/












































































































































































Автор: Сирота А.Г.
Теги: синтетические продукты полимеризации полимеризационные смолы синтетические каучуки химия
Год: 1974




Текст
л. Г. Сирота
с ' Ч (О
Модификация структуры и свойств полиолефинов
Издание второе, переработанное и дополненное
Издательство «Химия» Ленинградское отделение 1974
УДК 678.742 С 40
Сирота А. Г.
С40 Модификация структуры и свойств полиолефинов. Изд. 2-е, пер. и доп. Л., «Химия», 1974.
176 стр., 67 рис., 23 табл., список литературы 1180 ссылок.
В книге изложены методы модифицирования полиолефинов, позволяющие направленно изменять нх структуру и свойства, получать материалы с заранее заданными свойствами (сополимеризация, привитая и блоксополимеризацня, введение в макромолекулы функциональных групп, сшивание, создание композиций с различными полимерными и низкомолекулярными продуктами). Описаны важнейшие свойства получаемых материалов, области нх применения.
Книга предназначена для научных и инженерно-технических работников, занимающихся получением и применением полимерных материалов. Она может быть рекомендована преподавателям вузов н техникумов, а также студентам, специализирующимся в области химии полимеров.
20505-068
050Т0Т)-74 68~74
© Издательство «Химия», 1974.
Введение
Полиолефины в последние годы стали одним из основных типов синтетических полимерных материалов. Их мировое производство в 1970 г. составило около 8 300 000 т. Ожидается, что в 1975 г. будет произведено 14 700 000 т полиолефинов, а в 1980 г. — 23 000000 т [1]. Кроме полиэтилена и полипропилена все шире применяются более новые полиолефины: полибутен-1, поли-З-метилбутен-1, поли-4-метилпентен-1 и др. Тем не менее число выпускаемых промышленностью полиолефинов ограничено, в то время как количество материалов на их основе постоянно увеличивается. Это связано с использованием различных методов модифицирования полиолефинов, позволяющих значительно расширять области их применения и в известной мере решать проблему получения материалов с заданными свойствами.
По мере роста производства полиолефинов и удовлетворения потребностей в них различных отраслей техники модифицирование приобретает все большее значение. В частности, это подтверждается сведениями об основных направлениях патентования в области полиолефинов. Например, в США доля патентов на модифицированные полиолефины из общего числа патентов на полиолефины в 1954 г. составила около 17%, в 1967 г. — около 44% *.
Полиолефины обладают весьма ценным комплексом свойств: высокими диэлектрическими характеристиками, сохраняющимися в широком интервале температур, химической стойкостью, значительной теплостойкостью и в большинстве случаев морозостойкостью, прочностью, небольшим удельным весом и т. д. Однако зачастую те особенности структуры, которые обусловливают эти ценные свойства, оказываются одновременно причиной, препятствующей тому или иному специфическому применению материала. Например, незначительная полярность полиолефинов, с одной стороны, сообщает им прекрасные диэлектрические свойства и химическую стойкость, с другой — приводит к ограниченной адгезии к различным материалам и слабой восприимчивости к красителям. Высокая кристалличность и
* По данным Е. В, Монаховой, 3. Я. Новиковой и В. М. Демидовой.
1* 3
совершенство кристаллических образований линейных видов полиэтилена обусловливают их значительную прочность и теплостойкость, но одновременно ограничивают, например, их эластичность и стойкость к растрескиванию под влиянием длительных нагрузок и других факторов.
Модифицирование полиолефинов и призвано чаще всего некоторым изменением структуры воздействовать в заданном направлении на часть свойств материала при возможном сохранении комплекса остальных свойств.
Направленное изменение структуры и свойств полиолефинов осуществляется либо в процессе синтеза, либо воздействием на готовый полимер. К первой группе путей модифицирования можно отнести варьирование условий полимеризации и сополимеризацию а-олефинов с различными мономерами. Модифицирование готовых поли-а-олефинов достигается введением в их макромолекулы функциональных групп, образованием системы ковалентных или ионных связей между макроцепями, созданием композиций с различными полимерными и низкомолекулярными соединениями, а также способами, совмещающими отдельные методы модифицирования.
Пути модифицирования полиолефинов в значительной степени связаны с особенностями их строения и свойств. Так, химическая инертность ограничивает возможности использования реакций в цепях полимеров и заставляет прибегать к достаточно специфическим приемам преодоления малой химической активности. Значительная кристалличность полиэтилена позволяет, воздействуя на молекулярную структуру полимера или варьируя условия кристаллизации, изменять характер надмолекулярных образований, от которых в большой мере зависят свойства материала.
Различия в строении полиолефинов также определяют способы модифицирования, пригодные для отдельных полимеров. Например, наличие в каждом элементарном звене полипропилена третичного атома углерода позволяет в относительно мягких условиях (по сравнению с полиэтиленом) получать продукты окисления, пригодные как для непосредственного применения, так и для дальнейших химических превращений. Вместе с тем при радиационном модифицировании полипропилена необходимо учитывать, что третичные атомы углерода обусловливают его радиационную нестойкость, проявляющуюся в деструкции материала. Полиэтилен в тех же условиях гораздо менее подвержен деструкции. Это позволяет успешно применять радиационное воздействие для повышения его теплостойкости и для других целей. Таким образом, правильный выбор метода и условий модифицирования требует учета особенностей строения исходных полиолефинов.
Глава I
Структура и свойства полиолефинов
Полиэтилен
Сведения о структуре и свойствах полиэтилена приведены в ряде обширных трудов и руководств [4—10]. Это позволяет ограничиться кратким рассмотрением структуры и свойств полиэтилена и возможностей их регулирования изменением условий синтеза.
В технике применяются различные типы полиэтилена, получаемые при высоком, низком и среднем давлении. Полимеризация этилена разными методами приводит к получению продуктов, существенно отличающихся по структуре и свойствам. Но различные продукты могут быть получены при одном и том же способе полимеризации варьированием условий, определяющих структуру и свойства полиэтилена.
Полимеризация этилена при высоком давлении (от 1000 до 3000 кгс/см2) подчиняется обычным закономерностям реакции полимеризации винильных соединений, протекающей по свободнорадикальному механизму. Особенность полимеризации этилена, инициированной свободными радикалами, заключается в том, что полиэтилен с высоким молекулярным весом получается лишь при высоких концентрациях мономера. При малых концентрациях этилен присоединяется к свободным радикалам, но с реакцией их роста конкурируют реакции дезактивации свободных радикалов, и образующийся полимер имеет невысокий молекулярный вес [11, с. 7; 12, 13]. С повышением давления этилена, сопровождающимся увеличением его плотности, средний молекулярный вес продукта полимеризации (при постоянных температуре и концентрации инициатора) возрастает.
Плотность этилена может быть увеличена также понижением температуры. Нижний предел температуры определяется, выбором инициатора полимеризации. Температуры, при которых обычно проводится полимеризация, находятся в интервале от 80 до 300 °C.
Молекулярный вес полиэтилена обратно пропорционален температуре и концентрации инициатора и составляет обычно от 2000 до 40 000. Молекулярный вес (и молекулярно-весовое распределение) можно регулировать применением агентов
5
передачи цепи [93, 132, 153], например пропана. Константы передачи цепи для ряда различных агентов передачи цепи приведены в работе [132]. В случае пропана эта константа, найденная при давлении 1300 кгс/см2 и 130 °C, составляет 0,003.
От температуры полимеризации зависит разветвленность макромолекул, в значительной мере определяющая характер кристаллических образований и свойства полиэтилена. Степень разветвленности тем больше, чем выше температура полимеризации. Основной причиной образования ответвлений являются реакции передачи цепи [14], скорость которых возрастает с повышением температуры. Образующиеся в результате этих реакций ответвления могут иметь различную длину. Связь распределения длин ответвлений с условиями полимеризации в достаточной мере не изучена, однако предполагается, что чем выше температура полимеризации, тем больше длина боковых цепей [11, с. 7; 86].
С увеличением числа боковых ответвлений, определяемого методами ИК-спектроскопии, гель-хроматографии [136, 154], анализом продуктов радиолиза [135], снижаются кристалличность и связанные с нею физико-механические показатели полиэтилена: плотность, теплостойкость, прочность, жесткость и др. От длины боковых ответвлений также, несомненно, зависят свойства полиэтилена как в твердом состоянии [133], так и в расплаве [134]. Однако исследование этого интересного вопроса затруднено отсутствием надежного метода определения величины боковых цепей, в первую очередь сравнительно длинных (см., например, [104, 136, 137]). Относительно коротких ответвлений имеется указание, что в основном преобладают этильные и бутильные группы [15, 105, 135]. На примере модельных соединений, в качестве которых использовались сополимеры этилена с высшими а-олефинами, показано, что этильные и бутильные боковые группы особенно сильно препятствуют кристаллизации полимера [16, 17].
Проведение полимеризации этилена при сравнительно низких температурах (60 °C) и достаточно высоком давлении (1500 кгс/см2) позволяет резко снизить скорость реакций передачи цепи на полимер и получать полимер с малым числом ответвлений (3 группы СН3 на 1000 атомов С) и высокой плотностью (0,94 г/см3) [87]. Практически неразветвленный полиэтилен может быть получен при давлении около 7000 кгс/см2 и температуре 50—80 °C в присутствии динитрила азобисизомас-ляной кислоты [103]:
Число СНз-групп на 1000 атомов С . . . . <0,8 Плотность, г/см3...................... 0,955
Температура плавления, °C.............. 131,9
Содержание ненасыщенных групп на
1000 атомов С: винильных............................<0,01
винилиденовых...................... 0,02
транс-виниленовых..................<0,01
Такой полиэтилен по плотности и температуре плавления близок к линейным видам полиэтилена низкого и среднего давления (см. табл. 2). Для него характерно также пониженное содержание ненасыщенных групп.
Значительными возможностями регулирования свойств полиэтилена обладает также процесс радиационной полимеризации, в котором свободные радикалы генерируются действием у-лучей [18—23, 88, 89, ПО—114, 122]. В зависимости от условий проведения процесса могут получаться жидкие, воскоподобные или твердые полимеры. Молекулярный вес продукта тем больше, чем выше давление и ниже температура полимеризации. При относительно низких температурах (ниже 80 °C) процесс протекает с получением твердого малоразветвленного полиэтилена с высокой плотностью (до 0,975 г/см3) [90, 123]. При более высоких температурах образуются воскообразные и жидкие продукты.
Важной особенностью полиэтилена, полученного радиационной полимеризацией, являются весьма высокие диэлектрические свойства (тангенс угла диэлектрических потерь менее 1,6-10'4) [91, с. 231; 92, с. 356].
Полимеризация этилена при низком давлении (до 5 кгс/см2) в присутствии металлорганических комплексных катализаторов представляет совершенно иные возможности воздействия на структуру и свойства полиэтилена, определяющиеся анионным механизмом процесса. Полимеризация осуществляется в среде углеводородного растворителя при температурах 60—80 °C. Катализатор находится в растворителе в виде осадка или коллоидной дисперсии. В качестве катализаторов используются продукты взаимодействия алкилов металлов первой, второй или третьей группы (чаще всего алкилы алюминия) с солями металлов переменной валентности (обычно хлоридами титана или ванадия). Предполагается [24—26, 109], что в каталитическом комплексе сильно поляризованы связи между атомами металла и углерода в направлении карбаниона; молекулы мономера внедряются по месту связи металла с карбанионом, растущий анион координирован с катионом металла.
Основные пути влияния на свойства полиэтилена связаны с изменениями каталитического комплекса. Так, использование вместо алкила алюминия (например, триэтилалюминия) алкил-галогенида алюминия (например, диэтилалюминийхлорида) приводит к снижению молекулярного веса полимера [27]. В значительных пределах средний молекулярный вес (а также молекулярно-весовое распределение) можно регулировать варьированием соотношения алкила алюминия и хлорида титана [28, 29]. Так, при мольном соотношении триэтилалюминия и четыреххлористого титана 2:1 полиэтилен имеет молекулярный вес выше 1 000 000, в то время как при соотношении 1 :2 образуется хрупкий продукт с молекулярным весом менее 30000. Полиэтилен с молекулярным весом от 70 000 до 350 000,
7
Перерабатываемый в изделия литьем под Давлением или экструзией при относительно невысокой температуре (200—260°C), получают при близком к эквимолекулярному соотношении компонентов катализатора, содержащих алюминий и титан.
Снизить молекулярный вес полиэтилена можно также введением в реакционную среду водорода, вызывающего передачу и обрыв цепи. Полимеризация в присутствии добавок водорода позволяет также получать полиэтилен с повышенной плотностью [100, 101] п меньшим, чем обычно, содержанием непредельных группировок [102].
Для получения полимера с относительно узким молекулярно-весовым распределением рекомендуется проводить полимеризацию в присутствии небольшого количества окиси углерода [30], водорода [3], анизола [107, с. 27; 131] или подвергать полимеризации этилен, разбавленный инертным газом [31].
Отмечено повышение активности растворимой каталитической системы (C2H5)2TiCl2— A1R3 под влиянием добавок кислорода к этилену (до 0,06—0,1 объемн.%) [108, с. 193].
Возможности регулирования молекулярного веса полиэтилена введением различных добавок в реакционную среду систематизированы в работе [2]. К снижению молекулярного веса приводят, кроме водорода, добавки кислорода, спиртов, альдегидов, органических и неорганических перекисей, углекислого газа, четыреххлористого углерода, трехфтористого бора, ацетилена. Присутствие в сфере реакции воды, кислот Льюиса, органических соединений серы, силиконового масла, напротив, вызывает увеличение молекулярного веса. В случае некоторых добавок, способствующих повышению молекулярного веса, отмечается и улучшение физико-механических свойств. К таким добавкам относятся спирты, органические соединения серы, а также производные фенола, диэтилцинк, трихлоруксусная кислота.
Ионный механизм полимеризации на металлорганических катализаторах определяет очень незначительную разветвленность макромолекул. Линейная структура макроцепей является причиной высокой кристалличности полиэтилена низкого давления, с которой связаны его лучшие, чем у полиэтилена высокого давления, основные технические свойства (плотность, прочность, жесткость, теплостойкость).
Ответвления от основной цепи, содержащиеся в полиэтилене высокой плотности в сравнительно небольшом количестве, имеют, очевидно, значительный молекулярный вес [138]. Длинные ответвления не оказывают существенного влияния на кристалличность полимера и его свойства в твердом состоянии, однако проявляются в свойствах расплава и разбавленных растворов.
Особенно малой разветвленностью (содержание СН3-групп менее 1 на 1000 атомов С) в сочетании с высоким молекулярным весом и узким молекулярно-весовым распределением отличается полиэтилен, полученный с использованием гомогенных
8
катализаторов на основе соединений ванадия и алкилалюми-нийгалогенидов, например VO(OC2Hs)3— А1(С2Н5)2С1 [115].
Еще менее разветвленный и более кристалличный полиэтилен получают ионной полимеризацией этилена на твердых окиснометаллических катализаторах при среднем давлении (35—70 кгс/см2). Процесс осуществляется в среде углеводородного растворителя при 100—175°С. Катализаторами служат окислы хрома [32—34; 106, с. 387], молибдена, никеля, кобальта [35—40; 41, с. 82; 42, с. 90].
Окислы хрома наносятся чаще всего на пористый алюмосиликатный носитель. Носителями для окиси молибдена служат окись алюминия и окись титана, для окислов никеля и кобальта — активированный уголь. Значительная активность катализаторов достигается в результате специальной операции активирования, которая для окиснохромового катализатора, например, заключается в его нагревании при температуре 500—600 °C в токе сухого воздуха в течение нескольких часов.
Температура процесса полимеризации в большой мере определяет молекулярный вес и зависящие от него свойства полиэтилена. Повышение температуры полимеризации ведет к резкому снижению молекулярного веса и, соответственно, к возрастанию показателя текучести расплава [41]. Так, изменение температуры от 100 до 135 °C при полимеризации этилена на окиснохромовом катализаторе позволяет получать полимер с характеристической вязкостью от 2,4 до 0,8 дл/г [34, 43, 44].
Молекулярный вес зависит также и от температуры активации катализатора: по мере роста температуры активации молекулярный вес полиэтилена уменьшается [34; 41, с. 82].
Повышение давления в процессе полимеризации резко увеличивает скорость реакции. Возрастает при этом и молекулярный вес полиэтилена, но далеко не в той мере, как при повышении температуры [34, 43—45].
Механизм полимеризации этилена определяет и такую характеристику структуры полимера, как характер ненасыщенности. В табл. 1 представлены данные о содержании ненасыщенных групп в различных видах полиэтилена, полученные Гольденбергом и другими авторами [34, 46] и хорошо согласующиеся с данными Смита и др. [47, 132].
Из табл. 1 видно, что ненасыщенность полиэтилена высокого давления определяют в основном винилиденовые группы. Их образование связано с реакциями переноса цепи при участии молекул полимера, сопровождающими полимеризацию этилена по радикальному механизму [46]. Реакции переноса цепи, протекающие со значительной скоростью при повышенных температурах, ведут к образованию активных центров на атомах углерода макромолекул полиэтилена:
---СН2—СН2—С—СН2-----
R
9
Таблица 1
Содержание различных ненасыщенных групп в полиэтилене [34]
Полиэтилен Число групп ^с=«с^ па 1000 атомов С Содержание (в % от общего содержания групп /С=С\ ] групп
ванильных -СН=СН2 винилиденовых \-сн2 траяС’Вняи-леновых н\ /
Высокого давления . . . 0,3-0,4 17 71 12
Низкого давления . . . 0,3-0,4 52 31 17
Среднего давления . . . 1,1-1,3 87 7 6
Предполагается, что винилиденовые группы образуются в результате реакции:
••• СН2—СН2—С—СН2--—>-------СН2 + СН2=С—сн2---
R R
Механизм образования двойных связей в полиэтилене, получаемом на ионных катализаторах, менее ясен. Считают [46], однако, что в полиэтилене низкого давления двойные связи образуются при реакции диспропорционирования в результате переноса гидрид-иоиа от p-углеродного атома к металлу [48, с. 195; 49]:
м+----сн2—сн2—сн2— —> мн + сн2=сн—сн2--------
Такой механизм обрыва растущей цепи должен сопровождаться образованием в основном винильных групп, а при наличии заместителя у p-углеродного атома — винилиденовых групп. Как видно из табл. 1, в полиэтилене низкого давления действительно преобладают винильные группы.
В полиэтилене среднего давления также преимущественно содержатся винильные группы. Их образование можно представить, приняв предположение о катионном механизме полимеризации этилена [50] на окисных катализаторах, при котором обрыв цепи происходит вследствие отщепления протона от соседней с активным концом метиленовой группы:
Х(СН2—СН2)„—СН2-СН^ —> Х(СН2-СН2)„-СН=СН2 + н+
Связанные с механизмом и условиями полимеризации особенности молекулярной структуры, и в первую очередь разветвленность, естественно, в большой мере определяют важные свойства отдельных видов полиэтилена. В табл. 2 представлены основные сведения о структуре и свойствах полиэтилена высокого, низкого и среднего давления.
10
Таблица 2
Свойства различных видов полиэтилена
Полиэтилен Полиэтилен I юлиэтилен
Показатели высокого НИЗКОГО среднего
давления давления давления
Характеристическая вязкость в дека- 1,2-3,0
лине при 135 °C. дл/г 0.7-1,0 1,0-5,1
Число СНз-групп на 1000 атомов С . . 20-40 5—15 1,5-5,0
Степень кристалличности *, % .... 53-67 80-90 85—93
Плотность, г/см3 0,92—0,93 0,94—0,96 0,96—0,97
Температура плавления, °C Прочность, кгс/см2 108-110 120-134 127—130 270—330
при растяжении 120-160 220—350
при изгибе 120—170 200—380 250—400
Предел текучести, кгс/см2 Относительное удлинение при разрыве 110-100 150-600 200—250 250-300
(скорость растяжения 100 мм/мин), % 200—900 20—400
Модуль упругости, кгс/см2 1 500-2 500 5 500—8 000 8 000—10 500
Твердость по Бринеллю, кгс/Мм2 . . . Водопоглощение за 30 суток при 1,4-2,5 4,5—5,8 5,6—6,5
20 °C, % Тангенс угла диэлектрических потерь 0,04 0,03-0,04 0,01
при частоте 106 Гц и 20 °C, tgS-104 Диэлектрическая проницаемость при 2-4 2-5 2-4
Ю6 Гц 2,2—2,3 2,1-24 2,3
Удельное объемное электрическое сопротивление, Ом • см Электрическая прочность (переменный 1017 ю17 10”
ток), кв/мм, при толщине образца
1 мм 45—60 45-60 45—60
2 мм 28-35 28—36 28—35
• Понятие «степени кристалличности* в применении к кристаллизующимся полимерам в известной мере условно. Оно ие отражает отсутствия четких границ между кристаллическими и аморфными областями, существования промежуточных структур с разной степенью упорядоченности. Тем не менее это понятие бызает удобным при сравнении близких по строению полимеров.
В табл. 2 и далее, если это специально не оговаривается, приведены сведения о степени кристалличности, полученные рентгенографическим способом по методике» основанной иа сравнении интегральной интенсивности отражения рентгеновских лучей от аморфной и кристаллической областей полимера [99J.
Отличия в свойствах обычно являются следствием различной разветвленности макромолекул, поскольку именно эта величина в первую очередь определяет кристалличность полиэтилена. Наиболее разветвленный полиэтилен высокого давления относительно мало кристалличен, обладает пониженными значениями плотности, температуры плавления, прочности, жесткости, твердости. Наименее разветвленный полиэтилен среднего давления, напротив, кристалличен, значительно более плотен, жесток, прочен, тверд и обладает повышенной температурой плавления. Полиэтилен низкого давления занимает промежуточное положение по разветвленности и, соответственно, по указанным характеристикам.
С увеличением разветвленности степень кристалличности понижается вследствие нарушений кристаллической структуры в узлах разветвлений и близких к ним участках. Температура плавления, модуль упругости и твердость связаны с малыми смещениями молекул в твердом полиэтилене, что и определяет значительную зависимость этих свойств от степени кристалличности [51]. Свойства полиэтилена, связанные с большими деформациями (прочность при растяжении, относительное удлинение при разрыве и температура хрупкости), зависят не только от разветвленности и кристалличности, но в еще большей мере от молекулярного веса полиэтилена [52, с. 431; 145].
Влияние кристалличности и молекулярного веса на прочностные характеристики полиэтилена может быть пояснено следующим образом. Кристаллические образования способствуют диссипации напряжений в полиэтилене благодаря возможности сдвига отдельных элементов кристаллических структур (ламелей) [94, с. 464; 95, с. 424; 96]. Однако существенное повышение прочностных свойств в результате диссипации напряжений возможно лишь при условии вовлечения в этот процесс значительного числа кристаллических областей, что, в свою очередь, требует существования сил связи между ними. Связь между кристаллическими областями реализуется посредством цепей, проходящих через аморфные области и являющихся общими хотя бы для двух соседних кристаллических образований. Содержание таких цепей, называемых проходными, естественно, возрастает с увеличением молекулярного веса полиэтилена (или доли высокомолекулярной фракции). Таким образом, увеличение молекулярного веса и кристалличности приводит к возрастанию доли нагруженных при деформировании полимера цепей и доли сопротивляющегося деформации материала и к уменьшению различия между теоретической и практической прочностью полимера.
Предполагается, что на увеличение прочности при разрыве [126, 130], а также стойкости к растрескиванию [126] полиэтилена оказывают влияние, кроме проходных цепей, физические узлы зацепления молекул, образуемые главным образом длинными цепями. Физические узлы, сохраняющиеся при растяжении полиэтилена, связывают отдельные кристаллические участки фибрилл и тем самым способствуют сопротивлению нагрузке. Прочностные свойства полиэтилена определяются также характером его надмолекулярной структуры, зависящим от условий ее формирования [144].
На прочностные свойства полиэтилена значительное влияние оказывает и способность полимера к релаксации напряжений. Особенно существенно релаксационные характеристики влияют на прочность полиэтилена при длительных нагрузках, в частности на стойкость к растрескиванию [155]. Поскольку способность к релаксации напряжений определяется молекулярными и надмолекулярными характеристиками (молекулярным весом,
12
молекулярно-весовым распределением, разветвленностью, кристалличностью и пр.), влияние каждой из этих характеристик на прочностные свойства полиэтилена не является однозначным [146, с. 324]. Например, несмотря на повышение способности к диссипации напряжений при увеличении степени кристалличности полиэтилена, его стойкость к растрескиванию может снизиться из-за замедления скорости релаксации напряжений. Введение низкомолекулярных фракций в полиэтилен [147] приводит к снижению среднего молекулярного веса, уменьшая стойкость к растрескиванию под действием напряжения и окружающей среды [148]. Другим результатом наличия низкомолекулярных фракций в полиэтилене является снижение напряжений при деформации и увеличение скорости релаксационных процессов, что способствует повышению стойкости к растрескиванию [149—151].
Результирующий эффект того или иного изменения молекулярных или надмолекулярных характеристик полиэтилена в значительной мере определяется конкретными условиями эксплуатации. Это важно учитывать при оценке экспериментальных данных при определении прочностных свойств и выборе вида и марки полиэтилена [148].
Рентгенографическим методом показано, что разветвленность макромолекул оказывает влияние не только на степень кристалличности полиэтилена, но и на размеры элементарных кристаллических ячеек и плотность кристаллических областей. Возрастание числа ответвлений ведет к увеличению размеров ячеек в основном вдоль осей а и b (табл. 3). Размер элементарных ячеек по оси с (совпадающей с осью макромолекулы), составляющий в неразветвленном полиэтилене 2,53 А, практически не изменяется [53, 54, 128, 129].
Табл ица 3
Влияние разветвленности на размеры элементарных ячеек [54]
Число СНз-групп на 1000 атомов С Размеры, А Плотность кристаллической части полимера (теоретическая), г/см3
а ь
0 7,36 4,92 1,014
1 7,38 4,95 1,005
3 7,43 4,95 0,999
10 7,52 4,96 0,985
30 7,54 4,97 0,980
40 7,55 4,97 0,979
80 7,68 5,00 0,956
Увеличение объема элементарной ячейки при нагревании полиэтилена сопровождается ростом главным образом параметра а, достигающего при 100 °C величины 7,65 А [48, с. 57].
13
Разветвленность, характеризующаяся содержанием 40—80 СН3-групп на 1000 атомов углерода, приводит к такому же изменению объема ячейки, как повышение температуры неразветвлен-ного полиэтилена до 100°C.
Полипропилен
Полипропилен получают в процессе ионной полимеризации с использованием комплексных катализаторов, образующихся при взаимодействии соединений переходных металлов IV—VIII групп в состоянии низшей валентности (например, хлоридов двух- или трехвалентного титана, треххлористого ванадия) с алкилами, алкилгалогенидами или гидридами металлов (триэтилалюминием, диэтилалюминийхлоридом, гидридом лития и др.).
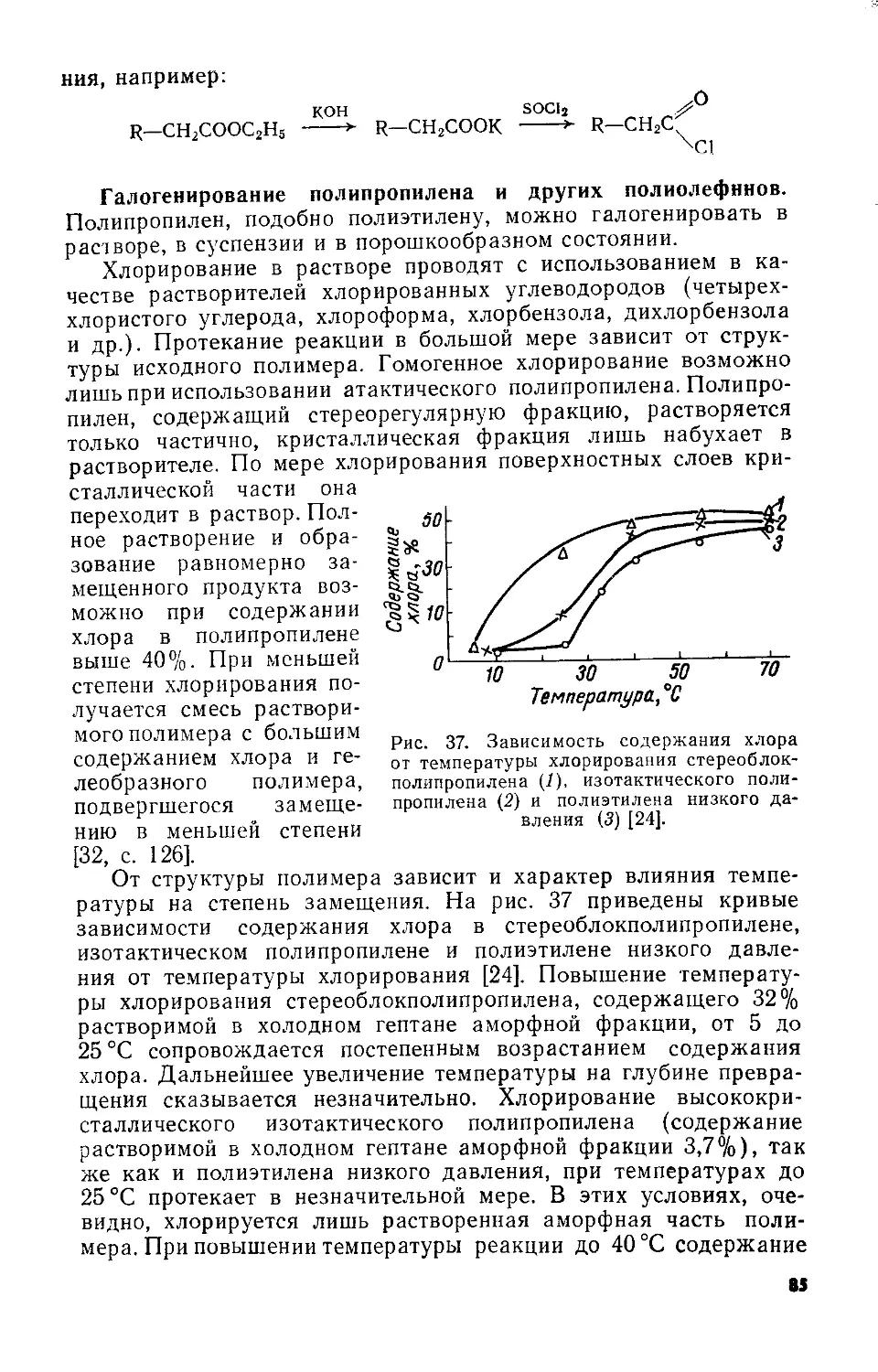
Важная особенность катализаторов полимеризации пропилена заключается в строении их кристаллической поверхности, способствующем определенной ориентации адсорбированных молекул мономера перед их присоединением к растущей цепи полимера [55—58; 59, с. 411]. Образующийся полимер может быть разделен на две фракции, различающиеся по растворимости в горячих углеводородных растворителях (например, в гептане). Нерастворимая кристаллическая фракция и растворимая аморфная фракция отличаются структурой макромолекул. Молекулы, составляющие кристаллическую часть полипропилена, имеют регулярное пространственное расположение боковых метильных групп по отношению к главной цепи. Метильные группы в макроцепях аморфной фракции, напротив, расположены хаотически. По терминологии, предложенной Натта, полимер с упорядоченным пространственным расположением звеньев называют стереорегулярным, в отличие от полимера с беспорядочным пространственным расположением звеньев, называемого атактическим.
Соотношение между количествами атактической и стерео-регулярной части полипропилена, оказывающее большое влияние на его свойства, определяется характером катализатора и условиями полимеризации пропилена. Высокой стереоспецифичностью, позволяющей получать почти полностью стереорегулярный полипропилен, обладают катализаторы на основе треххлористого титана и алкилгалогенида алюминия в присутствии электронодонорных добавок (например, пиридина) [60]. Увеличение стереорегулярности наблюдалось при добавлении кислорода к каталитической системе А1(С2Н5)3— TiCl3 [116].
Повышению содержания стереорегулярной фракции способствует также снижение температуры полимеризации.
Различают два вида стереорегулярного полипропилена: изотактический и синдиотактический. Для изотактического полипропилена характерно расположение метильных групп по одну сторону от плоскости зигзагообразной парафиновой цепи, для 14
синдиотактического — строго поочередное расположение боковых групп по одну и другую сторону этой плоскости.
Стереорегулярный полипропилен обладает значительной степенью кристалличности. Для изотактического полипропилена эта величина, составляющая 60—70% [61], в значительной мере зависит от содержания атактической фракции. Внутри кристаллических областей каждая макромолекула имеет форму спирали [62], в которой угол между осями заместителей соседних мономерных звеньев равен 120°. Период идентичности при этом включает в себя три мономерных звена и имеет величину 6,5 А.
Представляет интерес полипропилен, в макромолекулах которого чередуются участки изотактического строения с противоположным пространственным расположением метильных заместителей (стереоблокполимеры) [63, с. 78]. В зависимости от длины однотипных участков такой материал в большей или меньшей мере проявляет свойства эластомера. При достаточной длине таких участков происходит частичная кристаллизация. Стереоблочный полипропилен, оставаясь эластомером, вместе с тем проявляет некоторые свойства вулканизованных эластомеров. Кристаллические участки, препятствуя ползучести материала, оказывают влияние, подобное влиянию поперечных связей, образующихся при вулканизации.
Свойства полипропилена, как и полиэтилена, в значительной степени зависят от кристалличности и молекулярного веса.
Кристалличность определяется в первую очередь степенью тактичности, так как возможность плотной упаковки цепей с образованием кристаллических областей в стереорегулярном полимере значительно выше, чем в атактическом. Кристалличность в известной мере зависит также от молекулярного веса: при одинаковой степени изотактичности полипропилен с меньшим молекулярным весом может быть более кристалличным, чем полимер с большим молекулярным весом. Быстрое охлаждение расплава полипропилена с высокой степенью изотактичности может привести к получению материала с пониженной кристалличностью.
От кристалличности полипропилена зависят все его физические свойства. Варьируя степень изотактичности, можно получать гамму материалов от высококрпсталлических жестких, прочных и стойких к действию тепла до аморфных эластичных с невысокой теплостойкостью.
Прочность и способность к деформации в значительной мере определяются также размерами сферолитных образований в полипропилене, которые могут изменяться в значительном интервале в зависимости от условий формирования при сохранении практически постоянной степени кристалличности [141 — 143]. При изучении связи прочностных характеристик и размеров сферолитов в пленках изотактического полипропилена было показано [143], что прочность и относительное удлинение при разрыве уменьшаются с увеличением размеров сферолитов.
Ч
При достаточно больших размерах сферолитов в определенных условиях деформации полипропилен может подвергаться хрупкому разрушению. Разрушение может проходить по границам между сферолитами и по собственно сферолитам. Невысокие значения прочности и деформируемости полипропилена со сравнительно крупными сферолитами связывают с увеличением размеров дефектов в материале по мере возрастания размеров сферолитных образований.
Свойства различных типов полипропилена описаны в работах [63; 64, с. 95]. Отметим лишь, что для полипропилена характерно сочетание большой прочности, жесткости, высокой температуры размягчения с низкой плотностью:
Плотность, г/см3.............................. 0,90—0,91
Температура плавления, °C...................... 167—170
Температура хрупкости, °C.................. От —10 до +20
Прочность при растяжении, кгс/см2 .... 300—400
Относительное удлинение при разрыве, % не более.................................... 1 000
Модуль упругости при изгибе, кгс/см2 . . . 8 000—12 000
Теплостойкость по Вика, °C............... 145—155
Водопоглощение, %........................ 0,01—0,03
Диэлектрическая проницаемость при 5 • 107 Гц 2,2
Удельное объемное электрическое сопротивление, Ом -см........................... 3-10ls—8-Ю13
Существенные недостатки полипропилена — хрупкость при невысоких температурах и ограниченная стойкость к окисляющим воздействиям, теплу и ультрафиолетовому свету.
Атактический полипропилен может быть получен и в процессе свободнорадикальной полимеризации. Например, описана полимеризация пропилена с использованием в качестве инициатора перекиси бензоила при температуре от 80 до 100 °C и давлении от 2800 до 4000 кгс/см2 [127]. Был получен полипропилен невысокого молекулярного веса, атактического строения, хорошо растворимый в бензоле и циклогексане.
Полимеры высших а-олефинов
Металлорганические комплексные катализаторы полимеризации этилена и пропилена используются также для получения полимеров высших а-олефинов, в частности полибутена-1, поли-З-метилбутена-1 и поли-4-метилпентена-1. Эти полиолефины характеризуются значительной степенью кристалличности, зависящей от условий синтеза. Макромолекулы, входящие в состав кристаллических участков, имеют спиралевидную структуру.
В макромолекулах изотактического полибутена-1, так же как и в молекулах полипропилена, каждый виток спирали (период идентичности 6,50 А [65]) составляют три мономерных звена. По мере увеличения размеров заместителей период идентичности возрастает и конфигурация спиралей становится более 16
сложной. Период идентичности в спиральной структуре изотактического поли-З-метилбутена-1 равен 6,84 А, поли-4-метилпен-тена-1 13,85 А [66].
Изотактический полибутен-1 по свойствам близок к полиэтилену низкого давления [67—69, 118—120]:
Плотность, г/см3..........................0,912
Показатель текучести расплава, г/Ю мии 0,5
Температура плавления, °C..................135
Температура хрупкости, °C................—25
Прочность при растяжении, кгс/см2 .... 270
Предел текучести, кгс/см2..................155
Относительное удлинение при разрыве, % 350
Модуль упругости, кгс/см2 ............... 18 200
Теплостойкость по Вика, °C.................113
Диэлектрическая проницаемость при 10б Гц 2,2
Рис. 1. Зависимость температуры плавления поли-а-олефинов от длины боковых цепей [70].
Важнейшее отличительное свойство полибутена-1 заключается в весьма высокой сопротивляемости крипу в температурном интервале от —25 (температура хрупкости) до +90 °C. Полибутен-1 характеризуется также высокой стойкостью к растрескиванию под действием внутренних напряжений, внешних нагрузок и поверхностно-активных веществ.
X. Полипентен-1 обладает ограничен-
—>'нрй теплостойкостью, температура плавления изотактического полимера Xfc0°C [55].
Полимеризация следующих членов , (уомологического ряда а-олефинов •^^(гексена-! [139] и т. д.) приводит к получению каучукоподобных продук-ч^\тов.
J Зависимость температуры плавления от длины боковых цепей в макромолекулах изотактических продуктов полимеризации а-олефинов представлена на рис. 1. Переход от полипропилена к полимерам высших а-олефинов, вплоть до С7—С9, сопровождается
понижением температуры плавления из-за возрастающего несовершенства спиральной упаковки цепей [65, 70—72]. Повышение температуры плавления в ряду от полидецена-1 до полиоктадецена-1 является, очевидно, следствием совершенствования упаковки боковых цепей по мере увеличения их длины [70].
Полимеры с высокой теплостойкостью получают полимеризацией мономеров, образующих макромолекулы с разветвленными боковыми цепями, З-метилбутена-1 и 4-метилпентена-1. Температура плавления кристаллического поли-З-метилбутена-1 составляет 243 °C [66] (по другим данным [73, 121] — около
17
310°С), температура плавления кристаллического поли-4-метил-пентена-1 205°C [66] (по данным [73] около 240°C).
Поли-4-метилпентен-1 получается полимеризацией 4-метил-пентена-1 в присутствии металлорганических катализаторов, например комплексов, образованных алкилами или алкилгалоге-нидами алюминия и галогенидами титана или ванадия, в условиях, близких к используемым при получении полиэтилена или полипропилена. На примере каталитической системы А1(С2Н5)з—TiCls показано положительное влияние на процесс полимеризации третичных аминов [74]. Известна полимеризация 4-метилпентена-1 в присутствии окисла металла, способного к образованию галогенида, и органического соединения, содержащего не менее двух атомов галогена у одного углеродного атома, например смеси окиси алюминия и четыреххлористого углерода [75]. В качестве каталитической системы используется также смесь галогенидов металлов IV—VI групп и кремиийор-ганического соединения, например смеси трех- и четыреххлористого титана с фенилсилоксаном [76].
Поли-4-метилпентен-1 характеризуется специфическим ценным комплексом свойств [77—80, 97, 98, 140, 151, 152]. Ниже приведены свойства материала, полученного с применением последней из перечисленных каталитических систем [77]:
Плотность, г/см3................................. 0,83
Температура плавления, °C....................... 240
Прочность прн растяжении, кгс/см2 при 20 °C ..................................... 280
при 100 °C..................................... 70
Относительное удлинение, %........................ 50
Модуль упругости при растяжении (20 °C), кгс/см2 11000
Теплостойкость по Вика, °C........................ 179
Коэффициент линейного расширения, 1/°С...........11,5- 10~5
Тангенс угла диэлектрических потерь при 103, Гц 0,00015
Диэлектрическаи проницаемость при 25 °C......... 2,12
Удельное объемное электрическое сопротивление, Ом «см, не менее................................ 101в
Поли-4-метилпентен-1—самый легкий из пластиков, его плотность лишь 0,83 г/см3. Он почти так же прозрачен, как полиметилметакрилат,— пропускает около 90% света. Изделия из поли-4-метилпентена-1 сохраняют форму при нагревании до 200°C. Газо- и водопроницаемость поли-4-метплпентена-1 несколько выше, чем у других полиолефинов (полиэтилена, полипропилена).
Реологические свойства поли-4-метилпентена-1 в интервале температур 250—290°C позволяют перерабатывать его в изделия обычными для термопластов методами [124, 125].
Полимеры оптически активных высших а-олефинов (3-ме-тилпентена-1, 4-метилгексена-1, 4-метилгептена-1, 5-метилгеп-тена-1, 6-метилоктена-1 и др.) также обладают оптической активностью [81—85,117].
1»
Литература
1. Europ. Chem. News, 1970, v. 18, № 11, p. 8. — 2. Белов Г. П., Карпова Н. Д. Пласт, массы, 1967, X» 5, с. 8. — 3. Англ. пат. 966143, 1964.—• 4. Raff R. А. V., А 11 i s о n J. В. Polyethylene. N. Y. — L., Interscience, 1956. 551 р. — 5. Polyethylene. Ed. A. Renfrew, Р. Morgan. N. Y. — L., Interscience, 1960. — 6. Шифрина В. С., Самосатский Н. Н. Полиэтилен. Л., Гос-химиздат, 1961. 176 с. — 7. Hagen Н., Dornin inha us Н. Polyathylene und Andere Polyolefine. Hamburg, Verlag Brunke Garreis, 1961. — 8. Голдинг Б. Химия и технология полимерных материалов. М., ИЛ, 1963. 666 с. — 9. Николаев А. Ф. Синтетические полимеры и пластические массы на нх основе. Л., «Химия», 1967. 784 с. — 10. X у в и н к Р., С т а в е р м а н А. Химия и технология полимеров. Т. 11, ч. I. Л., «Химия», 1965. 508 с.
П.Хаитер Е. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Плата. М., «Мир», 1964. 595 с. — 12. Danby С. J., Hishelwood С. N. Proc. Roy. Soc., 1942, v. 179, Ser. A, p. 169.— 13. Landers L. C., Vo 1 m a n D. H. J. Am. Chem. Soc., 1957, v. 79, p. 2996.— 14. Roedel M. J. J. Am. Chem. Soc., 1953, v. 75, Ns 24, p. 6110.— 15. Willbourn A. H. J. Polymer Sci., 1959, v. 34, p. 569.— 16. Сирота А. Г., Рябиков E. П., Гольденберг А. Л. и др. Пласт, массы, 1965, Ns 11, с. 5.— 17. Гольденберг А. Л., Заплетняк В. М., Ильченко П. А. В ки.: Спектроскопия полимеров. Киев, «Наукова думка», 1967. — 18. В а 1-lantine D. S., Ma nowitz В. Nucl. Sci. Abs., 1953, v. 7, p. 3730.— 19. Lewis J. G., Martin J. J., Anderson L. C. Chem. Eng. Progr., 1954, v. 50, p. 249. — 20. H a у w о r d J. С,, В r e 11 о n R. H. Chem. Eng. Progr., 1954, v. 50, p. 73.
21. Laird R. K-, Morell A. G., Seed L. Disc. Faraday Soc., 1956, v. 22, p. 126. — 22. Бугаенко Л. T., Никитина T. С., Праведников A. H., Малинский Ю. M. Химическое действие ионизирующих излучений. М., ВИНИТИ, 1958. — 23. Munari S., Gastello G., Russo S„ Rossi C. Chim. e ind., Milan, 1965, v. 47, Ns 1, p. 20. — 24. Natta G. Expe-rienta Suppl., 1957, v. 7, p. 21. — 25. Julia M. Compt. rend., 1957, v. 245, p. 70. — 26. Боун. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ. М., «Мир», 1964. — 27. Полиэтилен низкого давления. Под ред. Н. М. Егорова, Л„ Госхимиздат, 1960. 96 с.—28. Бельг, пат. 533362, 1955. — 29. Burch G. N., Field G. В., М с Т i q u е Е. Н„ Spurlin Н. М. SPE J., 1957, V. 13, Ns 5, р. 34. — 30. Пат. ФРГ 1182827, 1964.
31. Пат. ФРГ 1183245, 1964.— 32. Бельг, пат. 530617, 1955.—33. Бельг, пат. 535082, 1955. — 34. Полиэтилен среднего давления. Под ред. С. В. Шуц-кого. Л., «Химия», 1965. 90 с. — 35. Пат. США 2658059, 1953, —36. Пат. США 2692261, 1954, —37. Пат. США 2692295, 1954.— 38. Англ. пат. 721046, 1954,— 39. Пат. США 2717888, 1955.—40. Пат. США 2717889, 1955.
41. Кларк, Хоган. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ. М., «Мир», 1964. — 42. Дувиль. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ, М., «Мир», 1964. — 43. Clark A., Hogan Р. Ind. Eng. Chem., 1956, v. 48, № 7, p. 1152. — 44. Архипова 3. В., Семенова А. С., Жба н ко-в а М. Г. и др. Пласт, массы, 1959, Ns 1, с. 17.—45. Ермаков Ю. И., Боресков Г. К., Дзисько В. А., Иванова Л. И. ДАН СССР, 1962, т. 145, No 4, с. 787. — 46. Гольденберг А. Л. Любецкий С. Г. Высо-комол. соед., 1963, т. 5, № 6, с. 905. — 47. Smith D. С. Ind. Eng. Chem. 1956, v. 48, Ns 7, p. 1161. — 48. Гейлорд H., Марк Г. Линейные и сте-реорегулярные полимеры. ИЛ, 1962. 565 с. — 49. Рекашева А. Ф., К и п-рнанова А. А. Высокомол. соед., 1961, т. 3, с. 1446. — 50. Curphey Е. С. Brit. Plast., 1958, v. 31, р. 63.
19
51. Richards R. В. J. Appl. Chem., 1951, v. 1, p. 370. — 52. Банн. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ. М., «Мир», 1964. — 53. Bunn С. W. Trans. Faraday Soc., 1939, v. 35, р. 482. — 54. Walter Е. R., Reding F. P. J. Polymer Sci., 1956, v. 21, № 99, p. 561. — 55. Natta G. Makromol. Chem., 1955, v. 16, p. 213.— 56. Natta G., Pino P., Mora gl io P. J. Am. Chem. Soc., 1955, v. 77, p. 1708. — 57. Natta G. J. Polymer Sci., 1956, v. 16, p. 143. — 58. Natta G., Mazzanti G., Grespi G., Moraglio G. Chim. e Ind., Milan, 1957, v. 39, № 4, p. 275; пер.: Химия и технология полимеров, 1957, № 6, с. 94.— 59. Бреслер С. Е., Е р у с а л и м с к и й Б. Л. Физика и химия макромолекул. «Наука», 1965. — 60. Natta G., Р a s q и о n J., Gatti Y. J. Polymer
Sci., 1961, v. 51, p. 387.
61. Cop pel J. M. Brit. Plast., 1959, v. 32, Ks 5, p. 207. — 62. Natta G„
Corradini P. J. Polymer Sci., 1959, v. 34, p. 529. — 63. К p e с с e p T.,
Полипропилен. ИЛ, 1963. 231 с. — 64. Ровнер И. Полипропилен. Под ред. В. И. Пилиповского и И. К. Ярцева. Л., «Химия», 1967. — 65. Natta G., Corradini Р. Bassi J. W. Macromol. Chem., 1956, v. 21, p. 240.— 66. Natta G. Angew. Chem., 1955, Bd. 68, S. 393. — 67. Hinds L. Rubber J. a Internet. Plast., 1960, v. 138, № 11, p. 382. — 68. Pract. Plast., 1960, v. 11, № 6, p. 29. — 69. Chem. Eng. News, 1960, v. 38, № 1, p. 24. — 70. Turner J. A. Makromol. Chem., 1964, v. 71, p. 1; пер.: Экспресс-информация. Серия «Синтетические высокомолекулярные материалы», 1964, № 22, с. 309.
71. Natta G., Corradini Р. Makromol. Chem., 1955, v. 16, 213.— 72. Natta G., Corradini P., Cesari M. Atti Accad. Line., 1956, Bd. 21, S. 365. — 73. Campbell T. W., Haven A. C. J. Appl. Polymer Sci., 1959, v. 1, p. 79. — 74. Англ. пат. 886093, 1962. — 75. Бельг, пат. 632378. — 76. Аигл. пат. 934119, 1963.—77. Beduneau Н. Rev. prod, chim., 1966, v. 69, № 1341, p. 9, пер.: Экспресс-информация, серия «Синтетические высокополимерные материалы», 1966, № 12, с. 145. — 78. Rubber Plast. Age, 1965, v. 46, № 4, p. 415. — 79. Rubber Plast. Age, 1965, v. 46, № 5, p. 499. — 80. Brit. Plast., 1965, v. 38, № 4, p. 213.
81. Pino P., Lorenzi G. P., Lardicci L. J. Polymer Set., 1961, v. 53, p. 340. — 82. Bayley W. J., Yates E. T. J. Org. Chem., 1960, v. 25, p. 1800. — 83. Nozakura S., Takeuchi S., Yuki H., Murahashi S. Bull. Chem. Soc. Japan, 1961, v. 34, p. 1673. — 84. Goodman M., Clarke K. G., Stake M. A., Abe A. Macromol. Chem., 1964, v. 72, p. 131.— 85.- Goodman M., Brandrup J., Mark H. F. High Polymers, v. 20, p. 1. Crystalline olefin polymers. Interscience, N. Y., 1965; пер.: Кристаллические полиолефины. T. I, Под ред. Б. Э. Давыдова, М., «Химия», 1970, с. 82. — 86. Murat а К., Kobayashi S. Kogyo Kagaku Zasshi, 1969, v. 72, № 12, p. 2517. — 87. Тумаркин H. Я., Ерусалимский Б. Л., Литвинова M. A. ДАН СССР, 1969, т. 184, № 3, с. 654; Тумаркин Н. Я. Автореф. каид. дисс. Л., 1968. — 88. Медведев С. С., Абкин А. Д., Хомиков-с к и й П. М. и др. Высокомол. соед., 1960, т. 2, с. 904. — 89. Иванов В. С. Радиационная полимеризация. Л., «Химия», 1967. 232 с. — 90. Пат. США 3533976, 1970.
91. Chapiro A., Radiation Chemistry of Polymeric Sistems. N. Y. — L., 1962. — 92. Бреге p A. X., Вайнштейн Б. И., Сыр кус Н. П. и др. Основы радиационно-химического аппаратостроения. М„ Атомиздат, 1967.— 93. П а с к в и т о в с к а я Е. С., Блинов Г. В., Гусев В. И. и др. Пласт, массы, 1972, № 12, с. 14. — 94. Б у нет р а Н. Усиление эластомеров. Под ред. Дж. Крауса. М., «Химия», 1968. 483 с. — 95. Джей л П. Разрушение твердых полимеров. Под ред. Б. Роузена. М., «Химия», 1971. 551 с.— 96. Патрикеев Г. А. «Механика полимеров», 1971, № 2, с. 221. — 97. Б у-ният-Заде А. А., Мамедов Э. Л., Авакян Л. Г. и др. Пласт, массы, 1972, № 3, с. 12. — 98. Хо У илем, Нечитайло Н. А., Гольд-
20
фар б Ю. Я. и др. Пласт, массы, 1972, № 3, с. 57.—99. Aggarwal S. L., Tilley G. Р. J. Polymer Sci., 1955, v. 18, p. 17.— 100. Англ. пат. 932231, 1963.
101. Пат. ГДР 26992, 1964. — 102. Lo Vullo A., Mon tan do G. Garz. Chim. Ital., 1964, v. 94, № 10, p. 1043. — 103. Hines R. A., Bryant W. M. D., Larchard A. W., Pease D. C. Ind. Eng. Chem., 1957, v. 49, № 7, p. 1071.— 104. Boyle D. A., Simpson W., Waldron J. D. Polymer, 1961, v. 2, № 3, p. 323. — 105. Boyle D. A., Simpson W., Waldron J. D. Polymer, 1961, v. 2, № 3, p. 335. — 106. Clark A. Addition and condensation Polymerization Process. Washington, 1969. — 107. Бадаев В. К., Архипова 3. В., Ерофеев Б. В. и др. Тезисы докладов Симпозиума по исследованию комплексных металлорганических катализаторов полимеризации. Л., 1970. — 108. Коновалов В. П., Махинько А. И. Физико-химия нефти и нефтехимический синтез. Алма-Ата, «Наука», 1970. — 109. Чирков Н. М. «Кинетика и катализ», 1970, т. II, № 2, с. 321.— ПО. Waichiro Kawakami, Takeshi Wada, Terutaka Watanabe, Sueo Ma chi, Tsutomu К a giy a. J. Appl. Polymer Sci., 1971, v. 15, № 6, p. 1507.
111. Hiroshi Mitsui, Fumio H о s о i, Tsutomu К a g i у a. J. Polymer Sci., 1969, v. 7, Ser. Al, № 9, p. 2575. — 112. Hiroshi Mitsui, Fumio Hosoi, Tsutomu Kagiya. J. Polymer Sci., 1970, v. 8, Ser. Al, № 2, p. 451. — 113. H a g i w a r a M., Okamoto H., Kagiya T. Bull. Chem. Soc. Japan, 1970, v. 43, Ns 1, p. 172.— 114. Смирнов, Гусев. Химия и технология полимеров, 1967, № 3, с. 16.— 115. Андреева И. Н., Варфоломеева Л. С., Заплетняк В. М. и др. Пласт, массы, 1970, № 5, с. 23. —116. Doi Y., Hattori Y., Oku г a J., Keii T., Kogyo Kagaku Zasshi. 1969, v. 72, № 12, p. 2621.— 117. Bacskai R. J. Polymer Sci., 1967, v. 5, Ser. Al, № 3, p. 619. — 118. Haas T. W., MacRae P. H. Polymer Eng. Sci., 1969, v. 9, Ns 6, p. 423. — 119. Haas T. W., Maxwell B. Polymer Eng. Sci., 1969, v. 9, № 6, p. 225.— 120. Yee R. Y., Stein R. S. J. Polymer Sci., 1970, v. 8, Ser. A2, № 10, p. 1661.
121. Quynn R. G., Sprague B. S. J. Polymer Sci., 1970, v. 8, Ser. A2, № 11, p. 1971,— 122. Sancho J. Rev. Plast, 1957, № 47, p. 273.— 123. Hosoi M., Kawai T., Kuriyama I. Polymer Report, 1970, № 143, p. 25.— 124. Авакяи Л. Г. Автореф. дисс. Баку, 1972. —125. Andreas F., Baier R. Plaste u. Kaut., 1969, Bd. 16, № 8, S. 561—568.— 126. Карасев A. H., Андреева И. H., До марева Н. М. и др. Высокомол. соед., 1970, т. 12, сер. А, № 5, с. 1127. — 127. Osugi J., На manoue К-, Tachi-bana Т. Nippon Kagaku Zasshi, 1969, v. 90, № 6, p. 549.— 128. Swan P. R. J. Polymer Sci., 1962, v. 56, p. 409. — 129. Swan P. R. J. Polymer Sci., 1962, v. 56, p. 439. — 130. Попов В. П., Бальтенас Р. А. «Физико-химическая механика материалов», 1971, т. 7, № 6, с. 41.
131. Ерофеев Б. В., Архипова 3. В., Бадаев В. К- и др. Becui АН БССР. Сер. хим. наук, 1971, № 3, с. 5. —132. Ehrlich Р., Mortimer G. A. Adv. in Polymer Sci., 1970, v. 7, № 3, p. 386.— 133. Des С 1 о i-zeaux J. J. Polymer Sci., 1970, v. 8, Ser. A2, Ns 10, p. 1773. — 134. R a-mesh N. Shroff, Mitsuzo Shida. J. Polymer Sci., 1970, v. 8, Ser. A2, № 11, p. 1917.— 135. Pandurang M., Kamath, Barlow A. J. Polymer Sci., 1967, v. 5, Ser. Al, № 8, p. 2023, — 136. С о t e J. A., Sci da M. J. Polymer Sci., 1971, v. 9, Ser. A2, Ns 3, p. 421.— 137. Otocka E. P., Roe R. J., Hellman M. Y., Mu glia P. M. Am. Chem. Soc. Polym. Prepr., 1971, v. 12, № 1, p. 274,— 138. Drott E. E„ Mendelson R. A. Am. Chem. Soc. Polym. Prepr., 1971, v. 12, Ns 1, p. 277.— 139. Tu C. F., Biesenberger J. A., Stivala S. S. Macromolecules, 1970, v. 3, № 2, p. 206.— 140. C amp-fa e 11 T. W. J. Appl. Polymer Sci., 1961, v. 5, Ns 14, p. 184.
141. Гуль В. E., Коврига В. В., Роговая Э. М., Громова Н. П. Высокомол. соед., 1964, т. 6, с. 1868. — 142. Гуль В. Е., Коврига В. В., Вассерман А. М. ДАН СССР, 1962, т. 146, с. 656.— 143. Гуль В. Е.,
21
Коврига В. В., Роговая Э. М., Громова Н. П. Изв. вузов. Химия и ХИм. технол., 1966, т. 9, № 3, с. 86. — 144. Каргин В. А., Слонимский Г. Л. Краткие очерки по физико-химии полимеров. М., «Химия», 1967. — 145. Кнебельман А. М„ Кантор Л. А., К а г а и Д. Ф. Высоко-мол. соед., 1970, т. 12, сер. А, № 12, с. 2746. — 146. Гуль В. Е. Структура и прочность полимеров. М„ «Химия», 1971. 344 с.— 147. Isaksen R. А„ Hewman S., Clark R. J. J. Appl. Polymer Sci., 1963, v. 7, p. 515.— 148. Howard J. В. В кн.: Конструкционные свойства пластмасс. Под ред. Э. Бэра. М., «Химия», 1967. —149. Веселовская Е. В., Левина А. А., Наливайко Е. И., Пукшанский М. Д. Пласт, массы, 1966, № 6, с. 43. — 150. Наливайко Е. И., Сирота А. Г. Пласт, массы, 1968, № 2, с. 13.
151. Yoshimichi Hase, Geil Р. Н. Polymer J„ Soc. Polymer Sci., Japan, 1971, v. 2, № 5, p. 550.— 152. Yoshimichi Hase, Geil P. H. Polymer. J., Soc. Polymer Sci., Japan, 1971, v. 2, № 5, p. 581.— 153. Kobayashi S. Kogyo Kagaku Zasshi, 1969, v. 72, № 12, p. 2511. — 154. Oto-c k a E. P., Roe R. J., Hell m a п M. Y., M u g 1 i a P. M. Macromolecules, 1971, v. 4, № 4, p. 507.— 155. Ок а мото Хироси, Кобунси Кагаку, 1971, т. 28, № 310, с. 97.
Глава II
Сополимеры а-олефинов
Сополимеры этилена с пропиленом и другими «-олефинами
Введение в макромолекулы полиэтилена звеньев, содержащих ответвления от основной полимерной цепи, путем сополимеризации этилена с пропиленом, «-бутиленом или другими а-олефинами — один из основных методов модифицирования структуры и свойств полиэтилена.
Разветвленность сополимеров обусловливает их меньшую по сравнению с полиэтиленом упорядоченность. Естественно, что поэтому сополимеризацию с а-олефинами используют для модифицирования полиэтилена с высокоупорядоченной структурой (полиэтилена низкого и среднего давления), хотя известна возможность сополимеризации этилена, например, с пропиленом, а-бутиленом, изобутиленом и по свободнорадикальному механизму при высоком давлении [180—183] (табл. 4).
Таблица 4
Константы свободнорадикальной сополимеризации этилена (г.) с различными мономерами ]г2]
Сомономер ri Г2 Давление, кгс/см2 Температура, °C Литература
Пропилеи . . а-Бутилен . . Изобутилен 3.1 ±0.2 3.4 ±0,3 2,6±0,2 0,77±0,55 0,86 ±0,02 0.56 ±0,01 1020-1700 1020-1700 1360 130-220 130—220 130—220 [181, 182] [181-182] [182]
Регулируя содержание сомономера в сополимере с этиленом, удается получать широкую гамму материалов, отличающихся в первую очередь по свойствам, связанным с упорядоченностью структуры. Сополимеры этилена с oc-олефинами представляют интерес и как модельные соединения [2, 3] для исследования связи структуры макромолекул и надмолекулярных образований. Модельный характер сополимерам придает практически полная однотипность ответвлений, число которых поддается регулированию и определению.
Основное следствие введения звеньев сомономеров в макромолекулы полиэтилена заключается в его аморфизации. По мере
23
увеличения в сополимере числа звеньев сомономеров, содержащих узлы ответвлений, кристалличность уменьшается. В каче стве примера на рис. 2 приведена зависимость степени Кристал-личности полученных на окиснохромовом катализаторе сополи-
меров этилена с пропиленом от групп [4].
содержания метильных боковых
0123^5678 Число СН3-групп на 100 атомов С
Рис. 2. Зависимость кристалличности сополимеров этилена с пропиленом от числа метильных боковых групп [4].
Содержание звеньев пропилена, мол. %
Рис. 3. Зависимость размеров элементарных кристаллических ячеек сополимеров этилена с пропиленом от содержания звеньев пропилена [5].
Кривые зависимости интенсивности рассеяния рентгеновых лучей от угла рассеяния, используемые для оценки степени кристалличности, позволяют обнаруживать с ростом числа ответвлений, кроме уменьшения кристалличности сополимеров, также смещение дифракционной картины. Это смещение свидетельствует об увеличении межплоскостных расстояний [4]. Увеличение размеров элементарных кристаллических ячеек происходит в основном вдоль оси айв меньшей степени вдоль оси b (рис. 3, табл. 5), размер вдоль оси с практически не изменяется.
Таблица 5
Размеры элементарных ячеек сополимеров этилена с а-опефинами [5]
Сомономер Содержание сомо номера, мол. % Размеры, А
а ь
Бутеи-1 3,6 7,521 4,964
Пентен-1 3,1 7,474 4,969
Гексеи-1 3,4 7,467 4,965
Гептен-1 1,4 7,479 4,956
Степень кристалличности сополимеров этилена определяется в первую очередь содержанием сомономера. Однако при по
24
стоянном составе сополимера обнаруживается также связь между степенью кристалличности и длиной углеводородной цепи сомономера [3, 6]. На рис. 4 представлена кривая зависимости степени кристалличности сополимеров этилена, полученных при среднем давлении, от числа атомов углерода в молекуле а-олефина. С увеличением длины цепи сомономера и соответственно длины ответвлений от макроцепи эффект снижения
кристалличности усиливается и достигает максимума при использовании в качестве сомономеров а-олефинов Ci—Св. Дальнейший рост длины цепи а-олефинов не вызывает значительного уменьшения степени кристалличности и даже приводит к некоторому снижению эффекта нарушения кристаллической структуры. По-видимому, это объясняется тем, что длина и гибкость боковых ответвлений, содержащих 4—5 и более атомов углерода, оказывается достаточной для их ориентации вдоль основной цепи. Такие достаточно длинные ответвле-
Рис. 4. Зависимость степени кристалличности сополимеров этилена от длиЯы цепи сомономера (при постоянном содержании звеньев сомономера в сополимере, равном 2,1 ± ±0,4 мол. %). Точка на оси ординат соответствует полиэтилену среднего давления [3].
ния способны размещаться в кристаллических образованиях сополимеров. К такому же выводу пришел Свен [5], изучая изменение размеров элементарных кристаллических ячеек сополимеров этилена с высшими а-олефи-нами (см. табл. 5).
Ассоциации кристаллических образований (сферолиты) в сополимерах менее совершенны и мельче, чем в по-
лиэтилене (рис. 5).
Пониженная степень кристалличности сополимеров по срав
нению с гомополпмерами этилена низкого и среднего давления проявляется в меньшей плотности сополимеров (рис. 6). Варьируя степень кристалличности сополимеров введением большего
или меньшего количества сомономера, можно получать материалы с различной величиной модуля упругости (рис. 7).
Сополимеры отличаются от полиэтилена низкого и среднего давления большей сопротивляемостью растрескиванию под влиянием внутренних напряжений, длительных нагрузок, поверхностно-активных сред. Повышенная сопротивляемость сополимеров растрескиванию согласуется с данными [9] электронно-микроскопического изучения структуры и процессов роста кристаллических образований в полиэтилене, показывающими, что уменьшение надмолекулярной структурной упорядоченности способствует увеличению стойкости к растрескиванию.
U
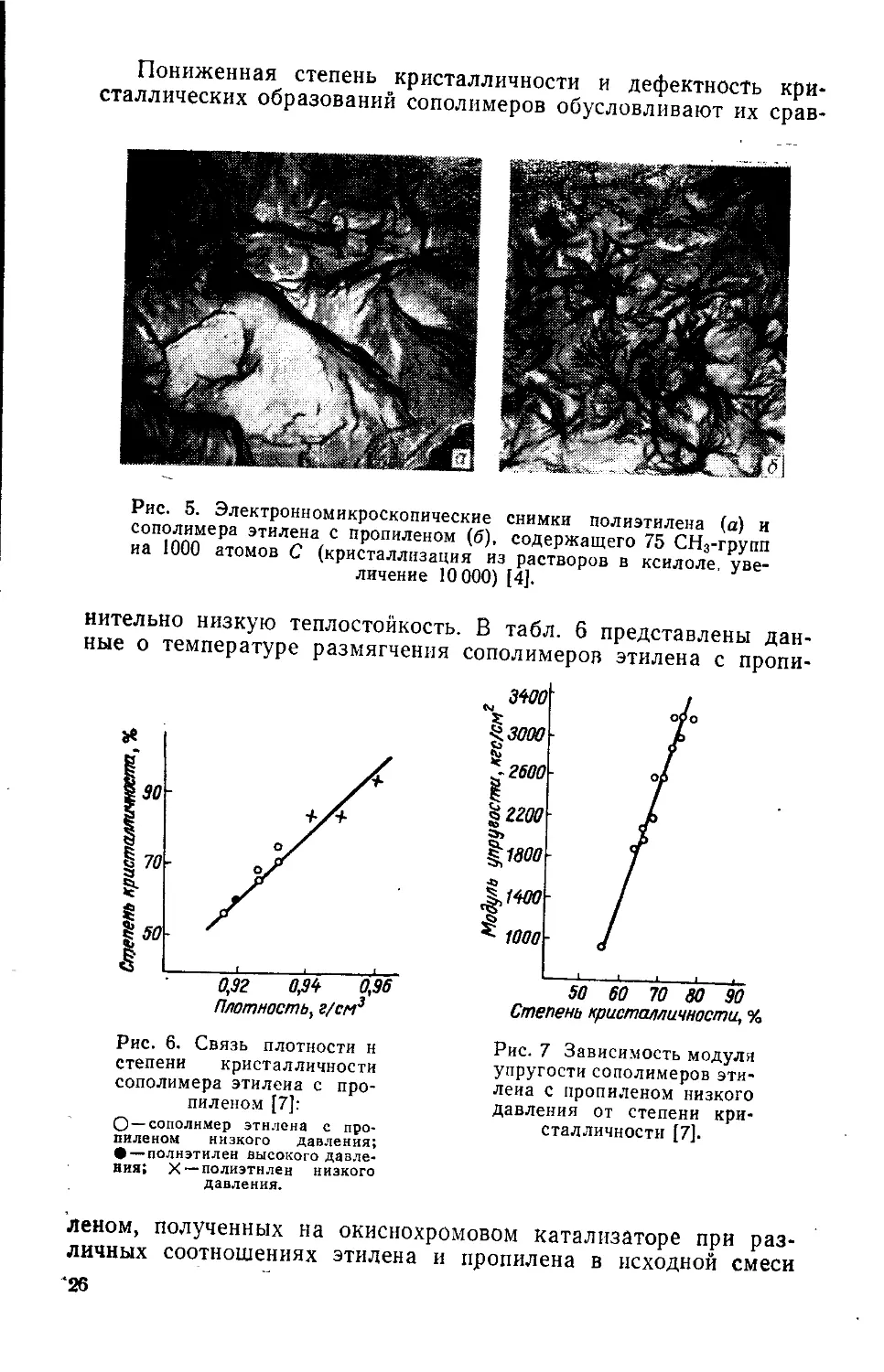
Пониженная степень кристалличности и дефектность кристаллических образований сополимеров обусловливают их срав-
Рис. 5. Электронномикроскопические снимки полиэтилена (а) и сополимера этилена с пропиленом (б), содержащего 75 СН3-групп иа 1000 атомов С (кристаллизация из растворов в ксилоле, увеличение 10 000) 14].
нительно низкую теплостойкость. В табл, б представлены данные о температуре размягчения сополимеров этилена с пропи-
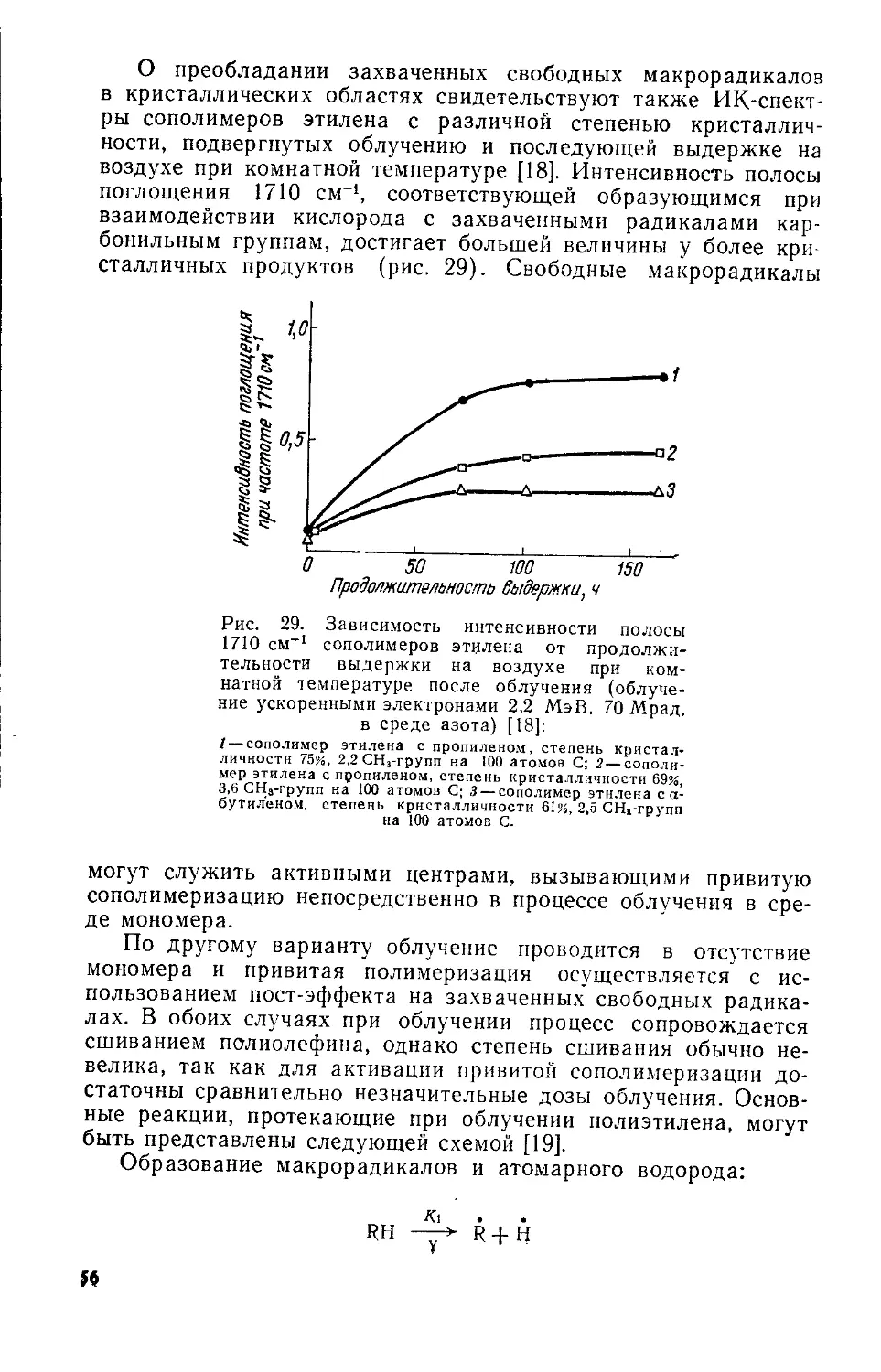
Рис. 7 Зависимость модуля упругости сополимеров этилена с пропиленом низкого давления от степени кристалличности [7].
Рис. 6. Связь плотности н степени кристалличности сополимера этилена с пропиленом [7]:
О—сополимер этилена с пропиленом низкого давления; S—полиэтилен высокого давления; X —полиэтилен низкого давления.
леном, полученных на окиснохромовом катализаторе при различных соотношениях этилена и пропилена в исходной смеси
‘26
Таблица 6
Свойства сополимеров этилена с пропиленом, полученных при различном соотношении мономеров [в]
Весовое соотношение С2/С3 Температура размягчения, °C Плотность, г/см3 Молекулярный вес
100/0 121 0.960 29 500
95/5 117 0,948 27 900
90/Ю 115 0.938 25 200
85/15 114 0,937 30 900
80/20 113 0.932 26 000
мономеров. По мере увеличения содержания пропилена одновременно снижаются температура размягчения и плотность сополимеров.
Эффективная вязкость расплава и энергия активации вязкого течения сополимеров этилена с пропиленом и особенно сополимеров этилена с а-бутиленом меньше этих показателей для полиэтилена (табл. 7) [173, с. 91]. Такое различие связывают, в частности, с пониженными межмолекулярными взаимодействиями в расплавах разветвленных полимеров.
Таблица 7
Свойства расплавов полиэтилена и сополимеров этилена [173]
Полимер Содержание сомономера, мол. % Характеристическая вязкость в декалине, при 135 °C, дл/г Эффективная вязкость трЮ4, П * Энергия активации вязкого течения [174], ккал/моль
при 190 °C прн 270 °C
Полиэтилен низкого давления 1,9 22.40 5,62 9,1
Сополимер этилена с пропиленом 10 2,2 10,72 3,27 7,5
Сополимер этилена с а-бутиленом 4 2,9 8.35 2,75 6.1
* Определено прн напряжении сдвига 105 днн/см2«
Таким образом, основные отличия сополимеров этилена с высшими а-олефинами от полиэтилена низкого и среднего давления являются следствием большей разветвленности макромолекул сополимеров и меньшей упорядоченности надмолекулярной структуры. Сочетание эластичности с высокими диэлектрическими свойствами, химической инертностью, способностью перерабатываться лнтьем под давлением и экструзией
27
делает сополимеры ценными материалами для получения кабельной изоляции, пленок и других изделий, для которых полиэтилен высокой плотности оказывается слишком жестким. Наибольшее использование в технике получили сополимеры этилена с пропиленом и с а-бутиленом [2—5, 10—24].
Сополимеризацию с использованием окиснометаллических катализаторов проводят в условиях, не отличающихся существенно от применяемых при полимеризации этилена. Обычно используют смеси этилена с небольшим количеством сомономера. Так, концентрация пропилена не превышает 20 объемн. % и чаще всего находится в интервале 3—10 объемн. % [25—27]. Это ограничение связано со значительным ухудшением прочностных показателей сополимеров, получаемых при больших концентрациях сомономера в смеси с этиленом.
Сополимеризации подвергают смеси заданного состава в среде углеводородного растворителя (декалин, бензол, гептан, толуол, изооктан). При использовании окиснохромовых катализаторов на носителях, например алюмосиликатных, процесс проводят при давлении 30—40 кгс/см2 и температуре 90—130 °C. Кроме окиснохромовых катализаторов можно применять также окислы и других металлов V и VI групп на носителях, в частности окислы молибдена на окиси алюминия [28—32], окислы ванадия на окиси алюминия [33, 34] и на силикагеле [35—37] и окислы вольфрама на окиси циркония [38—41]. Такие катализаторы активируют водородом при 350—480 °C и используют преимущественно в присутствии промоторов (гидридов натрия и кальция, гидроокиси натрия, металлического натрия, литийалю-минийгидрида, литийборгидрида). Сополимеризацию в этих случаях проводят при 150—240 °C и давлении 35—70 кгс/см2.
Хотя гомополимеризация пропилена и следующих членов ряда а-олефинов протекает с низкой скоростью [например, пропилен— 10 г/(г-ч), гексен-1—8 г/(г-ч)] [22], скорость сополимеризации этилена с этими мономерами высока и близка к скорости гомополимеризации этилена [1000 г/(г-ч)] [22, 24].
Интересный вариант синтеза сополимеров заключается в использовании катализатора (например, хромникельалюмомаг-нийсиликатного [179]), на котором часть этилена димеризуется в а-бутилен с последующей сополимеризацией последнего с этиленом.
Температура, при которой проводится сополимеризация, в большой мере определяет свойства получаемых продуктов. При повышении температуры понижается характеристическая вязкость сополимеров (рис. 8) и ухудшаются прочностные характеристики. Основным способом регулирования структуры и свойств сополимеров при среднем давлении является варьирование состава исходной смеси мономеров. Увеличение содержания сомономера в исходной смеси с этиленом ведет к повышению разветвленности (рис. 9) и к снижению степени кристалличности сополимера.
28
Содержание двойных связей в сополимерах близко к характерному для полиэтилена среднего давления (см. табл. 1). Основным типом ненасыщенных групп в макромолекулах сополимеров являются концевые винильные группы. Ниже показано
Рис. 8. Зависимость характеристической вязкости сополимеров этилена с пропиленом от температуры сополимеризации (содержание пропилена в исходной смеси с этиленом 10 объемн. %, окиснохромо-вый катализатор) [20].
Рис. 9. Зависимость разветвленности сспэлимеров среднего давления от содержания пропилена в смеси с этиленом [20].
содержание различных ненасыщенных групп в сополимере этилена с пропиленом среднего давления (25 СН3-групп на 1000 атомов С) [20]:
Число групп
иа 1000 атомов С
1.0-1,1
Содержание групп, % (от общего содержания групп ^>C=C<Q
вииильных — СН=СН2................. 65
винилиденовых...............\с=СН2 21
транс-вииилеиовых
14
По основным свойствам (плотность, прочность, теплостойкость, модуль упругости, водопоглощение) сополимеры среднего давления [20] занимают промежуточное положение между
29
полиэтиленом высокого давления и полиэтиленом среднего давления (см. табл. 2):
Характеристическая вязкость в декалине при
135 °C, дл/г................................. 1,0-2,5
Число СН3-групп на 1000 атомов С............... 15—30
Степень кристалличности, %........................ 75—80
Плотность, г/см3................................0,94—0,95
Температура плавления, °C........................ 114—125
Температура хрупкости, °C, не выше................. —70
Прочность при растяжении, кгс/см2 ............... 140—220
Относительное удлинение при разрыве, % .... 150—800
Модуль упругости при изгибе, кгс/см2 ........... 3000—6000
Твердость по Бринеллю, кгс/мм2................. 1,3—2,6
Водопоглощение за 30 суток при 20 °C, % . . . . 0,01—0,02
Тангенс угла диэлектрических потерь при 106 Гц, не более ........................................ 4 • 10~4
Диэлектрическая проницаемость при 10s Гц ... 2,3
Удельное объемное электрическое сопротивление, Ом-см.......................................... Ю17
Электрическая прочность (переменный ток) при толщине образцов 2 мм, кВ/мм................... 28—30
Введение одинаковых количеств звеньев пропилена и а-бути-лена в макромолекулярную цепь сополимеров этилена по-разному сказывается на их структуре и свойствах [11]. В соответ-
ствии с приведенными выше данными о влиянии длины от-
S ^7 / 2 j 4 ? 7 <3 Й7 я
* Содержание сомономера в сополимере, мол. %
Рис. 10. Зависимость степени кристалличности от состава сополимеров этилена с а-бутилеиом (У) и пропиленом (2), полученных на окиснохромовом катализаторе [20].
ветвлений на степень кристалличности этильные ответвления вызывают более значительное снижение степени кристалличности, чем метильные (рис. 10).
Сравнение свойств сополимера этилена с а-бу-
тиленом и полиэтилена (табл. 8) показывает, что сополимер весьма значительно превосходит полиэтилен по стойкости к
более рыве.
высокие показатели
Однако сополимер
растрескиванию и имеет относительного удлинения при раз-уступает полиэтилену в величи-
нах прочности при растяжении, жесткости и температуры размягчения. Вместе с тем температура размягчения сополимера достаточно высока для сохранения размеров изделий при стерилизации их водяным паром (при 121 °C в течение 20 мин).
Представляет интерес не только сополимеризация этилена с индивидуальными а-олефинами, но и с их смесями [3, 42]. Например, сополимеризации с этиленом на окиснохромовом ката-
30
Физико-механические свойства сополимера этиленй с а-бутиленом и полиэтилена [19]
Таблица 8
Показатели
Сополимер с плотностью О,§5 г/см3
Полиэтилен с плотностью 0,96 г/см3
Показатель текучести расплава, г/10 мин 0.3 1.2 4,0 6.5 0,2 0,9 3,5 5,0
Прочность при растяжении, кгс/см2 . . . 250 250 250 250 310 310 310 310
Жесткость, кгс/см2 . . 8 050 8 050 8 050 8 050 10 500 10 500 10 500 10 500
Температура размягчения по Вика, °C . . . 124 124 124 124 127 127 127 127
Твердость по Шору (шкала Д) 67 67 67 67 68 68 68 68
Растрескивание под влиянием внешней среды (игепал), ч . . 400 70 20 10 64 14 2 1
Температура хрупкости, °C -118 -118 -107 -96 -118 -118 -103 —73
лизаторе подвергались а-олефины С5 — С7, содержащиеся во фракции с Ткип 27—95 °C, выделенной из продукта высокоскоростного контактного крекинга мягкого парафина [43]. Эта фракция, кроме гептена-1 (52%), гексена-1 (17%) и пентена-1 (4%). содержала в основном пентан, гексан и гептан. По степени кристалличности и связанным с нею характеристикам (прочности при растяжении, модулю упругости, температуре плавления и др.) сополимеры этилена со смесью высших а-олефинов занимают промежуточное положение между полиэтиленом среднего давления и полиэтиленом высокого давления.
Эти сополимеры обладают большой стойкостью к растрескиванию под влиянием поверхностно-активных сред и длительных нагрузок. При испытаниях образцов сополимеров в напряженном состоянии в 5% водном растворе эмульгатора ОП-10 при 80 °C по методике, близкой к рекомендованной ASTM [44], растрескивания не наблюдалось при выдержке до 56 ч. Образцы полиэтилена среднего давления в тех же условиях подвергались растрескиванию через 9 ч.
Из сополимеров этилена, получаемых на металл органических комплексных катализаторах, наиболее полно изучены и получили широкое техническое применение сополимеры этилена с пропиленом. Соотношение звеньев этилена и пропилена в продуктах сополимеризации может колебаться в широком интервале. При содержании пропилена до 15—20% сополимеры обладают значительной кристалличностью и по свойствам относятся к пластомерам [45, 7]. Ниже приведены основные свойства сополимеров этилена с пропиленом низкого давления (содержание пропилена менее 20 мол.%) [7]:
31
Средневесовой молекулярный вес.............
Характеристическая вязкость в декалине при
135 °C, дл/г.............................
Показатель текучести расплава (груз 5 кгс, диаметр капилляра 2 мм), г/10 мин...........
Число СН3-групп на 1000 атомов С...........
Число групп на 1000 атомов С . . .
Содержание групп, % (от общего содержания
10 000—500 000
1.5-4,2
0,6-0,05 70-20
0,3-0,4
30
винильных —СН=СН2
винилиденовых
с=сн2...........
30
н\ /
транс-виниленовых ъ=С ...............
/ \н
Степень кристалличности, %..................
Плотность, г/см3............................
Температура плавления, °C...................
Прочность, кгс/см2
при растяжении (скорость растяжения 100 мм/мнн).............................
при изгибе .............................
Предел текучести, кгс/см2...................
Относительное удлинение при разрыве, % . . .
Модуль упругости при изгибе, кгс/см2........
Тангенс угла диэлектрических потерь при 10е Гц Диэлектрическая проницаемость при 10s Гц . . Удельное объемное электрическое сопротивление, Ом - см................................
Электрическая прочность (переменный ток, толщина образцов 2 мм), «В/мм..................
40
55-75 0,917—0,937 109—126
170—320
170-200
80—160
500—90Э 900—3 300 2-10-4—6-10~4
2,2-2,3
1017
30-36
При большем относительном количестве пропилена сополимеры аморфны и представляют собой типичные эластомеры. Таким продуктам посвящено значительное число работ, в частности работы Натта и его сотрудников [46—51]. Синтез, структура, свойства каучукоподобных сополимеров этилена с пропиленом и получение резин на их основе обстоятельно рассмотрены в монографии Сеидова [52].
Процесс сополимеризации этилена с пропиленом на комплексных металлорганических катализаторах подобен процессу полимеризации этилена при низком давлении. Сополимеризация осуществляется чаще всего при давлении менее 10 кгс/см2 и температурах до 80—100 °C в среде растворителей — предельных углеводородов, В качестве среды можно использовать также жидкий пропилен [53—62]. Изменяя концентрацию этилена в жидком пропилене, получают сополимеры с содержанием звеньев пропилена от 5 до 60%• Катализаторами служат продукты взаимодействия алкилов или алкилгалогенидов алюминия с хлоридами либо другими соединениями титана или ванадия. Пцименение ванадиевых катализаторов [163] предпочти-
32
тельнее при получении сополимеров со свойствами эластомеров, поскольку эти катализаторы способствуют образованию однородных по составу аморфных продуктов. Сополимеризация на титановых катализаторах сопровождается образованием частично кристаллических продуктов даже при больших содержаниях пропилена в исходной смеси с этиленом [63].
Фактором, оказывающим наибольшее влияние на состав сополимеров, является соотношение этилена и пропилена в исходной смеси. На рис. 11 представлена зависимость состава
сополимеров от соотношения концентраций мономеров. Характер этой зависимости указывает на значительно большую активность этилена в процессе сополимеризации по сравнению с пропиленом. Пониженная активность пропилена в реакции сополимеризации, протекающей по анионному механизму, связана с электронодонорным влиянием метильной группы пропи-
Рис. 11. Влияние мольного соотношения этилена и пропилена на состав сополимеров (ванадиевый катализатор, растворитель гептан, 30 °C, 1 кгс/cmj [63].
лена.
В отличие от констант
свободнорадикальной сополимериза-
ции, не зависящих от типа инициатора и связанных лишь со строением сомономеров, величины констант сополимеризации на комплексных металлорганических катализаторах зависят от характера катализатора. В частности, значения констант сополимеризации изменяются при переходе от одного металла переменной валентности в галогениде к другому (табл. 9).
Таблица 9
Константы сополимеризации этилена с пропиленом в присутствии различных каталитических систем
Каталитическая система rc2H, гс3нв гС2Н4*ГС3Н8 Литература
VCU+А1(С6Н13)3 7,08 0,088 0,62 [46]
VC13 +А1(С6Н1?)3 5,61 0,145 0,81 47
VOC13+ А1(С6Н|3)3 17,95 0,065 1,12 45
TiCl4+ А1(С6Н13)а 33,36 0,032 1,01 48
TiCl3 + А1(С6Н33)3 15,72 0,11 1,73 [48
При оценке каталитических систем учитывают известную из теории сополимеризации зависимость характера чередования звеньев сомономеров от соотношения констант сополимеризации: чем меньше величина произведения констант сополимеризации,
2 А. Г. Сирота
33
тем меньше и вероятность образования в цепях сополимеров блоков, состоящих из одинаковых звеньев. Понятно, что
чередование звеньев, исключающее
Мольное соотношение С3Н6-'(С2Н4+ С3Не)
Рис. 12. Зависимость состава сополимеров от соотношения этилена и пропилена в реакционной среде (сополимеризация в жидкой фазе на различных каталитических системах) [51]:
1—VC1, + AKCeHuh; '2-TiCl, + А1(СеН13)3
образование больших однородных блоков, благоприятно для получения аморфных сополимеров [166, 167].
Влияние характера катализатора на состав сополимеров при различном соотношении этилена и пропилена в исходной смеси показано на рис. 12.
Сополимеры обладают практически полностью аморфной структурой уже при содержании пропиленовых звеньев около 30 мол. % (рис. 13). Однако слишком большое содержание пропилена в сополимерах ведет к уменьшению их эластичности [51, 52]. Увеличение
содержания пропилена выше 60% сопровождается возрастанием температур стеклования сополимеров (рис. 14). В качестве каучуков обычно используются сополимеры, содержащие 30—40% пропилена [64]. Из
Содержание CjHg 6 сополимере,
мол. %
Рис. 13. Зависимость степени кристалличности сополимеров этилена с пропиленом от их состава [61].
Содержание С3Нв
6 сополимере, мол.%
Рис. 14. Зависимость температуры стеклования сополимеров этилена с пропиленом от их состава [52].
сополимеров, являющихся эластомерами, кроме сополимеров этилена с пропиленом, представляют интерес продукты сополимеризации этилена с а-бутиленом [51, с. 275; 54, 65—69, 158, 164]. Константы сополимеризации этилена и «-бутилена на ванадиевых катализаторах были определены Натта с сотрудниками [65]-.
VC14 + (C6H13)3AI
VC13 + (CeH13)3Al
rC2H4 ГС4Н8
29,60 0,019
29,96 0,043
34
Процесс сополимеризации этилена с а-бутиленом не отличается существенно от процесса сополимеризации этилена с пропиленом.
В работе [165] установлены отличия в константах сополимеризации этилена с различными а-олефинами и зависимость структурных характеристик и свойств полученных при сополимеризации эластомеров. Реакция сополимеризации этилена проводилась в среде жидкого сомономера в присутствии каталитической системы: соединения ванадия — диизобутилалюми-нийхлорид. По мере повышения молекулярного веса сомономера константы сополимеризации г% уменьшаются, в то же время константы сополимеризации этилена п возрастают (табл. 10).
Таблица 10
Константы сополимеризации этилена ) с а-олефинами (г2| [165]
Сомономер VC1-(C4Hs)2A1C1 У[СН(СОСНз)2)з-(и.эо-СЦ HsliAJCl
ri Г2 Г1 Г2
Пропилен 20,0 0,025 16 0,04
а-Бутилен 32,5 0,018 26 0,022
а-Амилен 42,1 0,015 32,2 0,014
Минимальное содержание (в мол. %) звеньев сомономера в цепи, необходимое для получения полностью аморфного сополимера, убывает в ряду пропилен, а-бутилен, а-амилен [165]:
Пропилен.....................27
а-Бутилен....................24
а-Амилен ....................20
Отметим, что этот факт хорошо согласуется с приведенными выше данными о влиянии длины ответвлений в цепях сополимеров этилена на кристалличность.
При одинаковом содержании сомономеров температура стеклования сополимеров этилена снижается по мере возрастания длины боковых ответвлений (содержание сомономера в полимере 30 мол. %) [165]:
Пропилен..................—52
а-Бутнпен..................—61
а-Амилен....................—70
Известны также тройные сополимеры этилена, пропилена и бутена-1 [51, с. 279; 70] или пентена-1 [167]. При содержании звеньев этилена менее 75 мол. % такие сополимеры полностью аморфны и обладают эластическими свойствами, подобными характерным для этилен-пропиленовых эластомеров. Описано получение эластомеров сополимеризацией этилена с пентеном !
2* 3$
в среде жидкого пентена-1 [52, 168, 169, 176]. Отсутствие ненасыщенных групп в каучукоподобных сополимерах этилена не позволяет использовать для вулканизации серу. Введение в сополимер этилена с пропиленом или другим сс-олефином небольшого количества (1—3%) звеньев третьего мономера, содержащего две или более двойные связи, позволяет применять обычные методы вулканизации [52, 71—99, 175].
Третьими мономерами служат алифатические и циклические диены, чаще всего с несопряженными двойными связями, хотя известны случаи применения и сопряженных диенов: изопрена, бутадиена-1,3, пентадиена-1,3 [76, 81]. Из циклических диенов используют дициклопентадиен, циклогептадиен, циклооктадиен, винилциклогексен-3, винилциклогексен-4, дивинилциклобутан и др. [52, 159, 170—172]. Из алифатических диенов наиболее пригодны соединения с различной активностью двойных связей, одна из которых находится в «-положении, а другая является внутренней. К таким соединениям относятся, например, гексадиен-1,4, гептадиен-1,5 [157]. В процессе сополимеризации раскрываются более активные двойные связи, находящиеся в а-положении. Внутренние двойные связи остаются в боковых ответвлениях от главной цепи сополимера, обеспечивая возможность вулканизации серой. Применение диенов, содержащих обе двойные связи в a-положении, менее желательно, так как имеется вероятность сополимеризации с раскрытием обеих двойных связей.
При сополимеризации этилена и пропилена с алифатическими диенами, кроме образования звеньев с двойной связью в боковой цепи, может протекать и нежелательная реакция, ведущая к образованию звеньев с предельными циклами. Это было показано на примерах гексадиена-1,5 и ^пс-формы гексадиена-1,4 [160—162]. Склонность к образованию циклов уменьшается при использовании диена с заместителем у двойной связи, например 2,5-диметилгексадиена-1,5. Содержание предельных циклов в сополимере зависит также от вида каталитической системы. Циклизация в большей мере протекает при сополимеризации с применением каталитической системы А12(С2Н5)зС1з — VOC13 и в меньшей мере — при использовании систем Al (u3o-C4H9)2Cl—VC14 и А1(С2Н5)2С1—V[CH (СОСН3)2]з.
Синтез тройных сополимеров осуществляют в тех же условиях, что и сополимеризацию этилена с пропиленом. Показано, что введение диена в реакционную среду влияет на соотношение этиленовых и пропиленовых звеньев в сополимере: содержание пропиленовых звеньев уменьшается, а этиленовых увеличивается (рис. 15). Считают [52], что это связано в первую очередь с изменениями в металлорганическом комплексном катализаторе в результате его взаимодействия с диеновым соединением.
Кроме сополимеров этилена с различными а-олефинами в последние годы уделяется внимание также сополимерам пропилена с этиленом. Введение в полипропиленовые цепи неболь-34
ших количеств этилена позволяет в значительной мере устранить важный недостаток полипропилена — хрупкость при низких температурах [145, 146].
Рекомендуется модифицировать сополимеризапиеи и дру= гие полиолефины, например поли-4-метилпентен-1. Сополимеризация 4-метилпентена-1 с этиленом [178], пропиленом, бутиленом [100—104], гексеном-1 [156], изобутиленом [105], бутадиеном-1,3 [106, 107] позволяет в той или иной мере уменьшить кристалличность материала, повысить его эластичность и растворимость. К нарушению кристаллической структуры, очевидно, приводит и сополимеризация 4-ме-тилпентена-1 с а-олефинами, создающими несимметричные ответвления от основной макроцепи: 3-метилпентеном-1, 3-метилгексе-ном-1, 4-метилгексеном-1 [108], 4-ме-тилпептеном-2 [109].
Описана радиационная сополимеризация этилена с пропиленом, tx-бутиленом, изобутиленом, стиролом [147, т. I, с. 121; 148], тетрафторэтиленом [149, т. I, с. 172; 151]. Сведения об условиях радиационной сополимеризации этилена и свойствах сополимеров систематизированы в монографии [152].
х э
S’§ з>
& &
0 О,Z 0,4 0,6
Концентрация третьего мономера, мопь/п гептана
15. Зависимость состава от концентрации
Рис.
сополимера
третьего мономера (циклооктадиена) в реакционной смеси [52].
Сополимеры этилена с полярными мономерами*
Большие возможности модифицирования полиэтилена с целью изменения свойств в нужном направлении представляет сополимеризация этилена с различными полярными мономерами: винилацетатом и другими виниловыми эфирами, винилхлоридом, тетрафторэтиленом и другими галогенсодержащими олефинами, с акриловой и метакриловой кислотами и их эфирами и т. д.
Введение в состав макромолекул полярных групп сопровождается изменением растворимости полимера, его температуры плавления, эластичности, адгезии и других свойств. Некоторые из сополимеров этилена с полярными мономерами являются промежуточными продуктами процессов модифицирования, в которых используются функциональные группы полярных звеньев. Например, сополимеры этилена, содержащие карбоксильные группы, применяются для синтеза иономеров.
Сополимеры этилена с полярными мономерами получают в основном методом свободнорадикальной сополимеризации.
* Раздел написан совместно с Е. П. Рябиковым.
37
Реакциям и продуктам такой сополимеризации посвящена обширная литература [110; 111, с. 345; 112, с. 256, 265; 183].
Сополимеризацию чаще всего проводят подобно полимеризации этилена блочным методом при высоких давлениях (1000— 2000 кгс/смг) и температурах (70—300 °C) в присутствии инициаторов — кислорода или перекисных соединений. Осуществление сополимеризации в углеводородных растворителях, воде и в эмульсиях позволяет применять менее высокое давление (до 300—500 кгс/см2). Константы сополимеризации этилена с рядом мономеров представлены в табл. 11.
Таблица 11 Константы сополимеризации этилена (rj с различными мономерами (г2)
Сомономер ri Г2 Давление, кгс/см2 Температура, °C Литература
Винилацетат 0,16 1.1 100 60 184
0,70 3,7 1200 60 184
1,0 ±0.4 1,3 +0,4 1000 90 185
1.0 + 0,1 09+0,1 300—400 130 186
Метилакрилат 0,12±0.03 13±5 820 150 187
Этилакрилат 0,19±0,04 2,2±0,7 2040 180 188
Бутилакрилат 0,034 + 0,008 14 + 4 1000 70 189
Метилметакрилат 0,2 17 820 150 187
Винилхлорид 0.23 + 0,02 1,9 + 0,02 300-1500 70 190
0.20 1,85 700-1300 140-150 191
Винилфторид 5,2 + 0,7 0,13 + 0,04 1000 160 189
Трифторпропилен 0.18 + 0,05 0,9±0,5 300—1250 70 192
Свойства сополимеров этилена зависят от строения сомономера, содержания и распределения его звеньев в макромолекулах [113]. Изменение свойств, связанное со строением звеньев сомономера, определяется прежде всего их полярностью и размером. Введение звеньев с полярными группами в полиэтиленовые цепи вызывает усиление межмолекулярных и внутримолекулярных взаимодействий. Проявляется это в повышении жесткости и температуры плавления сополимеров. Диэлектрические свойства сополимеров, естественно, ухудшаются, адгезия к различным материалам увеличивается.
Введение в цепи полиэтилена звеньев с боковыми группами, близкими по размеру к метиленовым, не сопровождается значительным нарушением кристаллической структуры. Наблюдается лишь некоторое изменение параметров кристаллических ячеек. Физические свойства, например плотность, меняются пропорционально содержанию полярных звеньев (рис. 16). Температура плавления в зависимости от состава сополимеров изменяется в интервале от температуры плавления полиэтилена до температуры плавления полярного гомополимера, 38
Сополимеризация этилена с полярными мономерами, которые имеют заместители сравнительно большого объема, не
укладывающиеся в кристаллическую решетку полиэтилена, со-
провождается снижением степени кристалличности. При этом изменяются связанные со степенью кристалличности характеристики, уменьшаются твердость и температура плавления, увеличивается эластичность.
Из двух противоположно влияющих факторов—полярности и размеров звеньев сомономеров — второй оказывается превалирующим при больших размерах звеньев сомономера. Например, сополимеры этилена с небольшим количеством винилацетата обладают большей, чем
Рис. 16. Зависимость
плотности сополимеров этилена с окисью углерода от содержания в" них звеньев ^СО (23 °C; по данным [114]).
полиэтилен, эластичностью и более низ-
кой температурой Плавления. Превращение ацетатных групп в сополимере при омылении в значительно меньшие по объему гидроксильные группы делает продукт более жестким и плавящимся
при более высокой температуре, чем полиэтилен. Это связано с изменением стерического фактора,
так как дипольные
Содержание винилацетата в сополимере, мол. %
Рис. 17. Зависимость степени кристалличности сополимеров этилена с винилацетатом от их состава (при 30 °C)
[113, 115].
моменты ацетатной и гидроксильной групп практически одинаковы [ИЗ].
Стерический фактор оказывает влияние также на зависимость свойств сополимеров от состава. Если звенья полярного сомономера имеют небольшие заместители, способные к упаковке в кристаллической решетке полиэтилена, то свойства сополимера изменяются постепенно по мере увеличения содержания полярных звеньев. При введении в полиэтиленовую цепь звеньев полярного мономера с крупными заместителями, например винилацетата, зависимость свойств от состава имеет более резко выраженный характер. Даже при небольшом содержании такого сомономера значительно
уменьшаются степень
кристалличности, жесткость и темпера-
тура плавления (рис. 17).
Важным фактором, влияющим на свойства сополимеров, яв-
ляется характер распределения звеньев этилена и сомономера в макроцепях. Известно, что это распределение связано с величиной произведения констант сополимеризации. Значениям произведения констант сополимеризации, близким к единице,
39
соответствует беспорядочное распределение, с уменьшением этой величины порядок в чередовании звеньев обоих типов в макроцепях увеличивается. Влияние различия в распределении иллюстрируется, например, рис. 18, на котором представлены кри-
П\__I_I--1--1-1--i-1--L
-60-W-20 0 20 40 60 80
Температура,°C
Рис. 18. Зависимость динамических потерь в сополимерах этилена с винилацетатом с беспорядочным (/) и частично упорядоченным (2) распределением звеньев от температуры [21].
вые температурной зависимости динамических потерь сополимеров этилена, содержащих 20% винилацетата. Максимум потерь для сополимера с беспорядочным распределением находится около —25°C, для сополимеров с частично упорядоченным распределением — около + 20 °C. Это различие связано с тем, что образование полиэтиленовых блоков, имеющих достаточные для кристаллизации размеры, более вероятно в сополимерах с упорядоченным распределением звеньев. (Минимальная длина кристаллизующихся последовательностей в сополимерах этилена с винилацета-
том, так же как и в разветвленном полиэтилене, составляет около 13 звеньев [116]). Переходы, связанные с перегруппировкой макроцепей в кристаллических областях, образованных полиэтиленовыми блоками, возможны
при сравнительно высоких температурах [21].
Наиболее изученными продуктами сополимеризации этилена с полярными мономерами являются сополимеры этилена с винилацетатом [197—218]. Сополимеризацию этилена с винилацетатом можно осуществлять в блоке, растворе и эмульсии. Кроме органических перекисей и азосоединений для инициирования можно использовать алкилы бора или ионизирующее излучение.
Уже небольшое количество винил-
1,02
Содержание винилацетата в сополимере, %
Рис. 19. Зависимость плотно-
ацетата в сополимере с этиленом сти сополимеров этилена с значительно уменьшает Кристал- винилацетатом от их состава личность (см. рис. 17). Это прояв- (ПР“ ’ 1 Г ляется, в частности, в зависимости
плотности сополимера от его состава (рис. 19). При относительно небольшом содержании винилацетата плотность
уменьшается, достигая минимума, затем с увеличением содержания винилацетата возрастает до величины 1,17 г/см3, соответствующей поливинилацетату. Температура плавления сопо
40
лимеров тем ниже, чем больше содержание в них винилацетата (рис. 20).
Кривая зависимости прочности от состава сополимера также имеет максимум (рис. 21), после прохождения которого прочность резко уменьшается по мере увеличения содержания винилацетата. Относительное удлинение при растяжении с увеличением количества винилацетата в сополимере значительно возрастает.
Варьируя соотношение сомономеров, можно получать различные материалы: и пластики, и эластомеры. Сополимеры этилена с содержанием винилацетата до 10—12 мол. % близки
Рис. 20. Зависимость температуры плавления сополимеров этилена с винилацетатом от их состава [121].
Содержание винилацетата В сополимере, вес. %
Рис. 21. Зависимость прочности при разрыве (7) и относительного удлинения (2) от состава сополимеров этилена с винилацетатом [120].
по свойствам к полиэтилену высокого давления, но отличаются большей эластичностью, прозрачностью и пониженной температурой плавления [122]. Сополимеры, содержащие 13—25% винилацетата, каучукоподобны. При содержании винилацетата 30—80% сополимеры мягки и клейки при комнатной температуре, при дальнейшем увеличении количества винилацетата продукты сополимеризации снова становятся жесткими, приближаясь по свойствам к поливинилацетату. Растворимость сополимеров в бензоле, ксилоле, четыреххлористом углероде возрастает по мере увеличения содержания винилацетата, при содержании его выше 40% сополимеры растворяются в спирте и ацетоне [121, 122]. Сополимеры этилена с винилацетатом легко сшиваются под действием перекисных соединений [120], поликарбоновых кислот [123, 124] и ионизирующего излучения [121, 125].
Гидролизом сополимеров этилена и винилацетата можно получать сополимеры этилена и винилового спирта, при частичном гидролизе — тройные сополимеры этилена, винилацетата и винилового спирта [126—130]. Продукты гидролиза обладают
41
значительной прочностью, жесткостью, важным их отличием
от поливинилового спирта является нерастворимость в воде.
Известны сополимеры этилена и со многими другими виниловыми эфирами, кроме винилацетата.
Из продуктов сополимеризации этилена с акриловыми мономерами промышленное значение имеют сополимеры этилена с метилакрилатом, этилакрилатом и метилметакрилатом [21, 131 —134, 194, 195]. Введение в молекулы полиэтилена звеньев этих мономеров, содержащих заместители большого объема, приводит к резкому снижению кристалличности даже при малом содержании сомономера. Уже при содержании 5 мол. % акри-
Содержание второго мономера, вес. Ч
Рис. 22. Зависимость проч-
лового мономера сополимеры практически полностью аморфны. Температура размягчения по Вика сополимеров на 30—50 °C ниже, чем у полиэтилена высокого давления того же молекулярного веса.
Плотность сополимеров не является такой определяющей характеристикой, как для полиэтилена, поскольку для сополимеров нет линейной зависимости плотности от кристалличности и разветвленности. Как правило, плотность сополимеров возрастает с увеличением содержания сомономера. В связи с этим свойства сополимеров
ности при разрыве от со-
става сополимеров этилена с метилакрилатом (О), метилметакрилатом (□) и этилакрилатом (©) (показатель текучести расплава 2 г/10 мин) [ 195].
этилена с акриловыми мономерами
обычно рассматриваются только в зависимости от строения и количества второго мономера.
Прочность сополимеров снижается по мере увеличения содержания вто-
рого мономера (рис. 22), хотя и остается в интервале, характерном для полиэтилена низкой плотности даже при значйтельном содержании сомономера (до
16—20 вес.%).
Пределы текучести сополимеров ниже пределов текучести
полиэтилена высокого давления того же молекулярного веса. Сополимеры этилена с акриловыми мономерами отличаются от полиэтилена более низкими температурами хрупкости, повышенными значениями ударной вязкости. Эти свойства сополимеров рекомендуется использовать для получения композиций
с полиэтиленом и полипропиленом, имеющих пониженную температуру хрупкости [1, 193].
Стойкость к растрескиванию сополимеров этилена с акриловыми мономерами при содержании более 15 вес. % сомономера при испытании по методу ASTM D 1693—60Т составляла более 340 ч, что намного превышает аналогичный показатель для полиэтилена со сравнимым показателем текучести расплава. Ди-
42
Содержание второго мономера, вес. %
Рис. 23. Зависимость диэлектрической проницаемости сополимеров этилена с метилакрилатом (О), метилметакрилатом (□) и этилакрилатом (®)от их состава [ 195].
электрические характеристики сополимеров этилена с метилакрилатом, этилакрилатом и метилметакрилатом определяются количеством введенных в макроцепь полярных звеньев. Как видно из рис. 23, величина диэлектрической проницаемости сополимеров линейно зависит от содержания второго мономера. Эта зависимость может быть использована для определения состава сополимеров по значениям диэлектрической проницаемости [196]. Тангенс угла диэлектрических потерь этих сополимеров в зависимости от их состава изменяется в пределах от 0,001 до 0,03.
Газопроницаемость сополимеров (табл. 12) значительно выше, чем у полиэтилена, хотя в отдельных случаях это свойство оказывается преимуществом сополимеров [195].
Сополимеры этилена с акрилатами могут перерабатываться в изделия в широком диапазоне температур всеми известными для полиэтилена методами на аналогичном оборудовании. Па
раметры процессов переработки сополимеров этилена с акрилатами сравнимы с параметрами переработки полиэтилена низкой плотности, хотя температура переработки на 14—17 °C ниже, чем для полиэтилена.
Таблица 12
Газопроницаемость сополимеров этилена [195]
Сополимер Содержание звеньев второго мономера в сополимере, вес. % Скорость диффузии при 30 °C, D-I03, М ? • М К сут*кгс/см2
кис тороДа углекислого газа
Этилен + метилакрилат 14,9 0,20 1,09
18,9 0,25 1,50
21,3 0,27 1,20
22,7 0,31 2,03
Этилен + этилакрилат 15,1 0,27 1,40
18,2 0,30 1,60
Этилен 4- метилметакрилат 16,1 0,17 0,58
Полиэтилен 0 0,04 0,08
43
Для свободнорадикальной сополимеризации с этиленом пригодны и соединения, не способные к гомополимеризации —
окись и двуокись углерода и двуокись серы.
Сополимеры этилена и окиси углерода обладают значительной кристалличностью. ИК-спектры сополимеров, содержащих 21% окиси углерода, имеют полосу поглощения 730 см-1, интенсивность которой связана с кристалличностью полиэтиленового типа [135—138]. Исчезновение этой полосы наблюдается при увеличении содержания звеньев окиси углерода в сополимере до 43% [139]. Температура плавления сополимеров этилена и окиси углерода находится в интервале 90—200 °C. Существенный недостаток этих сополимеров заключается в их хрупкости. При нагревании выше температуры плавления они ведут себя
как термореактивные полимеры, теряя плавкость и растворимость. Процесс сопровождается выделением воды. Считают [139], что сополимер приобретает трехмерную структуру в результате межмолекулярных реакций. Реакция конденсации сополимеров может осуществляться под влиянием щелочей [140, 141].
Сшивание сополимеров этилена с окисью углерода перекисными соединениями, хлорамином, азотистыми основаниями [114, 140, 142] сопровождается увеличением прочности, температуры размягчения и уменьшением растворимости.
Кетонные группы, входящие в макромолекулы сополимеров, создают возможности для проведения различных реакций в
макроцепях и получения полимерных продуктов разных классов. Схематически некоторые из таких реакций представлены ниже [114]:
он
I
---СН2—СН2—СН--- (1)
ОН
I
---СН2—СН2—С---- (2)
CN
---СН2—СН2—С---- (3)
NOH
Гидрирование сополимера этилена с окисью углерода (схема I) под давлением в присутствии хромита меди в качестве катализатора позволяет получать полимер, содержащий гидроксильные группы [114]. Полициангидрин может быть получен обработкой сополимера избытком синильной кислоты в присутствии цианистого калия (схема 2), превращению подвергается до 80% карбонильных групп. Полиоксим образуется при введении водного раствора гидроксиламина в раствор сополимера в углеводородном растворителе (схема 3), степень превращения гидроксильных групп достигает 78%.
44
Сополимеры этилена с двуокисью серы отвечают формуле:
Они имеют кристаллическую структуру и отличаются высокой температурой плавления. Сополимеры с эквимолекулярным, содержанием сомономеров имеют температуру плавления выше 300 °C. Температура плавления таких сополимеров совпадает с температурой разложения или превышает ее. Высокая температура плавления, очевидно, определяется сильным межмолекулярным взаимодействием полярных сульфоновых групп и регулярностью структуры сополимера [113]. Неплавкость сополимеров при температурах ниже температуры разложения и нерастворимость во всех известных растворителях весьма осложняют переработку. Сополимеры с содержанием двуокиси серы менее 50 мол. % можно перерабатывать литьем под давлением или экструзией при 180—220 °C [143, 144]. В работе [219] изучались свойства сополимеров высших а-олефинов (Се—Cie) с двуокисью серы. Максимальной деформируемостью и газопроницаемостью (по отношению к кислороду и углекислому газу) и минимальной плотностью обладал сополимер двуокиси серы с а-олефином Ci6H32.
Сополимеры пропилена с акрилонитрилом [153, 155], этилена с окисью углерода, двуокисью серы и рядом других мономеров (акрилонитрилом, винилацетатом, винилиденхлори-дом, винилпирролидоном, малеиновым ангидридом, метилакрилатом, метилметакрилатом, метилвинилкетоном, трифторхлор-этиленом) могут быть получены радиационной сополимеризацией [152, 154, 155].
Литература
1. Bonotto S., Junghans Р. R. Plast. Technol., 9, № 5, 34 (1963).— 2. Smith W. E., S toff er R. L., Hannan R. B. J. Polymer Sci., 1962, v. 61, Ns 171, p. 39. — 3. Сирота А. Г., Рябиков E. П., Гольден-берг А. Л., Ильченко П. А., Чопко Л. Ф. Пласт, массы, 1965, № 11, 5. — 4. Г о л ь д е н б е р г А. Л., Ильченко П. А., Сирота А. Г. н др. Пласт, массы, 1962, № 6, с. 8.—5. Swan Р. R. J. Polymer Sci., 1962, v. 56, р. 409. — 6. Гольденберг А. Л., Заплетняк В. М., И л ь ч е н-ко П. А. В кн.: Спектроскопия полимеров. Киев. «Наукова думка», 1967.— 7. Сополимер этилена с пропиленом низкого давления. Под ред. С. В. Шуц-кого. Л., «Химия», 1965, с. 16.—8. Гейлорд Н., Марк Г. Линейные и стереорегулярные полимеры. М., ИЛ, 1962. 565 с. — 9. Reding Е. Р., W alter Е. R. J. Polymer Sci., 1959, v. 38, р. 141.— 10. Clark A., Hogan J. P., Banks R. L., Lanning W. G. Ind. Eng. Chem., 1956, v. 48, Ns 7, p. 1152.
11. Smith D. C. Ind. Eng. Chem., 1956, v. 48, Ns 7, p. 1162.— 12. Б у н и я т-заде А. А., Касумов К. Г., Садыков Р. Б. Уч. зап. Азерб. гос. ун-та, 1959, Ns 3, с. 90. — 13. Далин М. А., Б у н и я т - з а д е А. А., П и с fa-ман И. И., Бахши-заде А. А. Азерб. хим. ж., 1959, Ns 4, р. 21.— 14. Далин М. А., Бахши-заде А. А., П и с ь м а н И. И., Б у н и я т-
45
заде А. А. Азерб. хим. ж., 1960, № 1, с. 25.— 15. Далин М. А., П и с fa-ман И. И., Б а х ш и - з а д е А. А., Б у н и я т - з а д е А. А. ДАН СССР, 1960, т. 133, № 5, с. 1084.— 16. Далин М. А., Шендерова Р. И., П и с fa-ман И. И. и др. Азерб. хим. ж., 1967, № 1, с. 17.— 17. Б у н и я т - з а де А. А., Автореф. дисс. Азерб. гос. университет, Баку, 1961. — 18. Далин М. А., П исьман И. И., Бахши-заде А. А. и др. Азерб. хим. ж., 1961, № 2, с. 9. —19. Pritchard J. Е., McClamery R. М., Boeke Р. J. Brit. Plast., 1960, № 2, с. 58.; пер.: Химия и технология полимеров, 1960, № 10, с. 70.—20. Полиэтилен среднего давления. Под ред. Шуцкого С. В. Л., «Химия», 1965. 90 с.
21. Takeshi К. Japan Plast. Age, 1965, v. 3, № 8, p. 34.; пер.: Экспресс-информация. Серия «Синтетические высокополимерные материалы», 1966, № 3, с. 30.—22. Hogan J. Р. In: Copolymerization. Ed. Ham G. E. N. Y., Interscience, 1964. пер.: Сополимеризация. Под. ред. В. А. Кабанова. М., «Химия», 1971. 616 с. — 23. Далин М А., Буният-заде А. А., Булатникова Э. Л. Пласт, массы, 1966, № 8, с. 4.-24. Камбаров Ю. Г., Сеидов Н. М., Буният-заде А. А. Полиолефины. Баку, Азерб. гос. изд., 1966 185 с. — 25. Бельг, пат. 535082, 1955. — 26. Инд. пат. 53618, 1956.— 27. Пат. США 2825721, 1958.—28. Пат. США 269257, 1954. — 29. Пат. ФРГ 1001003, 1957.— 30. Англ. пат. 734501, 1955.
31. Пат. США 2726231, 1955.— 32. Англ. пат. 748583, 1956.—32. Англ, пат. 748583, 1956. — 33. Бельг, пат. 545857, 1956. — 34. Англ. пат. 786014, 1957.—35. Пат. США 2727024, 1955.— 36. Англ. пат. 783744, 1957.— 37. Пат. США 2728757, 1955. — 38. Пат. США 2691647, 1954.— 39. Англ. пат. 748634, 1956.— 40. Пат. США 2731452, 1956.
41. Англ. пат. 753349, 1956.—42. Авт. свид. СССР 175658, 1965; Бюлл. изобр., 1965, № 20. — 43. Лавровский К. П., Бродский А. М., Мусаев И. А., Румянцев А. Н. Нефтехимия, 1962, т. 2, с. 487. — 44. ASTM, Standards on Plastics, Phil., 1958, p. 1049. — 45. Егоров H. M., Архипова 3. В., Андреева И. Н. и др. Пласт, массы, 1959, № 1, с. 10.— 46. Natta G., Mazzanti G., Valvassori A., Paiaro G. Chim. e ind., Milan, 1957, v. 39, № 9, p. 733. — 47. Mazzanti G„ Valvassori A., Paiaro G. Chim. e ind.. Milan. 1957. v. 39, № 9, p. 743. — 48. Mazzanti G„ Valvassori A., Paiaro G. Chim. e ind., Milan, 1957, v. 39, № 10, p. 825. — 49. Natta G., Valvassori A., Mazzanti G., Sartori G. Chim e ind., Milan, 1958, v. 40, № 9, p. 717. — 50. Natta G„ Valvassori A„ Mazzanti G., Sartori G. Chim. e ind., Milan, 1958, v. 40, № 11, p. 896.
51. C res pi G.. Valvassori A., Sartori G. In: Copolymerisation. N. Y., Interscience, 1964; пер.: «Сополимеризация». Под ред. В. А. Кабанова. М., «Химия», 1971. 616 с.—52. Сеидов Н. М. Новый синтетический каучук иа основе этилена и пропилена. Баку, Азерб. гос. изд., 1966. 126 с. — 53. Англ, пат. 898261, 1962.— 54. Венг. пат. 149767, 1962. — 55. Авт. свид. СССР 172989, 1965; Бюлл. изобр., 1965, № 14. — 56. Сеидов Н. М., Далин М. А., Камбаров Ю. Г. и др. Азерб. хим. ж., 1965, № 3, с. 73. — 57. Сеидов Н. М., Далин М. А., Абасов А. И. ДАН СССР, 1966, т. 166, № 6, с. 1376.— 58. Сеидов Н. М., Абасов А. И. Азерб. хим. ж., 1965, № 6, с. 54.— 59. Сеидов Н. М., Арутюнов И. А., Далин М. А., Б а х ш и - з а-д е А. А. Нефть и газ, 1965, № 7, с. 70. — 60. Сеидов Н. М., А р у т гонов И. А., Д а л и н М. А. Нефть и газ, 1965, № 10, с. 59.
61. Сеидов Н. М.. Д а л и н М. А., Камбаров Ю. Г. и др. Каучук и резина. 1966, № 6, с. 3. — 62. Сеидов Н. М., Арутюнов И. А., Абасов А. И. Уч. зап. Азерб. гос. ун-та, 1966, № 3, с. 78. — 63. Bier G., Gumboldt A., Schleitzer G. Macromol. Chem., 1962, v. 58, p. 43; nep.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ. М., «Мир», 1964. — 64. Amberg L. О., Robinson А. Е. Rubb. Plast. Age, 1961, v. 42, № 7, р. 875. — 65. Natta G., Mazzanti G., Valvassori A., Paiaro G. Chim. e ind., 1959, v. 41, p. 764. — 66. Англ. пат. 888986, 1962.— 46
67. Пат. США 2953592, 1960. — 68. Сеидов Н. М., Далин М. А., К я з и-мов С. М. ДАН СССР, 1965, т. 164, № 4, с. 826.-69. Сеидов Н. М„ Далин М. А., К язи мов С. М. и др. Азерб. хим. ж., 1966, № 3, с. 46.— 70. Valvassori A., Sartori G. Chim. е ind., Milan, 1962, v. 44, p. 1091.
71. Verbanc J. J., Fawcett M. S„ G о 1 d b e r g E. J. Ind. Eng. Prod. Res. a. Developm., 1962, v, 1, p, 70. — 72. Gladding E. K.. Fisher B. S., Collette J. W. Ind. Eng. Chem., Prod. Res. a Developm., 1962, v. 1, p. 65.— 73. Франц, пат. 1386600, 1965.— 74. Франц пат. 1370902, 1964. — 75. Sartori G., Valvassori A., Vaina S. Atti Accad. Line., 1963, Bd. 35. Ns 6, S. 565. — 76. Фин. пат. 32973, 1963. — 77. Франц, пат. 1389757, 1965.— 78. Франц, пат. 1392109, 1965. — 79. Англ. пат. 987734, 1965.—80. Англ. пат. 997885, 1965.
81. Пат. США 3208982, 1965.—82. Франц, пат. 1353179, 1964.— 83. Япон. пат. 22752, 1965.— 84. Франц, пат. 1362094, 1964. — 85. Франц, пат. 1375320, 1964, —86. Франц, пат. 1370358, 1964.— 87. Пат. ФРГ 1163551, 1964,— 88. Франц, пат. 1371435, 1964. — 89. Франц, пат. 1375319, 1964.—90. Франц, пат. 1377626, 1964.
91. Франц, пат. 1430560, 1966. — 92. Франц, пат. 1386600, 1965.— 93. Франц, пат. 1397652, 1965. — 94. Франц, хат. 1413234, 1965.—95. Франц, пат. 1416704, 1965. — 96. Франц, пат. 1424440, 1966. — 97. Франц, пат. 1428661( 1966. — 98. Natta G., Crespi G. J. Polymer Sci., 1962, v. 1, p. 83; nep.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ. М., «Мир», 1964. — 99. Франц, пат. 1345819, 1963. — 100. Англ. пат. 978101, 1964.
101. Англ. пат. 982716, 1965. — 102. Франц, пат. 1380649, 1964.— 103. Франц, пат. 1389013, 1965.— 104. Франц, пат. 1410245, 1965.— 105. Англ, пат. 987316, 1965,— 106. Англ. пат. 982708, 1965. — 107. Франц, пат. 1381689, 1964.- 108. Англ. пат. 973827, 1964. — 109. Пат. ФРГ 1189274, 1965.— НО. Raff R. А. V., Allison J. В. Polyethylene. N. Y. — London, Inter-seience, 1956. 551 p.
111. Pie ski E. D., Polythene. N. Y.— London, Interscience, 1960.— 112. Mod. Plast. Encyclopedia Issue for 1962. N. Y., Plastics Catalogue Corp., 1961. — 113. T e p т e p я н П. А., Б p а у д о E. E., Дипиес А. И. «Успехи химии», 1965, т. 34, № 4, с. 666. — 114. Brubaker М. М., Coffman D. D.,
Hoehn Н. Н. J. Am. Chem. Soc., 1952, v. 74, № 6, р. 1509. — 115. Nielsen L. В. J. Polymer Sci., 1960, v. 42, p. 357. — 116. Kilian H. G. Koll.
Z. u. Z. Polymere, 1963, Bd. 189, № 1, S. 23.— 117. Любецкий С. Г., E p y-
салимский Б. Л., Гольденберг А. Л. ДАН СССР, 1967, т. 172, № 6, с. 1372; Гольденберг А. Л., Зюзина Л. И., Любецкий С. Г. Высо-комол. соед., 1967, т. 9, сер. Б, № 7, с. 542.— 118. Любецкий С., Еруса-лимский Б., Гольденберг А. Доклад на международном симпозиуме по химии высокополимеров. Токио, 1966. — 119. Stokr J., Schneider В. Coll. Czech. Chem. Comm., 1963, d. 28, S. 1946.— 120. Bartl H„ Peter J. Kaut. u. Gummi, 1961, № 2, S. 23; пер.: «Химия и технология полимеров», 1961, № 5, с. 37.
121. Тертерян Р. А. Дипцесс А. И., Рысаков М. В. Журн. ВХО им. Менделеева, 1963, т. 8, с. 589. — 122. Тертерян П. А., Д и н ц е с А. И., Рысаков М. В. Нефтехимия, 1963, т. 3, с. 719. — 123. Бельг, пат. 591575, 1960.— 124. Пат. ФРГ 1055232, 1959. — 125. Пат. ФРГ 1116394, 1966 — 126. Англ. пат. 634140, 1950. — 127. Пат. США 2386347, 1943. — 128. Пат. США 2399653, 1946.— 129. Англ. пат. 607911, 1948. — 130. Пат. США 2467774, 1949.
131. Mod. Plast., 1961, v. 38, № 9, р. 41.— 132. Bonotto S., Krev-sky В. H. SPE J., 1962, v. 18, p. 555.— 133. Rubb. Age, 1962, v. 91, p. 806.— 134. Bonotto S., Krevsky В. H. Plast. World, 1962, v. 20, № 8, p. 32.— 135. King G. W.. Hainen R. M., McMahon H. O. J Appl. Phys., 1949, v. 20, p. 559. — 136. R u g g F. M.. Smith J. J.. W a r t m a n L. H. J. Polymer Sci., 1953, v. 11, p. 1.— 137. Stein R. S., Sutherland G. В. В. M.
47
J. Chem. Phys., 1953, v. 21, № 2, p. 370.— 138. Reding F. P., Brown A. J. Appl. Phys., 1954, v. 25, p. 848.— 139. Б pay до E. E., К а душив A. A., Динцес А. И. Нефтехимия, 1963, т. 4, № 3, с. 441.— 140. Пат. США 2495286, 1950.
141. Cur ph у Е. G. Can. Chem. Proc., 1956, v. 40, № 6, р. 78.— 142. Англ. пат. 597833, 1948. — 143. Пат. США 2943077, 1960. — 144. Пат. США 2976269, 1961, — 145. Bier G. Angew. Chem., 1961, Bd. 73, S. 186; nep.: Химия и технология полимеров, 1962, № 6, с. 3. — 146. Takagi К., I о s h i о-к a S. Japan Plast. Age, 1965, v. 3, № 9, p. 11; nep.; «Химия и технология полимеров», 1966, № 6, с. 109.— 147. Steinberg М., Colombo Р. Industrial Uses of Large Radiation Sources. Vienna, 1963.— 148. Colombo P., Steinberg M., Macchia D. J. Polymer Sci., 1963, v. I, Ser. B, p. 483.— 149. Tabata Y., Ishigure K., Shibano H., О shim a K-, Sobue H. Industrial Uses of Large Radiation Sources. Vienna, 1963.— 150. Пат. ФРГ 1813784, 1969; С. A., 1969, v. 71, № 22, p. 102441.
151. Hagiwara M., Miura T., Kagiya T. Bull. Chem. Soc. Japan, 1969, v. 42, № 12, p. 3380.— 152. Иванов В. С. Радиационная полимеризация. Л., «Химия», 1967. 232 с. — 153. Андреев Л. И., Крендель Б. А., Литманович А. Д., Полак Л. С., Топчиев А. В. Изв. АН СССР. Отд. хим. наук, 1959, № 8, с. 1507.— 154. Mitsui Н., Tsuneta К., Kagiya Т. Kogyo Kagaku Zasshi, 1971, v. 74, № 9, p. 1918.— 155. Kanae Hayashi, Koichiro Hayashi. Ann. Rep. of Osaka Laboratory for Radiation Chemistry, 1969, № 2, p. 45.— 156. C a pm be 11 T. W. J. Appl. Polymer Sci., 1961, v. 5, № 14, p. 184. — 157. Сеидов H. M„ К о п т е в Д. А., Далин М. А. и др. ДАН СССР, 1970, т. 195, № 3, с. 620. — 158. Громова В. Н., Мир он юк В. П., Покатило Н. А., Рейх В. Н. Высокомол. соед., 1967, т. 9, сер. А, № 5, с. 1123.— 159. Caywood S. W. Rubber Chem. TechnoL, 1971, v. 44, № 3, s. 653. — 160. Лившиц И. А., Коробова Л. M., Поддубный И. Я. и др. Высокомол. соед., 1970, т. 12, сер. А, № 8, с. 1794.
161. Лившиц И. А., Коробова Л. М., Соколов В. Н. и др. Высокомол. соед., 1971, т. 13, сер. Б, № 4, с. 304. — 162. V а 1 v a s s о г i A., Sartori G. Chem. е ind., Milan, 1962, v. 44, p. 1095.— 163. Карасев В. H., Минскер К- С. Высокомол. соед., 1971, т. 13, сер. А, № 7, с. 1468.— 164. Матковский П. Е., Белов Г. П., Руссиян Л. Н. и др. Высокомол. соед., 1970, т. 12, сер. A, А"» 10, с. 2286.— 165. Сеидов Н. И., Далин М. А. Высокомол. соед., 1971, т. 13, сер. Б, № 1, с. 80. — 166. С е и-д о в Н. М„ Алигулиев Р. М., Абасов А. И. Высокомол. соед., т. 11, сер. А, № 9, с. 2107. — 167. Сеидов Н. М., Абасов А. И„ Далин М. А., ДАН АзербССР, 1968, т. 24, № 4, с. 22.— 168. Сеидов Н. М„ Коптев Д. А. Азерб. хим. ж., 1967, № 5, с. 101.— 169. Сеидов Н. М„ Коптев Д. А., К у ко б а Л. Т. Уч. зап. Азерб. гос. ун-та, 1970, К» 4, с. 66.— 170. VahldieckJ. Plaste u Kaut., 1971, Bd. 18, № 9, S. 641.
171. Сеидов H. M., Далин M. А., Кязимов С. M., Кулиев T. M. Уч. зап. Азерб. гос. ун-та, 1971, № 2, с. 61.— 172. Сеидов Н. М„ Коптев Д. А., Далин М. А. и др. Высокомол. соед., 1969, т. 11, сер. А, № 4, с. 844.— 173. Аверьянов А. А., Фильберт Д. В., Баранова С. А. и др. В кн.: Волокна из синтетических полимеров. Под ред. А. Б. Пакшвера. М., «Химия», 1970. 324 с.— 174. Бартенев Г. М. Высокомол. соед., 1964, т. 6, № 2, с. 335.— 175. Лившиц И. А., Рейх В. Н. Каучук и резина, 1971, № 2, с. 27,— 176. Сеидов Н. М., Коптев Д. А., Далин М. А. и др. Высокомол. соед., 1969, т. 11, сер. А, № 4, с. 844.— 177. Causescu Е., LebSda Р„ Minti М. Mat. Plast., 1970, Bd. 7, № 11, S. 559,— 178. Б y-ният-заде А. А., Мамедов Э. Л., Авакян Л. Г. и др. Пласт, массы, 1972, № 3, с. 12.— 179. Буният-заде А. А., Акутин М. С., Эльдаров Э. Г. и др. Пласт, массы, 1970, № 4, с 12. — 180. Steiner R., S о-botta G., Schoenemann К. Kunststoffe, 1966, Bd. 56, № 3, S. 167.
181. Boghetich L., Mortimer G. A., Danes G. W. J. Polymer Sci., 1962, v. 61, p. 3.— 182. Mortimer G, A., J. Polymer Sci., 1965, v. 3, Ser. B, 4?
р. 343.— 183. Ehrlich Р., Mortimer G. A. Adv. Polymer Sci., 1970, v. 7, № 3, p 386.— 184. Erusalimskii B. L., Tumarkin N. Ya., Dun-tov F. I., Lyubetskii S. G., Goldenberg A. L. Makromol. Chem., 1967, v. 104, p. 288.— 185. Пат. США 3168456, 1965.— 186. Тер тер ян Р. А., Д и н цр с А- И., Русаков М. В. Нефтехимия, 1965, т. 5, с. 340. — 187. Brown F. Е.. Ham G. Е. J. Polymer Sci., '1964, v. 2. Ser. A, p. 3623.— 188. Пат. США 2953551, 1960.— 189. Burkhart R D„ Z u 11 у N. L„ J. Polymer Sci., 1963, v. 1, Ser. A, p. 1137.— 190. Дуятов Ф. И., Гольде н-берг А. Л., Литвинова М. А., Ерусалимский Б. Л. Высокомол. соед., 1967, т. 9, сер. А, с. 1920.
191. Erusalimskii В. L., Dun tov F. J., Tumarkin N. Ya. Macromol. Chem., 1963, v. 66, p. 205.— 192. Д унтов Ф. И., Ерусалимский Б. Л. Высокомол. соед., 1971, т. 13, сер. Б, № 7, с. 539.— 193. Пат. США 2953541, 1958.— 194. Bonotto S., Walton R. К. Mod. Plastics, 1963, v. 40, p. 143.— 195. Александер, Анспон, Браун и др. Химия и технология полимеров, 1967, № 4, с. 70.— 196. Steck N. S. SPE Trans., 1964, v. 4, р. 34. —197. Andrei С., Dina L. Mat. Plast., 1969, Bd. 6, № 3, s. 159.— 198. Rubini R. Nouva chim., 1970, v. 46, № 7, p. 27.— 199. Del
Gatto. J. Rubber World, 1970, v. 162, № 6, p. 58. — 200. Fisher E. T.,
Wired., 1971, v. 4, № 4, p. 45.
201. So Iyer I. O„ Leeper H. M. Rubber Age, 1971, v. 103, № 6, p. 37;
№ 7, p. 63. — 202. Cernia E. Nuova chim., 1971, v. 47, № 3, p. 39.—
203. Wisotsky M. J., Kober A. E., Zlochower J. A. Am. Chem. Soc. Polym. Prepr., 1970, v. 11, Ns 2, p. 1035. — 204. Sato Yoshiyasu, Saku-rai Masayuki, Kisnibe Masayuki, Kayama Itaru, Дзайре, J. Soc. Mater. Sci. Japan, 1971, v. 20, № 212, p. 591. — 205. Wisotsky M. T., Kober A. E., Zlochower I. A. J. Appl. Polymer Sci., 1971, v. 15, № 7, p. 1737.—206. Ratzsch M„ Schneider W., Musche D. J. Polymer Sci., 1971, v. 9, Ser. Al, № 3, p.’ 785. — 207. Д унтов Ф. И., Тертеряи P. A., Крундель В. X. и др., Пласт, массы, 1972, № 1, с. 19. — 208. Norimas а Okui, Tori Kawai. Makromol. Chem., 1972, v. 154, p. 161. — 209. Y a w a-ka Y., Tsuchihara T., Tanaka N. e. a. Kobunshi Kagaku, 1971, v. 28, № 314, p. 459, —210. Keller F. Plaste u. Kaut., 1971, Bd. 18, № 9, S. 657.
211. Sobottka J., Trettin R. Plaste u. Kaut., 1971, Bd. 18, № 9, S. 586. — 212. Barlow A., Young M. J. Appl. Polymer Sci., 1970, v. 14, Ns 7, p. 1731. — 213. Wu Ting Kai. Macromolecules, 1970, v. 3, № 5, p. 610. — 214. Wu Ting Kai. J. Polymer Sci., 1970, v. 8. Ser. A2, № 1, p. 167. — 215. Saito Mitsutaka, Tada Hideo, Kos aka Yujiro. J. Polymer Sci., 1970, v. 8, Ser. Al, Ns 9, p. 2555. — 216. Johnsen U., Nachtrab G., Zachmann H. G. Koll. Z. u. Z. Polymere, 1970, Bd. 240, Ns 1—2; S. 756. — 217. Кироки Хитоси, Мидзумо Тоёхиро, Сэт-т я к у. Adhesion and Adhesives, 1969, v. 13, № 11, p. 708. — 218. Косака Ю., Ко б у нс и, High Polymers, Japan, 1969, v. 18, Ns 5, p. 310; Ns 6, p. 386.— 219. Crawford T. E., Gray D. N. J. Appl. Polymer Sci., 1971v. 15, Ns 7, p. 1881.
Глава III
Привитые и блоксополимеры «-олефинов Привитые сополимеры
Важным методом модифицирования полиолефинов является прививка различных мономеров к макроцепям полиолефинов [1, 2, 64, 78].
Привитые сополимеры в зависимости от химического строения звеньев привитых цепей могут обладать различными специфическими свойствами: повышенными адгезией и теплостойкостью, способностью к ионному обмену и др.
Прививку можно проводить либо в массе полимера, находящегося, например, в состоянии суспензии, либо к поверхностному слою изделия (волокна, пленки и т. п.). В любом случае необходимое условие прививки заключается в создании в макромолекулах полиолефина активных центров, способных инициировать рост привитых ветвей.
К основным методам активации поли-а-олефинов относятся предварительное окисление с образованием гидроперекисных групп и радиационное воздействие на полимер. Для получения привитых сополимеров полиолефинов можно использовать также реакции передачи цепи и фотохимическое инициирование.
Получение привитых сополимеров с предварительным окислением полиолефинов. Окисление полиолефинов с образованием гидроперекисных групп позволяет при последующем термическом или ином разложении этих групп использовать образующиеся свободные радикалы как инициаторы роста привитых цепей.
Достаточной для последующей прививки концентрации гидроперекисных групп удается достичь в относительно мягких условиях при окислении полиолефинов, содержащих третичные углеродные атомы. В первую очередь это относится к полипропилену, суммарную схему образования гидроперекисных групп (гидропероксидации) в котором можно представить следующим образом:
О—ОН о2 |
—сн2—сн—сн,—сн— —>•---------СН2—С—СНг-СН----
II II
СН, СНз СН3 СН3
50
Окисляющим агентом может служить кислород воздуха или
озон. Натта с сотрудниками [3] подвергали гидропероксидации атактический полипропилен, растворенный в изопропилбензоле, при 70 °C. Для подавления побочных реакций в раствор добав-
ляли 3—5% метанола. Воздух барботировали через раствор
при атмосферном давлении. Достаточно глубокое окисление в
этих условиях сопровождается сравнительно небольшой деструкцией макроцепей (рис. 24). Скорость образования гидро-
перекисных групп может быть повышена увеличением давления воздуха, использованием вместо воздуха озона и добавкой в раствор перекисей, например, перекиси бензоила.
Гидропероксидация полипропилена в гетерогенных условиях протекает преимущественно в аморфных областях и на поверхностях кристаллических образований [3].
Зависимость содержания гидроперекисных групп от продолжительности окисления изучалась в работе [4] на при-
со
Продолжительность, мин
Рис. 24. Гидропероксидация атактического полипропилена, растворенного в изопропилбензоле (концентрация 15,5%, содержание метанола 4%.
70 °C) [3].
мере атактического полипропилена. Было показано, что содержание гидроперекисных групп после достижения максимума уменьшается вследствие их разложения (рис. 25)
[4; 5, с. 131]. Энергия активации образования гидроперекисных групп составляет 24—25 ккал/моль, энергия активации их распада 27 ккал/моль [4—6].
Прививка к поверхностному слою изделий из полиолефинов,
например полиэтиленовых или полипропиленовых волокон, может проводиться и без гидропероксидации как специальной предварительной операции, если продолжительность формирования изделия и температура расплава обеспечивают образование достаточной концентрации гидроперекисных групп на поверхности изделия [72] .
Реакция разложения гидроперекисных групп бимолекуляр-на и в основном может быть сведена к взаимодействию их с
атомами водорода в первую очередь при третичных углеродных атомах в макроцепях [7, с. 5]:
ROOH + RH —> r6 + r + h»o
Оба типа образующихся макрорадикалов (RO и R) способны инициировать привитую сополимеризацию. При изучении
51
прививки метилметакрилата к окисленному полипропилену было показано [8], что примерно половина привитых цепей присоединена к основным через атом кислорода, остальные привитые ветви связаны с главными цепями связью углерод — углерод. Это было установлено отщеплением полиметилметакрилата, привитого через атом кислорода, с помощью иодисто-водородной кислоты и определением убыли в весе.
Продолжительность, ч
Рис 25. Зависимость содержания гидроперекисных групп от продолжительности окисления атактического полипропилена при различных температурах [4, 5].
1 100
I 20
§ 10 0,001 0,01
Концентрация гидроперекисных групп, моль /кг полипропилена
Рис. 26. Зависимость скорости прививки акрилонитрила к полипропилену от концентрации гидроперекисных групп (температура ПО °C, давление паров акрилонитрила
2,34 кгс/см2) [9].
Процесс прививки можно осуществлять в блоке, растворе и из газовой фазы. Основными параметрами, определяющими скорость прививки, являются степень предварительного окисления полиолефина, температура, концентрация мономера и продолжительность процесса.
При исследовании прививки полиакрилонитрила на полипропиленовое волокно было найдено [9], что скорость прививки пропорциональна концентрации гидроперекисных групп в первой степени (рис. 26). Отклонение от обычного для свободнорадикальной полимеризации порядка по инициатору, равного 0,5, связано, по-видимому, с малой вероятностью бимолекулярного обрыва привитых цепей из-за ограниченной подвижности макрорадикалов в условиях гетерогенного процесса [10, с. 242].
С повышением температуры скорость разложения гидроперекисных групп возрастает и скорость привитой сополимеризации, естественно, увеличивается (рис. 27) [11]. Вместе с тем температура может оказать влияние не только на скорость
Рис. 27. Зависимость количества привитого полиакриламида к сополимеру этилена с пропиленом от температуры (продолжительность реакции 3 ч, содержание пропилена в исходном сополимере 7 мол. %, концентрация гидроперекисных групп 0,1 вес. %) [14-
Полимеризация, иници-
процесса, но и на структуру образующегося продукта. Обычно прививка протекает в аморфных участках и глубокого проникновения привитых цепей внутрь кристаллических областей не происходит [12]. Однако при температурах, достаточных для повышения подвижности элементов кристаллической структуры, прививка возможна не только в аморфных, но и в кристаллических областях. Полученные в таких условиях привитые сополимеры отличаются по характеру кристаллических образований. Это было показано на примере продуктов прививки полиакриламида к сополимеру этилена с 7 мол.% пропилена [11]. Продукт прививки, проведенной в гетерогенных условиях при 130 °C, т. е. выше интервала плавления исходного сополимера (116—120°C), отличался меньшей упорядоченностью, чем продукт прививки при более низкой температуре (100 °C).
Распад гидроперекиси может протекать не только по бимолекулярному механизму, но и по мономолекуляр-ному:
ROOH —► RO + OH
Образующиеся при этом свободные радикалы ОН наряду с полимерными свободными - радикалами способны инициировать полимеризацию мономера, в присутствии которого происходит разложение гидроперекисных групп,
ированная радикалами ОН, приводит к образованию гомополимера. К этому же ведет полимеризация на активных центрах, образованных передачей цепи на мономер или растворитель. Гомополимеризация, вызванная радикалами ОН, в значительной мере исключается с помощью окислительно-восстановительных систем, так как полимеризация в этих условиях инициируется только макрорадикалами:
Fe2’
---CH2—СН—СН2------> СН2—СН—СН2------1-OH’ + Fe3+
ООН О
м ---сн2—сн—сн2— —> —сн2-сн—сн2------- I I
О О—м—м—м-----
•
S3
Образование гомополимера было успешно предотвращено, например, в процессе прививки полиакриловой кислоты к полипропилену в присутствии сульфата железа [13]. В качестве метода подавления гомополимеризации на активных центрах, образованных передачей цепи на мономер или растворитель, значительные перспективы имеет процесс прививки мономеров из газовой фазы [10, с. 244; 14; 15, с. 131; 16, с. 181], поскольку скорость реакций передачи цепи в таких условиях очень мала.
Реакции передачи цепи, в частности на полимер, можно использовать для получения привитых сополимеров и без предварительного окисления. Если осуществлять полимеризацию какого-либо мономера под влиянием свободнорадикального инициатора в присутствии полиолефина, например полипропилена, то в результате актов передачи цепи на полимер образуются макрорадикалы:
---СН2—СН 4----СН—СН2-------------- —> ----------СН2—СН2 4-С—СН2- II-------------------------------------------------------II
X СН3 X сн3
Прививка протекает вследствие инициирования полимеризации макрорадикалом:
СН2—СН—X
I
---СН2—С-----|-СН2=СН —> —сн2—с— I------------I I
СН3 X СНз
Понятно, что продукт представляет собою смесь привитого сополимера и гомополимера. Этим методом к полиэтилену прививали поливинилацетат, поливинилхлорид, полистирол, по-ливинилформамид [17].
Получение привитых сополимеров с помощью радиационного воздействия. Активацию полимера для последующей прививки можно осуществлять радиационным воздействием. В качестве источников излучения используются чаще всего ускорители электронов и 60Со.
Характер активных центров, вызывающих привитую полимеризацию, зависит от условий облучения полимера. Радиационное воздействие на полимер в вакууме или -в среде инертного газа сопровождается распадом ковалентных связей (например, С—Н) и образованием свободных радикалов:
---СН2—СН2—СН2-------—>----СН2—СН—СН2-----
Часть свободных радикалов рекомбинирует с образованием поперечных связей между цепями:
---СН2—СН—СН2----- ----СН2-П1—СН2----
. + — I
---СН2—СН—СН2----- ----сн2—сн—сн2----
S4
Другая часть, так называемые «захваченные» свободные радикалы, сохраняется определенное время в зависимости от условий облучения и последующего хранения полимера и от его структуры. Большей продолжительности существования захваченных радикалов способствует отсутствие кислорода в среде, используемой для облучения и последующего хранения полимера. Пониженные температуры также позволяют увеличить время «жизни» свободных радикалов. Глубокое охлаждение может позволить сохранить некоторую концентрацию захваченных радикалов в течение многих суток даже при нахождении облученного полимера в воздушной среде.
Рис. 28. Зависимость интенсивности сигнала ЭПР сополимеров этилена от продолжительности выдержки на воздухе при температурах 25 °C (а). 45 °C (б) и 70 °C (в) после облучения (облучение ускоренными электронами 2,2 МэВ, 70 Мрад, в среде азота) [18]:
/ — сополимер этилена с пропиленом, степень кристалличности 75%, 2,2 СН3-групп на 100 атомов С; 2— сополимер этилена с а-бутиленом, степень кристалличности 66%,.
2,5 СН5-групп на 100 атомов С.
Концентрация свободных радикалов в значительной мере зависит от степени кристалличности полиолефина. Это связано, очевидно, с тем, что захваченные радикалы накапливаются преимущественно в кристаллических областях. Возможности для рекомбинации в кристаллических областях меньше, чем в аморфных, вследствие ограниченной подвижности участков макроцепей, входящих в кристаллические образования. На рис. 28 представлены зависимости интенсивностей сигналов ЭПР, характеризующих концентрацию свободных радикалов в сополимерах этилена, от продолжительности выдержки на воздухе при различных температурах после облучения ускоренными электронами в инертной среде. Концентрация свободных радикалов непосредственно после облучения и при последующей экспозиции на воздухе выше у сополимера с большей кристалличностью.
55
О преобладании захваченных свободных макрорадикалов в кристаллических областях свидетельствуют также ИК-спект-ры сополимеров этилена с различной степенью кристалличности, подвергнутых облучению и последующей выдержке на воздухе при комнатной температуре [18]. Интенсивность полосы поглощения 1710 см-1, соответствующей образующимся при взаимодействии кислорода с захваченными радикалами карбонильным группам, достигает большей величины у более кри сталличных продуктов (рис. 29). Свободные макрорадикалы
Рис. 29. Зависимость интенсивности полосы 1710 см-1 сополимеров этилена от продолжительности выдержки на воздухе при комнатной температуре после облучения (облучение ускоренными электронами 2,2 МэВ, 70 Мрад, в среде азота) [18]:
1 — сополимер этилена с пропиленом, степень кристалличности 75%, 2,2 СН3-групп на 100 атомов С; 2—сополимер этилена с пропиленом,степень кристалличности 69%, 3,6 СНа-групп на 100 атомоз С; 3— сополимер этилена с а-бутиленом, степень кристалличности 61%, 2,5 СН*-групп на 100 атомов С.
могут служить активными центрами, вызывающими привитую сополимеризацию непосредственно в процессе облучения в среде мономера.
По другому варианту облучение проводится в отсутствие мономера и привитая полимеризация осуществляется с использованием пост-эффекта на захваченных свободных радикалах. В обоих случаях при облучении процесс сопровождается сшиванием полиолефина, однако степень сшивания обычно невелика, так как для активации привитой сополимеризации достаточны сравнительно незначительные дозы облучения. Основные реакции, протекающие при облучении полиэтилена, могут быть представлены следующей схемой [19].
Образование макрорадикалов и атомарного водорода:
Кл
RH R+H
Образование макрорадикалов и молекулярного водорода:
. Кг
RH + H ---> R + H2
Образование виниленовых групп:
R + R R_ 4“ RH
Взаимодействие виниленовых групп с макрорадикалами:
Kt
R= + R ---> RR
Образование поперечных связей:
. Ks
RR + RH --> RRH + R
Здесь У — фактор, связанный с интенсивностью облучения и природой первичного акта; — макромолекула с виниленовыми группами.
В условиях квазистационарного состояния
d [R] d [Н] . d [RR]
dt dt dt
= Ki [RH] Y + Кг [RH] [H] - 2K3 [R]2 - Кд [RJ [R1 + Ks [RR] [RH] = 0 at
= K, [RH] Y — R2 [RH] [H] = 0
= Ki [RJ [R] - K5 [RR] [RH] = 0
Отсюда
\ Лз /
Отметим, что эта кинетическая схема отражает процесс в весьма упрощенном виде. В частности, ею предусматривается лишь бимолекулярный характер реакции, ведущей к образованию виниленовых групп:
---СН2—СН—СН2— —сн2—сн2—сн2- + —► +
---СН2—СН—СН2----- ----СН2—СН=СН—сн2—
Можно было бы предположить, что вероятность таких взаимодействий между мигрирующими свободными радикалами в аморфных областях выше, чем в кристаллических, вследствие различной подвижности участков цепей в областях с разной упорядоченностью. Однако содержание виниленовых групп не зависит от степени кристалличности полиолефинов и определяется лишь поглощенной дозой. Это видно из рис. 30, где представлена зависимость интенсивности полосы поглощения 965 см-1,
57
соответствующей rpawc-виниленовой группе, от поглощенной дозы для полиолефинов с различной степенью кристалличности. Этот факт, так же как и данные Лоутона с соавторами [20] о независимости содержания транс-виниленовых групп от физического состояния полиэтилена (в интервале температур от —150 до +150 °C), позволяет предположить, что основной механизм образования транс-виниленовых групп не связан с межцепными взаимодействиями. транс-Виниленовые группы могут возникать, очевидно, в участках макроцепей, содержащих неспаренные электроны в двух соседних атомах углерода:
—сн2—сн—сн—сн2— —> —сн2—сн=сн—сн2--------
Свободнорадикальное состояние соседних атомов углерода может быть результатом миграции двух свободных радикалов
Поглощенная дога, Мрад
Рис. 30. Зависимость интенсивности полосы 965 см-1 от поглощенной дозы при облучении ускоренными электронами (2 МэВ) [18]:
□ — полиэтилен низкого давления, степень кристалличности 83%; С? — полиэтилен низкого давления, степень кристалличности 78%; Д— сополимер этилена с пропиленом, степень кристалличности 75%; X — сополимер этилена с пропиленом, степень кристалличности 69%; ф— сополимер эти* лена с а-бутиленом, степень кристалличности 61%.
по длине макроцепи или, что менее вероятно, разрыва свя-зей С—Н у двух соседних атомов углерода при облучении.
Зависимость содержания макрорадикалов от поглощенной дозы и продолжительности облучения выражается уравнением [21]:
[R]=/GZr(l
где Ki — константа скорости образования макрорадикалов; I — доза облучения; т — продолжительность существования свободных радикалов; t— продолжительность облучения.
При большой продолжительности облучения это уравнение упрощается:
Рассмотрение прививки как обычной реакции цепной поли-
меризации без учета особенностей, связанных с гетерогенным характером процесса [10, с. 235], приводит к выражению, аналогичному уравнению скорости реакции свободнорадикальной
полимеризации:
И = И-^У/2[М]Ш+2 \ Аз /
где Ki — константа скорости инициирования; /С2—константа скорости роста привитых цепей; Кз — константа скорости обрыва цепей; [М] — концентрация мономера; [R] — концентрация макрорадикалов.
58
Прививка с использованием пост-эффекта, инициируемая захваченными свободными радикалами, выгодно отличается от прививки при совместном облучении полимера и мономера. Это отличие заключается в практически полном отсутствии гомополимеризации в первом случае. Образование гомополимера может быть, однако, исключено также и в случае прививки по второму варианту. Это возможно, если облучение полиолефина проводится в парах мономера [14—16].
Привитая радиационная сополимеризация из газовой фазы к полиэтилену и полипропилену описана для ряда мономеров: акрилонитрила, акриловой кислоты, винилацетата, стирола, метилметакрилата и др.
Подавлению гомополимеризации способствует невысокое давление паров прививаемого мономера. Вместе с тем понижение концентрации мономера, уменьшая скорость гомополимеризации, одновременно снижает и скорость прививки. Есть данные [65, 99], полученные при радиационно-химической прививке из газовой фазы поливинилхлорида на полиэтиленовую пленку, о возможности полного подавления гомополимеризации без снижения выхода привитого полимера, если прививку проводить в электрическом поле напряженностью 1000 В/см.
Экспериментально показано, что основными факторами, определяющими степень прививки из газовой фазы, инициируемой захваченными радикалами, являются доза облучения, величина поверхности образца полимерного материала, продолжительность контакта с мономером, давление паров мономера, температура при контакте паров и облученного полимера [22]. Скорость привитой сополимеризации, инициируемой макрорадикалами, пропорциональна дозе облучения в степени 0,5. Это было показано на примере прививки полистирола на полипропиленовое волокно [21]. Такая связь скорости привитой полимеризации и дозы облучения, как и половинный порядок по инициатору для обычных реакций свободнорадикальной полимеризации, свидетельствует о бимолекулярном механизме обрыва растущих привитых цепей.
При значительных степенях прививки пропорциональность скорости реакции дозе облучения в степени 0,5 может нарушаться в результате образования гомополимера, затрудняющего диффузию мономера к полимеру, к которому ведется прививка [66].
Скорость прививки при совместном облучении полимера и мономера может быть увеличена разбавлением мономера растворителем. Так, разбавление стирола или метилакрилата метанолом позволяет значительно увеличить скорость прививки к полиэтилену и полипропилену (табл. 13) [23, 24].
Повышение скорости прививки в этих условиях объясняется двумя причинами. Одна из них связана с гель-эффектом.
59
Таблица 13
Зависимость скорости привитой полимеризации от разбавления мономеров метанолом [24]
Полиолефин Мономер Содержание мономера, объемн. % Мощность ДОЗЫ, Мрад/ч Степень прививки, %/Мрад
Полиэтилен высокого Стирол 100 0.070 61
давления 50 0,070 117
Полиэтилен низкого да- Стирол 100 0,081 127
вления 50 0,081 414
Метил- 100 0,061 82
акрилат 50 0,061 124
Полипропилен Стирол 100 0,080 84
50 0,080 319
Метил- 100 0,021 119
акрилат 50 0.021 134
обусловленным уменьшением растворимости привитых цепей в системе мономер — полимер, другая заключается в увеличении скорости диффузии мономера в полимер [23, 68].
Возможность повышения скорости прививки из газовой фазы разбавлением паров мономера инертным газом показана в работе [25]. Авторы исследовали парофазную привитую сополимеризацию с полиэтиленом низкой плотности, предварительно подвергнутым у-облучению на воздухе. При относительно малом давлении паров стирола, когда скорость реакции существенно зависит от давления и определяется диффузией паров мономера в полимер, разбавление паров стирола аргоном уменьшало скорость реакции. Но при более высоком давлении паров стирола, когда скорость реакции мало зависит от давления и значительно больше от температуры, добавление аргона к парам мономера сопровождалось увеличением скорости процесса. Добавление аргона уменьшает концентрацию мономера в полиэтилене, поэтому повышение скорости реакции, очевидно, можно объяснить только гель-эффектом, так как прививка протекает в очень вязкой среде.
Повышение температуры привитой сополимеризации может сопровождаться уменьшением скорости реакции. Подобный эффект, наблюдавшийся [26] при получении продуктов прививки стирола на пленки полиэтилена при температурах от 0 до 53 °C, очевидно, связан с ростом скорости рекомбинации захваченных свободных радикалов при повышении температуры. Характер влияния температуры на скорость прививки зависит от степени кристалличности полиолефина. Скорость прививки к полиэтилену с высокой степенью кристалличности значительно возрастает с повышением температуры, в то же время изменение температуры в случае прививки к более аморфному полиэтилену существенного влияния не оказывает. Это объясняется тем, что большая часть захваченных свободных радикалов находится в кристаллических областях, диффузия мономера в кото-
60
рые возрастает по мере приближения температуры к интервалу плавления кристаллических образований. ✓
Облучение полиолефинов на воздухе также позволяет акта-вировять их для послепуюиIрй прививки. Воздействие ионизирующего излучения на полимер в присутствии кислорода воз-духа сопровождается образованием перекисных и гидроперекисных групп. При комнатной температуре эти группы устойчивы, при повышенных же температурах разлагаются с образованием макрорадикалов, способных инициировать привитую полимеризацию. Этим методом были получены продукты прививки акрилонитрила, метилметакрилата, ЛСвинилкарбазола к полиэтилену и акрилонитрилу, винилхлорида и винилиденхлорида к полипропилену [26—31, 84].
Концентрация перекисных и гидроперекисных групп, как показано [32, с. 145] на примере низкомолекулярных гомологов полиэтилена (н-гептана и изооктана), пропорциональна дозе облучения. Скорость прививки, однако, не пропорциональна дозе облучения в степени 0,5, как можно ожидать при бимолекулярном характере обрыва цепей. При изучении кинетики прививки акрилонитрила к полиэтилену [28] и акриловой кислоты к полипропилену [33] было найдено, что скорость реакции пропорциональна дозе облучения в первой степени. Это явление связано [28] с гетерогенным характером реакции прививки, когда обрыв растущих привитых цепей по бимолекулярному механизму затруднен из-за их малой подвижности в массе полимера. Скорости образования активных центров и роста цепей не зависят от гетерогенности процесса, степень прививки с увеличением (до определенного предела) дозы предварительного облучения на воздухе в связи с этим возрастает.
Сопоставление радиационной и инициированной перекисью бензоила привитой полимеризации стирола на полиэтиленовую пленку было проведено Ченом и Фридландером [34]. Прививка проводилась тремя методами: у-облучением пленки в жидком мономере при комнатной температуре, (3-облучением пленки на воздухе с последующим погружением ее в мономер с температурой 65 °C и обработкой пленки жидким мономером, содержащим перекись бензоила, при 65°C. Наиболее высокая степень прививки была получена при использовании предварительно облученной на воздухе пленки, минимальная степень прививки наблюдалась в случае инициирования реакции перекисью бензоила.
Радиационно-химическая привитая сополимеризация может протекать и по ионному механизму. Так, в работе [67] получены продукты прививки к полиэтилену мономеров, способных к полимеризации только по ионному механизму: а-метилстирола, винплбутилового эфира, изобутилена. Необходимое условие прививки при совместном облучении полиэтилена (пленки) и мономера заключалось в весьма тщательном предварительном высушивании реагентов.
61
К выводу об ионном механизме радиационно-химической газофазной прививки винилхлорида на полиэтиленовую пленку привело изучение кинетики этого процесса [81, 100]. В пользу этого вывода свидетельствовала, в частности, зависимость скорости реакции прививки от мощности дозы в первой степени, позволившая предположить мономолекулярный характер обрыва растущих привитых цепей. Кроме того, энергия активации процесса, которую определяли в интервале температур от 20 до 80 °C, имела отрицательное значение (—5,4 ккал/моль).
Получение привитых сополимеров фотохимическим методом. Инициирование привитой сополимеризации может осуществляться воздействием ультрафиолетового света на систему полимер — мономер в присутствии фотосенсибилизатора. Сенсибилизатор, поглощая энергию ультрафиолетовой радиации, передает избыточную энергию полимеру, что приводит (вследствие разрыва ковалентных связей, в частности связей С—Н) к образованию макрорадикалов, инициирующих привитую сополимеризацию.
Свободные радикалы, образующиеся при фотолизе сенсибилизатора, инициируют также гомополимеризацию. Образования гомополимера можно практически избежать, если облучению подвергать лишь полимер, содержащий сенсибилизатор, а затем проводить прививку, используя пост-эффект.
Фотохимическим методом на полиэтиленовые пленки прививался полиакриламид с использованием в качестве сенсибилизатора бензофенона [35, 36].
Ивакура и Такеда [37] исследовали прививку виниловых мономеров к полипропилену, используя фотопнициирующую систему бензофенон — соль двухвалентного металла переменной валентности. Добавка ацетата меди или кобальта увеличивает выход продукта прививки. Предполагается, что причина этого явления заключается в окислении семихинонного радикала бензофенона до бензофенона в присутствии соли.
В серии работ [102—106] проводили фотохимическую привитую сополимеризацию из газовой фазы виниловых мономеров (акрилонитрила, акриловой кислоты, винилизоцианата) на полиэтиленовые пленки, используя в качестве фотосенсибилизатора бензофенон. Как и при радиационной привитой сополимеризации из газовой фазы, процесс практически не сопровождался образованием гомополимера. Было установлено [102], что фотохимическая привитая сополимеризация, так же как и радиационная [15], протекает по радикальному механизму.
Интересной особенностью фотохимической привитой сополимеризации является увеличение порядка реакции до единицы по интенсивности облучения при уменьшении интенсивности УФ-света. Зависимость порядка реакции от интенсивности света объясняется [102, 103] тем, что с увеличением интенсивности УФ-облучения возрастает вероятность рекомбинации макрора-62
дикалов и устанавливается некоторая предельная их концентрация. Такое объяснение было подтверждено при проведении фотосенсибилизированной бензофеноном привитой пост-сополи-меризации акриловой кислоты. Содержащую бензофенон полиэтиленовую пленку облучали УФ-светом [/, 320 им, интен-
сивность 10"® Эйнштейн/(см2-с)], а затем в систему подавали пары мономера. Предельная концентрация макрорадикалов устанавливалась за 20 с облучения. Более продолжительное воздействие УФ-света не увеличивало скорости привитой пост
сополимеризации.
Для начальных линейных участков кинетических кривых скорость привитой сополимеризации акриловой кислоты с поли-
этиленом не зависела от интенсивности света в широком интервале интенсивностей [10-6—10-8 Эйнштейн/(см2-с)]. Этот факт позволил предположить [108], что инициирующие привитую сополимеризацию макрорадикалы полиэтилена образуются не во всем объеме полимера, а в дефектных областях поверхностного слоя полиэтиленовой пленки. В этих областях из-за высокой концентрации макрорадикалов быстро достигается стационарное состояние. Такое предположение было подтверждено электронно-микроскопическими снимками поверхности полиэтиленовой пленки, на которой осуществлялась привитая сополимеризация акриловой кислоты.
Если принять для привитой сополимеризации виниловых мономеров, протекающей по радикальному механизму, бимолекулярный обрыв цепей,
Продолжительность, мин
Рис. 31. Зависимость количества привитого поливинилизоцианата (в %) от продолжительности фото-сенсибилизированиой бензофеноном привитой сополимеризации вииилизоцианата к полиэтилену [106]:
1 — облучение источником с А = = 313 нм; 2 — облучение источ* никами с Л=313 и > 436 нм.
то порядок реакции по интенсивности, равный единице, свидетельствует о пропорциональности скоро-сти образования макрорадикалов квадрату интенсивности света и, следовательно, о двухквантовом механизме реакции. Участие двух квантов света при образовании макрорадикала было подтверждено опытами с двумя источниками света. Привитая сополимеризация акриловой кислоты [104] и винилизо-цианата [106] с полиэтиленом осуществлялась в присутствии бензофенона под влиянием света с А = 313 нм. При подключении второго источника, испускающего свет с А > 436 нм, скорость реакции возрастает (рис. 31), хотя действие самого света с А > 436 нм не вызывало полимеризацию и не продолжало ее после отключения источника света сХ = 313 нм.
Эти результаты позволили предположить триплет-триплет-ное поглощение [107, с. 226] квантов света бензофеноном с
63
последующей передачей энергии на полимер, приводящей к разрыву связей С—Н [102]:
БФСо + Av <=>: БФТ
БФТ + hv БФТ»
БФГ.+RH —> БФ + R + H
Н + RH —> R + Н2
где БФС., БФТ — молекула бензофенона в синглетном и триплетном состоянии соответственно.
Возможность последовательного поглощения двух квантов света обусловлена специфическими особенностями протекания фотохимических реакций в полимерной матрице, жесткость которой термозит дезактивацию возбужденных состояний.
В работе [69] проводилась инициированная УФ-светом привитая сополимеризация метилметакрилата и других виниловых мономеров к хлорированному изотактическому полипропилену. Предполагается, что привитая сополимеризация проходит по двум механизмам. Один из них заключается в прививке на макрорадикалах, образовавшихся при разрыве связей С—С, другой — в прививке, инициированной гидроперекисями, образующимися на третичных атомах углерода в аморфных областях полимера. Привитая сополимеризация сопровождалась побочными реакциями дегидрохлорирования, дехлорирования и сшивания.
Структура и свойства привитых сополимеров. В продуктах прививки к полиолефинам структурная упорядоченность, характерная для данного полиолефина, сохраняется даже при высоких степенях прививки, поскольку эта реакция обычно протекает в основном в аморфных областях и на поверхности кристаллических образований. Такая прививка сообщает неоднородность привитым сополимерам и обусловливает зависимость хода процесса привитой полимеризации от надмолекулярной структуры полимера. Геледжи и Одор [38], изучая прививку метилметакрилата на полипропиленовые волокна и пленки, подвергнутые предварительному у-облучению на воздухе, пришли к выводу, что характер надмолекулярной структуры полимера оказывает влияние на процессы рекомбинации, продолжительность существования свободных радикалов и образование перекисных и гидроперекисных групп.
От надмолекулярной структуры зависит скорость рекомбинации свободных радикалов с атомами водорода, так как диффузия водорода в материале усиливается с увеличением расстояний между макромолекулами. Кристаллическая структура полимера затрудняет диффузию мономера и способствует стабилизации радикалов до их взаимодействия с реакционной средой. Образование перекисных и гидроперекисных групп также связано с надмолекулярной структурой, поскольку об-
64
условлено диффузией кислорода к свободным радикалам, а полимеры с пониженной степенью кристалличности содержат большее количество кислорода, чем высококристаллические полимеры. Этими обстоятельствами авторы объясняют наблюдавшийся ими интересный факт пониженной скорости и степени прививки на ориентированное полипропиленовое волокно по сравнению со скоростью и степенью прививки на неориентированное волокно.
О гетерогенности привитых сополимеров и влиянии надмолекулярной структуры исходного полимера на ход сополимеризации свидетельствуют данные исследований прививки полимеров стирола, метилметакрилата и винилхлорида к изотактическому полипропилену [3] и стирола, винилацетата и акриловой кислоты к полиэтилену [39—41]. Подобные привитые сополимеры представляют собой смесь исходного кристаллического полимера и собственно привитого сополимера, полученного в результате реакции прививки на поверхности надмолекулярных структур [12, 82, 83]. Такое строение продуктов прививки к полиолефинам проявляется в их свойствах. При нагревании привитых сополимеров происходит плавление кристаллических образований при температурах, соответствующих интервалу плавления основного полимера. Прививка полярных полимеров — полиакрилонитрила, поливинилиденхлорида [42], полиакриламида [11] — может, однако, увеличить теплостойкость и прочность при повышенных температурах.
В работе [70] при изучении продукта прививки полиметилметакрилата к изотактическому полипропилену были обнаружены сферолиты, типичные для исходного полипропилена. Это связано с гетерогенным характером прививки, обусловливающим участие в привитой сополимеризации лишь части полимера. Удаление из продукта реакции изотактического полипропилена позволило установить, что привитой сополимер образует фибриллярные структуры, очевидно, в результате упорядочения основных цепей привитого сополимера.
Отличия в структуре привитых сополимеров в зависимости от способа сополимеризации показаны [74] с помощью метода радиотермолюминесценции [75, 76] на примере продуктов прививки полистирола и полиакрилонитрила к полиэтилену низкой плотности. Использование пост-эффекта позволяет получать продукт прививки со сравнительно равномерным распределением привитого сополимера в полиэтилене. Это объясняется тем, что радикалы, образовавшиеся в аморфных областях полиэтилена, быстро гибнут после окончания облучения. Молекулы мономера благодаря этому имеют возможность диффундировать в глубь облученного полимера, и прививка происходит на доступных для молекул мономера поверхностях кристаллических структур и в «пограничных» областях, где имеются условия для стабилизации свободных радикалов. В случае одновременного облучения полимера и мономера и при химически
3 А. Г. Сирота
65
инициированной прививке высокая концентрация свободных радикалов, инициирующих привитую сополимеризацию в аморфных областях, препятствует диффузии мономера во внутренние слои полимера. По мере уменьшения интенсивности облучения концентрация свободных радикалов снижается, возможность проникновения молекул мономера в глубь полимера возрастает, соответственно увеличивается и равномерность распределения привитого сополимера в полиэтилене.
Прививка позволяет значительно повысить адгезию полиолефинов к различным материалам. Содержание около 4 мол. % полиакриламида в виде ветвей, привитых к сополимеру этилена с пропиленом, почти в 5 раз по сравнению с исходным сополимером увеличивает его адгезию к алюминиевой фольге [И]. Поверхностной прививкой к полиэтилену, например, винилпирро-лидона улучшается восприимчивость полиэтилена к печатным краскам. Введение в состав полиолефинов функциональных групп, способных к взаимодействию с основными или кислотными красителями путем прививки полимеров акриловой и метакриловой кислот, акрилонитрила, акриламида, винилпиридина и других полимеров увеличивает сродство полимера к красителям. Для необходимого повышения способности к окрашиванию требуется небольшая степень прививки (4—8 вес. %), при которой механические свойства полимера существенно не изменяются [10, с. 257; 80].
Продукты прививки ряда полисилоксановых соединений к полиэтилену [100, 101] отличаются повышенной термостабильностью к действиям коронного разряда и длительных механических нагрузок.
Полиэтиленовые волокна с привитым слоем полиакриловой кислоты сохраняют прочностные характеристики при температурах, превышающих температуру плавления внутреннего слоя. Расплавленный полиэтиленовый слой сохраняет исходную ориентацию и после охлаждения восстанавливает свою первоначальную структуру и прочность [73].
Фотохимическая прививка 6—8% полиакриловой кислоты на полиэтиленовую пленку улучшает прочностные характеристики пленки на 50—60%, повышает ее адгезию и сообщает пленке способность к термоусадке [103]. Отличительной особенностью полиэтиленовых пленок с привитым поливинилизоцианатом является повышенная стойкость к термоокислительной деструкции. Как полагают [106], стабилизирующий эффект обусловлен продуктами термолиза привитого поливинилизоцианата, • представляющими собой циклические структуры с системой сопряженных связей.
Привитые сополимеры полистирола с полиолефинами (полиэтиленом, сополимерами этилена с пропиленом) после сульфирования или аминирования привитых цепей полистирола обладают ионообменными свойствами [43, с. 621; 44; 45, с. 179; 46, с. 501]. Обменная емкость таких катионитов и анионитов состав
66
ляет около 4 мг-экв/г. Ионообменные мембраны, полученные прививкой на полиэтиленовую пленку полистирола с последующим фосфонированием, обладают обменной емкостью до 5,5 мг-экв/г [45, с. 179].
Среди специфических свойств, проявляющихся у полиолефинов в результате прививки, отметим пониженную газо- и паро-проницаемость. Прививка небольших количеств полистирола, полиакрилонитрила на полиэтиленовую пленку сопровождается увеличением плотности упаковки в аморфных областях, что и приводит к уменьшению проницаемости пленки. При больших степенях прививки проницаемость возрастает, очевидно, вследствие аморфизации полимера [1, с. 162].
Радиационная привитая сополимеризация из газовой фазы на вытянутых полиолефиновых волокнах и пленках, протекающая в адсорбционном слое, благодаря матричному влиянию структуры ориентированного полимера приводит к образованию привитого слоя в ориентированном состоянии [14, 73]. Этот эффект наблюдался при полимеризации акрилонитрила, винил-иденхлорида, винилхлорида на вытянутых полиэтиленовых и полипропиленовых пленках и волокнах. Привитой слой может быть подвергнут химическим превращениям, например дегидрохлорированию, без нарушения ориентации.
Радиационная сополимеризация акриловой кислоты из газовой фазы на вытянутой полиэтиленовой подложке приводит к образованию привитой полиакриловой кислоты изотактического строения [109]. Показана также возможность получения методом радиационной газофазной сополимеризации на полиэтиленовой подложке привитого стереорегулярного полиакрилонитрила [110].
Таким образом, установлено, что при радиационной привитой сополимеризации на вытянутых полиэтиленовых пленках подложка обладает матричным влиянием, проявляющимся в ориентировании молекул мономера при их присоединении к растущей цепи и в задании геометрического направления роста привитых цепей. Возможность использования полимерной подложки в качестве матрицы, определяющей формирование привитого полимера, была показана и для фотохимической привитой сополимеризации [108].
Ориентирующее влияние полимерной подложки может проявляться и в случае прививки из жидкой фазы. Этот вывод был сделан в результате исследования радиационной прививки гли-цидилметакрилата на полиэтиленовую пленку по двум вариантам: с предварительным облучением пленки и при совместном ее облучении с мономером [77]. Ориентация в привитом слое объясняется протеканием прививки в микроканалах (межфибриллярных каналах) полиэтилена.
Значительный интерес представляют многослойные материалы (пемосоры), получаемые, в частности, методом газофазной привитой сополимеризации [71]. Описана двухкратная
3* 67
последовательная прививка на вытянутых полиэтиленовых и полипропиленовых волокнах и пленках нескольких пар мономеров: акрилонитрил — винилиденхлорид, винилиденхлорид — акриловая кислота, винилхлорид— винилиденхлорид, стирол — винилиденхлорид, акриловая кислота — акрилонитрил и др. Были получены волокна и пленки трехслойной структуры. В случаях, когда первый привитой слой обладал высокоориентированной структурой (поливинилиденхлорид, полиакрилонитрил, полиакриловая кислота, поливинилхлорид), наружный привитой слой также имел высокую ориентацию. Предполагается, что ориентированные структуры образуются при привитой сополимеризации из газовой фазы на вытянутых полимерных подложках благодаря тому, что начальная стадия прививки протекает в межфибриллярных каналах подложки, а на следующих стадиях реакции привитые цепи растут на поверхности фибриллярных структур «своего» полимера. При синтезе пемосоров образование третьего (внешнего) слоя начинается вероятно также в межфибриллярных каналах второго (промежуточного) слоя и подчиняется общим закономерностям прививки из газовой фазы на ориентированную подложку.
Блоксополимеры
Блоксополимеры различных типов могут быть получены либо в процессе полимеризации а-олефинов, либо модифицированием полимерных продуктов.
В первом случае блоксополимеры а-олефинов получают [47—51; 63, с. 129; 85] с применением каталитических систем, сохраняющих свою активность длительное время. При этом используют значительную продолжительность «жизни» растущих цепей, достигающую многих часов. В качестве таких катализаторов описаны системы TiCU — LiAlR2, VOC13 — A1R2C1 [50]. Для синтеза блоксополимеров сначала полимеризуют, например, этилен, образующий гомополимерный блок, затем, после удаления этилена, в сферу реакции вводят другой мономер, например пропилен или а-бутилен, или смесь мономеров. Продолжая подобные операции, можно получить макромолекулярные цепи, состоящие из чередующихся разных гомополимерных блоков или гомополимерных и сополимерных блоков. Возможен также синтез цепей, состоящих из сополимерных блоков разного состава. Характер продукта последовательной полимеризации в значительной мере зависит от продолжительности жизни растущих макроцепей. Если эта величина достаточно большая, получаются блоксополимеры, если малая — образуется смесь гомополимеров, в промежуточном случае — смесь блоксополиме-ра и гомополимеров [52].
Синтез блоксополимеров на катализаторах Циглера — Натта возможен и при одновременном присутствии в сфере реакции таких сомономеров, как пропилен и а-бутилен, пропилен и сти
68
рол, 4-метилгексен-1 и стирол, 5-метилгексен-1 и стирол [86— 88]. Продукты сополимеризации таких пар мономеров представляют собою блоксополимеры, в отличие от сополимеров этилена и пропилена, этилена и а-бутилена и др. (см. гл. II), для которых характерно хаотическое расположение звеньев в цепях [88, 89]. Различия в характере распределения звеньев для разных пар мономеров в работе [87] связываются с размерами заместителей в молекулах олефинов. При сополимеризации мономеров, у которых заместители при двойной связи невелики или отсутствуют (например, при сополимеризации этилена с пропиленом), произведение констант сополимеризации г\-г2 близко к единице [89—90]; блоки из мономерных звеньев каждого типа в цепях таких сополимеров имеют небольшую среднюю длину, а для самих сополимеров (в определенном интервале составов) характерна аморфная структура. В случае сополимеризации олефинов с крупными заместителями [88] значения произведения Г\-г2 обычно превышают единицу; однотипные звенья в цепях образуют длинные блоки; на рентгеновских дифракционных кривых сополимеров наблюдаются пики, соответствующие кристаллическим образованиям обоих гомополимеров или одного из них.
Образование блоксополимеров этилена или пропилена с ацетиленом [94—96] и этилена с фенилацетиленом [97] в присутствии комплексных катализаторов происходит, как полагают [97], благодаря преимущественной адсорбции на активных центрах одного из имеющихся в сфере реакции мономеров. Адсорбция молекул ацетилена активными центрами и вытеснение этилена приводят к образованию полиацетиленовых блоков, а после израсходования ацетилена образуются блоки из этиленовых звеньев.
Свойства блоксополимеров определяются составом, длиной и микроструктурой отдельных блоков [64, 92]. Табл. 14 содержит сравнительные данные о свойствах линейных полиэтилена, полипропилена, статистического сополимера и блоксополимера пропилена с этиленом, а также смеси полипропилена с полиэтиленом.
Блоксополимеры этилена и пропилена уступают линейным гомополимерам по величинам твердости и прочности (рис. 32), но превосходят их по ударной вязкости. Твердость и прочность блоксополимеров выше, чем у полиэтилена высокого давления [51]. Свойства описанных блоксополимеров объясняются их пониженной кристалличностью в сравнении с кристалличностью гомополимеров.
Высококристаллические блоксополимеры, в частности относящиеся к ним так называемые полиалломеры, — жесткие и прочные материалы. Полиалломеры получают сополимеризацией этилена и пропилена, пропилена и а-бутилена, пропилена и винилхлорида и других пар мономеров [53,54]. Исследование структуры полиалломеров методом ИК-спектроскопии показывает,
Таблица 14
Свойства блоксопопимеров пропилена с этиленом, гомопопимеров и смеси полипропилена и попиэтипена (по данным [51]J
Полимер Содержание растворимой фракции, % Относительная ВИЗ-КОСТЬ Твердость по вдавливанию шарика Ударная вязкость Температура кристаллизации, °C
20 °C о°с -20 °C
Полиэтилен . . . Полипропилен 1 4 3,5 6,9 440 730 9 7 8 2,3 6.5 1,9 127-131 160-165
Продукт статистической сополимеризации * 30,4 6.4 375 23,0 4,6 2,0 150-159
Смесь полипропилена и полиэтилена * . . . 4 6.4 708 7,3 2,8 1,9 157-163
Блоксополимер * 4,2—9,6 5,4-6,8 379—708 16,4—35,2 3,9-24,4 2,3-4,1 155-164
• Весовое соотношение сомономеров (компонентов смеси) пропилен : этилен (поли, пропилен: полиэтилеи)=90: 10.
Прочность на разрыв, кгс/см
Рис. 32. Зависимость прочности при разрыве (/) и твердости (2) блоксо-полимеров этилена с пропиленом от их состава (по данным [51]).
что сегменты их макроцепей, состоящие из звеньев одного из использованных мономеров, участвуют в образовании структур, характерных для соответствующего гомополимера. Степени кристалличности полиалломеров и соответствующих гомополимеров а-олефинов близки в отличие от продуктов статистической сополимеризации, имеющих значительно меньшие степени кристалличности (см. гл. II).
Полиалломеры по сравнению с другими полиолефинами обладают рядом ценных свойств. Полиалломеры пропилена и этилена отличаются от полипропилена более высокими величинами морозостойкости, ударной вязкости, устойчивости к разрастанию надреза. Линейный полиэтилен среднего давления они превосходят по способности к переработке,
показателям температуры размягчения, твердости, ударной вязкости (табл. 15). Полиалломеры характеризуются примерно вдвое меньшей величиной усадки, чем линейный полиэтилен. Важным преимуществом полиалломеров по сравнению с линей-
70
Таблица 15
Свойства попиалпомара пропилена и этилена, полипропилена и попиэтипена среднего давления [S3]
Показатели Полиалломер Полипропилен Полиэтилен Смеси полипропилена с различными количествами полиэтилена
5% 10% 15«
Плотность (после закалки), г/см3 0,906 0.910 0.972 0,913 0,916 0,921
Прочность, кгс/см2 при растяжении . . . 224 329 217 308 308 294
при изгибе 6300 10 000 7070 9400 9330 9100
Относительное удлинение, % >650 360 290 НО 200 150
Твердость по Роквеллу (шкала R) 70 93 54 91 88 83
Ударная вязкость при растяжении, кгс-см/см2 . . 176 70 138 77 83 97
Ударная вязкость по Изоду (с надрезом) при 20°С,-кгс-см/см надреза . . . 10,9 1.7 7,1 3,3 3,3 3,8
Ударная вязкость по Изоду (без надреза) при 23 °C, кг-см/см Не раз- 131 Не разрушается
Теплостойкость по Вика. °C рушается 132 145 122 143 139 139
Морозостойкость, °C . . . -22 +8 <-78 + 1 0 -3
ным полиэтиленом является также высокая стойкость к растрескиванию при воздействии нагрузок и поверхностно-активных веществ [91]. В табл. 16 приведены свойства полиалломеров пропилена с а-бутиленом и другими мономерами. Интересно, что применение в качестве второго мономера а-бутилена не повышает морозостойкости полиалломера, как это происходит при использовании эквивалентных количеств этилена.
Применение стереоспецифических катализаторов в условиях существования «долгоживущих» активных центров позволяет синтезировать блоксополимеры, макромолекулы которых состоят в основном из нерегулярных сополимерных блоков и гомополимерных стереорегулярных блоков. Такие сополимеры, названные стереоблочными, получены сополимеризацией этилена с пропиленом и этилена с а-бутиленом в присутствии катализаторов на основе <x-TiCl3, TiCU VOC13 [50, 57]. Эти продукты отличаются высокой эластичностью и большей растворимостью, чем гомополимеры.
Поведение стереоблочных сополимеров при растяжении отличается от поведения продуктов статистической сополимеризации и смесей гомополимеров того же состава. На рис. 33 представлены кривые растяжения аморфного нерегулярного
71
Таблица 16
Свойства полиалломеров пропилена [52, 53J
Сомономер
Показатели а-бути-лен изопрен стирол тетраме-тилбута-диен винил- хлорид
Плотность (после закалки), г/см3 Прочность, кгс/см2 0,909 0,912 0,918 0,912 0,921
при растяжении . . . 329 366 372 336 364
при изгибе 9 800 11 900 14 000 11 600 14 000
Относительное удлинение, % Твердость по Роквеллу, 225 75 — 250 —
(шкала R) 90 99 102 100 97
Ударная вязкость при растяжении, кгс • см/см2 . . Ударная вязкость по Изоду 121 66 — — —
(с надрезом) при 23 °C, кгс • см/см надреза . . . 3,3 3,3 — — —
Теплостойкость по Вика, °C 142 147 145 145 147
Морозостойкость, °C . . . —4 —2 >22 5 >20
сополимера этилена с пропиленом (кривая /) и стереоблоксопо-лимеров ( кривые 2 и 3) того же состава. Аморфный нерегулярный сополимер ведет себя подобно некристаллизующимся каучукам:
О -200 МО 600 800 1000 1200
Относительное удлинение, %
Рис. 33. Кривые растяжения сополимеров этилена с пропиленом (содержание этилена 50%) [50]:
1 — аморфный атактический сополимер; 2,3 — стереоблоксополимер ы.
имеет большие удлинения при относительно невысоких и почти не влияющих на величину удлинения напряжениях. Стереоблоксополимер, имеющий в исходном состоянии аморфную структуру, при растяжении ведет себя подобно кристаллизующемуся каучуку (кривая 2). Предполагается, что при достижении величины отно-
сительного удлинения 500— 600% происходит некоторая кристаллизация в результате ориентации макроцепей, увеличиваются силы межмолекулярного взаимодейст-
вия и сопротивление материала деформации. Рентгенографически показано, что возни-
кающая при растяжении ориентация полностью исчезает после снятия нагрузки. Кривая 3 характерна для стереоблоксополи-меров, имеющих в исходном состоянии до растяжения неболь
72
шую степень кристалличности (примерно 3—15%). Для сравнения стереоблоксополимеров и смесей гомополимеров отметим, что механические смеси полипропилена и полиэтилена имеют незначительные показатели удлинения, особенно при соотношениях компонентов, близких к 1. При содержании 50% полиэтилена его смеси с полипропиленом имеют удлинение от 50 до 100% [48].
При сополимеризации пропилена с а-бутиленом на высокостереоспецифических каталитических системах, содержащих галогенид алкилалюминия, хлорид трехвалентного титана и гексаметилтриамидфосфат, образуются сополимеры, макромолекулы которых состоят из чередующихся стереорегулярных блоков обоих мономеров [98]. Эти сополимеры обладают кристаллическими структурами, характерными для стереорегулярных полипропилена и поли-а-бутилена. Представление о цепях этих полимеров как о последовательностях стереорегулярных блоков согласуется с тем фактом, что произведение констант сополимеризации г^г2 больше единицы. При возрастании содержания звеньев ос-бутилена в сополимере происходит уменьшение твердости, сопротивления разрыву, жесткости и температуры плавления сополимеров; одновременно повышается ударная вязкость, морозостойкость и прозрачность материала.
К стереоблоксополимерам иногда относят [1, 55] также сте-реоблокгомополимеры — продукты стереоспецифической гомополимеризации пропилена, а-бутилена, пентена-1 [56, 57], а также некоторых диолефинов (бутадиена-1,3, изопрена) [58, 59], в макромолекулах которых чередуются стереорегулярные кристаллизующиеся участки с атактическими некристаллизую-щимися участками. Стереоблокгомополимеры представляют собою фракцию в продукте стереоспецифической полимеризации, отличающуюся по свойствам и от стереорегулярной кристаллической, и от атактической аморфной фракций. Стереоблокгомополимеры характеризуются более низкими по сравнению с изотактическими полимерами степенью кристалличности и температурой плавления. Стереоблокполимеры пропилена с малой степенью кристалличности (15—30%) высокоэластичны и обладают обратимыми относительными удлинениями до 200%. Дальнейшее удлинение сопровождается резким возрастанием прочности, очевидно, вследствие ориентации и кристаллизации изотактических сегментов. Подобно тому, что наблюдается при растяжении кристаллизующихся каучуков, образовавшиеся при ориентации кристаллические области играют ту же роль, что и поперечные связи в вулканизованных каучуках. Но в отличие от вулканизованных каучуков стереоблокполимеры могут плавиться и подвергаться формованию при температурах выше температуры плавления кристаллических образований. Увеличение степени кристалличности стереоблокполимера пропилена до 40—50% ведет к повышению жесткости, твердости, прочности; обратимые удлинения не превышают 10—20% [47].
75
Блоксополимеры а-олефинов могут быть получены также в процессе деструкции гомополимеров. Если происходит деструкция одновременно двух полимеров, то часть образующихся при разрывах цепей макрорадикалов рекомбинирует в новых сочетаниях, образуя блоксополимеры:
---А—А—А—А --А—А—А
—> . —> А—А—А—В—В—В
---В—В—В—В --В—В—В
Очевидно, что продукт содержит кроме блоксополимера оба гомополимера.
Получают блоксополимеры обычно механохимическим воздействием на смесь полимеров. Например, вальцеванием смеси полиизобутилена с полиэтиленом низкого давления при 165— 175 °C в течение 30 мин получают соответствующий блоксополи-мер [60]. Смесь блок- и привитого сополимеров полиэтилена высокой плотности и полиизобутилена была получена совместной экструзией полимеров, предварительно подвергнутых смешению на вальцах [93]. Количество химически связанного с полиэтиленом полиизобутилена определяли, удаляя непрореагировавшую часть полиизобутилена экстракцией гептаном. При температурах переработки 240—260 °C содержание химически связанного полиизобутилена составляло 10%. Продукт совместной экструзии превосходит полученную вальцеванием смесь полимеров по термостабильности.
Образование блоксополимеров возможно и при деструкции полимера в среде какого-либо мономера. Так, при воздействии ультразвука на высокомолекулярный атактический полипропилен в растворе, содержащем стирол, получается блоксополимер пропилена и стирола [61]. Подобный результат достигается и при воздействии электрической дуги на полипропиленовую пленку, находящуюся в эмульсии другого полимера [62].
Литература
1. Берлент У., Хофман А. Привитые и блок-сополимеры. М., ИЛ, 1963. — 2. Цереза Р. Блок- и привитые сополимеры. М., «Мир», 1964. 288 с. — 3. Natta G., Beat! Е., Sever! ni F. J. Polymer Sci., 1959, v. 34, p. 685. — 4. Манясек 3., Бе рек Д., Мичко М. и др. Высокомол. соед., 1961, т. 3, № 7, с. 1104.—5. Манясек 3., Беллуш Д. Полипропилен. Под ред. В. И. Пилиповского и И. К. Ярцева. Л., «Химия», 1967. — 6. Дудоров В. В., Самвел ян А. Л., Луковников А. Ф., Левин П. И Изв. АН АрмССР Хим. науки, 1962, т. 15, № 4, с. 311. — 7. Пудов В. С., Нейман М. Б. В кн.: Старение и стабилизация полимеров. Под ред. А. С. Кузьминского. М., «Химия», 1966. 210 с. — 8. Павлинеи И., Лазар М., Манясек 3. Хим. волокна, 1962, № 5, с. 21.—9. Манясек 3., Мичко М., Павлинец И., Лазар М. Хим. волокна, 1963, № 3, с. 20.— 10. Конкин А. А., Зверев М. П. Полиолефиновые волокна. М., «Химия», 1966. 280 с.
74
11. Сирота А. Г., Федотов Б. Г., Рябиков Е. П. и др. Пласт, •массы, 1967, № 7, с. 10.— 12. Платэ Н. А., Шибаев В. П. Жури. ВХО им. Менделеева, 1964, т. 9, № 6, с. 637. — 13. Жунжуй У., Роговин 3. А., Конкин А. А. Хим. волокна, 1961, № 5, с. 18. — 14. Власов А. В., Михайлов Н. В., Токарева Л. Г. и др., Хим. волокна, 1963, Ns 6, с. 24.— 15. Цетлиц Б. Л. и др. В кн.: Радиационная химия полимеров. М., «Наука», 1966. — 16. Рябчикова Г. Г. и др. В кн.: Радиационная химия полимеров. М., «Наука», 1966. — 17. Potts J., Bonner Е., Turbett В., Rugg F. Am. Chem. Soc., Meeting in Minature, Jan. 1957. — 18. Федотов Б. Г., Сирота А. Г., Рябиков Е. П. и др. Доклад на симпозиуме по синтезу, переработке и модификации полиолефинов. Баку, 1967.— 19. Пирсон Р. «Химия и технология полимеров», 1958, № 2, с. 20.. — 20. Lawton Е. J., Balwit J. S., Powell R. S. J. Polymer Sci., 1958, v. 32, p. 257.
21. Sh inohara Y., Tomioka K. J. Polymer Sci., 1960, v. 44, Xs 143, p. 195. — 22. Bevington J. C., Eaves D. E. Nature (London), 1956, v. 178, p. 1112.—23. Odian G„ Acker T., Sobel M. J. Appl. Polymer Sci., 1963, v. 7, № 1, p. 245.-24. Plast. Technol., 1963, v. 9, № 1, p. 53. — 25. Ода, X о ш и н о. Материалы международного симпозиума по полимерам. Токио — Киото, 1966; «Химия и технология полимеров», 1967, № 3, с. 5.—26. Ballantine D. S., G1 i п е s A., Adler G., Metz D. J. J. Polymer Sci., 1959, v. 34, p. 419. — 27. Chapiro A. J. Polymer Sci., 1958, v. 29, p. 321.— 28. Chapiro A. J. Polymer Sci., 1959, v. 34, p. 439. — 29. Chapiro А. Доклад на международном симпозиуме по макромолекулярной химии. М., 1960. — 30. Sobue Н., Tazima Y., Shimokawa Y. J. Appl. Polymer Sci., 1960, v. 4, № 11, p. 244
31. Miura M., Kawamatsu S. Chem. High Polymer Japan, 1962, v. 19, p. 175. — 32. Бах H. А. Сборник работ по радиационной химии. М., .Изд. АН СССР, 1955. — 33. О дор Л., Гелейн Ф. Хим. волокна, 1961, .№ 2, с. 18.—34. Чен, Фридландер. «Химия и технология полимеров», 1963, Xs 12, с. 46. — 35. Oster G., Moroson Н. J. Polymer Sci., 1957, v. 26, p. 233. — 36. Oster G., Oster G. K-, Moroson H. J. Polymer Sci., 1959, v. 34, Xs 127, p. 671. — 37. Ивакура H., Таке да К- Материалы международного симпозиума по полимерам. Токио — Киото, 1966; «Химия и технология полимеров», 1967, Xs 3, с. 17.—38. Геледжи Ф., О дор Л. «Химия и технология полимеров», 1963, Xs 12, с. 30. — 39. A. Chapiro. Материалы международного симпозиума по макромолекулярной химии. Прага, 1957.— 40. Липатова Т. Э., Липатов Ю. С., Тутаева Н. Л. Высокомол. соед., 1964, т. 3, с. 184.
41. Job С., Lebel Р. Материалы международного симпозиума по макромолекулярной химии. Париж, 1963. — 42. Pinner S. N., Wycherley V. Plastics, 1958, v. 23, Ns 244, p. 27. — 43. Mesrobian R. В. Труды второй международной конференции по мирному использованию атомной энергии. Женева, 1958, с. 621. — 44. Кочергинская Л. К-, Розеиблюм Н. Д., Стасюк X. А. Высокомол. соед., 1962, т. 4, Xs 5, с. 633. — 45. Розен-блюм Н. Д. и др. В кн.: Радиационная химия полимеров. М., «Наука», 1966.—46. Климанова Р. С., Серенков В. И., Тихомирова Н. С. Труды второго Всесоюзного совещания по радиационной химии. М., Изд. АН СССР, 1962, с. 501, —47. Natta G., J. Polymer Sci., 1959, v. 34, Хе 127, p. 531. — 48. Bier G., Gumboldt A., Lehmann G. Plastics Inst. Trans. J., 1960, v. 28, p. 98. — 49. Bier G., Lehmann G., Leugering H. J. .Macromol. Chem., 1961, v. 44—46, p. 347. — 50. Ko nt os E. G., E asterbrook E. K., Gilbert R. D. J. Polymer Sci., 1962, v. 61, p. 69; пер.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ. М., «Мир», 1964.
51. Бир Г. «Химия и технология полимеров», 1962, Хе 6, о. 3. — 52. Пи-липовский В. И., Ярцев И. К-, Шибаловская С. А. Доклад на
\ Я
симпозиуме по синтезу, переработке н модификации полиолефинов. Баку, 1967. — 53. Хагемайер. «Химия и технология полимеров», 1963, № 2, с. 88.—54. Франц, пат. 1359107, 1964. — 55. Натта Д., Маззанти Д., Креспи Д., Моральо Д. «Химия и технология полимеров», 1957, № 6, с. 94. — 56. Бельг, пат. 550093, 1956. — 57. Natta G. «Химия и технология полимеров», 1959, Ns 1, с. 98. — 58. Natta G. Chem. Ind. (London), 1957, Ns 47, p. 1520. — 59. Berger M., Buckley D. Chem. Eng. News, 1962, v. 40, p. 42. — 60. Авт. свид. СССР 156675, 1963; Бюлл. изобр., 1963, № 16.
61. Romanov A. R., Lazar M, Plaste u. Kaut., 1963, Ns 8, S. 470.— 62. Бельг, пат. 613561, 1962.—63. Bier G., Lehmann G. In: Copolymerization. Ed. G. E. Ham. N. Y., Interscience, 1964; пер.: Сополимеризация. Под ред. В. А. Кабанова. М., «Химия», 1971. 616 с. — 64. Баттерд Г., Трегер Д. У. Свойства привитых и блок-сополимеров. Пер. с англ, под ред. А. Г. Сироты. Л., «Химия», 1970. 216 с. — 65. Тихомиров В. С., Деев Ю. С., Серенков В. И. Высокомол. соед., 1971, т. 13, сер. Б, Ns 4, с. 256. — 66. Загорская 3. Г., Тихомирова Н. С., Серенков В. И. Высокомол. соед., 1971, т. 13, сер. Б, Ns 9, с. 688. — 67. Кабанов В. Я-, Сидорова Л. П., Спицнн Викт. И., ДАН СССР, 1971, т. 198, Ns 4, с. 883. — 68. Roloff Н., Swind А.-Е., Z il in ski Е. Plaste u. Kaut., 1970, Bd. 18, Ns 2, S. 90. — 69. Ohsika T. Kogyo Kagaku Zasshi, 1961, v. 64, p. 1864; C. A., 1962, v. 57, p. 4846. — 70. Муса A. M., Шибаев В. П., Плата Н. А., Козлов П. В. Высокомол. соед., 1967, т. 11, сер. А, № 2, с. 398.
71. Власов А. В., Комарова Л. И., Коршак В. В. и др. ДАН СССР, 1970, т. 193, № 3, с. 615. — 72. Костров Ю. А., С о л о в с к а я Н. Л. Хим. волокна, 1966, Ns 2, с. 25. — 73. Цетлин Б. Л. Высокомол. соед., 1968, т. 10, сер. A, Ns 12, с. 2611. — 74. Никольский В. Г., Данилов Е. П., Миронов Н. А., Карпов В. Л. Высокомол. соед., 1970, т. 12, сер. А, Ns 6, с. 1288. — 75. Никольский В. Г., Бубен Л. Я. ДАН СССР, 1960, т. 134, с. 134.—76. Бубен Н. Я., Гольданский В. И., Златк е-вич Л. Ю. и др. Высокомол. соед., 1967, т. 9, сер. А, с. 2275.-77. Кабанов В. Я., Казимирова Н. М., Чалых А. Е. Высокомол. соед., 1972, т. 14, сер. A, Ns 9, с. 2042. — 78. Odian G., Kruse R. L., Kho J. H. T. J. Polymer Sci., 1971, v. 9, Ser. Al, № 1, p. 91. — 79. LindemanJ, Czare-zynska H. Polimery, 1969, t. 14, Ns 12, str. 601. — 80. Zurakowska-Orszagh J., Olejniczak H. Polimery, 1969, t. 14, Ns 9, str. 440.
81. Тихомиров В. С., Попова А. И., Серенков В. И. Пласт, массы, 1969, Ns 3, с. 14.—82. Machi S., Silverman J. J. Polymer Sci., 1970, v. 8, Ser. Al, Ns 12, p. 3529. — 83. Machi S., Silverman J. J. Polymer Sci., 1969, v. 7, Ser. Al, Ns 9, p. 2737. — 84. Hayakawa K-, Kawa-se K. J- Polymer Sci., 1967, v. 5, Ser. Al, Ns 3, p. 439. — 85. Шибаев В. «Химия и технология полимеров», 1966, № 3, с. 3.—86. Coover Н. W., McDonnel L., Joyner F. В. е. a. J. Polymer Sci., 1966, v. 4, Ser. Al, p. 2563. — 87. Д анкович А., Кисси и Ю. В. Высокомол. соед., 1970, т. 12, сер. A, Ns 4, с. 802. — 88. Anderson I. Н., Barnett G. М., Geddes W.C. Europ. Polymer J., 1967, v. 3, p. 161. — 89. Кисс ин Ю. В., Цветкова В. И., Чирков Н. М. Высокомол. соед., 1969, т. 9, сер. А, с. 1374.— 90. Zerbi G., Gussoni М., Ciampelli F. Spectrochim. Acta, 1967, v. 23, Ser. A, p. 301.
91. Хагемейер, Эдвардс. «Химия и технология полимеров», 1964, Ns 1, с. 146. — 92. Clegg G. A., Gee D. R., Melia Т. Р. Makromol. Chem., 1970, v. 132, р. 203. — 93. Акутин М. С., Артеменко Б. Н. Пласт, массы, 1967, Ns 9, с. 64. — 94. Матковский П. Е., Заворохин Н. Д., Чирков Н. М. и др. Высокомол. соед., 1966, т. 8, с. 1712.—95. Матковский П. Е., Заворохин Н. Д., Чирков Н. М. Изв. АН КазССР. Сер. хим., 1966, с. 30. — 96. Ambroz J., Hamrik О. Coll. Czech. Chem. Comm., 1963, d. 28, s. 2560. — 97. Матковский П. E. Высокомол. соед., 1969, т. 11, сер. Б, Ns 3, с. 205. — 98. Кувер, Мак-Коннел, Джойнер и др. «Хи
76
мия и технология полимеров», 1967, № 1, с. 45. — 99. Авт. свид. СССР 256243; Бюлл. изобр., 1969, № 34. — 100. Андрианов К. А., Булгаков В. Я., ХанаиашвилиЛ. М. Пласт, массы, 1968, № 7, с. 12.
101. Андрианов К. А., Булгаков В. Я., Гуль В. Е. и др. «Труды ВНИИКП», 1972, вып. 18, с. 188.— 102. Качан А. А., Лебо Ю. Г., Ш р у-бович В. А. Высокомол. соед., 1970, т. 12, сер. А, № 1, с. 214. — 103. Костылева 3. А. Автореф. канд. дисс. Киев, 1971. — 104. Костылева 3. А., Качан А. А., Шрубович В. А. Укр. хим. ж., 1970, т. 36, № 10. с. 1055.— 105. Костылева 3. А., Шрубович В. А., Качан А. А. Пласт, массы, 1970, № 2, с. 14. —106. Лебо Ю. Г. Автореф. канд. дисс. Киев, 1972.— 107. Теренин А. И. Фотоника молекул красителей. Л., «Наука», 1967. 616 с. —108. Егоров Ю. П„ Качан А. А., Костылева 3. А. и др. Теор. и эксперим. химия, 1967, т. 3, № 6, с. 843. — 109. Власов А. В., Малахова Л. И., Цетлин Б. Л., Шаблыгин М. В. ДАН СССР, 1969, т. 184, с. 843.— ПО. Власов А. В., Малахова Л. И., Цетлин Б. Л., Шаблыгин М. В. Высокомол. соед., 1969, т. 11, сер. Б, № 8, с. 557.
Глава IV
Модифицирование с помощью реакций в цепях полиолефинов
Отличия реакций в цепях полиолефинов от аналогичных реакций в цепях других полимеров связаны прежде всего с химической инертностью полиолефинов и существованием в их структуре кристаллических и аморфных областей. Проведение реакций в растворах, когда кристалличность не имеет значения, затруднено, так как продукты реакции часто нерастворимы в растворителях исходных полимеров [1, с. 220]. На ход реакций в твердом полимере большое влияние оказывает скорость диффузии низкомолекулярного реагента, определяемая степенью кристалличности полимера. По мере уменьшения кристалличности в результате протекания реакции проницаемость полимера возрастает. Диффузия низкомолекулярного вещества, не растворяющего полимер, подчиняется закону Фика. Диффузия вещества, вызывающего набухание полимера, протекает значительно быстрее. Равномерное частичное превращение полимера возможно в случае, если скорость реакции меньше скорости диффузии низкомолекулярного реагента.
Реакции в цепях полиолефинов сопровождаются побочными процессами: сшиванием и деструкцией.
Из реакций, позволяющих ввести в макромолекулы полиолефинов различные функциональные группы, наиболее подробно исследованы галогенирование и сульфохлорирование. Эти процессы получили значительное практическое использование для получения модифицированных типов полиэтилена, имеющих самостоятельное применение и служащих промежуточными продуктами для дальнейших превращений, в первую очередь, вулканизации. В целях модифицирования полиолефинов могут использоваться также реакции их окисления и термической деструкции, которые обычно стремятся предотвратить.
Г алогенирование
Галогенирование полиэтилена. Наиболее глубоко исследован процесс хлорирования полиэтилена. Описано несколько способов проведения этой реакции. В первых публика
78
циях предлагалось хлорировать растворы или суспензии полиэтилена высокого давления в четыреххлористом углероде или уксусной кислоте газообразным хлором в присутствии катализаторов (хлорида алюминия или хлорного железа) [2]. Позднее было описано хлорирование в присутствии в качестве катализатора хлорида титана [3]. Скорость процесса, которая в значительной мере зависит от температуры, удовлетворительна при температуре около 45 °C на начальной стадии и выше 55 °C после начала повышения вязкости реакционной среды [4]. Увеличению скорости хлорирования способствует освещение реакционной массы светом с длиной волны до 4785 А [5, 6]. Механизм фотохимического хлорирования можно представить следующей схемой:
hv •
С12 ---> 2С1
СИ-----сн2----—>--------СН-----н НС1
----СН-----НС12 —>------СНС1------pci
Поскольку в макромолекулах полиолефинов в небольшом количестве имеются двойные связи, очевидно, что хлорирование происходит не только как реакция замещения, но частично и как реакция присоединения: hv .
С12 ---> 20
R2C = CR' + C1 —► r2c—cr', I Cl
r2c—cr2' + ci2 —> R2C—CR' + Cl
Cl Cl Cl
Фотохимическое хлорирование под действием видимого света при 20 °C протекает с повышенной скоростью в присутствии следов кислорода [5]. Это противоречит известным данным об обрыве цепей при низкотемпературном хлорировании парафиновых углеводородов. Предполагается, что повышению скорости хлорирования способствует образование свободных макрорадикалов в результате окисления полиэтилена [7]. Кислород в больших концентрациях, напротив, ингибирует реакцию хлорирования.
Известно использование в качестве инициаторов реакции хлорирования перекисных и азосоединений, образующих при нагревании свободные радикалы [8—10].
Хлорирование в суспензии может осуществляться в различных средах: в воде, уксусной кислоте, холодном четыреххлористом углероде [11, с. 328], смеси воды и четыреххлористого углерода, серной кислоте [12, 13]. Хлорирование может происходить также под давлением в среде жидкого хлора, растворяющего полиэтилен [14, 15].
79
Хлорирующими агентами, кроме хлора, могут также служить сульфохлорид [16] и фосген [8].
В качестве реакционной среды для хлорирования в растворе можно применять четыреххлористый углерод. При достаточной концентрации хлора в растворе (около 7%) реакция протекает с большой скоростью при невысоких температурах (до 50 °C).
При хлорировании так же, как и при бромировании [17], уменьшается степень кристалличности полиэтилена. После достижения степени галогенирования, соответствующей полной аморфизации, полимер становится растворимым при комнатной температуре и реакция продолжается в гомогенной среде при сравнительно мягком температурном режиме. В мягких температурных условиях в растворе хлорируются также аморфные сополимеры этилена с пропиленом.
Хлорирование в гетерогенных условиях сопровождается агрегацией частиц полимера, связанной с их размягчением в результате уменьшения степени кристалличности. Хлорирование в водной суспензии при 65 °C позволяет получить продукт с содержанием хлора около 40%. Более глубокое замещение осложняется агрегацией частиц полимера и требует повышения температур до 75 °C. Для уменьшения склонности частиц к агрегации рекомендуется [18, 19] насыщать воду хлористым водородом или добавлять хлорид кальция.
Строение продуктов хлорирования полиэтилена исследовали методом ИК-спектроскопии [20, 24, 136, 137]. При невысокой степени превращения (содержание хлора менее 35%) замещение проходит преимущественно у вторичных атомов углерода. По мере увеличения содержания хлора возрастает интенсивность поглощения при частоте 660 см-1, которая соответствует группе —СС12—, обнаруживаемой также в спектре поливинил-иденхлорида. С другой стороны, в спектрах хлорированного полиэтилена с содержанием хлора до 55,17% отсутствует полоса 1407 см'1, характеризующая звенья —СН2—СС12— [24]. При достижении содержания хлора около 65%, судя по исчезновению в спектрах полосы 1460 см-1, в полимере не остается нехлори-рованных мономерных звеньев. Последовательности из четырех незамещенных метиленовых звеньев (полоса 722 см'1) исчезают при содержании хлора более 50%.
На структуру продукта хлорирования существенное влияние оказывают условия реакции [135].
Продукты, полученные в суспензии в холодном четыреххлористом углероде или в воде, не теряют полностью кристалличности и содержат хлор в основном в аморфных областях. Продукты хлорирования в растворе утрачивают кристалличность уже при содержании 35% хлора, при хлорировании в суспензии кристаллические области еще сохраняются при содержании хлора 55%. .
80
Хлорирование до содержания 25—40% хлора понижает температуру размягчения полиэтилена (рис. 34), причем это понижение значительнее у продуктов реакции в растворе, чем у продуктов, полученных в суспензии [22]. Дальнейшее увеличение содержания хлора ведет к повышению температуры размягчения, несмотря на глубокую аморфизашгю, очевидно,
вследствие возрастания сил межмолекулярного взаимодействия. Через минимум проходят и кривые зависимости твердости и прочности хлорированного полиэтилена от содержания хлора (рис. 35). Увеличение содержа
Рис. 35. Зависимость прочности при разрыве и жесткости хлорированного полиэтилена от содержания хлора [25].
ние. 34. Зависимость температуры размягчения (теплостойкости по Вика, нагрузка 1 кге) полиэтилена, хлорированного в водной суспензии, от содержания хлора (по данным [22]).
ния хлора выше соответствующего минимальным величинам прочности и твердости (около 35—40%) вызывает повышение значений этих характеристик в результате преобладания роста сил межмолекулярного взаимодействия над влиянием уменьшения степени кристалличности [7]. Сохранение известной кристалличности при хлорировании полиэтилена в суспензии проявляется в его большей жесткости по сравнению с продуктом хлорирования в растворе до той же степени замещения [11, 19, 25]. Полиэтилен, хлорированный в суспензии, уступает по растворимости продуктам хлорирования в растворе.
Ценная особенность хлорированного полиэтилена — малая горючесть — также зависит от метода его получения. Продукты хлорирования в суспензии практически не горючи уже при содержании хлора около 25%, полиэтилен же, хлорированный в
81
растворе, теряет горючесть при содержании хлора около 40%. Это связано с образованием на поверхности полимерных частиц
при хлорировании в суспензии слоя с высоким содержанием
хлора.
Свойства хлорированного полиэтилена в значительной мере определяются типом исходного полимера [34]. Это естественно,
в частности, потому, что высококристалличный полиэтилен высокой плотности, подвергаясь аморфизации при хлорировании, достигает степени кристалличности, типичной для полиэтилена
Рис. 36. Термостабильность хлорированного полиэтилена (/), содержащего 60% хлора, и нестабилизированного поливинилхлорида (2) при 160 °C (по данным [26]).
Малая энергия активации реакции
низкой плотности, лишь при содержании хлора около 15%.
Термостабильность хлорированного полиэтилена оказывается несколько ниже термостабильности поливинилхлорида, если судить по суммарному количеству выделяющегося при нагревании хлористого водорода (рис. 36). Энергия активации термического разложения полиэтилена, содержащего 55,17% хлора, в интервале температур 135—168 °C составляет 7,1 ккал!моль [27]. дегидрохлорирования, а
также отсутствие влияния ингибиторов радикальных реакций на термостабильность позволили предположить ионный меха-
низм отщепления хлористого водорода в результате поляризующего действия атомов хлора, индуцирующих на а и 0-уг-леродных атомах положительные заряды, которые облегчают отщепление атомов водорода [27]:
Н Н
₽1 н
н н
---’С—С----+Н+
С1
Образовавшийся макроанион отщепляет ион хлора:
НН НН
II II
---'С—С--—> -----С=С-ь С1
С1
Межмолекулярное отщепление хлористого водорода сопровождается возникновением поперечных связей между цепями;
82
НН НН
II II
—с—с— —с—с---
I I I
Н С! С1
—> НС! +
Cl Н Н
I I I
---с—с---- ---с—с---
II--------II
НН НН
Повышение термостабильности хлорированного полиэтилена, как и поливинилхлорида, достигается введением стабилизаторов, нейтрализующих выделяющийся при нагревании хлористый водород [11].
Хлорированный полиэтилен имеет самостоятельные применения, например в качестве защитных покрытий, а также используется для получения композиций с поливинилхлоридом. Введение хлорированного полиэтилена даже в небольших количествах в поливинилхлорид уменьшает хрупкость последнего на холоду и расширяет интервал его рабочих температур.
Хлорирование полиэтилена повышает его совместимость с различными наполнителями. Хлорированный полиэтилен высокой плотности допускает наполнение, например, 400 вес. ч. двуокиси титана или 300 вес. ч. сажи на 100 вес. ч. полимера [134].
Эластичный продукт хлорирования полиэтилена высокой плотности с содержанием хлора 25% сравнивался по свойствам с пластифицированным поливинилхлоридом. Хлорированный полиэтилен превосходит поливинилхлорид по морозостойкости. Пленки из хлорированного полиэтилена отличаются более высокими значениями удлинения и сопротивления надрыву, чем пленки из поливинилхлорида [134].
Введение атомов хлора в полиэтилен открывает возможности для получения других производных в результате реакций в макроцепях. Так, при взаимодействии хлорированного полиэтилена низкого давления с анилином, дибутиламином или водным раствором аммиака были получены аминсодержащие продукты [27, 28]. Основными реакциями, протекающими при взаимодействии хлорированного полиэтилена с аминами или аммиаком, являются дегидрохлорирование с образованием двойных связей в цепях, замещение части атомов хлора на аминогруппы и межмолекулярное сшивание:
NH3
---СН2—СНС1— —►--------СН=СН----|-NH4C1
NH3
—сн=сн— —> ----------СН2—СН—
I nh2
---СН2—СН----1---СН=СН----—►------СН2—СН--
NH2 NH
I
---СН2—СН---
83
Бром реагирует с полиэтиленом с большой активностью. Предполагается, что бром присоединяется к макрорадикалам с большей скоростью, чем хлор [1, с. 226]. Соотношение необходимых для одинакового изменения свойств полиэтилена количеств брома и хлора близко к соотношению их атомных весов. Полиэтилен при содержании 55% брома — каучукоподобный материал, при содержании 71,5% брома — жесткий, при содержании 86% брома — твердый и хрупкий [2].
Весьма активно, с большим выделением тепла протекает фторирование полиэтилена. Для снижения скорости процесса и предотвращения деструкции макроцепей рекомендуют разбавлять фтор инертным газом. Обработка пленки или порошка фтором в смеси с азотом в темноте позволяет ввести в полиэтилен 10% фтора при ограниченной скорости реакции и без деструкции полимера [29, 30]. Избежать слишком бурного протекания фторирования, сопровождающегося обугливанием или горением полиэтилена, можно, проводя процесс в присутствии нереакционноспособных металлов: меди, никеля, фосфористой бронзы. При этом можно получать продукты с различной степенью замещения, вплоть до полного, соответствующего 76 %-ному содержанию фтора. Полностью фторированный полиэтилен по свойствам близок к политетрафторэтилену. По данным рентгеноструктурного анализа он подобен политетрафторэтилену с очень малой степенью кристалличности [31].
Описана [163] обработка поверхности полиэтилена и полипропилена дифторметиленом, получаемым при пиролизе хлордифторацетата натрия:
CC1F2COO’ —► CF2CF 4- СО2
CF2C1~ —> :СР2 + СГ
Природа взаимодействия дифторметилена с полимером не изучена. Однако на основе спектральных данных и сведений о повышении смачиваемости предполагается, что сорбированный поверхностным слоем полимера дифторметилен реагирует по схеме:
R—Н + : CF2 —► R—CF2H
Для модификации поверхности полиэтилена использовались также карбоэтсксикарбен (:СНСООС2Н5), нитрен (:NH) и карбоэтоксинитрен (:NCOOC2H5) [164]:
: chcooc2hs
R—Н --------->- R—СН2СООС2Н5
8 NH R—H -----> R—NH2
•NCOOC2Hs
R—H ---------> R— NHCOOC,H5
Введенные в макромолекулы поверхностного слоя функциональные группы позволяют проводить дальнейшие превраще-84
ния, например:
кон SOClj
R—СН2СООС2Н5 -----► R—СН2СООК -------> R—СН2СЧ
Галогенирование полипропилена и других полиолефинов.
Полипропилен, подобно полиэтилену, можно галогенировать в растворе, в суспензии и в порошкообразном состоянии.
Хлорирование в растворе проводят с использованием в качестве растворителей хлорированных углеводородов (четыреххлористого углерода, хлороформа, хлорбензола, дихлорбензола и др.). Протекание реакции в большой мере зависит от структуры исходного полимера. Гомогенное хлорирование возможно лишь при использовании атактического полипропилена. Полипропилен, содержащий стереорегулярную фракцию, растворяется только частично, кристаллическая фракция лишь набухает в растворителе. По мере хлорирования поверхностных слоев кри
сталлической части она переходит в раствор. Полное растворение и образование равномерно замещенного продукта возможно при содержании хлора в полипропилене выше 40%. При меньшей степени хлорирования получается смесь растворимого полимера с большим содержанием хлора и гелеобразного полимера, подвергшегося замещению в меньшей степени
Рис. 37. Зависимость содержания хлора от температуры хлорирования стереоблок-полипропилена (7), изотактического полипропилена (2) и полиэтилена низкого давления (5) [24].
[32, с. 126].
От структуры полимера зависит и характер влияния температуры на степень замещения. На рис. 37 приведены кривые зависимости содержания хлора в стереоблокполипропилене,
изотактическом полипропилене и полиэтилене низкого давления от температуры хлорирования [24]. Повышение температуры хлорирования стереоблокполипропилена, содержащего 32% растворимой в холодном гептане аморфной фракции, от 5 до 25 °C сопровождается постепенным возрастанием содержания хлора. Дальнейшее увеличение температуры на глубине превращения сказывается незначительно. Хлорирование высококристаллического изотактического полипропилена (содержание растворимой в холодном гептане аморфной фракции 3,7%), так же как и полиэтилена низкого давления, при температурах до 25 °C протекает в незначительной мере. В этих условиях, очевидно, хлорируется лишь растворенная аморфная часть полимера. При повышении температуры реакции до 40 °C содержание
85
хлора в кристаллических продуктах существенно увеличивается, по-видимому, в результате хлорирования и растворения поверхностных слоев кристаллических образований. Скорость хлорирования значительно повышается в присутствии перекисей [33] и азосоединений [34], а также при облучении светом с интервалом длин волн от 2000 до 6500 А. Кренцель с сотрудниками [35], изучая фотохимическое хлорирование полипропилена, содержащего около 60% изотактической фракции, в среде четыреххлористого углерода при 20—25 °C, получили продукты с содержанием хлора до 62%. Интересно, что хлорирование полиэтилена в аналогичных столь же мягких условиях позволяет ввести в полимер не более 20% хлора. Это может быть объяснено, вероятно, наличием в макроцепях полипропилена третичных углеродных атомов, водород при которых легко замещается ХЛОрОМ.”
В процессе хлорирования полипропилена так же, как и в случае полиэтилена [24], происходит некоторая деструкция полимерных цепей [34, 36]. Об этом свидетельствует уменьшение характеристической вязкости полимера по мере увеличения содержания в нем хлора. Степень деструкции может быть уменьшена при использовании в качестве катализатора четыреххлористого титана [3].
Хлорирование полипропилена в суспензии при температуре от 60 до 105 °C позволяет получать однородные продукты с содержанием хлора порядка 60%. В качестве среды используют воду, уксусную кислоту и ее водные растворы [37], соляную кислоту [18, 38]. Например, хлорирование полипропилена, а также полиэтилена и сополимеров этилена с пропиленом протекает эффективно при пропускании хлора через суспензию полимера в концентрированной соляной кислоте с одновременным облучением светом с длиной волны 2500—6000 А. Если нужно получить продукт, содержащий более 50% хлора, то реакцию ведут сначала при невысокой температуре (75—95°C), а затем, после достижения содержания хлора около 50%, хлорирование продолжают при 95—105 °C. Использование концентрированной соляной кислоты предотвращает агрегацию частиц полимера [18]. Суспензионный метод успешно применялся также для хлорирования поли-4-метилпентена-1 [37, 39]. Процесс, однако, сопровождался деструкцией этого полимера, значительно превышавшей деструкцию полиэтилена в аналогичных условиях.
С большой скоростью протекает хлорирование порошкообразного полипропилена сухим хлором [37, 40—42]. Энергия активации реакции составляет 4,8 ккал/моль [40, 42]. Степень деструкции может быть уменьшена, если процесс проводится в темноте в присутствии хлорида натрия [40]. В этих условиях содержание хлора достигает 25,8%, температура плавления и степень кристалличности существенно не изменяются.
Бромирование полипропилена протекает аналогично хлорированию на свету в растворе брома в хлорированных углево
86
дородах (четыреххлористый углерод, дихлорбензол и др.) [40, 43, 44]. Энергия активации процесса бромирования составляет 11,4 ккал/моль [40]. Бромированный полипропилен нестабилен и выделяет бромистый водород уже при комнатной температуре. Хлорированный полипропилен более стабилен, однако при нагревании отщепляет хлористый водород. При содержании хлора около 60% выделение хлористого водорода наблюдается при температуре 108 °C и выше. Стабилизаторы, используемые для стабилизации поливинилхлорида, значительно повышают температуру разложения хлорированного полипропилена. Например, добавка 4% стеарата кальция увеличивает температуру разложения до 171—173 °C, силиката свинца — до 188 °C [34].
Исследована термическая деструкция хлорированного полипропилена в зависимости от содержания хлора [27, 45]. Показано, что максимальная деструкция наблюдается при содержании хлора 45,7%. Такое содержание хлора близко соответствует замещению одного атома водорода в каждом мономерном звене. Аналогичные данные были получены при изучении термической деструкции поли-а-бутилена [27].
Было обнаружено [45], что дегидрохлорирование хлорированного полипропилена протекает во времени с переменной скоростью. В интервале температур 100—238 °C скорость разложения оставалась постоянной в течение 10—20 мин, затем она уменьшалась, и выделение хлористого водорода при данной температуре прекращалось. Для объяснения этого факта предполагается одновременное протекание дегидрохлорирования по двум механизмам:
внутримолекулярному
СН3 СН3
---СН2—С—СН2—С----—>
С1 Н
и межмолекулярному
СН3 СН3
---СН2—С—СН2—С----
di н
+ —>
Н С1
I I
—сн2—с—сн2—с------
I I
сн3 сн3
сн3 сн3 I I
НСЦ----СН2—С=СН -С----
н
НС1 +
СН3 СН3
—сн2—с—сн2—с----
I н
С1
—сн2—с—сн2—с----
СН3 СНз
Энергия активации межмолекулярного отщепления хлористого водорода, как известно, меньше, чем внутримолекулярного. Однако вероятность внутримолекулярного отщепления
$7
превышает вероятность межмолекулярного отщепления. Отсюда следует преобладание межмолекулярного механизма дегидрохлорирования при относительно низких температурах. При повышении температуры возрастает доля хлористого водорода, выделяющегося за счет внутримолекулярного отщепления. По мере структурирования полимера в результате актов межмолекулярного отщепления дальнейшее дегидрохлорирование затрудняется и после достижения определенной густоты поперечных связей между цепями разложение полимера при данной температуре прекращается. При повышенных температурах (около 250 °C) происходит не только выделение хлористого водорода, но и деструкция полимерных цепей с образованием газообразных углеводородных продуктов; суммарная скорость разложения полимера оказывается значительно более высокой, чем при сравнительно низких температурах (около 100°C).
Эти представления позволяют объяснить характер зависимости степени разложения хлорированного полипропилена от содержания хлора. Содержание хлора до 30% практически не оказывает влияния на термическую стабильность хлорированного полипропилена при относительно невысоких температурах вследствие достижения определенной степени структурирования. Увеличение содержания хлора выше 30% (т. е. более 1 атома хлора на 2 мономерных звена) сопровождается резким возрастанием скорости дегидрохлорирования, так как введение атомов хлора в мономерные звенья повышает подвижность атомов водорода в соседних звеньях [46].
Продолжение хлорирования после достижения содержания хлора 46,5% (т. е. после замещения водорода практически у всех третичных атомов углерода) ведет к замещению атомов водорода в метиленовых и метильных группах [46]. Число подвижных атомов водорода при этом уменьшается, термостойкость продукта хлорирования возрастает.
Энергия активации реакции дегидрохлорирования хлорированного полипропилена невелика и составляет около 8 ккал/моль [45], хлорированного поли-а-бутилена— 6,4 ккал/моль [27]. Величина энергии активации, значительно меньшая необходимой для разрыва связей С—С макроцепей и осуществления дегидрохлорирования по свободнорадикальному механизму, а также факт отщепления хлористого водорода при невысокой температуре (100 °C) позволили предположить ионную природу термической деструкции хлорированного полипропилена [46]. (Поливинилхлорид, деструктирующий по свободнорадикальному механизму, начинает отщеплять хлористый водород при температурах выше 140 °C, энергия активации распада составляет 36,5 ккал/моль [47, 48].) Возможно, что деструкция по ионному механизму происходит под влиянием остатков катализатора полимеризации, содержащихся в хлорированном полипропилене.
Хлорирование полипропилена протекает прежде всего в аморфных областях. Это следует, в частности, из большей ско-
ве
рости хлорирования полимеров с меньшей степенью кристалличности. Однако при достижении определенных степеней замещения реакция протекает и в кристаллических образованиях [34, 35]. С увеличением степени хлорирования уменьшается содержание изотактической фракции.
Температура плавления продуктов хлорирования, содержащих небольшое количество хлора, ниже чем у исходных полимеров [34, 35, 37]. Увеличение содержания хлора более 10—20% вызывает резкое повышение температуры плавления, а также плотности полипропилена.
Глубоко хлорированный полипропилен отличается повышенной хрупкостью, негорюч. Как показали термомеханические исследования [34], при содержании хлора более 45% полипропилен не обнаруживает высокоэластического состояния и при нагревании из стеклообразного состояния переходит непосредственно в вязкотекучее.
Из полипропилена с содержанием хлора более 20% в результате реакции с окислами магния, свинца, цинка получают стойкие к действию озона эластомеры [36, 41, 49, 50]. Хлорирование поверхности изделий из полипропилена повышает их восприимчивость к красителям, печатным краскам, клеям, фотоэмульсиям [51, 54]. Хлорированный полипропилен можно использовать для склеивания различных материалов, в частности поливинилхлорида и поливинилиденхлорида [55].
Сульфохлорирование
Хлорированные полиолефины имеют сравнительно ограниченные возможности использования для получения вулканизованных материалов. Значительно более пригодными для этой цели являются сульфохлорированные полиолефины [1, 7, 11, 56—58]. Это связано с высокой реакционной способностью суль-фохлоридных групп.
Сульфохлорирование полиэтилена. Сульфохлорирование полиэтилена осуществляется чаще всего воздействием смеси хлора и двуокиси серы и приводит к образованию продукта, в котором атомы водорода замещены на атомы хлора и сульфохлоридные группы:
---СН2—СН2—СН2—СН2-----р 2С12 + so2 —►
—> ----СН—СН2—СН2—СН------I-2HC1
Cl o=s=o
Cl
Условия реакции сульфохлорирования близки к условиям хлорирования. Цепной механизм процесса позволяет инициировать реакцию с помощью перекисей или азосоединений [59] и облучения [60, 61]. Обычно применяется фотохимическое инициирование сульфохлорирования полиэтилена, растворенного
89
в четыреххлористом углероде, при температуре 60—75 °C. Получающийся каучукоподобный продукт содержит 20—45% хлора и 0,4—3% серы. Соотношение между содержанием хлора и сульфохлоридных групп регулируется изменением соотношения между концентрациями сернистого ангидрида и хлора в исходной смеси газов [62] и температуры реакции [63—65]. Степень сульфохлорирования возрастает по сравнению со степенью хлорирования при увеличении отношения концентраций сернистого ангидрида и хлора в газовой смеси и понижении температуры сульфохлорирования.
В качестве сульфохлорирующего агента можно применять заранее приготовленный хлористый сульфурил [61]. Катализаторами могут служить пиридин, тиофенолы и другие подобные вещества, способные образовывать комплексы с хлористым сульфурилом и выделяющимся хлористым водородом [36]. Применение хлористого сульфурила в сочетании со свободнорадикальными инициаторами, например перекисями, приводит к преобладанию реакции хлорирования [7]. Хлористый сульфурил как сульфохлорирующий агент не имеет значительных преимуществ перед смесью хлора и сернистого ангидрида.
Сульфохлорирование можно проводить и в гетерогенных условиях. Например, обработка тонкого порошка полиэтилена низкого давления с молекулярным весом около 80 000 смесью хлора и сернистого ангидрида при температуре от 40 до 80 °C позволяет получать сульфохлорированный полиэтилен, содержащий 18% хлора и 3,2% серы [64].
Продукты сульфохлорирования могут разлагаться под действием тепла и света [140]. Разложение протекает с выделением хлористого водорода и сернистого ангидрида. В качестве стабилизаторов рекомендованы, например, а- и 0-пинены [66], фенил-глицидиловый эфир [67], окись пропилена в сочетании с желатиной и октилфенолом [68].
Один из наиболее важных типов сульфохлорированного полиэтилена, так называемый хайпалон, используют главным образом как способный к вулканизации эластомер [11, 25, 69]. Хайпалон получают из полиэтилена с молекулярным весом около 20 000. Содержание хлора в хайпалоне составляет 26—29%, серы 1,3—1,7%. Один атом хлора в этом продукте приходится примерно на 7 атомов углерода, одна сульфохлоридная группа — на 90 атомов углерода [25, 69, 70]. Сульфохлоридные группы присоединяются главным образом к вторичным углеродным атомам; атомы хлора, не входящие в состав сульфохлоридных групп, распределены по цепи беспорядочно и связаны с первичными, вторичными и третичными атомами углерода [71—74]. Например, в продукте сульфохлорирования полиэтилена высокого давления смесью хлора и сернистого ангидрида в среде четыреххлористого углерода, содержащем 31% хлора и 1,2% серы, к первичным атомам углерода присоединено 2,7%, к вторичным 89,8%, к третичным 3,5% атомов хлора и 4% входит в сульфохлоридные
90
группы [75]. Естественно, что при сульфохлорировании линейных типов полиэтилена, почти не имеющих ответвлений и метильных групп, получается продукт с еще большим относительным содержанием хлора у вторичных атомов углерода.
Сульфохлорированный полиэтилен при нагревании до 125— 150 °C разлагается с выделением сернистого ангидрида и хлористого водорода:
RCH2CHR' —> RCH=CHR' + SO2 + НС1
I
so2
Образующиеся при этом двойные связи могут использоваться для вулканизации серой.
Основное назначение сульфохлорированного полиэтилена определяется его способностью вулканизоваться с использованием реакций сульфохлоридных групп. При взаимодействии сульфохлоридных групп с окислами металлов образуются соли, при реакции с бифункциональными аминами — сульфамидные группы. В процессе вулканизации под влиянием диаминов, димеркаптанов и других соединений, при взаимодействии с которыми отщепляется хлористый водород, могут участвовать также атомы хлора, в первую очередь связанные с третичными атомами углерода.
Невулканизованный сульфохлорированный полиэтилен типа хайпалон — каучукоподобный материал, сохраняющий эластичность до температуры —45 °C. Он отличается весьма высокой стойкостью к действию озона. Важной его особенностью по сравнению с другими эластомерами является огнестойкость, связанная со значительным содержанием хлора. Растворимость хайпалона в ароматических и хлорированных углеводородах позволяет использовать его для получения лаков. Известно применение хайпалона в композициях с различными каучуками.
Высокая адгезионная и антикоррозионная способность — ценные свойства сульфохлорированного полиэтилена как материала для защитных покрытий [138]. Недостаточные для такого применения значения твердости и прочностных характеристик могут быть существенно повышены изменением надмолекулярных структур с помощью добавок фенолоформальдегидной смолы [139].
Модифицирование поверхности полиэтилена проводилось обработкой хлорсульфоновой и дымящей серной кислотами [159, 161, 162]. Образование сульфокислотных групп на поверхности резко увеличивало ее смачиваемость. Аналогичный результат наблюдался и при воздействии атомарной серы, полученной пиролизом карбонилсульфида, а также фотолизом карбонилсуль-фида, сероуглерода, паров серы [160]. Еще большую смачиваемость придают дальнейшие полимераналогичные превращения
91
в поверхностном слое с участием РС15, POCI3, CF3CH2OH и (CF3CO) 2О:
R—Н + S —► R—SH
KMnOlt НС1
H2SO4, SO3 C1SO3H
R—Н ----------> R—SO2OH •«-------- Н—R
РС15
(РОС13)
NH4OH CF3CH2OH
R—SO2NH2 -------- R—SO2C1 --------->- R-SO2OCH2CFj
|ch2o
(CFSCO)2O
R—SO2N(CH2OH)2 --------->- R—SO2N(CH2COCF3)2
Сульфохлорирование полипропилена. Полипропилен подвергается сульфохлорированию, подобно полиэтилену, при одновременном воздействии сернистого ангидрида и хлора. Реакцию можно проводить в гомогенных или гетерогенных условиях. В первом случае в качестве растворителей применяют хлористый метилен, четыреххлористый углерод, хлороформ, тетра-хлорэтан, гексахлорэтан [35, 76—78].
Повышение скорости процесса достигается воздействием света или у-излучения [76, 79]. Скорость сульфохлорирования зависит от степени регулярности полипропилена: с увеличением регулярности структуры скорость реакции уменьшается [80]. При определенной степени регулярности структуры гомогенность реакционной среды, очевидно, нарушается. Гомогенность нарушается также при глубоком сульфохлорировании в результате частичного сшивания макроцепей хлорсульфогруппами.
Сульфохлорирование полипропилена в отсутствие растворителя протекает преимущественно в аморфных областях. С увеличением кристалличности скорость реакции уменьшается. Энергия активации процесса существенно зависит от степени превращения полипропилена. При содержании непрореагировавшего водорода 93% энергия активации составляет 25 ккал/моль, при содержании водорода 82,9%—44 ккал/моль [81]. Проведение сульфохлорирования в гетерогенных условиях позволяет осуществлять процесс непрерывно по противоточной схеме [81] или в кипящем слое [82].
В процессе реакции происходит снижение характеристической вязкости полимера, тем большее, чем выше содержание хлора и особенно серы в продукте [76]. При содержании серы 1—2% сульфохлорированный полипропилен в интервале от температуры стеклования до температуры, соответствующей началу
92
сшивания, находится в высокоэластическом состоянии. Варьируя соотношение сернистого ангидрида и хлора в реакционной среде и условия процесса, можно получать различные продукты. Так, при постоянном содержании хлора (около 37%), изменяя содержание серы от 0 до 6%, можно получить либо кристаллический продукт, либо каучук с рабочей температурой 150— 190 °C, либо твердый сшитый материал. Сульфохлорированный полипропилен хорошо растворяется в хлорированных и ароматических углеводородах. При нагревании выше НО °C и при воздействии ультрафиолетового света он разлагается с выделением хлористого водорода и сернистого ангидрида. Для стабилизации можно применять стабилизаторы поливинилхлорида. Наибольший технический интерес сульфохлорированный полипропилен представляет в качестве исходного продукта для вулканизации. Известны возможности и самостоятельного использования продуктов сульфохлорирования [32]. Сульфохлорирование поверхности полипропиленовых пленок повышает их адгезионные свойства, сульфохлорирование волокон придает им восприимчивость к основным красителям.
Фосфонирование
При взаимодействии полиэтилена с треххлористым фосфором в присутствии кислорода в макромолекулы полиэтилена входят фосфонилхлоридные группы [83, 124—126, 133]
---СН2—СН2—СН2—СН2----Р РС13 + i/2O2 —>
—>-----СН2—СН—СН2—СН2----FHC1
Cl—Р—С1
Реакцию проводят при 50 °C и выше, поскольку при этих температурах треххлористый фосфор растворяет полиэтилен. Скорость фосфонирования зависит от содержания кислорода в сфере реакции. Инициаторами процесса, протекающего по радикальному механизму, служат перекисные соединения. Наличие двойных связей в полиэтилене способствует увеличению скорости фосфонирования, очевидно, вследствие реакции по схеме:
^С=С\ + РС13 + '/2Ог —> —с----с—
С1 С1—Р—С1
II о
Фосфонированный полиэтилен легко гидролизуется
---СН2—СН—СН2-----1- 2Н2О —>----СН,—СН—СН2-----I-2HC1
I ' I
С1-Р-С1 НО—Р—он
<5 !
93
ное окислительное
Степень кристалличности, %
Рис. 38. Влияние кристалличности на содержание фосфора в продуктах гетерогенного фосфонирова-ния (продолжительность реакции 15 мин; условия реакции см. в табл. 17; степень кристалличности определена по методике [129]) [127. 128]:
/ — полиэтилен высокого давления; 2 — сополимер этилена с пропиленом; 3 — полиэтилен низкого давления; 4—полиэтилен среднего давления.
При достаточном содержании фосфора (15,5% [84]) продукт гидролиза полностью растворим в воде. Продукты гидролиза способны к сшиванию при взаимодействии с окислами металлов, например с окисью свинца. В работе [83] получены различные производные фосфонированного полиэтилена (диэтиловый и другие эфиры, амидсодержащие продукты и др.).
В серии работ [126—128, 130, 131] исследовалось гетероген-ширование полиэтилена разных типов и сополимеров этилена с пропиленом в интервале температур от 20 до 60 °C. Обработке смесью паров треххлористого фосфора с кислородом подвергали достаточно тонкие (50 мкм) пленки, чтобы исключалось, как это было специально показано, влияние диффузионного фактора. При 20 °C порядок реакции по треххлористому фосфору оказался равным 1, а по кислороду — 0,5. Как и следовало ожидать, фос-фонирование протекает преимущественно в аморфной части полимера, что подтверждается зависимостью содержания фосфора в продуктах реакции от степени кристалличности
Сопоставлялось максимальное содержание фосфора, соответствовавшее завершению реакции фосфонирова-ния, с теоретически рассчитанным. При расчете исходили из предположения, что в реакции могут участвовать атомы водорода при третичных углеродных атомах и в a-положении по отношению к двойным связям, а также
ненасыщенные группировки. Как видно из табл. 17, экспериментально найденные и расчетные данные по содержанию фосфора совпадают с удовлетворительной точностью. Это позволяет считать, что при гетерогенном фосфонировании реакция протекает преимущественно по перечисленным реакционным центрам.
В зависимости от строения цепей полиолефинов преобладающая роль в реакции принадлежит различным типам реакционных центров. Так, для разветвленных полимеров (полиэтилен высокого давления, сополимер этилена с пропиленом) основную часть (около 96%) участвующих в реакции центров составляют атомы водорода при третичных углеродных атомах. В полиэтилене низкого давления в реакции почти в равной мере участвуют все три типа центров. Для практически линейного полиэтилена среднего давления характерно участие в реакции лишь
(рис. 38).
94
Зависимость содержания фосфора от количества способных к взаимодействию реакционных центров в цепях полиолефинов [131]
Условия реакции: [С>2) = 1,6-10~3 моль/л: [РС13] = 5,4-10~3 моль/л; температура 20 °C. продолжительность 20 мин.
Таблица 17
Полимер Число групп /с=с\ на 100 атомов С k Содержание групп (в % от общего содержания ненасыщенных групп) Общее количество групп СНз на 100 атомов С Число групп СНз па 100 атомов с, соответствующее ответвлениям / Количество атомов водорода в а-поло-жении по отношению к двойной связи на Ю0 атомов с fe(xl + 2xz + 2x3) Суммарное количество реакционноспособных центров иа 100 атомов С & +1 + m Экспериментально найденное содержание фосфора, мол. % (на поли-метнлено-вую цепь) п 1 Соотношение найденного и расчетного содержаний фосфора. % /г-100 & +1 4- m
виннль-ных сн2=сн- транс-внннлено-вых Н, . >С=С< 7 ХН Х2 винилиденовых сн2=с<^ хз
100
Полиэтилен высокого давления . . 0,044 21 12 67 2,6 2,6 0,079 2,7 2,6 96
Сополимер этилена с пропиленом 0,022 44 21 35 2.0 2,0 0,034 2,1 2,0 95
Полиэтилен низкого давления . . . 0,056 66 18 16 0,37 0,18 0,075 0,31 0,28 90
Полиэтилен среднего давления . . , 0,068 96 0 4 0,05 0 0.071 0,14 0.14 100
ненасыщенных группировок и атомов водорода в а-полоЖении к ним.
Уменьшение содержания реакционноспособных центров в цепях в результате гетерогенного окислительного фосфонирования определяет повышенную стабильность полиолефинов в условиях термоокислительной деструкции и атмосферного старения.
Термогравиметрические испытания показали, что фосфони-рованная пленка из полиэтилена высокого давления (содержание фосфора 2,8 вес.%) при нагревании до 400°C теряет 12% веса, в то время как убыль в весе исходной пленки в аналогичных условиях составляет 43%.
В процессе атмосферного старения накопление карбонильных и винильных групп в фосфонированной полиэтиленовой пленке происходит с меньшей скоростью, чем в исходной пленке. В условиях атмосферного старения, соответствующих почти полной (на 90%) потере способности к деформированию исходной полиэтиленовой пленки, фосфонированная пленка (содержание фосфора 1,7 вес.%) практически целиком сохраняет эту способность.
Эти данные находятся в соответствии с результатами работы [132], показывающими повышенную стойкость к атмосферному старению полиэтиленовой пленки, сшитой под действием УФ-света в присутствии сенсибилизатора — треххлористого фосфора. Одновременно с сенсибилизирующим действием (см. гл. V) треххлористый фосфор участвует и в гетерогенном фос-фонировании полиэтилена. Это и сообщает сшитому фотохимическим способом полиэтилену повышенную стойкость к атмосферному старению.
Очевидно, гетерогенное фосфонирование можно рассматривать как частный случай реакций в цепях, придающих полимерам повышенную стойкость в условиях термоокислительной деструкции и атмосферного старения.
Беллуш, Манясек и Лазар [85] исследовали фосфонирование атактического полипропилена. Реакцию осуществляли в растворе полимера в треххлористом фосфоре в присутствии кислорода при 50—55 °C. Авторы предполагали, что замещение происходит в основном по месту третичных атомов углерода. Содержание фосфора в гидролизованном продукте достигало 7,2% [11,7 групп —РО(ОН)2 на 100 элементарных звеньев]. По мере фосфонирования утрачивается растворимость полимера в горячих углеводородных растворителях (толуол и др.) и возрастает растворимость в полярных растворителях (этилацетат и др.). Гидрофильность фосфонированного полипропилена увеличивается с повышением степени превращения.
Возможно фосфонирование поверхности пленок, волокон и других изделий из полипропилена, набухших в треххлористом фосфоре или в его растворе в органическом растворителе [32, с. 139].
96
Продукты гидролиза фосфонированного полипропилена, подобно аналогичным продуктам модифицирования полиэтилена, способны сшиваться окислами двухвалентных металлов [86]. Получаемый таким образом вулканизат обладает высокой стойкостью к озону, способен к ионному обмену, отличается огнестойкостью.
Получение амидов и сложных эфиров в поверхностном слое с использованием групп —РО(ОН)2 [87] позволяет придавать изделиям из полипропилена повышенную термо- и фотоокпсли-тельную стабильность, окрашиваемость, способность к ионному обмену.
Для получения вулканизуемого продукта фосфонированию подвергали сополимер этилена с пропиленом [88, 89].
Фосфонирование полиизобутилена в условиях, аналогичных применявшимся при проведении реакции с полиэтиленом, приводит к глубокой деструкции полимера, степень фосфонирова-ния незначительна [1].
Окисление
Окислению полиолефинов посвящена обширная литература (см., например, [1, 90, 91, 149—151]). Основная часть исследований в этой области проводилась в связи с проблемой подавления процессов окислительной деструкции при переработке полиолефинов в изделия и при их эксплуатации. В последние годы окисление все шире используется в качестве метода модифицирования полиолефинов. Окисление при нагревании позволяет вводить в состав полиэтилена кислородсодержащие группы, основную доль которых составляют карбонильные, гидроперекисные и эфирные группы [92, 93]. Фотохимическое окисление сопровождается образованием в основном карбонильных групп и увеличением содержания випильных групп и внутренних двойных связей [21, 92]. Появление карбонильных групп резко повышает способность полиэтилена поглощать ультрафиолетовое излучение (в области 2800 А). При фотолизе карбонилсодержащих продуктов окисления образуются свободные радикалы, способные инициировать цепные окислительные процессы, результатом которых может быть возникновение гидро-перекисных групп [94]:
. о2 . RH
R ---> ROO —> ROOH + R
Гетерогенное окисление полиэтилена при температурах ниже температуры плавления кристаллической фазы протекает с тем большей скоростью, чем меньше степень кристалличности (рис. 39) [95]. Окисление в этих условиях происходит главным образом в аморфных областях, скорость диффузии кислорода в которых значительно выше, чем в кристаллических. На это указывает сохранение кристалличности после окисления
4 А. Г. Сирота
97
разветвленного полиэтилена, не
Продмкительнасть, ч
Рис. 39. Кинетические кривые окисления разветвленного (/, плотность 0,920 г/см3) и линейного (2, плотность 0,958 г/см3) полиэтилена при 80 °C [95].
полиэтилена при температуре, не достигающей температуры плавления полиэтилена [1]. Отличие в скоростях окисления, связанное с разницей в степенях кристалличности линейного и наблюдается при проведении процесса в расплаве (рис. 40). Следовательно, фактором, определяющим скорость окисления, является степень кристалличности, а не активность полимера, повышающаяся по мере возрастания разветвленности [95; 152, с. 273].
Роль разветвленности выявлена при высокотемпературном окислении расплава полиэтилена. При содержаниях 1, 10, 37 и 55 СНз-групп на 1000 атомов С константы начальной скоро
сти окисления при 300 °C соответственно равны 3,45-10-4; 5,1-Ю-4; 5,35-10 4; 5,6-10~4 с-1 [147].
Рис. 40. Кинетические кривые окисления полиэтилена при температурах выше и ниже температуры плавления [95]:
—---—разветвленный полиэтилен (плотность 0,920 г/см1);
---- линейный полиэтилен (плотность 0,958 г/см1).
По данным работы [155] по мере возрастания разветвленности полиэтилена уменьшается индукционный период поглощения кислорода при термоокислении (рис. 41). Для образцов полиэтилена, содержащих 0,5; 6,5 и И СН3-групп на 1000 атомов С, энергия активации термоокисления составляет соответ' ственно 34,8; 31,9 и 29,5 ккал/моль.
98
Изменения надмолекулярной структуры в результате окисления расплава линейного полиэтилена исследовали Бальтенас и Бальтенене [148]. Уже незначительные химические превраще-
ния в расплаве приводят к существенным изменениям надмолекулярной структуры в охлажденном после окисления полимере. Задолго до окончания индукционного периода процесса окисле-
ния эти изменения проявляются в укрупнении структур с образованием крупных кольцевых сферолитов. Это явление рассматривают как результат снижения во время индукционного периода вязкости расплава, что с одной стороны способствует повышению скорости линейного роста кристаллов, а с другой — уменьшает скорость образования центров кристаллизации благодаря накоплению полярных кислородсодержащих
надмолекулярных
Продолжительность^ мин
Рис. 41. Кинетика поглощения кислорода полиэтиленом при 160 °C и различном содержании СНз-групп на 1000 атомов С [155].
групп. При дальнейшем окислении с достижением автокаталитической стадии реакции крупные сферолиты, возникшие на предыдущей стадии, распадаются на мелкие сферолиты нерегулярной формы.
Продолжение окисления, сопровождающееся сшиванием макроцепей (в основном кислородсодержащими мостиками) и
накоплением труднокристаллизующихся продуктов окисления, приводит ко все большей аморфизации полиэти
лена.
При термоокислении в блоке первая стадия процесса — укрупнение сферолитов — начинаясь в поверхностном слое, захватывает постепенно слой толщиной до 500 мкм и прекращается с окончанием индукционного периода окисления. Дальнейшие изменения надмолекулярной структуры протекают лишь в тонком поверхностном слое (до 125 мкм), поскольку кислород не успевает диффундировать в глубинные слои блока из-за большой скорости связывания его в поверхностном слое. Таким образом, поверхностный слой оказывает защитное действие по отношению к глубинным слоям блока [148, 153].
Гомогенное окисление полиэтилена, растворенного в толуоле, в присутствии катализатора — ацетата кобальта — описано в качестве эффективного метода введения полярных групп (преимущественно карбонильных) в полиэтилен для повышения его адгезионной способности [146]. Кажущаяся энергия активации процесса равна 25,6 ккал/моль.
Значительный интерес представляет окисление полиэтилена для получения карбоксилсодержащих продуктов. Описана обработка расплава полиэтилена на смесительных вальцах при 160—200 °C на воздухе в течение нескольких часов [96].
4*
99
В результате такой обработки образуется карбоксилсодержащий полимер с содержанием кислорода от 0,1 до 0,5 вес.%.
Окисление можно проводить также воздействием окисляющих агентов, например азотной кислоты. Обработка полиэтилена водным раствором азотной кислоты с концентрацией 35— 85% при температурах 70—130 °C в течение нескольких часов позволяет получать в основном дикарбоновые кислоты с молекулярным весом от 250 до 1000 [8]. Эффективное окисление полиэтилена достигается при действии смеси озона и кислорода в растворе четыреххлористого углерода при 70—78 °C. При этом получаются главным образом оксидикарбоновые кислоты с молекулярным весом от 100 до 2000.
Основные виды применения (пропитывание тканей и т. п.) карбоксилированного полиэтилена связаны с его способностью диспергироваться в воде с образованием стойких эмульсий [11].
Полипропилен имеет значительно большую склонность к окислению по сравнению с полиэтиленом. Это обусловлено [97] большей реакционной способностью атомов водорода у третичных атомов углерода, чем в метиленовых и метильных группах.
Выше уже упоминались процессы гидропероксидации с целью создания активных центров для последующей прививки различных полимеров. Минскер с сотрудниками [98] использовали гидроперекисные группы для восстановления их до гидроксильных групп. По одному из методов гидропероксидированный полипропилен обрабатывали уксуснокислым раствором иодида калия при 20 °C в течение 6 ч. Все гидроперекисные группы при этом превращались в гидроксильные по следующей реакции:
СН3 СНЭ
I hi I
---с—сн2---------►-----С—СН,-----и Н2О + 12
I I
ООН он
Было изучено также взаимодействие гидропероксидирован-ного полипропилена с алюминийалкилами. Окисленные полипропиленовые волокна после обработки 3—15% раствором триэтилалюминия в гептане в течение 5 мин при комнатной температуре не содержали гидроперекисных групп. Появившаяся в ИК-спектрах продуктов такой обработки полоса поглощения 3330 см-1 была отнесена к гидроксильной группе. Предполагается [98], что превращение гидроперекисных групп в гидроксильные включает стадии образования металлорганических производных гидроперекисей и перегруппировку гидроперекисных солей алкплалюминия в его оксипроизводные:
100
СН3 СНз
I A1R3 | Перегруппировка
----С—СН2-----------►-----С—СН2------------------------
ZR
ООН О—О—Al
СНз I —с—сн2---
I /0R оа/
СНз
Гидролиз I
------->------С—СН2----
ОН
Замена гидроперекисных групп на гидроксильные уменьшает склонность полипропилена к процессам окисления.
Содержащий гидроксильные группы полипропилен способен окрашиваться азокрасителями. Введение гидроксильных групп в качестве способа придания восприимчивости к красителям имеет определенные преимущества по сравнению с привитой сополимеризацией с той же целью. Одним из таких преимуществ является исключение сложных процессов идентификации продуктов прививки.
Наличие в макромолекулах гидроксильных групп позволяет осуществлять различные превращения в цепях. Полипропилен, содержащий гидроксильные группы, способен сшиваться диизоцианатами [99].
Описаны процессы окислительной деструкции полиолефинов, в частности полипропилена, предназначенного для переработки в волокно, в спиртовых и водных суспензиях и в растворах при нагревании под действием воздуха [32, 100]. При глубокой окислительной деструкции атактического [101] и изотактического [102] полипропилена получаются воскоподобные продукты. Скорость деструкции повышается, если нагревание проводится в присутствии перекисного соединения [103, с. 198], например ди-трет-бутилперекиси [101]. Катализатором окисления атактического полипропилена может служить ацетилацетонат железа [145].
Весьма энергично, практически без индукционного периода протекает окисление полипропилена и полиэтилена под воздействием озона [32, 144]. Описано также использование таких окисляющих агентов, как бихромат калия, растворенный в серной кислоте перманганат калия [104], азотная кислота или двуокись азота [105, 158], перекиси [154]. Различные физические воздействия на полиэтилен и полипропилен в присутствии кислорода (деструкция под влиянием ультразвука, электрической дуги, коронного и тлеющего разрядов) также сопровождаются окислением [32]. •
Окисление под действием ионизирующих излучений в воздушной среде, уже упоминавшееся в связи с активацией полиолефинов для последующей привитой сополимеризации.
101
используется и в других целях, например для повышения адгезионной способности полиэтилена [156, с. 36; 157]. Условия процесса облучения могут быть подобраны таким образом, что значительное повышение адгезии в результате образования в поверхностном слое полярных кислородсодержащих групп достигается практически при сохранении остальных свойств полимера, в том числе и такой чувствительной к полярности характеристики, как тангенс угла диэлектрических потерь. Достижению этой цели способствует применение возможно малых доз излучения (до 1—5 Мрад) и высоких мощностей дозы. В этих условиях окисление ограничено лишь тонким поверхностным слоем, поскольку диффузия заметных количеств кислорода в глубь образца за короткое время облучения исключена.
При создании адгезионного шва полиэтилена с другим материалом при повышенных температурах, например горячим прессованием, зависимость адгезии от величины поглощенной дозы излучения имеет вид кривой с максимумом. Это объясняется одновременным протеканием при облучении двух противоположно влияющих на адгезию процессов: окисления и сшивания. Повышение дозы на участке кривой, соответствующем величинам доз ниже оптимальной, приводит к возрастанию адгезии благодаря накоплению полярных кислородсодержащих групп на поверхности полиэтилена. Дальнейшее увеличение дозы, однако, сопровождается снижением адгезии, несмотря на возрастание концентрации полярных групп в поверхностном слое из-за превышения оптимального значения вязкости полимера для заданных условий образования адгезионного шва (температура, давление, продолжительность, природа и характер поверхности другого материала, участвующего в адгезионном контакте). Высокая вязкость затрудняет изменение конфигурации цепей, необходимое для ориентации диполей полярных групп, введенных в поверхностный слой полиэтилена, и их эффективный контакт с поверхностью другого материала. В результате снижается доля полярных групп, способных к электростатическому взаимодействию и участвующих в создании адгезионного соединения. Повышенная вязкость полиэтилена может препятствовать также заполнению пор и дефектов в микрорельефе поверхности другого материала и тем самым снижать величину поверхности адгезионного контакта.
Примером могут служить представленные в табл. 18 сведения об изменении адгезии полиэтилена высокой плотности марки П-4007-ТК к медной оксидированной фольге и тангенса угла диэлектрических потерь в зависимости от поглощенной дозы у-излучения. Облучению в воздушной среде при комнатной температуре подвергались содержащие термостабилизатор (0,1 вес.% тиоалкофена БМ) и нестабилизированные прессованные образцы полиэтилена. Мощность дозы составляла 0,23 Мрад/ч. О степени окисления при облучении судили по интенсивности полосы 1720 см-1, соответствующей поглощению
Ю2
Таблица 18
Свойства облученного на воздухе стабилизированного и нестабилизированного полиэтилена высокой плотности *
Погло-щеяная доза, Мрад Оптическая п“о।ноеть полосы 1720 см-1 Адгезия к \ дированно ГС/ ОДНОЙ G1.CH-й фольге, см Тангенс у ТрИ ЧсСКИ s 10б Гц, Л"> т.иэлек-пот ерь при tg а-104
со стабилизатором без стабилизатора со стаби лизатором без стабилизатора со стаби- 1 лизатором без стабилизатора
0 0,1 0,1 800 800 2 о
2,5 0,53 1,02 2600 2600 2 5
5,0 0,65 1,24 2500 2500 2 5
8,0 0,76 2000 — 2
10 1,41 1,51 2000 2400 3 6
20 — 1,48 — 1100 — 6
30 1,50 1,81 1100 800 5 7
40 — 1,80 1000 800 6 8
* По данным Б. Г. Федотова, М.
П. Эйдельнант, А. Л. Гольденберга и
автора.
карбонильными группами. Величину адгезии определяли методом отрыва медной оксидированной фольги толщиной 35 мкм, предварительно нанесенной на полиэтиленовые пластины толщиной 2 мм прессованием (температура 210 °C, удельное давление 100 кгс/см2, продолжительность 10 мин, выдержка при 210 °C после снятия давления 10 мин, охлаждение со скоростью около 8°C в мин).
Как видно из табл. 18, облучение до 2,5 Мрад более чем втрое повышает адгезионную способность полиэтилена. При этом тангенс угла диэлектрических потерь стабилизированного полиэтилена не изменяется. Дальнейшее увеличение поглощенной дозы ведет к снижению адгезии, несмотря на возрастание содержания кислородсодержащих полярных групп. Тангенс угла диэлектрических потерь остается небольшим до дозы около 10 Мрад.
Для повышения адгезионной способности полиэтилена и полипропилена использовали обработку активированными газами (кислородом, гелием, азотом) в состоянии «холодной» плазмы [165].
Окисление полиэтилена различными способами с целью повышения его адгезионной способности обстоятельно рассмотрено в монографии Белого, Егоренкова, Плескачевского [166].
Термическая деструкция
В отсутствие кислорода полиэтилен отличается большой устойчивостью к термическому воздействию [106, с. 353; 107, с. ЮЗ]. Нагревание полиэтилена без доступа кислорода не сопровождается деструкцией вплоть до достижения
103
температур около 290 ГС [108, 109]. Дальнейшее повышение Температуры приводит к уменьшению молекулярного веса. При температуре выше 360 °C выделяются газообразные продукты деструкции, полное разложение полиэтилена происходит при температуре около 475 °C. Основной продукт деструкции представляет собою воскообразное вещество [108], молекулярный вес которого независимо от температуры пиролиза имеет величину 692 (средний молекулярный вес исходного полиэтилена 20 000). Кроме воскообразного продукта выделяются незначительные количества низкокипящих углеводородов и газов, в основном метана. В продуктах термической деструкции полиэтилена содержание этилена невелико.
Это отличает полиэтилен от многих полимеров (полистирола, полиизобутилена, полиакрилатов и др.), разлагающихся с образованием соответствующего мономера.
Процесс термической деструкции полиэтилена удовлетворительно объясняется с помощью механизма, предложенного Симха с сотрудниками [НО—112]. Полимерные цепи разрываются по связям С—С с образованием свободных радикалов (инициирование):
НН1НН НН НН
I I I I I II II
---с—С—:—С—С----—> -----С—С • + • с—с-
I I i I I II II
Н Н : Н Н НН НН
Макрорадикалы могут акцептировать атом водорода от другой или собственной цепи с образованием ненасыщенной группы на конце цепи и нового свободного радикала (передача цепи):
Н Н ! I —с—с
Н н
н н [н] н I н
I I T I [ I
+----С—С—С-г-С—•
' I I I | I
Н Н НгН
Н
Обрыв цепи осуществляется при рекомбинации свободных радикалов:
НН н н н н
II 1111
2-С—с. —> ---С—С—С—С-
II 1111
НН н н н н
Эти представления были дополнены указанием на возможность деструкции и без образования свободных радикалов [113,
104
114]. Напряжения, возникающие в макроцепях при их тепловом движении, могут приводить к разрыву связей С—С. При достаточном количестве атомов водорода (при незначительной разветвленности цепи) может происходить внутримолекулярный перенос атома водорода к месту разрыва:
н [н] нХн н н
। т । :\i । ।
-С—С —C-f-C—С—С....
I I I I I I
НН Н I н н н
Если часть атомов водорода замещена, например, на метиль-
ные группы, перенос водорода осуществляется лишь при части разрывов цепи, другая же часть разрывов приводит к образо
ванию свободных радикалов, продолжающих распад цепи вплоть до образования мономера. Так, образование мономера
при пиролизе полиизобутилена можно представить следующим образом [107]:
СН3 Н СН3! Н СН, Н
I Mill I
---С—С—С — i—с—с—с---—>
I I I i I I I
СН3 Н СН, j н сн3 н
сн3 н сн3 сн3 н сн3
III III
—с—с—с • + • с—с—с-
III III
сн3 н сн3 сн3 н н
При термической деструкции полиизобутилена действительно выделяется до 18% мономера.
Энергия активации термической деструкции возрастает с увеличением молекулярного веса полимера. Так, энергия активации термической деструкции полиэтилена с молекулярным весом 11000 равна 46 ккал/моль, при значениях молекулярного веса 16000 и 23 000 энергия активации составляла 52,6 и 66,1 ккал/моль соответственно [115]. Влияние степени разветвленности на скорость термической деструкции было показано при исследовании сополимеров этилена с различными количествами пропилена [116]. По мере увеличения числа метильных ответвлений скорость деструкции возрастает.
105
Снижение молекулярного веса полиэтилена и сополимеров этилена методом термической деструкции использовано для улучшения текучести и получения низкомолекулярных воскоподобных продуктов [117—120]. Например, полиэтилен или сополимер этилена с плотностью более 0,94 г/см3 и молекулярным весом выше 20 000 подвергают нагреванию в отсутствие кислорода при температурах 270—300 °C в течение времени, необходимого для уменьшения показателя текучести расплава не менее чем на 15%. Термическое воздействие может быть совме
щено с механическим, например, в экструдере непрерывного действия [119].
Полипропилен, в главной цепи которого каждый второй атом углерода третичный, характеризуется меньшей прочностью связей С—С в основной цепи, чем полиэтилен. Разрыв связей С—С при термической деструкции полипропилена протекает преимущественно с переносом атома водорода:
—С—С—С-+-С
I Д । I/1 н Гн, н /н
Н СН3Н1СН3Н сн3
-с—с-
I I
[ н н
н сн,н
I II
с—с=с +
н н
сн3 н сн3
I I I
с—с—с—
н н н
Повышенная подвижность атома водорода при третичном атоме углерода благоприятствует этому процессу [142, 143]. Разрыв цепей с образованием свободных радикалов, последующим их распадом и образованием пропилена значительно менее вероятен. Об этом свидетельствует весьма незначительное содержание пропилена в продуктах термической деструкции полипропилена [107].
Термическую деструкцию полипропилена, как и полиэтилена, используют для уменьшения молекулярного веса [120, 123]. Так, полипропилен с улучшенной способностью к переработке получают нагреванием полимера с высоким молекулярным весом при температуре до 400°C в вакууме или в среде инертного газа [122]. Снижения вязкости расплава можно достигнуть при одновременном воздействии тепла и механических колебаний, например вызванных ультразвуковым воздействием [123]. Отмечено повышение скорости термической деструкции полипропилена в присутствии добавок (2—5%) глицидилметакри-лата [141].
Литература
1. Джонс Г. В кн.: Химические реакции полимеров. Под ред. Е. Фет-теса. М., «Мир», 1967. 504 с. — 2. Англ. пат. 481515, 1938 —3. Пат. США 2849431, 1958, —4. Пат. США 2389803, 1946. —5. Пат. США 2422919, 1947. —
10$
6. Пат. США 2481188, 1949.—7. Кренцель Б. А., Ильина Д. Е., Ады-лов С А. Пласт, массы, 1963, № 6, с. 3. — 8. Пат. США 2405971, 1946.— 9. Пат. США 2503252, 1950, — 10. Michail R., Gherghel F., S fanes с u M., Kornbaum S. Kev. chim., 1961, v. 12. № b. c. 2/b.
11. Smook M. A., Remington W. J., Strain D. E. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Плата. М., «Мир», 1964. 594 с.— 12. Франц, пат. 1324506, 1962. — 13. Франц, пат. 1320413, 1963.— 14. Пат. США 2571901, 1951. — 15. Канад, пат. 471037, 1951.— 16. Myers С. S. Ind. Eng. Chem., 1952, v. 44, p. 1095.— 17. Пат. США 2183556, 1940.— 18. Пат. США 2926159, 1960.— 19. Пат. США 2592763, 1952.— 20. Thompson Н. W., Torkington Р. Trans. Faraday Soc., 1945, v. 41, p. 246.
21. Thompson H. W., Torkington P. Proc. Roy. Soc., 1945, v. 184, Ser. A, p. 2. — 22. Oakes W. G., Richards R. B. Trans. Faraday Soc., 1946, v. 42A, p. 197. — 23. Smook M. A., Pie ski F. T., Hammer C. F. Ind. Eng. Chem., 1953, v. 45, p. 2731. — 24. Ады лов С. А., Л еще в а И. Ф., Ильина Д. Е. н др. Нефтехимия, 1963, т.З, Ns 1, с. 82.—25. В г о о k s R. Е., Strain D. Е., McAlevy A. India Rubber World, 1953, v. 127, p. 791.— 26. Пат. США 2541492, 1951.—27. Ады лов С. А. Автореф. канд. дисс. М., 1963. — 28. Ады лов С. А., Ильина Д. Е., Кренцель Б. А., Шишкина М. В. Высокомол. соед., 1963, т. 5, № 3, с. 316. — 29. Пат. США 2497046, 1950.— 30. Пат. США 2811468, 1958.
31. Англ. пат. 710523, 1954. — 32. Манясек 3., Белл уш Д. Полипропилен. Под ред. В. И. Пилиповского и И. К- Ярцева. Л., «Химия», 1967, —33. Ми иске р К. С., Этлис В. С. ДАН СССР, 1958, т. 123, Ns 6, с. 1041. — 34. Кренцель Б. А., Топчиев А. В., Ильина Д. Е. ЖПХ, 1959, т. 32, № 6, с. 1404. — 35. Англ. пат. 843209, 1960. — 36. Англ. пат. 811848, 1959.— 37. Campbell Т. W., Lyman D. J. J. Polymer Sci, 1961, v. 55, p. 169. — 38. Англ. пат. 834905, 1960. — 39. Campbell T. W., Haven A. C. J. Appl. Polymer Sci., 1959, v. 1, p. 78. — 40. Shu К a m b a r a, Taka о Ohshika. Когё Кагаку Дзасси, 1959, в. 62, р. 1781; Chem. Abstr., 1962, v. 57, p. 13959.
41. Итал. пат. 591501, 1958. — 42. Shu Kambara, Takao Ohshika. J. Chem. Soc. Japan, Ind. Chem. Sec., 1959, v. 62, № 11, p. 1781. — 43. Англ, пат. 877880, 1961. — 44. Wasaburo Kawai, Shigeru Tsutsumi. Ниппон Кагаку Дзасси, 1959, v. 80, p. 780; Chem. Abstr., 1961, v. 55, p. 3410.— 45. Кренцель Б. А., Семен и до Г. Е., Ильина Д. Е. Высокомол. соед., 1963, т. 5, Ns 4, с. 558. — 46. Кренцель Б. А., Семе ни до Г. Е., Ильина Д. Е., Шишкина М. В. Высокомол. соед., 1963, т. 5, Ns 4, с. 564. — 47. Михайлов Н. В., Токарева Л. Г., Клименков В. С. Коллоидн. ж., 1956, т. 18, с. 578. — 48. Arlmann Е. J. J. Polymer Sci., 1954, v. 12, № 67, р. 543, 547.-49. Пат. США 2906743, 1960.—50. Итал. пат. 537429, 1955.
51. Австр. пат. 210135, 1960. — 52. Visser Р. J. Plastica, 1962, v. 15, р. 282. — 53. Япон. пат. 15277, 1961. — 54. Англ. пат. 868158, 1961. — 55. Итал. пат. 597560, 1959. — 56. Якубович А. Я., Зиновьев Ю. М. «Успехи химии», 1947, т. 17, № 5, с. 581. — 57. Gilbert Е. Е., Jones Е. Р. Ind. Eng. Chem., 1951, v. 43, Ns 9, p. 2028. — 58. Gilbert E. E., Jones E. P. Ind. Eng. Chem., 1954, v. 46, Ns 9, p. 1895. — 59. Пат. США 2640048, 1953.— 60. Пат. США 2416061, 1947.
61. Пат. США 2586363, 1952.—62. Schumacher Н. J., Stauff J. J. Chimie, 1942, v. 55, р. 341. — 63. Англ. пат. 815234, 1959. — 64. Пат. ФРГ 970578, 1958.— 65. Пат. США 2889259, 1959.—66. Пат. США 2556879, 1951 — 67. Пат. США 2658883, 1953.— 68. Пат. США 2578904, 1951,—69. Warner R. R. Rubber Age, 1952, v. 71, p. 205. — 70. Catton N. H. Kaut. u. Gummi, Bd. 9, Ns 11, S. 280.
71. Salomon G., Konigsberger C., Ultee A. J. Rubber Techn. Cont. Proc., 1948, p. 106. — 72. Salomon G., Konigsberger G. Rec. trav.
107
chim., 1950, v. 69, p. 711; 1951, v. 70, p. 545. — 73. Salomon G., Ultee A. Rec. trav. chim., 1950, v. 69, p. 95; 1951, v. 70, p. 537. — 74. Nersasian A., Andersen D. Proceeding of Intern. Rubber Conference. Washington, 1959, p. 537. — 75. Nersasian A., Anderson D. E. J. Appl. Polymer Sci., 1960, v. 4, p. 74. — 76. Ильина Д. E., Крендель Б. А., Топчиев А. В. Высокомол. соед., 1961, т. 3, с. 995. — 77. Пат. США 2972604, 1961. — 78. Пат. США 3050503, 1962. — 79. S о b u е Н., Т a j i m a Y„ Т a b a t a Y. Когё Катаку Дзасси, 1959, v. 62, р. 1774; Chem. Abstr., 1962, v. 57, р. 15332. — 80. О h s h i-k a T. Когё Катаку Дзасси, 1961, v. 64, № 7, p. 1299; РЖХим., 1962, Xs 19, реф. 19П30.
81. Итал. пат. 591501, 1958. — 82. Авт. свид. СССР 149773, 1962; Билл, изобр., 1962, Xs 17. — 83. Schroeder J. Р., Sopchak W. P. J. Polymer Sci., 1960, v. 47, p. 417. — 84. Пат. США 3008939, 1961. — 85. Беллуш Д., Маня сек 3., Лазар М. Высокомол. соед., 1963, т. 5, с. 145. — 86. Англ, пат. 849058, 1960.—87. Manasek Z., Bellus D., В ohm er В. Chem. zvesti, 1963, d. 17, s. 318. — 88. Leonard E. C., Loed W. E., Mason J. H., Wheel wight W. L. J. Appl. Polymer Sci., 1961, v. 5, p. 157. — 89. Leonard E. C., Loed W. E., Mason J. H., Stenstrom J. A. J. Polymer Sci., 1961, v. 55, p. 799.—90. Haywood С. К. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и Н. А. Платэ. М., «Мир», 1964. 594 с.
91. Гордон Г. Я. Стабилизация синтетических полимеров. М., Госхим-издат, 1963. 299 с. — 92. Rugg F. М., Smith J. J., Bacon R. С. J. Polymer Sci., 1954, v. 13, p. 535. — 93. Grafmiiller F., Husemann F. Mak-romol. Chem., 1960, v. 40, p. 161, 172. — 94. Pross A. V., Black R. M. J. Soc. Chem. Ind., 1950, v. 69, p. 113. — 95. Hawkins W. L., Matreyek W., Winslow F. H. J. Polymer Sci., 1959, v. 41, p. 1. — 96. Англ. пат. 581279, 1946. — 97. Natta G., Beati E., Sever ini F. J. Polymer Sci., 1959, v. 34, p. 685. — 98. Минскер К. С., Шапиро И. 3., Разуваев Г. А. Высокомол. соед., 1962, т. 4, № 3, с. 351. — 99. Англ. пат. 832068, 1960. — 100. Англ, пат. 910040, 1962.
101. Пат. США 2828296, 1958. — 102. Пат. США 2911384, 1959.— 103. Фреунд Л., Амброис Л. Полипропилен. Под ред. В. И. Пилипов-ского и И. К- Ярцева. Л. «Химия», 1967. — 104. Бельг, пат. 569129, 1958.— 105. Пат. США 2987501, 1961. — 106. Schwarz A., Cramer G. В кн.: Полиэтилен и другие полиолефины. Под ред. М. В. Козлова и Н. А. Платэ. М., «Мир», 1964. 594 с. —107. Мадорский С. Термическое разложение органических полимеров. «Мир», 1967. 328 с. — 108. Made г sky S. L., Straus S., Thompson D., Williamson S. J. Polymer Sci., 1949, v. 4, p. 639. — 109. Oakes W. G., Richards R. B. J. Chem. Soc., 1949, p. 2929.— 110. Sim ha R., Wall L. A., Blatz P. J. J. Polymer Sci., 1950, v. 5, p. 615.
111. Simha R., Wall L. A. J. Phys. Chem., 1952, v. 56, p. 707.— 112. Simha R., Wall L. A., Bram J. J. Chem. Phys., 1958, v. 28, p. 894.— 113. Ma dor sky S. L., Straus S. J. Res. Nat. Bur. Stand., 1954, v. 53, p. 361.— 114. Madorsky S. L. Soc. Plast. Eng. J., 1961, v. 17, p. 665,— 115. J e 11 i n e k H. H. G. J. Polymer Sci., 1949, v. 4, p. 13. — 116. Wall L. A., Straus S. J. Polymer Sci., 1960, v. 44, p. 313.— 117. Пат. США 2372001, 1942.— 118. Англ. пат. 569043, 1945.— 119. Англ. пат. 1000911, 1965,— 120. Пат. США 3230288, 1966.
121. Wisseroth К- Angew. Chem., 1960, Bd. 72, Xs 22, s. 866.— 122. Япон. пат. 15807, 1965. — 123. Япон. пат. 1654, 1965. —124. Рафиков С. Р., Ергебеков М. Е., Челнокова Г. Н., Ершова Г. В. Изв. АН СССР. Сер. хим., 1965, Xs 3, с. 526.— 125. Sander М., Steiniger Е. J. Macromol. Sci., 1968, v. 2, Ser. C, Xs 1, p. 57. — 126. Авт. свид. СССР 255901; Бюлл. изобр., 1969, Xs 34, с. 11. — 127. Каркозова Г. Ф., Любецкий С. Г., 3 ю з и н а Л. И. и др. Пласт, массы, 1970, Xs 5, с. 33. — 128. Каркозова Г. Ф. Автореф. канд. дисс. Л., 1973. — 129. Мартынов М. А., Вылегжанина К. А. Пласт, массы, 1965, Xs 8, с. 50. — 130. К а р к о-
108
зова Г. Ф., Сирота А. Г., Любецкий С. Г. и др. ЖПХ, 1973, т. 46, № 5, с. 1149.
131. К а р к о з о в а Г. Ф, С и р о т а А. Г., Любецкий С. Г. и др. Вы-сокомол. соед., 1973, т. 15, сео. Б. № 10. с. 730. — 132. П v кшзиский 51. Д., Ко л я сев а В. А., Зюзина Л. И. и др. Пласт, массы. 1970, Ns 11, с. 51.— 133. Phillips Р. J., MacKnight W. J. J. Polymer Sci., 1970, v. 8, Ser. В, № 2, p. 87.— 134. Кантерино, Кале. «Химия и технология полимеров», 1963, № 3, с. 91.— 135. Hein Ike W„ Keller F. Plaste u. Kaut., 1971, Bd. 18, № 10, S. 732.— 136. Oswald M. I., Kubu E. T. SPE Trans, 1963, v. 3, p. 168.— 137. Крендель Л. Б, Литманович А. Д, Пастухова И. В, А ч а с а и д я и В. А. Высокомол. соед, 1971, т. 13, сер. А, № 11, с. 2489.— 138. Лозовик Г. Я, Маркова Е. В, Донцов А. А, Клинов И. Я- «Лакокрасочные материалы и их применение», 1967, № 6, с. 45.— 139. Карякина Л1, И, Л а вен де ле С. М, Майорова Н. В. и др. «Механика полимеров», 1969, Ns 3, с. 387. — 140. Jobst К. Plaste u. Kaut, 1967, Bd. 14, Ns 3, S. 154.
141. Mizutani Y, Yamamoto K, Matsuoka S, Hisano S. Bull. Chem. Soc. Japan, 1967, v. 40, Ns 6, p. 1526. — 142. Буният - заде A. A, Андросова В. M, Булатникова Э. Л. ДАН АзербССР, 1970, т. 26, Ns 5, с. 34. — 143. Буният-заде А. А, Андросова В. М, Булатникова Э. Л. и др. Высокомол. соед, 1970, т. 12, сер. A, Ns 11, с. 2494.— 144. Кефели А. А, Разумовский С. Д, Зайков Г. Е. Высокомол. соед, 1971, т. 13, сер. A, Ns 4, с. 803. — 145. Stilava S. S, J a d г n i-cek В. R, Reich L. J. Appl. Polymer Sci, 1971, v. 15, Ns 9, p. 2185.— 146. Hara К, I mo to T. Koll. Z. u. Z. Polymere, 1969, Bd. 229, № 1, S. 4. — 147. E д e м с к а я В. В, Миллер В. Б, Шляпников Ю. А. ДАН СССР, 1971, т. 196, № 5, с. 1121.— 148. J5 а л ь т е п а с Р. А, Бальте-нене Я. Ю. Высокомол. соед, 1970, т. 12, сер. Б, № 5, с. 373. — 149. Старение и стабилизация полимеров. Под ред. М. Б. Неймана. М, «Наука», 1964. 232 с. — 150. Старение и стабилизация полимеров. Под ред. А. С. Кузьминского. М, «Химия», 1966. 210 с.
151. Брагинский Р. П, Финкель Э. Э, Лещенко С. С. Стабилизация радиационно-модифицированных полиолефинов. М, «Химия», 1973. 199 с. — 152. Березин И. В, Денисов Е. Т, Эмануэль Н. М. В кн.: Вопросы химической кинетики, катализа и реакционной способности. М, Изд. АН СССР, 1955.— 153. Biggs В. S, Hawkins W. D. Mod. Plast, 1953, v. 37, р. 121. — 154. Wunsch К, Stedtler L. Plaste u. Kaut, 1969, Bd. 16, Ns 9, S. 659.— 155. Стоцкая Л. Л, Хинькис С. С, Матвеева Е. Н, Кренцель Б. А. Пласт, массы, 1967, Ns 8, 14. — 156. Князев В. К, Сидоров Н. А. Применение облученного полиэтилена в радиоэлектронике. М, «Энергия», 1972. 64 с.— 157. Спицын Викт. И, Зубов П. И, Кабанов В. Я, Г роз и иска я 3. П. Высокомол. соед, 1966, т. 8, Ns 4, с. 604.— 158. Armond V. J, Atkinson J. R. J. Appl. Polymer Sci, 1970, v. 14, Ns 7, p. 1833.— 159. Olsen D. A, Os ter a as A. J. J. Polymer Sci, 1969, v. 7, Ser. Al, Ns 7, p. 1921, — 160. Olsen D. A, Osteraas A. J. J. Polymer Sci, 1969, v. 7, Ser. Al, Ns 7, p. 1913.
161. Пат. США 2832696, 1958; 2879177, 1959.— 162. Olsen D. A, Osteraas A. J. J. Polymer Sci, 1969, v. 7, Ser. Al, Ns 7, p. 1927. — 163. 0 1-s e n D. A, О s t e r a a s A. J. J. Appl. Polymer Sci, 1969, v. 13, Ns 7, p. 1523. — 164. Osteraas A. J, Olsen D. A. J. Appl. Polymer Sci, 1969, v. 13, Ns 7, p. 1537.— 165. Hall Y. R, Westerdahl C. A. L, Devine A. T, Bodnar M. J. J. Appl. Polymer Sci, 1969, v. 13, № 10, p. 2085. — 166. Белый В. A, E г о p e н к о в H. И, Плескачевский Ю. М. Адгезия полимеров к металлам. Минск, «Наука и техника», 1971. 288 с.
Глава V
Полиолефины с пространственной структурой
Получение полиолефинов с пространственной структурой составляет важное направление их модифицирования. В результате процессов сшивания значительно улучшаются такие ценные технические свойства, как теплостойкость, прочность, сопротивляемость растрескиванию, стойкость к действию растворителей и др. Для сшивания можно использовать реакции специально введенных функциональных групп, радиационно-химическое и фотохимическое воздействие и сшивающие агенты, главным образом перекисного типа.
Получение пространственных структур с помощью реакций специально введенных функциональных групп
Введение в цепи полиэтилена, полипропилена, сополимеров этилена с пропиленом и другими а-олефинами функциональных групп с помощью процессов сульфохлорирования, галогенирования, фосфонирования позволяет осуществлять сшивание в результате реакций с различными реагентами. Так, наиболее подробно исследованное и используемое в промышленности сшивание (вулканизация) сульфохлорированного полиэтилена [1, 2] может протекать при взаимодействии сульфохлоридных групп с окислами металлов с образованием солей или с диаминами с образованием сульфамидных групп. При взаимодействии с диаминами поперечные связи образуются также благодаря реакции их с атомами хлора, находящимися в 0-положении по отношению к сульфохлоридным группам и связанными с третичными атомами углерода. Реакция сопровождается выделением хлористого водорода. Образование двойных связей при отщеплении хлористого водорода и сернистого ангидрида от сульфохлорированного полиэтилена под влиянием тепла позволяет проводить сшивание полиэтилена серой в присутствии ускорителей, применяемых при вулканизации каучуков.
110
Основным используемым в технике способом является сшивание сульфохлорированного полиэтилена (с содержанием хлора 25—40%) с помощью окислов двух- и поливалентных металлов (магния, свинца и др.) [3, с. 328; 4]. Сшивание окислами металлов проводится при температуре 125—160"С. Процесс протекает только в присутствии влаги. Предполагается, что это связано с гидролизом сульфохлоридных групп, предшествующим образованию поперечных связей между цепями [5]:
RSO2C1 + Н2О —> НС1 + RSO2OH
МеО + 2НС1 —► МеС12 + Н2О
МеО + 2RSO2OH —> RSO2O—Me—OSO2R + H2O
Продукты сшивания отличаются от полиэтилена весьма высокой стойкостью к действию озона, кислорода, азотной и хромовой кислот и других окисляющих веществ. По комплексу механических свойств сшитый сульфохлорированный полиэтилен близок к вулканизованным синтетическим каучукам. Сшитый сульфохлорированный полиэтилен отличается от них большими значениями твердости, прочности на разрыв, модуля упругости, очень большой сопротивляемостью растрескиванию и истиранию и меньшей величиной удлинения.
Сопоставление свойств сшитых материалов, полученных из полимеров с разной степенью кристалличности, указывает на преимущества продуктов сульфохлорирования сравнительно аморфных полимеров. Например, продукты сшивания сульфо-хлорированного аморфного сополимера этилена с пропиленом, содержащего 20 вес.% хлора (I), имеют близкие или более высокие показатели при меньшей степени замещения, чем сшитый сульфохлорированный полиэтилен высокого давления, содержащий 30 вес. % хлора (II) [6, 7]:
I п
Прочность при разрыве, кгс/см2 . . 90—125 100—130
Относительное удлинение, % ... 300—400 350—400
Модуль упругости при 100%-ном
удлинении, кгс/см2........... 10—15 12—15
Твердость по Шору (шкала А) . . 50—60 60—65
Остаточное удлинение, %............... 10 20—30
Упругость, %........................ 35—45 25
Глубокая аморфпзация полипропилена при сульфохлорировании позволяет использовать для получения сшитых эластомеров продукт со значительно меньшим содержанием хлора, чем в случае полиэтилена.
Предпочтительно использование сульфохлорированного полипропилена с содержанием хлора до 20%, степенью кристалличности до 10% и молекулярным весом более 5000 [8, с. 138]. Так же как и при сшивании сульфохлорированного полиэтилена, сшитый продукт при использовании окислов металлов получается только в присутствии воды. Количество окисла металла обычно
111
находится в интервале 10—40 вес.%. Температура при сшивании составляет 150—170 °C. Кроме окислов металлов в качестве сшивающих агентов можно применять тпомочевину и диамины [9, 137], бензальдимины и циклогексилимины, получаемые конденсацией альдегидов и кетонов с аминами [138, с. 306], оксииминовые производные ароматических диаминов [139].
Продукты сшивания сульфохлорированного полипропилена имеют высокие показатели обратимости деформации, эластичности, прочности и весьма высокую озоностойкость [8, с. 139].
Гидролизованные продукты хлорфосфонирования
(—С—РОС12.), содержащие фосфоновые группы —РО(ОН)2, также способны сшиваться под влиянием окислов металлов (свинца, цинка и др.) [10, с. 220; 11]. Содержание фосфора в фосфонированном сополимере этилена с пропиленом (90% звеньев пропилена), позволяющее получать сшитый окисью цинка материал, составляет около 1% [12]. Сшитый материал на основе фосфопированного полипропилена [13] характеризуется высокой озоностойкостью, при содержании фосфора около 5% обладает свойством самозатухания [8, с. 139].
Известно сшивание хлорированного полипропилена [14—17] с помощью окислов цинка, свинца, магния. Образующиеся сшитые эластомеры отличаются хорошей озоностойкостью. Сшивание диаминами позволяет получать материал с высокими прочностными характеристиками [118].
Ненасыщенные группы в тройных сополимерах этилена, пропилена и какого-либо диена (см. гл. II) позволяют осуществлять сшивание этих сополимеров подобно вулканизации каучуков с помощью серы [19, с. 95; 20, с. 304]. Тройные сополимеры могут также сшиваться, взаимодействуя с дибензоилхинонди-оксимом в присутствии окиси свинца. Получаемый вулканизат имеет более высокую стойкость к старению, чем продукт сшивания серой. Сшитый эластомер с повышенной теплостойкостью и эластичностью может получаться при использовании в качестве агентов сшивания фенолоформальдегидных смол. В качестве фенольного сырья при синтезе этих смол используют н-ок-тилфенол, трет-бутилфенол, пщ'-диоксидифенилпропан.
Способность к образованию поперечных связей полиолефинам можно придавать введением в их макромолекулы звеньев, содержащих функциональные группы. Например, карбоксилсодержащие полимеры, получаемые сополимеризацией этилена с акриловой или метакриловой кислотой, могут подвергаться сшиванию, подобно карбоксилсодержащим каучукам [21—23], окислами и солями поливалентных металлов.
Карбоксилированным полиолефинам может придаваться способность сшиваться и без применения агентов отверждения. Это достигается введением в полимер путем полимераналогич-ных превращений функциональных групп, способных к обра-
112
зованию межмолекулярных связей при последующей термической обработке. Примером может служить получение термореактивного полимера из продукта прививки ненасыщенной карбоновой кислоты к полиолефину в результате взаимодействия части карбоксильных групп с эпихлоргидрином [107]. Процесс заключается в нейтрализации 60—80% карбоксильных групп спиртовым раствором щелочи и последующей обработке эпихлоргидрином с образованием эпоксидных групп:
---сн—сн2----
I о
I сн2
I
RC-COOH
сн2
RC—СООН
сн2
RC—СООН
I
+ кон ----->
• сн—сн2----
I о
I сн2
I
RC—COOK
сн2
RC—СООН
I сн2
RC—COOK
+ С1СН2—сн—сн2
-ко
---СН-СН2---- I
О
I
сн2 о о
I / / \
RC—С—О—СН2—СН—СН2
сн2
RC—СООН
сн2
RC—COOK
Непрореагировавшие карбоксильные группы в водородной и солевой формах ускоряют сшивание продукта под действием тепла.
Специфическими материалами с пространственной структурой являются так называемые иономеры, получаемые из сополимеров а-олефинов (этилена, пропилена, а-бутилена и др.) с моно-или дикарбоновыми ненасыщенными кислотами нейтрализацией части карбоксильных групп солями или гидроокисями металлов (лития, калия, натрия, магния, стронция, цинка, меди, свинца и др.) [24—27, 105, 106]. Содержание карбоксилсодержащих звеньев в сополимерах не превышает 25 мол.%. Замещению
113
на ионы металла, предпочтительно щелочного или щелочноземельного, подвергается не менее 10% атомов водорода карбоксильных групп. Оптимальные свойства иономеров достигаются при нейтрализации 50—80% карбоксильных групп.
В качестве карбоксилсодержащих полиолефинов кроме продуктов сополимеризации олефинов с непредельными кислотами могут быть использованы также продукты омыления сополимеров олефинов с эфирами ненасыщенных кислот и прививки ненасыщенных кислот к полиолефину.
Ионный характер межмолекулярных связей обусловливает особенности свойств таких материалов. Основная особенность иономеров заключается в обратимости процесса их сшивания. При повышенных температурах в процессах переработки происходит нарушение большей части ионных межмолекулярных связей и материал ведет себя как термопласт. После охлаждения ионные связи восстанавливаются и полимер приобретает свойства материала с пространственной структурой.
Иономеры обладают весьма ценным сочетанием свойств. Для них характерны высокие величины прочности и удлинения при разрыве; стойкость к действию растворителей, масел; сопротивляемость растрескиванию под влиянием внутренних напряжений; дугостойкость; большая, чем у полиэтилена, адгезия к различным материалам; восприимчивость к красителям. Свойства иономера Surlyn А [28, 29] приведены ниже:
Плотность, г/см3............................... 0,93—0,96
Показатель текучести расплава (при 190 °C), г/10 мин.................................... 0,1—4,0
Прочность при растяжении, кгс/см2 .......... 245—390
Прочность при ударном растяжении, кгс • см/см3 124—330
Относительное удлинение при разрыве, % . . . 300—400
Модуль упругости, кгс/см2 .................. 1900—2800
Ударная вязкость по Изоду, кгс • см/см2 .... 30—77
Теплостойкость по Вика, °C.................. 71—96
Максимальная температура переработки, °C . . 330
Температура эксплуатации (без нагрузки), °C . От —118 до +88
Тангенс угла диэлектрических потерь при
103 Гц................................... 0,0015
Диэлектрическая проницаемость при 103 Гц . . 2,5
Удельное объемное электрическое сопротивление, Ом-см.................................. 0,5 • 1017
Электрическая прочность, кВ/мм.............. 40
' Варьируя содержание карбоксильных групп, количество и природу ионов металлов, можно получать иономеры с различной структурой и свойствами. Кристаллическая структура при образовании ионных связей в сополимерах с малым содержанием карбоновой кислоты (до 1 мол.%) существенно не изменяется [26]. При увеличении содержания кислоты до 7— 10 мол.% происходит значительная аморфизация сополимера, и ионизация карбоксильных групп оказывает меньшее влияние на характер кристаллических образований.
Ш
Аморфизация структуры под влиянием ионогенных групп проявляется в повышении прозрачности иономеров. Это связано с уменьшением рассеяния света кристаллическими структурами, характерного для обычных полиолефинов. Несмотря на аморфизованную структуру, иономеры имеют значительную жесткость. Это объясняется существованием водородных связей между карбоксильными группами. По мере повышения содержания кислоты жесткость полимера, естественно, увеличивается. Разрывная прочность возрастает при увеличении содержания карбоксильных групп и степени их ионизации, однако относительное удлинение при разрыве становится меньше.
Получение пространственных структур радиационно-химическим и фотохимическим методами *
Радиационно-химическое сшивание. Процессам радиационно-химического сшивания, особенностям структуры и свойств сшитых полиолефинов, в первую очередь полиэтилена, посвящена весьма обширная литература, в частности ряд обзорных работ [30, с. 108; 31; 32, с. 184; 33; 34, с. 166].
Из различных видов излучений наиболее перспективны для сшивания полиолефинов в промышленности 0- и у-излучения. Применение ускоренных электронов (0-излучение) позволяет осуществлять сшивание с большой производительностью благодаря возможности получения с помощью ускорителей электронов излучений с высокой интенсивностью. Проникающая способность быстрых электронов, однако, сравнительно невелика. При энергии 2 МэВ равномерное воздействие достигается в толще материала глубиной примерно 1 см. Для сшивания материала толстостенных изделий можно использовать излучение 60Со (у-излучение), хотя интенсивность его обычно значительно уступает интенсивности потоков ускоренных электронов.
Образование редких поперечных связей на начальных стадиях облучения приводит к увеличению молекулярного веса полимера. При дальнейшем облучении с возрастанием числа поперечных связей образуется пространственная структура. Одновременно со сшиванием макроцепей протекает и процесс их деструкции. Соотношение скоростей этих основных процессов в большой мере определяет изменение структуры и свойств полимера в результате облучения и зависит от химического строения макроцепей и характера надмолекулярных образований. Результаты радиационного воздействия существенно связаны также с условиями облучения.
Для полиэтилена характерно преобладание реакций образования поперечных связей над реакциями деструкции. Число
* Раздел написан совместно с М. Д. Пукшанским.
115
актов разрыва связей, приходящееся на один акт образования поперечной связи p/а, определяется с использованием уравнения Чарлзби [31, с. 195]:
₽/а = s + ps
где s — содержание золь-фракции, равное I—g; g— содержание нерастворимой гель-фракции (отношение массы нерастворимой части образца к его массе до экстракции горячим растворителем, например ксилолом).
При одновременном протекании деструкции и сшивания с преобладанием процесса сшивания величина s с увеличением дозы облучения г стремится к некоторому пределу. Этот предел определяется соотношением скоростей деструкции и сшивания и не достигает нуля, если скорость деструкции не слишком мала по сравнению со скоростью сшивания. Экстраполируя зависимость (s + ]/s) от */г до 0, находят p/а (при г—►оо). Величина р/а для полиэтилена [35] составляет 0,18—0,20. Интересно, что в результате деструкции при облучении полиэтилена высокого давления образуется низкомолекулярных углеводородов примерно в шесть раз больше, чем при облучении полиэтилена низкого давления [36]. Хотя причины этого различия в деталях не известны, можно полагать, что оно связано с радиационной нестойкостью участков углеводородной цепи, содержащих третичные атомы углерода.
Отношение p/а, найденное для сшитых под действием р-излу-чения сополимеров этилена с пропиленом (7 мол.%) и этилена с а-бутиленом (3 мол.%), оказалось близким к характерному для полиэтилена и составило соответственно 0,24 + 0,03 и 0,14 ± 0,03 [37, 38].
При радиационно-химическом сшивании полипропилена деструкция основных цепей происходит со скоростью близкой к скорости образования поперечных связей: соотношение p/а составляет примерно 0,75—1,0 [39, 40; 41, с. 268].
Данных, которые позволили бы последовательно проследить связь между поведением при облучении и положением полимера в ряду продуктов полимеризации высших а-олефинов, не имеется. Известно, однако, что полипентен-1 и полигексен-1 под влиянием облучения сшиваются [42]. Экстраполяцией зависимостей содержания золь-фракции от дозы облучения ускоренными электронами показано, что гель-фракция в образцах полипентена-1 обнаруживается при достижении дозы около 24 Мрад, для полигексена эта величина составляет примерно 7 Мрад. При значениях доз, недостаточных для образования гель-фракции, установлено существенное уменьшение молекулярного веса.
Изучение спектров ЭПР облученных полибутена-1 и полипентена-1 позволило обнаружить свободные радикалы, образованные в результате разрыва связей С—Н и в основной цепи, и в ответвлениях [43]. С возникновением неспаренных электро
116
нов на атомах углерода в ответвлениях может быть связано разрушение боковых цепей с образованием виниленовых радикалов;
СН2 I СН2 СН
i II
---сн2-сн-сн2— —> сн4 + —сн2—с—сн2-
При облучении полиизобутилена основной реакцией является деструкция [44, 45]. Предполагается, что одна из причин радиационной нестойкости полиизобутилена заключается в пониженной энергии связей С—С в основной цепи из-за стерических напряжений, создаваемых метильными группами.
Важным, в частности для решения технических задач модифицирования полиолефинов радиационно-химическим методом, является вопрос о связи эффективности радиационнохимического сшивания и степени кристалличности исходного полимера. Имеющиеся по этому поводу данные довольно противоречивы. Чарлзби с сотрудниками и другие авторы [31, 34, 46—50] на основании анализа набухаемости и модуля эластичности облученных образцов полиэтилена высокой и низкой плотности пришли к выводу об одинаковой интенсивности процесса сшивания в кристаллической и аморфной фазах. Лоутон с соавторами [51, 52], напротив, исследуя сшивание полиэтилена при облучении ускоренными электронами при температурах, превышающих температуру плавления кристаллических образований, сделали заключение о протекании процесса сшивания исключительно в аморфных областях. Аналогичный взгляд высказан Шумахером [36]. Малая интенсивность сшивания в кристаллических областях связывается с недостаточной подвижностью свободных макрорадикалов [51—53; 159, с. 97; 160], хотя высказывалось мнение и о протекании процесса сшивания главным образом в кристаллической части полимера [152].
Изучение изменений степени кристалличности и размеров кристаллитов в процессе облучения также свидетельствует о большой скорости сшивания в аморфных областях [54]. Отсутствие однозначных доказательств преимущественности сшивания в аморфных областях все же позволяет, сопоставляя обе точки зрения, отдавать предпочтение мнению об одинаковой интенсивности сшивания в аморфных и кристаллических областях [34].
Между тем сравнение эффективности сшивания полиолефинов с различной степенью кристалличности и разветвленности под воздействием ускоренных электронов подтверждает пре-имущественность сшивания в аморфных участках [55]. Эффективность сшивания оценивалась с помощью величин модуля упругости и весового коэффициента набухания, определяемых, как известно [46], в основном расстоянием между поперечными
<17.
связями и не зависящих от молекулярного веса. Зависимость
весового коэффициента набухания от степени кристалличности (рис. 42) показывает, что эффективность сшивания при облучении сравнительно небольшими дозами (10 и 20 Мрад) выше у полиолефинов с меньшей степенью кристалличности. Увеличение дозы (до 100 Мрад) уже не позволяет обнаружить этой зависимости. Величины модуля упругости, рассчитанные по
Степень кристалличности, %
Рис. 42. Зависимость весового коэффициента набухания (в о-ксилоле при 120 °C) гель-фрак-ции облученных полиолефинов от степени кристалличности при различных поглощенных дозах [55]:
л — сополимер этилена с а-бутиленом; □ — сополимер этилена с пропиленом; X, О — полиэтилен низкого давления.
формуле Гесса по данным термомеханических испытаний выше температуры плавления полимера, находятся в зависимости от кристалличности облучаемых образцов [38, 55]. Представленные на рис. 43 результаты также указывают на большую интенсивность сшивания полимеров с меньшей степенью кристалличности.
Аналогичные выводы сделаны рядом авторов при сравнительном изучении радиационного сшивания аморфного и изотактического полипропилена. Радиационно-химический выход сшивания (количество сшитых цепей, приходящееся на 100 эВ поглощенной энергии излучения) для аморфного полипропилена в 2,7 раза [130] (по другим данным — в 1,5—1,7 раза [154, 155]) больше,
чем для изотактического полипропилена. Большая скорость радиационного сшивания в аморфных областях полипропилена подтверждена с помощью метода ЯМР [156, 157].
Упорядоченность структуры способствует процессу образования внутримолекулярных связей, который протекает в кристаллических областях полимера.
Сопоставляя радиационное сшивание в монокристалле полиэтилена и в блоке, Каваи с сотрудниками [153] нашли, что в
случае монокристаллов доза гелеобразования составила около 50 Мрад, в случае полиэтилена в блоке — около 4 Мрад; радиационно-химический выход структурирования для монокристалла и блочного образца соответственно составил 0,16 и 1,96. Такая резкая зависимость эффективности сшивания от упорядоченности полиэтилена привела к выводу об образовании различных видов поперечных связей — межмолекулярных («эффективных») и внутримолекулярных («неэффективных»), образующихся между складками одной и той же молекулы в ламелярных структурах и не приводящих к гелеобразованию. С помощью ИК-спектров было показано образование в про
118
цессе облучения полиэтилена и сополимеров этилена 1,2-диза-мещенных циклогексановых колец [150, 151]. Этот факт был истолкован как результат образования циклов. Наиболее интенсивное образование циклических структур наблюдалось в случае наиболее кристалличного из использованных полимеров — полиэтилена высокой плотности (0,960 г/см3). Циклические структуры появлялись в результате облучения при низких температурах, и «размораживание» молекулярного движения не влияло на образование внутримолекулярных связей. Следовательно, циклизация не связана с подвижностью макроцепей. Авторы работ
Рис. 43. Зависимость модуля упругости полиолефинов при 150 °C от поглощенной дозы (а) и от степени кристалличности при дозе 200 Мрад (б) [55]:
ф—сополимер этилена с а-бутиленом, степень кристалличности 61%;
А — сополимер этилена с пропиленом, степень кристалличности 63%;
□ — сополимер этилена с а-бутиленом, степень кристалличности 67%;
Q —сополимер этилена с пропиленом, степень кристалличности 72%;
И—полиэтилен низкого давления, степень кристалличности 78%.
[150, 151] считают, что процесс циклизации определяется особенностями надмолекулярной структуры и образование циклических структур происходит в ламелярных кристаллитах на поверхности складки внутри одного сгиба цепи. Что же касается вопроса о месте прохождения межмолекулярного сшивания, то авторы указанных работ установили повышение радиационного выхода межмолекулярного сшивания по мере уменьшения кристалличности и разделяют мнение о преимущественности образования поперечных связей в неупорядоченных (дефектных) областях полимера.
Таким образом, под действием облучения в полимере протекает межмолекулярное сшивание и циклизация. Первая реакция, определяющая изменения важных свойств полимера, зависит от подвижности сшиваемых участков макромолекул и
119
проходит преимущественно в неупорядоченных (аморфных) областях; вторая — не зависит от подвижности цепей и реализуется в упорядоченных (кристаллических) структурах.
На процесс сшивания оказывает влияние и морфология надмолекулярных образований в полиолефине. Морфологические изменения в полиэтилене, вызванные ультразвуковой обработкой перед радиационным сшиванием, привели к резкому уменьшению содержания гель-фракции. Если же обработанный
ультразвуком полиэтилен до сшивания подвергался рекристаллизации, то содержание гель-фракции было таким же, что и в необработаннохм образце [158].
Влияние морфологии на радиационно-химическое сшивание полиолефинов подробно рассмотрено в монографии [142].
На степень межмолекулярного сшивания из параметров излучения оказывает влияние величина дозы. Интенсивность излучения определяет скорость образования свободных радикалов, а следовательно, и скорость сшивания. От энергии излучения зависит глубина его проникновения. Используя излучения с относительно малой энергией, можно подвергать модифицированию лишь поверхностный слой материала. Вид излучения при прочих равных условиях облучения существенно на харак-
400
Рис.
поперечных
числа поли-г при
а юо 200 зоо Температура, К
44. Зависимость । связей в i этилене от температуры воздействии у-излучения [57].
тер радиационного эффекта не влияет [33].
Результат радиационного воздействия в значительной мере зависит от среды, в которой производится облучение. Облучение на воздухе, как уже упоминалось в связи с синтезом привитых сополимеров (см. гл. III), сопровождается окислением полимера в особенности в поверхностных слоях и в аморфных областях, где легче происходит диффузия кислорода. Кислород оказывает ингибирующее действие на
процесс сшивания, реагируя с активными промежуточными соединениями. Чем выше содержание гидроперекисных и карбонильных групп в полиэтилене, тем меньше количества поперечных связей между цепями и тра«с-виниленовых групп [56].
Важным параметром процесса облучения является температура [46, 53, 57]. Скорость сшивания с повышением температуры возрастает (рис. 44), очевидно, вследствие повышения подвижности макрорадикалов. Можно ожидать, что скорость деструкции с увеличением температуры не будет возрастать так, как скорость сшивания, поскольку реакции, ведущие к деструкции, не связаны с подвижностью полимерных цепей.
Варьируя температуру при облучении, можно существенно изменять свойства продуктов сшивания. Например, облучение
120
Полиэтилена при температуре, превышающей температуру плавления, позволяет получать резиноподобный материал [58]. Облучение при повышенных температурах можно использовать также для получения прозрачных изделий из полиэтилена [59—61].
Ушаков с сотрудниками [62], исследуя температуру плавления и степень кристалличности облученного при различных температурах полиэтилена низкого давления, пришли к выводу о зависимости характера пространственной сетки от фазового состояния материала при сшивании. Облучение расплавленного полиэтилена приводит к фиксированию поперечными связями неупорядоченного состояния цепей; степень кристалличности и температура плавления понижаются. Сшивание при температурах ниже температуры плавления также сопровождается разрушением кристаллических образований вблизи мест поперечных связей. При достаточно больших дозах это может проявиться в уменьшении степени кристалличности, размеров кристаллических образований и понижении температуры плавления. Специфическим результатом облучения при невысоких температурах, по данным работы [62], является фиксирование параллельной ориентации цепей поперечными связями, образующимися в кристаллических областях. Этим объясняется повышение температуры плавления оставшихся после облучения кристаллитов. Отметим, что эти интересные данные, очевидно, не противоречат мнению о преимущественности процесса сшивания в аморфных областях. Накопление значительного числа поперечных мостиков в кристаллической фазе при больших величинах применявшихся в работе [62] доз (от 150 до 1625 Мрад) не исключает значительно большей скорости сшивания в аморфной части материала.
Вместе с тем вывод о преимущественности межмолекулярного сшивания в аморфных областях [55], основанный на результатах определения свойств пространственной сетки, не учитывает различия в характере сеток, образованных при сшивании аморфных и кристаллических областей. Это различие, указанное Ушаковым с соавторами [62], означает, что вклад каждого из видов пространственных сеток в характеристики сшитых полимеров (коэффициент набухания и модуль упругости при температурах выше температуры плавления) неодинаков. Определение степени влияния обоих видов сетки могло бы позволить с большей строгостью и количественно показать разницу в скоростях сшивания аморфных и кристаллических областей.
Основным свойством, отличающим облученные изделия из полиэтилена и сополимеров этилена с пропиленом и другими а-олефинами, является повышенная теплостойкость. Пространственная структура обусловливает остаточную прочность материала и сохранение формы при температурах, превышающих температуру плавления. Облученные изделия из полиэтилена,
121
сополимеров этилена с небольшими количествами пропилена или а-бутилена могут эксплуатироваться без значительных механических нагрузок при температурах до 150—200 °C. Поведение сшитых полиолефинов при нагревании наглядно проявляется при термомеханических испытаниях. На рис. 45 представлены термомеханические кривые исходного и облученного быстрыми электронами сополимера этилена с пропиленом, полученные методом вдавливания шарика [63, с. 196].
Автором совместно с Е. И. Наливайко и Е. П. Рябиковым показана возможность расширения интервала рабочих температур полиэтилена низкой плотности в результате радиационной
Рис. 45. Термомеханические кривые сополимера этилена с 7°/о пропилена при различных поглощенных дозах [38].
обработки в область низких температур. Объектами исследования служили пластины из полиэтилена с различным молекулярным весом. Облучение осуществляли ускоренными электронами с энергией 2 МэВ при мощности дозы 1 Мрад/мин и температуре 20 °C. Облученные образцы подвергали термообработке при 100 °C в течение 30 мин. Отсутствие «захваченных» свободных радикалов в образцах после термообработки фиксировали методом ЭПР. Облучение и термообработку проводили в среде азота.
Температуру хрупкости определяли с помощью прибора ПХП-1 ( по ГОСТ 10995—64) как предельно низкую температуру, при которой материал не разрушается под ударной нагрузкой в заданных условиях нагружения.
Результаты, представленные на рис. 46, показывают значительное снижение температуры хрупкости полиэтилена по мере возрастания поглощенной дозы излучения. Температура хруп
122
кости резко снижается при облучении до поглощенной дозы около 10 Мрад. При дальнейшем облучении температура хрупкости продолжает снижаться, но не столь резко.
Существенно, что чем выше молекулярный вес исходного полиэтилена (ниже показатель текучести расплава), тем ниже температура хрупкости при тех же величинах поглощенной дозы. Это подтверждается данными для образцов облученного полиэтилена низкой плотности:
Поглощенная доза, Мрад..........
Температура хрупкости, °C
5,7 г/10 мин.................
8,0 г/10 мин.................
0 40 100
-65 -105 -130
-55 -95 -112
Повышение морозостойкости, по-видимому, имеет две основные причины. Одна из них заключается в увеличении числа «проходных» цепей уже при сравнительно малых поглощенных
дозах (примерно до 10 Мрад) вследствие образования в аморфных областях поперечных связей между
Рис. 46. Зависимость температуры хрупкости полиэтилена низкой плотности (показатель текучести расплава 1,6 г/10 мин) от величины поглощенной дозы.
Рис. 47. Схематическое изображение «проходных» цепей в структуре полиэтилена:
1—кристаллиты; 2—аморфная область; 3 — проходные цепи, существовавшие до облучения; 4—проходные цепи, образовавшиеся в результате сшивания двух цепей.
цепями, которые входят в эти области из соседних кристаллических образований (рис. 47). Отметим, что понижение температуры хрупкости с уменьшением показателя текучести расплава связано, очевидно, со сравнительно высоким содержанием проходных цепей в полиэтилене с большим молекулярным весом.
Второй причиной увеличения морозостойкости является образование каркаса поперечных связей между цепями, протекающее при значительных поглощенных дозах (более 10 Мрад) преимущественно в аморфных областях [55]. Повышение доли проходных цепей в аморфных областях и образование каркаса поперечных связей увеличивает способность полимера к делокализации напряжений.
123
Такое объяснение повышения морозостойкости при сшивании полиэтилена хорошо согласуется с представлениями о роли кристаллических областей в распределении напряжений благодаря возможности сдвига отдельных элементов кристаллических образований (ламелей) [143, с. 424]. Процесс распределения напряжений определяется концентрацией кристаллических образований и количеством проходных цепей между соседними кристаллическими областями. Таким образом, чем выше доля проходных цепей и плотнее сетка поперечных связей (больше «каркасная связанность» [144]), тем больше возможности делокализации напряжений.
При изучении влияния радиационно-химического сшивания на теплофизические свойства полиэтилена Самойлов [64] установил, что с увеличением дозы облучения теплопроводность и теплоемкость уменьшаются. Снижение теплопроводности связывают с дополнительным рассеянием и затуханием фононов на поперечных связях между макроцепями облученного полиэтилена. Сшивание макроцепей уменьшает число внутренних степеней свободы, что в свою очередь вызывает понижение удельной теплоемкости по мере облучения полиэтилена.
Изменения физико-механических свойств в результате облучения отражают сложность превращений в полимере. Процессы сшивания и деструкции, разрушения кристаллических структур и образования сравнительно низкомолекулярных продуктов деструкции по-разному влияют на поведение материала при деформациях. Сшивание уменьшает подвижность сегментов цепей. Деструкция и амортизация полимера ведут к противоположному эффекту. В связи с этим результирующее влияние облучения на прочность полимера при растяжении, относительное удлинение при разрыве, модуль упругости оптимально при определенных величинах доз.
Облучение полиэтилена до сравнительно небольших доз (десятки Мрад) не вызывает заметного изменения модуля упругости. При увеличении дозы кристалличность уменьшается. Влияние снижения кристалличности оказывается меньшим, чем влияние повышения степени сшивания. Модуль упругости при этом стремится к минимуму, соответствующему дозам, при которых достигается глубокая аморфизация (примерно 500— 600 Мрад). Дальнейшее возрастание степени сшивания при увеличении поглощенной дозы вызывает резкий рост модуля упругости [31].
Кривые зависимости прочности при растяжении и относительного удлинения от поглощенной дозы могут иметь максимумы (чаще всего при дозах 5—25 Мрад).
Механические свойства облученных полимеров в большой мере связаны с характеристиками исходных продуктов. Так, изменения предела текучести и прочности при растяжении находятся в зависимости от молекулярного веса исходного материа-124
ла (рис. 48). Сохранению свойств материалов способствует облучение в бескислородной среде (рис. 49).
Растяжение облученного полиэтилена при температуре, превышающей температуру плавления (или близкой к температуре плавления), с последующим охлаждением в растянутом состоянии позволяет получать материал, который при нагревании обладает способностью к усадке практически до первоначальных размеров [31, с. 247; 140, с. 241; 141, с. 101; 145—149]. Так, пленка из полиэтилена после облучения
Поглощенная доза Мрад
Рис. 48. Зависимость механических свойств полиэтилена низкого давления от поглощенной дозы при различных показателях текучести исходного материала [65].
низкой плотности (поглощенная доза
Рис. 49. Зависимость механических свойств полиэтилена высокого давления, облученного на воздухе (/) и в вакууме (2), от поглощенной дозы (источник излучения 60Со [66].
20 Мрад), растяжения в 2,5 раза при 80°C и охлаждения приобретает способность практически к полной усадке при нагревании до 105—ПО °C в течение 1—3 мин [148]. При более высоких температурах исходные размеры и форма могут восстанавливаться в течение нескольких секунд. Это явление, называемое эффектом «памяти», используется для получения различных термоусаживающихся изделий: пленок, труб, шлангов и др.
Подвергнутые облучению полиэтилен и другие сшивающиеся полиолефины отличаются стойкостью к действию углеводородных растворителей (рис. 50). Изменения физико-
12$
0 100 200 300 400
Поглощенная доза, Мрад
Рис. 50. Зависимость растворимости (в кипящем о-ксилоле) и набухаемости (в о-ксилоле при 120 °C в течение 2 ч) сополимера этилена с 3% а-бутилена от поглощенной дозы (облучение ускоренными электронами с энергией 2,0 МэВ в атмосфере гелия) [38].
механических свойств полиэтилена под воздействием ионизирующих излучений подробно описаны в монографии Чарлзби [31].
Весьма важным, наряду с повышением теплостойкости, является возрастание сопротивляемости растрескиванию полиэтилена в результате радиационно-химического сшивания [65, 67—69]. Сопротивляемость растрескиванию полиэтилена резко возрастает при достижении некоторых определенных доз облучения. Зависимости стойкости к растрескиванию от поглощенной дозы и молекулярного веса подобны. Согласно данным [161] стойкость к растрескиванию с повышением молекулярного веса резко увеличивается при определенной его величине.
Эффект значительного повышения сопротивляемости растрескиванию наблюдается уже при сравнительно небольших дозах облучения, которым соответствует незначительное содержание гель-фракции в облученном полиэтилене (3— 10%). Поглощенная доза, необходимая для значительного увеличения сопротивляемости растрескиванию, тем меньше, чем больше молекулярный вес полиэтилена (табл. 19).
Некоторые изменения в свойствах полиэтилена в результате сшивания (повышение прочности и деформируемости в определенном интервале поглощенных доз, снижение температуры хрупкости и увеличение стойкости к растрескиванию)
имеют, очевидно, общую причину, которая заключается в увеличении содержания проходных цепей, способствующих диссипации напряжений. Это подтверждается, в частности, влиянием молекулярного веса исходного полимера на изменение свойств полимера при облучении: чем выше молекулярный вес, тем меньшая доза требуется для достижения максимума прочности и деформируемости, повышения морозостойкости и стойкости к растрескиванию.
Поскольку молекулярный вес в большой мере определяет способность полиэтилена сопротивляться воздействию напряжений и окружающей среды [161], радиационно-химическое сшивание, так же как и другие методы сшивания, является одним из наиболее эффективных способов повышения стойкости к растрескиванию полиэтилена.
126
Таблица 19
Влияние поглощенной дозы на стойкость к растрескиванию полиэтилена низкого давления [65]
Облучение ускоренными электронами с энергией 2,0 МэВ, мощность дозы 0,4 Мрад/мин, в среде инертного газа.
Показатель текучести расплава, г/10 мин Поглощенная доза, Мрад Стойкость к растрескиванию ч
в 20% растворе ОП-7 при 50 °C в 20% ОП-Ю растворе при 80 °C
3,88 0 90 1.5
10 180 3,0
20 >6000 >4000
40 >6000 >4000
60 >6000 >4000
1,47 0 120 3,0
10 5500 28
20 >6000 865
40 >6000 >4000
60 >6000 >4000
0,55 0 570 7,0
10 3000 840
20 >6000 1180
40 >6000 >4000
60 >6000 >4000
0,16 0 1200 25
10 >6000 3000
20 >6000 2500
40 >6000 >4000
60 >6000 >4000
♦ Стойкость к растрескиванию определяли как время до разрушения 50% образцов, находящихся в напряженном состоянии в водных растворах поверхностно-активных веществ при заданной температуре. Данные табл. 19 позволяют предположить, что существенной причиной увеличения сопротивляемости растрескиванию облученного полиэтилена является повышение его молекулярного веса при облучении.
Разрушение полиэтилена под действием нагрузки начинается и развивается в аморфных областях, поскольку именно в этих областях, как более дефектных, происходит локализация и концентрация напряжений [161]. Поперечные связи, как уже отмечалось, также образуются преимущественно в аморфных областях. Можно полагать поэтому, что повышение стойкости к растрескиванию связано не только с возрастанием молекулярного веса при облучении, но и с возникновением в аморфных областях пространственной структуры, препятствующей развитию трещин.
Увеличение стойкости к растрескиванию облученного полиэтилена сопровождается потерей его перерабатываемое™. Поэтому облучению с целью повышения стойкости к растрескиванию подвергают изделия из полиэтилена [162, с. 196; 163].
Как отмечалось в гл. III, с увеличением степени кристалличности содержание «захваченных» свободных радикалов
127
в облученном полиэтилене возрастает, и послерадиационные процессы протекают более интенсивно. Взаимодействие захваченных радикалов с кислородом воздуха приводит к окислению полиэтилена и, как следствие [164, с. 73], к снижению его сопротивляемости растрескиванию. Для высококристаллического полиэтилена окисление после облучения оказывается столь существенным, что его стойкость к растрескиванию падает, несмотря на увеличение молекулярного веса при облучении (табл. 20). В случае менее кристаллического полиэтилена (полиэтилен высокого давления) окисление после воздействия радиации играет меньшую роль, поэтому в результате облучения даже в воздушной среде его сопротивляемость растрескиванию возрастает.
Таблица 20
Стойкость к растрескиванию полиэтилена, облученного в воздушной среде *
Источник излучения в0Со; мощность дозы 0,65 Мрад/ч.
Полиэтилен Поглощенная доза, Мрад Стойкость к растрескиванию **, ч Показатель текучести расплава 3*, г/10 мин
Полиэтилен низкого да- 0 12,0 3,5
вления (степень кристал- 0.75 6,0 0,5
личности 80%) 1,25 5,6 0,2
2,0 3,5 0,1
Полиэтилен высокого да- 0 2,0 1,1
вления (степень кристал- 0,5 2,5 0,6
личности 63%) 1,0 6,0 0,3
2,5 500 0,1
5,0 500 0,0
* По данным М. Д. Пукшанского, Л. А. Ирлиной и автора.
** Стойкость к растрескиванию определяли в соответствии с ГОСТ 13518—68.
3* Показатель текучести расплава определяли в соответствии с ГОСТ 11645 — 65.
Для достижения высокой сопротивляемости растрескиванию полиэтилена низкого и среднего давления следует избегать взаимодействия захваченных свободных радикалов с кислородом воздуха после облучения. С этой целью сшитый полимер прогревают в среде инертного газа или в вакууме при температуре, близкой к температуре плавления или несколько превышающей ее. Облученный и отожженный таким способом полиэтилен сохраняет, кроме того, высокие диэлектрические свойства исходного полиэтилена [31; 70, с. 325].
Фотохимическое сшивание. Эффективное сшивание полиолефинов может быть достигнуто фотохимическим воздействием, заключающимся в облучении УФ-светом в присутствии фото-
128
сенсибилизаторов. В результате такого воздействия под влиянием активированных УФ-светом молекул фотосенсибилизатора образуются макрорадикалы, рекомбинация которых приводит к возникновению поверенных связей [76]. При этом скорость сшивания весьма высока. Если для образования в полиэтилене значительного количества поперечных связей требуются сотни часов УФ-облучения [108], то в присутствии фотосенсибилизаторов полиэтилен эффективно сшивается уже после нескольких минут воздействия УФ-света той же интенсивности [165].
На рис. 51 представлены кривые зависимости выхода гель-фракций от продолжительности воздействия УФ-света на плен
ки из полиэтилена высокого давления,
содержащие различные фотосенсибилизирующие добавки. В течение первых минут облучения содержание гель-фракции быстро увеличивается и приближается к предельному значению уже примерно через 10 мин.
Для осуществления фотохимического сшивания используют, как правило, ртутные лампы низкого давления (испускающие в основном свет с длиной волны 253,7 нм) и среднего давления (спектр испускания в ближней ультрафиолетовой области и в области видимого света).
Выбор источника света определяется природой применяемого фотосенсибили
Рис. 51. Кинетические кривые процесса сшивания пленки из полиэтилена высокого давления, насыщенной различными сенсибилизаторами, под действием УФ-света (источники света—две лампы ПРК-2, расстояние между пленкой и лампами 2,5 см) [84]:
1 — тетрахлорэтилен; 2 —
хлороформ; 3 — четыреххлористый углерод; 4 — бензофенон.
[91; 92; 93, с. 48; 96;
затора, поглощающего свет в той или иной области спектра. Эффективность фотохимического процесса в значительной степени зависит от характера вводимого фотосенсибилизатора. В первых работах в качестве фотосенсибилизирующей добавки использовали бензофенон [71, 76]. Известно большое число соединений, обладающих подобным свойством. К ним относятся ряд кетонов и альдегидов [71—88; 109; ПО; 111, с. 134; 112— 117], некоторые ароматические углеводороды [73], хлорированные углеводороды [71, 73, 83, 84, 94], хлорангидриды кислот 118] и другие соединения [89, 90].
В зависимости от природы фотосенсибилизатора образова-
ние макрорадикалов происходит различными путями. Роль фотосенсибилизатора может сводиться к передаче им макромолекуле своей избыточной энергии в возбужденном состоя
нии. следствием чего оказывается разрыв связи С—Н и
5 А. Г. Сирота
129
последующая рекомбинация радикалов:
ФС + hv —> ФС’
ФС* + ЦН —> ФС + R • + Н •
R. + R. --► R-R
н • + н • —► н2 где ФС — молекула фотосенсибилизатора; RH — молекула углеводорода.
Из многих сенсибилизирующих добавок лишь некоторые участвуют в процессе фотохимического сшивания согласно этой схеме. По-видимому, таким путем осуществляется сшивание полиэтилена при использовании в качестве сенсибилизатора бензола [73]. Действительно, при облучении светом с /. = 253,7 нм в бензоле не происходит каких-либо химических изменений [119]. Поэтому можно полагать, что участие бензола в фотохимическом процессе сводится к передаче избыточной энергии макромолекуле.
При облучении полипропилена УФ-светом аналогичное фотосенсибилизирующее действие оказывают содержащиеся в полимере продукты разложения металлорганического комплексного катализатора (основные компоненты ТЮ2 и А12О3) [120].
Действие другой группы фотосенсибилизаторов заключается в отрыве под влиянием УФ-света атомов водорода от макромолекул с последующей рекомбинацией макрорадикалов. Типичным представителем этой группы является бензофенон, реакции фотовосстановления которого в среде низкомолекулярных углеводородов хорошо изучены [121]. Под действием УФ-света молекулы бензофенона переходят в возбужденное триплетное состояние, характеризующееся, как известно [122, с. 106], высокой реакционной способностью. Взаимодействие между возбужденной молекулой бензофенона и углеводородом приводит к образованию углеводородного и полубензпинакольного радикалов:
(С6Н5)2СО + hv —* (С6Н5)2СО*
(С6Н5)2СО* 4-RH —> (СеН5)2СОН + R •
Поперечные связи между углеводородными цепями возникают при рекомбинации радикалов R»; взаимодействие полу-бензпинакольных радикалов приводит к образованию бензпина-кола.
По-видимому, схема реакции, приведенная для бензофенона, верна для большинства кетонов, используемых в качестве добавок при фотохимическом сшивании. Так, установлено [115], что в процессе фотохимического сшивания полиэтилена используемые в качестве сенсибилизаторов а- и р-хлорантрахиноны восстанавливаются до хлорантрагидрохинонов.
Сенсибилизаторы третьей группы, к которой в первую очередь следует отнести хлорированные алифатические и арома-130
тические углеводороды и хлорангидриды кислот, под влиянием УФ-света распадаются на радикалы, способные акцептировать атомы водорода от макромолекул с последующей рекомбинацией образующихся макрорадикалов:
АБ + Av —> А • 4- Б .
A- + RH —> AH + R. Б • + RH —-> БН + R • R- + R- —* R-R
где АБ — сенсибилизатор, распадающийся под действием УФ-света на радикалы.
На ход фотохимического процесса сшивания существенное влияние оказывает природа полимерной среды. Качан и сотрудники показали [118], что при сшивании полиэтилена в присутствии хлористого оксалила начальная скорость гелеобразования пропорциональна квадрату интенсивности света. Это указывает на двухфотонный механизм инициирования процесса сшивания. Аналогичные результаты получены при использовании в качестве сенсибилизаторов хлористого бензоила [123], а- и 0-хлорантрахинона [115] и других соединений [124]. Таким образом, в жесткой полимерной среде время жизни возбужденного состояния молекулы фотосенсибилизатора столь значительно, что возможно поглощение еще одного фотона и достижение энергетического уровня, необходимого для оказания фотосенсибилизирующего действия. В случае а- и 0-хлорантрахинонов поглощение второго фотона делает возможным отрыв атомов водорода от макромолекул и восстановление семихинонных форм до хлорантрагидрохинонов [115, 123]. Возбужденные молекулы хлористого оксалила [118] и хлористого бензоила [123] под действием второго кванта света распадаются на радикалы, акцептирующие атомы водорода макромолекул.
По-видимому, фотосенсибилизирующее действие бензола [73] также проявляется после последовательного поглощения двух квантов света. Действительно, энергия первого триплетного состояния бензола равна 85 ккал/моль [125] и недостаточна для разрыва связи С—Н углеводородной цепи, энергия которой составляет 99,2 ккал/моль [31, с. 28].
Другого рода влияние полимерной среды проявляется при использовании фотосенсибилизаторов, распадающихся на радикалы. Оно заключается в ограничении подвижности образованных под влиянием УФ-света радикалов, вследствие чего при отрыве атомов водорода от макромолекул возникающие активные центры находятся близко друг к другу и могут непосредственно реагировать между собою [123, 126]. Взаимодействие активных центров, принадлежащих соседним макромолекулам, приводит к образованию поперечной связи. В случае образования активных центров путем акцептирования атомов водорода
5*
131
от одной макромолекулы возникает ненасыщенная группа. Такой ход фотохимического процесса имеет место, например, при использовании в качестве фотосенсибилизатора тетрахлорэтилена.
При облучении полиэтилена в присутствии тетрахлорэтиле
на основным продуктом его фотолиза в полимере является трихлорэтилен. Как видно из рис. 52, в ИК-спектре пленки, содер-
Пропускание
700 800 900 1000
О, см '
Рис. 52. ИК-спектры полиэтиленовой пленки, содержащей С2С14, до УФ-облучения (1), той же пленки после облучения (2) и пленки, содержащей С2НС13 (3) [126].
жащей тетрахлорэтилен, после облучения появляются новые полосы поглощения, соответствующие полосам поглощения трихлорэтилена. В этом процессе наряду с поперечными связями образуется значительное количество ненасыщенных групп транс-виниленового типа [126]. Эти группы полностью исчезают при взаимодействии с треххлористым фосфором [131], сорбирующимся в аморфных областях [128], следовательно, транс-виниле-новые группировки также располагаются лишь в этих областях. Такой результат указывает на незначительную роль актов миграции в рассматриваемом процессе фотохимического сшивания. Действительно, миграция активных центров
макромолекулы вдоль цепи и от одной цепи к другой приводила бы к образованию транс-виниленовых групп как в аморфных, так и в кристаллических областях.
Данные о фотолизе тетрахлорэтилена и строении сшитого полиэтилена позво-
ляют представить схему взаимодействия тетрахлорэтилена с полиэтиленом под влиянием УФ-света следующим образом [126]:
С2С14 + hv —> C2CIJ
—сн2—сн2—сн2---
—сн2—сн2—сн2---
c2ci:
---СН2—СН2—СН2- ---СН2—сн2—сн2-
—сн2—сн—сн2— —сн2—сн—сн2----
С2С!3Н, С2С13Н,
” НС! НС1
—сн2—сн—сн2— —сн2—сн—сн2----
132
и наряду с этим
------сн2—сн2—сн2—сн2-----—>
С1
—сн2—сн= сн—сн2--
НС1
Согласно этой схеме (квадратные скобки в ней обозначают нахождение фотопродуктов в «клетке» полимерной матрицы) образование поперечной связи не требует актов миграции свободной валентности, являющейся необходимой стадией процесса радиационно-химического сшивания [97; 127, с. 156].
Образование поперечной связи без предварительных актов миграции может быть причиной эффективного фотохимического сшивания полипропилена [128, 129]:
AB+Av —> А + Б ---СН2—СН—СН2----- ---СН2—СН—СН2-----
I I
сн3 сн2
А __
_ Б J
---сн2—СН—сн2----- ---сн2—с—сн2-----
I I
СН3 СН3
+ АН + БН
Радиационно-химическое сшивание полипропилена, у которого миграция активных центров из-за особенностей строения макроцепи затруднена [130], как известно [31, с. 307], протекает с весьма низким выходом гель-фракции.
Жесткость полимерной среды, обеспечивая возможность эффективного фотохимического сшивания, в то же время не должна быть чрезмерной. Рекомбинация макрорадикалов — заключительная стадия процесса сшивания — оказывается возможной лишь при достаточной подвижности макроцепей. Поэтому образование поперечных связей при фотохимическом сшивании [128], так же как при радиационно-химическом, происходит преимущественно в аморфных областях полимера.
Качан, Чернявский и Шрубович [84] определили отношение вероятностей процессов деструкции и сшивания полиэтилена по данным зависимости золь-фракции от обратной величины продолжительности облучения, пользуясь следующей формулой [95]:
S + ] ' S =-----п-
<7 о qoujt
1де s — содержание золь-фракции; Ро и q0 — число разрывов главной цепи и число поперечных связей, соответственно приходящихся на одну молекулу (в расчете на среднечисленный
133
молекулярный вес); Ut — среднечисленная степень полимеризации; I — интенсивность облучения; t — продолжительность облучения.
Найденные величины Polqo составляют при использовании в качестве сенсибилизатора бензофенона 0,40, тетрахлорэтилена — 0,42, четыреххлористого углерода — 0,71 и хлороформа — 0,86 (облучение лампой ПРК-2). В условиях преимущественного поглощения света сенсибилизатором и при малой продолжительности облучения трудно допустить возможность значительной деструкции полимерных цепей. В связи с этим полученные в работе [84] значения Polqa представляются несколько завышенными. Можно предположить, что одна из возможных причин сравнительно невысокой степени сшивания полиэтилена и, соответственно, относительно больших величин Polqa заключается в преобладании процесса сшивания в аморфных областях.
Ограниченная подвижность цепей в кристаллических областях затрудняет сшивание и приводит к сохранению растворимости части полимера [128]. В результате определяемые по кривым зависимости s + f (1/0 величины PJqa не отражают действительного соотношения скоростей деструкции и сшивания и требуют соответствующей поправки.
На образование поперечных связей преимущественно в аморфных областях указывает также полное совпадение ди-фрактограмм исходных и сшитых пленок. Различия во вторичных структурах рентгенографически обнаруживаются лишь при деформировании модифицированных и исходных пленок. Как видно из рис. 53, при растяжении сшитых пленок ориентация надмолекулярных образований отсутствует, в то время как у исходной пленки она четко выражена.
Важнейшим свойством сшитых фотохимическим методом полимеров является их повышенная теплостойкость. Пленка из полиэтилена высокого давления, полностью теряющая, как известно, сопротивляемость разрывному усилию уже при НО °C, после облучения УФ-светом в присутствии треххлористого фосфора при температуре 200 °C сохраняет сопротивление растяжению до 10 кгс/см2. При нагревании со скоростью 4 °С/мин и одновременном действии нагрузки 2 кгс/см2 модифицированная пленка не изменяет толщины вплоть до 180 °C. Исходная пленка в этих же условиях обнаруживает текучесть уже при 100 °C (рис. 54). Модуль упругости при 150 °C полиэтиленовых пленок, облученных УФ-светом в присутствии треххлористого фосфора, возрастает с увеличением глубины сшивания (рис. 55) [131].
Фотохимическое сшивание полиэтиленовых волокон также существенно повышает их теплостойкость. Так, волокно, сшитое до содержания гель-фракции 45%, теряет прочность при 125 °C, в то время как полная потеря прочности для исходного волокна наступает при 95 °C [132].
134
Сшитые фотохимическим способом пленки обладают пониженной газопроницаемостью. Например, сшивание пленки из
Рис. 53. Микрофотографии растянутых до 300% пленок сополимера этилена с 7% пропилена (увеличено в 300 раз) [96]:
а — исходная пленка; б — пленка, сшитая фотохимическим методом в присутствии треххлористого фосфора.
полиэтилена высокого давления толщиной 0,1 мм уменьшает коэффициент диффузии бензола примерно в 20 раз [72].
Сшитая полиэтиленовая пленка может эксплуатироваться
в условиях повышенных температур в контакте с органическими
135
растворителями, так как обладает повышенной теплостойкостью и малой растворимостью [128].
В ранних работах [72—76] сшивание и связанное с ним улучшение свойств рассматривалось как единственный практически важный результат фотохимического модифицирования. Однако в последние годы показано [118, 123, 131, 133], что продукты распада сенсибилизатора могут присоединяться к макромолекулам, взаимодействуя с их активными группами [126, 131] или рекомбинируя с макрорадикалами [118, 123]. что придает по-
лимеру ряд новых свойств.
Температура,°C
Рис. 54. Кривые текучести модифицированной фотохимическим методом (/) и исходной (2) пленок из полиэтилена высокого давления [96].
Содержание гель-фракции,-^
Рис. 55. Зависимость модуля упругости при 150 °C полиэтиленовой пленки, сшитой фотохимическим способом в присутствии
РС1з [131]?
При сшивании в присутствии треххлористого фосфора полиэтиленовые пленки по данным элементного анализа содержат до 1% фосфора [72]. Один из путей связывания фосфора полимером заключается во взаимодействии радикалов РС12 с макроцепями по местам двойных связей [98, 128].
Возможно также взаимодействие с макрорадикалом:
---СН2—СН—СН2------h РС12 —>----сн2—сн—сн2----
РС12
При дальнейшем взаимодействии с кислородом и влагой воздуха образуются фосфорнокислые группы:
о2, н2о
---сн2—сн—сн2------------> ---сн2—СН—сн2-----
I I /ОН
PCI: O=PZ
хон
Эта схема подтверждается спектральными данными. Спектры облученных пленок имеют полосы поглощения при 980 и 1180 см-1, соответствующие деформационным колебаниям
136
групп —Р—ОН и —Р=О [92]. Кроме того, при сшивании noli !
лиэтилсяй по снижению интенсивности полосы поглощения 909 см ’ было обнаружено [96] уменьшение содержания двойных связей.
Взаимодействие между треххлористым фосфором и полиэтиленом происходит уже в темноте по местам разветвлений и ненасыщенных групп (см. гл. IV). При УФ-облучении это взаимодействие протекает с повышенной скоростью [131]. По-види-мому, уменьшение числа активных групп в полимерной цепи приводит к значительному повышению стойкости к термоокислительному [131] и атмосферному [133] старению полиэтилена, подвергнутого фотохимическому сшиванию в присутствии треххлористого фосфора.
Сравнительные испытания в условиях атмосферного старения, проведенные в различных климатических зонах, показали, что относительное удлинение при разрыве полиэтиленовых пленок, модифицированных фотохимическим методом (сенсибилизатор РС13), так же как и стабилизированных бензоном ОА, фосфитом П-24, практически не изменяется в течение длительного времени, но резко уменьшается у нестабилизированных пленок (табл. 21) [133].
Таблица 21
Изменение относительного удлинения (в °/о| полиэтиленовых пленок в условиях атмосферного старения
Характеристика испытуемой полиэтиленовой пленки
В начале испытаний
Ленинград
Симферополь
Исходная...............
Модифицированная, гель-фракции—10% . . . .
Модифицированная, гель-
фракции — 47% . . . .
Стабилизированная бензоном ОА и фосфитом П-24
500
430
370
480
300 60
440 410
430 480
50 500
370 450
350 450
460 500
250 70 410 120
440 430
430 420
490 520
450 —
430 390
520 510
50
300
350
480
Присоединение продуктов распада сенсибилизатора к полимеру приводит к улучшению окрашиваемости [118, 126] и адгезии, повышению смачиваемости полярными агентами [126] и др.
Сопоставление процессов радиационно- и фотохимического модифицирования полиэтилена проведено в работе [99]. Различия в действии УФ-света и ионизирующего излучения на
137
полиэтилен проявляются уже на стадии образования макрорадикалов. Энергия связей С—Н и С—С в молекулах полиэтилена составляет, как известно, 4,28 и 3,44 эВ соответственно. Наиболее интенсивным линиям излучения ртутно кварцевых ламп в УФ-области спектра 2537, 3130 и 3650 А соответствуют величины квантов 4,88, 3,95 и 3,39 эВ. Следовательно, действие УФ-света с длиной волны 2537 А может вызывать разрывы связей обоих типов.
Полиэтиленовая пленка поглощает свет с /. < 200 нм. Это поглощение обусловлено наличием в полимере ненасыщенных и кислородсодержащих групп [134], а также примесей ароматических веществ [135, 136]. В результате такого избирательного поглощения под действием УФ-света полиэтилен преимущественно деструктирует. Спектр ЭПР [100] полиэтилена, облученного при 77 К УФ-светом, обнаруживая преимущественное образование радикалов —СН2—СН2 по сравнению с радикалами —СН2—СН—СН2—.указывает на преобладание актов деструкции основных цепей.
При воздействии ионизирующих излучений, напротив, энергия квантов значительно выше энергии химических связей и макрорадикалы могут образовываться в любых участках макроцепей со значительно большей скоростью, чем при облучении УФ-светом. Для образования одного макрорадикала в полиэтилене при 77 К требуется НО эВ энергии УФ-света [100] и лишь 30 эВ энергии ионизирующего излучения [101].
Несмотря на высокую эффективность радиационно-химического сшивания, дозы облучения, требуемые для достижения большого содержания гель-фракции (90—95%), все же велики (до 70—100 Мрад). Можно увеличивать эффективность радиационно-химического сшивания полиэтилена и полипропилена с помощью добавок полифункциональных мономеров (см., например, [102, с. 285; 103, с. 282]), действие которых осуществляется благодаря их способности образовывать поперечные связи между цепями:
---сн2—сн—сн2----
м
---СН2—СН—сн2----
где М — бифункциональный мономер.
Если с помощью указанных добавок можно лишь увеличить эффективность радиационно-химического сшивания, то введение в полимер фотосенсибилизаторов позволяет под действием УФ-света осуществить процесс, основным результатом которого оказывается не деструкция, а эффективное сшивание. Более того, как уже упоминалось, фотохимическое воздействие вызывает эффективное сшивание более широкого ряда полиолефинов, чем радиационно-химическое. Этим методом можно
138
сшивать такие полиолефины (например, полиизобутилен [76]), которые под действием частиц высокой энергии подвергаются деструкции.
Соединения, сенсибилизирующие процесс фотохимического сшивания, не только не повышаю! скорости радиационно-химического сшивания, но могут оказывать и ингибирующее влияние. Качан с сотрудниками [104] объясняют наблюдавшееся ими ингибирование рекомбинацией образованных под пучком ионизирующего излучения макрорадикалов с радикалами введенных добавок.
Выше уже обсуждались процессы, сопровождающие радиационное воздействие при осуществлении его в воздушной среде, и подчеркивалась необходимость бескислородной среды при сшивании полиолефинов и термической обработке сшитых полимеров для предотвращения взаимодействия «захваченных» радикалов с кислородом воздуха. В связи с тем что при облучении УФ-светом образуется сравнительно небольшое количество макрорадикалов, фотохимическое сшивание в отличие от радиационно-химического можно проводить на воздухе; оно не требует последующей термической обработки полимера.
Различие радиационного и фотохимического воздействия проявляется также в характере изменений в строении макроцепей. Наряду с возникновением поперечных связей при действии ионизирующего излучения увеличивается содержание двойных связей в полиэтилене. Основным типом образующихся непредельных группировок являются транс-виниленовые группы, которым в ИК-спектре соответствует полоса поглощения 965 см'1. Повышение содержания двойных связей по мере увеличения дозы радиационного воздействия вызывает появление коричневой окраски материала. При этом содержание концевых двойных связей, для которых характерна полоса поглощения 909 см-1, почти не изменяется [31] с увеличением поглощенной дозы.
Фотохимическое сшивание полиэтилена в присутствии распадающихся на радикалы сенсибилизаторов может приводить к уменьшению содержания ненасыщенных групп из-за взаимодействия этих групп с продуктами распада сенсибилизатора. Так, фотосенсибилизированное треххлористым фосфором сшивание сопровождается уменьшением интенсивности полосы поглощения 909 см-1.
Существенное различие между двумя методами сшивания связано с различной проникающей способностью ионизирующих излучений и УФ-света. Радиационно-химический метод позволяет подвергать сшиванию материал толстостенных изделий, фотохимическое сшивание, осуществимое лишь в тонких слоях (до 200—250 мк), можно использовать для модифицирования пленок или поверхностных слоев изделий. Вместе с тем оба метода сшивания имеют и общие черты. Изменения теплостойкости и основных физико-механических характеристик
139
полимеров в результате модифицирования обоими способами весьма близки. Сравнение фотохимического и радиационно-химического методов сшивания приведено в табл. 22.
Таблица 22
Сравнительная характеристика фотохимического и радиационно-химического методов сшивания полиолефинов [99]
Фотохимический метод Радиационно-химический метод
Источники излучения Ультрафиолетовые лампы Электронные ускорители, 60Со
Энергия излучения До 5 эВ До 1-3 МэВ
Глубина действия излучения в полиолефиновых материалах ~0.2 мм Для у-излучения с энергией 1,3 МэВ ~ 300 мм; для P-частиц с энергией 1,2 МэВ ~6 мм
Использование сенсибилизаторов (добавок) Необходимо Позволяет уменьшать необходимую поглощенную дозу
Виды сенсибилизаторов Кетоны, альдегиды, хлор- Полифункциональные
(добавок) ангидриды кислот и др. мономеры, закись азота, хлорид серы и др.
Действие сенсибилиза- Поглощение энергии В большинстве случаев
торов (добавок) УФ-света, отщепление атомов водорода от макромолекул, образование поперечных связей С—С между макроцепями являются сшивающими агентами при активировании макромолекул излучением
Среда при облучении (без значительной деструкции) Воздух или инертная среда Инертная среда *
Ненасыщенность Уменьшается в присутствии РС13 Увеличивается
Посг-эффект (на воздухе) Отсутствует Окисление «захваченных» макрорадикалов
* При умеренных дозах облучения.
Таким образом, фотохимический метод представляется более рациональным для модифицирования тонких изделий из полиэтилена или поверхностных слоев толстостенных изделий. Простота и малая стоимость аппаратуры, возможность осуществления процесса на воздухе являются преимуществами фотохимического метода по сравнению с радиационно-химическим.
Литература
1. Busse W. F„ Smook М. A. India Rubber World, 1953, v. 128, p. 348. — 2. Busse W. F., Bilmeyer F. W. J. Polymer Sci., 1954, v. 12, № 67, p. 599. — 3. Smook M. A., Remington W. J., Strain D. E. В кн.: Полиэтилен и другие полиолефины. Под ред. П. В. Козлова и
140
Н. А. Платэ. М., «Мир», 1964. 594 с. — 4. Пат. США 2416060, 1947.— 5. Smook М. A., Roche J. D., Clark W. В. India Rubber World, 1953, v. 128, р. 54. — 6. Итал. пат. 563508, 1956. — 7. Bier G. Angew. Chem., 1961, Bd. 73, S. 186; пер.; «Химия и технология полимеров», 1962. № 6, с. 3.— 8. М а н я с е к 3., Бел л v ш Д. Полипропилен. Под ред. В. И. Пплиповского и И. К. Ярцева. Л., «Химия», 1967. 316 с. — 9. N a t t a G., С г е s р i G.. В г u z-zone М. Chim. е ind., 1960, v. 42, р. 463.— 10. Джонс Г. Химические реакции полимеров. Т. 1. Под ред. Е. Феттеса. М.. «.Мир», 1967. 503 с.
11. Leonard Е. С., L о е d W. Е., Mason J. Н„ S tens from J. А. J. Polymer Sci., 1961, v. 55, p. 799.— 12. Leonard E. C., Loed W. E., Mason J. H., Wheelwright W. L. J. Appl. Polymer Sci., 1961, v. 5, p. 157.—13. Англ. пат. 849058, 1960. — 14. Пат. США 2906743, 1960.— 15. Итал. пат. 537429, 1955. — 16. Итал. пат. 591501, 1958. — 17. Англ. пат. 811848, 1959.— 18. Англ. пат. 867209, 1961. — 19. Сеидов Н. М. Новый синтетический каучук на основе этилена и пропилена, Баку, Азерб. гос. изд., 1966. 127 с. — 20. Амберг Л. О. В кн.; Вулканизация эластомеров. Под ред. Г. Аллигера, И. Сьетуна. М., «Химия», 1967. 428 с.
21. Пат. США 2626248, 1953.—22. Пат. США 2681327, 1954,— 23. Brown И. Р., Gibbs С. F. Ind. Eng. Chem., 1955, v. 47, p. 1006.— 24. Франц, пат. 1336464, 1964. — 25. Bonotto S., Purcell C. L. Mod. Plast., 1965, v. 42, № 7, p. 135, 140, 198, —26. Rees R, Vaughan D. Polymer Preprints, 1965, v. 6, № 1; пер.: «Химия и технология полимеров», 1966, № 6, с. 3, 9. — 27. Семенова А. С., Николаев А. Ф. Пласт, массы, 1967, Ks 10, с. 67.-28. Chem. Eng., 1964, v. 71, № 22, р. 90. — 29. Brit. Plast., 1965, v. 38, № 5, p. 262.— 30. Бовей Ф. Действие ионизирующих излучений на природные и синтетические полимеры. М., ИЛ, 1959.
31. Чарлз би А. Ядерные излучения и полимеры. М., ИЛ, 1962. 522 с.— 32. Своллоу А. Радиационная химия органических соединений. М., ИЛ, 1963. 408 с. — 33. Действие радиации на органические материалы. Под ред. Р. Болт и Дис Кэррол. М.. Атомиздат, 1965. 499 с. — 34. ШульцА. В кн.: Химические реакции полимеров. Т. 2. Под ред. Е. Феттеса. М., «Мир», 1967. 536 с. — 35. Basket А. С., Miller С. W. Nature, 1954, v. 174, р. 364.— 36. Schumacher К., Kolloid-Z., 1958, Bd. 157, S. 16. — 37. Сирота А. Г., Гольденберг А. Л., Ильиченко П. А. и др. Пласт, массы, 1966, К» 8, с. 58.—38. Сирота А. Г., Федотов Б. Г., Рябиков Е. П. и др. Пласт, массы, 1967, № 2, с. 3.—39. Black R. М., Lyons В. J. Nature, 1957, v. 180, р. 1346. — 40. Black R. М., Lyons В. J. Proc. Roy. Soc. (London), 1959, v. 253, Ser. A, p. 322.
41. В e с e л о в с к и й P. А., Лещенко С. С., Карпов В. Л. В кн.: Радиационная химия полимеров. М., «Наука», 1966. 408 с. — 42. Cooper G. D., G i lb е г t A. R. J. Polymer Sci., 1959, v. 38, p. 275. — 43. L о у В. R. J. Polymer Sci., 1963, v. 1, Ser. A, p. 2251. — 44. Law ton E. J., Bueche A. M., Balwit J. S. Nature, 1953, v. 172, p. 76. — 45. Alexander P., Black R. M., Charlesby A. Proc. Roy. Soc. London, 1955, v. 232, Ser. A, p. 31. — 46. Charlesby A., Davison W. H. T. Chem. a. Ind. (London), 1957, v. 8, p. 232. — 47. Charlesby A., Swallow A. J. Rev. Phys. Chem., 1959, v. 10, p. 289. — 48. Charlesby A., Arnim E., Callaghan L. Intern. J. Appl. Radiation and Isotopes, 1958, v. 3, p. 226.— 49. Kitamura R., Mandelkern L. J. Am. Chem. Soc., 1964, v. 86, № 17, p. 3529. — 50. Epstein L. M. J. Polymer Sci., 1957, v. 26, p. 399.
51. Law ton E. J., Balwit J. S., Powell R. S. J. Polymer Sci., 1958, v. 32, p. 257. — 52. L a w t о n E. J., Powell R. S., В a 1 w i t J. S. J. Polymer Sci., 1958. v. 32, p. 277. — 53. Williams T. F„ Dole M. J. Am. Chem. Soc., 1959, v. 81, p. 2919. — 54. Levy B. J. Appl. Polymer Sci., 1961, v. 5, p. 408; Trans. Am. Nucl. Soc., 1961, v. 4, p. 124. — 55. Сирота А. Г., Федотов Б. Г., Рябиков E. П. и др. ДАН СССР. Сер. хим., 1968, т. 183, № 2, с. 393.— 56. Black R. М„ Charlesby A. Intern. J. Appl. Radiation and Isotopes, 1959, v. 7, p. 126, —57. Black R. M. Nature, 1956, v, 178, p. 4528, 305,—
141
58. Англ. пат. 740899, 1955. — 59. Пат. США 2877500, 1959. — 60. Пат. США 2878174, 1959.
61. Пат. США 2904480, 1959.— 62. Ушаков Г. П., Гущо Ю. А., Л а-зуркин Ю. С., Казаков В. С. Высокомол. соед., 1960, т. 2, № 10, с. 1512. — 63. Майгельдинов И. А. В кн.: Химия и физико-химия высокомолекулярных соединений. М., Изд. АН СССР, 1952. 312 с. — 64. Самойлов А. В. Пласт, массы, 1967. № 5, с. 14. — 65. Наливайко Е. И., Сирота А. Г. Пласт, массы, 1967, № 9, с. 14. — 66. Houdret С., Lamm А. Rev. Gen. Caout, 1963, v. 40, № 9, p. 1323. — 67. L i e b 1 i n g G. J. Appl. Polymer Sci., 1962, v. 6, № 22, p. 461. — 68. Via gin G. Mater. Plast., 1966, v. 3, № 4, p. 209. — 69. Наливайко E. И., Сирота А. Г. Пласт, массы, 1968, № 2, с. 13. — 70. Власенко В. П., Карпов В. Л., Рашина Е. П., Финкель Э. Э. В кн.: Радиационная химия полимеров. М., «Наука», 1966. 408 с.
71. Charlesby A.. Grace С. S., Pilkington F. В. Proc. Roy. Soc., 1962, v. 268, Ser. A, № 1333, p. 205; nep.: «Химия и технология полимеров», 1963, № 2, с. 40. — 72. Чернявский Г. В. Автореф. канд. дисс. Киев, 1967.— 73. Wilski Н. Angew. Chem., 1959, Bd. 71, S. 612.— 74. Oster G. J. Polymer Sci., 1956, v. 22, p. 185. — 75. Англ. пат. 848414, 1960.—76. Oster G., Oster G. K-, Moroson H. J. Polymer Sci., 1959, v. 34, p. 671.— 77. Charlesby A., Grace C. S., Penh ale L. C. J. Polymer Sci., 1959, v. 34, p. 681. — 78. Ц я н ь Бао-гун, Чиан Пин-чен, Хау Ен-чиан. Высокомол. соед., 1959, т. 1, с. 635. — 79. Лян-Ян-цю, Ван С я-ю й, Фан Чуи-чан. Цзян Бин-чжен, Цянь Бао-гун. Scientia Sinica, 1962, т. 11, с. 903.—80. Цянь Бао-гун, Цзян Внн-чжен, Ляо Юй-чжень, Лян Ин-цю. Scientia Sinica, 1962, т. 11, с. 1513.
81. Wilski Н. Koll.-Z., 1963, Bd. 188, S. 4. — 82. Франц, пат. 1369014, 1964. — 83. Качан А. А., Чернявский Г. В., Шрубович В. А. «Допо-Bifli АН УРСР», 1966, т. 10, с. 1312. — 84. Качан А. А., Чернявский Г. В., Шрубович В А. Высокомол. соед., 1967, т. 9, № 1, с. 43.— 85. Англ. пат. 981255, 1965. — 86. Уэмацу Ититаро. J, Soc. Rubber Ind. Japan, 1959, v. 32, № 11, p. 902. — 87. Oster G. J. Polymer Sci., 1964, v. 32, Ser. B, p. 1181. — 88. Soumelis K-, Wilski H. Kunststoffe, 1962, Bd. 52, S. 471. — 89. Кауркова Г. К., Качан А. А., Корнев К. А. и др. Хи-мгчна промисловшть, 1965, № 2, с. 8.—90. Пат. ФРГ 1167018, 1964.
91. Авт. свид. СССР 191114. 1965; Бюлл. изобр., 1967, № 3. —92. Качан А. А., Чернявский Г. В., Шрубович В. А. Высокомол. соед., 1967, т. 9, сер. А, № 5, с. 1076. — 93. Качан А. А. и др. Тезисы докладов на Всесоюзном научно-техническом совещании по пластмассам, 1966. — 94. Качан А. А., Чернявский Г. В., Шрубович В. А. Высокомол. соед., 1967, т. 9, сер. Б, № 1, с. 40.—95. Charlesby A., Pinner S. Н. Proc. Roy. Soc., 1959, v. 249, Ser. A, p. 367. — 96. Качан А. А., Шрубович В. А., Чернявский Г. В. и др. Пласт, массы, 1968, № 6, с. 7.— 97. Бреслер С. Е., Казбеков Э. Н., Фомичев В. Н. и др. «Физика твердого тела», 1963, т. 5, № 2, с. 675. — 98. Kharasch М., Jensen Е., Urry U. Р. J. Am. Chem. Soc., 1945, v. 67, р. 1864. — 99. Качан А. А., Сирота А. Г., Чернявский Г. В., Шрубович В. А. Высокомол. соед., 1968, т. 10, сер. А, с. 471.— 100. Ranby В.. Joshi da Н. J. Polymer Sci., 1966, v. 10, Ser. С, p. 263.
101. Law ton E. J., В al wit J. S., Powell R. S. J. Chem. Phys., 1960, v. 33, p. 395, 405. — 102. Егорова 3. С., Карпов В. Л., Лещенко С. С. и др. В кн.: Радиационная химия полимеров. М„ «Наука». 1966. 408 с.— 103. Батычко С. В.. Брагинский Р. П., Ярмилко Е. Г., Кабак-чи А. М. Там же.— 104. Качан А. А., Чернявский Г. В., Шрубович В. А. «Доповщ! АН УРСР», 1967, № 7, с. 626.— 105. Sakamoto К-, МасК night W. J., Porter R. S. J. Polymer Sci., 1970, v. 8, Ser. A2, № 2, p. 277. — 106. MacKnight W. J., Kajiyama T., McKenna L.
142
Polymer Eng. Sci., 1964, v. 8, № 4, p. 267.— 107. Авт. свид. СССР 329186; Бюлл. изобр., 1972, № 7. — 108. Stephenson С. V., Moses В. С., Wilcox W. S. J. Polymer Sci., 1961, v. 55, p. 451.— 109. Англ. пат. 887950, 1961. — ПО. Франц, пат. 1369014, 1964.
111. Качан А. А., Чернявский Г. В., Шрубович В. А. В кн.: Синтез и физико-химия полимеров. Киев, «Наукова думка», 1966. 196 с. — 112. Англ. пат. 1087403, 1968.— 113. Лаврищев В. П., Павлов С. А., Боков Ю. С. Высокомол. соед. 1967, т. 9, сер. А, № 4, с. 894. — 114. Боков Ю. С., Лаврищев В. П., Павлов С. А. Изв. вузов. Технология легкой пром., 1968, № 4, с. 64. — 115. Андрущенко Д. А., Богдан Л. С., Качан А. А., Шрубович В. А. Высокомол. соед., 1972, т. 14, сер. А, № 3, с. 594, —116. Япон. пат. 41117, 1971. —117. Япон. пат. 32856, 1971.— 118. Андрущенко Д. А., Качан А. А., Чернявский Г. В., Шрубович В. А. Высокомол. соед., 1969, т. И, сер. Б, с. 600. — 119. Braun С. L., Kato S., Lip ski S. J. Chem. Phys., 1963, v. 39, p. 1645.— 120. Kuji-rai C., Ha shiga S., Fur uno H., Terada N. J. Polymer Sci., 1968, v. 6, Ser. A, № 3, p. 589.
121. Pitts J. N., Let sin ger R. L., Та у 1 er R. P. ea. J. Am. Chem. Soc., 1959, v. 81, p. 1068. — 122. Теренин A. H. Фотоника молекул красителей. Л., «Наука». 1967. 616 с. — 123. Андрущенко Д. А. Автореф. канд. дисс. Киев, 1971. — 124. Андрущенко Д. А., Качан А. А., Чернявский Г. В.. Шрубович В. А. Способы записи информации на бес-серебряных носителях. Вып. 2. Изд. Киевского гос. ун-та, 1970, с. 29. — 125. Ishikawa Н., Noyes W. A. J. Am. Chem. Soc., 1962, v. 84, р. 1502.— 126. Пукшанский М. Д.. Зюзина Л. И., Хайкип С. Я. и др. Высокомол. соед., 1972, т. 14, сер. А, № 10, с. 2096. — 127. Мяхлис Ф. А. Радиационная физика и химия полимеров. М., Атомиздат. 1972. 326 с. — 128. Пукшанский М. Д. Автореф. канд. дисс.. 1972. — 129. Павлов В. И., Шрубович В. О., Чернявский Г. В., Качан О. О. «Допов!д1 АН УРСР», 1969, т. 6, № 1, с. 36.— 130. Веселовский Р. А., Лещенко С. С.. Карпов В. Л. Высокомол. соед., 1968, т. 10, сер. А, с. 760.
131. Пукшанский М. Д., Сирота А. Г.. Качан А. А. и др. Пласт, массы, 1970, № 5, с. 50. — 132. Ткаченко А., Чернявский Г. В., Шрубович В. А., Качай А. А. Хим. волокна, 1970, № 2, с. 30. — 133. Пукшанский М. Д., Колясева В. А., Зюзина Л. И. и др. Пласт, массы. 1970, № 11, с. 51. —134. Pross A., Black Р. J. Soc. Chem. Ind., 1950. v. 69, р. 113.— 135. Пивоваров А. П., Луковников А. Ф. «Химия высоких энергий», 1968, т. 2, с. 220. — 136. П и в о в а р о в А. П., Г а к Ю В.. Луковников А. Ф. Высокомол. соед., 1971, т. 13, сер. А, № 9, с. 2110.— 137. Пат. США 2723257, 1955. — 138. Гофманн В. Вулканизация и вулканизующие агенты. Л., «Химия», 1968.— 139. Донцов А. А., Дроздова Г. П., Л оз овик Г. Я- и др. Высокомол. соед., 1972, т. 14, сер. Б, № 6, с. 433. —140. Финкель Э. Э., Лещенко С. С., Брагинский Р. П. Радиационная химия и кабельная техника. М.. Атомиздат, 1968. 312 с.
141. Пьянков Г. Н.. Мелешевич А. П., Ярмилко Е. Г. и др. Радиационная модификация полимерных материалов. Киев, «Техшка», 1969. 229 с. — 142. Брагинский Р. П., Финкель Э. Э., Лещенко С. С. Стабилизация радиационно-модифицированных полиолефинов. М., «Химия», 1973. 198 с. — 143. Дж ей л П. Разрушение твердых полимеров. Под ред. Б. Роузена. М., «Химия», 1971. 551 с. —144. Патрикеев Г. А. Механика полимеров. 1971, № 2. с. 221, — 145. Пат. США 2877500, 1959.— 146. Пат. США 302254, 1962. — 147. Брас л и ныл У. А., Калькис В. Я., Раявее Э. Л., Лауке в ип Я- Я- Пласт, массы. 1968, № 4. с. 39. — 148. Калькис В. Я. Автореф. канд. лисе. М.. 1972. — 149. Пат. США 3090735. 1963. — 150. Джиб-гашпили Г. Г., Словохотова Н. А.. Лещенко С. С., Карлов В. Л. Высокомол. соед., 1971, т. 13, сер. А, № 5, с. 1087.
143
151. Джибгашвили Г. Г. Автореф. каид. дисс. М., 1969. — 152. Abraham R., Whiffen D. Trans. Faraday Soc., 1958, v. 54, p. 1291.—
153. Kawai T., Keller A., Charlesby A., Orme rod M. G. Phil. Mag.,
1965, v. 12, p. 657.— 154. Sobue H„ Tasima I. Nature, 1960, v. 188, p. 315. — 155. Sobue H. e. а. Когё Кагаку Дзасси, 1969, v. 62, p. 136.— 156. Gupta R. P. Kolloid Z„ 1961, Bd. 174, S. 74,— 157. Gupta R. P. J.
Phys. Chem., 1962, v. 66, p. 849.— 158. Kawai T., Keller A. Phil. Mag.,
1965, v. 12, p. 673. — 159. Доуль M. В кн.: Свободнорадикальные состояния в химии. Новосибирск, «Наука», 1972. 250 с. — 160. Charlesby A., Libby D., О г me rod М. G. Proc. Roy. Soc., 1961, v. 262, Ser. A, p. 207.
161. Howard J. В. В кн.: Конструкционные свойства пластмасс. Под ред. Э. Бэра. М., «Химия», 1967. 464 с.— 162. Брагинский Р. П., Гаити В. Л., Наливай ко Е. И., Сирота А. Г., Финкель Э. Э. «Труды ВНИИК.П», 1972, вып. 16, с. 196.— 163. Никитский А. В., Г л у-ховская Г. И., С ош ко А. И., Кабакчи А. М. Пласт, массы, 1972, № 7, с. 33. — 164. Зуев Ю. С. Разрушение полимеров под действием агрессивных сред. М., «Химия», 1972. 229 с. — 165. Качан А. А., Ш р у б о-в и ч В. А. Фотохимическое модифицирование синтетических полимеров. Киев, «Наукова думка», 1972. 160 с.
Глава VI
Композиции но основе полиолефинов
Композиции полиолефинов с различными полимерами
Получение композиций полиолефинов друг с другом и с полимерами других классов позволяет существенно изменять свойства материалов в желаемом направлении. Так, композиции полипропилена с полиэтиленом низкой или высокой плотности [1—3] отличаются от полипропилена более низкой температурой хрупкости. Плохоцкий [4], изучая свойства смесей изотактического полипропилена и линейного полиэтилена, показал, что технологические свойства смесей лучше, чем свойства исходных полимеров. Показатель текучести расплава смесей в широком интервале весовых соотношений полипропилена и полиэтилена (15 : 85; 25 : 75; 50 : 50; 75 : 25) сохранялся более высоким, чем у отдельных компонентов.
Повышение ударной вязкости и физико-механических характеристик при пониженных температурах достигается одновременным введением в полипропилен полиэтилена и сополимеров этилена с пропиленом [5, 6].
Значительное понижение температуры хрупкости и повышение ударной вязкости происходит при смешении полипропилена с полиизобутиленом и различными каучуками [7, с. 196; 8; 9]. Улучшение указанных характеристик сопровождается, однако, уменьшением прочности материала (рис. 56). По мере повышения содержания полиизобутилена в смеси с полипропиленом понижается температура текучести [10]. В широком температурном интервале — между температурой стеклования полиизобутилена и температурой плавления полипропилена — смеси проявляют высокоэластические свойства. Высокоэластичность смесей тем выше, чем больше содержание полиизобутилена.
Немонотонный характер зависимости некоторых свойств от состава смеси обнаружен при изучении связи удлинения при разрыве с соотношением компонентов. При соотношении полипропилена и полиизобутилена близком к 1:1 наблюдался минимум удлинения при разрыве (рис. 57, кривая /). Существование этого минимума связывается с двумя различными механизмами деформации. Аморфный компонент смеси — полиизобутилен— способен к большим обратимым высокоэластическим
6 А. Г. Сирота 14S
деформациям. Добавление к полиизобутилену более жесткого кристаллического полипропилена увеличивает модуль упругости и уменьшает деформируемость. Высококристаллический компонент смеси — изотактический полипропилен — при достаточно высоких напряжениях способен к большим вынужденным высокоэластическим деформациям. Добавление полиизобутилена
понижает прочность полипропилена и препятствует созданию напряжений, достаточно больших для возникновения вынужден-
§ § го || 100 .1 § -11
is
15
10
5,
см
| § 400±
| 200 -
В’
§ 100'—i—1-------‘----L.
§. 5 10 15 20 25 30
Содержание бутилкаучука, %
ных высокоэластических деформаций. Это и приводит к уменьшению величин удлинения при разрыве. Изменение состава смеси вызывает смену одного механизма деформации другим. Минимум удлинения
1500 а? f § 1000
I
§ 500
§
О 50 100 Полипропилен
100 50 О Полиизобутилен
Содержание, %
х
+
О
Рис. 56. Зависимость свойств композиций полипропилена и бутилкаучука от содержании последнего [7].
Рис. 57. Зависимость относительного
удлинения при разрыве от состава смеси полипропилена и полиизобутилена при 23 °C (7) и 60’С (2) [10].
при разрыве соответствует промежуточной области составов, в которой ни один из механизмов не может проявиться в полной мере.
При повышенных температурах на кривой зависимости удлинения при разрыве от состава обнаруживаются две части (рис. 57, кривая 2), отвечающие малым и большим содержаниям аморфного компонента смеси. Это объясняется уменьшением деформируемости полиизобутилена при повышенных температурах вследствие текучести, затрудняющей развитие больших высокоэластических деформаций. По мере возрастания доли полипропилена в смеси текучесть оказывается подавленной, способность к высокоэластическим деформациям увеличивается. Эти явления, наблюдавшиеся также при изучении специально приготовленных смесей аморфного полипропилена с кристаллическим, рассматриваются [10] как следствие двухфаз-
146
ности смесей аморфного и кристаллического полимеров. С этой точки зрения изменение состава смеси означает переход от аморфной системы, наполненной кристаллическим компонентом, к кристаллической системе, наполненной аморфным компонентом. Этот переход и сопровождается скачкообразным изменением зависимости физико-механических свойств от состава смеси.
Подробному исследованию в ряде работ [11 —16; 17, с. 134; 18—20] подвергались композиции полиэтилена с полиизобутиленом или неполярными каучуками, находящие значительное практическое применение. Смешение обычно проводят в закрытых смесителях при 130—170 °C или на вальцах. Основным результатом введения полиизобутилена является повышение эластичности композиции и сопротивляемости растрескиванию под влиянием длительных нагрузок и поверхностно-активных веществ. С увеличением содержания полиизобутилена уменьшается прочность при растяжении, модуль упругости, твердость и возрастает удлинение при разрыве. По данным работ [11, 13, 14, 16] изменение физико-механических характеристик, в том числе и удлинения при разрыве, с изменением состава имеет плавный характер. Однако Слонимский с сотрудниками [18], в отличие от других исследователей применявшие композиции, полученные не на вальцах, а соосаждением из раствора в декалине, показали, что зависимость удлинения при разрыве ОТ состава проходит через минимум. Минимальное значение удлинения при разрыве при комнатной температуре наблюдается при соотношении полиэтилена и полиизобутилена в композиции около 3:1. Очевидно, что причина существования этого минимума аналогична описанной для композиции полипропилена с полй-изобутиленом. Отсутствие минимума удлинения при разрыве образцов, полученных механическим смешением на вальцах или смесителях, можно объяснить дополнительной гомогенизацией в результате химического взаимодействия компонентов композиции с образованием блок- и привитых сополимеров.
Особенности свойств смесей кристаллических полиолефинов, таких, как смеси полиэтилена с изотактическим полипропиленом, определяются неоднородностью их структуры [21]. Микроскопическое исследование смеси в поляризованном свете обнаруживает крупные сферолиты полипропилена и мелкие сферолиты полиэтилена. Температура текучести смеси повышается по мере увеличения содержания полипропилена. Прочность смесей полиэтилена с полипропиленом тем выше, чем больше доля полипропилена в смеси. В отличие от индивидуальных компонентов смеси, способных при комнатной температуре к значительным вынужденным высокоэластическим деформациям, смеси при 20 °C разрушаются при малых деформациях (при растяжении не более 10%). Однако при температурах выше температуры плавления более низкоплавкого компонента — полиэтилена—деформируемость смеси существенно увеличивается.
6*
<47
Полиэтилен, таким образом, выполняет в этом случае роль высокомолекулярного пластификатора полипропилена [21, 22]. Отметим, что введение в полиэтилен даже небольших количеств
полипропилена может существенно уменьшать сопротивляемость термоокислительным процессам [98] (рис. 58).
Особенностей поведения при деформировании можно ожидать,
очевидно, и при смешении других пар кристаллических поли-
Рис. 58. Влияние полипропилена на окисление полиэтилена, стабилизированного 4,4-тиобис(2-трег-бутил-5-метилфенолом), при 140 °C [98]:
1—полиэтилен с добавкой 5% полипропилена; 2 — исходный полиэтилен.
олефинов, смеси которых характеризуются неоднородностью в результате раздельной кристаллизации компонентов. Так, неоднородностью смеси объясняются пониженные величины относительного удлинения при растяжении и прочности при разрыве композиций, содержащих полиэтилен низкого и высокого давления (рис. 59) [23]. Пластифицирующим действием полиэтилена высокого давления в смеси с по-
лиэтиленом низкого давления, по-видимому, можно объяснить
пониженную хрупкость полиэтилена низкого давления при добавлении от 15 до 50 вес. ч. полиэтилена высокого давления [24].
Введение в кристаллические полиолефины добавок различ-
ных аморфных полимеров существенно изменяет важные технические свойства материала. Так, смешение полипропилена с аморфными полимерами повышает его окрашиваемость [25—28; 29, с. 189], морозостойкость [30, 267], светостойкость [31], термостойкость [32], уменьшает ползучесть [33]. Восприимчивость к красителям повышается особенно значительно при использовании в качестве добавок полярных полимеров, однако в этих случаях для смесей характерна большая неоднородность. Применение
неполярных и малополярных полимеров позволяет получать менее неоднородные смеси с полиолефинами, окрашивающиеся дисперсными красителями благодаря рыхлости структуры композиции.
Разрыхление структуры под влиянием небольших добавок полистирола и сополимера стирола с 20% акрилонитрила было показано при изучении волокна из изотактического полипропилена [29]. Уже при содержании 5% указанных добавок наблюдается уменьшение напряжения, требуемого для вытягивания волокна. Предполагается, что частицы второго компонента композиции, распределяясь в полипропилене, разрыхляют его структуру, уменьшают сопротивление движению одних элементов структуры относительно других при деформации. Рыхлость структуры смесей проявляется и в значительном увеличении поглощения дисперсных красителей волокнами из композиции по сравнению с волокнами из полипропилена.
141
Высокой сопротивляемостью истиранию обладает полиэтилен в смеси с полиамидами (полигексаметиленадипамидом, по-
лигексаметиленсебацамидом, поликапролактамом) [99]. Введение в порошкообразный полиэтилен 10% дисперсии полиамида с размером частиц до 20 мкм позволяет увеличить стойкость к истиранию в 30 раз.
Известны композиции полиолефинов с природными полимерами, в частности полиэтилена с включением целлюлозы [34], смеси полипропилена с лигнином [35—40]. Лигнин представляет интерес в качестве активного компонента смеси. Переработка смеси полипропилена со щелочным сульфатным лигнином при температурах около 200 °C сопровождается взаимодействием компонентов с образованием частично сшитой структуры. Максимальное образование гель-фракции достигается при содержании около 2% лигнина в полипропилене. Введение небольшого количества лигнина не изменяет кристаллической структуры материала, но повышает его теплостойкость, а также стабильность в условиях светового старения.
Важным вопросом, связанным с разработкой составов и условий получения композиций, является совместимость компонентов смесей на основе полиолефинов. Литературные данные по этому поводу весьма противоречивы. Так, различные авторы, изучая структуру и свойства смесей полиэтилена и пропилена, пришли к противоположным выводам об их со-
Рис. 59. Зависимость свойств от состава смеси полиэтилена высокого (ПВД) и низкого (ПНД) давления [23].
вместимости. Михайлов с сотрудниками [22] исследовали смеси полиэтилена низкого давления с изотактическим полипропиленом. Полученные ими кривые дифференциально-термического ана-
лиза обнаруживают один эндотермический пик при содержании
149
полиэтилена в смеси 75% и более. При меньших концентрациях полиэтилена наблюдается два эндотермических пика, соответствующих температурным областям плавления компонентов смеси. Этот факт наряду с повышенными значениями теплоемкости смесей с преобладанием полиэтилена и отклонение плотностей от аддитивных величин привели к выводу о совместимости полиэтилена с полипропиленом в определенной области их соотношений. Наиболее высокая степень совместимости соответствует концентрации полиэтилена 75% и выше. Явление совмещения связывается авторами с пластифицирующим действием полиэтилена на полипропилен; большая гибкость цепей полиэтилена позволяет в процессе смешения уменьшать жесткость полипропилена и способствует совмещению его с полиэтиленом. Этим объясняется, что совмещение происходит при сравнительно высоких концентрациях полиэтилена в смеси.
Слонимский с сотрудниками [21] также исследовали совмещение изотактического полипропилена с полиэтиленом. На основании структурно-оптических данных был сделан вывод о микронеоднородности смесей и несовместимости этих полимеров. Этот вывод согласуется и с данными других авторов [41, 42], изучавших сферолитные структуры смесей полипропилена и полиэтилена.
Гуль с соавторами [43] оценивали степень совместимости полиэтилена с полипропиленом по оптической плотности пленок из смеси этих полимеров (в соотношении 1:2), подвергнутых прогреву при 100 °C. Наблюдавшееся понижение оптической плотности с увеличением продолжительности прогрева указывало на уменьшение микронеоднородности в результате протекания диффузионных процессов. Снижение дисперсности кривых распределения прочности смеси по мере прогревания образцов также свидетельствовало о возрастании их однородности.
Фильберт с сотрудниками [44, с. 82] подробно исследовали смеси изотактического полипропилена с полиэтиленом низкого давления. Содержание полипропилена варьировалось от 5 до 25%. Кривые дифференциально-термического анализа имели два пика, соответствующих эндотермическим эффектам при температурах плавления каждого из полимеров. Степень кристалличности полипропилена, определяемая по спектральным данным (по полосам поглощения 845 и 1170 см-1), в присутствии полиэтилена не изменялась. Коэффициент поглощения при частотах, связанных с маятниковыми колебаниями метиленовых групп полиэтилена (полоса поглощения 731 см-1), оказался не зависящим от соотношения компонентов смеси. Эти факты объяснены несовместимостью полипропилена с полиэтиленом и раздельной их кристаллизацией в смеси. Отклонения от аддитивности плотностей и теплоемкостей смесей, которые ранее [22] интерпретировались в пользу представления о совместимости полипропилена и полиэтилена, авторами работы [44] связы
ваются с возможностью образования смешанных сферолитов, включающих более мелкие кристаллические образования и полиэтилена и полипропилена. Таким образом, отрицается совместимость на молекулярном уровне и допускается возможность совмещения на надмолекулярном уровне.
Противоположные заключения имеются и о совместимости полиэтилена с полиизобутиленом. Одни авторы [15, 17] считают полиизобутилен хорошо совмещающимся с полиэтиленом, другие [12, 16] отрицают их совместимость. Несовместимость полиэтилена с полиизобутиленом показана рентгенографически [12]. Уменьшение степени кристалличности смеси по мере увеличения содержания в ней полиизобутилена при сохранении среднего размера кристаллических областей позволяет считать, что полиизобутилен распределяется в аморфных областях полиэтилена, образуя отдельную аморфную фазу с ближним порядком расположения молекул (6,1 А), отличным от ближнего порядка аморфной фазы полиэтилена (4,55А). По данным работы [16], полное совмещение полиэтилена с полиизобутиленом не достигается даже выше температуры плавления полиэтилена, когда оба полимера аморфны. Расхождения в оценках совместимости в значительной мере связаны с отсутствием единого понимания этого свойства смесей полимеров.
Представляется полезным дифференцированное рассмотрение термодинамической и эксплуатационной совместимости [43; 45, 46, 267]. Обладающие термодинамической совместимостью смеси полимеров не изменяют своих свойств после повышения температуры и возвращения к исходной. При этом степень однородности смесей может повышаться. Степень термодинамической совместимости предложено оценивать интервалом соотношений компонентов, обеспечивающим термодинамическую устойчивость системы. Чем шире интервал соотношений, в котором один полимер полностью растворяется в другом, тем больше степень их совместимости. Учитывая сложность структур цепей и их ассоциатов, трудно предполагать термодинамическую Совместимость полиолефинов друг с другом и другими полимерами. Стабильность структур и свойств смесей, включающих полиолефины, может быть связана не с термодинамическими факторами, а с весьма малой скоростью изменения структур. Термодинамическая несовместимость не исключает, однако, эксплуатационной совместимости, которая зависит от допустимого предела изменения определенных свойств смеси во времени в условиях эксплуатации. Изменение свойств смеси термодинамически несовместимых полимеров при эксплуатации связано с возрастанием неоднородности системы.
В соответствии с этими представлениями полиэтилен и полипропилен относятся к термодинамически несовместимым, но обнаруживающим эксплуатационную совместимость полимерам. Эксплуатационной совместимостью с полиолефинами могут
151
обладать и полярные полимеры. Например, эксплуатационной совместимостью характеризуется стойкая к маслам смесь полиэтилена с поливиниловым спиртом в соотношении 95:5 [48]. Стойкая к растрескиванию композиция получается при добавлении к полиэтилену продукта поликонденсации бисфенола и эпихлоргидрина в количестве от 0,2 до 15 вес. ч. [261]. Такая композиция по сопротивляемости растрескиванию находится на уровне композиций полиэтилена с эластомерами и превосходит их по качеству поверхности (блеск, гладкость) и механическим свойствам.
Интересные результаты были получены при изучении связи между эффективностью ингибирования термоокисления антиоксидантами и их совместимостью с полиолефинами [248, с. 267; 249; 250]. Основными факторами, определяющими совместимость антиоксидантов и светостабилизаторов с полиэтиленом, являются: 1) наличие в молекуле стабилизатора алифатических или ароматических заместителей; для алифатических заместителей— длина цепи, разветвленность и равномерность распределения в молекуле; 2) способность молекул стабилизаторов к образованию межмолекулярных водородных связей; 3) дипольный момент молекулы. ‘ С учетом этих факторов можно повышать совместимость стабилизатора и полимера. При этом стабилизатор в меньшей мере будет мигрировать на поверхность полимера, улетучиваться, вымываться. Тем не менее наиболее эффективными стабилизаторами полиэтилена и полипропилена оказываются антиоксиданты, обладающие не максимальной, а меньшей, чем максимальная, совместимостью с полимером. Некоторое снижение совместимости, очевидно, ведет к повышению концентрации антиоксиданта в аморфных областях, различных дефектах структуры, на поверхностях надмолекулярных образований, т. е. в местах, наиболее подверженных окислению [251, 252].
Примером использования ограниченной совместимости полиолефинов может быть модифицирование полиэтилена введением в него небольших количеств продуктов химического превращения (фосфонирования, окисления, прививки полярных мономеров) того же полиэтилена. В процессе формования изделий при охлаждении расплава происходит миграция части добавки на поверхность изделия вследствие ограниченной совместимости полиэтилена и продукта его химического превращения. В результате полярный компонент композиции концентрируется в поверхностном слое и повышается адгезионная способность полимера. Этот эффект достигается при малых добавках полярного компонента и потому не сопровождается существенным изменением других свойств полимера.
Свойства композиций полиэтилена низкой плотности марки П-2003-К с добавкой окисленного у-облучением в воздушной среде порошкообразного полиэтилена той же марки приведены 152
ниже * (содержание карбонильных групп в радиационно-окисленном полиэтилене0,013 вес.%, в исходном — менее0,001 вес.%):
Содержание в композиции радиационно-окисленного полиэтилена.
вес. % 0 2 6 10
Адгезия к медной оксидированной
фольге, гс/см 312 1550 1800 2200
Предел текучести, кгс/см2 115 112 109 106
Прочность прн растяжении, кгс/см2 162 142 165 146
Относительное удлинение при раз-
рыве, % 640 636 592 610
Облучение проводили при мощности дозы до 200 рад/с, поглощенная доза составляла 0,5 Мрад. Из композиции горячим прессованием (120 °C) получили пластины, на которые затем напрессовывали медную оксидированную фольгу (150 °C, выдержка без давления 10 мин и затем под давлением 35 кгс/см2 10 мин, охлаждение под давлением со скоростью около 8°С/мин). Адгезионную способность композиции оценивали по усилию отрыва фольги от пластины по методике, близкой к описанной в работе [253]. Во всех четырех случаях композиции имели следующие диэлектрические характеристики;
Тангенс угла диэлектрических потерь при 10е Гц................................
Диэлектрическая проницаемость при 106 Гц................................
Удельное объемное электрическое сопротивление, Ом - см......................
2- 10~4
2,3
1017
По-видимому, эффект ограниченной совместимости полиолефина с более полярным продуктом его модификации лежит в основе ряда рекомендаций, имеющих целью изменение свойств поверхности. Например, способность изотактического полипропилена окрашиваться возрастает при введении в него продукта прививки к полипропилену цепей мономера, содержащего гидрофильные группы [72]. Такой же результат достигается с помощью добавок к полиолефинам сополимера этилена с акриловой кислотой [75], акриламидом [205], введением в полиэтилен небольших количеств термодеструктированного окисленного полиэтилена [206].
Наполненные полиолефины
Наполнение применяют для повышения жесткости (рис. 60), предела текучести и улучшения других свойств, важных при использовании полиолефинов в качестве конструкционных материалов. Наполнителями служат различные порошкообразные и волокнистые материалы; окиси алюминия, кремния, магния, алюмосиликат, порошкообразные металлы (железо,
* По данным автора, Б. Г. Федотова, Л. Ф. Кантровской, М. П. Эйдель-нант.
153
медь, никель, алюминий и др.), стекловолокно, асбест, сажа, мел, древесная мука и др. [17, с. 128; 47—52; 53, с. 77; 54, с. 171; 256, 262—266]. На свойства композиций кроме типа и содержания наполнителя могут влиять форма частиц, характер их поверхности. Так, наполнение полиэтилена порошком железа с дендритной формой частиц вызывает значительно большее повышение прочности, чем наполнение порошком с пластинчатой формой частиц [51].
Рис. 60. Зависимость модуля упругости при изгибе полиэтилена высокой плотности от содержания наполнителя — окиси алюминия [48].
Акутин с сотрудниками [55, с. 80], изучая ИК-спектры композиции полиэтилена с двуокисью титана, обнаружили полосы поглощения при частотах 1515 и 1430 см~‘, отсутствующие в спектрах отдельных компонентов, и увеличение поглощения в области 1000—1200 см-1. Эти особенности спектра, интерпретированные как результат появления связей Ti—О—R, послужили основанием для предположения о химическом взаимодействии двуокиси титана с полиэтиленом в процессе приготовления и переработки композиции при повышенных температурах с образованием поверхностных соединений типа
(О—Т1—О—)тН
---СН2—СН2—С—СН2—СН2-----
Н
Использование в качестве наполнителей (или армирующих материалов) металлов переменной валентности, в частности меди, может существенно снижать сопротивляемость полиолефинов термоокислительным процессам (рис. 61) [98].
Вследствие гетерогенности структуры полиолефинов необходимо учитывать взаимодействие поверхности частиц напод-
154
кителя не только с макроцепями, но прежде всего с надмолекулярными образованиями. От природы дисперсности и других характеристик наполнителя могут зависеть морфология и размеры надмолекулярных образований, а следовательно, и свойства композиции. Так, введение в полиэтилен при одинаковых условиях аминированного [56, с. 83] и необработанного аэросила приводит к различным результатам [54, с. 171]. Наполнение
исходным аэросилом не влияет на морфологию и размеры сферолитов. Частицы такого наполнителя, обладающего полимерофобной поверхностью, собираются в агрегаты. Аминированный аэросил, напротив, более равномерно распределяется в полиэтилене. Введение его даже в небольшом количестве (0,1%) разрыхляет структуру, расширяет границы раздела между сферолитами.
Поскольку стойкость полиэтилена к растрескиванию существенным образом зависит от структуры [207, 208], одной из npi
Продолжительность, ч
Рис. 61. Влияние меди на окисление полиэтилена, стабилизированного 4,4'-тиобис(2-грег-бутил-5-метилфенолом), при 140 °C [98]: 1—полиэтилен в контакте с медью;
2 —исходный полиэтилен.
характера надмолекулярной ши повышенной сопротивляе-
мости растрескиванию некоторых наполненных систем могут быть морфологические изменения, происходящие в полиэтилене при наполнении. При введении мела [209; 210, с. 39] или сажи [211, с. 6; 212, с. 28] стойкость полиэтилена к растрескиванию возрастает в некотором интервале содержаний наполнителя. Дальнейшее увеличение концентрации наполнителя делает полиэтилен хрупким. Добавлением к наполненному полиэтилену пластификаторов (низкомолекулярного углеводорода [213], минерального масла [214] и эластомеров [210]) удается преодолеть этот недостаток. Такие композиции обладают лучшей перера-батываемостью. Их стойкость к растрескиванию выше, чем у полиэтилена, содержащего только наполнитель.
Характер влияния наполнения на надмолекулярную структуру полимера различен при малых и больших степенях наполнения [254, с. 223]. Введение небольших количеств наполнителя обычно ведет к некоторому повышению упорядоченности полимера. Частицы наполнителя изменяют условия кристаллизации, в той или иной мере играя роль искусственных зародышей кристаллизации. Тем не менее основные особенности свойств композиции связаны с изменениями в аморфной части полимера, поскольку частицы наполнителя концентрируются преимущественно в аморфных областях [259, с. 25]. При высоких концентрациях наполнителя (до 40—50%) совершенство надмолекулярных образований снижается, наблюдается аморфизация полимера. Предполагается, что в случае высокого наполнения
1S5
полимер существует в виде тонких прослоек между твердыми частицами наполнителя, а в тонких слоях полимера специфические условия кристаллизации затрудняют образование хорошо организованных надмолекулярных структур [255].
Добавки, влияющие на структуру надмолекулярных образований и вводимые в небольших количествах, нельзя рассматривать с достаточным основанием как наполнители. Такими добавками — структурообразователями — могут быть тонкие минеральные дисперсии (окись кремния, силикаты, хлориды натрия, кальция, алюминия, сульфаты натрия, алюминия и др.) [58]. Структурообразователями могут быть также некоторые термостабилизирующие системы, которые, как показывают электронно-микроскопические исследования, способствуют образованию совершенных по форме и плотности упаковки надмолекулярных структур [260]. Твердые мелкие частицы (с диаметром менее 1 мкм), диспергированные в расплаве полимера, играют роль зародышей кристаллизации при его охлаждении. Быстрая кристаллизация на большом числе гетерогенных центров кристаллизации приводит к образованию более мелких и однородных сферолитных структур. Пленки из композиции полиэтилена высокой плотности и полипропилена, содержащей 0,25—3 вес.% твердого минерального вещества с диаметром частиц около 50 нм, отличаются прозрачностью благодаря мелко-сферолитной структуре и пониженной хрупкостью, также, очевидно, вследствие сравнительно малых размеров и повышенной однородности сферолитов [58].
Подробно исследовано влияние на надмолекулярную структуру и свойства полиолефинов искусственных зародышей кристаллизации, представляющих собою вещества, не взаимодействующие с полимером и имеющие температуру плавления выше температуры плавления полимера [59—66]. В качестве таких веществ использовали органические кислоты (адипиновую, себациновую) и соли тяжелых металлов и органических кислот (салицилат висмута, оксалат титана, ацетат, бензоат и пальмитат свинца, ацетат цинка, нафтионат кобальта). Введение искусственных зародышей кристаллизации этого типа (наиболее эффективное количество 0,15—0,2 вес.%) увеличивает прочность, деформируемость и напряжение рекристаллизации полиэтилена и полипропилена. Полиэтилен низкого давления и полипропилен, содержащие 0,4—1,5 вес. % солей органических кислот, обнаруживают повышенную устойчивость при деформационных, термических и световых воздействиях.
Одним из важных последствий изменения надмолекулярной структуры полиэтилена под влиянием искусственных зародышей кристаллизации является увеличение стойкости материала к растрескиванию [67, 68].
Существенное повышение стойкости к растрескиванию после введения в полиэтилен очень небольших добавок органических соединений с температурой плавления выше температуры плав
156
ления полимера (антраниловой, адипиновой, себациновой кислот и маннита [67—69]) связано, очевидно, с хорошим распределением вещества добавки в расплаве полимера. Возможно также, что эти соединения оказывают влияние на кинетику кристаллизации не только в качестве центров кристаллизации, но также проявляют и свои поверхностно-активные свойства, снижая величину поверхностного натяжения на границе расплав — твердая фаза.
Введение поверхностно-активных структурообразователей позволяет резко повышать стойкость к растрескиванию полиэтилена без заметного ухудшения его диэлектрических свойств.
С помощью наполнителей можно существенно изменить адгезионную способность полиолефинов. Наполнение полиэтилена низкой и высокой плотности окисью алюминия повышает адгезию к металлам [257; 258, с. 179]. Факторы, определяющие характер и степень влияния наполнителя на адгезию композиции, обсуждены в работе [258].
Сшивающиеся композиции *
Известно значительное число агентов химического сшивания, обеспечивающих создание в полиолефинах пространственной трехмерной структуры в момент формования изделий или непосредственно после него. Приготовление сшивающихся композиций, переработку в изделия и сшивание можно осуществлять на обычном оборудовании переработки пластмасс и каучуков, в то время как для радиационного и фотохимического сшивания необходимы специальные установки.
По величине энергии активации, необходимой для химического сшивания, полиолефины могут быть разделены на три группы [100]. К наиболее трудно сшивающимся полиолефинам относятся полиэтилен и сополимеры этилена с незначительным количеством пропилена или иного а-олефина. Значительно легче вступают в реакции химического сшивания полипропилен и сополимеры этилена с достаточным количеством сомономера (например, в сополимере этилена с пропиленом содержание пропилена должно превышать 30 мол.%). Легко сшивающимися полиолефинами считаются сополимеры пропилена с этиленом, этилена с диенами, сополимеры а-олефинов с мономерами, содержащими активные функциональные группы.
Наиболее часто сшиванию химическими агентами подвергают полиэтилен [76—78, 100], сополимеры этилена с пропиленом [70, с. 304; 71, 73, 74, 188—204, 215—223], винилацетатом [228] и другими мономерами [224—227]. Полипропилен, по данным Натта [188, 189], при сшивании перекисями более склонен к разложению, чем к сшиванию. Уменьшение скорости деструкции достигается использованием перекисных сшивающих агентов в сочетании с серой. Так, для полипропилена как наиболее
* Раздел написан совместно с Е. П. Рябиновым.
157
оптимальную рекомендуется использовать сшивающую систему, которая включает 4,5% перекиси дикумила и 0,5% серы [234, с. 154].
Для приготовления сшивающихся композиций используют также смеси полиэтилена с другими полимерами: полиизобутиленом, каучуками, полибутадиеном, сополимером изобутилена с изопреном, сополимерами этилена с а-олефинами [229—233].
Для полиолефинов известен ряд сшивающих агентов, используемых для получения отверждающихся композиций в промышленных условиях. К ним относятся в первую очередь органические перекиси, азосоединения, сера и ее соединения, дисульфиды, азиды, нитрозосоединения, алкилы металлов, аминоокиси, гидразин и его производные, хинон и его производные [100]. Наибольшее значение приобрели перекиси. В патентной литературе в качестве агентов сшивания упоминается более ста различных органических перекисей, но практически из них используется не более 20%. Подробно исследовано сшивание в присутствии перкислот, перацеталей, перкеталей, перэфиров, гидроперекисей, диалкил- и диацилперекисей. Наиболее пригодны для сшивания полиэтилена третичные диалкилперекиси и диацилалкилпереки-си [147].
Перекисные агенты сшивания должны отвечать определенным требованиям. Перекись должна равномерно распределяться в полимере, не образуя агломератов. В связи с этим перекиси следует вводить в материал при температурах выше его температуры размягчения (температура введения перекиси в полиэтилен— от 120 до 140 °C в зависимости от его типа и молекулярного веса). Перекись должна достаточно быстро разлагаться при температурах, на 30—40 °C превышающих температуру введения в композицию. Продукты распада перекиси должны активно акцептировать атомы водорода и в минимальной мере вызывать побочные реакции. Перекись должна быть малолетучей, а продукты ее разложения, наоборот, должны быть летучими и не иметь запаха.
Относительно полно удовлетворяют этим требованиям и наиболее часто используются для сшивающихся композиций на основе полиэтилена и его сополимеров 2,5-диметил-2,5-ди-трет-бутилпероксигексан, 1,3-ди-трет-бутилпероксиизопропилбен-зол, перекись дикумила, 1,1,4,4,7,7-гексаметил-5,6,8,9-перокси-циклононан.
В отсутствие добавок перекисное сшивание полиолефинов заключается исключительно в образовании углерод-углеродных поперечных связей между полимерными цепями без присоединения перекиси к полимеру. Механизм перекисного сшивания может быть представлен схематически следующим образом. Под действием тепла перекись претерпевает гомолитический распад с образованием двух свободных оксирадикалов:
R—О—О—R —► 2[RO •]
158
Часть этих радикалов акцептирует активные атомы водорода от полимерных молекул, приводя к образованию полимерных радикалов:
RO-4-----СН2—С—СН2----—► ROH 4-----сн2—с—сн2---
I I
R R
Возможность миграции радикала по полимерной цепи и появление пар полимерных радикалов в непосредственной близости друг от друга при распаде одной молекулы перекиси приводят к рекомбинации макрорадикалов с образованием поперечной углерод-углеродной связи между макроцепями:
R R
СН2—С—СН2—СН2 • —сн2—с—сн2—сн2
сн2-с—сн2—сн2 I —сн2—с—сн2—сн2
1 R R
Однако при взаимодействии оксирадикала с полимерной цепью также могут возникать неустойчивые макрорадикалы, деструктирующие с разрывом макроцепи (р-распад):
---СН2—СН—СН2—С—СН2---—► I I
R R
—► СН2—С + сн2=с—сн2---------
I I
R R
Соотношение скоростей реакций сшивания и деструкции и определяет суммарный эффект реакции.
Для увеличения степени сшивания и снижения продолжительности при перекисном сшивании полиолефинов рекомендуется использовать ускорители. В качестве ускоряющих сшивание добавок применяют соединения, содержащие ненасыщенные группировки: триаллилцианурат, триаллилфосфат, дитиокарбаматы, тиурамсульфид, меркаптобензтиазол, а-фенилпропио-новый альдегид, дибензилкетон, фенилацетон, ацетонитрил, бисбензоил-и-фенилендиамин [100, 114, 186]. Введение ускорителей в сшивающуюся композицию увеличивает число узлов сшивания, которыми служат ненасыщенные группировки, способные к дальнейшему участию в процессе:
—сн2
I N
—с. + CH2=CH—сн2—о— \с—о—сн2—сн=сн2
I I . II
сн2 ^,N
I с—о—сн2—сн=сн2
—сн2
I . N
—> R—С—СН2—СН—СН2—О—хс—о—сн2—сн=сн2
1„ I II
\н2 N4; /N
I С—О—сн2—сн=сн2
Содержание ускорителей обычно составляет 0,5—2 вес.%. С целью предотвращения спонтанного, нерегулируемого распада перекисей, уменьшения доли вторичных реакций, увеличения продолжительности жизни радикалов и возможности рекомбинации радикалов с образованием межмолекулярных связей в состав отверждающихся композиций вводят регуляторы процесса сшивания: серу, диоксимы, динитрозосоединения, хиноны, цианамиды, диаллильные соединения, динитрозопиперазин, ди-фенилгуанидин, 1,3-дигидро-2,2,4-триметилхинолин, 2,2'-метилен-бис (4-метил-6-трет-бутилфенол), 4,4'-тетраметиленбис (6-трет-бутил-м-крезол), 4,4'-тиобис(6-трет-бутил-л-крезол), А/-фенил-а-нафтиламин, А\Л^-дифенилэтилендиамин, диаллилфумарат, этилендиметакрилат, диаминобензол и др. [100, 147]. Регуляторы добавляют в эквимольном или несколько большем количестве по отношению к сшивающему агенту [100, 147].
Для предотвращения термоокислительной деструкции сшитых перекисями композиций в их состав вводят антиоксиданты. Только небольшая часть антиоксидантов, используемых для стабилизации полиолефинов, может быть использована в этом случае, так как многие антиоксиданты ингибируют реакцию сшивания. Для сшивающихся композиций рекомендуется использовать следующие антиоксиданты: 1,3-дигидро-2,2,4-триме-тилхинолин, ди-р-нафтил-и-фенилендиамин, фенил-р-нафтил-амин, 'цинкмеркаптобензимидазол, дилаурилтиодипропионат, 2,2'-метиленбис (4-метил-6-трет-бутилфенол) [100, 147, 181].
Кроме перечисленных добавок в сшивающиеся композиции вводят различные наполнители, в том числе сажу, мел, кремневую кислоту, окись кремния, каолин и силикаты кальция [100, 147]. Содержание наполнителей составляет чаще всего от 20 до 100 вес.%, но может быть и больше (до 400 вес.%).
Наиболее часто в сшивающиеся композиции в качестве наполнителя вводят сажу. Для композиций характерна значительно лучшая способность смешиваться с сажей, чем для несшитых полиолефинов. Обычное содержание сажи в композициях на основе полиэтилена колеблется в интервале от 20 до 70 вес.%, однако известны композиции, содержащие до 200 вес.°/о сажи,
1И
Оптимум свойств сшитых продуктов наблюдается при введении около 35 вес. % сажи. Повышенная «восприимчивость» сажи сшивающимися полиолефиновыми композициями объясняется способностью сажи реагировать с возникающими при распаде перекисей оксирадикалами с образованием собственных радикалов. Рекомбинация радикалов сажи с полимерными радикалами приводит к образованию химической связи между сажей и полимером [ИЗ]:
R R
Сажа—С—Н + О—С—R —> Сажа—С + НО—С—R
• R : R
СН2 R СН2 R
1-1 I I
R—СН2 + О-С—R —> R—С • + НО—С—R
СН2 R СН2 R
I I
СН2
I
Сажа—С + -С—R —>
I
СН2
I сн2
Сажа—С—С—R
СН2
Предполагается, что наиболее активные в реакции сшивания сажи содержат определенное количество высококонденсирован-ных ароматических углеводородных соединений [113].
В связи с тем что часть оксирадикалов, образующихся при распаде перекиси, расходуется на взаимодействие с сажей, в саженаполненные композиции необходимо вводить больше перекиси, чем в ненаполненные композиции, для достижения равных эффектов сшивания. На процесс перекисного сшивания оказывают влияние величина поверхности и полярность сажи. Сажи со значительной поверхностью частиц в большей мере ингибируют процесс сшивания, по-видимому, в результате увеличения количества реакций с оксирадикалами. Сажи с кислой реакцией поверхности вызывают ионный распад перекиси
R—О—О—R —> RO+ + RO*
не поставляющий необходимых для реакций сшивания оксирадикалов.
Сажи с основным и нейтральным характером поверхности не влияют на радикальный распад перекисей. Сажи с кислой реакцией поверхности можно использовать для получения сшивающихся композиций при одновременном введении в состав
U1
композиции соединений основного характера, например окислов металлов.
При промышленном изготовлении сшивающихся композиций для достижения равномерного распределения в полимере наполнителей и других компонентов смешение осуществляется на вальцах или в пластосмесителях (например, типа «Бенбери»). При этом рекомендуется вводить в расплав полимера сначала наполнители, затем, после тщательного перемешивания, остальные добавки и в последнюю очередь — перекиси. Очень важен температурный режим процесса, который подбирается так, чтобы предотвратить преждевременное начало сшивания в результате частичного распада сшивающего агента [70, 100, 147, 233].
С целью упрощения технологии изготовления сшивающихся полиолефиновых композиций предлагается вводить перекись в полимер диффузионным методом [100, 237]. В этом случае гранулы полиолефина пропитываются рассчитанным количеством раствора перекиси в углеводородном растворителе. После завершения пропитки и удаления растворителя полимер может перерабатываться так же, как и композиции, полученные смешением в расплаве. При таком способе введения перекиси отсутствует опасность преждевременного сшивания полиолефина вследствие местных перегревов на стадии приготовления композиций.
Формование изделий из сшивающихся композиций можно осуществлять методами, используемыми для переработки обычных полиолефинов. Однако и при этом также необходимо учитывать возможность преждевременного сшивания.
После формования изделия из композиций подвергают термической обработке для проведения сшивания. Температура обработки должна превышать температуру распада перекиси и в большинстве случаев колеблется в интервале от 150 до 250 °C в зависимости от вида применяемой перекиси.
При переработке сшивающихся полиолефиновых композиций прессованием и литьем под давлением формование изделий и сшивание могут осуществляться одновременно [156, 158—163]. В случае экструзии сшивание также можно проводить в экструдере, однако для этого необходимы специальные агрегаты [164—166]. В большинстве случаев композиции экструдируют в обычных экструдерах, а окончательное сшивание проводят в туннельных печах (штреках сшивания). Выходящее из экструдера изделие направляется в штрек для подогрева до температуры, при которой происходит быстрый распад перекиси. Необходимая глубина сшивания обеспечивается временем пребывания изделия в штреке, т. е. длиной штрека и скоростью продвижения материала. Сшивание чаще всего осуществляется при температурах 190—220 °C, температура полимера, выходящего из экструдера, 120—140 °C.
Быстрый прогрев еще не сшитого полимерного материала вызывает резкое уменьшение его вязкости, а наступающее при
этом размягчение может приводить к потере формы изделия. Указывается [166], что при достижении степени сшивания 10% для полиэтилена высокой плотности и 40% для полиэтилена низкой плотности материал выдерживает необходимый дальнейший прогрев без изменения формы. Для уменьшения опасности нарушения формы рекомендуется вводить большие (до 50 вес.%) количества сажи [238], подвергать экструзии смеси сшитых и несшитых полимеров [239], а также создавать в штреке более высокие температуры [238]. Обычная длина штрека сшивания составляет от 60 до 100 м. Прогрев полимера в штреке осуществляется водяным паром под давлением [240], инфракрасным излучением, горячим газом [239, 241], токами высокой частоты [236, 242].
О степени сшивания полиолефинов перекисями, так же как и при радиационном и фотохимическом сшивании, судят по изменению физико-механических свойств при низких и повышенных температурах, растворимости и набухании в органических растворителях, стойкости к истиранию. При перекисном сшивании можно также непрерывно наблюдать за глубиной протекания процесса, например, по изменению модуля кручения при проведении реакции в пластографе Брабендера [113, 158, 182, 235]
быть определена степень сшивания полиолефина в любой момент реакции. В работе [176] по значениям крутящего момента была определена константа скорости реакции сшивания полиэтилена перекисями, а также рассчитана энергия активации этого процесса. При этом предполагалось, что процесс перекисного сшивания протекает как реакция первого порядка.
На рис. 62 представлена температурная зависимость константы скорости К при сшивании полиэтилена высокой и
163
низкой плотности, значения К при температурах выше 200 °C
ниже ожидаемых, так как реакция сшивания завершается раньше, чем реакционная смесь достигает выбранной температуры. Для различных перекисей величина энергии активации отличалась незначительно и составляла около 40 ккал/моль. Однако по данным Брауна с сотрудниками для перекиси дикумила, 2,5-диметил-2,5-ди-трет-бутил перокси гексана, 1,3-бис-трет-бутил-пероксиизопропилбензола и ряда других перекисей энергия акти-
0,9т—1—।—।—।।—।____I—1
4 6 ~ а 10 12 14 16
Весовой коэффициент набухания
Рис. 63. Связь плотности сшитых перекисью дикумила полиэтилена высокой (/) и низкой (2) плотности и весового коэффициента набухания [203].
межмолекулярной связи во
вации реакции сшивания составляет 23—26 ккал/моль [235].
Влияние структуры полиолефинов на процесс перекисного сшивания мало изучено, и имеющиеся публикации содержат достаточно противоречивые сведения. Как уже отмечалось в гл. V, при радиационном и фотохимическом сшивании полиолефинов эффективность процесса зависит от соотношения содержания кристаллических и аморфных областей. При перекисном сшивании этот фактор не столь важен, так как процесс осуществляется при температурах, значительно превышающих температуру плавления полимера. В расплаве вероятность рекомбинации двух макрорадикалов с образованием всем объеме сшивающегося полиме
ра практически одинакова. Статистическое распределение межмолекулярных связей по всей длине полимерной цепи при перекисном сшивании приводит к снижению доли кристаллической части в полиолефинах после сшивания. Авторами работ [120, 122] показано, что при сшивании полиэтилена низкой плотно-
сти перекисью дикумила степень кристалличности снижается с 60% (исходный полимер) до 50—55% (сшитый полимер). Кристалличность полиэтилена высокой плотности снижается в еще большей степени. Пониженной кристалличности сшитого полиэтилена, естественно, соответствует пониженная плотность. На рис. 63 показано изменение плотности сшитого полиэтилена низкой и высокой плотности в зависимости от степени сшивания (о которой судили по весовому коэффициенту набухания). Из рисунка видно, что при увеличении степени сшивания (уменьшении коэффициента набухания) наиболее резкое изменение плотности наблюдается у полиэтилена высокой плот
ности.
При перекисном сшивании наблюдается также изменение размеров сферолитов (рис. 64) и увеличение степени их дефектности.
164
Снижение степени кристалличности, уменьшение размеров кристаллических образований обусловливают сдвиг интервала плавления сшитых полиолефинов к более низким температурам, увеличение прозрачности и газопроницаемости, понижение
Рис. 64. Влияние сшивания на размер сферолитов исходного (а) и сшитого перекисью дикумила (б) полиэтилена высокой плотности [132].
твердости и жесткости. Ниже приведены свойства полиэтилена низкой плотности до (I) и после (II) сшивания перекисью дикумила [163, 176]:
Показатель текучести расплава I II
(190 °C, 2,16 кгс), г/10 мин .... Весовой коэффициент набухания в 2 Не течет
ксилоле при 140 °C Растворился 4 4-6
Содержание гель-фракции, % . . . Растворился 95
Плотность, г/см3 0,924 0,916
Степень кристалличности, % ... Прочность при растяжении, кгс/см2 47 42
при 20 °C 150 200
при 190 °C — 5
Относительное удлинение при раз-
рыве, % 550 450
Твердость по Шору (шкала С) . . 85 77
Модуль эластичности, кгс/см2 . . . 2500 1700
Стойкость к растрескиванию, ч . . Газопроницаемость, 2 >1000
см3 • мм/(м2 • сут • кгс/см2)
азот 37 69
кислорэд 110 250
двуокись углерода 720 1800
Диэлектрическая постоянная при
106 Гц 2,3 2,4
Тангенс угла диэлектрических по-
терь при 106 Гц 2-10 5- 10 4
Возникновение в полиолефинах пространственной структуры при сшивании также вызывает значительные изменения в их свойствах: увеличиваются эластичность, прочность при
1Ы
повышенных температурах, ударная вязкость при низких температурах (рис. 65, 66), стойкость к старению, к действию кислорода, озона, органических растворителей, стойкость к растрескиванию, уменьшается хладотекучесть.
Рис. 65. Зависимость удлинения при разрыве (/, 2) и прочности при растяжении (3, 4) полиэтилена высокой плотности от температуры [113]:
.... исходный полиэтилен; --- полиэтилен сшитый и наполненный 35% сажи.
Рис. 66. Зависимость удельной ударной вязкости образцов с надрезом полиэтилена высокой плотности от температуры [ИЗ]:
/—исходный полиэтилен; 2—полиэтилен сшитый и наполненный
35% сажи.
Важным конструкционным свойством полимеров является текучесть под действием нагрузок. Как видно из табл. 23, этот показатель для сшитого полиэтилена высокой плотности приближается к значениям, характерным для полипропилена, а при высоких нагрузках значительно превосходит их.
Таблица 23
Долговременная прочность при 20 °C полиэтилена высокой плотности, полипропилена и сшитого попиэтипена высокой плотности (35 вес. % сажи) [113]
Нагрузка, кгс/см2 Время до разрыва, ч Нагрузка, КГС/СМ2 Время до разрыва» ч
Полиэтилен Полипропилен Сшитый полиэтилен Полиэтилен Полипропилен Сшитый полиэтилен
325 0 0 50 215 0 5 <5000
305 0 0 500 180 0 60 <5000
290 0 0,02 1500 145 4 2000 <5000
270 0 —. >5000 125 20 >5000 <5000
250 0 0,3 >5000 ПО 1000 >5000 <5000
Для сшитых композиций характерна очень высокая стойкость к действию растворителей. По данным [113], у сшитого полиэтилена высокой плотности не наблюдается заметного изменения физико-механических свойств и веса после длительного
166
воздействия (12 мес.) органических растворителей (бензина, ксилола, тетралина, хлороформа и др.), по отношению к которым исходный материал был значительно менее устойчивым. Сшитые саженаполненные композиции на основе полиэтилена
отличаются высокой стабильностью (рис. 67). Это можно объяснить, во-первых, присутствием в композиции сажи, во-вторых, тем, что во время сшивания перекисями образование поперечных связей происходит в первую очередь по «слабым» местам, обусловливающим начало термической деструкции.
Окрашивающиеся композиции
при термостарении
Рис. термостарении исходного (-----) и
сшитого наполненного 35% сажи (...) полиэтилена высокой плот-
ности при различных температурах [113].
67. Изменение свойств при
Отсутствие у полиолефинов сродства к красителям затрудняет их окрашивание. До настоящего времени основной способ окрашивания заключается во введении различных красителей и пигментов в массу полимера [17, с. 195; 17, с. 120; 79, с. 187; 80—82]. При этом возникают проблемы, связанные с миграцией красителя на поверхность изделий, ухудшением диэлектрических свойств (возрастание тангенса угла диэлектрических потерь),
термо- и светостойкостью красителей, необходимостью равномер-
ного распределения красителей в полимере.
Большой практический интерес представляют предлагаемые в последние годы способы окрашивания изделий на основе полиолефинов, основанные на взаимодействии красителей со специально введенным в композицию компонентом. Восприимчивость полипропилена к красителям может быть повышена введением небольшого количества полярных низкомолекулярных (например, стеариновой кислоты [83]) или полимерных веществ (например, поликарбоната [84]).
Эффективно происходит окрашивание в результате введения в полимер ионов металлов, способных к образованию комплексов с азокрасителями [85—97]. Такими металлами служат
W
никель, хром, кобальт, магний, марганец, железо, ванадий, медь, алюминий, цинк, стронций. В полиолефины вводят соли этих металлов (галогениды, сульфаты, оксалаты, фосфаты, бензоаты, салицилаты, цианиды, ацетаты, стеараты, тиоцианаты, цитраты и др.) в количествах от 0,1 до 6%. Окраска полиолефинов, достигаемая обработкой их горячей дисперсией азокрасителя, отличается стойкостью к действию света, растворителей, трения, температуры.
Литература
1. Англ. пат. 893540, 1962.—2. Пат. США 3153681, 1964. — 3. Франц, пат. 1350905, 1964. — 4. Plochocki A. Polimery tworzywa wielkoczastecz-kowe, 1965, t. 10, № 1, str. 23.—5. Пат. США 3137672, 1964. — 6. Япон. пат. 7345, 1966. — 7. Фреунд Л., Амбр о ж Л. Полипропилен. Под ред. Н. И. Пилнповского и И. К- Ярцева. Л., 1967. 316 с.—8. Австр. пат.
224190, 1962. — 9. Англ. пат. 856793, 1961. — 10. Слонимский Г. Л., Му-
саелян И. Н., Казанцева В. В. Высокомол. соед., 1964, т. 6, с. 219.
11. Newberg R. G., Jong D. W., Evan с H. C. Mod. Plast., 1948,
№ 12, p. 119. — 12. P ы л о в E. E„ К a p п о в В. Л. ЖФХ, 1953, т. 27, с. 579. —
13. Фельдман Р. И., Миронова А. К. Коллоидн. ж., 1957, т. 19, с. 654. —14. Делекторский Г. П. Вестник электропром., 1960, № 9, с. 9.— 15. Алексеенко В. И., Мишустин И. У. Пласт, массы, 1960, № 2, с. 8. — 16. Мартынов И. А., Южин В. М., М а л у ш и н А. И., Ткаченко Г. Ф. Пласт, массы, 1965, № 10, с. 6. — 17. Шифрина В. С., С а-мосатский Н. Н. Полиэтилен. Л., Госхимиздат, 1961. 176 с. —18. Слонимский Г. Л., Мусаелян И. Н., Казанцева В. В. Высокомол. соед., 1964, т. 6, с. 823. — 19. Railsback Н. Е., Wheat R. С. Rubber Age, 1958, v. 82, № 4, р. 664; пер.: «Химия и технология полимеров», 1958, № 5, с. 66.— 20. Пат. США 3123583, 1964.
21. Слонимский Г. Л.. Мусаелян И. Н., Казанцева В. В., Озеров Г. М. Высокомол. соед., 1964, т. 6, с. 818. — 22. Михайлов Н. В., Файнберг Э. 3., Горбачева В. О., Чен Цин-хай. Высокомол. соед., 1962, т. 4, с. 237. — 23. Шитов В. В. Электротехника, 1963, № 12, с. 13.—24. Пат. США 3231636, 1966.—25. Пат. ФРГ 1106450, 1961,— 26. Англ. пат. 893604, 1962. — 27. Австр. пат. 223813, 1962. — 28. Фин. пат. 32722, 1963. — 29. Фильберт Д. В., Муравьева В. П., Пакш-вер А. Б. В кн.: Карбоцепные волокна. М., «Химия», 1966. — 30. Пат. США 3018263, 1962.
31. Япон. пат. 12786, 1961. — 32. Франц, пат. 1284591, 1962.—33. Зверев М. П., Бычков Р, А., Костин Т. Ф. Хим. волокна, 1964, № 3, с. 15. — 34. Chem. Engng News, 1963, v. 41, Ns 25, p. 41. — 35. Гуль В. В., Л ю б е ш к и н а Е. Г. ДАН СССР, 1965, т. 165, № 1, с. 110. — 36. Гуль В. Е., Л юбешки на Е. Г., Шаргородский А. М. «Механика полимеров», 1965, № 6, с. 3. — 37. Гуль В. Е., Л юбешки на Е. Г. Пласт, массы, 1966, № 1, с. 68. — 38. Л юбешки на Е. Г., Т о р н е р Р. В., Гуль В. Е. «Механика полимеров», 1967, № 2, с. 200. — 39. Авт. свид. СССР 184432, 1966.— 40. Л юбешки на Е. Г., Торнер Р. В., Гуль В. Е. Пласт, массы, 1967, № 5, с. 60.
41. Masakazu I п о v е, J. Polymer Sci., 1963, v. 1, Ser. A, p. 3427.— 42. Barton J., Rak J. J. Appl. Polymer Sci., 1967, v. 11, p. 499.— 43. Гуль В. E., Пен ска я Е. А., Кулезнев В. Н., Арутюнова С. Г. ДАН СССР, 1965, т. 160, № 1, с. 154. — 44. Фильберт Д. В.. Муравьева В. П., Глазковский Ю. В. В кн.: Карбоцепные волокна. М., «Химия», 1966. — 45. Гуль В. Е., Пенская Е. А., Кулезнев В. Н. Коллоидн. ж., 1965, т. 27, № 3, с. 341. — 46. Пенская Е. А., Дубов О. Е., Гуль В. Е-
168
Доклад на симпозиуме по синтезу, модификации и переработке полиолефинов, Баку, воябрь 1967. — 47. Миролюбов И. Н., Сухарев М. Г. Пласт, массы, 1963, № 3, с. 62. — 48. Fur ter W. F. Canad. J. Chem. Engng, 1964, v. 42, № 2, p. 77; пер.: Экспресс-информация. Серия «Синтетические высокополимерные материалы». 1964. № 36. — 49. Сииегуб-Лаврен-ко А. А., Моргулис М. Л. Пласт, массы, 1966, № 2, с. 3.— 50. Южин В. М., Шишова И. С., Мартынов 54. А., Белова Р. И. Пласт, массы, 1967, № 4, с. 10.
51. Смирнова А. М., Ков а рек а я Л. Б., Райкова Т. В., Топоров Ю. П. Коллоидн. ж., 1964, т. 25, № 6, с. 683. — 52. А й н б и н д е р С. Б., Андреева Н. Г. «Механика полимеров», 1967. № 5, с. 873; № 6, с. 1070.— 53. Со ломко В. П„ Усков И. А. В кн.: Модификация свойств полимеров и полимерных материалов. Киев, «Наукова думка», 1965. 151 с.— 54. С о л о м к о В. П„ Мо л о к о е д о в а Т. А., Чуйко А. А,, Л а ш к о Т. Р. В кн.: Синтез и физико-химия полимеров. Киев, «Наукова думка», 1966. 196 с. — 55. Акутин М. С., Уваров А. В., Озеров Г. М. Доклад на симпозиуме по синтезу, модификации и переработке полиолефинов, Баку, ноябрь 1967. — 56. Чуйко А. А., Чуйко Е. А. В кн.: Синтез и физико-химия полимеров. Киев, «Наукова думка», 1964. — 57. Акутин М. С., Ш а-бадаш А. Н., Салина 3. И. и др. Высокомол. соед., 1972, т. 14, сер. Б, № 10, с. 769.-58. Пат. США 2991264, 1961, —59. Каргин В. А., С о г о-лова Т. И., Курбанова И. И. ДАН СССР, 1965, т. 162, № 5, с. 1092.— 60. Со го лов а Т. И. «Механика полимеров», 1965, № 1, с. 5.
61. Kuhre С. J., Wales М., Doyle М. Е. SPE J., 1964, v. 20, № 10, с. 1113; пер.: «Химия и технология полимеров», 1965, № 6. с. 86. — 62. Каргин В. А., С о г о л о в а Т. И., Шапошникова Т. К. Высокомол. соед., 1965, т. 7, № 2, с. 229. — 63. Каргин В. А., С о г о л о в а Т. И., Шапошникова Т. К. Высокомол. соед., 1965, т. 7, № 3, с. 385. — 64. Каргин В. А., Пашинии Б. П., Котрелев В. Н., Акутин М. С. Высокомол. соед., 1966, т. 8, № 12, с. 2097. — 65. Каргин В. А., Сото лова Т. И., Курбанова И. И. Высокомол. соед., 1966, т. 8, № 12, с. 2104.— 66. Карг и нВ. А., Соголова Т. И., Рубштейн В. М. Высокомол. соед., 1967, т. 9, сер. А, № 2, с. 288. — 67. Н а л и в а й к о Е. И., Сирота А. Г., Ильченко П. А., Шишова И. С. «Механика полимеров», 1968, № 1, с. 67. — 68. Н а л и-вайко Е. И., Сирота А. Г. Пласт, массы, 1968, № 2, с. 13. — 69. Авт. свид. СССР 203889, 1967.—70. Амберг Л. О. Вулканизация эластомеров. Под. ред. Г. Аллигера и И. Сьетуна. «Химия», 1967.
71. Сеидов Н. М. Новый синтетический каучук на основе этилена и пропилена. Баку, Азерб. гос. изд., 1966. 126 с. — 72. Англ. пат. 879195, 1961.— 73. Wei Р. Е., Renner J. J. Polymer Chem. Technol., 1962, № 1, p. 35.— 74. Robinson A. E., Marra J. V., Amberg L. O. Ind. Eng. Chem., 1962, v. 1, Ns 2, p. 78.-75. Пат. США 3410928, 1968.— 76. Пат. США 3123583, 1964.— 77. Пат. США 3046238, 1962.— 78. Франц, пат. 1337063, 1963. — 79. Кержковская Е. М. В кн.: Переработка пластических масс, 1966. — 80. D е s t a b 1 е A. Ind. Plast. Mod., 1957, v. 9, № 6, p. 41.
81. Kaufmann К. A. Mod. Plast., 1959, v. 36, № 6, p. 137.— 82. Баль-тенас P. А., Анкундинова P. К.., Бальтенене Я. Ю., К p я у ч гона с И. И. Доклад на симпозиуме по синтезу, модификации и переработке полиолефинов. Баку, ноябрь 1967. — 83. Пат. США 3231530, 1966. — 84. Япон. пат. 20376, 1961. — 85. Япон. пат. 17714, 1966. — 86. Япон. пат. 17715, 1966.— 87. Япои. пат. 18911, 1966. — 88. Япон. пат. 18912, 1966. — 89. Япон. пат. 18914, 1966.—90. Япон. пат. 20428, 1966.
91. Япои. пат. 20430, 1966. — 92. Япон. пат. 20431, 1966. — 93. Япон. пат. 20432, 1966.—94. Япон. пат. 20433, 1966. — 95. Япон. пат. 20434, 1966.— 96. Япон. пат. 829, 1967. — 97. Япон. пат. 830, 1967.—98. Hawkins W, L., Chan М. G„ Link G. L. Polymer Eng. Sci., 1971, v. 11, Ns 5, p. 377.— 99. Англ. пат. 871591. — 100. Wunsch K-, Kohlmann G. Plaste u. Kaut., 1966, № 5, S. 258.
16»
101. Carlson В. C. SPE J., 1961, v. 17, № 3, p. 265.-102. Tat* box N. E. Wire a. Wire Prod., 1961, v. 36, № 4, p. 465, 516, — 103. SubaM-M, Imhof L. Q. Wire a. Wire Prod., 1960, v. 35, № 2, p. 195, 246. — 104. F. A. de M e 1 i o. Wire a. Wire Prod., 1962, v. 37, X° 8, p. 1017,— 105. Werblinski W. Przemusl. Chem., 1961, t. 40, Xs 4, str. 188.— 106. R. J. Boot, Jordan T. J. Plastics Design Process, 1963, .Ns 9, p. 11. — 107. Suba M. M. Wire a. Wire Prod., 1959, v. 34, Xs 7, p. 841.— 108. Carlson В. C. Rubber World, 1960, v. 142, Xs 3, p. 91.— 109. Gregorian R. S., Bafford R. A. Ind. Engng Chem., Prod. Res. Developm., 1964, v. 3, Xs 4, p. 267. — 110. Rado R., SI-munkowa D. Chem. Prumysl., 1961, t. II, str. 657.
111. Rado R., Lazar M. Высокомол. соед., 1961, т. 3, с. 310.— 112. R a d о R., L a z а г М. Chem. Prumysl., 1960, t. 10, str. 496,— 113. Behr F. Kunststoffe, 1963, Bd. 53, Xs 8, S. 502. — 114. Nakamura Y., Saito Y. J. Chem. Soc. Japan, Ind. Chem. Sect., 1964, v. 67, N 4, p. 648.— 115. Rado R., Lazar M. J. Polymer Sci., 1960, v. 45, p. 257. — 116. T s u n о d о Y., Kato T., Hasegawa Sh. J. Chem. Soc. Japan, Ind. Chem. Sect., 1960, v. 63, p. 656. — 117. Rowley A. C. Wire a. Wire Prod., 1959, v. 34, Xs 7, p. 838, 880.— 118. Marra J. V., Amberg L. O. Rubber Chem. Technol., 1962, v. 35, p. 1083. —119. Christ T., Bosch A. G. Chem. Rdsch., 1960, Bd. 13, Xs 23, S. 643. — 120. D a n n e n b e r g E. M., Jordan M. E., Cole H. M. J. Polymer Sci., 1958, v. 31, p. 122, 127.
121. Догадкии Б. А., Доннов А. А. Высокомол. соед., 1964, т. 6, Xs 10, с. 1744. — 122. Ferch H. Kunststoffe, 1966, Bd. 52, № 6, S. 326.— 123. Simunkowa D., Rado R., Barton J. Plaste u. Kaut., 1965, Bd. 12, Xs 2, s. 75.— 124. Feytis J., Perrault J. Bull. Soc. Franz. Electriciens, 1961, v. 2, April, p. 212—216. — 125. Ogawa M., Kojima N. Kobunsi Kagaku Japan, 1962, v. 19, p. 370—375. — 126. Ogawa M. Kobunsi Kagaku Japan, 1962, v. 19, p. 204, 261. —127. Simunkowa D., Rado R. Chem. Prumysl., 1961, t. 11, str. 657. — 128. Ling T. A., Dates C. J. Rubber Age, 1962, v. 90, Xs 4, p. 634. — 129. Blyestein A. C. Rubber Age, 1964, v. 96, Xs 1, p. 57.— 130. R a d о R., L a z a r M. J. Polymer Sci., 1961, v. 53, p. 67.
131. Boonstra В. B., Jermyn T. E. Plastics, 1962, v. 27, p. 105; SPE J., 1962, v. 18, Xs 3, p. 315.— 132. Charlesby A., Hancock N. H. Proc. Roy. Soc. (London), 1953, v. 218, Ser. A, Xs 25/6, p. 245; v. 217, Xs 24/3, p. 122.— 133. Lakombe J., Wrobel W. Wire a. Wire Prod., 1963, v. 38, Xs 4, p. 505.— 134. Cook P. M„ Muchmore R. W., Rapp H. Rubber Plast. Age, 1961, v. 42, N 10, p. 522.— 135. Kerwin E. R. Rubber Plast. Age, 1959, v. 40, Xs 12, p. 1337.— 136. Kojima H. Plast. Age, 1972, v. 15, Xs 10, p. 87.— 137. S 1-munkova D., Rado R. Plast. Hmoty a. KauC, 1970, t. 7, Xs 7, s. 207.— 138. D a n n о A. At. Energy Rev., 1971, v. 9, Xs 2, p. 399. — 139. N a k s h i о Y. Plast. Age, 1969, v. 15, Xs 9, p. 103.— 140. Протасов В. Г., Барам-бойм Н. К. Пласт, массы, 1972, X» 8, с. 22.
141. Scheele W., Stefka L. Kaut. u. Gummi Kunststoffe, 1970, Bd. 23, Xs 12, s. 609.— 142. Narkis M., Miltzt J. Jsr. J. Chem., 1970, v. 8, Xs 1.— 143. Паушкин Я- M., Лосев Ю. П., Бриль Д. М. ДАН БССР, 1970, Т. 14, Xs 10, с. 912. — 144. Potter W. I. Plast. Technol., 1969, v. 15, Хв 7, p. 55.— 145. Максименко В. Н. В кн.: Нефть и газ и их продукты. М., 1971. — 146. Reichenbach D., Eckelmann W. Kaut. u. Gummi Kunststoffe, 1970, Bd. 24, Xs 9, S. 443,— 147. Arnaud P. Off Plast. Caout., 1969, V. 16, Xs 5, p. 361. — 148. Bonotto S. J. Appl. Polymer Sci., 1965, v. 9, p. 3819.— 149. Famechon R. Rev. Gen. Caout. Plast., 1971, v. 48, Xs 10, p. 1113.— 150. Simunkowa D. Elektroizol i Kabl. techn., 1970, t. 23, № 3, str. 136.
151. Manley T. R., Q а у у m M. M. Polymer, 1971, v. 12, № 3, p. 176. — 152. Schulze H. Industrianzeiger, 1970, Bd. 92, Xs 33, S. 726.— 153. Mat-subara H., Miyauchi H. Sumitomo Elec. Techn. Rev., 1970, Xs 14, p. 64,— 154. McManus M. S. J. IR1, 1971, v. 5, Xs 3, p. 109.— 155. Menges Q„ Reinfeld D. Plastverarbeiter, 1972, Bd. 23, № 5, S. 311.— 156. Ehren-170
traut P., Dalhoff W. Kunststoffe, 1967, Bd. 57, № 6, S. 439.— 157. Bowen S. T. C. A., 1972, v. 77, № 2, p. 6650. — 158. Menges G„ Dalhoff W„ Rhein f eld D. Plastverarb., 1970, Bd. 21, № 5, S. 111 — I—II. — 159. C arrow G. E. SPE J., 1972, v. 28, № 4, p. 48. — 160. Те n пеу K. S. SPE J. Techn. Papers, 1969, v. 15, p. 191.
161. Tenney K. S. SPE J., 1970, v. 26, № 3, p. 68. — 162. Lucches-si P. SPE J. Techn. Papers, 1968, v. 14, p. 616.— 163. Jacobi R., Flohr J. Kunststofftechnik, 1970, Bd. 9, № 3, S. 79.— 164. Baumgarten W. Gummi Asbest Kunststoffe, 1972, Bd. 25, № 8, S. 761. — 165. Europlastics, 1972, v. 45, № 7, p. 16.— 166. Benning C. J., Gregorian R. S., Kirk С. C., Werber F. X. Mod. Plast., 1966, v. 44, p. 131.— 167. Ha ger J. E. Plast. Technol., 1969, v. 15, № 7, p. 55.— 168. Benning C. J. SPE J., 1965, v. 21, p. 1083,— 169. Benning C. J. Mater. Design. Engng, 1965, v. 61, № 7, p. 1083.— 170. E n ge 1 T. Kunststoffe, 1967, Bd. 57, Xs 7, S. 536.
171. Кававата Ситар о. Хитати Хером, 1961, т. 43, № 8, с. 1043. — 172. Brit. Plast., 1970, v. 43, № 3, р. 8,—173. М a i г Н. S. Kunststoffe, 1970, Bd. 60, № 9, S. 630. — 174. Lacombe J., Wrobel W. Wire a. Wire Prod., 1963, v. 38, Xs 4, p. 508,— 175. Mod. Plast., 1970, v. 47, № 2, p. 25.— 176. Kohnlein E. Kunststoffe, 1970, Bd. 60, № 11, S. 883.— 177. Biigel H. Techn. Mitteilungen, 1965, Bd. 58, № 5, S. 227. — 178. К 6 s s 1 e r L., Mair H. S. Kunststoffe, 1972, Bd. 62, № 6, S. 359, 338,— 179. Nowak P, Saure M. Kunststoffe, 1966, Bd. 56, Xs 6, S. 390. — 180. Евдокимов E. И. и др. Пласт, массы, 1972, Xs 7, с. 29.
181. Романова М. В. и др. Пласт, массы, 1971, Xs 9, с. 34. — 182. N а г-kis М., Miltzt J. J. Appl. Polymer Sci., 1968, v. 12, № 5, p. 1031; 1970, v. 14, Xs 1, p. 65.— 183. Киртовская Г. И., Карливан В. П., А к м е-ие В. Я. В кн.: Модификация полимерных материалов. Сб. 2. Рига, 1969.— 184. Prat С., Negrel J. Р. Kaut. u. Gummi Kunststoffe, 1971, Bd. 24, Xs 8, S. 417.— 185. Van. Dine G. W., Shaw R. Am. Chem. Soc. Polym. Repr., 1971, v. 12, Xs 2, p. 713.— 186. Kerrut G. Kaut. и Gummi Kunststoffe, 1971, Bd. 24, № 8, S. 384,— 187. Gulpen N. Y. C.A., 1969, v. 71, Xs 26, p. 124932,— 188. Natta G., Crespi G., Bruzzone M. Kaut. u. Gummi, 1961, Bd. 14, s. 54.— 189. Natta G., Crespi G. и др. Trans. Faraday Soc., 1961, v. 57, Xs 3, p. 54. — 190. В a 11 i n i G., P о r t о 1 a n i A. Materie plast. ed Elastomeri, 1963, v. 29, Xs 9, 1450; 1964, v. 30, Xs 3, p. 266.
191. E. di G и i 1 i o, Ba Ilin i G. Kaut. u. Gummi, 1961, Bd. 15, Xs 1, S. 6; Plaste u. Kaut., 1961, Bd. 8, Xs 5, S. 247,— 192. E b у L. T„ F и s с о T. V. Rubber Age, 1962, v. 91, p. 949.—193. Lenas L. P. Ind. Engng Chem., 1964, v. 3, Xs 4, p. 269. — 194. ManaresiP. Chim. et. Ind., 1963, v. 45, 12, p. 1488.— 195. Morche K-, Eh rend H. Kaut. u. Gummi Kunststoffe, 1963, Bd. 16, S. 666. — 196. Loan L. D., S с a n 1 a n J. Rubber Plast. Age, 1963, v. 44, Xs 11, p. 1315.— 197. Amberg L. O., Robinson L. E. Ind. Engng Chem., 1961, v. 53, Xs 5, p. 368. — 198. M а г г a J. V., A m b e г g L. O. Rubber Chem. Technol., 1962, v. 35, p. 1083.— 199. Итал. пат. 612464, 1960.—200. Amberg L. O. Rubber Plast. Age, 1961, v. 42, Xs 10, p. 1210.
201. Amberg L. O. Proc. Technol. Conf. 4th. London, 1961 (44). 14pp.— 202. H a x о H. E., Bingham W. R., W h i t e h о и s e W. G. Rubber Age, 1963, v. 94, Xs 2, p. 255. — 203. Howarth I. T., Weinberg H. W. Rubber World, 1963, v. 149, Xs 1, p. 54. — 204. Loan L. D. J. Polymer Sci., 1964, v. 2, Ser. B, p. 59. —205. Франц, пат. 1486144, 1967.— 206. Пат. США 3222431, 1965.— 207. Menges G„ Alf E. Kunststoffe, 1972, Bd. 62, S. 259. — 208. Andrews E. H., Walker B. J. Proc. Roy. Soc., 1971, v. 325, Ser. А, X» 1560, p. 57. — 209. Лукин Д. M., Балыкина M. B., Ill и n а н С. Я., Несмер-чук И. С. Пласт, массы, 1971, № 4, с. 68. — 210. Вечен а Р. К., Карливан В. П. В кн.: Модифицированные и наполненные термопластичные материалы. Свойства, переработка и области применения. Л., ЛДНТП, 1968.
211. Лавенделе С. М., Карякина М. И., Карливан В. П. В ки.: Модификация полимерных материалов. Рига, 1969. 174 с. —212. Каган Д. Ф.,
171
Кантор Л. А., Багаудииова Л. А., К не бе льм а и А. М. В кн.: Модифицированные и наполненные термопластичные материалы. Свойства, переработка и области применения. Л., ЛДНТП, 1968. — 213. Пасквитов-с к а я С. Э., Медведева М. Д., Макарова О. А. н др. Пласт, массы, 1969, № 8, с. 46.—214. Larsen D. W. SPE J., 1971, v. 27, № 6, р. 40,— 215. Portolani A., Giandinoto G. V. Chim. e. Ind (Milano), 1963, v. 45, № 8, p. 927. — 216. Рейх В. H., Салнис К. Ю. Каучук и резина, 1961, т. 20, № 6, с. 1.—217. Robinson А. Е., Marra J. V. Rubber Age, 1961, v. 89, № 5, p. 803. — 218. Crespi G., Bruzzone M. Chim. e Ind., Milan, 1959, v. 41, № 2, p. 741. — 219. Lenas L. P. Ind. Engng Chem., 1963, v. 2, № 12, p. 202. — 220. G a 1 a n R. R. Rev. Plastics, 1962, v. 13, p. 182.
221. Tan Y. Y. P 1 a s t i c a, 1960, Bd. 13, S. 587. — 222. Stemmer H. D. Kaut u. Gummi, 1962, Bd. 15, Ks 1, S. 270. — 223. Rado R„ Gal E. Chem. Zvesti, 1970, d. 24, Ks 2, s. 107.— 224. Stemmer H. D. Kaut. u. Gummi, 1961, Bd. 14, № 5, S. 146.— 225. Wan less G. G„ Wei P. E., Rehner J. Rubber Chem. Technol., 1962, v. 35, p. 118. — 226. Донцов А. А., П p о й ч e в а А. Г., Новицкая С. П., Догадкин Б. А. Высокомол. соед., 1971, т. 13, сер. Б, № 9, с. 671.—227. Kaway W„ Kazuta S., J. Chem. Soc. Japan. Ind. Chem. Sect. 1971, v. 74, № 3, p. 532. — 228. Bartl H., Peter J. Kaut. u. Gummi, 1961, Bd. 14, S. 23, — 229. Howarth J. T„ Cornell J. A., Olson L. R. Rubber World, 1963, v. 148, № 5, p. 69. — 230. Пат. ФРГ 892240, 1953.
231. Coole R. W., Edwards D. C., Walker J. Trans. Proc. Instn. Rubber Ind., 1964, v. 40, № 2, p. 82. — 232. S i m u n k о w a D., Rado R., В a r-ton J. Plaste u. Kaut., 1965, Bd. 12, № 2, S. 75, —233. Пат. США 3073797, 1963; 3157564, 1964. — 234. Манясек 3., Беллуш Д. Полипропилен. Под ред. В. И. Пилиповского и И. К- Ярцева. Л., «Химия», 1967. 316 с.— 235. Braun D., Brendlein W. Kunststoffe. 1970, Bd. 9, № 8, S. 275.— 236. Англ. пат. 852371, 856833, 1960.— 237. Пат. ФРГ 1052681, 1959. — 238. Пат. США 2938083, 1960.— 239. Пат. ФРГ 1079145, 1960.— 240. Pathak V. В. Popular Plastics, 1966, т. 11, № 8, р. 26.
241. Fey t is Т., Perrault J. Bull. Soc. franz. Electriciens, 1961, v. 2, p. 212.— 242. Бельг, пат. 564394, 1958; 564395, 1960.— 243. Bartha Z„ Szor P., Ambus S. Plaste u. Kaut., 1964, Bd. 11, № 11, S. 670. — 244. P i e-p h о D. A., F i s c h e r J. SPE J. Techn. Papers, 1971, v. 18, p. 502. — 245. Goodrich J. E., Porter R. S. Polymer Engng, 1967, v. 7, № 1, p. 45. — 246. W i n-tergerst S., Bronner J. Kaut. u. Gummi Kunststoffe, 1969, Bd. 22, S. 683. — 247. Kempermann Th., Clamoth R., Schnittmann H. S. Kaut. u. Gummi Kunststoffe, 1969, Bd. 22, S. 609.— 248. Темчин Ю. И., Бурмистров E. Ф., Егидис Ф. М. и др. В кн.: Синтез и исследование эффективности химикатов для полимерных материалов. Вып. 4. Тамбов, 1970.— 249. Темчин Ю. И., Бурмистров Е. Ф.,.Д я дченко А. И. н др. Высокомол. соед., 1972, т. 14, сер. А, № 8, с. 1689.—250. Темчин Ю. И., Бурмистров Ё. Ф. Доклад на Международном симпозиуме по методам оценки и практическому применению стабилизаторов и синергических смесей. М., 1973.
251. Каргин В. А., Карякина М. И., Берестнева 3. Я., Майорова Н. В. ДАН СССР, 1966, т. 170, с. 369.—252. Кордунер Н. Е., Богаевская Т. А., Громов Б. А. и др. Высокомол. соед., 1970, т. 12, сер. Б, № 9, с. 693. — 253. Якубович С. В., Масленникова Н. Л. «Лакокрасочные материалы и их применение», 1962, № 4, с. 20.—254. СоломкоВ. П., Нижник В. В., Полудни В. И. и др. В кн.: Макромолекулы на границе раздела фаз. Киев, «Наукова думка», 1971. — 255. Малинский Ю. М., Орловская Т. Т., Каргин В. А. ДАН СССР, 1965, т. 160, с. 1128.— 256. Финкельштейн Б. А. Автореф. канд. дисс. М., 1972. — 257. К а л-нинь М. М. Автореф. канд. дисс. Рига, 1967.—258. Белый В. А., Егоренко Н. И., Плескачевский Ю. М. Адгезия полимеров к металлам. Минск, «Наука и техника», 1971. 288 с. — 259. Вайнштейн А. Б., Кут-нер Э. А., Карлнван В. П. В kjj.: Модификация полимерных материалов. Сб. 3. Рига, «Зинатне», 1972. 150 в. — 260. Берестнева 3. Я., Бра
172
гинский Р. П., Каргин В. А. н др. «Труды ВНИИКП», 1967, вып. 12, с. 115.
261. Япон. пат. 583, 1970. — 262. Акутнн М. С., Кербер М. Л., Стальнова И. О. «Механика полимеров», 1970, № 1, с 179 — 263. Акутин М. С., Кербер М. Л.. Стальнова И. О., Штейн С. А.. «Труды МХТИ», 1970, вып. 64, с. 236 — 264 Акутин М. С.. Кербер М. Л., Осипни к В. С., Стальнова И. О. Пласт, массы, 1970, № 12, с. 4.— 265. Акутнн М. С., Соколов С. И., Кербер М. Л. и др. «Механика полимеров», 1971, № 4, с. 722. — 266. Акутин М. С., Кербер М. Л., Стальнова И. О. н др. «Механика полимеров», 1972, № 6, с. 1048.— 267. Шварц А. Г., Дннзбург Б. Н. Совмещение каучуков с пластиками и синтетическими смолами. М., «Химия», 1972. 224 с.
Оглавление
Введение ........................................................ 3
Глава I. Структура и свойства полиолефинов...................5
Полиэтилен ............................................5
Полипропилен .......................................... 14
Полимеры высших а-олефинов...........................16
Литература............................................19
Глава II. Сополимеры а-олефинов..............................23
Сополимеры этилена с пропиленом и другими а-олефинами 23
Сополимеры этилена с полярными мономерами.............37
Литература............................................45
Глава III. Привитые и блоксополимеры а-олефииов...............50
Привитые сополимеры...................................50
Блоксополимеры .................................... . 68
Литература............................................74
Глава IV. Модифицирование с помощью реакций в цепях полиолефинов .............................................................78
Галогенирование ..................................... 78
Сульфохлорирование .................................. 89
Фосфонирование........................................93
Окисление ............................................97
Термическая деструкция...............................103
Литература...........................................106
Глава V. Полиолефины с пространственной структурой...............ПО
Получение пространственных структур с помощью реакций специально введенных функциональных групп............110
Получение пространственных структур радиационно-химическим и фотохимическим методами.......................115
Литература...........................................140
Глава VI. Композиции на основе полиолефинов.....................145
Композиции полиолефинов с различными полимерами . . 145
Наполненные полиолефины..............................153
Сшивающиеся композиции...............................157
Окрашивающиеся композиции ...........................167
Литература ..........................................168
Анатолий Георгиевич Сирота
Модификация структуры и свойств полиолефинов
Научный редактор Б. И. Тихомиров Редактор издательства А. Е. П и и ч у к Технический редактор Ф. Т. Черкасская Корректор Г. Я. Батракова
М-09527. Сдано в наб. 28/XI-73 г. Подп. в печ. 14/V-1974r.
Формат бумаги 60X90'/ie- Бумага тнп. №2. Усл.-печ. л. 11,0.
Уч.-изд. л. 12,38. Тираж 2600 экз- Зак. 881. Изд. № 263. Цена 1 р. 24 к.
Издательство «Химия*. Ленинградское отделение
191186, Ленинград, Д-186. Невский пр., 28
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени Евгении Соколовой Союзполиграфпрома при Государственном комитете Совета Министров СССР по делам издательств, полиграфии и книжной торговли.
198052, Ленинград, Л-52, Измайловский проспект, 29