных атомных контактов - которые устанавливаются с использованием молекулярно-механических силовых полей (Brooks etal. 1983). Затем в процессе минимизации нарушений всех ограничений строится модель. Минимизировать нарушения можно либо средствами метрической геометрии, либо путем оптимизации в вещественном пространстве. Так, изящным методом метрической геометрии является построение полноатомных моделей с использованием нижних и верхних границ для расстояний и двугранных углов (Havel and Snow 1991). Предпринимались дальнейшие попытки применения метрической геометрии в сравнительном моделировании, например, (Aszodi and Taylor 1996), однако в обсуждаемой области исследований преобладают более успешные, хотя и более консервативные подходы к моделированию в вещественном пространстве. Возможно, это связано с тем, что эволюция также оказалась весьма консервативна в сохранении структурных особенностей различных белков (Kihara and Skolnick 2003).
Сравнительное моделирование на основе критерия удовлетворения пространственным ограничениям применяется в компьютерной программе MODELLER (Fiser and Sali 2003a; Sali and Blundell 1993), которая в настоящее время является самой популярной программой компьютерного моделирования. На первом этапе построения модели на расстояния и двугранные углы последовательности мишени налагаются ограничения, полученные в ходе выравнивания мишени относительно пространственных структур шаблонов. Характер этих ограничений определяется с помощью статистического анализа связей между близкими белковыми структурами. В ходе сканирования базы данных выравниваний создаются таблицы, количественно отражающие различные взаимосвязи между белками, такие как корреляции между двумя эквивалентными расстояниями Са-Са или двугранными углами главной цепи двух близких белков (Sali and Blundell 1993). Эти взаимосвязи представляются затем в виде условных функций плотности вероятно
100
Глава 3. Сравнительное моделирование структуры белков
сти и могут использоваться непосредственно в качестве пространственных ограничений. Например, вероятность различных значений двугранных углов основной цепи рассчитывается с учетом типа рассматриваемого остатка, конформации главной цепи и соответствующего остатка шаблона и степени сходства последовательностей двух белков. Важная особенность метода состоит в том, что форма пространственных ограничений определяется эмпирически на основе информации из баз данных выравниваний белковых структур и не содержит субъективных пользовательских допущений. Наконец, в результате оптимизации целевой функции в пространстве декартовых координат создается модель. Оптимизация выполняется с использованием метода переменной целевой функции (Braun and Go 1985), в котором, в свою очередь, применяются методы сопряженного градиента и молекулярной динамики с имитацией отжига (Clore et al. 1986).
Сходные принципы заложены и в программный пакет NEST, с помощью которого можно строить модели по гомологии на основе одиночного выравнивания последовательности по шаблону или с использованием нескольких шаблонов. Метод также позволяет рассматривать различные структуры для различных участков мишени (Petrey et al. 2003).
Сочетание выравниваний и сочетание структур
Часто бывает сложно выбрать лучшие шаблоны или определить хорошее выравнивание. В таких случаях улучшить качество сравнительной модели можно за счет многократного повторения процесса отбора шаблонов, выравнивания и построения модели с использованием различных методов оценки моделей. Такое повторение можно продолжать до тех пор, пока не перестанет наблюдаться улучшение качества модели (Fiser etal. 2003; Guenther etal. 1997). Совсем недавно такие бессистемные подходы, требующие выполнения вручную, были автоматизированы (Petrey etal. 2003). Так, был представлен автоматизированный метод, в котором оптимизируются как выравнивание, так и предполагаемая модель (John and Sali 2003). Эта задача решается с применением протокола генетического алгоритма. На начальном этапе предполагается серия первичных выравниваний, за ними следуют многократные повторные выравнивания, построение модели и ее оценка. Целью является улучшение окончательной оценки модели. В ходе повторяющегося процесса с использованием нескольких процедур, таких как мутации и скрещивания выравниваний, создаются новые выравнивания; строятся соответствующие этим новым выравниваниям сравнительные модели, которые затем оцениваются по различным критериям, зависящим отчасти от атомного статистического потенциала. В другом методе генетический алгоритм применялся к шаблонам и выравниваниям, созданным автоматически. Для оценки полученных пробных
3.2. Этапы сравнительного моделирования структуры белков
101
сочетаний использовалась относительно простая оценочная функция, зависевшая от структуры. Несмотря на некоторые ограничения, показано, что метод устойчив к ошибкам выравнивания и успешно упрощает задачу отбора шаблонов (Contreras-Moreira et al. 2003).
Еще одной попыткой оптимизировать процедуру выравнивания мишени по шаблону является сервер Roberta, где выравнивания создаются с помощью динамического программирования при использовании оценочной функции, в которой сочетается различная информация о белковых свойствах, в том числе последние данные о том, насколько важна та или иная область последовательности для укладки белка. В ходе систематических изменений вклада различных свойств белка в оценку выравнивания создаются очень большие ансамбли различных выравниваний. Для отбора лучших моделей из ансамбля применялись разнообразные подходы, в том числе сочетание выравниваний, степень гидрофобной заглубленности, энергетические функции низкого и высокого разрешения и сочетание этих методов (Chivian and Baker 2006).
В рамках описанных метасерверных подходов осуществляются не только оценка и классификация различных моделей, полученных с помощью множества методов, но также дальнейшая их комбинация. Эти подходы можно также рассматривать в качестве методов исследования выравнивания и конформационного пространства данной последовательности мишени (Kolinski and Bujnicki 2005).
Еще одна альтернатива сложным серверам - программа М4Т. Для М4Т характерно наличие специфической внутренней оценочной функции, которая учитывает главным образом свойства структурного окружения шаблонов. С помощью этой функции автоматически определяются лучшие шаблоны, исследуются и оптимальным образом сращиваются альтернативные выравнивания (Fernandez-Fuentes et al. 2007b).
Метасерверы
В последнее время были разработаны метасерверные подходы, в которых используется множество существующих программ. В метасерверах осуществляется отбор моделей, созданных с помощью других методов, затем модели либо используются в качестве входных данных для создания новых моделей, либо рассматриваются в ходе поиска консенсусных решений. Например, в FAMS-ACE (Terashi et al. 2007) входные данные других серверов используются как стартовые точки для улучшения и перестройки, после чего для отбора наиболее точных решений используется Verify3D (Eisenberg et al. 1997). Еще одним консенсусным подходом является PCONS -метод на основе нейронной сети, в котором консенсусная модель создается
102
Глава 3. Сравнительное моделирование структуры белков
путем сочетания информации о точности и структурных сходствах моделей, полученных другими методами (Wallner et al. 2007). В 3D-JURY используется та же идея; основу отбора составляет согласованность сходства модельных структур (Ginalski et al. 2003).
3.2.4.2. Моделирование без использования шаблона: моделирование петель и вставок
В сравнительном моделировании последовательности мишени часто содержат остатки-вставки, которых нет в структурах шаблонов, или области, которые структурно отличаются от соответствующих областей шаблонов. Таким образом, из структур шаблонов нельзя получить информацию об этих сегментах-вставках. Такие области часто соответствуют петлям на поверхности. Петли обычно играют важную роль при определении функциональной специфичности данного белкового каркаса, образуя функциональные участки, такие как области, определяющие комплементарность антителам (Rudolph et al. 2006), участки связывания лигандов (например, АТФ (Saraste etal. 1990), кальция (Grabarek 2006) и НАД(Ф) (Lesk 1995)), участки связывания ДНК (Tainer et al. 1995), активные центры ферментов (например, серин-треониновых киназ (Johnson et al. 1998) или аспарагиновых протеаз (Wlodawer etal. 1989)). Точность моделирования петель -главный фактор, определяющий, насколько полезным окажется сравнительное моделирование в применении, например, к локированию лигандов или аннотированию функций (рис. 3.2). Моделирование петель можно рассматривать как миниатюрную проблему сворачивания белка, поскольку правильную конформацию данного сегмента полипептидной цепи требуется рассчитать главным образом на основе последовательности самого сегмента. Однако петли в большинстве своем имеют небольшой размер и не содержат достаточной информации о своей локальной укладке. (Исключение составляют случаи, когда в наличии имеется большое количество
Рис. 3.2. (Цветную версию рисунка см. на вклейке.)
Примеры петель (показаны желтым), которые отвечают за функциональную специфичность белковых надсемейств. Слева направо: флаводоксин, иммуноглобулин, нейроамидаза из белковых семейств с укладкой а+р бочонка, иммуноглобулина и антипараллельного Р-бочонка соответственно
3.2. Этапы сравнительного моделирования структуры белков
103
фрагментов известной конформации, соответствующих петлям по последовательности.) С другой стороны, окружение каждой петли уникальным образом определяется растворителем и белком, который обрамляет петлю. Для нескольких редких случаев показано, что даже идентичные по последовательности декапептиды в различных белках могут иметь различную конформацию (Fernandez-Fuentes and Fiser 2006; Mezei 1998).
Существует две основные группы методов моделирования петель: 1) подходы на основе поиска по базам данных, в рамках которых осуществляется поиск сегмента, соответствующего якорной области ядра, в базе данных всех известных белковых структур (Chothia and Lesk 1987; Jones and Thirup 1986); 2) подходы на основе конформационного поиска (Bruccoleri and Karplus 1987; Moult and James 1986; Shenkin et al. 1987). Существуют также методы, которые сочетают в себе названные подходы (de Bakker et al. 2003; Deane and Blundell 2001; van Vlijmen and Karplus 1997).
Моделирование петель, основанное на фрагментах
Моделирование на основе сканирования базы данных и поиска фрагментов является точным и эффективным методом моделирования петель, если используемая база данных содержит специфические петли того же класса, что и моделируемая петля. Так, применение метода эффективно при моделировании 0-шпилек (Sibanda etal. 1989) и петель специфических типов укладки, например, гипервариабельных областей в укладке иммуноглобулина (Chothia etal. 1989). Ранее представлялось маловероятным, что область исследований, связанная с созданием банков данных структур, когда-либо достигнет такого уровня развития, при котором подходы на основе фрагментов станут эффективным способом моделирования петель (Fidelis et al. 1994). Это привело к тому, что, начиная с 2000-ых годов, стремительно развивались методы конформационного поиска. Тем не менее, в последнее десятилетие были успешно исследованы многие уголки вселенной белковых структур, в связи с чем было экспериментально определено большое количество новых способов укладки, что в свою очередь оказало существенное влияние на количество известных структурных фрагментов. Последние исследования показали, что петлевые фрагменты не только полно представлены в современных банках данных структур - их более короткие сегменты, вероятно, уже полностью изучены (Du et al. 2003).
Сообщалось, что сегменты последовательностей размером до десяти остатков имеют близкий (т.е., не менее чем на 50% идентичный) сегмент известной конформации в базе данных PDB. Несмотря на шестикратное увеличение количества последовательностей в банках данных и удвоение PDB с 2002 года, не было обнаружено ни одного сегмента последователь
104
Глава 3. Сравнительное моделирование структуры белков
ности, который имел бы менее чем 50%-ную идентичность с уже известными последовательностями, а в PDB не появилось ни одной уникальной конформации петли. Это свидетельствует о том, что в области секвенирования белков продолжает циркулировать ряд уже известных коротких структурных сегментов. Все сегменты последовательностей размером 10-12 остатков имеют по крайней мере один соответствующий структурный сегмент, который имеет по меньшей мере 50% идентичности, что подтверждает структурное сходство. Исключение составляет очень небольшая группа сегментов, упомянутых выше (Fernandez-Fuentes and Fiser 2006). Как следствие, в последнее время были предприняты новые попытки классифицировать конформации петель с использованием более общих категорий, тем самым распространяя применимость подхода поиска по базам данных на более широкий круг случаев (Fernandez-Fuentes etal. 2006а; Michalsky et al. 2003). В одной из последних работ описаны преимущества использования профилей последовательностей СММ при классификации и предсказании петель (Espadaler et al. 2004). В другой недавно опубликованной работе для интересующей исследователя петли сначала делается предсказание конформации на основе последовательности, после чего осуществляются структурные выравнивания предсказанных структурных фрагментов относительно небольшого количества структурных шаблонов петель. Эти выравнивания последовательности петли относительно шаблонов затем количественно оцениваются с использованием модели искусственной нейронной сети, обученной на наборе предсказаний с известными результатами (Peng and Yang 2007).
ArchPred является, возможно, наиболее точным методом моделирования петель на основе баз данных. Подход коротко описан в работе (Fernandez-Fuentes et al. 2006а, b). В ArchPred используется иерархическая многомерная база данных, которая была создана для классификации около 300000 фрагментов петель и элементов вторичной структуры, располагающихся по флангах петель. Помимо длины петель и типов окружающих петли вторичных структур, база данных содержит четыре внутренние координаты - расстояние и три типа углов, - которые описывают геометрию соединительных областей (Oliva etal. 1997). В ходе поиска отбор фрагментов из библиотеки осуществляется на основе соответствия длины, типов окружающих вторичных структур и удовлетворения геометрическим ограничениям для соединений, после чего фрагменты встраиваются в каркас изучаемого белка, а затем степень их соответствия оценивается по СКО соединительных областей и числу наталкиваний твердых тел на окружающие атомы белка. На последней стадии отобранные петли оцениваются с использованием стандартизованной оценки, которая содержит ин
3.2. Этапы сравнительного моделирования структуры белков
105
формацию о подобии последовательностей и вероятных значениях предсказанных и наблюдаемых двугранных углов <рА|/ главной цепи белка. Для каждой длины петли установлены границы достоверности стандартизованной оценки, определяющие те фрагменты, предсказание которых является более качественным, чем полученное с помощью метода ab initio. Программной реализацией метода является веб-сервер, который как и производит расчеты, так и выполняет регулярное обновление библиотеки фрагментов. Предсказанные сегменты возвращаются в виде выходных данных или, по желанию, могут быть дополнены боковыми цепями, а затем подвергнуты отжигу в окружении исследуемого белка с использованием минимизации методом сопряженных градиентов.
Таким образом, последние сообщения о том, что конформации петель теперь более полно представлены в PDB, свидетельствуют, что методы на основе баз данных ограничиваются способностью распознавать соответствующие фрагменты, и причина этого кроется не в отсутствии сегментов, как полагали ранее.
Моделирование петель ab initio
Для преодоления ограничений, которые характерны для методов поиска по базам данных, были разработаны методы конформационного поиска. Существует множество таких методов, в которых используются различные способы представления белков, термы целевой функции и алгоритмы оптимизации или количественного учета. Стратегии поиска включают метод минимальных возмущений (Fine et al. 1986), моделирование методом молекулярной динамики (Bruccoleri and Karplus 1987), генетические алгоритмы (Ring and Cohen 1993), методы Монте-Карло и имитации отжига (Abagyan and Totrov 1994; Collura et al. 1993), одновременный поиск множественных копий (Zheng etal. 1993), оптимизация в самосогласованном поле (Koehl and Delarue 1995) и количественный учет на основе теории графов (Samudrala and Moult 1998). Предсказание петель с помощью оптимизации можно применять как при одновременном моделировании нескольких петель, так и для петель, взаимодействующих с лигандами - ни одна из этих задач не имела простого решения в методах поиска по базам данных, где собраны фрагменты из несвязанных структур в различных окружениях.
В модуле MODLOOP программы MODELLER применяется подход на основе оптимизации (Fiser et al. 2000; Fiser and Sali 2003b). Оптимизация петель в MODLOOP основана на сопряженных градиентах и молекулярной динамике с имитацией отжига. Псевдофункция энергии содержит множество термов, в том числе некоторые термы из молекулярно-механического силового поля CHARMM-22 (Brooks etal. 1983) и пространственные ограни
106
Глава 3. Сравнительное моделирование структуры белков
чения, в основу которых положения распределения расстояний (Melo and Feytmans 1997; Sippl 1990) и двугранных углов в известных белковых структурах. Для исследования проблем сравнительного моделирования процедура моделирования петель была оптимизирована с последующей оценкой качества. Использовалось большое количество петель известной структуры как в природном, так и в лишь приблизительно верном окружениях. Производительность метода позднее была улучшена за счет использования молекулярно-механического силового поля CHARMM и обобщенного потенциала сольватации Борна (Fiber et al. 2002). Включение потенциалов сольватации в оценочную функцию было ключевым вопросом нескольких работ, появившихся позже (Das and Meirovitch 2003; de Bakker et al. 2003; DePristo etal. 2003; Forrest and Woolf 2003). Повышение точности предсказания петель стало результатом включения в оценочную функцию потенциала, близкого к энтропии, «энергии колонии», который был разработан при сравнении геометрий и кластеризации отобранных конформаций петель (Fogolari and Tosatto 2005; Xiang et al. 2002). Постоянное совершенствование оценочных функций способствует улучшению качества методов моделирования петель. Недавно были представлены две процедуры моделирования петель, использующие эффективные статистические парные потенциалы, которые кодируются в DFIRE (Soto et al. 2008; Zhang et al. 2004). Еще один метод разработан для предсказания петель очень большой длины и использует подход ROSETTA, в котором по сути выполняется мини-упаковка сегментов петель (Rohl et al. 2004). В программе Prime выполняется процедура создания большого количества петель на основе двугранных углов, после чего следуют повторяющиеся циклы кластеризации, оптимизация боковых цепей и полная минимизация энергии отобранных структур петель с использованием полноатомного молекулярно-механического силового поля (OPLS) с неявно заданным растворителем (Jacobson et al. 2004).
3.2.4.3. Уточнение моделей
Сравнительные модели строятся с использованием лучшего из доступных наборов ограничений. Такой набор обычно представляет собой сочетание ограничений на расстояния и углы, определенных на основе различных структурных шаблонов, потенциалов молекулярно-механического силового поля и ограничений, налагаемые различными статистическими потенциальными функциями. Из-за большого количества доступных ограничений проблему можно назвать переопределенной. Этап построения модели является относительно простым и в первую очередь направлен на решение конфликта ограничений. В случае программы MODELLER эта цель достигается за счет сочетания минимизации методом сопряженных гради
3.2. Этапы сравнительного моделирования структуры белков
107
ентов и молекулярной динамики. Для создания модели обычно требуется несколько минут. Из-за преобладания ограничений на основе шаблонов часто сложно создать модель, на уровне основной цепи белка больше похожую на мишень, чем на фактический шаблон (если принято допущение о том, что выравнивание не содержит ошибок). Сложной задачей является также дальнейшее уточнение модели, поскольку наиболее точные ограничения и потенциалы силовых полей уже были использованы при построении модели. По сути, возникает та же проблема, что и при моделировании ab initio, так как любые уточнения на этом этапе должны осуществляться .без использования шаблона. Различные исследования и последние обзоры показывают, что большинство усовершенствований снижает точность моделей (Summa and Levitt 2007). Существовала лишь одна молекулярномеханическая энергетическая функция, которая улучшала исходную модель, однако улучшения были очень незначительными; с помощью статистических потенциалов также удавалось достичь очень небольшого повышения производительности.
В других перспективных методах уточнения моделей предпринимаются попытки разумно ограничить пространство конформационного поиска вокруг высококачественной исходной модели. Этого можно достичь путем простого определения максимального отклонения, которое допускается для смещений основной белковой цепи при отборе моделей (Kolinski etal. 2001). В последнее время появился еще один перспективный подход, в рамках которого определяется подпространство эволюционной и колебательной гармоник. Это сокращенное подпространство, которое состоит из сочетания эволюционно предпочтительных направлений, определяемых главными компонентами структурных вариаций в гомологичном семействе, и топологически предпочтительных направлений, полученных из анализа низкочастотных нормальных мод колебательной динамики - до 50 измерений. Такое подпространство является достаточно точным - настолько, что ядра большинства белков можно представить с точностью 1 А, и достаточно сокращенным - настолько, что можно применять эффективные методы оптимизации, например, метод моделирования Монте-Карло с обменом репликами (Han et al. 2008; Qian et al. 2004).
3.2.4.4. Моделирование белков и комплексов с дополнительными, экспериментальными, ограничениями
В некоторых методах сравнительного моделирования могут использоваться ограничения, полученные не на основе структуры гомологичного шаблона, а из других источников. Так, основу ограничений могут составлять
108
Глава 3. Сравнительное моделирование структуры белков
правила упаковки вторичной структуры (Cohen etal. 1989), данные анализа гидрофобности (Aszodi and Taylor 1994) и коррелированных мутаций (Taylor and Hatrick 1994), эмпирические потенциалы средней силы (Sippl 1995), эксперименты с использованием ядерного магнитного резонанса (Sutcliffe et al. 1992) или эксперименты по химическому перекрестному сшиванию, спиновому и фотоаффинному мечению (Отт etal. 1998), водородно-дейтериевый обмен в сочетании с масс-спектрометрией (Xiao et al. 2006), отслеживание гидроксильных радикалов (Kiselar et al. 2003), флуоресцентная спектроскопия, реконструкция изображений в электронной микроскопии (Topf etal. 2008), сайт-специфический мутагенез (Boissel et al. 1993) и т.д. Таким образом, сравнительную модель, особенно в сложных случаях, можно улучшить, добившись согласованности модели с доступными экспериментальными данными и более общими представлениями о структуре белка.
В прошлом основу сравнительного моделирования составляли сведения о шаблоне и статистические ограничения, разработанные на основе информации об известных белковых структурах и последовательностях. Однако ожидается, что с развитием крупномасштабных генетических методов и методов протеомики будет расти количество экспериментально установленных ограничений, доступных для автоматического включения в процесс моделирования. Помимо создания более точных моделей, это значительно поспособствует моделированию белковых комплексов и ансамблей.
Системный подход к моделированию больших белковых комплексов с помощью экспериментальных ограничений был разработан при моделировании комплекса ядерной поры - самого большого из известных комплексных белков клетки, который состоит из 456 белков (Alber et al. 2008). В подходе используется различная экспериментальная информация. Например, стехиометрия была определена с помощью количественного иммуноблоттинга; с помощью гидродинамических экспериментов удалось получить информацию о форме и исключенном объеме каждого нуклеопорина; иммунная электронная микроскопия (ЭМ) способствовала грубому определению локализации нуклеопоринов; на основе аффинной очистки был определен состав комплексов; анализ крио-ЭМ и биоинформатики выявил локализацию трансмембранных сегментов; в экспериментах с использованием наложения была получена информация о прямых парных взаимодействиях. Все эти входные данные были интегрированы в иерархический процесс, который объединял сравнительное моделирование, протягивание, методы жесткого и подвижного докинга. Конечной целью интеграции данных является преобразование всей доступной экспериментальной информации в пространственные ограничения, которые смогут направлять обобщенную процедуру моделирования. Процесс является гибким, что позволяет соче
3.2. Этапы сравнительного моделирования структуры белков
109
тать различные формы представлений, уровни разрешения (например, атомы, атомистические модели белков, элементы симметрии или целые ансамбли) и процедуры оптимизации (Alber et al. 2007а, b, 2008). В этом и близких к этому методах будут совместно использоваться результаты экспериментов по секвенированию генома, данные функциональной геномики, протеомики, системной биологии и структурной биологии.
3.2.5. Оценка моделей
После того, как модель построена, важно проверить ее на наличие возможных ошибок. Качество модели можно приблизительно оценить по степени сходства мишени и шаблона. Идентичность последовательностей выше 30% позволяет дать относительно хороший прогноз ожидаемой точности модели. Если идентичность последовательностей опускается ниже 30%, то этот критерий становится значительно менее надежным при оценке ожидаемой точности одиночной модели. Именно в таких случаях наиболее информативны методы оценки моделей.
Можно провести оценку двух типов. При «внутренней» оценке самосо-гласованности проверяется, удовлетворяет ли модель ограничениям, которые использовались для ее расчета, в том числе ограничениям, основанным на структуре шаблона или статистических наблюдениях. «Внешняя» оценка основана на информации, которая не используется при расчете модели.
Оценка стереохимии модели (например, длин связей, величин валентных и двугранных углов и расстояний несвязанных атомов) с помощью таких программ, как PROCHECK (Laskowski etal. 1993) и WHATCHECK (Hooft etal. 1996), - это пример внутренней оценки. Ошибки в стереохимии редки и менее информативны, чем ошибки, определяемые методами внешней оценки, однако кластер химических ошибок может свидетельствовать о том, что соответствующая область также содержит другие существенные ошибки (например, ошибки выравнивания).
С помощью внешней оценки можно как минимум ответить на вопрос, верный ли шаблон был использован при моделировании. К счастью, неверный шаблон можно легко определить с помощью доступных в настоящее время оценочных функций. Более сложная для оценочных функций задача состоит в предсказании ненадежных областей модели. Один из путей решения этой проблемы заключается в том, чтобы рассчитать «псевдоэнерге-тический» профиль модели, такой, например, как в методах PROSA (Sippl 1993) или Verify3D (Eisenberg etal. 1997). Профиль отражает энергию для каждого положения модели (рис. 3.3). Пики профиля часто соответствуют ошибкам модели. Существует несколько подводных камней использования
но
Глава 3. Сравнительное моделирование структуры белков
Рис. 3.3. (Цветную версию рисунка см. на вклейке.) Зависимость энергии остатков от положения остатков в последовательности для двух различных моделей одного и того же белка. Энергия рассчитана с использованием статистического потенциала парных взаимодействий. Отрицательные (синего цвета) и положительные (красного цвета) значения энергии указывают на энергетически благоприятное и неблагоприятное окружение остатков соответственно. Профили энергии соответствуют моделям, представленным справа. Менее точная модель располагается над более точной. Отдельные участки моделей показаны тем же цветом, что и соответствующие им профили энергии. Фактическая экспериментальная структура показана серым
энергетических профилей для определения локальных ошибок. Так, область может быть определена как ненадежная лишь потому, что она взаимодействует с неверно смоделированной областью (Fiser et al. 2000). В других подходах, разработанных в недавнем прошлом, для оценки моделей, либо целиком (Eramian etal. 2006), либо локально (Fasnacht etal. 2007), обычно используется сочетание различных входных данных. В лучших методиках оценки качества моделей при тестировании используется простой консенсусный подход, в рамках которого надежность модели оценивается на основе того, насколько она согласуется с альтернативными моделями, которые иногда получают различными методами (Wallner and Elofsson 2005а, 2007). Оценка моделей - важная, но сложная область, поскольку одна из основных ее проблем представляет собой замкнутый круг: эффективная оценка моделей требует использования таких термов оценочной функции, которые уже применялись для создания самих точных моделей.
3.3. Эффективность методов
сравнительного моделирования
3.3.1. Точность методов
Получить информативную оценку методов моделирования структуры белков, в том числе, сравнительного моделирования, можно на проходящем каждые два года совещании CASP (от «Critical Assessment of Techniques for
3.3. Эффективность методов сравнительного моделирования
111
Protein Structure Prediction») (Moult 2005). Перед специалистами по белковому моделированию стоит задача создать модель последовательности с неизвестной пространственной структурой и предоставить эту модель организаторам до начала совещания. Одновременно пространственные структуры мишеней, отобранных для предсказания, определяются методами рентгеновской кристаллографии или ЯМР. Структуры поступают в открытый доступ лишь после того, как модели рассчитаны и предоставлены для оценки. Таким образом обеспечивается оценка bona fide методов моделирования белковых структур, хотя в процессе такой оценки сложно определить, насколько успех того или иного метода связан с особенностями программы или квалификацией специалистов, создавших модель.
Существует альтернативный крупномасштабный длительный автоматизированный метод оценки, который используется в программе EVA («EValuation of Automatic protein structure prediction») (Eyrich etal. 2001). Каждую неделю EVA предоставляет серверам, участвующим в соревновании, последовательности, которые вскоре будут размещены в PDB. После сбора и обработки результатов моделирования метод предоставляет подробную статистику о предсказании вторичной структуры, предсказании типов укладки, сравнительном моделировании и предсказании пространственных контактов. В программе LiveBench существуют уникальные методы оценки, которые используются схожим образом (Bujnicki et al. 2001).
Строгая статистическая оценка (Marti-Renom etal. 2002) слепых прогностических экспериментов показала, что точность различных методов построения моделей, в которых используются сопоставление сегментов, сборка твердых тел, критерий удовлетворения пространственным ограничениям или любые сочетания этих родходов, в случае корректного применения методов приблизительно одинакова (Dalton and Jackson 2007; Wallner and Elofsson 2005b). Это также отражает тот факт, что такие ключевые обстоятельства моделирования, как отбор шаблона и точность выравнивания, существенно влияют на общую точность, а ядро белковых структур высококонсервативно. С практической точки зрения, следует оценивать, насколько модели полезны, т.е., насколько они способствуют пониманию белковых функций. Уникальная функциональная роль связана с уникальными структурными особенностями, что чаще встречается в изменчивых петлевых областях, в отличие от консервативного ядра. Однако описания функциональных участков не только создаются вручную, но и, в растущем количестве случаев, отсутствуют или являются неполными. Это в особенности справедливо для выходных данных проектов структурной геномики, которые часто конкретно и преднамеренно направлены на исследование белков неизвестной структуры. Таким образом, крупномасштабное тестирование методов моделиро
112
Глава 3. Сравнительное моделирование структуры белков
вания на основе оценки точности функциональных аннотаций создаваемых моделей является желательным, но пока еще сложно осуществимым на практике (Chakravarty and Sanchez 2004; Chakravarty et al. 2005).
3.3.2. Ошибки в сравнительных моделях
Общая точность сравнительных моделей варьирует в широких пределах. Вблизи нижнего предела находятся модели низкого разрешения, для которых единственным надежно определенным свойством является лишь их тип укладки. У верхнего предела расположены модели, точность которых сравнима со средним разрешением кристаллографических структур (Baker and Sali 2001). При рассмотрении биологических проблем часто полезны даже модели низкого разрешения, поскольку функции можно предсказать на основе грубых структурных свойств, как показано в последующих главах этой книги.
Ошибки, встречающиеся в сравнительных моделях, можно разделить на пять категорий: 1) ошибки в упаковке боковых цепей; 2) искажения или изменения области, для которой получено правильное выравнивание относительно структуры шаблона; 3) искажения или изменения области, которая не имеет соответствующих сегментов ни в одном из структурных шаблонов; 4) искажения или изменения области, для которой получено неправильное выравнивание относительно структур шаблонов; 5) неправильная упаковка структуры, полученная в результате использования неподходящего шаблона. Для решения каждой из этих проблем необходимы существенные методологические улучшения.
Ошибки 3-5 довольно редко встречаются при моделировании последовательностей, для которых идентичность с шаблоном составляет более 40%. Так, в подобных случаях для 90% атомов главной цепи СКО при моделировании будет составлять, вероятно, около 1 A (Sanchez and Sali 1998). В этом диапазоне сходства структур проще всего построить структурное выравнивание: разрывов немного, а структурные отличия между белками обычно ограничиваются петлями и боковыми цепями. Когда идентичность последовательностей составляет 30-40%, структурные различия становятся более выраженными, пропуски в выравниваниях встречаются чаще и имеют более крупные размеры, и основными проблемами становятся неправильные выравнивания и вставки в последовательность мишени. В результате СКО главной цепи возрастает примерно до 1,5 А примерно для 80% остатков. Моделирование остальных остатков осуществляется с большими ошибками, поскольку методы в целом не способны моделировать структурные искажения и изменение твердых тел, а также преодолеть проблему
3.3. Эффективность методов сравнительного моделирования
113
использования неверных выравниваний. Когда идентичность последовательностей опускается ниже 30%, главной проблемой становится определение близких шаблонов и их выравнивание относительно моделируемой последовательности. В целом, можно ожидать, что при таком уровне сходства последовательностей примерно для 20% остатков будет создано неверное выравнивание, что приведет к неправильному моделированию с ошибкой более 3 А. Такие неверные выравнивания представляют собой серьезное препятствие для сравнительного моделирования, поскольку, как оказывается, большинство структурно связанных белковых пар имеют менее 30% идентичности последовательностей (Rost 1999).
Чтобы оценить ошибки в сравнительных моделях в перспективе, приведем различия в структурах одного и того же белка, полученных экспериментально. Точность 1 А положения атомов главной цепи характерна для рентгеновской структуры с низким разрешением - около 2,5 А - и фактором достоверности около 25% (Ohlendorf 1994), а также для структур среднего разрешения, полученных методом ЯМР с 10 ограничениями межпротонного расстояния на остаток (рис. 3.4). Аналогичным образом, различия между рентгеновскими структурами и ЯМР-структурами высокого разрешения для одного и того же белка, как правило, составляют около 1 A (Clore et al. 1993). Изменения окружающей среды (например, олигомерное состояние, кристаллическая упаковка, растворитель, лиганды) также могут оказывать существенное влияние на структуру (Faber and Matthews 1990). В целом, сравнительное моделирование последовательностей, для которых идентичность с шаблоном составляет более 40%, позволяет получать модели почти такого же качества, что и экспериментальные структуры среднего разрешения, просто потому, что белки с таким уровнем сходства с высокой вероятностью похожи друг на друга так же, как и структуры одного белка, определенные различными экспериментальными методами в разных условиях. Тем не менее, потенциальный риск при сравнительном моделировании белков состоит в том, что некоторые области, главным образом петли и боковые цепи, могут содержать более существенные ошибки.
Производительность методов сравнительного моделирования иногда может быть завышенной. Это связано с тем, что показатели, обычно обсуждаемые в литературе, - это значения отклонений основной цепи. Тем не менее, одиночные ошибки в определенных остатках, важных для выполнения функций белка, даже при общем СКО основной цепи менее 1 А, все же могут быть достаточно велики, что не позволит сделать достоверные выводы относительно механизма действия, функций белка и разработки лекарственных препаратов.
114
Глава 3. Сравнительное моделирование структуры белков
Рис. 3.4. (Цветную версию рисунка см. на вклейке.) Демонстрация точности структурных моделей, полученных различными экспериментальными и вычислительными методами для одного и того же белка-аллергена Der Р 2. а) Наложение десяти различных структур Der Р 2, полученных методом ЯМР (код PDB 1A9V); усредненное СКО = 0,97 А. б) Наложение структур, полученных методом рентгеновской кристаллографии, двух изоформ белка Der Р 2, имеющих 87% идентичности последовательности: 2F08 (разрешение 2,20 А) и 1KTJ (разрешение 2,15 А). СКО = 1,33 А. в) наложение ЯМР- и рентгеновской структур белка Der Р 2 (1A9V и 1KTJ). СКО = 2,2 А. г) Наложение сравнительной модели, построенной для белка 1NEP с использованием 1KTJ в качестве шаблона, и рентгеновской структуры 1NEP. Структуры 1NEP и 1KTJ имеют идентичнные на 28% последовательности и представляют собой типичный пример объекта, сложного для моделирования. СКО = 1.66 А. Все значения СКО относятся к наложению атомов Са
3.4. Применение сравнительного моделирования
3.4.1. Моделирование одиночных белков
Сравнительное моделирование часто является эффективным способом получения полезной информации об интересующих белках. Так, сравнительные модели могут быть полезны при разработке генетических экспериментов, таких как создание мутантов для проверки гипотез функционирования белка (Vernal etal. 2002; G Wu etal. 1999), определение активных центров и сайтов связывания (Sheng et al. 1996). Модели полезны при изучении белок-белковых взаимодействий, а также взаимодействий белков с лигандами, разработке ингибиторов, например, поиске, разработке и усовершенствовании лигандов для данного участка связывания (Ring et al. 1993), моделирования субстратной специфичности (L. Z. Xu et al. 1996), предсказания эпитопов антигенов (Sali etal. 1993), моделирования белок-белкового докинга (Vakser 1995). Модели могут выявить физико-химические особенности, которые невозможно предположить на основе одной только информации о по
3.4. Применение сравнительного моделирования
115
следовательности. Например, выводы о функции белка можно сделать на основании рассчитанных значений электростатического потенциала вокруг белка (Sali et al. 1993); примерами также являются общее уточнение и объяснение известных экспериментальных данных (Fiser et al. 2003). Модели также весьма полезны при повышении качества структур, поскольку облегчают молекулярную замену при определении структур методом рентгеновской спектроскопии (Schwarzenbacher etal. 2008), уточнении моделей, основанных на ЯМР-ограничениях (Barrientos etal. 2001), подтверждении отдаленных структурных взаимосвязей (Guenther et al. 1997; G Wu et al. 1999).
3.4.2. Сравнительное моделирование и проект исследования структуры белков
Идея проектов по исследованию генома будет реализована полностью только тогда, когда мы определим и поймем функции новых закодированных белков. Такому пониманию значительно поспособствует структурная информация обо всех или почти всех белках. Множество структурных данных будет получено с помощью структурной геномики (Burley et al. 2008; Chance etal. 2002), крупномасштабного определения белковых структур методами рентгеновской кристаллографии и спектроскопии ядерного магнитного резонанса при эффективном сочетании с точными, автоматизированными и крупномасштабными методами сравнительного моделирования белковых структур. Учитывая производительность современных методов моделирования, представляется разумным в качестве результата экспериментов ожидать модели, построенные на основе по крайней мере 30%-ной идентичности последовательностей (Vitkup etal. 2001), соответствующие одной экспериментально определенной структуре на семейство последовательностей, а не на семейство укладки.
Чтобы масштабное сравнительное моделирование, необходимое для структурной геномики, стало возможным, этапы сравнительного моделирования сочетают в полностью автоматизированных разработках, таких как хранилища SWISS-MODEL или MODBASE (Корр and Schwede 2006; Pieper et al. 2006), каждое из которых содержит более миллиона моделей. Статистические данные в этих базах свидетельствуют о том, что приблизительно для 70% известных белковых последовательностей можно создать модели составляющих их доменов. Это можно утверждать на основании того факта, что почти 2000 структур были размещены в базах центрами структурной геномики, основными объектами исследования которых являются новые способы укладки и новые структуры. Эти пополнения составили 73% новых структурных свойств в базе PDB за последние 7 лет (Burley et al. 2008).
116
Глава 3. Сравнительное моделирование структуры белков
В то время как известное на сегодняшний день число белков, для которых получены хотя бы частичные модели, выглядит впечатляюще, как правило, модель построена только для одного белкового домена. В среднем же белки обычно имеют два или три домена. Например, средний размер открытой рамки считывания дрожжей составляет 472 аминокислотных остатка, а средний размер домена в САТН, базе структурных доменов, составляет 175 остатков. Средний размер модели в MODBASE, базе данных сравнительных моделей, лишь немногим выше - 192 остатка. Более того, две трети случаев моделирования характеризуются идентичностью последовательности менее 30% между ближайшими шаблонами.
3.5. Заключение
Уже доказано, что сравнительное моделирование является полезным инструментом во многих биологических приложениях. Ожидается, что его значение среди методов предсказания структуры в дальнейшем будет только расти, поскольку количество белковых структур, известных из экспериментов, постоянно увеличивается благодаря проектам исследования структуры белков и совершенствованию экспериментальных методик.
Средняя идентичность последовательностей для структурно связанных белков обычно составляет лишь 8-9%, для большинства из них характерно менее 15% идентичности (Rost 1997). Методы сравнительного моделирования сильно ограничены этим подмножеством последовательностей, для которых сходство последовательностей с белком известной структуры поддается распознаванию. В связи с этим можно с уверенностью предположить, что сравнительное моделирование является одним из первых шагов в области распознавания белковых структур и использования структурной информации. Методы распознавания способов укладки белка, которые обсуждались в главе 2, сыграют важную роль при расширении возможностей сравнительного моделирования даже для отдаленных гомологов и структурных аналогов.
Ключевые проблемы - усовершенствование существующих и создание новых методов уточнения моделей, полученных в ходе сравнительного моделирования, за счет добавления точных петель и боковых цепей; уточнение внутренней укладки элементов вторичной структуры; определение оценочных функций, с помощью которых можно сравнивать качество моделей; поиски оптимального сочетания фрагментов известных укладок и определение ошибок в пространственных моделях. Даже незначительное продвижение в решении этих проблем окажет большое влияние
Литература
117
на сравнительное моделирование, поскольку большинство связей между белковыми структурами являются слишком отдаленными для использования в сравнительном моделировании. С другой стороны, несмотря на то, что решение вышеназванных проблем, вероятно, не окажет существенного влияния на общую точность уже существующих белковых моделей, значение улучшений для получения более надежных в функциональном смысле пространственных моделей, т.е., моделей, которые можно с уверенностью использовать для создания функциональных аннотаций, сложно переоценить.
Упомянутые выше достижения методов сравнительного моделирования белковых структур являются необходимыми предпосылками для развития новых методов моделирования, которые относятся к области «структурной протеомики». Цель последних - соединение основных строительных блоков моделей упаковки в физиологически более релевантные четвертичные структуры и ансамбли. Это создаст возможности для моделирования взаимодействий множества различных белков с известной структурой.
Благодарности. Настоящий обзор отчасти основан на нашей ранней публикации (Fiser 2004).
Литература
Abagyan R, Totrov М (1994) Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol 235:983-1002
Alber F, Dokudovskaya S, VeenhoffLM, et al. (2007a) Determining the architectures of macromolecular assemblies. Nature 450:683-694
Alber F, Dokudovskaya S, Veenhoff LM, et al. (2007b) The molecular architecture of the nuclear pore complex. Nature 450:695-701
Alber F, Forster F, Korkin D, et al. (2008) Integrating diverse data for structure determination of macromolecular assemblies. Annu Rev Biochem 77:443—477
Al Lazikani B, Sheinerman FB, Honig В (2001) Combining multiple structure and sequence alignments to improve sequence detection and alignment: application to the SH2 domains of Janus kinases. Proc Natl Acad Sci USA 98:14796
Altschul SF, Madden TL, Schaffer AA, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389-3402
Andreeva A, Howorth D, Chandonia JM, et al. (2008) Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res 36:D419-425
Apostolico A, Giancarlo R (1998) Sequence alignment in molecular biology. J Comput Biol 5:173-196 Apweiler R, Bairoch A, Wu CH (2004) Protein sequence databases. Curr Opin Chem Biol 8:76-80 Aszodi A, Taylor WR (1994) Secondary structure formation in model polypeptide chains. Protein
Eng 7:633-644
Aszodi A, Taylor WR (1996) Homology modelling by distance geometry. Fold Des 1:325-334 Baker D, Sali A (2001) Protein structure prediction and structural genomics. Science 294:93-96
Barrientos LG, Campos-Olivas R, Louis JM, et al. (2001) 1H, 13C, 15N resonance assignments and fold verification of a circular permuted variant of the potent HIV-inactivating protein cyanovirin-N. J Biomol NMR 19:289-290
118
Глава 3. Сравнительное моделирование структуры белков
Battey JN, Корр J, Bordoli L, et al. (2007) Automated server predictions in CASP7. Proteins 69(Suppl 8):68-82
Becker OM, Dhanoa DS, Marantz Y, et al. (2006) An integrated in silico 3D model-driven discovery of a novel, potent, and selective amidosulfonamide 5-HT1A agonist (PRX-00023) for the treatment of anxiety and depression. J Med Chem 49:3116-3135
Berman H, Henrick K, Nakamura H, et al. (2007) The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 35:D301-303
Blake JD, Cohen FE (2001) Pairwise sequence alignment below the twilight zone. J Mol Biol 307:721-735
Blundell TL, Sibanda BL, Sternberg MJ, et al. (1987) Knowledge-based prediction of protein structures and the design of novel molecules. Nature 326:347-352
Boissel JP, Lee WR, Presnell SR, et al. (1993) Erythropoietin structure-function relationships. Mutant proteins that test a model of tertiary structure. J Biol Chem 268:15983-15993
Bonneau R, Baker D (2001) Ab initio protein structure prediction: progress and prospects. Annu Rev Biophys Biomol Struct 30:173-189
Bowie JU, Luthy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three- dimensional structure. Science 253:164—170
Braun W, Go N (1985) Calculation of protein conformations by proton-proton distance constraints. A new efficient algorithm. J Mol Biol 186:611-626
Brenner SE, Chothia C, Hubbard TJ (1998) Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc Natl Acad Sci USA 95:6073-6078
Brooks CL, Ш, Bruccoleri RE, Olafson BD, et al. (1983) CHARMM:A program for macromolecular energy minimization and dynamics calculations. J Comp Chem 4:187-217
Browne WJ, North ACT, Phillips DC, et al. (1969) A possible three-dimensional structure of bovine lactalbumin based on that of hen’s egg-white lysosyme. J Mol Biol 42:65-86
Bruccoleri RE, Karplus M (1987) Prediction of the folding of short polypeptide segments by uniform conformational sampling. Biopolymers 26:137-168
Bruccoleri RE, Karplus M (1990) Conformational sampling using high-temperature molecular dynamics. Biopolymers 29:1847-1862
Bujnicki JM, Elofsson A, Fischer D, et al. (2001) LiveBench-Г. continuous benchmarking of protein structure prediction servers. Protein Sci 10:352-362
Burley SK, Almo SC, Bonanno JB, et al. (1999) Structural genomics: beyond the human genome project. Nat Genet 23:151-157
Burley SK, Joachimiak A, Montelione GT, et al. (2008) Contributions to the NIH-NIGMS protein structure initiative from the PSI production centers. Structure 16:5-11
Bystroff C, Baker D (1998) Prediction of local structure in proteins using a library of sequence structure motifs. J Mol Biol 281:565-577
Chakravarty S, Sanchez R (2004) Systematic analysis of added-value in simple comparative models of protein structure. Structure 12:1461—1470
Chakravarty S, Wang L, Sanchez R (2005) Accuracy of structure-derived properties in simple comparative models of protein structures. Nucleic Acids Res 33:244-259
Chance MR, Bresnick AR, Burley SK, et al. (2002) Structural genomics: a pipeline for providing structures for the biologist. Protein Sci 11:723-738
Chinea G, Padron G, Hooft RW, et al. (1995) The use of position-specific retainers in model building by homology. Proteins 23:415-421
Chivian D, Baker D (2006) Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Res 34:el 12
Chothia C, Lesk AM (1986) The relation between the divergence of sequence and structure in proteins. EMBO J 5:823-826
Chothia C, Lesk AM (1987) Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 196:901-917
Chothia C, Lesk AM, Tramontane A, et al. (1989) Conformations of immunoglobulin hypervariable regions. Nature 342:877-883
Chothia C, Gough J, Vogel C, et al. (2003) Evolution of the protein repertoire. Science 300:1701-1703
Литература
119
Claessens М, Van Cutsem E, Lasters I, etal. (1989) Modelling the polypeptide backbone with ‘spare parts’ from known protein structures. Protein Eng 2:335-345
Clore GM, Brunger AT, Karplus M, etal. (1986) Application of molecular dynamics with interproton distance restraints to three-dimensional protein structure determination. A model study of crambin. J Mol Biol 191:523—551
Clore GM, Robien MA, Gronenbom AM (1993) Exploring the limits of precision and accuracy of protein structures determined by nuclear magnetic resonance spectroscopy. J Mol Biol 231:82-102
Cohen FE, Kuntz ID: Tertiary structure prediction, in Prediction of protein structure and the principles of protein conformations. Edited by Fasman GD. New York, Plenum, 1989, pp. 647-705
Collura V, Higo J, Gamier J (1993) Modeling of protein loops by simulated annealing. Protein Sci 2:1502-1510
Contreras-Moreira B, Fitqohn PW, Offinan M, et al. (2003) Novel use of a genetic algorithm for protein structure prediction: searching template and sequence alignment space. Proteins 53(Suppl 6): 424-429
Dalton JA, Jackson RM (2007) An evaluation of automated homology modelling methods at low target template sequence similarity. Bioinformatics 23:1901-1908
Das B, Meirovitch H (2003) Solvation parameters for predicting the structure of surface loops in proteins: transferability and entropic effects. Proteins 51470-483
Das R, Qian B, Raman S, et al. (2007) Structure prediction for CASP7 targets using extensive all atom refinement with Rosetta@home. Proteins 69(Suppl 8): 118-128
de Bakker PI, DePristo MA, Burke DF, et al. (2003) Ab initio construction of polypeptide fragments: accuracy of loop decoy discrimination by an all-atom statistical potential and the AMBER force field with the Generalized Bom solvation model. Proteins 51:21-40
Deane CM, Blundell TL (2001) CODA: a combined algorithm for predicting the structurally variable regions of protein models. Protein Sci 10:599-612
DePristo MA, de Bakker PI, Lovell SC, et al. (2003) Ab initio construction of polypeptide fragments: efficient generation of accurate, representative ensembles. Proteins 51:41—55
Dill KA, Chan HS (1997) From Levinthal to pathways to funnels. Nat Struct Biol 4:10-19
Do CB, Mahabhashyam MS, Brudno M, et al. (2005) ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res 15:330-340
Du P, Andree M, Levy RM (2003) Have we seen all structures corresponding to short protein fragments in the Protein Data Bank? An update. Protein Eng 16:407-414
Edgar RC, Batzoglou S (2006) Multiple sequence alignment. Curr Opin Struct Biol 16:368-373
Edgar RC, Sjolander К (2003) SATCHMO: sequence alignment and tree construction using hidden Markov models. Bioinformatics 19:1404—1411
Edgar RC, Sjolander К (2004) COACH: profile-profile alignment of protein families using hidden Maricov models. Bioinformatics 20:1309-1318
Eisenberg D, Luthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Method Enzymol 277:396-404
Eramian D, Shen MY, Devos D, et al. (2006) A composite score for predicting errors in protein structure models. Protein Sci 15:1653-1666
Espadaler J, Fernandez-Fuentes N, Hermoso A, et al. (2004) ArchDB: automated protein loop classification as a tool for structural genomics. Nucleic Acids Res 32:D 185-188
Evers A, Gohlke H, Klebe G (2003) Ligand-supported homology modelling of protein binding sites using knowledge-based potentials. J Mol Biol 334:327-345
Eyrich VA, Marti-Renom MA, Przybylski D, et al. (2001) EVA: continuous automatic evaluation of protein structure prediction servers. Bioinformatics 17:1242-1243
Faber HR, Matthews BW (1990) A mutant T4 lysozyme displays five different crystal conformations. Nature 348:263-266
Fasnacht M, Zhu J, Honig В (2007) Local quality assessment in homology models using statistical potentials and support vector machines. Protein Sci 16:1557-1568
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368-376
Fernandez-Fuentes N, Fiser A (2006) Saturating representation of loop conformational fragments in structure databanks. BMC Struct Biol 6:15
120
Глава 3. Сравнительное моделирование структуры белков
Fernandez-Fuentes N, Oliva В, Fiser А (2006а) A supersecondary structure library and search algorithm for modeling loops in protein structures. Nucleic Acids Res 34:2085-2097
Fernandez-Fuentes N, Zhai J, Fiser A (2006b) ArchPRED: a template based loop structure prediction server. Nucleic Acids Res 34:W173-176
Fernandez-Fuentes N, Madrid-Aliste CJ, Rai BK, et al. (2007a) M4T: a comparative protein structure modeling server. Nucleic Acids Res 35:W363-368
Fernandez-Fuentes N, Rai BK, Madrid-Aliste CJ, etal. (2007b) Comparative protein structure modeling by combining multiple templates and optimizing sequence-to-structure alignments. Bioinformatics 23:2558-2565
Fidelis K, Stem PS, Bacon D, et al. (1994) Comparison of systematic search and database methods for constructing segments of protein structure. Protein Eng 7:953-960
Fine RM, Wang H, Shenkin PS, et al. (1986) Predicting antibody hypervariable loop conformations. II: minimization and molecular dynamics studies of MCPC603 from many randomly generated loop conformations. Proteins 1:342-362
Finkelstein AV, Reva BA (1991) A search for the most stable folds of protein chains. Nature 351:497-499
Fiser A (2004) Protein structure modeling in the proteomics era. Expert Rev Proteomics 1:97—110
Fiser A, Sali A (2003a) Modeller: generation and refinement of homology-based protein structure models. Method Enzymol 374:461-491
Fiser A, Sali A (2003b) ModLoop: automated modeling of loops in protein structures. Bioinformatics 19:2500-2501
Fiser A, Do RK, Sali A (2000) Modeling of loops in protein structures. Protein Sci 9:1753-1773
Fiser A, Feig M, Brooks CL, III, et al. (2002) Evolution and physics in comparative protein structure modeling. Acc Chem Res 35:413—421
Fiser A, Filipe SR, Tomasz A (2003) Cell wall branches, penicillin resistance and the secrets of the MurM protein. Trends Microbiol 11:547-553
Fogolari F, Tosatto SC (2005) Application of MM/PBSA colony free energy to loop decoy discrimination: toward correlation between energy and root mean square deviation. Protein Sci 14:889-901
Forrest LR, Woolf ТВ (2003) Discrimination of native loop conformations in membrane proteins: decoy library design and evaluation of effective energy scoring functions. Proteins 52:492-509
Ginalski К (2006) Comparative modeling for protein structure prediction. Curr Opin Struct Biol 16:172-177
Ginalski K, Elofsson A, Fischer D, et al. (2003) 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics 19:1015-1018
Grabarek Z (2006) Structural basis for diversity of the EF-hand calcium-binding proteins. J Mol Biol 359:509-525
Greene LH, Lewis ТЕ, Addou S, et al. (2007) The CATH domain structure database: new protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Res 35:D291-297
Greer J (1981) Comparative model-building of the mammalian serine proteases. J Mol Biol 153:1027-1042
Greer J (1990) Comparative modeling methods: application to the family of the mammalian serine proteases. Proteins 7:317-334
Guenther B, Onrust R, Sali A, et al. (1997) Crystal structure of the alpha-subunit of the clamp loader complex of E. coli DNA polymerase III. Cell 91:335-345
Han R, Leo-Macias A, Zerbino D, et al. (2008) An efficient conformational sampling method for homology modeling. Proteins 71:175-188
Havel TF, Snow ME (1991) A new method for building protein conformations from sequence alignments with homologues of known structure. J Mol Biol 217:1-7
Henikoff JG, Pietrokovski S, McCallum CM, et al. (2000) Blocks-based methods for detecting protein homology. Electrophoresis 21:1700-1706
Henikoff S, Henikoff JG (1992) Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 89:10915-10919.
Литература
121
“fcdm L, Sander C (1991) Database algorithm for generating protein backbone and side-chain coordinates from a C alpha trace application to model building and detection of co-ordinate errors. J Mol Biol 218:183-194
4ooft RW, Vriend G, Sander C, et al. (1996) Errors in protein structures. Nature 381:272
-ecobson MP, Pincus DL, Rapp CS, et al. (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55:351-367
xiroszewski L, Rychlewski L, Zhang B, etal. (1998) Fold prediction by a hierarchy of sequence, threading, and modeling methods. Protein Sci 7:1431-1440
iiroszewski L, Rychlewski L, Godzik A (2000) Improving the quality of twilight-zone alignments. Protein Sci 9:1487-1496
iaroszewski L, Rychlewski L, Li Z, etal. (2005) FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res 33:W284-288
Jauch R, Yeo HC, Kolatkar PR, et al. (2007) Assessment of CASP7 structure predictions for template free targets. Proteins 69(Suppl 8):57-67
Jennings AJ, Edge CM, Sternberg MJ (2001) An approach to improving multiple alignments of protein sequences using predicted secondary structure. Protein Eng 14:227-231
John B, Sali A (2003) Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res 31:3982-3992
John B, Sali A (2004) Detection of homologous proteins by an intermediate sequence search. Protein Sci 13:54-62
Johnson LN, Lowe ED, Noble ME, et al. (1998) The Eleventh Datta Lecture. The structural basis for substrate recognition and control by protein kinases. FEBS Lett 430:1-11
Jones DT (1999) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol 287:797-815
Jones TA, Thirup S (1986) Using known substructures in protein model building and crystallography. EMBO J 5:819-822
Karchin R, Cline M, Mandel-Gutfreund Y, et al. (2003) Hidden Markov models that use predicted local structure for fold recognition: alphabets of backbone geometry. Proteins 51:504-514
Karplus K, Barrett C, Hughey R (1998) Hidden Markov models for detecting remote protein homologies. Bioinformatics 14:846-856
Karplus K, Katzman S, Shackleford G, et al. (2005) SAM-T04: what is new in protein-structure prediction for CASP6. Proteins 61 (Suppl 7): 135-142
Kihara D, Skolnick J (2003) The PDB is a covering set of small protein structures. J Mol Biol 334:793-802
Kiselar JG, Janiney PA, Almo SC, et al. (2003) Structural analysis of gelsolin using synchrotron protein footprinting. Mol Cell Proteomics 2:1120-1132
Koehl P, Delarue M (1995) A self consistent mean field approach to simultaneous gap closure and side-chain positioning in homology modelling. Nat Struct Biol 2:163-170
Kolinski A, Bujnicki JM (2005) Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins 61 (Suppl 7):84—90
Kolinski A, Betancourt MR, Kihara D, et al. (2001) Generalized comparative modeling (GENECOMP): a combination of sequence comparison, threading, and lattice modeling for protein structure prediction and refinement. Proteins 44:133-149
Kopp J, Schwede T (2006) The SWISS-MODEL Repository: new features and functionali-ties.Nucleic Acids Res 34:D315-318
Kopp J, Bordoli L, Battey JN, et al. (2007) Assessment of CASP7 predictions for template-based modeling targets. Proteins 69(Suppl 8):38-56
Krogh A, Brown M, Mian IS, et al. (1994) Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol 235:1501-1531
Laskowski RA, Moss DS, Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231:1049-1067
Lesk AM (1995) NAD-binding domains of dehydrogenases. Curr Opin Struct Biol 5:775-783
Lesk AM, Chothia C (1980) How different amino acid sequences determine similar protein structures: the structure and evolutionary dynamics of the globins. J Mol Biol 136:225-270
Levitt M (1992) Accurate modeling of protein conformation by automatic segment matching. J Mol Biol 226:507-533
122
Глава 3. Сравнительное моделирование структуры белков
Luthy R, McLachlan AD, Eisenberg D (1991) Secondary structure-based profiles: use of structure conserving scoring tables in searching protein sequence databases for structural similarities. Proteins 10:229-239.
Manjasetty BA, Shi W, Zhan C, et al. (2007) A high-throughput approach to protein structure analysis. Genet Eng (NY) 28:105-128
Marti-Renom MA, Stuart AC, Fiser A, et al. (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291-325
Marti-Renom MA, Madhusudhan MS, Fiser A, et al. (2002) Reliability of assessment of protein structure prediction methods. Structure(Camb) 10:435-440
Marti-Renom MA, Madhusudhan MS, Sali A (2004) Alignment of protein sequences by their profiles. Protein Sci 13:1071-1087
Melo F, Feytmans E (1997) Novel knowledge-based mean force potential at atomic level. J Mol Biol 267:207-222
Mezei M (1998) Chameleon sequences in the PDB. Protein Eng 11:411-414
Michalsky E, Goede A, Preissner R (2003) Loops In Proteins (LIP}-a comprehensive loop database for homology modelling. Protein Eng 16:979-985
Moretti S, Armougom F, Wallace IM, et al. (2007) The М-Coffee web server: a meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res 35:W645-648
Moult J (2005) A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr Opin Struct Biol 15:285-289
Moult J, James MN (1986) An algorithm for determining the conformation of polypeptide segments in proteins by systematic search. Proteins 1:146-163
Notredame C (2007) Recent evolutions of multiple sequence alignment algorithms. PLoS Comput Biol 3:e 123
Ohlendorf DH (1994) Accuracy of refined protein structures. Comparison of four independently refined models of human interleukin 1 beta. Acta Crystallogr D Biol Crystallogr D50:808-812
Oliva B, Bates PA, Querol E, et al. (1997) An automated classification of the structure of protein loops. J Mol Biol 266:814-830
Orr GA, Rao S, Swindell CS, etal. (1998) Photoaffinity labeling approach to map the Taxol-binding site on the microtubule. Method Enzymol 298:238-252
Pearson WR (2000) Flexible sequence similarity searching with the FASTA3 program package. Method Mol Biol 132:185-219
Pei J, Grishin NV (2007) PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 23:802-808
Pei J, Kim BH, Grishin NV (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36:2295-2300
Peng HP, Yang AS (2007) Modeling protein loops with knowledge-based prediction of sequence structure alignment. Bioinformatics 23:2836-2842
Petrey D, Honig В (2005) Protein structure prediction: inroads to biology. Mol Cell 20:811-819
Petrey D, Xiang Z, Tang CL, et al. (2003) Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins 53(Suppl 6): 430-435
Pieper U, Eswar N, Davis FP, et al. (2006) MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 34:D291-295
Pillardy J, Czaplewski C, Liwo A, et al. (2001) Recent improvements in prediction of protein structure by global optimization of a potential energy function. Proc Natl Acad Sci USA 98:2329-23233
Qian B, Ortiz AR, Baker D (2004) Improvement of comparative model accuracy by free-energy optimization along principal components of natural structural variation. Proc Natl Acad Sci USA 101:15346-15351
Rai BK, Fiser A (2006) Multiple mapping method: a novel approach to the sequence-to-structure alignment problem in comparative protein structure modeling. Proteins 63:644-661
Rai BK, Madrid-Aliste CJ, Fajardo JE, et al. (2006) МММ: a sequence-to-structure alignment protocol. Bioinformatics 22:2691-2692
Литература
123
•efciy BV, Li WW, Shindyalov IN, et al. (2001) Conserved key amino acid positions (CKAAPs) derived from the analysis of common substructures in proteins. Proteins 42:148-163
CS, Cohen FE (1993) Modeling protein structures: construction and their applications. FASEB J 7:783-890
i-ttf CS, Sun E, McKerrow JH, etal. (1993) Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc Natl Acad Sci USA 90:3583-3587
1л1 CA, Strauss CE, Chivian D, et al. (2004) Modeling structurally variable regions in homologous proteins with rosetta. Proteins 55:656-677.
Сдй В (1997) Protein structures sustain evolutionary drift. Fold Des 2:S19-S24
В (1999) Twilight zone of protein sequence alignments. Protein Eng 12:85-94
ijdolph MG, Stanfield RL, Wilson IA (2006) How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 24:419-466
<osch DB, Halpern AL, Sutton G, et al. (2007) The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5:e77
twhlewski L, Jaroszewski L, Li W, et al. (2000) Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci 9:232-241
fcykunov D, Fiser A (2007) Effects of amino acid composition, finite size of proteins, and sparse statistics on distance-dependent statistical pair potentials. Proteins 67:559-568
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779-815
Sali A, Matsumoto R, McNeil HP, et al. (1993) Three-dimensional models of four mouse mast cell chymases. Identification of proteoglycan binding regions and protease-specific antigenic epitopes. J Biol Chem 268:9023-9034
Sali A, Shakhnovich E, Karplus M (1994) How does a protein fold? Nature 369:248-251
Samudrala R, Moult J (1998) A graph-theoretic algorithm for comparative modeling of protein structure. J Mol Biol 279:287-302
Sanchez R, Sali A (1997) Evaluation of comparative protein structure modeling by MODELLER-3. Proteins(Suppl l):50-58
Sanchez R, Sali A (1998) Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc Natl Acad Sci USA 95:13597-13602
Sangar V, Blankenberg DJ, Altman N, et al. (2007) Quantitative sequence-function relationships
in proteins based on gene ontology. BMC Bioinformatics 8:294
Saraste M, Sibbald PR, Wittinghofer A (1990) The P-loop-a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci 15:430-434
Sauder JM, Arthur JW, Dunbrack RL, Jr. (2000) Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins 40:6-22
Schaffer AA, Aravind L, Madden TL, et al. (2001) Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 29:2994-3005
Schwarzenbacher R, Godzik A, Jaroszewski L (2008) The JCSG MR pipeline: optimized alignments, multiple models and parallel searches. Acta Crystallogr D Biol Crystallogr 64:133-140
Sheng Y, Sali A, Herzog H, et al. (1996) Site-directed mutagenesis of recombinant human beta 2-glycoprotein I identifies a cluster of lysine residues that are critical for phospholipid binding and anti-cardiolipin antibody activity. J Immunol 157:3744-3751
Shenkin PS, Yarmush DL, Fine RM, etal. (1987) Predicting antibody hypervariable loop conformation. I. Ensembles of random conformations for ringlike structures. Biopolymers 26:2053-2085
Shi J, Blundell TL, Mizuguchi К (2001) FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol 310:243-257
Sibanda BL, Blundell TL, Thornton JM (1989) Conformation of beta-hairpins in protein structures. A systematic classification with applications to modelling by homology, electron density fitting and protein engineering. J Mol Biol 206:759-777
Sippl MJ (1990) Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol 213:859-883
124
Глава 3. Сравнительное моделирование структуры белков
Sippl MJ (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17:355— 362
Sippl MJ (1995) Knowledge-based potentials for proteins. Curr Opin Struct Biol 5:229-235 Soto CS, Fasnacht M, Zhu J, et al. (2008) Loop modeling: Sampling, filtering, and scoring. Proteins 70:834—843
Srinivasan N, Blundell TL (1993) An evaluation of the performance of an automated procedure for comparative modelling of protein tertiary structure. Protein Eng 6:501-512
Summa CM, Levitt M (2007) Near-native structure refinement using in vacuo energy minimization. Proc Natl Acad Sci USA 104:3177-3182
Sutcliffe MJ, Haneef I, Carney D, etal. (1987) Knowledge based modelling of homologous proteins, Part I: three-dimensional frameworks derived from the simultaneous superposition of multiple structures. Protein Eng 1:377-384
Sutcliffe MJ, Dobson CM, Oswald RE (1992) Solution structure of neuronal bungarotoxin determined by two-dimensional NMR spectroscopy: calculation of tertiary structure using systematic homologous model building, dynamical simulated annealing, and restrained molecular dynamics. Biochemistry 31:2962-2970
Tainer JA, Thayer MM, Cunningham RP (1995) DNA repair proteins. Curr Opin Struct Biol 5:20-26
Taylor WR, Hatrick К (1994) Compensating changes in protein multiple sequence alignments. Protein Eng 7:341-348
Terashi G, Takeda-Shitaka M, Kanou K, et al. (2007) Fams-ace: a combined method to select the best model after remodeling all server models. Proteins 69(Suppl 8):98-107
Todd AE, Orengo CA, Thornton JM (2001) Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol 307:1113-1143
Todd AE, Orengo CA, Thornton JM (2002) Plasticity of enzyme active sites. Trends Biochem Sci 27:419-426
Topf M, Lasker K, Webb B, et al. (2008) Protein structure fitting and refinement guided by cryo-EM density. Structure 16:295-307
Topham CM, McLeod A, Eisenmenger F, et al. (1993) Fragment ranking in modelling of protein structure. Conformationally constrained environmental amino acid substitution tables. J Mol Biol 229:194—220
Unger R, Harel D, Wherland S, et al. (1989) A 3D building blocks approach to analyzing and predicting structure of proteins. Proteins 5:355-373
Vakser LA (1995) Protein docking for low-resolution structures. Protein Eng 8:371-377
van Gelder CW, Leusen FJ, Leunissen J A, et al. (1994) A molecular dynamics approach for the generation of complete protein structures from limited coordinate data. Proteins 18:174—185
van Vlijmen HW, Karplus M (1997) PDB-based protein loop prediction: parameters for selection and methods for optimization. J Mol Biol 267:975-1001
Venclovas C, Margelevicius M (2005) Comparative modeling in CASP6 using consensus approach to template selection, sequence-structure alignment, and structure assessment. Proteins 61:99-105.
Venter JC, Remington K, Heidelberg JF, et al. (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66-74
Vernal J, Fiser A, Sali A, etal. (2002) Probing the specificity of a trypanosomal aromatic alphahydroxy acid dehydrogenase by site-directed mutagenesis. Biochem Biophys Res Common 293:633-639
Vitkup D, Melamud E, Moult J, et al. (2001) Completeness in structural genomics. Nat Struct Biol 8:559-566
Wallner B, Elofsson A (2005a) Pcons5: combining consensus, structural evaluation and fold recognition scores. Bioinformatics 21:4248—4254
Wallner B, Elofsson A (2005b) All are not equal: a benchmark of different homology modeling programs. Protein Sci 14:1315-1327
Wallner B, Elofsson A (2007) Prediction of global and local model quality in CASP7 using Peons and ProQ. Proteins 69(Suppl 8): 184-193
Wallner B, Larsson P, Elofsson A (2007) Pcons.net: protein structure prediction meta server. Nucleic Acids Res 35:W369-374
Литература
125
Wlodawer A, Miller М, Jaskolski М, et al. (1989) Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 245:616-621
Wu CH, Apweiler R, Bairoch A, et al. (2006) The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res 34:D187-191
Wu G, Fiser A, ter Kuile B, et al. (1999) Convergent evolution of Trichomonas vaginalis lactate dehydrogenase from malate dehydrogenase. Proc Natl Acad Sci USA 96:6285-6290
Wu G, McArthur AG, Fiser A, et al. (2000) Core histones of the amitochondriate protist, Giardia lamblia. Mol Biol Evol 17:1156-1163
Xiang Z, Soto CS, Honig В (2002) Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc Natl Acad Sci USA 99:7432-7437
Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, et al. (2006) Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci USA 103:10166-10173
Xu J, Jiao F, Yu L (2007) Protein structure prediction using threading. Methods Mol Biol 413:91-122
Xu LZ, Sanchez R, Sali A, etal. (1996) Ligand specificity of brain lipid-binding protein. J Biol Chem 271:24711-24719
Yooseph S, Sutton G, Rusch DB, et al. (2007) The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 5:el6
Zhang C, Liu S, Zhou Y (2004) Accurate and efficient loop selections by the DFIRE-based all-atom statistical potential. Protein Sci 13:391-399
Zhang Y (2007) Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 69(Suppl 8): 108-117
Zheng Q, Rosenfeld R, Vajda S, et al. (1993) Determining protein loop conformation using scalingrelaxation techniques. Protein Sci 2:1242-1248
Zhou H, Pandit SB, Lee SY, et al. (2007) Analysis of TASSER-based CASP7 protein structure prediction results. Proteins 69(Suppl 8):90-97
Глава 4
Предсказание структуры мембранных белков
Тимоти Ньюджент, Дэвид Т. Джонс
В клетке трансмембранные (ТМ) белки выполняют множество крайне важных функций и составляют значительную часть протеома. Согласно имеющимся оценкам, до 30% всех человеческих генов могут кодировать а-спиральные ТМ-белки. Однако лишь для небольшого числа ТМ-белков известны структуры с высоким разрешением. В связи с этим возрастает значение методов исследования, которые позволяют извлечь максимум информации о структуре белка из имеющейся аминокислотной последовательности. Настоящая глава посвящена описанию современных методов предсказания топологии и структуры, основанных на анализе последовательностей и структур мембранных белков. Особое внимание уделено имеющимся в этой области «подводным камням», а также сложностям, которые еще только предстоит решить.
4.1. Введение
Трансмембранные (ТМ) белки принимают участие во многих важных биологических процессах, таких как клеточная передача сигналов, транспорт тех молекул, которые не проникают через мембраны самостоятельно, межклеточные взаимодействия, клеточное узнавание и клеточная адгезия. Многие трансмембранные белки также являются важными лекарственными мишенями. По имеющимся оценкам, более половины всех представленных сегодня на рынке лекарственных средств имеют своей мишенью
Timothy Nugent and David T. Jones* *
Bioinformatics Group, Department of Computer Science, University College, London, WC1E 6BT, UK
*e-mail: d.jones@cs.ucl.ac.uk
4.2. Структурные классы
127
трансмембранные белки (Klabunde and Hesler 2002). Однако поскольку получение кристаллических структур высокого качества сопряжено с рядом технических сложностей, этот класс белков крайне скудно представлен в структурных базах данных, составляя лишь 1% известных структур в базе данных PDB (White 2004). Трансмембранные белки имеют важное биологическое и фармакологическое значение. Изучить структуру и топологию трансмембранных белков - общее количество ТМ спиралей, их границы и ориентацию относительно мембраны - важно для анализа функций белков и определения направления дальнейшей экспериментальной работы. В отсутствие данных о структуре современные стратегии био информатики сводятся к методам предсказания на основе анализа последовательностей.
4.2. Структурные классы
4.2.1. Пучки альфа-спиралей
Выделяют два основных класса мембранных белков: а-спирали и р-бочонки. Альфа-спиральные мембранные белки представляют собой наиболее многочисленный класс ТМ белков и встречаются в биологических мембранах всех типов, включая наружные мембраны бактерий. Белки состоят из одной или нескольких альфа-спиралей, каждая из которых содержит тяж гидрофобных аминокислот, встроена в мембрану и связана с другими спиралями посредством внемембранных петлевых областей. Считается, что в состав альфа-спиральных мембранных белков может входить до 20 ТМ спиралей, что обеспечивает многообразие топологий. Известно, что петлевые области имеют сложное строение и содержат мембранные петли (re-entrant loops) и амфифильные спирали. Мембранные петли представляют собой короткие а-спирали, которые входят в мембрану и выходят из нее на одной и той же стороне. Амфифильные спирали располагаются параллельно плоскости мембраны и глобулярным доменам.
Альфа-спиральные ТМ белки делят на несколько подклассов. Белки подкласса I содержат одну ТМ а-спираль, N-конец которой расположен с наружной стороны мембраны, во внешней среде, а С-конец - с внутренней, цитоплазматической, стороны. Эти белки, в свою очередь, делятся на еще два подкласса. Подкласс 1а, к которому относится большинство эукариотических мембранных белков, содержит расщепляемые сигнальные последовательности, в то время как в подклассе 1b таких последовательностей нет. Мембранные белки подкласса II, как и белки подкласса I, пронизывают мембрану только один раз. Однако они имеют противополож-
128
Глава 4. Предсказание структуры мембранных белков
Рис. 4.1. (Цветную версию рисунка см. на вклейке.) а) Бактериородопсин Halobacterium salinarium. Семь трансмембранных спиралей рецептора, сопряженного с G-белком, окружены липидным бислоем. Рецептор функционирует как протонная помпа: энергия света используется для выведения из клетки протонов. PDB ID 1руб. Среди других рецепторов, сопряженные с G-белками, можно назвать галородопсин - хлорную помпу, которая активируется светом. PDB ID 1е12. б) Упрощенное представление топологии бактериородопсина
ную ориентацию: N-конец расположен на цитоплазматической стороне мембраны, а С-конец - на внешней стороне.
Мембранные белки подкласса III представляют собой одиночную по-липептидную цепь, которая на отдельных участках представлена множественными трансмембранными спиралями. Белки этого подкласса также делятся на подклассы а и Ь: белки подкласса Ша содержат расщепляемые сигнальные последовательности; N-концы белков подкласса ШЬ направлении к наружной поверхности мембраны, а сами белки не содержат расщепляемых сигнальных последовательностей. К числу мембранных белков подкласса III принадлежит семейство рецепторов, сопряженных с G-белками (G-protein-coupled receptors, или GPCR), представители которого имеют в своем составе семь трансмембранных спиралей (рис. 4.1). Рецепторы, сопряженные с G-белками, составляют большое семейство белков-рецепторов, которые распознают молекулы во внешней среде, активируют пути трансдукции сигнала и в конечном счете обеспечивают реализацию клеточного ответа.
4.2. Структурные классы
129
Мембранные белки IV подкласса содержат множество доменов, образующих ансамбль, элементы которого многократно пронизывают мембрану. Домены могут располагаться на одиночной полипетидной цепи, но часто трансмембранный белок представлен несколькими цепями. К числу белков этого подкласса относится фотосистема I, которая состоит из девяти различных цепей (PDB ID 1 jbO).
4.2.2. Бета-бочонки
Трансмембранные белки, имеющие структуру бета-бочонков, присутствуют во внешних мембранах грамотрицательных бактерий, клеточных стенках грамположительных бактерий, а также во внешних мембранах митохондрий и хлоропластов. Они состоят из рядов антипараллельных Р-тяжей, встроенных в мембрану. Каждый из тяжей связан с предыдущим и последующим тяжом в последовательности водородными связями. Ряды тяжей соединяются друг с другом с помощью внемембранных петель. Бета-тяжи содержат полярные и гидрофобные
аминокислоты, которые чередуются таким образом, что гидрофобные остатки направлены к внешней поверхности поры и контактируют с окружающими ее липидами, а гидрофильные остатки направлены к внутренней поверхности поры. Все трансмембранные белки, имеющие структуру р-бочонка, имеют простую топологию, организованную по принципу чередования подъемов и спадов, которая, вероятно, отражает их общее эволюционное происхождения и схожий механизм укладки. К числу ТМ белков, имеющих струк-
Рис. 4.2. (Цветную версию рисунка см. на вклейке.) Типичный Р-бочонок -мономерный порин OmpG из Escherichia coli, вид сбоку. Порины - трансмембранные белки, имеющие в центре полость, через которую осуществляется диффузия малых молекул. PDB ID 2f1c
туру р-бочонков, принадлежат порины. Эти Р-бочонки состоят из 16 или 18
Р-тяжей и представляют собой заполненные водой каналы. По таким каналам осуществляется пассивная диффузия питательных веществ и продуктов обмена через наружную мембрану (рис. 4.2). Потенциально токсичные вещества большего размера не могут попасть в клетку из-за ограничений, обусловленных размерами канала. Порины и схожие с ними структуры составляют 2-3% продукции генов грамотрицательных бактерий (Wimley 2003).
130
Глава 4. Предсказание структуры мембранных белков
4.3. Особенности кристаллизации мембранных белков
Выделять ТМ белки, поверхность которых содержит как гидрофобные, так и гидрофильные участки, гораздо сложнее, чем белки, растворимые в воде. В процессе выделения естественное мембранное окружение белка разрушается и замещается молекулами детергента, при этом важно не допустить денатурации белка. Несмотря на значительные усилия в этой области, кристаллографиические структуры с высоким разрешением сегодня получены для сравнительно небольшого количества ТМ белков. В то время как принято считать, что ТМ белки составляют около 30% протеома, они очень скудно представлены в структурных базах данных, таких как PDB (Bernstein etal. 2004), составляя лишь около 1% от общего числа представленных там структур (White 2004). В таблицах 4.1 и 4.2 приведены трансмембранные а-спирали и Р-бочонки, для которых на сегодняшний день получены кристаллографические структуры (Lomize etal. 2006b). С развитием и увеличением доступности высокотехнологичных методов исследования, таких как синхротронная рентгеновская микроскопия, появилась возможность изучать структуру белков методом рентгеноструктурного анализа, используя в качестве материала исследования мельчайшие белковые кристаллы. В сочетании с новейшими методами кристаллизации, такими как использование антител для улучшения растворимости белков, а также использование липидных фаз в качестве среды кристаллизации, эти исследования в ближайшие несколько лет, вероятнее всего, приведут к заметному прогрессу в области изучения структур ТМ белков.
Таблица 4.1. Надсемейства а-спиральных трансмембранных белков из базы данных ОРМ (Lomize et al. 2006b)
Функция Надсемейство
Светозависимые транспортеры Родопсиноподобные белки
Окислительно-восстановительные транспортеры Фотосинтетические реакционные центры и фотосистемы
Потенциал-зависимые транспортеры Светособирающие комплексы
Фосфатзависимые транс-тпортеры Белки, подобные трансмембранному цитохрому b
Портеры (унипортеры, сим-портеры, антипортеры) Цитохром с оксидазы
Каналы, включая ионные Многогемовые цитохромы
4.3. Особенности кристаллизации мембранных белков
131
Окончание таблицы 4.1
Функция Надсемейство
Ферменты Протон- или натрий-проводящие АТФазы F/V/A-типа
Белки с альфа-спиральными трансмембранными якорями АТФазы Р-типа
Кассетные транспортеры, подобные транспортеру витамина В12 Одно спиральные АТФазные регуляторы Кассетные транспортеры, подобные липидным флиппазам Кассетный транспортер захвата молибдата Общий секреторный путь (Sec) Митохондриальный переносчик Надсемейство главных посредников Resistance-nodulation-cell division Симпортер дикарбоновых/аминокислот и катионов Одновалентный катионный/протонный антипортер Нейротрансмиттер натриевый симпортер Аммониевый транспортер (Amt) Транспортер метаболитов Потенциал-зависимые каналы Механочувствительный ионный канал большой проводимости (MscL) Механочувствительный ионный канал малой проводимости (MscS) Транспортер ионов металлов СогА Лиганд-зависимый ионный канал нейротрансмиттерных рецепторов Хлорный канал Дополнительные белки внешней мембраны Эпителиальный натриевый канал Транспортер иона магния (MgtE) Главный внутренний белок (MIP) Метановая монооксигеназа Ромбоидные белки Оксидоредуктаза-В дисульфидных связей (DsbB) Трансмембранный домен димеризации Т клеточного рецептора Стерил-сульфат сульфогидролаза Станнин Гликофорин А Основной оболочечный белок иновируса Субъединицы пили Белок, связанный с легочным сурфактантом
132
Глава 4. Предсказание структуры мембранных белков
Таблица 4.2. Надсемейства трансмембранных белков, имеющих структуру 3-бочонков, из базы данных ОРМ (Lomize et al. 2006b)
Источник Надсемейство
Внешняя мембрана грам-отрицательных бактерий ОМРА-подобные
Олигомерные бета-бочонки грам-положительных бактерий ОМРТ-подобные
Аутотранспортер (АТ) Тримерный аутотранспортер ОМ фосфолипаза Нуклеозид-специфичный каналообразующий мембранный порин Внешнемембранный белок FadL (FadL) OmpG порин Тримерные порины Порины сахаров Omp85-TpsB транспортеры Лиганд-зависимые белковые каналы Внешнемембранный фактор (OMF) Лейкоцидиноподобные
4.4. Базы данных
На сегодняшний день существует довольно большое количество баз данных, в которых содержатся последовательности и структуры ТМ белков. ОРМ (Lomize et aL 2006b), PDBTM (Tusnady et al. 2005b), CGDB (Sansom et al. 2008), MPDB (Raman et al. 2006) и база данных Стивена Уайта (Stephen White’s database, http://blanco.biomol.uci.edu/) содержат ТМ белки, структуры которых были определены методами рентгеноструктурного анализа и электронной дифракции, ядерного магнитного резонанса и криоэлектронной микроскопии. ОРМ, PDBTM и CGDB также содержат информацию о предположительной ориентации белков относительно мембраны. Предположение делается на основе анализа данных об энергиях переноса из воды в липидный бислой (Lomize et al. 2006а), сведений о гидрофобности и структурных особенностях белков (Tusnady et al. 2005а), а также на основе результатов молекулярнодинамических исследований с использованием крупнозернистых моделей (Sansom et al. 2008). В ОРМ содержится информация о локализации N-концов белков. TOPDB (Tusnady et al. 2008) и Mptopo (Jayasinghe et al. 2001) содержат сведения о ТМ белках неизвестной пространственной структуры, топологии которых были подтверждены экспериментально с использованием методов низкого раз
4.5. Множественные выравнивания последовательностей
133
решения, таких как слияние генов, исследования с использованием антител и мутагенез. Некоторые базы данных ТМ белков содержат информацию о специфических семействах, в том числе, о калиевых ионных каналах (Li and Gallin 2004) и рецепторах, сопряженных с G-белками (Нот et al. 2003). В других базах данных, таких как LGICdb (Donizelli et al. 2006) и TCDB (Saier et al. 2006), представлены определенные структурные или функциональных классы белков.
Набор данных Мюллера (Moller etal. 2003) требует модификации с учетом последних аннотаций из SWISS-PROT (Boeckmann et al. 2003), однако представляет особый интерес. В нем содержатся разнообразные обучающие массивы и массивы для проверки достоверности результатов исследований. Поскольку имеющиеся на сегодня базы данных пространственных структур содержат информацию преимущественно о прокариотических белках, полученные с их использованием массивы данных для обучения и проверки достоверности результатов обладают определенной специфичностью. Массивы Мюллера лишены этого недостатка. При работе с базами данных по биоинформатике следует обращать особое внимание на частоту обновления информации. Так, интенсивность появления новых последовательностей и структур в базах данных Genbank и PDB (а также случаи их исчезновения - см., например, Pomillos et al. 2005) требуют от администраторов своевременной подготовки аннотаций новых последовательностей и структур, что не всегда выполнимо в силу больших объемов.
4.5. Множественные выравнивания последовательностей
Важную роль в предсказании структуры ТМ белков играют множественные выравнивания последовательностей. Гомологичные последовательности, найденные в базах данных, можно использовать для создания профилей последовательностей, которые могут существенно увеличивать точность предсказания ТМ топологии (Kall etal. 2005; Jones 2007), а найденные шаблоны можно использовать для моделирования по гомологии.
В ходе применения традиционных методов парного выравнивания подходящие для исследуемого белка последовательности отбирают на основе значений оценочной функции. Эти значения получают с использованием матриц аминокислотных замен, таких как РАМ (Dayhoff etal. 1978) или BLOSUM (Henikoff and Henikoff 1992). Такие матрицы разрабатывались для выравнивания глобулярных белков, и, поскольку глобулярные и
134
Глава 4. Предсказание структуры мембранных белков
ТМ белки значительно отличаются по аминокислотному составу, гидрофобности и характеру консервативных фрагментов (Jones et al. 1994а), эти матрицы в принципе непригодны для выравнивания ТМ белков. В связи с этим для трансмембранных белков были разработаны специальные матрицы замен, учитывающие их свойства. Так, ТМ матрица JJT (Jones et al. 1994b) была разработана с учетом того факта, что в трансмембранных белках полярные остатки характеризуются высокой консервативностью, а гидрофобные - взаимозаменяемостью. Матрица SLIM (Muller etal. 2001) характеризуется высокой точностью при определении отдаленных гомологов в курируемом экспертами массиве последовательностей GPCR. Матрица PHAT (Ng et al. 2000) по показателям производительности превосходит JJT, особенно в области поиска по базам данных. Тем не менее, на сегодняшний день не существует независимых исследований, в которых была бы проведена оценка ТМ матриц замен с использованием общего массива данных.
Некоторые новые методы характеризуются более высоким качеством выравнивания ТМ белков. В STAM (Sharif and Gut 2004) размер штрафа, налагаемого за вставки/делеции в ТМ сегменты, выше, чем аналогичное значение для вставок/делеций в петлевые области. Кроме того, в методе применяются различные матрицы замен. В результате этого модели, построенные по гомологии с применением метода, отличаются более высокой точностью. PRALINE™ (Pirovano et al. 2008) сочетает в себе новейшие методы предсказания последовательностей с использованием мембранспеци-фичных матриц замен. Было показано, что производительность метода выше в сравнении с такими стандартными методиками множественных выравниваний, как ClustalW (Higgins etal. 1994) или MUSCLE (Edgar 2004). В исследовании использовался контрольный массив данных для оценки качества трансмембранных выравниваний BaliBASE (Bahr et al. 2001). Последние изменения, введенные в BLAST и PSI-BLAST (Altschul and Koonin 1998) для отражения состава исследуемой последовательности, теоретически должны повышать качество поиска ТМ белков (Altschul et al. 2005), однако независимые исследования в этом случае также не проводились. Усовершенствованный метод выравнивания Т-Coffee (Notredame et al. 2000), несмотря на использование одной общей матрицы оценки, отличается качеством результатов в случае высокой идентичности последовательностей. Исследования проводились на контрольном массиве структур гомологичных мембранных белков. В методе HMAP (Tang et al. 2003) значительное улучшение качества выравниваний достигается за счет применения анализа и сравнения профилей, которые содержат информацию о структурах.
4.6. Предсказание топологии трансмембранных белков
135
4.6. Предсказание топологии трансмембранных белков
4.6.1. Альфа-спиральные белки
Как уже было отмечено ранее, ТМ белки крайне скудно представлены в структурных базах данных, что значительно осложняет их изучение. В то же время, ТМ белки имеют важное биологическое и фармакологическое значение, поэтому понимание их топологии - общего числа ТМ спиралей, их границ и ориентации относительно мембраны - одна из приоритетных задач теоретического предсказания. Существуют экспериментальные методы, такие как анализ гликозилирования, создание вставок, исследования с использованием антител и создание белков слияния, которые позволяют определить локализацию отдельных областей в топологии. Однако такие исследования требуют большого количества времени, их результаты часто противоречивы (Мао et al. 2003; Kyttala et al. 2004), и, кроме того, существует риск деформации природной топологии из-за изменения белковой последовательности.
В отсутствие данных о структуре современные стратегии биоинформатики сводятся к методам предсказания на основе анализа последовательностей. Задолго до появления первых кристаллографических структур стало возможным идентифицировать ТМ спирали как фрагменты последовательности, состоявшие из гидрофобных остатков и достаточно длинные, чтобы прошить мембрану насквозь. В основе ранних методов предсказания Kyte and Doolittle (1982) и Engelman etal. (1986), а позже и Wimley and White (1996) лежали экспериментально определенные индексы гидрофобности, которые использовались для создания графиков гидрофобности исследуемого белка. Процедура включала использование метода «скользящего среднего» с длиной окна 19-21 остаток и усреднение данных, которые образовывали пики на графике (области высокой гидрофобности) и соответствовали ТМ спиралям (рис. 4.3).
С увеличением числа проанализированных последовательностей обнаружилось, что ароматические остатки Тгр и Туг имеют тенденцию располагаться группами вблизи концов трансмембранных сегментов (Wallin etal. 1997). Возможно, такие группы действуют как физические буферы, которые стабилизируют ТМ спирали внутри липидного бислоя. В ходе более поздних исследований в составе трансмембранных спиралей были обнаружены специфические мотивы последовательностей, такие как мотив GxxxG (Senes et al. 2000), а также периодические элементы, которые участвуют в упаковке спиралей и формировании пространственной структуры
136
Глава 4. Предсказание структуры мембранных белков
Рис. 4.3. График гидрофобности по Kyte-Doolittle. Белковая последовательность исследована с помощью метода «скользящего среднего» с длиной окна 19-21 остаток. В каждом положении рассчитывается средний индекс гидрофобности аминокислот внутри окна, затем полученное значение наносится на график как средняя точка окна. Приведенный график соответствует ТМ белку с 4 ТМ спиралями
белка (Samatey et al. 1995). Однако одним из наиболее важных открытий в этот период было обнаружение того факта, что положительно заряженные остатки имеют тенденцию располагаться в области цитоплазматичекой петли - закономерность, известная как правило «положительное внутри», сформулированное фон Хейном (von Heijne 1992). Все эти сведения в сочетании с предсказаниями на основе данных о гидрофобности привели к появлению ранних методов предсказания топологии, таких как TopPred (Carlos and von Heijne 1994).
4.6.1.1. Подходы, основанные на машинном обучении
Ранние методы, в основе которых лежали физико-химический принцип скользящего окна гидрофобности и правило «положительное внутри», несмотря на свой первоначальный успех, позже были вытеснены методами, основанными на машинном обучении. Преимущество последних состояло в вероятностном представлении. Некоторые методы прогнозирования, в основе которых лежит машинное обучение, приведены в таблице 4.3.
4.6. Предсказание топологии трансмембранных белков
137
Таблица 4.3. Методы предсказания топологии трансмембранных альфа-спиралей, основанные на машинном обучении
Метод URL Алгоритм Особенности
MEMSAT3 http://bioinf.cs.ucl.ac.uk/psipred/ Нейронные сети Сигнальные пептиды, МВП*, ПАГ**
MINNOU http://minnou.cchmc.org/ Нейронные сети МВП
PHDhtm http://www.predictprotein.org/ Нейронные сети Сигнальные пептиды, МВП, ограниченный
Phobius http://phobius.sbc.su.se/ Скрытые марковские модели ПАГ
ТМНММ http://www.cbs.dtu.dk/services/ ТМНММ/ Скрытые марковские модели Повторно входящие области, ПАГ
PRODIV-ТМНММ http://www.pdc.kth.se/~hakanv/ prodiv-tmhmm/ Скрытые марковские модели Ограниченный
НММТОР http ://www. enzim.hu/hmmtop/ Скрытые марковские модели МВП
ENSEMBLE http://pongo.biocomp.unibo.it/pongo/ Нейронные сети и скрытые марковские модели Мембранные петли
OCTOPUS http://octopus.cbr.su.se/ Нейронные сети и скрытые марковские модели Консенсусный
SVMtop http://biocluster.iis.sinica.edu.tw/ -bioapp/SVMtop/ Метод опорных веторов Консенсусный
PONGO http://pongo.biocomp.unibo.it/pongo/ Множественный
BPROMPT http://www.jenner.ac.uk/bprompt/ Множественный
* - прогнозирование топологии осуществляется с использованием множественных выравниваний последовательностей (МВП).
** - метод пригоден для полного анализа генома (ПАГ)
Первыми попытками применения скрытых марковских моделей (СММ) (hidden Markov models, или HMMs) к предсказанию трансмембранных топологий стали методы ТМНММ (Krogh etal. 2001) и HMMTOP (Tusnady and Simon 1998), которые, как выяснилось впоследствии, оказались весьма успешными. В ТМНММ используются циклическая модель и семь состояний трансмембранной спирали, тогда как в НММТОР скрытые марковские модели используются для распознавания одного из пяти структурных состояний (ядро спирали, внутренняя петля, наружная петля, концы спирали (С и N) и глобулярные домены). Эти состояния связаны друг с другом через значения вероятностей перехода. С помощью динамического
138
Глава 4. Предсказание структуры мембранных белков
программирования осуществляется поиск модели, которая имеет наиболее вероятную для данной последовательности топологию. В НММТОР также существует возможность зафиксировать специфические остатки в определенных областях топологии, основываясь на данных экспериментов, и получить ограниченный прогноз топологии.
Искусственные нейронные сети (ИНС, neural networks, или NNs) применяются в таких методах, как PHDhtm (Rost etal. 1996) и MEMSTAT3 (Jones 2007). В PHDhtm для согласованного предсказания топологии ТМ спиралей выполняются множественные выравнивания последовательностей с использованием комбинации двух ИНС. Первой создается сеть «последовательность-структура», которая отражает структурную предрасположенность центрального остатка в окне считывания. Затем вторая сеть, «структура-структура», сглаживает эти предрасположенности, после чего к полученному результату применяется правило «положительное внутри», и формируется общая топология ТМ спиралей. Метод MEMSTAT3, использующий нейронную сеть и динамическое программирование, позволяет не только предсказывать топологию ТМ спиралей, но также оценивать качество полученной топологии и определять возможные сигнальные пептиды. Дополнительная эволюционная информация, получаемая из множественного выравнивания, позволила увеличить точность предсказания до 80% при использовании одного набора данных (Jones 2007).
В последнее время для предсказания топологии ТМ белков применяется метод опорных векторов (support vector machines, или SVMs) (Yuan et al. 2004; Lo et al. 2008). В отличие от нейронных сетей и скрытых марковских моделей, где в качестве результата исследования можно получить множественные наборы данных, метод опорных векторов является бинарным классификатором. Для классификации многочисленных предпочтений остатков метод необходимо применять многократно, после чего полученные данные можно скомбинировать в вероятностную рамку. Хотя метод допускает реализацию и многоклассового ранжирования, она считается ненадежным, поскольку во многих случаях не существует единой математической функции, с помощью которой можно было бы разделить данные на классы. Тем не менее, метод опорных векторов дает возможность обучить машину сложным взаимосвязям между аминокислотами внутри исследуемого окна, на примере которого идет обучение, в особенности, при использовании информации об эволюции белка. Метод также является более гибким по сравнению с другими методами машинного обучения в случае проблемы переобучения, хотя наличие множества настраиваемых параметров может привести к тому, что процедура оптимизации будет занимать очень большое количество времени.
4.6. Предсказание топологии трансмембранных белков
139
□ Цито- S Внекле- II Нераспоз- Трансмембран-плазма- точный нанный ная спираль тический
Рис. 4.4. Использование нескольких методов для согласованного предсказания топологии
4.6.1.2. Согласованные подходы
Некоторые современные методы сочетают в себе различные подходы, основанные на машинном обучении. В методе ENSEMBLE (Martelli et al. 2003) используются одна нейронная сеть и две скрытые марковские модели, в методе OCTOPUS (Viklund and Elofsson 2008) применяются два ряда из четырех нейронных сетей и одной скрытой марковской модели. Оба метода отличаются более высокой точностью прогнозов по сравнению с методами, в основе которых лежит одиночный алгоритм классификации. В методе BPROMPT (Taylor et al. 2003), где также используется согласованный подход, для получения окончательной топологии выходные данные пяти различных методов прогнозирования обрабатываются с помощью Байесовской сети доверия. Нильссон и соавторы (Nilsson et al. 2002) также использовали пять методов предсказания, но для выбора окончательной топологии применялось правило большинства. Результатом работы сервера PONGO (Amico et al. 2006) являются выходные данные пяти методов оценки топологии, представленные в графическом формате для прямого сравнения. В большинстве случаев необходимо принимать к рассмотрению несколько теоретических моделей, полученных разными методами прогнозирования (рис. 4.4). Это в особенности касается белковых молекул, отличающихся сложной топологией.
4.6.1.З. Сигнальные пептиды и мебранные спирали
Одной из задач, стоящих перед современными методами предсказания топологии, является умение отличать ТМ спирали от других элементов структуры, содержащих большое количество гидрофобных остатков. К таким
140
Глава 4. Предсказание структуры мембранных белков
Рис. 4.5. (Цветную версию рисунка см. на вклейке.) Субъединица калиевого канала из Streptomyces lividans, имеющая в своем составе мембраную спираль (в центре, сверху). PDB ID 1r3j
элементам относятся мотивы-мишени: сигнальные пептиды и сигнальные якоря, амфифильные спирали и мембранные спирали - спирали, проникающие в мембрану, но входящие и выходящие из нее на одной и той же стороне. Последние характерны для многих семейств ионных каналов (рис. 4.5).
Профили гидрофобности таких белковых структур и ТМ спиралей характеризуются высокой степенью сходства, что часто ведет к перекрыванию между предсказаниями различных типов. Если трактовать вышеперечисленные элементы как ТМ спирали, то последующее предсказание топологии, вероятнее всего, будет весьма отрывочным. Некоторые методы предсказания, такие как SignalP (Berendsen et al. 2004) и TargetP (Emanuelsson et al. 2007), эффективны при определении сигнальных пептидов. Их можно использовать в качестве предварительного фильтра, предшествующего анализу с использованием методов предсказания ТМ топологий. В методе Phobias (Kall et al. 2004) для решения проблемы сигнальных пептидов в предсказании топологии ТМ белков используется скрытая марковская модель. В PolyPhobius (Kall etal. 2005) точность предсказания увеличивается за счет включения информации о гомологии. В других методах, таких как TOP-MOD (Viklund et al. 2006) и OCTOPUS, были предприняты попытки включить в процесс предсказания ТМ топологии определение мембранных областей, однако этот подход нуждается в усовершенствовании. Основной проблемой, в особенности касающейся мембранных спиралей, является отсутствие надежных данных, которые можно использовать для машинного обучения.
4.6.2. Белки, имеющие структуру 0-бочонка
Количество а-спиральных ТМ белков как в полных протеомах, так и в базах данных пространственных структур относительно велико по сравнению с количеством белков, имеющих структуру Р-бочонков. В связи с этим методы предсказания структуры и топологии Р-бочонков развиваются менее интенсивно. Еще одна причина различного уровня развития методов, вероятно, состоит в том, что предсказывать структуру ТМ а-спи-ралей относительно легко ввиду большого количества входящих в их со
4.6. Предсказание топологии трансмембранных белков
141
став гидрофобных остатков. Антипараллельные р-тяжи ТМ р-бочонков содержат чередующиеся полярные и гидрофобные аминокислоты. Благодаря такому строению гидрофобные остатки обращены к мембране, а полярные - к поверхности, которая контактирует с растворителем. В основе ранних методов предсказания топологии р-тяжей лежал анализ данных скользящей рамки гидрофобности, который позволял выявить чередующиеся элементы структуры (Schirmer and Cowan 1993). В других методах использовались специальные эмпирические правила, разработанные на основе информации о предрасположенностях аминокислот, а также на основе данных о структурной природе белков (Gromiha and Ponnuswamy 1993). С ростом числа Р-бочонков, структура которых известна с атомарным разрешением, стали появляться методы, основанные на машинном обучении. К числу таких методов относятся нейронные сети (Jacoboni et al. 2001; Gromiha et al. 2004), скрытые марковские модели (Martelli et al. 2002; Liu etal. 2003; Bagos etal. 2004) и предсказание на основе метода опорных векторов (Park et al. 2005), где используются одиночные и множественные выравнивания последовательностей. Ряд методов предсказания структуры и топологии р-бочонков, в основе которых лежит машинное обучение, приведен в таблице 4.4.
Таблица 4.4. Методы предсказания топологии трансмембранных бета-бочонков, основанные на машинном обучении
Метод URL Алгоритм Особенности
B2TMR http://gpcr.biocomp.unibo.it/predictors/ ИНС МВП*
ТМВЕТА-NET http ://psfs. cbrc.j р/tmbeta-net/ ИНС МВП, ПАГ**
НММ- B2TMR http://gpcr.biocomp.unibo.it/predictors/ смм МВП
PROFtmb http ://www.rostlab. org/services/P ROF tmb/ смм ПАГ
PRED-ТМВВ http://biophysics.biol.uoa.gr/PREDTMBB/ смм ПАГ
ТМВЕТА-SVM http ://tmbeta-s vm. cbrc.jp/ SVM ПАГ
TMB-Hunt2 http://bmbpcu36.leeds.ac.uk/ СММ + SVM ПАГ
* - прогнозирование топологии осуществляется с использованием множественных выравниваний последовательностей (МВП).
** - метод пригоден для полного анализа генома (ПАГ)
142
Глава 4. Предсказание структуры мембранных белков
4.6.3. Полногеномный анализ
В ходе реализации крупномасштабных проектов по изучению геномов и протеомов часто обнаруживаются новые белки, клеточная локализация и функции многих из которых оказывается неизвестной. С помощью некоторых описанных выше методов можно довольно точно предсказать ТМ топологию белков. Однако арсенал методов, с помощью которых можно отличить ТМ белки от глобулярных, невелик. Для проведения подобной дифференцировки необходимо наличие специально настроенного метода и автономного программного пакета, поскольку методы предсказания, основанные на интернет-технологиях, непригодны для обработки больших объемов данных. Некоторые методы, пригодные для полногеномного анализа ТМ альфа-спиралей и бета-бочонков, приведены в таблицах 4.3 и 4.4. В целом, число ошибок существенно снижается благодаря использованию предварительного фильтрования. Из-за того, что многие глобулярные белки имеют в своем составе сигнальные последовательности, в ходе анализа их часто ошибочно относят к трансмембранным белкам. Предварительное фильтрование предполагает использование таких методов, как SignalP и TargetP, и позволяет убрать из рассмотрения сигнальные и транзитные пептиды. В настоящее время для лучших методов предсказания степень ошибки составляет менее 1% в случае а-спиральных ТМ белков (Jones 2007) и менее 6% в случае ТМ белков, имеющих структуру р-бочонков (Park et al. 2005). На рис. 4.6 приведены результаты применения метода дифференцировки а-спиральных ТМ белков к группе протеомов.
4.6.4. Наборы данных, гомологичность, точность и перекрестная проверка
При разработке любых методов предсказания крайне важно как для обучения, так и для оценки достоверности результатов использовать данные высокого качества. Извлечение из доступных баз данных обучающей выборки представляет собой весьма трудоемкую задачу и требует принятия большого количества важных решений. В качестве примера рассмотрим поиск по базе данных PDB с использованием ключевого слова «трансмембранный». Среди полученных в этом случае результатов будут как трансмембранные белки, закодированные в геноме, так и трансмембранные белки, не являющиеся нативными, например, пептид пчелиного яда 1ВН1, нарушающий структурную целостность бислоев, или бактериальный коли-цин 1CII, который используется для образования пор в наружных мембранах конкурирующих бактерий. Более того, в базах данных довольно часто
4.6. Предсказание топологии трансмембранных белков
143
Количество белков
Рис. 4.6. Одиннадцать протеомов проанализированы с помощью метода дифференцировки трансмембранных/глобулярных белков MEMSAT3. Для белков, которые определены как трансмембранные, выполнено полное предсказание ТМ топологии. Ось абсцисс - номер трансмембранной спирали. Ось ординат - количество белков
встречаются ошибки, которые привносят в метод искажения. В случае методов, основанных на машинном обучении, такие искажения не оказывают существенного влияния на результаты исследования. Для меньших по объему массивов данных эта проблема представляется более значимой.
Еще один вопрос, требующий рассмотрения, - это гомологичность последовательностей в выборках, которая для большинства случаев составляет 30-40% идентичности последовательностей. Поскольку данные о структурах ТМ белков сейчас вызывают повышенный интерес, для них этот показатель, возможно, несколько выше приведенного, который характерен, скорее, для выборок глобулярных белков. Несмотря на повышенный риск переобучения, или оверфиттинга, крайне важно использовать обучающие выборки достаточного размера. Для всех методов, основанных на машинном обучении, характерны множество свободных параметров и, как следствие, потенциальная возможность переобучения. Это значит, что
144
Глава 4. Предсказание структуры мембранных белков
вместо выявления некого паттерна последовательности алгоритм может заучить её «наизусть» со всеми возможными ошибками, которые она может содержать. Для переобученного метода характерно воспроизведение обучающих примеров с высокой точностью, в то время как в случае примеров, не встречавшихся ранее, метод будет малопригоден. Во избежание переобучения при оценке точности метода предсказания важно использовать обучающие и тестовые выборки с низкой гомологией.
Во всех случаях важно применять строгий скользящий контроль, или кросс-валидацию, достоверности данных. Скользящий контроль является статистическим методом и состоит в разделении исходной выборки на подвыборки меньшего размера. Каждая такая подвыборка проходит проверку на модели, настроенной с использованием остальных подвыборок. Процесс повторяется до тех пор, пока все подвыборки не пройдут проверку. При предсказании ТМ топологии наиболее часто используют два типа скользящего контроля. При контроле по К блокам (К-fold cross-validation) выборку делят на К подвыборок. Одну подвыборку из К, содержащую несколько последовательностей, принимают за тестовую выборку, а остальные К-1 подвыборок используют как обучающие. Процедуру повторяют К раз, при этом каждая из К подвыборок однократно используется в качестве тестовой. Затем полученные результаты (К штук) либо комбинируют, либо усредняют для получения общей оценки. Более строгий, хотя и более затратный с точки зрения вычислений тип скользящего контроля представляет собой контроль по отдельным объектам (LOOCV, leave-one-out cross-validation, кросс-валидация с исключенным элементом), также известный как критерий складного ножа (jack knife method). Этот метод предполагает использование одной последовательности из выборки в качестве тестовой, в то время как остальные последовательности составляют обучающую выборку. Процедура повторяется, пока каждая последовательность не пройдет однократную проверку. Этот метод представляет собой скользящий контроль по К блокам, где К равно числу последовательностей в выборке.
В некоторых исследованиях предпринимались попытки сравнить точность различных методов предсказания топологии трансмембранных белков (например, Melen et al. 2003), однако с тех пор сами методы были значительно усовершенствованы. В настоящее время принято считать, что при использовании лучших методов прогнозирования удается получить правильные топологии для 80-93% белков, хотя довольно сложно проводить оценку методов в отсутствие независимого скользящего контроля, в котором для проверки использовался бы общий набор данных. Методы могут отличаться высоким качеством прогнозов при тестировании на определенной выборке, например, на таком, который содержит небольшое
4.7. Предсказание пространственной структуры
145
количество сигнальных пептидов; при этом в случае массива данных, который содержит множество сигнальных пептидов, качество прогнозов может снижаться. Методы могут быть оптимизированы с использованием выборки, которая содержит множество слабо гидрофобных ТМ спиралей. В этом случае высоковероятна чрезмерная склонность методов к обнаружению ТМ спиралей в анализируемых массивах. На сегодняшний день наиболее качественные массивы данных, в которых топологии получены исключительно структурными методами, а степень гомологии снижена, содержат не более 150 последовательностей (Lomize etal. 2006b). Недостаточное согласование среди этих данных, а также нехватка необходимых данных скользящего контроля таким образом означают, что различия в точности используемых методов предсказания могут быть результатом различий в процессе обучения и проверки массивов данных, а не результатом заметных различий в качестве используемых подходов.
4.7. Предсказание пространственной структуры
Как и в случае глобулярных белков, для предсказания пространственной структуры ТМ белков используются два подхода: моделирование ab initio и моделирование по гомологии, описанные в главах 1 и 3 настоящей книги.
Моделирование по гомологии, известное также как сравнительное моделирование, предполагает использование близких структур в качестве шаблонов для построения пространственной модели исследуемого белка. В основу метода положено наблюдение, согласно которому структура белка более консервативна, чем аминокислотная последовательность. Следовательно, даже в случае выраженной дивергенции последовательностей белки могут обладать общими структурными свойствами, в частности, схожей укладкой, при условии, что последовательности обладают обнаруживаемым сходством (более 30% идентичности последовательности). Получение кристаллографических структур высокого разрешения сопряжено с рядом технических сложностей. В особенности это касается ТМ белков. В этой связи моделирование по гомологии представляется перспективным методом. Полученные с его помощью структурные модели могут быть полезны при разработке гипотез функционирования белков. Также этот метод исследования может помочь определить направление дальнейшей экспериментальной работы. Сам процесс моделирования по гомологии можно условно разбить на четыре этапа: выбор шаблона, выравнивание последовательностей исследуемого белка и шаблона, конструирование модели
146
Глава 4. Предсказание структуры мембранных белков
Таблица 4.5. Некоторые из наиболее часто используемых программ моделирования по гомологии (приведено с изменениями из Wallner and Elofsson 2005)
Программа Описание URL
Modeller Моделирование посредством удовлетворения пространственных ограничений. Включает в себя моделирование петель de novo. http://www.salilab.org/ modeller/
SegMod/ENCAD Моделирование посредством посредством подгонки сегментов с уточнением при помощи молекулярной динамики. http://csb.stanford.edu/ levitt/segmod/
SWISS-MODEL Удаленное моделирование посредством сборки из жестких фрагментов. http://swissmodel. expasy.org/
3D-JIGSAW Удаленное моделирование с минимизацией энергии в программе CHARMM. http://bmm. cancerresearchuk.org/ ~3djigsaw/
Nest Моделирование на основе множественных шаблонов с использованием методов искуственной эволюции. http://wiki.c2b2. columbia.edu/ honiglab_public/
Builder Моделирование петель и боковых цепей с помощью метода самосогласованного среднего поля (SCMF) (Koehl and Delarue 1996). On request: koehl@cs.ucdavis.edu
Jackal Модлирование с использованием различных программ. http ://wiki. c2b2. columb ia.edu/ honiglab_ public/index.php
SCWRL3 Моделирование боковых цепей на основе библиотеки ротамеров с учетом конформации основной цепи. http://dunbrack.fccc. edu/SCWRL3.php
и оценка модели. На каждом этапе можно осуществлять необходимое количество итераций для улучшения качества получаемой в итоге модели (Sanchez and Sali 1997; MartiRenom et al. 2000). Некоторые программы для моделирования по гомологии приведены в таблице 4.5.
Среди методов, приведенных в таблице 4.5, только SWISS-MODEL (Peitsch 1996), имеющий интерфейс 7TM/GPCR, был разработан специально для ТМ белков. В связи с этим особое внимание следует обращать на отсутствие в модели полярных боковых цепей, которые выдаются в гидрофобную область мембраны. Существуют инструменты моделирования, которые учитывают особенности структуры трансмембранных белков, связанные с боковыми цепями, например, SCWRL (Canutescu etal.
4.7. Предсказание пространственной структуры
147
2003). Такие инструменты не решают проблему полностью, однако их использование для построения модели повышает качество внемембранных областей. На сегодняшний день имеет место недостаток специфических инструментов моделирования ТМ белков. Тем не менее, последние исследования показывают, что методы биоинформатики, применяемые в настоящее время для изучения растворимых белков - от сопоставления профилей гидрофобности до предсказания вторичной структуры и моделирования по гомологии - с тем же успехом применяются для изучения ТМ белков (Forest et al. 2006). Действительно, важными областями моделирования ТМ белков являются определение и подтверждение лекарственных мишеней, а также определение и оптимизация состава лигандов. Методы разработки лекарств, в основе которых лежит моделирование по гомологии, на сегодняшний день применялись для исследования большого количества киназ, в том числе таких, как эпидермальный фактор роста-белок рецептора тирозин-киназы (Ghosh etal. 2001), тирозин-киназа Брутона (Mahajan et al. 1999), а также киназа 3 Януса (Sudbeck et al. 1999).
Моделирование ab initio, или de novo, предполагает создание пространственной модели в отсутствие какой бы то ни было информации о структуре белка или его гомологов. В данном случае существует три основных направления исследований: создание частично перекрывающихся белковых структур низкого разрешения, поиск методов точной оценки энергии, эффективные методы отбора проб. Большинство методов этой группы связаны с исследованием глобулярных белков, однако предпринимались также попытки предсказания структуры трансмембранных белков.
ROSETTA (Rohl et al. 2004) - сервер для моделирования ab initio, на котором для аминокислотной последовательности можно определить структуру с минимальной энергией. Определение осуществляется на основе анализа функций потенциальной энергии. В ходе формирования прогноза между отдельными звеньями метода постоянно поддерживается обратная связь, что позволяет оптимизировать функции потенциальной энергии и алгоритмы расчета. В модифицированной версии ROSETTA (Barth et al. 2007) используются функции, описывающие взаимодействия мембранной части белков с мембраной на атомарном уровне, взаимодействия мембранных белков с липидами в явном виде, а водородные связи -неявно. Полученные результаты свидетельствуют о том, что используемая модель отражает ключевые физические свойства системы, управляющие процессами растворения и отвечающие за стабильность мембранных белков. Метод позволяет предсказывать структуру малых ТМ белков (менее 150 остатков) с разрешением лучше 2,5 А. Такая точность сопоставима с предсказаниями для малых водорастворимых белковых доменов. Мем
148
Глава 4. Предсказание структуры мембранных белков
бранный метод ROSETTA в сочетании с моделированием по гомологии и методами сборки доменов применялся для моделирования структуры калиевых каналов Kvl.2 и KvAP. Полученные в результате модели отличались высокой степенью сходства с соответствующими кристаллографическими структурами. Моделирование открытого и закрытого состояний этих каналов способствовало объяснению механизма потенциал-зависимого открывания и закрывания каналов. Было показано, что работа воротного механизма опосредована конформационными изменениями в структуре каналов. На основе данных моделирования были выдвинуты гипотезы, которые можно было проверить в ходе дальнейшей экспериментальной работы (Yarov-Yarovoy et al. 2006).
FRAGFOLD (Jones 2007) - метод предсказания третичной структуры белков, в основе которого лежит сборка фрагментов супервторичной структуры с использованием алгоритма имитации отжига. Основная идея метода состоит в том, чтобы значительно сузить сканируемое конформационное пространство посредством предварительного отбора фрагментов из библиотек белковых структур высокого разрешения. В методе FILM (Pellegrini-Calace etal. 2003) к термам потенциальной энергии FRAGE-FOLD (парные взаимодействия, взаимодействия с растворителем, ковалентные и водородные связи) добавляется мембранный потенциал. Потенциал был получен в результате статистического анализа выборки, содержавшей 640 трансмембранных спиралей с экспериментально определенными топологиями. Спирали принадлежали 133 белкам, извлеченным из базы данных SWISS-PROT. Результаты, полученные после применения метода для предсказания заранее известной структуры белков небольшого размера, показывают, что метод характеризуется приемлемым уровнем точности как в случае предсказания топологии спиралей, так и в случае предсказания конформации белков.
4.8. Перспективы развития методов предсказания структуры мембранных белков
Несмотря на успешный опыт применения ROSETTA и FILM, в будущем предстоит устранить множество ограничений, которыми эти методы обладают. В настоящее время главным ограничением являются сложности при работе с большими трансмембранными структурами. Комбинаторная природа методов ab initio делает их громоздкими с вычислительной точки
Литература
149
зрения вычислений и непригодными для изучения структур, размер которых превышает 150 аминокислот. Существует несколько способов преодолеть это ограничение. Самый простой из них, пригодный для применения в FILM, состоит в создании меньших по объему библиотек фрагментов супервторичной структуры, в которых содержатся только структуры мембранных белков. В этом случае поиск фрагментов осуществляется среди конформаций, которые являются составными частями больших трансмембранных структур. Дальнейшего улучшения методов можно достичь, заменив простые суперспиральные мотивы структурными фрагментами большего размера. Усовершенствование ROSETTA, которое позволит применять метод для прогнозирования структуры доменов большого размера, предполагает развитие методов отбора конформаций и поиск способов более точного учета электростатических взаимодействий.
Литература
Altschul SF, Koonin EV (1998) Iterated profile searches with PSI-BLAST - a tool for discovery in protein databases. Trends Biochem Sci 23:444 447
Altschul SF, Wootton JC, Gertz EM, et al. (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272:5099-5100
Amico M, Finelli M, Rossi I, et al. (2006) PONGO: a web server for multiple predictions of all alpha transmembrane proteins. Nucleic Acids Res 34:169 172
Bagos PG, Liakopoulos TD, Spyropoulos IC, et al. (2004) A Hidden Markov Model method, capable of predicting and discriminating beta-barrel outer membrane proteins. BMC Bioinformatics 5:29
Bahr A, Thompson JD, Thierry JC, et al. (2001) BAliBASE (Benchmark Alignment dataBASE): enhancements for repeats, transmembrane sequences and circular permutations. Nucleic Acids Res 29:323-326
Barth P, Schonbrun J, Baker D (2007) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci USA 104:15682-15687
Bendtsen JD, Nielsen H, von Heijne G, et al. (2004). Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783-795
Bernstein FC, Koetzle TF, Williams GJB, et al. (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol 112:535-542
Boeckmann B, Bairoch A, Apweiler R, et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31:365-370
Canutescu AA, Shelenkov AA, Dunbrack RL (2003) A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci 12:2001-2014
Claros MG, von Heijne G (1994) TopPred П: an improved software for membrane protein structure predictions. Comput Appl Biosci 10:685-686
Dayhoff MO, Schwartz RM, Orcutt BC (1978) A model of evolutionary change in proteins. Atlas Protein Seq Struct 5:345-352
Donizelli M, Djite MA, Le Novere N (2006) LGICdb: a manually curated sequence database after the genomes. Nucleic Acids Res 34:267-269
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792-1797
Emanuelsson O, Brunak S, von Heijne G, et al. (2007) Locating proteins in the cell using Target P, SignalP and related tools. Nat Protoc 2:953-971
150
Глава 4. Предсказание структуры мембранных белков
Engelman DM, Steitz ТА, Goldman А (1986) Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu Rev Biophys Biophys Chem 15:321-353
Forrest LR, Tang CL, Honig В (2006) On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys J 91:508-517
Ghosh S, Liu XP, Zheng Y, et al. (2001) Rational design of potent and selective EGFR tyrosine kinase inhibitors as anticancer agents. Curr Cancer Drug Targets 1:129-140
Gromiha MM, Ponnuswamy PK (1993) Prediction of transmembrane beta-strands from hydrophobic characteristics of proteins. Int J Pept Protein Res 42:420-431
Gromiha MM, Ahmad S, Suwa M (2004) Neural network-based prediction of transmembrane betastrand segments in outer membrane proteins. J Comput Chem 25:762-767
Henikoff S, Henikoff JG (1992) Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA 89:10915-10919
Higgins D, Thompson J, Gibson T, et al. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673-4680
Hom F, Bettier E, Oliveira L, et al. (2003) GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res 31:294-297
Jacoboni I, Martelli PL, Fariselli P, et al. (2001) Prediction of the transmembrane regions of betabarrel membrane proteins with a neural network-based predictor. Protein Sci 10:779-787
Jayasinghe S, Hristova K, White SH (2001) MPtopo: a database of membrane protein topology. Protein Sci 10:455-458
Jones DT (2001) Predicting novel protein folds by using FRAGFOLD. Proteins 5:127-132
Jones DT (2007) Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23:538-544
Jones DT, Taylor WR, Thornton JM (1994a) A model recognition approach to the prediction of allhelical membrane protein structure and topology. Biochemsitry 33:3038-3049
Jones DT, Taylor WR, Thornton JM (1994b) A mutation data matrix for transmembrane proteins. FEBS Lett 339:269-275
KJabunde T, Hessler G (2002) Drug design strategies for targeting G-protein-coupled receptors. ChemBioChem 3:928-944
Koehl P, Delarue M (1996) Mean-field minimization methods for biological macromolecules. Curr Opin Struct Biol 6:222-226
Krogh A, Larsson B, von Heijne G, et al. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567-580
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105-132
Kyttala A, Ihrke G, Vesa J, et al. (2004) Two motifs target Batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol Biol Cell 15:1313-1323
Kall L, Krogh A, Sonnhammer E (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338:1027-1036
Kall L, Krogh A, Sonnhammer E (2005) An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 21:251-257
Li B, Gallin WJ (2004) VKCDB: voltage-gated potassium channel database. BMC Bioinformatics 5:3
Liu Q, Zhu YS, Wang BH, et al. (2003) A HMM-based method to predict the transmembrane regions of beta-barrel membrane proteins. Comput Biol Chem 27:69-76
Lo A, Chiu HS, Sung TY, et al. (2008) Enhanced membrane protein topology prediction using a hierarchical classification method and a new scoring function. J Proteome Res 7:487-496
Lomize AL, Pogozheva ID, Lomize MA, et al. (2006a) Positioning of proteins in membranes: a computational approach. Protein Sci 15:1318-1333
Lomize MA, Lomize AL, Pogozheva ID, et al. (2006b) OPM: orientations of proteins in membranes database. Bioinformatics 22:623-625
Mahajan S, Ghosh S, Sudbeck EA, etal. (1999) Rational design and synthesis of a novel antileukemic agent targeting Bruton’s tyrosine kinase (ВТК), LFM-A13. J Biol Chem 274:9587-9599
Mao Q, Foster BJ, Xia H, et al. (2003) Membrane topology of CLN3, the protein underlying Batten disease. FEBS Lett 541:40-46
Литература
151
Martelli PL, Fariselli P, Krogh A, et al. (2002) A sequence-profile-based HMM for predicting and discriminating beta barrel membrane proteins. Bioinformatics 18:46-53
Martelli PL, Fariselli P, Casadio R (2003) An ENSEMBLE machine learning approach for the prediction of all-alpha membrane proteins. Bioinformatics 19:205-211
Marti-Renom MA, Stuart AC, Fiser A, et al. (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291-325
Melen K, Krogh A, von Heijne G (2003) Reliability measures for membrane protein topology prediction algorithms. J Mol Biol 327:735-744
Moller S, Kriventseva EV, Apweiler R (2000) A collection of well characterised integral membrane proteins. Bioinformatics 16:1159-1160
Muller T, Rahmann S, Rehmsmeier M (2001) Non-symmetric score matrices and the detection of homologous transmembrane proteins. Bioinformatics 17:182-189
Ng PC, Henikoff JG, Henikoff S (2000) PHAT: a transmembrane-specific substitution matrix. Predicted hydrophobic and transmembrane. Bioinformatics 16:760-676
Nilsson J, Persson B, von Heijne G (2002) Prediction of partial membrane protein topologies using a consensus approach. Protein Sci 11:2974—2980
Notredame C, Higgins D, Heringa J (2000) Т-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205-217
Park KJ, Gromiha MM, Horton P, et al. (2005) Discrimination of outer membrane proteins using support vector machines. Bioinformatics 21:4223-4229
Peitsch MC (1996) ProMod and Swiss-Model: internet-based tools for automated comparative protein modelling. Biochem Soc Trans 24:274-279
Pellegrini-Calace M, Carotti A, Jones DT (2003) Folding in lipid membranes (FILM): a novel method for the prediction of small membrane protein 3D structures. Proteins 50:537-545
Pirovano W, Feenstra KA, Heringa J (2008). PRALINETM: a strategy for improved multiple alignment of transmembrane proteins. Bioinformatics 24:492-497
Pomillos O, Chen Y, Chen AP, et al. (2005) X-ray structure of the EmrE multidrug transporter in complex with a substrate. Science 310:1950-1953
Raman P, Cherezov V, Caffrey M (2006) The membrane protein data bank. Cell Mol Life Sci 63:36-51
Rohl CA, Strauss CE, Misura KM, et al. (2004) Protein structure prediction using Rosetta. Method Enzymol 383:66-93
Rost B, Fariselli P, Casadio R (1996) Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci 4:521-533
Saier MH, Tran CV, Barabote RD (2006) TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res 34:181-186.
Samatey FA, Xu C, Popot JL (1995) On the distribution of amino acid residues in transmembrane alpha-helix bundles. Proc Natl Acad Sci USA 92:4577-4581
Sansom SP, Scott KA, Bond PJ (2008) Coarse-grained simulation: a high-throughput computational approach to membrane proteins. Biochem Soc Trans 36:27-32
Schirmer T, Cowan SW (1993) Prediction of membrane-spanning beta-strands and its application to maltoporin. Protein Sci 2:1361-1363
Senes A, Gerstein M, Engelman DM (2000) Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with betabranched residues at neighboring positions. J Mol Biol 296:921-936
Shafrir Y, Guy HR (2004) STAM: simple transmembrane alignment method. Bioinformatics 20:758-769
Sanchez R, Sali A (1997) Advances in comparative protein-structure modelling. Curr Opin Struct Biol 7:206-214
Sudbeck EA, Liu XP, Narla RK, et al. (1999) Structure-based design of specific inhibitors of Janus kinase 3 as apoptosis-inducing antileukemic agents. Clin Cancer Res 5:1569-1582
Tang CL, Xie L, Koh IY, et al. (2003) On the role of structural information in remote homology detection and sequence alignment: new methods using hybrid sequence profiles. J Mol Biol 334:1043-1062
Taylor PD, Attwood TK, Flower DR (2003) BPROMPT: a consensus server for membrane protein prediction. Nucleic Acids Res 31:3698-3700
152
Глава 4. Предсказание структуры мембранных белков
Tusnddy GE, Simon I (1998) Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol 283:489-506
Tusnddy GE, Dosztanyi Z, Simon I (2005a) TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics 21:1276-1277
Tusnddy GE, Dosztanyi Z, Simon I (2005b) PDB TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 33:275-278
Tusnddy GE, Kalmar L, Simon I (2008) TOPDB: topology data bank of transmembrane proteins. Nucleic Acids Res 36:234—239
Viklund H, Elofsson A (2008) OCTOPUS: improving topology prediction by two-track ANNbased preference scores and an extended topological grammar. Bioinformatics 24:1662-1668
Viklund H, Granseth E, Elofsson A (2006) Structural classification and prediction of reentrant regions in alpha-helical transmembrane proteins: application to complete genomes. J Mol Biol 361:591-603
Wallin E, Tsukihara T, Yoshikawa S, et al. (1997) Architecture of helix bundle membrane proteins: an analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci 6:808-815
Wallner B, Elofsson A (2005) Pcons5: combining consensus, structural evaluation and fold recognition scores. Protein Sci 14:1315-1327
White S (2004) The progress of membrane protein structure determination. Protein Sci 13:1948-1949 Wimley WC (2003) The versatile beta-barrel membrane protein. Curr Opin Struct Biol 13:404-411 Wimley WC, White SH (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol 3:842-848
Yarov-Yarovoy V, Baker D, Catterall WA (2006) Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc Natl Acad Sci USA 103:7292-7297
Yuan Z, Mattick JS, Teasdale RD (2004) SVMtm: support vector machines to predict transmembrane segments. J Comput Chem 25:632-636
von Heijne G (1992) Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol 225:487-494
Глава 5
Методы биоинформатики для изучения структуры и функций неупорядоченных белков
Питер Томпа
Белки с присущей неупорядоченностью* 1 (БПН) (IDPs, от «intrinsically disordered proteins») существуют и функционируют, не обладая выраженной структурой, что требует пересмотра парадигмы «структура-функция». Все больше доказательств находится тому, что они выполняют важные функции при передаче сигналов и регуляции транскрипции, в первую очередь у эукариот. С помощью множества биофизических методов исследования была показана структурная неупорядоченность около 500 белков, и в основу классификации функций этих белков с использованием различных схем легли исследования функций. Косвенные данные свидетельствуют о том, что неупорядоченность структуры довольно широко распространена: только в человеческом протеоме насчитывается несколько тысяч белков с выраженной неупорядоченностью в структуре. Чтобы сократить разрыв между известными и предполагаемыми БПН, был разработан целый спектр алгоритмов биоинформатики, которые могут достоверно предсказывать неупорядоченное состояние в структуре белка на основе анализа аминокислотной последовательности. Также предпринимались попытки предсказывать функции БПН, хотя и с гораздо меньшим успехом. Поскольку эта группа белков эволюционировала довольно быстро, а в основе
Peter Тотра
Institute of Enzymology, Biological Research Center,
Hungarian Academy of Sciences,
1518 Budapest, Hungary
e-mail: tompa@enzim.hu
1 Как и для многих других терминов, для термина «intrinsically disordered proteins» в настоящее время нет общепринятого русского эквивалента. Нам показалось, что предлагаемый вариант является наиболее удачным. Прим, перев.
154 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
функционирования лежат, как правило, короткие мотивы, число фрагментов последовательностей, которые позволяли бы распознавать функции БПН, заметно ограничено. В настоящей главе мы приводим краткий обзор области исследований БПН, уделяя особое внимание их функциям и методам биоинформатики, разработанным для прогнозирования структуры и функций БПН. Также предлагаются и обсуждаются возможные перспективные направления исследований в этой области.
5.1. Идея неупорядоченности белков
Основная идея классической парадигмы, связавшей функционирование белков со стабильной пространственной структурой, широко и успешно применялась при интерпретации функций ферментов, рецепторов и структурных белков. Десятилетия попыток определить структуру белков и последние исследовательские программы в области структурной геномики привели к тому, что на сегодняшний день существует 50000 структур высокого качества, размещенных в базе данных белков the Protein Data Bank (PDB) (www.pdb.org), что лишь способствовало упрочнению традиционных взглядов в области. Однако появившиеся в последнее время сведения о том, что многие белки или отдельные их части не обладают легко определяемой пространственной структурой в природных, физиологических условиях поставили под сомнение универсальность этой парадигмы (Тотра 2002, 2005; Dyson and Wright 2005; Uversky et al. 2005). В условиях быстрого накопления данных в поддержку этого нового альтернативного взгляда на белки стала неоспоримой необходимость переоценки и расширения структурно-функциональной парадигмы (Wright and Dyson 1999).
С помощью ряда биофизических методов, главным образом рентгеновской кристаллографии, ЯМР, малоуглового рентгеновского рассеяния и кругового дихроизма, было показано, что белки с присущей неупорядоченностью, или неструктурированностью, (БПН) или отдельные области таких белков (ОПН) (IDRs, от «intrinsically disordered regions (of proteins)») в структурном отношении представляют особой не определенную конформацию, а флуктуирующий ансамбль различных структурных состояний (Тотра 2002, 2005; Dyson and Wright2005; Uversky et al. 2005). На первый взгляд они напоминают денатурированное состояние глобулярных белков. Однако детальный структурный анализ показывает, что различные БПН могут заселять любые конформационные состояния в диапазоне от полностью неупорядоченного (клубок) до компактного (расплавленная глобула), с характерным распределением переходных вторичных и третичных контактов (Uversky et al. 2000; Uversky 2002). В отличие от денату
5.2. Свойства последовательностей БПН
155
рированных глобулярных белков, функции БПН являются прямым следствием их неупорядоченного состояния и связаны преимущественно с процессами регуляции передачи сигналов и транскрипции генов (lakoucheva et al. 2002; Ward et al. 2004; Tompa et al. 2006). Схемы функциональной классификации БПН основаны либо на функции, являющейся прямым следствием неупорядоченности, либо на временном/постоянном связывании с молекулами-партнерами (Dunker et al. 2002; Tompa 2002, 2005).
БПН способны не только функционировать, несмотря на отсутствие у них стабильной структуры, - структурная неупорядоченность обеспечивает функциональные преимущества при выполнении регуляторных функций, таких как разделение специфичности и силы связывания (Wright and Dyson 1999), приспособляемость к различным партнерам (Tompa et al. 2005), увеличение скорости взаимодействия (Pontius 1993) и частое участие в посттрансляционных модификациях (lakoucheva et al. 2004). Эти преимущества позволяют БПН занимать уникальные функциональные ниши и объясняют успех неупорядоченности белков в эволюции, с критической разницей в частоте для эукариот и прокариот (lakoucheva etal. 2002; Ward et al. 2004; Tompa et al. 2006). Эти преимущества объясняют также высокий уровень неупорядоченности в функционально важных регуляторных белках, которые также играют важную роль в развитии заболеваний, таких как белки-прионы (Lopez Garcia et al. 2000), BRCA1 (Mark et al. 2005), белка tau (Schweers etal. 1994), p53 (Bell etal. 2002) и а-синуклеина (Weinreb etal. 1996). Самая полная на сегодняшний день коллекция БПН - база данных DisProt (www.disprot.org) - содержит около 500 неупорядоченных белков, обнаруженных преимущественно случайным образом (Sickmeier et al. 2007). Применение методов прогнозирования, основанных на таких коллекциях белков, однако, позволяет предположить, что в протеомах многоклеточных около 5-15% белков обладают полностью неупорядоченной структурой и 30-50% белков содержат хотя бы один протяженный неупорядоченный участок (lakoucheva etal. 2002; Ward etal. 2004; Tompa etal. 2006). Чтобы восполнить этот заметный пробел в знаниях, множество усилий тратится на разработку алгоритмов биоинформатики, которые бы позволяли предсказывать неупорядоченность и функции на основе анализа аминокислотной последоательности. Настоящий обзор посвящен принципам и последним разработкам в области исследования БПН.
5.2. Свойства последовательностей БПН
В основе современных методов прогнозирования неупорядоченности лежат различные принципы. Тем не менее, БНП присущи некоторые об
156 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
щие свойства - аминокислотный состав и аминокислотная последовательность, которая отличает их от упорядоченных белков.
5.2.1. Необычный аминокислотный состав БПН
Уверский (Uversky etal. 2000) и Дункер (Dunker etal. 2001) впервые отметили, что частоты встречаемости различных аминокислот в неупорядоченных белках заметно отличаются от аналогичных показателей в упорядоченных белках. Отличия не зависят от методов, которые используются для установления структурного принадлежности белка к определенной группе, поскольку белки всегда бедны аминокислотами с низким показателем гибкости (гидрофобные аминокислоты) и богаты аминокислотами с высоким показателем гибкости (полярные и заряженные аминокислоты). Первую группу (Тгр, Cys, Phe, Не, Туг, Vai и Leu) называют аминокислотами, способствующими упорядоченности, тогда как последнюю (Ala, Arg, Gly, Gin, Ser, Pro, Glu и Lys) - аминокислотами, способствующими неупорядоченности (Dunker etal. 2001). Аналогичные тенденции были обнаружены в других исследованиях (Uversky et al. 2000; Tompa 2002). В настоящее время принято считать, что два главных свойства, предопределяющих неупорядоченность, - низкий общий уровень гидрофобности, при котором невозможно формирование стабильного ядра глобулы, и высокий общий заряд, который способствует расширенному структурному состоянию за счет электростатического отталкивания.
5.2.2. Паттерны последовательностей БПН
Все упомянутые исследования, а также последующий успех алгоритмов простого прогнозирования на основе аминокислотной предрасположенности свидетельствуют о том, что основным фактором, определяющим неупорядоченность структуры, является аминокислотный состав. Превосходство методов прогнозирования на основе специфической информации о последовательности, однако, показывает, что последовательности БПН содержат дополнительную информацию о неупорядоченности структуры. Количество последовательностей, неупорядоченная структура которых подтверждена экспериментально, мало, и это ограничивает изучение их свойств более высокого порядка. Тем не менее, этот вопрос уже рассматривался в нескольких исследованиях. Описывая аминокислоты в таких терминах как гидрофобность, полярность, размер, алифатическая/ ароматическая природа, наличие гетероциклической компоненты (пролин) и заряда, на основе анализа неупорядоченных сегментов можно устано
5.2. Свойства последовательностей БПН
157
вить простые, но статистически достоверные паттерны последовательностей (Lise and Jones 2005). В исследованиях было показано преобладание паттернов, богатых пролином, а также паттернов, имеющих заряд благодаря присутствию положительно или отрицательно заряженных аминокислот (например, Положительный а.о.)-Пол-Х-Пол, Отрицательный а.о.)-Отр-Отр, а также Glu-Glu-Glu, Lys-X-X-Lys-X-Lys и Рго-Х-Рго-Х-Рго). Таким образом, локальные мотивы в последовательностях связаны с неупорядоченностью в белках; этот факт можно объяснить распространенностью повторов, которая широко известна среди механизмов эволюции БПН - процесса, который, очевидно, порождает в последовательностях такие повторяющиеся мотивы (Тотра 2003).
5.2.3. Низкая сложность последовательностей и неупорядоченность
Еще одним проявлением периодической природы БПН является низкая сложность последовательностей (low sequence complexity) их поли-пептидных цепей. Применение функции энтропии (Shannon 1948) к аминокислотным последовательностям белков (Wootton 1994а, Ь) показало, что глобулярные белки пребывают преимущественно в состоянии с высокой энтропией (сложностью), тогда как во многих других белках наблюдаются обширные области с низкой сложностью. До 25% всех аминокислот в базе данных SwissProt находятся в областях с низкой сложностью, а 34% всех белков имеют по меньшей мере один такой сегмент (Wootton 1994а, Ь). Характер связи между низким уровнем сложности и неупорядоченностью рассматривается в двух исследованиях. Прежде всего, связь размера алфавита (количества аминокислот) и сложность с емкостью способа укладки изучали Ромеро и соавт. (Romero etal. 1999). Было установлено, что белки базы данных SwissProt охватывают весь возможный диапазон размеров алфавита (1-20) и диапазон энтропии (К=0,0-4,5), в то время как глобулярные домены занимают лишь ограниченные области (алфавит= 10-20, К=3,0-4,2). Области с более низкими значениями (вплоть до размера алфавита=3 и К=1,5) соответствуют структурированным фибриллярным белкам, таким как скрученные спирали, коллагены и фиброины. Из полученных результатов следует, что минимальный размер алфавита, равный 10, и значение энтропии, составляющее около 2,9, являются необходимыми и достаточными условиями для определения последовательности, которая может сворачиваться в глобулярную структуру. При экстраполяции этих исследований на БПН (Romero et al. 2001) было показано, что распределение сложности в неупорядоченных белках смещается
158 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
в сторону меньших значений, но в значительной степени перекрывается с таковым для упорядоченных белков. В целом, неупорядоченность участков коррелирует с низкой сложностью; как неупорядоченные области, так и области с низкой сложностью широко представлены в протеомах; однако низкую сложность и неупорядоченность не следует рассматривать как синонимы.
5.3. Предсказание неупорядоченности
На основании указанных особенностей состава было разработано около 25 методов предсказания неупорядоченности (см. таблицу 5.1 (Ferron et al. 2006; Dosztanyi et al. 2007)). Лучшие методы предсказания по точности приближаются к лучшим алгоритмам предсказания вторичной структуры, а принципы сравнения их производительности уже были изложены.
Таблица 5.1. Программы предсказания неупорядоченности. В таблице перечислены наиболее часто используемые программы, их URL и принципы, на которых они основаны. Подробное описание таких программ можно найти в тексте и ссылках (Ferron et al. 2006; Dosztanyi et al. 2007)
Название URL Принцип действия
PONDR VSL2 http://www.ist.temple.edu/disprot/ predictorV SL2.php Метод опорных векторов с нелинейным ядром
DISOPRED2 http://bioinf.cs.ucl.ac.uk/disopred Метод опорных веторов, нейронные сети для сглаживания
lUPred http ://iupred. enzim.hu Оценочная энергия парных взаимодействий
DisEMBL http://dis.embl.de Нейронная сеть
GlobPlot http://globplot.embl.de Предпочтение аминокислотных остатков образовывать те или иные типы вторичной структуры
FoldUnfold http://skuld.protres.ru/~mlobanov/ ogu/ ogu.cgi Предпочтение аминокислотных остатков
Foldindex http ://bip. weizmann .ac.il/fldbin/ findex Предпочтение аминокислотных остатков
NORSp http://cubic.bioc.columbia.edu/ services/ NORSp Предпочтение типов вторичной структуры
PreLink http://genomics.eu.org/spip/ PreLink Предпочтение аминокислотных остатков + анализ гидрофобных кластеров
5.3. Предсказание неупорядоченности
159
5.3.1. Предсказание областей с низкой сложностью
Как показано в упомянутых выше исследованиях, низкая сложность последовательности отличается от неупорядоченности, хотя предсказание областей с низкой сложностью можно рассматривать как первый рациональный подход к оценке неупорядоченности или, по крайней мере, отсутствия глобулярности. Функция энтропии Шеннона (Shannon 1948), адаптированная для случая последовательностей белков (Wootton 1994а, Ь), лежит в основе программы SEG, которая обычно используется для определения фрагментов с низкой сложностью состава, последовательности которых предрасположены к формированию неупорядоченных областей. Эта процедура имеет большое значение при обнаружении неглобулярных областей белков.
5.3.2. Графики «заряд-гидрофобность»
Классический подход при оценке статуса белка в отношении неупорядоченности основан на наблюдении Уверского, согласно которому сочетание низкой средней гидрофобности и высокого общего заряда отличает БПН от других белков. Этот принцип просто применить, построив график зависимости общего заряда от общей гидрофобности (Uversky et al. 2000), либо в координатах «заряд/гидрофобность», либо в координатах Уверского. На графике БПН обычно располагаются в области с высоким общим зарядом и низкой общей гидрофобностью. Они отделены от глобулярных белков линейной функцией, которая задается формулой <заряд> = =2,743 ♦ <гидрофобность> - 1,109 (рис. 5.1) и была определена с высокой точностью в более раннем исследовании (Oldfield et al. 2005а). Ограничение графика «заряд/гидрофобность» состоит в том, что он лишь дает возможность бинарной классификации белков, не предоставляя информации на аминокислотном уровне разрешения. Чтобы решить эту проблему, Суссма-ни и его коллеги экстраполировали описанный принцип (Prilusky etal. 2005), применяя метод скользящего окна к белковой последовательности для расчета средней гидрофобности и общего заряда и предсказывая таким образом неупорядоченность среднего остатка (Индекс укладки, рис. 5.2).
5.3.3. Методы предсказания на основе предрасположенности
С описанными выше методами близки по концепции другие методы прогнозирования на основе предрасположенностей, которые позволяют оценить, насколько часто определенное аминокислотное свойство, связанное
160 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
♦ Набор неупорядоченных белков о Набор упорядоченных белков
Среднее нормализованное значение гидрофобности
Рис. 5.1. Диаграмма распределения неупорядоченности белков. Для белков с присущей неупорядоченностью (показаны черными ромбами) и упорядоченных белки (показаны белыми кружками) показаны их суммарный заряд и средняя гидрофобность. Эти группы белков разделены прямой < заряд > = 2.743 < гидрофобность > - 1.109. Стрелки указывают на линии, ограничивающие зону с точностью предсказания 95% для неупорядоченных белков и 97% для упорядоченных за счет исключения из рассмотрения 50% от общего количества белков (Перепечатано с разрешения Oldfield 2005а. Copyright 2005 American Chemical Society)
с неупорядоченностью, встречается в данном предопределенном сегменте белка. В GlobPlot (binding et al. 2003a), например, применяют показатели, полученные с использованием шкалы, которая отображает предрасположенность данной аминокислоты к нахождению в неупорядоченной области либо в области правильной вторичной структуры. В DisEMBL (binding et al. 2003b) аналогичным образом используются три дополнительных свойства: «петли-кольца» (в соответствии с классификацией метода DSSP), «горячие петли» (петли с высокими В-факторами в кристаллографической структуре) и значений поля «REMARK 465», которое описывает предрасположенность аминокислоты к отсутствию в рентгеновской структуре PDB.
Несколько иной подход для предсказания на основе предрасположенности применяется в Prelink (Coeytaux and Poupon 2005). В методе Prelink используются два свойства неупорядоченных областей (которые опреде-
5.3. Предсказание неупорядоченности
161
Номер остатка
Упорядоченное
Неупорядоченное
Рис. 5.2. Диаграмма величины Foldindex для неупоряченности белка р53. Неупорядоченность суппрессора опухоли р53 была предсказана по методу Foldindex (Prilusky et al. 2005). Темно-серым цветом показана предсказанная неупорядоченность, светло-серым - упорядоченность, что согласуется с биофизическими данными, которые предсказывают неупорядоченность в N-концевом транс-активаторном домене, С-концевом домене тетрамеризации и регуляторном домене (Bell et al. 2002; Dawson et al. 2003) (Перепечатано из Prilusky et al. 2005 с разрешения Oxford University Press)
лены как области, связывающие глобулярные домены): аминокислотный состав, предрасполагающий к образованию неупорядоченности, и незначительное количество либо полное отсутствие гидрофобных кластеров. Для количественной оценки этих двух свойств было рассчитано распределение аминокислот в упорядоченных белках и неупорядоченных областях. В исследуемой последовательности с помощью метода автоматического анализа гидрофобных кластеров (automated Hyrdophobic Cluster Analysis, HCA) вычисляется расстояние до ближайшего гидрофобного кластера. Метод основан на двумерном спиральном представлении белковых последовательностей. В основе оценки неупорядоченности лежит как аминокислотный состав, так и полученное значение расстояния.
5.3.4. Методы предсказания на основе отсутствия выраженной вторичной структуры
Другой, хотя и не имеющий принципиальных отличий, подход опирается на оценку склонности аминокислот образовывать элементы вторич-
162 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
ной структуры (а-спирали, р-тяжи, повороты). В его основе лежит предположение, согласно которому протяженные области (более 70 последовательных аминокислотных остатков), не обладающие прогнозируемой правильной вторичной структурой, являются структурно неупорядоченными (Liu and Rost 2003). Показатели производительности для метода прогнозирования NORSp сравнимы с аналогичными значениями для других методов. Однако следует отметить, что существуют белки, которые отличаются высокой упорядоченностью и при этом состоят исключительно из неповторяющихся локальных структурных элементов (их также называют петлевыми белками (Liu et al. 2002)), а также БПН, которые содержат переходные локальные структурные элементы (Fuxreiter et al. 2004). Тенденция последних образовывать упорядоченную структуру хорошо поддается прогнозированию, что составляет принципиальное ограничение для прогнозов на основе описанного выше принципа.
5.3.5. Алгоритмы машинного обучения
Вероятно, наиболее прогрессивными методами прогнозирования неупорядоченности структуры являются алгоритмы машинного обучения (МО) (ML, от «machine learning»), т.е., методы предсказания, «обученные» на определенных последовательностях, которые кодируют упорядоченные или неупорядоченные структуры. В отличие от более простых ранних методов, алгоритмы машинного обучения объединяют в себе учет нетривиальных свойств аминокислот и скрытых свойств последовательностей, чем, вероятно, объясняется их более высокая производительность. В то же время, в основе корректных прогнозов часто лежат принципы, неизвестные исследователям, т.е., методы машинного обучения не способствуют более глубокому пониманию процессов, лежащих в основе неупорядоченности структуры.
Классическим алгоритмом МО является PONDR (метод предсказания природных неупорядоченных областей, от «predictor of natural disordered regions»), который основан на анализе локального аминокислотного состава, гибкости и других свойств последовательностей (Romero et al. 1998). Он был разработан в нескольких вариантах и позволяет прогнозировать неупорядоченность в концевых областях белков (Li et al. 1999), области, которые с высокой вероятностью являются характеристическими мотивами, (VL-XT (lakoucheva et al. 2002)), а также сочетания коротких и протяженных областей с неупорядоченностью (VSL2 (Peng etal. 2006)). Поскольку короткие неупорядоченные области являются контекстными (т.е., отсутствие у них выраженной структуры определяется структурным окружением), а неупорядоченность протяженных областей - независимой,
5.3. Предсказание неупорядоченности
163
этот комбинированный подход составляет основу одного из наиболее производительных алгоритмов предсказания неупорядоченности структуры.
Другой подход, отличающийся по характеру вычислительной составляющей, заключается в применении метода опорных векторов (SVMs, от «support vector machines») и представлен алгоритмом DISOPRED2 (Ward et al. 2004). Этот алгоритм осуществляет в пространстве свойств поиск гиперплоскости, которая отделяет упорядоченные белки от неупорядоченных. Гиперплоскость может быть как линейной, так и нелинейной. Учитываются несбалансированные классовые частоты данных как упорядоченных (например, белки в PDB), так и неупорядоченных (например, как белки в DisProt (Sickmeier et al. 2007)) белков. В качестве входных данных также используются профили последовательностей, созданные с помощью PSI-BLAST.
5.3.6. Предсказание на основе потенциалов контакта
В основе работы некоторых методов предсказания лежит уникальный, отличный от описанных выше принцип. В соответствии с ним, БПН не могут сворачиваться в упорядоченные структуры, поскольку аминокислотные остатки в их последовательности взаимодействуют недостаточно для преодоления невыгодного уменьшения энтропии, сопровождающего процесс сворачивания. На этом принципе основано несколько методов предсказания, в которых применяются простые статистические закономерности (FoldUnfold (Galzitskaya et al. 2006)), сравнение потенциалов парных взаимодействий (Ucon (Schlessinger et al. 2007)) или оценка общей энергии взаимодействия между остатками цепи (lUPred (Dosztanyi et al. 2005a, b)). Опишем последний метод более подробно.
Для оценки общей энергии парных взаимодействий, имеющих место в полипептидной цепи, в lUPred используются силовые поля низкого разрешения (статистические потенциалы), полученные для глобулярных белков. Основная идея состоит в том, что вклад отдельного остатка в общую энергию определяется не только типом самого остатка, но и другими аминокислотными остатками, т.е. потенциальными партнерами, в последовательности. Поскольку вероятностное описание потенциальных взаимодействий всех остатков со всеми остальными остатками обработать сложно, проблему упрощают посредством введения квадратичного выражения для аминокислотного состава. Вклад отдельного аминокислотного остатка аппроксимируют матрицей предсказания энергии, которая связывает энергетический вклад аминокислотных остатков i и j. Параметры матрицы определяют выравниванием по методу наименьших квадратов относительно глобулярных белков. При использовании такого подхода средний уровень
164 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
О 100 200 300 400 500 600 700 800 900
Длина
Рис. 5.3. Оценочные энергии парного взаимодействия глобулярных белков и БПН. Общие парные энергии взаимодействия глобулярных белков (показаны серыми +) и неупорядоченных белков (показаны черными х) были оценены на основе их аминокислотного состава и приведены в зависимости от длины их последовательностей. Отрицательное направление соответствует увеличению стабильности благодаря парным аминокислотным взаимодействиям. Средние значения, выраженные в условных единицах энергии предполагают большую стабильность в случае глобулярных белков (-0,81 единицы), чем БПН (-0,07 единицы) (перепечатано из Dosztanyi et al. 2005b с разрешения Elsevier)
энергии, получаемый для неупорядоченных белков, (-0,07 условных единиц) является невыгодным по сравнению с аналогичным значением для глобулярных белков (-0,81 условных единиц). Это свидетельствует о том, что обсуждаемый подход является информативным в тех случаях, когда исследуемые белки по своему структурному статусу принадлежат к числу макроскопических (рис. 5.3). В случаях, когда в вычислениях рассматриваются лишь предопределенные локальные соседние аминокислотные остатки последовательности, с помощью обсуждаемого подхода можно получить специфическую для последовательности информацию о неупорядоченности структуры, что составляет основу алгоритма lUPred (Dosztanyi et al. 2005a, b).
5.3. Предсказание неупорядоченности
165
5.3.7. Для предсказания неупорядоченности достаточно сокращенного алфавита
Чтобы подчеркнуть ключевые аспекты физических принципов, лежащих в основе неупорядоченности, было показано, что упорядоченные белки можно отличать от неупорядоченных по сокращенному алфавиту, т.е. с помощью кластеризации 20-ти аминокислот в меньшее количество групп (Weathers et al. 2004). Было установлено, что алгоритм, основанный на методе опорных векторов, анализирующий аминокислотный состав, имеет точность прогнозов 87±2%. Это свидетельствует о том, что основной чертой, определяющей неупорядоченность, является аминокислотный состав. Последовательное значительное уменьшение области параметров за счет кластеризации физически/химически схожих аминокислот, однако, не оказывало влияния на точность прогнозов, вплоть до 4 векторов, описывающих 20 аминокислот (84±2%). В соответствии с описанными ранее положениями, состав и относительный вес векторов свидетельствуют о том, что первичными детерминантами неупорядоченности структуры являются не специфические аминокислоты, а общие физико-химические свойства белка.
5.3.8. Сравнение методов предсказания неупорядоченности
Как уже упоминалось ранее, методы предсказания отличаются по уровню производительности и качеству предсказаний, что было подтверждено в экспериментах по критической оценке алгоритмов прогнозирования структуры (CASP, от «critical assessment of structure prediction algorithms experiments») CASP6 (Jin and Dunbrack 2005) и CASP7 (Bordoli et al. 2007). Разумеется, производительность методов предсказания неупорядоченности зависит от критериев оценки и используемых наборов данных. По сути, существуют два фактора, ограничивающие прямое сравнение методов: 1) сравнение нельзя проводить на основе простых значений неупорядоченности, выраженных в процентах, поскольку количество положительных попаданий можно легко увеличить за счет ложноположительных результатов, т.е., прогнозирования неупорядоченности для упорядоченных областей; 2) количество доступных данных об упорядоченных и неупорядоченных структурах заметно отличается, что осложняет простое сравнение точности прогнозов. В соответствии с этим, производительность методов предсказания сравнивают различными способами, главным образом, определяя чувствительность и специфичность, т.е., коэффициент верно оп
166 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
ределенной неупорядоченности относительно коэффициента неверно определенных упорядоченных областей. Справедливости ради стоит отметить, что упомянутые выше методы предсказания по уровню производительности близки к лучшим алгоритмам предсказания вторичной структуры. Для получения надежной оценки неупорядоченности рекомендуется использовать несколько методов, основанных на различных принципах.
5.4. Функциональная классификация БПН
Предсказание функций БПН является гораздо более сложной задачей, чем предсказание их структуры, по нескольким причинам. Прежде всего, БПН очень быстро эволюционируют, и в то время как их основные структурные состояния часто остаются неизменными, о характере и масштабах изменения функций известно очень мало. Еще одна причина заключается в том, что функциональную классификацию белков/генов обычно осуществляют на уровне целого гена, и часто совершенно неясно, каким образом и до какой степени на этот процесс повлияла неупорядоченность области (ОПН). Кроме того, во многих случаях функции БПН нельзя включить в схемы функциональной классификации, разработанные для упорядоченных белков. Область функциональной классификации БПН находится в процессе активного развития, что к настоящему моменту привело к появлению двух принципиально различных подходов к классификации. Их ключевые аспекты обсуждаются ниже.
5.4.1. Функциональная классификация БПН на основе генной онтологии
Рассмотрению вопроса преобладания неупорядоченной структуры в некоторых функциональных классах белков посвящен ряд исследований (lakoucheva et al. 2002; Ward et al. 2004; Tompa et al. 2006; Xie et al. 2007). Как правило, в их основе лежит схема генной онтологии (ГО) (GO, от «Gene Ontology») (Ashbumer et al. 2000), а неупорядоченность рассматривается во во всех трех онтологических аспектах: молекулярные функции, биологические процессы и клеточная локализация. Авторы различных работ приходят к общим выводам: частота встречаемости неупорядоченной структуры значительно выше у эукариот по сравнению с прокариотами; неупорядоченность является общим свойством белков, выполняющих регуляторную и сигнальную функции. При рассмотрении в аспекте молекулярной функции наиболее высокие уровни неупорядоченности наблюдаются в таких катего
5.4. Функциональная классификация БПН
167
риях как регуляция транскрипции, белковые киназы, фактор транскрипции, связывание ДНК, тогда как низким уровнем отличаются категории оксидоредуктаз, катализа, лигаз, структурных молекул. В аспекте биологических процессов высоким уровнем неупорядоченности отличаются категории развития, фосфорилирования белков, регуляции транскрипции, передачи сигналов, в то время как в категориях биосинтеза и энергетических путей неупорядоченность встречается редко. В отношении аспекта клеточной локализации, неупорядоченность преобладает в ядерных и хромосомных белках, белках цитоскелета, низким уровнем характеризуются митохондриальные, цитоплазматические и мембранные белки.
Аналогичные закономерности были обнаружены в тех случаях, когда прогнозирование осуществлялось для обширных неупорядоченных областей, которые принято считать функционально значимыми (Xie et al. 2007). При анализе ключевых слов аспекта биологических процессов базы данных SwissProt была обнаружена выраженная корреляция с неупорядоченностью, как положительная (например, для дифференциации, транскрипции, регуляции транскрипции), так и отрицательная (например, для биосинтеза, транспорта, транспорта электронов, гликолиза) (Xie etal. 2007). При анализе ключевых слов аспекта молекулярной функции наибольшая положительная корреляция наблюдалась для рибонуклеопротеинов, рибосомальных белков, белков развития, в то время как отрицательной корреляцией отличались оксидоредуктазы, трансферазы, лиазы, гидролазы. Из 710 описывающих функции ключевых слов базы данных SwissProt 238 имели выраженную положительную и 302 - выраженную отрицательную корреляцию с неупорядоченностью структуры; 170 ключевых слов имели неясную принадлежность.
Все исследования, посвященные обсуждаемому вопросу, имеют общее положение, согласно которому для белков, выполняющих регуляторные функции, отмечается положительная корреляция с неупорядоченностью структур, тогда как для белков с каталитическими функциями характерна отрицательная связь с неупорядоченностью.
5.4.2. Классификация БПН на основе механизма действия
В другой системе, учитывающей молекулярные механизмы действия БПН, неупорядоченные белки относят к одной из пяти (Tompa 2002) и далее - к одной из шести (Tompa 2005) категорий. В ходе последних исследований в качестве самостоятельной категории были добавлены белки-прионы (Pierce et al. 2005). Эта схема классификации (таблица 5.2) позволяет
168 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
Таблица 5.2. Схема классификации БПН. Классификация БПН включает в себя семь функциональных категорий по типу их молекулярного механизма действия. Дается по два примера в каждой категории с указанием партнера (если применимо) и фактической клеточной функции белка
Белок Партнер Функция
Энтропийные цепи
Область повтора Nup2p FG неприменимо Пропускание в NPC
N-концевая область калиевого канала неприменимо Определение времени активации канала
Демонстрационные участки
CREB KID РКА Сайт фосфорилирования
N-концевой домен циклина В ЕЗ убиквитин лигаза Сайт убиквинирования
Шапероны
ERD 10/14 (напр.) Люцифераза Препятствование аггрегации
hnRNPAl (напр.) ДНК Отпуск нитей
Эффекторы
p27Kipl CycA-Cdk2 Ингибирование клеточного цикла
Секурин Сепараза Ингибирование анафазы
Ассемблеры
RNAP II CTD Фактор созревания мРНК Регуляция созревания мРНК
CREB рЗОО/СВР Инициация транскрипции
Скавенджеры
Казеин Фосфат кальция Стабилизация фосфата кальция в молоке
Слюнные белки PRP Таннин Нейтрализация растительных таннинов
Прионы
Ure2p Утилизация мочевины до азота
Sup35p NusA, мРНК Блокирование стоп-кодона, продолжение считывания
5.4. Функциональная классификация БПН
169
включить все известные механизмы действия БПН/ОПН, описанные к настоящему времени (Sickmeier et al. 2007).
5.4.2.1. Энтропийные цепи
Первая функциональная категория, уникальная для неупорядоченных белков, - энтропийные цепи, функция которых не связана с распознаванием молекулы-партнера, но прямо следует из неупорядоченности структуры. Внутри класса существуют следующие подкатегории: энтропийные пружины, щетинки/разделители, линкеры и стрелки. Основные механизмы функционирования наилучшим образом можно описать так: влияние на расположение присоединенных доменов, или создание сил, препятствующих движениям/структурным изменениям (Dunker et al. 2002). Наиболее полно описанные примеры из этой категории - функционирование энтропийного воротного механизма комплекса ядерной поры за счет неупорядоченных областей NUPs (Elbaum 2006), функционирование энтропийного разделителя/щетинки выступающих доменов белков, связанных с микротрубочками цитоскелета (Mukhopadhyay and Hoh 2001), а также действие энтропийной пружины области PEVK титина, которое обеспечивает пассивное напряжение мышц в состоянии покоя за счет эластичности (Trombitas et al. 1998).
5.4.2. Z. Функционирование посредством временного связывания
В остальных шести категориях функционирование БПН осуществляется за счет молекулярного распознавания, те., БПН связываются с другой макромолекулой/другими макромолекулами или лигандом/лигандами небольшого размера временно или постоянно. Демонстрационные участки предназначены главным образом для посттрансляционных модификаций. Так, для ферментативных модификаций требуется наличие в белках структурно гибких и легко адаптирующихся областей, как это показано для случаев ограниченного протеолиза, встречающегося в областях линкеров глобулярных белков (Fontana et al. 1997). Фосфорилирование (lakoucheva et al. 2004), убиквитирование (Cox etal. 2002) и деацетилирование (Khan and Lewis 2005) также встречаются в локально неупорядоченных областях. Общая корреляция неупорядоченности структуры с наличием таких участков была показана в ходе предсказания неупорядоченности в белках, которые содержат короткие элементы распознавания (известные также как линейные мотивы (Puntervoll etal. 2003)). Было установлено, что линейные мотивы в белке располагаются главным образом в окружении участков с локальной неупорядоченностью последовательности (Fuxreiter et al. 2007).
170 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
Посредством временного связывания также функционирует другая категория БПН - шапероны. Это показано с помощью статистического анализа на уровне неупорядоченности в белковых и РНК-шаперонах (Tompa and Csermely 2004). В РНК-шаперонах доля неупорядоченности очень высока: 40% остатков находятся в протяженных неупорядоченных областях. Белковые шапероны также принадлежат к числу наиболее неупорядоченных белков: в обширных неупорядоченных областях располагаются 15% остатков. Наличие неупорядоченных областей часто напрямую связано с функционированием шаперонов, что позволяет сформулировать модель «переноса энтропии», которая отражает роль структурной неупорядоченности в функционировании шаперонов (Tompa and Csermely 2004). Основные положения этой модели получили подтверждение в ходе недавних исследований полностью неупорядоченных белковых шаперонов (Kovacs et al. 2008).
5.4.2.3. Функционирование посредством постоянного связывания
В других четырех категориях БПН/ОПН функционируют за счет постоянного связывания молекулы-партнера. Белки, называемые эффекторами, связываются с молекулами-партнерами, главным образом, ферментами, и изменяют их активность (Tompa 2002). К этой группе относятся некоторые подробно описанные БПН, такие как p27Kipl, ингибитор цик-линзависимых киназ (Kriwacki etal. 1996; Lacy etal. 2004), секурин, ингибитор сепаразы (Waizenegger et al. 2002) и кальпастатин, ингибитор каль-паина (Kiss et al. 2008а, b). Интересно, что такие эффекторы иногда способны как ингибировать, так и активировать молекулы-партнеры, как это показано для p27Kipl (Olashaw etal. 2004) или для фрагмента С петли DHPR П-Ш (Haarmann et al. 2003). Эти и другие данные привели к появлению идеи участия элементов структурной неупорядоченности в реализации множества различных, иногда противоположных, функций белков, или совместительства (Tompa et al. 2005).
Следующая категория БПН, функционирующая за счет необратимого связывания партнера, - ассемблеры, или сборщики, которые либо управляют активностью прикрепленных доменов, либо собирают мультибелковые комплексы (Tompa 2002). Высокая степень неупорядоченности в некоторых белках апоптоза, таких как BRCA1 и Ste5 (Mark etal. 2005; Bhattacharyya et al. 2006), повышенная степень неупорядоченности в белках центра инте-рактомы (Dosztanyi et al. 2006; Haynes et al. 2006; Patil and Nakamura 2006), а также корреляция средней степени неупорядоченности с количеством молекул-партнеров в мультибелковых комплексах (Hegyi et al. 2007) подтверждают общий характер этой связи.
5.4. Функциональная классификация БПН
171
Третий класс обсуждаемой категории - скавенджеры, или мусорщики, -содержит неупорядоченные белки, которые хранят и/или нейтрализуют молекулы-лиганды небольшого размера. Питательное вещество молока, казеин(ы), например, функционирует в том числе и как хранилище фосфата кальция, обеспечивая высокую общую концентрацию фосфата кальция в молоке (Holt et al. 1996).
Последнюю функциональную категорию БПН - прионы - не включали в ранние схемы классификации (Тотпра 2002, 2005). Прионы обычно рассматривали как патогенные факторы, главным образом в связи с тем, что их невольно связывали с коровьим бешенством (Prusiner 1998). Многие публикации последних лет, однако, свидетельствуют о том, что автокаталитические конформционные изменения, лежащие в основе феномена прионов, встречаются также при нормальном физиологическом функционировании белков дрожжей (Tuite and Koloteva-Levin 2004) и даже высших организмов, например, D. melanogaster (Si et al. 2003a, b; Fowler et al. 2007). Такие прионы содержат неупорядоченные прионные домены, богатые глутамином/аспарагином (Pierce et al. 2005) и отвечающие в основном за автокаталитический конформационный переход, который оказывает влияние на функции соседних доменов.
5.4.3. Структурные элементы БПН, связанные с функционированием
Переходные структурные элементы БПН участвуют в молекулярном распознавании. Эта особенность их функционирования непосредственно связана с прогнозированием функций по последовательности. Наличие таких элементов, часто различимых как на уровне последовательности, так и на уровне структуры, можно использовать при прогнозировании функций. В процессе эволюции упорядоченных белков появилось большое количество доменов, связанных со специализированными функциями распознавания (Pawson and Nash 2003; Seet et al. 2006), тогда как их родственные партнеры, например, из домена SH3 (Hiroaki et al. 2001; Ferreon and Hilser 2004), домена 14-3-3 (Busto and Iglesias 2006) или домена PTB (Obenauer etal. 2003) чаще представляют собой короткие мотивы внутри гибких областей белков. Существует несколько отличающихся, но связанных между собой концепций, которые объясняют существование подобных коротких мотивов, акцентируя внимание на структуре или последовательности при обсуждении функций.
5.4.3.1. Предопределенные элементы структуры
Концепция предопределенных структурных элементов возникла на основе анализа структур БПН в комплексах с их молекулами-партнерами.
172 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
Ключевой вопрос, рассматривавшийся в ходе этого анализа, состоял в том, можно ли предсказать локальную структуру БПН в комплексе с его молекулой-партнером с помощью алгоритмов предсказания вторичной структуры (Fuxreiter etal. 2004). Было обнаружено, что точность предсказания этих элементов вторичной структуры БПН выше, чем аналогичный показатель прогнозов для их упорядоченных белковых партнеров. Это свидетельствует о том, что БПН свойственна выраженная предрасположенность к конформациям, приобретаемым в связанных состояниях: вероятно, для распознавания используются элементы, существование которых (частично) предопределено уже в растворе. Эта связь в наибольшей степени выражена для спиралей и в наименьшей - для колец. Она подтверждена в ходе изучения методом ЯМР ряда свободных БПН, для которых структура свободных образцов была схожа со структурой в связанном состоянии. К числу таких БПН принадлежат домен KID области CREB (Radhakrishnan et al. 1998), домен KID области Cdk ингибитора p27Kip2 (Kriwacki et al. 1996; Lacy et al. 2004), а также транс-акгиваторный домен супрессора опхолей р53 (Lee et al. 2000).
5.4.3.2. Элементы/свойства молекулярного распознавания
Упомянутые выше данные непосредственно связаны с тем, насколько успешно можно прогнозировать присутствие элементов распознавания в молекулах БПН, как это показано в рамках концепции элементов/свойств молекулярного распознавания (ЭМОР/СМОР) (MoREs/MoRFs, от «molecular recognition elements/features»). Целый ряд исследований посвящен анализу белок-белковых комплексов из базы данных PDB, в которых одна из молекул имела размер менее 30 аминокислотных остатков, а другая - более 30 (Oldfield et al. 2005b), либо одна из молекул включала от 10 до 70 аминокислотных остатков, а другая представляла собой глобулярный белок (Mohan et al. 2006; Vacic et al. 2007). В результате исследований были выявлены 372 свойства молекулярного распознавания (СМОР) (MoRFs), которые также называют элементами молекулярного распознавания (ЭМОР) (MoREs). В соответствии с преобладающими элементами вторичной структуры принято выделять четыре категории свойств молекулярного распознавания: а-СМОР, Р-СМОР, t-CMOP и смешанные СМОР (а-MoRFs, P-MoRFs, t-MoRFs и смешанные MoRFs). В целом, в СМОР 27% остатков пребывают в a-спиральной конформации, 12% - в конформации Р-тяжей и около 48% - в неупорядоченной конформации; оставшиеся 13% остатков не имеют атомных координат. Тесную связь концепции СМОР с предопределенными структурными элементами и неупорядоченностью подтверждает тот факт, что локальные структурные предрасположенности СМОР легко поддаются предсказанию, уступая в сложности предсказания глобулярным
5.5. Предсказание функций БПН
173
белкам, а также тот факт, что присутствие СМОР коррелирует с неупорядоченностью в несвязанном состоянии (Mohan et al. 2006).
Этот анализ СМОР привел к появлению идеи о том, что СМОР можно определять по характерной для них картине неупорядоченности. Как правило, нисходящий пик численных показателей неупорядоченности, в частности PONDR VL-XT (lakoucheva et al. 2002), является ярко выраженным свидетельством наличия функционально значимого элемента распознавания. В сочетании с мотивами последовательности и функциональным анализом белков, которые содержат СМОР, анализ этих элементов позволяет выдвигать обоснованные предположения относительно функций БПН/ОПН. Следует отметить, что большое количество белков, содержащих СМОР, было обнаружено среди молекул, выполняющих сигнальные функции (Mohan et al. 2006; Vacic et al. 2007).
5.4.3.3. Короткие линейные мотивы распознавания
Анализ последовательностей, участвующих в белок-белковых взаимодействиях, показал, что в некоторых белках элемент распознавания представляет собой короткий мотив с выраженной консервативностью, который часто обозначается как последовательность согласования. Такие последовательности, например, участвуют в модификации субстрата киназами или связывании с доменами SH3 (Neduvaand Russell 2005). Эти мотивы являются эволюционно изменчивыми и обычно состоят из нескольких консервативных определяющих специфичность остатков, которые рассеяны среди весьма изменчивых остатков, при этом общая длина мотива составляет от 5 до 25 остатков. Их часто называют линейными мотивами, линейными мотивами эукариот или короткими линейными мотивами. Анализ линейных мотивов, собранных в базе данных линейных мотивов эукариот (Puntervoll et al. 2003), с помощью предсказателей неупорядоченности показал, что линейные мотивы и их фланкирующие сегменты длиной около 20 остатков, как правило, располагаются в локально неупорядоченных областях (Fuxreiter et al. 2007). Предсказание линейных мотивов на основе последовательности в сочетании с предсказанием неупорядоченности и дополнительной информацией о функциях могут оказаться весьма полезными в предсказании функций БПН/ОПН.
5.5. Предсказание функций БПН
Как следует из соображений, изложенных выше, до всестороннего и достоверного предсказания функций БПН еще очень далеко, и для дости
174 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
жения этой цели сделаны лишь первые шаги. В следующем разделе будут рассмотрены те несколько методов, которые могут пролить свет на функции БПН, еще не описанные в экспериментах. Функциональная корреляция общей модели по неупорядоченности (Lobley et al. 2007), основанное на последовательности предсказание коротких линейных мотивов с помощью различных алгоритмов (Davey et al. 2006; Neduva and Russell 2006), прогнозирование СМОР в БПН/ОПН (Mohan et al. 2006; Vacic et al. 2007) и сочетание информации о последовательности с неупорядоченностью (lakoucheva et al. 2004; Radivojac et al. 2006) представляют собой примеры обоснованного подхода к оценке функций неизвестной области неупорядоченного белка.
5.5.1. Корреляция модели неупорядоченности и функции
Для поиска связи между общей моделью неупорядоченности и функцией белка (Lobley et al. 2007), описанной в стандартных категориях генной онтологии (ГО), Джонс и соавт. выбрали прямой подход. Впервые было обнаружено, что дескрипторы неупорядоченности, связанные как с месторасположением, так и с длиной, коррелируют с функциональными категориями, имеющими отношение к передаче сигналов и регуляции транскрипции. Использовались аннотации молекулярной функции и биологического процесса. Для дескрипторов месторасположения отмечен ряд тенденций, связанных с категориями ГО, например, повышенный уровень в середине белка в регуляторе транскрипции, при связывании ДНК и в факторе транскрипции РНК pol II, в С-конце активатора фактора транскрипции, репрессора фактора транскрипции и фактора транскрипции, а также в N-конце белков, аннотируемых с калиевыми каналами. Для дескрипторов длины показаны еще более выраженные связи с функцией, чем для дескрипторов месторасположения. Так, неупорядоченные области размером более 500 остатков в избытке представлены в категориях, связанных с транскрипцией, тогда как более короткие области длиной около 50 остатков или менее широко представлены в белках, связывающих ионы металлов, а также в белках, выполняющих функции ионных каналов и регуляторные функции ГТФаз. Обнаруженные связи можно использовать для улучшения качества прогнозов функций белков: в случае применения метода прогнозирования на основе метода опорных векторов к 26 категориям ГО прогнозы для 11 категорий биологических процессов и 12 категорий молекулярных функций были более высокого качества, что являлось результатом учета свойств неупорядоченности. В целом, учет неупорядо-
5.5. Предсказание функций БПН
175
ценности заметно улучшает качество прогнозов функций белков, при этом более значимые улучшения наблюдаются для категорий биологических процессов по сравнению с молекулярными функциями.
5.5.2. Предсказание коротких мотивов распознавания в БПН
Совершенной иной, но важный подход состоит в том, чтобы прогнозировать наличие в БПН/ОПН коротких мотивов последовательностей, которые затем можно непосредственно связать с определенными функциями, такими как посттрансляционные модификации или связывание с близкими молекулами-партнерами. Как уже было отмечено выше, функции БПН часто связаны с наличием коротких линейных мотивов, участвующих в белок-белковых взаимодействиях. Поскольку объем информации, содержащийся в этих коротких мотивах, ограничен, для распознавания таких белковых областей были разработаны специализированные методы, два из которых описаны ниже.
В одном из названных методов - DILIMOT (Discovery of Linear MOTifs) (Neduva and Russell 2006) - используется тот факт, что статистическую достоверность можно заметно повысить, если использовать для прогнозирования ряд последовательностей с общим функциональным свойством (таким как молекула-партнер для взаимодействия или локализация), которое обусловлено присутствием короткого мотива, с высокой вероятностью представленного в каждой из последовательностей ряда. Из рассмотрения исключаются те области входных последовательностей, которые с низкой вероятностью содержат примеры линейных мотивов (глобулярные домены, сигнальные пептиды, трансмембранные и биспиральные области). Затем среди оставшихся последовательностей при помощи алгоритма соединения с моделью осуществляется поиск мотивов. Обнаруженные мотивы ранжируются в соответствии с уровнем избыточности представления в последовательностях, а также с уровнем консервативности среди гомологов родственных видов. Производительность метода повышается в случае сравнения белков, принадлежащих различным биологическим видам, а также в случае рандомизации последовательностей. Предварительное применение метода к полученным высокопроизводительными методами наборам данных по взаимодействиям в последовательностях дрожжей, мухи, червя и человека привело к повторному открытию множества известных ранее примеров линейных мотивов, а также к обнаружению ряда новых мотивов. Прогнозы для двух предполагаемых новых мотивов получили подтверждение в экспериментах по прямому связыванию: мотив
176 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
DxxDxxxD связывает белковую фосфатазу 2 с Kd= 22рМ; мотив VxxxRxYS связывает траслин с K<i= 43цМ (Neduva and Russell 2005).
Концептуально близким методу DILIMOT является метод SlimDisc (Short Linear Motif Discovery) (Davey et al. 2006). В его основе лежит положение, согласно которому доказательство присутствия характеристического мотива в белке является тем более весомым, чем чаще данный мотив встречается в различных несвязанных между собой белках, эволюционирующих путем конвергенции. Обнаружению таких мотивов препятствует сходство в родственных белках, которое возникает вследствие общего происхождения. Принимая во внимание этот факт, поиск схожих мотивов ведут в группе белков с общим характерным свойством среди белков, обладающих незначительным сходством или полным отсутствием сходства в первичной последовательности. Общим характерным свойством в данном случае может быть биологическая функция белков, их субклеточная локализация или общая молекула-партнер, с которой белки взаимодействуют. Мотивы, обнаруженные с помощью основных алгоритмов распознавания паттернов, таких как TEIRESIAS, рассматриваются как более значимые, если они обнаружены в последовательностях, никак не связанных между собой, и как менее значимые, если очевидно, что они произошли от общего эволюционного предка. Проверка метода SlimDisc на калибровочном наборе белков, содержащих линейные мотивы, (Neduva and Russell 2005) показала значительное улучшение производительности.
5.5.3. Прогнозирование СМОР
Как было отмечено выше, ЭМОР/СМОР - это короткие функциональные мотивы, участвующие в связывании молекулы-партнера, наличие которых выражено коррелирует с локальной неупорядоченностью в белках (Mohan et al. 2006; Cheng et al. 2007; Vacic et al. 2007). В связи с этим обнаружение таких мотивов в белках имеет большое значение для предсказания функций белков. Показано, что часто такие области обнаруживаются в паттернах неупорядоченности, полученных с помощью методов предсказания, в особенности PONDR VL-XT: они распознаются как нерегулярные элементы и обычно характеризуются нисходящим пиком (см. рис. 5.4). Несмотря на то, что обсуждаемая связь структуры с функцией не проверена статистически, в некоторых случаях локальная область нерегулярной структуры в паттерне неупорядоченности и локальная функциональная область четко установлены. Связи такого рода обнаружены для ингибитора протеиназы IA3 и p21Cipl (Vacic etal. 2007), нуклеопротеина вируса кори (Bourhis et al. 2005) и многих других белков (Oldfield et al. 2005b; Uversky et al. 2005; Mohan et al. 2006; Vacic et al. 2007).
5.5. Предсказание функций БПН
177
Рис. 5.4. (Цветную версию рисунка см. на вклейке.) Предсказание ЭМОР/СМОР в белке р53. Паттерн неупорядоченности этого белка был предсказан с помощью программы PONDR VL-XT, предполагающей наличие элементов молекулярного распознавания функций связывания. Положение нерегулярностей (провалов) в графике неупорядоченности совпадает с областями, участвующими в связывании Mdm2 (а), ДНК (6) и тетрамеризации (в), а также с областью в регуляторном домене, которая связывает димер S1OOB(PP) (г). Связываемые молекулы показаны синим. (Перепечатано с разрешением Oldfield 2005b. Copyright 2005 American Chemical Society)
5.5.4. Сочетание информации о последовательности и неупорядоченности: участки фосфорилирования и мотивы связывания СаМ
Известны два случая, когда качество предсказания наличия в белках коротких характеристических мотивов было улучшено за счет учета информации о неупорядоченности, точнее, о участках фосфорилирования и связывания кальмодулина (СаМВТ). В ходе сравнения экспериментально определенных участков фосфорилирования с потенциальными участками, которые обычно не фосфорилированы (по Ser, Thr или Туг), Дункер и его коллеги (lakoucheva et al. 2004) установили, что области вблизи участков фосфорилирования характеризуются высоким содержанием аминокислотных остатков, способствующих появлению неупорядоченности структуры, и бедны аминокислотными остатками, наличие которых способствует упорядоченности (Dunker etal. 2001). Сочетая наборы положительных примеров и соответствующих отрицательных примеров с учетом локальной неупорядоченности, можно создать метод прогнозирования участков фосфорилирова
178 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
ния. Алгоритм DISPHOS (disorder-enhanced phosphorylation predictor) характеризуется повышенной точностью прогнозов по сравнению с другими алгоритмами предсказания участков фосфорилирования, такими как Net Phos (Blom et al. 1999) и Scansite (Obenauer et al. 2003).
Другой хорошо изученный пример - взаимодействие кальмодулина (СаМ) с его молекулами-мишенями, которое предполагает значительную гибкость со стороны обеих взаимодействующих молекул. Известно, что СаМ обычно обволакивает молекулу-мишень (СаМВТ) - спиральный пептид длиной около 20 аминокислотных остатков (Ikuraand Ames 2006). В ходе всестороннего анализа было отмечено, что для распознавания СаМ необходимо наличие неупорядоченности у молекулы-партнера (Radivojac et al. 2006). Так, СаМ-зависимые ферменты часто активируются при частичном протеолитическом переваривании (например, кальциневрин (Manalan and Klee 1983) или фосфодиэстераза циклических нуклеотидов (Tucker etal. 1981)), что предполагает локальную неупорядоченность в участках связывания. Учет информации о неупорядоченность использовался для разработки метода прогнозирования СаМВТ с повышенной производительностью (Radivojac et al. 2006).
5.5.5. Поддержание неупорядоченности
В завершение дискуссии о предсказании функций БПН по последовательности уместно будет упомянуть предложенный в некоторых работах подход, в рамках которого БПН на основе особенностей аминокислотного состава объединяют в кластеры, которые затем связывают с функциями. Данные, полученные на основе такого анализа, слишком ограничены для использования в предсказании функций, однако они могут оказаться полезны при выборе дальнейших направлений исследований.
Транс-активаторные домены факторов транскипции характеризуются выраженной склонностью к неупорядоченности (Sigler 1996; Minezaki etal. 2006); также их можно классифицировать по аминокислотному составу. Обычно факторы транскрипции классифицируют на основе таких особенностей аминокислотного состава их транс-активаторных доменов, как кислотные свойства, высокое содержание Pro и Gin (Triezenberg 1995). Несмотря на то, что такие различия недостаточно обоснованы статистически, принадлежность фактора транскрипции к той или иной категории можно подтвердить на том основании, что функции отдельной категории остаются нечувствительны к аминокислотным заменам до тех пор, пока сохраняется отличительное свойство транс-активаторного домена (Норе etal. 1988). С другой стороны, мутации, изменяющие это отличительное свойство, нарушают функционирование домена (Gill and Ptashne 1987).
5.6. Ограничения методов предсказания функций БПН
179
Таким образом, некоторые свойства, наблюдаемые на уровне аминокислотного состава, тесно связаны с выполняемыми функциями.
Существование таких общих связей исследовано непосредственно при кластеризации БПН в пространстве аминокислотного состава (Vucetic et al. 2003). Исходное положение этого исследования состоит в том, что методы прогнозирования неупорядоченности, обученные на одной группе белков, часто отличаются низкой производительностью при работе с другими группами, а это свидетельствует о значительных различиях в свойствах последовательностей неупорядоченных белков. Так, Дункер и его коллеги провели кластеризацию 145 БПН, используя различные методы прогнозирования и критерий точности предсказания в качестве разделителя единичных белков. Оказалось, что можно выделить три схожим образом заселенных группы неупорядоченных белков, которые отличаются аминокислотным составом. Группы были обозначены V, С и S. Группа С содержит большое количество His, Met и Ala; в группе S меньше His; группа V отличается повышенным содержанием наименее гибких аминокислот (Cys, Phe, Не, Туг). Для каждой из групп характерна связь с определенными функциями. Так, 9 из 10 рибосомальных белков Е. coli относятся к группе V. С другой стороны, среди БПН, отвечающих за связывание с РНК геномов вирусов, почти нет представителей группы V, как и среди белков, отвечающих за связывание с ДНК. БПН, принимающие участие в белок-белковых взаимодействиях, принадлежат преимущественно к группам V и S. Несмотря на известные ограничения такого анализа, он ясно показывает, что тип неупорядоченности белка, заложенный в аминокислотном составе, связан с выполняемыми функциями. Более глубокий анализ такого рода можно использовать при предсказании функций.
5.6. Ограничения методов предсказания функций БПН
Как уже ясно из предшествующих разделов, предсказание функций БПН по последовательности полно неясностей. Сложности, сопровождающие процесс предсказания, можно преодолевать различными способами, которые, однако, тесно переплетены: их основу составляют быстрая эволюция БПН и автономность последовательности от функций. В заключительном разделе они будут обсуждаться более подробно.
5.6.1. Быстрая эволюция БПН
Быстрая эволюция БПН/ОПН была продемонстрирована непосредственно путем сравнения частот аминокислотных замен в неупорядоченных и глобулярных областях тех белковых семейств, в которых одновременно
180 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
представлены оба типа областей (Brown et al. 2002). Для 26 таких семейств посредством сравнения каждой пары последовательностей во множественных выравниваниях был рассчитан статистический показатель изменчивости (среднее генетическое расстояние). Результаты показали, что в 19 семействах неупорядоченные области эволюционируют гораздо быстрее, чем упорядоченные; в 5 семействах скорость эволюции почти одинакова; и лишь в 2 семействах скорость эволюции для неупорядоченных областей заметно ниже, чем для упорядоченных. В отношение функций установить простые правила нельзя, поскольку к числу быстро эволюционирующих областей принадлежат участки связывания белков, ДНК и РНК, эти области также могут служить гибкими линкерами. В случае медленно эволюционирующих неупорядоченных областей наблюдается более четкая картина: большинство из них участвуют в связывании ДНК, а также составляют крупные участки взаимодействия с молекулами-партнерами. Эти участки, в которых осуществляются взаимодействия, вероятно, служат ограничивающим фактором, определяющим приемлемые изменения в последовательности (Brown et al. 2002).
Обсуждаемый вопрос был рассмотрен в двух исследованиях. Холт и Сойер сравнили частоты замен в транслируемых и нетранслируемых участках гена казеина (Holt and Sawyer 1988). Было установлено, что участок, кодирующий аминокислотную последовательность, эволюционирует быстрее, т.е., по-видимому, подвергается меньшим эволюционным ограничениям, чем нетранслируемый участок, отвечающий за регуляцию трансляции. В другом исследовании Додрилл и его коллеги (2007) анализировали процесс эволюции и функции неупорядоченной области линкера, соединяющего два глобулярных домена в субъединицу А белка репликации RPA70 размером 70 кДа (Olson et al. 2005). Изучение темпов эволюции показало, что область линкера отличается большой изменчивостью, при этом множество участков эволюционируют со средней скоростью. Гибкость линкера изучали с помощью ЯМР-спектроскопии. Прямые способы измерения гибкости основной цепи, такие как дипольное взаимодействие остатков и время броуновской переориентации, показали, что характер гибкости основной цепи сохраняется, несмотря на большую изменчивость последовательности. Эти результаты подчеркивают, что выраженная изменчивость последовательности совместима с сохранением функций, что, в свою очередь, сильно осложняет попытки прогнозирования функций.
5.6.2. Независимость последовательности и функции и неопределенность
В соответствии с вышеизложенными положениями некоторые последние исследования с использованием мутагенеза были направлены на изу
5.6. Ограничения методов предсказания функций БПН
181
чение нетрадиционной связи между последовательностью и функциями БПН. В этих исследованиях последовательности функциональных областей шифровались и смешивались; при этом было обнаружено, что функции белков нечувствительны к рандомизации. Явление получило название автономности последовательности (Ross etal. 2005; Тотра and Fuxreiter 2008). Полученные результаты подчеркивают, насколько ограничены наши представления о связи между последовательностью и функциями БПН.
Классическим примером служат факторы транскрипции, в которых кислый транс-активаторный домен (ТАД) Gen4p можно заменить случайными кислыми сегментами без заметной потери биологической активности (Hope etal. 1988). Эта закономерность, вероятно, имеет всеобщий характер, что привело к появлению предположения, согласно которому сборка комплекса преинициации транскрипции, возможно, не требует характерной строгой геометрической комплиментарности, которая необходима при специфическом белок-белковом распознавании (Sigler 1988). В более позднем и весьма подробном исследовании для химерного фактора транскрипции EWS белка слияния (EPF, от «EWS fusion protein») было обнаружено похожее поведение (Ng et al. 2007). В ТАД EFP с высоким содержанием повторяющихся последовательностей одиночные повторы можно свободно чередовать, располагая последовательности в случайном или даже в обратном порядке; функции EFP при этом сохраняются.
Автономность последовательностей также показана для двух других систем - гистонов-линкеров и прионов. В случае гистонов-линкеров исследовали область связывания фактора фрагментации ДНК 40 (DFF40) апоптической нуклеазы. Было установлено, что любой участок С-конце-вого домена (CTD, от «С-terminal domain»), имеющий достаточную длину, может связывать и активировать фермент, независимо от его первичной последовательности и положения в интактном CTD (Hansen et al. 2006). В случае прионов дрожжей Ure2p и Sup35p образование амилоида является еще одним общим примером автономности последовательности распознавания (Ross et al. 2005). Эти физиологические прионы, вероятно, обеспечивают клеткам хозяина преимущество при отборе (Wickner etal. 1999). Они содержат богатые Q/N неупорядоченные прионные домены, которые можно располагать в случайном порядке без потери прионоподобных свойств белка (Ross et al. 2005).
Отчасти на этих данных основана концепция неопределенности (Тотра and Fuxreiter 2008). Неопределенность - представление о неупорядоченности связанного БПН, для которого характерны два, возможно, взаимосвязанных, состояния. В некоторых случаях молекулярное распознавание и связывание молекулы-партнера не сопровождаются укладкой, или упорядочиванием,
182 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
БПН, как это описано для ^-цепей рецептора Т-клеток (Sigalov et al. 2004) и белка гена umuD (Simon et al. 2008). В других случаях при связывании белок не приобретает единственную доминирующую структуру, а имеет множество состояний, которые можно рассматривать как полиморфизм в связанном состоянии. Такое поведение наблюдается в случае фактора 4 Т-клеток (Tcf4) при связывании с р-катенином (Graham etal. 2001) и в случае сигнального белка ядерной локализации (NLS) при связывании с а-импортином (Fontes etal. 2000). Несомненно, описанное явление ограничивает возможности предсказания функций БПН, поскольку оно противоречит идее строгого со-отвествия между последовательностью белка и его функциями.
5.6.3. Консервативность и неупорядоченность
В качестве последнего замечания следует отметить, что неупорядоченность не является строгой противоположностью консервативности последовательностей, поскольку некоторые неупорядоченные области эволюционно консервативны (Chen et al. 2006а, b). Всестороннее исследование семейств доменов, проведенное Дункером и его коллегами, показало, что многие участки исследованных белков, содержащие не менее 20 аминокислотных остатков, характеризуются значительной консервативностью. Такие области получили название консервативных прогнозов неупорядоченности (CDP, от «conserved disorder predictions») и были обнаружены почти в 30% семейств доменов. Большинство CDP короткие, лишь 9% из них содержат более 30 остатков; обычно они покрывают менее 15% соответствующего домена. Самый длинный CDP, однако, содержит 171 аминокислотный остаток (в белке дентинового матрикса). Возможно, еще более важным является тот факт, что 8,7% CDP покрывают более половины соответствующих им доменов, а 16 CDP покрывают домен целиком. Функции доменов, содержащих CDP, соответствуют общим функциональным особенностям БПН, таким как связывание ДНК/РНК, образование структуры рибосом, связывание белков (как сигнальные функции/регуля-ция, так и образование комплексов). Несмотря на то, что приведенные данные до сих пор не используются при предсказании функций, они могут лечь в основу важных приложений в будущем.
5.7. Заключение
В целом, предсказание функций - вероятно, более сложная задача, чем предсказание структуры, поскольку сходные структуры могут выпол
Литература
183
нять совершенно разные функции (см. также главу 6). Это в особенности справедливо в случае БПН, для которых структура - не просто отсутствие определенной пространственной укладки. Структурные элементы БПН различных размеров представляют собой ансамбль взаимопревращаю-щихся конформационных состояний, характеризующихся различными временами существования и связанных с выполняемой функцией. Существует ряд методов прогнозирования, которые надежно предсказывают неупорядоченное состояние на основе анализа аминокислотной последовательности, а также способны с приемлемой точностью определять структурные элементы, для которых вероятна связь с определенной функцией. Попытки предсказывать функции БПН по последовательности, однако, в своем развитии значительно отстают от методов предсказания структуры. Кроме того, успешность таких попыток сильно ограничивается осложняющими факторами, такими как быстрая эволюция и часто встречающаяся автономность последовательности от функций. Учитывая важности функций многих БПН, можно ожидать значительной активности в этой области исследований в ближайшем будущем.
Литература
Ashbumer М, Ball СА, Blake JA, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25-29
Bell S, Klein C, Muller L, et al. (2002) p53 contains large unstructured regions in its native state. J Mol Biol 322:917-927
Bhattacharyya RP, Remenyi A, Good MC, et al. (2006) The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 311:822-826
Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294:1351-1362
Bordoli L, Kiefer F, Schwede T (2007) Assessment of disorder predictions in CASP7. Proteins 69(Suppl 8): 129-136
Bourhis JM, Receveur-Brechot V, Oglesbee M, et al. (2005) The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci 14:1975-1992
Brown CJ, Takayama S, Campen AM, et al. (2002) Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol 55:104-110
Bustos DM, Iglesias AA (2006) Intrinsic disorder is a key characteristic in partners that bind 14-3-3 proteins. Proteins: Struct, Funct, Bioinformatics 63:35-42
Chen JW, Romero P, Uversky VN, et al. (2006a) Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J Proteome Res 5:879-887
Chen JW, Romero P, Uversky VN, et al. (2006b) Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder. J Proteome Res 5:888-898
Cheng Y, Oldfield CJ, Meng J, et al. (2007) Mining alpha-Helix-Forming Molecular Recognition Features with Cross Species Sequence Alignments. Biochemistry 46:13468-13477
Coeytaux K, Poupon A (2005) Prediction of unfolded segments in a protein sequence based on amino acid composition. Bioinformatics 21:1891-1900
Cox CJ, Dutta K, Petri ET, et al. (2002) The regions of securin and cyclin В proteins recognized by the ubiquitination machinery are natively unfolded. FEBS Lett 527:303-308
184 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
Daughdrill GW, Narayanaswami Р, Gilmore SH, et al. (2007) Dynamic behavior of an intrinsically unstructured linker domain is conserved in the face of negligible amino Acid sequence conservation. J Mol Evol 65:277-288
Davey NE, Shields DC, Edwards RJ (2006) SLiMDisc: short, linear motif discovery, correcting for common evolutionary descent. Nucleic Acids Res 34:3546-3554
Dawson R, Muller L, Dehner A, et al. (2003) The N-terminal domain of p53 is natively unfolded. J MolBiol 332:1131-1141
Dosztanyi Z, Csizmok V, Тотра P, et al. (2005a) lUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21:3433-3434
Dosztanyi Z, Csizmok V, Тотра P, et al. (2005b) The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol 347:827-839
Dosztanyi Z, Chen J, Dunker AK, et al. (2006) Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res 5:2985-2995
Dosztanyi Z, Sandor M, Тотра P, et al. (2007) Prediction of protein disorder at the domain level. Curr Protein Pept Sci 8:161-171
Dunker AK, Lawson JD, Brown CJ, etal. (2001) Intrinsically disordered protein. J Mol Graph Model 19:26-59
Dunker AK, Brown CJ, Lawson JD, et al. (2002) Intrinsic disorder and protein function. Biochemistry 41:6573-6582
Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 6:197-208
Elbaum M (2006) Materials science. Polymers in the pore. Science 314:766-767
Ferreon JC, Hilser VJ (2004) Thermodynamics of binding to SH3 domains: the energetic impact of polyproline II (P(II) ) helix formation. Biochemistry 43:7787-7797
Ferron F, Longhi S, Canard B, et al. (2006) A practical overview of protein disorder prediction methods. Proteins: Struct, Funct, Bioinformatics 65:1-14
Fontana A, Polverino de Laureto P, De Filippis V, et al. (1997) Probing the partly folded states of proteins by limited proteolysis. Fold Des 2:R17-26
Fontes MR, Teh T, Kobe В (2000) Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-alpha. J Mol Biol 297:1183-1194
Fowler DM, Koulov AV, Balch WE, et al. (2007) Functional amyloid-from bacteria to humans. Trends Biochem Sci 32:217-224
Fuxreiter M, Simon I, Friedrich P, et al. (2004) Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol 338:1015-1026
Fuxreiter M, Тотра P, Simon I (2007) Structural disorder imparts plasticity on linear motifs. Bioinformatics 23:950-956
Galzitskaya OV, Garbuzynskiy SO, Lobanov MY (2006) FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics 22:2948-2949
Gill G, Ptashne M (1987) Mutants of GAL4 protein altered in an activation function. Cell 51:121—126.
Graham TA, Ferkey DM, Mao F, et al. (2001) Tcf4 can specifically recognize beta-catenin using alternative conformations. Nat Struct Biol 8:1048-1052
Haarmann CS, Green D, Casarotto MG, etal. (2003) The random-coil ‘C’ fragment of the dihydropyridine receptor П-III loop can activate or inhibit native skeletal ryanodine receptors. Biochem J 372:305-316
Hansen JC, Lu X, Ross ED, et al. (2006) Intrinsic protein disorder, amino acid composition, and histone terminal domains. J Biol Chem 281:1853-1856
Haynes C, Oldfield CJ, Ji F, et al. (2006) Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol 2:el00
Hegyi H, Schad E, Тотра P (2007) Structural disorder promotes assembly of protein complexes. BMC Struct Biol 7:65
Hiroaki H, Ago T, Ito T, et al. (2001) Solution structure of the PX domain, a target of the SH3 domain. Nat Struct Biol 8:526-530.
Holt C, Sawyer L (1988) Primary and predicted secondary structures of the caseins in relation to their biological functions. Protein Eng 2:251-259.
Литература
185
Holt С, Wahlgren NM, Drakenberg T (1996) Ability of a beta-casein phosphopeptide to modulate the precipitation of calcium phosphate by forming amorphous dicalcium phosphate nanoclusters. Biochem J 314:1035-1039.
Hope IA, Mahadevan S, Struhl К (1988) Structural and functional characterization of the short acidic transcriptional activation region of yeast GCN4 protein. Nature 333:635-640.
lakoucheva L, Brown C, Lawson J, et al. (2002) Intrinsic Disorder in Cell-signaling and Cancer associated Proteins. J Mol Biol 323:573-584
lakoucheva LM, Radivojac P, Brown CJ, et al. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 32:1037-1049
Ikura M, Ames JB (2006) Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: two ways to promote multifunctionality. Proc Natl Acad Sci USA 103:1159-1164
Jin Y, Dunbrack RL, Jr. (2005) Assessment of disorder predictions in CASP6. Proteins 61 (Suppl 7): 167-175
Khan AN, Lewis PN (2005) Unstructured conformations are a substrate requirement for the Sir2 family of NAD-dependent protein deacetylases. J Biol Chem 280:36073-36078
Kiss R, Bozoky Z, Kovacs D, et al. (2008a) Calcium-induced tripartite binding of intrinsically disordered calpastatin to its cognate enzyme, calpain. FEBS Lett 582:2149-2154
Kiss R, Kovacs D, Tompa P, et al. (2008b) Local structural preferences of calpastatin, the intrinsically unstructured protein inhibitor of calpain. Biochemistry 47:6936-6945
Kovacs D, Kalmar E, Torok Z, et al. (2008) Chaperone activity of ERD10 and ERD14, two disordered stress-related plant proteins. Plant Physiol 147:381-390
Kriwacki RW, Hengst L, Tennant L, etal. (1996) Structural studies of p21 Wafl/Cipl/Sdil in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci USA 93:11504-11509
Lacy ER, Filippov I, Lewis WS, et al. (2004) p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol 11:358-364
Lee H, Mok KH, Muhandiram R, et al. (2000) Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem 275:29426-29432
Li X, Romero P, Rani M, et al. (1999) Predicting protein disorder for N-, C-, and internal regions. Genome Inform Ser Workshop Genome Inform 10:30-40
Linding R, Russell RB, Neduva V, et al. (2003 a) GlobPlot: Exploring protein sequences for globu-larity and disorder. Nucleic Acids Res 31:3701-3708
Linding R, Jensen LJ, Diella F, et al. (2003b) Protein disorder prediction: implications for structural proteomics. Structure 11:1453-1459
Lise S, Jones DT (2005) Sequence patterns associated with disordered regions in proteins. Proteins 58:144-150
Liu J, Rost В (2003) NORSp: Predictions of long regions without regular secondary structure. Nucleic Acids Res 31:3833-3835
Liu J, Tan H, Rost В (2002) Loopy proteins appear conserved in evolution. J Mol Biol 322:53-64
Lobley A, Swindells MB, Orengo CA, et al. (2007) Inferring function using patterns of native disorder in proteins. PLoS Comput Biol 3:el62
Lopez Garcia F, Zahn R, Riek R, et al. (2000) NMR structure of the bovine prion protein. Proc Natl Acad Sci USA 97:8334-8339
Manalan AS, Klee CB (1983) Activation of calcineurin by limited proteolysis. Proc Natl Acad Sci USA 80:4291—4295
Mark WY, Liao JC, Lu Y, et al. (2005) Characterization of segments from the central region of BRCA1: an intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J Mol Biol 345:275-287
Minezaki Y, Homma K, Kinjo AR, et al. (2006) Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J Mol Biol 359:1137-1149
Mohan A, Oldfield CJ, Radivojac P, et al. (2006) Analysis of molecular recognition features (MoRFs). J Mol Biol 362:1043-1059
Mukhopadhyay R, Hoh JH (2001) AFM force measurements on microtubule-associated proteins: the projection domain exerts a long-range repulsive force. FEBS Lett 505:374-378.
Neduva V, Russell RB (2005) Linear motifs: evolutionary interaction switches. FEBS Lett 579:3342-3345
186 Глава 5. Методы биоинформатики для изучения неупорядоченных белков
Neduva V, Russell RB (2006) DILIMOT: discovery of linear motifs in proteins. Nucleic Acids Res 34:W350-355
Neduva V, binding R, Su-Angrand I, et al. (2005) Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol 3:e405
Ng KP, Potikyan G, Savene RO, et al. (2007) Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc Natl Acad Sci USA 104:479-484
Obenauer JC, Cantley LC, Yaffe MB (2003) Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31:3635-3641
Olashaw N, Bagui TK, Pledger WJ (2004) Cell cycle control: a complex issue. Cell Cycle 3:263-264
Oldfield CJ, Cheng Y, Cortese MS, et al. (2005a) Comparing and combining predictors of mostly disordered proteins. Biochemistry 44:1989-2000
Oldfield CJ, Cheng Y, Cortese MS, etal. (2005b) Coupled folding and binding with alphahelixforming molecular recognition elements. Biochemistry 44:12454-12470
Olson KE, Narayanaswami P, Vise PD, et al. (2005) Secondary structure and dynamics of an intrinsically unstructured linker domain. J Biomol Struct Dyn 23:113-124
Patil A, Nakamura H (2006) Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett 580:2041-2045
Pawson T, Nash P (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300:445-452
Peng K, Radivojac P, Vucetic S, et al. (2006) Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 7:208
Pierce MM, Baxa U, Steven AC, et al. (2005) Is the prion domain of soluble Ure2p unstructured? Biochemistry 44:321-328
Pontius BW (1993) Close encounters: why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem Sci 18:181-186.
Prilusky J, Felder CE, Zeev-Ben-Mordehai T, etal. (2005) Foldindex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21:3435-3438
Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363-13383
Puntervol 1 P, binding R, Gemund C, et al. (2003) ELM server: a new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res 31:3625-3630
Radhakrishnan I, Perez-Alvarado GC, Dyson HJ, etal. (1998) Conformational preferences in the Serl33-phosphorylated and non-phosphorylated forms of the kinase inducible transactivation domain ofCREB. FEBS Lett 430:317-322
Radivojac P, Vucetic S, O’Connor TR, et al. (2006) Calmodulin signaling: analysis and prediction of a disorder-dependent molecular recognition. Proteins 63:398-410
Romero P, Obradovic Z, Kissinger CR, et al. (1998) Thousands of proteins likely to have long disordered regions. Рас Symp Biocomputing 3:437-448
Romero P, Obradovic Z, Dunker AK (1999) Folding minimal sequences: the lower bound for sequence complexity of globular proteins. FEBS I^ett 462:363-367
Romero P, Obradovic Z, Li X, et al. (2001) Sequence complexity of disordered protein. Proteins 42:38-48
Ross ED, Edskes HK, Terry MJ, et al. (2005) Primary sequence independence for prion formation. Proc Natl Acad Sci USA 102:12825-12830
Schlessinger A, Punta M, Rost В (2007) Natively unstructured regions in proteins identified from contact predictions. Bioinformatics 23:2376-2384
Schweers O, Schonbrunn-Hanebeck E, Marx A, et al. (1994) Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem 269:24290-24297.
Seet ВТ, Dikic I, Zhou MM, et al. (2006) Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol 7:473-483
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379-423,623-656
Si K, Giustetto M, Etkin A, et al. (2003a) A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell 115:893-904
Si K, Lindquist S, Kandel ER (2003b) A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115:879-891
Sickmeier M, Hamilton JA, LeGall T, et al. (2007) DisProt: the Database of Disordered Proteins. Nucleic Acids Res 35:D786-793
Литература
187
Sigalov A, Aivazian D, Stem L (2004) Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 43:2049-2061
Sigler PB (1988) Transcriptional activation. Acid blobs and negative noodles. Nature 333:210-212
Simon SM, Sousa FJ, Mohana-Borges R, et al. (2008) Regulation of Escherichia coli SOS mutagenesis by dimeric intrinsically disordered umuD gene products. Proc Natl Acad Sci USA 105:1152-1157
Тотра P (2002) Intrinsically unstructured proteins. Trends Biochem Sci 27:527-533
Тотра P (2003) Intrinsically unstructured proteins evolve by repeat expansion. Bioessays 25:847-855
Тотра P (2005) The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett 579:3346-3354
Тотра P, Csermely P (2004) The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 18:1169-1175
Тотра P, Fuxreiter M (2008) Fuzzy complexes: polymorphism and structural disorder in proteinprotein interactions. Trends Biochem Sci 33:2-8
Тотра P, Szasz C, Buday L (2005) Structural disorder throws new light on moonlighting. Trends Biochem Sci 30:484-489
Тотра P, Dosztanyi Z, Simon I (2006) Prevalent structural disorder in E. coli and S. cerevisiae pro-teomes. J Proteome Res 5:1996-2000
Triezenberg SJ (1995) Structure and function of transcriptional activation domains. Curr Opin Genet Dev 5:190-196
Trombitas K, Greaser M, Labeit S, etal. (1998) Titin extensibility in situ: entropic elasticity of permanently folded and permanently unfolded molecular segments. J Cell Biol 140:853-859.
Tucker MM, Robinson JB, Jr., Stellwagen E (1981) The effect of proteolysis on the calmodulin activation of cyclic nucleotide phosphodiesterase. J Biol Chem 256:9051-9058
Tuite MF, Koloteva-Levin N (2004) Propagating prions in fungi and mammals. Mol Cell 14:541-552
Uversky VN (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci 11:739-756
Uversky VN, Gillespie JR, Fink AL (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41:415-427
Uversky VN, Oldfield CJ, Dunker AK (2005) Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit 18:343-384
Vacic V, Oldfield CJ, Mohan A, et al. (2007) Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res 6:2351-2366
Vucetic S, Brown CJ, Dunker AK, et al. (2003) Flavors of protein disorder. Proteins 52:573-584
Waizenegger I, Gimenez-Abian JF, Wemic D, et al. (2002) Regulation of human separase by securin binding and autocleavage. Curr Biol 12:1368-1378
Ward J J, Sodhi JS, McGuffin LJ, et al. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337:635-645
Weathers EA, Paulaitis ME, Woolf ТВ, et al. (2004) Reduced amino acid alphabet is sufficient to accurately recognize intrinsically disordered protein. FEBS Lett 576:348-352
Weinreb PH, Zhen W, Poon AW, et al. (1996) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35:13709-13715
Wickner RB, Edskes HK, Maddelein ML, et al. (1999) Prions of yeast and fungi. Proteins as genetic material. J Biol Chem 274:555-558
Wootton JC (1994a) Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput Chem 18:269-285
Wootton JC (1994b) Sequences with “unusual” amino acid compositions. Curr Opin Struct Biol 4:413-421
Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re-assessing the protein structurefunction paradigm. J Mol Biol 293:321-331
Xie H, Vucetic S, lakoucheva LM, et al. (2007) Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res 6:1882-1898
Глава 6
Функциональное разнообразие в элементах упаковки и надсемействах
Бенуа X. Дессейли, Кристина А. Оренго
С первыми успехами структурной геномики стало доступно всё увеличивающееся число пространственных структур белков с неизвестными функциями. Тем не менее, по-прежнему спорным остается вопрос, насколько информация о структуре способствует пониманию функций. В настоящей главе рассматривается значение методов, которые позволяют на различных уровнях (обычно элемент укладки и надсемейство) устанавливать структурные связи между белками, с тем, чтобы затем осуществлять перенос функциональных аннотаций. Прежде всего, исследовано разнообразие функций белков, имеющих общий элемент укладки. Определение элемента укладки в некоторых случаях может служить подсказкой при определении функциональных свойств. Однако показано, что разнообразие функций, соответствующих одному элементу укладки, может быть весьма широко, и для некоторых многообразных элементов укладки (например, суперфолдов) получаемая таким образом информация о функциях оказывается очень скудной. Далее, проанализировано разнообразие функций среди белков одного надсемейства (гомологичных белков), поскольку данные о структуре могут помочь при определении гомологии в отсутствие сходства последовательностей. Обсуждаются эволюционные основы и механизмы, породившие существующее функциональное многообразие связанных между собой белков. Рассмотрены полезные инструменты для взаимосвязанного анализа структуры, функций и эволюции белков.
В. Н. Dessailly and С. A. Orengo* *
Department of Structural and Molecular Biology, University College London,
London WC1E 6BT, UK
*e-mai 1: orengo@biochemistry. ucl. ac. uk
6.1. Определение функций
189
6.1. Определение функций
Прежде, чем обсуждать, каким образом определение связей на уровне элемента упаковки или надсемейства может помочь при определении функций белка, необходимо дать четкое определение термину «функция» в настоящей главе. Это необходимо в том числе и для того, чтобы наметить аспекты функций, наличие которых можно наилучшим образом предполагать на основе структурной информации.
Функция - относительно расплывчатое понятие, охватывающее множество различных аспектов активности белка. Более того, аспекты, подразумеваемые под этим термином, изменяются в зависимости от конкретной области науки о белках. Так, физиолог, вероятно, описал бы функцию белка в терминах его влияния на глобальный фенотип (например, «инициатор гибели клеток»), тогда как биохимик, как правило, определяет функцию исследуемого белка на основе характерных для него молекулярных взаимодействий или каталитической активности (например, «серин-треониновая протеин-киназа, взаимодействующая с рецептором»). Из-за этих различий в использовании термина весьма сложно дать универсальное и широко применимое определение функции белка.
Однако разрабатывать универсальную формулировку вовсе не обязательно. Консорциумом по генной онтологии (ГО) предложена общая схема, с помощью которой можно определить и, что более важно, классифицировать функции белков универсальным образом (The Gene Ontology Consortium 2000). В ГО функции белка рассматриваются с трех различных позиций, а затем с каждой из позиций дается независимое определение. Согласно ГО, клеточный компонент описывает биологические структуры, к которым принадлежит белок (например, ядро или рибосома); биологическая цель отвечает целям или путям, в которых принимает участие белок (например, метаболизм, передача сигналов или дифференцировка клеток); молекулярная функция белка - совокупность функций, которые он может выполнять (например, катализ или транспорт).
Информация о пространственной структуре белков преимущественно помогает понять каталитические механизмы и предположить возможные взаимодействия с другими молекулами - оба эти аспекта принадлежат к категории молекулярных функций. В результате, в случаях, когда речь идет о связях между структурой и функциями (как в этой главе), рассматривается главным образом категория молекулярных функций.
Для описания молекулярных функций белков существует ряд баз данных и систем аннотации (см. таблицу 6.1), которые весьма полезны
190
Глава 6. Разнообразие в элементах упаковки и надсемействах
Таблица 6.1. Ссылки и краткие описания интересных баз данных и инструментов, упомянутых в тексте
Название URL Описание
САТН http ://cath www. biochem. ucl. ac.uk/ Структурная классификация белков
SCOP http://scop.mrc-lmb.cam.ac.uk/scop/ Структурная классификация белков
SFLD http://sfld.rbvi.ucsf.edu/ Функциональная классификация надсемейств ферментов
PROCOGNATE http ://www. ebi. ac.uk/thomton-srv/databases/procognate/index. html Картирование доменов по их известным лигандам
Gene Ontology http ://www. geneontology.org Регулируемый словарь белковых функций
EC http ://www. chem. qmul. ac.uk/ iubmb/ enzyme/ Классификация ферментативных реакций
EzCatDB http ://mbs.cbrc.jp/EzCatDB/ База данных каталитических механизмов ферментов
MACiE http ://www. ebi. ac. uk/ thomton-srv/databases/MACiE/ База данных ферментативных реакций
KEGG http ://www. genome.jp/kegg/ Объединенное представление генов, генных продуктов и метаболических путей
FUNCAT http ://mips.gsf.de/proj ects/funcat Схема аннотирования функций белков
DALI http://ekhidna.biocenter.helsinki.fi/ daliserver Структурное выравнивание
FATCAT http ://fatcat.bumham.org/ Гибкое структурное выравнивание
SSM http ://www. ebi. ac. uk/msd-srv/ssm/ Структурное выравнивание по совпадению вторичной структуры
CATHEDRAL http ://www.cathdb. info/ cgi-bin/CathedralServer.pl Алгоритм выявления известных типов укладки в структурах белков
6.1. Определение функций
191
при изучении структурно-функциональных связей, в особенности, на автоматической основе. Вероятно, старейшим методом описания молекулярных функций белков является схема нумерации Комиссии по ферментам (КФ, ЕС, от «Enzyme Commission»), в которой ферментативные реакции классифицированы иерархически. Используется четырехразрядная система, в которой каждый уровень все более подробно описывает особенности реакции, от общего типа каталитической активности (оксидоредуктаза, гидролаза и т.д.) до специфических молекул, выступающих в роли субстрата (Nomenclature Committee of the IUBMB 1992). Для преодоления давно известного ограничения классификации КФ в последнее время были созданы две новые базы данных для классификации ферментов и их реакций: EzCatDB (от «Enzyme catalytic-mechanism Database») (Nagano 2005) и MACiE (от «Mechanism, Annotation and Classification in Enzymes») (Holliday et al. 2007). Обе базы данных содержат описание и классификацию механизмов ферментативных реакций, а не информацию о реакциях как таковых. Это связано с тем, что в последнее время все большее одобрение получает идея, согласно которой классификацию, в основу которой положены реакции, (такую как система КФ) не всегда можно использовать в качестве классификации соответствующих ферментов (О’Boyle et al. 2007). В дополнение к этим базам данных, Атлас каталитических сайтов содержит подробную информацию о специфических аминокислотных остатках, непосредственно задействованных в механизмах катализа, для ферментов известной структуры (Porter et al. 2004). Некоторые базы данных предоставляют дальнейшее описание всех белковых остатков, участвующих в связывании биологически важных молекул, таких как субстраты и кофакторы (Lopez et al. 2007; Dessailly et al. 2008). К числу других широко используемых систем аннотации белковых функций принадлежат KEGG и FUNCAT. KEGG первоначально был нацелен на описание метаболических путей и сетей биологических реакций, а в настоящее время представляет собой более широкую систему классификации биологических функций (Kanehisa et al. 2008). FUNCAT (от «the Functional Catalogue») классифицирует белковые функции и строит уникальное иерархическое дерево (Ruepp et al. 2004).
Как и большинство баз данных такого рода, и KEGG, и FUNCAT содержат информацию преимущественно о биологических процессах, в которых участвуют описываемые белки, а не о разновидностях их молекулярной активности, однако обе эти базы данных содержат сведения, которые могут оказаться весьма полезны при исследовании свойства, которое в системе ГО принято называть молекулярной функцией.
192
Глава 6. Разнообразие в элементах упаковки и надсемействах
6.2. От способа укладки к функции
6.2.1. Определение способа укладки
6.2.1.1. Общие положения
Под способом укладки в общем случае понимают расположение главных элементов вторичной структуры белка, которое учитывает их взаимную ориентацию и топологические связи. Основная проблема, непосредственно вытекающая из этого общего определения, состоит в том, что не существует объективных правил, определяющих главные элементы вторичной структуры, которые необходимо рассматривать при определении способа укладки (Grishin 2001).
Одна из целей этой главы состоит в описании того, каким образом сведения о связях между белками, такие как информация об общем способе укладки, помогают распространять функциональные аннотации детально описанных белков на белки с неизвестными функциями. Как будет обсуждаться далее в разделе 6.3, процесс переноса аннотаций с одних белков на другие основан на предположении, согласно которому связанные в процессе эволюции (т.е., гомологичные) белки, как правило, имеют общие функциональные свойства. Однако белки, имеющие общий способ укладки, не обязательно являются гомологичными. В последнее время обсуждается вопрос о том, что различные белки могут приобретать один и тот же способ укладки независимо друг от друга в ходе конвергентной эволюции, поскольку количество способов укладки, приемлемых с физической точки зрения, ограничено (Russell et al. 1997).
Так, не установлено, связаны ли между собой эволюционно все белковые надсемейства, для которых характерен способ укладки, напоминающий структуру бочонка TIM (р/а)8, поскольку четких доказательств в этом вопросе пока не обнаужено (Nagano et al. 2002).
6.2.1.2. Практические подходы
Существуют базы данных, в которых классификация белков представляет собой всеобъемлющую схему структурных связей. Ниже приводится практическое определение способа упаковки белковых структур, которое широко используется в некоторых базах данных. Как следует из определения, понятие способа упаковки применимо к доменам в большей степени, чем к полноразмерным белкам, однако определения домена для различных баз данных могут отличаться.
6.2. От способа укладки к функции
193
База данных САТН представляет собой иерархическую систему классификации структур белковых доменов (Orengo etal. 1997; Greene etal. 2007). Самый высокий уровень классификации определяет принадлежность белкового домена к одному из трех классов на основе его общего содержания во вторичной структуре. В пределах классов САТН домены относят к различным типам архитектуры, которые описывают взаимное расположение элементов вторичной структуры без учета возможностей их взаимодействия. Далее домены определенной архитектуры классифицируют по типу топологии в зависимости от того, каким образом элементы вторичной структуры соединены друг с другом. Именно этот топологический уровень наиболее близок к общему определению способа укладки, приведенному выше. На практике определение принадлежности домена к той или иной топологии САТН осуществляется автоматически с помощью программы выравнивания структур SSAP (Orengo and Taylor 1996) и разработанных опытным путем отсечек.
SCOP (от «the Structural Classification of Proteins»), так же как и САТН, является иерархической системой классификации структур белковых доменов (Murzin et al. 1995; Andreeva et al. 2008), однако уровни классификации в этих базах данных отличаются. Как и в САТН, самым высоким иерархическим уровнем в SCOP является структурный класс, однако в SCOP существует четыре различных класса, тогда как в САТН - три. Следующий уровень классификации - способ укладки', два белковых домена имеют один и тот же способ укладки, если им свойственны схожие элементы вторичной структуры, расположенные друг относительно друга схожим образом и связанные схожими топологическими связями. Это определение хорошо согласуется с определением топологии в САТН, но на практике принадлежность отдельных доменов к определенным иерархическим уровням в двух базах данных может отличаться, поскольку в каждом из определений присутствует элемент субъективности (а именно, какие элементы вторичной структуры считать основными), а также из-за разных протоколов, используемых для классификации доменов (автоматизированных в САТН и преимущественно настраиваемых вручную в SCOP).
Исключительно объективный метод определения способов укладки предложен в FSSP (от «families of structurally similar proteins» (Holm and Sander 1996b). В FSSP для ряда представительных и неизбыточных структур PDB выполнялись парные выравнивания, при этом использовалась программа для структурного выравнивания DALI (Holm and Sander 1993). Полученные таким образом численные показатели парного выравнивания структур использовали при иерархической кластеризации, с помощью ко-
194
Глава 6. Разнообразие в элементах упаковки и надсемействах
торой создавали так называемое дерево структурной укладки. Семейства структурной укладки определяли автоматически, разделяя полученное дерево на различных уровнях подобия.
6.2.1.3. Изменение парадигмы
В общем случае в процессе эволюции структура белка является более консервативной, чем его последовательность, что отражено во множестве общих структурных характеристик белков. По мере того, как в середине 1990-ых годов росло количество пространственных структур белков, всё острее становилась необходимость появления систем структурной классификации, с помощью которых можно было извлекать из анализа структурных данных значимую информацию. Такая ситуация привела к появлению описанных выше иерархических систем классификации белковых структур. Общие структурные мотивы, такие как бочонки (р/а)8 или четырехспиральные пучки, встречаются в белках, последовательности которых не имеют общих черт. Осмысление этого факта привело к формированию представлений о способах пространственной укладки в белках, которые изложены выше. До недавнего времени под способами пространственной укладки белков понимали периодически повторяющиеся общие структурные мотивы, которые случайным образом разделяют структурное пространство белков. В рамках этой теории подразумевается, что пространство укладки дискретно, т.е.: а) каждый белок характеризуется уникальным способом укладки, который объединяет его со схожими белками и отделяет от большинства несвязанных белков (несмотря на объяснение существования аналогичных способов укладки, см. раздел 6.2.2.1); б) каждый способ укладки обладает собственными структурными характеристиками и представляет собой отдельную структурную группу, не перекрывающуюся с другими группами (Kolodny et al. 2006).
Однако по мере увеличения количества доступных структурных данных, в особенности за счет успехов структурной геномики, представления о способах укладки белков меняются, и пространство укладки представляется скорее непрерывным, чем дискретным (Harrison et al. 2002). В последнее время всё более широкое распространение получает идея, согласно которой гомологичные белки могут сворачиваться различными способами (Grishin 2001; Kolodny et al. 2006), а для некоторых белков характерно множество непостоянных мотивов укладки, при этом мотив в данный момент времени определяется условиями, в которых существует белок (Andreeva and Murzin 2006). Все это имеет свои последствия при учете способов укладки в ходе исследования функций белков. Главный аргумент
6.2. От способа укладки к функции
195
в пользу учета способов укладки при исследовании функций состоит в том, что белки, обладающие схожей укладкой, зачастую обладают также отдаленной гомологией, которую нельзя установить другими методами исследования, а также в том, что гомологичные белки, как правило, выполняют схожие функции (Moult and Melamud 2000). Это неизбежно следует из того факта, что при неопределенной связи между способом укладки и гомологией связь между способом укладки и функциями, вероятно, также будет неопределенной. Однако результаты последних исследований, полученные при использовании совокупности структурных данных, доступных в САТН, свидетельствуют о том, что большинство способов укладки структурно согласованы и заметно отличаются от остальных способов укладки (Cuff et al., рукопись в стадии подготовки). Действительно, как будет продемонстрировано позднее, подобие способов укладки может способствовать установлению подобия функций изучаемых белков (Martin etal. 1998).
6.2.2. Связь между способами укладки и предсказание функций
В этом разделе рассматриваются свойства функций, существование которых можно предположить без использования данных о гомологии, т.е., таких функций, которые возникли в ходе конвергентной эволюции. Вопросы, связанные с изучением функций, установленных на основе информации о гомологии, рассматриваются в разделе 6.3 настоящей главы.
В общем случае, определение структуры и способа укладки белка предоставляет исследователю возможность использовать множество методов предсказания белковых функций на основе структурного анализа, которые были бы недоступны в отсутствие известной белковой структуры. В основе некоторых из этих методов лежит принцип, согласно которому информация о структуре позволяет установить общие гомологии, не очевидные на уровне последовательности (Lee et al. 2007). В других методах структурная информация используется лишь на основе предположения о том, что она является значимой при выполнении белком его молекулярных функций, при этом эволюционный контекст структурных свойств не принимают во внимание. Многие из этих методов описаны в других главах книги (см. главы 7, 8, 10 и 11). В настоящей главе обсуждаются лишь случаи, имеющие непосредственное отношение к информации о способах укладки.
196
Глава 6. Разнообразие в элементах упаковки и надсемействах
6.2.2.1. Способы укладки с единственной функцией
Структуру белка, определенную недавно, можно использовать для поиска схожих способов укладки среди уже известных белковых структур. При этом используются программы сравнения структур, которые обычно оценивают значимость обнаруженных структурных сходств, используя специфические схемы оценки. Некоторые из этих программ являются общедоступными и в последнее время были протестированы с использованием большого набора данных известных структурных сходств, созданного САТН (Kolodny et al. 2005; Redfern et al. 2007). К числу таких программ принадлежат DALI (Holm and Sander 1996a), FATCAT (Ye and Godzik 2004), SSM (Krissinel and Henrick 2004), CE (Shindyalov and Bourne 1998) и CATHEDRAL (Redfern et al. 2007). В случае, когда новая структура обнаружена в белке с неизвестной функцией, на следующем шаге исследования необходимо оценить, можно ли на основе подобия белковых структур осуществить перенос функциональной аннотации.
Некоторые способы укладки свойственны лишь гомологичным белкам, тогда как другие могут встречаться у белков, эволюционировавших конвергентно, - их принято обозначать гомологичными и аналогичными способами укладки, соответственно (Moult and Melamud 2000). Аналогичным образом, некоторые способы укладки являются однородными с точки зрения функций, тогда как другие характерны для белков, выполняющих весьма разнообразные функции. Принято считать, что гомологичные способы укладки более однородны в функциональном смысле по сравнению с аналогичными (Moult and Melamud 2000). Очевидно, что если способ укладки связан с уникальной функцией X, обнаружение этой укладки в белке с неизвестной функцией прямо приведет к описанию белка с функцией X. Однако на практике ситуация несколько сложнее, поскольку многообразные в функциональном отношении способы укладки можно ошибочно идентифицировать как связанные с единственной функцией из-за специфической выборки.
Так или иначе, известны случаи, когда определение способа укладки способствовало предсказанию функции белка (Moult and Melamud 2000). Например, пространственная структура продукта гена усаС Escherichia coli содержит укладку, которая близка к укладке белков семейства амидогидролаз. Дальнейшие исследования показали, что этот белок имеет каталитический аппарат, близкий к имеющемуся у белков с таким же способом укладки (Colovos et al. 1998; Moult and Melamud 2000). Растет количество случаев успешного предсказания функций белков на основе определения способа укладки с помощью средств структурной геномики, общей целью которой является определение структуры как можно большего числа белков
6.2. От способа укладки к функции
197
(Adams et al. 2007). В большинстве случаев, однако, успешное предсказание функции белка не является результатом простого определения способа укладки, а связано с сочетанием способа укладки с каким-либо другим свойством, например, мотивом распознавания последовательности или сходством функциональных участков исследуемых белков.
6.2.2.2. Суперучастки
В целом, данные о пространственной структуре весьма полезны при определении функционального участка - подмножества остатков, которые имеют решающее значение для выполнения белком его молекулярной функции. Функциональные участки представлены главным образом участками связывания (совокупность белковых остатков, которые взаимодействуют с лигандами) (Dessailly et al. 2008) или каталитическими участками (совокупность остатков, которые принимают непосредственное участие в ферментативной реакции) (Porter et al. 2004).
Одна из причин, по которым структуры полезны при определении функциональных участков, состоит в том, что последние, как правило, располагаются в наиболее консервативных топологических областях структур. Более того, даже в тех случаях, когда нет убедительных доказательств гомологии белков, имеющих общий способ укладки, функциональные участки располагаются в одних и тех же областях их пространственной структуры. Такие функциональные участки называют суперучастками. Показано, что суперучастки в большом количестве встречаются при аналогичной упаковке (или суперупаковке, см. раздел 6.2.3.1), т.е., при упаковке, которая является общей для негомологичных белков (Russell etal. 1998). На рис. 6.1 представлен хорошо известный пример суперучастка: это каталитический участок белков, для которых характерна упаковка в форме бочонка (р/а)8. Остатки, принимающие участие в катализе, всегда располагаются на С-концах р-тяжей центрального параллельного p-листа, хотя структура самих р-тяжей может изменяться (Nagano et al. 2002).
6.2.2.3. Суперукладка
Способ укладки, распространенный среди множества самых разных надсемейств и отличающийся выраженным функциональным разнообразием, называют суперукладкой (Orengo etal. 1994). Элементы суперукладки входят в состав белков, выполняющих большое количество различных функций. Замечательными примерами суперукладки являются способ укладки, напоминающий структуру бочонка TIM (р/а)8, который встречается у представителей более 25 различных надсемейств (Nagano et al. 2002), а
198
Глава 6. Разнообразие в элементах упаковки и надсемействах
также укладка Россмана, которую можно обнаружить у белков 114 надсемейств CATH (CATH v3.1), многие из которых отличаются функциональным разнообразием. Элементы суперукладки составляют весьма незначительную часть известных способов укладки, однако именно они, по-видимому, являются продуктами большой части известных геномов (Lee etal. 2005). Также элементы суперукладки являются одной из основных проблем при предсказании функций белков на основе определения и анализа способов укладки, поскольку белки, обладающие схожими элементами суперукладки, далеко не всегда обладают схожими функциями. Существование таких способов укладки и их широкое распространение среди белковых молекул вынуждает исследователей с осторожностью использовать информацию о связях между известными способами укладки при предсказании функций белков.
Рис. 6.1. (Цветную версию рисунка см. на вклейке.) Суперучастки укладки, напоминающей структуру бочонка TIM (р/а)8. Схематичные изображения четырех белков, имеющих укладку бочонка (р/а)8> представленные в различных надсемействах САТН (и SCOP): а) дегидроптероатсинтаза Е. coli (идентификационный номер (ИН) домена в CATH: lajOAOO); б) альфа-субъединица триптофан-синтазы Р. furiosus (ИН домена в CATH: IgeqBOO); в) эндо-1,4-бета-ксиланаза Z С. thermocellum (ИН домена в CATH: IxyzAOO); г) альдегидредуктаза Н. sapiens (ИН домена в САТН: 2а1гА00). Структуры совмещались с помощью CORA (Orengo 1999). Молекулы ориентированы одинаково. Общие элементы четырех структур показаны красным цветом. Положение каталитических остатков (согласно Атласу каталитических центров) показано зеленым цветом. Несмотря на значительные структурные различия и отсутствие доказательств гомологии между этими белками, каталитические центры всегда находятся вблизи С-конца центральных Р-тяжей. Изображения пространственных структур были созданы с помощью программы Molscript (Kraulis 1991), рендеринг изображений осуществлялся с помощью программы Raster3D (Merritt and Bacon 1997)
6.3. Разнообразие функций гомологичных белков
199
6.3. Разнообразие функций гомологичных белков
В целом, выявление гомологии (связей между надсемействами) является гораздо более полезным при предсказании функций, чем выявление лишь структурного сходства (связей между способами укладки). В этом разделе рассматривается связь между гомологией структур и функциональным разнообразием. Показано, что даже в случаях определения гомологии остается множество препятствий при попытках использовать функциональные аннотации одного белка для описания другого.
6.3.1. Определения
Прежде, чем объяснять, каким образом функция в надсемействах ди-вергирует, необходимо дать четкое определение термину надсемейство и представление о том, как он используется на практике. Также вводится термин семейство, который используется в настоящем разделе.
6.3.1.1. Общие представления
Надсемейство - это группа белков, которые считаются эволюционно связанными друг с другом. Связи между белками в надсемействе можно установить по сходству последовательностей, которое определяется с помощью традиционных методов выравнивания последовательностей или более чувствительного поиска с использованием СММ (Reid et al. 2007). В отсутствие сходства последовательностей на основе анализа структуры можно также выявить наличие отдаленной гомологии и/или сходства функций. Однако, в отличие от сходства последовательностей, в последнем случае не существует широко признанных способов оценки уровня статистической значимости структурного или функционального подобия. По этой причине отсечки, используемые для определения связей в рамках надсемейства, могут быть произвольными и до некоторой степени субъективными. На сегодняшний день некоторые базы данных, такие как САТН и SCOP, разработали стандартные и широко принятые определения надсемейств (см. раздел 6.3.2.1). Однако во всех этих базах данных до сих пор присутствует определенная степень субъективности при отнесении белка к надсемейству, что подтверждается рядом фактов: во-первых, при определении принадлежности белка к тому или иному семейству по-прежнему требуется ручная проверка достоверности, а во-вторых, для некоторых доменов различные базы выдают несовместимые результаты (Greene et al.
200
Глава 6. Разнообразие в элементах упаковки и надсемействах
2007; Andreeva et al. 2008). Следует отметить, что в настоящее время как в САТН, так и в SCOP предварительная классификация новых белковых структур осуществляется с помощью автоматических протоколов, однако окончательное отнесение белка к надсемейству по-прежнему включает ручную обработку данных.
Понятие о семействе более расплывчато. В настоящее время под семейством в общем случае понимают подсистему классификации гомологичных белков, соответствующую ряду критериев. Так, к семейству последовательностей с определенным уровнем сходства относятся все белки, которые характеризуются по меньшей мере этим уровнем сходства; к функциональному семейству относятся гомологи, которые имеют общую функцию; к ортоло-гичному семейству относятся все ортологи и т.д. В зависимости от направленности базы данных, определение семейства будет меняться.
6.3.1.2. Практические подходы
В настоящем разделе описаны только базы данных, содержащие сведения о структурах.
САТН и Gene3D. В классификации САТН домены данной топологии (см. раздел 2.1.2) затем относят к одному и тому же надсемейству гомологов (Н-уровенъ, от «Homologous»), если считается, что они имеют общего предка. Два домена считаются гомологичными, если они удовлетворяют по меньшей мере двум из следующих критериев: а) структурное сходство, определенное с помощью отсечек, разработанных опытным путем; б) сходство последовательностей, определенное с помощью стандартных методов сравнения последовательностей и поиска СММ; в) функциональное сходство, определенное с помощью ручного анализа. С помощью Gene3D эта классификация распространяется на белки неизвестной структуры: поиск последовательностей осуществляют среди профилей СММ библиотеки САТН и таким образом устанавливают принадлежность частей последовательностей к гомологичным надсемействам САТН (Yeats et al. 2008). Надсемейства САТН далее делят на семейства последовательностей, для каждого из которых определены отсечки идентичности последовательности. Для определения неизбыточных групп белков используется отсечка, составляющая 35% идентичности последовательности (семейства s35).
SCOP и Superfamily. Для надсемейств SCOP гомология определяется на основании сходства последовательностей или сравнения структурных и функциональных свойств, выполненного вручную (Andreeva et al. 2008). Такой подход, подразумевающий распределение белков по группам вручную, предоставляет сообществу исследователей возможность использовать классификацию доменных структур, которую постоянно курируют
6.3. Разнообразие функций гомологичных белков
201
эксперты, однако и не лишен свойственного всем ручным процессам недостатка - неизбежного наличия субъективных решений. Домены относят к одному семейству SCOP, если между ними установлена «четкая эволюционная связь». На практике это определение в общем случае означает, что белковые домены относят к одному семейству, если идентичность парных остатков составляет для них более 30%. Однако иногда домены относят к одному семейству и в отсутствие высокой степени идентичности последовательностей - в том случае, если сходство структуры и функций являются однозначным доказательством общего происхождения. Это свойство наделяет систему классификации определенной гибкостью при установлении гомологичных связей, но также повышает степень субъективности процесса. С помощью базы данных Superfamily можно классифицировать белки неизвестной структуры, используя информацию SCOP для описания последовательностей на уровне семейств и надсемейств (Wilson et al. 2007). Как и в случае Gene3D, для оценки соответствия между последовательностями в Superfamily используются профили СММ, основанные на данных SCOP.
SFLD (от «The Structure-Function Linkage Database») - недавно разработанная база данных, специфическая цель которой - исследование связей между структурой и функциями гомологичных ферментов. В настоящее время база содержит относительно небольшое количество надсемейств по сравнению с САТН и SCOP, однако предоставляет для них подробное описание эволюции функций. В SFLD ферменты, принадлежащие к одному надсемейству, должны обладать не только гомологией, но и общей отличительной чертой механизма каталитической реакции с участием консервативных структурных элементов (Pegg et al. 2006). Семейства SFLD состоят из ферментов, выполняющих одну и ту же общую реакцию в данном надсемействе.
6.3.2. Эволюция белковых надсемейств
В конечном счете, основным критерием группы белков, принадлежащих одному надсемейству, является тот факт, что гены, кодирующие эти белки, происходят от одного общего гена-предка. Процесс, в ходе которого ген-предок дает начало двум (или более) копиям самого себя, обычно обозначают термином дупликация.
Согласно определению, событие дупликации дает начало гомологичным генам. Но для дальнейших процессов следует провести различие. Гены, которые происходят от общего гена-предка посредством дупликации генома в отсутствие сопутствующего процесса видообразования, известны как паралоги. Гены, которые происходят от общего гена-предка посредст
202
Глава 6. Разнообразие в элементах упаковки и надсемействах
вом дупликации генома в процессе видообразования, известны как ортологи. Принято считать, что из-за сильного давления отбора в ортологич-ных генах обычно сохраняется функция гена-предка и оба вида-потомка по-прежнему способны осуществлять функцию предка (Tatusov et al. 1997). На основании этого предположения некоторые авторы даже определяют ортологи как гомологи, которые выполняют одну и ту же функцию у различных видов. Существует несколько баз данных, которые позволяют определять ортологичные гены у ряда организмов (Dolinski and Botstein 2007). С другой стороны, наличие в геноме большого количества копий данного гена, т.е., паралогов, вероятно, привело бы к сильному давлению отбора на одну из копий и сохранению ее первоначальной функции, что, в свою очередь, обеспечило бы больше возможностей для дивергенции других копий гена. Процесс, в ходе которого одна копия дуплицированного гена сохраняет функцию гена-предка, а остальные копии эволюционируют и приобретают новые функции, известен под названием неофункционализации. В результате отсутствия давления отбора в ходе эволюционной дивергенции эти дополнительные копии часто превращаются в псевдогены, которые представляют собой реликты генов и не экспрессируются (Harrison and Gerstein 2002). Этот эволюционный процесс называется бес-функционализацией. Субфункционализация - третий эволюционный процесс, обозначающий случаи, когда множество функций гена-предка распределяется между паралогами. Так или иначе, принято считать, у паралогов больше возможностей дивергенции по сравнению с ортологами, и поэтому они более разнообразны в функциональном отношении.
События, последовавшие за дупликацией и появлением ортологов и паралогов, в каком бы порядке они ни следовали в ходе биологической истории, привели к появлению современных надсемейств белков. По-видимому, не для всех надсемейств этот процесс был в равной степени успешным, поскольку, как известно, некоторые надсемейства являются результатом экспрессии непропорционально большого числа генов в полностью секвенированных геномах (Marsden etal. 2006). На сегодняшний день причины неодинакового эволюционного успеха различных семейств не ясны. Выдвигались аргументы, связанные со структурными и функциональными свойствами, а также с эволюционной динамикой (Goldstein 2008). Можно ожидать, что более старые надсемейства, имевшие больше времени для дивергенции и приобретения различных функций, как правило, более многочисленны в настоящее время. Так, надсемейство HUP (код САТН 3.40.50.620) восходит к миру РНК, как принято считать, основываясь на филогенетических данных. Оно отличается чрезвычайно широким спектром на первый взгляд не связанных между собой функций (Aravind
6.3. Разнообразие функций гомологичных белков
203
et al. 2002). В то же время, некоторые современные надсемейства, встречающиеся исключительно у эукариотических видов, часто ограничены весьма специфическим набором функций. Однако возраст, по-видимому, не основной фактор, объясняющий различные размеры надсемейств. В недавнем исследовании проанализирована эволюционная динамика различных семейств генов: одни семейства содержали гены, связанные с основными функциями (Е-семейства), а другие семейства не содержали таких генов (N-семейства). Предполагается, что паралоги в Е-семействах с большей вероятностью эволюционируют и приобретают новые функции, чем те же гены в N-семействах. Это свидетельствует о том, что функции генов-предков семейства являются ключевым фактором, определяющим успех в эволюции (Shakhnovich and Koonin 2006). Как будет показано в следующем разделе, другие аргументы, объясняющие изменчивость успеха белковых семейств в эволюции, можно извлечь из механизмов, которые были предложены для объяснения эволюции функций.
6.3.3. Дивергенция функций в ходе эволюции белков
Традиционный подход при описании белка с неизвестной функцией состоит в поиске гомологии между этим исследуемым белком и другими белками, функции которых хорошо изучены, с тем, чтобы затем перенести функциональные аннотации последних на первый, предполагая, что белки, которые произошли от общего предка, должны обладать определенном функциональным сходством (Whisstock and Lesk 2003). Однако на сегодняшний день однозначно установлено, что этот подход предрасполагает к возникновению ошибок и его неосторожное применение приводит к неуправляемому распространению ошибочных аннотаций в базах данных (Devos and Valencia 2001).
Основной источник ошибок в этом процессе - предположение о том, какие гомологичные белки имеют сходные функции, которое часто является неточным (Devos and Valencia 2000). В настоящее время известно множество примеров близких белков, обладающих совершенно разными функциями. Так, лизоцим белка куриных яиц и а-лактальбумин млекопитающих имеют более 35% идентичности последовательности и весьма схожие структуры. Тем не менее, разумно предположить, что чем больше эволюционное расстояние между двумя гомологичными белками, тем меньше вероятность того, что эти белки будут обладать одной и той же функцией. В ряде исследований предпринимались попытки определить отсечки идентичности последовательностей, применение которых явилось бы гарантией сохранения функции для пары гомологов, однако полученные результаты
204
Глава 6. Разнообразие в элементах упаковки и надсемействах
оказались противоречивы, и проблема все еще находится в стадии обсуждения (Todd etal. 2001; Rost 2002; Tian and Skolnick 2003; Sangar etal. 2007). Одно из возможных объяснений сложности получения универсальных отсечек состоит в том, что, как уже упоминалось выше, образцы последовательностей и характер дивергенции функций для разных надсемейств очень сильно отличаются. Таким образом, многие последние исследования сосредоточены на анализе связей между последовательностью, структурой и функцией в рамках отдельных надсемейств или их подмножеств. Такой подход может оказаться полезным при выявлении закономерностей, которые определяют, каким образом вариации в последовательности и структуре связаны с вариациями функций.
В следующем разделе вариации функций в пределах надсемейств будут описаны более подробно, при этом особое внимание будет уделено механизмам, которые считаются причиной, вызывающий функциональные изменения.
6.3.3.1. Разнообразие функций на уровне надсемейств
Последовательности белков, принадлежащих к одному и тому же семейству, иногда настолько далеко дивергируют в ходе эволюции, что связь между ними невозможно установить с помощью стандартных методов выравнивания последовательностей. Несмотря на то, что пространственные структуры, как правило, считаются гораздо более консервативными, чем последовательности, структуры отдаленных гомологов все же весьма значительно отличаются друг от друга. Такие структурные различия, вероятно, являются следствием вставок/делеций (инделов, от «insertion/deletion») крупных элементов вторичной структуры или даже их сочетаний. Недавнее исследование инделов гомологичных структур показало, что нередко вставки вторичной структуры, которые впоследствии оказываются удачными в ходе эволюции, располагаются в одних и тех же местах белковой укладки, т.е., формируются так называемые встроенные инделы (Jiang and Blouin 2007). Другое исследование вставок в надсемействах САТН показало, что встроенные элементы вторичной структуры обычно не только занимают одно и то же положение в определенных участках белковой укладки, но также формируют элементы, которые располагаются вблизи функционально важных областей, таких как каталитические участки ферментов или области контактов при белок-белковых взаимодействиях (Reeves et al. 2006). Эти данные указывают на связь между структурными и функциональными изменениями.
Вставка новых элементов вторичной структуры вблизи активного центра, скорее всего, приведет к изменению функции, однако более тонкие
6.3. Разнообразие функций гомологичных белков
205
Рис. 6.2. Мультидоменная архитектура (а) периплазматического глутамат-связывающего белка грамотрицательных бактерий и (б) субъединицы NR2 глутаматного рецептора [NMDA] крысы. Отдельные домены показаны в виде прямоугольников. N- и С-концы показаны заглавными буквами «N» и «С» соответственно. Лиганд L-глутамат показан в виде сферы. Клеточная мембрана (б) показана двойной пунктирной линией. Домены, между которыми связывается L-глутамат, окрашены в белый цвет. Они гомологичны как между собой, так и для двух рассматриваемых белков (надсемейство САТН 3.40.190.10). Приведенные два белка выполняют совершенно разные функции что отражено в из совершенно разной мультидоменной архитектуре, а) Бактериальный периплазматический глутамат-связывающий белок. Он содержит всего два домена, которые участвуют в связывании глутамата и свободно перемещают последний через периплазму (Takahashi et al. 2004). б) Рецептор глутамата [NMDA] (субъединица NR2) является частью трансмембранного канала, который играет важную роль в передаче возбуждающих нервных импульсов. Он содержит пять глобулярных доменов. Его связывание с L-глутаматом является частью процесса открывания канала для потока катионов (Furukawa et al. 2005). Несмотря на то, что пара доменов этих двух белков, показанная белым цветом, гомологична и имеет общую способность связывать L-глутамат в одной и той же области структуры, домены, вне всяких сомнений, выполняют очень разные функции
изменения, такие как аминокислотные замены остатков, важных для осуществления катализа, также приведут к возникновению функциональных различий, хотя на уровне надсемейства, обсуждаемом здесь, вероятность определить близкие гомологи выше, чем та же вероятность для отдаленных гомологов. Изменения на уровне домена могут также привести к радикальным изменениям роли белков, так, что, даже если какой-либо аспект молекулярной функции остается неизменным, о белках нельзя сказать, что они выполняют одну и ту же функцию (Hegyi and Gerstein 2001; Todd et al. 2001). Примером такой ситуации служат РВР-подобные домены глутаматных рецепторов эу- и прокариот, которые связываются с одним и тем же лигандом в одном и том же месте топологии, но значительно отличаются по своим функциям, выполняемым на уровне клетки (см. рис. 6.2).
206
Глава 6. Разнообразие в элементах упаковки и надсемействах
Существует множество длительных эволюционных процессов, в ходе которых функция дивергирует в разных гомологах. Дать им всеобъемлющее описание довольно сложно. Тем не менее, Бэштон и Чотиа в своем последнем исследовании описали и привели примеры множество таких процессов, попытавшись понять, как функция гомологичных доменов может меняться в зависимости от того, является ли домен единственным в белке или составляет часть мультидоменного белка (Bashton and Chothia 2007). К числу описанных процессов принадлежат случаи, когда функция домена изменяется при его комбинации с другими доменами, изменяющими субстратную специфичность белка, а также случаи мультифункциональных белков, в которых каждый домен отвечает за определенную функцию.
Вышеупомянутые структурные изменения, часто встречающиеся вблизи функциональных областей, указывают на разнообразие функций, которого можно ожидать от членов отдельного надсемейства. И действительно, результаты некоторых исследований свидетельствуют о том, что отдаленные гомологи в пределах надсемейств часто выполняют очень разные функции (Todd etal. 2001). В большинстве случаев эти исследования направлены на изучение эволюции функций в пределах надсемейств, которые, как правило, демонстрируют исключительное функциональное многообразие. К числу известных примеров принадлежат дегалогеназы галогенокисл от (Burroughs et al. 2006), дегидрогеназы/редукгазы с короткой цепью (Favia etal. 2008), енолазы (Gerlt and Babbitt 2001), домены HUP (Aravind et al. 2002) и флавопротеины с доменами, связывающими два динуклеотида (tDBDF, от ««Two dinucleotide binding domains» flavoproteins») (Ojha et al. 2007). При исследовании этих различных групп белков было выявлено большое количество различных процессов, посредством которых функция дивергирует среди родственных белков. Эти процессы и соответствующие примеры будут рассмотрены отдельно.
Разнообразие механизмов в надсемействах
Совокупность хорошо изученных надсемейств составляет основную массу сведений в базе данных SFLD (Pegg et al. 2006). Несмотря на функциональное разнообразие, согласно критериям включения в SFLD (см. раздел 6.3.1.2), все члены надсемейств обладают общими свойствами механизма действия в различных реакциях, которые они катализируют.
По сути, база SFLD создана специально для описания таких надсемейств ферментов с различиями в механизме действия. Она обеспечивает классификацию эволюционно связанных ферментов, в основе которой лежит сходство их механизмов функционирования. Так, семейство дегалогеназ галогенокислот из базы SFLD составляют ферменты, которые могут
6.3. Разнообразие функций гомологичных белков
207
катализировать реакции с огромным количеством разнообразных субстратов, но всегда действуют через образование промежуточного фермент-субстратного соединения, в котором компоненты связаны ковалентно в области консервативного аспартата (Glasner et al. 2006), и последующего расщепления связей С-С1, Р-С или Р-О.
Надсемейство дегалогеназ галогенокислот содежит 1285 уникальных аминокислотных последовательностей, которые относятся к 20 семействам, каждое из которых катализирует уникальную реакцию (например, фосфатазы гистидинола, номер КФ 3.1.3.15, или фосфатазы трегалозы, номер КФ 3.1.3.12). Некоторые семейства объединены в подгруппы, которые представляют собой удобный промежуточный уровень классификации, определение которого варьирует от одного надсемейства к другому.
В настоящее время в SFLD содержится лишь шесть надсемейств. Однако общность черт каталитической реакции весьма распространена в пределах надсемейств и наблюдается в 22 из 31 надсемейства ферментов, изученных Тоддом и соавт. (2001). Субстратная специфичность, напротив, не сохранялась в 20 из этих надсемейств (см. ниже).
Распространенность общего шага в механизме реакции семейств, весьма разнообразных в отношении механизма, свидетельствует о том, что ферменты этих надсемейств сохранили аспекты каталитического механизма в ходе эволюционной диверсификации. Такие ситуации дают основание предполагать такой сценарий эволюции, при котором ферменты приобретают новые функции за счет дупликации и увеличения численности при частичном сохранении механизмов реакции (а не субстратной специфичности, см. ниже), что в итоге привело к разнообразию механизмов реакций в надсемействах, которое наблюдается в наши дни (Gerlt and Babbitt 2001).
Разнообразие специфичности в надсемействах
Иной сценарий для дивергентной эволюции функций ферментов в пределах надсемейств представляет собой последовательность событий, при которой предковые формы ферментов проходят через процесс дупликации, и появившиеся копии потомков адаптируются к связыванию более специфичных субстратов. При таком сценарии субстратная специфичность - доминирующий фактор эволюции функции в надсемействах. В ходе детального анализа надсемейств ферментов Тодд и соавт. (2001) показали, что в большинстве случаев среди гомологичных ферментов механизмы реакций были более консевативны, чем субстратная специфичность. Из 28 надсемейств, среди которых проводили сравнение процесса связывания с
208
Глава 6. Разнообразие в элементах упаковки и надсемействах
субстратом, десять не обладали субстратной специфичностью ни при каких условиях и еще для десяти наблюдалось весьма широкое разнообразие субстратов, которые обладали лишь незначительным сходством химического строения, таким, например, как пептидная связь (Todd et al. 2001).
Предположение о том, что субстратная специфичность для гомологичных ферментов в пределах надсемейства, вероятнее всего, сохраняется, вытекает из предложенной Хоровицем гипотезы об обратной эволюции метаболических путей (Horowitz 1945). Согласно этой гипотезе, в случае недостатка субстрата для данного фермента преимущество при отборе по сравнению с другими будет иметь тот организм, у которого есть новый фермент, способный производить необходимый субстрат из имеющегося в наличии предшественника. Такой новый фермент закрепляется в эволюции, тем самым давая начало первичному двухступенчатому метаболическом пути. Аналогичный эволюционный процесс может иметь место и для других ступеней существующего пути. Согласно этому сценарию, эволюция метаболического пути идет в обратном направлении по отношению к направлению метаболического потока (Rison and Thornton 2002). Поскольку исходный фермент способен связываться с молекулой субстрата, которая является продуктом в реакции, катализируемой новым ферментом, полагают, что описанное свойство лежит в основе появления в процессе эволюции новых ферментов. Следуя этой идее, все ферменты в пределах одного метаболического пути должны был» гомологами, а фермент, катализирующий последнюю ступень пути, - наиболее древним. Кроме того, движущим фактором эволюции ферментов в этом случае должна был» избирательность по отношению к субстрату. В конечном счете это привело бы к появлению общих черт в субстратной специфичности существующих надсемейств. Существует ряд исследований, в которых были собраны и систематизированы возможные примеры обратной эволюции ферментов, в том числе путь биосинтеза триптофана, в котором некоторые ферменты, катализирующие последовательные шаги, обладают ярко выраженной гомологией (Todd et al. 2001; Gerlt and Babbitt 2001).
Однако результаты нескольких исследований позволяют предположить, что этот гипотетический процесс в действительности играл незначительную роль в эволюции метаболизма, который, на самом деле, скорее мог бы возникнуть из химически обусловленного распределения ферментов между метаболическими путями (Rison and Thornton 2002). Действительно, надсемейства, в которых консервативна селективность к субстрату, кажутся более редкими по сравнению с теми, где консервативен каталитический механизм. Интересно, что надсемейство фосфоенолпируват-связывающих ферментов TIM-бочонков, которое являлось единственным надсемейством с абсолютно
6.3. Разнообразие функций гомологичных белков
209
консервативной специфичностью к субстрату по данным Тодда с сотр. (Todd etal., 2001), оказалось в числе надсемейств с максимально широким спектром лигандов в более позднем исследовании (Bashton et al. 2006), позволяя предположить, что полученное в раннем анализе ложное заключении имеет своей причиной малочисленность использованных данных.
База данных PROCOGNATE (Bashton et al. 2008) является очень полезным средством для анализа разнообразия лигандов, связанных с различными ферментами внутри надсемейства. Эта база данных картирует ферменты по известным для них лигандам, т.е. лигандам, которые ферменты связывают in vivo. Действительно, данные о лигандах, содержащихся в структурах PDB, имеют один недостаток: зачастую в активных центрах ферментов связываются неспецифические лиганды, мимикрируя таким образом под истинные лиганды, связывающиеся in vivo (Dessailly et al. 2008). Эти примеси делают затруднительным автоматическое изучение разнообразия лигандов в белках с известной структурой, поскольку неочевидно, как отличать их от биологических лигандов. PROCOGNATE предназначена для работы с надсемействами (по классификации САТН, SCOP или PFAM), к которым принадлежат ферменты, и, таким образом, полезна для определения разнообразия лигандов в любом интересующем семействе. Например, поиск в структурно неоднородном надсемействе дегалогеназ галогенокислот (код в САТН 3.40.50.1000) возвращает список из 57 известных лигандов из PDB и 17 известных соединений из KEGG, которые связываются с ферментами этого надсемейства. Древнее и разнородное надсемейство HUP-домена (код в САТН 3.40.50.620) ассоциировано в PROCOGNATE с 92 лигандами в PDB и 29 лигандами из KEGG Эти 29 лигандов показаны на рис. 6.3. и иллюстрируют разнообразие молекул, которые могут связываться с эволюционно родственными белками.
Изменения функции, вызванные изменением в окружении
Функциональные изменения между дуплицированными копиями белка также могут появиться не столько из-за изменений в самих белках, сколько из-за изменений условий окружающей среды, в которых эти копии активны. Например, распространение белка в новые области организма может теоретически привести к встрече этого белка с низкомолекулярными соединениями, не характерными для исходной окружающей среды белка-предка, и новый белок может проявить неожиданную активность в связывании этих новых лигандов. Кроме того, молекулярная функция белка может измениться, если другие белки в его окружении претерпевают мутации, которые делают возможными новые взаимодействия или, напротив, какие-то белок-белковые взаимодействия становятся далее невозможными.
210
Глава 6. Разнообразие в элементах упаковки и надсемействах
L-аспартат
L-глутамат
L-аргинин
L-метионин
L-глутамин
Рис. 6.3. Лиганды из базы данных KEGG, распознанные с помощью PROCOGNATE как связывающиеся с HUP-доменами (суперсемейство САТН 3.40.50.620). Для ясности лиганды разбиты на три основных категории: а) аденин-содержащие лиганды и их производные; б) аминокислоты и их производные; в) различные лиганды, которые не относятся ни к одной из двух обозначенных выше категорий. Гораздо большее количество молекул (92), связывающихся с доменами HUP, было обнаружено в базе данных PDB, однако на рисунке они не приведены. Рисунок иллюстрирует идею о том, что эволюционно близкие домены способны связывать широкий круг молекул
Селенмет ионин
L-триптофан
L-N2-(2-Kap6oKCH3THn) аргинин
Дезокси гуанидино-проклаваминовая кислота
Пирофосфат Бета-аланин (Я>пантоат
в) Сульфат
Пантетеин 4'-фосфат
6.3. Разнообразие функций гомологичных белков
211
Известный пример функциональных изменений гомологичных ферментов, т.е., изменений, связанных с изменениями в окружающей среде, описан в литературе для «связывающих два динуклеотида доменов» флавопротеинов. Диверсификация функции внутри семейства в данном случае стала результатом большого количества различных белковых молекул-партнеров, обладающих свойствами акцепторов электронов, белок-белковые взаимодействия с которыми носят консервативный характер (Ojha et al. 2007).
Ферменты и молекулы, не обладающие ферментативной активностью
Одним из источников функционального разнообразия надсемейств, который редко обсуждается в литературе, является утрата/приобретение каталитических свойств гомологами. Действительно, анализ белков неферментной природы - более сложная задача, чем анализ ферментов, для которых существуют системы аннотации и инструменты анализа (например, КФ, KEGG и CSA, см. раздел 6.1) (таблица 6.1). Тем не менее, белки неферментной природы часто обнаруживаются в так называемых фермен-тых семействах. Процессы, в ходе которых происходит утрата каталитических свойств, довольно просты: замена лишь одного аминокислотного остатка, имеющего решающее значение для проявления каталитических свойств, обычно приводит к утрате ферментативной активности (Todd et al. 2002). Надсемейство доменов HUP (код САТН 3.40.50.620) состоит в основном из ферментов, но содержит несколько отдельных примеров белков, для которых каталитическая активность не выявлена. Так, субъединицы электронтранспортных флавопротеинов составляют отдельное функциональное семейство, которое значительно отличается от других членов надсемейства по последовательности, структуре и функциям (Aravind et al. 2002). Пример другого уровня в пределах этого надсемейства - криптохром DASH. Он имеет не ферментную природу и демонстрирует поразительное сходство с эволюционно близкими фотолиазами репарации ДНК при связывании с ДНК и выполнении редокс-зависимой функции, однако имеет также существенные отличия, в основном в активном центре (Brudler et al. 2003). Существуют также примеры надсемейств, в которых преобладают молекулы неферментной природы. Так, домены, схожие с периплазматическим связывающим белком, (код САТН 3.40.190.10) - надсемейство, в котором принадлежность ко многим функциональным семействам определяется на уровне молекул, с которыми связываются члены семейства, или на уровне функций, которые они выполняют в клетке (например, транспортеры или поверхностные рецепторы).
212
Глава 6. Разнообразие в элементах упаковки и надсемействах
Особые примеры функционально разнообразных надсемейств
Из приведенного выше обсуждения надсемейств, разнообразных с точки зрения механизмов реакции и специфичности, следует, что для большинства надсемейств, несмотря на их дивергенцию, характерно сохранение определенной степени функциональной общности членов. Этого можно ожидать, поскольку, согласно определению, надсемейства образованы белками, которые эволюционно связаны друг с другом. Исходя из правил экономии, разумно предположить, что гомологичные белки в ходе эволюции могут сохранять по крайней мере некоторые аспекты функции. Тем не менее, известны примеры надсемейств, для которых такие функциональные общности до сих пор не выявлены. Так, в уже упоминавшемся ранее исследовании многочисленных и разнобразных надсемейств, проведенном Тоддом и соавт., члены одного надсемейства - белки из повторяющихся гексапептидов - не имели общих черт ни в отношении механизма катализа, ни в отношении субстратной специфичности (Todd et al. 2001). Другой пример надсемейства, для членов которого не выявлены общие функциональные свойства, - домены HUP. На рис. 6.4 приведены обобщенная схема, отражающая функциональное разнообразие этого надсемейства, а также типичные структуры функциональных групп. Тем не менее, вполне вероятно, что общие функциональные свойства этих весьма разнообразных надсемейств, не очевидные в настоящее время в силу крайней сложности изучения функций, будут установлены по мере накопления и изучения большего количества данных.
6.3.3.2. Функциональное разнообразие близких гомологов
В предыдущих разделах при описании функционального разнообразия белковых надсемейств внимание акцентировалось на отдаленных гомологах. Однако разнообразие функций также наблюдается среди близких гомологов (например, между лизоцимом белка куриных яиц и а-лакталь-бумином млекопитающих, см. раздел 6.3.3), а иногда даже между совершенно идентичными белками, если рассматривать их в различных контекстах. Обычно такие белки приобретают совершенно новые функции в связи с существованием в новой среде. Хорошо известным примером таких белков являются кристаллины хрусталика глаза утки, которые идентичны по последовательности енолазе и лактатдегидрогеназе печени (Piatigorsky etal. 1994; Whisstock and Lesk 2003). На сегодняшний день описано несколько случаев такого поведения, а сами молекулы получили общее название «белки-совместители» (Jeffery 2003). Более того, все больше данных указывают на то, что ферменты потенциально способны к функциональным изменениям. Суть этой способности заключается в том, что ферменты могут катализировать самые различные реакции в дополнение к основной,
6.4. Заключение
213
а) Фотолиаза репарации ДНК
в) Пирофосфатаза АТФ N-типа
г) Амииояпип-тРНК синтетаза I класса
Рис. 6.4. {Цветную версию рисунка см. на вклейке.) Разнообразные структуры и функции надсемейства доменов HUP (код САТН 3.40.50.620). Для доменов HUP характерен способ укладки, близкий к укладке Россмана. Показано, что домены HUP -очень древние белки (Aravind et al. 2002). Вместе они образуют большое семейство и выполняют множество различных функций. На рисунке схематично представлены репрезентативные структуры основных функциональных групп семейства. Структуры подверглись множественному выравниванию с помощью алгоритма CORA (Orengo 1999). Также множественное выравнивание использовалось для получения общего ядра домена. Во всех структурах остатки, входящие в состав ядра, окрашены красным. В качестве типичных представителей основных функциональных групп были использованы следующие домены САТН: a) 1dnpA01 для фотолиаз репарации ДНК; б) 1ej2A00 для нуклеотидилтрансфераз; в) 1gpmA02 для пирофосфатаз АТФ N-типа; г) 1п31А01 для аминоацил-тРНК синтетаз I класса; д) 1o97D01 для электронтранспортных флавопротеинов
которая, как правило, является весьма специфичной (Khersonsky et al. 2006). Эти исключительные случаи функционального разнообразия белков, которые характеризуются весьма незначительными различиями в последовательности и структуре или полным их отсутствием, приведены здесь для того, чтобы подчеркнуть, что связь между последовательностью, структурой и функциями носит комплексный характер, а значит, разработать простые и надежные правила прогнозирования функций на основе последовательности и структуры довольно сложно.
6.4. Заключение
В этой главе рассматривались взаимосвязи между структурным сходством и функциями белков. Прежде всего, было показано, что белки с об
214
Глава 6. Разнообразие в элементах упаковки и надсемействах
щим способом укладки не обязательно имеют общие функции, однако информация о структуре и способе укладки часто оказывается полезной при описании функций. Обсуждалась концепция способа укладки, при этом особое внимание было уделено последним изменениям в определении этого понятия, согласно которым пространство укладки является непрерывным, а не дискретным.
Белки, имеющие общий способ укладки, не всегда являются гомологичными. Надсемейства, напротив, представляют собой группы эволюционно связанных белков. Однако даже в пределах надсемейств белки могут выполнять различные функции. Разнообразные процессы, объясняющие эволюцию надсемейств и функций, выполняемых белками в пределах надсемейств, описанные в литературе, рассмотрены в настоящей главе с комментариями. Показано, что даже в тех случаях, когда эволюционно связанные белки не имеют общих функций, общие функциональные элементы обычно сохраняются. Так, надсемейства, белки которых отличаются по механизму действия, содержат ферменты, имеющие общие черты в механизме ферментативных реакций, которые они катализируют.
Взаимосвязь между функциями белка, структурой и гомологией является комплексной, и предсказание одного из этих свойств на основе любых других все еще невозможно без ошибок. Однако, выявление сходства способов укладки и структурной гомологии весьма полезно при прогнозировании функций. Увеличение количества данных о структуре, последовательностях и функциях белков за счет успехов различных -омик будет способствовать более глубокому пониманию связей между этими свойствами белков.
Литература
Adams МА, Suits MD, Zheng J, et al. (2007) Piecing together the structure-function puzzle: experiences in structure-based functional annotation of hypothetical proteins. Proteomics 7:2920-2932
Andreeva A, Murzin AG (2006) Evolution of protein fold in the presence of functional constraints.
Curr Opin Struct Biol 16:399-408
Andreeva A, Howorth D, Chandonia JM, et al. (2008) Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res 36:D419-D425
Aravind L, Anantharaman V, Koonin EV (2002) Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: implications for protein evolution in the RNA. Proteins 48:1-14
Bashton M, Chothia C (2007) The generation of new protein functions by the combination of domains. Structure 15:85-99
Bashton M, Nobeli 1, Thornton JM (2006) Cognate ligand domain mapping for enzymes. J Mol Biol 364:836-852
Bashton M, Nobeli I, Thornton JM (2008) PROCOGNATE: a cognate ligand domain mapping for enzymes. Nucleic Acids Res 36.D618-D622
Brudler R, Hitomi K, Daiyasu H, et al. (2003) Identification of a new cryptochrome class. Structure, function, and evolution. Mol Cell 11:59-67
Литература
215
Burroughs AM, Allen KN, Dunaway-Mariano D, et al. (2006) Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol 361:1003-1034
Colovos C, Cascio D, Yeates TO (1998) The 1.8 A crystal structure of the ycaC gene product from Escherichia coli reveals an octameric hydrolase of unknown specificity. Structure 6:1329-1337
Dessailly BH, Lensink MF, Orengo CA, et al. (2008) LigASite-a database of biologically relevant binding sites in proteins with known apo-stinctures. Nucleic Acids Res 36:D667-D673
Devos D, Valencia A (2000) Practical limits of function prediction. Proteins 41:98-107
Devos D, Valencia A (2001) Intrinsic errors in genome annotation. Trends Genet 17:429-431
Dolinski K, Botstein D (2007) Orthology and functional conservation in eukaryotes. Annu Rev Genet 41:465-507
Favia AD, Nobeli I, Glaser F, et al. (2008) Molecular docking for substrate identification: the shortchain dehydrogenases/reductases. J Mol Biol 375:855-874
Furukawa H, Singh SK, Mancusso R, et al. (2005) Subunit arrangement and function in NMDA receptors. Nature 438:185-192
Gerlt JA, Babbitt PC (2001) Divergent evolution of enzymatic function: mechanistically diverse uperfamilies and functionally distinct suprafamilies. Annu Rev Biochem 70:209-246
Glasner ME, Gerlt JA, Babbitt PC (2006) Evolution of enzyme superfamilies. Curr Opin Chem Biol 10:492—497
Goldstein RA (2008) The structure of protein evolution and the evolution of protein structure. Curr Opin Struct Biol 18:170-177
Greene LH, Lewis ТЕ, Addou S, et al. (2007) The CATH domain structure database: new protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Res 35:D291-D297
Grishin NV (2001) Fold change in evolution of protein structures. J Struct Biol 134:167-185
Harrison A, Pearl F, Mott R, et al. (2002) Quantifying the similarities within fold space. J Mol Biol 323:909-926
Harrison PM, Gerstein M (2002) Studying genomes through the aeons: protein families, pseudogenes and proteome evolution. J Mol Biol 318:1155-1174
Hegyi H, Gerstein M (2001) Annotation transfer for genomics: measuring functional divergence in multi-domain proteins. Genome Res 11:1632-1640
Holliday GL, Almonacid DE, Bartlett GJ, et al. (2007) MACiE (Mechanism, Annotation and Classification in Enzymes): novel tools for searching catalytic mechanisms. Nucleic Acids Res 35:D515-D520
Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233:123-138
Holm L, Sander C (1996a) Mapping the protein universe. Science 273:595-603
Holm L, Sander C (1996b) The FSSP database: fold classification based on structure-structure alignment of proteins. Nucleic Acids Res 24:206-209
Horowitz NH (1945) On the evolution of biochemical syntheses. Proc Natl Acad Sci USA 31:153-157
Jeffery CJ (2003) Moonlighting proteins: old proteins learning new tricks. Trends Genet 19:415-417
Jiang H, Blouin C (2007) Insertions and the emergence of novel protein structure: a structurebased phylogenetic study of insertions. BMC Bioinformatics 8:444
Kanehisa M, Araki M, Goto S, et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480-D484
Khersonsky O, Roodveldt C, Tawfik DS (2006) Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol 10:498-508
Kolodny R, Koehl P, Levitt M (2005) Comprehensive evaluation of protein structure alignment methods: scoring by geometric measures. J Mol Biol 346:1173-1188
Kolodny R, Petrey D, Honig В (2006) Protein structure comparison: implications for the nature of ‘fold space’, and structure and function prediction. Curr Opin Struct Biol 16:393-398
Kraulis PJ (1991) Molscript: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 24:946-950
216
Глава 6. Разнообразие в элементах упаковки и надсемействах
Krissinel Е, Henrick К (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256-2268
Lee D, Grant A, Marsden RL, et al. (2005) Identification and distribution of protein families in 120 completed genomes using Gene3D. Proteins 59:603-615
Lee D, Redfern O, Orengo C (2007) Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol 8:995-1005
Lopez G, Valencia A, Tress M (2007) FireDB-a database of functionally important residues from proteins of known structure. Nucleic Acids Res 35:D219-D223
Marsden RL, Ranea JA, Sillero A, et al. (2006) Exploiting protein structure data to explore the evolution of protein function and biological complexity. Philos Trans R Soc Lond В Biol Sci 361:425-440
Martin AC, Orengo CA, Hutchinson EQ et al. (1998) Protein folds and functions. Structure 6:875-884
Merritt EA, Bacon DJ (1997) Raster3d version 2: photorealistic molecular graphics. Method Enzymol 277:505-524
Moult J, Melamud E (2000) From fold to function. Curr Opin Struct Biol 10:384-389
Murzin AG, Brenner SE, Hubbard T, et al. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536-540
Nagano N (2005) EzCatDB: the Enzyme Catalytic-mechanism Database. Nucleic Acids Res 33: D407-D412
Nagano N, Orengo CA, Thornton JM (2002) One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol 321:741-765
Nomenclature Committee of the International Union of Biochemistry and Molecular Biology and Webb EC (1992) Enzyme Nomenclature: Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes. Academic, San Diego, CA
O’Boyle NM, Holliday GL, Almonacid DE, et al. (2007) Using reaction mechanism to measure enzyme similarity. J Mol Biol 368:1484-1499
Ojha S, Meng EC, Babbitt PC (2007) Evolution of function in the “Two Dinucleotide Binding Domains” flavoproteins. PLoS Comput Biol 3(7):el21
Orengo CA (1999) CORA-topological fingerprints for protein structural families. Protein Sci 8:699-715
Orengo CA, Taylor WR (1996) SSAP: sequential structure alignment program for protein structure comparison. Method Enzymol 266:617-635
Orengo CA, Jones DT, Thornton JM (1994) Protein superfamilies and domain superfolds. Nature 372:631-634
Orengo CA, Michie AD, Jones S, et al. (1997) CATH-a hierarchic classification of protein domain structures. Structure 5:1093-1108
Pegg SC, Brown SD, Qjha S, et al. (2006) Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry 45:2545-2555
Piatigorsky J, Kantorow M, Gopal-Srivastava R, et al. (1994) Recruitment of enzymes and stress proteins as lens crystallins. EXS 71:241-250
Porter CT, Bartlett GJ, Thornton JM (2004) The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 32:D129-D133
Redfern ОС, Harrison A, Dallman T, et al. (2007) CATHEDRAL: a fast and effective algorithm to predict folds and domain boundaries from multidomain protein structures. PLoS Comput Biol 3(1 l):e232
Reeves GA, Dallman TJ, Redfern ОС, et al. (2006) Structural diversity of domain superfamilies in the CATH database. J Mol Biol 360:725-741
Reid AJ, Yeats C, Orengo CA (2007) Methods of remote homology detection can be combined to increase coverage by 10% in the midnight zone. Bioinformatics 23:2353-2360
Rison SC, Thornton JM (2002) Pathway evolution, structurally speaking. Curr Opin Struct Biol 12:374-382
Литература
217
Bost В (2002) Enzyme function less conserved than anticipated. J Mol Biol 318:595-608
Rucpp A, Zollner A, Maier D, et al. (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32:5539-5545
Russell RB, Saqi MA, Sayle RA, et al. (1997) Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation. J Mol Biol 269:423-439
Russell RB, Sasieni PD, Sternberg MJ (1998) Supersites within superfolds. Binding site similarity in the absence of homology. J Mol Biol 282:903-918
Sangar V, Blankenberg DJ, Altman N, et al. (2007) Quantitative sequence-function relationships in proteins based on gene ontology. BMC Bioinformatics 8:294
Shakhnovich BE, Koonin EV (2006) Origins and impact of constraints in evolution of gene families. Genome Res 16:1529-1536
Shindyalov IN, Bourne PE (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 11:739-747
Takahashi H, Inagaki E, Kuroishi C, et al. (2004) Structure of the Thermus thermophilus putative periplasmic glutamate/glutamine-binding protein. Acta Crystallogr D Biol Crystallogr. 60:1846-1854
Tatusov RL, Koonin EV, Lipman DJ (1997) A genomic perspective on protein families. Science 278:631-637
The Gene Ontology Consortium (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25-29
Tian W, Skolnick J (2003) How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol 333:863-882
Todd AE, Orengo CA, Thornton JM (2001) Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol 307:1113-1143
Todd AE, Orengo CA, Thornton JM (2002) Sequence and structural differences between enzyme and nonenzyme homologs. Structure 10:1435-1451
Whisstock JC, Lesk AM (2003) Prediction of protein function from protein sequence and structure. Q Rev Biophys 36:307-340
Wilson D, Madera M, Vogel C, etal. (2007) The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res 35:D308-D313
Ye Y, Godzik A (2004) FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res 32:W582-W585
Yeats C, Lees J, Reid A, et al. (2008) Gene3D: comprehensive structural and functional annotation of genomes. Nucleic Acids Res 36:D414—D418
Глава 7
Предсказание функции белка по свойствам его поверхности
Николас Дж. Бургойн, Ричард М. Джексон
Ситуация, когда для некого белка известна его структура, но неизвестна функция, является частым следствием современных проектов по структурной геномике. Осмысление свойств поверхности белка - лишь один из нескольких подходов, которые могут быть использованы при описании его функций. Анализ этой поверхности может быть полезен для понимания взаимодействий белка как с нативными биологическими партнерами, так и с лекарственными препаратами. В этой главе мы введем понятие поверхности белка и вкратце расскажем о широко применяемых свойствах этой поверхности, а также использовании этих свойств при введении производных понятий и специальных функций для оценки взаимодействий белков с нативными лигандами, лекарственными препаратами и другими белками
7.1. Способы представления поверхности
Прежде чем рассматривать вопрос о том, что может нам рассказать поверхность белка о его функциях и взаимодействиях с другими молекулами, стоит определить различные представления поверхности и их смысл. С точки зрения структурной биологии можно дать несколько определений поверхности белка, каждое из которых имеет различные особенности, определяющие их применение. Наиболее используемые способы представления поверхности рассмотрены ниже.
7.1.1. Поверхность ван-дер-Ваальса
Несмотря на то, что поверхность Ван-дер-Ваальса редко используется для описания поверхности сама по себе, важно упомянуть о ней, посколь-
Nicholas J. Burgoyne and Richard M. Jackson
Institute of Molecular and Cellular Biology, Faculty of Biological Science, University of Leeds, Leeds, LS2 9JT, UK
*e-mai 1: r. m.jackson@leeds. ac. uk
7.1. Способы представления поверхности
219
ку она является основой для последующих определений. Атомы в молекуле представляются перекрывающимися сферами, где радиус каждой сферы равняется ван-дер-ваальсовому радиусу соответствующего атома. Ван-дер-ваальсова поверхность молекулы, таким образом, представляет собой внешнюю поверхность атомов этой молекулы. Такое представление поверхности приводит к появлению кажущихся пустот внутри белковой структуры и поэтому редко используется при рассмотрении поверхности белка (Рис. 7.1), однако оно служит основой для распространенного графического представления пространственного строения молекул, называемого моделью СРК (по имени впервые предложивших её Кори, Полинга и Колтуна, англ. Corey-Pauling-Koltun) или объемной моделью.
7.1.2. Молекулярная поверхность (поверхность без растворителя)
Более практичное представление поверхности - это так называемая молекулярная поверхность, известная также как поверхность Коннолли (Connolly, 1984). Эта поверхность образована точками контакта молекулы растворителя (обычно воды), которая моделируется сферой радиуса 1.4 А, катящейся по поверхности Ван-дер-Ваальса. Молекулярная поверхность аналогична ван-дер-ваальсовой поверхности в том, что области, недоступные растворителю, скрыты вогнутыми поверхностями (рис 7.1). Как следствие, молекулярная поверхность представляет собой непрерывную функциональную поверхность молекулы, т.е. поверхность, доступную для взаимодействия.
Поверхность, доступная растворителю
Рис. 7.1 Варианты представления поверхности: ван-дер-ваальсова поверхность; молекулярная поверхность; поверхность, доступная растворителю (ПДР)
220
Глава 7. Предсказание функции белка по свойствам его поверхности
7.1.3. Поверхность, доступная растворителю
В структурной биологии в качестве поверхности молекул наиболее широко используется поверхность, доступная растворителю (ПДР, англ. Solvent accessible surface, SAS) (Lee & Richards, 1971). Как и молекулярная поверхность, она представляет собой поверхность контакта белка с окружающим растворителем, но, в отличие от прежде рассмотренного варианта, она образована центром молекулы растворителя, катящейся по ван-дер-ваальсовой поверхности. Таким образом, способ построения ПДР какого-либо атома обеспечивает пропорциональность между площадью этой поверхности и числом молекул растворителя, которые одновременно могут контактировать с этим атомом. Этот признак является ключевым при установлении типа поверхности по ее свойствам. По определению, вогнутая область молекулярной поверхности может быть отнесена не одному, а к нескольких атомам, в то время как участок, доступный растворителю, связан только с одним атомом белка (рис. 7.1). Это различие является существенным при определении свойств области поверхности, например, при является ли она полярной или неполярной. Таким образом, для определения свойств отдельных атомов, образующих поверхность белка, использование поверхности, доступной растворителю, более предпочтительно по сравнению с использованием молекулярной поверхности, и поэтому именно ПДР широко используют при описании биологических и физических свойств поверхности белка. Эти свойства будут рассмотрены в последующих разделах.
7.2. Свойства поверхности
После введения определений поверхности белка важно установить, какие ее свойства являются существенными для исследований. Это могут быть химические, биологические или физические свойства. Здесь вкратце представлены наиболее широко используемые свойства поверхности.
7.2.1. Гидрофобность
Полярные и неполярные атомы на поверхности белка будут по-разному взаимодействовать с окружающим их растворителем. Стремление полярных молекул воды к образованию водородных связей в основном удовлетворяется полярными атомами на поверхности белка. Неполярные атомы не могут образовывать водородных связей, и это различие является причиной гидрофобного эффекта в водных растворах (Chothia and Janin
7.2. Свойства поверхности
221
1975). Взаимодействие молекул воды с неполярной поверхностью белка невыгодно из-за того, что те молекулы, которые располагаются рядом с такой поверхностью, не могут образовать такое же число водородных связей, что и молекулы воды в объеме раствора. Как результат, приповерхностные молекулы воды будут формировать полуустойчивую сеть для оптимизации числа водородных связей между собой. Такое упорядочивание является той движущей силой, под действием которой вода стремится уменьшить контакт с неполярной поверхностью, в результате чего изменения в площади неполярной поверхности молекул растворенного вещества имеют прямое отношение к свободной энергии процессов в растворе. Важно заметить, что использование молекулярной поверхности из-за особенностей её построения вызывает искажения при моделировании гидрофобного эффекта по сравнению с использованием поверхности, доступной растворителю. Свободная энергия, связанная с этим процессом, является самым главным стабилизирующим фактором при определении структуры глобулярных белков, которые обычно имеют гидрофобное ядро и полярную поверхность. Гидрофобный эффект также важен как движущая сила при взаимодействии молекул. Гидрофобность поверхности белка является, таким образом, ключевым свойством для предсказания его функций.
Самой простой мерой гидрофобности является сумма экспонированных полярных и неполярных площадей поверхности, но существуют и другие, более сложные, ее определения. Они приравнивают гидрофобность отдельных типов атомов к энергии сольватации, полученной из экспериментальных источников или баз данных характерных белковых структур. Общепринятый подход состоит в рассмотрении энергии сольватации как функции двух переменных: уменьшения площади ПДР и наблюдаемой энергии переноса соответствующих атомов. Энергия переноса данной молекулы представляет собой изменение свободной энергии при перемещении ее между двумя различными средами. При рассмотрении белковых взаимодействий в качестве такого перемещения наиболее подходит перенос аминокислот из воды в октанол, имитирующий внутренний объем белка (Fauchere and Pliska 1983). Также существуют значения энергии переноса и между другими различными средами, которые выступают в качестве конечных состояний в других физических процессах: пар-вода (Wolfenden etal. 1981), циклогексан-вода (Radzicka etal. 1988). В качестве альтернативы выступает атомный потенциал сольватации (Atomic Solvation Potential, ASP), значения которого подобраны при сопоставлении разниц в энергиях переноса нативных структур белков и моделей с заведомо неправильной укладкой (Wang et al. 1995). Полученные значения часто используют в расчетах сворачивания белка и методе молекулярного докинга.
222
Глава 7. Предсказание функции белка по свойствам его поверхности
7.2.2. Электростатические свойства
Особенности распределения электростатического потенциала на молекулярной поверхности определяют специфичность многих происходящих на ней взаимодействий. Эти особенности формируются под влиянием всех атомов молекулы, а не только тех, что лежат на ее поверхности. Взаимодействие разноименных зарядов представляет собой одну из движущих сил специфичных взаимодействий. Например, белки, которые связываются с ДНК и РНК, обычно имеют несколько положительно заряженных аминокислот, которые могут связываться с отрицательно заряженными фосфатными группами основной цепи нуклеиновых кислот. Фактор транскрипции MetJ из E.coli также использует дипольный момент положительно заряженной а-спирали для улучшения своего связывания с ДНК (Garvie and Phillips 2000). Фермент супероксиддисмутаза имеет значительный градиент заряда в приповерхностном объеме, что увеличивает скорость образования продуктивного комплекса. В отличие от большинства ферментов, скоростьлимитирующей стадией перевода супероксидного радикала в безопасную форму является его нековалентное связывание с ферментом, а не следующая за этим химическая реакция (Getzoff et al. 1983).
Самая простая мера электростатического поверхностного потенциала определяется законом Кулона, который определяет потенциал в точке поверхности как сумму зарядов окружающих ее атомов белка, нормированную на расстояние до них и диэлектрическую проницаемость среды. Более сложное моделирование на атомарном уровне без учета молекул воды в явном виде может быть осуществлено с помощью уравнения Пуассона-Больцмана (Fogolari et al. 2002). Оно учитывает воду неявно, используя аппроксимацию эффектов растворителя на взаимодействия биомолекул в растворах с различной ионной силой. Существует несколько компьютерных программ, таких, например, как DelPhi, которые могут быть использованы для численного решения этого уравнения для биомолекул (Rocchia et al. 2002). Результирующий электростатический потенциал может быть отображен на поверхности белка с помощью программ молекулярной графики.
7.2.3. Консервативность поверхности
Псевдослучайный процесс дивергентной эволюции означает, что большинство остатков в белках, имеющих консервативную функцию, будут консервативными, так как они существенны либо для структуры, либо для функционирования белка. Профиль консервативности остатков создается с помощью анализа множественного выравнивания последовательностей
7.2. Свойства поверхности
223
семейства белков, используя, например, метод Scorecons (Valdar 2002), и затем наносится на поверхность белка. Карта консервативности (не несущая физической или химической информации) позволяет идентифицировать консервативные области на поверхности белка. Такая карта может быть полезна при определении сайта связывания и активного центра для рассматриваемого семейства белков.
Обычно только малая часть поверхности белка вовлечена в его функционирование. Только несколько остатков являются ключевыми для катализа в активном центре. Даже для тех функций белка, которые задействуют большой участок поверхности, как, например, белок-белковые взаимодействия, только несколько остатков вносят значительный вклад в образование стабильного комплекса (см. Раздел 7.5.2). Эти “функциональные остатки” достаточно часто консервативны у эволюционно родственных белков, имеющих схожие функции. Таким образом, сравнение консервативных остатков на поверхности белка позволяет сделать вывод о его функциях. Интерпретация консервативности зачастую сложна, поскольку, если только белки не являются очевидными ортологами, может случиться такая мутация белка, которая полностью изменит его функцию, что приведет к смеси паралогов и ортологов в анализируемом семействе белков. Пока существуют только способы исключения паралогов из множественного выравнивания (O’Brienetal. 2005), поэтому надо быть осторожным при сравнении последовательностей с 30% идентичностью, так как в таких случаях только половина сайтов связывания предсказывается правильно (Devos and Valencia 2000). Однако высокое сходство последовательностей не обязательно подразумевает схожесть функций (Todd et al. 2002). Пример белков семейства TIM-бочонков (англ, triosephosphateisomerase (TIM) barrel) показывает, что одна и та же структура может сочетать в себе огромное разнообразие функций, о чем более подробно рассказано в Главе 6.
Другой проблемой является определение, какой из консервативных остатков наиболее функционально значим. Метод эволюционного следа (Lichtarge etal. 1996) использует филогенетическое дерево для аппроксимации эволюционного процесса, в результате получая набор последовательностей, обладающих некоторым сходством с исследуемой последовательностью. Иерархические ветви этого дерева представляют собой группы последовательностей с консервативными функциями, где наиболее близкие группы, находящиеся на концах ветвей, имеют наиболее близкие функции. Остатки, которые консервативны в одном и том же положении в последовательностях одной ветви, и в меньшей степени представленные в других ветвях, называются остатками “эволюционного следа”. Эти остатки являются определяющим признаком ветви и можно полагать, что они
224
Глава 7. Предсказание функции белка по свойствам его поверхности
существенны для функционирования белков этой ветви. Сервер ConSurf (Pupko et al. 2002) использует модификацию базового метода эволюционных следов при окрашивании структуры белка, выделяя локализацию консервативных остатков на поверхности белка.
7.3. Предсказание функций по свойствам поверхности
Одной из главных целей проектов по структурной геномике является пополнение пространства всевозможных белковых укладок, в частности для того, чтобы определять структуры белков с неизвестными функциями, либо имеющих важное медицинское значение. В случае белков с неизвестными функциями выбор мишени часто определяется отсутствием заметной гомологии с белками, чья функция известна, поскольку такой выбор может привести к обнаружению новых типов укладки. Основные методы определения функции белка, основывающиеся на его струкуре, включают в себя сравнение локальных мотивов в последовательности или структуре. Этот вопрос подробно обсуждается в Главах 8 и 11. Есть примеры близких подходов, основанных лишь на консервативности поверхности белка, а не всей его структуры. Эти примеры рассматриваются ниже в том же порядке, что и свойства поверхностей, описанные в п.7.2.
7.3.1. Гидрофобная поверхность
Обширная площадь гидрофобной поверхности нестабильна по своей природе и не характерна для белок-белковых интерфейсов в необлигатных комплексах, компоненты которых должны быть стабильны в воде сами по себе. Ранние методы для предсказания интерфейса белковых взаимодействий включали в себя определение гидрофобных областей на их поверхности (Lijnzaad etal. 1996). Однако эти методы обычно применялись для предсказания облигатных интерфейсов, таких, например, как наблюдаются в олигомерных комплексах. В то же время эта проблема в некотором смысле искусственна, так как облигатные интерфейсы формируются в процессе сворачивания белка и предсказание их по своей природе отличается от предсказания необлигатных интерфейсов. Кроме того, необлигатные интерфейсы обладают большей гидрофобностью по сравнению поверхностью белка в целом, и эта особенность используется при предсказании структуры белок-белковых комплексов из отдельных мономеров (Berchanski et al. 2004).
7.3. Предсказание функций по свойствам поверхности
225
7.3.2. Электростатическая поверхность
Для активных центров ферментов часто характерно наличие области с высоким электростатическим потенциалом, что можно рассматривать как компромисс между биологической функцией белка и его стабильностью (Beadle and Shoichet 2002). Таким образом, картирование электростатического потенциала на поверхности может иногда быть полезным для предсказания функциональных сайтов, которые могут оказаться активными центрами (Elcock 2001) или сайтами связывания ДНК и РНК (Tsuchiya et al. 2005). Например, при использовании программы PatchFinderPlus на поверхности белка отображается самый большой участок с положительным электростатическим потенциалом (Рис. 7.2), который часто соответствует сайту связывания белков, взаимодействующих с нуклеиновыми кислотами (Stawiski et al. 2003).
Далее приведены примеры, где консервативность функции связана скорее с консервативностью поверхности, чем с консервативностью укладки белка или с консервативными остатками. Наиболее известным примером является каталитическая триада сериновых протеаз, где даже три основные аминокислоты активного сайта (как правило, His, Asp и Ser) могут варьировать. Единственное, что остается постоянным - это поверхность активного сайта и электростатическая природа его каталитической активности. Как следствие, важным оказывается сравнение молекулярных поверхностей неизвестных белков с базами данных поверхностей, определяемых аналогичным способом.
Например, eF-Site (Kinoshita and Nakamura 2003) выполняет поиск по базе данных поверхностей белков, описанных с точки зрения их электроста-
Рис. 7.2. Предсказание функциональных сайтов с помощью программы PatchFinderPlus. Показана кристаллографическая структура эндонуклеазы рестрикции Bgll (атомы белка показаны сферами ван-дер-Ваалься), связанной со специфичной для нее последовательностью ДНК (Newman et al. 1998). Наибольший участок с положительным электростатическим потенциалом показан темно-серым
Л*
226
Глава 7. Предсказание функции белка по свойствам его поверхности
тической и гидрофобной природы. Запрос, касающийся изучаемого белка, может быть адресован к наиболее подходящей базе данных (базе данных антител, активных центров, фосфат-связывающих сайтов или базы данных, полученной из определений, данных в PROSITE), и в ответ будет получен набор белков, сходных с изучаемым по свойствам поверхности.
7.3.3. Консервативность поверхности
Консервативность поверхности, оцениваемая с помощью алгоритма ConSurf, оказалась полезной при предсказании функций некоторых белков. Одним из примеров ее использования является анализ белков лектина С-типа (англ. C-type lectin (CTLs) proteins), находящихся на поверхности клетки и связывающих гликопротеины, вовлеченные в иммунитет и эндоцитоз (Ebner et al. 2003). Эти белки имеют структуру, очень похожую на структуру доменов, подобных лектину С-типа (англ. C-type lectin like domains, CTLDs), но последние не могут связывать функциональные группы углеводов. Анализ всех субъединиц домена с помощью программы ConSurf оказался полезным при выявлении остатков, составляющих консервативную поверхность и определяющих активные центры белков CTL. Такие остатки совершенно отсутствует у доменов, подобных CTL, и это различие было использовано при классификации CTL/CTLD доменов с неизвестной функцией.
7.3.4. Сочетание свойств поверхности для предсказания функций
Помимо электростатики и гидрофобности влиять на функцию белка могут и другие свойства его поверхности. Например, в программе HotPatch для предсказания функции белка на базе рассмотрения его поверхности использован ряд других ее свойств (Pettit etal. 2007). В данном случае функция белка может быть установлена путем сравнения его с базами данных белков со схожими функциями. Одиннадцать характеристик (включая четыре различные электростатические характеристики, три меры вогнутости и шероховатости и четыре меры гидрофобности и гидрофильности) используются для описания пятнадцати различных иерархически классифицированных свойств поверхности белка. Они определяют функции, включающие общую функцию (каждый функциональный участок поверхности имеет свойство), которая разбивается на функцию связывания (включая белок-белковые, олигомерные, ДНК-РНК, углеводные интерфейсы, а также интерфейсы небольших молекул), ферментативную функцию (включая протеазы, гидролазы, трансферазы, оксидоредуктазы и киназы) и функ
7.4. Взаимодействие лиганда с белком
227
ции связывания малых ионов (анионов и катионов). При этом определяется соотношение между свойствами каждой из функциональных групп, и, основываясь на их сходстве, могут быть установлены функциональные участки исследуемого белка. Участки, которые не соответствуют ни одной из категорий, могут быть определены как имеющие неизвестную функцию.
7.4. Взаимодействие лиганда с белком
Возможно, самыми важными функциями белка, имеющими непосредственное отношение к его поверхности, являются взаимодействия с другими молекулами. Эти взаимодействия являются фундаментальными при рассмотрении любого аспекта жизни, от метаболизма до передачи сигнала и так далее. В этом разделе рассмотрены взаимодействия белков с небольшими молекулами лиганда. Другие виды взаимодействий будут рассмотрены позже в главе 9. Небольшие молекулы лиганда важны не только как биологические молекулы, но и как лекарства, используемые для контроля нарушенного функционирования белков при заболеваниях.
7.4.1. Свойства взаимодействий лиганда с белком
Интерфейсы взаимодействия лиганда с белком составляют две обширные группы. Это область активного центра фермента, которая связывает небольшие молекулы для дальнейшей химического превращения, и карманы связывания, которые присоединяют молекулу без последующего катализа. В области активного центра аминокислотные остатки находятся под давлением эволюционного отбора для сохранения его каталитической активности, а также способности специфически связывать требуемые молекулы. Семейства белков, которые транспортируют или реагируют на небольшие молекулы лиганда без дальнейшего катализа, не подвержены явно действию эволюционного отбора, сохраняющего каталитическую активность белка, и свойства сайтов связывания часто отражают разнообразие небольших молекул, которые могут связываться с белками различных семейств. Сложности и затраты, связанные с разработкой достаточно селективных лекарств для данного класса взаимодействий подтверждают это разнообразие.
7.4.2. Предсказание расположения активного центра
Единственное, что, похоже, действительно является общим для всех лиганд-связывающих сайтов белка, - это наличие карманов, содержащих в
228
Глава 7. Предсказание функции белка по свойствам его поверхности
себе эти сайты. Когда одна молекула меньше другой, самым простым путем обеспечения обширного контакта является окружение лиганда карманом. Кроме того, в случае ферментов это выгодно еще и потому, что субстрат изолируется от раствора и, таким образом, снижается высокая энергия переупорядочивания, связанная с реакциями в растворе (Yadav etal. 1991). Существует два основных подхода для определения свойств поверхности белка, геометрический и энергетический; эти два подхода описаны в последующих разделах.
7.4.2.1. Геометрическое определение центров связывания лиганда
Основная идея, лежащая в основе геометрического подхода, состоит в том, что небольшие молекулы предпочитают связываться с самым большим углублением на поверхности белка (Laskowski etal. 1996). Есть много различных методов для определения этих углублений (Laurie and Jackson 2006), некоторые из них описаны в этом разделе. Простейшие методы сначала окружают структуру белка пространственной решеткой. В программе Pocket (Levitt and Banaszak 1992) узлы решетки располагаются вдоль осей х, у, z, а углубления определяются как пустое пространство, окруженное атомами белка. Программа LIGSITE (Hendlich etal. 1997) делает этот подход менее чувствительным к ориентации белка, проводя поиск углублений не только вдоль осей кубической решетки, но и вдоль диагоналей. Повторно этот же прием был реализован в веб-сервисе Pocket-Finder (Laurie and Jackson 2005). В программе PASS (Brady and Stouten 2000) узлы решетки располагаются в каждой точке белка, где они могут примыкать к трем атомам белка, но не пересекаться с ними. Эти узлы покрывают фактически всю поверхность белка, и отбираются в зависимости от числа атомов белка в пределах заданного расстояния от них - узлы, оказавшиеся в углублениях, будут иметь больше атомов белка поблизости, чем те узлы, которые оказались вне углублений. Такие циклы размещения и отбора узлов решетки в конечном счете приведут к заполнению ими всего объема углублений.
В программе SurfNet (Laskowski 1995) сферы размещаются между парами атомов в белке, причем диаметр сфер уменьшается до тех пор, пока не будет устранено перекрывание с остальными атомами белка (это не всегда возможно, и в этом случае сфера удаляется). Оставшиеся сферы оказываются собранными в полостях белка. Эти полости можно увидеть в реальном времени для любого структуру из PDB, используя опцию “Clefts” в программе PDBsum (Laskowski et al. 2005). В программе CASTp (Binkowski et al. 2003) внешняя поверхность атомов определяется с помощью так называемого представления Делоне. Это геометрический подход, который присваивает
7 А. Взаимодействие лиганда с белком
229
Рис. 7.3. Схематичное изображение теории дискретного потока. Один треугольник Делоне действует как сток для потока. В программе CASTp считается, что это истинный карман
каждому атому в молекуле многогранник максимально возможного объема. Грани возникают при соприкосновении атомов, и если в каком-то направлении такую грань построить не удается, то значит в этом направлении у атома нет соседей и он является атомом поверхности. Соединение центров таких атомов дает поверхность, ограничивающую многогранник с треугольными гранями. Часть этих граней может быть сокращена с использованием теории дискретного потока (см. Рис. 7.3), а результирующий центральный многогранник определяет полость в белке.
Общей процедурой, принятой во всех этих методах, является определение границы между карманом и остальной поверхностью белка. Кроме того, должна быть определена и дополнительная граница - между карманом и пространством, окружающим белок. В некоторых геометрических определениях эти границы зависят от структуры белка и объем предсказанных сайтов связывания имеет тенденцию расти с увеличением размера белка. При определении сайта связывания с помощью энергетических характеристик (см.ниже) его объем оказывается примерно равным размеру лиганда независимо от размера всего белка, что согласуется с предположением, что размер кармана для связывания лиганда соответствует размеру последнего и не зависит от размеров белка (Laurie and Jackson 2005).
7.4.2.2. Энергетическое определение активного центра
Помимо геометрического подхода возможно использовать энергетический подход, при котором карманы определяются как области, наиболее предпочтительные для взаимодействия с другими молекулами (Laurie and Jackson 2005). В программе Q-SiteFinder белки, во-первых, окружаются пространственной решеткой, во все непересекающиеся с белком узлы которой помещается пробная метильная (-СНЗ) группа для оценки взаимодействий.
230
Глава 7. Предсказание функции белка по свойствам его поверхности
Рис.7.4. (Цветную версию рисунка см. на вклейке.} Предсказание сайтов связывания лигандов с помощью программы Q-SiteFinder. Приведен пример активного сайта молекулярной поверхности ацетилхолинстеразы (код PDB 1EVE), где сайт связывания небольшой лекарственной молекулы - ингибитора этого фермента -арисепта, хорошо определяется наилучшим предсказанием (прозрачная серая поверхность)
Оценочная функция узлов рассчитывается как потенциальная энергия ван-дер-Ваальсова взаимодействия этой группы в каждом узле. Те узлы решетки, которые получили достаточно высокую оценку, оставляются для дальнейшей кластеризации, а полученные кластеры затем ранжируются, причем предполагается, что карманы с наиболее подходящей энергией (обычно самой большой) и являются сайтами связывания лигандов. Так было установлено, что 90% рассмотренных реальных сайтов связывания оказывались в числе трех наиболее вероятных карманов, полученных предсказанием (см. рис.7.4).
7.4.2.3. Теоретические кривые микроскопического титрования
Электростатические свойства поверхности белка влияют на поведение ионизируемых групп в её окрестности. Во многих случаях ионизируемые боковые цепи, тесно вовлеченные в процессы химического катализа, имеют окружение, которое значительно влияет на их состояние ионизации. Например, они зачастую оказываются способными поддерживать конкретное частично протонированное состояние в аномально большом диапазоне изменения pH. В результате получающиеся значения константы кислотности рКа и кривые титрования изменяются, соответственно, по величине и форме от средних значений для тех же остатков. Поскольку электростатические свойства и состояния ионизации белка могут быть вычислены, то можно предсказывать остатки каталитического центра на основе их аномальных теоретических кривых микроскопического титрования (например, Antosiewicz etal. 1994; Elcock, 2001). Применение этого метода, называемого THEMATICS (англ, theoretical microscopic titration curves, теоретические кривые микроскопического титрования), позволило успешно идентифицировать остатки каталитического центра в семи структурах различных ферментов с небольшой долей ложно-положительных предсказаний, но не выявило каталитической активности в некаталитических белках, используемых в качестве контроля. С тех пор этот метод был улучшен за счет разработки статистических показателей для классификации теоретических
ПА. Взаимодействие лиганда с белком
231
кривых титрования (Ко et al. 2005) и внедрения метода опорных векторов для повышения чувствительности предсказаний (Tong et al. 2008). Совсем недавно было показано, что производительность метода слегка снижается, если анализируются апо-структуры вместо голо-структур (которые имеют существенные различия в конформации) (Murga et al. 2008). Наряду с тем, что сильной стороной этого метода служит его независимость от наличия или отсутствия гомологичных последовательностей, следует подчеркнуть, что он подходит только для предсказания каталитических сайтов, а не активных центров в целом.
7.4.3. Предсказание чувствительности к лекарствам
Нынешнее разнообразие белков, которые имеют отношение к тому или иному заболеванию и могут успешно выступать в качестве мишени для небольших лекарственных молекул, ограничивается только небольшим числом белковых семейств, и благодаря сходству последовательностей это число может быть расширено лишь до 5% от всех белков проте-ома человека (Hopkins and Groom 2002). Поиск других потенциальных лекарственных препаратов экспериментальными методами, такими, как высокопроизводительный скрининг лигандов, требует большого количества времени и материальных затрат при том, что в 60% проектов поиска нового препарата мишень в итоге оказывается невосприимчивой к лекарствам (Brown and Superti-Furga 2003). Предсказание, может ли активный центр связывать молекулы, подобные лекарственным, представляет собой одну из новейших проблем в структурной биологии белка (Cheng et al. 2007).
В одной из недавних работ описывается подход, расширивший вышеописанные геометрические алгоритмы нахождения карманов, добавив к ним оценку десольватации поверхности белка и показатели кривизны этой поверхности (Cheng etal. 2007). Эти нововведения были объединены с оценкой таких типичных характеристик взаимодействий белок-лиганд, как, например, корреляция между молярной массой лиганда и площадью заглубленной поверхности белка, в единый эмпирический показатель, позволяющий судить о возможности связывания лекарственных молекул. Аналогичный показатель может быть сформулирован на основе анализа связывающих карманов, выполненного с помощью ядерного магнитного резонанса (Hajduk et al. 2005). Оба этих показателя сходятся в том, что связывание лекарственных препаратов и их аналогов происходит преимущественно в больших, легко де сольватируемых карманах сложной формы.
232 Глава 7. Предсказание функции белка по свойствам его поверхности
7.4.4. Аннотация сайтов связывания лигандов
При разработке новых лекарств первым важным шагом является предсказание активных центров белка и их способности связывать небольшие молекулы гипотетических лекарственных препаратов. Вторым таким шагом является скрининг библиотек с десятками тысяч соединений и отбор тех из них, которые с большой вероятностью окажутся селективными и сильнодействующими и приведут к созданию эффективного лекарственного препарата. Этот отбор представляет собой другую задачу, где также может помочь понимание свойств поверхности белка. Как правило, эти задачи решаются сначала с помощью виртуального скрининга лигандов, включающиего методы молекулярного докинга или поиска фармакофора (Oledzki et al. 2006).
Однако аннотации сайтов связывания лигандов с энергетической точки зрения могут помочь визуализовать наиболее энергетически значимые точки поверхности и, таким образом, направить к ним применение вычислительных методов. Начало таким энергетическим подходам положил Питер Гудфорд (Peter Goodford) с его широко используемой программой GRID (Goodford 1985). В этом методе, как и в других методах, последовавших за
Aigl58
Рис. 7.5. Пространственная решетка, построенная программой Q-fit (Jackson, 2002) для активного центра связывающего белка периплазмы (PDB код 2GBP). Показаны наиболее предпочтительные точки взаимодействия на сетке для гидроксилов кислорода (выделены белым). Связанный лиганд - глюкоза -не был задействован в расчетах и показан для сравнения. Видно, что четыре из пяти гидроксильных группы глюкозы оказались в областях наиболее благоприятного положения пробных атомов
Asp236
7.5. Белок-белковый интерфейс
233
ним, взаимодействие белка с пробным атомом в узле пространственной решетки на поверхности белка представляется в виде взаимодействия ван-дер-Ваальса, водородных связей и электростатического взаимодействия. Особенно подходящие точки взаимодействия могут быть визуализованы (Рис. 7.5) и затем использованы при разработке новых лигандов. Другой похожий подход заключается в применении потенциала, использующего уже известные данные. Например, программа SuperStar (Nissink et al. 2000) основана на статистическом анализе большого числа взаимодействий лиганда с белком, рассмотренных в структурных базах данных. Таким образом, оба подхода могут оказаться полезными в аннотировании потенциальных активных центров, определяя области, где возможные взаимодействия между химическими группами являются более предпочтительными.
7.5. Белок-белковый интерфейс
В период геномной эпохи был достигнут значительный прогресс в понимании количества генов в геноме и их экспрессии. Но продукты этих генов гораздо менее понятны, особенно если рассматривать их в целом. Существуют экспериментальные методы определения сетей белок-белковых взаимодействий, и целые разделы биоинформатики посвящены их анализу. Однако тема этого раздела ограничивается анализом и предсказанием отдельных интерфейсов белков, используя свойства их поверхностей. В частности, мы сосредоточимся на тех взаимодействиях, которые появляются в результате ассоциаций белков, стабильных в воде самих по себе. Такие взаимодействия, формирующие переходные комплексы, известны как необлигатные белок-белковые взаимодействия в противоположность облигатным взаимодействиям, характерным для белков, которые могут существовать только в олигомерном состоянии.
7.5.1. Свойства белок-белкового интерфейса
Исходя из наблюдений большого количества взаимодействий, мы можем выявить статистические закономерности, которые обычно определяют белок-белковый интерфейс. Стоит отметить, что для всех свойств, упомянутых здесь, будут приведены примеры комплексов, в которых свойства имеют значения параметров либо гораздо выше, либо гораздо ниже, чем средние. Интерфейс между двумя глобулярными белками чаще всего представляет собой плоский круглый участок поверхности белка, размер которого обычно прямо пропорционален размеру белков (Jones and Thornton 1996).
234
Глава 7. Предсказание функции белка по свойствам его поверхности
Небольшие мономеры, например, супероксиддисмутаза, могут иметь площадь интерфейса всего 700 А2, в то время как мономеры гораздо более крупной тетрамерной каталазы имеют площадь интерфейса 10 500 A2 (Janin etal. 1988). Среднее значение площади интерфейса одного мономера составляет 800 А2, что составляет около 10% поверхности типичного глобулярного белка (Janin and Chothia 1990; Jones and Thornton 1995).
Обычно 55% поверхности интерфейса является неполярной, 25% полярной, а оставшиеся 20% поверхности относятся к заряженным атомам (Janin and Chothia 1990). В итоге средний интерфейс оказывается менее гидрофобным, чем внутренний объем белка, но более гидрофобным, чем оставшаяся поверхность белка. Кроме того, интерфейс, как правило, менее заряжен, чем остальная часть поверхности белка. Для образования стабильных комплексов взаимодействующие белки должны иметь соответствие между заглубленными полярными и заряженными группами интерфейса. Действительно, считается, что именно комплементарность между такими группами является причиной специфичности взаимодействий (Chothia and Janin 1975). Также высока комплементарность между группами, склонными к образованию водородных связей, причем на любом интерфейсе 80% таких групп одного из белков образуют связи с аналогичными группами другого белка (Xu etal. 1997). В среднем, белковый интерфейс будет иметь одну группу, образующую водородную связь, на 80 А2 заглубленной области поверхности (Lo Conte etal. 1999). Заряженные группы на интерфейсе одного белка, как правило, не будут взаимодействовать с группой противоположного заряда другого белка, но вместо этого они могут быть окружены его комплементарно полярными группами (Lo Conte et al. 1999).
7.5.2. Активные точки белковых интерфейсов
Показано, что определенные остатки интерфейса при мутации на аланин (процедура, называемая аланин-сканирующим мутагенезом) гораздо сильнее влияют на стабильность комплекса, чем другие остатки интерфейса белка (Clackson and Wells 1995). Наиболее важные аминокислоты, называемые “активными” остатками, стремятся расположиться в глубине карманов на поверхности белка (Bogan and Thom 1998). Остальная поверхность карманов выстлана аминокислотами средней важности. Большинство активных остатков, определенных таким образом, хранится в базе данных ASEdb (Thom and Bogan 2001). Анализ этих остатков предполагает, что чаще активный остаток является большим и ароматическим (как триптофан или тирозин) или положительно заряженным аргинином (но не лизином). Менее
7.5. Белок-белковый интерфейс
235
Рис. 7.6. Поверхность ДНКазы I (обозначена белым, PDB код 1ATN) в комплексе с актином (не показан), со всеми предсказанными с помощью программы Q-SiteFinder карманами (серые/черные). Карманы, окрашенные в черный, соответствуют тем, в которых находятся атомы актина
вероятно, что активные остатки будут небольшими амфифильными (серин, треонин), либо гидрофобными (валин и лейцин) (Bogan and Thom 1998).
Анализ карманов на интерфейсах позволяет предположить, что наиболее существенное различие между такими карманами и карманами в остальной части белка заключается в том, что первые заметно легче десоль-ватируются (Burgoyne and Jackson 2006). Авторы использовали программу обнаружения карманов Q-SiteFinder (Раздел 7.4.2.3, Рис. 7.6) для определения всех карманов на поверхности. Затем они были ранжированы в соответствии с различными свойствами поверхности.
Ранжирование по десольватации с наибольшим успехом позволяет явно выделить карманы на интерфейсах среди остальных карманов белка. По легкости десольватации ароматические и алифатические поверхности превосходят полярные и, что более важно, заряженные поверхности, которые являются наименее легко десольватируемыми. Это позволяет предположить, что активные остатки, вероятно, необходимы для облегчения десольватации при связывании или для удаления воды из карманов. Может показаться странным включение аргинина в число активных остатков, поскольку он должен бы быть трудно дельсоватируем из-за своего заряда, однако если для молекулярного распознавания на интерфейсе необходим положительно заряженный остаток, то его гуанидиновая группа намного проще десольватируема по сравнению с аминогруппой лизина. Также было предположено, что гидрофобное окружение может уменьшать эффективную диэлектрическую константу в области локализации важных водородных связей, усиливая тем самым взаимодействие (Bogan and Thom 1998). Стоит также отметить, что активные остатки могут быть в числе наиболее консервативных остатков в семействе белков (Ma et al. 2003), но эта консервативность, как правило, сохраняется только при множествен
236
Глава 7. Предсказание функции белка по свойствам его поверхности
ном выравнивании последовательностей, когда взаимодействующие белки в выравнивании являются интерлогами (интерлогами называются взаимодействующие белки, чьи гомологи из организмов других видов также могут взаимодействовать).
7.5.3. Предсказание расположения интерфейса
Как уже упоминалось ранее, необлигатные интерфейсы белков не имеют однозначной характеристики, поэтому использование одного свойства для поиска интерфейса белка чаще всего оказывается неудачным. Комбинирование многочисленных свойств, ни одно из которых независимо от других не указывает на активный центр, может достаточно точно предсказать интерфейс белка. Некоторые методы строят свои предсказания на выборе наиболее похожей на интерфейс области поверхности среди круглых областей, покрывающих всю поверхность белка. Для каждой такой области рассчитывают ряд характеристик, которые сравнивают с типичными характеристиками, наблюдаемыми в известных интерфейсах.
В простейшем алгоритме, реализованном в виде программы Sharp2 (Murakami and Jones 2006), используется одна формула, объединяющая шесть свойств (Jones and Thornton 1997). Этими свойствами являются: (1) гидрофобность, измеренная с помощью экспериментально полученных величин, (2) сольватация, измеренная с использованием аналогичных значений, (3) мера вероятности нахождения каждого остатка на интерфейсе, (4) плоскостность участка, (5) неровность участка и (6) площадь ПДР участка. Применение этого алгоритма оказалось успешным для примерно 65% тестируемых комплексов. Качество предсказания может быть улучшено благодаря использованию различных методов машинного обучения (процедур, которые могут выявить взаимосвязи между свойствами). В программе
Рис. 7.7. Предсказание интерфейса с использованием программы PPI-Pred. Показаны предсказанные активные центры на поверхности ферредоксинредуктазы (PDB кед 1EWY, цепь А). Метка А указывает на активный центр, находящийся наверху списка ранжированных участков, который достаточно точно соответствует настоящему активному центру ферредоксина, В и С -участки, стоящие в списке на втором и третьем местах, соответственно, и покрывающие другие области поверхности белка
7.5. Белок-белковый интерфейс
237
Таблица 7.1. Интернет-ресурсы и сервисные программы, имеющие отношение к функции предсказания на основе анализа поверхностей белков.
Метод URL
Предсказание функции на основе анализа поверхности белка
ef-Site http://ef-site.hgc.jp
PatchFinderPlus http://pfp.technion.ac.il/
Scorcons http://www.ebi.ac.uk/thomton-srv/databases/cgi-bin/valdar/scorecons_server.pl
ConSurf http://consurf.tau.ac.il/
hotpatch http://hotpatch.mbi.ucla.edu
Предсказание сайтов связывания лигандов
PASS http://www.ccl.net/cca/software/UNIX/pass/overview.shtml
CASTp http://sts-fw.bioengr.uic.edu/castp/
SurfNet http://www.biochem.ucl.ac.uk/~roman/surfhet/surfhet.html
PDBsum http ://www.ebi. ac.uk/pdbsum/
Pocket-Finder http://www.bioinformatics.leeds.ac.uk/pocketfinder
LIGSITEcsc http ://scoppi .biotec. tu-dresden. de/pocket/
PocketPicker http://gecco.org.chemie.uni-ffankfurt.de/pocketpicker/index.html
Q-SiteFinder http://www.bioinformatics.leeds.ac.uk/qsitefmder
THEMATICS http://pfweb.chem.neu.edu/thematics/submit.html
Предсказание интерфейсов белок-белковых взаимодействий
Sharp2 http://www.bioinformatics.sussex.ac.uk/SHARP2/sharp2.html
PPI-PRED http://www.bioinformatics.leeds.ac.uk/ppi_pred/
InterProSurf http://curie.utmb.edu/
Cons-PPISP http ://pipe. scs. fsu. edu/ppisp. html
ProMate http://bioportal.weizmann.ac.il/promate/
PPI-Pred (Bradford and Westhead 2005) реализован очень похожий метод, применяющий машинное обучение для тех же свойств, которые описаны в программе Sharp2 (Рис.7.7). В других методах используются иные параметры остатков - так, например, успешное применение нашли гидрофобность, атомная энергия сольватации, доступность поверхности остатка и его консервативность (Bordner and Abagyan 2005).
В другом методе, реализованном в программе ProMate (Neuvirth et al. 2004), схожий успех достигнут при использовании гидрофобности, распределения атомов, предпочтений с точки зрения свойств химических групп и соседних остатков, консервативности остатков, типа вторичной
238
Глава 7. Предсказание функции белка по свойствам его поверхности
структуры, удаленности остатков в последовательности, длины петель и локализации кристаллографической воды. Некоторые другие аналогичные методы также представлены в Таблице 7.1. Кроме того, было сделано сравнение производительности этих методов (Zhou and Qin 2007).
7.6. Заключение
В этой главе в деталях описаны современные подходы, использующие свойства поверхности в ряде важных приложений, включающих предсказание функции белка, предсказание локализации активного центра и чувствительности к лекарствам, аннотацию сайта связывания лиганда для конструирования лекарств, основывающиеся на структуре белка, и предсказание интерфейса белок-белковых взаимодействий. Кроме того, были представлены различные методы и веб-сервисы, и для заинтересовавшегося читателя приведены ссылки на опубликованные статьи для уточнения деталей. Мы попытались дать возможно широкий обзор методов в этой важной и новой области и их практического применения, однако этот обзор нельзя назвать всеобъемлющим.
Литература
Antosiewicz J, McCammon J A, Gilson МК (1994) Prediction of pH-dependent properties of proteins. J Mol Biol 238:415-436.
Beadle BM, Shoichet BK (2002) Structural bases of stability-function tradeoffs in enzymes. J Mol Biol 321:285-296.
Berchanski A, Shapira B, Eisenstein M (2004) Hydrophobic complementarity in protein-protein docking. Proteins 56:130-142.
Binkowski TA, Naghibzadeh S, Liang J (2003) CASTp: computed atlas of surface topography of proteins. Nucleic Acids Res 31:3352—3355.
Bogan AA, Thom KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280:1-9.
Bordner AJ, Abagyan R (2005) Statistical analysis and prediction of protein-protein interfaces. Proteins 60:353-366.
Bradford JR, Westhead DR (2005) Improved prediction of protein-protein binding sites using a support vector machines approach. Bioinformatics 21:1487-1494.
Brady GP Jr, Stouten PF (2000) Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des 14:383-401.
Brown D, Superti-Furga G (2003) Rediscovering the sweet spot in drug discovery. Drug Discov Today 8:1067-1077.
Burgoyne NJ, Jackson RM (2006) Predicting protein interaction sites: binding hot-spots in proteinprotein and protein-ligand interfaces. Bioinformatics 22:1335-1342.
Cheng AC, Coleman RG, Smyth KT, et al. (2007) Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol 25:71-75.
Chothia C., Janin J (1975) Principles of protein-protein recognition. Nature 256: 705-708.
Clackson T, Wells J A (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267:383-386.
Литература
239
Connolly ML (1984) Analytical molecular surface calculation. J Appl Cryst 16:548-558.
Devos D, Valencia A (2000) Practical limits of function prediction. Proteins 41:98-107.
Ebner S, Sharon N, Ben-Tai N (2003) Evolutionary analysis reveals collective properties and specificity in the C-type lectin and lectin-like domain superfamily. Proteins 53:44-55.
Elcock AH (2001) Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol 312:885-896.
Fauchere JL, Pliska V (1983) Hydrophobic paramaters-pi of amino-acid side-chains from the partitioning of N-acetyl-amino-acid amides. Eur J Med Chem 18:369-375.
Fogolari F, Brigo A, Molinari H (2002) The Poisson-Boltzmann equation for biomolecular electrostatics: a tool for structural biology. J Mol Recognit 15:377-392.
Garvie CW, Phillips SE (2000) Direct and indirect readout in mutant Met repressor-operator complexes. Structure 8:905-914.
Getzoff ED, Tainer J A, Weiner PK, etal. (1983) Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature 306:287-290.
Goodford PJ (1985) A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 28:849-857.
Hajduk PJ, Huth JR, Fesik SW (2005) Druggability indices for protein targets derived from NMR-based screening data. J Med Chem 48:2518-2525.
Hendlich M, Rippmann F, Bamickel G (1997) LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model 15:359-363.
Hopkins AL, Groom CR (2002) The druggable genome. Nat Rev Drug Discov 1:727-730.
Jackson RM (2002) Q-fit: a probabilistic method for docking molecular fragments by sampling low energy conformational space. J Comput Aided Mol Des 16:43-57.
Jackson RM, Sternberg MJE (1993) Protein surface area defined. Nature 366:638.
Janin J, Chothia C (1990) The structure of protein-protein recognition sites. J Biol Chem 265:16027-16030.
Janin J, Miller S, Chothia C (1988) Surface, subunit interfaces and interior of oligomeric proteins. J Mol Biol 204:155-164.
Jones S, Thornton JM (1995) Protein-protein interactions: a review of protein dimer structures. Prog Biophys Mol Biol 63:31-65.
Jones S, Thornton JM (1996) Principles of protein-protein interactions. Proc Natl Acad Sci USA93:13-20.
Jones S, Thornton JM (1997) Prediction of protein-protein interaction sites using patch analysis. J Mol Biol 272:133-143.
Kinoshita K, Nakamura H (2003) Identification of protein biochemical functions by similarity search using the molecular surface database eF-site. Protein Sci 12:1589-1595.
Ko J, Murga LF, Andre P, et al. (2005) Statistical criteria for the identification of protein active sites using Theoretical Microscopic Titration Curves. Proteins 59:183-95.
Laskowski RA (1995) SURFNET: a program for visualizing molecular surfaces, cavities, and in-termolecular interactions. J Mol Graph 13:323-330.
Laskowski RA, Luscombe NM, Swindells MB, et al. (1996) Protein clefts in molecular recognition and function. Protein Sci 5:2438-2452.
Laskowski RA, Chistyakov W, Thornton JM (2005) PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res 33:D266-268.
Laurie AT, Jackson RM (2005) Q-SiteFinder: an energy-based method for the prediction of proteinligand binding sites. Bioinformatics 21:1908-1916.
Laurie AT, Jackson RM (2006) Methods for the prediction of protein-ligand binding sites for structure-based drug design and virtual ligand screening. Curr Protein Pept Sci 7:395-406.
Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55:379-400.
Levitt DG, Banaszak LJ (1992) POCKET: a computer graphics method for identifying and displaying protein cavities and their surrounding amino acids. J Mol Graph 10:229-234.
Lichtarge O, Bourne HR, Cohen FE (1996) Evolutionarily conserved Galphabetagamma binding surfaces support a model of the G protein-receptor complex. Proc Natl Acad Sci USA 93:7507-7511.
240
Глава 7. Предсказание функции белка по свойствам его поверхности
Lijnzaad Р, Berendsen HJ, Argos Р (1996) Hydrophobic patches on the surfaces of protein structures. Proteins 25:389-397.
Lo Conte L, Chothia C, Janin J (1999) The atomic structure of protein-protein recognition sites. J Mol Biol 285:2177-2198.
Ma B, Elkayam T, Wolfson H, et al. (2003) Protein-protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc Natl Acad Sci USA 100:5772-5777.
Murakami Y, Jones S (2006) SHARP2: protein-protein interaction predictions using patch analysis. Bioinformatics 22:1794-1795.
Murga LF, Ondrechen MJ, Ringe D (2008) Prediction of interaction sites from apo 3D structures when the holo conformation is different. Proteins 72:980-992.
Neuvirth H, Raz R, Schreiber G (2004) ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J Mol Biol 338:181-199.
Newman M, Lunnen K, Wilson G, etal. (1998) Crystal structure of restriction endonuclease Bgll bound to its interrupted DNA recognition sequence. EMBO J 17:5466-5476.
Nissink JWM, Verdonk ML, Klebe G (2000) Simple knowledge-based descriptors to predict protein-ligand interactions, methodology and validation. J Comput Aided Mol Des 14:787-803.
O’Brien KP, Remm M, Sonnhammer EL (2005) Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res 33:D476-480.
Oledzki PR, Laurie AT, Jackson RM (2006) Protein-ligand docking and structure-based drug design. Edited by Westhead DR, Dunn MJ In, Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics, 1-17.
Ondrechen MJ, Clifton JG, Ringe D (2001) THEMATICS: a simple computational predictor of enzyme function from structure. Proc Natl Acad Sci USA 98:12473-12478.
Pettit FK, Bare E, Tsai A, et al. (2007) HotPatch: a statistical approach to finding biologically relevant features on protein surfaces. J Mol Biol 369:863-879.
Pupko T, Bell RE, Mayrose I, et al. (2002) Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18(Suppl l):S71-77.
Radzicka A, Pedersen L, Wolfenden R (1988) Influences of solvent water on protein folding: free energies of solvation of cis and trans peptides are nearly identical. Biochemistry 27:4538-4541.
Rocchia W, Sridharan S, Nicholls A, et al. (2002) Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field eneigies: applications to the molecular systems and geometric objects. J Comput Chem 23:128-137.
Stawiski EW, Gregoret LM, Mandel-Gutfreund Y (2003) Annotating nucleic acid-binding function based on protein structure. J Mol Biol 326:1065-1079.
Thom KS, Bogan AA (2001) ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics 17:284-285.
Todd AE, Orengo CA, Thornton JM (2002) Sequence and structural differences between enzyme and nonenzyme homologs. Structure 10:1435-1451.
Tong W, Williams RJ, Wei Y, et al. (2008) Enhanced performance in prediction of protein active sites with THEMATICS and support vector machines. Protein Sci 17:333-341.
Tsuchiya Y, Kinoshita K, Nakamura H (2005) PreDs: a server for predicting dsDNA-binding site on protein molecular surfaces. Bioinformatics 21:1721-1723.
Valdar WS (2002) Scoring residue conservation. Proteins 48:227-241.
Wang Y, Zhang H, Li W, et al. (1995) Discriminating compact nonnative structures from the native structure of globular proteins. Proc Natl Acad Sci USA 92:709-713.
Wolfenden R, Andersson L, Cullis PM, et al. (1981) Affinities of amino acid side chains for solvent water. Biochemistry 20:849-855.
Xu D, Tsai CJ, Nussinov R (1997) Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng 10:999-1012.
Yadav A, Jackson RM, Holbrook JJ, et al. (1991) Role of solvent reorganization energies in the catalytic activity of enzymes. J Am Chem Soc. 113:4800-4805.
Zhou H-X, Qin S (2007) Interaction-site prediction for protein complexes: a critical assessment. Bioinformatics 23:2203-2209.
Глава 8
Пространственные мотивы
Элейн К. Менг, Бенджамин Дж. Полакко, Патрисиа К. Баббит
Структурные мотивы - это паттерны локальной структуры белка, связанные с его функционированием, и обычно представляющие собой аминокислотные остатки сайта связывания или каталитического центра. Структуры белков с неизвестной функцией могут быть аннотированы после их сравнения с известными структурными мотивами. Для выявления структурных мотивов и поиска их в структурах разработано большое число методов. Эти методы различаются по типу и количеству входных данных, по описанию мотивов и их соответствия, по тому, учитывается ли в результатах статистическая значимость, и по тому, как проводится сопоставление мотивов и функций. Меньший прогресс по сравнению с разработкой алгоритмов был достигнут в создании общедоступных баз данных структурных мотивов, которые одновременно были бы и функционально специфичны, и охватывали широкий диапазон функций. Препятствием стали трудности в создании подробных структурно-функциональных классификаций; масштабные автоматизированные исследования опирались вместо них на уже существующие структурные или функциональные классификации. Дополнением к методам определения структурных мотивов являются методы, ориентированные на описание молекулярной поверхности, сравнение глобальной структуры (типа укладки), предсказание взаимодействий с другими макромолекулами, а также на идентификацию физиологических субстратов путем локирования небольших молекул из соответствующих баз данных.
Elaine С. Meng, Benjamin J. Polacco, and Patricia C. Babbitt University of California San Francisco (UCSF) Department of Pharmaceutical Chemistry, 600 16th Street, San Francisco, CA 94158-2517
Patricia C. Babbitt
UCSF Department of Biopharmaceutical Sciences, 1700 4th Street, San Francisco, CA 94158-2330
e-mail: babbitt@cgl.ucsf.edu
242
Глава 8. Пространственные мотивы
Сокращения
3D - пространственный, структурный,
CSA - атлас каталитических центров (англ. CSA: Catalytic Site Atlas),
DRESPAT - обнаружение повторяющихся участков боковых цепей (англ. DRESPAT:
Detection of REcurring Sidechain PATtems),
EC - классификация ферментов (англ. EC: Enzyme Commission),
FFF- нечеткая функциональная форма (англ. FFF: Fuzzy Functional Form),
GASPS - генетический алгоритм поиска паттернов в структурах (англ. GASPS: Genetic Algorithm Search for Patterns in Structures),
GO - генная онтология (англ. GO: Gene Ontology),
PAR-3D - выявление остатков активного центра белка при помощи пространственных структурных мотивов (англ. PAR-3D: Protein Active site Residues using
3-Dimensional structural motifs),
PDB - база данных PDB (англ. PDB: Protein Data Bank),
PINTS - паттерны в негомологичных третичных структурах (англ. PINTS: Patterns in Non-homologous Tertiary Structures),
S-BLEST - инструмент поиска локального окружения на основе структры
(англ. S-BLEST: Structure-Based Local Environment Search Tool),
SCOP - структурная классификация белков (англ. SCOP: Structural Classification of Proteins),
SOIPPA - независимый порядок следования при выравнивании двух профилей
(англ. SOIPPA: Sequence Order-Independent Profile-Profile Alignment),
SPASM - пространственное расположение боковых и главных цепей (англ. SPASM: SPatial Arrangements of Sidechains and Mainchains),
TESS - поиск шаблона и суперпозиция (англ. TESS: TEmplate Search and Superposition), БД - база данных,
CKO - среднеквадратичное отклонение,
КФ - классификация ферментов,
МД - молекулярно-динамический
8.1. Предыстория и значение
Применение геномного подхода к биологии стало причиной появления не только обширных данных о последовательностях и структурах, но и перспективы получения полного «списка деталей» для многих организмов. Однако от такого списка мало пользы, если нет некоторого понимания того, для чего каждая деталь предназначена. Даже имея на руках целые последовательности генома, были идентифицированы не все гены, а значительному числу идентифицированных генов не была аннотирована какая-либо функция. Количество последовательностей значительно превосходит количество имеющихся структур, поэтому присваивание функции (функциональное аннотирование) в значительной степени выполня
8.1. Предыстория и значение
243
лось с использованием масштабного поиска в пространстве последовательностей, и переносом на рассматриваемый белок функциональной информации о любых достаточно сходных последовательностях (аннотирование по аналогии). Многие структурные мотивы были выявлены в определенных наборах белков и связаны с каким-то аспектом функционирования белка или его структуры. Однако надежность и функциональная специфичность аннотирования по аналогии уменьшается по мере того, как последовательности становятся менее схожими (Devos and Valencia 2001; Rost 2002). Говоря о функциональной специфичности, мы имеем в виду узость нашего суждения; например, термин «лейциновая аминопептидаза» является более специфическим, чем «пептидаза».
Рассмотрение структур белков может выявить важные сходства или возможные эволюционные связи, которые не видны при рассмотрении одних лишь их последовательностей. Белки могут разойтись в процессе эволюции настолько далеко, что их последовательности не могут быть достоверно выровнены, но сходство общей структуры, или фолда, все еще сохранится (Chothia and Lesk 1986; Rost 1997). Использование сходства фол-дов для аннотирования по аналогии (см. Главу 6) обладает той же ограниченностью, что и использование сходства последовательностей: с одной стороны, надежность аннотирования по аналогии уменьшается с ростом удаленности между родственными белками, а с другой стороны, белки с очень схожими фолдами могут выполнять различные функции (Babbitt and Gerlt 1997; Todd et al. 2001). Таким образом, для точного описания и предсказания функции белка нужно рассматривать детали его структуры, примером которых могут служить структурные мотивы, представляющие собой паттерны локальной структуры. В процессе эволюции идентичные белки могут дивергировать путем накопления случайных нейтральных изменений, которые не изменяют его функции (нейтральный дрейф), и сохраняют структурные компоненты, которые важны для выполнения этой функции. В идеале, эти функционально необходимые структурные компоненты и будут описываться структурными мотивами, которые также будут служить как чувствительные и специфичные признаки функции. Наличие общего структурного мотива может также отражать конвергентную эволюцию между различными фолдами, фиксируя аналогичное расположение боковых цепей, связанное со схожей функцией. Хорошо известным примером является каталитическая триада сериновой протеазы Asp-His-Ser, используемая механически сходным образом структурно разнородными протеазами (см. ниже).
Проекты по структурной геномике направлены на определение структуры всех белков, что является признанием важности этой информации для
244
Глава 8. Пространственные мотивы
аннотирования и других приложений - таких, например, как разработка лекарств. Огромный объем этой задачи может быть несколько уменьшен за счет группировки схожих последовательностей и выбора представительной мишени в каждой группе, что позволит построить сравнительные модели для оставшихся последовательностей. На протяжении нескольких последних лет общее число структур в Protein Data Bank (PDB) (Berman et al. 2000) и в проектах по структурной геномике растет все более быстрыми темпами, причем функция для многих из этих структур неизвестна. Такая тенденция позволяет предположить, что методы, связанные с структурными мотивами, станут более распространенными и полезными, поскольку появляется все больше расшифрованных и смоделированных структур.
8.1.1. Что такое функция?
Понятие функции может быть описано на многих уровнях и с разных точек зрения. Для обучения и тестирования различных методов функционального аннотирования необходима единая система классификации функций. Система генной онтологии (ГО, англ. Gene Ontology, или GO) (Ashbumer et al. 2000) представляет собой иерархический набор функциональных дескрипторов, ранжированных от общего к частному в каждой из трех категорий: биологические процессы, клеточные компоненты и молекулярные функции. Для описания специфичных молекулярных функций ферментов система ГО включает в себя систему классификации ферментов (КФ, англ. Enzyme Commission, или ЕС2), которая также иерархична: катализируемые реакции описываются с помощью четырех индексов, где первый отражает номер широкого класса реакций, а последний присваивается, исходя из субстратной специфичности фермента. Система ГО также включает термы молекулярной функции для стабильных химических взаимодействий (где связывание субстрата не является функционально связанным с мембранным транспортом или каталитической активностью).
Поскольку структурные мотивы описываются с помощью атомных координат, наиболее естественно их использовать для детализации функций молекул, таких как катализ конкретной реакции или связывание конкретного лиганда. Однако ни КФ, ни ГО не содержат каких-либо деталей о ферментативных механизмах или о том, какие области структуры ответственны за функционирование белка (Babbitt 2003). Например, двум ферментам, катализирующим одинаковые полные реакции, будет присвоен
2 Последняя редакция составлена в 1992 г. проф. Уэббом и номенклатурным комитетом Международного союза биохимии и молекулярной биологии. Прим, автора.
8.1. Предыстория и значение
245
одинаковый шифр КФ, даже если их структуры и каталитические механизмы сильно различаются. И наоборот, ферменты, которые явно гомологичны и имеют общие механистические особенности (такие как одинаковая частичная реакция), могут катализировать разные полные реакции, отличающиеся всеми четырьмя индексами КФ.
Помимо классификации по функциям, часто для обучения и тестирования методов аннотации используются структурные классификации. Классификация SCOP (англ. Structural Classification of Proteins, Структурная классификация белков) (Murzin etal. 1995), и классификация САТН (англ. Class, Architecture, Topology, and Homologous superfamily, Класс, архитектура, топология и гомологичные надсемейства) (Orengo etal. 1997) представляют собой иерархические классификации белковых доменов, или компактных структурных блоков (Richardson 1981), которые, согласно наблюдениям, были скомбинированы в процессе эволюции (Chothia et al. 2003). По классификации SCOP, домены объединены в семейства, надсемейства, фолды и классы. Аннотирование семейств зачастую часто бывает очевидным уже из одних лишь данных о последовательностях, поэтому представляет собой простую задачу. Большая часть анализа в SCOP сосредоточена на определении принадлежности к надсемейств. Принадлежность к тому или иному надсемейству дает много подсказок к разгадыванию функции белка, но она не является функционально специфичной. Конкретная структура может выполнять любую из функций, известных для других членов семейства, или какую-нибудь сходную функцию, прежде в этом семействе не наблюдавшуюся. Также как и в случае функциональной классификации, структурная классификация не дает прямой информации о специфических структурных особенностях, связанных с функцией.
8.1.2. Структурные мотивы: определение и область действия
Структурные мотивы представляют собой наборы точек пространства, основанные на расположении некоторых остатков (обычно их не больше десятка), которые связаны с некоторой функцией белка или интересующей классификацией. Иногда их называют шаблонами активного центра, поскольку остатки могут участвовать в образовании кармана для каталитического центра или сайта связывания, или структурными шаблонами. При подборе мотивов используется информация о расположении одного или нескольких атомов на остаток, и эти точки маркируются дополнительной информацией, такой как тип атома и остатка. Структурные мотивы локальны и дискретны, остатки соседствуют в пространстве, но нс
246
Глава 8. Пространственные мотивы
обязательно находятся рядом в аминокислотной последовательности, и расположением остатков в последовательности при подборе чаще всего пренебрегают. Мотивы не описывают конформацию пептидной цепи или доменов целиком.
8.2. Обзор методов
8.2.1. Поиск мотивов
Структурные мотивы выявляют, просматривая структуры на предмет наличия в них паттернов атомов или остатков, которые представляются функционально значимыми. Методы поиска мотивов могут быть объединены в несколько общих категорий:
1. Литература. Информация об остатках, важных для определенной функции, извлекается из литературы и других источников экспериментальных данных. Такой «экспертный» подход хоть и позволяет выявлять мотивы высокого качества, для которых подтверждается экспериментально, что образующие их остатки являются функционально важными, но требует затрат большого количества времени и не пригоден к автоматизации.
2. Ненаправленный поиск. Исследуется выборка структур на наличие статистически аномальных участков без предположения об их функциональной роли.
3. Единичные структуры. Структуры рассматриваются последовательно. Остатки, расположенных возле связанного лиганда, или те остатки, которые указаны в строке SITE в PDB-файле, просто принимаются как мотив. При этом не делается попыток сгруппировать структуры или создать консенсусный паттерн.
4. Положительные примеры. Выявление мотивов происходит путем сравнения гарантированно положительных структур (т.е. структур тех белков, которые либо выполняют интересующую функцию функции, либо принадлежат к интересующей категории по структурной классификации). Остальные структуры не рассматриваются.
5. Положительные и отрицательные примеры. Отбор мотивов происходит по их способности выявлять гарантированно положительные структуры и исключить остальные.
Для улучшения соотношения «сигнал/шум» при определении функционально важных мотивов часто также рассматривают консервативность остатков в выравнивании последовательностей и пространственную близость остатков в данном мотиве.
8.2. Обзор методов
247
8.2.2. Определение и подбор мотивов
Точки, образующие структурный мотив, определяются как атомы или псевдоатомы, полученные непосредственно из положения атомов в структуре. Геометрический центр боковой цепи, например, представляет собой псевдоатом с координатами, равными среднему арифметическому координат атомов в боковой цепи. При описании мотива используется до нескольких точек от каждого остатка, входящего в мотив, и все точки маркируются дополнительной информацией, такой как тип атома, тип остатка или физико-химические характеристики.
При проверке структуры на наличие совпадений со структурным мотивом используются количественные правила, указывающие, какая точка структуры с какой точкой мотива может быть сопоставлена, и геометрические пороговые значения, определяющие, какой набор точек обладает достаточным пространственным сходством, чтобы считаться совпадением (хитом). Степень точности этого совпадения также зависит от числа остатков и точек в мотиве. Существует компромисс между точностью совпадения и допускаемой величиной отклонения: желательно учитывать замену остатков, конформационную подвижность и низкое разрешение структуры, однако такой учет увеличит число биологически бессмысленных хитов и затруднит выделение среди них осмысленных. Включение определенных атомов в структурные мотивы призвано подчеркнуть локальные взаимодействия, например, водородные связи, в то время как использование геометрических центров функциональных групп или боковых цепей лучше приспособлено к подвижности и изменению типа остатка (Рис. 8.1). Представление симметричной боковой цепи, например, ароматического кольца в Phe, в виде одной точки также исключает необходимость сравнивать их различными способами (Oldfield 2002).
Поиск мотивов может быть трудоемок с вычислительной точки зрения, особенно если учесть, что может понадобиться сравнение тысяч структур с тысячами мотивов. Поиск структурных мотивов основывается на разработке эффективных алгоритмов, часто включающих одно или более из следующих действий:
1) Геометрическое хэширование. Хэширование - это широкий термин, описывающий приведение сложных данных к более простой форме, для которой сравнение может быть выполнено быстрее. Многочисленные величины, такие как расстояния, углы и типы атомов, могут быть редуцированы с помощью некой функции до нескольких чисел или даже до одного числа. Другие наборы значений, которые сводятся к такому же результату, соответствуют потенциально совпадающим подструктурам. Геомет
248
Глава 8. Пространственные мотивы
рическое хэширование кодирует пространственные соотношения между точками (Fischer et al. 1994), но также может включать и другие типы информации, например, физико-химические дескрипторы (Shulman-Peleg et al. 2004). Перед выполнением вычислительно затратных шагов по преобразованию групп и вычислению значений оценочных функций отдельные совпадения подструктур, которые предполагают сходные преобразования (перенос/вращение для совмещения соответствующих точек), могут быть объединены в более широкие группы (Реплее and Ayache 1998). Хэширование, или предварительная обработка данных, требует времени, но выполняется лишь один раз для каждой структуры и может сильно ускорить выполнение сравнений в целом.
Рис. 8.1. (Цветную версию рисунка см. на вклейке.) Остатки каталитического центра членов надсемейства енолаз, иллюстрирующие аспекты представления мотивов и их специфичность. Для каждого из последующих белков показаны наложенные друг на друга боковые цепи двух основных и трех кислотных остатков: рацемаза миндальной кислоты (желтый, PDB 2тпг),енолаза (оранжево-розовый, PDB 4enl), метиласпартат-аммиак-лиаза (синий, PDB 1kcz). В виде шаров изображены положения альфа-углеродов и центра масс боковых цепей. Однобуквенный код, изображенный рядом с альфа-углеродами, обозначает различные типы остатков: Н - гистидин, К - лизин, D - аспарагиновая кислота, Е - глутаминовая кислота. Несмотря на то, что представленные в левом нижнем углу аминокислотные остатки высоко консервативны по типу и конформации, активный центр включает следующие изменения:
(1) различные (хотя подобные) типы остатков на трех других позициях; (2) различные конформации боковой цепи, проиллюстрированные двумя лизинами справа;
(3) различные положения в первичной структуре: основной остаток, изображенный вверху слева, является С-концевым у енолазы, но N-концевым в последовательности двух других белков. Использование центра масс боковой цепи, а не позиции функциональных атомов, обычно уменьшает чувствительность к изменениям в конформации и типах остатков. Учет атомов боковых цепей или центра масс (только не основной цепи) уменьшает допустимое отклонение в подвижности боковой цепи, но наоборот, обеспечивает большую специфичность в отношении точности расположения атомов в функциональном сайте. Учет атомов боковой цепи уменьшает степень чувствительности по отношению к случаям миграции функционального остатка, где важная боковая цепь может принадлежать аминокислотам, расположенным в разных местах первичной последовательности (Todd et al. 2002). Рисунок создан с помощью программы визуализации IICSF Chimera (Pettersen et al. 2004) (http://www.cgl.ucsf.edu/chimera)
8.2. Обзор методов
249
2) Метод, основанный на теории графов. Граф состоит из вершин (точек) и ребер (линий, соединяющих пары вершин). Структура молекулы или структурный мотив может быть рассмотрен как маркированный граф. Например, атомы можно представить в виде вершин с промаркированным типом остатка и ребер, соединяющих каждую пару вершин, для каждого из которых приписываются соответствующие межатомные расстояния. С помощью алгоритма изоморфных подграфов производится поиск меньших графов и всех их ребер внутри более крупного графа. Такой алгоритм может применяться совместно с маркированием для поиска набора атомов в структурах, которые совпадают с типами атомов и межатомными расстояниями в структурных мотивах (Artymiuk et al. 1994; Spriggs et al. 2003). Допустимые отклонения в значениях позволяют сопоставлять похожие, но не идентичные расстояния. Определение клики графа (Schmitt etal. 2002) в конечном счете представляет собой аналогичную процедуру, но граф в этом случае описывает геометрию обеих структур вместе. В этом случае вершины графов представляют собой пары атомов или псевдоатомов, один из которых принадлежит структуре А, а другой - структуре В («структурой» также может быть и структурный мотив). Образовать пары могут только одинаковые типы атомов. Две вершины соединяются ребром, если расстояние между двумя атомами в структуре А совпадает с расстоянием между атомами в структуре В в рамках допустимых отклонений. Клика представляет собой граф, в котором каждая вершина соединена со всеми другими вершинами. Таким образом, выявлением клик можно определить набор атомов структуры А с внутренними расстояниями, полностью совместимыми с теми, которые образованы парами атомов структуры В.
3) Поиск в глубину (англ. Depth-first search). Все структуры исследуются полностью на наличие структурного мотива. Пространство поиска определено ограничениями на типы атомов или остатков, которые можно совмещать, и геометрическими пороговыми значениями, такими как допустимые отклонения и верхняя граница суммарного среднеквадратичного отклонения (СКО).
Расширение совпадения. Сначала определяются частичные или затравочные совпадения с структурным мотивом, а затем предпринимаются попытки распространить совпадение на весь мотив целиком.
Все эти методы будут искать мотивы в данной статической структуре. Последние результаты показывают, что комбинирование алгоритмов поиска мотивов с МД расчетами, сэмплирующими конформационное пространство (см. тж. Главу 9), является многообещающим, хотя и вычислительно дорогим, путем к улучшению результатов поиска (Glazer et al. 2008).
250
Глава 8. Пространственные мотивы
8.2.3. Интерпретация результатов
Существует большое число веб-серверов, позволяющих сравнить рассматриваемую структуру с базами данных структурных мотивов (Табл. 8.1). Какие выводы можно сделать о функции белка, если в его структуре будет обнаружен какой-либо мотив? При определении, является ли это совпадение значимым, и если да, какой смысл оно за собой несет, необходимо рассмотреть несколько вопросов.
Предполагаемая функция зависит от того, как был получен мотив. Если мотив представляет собой набор остатков, для которых экспериментально показано, что они важны для выполнения белком конкретной функции, то обнаружение в структуре рассматриваемого белка такого мотива позволяет предположить, что этот белок также может выполнять эту функцию. Аналогично, обнаружение мотива, полученного на основании структур многочисленных белков, выполняющих схожую функцию (положительные примеры), позволяет предположить о выполнении этой же функции, особенно если мотив был создан для исключения отрицательных примеров. Однако то, какая функция будет приписана структуре, зависит от критериев принадлежности структур к положительным примерам. Например, если все положительные примеры связывают аденозин, но катализируют разнообразные реакции, то обнаружение в рассматриваемой структуре их общего мотива будет подразумевать связывание аденозина, но не обязательно аденозиндеаминазную активность. Если набор структур, являющихся положительными примерами, состоит из представителей надсемейства по классификации SCOP, то обнаружение мотива в рассматриваемом белке будет означать, что он также принадлежит к этому надсемейству, однако нельзя будет предположить что-то более конкретное о его функции.
В идеале набор положительных примеров для поиска мотива должен быть по возможности разнообразным при сохранении у его членов некого общего свойства, а отрицательные примеры должны быть по возможности схожи со положительными примерами, но при отсутствии этого свойства. На практике наборы положительных и отрицательных примеров могут быть не идеальны, и часть или все полученные структурные мотивы могут все-таки отражать общее происхождение или совпадение, а не общую функцию.
Интерпретация обнаруженных в структуре мотивов, полученных с помощью метода «единичных структур», довольна субъективна. Как и в случае последовательностей, аннотирование рассматриваемой структуры осуществляется по аналогии с белком, который является источником мотива, совпавшего наилучшим образом. Обнаружение мотива, соответствующего
8.2. Обзор методов
251
Таблица 8.1. Веб-серверы для поиска и сравнения структурных мотивов
Название и URL Функция сервера База данных структурных мотивов11
Catalytic Site Atlas (CSA) www.ebi.ac.uk/thomton-srv/ Databases/cgi-bin/CS S /makeEbiHtml.cgi?file =form.html Сравнивает рассматриваемую структуру с БД мотивов с помощью программы JESS, оценивает значимость Мотивы из альфа углерод-ных/бета-углеродных и функциональных атомов для 147 хорошо изученных семейств ферментов; также доступна для скачивания
funClust www.pdbfun.uniroma2.it /Funclust С помощью программы Query3D определяет мотивы, общие для трех или более структур, при общем числе загружаемых структур до 20 Нет баз данных
GASPSdb www.gaspsdb.rbvi.ucsf.edu Используя программу RIGOR6, сравнивает рассматриваемую структуру со структурными мотивами, присутствующими в SCOP и ГО, оценивает значимость Мотивы из альфа углеродов и геометрических центров боковых цепей: 4385 мотивов, представляющих 272 молекулярные функции по системе ГО, 3599 мотивов, представляющих 186 надсемейств и 137 семейств по классификации SCOP, 4581 мотивов, представляющих 376 групп, в которых белки как входят в одинаковые надсемейства по классификации SCOP, так и имеют одинаковую молекулярную функцию по системе ГО
PAR-3D www. sunserver, cdfd.org. in: 8080 /protease/PAR_3D Сравнивает рассматриваемую структуру с мотивами, представленными в виде расстояний между точками и диапазонами углов. Мотивы из альфа-углерод-ных и бета-углеродных атомов для 6 классов протеаз и 10 ферментов гликолитического пути; Мотивы из аль-фа-углеродных, бета-угле-родных атомов и псевдоатомов боковых цепей для металл-содержащих сайтов связывания, состоящих из трех или четырех остатков.
252
Глава 8. Пространственные мотивы
Продолжение таблицы 8.1
Название и URL Функция сервера База данных структурных мотивов*
pdbFun www.pdbfun.uniroma2.it Сравнивает конкретный образец и целевые наборы остатков, используя программу Query3D более 12 млн отдельных остатков, выборки из которых могут быть заданы с помощью булевских комбинаций дескрипторов
PDBSiteScan www.mgs.bionet.nsc.ru/ cgi-bin/mgs/fastprot /pdbsitescan.pl?stage=O Сравнивает рассматриваемую структуру со всеми мотивами в БД PDBSite или их частью 36273 мотива, образованные атомами основной цепи и основанные на аннотации в строке SITE единичных pdb-структур или интерфейсах этих структур с другими белками, РНК или ДНК
PINTS www.russell.embl.de/pints Результаты недельной давности: www.russell.embl.de/pints-weekly Сравнивает рассматриваемую структуру с мотивом из базы данных, задаваемый пользователем мотив с БД белковых стуктур, или два белка друг с другом; оценивает значимость Лиганд-связывающие и аннотированные в строке SITE мотивы, состоящие из точек боковой цепи полярных остатков
ProFunc www. ebi. ac. uk/thomton-srv/databases/profunc Множественный поиск, включающий поиск мотива с использованием программы JESS: рассматриваемая структура целиком в БД мотивов, фрагменты рассматриваемой структуры в БД полных цепей; оценивает значимость Активные центры мотивов, взятых из БД CSA, 13057 лиганд-связывающих и 1200 ДНК-связывающих мотивов из единичных структур, 11750 полных цепей; остатки представлены атомами боковых цепей, небольшие остатки также в виде одного или нескольких атомов основной цепи.
ProKnow www. Proknow, mbi. ucla. edu Множественный поиск, включающий поиск мотива с помощью программы RIGOR, результатом является аннотация по системе ГО 10230 мотивов из структур, аннотированных по системе ГО, или 7819 мотивов, если исключить электронные аннотации
8.2. Обзор методов
253
Окончание таблицы 8.1
Название и URL Функция сервера База данных структурных мотивов*
Protemot www.protemot.csbb.ntu. edu.tw Сравнивает рассматриваемую структуру со всеми лиганд-связываю-щими мотивами или каким-то их подклассом (например, только ферменты) 2362 мотива сайтов связывания из альфа-углеродных атомов, 1051 из которых относятся к ферментам
SuMo www. sumo-pbi 1. ibcp. fr Коммерческий сайт: www. medit. fr/products-page2-med- sumo.html Сравнивает рассматриваемую структуру, цепь или лиганд-связываю-щий сайт с БД структур или только их лиганд-связывающих сайтовб 34210 лиганд-связывающих сайтов в добавление к полным структурам, описываемым как пространственные паттерны функциональных групп
а Из публикаций, веб-сервисов или информация от авторов; ссылки могут быть устаревшими.
6 Только для некоммерческого использования.
сайту связывания, может быть традиционно интерпретировано как предсказание специфичности связывания, а кроме того, можно судить (верно или неверно) и о других аннотациях - каталитической активности, принадлежности к какому-либо семейству и надсемейству. Следует отметить, что само по себе близкое расположение к лиганду не гарантирует, что остаток важен для связывания или катализа, поскольку он мог появиться там в результате мутации. Помимо использования остатков, расположенных рядом с лигандом, другим способом получить мотив из единичной структуры является использование аннотаций, которые приведены в PDB файле (строки SITE). Хотя эти аннотации предназначены для перечисления остатков, образующих сайты связывания, смысл обнаружения такого мотива также неясен, поскольку нет строгих критериев того, какие остатки должны быть перечислены. Кроме того, многие PDB файлы лиганд-связывающих структур не содержат строк SITE вообще.
Другим обстоятельством, заслуживающим рассмотрения, является строгость соответствия между мотивами, которая зависит от размера мотива и его представления, а также от количественных критериев и численных пороговых значений, используемых при поиске соответствий. Представление мотивов, опирающееся лишь на альфа-углеродные атомы, менее специфично, чем то, которое включает в себя и атомы боковых цепей. Структурные мотивы, учитывающие небольшое число остатков, также менее
254
Глава 8. Пространственные мотивы
специфичны. Строгие параметры соответствия (сопоставление остатков только идентичных типов, низкое пороговое значение для СКО) могут ограничить результаты близкородственными белками, даже если при использовании более мягких критериев могли бы быть получены осмысленные соответствия с более удаленными белками.
Большинство методов имеет оценочные функции для ранжирования хитов и определения качества соответствия. Например, значения СКО указывают на степень геометрической близости между точками структурного мотива и соответствующими точками в структуре. СКО является подходящей мерой для ранжирования мотивов по степени соответствия с эталонным мотивом, но этот критерий неприменим при рассмотрении мотивов различных размеров. Кроме того, некоторые мотивы более склонны к соответствию друг с другом просто потому что они включают большее число одинаковых остатков. Для учета этих вопросов и обеспечения лучшего ранжирования хитов, некоторые методы рассчитывают величины статистической значимости (англ. P-value) или математического ожидания (англ. E-value). Однако необходимо иметь ввиду некоторые ограничения в их применимости, так как эти величины зависят от имеющихся фундаментальных предположений о статистической модели и от данных, используемых при параметризации этой модели.
Независимо от того, каким образом был получен структурный мотив, он может быть протестирован на некоторой выборке структур. Когда эта выборка включает в себя достоверные положительные и отрицательные примеры, результаты тестирования могут быть выражены в терминах чувствительности, т.е. способности отбирать положительные примеры, и специфичности, т.е. способности исключать отрицательные примеры. Когда выборка включает лишь отрицательные примеры, то результирующее распределение по СКО может быть использовано для оценки статистической значимости соответствий этому мотиву. Применимость этих производных величин зависит от в достаточной степени обширной и представительной выборки.
Может быть полезен консенсусный подход, когда несколько хитов к родственным мотивам или аналогичные результаты, полученные с помощью различных программ и баз данных, могут приводить к общему предсказанию.
Наконец, необходимо обращаться к здравому смыслу. Например, возможен случай, когда есть хорошее соответствие мотиву какого-нибудь активного центра, но при этом нет кармана для связывания субстрата. Кроме того, статистическая значимость и биологическая значимость - это не одно и то же; биологически значимый мотив может не быть оценен как статистически значимый по сравнению с мотивом, который не имеет биоло
8.3. Специфичные методы
255
гической ценности. Таким образом, перед применением мотивов для суждения о функции или какой-либо другой характеристике белка, целесообразно проверять соответствия визуально и оценивать их с помощью биологических значимых критериев.
8.3. Специфичные методы
Работы по изучению структурных мотивов могут быть описаны с точки зрения того, как в них решается проблема выбора этих мотивов. Первая группа - это работы, которые сосредоточены на оценке методов нахождения соответствий с каким-либо мотивом, заданным пользователем, и оставляющие проблему выбора подходящего мотива пользователям метода или дальнейшим исследованиям. Вторая группа - это работы, в которых проблема нахождения мотива или создания библиотек мотивов рассматривается как важная, если не первостепенная, цель для методов, развиваемых в этих работах.
8.3.1. Мотивы, заданные пользователем
Методы для выявления определяемых пользователем структурных мотивов, как правило, демонстрируются на нескольких мотивах, известных из литературы. Самым распространенным примером является каталитическая триада Ser-His-Asp, впервые обнаруженная в сериновых протеазах (Blow et al. 1969; Wright et al. 1969), а затем и в других гидролазах, таких как эстеразы и липазы. Являясь хорошей тест-системой благодаря очень хорошей изученности и наличию в базе данных PDB большого числа её структур, эта каталитическая триада часто используется для оценки производительности методов создания и оценки структурных мотивов. Эта триада встречается в различных типах укладки, и, таким образом, охватывает случаи как дивергентной, так и конвергентной эволюции (Рис. 8.2).
При рассмотрении сериновых протеаз геометрическое хэширование с использованием минимальной структурной информации - только расположение альфа-углеродов (а не типов остатков или их порядка в последовательности) - позволило выявить не только другие сериновые протеазы, но также и сходные подструктуры в субтилизинах, которые содержат каталитическую триаду в ином типе укладки (Fischer etal. 1994). Независимость от порядка остатков в последовательности является важной особенностью многих методов, связанных с структурными мотивами; однако в обсуждаемой работе эти подструктуры были относительно большими (>50 остатков) и были выявлены из целых структур, а не заданы заранее.
256
Глава 8. Пространственные мотивы
Рис. 8.2. {Цветную версию рисунка см. на вклейке.) Две сериновые протеазы, совмещенные по их каталитическим триадам, показывают близкое сходство остатков в активном центре, несмотря на разницу в общей укладке, а) Из ленточного представления трипсина (синий/голубой, PDB код 1 sgt) и протеиназы К, гомолога субтилизина, (красный/розовый, PDB код 2ркс), видно, что два белка имеют различный тип укладки без соответствующих друг другу элементов вторичной структуры, кроме каталитических триад (стержневое представление на рисунке), которые частично накладываются. Считается, что у них нет общего предшественника, б) Боковые цепи остатков каталитической триады изображены увеличенными, чтобы показать сходство в их ориентации (1 sgt: Asp102, His57, Ser195; и 2pkc: Asp39, His69, Ser224). Сходство каталитической триады в этих негомологичных структурах говорит о способности структурных мотивов выявлять схожие функции белков в тех случаях, когда методы, основанные на использовании гомологии, потерпели бы неудачу. Рисунок создан с помощью программы визуализации UCSF Chimera (Pettersen et al. 2004) (http://www.cgl .ucsf.edu/chimera)
В другой ранней работе в этой области группа Торнтон классифицировала структуры протеаз и липаз, содержащих каталитические триады, на четыре группы по типу укладки (Wallace et al. 1996). Было замечено, что атомы кислорода серина и аспарагиновой кислоты занимают приблизительно постоянное положение относительно гистидинового кольца во всех четырех группах, тогда как остальные атомы боковых цепей хорошо совмещаются друг с другом только в пределах каждой группы. Был составлен общий структурный мотив, или шаблон, содержащий только гистидиновое кольцо и два атома кислорода, а также шаблоны, специфичные для каждой группы и содержащие боковые цепи целиком. Для ускорения процесса сравнения был разработан метод геометрического хэширования TESS (англ. TEmplate Search and Superposition, поиск и суперпозиция шаблона) (Wallace et al. 1997).
В этом методе один остаток шаблона представляет собой систему отсчета, а окружающие его атомы заключаются в ячейки пространственной решетки, и информация хэшируется. Исследуемые структуры требуют аналогичной предварительной обработки, при которой каждый остаток то
8.3. Специфичные методы
257
го же типа, что и эталонный остаток шаблона (например, гистидин каталитической триады), используется для определения пространственного паттерна для хэширования. Помимо необходимости предварительной обработки и хранения файла, программа TESS накладывает некоторые ограничения на определение мотивов и их поиск в структурах. Для решения этих вопросов без принесения в жертву скорости расчета был разработан алгоритм JESS (не аббревиатура), реализующий поиск с возвратом при решении задач с ограничениями (Barker and Thornton 2003); алгоритм выполняет поиск в глубину среди эффективно упорядоченных дескрипторов структур. В работе также описывается получение математического ожидания путем сравнения каждого структурного мотива с эталонным набором структур и моделированием результирующего диапазона значений СКО как суперпозиции нормальных распределений (Barker and Thornton 2003).
Было показано, что «нечеткие функциональные формы» (англ. “Fuzzy functional forms” (FFF)), состоящие из альфа-углеродов важных остатков, могут быть использованы для отбора как экспериментально определенных, так и смоделированных структур низкого или среднего разрешения (Fetrow and Skolnick 1998; Di Gennaro et al. 2001). Глутаредоксины и тио-редоксины были распознаны по мотиву, включающему два цистеина и пролин, с дополнительным ограничением, что пролин должен быть в цисформе, а цистеины должны образовать мотив СххС возле N-конца спирали. Рибонуклеаза Т1 была распознана по мотиву, содержащему шесть остатков. В последующих работах использование нечетких функциональных форм для опознавания больших семейств с целью более тонкой классификации сочетали с рассмотрением профилей активных центров, основанных на последовательности (Cammer et al. 2003). Нечеткая функциональная форма мотива активного центра дисульфид окидоредуктазы, найденного во многих белках, показана на рис. 8.3.
В программе ASSAM для поиска заданного пользователем паттерна остатков используется изоморфизм подграфов (Artymiuk et al. 1994). Каждая из функциональных групп боковых цепей представлена двумя или тремя псевдоатомами, и расстояния между этими точками в мотиве сравниваются с соответствующими расстояниями в структуре. Остатки могут быть промаркированы либо по типу, либо согласно химической классификации (например, по гидрофобности). Для каталитических триад был продемонстрирован компромисс между специфичностью и степенью допустимого отклонения расстояний, а также представлены и рассмотрены дополнительные примеры. Улучшения в оригинальной программе включают в себя возможность использовать атомы основной цепи и маркировать остатки по типу вторичной структуры и степени доступности растворителю (Spriggs et al. 2003).
258
Глава 8. Пространственные мотивы
Глутаредмсмн бактериофага T4(T4-GRX)
Тиоредою» человека (TRX)
Дисульфид оковдоредукгаза
Ecoi(DSB)
«4-сжх .................... .вгптхмяжбулхжкхакь
SSS AQ£WGKQrrTiKKS4n£9KPQVLfiyr&rz£FS^t< (УШжЗО: ’BOVKMTKXMVBntOGgaLeK >
ТЮГ KQTBSXT&FQEALfiAMDiKZ.ЛЛЛЖвХПГТЙфаKPPTHBbS KY3M VXfUVWD . . D
w-ей ьяалютогвсэд^очтждожкгобюдыкхгк ......................................
в$а мсмгммм»юк^г<л>илю<<^тдпк8*бР вр^лиохкоптвммгаггакз ;
ТМГ CQIW34S8CKVlEeT_JTVgrncX .......гэве. «кккшххяхэт................
«4-СШХ . . ............................
вжв 4ягжмапжмогй1Я№йг мт» iwwuvrraarwiTvirn iwkk
«а ............... ........................
Рис. 8.3. (Цветную версию рисунка см. на склейке.) Нечеткая функциональная форма мотива активного центра дисульфид окидоредуктазы, обнаруженного во многих белках. На рисунке представлены глутаредоксин бактериофага Т4, laaz, цепь А (слева), человеческий тиоредоксин, 4trx (посередине) и дисульфидоксидоредуктаэа, 1dsb, цепь А (оправа). Нечеткую функциональную форму определяют три ключевых остатка - два цистеина (боковые цепи показаны красным) и пролин (боковые цепи показаны голубым). Структура активного центра этих белков консервативна, хотя в остальной части белков проявляются некоторые различия.
С использованием этих трех ключевых остатков были определены характерные черты активного центра белка (в каждом белке фрагменты показаны синими лентами).
На глобальном выравнивании последовательностей этих трех белков, выполненном с помощью программы ClustalW, показана локализация ключевых остатков (подчеркнуты и выделены красным и голубым) и характерные фрагменты активного центра (синие). Выравнивание иллюстрирует отсутствие общего сходства в последовательностях между тремя белками, даже несмотря на то, что структура активного сайта высоко консервативна
В программе SPASM (англ. SPatial Arrangements of Sidechains and Main-chains, пространственное расположение боковых и главных цепей) каждый остаток структурного мотива представлен альфа-углеродом (СА) и/или центром масс боковой цепи (SC) (Kleywegt 1999). Пользователь определяет, какие типы остатков могут быть сопоставлены каждому остатку мотива, а для выявления хитов используется алгоритм полного поиска в глубину. Совмещению паттернов для вычисления СКО предшествует отбор по расстояниям между псевдоатомами СА-СА и SC-SC внутри каждого вероятного мотива. Дополнительно могут быть учтены ограничения на порядок остатков в последовательности. Среди примеров можно назвать использование паттерна активного центра из трех кислотных остатков для распознавания семейства глюканаз. Исполняемые и прочие файлы программы SPASM могут быть загружены с сайта Uppsala Software Factory (см. Таблицу 8.2).
8.3. Специфичные методы
259
Таблица 8.2. Веб-серверы для загрузки программного обеспечения по структурным мотивам
Название и URL Описание Скачиваемые файлы
Nestor3D www.staflfiiet.kingston.ac.uk/ ~ku33185/Nestor3D.html Программа Nestor3D создает консенсусный мотив исходя из входных структур и инструкцию по их совмещению Файлы Nestor3D Java jar, требующие Java 1.5 или более поздние версии, протестировано только для Windows
PAR-3D www. sunserver, cdfd. org. in: 8080/ protease/PAR 3D Программа PAR-3D проверяет структуру на соответствие диапазонам расстояний между точками и углов для заранее опред-ленных мотивов Скрипт PAR-3D и геометрическое описание мотивов двух металл-содержащих сайтов, шести протеаз и десяти ферментов гликолитического пути
Uppsala Software Factory www.alpha2.bmc.uu.se/usf Программа SPASM сравнивает определяемые пользователем структурные мотивы с базой данных структур, программа RIGOR сранивает рассматриваемую структуру с базой данных структурных мотивов. Исполняемые файлы программ SPASM and RIGOR для Unix-платформ, включая Mac OS X; индексируемые БД для SPASM и RIGOR. БД RIGOR включает 73164 мотива из единичных структур, из которых 57719 имеют маркировку типов остатков и 15445 - не имеют.
В группе под руководством Баббитт использовали программу SPASM не только с мотивами семейств, каждый из которых связан с одной функцией (катализируемой реакцией), но и с мотивами надсемейств, связанными с общим механизмом одной из ступеней различных полных реакций (Meng et al. 2004). Мотивы, полученные на основе единичных структур, позволяли определять надсемейство с большей чувствительностью и специфичностью, чем консенсусные мотивы, подтверждая предположение, что усреднение координат может оказаться вредным, когда структуры слишком дивергенты.
Для обнаружения всех мотивов со значениями СКО меньше порогового алгоритм Match Augmentation («Увеличение совпадения») выполняет приоритетный поиск, стартуя с трех остатков в структурном мотиве, совпавших наилучшим образом, и постепенно включая остатки, совпавшие менее удачно (Chen et al. 2005). Остатки были представлены в виде альфа-углеродов с промаркированным типом остатка и отранжированы по сте
260
Глава 8. Пространственные мотивы
пени эволюционной важности, которая следовала из выравнивания последовательностей (Kristensen et al. 2006), хотя также могут быть использованы и иные методы ранжирования. Указание приоритетов уменьшает пространство поиска, а дальше производительность улучшается за счет эффективного сравнения расстояний. Увеличение совпадений (расширение списка совпадающих остатков) происходит за счет метода поиска в глубину, основанного на использовании стека. Наконец, статистическая значимость оценивается с помощью непараметрической модели, основанной на распределении значений СКО для случая, когда мотив сравнивается с выборкой белковых цепей, взятых из базы данных PDB. Для этой цели оказалось достаточным уже 5%-ных случайных выборок (Chen et al. 2005). Было показано, что для мотивов, состоящих из 5-8 точек, представляющих изофункциональные семейства (члены которых катализируют идентичные реакции), усредненные координаты мотива обеспечивают ту же чувствительность, что и наиболее чувствительные мотивы единичных структур, и специфичность, схожую со средней специфичностью мотивов единичных структур (Chen et al. 2007b).
8.3.2. Обнаружение мотива
8.3.2.1. Литература
Пожалуй, самый надежный, но наименее автоматизируемый подход к обнаружению мотивов состоит в изучении опубликованной литературы в поиске экспериментальных данных о том, какие остатки важны для функционирования белка. В случае структурных мотивов акцент делается на остатки, которые обеспечивают специфическое связывание или каталитическую способность, а не поддержание стабильности структуры, хотя разделить эти аспекты функционирования не всегда возможно.
Атлас каталитических центров (Catalytic Site Atlas, CSA) (Таблица 8.1) содержит несколько сотен семейств ферментов, для каждого из которых приводится структура с аннотациями остатков каталитического центра, полученными из литературы, и набор родственных последовательностей (Porter et al. 2004). Представительные структурные шаблоны (структурные мотивы), основанные на функциональных атомах боковых цепей или на а- и р-атомах углерода, доступны для ряда семейств (Torrance et al. 2005). Можно выполнить поиск этих мотивов в интересующей структуре или скачать БД мотивов с сайта Атласа (Таблица 8.1). Поиск осуществляется с помощью программы JESS (Barker and Thornton 2003); допускается соответствие между химически схожими типами остатков, такими как аспартат и глутамат. Статистическая значимость оценивается по формуле,
8.3. Специфичные методы
261
которая включает число остатков в мотиве, число точек на один остаток, распространенность остатка и параметры, эмпирически подобранные при рассмотрении распределений СКО как экспонент от степенных функций (Stark etal. 2003). Данная формула оценивает фоновые распределения СКО априори, поэтому нет необходимости сравнивать каждый мотив со случайным или эталонным набором структур.
8.3.2.2. Ненаправленный поиск
Ненаправленный поиск означает обнаружение общих паттернов в случайном наборе структур, где «случайный» означает, что выбор не был основан на наличии каких-либо общих черт или функций. На практике оказывается, что есть слишком много возможных комбинаций аминокислотных остатков в структурах, чтобы можно было рассмотреть их все, поэтому пространство поиска должно быть ограничено.
Рассел провел всевозможные попарные сравнения структур в представительном наборе структур (Russell 1998). Пространство поиска было ограничено условиями на расстояния и исключением из рассмотрения неполярных остатков, цистеинов, связанных дисульфидными мостиками, и остатков, недостаточно консервативных в выравниваниях последовательностей. Для выявления случаев конвергентной эволюции не учитывались совпадения между белками со схожими фолдами. В результате были найдены несколько металл-содержащих сайтов связывания и паттернов активных центров, включая каталитическую триаду.
Программа TRILOGY также не учитывает остатки, которые недостаточно консервативны в выравниваниях последовательностей (Bradley ct al. 2002). Необходимо, чтобы паттерны присутствовали как минимум в трех разных надсемействах по классификации SCOP. Выявляются тройки потенциально совместимых остатков, включая консервативные замены, и объединяются в более широкие паттерны. Однако эта программа предназначена для определения паттернов в последовательности и структуре одновременно, а не просто структурных мотивов; паттерны остатков должны быть сходными в пространстве последовательностей также как в трехмерном пространстве.
Олдфилд проанализировал представительный набор структур посредством исключения небольших неполярных остатков, представления остальных остатков в виде одиночных точек, объединения троек остатков в группы похожих типов и сортировки расстояний между остатками в этих группах по интервалам шириной 0,5 A (Oldfield 2002). В получившейся трехмерной гистограмме (в каждой тройке есть три расстояния между остатками) интервалы с высокой заселенностью представляют собой рас
262
Глава 8. Пространственные мотивы
пространенные паттерны таких троек остатков. По мере возможности такие паттерны объединяли между собой для включения в них более чем трех остатков. С помощью такой процедуры были выявлено несколько известных структурных мотивов, таких как сайты связывания и каталитические триады. В рассматриваемой работе описаны также программы для поиска мотивов в структурах белков (Oldfield 2002).
Другое исследование включало в себя всевозможные парные сравнения вероятных функциональных сайтов, образованных такими остатками внутренней поверхности углублений, которые либо расположены рядом с лигандом, либо консервативны в выравнивании последовательностей (Ausiello etal. 2007). Несмотря на направленность на выявление сайтов, потенциально важных для функционирования белка, этот поиск был все же ненаправленным, так как структуры не были сгруппированы по какими-либо структурным или функциональным критериями. Для определения случаев конвергентой эволюции авторы сосредоточились на тех случаях, в которых совпавшие остатки имели различный порядок в соответствующих последовательностях. Были обнаружены как известные примеры таких перестановок, так и новые примеры. Совпадения нескольких остатков были найдены с помощью программы Query3D (Ausiello etal. 2005а), которая выполняет полный поиск в глубину, используя двухточечное представление остатков - а-атом углерода и геометрический центр боковой цепи. Программа Query3D определяет совпадения до десяти пар остатков, где соответствующие остатки принадлежат к сходным типам и мотивы совмещаются со значением СКО ниже порогового.
8.3.2.3. Индивидуальные структуры
Некоторые базы данных структурных мотивов были созданы с использованием только информации о каждой структуре отдельно. Например, мотивы сайтов связывания могут быть составлены путем рассмотрения остатков, расположенных на определенном расстоянии от лигандов, нуклеиновых кислот или даже цепей других белков. Часто эти исследования сосредотачиваются на поиске методов, а не на создании баз данных, а некоторые из них также представляют результаты исследований других типов, в частности, поиск мотивов, описанных в литературе.
Сервер PINTS (Patterns in Non-homologous Tertiary Structures, шаблоны в негомологичных третичных структурах) (Stark and Russell 2003) (Таблица 8.1) сравнивает рассматриваемую структуру с базой данных структурных мотивов, представляющих собой либо сайты связывания, определяемые как остатки, расположенные на расстоянии 3 А от лиганда, либо мотивы, аннотированные в строке SITE структурного файла белка из
8.3. Специфичные методы
263
PDB. С другой стороны, можно сравнить мотивы, определяемые пользователем, с базами данных белков (например, представленными на различных уровнях по классификации SCOP), или две конкретные структуры между собой. Как и в более ранней работе Рассела по ненаправленному поиску, PINTS производит поиск в глубину, не рассматривает неполярные остатки, использует атомы боковых цепей и допускает совпадение некоторых схожих типов атомов. Статистическая значимость оценивается по разработанному авторами методу (Stark et al. 2003), вышеописанному для CSA. На веб-сайте PINTS также доступны результаты еженедельного сравнения структур, только что депонированных в PDB, с базами данных мотивов (Stark et al. 2004) (Таблица 8.1).
В дополнение к мотивам, записанным в строке SITE, база данных PDBSite (Ivanisenko etal. 2005) (Таблица 8.1) включает сайты взаимодействия с другими белками, РНК и ДНК. В сайт взаимодействия включены остатки, имеющие как минимум три атома, расположенных на расстоянии в пределах 5 А от другой цепи. Все сайты в базе данных или их выборку можно сравнить с рассматриваемой структурой с помощью программы PDBSiteScan (Ivanisenko и др. 2004) (Таблица 8.1), которая использует данные о типах остатков, положении атомов основной цепи и задаваемые пользователем пороговые значения. Результат совмещения рассматриваемой структуры и найденных структурных мотивов может быть загружен в PDB формате.
Программа RIGOR по существу не отличается от программы SPASM, за тем лишь исключением, что выполняет обратный процесс: сравнивает структуру с базой данных структурных мотивов, а не мотив с базами данных структур (Kleywegt 1999). Исполняемые файлы программы RIGOR и связанные с ней базы данных мотивов доступны для скачивания на вебсайте Uppsala Software Factory (Таблица 8.2). База данных включает сайты вокруг связанных лигандов, фрагменты, составленные из следующих друг за другом одинаковых остатков, и некоторые другие группы остатков. Каждый сайт связывания лиганда включен дважды - с маркировкой типа остатков и без таковой. Совпадение с немаркированным мотивом может означать, что такой мотив может быть включен в рассматриваемую структуру методами белковой инженерии.
Обнаружение структурных мотивов занимает центральное место в предсказаниях функции, которые выполняются двумя серверами, объединяющими результаты, полученные с использованием сторонних данных. Эти серверы детально обсуждаются в Главе 11, здесь же вкратце упомянуты для полноты картины. Сервер ProKnow (Pal and Eisenberg 2005) (Таблица 8.1) выполняет для рассматриваемой структуры множественный по
264
Глава 8. Пространственные мотивы
иск на основе последовательностей или структур, включая поиск структурных мотивов единичных структур с помощью программы RIGOR. Каждая база данных, по которой сервер ProKnow выполняет поиск, содержит аннотации по системе ГО, и конечным результатом является список возможных аннотаций для рассматриваемой структуры и их байесовские оценки (оценки вероятности). Однако многие из термов системы ГО носят достаточно общий характер. Второй интегральный метод, сервер ProFunc (Laskowski etal. 2005b) (Таблица 8.1) также выполняет множественный поиск на основе последовательностей и структур.
Программа JESS (Barker and Thornton 2003) используется для поиска шаблонов активных центров ферментов в базе данных CSA и поиска троек остатков, связывающих лиганды или нуклеиновые кислоты, в невырожденной выборке из базы данных PDB. Для более полного охвата пространства структур, также осуществляется поиск «обратного шаблона», когда рассматриваемая структура разбивается на структурные мотивы, которые сравниваются с представительным набором исходных структур базы данных PDB (Laskowski et al. 2005а). Хиты программы JESS затем оцениваются расширением области сравнения остатков до сферы радиусом 10 А с центром в найденном мотиве. Совпадения сортируются с помощью оценочной функции, которая положительно оценивает наложение пар остатков схожих типов со схожим расположением в последовательности и порядком следования. Таким образом, поиск мотива сделан более специфичным, но менее локальным, и, следовательно, хуже подходящим для определения примеров конвергентной эволюции. Выполняется поиск каждого мотива в выборке структур из базы данных PDB, и математические ожидания вычисляются из предположения о распределении экстремальных значений оценочных функций (Laskowski et al. 2005а).
Сервер SuMo (Jambon etal. 2005) (Таблица 8.1) сравнивает рассматриваемую структуру либо с базой данных «полных структур из PDB» (все структуры, но повторно встречающиеся цепи удалены), либо только с ли-ганд-связывающими сайтами из той же базы данных. Рассматриваемой структурой может быть либо вся структура целиком, либо её цепь, либо только её лиганд-связывающий сайт. Сервер представляет структуры как графы треугольников химических групп, среди которых различные доноры и акцепторы водородных связей, ароматические кольца и так далее (Jambon et al. 2003). При сравнении пары структур в первую очередь выявляются пары схожих треугольников, а затем согласующиеся наборы пар, или патчи. Патчи затем уточняются удалением пар химических групп, которые относительно плохо накладываются либо значительно отличаются по степени заглубленное™.
8.3. Специфичные методы
265
Веб-сервер программы Protemot (Chang et al. 2006) (Таблица 8.1) сравнивает структуру с базой данных сайтов связывания мотивов, определяемых как остатки, в которых по крайней мере один атом находится в пределах 4,5 А от лиганда. Из базы данных были исключены вырожденные цепи с идентичностью последовательностей 60% и сайты связывания биологически малоинтересны лигандов. Возможно выполнение поиска либо всех мотивов, либо тех, которые встречаются в структуре ферментов, либо тех, встречаются в структуре определенных классов ферментов. При выполнении поиска рассматриваемая структура редуцируется до альфа-атомов углерода с промаркированным типом остатка, находящихся возле углубления сайта связывания. Эта информация хэшируется и сравнивается с хэшами элементов базы данных. Для сопоставления остатков пользователь устанавливает пороговое значение сходства между ними. Из этих грубых совпадений сто лучших затем уточняются для учета большего числа остатков, и в итоге остаются только те совпадения, у которых наблюдается одинаковая направленность углублений и значение СКО лежит в пределах 1,5 А. Эти совпадения изображаются графически, однако списка сопоставленных остатков не приводится, выдается лишь информация о PDB кодах хитов.
Веб-сервер pdbFun (Ausiello etal. 2005b) (Таблица 8.1) позволяет сравнивать наборы пробных и целевых остатков с помощью программы Query3D (Ausiello et al. 2005a) (описана в предыдущем разделе). Остатки могут быть либо указаны вручную по отдельности, либо могут быть использованы их заранее заданные наборы или булевские комбинации таких наборов. Один из типов заранее заданных наборов представляет собой сайт связывания, т.е. остатки, находящиеся в пределах 3,5 А от лиганда. Также возможно использовать активные центры базы данных CATRES, определенные из литературных данных (Bartlett et al. 2002). Пробный набор может содержать остатки только одной цепи, тогда как целевой набор может содержать вплоть до всей базы pdbFun (-50000 цепей). Процесс указания наборов пробных и целевых остатков очень удобен для применения, но может и ввести в заблуждение. Для обеспечения быстрого поиска, пороговые значения СКО задаются очень жесткими и не могут быть скорректированы. Однако программа Query3D может быть получена от разработчиков для локального использования (на Unix-платформах), и этом случае пользователь может задать желаемые пороговые значения.
8.3.2.4. Положительные примеры
Локальные структурные особенности, общие для всех белков, выполняющих определенную функцию или входящие в состав конкретного структурного класса, могут трактоваться как структурные мотивы. При
266
Глава 8. Пространственные мотивы
таком подходе используются разнообразные положительные примеры для определения того, какие атомы или остатки могут быть включены в мотив, хотя координаты мотива могут быть взяты из единичной структуры, а не из усредненных данных. В процессе получения мотивов отрицательные примеры не рассматриваются, хотя они часто используются при оценке этих мотивов.
Некоторые лиганд-ориентированные исследования используют жесткую часть лиганда для совмещения сайтов связывания. Например, для сравнения различных сайтов связывания аденинмононуклеотида было использовано их совмещение по аденину. Одно из исследований включало всевозможные сравнения доступного на тот момент 121 аденинмононукле-отидного комплекса (38 комплексов после отсева повторов) (Kobayashi and Go 1997). Для каждой пары структур было оценено число соответствующих пар атомов (на основе элементов и соседнего расположения) возле аденина и степень их совмещения. Было обнаружено высокое сходство между структурами с различной укладкой: они имели общий структурный мотив из атомов сегментов основной цепи длиной по четыре остатка и трех остатков, разнесенных в последовательности (Kobayashi and Go 1997).
Аналогичный подход был использован при создании консенсусных мотивов сайтов связывания (Nebel et al. 2007). Так как к моменту этого исследования стало доступно гораздо больше структур, то комплексы с аденинмоно-, -ди- и -трифосфатами были рассмотрены отдельно. Сходство между парой структур оценивали как долю таких атомов в окружении лиганда, которые присутствуют в обеих структурах. Структуры были сгруппированы согласно этим значениям сходства, а неподходящие структуры были исключены из рассмотрения. В пределах каждой группы были сохранены только общие во всех парных сравнениях атомы, и их расположение было усреднено для создания структурного мотива. Наконец, очень похожие мотивы были объединены. Результирующие 13 мотивов, полученные на основе анализа от 3 до 20 структур, содержат от 6 до 71 атома и в большинстве случаев соответствуют некоторым известным классификациям структур или функций. Координаты мотивов доступны в качестве дополнительной информации к публикации (Nebel et al. 2007).
С помощью программы Nestor3D (Nebel 2006) были разработаны консенсусные шаблоны для порфирин-связывающих сайтов. Шаблоны, созданные с помощью этой программы, могут включать атомы, функциональные группы в виде псевдоатомы и «растворитель» (фактически он представляет собой точки на решетке для представления объема углубления). Программа Nestor3D также включает графический интерфейс и доступна для скачивания (Таблица 8.2). Пользователи должны указать список
8.3. Специфичные методы
267
файлов PDB и подходящие для совмещения структур атомы; некоторые другие параметры могут быть настроены дополнительно.
Всевозможные сравнения 3737 фосфатных окружений из белок-нуклеотидных комплексов позволили классифицировать их на 476 компактных кластеров и 10 более широких групп (Brakoulias and Jackson 2004). Полученное разделение на кластеры в целом согласуется с классификациями, в основе которых лежат глобальная структура или функция белка. Для выявления соответствующих наборов атомов использовался эффективный метод обнаружения клик, поэтому не было необходимости использовать атомы лиганда для совмещения структур.
Программа SOIPPA (англ. Sequence Order-Independent Profile-Profile Alignment, Выравнивание профилей, независящее от порядка следования) находит общие паттерны локальной структуры при парных сравнениях (Xie and Bourne 2008). Структура белка редуцируется до его а-атомов углерода, каждому из которых ставится в соответствие значение геометрического потенциала и профиль возможных замен, полученный из автоматического выравнивания последовательностей. Геометрический потенциал а-атома углерода рассчитывается исходя из расстояния от него до поверхности белка и расположения соседних а-атомов углерода (Xie and Bourne 2007). Возможное совпадение между двумя структурами начинается с пары точек со схожими геометрическими потенциалами; к ним могут быть добавлены соседние пары, если они согласуются по расстояниям и углам с нормалью к поверхности.
Каждой паре а-атомов углерода присваивается её вес исходя из схожести их профилей замен, и затем находится подграф с максимальным общим весом. Оценочная функция выравнивания после совмещения атомов представляет собой сумму по всем парам, включающую вес пары, степень совпадения атомов пары в пространстве и угол между нормалями к поверхности. Статистическая значимость оценивается с помощью непараметрической модели распределения значений оценочной функции, когда паттерн сравнивается с представительным набором структур. Программа SOIPPA использовалась для сравнения разнообразных аденин-связывающих структур и для поиска репрезентативного набора структур для совмещения с известными функциональными сайтами; программа была способна выравнивать сайты связывания и выявлять локальные сходства лучше, чем это позволяли делать глобальные сравнения последовательностей или структур. Эта работа была в большей степени ориентирована на определение взаимосвязей, чем на обнаружение мотива.
Для определения функционально важных атомов в структурах, имеющих общую функцию, но эволюционно не связанных между собой или на-
268
Глава 8. Пространственные мотивы
холящихся в отдаленном родстве, был предложен метод общих структурных клик (англ. Common Structural Cliques method) (Milik etal. 2003). Каждый белок сводится к графу, который включает в себя только репрезентативные атомы каждой боковой цепи. Затем для определения общих структурных клик, т.е. наборов атомов с эквивалентными типами и межатомными расстояними в обоих структурах, извлекаются и сравниваются между собой наборы из четырех атомов. Такие клики объединяются в более крупные наборы соответствующих друг другу атомов.
Примечательно, что получающиеся структурные мотивы могут иметь различный вес, даже равный нулю, для различных межатомных расстояний, что позволяет говорить о совпадении даже тогда, когда определенные расстояния значительно варьируются из-за конформационной подвижности. Например, мотив может включать атомы, расположенные в одном из шарнирно соединенных доменов. Малый или нулевой вес междоменных расстояний позволяет выявить мотив в структурах с различными конформациями шарнира, тогда как веса внутридоменных расстояний могут сохраняться высокими для определения точных геометрических связей в пределах каждого домена. Ограничением этого метода является невозможность автоматического сочетания результатов, полученных из попарных сравнений.
Программа DRESPAT (англ. Detection of REcurring Sidechain PATtems, обнаружение повторяющихся паттернов боковых цепей) извлекает общий мотив из набора структур, являющихся положительными примерами (Wangikar et al. 2003). Каждый белок сводится к графу из функциональных атомов (по одному на остаток), исключая остатки с неполярными боковыми цепями и цистеины, связанные дисульфидными мостиками. Затем выделяются паттерны из трех или более остатков и сравниваются с паттернами других структур, состоящими из остатков того же типа, при этом в дополнение к функциональным атомам учитываются а- и Р-атомы углерода, и не рассматриваются те совпадения, у которых отклонения в расстояниях между точками и/или значения СКО больше, чем заданные пороговые значения. Другим настраиваемым параметром является размер паттерна (по умолчанию от трех до шести остатков) и число входящих структур, которые должны содержать этот паттерн.
На основе встречаемости паттернов в случайно выбранном наборе структур были получены эмпирические соотношения для расчета статистической значимости обнаруженных паттернов, исходя из их размера, общего числа структур и числа структур, которые должны содержать этот участок. Результаты были представлены для невырожденных наборов из 17 надсемейств по классификации SCOP. Было обнаружено, что мотивы,
8.3. Специфичные методы
269
состоящие как минимум из четырех остатков и полученные из наборов, содержащих 5 и более структур, обычно соответствуют функциональным сайтам. При рассмотрении только попарных сравнений эволюционно родственных структур было получено слишком много дополнительных паттернов. Программу DRESPAT можно получить от ее разработчиков в виде кода на C++ (Wangikar et al. 2003).
Сервер funClust (Ausiello et al. 2008) (Таблица 8.1) определяет структурные мотивы, общие для различных входных структур, которых может быть до 20. Структуры в дальнейшем отбираются по степени идентичности последовательностей и затем попарно сравниваются с помощью программы Query3D (Ausiello etal. 2005а). Программа Query3D использует представление остатка в виде двух точек: альфа-углерод и геометрический центр боковой цепи. Кроме максимальной идентичности последовательностей, пользователь может указать, должны ли пороговые значения для СКО и близости боковых цепей быть низкими, средними или высокими; должны ли быть исключены из рассмотрения гидрофобные или заглубленные остатки; и можно ли разрешить сопоставление остатков сходных типов, а не только идентичных. Сервер сообщает о мотивах из трех или более остатков, обнаруженных в трех или более входных структурах.
Сервер PAR-3D (англ. Protein Active site Residues using 3-Dimensional structural motifs, остатки активного центра белка, выявленные с помощью структурных мотивов) (Goyal et al. 2007) (Таблица 8.1) сравнивает загруженную структуру с мотивами шести классов протеаз, десяти ферментов гликолитического пути и металл-содержащих сайтов, состоящих их 3 или 4 остатков (Goyal and Mande 2008). Мотивы, каждый из которых был получен из обучающего набора структур, представлены в виде допустимых интервалов межатомных расстояний или других геометрических скаляров, а не как пространственные координаты. Значения чувствительности и специфичности для мотивов доступны на веб-сайте, и сама программа (Perl-сценарии и связанные с ними файлы данных) также может быть загружена с этого сайта.
8.3.2.5. Положительные и отрицательные примеры
Основное различие между подходом с «положительными и отрицательными примерами» и подходом с «положительными примерами» состоит в том, что в процессе обнаружения мотивов первый подход в явном виде рассматривает структуры, не принадлежащие интересующему классу. Другими словами, создание мотива и оценка его специфичности взаимосвязаны.
Отсеивание по геометрическим параметрам уточняет либо существующий мотив, либо список потенциально важных остатков, основанный на их геометрической уникальности (Chen etal. 2007а). Распределения
270
Глава 8. Пространственные мотивы
СКО для кандидатов в мотивы (подгрупп из входного списка), получаются сравнением их с репрезентативной выборкой структур. Отсеивание по геометрическим параметрам не требует разделения на положительные и отрицательные примеры; вместо этого предполагается, что хвост кривой распределения с низким СКО отображает истинно положительные примеры, а остальная часть - ложноположительные примеры. Среди мотивов с определенным числом остатков выбирается обладающий самым высоким значение медианы СКО, и он рассматривается как наиболее геометрически уникальный, поскольку он обеспечивает лучшее разделение между основной частью распределения и хвостом с низким СКО. Главное ограничение такого подхода состоит в том, что «правильные» остатки должны быть включены в исходный мотив.
Рассматривая положительные и отрицательные примеры структур, алгоритм GASPS (англ. Genetic Algorithm Search for Patterns in Structures, генетический алгоритм поиска паттернов в структурах) находит паттерны остатков, которые позволяют лучше всего разделить эти две группы (Polacco and Babbitt 2006). При этом предварительный список остатков не требуется, и от метода не зависит, как определены группы с положительными и отрицательными примерами. Основной инструмент поиска представляет собой программу SPASM (Kleywegt 1999), использующей представление остатков в виде альфа-углеродов и геометрических центров боковых цепей и допускающей совмещение остатков только одинакового типа. Для ограничения пространства поиска, программа GASPS рассматривает только 100 наиболее консервативных остатков структуры, которые определяются по автоматически построенному выравниванию последовательностей. Первоначальный кандидат в мотивы строится случайным выбором одного остатка и затем также случайным выбором еще четырех остатков, которые находятся вдали от первого остатка.
Каждый из 50 первоначальных кандидатов оценивается с точки зрения того, насколько хорошо он позволяет разделить положительные и отрицательные примеры структур в терминах значений СКО для наилучших совпадений. В каждом цикле генетического алгоритма 16 мотивов с наивысшей оценкой становятся родителями 36 новых мотивов, и после 50 циклов лучший мотив объявляется победителем. Мотивы могут содержать от трех до десяти остатков. Для разных надсемейств (Babbitt and Gerlt 2000) и сериновых протеаз были получены чувствительные и специфичные мотивы. Было обнаружено, что большая часть остатков в мотивах функционально важна, но в некоторых случаях оказалось, что остатки, не имеющие известной функциональной роли, имеют такое же прогностическое значение (Polacco and Babbitt 2006).
8.4. Аналогичные методы
271
Сервер GASPSdb (Polacco and Babbitt, статья готовится к печати) (Таблица 8.1) сравнивает рассматриваемую структуру с базой данных структурных мотивов, ранее созданной с помощью программы GASPS для некоторых схем классификации белков: надсемейств и семейств по классификации SCOP, белков, имеющих общие молекулярные функции по системе ГО, и белков, входящих в состав надсемейств по классификации SCOP и являющихся молекулярной функцией по системе ГО. Использовался невырожденный набор структур. Мотив создавался на основе каждой структуры из группы положительных примеров, структуры из остальных групп рассматривались как отрицательные примеры. Мотивы создавались только для групп, содержащих не менее 6 невырожденных структур. Для поиска на сервере используется программа RIGOR (Kleywegt 1999). Для коммерческого использования этого продукта нужно связаться с разработчиками для получения лицензии (см. веб-сайт Uppsala Software Factory, Таблица 8.2). Статистическая значимость оценивается с помощью функции, разработанной авторами программы PINTS (Stark et al. 2003).
8.4. Аналогичные методы
Гибридные описания «точка-поверхность» и одноточечные описания локальной структуры не могут быть, строго говоря, отнесены к методам структурных мотивов, но имеют с ними много общего. Методы, основанные на описании поверхности, рассмотрены в Главе 7.
8.4.1. Гибридные описания «точка-поверхность»
Программы Cavbase (Schmitt et al. 2002; Kuhn et al. 2006) и SiteEngine (Shulman-Peleg et al. 2004) описывают сайты связывания, как наборы псевдоатомов и связанных с ними лоскутов поверхности. Псевдоатомы соответствуют различным экспонированным на поверхности функциональным группам, таким как доноры или акцепторы водородных связей. Сравнение включает в себя поиск геометрически и физико-химически совместимых наборов псевдоатомов, пространственное совмещение структур на основе этих псевдоатомов и последующую оценку, основанную на перекрытии лоскутов и физико-химическом сходстве. Число точек поверхности обычно сильно превосходит число псевдоатомов, поэтому оценка является относительно ресурсоемкой. Два упомянутых метода отличаются некоторыми деталями описания псевдоатомов, выполнения оценки и того, как производится поиск совпадений: Cavbase осуществляет
272
Глава 8. Пространственные мотивы
поиск клик, в то время как SiteEngine использует геометрическое хэширование. Cavbase и связанная с ним база данных сайтов доступна как часть коммерческого программного пакета Relibase+ по лицензии Кембриджского кристаллографического центра обработки данных. Веб-сервер SiteEngine (Shulman-Peleg et al. 2005) (Таблица 8.3) выполняет попарные сравнения, но не поиск по базе данных. Для некоммерческого использования может быть загружен исполняемый файл для ОС Linux (Таблица 8.3).
8.4.2. Одноточечные описания
Программа FEATURE (Bagley and Altman 1995) описывает локальную структуру как набор свойств в концентрических оболочках, исходящих из одной точки. Свойства включают в себя дескрипторы атомов, функциональных групп, остатков, вторичной структуры и простые биофизические характеристики. Радиальное распределение свойств вокруг точек, представляющих интерес с точки зрения функции, сравнивается с таким распределением вокруг контрольных точек и оценивается статистическая значимость различий. Однако при этом теряется информация по направлениям, поскольку величины суммируются по сферическим оболочкам. Всб-ссрвср FEATURE (Liang et al. 2003) (Таблица 8.3) выполняет сравнение структуры с любым из своих заранее вычисленных паттернов, представляющих различные типы сайтов. Имеется свыше сотни паттернов, но этот набор весьма ограничен в пространстве функций; многие из паттернов просто центрированы на разных атомах сайтов одного типа. Также доступны заранее вычисленные совпадения для конкретных паттернов, конкретных структур из PDB или наборов белков, полученных в проектах по структурной геномике.
Веб-сервер S-BLEST (Structure-Based Local Environment Search Tool, Инструмент поиска локального окружения, основанный на структуре) (Mooney et al. 2005; Peters et al. 2006) (Таблица 8.3) выполняет сравнение предлагаемых программой FEATURE паттернов, центрированных на каждом остатке в рассматриваемой структуре, с базой данных таких паттернов, которая содержит по одному паттерну для каждого остатка из невырожденной выборки структур PDB. Распределение значений сходства для рассматриваемого остатка дает стандартизованные оценки для каждого конкретного остатка из базы данных. Общая оценка совпадения белковой цепи в базе данных представляет собой среднее значение стандартизованных оценок по К лучшим остаткам, где К - это число, определяемое пользователем. Цепи с оценками лучшими, чем некая пороговая, выводятся в список наряду с их аннотациями по ГО, КФ и SCOP и аналогичными
8.5. Использование молекулярного докинга
273
Таблица 8.3. Веб-серверы с аналогичными подходами
Название и URL Функция сервера Загрузки
S-BLEST www.sblest.org Выполняет сравнение остаток-центрирован-ных конфигураций в рассматриваемой структуре с конфигурациями в невырожденном наборе структур из PDB, выдает список наиболее похожих цепей и их аннотации S-BLEST также может быть использован удаленно с визуализацией в программе UCSF Chimera (www.cgl.ucsf.edu/chimera). Загрузить плагины Chimera для Windows, Linux, Mac OSX можно на сайте: www.lifescienceweb.org
SiteEngine bioinfo3d.cs.tau.ac.il/ SiteEngine Выполняет сравнение сайта связывания в структуре со связанным лигандом со всей поверхностью другой структуры Доступен исполняемый файл для Linux исключительно для некоммерческого использования
WebFEATURE feature, stan ford, edu/ webfeature Выполняет сравнение рассматриваемой структуры с заранее рассчитанными точечно-центрированными конфигурациями, представляющими несколько десятков функциональных сайтов Загрузить исходный код программы FEATURE можно по ссылке: simtk.org/home/feature
результатами, полученными из сравнения последовательностей. Веб-сервер S-BLEST также может быть использован удаленно через молекулярнографический интерфейс программы UCSF Chimera (Pettersen et al. 2004). Программа-клиент может быть загружена из сети Life Science Web (см. Таблицу 8.3).
8.5. Использование молекулярного докинга при аннотировании функции
В конце концов, специфичность к лиганду и каталитические возможности белка зависят от расположения атомов в его сайте связывания или активном центре. В то время как методы структурных мотивов стремятся
274
Глава 8. Пространственные мотивы
напрямую установить связь между пространственными паттернами и функциями, можно использовать структуру белка для подбора вероятных лигандов, а затем с помощью получившегося предсказания специфичности связывания сделать выводы о молекулярной функции белка. В вычислительном докинге для каждого из большого числа низкомолекулярных органических соединений выполняется встраивание его структуры в сайт связывания белка и оценка комплементарности получившегося комплекса. Для каждого соединения могут быть рассмотрены сотни тысяч вариантов такого встраивания. Молекулы, получившие наивысшие оценки, считаются наиболее вероятными лигандами белка и является первыми кандидатами на экспериментальное тестирование.
Такой процесс вычислительного молекулярного распознавания может показаться очевидным, но на практике встречается со многими трудностями. Одна из них состоит в большом количестве метаболитов, которые может оказаться необходимым проверить в качестве возможных лигандов. Другая заключается в сложности быстрой, но достаточно точной оценки структурной комплементарности, позволяющей отличить истинные лиганды от схожих, но не связывающихся соединений. Для точной сортировки может понадобиться учет конформационной подвижности в процессе докинга, который еще больше увеличивает вычислительную нагрузку. Традиционно локирование большого числа соединений применялось для обнаружения соединений-лидеров, являющихся потенциальными лекарственными веществами. При аннотировании функции необходимость точной сортировки оказывается еще большей, чем в поиске лидеров, поскольку экспериментальный скрининг неизвестной каталитической активности гораздо более сложен, чем скриниг связывания, и поскольку цель состоит в аннотировании тысяч белков в автоматическом или полуавтоматическом режимах. Поиск лидеров же, напротив, сосредотачивается на одной или нескольких хорошо изученных мишенях.
Несмотря на эти трудности, такому подходу с недавних пор начали уделять значительное внимание (Macchiarulo et al. 2004; Paul et al. 2004; Kalyanaraman et al. 2005; Tyagi and Pleiss 2006; Favia et al. 2008). В двух опубликованных примерах предсказания функции с помощью молекулярного докинга (Hermann et al. 2007; Song et al. 2007), рассматриваемый белок был сначала опознан как представитель функционально разнородного надсемейства ферментов, что позволило сузить область поиска потенциальных субстратов. Однако в каждой работе использовались различные методы для получения точной сортировки продокированных молекул.
В первой работе рассматривалось семейство белков, которые, судя по информации об их последовательностях, могли быть отнесены к надсе
8.5. Использование молекулярного докинга
275
мейству енолаз, но их детальная функция надежно определена не была (Song et al. 2007). Все представители этого надсемейства катализируют отделение протона от углерода, примыкающего к карбоксильной группе, но их субстраты и реакции в целом сильно варьируются (Babbitt and Gerlt 2000). Поскольку для белков этого семейства нет экспериментально полученных структур, то для одного из представителей была построена модель на основании гомологии с наиболее схожим белком с известной структурой (идентичность последовательностей 35%) - аланинглутаматэпимера-зой. N-сукцини л аминокислотные рацемазы также локализованы возле неизвестного семейства в пространстве сходства последовательностей, поэтому в библиотеки для вычислительного и экспериментального скрининга были включены дипептиды и N-сукциниламинокислоты. Несмотря на в целом гораздо большее сходство с эпимеразами, чем с рацемазами, в параллельно выполненных экспериментальном изучении и in silico докинге было обнаружено, что белок катализирует рацемизацию N-сукцинилар-гинина и -лизина. Более того, хотя основанная на гомологии модель была построена по шаблону структуры аланинглутаматэпимеразы, докинг и оценка его решений с помощью подвижности боковых цепей и функций, основанных на физических принципах, подтвердила найденные экспериментально предпочтения среди N-сукциниламинокислот. Позже были получены кристаллографические структуры белка с обоими субстратами, которые показали значительное согласие с решениями докинга. Однако если бы в процессе докинга боковые цепи оставались неподвижными, то идентификация этих субстратов при докинге оказалась бы гораздо менее успешной (Song et al. 2007).
Во второй работе структура, определенная в рамках проекта по структурной геномике, могла быть отнесена к надсемейству амидогидролаз на основе типа укладки и наличия определенных высококонсервативных остатков в активном центре (Hermann et al. 2007). Однако эти остатки не позволяли понять, какая (если вообще какая-нибудь) из десятков реакций гидролиза, известных для данного надсемейства, могла бы катализироваться именно этим ферментом. Для докинга были отобраны только метаболиты, содержащие поддающиеся гидролизу фрагменты, такие как амидные, сложноэфирные и фосфоэфирные группы. Поскольку ферменты участвуют в катализе реакций, а не просто в связывании субстрата, авторы решили, что использование в докинге структур переходного состояния таких молекул должно дать лучшие результаты. Таким образом, для каждого метаболита были созданы структуры, имитирующие его переходное состояние: амиды и сложные эфиры были преобразованы в тетраэдрическую форму, фосфоэфиры в треугольно-бипирамидальную форму и так далее.
276
Глава 8. Пространственные мотивы
Предварительные проверочные исследования показали, что использование в докинге таких высокоэнергетических форм улучшает результаты сортировки известных лигандов (Hermann et al. 2006).
В прогностическом исследовании (Hermann et al. 2007), было замечено, что многие соединения с высокой оценкой содержат адениновый фрагмент, в котором внециклический азот был преобразован в тетраэдрическую форму, как это происходило бы при дезаминировании. Экспериментально было установлено, что фермент катализирует дезаминирование трех из четырех протестированных аденин-содержащих метаболитов, но не производные цитозина, хотя наибольшее сходство последовательностей наблюдалось с хлоргидролазами и цитозиндезаминазами. Кристаллограффиче-ская структура фермента в комплексе с одним из продуктов дезаминирования показала наличие тех взаимодействий, которые были предсказаны в докинге. Подтвержденное функциональное описание этого фермента позволило аннотировать еще несколько последовательностей, также содержащих характерные для такого активного центра остатки.
Следует заметить, что сужение пространства реакций до реакций гидролиза помогло сделать задачу решаемой, поскольку создание структур, имитирующих переходное состояние, значительно увеличило число молекул, предназначенных для локирования. При оценке учитывались стерический, электростатический и де сольватационный вклады, но подвижность белка не рассматривалась.
Как методы, использующие структурные мотивы, так и молекулярный докинг при функциональном аннотировании сосредотачиваются скорее на локальной структуре, чем на структуре глобальной или на сходстве последовательностей. Будучи с вычислительной точки зрения более ресурсоемким, чем поиск совпадений структурных мотивов, докинг обладает возможностью экстраполяции на функции, не связанные с ранее описанными структурами; для рассматриваемого белка может быть предсказан «новый» субстрат.
8.6. Обсуждение
Основной причиной использования структурных мотивов, а не сравнения типов укладки, является то, что непосредственно причастные к функционированию белка структурные особенности служат наиболее точными и эффективными признаками этого функционирования. В то же время определить, какие именно остатки действительно важны для функционирования, сложнее, чем использовать тип укладки целиком. Хотя для
8.6. Обсуждение
277
остатков в близком окружении каталитического центра или сайта связывания можно сделать предположения об их важности, это не является отличным показателем. Однако с ростом числа структур методы выявления мотивов в разнообразных наборах положительных примеров становятся более жизнеспособными.
Такой подход переносит нагрузку с прямого определения остатков на выбор подходящих наборов структур, позволяющих составить представление о функциях.
Какая классификация белков является наиболее естественной с точки зрения этих структурных мотивов «тонкой структуры»? В ферментах индивидуальные остатки или функциональные группы играют различную роль в процессе реакции: распознавание субстрата, катализ отдельных стадий реакции, стабилизация интермедиата или какая-то комбинация из названного. Поскольку белки эволюционируют для выполнения новых функций, то, чтобы усложнить процесс, они могут использовать имеющиеся локальные структурные особенности, которые ответственны за выполнение частичной функции как в новой, так и в прежней функции (Babbitt and Gerlt 2000; Bartlett et al. 2003). Это отчасти позволяет понять, почему представители гомологичных, но различных групп ферментов часто имеют одинаковую конфигурацию небольшого числа аминокислот, несмотря на в целом различающиеся катализируемые реакции. В случае ферментов можно представить себе иерархию мотивов, включающую паттерны, связанные с частичными функциями, общими для всех представителей разнородной группы, и более сложные паттерны, относящиеся к полной реакции, которую катализируют более близкие белки. Тогда естественная классификация должна включать уровни для гомологичных групп с общей частичной функцией и связанными с этой частичной функцией специфическими структурными особенностями (структурным мотивом) (Babbitt 2003). Возможно, таким же образом можно описать и функционирование, включающее в себя связывание небольших молекул, причем подструктуры лиганда будут играть ту же роль, что и частичные функции. Более того, связывание лиганда редко является исчерпывающим описанием функции, и дополнительные элементы структуры могут быть связаны с конформационными превращениями или распознавание других молекул при связывании лиганда.
Трудность в создании такой функциональной классификации, опирающейся на структуру, говорит о необходимости получения более детальной информации о структурах и ферментативных механизмах для определения, какие из частичных функций в процессе эволюции оказались более консервативными, чем другие.
278
Глава 8. Пространственные мотивы
8.7. Заключение
Структурные мотивы - это паттерны локальной структуры белка, которые обычно связаны с его функцией и опираются на остатки в сайтах связывания или каталитических центрах. Со временем в течении эволюции белки могут расходиться, накапливая случайные изменения, которые не затрагивают их функцию, и оставляют неизменными структурные компоненты, критически важные для этой функции. В идеале структурный мотив должен включать в себя ровно эти функционально критические структурные компоненты и служить чувствительным и специфичным индикатором функции. Для выявления структурных мотивов и поиска их в структурах было разработано много методов. Но по сравнению с разработкой алгоритмов меньший успех был достигнут в создании доступных и индексируемых баз данных структурных мотивов, которые были бы одновременно и функционально специфичными, и охватывали широкий спектр функций. Число еще не аннотированных структур может только возрастать, особенно если включить те, что получены сравнительным моделированием (см. Главу 3). Среди трудностей можно назвать создание дескрипторов функции и подробной структурно-функциональной классификации в такой машинно-читаемой форме, которая не приводила бы к потере в точности или смысле, и разработку полуавтоматических и автоматических методов для включения в эти базы данных непрерывного потока новых последовательностей и структур.
Благодарности. Мы благодарим за поддержку NIH GM60595 и NSF DBI-0234768. Молекулярная графика была получена с использованием пакета UCSF Chimera от Ресурса по биовычислениям, визуализации и информатике в Университете Калифорнии, Сан-Франциско (поддерживается NIH Р41 RR-01081). Мы благодарим Жаклин Фетроу и Стэйси Натсон (Университет Вейк Форест) за предоставление рисунка 8.3 в качестве примера результата из программы анализа мотивов FFF/DASP/PASSS
Литература
Artymiuk PJ, Poirrette AR, Grindley HM, et al. (1994) A graph-theoretic approach to the identification of three-dimensional patterns of amino acid side chains in protein structures. J Mol Biol 243:327-344
Ashbumer M, Ball CA, Blake JA, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25-29
Ausiello G, Via A, Helmer-Citterich M (2005a) Query3d: a new method for high-throughput analysis of functional residues in protein structures. BMC Bioinformatics 6(Suppl 4):S5
Ausiello G, Zanzoni A, Peluso D, et al. (2005b) pdbFun: mass selection and fast comparison of annotated PDB residues. Nucleic Acids Res 33:W133-137
Литература
279
Kusiello G, Peluso D, Via A, et al. (2007) Local comparison of protein structures highlights cases of convergent evolution in analogous functional sites. BMC Bioinformatics 8(Suppl 1):S24
Kusiello G, Gheraidini PF, Marcatili P, et al. (2008) FunClust: a web server for the identification of structural motifs in a set of non-homologous protein structures. BMC Bioinformatics 9(Suppl 2):S2
Babbitt PC (2003) Definitions of enzyme function for the structural genomics era. Curr Opin Chem Biol 7:230-237
Babbitt PC, Gerlt JA (1997) Understanding enzyme superfamilies. Chemistry as the fundamental determinant in the evolution of new catalytic activities. J Biol Chem 272:30591-30594
Babbitt PC, Gerlt JA (2000) New functions from old scaffolds: how nature reengineers enzymes for new functions. Adv Protein Chem 55:1-28
Bagley SC, Altman RB (1995) Characterizing the microenvironment surrounding protein sites. Protein Sci 4:622-635
Barker J A, Thornton JM (2003) An algorithm for constraint-based structural template matching: application to 3D templates with statistical analysis. Bioinformatics 19:1644—1649
Bartlett GJ, Porter CT, Borkakoti N, et al. (2002) Analysis of catalytic residues in enzyme active sites. J Mol Biol 324:105-121
Bartlett GJ, Borkakoti N, Thornton JM (2003) Catalysing new reactions during evolution: economy of residues and mechanism. J Mol Biol 331:829-860
Berman HM, Westbrook J, Feng Z, et al. (2000) The Protein Data Bank. Nucleic Acids Res 28:235-242
Blow DM, Birktoft JJ, Hartley BS (1969) Role of a buried acid group in the mechanism of action of chymotrypsin. Nature 221:337-340
Bradley P, Kim PS, Berger В (2002) TRILOGY: Discovery of sequence-structure patterns across diverse proteins. Proc Natl Acad Sci USA 99:8500-8505
Brakoulias A, Jackson RM (2004) Towards a structural classification of phosphate binding sites in protein-nucleotide complexes: an automated all-against-all structural comparison using geometric matching. Proteins 56:250-260
Cammer SA, Hoffman ВТ, Speir JA, et al. (2003) Structure-based active site profiles for genome analysis and functional family subclassification. J Mol Biol 334:387 401
Chang DT, Weng YZ, Lin JH, et al. (2006) Protemot: prediction of protein binding sites with automatically extracted geometrical templates. Nucleic Acids Res 34:W303-309
Chen BY, Fofanov VY, Kristensen DM, et al. (2005) Algorithms for structural comparison and statistical analysis of 3D protein motifs. Рас Symp Biocomput 10:334-345
Chen BY, Fofanov VY, Bryant DH, et al. (2007a) The MASH pipeline for protein function prediction and an algorithm for the geometric refinement of 3D motifs. J Comput Biol 14:791-816
Chen BY, Biyant DH, Cruess AE, et al. (2007b) Composite motifs integrating multiple protein structures increase sensitivity for function prediction. Comput Syst Bioinformatics Conf 6: 343-355
Chothia C, Lesk AM (1986) The relation between the divergence of sequence and structure in proteins. EMBO J 5:823-826
Chothia C, Gough J, Vogel C, et al. (2003) Evolution of the protein repertoire. Science 300:1701-1703
Devos D, Valencia A (2001) Intrinsic errors in genome annotation. Trends Genet 17:429-431
Di Gennaro J A, Siew N, Hoffman ВТ, et al. (2001) Enhanced functional annotation of protein sequences via the use of structural descriptors. J Struct Biol 134:232-245
Favia AD, Nobeli I, Glaser F, et al. (2008) Molecular docking for substrate identification: the shortchain dehydrogenases/reductases. J Mol Biol 375:855-874
Fetrow JS, Skolnick J (1998) Method for prediction of protein function from sequence using the se-quence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J Mol Biol 281:949-968
Fischer D, Wolfson H, Lin SL, et al. (1994) Three-dimensional, sequence order-independent structural comparison of a serine protease against the crystallographic database reveals active site similarities: potential implications to evolution and to protein folding. Protein Sci 3:769-778
Glazer DS, Radmer RJ, Altman RB (2008) Combining molecular dynamics and machine learning to improve protein function recognition. Рас Symp Biocomput 2008:332-343.
Goyal K, Mande SC (2008) Exploiting 3D structural templates for detection of metal-binding sites in protein structures. Proteins 70:1206-1218
Goyal K, Mohanty D, Mande SC (2007) PAR-3D: a server to predict protein active site residues. Nucleic Acids Res 35:W503-505
280
Глава 8. Пространственные мотивы
Hermann JC, Ghanem Е, Li Y, et al. (2006) Predicting substrates by docking high-energy intermediates to enzyme structures. J Am Chem Soc 128:15882-15891
Hermann JC, Marti-Arbona R, Fedorov AA, et al. (2007) Structure-based activity prediction for an enzyme of unknown function. Nature 448:775-779
International Union of Biochemistry and Molecular Biology: Nomenclature Committee, Webb EC (1992) Enzyme nomenclature 1992: recommendations of the Nomenclature Committee of the
International Union of Biochemistry and Molecular Biology on the nomenclature and classification of enzymes. Academic, San Diego, CA
Ivanisenko VA, Pintus SS, Grigorovich DA, et al. (2004) PDBSiteScan: a program for searching for active, binding and posttranslational modification sites in the 3D structures of proteins. Nucleic Acids Res 32:W549-554
Ivanisenko VA, Pintus SS, Grigorovich DA, et al. (2005) PDBSite: a database of the 3D structure of protein functional sites. Nucleic Acids Res 33:D183-187
Jambon M, Imberty A, Deleage G, et al. (2003) A new bioinformatic approach to detect common 3D sites in protein structures. Proteins 52:137-145
Jambon M, Andrieu O, Combet C, et al. (2005) The SuMo server: 3D search for protein functional sites. Bioinformatics 21:3929-3930
Kalyanaraman C, Bemacki K, Jacobson MP (2005) Virtual screening against highly charged active sites: identifying substrates of alpha-beta barrel enzymes. Biochemistry 44:2059-2071
Kleywegt GJ (1999) Recognition of spatial motifs in protein structures. J Mol Biol 285:1887-1897
Kobayashi N, Go N (1997) A method to search for similar protein local structures at ligand binding sites and its application to adenine recognition. Eur Biophys J 26:135-144
Kristensen DM, Chen BY, Fofanov VY, et al. (2006) Recurrent use of evolutionary importance for functional annotation of proteins based on local structural similarity. Protein Sci 15:1530-1536
Kuhn D, Weskamp N, Schmitt S, et al. (2006) From the similarity analysis of protein cavities to the functional classification of protein families using cavbase. J Mol Biol 359:1023-1044
Laskowski RA, Watson JD, Thornton JM (2005a) Protein function prediction using local 3D templates. J Mol Biol 351:614-626
Laskowski RA, Watson JD, Thornton JM (2005b) ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res 33:W89-93
Liang MP, Banatao DR, Klein ТЕ, et al. (2003) WebFEATURE: an interactive web tool for identifying and visualizing functional sites on macromolecular structures. Nucleic Acids Res 31:3324—3327
Macchiarulo A, Nobeli I, Thornton JM (2004) Ligand selectivity and competition between enzymes in silico. Nat Biotechnol 22:1039-1045
Meng EC, Polacco BJ, Babbitt PC (2004) Superfamily active site templates. Proteins 55:962-976
Milik M, Szalma S, Olszewski KA (2003) Common Structural Cliques: a tool for protein structure and function analysis. Protein Eng 16:543-552
Mooney SD, Liang MH, DeConde R, et al. (2005) Structural characterization of proteins using residue environments. Proteins 61:741-747
Murzin AG, Brenner SE, Hubbard T, et al. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536-540
Nebel JC (2006) Generation of 3D templates of active sites of proteins with rigid prosthetic groups. Bioinformatics 22:1183-1189
Nebel JC, Herzyk P, Gilbert DR (2007) Automatic generation of 3D motifs for classification of protein binding sites. BMC Bioinformatics 8:321
Oldfield TJ (2002) Data mining the protein data bank: residue interactions. Proteins 49:510-528
Orengo CA, Michie AD, Jones S, et al. (1997) CATH-a hierarchic classification of protein domain structures. Structure 5:1093—1108
Pal D, Eisenberg D (2005) Inference of protein function from protein structure. Structure 13:121-130
Paul N, Kellenberger E, Bret G, et al. (2004) Recovering the true targets of specific ligands by virtual screening of the protein data bank. Proteins 54:671-680
Pennec X, Ayache N (1998) A geometric algorithm to find small but highly similar 3D substructures in proteins. Bioinformatics 14:516-522
Peters B, Moad C, Youn E, et al. (2006) Identification of similar regions of protein structures using integrated sequence and structure analysis tools. BMC Struct Biol 6:4
Литература
281
Pettersen EF, Goddard TD, Huang CC, et al. (2004) UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 25:1605-1612
Polacco В J, Babbitt PC (2006) Automated discovery of 3D motifs for protein function annotation. Bioinformatics 22:723-730
Porter CT, Bartlett GJ, Thornton JM (2004) The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 32:D129-133
Richardson JS (1981) The anatomy and taxonomy of protein structure. Adv Protein Chem 34:167-339
Rost В (1997) Protein structures sustain evolutionary drift. Fold Des 2:S 19-24
Rost В (2002) Enzyme function less conserved than anticipated. J Mol Biol 318:595-608
Russell RB (1998) Detection of protein three-dimensional side chain patterns: new examples of convergent evolution. J Mol Biol 279:1211-1227
Schmitt S, Kuhn D, Klebe G (2002) A new method to detect related function among proteins independent of sequence and fold homology. J Mol Biol 323:387-406
Shulman-Peleg A, Nussinov R, Wolfson HJ (2004) Recognition of functional sites in protein structures. J Mol Biol 339:607-633
Shulman-Peleg A, Nussinov R, Wolfson HJ (2005) SiteEngines: recognition and comparison of binding sites and protein-protein interfaces. Nucleic Acids Res 33:W337-341
Song L, Kalyanaraman C, Fedorov AA, et al. (2007) Prediction and assignment of function for a divergent N-succinyl amino acid racemase. Nat Chem Biol 3:486-491
Spriggs RV, Artymiuk PJ, Willett P (2003) Searching for patterns of amino acids in 3D protein structures. J Chem Inf Comput Sci 43:412-421
Stark A, Russell RB (2003) Annotation in three dimensions. PINTS: Patterns in Non-homologous Tertiary Structures. Nucleic Acids Res 31:3341-3344
Stark A, Sunyaev S, Russell RB (2003) A model for statistical significance of local similarities in structure. J Mol Biol 326:1307-1316
Stark A, Shkumatov A, Russell RB (2004) Finding functional sites in structural genomics proteins. Structure 12:1405-1412
Todd AE, Orengo CA, Thornton JM (2001) Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol 307:1113-1143
Todd AE, Orengo CA, Thornton JM (2002) Plasticity of enzyme active sites. Trends Biochem Sci 27:419-426
Torrance JW, Bartlett GJ, Porter CT, et al. (2005) Using a library of structural templates to recognize catalytic sites and explore their evolution in homologous families. J Mol Biol 347:565-581
Tyagi S, Pleiss J (2006) Biochemical profiling in silico-predicting substrate specificities of large enzyme families. J Biotechnol 124:108-116
Wallace AC, Laskowski RA, Thornton JM (1996) Derivation of 3D coordinate templates for searching structural databases: application to Ser-His-Asp catalytic triads in the serine proteinases and lipases. Protein Sci 5:1001-1013
Wallace AC, Borkakoti N, Thornton JM (1997) TESS: a geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases. Application to enzyme active sites. Protein Sci 6:2308-2323
Wangikar PP, Tendulkar AV, Ramya S, et al. (2003) Functional sites in protein families uncovered via an objective and automated graph theoretic approach. J Mol Biol 326:955-978
Wright CS, Alden RA, Kraut J (1969) Structure of subtilisin BPN’ at 2.5 angstrom resolution. Nature 221:235-242
Xie L, Bourne PE (2007) A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites. BMC Bioinformatics 8(Suppl 4):S9
Xie L, Bourne PE (2008) Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc Natl Acad Sci USA 105:5441 5446
Глава 9
Динамика белков: от структуры к функционированию
Маркус Б. Кубицки, Берт Л. де Грут, Дэниэл Силиджер
Для понимания функционирования белка требуются подробные сведения о динамике белка, т.е. о различных конформационных состояниях, которые может принимать система. Несмотря на значительный прогресс в экспериментальных методах, такие вычислительные методы, как молекулярная динамика (МД), в настоящее время оказываются единственным общепринятым инструментом для получения динамической информации на атомарном уровне и временах от нано- до микросекунд. Даже при современном уровне вычислительных мощностей для улучшения сэмплирования больших белков и их ансамблей необходимы методы получения конформаций (сэмплирования), отличные от МД. В связи с этим использование коллективных координат оказалось многообещающим подходом либо в качестве средства для анализа, либо в качестве новых алгоритмов сэмплирования. В этой главе рассмотрено несколько улучшенных алгоритмов сэмплирования для биомолекулярных вычислений, начиная с расчетов МД. Приводятся многочисленные примеры, показывающие, как рассмотрение динамических свойств белка позволяет пролить свет на его функционирование.
9.1. Молекулярно-динамические расчеты
За последние десятилетия экспериментальные методы достигли значительного успеха в расшифровке пространственной структуры белков -особенно это относится к рентгеновской кристаллографии, спектроскопии
Marcus В. Kubitzki* *, Bert L. de Groot, and Daniel Seeliger Computational Biomolecular Dynamics Group, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, 37077, Goettingen, Germany
*e-mail: mkubitz@gwdg.de
9.1. Молекулярно-динамические расчеты
283
ядерного магнитного резонанса (ЯМР) и криоэлектронной микроскопии. В то же время выход за рамки статического изображения структуры одиночного белка оказался более проблематичным, хотя и появился ряд таких методик, как релаксация ЯМР, флуоресцентная спектроскопия или время-разрешающая рентгеновская кристаллография (Kempf and Loria 2003; Weiss 1999; Moffat 2003; Schotte etal. 2003), которые позволяют получить информацию о внутренней конформационной подвижности белков. Несмотря на это огромное разнообразие, рядовому исследователю не доступны экспериментальные методики с пространственно-временным разрешением в нано- и микро секундном и нанометровом диапазоне, и поэтому информация о конформационном пространстве, доступном белку in vivo, часто остается неполной. Например, детали на пути перехода между различными известными конформациями обычно остаются неизвестными, хотя часто оказываются важны для функционирования белка. В этом случае вычислительные методы предоставляют заманчивую возможность получить информацию о динамике белка с атомарным разрешением в диапазоне от наносекунд до микросекунд. Из всех способов расчета движения белков (Adcock and McCammon 2006) наиболее распространены молекулярно-динамические (МД) методы. Со времени первого сообщения о МД расчете белка более 30 лет назад (McCammon etal. 1977), такие расчеты стали общепринятым инструментом при изучении биомолекул. Как и все вычислительные отрасли науки, область МД расчетов находится в постоянном развитии благодаря все возрастающей производительности вычислительной техники. Это развитие обеспечивается также улучшениями в методологии, которые привели к большому числу алгоритмов для таких разнообразных задач как клеточный транспорт, передача сигнала, аллостерия, клеточное распознавание, молекулярный докинг, моделирование атомной силовой микроскопии и ферментативного катализа.
9.1.1. Принципы и приближения
Несмотря на значительный прогресс в алгоритмах, основная теория, на которую опираются МД расчеты, довольно проста. Для биомолекуляр-ных систем, имеющих N частиц, численное решение временного уравнения Шредингера
ih—'¥(r,t) = H'¥(r,t) dt
для N-частичной волновой функции \|/(r,t) системы невозможно. Поэтому необходимы некоторые приближения, позволяющие моделирование сольватированной биомолекулы на временных масштабах порядка наносекунд.
284
Глава 9. Динамика белков: от структуры к функционированию
Первое из них относится к расположению ядер и электронов: благодаря гораздо меньшей массе и, следовательно, гораздо большей скорости электронов по сравнению с ядрами, электроны часто можно считать мгновенно приспосабливающимися к движению ядер. Таким образом, в приближении Борна-Оппенгеймера нужно рассматривать только движение ядер, влияние на которое электронных степеней свободы описывается поверхностью потенциальной энергии V(r). Второе существенное приближение, используемое в МД, состоит в классическом описании движения ядер с помощью законов Ньютона
d1 г dt
где m. и г, - это масса и положение i-того ядра. Для получения потенциальной энергии V(r) нужно решить уравнение Шредингера для электронных степеней свободы при классическом описании движения ядер. Однако ввиду большого числа участвующих электронов необходимо дальнейшее упрощение, состоящее во введении полуэмпирических силовых полей, которые аппроксимируют V(r) большим числом функционально простых энергетических термов для валентных и невалентных взаимодействий. В общем виде
K(r) - + Vdihedrais + У improper + ^Coul +
= Z + Z -3,o)2 +
bonds angles
+ Z y(l + COS(n(!>-£))+ £ +
dihedrals improper
Эти простые термы часто имеют гармонический вид (например, Vbonds^VangiesjVimproper) или обусловлены физическими законами (например, законом Кулона VCoul и законом Леннарда-Джонса Уи). Термы описываются своей функциональной формой и небольшим числом параметров, таких как атомный радиус в случае ван-дер-ваальсовых взаимодействий. Все параметры определяются либо из ab initio квантово-химических расчетов, либо из сравнения структурных или термодинамических данных с подходящими средними величинами для молекулярно-динамических ансамблей малых молекул. Число энергетических термов, их функциональная форма и индивидуальные параметры могут сильно варьироваться между различ
9.1. Молекулярно-динамические расчеты
285
ными силовыми полями (Brooks etal. 1983; Weiner etal. 1986; Van Gunsteren and Berendsen 1987; Jorgensen et al. 1996).
Исходя из вышеприведенного описания белка как системы точечных масс (с координатами Tj и скоростями Vj), движущихся в классическом потенциальном поле под действием внешних сил Fi? стандартный МД расчет с дискретным шагом по времени At, лежащем в фемтосекундном диапазоне, интегрирует ньютоновские уравнения движения по некоторой численной схеме, например, используя алгоритм с перескоками (leap-frog algorithm (Hockney etal. 1973)):
z Ar z Ar F^t) A
2 2 mi
r. (t + A/) = r (t) + v. (t + ““) Af
Несмотря на взаимодействие с мембранами и другими макромолекулами, основной окружающей средой белков является вода. Для расчета модельной системы, которая приближена к in vivo системе насколько это возможно, белок должен быть сольватирован путем добавления молекул воды и ионов в физиологической концентрации. В расчетной ячейке, заполненной белком и растворителем, могут возникнуть такие артефакты, связанные с границей раздела сред, как испарение, высокое давление из-за поверхностного натяжения и ориентация молекул растворителя на поверхности. Чтобы избежать этих артефактов, часто используют периодические граничные условия. В этом случае в модельной системе исчезают поверхности вообще, что, в свою очередь, может привести к новым артефактам, если молекула оказывается искусственно взаимодействующей со своим изображением из-за, например, дальнодействующих электростатических взаимодействий. Эти присущие периодичности артефакты могут быть сведены к минимуму увеличением размера расчетной ячейки. Различные формы ячеек, например, куб, додекаэдр или усеченный октаэдр позволяют наилучшим образом подобрать ячейку для конкретной формы белка и, таким образом, достичь компромисса между числом молекул растворителя и расстоянием между молекулой белка и её изображением.
Поскольку среда растворителя сильно влияет на структуру и динамику белков, то параметры воды должны быть аккуратно подобраны. Несмотря на появление моделей неявного растворителя, где вода представляется непрерывной средой вместо индивидуальных «явных» молекул (Still et al. 1990; Gosh et al. 1998; Jean-Charles et al. 1991; Luo et al. 2002), в наше время используются и разнообразные явные модели (e.g. Jorgensen et al. 1983). Эти модели различаются числом частиц, используемых для пред
286
Глава 9. Динамика белков: от структуры к функционированию
ставления молекулы воды и размещения статических парциальных зарядов, отражающих полярность и, по сути, в большинстве силовых полей, поляризацию. Поскольку эти заряды в процессе расчета остаются постоянными, эффекты явной поляризации исключаются. В настоящее время есть несколько моделей воды (и силовых полей) с поляризуемостью (см. недавний обзор Warshel et al. (2007)).
При решении ньютоновских уравнений движения общая энергия системы сохраняется, приводя к микроканоническому ансамблю NVE с постоянным числом частиц N, объемом V и энергией Е. Однако реальные биологические системы с доступными для моделирования размерами постоянно обмениваются энергией со своим окружением. Кроме того, имеется постоянное давление Р, обычно равное атмосферному (1 бар). Чтобы учесть эти особенности, были созданы алгоритмы, позволяющие поддерживать постоянные температуру и давление (Anderson 1980; Nose 1984; Berendsen et al. 1984), что приводит к каноническому ансамблю NPT.
9.1.2. Приложения
Молекулярно-динамические расчеты стали стандартной методикой при изучении белков и повседневно применяются при решении широкого круга задач. Конформационная динамика белков, однако, остается трудной задачей для МД расчетов, поскольку функционально значимые конформационные переходы часто совершаются за времена от микросекунд до секунд, что выходит за рамки общедоступных вычислительных алгоритмов и мощностей.
9.1.2.1. Рецепторы ядерного транспорта
Несмотря на потребность в вычислительных ресурсах, МД расчеты были успешно использованы при изучении функционирования белков. В качестве иллюстрации мы несколько глубже обсудим недавнюю работу (Zachariae and Grubmuller 2006), в которой был обнаружен поразительно быстрый конформационный переход экспортина С AS (Cselp в дрожжах) между открытым и закрытым состоянием. Белок CAS/Cselp - это состоящий из 960 аминокислот рецептор ядерного транспорта, который связывает в ядрах импортин-а и белок RanGTP. Гетеротримерный комплекс (Рис. 9.1) может проходить через ядерные поры и диссоциирует с помощью каталитического гидролиза ГТФ в цитоплазме, представляя, таким образом, важную часть нуклеоцитоплазматического траспортного цикла в клетках.
Для функционирования системы импортин-о/CAS необходимо, чтобы после диссоциации комплекса в цитоплазме белок CAS/Cselp претерпевал
9.1. Молекулярно-динамические расчеты
287
Рис. 9.1. (Цветную версию рисунка см. на вклейке.) Гетеротримерный комплекс Cselp (показан голубым), RanGTP (показан желтым) и импортина-а (показан красным). Cselp принимает сверхспиральную конформацию и связывает RanGTP и импортин-а. Комплекс может проникать сквозь ядерные поры и диссоциирует с помощью каталитического гидролиза ГТФ в цитоплазме
Рис. 9.2. (Цветную версию рисунка см. на вклейке.) Нуклеоплазматическая (слева) и цитоплазматическая (справа) формы Cselp. В нуклеоплазматической форме Cselp связан с RanGTP и импортином-а (оба не показаны). После диссоциации в цитоплазме Cselp претерпевает серьезные конформационные изменения и образует кольцевую конформацию, которая закрывает сайт связывания RanGTP и препятствует обратному образованию комплекса. Структуры окрашены по спектру от голубого (N-конец) до красного (С-конец)
сильное конформационное изменение, которое препятствовало бы обратному образованию комплекса. Рентгеновская структура белка Cselp показывает, что его загруженная конформация обладает сверхспиральной структурой, охватывая связанный RanGTP (Рис. 9.2 слева), в то время как цитоплазматическая форма этого белка имеет конформацию замкнутого кольца, что приводит к закрытию сайта связывания RanGTP (Рис. 9.2 справа). Чтобы понять механизм этого конформационного переключения, авторы провели МД расчет белка Cselp, стартовав с его загруженной конформации. Они обнаружили, что, в основном благодаря электростатическим взаимодействиям, структура белка внезапно сворачивается, и относительно быстро, в течение 10 нс, принимает конформацию, близкую к экспериментально наблюдаемой цитоплазматической форме. Расчеты МД для мутантных форм белка с различными значениями электростатического потенциала на поверхности не выявили в них значительных конформационных изменений. Напротив, эти формы сохранили открытую конформацию, что находится в хорошем согласии с экспериментальными наблюдениями (Cook et al. 2005). Этот пример показывает, что функционально значимые конформационные изменения, которые происходят на малых временных
288
Глава 9. Динамика белков: от структуры к функционированию
масштабах могут быть изучены с помощью МД. Однако в этом конкретном случае расчет стартовал не из равновесной конформации, поскольку им-портин-а и RanGTP были удалены, и поэтому, вероятно, не было необходимости в преодолении значительного энергетического барьера для достижения закрытой конформации. Когда же расчет начинается с минимума свободной энергии, как это обычно бывает, достижимые времена расчета часто оказываются слишком малы для преодоления высоких энергетических барьеров и, следовательно, наблюдения функционально значимых конформационных переходов. Это называется проблемой сэмплирования и представляет собой общую проблему всех МД расчетов.
9.1.2.2. Лизоцим
МД моделирование лизоцима из бактериофага Т4 (T4L), фермента, в шесть раз меньшего, чем Cselp, наглядно иллюстрирует проблему сэмплирования для относительно длинных МД траекторий. Лизоцим T4L всесторонне изучался с помощью рентгеновской кристаллографии (Faber and Matthews 1990; Kuroki et al. 1993) и, будучи закристаллизован в различных конформациях, представляет собой один из редких случаев, когда информация о функционально важных состояниях может быть получена с атомарным разрешением напрямую из экспериментальных данных (Zhang etal. 1995; de Groot etal. 1998). Доменное строение этого фермента ярко выражено (Matthews and Remington 1974) и на основании различий между кристаллографическими структурами различных мутантных форм T4L было предположено, что шарнирный характер движения T4L (Рис. 9.3) является внутренне присущей чертой этой молекулы (Dixon et al. 1992).
Более того, предсказывается, что доменные флуктуации являются необходимыми для функционирования фермента, позволяя в случае открытой конформации субстрату входить в активный центр, а продуктам покидать его, в то время как каталитические превращения протекают при закрытой конформации.
Богатство экспериментальных данных предоставляет также возможность оценить надежность вычислительных методов и эффективность сэмплирования. Было выполнено два МД расчета с использованием в качестве стартовых закрытой (расчет 1) и открытой конформации (расчет 2). Для оценки эффективности сэмплирования был проведен анализ главных компонент (principal components analysis, РСА, см. Раздел 9.2 ниже) ансамбля экспериментально определенных структур, и этот ансамбль, а также две МД траектории были спроецированы на два первых собственных вектора.
Первый собственный вектор соответствует шарнирному движению, в то время как второй собственный вектор соответствует скручиванию доменов
9.1. Молекулярно-динамические расчеты
289
Рис. 9.3. Шарнирное движение лизоцима из бактериофага Т4. Доменные флуктуации (домены показаны разным цветом) необходимы для функционирования фермента, позволяя в случае открытой конформации субстрату входить в активный центр, а продуктам покидать его
T4L. Проекции показаны на Рис. 9.4. Ансамбль рентгеновских структур представлен точками, каждая из которых соответствует одной конформации. Движение вдоль первого собственного вектора (ось абсцисс) описывает коллективное движение от закрытого к открытому состоянию. Легко заметить, что ни одна из МД траекторий, показанных линиями, сама по себе не охватывает все конформационное пространство, содержащее в себе ансамбль рентгеновских структур, хотя времена расчета (184 нс для расчета 1 и 117 нс для расчета 2) на порядок больше ранее обсуждавшегося времени МД расчета белка Cselp. Из плотности фазового пространства можно предположить, что существует энергетический барьер между закрытым и открытым состояниями, и ни один из расчетов не позволяет увидеть полного перехода из закрытого состояния в открытое или наоборот.
9.1.2.З. Аквапорины
Аквапорины являются лучшим примером того, какой вклад внесли МД расчеты в понимание функционирования белка с динамической и энергетической точки зрения. Аквапорины способствуют эффективному и селективному проникновению воды через биологические мембраны. Родственные им акваглицеропорины пропускают, кроме того, и небольшие нейтральные молекулы растворителей вроде глицерина. Доступные структуры высокого разрешения дали бесценную возможность взглянуть на молекулярные механизмы, действующие в аквапоринах (Fu et al. 2000; Murata et al. 2000; de Groot et al. 2001; Sui et al. 2001). Однако такие структуры несут в основном статическую информацию, и поэтому мы не можем напрямую наблюдать аквапорины «за работой». К тому же, до сих пор нет экспериментального метода, который давал бы достаточное пространственное и временное разрешение, чтобы отслеживать проникновение через
290 Глава 9. Динамика белков: от структуры к функционированию
-5 0 5
Проекция первого собственного вектора (нм)
Рис. 9.4. Анализ главных компонент для лизоцима из бактериофага Т4. Ансамбль рентгеновских структур показан точками, МД траектории линиями. Движение вдоль первого собственного вектора (ось абсцисс) соответствует шарнирному движению. Ни расчет 1, стартовавший из закрытой конформации, ни расчет 2, стартовавший из открытой, не демонстрируют полного перехода по причине энергетического барьера, который разделяет конформационные состояния
аквапорины на молекулярном уровне. Таким образом, расчеты МД дополняют экспериментальные результаты, показывая движение биомолекуляр-ной системы с атомарным разрешением. Поскольку известно, что проникновение происходит в наносекундном диапазоне, то можно ожидать, что спонтанное проникновение будет иметь место в процессе многонаносе-кундных расчетов, позволяя прямое наблюдение динамики при функционировании. По этой причине такие расчеты были названы «расчетами реального времени» (de Groot and Grubmuller 2001).
В самом деле, случаи спонтанного проникновения наблюдались в МД расчетах аквапорина-1 и акваглицеропорина GlpF. Эти расчеты показали, что эффективность в проникновении молекулы воды объясняется компле-ментарностью водородных связей внутри канала, сравнимой с таковой в объеме воды, что обуславливает низкий энергетический барьер этого события (de Groot and Grubmuller 2001; Tajkhorshid etal. 2002). Расчеты прояснили
9.1. Молекулярно-динамические расчеты
291
Энергия водородных связей в расчете на одну молвсулу воды (ДОмоль)
Рис. 9.5. (Цветную версию рисунка см. на вклейке.) а) Молекулы воды сильно упорядочены в канале аквапорина-1, причем их дипольные моменты направлены от центральной NPA-области (de Groot and GrubmOller 2001). Водные диполи (показаны желтыми стрелками) поворачиваются приблизительно на 180 градусов, проходя по каналу AQP1. Красным и синим показаны локальные электростатические потенциалы, отрицательный и положительный, соответственно, б) Энергия водородных связей в расчете на молекулу воды (показана черными линиями) в AQP1 (слева) и GIpF (справа). Водородные связи белок-вода (показаны зеленым) компенсируют потерю водородных связей вода-вода (показано голубым). Главными центрами взаимодействия белок-вода являются ar/R-область и NPA-область
также, что селективность этих каналов объясняется двухступенчатым фильтром. Первая ступень фильтра локализована в центральной части канала в консервативной аспарагин-пролин-аланиновой (NPA) области; вторая ступень расположена на внеклеточной поверхности канала в ароматическо-аргининовой (ar/R) области сужения (Рис. 9.5) Поскольку проникновение воды происходит в наносекундном масштабе, то коэффициенты проникновения могут быть вычислены непосредственно из МД расчетов и сопоставлены с экспериментальным значением. Количественное согласие, полученное при таком сопоставлении, свидетельствует о достоверности расчетов.
Долгое время в изучении аквапоринов оставался нерешенным вопрос о механизме, по которому протоны выводятся из водной поры. МД расчеты, посвященные проникновению воды, выявили отчетливую картину ориентации водных диполей в канале с центром симметрии в NPA-области (de Groot and Grubmuller 2001). Обнаружилось, что в процессе динамики молекулы воды переворачиваются на 180 градусов на своем пути по каналу (Рис. 9.5а). В ряде расчетов, посвященных выяснению механизма выведения протона, было обнаружено, что такая ориентация молекул воды вызвана электрическим полем в канале с центром симметрии в NPA-области (de
292
Глава 9. Динамика белков: от структуры к функционированию
Groot et al. 2003; Chakrabarti et al. 2004; Ilan et al. 2004). Таким образом, основу для выведения протона создают электростатические эффекты. Об источнике этого электростатического барьера продолжаются споры, причем были предложены как прямые электростатические эффекты, обусловленные дипольными моментами спиралей (de Groot etal. 2003; Chakrabarti etal. 2004), так и специфические эффекты десольватации (Burykin and Warshel 2003). Последние результаты позволяют предположить, что и те, и другие эффекты вносят примерно одинаковый вклад (Chen et al. 2006).
Также МД расчеты позволили прояснить механизм селективности ак-вапоринов и акваглицеропоринов к нейтральным растворителям. Было обнаружено, что аквапорины проницаемы только для небольших полярных молекул, подобных воде, за редким исключением в виде иона аммония, в то время как акваглицеропорины проницаемы также для неполярных молекул, подобных СО2, и более крупных молекул, подобных глицерину, но не для мочевины (Hub and de Groot 2008). Для аквапоринов обнаружилось обратное соотношение между проникающей способностью растворенного вещества и его гидрофобностью - молекулы этих веществ, конкурирующие с проникающими молекулами воды за образование водородных связей с каналом, обуславливают барьер проникновения. Таким образом, в основе селективности аквапоринов и акваглицеропоринов лежит отбор по размеру и гидрофобности.
9.1.3. Ограничения и улучшенные алгоритмы сэмплирования
Несмотря на то, что молекулярно-динамические расчеты стали цельной частью структурной биологии и неоднократно представляли бесценную возможность взглянуть на биологические процессы на атомарном уровне, в этой области остаются ограничения как методологического характера, так и связанные с вычислительными ресурсами. Методологические ограничения возникают из классического описания атомов и приближения взаимодействия простыми энергетическими термами вместо уравнение Шредингера. Это означает, что химические реакции (разрыв и образование химических связей) не могут быть описаны. Поляризационные эффекты и туннелирование протона также оказываются вне области классических МД расчетов.
Вторая группа ограничений возникает из вычислительных потребностей МД расчетов. Несмотря на то, что связи обычно рассматриваются в качестве пространственных ограничений, налагаемых на атомы, что позволяет исключить наиболее высокочастотные движения, длина шага по
9.1. Молекулярно-динамические расчеты
293
времени в МД расчетах, как правило, не может быть выбрана более 2 фс. Наносекундный расчет, следовательно, требует 500 000 кратного вычисления сил и такого же количества шагов интегрирования. При нынешних алгоритмах и вычислительной мощности времена порядка 100 нс оказываются достижимыми после 3-4 недель расчетов для сольватированного белка, имеющего 200 остатков.
Однако биологически важное движение белков, такое как масштабные конформационные переходы, сворачивание или денатурация происходят за времена от микро- до миллисекунд. Таким образом, становится очевидным, что несмотря на все возрастающую производительность вычислительной техники, чей рост можно охарактеризовать как примерно стократный за 10 лет, МД расчеты в обозримом будущем не смогут решить проблему сэмплирования исключительно за счет быстродействия компьютеров. Поэтому специально для решения проблемы сэмплирования конформаций и предсказания функционально значимых движений белков были предложены альтернативные методы, отчасти основанные на МД.
Один из подходов состоит в уменьшении числа частиц. Поскольку белки, как правило, рассматриваются в окружении растворителя, и большая часть моделируемой системы представлена молекулами воды, то создание неявных моделей растворителя является многообещающим способом уменьшения ресурсоемкое™ вычислений (Still etal. 1990; Gosh etal. 1998; Jean-Charles etal. 1991; Luo etal. 2002). Другим способом уменьшения числа частиц является использование так называемых крупнозернистых (coarse-grained) моделей (Bond et al. 2007), в которых атомы объединены в группы, называемые псевдоатомами (зернами). Например, обычно четыре молекулы воды рассматриваются как один псевдоатом. Такое объединение приводит к двум эффектам: во-первых, сокращается число частиц, а во-вторых, может быть увеличен шаг по времени, который зависит от частоты наиболее быстрых колебаний в системе. Но крупнозернистое представление не ограничивается молекулами воды. Например, аминокислоты могут быть также представлены одним или несколькими зернами. Это позволяет резко снизить требования к ресурсам, делая возможным расчет крупных макромолекулярных агрегатов на временах до микро- и миллисекунд. Этот выигрыш в эффективности, однако, невольно сопровождается проигрышем в точности по сравнению с полноатомным описанием белков, что позволяет получать лишь полуколичественныс результаты. Принципиально важным для успешности крупнозернистых моделей является параметризация силовых полей, которые длжны быть одновременно и точными, и универсальными, то есть подходить для описания систем различного состава и конфигурации. Чем более крупными являют
294
Глава 9. Динамика белков: от структуры к функционированию
ся зерна, тем сложнее процесс параметризации, поскольку более специфические взаимодействия должны быть эффективно учтены в небольшом числе параметров и термов. Это привело к множеству моделей белков, липидов и воды, являющихся различными компромиссами между точностью и универсальностью (см., например, Marrink et al. 2004).
Другие улучшенные методы сэмплирования, основанные на МД расчетах и сохраняющие атомарное представление структуры, включают в себя метод обмена репликами (replica exchange molecular dynamics, REMD) и коллективную динамику (essential dynamics, ED), которые обсуждаются в следующих разделах. Кроме того, обсуждается и ряд не имеющих отношения к МД методов, направленных на предсказание того, каким образом функционируют белки.
9.1.З.1. Метод обмена репликами
Целью большинства работ по компьютерному моделированию био-молекулярных систем является расчет макроскопического поведения исходя из микроскопических взаимодействий. Согласно равновесной статистической механике, любая наблюдаемая величина, которая может быть связана с макроскопическим экспериментом, определяется как среднее по ансамблю всех возможных состояний системы. Однако из-за ограниченных возможностей вычислительного оборудования полностью сошедшееся сэмплирование всех возможных конформационных состояний с соответствующими статистическими весами по Больцману достижимо только для простых систем, содержащих небольшое число аминокислот (см., например, Kubitzki and de Groot 2007). Для белков же, состоящих из сотен и тысяч аминокислот, традиционные МД расчеты зачастую не обладают сходимостью, и по этой причине не может быть проведена надежная оценка экспериментальных величин.
Неэффективность сэмплирования является результатом холмистой поверхности свободной энергии системы - понятия, введенного Фрауэн-фельдером (Frauenfelder et al. 1991; Frauenfelder and Leeson 1998). Предполагается, что в целом поверхность является воронкообразной, причем нативные состояния системы заселяют глобальный минимум свободной энергии (Anfinsen 1973).
При более пристальном взгляде видно, что сложная многомерная поверхность свободной энергии характеризуется множеством локальных минимумов с почти одинаковой энергией, которые разделены барьерами различной высоты. Каждый из этих минимумов соответствует одному конкретному конформационному подсостоянию, причем соседние минимумы соответствуют схожим конформациям. В терминах этого наглядного пред
9.1. Молекулярно-динамические расчеты
295
ставления структурные переходы являются преодолением барьеров, причем скорость перехода зависит от высоты барьера. Для МД расчетов при комнатной температуре возможно легкое преодоление только тех барьеров, которые меньше или сравнимы с тепловой энергией квТ, что соответствует лишь небольшим внешним структурным изменениям, например переупо-рядочиванию боковых цепей. По этой причине система будет проводить большую часть времени в локально стабильных состояниях (кинетический захват, kinetic trapping) вместо перемещения по различным конформационным состояниям. Такое перемещение представляет огромный интерес, поскольку связано с биологической функцией, но требует от системы способности преодолевать высокие энергетические барьеры. К сожалению, поскольку МД расчеты в основном ограничены наносекундным диапазоном, функционально значимые конформационные переходы наблюдаются редко.
Чтобы попытаться найти решение этой проблемы многих минимумов было предложено множество улучшенных методов сэмплирования (см., например, Van Gunsteren and Berendsen 1990; Tai 2004; Adcock and McCammon 2006 и ссылки в них), среди которых можно упомянуть алгоритмы обобщенного ансамбля, широко используемые в последние годы (см. например, обзоры Mitsutake et al. 2001; Iba 2001). Эти алгоритмы сэмплируют искусственный ансамбль, который создается путем комбинаций или вариаций исходного ансамбля. Алгоритмы второй категории (например, Berg and Neuhaus 1991) в основном изменяют исходное колоколообразное распределение импульсов p(V) в системе, вводя так называемый мультиканониче-ский весовой фактор w(V), так что итоговое распределение оказывается постоянным p(V)w(V) = const. Затем это плоское распределение может быть широко сэмплировано методами МД и Монте-Карло, поскольку барьеров потенциальной энергии больше нет. Из-за введенных модификаций оценки средних значений физических величин по каноническому ансамблю должны быть получены с помощью перенормировки (Kumar etal. 1992; Chodera et al. 2007). Основной недостаток этих алгоритмов, однако, состоит в нетривиальном определении различных мультиканонических весовых факторов с помощью итеративного процесса, использующего короткие пробные расчеты. Для сложных систем эта процедура может оказаться очень громоздкой, в связи с чем были предприняты попытки улучшить сходимость итеративного процесса (Berg and Celik 1992; Kumar etal. 1996; Smith and Bruce 1996; Hansmann 1997; Bartels and Karplus 1998).
Метод обмена репликами (replica exchange (REX) algorithm), разрабо тайный как расширение метода имитации отпуска (simulated tvinpci iiih> (Marinari and Parisi 1992), устраняет проблему нахождения м>ррсмпмх m совых множителей. Он относится к первой катеюрии а л ориiмои »
296
Глава 9. Динамика белков: от структуры к функционированию
рых сэмплируется обобщенный ансамбль, построенный на основе нескольких копий исходного ансамбля. Благодаря простоте и легкости в реализации, этот алгоритм широко использовался в последнее время. Чаще всего используется формулировка алгоритма REX при стандартной температуре (Sugita and Okamoto 1999), причем структура общего гамильтониана в этом алгоритме привлекает все большее внимание (Fukunishi et al. 2002; Liu et al. 2005; Sugita et al. 2000; Affentranger et al. 2006; Christen and van Gunsteren 2006; Lyman and Zuckerman 2006).
В МД расчетах по методу обмена репликами при стандартной температуре (Sugita and Okamoto 1999), обобщенный ансамбль создается из М + 1 невзаимодействующей копии, или реплики; системы в диапазоне температур {Т0,...,Тм} (Тт <Тю+1;т = 0,...,М), что может быть сделано, например, распределением вычислений по М + 1 узлу на компьютере с параллельной архитектурой (Рис. 9.6 слева). Состояние этого обобщенного ансамбля описывается набором состояний S = {...,sm,...}, где sm описывает состояние реплики гл, имеющей температуру Тт. Теперь алгоритм состоит из двух последовательных шагов: (а) независимый расчет каждой реплики при постоянной температуре, и (Ь) обмен репликами S= {...,sm,...,sn,...} —► —► S'= {...,sns™...} согласно критерию, подобному критерию Метро-полиса. Вероятность принятия обмена задается выражением
Р(5^5’)=т1п{1,ехр{(Д-Д)[Гт-Г„]}} (9.1)
где Vm является потенциальной энергией, а рт 1 = квТт. Чередуя шаги (а) и (Ь) траектория обобщенного ансамбля блуждает в пространстве температур, что в свою очередь приводит к блужданию в пространстве энергий. Это облегчает эффективное и статистически обоснованное конформационное сэмплирование на энергетической поверхности системы даже при наличии большого количества локальных минимумов.
Выбор температуры сильно влияет на производительность алгоритма. Температуры реплик должны быть выбраны так, чтобы а) низшая температура была достаточно малой для эффективного сэмплирования низкоэнергетических состояний, б) высшая темература была достаточно большой для преодоления энергетических барьеров в рассматривамой системе и в) вероятность принятия P(S—>S’) была досточно высока, что требует подходящего перекрывания распределений потенциальной энергии для соседних реплик. Для больших систем с явно заданным растворителем это последнее условие является наиболее затруднительным. Простая оценка (Cheng et al. 2005; Fukunishi et al. 2002) показывает, что наибольший вклад в разность свободных энергий AV ~ NdfAT вносит растворитель, число степеней свободы которого Ndf 801 составляет большую долю общего числа
9.2. Анализ главных компонент
297
Рис. 9.6. Схематическое сравнение алгоритма REX при стандартной температуре (слева) и алгоритма TEE-REX (справа) для случая расчета с тремя репликами. Температуры расположены по возрастанию, Ti +1 > Т. Попытки обмена (<-►) предпринимаются (...) с частотой vex. В отличие от REX, в методе TEE-REX возбуждению подвергается только коллективное подпространство {es} (essential subspace) (серые квадраты), содержащее несколько коллективных мод в каждой реплике. Референсная реплика (То, То), содержащая приблизительно больцмановский ансамбль, используется для анализа
степеней свободы системы Ndf. Таким образом, получения разумной вероятности принятия можно достичь лишь сохраняя температурные интервалы AT = Tm+i-Tm малыми (обычно несколько градусов), что резко увеличивает вычислительные требования для систем, имеющих несколько тысяч атомов и более. Несмотря на это жесткое ограничение, алгоритмы REX стали общепризнанным методом для изучения сворачивания и денатурации пептидов (Zhou etal. 2001; Rao and Caflisch 2003; Garcia and Onuchic 2003; Pitera and Swope 2003; Seibert etal. 2005), предсказания структуры (Fukunishi et al. 2002; Kokubo and Okamoto 2004), фазовых переходов (Berg and Neuhaus 1991) и вычисления свободной энергии (Sugita et al. 2000; Lou and Cukier 2006).
Для решения задачи выяснения функционирования белка успешно применяется и другой класс методов улучшенного сэмплирования, который выходит за рамки традиционной МД. Эти алгоритмы используют то наблюдение, что флуктуации в белках в общем случае скоррелированы. Выделение таких коллективных мод движения и их применение к новым алгоритмам сэмплирования будет предметом рассмотрения в двух следующих разделах.
9.2. Анализ главных компонент
Анализ главных компонент (Principal component analysis, РСА) является хорошо разработанной методикой для получения низкоразмерных описаний высокоразмерных данных. Его приложения включают сжатие данных, обработку изображений, визулизацию данных, научный анализ данных, распо
298
Глава 9. Динамика белков: от структуры к функционированию
знавание образов и предсказание временных рядов (Duda et al. 2001). В контексте биомолекулярных расчетов РСА стал важным инструментом в извлечении и классификации значимой информации о крупных конформационных изменениях в ансамблях белковых структур, полученных экспериментально или теоретически (Garcia 1992; Go- etal. 1983; Amadei etal. 1993). Кроме РСА в настоящее время используется и ряд других сходных методов, среди которых стоит упомянуть анализ нормальных мод (NMA) (Brooks and Karplus 1983; Go etal. 1983; Levitt etal. 1983), квазигармонический анализ (Karplus and Kushick 1981; Levy etal. 1984a, b; Teeter and Case 1990) и сингулярное разложение (Romo et al. 1995; Bahar et al. 1997).
Анализ главных компонент основан на наблюдении, что подавляющая часть пространственных флуктуаций в белках происходит вдоль небольшого числа осей, связанный с коллективными степенями свободы. Это было впервые осознано при анализе нормальных колебаний небольших белков (Brooks and Karplus 1983; Go- etal. 1983; Levitt etal. 1983). При таком анализе (см. раздел 9.4.1) поверхность потенциальной энергии полагается гармонической и коллективные переменные определяются диагонализацией гессиана1 в локальном минимуме энергии. Квазигармонический анализ, анализ главных компонент и сингулярное разложение молекулярно-динамических траекторий, которые не предполагают гармоничности колебаний, показали, что и в самом деле среди колебаний потенциальной энергии в прочессе динамики преобладает ограниченное число коллективных координат, причем основные моды зачастую оказываются сильно ангармоничны-ми. Эти методы позволили выявить те коллективные степени свободы, которые наилучшим образом аппроксимируют все наблюдаемые колебания.
Наиболее сильно изменяющиеся переменные образуют набор обобщенных внутренних координат, которые можно использовать для эффективного описания динамики белка. Часто использование 5-10% от общего числа степеней свободы дает удивительно точное приближение. В отличие от внутренних координат в виде торсионных углов, эти коллективные внутренние координаты не известны заранее, а должны быть определены с использованием либо экспериментальных структур, либо ансамбля модельных структур. Как только эти коллективные степени свободы найдены, эта информация может быть использована для анализа расчетов, а также для создания улучшенных протоколов динамики с целью улучшения конформационного сэмплирования (Grubmuller 1995; Zhang etal. 2003; Не et al. 2003; Amadei et al. 1996).
1 Матрица вторых производных потенциальной энергии д2У / дхдху . Прим, автора.
9.2. Анализ главных компонент
299
Рис. 9.7. Иллюстрация метода главных компонент (РСА) для двумерного случая. Для определения положения точки из ансамбля нужны две координаты (х, у) (б), в то время как для приблизительного определения положения достаточно одной координаты х' (а)
В сущности, метод главных компонент - это многоразмерный метод наименьших квадратов в пространстве конфигураций. Ансамбль структур молекулы, имеющей N атомов, может быть представлен в 314-мерном пространстве конфигураций, как облако точек, где каждая конфигурация представлена одной точкой. Для такого облака всегда может быть определена ось, вдоль которой наблюдается максимальный разброс точек. Как показано для двумерного случая (Рис. 9.7), если такая линия хорошо соответствует данным, то положение каждой точки может быть аппроксимировано одной лишь проекцией на эту ось, что дает разумную аппроксимацию даже при отбрасывании проекций на остальные направления, перпендикулярные этой оси. Если выбрать эту ось в качестве координатной, то положение точки может быть описано одной координатой. В общем 3N-мерном случае процедура выполняется аналогично.
Зная ось, которая в первом приближении описывает данные наилучшим образом, можно выбрать ортогональные направления для второго приближения, третьего приближения и так далее {главные компоненты). Все вместе эти направления охватывают 31Ч-мерное пространство. Математически эти направления задаются собственными векторами ковариационной матрицы флуктуаций атомов
с=^(х(0 - - {х))Т,
где угловые скобки <•> означают усреднение по ансамблю. Собственные числа Xj соответствуют средним квадратам пространственных флуктуаций вдоль соответствующих собственных векторов и, таким образом, показывают вклад каждого главного компонента в общую флуктуацию (Рис. 9.8)
300
Глава 9. Динамика белков: от структуры к функционированию
Номер собственного числа
Номер собственного числа
Рис. 9.8. Типичный спектр собственных значений в методе главных компонент (МД ансамбль структур основной цепи гуанилина). Первые пять собственных векторов (панель а) покрывают 80% всех наблюдаемых флуктуаций (панель б)
Применение таких процедур многоразмерной подгонки к конфигурациям белков из МД расчетов показало на нескольких примерах, что обычно первые 10-20 главных компонент ответственны за 90% флуктуаций белка (Kitao etal. 1991; Garcia 1992; Amadei etal. 1993). Эти главные компоненты соответствуют коллективным координатам, содержащим вклад от каждого атома белка. В ряде случаев было показано, что эти главные моды являются частью функциональной динамики изучаемых белков (Amadei et al. 1993; Van Aalten et al. 1995a, b; de Groot et al. 1998). По этой причине подпространство, отвечающее за большую часть флуктуаций, было названо коллективным подпространством (essential subspace) (Amadei et al. 1993).
Тот факт, что небольшая часть от общего числа степеней свободы (основное подпространство) доминирует в молекулярной динамике белков, имеет место из-за большого количества внутренних ограничений, которые определяются взаимодействием атомов в биомолекуле. Такими взаимодействиями являются как сильные ковалентные связи, так и слабые невалентные взаимодействия, в то время как ограничения задаются плотной упаковкой атомов в нативной структуре.
В целом, динамика белков при физиологических температурах была описана как диффузия среди множества минимумов (Kitao etal. 1998; Amadei etal. 1999; Kitao and Go- 1999). В динамике на малых временах преобладают колебания возле локального минимума, соответствующие собственным векторам с низкими собственными значениями. В больших колебаниях на больших временах преобладает ангармоническая диффузия между многочисленными потенциальными ямами. Такие медленные динамические переходы в методе главных компонент обычно представлены модами с большими амплитудами. В отличие от анализа нормальных мод, примене
9.3. Алгоритмы сэмплирования коллективных координат
301
ние метода главных компонент к МД траектории не основывается на предположении о гармоническом потенциале. В самом деле, метод главных компонент может быть использован для изучения степени ангармоничности в молекулярной динамике моделируемой системы. Было показано, что для белков при физиологической температуре среди главных мод коллективных флуктуаций, зачастую функционально значимых, преобладают ангармонические флуктуации (Amadei et al. 1993; Hayward et al. 1995).
9.3. Алгоритмы сэмплирования коллективных координат
Анализ МД расчетов в терминах коллективных координат (получаемых, например, по методу главных компонент или при анализе нормальных мод) показывает, что только некоторые из общего числа степеней свободы преобладают в молекулярной динамике биомолекул. Поскольку функция белка во многих случаях может быть связанной с этими модами основного подпространства, динамика в этом низкоразмерном пространстве была названа «коллективной динамикой» (essential dynamics, ED) (КД). Она не только помогает в анализе и интерпретации МД траекторий, но также открывает пути к улучшенным алгоритмам сэмплирования, которые проводят поиск в основном подпространстве либо систематическим, либо зондирующим образом (Grubmuller 1995; Amadei et al. 1996).
9.3.1. Коллективная динамика
Первые попытки в этом направлении имели своей целью создание вычислительной схемы, в которой уравнения движения интегрировались бы исключительно вдоль выбранных первичных главных мод, резко уменьшая таким образом число степеней свободы (Amadei etal. 1993). Однако эти попытки оказались затруднительными из-за нетривиальных связей между высоко- и низкоамплитудными модами, несмотря даже на то, что после диагонализации моды стали линейно независимыми (ортогональными). Поэтому вместо них предпочтение было отдано ряду методик, в которых принимается во внимание полноразмерная расчетная система и улучшается движение вдоль выбранных главных мод. Самыми распространенными из этих методик являются конформационное затопление (conformational flooding) (Grubmuller 1995) и КД сэмплирование (Amadei etal. 1996; de Groot etal. 1996a, b). В конформационном затоплении для выбранных главных компонент в расчет потенциальной энергии вводится
302
Глава 9. Динамика белков: от структуры к функционированию
дополнительный член, который толкает моделируемую систему к перемещению в новые области фазового пространства, в то время как в КД сэмплировании аналогичная цель достигается геометрическими ограничениями для выбранных главных мод. Используя эти методики, можно получить на порядок более эффективное сэмплирование при условии что из традиционного расчета было получено разумное приближение главных мод. Однако из-за используемого пространственного или энергетического искажения системы, ансамбли, получаемые при ОД сэмплировании и конформационном затоплении, не являются каноническими, что ограничивает анализ только структурными вопросами.
9.3.2. TEE-REX
Улучшенные методы сэмплирования, такие как ОД (Amadei et al. 1996) достигают своей эффективности (Amadei etal. 1996; de Groot etal. 1996a, b) в основном благодаря тому, что в конфигурационной динамике белков преобладает лишь небольшое число внутренних коллективных степеней свободы. К тому же, системы, рассчитываемые с помощью таких методов, всегда находятся в неравновесном состоянии, что затрудняет расчет термодинамических, т.е. равновесных, свойств этих систем. С другой стороны, алгоритмы обобщенных ансамблей, такие как REX, не только улучшают сэмплирование, но и дают правильные статистические ансамбли, необходимые для расчета равновесных свойств, которые могут быть подвергнуты экспериментальной проверке. Однако REX быстро становится вычислительно невозможным для систем, имеющих больше, чем несколько тысяч частиц, что ограничивает его нынешнее применение расчетом небольших пептидов (Pitera and Swope 2003; Cecchini etal. 2004; Nguyen et al. 2005; Liu et al. 2005; Seibert et al. 2005). Новый алгоритм обмена репликами в коллективной динамике с улучшенной температурой TEE-REX (Temperature Enhanced Essential dynamics Replica Exchange) (Kubitzki and de Groot 2007) совмещает в себе привлекательные свойства REX со свойствами, возникающими из индивидуального возбуждения функционально значимых мод, не имея при этом недостатков обоих подходов.
На рисунке 9.6 показано схематическое сравнение алгоритма REX при стандартной температуре (слева) и алгоритма TEE-REX (справа). Алгоритм TEE-REX построен по той же схеме, что и REX: параллельно и независимо рассчитывается ряд реплик системы и периодически делаются попытки обмена между соседними репликами. Но в отличии от REX, во всех репликах, кроме референсной, термически возбуждаются только те степени свободы, которые дают значительный вклад в общие флуктуации
9.3. Алгоритмы сэмплирования коллективных координат
303
Рис. 9.9. Сравнение двумерных поверхностей свободной энергии (в кДж/моль) диаланина, вычисленная с помощью МД (панель а) и TEE-REX (панель б). Отклонение AGtee-rex - Gmd соизмеримо со статистической ошибкой, составляющей -0.1 квТ
системы (коллективное подпространство {es}). Это позволяет объединить несколько преимуществ при устранении недостатков. В отличие от стандартного алгоритма REX, индивидуальное возбуждение коллективных координат способствует сэмплированию по этим функционально важным модам движения, т.е. используется преимущество КД. Для компенсации недостатков, связанных с таким индивидуальным возбуждением, т.е. искажением ансамбля, схема встроена в протокол REX. В связи с этим получаемые ансамбли имеют приблизительно больцмановское распределение и используются улучшенные сэмплирующие свойства REX. Вероятность обмена (уравнение 9.1) между двумя репликами критически зависит от числа возбужденных степеней свободы системы. Поскольку такие степени свободы
304
Глава 9. Динамика белков: от структуры к функционированию
составляют лишь незначительную часть от общего числа степеней свободы системы, то обходится узкое место, которым являются низкие вероятности обмена в полноатомных расчетах по алгоритму REX. Таким образом, при заданных вероятностях обмена может быть использована настолько большая разница в температурах АТ, что потребуется лишь несколько реплик.
На рисунке 9.9 показана двумерная проекция ландшафта свободной энергии диаланина, вычисленная с помощью МД (панель А) и TEE-REX (панель В). Термодинамическое поведение системы можно считать полностью известным, если известен какой-нибудь термодинамический потенциал, такой как относительная свободная энергия Гиббса AG. Сравнение свободных энергий позволяет нам, таким образом, решить, до какой степени совпадают ансамбли, созданные разными вычислительными методами. При этом абсолютно необходимо, чтобы ансамбли имели идентичный состав. Для тестового случая диаланина это требование выполняется. Детальный анализ формы поверхностей свободной энергии, полученных из МД и TEE-REX, показывает, что максимальное абсолютное отклонение составляет 1.5 kJ/mol ~ 0.6 квТ от идеального случая AGTee-rex _ GMd= 0, что соизмеримо с максимальной статистической ошибкой, составляющей 0.15 квТ для каждого из методов. Небольшие отклонения, обнаруженные в ансамбле TEE-REX, вызваны, вероятно обменом неравновесных структур в референском ансамбле.
Для гуанилина, небольшого пептидного гормона, состоящего из 13 аминокислот, была оценена эффективность сэмплирования алгоритмом TEE-REX по сравнению с МД (Currie etal. 1992). Траектории, рассчитанные обоими методами при одинаковых затратах вычислительных ресурсов, были спроецированы на пространство (ср, у), а также на различные двумерные подпространства, которые задаются главными компонентами, вычисленными по МД ансамблю структур гуанилина. По этим проекциям было измерена временная эволюция сэмплированного объема конфигурационного пространства. В итоге оказалось, что производительность сэмплирования у МД довольно ограничена по сравнению с TEE-REX, который по этому показателю превосходит МД в среднем в 2,5 раза в зависимости от подпространства, использованного для проецирования.
9.3.2.1. Приложения: поиск переходного пути в аденилаткиназе
Понимание функциональных основ функционирования многих белков (Gerstein etal. 1994; Berg etal. 2002; Karplus and Gao 2004; Xu etal. 1997) требует подробного знания переходов между функционально значимыми конформациями. В последние годы рентгеновская кристаллография
9.3. Алгоритмы сэмплирования коллективных координат
305
и спектроскопия ЯМР предоставляли в основном статические картины различных конформационных состояний белков, оставляя без ответа вопросы, касающиеся переходов между этими состояниями. Для расчетов МД атомарного разрешения прояснение путей и механизмов конформационной динамики белков представляет собой нетривиальную задачу из-за больших характерных времен. В этом отношении хорошим примером является аденилаткиназа из Е. coli. Это мономерный фермент, играющий ключевую роль в энергетическом балансе клетки, поскольку контролирует уровень АТФ в клетке, катализируя реакцию (Mg2+:AT<D) + АМФ <-► <-► (Mg2+:AДФ) + АДФ. Структура фермента состоит из трех доменов (Рис. 9.10): большого центрального домена “CORE” (показан светлосерым), АМФ-связывающего домена, называемого “AMPbd” (показан черным) и похожего на крышку АТФ-связывающего домена, называемого “LID” (показан темносерым), который закрывает фосфатные группы в активном центре (Miiller etal. 1996). В отсутствии лиганда домены LID и AMPbd принимают открытую конформацию, если считать, что закрытая конформация наблюдается в структуре, закристаллизованной с ингибитором переходного состояния Ар5А (Muller and Schulz 1992). Здесь лиганды находятся в высокоспецифичном окружении, необходимом для катализа. Недавние ЯМР исследования по спиновой релаксации ядер 15N (Shapiro and Meirovitch 2006) показали существование движений каталитического домена в подвижных доменах AMPbd и LID в наносекундном диапазоне, в то время как релаксация домена CORE происходит в пикосекундном диапазоне (Tugarinov etal. 2002; Shapiro etal. 2002). Конформационная подвижность аденилаткиназы была рассмотрена в нескольких вычислительных работах (Temiz et al. 2004; Maragakis and Karplus 2005; Lou and Cukier 2006; Whitford et al. 2007; Snow et al. 2007) Однако из-за большой амплитуды движений и больших времен, на которых они происходят, в полноатомной МД до сих пор не удалось наблюдать самопроизвольных переходов между открытой и закрытой конформациями. Применение алгоритма TEE-REX облегчает наблюдение таких переходов и делает возможным получение полноатомного описания пути перехода и лежащих за ним механизмов (Kubitzki and de Groot 2008). Для осуществления такого описания из коротких МД расчетов произвольной конформации были сконструированы коллективные подпространства {es}, включившие в себя также структуры в открытой и закрытой конформации. В последнем случае моды {es}, включая разностную моду, связывающую открытые и закрытые экспериментальные структуры, были возбуждены.
Наблюдаемый путь перехода можно представить в виде двух фаз. Стартуя из закрытого состояния (Рис. 9.10 слева), домен LID остается закрытым,
306
Глава 9. Динамика белков: от структуры к функционированию
Рис. 9.10. Закрытая (слева) и открытая (справа) кристаллографические структуры аденилаткиназы из Е. coli вместе с промежуточными структурами, описывающими две фазы перехода «закрыто-открыто». Аденилаткиназа состоит из домена CORE (показан светло-серым), домена AMPbd (показан черным) и домена LID (показан темно-серым). В закрытой структуре (слева) ингибитор переходного состояния Ар5А удален
в то время как домен AMPbd, содержащий спирали а2 и аЗ, принимает полуоткрытую конформацию. В процессе этого спираль а2 изгибается к спирали а4 из домена CORE на 15 градусов по отношению к спирали аЗ. Такое открывание сайта связывания АМФ могло бы облегчить эффективное высвобождение образовавшегося продукта. Во время второй фазы наблюдается открывание домена L1D, частично скоррелированное с открыванием домена AMPbd. По сравнению с крупнозернистым подходом, полноатомный расчет по алгоритму TEE-REX делает возможным детальное рассмотрение взаимодействий между остатками. В случае аденилаткиназы это позволило выявить образование в первой фазе стабильного солевого мостика между остатками Aspll8 и Lysl36, связывающего домены LID и CORE. При оценке всех невалентных взаимодействий между этими доменами оказалось, что этот солевой мостик вносит значительный вклад во взаимодействие между доменами. Таким образом, разрыв мостика с помощью мутации, например, Aspll8Ala должен уменьшить стабильность открытого состояния. При сравнении четырнадцати PDB-структур аденилаткиназы из дрожжей, кукурузы, человека и бактерий для одиннадцати из них оказался характерен такой солевой мостик на интерфейсе LID-CORE. Представляется возможным и альтернативный переходный путь, но анализ всех расчетов по алгоритму TEE-REX позволяет предположить наличие высокоэнергетического барьера, препятствующего полному открыванию домена AMPbd после того, как открывается домен LID. Наряду с наблюдаемыми значительными флуктуациями в элементах вторичной структуры, говорящими о высокой энергии внутренних деформаций, энтальпийные ограничения на этом пути, вероятно, делают его неподходящим на роль переходного пути в аденилаткиназе.
9.4. Методы предсказания функциональных мод
307
9.4. Методы предсказания функциональных мод
Как обсуждалось в предыдущем разделе, функциональными модами в белках обычно являются самые низкочастотные. Кроме методик, основанных на молекулярной динамике, есть несколько альтернативных методов, которые сосредоточены на предсказании таких основных степеней свободы, основываясь на единственной структуре.
9.4.1. Анализ нормальных мод
Анализ нормальных мод (Normal mode analysis, NMA) является одной из главных расчетных методик, импользуемых при изучении масштабных движений, изменяющих форму биологических молекул (Go- etal. 1983; Brooks and Karplus 1983; Levitt etal. 1983). Эти движения зачастую связаны с функцией и являются следствием связывания других молекул, таких как субстраты, лекарственные средства или другие белки. При анализе нормальных мод неявно предполагается, что моды с наибольшей амплитудой (самые низкочастотные моды) - это как раз функционально значимые моды, поскольку, как и функции, они обязаны своим существованием «дизайнерской мысли» эволюции, а не случайности.
Анализ нормальных мод - это гармонический анализ. За ним стоит предположение, что поверхность конформационной энергии может быть аппроксимирована параболой, несмотря на то, что функциональные моды при физиологических температурах сильно ангармоничны (Brooks and Karplus 1983; Austin et al. 1975). Для выполнения анализа нормальных мод необходимы набор координат, силовое поле, описывающее взаимодействия между атомами, и компьютерная программа для выполнения требуемых расчетов. Выполнение анализа нормальных мод в декартовых координатах включает три главных вычислительных шага.
1. Минимизация конформационной потенциальной энергии как функции координат атомов.
2. Вычисление так называемого гессиана
dqftlj ’
который является матрицей вторых производных потенциальной энергии по массовзвешенным атомным координатам2.
2 Переход к массовзвешенным координатам (qt = ^Jrn,xl) возникает при упрощении записи гамильтониана системы атомов. Прим, перев.
308
Глава 9. Динамика белков: от структуры к функционированию
3. Диагонализация гессиана. Это завершающий шаг дает собственные значения и собственные векторы («нормальные моды»).
Минимизация энергии может потребовать довольно много процессорного времени. Более того, поскольку гессиан является матрицей 3N х 3N, где N - число атомов, то последний шаг также может быть затратным с вычислительной точки зрения.
9.4.2. Модели эластичных сетей
Модели эластичных или гауссовых сетей (Elastic or Gaussian network models, ENM) (Tirion 1996)- это, по сути, упрощение анализа нормальных мод. Обычно вместо полноатомного представления белка учитывают только его Са-атомы. Это означает десятикратное уменьшение числа частиц, что резко снижает вычислительные затраты. Более того, поскольку считается, что исходные координаты соответствуют основному состоянию, то минимизация энергии не требуется.
Потенциальная энергия рассчитывается согласно формуле
где у обозначает константу жесткости, a Rc - радиус отсечки. По сравнению с сильными допущениями, присущими анализу нормальных мод, такие упрощения не приводят к значительной потере качества. Это обстоятельство вместе с относительно низкой вычислительной стоимостью объясняет популярность таких моделей эластичных сетей в настоящее время. Реализации алгоритмов ENM также представлены на веб-серверах, таких как ElNemo (Suhre and Sanejouand 2004a, b) (http://www.igs.cnrs-mrs.fr/ elnemo/) и AD-ENM (Zheng and Doniach 2003; Zheng and Brooks 2005) (http://enm.lobos.nih.gov/).
9.4.3. CONCOORD
Программа CONCOORD (de Groot etal. 1997) использует геометрический подход к предсказанию подвижности белков. Пространственная структура белка определяется различными взаимодействиями, такими как ковалентные связи, водородные связи и неполярные взаимодействия. Большая часть этих взаимодействия остается неизменной во время функционально значимых конформационных перестроек. Это наблюдение лежит в основе метода CONCOORD: на основе данных об исходной структуре генерируются
9.4. Методы предсказания функциональных мод
309
«План сборки»
о о 2 О О о
S
Случайные координаты
Для каждого ограничения: правильная геометрия
Для каждого атома: проверка на наталкивание с другими атомами
Иначе
Если достигнуто максимальное число итераций
Структура отклоняется
Выходная структура
Иначе
Ансамбль структур
Структура принимается
Если все ограничения выполнены и наталкивания отсутствуют
Если достигнуто максимальное число структур
(возвращение на шаг генерации случайных координат)
Конец работы
Ансамбль выходных структур
&
Рис. 9.11. Схематическое представление метода CONCOORD для создания структурных ансамблей из единственной исходной структуры. На первом шаге (программа dist) исходная структура анализируется и превращается в геометрическое описание белка. На втором шаге (программа disco), стартовав из случайных координат, структура воссоздается на основе определенных ранее ограничений
310
Глава 9. Динамика белков: от структуры к функционированию
альтернативные структуры, в которых подавляющее большинство взаимодействий остается тем же, что и в исходной структуре. Для этого на первом шаге расчета методом CONCOORD анализируются взаимодействия в исходной структуре и на их основе создаются геометрические ограничения - в основном ограничения по расстоянию между атомами с верхней и нижней границами, а также ограничения по углам и информация о плоских и хи-ральных группах. Такое геометрическое описание структуры можно сравнить с планом сборки белка. На втором шаге, стартовав из случайного расположения атомов, структура итеративно перестраивается на основе ранее составленного плана - обычно это происходит несколько сотен раз. Поскольку каждый запуск стартует со случайного расположения атомов, то, в отличие от МД, этот метод не страдает от проблемы сэмплирования и приводит к получению ансамбля, охватывающего все конформационное пространство, которое доступно с учетом ранее полученных ограничений. Однако метод не дает информации ни о пути между двумя подсостояниями, ни о времени перехода, ни о его энергии (Рис. 9.12).
9.4.3.1. Приложения
CONCOORD и его недавно разработанное расширение tCONCOORD (Seeliger et al. 2007) были применены к рассмотрению различных белков. Аденилаткиназа демонстрирует отчетливое закрывающее движение доменов при связывании субстрата (АТФ/АМФ) или ингибитора (см. Рис. 9.13 вверху), причем СКО по Са-атомам между конформацией со связанным лигандом и без него составляет 7.6 А. С использованием программы tCONCOORD было выполнено два расчета, исходной конформацией для которых была выбрана закрытая (PDB код 1 АКБ). В одном из расчетов лиганд (Ар5А) был удален. На Рис. 9.13 внизу, показан результат анализа главных компонент для экспериментальных структур. Первый собственный вектор (ось абсцисс) соответствует домен-открывающему движению, показанному стрелкой на рис. 9.13 (внизу). Каждая точка на графике представляет одну структуру. Красные точки представляют ансамбль, который был получен исходя из закрытой конформации аденилаткиназы без лиганда. Зеленые точки представляют ансамбль, который был получен исходя из лиганд-связывающей конформации. Несмотря на то, что расчет с ингибитором позволил просэмплировать, главным образом, закрытые конформации в окрестности структуры с лигандом, расчет структуры без лиганда позволил просэмплировать как закрытую, так и открытую конформации, что дало возможность приблизиться к экспериментально полученным открытым конформациям с СКО 2.4, 2.6 и 3.1 А для структур 1DVR, 1АК2 и 4АКЕ, соответственно. В создании лекарственных препаратов, которое
9.4. Методы предсказания функциональных мод
311
Рис. 9.12. Сравнение сэмплирующих свойств молекулярной динамики и метода CONCOORD на гипотетической энергетической поверхности. МД-траектория (слева) «гуляет» по поверхности, позволяя, таким образом, получить информацию о времени и пути между конформационными подсостояниями. CONCOORD (недегерминистически) «скачет» по поверхности, позволяя, таким образом, лучше просэмплировать конформационное пространство
Рис. 9.13. (Цветную версию рисунка см. на вклейке.) Вверху: наложение рентгенографических структур аденилаткиназы. Внизу: анализ главных компонент. Два ансамбля структур, полученных по методу tCONCOORD, спроецированы на два первых собственных ветора, полученных при анализе главных компонент ансамбля рентгеновских структур. Ансамбль, представленный красными точками, построен исходя из закрытой конформации (1 АКБ) с удаленным ингибитором. Построенный ансамбль сэмплирует как закрытые, так и открытые конформации. Ансамбль, представленный зелеными точками, также построен исходя из закрытой конформации (1 АКБ), но с ингибитором. Этот ансамбль сэмплирует только закрытые конформации в окрестности исходной лиганд-содержащей
опирается на знание структуры рецептора, часто возникает обратная задача -предсказание структуры рецептора с лигандом исходя из структуры свободного рецептора. Расчет, стартовавший из открытой конформации (4АКЕ), привел к получению структур, приближающихся к закрытой конформации с СКО 2.5, 2.9 и 3.3 А для 1DVR, 1АК2 и 1АКЕ, соответственно. Таким образом, функциональное домен-открывающее движение было предсказано в обоих случаях: при использовании в качестве исходной как закрытой, лиганд-содержащей, конформации, так и открытой конформации, не содержащей лиганда.
312
Глава 9. Динамика белков: от структуры к функционированию
Связывание
GroES
10,0
5,0
0,0
-5,0
-10,0 — -100,0
15,0
10,0
5,0
0,0
-5.0 — -100,0
5,0
0,0
-5,0
-10,0
-5,0
PDB
-50,0
Двойное кольцо
Одинарное*-кольцо С = 0,09 , 0,0
30,0
Рис. 9.14. Асимметричный комплекс GroEL-GroES (слева) вместе с результатами расчетов CONCOORD (справа). Комплекс GroEL-GroES состоит из двух кошаперонинов GroES (показаны черным), транс-кольца GroEL, связанного с GroES, (показано темно-серым) и цис-кольца (показано светло-серым). Анализ главных компонент выявил два главных структурных перехода для кольца GroEL: после связывания нуклеотида (вертикальная ось на правой панели) и после связывания GroES (горизонтальная ось), соответственно. В расчетах двойного кольца, но не одинарного, эти моды оказались связанными, предполагая наличие связи между внутрикольцевой и межкольцевой кооперативностью
Благодаря своей вычислительной эффективности CONCOORD может быть широко использован для выявления функционально значимых мод подвижности для таких молекулярных систем, которые лежат за пределами ограничений по размеру, свойственными другим методикам расчетов на атомарном уровне, например, молекулярной динамике. Применение CONCOORD к комплексу шаперонинов GroEL-GroES, содержащему более 8 000 остатков, выявило новую форму связи между внутрикольцевой и межкольцевой кооперативностью (de Groot etal. 1999). Каждое кольцо GroEL продемонстрировало две основных моды коллективного движения: основной конформационный переход при связывании с кошаперонином GroES и вторичный переход при связывании АТФ (Рис. 9.14 вверху справа). Расчет с помощью CONCOORD одного лишь кольца GroEL не показал какой-либо связи между этими модами, в то время как расчет системы
9.5. Итоги и перспективы
313
двух колец оказал четкую корреляцию между двумя этими модами, объясняя, таким образом, как связывание нуклеотидов влияет на аффинность к GroES в двойном кольце, но не в одинарном.
9.5. Итоги и перспективы
Вычислительные методы приобретают все большее признание в структурной биологии и исследовании белков. Функционирование белков -это обычно динамический процесс, включающий структурные перестройки и конформационные переходы между стабильными состояниями. Поскольку такие динамические процессы трудны для экспериментального изучения, методы in silico могут внести значительный вклад в понимание функционирования белков с атомарным разрешением.
Наиболее известным методом изучения динамики белков является молекулярная динамика (МД), в которой атомы рассматриваются, как классические частицы, а их взаимодействия аппроксимируются эмпирическими силовыми полями. На каждом дискретном шаге по времени решаются ньютоновские уравнения движения, что приводит к получению траектории, описывающей динамическое поведение системы. Несмотря на растущую популярность МД, сфера её применимости ограничена вычислительными требованиями. В течение ближайших 10 лет доступные времена расчета белков среднего размера, по всей вероятности, не достигнут микросе-кундного диапазона для большинства биомолекулярных систем. Однако поскольку функционально значимая динамика белков обычно представлена низкочастотными движениями, происходящими в диапазоне от микро- до миллисекунд, стандартные МД расчеты плохо подходят для широкого применения в изучении конформационной динамики больших биомолекул.
Для смягчения этой проблемы сэмплирования, от которой страдает стандартная МД, были предложены различные методы. Один из подходов состоит в уменьшении числа частиц либо объединением групп атомов в псевдоатомы (переход к крупнозернистому представлению), либо заменой явных молекул растворителя на его неявную континуальную модель. В обоих случаях число частиц значительно уменьшается, способствуя достижению гораздо больших времен, чем в полноатомных расчетах с явным растворителем. Однако, ухудшение «разрешения», присущее обоим методам, может ограничить их точность и, следовательно, применимость. Другие подходы сохраняют атомарное описание и реализуют различные стратегии сэмплирования.
Алгоритмы обобщенного ансамбля, такие как обмен репликами (Replica Exchange, REX) используют тот факт, что конформационные пере
314
Глава 9. Динамика белков: от структуры к функционированию
ходы при высоких температурах происходят чаще. В методе REX при стандартной температуре расчет выполняется для нескольких копий (реплик) системы с помощью МД при различных температурах с частым обменом между репликами, посредством чего низкотемпературные реплики используют повышенные способности высокотемпературных реплик к преодолению барьеров. Хотя динамическая информация при такой постановке расчета теряется, каждая реплика по-прежнему представляет собой больцманов-ский ансамбль при соответствующей температуре, позволяя получить ценную информацию о термодинамике и стабильности различных конформационных подсостояний. Хотя расчеты по методу обмена репликами в полноатомном представлении часто используются в контексте сворачивания белков, с вычислительной точки зрения они быстро становятся очень требовательными для систем, содержащих более, чем несколько тысяч атомов.
В то время как метод REX является неискажающим методом сэмплирования, существует несколько методов, которые искажают систему, чтобы улучшить сэмплирование по определенным коллективным степеням свободы. Функционально значимые белковые движения зачастую соответствуют тем собственным векторам в матрице ковариации атомных флуктуаций, которые имеют наибольшие собственные значения. Если эти векторы известны из анализа главных компонент (a principal component analysis, РСА), или из экспериментальных данных или из предыдущих расчетов, то их можно использовать в таких протоколах расчета как конформационное затопление (Conformational Flooding) или коллективная динамика (КД, Essential Dynamics, ED). Однако в обоих методах за улучшение сэмплирования приходится платить утратой канонических свойств итоговой траектории.
Недавно разработанный протокол TEE-REX совмещает в себе положительные свойства алгоритма REX с положительными свойствами, возникающими вследствие специфического возбуждения функционально значимых мод (как, например, в КД), в то же время избегает вышеупомянутых недостатков обоих методов. В частности, поддерживается приблизительно каноническая целостность референсного ансамбля и значительно улучшено сэмплирование по главным коллективным модам движения. Таким образом, итоговый референсный ансамбль может быть использован для вычисления равновесных свойств системы, что позволяет провести сравнение с экспериментальными данными.
Хотя в развитии методов улучшенного сэмплирования был достигнут значительный успех, вычислительные требования методов, основанных на МД, по-прежнему остаются высокими, и вычисления обыкновенно занимают недели и месяцы расчетного времени на современных многопроцессорных кластерах. Однако для многих вопросов в структурной биологии оказывается достаточным просто иметь представление о возможных кон
Литература
315
формациях белка и его функциональных модах и нет необходимости в детальной информации о характерных временах и энергиях. В этих случаях эластичные модели предлагают простой путь для оценки возможных функциональных движений белка. Хотя при этом делаются значительные упрощения и не получается картины движения на атомарном уровне, предсказанные коллективные движения зачастую оказываются в хорошем качественном согласии с экспериментальными результатами. Другим вычислительно эффективным путем, сохраняющим атомарное представление структуры, является метод CONCOORD, в котором белок описывается посредством геометрических ограничений. На основе плана сборки, полученного из исходной структуры, создается ансамбль структур, который представляет собой исчерпывающее сэмплирование конформационного пространства, возможное при заданных ограничениях. Однако не дается никакой информации о характерных временах и энергиях.
В настоящий момент нет единого метода, готового к повсеместному использованию для предсказания функционально значимых белковых движений на основе пространственной структуры. Однако есть большое число методов, охватывающих различные стороны этой задачи и вносящих вклад в наше понимание функции белков. Таким образом, комбинирование существующих методов станет, вероятно, наиболее прямым путем увеличения предсказательной силы методов in silico.
Литература
Adcock SA, McCammon JA (2006) Molecular dynamics: survey of methods for simulating the activity of proteins. Chem Rev 106:1589-1615
Affentranger R, Tavemelli I, di Iorio E (2006) A novel Hamiltonian replica exchange MD protocol to enhance protein conformational space sampling. J Chem Theory Comput 2:217-228
Amadei A, Linssen ABM, Berendsen HJC (1993) Essential dynamics of proteins. Proteins 17:412-425 Amadei A, Linssen ABM, de Groot BL, et al. (1996) An efficient method for sampling the essential
subspace of proteins. J Biom Str Dyn 13:615-626
Amadei A, de Groot BL, Ceruso M-A, et al. (1999) A kinetic model for the internal motions of proteins: diffusion between multiple harmonic wells. Proteins 35:283-292
Anderson HC (1980) Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys 72:2384-2393
Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181:223-230 Austin RH, Beeson KW, Eisenstein L, et al. (1975) Dynamics of ligand binding to myoglobin. Biochemistry 14(24):5355-5373
Bahar I, Erman B, Haliloglu T, et al. (1997) Efficient characterization of collective motions and interresidue correlations in proteins by low-resolution simulations. Biochemistry 36:13512-13523
Bartels C, Karplus M (1998) Probability distributions for complex systems: Adaptive umbrella sampling of the potential energy. J Phys Chem В 102:865-880
Berendsen HJC, Postma JPM, di Nola A, et al. (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684—3690
Berg BA, Celik T (1992) New approach to spin-glass simulations. Phys Rev Lett 69:2292-2295 Berg BA, Neuhaus T (1991) Multicanonical algorithms for first-order phase transitions. Phys Lett 267:249-253
316
Глава 9. Динамика белков: от структуры к функционированию
Berg JM, Tymoczko JL, Stryer L (2002) Biochemistry, fifth edition. WH Freeman, New York
Bond PJ, Holyoake J, Ivetac A, et al. (2007) Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J Struct Biol 157:593-605
Brooks B, Karplus M (1983) Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc Natl Acad Sci USA 80:6571-6575
Brooks BR, Bruccoleri RE, Olafson BD, et al. (1983) CHARMM: a program for macromolecular energy minimization and dynamics calculations. J Comp Chem 4:187-217
Burykin A, Warshel A (2003) What really prevents proton transport through aquaporin? Charge self-energy versus proton wire proposals. Biophys J 85:3696-3706
Cecchini M, Rao F, Seeber M, et al. (2004) Replica exchange molecular dynamics simulations of amyloid peptide aggregation. J Chem Phys 121:10748-10756
Chakrabarti N, Tajkhorshid E, Roux B, et al. (2004) Molecular basis of proton blockage in aquaporins. Structure 12:65-74
Chen H, Wu Y, Vbth GA (2006) Origins of proton transport behavior from selectivity domain mutations of the aquaporin-1 channel. Biophys J 90:L73-L75
Cheng X, Cui G, Homak V, et al. (2005) Modified replied exchange simulation for local structure refinement. J Phys Chem В 109:8220-8230
Chodera JD, Swope WC, Pitera JW, et al. (2007) Use of the weighted histogram analysis method for the analysis of simulated and parallel tempering simulations. J Chem Theory Comput 3:26-41
Christen M, van Gunsteren WF (2006) Multigraining: an algorithm for simultaneous fine-grained and coarse-grained simulation of molecular systems. J Chem Phys 124:154106
Cook A, Fernandez E, Lindner D, et al. (2005) The structure of the nuclear export receptor csel in its cytosolic state reveals a closed conformation incompatible with caigo binding. Mol Cell 18:355-357
Currie MG, Fok KF, Kato J, et al. (1992) Guanylin: an endogenous activator of intestinal guanylate cyclise. Proc Natl Acad Sci USA 89:947-951
de Groot BL, Grubmuller H (2001) Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF. Science 294:2353-2357
de Groot BL, Amadei A, Scheek RM, et al. (1996a) An extended sampling of the configurational space of HPr from E coli. Proteins 26:314-322
de Groot BL, Amadei A, van Aalten DMF, et al. (1996b) Towards an exhaustive sampling of the configurational spaces of the two forms of the peptide hormone guanylin. J Biomol Str Dyn 13:741-751
de Groot BL, van Aalten DMF, Scheek RM, et al. (1997) Prediction of protein conformational freedom from distance constraints. Proteins 29:240-251
de Groot BL, Hayward S, van Aalten DMF, et al. (1998) Domain motions in bacteriophage T4 lysozyme: a comparison between molecular dynamics and crystallographic data. Proteins 31:116-127
de Groot BL, Vriend G, Berendsen HJC (1999) Conformational changes in the chaperonin GroEL: new insights into the allosteric mechanism. J Mol Biol 286:1241-1249
de Groot BL, Engel A, Grubmuller H (2001) A refined structure of human Aquaporin-1. FEBS Lett 504: 206-211
de Groot BL, Frigato T, Helms V, et al. (2003) The mechanism of proton exclusion in the aqua-porin-1 water channel. J Mol Biol 333:279-293
Dixon MM, Nicholson H, Shewchuk L, et al. (1992) Structure of a hinge-bending bacteriophage T4 lysozyme mutant Ile3 —► Pro. J Mol Biol 227:917-933
Duda RO, Hart PE, Stork DG (2001) Pattern Classification, second edition. Wiley, New York
Faber HR, Matthews BW (1990) A mutant T4 lysozyme displays five different crystal conformations. Nature 348:263-266
Frauenfelder H, Leeson DT (1998) The energy landscape in non-biological and biological molecules. Nat Struct Biol 5:757-759
Frauenfelder H, Sligar SG, Wolynes PG (1991) The energy landscapes and motions of proteins. Science 254:1598-1603
Fu D, Libson A, Miercke LJ, et al. (2000) Structure of a glycerol-conducting channel and the basis for its selectivity. Science 290: 481-486
Fukunishi H, Watanabe O, Takada S (2002) On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: application to protein structure prediction. J Chem Phys 116:9058-9067
Garcia AE (1992) Large-amplitude nonlinear motions in proteins. Phys Rev Lett 68:2696-2699
Литература
317
Garcia AE, Onuchic JN (2003) Folding a protein in a computer: An atomic description of the fold-ing/unfolding of protein A. Proc Natl Acad Sci USA 100:13898-13903
G N, Noguti T, Nishikawa T (1983) Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc Natl Acad Sci USA 80:3696-3700
Gerstein M, Lesk AM, Chothia C (1994) Structural mechanisms for domain movements in proteins. Biochemistry 33:6739-6749
Gosh A, Rapp CS, Friesner RA (1998) Generalized Bom model based on a surface integral formulation. J Phys Chem В 102:10983-10990
Grubmuller H (1995) Predicting slow structural transitions in macromolecular systems: Conformational flooding. Phys Rev E 52:2893-2906
Hansmann UHE (1997) Effective way for determination of multicanonical weights. Phys Rev E 56:6200-6203
Hayward S, Kitao A, Go N (1995) Harmonicity and anharmonicity in protein dynamics: a normal mode analysis and principal component analysis. Proteins 23:177-186
He J, Zhang Z, Shi Y, et al. (2003) Efficiently explore the energy landscape of proteins in molecular dynamics simulations by amplifying collective motions. J Chem Phys 119:4005-4017
Hockney RW, Goel SP, Eastwood JW (1973) 10000 particle molecular dynamics model with longrange forces. Chem Phys Lett 21:589-591
Hub JS, de Groot BL (2008) Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc Natl Acad Sci USA105: 1198-1203
Iba Y (2001) Extended ensemble Monte Carlo. Int J Mod Phys C 12:623-656
Пап B, Tajkhorshid E, Schulten K, et al. (2004) The mechanism of proton exclusion in aquaporin channels. Proteins 55:223-228
Jean-Charles A, Nicholls A, Sharp K, et al. (1991) Electrostatic contributions to solvation energies: comparison of free energy perturbation and continuum calculations. J Am Chem Soc 113:1454-1455
Jorgensen WL, Chandrasekhar J, Madura JD, etal. (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926-935
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225-11236
Karplus M, Gao YQ (2004) Biomolecular motors: the Fl-ATPase paradigm. Curr Opin Struct Biol 14:250-259
Karplus M, Kushick JN (1981) Method for estimating the configurational entropy of macromolecules. Macromolecules 14:325-332
Kempf JG, Loria JP (2003) Protein dynamics from solution NMR theory and applications. Cell Biochem Biophys 37:187-211
Kitao A, Go- N (1999) Investigating protein dynamics in collective coordinate space. Curr Opin Struct Biol 9:143-281
Kitao A, Hirata F, Go N (1991) The effects of solvent on the conformation and the collective motions of proteins - normal mode analysis and molecular-dynamics simulations of melittin in water and vacuum.Chem Phys 158:447-472
Kitao A, Hayward S, Go- N (1998) Energy landscape of a native protein: Jumping-among-minima model. Proteins 33:496-517
Kokubo H, Okamoto Y (2004) Prediction of membrane protein structures by replica-exchange Monte Carlo simulations: case of two helices. J Chem Phys 120:10837-10847
Kubitzki MB, de Groot BL (2007) Molecular dynamics simulations using temperature-enhanced essential dynamics replica exchange. Biophys J 92:4262-4270
Kubitzki MB, de Groot BL (2008) The atomistic mechanism of conformational transition in adenylate kinase: a TEE-REX molecular dynamics study. Structure 16:1175-1182
Kumar S, Bouzida D, Swendsen RH, etal. (1992) The weighted histogram analysis method for free-energy calculations on biomolecules. I. the method. J Comp Chem 13:1011-1021
Kumar S, Payne PW, Vasquez M (1996) Method for free-energy calculations using iterative techniques. J Comput Chem 17:1269-1275
Kuroki R, Weaver LH, Matthews BW (1993) A covalent enzyme-substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science 262:2030-2033
318
Глава 9. Динамика белков: от структуры к функционированию
Levitt М, Sander С, Stem PS (1983) Normal-mode dynamics of a protein: Bovine pancreatic trypsin inhibitor. Int J Quant Chem: Quant Biol Symp 10:181-199
Levy RM, Karplus M, Kushick J, et al. (1984a) Evaluation of the configurational entropy for proteins: application to molecular dynamics of an a-helix. Macromolecules 17:1370-1374
Levy RM, Srinivasan AR, Olsen WK, et al. (1984b) Quasi-harmonic method for studying very low frequency modes in proteins. Biopolymers 23:1099-1112
Liu P, Kim B, Friesner RA, et al. (2005) Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc Natl Acad Sci USA 102:13749-13754
Lou H, Cukier RI (2006) Molecular dynamics of apo-adenylate kinase: a distance replica exchange method for the free energy of conformational fluctuations. J Phys Chem В 110:24121-24137
Luo R, David L, Gilson ML (2002) Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J Comput Chem 23:1244-1253
Lyman E, Zuckerman DM (2006) Ensemble-based convergence analysis of biomolecular trajectories. Biophys J 91:164-172
Maragakis P, Karplus M (2005) Large amplitude conformational change in proteins explored with a plastic network model: adenylate kinase. J Mol Biol 352:807-822
Marinari E, Parisi G (1992) Simulated tempering: a new Monte Carlo scheme. Europhys Lett 19:451-458
Marrink SJ, de Vries AH, Mark AE (2004) Coarse grained model for semiquantitative lipid simulations. J Phys Chem В 108:750-760
Matthews BW, Remington SJ (1974) The three dimensional structure of the lysozyme from bacteriophage T4. Proc Natl Acad Sci USA 71:4178-4182
McCammon JA, Gelin BR, Karplus M (1977) Dynamics of folded proteins. Nature 267:585-590
Mitsutake A, Sugita Y, Okamoto Y (2001) Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 60:96-123
Moffat К (2003) The frontiers of time-resolved macromolecular crystallography: movies and chirped X-ray pulses. Faraday Discuss 122:65-77
Murata K, Mitsuoka K, Walz T, et al. (2000) Structural determinants of water permeation through Aquaporin-1. Nature 407: 599-605
Muller CW, Schulz GE (1992) Structure of the complex between adenylate kinase from Eschericia coli and the inhibitor Ap5A refined at 19 A resolution: a model for a catalytic transition state. J Mol Biol 224:159-177
Muller CW, Schlauderer G, Reinstein J, et al. (1996) Adenylate kinase motions during catalysis: an energetic counterweight balancing substrate binding. Structure 4:147-156
Nguyen PH, Mu Y, Stock G (2005) Structure and energy landscape of a photoswitchable peptide: a replica exchange molecular dynamics study. Proteins 60:485-494
Nose S (1984) A unified formulation of the constant temperature molecular dynamics method. J Chem Phys 81:511-519
Pitera JW, Swope W (2003) Understanding folding and design: replica-exchange simulations of “Trp-cage” miniproteins. Proc Natl Acad Sci USA 100:7587-7592
Rao F, Caflisch A (2003) Replica exchange molecular dynamics simulations of reversible folding. J Chem Phys 119:4035-4042
Romo TD, Clarage JB, Sorensen DC, et al. (1995) Automatic identification of discrete substates in proteins: singular value decomposition analysis of time-averaged crystallographic refinements. Proteins 22:311-321
Schotte F, Lim M, Jackson TA, etal. (2003) Watching a protein as it functions with 150 ps timeresolved X-ray crystallography. Science 300:1944-1947
Seeliger D, Haas J, de Groot BL (2007) Geometry-based sampling of conformational transitions in proteins. Structure 15:1482-1492
Seibert MM, Patriksson A, Hess B, et al. (2005) Reproducible polypeptide folding and structure prediction using molecular dynamics simulations. J Mol Biol 354:173-183
Shapiro YE, Meirovitch E (2006) Activation energy of catalysis-related domain motion in E coli adenylate kinase. J Phys Chem В 110:11519-11524
Shapiro YE, Kahana E, Tugarinov V, et al. (2002) Domain flexibility in ligand-free and inhibitor bound Eschericia coli adenylate kinase based on a mode-coupliqg analysis of 15N spin relaxation. Biochemistry 41:6271-6281
Smith GR, Bruce AD (1996) Multicanonical Monte Carlo study of solid-solid phase coexistence in a model colloid. Phys Rev E 53:6530-6543
Литература
319
Snow С, Qi G, Hayward S (2007) Essential dynamics sampling study of adenylate kinase: comparison to citrate synthase and implication for the hinge and shear mechanisms of domain motion. Proteins 67:325-337
Still WC, Tempczyk A, Hawley RC, et al. (1990) Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc 112:6127-6129
Sugita Y, Okamoto Y (1999) Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 314:141-151
Sugita Y, Kitao A, Okamoto Y (2000) Multidimensional replica-exchange method for free-energy calculations. J Chem Phys 113:6042-6051
Suhre K, Sanejouand YH (2004a) ElNemo: a normal mode web-server for protein movement analysis and the generation of templates for molecular replacement. Nucl Acids Res 32:610-614
Suhre K, Sanejouand YH (2004b) On the potential of normal mode analysis for solving difficult molecular replacement problems. Act Cryst D 60:796-799
Sui H, Han B-G, Lee JK, et al. (2001) Structural basis of water-specific transport through the AQP1 water channel. Nature 414: 872-878
Tai К (2004) Conformational sampling for the impatient. Biophys Chem 107:213-220
Tajkhorshid E, Nollert P, Jensen M0, et al. (2002) Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 296: 525-530
Teeter MM, Case DA (1990) Harmonic and quasi harmonic descriptions of crambin. J Phys Chem 94:8091-8097
Temiz NA, Meirovitch E, Bahar I (2004) Eschericia coli adenylate kinase dynamics: comparison of elastic network model modes with mode-coupling 15N-NMR relaxation data. Proteins 57:468-480
Tirion MM (1996) Large amplitude elastic motions in proteins from a single-parameter atomic analysis. Phys Rev Lett 77:186-195
Tugarinov V, Shapiro YE, Liang Z, et al. (2002) A novel view of domain flexibility in E coli adenylate kinase based on structural mode-coupling 15N NMR spin relaxation. J Mol Biol 315:155-170
Van Aalten DMF, Amadei A, Vriend G, et al. (1995a) The essential dynamics of thermolysin - confirmation of hinge-bending motion and comparison of simulations in vacuum and water. Prot Eng 8:1129-1136
Van Aalten DMF, Findlay JBC, Amadei A, et al. (1995b) Essential dynamics of the cellular retinol binding protein - evidence for ligand induced conformational changes. Prot Eng 8:1129-1136
Van Gunsteren WF, Berendsen HJC (1987) Groningen Molecular Simulation (GROMOS) Library Manual. Biomos, Groningen
Van Gunsteren WF, Berendsen HJC (1990) Computer-simulation of molecular-dynamics - methodology, applications, and perspectives in chemistry. Angew Chem Int Edit Engl 29:992-1023
Warshel A, Kato M, Pisliakov AV (2007) Polarizable force fields: history test cases and prospects. J Chem Theory Comput 3:2034-2045
Weiner SJ, Kollman PA, Nguyen DT, et al. (1986) An all atom force field for simulations of proteins and nucleic acids. J Comp Chem 7:230-252
Weiss S (1999) Fluorescence spectroscopy of single biomolecules. Science 283:1676-1683
Whitford PC, Miyashita O, Levy Y, et al. (2007) Conformational transitions of adenylate kinase: switching by cracking. J Mol Biol 366:1661-1671
Xu Z, Horwich AL, Sigler PB (1997) The crystal structure of the asymmetric Gro-EL-GroES-(ADP)7 chaperonin complex. Nature 388:741-750
Zachariae U, Grubmuller H (2006) A highly strained nuclear conformation of the exportin Cselp revealed by molecular dynamics simulations. Structure 14:1469-1478
Zhang X-J, Wozniak JA, Matthews BW (1995) Protein flexibility and adaptability seen in 25 crystal forms of T4 lysozyme. J Mol Biol 250:527-552
Zhang Z, Shi Y, Liu H (2003) Molecular dynamics simulations of peptides, and proteins with amplified collective motions. Biophys J 84:3583-3593
Zheng W, Brooks BR (2005) Probing the local dynamics of nucleotide-binding pocket coupled to the global dynamics: myosin versus kinesin. Biophys J 89(1): 167-178
Zheng W, Doniach S (2003) A comparative study of motor-protein motions by using a simple elastic-network model. Proc Natl Acad Sci USA 100(23): 13253-13258
Zhou R Berne BJ, Germain R (2001) The free energy landscape for P-hairpin folding in explicit water. Proc Natl Acad Sci USA 98:14931-14936
Глава 10
Интегральные серверы для предсказания функции по структуре
Роман А. Ласковски
Сам по себе ни один метод предсказания функции белка по его пространственной структуре не совершенен; какие-то методы хорошо работают в одних случаях, какие-то оказываются лучше в других. Поэтому имеет смысл применить несколько различных методов предсказания к данной белковой структуре и получить от них либо согласованное предсказание, либо наиболее правдоподобное. В этой главе мы опишем два веб-сервера ProKnow (http://proknow.mbi.ucla.edu) и ProFunc (http://www.ebi.ac.uk/profunc), которые используют сочетание методов предсказания функции белка по его пространственной структуре.
10.1. Введение
Предсказание функции только что расшифрованной структуры белка слегка похоже на разгадывание привлекательной детективной загадки. Пространственная структура белка несомненно хранит ключи к его функции; но как разглядеть эти ключи, как оценить их надежность, как распознать и отбросить тупиковые пути и как собрать оставшиеся ключи воедино, чтобы достичь окончательного решения этой загадки.
На самом деле эта проблема возникла совсем недавно, став прямым следствием различных проектов по структурной геномике, которые стартовали в начале этого десятилетия. До них экспериментаторы уже многое
Roman A. Laskowski
European Bioinformatics Institute,
Wellcome Trust Genome Campus, Hinxton, Cambridge,
CB10 1SD, UK
e-mail: roman@ebi.ac.uk
10.1. Введение
321
знали о своих белках перед тем, как погрузиться в определение их пространственной структуры, и могли выбрать белки, исходя из интереса с точки зрения биологии. Основная цель расшифровки структуры белка заключалась в прояснении вопроса, как белок реализует свою биологическую функцию на атомарном уровне. У проектов по структурной геномике с их высокопроизводительными методами определения структуры цели совершенно иные. Теперь белок требует расшифровки структуры, если он принадлежит семейству, в котором еще нет представителей с известной структурой, или предполагается, что этот белок будет иметь новый тип укладки, или этот белок имеет важное значение для какого-нибудь заболевания. Определение функции белка больше в эти цели не входит.
Как следствие, стало появляться много структур таких белков, чья функция неизвестна. Действительно, около одной трети структур, полученных благодаря структурной геномике, соответствуют белкам с неизвестной или не полностью известной функцией. Это серьезно ограничивает пользу от этих структур, которые больше не объясняют, как белок реализует свою функцию, поскольку сама функция фактически неизвестна.
Впрочем, действительно ли структура, будучи известной, сможет дать ответ на все наши вопросы? В конце концов, история структурной биологии говорит нам, что пространственная структура объясняет функцию. Фактически каждая из прежде расшифрованных структур помогла объяснить какой-нибудь биологический или биохимический процесс. Таким образом, имея структуру, мы - вуаля! - получаем функцию.
К сожалению, в жизни - или в биоинформатике - не все так просто. Структура может объяснить функцию, но только если вы эту функцию уже знаете. Несмотря на доступность множества различных методов, обсуждавшихся в этой книге ранее, оказывается на удивление трудным определить функцию на основе одной лишь структуры.
10.1.1. Задача предсказания функции по структуре
Почему это так? Во-первых, если есть белок с неизвестной функцией, то это означает, что не только нет экспериментальной информации о его функции, но и потерпели неудачу стандартные методы анализа последовательности для функциональной аннотации. Эти методы, в особенности различные методы профилей, такие как методы скрытых марковских моделей, стали в последние годы достаточно изощренными, и теперь в состоянии обнаружить сходство функций при весьма низком уровне идентичности последовательностей. Так что если и эти методы потерпели неудачу, то нам остается положиться исключительно на пространственную структуру.
322
Глава 10. Интегральные серверы для предсказания функции
Структура белка содержит разного рода ключи к его функции, которые имеют и разную степень надежности, как это было описано в предыдущих главах. В Главе 6 было показано, что на глобальном уровне тип укладки белка очень часто может дать ключи к его функции, поскольку некоторые типы укладки прочно связаны с определенными функциями. Поэтому первым шагом на пути определения функции по структуре неизменно будет поиск белка с известной функцией и схожей укладкой. Сделать это можно с помощью большого числа предназначенных для сравнения укладок веб-серверов, для которых опубликовано несколько сравнительных обзоров (Sierk and Pearson 2004; Novotny et al. 2004; Carugo 2006). Однако вам следует иметь в виду, что сходство укладок не обязательно означает сходство функций. Например, так называемые суперфолды (Orengo etal. 1994; см тж. Главу 6), такие как семейство Т1М-бочонков, могут иметь представителей с большим разнообразием функций (Nagano et al. 1999; Anantharaman et al. 2003). А если белок имеет новый тип укладки -что, по мнению некоторых групп, является успешным результатом - то схожих укладок не будет найдено вовсе.
Если рассуждать не столь глобально, то важные ключи к функции могут лежать на поверхности белка, особенно в её углублениях и карманах (Глава 7), которые могут обеспечивать особое локальное расположение остатков, необходимое для катализа, распознавания ДНК и т.д. (Глава 8). Так, возможно, вы сможете идентифицировать, скажем, гипотетический сайт связывания АТФ. Это будет важным ключом к функции, но история на этом не заканчивается.
Есть еще различные обстоятельства, которые вставляют палки в колеса. Во-первых, часто бывает трудно получить нативную структуру всего белка. В этих случаях можно получить структуру части белка - скажем, всего лишь единственного домена. Сам по себе этот домен может мало сказать о функции целого белка. Во-вторых, даже если получена структура белка целиком, это может быть всего один компонент из многобелкового комплекса. И снова структура оказывается лишь частью истории. Еще более неприлично ведут себя так называемые белки-совместители, которые на самом деле могут иметь несколько функций в зависимости от контекста: расположения в клетке, окружения и так далее (Jeffery 1999). А некоторые белки могут изменять свою функцию в зависимости от того, какой из вариантов альтернативного сплайсинга экспрессирован в данный момент времени (Stamm et al. 2005).
Другая проблема в предсказании функции заключается в сложности оценки успеха или неудачи данного метода предсказания, и, на самом деле, даже в определении того, что понимать под функцией. Функция может быть
10.1. Введение
323
описана на разных уровнях, начиная от биохимической функции, переходя к биологическим процессам и путям и достигая уровня органов или организмов (Shrager 2003). Поэтому конкретный белок может быть аннотирован на нескольких различных уровнях функциональной специфичности: например, убихитиноподобный домен, сигнальный белок, предсказываемая сериновая гидролаза, вероятная эукариотическая D-аминокислотная тРНКаза и так далее. Таким образом, трудно судить о точности любого такого описания, особенно если это одно из еще более нечетких описаний.
Общепринятая стратегия при оценке методов предсказания функции состоит в использовании генной онтологии (ГО, Gene Ontology, GO) (The Gene Ontology Consortium 2000; Camon etal. 2004). Это открытая классификация для функционального аннотирования белковых последовательностей. Она представляет из себя машинно-читаемую онтологию, основанную на контролируемом словаре функциональных дескрипторов, и многие методы предсказания функции представляют свои результаты в терминах классификации ГО. Хотя и не строго иерархические, функциональные ГО-дескрипторы варьируются от совершенно неспецифических (например, фермент) до высокоточных (например 1 -пирролидин-4-гидрокси-2-карбоксилатдеаминаза).
10.1.2. Методы предсказания структура-функция
Как было показано в предыдущих главах, есть очень большое количество различных методов предсказания функции белка на основе его структуры. Их описание и рассмотрение пригодности приведены в нескольких обзорах (Kim et al. 2003; Watson et al. 2005; Rigden 2006). Ни один из методов не является совершенным и ни от одного из них нельзя ожидать успешного применения во всех случаях. Например, некоторые методы подходят только для ферментов и совершенно не в силах помочь, если рассматриваемый белок таковым не является. Другие методы сильно полагаются на некое совпадение, - будь то укладка, или мотив, или сайт связывания и так далее, - с белком, чья структура известна. Поэтому если такое совпадение найти нельзя, или оно оказывается совпадением с другим гипотетическим белком, то эффективность такого метода обращается в ноль.
Как следствие, разумный подход состоит в том, чтобы направить большое число этих методов на структуру белка и посмотреть, что выпадет. Именно так и поступают два сервера, описываемые в этой главе. Это ProKnow из Университета Калифорнии (UCLA) (http://proknow.mbi.ucla.edu) и ProFunc из Европейского института биоинформатики (European Bioinformatics Institute, EBI) (http://www.ebi.ac.uk/profimc). Оба они используют предсказания, основанные как на последовательности белка, так и на его
324
Глава 10. Интегральные серверы для предсказания функции
ProKnow
Сканы Тип упаковки Функциональные
последовательности и структурные мотивы связи
Поиск последовательностей с помощью PSI-BLAST в БД Uniprot
Поиск типа укладки с помощью Dali
База данных взаимодействующих белков (DIP)
Мотивы в последовательностях согласно PROSITE
Пространственные мотивы согласно RIGOR
База данных Prolinks
Рис. 10.1. Схематическая диаграмма основанных на структуре и последовательности методов, применяемых к пространственной структуре любого белка, загруженной на сервер предсказания функции ProKnow. Основанные на последовательности методы -это PSI-BLAST (Altschul et al. 1997) и PROSITE (Hulo et al. 2004). Основанные на структуре методы - это поиск по фолду Dali (Holm and Sander 1998) и поиск по пространственным мотивам RIGOR (Kleywegt 1999). Последние два метода для определения интересных функциональных связей для лучших результатов программы PSI-BLAST используют результаты базы данных взаимодействующих белков DIP (Database of Interacting Proteins, Xenarios et al. 2002) и базы данных Prolinks (Bowers et al. 2004). Обобщая все результаты, получают функциональные аннотации генной онтологии (GO) и комбинируют их, используя байесовское присвоение весов, для получения набора предсказаний функции и соответствующих оценок надежности
структуре, и широко автоматизированы: загружаешь файл в формате PDB и терпеливо дожидаешься результатов.
Для иллюстрации двух этих методов в действии в качестве примера мы взяли только что полученную пространственную структуру. Это структура предполагаемой ацетилтрансферазы из Vibrio cholera, полученная в 2005 году в Центре по структурной геномике на Среднем Западе (MCSG). Она была опубликована в базе данных PDB 28 февраля 2006 года с кодом 2fck (Cuff et al. 2007). На момент появления структуры функция белка была известна только предварительно; последовательность имела более 50% идентичности с серин-ацетилтрансферазой рибосомальных белков (ribosomal-protein-serine acetyltransferase) и содержала несколько мотивов, характерных для ацетилтрансферазной активности. Раз структура была известна, эти предварительные описания функции получили мощную поддержку, поскольку оказалось, что есть сильное структурное сходство, как глобальное, так и локальное, с
10.2. ProKnow
325
другими - удаленными - ацетилтрансферазами. Наиболее сильные сходства были обнаружены в вероятном сайте связывания, где мог бы связаться коэнзим А (соА). Некоторые из этих сходств будут рассмотрены ниже.
10.2. ProKnow
Первый из двух описываемых тут интегральных серверов называется ProKnow (Pal and Eisenberg 2005) и создан в Калифорнийском университете в Лос-Анджелесе (UCLA) (http://proknow.mbi.ucla.edu). Текущая версия сервера, ProKnow 2.0, использует шесть главных методов предсказания для любой загруженной пространственной структуры (Рис. 10.1). На самом деле, на сервер также можно загрузить всего лишь последовательность белка, но в этом случае один из шести методов отпадает. Характерными особенностями, которые анализируются этими методами, являются: общая укладка белка, различные структурные мотивы (не используется, если загружена только последовательность), сходство последовательностей, мотивы в последовательности и функциональные связи базы данных взаимодействующих белков (Database of Interacting Proteins, DIP) и базы данных Prolinks. Каждый метод может дать один или несколько ключей к функции белка с разной степенью надежности. Этим ключам с использованием теоремы Байеса присваиваются веса, а затем ключи комбинируются для получения наиболее вероятной общей функции, которая выражается в терминах ГО, и степенью достоверности каждого из них. Результатом работы сервера является карта взаимосвязей между наиболее вероятными предсказаниями по классификации ГО (Рис. 10.2), которая позволяет пользователю более уверенно трактовать предсказания. На итоговой вебстранице представлена также детальная информация о наилучших вариантах и их оценки. Лучшие варианты для нашего примера, структуры 2fck, представлены на рис. 10.3. В сущности, здесь сервер выдает лишь один наилучший результат: N-ацетилтрансфераза, которая предсказана с высокой надежностью и согласуется с вероятной функцией белка.
10.2.1. Подбор типа укладки
Первая стадия в ProKnow состоит в поиске других белков, имеющих такую же укладку, что и рассматриваемый белок, или максимально похожую на неё. Конечно, это в некоторой степени жульничество, поскольку ProKnow требует от пользователя сперва использовать программу по распознаванию сходных укладок DALI (Holm and Sander 1998), а затем её результат в формате FSSP и загружать на сервер.
326
Глава 10. Интегральные серверы для предсказания функции
Рис. 10.2. Схема генной онтологии, сгенерированная для PDB-структуры 2fck, которая показывает иерархию функциональных термов от общих к специфическим. В тех случаях, когда ProKnow предсказывает более одного функционального варианта, на схеме показывается их сеть, где каждый из вариантов связан с остальными сходными вариантами линиями, раскрашенными в зависимости от сходства
Совпадения, полученные с помощью Dali, и являются первыми ключами к функции белка, которые используются в ProKnow.
Любопытно, но если загрузить на сервер одну лишь последовательность, то всю работу ProKnow делает сам: определяет укладку, совместимую с последовательностью, и использует её как ключ к функции. Для определения наиболее вероятной укладки сервер ProKnow использует результаты сервера по распознаванию фолда, созданному в UCLA. Этот сервер также реализует стратегию из нескольких шагов. Сначала, используя программу BLAST, он пытается найти совпадения рассматриваемой последо-
10.2. ProKnow
327
Evidence Number of
Rank Clues
. ir FuneUen
К
0008152 010264
0009405 0.01901
0006807: 0.0184
0006508 0.0172
0005975 0.0172
Description
19 4
Е'.оД| .4’
243 4
1.8 ”4
2-0 4
pathogenesis
Btotogfcat Process
5.00000
PROUNKS
Guo 10
Ж1б8эв мт от» ир 1шйиэ ю от»
« ат» йгйотт 1»лэваз *т» qotqqq
IMal No
Ctaes
Э.росхХ
400000
4.00000
4.000001
Рис. 10.3 а) Лучшие предсказания функции сервера ProKnow для PDB структуры 2fck. Наиболее достоверное предсказание говорит об N-ацетилтрасферазной активности белка, б) Сводная таблица ключей, использованных в предсказании каждого ГО-терма для 2fck. Кликнув на любое число, можно увидеть детальную информацию по данному ключу
вательности с последовательностями структур PDB. Затем он пробует получить результат с помощью итеративной программы PSI-BLAST. Если обе попытки заканчиваются неудачей, то сервер использует результат предсказания вторичной структуры белка, полученный от сервера PSIPRED, поддерживаемого Университетским колледжем в Лондоне (Bryson et al. 2005). Это предсказание подается в программу SDP (Sequence Derived Properties, Fischer and Eisenberg 1997), которая пытается подобрать подходящий тип упаковки. Наконец, если даже это ничего не дает, то в ход идет метод, названный DASEY (Directional Atomic Solvation EnergY, Mallick et al. 2002).
Полагаясь на результаты любого метода по распознаванию фолда, или протягивания, нужно помнить одно - эти методы представляют что-то вроде черной магии и требуют осторожной интерпретации. Порой они могут давать примерно правильный ответ - обычно это бывает в случае небольших однодоменных белков, где получаются топологически почти правильные модели (Moult 2005); но в общем случае точность широко варьируется. Если же последовательность белка очень длинная, то шансы на успех становятся еще меньше, поскольку белок почти наверняка состоит
328
Глава 10. Интегральные серверы для предсказания функции
из нескольких структурных доменов, границы которых было бы идеальным определить вручную. Тогда нужно будет распознать укладку каждого домена, но даже если эти стадии оказались успешными, пространственное расположение доменов может стать принципиально важным для функционирования белка, а методы предсказания упаковки доменов еще не достигли своей зрелости (Wollacott et al. 2007; Berrondo et al. 2008).
10.2.2. Структурные мотивы
После стадии поиска совпадающих укладок пространственная структура белка просматривается на предмет каких-либо структурных мотивов, имеющих прямое отношение к функционированию. Такие мотивы содержатся в базе данных автоматически созданных структурных мотивов RIGOR (Kleywegt 1999). Каждый мотив состоит из «интересного» расположения остатков в PDB-структуре. Для отличия интересных остатков от неинтересных используются три правила: (а) белок содержит и последовательных остатков одного типа (например, четыре последовательных остатка аргинина), (б) ряд соседних остатков целиком гидрофобен, или поля-рен/заряжен, или является сочетанием гидрофобных и полярных/заряжен-ных, и (в) все остатки, находящиеся в контакте с одним гетеросоединением. ProKnow использует более 10 000 мотивов из базы RIGOR, с каждым из которых связан ГО-терм для соответствующей цепи белка.
10.2.3. Гомология последовательностей
Программа PSI-BLAST (Altschul et al. 1997) используется для определения таких белков в базах данных последовательностей UniProt/SWISS-PROT и UniProt/TrEMBL, которые гомологичны рассматриваемому белку. Любые совпадения, которые имеют аннотации по системе ГО, добавляют свои ключи в общую корзину.
10.2.4. Мотивы в последовательности
Затем последовательность рассматриваемого белка проверяется на наличие в ней мотивов с помощью базы данных по функциональным мотивам PROSITE (Hulo et al. 2004). И снова каждый мотив имеет ряд связанных с ним индексов по классификации ГО.
10.2.5. Взаимодействия белков
Последний набор извлекаемых сервером ProKnow признаков относится к белок-белковым взаимодействиям, взятым из базы данных взаи
10.2. ProKnow
329
модействующих белков (Database of Interacting Proteins, DIP) (Xenarios et al. 2002), и функциональным аннотациям из базы данных Prolinks (Bowers et al. 2004). Любая последовательность, совпадение с которой установлено с помощью программы PSI-BLAST, может дать функциональную связь, если присутствует в DIP или Prolinks.
10.2.6. Объединение предсказаний
Когда все процессы завершены, функции (т.е. термы по системе ГО), связанные с неоднократно встречающимися выявленными особенностями, объединяются с присвоением весов по теореме Байеса, что позволяет сделать оценку значимости каждого предсказанного терма. Рассматриваются только термы относящиеся к молекулярной функции и биологическим процессам, т.е. не принимаются во внимание термы, относящиеся к внутриклеточной локализации. Значимость каждого предсказанного терма отображается тремя числами. Первое - это байесовский вес, который соответствует вероятности, - от нуля до единицы, - того, что предсказание терма сделано правильно. Второе число - это ранг признака, показывающий, насколько надежным считается конкретное ГО-присваивание, чтобы быть на первом месте. Дело в том, что такие присваивания имеют разное происхождение: они могут быть сделаны куратором, получены прямым наблюдением, выведены на основании сходства структуры или последовательности и так далее. Этим вызвана и различная надежность, наиболее высокая для тех присваиваний, которые имеют прямые экспериментальные свидетельства в свою поддержку. Источник аннотации указан как код признака в данных ГО. Сервер ProKnow переводит каждый код признака в ранг для численной оценки его надежности, а ранги нескольких предсказаний при усреднении дают ранг признака. Третьим показателем значимости является число ключей, которое равно числу весов, использованных для расчета байесовского веса, и имеет отношение к тому, какое число методов сервера ProKnow внесло вклад в данное предсказание терма ГО.
10.2.7. Успешность предсказания
На рисунке 10.3 показана часть результатов для нашего примера, структуры 2fck. Тип укладки по данным Dali совпадает почти исключительно с ацетилтрансферазами. Поиск с помощью BLAST в базе данных UniProt также обнаруживает ряд хороших совпадений с ацетилтрансферазами. Поиск с помощью RIGOR предлагает нам несколько тупиковых путей в виде фактора роста фибробластов, липид-связывающего белка липо-вителлина и интегразы. Среди результатов PROSITE тупиковых путей
330
Глава 10. Интегральные серверы для предсказания функции
больше, и они выводят нас на короткие мотивы, два из которых являются центрами фосфорилирования и один центром миристоилирования (все они на веб-сайте PROSITE имеют комментарий: «В некоторых случаях эта запись может не приниматься во внимание программой, поскольку является слишком неспецифичной»). Поиск с помощью DIP ничего не дал. Тем не менее, ошеломляюще сильным предсказанием было то, которое оказалось правильным; а именно, что белок является ацетилтрансферазой. Поэтому в этом случае общее предсказание выглядит правильным.
В общем, сервер ProKnow работает достаточно хорошо. Авторы протестировали его на невырожденном наборе белков с известной функцией и обнаружили, что около 70% функциональных аннотаций оказались правильными (Pal and Eisenberg 2005). Менее специфичные предсказания (например, гидролаза) являются более точными, чем более специфичные (например, лейциламинопептидаза). Точность предсказания была слегка увеличена за счет недавнего включения базы данных Prolinks, которая не использовалась в первоначальной версии, и увеличится еще больше, поскольку размер Prolinks растет.
10.3. ProFunc
Вторым интегральным сервером, описываемым тут, является ProFunc (Laskowski et al. 2005b) (http://www.ebi.ac.uk/profunc), созданный в Европейском институте биоинформатики (EBI) в рамках сотрудничества с Центром по структурной геномике на Среднем Западе (Midwest Center for Structural Genomics, MCSG). Сервер ProFunc позволяет пользователю либо самому загрузить структуру белка, либо ввести PDB-код структуры, уже имеющейся в базе данных белковых структур. В последнем случае, если сервер однажды уже выполнял расчет для этой структуры, то результат появится незамедлительно. Когда ProFunc выполняет расчет, то, как показано на рисунке 10.4, он использует ряд методов, основанных на последовательности белка и его структуре, причем производится такое распараллеливание расчетов, что разные методы выполняются на разных процессорах. Некоторые из ресурсоемких методов сами тоже запускаются параллельно на множестве процессоров. Весь расчет заканчивается обычно в течение часа.
Затем результаты всех методов суммируются, причем детали результатов каждого метода остаются доступными. Однако здесь результаты не объединяются таким изощренным образом, как это делается в ProKnow. Вместо этого наверху страницы с результатами приводится краткий итог, показывающий наиболее общие термы по системе ГО и названия белков,
Сканы последовательностей
Тип упаковки и структурные мотивы
Шаблоны
из л остатков
Поиск последовательностей с помощью FASTA вБДРОВ
Поиск типа укладки с помощью SSM
Анализ активных центров ферментов
Поиск последовательностей с помощью BLAST в БД Uniprot
Анализ углублений на поверхности
Анализ сайтов связывания лигандов
Мотивы в последовательностях согласно InterProScan (PROSITE, BLOCKS, SMART, Pfam и т.д.)
Анализ консервативности остатков
Анализ сайтов связывания ДНК
Библиотека СММ надсемейств
ДНК-связывающие мотивы «спираль-поворот—спираль»
Анализ обратных шаблонов
Соседствующие гены
Анализ ячеек
Рис. 10.4. Схематическая диаграмма используемых в ProFunc методов, основанных на последовательности и структуре. Методы анализа последовательности, перечисленные в левом столбце, включают поиск последовательности белка в базах PDB и Uniprot. Поиск с помощью InterProScan и Superfamily выявляет любые мотивы в последовательностях из соответствующих им баз данных, которые есть в последовательности рассматриваемого белка. Для каждого подходящего варианта из Uniprot, отобранного программой BLAST, по возможности выполняется локализация соответствующего ему гена в геноме и определение всех соседних генов. В среднем столбце первый из методов поиска, основанных на структуре, использует программу SSM для определения структур, общая укладка которых наиболее сходна с укладкой рассматриваемого белка. Затем рассчитываются углубления на поверхности, которые могут быть визуализованы с раскраской по типу образующих эти углубления остатков или их консервативности. Затем определяется два типа структурных мотивов: мотивы «спираль-поворот-спираль» (helix-tum-helix, НТН), характерные для многих ДНК-связывающих белков, и ячейки, которые часто обнаруживаются в функционально важных местах. Наконец, в правом столбце представлены различные методы шаблонов для поиска локальных пространственных совпадений с известными структурами белков
но это следует рассматривать лишь как краткую инструкцию. Основная цель сервера состоит в представлении результатов в легкодоступном виде, чтобы дать исследователям возможность интерпретировать их, используя свой собственный опыт и информацию о рассматриваемом белке.
332
Глава 10. Интегральные серверы для предсказания функции
Теперь, хотя ProFunc и использует ряд методов, основанных на последовательности, включая хорошо известные методы, такие как FASTA и InterProScan (Quevillon et al. 2005), мы опишем только методы, основанные на структуре, поскольку большинство из них уникальны.
10.3.1. Основанные на структуре методы, используемые ProFunc
10.3.1.1. Поиск совпадений типа укладки
Первым из методов, основанных на структуре, является поиск в представительной выборке из базы данных PDB структур с таким же, или схожим, типом укладки, что и у рассматриваемой структуры. Для этого используется программа SSM (Secondary Structure Matching, Сопоставление вторичной структуры) (Krissinel and Henrick 2004). Эта программа производит быструю процедуру сопоставления графов для сравнения элементов вторичной структуры рассматриваемой структуры с такими элементами структур в базе данных. Хорошо совпавшие структуры накладываются й для них рассчитывается СКО по эквивалентным Са-атомам, а также стандартизованная мера значимости и собственная мера значимости SSM, называемая Q-показатель. ProFunc показывает десять лучших совпадений, упорядоченных по Q-показателю, и любое из них, или все сразу, могут быть наложены на рассматриваемую структуру при помощи программы молекулярной графики RasMol (Sayle and Milner-White 1995).
Лучшее совпадение типа укладки для нашего примера, структуры 2fck, показано на рисунке 10.5. Этим совпадением является структура из PDB с кодом 1 s7f, представляющая собой RimL 1Ч(а)-ацетилтрансферазу из Salmonella typhimurium (Vetting et al. 2005). Этот белок отвечает за превращении прокариотического рибосомального белка из L12 в L7 путем ацетилирования его N-концевой аминогруппы. Белок образует гомодимер и интерфейс димера образует большой желоб, способный к связыванию N-концевой спирали L12. На некотором удалении от сайта связывания субстрата белок связывает также кофермент А (Ко А).
10.3.1.2. ДНК-связывающий мотив «спираль-поворот-спираль»
Вторым основанным на структуре методом является поиск каких-либо мотивов «спираль-поворот-спираль» (helix-tum-helix, НТН), которые совпадают с мотивами, извлеченными из участвующих в связывании ДНК структур PDB (Jones et al. 2003; Aravind et al. 2005). Ложноположительные
10.3. ProFunc
333
Рис. 10.5. (Цветную версию рисунка см. на вклейке.) Наиболее близкий к структуре 2fck тип укладки, обнаруженный с помощью программы поиска совпадающих типов укладки SSM в структуре 1s7f, RimL Ы(а)-ацетилтраснферазе из Salmonella typhimurium. а) Общая пространственная структура 2fck и б) общая структура 1s7f в той же ориентации, в) Структуры совмещены и показаны в виде линии Са-атомов - 2fck желтым, a 1s7f фиолетовым. Совпадающие области выделены более толстыми линиями в каждой структуре
результаты отсеиваются по ряду параметров, основанных на сочетании доступности растворителю и электростатического потенциала (Shanahan etal. 2004), поэтому любой положительный результат может указывать, что рассматриваемый белок связывает ДНК, хотя, конечно, это сообщает мало нового о функции белка. В нашем примере структура 2fck не имеет мотивов НТН, как мы и ожидали.
10.3.1.3. Ячейки
Третий метод выявляет в структуре мотивы типа «ячейка», которые часто связаны с функциональными сайтами. Ячейка - это сайт связывания аниона или катиона, образованный тремя или более аминокислотными остатками, у которых двугранные углы основной цепи (у-(р) изменяются между а- и у-областями на карте Рамачандрана, соответствующими право-и левозакрученным спиралям (Watson and Milner-White 2002а, b). Как и прежде, визуализация в программе RasMol показывает расположение ячеек в контексте всей пространственной структуры. Сервер ProFunc присваивает каждой ячейке оценку, исходя из следующих критериев: количество доступных растворителю атомов NH, консервативность составляющих ячейку остатков и того, находится ли ячейка в более крупном углуб
334
Глава 10. Интегральные серверы для предсказания функции
лении поверхности. Ячейки могут оказаться полезными, когда ни один из других методов ничего не может сказать о функции белка. В таких случаях расположение ячеек может указать на предположительно функционально важные места в пространственной структуре белка.
Структура 2fck содержит несколько таких ячеек, три из которых имеют достаточно высокую оценку, что говорит об их потенциальной функциональной значимости. И в самом деле, ячейка, имеющая наиболее высокую оценку, расположена в вероятном сайте связывания субстрата (по аналогии со сходной структурой ls7f), в то время как вторая и третья ячейки обнаружены возле ворот сайта связывания соА.
10.3.1.4. Углубления на поверхности
Затем с помощью программы SURFNET (Laskowski 1995) рассчитываются все углубления на поверхности белка. Углубления ранжируются по размеру и могут быть визуализованы с помощью RasMol. Опции визуализации позволяют раскрасить углубления по их специфическим свойствам: размеру углубления, типу остатков или их консервативности. Размер важен, поскольку самое большое углубление на поверхности белка обычно находится в месте расположения его активного центра (Laskowski et al. 1996). Консервативность остатков также важна, поскольку группа высококонсервативных остатков, особенно расположенная в большом кармане, с большой вероятностью указывает на функциональный сайт (Lichtarge and Sowa 2002; Madabushi et al. 2002; Glaser et al. 2003). Как и анализ ячеек, изучение углублений приносит наибольшую пользу, когда остальные методы потерпели неудачу или предлагают только неопределенные варианты. В нашем случае наибольшее углубление действительно соответствует предполагаемому сайту связывания белка, совпадающему по расположению со связанным коА в родственных структурах, выявленных по совпадению типа укладки методами, рассмотренными выше, и методами шаблонов, которые описаны ниже.
10.3.1.5. Методы шаблонов
Последние методы, используемые сервером ProFunc, включают в себя четыре различных типа поиска по шаблону из остатков (Laskowski et al. 2005с). По определению, шаблоны - это особые пространственные конформации, как правило, трех аминокислотных остатков. Поиск по шаблонам выполняется быстрым алгоритмом пространственного поиска, называемым JESS (Barker and Thornton 2003), который запускается параллельно на нескольких процессорах.
10.3. ProFunc
335
Шаблоны ферментов
Первую группу шаблонов составляют шаблоны активных центров ферментов, которые взяты из составленного вручную Атласа каталитических центров (Catalytic Site Atlas, CSA) (Porter et al. 2004). Здесь каждый шаблон имеет от двух до пяти остатков, которые либо описаны в литературе, как каталитические, либо являются высококонсервативными и лежат в непосредственной близости к каталитическим остаткам. Хорошее совпадение (см. ниже) с одним из таких шаблонов может оказаться явным указанием на функцию белка.
Шаблоны связывания лигандов и ДНК
Две следующие группы шаблонов - это шаблоны связывания лигандов и ДНК, которые автоматически генерируются раз в неделю, чтобы учитывать все обновления, произошедшие в базе данных PDB. Шаблоны связывания лигандов генерируются поочередным рассмотрением каждого типа гетерогрупп (согласно словарю гетерогрупп (Het Group Dictionary) в PDB) и составлением списка негомологичных PDB-структур, содержащих эти гетерогруппы. Сначала отмечаются остатки, взаимодействующие с гетерогруппами в каждой из выбранных структур. Затем определяются шаблоны, состоящие из групп по три остатка из ранее отмеченных, и сохраняются как шаблоны для соответствующих гетерогрупп. Есть следующие критерии отбора, определяющие, какая тройка остатков может считаться шаблоном: каждый остаток должен находиться не далее 5 А от других остатков шаблона, каждый шаблон может иметь не более одного гидрофобного остатка (т.е. Ala, Phe, Не, Leu, Met, Pro или Vai), - это необходимо, чтобы настроить шаблоны на остатки поверхности, - и не должно быть двух шаблонов из одной структуры, имеющих более одного общего остатка. Порядок рассмотрения потенциальных шаблонов определяется их относительной значимостью. Так, шаблон, содержащий остатки, образующие несколько водородных связей с данной гетерогруппой, более значим, чем те шаблоны, в которых остатки лишь слегка взаимодействуют с гетерогруппой. Шаблоны связывания ДНК генерируются точно таким же образом, за исключением того, что все молекулы ДНК и РНК рассматриваются как единая гетерогруппа. По состоянию на май 2008 года в этой базе данных насчитывалось 584 шаблона каталитических центров, 97,534 шаблонов связывания лигандов и 3,390 шаблонов связывания ДНК. На рисунке 10.6 показано совпадение шаблона в структуре 2fck с шаблоном связывания ко А в структуре ls7n, RimL М(а)-ацетилтрансферазе из Salmonella typhimurium.
336
Глава 10. Интегральные серверы для предсказания функции
Обратные шаблоны
Четвертая группа шаблонов посвящена поиску любых совпадений, которые могли ускользнуть от первых трех групп. Это обратные шаблоны, которые вычисляются по самой рассматриваемой структуре. Они генерируются с использованием в основном тех же самых правил, что шаблоны связывания лигандов и ДНК. Главное различие состоит в том, что, во-первых, рассматривается вся структура белка целиком, а не только остатки, контактирующие с лигандами или ДНК, и, во-вторых, каждому шаблону присваивается вес в зависимости от консервативности входящих в него остатков (которая рассчитывается по сделанному с помощью программы BLAST множественному выравниванию последовательностей, взятых из базы данных последовательностей UniProt). Шаблоны выбираются таким образом, что, в идеале, каждый остаток белка представлен по крайней мере в одном шаблоне, хотя если шаблонов получается слишком много, то их число сокращается до удвоенного числа остатков в последовательности.
Лучший обратный шаблон к структуре 2fck показан на рис. 10.7. Совпадением является структура ls7f, RimL Ы(а)-ацетилтрансфераза из S. typhimurium. Это апо-форма структуры ls7n, совпадение с которой было выявлено с помощью вышеописанных программы поиска совпадений вторичной структуры SSM и шаблона связывания лигандов.
Поиск по шаблонам и их оценка
Поиск по шаблону может дать сотни, тысячи и даже десятки тысяч совпадений, особенно в случае обратных шаблонов. Поэтому задача состоит в том, чтобы отбросить случайные совпадения и оставить только значимые, отсортировав их по значимости. ProFunc делает это, сравнивая окружение остатков шаблона в его родительской структуре с окружением совпадающих с ним остатков в структуре рассматриваемой. Остаткам родительской структуры в радиусе 10 А от геометрического центра шаблона согласно степени сходства и перекрывания ставятся в соответствие остатки из такой же области рассматриваемой структуры. В случаях альтернативных вариантов создания таких пар применяется процедура оптимизации, направленная на максимизацию числа пар идентичных или схожих остатков с эквивалентным положением в пространстве. Число пар остатков дает грубую оценку локального сходства совпадающих сайтов в двух белках (Рис. 10.6b и 10.7b). Однако и эта грубая оценка все еще оставляет слишком много ложно-положительных совпадений. Поэтому применяемая в действительности оценка учитывает относительное расположение составляющих пару остатков в соответствующих аминокислотных последовательностях. Если составляющие пару остатки следуют в одинаковом
10.3. ProFunc
337
Рис. 10.6. (Цветную версию рисунка см. на вклейке.) Совпадение шаблонов связывания лиганда по данным сервера ProFunc. а) Три остатка, показанных фиолетовым, соответствуют шаблону связывания лиганда для лиганда из кофермента А (коА), показанному раскраской по типу атомов (углерод серый, азот синий, кислород красный, сера желтая и фосфор оранжевый). Шаблон образован остатками Asn138, Ser141 и Cys134 из PDB-структуры 1s7n, RimL N(a^ацетилтрансферазы из Salmonella typhimurium. Три остатка, показанные желтым, - это остатки из рассматриваемой структуры 2fck, совпадающие с остатками шаблона. Это Asn140, Ser143 аи Cys136, соответственно. СКО по 14 атомам боковых цепей составляет 1.18 А. б) Как и на рисунке (а), но дополнительно показаны совпадающие остатки, лежащие в радиусе 10 А от центра шаблона. Это остатки того же типа, которые совмещаются при наложении рассматриваемой структуры и шаблонной структуры. Фиолетовым показаны остатки из шаблонной структуры (1s7n), желтым - из рассматриваемой структуры (2fck)
порядке в обоих последовательностях, тогда высока вероятность гомологичности последовательностей.
Чтобы увидеть, почему это так, рассмотрим две имеющие общего предка последовательности, которые разошлись настолько далеко, что их родство не может быть установлено методами анализа последовательностей. Однако если обе они сохранили одну и ту же функцию, то областью, которая претерпела наименьшие изменения, вероятно будет активный центр, поскольку любое изменение в нем изменило бы и функцию. Сухой остаток этого рассуждения состоит в том, что самый высокий уровень сходства между двумя белками будет среди остатков в окрестности активного центра. Эти остатки будут близки в пространстве, но могут быть разбросаны по последовательности этих белков. Вот почему можно обнаружить сходство в пространственной структуре, но практически невозможно уловить его при сравнении последовательностей.
Иллюстрация этому приведена на рисунке 10.7с, где представлено выравнивание последовательностей между структурой 2fck и наиболее сходной с ней по обратному шаблону структурой 1 s7f. Выравнивание задавалось остатками, которые были определены как эквивалентные в процедуре поиска локального совпадения, описанной выше. Эти остатки отмечены двумя точками между последовательностями. (Одна точка соответствует остаткам, которые утратили своих пространственно-эквивалентных партнеров в
338
Глава 10. Интегральные серверы для предсказания функции
Рис. 10.7. (Цветную версию рисунка см. на вклейке.) Совпадение обратных шаблонов между структурами 2fck и 1 s7f, соответствующими RimL М(а)-ацетилтрансферазе из Salmonella typhimurium. а) Желтым показаны остатки шаблона из 2fck (Gly99, ТугЮО и Leu 116), которые соответствуют остаткам из 1s7f (Gly97, Туг98 и Leu 114, соответственно), показанным фиолетовым, с СКО 0.62 А по 17 совпадающим атомам, б) Эквивалентные остатки одинаковых типов в радиусе 10 А от центра шаблона. 16 таких остатков (из всего 44 на удалении 10 А и менее) дают 36.4% локальной идентичности. Следующие 20 остатков (не показаны) имеют сходный тип (например, Не соответствует Vai), в) Выравнивание структур, полученное из наложения структур. Верхняя строка соответствует вторичной структуре в 2fck, а нижняя - в 1 s7f; спирали показаны зубчатыми элементами, р-тяжи стрелками. Три выделенных остатка в выравнивании последовательностей соответствуют остаткам шаблона. Двойные точки между последовательностями обозначают остатки, содержащиеся внутри сферы с радиусом 10 А и центром в центре шаблона, т.е. те остатки, которые были использованы для выполнения выравнивания. Области в рамках представляют сегменты выравнивания, где идентичность последовательностей превышает 35%. Длинные тонкие стрелки снизу показывают структурно подходящие области, т.е. сегменты обоих белков, где Со-атомы могут быть совмещены по структуре с СКО менее 3.0 А
выравнивании). Легко видеть, что составляющие пары остатки, лежащие в компактной области пространства, распределены почти по всей длине обеих последовательностей.
Еще более интересно, что в то время как выравнивание в целом дает 24,7% идентичности последовательностей двух белков, идентичными оказываются 16 из 44 остатков в радиусе 10 А от центра шаблона, что дает локальную идентичность 36,4%. Поскольку эта область соответствует значительной части сайта связывания ко А в структуре ls7f, это является
10.3. ProFunc
339
сильным структурным аргументом в пользу того, то структура 2fck также связывает коА. Область также захватывает и часть предполагаемого сайта связывания субстрата, но её недостаточно, ни чтобы сделать вывод об одинаковом субстрате для обоих белков, ни чтобы предположить у них одинаковую функцию.
В дополнение к оценке локального сходства, ProFunc использует и другие статистические показатели. Одним из них является математическое ожидание Е (E-value), связанное с этой оценкой. Для обратных шаблонов Е вычисляется из распределения всех оценок, полученных в данном поиске, по той же процедуре, что используется в программе FASTA (Pearson 1998). При поиске по другим шаблонам математическое ожидание Е вычисляется с использованием заранее рассчитанных параметров. Лучшие результаты сортируются по значению Е на четыре группы: достоверное совпадение (Е < 10 6), весьма вероятное совпадение (10~6 < Е < 0.01), вполне вероятное совпадение (0.01 < Е < 0.1) и маловероятное совпадение (0.1 < Е < 10.0).
Также используется общее структурное сходство и наибольшая протяженность фрагментов последовательностей, которые еще можно наложить с СКО 3.0 А по Са-атомам. Последний показатель может быть полезным, когда есть длинное перекрывание, предполагающее значительное структурное совпадение даже в случае маловероятных совпадений.
10.3.1.6. Структурный анализ с помощью PDBsum
Не очень существенным для предсказания функции, но полезным побочным эффектом загрузки структуры на сервер ProFunc является создание для этой структуры нескольких страниц в атласе PDBsum. Это красочно иллюстрированный атлас белковых структур (http://www.ebi.ac.uk/pdbsum), выполняющий ряд анализов структуры для загруженных белков и представляющий результаты с помощью различных схематических диаграмм (Laskowski et al. 2005а). Пара примеров приведена на рис. 10.8.
10.3.2. Оценка структурных методов
Насколько хороши структурные методы в предсказании функции белка? Авторы ProFunc попытались ответить на этот вопрос, применив его к 92 структурам белков с известной функцией, полученным в MCSG (Watson et al. 2007). В каждом случае предсказания сервера были настроены таким образом, чтобы исключить информацию о структурах, полученных после выхода в свет рассматриваемой структуры, для лучшего понимания того, что могло бы быть предсказано на тот момент.
340
Глава 10. Интегральные серверы для предсказания функции
MTPDFQIVTQRLQLRLITADEAEELVQCI RQSQTLH^WW FSQQEAEQFIQATRLNW 15Ю15202530354О 505560
VKAEAYGFGVFERQTQTLVGMVA INEFYHTFI^ASLGYWIGDRYQRQGYGKEALTAL 1LF
63 70 75 80 85 90 95 Ю0 105 1Ю 115 120
CFERLELTRLEiyCDPENVPSQALALRCGANREQLAPNRFLYAGEPKAGl VFSLIP 123 130 135 140 145 150 155 160 165 170 175
a)
Рис. 10.8. Пример анализа co страниц PDBsum, выполняемого при загрузке любой структуры на сервер ProFunc. а) Схематическая а
диаграмма белковой цепи, представляющая элементы вторичной структуры ~ белка (а-спирали и р-тяжи) вместе с различными структурными мотивами, такими как Р- и у-повороты и Р-шпильки. В этом примере остатки, взаимодействующие со связанным лигандом, отмечены точками над однобуквенным аминокислотным кодом. В структуре 2fck лигандами являются 12 ниграт-ионов и одна молекула глицерина, которые не представляют особенного интереса или функциональной информации, будучи элементами кристаллизационного раствора. б) Диаграмма топологии белковой цепи в 2fck. Диаграмма показывает, как р-тяжи, показанные широкими серыми стрелками, объединяются сторона к стороне с образованием центрального p-листа домена. Также диаграмма показывает относительное взаиморасположение а-спиралей, показанных здесь цилиндрами. Маленькие стрелки показывают направление белковой цепи от N- к С-концу. Числа при элементах вторичной структуры соответствуют нумерации остатков в PDB-файле. Диаграмма создана на основе результатов программы Нега (Hutchinson and Thornton 1990)
Анализ показал, что 70% структур имели бы правильное описание своей функции, если бы сервер ProFunc был доступен на тот момент, причем три четверти из этих 70% получили правильное предсказание более чем одним методом.
Из методов, основанных на структуре, двумя наиболее успешными стали сравнение типов укладки с помощью программы SSM и обратные шаблоны. Доля успешных предсказаний для у обоих составила 50-60%. Действительно, в большинстве случаев оба метода давали одинаковые лучшие результаты, хотя однажды один из методов обнаружил правильное совпадение там, где другой не смог этого сделать. Это может означать, что, поскольку
10.3. ProFunc
341
342
Глава 10. Интегральные серверы для предсказания функции
два метода дают настолько схожие результаты, все, что нам действительно нужно, это метод сравнения типов укладки, такой как SSM. Однако будучи далеко не вырожденным метод обратных шаблонов дает гораздо больше специфической информации о сходстве между двумя любыми структурами. Более того, он выявляет области, имеющие наиболее высокое сходство, и, следовательно, с наибольшей вероятностью являющиеся функциональными сайтами. Кроме того, метод дает весомые свидетельства в пользу вероятной функции, показывая, как он это делает, ключевые остатки.
Конечно, единственным истинным путем проверки правильности предсказания является его экспериментальное подтверждение. Это трудно, требует затрат времени и ресурсов, хотя на этом пути наметился некоторый прогресс в направлении создания высокопроизводительных функциональных тестов (Yakunin et al. 2004).
10.4. Заключение
Мы рассмотрели здесь ProKnow и ProFunc, два интегральных сервера, использующие сочетание основанных на совпадении последовательности и структуры методов для того, чтобы попытаться предсказать функцию белка по его пространственной структуре, загружаемой на эти серверы. В большинстве случаев они оказываются в состоянии сделать некоторые предположения относительно возможной функции, хотя в некоторых случаях они могут быть весьма расплывчатыми (например, ДНК-связывающая активность). В других случаях, однако, все их методы ничего не находят и терпят полную неудачу. Наиболее интригующими являются случаи структур принадлежащих к ^охарактеризованным семействам, обладающим новыми типами укладок. Так, все, с чем может остаться любой исследователь, это понимание того, что структура имеет интересного вида углубление на своей поверхности, выложенное высококонсервативными остатками, но без каких-либо идей относительно того, что в этом углублении может связываться. Подобные случаи нуждаются в создании новых методов и включении их в существующие серверы. Наиболее полезными стали бы методы, которые могут предсказывать вероятный субстрат для данного белка на основе анализа одной лишь его структуры. То есть методы не должны полагаться на совпадение с существующими структурами, поскольку, как в случае новых типов укладки, таких совпадений нет по определению. В настоящее время такие методы очень ресурсоемки и обычно стартуют по меньшей мере с каких-либо соображений по поводу класса субстрата (например, Hermann et al. 2006). Итак, еще какое-то время предсказание функции белков продолжит полагаться на искусное выслеживание и логические выводы.
Литература
343
Благодарности. Автор хотел бы поблагодарить Дебнат Пал за помощь с сервером ProKnow и за его полезные замечания по этой главе.
Литература
Altschul SF, Madden TL, Schaffer AA, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
Anantharaman V, Aravind L, Koonin EV (2003) Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins Curr Opin Chem Biol 7:12-20.
Aravind L, Anantharaman V, Balaji S, et al. (2005) The many faces of the helix-tum-helix domain: transcription regulation and beyond. FEMS Microbiol Rev 29:231-262.
Barker J A, Thornton JM (2003) An algorithm for constraint-based structural template matching: application to 3D templates with statistical analysis. Bioinformatics 19:1644-1649.
Berrondo M, Ostermeier M, Gray JJ (2008) Structure prediction of domain insertion proteins from structures of individual domains. Structure 16:513-527.
Bowers PM, Pellegrini M, Thompson MJ, et al. (2004) Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol 5:R35.
Bryson K, McGuffin U, Marsden RL, et al. (2005) Protein structure prediction servers at University College London. Nucleic Acids Res 33:W36-W38.
Camon E, Magrane M, Barrell D, et al. (2004) The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res 32:D262-D266.
Carugo О (2006) Rapid methods for comparing protein structures and scanning structure databases. Curr. Bioinformatics 1:75-83.
Cuff ME, Li H, Moy S, et al. (2007) Crystal structure of an acetyltransferase protein from Vibrio cholerae strain N16961. Proteins 69:422-427.
Fischer D, Eisenberg D (1997) Assigning folds to the proteins encoded by the genome of Mycoplasma genitalium. Proc. Natl Acad Sci USA 94:11929-11934.
Glaser F, Pupko T, Paz I, et al. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19:163-164.
Hermann JC, Ghanem E, Li Y, et al. (2006) Predicting substrates by docking high-energy intermediates to enzyme structures. J. Am. Chem. Soc. 128:15882-15891.
Holm L, Sander C (1998) Touring the fold space with DALI/FSSP. Nucleic Acids Res. 26:316-319.
Hulo N, Sigrist CJ, Le Saux V, et al. (2004) Recent improvements to the PROSITE database. Nucleic Acids Res. 32:D134-D137.
Hutchinson EQ Thornton JM (1990) HERA: a program to draw schematic diagrams of protein secondary structures. Proteins 8:203-212.
Jeffery CJ (1999) Moonlighting proteins. Trends Biochem. Sci. 24:8-11. Jones S, Barker JA, Nobeli I, et al. (2003) Using structural motif templates to identify proteins with DNA binding function. Nucleic Acids Res. 31:2811-2823.
Kim SH, Shin DH, Choi IQ et al. (2003) Structure-based functional inference in structural genomics. J. Struct. Funct. Genomics 4:129-135.
Kleywegt GJ (1999) Recognition of spatial motifs in protein structures. J. Mol. Biol. 285:1887-1897.
Krissinel E, Henrick К (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D60:2256-2268.
Laskowski RA (1995) SURFNET: a program for visualizing molecular surfaces, cavities and inter-molecular interactions. J. Mol. Graph. 13:323-330.
Laskowski RA, Luscombe NM, Swindells MB, et al. (1996) Protein clefts in molecular recognition and function. Protein Science 5:2438-2452.
Laskowski RA, Chistyakov VV, Thornton JM (2005a) PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 33: D266-D268.
344
Глава 10. Интегральные серверы для предсказания функции
Laskowski RA, Watson JD, Thornton JM (2005b) ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 33.W89-W93.
Laskowski RA, Watson JD, Thornton JM (2005c) Protein function prediction using local 3D templates. J. Mol. Biol. 352:614-626.
Lichtarge O, Sowa ME (2002) Evolutionary predictions of binding surfaces and interactions. Curr. Opin. Struct. Biol. 12:21-27.
Madabushi S, Yao H, Marsh M, et al. (2002) Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J. Mol. Biol. 316:139-154.
Mallick P, Weiss R, Eisenberg D (2002) The directional atomic solvation energy: an atom-based potential for the assignment of protein sequences to known folds. Proc. Natl. Acad. Sci. USA 99:16041-16046.
Moult J (2005) A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol. 15:285-289.
Nagano N, Hutchinson EQ Thornton JM (1999) Barrel structures in proteins: automatic identification and classification including a sequence analysis of TIM barrels. Prot. Sci. 8:2072-2084.
Novotny M, Madsen D, KJeywegt GJ (2004) Evaluation of protein fold comparison servers. Proteins 54:260-270.
Orengo CA, Jones DT, Thornton JM (1994). Protein superfamilies and domain superfolds. Nature 372:631-634.
Pal D, Eisenberg D (2005) Inference of protein function from protein structure. Structure 13:121— 130.
Pearson WR (1998) Empirical statistical estimates for sequence similarity searches. J. Mol. Biol. 276:71-84.
Porter CT, Bartlett GJ, Thornton JM (2004) The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 32: D129-D133.
Quevillon E, Silventoinen V, Pillai S, et al. (2005) InterProScan: protein domains identifier. Nucleic Acids Res. 33:W116-W120.
Rigden DJ (2006) Understanding the cell in terms of structure and function: insights from structural genomics. Curr. Opin. Biotechnol. 17:457-464.
Sayle RA, Milner-White EJ (1995) RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 20:374-376.
Shanahan HP, Garcia MA, Jones S, et al. (2004) Identifying DNA-binding proteins using structural motifs and the electrostatic potential. Nucleic Acids Res. 32:4732-41.
Shrager J (2003) The fiction of function. Bioinformatics 19:1934-1936.
Sierk ML, Pearson WR (2004) Sensitivity and selectivity in protein structure comparison. Protein Sci. 13:773-785.
Stamm S, Ben-Ari S, Rafalska 1, et al. (2005) Function of alternative splicing. Gene 344:1-20.
The Gene Ontology Consortium (2000) Gene Ontology tool for the unification of biology. Nat. Genet. 25:25-29.
Watson JD, Milner-White EJ (2002a) A novel main-chain anion-binding site in proteins: the nest. A particular combination of phi,psi values in successive residues gives rise to anion-binding sites that occur commonly and are found often at functionally important regions. J. Mol. Biol. 315:171-182.
Watson JD, Milner-White EJ (2002b) The conformations of polypeptide chains where the mainchain parts of successive residues are enantiomeric. Their occurrence in cation and anion-binding regions of proteins. J. Mol. Biol. 315:183-191.
Watson JD, Laskowski RA, Thornton JM (2005) Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 15:275-284.
Watson JD, Sanderson S, Ezersky A, et al. (2007) Towards fully automated structure-based function prediction in structural genomics: a case study. J. Mol. Biol. 367:1511-1522.
Wollacott AM, Zanghellini A, Murphy P, et al. (2007) Prediction of structures of multidomain proteins from structures of the individual domains. Protein Sci. 16:165-175.
Xenarios 1, Salwinski L, Duan XJ, et al. (2002) DIP, database of interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 30:303-305.
Yakunin AF, Yee A A, Savchenko A, et al. (2004) Structural proteomics: a tool for genome annotation. Curr. Opin. Chem. Biol. 8:42-48.
Глава 11
Примеры: предсказание функции структур, полученных в проектах по структурной геномике
Джеймс Д. Уотсон, Джанет М. Торнтон
Развитие технологий высокопроизводительного определения структур белков в различных проектах по структурной геномике по всему миру привело к появлению в базе данных PDB нескольких тысяч таких структур. Однако из-за природы отбора объекта для кристаллизации и необходимости быстрого опубликования получаемых данных значительная доля этих структур почти или совсем не имеет информации о функции. Чтобы решить эту проблему было разработано огромное множество вычислительных методов, выполняющих предсказание функции белка исходя из его пространственной структуры. Диапазон этих методов простирается от масштабных сравнений укладок белков до высокоспецифичного моделирования отдельных остатков, а сами методы имеют свои преимущества и недостатки. Здесь мы рассматриваем применение этих методов в структурной геномике и делаем обзор попыток определить, насколько успешным оказалось предсказание функции белка исходя из его структуры, иллюстрируя успешные случаи конкретными примерами.
11.1. Введение
Проекты по секвенированию генома, реализуемые по всему миру, уже дали огромное количество информации о генах, существенных для ряда организмов, и количество этой информации быстро растет благодаря
James D. Watson and Janet M. Thornton
European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, UK
*e-mail: thornton@ebi.ac.uk
346
Глава 11. Структуры, полученные в проектах по структурной геномике
масштабным проектам по метагеномике, реализуемым в настоящее время (Yooseph et al. 2007). По сравнению с таким бурным ростом число имеющихся белковых структур остается далеко позади. Целью структурной геномики является устранение этого отставания путем высокопроизводительного получения большого числа новых структур, которые можно использовать для сравнительного моделирования еще большего числа белков (Fox et al. 2008; Service 2005). Следствием такого подхода стало получение большого числа структур, содержащих мало информации об их функции или вовсе её не содержащих. Это находится в прямом противоречии с традиционной структурной биологией, где функция белка зачастую заранее известна, а расшифровку структуры проводят для определения биохимических механизмов функционирования белка и их возможных нюансов. Экспериментальное определение функции - это очень ресурсоемкий процесс, поэтому, столкнувшись с большим числом структур с неизвестной функцией, современная биоинформатика поставила себе целью точное и автоматическое предсказание функции белков. В настоящее время существует множество вычислительных методов, направленных на предсказание функции, многие из которых подробно обсуждались в предыдущих главах, но все их в действительности можно раделить на две большие категории: методы, основанные, главным образом, на структуре изучаемого белка и методы, основанные на его последовательности.
Анализ последовательности обычно является первым шагом на пути предсказания функции белка, поскольку высокое сходство последовательностей по-прежнему является наиболее надежной основой для такого предсказания. В ряде работ было показано, что гомологичные белки, имеющие последовательности, идентичные на 40% и больше, скорее всего имеют и одинаковую функцию (Todd et al. 2001). Однако при предсказании функции следует быть осторожным, поскольку есть ряд исключений из этого правила, когда почти идентичные белки имеют разные функции или, напротив, одинаковой функцией обладают белки без явного сходства последовательностей (Whisstock and Lesk 2003). Развитие мощных и чувствительных методов, использующих профили и мотивы, расширило наши возможности по распознаванию схожих функций у все более далеких последовательностей. Среди других методов, призванных помочь нам в определении функции, можно назвать учет консервативности остатков, филогенетические профили, расположение генов и масштабная организация генома. Когда сама последовательность недостаточно четко указывает на возможную функцию или базы данных не содержат гомологов, на помощь приходит структура белка. Поскольку элементы структуры часто консер
11.2. Примеры масштабного предсказания функции белков
347
вативны из-за консервативности его функции, то методы, использующие структурные данные, могут распознать более удаленное родство, чем методы, использующие только данные о последовательности. Диапазон этих методов простирается от масштабных сравнений укладки белков (Krissinel and Henrick 2004; Holm and Sander 1995) или их биологически активных комплексов (Krissinel and Henrick 2007) (см. тж. Главу 6), через сравнение рельефа поверхности белков (Laskowski 1995; Glaser et al. 2006; Binkowski et al. 2004) (см. тж. Главу 7) к сравнению высокоспецифичных пространственных групп функциональных остатков (Laskowski etal. 2005а; Stark and Russell 2003; Kristensen et al. 2008) (см. тж. Главу 8).
Ни один из методов не гарантирует 100% успеха, поэтому нужно поступать более осторожно и использовать максимально возможное число методов для предсказания функции: чем более независимые методы сойдутся в своих предсказаниях, тем более надежным будет результат. Как следствие, появился ряд серверов, реализующих различные методы предсказания функции. Некоторые из них, такие как сервер ProKnow (Pal and Eisenberg 2005), пытаются сделать консенсусное предсказание, в то время как другие, например, сервер ProFunc (Laskowski et al. 2005b) предоставляют пользователю возможность самому интерпретировать результаты различных методов (см. Главу 10). Однако возникает вопрос, насколько успешными попытки предсказания функции белков на основе их структуры были на самом деле? В этой главе на примере проектов по структурной геномике мы рассмотрим различные попытки ответить на этот вопрос и встретившиеся при этом трудности.
11.2. Примеры масштабного предсказания функции белков
Несмотря на то, что проекты по структурной геномике привели к получению огромного количества структур за последние годы, насчитывается неожиданно мало примеров использования этих структур для предсказания их функции. Здесь мы рассмотрим различные попытки улучшить эффективность предсказания функции белков на основе их структуры, используя некоторые мишени из структурной геномики. Краткое описание этих примеров и методов приведено в Таблице 11.1.
Обзор 15 гипотетических белков с известной пространственной структурой и процедуры предсказания их функций (Teichmann etal. 2001) дает некоторое представление о качестве таких предсказаний. Структуры в совокупности с выравниванием гомологичных последовательностей были
348
Глава 11. Структуры, полученные в проектах по структурной геномике
Таблица 11.1. Примеры предсказания функции для масштабного анализа и их источники.
В таблицу сведены рассматриваемые в обзорах примеры, в которых была предпринята попытка улучшить эффективность предсказания функции белков на основе их структуры, используя некоторые мишени из структурной геномики. Для каждого из белков в публикациях, рассмотренных в Разделе 11.3, дается краткое описание проведенного анализа вместе с отметкой в соответствующем столбце, показывающей, какой из методов анализа структуры оказался наиболее информативным
Исследование Белок Описание Ключевой метод анализа, позволивший определить функцию
Укладка/ структура (см. Главу 6) Поверхность/ расселина (см. Главу 7) Шаблон (см. Главу 8) Связанный лиганд
Kim et al. (2003) MJ0882 Определение потенциальной метилтрансферазной активности благодаря сходству в укладке - впоследствии экспериментально подтвержденное. X
MJ0577 Обнаружение связанной АТФ позволило предположить АТФ-гидролизную активность. X
TM841 Обнаружение связанного пальмитата показало возможность связывания жирных кислот. X
MJ0226 Обнаружение нового фолда со слабым сходством с нуклеотид-связывающими белками и белком НАМ 1. X
MJ0285 Многомерная структура, образующая полую сферу с окошками, вызвала вопросы о принципе действия. X
MPN625 Обнаружение двух консервативных цистеинов, лежащих в углублении потенциального активного центра, схожего с активным центром в семействе 2-цистеин пероксиредоксинов. X
Watson et al. (2007) BioH Новая карбоксилэстераза. Поиск по шаблону ферментативного активного центра выявил каталитическую триаду X
IsdG Сравнение укладок и методы обратного шаблона указали на монооксигеназную активность X X
Adams et al. (2007) ChuS Три из четырех консервативных гистидинов оказались примыкающими к одной из двух больших расселин или смотрящими в неё, демонстрируя необычную координацию гема. X
11.2. Примеры масштабного предсказания функции белков
349
Окончание таблицы 11.1
Исследование Белок Описание Ключевой метод анализа, позволивший определить функцию
Укладка/ структура (см. Главу 6) Поверхность/ расселина (см. Главу 7) Шаблон (см. Главу 8) Связанный лиганд
YgiN Сходство укладки с белком ActVA-Orf6, принадлежащим семейству монооксигеназ. Обнаружение двух более ранних сообщений, предоставивших дополнительные аргументы в пользу этого сходства. X
YjjX Сходство укладки с нуклеотид-связываю-щими белками. Пристальное изучение активного центра выявило значительные сходства, что позволило предположить новую ИТФ/КТФ*-азную активность. X X
YhhW Сходство укладки с многофункциональным семейством. Локальные сходства поверхности указали на глубокий заряженный карман рядом с металл-связывающим центром. Имеет значительное сходство с кверцитин 2,3-диоксигеназой. X X
z3393 Общее структурное сравнение и локальное сравнение молекулярной поверхности позволяет предположить гентизат 1,2-диоксигеназную активность. X X
* ИТФ - инозинтрифосфат, ITP; КТФ - ксантозинтрифосфат, ХТР)
использованы для определения углублений на поверхности белка, и если углубления были образованы консервативными остатками, то это указывало на активный центр. Используя информацию о всех возможных кофакторах в структурах и доступные экспериментальные данные для рассматриваемого белка или родственных последовательностей, было сделано настолько точное предсказание функци, насколько это позволяла существующая информация. Оказалось, что из 15 белков подробное предсказание было выполнено для четверти, еще для половины удалось получить хоть какую-то информацию о функции, а для оставшейся четверти не удалось получить вообще никакой информации.
В 2003 Kim с сотр. опубликовали анализ восьми структур, часть из которых была получена в Берклневском Центре по Структурной Геномике. Этот анализ позволил взглянуть на структуры с функциональной или эволюционной точки зрения и разделить их на пять категорий:
350
Глава 11. Структуры, полученные в проектах по структурной геномике
1. Удаленные гомологи. Здесь вывод о функции белка делается на основе структурного сходства, которое, однако, не вытекает из сравнения последовательностей. В качестве примера авторы приводят белок MJ0882, который сначала был отнесен к метилтрансферазам на основе сходства укладки, а затем это было экспериментально подтверждено (Huang et al. 2002).
2. Белки с неожиданно присутствующими лигандами. Здесь вывод о функции делается из случайно обнаруживающегося лиганда или кофактора. Первый пример, анализ белка MJ0577 из Methanococcus jannaschii, включил в себя обнаружение в структуре связанной АТФ, что позволило предположить АТФ-гидролитическую функцию. Более тщательный анализ АТФ-свя-зывающего кармана в MJ0577 показал наличие нескольких мотивов, характерных для нуклеотид-связывающих белков, но их взаимное расположение отличалось от существующих аналогов, поэтому традиционными методами обнаружения мотивов их выявить не удалось. Последующая экспериментальная проверка подтвердила АТФ-гидролитическую функцию, но только в присутствии клеточного экстракта, что означает, что белок является молекулярным переключателем и требует для своего функционирования одного или нескольких белков-партнеров. Второй пример, анализ белка ТМ841 из Thermotoga maritime, показал, что этот белок принадлежит большому семейству DegV по классификации Pfam и группе белков COG 1307, функция которых не известна. Получение структуры ТМ841 показало наличие связанной молекулы пальмитиновой кислоты, т.е. способность белка связывать жирные кислоты. Сравнение с другими членами семейства DegV и группы COG 1307 выявило большую консервативность в области связывания карбоксильной группы кислоты и меньшую в области связывания хвоста, позволяя предположить, таким образом, что различные члены этих семейств могут селективно связывать жирные кислоты с различной длиной хвоста.
3. «Сумеречные белки». Здесь ни последовательность, ни структура не позволяют сделать однозначный вывод о функции белка. В представленном примере структура белка MJ0226 имела новую укладку, но была слегка похожа на нуклеотид-связывающие белки (Hwang etal. 1999). Экспериментальный анализ позволил определить биохимическую функцию как новую нуклеотид-трифосфатазу. В совокупности с небольшим сходством с белком НАМ1 (Noskov etal. 1996) это позволило авторам предположить, что роль белка состоит в предотвращении мутаций посредством удаления нестандартных нуклеотид-трифосфатов. Это предсказание позже было подтверждено в эксперименте по комплементации (Stepchenkova et al. 2005).
4. Новая молекулярная функция при известной клеточной функции. Здесь общая функция белка известна, но биохимические детали механизма раскрываются при изучении его структуры. В первом цитированном
11.2. Примеры масштабного предсказания функции белков
351
примере, анализе белка MJ0285 из М. Jannaschii, белок был аннотирован как небольшой белок теплового шока, появляющийся при внутриклеточном стрессе. Структура показала, что 24 молекулы белка образуют полую сферу с восемью треугольными «окошками» и шестью квадратными (Kim etal. 1998). Исходя из этих данных был поставлен вопрос, захватываются ли частично денатурированные белки внутрь сферы или прикрепляются к ней снаружи? Результаты биохимических экспериментов дали веские аргументы в пользу того, что частично денатурированные клеточные белки в случае стресса прикреплены к сфере снаружи, что предохраняет их от агрегации и инактивации. Во втором примере анализируется белок MPN625, являющийся членом семейства OsmC. В это семейство входят довольно несхожие последовательности, но множественное выравнивание позволяет выявить два консервативных цистеина. Кристаллографическая структура MPN625 позволила увидеть, что два эти цистеина лежат в углублении потенциального активного центра, напоминающего активный центр в семействе 2-цистеин пероксиредоксинов, чья функция состоит в инактивации активных форм кислорода (Schroder et al. 2000). Таким образом, сравнение двух активных центров вместе с данными о клеточной функции позволило понять молекулярную функцию белков этого семейства и объяснить различие в специфичности к субстратам.
5. Белки, функция которых остется неизвестной. Здесь приводится два примера: белок Aql575 из Aquifex aeolicus и белок MPN314 из Mycoplasma pneumonia, которые оба являются гипотетическими белками, принадлежащими доменам с неизвестной функцией по классификации Pfam. Данные о консервативности остатков позволяют предположить возможные активные центры для обоих белков, но изучение баз данных как мотивов, так и функций не смогло дать каких-либо ключей к пониманию молекулярной функции этих белков. Возможно самым широким анализом, выполненным к настоящему времени, стал анализ, выполненный Watson et al. (2007), которые оценивали эффективность сервера для предсказания функции белков по их структуре ProFunc, используя структуры, полученные в Центре по структурной геномике на Среднем Западе (Midwest Center for Structural Genomics, MCSG). В этой работе все 319 белков, полученных в MCSG на первой стадии проекта по структурной геномике PSI-1 (N1H/NIGMS Protein Structure Initiative), были классифицированы на те белки, функция которых известна, те белки, о функции которых можно сделать какие-либо предпололожения, и те белки, о функции которых ничего не известно. Дальнейшей оценки подвергались только белки с известной функцией, поскольку целью работы являлась оценка того, насколько успешно алгоритмы ProFunc смогут эту функцию предсказать. В итоге серверу было предложено 93 белка с извест
352
Глава 11. Структуры, полученные в проектах по структурной геномике
ной структурой, а в ответ были получены и сохранены совпадения с наивысшими оценками, предсказанные каждым методом. Затем результаты были соотнесены с датами размещения каждой структуры, чтобы убедиться в том, что рассчитанные оценки действительно показывают, насколько успешным мог бы быть сервер, будь он использован для априорного предсказания функции. Наконец, наилучшие итоговые предсказания каждого из методов были сопоставлены с известной функцией каждого из белков и на основе этого было сделано заключение о правильности предсказания.
Результаты исследования показали, что из всех методов, являющихся компонентами сервера ProFunc, метод распознавания фолда и метод «обратного шаблона» оказываются самыми успешными, правильно предсказывая функцию приблизительно в 60% случаев. Подробное рассмотрение показало, что оба эти метода часто верно предсказывают функцию для одного и того же белка, но есть и примеры, где лишь один из методов оказывается успешным. Причина этого кроется в природе методов: в то время, как распознавание фолда нацелено на узнавание общего сходства в сравниваемых белках, метод «обратного шаблона» выполняет локальные сравнения. Одним из главных недостатков этого исследования стала его неспособность ответить на вопрос, что является более точным: предсказание функции белка по его структуре или по его последовательности? Однако это является общей проблемой, приемлемого решения которой в литературе к настоящему моменту не описано из-за внутренних трудностей, возникающих при аккуратном возврате к конкретной дате состояния баз данных последовательностей, равно как и построенных на их основе мотивов и профилей.
Кроме общего сравнения методов Уотсон с сотр (Watson et al. 2007) представили несколько специфических примеров предсказания функции, часть из которых была проверена. Примером успешного предсказания функции по структуре стал белок BioH из Escherichia coli (Sanishvili et al. 2003). Было известно, что этот белок принимает участие в синтезе биотина, но его биохимическая роль не была установлена. Анализ структуры с помощью ProFunc выявил высокозначимое соответствие (СКО 0,28 А) между шаблоном активного центра этого фермента и каталитической триадой липаз Ser-His-Asp. Сопоставление типов укладки с помощью DALI выявило структурное сходство белка BioH с множеством белков, имеющих различные ферментативные функции, хотя идентичность последовательностей этих белков оказалась низкой - 15-25%. Примерами наиболее полного соответствия стали бромопероксидаза (КФ 1.11.1.10), аминопептидаза (КФ 3.4.11.5), две эпоксидгидролазы (КФ 3.3.2.3), две галоал-кандегалогеназы (КФ 3.8.1.5) и лигаза (КФ 4.2.1.39). Лишь тщательный анализ этих ферментов вручную и обзор литературы мог бы показать с та
11.2. Примеры масштабного предсказания функции белков
353
кой же ясностью, что все эти ферменты имеют каталитическую триаду Ser-His-Asp в своих активных центрах. В то же время поиск по шаблону ферментативного активного центра позволил обнаружить наличие таких триад мгновенно. Экспериментальное изучение белка BioH показало, что он является новой карбоксилэстеразой, действующей на субстраты с короткой ацильной цепью (Sanishvili et al. 2003).
Другим примером, показывающим, как сведения о функции могут быть получены путем анализа структуры, является гипотетический белок (IsdG) из Staphylococcus aureus. Анализ последовательности с помощью ProFunc выявил множество функций, включая монооксигеназную, цистеинпептидазную, оксидоредуктазную, метилтрансферазную, эпимеразную, транспортную, потенциальную РНК-связывающую и другие. После того, как структура была проверена с помощью сервиса MSDfold/SSM, оказалось, что все наиболее подходящие типы укладки соответствуют гипотетическим белкам без функциональной аннотации, а остальные, менее подходящие типы, различным монооксигеназам. Не было выявлено никакого значительного соответствия ни с ферментами, ни с ДНК или лигандами, но сканирование по обратным шаблонам дало большое число соответствий. Опять же большая часть этих соответствий пришлась на белки с неустановленной функцией, но первым содержательным совпадением стала монооксигеназа из Streptomyces coelicolor (PDB код Hq9). Таким образом, результаты как сравнения фолдов, так и методов «обратного шаблона» указывают на монооксигеназную функцию. Последующий экспериментальный анализ позволил охарактеризовать белок как гем-разрушающий фермент, структурно схожий с монооксигеназами (Wu etal. 2005). Это прекрасный пример того, как анализ структуры дал дополнительные свидетельства в пользу одного из многих равноценных предсказаний, полученных из анализа последовательности.
В более поздней работе Адамса с сотр. (Adams et al. 2007) обсуждается аннотация функции гипотетических белков на основе их структуры. В работе приводится пять примеров, когда несколько методов в сочетании с биохимическими анализами позволили описать функцию белка. Первый пример - это белок (ChuS) с новым типом укладки, для которого исследования оперона и нокаут гена позволили предположить, что белок участвует в захвате и утилизации гема. Структура была получена для апо-формы белка и, поскольку тип укладки оказался совершенно новым, первоначальное предсказание функции на основе структуры не принесло конкретных результатов, но последующий биохимический анализ позволил предположить гемоксигеназную функцию. Множественное выравнивание последовательности ChuS с его гомологами выявило четыре консерватив-
354
Глава 11. Структуры, полученные в проектах по структурной геномике
ных гистидина, три из которых со структурной точки зрения примыкали или были направлены в одну из двух широких расселин на противоположных сторонах белковой глобулы. Это наблюдение подтолкнуло дальнейшие структурные исследования по совместной кристаллизации белка ChuS с гемом и мутагенезу консервативных гистидинов, в результате чего было обнаружено, что координация гема происходит здесь иначе, чем в других расщепляющих гем ферментах, и что ChuS является первой гемо-ксигеназой идентифицированной в Е. coli.
Во втором примере рассматривается случай белка YgiN. С помощью веб-сервера MSDfold (SSM) его структура была сопоставлена с типичными структурами по классификации SCOP и показала сходство укладки с белком ActVA-Orf6 - монооксигеназой из S. coelicolor.
Представители семейства этой монооксигеназы участвуют в синтезе больших поликетидных соединений при биосинтезе антибиотиков в Грам-пол ожительных бактериях. Белок ActVA-Orf6 действует как фермент на поздних стадиях процесса, подстраивающего противогрибковое соединение - дигидрокалафунгин - для придания ему специфичной активности (Sciara et al. 2003). Поскольку для £. coli продукция такого соединения не была описана, то ожидалось, что природный субстрат для YgiN будет отличаться. Первые попытки биохимически охарактеризовать фермент оставались бесплодными до тех пор, пока в литературе не были обнаружены два более ранних сообщения, на самом деле относящихся к белку YgiN. Эти дополнительные экспериментальные данные, а также уже использо-вавшайся структурная информация о различных субстратах, позволила авторам сделать предположение об участии белка YgiN в метаболизме менадиона. Дальнейшая экспериментальная работа с этим белком позволила закристаллизовать его как в апо-форме, так и с менадионом и с флавина-дениндинуклеотидом (Adams and Jia 2006).
Третим примером является белок YjjX, для которого предположения о функции нельзя сделать ни на основе его расположения в геноме, ни на основе мотивов в последовательности. Структура этого белка имеет укладку, сходную с рядом нуклеотид-связывающих белков (включая ранее рассмотренный белок MJ0226 из работы Kim et al. 2003). Детальное рассмотрение активных центров YjjX и обнаруженных структурных совпадений позволило выявить значительное сходство в ряде консервативных и полуконсер-вативных остатков. Дальнейший биохимический анализ дал основания классифицировать белок YjjX как новую инозинтрифосфатазу/ксантозин-трифосфатазу, которая действует в Е. coli как служебный фермент во время окислительного стресса для предотвращения накопления неканонических оснований и их последующего встраивания в нуклеиновые кислоты.
11.3. Несколько особых примеров
355
Четвертый пример отличается от предыдущих тем, что включает аннотацию белка внутри надсемейства. В этом примере была определена структура белка YhhW (ранее аннотированного как принадлежащего надсемейству купинов) и, как и ожидалось, в ней был обнаружен белковый остов, аналогичный имеющемуся у известных купинов, однако анализ последовательности давал сильные аргументы в пользу близкого родства с пиринами. Большое разнообразие функций в надсемействе купинов не позволяет составлять аннотации на основе общего структурного сходства, поэтому авторы обратились к локальным сходствам на поверхности белков. Обнаружение глубокого заряженного кармана рядом с металл-связы-вающим сайтом в YhhW и одним из его гомологов h-пирином позволило предположить, что он является активным центром. Рассмотрение этого кармана выявило значительное сходство с карманом в кверцитин-2,3-диоксигеназе, которое было дополнительно подтверждено успешным докингом кверцитина в гомологи пирина. Кверцитин-2,3-диоксигеназная активность была подтверждена затем и биохимическими тестами и стала первой ферментативной функцией, определенной для белков пиринового семейства. Этот пример иллюстрирует также проблемы, возникающие при работе с большими белковыми надсемействами. Часто общее сходство, такое, как сходство укладки, оказывается недостаточным для определения функции и необходим более детальный анализ.
Последний пример из работы Адамса касается другого представителя надсемейства купинов - продукта гена z3393 из Е. coli. Его последовательность показывает, что он ближе к гентизат-1,2-диоксигеназе, чем к остальным купинам. Сравнение общей структуры и локальных свойств молекулярной поверхности также выступает в пользу гентизат-1,2-диокси-геназной активности, и авторы надеятся, что полученная ими структура z3393 поможет будущим исследованиям фермента с механистической точки зрения и последующему пониманию того, как гентизат-оперон может быть связан с патогенными штаммами Е. coli.
11.3. Несколько особых примеров
Хотя имеется относительно немного масштабных работ, есть большое число отдельных интересных структур, опубликованных различными консорциумами по структурной геномике или в сотрудничестве с ними, где наличие структуры белка оказывалось принципиально важным для описания его функции. Одним из таких примеров является белок Тт0936 из Thermotoga maritima (имеющий PDB код 2р1т), который был получен в
356
Глава 11. Структуры, полученные в проектах по структурной геномике
сторонней лаборатории с использованием клонов, предоставленных Объединенным Центром по Структурной Геномике. Белок был аннотирован в базах данных, как белок с неизвестной функцией, принадлежащий семейству амидогидролаз по классификации Pfam (PF01979), которое содержит ряд деаминаз и, в свою очередь, является частью более широкого надсемейства амидогидролаз. Как пример белка с неизвестной функцией Тт0936 был отобран для анализа с помощью сервера ProFunc, и в результате обнаружилось возможное сходство с одним из 189 используемых сервером известных шаблонов ферментативных активных центров.
Схожим активным центром (с математическим ожиданием случайности сходства Е = 2.45 х 104) оказался шаблон активного центра аденозиновой деаминазы (КФ 3.5.4.4), полученный из структуры с PDB кодом 1а41, которая участвует в пуриновом метаболизме. Общая идентичность последовательностей между этой структурой и Тт0936, вычисленная по парному выравниванию с помощью программы FASTA, составила лишь 24%, хотя структурное сходство (рассчитанное для структурного выравнивания сервером ProFunc, как доля пар остатков, лежащих в одном или нескольких совмещаемых сегментах, в общем числе эквивалентных остатков в выравнивании) достигает 95%, что говорит само за себя. Внутри сферы с радиусом 10 А вокруг шаблона активного центра локальная идентичность последовательностей составляет 27,7%, что выше среднего по структуре и свидетельствует о большем сходстве между последовательностями в области вокруг активного центра. Кроме сильного сходства с аденозиновой деаминазой, было обнаружено и несколько совпадений обратных шаблонов с другими деаминазами и амидогидролазами.
Авторы структуры Тт0936 опубликовали работу, в которой предсказывают для этой структуры функцию адениндеаминазы (Hermann et al. 2007). Использованный ими подход включал проведение молекулярного докинга высокоэнергетических метаболических интермедиатов в структуру белка на том основании, что докинг исходных субстратов или продуктов может быть не столь эффективным, как докинг интермедиатов, стабилизированных ферментом. Получившийся список потенциальных лигандов состоял преимущественно из аналогов аденина, которые оказались хорошо подходящими для Сб-деаминирования. Четыре из этих лигандов были протестированы в качестве субстратов, причем три из них показали существенную каталитическую константу скорости. Была определена структура комплекса между Тт0936 и продуктом (S-инозилгомоцистеином), образовавшимся при деаминировании S-аденозилгомоцистеина, и обнаружено очень точное соответствие между этим лигандом, связанным с Тт0936, и дезоксикоформицином, аналогом инозина, связанным со струк
11.3. Несколько особых примеров
357
турой, послужившей шаблоном при определении структуры Tm0936 (PDB код 1а41) (Рис. 11.1)
Интересно, что анализ фолда с использованием программы MSDfold показывает сходство с различными амидогидролазами и гуанин/цитозин-деаминазами. Причина, по которой этот сервер не выделяет структуру аденозиндеаминазы, как наиболее похожую, кроется в том, что структура самого Тт0936 имеет некоторые украшения за пределами совпадающей области, и структурное сходство падает ниже критического уровня в 70% по числу совпадающих элементов вторичной структуры (Рис. 11.2). Это наблюдение подчеркивает эффективность локальных сравнений и служит хорошим примером, когда функция может быть точно определена исходя из совпадения с шаблоном фермента, которое осталось бы незамеченным при использовании одних лишь методов анализа последовательности. Другой интересный пример можно найти в недавней публикации Центра по структурной геномике на Среднем Западе (MCSG), где описываются структуры открытого (R) и закрытого (Т) состояний префенатдегидратазы (PDT) (Tan et al. 2008), проясняющие наше понимание аллостерической регуляции этого фермента с помощью L-фенилаланина и других аминокислот. Префенатдегидратаза (КФ 4.2.1.51) превращает префенат в фенилпируват при биосинтезе L-фенилаланина и играет ключевую роль в этом процессе у организмов, использующих шикиматный метаболический путь, что делает этот фермент незаменимым для микроорганизмов. У человека этот фермент не обнаружен, и это означает, что он может быть выбран в качестве возможной мишени при разработке противомикробных препаратов.
Структуры префенатдегидратазы, размещенные в PDB (коды 2qmw и 2qmx), происходят из двух различных организмов и являются первыми кристаллографическими структурами PDT в расслабленном (R) и возбужденном (Т) состояниях (из Staphylococcus aureus и Chlorobium tepidum, соответственно). Эти ферменты демонстрируют низкую идентичность последовательностей (27,3%), но одинаковую общую архитектуру и доменную организацию: оба фермента являются тетрамерами (образуя димеры димеров - см. изображения индивидуальных димеров на Рис. 11.3) и состоят из каталитического домена (домена PDT) и регуляторного домена (домена ACT). Опираясь на эти структуры префенат дегидратаз, авторы предположили, что активный центр этого фермента располагается в зазоре между двумя доменами PDT.
Это предсказание, сделанное на основе структурного сходства, подтверждается так же анализом последовательности и данными по мутагенезу. Множественное выравнивание последовательностей и картирование выявленных консервативных остатков показало, что эти остатки локализованы
358
Глава 11. Структуры, полученные в проектах по структурной геномике
Рис. 11.1. (Цветную версию рисунка см. на вклейке.) Соответствие ферментативного активного центра шаблону, иллюстрирующее перекрывание между связанным S-инозитол-гомоцистеином в Тт0936 (показан оранжевым; PDB код 2plm) и дезоксикоформицином, аналогом инозина, связанным с шаблоном, присутствующим в аденозицдеаминазе (показан фиолетовым, PDB код 1а41). Связанные атомы цинка показаны перекрывающимися сферами тех же цветов, что и соответствующие лиганды. Остатки Тт0936 и аденозиндеаминазы показаны синим и красным, соответственно
Рис. 11.2. Стереоизображение наложения рассматриваемой структуры Tm0936 (PDB код 2plm - показана черным) на структуру, использовавшуюся при создании шаблона ферментативного активного центра (PDB код 1а41 - показана серым). Дополнительные элементы вторичной структуры в рассматриваемом белке можно легко увидеть в левой части изображения
на дне расселины между субдоменами. Остатки, по данным мутагенеза критичные для активности префенатдегидратазы в Е. coli, оказались эквивалентными остаткам в расселине между двумя PDT субдоменами (Zhang et al. 2000). Дополнительные данные мутагенеза для префенатдегидратазы в Corynebacterium glutamicum подтвердили, что эквивалентные остатки участвуют в связывании субтрата и/или каталитической активности (Hsu et al. 2004). В итоге, данные подтверждают, что и расселина, и консервативные остатки в ней образуют активный центр префенатдегидратазы, при этом Т168 является наиболее вероятным ключевым каталитическим остатком.
За определением вероятного активного центра последовало определение аллостерического сайта. Расположение этого сайта связывания L-фенил-аланина в префенатдегидратазе сходно с расположением сайта связывания эффектора в некоторых других ферментах, имеющих домен ACT и участвующих в связывании аминокислот или других небольших молекул. Наличие
11.3. Несколько особых примеров
359
Димер PDT-домена
Рис. 11.3. Структуры префенатдегидратазы, полученные в Центре структурной геномики на Среднем Западе, иллюстрируют сходства и отличия: а) структура R-состояния из Staphylococcus aureus (PDB код 2qmw); б) структура Т-состояния из Chlorobium tepidum (PDB код 2qmx). Идентичность последовательностей этих ферментов составляет лишь 27,3%
Димер АСТ-домена
PDT-домен АСТ-домен
Рис. 11.4. Мономер префенатдегидратазы из Staphylococcus aureus. Каждый домен окрашен в свой цвет. Предполагаемый активный центр расположен в расселине между двумя доменами (обведен)
связанного L-фенилаланина в структуре позволило визуализовать его взаимодействия с доменами ACT в структуре белка из Chlorobium tepidum. Рассмотрение связывающих остатков показало, что по большей части взаимодействия, вероятно, не являются специфическими, что служит объ
360
Глава 11. Структуры, полученные в проектах по структурной геномике
яснением тому факту, что и другие аминокислоты, такие, как метионин, также могут связываться в этом сайте и регулировать каталитическую активность (Liberies et al. 2005).
Сравнение структур префенатдегидратазы из Staphylococcus aureus и Chlorobium tepidum показывает, как связывание L-фенилаланина изменяет конформацию димера ACT. В работе Тан с сотр. (Tan et al. 2008) подробно обсуждается вопрос распространения этих изменений на активный центр, что приводит к блокированию ферментативной активности. Авторы предполагают, что связывание L-фенилаланина вызывает ряд крупных конформационных перестроек в префенатдегидратазе (локальных и глобальных), которые изменяют относительную ориентацию доменов в белке в целом, что приводит к изменению доступности активного центра. Подробнее, эти перестройки приводят к разделению одного широкого просвета, направленного к каталитическому центру в середине димера PDT, на два меньших просвета, что затрудняет доступ префената к каталитическому центру и высвобождение из него фенилпирувата. Этот пример показывает, как анализ структур, полученных в рамках проекта по структурной геномике может иметь значение, выходящее за пределы простого предсказания функции.
Еще одним примером из работ Центра по структурной геномике на Среднем Западе служит белок AF0491 из A. fulgidus (Savchenko etal. 2005), гомологичный белку синдрома Швахмана-Даймонда (СШД) человека. СШД - это редкое аутосомальное рецессивное заболевание, вызванное мутациями в гене SBDS седьмой хромосомы и характеризующееся ненормальной экзокринной функцией поджелудочной железы, дефектами скелета и гематологической дисфункцией (Boocock et al. 2003). Белок AF0491 является архейным гомологом и определение его структуры позволило выявить у него трехдоменное строение (Рис. 11.5).
С-концевой домен имеет широко распространенную укладку, что затрудняет определение его функции. Известно, однако, что такие домены обнаружены во многих РНК- и ДНК-связывающих белках. Центральный домен также имеет распространенную укладку - крыловидный мотив «спираль-поворот-спираль» (winged helix-tum-helix, wHTH). Такой домен часто используется при связывании ДНК (Aravind et al. 2005), а также встречается в РНК-связывающих белках (Schade etal. 1999). В данном случае, однако, поверхность AF0491 не имеет ожидаемого основного характера, поэтому маловероятно, что функция домена состоит в связывании нуклеиновых кислот. Скорее, как это предположили авторы, домен может участвовать в белок-белковых взаимодействиях.
N-концевой домен имеет новый тип укладки, и именно в этом домене локализована большая часть связанных с заболеванием мутаций, которые
11.3. Несколько особых примеров
361
Рис. 11.5. Мономер белка AF0491 из A. fulgidus, гомологичный белку синдрома Швахмана-Даймонда (СШД) человека. Три домена этого белка показаны светло-серым, темно-серым и черным в порядке от N- к С-концу
выявлены у пациентов с СШД. При последующем структурном поиске этот же тип укладки был найден в дрожжевом белке YHR087W. Обнаружение этого структурного гомолога дает возможность провести дополнительные эксперименты, неосуществимые с человеческим белком. Экспериментальное изучение гомологов белка СШД по структуре и последовательности (YHR087W и YLR022C соответственно) указывает на их связь с метаболизмом РНК. Штаммы с удаленным геном YLR022C оказались нежизнеспособными, но белки, помеченные TAP-тагом, выделялись совместно с многочисленными рибосомальными белками и белками, участвующими в процессинге рРНК. Штаммы с удаленным геном YHR087W оказались жизнеспособными, поэтому такой штамм был скрещен с другими 383 штаммами, у каждого из которых был удален какой-либо ген из тех, что отвечают за белки, участвующие в метаболизме РНК, и в ряде таких комбинация наблюдалась заметная летальность. Это наблюдение и все генетические взаимодействия, выявленные для YHR087W, говорят в пользу участия этого белка в процессинге РНК. Несмотря на то, что эти данные указывают на связь белка СШД с рибосомальным биогенезом, конкретная роль этого белка в метаболизме остается неизвестной, и фундаментальные отличия между рибосомальным биогенезом в бактериях (Lecompte et al. 2002), эукариотах и археях означают, что любые выводы о функции должны внимательно анализироваться. Однако белок СШД является примером, когда определение структурного бактериального гомолога для человеческого белка помогло найти также гомолог и в дрожжах, что оказалось полезным для определениях их функции.
362
Глава 11. Структуры, полученные в проектах по структурной геномике
11.4. Коллективное аннотирование
Большому количеству белковых структур, расшифрованных в проектах по структурной геномике, не соответствует, к сожалению, столь же большое количество публикаций. Фактически, в настоящий момент опубликование структуры видится как одно из самых узких мест на отлаженном пути высокопроизводительной расшифровки структур (Rigden 2006). Это не говорит об отсутствии заинтересованности в публикациях, плохом выборе белка или отсутствии интересных структур. Скорее это говорит о том, что многим проектам не хватает такой неотъемлемой стадии, как быстрое опубликование своих результатов и предоставление доступа к ним для общественности. Это означает, что предсказание или экспериментальное определение функции, что часто бывает сопряжено с большими затратами времени, выполняется после появления структуры и часто в сотрудничестве с другими лабораториями, специализирующимися на конкретном изучаемом белке. Одним из способов исправления ситуации является сокращение времени, затрачиваемого на эти эксперименты, путем разработки высокопроизводительных тестов для скрининга ферментативной активности (Kuznetsova et al. 2005). Такие тесты оказались успешными (Proudfoot et al. 2004), но ограничения, связанные с реализацией ряда ключевых ферментативных реакций, означают, что предлагаемый способ пока не может быть расширен для определения любой функции. Другой подход к улучшению аннотации белков, являющихся объектами изучения структурной геномики, состоит в изучении возможности коллективного аннотирования этих белков с использованием технологии wiki (Giles 2007; Mons et al. 2008).
Одной из первых попыток реализации этого подхода стал проект TOPSAN (The Open Protein Structure Annotation Network, Открытая сеть по аннотированию белковых структур), стартовавший в Объединенном центре по структурной геномике (JCSG) и объединяющий сейчас данные для структур, полученных в Центре по структурной геномике на Среднем Западе (MCSG) и Исследовательском центре по структурной геномике в Нью-Йорке (New York Structural GenomiX Research Center, NYSGXRC). Проект основан на технологии wiki (http://www.topsan.org/), то есть доступен для просмотра любому пользователю, но заполнять страницы могут только зарегистрированные пользователи. Идея, заложенная в TOPSAN, состоит в том, что коллективное аннотирование глобальным сообществом экспертов, каждый из которых специализируется в своей конкретной области, может обеспечить гораздо более разностороннюю информацию обо всех доступных белках с известной структурой, чем та, которую смог бы когда-либо обеспечить отдельный специалист или небольшая их группа. Страницы с аннотациями будут, таким образом, предлагать широкой об
11.5. Заключение
363
щественности сочетание аннотаций, созданных автоматически, и тех, что составлены экспертами. Начальная версия вышла за рамки прототипа и теперь расширяется с намерением включить все мишени из проекта по белковым структурам (Protein Structure Initiative, PSI).
Более масштабным проектом является PDBWiki (http://pdbwiki.org/), который был создан в августе 2007 года в Группе структурной протеомики в Институте молекулярной генетики имени Макса Планка. В настоящее время проект охватывает все структуры в базе данных PDB, а на каждой странице наряду с основной информацией о белке представлены ссылки на другие базы данных и ряд средств для анализа последоваельности и структуры.
Следующим шагом стала новая энциклопедия, основанная на технологии wiki и базе данных PDB, и названная Proteopedia (http://www. proteopedia.org/). Первостепенная цель этой энциклопедии состоит в представлении структурной и функциональной информации о макромолекулах в таком виде, чтобы она была легко доступна студентам, ученым и общественности. Каждая структура в PDB имеет свою собственную страницу, заполняемую с использованием информации из базы данных ОСА (Prilusky 1996) и других источников. Есть ряд существенных различий между Proteopedia и PDBWiki, важнейшим из которых является уникальная система ссылок в тексте на сцены, создаваемые в апплете Jmol viewer (Proteopedia представляет полностью интерактивное изображение структуры белка, а не статическое). Используя эту систему, любой редактирующий страницу может легко создавать сцены, чтобы выделить ключевые области белка или ограничить взгляд областью, обсуждаемой в тексте. Это делает Proteopedia ресурсом большим, нежели простой набор статических страниц для отображения информации, создавая дополнительный уровень взаимодействия, который можно использовать для более наглядного представления идей и освещения интересных областей в структуре белка. Другой уникальной чертой сайта является отсутствие анонимных правок и полное имя пользователя сохраняется в истории правок страницы. У такого подхода есть еще одно преимущество: у пользователей может быть свои доступные для просмотра, но не доступные для редактирования области в системе. Это позволяет создание тематических статей или статей-примеров, которые остаются постоянными и таким образом могут использоваться как в обучающих целях, так и в целях коллективного аннотирования.
11.5. Заключение
Группами по структурной геномике было определено огромное количество структур, которые обогатили нашу сокровищницу пространственных структур белков. Однако из-за высокой скорости получения и опубли
364
Глава 11. Структуры, полученные в проектах по структурной геномике
кования этих структур, что являлось требованием проекта, большая доля структур почти или вовсе не имеет информации о функции. Возможность предсказания функции белка по его последовательности и структуре являлось для специалистов по биоинформатике чем-то вроде Святого Грааля, поэтому за годы работы в этом направлении было разработано большое разнообразие методов. Каждый из этих методов имеет свои «за» и «против», и многочисленные примеры исследовательских работ ярко показывают, как один подходящий метод может дать биологическое понимание там, где другие методы потерпели неудачу. В настоящее время ни один из методов не является успешным на 100%, поэтому для дальнейшего прогресса в нашем понимании необходимо продолжать разработку новых средств структурного анализа и предсказания функции.
Несколько попыток провести масштабный анализ возможности предсказания функции белка по его структуре окончились с переменным успехом, но есть ряд важных задач в этой области, которые требуют решения. Одной из главных задача, возникающих при разработке методов предсказания функции по структуре, является отсутствие набора данных, которые могли бы стать «золотым стандартом» при проверке этих методов. Как следствие, каждая группа разработчиков вынуждена создавать свой набор данных для тестирования, что приводит к сложностям при сравнении различных методов. Другим важным препятствием, которое должно быть преодолено, является задача сравнения методов предсказания по структуре и методов предсказания по последовательности. Хотя и не всегда невозможное, удаление информации из баз данных последовательностей и профилей, полученных на их основе, является гораздо более сложной задачей. Это применимо лишь к апостериорным видам анализа (как, например, выполненный Watson etal. 2007), но если бы были сохранены все предсказания функции белка, сделанные на момент появления его структуры, и в будущем проанализированы, то проблема была бы решена. Такая работа уже начата с использованием сервера ProFunc, результаты предсказания которого для структур, полученных в MCSG, сохраняются для дальнейшего анализа и сравнения. Все эти вопросы нетривиальны и потребуют внимания, если создаваемые наборы данных должны будут использоваться в будущем для оценки и сравнения методов. Чтобы увеличить вероятность обнаружения правильной функции, упор делается на создание комбинированных методов и использование всей доступной информации. Объединение существующих биоинформатических методов стало прямым ответом на большое число белков, почти или совсем не имеющих функциональной аннотации. Один из трудных вызовов, возникающих на пути предсказания функции белков, состоит в учете информации из всех облас
Литература
365
тей биологической науки, и поскольку количество доступной информации растет, то наметившийся уклон в сторону проектов коллективного аннотирования, вероятно, окажет главныю помощь в предоставлении более глубокого биологического понимания.
Благодарности. Авторы хотели бы поблагодарить Романа Ласковски (Roman Laskowski) и Вики Шнайдер (Vicky Schneider) за их ценные замечания к этой главе.
Литература
Adams МА, Jia Z (2006) Modulator of Drug Activity В from Escherichia coli: crystal structure of a prokaryotic homologue of DT-diaphorase. J Mol Biol 359:455-465
Adams MA, Suits MD, Zheng J, et al. (2007) Piecing together the structure-function puzzle: experiences in structure-based functional annotation of hypothetical proteins. Proteomics 7:2920-2932
Aravind L, Anantharaman V, Balaji S, et al. (2005) The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev 29:231-262
Binkowski, TA, Freeman P, Liang J (2004) pvSOAR: detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res 32: W555-W558
Boocock GR, Morrison JA, Popovic M, etal. (2003) Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet 33:97-101
Fox BQ Goulding C, Malkowski MG, et al. (2008) Structural genomics: from genes to structures with valuable materials and many questions in between. Nat Methods 5:129-132
Giles J (2007) Key biology databases go wiki. Nature 445: 691
Glaser F, Morris RJ, Najmanovich RJ, et al. (2006) A method for localizing ligand binding pockets in protein structures. Proteins 62:479-488
Hermann JC, Marti-Arbona R, Fedorov AA, et al. (2007) Structure-based activity prediction for an enzyme of unknown function. Nature 448:775-779
Holm L, Sander C (1995) Dali: a network tool for protein structure comparison. Trends Biochem Sci 20:478-480
Hsu SK, Lin LL, Lo HH, et al. (2004) Mutational analysis of feedback inhibition and catalytic sites of prephenate dehydratase from Corynebacterium glutamicum. Arch Microbiol 181:237-244
Huang L, Hung LW, Odell M, et al. (2002) Structure-based experimental confirmation of biochemical function to a methyltransferase, MJ0882, from hyperthermophile Methanococcus jannaschii. J Struct Funct Genomics 2:121-127
Hwang KY, Chung JH, Kim S-H, etal. (1999) Structure-based identification of a novel NTPase from Methanococcus jannaschii. Nat Struct Biol 6:691-696
Kim KK, Kim R, Kim S-H (1998) Crystal structure of a small heat shock protein. Nature 394:595-599 Kim SH, Shin DH, Choi IG, et al. (2003) Structure-based functional inference in structural genomics. J Struct Funct Genomics 4:129-135
Krissinel E, Henrick К (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1): 2256-2268
Krissinel E, Henrick К (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774—797
Kristensen, DM, Ward RM, Lisewski AM, et al. (2008) Prediction of enzyme function based on 31) templates of evolutionarily important amino acids. BMC Bioinformatics 9:17
Kuznetsova E, Proudfoot M, Sanders SA, et al. (2005) Enzyme genomics: application of general enzymatic screens to discover new enzymes. FEMS Microbiol Rev 29:263 279
Laskowski RA (1995) SURFNET: a program for visualizing molecular surfaces, cn\ii»cs. aimI m termolecular interactions. J Mol Graph 13:323-328
366
Глава 11. Структуры, полученные в проектах по структурной геномике
Laskowski RA, Watson JD, Thornton JM (2005a) Protein function prediction using local 3D templates. J Mol Biol 351:614-626
Laskowski RA, Watson JD, Thornton JM (2005b) ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res 33:W89-W93
Lecompte O, Ripp R, Thierry JC, et al. (2002) Comparative analysis of ribosomal proteins in complete genomes: an example of reductive evolution at the domain scale. Nucleic Acids Res 30:5382-5390
Liberies JS, Thaoraolfsson M, Martainez A (2005) Allosteric mechanisms in ACT domain containing enzymes involved in amino acid metabolism. Amino Acids 28:1-12
Mons B, Ashbumer M, Chichester C, et al. (2008) Calling on a million minds for community annotation in WikiProteins. Genome Biol 9:R89
Noskov VN, Staak K, Shcherbakova PV, etal. (1996) HAM1, the gene controlling 6-N-hydroxylaminopurine sensitivity and mutagenesis in the yeast Saccharomyces cerevisiae. Yeast 12:17-29
Pal D, Eisenberg D (2005) Inference of protein function from pwtein structure. Structure 13:121-130
Prilusky J (1996) OCA, a browser-database for protein structure/function. URL http://bip.weizmann. ac.il/oca and mirrors worldwide
Proudfoot M, Kuznetsova E, Brown G, et al. (2004) General enzymatic screens identify three new nucleotidases in E. coli: biochemical characterization of SurE, YfbR, and YjjG J Biol Chem 279:54687-54694
Rigden DJ (2006) Understanding the cell in terms of structure and function: insights from structural genomics. Curr Opin Biotechnol 17:457-464
Sanishvili R, Yakunin AF, Laskowski RA, et al. (2003) Integrating structure, bioinformatics, and enzymology to discover function: BioH, a new carboxylesterase from Escherichia coli. J Biol Chem 278:26039-26045
Savchenko A, Krogan N, Cort JR, et al. (2005) The Shwachman-Bodian-Diamond Syndrome protein family is involved in RNA metabolism. J Biol Chem 280:19213-19220
Schade M, Turner CJ, Lowenhaupt K, et al. (1999) Structure-function analysis of the Z-NAbinding domain Zalpha of dsRNA adenosine deaminase type 1 reveals similarity to the (alpha+ beta) family of helix-tum-helix proteins. EMBO J 18:470—479
Schroder E, Littlechild JA, Lebedev AA, et al. (2000) Crystal structure of decameric 2-Cys Peroxiredoxin from himan erythrocytes at 1.7 A resolution. Structure 8:605-615
Sciara G, Kendrew SG, Miele AE, et al. (2003) The structure of ActVA-Orf6, a novel type of monooxygenase involved in actinorhodin biosynthesis. EMBO J 22:205-215
Service R (2005) Structural biology. Structural genomics, round 2. Science 307:1554-1558 Stark A, Russell RB (2003) Annotation in three dimensions. PINTS: patterns in non-homologous tertiary structures. Nucleic Acids Res 31:3341-3344
Stepchenkova El, Kozmin SG, Alenin VV, et al. (2005) Genome-wide screening for genes whose deletions confer sensitivity to mutagenic purine base analogs in yeast. BMC Genet 6:31
Tan K, Li H, Zhang R, et al. (2008) Structures of open (R) and close (T) states of prephenate dehydratase (PDT) - implication of allosteric regulation by L-phenylalanine. J Struct Biol 162:94-107
Teichmann SA, Murzin AG, Chothia C (2001) Determination of protein function, evolution and interactions by structural genomics. Curr Opin Struct Biol 11:354-363
Todd A, Orengo C, Thornton JM (2001) Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol 307:1113-1143
Watson JD, Sanderson S, Ezersky A, et al. (2007) Towards fully automated structure-based function prediction in structural genomics: a case study. J Mol Biol 367:1511-1522
Whisstock JC, Lesk AM (2003) Prediction of protein function from protein sequence and structure. Q Rev Biophys 36:307-340
Wu R, Skaar EP, Zhang R, et al. (2005) Staphylococcus aureus IsdG and Isdl, heme-degrading enzymes with structural similarity to monooxygenases. J Biol Chem 280:2846-2846
Yooseph S, Sutton G, Rusch DB, et al. (2007) The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 5:el6
Zhang S, Wilson DB, Ganem В (2000) Probing the catalytic mechanism of prephenate dehydratase by site-directed mutagenesis of the Escherichia coli P-protein dehydratase domain. Biochemistry 39:4722-4728
Глава 12
Предсказание функции белков на основе их теоретических моделей
Ивона А. Симерман, Дэниэл Дж. Ригден, Януш М. Бужницки
В настоящее время моделирование на основании гомологии уже является сложившейся методикой, а моделирование de novo ценную информацию для небольших белков с не встречавшейся ранее укладкой. Успехи в области предсказания функции белка на основе его структуры, получившие развитие благодаря проектам по структурной геномике, привели к появлению целого набора методов, применимых к моделям любого происхождения. Есть, однако, и важные ограничения в точности моделей и, отчасти как следствие, в производительности алгоритмов по предсказанию функции, которые эти модели анализируют. Тем не менее, эта глава показывает, как функция белка может быть освещена с разных сторон различными методами моделирования, что зачастую облегчает планирование и объяснение результатов эксперимента. В то же время в установлении плодотворного диалога между моделистами и биологами-экспериментаторами остаются важные вопросы, решение которых позволит расширить практическое применение результатов моделирования. Базы данных, содержащие как сами модели белков, так и индикаторы их точности и надежности, в будущем могут иметь важное значение.
IwonaA. Су merman
International Institute of Molecular and Cell Biology, Trojdena4, 02-109 Warsaw, Poland
Daniel J. Rigden
School of Biological Sciences, University of Liverpool,
Liverpool L69 7ZB, UK
Janusz M. Bujnicki
Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University, Umultowska 89, 61-614 Poznan', Poland
e-mail: iamb@genesilico.pl
368 Глава 12. Предсказание функции белков на основе теоретических моделей
12.1. Введение
Стремительный прогресс вычислительной техники и рост возможностей по обмену данными, наблюдаемые в последние десятилетие, сильно повлияли на направление и методологию биологических исследований. Этот прогресс сделал возможным создание масштабных проектов, примерами которых являются проект по секвенированию генома, разработка микрочипов и структурная геномика. Это, в свою очередь, изменило направление отдельных исследований. Таким образом, вместо выявления генов и белков, определяющих наблюдаемый фенотип, ученые часто сосредотачиваются на поиске функций огромного числа последовательностей, содержащихся в базах данных. Очевидно, что изменение описательных подходов на предсказательные требует разработки новых методов. Самой важной информацией о конкретном гене или белке является связанная с ним функция. Наиболее общий подход в предсказании функции основывается на наблюдении, что имеющие сходную последовательность белки часто имеют и сходную функцию. Все возрастающее число доступных последовательностей способствует тому, что схожие последовательности могут быть выровнены и объединены в семейства. Если функция одного из членов семейства известна, то предполагается, что и остальные последовательности в семействе «наследуют» эту функцию. Это предположение ставит вопрос о том, нужно ли знание пространственной структуры белка для предсказания его функции или достаточно информации, содержащейся в последовательности. На первый взгляд можно ответить, что это зависит от сходства между сравниваемыми белковыми последовательностями. Общепринято, что идентичность последовательностей, превышающая 30%, является сильным аргументом в пользу того, что белки будут иметь очень сходную структуру, которая может быть предсказана методами, основанными на гомологии (Глава 3), давая в целом точные модели. Ниже этого порога, однако, предсказание структуры белка требует более изощренных подходов (таких, например, какие были описаны в Главах 1 и 2) и не является столь точным. Поскольку функция зависит от структуры, можно было бы подумать, что схожесть последовательностей определяет и схожесть функций, однако это необязательно, поскольку значительные вариации функции наблюдаются даже для белков с очень сходными последовательностями и структурами. Например, аннотации функций, основанные на данных генной онтологии (ГО, Gene Ontology) консервативны лишь для 80% белковых пар даже в тех случаях, когда белки имеют 90-100% идентичность последовательностей; если их идентичность ниже 30%, это консервативность аннотаций опускается ниже 50%. Некоторые аспекты
12.1. Введение
369
функции является более консервативными, чем остальные, например, если функция ферментов рассматривается согласно системе КФ, то все четыре индекса будут такими же с вероятностью почти 100% при идентичности последовательностей выше 70%, в то время как для последовательностей с идентичностью ниже 30% вероятность сохранения всех четырех индексов КФ падает ниже 50% (Tress et al. 2008).
Сохранение функции является более сложным явлением, чем сохранение структуры, поскольку перекрывание функций (например, идентичная функция двух копий гена после дупликации) подвержено увеличению скорости эволюции, которое, однако, зависит от полезности той функции белка, которая была закодирована в гене, позже подвергшемся дупликации (Jordan et al. 2004). Таким образом, дупликация, которая дает появление па-ралогичных белков с почти идентичными последовательностями и структурами, приводит либо к потере одной из копий из-за инактивирующей мутации (т.е. возвращению к предшествующему состоянию), либо к изменению функций одной или обеих копий, что уменьшает перекрывание.
С другой стороны, ортологичные последовательности имеют тенденцию к сохранению идентичных функций, зачастую несмотря на значительное различие в них самих. Однако парное сравнение последовательностей не позволяет нам различить ортологи и паралоги, и, таким образом, оказывается неподходящим для аннотирования функции. Существует ряд методов, которые выполняют предсказание функции на основе эволюционного анализа и различают паралоги и ортологи (напр. FlowerPower (Krishnamurthy et al. 2007)), но применение этих методов требует наличия большого числа последовательностей с относительно равномерной скоростью расхождения, по которым и восстанавливается предполагаемая картина дупликации. Кроме того, эти методы наталкиваются на трудности в тех случаях, когда последовательности утрачивают общую функцию, несмотря на то, что являются ортологами.
Анализ консервативности функции может быть значительно облегчен, если рассматривать последовательность не только как линейную цепочку аминокислотных остатков, но и в контексте её пространственной структуры. Поскольку обычно функция белка обеспечивается аминокислотными остатками, которые расположены близко в пространстве, но не обязательно находятся рядом в последовательности, то рассмотрение этой функции может быть ограничено лишь анализом конкретного функционального сайта. Таким образом, для консервативности функции обычно необходима лишь консервативность пространственного расположения ключевых аминокислотных остатков, а не полная идентичность последовательностей. Это может быть проиллюстрировано простым примером:
370 Глава 12. Предсказание функции белков на основе теоретических моделей
исчезновение всего лишь одного остатка из каталитического центра почти не сказывается на сходстве последовательностей в целом, но обычно приводит к полной утрате одной из функций белка (например, белок по-прежнему может связывать субстрат, но уже не катализирует его превращение, для которого была необходима функциональная группа исчезнувшего остатка). Таким образом, сравнение остатков в функциональном сайте и анализ таких нечетко определенных свойств, как различные особенности поверхности белка (Главы 7 и 8), больше подходят для сравнительного анализа функций, чем рассмотрение последовательностей. Но такой анализ, очевидно, требует наличия пространственной структуры белка, и в этой главе мы обсудим, какой вклад компьютерное моделирование может внести в получение таких структур и, в особенности, покажем, как модели позволяют улучшить наше понимание функционирования белка.
12.2. Модели белков как общедоступный ресурс
Как ранее было упомянуто в Главе 3, одной из целей структурной геномики и, в частности, проекта PSI (Protein Structure Initiative), является экспериментальное получение таких белковых структур, которые позволили бы максимально охватить пространство белковых укладок. За прошедшие 7 лет в научных центрах, участвующих в этом проекте, было определено около 3 тысяч структур, что составляет примерно 40% от общего числа новых структур с ранее неизвестной укладкой, размещенных в базе данных PDB (Service 2008а). В то же время в области предсказания белковых структур было приложено много усилий для улучшения алгоритмов и программ позволяющих теоретическим моделям приблизиться как можно ближе к структурам, полученным экспериментально. Успешные этапы мероприятия CASP (англ. Critical Assessment of techniques for protein Structure Prediction - Критическая оценка методик предсказания белковых структур), являющегося индикатором успехов в данной области (Kryshtafovych etal. 2005), показали, что точность предсказания структуры непрерывно повышается. Если бы модели были плохого качества, то от них было бы мало практической пользы, но на самом деле в среднем для 80% белков, предлагаемых в качестве мишеней в CASP, построенные модели оказываются достаточно близки и содержат больше полезной информации, чем содержалось в шаблоне (Kryshtafovych et al. 2007). (Добавленная ценность моделей по отношению к последовательности и шаблону обсуждается ниже более детально.) Растущее число новых описанных типов укладки,
12.2. Модели белков как общедоступный ресурс
371
полученное благодаря структурной геномике и росту точности компьютерных методов, приводит к возможности создания возрастающего числа моделей белков. Согласно недавним оценкам, новые структуры, расшифрованные в рамках проекта PSI, позволяют получить около 40 тысяч моделей на основании гомологии, которые иначе не могли быть получены (Service 2008b). Однако для того, чтобы воспользоваться этим множеством моделей в полной мере, они, наряду с информацией об их надежности, должны быть свободно доступны биологам.
12.2.1. Качество моделей
От качества структуры белка зависит, подходит ли она для предсказания его функции. Большая часть экспериментально расшифрованных структур имеют достаточное разрешение для того, чтобы судить о химическом механизме их действия, но про модели белков сказать такое можно не всегда. В то же время именно от качества структуры или модели зависит, насколько верной будет полученная на их основе информация о функции белка. Например, модель среднего качества лишь с осторожностью может быть использована для таких расчетов, как молекулярных докинг лекарственных соединений и их аналогов. Как уже упоминалось выше, точность моделей на основании гомологии падает с уменьшением сходства между последовательностями белка-мишени и белка-шаблона (см. тж. Главу 3) и можно сформулировать эмпирическое правило: модели, построенные по близкородственному шаблону, как правило, «хорошие», в то время как модели, построенные по удаленному шаблону, обычно «плохие». Это правило, однако, подразумевает, что при построении моделей используются наилучшие выравнивания и не производятся никакие улучшения стартовой конформации в направлении истинной структуры. Кроме того, степень близости к шаблону нельзя рассматривать как меру качества модели при оценке de novo моделей (Глава 1), так как они вовсе не используют шаблон.
В последнее время было создано несколько программ для оценки качества моделей белков без использования информации об их нативной структуре. Некоторые из этих методов (например, PROQ (Wallner and Elofsson 2003)) нацелены на различение нативных структур, структур, близких к нативным, и ненативных структур, в то время как другие методы (например, PROQres (Wallner and Elofsson 2006), ModFold (McGuffin 2008) или MetaMQAP (Pawlowski etal. 2008)) сосредоточены на предсказании того, насколько сильно различные фрагменты модели могут отклоняться от соответствующих фрагментов нативной структуры. В настоящее время нет универсальной функции, с помощью которой можно было бы уверенно оценить
372 Глава 12. Предсказание функции белков на основе теоретических моделей
общую и локальную точность структурных моделей. Однако предположения о функциях белков, основанные на их теоретических моделях, должны учитывать предсказываемое качество модели в целом, если рассматриваются её общие свойства (заряд, форма поверхности и т.д.), и локальное качество, если речь идет об активных центрах или других небольших областях белка. В последнем случае особенно полезными оказываются именно те методы, которые предсказывают отклонение различных фрагментов конкретной модели белка от его неизвестной нативной структуры.
Учет таких оценок качества может, по меньшей мере, помочь избежать излишне детального рассмотрения модели, поскольку, например, очевидно, что нет смысла анализировать геометрию взаимодействий между боковыми цепями остатков в потенциальном каталитическом центре, если основная цепь этих остатков предсказана с точностью лишь 5 А.
12.2.2. Базы данных моделей
Для эффективного использования структурных моделей белков необходимо представить их научному сообществу в таком виде, чтобы они могли быть доступны отдельным исследователям. Создание публичных хранилищ могло бы стимулировать использование моделей при планировании экспериментальных работ. В настоящее время общедоступны два типа баз данных моделей пространственных структур белков.
Такие базы данных, как MODBASE (Pieper etal. 2006) и SWISS-MODEL Repository (Корр and Schwede 2004) (см. Таблицу 12.1), содержат модели, полученные полностью автоматизированными методами. Другие базы данных, как PMDB (Protein Model Database) (Castrignano et al. 2006), разработаны для хранения и работы с моделями, построенными вручную.
Вне зависимости от принципов устройства все базы данных имеют веб-интерфейс для доступа к интересующим моделям, а также предоставляют дополнительную информацию, включая оценку надежности модели и аннотацию функции белка.
Для того, чтобы обеспечить точность моделей, достаточную для их использования в экспериментальных исследованиях, заложенные в SWISS-MODEL Repository и MODBASE подходы основываются на сравнительном моделировании, которое в настоящее время является наиболее надежным методом при реализации масштабных проектов. Обе базы данных отображают степень идентичности между последовательностью белка-мишени и шаблона, a SWISS-MODEL Repository вообще содержит лишь модели с идентичностью, превышающей 40%. Последовательности белков-мишеней выбираются из базы данных UniProt (http://www.expasy.uniprot.org/) и оба
12.2. Модели белков как общедоступный ресурс
373
упомянутых хранилища регулярно обновляются вслед за базами данных последовательностей и структур, включая в себя новые или модифицированные последовательности белков-мишеней и используя новые структуры в качестве шаблонов. Оба хранилища предоставляют также доступ и к выравниваниям, согласно которым были построены модели, но их подходы к оценке качества моделей отличаются. Качество упаковки моделей, содержащихся в хранилище SWISS-MODEL, оценивается с помощью метода ANOLEA (Melo and Feytmans 1998) и вычисления энергии согласно силовому полю GROMOS96 (van Gunsteren 1996), в то время как MODBASE оценивает надежность моделей по статистическим критериям (Melo et al. 2002). Визуализация моделей, реализованная в SWISS-MODEL Repository, дает также графическое представление аннотации на доменном уровне и аннотации функции белка согласно InterPro, а визуализация в MODBASE позволяет отобразить также вероятное связывание лиганда и сайты аннотации однонуклеотидных полиморфизмов (SNP).
Таблица 12.1. Базы данных рассчитанных моделей, доступные через веб-интерфейс
База данных URL http://
SWISS-MODEL Repository (Корр and Schwede 2004) MODBASE (Pieper et al. 2006) PMDB Protein Model Database (Castrignano et al. 2006) Protein Model Portal DBMODELING (Silveira et al. 2005) swissmodel.expasy.org/repository/ sal i I ab. org/modbase www.caspur.it/PMDB www. proteinmodelportal. org/ laboheme.df.ibilce.unesp.br/ dbmodeling/
Коллекция моделей в базах данных MODBASE и SWISS-MODEL Repository, а также материалы, собранные в исследовательских центрах, участвующих в проекте PS1, доступны через единый интерфейс, предоставляемый порталом Protein Model Portal. Этот портал разработан для одновременного опроса всех включенных в него баз данных при поиске уже рассчитанных моделей для данной аминокислотной последовательности, а на странице с результатами поиска представлен список моделей, размещенных в конкретной базе данных, и обеспечен доступ к ним.
Модели, создаваемые с участием человека (обычно это трудные случаи, которые требуют нетривиальных методов при построении моделей и выборе наиболее надежной структуры среди нескольких альтернатив), размещены в базе данных PMDB. Эта БД призвана обеспечить доступ к моделям, опубли
374 Глава 12. Предсказание функции белков на основе теоретических моделей
кованным в научной литературе, вместе с подтверждающими их экспериментальными данными, но в настоящий момент большая часть моделей - это предсказания, полученные в рамках С ASP. PMDB предоставляет возможность отдельным моделистам пополнять базу своими моделями вместе с подтверждающими их экспериментальными свидетельствами, а пользователи могут свободно скачивать различные модели одного и того же белка-мишени или модели различных областей такого белка. В случаях, когда это возможно, предоставляется также ссылка на БД последовательностей SwissProt (http://www.ebi.ac.uk/swissprot/). После расшифровки структуры белка к соответствующей записи в базе данных добавляется также ссылка на экспериментальную структуру в PDB (http://www.rcsb.org/pdb/home/home.do).
Помимо масштабных хранилищ моделей есть и те, которые посвящены отдельным организмам. В качестве примера можно привести DBMODEL-ING (Silveira et al. 2005) - это база данных, предназначенная для создания лекарственных средств для борьбы с различными инфекциями. К настоящему моменту эта БД содержит сравнительные модели белков, закодированных в геноме только двух патогенных организмов Mycobacterium tuberculosis и Xylella fastidiosa, являющихся возбудителями пестрого хлороза цитрусовых.
12.3. Точность и добавленная ценность основанных на моделях предсказаний
Хотя идеальный алгоритм для определения точности модели в целом еще только предстоит разработать, уже сейчас можно определить среднюю точность предсказания конкретных свойств, сделанных на основе структурных моделей. Большинство белков используют межмолекулярное узнавание для выполнения своих функций, причем лигандами могут быть как малые молекулы, так и многосубъединичные белковые комплексы. Таким образом, свойства поверхности, в особенности такие, как экспониро-ванность остатков, площадь доступной поверхности, карманы и электростатический потенциал, напрямую относятся к выполняемой белком функции.
Точность расчета основанных на моделях предсказаний свойств была изучена при масштабном анализе простых сравнительных моделей (Chakravarty et al. 2005). Было показано, что общая точность всех структурных свойств, которые были включены в анализ, падает в зависимости от сходства последовательностей белка-мишени и белка-шаблона, но для разных свойств это падение в разной степени влияет на их точность (Таблица 12.2).
12.3. Точность и добавленная ценность предсказаний
375
Таблица 12.2. Точность и добавленная ценность структурных свойств при сравнительном моделировании по одному шаблону (Chakravarty and Sanchez 2004; Chakravarty et al. 2005)
Свойство Точность Добавленная ценность
В целом Возрастает с увеличением идентичности между мишенью и шаблоном Возрастает с падением идентичности между мишенью и шаблоном
Экспонированность остатков Уменьшается с увеличением размера белка; зависит от ошибок в выравнивании при идентичности меньше 30% Отсутствует
Окружение заглубленных остатков Нет четкой зависимости от размера белка, но выше, чем для экспонированных остатков; зависит от ошибок в выравнивании при идентичности меньше 30% Отсутствует
Окружение экспонированных остатков Нет четкой зависимости от размера белка, но ниже, чем для заглубленных остатков; зависит от ошибок в выравнивании при идентичности меньше 30% Умеренная
Площадь доступной поверхности (ASA) Ошибка в общей площади возрастает с увеличением размера белка и очень мало зависит от ошибок в выравнивании Умеренная
Идентификация карманов на поверхности Ложные карманы; повышенное число карманов по сравнению со структурой шаблона и мишени; нет четкого влияния ошибок в выравнивании на число карманов Отрицательная
Состав карманов на поверхности Высокая
Электростатический потенциал Зависит от ошибок в выравнивании при идентичности ниже 50% Высокая
Например, ошибки в выравнивании оказывают незначительное влияние на правильность предсказания площади доступной поверхности (ASA), в то время как правильность предсказания электростатического потенциала начинает страдать, когда идентичность последовательностей падает ниже 50% (Таблица 12.2). Следует особо отметить, что анализ был выполнен на
376 Глава 12. Предсказание функции белков на основе теоретических моделей
основе набора сравнительных моделей, построенных по одному шаблону, а моделирование петель не применялось. Таким образом, эти результаты являются репрезентативными по отношению к методам масштабного автоматизированного моделирования. Более аккуратные методики моделирования, состоящие в использовании нескольких шаблонов и оптимизации моделей, могут дать лучший результат и повысить точность предсказания конкретных свойств при заданной идентичности последовательностей.
Зная, насколько надежными являются те или иные свойства модели, интересно выяснить, какую дополнительную информацию (добавленную ценность) они имеют по отношению к структуре шаблона, по которому была построена модель. Как и прежде, систематический анализ добавленной ценности был выполнен на большом наборе моделей, построенных по одному шаблону (Chakravarty and Sanchez 2004), и позволил сделать ценные выводы о том, какие конкретно свойства моделей могут быть информативными (Table 12.2).
В целом, чем больше различие между последовательностями шаблона и мишени, тем более значимой становится добавленная ценность. Это следует из того, что в случаях с низким сходством шаблон содержит меньше информации о размере и физико-химических свойствах конкретного остатка в мишени. Однако, не все структурные свойства получают дополнительную информацию по сравнению с шаблоном. Для тех из них, которые зависят, главным образом, от положения остатка - экспонированность, соседство с заглубленными остатками и число карманов на поверхности, - модели не привносят добавленной ценности. Вероятно, это вызвано тем фактом, что заглубленные остатки, образуя структурное ядро, которое отвечает за целостность белка, являются более консервативными, чем экспонированные.
Для других структурных свойств, таких как соседство с экспонированными остатками или общая площадь доступной поверхности, модели позволяют получить некую добавленную ценность. Это имеет важное значение, когда остатки, доступные растворителю, ответственны за взаимодействие с другими молекулами, определяя, таким образом, биологическую функцию белка.
Наконец, для свойств белка, которые сильно зависят от физико-химических свойств его аминокислотной последовательности, например, состав карманов и электростатический потенциал, модели предоставляют значительную добавленную ценность. Действительно, идентификация заряженных областей имеет большое значение, поскольку эти области могут входить в сайт связывания или активный центр белка (см. Главу 7).
Подводя итог, можно сказать, что работы Чакраваты с сотр. показали, что за исключением обнаружения карманов большая часть структурных свойств
12.3. Точность и добавленная ценность предсказаний
377
моделей имеет некую добавленную ценность, и чем больше какое-либо свойство зависит от аминокислотной последовательности белка, тем более полезной оказывается модель при оценке величины этого свойства. Обнадеживает, что, в зависимости от рассматриваемого свойства, идентичность последовательностей шаблона и мишени на уровне 25-40% дает такую точность в оценке этого свойства, которая может быть получена и из ЯМР-структуры.
12.3.1. Реализация
Знание добавленной ценности конкретного свойства структуры или поверхности моделей ставит вопрос, будут ли они полезными при предсказании функции. В 1998 Фетроу и Сколник предложили многошаговую процедуру, которая позволяет идентифицировать функциональные сайты белков в моделях низкого и среднего разрешения (Fetrow and Skolnick 1998). Основываясь на геометрии, идентичности остатков, расстоянии между атомами Са и конформации, остатки активного центра стали трехмерным дескриптором, названным нечеткой функциональной формой (англ. Fuzzy Functional Form, FFF). С помощью таких форм были проанализирован набор пространственных моделей с целью отобрать те из них, которые содержат сходные пространственные мотивы. Применимость метода была подтверждена идентификацией новых членов семейства дисульфидных глутаредоксин/тиоредоксин белков в геномах дрожжей (Fetrow and Skolnick 1998) и Е. coli (Fetrow et al. 1998), функции которых не могли быть определены ранее на основе сравнения последовательностей. Большим достижением метода FFF и ему аналогичных стало то, что они позволили отделить пары белков, имеющие сходные активные центры, от тех, которые могут иметь сходные укладки, но необязательно сходные центры.
Дальнейшим развитием методики FFF стал метод построения профиля активного центра (Cammer et al. 2003), который был успешно объединен с экспериментальными методиками для выявления новых сериновых гидролаз в дрожжах (Baxter et al. 2004). Особенностью этого метода стало то, что упор был сделан не на остатки, консервативные во всем семействе, а на ключевые функциональные остатки, которые были специально выявлены среди всех белков с заданной функцией безотносительно к сходству их последовательностей. Таким образом, метод может применяться для идентификации и аннотации различных функциональных центров, включая каталитические центры, регуляторные центры и сайты связывания кофакторов.
Стоит упомянуть также и гибридный подход, совмещающий анализ поверхности белка с эволюционными методами, который был предложен Павловски с сотр. (Pawlowski and Godzik 2001). Они создавали карту по
378 Глава 12. Предсказание функции белков на основе теоретических моделей
верхности белковой молекулы нанесением распределения различных свойств (таких, как заряд или гидрофобность) на сферу, аппроксимирующую поверхность белка. Этим способом можно сравнить белковые молекулы целиком и сделать вывод об их глобальном функциональном сходстве, например, согласно какой-либо численной мере сходства между их картами. Было показано, что сравнение таких карт поверхности позволяет улучшить предсказание функции белка по сравнению с общими методами анализа последовательности и способно воспроизводить известные примеры варьирования функции с разнородной группе белков, включая выявление неожиданных наборов общих функциональных свойств для казалось бы удаленных паралогов. Было показано, что этот метод, теперь имеющий вебинтерфейс (Sasin et al. 2007), является достаточно устойчивым и допускает использование моделей по гомологии вместо экспериментальных структур.
Другие исследования были посвящены вопросу, могут ли более специфичные предсказания функций быть выполнены столь же точно для моделей, как и для экспериментальных структур. Результаты метода MetSite, который объединяет информацию о последовательности и структуре, для металл-связывающих сайтов оказались воодушевляющими (Sodhi etal. 2004). Хотя производительность при анализе моделей была ниже, чем при анализе экспериментальных структур, правильные предсказания металлсвязывающих сайтов были выполнены примерно для половины достоверных моделей, полученных с помощью mGenTHREADER. Примечательно, что эти модели содержали лишь атомы основной цепи, поэтому ошибки в расположении боковых цепей никак не сказывались на производительности. Аналогичный метод предсказания способности к связыванию ДНК также был разработан как для экспериментальных структур, так и для моделей, и использовал информацию о последовательности белка, пространственную асимметрию в распределении некоторых остатков и дипольных моментов (Szilagyi and Skolnick 2006). Этот метод также расчитан на структуры, содержащие только Са-атомы. При анализе моделей с СКО менее 6 А от природной структуры производительность этого метода была лишь чуть ниже, чем при анализе экспериментальных структур. Таким образом, метод можно использовать для моделей любого происхождения, включая модели ab initio и модели, полученные методами распознавания фолда, для которых, правда, следует ожидать более низкой точности.
Одно из важных практических применений моделей белков состоит в виртуальном скрининге баз данных низкомолекулярных соединений в поисках подходящих ингибиторов для детальной разработки и создания соединений-лидеров (Jacobson and Sali 2004). Поскольку наша книга посвящена функциям белков, то такие применения в этой главе обсуждаться не будут.
12.4. Практическое применение
379
Тем не менее, докинг небольших молекул, точно такой же, как и в фармацевтическом сценарии, начинает использоваться для предсказания функций белка. Этот способ, как обсуждалось в Главе 8, подразумевает, что соединения, наилучшим образом подошедшие по сравнению с другими кандидатами, предположительно и являются истинными лигандами (Hermann etal. 2007; Song et al. 2007). В связи с этим здесь следует упомянуть работы по изучению пригодности белковых моделей для низкомолекулярного докинга по сравнению с экспериментальными структурами. Вот две работы, в каждой из которых делается вывод о пригодности моделей, но используются разные критерии сравнения с экспериментальными структурами. Макговерн и Суаше (McGovern and Shoichet 2003) сравнивали обогащение известных лигандов по сравнению с пустышками в решениях докинга для голо- и апо-форм девяти ферментов и их моделей. Шаблоны, которые были выбраны для моделирования, имели общую идентичность последовательности с мишенями на уровне 34-87% и до 45-100% в активном центре. Наилучшее обогащение было достигнуто для восьми голо-форм, двух апо-форм и трех моделей, подтверждая, таким образом, преимущество экспериментальных структур. Тем не менее, почти все модели показали результат, лучший, чем случайная выборка активных соединений. Модели, построенные по более близким шаблонам, в целом оказались более эффективными, но небольшие конформационные искажения в активном центре могли испортить эту картину в отдельных случаях. Позднее Оширо с сотр. (Oshiro et al. 2004) сравнили обогащение заведомо активных соединений в результатах докинга для нескольких экспериментальных структур и моделей CDK2 и фактора Vila. Выбранные для создания моделей шаблоны имели идентичность последовательностей в окрестности активного центра на уровне 37-77%. Примечательно, что эффективность моделей оказалась сравнимой с эффективностью экспериментальных структур в тех случаях, когда идентичность была выше 50%, и существенно снижалась в противном случае. Суммируя результаты двух этих работ, можно сделать вывод, что использование моделей для проведения докинга оправдано только в случае отсутствия экспериментальной структуры. Будет заманчивым оценить эффективность моделей в предсказаниях функции непосредственно по результатам докинга, подобных тем, которые были описаны выше.
12.4. Практическое применение
После того, как модель построена, оценена и, возможно, размещена в базе данных, она должна стать инструментом для лучшего понимания взаимосвязей между структурой и функцией белков. Обычно такой анализ
380 Глава 12. Предсказание функции белков на основе теоретических моделей
выполняется специалистами по биоинформатике, но потенциально использование БД моделей позволит любому исследователю выяснить, что структура модели белка говорит о его функции. Предыдущие главы этой книги продемонстрировали много различных способов, которые позволяют сделать вывод о функции белка, основываясь на его структуре. Ниже будут представлены примеры, иллюстрирующие применение многих из этих методик к структурной информации, полученной методами моделирования ab initio, распознавания фолда и сравнительного моделирования (Главы 1-3, соответственно).
12.4.1. Пластичность остатков каталитического центра
Несмотря на усилия, предпринятые при выполнении проектов по структурной геномике, по охвату всего пространства укладок белков и получению шаблонов для всех ныне существующих семейств, есть примеры, когда критерий близости последовательностей оказывается недостаточным для того, чтобы определить наличие у анализируемого семейства уже описанной функции. Во многих случаях, однако, выводы о структуре белка могут быть сделаны с помощью методов распознавания фолда - как самих по себе, так и в сочетании с моделированием ab initio (Kolinski and Bujnicki 2005), - а затем применены к поиску потенциального активного центра, который позволит предположить возможную функцию белка. Это может быть проиллюстрировано опубликованным анализом (Feder and Bujnicki 2005) семейства последовательностей, объединенных в кластер COG4636 в базе данных кластеров ортологичных групп (англ. Clusters of Orthologous Groups, COG) (Tatusov et al. 2003) и аннотированного как «не-охарактеризованный белок, консервативный в цианобактериях». Детальный анализ консервативности последовательностей внутри семейства COG4636, дополненный предсказанием вторичной структуры, выявил характерную последовательность а-спиралей и Р-тяжей, связанную с консервативным карбоксильным остатком, которая ранее была идентифицирована в надсемействе нуклеаз PD-(D/E)XK (Bujnicki 2003). Это сходство позволяет предположить, что и члены семейства COG4636 могут принадлежать к этому надсемейству (Рис. 12.1а, Ь).
Однако множественное выравнивание показало, что только «полумотив» PD является почти идеально консервативным, в то время как ключевой остаток лизина во втором полумотиве (D/E)XK отсутствует. Более точно, на его месте в большинстве членов семейства COG4636 расположен гидрофобный остаток лейцина или валина. Этому факту можно предложить
12.4. Практическое применение
381
Рис. 12.1. (Цветную версию рисунка см. на вклейке.) Пространственная консервативность активного центра в PD-(D/E)XK. Для наглядности показаны только структурные ядра белков в сходной ориентации, терминальные области и вставки опущены. В верхнем ряду показаны истинные нуклеазы PD-(D/E)XK - а) резольваза структуры Холидея Hje (PDB код 1 оЬ8) и б) рестриктаза Ngo-MIV (PDB код 1fiu).
В нижнем ряду показаны структуры COG4636 - в) теоретическая модель (Feder and Bujnicki 2005) и г) кристаллографическая структура другого члена семейства (PDB код 1wdj). Боковые цепи типичного для PD-(D/E)xK активного центра и альтерантивного варианта показаны оранжевым (кроме остатков лизина, показанных синим). Этот рисунок, а также все остальные рисунки в данной главе, подготовлен с использованием программы PyMOL (http://pymol.sourceforge.net)
три возможных объяснения. Во-первых, семейство COG4636 может вовсе не относиться к надсемейству PD-(D/E)XK. Во-вторых, оно может относиться к этому надсемейству, но утратило остаток в активном центре и вместе с ним каталитическую активность. И в-третьих, роль утраченного лизина мог взять на себя какой-то другой остаток, но выявить его на основе лишь выравнивания последовательностей не представляется возможным. Будет ли тут полезным предсказание структуры и сможет ли оно помочь определить истинную функцию белков семейства COG4636?
Прежде всего, распознавание фолда для последовательностей семейства COG4636 подтвердило предсказание об их принадлежности к надсемейству ферментов PD-(D/E)XK. Затем по шаблону истинной нуклеазы этого надсемейства была построена сравнительная модель, которую проверили на наличие пространственно близкого консервативного остатка. Оказалось, что в белках семейства COG4636 утраченный лизин замещен другим лизином, заметно удаленным в аминокислотной последовательности (Рис. 12.1с). Функциональная группа этого замещающего лизина может занимать то же пространственное положение, что и группа каталити
382 Глава 12. Предсказание функции белков на основе теоретических моделей
ческого лизина в шаблонах, создавая, таким образом, полноценный мотив PD-(D/E)XK в пространстве, несмотря на отсутствие консервативности последовательности. Это наблюдение и позволило сделать нетривиальное предсказание, неосуществимое при анализе одной лишь последовательности, о том, что семейство COG4636 включает в себя активные нуклеазы.
Позднее правильность предсказания необычной конфигурации активного центра была подтверждена кристаллографическими исследованиями другого члена семейства COG4636 (Fig. 12.Id; PDB код Iwdj), а также обнаружением других истинных нуклеаз со сходной конформацией активного центра (Tamulaitiene et al. 2006).
12.4.2. Картирование мутаций
Редкие мутации, встречающиеся в ключевых белках, лежат в основе многих наследственных заболеваний. Другие различия в последовательности белков, например, аллельные варианты белков-мишеней лекарственных препаратов, могут привести к различному связыванию этих препаратов и, следовательно, различной реакции пациентов. Таким образом, структурное картирование мутаций, которое является ключевым применением молекулярных моделей, оказывается полезным для понимания молекулярных механизмов заболеваний и предсказанию реакции пациентов, составляя шаг на пути к персонифицированной медицине.
АТФ-чувствительные калиевые каналы играют ключевую роль во многих тканях, связывая клеточный метаболизм и электрическую активность. Эти каналы представляют собой октамерные комплексы двух различных белков Kir6.2 и SUR. Связывание АТФ или АДФ с каналами приводит к их ингибированию. Обнаружение ряда мутаций в Kir6.2, приводящих к понижению чувствительности канала к АТФ, дало понимание причины сахарного диабета новорожденных (Hattersley and Ashcroft 2005). Ингибированные каналы вызывают гиперполяризацию мембран в панкреатических Р-клетках, что, в свою очередь, приводит к уменьшению секреции инсулина и, следовательно, к диабету. Осознание генетической этиологии заболевания произвело революцию в терапии пациентов с диабетом новорожденных, являющимся следствием мутаций в Kir6.2, поскольку эти каналы могут оставаться закрытыми под действием таких препаратов, как производные сульфонилмочевины или глиниды, и инсулиновая терапия может быть ограничена или приостановлена.
Сравнительная модель субъединицы Kir6.2 позволила выполнить пространственное картирование остатков, подвергшихся мутации при диабете новорожденных, и тем самым проиллюстрировала молекулярный меха
12.4. Практическое применение
383
низм, лежащий в основе пониженной чувствительности АТФ-чувствитель-ных калиевых каналов (Hattersley and Ashcroft 2005). Пациенты с мутацией в Kir6.2 имеют ряд фенотипов, которые однозначно коррелируют с природой мутации. Например, пациенты с нейрологическими симптомами имеют мутации, которые не имеют непосредственного отношения к связыванию АТФ, но делают сильно предпочтительным открытое состояние канала и таким образом уменьшают способность АТФ его блокировать (АТФ стабилизирует закрытое состояние канала).
Недавние исследования показали, что можно выделить группу пациентов с постоянным диабетом новорожденных, имеющих мутацию L164P в Kir6.2 и невосприимчивых к терапии сульфонилмочевиной (Tammaro etal. 2008). Анализ пространственного положения L164 показал, что этот остаток лежит глубоко внутри структуры белка на удалении 35 А от сайта связывания АТФ, и поэтому его непосредственное участие в снижении связывания АТФ маловероятно. Вместо этого, мутация L164P, вероятно, дестабилизирует закрытое состояние канала, с которым преимущественно и связываются производные сульфонилмочевины и которое редко достигается в каналах с повышенной вероятностью открытого состояния. Все вместе эти результаты показывают, что восприимчивость к лекарственным препаратам зависит от природы конкретной мутации, но она может быть предсказана путем детального анализа модели белка.
12.4.3. Комплексы белков
Если системная биология, в соответствии с которой те или иные масштабные наборы данных объединяются в смысловое целое, стремится однажды достичь успеха, то необходимо полное понимание сложных сетей белок-белковых взаимодействий, которые существуют в клетке. Таким образом, с её стороны есть большая выгода в добавлении предсказаний, полученных сравнительным моделированием, в арсенал экспериментальных и вычислительных методов для предсказания белок-белковых взаимодействий (Aloy and Russell 2006). Принцип очень простой: при заданной известной структуре комплекса белка А с белком X, именно анализ потенциального комплекса белка В с белком Y (где В гомологичен A, a Y гомологичен X) позволяет предположить, что это взаимодействие будет иметь место in vivo (Aloy and Russell 2002). Ранние методы в этой области оценивали пригодность интерфейса, заимствуя потенциалы парных взаимодействий из методов протягивания и анализируя известные контакты в структуре А-Х после выравнивания последовательностей В и А и последовательностей Y и X. В настоящее время анализ такого рода можеть быть
384 Глава 12. Предсказание функции белков на основе теоретических моделей
выполнен с помощью ряда веб-серверов, включая InterPreTS (Aloy and Russell 2003) и MULTIPROSPECTOR (Lu et al. 2002). В последующих работах моделирование белкового комплекса выполняли явным образом и снова использовали потенциалы взаимодействия, чтобы провести различие между истинными и ложными взаимодействиями (Davis et al. 2006). Заслуживает внимания интересное масштабное приложение такого типа предсказаний (Davis et al. 2007), которое было выполнено для белков человека и белков из геномов десяти патогенных организмов, ответственных за запущенные заболевания. Геномы патогенов и хозяина были сперва просканированны в поисках белков, гомологичных тем белкам, чьи взаимодействия известны. Если такая структурная информация о взаимодействии была недоступна, то процесс продолжали, применяя простые функции для оценки сходства последовательностей. Однако, этот подход позволил сделать лишь небольшое число предсказаний, поскольку были использованы строгие критерии для их надежности.
Более интересным и значительным было явное моделирование потенциально взаимодействующих партнеров по шаблону белкового комплекса. Полученные модели комплексов были оценены с использованием статистических потенциалов, и, получившие положительную оценку, были допущены к следующему хитроумному фильтру. Этот фильтр использовал известную информацию о тканевой и внутриклеточной локализации и функции взаимодействующих белков, чтобы исключить из рассмотрения те взаимодействия, которые не могли бы произойти in vivo. Таким образом, в качестве кандидатов для взаимодействия с белками Mycobacterium leprae были отобраны только белки, экспрессируемые в коже, лимфатических узлах или легких хозяина. Патогенные белки также должны были подходить под специфические биологические критерии. Например, белки М. leprae должны были иметь подходящую аннотацию в системе ГО (например, указание на патогенность) или быть аннотированными как внеклеточные или расположенные на поверхности. После этого фильтра число предсказываемых взаимодействий оказалось в пределах от 0 до 1 501 в зависимости от патогена.
И хотя в распоряжении авторов методики было не так много известных взаимодействий для её тестирования, им удалось предсказать 4 из 33 взаимодействий, описанных к настоящему времени. В остальных случаях для моделирования взаимодействий не нашлось подходящего шаблона, что дает повод считать недостаток шаблонов виновным в малом количестве описанных взаимодействий (Davis et al. 2007). Интересно, что одно предсказание было экспериментально подтверждено: метод предсказал взаимодействие фальципаина-2 и цистатина (PDB код lyvb), основываясь
12.4. Практическое применение
385
на более ранней структуре катепсина-Н в комплексе со стефином A (PDB код 1пЬЗ) (Рис. 12.2). Два эти фермента имеют около 24% идентичности, их ингибиторы идентичны лишь на 11 %, поэтому успех предсказания при столь низкой схожести последовательностей и заметном различии в структурах (Рис. 12.2) хорошо характеризует возможности метода.
12.4.4. Предсказания функции на основе моделей ab initio
До недавнего времени, пока не были разработаны более мощные, но ресурсоёмкие алгоритмы (Bradley et al. 2005), разумной целью моделирования без использования шаблона (ab initio или de novo) было лишь получение правильной укладки, нежели более точные предсказания (см. Главу 1). Это ограничивало диапазон применимых методов предсказания функции белков, и означает, что большинство предсказаний, описанных в литературе, основаны, главным образом, на предсказаниях укладки белка и её корреляции с его функцией (обсуждалось в Главе 6).
В одном из первых масштабных применений сервера ROSETTA, Бонно с коллегами (Bonneau et al., 2002) создал модели для 510 белковых семейств по классификации Pfam со средней длинной менее 150 остатков. Это были неизвестные к тому времени структуры белков, но для некоторых из них функция была известна или предполагалась. В нескольких случаях умозрительные предсказания могли быть подкреплены результатами моделирования. Например, предполагали, что PF01938, домен белка TRAM, может связываться с нуклеиновыми кислотами. Это предположение строилось, в основном, на сходстве его ab initio модели со структурами в надсемействах базы данных SCOP, содержащих различные белки, связывающие нуклеиновые кислоты. Теперь мы можем судить о точности модели по двум неопубликованным результатам проекта по структурной геномике (lyez и lyvc). Примером предсказания функции для совершенно не охарактеризованного белка служит домен с неизвестной функцией 37 (PF01809). Его модель подошла в качестве структуры NK-лизина - гемолитического белка, экспрессируемого в естественных киллерах. Хотя структура белка PF01809 остается неизвестной, база данных Pfam на момент написания этих строк сообщает неопубликованные свидетельства, что такой белок из Aeromonas hydrophila действительно демонстрирует гемолитическую активность.
Примечательно, что одна ab initio модель не обязана в точности соответствовать известному типу укладки, чтобы дать ключ к пониманию функции; напротив, иногда на решение наталкивает весь широкий класс структур, которому принадлежит конкретная модель. Примером этого
386 Глава 12. Предсказание функции белков на основе теоретических моделей
Рис. 12.2. Предсказание белок-белковых взаимодействий на основе моделирования структуры белков. Подход, основанный на сравнительном моделировании структуры белковых комплексов (Davis et al. 2008), позволил, используя структуру катепсина А в комплексе со стефином A (PDB код 1 пЬЗ; а) предположить возможное взаимодействие между фальципаином-2 и цистатином, что и было подтверждено кристаллографически (PDB код 1yvb; б). На обеих панелях ферменты показаны сверху, ингибиторы снизу
является модель, построенная для муцин-связывающего домена (Bumbaca etal. 2007). Предпочтительная модель содержала укладку типа «бета-сэндвич» такого типа, который сильно коррелирует со связыванием углеводов, - на момент публикации половина семейств углевод-связывающих доменов с известной структурой имела такую укладку. Это скорее могло бы согласовываться со связыванием домена с углеводородной частью его мишени, высокогликозилированного муцина, нежели чем с его белковой частью. Более того, эта ab initio модель имела три экспонированных ароматических остатка такого типа, который считается характерным для связывания углеводородов (Quiocho and Vyas 1999).
Недавний пример показал, как функция, предсказанная по укладке ab initio модели может быть подтверждена другими методами (Rigden and Galperin 2008). Известно, что белок SpoVS необходим для спорообразования у спорообразующих бактерий, но на самом деле распространен гораздо шире. Фенотипическое описание организмов, имеющих мутированный SpoVS, мало что говорит о его молекулярной роли. Однако лучшие модели, полученные с помощью серверов ROSETTA и I-TASSER, хорошо соответствуют укладке белка Alba, содержащегося в хроматине архей (Рис. 12.3а). Такая укладка тесно связана со связыванием нуклеиновых кислот в различных
12.4. Практическое применение
387
Рис. 12.3. (Цветную версию рисунка см. на вклейке.) Анализ ab initio моделей белка SpoVS позволил предположить наличие у него функции связывания нуклеиновых кислот (Rigden and Galperin 2008). а) Модели, полученные как с помощью ROSETTA (показано серым) так и с помощью I-TASSER (показано черным) сильно схожи со структурой белка Alba, содержащемся в хроматине архей (PDB код 1 nfj: окрашен по спектру от синего N-конца до красного С-конца). б) Электростатический потенциал возможного димера SpoVS, построенного по модели ROSETTA (синим показаны области положительного потенциала, красным - отрицательного)
Рис. 12.4. (Цветную версию рисунка см. на вклейке.) Подтвержденное предсказание структуры, выполненное Мальмстрем с коллегами (2007). Модель белка TRS20/YBR254C (а) соответствовала надсемейству SNARE по классификации SCOP; это соответствие позже было подтверждено получением экспериментальной структуры (PDB код 1h3q) схожего белка (б). Цветом выделены структурно схожие элементы, остальное показано серым
случаях и, более того, картирование электростатического потенциала на поверхности моделей выявило отчетливую положительно заряженную область характерную для белков, связывающихся с нуклеиновыми кислотами (Рис. 12.3b; см. Главу 7). Суммируя результаты этих методов можно предположить, что белок SpoVS является новым фактором транскрипции, который вовлечен в контроль запутанной схемы экспрессии генов, происходящей при споруляции (Rigden and Galperin 2008).
Недавнее масштабное применение ab initio моделирования в рамках подхода, включавшего также использование PSI-BLAST и методов предсказания структуры, основанных на протягивании, было направлено на анализ генома дрожжей (Malmstrom et al. 2007). Авторы применили новую стратегию по использованию известной информации о функции, чтобы облегчить отбор правильных совпадений между потенциальными структурами ab initio моделей и надсемейств по SCOP. С этой целью вдобавок к сравнению структур было оценено совпадение по классификации ГО между белком-мишенью и белками рассматриваемых надсемейств. Эти допол
388 Глава 12. Предсказание функции белков на основе теоретических моделей
няющие друг друга источники информации были объединены с применением байесовской статистики. На рисунке (Рис. 12.4) показан пример предсказания принадлежности белка TRS20/YBR254C к надсемейству SNARE по классификации SCOP, которое позже было подтверждено экспериментальной расшифровкой структуры. Совпадение между моделью и кристаллографической структурой частичное и умеренное (Рис. 12.4), что иллюстрирует ценность информации, относящейся к белку-мишени и надсемействам потенциально совпадающих структур, которая содержится в БД ГО. В рассматриваемом случае мишень TRS20/YBR254C являлась одной из субъединиц комплекса переноса белковых частиц (англ, transport protein particle (TRAPP)), участвующем в состыковке и слиянии везикул. Её совпадение со структурой из надсемейства SNARE по классификации SCOP было, таким образом, надежно подтверждено, поскольку везикулярный транспорт является одной из главных функций белков этого надсемейства.
12.4.5. Предсказание специфичности к лигандам
Одно из самых основных предсказаний функции, которое может быть получено из модели белка, - это специфичность к лигандам. Зачастую если известна структура белка А связанного с лигандом X, интересно предсказать, будет ли белок В, гомологичный белку А, также специфичен к X или, на самом деле, он связывает другой лиганд У. Такой анализ основывается на предположении, что связывающий сайт в белках А и В расположен одинаково. Обычно это верно для гомологичных белков и, в случае ферментов, подтверждается наличием поблизости ключевых каталитических остатков. Затем по шаблону структуры А создают структуру В, и изучение этой структуры, в частности, сравнение её со структурой А, показывает, появились ли изменения в сайте связывания. Уменьшение его размера, например, будет трактоваться, как уменьшение размера связываемого лиганда.
Примером одной из первых работ в этой области стало моделирование липид-связывающего белка мозга (англ, brain lipid binding protein, BLBP), основанное на структуре родственного белка, связывающего жирные кислоты (Xu et al. 1996). Взаимодействия известных жирных кислот с белком BLBP были смоделированы в исследовательских работах, направленных на установление молекулярных основ в двадцать раз более сильного связывания докозагексоеноевой кислоты по сравнению с более короткими олеиновой и арахидоновой кислотами. Модель показала, что два дополнительных атома у первой из этих кислот могут располагаться в кармане белка, усиливая благоприятное гидрофобное взаимодействие.
12.4. Практическое применение
389
Вычисленная дополнительная энергия связывания, основанная на величине дополнительной площади гидрофобного контакта, составляет около 2 ккал/моль и хорошо коррелирует с различием в аффинности этих кислот. Исходя из проверенной таким образом модели авторы смогли предсказать, что более длинные жирные кислоты не будут образовывать дополнительных контактов, и не будут поэтому иметь большую аффинность.
Молекулярные основы различной специфичности иногда могут быть на удивление простыми. Это как раз имеет место в случае с донорами фосфата в некоторых 6-фосфофруктокиназах - гликолитических ферментах, катализирующих перенос фосфогруппы донора, которыми могут быть АТФ, АДФ или неорганические пирофосфаты. АТФ- и пирофосфат-зависимые ферменты имеют эволюционную близость, в то время как АДФ-зависимые фосфофруктокиназы относятся к иному структурному классу. Ранее было отмечено, что АТФ-зависимые ферменты трипаносоматид более близки к определенным пирофосфат-зависимым ферментам, чем к хорошо известным АТФ-зависимым ферментам бактерий и млекопитающих (Michels et al. 1997). Позже моделирование позволило установить, что причина специфичности к АТФ или пирофосфату может быть указана с точностью до одного аминокислотного остатка, которым является Gly в АТФ-зависимых ферментах и Asp в пирофосфат-зависимых (Lopez et al. 2002). Как показано на Рис. 12.5, аспартат в этом положении имеет стерические и электростатические наталкивания с а-фосфатом связанной АТФ или АДФ, что уменьшает сайт связывания до размеров, подходящих только пирофосфату. Подтверждением этой впечатляюще простой причины специфичности стало превращение пирофосфат-зависимого фермента в АТФ-зависимый одной лишь заменой соответствующего аспартата на глицин (Chi and Kemp 2000).
Рис. 12.5. (Цветную версию рисунка см. на вклейке.) Каталитический центр 6-фосфофруктокиназы Е. coli со связанным 6-фосфатом фруктозы и АДФ (PDB код 4pfk). Лиганды показаны цветными стержнями (6-фосфат фруктозы слева и АДФ справа). АТФ-зависимые ферменты, такие как этот, имеют глицин в каталитическом центре (не показан). Моделирование расположения аспартата в этом же положении (показан розовым), аналогично наблюдаемому в пирофосфат-зависимых ферментах, показывает, что этот остаток ответственен за изменение специфичности к донору фосфата (см. текст)
390 Глава 12. Предсказание функции белков на основе теоретических моделей
12.4.6. Моделирование структуры изоформ, полученных альтернативным сплайсингом
Многие, если не большинство, эукариотических генов допускают альтернативный сплайсинг, резко увеличивающий разнообразие зрелых молекул. Часто бывает трудно предсказать по последовательности альтернативного транскрипта, сохранится ли его функция или изменится. Моделирование структуры, где это возможно, может пролить свет на связь между структурой и функцией у альтернативных транскриптов одного гена.
В одной из первых работ в этом направлении (Fumham et al., 2004), охватившей модели 14 белков и 40 вариантов сплайсинга, было показано, что утрата экзона чаще включает потерю целиком структурных единиц, нежели более мелких фрагментов. Авторы показали, что согласно программам оценки надежности моделей, в случае моделирования делеций и вставок результаты моделирования первых более надежны. Для четырех белков с биомедицинским применением авторы смогли установить корреляцию между известными функциональными свойствами альтернативных вариантов и моделями их структур. Позднее было показано (Wang et al., 2005), что места альтерации (splicing events) обычно находятся в петлевых участках на поверхности белка, а не в элементах упорядоченной вторичной структуры. У конкретного гена в общем случае есть лишь одно-два места альтерации, охватывающие в 60% случаев не более 50 остатков. Эти результаты позволили предположить, что сплайсинг обычно происходит в тех местах и таким способом, что третичная структура белка по возможности не затрагивается. Это согласуется с тем, что большая часть альтернативных изоформ имеют близкие к оригиналу свойства укладки и, поэтому, близкую функцию. Однако позднее при анализе нескольких транскриптов со структурной точки зрения обнаружилось (Tress et al. 2007), что многие изоформы должны были бы иметь резко отличающиеся структуры по сравнению с уже известными структурами остальных изоформ. Для 49 из 85 транскриптов, сопоставленных с гомологичными им структурами, авторы пришли к выводу, что изоформы и главные транскрипты должны образовывать существенно различные структуры. Пример из работы Тресс с сотр. (Tress et al. 2007) (Рис. 12.6) иллюстрирует изоформу интерлейкина 4 без экзона 2. Структурный фрагмент, кодируемый этим экзоном, входит в ядро белка и участвует в образовании дисульфидного мостика, позволяя предположить, что пространственная структура изоформы будет существенно отличаться от известной структуры полного белка. Пока еще мы имеем далеко не полную картину влияния на функцию белка структурных
12.4. Практическое применение
391
Рис. 12.6. (Цветную версию рисунка см. на вклейке.) Структура интерлейкина 4 с фрагментов, кодируемым экзоном 2. Экспериментальная структура (PDB код lilt) показана в ленточном представлении, фрагмент, кодируемый экзоном 2, выделен пурпурным. Дисульфидные мостики показаны в стержневом представлении; мостик во фрагменте, кодируемом экзоном 2, показан в шаростержневом представлении
изменений - больших или малых, - вызванных альтернативным сплайсингом. Так, лишь для 4 из 214 локусов оказалось возможным найти экспериментальные данные, иллюстрирующие различие в функции между альтернативными изоформами Tress et al. (2007).
12.4.7. От общей функции к молекулярным деталям
Функцию белка может рассматривать на разных уровнях сложности -от участия в клеточных процессах до понимания механизма действия на молекулярном уровне. Лизосомальная дезоксирибонуклеаза II а (ДНКаза Па) стала одной из первых обнаруженных эндонуклеаз (1947) и была хорошо описана с биохимической точки зрения уже в 1960х. Этот фермент необходим для развития организма, поскольку отвечает за удаление лишней ДНК и дополнительную апоптическую фрагментацию ДНК у высших эукариот - нокаут лизосомальной ДНКазы Па у мышей оказался летальным. Несмотря на интенсивные исследования в течение последних 50 лет и бесспорную важность ДНКазы Па, не удалось выявить схожесть этого фермента с каким-либо другим семейством белков, что затрудняет исследование функции этого белка на молекулярном уровне. Ни один из методов распознавания фолда не позволил построить достоверное выравнивание мишени с шаблоном, но анализ их результатов показывает, что некоторые из этих мишеней них имеют сходство с укладкой фосфолипазы D в области активного центра, образуя так называемый мотив НхК (Cymerman et al. 2005). Известные члены надсемейства фосфолипаз D имеют двудоменную структуру с одним активным центром, состоящим из двух мотивов HxK-Xn-N-Xn-(E/Q/D), которые расположены на интерфейсе между доменами. Установить остальные остатки, образующие активный центр, основываясь на выравнивании, было невозможно. Однако анализ расположения конкретных остатков в структуре модели позволил получить эту существенную информацию и определить аминокислотные остатки, которые
392 Глава 12. Предсказание функции белков на основе теоретических моделей
Рис. 12.7. (Цветную версию рисунка см. на вклейке.} Структурная модель ДНКазы Па человека. Вычислительный анализ позволил установить принадлежность ДНКазы Па к семейству фосфолипаз D. Фермент имеет мономерную структуру с псевдодимерной архитектурой. Два мотива НхК в N- и С-концевых доменах (показаны в ленточном представлении светлоголубым и серым соответственно) имеют каталитически важные аминокислотные остатки показаны в стержневом представлении (красным и зеленым), которые вместе образуют единый активный центр. Кроме идентификации потенциальных каталитических остатков модель объяснила близкое расположение остатков цистеина, образующих дисульфидные связи (показаны оранжевыми и темно-синими шариками), и экспонированный характер N-гликозилированных остатков (показаны зелеными шариками). Петли, потенциально связывающие ДНК, показаны лиловым. Определение функционально важных остатков в теоретической модели фермента может значительно облегчить процесс его инженерии
потенциально могут участвовать в образовании активного центра (Рис. 12.7). Обнаружение того, что ДНКаза Па является отдаленным родственником фосфолипазы D позже было подтверждено экспериментальным изучением (Schafer et al. 2007) и объяснило необычные свойства этой нуклеазы, такие как устойчивость к ЭДТА. По аналогии с фосфолипазой D, чей механизм был известен, стало возможным сделать вывод, что реакция гидролиза фосфодиэфирной связи выполняется ДНКазой Пас участием ковалентно связанного промежуточного веещества реакции.
Случай ДНКазы Па наглядно показывает, как биоинформатика помогает обойти некоторые экспериментальные ограничения (ДНКаза Па не поддается кристализации, поскольку гликозилируется с трудом) и, таким образом, делает возможным дальнейшее изучение свойств белка.
12.5. Что дальше?
Несмотря на пользу методов моделирования белков, преимущества предлагаемого ими взгляда на функцию белков доступны не всем биологам. Простой доступ к надежным моделям, обеспечиваемый различными базами данных, является первым шагом к установлению диалога между
Литература
393
моделистами и биологами-экспериментаторами. Взаимодействие двух сообществ не только взаимовыгодно само по себе, но также необходимо для эффективного развития обеих дисциплин. Структурные модели могут значительно облегчить планирование и интерпретацию реальных экспериментов, поскольку они ограничивают число проверяемых гипотез, а иногда вносят и очень точные предложения к намечаемым экспериментам. Таким образом, мы предполагаем, что пропагандируемые усилия по идентификации все еще неизвестных белков, которые имеют известную ферментативную активность (Roberts 2004), должны быть интегрированы с усилиями по моделированию, которые позволят закрыть брешь между функциями, выполняемыми неизвестными белками, и белковыми последовательностями, функция которых должна быть предсказана.
С другой стороны, следует помнить, что существующие каталоги функций ограничены экспериментально обнаруженными проявлениями. Другими словами, предсказание функции обычно подразумевает поиск подходящей функции среди уже известных, а предположения о возможных, но еще не описанных функциях, делаются редко. Иначе говоря, нет методов предсказания функции de novo. Таким образом, хотя развитие таких новых вычислительных подходов должно воодушевлять, существующие методы, основанные на известных закономерностях, нуждаются в экспериментальной поддержке для идентификации новых реакций и процессов, которые затем могут быть добавлены в базы данных белковых структур и последовательностей. Экспериментальный анализ становится даже еще более важным в свете недавних результатов, показывающих, что скорость обнаружения новых белковых семейств является практически постоянной (Yooseph et al. 2007) и мы, таким образом, далеки от возможности составить разумное представление о размерах пространства разнообразных функций существующих на Земле белков (Raes et al. 2007).
Литература
Aloy Р, Russell RB (2002) Interrogating protein interaction networks through structural biology. Proc Natl Acad Sci USA 99:5896-5901
Aloy P, Russell RB (2003) InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics 19:161-162
Aloy P, Russell RB (2006) Structural systems biology: modelling protein interactions. Nat Rev Mol Cell Biol 7:188-197
Baxter SM, Rosenblum JS, Knutson S, et al. (2004) Synergistic computational and experimental proteomics approaches for more accurate detection of active serine hydrolases in yeast. Mol Cell Proteomics 3:209-225
Bonneau R, Strauss CE, Rohl CA, et al. (2002) De novo prediction of three-dimensional structures for major protein families. J Mol Biol 322:65-78
394 Глава 12. Предсказание функции белков на основе теоретических моделей
Bradley Р, Misura КМ, Baker D (2005) Toward high-resolution de novo structure prediction for small proteins. Science 309:1868-1871
Bujnicki JM (2003) Crystallographic and bioinformatic studies on restriction endonucleases: inference of evolutionary relationships in the “midnight zone” of homology. Curr Protein Pept Sci 4:327-337
Bumbaca D, Littlejohn JE, Nayakanti H, et al. (2007) Genome-based identification and characterization of a putative mucin-binding protein from the surface of Streptococcus pneumoniae. Proteins 66:547-558
Cammer SA, Hoffman ВТ, Speir J A, et al. (2003) Structure-based active site profiles for genome analysis and functional family subclassification. J Mol Biol 334:387-401
Castrignano T, De Meo PD, Cozzetto D, et al. (2006) The PMDB Protein Model Database. Nucleic Acids Res 34:D306-309
Chakravarty S, Sanchez R (2004) Systematic analysis of added-value in simple comparative models of protein structure. Structure 12:1461—1470
Chakravarty S, Wang L, Sanchez R (2005) Accuracy of structure-derived properties in simple comparative models of protein structures. Nucleic Acids Res 33:244-259
Chi A, Kemp RG (2000) The primordial high energy compound: ATP or inorganic pyrophosphate? J Biol Chem 275:35677-35679
Cymerman LA, Meiss G, Bujnicki JM (2005) DNase II is a member of the phospholipase D super-family. Bioinformatics 21:3959-3962
Davis FP, Braberg H, Shen MY, et al. (2006) Protein complex compositions predicted by structural similarity. Nucleic Acids Res 34:2943-2952
Davis FP, Barkan DT, Eswar N, et al. (2007) Host pathogen protein interactions predicted by comparative modeling. Protein Sci 16:2585-2596
Feder M, Bujnicki JM (2005) Identification of a new family of putative PD-(D/E)XK nucleases with unusual phylogenomic distribution and a new type of the active site. BMC Genomics 6:21
Fetrow JS, Skolnick J (1998) Method for prediction of protein function from sequence using the se-quence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J Mol Biol 281:949-968
Fetrow JS, Godzik A, Skolnick J (1998) Functional analysis of the Escherichia coli genome using the sequence-to-structure-to-function paradigm: identification of proteins exhibiting the glutaredoxin/thioredoxin disulfide oxidoreductase activity. J Mol Biol 282:703-711
Fumham N, Ruffle S, Southan C (2004) Splice variants: a homology modeling approach. Proteins 54:596-608
Hattersley AT, Ashcroft FM (2005) Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes 54:2503-2513
Hermann JC, Marti-Arbona R, Fedorov AA, et al. (2007) Structure-based activity prediction for an enzyme of unknown function. Nature 448:775-779
Jacobson M, Sali A (2004) Comparative protein Structure Modelling and its applications to drug discovery. Annu Rep Med Chem 39:259-274
Jordan IK, Wolf YI, Koonin EV (2004) Duplicated genes evolve slower than singletons despite the initial rate increase. BMC Evol Biol 4:22
Kolinski A, Bujnicki JM (2005) Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins 61 (Suppl 7):84-90
Kopp J, Schwede T (2004) The SWISS-MODEL Repository of annotated three-dimensional protein structure homology models. Nucleic Acids Res 32:D230-234
Krishnamurthy N, Brown D, Sjolander К (2007) FlowerPower: clustering proteins into domain architecture classes for phylogenomic inference of protein function. BMC Evol Biol 7(Suppl 1 ):S 12
Kryshtafovych A, Venclovas C, Fidelis K, et al. (2005) Progress over the first decade of CASP experiments. Proteins 61 (Suppl 7):225-236
Kryshtafovych A, Fidelis K, Moult J (2007) Progress from CASP6 to CASP7. Proteins 69(Suppl 8): 194-207
Lopez C, Chevalier N, Hannaert V, et al. (2002) Leishmania donovani phosphofructokinase. Gene characterization, biochemical properties and structure-modeling studies. Eur J Biochem 269:3978-3989
Lu L, Lu H, Skolnick J (2002) MULTIPROSPECTOR: an algorithm for the prediction of proteinprotein interactions by multimeric threading. Proteins 49:350-364
Литература
395
Malmstrom L, Riffle M, Strauss CE, et al. (2007) Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol 5:e76
McGovern SL, Shoichet BK (2003) Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J Med Chem 46:2895-2907
McGuffln U (2008) The ModFOLD server for the quality assessment of protein structural models. Bioinformatics 24:586-587
Melo F, Feytmans E (1998) Assessing protein structures with a non-local atomic interaction energy. J Mol Biol 277:1141-1152
Melo F, Sanchez R, Sali A (2002) Statistical potentials for fold assessment. Protein Sci 11430-448
Michels PA, Chevalier N, Opperdoes FR, et al. (1997) The glycosomal ATP-dependent phospho-fructokinase of Trypanosoma brucei must have evolved from an ancestral pyrophosphatedependent enzyme. Eur J Biochem 250:698-704
Oshiro C, Bradley EK, Eksterowicz J, et al. (2004) Performance of 3D-database molecular docking studies into homology models. J Med Chem 47:764-767
Pawlowski K, Godzik A (2001) Surface map comparison: studying function diversity of homologous proteins. J Mol Biol 309:793-806
Pawlowski M, Gajda MJ, Matlak R, et al. (2008) Meta-MQAP: a meta-server for the quality assessment of protein models. BMC Bioinformatics in press
Pieper U, Eswar N, Davis FP, et al. (2006) MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 34:D291-295
Quiocho F, Vyas N (1999) Atomic interactions between proteins/enzymes and carbohydrates. In: Hecht SM (ed.), Bioinorganic Chemistry: Carbohydrates. Oxford University Press, New York, NY
Raes J, Harrington ED, Singh AH, et al. (2007) Protein function space: viewing the limits or limited by our view? Curr Opin Struct Biol 17:362-369
Rigden DJ, Galperin MY (2008) Sequence analysis of GerM and SpoVS, uncharacterised bacterial ‘sporulation’ proteins with widespread phylogenetic distribution. Bioinformatics, accepted DOI 10.1093/bioinformatics/btn314
Roberts RJ (2004) Identifying protein function-а call for community action. PLoS Biol 2:E42
Sasin JM, Godzik A, Bujnicki JM (2007) SURF’S UP! - protein classification by surface comparisons. J Biosci 32:97-100
Schafer P, Cymerman LA, Bujnicki JM, et al. (2007) Human lysosomal DNase Ilalpha contains two requisite PLD-signature (HxK) motifs: evidence for a pseudodimeric structure of the active enzyme species. Protein Sci 16:82-91
Service RF (2008a) Structural biology. Protein structure initiative: phase 3 or phase out. Science 319:1610-1613
Service RF (2008b) Structural biology. Researchers hone their homology tools. Science 319:1612
Silveira NJ, Uchoa HB, Pereira JH, et al. (2005) Molecular models of protein targets from Mycobacterium tuberculosis. J Mol Model 11:160-166
Sodhi JS, Bryson K, McGuffln U, et al. (2004) Predicting metal-binding site residues in low-resolution structural models. J Mol Biol 342:307-320
Song L, Kalyanaraman C, Fedorov AA, et al. (2007) Prediction and assignment of function for a divergent N-succinyl amino acid racemase. Nat Chem Biol 3:486-491
Szilagyi A, Skolnick J (2006) Efficient prediction of nucleic acid binding function from low-resolution protein structures. J Mol Biol 358:922-933
Tammaro P, Flanagan SE, Zadek B, et al. (2008) A Kir6.2 mutation causing severe functional effects in vitro produces neonatal diabetes without the expected neurological complications. Diabetologia 51:802-810
Tamulaitiene G, Jakubauskas A, Urbanke C, et al. (2006) The crystal structure of the rare-cutting restriction enzyme Sdal reveals unexpected domain architecture. Structure 14:1389-1400
Tatusov RL, Fedorova ND, Jackson JD, et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41
Tress M, Bujnicki JM, Valencia A (2008) Integrating structures, functions, and interactions. In: Bujnicki JM (ed.), Prediction of Protein Structures, Functions and Interactions. Wiley.
Tress ML, Martelli PL, Frankish A, et al. (2007) The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci USA 104:5495-5500
396 Глава 12. Предсказание функции белков на основе теоретических моделей
van Gunsteren W (1996) Biomolecular Simulations: The GROMOS96 Manual and User Guide. Biomos : Groningen
Wallner B, Elofsson A (2003) Can correct protein models be identified? Protein Sci 12:1073-1086
Wallner B, Elofsson A (2006) Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci 15:900-913
Wang P, Yan B, Guo JT, et al. (2005) Structural genomics analysis of alternative splicing and application to isoform structure modeling. Proc Natl Acad Sci USA 102:18920-18925
Xu LZ, Sanchez R, Sali A, etal. (1996) Ligand specificity of brain lipid-binding protein. J Biol Chem 271:24711-24719
Yooseph S, Sutton G, Rusch DB, et al. (2007) The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 5:el6
Указатель основных сокращений и наименований*
А AMBER 19 ASP 221 FUNCAT 191 G GB 20, 34
В BLAST 60, 91, 326, 336 BLOSUM 47, 55, 62, 133 GeneJD 200 GO 166,242, 323 GPCR 128, 134 GROMOS96 19
С CASP 11, 20, 22, 45, 64, 68, 75, 110, 165, 370 САТН 116, 193, 245 CHARMM 19, 105 ClustalW 258 COG 380 COMPOSER 98 CS\23,31,211,242, 260, 335 н HMM 64 HMMTOP 137 HTH 331, 332, 360 I IDP 153 I-TASSER 27, 75 К
D Dali 325 DALI 193, 352 DIP 324, 329 DSSP 160 KEGG 191,209,211 L LOOCV 144 M
E EBI 330 EC 84, 191,242 ED 294, 301,314 ENM 308 ENSEMBLE 139 E-value 254, 339 MCSG 330, 351 MODBASE 115 MODELLER 99, 105, 106 MQAP 33, 35 N NMA 307 NMDA 205
F FASTA97.332, 339, 356 FFF 242, 257,377 FSSP 193, 325 0 OCTOPUS 139 OPLS 19, 106 OPM 132
* В указателе представлены сокращения и наименования, которые относительно широко распространены в русскоязычной литературе по молекулярному моделированию. P PAM 60, 133 PCA2SS, 297,314 Peons 35, 69, 75, 101
398
Указатель основных сокращений и наименований
PDB 15
PDBsum 339
PHS 30
Prime 106
PROCHECK 109
ProFunc 264, 330, 347, 352
ProKnow 263, 325, 347
PROSA 109
PSA 87, 370
PSI-BLAST 63, 64, 91,387
PSIPRED 62
PSSM 55, 63
P-value 254
R
REM 30
REMD 294
REX 295,313
RMSD 20
Roberta 101
ROSETTA20, 24, 37, 106, 147, 385
s
SAS 220
SCOP 48, 193, 200, 242, 245, 385
SFLD 201,206
SSM 332
SVM 68, 138, 163
SWISS-MODEL 115, 146
T
TASSER 25
T-Coffee 92, 134
THREADER 57
TIM 192, 223
TIP3P 20
ТМНММ 137
TopPred 136
u
UCLA 325
UniProt 372
UniProtKB 15
UNRES 21
V
Verify 3D 36, 94, 101, 109
w
WHATCHECK 109
3D 242
aBB 23, 32
Б
БД 242
БПН 153
Г
ГО 166, 174, 189, 323,368
И
ИНС 138
ИО 29
К
КД 301, 314
КФ 84, 191,211,242
м
МД 19, 242, 282
МК20
ММ 21
МО 162
О
ОПН 154
п
ПДР 220
С
СКО 20, 242
СММ 64, 73, 91, 104, 137, 199
СШД 360
т
ТМ 126
Э
ЭМ 108
ЭМОР/СМОР/ 72, 176
Приложение
Цветная версия иллюстраций
Рис. 1.3
Рис.1.4
400
Приложение. Цветная версия иллюстраций
Известная последовательность
Исследуемая последовательность
LFDLCDLIPV--CGFA ARDL--VIPMIYCAHG
Рис. 2.1
«Замороженное» приближение
Последовательность
шаблона LFDLCDLIPV - - CGFA
Исследуемая Чк, .и.....
последовательность
LFDLCDLIPV--CGFA
ARDL--VIPMIYCAHG _________A___/
Рис. 2.3
Иллюстрации к главе 2
401
а)
if И I -I i 1-11!
Рис.2.4
402
Приложение. Цветная версия иллюстраций
Рис. 3.2
0 50 100
Номер остатка в последовательности
Рис. 3.3
Рис. 3.4
Рис. 4.1
Рис. 4.2
404
Приложение. Цветная версия иллюстраций
Рис. 4.5
Рис. 5.4
Иллюстрации к главе 6
405
а) Фотолиаза репарации ДНК
в) Пирофосфатаза АТФ N-типа
г) Аминоацил-тРН К синтетаза I класса
Рис. 6.4
406
Приложение. Цветная версия иллюстраций
Рис. 7.4
Рис. 8.1
Иллюстрации к главе 8
407
Рис. 8.2
Глутаредоксин бактериофага Т4 (T4-GRX)
Дисульфид оксидоредуктаза E.coli (DSB)
T4-GRX .... .... .MFKVYGYDSNIHKCVYCDNAKRLLTVKKQPF. . . .EFINIMPEKGVFDDEKIAEL
DSB AQYEDGKQYTTLEKPVAGAPQVLEFFSFFCPHCYQFEEVLHISDNVKKKLPEGVKMTKYHVNFMGGDLGKDLTQ
TRX KQIESKTAFQEALDAAGDKLVWDFSATWCGPCKMIKPFFHSLSE . . . KYSN. VIFLEVDVD......D
T4-GRX LTKLGRDTQIGLTM_QVFAPDGSHIGGFDQLREYFK.......................................
DSB AWAVAMALGVEDKVTVPLFEGVQKTQTIRSASDIRDVFINAGIKGEEYDAAWNSFWKSLVAQQEKAAADVQLR
TRX CQDVASECEVKCT_TFQFFKKGQKVGE...............FSGA. NKEKLEATINELV.................
T4-GRX ..........................................
DSB GV AMFVN KY LNPQGMDTSNMDVFVQQYADTVKYLSEKK
TRX .............................................
Рис. 8.3
408
Приложение. Цветная версия иллюстраций
Рис. 9.1
Рис. 9.2
Иллюстрации к главе 9
409
Внутриклеточная сторона
Ось поры Z (А)
Энергия водородных связей в расчете на одну молекулу воды (кДж/моль)
Рис. 9.5
Проекция первый собственный вектор (нм)
Рис. 9.13
410
Приложение. Цветная версия иллюстраций
Рис. 10.5
Рис. 10.6
Иллюстрации к главе 10
411
Chain А
75
IRQSVT LlKjMJN tbuULAEOF I QATfc JWYKAEAYOl OVILRQTVT VUMVAIXEI'YHTINM
LKNMMlI IRKHUJ.NlJJIQWY\KM>L1IGQ NEMAt.VLSFXAI fcP 1NKA
l«7pA)
30____53
30______35
MTf
DBQ1YTQ
Gt Vl**GSMXKE 11PVST
Chain A -^W—= =>-=>-
96 100 105 IIP IIS IM 125 130 135 140 145 150 155 160 165 170 175
5SI^I<a«n><X.VGKI AlltAl ILKIEKLI I TRIIE l V<»Pl NVPSQM AlMTANIll<QLAP NRI YA< PKA0IVF4L P pj^LDLSFyaa,IMfr' 1К^У1К^У1^МУАКЙ>0^каМС0^’ » ;YHPVN<ftARll
04 100 IOS 110 115 120 125 130 135 140 145 150 155 160 165 170 I75~
l«7ftA) | 1 > —*
e)
Рис. 10.7
412
Приложение. Цветная версия иллюстраций
Рис. 11.1
Рис. 12.1
Иллюстрации к главе 12
413
Рис. 12.3
Рис. 12.4
Рис. 12.5
414
Приложение. Цветная версия иллюстраций
Рис. 12.6
Рис. 12.8