/

























































































































































































































































































































































































































Текст
ЗДРШЕ.П.ФРАНЦ.ШМКЕ
ХИМИЯ
ОТРАВЛЯЮЩИХ
ВЕЩЕСТВ
S.Franke, P. Franz, W.Warnke
Lehrbuch
der Militarchemie
Band 2
Dcutscher Militarverlag Berlin 1967
З.ФРАНКЕ, П.ФРАНЦ.ВВАРНКЕ
ХИМИЯ
ОТРАВЛЯЮЩИХ
ВЕЩЕСТВ
--------2-----------
Перевод с немецкого
кандидата химических иаук И. Т. ПЕНЗУЛАЕВА
под редакцией академика И. Л. КНУНЯНЦА
и доктора химических наук Р. Н. СТЕРЛИНА
МОСКВА ИЗДАТЕЛЬСТВО «ХИМИЯ» 1973
355.728
УДК 623.459.5+623.459.8
Ф 83
Ф83 Франке 3., Франц П., Варнке В.
Химия отравляющих веществ. Т. 2. Пер. с нем., под
ред. акад. И. Л. Кнунянца и д-ра хим. наук.Р. Н. Стер-
лина. М., «Химия», 1973. 404 стр., 45 табл., 33 рнс.,
список литературы 882 ссылки
ААонографня, вышедшая в ГДР в 1967—1969 гг.,
представляет собой одно из наиболее полных совре-
менных руководств по химии отравляющих веществ.
Второй том этого двухтомного издания посвящен
вопросам индикации и дегазации. В нем подробно осве-
щены методы и средства индикации отравляющих ве-
ществ в различных средах, приведены методики эле-
ментного и количественного анализа, систематизированы
сведения об обнаружении и количественном определе-
нии фосфорорганических отравляющих веществ, неко-
торых фосфорсодержащих инсектицидов, дефолиантов,
алкалоидов и др.; подробно разбираются теоретические
основы превращений отравляющих веществ в нетоксич-
ные соединения.
Книга представляет интерес для преподавателей и
студентов высших учебных заведений, личного состава
подразделений гражданской обороны, а также может
быть полезна широкому кругу химиков, деятельность
которых связана с химией физиологически активных
веществ.
л 0251-155
Ф 050(01 )-73
15-73
355.728+543
© Перевод на русский язык, Издательство «Химия», 1973.
СОДЕРЖАНИЕ
АНАЛИЗ БОЕВЫХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ
1. Задачи анализа боевых отравляющих веществ (БОВ).................17
Контрольные вопросы ...............................................18
2. Основы и особенности анализа ОВ ................................19
2.1. Организация работы............................................19
2.2. Рабочее место и оборудование..................................21
2.3. Рабочие записи................................................22
2.4. Техника безопасности .........................................23
2.5. Чувствительность определений..................................25
Контрольные вопросы................................................26
3. Элементный анализ ОВ............................................27
3.1. Качественный элементный анализ.....................27
3.1.1. Обнаружение азота, серы и галогенов.....................27
3.1.1.1. Минерализация образца сплавлением с щелочными металла-
ми для последующего обнаружения азота, серы и галогенов
(проба по Лассеню).........................................27
3.1.1.2. Обнаружение азота..................................28
3.1.1.3. Обнаружение серы . ,...............................28
3.1.1.4. Обнаружение галогенов (за исключением фтора) .... 29
3.1.1.5. Обнаружение фтора .................................29
3.1.2. Обнаружение фосфора и мышьяка...........................30
З.1.2.1. Минерализация пробы................................30
3.1.2.2. Обнаружение фосфора................................31
3.1.2.3. Обнаружение мышьяка ...............................32
3.1.3. Элементный микроанализ ................................ 32
3.1.4. Оценка результатов качественного элементного анализа .... 33
3.2. Количественный элементный анализ............................ 33
3.2.1. Определение углерода и водорода.........................34
3.2.2. Способы минерализации для последующего определения Р, S, N,
F, С1 н As.....................................................35
3.2.3. Определение фосфора.....................................37
3.2.4. Определение серы........................................38
3.2.5. Определение азота ......................................40
3.2.6. Определение фтора ......................................40
3.2.7. Определение хлора, брома и иода.........................43
3.2.8. Определение мышьяка.....................................43
Контрольные вопросы................................................43
Литература ........................................................44
5
4. Определение некоторых функциональных групп......................46
4.1. Обнаружение групп О-метил и N-метнл...........................
4.2. Обнаружение группы N-этил.....................................
4.3. Обнаружение группы О-этил.....................................
4.4. Обнаружение группы О-изопропил................................
4.5. Обнаружение группы О-пинаколил................................
4.6. Обнаружение фенильной группы .................................
4.7. Обнаружение сложных эфиров карбоновых кислот..................
4.8. Обнаружение третичных аминов..................................
4.9. Обнаружение этиленовой группы (—СН2—СН2—).....................
4.10. Оценка способов обнаружения функциональных групп.............
4.11. Количественное определение функциональных групп..............
Контрольные вопросы ...............................................
Литература ........................................................
46
46
47
47
47
47
48
49
49
49
50
50
51
5. Химические методы анализа ОВ........................................52
5.1. Фосфорорганические соединения....................................52
5.1.1. Методы обнаружения ........................................ 52
5.1.1.1. Реакция Шёнеманиа.....................................52
5.1.1.2. Реакция с изонитрозокетоиами..........................61
5.1.1.3. Реакция с 4-(л-нитробензил) -пиридином................62
5.1.1.4. Реакции с гидроксамовыми кислотами....................63
5.1.1.5. Идентификация фосфорорганических ОВ............... . 64
5.1.1.6. Индикация паратиона (тиофоса).........................65
5.1.1.7. Индикация изосистокса ................................66
5.1.1.8. Индикация тетраэтилпирофосфата (ТЭПФ) ................66
5.1.1.9. Индикация тетрама (амитона) и родственных соединений 67
5.1.2. Количественное определение . . . . -........................69
'5.1.2.1 . Методы, основанные на реакции Шёнеманиа.............69
5.1.2.2. Объемное определение фтор а и гидридов фосфоновой кислоты
и эфиров пирофосфориои кислоты (гидролизный метод) . . 70
5.1.2,3. Объемное определение фтораигидридов эфиров метилфосфо-
новой кислоты и пирофосфонатов (перекисный метод) . . 71
5.1.2.4. Колориметрическое определение при помощи 4-(л-нитробеи-
зил)-пиридина (экспресс-метод) ...........72
5.1.2.5. Колориметрическое определение при помощи'" гидроксамовой
кислоты ...................................................... 73
5.1.2.6. Колориметрические способы определения паратиона и пара-
оксона ........................................................73
5.1.2.7. Колориметрическое определение систокса................74
5.1.2.8. Объемное определение изосИстокса................. . 75
5.1.2.9. Фотометрическое определение фосфорилтиохолинов .... 75
5.2. Галогенированные тиоэфиры (серные-иприты)........................76
5.2.1. Методы индикации ............................................76
5.2.1.1. Реакция с тиомочевиной и солью никеля.................76
5.2.1.2. Реакция со щелочным раствором тимолфталеина .... 78
5.2.1.З. Реакция с реагентом Гриньяра.........................79
5.2.1.4. Реакция с хлорным золотом............................. 79
5.2.1.5. Другие методы индикации иприта.........................80
5.2.1.6. Идентификация иприта получением производных .... 80
5.2.2. Количественное определение ..................................81
5.2.2.1. Фотометрическое определение с иодоплатинатом .... 81
5.2.2.2. Титрование бромид-броматным раствором..........82
5.2.2.3. Объемное микроопределенне .............. 82
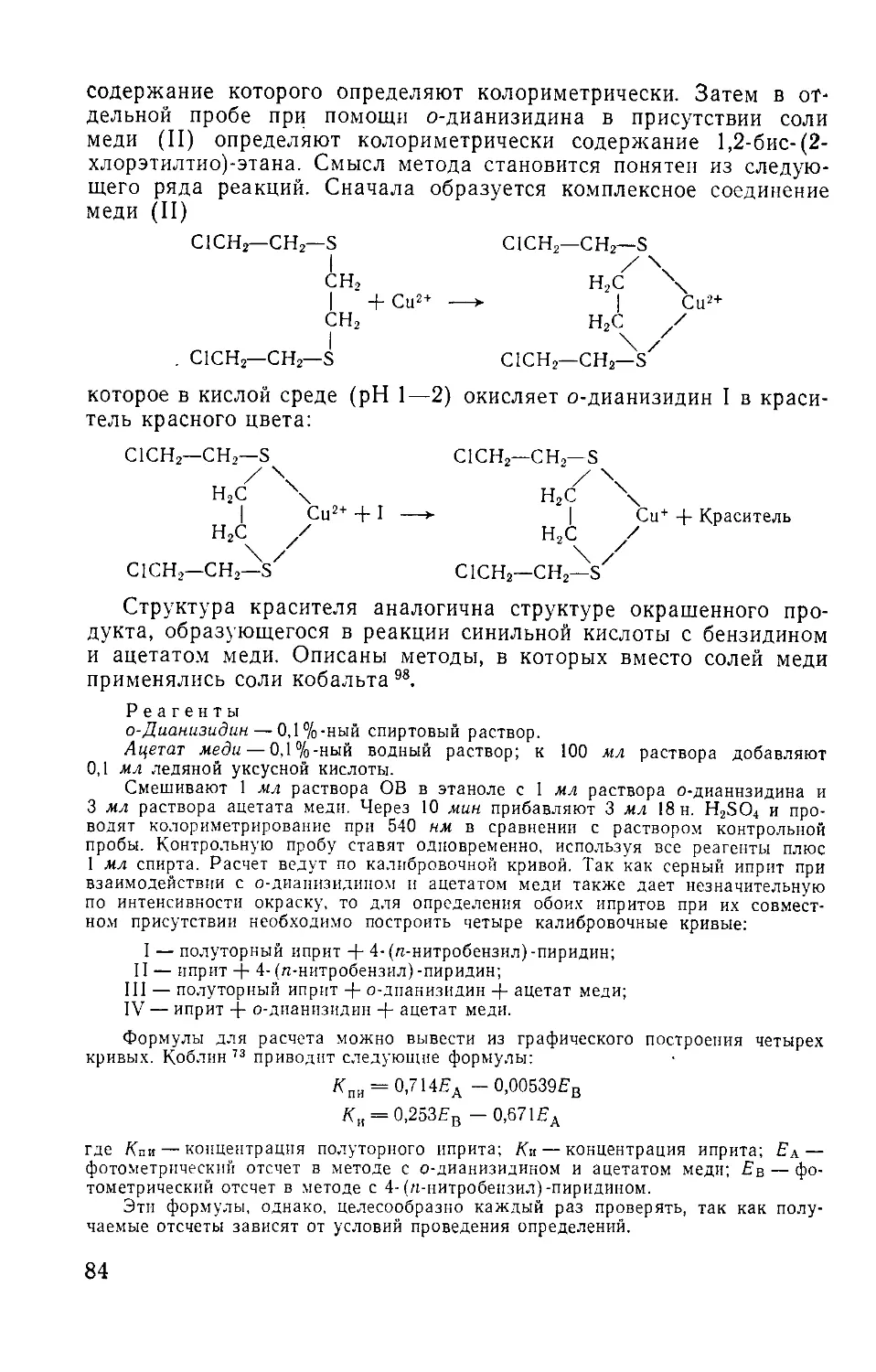
5.2.2.4. Колориметрическое определение 1,2-бис- (2-хлорэтилтио) -эта-
на (сесквииприта) при совместном присутствии с ипритом . 83
5.2.2.5. Другие методы определения бис-2-хлбрЭтилового тиоэфира 85
6
5.3. ГалогензаМещенные алифатические Третичные амины1 (азотистые иприты) 85
5.3.1. Качественное обнаружение...................................85
5.3.1.1. Осаждение калнйвисмутиодидом (реагент Драгендорфа —
Краута) ..................................................... 85
5.З.1.2. Осаждение фосфорновольфрамовой кислотой ...... 86
5.З.1.З. Другие реагенты, образующие осадки с азотистым ипритом 86
5.3.1.4. Обнаружение азотистого иприта по полученным производным 86
5.3.2. Количественное определение.................................86
5.3.2.1. Колориметрическое определение ипритов при помощи брокси-
хинолина (оксина) . ...........................................86
5.3.2.2. Колориметрическое определение при помощи 4-(п-нитробен-
зил)-пиридина .................................................87
5.3.2.3. Другие методы определения ............88
5.4. Мышьяксодержащие ОВ..............................................90
5.4.1. Качественное обнаружение...................................90
5.4.1.1. Общие методы обнаружения..............................90
5.4.1.2. Специфические методы индикации...................... 93
5.4.2. Количественное определение.................................96
5.4.2.1. Определение в виде мышьяковистого водорода............96
5.4.2.2. Йодометрическое определение мышьяксодержащи'х ОВ . . 98
5.4.2.3. Йодометрическое определение метил-, этил- и фенилдихлор-
арсииов .......................................................99
5.4.2.4. Йодометрическое определение дифеиилхлорарсина .... 99
5.4.2.5. Определение галогена и циангруппы в мышьяксодержа-
щих ОВ .......................................................100
5.4.2.6. Колориметрическое определение адамсита (10-хлор-9,10-ди-
гидрофенарсазинхлорида) ......................................100
5.4.2.7. Определение люизита..................................101
5.5. Синильная кислота и галогенцианы................................101
5.5.1. Обнаружение синильной кислоты и цианидов..................101
5.5.1.1. Образование берлинской лазури .......................101
5.5.1.2. Реакция с сульфидом меди.............................101
5.5.1.3. Образование тиоцианата...............................102
5.5.1.4. Реакция с ацетатами меди и бензидином................102
5.5.1.5. Другие методы обнаружения цианидов путем окисления
различных соединений в присутствии солей медн(И) . . 103
5.5.1.6. Реакция с пикратом натрия ...........................103
5.5.1.7. Образование недиссоциирующих цианидов тяжелых ме-
таллов ..................................................... 103
5.5.1.8. Разложение виутрикомплексиых солей металлов . . . .104
5.5.1.9. Каталитическое ускорение бензоиновой конденсации . . . 104 .
5.5.1.10. Образование галогенциаиа............................106
5.5.2. Обнаружение галогенциаиов.................................106
5.5.2.1. Важнейшие методы обнаружения и определения галоген-
цианов........................................................106
5.5.2.2. Идентификация различных галогенциаиов................108
5.5.3. Количественное определение синильной кислоты и цианидов . .108
5.5.3.1. Титрование нитратом серебра..........................108
5.5.3.2. Титрование хлорной ртутью............................109
5.5.3.3. Титрование сульфатом никеля..........................109
5.5.3.4. Йодометрическое титрование...........................109
5.5.3.5. Колориметрическое определение после превращения циани-
дов в хлорциан................................................10Э
5.5.3.6. Фотометрические методы определения...................110
5.5.3.7. Другие колориметрические методы ......................ПО
5.5.4. Количественное определение галогенцианов....................111
5.5.4.1. Определение хлорциана реакцией со щелочью.............111
5.5.4.2. Определение бромциана.................................111
5.6. Галогенпроизводные угольной кислоты...............................111
5.6.1. Качественное обнаружение фосгена............................111
5.6.1.1. Обнаружение фосгена анилиновой водой..................111
5.6.1.2. Реакция с п-диметиламннобензальдегидом н диметнланили-
ном ...........................................................111
5.6.1.3. Реакция с феннлгидразином.............................112
5.6.2. Количественное определение ..................................ИЗ
5.6.2.1. Реакция с анилином ....................................ИЗ
5.6.2.2. Йодометрическое определение............................ИЗ
5.6.2.3. Колориметрическое определение 4-(n-нитробензил)-пиридином 114
5.6.2.4. Другие методы определения фосгена.....................114
5.6.2.5. Колориметрическое определение дифосгена . ............114
5.6.2.6. Другие методы определения дифосгена и трифосгена . . .115
5.7. Галогенированные кетоны..........................................116
5.7.1. Хлор- и бромацетон.........................................116
5.7.2. Хлорацетофенон ............................................116
5.7.2.1. Качественное обнаружение..............................116
5.7.2.2. Количественное определение............................117
5.8. Бромбензилцнанид.................................................118
5.8.1. Качественное обнаружение ..................................118
5.8.1.1. Сплавление с щелочами.................................118
5.8.1.2. Омыление с образованием аммиака.......................118
5.8.1.3. Другие методы обнаружения.............................119
5.8.2. Количественное определение.................................119
5.8.2.1. Реакция с сульфидом натрия............................119
5.9. Трихлорннтрометан (хлорпикрин)...................................119
5.9.1. Качественное обнаружение.................................. 119
5.9.1.1. Восстановление до нитрита . . . 119
5.9.1.2. Проба Лабатша.........................................120
5.9.1.3. Реакция с образованием бромциана......................121
5.9.1.4. Другие методы обнаружения...............;.............121
5.9.2. Количественное определение.................................122
5.9.2.1. Аргентометрическое определение хлора в растворе после раз-
ложения хлорпикрина............................................122
5.Э.2.2. Колориметрическое определение в виде галогенциана . . .122
5.9.2.3. Колориметрическое определение хлорпикрина по образую-
щемуся нитриту.................................................123
5.10. Современные раздражающие ОВ................................ . . 123
5 10.1. о-Хлорбензилнденмалонодинитрил (2-хлорбензальмалонодинитрнл,
Си Эс)........................................................123
5.10.1.1. Качественное обнаружение.............................123
5.10.2. Морфолнд пеларгоновой кислоты.............................124
5.11. Свинецорганическне соединения...................................124
5.11.1. Тетраэтилсвинец...........................................125
5.11.1.1. Разложение УФ-облучением ...........................125
5.11.1.2. Разложение азотной кислотой..........................125
5.11.1.3. Разложение хлором ...................................125
5.11.1.4. Количественное определение тетраэтилсвинца...........125
5.11.2. Другие свинецорганическне соединения......................126
8
5.12. Окись углерода и карбонилы металлов........................
5.12.1 . Качественное обнаружение ...............................126
5.12.1.1 . Восстановление хлорида палладия....................126
5.12.1.2 . Восстановление нитрата серебра ....................127
5.12.1.3 . Восстановление пятиокиси иода......................127
5.12.1.4 . Спектрофотометрическое обнаружение ................127
5.12.1.5 . Обнаружение с помощью мышьяковистой кислоты и хлор-
ного золота ...................................................128
5.12.1.6 . Обнаружение карбонила железа........................128
5.12.1.7 . Обнаружение карбонила никеля .......................128
5.12.2 . Количественное определение ..............................128
5.12.2.1 . Определение при помощи пятиокиси иода...............128
5.12.2.2 . Гопкалнтовый метод..................................128
5.12.2,3 . Восстановление окиси ртути.................•• . . • 129
5.12.2.4 . Определение карбонила никеля......................129
5.13. Фторкарбоновые кислоты..........................................129
5.13.1. Качественное обнаружение..................................129
5.13.1.1. Отщепление и обнаружение фтора......................129
5.13.1.2. Обнаружение фторацетатов по реакции с солями лантана 130
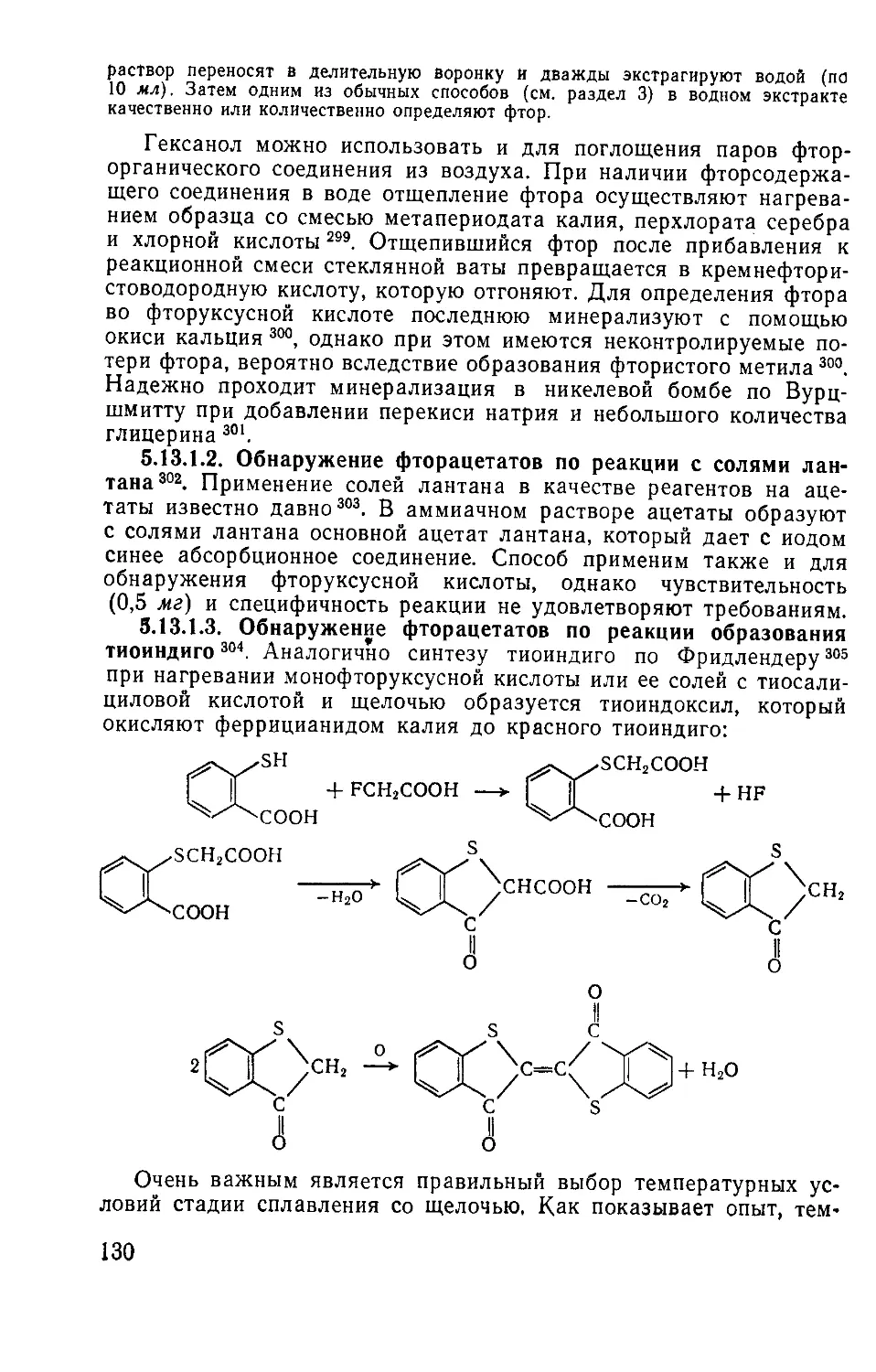
5.13.1.3. Обнаружение фторацетатов по реакции образования тио-
индиго ................................................... . . 130
5.13.1.4. Обнаружение при помощи концентрированной серной и
хромотроповой кислот ............................ 131
5.13.2. Количественное определение ...............................131
5.14. Алкалоиды ................'.....................................132
5.14.1. Подготовка к анализу .....................................132
5.14.2. Методы обнаружения........................................133
5.14.2.1. Определение температуры плавления....................133
5.14.2.2. Общие цветные реакции .........................134
5.14.2.3. Реакции группового осаждения.........................135
5.14.2.4. Характерные реакции обнаружения некоторых алкалоидов 139
5.15. Животные и бактериальные яды ...................................140
5.15.1. Кантаридин ..............................................140
5.15.2. Токсин ботулизма ........................................140
5.16. Фитотоксические ОВ ...............................................141
5.16.1. Хлорированные феноксиуксусные кислоты.....................141
5.16.1.1. Обнаружение хлорфеноксиуксусиых кислот.............141
5.16.1.2. Бумажная и тонкослойная хроматография................142
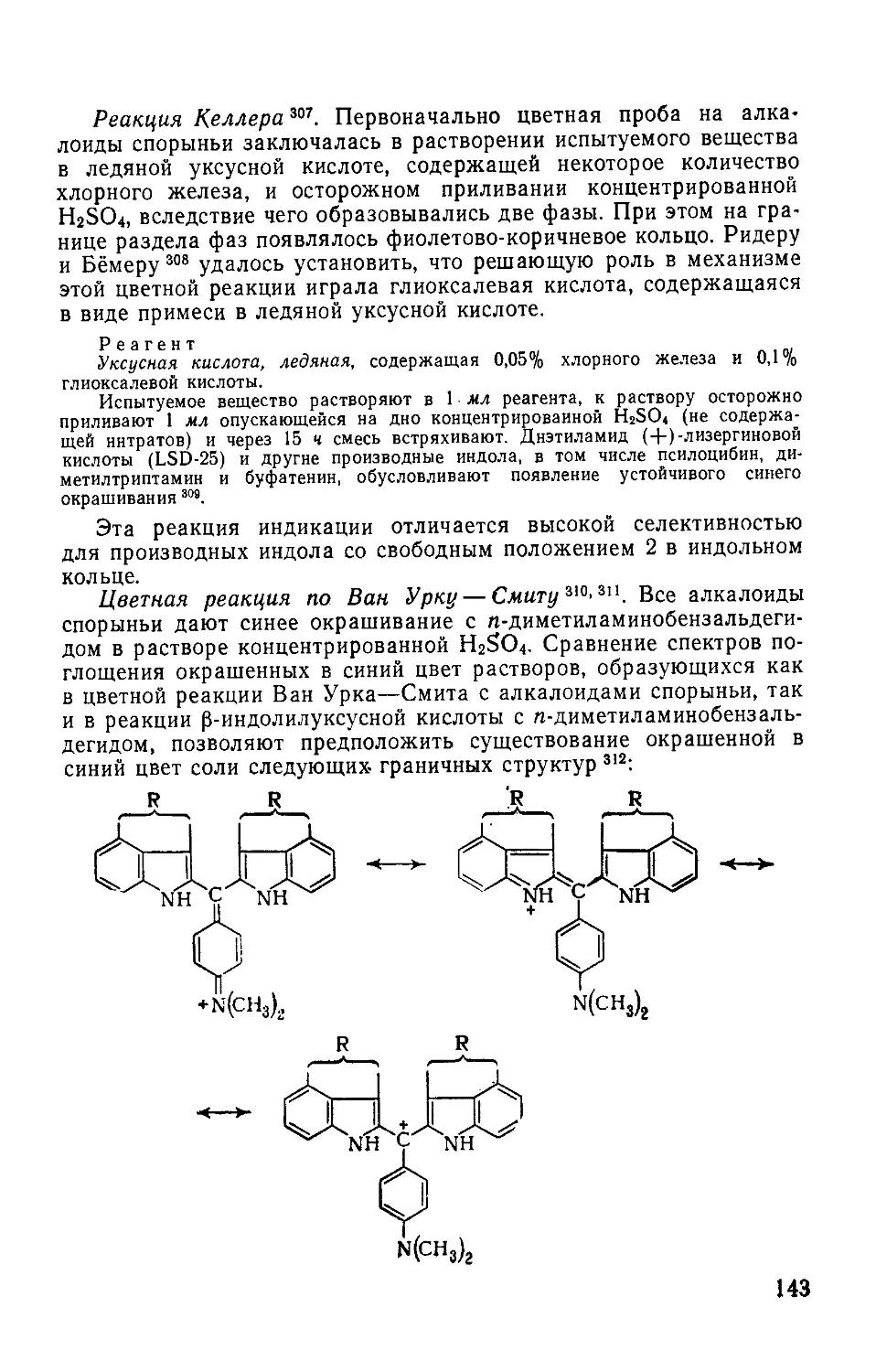
5.16.1.3. Колориметрическое определение .......................142
5.16.2. Динитро-о-крезол.....................•....................142
5.17. Психохимические ОВ.......................................... 142
5.17.1. Производные индола.......................................142
5.17.1.1. Методы обнаружения . .,...................142
5.17.1.2. Количественное определение.........................144
5.17.2. Фениламиноалканы.........................................145
5.17.2.1. Методы обнаружения.................................145
5.18. Неорганические яды............................................145
5.18.1. Токсичные катионы........................................146
5.18.1.1. Групповое обнаружение токсичных катионов...........146
5.18.1.2. Обнаружение бария .................................146
5.18.1.3. Обнаружение бериллия...............................147
9
5.18.1.4. Обнаружение свинца...............................148
5.18.1.5. Обнаружение таллия...............................148
5.18.1.6. Обнаружение ртути ...............................148
5.18.1.7. Обнаружение кадмия............................. 149
5С18.1.8. Обнаружение сурьмы фосфорномолнбденовой кислотой . . 149
5.18.2. Характерные реакции...................................149
5.18.2.1. Обнаружение фтор-иона............................149
5.18.2.2. Обнаружение нитрнт-иона..........................149
5.18.2.3. Обнаружение арсенит- и арсенат-ионов...........150
5.18.2.4. Обнаружение селенита.............................150
Контрольные вопросы...............................................150
Литература .......................................................151
6. Биохимические методы ..........................................158
6.1. Преимущества применения биохимических методов................158
6.2. Действие ОВ на ферменты......................................160
6.3. Угнетение холинэстеразы фосфорорганическими соединениями .... 160
6.3.1. Определение ннгнбнрующего действия.....................164
6.3.2. Определение активности холинэстеразы...................165
6.3.2.1. Определение активности холинэстеразы сыворотки в аппа-
рате Варбурга..............................................171
6.3.2.2. Определение активности холинэстеразы электрометрическим
Д рН-методом ..............................................172
6.3.2.3. Фотометрическое определение активности холинэстеразы . 173
6.3.3. Определение ингибиторов холинэстеразы..................173
6.3.3.1. Индикация и полуколнчественное определение фосфорорга-
нических ингибиторов в воде при помощи субстрат-индика-
торной бумаги ............................................ 178
6.3.3.2. Фотометрическое определение с индолфенилацетатом в каче-
стве субстрата..........................................179
6.4. Биохимическое определение фосфорорганических соединений при помо-
щи других ферментов..................................... . 180
Контрольные ' вопросы .................... 180
Литература ..................................................... 181
7. Биологические методы.......................................... 184
7.1. Определение значения ЛД и ЛК..............................184
7.2. Использование животных для индикации ОВ...................185
Контрольные вопросы . . . ...................................... 187
Литература . :.................................................187
8. Физические и физико-химические методы..........................189
8.1. Определение плотности .......................................189
8.2. Определение температуры плавления.......................... 192
8.3. Определение температуры кипения..............................193
8.4. Определение давления насыщенного пара........................194
8.5. Определение показателя преломления...........................196
8.6. Спектроскопические методы....................................197
8.6.1. УФ-Спектры.............................................199
8.6.2. ИК-Спектры........................................... 200
10
8.7. Разделение и идентификация при помощи хроматографических методов 203
8.7.1. Хроматография в колонках...................................203
8.7.2. Бумажная хроматография.....................................204
8.7.2.1. Исследование фосфорорганических соединений методом бу-
мажной хроматографии .........................................204
8.7.3. Тонкослойная хроматография.................................207
8.7.3.1. Исследование фосфорорганических соединений ..... 207
8.7.3.2. Обнаружение алкалоидов ...............................208
8.7.3.3. Обнаружение токсичных катионов и анионов..............209
8.7.3.4. Разделение других ОВ и гербицидов.....................209
8.7.4. Газовая хроматография .....................................212
8.7.4.1. Анализ фосфорорганических соединений..................213
8.7.4.2. Анализ других ОВ......................................215
8.8. Электрохимические методы ........................................215
8.8.1. Полярографические методы...................................215
Контрольные вопросы...................................................217
Литература ...........................................................217
9. Индикация боевых отравляющих веществ в полевых условиях .... 221
9.1. Субъективное восприятие........................................221
9.2. Простейшие средства разведки...................................223
9.2.1. Индикаторный порошок......................................223
9.2.2. Индикаторные карандаши ...................................226
9.2.3. Индикаторные бумаги.......................................227
9.2.З.1. Неспецифические индикаторные бумаги.................228
9.2.3.2, Индикаторные бумаги на хлор...................228
9.2.3.3. Индикаторные бумаги на фосген..................229
9.2.3.4. Индикаторные бумаги иа синильную кислоту............229
9.2.3.5. Индикаторные бумаги иа хлорпикрин . ................231
9.2.3.6. Индикаторные бумаги иа серный иприт.................231
9.2.3.7. Индикаторные бумаги на галогеналкиламины (азотистые
иприты) .....................................................231
9.2.3.8. Индикаторная бумага дли обнаружения фосфорсодержа-
щих ОВ в воде............................................... 232
9.2.3.9. Индикаторная бумага для обнаружения фосфорсодержащих
ОВ в воздухе . ..............................................232
9.2.3.10. Индикаторные бумаги иа этилднхлорарсии............. 232
9.2.3.11. Индикаторная бумага иа окись углерода..............232
9.2.4. Индикаторные трубки.....................................232
9.3. Автоматические приборы.........................................239
9.3.1. Приборы, действие которых основано на химических реакциях . 241
9.3.2. Приборы, основанные на физических принципах индикации . . 244
9.3.3. Приборы, действие которых осиоваио иа биохимическом принципе 245
9.4. Индикаторные наборы и носимые полевые лаборатории..............246
9.5. Передвижные полевые лаборатории................................248
9.6. Отбор проб для анализа....................................... 249
9.6.1. Отбор пробы почвы . . ................................. 250
9.6.2. Отбор пробы воды........................................251
9.6.3. Отбор проб ОВ с военной техники, обмундирования и снаряжения 252
9.6.4. Отбор проб продовольствия и фуража......................252
11
9.6.5. Отбор проб воздуха ........................................253
9.6.5.1. Адсорбция ...........................•................254
9.6.5.2. Абсорбция ........................................... 254
9.6.5.3. Фильтрация . . . .....................................256
9.6.6. Маркировка проб ......................................... 257
9.7, Подготовка проб к анализу..................................... . 257
9.8. Систематизация качественного анализа проб ОВ в полевых лабораториях 260
9.9. Специальные задачи полевого анализа ОВ............................264
9.9.1. Анализ смесей ОВ...........................................264
9.9.2. Анализ загущенных ОВ ................................ . £68
9.9.3. Исследование проб пищевых продуктов........................268
9.9.4. Исследование проб воды................................... 268
9.9.5. Контроль полноты дегазации................................ 269
Контрольные вопросы....................................................272
Литература ............................................................273
ДЕГАЗАЦИЯ И СРЕДСТВА ДЕГАЗАЦИИ
10. Роль дегазации в системе мероприятий защиты от химического оружия 277
10.1. Причины необходимости дегазации.............................277
10.2. Определение понятия «дегазация».............................278
Контрольные вопросы...............................................279
11. Комбинированная дегазация................................... 280
Контрольные вопросы ..................... 282
12. Химическая дегазация........................................ 283
12.1. Общие положения ........................................... 283
12.1.1 Некоторые вопросы теории процессов дегазации...........285
12.1.2. Каталитическое ускорение реакций дегазации............293
12.1.3. Влияние растворителей на реакции дегазации . . .... 294
12.1.4. Влияние физических факторов на химические способы дегазации 295
12.2. Дегазация нуклеофильными реагентами.........................298
12.2.1. Общие сведения....................................... 298
12.2.2. Дегазация едким натром .............................. 300
12.2.3. Дегазация содой...................................... 303
12.2.4. Дегазация другими кислородсодержащими нуклеофильными ре-
агентами .....................................................304
12.2.5. -Сульфид натрия и другие сернистые щелочные соединения , , . 309
12.2.6. Аммиак и амины .......................................311
12.2.6.1. Общие сведения..................................311
12.2.6.2. Аммиак .........................*...............312
12.2.6.3. Бикарбонат аммония............................ 314
12.2.6.4. Продукты конденсации аммиака и гидроксиламина . . . 314
12.2.6.5. Общие свойства алкиламинов .....................315
12.2.6.6. Комплексные соединения аминов...................317
12.3. Дегазация окислением и хлорированием........................318
12.3.1. Общие сведения........................................318
12.3.2. Гипохлориты 320
12.3.2.1. Общие сведения..................................320
12
12.3.2.2. Хлор ...............................................323
12.3.2.3. Хлорная известь.....................................324
12.3.2.4. Гипохлорит кальция..................................326
12.3.2.5. Гипохлорит натрия ..................................330
12.3.2.6. Дегазация гипохлоритами........................... 333
12.3.3. Хлориты ..................................................336
12.3.4. Хлорамины ................................................337
12.3.4.1. Общие сведения.................................... 337
12.3.4.2. Хлорамин Б..........................................338
1213.4.3. Хлорамин Т..........................................338
12.3.4.4. Дихлорамин Б и дихлорамин Т.........................340
12.3.4.5. Гексахлормеламин (ДТ-6).............................342
12.3.4.6. Изоцианурхлорид.....................................342
12.3.4.7. Дихлорамид Метансульфокислоты.......................343
12.3.4.8. 1,3-Дихлор-5,5-диметилгидантоин ....................343
12.3.4.9. N-Хлоргликольурил...................................344
12.3.5. Сульфурилхлорид ..................................... • 344
12.3.6. Дегазирующие вещества, разлагающиеся с выделением кислорода 345
12.3.6.1. Общие сведения...................'..................345
12.3.6.2. Озон ...............................................346
12.3.6.3. Перекись водорода ..................................347
12.3.6.4. Другие перекисные соединения........................348
12.3.6.5. Персульфаты.........................................343
Контрольные вопросы ..................................................349
Литература ...........................................................350
13. Физические способы дегазации......................................352
13.1. Общие сведения ................................................ 352
13.2. Дегазация растворителями........................................352
13.2.1. Общие сведения............................................352
13.2.2. Свойства важнейших растворителей..........................354
13.2.2.1. Дихлорэтан . .......................................354
13.2.2.2. Четыреххлористый углерод (тетрахлорметан)...........355
13.2.2.3. Метилрвый и этиловый спирты . . .............355
13.2.2.4. Прочие растворители............................... 356
13.2.3. Дегазация зараженных поверхностей растворителями .... 356
13.2.4. Экстракционные способы дегазации обмундирования...........357
13.3. Дегазация стиркой...............................................358
13.3.1. Общие сведения ......... 358
13.3.2. Важнейшие поверхиостио-активные вещества..................360
13.3.3. Дегазация моющими растворами..............................364
13.4. Дегазация адсорбцией.......................................... 365
13.4.1. Общие сведения............................................355
13.4.2. Основные адсорбенты.................................... . 365
13.4.3. Дегазация воздуха........................................ 367
13.4.4. Дегазация воды........................................... 358
Контрольные вопросы ............. ................................... 369
Литература .......................................................• . . 370
14. Испытание средств дегазации.......................................371
14.1. Общие сведения..................................................371
14.2. Отбор проб . 371
14.3. Испытание дегазирующих средств перегонкой.......................372
13
14.4. Определение плотности и насыпной массы........... '..........372
14.5. Определение температуры плавления.............................373
14.6. Определение нерастворимого остатка твердых дегазирующих веществ 373
14.7. Определение содержания влаги ............................... 373
14.8. Анализ щелочных дегазирующих веществ....................... 374
14.9. Испытание дегазирующих веществ, содержащих активный хлор . . 37S
14.10 Испытание моющих средств......................................376
14.11. Исследование неизвестного дегазирующего вещества.............377
Контрольные вопросы.................................................378
Литература......................................................... 378
Приложение. Меры первой помощи при поражениях и отравлениях дегази-
рующими веществами и растворителями............................... 379
Предметный указатель ............................................ 381
АНАЛИЗ
БОЕВЫХ
ОТРАВЛЯЮЩИХ
ВЕЩЕСТВ
1. ЗАДАЧИ АНАЛИЗА БОЕВЫХ
ОТРАВЛЯЮЩИХ ВЕЩЕСТВ (БОВ)
Индикация боевых отравляющих веществ является частью общей
задачи защиты от химических средств массового уничтожения.
В случае применения химического оружия различные мероприятия
военной и гражданской обороны должны начинаться с обнаруже-
ния и количественного определения отравляющих веществ. Эти же
методы используются для контроля эффективности тех или иных
мер защиты. Следовательно, индикация ОВ в основном должна
решать две главные задачи.
1. Своевременно предупреждать о применении химического
оружия путем постоянного наблюдения за воздухом средствами
химической разведки, для того чтобы в случае обнаружения бое-
вых отравляющих веществ и после установления вида ОВ и места
его применения можно было в кратчайшие сроки предпринять не-
обходимые меры по защите людей и животных, а также устранить
последствия химического нападения.
2. Контролировать работу по ликвидации последствий химиче-
ского нападения.
Следующей задачей, которая уже вторгается в область меди-
цинской службы, является диагноз типа поражения химическими
или биохимическими способами исследования. Выбор способа ана-
литического контроля должен зависеть от поставленной задачи.
Так, для своевременного обнаружения ОВ имеют большое значение
автоматические газосигнализаторы, работающие на физико-хими-
ческих или чисто физических принципах, подающие сигналы о на-
личии в воздухе ничтожных концентраций ОВ. При подозрении хи-
мического нападения для первичного обнаружения зараженной
местности и источников водоснабжения существуют такие простые
в обращении и надежные средства индикации, как индикаторные
бумаги и трубки. Более точные исследования образцов зараженных
материалов, заключающиеся в количественном определении отрав-
ляющих веществ, осуществляются в передвижных и стационарных
полевых лабораториях. В обязанности этих лабораторий входит
также идентификация неизвестных, впервые примененных против-
ником ОВ. Для этого вместо обычных высокоспецифических мето-
дов определения приходится пользоваться элементным анализом,
методами определения функциональных групп, установлением
физических констант — температуры плавления, температуры кипе-
ния, относительной плотности, показателя преломления.
17
Полевые способы анализа отравляющих и дегазирующих ве-
ществ, а также материалов средств индивидуальной защиты дол-
жны постоянно совершенствоваться путем использования эффек-
тивных химических, биохимических и физико-химических методов
макро- и микроанализа. Специальной областью является получение
в лаборатории малых количеств чистых отравляющих веществ, не-
обходимых для разработки точных аналитических методов. Для
идентификации и определения действующего начала, а также
характера и степени загрязнения используются методы разделения,
такие, как адсорбционная, бумажная, тонкослойная и газовая хро-
матография.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какие задачи можно решать при помощи анализа отравляющих веществ?
2. ОСНОВЫ И ОСОБЕННОСТИ АНАЛИЗА ОВ
Анализ ОВ имеет много общего с анализом промышленных ядов.
Многие способы и применяемые приборы удовлетворяют требова-
ниям обеих этих областей. Примерами ядовитых веществ, с кото-
рыми приходится встречаться как в качестве БОВ, так и в качестве
промышленных ядов, могут служить — фосген, синильная кислота
и некоторые фосфорорганические инсектициды. Благодаря этому
аналитические методы, разработанные для промышленных целей,
используются при анализе ОВ и — наоборот. Как для той, так и
для другой области очень важно, чтобы чувствительность метода
соответствовала токсичности индицируемого или определяемого
ядовитого вещества.. Особенность полевых методов анализа ОВ за-
ключается главным образом в условиях проведения работы и в том,
что должны быть предусмотрены аналитические методы, пригодные
для исследования проб различных материалов. Несмотря на отра-
ботку методов химической разведки в мирное время, трудности,
которые приходится решать при выполнении задач в боевых усло-
виях и при работе в подвижных полевых лабораториях и лабора-
ториях гражданской обороны, несравнимы с' встречающимися в
обычных условиях проведения анализов промышленных ядов.
Необходимость в возможно более короткий срок получить абсо-
лютно достоверный результат ставит перед, аналитиком, работаю-
щим с ОВ, чрезвычайно ответственную задачу. Меры, которые
будут приняты на основании результатов его исследования, имеют
жизненно важное значение для многих людей. В качестве примера
можно привести решение о возможности использования воды для
питья и продуктов,в пищу. Своевременные сведения, подтверждаю-
щие отсутствие ОВ в атмосфере, дают возможность уменьшить
физическую нагрузку, связанную с ношением противогаза и защит-
ной одежды.
2.1. ОРГАНИЗАЦИЯ РАБОТЫ
Работы, проводимые в мирное время в лаборатории по анализу ОВ,
отличаются от работ в обычных аналитических лабораториях в ос-
новном повышенными требованиями техники безопасности. Совсем
иначе строится работа в полевых лабораториях в случае сигнала
о применении химического оружия. Лаборант должен за короткий
срок проанализировать большое количество разнообразных проб.
19
Работа должна быть организована так, чтобы, несмотря на ограни-
ченные технические и пространственные возможности, суметь
быстро проанализировать полученные образцы. Любая задержка
анализа образцов в лаборатории приводит к задержке принятия
важных решений. Кроме того, ОВ, находящиеся в пробах, при стоя-
нии могут претерпевать ряд превращений в результате гидролиза,
окисления, возможных взаимодействий с веществом материала
пробы и т. п. Навыки по умению работать в такой обстановке при-
обретаются только длительной практикой и тренировкой в усло-
виях, приближающихся к реальным. В основу таких практических
занятий должен быть положен предварительный расчет возможного
числа проб и их распределения по различным группам, в зависи-
мости от условий боевой обстановки. Главной предпосылкой быст-
рой и хорошей работы в полевых условиях является надежное
овладение теоретическими и практическими основами применяемых
методик. Подготовка проб к анализу и сами анализы следует вы-
полнять по памяти, не заглядывая в прописи. Таково основное тре-
бование, предъявляемое к квалификации полевого лаборанта.
При анализе ОВ как в полевых условиях, так и в стационарных
лабораториях все поступающие на анализ пробы должны быть
тщательно и единообразно надписаны. Если это требование не было
выполнено разведчиком (лицом, отбиравшим пробы), маркировку
следует уточнить. Такая предварительная подготовка исключает
возможность ошибок со всеми вытекающими отсюда последствиями.
Для проведения анализа пробу тщательно измельчают. При нали-
чии только одной пробы, на случай необходимости повторного ана-
лиза, следует часть ее сохранить.
Чтобы исключить перенесение искомого вещества из одного об-
разца в другой, важно уделять особое внимание чистоте посуды,
колб, пипеток, шпателей и т. п. Так как некоторые ОВ определяют
микрометодами, огромное значение имеет также и чистота приме-
няемых реагентов. Так, например, серная кислота и цинк содержат
небольшие примеси мышьяка. Если содержание примеси не превы-
шает чувствительности метода, то вводимое с кислотой и цинком
количество мышьяка не будет мешать определению при условии
точного соблюдения указанных в методике норм добавления ре-
агентов. Превышение указанных норм может привести к искаже-
нию результатов анализа. Разумеется, что для определения мышья-
ка допустимо применение серной кислоты только квалификации
«ч.д. а.» и цинка, используемого для целей судебной экспертизы.
Проведение контрольной пробы обеспечивает как в этом, так и в
других подобных случаях необходимую уверенность в том, что при-
мененные реагенты имели требуемую степень чистоты. Понятие
чистоты относительно, и вряд ли имеются абсолютно чистые ве-
щества.
Нередко существует несколько методов обнаружения ОВ. В та-
ком случае следует выбрать (если, конечно, количество испытуе-
мого вещества это позволяет) метод, основанный на самой специ-
20
фичной для данного ОВ реакции, хотя он может быть и не самым
чувствительным. Более чувствительные методы, однако менее спе-
цифичные в силу своей чувствительности и к примесям, используют
в тех случаях, когда выбор аналитического метода ограничен на-
личием малого количества веществд для исследования. Не вызы-
вающая сомнения реакция более убедительна,"чем ряд неспецифи-
ческих реакций, которые можно было бы предпочесть из-за их про-
стоты и удобства осуществления.
2.2. РАБОЧЕЕ МЕСТО И ОБОРУДОВАНИЕ
Оборудование рабочего места для анализов в полевых условиях
будет более подробно описано в следующей главе.
Минимальное требование, которое предъявляют к лаборатории,
пригодной для анализа ОВ, является наличие вытяжного шкафа и
покрытых плиткой рабочих столов. Идеальными являются лабора-
тории, пол и стены которых также покрыты плиткой. Лаборатории
должны располагать кранами с душевой насадкой и водостоками.
В канализационной системе лаборатории перед ее присоединением
к общей канализационной системе должна находиться ловушка, за-
полняемая щелочью.
Особые требования предъявляются к мощности вытяжной вен-
тиляции главным образом при работе с фосфорорганическими
веществами. По возможности скорость воздушного потока должна
быть не менее 0,7 м/сек. Во избежание заражения атмосферы вы-
тяжная вентиляция должна иметь фильтр, поглощающий пары ОВ.
На случай прекращения подачи электрической энергии из сети
лаборатория должна быть оборудована автоматически включаю-
щейся аварийной установкой, обеспечивающей бесперебойную ра-
боту вытяжки и аппаратуры. Это важно не только в военное время,
но и вообще исключает аварии, которые, например, могут произойти
хотя бы вследствие отсутствия освещения. Целесообразно также
иметь подсобные источники тепла — спиртовые горелки, запасные
баллоны с пропаном и, кроме того, запас воды.
Если рабочая поверхность стола представляет собой ничем не
покрытые доски, то на рабочих местах должны быть положены
легко дегазирующиеся пластины пластика или стекла. По этой же
причине предпочтительны штативы из пластмассы для пробирок,
колб и фильтров. Все общеупотребительные в обычных лаборато-
риях стеклянные приборы, кроме обычных пипеток, применимы и
в анализе ОВ (даже в том случае, если работа проводится без про-
тивогазов). Растворы проб и реагенты отмеривают специальными
пипетками с поршнем (безопасные пипетки), которые можно из-
готовить из обычных пипеток, соединяя их короткими кусками рези-
новых трубок с медицинским шприцем, или из микропипеток, снаб-
див их резиновой грушей или резиновым отсосом.
Особое внимание следует уделить также организации рабочего
места; сосуды с пробами ОВ или их растворами должны быть
21
четко надписаны и поставлены в специально предназначенное для
них место. Сосуды с эталонными образцами ОВ и их концентри-
рованными растворами должны храниться в вытяжном шкафу в
больших сосудах, заполненных активированным углем. Дегазирую-
щие средства должны быть приготовлены в достаточном количестве
и помещаться в удобном для пользования месте, вблизи от отрав-
ляющих веществ.
По окончании работы все стеклянные приборы, находившиеся
в контакте с ОВ, подлежат дегазации. Это производится обработ-
кой соответствующим дегазирующим раствором в течение ночц,
причем при погружении посуды в дегазирующий раствор следует
обращать внимание на равномерность смачивания всей ее поверх-
ности. После дегазации все приборы должны быть тщательно про-
мыты водой или другими очищающими средствами для удаления
остатков дегазирующих веществ, чтобы в дальнейшем их следы не
искажали результаты последующих анализов, особенно при опре-
делении малых количеств ОВ. Зараженные каучуковые трубки, ре-
зиновые и корковые пробки после работы сжигают.
Так как большая часть дегазирующих средств выделяет в атмр-
сферу корродирующие газы или пары, все чувствительные при-
боры— весы, фотометры и др., находящиеся в том же помещении,
следует защищать колпаками или футлярами из поливинилхлорида
или метилметакрилата.
2.3. РАБОЧИЕ ЗАПИСИ
Обработка проб в лаборатории начинается с записи в рабочий
журнал информации от лица, отбиравшего пробу (разведчика),
содержащей данные о типе ОВ и получаемой на основании показа-
ний использованных им средств первичной индикации. После этого,
сообразно имеющимся рабочим прописям и оборудованию, наме-
чается план исследований, обеспечивающий наиболее быстрое полу-
чение результатов. В рабочий журнал записывают использованные
методы исследования, реагенты, все наблюдения и, наконец, резуль-
таты, а при количественном анализе и все проводившиеся расчеты.
После окончания исследования эти записи являются основанием
для составления донесения или написания протокола при возмож-
ной передаче пробы для дальнейшего обследования в вышестоящие
инстанции. Рабочий журнал должен заботливо храниться на случай
необходимости повторного представления сведений.
Что касается точности результатов количественных определений
в полевых лабораториях, то в связи со стремлением упростить
работу этих лабораторий они несомненно уступают в этом смысле
стационарным лабораториям, располагающим большим запасом
времени для проведения работы и лучшим техническим оснаще-
нием. Это должно найти отражение и в точности расчетов. Бес-
смысленно, например, данные по угнетению холинэстеразы, полу-
ченные при биохимическом анализе на бумаге, рассчитывать с
22
точностью до десятой доли, или содержание иприта в воде после
адсорбции на активированном угле, экстракции и колориметриче-
ского определения — с точностью до тысячной. Такая излишняя
точность в расчетах вызывает лишь ложное представление о точ-
ности методов, которые в остальном вполне удовлетворяют требо-
ваниям к анализам в полевых условиях.
2.4. ТЕХНИКА БЕЗОПАСНОСТИ
При анализе ОВ следует в соответствии с их токсичностью, родом
работы и условиями пользоваться средствами защиты. Работа
с небольшими количествами ОВ (по сравнению с теми, которые
используются в препаративных работах) не должна являться пово-
дом к пренебрежению средствами защиты. Токсичность современ-
ных отравляющих веществ, в особенности эфиров фосфорной кис-
лоты, настолько высока, что даже контакт с минимальными их ко-
личествами и разбавленными растворами или вдыхание ничтожных
концентраций могут вызвать тяжелые или смертельные отравления.
Кроме того, следует помнить, что при повторном воздействии ОВ в
концентрациях даже ниже пороговых проявляется и кумулятивный
эффект. Множество операций, выполняемых при аналитических ра-
ботах, а также распределение ОВ по большому количеству посуды
при недостаточном соблюдении техники безопасности создают опас-
ность отравления.
При работе с выделенным в чистом виде ОВ или высококон-
центрированными растворами, которые могут вызывать поражение
как при контакте с кожей, так и при действии через органы дыха-
ния, следует пользоваться защитной одеждой, перчатками и проти-
вогазами. При работе с разбавленными растворами или при опера-
циях, проводимых в вытяжном шкафу, эффективность вентиляции
которого может быть установлена по дымовой пробе, ношение про-
тивогаза не обязательно, но он должен находиться в положении
«наготове». Исследуя образцы неизвестных веществ, работу в про-
тивогазе следует проводить до тех пор, пока не станет ясным, что
исследуемое вещество не является фосфорорганическим ОВ.
При всех работах, связанных с анализом ОВ, необходимо поль-
зоваться защитными перчатками (хирургическими или секцион-
ными). Поскольку жидкие ОВ способны через какое-то время
проникать через тонкие перчатки, вне зависимости от вида ОВ и
качества и толщины материала перчаток, необходимо избегать
любого контакта с ОВ. Если при попадании ОВ на перчатки есть
возможность их сразу сменить, то после дегазации их можно вновь
употребить; при более длительном воздействии ОВ на перчатки
их сжигают.
До начала работы всегда проверяют состояние средств защиты,
причем найденные дефекты устраняют, а при невозможности —
заменяют средства защиты новыми. Герметичность противогаза
проверяют по соответствующим инструкциям. Перчатки перед
употреблением проверяют на герметичность надуванием.
23
В подготовку к работе входит также приготовление достаточ-
ных количеств эффективных дегазирующих средств. Для удаления
отдельных капель и брызг растворов ОВ заготавливают тампоны
из целлюлозы, помещаемые на видном месте. Тампоны после упо-
требления опускают в ведро с дегазирующим раствором. Более зна-
чительные количества разбрызганных или разлитых ОВ засыпают
зерненым активированным углем, после чего эту поверхность до-
полнительно обрабатывают дегазирующим раствором.
Так как в защитной одежде и противогазе переговоры очень
затруднены, а поле зрения ограничено, план работы, во избежание
недоразумений, должен быть согласован заранее. Не рекомендует-
ся в одном и том же помещении проводить не связанные друг с дру-
гом работы. Число сотрудников, работающих в одной комнате,
должно быть не менее 2 и не более 3 или 4 человек (кроме учебных
лабораторий). По окончании работы следует убедиться в том, что
все продегазировано. Если работа будет позднее продолжена, то на
всех содержащих ОВ сосудах должны быть сделаны четкие
надписи. Для транспортирования стеклянных сосудов с ОВ внутри
лабораторных помещений пригодны ящики из пластмассы или
жести с ручками и герметичными крышками; сосуды с ОВ окру-
жены в этих ящиках слоем зерненого активированного угля.
В лабораториях, где проводится работа с ОВ, категорически
запрещается прием пищи и курение. Кроме того, желательно (пред-
полагается мирное время), чтобй работа с отравляющими веще-
ствами проводилась людьми неутомленными и находящимися в
хорошем физическом состоянии. При плохом самочувствии, воз-
никшем во время работы, о нем следует сразу доложить врачу,
который при работе с фосфорорганическими ОВ должен присутст-
вовать в лаборатории. Врач контролирует состояние здоровья со-
трудников до и после работы. Каждый работающий с ОВ должен
быть обучен оказанию мер первой помощи при поражении.
После .окончания работы следует снять сначала защитную
одежду и перчатки-и только после этого — противогаз. Перчатки
кладут в ведро со слабым раствором дегазирующего средства; за-
щитную одежду чистят и, если нужно, дегазируют. При пользова-
нии защитной одеждой во избежание перегрева и, как следствие
этого, теплового удара следует придерживаться максимально до-
пустимых сроков ее ношения, установленных в зависимости от тем-
пературы помещения:
Температура, Время работы в
воздуха, °C защитной одежде,
. мин
30 15-20
25—29 30
20—24 40-50
15—19 180 и более
При ношении противогаза следует глубоко дышать даже при
длительной работе и начинающемся утомлении. Поверхностное бы-
24
Строе дыхание в значительной степени увеличивает вредное влия-
ние мертвого пространства лицевой части маски, в котором содер-
жание двуокиси углерода повышено, а содержание кислорода —
понижено. Результатом этого может быть кризис дыхания, кончаю-
щийся срывом маски или обмороком.
2.5. ЧУВСТВИТЕЛЬНОСТЬ ОПРЕДЕЛЕНИЙ
Характеристикой эффективности реакции обнаружения, особенно
при индикации следов вещества, служат, наряду с такими факто-
рами, как, например, специфичность, также и данные по чувстви-
тельности. Эти данные очень важны для анализов ОВ, так как
они позволяют сравнивать между собой различные методы. Вместе
с тем приводимые в литературе определения понятия чувствитель-
ности весьма разноречивы и не всегда достаточно ясно сформули-
рованы. Наиболее часто под чувствительностью подразумевают ми-
нимальную концентрацию вещества (которую можно обнаружить
данным методом), выраженную в граммах, миллиграммах или
микрограммах в миллилитре или литре растворителя или же (при
определении газов и паров) в миллиграммах или микрограммах
в 1 л или 1 м3 воздуха. При обозначении в миллиграммах или
микрограммах без указания объема растворителя или воздуха
подразумевается количество ОВ, которое при данном методе ана-
лиза еще можно определить во взятом объеме пробы. Более чет-
кая характеристика понятия чувствительности приведена Файглем
в его книге по капельному анализу. Файгль ввел понятия откры-
ваемого минимума и предельной концентрации (предельного раз-
бавления).
Под открываемым минимумом понимают количество вещества
(в мкг), которое еще можно обнаружить безошибочно данным
методом. Под предельной концентрацией понимают такую мини-
мальную концентрацию (разбавление вещества), при которой еще
возможно достоверное обнаружение его в растворе. Между этими
двумя понятиями существует простая зависимость:
Предельная концентрация = 1 :
Объем раствора пробы • 106 (мл)
Открываемый минимум (мкг)
Приведенные термины можно уточнить на следующем примере:
для капельной пробы, проводимой с 1 каплей раствора (0,05 мл),
открываемый минимум равен 0,2 мкг, отсюда предельная концен-
трация будет равна:
1:-°^= 1:250 000
Если для анализа взят 1 мл испытуемого раствора, то предель-
ная концентрация составит 1:5000000 или, иначе говоря, 1 г ве-
щества можно определить в 5000 л раствора пробы. Чувствитель-
ность такого высокого порядка достигается весьма часто. Строго
25
говоря, для Полноты оценки характерной реакции необходимо в
каждом отдельном случае устанавливать открываемый минимум
и предельную концентрацию (или предельное разбавление).
На практике часто не удается получить величину открываемого
минимума соответствующей реакции несмотря на точное соблю-
дение всех условий анализа, т. е. концентраций реагентов, объема
проб, продолжительности реакции, температуры и др. Это объяс-
няется в основном двумя причинами. Во-первых, чувствительность
реакции может сильно понижаться за счет наличия в пробе испы-
туемого вещества примесей, которые не были учтены при разра-
ботке реакции обнаружения; во-вторых, возможность наблюдения
слабой окраски или небольшого осадка зависит от .внешних усло-
вий проведения реакции — освещения, выбора соответствующего
фона и т. п. Эти факторы, в условиях возможного неблагоприят-
ного освещения полевых лабораторий, следует учитывать при вы-
боре метода анализа. В некоторых литературных источниках ча-
сто данные по чувствительности обозначаются в единицах р. р. т.
(части на миллион) и р. р. Ь. (части на миллиард). Если р. р. т.
и р. р. Ь. относятся к концентрации пара или газа в воздухе, то их
можно привести к более общепринятым единицам измерения, поль-
зуясь следующей формулой пересчета:
Мр х р- р- т. , , ,
62 360Г мг л или гм -
где М — молекулярный вес вещества; д — давление, мм рт. ст.; Т — темпера-
тура, К.
При использовании р. р. т. для обозначения концентрации рас-
творов их можно безошибочно приравнять к мг/л или мкг/мл.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. На что следует обращать внимание при выборе реактивов для проведе-
ния анализов?
2. Охарактеризуйте оборудование и организацию рабочего места для про-
ведения анализов ОВ.
3. Какие возможности существуют для описания чувствительности анали-
тических методов? Дайте определение понятий открываемый минимум и предель-
ная концентрация.
4. Какая зависимость существует между открываемым минимумом и пре-
дельной концентрацией?
5. Каковы причины, обусловливающие невозможность достижения заданной
прописью чувствительности метода?
6. Что обозначает принятая в англосаксонской литературе величина р. р. т.
и как пересчитать эти данные для концентрации пара вещества в мг/л нли а/ж3?
3. элементный анализ ОВ
Химическое исследование неизвестного вещества после его очи-
стки перегонкой, перекристаллизацией или возгонкой начинается
с качественного обнаружения содержащихся в нем элементов, т. е.
с качественного элементного анализа, который далее дополняют
установлением определенных функциональных групп. В результате
этих исследований создается представление о типе данного ОВ.
Полная идентификация и подтверждение осуществляются затем
с помощью специальных химических методов индикации предпо-
лагаемого ОВ, определения физических констант, спектрального
исследования и, наконец, количественного элементного анализа ве-
щества, подвергнутого высокой степени очистки.
3.1. КАЧЕСТВЕННЫЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ
Установление элементного состава органического соединения отно-
сительно просто, так как речь идет о небольшом числе элементов.
В состав ОВ, в частности, входят такие элементы, как фосфор,
сера, азот, галогены, и некоторые металлы — мышьяк, свинец;, при-
сутствие этих элементов в определенных комбинациях указывает
на тип яда.
3.1.1. Обнаружение азота, серы и галогенов
3.1.1.1. Минерализация образца сплавлением с щелочными металлами -
последующего обнаружения азота, серы и галогенов (проба по Лассеню) Не-
сколько миллиграммов исследуемого вещества помещают в узкую пробирку или
в запаянную с одного конца стеклянную трубку длиной 10 см и диаметром
0,5 см, запаянный конец которой раздут в маленький шарик. При анализе спир-
товых растворов ОВ отбирают 0,5 мл этого раствора и, осторожно нагревая,
испаряют растворитель.
К веществу или сухому остатку добавляют равное или удвоенное количе-
ство глюкозы Затем при помощи стеклянной палочки в трубку до поверхности
вещества проталкивают кусок (величиной с горошину) чистого сухого металли-
ческого натрия (еще лучше калия) и начинают ее прогревать с открытого конца
ца микрогорелке до расплавления щелочного металла и его полного смешения с
ясследуемым веществом. Для завершения минерализации нижний конец трубки
нагревают до красного каления. Затем еще горячую трубку погружают в ста-
кан или фарфоровую чашку с 5 мл холодной воды (надевать защитные очки!).
При соприкосновении^ с водой шарик трубки разрывается. Так как и на стенках
трубки может оставаться исследуемое вещество, то ее измельчают пинцетом и
Тоже погружают в воду. Содержимое стакана или чашки нагревают до кипения
и после удаления горелкн несколько минут перемешивают. Затем остатки стекла
27
и обуглившиеся частицы отделяют фильтрованием. В фильтрате определяемые
элементы находятся в виде анионов — цианида, сульфида и галогенидов.
При таком способе минерализации можно провести предварительное обна-
ружение фосфора. Для этого нужно в момент соприкосновения испытуемого ве-
щества с щелочным металлом поднести к отверстию стеклянной трубки филь-
тровальную бумагу, пропитанную раствором AgNO3. При наличии фосфора па
бумаге появляется черное пятно, а при наличии серы — серое3.
3.1.1.2. Обнаружение азота. Обнаружение образовавшегося при
сплавлении со щелочным металлом цианид-иона осуществляется
специфической реакцией образования берлинской лазури.
Для этого к части щелочного фильтрата (в случае кислой среды добавляют
NaOH) приливают 2—3 капли 1%-ного раствора FeSO4, нагревают смесь до
кипения и после добавления 2—3 капель 10%-ного раствора FeCl3 подкисляют
НС1. В зависимости от содержания азота раствор приобретает окраску от зеле-
ной до синей или образуется синий осадок берлинской лазури (при малом со-
держании азота — только после длительного стояния). Положительный резуль-
тат этой реакции является доказательством присутствия азота.
Чувствительное обнаружение CN* возможно, если часть фильтрата после
щелочного сплавления подкислить разбавленной H2SO4, нагреть и к отверстию
пробирки поднести полосу фильтровальной бумаги, смоченной раствором о-толи-
дина и ацетата меди(Н); в результате выделения свободной синильной кислоты
бумага окрашивается в синий цвет ".
Если испытуемое вещество содержит наряду с азотом и серу,
то, проводя минерализацию пробы с недостаточным количеством
щелочного металла, можно получить тиоцианат. Ион CNS- обна-
руживают по образованию красной окраски при добавлении к под-
кисленному фильтрату раствора FeCK.
3.1.1.3. Обнаружение серы. К части щелочного фильтрата при-
бавляют несколько капель свежеприготовленного 1%-ного водного
раствора нитропруссида натрия Na2[Fe(CN)5NO]-2H2O. Красно-
фиолетовая окраска указывает на присутствие в испытуемом рас-
творе серы в виде сульфида.
При значительном содержании серы ее можно обнаружить до-
бавлением раствора ацетата свинца к подкисленному уксусной
кислотой фильтрату. При этом образуется черный осадок PbS.
Чувствительное обнаружение минимальных количеств серы мо-
жно осуществить каталитическим ускорением иод-азидной реакции:
2NaN3 + 12 —> 2NaI 4- 3N2
Обычно эта реакция идет очень медленно, но в присутствии
следов сульфида она протекает быстро и до конца, что можно об-
наружить по ослаблению или исчезновению окраски иода или по
выделению газообразного азота Добавление ацетата кадмия в
кислой среде препятствует образованию сероводорода.
Реагенты
Иод-азидный реагент — раствор 3 г азида натрия в 100 мл 0,1 н. раствора
иода; раствор устойчив.
Ацетат кадмия — 20%-ный водный раствор.
* Ионы Си2+ служат катализатором реакции. — Прим. ред.
28
К 0,5 мл щелочного фильтрата прибавляют 1 каплю раствора ацетата кад-
мия и подкисляют разбавленной уксусной кислотой. После прибавления 2 капель
иод-азидного реагента при наличии серы наблюдается выделение пузырьков
азота.
Реакция с нитропруссидом натрия пригодна для индикации
иприта.
Для этого к 2 мл спиртового раствора ОВ добавляют кусочек металличе-
ского натрия, чтобы перевести серу в сульфид-ион. После завершения реакции,
т. е. после полного растворения натрия, к раствору приливают 2 мл воды и не-
сколько капель свежеприготовленного 1%-ного раствора нитропруссида натрия.
Красно-фиолетовое окрашивание указывает на присутствие сульфид-иона и тем
самым иприта. Чувствительность реакции 0,1 мг иприта. Помехой являются
фосфорилтиохолнновые соединения, из которых металлический натрий в спир-
товом растворе выделяет тиохолин, образующий с нитропруссидом натрия ана-
логичное окрашивание.
3.1.1.4. Обнаружёние галогенов (за исключением фтора). Перед
определением галогенов из фильтрата должны быть удалены воз-
можно присутствующие цианид- и сульфид-ионы.
Для этого подкисляют HNO3 фильтрат после минерализации и выпаривают
раствор до половины первоначального объема.
Удаление цианид- н сульфид-ионов возможно также добавлением к щелоч-
ному фильтрату нескольких капель 5%-ного раствора Ni(NO3)2. Выпадающие в
осадок цианид и сульфид отделяют фильтрованием.
К азотнокислому фильтрату добавляют несколько капель раствора AgNO3.
Белый осадок указывает на присутствие хлора, желтоватый — брома и жел-
тый— иода. Если осадок полностью растворяется в растворе (NH4)2CO3, то
испытуемое соединение содержит только хлор.
Обнаружение хлора в присутствии брома и иода может быть осуществлено
путем осаждения галогенидов серебра, отделения осадка фильтрованием, про-
мывки, суспендирования в воде и прибавления к этой суспензии I мл разбав-
ленного раствора феррицианида калия и нескольких капель примерно 3°/о-ного
раствора NH4OH. В присутствии хлора осадок покрывается слоем коричневого
феррицианида серебра.
Бром обнаруживают при помощи флуоресцеиновой бумаги. При нагревании
подкисленного H2SO4 испытуемого раствора с бихроматом калия выделяется
свободный бром, в присутствии которого на индикаторной бумаге образуется
розовое окрашивание (получается тетрабромфлуоресцеин).
Проба по Бельштейну. Для установления наличия галогенов в органических
соединениях нагревают кусок медной проволоки диаметром около 1 мм, чтобы
он покрылся пленкой окиси меди. Затем погружают конец проволоки в испытуе-
мое вещество или его раствор и вносят проволоку в бесцветное пламя бунзенов-
ской горелки. В присутствии галогенов пламя горелки окрашивается летучими
галогенидами медн в синий или зеленый цвет
Некоторые не содержащие галогенов соединения азота, а так-
же серусодержащие соединения дают аналогичную реакцию. Со-
единения фтора пламя не окрашивают, так как фторид меди не
летуч.
3.1.1.5. Обнаружение фтора. Ион фтора, образовавшийся в ис-
ходном щелочном растворе, обнаруживают реакцией с красным
цирконализариновым лаком. В присутствии фтор-иона катион цир-
кония превращается в бесцветный комплексный анион [ZrF6]2- и
освобождается окрашенный в желтый цвет ализаринсульфонат
натрия.
29
Реагенты
Цирконализаринввый лак — смесь раствора 0,05 г азотнокислого циркония
в 50 мл разбавленной HCJ (1:5) с'раствором 0,05 г ализаринсульфоната натрия
в 50 мл воды.
Цирконазоарсоновая индикаторная бумагаг — пропитывают фильтроваль-
ную бумагу в течение 2—3 мин 0,025%-ным раствором п-диметиламиноазофенил-
арсоновой кислоты в смесн спирта н концентрированной НС1 (9: 1). После суш-
ки на воздухе эту светло-красную бумагу погружают на 10 мин в 0,01%-ный
раствор хлорокиси циркония в 1 н. растворе НС1, после чего она тотчас при-
обретает коричневую окраску. Затем бумагу последовательно промывают холод-
ным и горячим (55 °C) 2 и. раствором НС1 (по 5 мин), водой и? наконец, спир-
том или эфиром и сушат в вакууме.
К 0,5 мл подкисленного HNO3 фильтрата после минерализации прибавляют
несколько капель реагента. В присутствии фтора красная окраска лака исчезает,
появляется желтое окрашивание, обусловленное присутствием ализарннсульфо-
ната натрия.
Определению мешают большие количества РО^-, AsOl-,
которые реагируют аналогично. Специфичное обнаружение фтора
в присутствии этих анионов удается после его выделения в виде
фтористоводородной кислоты и идентификации последней индика-
торной цирконазоарсоновой бумагой.
Для этого часть фильтрата выпаривают досуха в свинцовом тигле. К остат-
ку прибавляют несколько капель концентрированной H?SO4, закрывают тигель
крышкой с отверстием и нагревают. К отверстию подносят стеклянную палочку
с каплей разбавленной НС1. Через 1—2 мин эту каплю опускают на коричневую
цнрконазоарсоновую бумагу. Появление красного окрашивания указывает на
наличие нона фтора.
Для обнаружения фтора в фосфорорганических соединениях,
например в зарине, зомане и ДФФ, по реакции с цирконализйри-
новым лаком нужно исходные соединения предварительно обрабо-
тать алкоголятом.
К 1—2 мл спиртового раствора пробы добавляют маленький кусочек нат-
рия, по окончании реакции несколько минут нагревают, подкисляют раствор и
проводят обнаружение, как описано выше.
Фтор в органических соединениях можно обнаружить и по
травлению стекла (см. раздел. 5.18.2.1).
3.1.2. Обнаружение фосфора и мышьяка
3-1.2.1 Минерализация пробы. В никелевый тигель помещают смесь 0,2 г
безводного Na2COs с 0.5 г Na2O2 и добавляют некоторое количество испытуе-
мого вещества или 1—2 капли его экстракта и дают ему впитаться в карбонат.
Тигель осторожно нагревают на горелке до воспламенения смеси и затем массу
плавят. После охлаждения тигель погружают в стакан с 10 мл воды и выщела-
чивают плав. В раствор переходят: фосфор в виде фосфат-нонов, мышьяк в виде
арсенат-ионов, сера в виде сульфат-ионов и ионы галогенов.
Минерализацию можно также провести по Файглю смешением пробы веще-
ства с окисью кальция и нагреванием смесн до красного каления. При этом
получают термостойкий трехзамещенный фосфат (или арсенат) кальция. Метод
применим только для малолетучих соединений.
Фосфорорганические соединения превращаются также в ортофосфаты путем
10—15 мин кипячения их с 0,25н. раствором (NH4)2S20s- Этот метод4 особенно
пригоден для обнаружения фосфора в вырезанных пятнах бумажных хромато-
грамм.
30
З.1.2.2. Обнаружение фосфора. Большие количества фосфора
осаждают в виде фосфоромолибдата аммония.
Для этого к раствору молибдата аммония приливают по каплям концентри-
рованную HNOj до полного растворения образовавшегося вначале белого
осадка молибденовой кислоты. Затем добавляют несколько капель подкислен-
ного HNO3 раствора минерализованного вещества. Желтое окрашивание, а после
слабого нагревания — образование желтого осадка указывают на присутствие
фосфора. Чувствительность этого способа может быть значительно повышена,
если к реакционной смеси добавить несколько капель насыщенного уксуснокис-
лого раствора бензидина и раствора NH«OH до щелочной среды. Синее окра-
шивание, возникающее вследствие окислительного действия гетерополисоедине-
иия, указывает на присутствие фосфора.
Очень точным и надежным способом обнаружения фосфора яв-
ляется реакция Цинцадзе5.
Реагент Цинцадзе— нагревают 50 мл концентрированной H2SO4 (ч. д. а.)
до появления белого тумана, затем прибавляют 3 г чистой порошкообразной
окиси молибдена (МоО3) и вновь нагревают 5—10 мин-до растворения окнси.
Раствор охлаждают и вливают в 50 мл воды. К еще горячей смесн прибавляют
0,15 г растертого металлического молибдена и нагревают 3—5 мин до кипения.
Восстановление можно считать законченным, если 0,2 мл 1 и. раствора КМпО4
обесцвечиваются 2,5 мл полученного раствора. Через 10—20 мин синий раствор
декантируют с осадка. Реагент сохраняется около 3 лет.
Дли проведения определения к 2 мл слабо подкисленного H2SO4 минерали-
зованного раствора испытуемого вещества прибавляют 1 мл предварительно
обесцвеченного разбавлением водой реагента Цинцадзе и нагревают смесь 20—
30 мин на кипящей водяной бане. Синее окрашивание указывает на присутствие
фосфора.
Во всех приведенных выше реакциях арсенат-ион реагирует
аналогично, поэтому при одновременном присутствии фосфат- и
арсенат-ионов последний должен быть удален либо осаждением
сероводородом из солянокислого раствора в виде сульфида, либо
восстановлен до арсенита нагреванием пробы с сернистой кис-
лотой.
Быстрое и чувствительное обнаружение фосфора во многих
гидролизующихся фосфорорганических соединениях, в том числе
и в зарине, было разработано Велчем и Вестом6.
Реагенты
Перборат натрия—1%-иый раствор в воде.
о-Дианизидинмолибдатный реагент32 — смешивают раствор 2,5 г гидрата
молибдата натрия в 15 мл воды и 5 мл концентрированной НС1 с раствором
0,125 г о-дианизидина в 2 мл уксусной кислоты. Через 12 ч смесь фильтруют.
Полученный реагент устойчив в течение 6—12 месяцев.
Для проведения реакции помещают в небольшую пробирку 2 капли свеже-
приготовленного раствора пербората натрия и несколько капель раствора испы-
туемого вещества в летучем растворителе (вода, изопропиловый спирт или че-
тыреххлористый углерод). Для удаления растворителя пробирку нагревают в
вертикальном положении. Образующийся незначительный белый осадок хорошо
прогревают в пламени горелки до его исчезновения. После этого пробирку охла-
ждают сначала на воздухе, а затем в воде и прибавляют к пробе 2 капли
о-дианизидинмолибдатиого реагента, предварительно вдвое разбавленного водой.
Образование красновато-коричневого осадка указывает на присутствие фосфора.
На проведение испытания, требуется не более 2 мин. Метод дает возможность
обнаружить 0,5 мкг зарина. Гидролизующиеся мышьяк, содержащие соединения
реагируют аналогично,
31
Другой широко применяемый способ обнаружения фосфора в
фосфорорганических соединениях основан на восстановлении этих
соединений алюмогидридом лития до фосфинов различной степени
замещения 7 и обнаружении их по реакции с хлорной ртутью.
Реагенты
Алюмогидрид лития — непосредственно перед употреблением встряхивают
50 мг порошкообразного алюмогидрида лития в 2 мл абсолютного эфира.
Индикаторная бумага — подучают погружением фильтровальной бумаги в
5%-ный раствор HgCl2 в 95%-ном спирте. Бумагу применяют слегка влажной.
Раствор алюмогидрида лития помещают в пробирку, добавляют 1 мл рас-
твора испытуемого соединения в эфире или дибутилфталате н тотчас закрывают
п резиновой пробкой с двумя отверстиями. Через одно отвер-
стие вставлена доходящая до дна пробирки изогнутая под
углом стеклянная трубка, а через другое — прямая сужаю-
щаяся книзу стеклянная короткая трубка диаметром 6 мм
и длиной 140 мм (рис. 1), в которой находится полоса инди-
каторной бумаги шириной 4 мм и длиной 150 мм. Смеси
дают постоять примерно 5 мин, после чего очень осторожно
при помощи резиновой груши, присоединяемой к отводу изо-
гнутой стеклянной трубки, вытесняют образовавшийся фос-
х фин из пробирки, причем часть эфира испаряется. При нали-
[[ чии фосфора образуемое с HgCl2 соединение дает на бумаге
£-4 желтое окрашивание. Если бумагу на короткое время по-
ВтГ местить в 10%-ный раствор КТ, то она приобретает оранже-
вую окраску. Эта обработка делает определение более чув-
ствительным.
Мышьяксодержащие соединения реагируют ана-
логично; образовавшиеся арсины окрашивают инди-
каторную бумагу в цвета от желтого до коричневого,
при обработке раствором KI окраска становится
г;) светло- или темно-коричневой. При не слишком
низких концентрациях идентифицируемых соедине-
ние. 1. Прибор ний разная окраска индикаторной бумаги дает воз-
для восстало- можность хорошо различить фосфор- и мышьяк-
вления при ПО- содержащие соединения,
мощи алюмо- тт
гидрида лития. Чувствительность реакции для отдельных соеди-
нений различна и варьирует от 1 до 100 мкг/мл.
В этих условиях содержащаяся в иприте сера не восстанавливается
до H2S, который также мешает определению.
3.1.2.3. Обнаружение мышьяка. Обнаружение мышьяка прово-
дится после минерализации образца при помощи пробы по Гут-
цейту (см. стр. 97).
3.1.3. Элементный микроанализ
Описание систематического хода микроанализа и, особенно, обна-
ружения элементов в ОВ приведено в работах Видмарка8 и Бен-
нета с сотр.9. Видмарк минерализовал пробы при помощи щелоч-
ных металлов, а Беннет и ертр. — по способу Эмиха, используя
окись кальция и цинк. Для этих анализов достаточно несколь-
ких миллиграммов испытуемого вещества, Такие количества ОВ
32
получают путем адсорбции их пропусканием отравленного воздуха
через трубки, заполненные активированным углем и силикагелем,
и последующей экстракцией. Обнаруживать гетероэлементы (Р, N,
S, As и др.) в растворах, полученных после обработки щелочным
металлом, можно с большой чувствительностью, используя также
метод зонной плавки 10.
3.1.4. Оценка результатов качественного элементного анализа
Чтобы установить по данным качественного элементного анализа
возможное ОВ, удобно полученные результаты записывать в виде
таблицы (табл. 1).
Таблица 1. Сопоставление результатов элементного
качественного анализа
Возможное ОВ
Зарин, зоман, ДФФ и другие заме-
щенные фторфосфаты
Табун, параоксон (Е600)
Амитон, фосфорилтиохолин, пара-
тион (Е 605)
Систокс, изосистокс
Иприт
Азотистый ипрнт, фосгенокенм, хлор-
пикрин. о-хлорбензилиденмалоноди-
нитрил
Люизит, метил-, этил-и фенилдихлор-
арсин, дифенилхлорарсин (кларк I)
Дифеиилцианарсин (кларк II)
Адамсит
Фторацетат, фторэТанол
Хлорацетофенон, 2,4-дихлорфеноксн-
уксусная кислота (2,4-D),
2,4,5-трнхлорфеноксиуксусная кис-
лота (2,4,5-Т)
Бромбензилцианид
Бромацетон, ксилилбромнд
Алкалоиды, LSD
3.2. КОЛИЧЕСТВЕННЫЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ*
В отравляющих веществах гетероэлементы определяют преимуще-
ственно после окислительного разложения этих соединений. В ре-
зультате разложения фосфор превращается в РО?-, сера — в
* С микрометодами анализа органических соединений можно познакомиться
в книге: Климова В. А., Основные микрометоды анализа органических со-
единений. М., «Химия», 1967. — Прим. ред.
2 Зак. 677
33
SOl-, азот — в CN~ или NH3 и галогены — в соответствующие
ионы, обычно определяемые методами количественного неоргани-
ческого анализа. При определении содержания углерода и водоро-
да в молекулах ОВ, содержащих фосфор и фтор, следует обра-
тить внимание на некоторые особенности методов, отличающие
их от обычных методов элементного анализа. Вообще, элементный
анализ имеет смысл только тогда, когда исследуемый образец яв-
ляется индивидуальным веществом высокой степени чистоты. Для
очистки пользуются многократной перегонкой или перекристалли-
зацией образца, например, до достижения постоянной температуры
плавления. При точном соблюдении методики количественный эле-
ментный анализ является надежным методом установления иден-
тичности соединения.
3.2.1. Определение углерода и водорода
Принцип определения заключается в том, что навеску испытуемо-
го вещества окисляют кислородом в трубке для сожжения, в ко-
торую помещен платиновый катализатор, окись меди и хромат
свинца. Продукты окисления — двуокись углерода и вода погло-
щаются количественно в поглотительных трубках, что устанавли-
вают взвешиванием их до и после сожжения. Присутствие азота,
серы, хлора и брома в соединениях не мешает определению, так
как обычно в трубку для сожжения помещают универсальный
наполнитель. Так, образовавшаяся окись азота восстанавливается
в слое раскаленной меди, галогениды связываются серебряной ва-
той, а сера — серебряной ватой и хроматом свинца. При сожжении
фторорганических соединений пользуются наполнителем, содержа-
щим также окись магния11’12 или сурик (РЬ3О4), нанесенный на
пемзу13, который нагревают до 550 °C. В качестве катализатора
сожжения при определении углерода и водорода в фторсодержа-
щих органических соединениях хорошо зарекомендовал себя про-
дукт разложения перманганата серебра 13, получающийся при на-
гревании этого соединения при 100—120 °C и последующего двух-
часового нагревания при 550 °C. Этот продукт может быть также
использован в качестве катализатора сожжения и фосфороргани-
ческих соединений14. Кроме оказываемого каталитического дей-
ствия он связывает образующиеся при сожжении и мешающие
определению окисли фосфора, серы и галогены, включая фтор.
Изящный способ одновременного .определения углерода, водо-
рода и фтора разработан Мазором 15. Вместо обычного наполни-
теля в гильзу для сожжения помещают две платиновые звездочки
и трубку из спекшейся окиси алюминия, внутренняя поверхность
которой покрыта суриком. Нагретый до 550°C слой сурика спо-
собствует полному окислению углерода и водорода, фтор связы-
вается во фторид свинца PbF2. По окончании сожжения слой су-
рика вынимают и определяют в нем фтор в виде фторхлорида
свинца.
34
При определении углерода и водорода в органических соеди-
нениях, содержащих фосфор и другие элементы, исследуемый об-
разец перемешивают 12’16 в лодочке для сожжения с окисью воль-
фрама WO3, а в качестве окислителя помещают в трубку для
сожжения слой окиси кобальта Со3О4. Мешающие определению
окислы фосфора поглощаются слоем пемзы, покрытой серебром 17.
При определении кислорода в соединениях, содержащих фос-
фор и фтор, образец смешивают с порошкообразным никелем и
хлоридом серебра 18. Пиролиз можно проводить в никелевой
трубке при 900 °C над катализатором — платиной на газовой саже.
В качестве инертного газа через систему пропускают смесь, со-
стоящую из-98% N2 и 2% Н2. Фторсодержащие продукты после
сожжения поглощают в трубке, наполненной аскаритом (едкий
натр на асбесте).
3.2.2. Способы минерализации для последующего
определения Р, S, N, F, С1 и As
Из большого числа способов, пригодных для минерализации орга-
нических соединений, здесь будут описаны только некоторые са-
мые простые и быстро осуществляемые, что особенно важно для
анализа ОВ. Наиболее упо-
требляемым и современным яв-
ляется метод Шёнигера 19~20,
развитый в дальнейшем други-
ми авторами.
Согласно методу сожжение про-
водят в колбе, наполненной кислоро-
дом. На рис. 2 показано устройство
колбы Шёнигера. Она представляет со-
бой колбу Эрленмейера емкостью 300
или 500 мл, закрываемую пришлифо-
ванной пробкой, в оттянутый конец
которой впаяна платиновая сетка
или спираль. Навеску вещества по-
мещают на специально вырезанный
кусок беззольной фильтровальной
бумаги (рис. 3), который затем скла-
дывают в соответствии с пунктир-
ными линиями. Для анализа жидких
веществ ими пропитывают кусок
фильтровального картона или берут
навеску в небольших желатиновых
Рис. 2. Кислород-
ная колба Шёни-
гера.
Рис. 3. Филь-
тровальная бу-
мага для сож-
жения по Шё-
нигеру.
или полиэтиленовых капсулах, которые заворачивают в фильтровальную бумагу
и прикрепляют к платиновой сетке или спирали. В колбу вливают примерно
10 мл используемого в определении поглощающего раствора и в течение не-
скольких секунд пропускают кислород для вытеснения воздуха. Узким пламенем
бунзеновской горелки поджигают свободный конец фильтровальной бумаги и
быстро вставляют пробку в колбу. Вследствие возникающего избыточного дав-
ления пробку следует сильно прижимать рукой (работа в защитных очках обя-
зательна!). Во время протекающего очень быстро сожжения (примерно 20 сек)
колбу держат наклонно, чтобы могли сгореть опадающие на сухие стенки
колбы кусочки фильтровальной бумаги. Для полного поглощения продуктов
2*
35
Рис. 4. Универ-
сальная бомба
Вуртцшмитта.
сгорания колбу оставляют на 10—15 мин, периодически ее встряхивая. За-
тем на края горла колбы наливают некоторое количество воды и вынимают
пробку. Образовавшееся вследствие поглощения продуктов сгорания разреже-
ние способствует всасыванию воды в колбу; при этом обмывается пробка и
ее шлиф. В полученном растворе соответствующими методами определяют на-
ходящиеся в виде анионов фосфор, серу, галогены или
мышьяк. Для жидкостей, кипящих’ниже ТОО °C, этот спо-
соб минерализации непригоден.
Примерно для тех же целей, что и описанный
выше способ, применима и минерализация в уни-
версальной бомбе Вуртцшмитта21, устройство
которой понятно из рис. 4.
Навеску испытуемого вещества перемешивают в бомбе
с перекисью натрия; в качестве горючего в бомбу добав-
ляют некоторое количество этиленгликоля. При нагрева-
нии закрытой бомбы смесь загорается уже при 56 °C; раз-
виваемая при сгорании высокая температура приводит к
полной минерализации вещества в течение 1 мин.
Для минерализации органических соединений
очень часто используют разлагающее действие
металлического калия. При этом сера количе-
ственно превращается в сульфид калия, а галоге-
ны — в соответствующие галогениды калия.
Для микроопределений пригодно разложение по Циммерману22, осущест-
вляемое в запаянной стеклянной трубке длиной 8 см и диаметром 6 мм (рис. 5).
Металлический калий отделен от навески вещества стеклянной палочкой. Трубку
нагревают в горизонтальном положении до расплавления калия и дают ему
стечь на вещество. Минерализа-
ция заканчивается в течение не-
скольких минут. Определение
можно проводить с несколькими
миллиграммами вещества.
Для определения фосфо-
ра, мышьяка и азота в орга-
нических соединениях при-
годны также способы мине-
рализации различными жид-
кими окисляющими смесями.
Наряду с такими давно из-
вестными окислителями, как
смеси серной кислоты и пере-
киси водорода, серной и
азотной кислот, серной кислоты и перманганата калия и другими,
очень эффективна смесь серной и хлорной кислот23 и хлорная кис-
лота в момент ее выделения из перхлората аммония действием
смесью азотной и соляной кислот24. Чаще всего минерализация
сводится к тому, что исследуемый образец нагревают в открытой
колбе Кьельдаля с окисляющей смесью. Применение прибора, пред-
ложенного Бетге25, в котором окисляющая смесь конденсируется
в обратном холодильнике, имеет то преимущество, что в нем воз-
Калий
Рис. 5. Трубка для минерализации органи-
ческих соединений металлическим калием
по Циммерману22 (верхняя трубка предназ-
начена ' для разложения жидкостей, ниж-
няя— для твердых веществ).
36
можно разложение и летучих компонентов, кроме того, расход окис-
ляющей смеси невелик.
Для сожжения азотсодержащих соединений пригоден метод
Кьельдаля, при котором образуется аммиак. Предложено много
различных минерализующих смесей. Наиболее действенной явля-
ется смесь концентрированной серной кислоты, сульфата калия,
окиси ртути и селена 26-27, с помощью которой минерализация за-
нимает примерно 20 мин. Способ Кьельдаля с успехом применяется
для веществ, содержащих аминогруппы. Весьма медленно минера-
лизуются гетероциклы; вещества, содержащие нитрогруппы, коли-
чественно разлагаются только после предварительного восстанов-
ления в бомбе иодистым водородом28 или цинком с соляной кис-
лотой 29.
Из специальных методов минерализации фторорганических со-
единений следует упомянуть универсальной кварцевый прибор для
сожжения Викболда30-31, в котором сожжение идет в токе грему-
чего газа. Работа с этим прибором или со значительно более де-
шевой колбой Шёнигера в присутствии кислорода имеет то пре-
имущество, что минерализованный раствор содержит незначитель-
ные количества посторонних ионов, присутствие которых может
понижать точность определения интересующих элементов.
3.2.3. Определение фосфора
При сожжении фосфорорганических соединений в колбе Шёни-
гера при недостаточном количестве окислителя на платиновой сет-
ке могут оставаться частички углерода, содержащие фосфор, что
приводит к искажению результатов анализа. Чтобы избежать это-
го, рекомендуется работать в колбе Эрленмейера емкостью 500 мл
и добавлять к навеске исследуемого вещества 10—20 мг персуль-
фата аммония33. Так как обычная платиновая сетка быстро раз-
рушается фосфором, то лучше пользоваться спиралью из платино-
вой проволоки34. Поглотительным раствором обычно служит смесь
из 10 мл воды и 2 мл серной или азотной кислоты, часто с добав-
кой перекиси водорода. После полного поглощения продуктов
сожжения и смывания со стенок колбы минерализованного рас-
твора его кипятят не менее 10 мин для гидролиза образовавшихся
пиро- и метафосфатов.
Для определения фосфора имеется большое число весовых, объ-
емных и фотометрических методов. При одновременном присут-
ствии в соединении и фтора для полумикроопределения удобно
осаждать фосфор в виде хинолинмолибденофосфата и завершать
определение титрованием растворенного осадка 35-36-74. Для мик-
роопределения целесообразно пользоваться фотометрическим ме-
тодом, основанным на образовании фосфорномолибденовой кис-
лоты и ее восстановлении в молибденовую синь72. Очень быстрым
и простым методом 37 является экстракция желтой фосфорномолиб-
деновой кислоты амилацетатом’ и ее фотометрическое определение
37
при 400 или 430 нм. Если требуется более высокая чувствитель-
ность, например при навеске вещества менее 10 мг, то, прибавляя
ванадат аммония 33’38’73, получают желтый фосфатованадатомолиб-
датный комплекс, содержание которого определяют фотометриче-
ски. В присутствии фтора фосфат может быть также определен
комплексометрическим титрованием 34’39. При этом в соответствии
с прописью 34-39 к раствору при pH 10 добавляют избыток MgCl2,
который затем обратно оттитровывают комплексоном III в присут-
ствии метилтимолового синего в качестве индикатора. Сера, хлор
и бром определению фосфора этим полумикрометодом не пре-
пятствуют.
Определение фосфора во фторсодержащих соединениях в виде
хинолинмолибденофосфата производят следующим образом 36.
Реагенты
Молибдат натрия — раствор 15 г молибдата натрия в 100 мл воды (приго-
товлять и хранить раствор следует в полиэтиленовой посуде).
Гидрохлорид хинолина — раствор 20 мл перегнанного хинолина в смеси
800 мл горячей воды и 25 мл концентрированной НС1. Полученный раствор
охлаждают, фильтруют и доводят объем до 1 л.
Минерализованный раствор, содержащий приблизительно 2—3 мг фосфора,
подкисляют НС1 и кипятят несколько минут для удаления СО2. После упарива-
ния раствора до 10 мл прибавляют 0,5 г бориой кислоты, фильтруют и промы-
вают фильтр примерно 20 мл горячей воды. К фильтрату прибавляют 5 мл
профильтрованного раствора молибдата натрия и 5 мл концентрированной НС1.
Раствор нагревают до кипения и для осаждения добавляют к нему при встря-
хивании, по каплям, сначала медленно, а затем быстро 5 мл раствора гидро-
хлорида хииолииа. Для образования осадка смесь нагревают 15 мин на водя-
ной баие, охлаждают и декантируют через фритту с толщиной фильтрующего
слоя примерно 0,5 см. Осадок дважды промывают декантацией, приливая каж-
дый раз по 4 мл разбавленной НС1 (1:9), затем холодной водой переносят
его на фильтр. Промывку холодной водой продолжают до исчезновения следов
НС1 в фильтрате. Затем осадок и фильтрующий слой количественно переносят
в колбу Эрленмейера, разбавляют 20 мл воды и при встряхивании растворяют
в 10 мл 0,5 н. раствора NaOH. Прибавляют 3 капли смешанного индикатора
(смесь 0,1%-ных спиртовых растворов фенолфталеина и тимолового синего в
соотношении 2:3) и оттитровывают избыток щелочи 0,5 и. раствором НС1 до
изменения окраски от фиолетовой через светло-зеленую до бледно-желтой; 1 мл
0,5 н. раствора NaOH соответствует 0,5958 мг фосфора.
Определение можно также завершать весовым способом путем
взвешивания осадка 76,77, состав которого отвечает следующей фор-
муле
(C9H7N)3-H3PO4- 12МоО2
Осаждение фосфора в виде хинолинмолибденофосфата может
быть использовано также для определения фосфора в серусодер-
жащих соединениях75.
3.2.4. Определение серы
При определении серы путем сожжения испытуемого вещества
по Шёнигеру в качестве поглощающей жидкости в колбу нали-
вают 4 мл воды и 0,2—0,5 мл Н2О2. При больших навесках ис-
пытуемого вещества и значительном содержании серы образовав-
38
шуюся при минерализации серную кислоту определяют весовым
способом — осаждением в виде сульфата бария при помощи BaClj.
Повышая содержание НС1 в осаждающем растворе примерно до
1%, можно уменьшить помехи, вызываемые присутствием фос-
фата 39.
Весьма чувствительным является объемное определение суль-
фата по Фритцу и Ямамура 40, которое было разработано Вагне-
ром 41 в качестве микрометода. Титрование проводят перхлоратом
бария в присутствии торйна — натриевой соли о-[(2-окси-3,6-
дисульфо-1-нафтил)-азо]-фениларсоновой кислоты. Катионы, ме-
шающие определению, удаляют перед титрованием пропусканием
раствора через катионообменную колонну, фосфат — кипячением с
карбонатом магния40 или прибавлением окиси серебра к получен-
ному кислому раствору перед обработкой на катионообменнике 42.
Для микроопределения серы пользуются следующей методи-
кой 41.
Реагенты
Торин — 0,2%-ный водный раствор.
Метиленовый синий — 0,0125%-ный водный раствор.
Перхлорат бария — 0,02 н. раствор. Растворяют 3,3627 г Ва(С1О4)2 в 200 мл
воды, доводят объем раствора изопропиловым спиртом примерно до 1 л и до-
бавлением НСЮ4 устанавливают в растворе pH 2,5—4. После этого изопропи-
ловым спиртом доводят объем точно до 1 л и титруют аликвотную часть рас-
твора 0,01 н. раствором H2SO4; 1 мл 0,02 н. раствора Ва(С1О4)2 соответствует
0,6413 мг серы.
После минерализации и поглощения продуктов сожжения на края горла
колбы наливают несколько капель изопропилового спирта (всего на промывку
расходуют 16 мл), вынимают пробку и обмывают спиртом платиновую сетку и
стенки колбы. Разбавленной НСЮ4 доводят кислотность раствора до pH 2,5—4,
что проверяют по индикаторной бумаге. После прибавления 1 капли 0,2%-ного
водного раствора торйна и 1 капли 0,0125%-ного водного раствора метиленового
синего титруют 0,02 н. раствором Ва(С1О4)г До изменения окраски от светло-
зеленой до розовой.
Таким способом можно одновременно определять серу и хлор43,
но при этом не нужно прибавлять метиленовый синий.
Реагенты
Дихлорфлуоресцеин — О,\а1о-кый раствор в изопропиловом спирте.
Перхлорат серебра — 0,01 и. раствор. Растворяют 1,378 е Ag2CO3 (или
1,159 г AgO) в эквимольном количестве НС1О4 и доводят водой объем до 50 мл,
быстро нагревают до кипения и после охлаждения разбавляют изопропиловым
спиртом до 1 л. Титр раствора устанавливают по хлористому натрию.
После определения серы титрованием Ва(С1О4)г прибавляют 2—3 капли рас-
твора дихлорфлуоресцеина, 0,1 н. раствор NaOH до светло-розовой окраски и
титруют 0,01 н. раствором AgC104 до перехода окраски в фиолетово-розовую.
Серу в фосфорорганических соединениях можно определять
сожжением по ГТреглю на платиновом катализаторе44 при 930°C.
Окись фосфора поглощается слоем окиси цинка, а образовавшаяся
трехокись серы — раствором перекиси водорода, после чего обра-
зовавшийся сульфат определяют титрованием раствором Ва(С1О4) 2
39
3.2.5. Определение азота
Образующийся при минерализации по Кьельдалю аммиак отгоняют
и улавливают в колбе с определенным объемом титрованной кис-
лоты, после чего содержание его определяют ацидиметрически
(обратным титрованием избытка кислоты) или колориметриче-
ски — реагентом Несслера.
При малом содержании азота его можно иногда определять не-
посредственно в колбе для минерализации путем окисления гипо-
хлоритом и последующим взаимодействием с фенолом с образова-
нием индофеноловых красителей 45’ 46. Вместо гипохлорита можно
применять хлорамин Т, при этом получается хлорамин NH2C1, кото-
рый при действии пиридин-пиразолонового реагента дает окрашен-
ную рубеановую кислоту 47.
При действии гипобромита на аммониевые соединения выде-
ляется азот:
2NHJ + 4ОВг" —► N2 4-4ВГ +4Н2О
Избыток гипобромита может быть определен иодометрически.
Этот метод может быть использован для определения азота после
минерализации48.
Реагенты
Раствор гипобромита —к раствору 16 г NaOH в 40 мл воды приливают
4 мл брома и доводят водой объем до 1000 мл.
Йодистый калий— 10%-иый водный раствор.
Тиосульфат натрия — 0,1 и. раствор.
Навеску вещества ~0,1 г, содержащую 2—10 мг азота, минерализуют в
колбе Кьельдаля емкостью 250 мл смесью, состоящей из 20 мл концентрирован-
ной H2SO4, 7 г K2SO4 и 0,1 г окиси ртути. После охлаждения реакционную
смесь нейтрализуют в присутствии метилового красного 10 н. раствором NaOH
и затем для создания избытка щелочи добавляют еще 2 капли щелочи. Прили-
вают 75 мл 0,1 н. раствора гипобромита и через 5 мин — 20 мл 10%-ного рас-
твора KI и 50 мл 2 н. раствора H2SO4. Выделившийся иод титруют 0,1 н. рас-
твором ИагЗгОз в присутствии крахмала в качестве индикатора. Параллельно
проводят контрольную пробу с 75 мл гипобромита.
Если применявшаяся для минерализации смесь содержала се-
лен, то после разбавления раствора до 500 мл и прибавления 10 мл
5%-ной H2SO4 минерализованный раствор нагревают до обесцве-
чивания, фильтруют и после нейтрализации NaOH анализируют,
как указано выше.
3.2.6. Определение фтора
Количественная минерализация фторорганических соединений до-
статочно трудна из-за прочности связи С—F. Для всех фтороргани-
ческих соединений удобным является сожжение в пламени грему-
чего газа по Викболду30. При сожжении в колбе Шёнигера реко-
мендуется добавлять к навеске испытуемого вещества перекись
натрия, способствующую сожжению. Для поглощения продуктов
сгорания в колбу наливают 10 мл воды и 1 мл 2 н. раствора
NaOH.
40
Фторорганические соединения можно количественно разложить,
нагревая их с металлическим калием в запаянных трубках49. Для
разложения 3—4 мг вещества требуется 50—60 мг калия. Мине-
рализация заканчивается в течение 3 мин. Чтобы избежать дей-
ствия фтора на некоторые сорта стекла, трубки для сожжения
предварительно кипятят с соляной кислотой.
Минерализовать фторорганические соединения удобно также
в бомбе Вуртцшмитта. Применение различных жидких окисли-
тельных смесей не всегда приводит к количественному разложению.
Известно большое число весовых, объемных и колориметриче-
ских методов определения фторид-ионов в растворах после минера-
лизации. В большинстве способов нужно предварительно отделять
мешающие определению, сопутствующие ионы, особенно фосфат-
ионы, образующиеся при минерализации фторфосфорных соедине-
ний. Фосфат-ионы не мешают определению фтор-иона при осажде-
нии его в виде хлорфторида или бромфторида свинца, количество
которых можно определять как весовыми50, так и объемными спо-
собами. Вследствие сравнительно большой растворимости PbCIF
точность весового определения мала. При объемном анализе можно
определять в осадке свинец51 или хлор 52~54, а в растворе — избы-
точные ионы свинца55. Более трудоемким способом определения
фтора в присутствии фосфатов является осаждение его при pH
4,0—4,5 в виде фторида кальция 56, добавленный избыток кальция
определяют комплексометрически. Для микро- и полумикроопреде-
лений наиболее удобен метод с применением нитрата тория, который
будет описан ниже, а также объемное определение57 хлоридом це-
рия (III). Однако в этих способах приходится отделять фтор от фос-
фат-ионов. Основным способом отделения фтора является отгонка
фтористого водорода с паром из кислотного раствора, впервые опи-
санная Виллардом и Винтером 58. .К перегоняемой смеси для свя-
зывания галоидоводородных кислот добавляют сульфат серебра,
сероводород связывают серебряной спиралью, находящейся в ло-
вушке, помещенной по пути следования пара. Разработаны различ-
ные типы перегонных аппаратов, из которых прибор Питцка и
Эрлиха59 имеет то преимущество, что при работе с ним не нужно
наблюдать за процессом отгонки, так как требуемая для образова-
ния пара вода находится в цикле. Отгонку ведут из раствора, со-
держащего серную и хлорную кислоты. Кроме того, к смеси добав-
ляют немного битого стекла, для того чтобы фтор отгонялся в виде
кремнефтористоводородной кислоты. Последняя поглощается в
приемнике со слабым раствором карбоната натрия.
Разработанный также Виллардом и Винтером58 метод опреде-
ления фторидов титрованием нитратом тория многократно подвер-
гался видоизменениям:
4NaF + Th(NO3)4 —> ThF4 + 4NaNO3
В то время как в ранее рекомендованных буферных системах
реакция протекала нестехиометрически, вследствие чего требовалось
41
прибегать к построению калибровочной кривой, Капицу и Шол-
леру49 удалось, используя буферную смесь глицин — перхлорат
натрия — хлорная кислота, достигнуть строгого стехиометрического
течения реакции. В качестве индикаторов для установления конца
титрования нитратом тория пригодны смешанный индикатор Баллс-
цо60, состоящий из ализаринсульфоната натрия, метиленового си-
него или водяного голубого 49, и предложенный Шмидтом иОртлоф-
фом 61 смешанный индикатор, содержащий ксиленол оранжевый и
Чикаго синий.
Определение фтора после минерализации вещества нагрева-
нием с калием осуществляется следующим образом 49.
Реагенты
Нитрат тория — 0,02н. раствор (титр устанавливают по фториду натрия).
Ализариновый красный S — 0,05%-иый водный раствор.
Буферная смесь pH 3,3 — смесь 6.7 г глицина (гликокола), 11 г NaC104,
И мл I н. раствора НС1О4 разбавляют водой до 100 мл.
Разбивают трубку, в которой проводилась минерализация, и переносят
осколки и содержимое в колбу Эрленмепера емкостью 100 мл. Прибавлением
2 мл метанола разлагают избыток калия, затем отдельными порциями добав-
ляют 5 мл воды. Раствор фильтруют через волокнистый фильтр, который че-
тыре раза промывают кипящей водой (каждый раз по 2,5 мл). После охлажде-
ния к нему добавляют 0,5 мл раствора ализаринового красного S и нейтрали-
зуют 10%-ной хлорной кислотой до перехода красной окраски в желтую. Затем
добавляют 2 мл буферной смеси и по каплям 0.01 %-пый раствор метиленового
синего, водяного голубого или чпкаго синего до появления зеленого окрашива-
ния. Титрование проводят 0.02 н. раствором нитрата тория до перехода окраски
в красно-фиолетовую.
После минерализации соединений, содержащих кроме фтора
также фосфор, фтор следует отделять перегонкой. Если фториды
пятикоординированного фосфора подвергаются обработке этилатом
натрия с образованием фторида натрия62-63
R(Ok ,О Р(О)Х ,О
+ NaOC2H5 —> 'Р^ + NaF
ROZ XF ROZ ^ОСПН-,
то для определения фтора предварительная его отгонка не нужна.
Образующиеся сложные диэфиры не препятствуют определению
фтора титрованием нитратом тория.
Для проведения определения навеску фторфосфата (пли фторфосфоната),
содержащую 1 —10 мг фтора, обрабатывают 10 мл раствора 1 г металлического
натрия в 20 мл этанола, не содержащего альдегида. Реакция обычно заканчи-
вается в течение 5—15 мин. Однако некоторые вещества следует предваритель-
но растворять в 25 мл этанола и после добавления 1 г металлического натрия
кипятить с обратным холодильником 15 мин. Очень медленно реагирующие со-
единения предварительно растворяют в н-гексаполе п после прибавления натрия
также кипятят с обратным холодильником. Титрование выделившегося в резуль-
тате алкоголиза фтор-иона нитратом тория проводят по Хоскинсу и Феррису64
или методом, описанным Каинцем и Шоллером 49.
Так как фторфосфаты в применяемой для титрования буферной
смеси (pH 3) не гидролизуются, то ионный фтор из соответствую-
щего HF и гидролизуемый фтор дифторидов могут быть определе-
42
ны прямым титрованием нитратом тория в присутствии монофтор-
фосфатов. Моноалкилфосфаты, образующиеся при гидролизе легко
гидролизуемых алкилдихлорфосфатов, мешают титрованию. Раз-
личие между ионом фтора из HF и фтор-ионом, полученным при
гидролизе алкилдифторфосфатов, может быть установлено коли-
чественно двумя ацидиметрическими титрованиями—едким нат-
ром па холоду и раствором трибутиламина в этаноле с примене-
нием индикатора бромкрезолового зеленого62.
3.2.7. Определение хлора, брома и иода
При сожжении по методу Шёнигера в присутствии кислорода для
поглощения продуктов сгорания наливают в колбу 10 мл воды
и 1 мл 1 н. раствора КОН. Хлор определяют по Фольгарду или
ацидиметрически устанавливают количество гидроксильных групп,
эквивалентное содержанию галогенида, выделяющегося при доба-
влении к нейтральному раствору пробы нейтрального раствора
основного цианида ртути65. Можно пользоваться также меркуро-
метрическим титрованием с дифени.ткарбазоном в качестве индика-
тора66’78 или аргентометрическим титрованием с индикатором ди-
хлорфлуоресцеином67. Бром определяют аргентометрически по
Фольгарду или иодометрически по Кольтгоффу68. Определение
иодидов производится очень точным методом Лейперта69, в кото-
ром иодид окисляют бромом в иодат и после прибавления йоди-
стого калия выделяющийся иод титруют раствором тиосульфата
натрия. Для микроопределения хлора и брома в органических сое-
динениях удобной является минерализация калием в запаянных
трубках с последующим титрованием по Фольгарду70.
3.2.8. Определение мышьяка
Наряду с различными мокрыми способами минерализации, приме-
нение которых для определения мышьяка в ОВ описано в разделе
5.4.2, удобным является также сожжение в колбе Шёнигера71. Так
как платина с мышьяком образует сплав, то для прикрепления на-
вески вещества, завернутой в фильтровальную бумагу, к пробке
колбы припаивают кварцевую спираль. Продукты сгорания погло-
щаются 10 мл 0,1 н. раствора иода, в котором образовавшийся
трехвалентный мышьяк окисляется в пятивалентный. В дальней-
шем проводят фотометрическое определение молибденовой сини,
образующейся в результате восстановления мышьякмолибденовой
кислоты.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какие продукты образуются при минерализации органических соединений
щелочным металлом?
2. Какие продукты образуются при минерализации органических соединений
окислительным сплавлением с перекисью натрия?
43
3. Какие сочетания гетероэлементов, галогенов и металлов, обнаруживаемые
качественным элементным анализом, характерны для отравляющих веществ?
4. Какие способы минерализации употребимы в количественном элементном
анализе?
5. Как определяют фтор в фторфосфатах и фторфосфонатах?
ЛИТЕРАТУРА
1. Fei gl F., Z. anal. Chem., 74, 369 (1928).
2. F e i g 1 F., R a j m a п E., Mikrochem., 12, 133 (1932).
3. Ketcham R., L о w - В e e г A., J. Chem. Educ., 38, 414 (1961).
4. GetzM. E„ J. Assoc. Off. Agric. Chem., 47, 1103 (1964).
5. Zinzadze S. R., Z. Pflanzenernahr. Diing. Bodenkunde, A16, 129 (1930).
6. Welch C. M„ West P. W„ Anal. Chem., 29, 874 (1957).
7. Eggert sen F. T., Weiss F. T., Anal. Chem., 29, 453 (1957).
8. Widmark G., Akta Chem. Scand., 7, 1395 (1953).
9. В e n n e 11 E. L. et al., Anal. Chem., 19, 1035 (1947).
10. M e i s e 1 T. et al., Mikrochim. Acta, 1961, 874.
11. I n gr a m G., Analyst, 86, 539 (1961).
12. Campbell A. D., McDonald A. M. G., Anal. Chim. Acta, 26, 275 (1962).
13. Horacek J., Korb 1 J., Chem. Listy, 51, 2132 (1957).
14. L у s s у j I., Z а г e m b о J. E., Microchem. J., 2, 245 (1958).
15. Ma z or L., Mikrochim. Acta, 1957, 113.
16. G a w a r g io u s Y. A., McDonald A. M. G., Anal. Chim. Acta, 27, 842
(1962).
17. Binkowski J., Vecera M., Mikrochim. ichnoanalyt. Acta, 1965, 842.
18. E h г e n b e r g e r F. et al., Z. anal. Chem., 198, 242 (1963).
19. Schoniger W., Mikrochim. Acta, 1955, 123.
20. Schoniger W., Mikrochim. Acta, 1956, 869.
21. Wurtz schmitt B., Mikrochem. verein. Mikrochim. Acta, 36/37, 769 (1951).
22. Zimmermann W., Mikrochem. verein. Mikrochim. Acta, 31, 15 (1943).
23. Diehl H„ Smith G. F„ Taianta, 2, 209 (1959).
24. Smith G. F„ Taianta, 11, 633 (1964).
25. Bethge P. O„ Anal. Chim. Acta, 10, 317 (1954).
26. Milbauer J., Z. anal. Chem., Ill, 397 (1938).
27. Kala H., Pharmazie, 18, 29 (1963).
28. Friedrich A. et al., Z. physiol. Chem., 216, 68 (1933).
29. Fish V. B„ Anal. Chem., 24, 760 (1952).
30. Wickbold R„ Angew. Chem., 64, 133 (1952); 66, 173 (1954).
31. E h r e n b e r g e r F., Mikrochim. Acta, 1959, 192.
32. Robinson J. W., West P. W„ Microchem. J., 1., 93 (1957).
33. D i г s c h e r 1 A., Erne F., Mikrochim. Acta, 1960, 755.
34. В e n n e w i t z R., Mikrochim. Acta, 1963, 1094.
35. В e 1 ch er R„ Me D о n a 1 d A. M. G., Taianta, 1, 185 (1958).
36. Fennell T. R. et al., Analyst, 82, 639 (1957).
37. Kirsten W. J., Carlsson M. E., Mikrochem. J., 4, 3 (1960).
38. De ba 1 E., Chim. Anal., 45, 66 (1963).
39. Kramer N., Mikrochim. Acta, 1965, 144.
40. Fritz J. S., Yamamura S. S., Anal. Chem., 27, 1461 (1955).
41. W a gne r H., Mikrochim. Acta, 1957, 19.
42. С о 1 s о n A. F., Analyst, 88, 26 (1963).
43. Giesselmann G., Hagedorn I., Mikrochim. Acta, 1960, 390.
44. Corliss J. M„ Rhodes E. J. W„ Anal. Chem., 36, 394 (1964).
45. В d 11 c h e г C. F. J, et al., Rec. trav. chim., 80, 1157 (1960).
46. Stegemann H. et al., Hoppe-Seylers Z. physiol. Chem., 329, 241 (1962),
47. Kala H., Pharmazie, 18, 29 (1963).
48. Hashmi M. H. et al., Anal. Chem., 34, 988 (1962).
49. К a i n z G., S c h 6 11 e r F., Mikrochim. Acta, 1956, 843.
50. Chapman N. B. et al., Analyst, 73, 434 (1948).
44
51. L a s z 1 о v s z к у I., Magyar Kem. Fol., 60, 209 (1954).
52. E r d e у L. et al., Mikrochim. Acta, 1958, 432.
53. Belcher R., Brewer P. F., Anal. Chim. Acta, 8, 235 (1953).
54. В о g n a r I., Nagy L„ Magyar Kem. Fol., 65, 335 (1959).
55. V t e s t a 1 I. et al., Coll. Czech. Chem. Conim, 23, 886 (1958).
56. В e 1 c h e r R., С1 а г к S. I., Anal Chim. Acta, 8, 222 (1953),
57. Brunisholz G., Michod J., Helv. Chim. Acta, 37, 598 (1954).
58. Willard H. H., Winter О. B, Ind. Eng. Chem., Anal. Ed., 5, 7 (1933).
59. P i e t z к a G., Ehrlich P., Angew. Chem., 65, 131 (1953).
60. В a 11 c z о H., Kaufmann O., Mikrochem. verein. Mikrochim. Acta, 38, 237
(1951).
61. Schmidt J„ Ort loff R„ Z. Chem, 5, 236 (1965).
62. Sass S. et al. Anal. Chem, 31, 1970 (1959).
63. S a 1 s b u г у J. M. et a 1, Ana 1. Chem, 23, 603 (1951).
64. Hoskins W. M, Ferris G. A, Ind. Eng. Chem, Anal. Ed, 8, 6 (1936).
65. V i e b б с к F, Ber, 65, 248 (1932).
66. P r a e g e r K, F fl r s t H, Chem. Techn, 10, 537 (1958).
67. Wagner H, Bflhler F, Mikrochem. verein. Mikrochim. Acta, 36/37, 641
(1951).
68. К о 11 h о f f I. M, Ind. Eng. Chem, Anal. Ed, 9, 75 (1937).
69. L e i p e r t T, Biochem. Z, 261,436 (1933).
70. К a i n z G, R e s c h A, Mikrochemie, 39, 1 (1952).
71. Merz W, Mikrochim. Acta, 1959, 640.
72. Merz W, Mikrochim. Acta, 1959, 546.
73. M a T. S, M с К i n 1 e у A. 14. G, Mikrochim. Acta, 1953, 4.
74. W i 1 s о n H. H, Analyst, 76, 65 (1951).
75. McDonald A. M. G, Stephen W. I, J. Chem. Educ, 39, 528 (1962).
76. Perrin С. H, J. Assoc. Off. Agric. Chem, 41, 758 (1958),
77. F e n n e 11 T. R. F. W, W e b b J. R, Taianta, 1959, 105.
78. White D. C, Mikrochim. Acta, 1962, 807.
4. ОПРЕДЕЛЕНИЕ НЕКОТОРЫХ
ФУНКЦИОНАЛЬНЫХ ГРУПП
Наличие определенных группировок атомов в молекуле служит
основой классификации органических соединений. Поэтому иден-
тификация функциональных групп методами качественного органи-
ческого анализа является важным вспомогательным средством
установления структуры неизвестного вещества. В этом разделе
будут описаны методы обнаружения некоторых важных функцио-
нальных групп, часто встречающихся в отравляющих веществах.
При использовании этих методов следует помнить, что реакционная
способность функциональных групп зависит от структуры всей
молекулы, вследствие чего методы обнаружения одинаковых функ-
циональных групп в разных соединениях могут протекать иначе.
Это может выражаться в разных скоростях и чувствительности
реакций. Большая часть методов обнаружения ОВ, описанных в
гл. 5, основана на идентификации функциональных групп, харак-
терных для соединений определенной структуры.
4.1. ОБНАРУЖЕНИЕ ГРУПП О-МЕТИЛ И N-МЕТИЛ
Для обнаружения этих групп исследуемое вещество окисляют до
формальдегида нагреванием с перекисью бензоила, присутствие ко-
торого обнаруживают по реакции с хромотроповой кислотой
В пробирку, обрезанную на половину своей высоты, помещают некоторое
количество испытуемого вещества или несколько капель его раствора в эфире
или петролейном эфире, прибавляют 1—2 капли 10%-ного раствора перекиси
бензоила в бензоле и испаряют растворитель. При помощи резинового кольца в
пробирку вставляют микропробирку так, чтобы ее дно на 2 см не доходило до
дна большой пробирки. Микропробирка наполнена водой, а дно ее с внешней
стороны смочено свежеприготовленным раствором 10 мг хромотроповой кислоты
в 2—3 мл концентрированной H2SO4. Собранный прибор опускают в глицерино-
вую баню с температурой 120 °C. При положительной реакции капля, свисающая
со дна микропробирки, через несколько минут окрашивается в фиолетовый цвет.
Производные альдегидов — алифатические оксимы — при нагре-
вании с перекисью бензоила до 120—130 °C образуют азотистую
кислоту, которую можно обнаружить реагентом Грисса8.
4.2. ОБНАРУЖЕНИЕ ГРУППЫ N-ЭТИЛ
Группа N-этил при нагревании с перекисью бензоила окисляется
и образуется уксусный альдегид, который с пиперидином (или мор-
фолином) и нитропруссидом натрия дает синее окрашивание
46
Окисление проводят так же, как и в случае обнаружения группы
N-метил (ср. раздел 4.1).
Накрывают отверстие пробирки куском фильтровальной бумаги, пропитан-
ной приготовленной непосредственно перед употреблением смесью равных объе-
мов 20%-иого раствора морфолина и 5%-ного раствора нитропруссида натрия.
Синее пятно указывает на присутствие группы N-этил.
4.3. ОБНАРУЖЕНИЕ ГРУППЫ О-ЭТИЛ
Для окисления испытуемого вещества в уксусный альдегид его на-
гревают с кислым раствором бихромата калия 1>3.
В микропробирку вносят некоторое количество вещества или несколько ка-
пель его раствора в эфире или в петролейном эфире и испаряют на водяной
бане растворитель. Затем прибавляют несколько капель раствора 1 г КгСггО?
в 6 мл воды и 7,5 мл концентрированной H2SO4. Отверстие пробирки прикры-
вают индикаторной бумагой (см. раздел 4.2) и нагревают реакционную смесь
в кипящей водяной бане. Синее окрашивание указывает на присутствие соеди-
нений, содержащих группу О-этил. Соединения, содержащие группу N-этил,
можно отделить встряхиванием эфирного раствора пробы с разбавленной НС1.
4.4. ОБНАРУЖЕНИЕ ГРУППЫ О-ИЗОПРОПИЛ
Изопропанол реагирует с «-диметиламинобензальдегидом в при-
сутствии концентрированной H2SO4 с образованием красителя 2.
К 1 мл раствора испытуемого вещества в дихлорэтане или воде осторожно
приливают концентрированную H2SO4, содержащую 1 % п-диметиламнноб?нзаль-
дегида. Красное кольцо на границе раздела фаз указывает на наличие О-изо-
пропильиой группы. Реакция весьма чувствительна. Проведение контрольной
пробы с применяемыми реактивами и растворителем обязательно.
4.5. ОБНАРУЖЕНИЕ ГРУППЫ О-ПИНАКОЛИЛ
Реагентом служит раствор ванилина в серной кислоте (0,1 г вани-
лина в 20 мл концентрированной H2SO4).
Смешивают 2 мл этого реагента с 3—4 каплями водного раствора испытуе-
мого вещества, прибавляют по каплям воду, каждый раз перемешивая, и наблю-
дают изменение окраски. В присутствии группы О-пинаколил появляется красно-
коричневая до красно-фиолетовой окраска; О-изопропильная группа дает четки
различимую сине-фиолетовую окраску.
4.6. ОБНАРУЖЕНИЕ ФЕНИЛЬНОЙ ГРУППЫ
Большое число ароматических соединений со свободным «-положе-
нием или с гидроксильной группой в «-положении реагирует с ра-
створом формальдегида в концентрированной H2SO4 с образова-
нием окрашенных «-хиноидных соединений 3. Следует иметь в виду,
что некоторые органические соединения дают окрашенные продукты
уже с концентрированной H2SO4. Поэтому целесообразно всегда
проводить параллельный опыт с добавлением только серной кис-
лоты. Если при этом будет появляться окрашивание, то смотрят,
меняется ли его цвет от прибавления формальдегида или его смеси
47
с серной кислотой. Этот контрольный опыт дает возможность более
достоверно доказать наличие ароматического соединения.
Реагент
Формальдегид-сернокислотный реагент — смесь 0,2 мл 37%-ного формальде-
гида с 10 мл концентрированной H2SO4. Срок годности реагента 1—2 суток.
Для проведения реакции смешивают небольшое количество испытуемого
вещества или каплю его раствора в неводном растворителе с каплей реагента и
прн необходимости слабо нагревают. Для анализа летучих веществ целесообраз-
но пользоваться простым устройством, описанным в разделе 4.1 для обнаруже-
ния группы О-метил. Пробу вещества испаряют в большой пробирке, погружен-
ной в водяную баню, а дно мнкропробиркн смачивают формальдегид-сернокис-
лотным реагентом. При этом появляется преимущественно красная окраска, но
может появиться также и желтая, зеленая, синяя, коричневая или черная. Для
проведения реакции достаточно микрограммовых количеств исследуемого ве-
щества.
Ароматические соединения образуют с хлороформом в присут-
ствии А1С13 окрашенные соединения (реакция Фриделя—Крафтса).
Эту реакцию можно использовать для обнаружения ароматиче-
ских соединений в следующем исполнении4.
Нагревают в пробирке на сильном пламени 100 мг А1С13 до сублимации его
на стенке. Пробирку охлаждают и вливают по стенке раствор 10—20 мг испы-
туемого вещества в 5—8 каплях хлороформа. При соприкосновении раствора
с сублимированным А1С13 появляется желто-оранжевая до красной окраска. Не-
ароматические соединения, содержащие бром нли иод, дают в этих условиях
желтое или фиолетовое окрашивание.
4.7. ОБНАРУЖЕНИЕ СЛОЖНЫХ ЭФИРОВ
КАРБОНОВЫХ КИСЛОТ
Сложные эфиры карбо'новых кислот реагируют с гидроксиламином
в присутствии щелочи с образованием гидроксаматов щелочных
металлов:
R—+ NaOH + NH2OH —► R—+ R'OH + Н2О
\)R' ^NHONa
Образующаяся при подкислении гидроксамовая кислота дает с
FeCh окрашенную в фиолетовый цвет внутрикомплексную соль:
,О...Fe/3
R—С/ + >/3Fe3+ —> R—| + Н+
NHOH XNH—О
Ангидриды карбоновых кислот и их галогенангидриды реаги-
руют непосредственно с гидроксиламином, образуя гидроксамовые
кислоты. Следует иметь в виду, что хлорное железо с фенолом так-
же дает окрашенные продукты, поэтому рекомендуется предвари-
тельно проводить пробу с FeCl3, чтобы убедиться в отсутствии в
пробе вещества подобных соединений.
Для проведения реакции смешивают 1 мл спиртового или эфирного рас-
твора испытуемого вещества с 5—6 каплями насыщенного спиртового раствора
48
гидрохлорида гидроксиламина и 1—2 каплями насыщенного спиртового раствора
КОН и слегка нагревают. Затем добавляют 5—6 капель 0,5 и. НС1 и 1 каплю
1%-ного водного раствора FeCU. Красно-фиолетовое окрашивание свидетель-
ствует о наличии сложного эфира карбоновой кислоты.
4.8. ОБНАРУЖЕНИЕ ТРЕТИЧНЫХ АМИНОВ
Третичные алифатические и ароматические амины дают при нагре-
вании с 2%-ным раствором лимонной кислоты в уксусном ангидри-
де окрашенный продукт5.
Смешивают 1 мл спиртового раствора испытуемого вещества с 5 каплями
2%-ного раствора лимонной кислоты и нагревают на кипящей водяной бане;
одновременно проводят контрольную пробу с 1 мл этанола. Желто-зеленая или
красная окраска свидетельствует о наличии третичного амина.
Реакция весьма чувствительна и в модифицированном виде10’’1
пригодна также для обнаружения и идентификации некоторых вы-
сокотоксичных соединений, содержащих третичные аминогруппы,
например для идентификации алкалоидов — стрихнина, бруцина,
атропина, морфина, а также LSD.
Менее чувствительной является видоизмененная проба Хин-
сберга 6 с бензолсульфохлоридом.
Растворяют Г00 мг испытуемого вещества в 1—2 мл пиридина, прибавляют
0,5 мл 10%-ного NaOH и встряхивают; затем прибавляют 1 каплю беизолсульфо-
хлорида и смесь снова встряхивают. В присутствии первичных аминов появ-
ляются окраски от бледно- до светло-желтой, в присутствии вторичных — корич-
неватая до темно-коричневой, в присутствии третичных — от розовой до глу-
бокой пурпурно-красной.
При нагревании аминов с раствором хлоранила (тетрахлор-п-
бензохинон) в эпихлоргидрине (1-хлор-2,3-эпоксипропа'н) или то-
луоле образуются различные окраски, а именно: первичные амины
дают красную окраску, вторичные — пурпурную, третичные — сма-
рагдово-зеленую 7.
4.9. ОБНАРУЖЕНИЕ ЭТИЛЕНОВОЙ ГРУППЫ (-СН2—СН2-)
При нагревании соединений, содержащих этиленовые группы, с без-
водным ZnCh образуется уксусный альдегид, который обнаружи-
вают по посинению смеси нитропруссида натрия с морфолином 8.
Немного испытуемого вещества нагревают до 250 °C в пробирке со взятым
иа кончике шпателя небольшим количеством безводного ZnCl2. Отверстие про-
бирки прикрывают куском фильтровальной бумаги, пропитанной реагентом. По-
явление синего окрашивания свидетельствует о наличии соединения с одной или
несколькими этиленовыми группами.
4.10. ОЦЕНКА СПОСОБОВ ОБНАРУЖЕНИЯ
ФУНКЦИОНАЛЬНЫХ ГРУПП
В табл. 2 указано, в каких ОВ (или инсектицидах) имеются функ-
циональные группы, обнаруживаемые описанными выше методами.
49
Таблица 2. Функциональные группы, встречающиеся в ряде ОВ
и инсектицидов (звездочкой отмечены инсектициды)
Функциональная группа Вещество
О-Метил N-Метнл Различные фосфорорганические соединения М,М-Диметнламидо-О-этилцианфосфат (табун), N-метил-М,М-бис-(2-хлорэтил)-амии, метнлфтор-
О-Этил фосфорилхолии и фосфорилтиохолин Табун, тетрам *, параоксон *, паратион *, систокс
N-Этил О-Изопропил О-Пинаколил Фенильная группа и изосистокс, диэтоксифосфорилтиохолин N-Этил-М,И-бис-(2-хлорэтил)-амин, тетрам * ДФФ, зарин, изопропоксиметилфосфорилтиохолин Зоман (втор-О-неогексилметилфторфосфэнат) Хлорацетофенон, бромбеизилцианид, фенилдихлор- арсии, днфенилхлорарсин, дифеиилцианарсин,
Эфир карбоновой кислоты некоторые гербициды Эфир бромуксусной кислоты, дифосген (эфир
Третичная аминогруппа угольной кислоты) Азотистый иприт, табун, фосфорилтиохолины, тет- рам * и аналогичные соединения, алкалоиды (атропин, стрихнин, бруцин, вератрин, никотин
Этиленовая группа а ДР’/ Сериый и азотистый иприты, тетрам * и аналогич- ные соединения
4.11. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
ФУНКЦИОНАЛЬНЫХ ГРУПП
Количественное определение алкоксигрупп проводится по методу
Цейзеля. Действием иодистоводородной кислоты их превращают в
алкилиодиды, которые, реагируя с избыточным количеством брома,
образуют йодноватую кислоту:
RI + Вг2 —> RBr + IBr
1Вг + ЗН2О + 2Вг2 —> НЮ3 + 5НВг
При взаимодействии йодноватой кислоты с KI выделяется свобод-
ный иод, количество которого определяют титрованием раствором
NajS^Oj; избыточный бром предварительно связывают муравьиной
кислотой. Этот метод в модификации Рекендорфера 9 применим для
определения метоксильных и этоксильных групп в эфирах фосфор-
ной кислоты.
Серу в эфирах тиофосфорной кислоты окисляют бромом до суль-
фат-иона, после чего она определению не мешает.
КОНТРОЛЬНЫЕ ВОПРОСЫ
Назовите функциональные группы, обычно встречающиеся в молекулах бое-
вых отравляющих веществ, и отравляющие вещества, для которых эти группы
характерны.
50
ЛИТЕРАТУРА
1. Файгль Ф. Капельный анализ органических веществ. Пер. с англ., под
ред. В. И. Кузнецова. М., Госхимиздат, 1962.
2. Au terh of f H„ Pharmaz. Zhalle Dtschld., 89, 300 (1950).
3. LeRosen A. L?. et al., Anal. Chem., 24, 1335 (1952).
4. C h e г о n i s N. D., E n t г i k i n J. B., Semimicro Qualitative Organic Analysis,
New York, Interscience Publ. Corp., 1957.
5. Okhuma S. J., Pharm. Soc. Japan, 75, 1124 (1955).
6. Lacy et al., Proc. Lousiana Acad. Sciences, 18, 94 (1955).
7. Sivadjian J., Bull. Soc. Chim. France, 2, 623 (1935).
8. Feigl F„ Silva E„ Analyst, 82, 582 (1957).
9. Rekcendorfer P., Pflanzenschutzber., 5, 287 (1950).
10. Groth A. B., Wallenberg G., Acta Chem. Scand., 20, 2628 (1966).
11. Groth A. B., Dahlen M. E., Acta Chem. Scand., 21, 291 (1967).
5. ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА ОВ
Эта глава посвящена различным химическим методам анализа, при
помощи которых можно обнаруживать и количественно определять
содержание отравляющих веществ в различных материалах. Мето-
ды эти различаются по эффективности (специфичность, чувстви-
тельность, точность, область применения) и рассчитаны на химиков-
аналитиков разной квалификации и на лаборатории с различным
оборудованием.
Описания методов индикации и количественных определений
ОВ будут изложены в этой книге в основном в том порядке, кото-
рый соответствует практике работы полевых лабораторий. Такая
система изложения материала отличается от системы изложения,
использованной в части 1 этой книги. Однако она целесообразна не
только потому, что так принято работать в полевых лабораториях,
но и из других более общих соображений. Например, в соответ-
ствии с систематикой книги1 анализ кожно-нарывных и раздра-
жающих мышьякорганических ОВ пришлось бы излагать раздель-
но, что сказалось бы на возможности общего обозрения методов.
5.1. ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Принадлежность к этой группе соединений наиболее токсичных ОВ
и многих инсектицидов является причиной постоянных поисков все
более чувствительных методов их индикации и анализа. Это находит
отражение и в обширной литературе, посвященной этим вопросам.
5.1.1. Методы обнаружения
5.1.1.1. Реакция Шёнеманиа. Эта до сих пор широко приме-
няемая селективная реакция обнаружения некоторых производных
кислот пятивалентного фосфора, была открыта Шёнеманном1 в
1944 г. при работах с отравляющими веществами типа трилонов.
Она основана на том, что при реакции галогенангидридов метилфос-
фоновых кислот с перекисью водорода в щелочной среде образу-
ются промежуточные гидроперекисные соединения, которые быстро
окисляют некоторые ароматические амины в окрашенные продукты.
По исследованиям Ларсона 2, это превращение, по аналогии с
щелочным гидролизом^объясняется нуклеофильной атакой атома
фосфора ионом гидроперекиси, Образование надкислоты было до-
52
казано Гехауфом 3 путем окисления с помощью такой реакционной
смеси тиосульфата в сульфат, а также Хильгетагом, который выде-
лил эфир надфосфорной кислоты. Дальнейшее течение реакции в
отсутствие окисляющегося амина трактуется по-разному:
R(Ok л
4-НОО' —>
R07 \Х
R(Ok .О
+Н2О2 —>
ro7 ЮО~
R(Ok Л
+ н+ + X"
RO7 \ОО’
R(Ok X).
;р^ + о2 + н2о
R0-7 XT
(О
(2)
Гехауф3 и Эпштейн4 считают последнюю реакцию бимолеку-
лярной и определяющей скорость всего процесса. Однако если бы
разложение надфосфорной (или надфосфоновой) кислоты явля-
лось в основном бимолекулярной реакцией, то мольное соотноше-
ние между разлагающейся перекисью водорода и галогенангидри-
дом алкилфосфоновой кислоты, независимо от концентрации пере-
киси водорода, должно было бы равняться 2, а скорость образова-
ния кислорода должна была быть пропорциональной концентрации
перекиси водорода. Такой вывод не отвечает экспериментальным
данным, вследствие чего Ларсон допускает, что между надфосфор-
ной (или надфосфоновой) кислотой и еще не прореагировавшим
галогенангидридом происходит конкурирующая реакция:
R(OR X) R(OK .О R(Ok
/РС + /Р\ +он" —► 2 /р\ +1/гО2 + Н+ + Х’
RO-7 4)0' . R0-7 \Х R0-7 ХГ
Акснес5, наоборот, трактует разложение галогенангидрида как
мономолекулярную реакцию, определяющую скорость всего про-
цесса, которая осуществляется возможно под влиянием раствори-
теля или стеклянной поверхности сосуда, а окисление перекиси во-
дорода — как быструю реакцию «активного» кислорода с перекисью
водорода. Наблюдающееся уменьшение расхода перекиси водоро-
да при уменьшении ее концентрации он объясняет превращением
«активного» кислорода в молекулярный:
R(Ok X) R(Ok хО
+ О
RO-7 N)0' rcZ XT
О Н2О2 —► Н20 02
0 4-0 —► 02
Установлено, что реакция галогенангидрида алкилметилфосфо-
новой кислоты с перекисью водорода протекает в 50 раз быстрее,
чем его щелочный гидролиз. Этот факт можно объяснить проме-
жуточным образованием водородной связи между ионом гидро-
перекиси и связью Р=.О, что облегчает диссоциацию связи Р—X.
53
Эту разницу в скорости легко проследить по данным Марша и
Нила6, представленным в табл. 3 для зарина (О-изопропилметил-
фторфосфоната).
Таблица 3. Стойкость зарина при различных значениях pH
в отсутствие и в присутствии Н2О2
Буферная смесь pH Время полураспада при 25 °C, ч
без Н,О, 0,1% Н,Оа
НС1—КС1 в растворе вода — ацетон (1:1) 1,2 1,7 1,7
Водный раствор
ацетата 5 6,7 6,3
бикарбоната 7,4 8,0 1,4
фосфатов * 8,4 1,4 0,2
Фосфатный буфер * в растворе вода — аце- 8,4 2,5 0,5
тон (1:1)
То же 11,0 6,5 1,8 сек
* Состав фосфатного буфера: 0,5 моль К3РО4 + 0,01 моль КН2РО4.
Из данных табл. 3 видно, что перекись водорода в кислой среде
инертна.
Аналогично изменению гидролитической устойчивости фосфор-
органических соединений изменяется и скорость реакций этих со-
единений с ионом гидроперекиси. Это можно установить по приве-
денным Маршем и Нилом 6 для трех разных фосфорорганических
соединений константам скоростей второго порядка, измеренным при
25 °C (в л-моль'1 -мин-1):
О-Изопропилметилфторфосфоиат (зарин)................0,94 • 105
Тетраэтилпирофосфат (ТЭПФ) .......................... 2-10’
Диэтил-4-нитрофенилфосфат (параоксон).............. 14
На основании этих данных можно считать, что чувствительность
реакции в случае параоксона невелика, что и соответствует дей-
ствительности. Вместе с тем, для соединений с низкой гидролитиче-
ской устойчивостью или при проведении реакции в более щелочной
среде (выше pH 12) гидролиз становится конкурирующей реакцией.
Так, например, легко гидролизуемые дигалогенангидриды кислот
пятивалентного фосфора или фторфосфорилхолины нельзя обнару-
жить по реакции Шёнеманиа. И, наоборот, — для медленно гидро-
лизующихся фосфорилтиохолинов приходится принимать опреде-
ленные меры для ускорения реакции.
Короче говоря, для достижения максимальной чувствительности
реакции нужно создавать условия, соответствующие свойствам
испытуемого фосфорорганического соединения. Для подавления
гидролиза можно снизить pH, но при этом будет увеличиваться
время развития максимальной окраски. Увеличение продолжитель-
ности реакции, особенно для таких очень медленно реагирующих
веществ, как ДФФ, можно отчасти компенсировать увеличением
54
концентрации перекиси водорода, однако не на столько, чтобы это
могло вызвать заметное окисление амина. Количество добавляемо-
го к реакционной смеси амина должно составлять 4 моль на I моль
фосфорорганического соединения.
Рассмотрим теперь вторую часть реакции Шёнеманна — окисле-
ние амина. О кинетике этой реакции в литературе нет каких-либо
сведений; встречаются различные суждения по поводу образующих-
ся продуктов реакции. Так, Марш и Нил6 установили, что спектры
поглощения продуктов.окисления о-дианизидина, полученных как в
реакции Шёнеманна, так и при взаимодействии со щелочным рас-
твором феррицианида калия или с перманганатом калия, идентич-
ны, но вместе с тем некоторые свойства этих окрашенных соеди-
нений различны, например экстрагируемость, изменение окраски
при подкислении. Авторы считают, что образовавшиеся окрашенные
соединения представляют собой комплексные соединения хинонди-
имина и непрореагировавшего амина (реакция 2). Образование хи-
нондиимина IV объясняется предварительным образованием се-
михинонимина II за счет отдачи одного протона амина-основания
фосфорсодержащему ион-радикалу I (реакция 1):
R(Ok хО R(Ok ,0
+ H2N—R—R—NH2 —► + H2N—R—R—NH’ (1)
RQ/ ^О" R(y Х)Н
Неустойчивый в щелочном растворе ион-радикал семихиноними-
на II образует, вследствие утраты второго протона и передачи его
другому фосфорсодержащему ион-радикалу, хинондиимин IV или
же происходит диспропорционирование двух молекул семихинон-
имина, в результате чего может получиться диимин и исходный
амин.
Появление окрашенных веществ объясняют также образованием
азокрасителей:
R(0)x ,0
;Р^ + 2HjN—R—R—NH2 —►
RO/ ХЮ"
R(Ok Ю
—»- )p^ + H2N—R—R—N=N—R—R—NH2 + 2H2O
RQ/ 4'
55
Установлено3, что спектр поглощения продуктов окисления бенз-
идина очень похож на спектр диаминоазобензола, но не идентичен
ему. Акснес и Зандберг7 окисляли бензидин и о-дианизидин над-
уксусной кислотой и исследовали полученные продукты окисления.
Продуктом окисления бензидина оказался 4-амино-4'-нитродифе-
нил. Из о-дианизидина было получено три продукта окисления:
3,3'-диметокси-4-амино-4'-нитродифенил (желтое вещество, ХмаКс
370 нм)-, 3,3'-диметокси-4-амино-4'-нитрозодифенил VI (коричневое
вещество, 7.Макс 445 нм)-, бис-(3,3'-диметокси-4-амино)-азобифе-
нил VII (красное вещество, Хмакс 435 нм).
Вещество VII, возможно, является промежуточным соединением,
получающимся из нитрозосоединения VI и о-дианизидина:
Наряду с названными ароматическими диаминами — бензиди-
ном и о-дианизидином для выполнения колориметрической реакции
Шёнеманна можно применять и о-толидин. В качестве окислителя
в простейшем случае используют щелочной раствор перекиси водо-
рода. Более устойчивыми являются такие перекисные соединения,
как перборат натрия, перекись пирофосфата натрия и комплексное
соединение перекиси водорода с мочевиной, при применении кото-
рых вследствие их щелочного характера не требуется водить буфер-
ные смеси.
Для стационарных лабораторий в качестве растворителей при-
годны ацетон и изопропиловый спирт; для работы в полевых лабо-
раториях был рекомендован 1,6-гександиол8. При осуществлении
реакций следует помнить, что последовательность и способ смеше-
ния реагентов, естественно, оказывают значительное влияние на
чувствительность реакций. Лучшие результаты при качественных
реакциях достигаются путем приливания по каплям раствора испы-
туемого вещества к свежеприготовленной смеси реагентов. В слу-
чае прибавления реагентов к раствору испытуемого вещества нуж-
но придерживаться следующего порядка: в первую очередь доба-
вляют амин, затем окислитель и в конце — буферную смесь. Ни в
коем случае нельзя прибавлять окислитель до прибавления амина.
Это может привести к значительной потере чувствительности, что
следует из уравнения (2), приведенного на стр. 53.
56
Повышение чувствительности достигается в том. случае, если
вместо названных аминов используют индол9, окисление которого
при pH 9 приводит к образованию сначала сине-зеленого сильно
флуоресцирующего индоксила, затем белого индиго и, наконец,
синего индиго. Пероксисоединения вызывают окисление до инд-
оксила; дальнейшее окисление протекает легко за счет кислорода
остаточной перекиси.
ИНДОЛ
+ Ог
-2Н2О
белое индиго
индиго
При этой модификации реакции Шёнеманиа наряду с измере-
нием флуоресценции определяют также интенсивность синей окрас-
ки индиго, например, при помощи индикаторной трубки, используе-
мой для индикации нервно-паралитических ОВ, которой оснащены
приборы 10 химической разведки американской армии М9 и М18.
Для стабилизации флуоресцирующих продуктов рекомендуется
прибавлять некоторое количество ацетона или глицерина. Присут-
ствие индоксила можно также обнаружить, добавляя к реакцион-
ной смеси изатин и наблюдая за образованием окрашенного инди-
рубина:
изатин
кетоформа
нидоксила
иидирубнн
Такой способ получения индирубина описан в литературе.
В последнее время была подробно исследована еще одна моди-
фикация реакции Шёнеманиа, Метод заключается в измерении
57
сине-зеленой хемолюминесценции, наблюдаемой при окислении лю-
минола (гидразид 3-аминофталевой килоты) п>12’16. Хемолюмине-
сценция вызывается следующим окислительным процессом:
Исследованиями, проведенными одной югославской исследова-
тельской группой, было установлено, что в случае зарина хемолю-
минесценция имеет два максимума, из которых первый, кратковре-
менный, обусловлен соединением, образующимся по приведенной
выше реакции, а второй, более продолжительный, вызван окисле-
нием люминола выделяющимся кислородом (см. реакцию 2, стр. 53).
Добавление галогенидов щелочных металлов повышает чувстви-
тельность метода, по-видимому, за счет промежуточного образо-
вания гипогалогенидов 17-19. Чувствительность модификации, прово-
димой с люминолом (около 0,5 мкг!мл), правда, меньше, чем в
методе, основанном на наблюдении флуоресценции, однако эта
реакция может быть использована в автоматическом приборе хими-
ческой разведки при небольшом расходе энергии. Применяя хемо-
люминесцентный индикатор люцигенин (динитрат N.N'-диметил-
диакридиния), можно с большой чувствительностью обнаружи-
вать20 табун (0,1 мкг/мл). Измерение слабой хемолюминесценции
для количественного определения ОВ возможно с помощью вторич-
ного электронного умножителя.
Несмотря на то, что такой способ до сих пор еще не нашел зна-
чительного практического применения, заслуживает внимания рабо-
та одной канадской исследовательской группы 13~15, которая в по-
исках соответствующего ред-окс-индикатора для реакции Шёнеман-
на обнаружила группу соединений производных триарилметана
типа
обладающих высоким коэффициентом экстинкции и нужным значе-
нием ред-окс-потенциала (от —0,7 до —1,0 в). Наиболее подходя-
щим оказалось соединение, у которого R=R/=CH3, a R"=H, а
именно: 4,5-диметил-бис- (4',4"-М,М-диметиланилино) -тенилиден.
В отличие от условий проведения реакции Шёнеманна окисление
58
этих индикаторов должно осуществляться в кислой среде в присут-
ствии хлор-иона. Полагают, что индикатор окисляется хлорновати-
стой кислотой, которая образуется в результате реакции надкис-
лоты с хлор-ионом в кислой среде (реакции 1 и 2):
R(Ok Ю R(Ok Ю
+ н+ + сг —> + нею (1)
RCr \00‘ RCr \Q'
Реакцию проводят следующим образом: дают фосфорорганиче-
скому соединению реагировать около 30 сек с щелочным раствором
перекиси (pH 10,4), после чего прибавляют ряствор лейкосоедине-
ния индикатора в разбавленной H2SO4, содержащей NaCl. При
значении pH 1,6 окрашивание появляется уже при содержании за-
рина 0,1 MKZjMA.
Табун не дает этой реакции, возможно, вследствие восстановле-
ния ионов надкислоты ионами цианида в щелочном растворе.
Реакция Шёнеманиа является групповой реакцией и проходит
не только с соответствующими фосфорорганическими соединениями,
но также и с другими веществами, образующими с Н2О2 перекис-
ные соединения. Так, альдегиды, ангидриды и галогенангидриды
кислот, а также галогенангидриды арилсульфокислот реагируют
аналогично. Ее можно использовать для определения таких ве-
ществ, как, например, ацилхолины или дифосген 167. Последние мо-
гут быть применены в качестве модельных веществ. Марш и Нил 6
применяли для построения калибровочных кривых мало токсичный
л-хлорбензолсульфофторид; бензолсульфохлорид является хорошим
имитатором зарина.
Подобные соединения мешают обнаружению или определению
фосфорорганических соединений. Особенно опасно присутствие
альдегидов в спиртах и ацетоне, применяемых в качестве раство-
рителей. Перед использованием таких растворителей их следует
очищать от альдегида специальными методами.
59
Мешают определению также и некоторые катионы, например
медь, железо, марганец, встречающиеся иногда в воде. Катионы
следует связывать добавлением комплексообразователей, напри-
мер гексаметафосфата натрия. Вредное влияние дегазирующих ве-
ществ, являющихся сильными окислителями или восстановителями,
следует устранять предварительной обработкой .исследуемого образ-
ца, специфичной для каждого конкретного случая.
Реакция Шёнеманна находит применение в индикаторных труб-
ках на зарин, имеющихся в американских детекторах нервно-пара-
литических ядов (детекторы Е21, В17, В25 и В38), в газосигнали-
заторе ГСП-1 и в различных методах индикации и определения в
носимых и передвижных полевых лабораториях. Работами Лоса и
сотр. применение реакции Шёнеманна распространено и для инди-
кации и определения инсектицидов типа сложных эфиров фосфор-
ной .кислоты, в том числе систокса (меркаптофос), дихлофоса,
хлорофоса (диптерекс) и фосфамида (диметоат) 322’323.
Мы ограничимся описанием двух методов обнаружения, однако
естественно, что любые другие методы количественного определе-
ния (см. раздел 5.1.2.1) могут быть использованы также и для
индикации.
I. Цветная реакция’
Реагенты
Реагент I — 2,5%-ный ацетоновый раствор бензидина, перекристаллизован-
ного из бензола.
Реагент II — 0,25%-иый водный раствор пербората натрия (срок хране-
ния— не больше суток).
К смеси 0,5 мл реагента I и 2 мл реагента II прибавляют 2 мл раствора
испытуемого вещества в изопропиловом спирте или воде. При наличии фосфор-
органических ОВ возникает желтая окраска, достигающая максимума примерно
через 20 мин. Чувствительность реакции около 1—2 мкг!мл. Ее можно повысить,
экстрагируя окрашенное соединение бензолом, толуолом или ксилолом. При при-
менении спиртовых растворов ОВ их следует перед экстракцией образовавше-
гося красителя разбавить небольшим количеством воды. Целесообразно одно-
временно с основной реакцией проводить контрольную пробу с такими же
количествами реактивов и изопропилового спирта или воды.
II. Ф л у о р е с ц е н тн а я р е а к ц и я9
Реагенты
Реагент I— 1%-ный ацетоновый раствор индола.
Реагент II — 0,25%-ный водный раствор пербората натрия.
Реагент III — 1%-ный водный раствор глицерина.
Смесь 2 мл реагента I, 1 мл реагента II, 3 мл реагента III и 3 мл ацетона
приливают к 1 мл раствора испытуемого вещества и тотчас подвергают-облуче-
нию УФ-лампой. Сине-зеленая флуоресценция указывает на присутствие в рас-
творе фосфорорганических ОВ. Чувствительность реакции 0,05—0,1 мкг/мл. При
более высоких концентрациях (выше 1 мкг/мл) раствор вскоре окрашивается в
сине-зеленый цвет.
Методы очистки требуемых для реакции Шёнеманна рас-
творителей и аминов. Ацетон. Для окисления примеси альдегидов ацетон
обрабатывают КМпО4. Для этого к 1 л ацетона прибавляют примерно 4—5 г
КМпО4 и дают стоять несколько суток. Затем ацетон фильтруют и перегоняют,
отбрасывая первые 5% дистиллята и оставляя 5% остатка.
Спирты. Для удаления альдегидов к 1 л спирта добавляют 1 г бензидина
или о-дианизидина, кипятят 1 ч с обратным холодильником и перегоняют. Затем
прибавляют 1 г щавелеиой кислоты и еще раз перегоняют.
60
Все применяемые растворители в каждом случае испытывают на их при-
годность для реакции Шёнеманна по отсутствию окраски в контрольной пробе.
Амины21. Растворяют 50 г технического амина (бензидина, о-толидина или
о-дианизидина) в смеси 35 мл 85%-ного гидрата гидразина и 500 мл 95%-ного
этанола. Раствор нагревают при перемешивании, затем прибавляют 10 г по-
рошкообразного активированного угля и 6 г бисульфита натрия и нагревают
при перемешивании еще 2 мин. Горячий раствор фильтруют и фильтрат разбав-
ляют 1,5 л холодной воды. Выпавший белый осадок амина отфильтровывают,
промывают 100 мл воды и сушат в вакуум-эксикаторе над Р2О5. Полученный
амин должен быть чисто белым, в противном случае очистку повторяют.
Дигидрохлорид о-дианизидина22. Очищают технический о-дианизидин (сво-
бодное основание) по приведенной выше методике или подвергают его много-
кратной перекристаллизации из спирта с добавлением активированного угля до
получения белого вещества. Затем готовят насыщенный его раствор в охлажден-
ном льдом ацетоне и при постоянном охлаждении пропускают через него ток
сухого НС1. Выпавший гидрохлорид отфильтровывают, промывают холодным
ацетоном и сушат. Затем его измельчают в порошок и еще раз промывают аце-
тоном. Вещество должно быть бесцветным. Его хранят в склянке из оранжевого
стекла или в темном месте.
5.1.1.2. Реакция с изонитрозокетонами. В проводившихся в США
и Англии поисковых работах по нахождению дегазирующих и за-
щитных средств при изучении реакций между различными кетокси-
мами и фосфорорганическими соединениями было установлено,
что некоторые из них представляют интерес также и для аналити-
ческих целей. Сэвиль23 нашел, что моноизонитрозоацетон в резуль-
тате нуклеофильной атаки на атом фосфора быстро и количествен-
но гидролизует некоторые фосфорорганические соединения, обра-
зуя при этом цианистоводородную и карбоновую кислоты24:
R(Ok .0 СО-СН3 R(Ok Л
+1 +н2о —> х + нх + hcn + снзсоон
RCK \х CH=NOH RCr \он
Аналогично реагируют также и такие соединения, как галоген-
ангидриды кислот, и другие ацилирующие соединения. При этом
способе анализа определение осуществляется колориметрически по-
следующим превращением образовавшейся синильной кислоты по
реакции с хлорамином Т в хорциан, который с пиридинпиразолоном
образует окрашенное соединение25. По данным Засса и сотр. при
реакции между ацилфосфатами (или ацилфосфонатами) с диизо-
нитрозоацетоном образуется красно-фиолетовое полиметиновое со-
единение, структура которого до настоящего времени не установ-
лена 26-27.
~ CH=NOH I
R<0V + Jo — "V 9°
R°Z Xx (Jh-noh ~HX rq/ on ~ch
R(Ok л
—► P. + HCN + Краситель
RCK XOH
61
Для этой реакции оптимальное значение pH 8,3—8,6; макси-
мальная окраска при определении зарина достигается за 7 мин.
Применение одноосновных солей диизонитрозоацетона (натриевой
или солей дибутиламина или гуанидина) делает излишним прибав-
ление буферной смеси для создания желаемого pH. Преимущество
применения этих солей заключается в удобстве работы и возмож-
ности самого различного их применения. Так, соли аминов или по-
лизамещенные натриевые соли, спрессованные в таблетки или по-
мещенные в желатиновые капсули, применяют для обнаружения
фосфорорганических веществ в воде. Из полинатриевой соли могут
быть изготовлены штифты для обнаружения ОВ в воздухе. При ко-
личественном определении измеряют интенсивность окраски в ко-
лориметре при 486 или 580 нм и для оценки пользуются калибро-
вочной кривой, построенной для концентраций фосфорорганиче-
ского соединения в пределах 5—60 мкг.
Pear е н т ы
Диизончтрозоаистон —0.4%-ный водный раствор.
Буферная смесь pH 8.4 — смесь 500 мл 0.1 М раствора Н3ВО3 п 17 мл 0,5 М
раствора ХаОН пли свежеприготовленного 2%-ного раствора КаНСОз.
К 1 мл водного или спиртового раствора испытуемого вещества прибавляют
1 мл раствора диизопитрозоацетопа и 3 мл буферной смеси. Появление красно-
оранжевой до красно-фиолетовой окраски свидетельствует о наличии фосфор-
органических ОВ; чувствительность реакции для зарина, зомана, табуна и ДФФ
составляет 0.25— 1 мкг'мл.
Обнаружению мешают дихлорангидрид метилфосфоновой кислоты, хлорокись
фосфора и аналогичные соединения: уксусный ангидрид, бензолсульфохлорид и
бензоилхлорид дают подобные окраски. Присутствие этих веществ можно не
принимать во внимание при проведении индикации в полевых условиях. При
анализе проб водь: следует обращать внимание также и па присутствие же-
леза (II), меди и свободных галогенов. Эти примеси нужно либо связывать, либо
удалять.
5.1.1.3. Реакция с 4-(п-нитробензил)-пиридином. Это вещество
характеризуется весьма высокой реакционной способностью по от-
ношению к аткилируюшим соединениям, в том числе ко многим
ОВ, а также и к фосфорилирующим соединениям28. В результате
реакции29 с некоторыми фосфорорганическими ОВ, такими, как
зарин, зоман и табун, образуется окрашенное в синий цвет соеди-
нение, максимум поглощения которого лежит при 625 нм:
62
Несмотря на недостаточную специфичность и малую чувстви-
тельность (25—75 мкг/мл) этой реакции, она используется в поле-
вом анализе для предварительного обнаружения фосфорорганиче-
ских ОВ. При проведении испытания следует пользоваться методи-
кой, описанной для определения азотистого иприта (см. раздел
5.3.2.2).
5.1.1.4. Реакция с гидроксамовыми кислотами. В этой реак-
ции, проходящей в слабощелочной среде, вначале вследствие поля-
ризации связи Р = О анион гидроксамовой кислоты вызывает ну-
клеофильное замещение:
О
R(O)X ^0 ||
+ О—NH—С—R'
RCP \Х
R(0) О 0
II + Х~
R0Z ХО—\Н—С—R'
Образующееся неустойчивое соединение, претерпевая перегруп-
пировку Лоссена30, разлагается с выделением изоцианата
К(°)\ .0 0 R(Ok Л
Рх у + ОН' —> + O=C=N—R' + Н2О
ROZ \0—NH—С—R' R0Z Х0’
который, реагируя еще с одним анионом гидроксамовой кислоты,
образует карбамилгидроксамат:
О г о о
II II II
R'—С—NHO + O=C==N—R —> |_R'—С—N-О—С—NH—R'J
В реакции с бензгидроксамовой кислотой получается фенилкарб-
амилбензгидроксамат, который термически разлагается до анили-
на 32. В кислой среде в результате взаимодействия с п-диметилами-
нобензальдегидом получается азометиновый краситель, что дает
возможность, например, обнаруживать зарин31 в концентрации
0,04 мг/мл.
В результате взаимодействия фосфорсодержащего соединения с
N-оксиарилкарбаматом при pH 8 в водной среде образуется устой-
чивый продукт фосфорилирования 33
О
R(O)\ ,О у
+ 'О—NH—С—OR' —>
roz \х
R(O)X ^0 0
X || +х-
ROZ XO-NH—С—О—R'
который осаждается при подкислении реакционной смеси. Эта
реакция может быть использована для идентификации фосфор-
органических соединений по температуре плавления образующихся
продуктов.
Реакцию с легко гидролизуемыми соединениями проводят в пи-
ридине.
К раствору 1 г N-оксифенилкарбамата (0,0065 моль) в 50 мл воды прибав-
ляют 0,045 моль фосфорорганического соединения. Добавлением 0,2 и. раствора
NaOH поддерживают pH равным 8. По завершении реакции, в чем убеждаются
63
по прекращению образования кислоты, реакционную смесь подкисляют НС1 до
pH 3 и отфильтровывают выделяющийся белый осадок. После сушки и перекри-
сталлизации определяют температуру плавления.
При образовании легко растворимых в воде продуктов фосфори-
лирования их после подкисления экстрагируют хлороформом,
экстракт сушат над M.gSC>4 и после концентрирования осаждают
прибавлением лигроина (т. кип. 60—80 °C).
Приводим температуры плавления продуктов фосфорилирова-
ния N-гидроксифенилкарбамата со следующими фосфорорганиче-
скими ОВ (в °C):
с зарином (перекристаллизация из смеси метиленхло-
рид —лигроин).......................................137—139
с ДФФ (перекристаллизация из смеси хлорфор.м — лигро-
ин)................................................. 122-124
с ТЭПФ (перекристаллизация из смесн хлороформ — пет-
ролейный эфир) ...................................... 65—66
5.1.1.5. Идентификация фосфорорганических ОВ. Идентифика-
ция фосфорорганических ОВ возможна при помощи описанных
ранее способов определения функциональных групп и элементов.
Подробное описание этих методов приведено в разделе 3.1 и гл. 4.
Прежде всего присутствие фосфорорганического ОВ должно
быть установлено более общим способом, например реакцией Шё-
неманна. Далее могут быть проведены отдельные характерные ис-
пытания, на основании которых могут быть сделаны определенные
выводы о присутствии ОВ.
Обнаружены следующие ионы или группы Возможно присутствуют
1) Фтор-ион после разложения C2H5ONa; 1
2) фосфат-ион после разложения коицентриро- |
ванным раствором NaOH; } ДФФ
3) О-изопропильная группа по реакции с H2SO4 |
и га-диметиламинобензальдегидом. j
1) Цианид-ион после разложения NaOH; 1 Tb6vh
2) диметиламиногруппа по реакции Драгендорфа. J '
1) Фтор-ион после разложения C2H5ONa; ]
2) О-изопропильная группа по реакции с H2SO4 ? Зарин
и п-диметиламинобензальдегидом. )
1) Фтор-ион после разложения C2H5ONa; 1
2) О-пинаколильная группа по реакции с H2SO4 > Зоман
и ванилином. J
1) Дйалкиламиногруппы по реакции Драгендор-
фа;
2) меркаптогруппы после щелочного разложе-
ния с помощью нитропруссида натрия;
3) соответствующие алкоксигруппы (RO или
R'O)
Р-Диалкиламиноэтилтиол-
фосфаты (или фосфонаты)
R(Ok zO
р7
R'O7 4S-(CH2)„-N(R")2
Подобное сопоставление полученных данных может облегчить
принятие соответствующих выводов.
64
5.1.1.6. Индикация паратиона (тиофоса). Приводимые ниже ме-
тоды обнаружения могут быть использованы и для индикации кис-
лородного аналога паратиона — параоксона. Простейший способ
индикации основан на ускорении гидролиза этих соединений ще-
лочью:
+ NaOH
------>
С2Н5ОХ ^S(O) .
+ NaO—/ —NO2
с2н5сг \он
Интенсивно желтая окраска образующейся натриевой соли
л-нитрофенола свидетельствует о наличии этих соединений. Для
повышения чувствительности реакции можно воспользоваться ре-
агентом Миллона, который с фенолами и фенолкарбоновыми кис-
лотами дает малиново-красное окрашивание.
Реагент Миллона — растворяют 16 вес. ч. ртути в 2 вес. ч. HNO3 (d 1,42)
сначала на холоду, затем при нагревании, разбавляют двукратным количеством
воды и через несколько часов сливают раствор с осадка.
К 5 мл водного или спиртового раствора испытуемого вещества добавляют
0,5 мл 20%-ной NaOH и некоторое время нагревают смесь на кипящей водяной
бане, по возможности с обратным холодильником. При применении реагента
Миллона удается обнаружить 2—3 мкг паратиона 35.
Весьма чувствительный способ, разработанный для проявления
бумажной хроматограммы36, может быть использован и в виде
капельной реакции. Эта реакция положена в основу колориметри-
ческого метода, разработанного Авереллом и Норрисом (см. раз-
дел 5.1.2.6).
Реагенты
Хлорид титана — свежеприготовленный 5%-ный раствор.
}^-(1-Нафтил)-этилендиамин, гидрохлорид — 0,2%-иый раствор в 50%-ном
этаноле.
Аммиак — 25%-ный раствор.
Наносят 1 каплю раствора испытуемого вещества на полосу фильтроваль-
ной бумаги. После того как пятно высохнет, на то же место опускают 1 каплю
раствора TiCl3. Затем бумагу помещают на 3 мин в закрытый сосуд, на дне
которого находится несколько миллилитров раствора аммиака. Бумагу сушат
и укрепляют в закрытом стеклянном цилиндре, в котором находится чашка с
амилнитритом. После этого на пятно наносят 1 каплю раствора гидрохлорида
N-(l-нафтил)-этилендиамина. Паратион и параоксон вызывают фиолетовое окра-
шивание.
Паратион и параоксон (так же, как и инсектициды хлортион й
ЭПН) дают с дифениламином в растворе H2SO4 интенсивную си-
нюю окраску37. Эта реакция аналогична известной реакции обна-
ружения нитратов.
з Зак. 677
65
5.1.1.7. Индикация изосистокса. Метод обнаружения изосисток-
са основан на способности серы тиоэфира образовывать с солями
благородных металлов, в частности с хлоридом золота, продукты
присоединения. Эти аддукты слабо окрашены, однако при взаимо-
действии их с хлорамином или анти-арилдиазотатом получаются
интенсивно окрашенные продукты. Этот способ, который также мо-
жет быть использован для обнаружения серного иприта, исполь-
зован фирмой Дрэгер в индикаторных трубках для обнаружения
систокса. Простое осуществление этого способа в виде капельной
реакции описано для серного иприта.
Изосистокс реагирует аналогично серному иприту и в реакции
с реагентом Майера (HgI2-f-KI). После нагревания со щелочью
раствор подкисляют и добавляют реагент Майера; появляющееся
помутнение различимо вплоть до концентраций 0,02 мг)мл.
Более чувствительные методы обнаружения изосистокса осно-
ваны на использовании цветных реакций для идентификации мер-
каптидов, образующихся в результате его щелочного гидролиза42:
С2Н5ОЧ Х> •
+ NaOH —>
C2H5OZ ZS—СН2—СН2—S—С2Н5
С2Н5О, .0
—► + NaS—СН2—СН2—S—С2Н5
C2H5OZ ZOH
Реакция с нитропруссидом натрия. Смешивают 3 мл раствора испытуемого
вещества с 0,5 мл 10%-ного NaOH и нагревают до кипения. После охлаждения
добавляют 3 капли 10%-ного водного раствора нитропруссида натрия. Красно-
фиолетовая окраска возникает уже при наличии изосистокса 0,1 мг!мл.
Реакция с реагентом Фолина — Циокалтё
Реагент Фолина — Циокалтё — растворяют 50 г Na2WO4 • 2Н2О и 12,5 г
Na2MoO4 2Н2О в 350 мл воды и после прибавления 25 мл 85%-ной Н3РО4 на-
гревают 10 мин с обратным холодильником до слабого кипения. Здтем добав-
ляют 75 г Li2SO4, 25 мл воды и 2 капли брома. Для удаления избытка брома
смесь нагревают без обратного холодильника и после охлаждения титруют в
присутствии 1 капли фенолфталеина разбавленной щелочью до слабой розовой
окраски. Реактив не должен приобретать зеленоватой окраски; его хранят в
склянке из оранжевого стекла.
Гидролизованную щелочью пробу раствора испытуемого вещества нейтра-
лизуют разбавленной H2SO4 и смешивают с 0,5 мл реагента. Синее окрашивание
возникает уже в присутствии изосистокса 0,1 мг/мл.
5.1.1.8. Индикация тетраэтилпирофосфата (ТЭПФ). При нагре-
вании сернокислого раствора ТЭПФ в присутствии персульфата
аммония (NH4)2S2O8 происходит разложение ТЭПФ до ортофос-
форной кислоты, которую можно обнаруживать способами, описан-
ными в элементном качественном анализе, например осаждением
в виде фосформолибдата аммония. Количественно фосфаты мож-
но определять колориметрически по интенсивности окраски молиб-
деновой сини, образующейся после восстановления молибдена 52.
66
5.1.1.9. Индикация тетрама (амитона) и родственных соедине-
ний. Для обнаружения фосфорных соединений типа тетрама
R(Ok Л)
Р
R'0Z \g_(CH2)rt—NR"
пригодны известные уже общие методы. Кроме того, их можно
обнаруживать: 1) установливая присутствие тиольной серы в ос-
новном после гидролитического расщепления молекулы, 2) уста-
навливая присутствие третичного или, в случае тиохолиновых со-
единений, четвертичного атома азота.
Для соединений, у которых радикалы алкоксигрупп RO и R'O
имеют длинные углеводородные цепи, существует также и другая
возможность обнаружения — по цветной реакции, подобной той,
которая была описана в гл. 4 для обнаружения О-изопропильной
группы.
Для обнаружения тиольной серы с высокой степенью чувстви-
тельности особенно пригодны два описываемых ниже метода. Пер-
вый метод представляет собой модификацию разработанного Се-
виллем 53 колориметрического микроопределения тиола. Механизм
образования окрашенного продукта следующий: первая стадия
реакции является известным, превращением тиола в нитрозилмер-
каптан под воздействием азотистой кислоты54:
R—SH + HONO —> R—8—NO + H2O
Избыточное количество азотистой кислоты разрушают аммоние-
вой солью сульфаминовой кислоты, в присутствии которой нитро-
зилмеркаптан достаточно устойчив:
HONO + NH4SO3NH2 —> NH4HSO4 + N2 + Н2О
Быстрый гидролиз нитрозилмеркаптана, осуществляемый добав-
лением соли ртути (II), объясняется образованием комплексного
соединения со ртутью, вследствие чего связь N—S становится не-
устойчивой к нуклеофильной атаке молекулы воды и разрушается
(реакции I и 2):
О О +
II II + Hg
N—S + ^=*Г N-S^ (1)
I R
R
О + о и +
ll н у ng
In—s -------*- ^o—n + (2)
Hl H || R
H* + HONO
3*
67
Выделяющаяся азотистая кислота диазотирует вводимый суль-
фаниламид А. При этом образуется соль диазония, которую соче-
тают с а-нафтиламином .или другим ароматическим амином и по-
лучают азокраситель:
HONO + A —> [Ar—feN]* X’ + Н2О
ArNHj |-х Ar_N==N_R_^Ha
краситель
Одновременно протекает конкурентная реакция азотистой кис-
лоты с присутствующей в реакционной смеси аммониевой солью
сульфаминовой кислоты:
HONO + NH4SO3NH2 —> NH4HSO4 + N2 + Н2О
Эта реакция идет значительно медленнее и не мешает образова-
нию азокрасителя.
Реагенты
Аммоний сульфаминовокислый— 0,5%-ный раствор в воде.
Реагент I — смесь 1 части 1 %-кого водного раствора HgCl2 и 4 частей
3%-ного раствора сульфаниламида в 2%-ной НС1.
Реагент II — 0,05%-ный раствор а-нафтиламина или дигидрохлорида
N-(l-нафтил)-этилендиамина в 2%-ной НС1.
Нитрит натрия — 0,015%-ный водный раствор.
В пробирку с пришлифованной пробкой помещают 5 мл водного раствора
испытуемого вещества или 4 мл спиртового его раствора и 1 мл воды. До-
бавляют 0,5 мл 5н. раствора NaOH и нагревают 10 мин в кипящей водяной
бане. Затем добавляют 1 мл 5 н. раствора H2SO4, быстро охлаждают и смеши-
вают с 1 мл раствора NaNO2. По прошествии 5 мин добавляют 1 мл раствора
NH4SO3NH2 и, закрыв пробирку пробкой, энергично встряхивают и помещают
ее на 2 мин в водяную баню. Затем содержимое пробирки еще раз энергично
встряхивают, пробку приоткрывают и немного поворачивают так, чтобы горячий
раствор полностью смочил поверхность шлифа пробирки и пробку. Затем про-
бирку охлаждают и к реакционной смеси тотчас последовательно приливают по
2 мл реагентов I и II. Различимое через несколько минут красно-фиолетовое
скрашивание указывает на присутствие тиолового эфира фосфоновой (или фос-
форной) кислоты. Реакция дает возможность обнаруживать эфир в количестве
примерно 0,1 мкг)мл.
Очень простым в выполнении, но менее чувствительным являет-
ся обнаружение тиола при помощи 5,5'-дитио-бис- (2-нитробензой-
ной) кислоты 55’56, разработанное для гистохимического обнаруже-
ния SH-групп. Это водорастворимое вещество реагирует с тиолами
с образованием смешанных дисульфидов и аниона 5-тиол-2-нитро-
бензойной кислоты, который в щелочной среде окрашен в интенсив-
ный желтый цвет:
68
Таким же образом реагирует бис-(n-нитрофенил)-дисуль-
фид57’58. Вследствие малой растворимости этого соединения в воде
реакцию проводят в водноацетоновом растворе. Идентификация
гидролитически отщепленного тиола возможна также при помощи
других цветных реакций, например: с нитропруссидом натрия59,
фосфор-18-вольфрамовой кислотой60, 2,6-дихлорфенолиндофено-
лом 61’62 и хлоридом трифенилтетразолия.
Определение третичного или четвертичного атома азота в со-
единениях типа тетрама может быть осуществлено реагентом Дра-
гендорфа (см. стр. 86) или фосфорновольфрамовой кислотой.
5.1.2. Количественные определения
5.1.2.1. Методы, основанные на реакции Шёнеманиа. Для коли-
чественного определения фосфорорганических ОВ пригодны раз-
личные модификации реакции Шёнеманиа, механизм которой был
подробно рассмотрен в разделе 5.1.1.1. Некоторые из этих моди-
фикаций, отличающиеся по применяемым реагентам, описываются
ниже.
I. Колориметрические методы определения
Реакция с о-дианизидинам 34
Реагенты
Буферная смесь — pH 11,5 —раствор 16 г КН2РО4 и 9 г КОН в 1000 мл
дистиллированной воды, не содержащей СО2, смешивают в соотношении 1 : 1
с очищенным ацетоном; добавлением разбавленной щелочи (КОН) устанавли-
вают нужное значение pH.
Дигидрохлорид о-дианизидина—1,3%-ный водный раствор; сохраняется в
темном месте не более 1 недели.
Смешанный реагент — смесь из 23 ч. буферной смеси, 1 ч. раствора гидро-
хлорида о-дианизидина и 1ч. 1%-ного раствора Н2О2. Применяется для опре-
деления зарина и зомана. При определении ДФФ или ТЭПФ (тетраэтилпирофос-
фата) в реагент добавляют 3 ч. 1%-ного раствора Н2О2. Смешанный реагент
годен в течение не более 30 мин.
Построение калибровочной кривой производится непосредственно перед
определением. Для этого приготовляют стандартный раствор фосфорсодержа-
щего ОВ в изопропиловом спирте и используют его в объеме, не превышающем
2 мл. Степень разбавления должна быть подобрана так, чтобы в случае зарина
в 2 мл или в аликвотной части содержалось от 0 до 300 мкг вещества. Аликвот-
ные части доводят до объема 2 мл добавлением из микробюретки изопропило-
вого спирта. Это требуется для того, чтобы в реакционной смеси всегда нахо-
дилось одно и то же количество спирта.
В мерную колбу емкостью 25 мл наливают 20 мл смешанного реагента и
по каплям в течение 60—90 сек при постоянном помешивании добавляют стан-
дартный раствор ОВ. Затем, если это необходимо, добавляют изопропиловый
спирт и доводят объем до метки смешанным реагентом. При комнатной темпе-
ратуре интенсивность окраски можно измерять в колориметре с фильтром, имею-
щим максимум поглощения при 450 нм. Для зарина и зомана ее измеряют че-
рез 10 мин, для ДФФ — через 30 мин, для ТЭПФ — через 20 мин. Колориметри-
рование проводят по сравнению с водой и, если это необходимо, с учетом
экстинкции контрольной пробы, к которой вместо стандартного раствора добав-
ляют 2 мл изопропилового спирта. Полученные для различных концентраций ОВ
значения экстинкций переносят на калибровочную кривую. Затем проводят опре-
деление с образцом исследуемого вещества, результаты которого оценивают по
калибровочной кривой.
69
В применении к аминоалкилтиофосфорным соединениям мето-
дика несколько изменена. Удовлетворительная чувствительность
достигается либо при некотором увеличении концентрации переки-
си водорода и продолжительности развития окраски либо путем
кратковременного нагревания реакционной смеси в воспроизводи-
мых условиях. Естественно, особое внимание следует обратить на
проведение контрольной пробы.
Реакция Шёнеманна не применима для определения иодмети-
латов этих соединений (тиохолинового эфира). С незначительными
изменениями метод может быть использован для определения фос-
форорганических ОВ в воде или в воздухе после поглощения в
подходящем растворителе. Очень важно, чтобы построение кали-
бровочной кривой по возможности проводилось со стандартным
раствором примерно того же состава, что и раствор испытуемого
вещества. Количество прибавляемого к смешанному реагенту стан-
дартного раствора (или раствора испытуемого вещества) не должно
превышать 12,5 мл (50%), однако при этом увеличивается время
развития максимальной окраски, которое в этом случае нужно за-
ранее установить.
Реакция с о-толидином45-47
Реагенты
Буферная смесь, pH 8,7 — смешивают 100 мл 0,1 М раствора КНгРО*, 100 мл
1 М раствора NaOH и 1000 мл воды и добавлением 1 М раствора NaOH уста-
навливают нужное значение pH, после чего водой доводят объем раствора
до 2 л.
Гидрохлорид о-толидина— 1%-ный водный раствор.
Перборат натрия — свежеприготовленный 1,25%-ный водный раствор.
К приготовленному непосредственно перед употреблением смешанному ре-
агенту, состоящему из 40 мл очищенного ацетона, 4 мл буферной смеси, 2 мл рас-
твора гидрохлорида о-толидина и З'мл раствора пербората натрия, прибавляют
10 мл водного раствора испытуемого вещества. Через 20 мин окраску колори-
метрируют при 420 нм. Содержание ОВ определяют по заранее приготовленной
калибровочной кривой.
II. Флуоресцентное определение48
Реагенты
Реагент I — раствор 2 г индола, 3,75 г NaHCO3 и 0,43 г Na2CO3 в смеси
1000 мл воды, 100 мл ацетона и 100 мл изопропилового спирта.
Реагент II — смесь 1000 мл воды и 250 мл изопропилового спирта, к кото-
рой добавлено 3,5 мл 13,3 %-ной Н2О2.
К 24 мл смеси реагентов I и II (1 : 1) с pH 10,6 приливают 1 мл раствора
испытуемого вещества в изопропиловом спирте и тотчас измеряют максимум
флуоресценции на флуорометре. Возбуждение осуществляется УФ-облучением
в области от 350 до 400 нм, максимум сине-зеленой флуоресценции располагает-
ся у 500 нм. Содержание вещества определяют по калибровочной кривой.
5.1.2.2. Объемное определение фторангидридов фосфоновой ки-
слоты и эфиров пирофосфорной кислоты (гидролизный метод)49 145.
Этот простой макрометод применяется для контроля чистоты
названных фосфорных соединений. Он основан на различии в ско-
рости гидролиза этих веществ и таких примесей, как хлор- и ди-
фторангидриды. Для того чтобы отделить зарин от возможной при-
меси соответствующего пирофосфоната, обладающего той же спо-
70
собностью к гидролизу, смесь этих веществ можно пропустить через
колонку, заполненную насыщенным водой силикагелем. При этом
пирофосфонат абсорбируется силикагелем, а зарин элюируют изо-
пропиловым спиртом и затем определяют титрованием. Содержа-
ние пирофосфоната определяют по разности величин, полученных
при титровании пробы до и после разделения на колонке. Содер-
жание пирофосфоната может быть также определено отдельно в
водном элюате. В последнем случае продолжительность щелоч-
ного гидролиза после прибавления NaOH должна быть увеличена
до 10 мин по сравнению с 2—5 мин при определении зарина.
Другим упрощенным способом разделения зарина и пирофос-
фоната является воспроизводимое распределение этих веществ
(80:20) между органическим растворителем (изопропиловый эфир)
и водным 3,6%-ным раствором NaCl. Это разделение менее точно,
чем разделение на колонке с силикагелем.
Количество раствора, в котором содержится 0,13—0,18 г фосфорорганиче-
ского вещества, переносят при помощи микропипетки с поршнем в предвари-
тельно взвешенную колбу Эрленмейера и повторным взвешиванием ее опреде-
ляют взятое для анализа количество вещества. В колбу приливают 15 мл воды
и 4—5 капель смешанного индикатора, состоящего из 0,05%-ного раствора ме-
тилового красного и 0,1%-ного -раствора тимолфталеина в метаноле. Более
точно можно взвесить раствор испытуемого вещества в стеклянной ампуле, ко-
торую затем помещают в колбу Эрленмейера, содержащую воду и индикатор, и
разбивают стеклянной палочкой. После растворения вещества раствор сразу
титруют 0,1 и. раствором NaOH до перехода красной окраски в желтую. По-
шедшее на титрование количество щелочи записывают, так как оно эквива-
лентно количеству свободной кислоты и кислых продуктов, образовавшихся в
результате быстрого гидролиза примесей. Затем продолжают титрование до
перехода окраски раствора в синюю. Этот расход щелочи также записывают.
По прошествии 2 или максимально-5 мин проводят обратное титрование 0,1 н.
раствором НС1 до перехода окраски метилового красного из желтой в желто-
розовую. Содержание зарина (в %) рассчитывают по формуле:
7,005 (а^иаон 6/гнс1) 100
А .
где а — количество 0,1 н. раствора NaOH, пошедшее на обратное титрование, мл;
Ь — количество 0,1 н. раствора НС1, мл; А — навеска, мг.
Фосфорорганические соединения, плохо растворимые в воде
(например, зоман), растворяют в этаноле, а затем этот спиртовой
раствор так разбавляется водой, чтобы содержание в нем спирта
не превышало 20 объемн. %. Большее содержание спирта может
привести к снижению скорости гидролиза фторфосфоната.
5.1.2.З. Объемное определение фторангидридов эфиров метил-
фосфоновой кислоты и пирофосфонатов (перекисный метод). При
помощи этого метода50 можно определять меньшие количества
фосфорорганических веществ и особенно фторангидридов эфиров
алкилфосфоновых кислот, нежели это удается осуществить гидро-
лизными способами. Основой метода является реакция фосфор-
содержащих соединений с щелочным раствором перекиси водорода,
71
описываемая уравнениями 1 и 2 (стр. 53), и последующее иодо-
метрическое определение избыточной перекиси водорода:
Н2О2 + 2HI —> 12 + 2Н2О
Реагенты
Раствор перекиси (pH 10)—растворяют 8,5—9,0 г перекиси пирофосфата
натрия Na4P2O7 • 2Н2О2 и 5,68 г пербората натрия Na2B4O7 • ЮН2О в 500 мл
воды и 10%-ным раствором NaOH (примерно 12 мл) устанавливают значение
pH 10, после чего доливают воду до объема 1 л.
Тиосульфат натрия — 0,1 н. раствор.
В мерную колбу емкостью 100 мл помещают навеску испытуемого вещества
0,18—0,24 г. Это производят либо внесением ОВ пипеткой во взвешенную колбу
и последующим определением точной навески вещества повторным взвешива-
нием колбы, либо внесением в колбу стеклянной ампулы с навеской вещества:
ампулу затем разбивают стеклянной палочкой под слоем 20 мл 50%-ного изо-
пропилового спирта, предварительно налитого в колбу. Объем жидкости в колбе
доводят до метки также этим спиртом. В две колбы Эрленмейера с притертыми
пробками емкостью 500 мл, одна из которых предназначается для контрольной
пробы, наливают по 50 мл раствора перекиси (pH 10). В одну из колб пипеткой,
при постоянном перемешивании, приливают 20 мл раствора испытуемого веще-
ства, в другую колбу — 20 мл 50%-ного изопропилового спирта. Затем обе сме-
си периодически перемешивают 2—4 мин. Прибавив к обоим растворам по 30 мл
воды, 10 мл 18 н. раствора HzSOt и 3 г KI в указанной последовательности и
перемешав содержимое, колбы выдерживают в темном месте 10 мин. Выделив-
шийся иод титруют 0,1 н. раствором Na2S20s. Незадолго до исчезновения желтой
окраски иода в качестве индикатора добавляют некоторое количество раствора
крахмала и титруют до обесцвечивания; 1 мл 0,1 и. раствора Na2S20s соответ-
ствует 1/4000 моль фосфорсодержащего вещества. Так, содержание зарина (в %)
рассчитывают по формуле;
3,502 (а - b) FNa2S2O3 • 5 • 100
А
где а — количество 0,1 н. раствора Na2S2O3, пошедшее иа титрование контроль-
ной пробы, мл; b — количество 0,1 н. раствора Na2S2O3, пошедшее на титрова-
ние испытуемого вещества, мл; А — навеска, мг.
Хлорангидриды и дифторангидриды не мешают определению,
так как они гидролизуются в водноспиртовом растворе еще до при-
бавления перекиси. Точность определения примерно 1%. Хлор-
ангидриды могут быть определены этим методом после предвари-
тельного их перевода при помощи фтористоводородной кислоты в
соответствующие фторангидриды.
5.1.2.4. Колориметрическое определение при помощи 4-(л-нитро-
бензил)-пиридина51 (экспресс-метод). Принцип метода описан в
разделе 5.1.1.3.
Реагенты
Реагент I—2%-ный раствор 4-(4'-нитробензил)-пиридина в ацетоне.
Реагент II — 2%-ный раствор циклогексиламина в очищенном ацетоне.
Очистку ацетона производят кипячением ацетона в течение 1 ч с КМпО4, до-
бавляемым из расчета 1 г KMnOt на 1 л ацетона, и последующей перегонкой.
Смешивают 0,05 мл раствора отравляющего вещества в ацетоне с 0,2 мл
раствора реагента I и 0,2 мл реагента II и нагревают 3 мин иа масляной бане
с обратным холодильником. После охлаждения объем смеси доводят дважды
перегнанным этилацетатом до 3 мл и через 10 мин колориметрируют при 520 нм
(сравнение с контрольной пробой). Для расчета пользуются калибровочной
кривой, которую строят для концентраций фосфорорганического вещества
2—15 мкг.
72
5.1.2.5. Колориметрическое определение при помощи гидрокса-
мовой кислоты. Очень интересный способ обнаружения и опреде-
ления сложных эфиров фосфорных кислот 320 основан на свойстве
ионов гидроксамовой кислоты катализировать гидролиз некоторых
ацилирующих соединений:
О О о о
II II (он-) II II
R—С—X + R'~С—NHOH -------► R—С—NHOC—R' + НХ
О О
II II (он-)
R—С—NHOC—R 4- Н2О -----
Суммарная реакция:
О О
II II
R'—С—NHOH + R—С—ОН
М || м
II R'—С—NHOH II
R—С—Х + Н2О --------------* R—С—ОН + НХ
По этому способу добавляют к сложному эфиру фосфорной
кислоты избыточное количество гидроксамовой кислоты при pH 9
(см. раздел 5.1.1.4); неизрасходованная гидроксамовая кислота ка-
тализирует затем гидролиз добавляемого к реакционной смеси
окрашенного в желтый цвет 2-азобензол-1-нафтилацетата, в ре-
зультате чего образуется красный 2-азо-бензол-1-нафтол. По исте-
чении определенного времени прибавлением кислоты гидролиз оста-
навливают и измеряют фотометрически интенсивность окраски про-
дуктов гидролиза, количество которых эквивалентно количеству
введенного в реакцию фосфорорганического вещества. Аналогия с
описанным в гл. 6 биохимическим способом, основанным на угне-
тении холинэстеразы, очевидна. Химический способ, однако, значи-
тельно менее чувствителен. Оптимальные условия для каждого
фосфорорганического вещества различны и зависят от скорости
взаимодействия данного соединения с гидроксамовой кислотой и
гидроксил-ионом. Ниже приведена методика определения зарина.
Реагенты
Гидроксамовая кислота — 2 • 10-3 М водный раствор.
Буферная смесь — 0,001 М раствор Na2B4O7, в которой pH 9 устанавливают
добавлением 0,05 н. раствора НС1.
2-Азобензол-1-нафтилацетат (субстрат)—2,5-1О-3Л1 раствор в ацетоне.
Смешивают 4 мл водного раствора испытуемого вещества с 1 мл раствора
гидроксамовой кислоты и 1 мл буферной смеси и выдерживают 10 мин при
25 °C. Затем добавляют 4 мл раствора субстрата, перемешивают и через 5 мин
добавляют 0,5 мл раствора 0,05 н. НС1. Через 2 мин в фотометре при 540 нм
определяют экстинкцию. Для расчета пользуются калибровочной кривой. Мини-
мально определяемая концентрация зарина составляет 0,5 мкг! мл
5.1.2.6. Колориметрические способы определения паратиона и
параоксона. Образующуюся в результате щелочного гидролиза
этих соединений (см. раздел 5.1.1.6.) окрашенную в желтый цвет
натриевую соль n-нитрофенола можно определять колориметри-
чески 38.
73
Смешивают 5 мл спиртового раствора испытуемого вещества с 5 мл 1 и.
раствора КОН в 50%-ном этаноле и кипятят 30 мин с обратным холодильни-
ком. После быстрого охлаждения раствор переносят в мерную колбу емкостью
100 мл, доливают до метки и колориметрируют при 405 нм по сравнению с
50%-ным спиртом.
Другие методы определения выделяющейся при гидролизе соли
«-нитрофенола основаны на восстановлении нитрогруппы в амино-
группу цинком в кислом растворе или TiCU и в превращении ее в
краситель. Диазотирование аминогруппы и образование красителя
впервые было применено Авереллом и Норрисом39. Впоследствии
этот метод многократно модифицировался.
Реагенты40
Хлористый титан — раствор, приготовленный смешением 1 объема 15%-ного
раствора Tide, 1 объема уксусной кислоты (2 М) и 2 объемов раствора ацетата
натрия (2 М).
Нитрит натрия — 3,2%-ный водный раствор.
Сульфаминовая кислота — 2%-ный водный раствор.
Гидрохлорид И-(1-нафтил)-этилендиамцна—1%-ный водный раствор (азо-
составляющая компонента).
К 5 мл спиртового раствора пробы прибавляют 0,05 мл раствора ТЮз и
через 2 мин — 0,2 мл 8 н. раствора НС1. Еще спустя 2 мин к смеси добавляют
0,1 мл раствора NaNO2 и через 4 мин — 0,5 мл раствора сульфаминовой кислоты.
Затем через 3 мин прибавляют 0,5 мл азосоставляющей компоненты н после
доведения объема раствора спиртом или водой до 10 мл (доля спирта должна
составлять 30—50%) колориметрируют при 560 нм. Расчет проводят по предва-
рительно построенной калибровочной кривой.
Другая возможность определения образовавшегося после вос-
становления п-аминофенола состоит в превращении его под воздей-
ствием фенола в индофенольный краситель. Для определения содер-
жания паратиона и параоксона в воздухе разработана следующая
методика 4I.
Реагенты
Реагент А — раствор 38,1 г Na2B4O7 • ЮН2О в 700 мл воды, содержащий
50 г фенола н доведенный водой до 1000 мл.
Хлористый титан — раствор, приготовленный разбавлением — 15 мл 15%-ного
раствора TiCls 100 мл воды (применяют свежеприготовленный раствор).
Поглощают паратион пропусканием пробы воздуха через смесь из 10 мл
метилцеллозольва (монометиловый эфир этиленгликоля) и 1 н. раствора NaOH
(1 : 1). Поглотитель переносят в мерную колбу емкостью 50 мл и нагревают
30 мин в кипящей водяной бвие. Затем к еще горячей смеси приливают 10 мл
реагента А, 1 мл раствора Т1С13 и встряхивают до тех пор, пока черная гидро-
окись титана(III) не превратится полностью в белую двуокись. Смесь охла-
ждают, доливают водой до 50 мл и фильтруют (первые 10 мл фильтрата
отбрасывают). Через 6 ч раствор колориметрируют при 625 нм. При толщине
слоя в 1 см содержание паратиона равно Е-10,32 мкг/мл, параоксона —
Е • 9,94 мкг/мл.
Метод дает возможность определять паратион уже при концентрации
1,5 • 10'3 р. р. т.
5.1.2.7. Колориметрическое определение систокса. Это опреде-
ление основано на свойстве сульфидной серы образовывать комп-
лексное соединение с иодом43.
74
Смешивают 4 мл раствора испытуемого вещества в четыреххлористом угле-
роде с 1 мл 1%-ного раствора иода в том же растворителе и через 15 мин
колориметрируют при 305 нм, сравнивая окраску с окраской контрольной пробы
(4 мл четыреххлористого углерода и 1 мл 1%-ного раствора иода). Расчет про-
водят по калибровочной кривой, построенной для концентрации систокса 5—
70 мкг/мл.
5.1.2.8. Объемное определение изосистокса. Образующийся при
щелочном гидролизе изосистокса меркаптид натрия (см. раздел
5.1.1.7) весьма неустойчив и быстро окисляется до дисульфида.
Если для омыления изосистокса пользоваться щелочным раствором
плюмбита, то образуется устойчивое к окислению внутрикомплекс-
ное соединение44
С2Н5 C2HS
н2с—s4 zs—сн2
I X I
H2C—s/ ^S—CH2
в котором содержание свинца может быть определено йодометри-
ческим или комплексометрическим титрованием.
Реагенты
Ацетат свинца — раствор 189,7 г РЬ(СН2СОО)2 • ЗН2О в 1 л воды (стан-
дартный раствор).
Щелочной раствор плюмбита — к 200 мл нагретого до кипения 0,5 н. рас-
твора NaOH прибавляют по каплям стандартный раствор ацетата свинца до
образования устойчивого помутнения, затем его охлаждают, фильтруют и до-
ливают 0,5 н. раствором NaOH до 1 л; реагент хранят в полиэтиленовом со-
суде.
В колбе с притертой пробкой емкостью 500 мл смешивают навеску испытуе-
мого вещества (0,2—0,6 г) с 10 мл этанола. После прибавления 60 мл щелоч-
ного раствора плюмбита реакционной смеси дают стоять 1 ч, при этом при-
мерно через 10 мин выпадают^рыхлые желтые хлопья меркаптида свинца. За-
тем прибавляют 50 мл хлороформа и 70 мл раствора ацетата свинца и после
энергичного встряхивания раствор переносят в делительную воронку емкостью
500 мл. После трехкратного встряхивания с хлороформом (каждый раз по
30 мл) отделяют органический слой,, а водный — двукратно экстрагируют хло-
роформом. Объединенные хлороформные вытяжки фильтруют через смоченный
хлороформом двойной бумажный фильтр, который дополнительно дважды про-
мывают тем же растворителем. К фильтрату прибавляют 30 мл 20%-ной H2SO«
и титруют в присутствии крахмала при энергичном перемешивании 0,1 н. рас-
твором иода до синего окрашивания. Содержание изосистокса в пробе рас-
считывается по следующей формуле:
2,58aF-100
-----3------ % изосистокса
где а — количество пошедшего на титрование 1 н. раствора иода, мл; F—по-
правка к титру раствора иода; А — иавеска ОВ, мг.
5.1.2.9. Фотометрическое определение фосфорилтиохолинов 63' 64.
Определение основано на быстром разрыве связи Р—S при дей-
ствии хлорида палладия (II) с образованием соответствующего
75
хлорангидрида алкилфосфоновой кислоты и соединения палладия
с тиолом:
R(Ok ,О
/R" + PdCl2 —>
R'OZ XS—(СН2)„—г/
\r”'
R(°)\ A) R"x
-> /P\ + ^N—(CH2)„—SPdCl
R'OZ XC1 R"'Z
Содержание палладийтиолового соединения определяют фото-
метрически в области УФ-спектра.
Реагенты
Хлорпалладит аммония (NII4)2PdCl4 — 0,05 М раствор соли в 0,3 М рас-
творе НС1.
Раствор сравнения — разбавляют 0,5 мл раствора хлорпалладита до 50 мл
соляной кислотой (0,3 М).
Растворяют испытуемое ОВ в воде или в смешивающемся с водой раство-
рителе, не дающем поглощения в применяемой области длин волн. Хорошо
подходит изопропиловый спирт. В мерной колбе емкостью 50 мл смешивают
0,5 мл раствора хлорпалладита с определенным объемом раствора испытуе-
мого ОВ и доводят до метки соляной кислотой (0,3 М). Поглощение измеряют
при 250 или 310 нм, сопоставляя с раствором для сравнения. Расчет ведут по
калибровочной кривой, построенной по стандартному раствору соответствую-
щего тиольного соединения. Время достижения максимального поглощения,
зависящее от вида тиольного соединения, следует каждый раз специально
определять.
Этот метод применим для определения 2—200 мкг]мл любых
тиолсодержащих соединений, образующих водорастворимые комп-
лексы с палладием.
Тиолы типа диметиламнноэтилмерк.аптана можно определять
титрованием раствором хлористого палладия, пользуясь п-нитро-
зодиметиланилином в качестве индикатора64.
5.2. ГАЛОГЕНИРОВАННЫЕ ТИОЭФИРЫ (СЕРНЫЕ ИПРИТЫ)
Во время первой мировой войны бис-2-хлорэтиловый тиоэфир имел
большое значение; поэтому в то время были разработаны много-
численные методы его обнаружения. Однако в результате появле-
ния новых чувствительных цветных реакций на иприт старые ме-
тоды представляют теперь лишь ограниченный интерес и поэтому
от описания их можно отказаться.
5.2.1. Методы индикации
5.2.1.1. Реакция с тиомочевиной и солью никеля. В этой реак-
ции, описанной Харли-Мэзоном65, атомы хлора серного иприта при
76
взаимодействии с тиомочевиной замещаются тиольными группами:
/NH2
S(CH2—СН2С1)2 + 2S=X
xnh2
/ ZNH3\ 12+
S CH2—CH2—S—с; 2СГ
. \ Чн/J
Полученное соединение разлагают нагреванием со щелочью до
дитиола — димеркаптодиэтилсульфида
/ ZNH3\ р
S СН2—СН2— S—X 2СГ + 6NaOH —►
—> S(CH2—СН2—SH)2 + 4NH3 + 2Na2CO3 + 2NaCl
который при добавлении аммиачного раствора соли никеля обра-
зует окрашенное в красный цвет комплексное соединение:
СН2--СН2
I I
СН2—S. /S—сн2
I >< >< ।
СН2—Sz xs—сн3
<Lh2—сн2
Проведение цветной реакции с солью никеля разработано Мато-
ушеком и Томечеком31.
Реагенты
Тиомочевина — 5%-ный раствор в этаноле.
Сульфат никеля — раствор 5 г NiSO4 в 50 мл воды, доведенный до 100 мл
20%-ным раствором аммиака.
Уксусная кислота — 50%-ный раствор.
Приливают к 1 мл спиртового раствора испытуемого вещества 1 мл рас-
твора тиомочевины и в течение 2—3 мин доводят до кипения нагреванием
в водяной бане. Затем добавляют 0,5 мл 30%-ного NaOH и еще раз быстро
доводят раствор до кипения. После охлаждения, смесь нейтрализуют или слегка
подкисляют уксусной кислотой. После прибавления 0,5 мл раствора NiSO4
в присутствии иприта появляется красное окрашивание. Затем раствор раз-
бавляют 1—2 мл воды, добавляют 2—3 мл хлороформа или дихлорэтана для
перевода окрашенного продукта в органический слой. Реакция дает возмож-
ность обнаруживать 0,04 мг серного или азотистого иприта.
Вместо реакции с сульфатом никеля можно также использовать
реакцию с 0,5%-ным раствором нитропруссида натрия, который в
щелочной среде образует с ипритом продукт, окрашенный в фиоле-
товый цвет.
Аналогичное окрашивание возникает спустя некоторое время в
присутствии более высоких концентраций бромацетона, хлорпик-
рина, хлорацетофенона, метил- и этилдихлорарсинов.
77
5.2.1.2. Реакция с щелочным раствором тимолфталеина. Как
для серного, так и для азотистого ипритов в равной степени спра-
ведлив следующий механизм реакции:
NaOH
-------->
кислота
желтый синий
+ S(CH2—СН2С1)2
| —NaCl
♦
желто-оранжевый
где R —СН3, R' —СН(СНз)2.
Строение этого красителя, образующегося в кислой среде, уста-
новлено Матоушеком и сотр,31 с помощью инфракрасной спектро-
скопии.
Реагент
Тимолфталеин — раствор 0,4 г тимолфталеина в 60 мл чистого спирта, к ко-
торому добавлено 20 мл 0,6%-ного КОН; раствор должен иметь глубокую си-
нюю окраску.
К 2 мл спиртового или водного раствора испытуемого вещества приливают
равный объем раствора тимолфталеина и нагревают смесь 20 мин в водяной
бане при 60—80° С. Во время нагревания смесь должна сохранять синюю
окраску, в противном случае к ней добавляют несколько капель разбавлен-
ного раствора КОН до появления синей окраски. После охлаждения к смеси
добавляют 1—2 капли уксусной кислоты, после чего синяя окраска исчезает
и в присутствии серного или азотистого ипритов появляется желтое или оран-
жевое окрашивание.
Слабая окраска надежно обнаруживается, если окрашенное соединение
экстрагировать встряхиванием с 1 мл толуола или бензола. При использова-
78
нии спиртового раствора испытуемого вещества к нему перед экстракцией сле-
дует добавить некоторое количество воды. Метод дает возможность обнару-
живать 0,5 мкг/мл серного или азотистого иприта. Он может быть использо-
ван также и для фотометрического определения ипритов, причем экстинкцию
измеряют при 450 нм\ расчет проводят по калибровочной кривой.
Галогеисодержащие алифатические соединения, такие, как дихлорэтан,
бромацетат, хлорпикрин и др., дают аналогичную окраску и мешают проведе-
нию реакции.
5.2.1.З. Реакция с реагентом Гриньяра. Описанный Гриньяром66
реагент представляет собой комплексное соединение, образующееся
в водном растворе при взаимодействии сульфата меди с избытком
иодида натрия:
CuSO4 + 2NaI —► Сц12 + Na2SO4
2CuI2 —► 2CuI + I2
2CuI + 2NaI —Na2Cu2I4
При взаимодействии этого соединения с серным ипритом обра-
зуется нерастворимое молекулярное соединение бис-2-иодэтилового
тиоэфира с иодидом меди:
Na2Cu2I4 + 3(СН2—СН2С1)2 —> S(CH2—CH2I)2Cu2I2 + 2NaCl
Образующуюся опалесценцию стабилизуют добавлением гум-
миарабика.
Реагент Гриньяра — к раствору 20 г Nal в 50 мл воды добавляют 1 мл
7,5%-ного раствора CuSO4 и затем 2 мл раствора 35 г гуммиарабика в 100 мл
воды. Общий объем смеси доводят водой до 200 мл. Профильтрованный ре-
агент следует хранить в. склянке из коричневого стекла.
Можно пропускать пробу воздуха, содержащего ОВ, через поглотительную
склянку, содержащую реагент Гриньяра, или же приливать реагент к водному
раствору испытуемого вещества. Присутствие серного иприта обнаруживается
по появлению желтого помутнения. Чувствительность реакции 0,05 мг/мл.
Обнаружению иприта мешают арсины, также вызывающие по-
мутнение 68'136.
Эта реакция может быть осуществлена в виде капельной67.
Каплю спиртового раствора испытуемого вещества наносят на фильтро-
вальную бумагу. Испаряют растворитель, на то же место опускают кацлю
5%-ного спиртового раствора Nal и затем на край пятна — каплю 10%-ного
раствора CuSO4. В присутствии сериого иприта в УФ-свете в зоне соприкосно-
вения капель появляется желто-зеленая флуоресценция; чувствительность реак-
ции 0,1 мг/мл.
5.2.1.4. Реакция с хлорным золотом. В этой реакции, впервые
использованной для индикации серного иприта Шротером69 и
Обермюллером70, образуется нерастворимое комплексное соеди-
нение
zCH2—СН2С1
AuCl3- S
ХСН2—СН2С1
Подобные соединения образуются со многими солями тяжелых
металлов, однако наиболее удобны для индикации соли золота и
79
палладия. Метод применим в виде капельной реакции на бумаге, а
в сочетании с тиосульфатом натрия, хлорамином Т или антм-арил-
диазотатами, образующими с комплексным соединением окрашен-
ные продукты, часто используется в индикаторных трубках. При
индикации иприта в водном растворе образующееся желтое помут-
нение можно стабилизовать добавлением некоторого количества
раствора какой-либо гетерополикислоты (фосфорномолибденовой,
фосфорновольфрамовой, кремневольфрамовой), что обусловлено,
очевидно, образованием агрегатов молекул71’72.
Реагенты
Хлорид золота — 5%-ный водный раствор.
Хлорамин Т — 1°/о-ный водный раствор.
Для осуществления капельной реакции на кусок фильтровальной бумаги
помещают каплю' раствора испытуемого вещества, затем, последовательно, —
каплю раствора хлорида золота и каплю раствора хлорамина Т. Красно-корич-
невое окрашивание пятна указывает на присутствие серного иприта; чувстви-
тельность реакции 0,1 мг/мл.
5.2Л.5. Другие методы индикации иприта. Для обнаружения
серного иприта могут быть использованы при незначительном из-
менении также методы, описанные для индикации или определения
азотистого иприта при помощи 4-(n-нитробензил)-пиридина и 8-ок-
сихинолина (см. разделы 5.3.2.1 и 5.3.2.2).
Ниже приведены некоторые другие менее чувствительные и
менее специфичные методы обнаружения серного иприта.
1) Обесцвечивание 0,003%-ного раствора перманганата калия74;
чувствительность реакции — около 0,15 мг/мл.
2) Образование помутнения при добавлении спиртового щелоч-
ного раствора р-нафтола75; чувствительность реакции 0,06 мг/мл.
3) Образование желтовато-белого осадка с дииодмеркуратом
калия (10 г КД и 14 г Hgl2 в 70 мл воды) 7е;
K2HgI4 + S(CH2—СН2С1)2 —> S(CH2—CH2I)2 • Hgl2 + 2KC1
Чувствительность реакции 0,05 мг/мл.
4) Образование оранжевой суспензии или желтого осадка
селена при нагревании иприта до 85°C с селенистой кислотой77
[раствор 1 г SeO2 в 100 мл разбавленной H2SO4 (1:1)]. Арсины
дают аналогичную реакцию.
5) Синий ред-окс-индикатор 2,6-дихлорфенол — индофенол в
присутствии иприта при pH 7 превращается в бесцветное веще-
ство78; чувствительность реакции 0,001 мг/мл.
6) Образование комплексных соединений из солей меди (II) и
иприта79; чувствительность реакции 0,1 мг/мл.
7) Образование с реагентом Несслера желтовато-белого
осадка81.
8) Кислые растворы йодата калия и крахмала в присутствии
иприта окрашиваются в синий цвет.
5.2.1.6. Идентификация иприта получением производных. Если
в распоряжении исследователя имеется большое количество ОВ,
80
например из трофейных боеприпасов, то его можно идентифициро-
вать переводом в производные, обладающие определенной темпе-
ратурой плавления.
Получение сульфимина113 [CH3C6H4SO2—N = S(CH2CH2C1)2]. При взаимо-
действии серного иприта с избытком водного раствора хлорамина Т через 1 ч
выпадают белые кристаллы сульфимина; т. пл. 144,6° С.
Получение диэтилендисульфида (дитиана)31
ZCH2— CH2x
s\ >
хсн2—сн/
При прибавлении иприта к 20%-ному раствору Na2S выпадает диэтилен-
дисульфид; т. пл. 111—112° С.
Получение сульфоксида85. При прибавлении по каплям серного иприта к
концентрированной HNO3 (d 1,40) образуется светло-зеленая жидкость, из ко-
торой при разбавлении ее водой выпадает белый осадок сульфоксида. После
перекристаллизации из 60%-ного спирта получаются бесцветные чешуйки;
т. пл. 110° С.
Получение сульфоксида86 возможно также растворением серного иприта
в ледяной уксусной кислоте и добавлением при охлаждении 30%-ной Н2О2.
5.2.2. Количественное определение
5.2.2.1. Фотометрическое определение с иодоплатинатом. Как
было установлено Чугаевым 87, органические сульфиды реагируют
с иодплатинатом по двум направлениям, приводящим в каждом
случае к выделению иода:
Pti;+4R2S —> Pt(R2S)*+ + 41” + 12
Pti; + 2R2S —> Pt(R2S)2I2+21" + 12
Эта реакция может быть использована для определения бис-2-
хлорэтилового тиоэфира по выделению иода, образующего с крах-
алом окрашенный в синий цвет продукт. На этом принципе осно-
ван ряд визуальных и фотометрических методов анализа 75-88-91.
По сравнению с другими эти способы имеют то преимущество,
что они меньше зависят от находящихся в исследуемом веществе
полисульфидов.
Реагенты
Иодоплатинат—1 мл 5%-иого раствора хлорида платины и 3,5 мл свеже-
приготовленного, не содержащего иода раствора Nal разбавляют дистиллиро-
ванной водой до 180 мл.
Раствор крахмала—1 г растворимого крахмала растирают с некоторым
количеством воды и выливают в 100 мл кипящей воды; применяют свежепри-
готовленный раствор.
Уксусная кислота — 50 мл уксусной кислоты разбавляют водой до 1000 мл.
К 10 мл водного раствора испытуемого вещества, содержащего 5% уксус-
ной кислоты, добавляют 2 мл раствора иодплатината и после перемешивания —
2 мл раствора крахмала. Через 4—5 мин колориметрируют смесь при 650—
70 нм в сравнении с 10 мл 5%-ной уксусной кислоты. Расчет проводят по
калибровочной кривой, для построения которой используют раствор серного
иприта в 5 %-ной уксусной кислоте, содержащий от 0,005 до 0,05 мг]мл ве-
щества.
81
5.2.2.2. Титрование бромид-броматным раствором. Разбавлен-
ный водный раствор тиоэфира количественно окисляется бромом
до сульфоксида:
R2S4-Br2 + H2O —> R2SO + 2HBr
Бис-2-хлорэтиловый тиоэфир можно определять титрованием
раствором брома в присутствии индикатора метилового красного92.
Вместо неустойчивого раствора брома более целесообразно исполь-
зовать получающийся электрохимически бром в момент выделения
или бромид-броматный раствор93. Метод применим только для
чистого иприта, так как полисульфиды и тиодигликоль реагируют
с бромом аналогично и мешают определению. Для исключения
ошибок, обусловленных воздействием брома на индикатор, удобнее
прибавлять избыточное количество бромид-бромата с последующим
йодометрическим его определением102.
Реагенты
Бромид-броматный раствор — раствор 11,9016 г КВг и 2,784 г КВгОз в
1000 мл воды.
Йодистый калий— 10%-ный водный раствор.
Тиосульфат натрия — 0,1 н. раствор.
В колбе Эрленмейера с притертой пробкой емкостью 250 мл отвешивают
0,1—0,3 г ОВ и растворяют в 40 мл уксусной кислоты. После прибавления 5 мл
воды и 3 мл концентрированной НС1 смесь медленно титруют бромид-броматным
раствором до появления желтого окрашивания. Затем, после прибавления 5 мл
раствора К! и выдерживания в темноте в течение 5 мин, титруют выделивший-
ся иод 0,1 н. раствором Na2S2O3 в присутствии индикатора крахмала. Расчет
проводят по следующей формуле:
7,952 (а - 6) 100
-----—j—------= % серного иприта
Л
где а — количество пошедшего на титрованве 0,1 н. раствора бромид-бромата, мл\
b — количество пошедшего на обратное титрование 1 н. раствора Na2S2O3, мл-,
А — навеска отравляющего вещества, мг.
5.2.2.3. Объемное микроопределение. В реакции бис-(2-хлор-
этилового)-'тиоэфира с водным раствором хлорамина Т наряду с
сульфимином (см. раздел 5.2.1.6) могут также получаться сульфок-
сид (реакция 1) и сульфон (реакция 2):
/Na
CH3C6H4SO2N^ +R2S + H2O —> CH3C6H4SO2NH2 + R2SO + NaCl (1)
'Cl
.Na
2CH3C6H4SO2N^ + R2S + 2H2O —> 2CH3CeH4SO2NH2 + R2SO2 + 2NaCl (2)
'Cl
Тип образующегося соединения зависит от кислотности раствора
и добавляемой кислоты. В уксуснокислом (до 50%) и сернокислом
(не выше 3 н.) растворах в реакцию вступает 1 моль хлорамина Т
и содержание сульфоксида в продуктах реакции составляет 90—
82
100%. В 2 н. растворе по НС1 расходуется 2 моль хлорамина Т и
образуется сульфон. В этом случае окисление осуществляется за
счет промежуточного образования хлора.
Колориметрический способ определения иприта основан на вза-
имодействии о-толидина с избытком взятого для проведения реак-
ции хлорамина Т в уксуснокислом растворе 94’95. Более высокая
чувствительность достигается в методе, разработанном Кинсейем
и Грантом96. Авторы показали, что в реакции бис-2-хлорэтилового
тиоэфира с дихлорамином Т в безводной среде в результате слож-
ного процесса хлорирования расход дихлорамина составляет 5моль
на 1 моль сульфида. Количество израсходованного дихлорамина Т
зависит от растворителя. Подходящим является циклогексан; до-
бавление циклогексанола сенсибилизирует каталитическое хлори-
рование иприта.
Подвергаемое анализу ОВ растворяют в смеси 80% циклогексана и 20%
очищенного керосина (фракция 175—280 °C) или экстрагируют этой смесью ОВ
из зараженной воды. В колбе Эрленмейера с притертой пробкой емкостью 50 мл
смешивают 1 мл раствора испытуемого вещества с 1 мл 0,2%-ного раствора
дихлорамина Т в четыреххлорнстом углероде и выдерживают 20 мин в термо-
статированной водяной бане при 27 ±0,1 °C. Затем прибавляют 4 Капли насы-
щенного раствора KI и 4 капли ледяной уксусной кислоты и после пере-
мешивания титруют реакционную смесь до обесцвечивания 0,01 и. раствором
Na2S2Qs, ограждая от попадания солнечного света. Метод удобен для опреде-
ления от 5 до 200 мкг серного иприта. Расчет содержания иприта по данным
титрования невозможен, так как расход дихлорамина всегда колеблется в за-
висимости от чистоты растворителя. Вычисления проводят по калибровочным
кривым, полученным титрованием стандартных растворов ОВ.
Для определения меньших количеств серного иприта от 0,2 до
0,5 мкг поступают следующим образом.
К 1 мл раствора ОВ в циклогексане прибавляют 1 мл 5%-ного раствора
циклогексанола в керосине. Смесь термостатируют в водяной бане при 27 ±0,1 °C
и после добавления 1 мл 0,1 %-кого раствора дихлорамина Т в четыреххлори-
стом углероде выдерживают ее при указанной температуре точно 20 мин. После
прибавления K.I и уксусной кислоты выделившийся иод титруют 0,1 н. раствором
Na2S20s. При построении калибровочной кривой нужно учитывать количество
тиосульфата, пошедшее на титрование контрольной пробы; 1 мл 0,01 н. раствора
Na2S2O3 соответствует примерно 5 мкг серного иприта.
В этом методе, как и в предыдущем, очень важно точно выдер-
живать Температуру и-продолжительность реакции.
5.2.2.4. Колориметрическое определение 1,2-бис-(2-хлорэтил-
тио)-этана (сесквииприта) при совместном присутствии с ипри-
том 73. Способ удобен для определения полуторного иприта (се-
сквииприта) в. смеси с ипритом. Содержание суммы ипритов пред-
варительно колориметрически определяют при помощи 4-(и-нитро-
бензил)-пиридина. Для этого спиртовой раствор смеси ОВ нагре-
вают 15 мин при 70 °C с 4- (n-нитробензил)-пиридином. В качестве
основания добавляют пиперидин (см. раздел 5.3.2.2). При этом
образуется окрашенное в фиолетовый цвет соединение
cicH2—сн2—s—сн2—сн2——NO2
83
содержание которого определяют колориметрически. Затем в от-
дельной пробе при помощи о-дианизидина в присутствии соли
меди (II) определяют колориметрически содержание 1,2-бис-(2-
хлорэтилтио)-этана. Смысл метода становится понятен из следую-
щего ряда реакций. Сначала образуется комплексное соединение
меди (II)
С1СН2—СН2—S
I
сн2
| + Си2+ —►
сн2
. С1СН2—СН2— S
С1СН2—сн2—
С1СН2—сн2—sz
которое в кислой среде (pH 1—2) окисляет о-дианизидин I в краси-
тель красного цвета:
С1СН2—СН2—С1СН2—СН2—S
Н2С \ \
| Cu2+ +1 —► | Си+ + Краситель
Н2С / Н2С /
С1СН2—СН2—SZ С1СН2—сн2—sz
Структура красителя аналогична структуре окрашенного про-
дукта, образующегося в реакции синильной кислоты с бензидином
и ацетатом меди. Описаны методы, в которых вместо солей меди
применялись соли кобальта 98.
Реагенты
о-Дианизидин— 0,1%-ный спиртовый раствор.
Ацетат меди — 0,1%-ный водный раствор; к 100 мл раствора добавляют
0,1 мл ледяной уксусной кислоты.
Смешивают 1 мл раствора ОВ в этаноле с 1 мл раствора о-дианнзидина и
3 мл раствора ацетата меди. Через 10 мин прибавляют 3 мл 18 н. H2SO4 и про-
водят колориметрирование при 540 нм в сравнении с раствором контрольной
пробы. Контрольную пробу ставят одновременно, используя все реагенты плюс
1 мл спирта. Расчет ведут по калибровочной кривой. Так как серный иприт при
взаимодействии с о-диапизидипом и ацетатом меди также дает незначительную
по интенсивности окраску, то для определения обоих ипритов при их совмест-
ном присутствии необходимо построить четыре калибровочные кривые:
I — полуторный иприт + 4-(га-нитробензил)-пиридин;
II — иприт + 4-(га-нитробензил)-пиридин;
III — полуторный иприт + о-дпанизидин ацетат меди;
IV — иприт + о-дианизидин + ацетат меди.
Формулы для расчета можно вывести из графического построения четырех
кривых. Коблин 73 приводит следующие формулы:
Кпи = 0,71 4£а - 0,005395ц
Кн = 0,253£в - 0,671£а
где Кпи — концентрация полуторного иприта; Ки— концентрация иприта; 5 а—
фотометрический отсчет в методе с о-дианизидином и ацетатом меди; Ев — фо-
тометрический отсчет в методе с 4-(п-нитробепзил)-пиридином.
Эти формулы, однако, целесообразно каждый раз проверять, так как полу-
чаемые отсчеты зависят от условий проведения определений.
84
5.2.2.5. Другие методы определения бис-2-хлорэтилового тио-
эфира. Колориметрическое количественное определение серного ип-
рита может быть выполнено теми же методами, которыми поль-
зуются для его обнаружения, например при помощи тимолфталеи-
на или тиомочевиной в присутствии соли никеля. Методы, опи-
санные для обнаружения и определения азотистого иприта 8-окси-
хинолином, в соответствующей модификации31 пригодны и для
серного иприта. Для этой цели применим и весьма употребитель-
ный способ с 4- (га-нитробензил)-пиридином.
Из других способов назовем следующие.
1) Косвенное определение иприта по данным количественного
элементного анализа, например по количеству хлора или серы в
веществе после его минерализации в токе кислорода по Гротте и
Крекелеру или в кислородной колбе по Шёнигеру. Серу можно
также определять после окисления образца перманганатом калия
в виде сульфата бария97, а хлор — титрованием по Фольгарду
после гидролитического разложения испытуемого образца в при-
сутствии триэтиламина 9Э.
2) Титрование исследуемого вещества раствором гипохлорита
натрия с установленным титром в присутствии индикаторов мети-
лового или нейтрального красного 10°.
3) Осаждение в виде двойной соли с хлоридом меди (I) и по-
следующее йодометрическое определение избытка соли меди ,01.
4) Нефелометрическое определение суспензии металлического
селена, образующегося при нагревании иприта с селенистой кис-
лотой 77.
5) Реакция с сульфгидрильными группами цистеина, уреазы,
папаина или денатурированного овальбумина и последующее опре-
деление остаточных SH-групп юз—105 Метод пригоден для опреде-
ления не только серного иприта, но и всех ОВ, действующих на
сульфгидрильные группы, в частности, таких, как бромацетон, хлор-
пикрин, эфиры иод- и бромуксусной КИСЛОТЫ.
5.3. ГАЛОГЕНЗАМЕЩЕННЫЕ АЛИФАТИЧЕСКИЕ
ТРЕТИЧНЫЕ АМИНЫ (АЗОТИСТЫЕ ИПРИТЫ)
5.3.1. Качественное обнаружение
Для обнаружения азотистого иприта наряду со способами, при-
менимыми для индикации серного иприта (действием щелочного
раствора тимолфталеина или тиомочевиной в присутствии соли ни-
келя), пригодны способы, основанные на применении реагентов, оса-
ждающих третичные амины. Большая часть алкалоидов, а также
холин- и тиохолинфосфорильные соединения способны осаждаться
этими реагентами и потому мешают индикации азотистого иприта.
5.3.1.1. Осаждение калийвисмутиодидом (реагент Драгендор-
фа— Краута) ,06. При взаимодействии азотистого иприта с калий-
висмутиодидом образуется осадок от оранжевого до красного цвета
следующего состава: ВПз-М(СН2СН2С1)з-Н1.
85
Реагент Драгендорфа — смеси раствора 8 г Основного нитрата висмута в
20 мл HNO3 (d 1,18) и раствора 27,2 г KI в 40 мл воды дают постоять 1—2 су-
ток, после чего раствор сливают с выделившегося осадка KNO3 и доводят объем
водой до 100 мл.
Смешивают 5 мл водного раствора испытуемого вещества с 0,5 мл разбав-
ленной НС1 (1:1) и 1 мл реагента Драгендорфа. Появление красно-оранжевого
осадка указывает на присутствиё азотистого иприта; чувствительность реакции
0,01 мг/мл. Обнаружению мешает серный иприт при большом его содержании
(больше 1 мг), который дает аналогичную реакцию.
5.3.1.2. Осаждение фосфорновольфрамовой кислотой. Для сни-
жения мешающего действия люизита, метил- и этилдихлорарсинов,
адамсита и бромбензилцианида реакцию следует проводить в со-
лянокислом растворе.
Реагент
Фосфорновольфрамовая кислота — 5%-иый водный раствор.
Смешивают 5 мл водного раствора испытуемого вещества с 0,5 мл НС1 и
1 мл раствора фосфорновольфрамовой кислоты. Появление помутнения или сту-
денистого осадка указывает на присутствие азотистого иприта; чувствитель-
ность реакции 0,1 мг/мл.
5.3.1.3. Другие реагенты, образующие осадки с азотистым ип-
ритом. Кремневольфрамовая кислота 107 дает осадок состава
12WO3-SiO2-[N(CH2CH2Cl)3]4, что может быть использовано для
весового определения азотистого иприта.
Соль Ре'йнекг NH^Cr (NH3)2(SCN)4] и другие комплексные соли
хрома образуют с азотистым ипритом осадки розового цвета, даю-
щие возможность обнаруживать 0,005 мг/мл вещества 108
Реагент Несслера 109 на холоду приводит к образованию желто-
го, а при нагревании — коричневого осадка. Открываемый мини-
мум примерно 0,2 мг/мл.
5.3.1.4. Обнаружение азотистого иприта по полученным произ-
водным. В простейшем случае идентификацию можно проводить
определением температур плавления перекристаллизованных из
ацетона гидрохлоридов. Гидрохлорид бис- (2-хлорэтил)-метиламина
плавится при 110°С, а трис-(2-хлорэтил)-амина — при 130—131 °C.
Для этой же цели пригодны соответствующие пикраты. Водный
раствор испытуемого вещества смешивают с насыщенным водным
раствором пикриновой кислоты. При достаточном количестве хлор-
этиламина выпадающие желтые пикраты можно перекристаллизо-
вывать из этанола, слегка разбавленного водой, и определить их
температуру плавления. Пикрат бис-(2-хлорэтил)-амина плавится
при 133 °C, а пикрат трис-(2-хлорэтил)-амина — при 135 °C. При
такой же температуре плавится и пикролонат трис-(2-хлорэтил)-
амина, получаемый при действии пикролоновой кислоты.
5.3.2. Количественное определение
5.3.2.1. Колориметрическое определение ипритов при помощи
8-оксихинолина (оксина)31’110'111. В основе этого специфического
метода лежит образование в щелочной среде окрашенного про-
86
дукта взаимодействия азотистого или серного ипритов с 8-окси-
хинолином неустановленной структуры.
Реагенты
8-Оксихинолин— 5%-ный раствор в 95%-ном этаноле.
Бикарбонат натрия— 10%-ный водный раствор.
Смешивают 2,5 мл водного раствора испытуемого вещества с 2,5 мл
95%-ного этанола и слегка подкисляют несколькими каплями 5 %-ной уксусной
кислоты. Добавляют 0,5 мл раствора 8-оксихинолина и встряхивают реакцион-
ную смесь. После добавления 0,5 мл раствора Па2СОз продолжают встряхивать
еще 30 сек и затем оставляют стоять 90 мин при комнатной температуре. Рас-
твор колориметрируют при 486 нм в сравнении с контрольной пробой. Расчет
проводят по калибровочной кривой, построенной для концентраций ОВ 0,01 —
1 мг!мл. При определении серного иприта31 реакционную смесь выдерживают
90 мин при 45 °C и колориметрируют при 500 нм.
В качественной пробе присутствие азотистого иприта устанав-
ливают по ярко-оранжевому окрашиванию реакционной смеси;
окраска контрольной пробы должна быть желто-зеленой.
5.3.2.2. Колориметрическое определение при помощи 4-(п-нитро-
бензил)-пиридина 114. Этот метод весьма многосторонне использу-
ется для обнаружения и определения алкилирующих соединений.
Он основан на наблюдении Кёнига112, получившего в реакции
йодистого метила с 4-(n-нитробензил)-пиридином в щелочной сре-
де синий кррситель, которому была приписана следующая струк-
тура
СН3—N
СН
2
В период второй мировой войны американские исследователи
(Броун, Гехауф и др.) установили общую применимость этой цвет-
ной реакции и разработали различные методы обнаружения таких
ОВ, как серный иприт, дифенилхлорарсин и др. Эпштейн 113 про-
должил эти работы и разработал способы обнаружения этилен-
иминов и других алкилирующих соединений, в том числе многих
ОВ. Путем сравнения интенсивности окраски продуктов, получае-
мых в результате взаимодействия различных соединений с 4-(п-нит-
робензил)-пиридином, была определена их активность или актив-
ность содержащихся в них функциональных групп по отношению
к этому реагенту, что иллюстрируется следующим рядом (X — га-
логен) :
rch2ch2x > rch2chx2 > rch2cx3
АгСН2Х
roch2x >
С\сн2х
> АгХ
ROCH2CH2X
> -с\
\х
Установлено, что в ряду галогенпроизводных активность убы-
вает от иод- к хлорсодержащим соединениям 115. Реакция проходит
через образование галогенидов n-нитробензилпиридиння, которые
87
в присутствии органических или неорганических оснований (пипе-
ридин, триэтиламин, карбонаты и гидроокиси щелочных металлов),
в результате потери протона, превращаются в окрашенные соеди-
нения:
RX + N
Наряду с алкилирующими соединениями эту цветную реакцию
дают следующие ОВ: фторангидриды эфиров О-алкилметилфосфо-
новых кислот (зарин и зоман)29-51, серные иприты113, вторичные и
третичные азотистые иприты114’116-119, мышьяксодержащие ОВ
(люизит, метил-, этил- и фенилдихлорарсины и дифенилхлорар-
син) 113, метилфторацетат из, хлорацетофенон120.
Оптимальные условия образования окраски для каждого соеди-
нения различны. Минимальные обнаруживаемые количества для
некоторых соединений, например для серного иприта, составляют
несколько микрограммов в 1 мл, для других ОВ, таких, как фос-
фонаты, чувствительность еще меньше. Савицкий и его сотр. 115 про-
должили работу по применению других производных пиридина.
Исключительно высокая чувствительность, являющаяся следствием
аутокаталитического эффекта, достигается при обнаружении неко-
торых соединений 4-пиридинкарбоксиальдегид-2-бензтиазолилгидр-
азоном (т. пл. 70—71 °C). Этим реагентом можно обнаружить
0,05 мкг!мл 1-иодбутана; он с успехом использовался также для
проведения капельных реакций на бумаге.
Реагенты
4-(п-Нитробензил)-пиридин (т. пл. 70—7ГС)—5%-ный раствор в ацетоне.
Ацетатный буфер — 0,1 н. раствор, pH 4,62.
В пробирку с пришлифованной пробкой вливают пипеткой 3 мл водного
раствора ОВ, 1 мл ацетатного буфера, 0,4 мл раствора 4- (n-нитробензил)-пири-
дина и на 20 мин помещают пробирку в кипящую водяную баню. Затем ее
охлаждают в ледяной] бане и последовательно прибавляют 2 мл ацетона, 4 мл
этилацетата и 1,5 мл 0,25 н. раствора NaOH. Смесь встряхивают примерно 20 раз,
содержимое переносят в пробирку для центрифугирования и центрифугируют
2 мин. Соответствующее количество верхнего слоя жидкости пипеткой переносят
в кювету и колориметрируют при 540 нм, сравнивая с пробой, приготовленной
на воде. Вследствие неустойчивости образовавшейся окраски все операции, начи-
ная от прибавления едкого натра и до колориметрирования, должны проводить-
ся в течение 3—5 мин при затемнении. Расчет проводят по калибровочной кри-
вой, которую строят, пользуясь приведенной методикой, добавляя азотистый
иприт в количестве 0,5—10 мкг.
5.3.2.3. Другие методы определения. 1) При гидролизе азоти-
стого иприта, содержащего третичный атом азота (C1CH2CH2)3N,
быстро возникают ионы этиленимония, скорость образования ко-
торых растет с увеличением концентрации гидроксильных ионов114;
88
при pH 6 реакция в основном заканчивается в течение 5 мин. Го-
ломбик, Фрутон и Бергманн 121 установили, что ион этиленимония
может быть определен обменной реакцией с тиосульфатом нат-
рия 121:
R4 + /СН2 R\
N I "Ь Na2S2O3 —> N—СН2—СН2 S2O3N<i-}- Na*
RZ \сн2 rz
Оказалось, однако, что при прибавлении избыточного количе-
ства тиосульфата к раствору азотистого иприта и обратном его
титровании раствором иода процесс протекает не количественно.
Это объясняется наличием побочных реакций, например гидролиза
ионов этиленимония или их димеризации. Иначе гидролизуется азо-
тистый иприт, содержащий вторичный атом азота (ClCHzCHs^NH.
Между ионом этиленимония и этиленимином устанавливается рав-
новесие
С1СН2—СН2Х + /СН2 уСН2
I <=* С1СН2—СН2—NZ | + Н+
HZ ZCH2 ZCH2
зависящее от pH среды.
Так, начиная от pH 3 до pH 5 скорость превращения вторич-
ного амина в p-хлорэтильное производное этиленимина возрастает,
после чего она снижается и уже при pH 8 становится равной
нулю ш. Этим можно объяснить устойчивость иона этиленимония
в этих условиях к нуклеофильной атаке. Аллен и Зиман 122 иссле-
довали реакцию этиленимина с тиосульфатом:
+н2о
—I + Na2S2O3 —N—СН2—СН2—S2O3Na + NaOH
\cHl ,1
Они установили, что при pH 4 этилениминные соединения мо-
жно количественно определить при проведении реакции с большим
избытком тиосульфата по расходу кислоты, идущей на титрование
щелочи. В бикарбонатном буферном растворе разложение вторич-
ных азотистых ипритов идет нестехиометрично, с образованием ок-
сазолидоновых производных 119, поэтому расход тиосульфата не мо-
жет быть установлен.
2) Определение трис-(2-хлорэтил)-амина возможно осаждением
калийвисмутиодидом, растворением осадка при прибавлении 8-ок-
сихинолина и бромометрическим определением висмутоксихинолята
после растворения его в соляной кислоте 106. Количества иприта до
0,1 мг определяются этим методом с хорошей точностью.
3) М-Метил-бис-1Ч,М-(2-хлорэтил) -амин можно определять реак-
цией обменного разложения с n-фенилфенолом в щелочном рас-
творе и измерением флуоресценции продукта реакции 123,
89
4—5) Засс и corp.124 описали два колориметрических способа
определения третичных аминов (солей и оснований) и солей тет-
раалкил (арил) аммония.
Первый метод представляет собой модификацию известного спо-
соба определения аминов при помощи хлоранила 125, по которому
первичные амины образуют продукты, окрашенные в красный
цвет, вторичные амины — продукты, окрашенные в фиолетовый
цвет, а третичные амины (соли и основания) —продукты, окрашен-
ные в смарагдово-зеленый цвет:
Последние определяют фотометрированием окраски при 610 нм.
Этим методом можно определять до 50 мкг]мл третичных аминов.
По второму способу для определения свободных оснований и
солей третичных аминов, а также солей тетраалкил (арил) аммониев
используют образование окрашенных в красный цвет продуктов
их взаимодействия с ангидридом х{ыс-аконитовой кислоты в среде
уксусного ангидрида:
/°
Н2С—С" н2с—сх
| \он | \он
С—С=О + NR3 —► С=С—О" .
I > I >.
НС—-С=О НС=С—ONR3
Чувствительность определения 3 мкг/мл; фотометрирование
окраски проводят при 500 нм.
6) Определение азотистого иприта щелочным раствором тимол-
фталеина, а также тиомочевиной с образованием солей тиурония
см. разделы 5.2.1.1 и 5.2.1.2.
5.4. МЫШЬЯКСОДЕРЖАЩИЕ ОВ
5.4.1. Качественное обнаружение
При анализе мышьяксодержащих ОВ следует наряду с обнаруже-
нием собственно ОВ уделять внимание также и обнаружению ток-
сических продуктов их разложения. Это имеет значение, в частно-
сти, при анализе питьевой воды и продуктов питания.
5.4.1.1. Общие методы обнаружения. Восстановление до мышья-
ковистого водорода. Водород в момент выделения восстанавливает
первичные и вторичные, алифатические арсины, такие, как метил-
и этилдихлорарсины, 2-хлорвинилдихлорарсин (а-люизит) и ди-
90
(2-хлорвинил)-хлорарсин (р-люизит), до мышьяковистого водоро-
да; для трис- (2-хлорвинил)-арсина (у-люизит) и ароматических
арсинов — фенилдихлорарсина, дифенилхлорарсина (кларк-I), ди-
фенилцианарсина (кларк-П), дифениламинхлорарсина (адамсит)
такое прямое восстановление невозм,ожно. Эти соединения должны
быть предварительно минерализованы методами, приведенными в
разделе 5.4.2.1, посвященном количественному определению мышь-
яксодержащих соединений. Образующуюся при этом трехокись
мышьяка затем восстанавливают.
Обнаружение образующегося мышьяковистого водорода прово-
дится при пом'ощи следующих характерных реакций.
Реакция с нитратом серебра ,2в.
Реагент
Индикаторная бумага — пропитывают 20%-ным раствором AgNO3 фильтро-
вальную бумагу; приготавливается непосредственно перед употреблением.
В пробирку вливают несколько миллилитров подкисленного 20%-ной H2SO4
испытуемого вещества или раствора после минерализации образца, добавляют
кристалл CuSO4 и 2 гранулы цинка. Затем в верхнюю часть пробирки вклады-
вают ватный тампон, пропитанный ацетатом свинца, н на отверстие пробирки
накладывают индикаторную бумагу. Образующееся через некоторое время на
бумаге лимонно-желтое пятно состава AsAg3 • AgNO3 после обработки водой
чернеет вследствие выделения металлического серебра.
Применяемые серная кислота и цинк не должны содержать мышьяка.
Реакция с бромидом ртути(П)12Т. Пропитанная раствором бромида рту-
ти(П) индикаторная "бумага в присутствии и мышьяка окрашивается в желтый
(до коричневого) цвет, который при обработке раствором иодида калия пере-
ходит в красно-коричневый:
3HgBr2 + AsH3 —► As(HgBr)3 + ЗНВг
As(HgBr)3 + AsH3 —► As2Hg3 + 3HBr '
Реакция с хлоридом золота™. На пропитанной 1%-ным раствором хлорида
золота бумаге в присутствии мышьяка в результате восстановления золота по-
является синее до сине-красиого пятно:
2AuCl3+ AsH3 + 3H2O —► 2Au 4-6НС1 + H3AsO3
Эта реакция наиболее чувствительна.
Реакции с сероводородом129. При взаимодействии мышьяк-
содержащих ОВ с H2S выпадает осадок арсйнсульфидов.
К 2—3 мл водного раствора испытуемого вещества приливают свежеприго-
товленный подкисленный НС1 раствор сероводорода до прекращения образова-
ния осадка. При исследовании ароматических арсииов готовят спиртовые рас-
творы вещества и приливают насыщенный спиртовой раствор сероводорода.
Иногда при осаждении сульфидов испытуемый раствор охлаж-
дают ледяной водой. Удобно применять так называемый «твер-
дый сероводород», т. е. такие соединения, как тиоацетамид или тио-
карбамат аммония, выделяющие в кислой среде сероводород.
Приводим характерные окраски осадков, образуемых некото-
рыми мышьяксодержащими ОВ:
а-люизит образует белый- осадок С1СН=СН—As=S, чувстви-
тельность реакции 0,02—0,05 мг!мл\
91
метилдихлорарсин — желтоватый осадок СНз—As=S, т. пл.
110°С;
этилдихлорарсин— желтоватая масляная эмульсия, чувстви-
тельность реакции 0,02—0,05 мг/мл;
фенилдихлорарсин— белый осадок С6Н5—As = S, т. пл. 152°С;
дифенилхлор- и дифенилцианарсины — белый осадок
(C6H5As)2S, т. пл. 67 °C.
Реакции с нитратом ртути I. При прибавлении к водному рас-
твору исследуемого образца нескольких капель слабо подкислен-
ного 5%-ного водного раствора нитрата ртути(1) некоторые мышь-
яксодержащие ОВ образуют следующие осадки 135:
при взаимодействии с люизитом образуется белый осадок кало-
мели Hg2Cl2, который через 12 ч сереет, чувствительность реакции
1 мг/мл;
с метилдихлорарсином — серый осадок металлической ртути,
чувствительность 1 мг/мл;
с этилдихлорарсином — быстро сереющий белый осадок, чув-
ствительность 2 мг/мл;
с фенилдихлорарсином — белый осадок;
дифенилхлор- и дифенилцианарсины через некоторое время об-
разуют белый осадок.
Реакции с фосфорноватой кислотой. Все мышьяксодержащие
ОВ, за исключением адамсита, образуют с фосфорноватой кислотой
характерные осадки.
Реагент
Фосфорноватая кислота 130 — растворяют 10 г гипофосфата натрия Na4P20e
при нагреваний в 10 мл воды и доводят объем концентрированной НО до
100 мл. После декантации (выпадает осадок NaCl) на каждые 10 мл раствора
добавляют 1—2 капли 0,1 н. раствора иода в KI.
Для проведения реакции к 1—2 мл водного испытуемого раствора прили-
вают 5 мл реагента и наблюдают за образованием осадка (при необходимости
нагревают 15—30 мин на водяной бане).
В присутствии мышьяксодержащих ОВ образуются следующие
осадки:
люизит дает белый, в последствии желтеющий осадок;
метил- и этилдихлорарсины — желтый или желтовато-коричне-
вый осадок;
фенилдихлорарсин — осадки от белого до желто-зеленого цвета;
дифенилхлор- и дифенилцианарсин — после их нагревания с ре-
агентом возникает белое помутнение.
Иприт в больших концентрациях образует с фосфорноватой
кислотой эмульсию, при нагревании которой, вследствие слияния
капелек, получается прозрачный раствор.
Другие общие способы индикации. 1) Первичные алифатиче-
ские арсины, например люизит, метил- и этилдихлорарсины, вос-
станавливают четырехокись осмия до черной двуокиси осмия131:
OsO4 + 2RAsC12 + 4Н2О —> OsO2 + 2RAsO(OH)2 + 4НС1
92
Для индикаторных трубок может быть использован силикагель,
пропитанный 1%-ным раствором OsC>4.
2) Реакция первичных алифатических арсинов с раствором хло-
ристого олова в концентрированной НС1 (реакция Беттендорфа) 132.
При просасывании воздуха, содержащего люизит, через индикатор-
ные трубки, наполненные силикагелем, пропитанным солянокис-
лым раствором SnCl2, появляется желтое кольцо.
3) При просасывании воздуха, содержащего алкилдихлорарси-
ны, через силикагель, пропитанный молибденовой кислотой и суль-
фатом цинка, возникает синее окрашивание вследствие восстанов-
ления молибденовой кислоты 133. Реакция очень чувствительна.
4) Продукты, полученные при окислении раствором иода клар-
ка I или II и адамсита, образуют с уксуснокислым раствором нит-
рата уранила осадки. Например, при наличии адамсита образуется
зеленый осадок 134.
5) Мышьяксодержащие ОВ' (за исключением адамсита) можно
обнаружить по реакции Гриньяра136 (см. раздел 5.2.1.3); реакция
очень чувствительна.
Обнаружение мышьяксодержащих ОВ получением кристалли-
ческих производных. Можно идентифицировать мышьяксодержа-
щие ОВ по температурам плавления соответствующих продуктов
гидролиза или окисления, а также по температурам плавления ар-
синсульфидов. Ниже в таблице приведены температуры плавления
таких производных.
Отравляющее вещество Продукт гидролиза T. пл., °C Продукт окисления T. пл. °C
Метилдихлорар' СИН CH3As=O 95 CH3AsO(OH)2 159
Этилдихлорар- сии C2HsAsO(OH)2 99-100
2-Хлорвинилди- хлорарсин (а-люизит) . . CHCl=CHAsO 143 CHC1=CH—AsO(OH)2 130
Ди-(2-хлорви- нил)-хлорар- син (Р-люизит) [(CHC1=CH)2As]2O 62-63 (CHC1=CH)2AsO(OH)2 120-122
Дифенилхлор- и дифенил- цианарсины (кларк I и II) [(C6H5)2As]2O' 92,5-93,5 (CsHs)2AsO(OH)2 175
10-Хлор-Э, 10-ди- гидрофенар- са зинхлорид (адамсит) . . (NH(C6H4)2As]2O 350 — —
5.4.1.2. Специфические методы индикации. Индикация люизита.
1) Весьма чувствительной и специфичной реакцией обнаружения
люизита является его индикация по ацетилену, образующемуся при
93
взаимодействии с сильными щелочами и определяемому реагентом
Илосвая 137 в виде ацетиленида меди:
C1CH=CHAsC12 + 6NaOH —► 2HCs=CH + Na3AsO3 + 3H2O + 3NaCl
HC=CH + 2Cu+ —► CuC=s=CCu + 2H+
В пробирке смешивают 2 мл водного или 1 ял спиртового раствора испы-
туемого вещества с 1 ял воды и 0,5 ял 30%-ной щелочи. Пробирку закрывают
и легким ее покачиванием перемешивают содержимое. Через 5 мин при периоди-
ческом охлаждении водой раствор слабо подкисляют разбавленной уксусной
кислотой. Красная окраска, а При высоких концентрациях красный осадок, по-
являющиеся тотчас или через короткое время после прибавления 1 ял реагента
Илосвая, указывают на присутствие а-люизита. Реакция специфична. Чувстви-
тельность реакции 0,005 мг!мл а-люизита.
Прибавление 2%-ного раствора желатины стабилизует коллоид-
ный раствор ацетиленида меди, что дает возможность проводить
количественное определение путем сравнения со стандартным рас-
твором.
Несколько модифицированный реагент138 , содержащий 0,2 г
СиСОз, 12 г Аз20з, 12 г NaOH и 1 мл пиперидина в 100 мл воды,
дает с а-люизитом яркое красное окрашивание. Кроме того, воз-
можно осаждение желтого ацетиленида серебра добавлением рас-
твора AgNO3.
2) Силикагель, пропитанный раствором эргостерина * в хлоро-
форме, при соприкосновении с парами люизита окрашивается в
фиолетовый, а при высоких концентрациях в глубокий зеленый
цвет 139. Серный иприт в больших концентрациях при длительном
воздействии вызывает только слабое окрашивание; пары соляной
кислоты при взаимодействии с эргостерином вызывают буро-корич-
невую окраску.
Индикация дифенилцианарсина (кларк П>. Присутствие дифе-
нилцианарсина в испытуемом веществе может быть установлено
нагреванием пробы со спиртовым раствором КОН и определением
циангруппы одним из обычных методов.
Индикация адамсита (10-хлор-9,10-дигидрофенарсазинхлорида).
1) Простой способ обнаружения основан на образовании окра-
шенного в интенсивно красный цвет соединения при действии кон-
центрированной H2SO4 на кристалл испытуемого вещества (или
на остаток после выпаривания экстракта). Природа окрашенного
продукта не установлена. Бромбензилцианид в этих условиях вы-
зывает коричнево-красное окрашивание.
2) Высокочувствительным и специфичным методом обнаруже-
ния адамсита является реакция получения интенсивно окрашенной
* СгвНцО, провитамин, при УФ-облучении превращается в витамин Д2.
94
в красный цвет натриевой соли его продукта нитрования:
К нескольким кристаллам испытуемого ОВ или к остатку, полученному по-
сле выпаривания 5—10 мл экстракта, прибавляют 1 каплю концентрированной
HNO3 и выпаривают досуха на водяной бане. После охлаждения к остатку при-
бавляют 1 каплю 30%-ного раствора NaOH.
Появление вишнево-красного окрашивания указывает на присутствие адам-
сита; чувствительность реакции 0,5—1,0 мкг/мл.
Таблица 4. Идентификация мышьяксодержащих ОВ
Отравляющее вещество Фосфорноватая кислота (реагент Б у го) Сероводород Нитрат ртути (I) Реактив Илосвая Концен- триро- ванная H2SO4
Люизит Белый осадок, постепенно становящийся желто-корич- иевым Белый осадок Белый осадок, постепенно сереющий Крас- ное окра- шива- ние —~
Метилдихлор- арсин Желтый до жел- то-коричие- вого осадок Желтоватый осадок Серый осадок — —
Этилдихлор- арсии То же Желтоватое масло Белый осадок, быстро сереющий — —
Фенилдихлор- арсин Белый до жел- то-зеленого осадок Белый осадок Белый осадок — —•
Дифенилхлор- арсин После нагре- вания белое Белое помутне- ние * Белый осадСж * — —
Днфенилцианар- син То же Белое помут- нение * Белый осадок * — —
Адамсит Крас- нее окра- шива- ние
* Возникает через некоторое время.
95
3) Более сложной по выполнению является реакция, основан-
ная на образовании дифениламина при нагревании адамсита с
иодистоводородной кислотой 140:
HN(C6H4)2AsC1 + 2HI —> (CsH5)2NH+ AsC1I2
Дифениламин можно отогнать с водяным паром и обнаружить
по появлению синего окрашивания при прибавлении нитрата и
концентрированной H2SO4. Для индикации адамсита может быть
также использовано появление синего окрашивания при действии
на него концентрированной H2SO4, содержащей некоторое количе-
ство HNO3, т. е. по сути используется реакция, характерная для
дифениламина.
4. Реагент, состоящий из равных объемов 10%-ного раствора
AgNO3 и ледяной уксусной кислоты, является специфическим для
адамсита. При нагревании пробы на водяной бане образуется
устойчивое зелено-желтое окрашивание, различимое уже при на-
личии 0,02 мг адамсита 141.
Идентификация мышьяксодержащих ОВ. Метил-, этил- и фе-
нилдихлорарсины, а также кларк I, для которых нет специфи-
ческих реакций, обнаруживают по некоторым различиям при вза-
имодействии их с H2S, Н4Р2О6, HgNO3 (табл. 4). Предварительно
удостоверяются в присутствии мышьяка в исследуемом ОВ восста-
новлением его до AsH3 (см. раздел 5.4.1.1).
5.4.2. Количественное определение
5.4.2.1. Определение в виде мышьяковистого водорода. Общий
принцип этого способа приведен в разделе 5.4.1.1 при описании
качественных реакций. Во многих случаях восстановлению до
AsH3 должна предшествовать минерализация испытуемого веще-
ства. Это относится к неподдающимся непосредственному вос-
становлению ароматическим арсинам и к определению мышьяк-
содержащих ОВ в продуктах питания и, прежде всего, в жирах.
Минерализация осуществима при помощи ряда способов. Для
исследования продуктов питания был выбран следующий способ
минерализации.
Измельчают примерно 100 г исследуемого материала, основательно его пере-
мешивают и отвесив 2—3 г помещают в колбу Кьельдаля. Добавляют 5 мл
30%-ной Н2О2 (пергидрол), примерно через 10 мин (по каплям) 10 мл H2SO4
(d 1,84) и осторожно нагревают колбу на малом пламени. При побурении реак-
ционной массы нагревание прекращают, колбу охлаждают и, после прибавления
нескольких миллилитров перекиси водорода, снова нагревают до выделения па-
ров. Это повторяют до тех пор, пока после 15 мин кипячения жидкость больше
не темнеет. Процесс минерализации жиров может продолжаться несколько ча-
сов. Убедившись, что в горловине колбы нет прилипших твердых частиц, при-
бавляют 0,5 г гидразинсульфата для восстановления As (V) до As (III). После
повторного нагревания в течение 30 мин и последующего охлаждения содержи-
мое колбы может быть использовано для определения мышьяка.
Подобным образом можно обрабатывать небольшие количества
выделенных из продуктов питания 142 или из жиров143 отравляю-
96
щих веществ или Их спиртовых экстрактов. Следует заметить, что
без предварительного восстановления гидразинсульфатом получае-
мого при минерализации As (V) образование мышьяковистого
водорода настолько замедляется, что продол-
жительность определения увеличивается более
чем вдвое.
Реагенты
Индикаторная бумага — фильтровальная бумага,
пропитанная примерно в течение 1 ч 5%-ным спирто-
вым раствором HgBr2 и высушенная на воздухе (бу-
магу хранят в эксикаторе над P2Os и предохраняют от
соприкосновения с воздухом).
Цинк гранулированный, не содержащий мышьяка —
перед употреблением его помещают на 1—2 мин в
0,05%-ный раствор CuSO< и промывают водой.
Свинцово-ацетатная вата — вата, пропитанная
5%-ным раствором ацетата свинца' и высушенная на
воздухе.
Серная кислота — 20%-ная, не содержащая мышьяка.
В колбу аппарата Гутцейта (рис. 6) наливают
10 мл минерализованного раствора, предварительно до-
веденного до определенного объема, 20 мл H2SO< и до-
водят водой объем до 50 мл. Поместив в нижнюю
часть трубчатой насадки неплотный тампон свинцово- Рис. 6. Приборы для
ацетатной ваты, а в верхнюю часть — полосу индика- определения мышьяка
торной бумаги, в колбу вносят 5 г цинка, быстро закры- по Гутцейту.
вают ее насадкой и помещают на 1 ч в темное место.
Содержание мышьяка определяют, сравнивая окраску
индикаторной бумаги с бумажными эталонами, полученными восстановлением
раствора трехокиси- мышьяка определенной концентрации. В течение некоторого
времени (при хранении в темноте) эти эталоны могут быть использованы в ка-
честве шкалы сравнения.
При применении бумаги определенного размера длина окрашенной зоны
является мерой содержания мышьяка 1П. При ширине бумаги 2 мм содержание
мышьяка соответствует следующим количествам:
Длина бумаги, JHJH Мышьяк, мг Длина бумаги, JHJH Мышьяк, ма
2 0,02 14,5 0,25
5 0,05 16 0,30
9 0,1 18 0,35
11 0,15 20 0,40
13 0,2 22 0,50
При других модификациях насадки к аппарату Гутцейта инди-
каторную бумагу вставляют в калиброванное отверстие. Окраску
круглого пятна сравнивают с окраской независимо изготовленных
бумажных эталонов или со специально отпечатанной цветной шка-
лой. В каждом случае для контроля реагентов (цинк, H2SO4) на
присутствие мышьяка желательно проведение контрольной пробы.
Этим способом можно определять до 1 мкг мышьяка.
Весьма чувствительным и хорошо воспроизводимым является
колориметрическое определение выделяющегося мышьяковистого
водорода диэтилдитиокарбаматом серебра 318>319.
4 Зак, 677
Реагент
Диэтилдитиокарбамат серебра — соль готовят растворением 2,25 г диэтил-
дитиокарбаминовой кислоты или эквивалентного количества ее натриевой соли
в 100 мл воды и к раствору отдельными порциями приливают раствор нитрата
серебра (1,7 г AgNO3 в 100 мл воды). При этом осаждается лимонно-желтая
серебряная соль. Ее отфильтровывают и после тщательной промывки водой су-
шат в вакуумэксикаторе. Соль хранят в темном месте.
Для анализа применяют 5%-ный раствор диэтилдитиокарбамата серебра в
свежеперегнанном пиридине. При хранении в темном, прохладном месте раствор
годен в течение нескольких месяцев.
Мышьяковистый водород выделяют обычным путем. Колбу, в которой про-
исходит выделение, соединяют трубкой с сосудом для поглощения, в котором
находится определенное количество (5 мл) раствора реагента. Через 30 мин
измеряют экстинкцию появившегося красного окрашивания при 540 нм. Расчет
проводят по калибровочной кривой, построенной с применением трехокиси
мышьяка; чувствительность метода — 0,05 мкг мышьяка.
Определению мешает сурьмянистый водород, образующий с реагентом та-
кое же красное окрашивание, сероводород вызывает черное окрашивание. Дей-
ствие сероводорода можно исключить, если в трубку, соединяющую колбу с по-
глотителем, поместить тампон свиицово-ацетатной ваты для связывания серо-
водорода в сульфид свинца.
5Л.2.2. Йодометрическое определение мышьяксодержащих ОВ.
В этом способе, применимом для определения всех мышьяксо-
держащих ОВ, испытуемое вещество минерализуют и количество
образовавшейся Мышьяковой кислоты определяют йодометриче-
ским титрованием. В некоторых случаях мышьяковую кислоту в
кислой среде восстанавливают иодидом калия до мышьяковистой
(реакция 1). Образовавшийся иод удаляют кипячением и мышья-
ковистую кислоту титруют раствором иода (реакция 2):
(П .
As20s -f- 4HI * AS2O3 “h 2Н2О 212
(2)
При количественном определении мышьяковистой кислоты
избыток иодистоводородной кислоты связывают добавлением би-
карбоната натрия:
HI + NaHCOj —► Nal + Н2О + СО2
1) Определение по немецкой фармакопее (DAB6). Взвешивают 0,2—0,3 г
испытуемого вещества в колбе Кьельдаля и после прибавления 10 мл концен-
трированной H2SO4 и 1 мл дымящей HNO3 кипятят смесь 1 ч. После охлажде-
ния к массе добавляют 50 мл воды, содержимое выпаривают и вторично подвер-
гают действию смесн кислот. После повторного охлаждения прибавляют после-
довательно 10 мл воды, 2 г KI и, если имеется осадок, воду до растворения по-
следнего. Колбу помещают на 30 мин в темное место, после чего выделившийся
иод титруют 0,1 и. раствором Na2S2O3.
Факторы пересчета (F): I мл 0,1 н. раствора Na2S2O3 соответствует 8,043 мг
метилдихлорарсина, 8,744 мг этилдихлорарсина, 11,15 мг фенилдихлорарсина,
13,228 мг дифеиилхлорарсина, 12,756 мг дифенилцианарсина, 13,878 мг дигидро-
фенарсазин хлорида.
2) Метод Руппа 148. К навеске 0,2—0,3 г испытуемого вещества, находя-
щейся в колбе Эрленмейера, прибавляют около 5 мл концентрированной H2SO4
и умеренно нагревают. Затем в течение 30 мин малыми порциями при постоян-
ном перемешивании вносят 1—1,5 г тонкорастертого КМпО4. Примерно через
15 мин содержимое колбы нагревают 1 ч на кипящей водяной баие. По охла-
ждении добавляют 20 мл воды и 0,5 г щавелевой кислоты, закрывают колбу
98
воронкой и раствор кипятят 10—15 мин. Если раствор не обесцвечивается, до-
бавляют еще щавелевой кислоты. После охлаждения воронку обмывают, добав-
ляют в колбу 50 мл воды и 5 г KI и помещают на 30 мин в темное место. Вы-
делившийся иод титруют 0,1 н. раствором Na2S2Os. Содержание мышьяка рас-
считывают по формуле:
л• Л.,,. п-0,003748•100
----Na2Sa°3 ----------= % мышьяка
где а — пошедшее на титрование количество 0,1 н. раствора Na2S20j, мл\ А —
навеска испытуемого вещества, г.
Значения факторов пересчета количеств отравляющих веществ приведены
в предыдущем методе.
3) Метод Робертсона 14в. Взвешивают в колбе Кьельдаля 0,02—0,03 г испы-
туемого вещества, прибавляют 5 мл концентрированной H2SO4, 1 мл дымящей
HNO3 и нагревают 1 ч на прямом пламени. После охлаждения добавляют еще
0,5 мл HNO3 и нагревают колбу до прекращения выделения бурых паров окис-
лов азота. Для полного удаления окислов азота в колбу осторожно прибавляют
1 г (NH4)2SO4 и после энергичного встряхивания нагревают 2—3 мин до пол-
ного прекращения выделения окислов азота. Затем содержимое колбы осто-
рожно разбавляют водой до 40—50 мл и переводят в колбу Эрленмейера, расхо-
дуя на это еще 30 мл воды. После добавления 1 г KI и нескольких стеклянных
шариков в горло колбы помещают насадку для кипения н выпаривают раствор
до объема примерно 40 мл. После охлаждения окраску, вызванную выделившим-
ся иодом, обесцвечивают добавлением 0,1 н. раствора Na2S2Os. Раствор разбав-
ляют водой до 100 мл, добавляют 1—2 капли фенолфталеина и нейтрализуют
реакционную смесь концентрированным раствором Na2CQa до появления розо-
вого окрашивания. Затем добавлением нескольких капель 1 н. раствора H2SO4
его обесцвечивают и, после внесения 1—2 г Na2COs и 1 мл раствора крахмала,
титруют 0,1 н. раствором иода до появления синего окрашивания. Для расчета
пригодны факторы пересчета, приведенные в методе (1).
Для определения мышьяксодержащих соединений можно поль-
зоваться также методами Эвинса 160 — с серной кислотой и суль-
фатом калия и Роджерса181 — с азотной кислотой в персульфатом
аммония.
5. 4.2.3. Йодометрическое определение метил-, этил- и фенилди-
хлорарсинов. Определение основано на окислении иодом продуктов
гидролиза мышьяксодержащих ОВ до алкилмышьяковой кислоты!
RAsC12 + 4NaOH —► RAs(ONa)2 + 2NaCl + 2Н2О
RAsO + I2 + 2H2O —> RAsO(OH)2 + 2HI
Взвешивают в мерной колбе емкостью 100 мл 1 г испытуемого вещества,
добавляют 20 мл воды, 15 мл 10%-ного NaOH и встряхивают до полного рас-
творения. После доведения объема до 100 мл, отбирают пипеткой 25 мл рас-
твора и переносят его в колбу Эрленмейера, где смешивают с 30 мл воды,
1—2 каплями фейолфталеина и нейтрализуют разбавленной H2SO4 до исчезно-
вения розовой окраски. После охлаждения добавляют 50 мл насыщенного рас-
твора NaHCOs, 1 мл раствора крахмала и титруют 0,1 н. раствором иода до по-
явления синего окрашивания.
Факторы пересчета-. 1 мл 0,1 н. раствора иода соответствует 8,043 мг ме-
тилдихлорарсина, 8,744 мг этилдихлорарсина, 11,15 мг фенилдихлорарсина.
В. 4.2.4. Йодометрическое определение дифенилхлорарсина f46>147.
Метод, как и предыдущий, основан на гидролизе и последующем
окислении иодом продукта гидролиза до дифенилмышьяковой кис-
лоты:
(С6Н5)2АзС1 -На + 2НгО —► (C6H5)2AsO(OH) + 2HI + НС1
4*
99
Взвешивают в колбе Эрлеимейера 0,2—0,4 г испытуемого вещества и рас-
творяют в 10—15 мл бензола или хлороформа. После прибавления 20 мл насы-
щенного водного раствора NaHCO3 реакционную смесь при энергичном встря-
хивании титруют 0,1 и. раствором иода до фиолетового окрашивания слоя
растворителя; 1 мл 0,1 и. раствора иода соответствует 13,228 мг дифенилхлор-
арсина.
5.4.2.5. Определение галогена и циангруппы в мышьяксодер-
жащих' ОВ. Разложение люизита и метил-, этил- и фенилдихлорар-
синов происходит на холоду при обработке водой или едкой ще-
лочью (стр. 99); для омыления ароматических арсинов, например
дифенилхлорарсина и дифенилцианарсина, требуется нагревание
со спиртовой щелочью:
(C6H6)2AsX + 2NaOH —> (C6He)2AsONa + NaX + Н2О
где X — Cl или CN.
Выделившийся при гидролизе хлор определяют аргентометри-
чески по методу Фольгарда, а цианид — титрованием по методу
Либиха. Разложение алифатических арсинов и фенилдихлорарсина
уже описано в разделе 5.4.2.3. Для гидролиза дифенилхлорарсина
и дифенилцианарсина поступают следующим образом.
Взвешивают в колбе Эрлеимейера 0,3 г испытуемого ОВ и растворяют в
5 мл этанола. После прибавления 7 мл 10%-ного NaOH содержимое колбы ки-
пятят с обратным холодильником на водяной бане. По охлаждении разбавляют
25 мл воды и фильтруют в другую колбу Эрлеимейера. Фильтр тщательно про-
мывают водой.
Для определения хлор-иона полученный таким образом раствор или 25 мл
раствора, полученного разложением едким натром (см. раздел 5.4.2.3) и раз-
бавленного до 100 мл, подкисляют 2 и. раствором HNO2 и для осаждения хлор-
иона добавляют избыточное количество 0,1 и. раствора AgNO3. Реакционную
смесь фильтруют в колбу Эрлеимейера. Фильтр, содержащий осадок хлорида
серебрв, тщательно промывают и в фильтрате определяют избыточное количе-
ство AgNO3 титрованием 0,1 и. раствором NH4SCN. В качестве индикатора до-
бавляют 2 мл насыщенного на холоду раствора железоаммиачных квасцов, в
который прибавляют азотную кислоту до исчезновения коричневой окраски. Ко-
нец титрования определяют по появлению устойчивого слабого красно-коричне-
вого окрашивания. Разница между количеством прибавленного 0,1 и. раствора
AgNO3 и расходом 0,1 н. раствора NH«SCN, умноженная на 0,003545, эквивалент-
на содержанию хлора.
Для определения циа'н-иона щелочной раствор после гидролиза смешивают
с несколькими кристаллами KI и титруют 0,1 н. раствором AgNO3 до появления
опалесценции; 1 мл 0,1 н. раствора AgNO3 соответствует 0,005203 г цианида.
5.4.2.O. Колориметрическое определение адамбита (10-хлор-
9,10-дигидрофенарсазинхлорида)162. В основу этого метода поло-
жена цветная реакция, описанная на стр. 94.
Реагенты
Смесь концентрированной азотной и уксусной кислоты 1 :9.
Смешивают от 0,2 до 1,0 мл раствора испытуемого ОВ с 0,5 мл смеси кислот
и 10 мл 20%-ного раствора NaOH. После разбавления водой до 20 мл появив-
шуюся фиолетовую окраску колориметрируют при 530 нм. Расчет ведут по ка-
либровочной кривой,- построенной на адамсите (от 1 до 8 мкг).
По другому способу адамсит в количестве 1—5 мкг может быть определен
колориметрически а ацетоновом растворе ОВ по оранжево-желтой суспензии 15\
100
образующейся при прибавлении концентрированной H2SO4 в присутствии эмуль-
гатора (эфира полиалкиленгликоля).
5. 4.2.7. Определение люизита. 1) Люизит можно определять по
количеству ацетилена, выделяющегося после воздействия 15%-иого
раствора NaOH. Объем ацетилена измеряют в газовой бюретке
соответствующего прибора154. В качестве запорной жидкости для
газовой бюретки используют насыщенный водный раствор хлори-
стого натрия. Так как при температуре ниже 37 °C щелочью разла-
гается только а-люизит, метод может найти применение также
и для анализа смесей ОВ.
2) Чувствительный колориметрический метод определения лю-
изита основан на определении p-хлорвинилмышьяковой кислоты,
которая получается при окислении иодом мышьяковистой кислоты,
образующейся при гидролизе люизита. Мышьяковую кислоту пре-
вращают затем в молибдат мышьяка, который восстанавливают до
молибденовой сини и определяют колориметрически 155.
5.5. СИНИЛЬНАЯ КИСЛОТА И ГАЛОГЕНЦИАНЫ
Соответственно их большому значению как отравляющих веществ
и их многостороннему применению в химической промышленности
и гальванотехнике для синильной кислоты, ее солей и других про-
изводных разработано большое число методов обнаружения и ко-
личественных методов определения. Ббльшая часть способов, опи-
санных для синильной кислоты и цианидов, применима и для га-
логеноцианов.
5.5.1. Обнаружение синильной кислоты и цианидов
5.5.1.1. Образование берлинской лазури. Эта реакция является
специфической для обнаружения циан-ионов.
Fe2+ + 2CN" —> Fe(CN)2
Fe(CN)2 + 4CN" —> [Fe(CN)«]«-
3[Fe(CN)«]«-+ 4Fe’+ —> FeJFe(CN)e]3
К нескольким миллилитрам раствора испытуемого вещества добавляют рас-
твор NaOH до слабощелочной реакции, 2—3 каплиц 1%-иого раствора FeSO< и
нагревают. Затем добавляют 2—3 капли 10%-ного раствора FeCl3. Появляющее-
ся при подкислении раствора разбавленной НС1 сине-зеленое или синее окра-
шивание, или образование синего осадка указывают иа присутствие цианидов;
чувствительность реакции 0,02 мг1мл.
5.5.1.2. Реакция с сульфидом меди189. Сульфид меди растворя-
ется в растворе цианида калия с образованием калийтетрациано-
купрата (I) :
2CuS + 4KCN —> 2Cu(CN)2 4-2KsS
2Cu(CN)2 —► 2CuCN 4- (CN)a
2CuCN + 2K.CN —> KjlCuHCNh]
101
Реакция специфична.
Индикаторная бумага — фильтровальная бумага, пропитанная 0,1%-ным
аммиачным раствором сульфата меди и высушенная.
Незадолго до применения через индикаторную бумагу пропускают сероводо-
род до тех пор, пока бумага равномерно ие окрасится в коричневый цвет. По-
явление белого кольца и пятна при нанесении капли испытуемого раствора сви-
детельствует о наличии цианида; чувствительность реакции 0,03 мг/мл.
5.5.1.З. Образование тиоцианата169. При нагревании цианида
с желтым сульфидом аммония образуется тиоцианат, обнаружи-
ваемый по реакции с солью трехвалентного железа:
KCN+ (NH4)2S2 —> KSCN 4-(NH4)2S
Несколько миллилитров раствора испытуемого вещества смешивают с не-
сколькими каплями 10—20%-ного раствора сульфида аммония и выпаривают
на водяной бане до обесцвечивания. Остаток слабо подкисляют разбавленной
НС1 и смешивают с одной каплей 10%-иого раствора FeCl3. Красное окраши-
вание указывает на присутствие цианида; чувствительность реакции 0,01 мг/мл.
Слабая окраска становится 'ясноразличимой после экстракции раствора эфиром.
5.5.1.4. Реакция с ацетатами меди и бензидина190. Этот часто
используемый метод основан на повышении окислительного по-
тенциала солей меди (II) по отношению к бензидину за счет обра-
зования нерастворимого цианида меди(1):
Cu(CH3COO)2 4- HCN —> Cu(CN)2 4-2СН3СООН
2Cu(CN)2 4-Н2О —>• 2CuCN 4-2HCN 4-’/2O2
Бензидин при этом окисляется в мерихиноидное соединение,
окрашенное в синий цвет
H2N——NH2 • Н№=.
NH • 2НХ
Этот метод преимущественно осуществляют при помощи инди-
каторных бумаг (см. раздел 9.2.3) или индикаторных трубок191.
Применение о-толидина вместо бензидина имеет то преимущество,
что его раствор и образовавшееся окрашенное соединение стабиль-
ны; к тому же о-толидщт легче окисляется. Аналогичными преиму-
ществами обладает тетраметилдиаминодифенилметан !92-!94. В этом
случае образуется окрашенное в синий цвет хиноидное соедине-
ние I цли карбонийкатионП:
(CH 3)2N —ЛЛ—CH=/~^=N (СН3 )2
(CH3)2N
>—СН—N(CH3)2
И
102
Эти амины предпочтительны еще и потому, что бензидин обла-
дает сильно выраженным канцерогенным действием. Очень чувстви-
тельна также реакция с замещенным бензидином — 2,7-диамиио-
дифениленоксидом 204.
Общим недостатком этого метода является то, что многие окис-
лители мешают определению.
5.5.1.5. Другие методы обнаружения цианидов путем окисления
различных соединений в присутствии солей меди(П). 1) Реагент,
содержащий гваяковую смолу и сульфат меди, образует с циани-
дом окрашенное в синий цвет соединение (реакция Шёнбейна).
2) В уксуснокислом растворе пирамидон окисляется сульфатом
меди и цианидом в соединение, имеющее синюю окраску 195.
3) Бумага, пропитанная сульфатом меди и 4,4'-диокси-3,5,3',
5'-тетраметоксидифенилом
СН3О. _ ,ОСН3
но——он
СН3О'
осн3
приобретает в присутствии синильной кислоты пурпурное окраши-
вание 196.
4) Фенолфталин окисляется сульфатом меди и цианидом в фе-
нолфталеин, дающий в щелочной среде красно-фиолетовое окра-
шивание205’206. Крезолфталин дает аналогичную реакцию197.
5) В реакции с люминолом 198’208 хемолюминесценция, возни-
кающая при его окислении перекисью водорода в присутствии соли
меди(II) в качестве катализатора, подавляется цианид-ионами.
Используя это ингибирующее действие, можно обнаружить до
0,5 мкг цианида.
5.5.1.6. Реакция с пикратом натрия 199’ 20°. Пикрат натрия обра-
зует с синильной кислотой изопурпуровую кислоту (2-гидрокси-
амино-3,5-дициан-4,6-динитрофеиол). Индикация проводится на бу-
маге или в пробирке в присутствии щелочи. Окрашенный в желтый
цвет раствор приобретает красно-коричневую или оранжево-крас-
ную окраску; чувствительность реакции 0,3 мг[мл. Восстановители
мешают определению.
5.5.1.7. Образование иедиссоциирующих цианидов тяжелых ме-
таллов. При реакции синильной кислоты с хлоридом ртути(II)
образуется эквивалентное количество соляной кислоты и недис-
социированный цианид ртути:
HgCl2 + 2HCN —> Hg(CN)2 + 2HCl
Соляную кислоту обнаруживают индикаторами: конго красным,
бромтимоловым синим или метиловым оранжевым. Этот способ, в
котором могут быть использованы также нитрат серебра, соли меди
и палладия, удобен для обнаружения цианидов на бумаге и в
трубках 207.
103
5.5.1.8. Разложение внутрикомплексных солей металлов. 1) Ионы
цианидов выделяют201 из щелочного раствора диметилглиоксиМата
палладия PdDj диметилглиоксим D
[PdD2]2++ 4CN' —> [Pd(CN)4]2" + 2D
присутствие которого обнаруживают по известной реакции с солью
никеля, в результате которой выпадает диметилглиоксимат никеля,
чувствительность реакции 1 мкг!мл цианида.
2) Образующийся в реакции циаи-иона с оксинатом меди(П)
8-оксихинолин при взаимодействии с солями алюминия дает флуо-
ресцирующий оксинат алюминия. Реакция пригодна для обнару-
жения 2,5 мкг/мл цианида 203.
5.5.1.9. Каталитическое ускорение бензоиновой конденсации 209.
Известная реакция бензоиновой, конденсации, катализируется в
присутствии следов цианидов. Образующийся бензоин может быть
обнаружен чувствительной цветной реакцией с о-динитробензолом:
+2ОН‘
2С6Н5—СО—СН(ОН)—С6Н6 + C6H4(NO2)2 ---»
+ 2CeHe—СО—СО—С6Н6 + ЗН2О
Реагенты
Бензальдегид — смесь 0,5 мл бензальдегида с 0,5 мл 25%-ного NaOH н 4 мл
этанола; раствор должен быть бесцветным.
о-Динитробензол— 0,5%-ный раствор в бензоле.
В микропробирке смешивают 1 каплю раствора испытуемого вещества с
1—2 каплями свежеприготовленного раствора бензальдегида и нагревают 2—
5 мин на водяной бане. Прибавляют 1 каплю раствора о-динитробензола и вновь
нагревают. В зависимости от концентрации цианида через 1—3 мин появляется
более или менее интенсивное фиолетовое окрашивание. Окраску сравнивают с
окраской контрольной пробы; чувствительность реакции 0,01 мкг!мл. Реакции
мешают сульфиды и соединения, действующие в щелочных растворах как доно-
ры водорода.
Аналогичным путем в результате каталитического действия
цианид-ионов из п-нитробензальдегида 210 в присутствии щелочи об-
разуется окрашенный продукт ацилоиновой конденсации:
Еще большей чувствительности достигли Гилбо и Крамер211,
применив смесь n-нитробензальдегида и о-динитробензола. Обра-
зующийся при взаимодействии цианида и п-нитробензальдегида
104
циангидрин восстанавливает о-динитробензол в синий дианион аци
формы о-нитрофенилгидроксиламина:
4- CN' и т. д.
Высокая чувствительность определения достигается благодаря
тому, что циан-ион остается в сфере реакции и может реагировать
повторно.
Добавление к реакционной смеси изонитрозобензоилацетона
приводит к дальнейшему повышению чувствительности. Этот эф-
фект основан на образовании бензоил- и ацетилцианида (реак-
ция 1), каждый из которых при гидролизе отщепляет циангруппу
(реакции 2 и 3), так что в конечном счете число первоначально
имеющихся циангрупп удваивается:
С6Н5. у /СН3 yCN ZCN
/С—С--IT—> С6Н6-(/ +СН3—(1)
СГ %О ^0^0
XN +H2oi—> CeH5COOH + HCN
2С6Н5-С; ---- (2)
Ч0 — > C6H5COCONH2
,CN +Н2оГ^ CHjCOOH + HCN
2СН3—------- (3)
*—> ch3coconh2
Реагенты
п-Нитробензальдегид— 0,1 М раствор в метилцеллозольве.
о-Динитробензол— 0,1 М раствор в метилцеллозольве.
Изонитрозобензоилацетон— 0,02М раствор в 0,1 М буферном растворе бор-
ной кислоты pH 8,5.
Едкий натр — 0,5 М раствор.
К смеси, составленной из 1 мл раствора п-иитробензальдегида, 0,1 мл рас-
твора динитробензола и таких же количеств растворов изонитрозобензоилаце-
тона и щелочи, приливают 0,1 мл раствора испытуемого вещества. Пурпурное
окрашивание указывает иа присутствие цианида; чувствительность реакции
0,003 мгк цианида.
105
Если вводить в реакцию вместо п-нитробензальдегида его про-
дукт присоединения к бисульфиту, то реакционная смесь становится
водорастворимой211. Прибавляемый к смеси трифенилтетразолий-
хлорид восстанавливается циангидрином в красный трифснилформ-
азан, чувствительность реакции 0,03 мкг/мл.
Описанные в этом разделе методы могут быть использованы для
фотометрического определения цианида.
5.5.1.10. Образование галогенциана. Синильная кислота реаги-
рует с иодом, образуя иодциан:
hcn + i2 —> HI + с.\1
Если подкислить раствор цианида, содержащий окрашенный в
синий цвет продукт взаимодействия крахмала с иодом, то в резуль-
тате выделения синильной кислоты и связывания иода синее окра-
шивание исчезает.
Значительные возможности обнаружения синильной кислоты
открывает ее превращение под воздействием хлораминов (или
бромной воды) в хлорциан (или бромциан)
который можно открыть, пользуясь методами, описанными в раз-
деле 5.5.2.1. При применении бромной воды избыток ее через 1 —
2 мин разлагают прибавлением мышьяковистой кислоты или рас-
твора фенола.
5.5.2. Обнаружение галогенцианов
Часть способов, описанных для синильной кислоты, может быть
использована также и для индикации галогенцианов.
5.5.2.1. Важнейшие методы обнаружения и определения гало-
генцианов. Важнейшие методы качественного и количественного
определения галогенцианов, а после соответствующего превраще-
ния и синильной кислоты основаны на реакции Цинке — Кёнига212.
Галогенцианы реагируют с пиридином и подобными соединениями,
содержащими третичный атом азота, с образованием глугаконо-
вого альдегида, который конденсируется с различными первичными
ароматическими аминами в полиметиновые красители (шиффовы
106
основания). Механизм этой реакции заключается в разрыве пирй
динового цикла:
+ XCN
сн
STX. НС^ ХСН
1 + 2Н2О —► °Ч | II + NH2CN + НХ
5 J ^С снон
CN X'
При взаимодействии глутаконового альдегида с первичным
ароматическим амином образуется шифово основание:
СН СН
НС^ ХСН НС^ ХСН
О. | ]| + 2NH2R — | ||
СНОН RN=HC СН—NHR
W
глутаконового диальдегида с <5-метил-1-сре-
На этом принципе разработано большое число методов, в кото-
рых вместо пиридина применяли пиколин, анабазин213, 4-бензилпи-
ридин, а в качестве аминов — анилин, бензидин, о-толидин, толу-
идин, n-фенилендиамин218, резорцин, диметилдигидрорезорцин
(димедон), барбитуровую кислоту.
Эпштейн25 применял окрашенный в синий цвет реагент, полу-
чаемый конденсацией
нил-5-пиразолоном:
О
II
CeHj—N—С-. ......... k, ............ а,—iv
| ;cHj + or No + hJc^ I
N=C/ .......................... 'C=N
I I
CH3 CH3
СН
НС^ ХСН2 о
н\| |/н II
Существует много модификаций этой реакции214-217, в резуль-
тате которых ее чувствительность была повышена, и стало возмож-
ным определять 0,2 мкг/мл галогенциаиов. Вместо З-метил-1-фе-
нил-5-пиразолона можно применять этилацетондикарбонат или
ацетоуксусный эфир219.
Из названных выше аминов в сочетании с пиридином преиму-
щественно использовались бензидин 220-229, димедон 230> 231 и барби-
туровая кислота 232^234. Применение последней вместо бензидина
приводит к повышению чувствительности метода и к образованию
107
более устойчивого окрашенного соединения. Замена бензидина бар-
битуровой кислотой или другими подходящими соединениями обус-
ловлена в основном легкой окисляемостью бензидина и его канце-
рогенным действием. При взаимодействии 2 моль барбитуровой
кислоты с глутаконовым диальдегидом образуется красно-фиоле-
товый продукт конденсации235
HN—СО ОС—NH
II II
ОС с=сн—сн=сн—сн=сн—сн со
II II
UN—СО ОС—NH
Эта качественная реакция может быть использована и для
фотометрического определения (см. раздел 5.5.3.5).
5.5.2.2. Идентификация различных галогенцианов. Возможность
идентификации галогенцианов основана на их различном отноше-
нии к восстановителям. В кислом растворе хлорциан не восстанав-
ливается иодидом и тиосульфатом, в то время как бромциан быстро
восстанавливается тиосульфатом и медленно иодидом и сульфи-
том. Иодциан моментально восстанавливается до цианида всякими
восстановителями.
5.5.3. Количественное определение
синильной кислоты и цианидов
5.5.З.1. Титрование нитратом серебра. Цианид можно определять
аргентометрически: в нейтральном растворе по Мору, в кислом —
по Фольгарду, в щелочном — по Либиху 245. В способе Либиха бла-
годаря существующему вначале избытку цианида получающийся
цианид серебра переходит в раствор вследствие образования комп-
лексного соединения K[Ag(CN)2]. Первая избыточная капля AgNO3
разлагает это соединение и появляется опять нерастворимый AgCN,
о чем можно судить по возникающему помутнению, указывающему
таким образом на конец титрования:
AgCN + KCN K[Ag(CN)2]
K[Ag(CN)2] + AgNO3 2AgCN + KNO3
Классический способ улучшен применением в качестве инди-
катора избытка AgNO3 n-диметиламинобензилиденроданина 244> 246,
предложенного Файглем 243 для обнаружения серебра. Кроме того,
в качестве индикаторов аргентометрического титрования приме-
нимы дифенилкарбазид 247, дитизон 248, тиофлуоресцеин 249 и вари-
аминовый синий 250.
Реагенты
Индикатор — раствор 30 мг диметиламинобензилиденроданина в 100 мл
ацетона.
Нитрат серебра—0,1 н. раствор.
К 30—50 мл щелочного (по фенолфталеину) раствора цианида прибавляют
0,5 мл индикатора — диметиламинобензилиденроданина и титруют из микробю-
ретки 0,01 н. раствором AgNO3 до тех пор, пока начальная желтая окраска не
108
приобретет красноватый тон; 1 мл 0,01 н. раствора AgNO3 соответствует 0,52 мг
цнан-иона. Наименьшее определяемое количество составляет 0,02 мг цианида
в пробе244.
5.5.3.2. Титрование хлорной ртутью. При взаимодействии циа-
нидов с раствором хлорной ртути образуется цианид ртути; избы*
ток хлорной ртути можно - определять по индикатору дитизону.
Если к титруемому раствору добавить ферроцианид калия и нитро-
зобензол 251, то при избытке соли ртути(П) образуется комплексное
соединение, окрашенное в фиолетовый цвет:
K4[Fe(CN)6] + Hg++ + Н2О —> K3[Fe(CN)5 • Н2О] + Hg(CN)+
4-нитрозобензол
K3[Fe(CN)s • C6H5NO]
Этот метод может быть использован для фотометрического оп-
ределения цианидов 252. Конец титрования можно также определять
по обесцвечиванию добавляемого раствора диэтилдитиокарбами-
ната меди(II) в четыреххлористом углероде 255.
5.5.3.3. Титрование сульфатом никеля. Раствор цианида смеши-
вают с избытком аммиачного раствора сульфата никеля, причем
образуется весьма устойчивое комплексное соединение K2[Ni(CN)J.
Избыток сульфата никеля определяют обратным титрованием эти-
лендиаминтетрауксусной кислотой в присутствии мурексида (ам-
мониевая соль пурпуровой кислоты) в качестве индикатора 253;
возможно также и прямое титрование цианида сульфатом никеля
с применением этого же индикатора 254.
5.5.3.4. Йодометрическое титрование. Цианид окисляется рас-
вором иода с образованием иодциана:
HCN + 12 —► ICN + НГ
Реакцию проводят в щелочной среде, создаваемой добавлением
0,5 г NaHCO3, и титруют 0,1 н. раствором иода до слабо-желтого
окрашивания.
5.5.3.5. Колориметрическое определение после превращения
цианидов в хлорциан232. Приводимый метод является одним из
вариантов способа, описанного в разделе 5.5.2.1.
Реагенты
Хлорамин— 1%-иый водный раствор хлорамина Т или Б.
Пиридин-барбитуровый реагент—в мерную колбу емкостью 50 мл отвеши-
вают 3 г барбитуровой кислоты и суспендируют ее примерно в 30 мл воды, за-
тем прибавляют 15 мл пиридина и встряхивают смесь до растворения барбиту-
ровой кислоты. После добавления 3 мл концентрированной НС1 объем раствора
доводят водой до метки. Раствор должен быть бесцветным.
Смешивают 20 мл водного раствора пробы испытуемого вещества (pH 2—10)
с 1 мл раствора хлорамина и после 1—2 мин встряхивания добавляют 3 мл
раствора реагента. Через 8 мин красно-фиолетовое окрашивание фотометрируют
при 570 нм. Расчет проводят по калибровочной кривой. Качественно можно
обнаружить (в небольшом объеме) до 0,01 мкг/мл цианида. Тиоцианаты и окси-
мы мешают определению.
109
5.5.3.6. Фотометрические методы определения. Минимальные ко-
личества (до 0,05 мкг\ синильной кислоты можно определять об-
менной реакцией с хлорамином Т, в результате которой получается
хлорциан. Последний, реагируя с никотинамидом в щелочном рас-
творе, дает соединение, обладающее интенсивной флуоресцен-
цией 256. Для определения до 0,02 мкг цианида используют способ-
ность циан-ионов разлагать хелатный комплекс палладия с 8-окси-
5-хинолинсульфокислотой, который с солями магния образует
хелатное соединение, обладающее сильной флуоресценцией 257:
SO3K
Флуоресцирующее соединение образуется также в реакции
циан-ионов с n-бензохиноном (Хех 400—420 нм; Хеш 480—490 нм):
NCk ,CN
0=^ ^=0 + 2HCN —> НО—ОН
и с различными другими производными хинона 258.
При помощи п-бензохинона удается обнаружить 0,2 мкг циа-
нида, а при помощи эфира бензохинонмонооксимсульфокислоты 259—
0,5 мкг,
5.5.3.7. Другие колориметрические методы. При разложении
циан-ионами окрашенного в желтый цвет хелатного комплекса
палладия с 8-окси-7-иод-6-хинолинсульфокислотой в присутствии
ионов Fe(III) образуется окрашенное в синий цвет комплексное
соединение с железом 257, максимум поглощения которого находится
при 650 нм. Этим способом можно обнаружить до 0,2 мкг цианида.
Сульфид-ионы дают аналогичную окраску.
Косвенно цианид-ион можно определять при помощи хлорной
ртути, для чего избыток ее прибавляют к раствору дианида. Не
110
связываемые в комплекс ионы ртути образуют с /г-диметиламино-
бензилиденроданином вещество, окрашенное в красный цвет 260.
Удобное для колориметрического определения окрашенное сое-
динение с максимумом поглощения при 540 нм образуется в реак-
ции цианидов с хлораниловой кислотой261.
5.5.4. Количественное определение галогенцианов
Для колориметрического определения удобны способы, основанные
на образовании полиметиновых красителей (см. раздел 5.5.3.5).
5.5.4.1. Определение хлорциана реакцией со щелочью. Раствор
исследуемого вещества разлагают избытком титрованного раствора
едкого натра, при этом, согласно стехиометрии, на 1 моль хлор-
циана расходуется 2 моль едкого натра:
C1CN + 2NaOH —>- NaCl + NaOCN + Н2О
Избыток едкого натра определяют титрованием серной кислотой
в присутствии фенолфталеина.
5.5.4.2. Определение бромциана. Метод основан на восстановле-
нии бромциана тиосульфатом натрия в кислой среде 262:
BrCN + 2SiOr + Н+ —>- ВГ + HCN + S4O62-
Раствор бромциана (не более 0,05 Л-f) в 0,5 и. растворе H2SO4 быстро сме-
шивают с равным объемом 0,1 н. раствора Na2S2O3. После внесения раствора
крахмала до образования синего окрашивания прибавляют 0,1 н. раствор иода,
после чего титруют 0,1 н. раствором Na2S2O3 до обесцвечивания.
5.6. ГАЛОГЕНПРОИЗВОДНЫЕ УГОЛЬНОЙ КИСЛОТЫ
Наибольший интерес представляет проблема обнаружения в воз-
духе важнейшего представителя этой группы отравляющих ве-
ществ— фосгена. Некоторые индикаторные бумаги, карандаши и
трубки, применяемые для этой цели в полевых условиях, описаны
в разделе 9.2.
5.6.1. Качественное обнаружение фосгена
5.6.1.1. Обнаружение фосгена156 анилиновой водой. При взаи-
модействии фосгена с анилином образуется дифенилмочевина
(т. пл. 235°C):
СОС12 + 4C6H6NH2 —>- CO(NHC6H6)2 + 2C6H5NH2-HC1
Реагенты
Анилиновая вода — встряхивают 4 г аиилииа со 100 мл воды; через 1 ч
эмульсию фильтруют через фильтр, смоченный водой, и смешивают фильтрат
с некоторым количеством дифенилмочевины, встряхивают и снова фильтруют.
Дифенилмочевина.
Воздух, содержащий фосген, просасывают через поглотительную склянку
с раствором реагента. Появление помутнения или образование осадка указы-
вает на присутствие фосгена; чувствительность реакции — 0,05 мг]л.
111
5.6.1.2. Реакция с л-диметиламинобензальдегидом и диметил-
анилином 157. В этой реакции, применимой для осуществления в
индикаторных трубках, вероятно, происходит образование сине-
зеленого дифенилметанового красителя:
(CH3)2n——cho + coci2 —> (ch3)2n——chci2 + co2
Очевидно, одновременно в результате побочной реакции обра-
зуется бесцветное лейкооснование кристаллического фиолетового,
которое при окислении превращается в кристаллический фиоле-
товый:
5.6.1.3. Реакция с фенилгидразином 158. В этой капельной реак-
ции из фосгена и фенилгидразина образуется дифенилкарбазид,
дающий с солями меди внутрикомплексное соединение, окрашен-
ное в фиолетовый цвет:
С6Н5—NH—NHX
2CeH5—N’H-NH2 + COClj —> Vo + 2HC1
С6Н5—NH— NH/
|+'/2Cu2+
CeH5—N---NH.
I \
Cu/2 /CO + H+
C6Hj—NH—NHZ
112
Реагенты
Фенилгид разина соль с коричной кислотой.
Сульфат меди — 1%-ный водный раствор.
В углублении пластинки для капельного анализа смешивают 1 каплю рас-
твора фосгена (в эфире, хлороформе, четыреххлористом углероде) с маленькой
горошиной солн фенилгидразина н через 5 мин прибавляют 1 каплю раствора
сульфата меди. В присутствии фосгена появляется красно-фиолетовое или розо-
вое окрашивание; чувствительность реакции 0,01 мг[мл раствора пробы.
5.6.2. Количественное определение
5.6.2.1. Реакция с анилином. Фосген реагирует с анилиновой во-
дой количественно. При достаточной концентрации фосгена коли-
чество дифенилмочевины, полученной при пропускании пробы
испытуемого воздуха через анилиновую воду (см. раздел 5.6.1.1),
можно определять весовым путем.
Для этого реакционную смесь фильтруют через небольшой бумажный
фильтр, осадок промывают небольшим количеством воды, насыщенной дифенил-
мочевиной, и для удаления анилина сушат 2 ч при 50—60 °C. Затем осадок рас-
творяют кипящим спиртом во взвешенном тигле, спирт выпаривают на водяной
бане и остаток сушат 2 ч при 50—60 °C. Вес остатка, умноженный на 0,4661,
дает количество фосгена.
Этим способом можно определять фосген в боеприпасах или в
заводских продуктах. Ампулу с жидким ОВ помещают в колбу с
анилиновой водой и, закрыв колбу, разбивают ампулу. Навеску ОВ
берут во взвешенную ампулу'путем погружения ее капилляра в
предварительно охлажденный смесью льда и поваренной соли фос-
ген, затем охлаждают ампулу смесью эфира и твердой углекис-
лоты, запаивают капилляр и снова взвешивают.
При незначительном количестве образовавшейся дифенилмоче-
вины ее можно разложить по способу Кьельдаля до аммиака, кото-
рый затем определяют реагентом Несслера или ацидиметрически.
Другим вариантом способа 159 является экстракция н-гексан —
пентанольной смесью (1:9) дифенилмочевины из подкисленного
серной кислотой раствора. В экстракте измеряют поглощение в ин-
тервале 215—290 нм.
5.6.2.2. Йодометрическое определение 16°-162. При поглощении
или растворении фосгена в насыщенном ацетоновом растворе Nal
выделяется иод
COCl2 + 2NaI —► 2NaCl + 12 + СО
количество которого определяют титрованием. Применяемый аце-
тон следует сушить несколько дней над хлористым кальцием, так
как реакция фосгена с Nal проходит количественно только в от-
сутствие воды.
Ампулу, содержащую приблизительно 0,4 г жидкого фосгена, помещают в
тщательно высушенную колбу емкостью 500 мл. Затем приливают 30 мл ацето-
на и добавляют 5 г Nal. Закрыв колбу, быстрым круговым движением разбивают
ампулу н энергично встряхивают колбу в течение 15 мин. После некоторого
пребывания колбы в темноте выделившийся иод титруют 0,1 н. раствором
Na2S2O3. Параллельно устанавливают, какое количество раствора Na2S2O3 идет
ИЗ
на титрование контрольной пробы, проводимой с теми же количествами иодида
и ацетона. Содержание фосгена рассчитывают по формуле:
(X - Y) • 4,95 • 100 ,
-------д-------= % фосгена
где X н Y — количество 0,1 н. раствора Na2S2O3, пошедшее на титрование на-
вески испытуемого вещества и контрольной пробы, мл; А — навеска, г.
Присутствие в фосгене растворенного свободного хлора иска-
жает результаты анализа. Свободный хлор можно определить163
растворением навески фосгена в растворе ЫагСОз с последующим
добавлением Nal и титрованием иода, выделившегося при подкис-
лении 0,1 н. раствором NajSsOs. При анализе парообразного фос-
гена можно, перед поглощением пара фосгена ацетоном, удалить
свободный хлор и хлористый водород, пропуская газовый поток
сначала через промывалку с концентрированной H0SO4 и затем
через трубку, наполненную металлической сурьмой.
5.6.2.3. Колориметрическое определение 4-(п-нитробензил)-пИ’
ридином. При взаимодействии фосгена с 4- (n-нитробензил) -пири-
дином 164 образуется окрашенная в желтый или оранжевый цвет
бисчетвертичная соль.
Определенный объем воздуха, содержащего фосген, пропускают в течение
1 мин через 6 мл раствора, приготовленного растворением 20 мг 4-(п-ннтробен-
зил)-пиридина в 6 мл метилизобутилкарбинола, и через 5 мин колориметрируют
смесь при 415 нм. Калибровочная кривая прн концентрациях от 0,5 до 5 мкг/мл
фосгена представляет собой прямую. Чувствительность способа от 0,1 до0,2ль?/л3
фосгена в воздухе.
S.6.2.4. Другие методы определения фосгена. При пропускании
тока воздуха, содержащего фосген, при 800 °C через серебряную
вату наряду с хлоридом серебра образуется окись углерода, кото-
рую собирают в бюретку азотометра, заполненную раствором КОН,
и измеряют объем выделившегося газа 165.
В реакции фосгена с гексаметилентетрамином (уротропином)
количественно образуется комплексное соединение
2 (CH2)eN4 + СОС12 —> [(CH2)6N4]2-COC12
После отгонки с паром из щелочного раствора избытка гекса-
метилентетрамина его содержание определяют титрованием
щелочью в присутствии смешанного индикатора метилового крас-
ного и метилового синего 166. Хлор и соляная кислота не мешают
определению.
Некоторые старые методы определения фосгена основывались
на титровании нитратом серебра иона хлора, образующегося при
щелочном гидролизе фосгена 163, или на титровании щелочью соля-
ной кислоты, образующейся при гидролизе фосгена водой.
5.6.2.5. Колориметрическое определение дифосгена167. При под-
робном описании реакции Шёнеманна (раздел 5.1) было указано,
что хлорангидриды кислот также вступают в эту реакцию, и, таким
114
образом, она может быть использована для определения дифос-
гена. Воспроизводимые результаты получаются, если ОВ раство-
римо в изобутиловом спирте и этот раствор устойчив в течение
многих часов. При этом с количественным выходом образуется
изобутилтрихлорметилкарбонат, при действии на который пере-
кисью водорода в щелочной среде, вероятно, происходит замещение
одного или большего числа атомов хлора с образованием гидропе-
рекисного соединения:
ZC1 ЮСНг-СЩСНзЬ
ОС + (СН3)2СН—сн2он —> ос/
\occi3 . \ОСС13
/ОСН2-СН(СН3)2 ,ОСН2—СН(СН3)2
ос/ -|-Н00‘ —> ос/ /С1 4-СГ
\)СС13 Ч)С—С1
/оон
Образовавшееся гидроперекисное соединение при действии на
него каким-либо амином дает окрашенное соединение.
Реагенты
о-Дианизидин—1,2%-ный раствор в ацетоне; раствор готовят ежедневно.
Перборат натрия — 0,25%-ный водный раствор.
К смеси 2 мл о-дианнзидина и 3 мл раствора Na2B4O7 прибавляют 3 мл
раствора испытуемого ОВ в абсолютном изобутиловом спирте, перемешивают
5 мин и измеряют экстинкцию прн 450 нм по сравнению с контрольной пробой.
Расчет проводят по калибровочной кривой, которая для концентраций от 2 до
10 мкг удовлетворяет закону Бера. Для определения более высоких концентра-
ций ОВ (40—120 мкг) в качестве окисляемого реагента используют 2 мл
0,25%-ного раствора индола в ацетоне. Через 10 мин для растворения образо-
вавшегося индиго добавляют 15 капель анилина н измеряют экстинкцию при
630 ни.
5.6.2.6. Другие методы определения дифосгена и трифосгена.
Дифосген может быть определен аналогично фосгену по выделе-
нию иода из ацетонового раствора иодида натрия:
,С1
ос/ + 4NaI —► 4NaCl + 2I2 + 2СО
/ОСС13
С трифосгеном реакция протекает нестехиометрически.
И ди-, и трифосген реагируют с анилиновой водой с образова-
нием дифенилмочевины, количество которой можно определить спо-
собом, описанным в разделе 5.6.2.1. В соответствии с уравнением
реакции фактор пересчета для обоих соединений равен 0,4661. Гид-
ролиз этих эфиров проходит медленно в холодной воде и быстро в
горячей. При этом из дифосгена выделяется 4 экв соляной кислоты
а из трифосгена — 6 экв.
115
5.7. ГАЛОГЕНИРОВАННЫЕ КЕТОНЫ
5.7.1. Хлор- и бромацетон
Для индикации этих ОВ, а также бромметилэтилкетона удобной
является реакция с ванилином, в результате которой образуются
окрашенные соединения. Так, бромМетилэтилкетон образует крас-
ное, постепенно синеющее вещество; хлорацетон — красное, посте-
пенно приобретающее от зелено-синей до зелено-черной окраски
вещество; бромацетон — зеленое вещество.
Реагент
Ванилин—серная кислота — растворяют 0,1 г ванилина в 7,5 мл концен-
трированной НС1 и к раствору осторожно прибавляют 2,5 мл концентрирован-
ной H2SO4. Реагент готовят перед употреблением.
Смешивают 1 мл раствора ОВ в четыреххлористом углероде с 1 мл рас-
твора ванилина, встряхивают и наблюдают появление окраски. При нагревании
пробы на водяной бане хлорацетон и бромметилэтилкетон быстро вызывают по-
явление синего окрашивания. Отрицательный результат или появление слабо
желтоватого окрашивания с достоверностью свидетельствуют об отсутствии
всех трех раздражающих ОВ.
5.7.2. Хлорацетофенон
5.7.2.1. Качественное обнаружение. Реакция с м-динитробензо-
лом. Общей реакцией обнаружения соединений с активной метиле-
новой группой, которую содержат хлорацетофенон и бромбензил-
цианид, является цветная реакция с .и-динитробензолом в присут-
ствии щелочи 168. Весьма вероятно, что возникновение окраски мож-
но объяснить образованием соли нитроловой кислоты в хиноидной
форме:
Смешивают 1 мл спиртового раствора испытуемого вещества с 0,5 мл рас-
твора м-динитробеизола (1%-ный раствор в этаноле) и 3—5 каплями спирто-
вого раствора КОН. Появление красно-фиолетового окрашивания указывает на
присутствие хлорацетофенона; чувствительность реакции 0,001 мг/мл.
Бромбензнлцианид дает красно-коричневую окраску, а хлорацетон — крас-
ную. Отрицательный результат свидетельствует об отсутствии этих ОВ.
116
Нитрование до динитробензойной кислоты. При помощи нит-
рующей смеси хлорацетофенон нитруется до динитробензойной
кислоты, которую после восстановления можно обнаружить в виде
окрашенной аммониевой соли диаминобензойной кислоты169.
Помещают несколько кристаллов испытуемого вещества в пробирку и об-
ливают 0,5—1,0 мл концентрированной H2SO4, к которой добавлено несколько
кристаллов KNO3. Непродолжительное время нагревают смесь на горелке до
выделения бурых паров и после охлаждения переводят ее в небольшой стакан
при помощи 2—3 мл воды и смешивают с 10 мл 15%-иого раствора аммиака и
небольшим количеством гидрохлорида гидроксиламина (взятого на кончике шпа-
теля). Появление красно-коричневой или красной окраски свидетельствует о
присутствии хлорацетофенона. Иногда пробу следует немного нагреть на водя-
ной бане.
Образование индиго 169> 17°. Хлорацетофенон образует с крепким
спиртовым раствором аммиака после длительного стояния при
обычной температуре индол, который окисляется перекисью водо-
рода в синее индиго. Образование индиго происходит также при
кипячении хлорацетофенона со спиртовым раствором сульфида
аммония.
Индикация при помощи 4-(п-нитробензил)-пиридина Хлор-
ацетофенон в спиртовом растворе в присутствии диметилформ-
амида реагирует с 4-(п-нитробензил)-пиридином с образованием
окрашенного в красный цвет продукта дегидрохлорирования — хло-
рида фенацил-4- (n-иитробензил) -пиридиния:
-НС1 *
5.7.2.2. Количественное определение. Колориметрическое опре-
деление при помощи пиридина171. Хлорацетофенон реагирует с
пиридином с образованием окрашенного в желтый цвет фенацил-
пиридиния:
[СЗ*—со~сн2—nO]
сг
Содержащийся в воздухе в виде пара или аэрозоля хлорацето-
фенон поглощают смесью пиридина и диметнлформамида. Образо-
вание окраски наступает после нагревания. Прибавление пипери-
дина стабилизует окраску, которую колориметрируют через 15 мин
при 450 нм. Калибровочная кривая в пределах концентраций 1—
25 мкг!мл представляет собой прямую. При поглощении из 10 л
воздуха можно определять хлорацетофенон в количестве 0,6 .чг/лг3.
117
Определение при помощи тиомочевины 171. При взаимодействии
хлорацетофенона с тиомочевиной образуется гидрохлорид 4-фенил-
2-аминотиазола
С6Нб—С---N
„II II
НСх /С—NH2-HC1
S
Реакция протекает количественно и может быть завершена либо
объемным определением соляной кислоты либо колориметрическим
определением окрашенного соединения, образующегося при соче-
тании фениламинотиазола с диазотированным п-нитроанилином.
Отщепление хлора сульфидом натрия 173. При нагревании спир-
тового раствора хлорацетофенона с сульфидом натрия образуется
хлорид натрия
2С6Нб—СО—СН2С1 + Na2S —> (CeH5—СО—CH2)2S + 2NaCl
содержание которого можно определить по Фольгарду.
Мешающий определению избыток сульфида необходимо перед
титрованием удалить окислением перекисью водорода в кислой
среде и кипячением раствора.
Реакция с фенолятом натрия. В реакции хлорацетофенона с
фенолятом натрия стехиометрически образуется хлорид натрия
С6Н5—СО—СН2С1 + NaOC6H5 —> С6Н6—СО—СН2—ОС6Н5 + NaCl
который, так же как и в предыдущем способе, можно определять
аргентометрически по Фольгарду ,72.
5.8. БРОМБЕНЗИЛЦИАНИД
5.8.1. Качественное обнаружение
5.8.1.1. Сплавление с щелочами. При сплавлении бензилцианида
с гидроокисями щелочных металлов или при обработке спиртовой
щелочью на холоду от него отщепляется циангруппа, которую
можно обнаружить обычными способами (см. раздел 5.5). Весьма
удобной для этого является реакция образования берлинской
лазури.
5.8.1.2. Омыление с образованием аммиака. При нагревании со
спиртовой щелочью (реакция 1) или с кислотами (реакция 2)
бромбензилцианид омыляется с образованием аммиака:
с6н6—с—соок
2C6H6CHBrCN + 4КОН —► || + 2KBr + 2NH3 (1)
С6Н6—С—COOK
C3H6CHBrCN + 2Н2О + НС1 —> C6H5CHBr—СООН + NH4C1 (2)
Аммиак может быть обнаружен реагентом Несслера. Так как
в щелочной среде аммиак летуч, рекомендуется омылять пробу кис-
118
лотой, для чего к спиртовому раствору испытуемого вещества до-
бавляют несколько капель концентрированной НС1 и выпаривают
раствор досуха. Остаток обрабатывают небольшим количеством
разбавленного раствора NaOH и после прибавления реагента Нес-
слера обнаруживают бромбензилцианид либо по появлению светло-
коричневой окраски, либо по образованию осадка того же цвета.
Можно также обнаруживать бромбензилцианид, подкисляя
реакционную смесь после омыления пробы спиртовой щелочью
(см. выше реакцию 1). Выпадающий кристаллический осадок ан-
гидрида дифенилмалеиновой кислоты можно идентифицировать по
температуре плавления (156°С).
5.8.1.3. Другие методы обнаружения. При нагревании несколь-
ких кристаллов испытуемого ОВ с концентрированной H2SO< по-
является коричнево-красное окрашивание неизвестного происхож-
дения; адамсит в этих условиях дает карминово-красное окраши-
вание, а раздражающее ОВ ксилилбромид — красное окрашива-
ние. Описанная выше (см. раздел 5.7.2.1) реакция обнаружения
хлорацетофенона при помощи л/-динитробензола дает с бромбензил-
цианидом аналогичное красно-фиолетовое окрашивание. При на-
гревании бромбензилцианида с раствором пикрата натрия, получае-
мым растворением 0,5 г пикриновой кислоты в 100 мл 2 н. раствора
NaOH, появляется красное окрашивание, обусловленное образова-
нием натриевой соли изопурпуровой кислоты. Реакцию дают и дру-
гие ОВ, разлагающиеся с отщеплением циангруппы, в том числе
дифенилцианарсин и табун.
5.8.2. Количественное определение
5.8.2.1. Реакция с сульфидом натрия. Взаимодействие бромбен-
зилцианида со спиртовым раствором сульфида натрия приводит к
образованию бис-(фенилцианметил)-сульфида и к количественному
отщеплению бром-иона
2CeH6—CHBr—CN + Na2S —►
'СвН5—CH—I S + 2NaBr
CN J2
который определяют аргентометрическим титрованием по Фоль-
гарду. Перед титрованием избыток сульфида должен быть удален
кипячением раствора, подкисленного серной кислотой, и окисле-
нием остаточного сероводорода перекисью водорода.
5.9. ТРИХЛОРНИТРОМЕТАН (ХЛОРПИКРИН)
5.9.1. Качественное обнаружение
5.9.1.1. Восстановление до нитрита. По Алексеевскому 174 хлор-
пикрин при обработке металлическим кальцием или амальгамой
натрия восстанавливается до нитрита:
CCljNOj + SH —► HNO2 + 3HC1 + CH4
119
Нитрит образуется также в реакции хлорпикрина с этилатом
натрия (реакция 1), с метанольным раствором иодида калия (реак-
ция 2) или с раствором едкой щелочи в присутствии перекиси водо-
рода 185 (реакция 3):
CC13NO2 + 4C2HsONa —* C(OC2HS)4 + 3NaCl + NaNO2 (1)
CC13NO2 + 4KI —► CI4 + 3KC1 + KNO2 (2)
(H2O2)
CC13NO2 + 6NaOH --------->- Na2CO3 + 3H2O + 3NaCl + NaNO2 (3)
Образующийся нитрит обнаруживают реагентом Грисса, содер-
жащим а-нафтиламин и сульфаниловую кислоту. Последняя в кис-
лой среде диазотируется нитритом, а образовавшаяся соль диазо-
ния сочетается с а-нафтиламином в азокраситель красного цвета:
Реагенты
Восстановители — 8%-ная амальгама натрия, металлические кальций или
иатрий, порошкообразный сплав Деварда, насыщенный раствор KI в метаноле.
Реагент Грисса — готовят смешением двух растворов: раствора 0,1 г сульф-
аниловой кислоты в 100 мл 30%-ной уксусной кислоты и раствора а-нафтил-
амина, который готовят нагреванием 0,03 г а-нафтиламина с 70 мл воды, затем
смесь фильтруют или сливают раствор с нерастворимого осадка и смешивают с
30 мл ледяной уксусной кислоты. Перед употреблением смешивают равные ча-
сти обоих растворов.
К 5 мл спиртового раствора испытуемого ОВ добавляют какой-либо восста-
новитель. По окончании восстановления реакционную смесь подкисляют уксус-
ной кислотой. Образовавшееся после прибавления 1—2 мл реагента Грисса
красное окрашивание свидетельствует о присутствии хлорпикрина.
5.9.1.2. Проба Лабатша175. Несколько капель испытуемого ОВ
или спиртового экстракта кипятят с 2 мл 5%-ного спиртового рас-
твора КОН и прибавляют кристаллик тимола или резорцина. В при-
сутствии хлорпикрина появляется с тимолом — желтое, а с резор-
цином— красное окрашивание. Желтое окрашивание, характерное
для реакции с тимолом, от прибавления H$SO4 переходит в красно-
фиолетовое.
120
5.9.1.3. Реакция с образованием бромциана176. Хлорпикрин при
действии цианида калия в присутствии бромида калия в спиртовом
растворе превращается в тетрахлординитроэтан и бромциан. Об-
разовавшийся бромциан можно обнаружить по его реакции с пи-
ридином и анилином, с которыми он образует полиметиновый кра-
ситель красного цвета:
СН
НО" ^сн
II | + BrCN + 2C6H5NH2 —>
НС\ ^СН
N
CH
НС/ чсн
—> || I + NH2CN + HBr
C6H5—NH-CH CH=N—C6H5
Подробности о механизме этой реакции были приведены при опи-
сании способов обнаружения галогенцианов (см. раздел 5.5.2.1).
Реагенты
Анилин солянокислый— 10%-ный водный раствор.
Раствор А — смесь раствора 1 г КВг в 4 мл воды и такого же раствора KCN,
доведенная до 10 мл.
Раствор Б — свежеприготовленная смесь 2 мл раствора А, 1 мл пиридина
н 1 мл анилина.
Для обнаружения ОВ в воздухе последний просасывают через поглотитель-
ную склянку с раствором Б. В зависимости от концентрации хлорпикрина по-
является желтое, оранжевое нли красное окрашивание; чувствительность реакции
0,3—0,4 мг/л воздуха. Аналогичный реагент, состоящий нз KCN, пнрнднна и
флороглюцина, был предложен Моро18в.
5.9.1.4. Другие методы обнаружения. При пропускании испы-
туемого воздуха через спиртовой раствор тиофенола в присутствии
хлорпикрина вследствие образования дифенилдисульфида наблю-
дается белое помутнение или опалесценция 177. При пропускании 2 л
воздуха в течение 3—4 мин можно обнаружить ОВ в концентра-
ции 0,06 мг]л.
При просасывании воздуха, содержащего хлорпикрин, через
10%-ный бензольный раствор диметиланилина первоначальная
желтая окраска раствора переходит в красную. Эта окраска уси-
ливается при добавлении нескольких капель перекиси водорода
и нагревании 178.
Хлорпикрин реагирует с меркаптидами калия с образованием
нерастворимых продуктов конденсации 179. Эта реакция может быть
использована для индикации хлорпикрина: испытуемый воздух про-
пускают через спиртовой раствор дитиогликолята калия. В при-
сутствии хлорпикрина образуется желтоватый осадок.
При термическом разложении хлорпикрина в нагретых докрас-
на кварцевых или фарфоровых трубках среди прочих продуктов
образуется также хлор, присутствие которого обнаруживают иод-
крахмальным реагентом 180>,81.
121
5.9.2. Количественное определение
5.9.2.1. Аргентометрическое определение хлора в растворе после
разложения хлорпикрина. Разложение хлорпикрина может быть
осуществлено многими методами. Наряду с реакциями со спирто-
выми растворами едких щелочей и щелочных алкоголятов хлор-
ион в стехиометрическом количестве образуется также при дей-
ствии перекиси 183 или сульфита натрия 182.
CCl3NO2 + 3Na2SO3 + H2O —► CHNO2(SO3Na)2 + NaCl + NaHSO4
Применение перекиси натрия имеет то преимущество, что в
процессе окисления она обесцвечивает все образующиеся окрашен-
ные органические соединения, что облегчает установление конца
титрования по Фольгарду.
В раствор 1,5 г перекиси натрия в 50 мл 50%-кого этанола вносят навеску
0,1—0,2 г испытуемого ОВ. Реакционную смесь кипятят с обратным холодиль-
ником до обесцвечивания (примерно 1,5—2 ч). После прибавления 20 мл 2 н.
раствора NaOH смесь без холодильника дополнительно кипятят еще 30 мин.
Охлажденный раствор подкисляют 30 мл 2 н. раствора HNO3, после чего хлор
определяют по Фольгарду.
5.9.2.2. Колориметрическое определение в виде галогенциана.
Кроме описанных в разделе 5.9.1.3 качественных способов обнару-
жения разработаны колориметрические методы определения хлор-
пикрина в воде и в воздухе 184.
Реагенты
Пиридин — флороглюцин (А) — смесь 50 мл пиридина, 50 мл метанола и
0,6 г KCN встряхивают 30 мин при охлаждении льдом. Отфильтровывают остаток
нерастворившегося KCN и прибавляют 0,1 е флороглюцина; реагент готовят по
мере необходимости и хранят в склянке из коричневого стекла.
Пиридин — 5,5-диметилдигидрорезорцин (Б) — смесь 30 мл пиридина, 70 мл
метанола и 0,4 г KCN встряхивают в течение 30 мин при охлаждении льдом,
после чего отфильтровывают остаток нерастворившегося цианида калия и при-
бавляют 0,8 г 5,5-диметилдигндрорезорцина.
Определение в воде. Встряхивают дважды 5 мл пробы анализируемой воды
с 3 мл лигроина и к объединенному экстракту прибавляют 2 мл реагента А. Че-
рез 5 мин раствор, окрасившийся в красно-фиолетовый цвет, колориметрируют
при 535 нм, сравнивая с контрольной пробой; чувствительность реакции 0,004 мг
хлорпикрина в Г мл воды.
Определение в воздухе. Испытуемый воздух медленно пропускают (2 л в
течение 15 мин) через поглотительный сосуд, заполненный 5 мл монометилового
эфира этиленгликоля. После прибавления 2 мл реагента Б раствор нагревают
5 мин на кипящей водяной бане и охлаждают. Затем оранжевый до крас-
ного раствор колориметрируют при 490 нм; чувствительность реакции 0,4 мкг/мл
раствора.
Из других цветных реакций, пригодных для обнаружения хлор-
пикрина, следует назвать измерение экстинкции при 405 нм окра-
шенного в желтый цвет соединения, образующегося в результате
взаимодействия в щелочном растворе хлорпикрина, пиридина и
цианида калия 187; метод может быть использован для определения
от 2,5 до 25 мкг хлорпикрина в 1 мл раствора. При прибавлении
барбитуровой кислоты к слабокислому раствору образуется поли-
122
метановый краситель, максимум поглощения которого находится
при 578 нм.
5.9.2.3. Колориметрическое определение хлорпикрина по обра-
зующемуся нитриту. Образующийся при восстановлении хлорпик-
рина (см. раздел 5.9.1.1) нитрит дает при взаимодействии с реаген-
том Грисса азокраситель, по экстинкции которого колориметри-
чески может быть определено содержание хлорпикрина. Для этой
же цели может быть использован предложенный Браттоном и
Маршаллом модифицированный реагент Грисса, состоящий из
сульфаниламида и гидрохлорида N-(1-нафтил)-этилендиамина 195.
В аналогичном способе хлорпикрин, поглощенный из воздуха изо-
пропиловым спиртом, нагревают с перекисью водорода. После
прибавления солянокислого раствора сульфаниламида, раствора
гидрохлорида л-фенилендиамина и концентрированного едкого нат-
ра колориметрируют перешедший в слой спирта краситель (крас-
ный стрептоцид)188.
5.10. СОВРЕМЕННЫЕ РАЗДРАЖАЮЩИЕ ОВ
5.10.1. о-Хлорбензилиденмалонодинитрил
(о-хлорбензальмалонодинитрил, Си Эс)
Это белое кристаллическое вещество, легко растворимое в ацето-
не, бензоле и хлороформе, хуже — в спирте, четыреххлористом уг-
лероде и эфире и почти нерастворимое в воде, петролейном эфире
и сероуглероде. Оно медленно гидролизуется водой; добавление
щелочей значительно ускоряет гидролиз:
aCH=C(CN)2
+ Н2О —> Г ]Г +CH2(CN)2
Cl
Образовавшиеся о-хлорбензальдегид и динитрил обнаруживают
известными методами.
5.10.1 .1. Качественное обнаружение. Реакция с м-динитробензо-
лом.
Смешивают 1 мл спиртового раствора испытуемого вещества, 0,1 мл 1%-ного
спиртового раствора ж-динитробензола н 0,2 мл 30%-ного водного раствора КОН
и полученную смесь нагревают. Появляющееся при высоких концентрациях ОВ
красно-коричневое, а при низких — вначале красно-фиолетовое, переходящее
затем в оранжевое окрашивание указывает на присутствие о-хлорбензилиден-
малоноднннтрила; чувствительность реакции 1 мкг! мл.
Хлорацетофенон, бромбензилцианид и хлорацетон дают анало-
гичные окрашивания (см. раздел 5.7.2.1).
Обнаружение динитрила малоновой кислоты. При нагревании
нитрилов со смесью окиси и карбоната кальция (1:1) сначала об-
разуется цианид кальция, разлагающийся затем в результате гид-
ролиза при высокой температуре до цианистого водорода и окиси
кальция (см. сс.263, стр. 233):
Ca(CN)2 + Н2О (пар) —СаО + 2HCN
123
Смешивают в микропробирке 1 каплю раствора испытуемого вещества или
несколько миллиграммов ОВ с 0,2—0,3 г смеси окиси и карбоната кальция и
нагревают на масляной бане до 250 °C. Выделяющийся цианистый водород об-
наруживают по синему окрашиванию помещенной на отверстие пробирки инди-
каторной бумаги, пропитанной ацетатом меди и ацетатом о-толидина. Этой
реакцией можно обнаружить 10 мкг HCN.
Синильная кислота образуется также при нагревании пробы
с двуокисью марганца до 130—140°C (см. сс. 263, стр. 235). Другой
способ обнаружения нитрила321 заключается в нагревании с серой.
Нагревают в микропробирке на малом пламени несколько миллиграммов
вещества примерно со 100 мг порошка серы. Выделяющуюся в случае присут-
ствия соединений, содержащих CN-группы, роданистоводородную кислоту обна-
руживают по покраснению помещаемой в пробирку индикаторной бумаги, про-
питанной подкисленным 1%-ным раствором нитрата железа(Ш).
Спектрофотометрическое обнаружение в ультрафиолетовом
свете264. Аэрозоль динитрила малоновой кислоты, задержанный
бумагой при фильтрации воздуха, может быть обнаружен спектро-
фотометрически в ультрафиолетовом свете при 260 нм.
15.10.2. Морфолид пеларгоновой кислоты
При нагревании в кислой среде морфолид гидролизуется
н2о
О=С—(СН2)т—СНз
О
Q+ CH3-(CH2)r-cf
\он
4
Выделяющийся морфолин может быть обнаружен чувствитель-
ной цветной реакцией с уксусным альдегидом и нитропруссидом
натрия по образованию окрашенного продукта неизвестной струк-
туры (сс.236, стр. 264) или по температуре плавления пикрата
(145—147°C). Очень чувствительным является обнаружение мор-
фолина по образованию дитиокарбамината меди (сс.23в, стр. 266).
В микропробнрку помещают 1 каплю кислого гидролизата, 1 каплю 5%-ного
раствора CuSO4 и добавляют водный NH4OH до образования прозрачного си-
него раствора, который встряхивают с 2 каплями смеси, состоящей из 1 части
сероуглерода и 3 частей бензола. Коричневая или желтая окраска бензольного
слоя указывает на присутствие морфолина. Чувствительность реакции примерно
5 мкг морфолина.
5.11. СВИНЕЦОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Перед анализом органические соединения свинца следует подвер-
гнуть минерализации.
124
5.11.1. Тетраэтилсвинец
Так как весьма ядовитый применяемый в качестве антидетонатора
тетраэтилсвинец не используется как отравляющее вещество и как
таковое в литературе не описано, мы ограничимся изложением не-
которых простых способов его обнаружения. Абсорбировать тет-
раэтилсвинец из воздуха можно, пропуская воздух через поглоти-
тельную склянку, содержащую раствор иода в KI или четыреххло-
ристом углероде или других растворителях. Пригодны также по-
глотительные трубки, заполненные кристаллическим иодом 265.
В бензине, содержащем тетраэтилсвинец, можно обнаруживать
тетраэтилсвинец без предварительной экстракции.
5.11.1.1. Разложение УФ-облучеиием 266. Этилированный бензин
при облучении его в течение некоторого времени светом ртутной
лампы вследствие выпадения двуокиси свинца мутнеет. Образец
бензина следует помещать в сосуд из увиолевого стекла, прони-
цаемого для УФ-облучения.
Очень быстро осуществляется разложение, если 1 каплю этилированного
бензина или раствора тетраэтилсвинца в органическом растворителе нанести на
фильтровальную бумагу и подвергнуть её облучению светом УФ-лампы. Обра-
зовавшуюся через несколько минут двуокись свинца растворяют нанесением
1 капли разбавленной уксусной кислоты и обнаруживают свинец по образова-
нию желтого иодида свинца при прикапывании раствора KI или по образованию
черного сульфида свинца после обработки сероводородом или раствором суль-
фида аммония.
Весьма чувствительна реакция обнаружения свинца дитизоном. Для этого
помещенный на фильтровальную бумагу бензин испаряют под УФ-лампой и
на то же место на бумаге наносят 1 каплю 0,01%-ного раствора дитизона в
четыреххлористом углероде. Появление красного окрашивания указывает на
присутствие свинца; в отсутствие свинца пятно остаетси зеленым.
5.11.1.2. Разложение азотной кислотой168. Для этого поступают
следующим образом.
К 1 капле этилированного бензина или эфирного экстракта добавляют 1 мл
концентрированной HNO3 (d 1,4) и, периодически встряхивая, доводят смесь -в
течение примерно 5 мин до кипения. Затем путем сильного нагревания удаляют
азотную кислоту, к остатку приливают 2 мл воды и после подкисления раствора
уксусной кислотой обнаруживают свинец по реакции с К1 илн H2S. Способ
становится более надежным, если обработке азотиой кислотой подвергают боль-
шие объемы бензина или экстрактов в четыреххлористом углероде или эфире и
операцию осуществляют в делительной воронке. После отделения азотной кис-
лоты ее смешивают с концеитрированиой H2SO< и нагревают. При этом оса-
ждается сульфат свинца. Раствор декантируют, растворяют осадок PbSO< в
аммиачном растворе ацетата аммония и обнаруживают свинец в виде хромата.
5.11.1.3. Разложение хлором267. Для разложения тетраэтилсвин-
ца бензин обрабатывают хлоратом калия.
В пробирке смешивают 0,1—0,2 г КС1О3 и 5 мл испытуемого бензина и, не
встряхивая, прибавляют к смеси 3—5 капель концентрированной НС1. В при-
сутствии этилированного бензина через несколько минут появляется белое по-
мутнение, обусловленное выпадением хлорида свинца.
5.11.1.4. Количественное определение тетраэтилсвинца. После
Минерализации образца свинец определяют весовым методом в виде
125
хромата свинца или комплексонометрическим титрованием. Можно
непосредственно титровать этилированный бензин раствором иода:
рь(С2н6)4 + 12 —* Pb(C2Hs)3I + С2Н61
5.11.2. Другие свинецорганические соединения
В литературе нет указаний о специальных способах индикации
соединений, синтезированных в Англии во время второй мировой
войны. К числу таких соединений относятся соли триэтил- и три-н-
пропилсвинца, а также триалкилсвинецсульфамиды.
Их индикация и определение могут быть выполнены (после раз-
личйого рода «мокрой» минерализации) качественными и количе-
ственными аналитическими методами, разработанными для опре-
деления свинца.
5.12. ОКИСЬ УГЛЕРОДА И КАРБОНИЛЫ МЕТАЛЛОВ
В большей части способов обнаружения и количественного опре-
деления этих соединений используется высокая восстановительная
способность окиси углерода.
5.12.1. Качественное обнаружение
5.12.1.1. Восстановление хлорида палладия. При действии окиси
углерода хлористый палладий восстанавливается до палладия:
Pd++ + СО + Н2О —► Pd + СО2 + 2Н+
Индикаторная бумага, пропитанная хлористым палладием,
окрашивается в серый или черный цвет в присутствии уже от 0,01
до 0,03% окиси углерода в воздухе (ср. раздел 9.2.3.11). Реакция
становится значительно более чувствительной при добавлении фос-
форномолибденовой кислоты. В то время как сама окись углерода
весьма медленно реагирует с фосфорномолибденовой кислотой, в
присутствии хлорида палладия тотчас образуется молибденовая
синь. Это объясняется адсорбцией окиси углерода металлическим
палладием, образовавшимся в результате восстановления, что при-
водит к активации окиси углерода. В результате гетерогенного
катализа происходит ускорение реакции между окисью молибдена
и СО:
2Мо03 + СО —> Мо205 + СО2
Этот принцип может быть использован в индикаторных труб-
ках268 или в капельной реакции для обнаружения окиси углерода,
образующейся при разложении определенных органических соеди-
нений сиропообразной Н3РО4 или карбонилов металлов концентри-
рованной H2SO4.
Реагент
Фосфорномолибденовая кислота + хлорид палладия—раствор 0,02 г хло-
рида палладия в 2 каплях концентрированной H2SO4 разбавляют водой до 10 лм.
126
Реагент готовят смешением 2 мл этого раствора с 8 мл насыщенного на холоду
водного раствора фосфорномолибденовой кислоты (сс.23в, стр. 331).
Вносят на стеклянной палочке или стеклянной пробке в сосуд, где проводи-
лось разложение исследуемого вещества, 1 каплю реагента и затем 1 каплю
полученной реакционной смеси наносят на бумагу и дают ей соединиться с на-
несенной рядом каплей воды. Синее окрашивание указывает на присутствие
окиси углерода. Двуокись серы, сероводород и цианистый водород мешают об-
наружению.
На том же принципе основано обнаружение СО при помощи
индикаторного геля NBS. На силикагеле, пропитанном сульфатом
палладия и молибдатом аммония, образуется желтый кремние-
молибдат, который под воздействием окиси углерода восстанав-
ливается в зеленовато-синее гетерополисоединение 268. Чувстви-
тельность реакции 0,001 % ' окиси углерода. Восстановление фос-
форномолибденовой кислоты может быть также использовано для
фотометрического определения окиси углерода.
5.12.1.2. Восстановление нитрата серебра. При пропускании воз-
духа, содержащего окись углерода, через аммиачный раствор нит-
рата серебра последний мутнеет вследствие выделения металли-
ческого серебра:
2AgNO3 + СО + Н2О —► 2Ag + СО2 + 2HNO3
5.12.1.3. Восстановление пятиокиси иода.
I20g 4“ 5СО —► 5СО2 4~ 12
Эта реакция используется для обнаружения и полуколичествен-
ного определения окиси углерода в индикаторных трубках, для
чего пятиокись иода вместе с дымящей серной кислотой (олеумом)
наносят на силикагель или пемзу. Такой наполнитель известен под
названием хуламит 269. При наличии СО он окрашивается в резуль-
тате выделения иода в сине-зеленый, серо-синий или черный цвет.
Если объем пропущенного воздуха известен, длина окрашенной
зоны является мерой концентрации окиси углерода.
5.12.1.4. Спектрофотометрическое обнаружение. В основе этого
специфического метода индикации лежит характерное отношение
оксигемоглобина НЬО2 и карбоксигемоглобина НЬСО к восстанови-
тельному действию сульфида аммония. Испытуемый воздух про-
пускают через разбавленную кровь (1:10 или 1:100) и спектро-
фотометрически наблюдают поглощение. Для нормальной крови
НЬО2 характерны две полосы поглощения между фраунхоферскими
линиями D и Е (Х577 и 537 нм)', в присутствии в крови НЬСО
также наблюдаются две полосы примерно в тех же границах
(Л 576 и 542 нм). Если к крови прибавить несколько капель рас-
твора (NH4)2S, то через 6—8 мин НЬО2 восстанавливается и обе
полосы, соответствующие НЬО2, исчезают, а при 559 нм появляется
слабая полоса гемоглобина НЬ. Полосы НЬСО под действием вос-
становителя не изменяются. Таким способом можно обнаружить до
10% НЬСО в нормальной крови,
127
При использовании раствора гемоглобина, к которому для пред-
отвращения образования НЬО2 прибавлен тиосульфат натрия, мо-
жно обнаружить и определить до 0,001% СО в воздухе281.
5.12.1.5. Обнаружение с помощью мышьяковистой кислоты и
хлорного золота 270. Окись углерода каталитически ускоряет реак-
цию между мышьяковистой кислотой и хлорным золотом. Бумага,
пропитанная 0,1%-ным раствором As2O3 в фосфатном буфере (pH 4)
и 2%-ным раствором хлорного золота, при воздействии воздуха,
содержащего окись углерода, окрашивается в черный цвет.
5.12.1.6. Обнаружение карбонила железа. При пропускании под-
лежащего исследованию воздуха через стеклянную трубку, нагре-
ваемую на газовой горелке, карбонил железа термически разла-
гается. Окислы железа осаждаются в холодной зоне трубки, а в
выходящем из трубки газе можно обнаруживать окись углерода.
Можно так же подлежащую исследованию газовую смесь про-
пускать через промывную склянку с концентрированной серной
кислотой. Затем содержимое склянки выпарить досуха, остаток
обработать водой и определить в растворе железо с помощью из-
вестных реакций.
Минерализовать пентакарбонил железа можно и обрабатывая
метанольный раствор карбонила перекисью водорода и раствором
аммиака. Образовавшуюся гидроокись железа(III) растворяют в
соляной кислоте; при добавлении роданида калия образуется окра-
шенный в красный цвет роданид железа (III).
5.12.1.7. Обнаружение карбонила никеля271. Удобным реагентом
для индикаторных трубок является окрашенная в желтый цвет
нанесенная на силикагель фосфорномолибденованадиевая кисло-
та. Появление синей окраски восстановленного гетерополисоедине-
ния при пропускании 1 л воздуха свидетельствует о наличии кар-
бонила никеля. Метод пригоден для обнаружения 1 мкг/л карбо-
нила никеля.
5.12.2. Количественное определение
5.12.2.1. Определение при помощи пятиокиси иода. Исследуемый
воздух пропускают в соответствующей аппаратуре через трубку
с пятиокисью иода, нагреваемой до 150 °C. Образующийся при
этом иод (см. раздел 5.12.1.3) может быть определен титрованием
тиосульфатом натрия 272>273; в равной степени пригодно весовое
или объемное определение двуокиси углерода 274.
5.12.2.2. Гопкалитовый метод. Гопкалитом, разработанным для
фильтров противогаза, называют смеси различных соотношений
окислов марганца и меди с добавками окислов кобальта и серебра.
Примером гопкалита является смесь 50% МпО2, 30% СиО, 15%
Со2О3 и 5% Ag2O; другая смесь состоит из 60% МпО2 и 40% СиО.
Гопкалит уже при комнатной температуре каталитически окисляет
окись углерода в двуокись:
2СО + О2 —> 2СО2 + 67,9 ккал
128
Тепло, выделяющееся в реакции, может быть измерено. Этот
принцип положен в основу устройства прибора Дрегера для оп-
ределения концентрации окиси углерода и других автоматических
газоанализаторов этого типа 275’276.
5.12.2.3. Восстановление окиси ртути. Воздух, содержащий
окись углерода, пропускают через нагреваемую до 175—200 °C взве-
шенную трубку, содержащую красную окись ртути. При этом
окись ртути восстанавливается с образованием эквивалентного ко-
личества паров ртути и выделяется COg:
HgO + CO —> Hg + CO2
Потери в массе трубки определяют повторным взвешиванием 282.
Определение может стать более чувствительным, если пары ртути
спектрофотометрировать 283 или использовать реакцию с сульфидом
селена. Пропитанная раствором сульфида селена фильтровальная
бумага вследствие образования сульфида ртути приобретает чер-
ную окраску:
3Hg + SeS2 —► 2HgS + SeHg
5.12.2.4. Определение карбонила никеля. Исследуемый воздух
пропускают через раствор иода в четыреххлористом углероде277
или через солянокислый раствор хлорамина Т 278. При прибавле-
нии диметилглиоксима в щелочной среде появляется красная окра-
ска, интенсивность которой измеряют либо в компараторе либо
фотометрируют при 496 нм. В другом способе 279 воздух пропускают
через насыщенный коллоидной серой раствор трихлоруксусной
кислоты и образовавшийся сульфид никеля определяют в УФ-спек-
трофотометре; чувствительность метода 2 мкг карбонила в 1 м3
воздуха.
5.13. ФТОРКАРБОНОВЫЕ КИСЛОТЫ
Характерным для этих ОВ, обладающих высокой пероральной ток-
сичностью и являющихся особенно опасными для отравления пить-
евой воды, является прочность связи С—F.
Малая реакционная способность монофторуксусной кислоты и
ее производных затрудняет дегазацию и индикацию этих соеди-
нений.
5.13.1. Качественное обнаружение
5.13.1.1. Отщепление и обнаружение фтора. Общепринятым спо-
собом разложения фторорганических соединений является нагрева-
ние их с алкоголятами натрия, причем для того чтобы проводить
реакцию при более высокой температуре, способствующей более
быстрому разложению ОВ, применяют спирты с 6—8 атомами угле-
рода, предпочтительно гексанол 298.
Навеску фторсодержащего соединения (10—20 мг) растворяют в 15 мл
гексанола в колбе, снабженной обратным холодильником, и после прибавления
0,2 г металлического натрия нагревают смесь 15 мин до кипения. Еще горячий
5 Зак. 677
129
раствор переносят в делительную воронку и дважды экстрагируют водой (по
10 мл). Затем одним из обычных способов (см. раздел 3) в водном экстракте
качественно или количественно определяют фтор.
Гексанол можно использовать и для поглощения паров фтор-
органического соединения из воздуха. При наличии фторсодержа-
щего соединения в воде отщепление фтора осуществляют нагрева-
нием образца со смесью метапериодата калия, перхлората серебра
и хлорной кислоты2". Отщепившийся фтор после прибавления к
реакционной смеси стеклянной ваты превращается в кремнефтори-
стоводородную кислоту, которую отгоняют. Для определения фтора
во фторуксусной кислоте последнюю минерализуют с помощью
окиси кальция 30°, однако при этом имеются неконтролируемые по-
тери фтора, вероятно вследствие образования фтористого метила 30°.
Надежно проходит минерализация в никелевой бомбе по Вурц-
шмитту при добавлении перекиси натрия и небольшого количества
глицерина 301.
5.13.1.2. Обнаружение фторацетатов по реакции с солями лан-
тана302. Применение солей лантана в качестве реагентов на аце-
таты известно давно 303. В аммиачном растворе ацетаты образуют
с солями лантана основной ацетат лантана, который дает с иодом
синее абсорбционное соединение. Способ применим также и для
обнаружения фторуксусной кислоты, однако чувствительность
(0,5 мг) и специфичность реакции не удовлетворяют требованиям.
5.13.1.3. Обнаружение фторацетатов по реакции образования
тиоиндиго 304. Аналогично синтезу тиоиндиго по Фридлендеру305
при нагревании монофторуксусной кислоты или ее солей с тиосали-
циловой кислотой и щелочью образуется тиоиндоксил, который
окисляют феррицианидом калия до красного тиоиндиго:
Очень важным является правильный выбор температурных ус-
ловий стадии сплавления со щелочью, Как показывает опыт, тем-
130
пература должна быть равной примерно 200 °C, а не 100°C, как
это указывали Рамзей и Паттерсон 304. Реагенты, необходимые для
проведения определения, приведены в разделе 5.13.2.
В пробе воды при помощи разбавленных NaOH или H2SO4 устанавливают
значение pH от 5 до 7. Помещают в небольшую фарфоровую чашку 1 мл этого
нейтрализованного раствора и смешивают с 1 мл раствора тиосалициловой кис-
лоты и 2 каплями NaOH (1 : 1). Смесь выпаривают досуха на асбестовой сетке
на газовой горелке. Остаток дополнительно нагревают в течение 5—10 мин, при
этом он может окраситься в желтый, но ни в коем случае не в красный цвет.
После охлаждения остаток растворяют в 1 мл раствора K3Fe(CN)6. В присут-
ствии фторацетатов, в зависимости от их концентрации, образуется более или
менее значительный красно-фиолетовый осадок тиоиндиго. В случае малого со-
держания фторацетатов осадок становится видимым только после длительного
стояния.
Этот метод дает возможность обнаружить до 0,02 мг фторацетата натрия.
Чувствительность метода можно повысить, если взять для анализа больший
объем воды, добавить NaOH для создания щелочной среды (по лакмусу) и по-
сле прибавления 1 мл тиосалицилового реагента выпарить досуха.
5.13.1.4. Обнаружение при помощи концентрированной серной
и хромотроповой кислот. Как все моногалогенированные производ-
ные уксусной кислоты, фторуксусная кислота при нагревании с
концентрированной серной кислотой гидролизует с образованием
гликолевой кислоты, которая далее под действием серной кислоты
разлагается на формальдегид и окись углерода. Формальдегид об-
наруживают чувствительной цветной реакцией с хромотроповой
кислотой (см. раздел 5.16.1).
Аналогичную реакцию дают применяемые в качестве гербици-
дов хлорированные феноксиуксусные кислоты. Реакцию проводят
по методу, аналогичному изложенному в разделе 6.16.1.1; чувстви-
тельность метода 0,1 мкг фторуксусной кислоты.
5.13.2. Количественное определение
После отщепления фтора или минерализации образца количествен-
ное определение фтор-иона возможно одним из обычных объемных
колориметрических или энзиматических способов (см. раздел 3).
Фотометрическое определение монофторуксусной кислоты 306 осно-
вано на цветной реакции с тиосалициловой кислотой (см. выше,
раздел 5.13.1.3).
Реагенты
Тиосалициловая кислота — раствор 1 г тиосалициловой кислоты в 7 мл
4%-ного NaOH, доведенный водой до объема 50 мл.
Феррицианид калия — 2%-ный водный раствор.
К пробе воды или водному экстракту прибавляют 1 н. раствор NaOH до
щелочной реакции по лакмусу, после чего выпаривают в фарфоровом тигле до
объема, равного примерно 5 мл. С помощью 1 н. раствора H2SO4 устанавливают
pH 5—7 и после прибавления 1 мл раствора тиосалициловой кислоты и 2 капель
раствора NaOH (1:1) нагревают смесь 20—30 мин на водяной бане, а затем
выпаривают досуха на масляной бане при 170—200 °C, после чего нагревают при
этой же температуре еще 30 мин. После охлаждения остаток растворяют в 2 мл
воды и приливают раствор феррицианида калия до появления желтого
окрашивания. Содержимое тигля количественно переносят в делительную
5*
131
воронку и образовавшееся тиоиндиго дважды экстрагируют толуолом. Толуоль-
ный экстракт доводят до соответствующего объема и фотометрируют при 540 нм,
сравнивая с контрольной пробой. Расчет осуществляют по калибровочной
кривой.
5.14. АЛКАЛОИДЫ
5.14.1. Подготовка к анализу
Алкалоиды могут быть использованы в качестве диверсионных
ядов для отравления продуктов питания, фуража и питьевой воды.
Поэтому пробы, подлежащие исследованию, могут представлять
собой: продукты питания, фураж, воду, водные экстракты, инди-
видуальные алкалоиды.
Алкалоиды представляют собой азотистые основания, извлекае-
мые из растений и содержащие в подавляющем числе случаев ге-
тероциклические системы, например остатки пиридина, пиперидина,
хинолина, пирролидина и др. Большая часть алкалоидов является
третичными основаниями, меньшая часть представляет собой
первичные, вторичные и четвертичные основания. За небольшим
исключением алкалоиды — твердые кристаллические бесцветные
вещества, обладающие незначительными основными свойствами.
Для идентификации этих веществ имеются многочисленные
реагенты, применимые для группового осаждения, и многие общие
цветные реакции. После подобных предварительных исследований
используют различные специальные способы обнаружения отдель-
ных алкалоидов. Хроматографические методы разделения и обна-
ружения применяются в лабораториях, имеющих соответствующее
оборудование.
Подготовка к анализу зависит от вида материала пробы и тре-
буемой подробности исследования. При работе с определенным
веществом проводят общие цветные реакции и после того, как
этим путем будут получены некоторые данные о наличии опреде-
ленного алкалоида, осуществляют специальные реакции обнаруже-
ния данного алкалоида. Аналогично поступают при анализе проб
воды и водных экстрактов, применяя групповые реагенты для
осаждения.
Помощь в оценке результатов предварительных определений ока-
зывают сведения о температурах плавления алкалоидов (стр. 133)
и результаты цветных реакций, проведенных с заведомо чистыми
алкалоидами (см. табл. 5, стр. 136). Алкалоиды, обнаруживаемые
в продуктах питания и фураже, приходится экстрагировать и после
отделения мешающих определению примесей выделять. Затем при-
меняют весьма распространенный в токсикологическом анализе
способ Стаса — Отто. *Этот метод разделения алкалоидов на три
группы зиждется на различной их основности. Материал пробы из-
мельчают, суспендируют в спирте и после прибавления некоторого
количества винной или небольшого количества серной кислоты ки-
пятят 15 мин с обратным холодильником. Смесь после охлаждения
фильтруют и фильтрат значительно упаривают на водяной бане.
132
При разбавлении фильтрата холодной водой выпадают жиры и
смолы. Их отфильтровывают и вновь выпаривают фильтрат. Си-
ропообразный остаток обрабатывают абсолютным спиртом, при
этом выпадают неорганические соли, белки, пептоны и декстрины.
После повторного фильтрования, испарения спирта и растворения
остатка некоторым количеством воды получают раствор виннокис-
лых или сернокислых алкалоидов.
Затем в этом растворе проводят групповое разделение без вся-
кой предварительной обработки попеременной экстракцией из кис-
лой и щелочной среды. Первую группу составляют алкалоиды,
экстрагирующиеся из слабокислого раствора. Их выделяют из по-
лученного выше раствора или водных проб, смешанных с винной
кислотой, встряхиванием сначала с эфиром, затем с хлорформом,
содержащим 10% этанола. После испарения растворителя в остат-
ке эфирного экстракта обнаруживают среди прочих алкалоидов
пикротоксин, а в остатке хлороформного экстракта — колхицин
и вератрин. Вторую группу составляют алкалоиды более основ-
ного характера, которые только после обработки водного раствора
едким натром можно извлечь эфиром. После испарения эфира в
остатке среди прочих находят атропин, никотин, стрихнин, бруцин,
аконитин, физостигмин, кониин и кокаин. Третью группу состав-
ляют алкалоиды со свободными фенокси-группами, образующие
с едким натром растворимые в воде феноляты, не экстрагируемые
эфиром. Их можно извлечь хлороформом из первоначального под-
кисленного водного раствора после подщелачивания его аммиа-
ком до слабощелочной реакции.
Разумеется, что при проведении этого группового разделения
в различных экстрактах могут быть обнаружены (в зависимости
от их основности или кислотности) и присутствующие в исследуе-
мых образцах отравляющие вещества, такие, например, как азо-
тистый иприт (третичное основание). Это обстоятельство следует
принимать во внимание при работе с групповыми реагентами.
5.14.2. Методы обнаружения
5.14.2.1. Определение температуры плавления. При работе с ин-
дивидуальным веществом имеется возможность идентифицировать
вещество по температуре плавления, пользуясь микроаппаратом
и микроскопом. Ниже приводятся температуры плавления некото-
рых важнейших алкалоидов (в °C):
Аконитин................................... 187—188
Атропин.................................... 115—116
Бруцин........................................ 178
Колхицин....................................... 155
Морфии.............. ...................... 230 (разл.)
Пикротоксин
Стрихнин .
200
265-266
Перед определением рекомендуется вещество очистить, что
осуществляется его растворением в спирте, фильтрованием,
133
упариванием фильтрата до сиропообразной косистенции, растворе-
нием остатка в воде, подщелачиванием едким натром и экстракцией
хлороформом. После этого хлороформ испаряют и остаток исполь-
зуют для определения температуры плавления.
5.14.2.2. Общие цветные реакции. Использование общих цветных
реакций имеет смысл только для чистых алкалоидов. Для про-
ведения цветной реакции несколько кристалликов испытуемого ве-
щества смешивают в небольшой фарфоровой чашке или в углуб-
лении капельной пластинки с несколькими каплями реагента. На-
блюдают за течением и результатом цветной реакции и, используя
данные табл. 5, делают выводы о том, какие алкалоиды могут
дать вызванную окраску.
Ниже приведены реагенты, наиболее часто применяемые для
проведения общих цветных реакций на алкалоиды.
1. Серная кислота, концентрированная, ч. д. а.
2. Азотная кислота, концентрированная, ч. д. a. (d 1,40).
3. Реагент Фрёде — раствор 0,1 г молибдата натрия или 0,5 г
молибдата аммония в 10 мл концентрированной H2SO4, получен-
ный при умеренном нагревании; сохраняется на холоду в течение
примерно 10 суток.
4. Реагент Манделина — раствор 0,1 г мелко растертого мета-
ванадата аммония, не содержащего азотной и хромовой кислот,
в 20 мл концентрированной H2SO4; сохраняется примерно в тече-
ние 10 суток.
5. Реагент Эрдманна — смесь 20 .мл концентрированной H2SO4
с 10 каплями раствора концентрированной HNO3 (10 капель
кислоты в 100 мл воды), применяют свежеприготовленным. Ре-
агент дает с фенолом и его производными также цветные реак-
ции.
6. Реагент Мекке — раствор 0,1 г селенистой кислоты в 20 мл
концентрированной H2SO4; на холоду сохраняется примерно в те-
чение 10 суток.
7. Реагент Марки — смесь 2 мл 30%-ного раствора формальде-
гида со 100 мл концентрированной H2SO4; срок хранения — около
недели. Для обнаружения следов алкалоида применяют 3 мл реа-
гента.
8. Реагент Васицкого — раствор 2 г п-диметиламинобензальде-
гида в 6 мл концентрированной H2SO4, к которому прибавлено
0,4 мл воды; сохраняется примерно в течение 1 суток. Для прове-
дения реакции несколько кристаллов алкалоида на часовом стекле
смешивают с каплей реагента и осторожно нагревают на асбесто-
вой пластинке.
9. Реагент Витали — раствор Са(ОН)2 в абсолютном этаноле.
Для проведения реакции несколько кристаллов алкалоида смеши-
вают с несколькими каплями дымящей HNO3 и выпаривают на во-
дяной бане досуха. Остаток после охлаждения смешивают с кап-
лей реагента, после чего наблюдают появление характерного окра-
шивания.
134
Окраски, получаемые при взаимодействии этих реагентов с раз-
личными алкалоидами, приведены в табл. 5.
5.1 4.2.3. Реакции группового осаждения. Для их проведения к
осаждающему реагенту по каплям прибавляют подкисленный рас-
твор алкалоида и наблюдают образование и внешний вид осадка.
Ниже приведены наиболее употребимые осаждающие реагенты
и получаемые с их помощью осадки.
1. Реагент Майера — раствор 1,35 г хлорной ртути (II) в 100 мл
5%-ного раствора KI. Осаждение проводят из солянокислого или
сернокислого раствора. Получаются аморфные или кристалличе-
ские осадки. Не осаждаются колхицин и соланин. Осадки образуют
атропин (белый, творожистый), берберин (желто-зеленый), бруцин
(1:50ООО), кониин (аморфный), наркотин, никотин, морфин, физо-
стигмин, стрихнин (1:15000), вератрин.
2. Реагент Драгендорфа — раствор 8 г азотнокислого висмутила
(BiONO3) в 20 мл HNO3 (d 1,18) вливают в раствор 27 г KJ в 40 мл
воды. Через 2—3 суток раствор сливают с выпавшего осадка
KNO3 и доводят объем до 100 мл\ реагент хранят в коричневой
склянке. № сернокислого раствора осаждаются следующие алка-
лоиды: аконитин (хромово-желтый), атропин (порошкообразный
красно-желтый, со временем канареечно-желтый), берберин
(оранжево-красный), колхицин, морфин (желто-красный, посте-
пенно растворяющийся при стоянии), наркотин,никотин (1 :40000),
бруцин, физостигмин, стрихнин (светло-желтый, постепенно тем-
неет), вератрин (светло-желтый, переходящий в канареечно-
желтый).
3. Реагент Вагнера — раствор 5а иода в 100мл 10%-ного рас-
твора KI. При осаждении из солянокислого раствора получаются
светло- и темно-коричневые аморфные осадки. Осаждаются
атропин, бруцин, колцихин, кониин, героин, наркотин, папаверин
(по прошествии некоторого времени темно-красные иглы) и
стрихнин.
4. Реагент Зонненшейна — раствор 10 г фосфорномолибденовой
кислоты в 100 мл воды. Реагент осаждает из соляно-, азотно- или
сернокислого растворов аморфные желтые до желто-коричневых
осадки, которые по прошествии некоторого времени принимают
синюю или зеленую окраску. Осаждаются следующие алкалои-
ды— аконитин, бруцин, кониин (кристаллический осадок), колхи-
цин, наркотин, никотин (из солянокислого раствора 1:40 000), фи-
зостигмин, вератрин.
5. Реагент Шейблера — раствор 10 г фосфорновольфрамовой
кислоты в 100 мл воды. Хлопьевидные, в большинстве случаев жел-
тые осадки дают алкалоиды — берберин, кониин (кристалличе-
ский), морфин, никотин (1:100000), стрихнин (1:200000).
6. Реагент Марме — раствор 2 г Cdl2 и 4 г KI в 12 мл воды.
Белые, в большинстве случаев аморфные осадки дают берберин
(желтый), бруцин, кониин (кристаллический), морфин, папаве-
рин, физостигмин, вератрин.
135
136
Таблица 5. Окраски, образуемые алкалоидами с различными реагентами
Алкалоид Концентри- рованная H2SO4 Концентри- рованная HNO3 d 1,4 Реагенты
Фрёде Эрдманна Мекке Васицкого Витали Манделина
на холоду при нагре- вании
Аконитин Желтая — Желтая Снне- желтая — желтая — — — — Коричневая
Атропин — — —- — Красно- фиолето- вая — красная Фиолето- вая — красная Красная — желтая
Берберии Оливково- зеленая — желтая Красно- коричневая Коричнево- зеленая Оливково- зеленая — желто- коричневая —- — — — —
Бруцин Кроваво- красная — желтая Малиново- красная — коричне- ватая — Желто- красная Лимони 0- желтая — — Красная — желтая
Коннин — —* Бесцвет- ная — жел- товатая — — — — — —
Колхнцнн Желтая Фиолето- вая — жел- тая Фиолето- вая — жел- тая Фиолето- вая — жел- тая Лимонно- желтая Желто- коричне- вая — — —
Кураре Коричне- вая — красная Фиолетовая Фиолетовая Желтая
Дигиталин (D. verum) Дигитонин Желтая Красная — Красно- фиолетовая
Дигитоксин Коричнево- красная — — —
Морфин — Красио- желтая Фиолетово- красная — зеленая —
Никотин — Желтова- тая—крас- ная — —
Физостиг- мин Желтая — зеленая Желтая Слабо краснова- тая — желтая Слабо красно- ватая — желтая
Пикроток- син Оранжево- красная — — —
Скополамин — — Глубоко синяя —
Соланин Коричневая, по краю красно- фиолетовая — Сине-фио- летовая — зелено- желтая —
Стрихнин — Желтая — —
Вератрин оо Желтая — оранжево- зеленая — карминово- красная, флуорес- цирующая Слабо- желтая Желтая — оранжево- вишнево- красная Желтая — оранжево- зеленая — красная — карминовая флуорес- ценция
— — — — —
— — — — —
— — — — —
Синяя — Коричне- Светло- — Красная —
сине- вая красная сине-фиоле-
зеленая товая
— — Красно- — —
коричневая
Коричне- Слабо Яалрнйя — Желто-
ватая — красно- зелеиая
желтая коричне-
вая
— — — — —
— — — Красная —
— — * — —. Оранжево-
красная
-— Красно- Оранжево-
оранжевая красная
— — Глубоко Красно- Желтая —
зеленая — фиолето- вишнево-
коричневая вая до красиая
оранжевой
7. Золотохлористоводородная кислота — раствор 6—Юг хло-
рида золота в 100 мл разбавленной НС1. Белые или золотисто-жел-
тые, в большинстве случаев кристаллические осадки дают алкалои-
ды — атропин (после перекристаллизации из соляной кислоты т. пл.
138°C), аконитин (аморфный), берберин (оранжево-красный), кол-
хицин, кокаин (светло-желтый), физостигмин, папаверин (т. пл.
196°C), стрихнин (оранжевый), вератрин [после перекристалли-
зации из спирта иглы т. пл. 182 °C (разл.)].
8. Платинохлористоводородная кислота — раствор 10 г хлорида
платины в 100 мл разбавленной НС1. При осаждении из соляно-
кислых растворов почти все алкалоиды образуют светло-желтые до
оранжевых кристаллические осадки, имеющие характерные темпе-
ратуры плавления.
Таблица 6. Осаждение алкалоидов групповыми реагентами
Алкалоиды
Реагенты
Аконитин
Апоморфин
Атропин
Хинин
Кокаин
Кофеин
Кониин
Колхицин
Кураре
Кодеин
Бруцин
Эметин
Гиосциамин
Гидрастин
Берберин
Морфин
Наркотин
Наркеин
Никотин
Папаверин
Физостигмин
Стрихнин
Вератрин
+
+
+
+
+
9. Пикриновая кислота — насыщенный на холоду водный рас-
твор. В виде пикратов из сернокислых растворов осаждаются нар-
138
котин и вератрин (аморфные), бруцин (кристаллический, т. пл.
220°С), папаверин, стрихнин и никотин (т. пл. 218°С). Пикрат
атропина выпадает из концентрированного раствора (т. пл. 175—
176 °C).
10. Пикролоновая кислота — раствор 2,64 г пикролоновой кис-
лоты в 100 мл этанола. Большая часть алкалоидов образует жел-
тые до красных кристаллические осадки.
11. Таннин — свежеприготовленный 5%-ный водный раствор тан-
нина. Образует с алкалоидами аморфные, белые до желтоватых
осадки.
12. Реагент Годффройя — раствор 5 г кремневольфрамовой кис-
лоты в 100 мл воды. Некоторые алкалоиды дают осадки в очень
разбавленных растворах, например атропин (1 : 15 000).
В табл. 6 приведены результаты осаждения наиболее распро-
страненных алкалоидов групповыми реагентами.
5.14.2.4. Характерные реакции обнаружения некоторых алкалои-
дов. Этими реакциями пользуются после того, как при помощи
групповых осаждающих реагентов или общих цветных реакций
получены данные, свидетельствующие о наличии в пробе опреде-
ленного алкалоида.
Обнаружение аконитина. Смешивают в небольшой фарфоровой чашке не-
сколько миллиграммов испытуемого вещества с 4 каплями 80%-ной H2SO4 и на-
гревают 5 мин на водяной бане. После прибавления нескольких кристаллов
резорцина нагревание продолжают. Появление красного окрашивания, дости-
гающего максимума через 20 мин, указывает на присутствие аконитина; чув-
ствительность реакции 0,1—0,5 мг.
Обнаружение атропина. С реагентом Витали (стр. 134) сначала образуется
фиолетовое окрашивание, переходящее затем в красное. Другие алкалоиды сразу
вызывают красное окрашивание.
Обнаружение колхицина. Растворяют около 10 мг испытуемого вещества в
2 мл разбавленной НС1 (1:1) и смешивают с 2 каплями 5%-ного раствора FeCh.
В присутствии колхицина при нагревании на водяной бане образуется оливково-
зеленое окрашивание, которое при охлаждении становится более интенсивным.
Экстракт окрашенного соединения хлороформом окрашен в гранатово-красный
цвет, при незначительных количествах колхицина — в оранжевый; чувствитель-
ность реакции 2—5 мг.
Обнаружение никотина. Смешивают 3 мл водного раствора испытуемого
вещества с 0,1 мл 1%-ного раствора KCN и 0,5 мл 1%-ного раствора моно-
хлорамина. Через 1 мин прибавляют 1 мл 1%-ного раствора барбитуровой кис-
лоты. Никотиновые основания вызывают красное до красно-оранжевого окра-
шивание раствора; чувствительность реакции 0,005 мг.
Обнаружение пикротоксина. К нескольким миллиграммам испытуемого ве-
щества прибавляют 1—2 капли концентрированной HNO3 и выпаривают на
водяной бане досуха, затем остаток растворяют в 2—3 каплях H2SO4. Обра-
зующееся при осторожном прибавлении нескольких капель 30%-ного рас-
твора NaOH кирпично-красное окрашивание указывает на присутствие пикро-
токсина.
Обнаружение стрихнина. В небольшой фарфоровой чашке растворяют в 1 мл
H2SO4 несколько кристаллов испытуемого вещества, прибавляют маленький
кристалл К2СГ2О7 и осторожно покачивают чашку. При наличии стрихнина на-
блюдаются в стекающей с бихромата калия серной кислоте фиолетовые до
сине-фиолетовых полосы. При помощи этой реакции можно обнаружить до
0,001 мг стрихнина; азотная кислота или нитраты мешают определению. Акони-
тин дает синее до сине-зеленого окрашивание.
139
5.15. ЖИВОТНЫЕ И БАКТЕРИАЛЬНЫЕ ЯДЫ
5.15.1. Кантаридин
Для обнаружения кантаридина используют его нерастворимость
в разбавленных кислотах. При прибавлении серной кислоты к
раствору испытуемого вещества в едком натре кантаридин выпа-
дает в виде аморфного осадка по достижении кислой среды.
Специальная реакция обнаружения кантаридина. Некоторое количество ис-
пытуемого вещества растворяют в концентрированной H2SO4, часть раствора
смешивают с несколькими кристаллами КаСгОч и нагревают. Зеленое окрашива-
ние указывает на наличие кантаридина. К другой части раствора прибавляют
кристалл селенистой кислоты; в присутствии кантаридина появляется пурпурно-
красное окрашивание.
5.15.2. Токсин ботулизма
В общем случае наличие бактериальных токсинов определяют
биологическим испытанием на подопытных животных, чаще на бе-
лых мышах. Контрольные опыты проводят на животных, которым
одновременно делают соответствующую прививку. В случае токси-
на ботулизма сыворотку крови пораженных лиц или раствор ток-
сина впрыскивают в брюшную полость. Чувствительность этих био-
логических проб очень высока 240. Так, в зависимости от типа ток-
сина ботулизма при помощи белых мышей определяют его коли-
чества от 0,0009 до 0,2 мкг. Недостатком этого метода является
большая продолжительность испытания, обусловленная сроком от
начала латентного периода до появления симптомов отравления,
который для токсина ботулизма достигает нескольких суток.
Установить в более короткое время присутствие и вид токсина
можно при помощи агглютинационных проб, под которыми пони-
мают наблюдаемую в пробирке реакцию (флокуляцию) между
антигеном (токсином) и гомологическими антителами (вакциной).
От 0,006 до 0,19 мкг токсина ботулизма типов А, В, С и Е могут
быть определены в стекле по способу Кониковой241 при использо-
вании специально модифицированной для этой цели пассивной
гемагглютинационной пробы Биодена 242. Чувствительность спо-
соба в значительной мере зависит от специфичности и качества
вакцин, применяемых для сенсибилизации эритроцитов. При по-
мощи гемагглютинационной пробы обнаруживают также токсоиды,
так что чувствительность в случае токсина, содержащего токсоид,
более высокая по сравнению с биологической пробой.
Другим способом Ьбнаружения токсина ботулизма типа А в
воде является определение фагоцитарного числа лейкоцитов крови
человека 280. Он основан на том, что токсин в значительной мере
угнетает фагоцитную функцию лейкоцитов, иными словами, их
способность «пожирать» бактерии. В данном случае фагоцитное
число является отношением числа бактерий, уничтоженных угне-
тенными лейкоцитами, к числу бактерий, уничтожаемых неугне-
тенными лейкоцитами.
140
5.16. ФИТОТОКСИЧЕСКИЕ ОВ
Из большого числа возможных соединений особый интерес пред-
ставляют хлорированные производные феноксиуксусной кислоты и
динитро-о-крезол, применявшиеся американской армией в Южном
Вьетнаме в качестве дефолиантов.
5.16.1. Хлорированные феноксиуксусные кислоты
2,4-дихлорфенокси- 2,4,5- трихлорфенокси-
уксусная кислота, уксусная кислота
2,4-D 2,4,5-Т
Из препаратов 2,4-D и 2,4,5-Т и аналогичных соединений при
нагревании с концентрированной H2SO4 образуется формальдегид,
который можно обнаружить чувствительными цветными реак-
циями с различными нафтолсульфокислотами и таким образом
установить присутствие этих фитотоксических соединений. Реак-
ция формальдегида с хромотроповой кислотой (1,8-диоксинафта-
лин-3,6-дисульфокислотой) в растворе концентрированной серной
кислоты может быть представлена уравнением 236:
5.16.1.1. Обнаружение хлорфеноксиуксусных кислот. Отделение
хлорированных феноксиуксусных кислот возможно предваритель-
ной экстракцией бензолом.
В фарфоровой чашке осторожно выпаривают досуха несколько миллилитров
испытуемого вещества, к остатку добавляют 2 мл концентрированной H2SO4,
несколько миллиграммов хромотроповой кислоты и нагревают примерно до
141
150 °C. Фиолетовое окрашивание указывает на образование формальдегида,
а следовательно, на присутствие и хлорфеноксиуксусных кислот. Чувствитель-
ность реакции 1 мкг.
Аналогичную реакцию дает фторуксусная кислота (см. раздел
5.13.1.4).
5.16.1.2. Бумажная и тонкослойная хроматография. Разделение
смесей гербицидов хорошо удается применением бумажной и тон-
кослойной хроматографии (см. раздел 8.7.2).
5.16.1.3. Колориметрическое определение 237. При взаимодейст-
вии формальдегида, выделяющегося при обработке испытуемого
вещества концентрированной H2SO4, с 6-амино-1-нафтол-3-сульфо-
кислотой (И-кислота) образуется окрашенное соединение хиноид-
ной структуры.
В маленькой колбе выпаривают досуха на водяной бане определенный
объем экстракта испытуемого вещества. Остаток смешивают с 3 мл 0,3%-ного
раствора И-кислоты в концентрированной H2SO4 и точно 5 мин нагревают на
масляной бане до 165 °C. Смесь быстро охлаждают до комнатной температуры
и переносят в мерную колбу емкостью 25 мл, содержащую 15 мл 20%-ной уксус-
ной кислоты. Для переноса смеси и доливания мерной колбы до метки также
пользуются уксусной кислотой. Появляющееся синее окрашивание фотометри-
руют при 580 нм. Метод дает возможность определить от 10 до 100 мкг 2,4-D.
Эфирные и бензольные экстракты из продуктов питания и растений целесооб-
разно очистить пропусканием через колонку с окисью алюминия 238 или флори-
силом 239. *
5.16.2. Динитро-о-крезол
Интенсивно желтую окраску аниона этого соединения в бикарбо-
натом растворе (Хмакс 440 нм) используют для его обнаружения
и фотометрического определения. Для проведения определения
экстракт, полученный извлечением вещества из растений или про-
дуктов петролейным эфиром, подвергают обработке 0,1%-ным рас-
твором NaHCOs и затем колориметрируют.
5.17. ПСИХОХИМИЧЕСКИЕ ОВ
5.17.1. Производные индола
Интенсивные исследования в области физиологически активных
веществ позволяют отнести производные индола к группе соеди-
нений, представляющей большой интерес в биологическом отно-
шении. Чрезвычайно малые дозы этих веществ, действующих как
галлюциногены, среди которых, в частности, должны быть назва-
ны производные лизергиновой кислоты и псилоцибин, а также ди-
метилтриптамин, буфотенин и адренохром, вызывают необходи-
мость в разработке чувствительных методов их обнаружения и
определения.
5.17.1.1. Методы обнаружения. Цветные реакции, описанные для
обнаружения алкалоидов спорыньи, могут быть использованы и
для обнаружения представляющих военный интерес индольных
соединений.
142
Реакция Келлера 307. Первоначально цветная проба на алка-
лоиды спорыньи заключалась в растворении испытуемого вещества
в ледяной уксусной кислоте, содержащей некоторое количество
хлорного железа, и осторожном приливании концентрированной
H2SO4, вследствие чего образовывались две фазы. При этом на гра-
нице раздела фаз появлялось фиолетово-коричневое кольцо. Ридеру
и Бёмеру 308 удалось установить, что решающую роль в механизме
этой цветной реакции играла глиоксалевая кислота, содержащаяся
в виде примеси в ледяной уксусной кислоте.
Реагент
Уксусная кислота, ледяная, содержащая 0,05% хлорного железа и 0,1%
глиоксалевой кислоты.
Испытуемое вещество растворяют в 1 мл реагента, к раствору осторожно
приливают 1 мл опускающейся на дно концентрированной H2SO4 (не содержа-
щей нитратов) и через 15 ч смесь встряхивают. Днэтиламид (+)-лизергиновой
кислоты (LSD-25) и другие производные индола, в том числе псилоцибин, ди-
метилтриптамин и буфатенин, обусловливают появление устойчивого синего
окрашивания ЗО9.
Эта реакция индикации отличается высокой селективностью
для производных индола со свободным положением 2 в индольном
кольце.
Цветная реакция по Ван Урку — Смиту 310>311. Все алкалоиды
спорыньи дают синее окрашивание с п-диметиламинобензальдеги-
дом в растворе концентрированной H2SO4. Сравнение спектров по-
глощения окрашенных в синий цвет растворов, образующихся как
в цветной реакции Ван Урка—Смита с алкалоидами спорыньи, так
и в реакции 0-индолилуксусной кислоты с п-диметиламинобензаль-
дегидом, позволяют предположить существование окрашенной в
синий цвет соли следующих- граничных структур 312:
143
Цветной эффект дают только те производные индола, у кото-
рых положение 2 свободно и имеется алкильная группа в поло-
жении 3. Состав реагента приведен ниже, при описании количест-
венного определения.
Для проявления LSD-25 на тонкослойных хроматограммах удобен несколько
модифицированный реагент, приготовляемый растворением313 0,5 г п-диметил-
аминобензальдегида в 5 мл 37%-ной НС1 и 95 мл этанола. При обрызгивании
хроматограммы этим раствором в присутствии LSD-25 появляется сине-фиоле-
товое окрашивание, достигающее максимума через 10 мин, чувствительность
реакции составляет 0,05 мкг LSD-25.
Обнаружение в УФ-свете. Все производные лизергиновой кис-
лоты можно обнаружить в растворах и на хроматограммах по
синей флуоресценции в УФ-свете. Характерный для этих соедине-
ний спектр поглощения, имеющий слабый максимум при 316—
318 нм и минимум при 268 нм, не разрешает дифференцировать
различные производные.
5.17.1.2. Количественное определение. Фотометрическое опреде-
ление при помощи реагента Келлера. Для определения приме-
няется реагент, модифицированный Ридером и Бёмером 314'315.
Реагент Келлера — приготовленная при охлаждении смесь 40 мл 0,25%-ного
водного раствора глиоксалевокислого натрия, 0,165 мл 2%-ного раствора
FeCls • 6Н2О и 60 мл концентрированной H2SO4. В темной склянке реагент со-
храняется 2 недели.
К 0,5 мл водного раствора производного индола (1 —3 -10~4 Л4) прибавляют
3 мл реагента. После перемешивания реакционную смесь помещают на 3 мин
в кипящую водяную баню, дают охладиться до комнатной температуры и из-
меряют экстинкцию при 650 нм (для LSD-25) или при 540 нм (для псилоциби-
на). Расчет ведут по калибровочной кривой.
Фотометрическое определение по реакции Ван Урка — Смита.
В этом определении используют реагент, стандартизованный Олл-
портом и Кокингом316.
Реагент — раствор 125 мл п-диметиламинобензальдегида в 100 мл 65 %-ной
H2SO4 смешивают с 0,1 мл 5%-ного раствора FeCU- В темноте реагент сохра-
няется в течение 2 недель.
Смешивают 0,5 мл водного или спиртового раствора испытуемого производ-
ного нндола с 2 мл реагента; реакционной смеси дают постоять точно 5 мин,
добавляют 1 мл этанола и измеряют экстинкцию при 540 нм для LSD-2S и
псилоцибина или в зависимости от положения максимума поглощения для
алкалоидов спорыньи в пределах от 550 до 610 нм.
Флуорометрическое определение LSD-25. Это высокочувстви-
тельное определение следует проводить со специально приготов-
ленной вытяжкой испытуемого вещества317.
Пробу воды (20 мл) или водного экстракта после установления pH 8,5—9
экстрагируют тремя порциями хлороформа по 15 мл каждая. Объединенный
хлороформный экстракт 5 мин встряхивают с 5 мл 0,01 н. НС1, отделяют вод-
ный слой, подщелачивают разбавленным NaOH и вновь экстрагируют 10—15 мл
хлороформа. Растворитель испаряют на водяной бане и растворяют остаток в
метаноле. Этот раствор переносят на пластину со слоем силикагеля, на которой
вещества, содержащиеся в растворе, разделяются на тонкослойной хромато-
грамме. В качестве растворителя используют смесь трихлорэтана и метанола
144
(9: 1). Пятна LSD, которые можно обнаружить в УФ-свете по синей флуорес-
ценции, вымывают водой. Для количественного флуориметрического измерения
флуоресценцию возбуждают монохроматическим УФ-светом с длиной волны
325 нм, свечение флуоресценции измеряют при 445 нм. Линейная зависимость
свечения флуроресценции от концентрации LSD находится в пределах 0,01—
0,1 мкг! мл.
5.17.2. Фениламиноалканы
Из соединений этой группы особый интерес представляет весьма
психотоксически активный 3,4,5-триметоксифенил-2-аминоэтан
(мескалин).
5.17.2.1. Методы обнаружения. В основу реакций обнаружения
мескалина положено его характерное отношение к некоторым ре-
агентам на алкалоиды.
С концентрированной H2SO4 он дает лимонно-желтое окрашивание, перехо-
дящее при нагревании в фиолетовое.
При осторожном прибавлении к раствору мескалина в H2SO4 смеси
H2SO4—HNO3 появляется зеленое окрашивание, быстро переходящее в корич-
нево-фиолетовое и затем в коричневое.
С реагентом Марки (см. раздел 5.14.2.2) получается оранжевое окрашивание.
С реагентом Витали (см. там же) мескалин дает фиолетовое, переходящее
в коричневое окрашивание.
При добавлении раствора пикриновой кислоты к спиртовому раствору ме-
скалина выделяются желтые кристаллы пикрата, т. пл. 219—220 °C.
Платинохлорнстоводородная кислота осаждает из водного раствора меска-
лина светло-желтые иглы хлороплатината, т. пл. 187—188 °C (разл).
Реагент Драгендорфа (см. раздел 5.14.2.3) осаждает мескалин в виде плот-
ных розеток, реагент Майера дает белый аморфный осадок, реагенты Зоннен-
шейна (фосфорномолибденовая кислота) и Шейблера (фосфорновольфрамовая
кислота) дают аморфные, желтовато-белые осадки.
Обнаружение мескалина по флуоресценций основано на кон-
денсации его с формальдегидом, причем, очевидно, образуется
главным образом 6,7,8-триметоксиизохинолин.
Несколько капель раствора испытуемого вещества, который должен содер-
жать примерно 0,01—10 мкг мескалина, наносят на хроматографическую бумагу.
После высыхания бумагу обрызгивают 5%-ным раствором гликоля, pH которого
доведен соляной кислотой до 3. Бумагу сушат при 80 °C и помещают в закрытый
стеклянный сосуд, содержащий 20 г параформальдегида и 6 мл воды, и нагре-
вают 3 ч при 80 °C. При последующем наблюдении в свете УФ-лампы мескалин
обнаруживают по возникновению отчетливой флуоресценции.
Для количественного определения флуоресцирующее соединение
можно экстрагировать 0,1 н. раствором НС1 и измерять свечение
флуоресценции при 515 нм.
5.18. НЕОРГАНИЧЕСКИЕ ЯДЫ
При исследовании зараженных продуктов питания, фуража и воды
необходимо проверять, содержатся ли в этих объектах токсиче-
ские неорганические соединения (катионы и анионы). Некоторые
145
из этих неорганических токсичных соединений образуются из ОВ
при разложении их в материале пробы, например арсенит- и ар-
сенат-ионы из мышьяксодержащих ОВ, свинец из тетраэтилсвин-
ца, железо и никель из соответствующих карбонилов. Неорганиче-
ские яды обнаруживают в водных вытяжках. Для полевых лабо-
раторий особенно большое значение имеют капельные реакции,
осуществляемые с помощью возможно более специфичных реаген-
тов, что исключает необходимость предварительного разделения
катионов или анионов. В.хорошо оборудованных специальных ла-
бораторих, пользуясь различными способами разделения, такими,
например, как распределительная и адсорбционная хроматогра-
фия, метод зонной плавки, можно в какой-то степени преодолеть
трудности, связанные с присутствием других веществ. Вполне ве-
роятно, что какой-либо из двух последних способов станет доступ-
ным в модифицированном виде и для полевых аналитических ла-
бораторий.
Такая перспектива, например, открывается в быстром развитии
методов тонкослойной хроматографии (ср. раздел 8.7.2).
Дальнейшее развитие пригодных для полевых лабораторий ме-
тодов обнаружения должно идти в основном по пути использова-
ния стабильных индикаторных бумаг, пропитанных соответствую-
щими реагентами. Для того, чтобы локализовать продукты реакции
в местах их образования и тем самым облегчить их обнаружение,
следует применять преимущественно реагенты, трудно или совсем
нерастворимые в воде. Совершенно очевидно, что при наличии до-
статочно чувствительных реагентов и индикаторных бумаг в поле-
вых лабораториях рекомендуется применять преимущественно ка-
пельные реакции вследствие простоты и быстроты их выполнения.
5.18.1. Токсичные катионы
5.18.1.1. Групповое обнаружение токсичных катионов. Часть
представляющих интерес катионов можно осадить в виде сульфи-
дов. Чтобы не прибегать к малоприятной работе с сероводородом,
пользуются так называемым «твердым сероводородом», т. е. соеди-
нениями, которые в кислом растворе разлагаются с выделением
сероводорода. Для этой цели пригодны, тиоацетамид 292, тиокарб-
аминат аммония 287, этилдитиокарбаминат 288, тиомочевина 289, тио-
форманилид290 и другие вещества291.
К 5 мл испытуемого раствора, кислотность которого должна соответствовать
кислотности 0,25—0,4 н. раствора НС1, прибавляют на кончике шпателя тио-
ацетамид (20—30 мг) и нагревают 10 мин на кипящей водяной бане. Если оса-
док не выпадает, то добавляют раствор аммиака до pH 5 и повторно нагревают
10 мин до образования осадка. Осадок может содержать следующие сульфиды:
HgS, PbS, PbS2 (черные), CdS, As2S3, As2S3 (желтые) и Sb2S3, Sb2Ss (оран-
жевые) .
5.18.1.2. Обнаружение бария. Реакция с родизонатом натрия2М.
Это вещество с солями двухвалентных тяжелых металлов образует
146
окрашенные осадки по следующей реакции:
На фильтровальную бумагу наносят вначале каплю нейтрального или сла-
босолянокислого раствора испытуемого вещества и затем на то же место каплю
0,2%-него раствора родизоната натрия. Появление красно-коричневого окраши-
вания указывает на присутствие бария; чувствительность реакции 2 мг[л.
Так как свинец, серебро, кадмий, таллий и стронций образуют
с родизонатом натрия аналогично окрашенные соединения, то об-
наружение бария этим путем имеет смысл только в отсутствие
названных катионов.
Осаждение в виде сульфата 285'286. В отличие от сульфата свинца
осаждаемый в присутствии перманганата калия сульфат бария
столь прочно обволакивает перманганат, что на него не действуют
восстановители.
Смешивают 1 каплю испытуемого раствора с 3 каплями насыщенного на
холоду раствора КМпСЦ и несколькими1 каплями разбавленной H2SO4. При до-
бавлении перекиси водорода, щавелевой или сернистой кислот раствор обесцве-
чивается, а выделяющийся фиолетовый осадок указывает на присутствие бария.
Чувствительность реакции 100 мг!л.
5.18.1.3. Обнаружение бериллия. Реакция с хинализарином293.
Соли бериллия образуют с хинализарином (1,2,5,8-тетраоксиантра-
хиноном) хелатные соединения, окрашенные в сине-фиолетовый
цвет, следующей предполагаемой структуры:
Реагент
Хинализарин — 0,05%-ный раствор в 2 н. растворе NH4OH.
На капельную пластинку помещают 1 каплю раствора испытуемого вещества
и 1 каплю свежеприготовленного раствора реагента. В присутствии незначитель-
ных количеств бериллия появляется синее окрашивание, а при больших количе-
ствах — выпадает синий осадок; чувствительность реакции 1 мг/л. Образующаяся
в присутствии магния аналогичная окраска полностью обесцвечивается от при-
бавления насыщенной бромной воды. Соли железа (III) следует маскировать
прибавлением тартрата.
147
5.18.1.4. Обнаружение свинца. Соли свинца образуют с дити-
зоном красное комплексное соединение
/С6НГ
ZNH—Nx
S=C^ ,Pb/2
\N=NZ
\c6Hs
Так как многие другие катионы образуют с дитизоном аналогично
окрашенные комплексные соли, то их следует маскировать добав-
лением цианида и тартрата.
Реагент
Дитизон — раствор 1—2 мг дитизона в 100 мл четыреххлористого углерода.
Цианистый калий — 0,05%-ный водный раствор.
Сегнетова соль.
В микропробирке смешивают 1 каплю нейтрального раствора испытуемого
вещества последовательно с 1 каплей растворов KCN, 1 каплей кислого винно-
кислого калия натрия (сегнетовой соли), затем с 1 каплей раствора дитизона
и энергично встряхивают. В присутствии свинца окрашенный в зеленый цвет
раствор реагента приобретает кирпично-красную окраску; чувствительность реак-
ции 1—2 мг/л.
Реакция с бихроматом калия. Из нейтрального или уксусно-
кислого испытуемого раствора при прибавлении бихромата ще-
лочного металла свинец выпадает в виде желтого РЬСгО4.
5.18.1.5. Обнаружение таллия в виде иодида таллия.
Реагенты
Йодистый калий — 10%-ный водный раствор.
Тиосульфат натрия — 2%-иый водный раствор.
Смешивают на часовом стекле 1 каплю слабокислого испытуемого раствора
с 1 каплей раствора KI и 2 каплями раствора Na2S2O3. Образующийся желтый
осадок иодида таллия(П), хорошо видимый на темном фоне, указывает на
присутствие таллия; чувствительность реакции 10 мг/л.
Ионы таллия(Ш) также осаждаются в виде иодида, но при
этом происходит выделение иода. Ионы свинца и ртути(II) ме-
шают осаждению ТП2. Этого можно избежать добавлением раство-
ров Na2S2O3 и избыточного количества раствора KI- При этом об-
разующийся по реакции РЫ2 превращается в растворимый комп-
лексный анион, a Hgl2— в растворимую комплексную соль K^HgU
5.18.1.6. Обнаружение ртути. Реакция с дитизоном. Дитизон об-
разует с солями Hg(II) внутрикомплексное соединение.
Реагенты
Дитизон — 0,01%-ный раствор в хлороформе.
Комплексон III — 10%-ный водный раствор.
В микропробирку помещают 1 каплю испытуемого раствора и добавляют
1 каплю раствора комплексона III, 1 каплю 0,5 н. раствора НС1 и 2 капли рас-
твора дитизона. Реакционную смесь встряхивают, в присутствии ртути хлоро-
формный слой приобретает желто-оранжевое окрашивание; чувствительность
реакции 10 мг/л.
148
Реакция с алюминием. Весьма простое обнаружение ртути
можно провести при помощи куска алюминиевой фольги, предвари-
тельно обработанной NaOH.
На промытую и высушенную фольгу наносят 1 каплю испытуемого раствора,
которую через 5 мин удаляют кусочком фильтровальной бумаги. В присутствии
ртути через несколько минут образуется белый снарост»; чувствительность реак-
ции 2 мг/л.
5.18.1.7. Обнаружение кадмия по реакции с сульфидом натрия294.
Осаждение желтого CdS можно считать специфичной реакцией при
условии, что катионы, образующие аналогично окрашенные суль-
фиды, будут связаны при помощи цианистого калия.
Реагенты
Цианистый калий — 20%-ный водный раствор.
Сульфид натрия— 10%-ный водный раствор.
В микропробирке смешивают 1 каплю испытуемого раствора с 1 каплей
концентрированного раствора NH«OH и 1 каплей раствора цианистого калия.
Полученный раствор должен быть прозрачным и бесцветным, в противном слу-
чае следует добавить еще KCN. Наносят на фильтровальную бумагу 1 каплю
полученного раствора и обрабатывают раствором Na2S. Желтое пятно или коль-
цо указывают на присутствие кадмия; чувствительность реакции 100 мг/л.
5.18.1.8. Обнаружение сурьмы фосфорномолибденовой кисло-
той 296. Соли трехвалентной сурьмы в кислом растворе восстанавли-
вают фосфорномолибденовую кислоту до молибденовой сини. Реак-
ция специфична в отсутствие солей двувалентного олова. При про-
ведении реакции с солянокислым раствором сульфидов металлов
олово, превращаясь в четыреххлористое, определению не мешает.
Реагент
Индикаторная бумага — пропитанная раствором фосфорномолибденовой кис-
лоты и высушенная фильтровальная бумага.
На индикаторную бумагу наносят 1 каплю солянокислого раствора испы-
туемого вещества и некоторое время выдерживают ее в парах воды. Появляю-
щееся через несколько минут синее окрашивание указывает на присутствие
сурьмы; чувствительность реакции 1 мг/л.
5.18.2. Характерные реакции
5.18.2.1. Обнаружение фтор-иона. Способы обнаружения фтор-
иона описаны в разделе 3.1. Очень простой по выполнению являет-
ся проба на травление стекла 296.
В пробирке растворяют несколько кристаллов К2СГ2О7 в 1—1,5 мл концен-
трированной H2SO4. Покачиванием пробирки ее стенкн омывают хромовой
смесью до тех пор, пока они полностью не обезжирятся и смесь не будет равно-
мерно их смачивать. После этого в пробирку вносят несколько зернышек твер-
дого испытуемого вещества или 1 каплю его раствора и нагревают. Прн повтор-
ном покачивании пробирки присутствие фтора - обнаруживается пр неравномер-
ному стеканию серной кислоты вследствие образования несмачиваемых зон.
Этим способом можно обнаружить 0,5 мкг фтор-иоиа.
5.18.2.2. Обнаружение нитрит-иона. Для этого пользуются ре-
агентом Грисса 297, описанным в разделе 5.9 для определения
хлорпикрина. При его помощи можно обнаружить 0,01 мкг нитрита.
149
5.18.2.3. Обнаружение арсенит- и арсенат-ионов. В общем слу-
чае арсенит- и арсенат-ионы обнаруживают пробой Гутцейта, опи-
санной в способах индикации мышьяксодержащих ОВ. В кислой
среде и арсениты и арсенаты восстанавливаются до мышьяковисто-
го водорода, а при нагревании щелочного раствора этих анионов с
металлическим алюминием восстанавливается до мышьяковистого
водорода только арсенит-ион. При этом арсенаты, а также и соеди-
нения сурьмы, которые в кислой среде при действии водорода в мо-
мент выделения образуют SbH3, остаются неизменными. В том слу-
чае, когда соединения мышьяка приходится определять в присут-
ствии сурьмы, переводят арсенат-пон в кислой среде при помощи
МаоБгОз в арсенит-ион, который затем восстанавливают в щелочной
среде. В отсутствие других ионов, реагирующих с иодом, обесцве-
чивание раствора иода или синего иод-крахмального раствора ха-
рактерно для арсенит-иона. В уксуснокислом растворе арсенат-ион
при взаимодействии с 1%-ным раствором AgNO3 дает красно-ко-
ричневый осадок арсената серебра:
AsO4“ + 3Ag+ —> Ag3AsO4
5.18.2.4. Обнаружение селенита. При нагревании или выпари-
вании с концентрированной НС1 селеновая кислота и селениты ко-
личественно восстанавливаются до селена. Способность селенитов
каталитически ускорять восстановление метиленовой сини щелоч-
ными сульфидами используют для чувствительного их обнаруже-
ния 298.
В углубление капельной пластинки помещают 1 каплю раствора испытуе-
мого вещества, а в соседнее углубление— 1 каплю воды. Затем в каждое углуб-
ление прибавляют по 1 капле 0,2 Л/ раствора Na2S и по 1 капле 0,01%-ного
раствора метиленовой сини. Более быстрое обесцвечивание пробы с испытуемым
веществом по сравнению с контрольной пробой указывает на присутствие селе-
нита; чувствительность реакции 1 мг/л.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Представьте схему первой стадии реакции Шёнеманна, т. е. взаимодей-
ствие фосфорорганического соединения с перекисью водорода в щелочной среде.
2. Какие амины употребляют для колориметрического осуществления реак-
ции Шёнеманна?
3. В какой последовательности добавляют реагенты при проведении реак-
ции Шёнеманна?
4. Чем объясняется влияние последовательности прибавления реагентов
на чувствительность реакции Шёнеманна?
5. Какие флуоресцирующие продукты образуются при окислении индола?
6. Приведите известные вам реакции обнаружения фосфорорганических ОВ.
7. Какими способами идентифицируют фосфорсодержащие ОВ?
8. Какие возможности имеются для аналитической характеристики фосфор-
органических соединений типа тетрама?
9. Поясните принцип гидролитического метода объемного определения
фторангидридов эфиров метилфосфоновой кислоты и эфиров пирофосфоновой
кислоты.
10. Объясните механизм цветной реакции иприта с щелочными растворами
тимолфталеина.
150
11. Как реагирует серный иприт с некоторыми солями тяжелых металлов
и, в частности, с хлоридом золота?
12. Какие производные серного иприта можно использовать для его иден-
тификации?
13. Как обнаруживают присутствие 1,2-бис-(2-хлорэтилтио) -этана (сескви-
ипрпта) в серном иприте?
14. Назовите реагенты, образующие с азотистым ипритом характерные
осадки.
15. Какие ОВ образуют с 4-(n-нитробензил)-пиридином окрашенные про-
дукты и каково общее уравнение этой цветной реакции?
16. Напишите уравнение реакции мышьяковистого водорода с бромидом
ртути.
17. Какие продукты образуются в реакции мышьяксодержащих ОВ с серо-
водородом?
18. Почему а-люизит обрабатывают раствором NaOH перед обнаружением
его реагентом Илосвая?
19. Назовите специфические реакции обнаружения адамсита.
20. Какая реакция является специфической для циан-иона?
21. На чем основано образование синего окрашивания в реакции цианидов с
солями меди(II) и бензидином?
22. Напишите общее уравнение реакции галогенцианов с пиридином и пер-
вичными ароматическими аминами.
23. Какой реагент применим для обнаружения соединений с активной мети-
леновой группой, к которым относятся такие ОВ, как хлорацетофенон и бром-
бензилцианид?
24. Какое окрашенное соединение образуется в реакции фторацетатов с тпо-
салипиловой кислотой и щелочью после прибавления феррицианида калия?
25. Как осуществляется групповое разделение алкалоидов?
26. Какие соединения называют «твердым сероводородом» и для чего они
применяются?
ЛИТЕР АТ УРА
1. Schonemann, Eine neue Reaktion fiir den Nachweis von labilen Nicht-
metall-Halogen-Bindungen, P. B. 119887, U. S. Department of Commerce.
(Результаты неопубликованных работ Военно-химических лабораторий гер-
манского вермахта. Берлин, 1944).
2. L а г s s о n L., Acta Chem. Scand.. 12. 273 (1958).
3. Gehauf В. et al.. Anal. Chem., 29. 278 (1957).
4. E p s t e i n J. et al., J. Org. Chem., 21. 796 (1956).
5. Aksnes G., Acta Chem. Scand., 14. 2075 (1960).
6. March D. J.. Neale r., J. Appl. Chem., 8, 394 (1958).
7. A k s n e s G., Sandberg K-, Acta Chem. Scand., 11. 876 (1957''
8. К о bl in A., Epstein J., Armed Forces Chem. J., 9/10, 24 (1957)
9. G e h a u f B., G о 1 d e n s о n J., Anal. Chem., 29, 276 (1957).
10. KondritzerA., Armed Forces Med. J., 7, 791 (1956).
11. Golden son J.. Anal. Chem., 29, 877 (1957).
12. Weber K., Arh. hig. rada I toksicol., 12, 169 (1961).
13. G г a n t G. A. et al., Canad. J. Chem., 35, 40 (1957).
14. S m i t h D. M. et al„ Canad. J. Chem., 35, 156 (1957).
15. G r a n t G. A. et al., Canad. J. Chem., 36, 242 (1958).
16. Matkovic J., Weber K-, Arh. hig. rada i toksicol., 15, 141 (1964).
17. W e b e r K. et al., Croat, chem. Acta, 28, 25 (1956).
18. Weber K. et al., Arh. hig rada i toksicol., 9, 325 (1958).
19. Weber K-, Matkovic J., Arh. hig. rada i toksicol., 15, 151 (1964).
20. Web er К., M a tko vic J., Arch. Toxiko]., 21, 38 (1965).
21. P г о p e r R., Rosenthal R. W., Chemist-Analyst, 45, 79 (1956).
22. Grant G. A., McEwen К. L.. Canad. J. Techn., 30, 66.
23. S a ville B., Analyst, 82; 269 (1957),
151
24. Freon P„ Ann. Chim., 11, 458 (1939).
25. E p s t e i n J., Anal. Chem., 19, 272 (1947).
26. Sass S. et al., Anal. Chem., 29, 1346 (1957).
27. Пат. США 2867509 (1959).
28. E p s t e i n J. et al., Anal. Chem., 27, 1435 (1955).
29. Kramer D. N., Gamson R. M., Anal. Chem., 29, 21A (1957).
30. Swidler R., Steinberg G. M., J. Am. Chem. Soc., 78, 3594 (1956)
31. Matousek J., Tomecek I., Analyse synthetischer Gifte, Berlin, 1965.
32. S t о 1 b e r g M. A. et al., J. Am. Chem. Soc., 77, 765 (1955).
33. Steinberg G. M., Bolger J., J. Org. Chem., 21, 660 (1956).
34. Marsh D. J., N e a 1 e E., Chem. a. Ind., 1956, 494.
35. S c h г e i b e r H., Arch. Toxikol., 16, 129 (1956).
36. Sperlich H., Dtsch. Apoth.-Ztg., 100, 774 (1960).
37. PuranenU. H., Suomen Kemistilehti, 37, 16 (1964).
38. He 1 rich K-, J. Assoc. Off. Agric. Chem., 43, 344 (1960).
39. A v e r e 11 P. R,, N о r r i s M. V., Anal. Chem., 20, 753 (1948).
40. Lausen H. H., Nature, 194, 1174 (1962).
41. P i 1 z W., Mikrochim. Acta, 1958, 383.
42. Volksen W., Dtsch. Apoth.-Ztg., 95, 865 (1955).
43. Kisser W., Ma ch ata G., Z. anal. Chem., 213, 349 (1965).
44. Pilz W., Z. anal. Chem., 164, 241 (1958).
45. E p s t e i n J. et al., J. Am. Chem. Soc., 78, 341 (1956).
46. R о s e n t h a 1 R. W. et al., J. Phys. Chem., 60, 1596 (1956).
47. Oksne S., Acta Chem. Scand., 13, 1814 (1959).
48. C h e г г у R. H. et al., Anal. Chem., 30, 1239 (1958).
49. В e a c h L. R., S a s s S., Anal. Chem., 33, 901 (1961).
50. S a s s S. et al., Anal. Chem., 32, 285 (I960).
51. Getz M. E., Watts R. R., J. Assoc. Off. Agric. Chem., 47, 1094 (1964).
52. F о u r n i e r R. M., Chim. et ind., 76, 246 (1956).
53. Saville B„ Analyst, 83, 670 (1958).
54. Rheinboldt H., Ber., 60, 184 (1927).
55. E 11 m a n n G. L., Arch. Biochem. Biophys., 82, 70 (1959).
56. E 11 m a n n G. L., Biochem. Pharmakoi., 7, 88 (1961).
57. E 11 m a n n G. L., Arch. Biochem. Biophys., 74, 443 (1958).
58. Пат. США 3119668 (1959).
59. Grunert R. R., Phillips P. H., Arch. Biochem. Biophys., 30, 217 (1951).
60. F о 1 i n О., M a r e n z i A. L., J. Biol. Chem., 83, 103 (1929).
61. Ba sf ord R. E., Huennekens F. M., J. Am. Chem. Soc., 77, 3873
(1955).
62. A к e r f e 1 d S., Acta Chem. Scand., 17, 319 (1963).
63. A к e r f e 1 d S., L 6 v g r e n G., Anal. Biochem., 8, 223 (1964).
64. H a g 1 u n d H., L i n d g r e n I., Taianta, 12, 499 (1965).
65. H a r 1 e у - M a s о n J., J. Chem. Soc., 1952, 146.
66. G r i g n a r d V. et aL, Ann. Chim., 15, 5 (1921).
67. Marbot C., Dissertation. Universitat Basel, 1942.
68. Dre von B., J. Pharm. Chem., 9, 11 (1942).
69. S c h r 6 t e r G. A., Angew. Chem., 49, 164 (1936).
70. Obermiiller M., Angew. Chem., 49, 162 (1936).
71. Нем. пат. 712211 (1941).
72. Ma r q u e s R. J., Teresa J. P., Ion, 4, 7378 (1944).
73. К о bl i n A., Anal. Chem., 30, 430 (1958). .
74. Spica P., Gazz. chim. ital., 49, 299 (1919).
75. S t a i n s b у W. J., T а у 1 о r A., Analyst, 66, 44 (1941).
76. В u r u i a n a L., Z: anal. Chem., 109, 107 (1937).
77. Y s bl ic h M„ J. Am. Chem. Soc., 42, 266 (1920).
78. Me у e r - D 6 r m i n g H. H., Z. anal. Chem., 130, 232 (1950).
79. Poly a J. B., Ind. Eng. Chem., Anal. Ed., 15, 360 (1943).
80. J e 1 i n e к B., Bull. soc. chim. France, 4, 1813 (1937).
81. D e 1 g a J., J. Pharm. chim., 1, 5 (1940).
82. Нем. пат. 721094 (1942).
152
83. M a n n, Р о p e, J. Chem. Soc., 121, 1052 (1922).
84. Johnson J., J. Chem. Soc., 132, 1530 (1933).
85. G i b s о n, P о p e, J. Chem. Soc., 117, 271 (1920).
86. Steinkopf, Ber., 53, 1007 (1920).
87. T s c h u g a j e f f L., Fraenkel D., Compt. rend., 154, 33 (1912).
88. Lockwood H. G, Analyst, 66, 480 (1941).
89. SeaseJ.W. et al., Anal. Chem., 20, 431 (1948).
90. A 11 s о p p С. В., Analyst, 75, 281 (1950).
91. Rieman W, Ind. Eng. Chem., Anal. Ed., 15, 411 (1943).
92. N о r t h г о p J. H., Proc. Soc. Exp. Biol. Med., 67, 15 (1948).
93. Sease J. W. et ah, Ind. Eng, Chem., Anal. Ed., 19, 197 (1947).
94. C h a m b e r 1 i n N. S., Glass J. R., J. Am. Waterworks Assoc., 35, 1065
(1943).
95. G r i f f i n A. E. C h a m b e г 1 i n N. S., J. Am. Waterworks Assoc., 35, 571
(1943).
96. Kinsey V. E., Grant W. M., Ind. Eng. Chem., Anal. Ed., 18, 794
(1946).
97. E 1 c h 1 e r H„ Oster. Chem.-Ztg., 40, 80 (1937).
98. H a n к e r J. S. et al., Anal. Chem., 29, 82 (1957).
99. Ma la testa P., Lorenzini A, Ric. sci., 28, 1874 (1958).
100. Leitch J. L, J. Franklin Inst., 239, 334 (1945).
101. Hollely W. F„ J. Chem. Soc., H7, 898 (1920).
102. Houf f H„ Schnetz R. D„ Anal. Chem., 25, 1258 (1953).
103. Bacq Z. M, Fischer P., Bull. soc. chim. biol., 28, 234 (1946).
104. Fischer P., Bull. soc. chim. biol., 28, 240 (1946).
105. F i s c h e r P., J. Pharm. Belg., 2, 225 (1947). .
106. Dre von B., Bull. soc. chim.. France, 1947, 330.
107. Dre von B., Bull. soc. chim. France, 1949, 327,
108. Graham R. P., Burke K. A., Canad. J. Research 24B, 280 (1946).
109. Mohler H., Hammerle W., Helv. chim. Acta, 23, 1211 (1940).
110. Trams E. G„ Anal. Chem., 30, 256 (1958).
111. J. Pharmac. Sci., 53, 1232 (1964).
112. Koenigs E. et al., Ber., 58B, 933 (1925).
113. E p s t e i n J. et al., Anal. Chem., 27, 1435 (1955).
114. Friedman О. M, Boger E., Anal. Chem., 33, 906 (1961).
115. S a w i с к i E. et al., Anal. Chem., 35, 1479 (1963).
116. К 1 a 11 O. et al., Proc. Soc. Exp. Biol. Med., 104, 629 (1960).
117. Truhaut R. et al., Clin. Chim. Acta, 8, 235 (1963).
118. Obrecht P. et al., Z. Krebsforsch., 66, 151 (1964).
119. Rauen H. M., Arzneimittel-Forschg., 14, 855 (1964).
120. Lamouroux A., LeDuigou Y., Mem. Poudres, 40, 377 (1958).
121. Golumb ic C. et al., J. Org. Chem., 11, 418, 536, 550 (1946).
122. A 11 e n E„ S e a m a n W„ Anal. Chem., 27, 540 (1955).
123. Mel left L. B, Woods L. A', Cancer. Res., 20, 518 (1960).
124. S a s s S. et al. Anal. Chem, 30, 529 (1958).
125. S i v a d j i a n J. S, Bull. soc. chim. France, 2, 623 (1935).
126. G u t z e i t M, Pharmaz. Ztg, 24, 263 (1879).
127. S a n g e г, В 1 a c k, J. Chem. Soc, 26, 1115 (1917).
128. Winkler L. W, Angew. Chem, 30, 114 (1917).
129. Nametkin S, Nekrassow W, Z. anal. Chem, 77, 285 (1929).
130. В о u g a u 11, J. Pharm. chim, 36, 193 (1907).
131. F г о g e r Ch, Compt. rend, 1939, 209.
132. Нем. пат. 742689 (1944).
133. Пат. США 2611748 (1952).
134. Нем. пат. 663907 (1938).
135. Peronnet М, Remy R. Н, J. Pharm. chim, 30, 353 (1939).
136. Dre von В, J. Pharm. chim, 1942, 54.
137. 11 о s v a у L, Ber, 32, 2697 (1899).
138. Пат. США 2689831 (1954).
139. M a s о n S. H, J. Am. Chem. Soc, 67, 2267 (1945).
153
140. Rasuwajew G. A., Malinowski W. S., Ber., 64, 120 (1931).
141. Delga J., J. Pharm. chim., 9, 73 (1940).
142. Va s t a gh G., Pharmaz. Zt. Dtsch., 81, 265, 277 (1940).
143. Watz in ger F., Z. Lebensmittel-Unters. u. Forsch., 95, 313 (1952).
144. de W о 1 f f C. J., Pharmac. Weekbl., 76, 1612 (1939).
145. Пат. США 2991302 (1961).
146. Fleury P., Bull. soc. chim. France, 27, 490, 699 (1920).
147. S i e v e r t s A., Z. Angew. Chem., 35, 17 (1922).
148. Rupp E., Arch. Pharm., 256, 194 (1918).
149. Robertson, J. Am. Chem. Soc., 43, 182 (1921).
150. E wins, J. Chem. Soc., 109, 1355 (1916).
151. R о ge r s, Canad. Chem., 3, 398 (1919).
152. Bruckner G., Parkany M., Magyar Kem. Fol., 68, 164 (1962).
153. Andriska V., Bruchner G., Magyar Kem. Fol., 67, 257 (1961),
154. Lewis, Perkins, J. Ind. Eng. Chem., 15, 290 (1923).
155. Fournier R. M., Mem. Poudres, 40, 385 (1958).
156. К 1 i n g A., S c h m u t z R., Compt. rend., 168, 773 (1919).
157. Moureu H. et al., Compt. rend., 228, 1954 (1949).
158. Anger V., Mikrochim. Acta, 3, 24 (1938).
159. Grummett W. B., McLean J. D., Anal. Chem., 37, 424 (1965).
160. Jahresber. der Chem.-Techn. Reichsanstalt, 5, 11 (1926).
161. M a t u s z а к M., J. Ind. Eng. Chem., Anal. Ed., 1934, 374.
162. R u s h C. A., D a n n e г С. E., Anal. Chem., 20, 644 (1948).
163. D e 1 e p i n e, Bull. Soc. Chim. France, 27, 288 (1920).
164. Lamouroux A., Mem. Poudres, 38, 383 (1956).
165. M e i s e 1 T., M a z о r L., Magyar Kem. Lapja, 17, 421 (1962).
166. Терентьев А. П. и др., жАх, 16, 743 (1961).
167. M a 1 a t e s t a P. et al., Ric. sci., 28, 1683 (1958).
168. R e i s s e г t A., Ber., 37, 831 (1904).
169. Studinger J., Mitt. Geb. Lebensmittelunters., 27, 8 (1936).
170. Cox H. E., Analyst, 64, 807 (1939).
171. Lamourous A., LeDuigon Y., Mem. Poudres, 40, 377 (1958).
172. I у e n g a r N. K., R a j u P. S., Sci. a. Cult., 26, 236 (1960).
173. Kretov et al., Z. allg. Chem., 63, 419 (1931).
174. Алексеевский E. В., ЖПХ, 8, 50 (1931).
175. Quillemard, Labat, Bull. Soc. Pharm., (1919).
176. Daecke H., К r a u 1 K., Z. anal. Chem., 178, 412 (1961).
177. Nekrassow W., Melnikow N. N., Ber., 62, 2991 (1929).
178. D e c k e r t W., Z. anal. Chem., 113, 183 (1938).
179. Некрасов В., Война н техника, 275, 32 (1926).
180. Engel, Z. ges. Sch.- u. Sprengst., 24, 451 (1926).
181. Дубинин M. M„ ЖПХ, 4, 1109 (1931).
182. Thompson, Black, Ind. Eng. Chem., 12, 1067 (1920).
183. F i e 1 d n e r, Ind. Eng. Chem., 11, 519 (1919).
184. Fournier R. M., Person M., Chim. analyt., 29, 155, 263 (1957),
185. F e i n s i 1 v e r L., О b e r s t F. W., Anal. Chem., 25, 820 (1953).
186. Morau H. et al., Arch. Malad. Prof., 11, 445 (1950).
187. As mu s E., К u c h e n b e c k e r‘ H., Z. anal. Chem., 213, 266 (1965).
188. J о a n i d N. et al., Farmacia, 11, 349 (1963).
189. В a r n e b e у O. L., J. Am. Chem. Soc., 36, 1092 (1914).
190. P e r t u s i C., G a s t a 1 d i G., Chem.-Ztg., 37, 609 (1913).
191. Янков С. П„ ЖАХ, 12, 759 (1957).
192. Пат. США 2534229 (1950).
193. Пат. США 2855289 (1958).
194. F е i g 1 F„ A n g е г V., Analyst, 91, 282 (1966).
195. R о z e n d a a 1 N. A., J. pharm. chim., 7, 20 (1919).
196. G u t z e i t G., Helv. chim. Acta, 12, 829 (1929).
197. Nicholson R. J., Analyst, 66, 189 (1941).
198. S t e i gm a n A., J. Soc. Chem. Ind., 61,'36 (1942).
199. S t e у n D о u w, J. S. afric. vet. med. Assoc., 10, 65 (1939),
154
200. Kubista Z., Korose ochrana materialu, 1, 26 (1957).
201. Feigl F., Feigl H. E, Anal. Chim. Acta, 3, 93 (1949).
202. Hanker J. S. et al., Anal. Chem., 30, 93 (1958).
203. F e i g 1 F., H e i s i g G. B., Anal. Chim. Acta, 3, 561 (1949).
204. Cullinane N. H„ Chard S. J., Analyst, 73, 95 (1948).
205. Weehuizen F., Pharmak. .Weekbl., 42, 271 (1905).
206. R о b b i e W. A., Arch. Riccham, 5, 49 (1944).
207. Пат. ФРГ 1105199 (1961).
208. Mush a S. et al., J. Chem. Soc. Japan, 80, 1285 (1959).
209. F e i g 1 F., С a 1 d a s A., Mikrochim. Acta, 1955, 992.
210. Пат. США 2753248 (1956).
211. Guilbaut G. G., Kramer D. N., Anal. Chem., 38, 834 (1966).
212. К 6 n i g W., J. prakt. Chem., 69, 105 (1904).
213. Федотов В. П„ ЖАХ, 11, 250 (1956).
214. Пат. США 2678260 (1954).
215. К г u s е J. М., Mellon М., Anal. Chem., 25, 446 (1953).
216. Jorgensen К-, Acta Chem. Scand., 9, 548 (1955).
217. Даниленко Г., Субенко В. Г., Фармак. ж., 15, 17 (1960).
218. В а г k L. S., Н i gs о n Н. G„ Taianta, 11, 471, 621 (1964).
219. Ma latest а Р„ Dubini М„ Ric. Sci., 27, 3649 (1957).
220. Aldridge W. N„ Analyst, 69, 252 (1944).
221. A 1 d г i d ge W. N., Analyst, 70, 474 (1945).
222. R u s s e 1 F. R., W i к i n s о n N. T., Analyst. 84, 751 (1959).
223. Zymny E., Prakt. Chem., 5, 256 (1954).
224. В a ke r M. O. et al., Anal. Chem., 27, 448 (1955).
225. P j e к a c z H., Mazur H., Roczn. Zakl. Hig., 12, 481 (1961).
226. T г u h a u t R. et al., Ann. Biol. Clin., 22, 901 (1964).
227. Saltzman В. E., Anal. Chem., 33, 1100 (1961).
228. H i g s о n H. G., В а г к L. S„ Analyst, 89, 338 (1964).
229. Mirsch E., Karsch U., Wasserwirtsch.-Wassertechn., 15, 207 (1965).
230. К r a t о c h v i 1 V., Coll. Czech. Chem. Comm., 25, 299 (1960).
231. S p ё v a к A. et al., Там же, 26, 887 (1961).
232. Asmus E., Garschhagen H., Z. anal. Chem., 138, 414 (1953).
233. Murty G. V., Viswanathan T. S., Anal. Chim. Acta, 25, 293 (1961).
234. Grigorescul., TobaGh., Rev. Chim., 15, 572 (1964).
235. Asmus E., Pa penfuse D., Z. anal. Chem., 185, 201 (1962).
236. Feigl F., Tflpfelanalyse. B. 2. Frankfurt a. M., T960. S. 333.
237. A1 у D. M„ F a u s t S. D„ Anal. Chem., 36, 2200 (1964).
238. Daond N. H., Luh B. S., Fruchtsaft. Ind., 7, 33 (1962).
239. Coakley J. E. et al., J. Agric. Chem., 12, 262 (1964).
240—241. Ко Нико в a P. E., Военно-медиц. ж., 2, 46 (1962).
242. В о у d e n S. V., J. Exper. Med., 92, 107 (1951).
243. Fei gl F., Z. anal. Chem., 74, 380 (1928).
244. Gad G., S c h 1 i c h t i n g H., Gesundheits-Ing., 76, 373 (1955).
245. Liebi g J. V., Ann., 77, 102 (1951).
246. Ryan J. A., Culshaw G. W., Analyst, 69, 370 (1944).
247. S a 1 к a A., Metal Finishing, 58, 59 (1960).
248. Archer E. E., Analyst, 83, 571 (1958).
249. W г о n s к i M., Chem. analyt., 5, 293 (1960).
250. Erdey L. et al., Taianta, 1, 377 (1958).
251. К г a 1 j i c J., Acta Pharm. Jugosl., 19, 37 (1960).
252. К r a 1 j i c J., Mate M., Croat, chem. Acta, 28, 249 (1956).
253. d e S о u s a A., Taianta, 8, 782 (1961).
254. W г о n s к i M., Analyst, 84, 668 (1959).
255. Tanaka Y„ Yamamoto S„ Jap. Analyst, 9, 8 (1960); C., 176, 440
(I960).
256. Hanker J. S. et al., Anal. Chem., 29, 879 (1957).
257. Hanker J. S. et al., Anal. Chem., 30, 93 (1958).
258. Guilbaut G. G., Kramer D. N., Anal. Chem., 37, 1395 (1965).
?59. Guilbaut G. G„ Kramar p. N„ Anal. Chem., 37, 918 (1965).
155
260. Ohlweiler О. A., Med it sc h J. O., Anal. Chem., 30, 450 (1958),
261. M e d i t s c h J. O., Engenh. Quim., 17, 15 (1964).
262. Berg R., Fridlander, 69, 9 (1926).
263. Файгль Ф. Капельный анализ органических веществ. Пер. с англ., под
ред. В. И. Кузнецова.. М., Госхимиздат, 1962.
264. Owens Е. J., Punte С. L., Am. Ind. Hyg. Assoc. J., 24, 202 (1963).
265. Snyder L. J., Henderson S. R., Anal. Chem., 33, 1175 (1961).
266. К i e m s t e d t H., Z. angew. Chem., 1929, 1107.
267. Rompp H., Chemie-Lexikon. Stuttgart, 1952, S. 82.
268. S h e p h e r d M. et. al., Anal. Chem., 19, 77 (1947).
269. Hoover, Ind. Eng. Chem., 13, 770 (1921).
270. Paulin P., Bull. Soc. Chim. France, 1959, 1845.
271. П e p e г у д И. А., Б о й к и и а Б. С., Зав. лаб., 29, 674 (1963).
272. N i с 1 о и х М., Compt. rend., 126,746 (1898).
273. Gutier A., Compt. rend., 126, 293 (1898).
274. Froboese, Z. anal. Chem., 54, 1 (1915).
275. Англ. пат. 343724 (1930).
276. Frevert H. W., Francis E. H., Ind. Eng. Chem., Anal. Ed., 6, 226
(1934).
277. Фихтенгольц В. C„ Козлова H. П., Зав. лаб., 23, 917 (1957).
278. Беляков А. А., Зав. лаб., 26, 158 (1960).
279. Р i tet G., Arch. Malad. prof. Med., 21, 674 (1960).
280. Мин ер вин С. M„ Стояновский О. Ф., Ж. микробиол., 26, 13
(1964).
281. Pieters Н. A. J., Safety in . the Chemical Laboratory, London, 1951,
P. 156.
282. M с C u 11 о u gh J. D. et aL, Anal. Chem., 19, 999 (1947).
283. Пат. СССР 135684 (1961).
284. Fe i g 1 F., Mikrochem., 2, 188 (1924).
285. Feigl F., Aufrecht W., Rec. trav. chim., 58, 1127 (1939).
286. Feigl F. Ttipfelanalyse. В. 1. Frankfurt a. M„ 1960.
287. Wiberg E., Bauer E., Angew. Chem., 64, 270 (1952).
288. Sen B. N., Anal. Chim. Acta, 24, 386 (1961).
289. Bauer R„ W e h 1 i n g J., Z. anal. Chem., 199, 171 (1963).
290. Antia M. B. et al., Analyst, 86, 202 (1961).
291. P о p p e г E. et al„ Z. anal. Chem., 184, 184 (1961).
292. F 1 a s c h k a H., Chemist-Analyst, 44, 2 (1955).
293. F i s c h e r H., Z. anal. Chem., 73, 54 (1928).
294. D a 1 e n E., de V r 1 e s G., Anal. Chim. Acta, 3, 567 (1949).
295. F e i g 1 F., N e u b e r F., Z. anal. Chem., 62, 382 (1923).
296. K0hnel-Ha gen S., Mikrochem., 15, 313 (1934).
297. Griess P., Ber., 12, 427 (1879).
298. Fei gl F„ West P. W., Anal. Chem., 19, 351 (1947).
299. S a 1 s b u г у J. M. et al., Anal. Chem., 23, 603 (1951).
300. Ramsey L. L., С 1 i f f о r d P. A., J. Assoc. Off. Agric. Chem., 32, 788
(1949).
301. D i r s c h e r 1 A., Z a c h e r 1 M. K., Mikrochim. Acta, 1954, 340.
302. Hutchens J. C., Kass В. M., J. Biol. Chem., 177, 571 (1949).
303. Kruger D., Tschirch E., Ber., 62, 2776 (1929).
304. Ramsey L. L., Patterson W., J. Assoc. Off. Agric. Chem., 34, 827
(1951).
305. Friedlander P., Ber., 39, 1060 (1908).
306. Mai use k J., Matousek J., Pracovni lekarstvl, 15, 245 (1963).
307. Keller С. C., Schweiz. Wschr. Chem. Pharm., 34, 65 (1896).
308. Rieder H. P., В 6 h m e r M., Experienta, 14, 463 (1958).
309. Hofmann A., Die Mutterkornalkaloide, Stuttgart, 1964, S. 115.
310. Van U r k H. W„ Pharm. WeekbL, 66, 473 (1929).
311. Smith M. J., U. S. Publ. Health Rep., 45, 1466 (1930).
312. PohmM., Arch. Pharm., 286, 509 (1953).
156
313. Genest К., Farmilo C. G., J. Pharmacy a. Pharmacology, 16, 250
(1964).
314. Rieder H. P., Bohmer M., Helv. Chim. Acta, 43, 638 (1960).
315. Helv. Chim. Acta, 42, 1793 (1959).
316. Allport N. L., Cocking T. T., Quart. J. Pharmac. Pharmacol., 5, 341
(1932).
317. Da 1 Corti vo L. A., Anal. Chem., 38, 1959 (1966).
318. Fresenius W., Schneider W., Z. anal. Chem., 203, 417 (1964).
319. Kroller E., Dtsch. Lebensmittel-Rdsch., 61, 115 (1965).
320. Epstein J., Demek M. M., Anal. Chem., 39, 1136 (1967).
321. F e i g 1 F. und Mitarb., Mikrochim. Acta, 1959, 47.
322. Lohs Kh., Donner R., Z. Chem., 6, 224 (1966).
323. Lohs Kh., D 6 p e 1 W„ Z. Chem., 7, 106 (1967).
6. БИОХИМИЧЕСКИЕ МЕТОДЫ
6.1. ПРЕИМУЩЕСТВА ПРИМЕНЕНИЯ БИОХИМИЧЕСКИХ
МЕТОДОВ
В аналитической химии во все увеличивающемся числе начали при-
менять ферменты в качестве реагентов.
Это стало возможным благодаря успехам, достигнутым в иссле-
дованиях механизма действия и структуры этих биокатализаторов.
В настоящее время ферменты высокой степени чистоты поступают
в распоряжение аналитиков во все возрастающем количестве и
применяются для проведения реакций in vitro, используемых в ана-
лизе. Преимущества аналитического применения ферментов заклю-
чаются в их высокой функциональной специфичности и чувствитель-
ности. Способность ферментов специфически взаимодействовать с
отдельными веществами в различных смесях исключает, в боль-
шинстве случаев, продолжительные операции предварительного раз-
деления смесей и сокращает время анализа. Каталитическое дей-
ствие ферментов и высокое число оборотов при разложении суб-
страта позволяют достигать чувствительности, значительно прево-
сходящей чувствительность обычных химических аналитических ме-
тодов.
Числом оборотов * •• называют число молекул субстрата, которое подвергает-
ся превращению одной молекулой фермента за 1 мин при температуре О °C. Так,
например, 1 молекула ацетилхолинэстеразы в течение 1 мин расщепляет 1,8 107
молекул ацетилхолина вместе с тем одна молекула ингибитора может вы-
звать полное угнетение молекулы фермента. 'Все это позволяет сделать вывод
о высокой чувствительности биохимических способов определения фосфорорга-
нических ингибиторов.
Биохимические методы находят применение для определения
активности ферментов, для определения субстратов, для определе-
ния ингибиторов ферментов.
Особый интерес для аналитиков, исследующих ОВ, представ-
ляют биохимические способы определения ингибиторов фермен-
тов — веществ, угнетающих ферменты.
* Согласно последним рекомендациям Международного Биохимического
союза, вместо термина число оборотов введен термин молекулярная активность,
обозначающий число молекул субстрата, подвергающихся превращению на од-
ном активном центре фермента в 1 мин при 25 °C. — Прим. ред.
•• Молекулярная активность ацетилхолинэстеразы эритроцитов человека
равна 6- 105 молекул ацетилхолина на одном активном центре. — Прим. ред.
158
Таблица 7. Ингибирующее действие ОВ на ферменты
(АХЭ — ацетилхолинэстераза, ХЭ — холинэстераза, см. стр. 160)
Фермент Источник Ингибитор Ингибирую- щее дей- ствие р15о Субстрат
АХЭ Из разных Органические До 10-11 Ацетилхо-
ИСТОЧНИКОВ соединения лиихло-
фосфора РИД
ХЭ То же Органические Выше 9 Бутирилхо-
соединения линиодид и
фосфора Другие
Химотрипсин (Р‘ и Бактерии ДФФ Около 5 —
Y-)
Протеиназа » ДФФ 4,5 —
Липаза Молоко ДФФ 5,3
Пирофосфатаза Дрожжи ДФФ 3,0 —
Липаза Поджелудоч- Параоксон 5,2 —
ная железа
Алиэстераза Печень ДФФ Около 6,5 —
Эстераза Сыворотка Параоксон Около 8 —
крови
Дегидрогеназа Мозг ДФФ Около 4
Фосфатаза Почки ДФФ Около 3 —.
Тромбин — Зарин, табун — —
Химопапаин, ак- — ДФФ — —>
тивированный CN
Каталаза ) — Фосфороргани- — Н2О2
Уреаза \ ческие инсек- Мочевина
Карбоксилаза J тициды Пировиио-
градная
кислота
Пируватоксидаза Мозг Люизит —
Сукциноксидаза — Люизит — —
Пируватоксидаза — Сериый нприт — —
Гексокиназа Дрожжи Серный иприт — —
Креатинфосфоки- — Серный иприт — —
наза
Пепсин Свиной Серный иприт —
Пирофосфатаза Почки Серный иприт —
Холиноксидаза — Серный иприт 6 —
Серумпептидаза — Сериый иприт — —
Сукцинилдегид- — Сериый иприт — —
рогеназа
Липаза — Сериый иприт — —
Ксантинокси-
даза
Гексокиназа
Уреаза Раздражающие
Пируватокси- вещества
даза
159
6.2. ДЕЙСТВИЕ ОВ НА ФЕРМЕНТЫ
Исследования физиологического действия ОВ показали, что в ряде
случаев причиной их специфической физиологической активности
является угнетение различных ферментных систем. Часть этих эф-
фектов, протекающих in vivo (в живом организме), может быть
воспроизведена в исследованиях in vitro (вне живого организма,
дословно в стекле) и является основой высокочувствительных ме-
тодов обнаружения и определения ингибиторов. В табл. 7 приве-
дены некоторые данные, заимствованные из литературных источ-
ников, об ингибирующем действии ОВ на ферменты (см. также
раздел 6.3.1).
6.3. УГНЕТЕНИЕ ХОЛИНЭСТЕРАЗЫ
ФОСФОРОРГАНИЧЕСКИМИ СОЕДИНЕНИЯМИ
Следует различать ацетилхолинэстеразу АХЭ и холинэстеразу ХЭ.
Ацетилхолинэстераза— специфическая или истинная холин-
эстераза — содержится главным образом в эритроцитах (ЭХЭ),
головном мозге, окончаниях нервных волокон; холинэстераза — не-
специфическая, или псевдохолинэстераза, содержится в сыворотке
крови (СХЭ). Действие этих ферментов характеризуют следующие
показатели: оптимум pH — область концентрации водородных ио-
нов, в которой соответствующий фермент обнаруживает наивысшую
активность; изоэлектрическая точка (у амфотерных электролитов
изоэлектрической точкой, представляющей специфическую кон-
станту, является значение pH, при котором число положительно
заряженных ионов равно числу отрицательно заряженных); угне-
тение субстратом-, влияние на гидролиз.
АХЭ представляет собой фермент высокой специфичности по
отношению к субстрату и гидролизует в основном ацетилхолин;
ХЭ — фермент, представляющий собой смесь различных эстераз,
гидролизует наряду с ацетилхолином также и ряд других субстра-
Таблица 8. Отличительные признаки ацетихолииэстеразы
и холинэстеразы
(по данным Аугустинсона)
Характерные признаки АХЭ ХЭ
Оптимум pH 7,5-8,0 8,2-8,5
Изоэлектрическая точка 4,65-4,70 4,36
Устойчивость к смещению pH Меньше Больше
Угнетение избытком субстрата + —
Вызывает гидролиз
трибутирина — +
ацетилхолина + +
ацетилф-метилхолнна + ——
бензоилхолина — +
160
тов, кроме приведенных в табл. 8, в частности бутирилхолин, бути-
рилтиохолин и некоторые хромогенные субстраты. Характерным
для АХЭ является то, что наивысшая активность этого фермента
проявляется только при определенной оптимальной концентрации
субстрата (10-3 моль/л ацетилхолина); более высокие концентра-
ции субстрата ее ингибируют.
В сыворотке крови была обнаружена другая специфическая хо-
линэстераза, отличающаяся от находящейся в эритроцитах АХЭ
наряду с другим также и тем, что ее наивысшая активность про-
является при иной концентрации субстрата’. Основная функция
АХЭ, как это установлено, заключается в быстром снижении кон-
центрации ацетилхолина, образовавшегося при передаче нервного
импульса; физиологическая функция ХЭ еще полностью не выясне-
на. При отравлении фосфорорганическими соединениями может
происходить снижение активности ХЭ, которое, однако, не находит-
ся в определенной связи с тяжестью клинических симптомов по-
ражения. Так, снижение активности ХЭ на 85% от начальной было
установлено при отсутствии симптомов поражения. С другой сто-
роны, при ингаляционном отравлении появление тяжелых призна-
ков поражения наступало без заметного изменения активности фер-
мента 2. Однако, в общем, определение активности ХЭ в сыворотке
крови дает хорошую возможность диагностировать отравления
фосфорорганическими веществами для профилактических или те-
рапевтических целей. Угнетение активности фермента на 40% мо-
жет служить признаком начала поражения, естественно, если иск-
лючены другие факторы, снижающие его активность, такие, как
анемия, гепатит, или разные другие инфекционные заболевания.
Другим, более надежным, однако мене чувствительным, пока-
зателем отравления фосфорорганическими веществами является
определение активности АХЭ. Меньшая чувствительность делает
его менее пригодным для профилактического обследования лиц,
работающих с фосфорорганическими соединениями. К этому сле-
дует добавить, что измерить активность ХЭ проще, так как имеется
больший выбор субстратов.
Осуществляемый при помощи фермента гидролиз субстрата мо-
жет быть описан уравнением Михаэлиса — Ментена:
Ki Кг Кз к<
Е + S ——► ES ► Е' + Р, -----► ЕРа ----> Е + Р2
где ES — промежуточный комплекс фермента и субстрата.
Для наглядного объяснения этой реакции может быть исполь-
зована упрощенная схема ориентирования молекулы ацетилхолина
на поверхности АХЭ. На внешней поверхности молекул фермента,
сложная белковая структура которого детально еще не изучена, на-
ходится активный центр, содержащий отрицательно заряженный
анионный участок и эстеразный участок.
Считается, что анионный участок представляет собой карбок-
сильную группу, а в эстеразном участке координированно высту-
6 Зак. 677
161
пают атом азота имидазольного цикла и гидроксильные группы се-
рина и тирозина. Во всяком случае при омылении ингибирован-
ной ХЭ обнаружен серин 3, происхождение которого может быть
объяснено гипотезой о трансфосфорилировании. Приводим схему
ориентирования ацетилхолина на поверхности фермента:
Н3С
I сн2 о
"'"СН2 /9=9
н3с :_сн3 н3с ; :
— О--------------— N—Н
Следующей стадией является образование на эстеразном участ-
ке ацетилированного фермента Е', что сопровождается отщепле-
нием холина Pi. Ацетилированный фермент очень быстро гидроли-
зуется водой с образованием уксусной кислоты Р2 и исходного
фермента Е.
Аналогично действуют на фермент органические соединения фос-
фора с той лишь разницей, что промежуточно образовавшийся
комплекс фермент-ингибитор EI после отщепления ацильного остат-
ка превращается в весьма устойчивый фосфорилированный фер-
мент Е', исключительно медленно гидролизующийся в условиях ор-
ганизма:
Ki Kt Кз Kt
E + I > EI -------> E' 4-Pj ----> EP2 ---> E + P2
k-i
Отщепляемый в результате реакции фосфорилирования (К2)
продукт Pi различен и зависит от структуры фосфорорганического
соединения. Так, например, в случае фосфорилирования фермента
зарином образуется HF:
н3с сн3 н3с | 9
--Q------------—N —Н
+ HF
То же происходит в случае фосфорилирования зоманом и фтор-
фосфорилхолином. Табун при взаимодействии с ферментом отщеп-
ляет HCN, а фосфорилтиохолин — тиохолин.
Скорость реакции с ферментом и устойчивость продуктов фос-
форилирования, определяющая обратимость токсического воздей-
ствия, в значительной степени зависят от структуры исходного фос-
форорганического соединения. Наиболее сильные из известных до
настоящего времени ингибиторов холинэстеразы — фосфорилхоли-
новые и фосфорилтиохолиновые производные связываются как с
эстеразным, так и с анионным участками активного центра, как это
162
схематично показано на примере ингибирования фермента метил-
фторфосфорилхолином:
н,с
СН, /Р=с
н3с ;^сн3 Н3с I I
-----О------------N— Н----
Структурное соответствие ингибитора и субстрата становится
ясным из рассмотрения первой и третьей схем. Для названных ра-
нее фосфорорганических соединений становится также понятной и
связь между структурой и ингибирующим действием. Диизопроп-
оксифосфорилтиохолин по сравнению с диэтоксифосфорилтиохоли-
ном является более слабым ингибитором АХЭ. Так как расстояние
между анионным и эстеразным участками активного центра у АХЭ
равняется 4 50 нм, то слабое ингибирующее действие первого соеди-
нения легко объясняется пространственными затруднениями, вызы-
ваемыми наличием изопропоксигруппы. Взаимная «подгонка», су-
ществующая между молекулами субстрата и ингибитора, с одной
стороны, и фермента, с другой (см. табл. 7), проявляется, очевид-
но, в специфичности действия последнего на субстрат, что может
быть проиллюстрировано следующими примерами селективного
ингибирования. Так, метилэтоксифосфорилтиохолин является селек-
тивным ингибитором АХЭ, в то время как диэтоксифосфорилтио-
хоЛин более сильно ингибирует ХЭ5. Фосфорорганические соедине-
ния являются конкурирующими ингибиторами и, аналогично суб-
страту, находятся во взаимодействии по меньшей мере с одним из
одинаковых участков фермента. Из этого следует, что в присутствии
субстрата с увеличением его концентрации скорость реакции инги-
битора с ферментом уменьшается. Следовательно, при определении
ингибитора вначале необходимо фермент инкубировать с ингиби-
тором, и после этого ввести субстрат для определения остаточной
активности фермента. Следующим, также важным для аналитиче-
ских методов обстоятельством является различная скорость взаимо-
действия фосфорорганических соединений с ферментом, что следует
учитывать при установлении продолжительности инкубации. Метил-
фторфосфорилхолины весьма быстро реагируют с ферментом, ме-
тилалкоксифтор (или циан-)фосфонаты (зарин, зоман)—несколь-
ко медленнее, а фосфорилтиохолиновые соединения еще более мед-
ленно.
Установлена зависимость между гидролитической стабиль-
ностью соединений и константой скорости бимолекулярной реак-
ции ингибирования холинэстеразы (Кг). Характер распределения
электронной плотности на центральном и других атомах (F, S, О)
молекулы фосфорорганических соединений оказывает определенное
влияние на их реакционную способность по отношению к нуклео-
фильным реагентам как при гидролизе, так и при взаимодействии с
ферментом,
б* 163
6.3.1. Определение ингибирующего действия
Мерой ингибирующего действия является концентрация ингибитора,
при которой фермент в определенных условиях угнетается на 50%.
Эту концентрацию обозначают I50, часто ее приводят в форме от-
рицательного логарифма plgo:
plso = — 1g I50
Рис. 7. Кривые для определения5
plso-
1 — угнетение АХЭ метнлфторфосфорилтио-
холином; 2—угнетение АХЭ метилэтокси-
фосфорилхолином; 3 — угнетение ХЭ метил-
фторфосфорилхолнном; 4—угнетение ХЭ
метилэтоксифосфор илхолииом-
ным логарифмом концентрации
plso представляет собой константу, характерную для данного
ингибитора (см. табл. 9, стр. 165).
Значения констант можно сравнивать только в том случае, если
они устанавливались в точно соблюдаемых условиях, т. е. при опре-
деленной температуре, pH, про-
должительности инкубации и ре-
акции, концентрации фермента и
субстрата. Так, например, в элек-
трометрическом методе6 фермент
инкубируют 30 мин при 25°C,
исходное значение pH 8,0; реак-
цию с ацетилхолином проводят в
течение 60 мин при концентрации
последнего 7,3-10~3 моль/л. Для
определения значения I50 изме-
ряют активность ингибитора при
различных его концентрациях. По
полученным результатам строят
график, в котором процент угне-
тения (отношение ингибированно-
го фермента к неингибированно-
му) сопоставляют с отрицатель-
ингибитора. Пример такой обра-
ботки опытных данных представлен на рис. 7. Истинное значение
plso искажается побочными реакциями, протекающими в зависимо-
сти от свойств ингибитора и фермента, а также условий опыта. Та-
кими побочными реакциями, конкурирующими с ингибированием
фермента, являются спонтанный гидролиз субстрата, гидролиз инги-
битора, вызываемый содержащимися в ферменте фосфорилфосфа-
тазами, адсорбция на стенках стеклянного сосуда при работе с
очень разбавленными растворами, реакция с другими активными
группами присутствующих белков. Кроме того, в случае слабого
ингибитора одновременно протекают и угнетение, и реактивация
фермента. Некоторые авторы считают, что для бимолекулярной
реакции между ингибитором и ферментом
ЕН4-РХ —► ЕР + НХ
более специфична константа скорости К.2, особенно для медленно
реагирующих ингибиторов, так как она больше зависит от свойств
вещества. Однако значения I50 или plso можно рассматривать как
более четкую характеристику токсичности ингибиторов.
164
В табл. 9 приведены константы скорости и plso некоторых реак-
ций.
Таблица 9. р153 и константы скорости Кг реакций ингибирования ХЭ
и щелочного гидролиза (В скобках для сравнении приведено р150 АХЭ)
Ингибитор plso Ха при 25 °C, л моль 1 «сек 1
ингибирования ХЭ гидролиза
Зарин 8,4 (8,8) 7- 103 * 26
Табун -(8 4) — 7,5
ДФФ 7,8(6,1) — 0,83
ТЭПФ 8,3 (7,5) — 2,6
Паратион (тнофос) 4,6 (7,7) — 2,5 (при 37 °C)
Метилфторфосфорилхолии 8,4(10,0) — 935
Метил фторфосфорилгомохолин 8,4(11,0) 1,9- 105* 305
Диэтоксифосфорилтиохолии 8,9 (8,4) 2- 10* 0,7
Метилэтоксифосфорилтиохолин 7,9 (9,1) 2- 103 0,2
Изосистокс - (6.0) — 0,81
* При концентрации субстрата 1,87.10~г моль/л.
Как видно из табл. 9, медленно реагирующие с ферментом фос-
форилтиохолины имеют почти такую же высокую токсичность, что и
весьма быстро реагирующие метилфторфосфорилхолины.
6.3.2. Определение активности холинэстеразы
Можно определять активность АХЭ ’или ХЭ. Для определения
активности АХЭ в большинстве случаев подвергают гемолизу эри-
троциты; ХЭ определяют в сыворотке крови. При использовании
цельной крови можно путем выбора специфического субстрата
(ацетил-р-метилхолина для АХЭ и бутирилхолина для ХЭ) или
применяя определенные концентрации субстрата достигнуть диф-
ференцирования ферментов. Последний из указанных способов
основан на уже описанном ингибировании АХЭ более высокими
концентрациями субстрата. Так, при концентрации ацетилхолина
10‘3М определяется преимущественно АХЭ, при концентрации же
Ю-1 М — ХЭ, однако в каждом случае в определенной степени
(примерно на Vs) проявляет активность и другой фермент. Опре-
деление активности осуществляют либо по установлению скорости
ферментативного расщепления субстрата (кинетический метод) или
путем определения конечных продуктов и не вступившего в реак-
цию субстрата.
В зависимости от метода активность фермента определяют по
разным критериям. В классическом манометрическом способе ме-
рой активности фермента является количество выделяющейся дву-
окиси углерода.
165
Рис. 8 Аппарат Варбурга (изго-
товитель — стекольный завод
- Штютцербах):
/ — стеклянный резервуар; термо
стат; 3 — манометр; 4 — регулятор ча-
стоты встряхивания.
Аммон12 ввел холинэстеразную единицу, которая обозначает
количество фермента, требующееся для расщепления 10 6 М аце-
тилхолина в течение 1 ч при 37 °C и pH 7,4. Такое же количество
ацетилхолина требуется для выделения 10-6 М двуокиси углерода
(22,4 мм3).
Для оценки активности фермента в крови контролируемого, лица
важно знать нормальную активность фермента и возможные ее от-
клонения от нормы. При работе с
0,05 мл сыворотки крови и соблюде-
нии условий манометрического ме-
тода нормальные значения активно-
сти равны 7—10 холинэстеразным
единицам12. В колориметрическом
методе, в котором \w_-
прореагировавший ацетилхолин 13, в
качестве стандартной холинэстераз-
ной единицы принято число микро-
молей ацетилхолина, которое гидро-
лизуется 1 мл сыворотки за 1 ч при
37 °C. Эта холинэстеразная единица
в 20 раз больше единицы, принятой
Аммоном. Нормальной считается ак-
тивность, равная 130—310 таким
единицам.
В электрометрическом методе
активность холинэстеразы опреде-
ляют по снижению pH через 60 мин
после начала реакции и. При этом
нормальным значением активности
фермента считается изменение ДрН
в течение 1 ч от 0,70 до 1,05 при ра-
боте с 0,2мл сыворотки при 25 °C.
Для определения активности хо-
линэстеразы разработано много спо-
собов. Надежные результаты дают
методы, основанные на определении
количества образующегося продукта, в которых соблюдается ли-
нейная зависимость между активностью фермента и количеством
образующегося продукта. Несмотря на то, что манометрический ме-
тод довольно сложен в выполнении, он в этом отношении (соблюде-
ние линейной зависимости) еще не превзойден. Более простыми
являются потенциометрические методы, которые дают весьма точ-
ные результаты, особенно при использовании автотитратора, авто,
матически поддерживающего постоянный pH, вследствие чего
исключается влияние pH на активность фермента. Ниже приведен
(см. также табл. 10) обзор различных способов определения актив-
ности фермента. Подробно описаны манометрический и потенцио-
метрический способы как наиболее пригодные.
166
Таблица 10. Обзор методов определения активности
холинэстеразы сыворотки7
Субстрат Продолжи- тельность инкубации, мин Нормальная активность, мкмоль/мл Ссылка на литературу
Манометрические методы
Трибутирин 60 60—150 9
Ацетилхолин 10 30(лкл СО2/0,2 мл) 8
Ацетилхолин 60 90-240 9
Ацетилхолин 60 140—200 [Н, 12]
Электрометрические методы
Ацетилхолин | 60 | 0,703 (ДрН/0,02 мл) | [10]
Титриметричес к ие методы
Трибутирин 10 10-12 88]
Трибутирин 60 100-200 89
Трибутирин 60 75 90
Ацетилхолин 10 40-80 91
Ацетилхолин 10 30-40 88
Колориметрические методы
Нитрофеиилпропионат 20 1,5-10,5 [83
Нитрофенилпропионат 60 1,5-4 90
Ацетилсалициловая кислота 60 21 92
Прокаин 30 100 93
Фенилбензоат 60 14-41 94
Феиилбензоат 60 1,6-3,0 95
а-Нафтил ацетат 60 3,5—6,5 (мг/мл) 87
а-Нафтилбутират 60 90—250 [7
а-Нафтилаурат 306 0,03—0,08 (мг/0,2 мл) 96
Карбонафтоксихолин 60 L8—4,6 (мг/мл) 87
Ацетилхолин 60 1W—280 13
I. Определение кислоты, образующейся при
разложении карбоксилхолина. Манометрические мето-
ды 15-17. Эти методы основаны на измерении объема СОг, вы-
деляющейся при действии на бикарбонатный буфер уксусной кис-
лоты, которая образуется в результате гидролиза ацетилхолина.
Определение проводят в аппарате Варбурга (рис. 8). Предложены
новые колбы Варбурга, особенно удобные для измерения угнетения
ХЭ27.
Титриметрические методы. Выделяющуюся кислоту титруют в
присутствии соответствующего индикатора (бромтимоловый синий,
феноловый красный) или проводят в pH-метре потенциометриче-
ское титрование едким натром 18’28-32 с интервалами в 5 мин в тече-
ние 1 ч до достижения неизменяющегося pH.
167
Весьма удобно также титрование при помощи автотитратора,
автоматически поддерживающего pH, в результате чего активность
фермента во времени не меняется IS~21.
Электрометрические методы (измерение ЛрН)22 ~26-29. В этих
методах изменение pH, вызываемое выделяющейся кислотой, изме-
ряется по истечении определенного времени pH-метром. При этом
используется специально разработанный Михелем 22 буфер с исход-
ным pH 8,0, буферные свойства которого обеспечивают линейную
зависимость между активностью фермента и изменением pH. Дру-
гим преимуществом метода является возможность параллельно
проводить измерения в нескольких пробах 30 31. По полученным
значениям ДрН можно определить число микромолей разложив-
шегося ацетилхолина. Для этого прибавляют стандартный раствор
кислоты к смеси буфер — фермент и измеряют уменьшение значения
pH при определенных количествах кислоты. Используя эти величи-
ны, строят калибровочную кривую, по которой устанавливают ко-
личество разложившегося субстрата.
Колориметрические методы с pH-индикатором. Пробу, содер-
жащую фермент, смешивают с субстратом и соответствующим
индикатором и определяют время, в течение которого окраска пробы
становится одинаковой с окраской эталона. Значение pH, устанав-
ливаемое прибавлением соответствующих количеств стандартного
раствора кислоты, целесообразно выбирать в границах наиболее
заметного изменения окраски индикатора, например, в случае
бромтимолозого синего при pH 7—7,2. Другими удобными индика-
торами являются феноловый красный36 и л-интрофенол Д Окраски
сравнивают визуально33 либо фотометрически и отмечают время
выравнивания окрасок пробы и эталона.
Методы с использованием субстрат-индикатооной бумаги3S.
Эти методы, являющиеся модификацией колориметрического мето-
да, основаны на использовании индикаторной бумаги и применяют-
ся для быстрого клинического определения АХЭ и ХЭ 39~
Рекомендуется использовать индикаторные бумаги, выпускаемые под тор-
говым названием «ахолест» 42 и «биофан С» 43, при изготовлении которых в ка-
честве субстрата выбран ацетилхолин, а в качестве индикатора — бромтимоло-
вый синий.
Более быстрым и специфичным для определения ХЭ является
метод с использованием индикаторной бумаги, описанной Матоу-
шеком и Церманом 44, при изготвлении которой в качестве субстра-
та применяется бутирилхолин. После инкубации этой бумаги с кап-
лей сыворотки (0,05 мл) измеряют время выравнивания окраски
испытуемого вещества с окраской эталона (pH 6,47).
II. Определение нераз ложившегося ацетилхо-
л и н а. Через 30—60 мин взаимодействия фермента с субстратом
прибавляют щелочной раствор солянокислого гидроксиламина для
превращения избыточного ацетилхолина в ацетгидроксамовую кис-
лоту, которая при pH 1,0 —1,4 образует с хлорным железом крас-
168
но-коричневое внутрикомплексное соединение, максимум поглоще-
ния которого лежит при 520 нм-.
О
(CH3)3N—СН2—СН2—О—С—СН3 + NaOH + NH2OH —►
О
II
—>• [(CH3)3N—СН2—СН2ОН] (ОН]’ + Na + СН3—С—NHOH + Н2О
О
3+ / \
СНз—+73Fe3+ СН3—С' 'Fe/З + Н+
\nhoh \n-oz
н
Применяемый весьма часто метод, разработанный Хестриным 45
и выполняемый, в частности, по методике, описанной Пилцем 46-48,
дает весьма надежные результаты 49-53.
Неразложившийся ацетилхолин можно также определить по
реакции Шёнеманна54 (см. раздел 5.1).
III. О п р е д е л е н и е продуктов разложения или и е-
разл о ж и вшихся карбокситиохолинов 60’ 122’ 123. Карб-
окситиохолины расщепляются быстрее, чем карбоксихолины. Они
находят широкое применение для определения угнетающего дей-
ствия фосфорорганических соединений на субстраты еще и из-за
того, что имеется много способов определения тиохолина — про-
дукта расщепления карбокситиохолинов.
В простейшем методе определяют убыль субстрата спектрофо-
тометрически, измерением в УФ-свете (максимум поглощения
ацетилтиохолина лежит при 226, а бутирилтиохолина — при
250 нм) 55’56-97.
Колориметрические методы определения тиохолина. Тиохолин
образует с нитропруссидом натрия окрашенный продукт57 (ХМакс
520 нм). Другой фотометрический метод основан на измерении
интенсивности желтого окрашивания, образующегося при реак-
ции тиохолина с 5,5'-дитио-бис-(2-нитробензойной) кислотой
(ДТНБ) 58’59. Этот метод весьма прост и требует незначительной
затраты времени. Гистохимическое обнаружение холинэстеразы
основано на реакции с солью меди и сульфидом аммония, в ре-
зультате которой образуется черный сульфид меди60’61.
Электрохимические методы определения тиохолина 62-65. Совре-
менный электролитический способ основан на деполяризации пла-
тинового анода тиохолином, на котором он окисляется в дисульфид.
Определение проводят в тройной буферной смеси (pH 7,4) с кон-
центрацией субстрата (иодистых бутирилтиохолина для ХЭ и
ацетилтиохолина для АХЭ) 2 • 10~3 М.
В раствор субстрата, содержащего буфер, погружают один каломельный и
два платиновых электрода. К платиновым электродам подают постоянный ток
в 25 мка. После прибавления раствора фермента изменение напряжения между
Платиновым анодом и каломельным катодом регистрируется во времени авто-
169
магическим регистрационным прибором (самописцем). По полученной кривой
определяют наклон ЬЕ/Ы (в мв/сек) и сопоставив с данными калибровочной
кривой, построенной по данным изменений известной активности фермента, вы-
числяют активность испытуемого фермента.
Способ применим для определения активности фермента и кон-
центраций субстрата и ингибитора. Для возникновения начального
потенциала необходимо присутствие в растворе иона иода:
21" —> 12 + 2е"
Быстрый полярографический метод68 определения активности
холинэстеразы основан на том, что ацетилтиохолин полярографи-
чески неактивен, а образующийся тиохолин, напротив, дает ти-
пичную для SH-групп анодную ступень. Рост анодной ступени в
единицу времени служит мерой активности холинэстеразы 66’67.
Определение тиохолина, образующегося при распаде субстрата,
можно также осуществить амперометрическим титрованием в
присутствии соединений ртути69. Резкое возрастание силы тока
свидетельствует о конце титрования, т. е. об избытке CH3HgI в
реакционной среде.
RSH + CH3HgI —> CHjHgSR + Hl
IV. Определение с помощью хромогенных и флуо-
рогенных субстратов*. Применение субстратов, образую-
щих при отщеплении карбоксильной группы окрашенное или флуо-
ресцирующее соединение, удобно при гистохимической локализации
и для колориметрического или флуорометрического определения
активности эстераз. Некоторые из этих субстратов специфически
расщепляются определенными эстеразами. Для гистохимических
исследований наиболее удобен индоксилацетат и некоторые его
галогензамещенные, преимущественно 5-бром-4-хлориндоксил-
ацетат. Под влиянием эстеразы из индоксилацетата образуется
обладающий флуоресценцией индоксил, а при дальнейшем окис-
лении индоксила — синее индиго:
* Субстраты, дающие при ферментативном расщеплении окрашенные иди
флуоресцирующие продукты.
170
Активность эстеразы можно определять либо измеряя флуорес-
ценцию индоксила 119, либо интенсивность окраски ингидо 70-74. При
расщеплении резоруфинацетата холинэстеразой (pH 7,4) или фос-
фатазой и а- или у-химотрипсином образуется интенсивно флуорес-
цирующий резоруфин 12°, флуоресценцию которого можно измерить
уже при концентрации его 10~8 М.
Флуоресценция нафтола, образующегося при расщеплении а-
и р-нафтилацетатов, использована в очень чувствительном методе
определения активности фермента 75>76. При гистохимических иссле-
дованиях эстераз нафтол обнаруживают в виде окрашенных соеди-
нений, получаемых в результате различных реакций сочетания 77,
например с а-нафтилдиазо- 1,5-нафталиндисульфокислотой78 или
диазотированным розанилином79.
Некоторые методы 80-82 основаны на расщеплении ферментом
при pH 8,0 индофенилацетата:
АХЭ
О_=/ ^=N——°СОСН3 О=<^ N—\ /—°'
индофеннлацетат индофенол
Содержание образующегося окрашенного в синий цвет индо-
фенола определяют фотометрически при 625 нм. Способ дает точ-
ные и воспроизводимые результаты в пределах 25—150 холинэсте-
разных единиц Аммона.
Другими хромогенными субстратами являются различные слож-
ные эфиры п- или о-нитрофенола 83-85 и фенилбензоат86. Образую-
щийся фенол О11ределяют цветной реакцией Фолина и Циокалтё,
а 2-карбонафтоксихолин 87 — по окраске азокрасителя, получающе-
гося после гидролитического расщепления и сочетания с диазоти-
рованным о-дианизидином.
6.3.2.1. Определение активности холинэстеразы сыворотки в
аппарате Варбурга ". Более подробно с условиями работы с аппа-
ратом Варбурга, требующей некоторых практических навыков,
можно познакомиться в статье Буго (см. раздел 5, сс. 13°).
Реагенты
Раствор А (бикарбонатный раствор Рингера) —смесь 30 мл 1,26%-ного рас-
твора NaHCO3, 100 мл 0,9%-ного раствора NaCl, 2 мл 1,2%-ного раствора КС1
и 2 мл 1,76%-ного раствора СаС12 6Н2О.
Раствор субстрата — раствор 50 мг хлористого ацетилхолина в 10 мл рас-
твора А; ежедневно готовят свежий раствор.
Сыворотка крови.
Для работы используют точно отмеренный объем жидкости, равный 2 мл,
измерения проводят при 37 °C, газовая фаза — азот, содержащий 5 объемн.%
СО2. В основную часть резервуара сосуда Варбурга наливают 1,5 мл раствора
субстрата, а в боковой отросток — 0,5 мл сыворотки, разбавленной раствором А
в отношении I : 50. После 10 мин выравнивания температуры перекрывают кран
манометра и переводят раствор сыворотки в основной резервуар. В течение 1 ч
через каждые 10 мин включают манометр и записывают его показания. Для
каждого определения нужно устанавливать контрольную величину спонтанного
171
гидролиза субстрата. Для этого измерение проводят с субстратом, к которому
вместо сыворотки добавлен только раствор А.
Измеренное приращение давления (в миллиметрах раствора Броди) корри-
гируют по контрольным величинам спонтанного гидролиза. Кроме того, резуль-
таты умножают на поправку, полученную при калибровке сосудов. Строят
график, откладывая на одной оси объем двуокиси углерода (мкл), а на другой
время (мин). Определенную графически величину, соответствующую 60 мин,
делят на 22,4 мкл и получают число микромолей уксусной кислоты, которое
равно числу микромолей ацетилхолина. Так как для определения брали 0,01 мл
сыворотки, то для пересчета в единицы холинэстеразы полученный выше ре-
зультат умножают на 100. Нормальное значение активности должно составлять
140—200 единиц.
6.3.2.2. Определение активности холинэстеразы электрометриче-
ским АрН-методом 10. Способ может быть использован для опреде-
ления Igo и концентрации ингибитора96.
Реагенты
Раствор А (буферная смесь Михеля) — 1,2371 г диэтилбарбитурата натрия
(0,006М), 0,1361 г КН2РО4 (0,001 Af) и 17,535 г NaCl (0,3 М) растворяют в
900 мл воды, не содержащей СО2, и смешивают с 10 мл 0,1 н. раствора НС1.
Общий объем раствора доводят до 1 л и при 25 °C измеряют pH, который дол-
жен быть равен 8,0 (при необходимости добавляют еще 0,1 н. раствор НС1 точно
до pH 8,00). Стабилизуют раствор добавкой 2 капель толуола.
Раствор субстрата — раствор 3 г бромида ацетилхолина в 100 мл воды, к
которому добавлены 2 капли толуола; хранят в холодильнике.
В колбу Эрлеимейера емкостью 50 мл пипеткой Вносят 10 мл дистиллиро-
ванной воды, 0,2 мл сыворотки и 10 мл раствора А. Эту смесь помещают на
10 мин в термостат при 25 °C, после чего определяют pH с точностью до 0,01,
пользуясь pH-метром. Затем прибавляют 2 мл раствора субстрата, содержимое
колбы быстро перемешивают и отмечают время. Точно через 60 мин снова из-
меряют pH. При анализе большого числа проб их обрабатывают последователь-
но через короткие промежутки времени. Для выравнивания температуры си-
стемы электроды перед измерением погружают в испытуемый раствор на 30—
60 сек. В работе 23 описано устройство, обеспечивающее параллельную обработку
6 проб.
Для расчета пользуются следующей формулой:
/ pH, - рН2 Л р =
\ *2 ~ *1 /
где pH, и РН2 — начальное и конечное pH; — время прибавления субстрата;
t2— время отсчета рНг! h— время, ч; Ь — поправочный коэффициент, учитываю-
щий спонтанный гидролиз, соответствующий значению рН2; F— поправочный
коэффициент, учитывающий погрешности при определении величины Д pH//i.
Ниже приведены поправочные коэффициенты для разных величин рН2.
рн2 ь F рн2 Ь F
7,9 0,09 0,98 7,2 0,02 1,0
7,8 0,07 1,0 7,0 0,01 1,0
7,7 0,06 1,01 6,8 0,01 1,0
7,6 0,05 1,02 6,6 0,01 1,01
7,5 0,04 1,02 6,4 0,01 1,02
7,4 0,03 1,01 6,2 0,01 1,04
7,3 0,02 1,01 6,0 0,01 1,09
При определении активности сухих или обогащенных сывороток
исходная проба должна содержать 12—13 мг белка. Это необхо-
172
димо для сохранения такого содержания белка в буферной ем-
кости системы, которое бы обеспечивало сравнимые результаты.
Для определения концентрации ингибитора смесь сыворотки
и буфера инкубируют 30 мин с раствором ингибитора перед при-
бавлением субстрата. Параллельно проводят контрольный опыт
без ингибитора. По pH, измеренным по окончании реакции, рас-
считывают процент угнетения фермента по следующей формуле:
-------P(pH, Z^H2K)--------100=% угнетения
где pH, — начальное pH, рН2К — конечное pH контрольного опыта, рН2П — ко-
нечное pH пробы.
По калибровочной кривой, построенной по известным концен-
трациям ингибитора, определяют искомую концентрацию ингиби-
тора, соответствующую угнетению, рассчитанному поданным опыта.
Работают со стандартизованными растворами сухой сыворотки.
6.3.2.3. Фотометрическое определение активности холинэстера-
зы59. Этот быстро осуществляемый микрометод основан на реак-
ции тиохолина, образующегося при ферментативном расщепле-
нии ацетилтиохолина 5,5'-дитио-бис-(2-нитробензойной) кислотой
(ДТНБ). Механизм этой цветной реакции обнаружения тиола опи-
сан в разделе 5.1.1.9.
Реагенты
Раствор ДТНБ с буфером — в мерную колбу емкостью 250 мл вносят 25 мг
ДТНБ, 1,66 г NaCl, 62,5 мл 0,2 М раствора трис-(гидроксиметил)-аминометана,
100 мл 0,1 н. раствора НС1 и доводят раствор водой до метки. Прн 37°С этот
буферированный раствор имеет pH 7,4; в холодильнике сохраняется 2 недели.
Раствор субстрата — раствор 0,52 г йодистого ацетилтиохолина в 100 мл
воды (0,018 М раствор).
Сульфат хинидина — 0,5%-иый раствор в воде или в ДФФ.
6 каждую из двух пробирок, из которых одна предназначена для работы
с испытуемым веществом, а другая — для контрольного опыта, вносят по 4 мл
раствора ДТНБ и в течение 5 мин термостатируют при 37 °C на водяной бане.
В каждую пробирку прибавляют по 0,02 мл сыворотки, а в пробирку для кон-
трольного опыта кроме того 1 мл раствора хинидина. Затем в обе пробирки
вносят по 0,5 мд раствора субстрата и содержимое хорошо перемешивают.
Точно через 3 мин реакцию в пробирке останавливают прибавлением 1 мл рас-
твора хинидина и последовательно фотометрически измеряют при 412 нм экстин-
кцию в испытуемом веществе и. в контрольном опыте.
Разницу в значениях экстинкции пробы и контрольного опыта выражают в
микромолях прогидролизовавшегося ацетилтиохолина; полученные данные от-
носят к 1 мл сыворотки и ко времени 3 мин. Для этой цели служит коэффи-
циент, определяемый по калибровочной кривой, для построения которой исполь-
зуют глутатион или другое вещество с известным содержанием SH-групп. Нор-
мальными считаются: для мужчин 7,8—16,6 мкмоль SH (иа 1 мл за 3 мин), для
женщин 5,8—13,8 мкмоль SH.
6.3.3. Определение ингибиторов холинэстеразы
Все приведенные выше способы определения активности холинэсте-
разы, соответственно модифицированные, могут быть использованы
и для определения ингибиторов. Исключением является маноме-
173
трическпй метод, который вследствие малой чувствительности не-
пригоден для определения следов веществ.
Для определения ингибиторов раствор фермента известной кон-
центрации заданное время инкубируют с ингибитором. Затем
вводят субстрат и определяют обычным путем остаточную актив-
ность. Период инкубации существенно зависит от скорости взаимо-
действия ингибитора с ферментом. Реакцию фосфорилтиохолинов
с ферментом можно считать законченной только через 2 ч. Однако
преимуществом этих реакций является то, что эти соединения
весьма мало подвержены гидролизу, а также возникновению дру-
гих побочных реакций. В противоположность фосфорилтиохолинам
метилфторфосфорилхолины быстро реагируют с ферментом, поэ-
тому продолжительность инкубации очень мала.
Для чувствительного определения галоген- и псевдогалоген-
ангидридов кислот фосфора в общем достаточно 10 мин инкуба-
ции, для фосфорорганических инсектицидов продолжительность
принята'-'8 равной 30 мин. Поскольку как ингибирование, так и
расщепление субстрата зависит от pH среды, необходимо во время
инкубации ферментов с ингибиторами поддерживать соответствую-
щее каждому ферменту оптимальное pH. В случае тетрама и ана-
логичных соединений, содержащих в молекуле третичную амино-
группу, ингибирование зависит от степени ионизации соединения.
Так, при pH более 8,5 доля ненонизированного соединения возра-
стает и угнетение уменьшается. Для получения воспроизводимых
результатов необходимо применять ферменты со стандартизован-
ной активностью. Наиболее широко используется лиофилизиро-
ванная сыворотка крови лошади. При отсутствии готового препа-
рата его можно относительно просто приготовить в лаборатории
сублимационной сушкой жидкой сыворотки (сушка выморажива-
нием) .
Для этого жидкую сыворотку замораживают в стеклянной чаш-
ке и помещают в вакуум-эксикатор, который при помощи стеклян-
ной трубки по возможности большого диаметра соединяют через
охлаждаемую ловушку с масляным вакуум-насосом. В зависимости
от вакуума, который должен достигать' не менее Ю-2 л/л/ рт. ст.,
через несколько часов (или дней) сыворотка полностью высыхает.
Активность полученного препарата перед использованием его для
аналитических целей определяют любым стандартным методом,
например электрометрическим ДрН-методом. Построение калибро-
вочной кривой и само определение проводят с растворами фермен-
тов равной активности.
Рекомендуется применять очищенные препараты ферментов,
для чего пользуются фракционным осаждением части белкового
балласта сульфатом аммония при разных pH. После отделения
осадка на центрифуге, диализа и лиофилизации полученные сухие
вещества обладают по сравнению с нормальной сывороткой повы-
шенной, в зависимости от степени очистки, активностью и пони-
женным буферным действием и склонностью к денатурации 28’99’ |0°.
174
Активность очищенных и обогащенных препаратов в большин-
стве случаев выражают в микромолях ацетилхолина на 1 мг су-
хого вещества или, после определения белка по способу Кьельдаля,
в микромолях ацетилхолина на 1 мг белка в час.
Другими часто используемыми для аналитических целей источ-
никами фермента являются сыворотка крови человека, гемолизи-
рованные эритроциты крови человека, крупного рогатого скота или
лошади, или при работе с неспецифическими субстратами — гомо-
генаты печени. По возможности выбирают препарат фермента, ак-
тивность которого наиболее чувствительна к действию определяе-
мого ингибитора.
В разных способах определения температурные условия раз-
личны, однако обычно не ниже 25 и не выше 37°C, что отвечает
физиологическим условиям. Перед анализом проб биохимическими
методами необходимо построить калибровочные кривые или состав-
лять таблицы на основе данных, полученных при работе с чистым
ингибитором. При точном соблюдении условий, к которым также
относится и строгое соблюдение времени сливания растворов ре-
агентов, получают результаты, которые после пересчета в проценты
угнетения выражают как функцию концентрации или ее отрица-
тельного логарифма.
Способ служит для определения значений 150 или plgo- Следует
стремиться подбирать такие условия, чтобы угнетение фермента
соответствовало 40—60%, ио ни в коем случае не ниже 20 и не
выше 80% •
Серусодержащие фосфорорганические инсектициды, окисляются
в организме в их кислородные аналоги, сульфоксиды и сульфоны,
которые являются сильными ингибиторами холинэстеразы. По-
этому перед проведением биохимического определения их спе-
циально окисляют бромной водой или N-бромсукцинямидом 101 или
же смесью перекиси водорода и уксусной кислоты 102. Избыток
брома связывают добавлением некоторого количества водного рас-
твора фенола.
Ниже приведены наиболее употребительные способы определе-
ния фосфорорганических соединений, ингибирующих холинэсте-
разу.
I. Методы, в которых в качестве субстрата ис-
пользуется карбокси л хол ин. Весьма чувствительным
является визуальный колориметрический метод определения обра-
зующихся уксусной или масляной кислот при помощи бромтимоло-
вого синего 103- 104. При этом измеряют время, нужное для вырав-
нивания окрасок испытуемого вещества и эталона. Угнетение рас-
читывают по формуле:
inn 'к • 100
100--------= % угнетения
п
где tK — продолжительность выравнивания окраски контрольной пробы; tn — про-
должительность выравнивания окраски пробы испытуемого вещества.
175
Титриметрическое определение уксусной кислоты, образующей-
ся при расщеплении ацетилхолина, с феноловым красным в каче-
стве индикатора используется для определения табуна и зарина 105
в концентрации 0,1 мкг!мл, а с крезоловым красным — для опре-
деления инсектицидов106. Многие авторы 96’107-109 применяют
АрН-метод. Наиболее подходящим для контролирования проте-
кающих очень быстро процессов ингибирования действия фермен-
тов, например в случае расщепления метилфторфосфорилхолинов,
является потенциометрический метод, при котором постоянное зна-
чение pH поддерживается автотитратором. Очень важно, что по
этому методу можно работать без добавления буфера. Таким об-
разом исключается присутствие различных ионов, которые в из-
вестной мере влияют на действие ферментов.
Для анализа фосфорсодержащих ОВ в полевых условиях весь-
ма удобны индикаторные бумаги, содержащие смесь субстрата
и индикатора44’110. Преимущества этих методов заключаются в их
простоте, быстроте проведения, потреблении незначительных коли-
честв испытуемых веществ и реагентов, возможности работы в ши-
роком интервале температур. Дальнейшее совершенствование ме-
тодов состоит в том, что на бумагу, содержащую субстрат и ин-
дикатор, наносят также лиофилизированную сыворотку111. При
использовании очищенных ферментов и хранении в сухом месте
такие индикаторные бумаги хорошо сохраняются. Отличным спо-
собом для проведения большого числа параллельных определений
является модифицированный диффузионный метод с агаром 112.
В плоском слое нанесенного на стеклянную пластинку агаро-
вого геля, содержащего фермент и 'индикатор бромтимоловый
синий, проделывают отверстия, в которые заливают испытуемый
раствор. Для прохождения диффузии пластинку оставляют на оп-
ределенное время (по возможности, на ночь), затем заливают слой
агара раствором ацетилхолина, в результате чего на желтом фоне
появляются синие круги, диаметр которых пропорционален концен-
трации ингибитора. Это дает возможность построить также и ка-
либровочную кривую. Метод пригоден для определения паратиона
в концентрации 0,03 мкг/мл.
Фотометрическое определение непрогидролизовавшегося ацетил-
холина в виде гидроксамата железа используется при биохимиче-
ском анализе различных фосфорсодержащих инсектицидов113’114.
II. Методы, в которых в качестве субстрата ис-
пользуется карбокситиохолин. Упоминавшийся ранее
(етр. 169) электрохимический метод62, основанный на измерении
степени деполяризации платинового электрода, был использован
для определения фосфорорганических ингибиторов холинэсте-
разы 115. При продолжительности инкубации 10 мин было опреде-
лено 2 • 10“4 мкг/мл зарина и 1 - 10-2 мкг/мл систокса (меркап-
тофос).
Интересные возможности для развития биохимической инди-
кации нервно-паралитических ОВ этим электрохимическим мето-
176
дом возникают при применении нерастворимой холинэстеразы 116’117.
Для получения фермента в нерастворимом состоянии пропитывают
его раствором в крахмале подушечку из пористого полиуретана и
затем сушат вымораживанием. Фермент сохраняет в подушечке
свою устойчивость и активность 12 ч. Через подушечку, помещен-
ную между двумя платиновыми электродами, пропускают раствор
йодистого бутирилтиохолина, в результате чего в системе при при-
ложении тока, равного 2 мка, возникает низкий потенциал, являю-
щийся следствием окисления на аноде тиохолина в дисульфид.
Если раствор субстрата содержит ингибитор или последний при-
сутствует в токе воздуха, просасываемого через подушечку, то
вследствие снижения активности холинэстеразы потенциал системы
возрастает.
III. Методы, использующие хромогенные и ф л у-
орогенные субстраты. Описанные полиуретановые поду-
шечки с ферментом при соответствующем аппаратурном оформле-
нии и применении в качестве субстрата р-нафтилацетата в буфер-
ном растворе (pH 7,4) дают возможность непрерывно контролиро-
вать содержание фосфорорганических ингибиторов в воздухе или
воде И8.
Постоянная флуоресценция р-нафтола, которая возникает при
равномерном течении раствора субстрата и воды через подушечку
с ферментом снижается или гасится при наличии в воде фосфор-
органического ОВ.
Хромогенный субстрат, представляющий собой 2-азобензол-1-
нафтилацетат, который вместе с сывороткой крови лошади приме-
няется для определения зарина 121, образует в результате фермента-
тивного гидролиза при pH > 7 красный 2-азобензол-1-нафтол:
ХЭ; н2о
По истечении некоторого времени ферментативный распад оста-
навливают прибавлением раствора соляной кислоты в ацетоне и
окрашенный продукт определяют фотометрически.
Работа некоторых простых приборов, применяемых для инди-
кации нервно-паралитических ОВ в полевых условиях, основана
на расщеплении ацетилхолинэстеразой эритроцитов 6-бром-2-
нафтилацетата до уксусной кислоты и 6-бром-2-нафтола, который
177
при сочетании его со стабилизованной солью диазония дает азо-
краситель 127.
6.3.3.1. Индикация и полу количественное определение фосфорорганических
ингибиторов в воде при помощи субстрат-индикаторной бумаги44.
Реагенты
Субстрат-индикаторная бумага — растворяют 1,70 г бутирилхолиниодида и
0,30 г бромтимолового синего в 100 мл 96%-иого спирта и пропитывают этим
раствором бумагу, применяемую для хроматографирования (1 м2 бумаги дол-
жен весить 100 г). Для этого бумагу на 30 сек погружают в раствор, дают ему
стечь и в подвешенном состоянии сушат бумагу на воздухе. После чего ее раз-
резают (отбрасывая с краю полосы шириной 5 мм) на прямоугольники разме-
ром 15 X 20 мм.
Для приготовления примерно 1500 кусков достаточно 100 мл раствора.
Индикаторная бумага хранится неограниченно долго. Для серии определений
следует использовать бумагу одного приготовления.
Раствор фермента — 2%-ный раствор лнофилизоваииой сыворотки крови
лошади в буфере Михеля (pH 8,0); хранится в холодильнике.
Буферная смесь Михеля—растворяют 0,742 г диэтилбарбитурата натрия,
0,082 г КН2РО4 и 17,535 г NaCl в 1 л воды, не содержащей СОг, и добавлением
0,1 и. раствора НС1 устанавливают pH 8,0.
Буфер сравнения (фосфатный буфер Зёренсеиа, pH 7,0) — смесь 39 мл рас-
твора 9,078 г КН2РО4 в'1 л воды и 61 мл раствора 11,876 г Na2HPC>4 в 1 л воды.
Сначала проверяют пригодность бумаги. Для этого наносят на одно и то же
место предметного стекла по 0,05 мл воды и раствора фермента и рядом поме-
щают 0,1 мл буфера сравнения. На оба эти участка накладывают кусочки суб-
страт-иидикаторной бумаги, накрывают их стеклом и определяют время вырав-
нивания окрасок обоих отрезков индикаторной бумаги, которое должно равняться
3—7 мин. Если выравнивание окраски не укладывается в указанный проме-
жуток времени, то причиной этого может быть повышенное содержание кислоты
в бумаге илн индикаторе. В этом случае приходится готовить свежую индика-
торную бумагу, используя другой сорт бумаги, либо повторить пропитку бумаги
раствором субстрата и индикатора, который предварительно нейтрализуют при-
бавлением 0,1 н. раствора NaOH до появления зеленовато-снней окраски.
Перед определением пробу воды нужно нейтрализовать. Для этого к 10 мл
исследуемой воды прибавляют 5 капель 0,1%-ного раствора бромтимолового
синего в 20%-ном спирте и осторожным прибавлением 0,01 и. раствора NaOH
или НС1 доводят окраску до зеленовато-синей. Если жесткость испытуемой воды
велика, то могут наблюдаться различия в изменении окраски по сравнению с
контрольной пробой, что объясняется буферным действием содержащихся в
воде солен и может явиться причиной ошибочных результатов. Целесообразно
поэтому вместо дистиллированной воды использовать для контрольной пробы
испытуемую воду, предварительно продегазированную активированным углем.
К 5 мл зараженной воды добавляют набранное на кончик шпателя достаточное
количество порошкообразного активированного угля. После многократного встря-
хивания уголь отфильтровывают и фильтрат нейтрализуют.
Обнаружение ингибитора. На предметное стекло пипеткой наносят 2 капли
по 0,05 мл раствора фермента. Левую каплю смешивают с 0,05 мл испытуемой
воды, а правую — с 0,05 мл чистой " воды, приготовленной для контрольной про-
бы. Перемешивание осуществляют двумя тонкими стеклянными палочками,
следя за тем, чтобы всегда употреблялась соответствующая палочка. После инку-
бирования (5 мин для зарина, зомана, табуиа, ДФФ и 15 мин — для более
медленно реагирующих ингибиторов) на оба участка накладывают кусочки инди-
каторной бумаги и покрывают покровным стеклом.' В то время как индикаторная
бумага па правой капле (контрольная проба) постепенно изменяет свою окраску
от сине-зеленой через зеленую, лимонно-желтую до светло-желтой, бумага иа ле-
вой капле при наличии фосфорорганических ингибиторов остается сине-зеленой
или изменяет свой цвет значительно медленнее.
Полуколичественное определение ингибитора. Поступают так же, как при
качественном обнаружении, с той только разницей, что посередине между кап-
178
лей испытуемой воды и контрольной пробой дополнительно помещают 0,1 мл
буфера сравнения и по истечении инкубации Это пятно так же накрывают инди-
каторной бумагой. При этом одновременно накрывают каплю испытуемой воды
и контрольной пробы, а затем каплю буфера сравнения и отмечают продолжи-
тельность выравнивания окраски контроля и пробы с буфером. По полученным
данным рассчитывают угнетение по следующей формуле:
100—% угнетения
•п
где tK — продолжительность выравнивания окраски контрольной пробы; /п —
продолжительность выравнивания окраски испытуемой воды.
Для отдельных ингибиторов строят калибровочную кривую зависимости
процента угнетения для определенных концентраций -от отрицательного лога-
рифма концентрации. При точном соблюдении условий средняя область кривой
(20—80% угнетения) позволяет получать достаточно точные результаты. Пре-
имуществом этого метода (качественного и полуколичественного) является то,
что получаемые результаты почти ие зависят от температуры; обычно изменяет-
ся только продолжительность определения, что связано с зависимостью расщеп-
ления субстрата от температуры. Чувствительность метода составляет для зари-
на и зомана 1 • 10~® мг/мл, для табуна — 5 10"® мг/мл.
Средняя часть калибровочной кривой (20—80% угнетения) охватывает об-
ласть концентраций фосфорорганических ингибиторов указанного выше порядка.
При большем содержании ОВ исследуемые образцы приходится дополнительно
разбавлять. Так, если индикаторная бумага не изменяет своей окраски или на
это требуется 30—40 мин, то образец следует последовательно разбавлять чи-
стой водой каждый раз в отношении 1 : 10 и проводить определение для каждо-
го разбавления.
Проведение расчетов покажем на примере определения концентрации зарина
в образце воды. При определении, проведенном с неразбавленным образцом
воды, индикаторная бумага не изменила свою окраску. После первого десяти-
кратного разбавления испытуемой воды также нельзя было установить время
выравнивания окраски, после второго разбавления — время выравнивания окра-
ски составляло 10 мин. Время выравнивания окраски контрольной пробы равня-
лось 4 мин. Подставив эти данные в расчетную формулу, получаем:
100—* = 60 % угнетения
По калибровочной кривой, построенной Для зарина, этому угнетению соот-
ветствует значение pl (отрицательный логарифм концентрации зарина в мг/мл),
равный 5,2. Чтобы определить содержание зарина в исследуемой воде, проводят
следующий пересчет.
Концентрация зарина равна 10_pI или 10-5'2; 1g концентрации зарина равен
0,8^- 6,0, откуда концентрация зарина равна 6,3 10~6 мг/мл.
Так как проба воды была разбавлена в отношении 1 : 100, то содержание
зарина в пробе составляет 6,3 • 10*® • 100, т. е. 6,3 > 10-4 мг/мл или в пересчете
на 1 л — 0,63 мг/л.
6.3.3.2. Фотометрическое определение с индофенилацетатом в качестве суб-
страта 125,12в.
Реагенты
Раствор фермента — раствор лиофилизованной сыворотки крови лошади в
0,05 М растворе фосфатного буфера (pH 8,0); активность — 40 холинэстеразных
единиц в 1 мл (по Аммону).
Раствор субстрата (3,3-10~3Л1 раствор) 124 — раствор 0,08 г цндофенилаце-
тата (т. пл. после перекристаллизации из петролейного эфира 115—116 °C) в
100 мл абсолютного спирта.
Фосфатный буфер (по Кларку и Лабсу, pH 8,0) —смесь 46,8 мл 0,1 н. рас-
твора NaOH и 50 мл 0,1 н. раствора КН2РО4, доведенная водой до объема
100 мл.
179
В пробирке с притертой пробкой смешивают 1 мл водного раствора ОВ с
4 мл раствора фермента и инкубируют в термостате при 30 °C в случае зарина,
зомана, табуна, ТЭПФ 10 мин, в случае фосфорилтиохолинов или инсектицидов
типа эфиров фосфорной кислоты — 30 мин. По истечении инкубации в пробирку
добавляют 0,15 мл раствора субстрата и оставляют в термостате еще на 30 мин,
затем ферментативную реакцию останавливают прибавлением капли раствора
ДФФ (5 мг!мл) и измеряют экстинкцию сразу или по истечении точно 30 мин.
Измерение проводят в спектрофотометре при 625 нм по сравнению с контроль-
ной пробой, выполняемой параллельно с основной и содержащей 1 мл воды и
4 мл фосфатного буфера. Расчет концентрации ингибитора в пробе осущест-
вляют по калибровочной кривой, построенной на чистом веществе и представ-
ляющей собой функцию экстинкции от концентрации.
В этом способе можно использовать эстеразу эритроцитов (АХЭ), которая
более удобна для определения некоторых фосфорорганических ингибиторов;
расщепление субстрата осуществляют примерно с одинаковой скоростью.
6.4. БИОХИМИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ФОСФОРОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ ПРИ ПОМОЩИ ДРУГИХ ФЕРМЕНТОВ
Угнетение стеапсинлипазы зарином и систоксом дает возможность
флуорометрически определить эти ингибиторы в присутствии ди-
бутирилфлуоресцеина в качестве субстрата 128. Определение осуще-
ствляют с концентрацией субстрата 5 - 10-5 М (pH 8,0); при вре-
мени инкубации 2 мин можно определить от 3 • 10~5 до 3 • 10-4
мг!мл ингибитора.
Некоторые ферменты, расщепляющие субстрат с выделением
газообразных продуктов, угнетаются фосфорорганическими инги-
биторами. При этом мерой концентрации ингибитора служит
уменьшение выделяющегося в единицу времени количества газа,
измерить который можно с достаточной чувствительностью в бро-
дильном фильтре с добавкой пенообразователя. Для таких опре-
делений удобны — каталаза с перекисью водорода в Качестве суб-
страта, уреаза с мочевиной и карбоксилаза с пировиноградной кис-
лотой в качестве субстратов ш.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какими преимуществами обладают биохимические методы обнаружения
и количественного определения ОВ?
2. Какие вы знаете типы эстераз, расщепляющих эфиры холина и каковы
их основные отличительные признаки?
3. Какое значение имеет определение активности холинэстеразы сыворотки
крови человека?
4. На чем основано исключительно сильное угнетающее действие фосфорил-
холиновых и фосфорилтиохолиновых соединений на холинэстеразы?
5. Почему при определении ингибиторов сначала инкубируют фермент с
ингибитором, а потом прибавляют субстрат?
6. Что означают показатели Ко и рКо?
7. Как определяют pho ингибитора?
8. В каких единицах можно выражать активность холинэстераз?
9. На чем основаны важнейшие способы определения активности холин-
эстеразы?
10. На каком принципе основано биохимическое определение фосфороргани-
ческих ОВ?
180
11. Почему перед биохимическим определением паратиои должен быть об-
работан бромной водой?
12. Как осуществляется полуколичественное определение ингибиторов хо-
линэстеразы при помощи субстрат-индикаторной бумаги?
ЛИТЕРАТУРА
1. Р i 1 z W„ Z. ges. exp. Med., 132, 310 (1959).
2. Holmstedt В., Pharm. Rev., 11, 567 (1959).
3. Cohen J. A. et al., Biochim. Biophys. Acta, 20, 402 (1956).
4. К r u p k a R. M., L a i d 1 e r K. J-, Nature, 190, 916 (1961).
5. T a m m e 1 i n L. E., Ark. Kemi, 12, 287 (1958).
6. T a m m e 1 i n L. E., Acta Chem. Scand., 11, 1340 (1957).
7. Gomori G., Amer. J. Clin. Path., 24, 99 (1954).
8. Grob D. et al., Bull. John Hopkins Hosp., 81, 217 (1947).
9. R i c h t e r D., С г о f t P. G., Biochem. J., 36, 746 (1942).
10. M ich e 1 H. 0., J. Lab. Clin. Med., 34, 1564 (1949).
11. Ammon R., Voss G., Pflugers’ Arch. ges. Physiol., 235, 393 (1935).
12. Ammon R., Z a p p F. J., Klin. Wchschr., 33, 759 (1955).
13. H u e r g a J. et al., Amer. J. Clin. Path., 22, 1126 (1952).
14. H e i n e c k e r R., Mayer I., Klin. Wschr., 35, 340 (1957).
15. Ammon R„ Pfliigers’ Arch. ges. Physiol., 239, 486 (1939).
16. A u gu s t i n s s о n К. B., Heimbiirger G., Acta Physiol. Scand., 30, 45
(1953).
17. Wirth W., Arch, exptl. Pathol. Pharmakoi., 207, 545 (1949).
18. Boulliat G., Duperray J.-N., Arch. Malad. prof. Med., 25, 589 (1964).
19. Larsson L., Hansen B., Svensk Kem. Tidskr., 68, 521 (1956).
20. Jorgensen K., Scand. J. Clin. Lab. Invest., 11, 282 (1959).
21. Jensen-Holm J. et al., Acta Pharmacol., 15, 384 (1959).
22. Michel O. H„ J. Lab. Clin. Med., 34, 1564 (1949).
23. T a m m e 1 i n L. E., Scand. J. Clin. Lab. Invest., 5, 267 (1953).
24. T a m m e 1 i n L. E., Strindberg B., Acta Chem. Scand., 6, 1041 (1952).
25. Дмитриева H. В, Лаб. дело, 9, 34 (1963).
26. M e i n e с k e К. H., О e 11 e 1 H., Arch. Toxikol., 21, 321 (1966).
27. Adie P. A., T u b a J., Biochim. Biophys. Acta, 50, 70 (1961).
28. Stedman E., EassonL. H., Biochem. J., 26, 2056 (1932).
29. Glick D„ Biochem. J., 31, 521 (1937).
30. G r e en b e r g S., Calvert C., Med. Techn. Bull., 8, 59 (1957).
31. Me 1 ich a r B., Pracovni Lekarstvi, 13, 355 (1961).
32. F 1 e i s h e r J. H. et aL, Anal. Chem., 27, 1080 (1955).
33. F 1 e i s h e r J. H. et al., Arch. Ind. Health, 14, 510 (1956).
34. Ackermann H., Dtsch. Ges. Wesen, 19, 1213 (1964).
35. Gerarde H. W. et al., J. Occupat. Med., 7, 303 (1965).
36. Takahashi T. H., Shibata S., Chem. Abstr., 46, 9647 (1952)
37. Methods of biochemical Analysis., V. 5., Intersc. Publ. Corp., 1957. P, 29.
38. Herzfeld E., Stumpf Ch., Wiener klin. Wchschr., 67, 874 (1955).
39. Sailer S., Braunsteiner H., Klin. Wchschr., 37, 986 (1959).
40. R i c h t e r i c h R., Schweiz, med. Wschr., 92, 263 (1962).
41. Brzozowski J. et aL, Pol. Tyg. Lek., 19, 1607 (1964).
42. Поставщик Osterreich. Stickstoffwerke AG, Linz.
43. Поставщик К. H. Kallies KG, Sebnitz (Sachsen).
44. Matousek J., German J., Pracovni Lekarstvi, 16, 13 (1964).
45. H e s t r i n S., J. Biol. Chem., 180, 249 (1949).
46. P ilz W„ Klin. Wchschr., 36, 1017 (1958).
47. P i 1 z W., Z. anal. Chem., 162, 81 (1958).
48. P i 1 z W. et al., Klin. Wchschr., 43, 1227 (1965).
49. Vincent D., Segonzac G., Clin. Chim. Acta, 3, 104 (1958).
50. Fleisher J. H., Pope E. J., Ind. Hyg. Occupat. Med., 9, 323 (1954).
51. M e 1 c h i о г г i P., Maffei F., Arch. ital. Sci. farmacol., 13, 217 (1963).
181
52. Эдельман М. М., Лаб. дело, 9, 29 (1963).
53. Wetstone Н. J., Bowers G. N., Stand Methods Clin. Chem 4, 47
(1963).
54. H a n к e r J. S. et al., J. Am. Pharm. Assoc., 157, 728 (1958).
55. T a b a c h n i к I. A., Biochim. Biophys. Acta, 21, 580 (1956).
56. G a I E. M., R о t h E., Clin. Chim. Acta, 2, 316 (1957).
57. McOsker D. E., Daniel L. J., Arch. Biochem. Biophys., 79, 1 (1959).
58. E 11 m a n n G. L. et al., Biochem. Pharmakoi., 7, 88 (1961).
59. Carry P. J., Routh J. I., Clin. Chem., 11, 91 (1965).
60. Koelle G. B., Friedenwald S., Proc. Soc. Exp. Biol., 70, 617 (1949).
61. G e r e b t z о f f M. A., Acta Anatom., 19, 366 (1953).
62. К г a m e r D. W. et al., Anal. Chem., 34, 842 (1962).
63. G u i 1 b a u t G. G. et al., Anal. Chem., 34, 842 (1962).
64. G u i 1 b a ti t G. G. et al., Anal. Biochem., 5, 208 (1963).
65. G u i 1 b a u t G. G., Bacter. Reviews, 30, 94 (1966).
66. Fischerova-Bergerova V., Coll. Czech. Chem. Comm., 28, 3311
(1963).
67. R i d gr а у T. H., M а г к H. В., Anal. Biochem., 12, 357 (1965).
68 Fischerova-Bergerova V., Pracovni Lekarstvi, 16, 8 (1964).
69. H о A. K. S., Paddle В. M., Freeman S. E., Biochem. Pharmakoi, 14,
151 (1964).
70. Barnet R. J., Seligman A. M., Science, 114, 579 (1951).
71. HoltS. J., J. Histochem. Cytochem., 5, 541 (1957).
72. Pepler W. J., Pearse A. G. E., J. Neurochem., 1, 193 (1957).
73. Holt S. J., Sadler P. W., Proc. Roy. Soc., 148, 495 (1958).
74. А с к e r m a n G. A., Lab. Invest., 9, 298 (1960).
75. Lehrer G. M., Siegel G. L, J. Histochem. Cytochem., 13, 8 (1965).
76. C h m e 1 a г о v a M. et al., Coll. Czech. Chem. Comm., 31, 1886 (1966).
77. Ravin H. A. et al., J. Pharmacol. Exp. Therap. 107, 37 (1953).
78. Nachlas M. M„ Seligman A. M., J. Nat. Cancer Inst., 9, 415 (1949)
79. D a v i s B. J., Proc. Soc. Exp. Biol., 101, 90 (1959).
80. Kramer D. N., Gamson R. M., Anal. Chem., 30, 251 (1958).
81. Яковлев В. А., Фруентова В. А., Биохимия. 28, 850 (1963).
82. Sala f ski B., Arch. Int. Pharmakodyn. Therap., 154, 184 (1965).
83. Huggins C., L a p i d e s J., J. Biol. Chem., 170, 467 (1947).
84. Aldridge W. N., Biochem. J., 53, 110 (1953); 57, 692 (1954).
85. Main A. R., Miles К- E., Braid P. E., Biochem. J., 78, 769 (1961)
86. S mi th R. L. et al., Clin. Chim. Acta, 4, 384 (1959).
87. R a v i n A. H. et al., J. Biol. Chem., 191, 843 (1951).
88. V a h 1 q u i s t B., Scand. Arch. Physiol., 71, 133 (1935).
89. Goldstein N. P. et al., J. Lab. Clin. Med., 33, 1047 (1948).
90. V i 11 e 1 a G. G., M e 11 о M. I., Hospital, 36, 177 (1949).
91. H a 11 G. E., L u c a s С. C., J. Pharmacol. Exp. Then, 59, 34 (1937).
92. H о f s t e e В. H. J., Science, 114, 128 (1951).
93. Hazard R, Presse med., 56, 529 (1948).
94. G omori G., J. Lab. Clin. Med., 34, 275 (1949).
95. R i d e r J. A. et al., Proc. Soc. Exp. Biol., 76, 427 (1951).
96. G la n g P. A., H a 11 S. A., Anal. Chem., 23, 1830 (1951).
97. Tabachnik J. A. et al., Arch. Int. Pharmacodyn., 114, 351 (1958).
98. Yip G., Cook J. W., J. Assoc. Off. Agric. Chem., 42, 194 (1959).
99. S t re 1 i t z F., Biochem. J., 38, 86 (1944).
100. Jansz H. S„ Cohen J. A., Biochim. Biophys. Acta, 56, 531 (1962).
101. Cook J. W., J. Assoc. Oil. Agric. Chem., 37, 984, 987 (1954).
102. P a t c h e 11 G. G., В a t c h e i d e r G. H„ J. Agric. Food Chem., 8, 54 (1960).
103. Покровский А. А., Вопросы мед. хим., 4, 292 (1958).
104. Покровский А. А., Пономарева Л. Г., Гигиена и санитария, 29,
53 (1964).
105. Fournier R. М., Mem. Poudres, 40, 403 (1958).
106. Z u г 1 о N. et al., Med. lavoro, 45, 533 (1954).
107. В о у d G. R., J. Agric. Food Chem., 7, 615 (1959).
182
4
108. Deg re lie H., Tillement J. P., Ann. Pharm. franc., 22, 511 (1964).
109. Archer T. E. et al., J. Agric. Food Chem., 11, 471 (1963).
110. van Oudheusden A. P. M., Pharmac. Weekbl., 97, 606 (1962).
111. Пат. ГДР 34318 (1961).
112. Sandi E., Wight J., Chem. Ind., 30, 1161 (1961); Sandi E., Nahrung,
6, 57 (1962).
113. Nesheim E. D., Cook J. W., J. Assoc. Off. Agric. Chem., 42, 187 (1959).
114. В 1 u m a n N., J. Assoc. Off. Agric. Chem., 47, 272 (1964).
115. Guilbaut G. G. et al., Anal. Chem., 34, 1437 (1962).
116. В a u m a n E. K. et al., Anal. Chem., 37, 1378 (1965).
117. Hicks G. P., Up dike S. J, Anal. Chem., 38, 726 (1966).
118. Guilbaut G. G., Kramer D. N., Anal. Chem., 37, 1675 (1965).
119. Anal. Chem., 37, 120 (1965).
120. Kramer D. N., Guilbaut G. G., Anal. Chem., 36, 1662 (1964).
121. E p s t e i n J. et al., Anal. Chem., 29, 1050 (1957).
122. HansenB., Acta Chem. Scand., 11,537 (1957).
123. Gillis R. G„ Chem. Ind., 1957, 111.
124. Heller G„ Ann., 392, 16 (1912).
125. Archer T. E., Zweig G„ J. Agric. Food. Chem., 7, 178 (1959).
126. К a w a i M., Nat. Def. Med. J. Japan, 11, 194 (1964).
127. Zacks S. I., Blumberg J. M., Military Medizine, 129, 1084 (1964).
128. Guilbaut G. G„ Kramer D. N„ Anal. Chem., 36, 409 (1964).
129. Пат. ФРГ 1126650 (1955).
7. БИОЛОГИЧЕСКИЕ МЕТОДЫ
7.1. ОПРЕДЕЛЕНИЕ ЛД И ЛК
В последние годы для характеристики и сравнения токсичности
стали приводить данные о смертельных (летальных) дозах или
смертельных концентрациях, которые оказались более точными
характеристиками, чем ранее употреблявшееся произведение кон-
центрации на время (с/)- В анализах ОВ эти величины могут до-
полнять результаты определений химическими методами.
Под средней смертельной дозой ЛД50 понимают дозу ОВ, вы-
зывающую гибель (смертность*) 50% подопытных животных.
Соответственно этому смертельной дозе ЛДю0 отвечает 100%-ная
смертность. Наряду с этим, но реже, употребляется также понятие
средней эффективной дозы — ЭД50, т. е. дозы, вызывающей опреде-
ленный физиологический эффект у 50% подопытных животных.
При экспериментальном определении ЛД50 в гражданских ток-
сикологических исследованиях используют по возможности группы
с одинаковым числом подопытных животных (не менее 4—6 еди-
ниц), которым одинаковым способом вводят ОВ в дозах, возра-
стающих примерно на 10—20%. Затем в каждой группе опреде-
ляют число павших животных, на основании этого рассчитывают
среднюю смертельную дозу 15 по следующей формуле:
лд50 = дт-У-^-
m
где Дт — смертельная доза, при которой пали все животные (при расчете эф-
фективной дозы ЭД — это доза, при которой у всех животных проявился опре-
деленный физиологический эффект); г—половина суммы павших животных в
двух группах или при двух дозах, следующих друг за другом; d — разница
между двумя дозами, вводимыми следующим друг за другом группам; m —
число животных в каждой группе.
Значение г и d определяют по возможности для следующих
друг за другом групп, начиная с группы, в которой пало одно
животное, и кончая группой, где пали все животные. Эти величины
перемножают и используют произведение для расчета ЛДзо (см.
выше).
ЛД50 можно также определять графически или расчетным пу-
тем, при этом одновременно могут быть определены ошибки и до-
верительный интервал 16. Аналогичным путем определяют средние
смертельные концентрации ЛКэо и эффективно действующие кон-
центрации ЭКбо- Определяемая величина зависит от типа ОВ, вида
подопытного животного и возможного вида аппликации. В случае
* В данном случае смертность — это отношение числа павших животных к
общему числу подопытных животных.
184
кожно-нарывных или нервно-паралитических ОВ преимущественно
определяют значения ЛД50 или ЛДюо, а для раздражающих ОВ и
веществ, действующих соответственно их физическим свойствам
только как ингаляционные яды (фосген, синильная кислота), — оп-
ределяют ЛК50 и ЛКюо- Смертельную концентрацию устанавливают
и тогда, когда в качестве подопытных биологических объектов ис-
пользуются черви или насекомые (например, проба на мухах И7) и
когда никакие другие виды аппликации невозможны. В опытах с
млекопитающими можно вводить ОВ разными способами. Чаще
применяются следующие способы: через рот (р. о)., подкожно (s. с.),
внутримышечно (i. пт), внутрибрюшинно (i. р.), внутривенно (i.v.),
а также через кожу — в результате кожной резорбции.
Летальные дозы ЛД выражают в миллиграммах на килограмм
живой массы. Эти величины имеют смысл только при использова-
нии подопытных животных одной и той же линии, одинаковых по
возрасту, массе и полу. В дополнение к этим данным указывают
также экспозицию, приводящую к смерти 50 или 100% животных.
Приведение экспозиции совершенно необходимо при указании ЛК.
Кроме того, отмечают также объем легочной вентиляции в единицу
времени и частоту дыхания. Приводимые в литературе величины
ЛД и ЛК можно сравнивать только в том случае, если одновремен-
но приведены и указанные выше данные.
7.2. ИСПОЛЬЗОВАНИЕ ЖИВОТНЫХ ДЛЯ ИНДИКАЦИИ ОВ
Подобно тому, как во время первой мировой войны для индикации
использовали субъективное восприятие человеком определенных
ОВ, для этой же цели изучали или использовали высокую чувстви-
тельность органов чувств и физиологическую реакцию некоторых
животных. Чрезвычайно высокой чувствительностью обладает ор-
ган обоняния собак, которые, например, в состоянии воспринимать
пары серного иприта в концентрациях ниже 0,1 мкг/л. Удалось
также, используя обоняние собак и их реакцию, обучить их диффе-
ренцированно выделять запахи ОВ из разных других запахов '~3.
Повышенной чувствительностью по отношению к фосгену обладают
кошки. Еще более чувствительны к малейшим следам фосгена
куры; вообще можно утверждать, что птицы наиболее чувствитель-
ны к присутствующим в атмосфере ОВ. Канарейки могут быть ис-
пользованы в качестве живых индикаторов присутствия в воздухе
синильной кислоты и окиси углерода. Во время первой мировой
войны в американских войсках использовали улиток, которые в
присутствии незначительных концентрацйй иприта в воздухе выде-
ляют молочный секрет.
Несмотря на то, что применение животных для индикации ОВ
связано со многими недостатками, обусловленными либо не всегда
правильным пониманием отклонений от нормального поведения
животных либо трудностями их оценки, этот метод индикации изу-
чается и в настоящее время. Как явствует из литературы по общей
185
токсикологии и иностранной военно-химической литературы, этот
метод пригоден для обнаружения фосфорсодержащих ОВ и инсек-
тицидов в воде и продуктах питания. Наряду с использованием
животных (мышей) для установления токсичности тех или иных
соединений, о чем сообщалось в разделе 7.1, животные исполь-
зуются также для установления факта отравления продуктов пи-
тания путем скармливания им этих продуктов или инъекции экс-
трактов из них. Некоторые породы декоративных рыб, обладаю-
щие явновыраженной высокой чувствительностью по отношению к
Рис. 9. Установка для контроля качества воды
с использованием рыб4:
/—резервуары; 2—датчик температуры; з—дозатор хлора.
фосфорорганическим ОВ и инсектицидам, используют для индика-
ции этих ядов в питьевой воде. Испытания проводят в соответ-
ствующей установке (рис. 9), дающей возможность постоянно кон-
тролировать воду в системе водоснабжения4-7’18. В табл. 11 при-
ведены ЛК50 для зарина, установленные для разного типа рыб при
различных экспозициях.
Так как рыбы в воде с незначительной жесткостью особенно
чувствительны к действию хлора, его восстанавливают добавле-
нием тиосульфата натрия из расчета 7 мг Na2S2O3 на 1 мг хлора.
Температуру поддерживают в интервале 20—28 °C, содержание
кислорода должно быть выше 4 мг!л, а pH между 5 и 9. Наибо-
лее удобно работать с рыбами породы Pimephales promelas, реак-
ция которых на незначительные концентрации яда весьма харак-
терна (выпячивание вперед грудных плавников, растопыривание
жаберных створок). При концентрации зарина 0,5 мг!л и экспо-
зиции 6 мин у 50% этих рыб наблюдается потеря равновесия, а че-
рез 12 мин наступает смерть. С помощью Lepomis macrochirus при
более длительной экспозиции (30 суток) удается обнаружить кон-
центрацию паратиона (тиофоса), равную 0,1 мкг[л, Гуппии можно
186
Таблица 11. JlKso зарлна для разного типа рыб при разных экспозициях
Порода рыб Вода ЛК50 (мкг/л) при экспозиции
24 ч 48 ч 96 ч
Pimephales promelas Мягкая 6,5 5,3 4,4
Жесткая 32,1 31,9 31,9
Lepomis cyanellus Мягкая 4,6 4,2 4,2
Жесткая 15,2 15,2 15,2
Lebistes reticulalus (гуппии) Мягкая 8,3 7,2 7,2
Жесткая 21,0 14,5 13,8
Lepomis macrochirus Мягкая 7,5 3,2 3,2
Жесткая 23,5 23,5 23,5
Carassus auratus (золотые рыбки) Мягкая 16,1 11,8 9,8
использовать 8 для обнаружения следов растворенного в воде сер-
ного иприта (5 мг/л).
Для обнаружения и определения остаточных количеств фосфор-
содержащих инсектицидов в растениях могут быть использованы
мухи9~и’19~21, кровяные пиявки12’13 и водяные блохи21. Для этого
мух помещают в плоские прикрытые стеклянные чашки, где они со-
прикасаются с экстрактом, полученным из испытуемого материала;
пиявок помещают в водную вытяжку. Чувствительность, достигае-
мая в этих определениях, очень высока. Так как получаемые ре-
зультаты зависят от условий, последние должны строго соблю-
даться и фиксироваться. Так, например, можно обнаружить уже
5 мкг паратиона (после предварительного окисления) по гибели
пиявок в 50 мл водной вытяжки при экспозиции 24 ч. Интерес
представляет обнаружение токсических фосфорорганических сое-
динений на бумажных хроматограммах посредством контакта ли-
чинок комаров с отдельными участками хроматограммы, разрезан-
ной на равные части14.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Что такое средняя смертельная доза ЛД50 н средняя смертельная кон-
центрация JlKso?
ЛИ ТЕРА ТУРА
1. Northrop J. Н., J. Gen. Physiol., 30, 475 (1947).
2. R i с h t е г s С. Е„ Berliner Tierarztl. Wschschr., 1933, 477.
3. Richters С. E., Die Tiere im chemischen Kriege, Berlin, 1932.
4. Weiss Ch. M., Botts J. L., Sewage Ind. Wastes, 29, 810 (1957).
5. A m m о n R., Bockendahl H., Arzneimiffel-Froschg., 12. 812 (1962)
6. Weiss Ch. M., G a k s t a 11 e r J. H., J. Water Pollut. Control. Fed., 36, 240
(1964).
7. Weiss Ch. M., J. Water Pollut. Control Fed., 37, 647 (1965).
8. Ц и т о в и ч И. С. и др., Фарм. и токсикол.. 7, 44 (1944).
9. К 1 i m m е г О. R., Р f a 11 W., Arzneimittel-Forschg., 5, 3 (1955).
187
10. Hoffmann R. A., Cohen N. W., J. Econ. Entom., 47, 701 (1954).
11. McCaulley D. F., Cook J. W., J. Assoc. Off. Agric. Chem., 42, 200 (1959).
12. S trailer R., Dissertation, Naturw. Fakultat Univ. Erlangen, 1957.
13. D a n n 0., S t r a 11 e r R., Arch. Pharmaz., 292, 723 (1959).
14. T a p i о S., Suomen Kem., 35, A6 (1962).
15. Behrens, К a r b e r, Arch. Exper. Path. Pharmak., 177, 379 (1935).
16. van der Waerden, Arch. Exper. Path. Pharmak., 195, 389 (1940).
17. Henderson C., Pickering H., Trans. Amer. Fish. Soc., 87, 39 (1957)-
18. T i gh e J. F., J. Assoc. Off. Agric. Chem., 43, 82 (1960).
19. Sanjean J. et al., J. Assoc. Off. Agric. Chem., 44, 163 (1961).
20. Sun Y. P., Johnson E. R., J. Assoc. Off. Agric. Chem., 46, 524 (1963).
21. Di em a i r W., К n о p f K., Z. Ernahrungswiss., 3, 63 (1963).
8. ФИЗИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ
МЕТОДЫ
Определение физических констант используют для идентификации
соединений при анализе неизвестных веществ, в лабораторных
исследованиях известных веществ оно служит контролем чистоты
соединений, а в синтетических работах — способом установления
идентичности полученных продуктов. Для твердых веществ опреде-
ляют температуру плавления, для жидкостей — температуру ки-
пения, плотность, давление пара и показатель преломления. Перед
определением констант вещество подвергают тщательной очистке
путем перекристаллизации, сублимации, перегонки или методами
препаративной хроматографии. Спектральные исследования, осо-
бенно в области ИК-спектров, предоставляют более широкие воз-
можности для характеристики уже известных и идентификации не-
известных веществ. ИК-спектр чистого вещества по своей специфич-
ности сравним со значением отпечатка пальца в криминалистике.
Несмотря на то, что физические методы уступают по точности
химическим методам анализа, они имеют значительные преимуще-
ства, заключающиеся в относительном сокращении времени ана-
лиза, уменьшении ручных операций и большей чувствительности.
Данные табл, 12, заимствованные из работы Кинитца, дают воз-
можность сравнить некоторые химические и физические методы,
применяемые для количественного анализа ,60.
8.1. ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ
Для определения плотности ОВ удобны пикнометры. Определив
массу пикнометра с веществом (Д), массу пикнометра с водой
(Б) и массу пустого пикнометра (В), рассчитывают плотность (d)
по следующей формуле:
. А —В
d==~B=B
Обычные пикнометры обладают тем существенным недостат-
ком, что при погружении капилляра, как правило, наружу через
шлиф вытесняется некоторое количество жидкости. Это делает
их малопригодными для работы с ОВ. Значительно более удобны
пикнометры, предложенные Шпренгелем — Оствальдом (рис. 10).
Жидкость засасывают через отверстие А (при помощи резино-
вой груши или медицинским шприцем с резиновым шлангом),
189
Таблица 12. Сравнительная оценка некоторых химических и физических методов анализа
О Что подлежит определению Метод Количество вещества Р, мг Границы опреде- ления Е *, мг Эффектив- ность метода 3* Р/Е Точность результатов, относ. % Продс (без подго ручные операции, мин >лжительно товительны расчеты, мин :ть анализа х мероприятий) общая продолжи- тельность анализа, мин
Анализ неорганических соединений
Элементы, катио- ны, анионы Весовой макроопределе- ние микроопределе- нне 100—500 3-10 0,1 0,002 103 ю3—ю4 0,2-0,5 0,5 20—120 30—120 2-5 2-5 60—300 60—300
То же Объемный макроопределе- ние 20-500 0,1 103-104 0,2-0,5 2-20 2-5 5—25
мнкроопределе- ние 3-10 0,001 103-104 0,2—0,5 2-20 2-5 5—25
> Колориметрия 0,1-1 0,0001 ю3—ю4 1-2 2-5 2-5 5-10
Анализ органических соединений
Функциональные группы, молеку- Общий анализ макроопределе- 200—1000 0,1 2- 103—104 0,2—0,5 10-240 5-10 15-250
лы ние
микроопределе- ние 3—10 0,002 103—104 0,2—0,5 5-150 5—10 10-160
С, Н, N, О, S, С1, неорганические соединения Элементы, катио- ны, функцио- нальные группы Элементный анализ, микрометод Капельный анализ органический и не- органический 3-5 0,01—0,1 0,01 0,00001 103 108—104 0,3-1,0 качественно 20-40 1-2 2-5 25-45 1-2
Ф и з и ч е ские методы анализа
Молекулы Газовая хроматогра- фия 0,5-10 0,0001 10s—106 1-3 5-60 5-15 10-75
Функциональные группы, молеку- лы ИК-спектроскопия 0,5-5 0,005 102-103 1—2 5-10 10-30 15-45
Элементы Пламенная фотомет- рия 1-10 0,0005 10s—ю4 1-2 1-2 2-5 3-10
То же Эмиссиониаи спек- трометрия 0,1-1,0 0,00005 104—10s 2-3 1—2 2-10 3-15
» Рентгеио-флуорес- центный спектраль- ный анализ 100 0,01 ю4 2-5 1-2 2-10 3-15
Элементы, моле- кулы Масс-спектрометрня 0,1-10 0,0001 103-106 1-2 10-20 10-50 15-60
Элементы Нейтронный актива- ционный анализ 1-100 10“8 106—10s 5-10 10-60 15 25-1000
• Без очистки вещества.
* * Продолжительность облучения 10—1000 мин.
на отростке В имеется метка, до которой нужно доводить объем
жидкости. Избыток жидкости удаляют тампоном из целлюлозы.
Рис. 11. Самонаполняющийся
пикнометр Хенниона124.
Рис. 10. Пикнометр Шпренге-
ля — Оствальда.
Очень удобен для работы с токсичными жидкостями самонапол-
няющийся пикнометр Хенниона 124, который легко может быть из-
готовлен в лаборатории из обычной пипетки соответствующего раз-
мера (рис. 11). Отросток капилляра А погружают в жидкость, и
после того как пикнометр заполнится до кали-
бровочной метки В, отверстие С закрывают
пальцем, удаляют остатки вещества с поверх-
ности отростка А и взвешивают пикнометр.
8.2. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ
ПЛАВЛЕНИЯ
Для определения температуры плавления ис-
пытуемое вещество помещают в запаянный с
одного конца стеклянный капилляр, опускают
в аппарат Тиле (рис. 12) и нагревают в бане
с серной кислотой или, в случае ОВ, в более
удобном для этой цели медном или алюминие-
вом блоке. Наполнение капилляра проводят
на стеклянной пластинке, которую затем де-
газируют.
Для быстрого определения температуры
Рис. 12. Стеклянный ПЛавления ОВ, разлагающихся при длительном
ления температуры нагревании, очень удобна нагревательная баня
плавления по Тнле. Кофлера. Она представляет собой металличе-
ское тело шириной 4 см и длиной 37 см, в ко-
тором вследствие одностороннего электрического нагрева возникает
температурный перепад с градиентом, близким к линейному. Веще-
ство насыпают непосредственно на хромированную поверхность на-
гревательного элемента. В зависимости от чистоты вещества тотчас
192
обнаруживается более или менее резкая граница между жидкой и
твердой фазами.
Определение температуры плавления в капиллярах может быть
выполнено очень точно. Для этого определяют в предварительном
опыте приблизительную температуру плавления, а затем при вто-
ром определении капилляр помещают в прибор после того, как
последний будет нагрет на несколько градусов ниже температуры,
установленной в предварительном определении, и вновь определяют
температуру плавления.
Если испытуемое вещество имеется в очень ограниченном коли-
честве, то температуру плавления можно определять под микро-
скопом в микронагревателе, для чего требуется всего несколько
кристаллов вещества. Метод дает возможность обнаруживать при-
меси значительно лучше, чем в капилляре, и наблюдать за обра-
зованием эвтектических смесей с основным веществом. Вещество
считается чистым, если после перекристаллизации его температура
плавления изменяется не больше чем на 1 °C.
Точным критерием идентичности вещестра является температура
плавления смеси испытуемого и известного чистого вещества с оди-
наковой температурой плавления. Для этого тщательно перемеши-
вают при растирании равные части испытуемого и известного веще-
ства и помещают в капилляр. Если температура плавления смеси
соответствует температуре плавления известного вещества, то это
означает, что испытуемое вещество идентично ему.
8.3. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ КИПЕНИЯ
Выбор метода определения зависит от имеющегося в распоряжении
количества исследуемого вещества. При исследовании ОВ в поле-
вых условиях количество вещества будет в общем незначительно, и
потребуется определять температуру кипения микрометодом. Для
этого хорошо подходит стеклянная трубка диаметром примерно
6 мм, прикрепляемая при помощи резинового кольца к термометру
(рис. 13). В трубку, содержащую некоторое количество испытуе-
мой жидкости, опускают запаянный с одного конца капилляр так
чтобы его открытый конец был погружен в испытуемую жидкость.
При нагревании на горелке или в бане по достижении температуры
кипения испытуемого вещества по стенке капилляра идет равномер-
ный ток пузырьков.
Иную возможность определения температуры кипения пред-
ставляют трубки, предложенные Эмихом (рис. 14). У капилляра
длиной примерно 8 см и внутренним диаметром 1 мм оттягивают
при помощи микрогорелки отросток длиной 2 см, через который
засасывают испытуемое вещество. Затем отросток запаивают так,
чтобы между запаянным концом и жидкостью остался небольшой
пузырек воздуха. Капилляр укрепляют на термометре и вносят в
прибор для определения температуры плавления. При достижении
температуры кипения воздушный пузырек поднимается до зеркала
жидкости бани (см. рис. 14); в этот момент отмечают температуру.
7 Зак. 677
193
Приведенные выше методы дают надежные результаты только
для чистых веществ, кипящих без разложения при нормальном дав-
лении. Для смесей веществ температуру кипения компонентов опре-
деляют в процессе фракционной перегонки.
Температуру кипения ОВ, которые при нормальном давлении
кипят при высокой температуре (>200°C) и с разложением, уста-
навливают путем перегонки в вакууме в соответствующей микро-
аппаратуре. Для оценки чистоты вещества достаточно установить,
что при постоянном давлении большая часть вещества перегоняется
Рис. 13. Прибор для
определения темпера-
туры кипения.
Рис. 14. Трубки Эмиха для
определения температуры ки-
пения.
в узком интервале температур. Установление точной температуры
кипения этого вещества при нормальном давлении осуществляется
перегонкой его при двух различных пониженных давлениях с по-
следующим расчетом температуры, основанном ga зависимости
давления пара от давления. Обзор различных способов расчета и
требуемые для этого константы можно найти в справочнике Крелля 8.
8.4. ОПРЕДЕЛЕНИЕ ДАВЛЕНИЯ НАСЫЩЕННОГО ПАРА
Различают два типа методов определения давления пара — стати-
ческий и динамический.
В статических методах вещество, подлежащее исследованию,
доводят до определенной, поддерживаемой постоянной, температу-
ры и измеряют устанавливающееся при термическом равновесии
между обеими фазами давление пара каким-либо прибором.
В динамических методах при помощи маностата поддерживают
постоянное давление и доводят испытуемое вещество до кипения.
Давление пара вещества при температуре кипения будет равно
установленному давлению.
К динамическим способам относится метод переноса, в котором
определяют количество газообразной фазы, образующейся при со-
ответствующей измеряемой температуре и находящейся в равнове-
194
сии с жидкой или твердой фазами исследуемого вещества. Через
исследуемое вещество пропускают ток инертного газа. При соот-
ветствующей малой скорости газового потока давление насыщен-
ного пара вещества становится равным его парциальному давлению
в смеси. Увлеченный газообразным потоком пар исследуемого ве-
щества выделяют из смеси конденсацией или вымораживанием и
определяют взвешиванием, либо его связывают каким-либо реаген-
том и после этого определяют соответствующим количественным
методом. Последний способ удобен для определения весьма низких
давлений пара, ниже 10~4 мм рт.ст. Такой способ был использован
для определения давления пара серного иприта 125.
Недостаточно точным, но вместе с тем простым и удовлетворяю-
щим многим целям является способ определения температуры ки-
пения по крайней мере при двух различных давлениях. Искомое
давление пара интерполируют по калибровочной кривой, построен-
ной как функция lg Р (мм рт. ст.) от обратной температуры 1/Т
(°К-1) для любого значения температуры.
В литературе описаны методы точного определения весьма
низких давлений пара, в которых используется сложная аппарату-
ра 66>67.
Рис. 15. Прибор для определения давления насыщенного
пара по Дрюсу:
/—термостат; 2—измерительный сосуд; 3—манометр; 4, 5—краны; 6 —
промежуточная емкость.
Весьма удобное приспособление, разработанное Дрюсом, дает
возможность определять давления пара жидкостей от 1 мм рт. ст.
при любой температуре. Для этого требуется немного исследуемого
вещества и ртути, а сам измерительный сосуд так дешев в изготов-
лении, что его можно использовать однократно. Способ может быть
осуществлен в любой лаборатории, имеющей масляный вакуум-
насос. На рис. 15 показано устройство установки.
При проведении определения измерительный сосуд 2 находится в термо-
стате 1. Как это показано на рисунке, он заполнен ртутью. После включения его
всю систему эвакуируют. После того как уровень ртути в запаянном колене из-
мерительного сосуда слегка опустится, впускают через трехходовой кран 5 воздух.
Осторожным опрокидыванием сосуда 2 из него удаляют возникший над ртутью
Воздушный пузырек н вводят в него несколько капель исследуемого вещества.
7* 195
Опрокидыванием переводят его в запаянное колено — над ртутью. После по-
вторного эвакуирования системы и заполнения ее воздухом возникший снова
пузырек воздуха удаляют. (Не имеет значения, что при этом удаляется и часть
испытуемого вещества). По окончании этих операций измерительный сосуд тер-
мостатируют и установку снова эвакуируют. При помощи крана 4, конец кото-
рого оттянут в капилляр, регулируют вакуум так, чтобы ртуть в обоих коленах
сосуда находилась иа одном уровне. Давление, отмечаемое манометром 3, соот-
ветствует давлению пара вещества при температуре термостата. Ступенчатым
повышением температуры и соответствующим выравниванием вакуума можно
установить зависимость давления пара от температуры. Если при весьма низ-
ком давлении пара разрежение, создаваемое вакуум-насосом, оказывается не-
достаточным для выравнивания уровня ртутн в измерительном сосуде, то к
измерительному сосуду прикрепляют шкалу, по которой отмечают наблюдаемый
уровень ртути и устанавливают давление пара вещества в миллиметрах по раз-
ности показаний манометра 3 и шкалы измерительного сосуда 2.
8.5. ОПРЕДЕЛЕНИЕ ПОКАЗАТЕЛЯ ПРЕЛОМЛЕНИЯ
Если показатель преломления измеряется при постоянной темпе-
ратуре и определенной длине волны падающего луча света, то в
известных пределах он может рассматриваться как константа ве-
щества и служить характеристикой органического соединения. Кро-
ме того, показатель преломления может быть использован для ко-
личествененого определения концентрации раствора или для ана-
лиза бинарных жидких смесей. Связав показатель преломления с
другой величиной, зависящей от температуры, а именно с плот-
ностью,. Лорентц и Лоренц ввели понятие молекулярной рефракции,
представляющей собой абсолютную константу вещества, которая
очень мало зависит от температуры и выражается следующей фор-
мулой:
где п — показатель преломления определенной длины волны при определенной
температуре; М — определенный или принятый молекулярный вес; d — плотность
при той же температуре.
Молекулярная рефракция, в соответствии с ее первоначальным
теоретическим толкованием, должна быть величиной аддитивной,
складывающейся из атомных рефракций. Однако многочисленные
исследования показали, что аддитивность осуществляется только в
первом приближении, так как при вычислении ее не были приняты
во внимание взаимное влияние связей и строение молекулы. По-
этому в последнее время приняло вычислять молекулярную рефрак-
цию исходя из суммы рефракций связей. Величины рефракций свя-
зей, рассчитанные преимущественно Фогелем 1 и Сайром 2, приве-
дены в табл. 13. Рефракции связей для фосфорорганических со-
единений были рассчитаны и другими авторами 3-4’7-8. Эйзенлор и
Вёлиш5 показали, что так называемый молекулярный показатель
преломления Мп2£, представляющий собой произведение молеку-
лярного веса на показатель преломления, вычисленный для линии
D при 20 °C, обладает такими же аддитивными свойствами, что и
молекулярная рефракция. Константы рефракций связей, приведен-
196
Таблица 13. Рефракции связей и их константы
(при 20° С для £>-лниии натрия)
Связь Рефракция связи Константы рефракций связей (рефракции связей Эйзенлора) Связь Рефракция связи Константы рефракций связей (рефракции связей Эйзеилора)
Р-С 3,575 25,57 С—О (ацета- 1,46
Р-С1 8,856 68,57 ли)
р-н 4,010 16,68 <=О 3,32 29,39
Р-0 3,102 28,11 С=О (метил- 3,49 —
Р->О -1,032 22,17 кетоны)
P-S 7,583 47,07 C-S 4,61 32,84
P->S 6,866 54,26 C=S 11,91 —
P-F — 34,98 С—N 1,57 —
P-N — 29,28 C=N 3,76 —
С-Н 1,676 3,87 О—Н (спирты) 1,66 13,15
С-С 1,296 12,86 О—Н (кисло- 1,80 10,54
С=С 4,17 9,39 ты)
С—С (шестичлен- 1,27 S-H 4,80 —
ных циклов) S-S 8,11 —
С—С (в аромати- 2,688 — S-O 4,94 37,1
ческих систе- s->o -0,20 20,84
мах) N-H 1,76 7,26
C-F 1,55 N—О 2,43 —
С-С1 6,51 56,80 N->O 1,78 —
С-Вг 9,39 124,51 N=O 4,0 32,26
С—О (эфир) 1,54 — N-N 1,99 —
N=N 4,12 —
ные в графе 3 табл. 13, вычислялись на этой основе. Исходя из них
и используя выражение
20 S
п°~~м
(где S — сумма констант рефракций связей), можно вычислить по-
казатель преломления.
Показатель преломления ОВ определяют в рефрактометре Аббе,
устанавливаемом в вытяжном шкафу. Чистку прибора осуще-
ствляют осторожным, но тщательным промыванием призм ватными
тампонами, смоченными спиртом. Тампоны после употребления
собирают в отдельный сосуд и затем сжигают. Очень удобен для
работы с малыми количествами жидкости микрорефрактометр
Джелли. В этом рефрактометре можно определять показатели пре-
ломления в интервале от 1,33 до 1,92 с точностью до третьего зна-
ка после запятой.
8.6. СПЕКТРОСКОПИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Абсорбционная спектроскопия в видимой и ультрафиолетовой об-
ласти спектра является одним из старейших физических спосо-
бов изучения строения органических соединений, а визуальная
197
колориметрия и фотометрия представляют собой наиболее употре-
бительные методы определения концентрации окрашенных илн
флуоресцирующих соединений.
Поглощение света в этих областях спектра, т. е. от 400 до
1000 Нм (видимая область) и от 200 до 400 нм (УФ-спектр), при-
водит к возбуждению слабо связанных валентных электронов и
вызывает их колебание. Такое поглощение обнаруживается у мо-
лекул, содержащих хромофорные группы *, причем оно существен-
но усиливается конъюгацией большого числа этих грудп и одновре-
менным присутствием ауксохромных групп**. Прочно связанные
валентные электроны возбуждаются в области ниже 200 нм так на-
зываемым вакуумным или УФ-излучением Шумана, а возбужде-
ние еще более прочно связанных, близких к ядру внутренних элек-
тронов происходит только в области рентгеновского излучения.
Спектры в видимой и УФ-областях йе очень четко выражены
и обычно имеют мало максимумов и минимумов. Поэтому их ис-
пользование для характеристики органических соединений весьма
ограничено. Значительный успех молекулярной спектроскопии был
обусловлен открытием ИК-области спектра. Благодаря дальней-
шему развитию термоэлементов, явившихся детекторами термо-
излучения, эта область спектроскопии стала более чувствительной;
последнее увеличило разрешающую способность монохроматоров.
Инфракрасная область простирается от красного конца видимой
части спектра до области коротких электромагнитных волн. Интер-
вал от 0,75 до 3-10~6 м обозначают как область близкого ИК-излу-
чения, интервал от 3 до 40 • 10~8 м как область среднего, а выше
этих величин волн — область далекого ИК-излучения. Поглощение
в близкой и средней областях основано на совпадении частоты ко-
лебания молекул с частотой колебания волн этой области. В интер-
вале дальнего ИК-излучеиия вращательное движение молекул про-
исходит вокруг осей главного момента инерции.
Различают в основном два -вида колебаний — валентные коле-
бания, при которых атомы колеблются вдоль осей, соединяющих
их центры, и такие колебания, при которых атомы колеблются по-
перек связей, т. е. колебания, приводящие к изгибу или деформации
молекулы.
В тех же областях спектра обнаруживают полосы, отвечающие
комбинационному рассеянию света (спектр Рамана). Их возникно-
вение основано на поляризации молекул при колебаниях и враще-
нии. Эффект Рамана обнаруживается в спектре рассеянного света
* Присутствие хромофорных групп сообщает соединению окраску. Приме-
ром таких групп являются нитрогруппа (—NOa), азогруппа (—N=N—) или
система с сопряженными двойными связями.
*♦ В теории цветности ауксохромными группами называют заместители со
свободными электронными парами, которые при замещении вызывают в хромо-
генах (ароматических радикалах, окрашенных благодаря присутствию хромо-
форной группы) усиление интенсивности окраски и ее углубление. Примерами
таких групп являются группы —МНг и —N(CHj)j.
198
наряду с линиями источника света. Для Исследования структуры
молекул метод спектров комбинационного рассеяния имеет такое
же большое значение, как и метбд ИК-Спектров, однако из-за не-
значительной интенсивности излучения для измерения спектра ком-
бинационного рассеяния требуется сложная аппаратура, поэтому
этот метод еще не находит широкого применения. По сравнению
со спектрами в видимой и УФ-областях ИК-спектры даже веществ
с простой структурой изобилуют большим числом максимумов и
минимумов, отражающих многообразие теоретически возможных
колебаний и вращений, что требует для их оценки большого опыта.
Абсорбционная спектроскопия может быть- использована для
идентификации неизвестных и определения уже известных соеди-
нений. В последнем случае измеряют интенсивность характерного
для определяемого вещества максимума, зависящего от концентра-
ция вещества. УФ-Спектры снимают в разбавленных растворах ве-
щества в растворителе, не поглощающем в этой области, например
в н-гексане, или в кварцевых кюветах, пропускающих УФ-излуче-
ние. Можно снимать ИК-спектры как твердых, так и жидких и га-
зообразных веществ. При этом стенки кювет должны быть выпол-
нены из определенных материалов, например из галогенидов ще-
лочных или щелочноземельных металлов.
Наряду с высокой специфичностью спектроскопические методы
анализа имеют еще и то преимущество, что при этом вещество
не изменяется и может быть использовано для других исследова-
ний. Разумеется, снятие спектрограммы для идентификации и ха-
рактеристики соединения только тогда имеет смысл, когда удается
получить вещество высокой степени чистоты.
8.6.1. Уф-Спегфы
Фундаментальные работы по идентификации и-определению ОВ
при помощи УФ-спектроскопии проводились Молером и сотр.9-15
в период с 1936 по 1944 г. Различными авторами разработано
Таблица 14. Максимумы поглощения ряда ОВ в области обычного
и вакуумного УФ-излучения
Отравляющее вещество X, нм Ссылки на лите- ратуру । Отравляющее вещество X, нм Ссылки на лите- ратуру
Адамсит 198 [12, 14] 0 Ксилилбромид 199 12, 14]
Бис-2-хлорэтиловый тио 202 [12, 14 | Параоксон 270 50]
эфир В Паратион 274 50]
Бромацетон 209 12, 14 1 ш-Хлорацетофенон 198 12, 14]
Бромбензилцианид 244 12, 14 12-Хлорвинилдихлор- 214 12, 14]
Бром метилатилкетои 212 12, 14 I арсин
Дифенилхлорарсин 271 12, 14 | Хлорпикрин 205 [12. 14]
Дифенилциан арсин 226 12, 14 | Этилдихлорарсин 249 [12. 14]
ДФФ 261 52
199
применение УФ-спектроскопии для идентификации ядов 16, эфиров
фосфорной кислоты 17, зарина 18 и для определения паратиона 19>68.
Обзор приведенных в литературе характерных максимумов погло-
щения в областях обычного и вакуумного УФ-излучения для неко-
торых ОВ и фосфорорганических соединений представлен в табл. 14.
8.6.2. ИК-Спектры
Большое значение, которое имееют фосфорорганические соедине-
ния как инсектициды и пластификаторы^ а также их применение в
медицине, фармакологии и военной химии послужило поводом для
проведения весьма обширных исследований их ИК-спектров. После
1500 Ш)0 1300 1200 1100 1000 900 800 700
Частота, см-1
Рис. 16. ИК-Спектрограмма зарина21.
того как наблюдаемые максимумы (полосы) были связаны с от-
дельными колебаниями, т. е. была установлена связь между струк-
турой вещества и его ИК-спектром, в дальнейшем с помощью
ИК-спектроскопии были решены различные практические задачи,
как, например: исследован гидролиз табуна20, осуществлен кон-
троль чистоты и исследована устойчивость зарина 21, исследована
изомеризация со-диметиламиноэтилдиэтилтионфосфата22 и систок-
са23, разработан контроль чистоты различных фосфорных соедине-
ний 24, исследовано различие в поляризуемости фосфорильной груп-
пы при нахождении ее рядом с алкилтиогруппой, фтором и алк-
оксигруппой 35, разработано количественное определение ряда ин-
сектицидов22-29, паратиона30-31. Были разработаны также методы,
сочетающие ИК-спектроскопию с тонкослойной32 и газовой хрома-
тографией 28-33.
Положение важнейших полос ИК-поглощения фосфорорганиче-
ских соединений представлено в табл. 15.
Само собой разумеется, что в спектрах фосфорорганических
соединений находятся полосы, соответствующие валентным и де-
формационным колебаниям связи С—Н, и многие другие. Пред-
ставление о распределении полос в ИК-спектре зарина дает рис. 16
и относящаяся к нему табл. 16, заимствованные из работы Лорке
и Вассара21.
200
Таблица 15. Полосы ИК-поглощеиия фосфорганических соединений
Структура Колеба- ние связи Вид колебания Интервалы частот. Интенсив- ность полосы Ссылки на литературу (примечание)
Р=О Р=О Валентное 1170—1310 Сильная [34-38]
Р—О—алкил о-с Валентное 995-1050 Очень [37—39]
сильная
Р-0 Сим. валент- 720-750 — [41]
ное
Р-0 Асим. ва- 970-1025 — [42] (не надеж-
лентное на)
Р-0 Валентное 788-820 — 40
Р—О—арил — — 1190-1240 Сильная 43
— 1030 Средняя 43
р-О-СНз — 1190 Слабая 37
Р—О—С2Н6 — —. 1143-1163 Слабая 37
Р—О—С3Н7-пзо —. —. 1109—1114 — —
1146—1149 — —
1184 — —
Р-0 990-1012 — —
Р-О-Р — — 909—971 — [44]
Р-О-Н о-н Валентное 2560-2700 — [37, 43, 42]
(широкая,
плоская)
Р-Н о-н Валентное 2320-2457 Средняя [37, 40, 43, 47]
Р—арил — — 1435—1450 Средняя [37]
Р—алкил Р-С — 650-750 Средняя [45] (не надеж-
на)
Р-СН3 — Деформа- 1310-1320 — [41]
ционное
Р-С2Н8 — — ~ 1228 —- 40, 44
P=S P=S Валентное 685-862 Сильная 44, 46
550—730 Сильная 44, 46
(в присутствии
галогена,
HS-или
RS-rpynn)
P-S-C С—S Валентное 625-645 — [44, 46]
554-613 — (в соединениях
с группой
Р=О)
510-574 — [46] (в соеди-
нениях с труп-
пой Р=О)
S-P-S-C P-S Валентное, -548 — 44
-515 — 44
P-S-H P-S Валентное 526-649 — 44
493-617 — 44
P-S-C2H8 S—с — 1259-1267 — 46
P-S-CHj S-C — 1325-1420 — 46
1292—1305 — 46
Р —F P-F Валентное 740—800 — [37, 47] (Р3+)
— — Валентное 800—900 Сильная [37, 47] (Ps+)
201
Продолжение табл. 15
Структура Колеба- ние связи Вид колебания Интервалы частот. с*-1 Интенсив- ность полосы Ссылки на литературу (примечание)
ДФФ P-F 864 14Ц
805-871 —- [47] (в соеди-
нениях со свя-
зямн Р—N и
P-S)
Р-С1 — 420-587 —- 47
p-N(R)2 P-N Валентное 702-730 Сильная 39
P-N-C N—С Валентное 989-1006 Сильная [39
P-C^N C = N Валентное 2232 — [20, 39] (у та-
P-N(CH3)2 CHS Деформа- — 1320 — бу на) [48]
цнонное
Точное положение полос в границах частот, указанных в табл.
15, зависит от соответствующей структуры соединения; для неко-
торых полос их связь со структурой еще спорна. Это относится
также и к весьма интенсивной полосе, наблюдаемой при частоте
порядка 980 см~' у всех органических соединений пятивалентного
фосфора, которую Беллами42 на основании экспериментальных
Таблица 16. ИК-спектры зарина
Частота колебания, см-1 Интенсивность полосы Колеба- ние связи Вид колебания Структура
720 Сильная Р-С Сим. валентное р-о-с
777 Средняя Р-0 Валентное Р-О-С
840 Очень сильная P-F Валентное P-F
880 Слабая — —
905 Очень сильная — Деформационное О—С3Н7-изо
925 Очень сильная — Деформационное О—С3Н7-изо
1015 Очень сильная о-с Валентное Р-О-С
1105 Сильная — Деформационное О—С3Н7-изо
1145 Средняя — Скелетное С(СН3)2
1180 Средняя — Деформ ационное О—С3Н7-изо
1280 Очень сильная Р=О Валентное Р=О
1320 Очень сильная с-н Аснм. деформационное Р-СН3
1360 Слабая с-н Деформ ацнонное с-н
1380 Средняя — — ( СН3 изопро-
{ поксигруппы
1390 Средняя —- 1 То же
1420 Средняя с-н Деформационное сн3
1455 Средняя — — сн3
1470 Широкая полоса —— — сн3
2854 Очень сильная — —— сн3
2922 Слабая с-н Валентное сн3
2955 Сильная — — СН3
202
данных приписывает связи Р—О. Вследствие ее высокой интен-
сивности она предпочтительно используется для количественного
ИК-спектрометрического определения фосфорорганических соеди-
нений. Несомненно, что в области ИК-спектроскопии органических
соединений фосфора решены далеко не все вопросы, что находит
свое отражение в непрерывном появлении ряда новых работ 49'63-®5.
8.7. РАЗДЕЛЕНИЕ И ИДЕНТИФИКАЦИЯ ПРИ ПОМОЩИ
ХРОМАТОГРАФИЧЕСКИХ МЕТОДОВ
Хроматография является важным физико-химическим способом
разделения веществ, который в общем виде основывается на раз-
личиях сорбционного равновесия на твердой фазе или на различ-
ном распределении вещества между двумя жидкими или между
газообразной и жидкой фазами. Во многих случаях одновременно
эффективны как адсорбция, так и распределение. Распределение
веществ осуществляется между подвижной н стационарной фа-
зами. В качестве подвижных или движущихся фаз используются
растворители или, в специальном случае Газовой хроматографии,
газ-носцтель; в качестве стационарных фаз — твердые адсорбенты
или жидкости, фиксированные на твердом носителе. Особыми ви-
дами хроматографии являются ионообменная хроматография н
способы разделения, основанные на использовании молекулярных
сит или на фильтрации через гели. До сих пор широкое примене-
ние находит адсорбционная или распределительная хроматогра-
фия в колонках, на бумаге, тонкослойная и газовая.
8.7.1. Хроматография в колонках
Помещают в колонку неподвижную фазу, в качестве которой при-
меняются такие адсорбенты, как силикагель, окись алюминия, по-
рошкообразная целлюлоза или иониты. В эту колонку вначале вво-
дят раствор смеси веществ, подлежащих разделению, а затем по-
движную фазу. В результате сорбции (или распределения) обра-
зуются пространственно разделенные зоны отдельных компонентов
смеси, которые извлекают либо после удаления адсорбента из ко-
лонны, либо используют различную растворимость компонентов в
подвижной фазе. Хроматография на колонке дает возможность
очищать и разделять препаративные количества вещества.
Содержащиеся в техническом зарине примеси (пирофосфонат,
фтористоводородная и метилфосфоновая кислоты и другие фосфор-
органические соединения) могут быть удалены хроматографическн
на колонке, содержащей в качестве неподвижной фазы насыщен-
ный водой силикагель84. Для этого через колонку пропускают рас-
твор зарина в диизопропиловом эфире. Примеси удерживаются
сорбентом, после чего их можно элюировать водой.
Разделение различных инсектицидов — эфиров тиофосфорной
кислоты, таких, как паратион, параоксон, хлортион и бромтион.
203
было осуществлено на колонке, в которой в качестве неподвижной
фазы использовался циклогексан на полиэтилене низкого давле-
ния, а в качестве подвижной фазы — смеси этанола и ацетатного
буфера 50.
Для разделения смесей различных органических кислот фос-
фора и их эфиров удобной оказалась ионообменная хроматография
на катионитах 85’86; колонка с анионитом дауэкс 1x8 при примене-
нии соляной кислоты в качестве элюента использовалась для разде-
ления продуктов гидролиза фосфорорганических соединений154.
8.7.2. Бумажная хроматография
В общих чертах метод представляет собой следующее: незначи-
тельное количество раствора исследуемого вещества наносят пят-
нами на определенное место (линию старта) нижнего конца по-
лосы специальной фильтровальной бумаги, затем эти полосы под-
вешивают в камерах так, чтобы их нижние концы, у которых на-
несены капли веществ, были погружены в насыщенный водой, но
не смешивающийся с ней растворитель. Подвижная фаза (раство-
ритель) поглощается бумагой и передвигается кверху, проходя че-
рез пятна вещества. Вещества, в зависимости от их распределения
между подвижной и неподвижной фазами, в данном случае влаж-
ной целлюлозой, передвигаются с различной скоростью вместе с
растворителем. Через какое-то время, как только фронт раствори-
теля почти достигнет верхнего среза бумаги, бумажную полосу вы-
нимают из камеры, отмечают положение фронта растворителя и
при помощи цветной реакции проявляют (делают видимыми) пят-
на вещества. Мерой скорости передвижения веществ является по-
казатель Rf, определяющийся по следующей формуле
__ Расстояние между линией старта и положением пятна вещества
' Расстояние между линией старта и фронтом растворителя
R; зависит от подвижного растворителя и сорта бумаги и яв-
ляется величиной, характерной для вещества. Бумажная хромато-
графия связана со сравнительно большой затратой времени. Так,
получение хроматограммы длиной 40 см требует, в зависимости
от бумаги, растворителя и аппаратуры, от 6 до 20 ч.
8.7.2.1. Исследование фосфорорганических соединений методом
бумажной хроматографии. Бумажная хроматш рафия является од-
ним из важнейших методов химического и биохимического иссле-
дования фосфорорганических соединений. По поведению веществ
на хроматограммах можно сделать выводы о структуре этих сое-
динений, особенно о характере связи серы. Так, при применении
полярной стационарной фазы и неполярной подвижной фазы Rf
соединений с тионной серой (P=S) имеет высокое значение, а
переход серы в тиольную форму (Р—S—) знаменуется уменьше-
нием /?,. Понижение Rf наблюдается также при замещении серы
кислородом.
204
Системы растворителей. Применение воды в качестве стацио-
нарной фазы ограничивается сравнительно малым числОхМ водо-
растворимых веществ. Поэтому для ряда фосфорорганических со-
единений в качестве стационарной фазы применяют полярные
растворители или работу проводят так, что на бумаге находится
неполярная жидкость (например, полиалкилсилоксаны, оливковое
или минеральные масла), а в качестве подвижного растворителя
используют полярный растворитель. В табл. 17 приведены примеры
некоторы.х употребительных систем растворителей.
Таблица 17. Системы растворителей, применявшиеся при изучении некоторых
фосфорорганических соединений методом бумажной хроматографии
Соединение Система растворителей Ссылки на литературу
Диэтоксифосфорилтио- холин, метилэтокси- фосфорилтиохолин и продукты гидролиза н-Бутанол — этанол — ледяная уксусная кислота — вода (8 :2 : 2 : 3) [87]
ш-Диметиламиноэтил- диэтилтионфбсфат и изомерные соединения продукты гидролиза н-Бутанол — этанол — ледяная уксусная кислота — вода (8 : 2 : 2 : 3) То же в соотношении 8 : 2 : 1 : 3 [88]
Продукты гидролиза зарина 1) w-Бутанол — этанол — ледяная уксус- ная кислота — вода (8:2:1: 3) 2) Изобутанол — изопропанол — 2 М рас- твор NH4OH (2:2; 1) [89]
Паратион, параоксон Стационарная фаза — пропиленгликоль, подвижная фаза — смесь петролейного эфира и бензола (4:1) [90]
Паратион и аналогич- ные соединения Стационарная фаза — полналкилсилок- саны, подвижная фаза — смесь воды, этанола и хлороформа (6: 10: 10) [93, 96]
Паратион, систокс и изомеры Стационарная фаза—минеральное масло, подвижная фаза — смесь этанола, аце- тона, воды (1:1:2) Стационарная фаза — пропиленгликоль, подвижная фаза — смесь петролейного эфира и толуола (4:1) [94]
Систокс, изосистокс [91]
Систокс н-Бутанол — вода (84:14) [92]
Изосистокс и продукты окисления Толуол — ацетонитрил — метанол — вода (8:2: 5: 5) [98]
Эфиры органотиофос- форных кислот 1) Стационарная фаза — диметилформ- амид, подвижная фаза — керосин (фрак- ция 100-150 °C) 2) Стационарная фаза — жидкий пара- фин, подвижная фаза — смесь диметил- формамида, н-бутанола, воды (14: 1 : 5) 3) н-Бутанол — вода — 10%-ный NH4OH (24:6: 1) [95] [997] [112, 114]
Продукты гидролиза эфиров тиофосфорных кислот н-Бутанол — 25%-ный аммиак — ацетон— вода (4 : 1 : 1 : 1) [95]
205
Обнаружение фосфорорганических соединений на хроматограм-
мах. Широко используемый метод обнаружения Хейнса и Ишер-
вуда" основан на получении из фосфорорганического соединения
фосфорномолибденовой кислоты и восстановлении ее в молибдено-
вую синь.
Для этого после получения хроматограммы ее сушат и затем обрызгивают
раствором, представляющим собой смесь 5 мл 60%-иой НС1О4, 10 мл 1 и. рас-
твора НС1 и 25 мл 4%-ного раствора молибдата аммония, доведенного водой
до объема 100 мл. Обработанную этим раствором хроматограмму нагревают
7 мин при 85 °C и подвергают действию сероводорода или обрызгивают раз-
бавленным солянокислым раствором SnCl2. При наличии фосфатов появляются
синие пятна.
Обнаружение трудно гидролизующихся тиол- и тионфосфатов
облегчается предварительным обрызгиванием хроматограммы рас-
твором N-бромсукцинимида 10°.
Таблица 18. Некоторые методы обнаружения фосфорорганических соединений
на бумажных хроматограммах
Соединение
Способ обнаружения
Ссылки
на литературу
Эфиры оргаиотио-
фосфорных кнслоГ
Эфиры оргаиотион-
фосфорных кислот
Изосистокс
Эфиры органофос-
форных кислот
Четвертичный азот
в фосфорилтиохо-
линах
Группа SH в про-
дуктах гидролиза
Холни и сложные
эфиры холина, тре-
тичная аминогруппа
Гексаиодоплатинат калия: смесь рас-
твора 1 г PtCl2 в 10 мл воды н рас-
твора 10 г KI в 250 мл воды, раз-
бавленная водой 1 ; 6,—Через несколько
минут или часов — желтые пятна па
розовом фоне; более чувствителен после
нагревания 1 ч при 80 °C
№:Бромсукцинимид: 0,002 М раствор
в этаноле. — На красном фоне желто-
зеленые, флуоресцирующие пятна
НС1О4—НЮ4 — раствор крахмала —
на белом фоне синие пятна
Иодазидный раствор — иа коричневом
фоне белые пятна
2,6-Дибром-М-хлор-п-хинонимин. — жел-
тые до коричневых пятна
Т рифенилтет разолийхлорид — на белом
фоне красные пятна
Тетразолий синий — на белом фоне си-
ние пятна
Пары брома, растворы FeCl3 и сульфо-
салициловой кислоты — тиофосфаты
дают на фиолетовом фоне желтые
пятна, фосфаты — белые
Дипикриламин — иа светло-желтом фоне
желтые пятна
Реагент Гроте120: нагретый в течение
5 мин при 100 °C N-этилмалеинимид121
Реагент Шарграфа: фосфорномолибде-
новая кислота, SnCl2
Реагент Драгендорфа, модифицирован-
ный Брегоффом и Дельвнчем — на
желтом фоне красные пятна
[92]
[98]
ЦП]
[112-114]
[95, 115, 116]
[117]
[98, 118]
[П9]
[87]
[88]
[87]
[153]
206
Более чувствительное обнаружение фосфорорганических соеди-
нений, угнетающих in vitro холинэстеразу, осуществляется моди-
фицированными биохимическими методами. Опубликовано боль-
шое число методов с различными субстратами93> 101~107. Весьма
удобен в работе способ, в котором используется сыворотка крови
лошади и индоксилацетат108. Такие соединения, как, например, па-
ратион, угнетающий фермент только in vivo, окисляют путем об-
работки хроматограммы парами брома или обрызгиванием раство-
ром N-бромсукцинимида в ингибиторы, действующие in vitro.
Имеется много реагентов, пригодных для обнаружения серусо-
держащих фосфорорганических соединений. Особенно удобен рас-
твор 0,5 г PdCl2 в 2 мл концентрированной H2SO4 и 98 мл воды 109,
который в зависимости от вида связи серы дает желтые (—S—С,
—SO—С, —SO2—С) до коричневых (Р -> S, —SH) пятна.
Обзор других способов обнаружения различных органических
фосфатов и некоторых важных функциональных групп приведен
в табл. 18.
По размерам пятен нельзя определить количество вещества.
Для этого пятно вырезают и подвергают бумагу минерализации
мокрым способом 122 или сожжению в кислородной колбе 123, после
чего фосфат определяют фотометрически.
8.7.3. Тонкослойная хроматография
В общем виде методы тонкослойной хроматографии аналогичны
описанным в бумажной хроматографии, за исключением того, что
вместо бумаги используют слои адсорбентов толщиной примерно
250 • 10-6 м, нанесенные на стеклянные пластинки. В качестве ад-
сорбентов наиболее часто применяются силикагель и окись алю-
миния с добавками связующих агентов (гипс, крахмал). После
перемешивания с водой до кашицеобразной консистенции их на-
носят при помощи подходящего приспособления на стеклянные
пластины. В последнее время путем добавления к адсорбентам по-
ливинилового спирта научились создавать устойчивые слои на по-
лиэфирных пластинках 152.
Преимуществами тонкослойной хроматографии являются: спо-
собность хорошо разделять липидофильные вещества *, быстрота
определения, высокая чувствительность, отличная разрешающая
способность и широкий выбор стационарных фаз и проявителей.
8.7.3.1. Исследование фосфорорганических соединений. Хоро-
шей системой растворителей для разделения эфиров органотио-
фосфорной кислоты на силикагелевых слоях126 является н-гексан —
ацетон (4:1). Другие системы растворителей приведены в рабо-
тах, в которых изучались способы выделения и разделения остат-
ков инсектицидов — эфиров органотиофосфорных кислот — из
* Вещества, обладающие сродством к жирам, особенно хорошо растворяю-
щиеся в жирах, маслах, углеводородах и жироподобных соединениях и плохо рас-
творимые в воде.
207
растений и продуктов питания 127~135 или способы разделения раз-
личных замещенных органофосфорных кислот 136’137.
Для локализации пятен используют способы обнаружения, опи-
санные для бумажной хроматографии органических фосфатов, та-
кие, например, как хлорид палладия, гексаиодоплатинат калия,
иодазид114, HCIO4—НЮ4— крахмал138, 2,6-дибром-М-хлор-л-хинон-
имин139. Можно применять также КМПО4 в концентрированной
серной кислоте 129 или ацетоне 140 и раствор СоС12 в ацетоне *41.
Обнаружение фосфорорганических соединений, угнетающих хо-
линэстеразу, удается осуществить с высокой чувствительностью
при помощи модифицированных биохимических методов 108’142’ г01.202,
из которых будет описан метод с применением в качестве суб-
страта индоксилацетата 108.
Реагенты
Раствор фермента — смесь равных частей 10%-ного водного раствора лио-
филизированной сыворотки крови лошади и буферной смеси с pH 8,5.
Раствор субстрата — раствор 0,5% индоксилацетата (т. пл. 127—128 °C) в
смеси, состоящей из 2 ч. ацетона, не содержащего альдегида, и 3 ч. воды.
Вынутые из разделительной камеры пластины, после испарения раствори-
теля, помещают на 1 мин для окисления ингибиторов, действующих in vivo, в
атмосферу паров брома (для in vitro-ингибиторов эта обработка парами брома
отпадает). Пластины оставляют на 5—10 мин на воздухе для удаления избы-
точного брома и обрызгиванием равномерно пропитывают их раствором фер-
мента. Выдерживают 30 мин в сосуде в атмосфере насыщенного водяного пара,
после чего тотчас обрызгивают раствором субстрата и рассматривают в свете
УФ-лампы. Через 1-—2 мин ингибиторы обнаруживаются в виде темных пятен
на сильно флуоресцирующем сине-зеленом фоне. По мере ослабления флуорес-
ценции в результате окисления индоксила в индиго фон окрашивается в синий
цвет, на котором выделяются белые пятна, образованные ингибитором. Этим
способом можно обнаружить до 0,005 мкг ингибитора.
При использовании хромогенного субстрата, представляющего
собой 0,05%-ный раствор 2-азобензол-1-иафтилацетата в ацетоне,
и 2%-кого раствора сухой сыворотки в буферной смеси (pH 7,5)
достигается несколько меньшая чувствительность. При этом инги-
битор обнаруживают по белым пятнам на красном фоне.
8.7.3.2. Обнаружение алкалоидов. Тонкослойная хроматография
дает возможность провести систематический анализ алкалоидов
по методу, разработанному Валди и сотр.143. При этом алкалоиды
разделяются на две группы. Принадлежность неизвестного веще-
ства к той или иной группе устанавливается предварительным ис-
пытанием при помощи растворителя III (см. табл. 19) на слое
силикагеля G *.
Оценку результатов осуществляют рассматриванием хромато-
граммы в УФ-свете и обрызгиванием раствором гексаиодоплати-
ната калия. Одновременно хроматографируют вещество сравне-
ния — родамин С. Алкалоиды со значением Rf ниже 0,30 относят к
группе А, а выше 0,30 — к группе Б.
* Силикагель G — это силикагель, к которому в качестве связующего сред-
ства добавляют 5—15% гипса. В ГДР этот продукт выпускается народным хи-
мическим предприятием Грейц-Дёлау под маркой силикагель D.
208
Таблица 19. Растворители, применяемые для разделения алкалоидов
методом тонкослойной хроматографии
Растворитель Вещество сравнения Rf
I. Хлороформ — ацетон — диэтиламин (50:40: 10) Родамин С 0,58
II. Хлороформ — диэтиламин (90 : 10) Родамин С 0,49
III. Циклогексан — хлороформ — диэтиламин (50 : 40 : 10) Родамин С 0,20
IV. Циклогексан — диэтиламин (90 : 10) Масляный желтый 0,45
V. Бензол — этилацетат — диэтиламин (70:20: 10) Масляный желтый 0,44
VI. Хлороформ Масляный желтый 0,85
VII. Циклогексан — хлороформ (30: 70) + 3 кап- ли диэтиламина Масляный желтый 0,85
VIII. Метанол Родамин С 0,53
Для идентификации алкалоидов группы А их разделяют на си-
ликагелевых слоях растворителями I и П, а алкалоидов группы
Б — растворителями HI, IV и V на тех же слоях, после чего обра-
батывают раствором иодоплатината калия и наблюдают окраску
пятен в УФ-свете. В сомнительных случаях можно дополнительно
провести хроматографирование растворителями VI, VII и VIII на
окиси алюминия G(D) и щелочном силикагеле (для замешивания
наносимой на пластину массы силикагеля вместо воды используют
0,1 н. раствор NaOH). Обычно для анализа используют 50 мкг
исследуемого вещества. Оценку результатов производят по табл. 20.
Хроматографирование широкого спектра токсичных соединений,
в том числе и алкалоидов, удобно проводить растворителем, содер-
жащим метанол — ацетон — триэтаноламин (50:50:1,5) на слоях
силикагеля144. При этом целесообразно проводить предваритель-
ное разделение по методу STAS-Otto.
8.7.3.3. Обнаружение токсичных катионов и анионов. Обычный
силикагель с гипсом в качестве связующего средства не подходит
для разделения и последующего обнаружения неорганических
ионов. Для этой цели силикагель обрабатывают НС1, промывают
водой до нейтральной реакции и после сушки добавляют к нему
крахмал в качестве связующего. Другими употребительными ма-
териалами для тонкослойной хроматографии являются порошкооб-
разная целлюлоза и иониты. В табл. 21 приведены примеры раз-
деления и обнаружения некоторых токсичных катионов и анионов
методами тонкослойной хроматографии.
8.7.3.4. Разделение других ОВ и гербицидов. Различные хлорсо-
держащие гербициды, такие, как 2,4-дихлорфеноксиуксусная кис-
лота (2,4-D), 2,4,5-трихлорфеноксиуксусная кислота (2,4,5-Т), 2,4-
дихлорфеноксимасляная кислота (2,4-DB), 4-хлор-2-метилфенокси-
уксусная кислота, у-(4-хлор-2-метилфенокси)-масляная кислота,
а,а-дихлорпропионовая кислота (далапон), можно разделять
209
Таблица 20. Значения и цветные реакции алкалоидов с иодоплатннатом калия
Алкалоиды Силикагель AljOj Щелочной силикагель Флуорес- ценция в УФ-свете (365 нм) Окраска пятна Форма пятна Число дополни- тельных пятен
растворители растворители растворитель УШ
1 11 III | IV V VI VII
Группа А
Морфии 0,10 0,08 0,0 0,0 0,03 0,03 0,0 0,34 — Глубоко синяя Круглая —
Атропин 0,38 0,40 0,16 0,05 0,12 0,0 0,10 0,17 — Фиолетово-синяя Круглая —
Колхицин 0,47 0,41 0,04 0,0 0,04 0,11 0,0 0,57 — Светло-серая Круглая —
£оматропнн 0,37 0,45 0,15 0,05 0,23 0,04 0,24 0,15 — Фнолетово-синяя Круглая —
Бруцин 0,42 0,63 0,18 0,0 0,19 0,50 0,54 0,12 — Фиолетово-корич- невая Круглая 1
Стрихнин 0,53 0,76 0,28 0,05 0,38 0,57 , 0,60 0,22 — Желтая Вытянутая —
Г руппа Б
Физостигмин 0,65 0,9 0,32 0,04 0,44 0,59 0,50 0,46 — Розовая Круглая 1
Аконитин 0,68 0,9 0,35 0,03 0,49 0,36 0,60 0,65 — Красно-коричне- вая Круглая 3
Эметин 0,67 0,9 0,40 0,06 0,45 0,38 0,58 0,50 Синяя Крдсно-корнчне- вая Круглая 3
Папаверин 0,67 0,9 0,42 0,03 0,47 0,85 0,84 0,70 Желтоватая Желтая Круглая —
Наркотин 0,72 0,9 0,51 0,10 0,57 0,81 0,79 0,72 Синяя Светло-желтая Круглая —
Кокаин 0,73 0,9 0,65 0,36 0,58 0,84 0,77 0,62 — Фиолетовая Круглая —
Таблица 21."Разделение и обнаружение токсичных катионов и Анионов методом тонкослойной хроматографии
Ионы Адсорбент Растворитель Реагенты Ссылки иа литературу
Ni, Со, Си, Fe, Pb, Мп, Сг, As Силикагель — крах- мал Ацетон — 3 н. НС1 (99:1) Различные [145]
Ba, Sr, Са, Mg, Al, NH4, Na, К, Li Силикагель — крах- мал Метанол — н-бутанол (80 : 20) Различные [145]
Sb, As, Си, Cd, Sn, Bi, Zn, Hg Силикагель — крах- мал н-Бутанол — бензол — 1 и. HNO3 — 1 н. НС1 (50:46:26: 1,4) Различные [145]
Катионы после разделений сероводородом Силикагель, А13О3 Различные Различные [146]
Pb, Си, Bi, Cd, Hg, As, Sb, Sn Маисовый крахмал Ацетон — 3 и. НС1 (1:1) (NH4)2S, дитизон [147]
Токсичные катионы, в том числе As, Ba, Be, Cd, Hg,' Pb, Sb, SeO3, T1 Целлюлоза ' MN 300 । Ацетон — 4 н.НС1 (или 24%-ная HNO3) (7:3) Различные [148]
Катионы группы сероводорода Силикагель — гипс н-Бутанол — 1,5 н. НС1 — ацетонил- ацетон (100 : 20: 0,5) KI, (NH4)2S
Катионы группы сульфида аммония Силикагель — гипс Ацетон — НС1 (коиц.) — ацетоиилаце- тон (100:1:0,5) NH3, оксихинолин [149, 150]
Диэтилдитиокарбаматы Hg, Си, Pb, Bi, Cd Силикагель D н-Гексаи — хлороформ — диэтнламии (20:2:1) CuSO4 (5%-иый) [151]
Tl, Hg, As, Pb, Cd, Си, Bi, Sb Силикагель Ацетон — бензол — HNO3 — винная кислота Дитизон, (NH4)2S [155]
Cl, Br, I, F, CN, SCN, Fe(CN)j+, Fe(CN)J+ Силикагель — гипс Изобутанол — пропанол — дибутил- амин (9:9:2); пропанол — хлоро- форм — бензиламин (6:3: 1) Различные [161]
методом тонкослойной хроматографии на слоях, состоящих из 60%
кизельгура и 40% силикагеля с добавкой крахмала в качестве
связующего. Растворителем служит смесь парафинового масла,
бензола, ледяной уксусной кислоты, циклогексана (1:3:2:20).
Обнаружение осуществляется обрызгиванием хроматограммы спир-
товым раствором AgNO3 и облучением УФ-светом; на белом фоне
становятся видимыми коричнево-черные пятна 156.
Для разделения хлорированных алкилфеноксикарбоновых кис-
лот пригодны также слои из силикагеля G с добавкой фосфорной
кислоты157, а в качестве растворителя смесь диизопропилового и
петролейного эфиров (1:1). Для обнаружения обрызгивают хро-
матограмму 0,05%-ным водным раствором родамина С, который
проявляет гербициды в виде белых пятен на розовом фоне; метод
весьма чувствителен.
Диэтиламид лизергиновой кислоты (LSD) и родственные ве-
щества 158 могут быть разделены на слоях силикагеля G, раство-
ритель хлороформ — метанол (9:1), или на окиси алюминия G,
растворитель трихлорэтан — метанол (9:1). Обнаружение осуще-
ствляется по синей флуоресценции в УФ-свете или обрызгиванием
модифицированным реагентом Ван Урка (раствор 0,8 г п-диметил-
аминобензальдегида в 100 мл этанола, содержащего 10 объемн.%
концентрированной H2SO4), проявляющего эти соединения в виде
сине-фиолетовых пятен; максимум обнаружения 0,05 мкг LSD.
8.7.4. Газовая хроматография
Газовая хроматография является самой молодой областью хро-
матографии. За последние два десятилетия, прошедших с начала
ее бурного развития, она превратилась в важнейший метод ана-
лиза органических соединений, перед которым открываются все
новые области применения. Особым преимуществом газовой хро-
матографии по сравнению с до сих пор описанными способами хро-
матографии является то, что этим методом могут быть осуществ-
лены процессы как качественного, так и количественного разде-
ления.
Газовая хроматография подобна разделительной хроматографии
в колонке, с той разницей, что вместо жидкого растворителя в ней
используется подвижная газовая фаза-, газ-носитель. Тем самым
область ее применения распространяется на газообразные или по
возможности испаряющиеся без разложения вещества. Этим спо-
собом можно разделять также и летучие продукты пиролиза, что
дает возможность судить о составе исходного вещества.
Пробу исследуемого газа или пара вводят в ток газа-носителя,
который пропускают через термостатированную колонку, напол-
ненную стационарной фазой. В качестве последней используют
твердый сорбент — силикагель или активированный уголь, или
жидкость с малым давлением пара, которая удерживается инерт-
ным пористым носителем. Наряду с этими наполненными колон-
ками в последнее время применяются капиллярные колонки, пред-
212
ставляющие собой капилляры (внутренний диаметр 0,2—0,5 мм,
длина 150 м), на внутреннюю стенку которых наносят жидкую
пленку стационарной фазы. Эти капиллярные колонки обладают
исключительно высокой разделительной способностью, достига-
ющей 100000—500 000 теоретических тарелок (напомним, что в хо-
рошей ректификационной колонне их число не превышает 100). При
использовании коротких капиллярных колонн сильно сокращается
продолжительность анализа, до долей секунды.
Конечно, газовая хроматография имеет очень сложное аппара-
турное оформление. Регистрация компонентов, появляющихся в
газе-носителе в определенный интервал времени после прохожде-
ния ими разделительной колонки, осуществляется детекторами,
действие которых чаще всего основано на измерении физических
параметров, например теплопроводности и плотности газа, на пла-
менной и лучевой ионизации или масс-спектрометрии. Показания
детекторов фиксируется автоматическим самописцем, причем полу-
чаются кривые, на которых соответственно числу компонентов име-
ются пики. Расстояния этих пиков от точки старта выражают время
удерживания, что дает возможность идентифицировать компонен-
ты. В количественных определениях рассчитывают площадь, огра-
ниченную пиком.
Газовая хроматография позволяет анализировать сложные сме-
си за значительно более короткое время, чем это требуется при
анализах обычными способами. Она является наиболее чувстви-
тельным методом качественного и количественного определения ток-
сичных веществ.
8.7.4.1. Анализ фосфорорганических соединений. Силиконовые
масла и высоковакуумные смазкн широко применяются в газовой
хроматографии в качестве стационарного слоя, а крупнозернистый
кизельгур (хромосорб) в качестве носителя. Использование газо-
вой хроматографии для разделения и определения фосфороргани-
ческих соединений привело к некоторым интересным достижениям
в области детекторов. Из числа обычно применяемых детекторов
для регистрации фосфорорганических соединений пригодными ока-
зались детекторы, измеряющие теплопроводность 162-,65, и пламенно-
ионизационные детекторы 166>167. Особое значение для весьма чув-
ствительного обнаружения соединений, обладающих большим срод-
ством к электронам, к которым относятся также различные токсич-
ные фосфорорганические и хлорсодержащие инсектициды и герби-
циды, имеет модификация лучевого ионизационного детектора, так
называемого электронного накопителя или электронной ловушки
(электроннозахватного детектора) 168-171.
Молекулы азота, служащего газом-носителем, ионизируются
при облучении тритиевым источником; при этом образуются сво-
бодные электроны, которые под действием прилагаемого к камере
детектора определенного потенциала движутся к аноду, служаще-
му измерительным электродом, и дают устойчивый фоновый ток.
213
Если в испытуемом веществе присутствуют молекулы, обладаю-
щие сродством к электрону, то образуются отрицательные ионы,
которые медленнее передвигаются в электрическом поле и почти
все претерпевают рекомбинацию с положительными ионами, пре-
жде чем они достигают анода. Измеренный вначале при пропуска-
нии чистого газа-носителя ионизационный ток уменьшается на ве-
личину, пропорциональную числу образовавшихся в единицу вре-
мени отрицательных ионов.
В определенном интервале существует пропорциональная зави-
симость между сигналом этого типа детекторов и произведением
концентрации на способность соединений захватывать электроны.
Измеряемая таким детектором различная способность веществ к
захвату электронов может быть использована для идентификации
неизвестных соединений.„ Средняя чувствительность детектора по
фосфорорганическим соединениям находится в области 10*9г.
Широкое применение для определения хлор-, серу- и фосфорсо-
держащих соединений находит устройство, в котором содержащие-
ся в газе-носителе вещества после прохождения через разделитель-
ную колонку сжигают в кварцевой трубке в токе кислорода или
же восстанавливают водородом при высокой температуре; про-
дукты реакции затем непрерывно определяют в кювете кулономет-
рическим титрованием 172-174. Хотя по сравнению с определением
при помощи электронозахватного детектора этот способ менее чув-
ствителен, его показания элементспецифичны и испытывают мень-
шее влияние других присутствующих органических соединений.
Этот метод пригоден для определения органических фосфатов в
различных материалах 17S~178. При комбинации этого метода с эк-
стракционным способом можно было определять фосфаты 179 в кон-
центрации 25—50 нг в 1 л воды.
Наибольшей специфичностью и чувствительностью при опреде-
лении фосфорорганических соединений обладает натрий-термоион-
ный детектор180-181, который был впервые описан в 1964 г. Детектор
состоит из покрытой солью натрия проволочной спирали, располо-
женной непосредственно над стандартным водородным пламенно-
ионизационным детектором. Соль, нагреваемая пламенем, генери-
рует поток ионов, значительно усиливающийся при прохождении
через детектор галоген- или фосфорсодержащих соединений. Впо-
следствии это устройство многократно совершенствовалось. В ча-
стности, измерение тока ионов натрия производилось во вто-
ром последовательно включенном пламенноионизационном детек-
торе *82-185, а проволочная спираль, покрытая солью щелочного
металла, была заменена наконечником, выполненным из бромида
цезия, спрессованного с соответствующим наполнителем, насажен-
ным на кварцевую горелку пламенноионизационного детектора 18в.
Все это привело к значительному увеличению продолжительности
работы детектора при сохранении чувствительности. Этим детекто-
ром можно обнаружить до 10“12г фосфорсодержащего соединения
и до 10*9 г галогензамещенных.
214
Высокая специфичность показаний достигается также комои-
нацией газовой хроматографии как с эмиссионной спектрометри-
ей 187-189 и измерением интенсивности эмиссионных полос фосфора
(2535, 65А), серы (2576А) и фтора (2516А), так и с инфракрасной
спектроскопией 28 и масс-спектрометрией190.
Исследование высококипящих органических фосфатов облегча-
ется применением газовой хроматографии для разделения и иссле-
дования продуктов пиролиза 190>191, отделения алкилиодидов после
реакции Цейзеля 192, сложных метиловых эфиров — после обработ-
ки диазометаном 193 или, в случае аналогов систокса, отделения ле-
тучих тиоэфиров после щелочного гидролиза или реакции с алко-
голятами 194. Сочетание тонкослойной хроматографии с газовой 195
дает возможность разделять смеси веществ, содержащих нелету-
чие компоненты.
8.7.4.2. Анализ других ОВ. В одном общеупотребительном спо-
собе обнаружения токсичных газов и паров в атмосфере198 испы-
туемый воздух пропускают через короткую газохроматографиче-
скую колонку до установления равновесия. Затем эту трубку, со-
держащую пробу воздуха, присоединяют к разделительной колонке
газового хроматографа и нагреванием переводят адсорбированные
соединения в хроматограф, где они разделяются и определяются.
Определение фосгена в воздухе в пределах концентраций 1 р.р. Ь.—
2 р. р т. осуществляется при помощи электроннозахватного детек-
тора 197.
Газовая хроматография во все возрастающем масштабе нахо-
дит применение в аналитической токсикологии 198’199, в том числе
и для определения алкалоидов и LSD203.
8.8. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Особый интерес представляют те методы полярографии, при по-
мощи которых возможны чувствительное определение и кинетиче-
ские исследования и которые можно комбинировать с методами
разделения при помощи распределительной или газовой хромато-
графии. 1
Применение других электрохимических методов и, в частности,
потенциометрического титрования приводилось при описании мно-
гих способов химического, биохимического и элементного анали-
зов ОВ.
8.8.1. Полярографические методы
Так как в общем полярографически определять можно только та-
кие ионы или молекулы, которые в результате приема или отдачи
электрона могут восстанавливаться или окисляться на электроде,
применение этого электрохимического способа для анализа орга-
нических соединений ограничено. Однако некоторые ОВ могут
быть определены полярографически.
215
Серный иприт полярографически пассивен по отношению к ртут-
ному капельному электроду. Однако при потенциале +0,9 в в паре
с насыщенным каломельным электродом сравнения он окисляется
на вращающемся сетчатом платиновом электроде, с которым ра-
ботают в этой области напряжения, и может быть определен ка-
чественно и количественно по высоте образующейся полярографи-
ческой волны69.
При исследовании каталитических волн, под которыми пони-
мают каталитическое смещение выделения водорода в область бо-
лее низкого потенциала под воздействием веществ даже поляро-
графически пассивных, Брдичка70 установил, что серный иприт в
присутствии аммиачного раствора C0CI2 вызывает аналогичный эф-
фект. Это каталитическое действие приписывается не самому ип-
риту, а продукту его реакции с аммиаком.
Из числа соединений, возможно обладающих этим свойством,
полярографическому исследованию был подвергнут р-аминоэтил-
меркаптан 71, для которого это предположение подтвердилось.
Количественное определение серного иприта возможно путем
измерения полярографической волны, которая следует за «кобаль-
товой ступенью».
Азотистые иприты полярографически пассивны, однако четвер-
тичный атом азота (ион этиленимония), промежуточно образую-
щийся при гидролизе в результате циклизации, полярографически
восстанавливается по следующей схеме72:
R\+2е- + н+
N( I --------;n—сн2—сн3
R'/ \СН2 R'/
На этом основании полярографические методы могут быть ис-
пользованы для кинетического исследования превращения азо-
тистых ипритов в этиленимониевые соединения 72’73.
Обнаружение и определение таких мышьяксодержащих ОВ,
как дифенил-, хлор- и цианарсины, адамсит, а-люизит, возможно
как методами классической полярографии с ртутным электродом,
так и осциллографической полярографией74. В этом методе на-
званные ОВ можно определять качественно по различному поло-
жению максимумов иа обеих полуволнах осциллограммы.
Применение классической полярографии для определения фос-
форорганических соединений, не говоря о косвенных методах, в
каждом случае определяется либо наличием восстанавливаемых
функциональных групп, таких, как в паратионе 75’76 и параоксоне77,
либо образованием при гидролитическом разложении полярогра-
фически активных продуктов. К таким веществам относятся мала-
тион (определяется фумаровая кислота78), систокс79 и тинокс80,
при определении которых измеряют каталитические волны водо-
рода, вызываемые образовавшимися при гидролизе тиольными
группами. Успехи в развитии осциллографической полярографии
привели к тому, что при помощи ее методов можно анализировать
216
и определять с высокой чувствительностью такие полярографиче-
ски пассивные вещества, как сложные эфиры фосфорной кис-
лоты 81-83.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Как осуществляется определение плотности ОВ при помощи пикнометра?
2. Как по температуре плавления можно идентифицировать неизвестное ве-
щество?
3. Какие существуют методы определения давления пара?
4. Какое значение имеет показатель преломления для характеристики орга-
нических соединений?
5. Какие аналитические возможности дает ИК-спектроскопия?
6. Охарактеризуйте различные виды хроматографии.
7. Как определяется величина R/ и какое значение она имеет?
8. Какие преимущества имеет газовая хроматография по сравиенню с дру-
гими видами хроматографии?
ЛИТЕРАТУРА
1. Vogel A. I. et al., J. Chem. Soc., 1952, 514, 531, 535.
2. Sayre R., J. Am. Chem. Soc., 80, 5438 (1958).
3. Jones W. C. et al., J. Phys. Chem., 37, 583 (1933).
4. Разумов А., Макашева О., ЖОХ, 26, 1436 (1956).
5. EisenlohrF., Wohlisch E., Ber., 53, 1746 (1920).
6. К г e 11 E., Handbuch der Laboratoriumsdestillation. Berlin, 1958.
7. F a ge г 1 i n d L. G., Acta Chem. Scand., 7, 701 (1953).
8. G i 11 i s R. G. et al., J. Am. Chem. Soc., 80, 2999 (1958).
9. Mohler H., Polya J., Helv. Chim. Acta, 19, 283, 1222, 1239 (1936).
10. Mohler H„ Helv. Chim. Acta, 20, 287, 1188 (1936).
11. Mohler H., S о г ge J., Helv. Chim. Acta, 22, 235 (1938).
12. Mohler H. et al., Helv. Chim. Acta, 23, 100, 104, 1211 (1940).
13. Moh ler H., Helv. Chim. Acta, 24, 571 (1941).
14. M о h 1 e r H., Protar, 7, 78 (1941).
15. Mohlefr H„ Protar, 10, 27, 60 (1944).
16. Bradford L. W., Brackett J. W., Mikrochim. Acta, 1958, 353.
17. Gershmann H. R., Ke tela ar J. A., Rec. trav. chim., 77, 1018 (1958).
18. .S p u г г R. A., C h u b b T. A., Spektrochim. Acta, 10, 431 (1958).
19. Imparato-Gargano E., Plamieri F., Ann. Fac. Sci. Agrar., 28, 239
(1962/1963).
20. Larsson L., Acta Chem. Scand., 6, 1470 (1952).
21. Lorquet J. C., Va ssart S., Bull. Soc. Chim. Belg., 68, 336 (1959).
22. T a m m e 1 i n L. E., Acta Chem. Scand., 11, 1738 (1957).
23. Henglein A., Schrader G., Mtihlmann R., Z. anal. Chem., 141, 276
(1954).
24. Goldenson J., Appl. Spectroscopy, 18, 155 (1954).
25. McCaulley D. F., Cook J. W., J. Assoc. Off. Agric. Chem., 43, 710
(1960).
26. Frehse H., Pflanzenschutz-Nachr., 16, 182 (1963/64).
27. В 1 i n n R. C., G й n t h e г F. A., Residue Rev., 2, 99 (1963).
28. С г о s b у N. T., Laws E. Q., Analyst, 89, 319 (1964).
29. S t a n es cu G. et aL, Rev. Chim. Bukarest, 15, 416 (1964).
30. Derkos'ch J. et al., Monatsheft fur Chemie, 85, 684 (1957).
31. White T. T., McKinley G. G., J. Assoc. Off. Agric. Chem., 44, 589
(1961).
32. BlinnR. C., J. Assoc. Off. Agric. Chem., 46, 952 (1963).
33. G i и f f г i d a L., J. Assoc. Off. Agric. Chem., 48, 354 (1965).
34. Aksnes D., Aksnes G., Acta Chem. Scand., 17, 1262 (1963).
217
35. В е 11 J. V. et al., J. Am. Chem. Soc., 76, 5185 (1954).
36. Thomas L. C„ Chittenden R. A„ Spectrechim. Acta, 20, 467 (1964)
37. D a a s c h L. W., S m i t h D. C„ Anal. Chem.. 23, 853 (1951).
38. Ketelaar J. A. A., Gersmann H. R., Rec. trflv. cflim., 78, 190 (1959).
39. Holmstedt B., Larsson L., Acta Chem. Scand., 5, 1179 (1951).
40. Meyrick С. I., Thompson H. W„ J. Chem. Soc., 1950, 225.
41. Ma ar sen J. W. et al., Rec. trav. chim., 76, 713 (1957).
42. В e 11 a m у L. J., В e e c h e г L„ J. Chem. Soc., 1953, 728.
43. Bellamy L. J., Beecher L., J. Chem. Soc., 1952, 475, 1701.
44. Mclvor R. A. et al„ Canad. J. Chem., 34, 1611 (1956).
45. D a a s ch L. W., S m i t h D. C., J. Chem. Phys., 19, 22 (1951).
46. Chittenden R. A., Thomas L. C„ Spectrochim. Acta, 20, 1679 (1964).
47. Chittenden R. A., Thomas L. C. Spectrochim. Acta, 21, 861 (1965).
48. Mayhood J. E„ Harvey. R. B., Canad. J. Chem., 33, 1552 (1955).
49. Q u i n ch о n J. et al., Bull. Soc. Chim., 1961, 1086.
50. S a n d i E„ Z. anal. Chem., 167, 241 (1959). .
51. S p e nсеr E. Y. et al., J. Chem. Soc., 1958, 2968.
52. S e r r a M„ M a 1 a t es t a P., Ann. Chim., 43, 569 (1953).
53. Thomas L. C., Chittenden R. A., Spectrochim. Acta, 20, 489 (1964).
54. Nyquist R. A., Appl. Spectroscopy, 11, 161 (1957).
55. G i 11 i s R. G. et al., J. Am. Chem. Soc., 80, 2999 (1958).
56. P e p p a r d D. F. et al., J. Inorg. Nucl. Chem., 12, 60 (1959).
57. Mclvor R. A., Hub ley С. E., Canad. J. Chem., 37, 869 (1959).
58. HouallaD., Wolf R., Bull. Soc. Chim. France, I960, 129.
59. T h о m a s L. С., C h i 11 e n d e n R. A., Chem. Ind., 1961, 1913.
60. Gryszkiewicz-Trochimowski E. et al„ Bull. Soc. Chim. France,
1961, 739.
61. Q u i n ch о n J. et al., Bull. Soc. Chim., France, 1961, 735.
62. Quinchon J. et al., Bull. Soc. Chim. France, 1962, 169.
63. Q u i n c h о n J. et al., Compt. rend., 225, 858 (1962).
64. Nyquist R. A., Spectrochim. Acta, 19, 713 (1963).
65. Stolz er C., Simon A., Z. anorg. allg, Cnem., 339, 30, 38 (1965).
66. К i r ch n e r F., Z. angew. Phys., 8, 478 (1956).
67. M о 111 a u A. Y„ Anal. Chem., 29, 1196 (1957).
68. Riggs A. J., Analyst, 80, 279 (1955).
69. Нем. пат. 741368 (1943).
70. В r d i с к a R., Coll. Czech. Chem. Comm., 9, 76 (1937).
71. Trkal V., Chem. Listy, 49, 1499 (1955).
72. Man t savin os R., Christian J. E., Anal. Chem., 30, 1071 (1958).
73. Zallen H. et al„ J. Pharm. Science, 50, 783 (1961).
74. К a 1 v о d a R., Sbornik praci, 1954, 58.
75. Bowen С. V., Edwards F., Anal. Chem., 22, 706 (1950).
76. G a j a n R. J., J. Assoc. Off. Agric. Chem., 46, 216 (1963).
77. В 1 a z e к J., Cs. farm., 7, 455 (1958).
78. Jur a H. W„ Anal. Chem., 27, 525 (1955).
79. G a j a n R. J., J. Assoc. Off. Agric. Chem., 45, 401 (1962).
80. Wo ggo n H. et al., Z. anal. Chem., 211, 113 (1965).
81. G'a j a n R. J., J. Assoc. Off. Agric. Chem., 45, 401 (1962).
82. G a j a n R. J., Residue Rev., 6, 75 (1964).
83. N a n g n i о t P., Anal. Chim. Acta, 31, 166 (1964).
84. Пат. США 2991302 (1961).
85. Jakob F. et al.. Taianta, 8, 431 (1961).
86. V a г о n A. et al., Taianta, 9, 573 (1962).
87. T a m m e 1 i n L. E„ Arkiv for Kemi, 12, 287 (1958).
88. Tammelin L. E., Acta Chem. Scand., 11, 1738 (1957)
89. Larsson L., Acta Chem. Scand., 11, 1131 (1957).
90. Gage J. C., Biochem. J., 54, 426 (1953).
91. March R. B. et al., J. Agric. Food Chem., 2, 732 (1954).
92. M 0 h 1 m a n n R., T i e t z H., Hofchen-Briefe, 2, 116 (1956).
93. Augustinsson К. B., Jonsson G., Acta Chem. Scand., 11, 275 (1957).
218
94. С о о к J. W„ J. Assoc. Off. Agric. Chem., 37, 987 (1954).
95. R ah n H. W., Urban G., Pharmazie, 19, 597 (1964).
96. M e t с a 1 f R. L, M a r c h R. B, Science, 117, 527 (1953) .
97. Irudayasamy A., Nat a raj an A. R., Analyst, 90, 503 (1965).
98. Eichenberger J., Gay L., Mitt. Geb. Lebensmitt. Hyg., 51, 423 (1960).
99. H a n e s C. S., I s h e r w о о d F. A., Nature, 164, 1107 (1949).
100. Ottet J. К. H, Nature, 176, 1078 (1955).
101. G a ge J. C., Biochem. J., 54, 426 (1953).
102. С о о к J. W„ J. Assoc. Off. Agric. Chem., 38, 150 (1955).
103. McKinley W. P., Read S, J. Assoc. Off. Agric. Chem., 45, 467 (1962).
104. Getz M. E, Friedman S. I., J. Assoc. Off. Agric. Chem., 46, 707 (1963).
105. McKinley W. P., J oh al P. S., J. Assoc. Off. Agric. Chem., 46, 840 (1963).
106. M с К i n 1 e у W. P., Proc. Canad. Soc. Forensic. Sci., 2, 433 (1964).
107. Menn J. J. et al., Nature, 202,697 (1964).
108. О r 11 о f f R, F r a ti z P, Z. Chem., 5, 388 (1965).
109. Nies sen H. et al., J. Chromatorg., 9, 111 (1962).
110. Cook J. W., J. Assoc. Off. Agric. Cheni., 37, 984 (4954).
111. Petsch ik H., Steger E. J., Chromatogr, 7, 135 (1962).
112. F i s ch e r R., О 11 e r b e с к N., Sci. Pharm., 27, 1 (1959).
113. Fischer R., Dtsch. Z. ges. gerlchtl. Med., 51, 537 (1961).
114. Fischer R, К 1 i n ge 1 h 6 11 e r W., Arch. Toxikol, 19, 119 (1961).
115. Menn J. J. et al., J. Agric. Food Chem., 5, 601 (1957).
116. Adams J. M. et al., J. Agric. Food Chem., 11, 178 (1963).
117. Ba tor a V. Dissertation. Bratislava. 1955.
118. Anliker R., Menzer R. E., J. Agric. Food Chem., 11, 291 (1963).
119. McRay H. F., McKinley W. P., J. Assoc. Off, Agric. Chem., 44, 207
(1961).
120. Grote J., J. Biol. Chem., 93, 25 (1931).
121. Benesch R. et al., Science, 123, 981 (1956).
122. Gerlach E., Deuticke B, Biochem. Z, 337, 477 (1963).
123. Fa derl N„ Mitt. Geb. Lebensmitt. Hyg., 53, 154 (1962).
124. Henn ion G. F., Ind. Eng. Chem., Anal. Ed., 9, 479 (1937).
125. Bent H. E, Krinbill C. A., J. Am. Chem. Soc., 72, 2757 (1950).
126. Baumler. J, Ripstein S., Helv. Chim. Acta, 44, 1062 (1961).
127. S a lo T. et al., Z. Lebensmitt. Unters, 117, 369 (1962).
128. Walker К. C, Beroza M., J. Assoc. Off. Agric. Chem, 46, 250 (1963).
129. Мастрюкова T. А. и др, Изв. АН СССР, ОХН, 1963, 2211.
130. S t a n 1 е у С. W, J. Chromatogr, 16, 467 (1964).
131. С о п к i п R. A, Residue Rev, 6, 136 (1964).
132. Eder F. et al, Mitt. Geb. Lebensmitt. Hyg, 55, 98 (1964).
133. Kovacs M. F, J. Assoc. Off. Agric. Chem, 47, 1097 (1964).
134. Licht L, Mitt.-Bl. GDCh. Lebensmittelch. gerichtl. Chem, 18, 190 (1964).
135. S a 1 a m ё M, J. Chromatogr, 16, 476 (1964).
136. К1 e m e n t R, W i 1 d A, Z. anal. Chem, 195, 180 (1963).
137. LamotteR. et al. Bull. Soc. Chim. France, 1965, 919.
138. P e t s c h i к H, S t e g e r E, J. Chromatogr, 9, 307 (1962).
139. В r a i t h w a i t e D. P, Nature, 200, 1011 (1963).
140. Ackermann H, Sp ran ger D, J. Chromatogr, 17, 608 (1965).
141. Donner R, Lohs K, J- Chromatogr, 17, 349 (1965).
142. В uny an B. J, Analyst, 89, 615 (1964).
143. W a 1 d i D. et al, J. Chromatogr, 6, 61 (1961).
144. Baumler J,. Ripstein S, Pharmac. Acta Helv, 36, 382 (1961).
145. Ta kit a ni S. et al, Jap. Analyst, 12, 1156 (1963); Z. anal. Chem, 211, 364
(1965).
146. Hashmi M. H. et al. Taianta, 12, 713 (1965).
147. Canic V. D. et al, Z. anal. Chem, 213, 251 (1965).
148. Merkus F. W. H. M, Pharmac. Weekbl, 98, 947 (1963).
149. S e i 1 e r H, S e i I e r M, Helv. Chim. Acta, 43, 1939 (1960).
150. Seiler H, Helv. Chim. Acta, 45, 381 (1962).
151. Sent H. J, Z. Chem, 6, 102 (1966).
219
152. Przybylowicz E. P. et al., J. Chromatogr., 20, 506 (1965).
153. В r e g о f f H. M., J. Biol. Chem., 205, 565 (1953).
154. P 1 a p p F. W„ C a s i d a J. E„ Anal. Chem., 30, 1622 (1958).
155. Kunzi P. et al., Dtsch. Z. ges. gerichtl. Med., 52, 605 (1962).
156. Abbott D. C. et al., Analyst, 89, 480 (1964).
157. Henkel H. G„ Chemia, 19, 128 (1965).
158. Genest K-, Farmilo C. G., J. Pharm. Pharmacol., 16, 250 (1964).
159. d a 1 С о r t i v о L. A. et al., Anal. Chem., 38, 1959 (1966).
160. К i e n i t z H., Chemie-Ing.-Techn., 32, 643 (1960).
161. Gagliarde E., Pokorny G., Mikrochim. ichnoanalyt. Acta, 1965, 699.
162. Shipotofsky S. H., Moser H. C., Anal. Chem., 33, 521 (1961).
163. Fe i n 1 a n d R. et al., Anal. Chem., 35, 920 (1973).
164. D a v i s A. et al., J. Gaschrom. 1963, 23
165. Kanazawa J. et al., Agric. Biol. Chem. Tokio, 29, 56 (1965); Z. anal.
Chem., 218, 224 (1966).
166. Barette J. P., Pay! er R., J. Assoc. Agric. Chem., 47, 259 (1964).
167. Ber 1 i n K. D. et al., J. Gaschrom., 3, 256 (1965).
168. Moore A. D„ J. Econ. Entom., 55, 271 (1962).
169. E ga n H. et al., Analyst, 89, 175 (1964).
170. J a i n N. C. et al., J. Pharm. Pharmakoi., 17, 362 (1965).
171. Dimick К. B., Hartmann H., Residue Rev., 4, 150 (1963)
172. Coulson D. M. et al., J. Agric. Food Chem., 7, 250 (1959).
173. Coulson D. M. et al., J. Agric Food Chem., 8, 399 (1960).
174. Coulson D. M., Cavanagh L. A., Anal. Chem., 32, 1245 (1960).
175. В о s i n W. A., Anal. Chem., 35, 833 (1963).
176. Burke J., Holswade W., J. Assoc. Off. Agric. Chem., 47, 1245 (1964).
177. N e 1 s о n R. C., J. Assoc. Off. Agric. Chem., 47, 284 (1964).
178. В u r c h f i e 1 d H. P. et al., J. Gaschrom., 3, 28 (1965).
179. Teasley J. I., Cox W. S., J. Am. Water Works Assoc., 55 (1963).
180. G i u f f r i d a L., J. Assoc. Off. Agric. Chem., 47, 293 (1964).
181. Giuffrida L., Ives F., J. Assoc. Off. Agric. Chem., 47, 1112 (1964)
182. Karmen A., Giuffrida L., Nature, 201, 1204 (1964).
183. Karmen A., Anal. Chem., 36, 1416 (1964).
184. Karmen A., J. Gaschrom., 3, 336 (1965).
185. Jentzsch D. et al., Z. Anal. Chem., 221, 377 (1966).
186. Hartman С. H., Aerograph Research Notes, 1966 (Summer), p. 1.
187. M с С о r m а с к A. J., Anal. Chem., 37, 1470 (1965).
188. В a ch e C. A., L i s к D. J., Anal. Chem., 37, 1477 (1965).
189. В г о d у S. S., C h a n e у J. E., J. Gaschrom., 4, 42 (1966).
190. Legate С. E., Burnham H. D., Anal. Chem., 32, 1042 (1960).
191. Hanneman W. W., Porter R. S., J. Org. Chem., 29, 2996 (1964).
192. Gutenman W. H., Lisk D. J., J. Agric. Food. Chem., 11, 470 (1963)
193. H a r d у C. J., J. Chromatogr., 13, 372 (1964).
194. Wei n i g E. et al., Dtsch. Z. gerichtl. Med., 51, 566 (1961).
195. Kaiser R., Z. anal. Chem., 205, 284 (1964).
196. N о v a к J. et al., Anal. Chem., 37, 660 (1965).
197. P r i es t le у L. 1. et al., Anal. Chem., 37, 70 (1965).
198. Goldbaum L. R., S chloe gel E. L., Proc. Chem. Toxicol., 1, 11 (1963).
199. К a z у a к L., К n о b 1 о с к E. C., Anal. Chem., 35, 1448 (1963).
200. Soh r H., Chem. Zvesti, 16, 316 (1962).
201. M e n d о z a С. E. et al., Analyst, 93, 34 (1068).
202. Mendoza С. E. et al., Analyst, 93, 173 (1968).
203. R a d e с к a C., N i g a m I. C., J. Pharm. Sci., 55, 861 (1966).
9. ИНДИКАЦИЯ БОЕВЫХ ОТРАВЛЯЮЩИХ
ВЕЩЕСТВ В ПОЛЕВЫХ УСЛОВИЯХ
9.1. СУБЪЕКТИВНОЕ ВОСПРИЯТИЕ
Даже в условиях современной войны большое значение для целей
разведки имеет субъективное восприятие различных форм и видов
применения химического оружия. Для создания полной картины
военнослужащие, призванные осуществлять химическую разведку
и наблюдение, могут существенно дополнить результаты объектив-
ных методов данными субъективного восприятия.
Различают следующие субъективные восприятия* — это ощу-
щение раздражения органов дыхания, глаз и кожи; ощущение за-
паха или вкуса; обнаружение отличающейся от нормального вида
растительности, удушливого тумана в воздухе или капель ОВ на
растительности и почве при слабом глухом разрыве снарядов; об-
наружение осколков или неразорвавшиХся боеприпасов, по мар-
кировке которых можно судить о типе отравляющего вещества.
Во время первой и даже второй мировых войн большое внима-
ние уделялось обучению химиков-разведчиков определять ОВ по
запаху 1> 2. Для этогб предварительно отбирались люди, обладаю-
щие очень чувствительным обонянием, что устанавливалось про-
бой на растворах уксусной кислоты (0,1 н.; 0,2 н.; 0,5 н. растворы)
или аммиака (0,1 н.; 0,15 н.; 0,2 н.; 0,5 н. растворы). Для развития
повышенной способности различать и запоминать определенные за-
пахи, для тренировки избирательной способности обоняния и спо-
собности ориентироваться на местности отобранный персонал под-
вергался обучению по специальной программе, в том числе с так
называемым набором запахов. Для создания набора запахов ис-
пользовались разные, часто употребляемые технические продукты,
обладающие характерными запахами, а также ОВ. При этом обу-
чении руководствовались такими основными правилами, которые
еще и теперь имеют значение при определении запаха токсичных
веществ.
1. Не делать глубокого вдоха, легкий вдох должен ограни-
читься областью цоса.
* Субъективные методы обнаружения ОВ, токсинов и т. п., в связи с по-
явлением высокотоксичных веществ и тем более токсинов, сейчас практически
уже теряют свое значение. Обнаружение ОВ по запаху невозможно, так как
в боевых концентрациях эти вещества почти лишены запаха; современные спо-
собы применения ОВ таковы, что не позволяют обнаружить на местности капли
веществ; звуки разрыва химических боеприпасов очень трудно отличимы от
звуков разрыва обычных бризантных средств, и, наконец, попытка органолепти-
ческого обнаружения ОВ может привести к серьезным отравлениям. — Прим. ред.
221
2. Понюхать нужно только один раз; при многократном повто-
рении испытания обоняние притупляется.
3. Ощутив запах, надо подумать, чем он вызван; память на за-
пахи можно развить тренировкой.
4. Каждое ощущение запаха или полное отсутствие запаха сле-
дует зафиксировать.
I
Таблица 22. Запахи ОВ и количества их, обнаруживаемые по этому признаку
Отравляющее вещество Характерный запах и раздражающее действие Минимальные кон- центрации вещества, обнаруживаемые по запаху, мг/л Минимальные опасные концентрации при ЭКСПОЗИЦИИ 10 MUHt мг/л
Зоман Эфирный, похожий на! камфору Концентрации, 0,0001 (через 2 мин миоз)
Зарин Табун Эфириый, слабый распознавае- мые по запаху, 0,0005 (через 2— 3 мин миоз)
чистый техииче- Фруктовый или горь- кого миндаля Рыбный вызывают силь- нейшие отра- вления 0,005 (через 2 мин сильный миоз)
с кий
Серный иприт Свежего Лука 0,0013 0,001
ЧИСТЫЙ
техниче- ский Хрена, горчицы или чеснока — —
Азотистый иприт Рыбы (по амину), в большом разведе- нии герани —
Люизит Герани, резкий и не- приятный - 0,014 0,0008
Этилдихлор- арсии Чеснока, резко раз- дражающий 0,001 0,001
Метилдихлор- То же 0,0008 0,002
Дифеиилхлор- арсин Фруктовый, раздра- жающий верхние ды- хательные пути 0,0003 0,0005
Дифен илциан- арсин Горького миндаля, сла- бый 0,0003 0,0001
Адамсит Без запаха 0,0003 0,00038
Хлорацетофе- Фруктовый 0,0002 0,0003
Бромбензил- цианид Горького миндаля, сла- бый 0,0001 0,00015
Хлорпикрин Картофельной ботвы, затхлый, режущий 0,0073 0,009
Фосген Гнилых фруктов, гние- ния, прелой листвы иля мокрого сеиа, сладковатый’ 0,0044 0,005
Синильная Горького миндаля 0,001 0,020
кислота
Хлорацетон Похожий на запах НС1, резкий 0,01
222
5. После каждой пробы на запах перед новой пробой нужно не-
сколько раз вдохнуть носом чистый воздух, пока не исчезнет ощу-
щение предыдущего запаха.
6. Не курить при испытаниях на запах; курение притупляет обо-
няние. '
Противогазы химиков-разведчиков обладали специальными кла-
панами, при помощи которых воздух мог поступать в подмасочное
пространство, минуя коробку противогаза. Эти клапаны монтиро-
вались между коробкой и лицевой частью или в саму лицевую
часть и открывались нажимом на кнопку или при помощи вытяж-
ного кольца3. Так как по сравнению с техническими средствами
разведки этд приспособление было весьма простым и очень быстро
приводилось в действие, то в то время снаряжение солдат объек-
тивными средствами обнаружения ОВ считалось излишним и бес-
полезным 4.
В связи с появлением к концу второй мировой войны фосфор-
органических ОВ, обладающих высокой токсичностью при инга-
ляции, обучение специалистов распознавать ОВ по запаху утра-
тило свое первоначальное значение. Такие вещества, как зоман,
зарин и V-газы, недьзя определять по запаху. Малейшие их коли-
Обладающие в оргадыдыхания, уже токсичны.
^^ыми симптдййми, предупреждающими о действии этих ве-
ществ, являются миоз и загрудинный эффект. Поэтому для обна-
ружения таких ОВ пользуются только объективными методами
индикации, которые соответствуют высокой токсичности ОВ.
Вместе с тем нельзя не отметить, что возможности человеческо-
го обоняния по отношению к некоторым химическим соединениям
необычайно велики. Так, например, меркаптан ощущается по за-
паху уже при концентрации 0,000044 мг)м3. Из данных табл. 22
видно, что для некоторых ОВ область органолептического их об-
наружения достигает количеств, обнаруживаемых аналитическими
методами.
Об использовании высокоразвитых способностей обоняния не-
которых животных для обнаружения ОВ было указано в раз-
деле 7.
9.2. ПРОСТЕЙШИЕ СРЕДСТВА РАЗВЕДКИ
В последующем будут рассмотрены простые в обращении средства
индикации, используемые для быстрого обнаружения в полевых
условиях стойких ОВ на поверхностях и паров ОВ в воздухе.
Эти средства, так называемые средства массовой индикации, на-
ходят применение в армиях и службе гражданской обороны мно-
гих стран.
9.2.1. Индикаторный порошок
Для того чтобы отличить заражеинйй стойкими ОВ участок ме-
стности от незараженного, что необходимо для дегазации, обхода
223
или преодоления этого участка, его можно обработать так назы-
ваемыми индикаторными порошками. Обычно они представляют
собой порошкообразную основу, наполнитель или носитель с не-
значительной добавкой реагента. В зависимости от реагента раз-
личают два типа индикаторных порошков.
В первом типе порошков краситель или смесь красителей до-
бавлена в таком количестве, что основа порошка остается совсем не
окрашенной или окрашена очень слабо. Для этой цели используют
такие красители, которые не растворяются в воде, но хорошо рас-
творяются в индицируемых ОВ. При обработке индикаторным по-
рошком поверхности, на которой имеются капли ОВ, краситель
переходит в раствор. Появляющаяся интенсивная окраска создает
хорошо заметный контраст с исходной окраской порошка или по-
верхности.
Специфичность этого типа индикаторных порошков очень мала,
так как и брызги от горючего, и применяемые при дегазации рас-
творители могут также явиться причиной изменения окраски. По-
этому такие порошки в основном используют в тех случаях, когда
факт применения противником ОВ и тип его установлены другим
путем, а порошок дает возможность наиболее простым и дешевым
способом установить границы зараженной поверхности.
В качестве примера можно привести рецептуры некоторых та-
ких порошков.
Индикаторный порошок 1
Кварцевая мука....................................... 950 г
Пнразолоновый пигмент (осветитель)..................... 5 г
Перманентный красный R......................... 30 г
Судан красный 3R...................................... 10 г
При контакте с серным ипритом порошок приобретает кроваво-
красную окраску5.
Индикаторный порошок 2
Тонко измельченный силикагель............ 1000 г
Вофаноловый прочный черный................. 10 г
Жирорастворимый желтый.................... 0,5 г
При контакте с серным ипритом и фосфорсодержащими ОВ
типа зарина порошок приобретает темно-зеленую окраску.
Индикаторный порошок 3
Жирорастворимый красный................... I г
Мел в порошке......................... 100 г
Морской песок......................... 3000 г
При контакте с серным ипритом порошок приобретает темно-
красную окраску6. Если на капли, окрашенные таким образом, на-
нести смесь из 1 вес. ч. FeCK и 7 вес. ч. мела, то при наличии сер-
ного иприта или бромбензилцианида окраска изменится на зеле-
ную. Капли углеводородов, масел и хлорпикрина приобретают
ржавый оттенок и только после продолжительного времени зе-
ленеют.
224
Индикаторный порошок 4
Тальк............................ 100 г
Розанилин.......................... 5 г
Минеральное масло.................. 3 г
Тальк и минеральное масло перемешивают 30 мин на шаровой
мельнице, а затем смешивают с розанилином. Прибавление ми-
нерального масла способствует гидрофобизации порошка7. При
контакте с серным ипритом порошок приобретает темно-красную
окраску.
В качестве других красителей при изготовлении индикаторных
порошков используют ацетатнорастворимый красный, кристалли-
ческий фиолетовый —основание, церезиновый синий, церезиновый
фиолетовый или его основание и др.; в качестве осветлителя можно
добавлять хромовый желтый.
Основой обычно служат кварцевая мука, измельченный сили-
кагель, тальк, тонкий морской песок, карбонаты щелочноземель-
ных металлов и сульфат бария.
Вторым типом порошков являются так называемые реактивные
индикаторные порошки. В этом случае вещества, добавленные к
носителю, вступают с ОВ в химическое взаимодействие, в резуль-
тате которого образуются окрашенные продукты. Используя соот-
ветствующие реагенты, можно получить порошок, специфичный по
отношению к определенному ОВ. В простейшем случае, например
применяя кислотно-основные индикаторы, можно определять кис-
лотные ОВ или ОВ, отщепляющие при гидролизе кислоты38. При-
мером такого типа индикаторного порошка8 может служить поро-
шок следующего состава (в %):
Кварцевая мука (pH 7,1—7,5).............. 88,5
Кизельгур (pH 7,5—8,0)................... 5
NaHCO3 .................................. 5
Метиловый оранжевый...................... 0,5
Метиловый красный........................ 0,6
При наличии серного и азотистого ипритов, люизита и фос-
форорганических ОВ типа зарин порошок окрашивается в красный
цвет.
Индикаторные порошки готовят измельчением и перемешива-
нием компонентов в шаровой мельнице и применяют рассыпанием
из барабанов.
Индикаторные порошки наиболее удобны для обнаружения
стойких ОВ на местности, особенно при низких температурах, ко-
гда давление паров ОВ настолько мало, что определение их кон-
центраций в воздухе с помощью индикаторных трубок невозможно.
На окрашенных в различные цвета поверхностях часто бывает
очень трудно быстро обнаружить капли ОВ, вследствие чего при-
менение индикаторных бумаг становится возможным только пос-
ле затраты значительного времени на выявление капель. Нанесение
8 Зак. 677 225
же индикаторного порошка сразу на большую поверхность позво-
ляет быстро и просто обнаружить ОВ. Недостатком индикаторных
порошков является малая специфичность известных до сих пор по-
рошков, а также невозможность их применения при аэрозольном
состоянии О В. При оседании аэрозоля на поверхностях образуется
очень тонкая «пленка» ОВ, в которой вещество может быть обна-
ружено только при помощи реактивного индикаторного порошка, в
состав которого входил бы очень чувствительный реагент. В этих
условиях более применима индикаторная бумага, при помощи ко-
торой можно взять пробу мазком.
9.2.2. Индикаторные карандаши
Пропитанные специальным реагентом наполнители можно прессо-
вать в штифты — карандаши. При помощи таких карандашей мож-
но наносить штриховку на твердые материалы: камни, металлы,
дерево и бумагу, которые в присутствии паров ОВ будут изменять
окраску. Защищенные футляром карандаши сохраняются продол-
жительное время.
Ниже приводятся некоторые рецепты изготовления индикатор-
ных карандашей.
Карандаш для индикации галогенциаиов 9. Смесь из 10% 4-бензилпиридииа,
4% барбитуровой кислоты, 86% BaSO4 тщательно перемешивают до полного
поглощения 4-бензилпиридина и из полученной массы прессуют штифты. Белые
штрихи, нанесенные таким карандашом, в присутствии паров C1CN и BrCN
окрашиваются сначала в красный, а потом в синий цвет. Такие карандаши мо-
гут сохраняться при комнатной температуре в течение трех лет.
Карандаш для индикации синильной кислоты. Его готовят смешением и по-
следующим прессованием в штифты 14% хлорамина Т и 86% сульфата бария.
Поверх белых штрихов, нанесенных этим карандашом, наносят штриховку ка-
рандашом на галогенцианы. Этим способом через 1 мин можно обнаруживать
HCN, начиная от концентраций 0,005 мг!л, и C1CN — начиная от 0,001 мг/л.
Карандаш для индикации фосгена*. В его состав входят 2% 4-(п-иитробен-
знл)-пиридина, 5% N-фенилбензиламина, 5% Na2CO3 и 88% аморфного, ней-
трального BaSO4.
Растворяют в бензоле 4- (n-нитробензил)-пиридин и N-фенилбензиламин и
смешивают раствор с сульфатом бария. Количество бензола подбирают так,
чтобы весь раствор был поглощен сульфатом бария и последний был пол-
ностью им смочен (на 100 г смеси берут 60—80 мл бензола). Бензол испаряют
или лучше отгоняют в вакууме, после чего смесь тщательно растирают с водным
раствором карбоната натрия. Необходимое количество воды определяют пред-
варительно. Для этого к 1 г смеси из градуированной пипетки постепенно при-
бавляют воду до тех пор, пока она вся не впитается в смесь. К остальной части
смеси прибавляют воду по расчету, предварительно растворив в ней карбонат
натрия. Перед прессованием воду удаляют сушкой в вакууме. Срок хранения —
1 год.
Пары фосгена, начиная от 0,00003 мг)л, через 1 мин окрашивают светло-
серые полосы, нанесенные этим карандашом, в красный цвет; пары соляной
кислоты при более высоких концентрациях мешают индикации.
Индикаторный карандаш для фосгена, обладающий в два раза меиьшей
чувствительностью, можно приготовить, если в раствор из 1 г бис-(п-диметил-
амино-о-амннофенил)-кетона, 0,5 г кетона Михлера и 5 г М-фенил-1-нафтиламина
в 80 мл ацетона погрузить на 24 я для пропитки стержни школьного мела.
Карандаш для индикации люизита*. Состав — 5% тиокетона Михлера, 95%
BaSO4.
226
Растворяют тнокетон Михлера в бензоле и полученный раствор перемеши-
вают с сульфатом барня. Бензол удаляют из смеси сначала проветриванием на
воздухе, а потом испарением в вакууме; из оставшейся массы прессуют штифты.
Бледно-коричневая окраска этих карандашей при соприкосновении с жидким
люизитом или при наличии в воздухе высокой концентрации его паров изме-
няется на интенсивно сине-зеленую. Такое же изменение окраски происходит в
присутствии этилдихлорарсина; фосген вызывает пурпурную окраску.
Карандаши для индикации нервно-паралитических ОВ 10. Состав 1—80%
4,4'-бис-(диэтиламино)-бензофеноиоксима, 10% NaCN, 10% BaSO4. Из смеси
прессуют штифты. В присутствии паров табуна и зарина, при концентрациях
менее 1 мкг/л, штрихи, нанесенные этими карандашами, за 1 мин окрашиваются
в цвета от оранжевого до красного.
Состав 2 — натриевая соль диизонитрозоацетона11. Эта соль получается при
взаимодействии 5 же этилата натрия с 1 моль динзонитрозоацетона в этиловом
спирте. Из соли прессуют штифты. Штрихи, нанесенные этими карандашами в
присутствии незначительных количеств паров ДФФ, табуна, зарина и с мень-
шей чувствительностью ТЭПФ, параоксона и паратиона, приобретают красно-
фиолетовое окрашивание.
9.2.3. Индикаторные бумаги
Преимущество различных видов индикаторных бумаг состоит не
только в том, что ими очень просто пользоваться, но еще и в том,
что их очень легко изготовлять. В малых количествах они могут
быть изготовлены даже в лаборатории, без применения специаль-
ного оборудования. В качестве основы используется как фильтро-
вальная бумага, так и фильтровальный картон. Поскольку некото-
рые свойства бумаги, как-то: прочность, впитывающая способность,
содержание щелочных, щелочноземельных и следов тяжелых ме-
таллов, могут оказать существенное влияние на качество инди-
кации, рекомендуется применять специальные сорта бумаги, на-
пример бумагу народного предприятия в Эрцгебирге (ГДР) или
бумагу для хроматографии высокой степени очистки.
Можно назвать три типа индикаторной бумаги: для индикации
жидкого ОВ на поверхностях, паров ОВ в воздухе, ОВ, раство-
ренных в воде.
Бумагой для индикации ОВ в каплях промокают подозритель-
ные капли на поверхностях, защитной одежде и т. п. или наносят
на нее капли исследуемого вещества, например, в лаборатории при
установлении типа неизвестного ОВ.
Для индикации паров ОВ или ядовитых газов в воздухе суще-
ствуют две возможности: либо бумагу подвешивают, укрепляют
или размещают в определенных местах так, чтобы она подверга-
лась естественному воздействию отравленной атмосферы, и затем
контролируют полученные результаты, либо отравленную атмо-
сферу при помощи насоса, резинового баллона или велосипедного
насоса принудительно просасывают через бумагу, укрепленную в
специальном держателе (рис. 17). При просасывании воздуха повы-
шается чувствительность и быстрота выполнения анализа.
Некоторые индикаторные бумаги, описанные ниже, пригодны
для индикации ОВ в различных средах. Концентрацию ОВ в
воздухе часто можно оценить по окраске, которая появляется на
81
227
индикаторной бумаге после определенного времени воздействия,
или, наоборот, по времени, которое требуется для возникновения
определенной окраски. Известная точность может быть достигнута
путем установления объема воздуха, просасываемого с помощью
насоса. Большое влияние на чувствительность индикации, 3 также
на точность таких количественных определений наряду с качеством
бумаги оказывает и влажность воздуха.
fiuniititmntmnntic/7
Рис. 17. Держатель для индикаторной бумаги.
9.2.3.1. Неспецифичные индикаторные бумаги. Индикация при
помощи таких бумаг ОВ в каплях на поверхности основана, как
и у некоторых порошков, на углублении окраски в результате пе-
рехода красителя в раствор 33.
Индикаторную бумагу для серного иприта приготовляют пропитыванием бу-
маги суспензией из 10 г судаиа красного в 1 л воды, перемешивавшейся на
шаровой мельнице в течение 30 мин. В присутствии капель серного иприта и
некоторых других ОВ на бумаге появляются ярко-красные пятиа.
Для этой же цели можно приготовить фильтр-картон34 путем смешения
10 г целлюлозы с 200 мл воды и добавления в эту массу 10 мл суспензии, при-
готовленной из 0,5 г Судана красного В и 1 л воды. После фильтрования полу-
ченной смеси на воронке Бюхнера под вакуумом и сушки получают диск кар-
тона, окрашенный в бледно-розовый цвет, на котором капли ОВ оставляют
красные, интенсивно окрашенные пятиа или кольца. Этому картону можно при-
дать гидрофобные свойства, если после сушки его пропитать раствором 10 г
сульфата алюминия и 10 г ацетата свинца в 100 мл воды и снова высушить.
Разумеется, что индикация этими, так же как и пропитанными
другими красителями, бумагами связана с такими же помехами,
какие упоминались при описании индикаторных порошков. Индика-
ция кислотных или отщепляющих кислотные продукты ОВ возмож-
на с помощью бумаг, пропитанных растворами кислотно-основных
индикаторов, например растворами метилового красного, метило-
вого оранжевого и конго красного.
9.2.3.2. Индикаторные бумаги на хлор. Для обнаружения при-
годны следующие индикаторные бумаги.
1) Иодкрахмальная бумага. Ее готовят пропитыванием бумаги раствором
1 г растворимого крахмала в 100 мл воды, к которому добавлено 5 г KI и
немного NaHCO3, с последующей сушкой. В присутствии хлора индикаторная
бумага приобретает окраску от синей до сине-фиолетовой; чувствительность
пробы 0,006 мг/л. Такую же окраску вызывают бром, озон и окислы азота. При
хранении в закрытых темных сосудах бумага не утрачивает своих свойств
в течение неограниченно долгого срока.
2) Флуоресцеиновая бумага. Готовят раствор 0,2 г флуоресцеина, 30 г КВг,
2 г NajCOs, 3 мл 10%-ного раствора NaOH и 10 мл глицерина в 100 мл воды
и пропитывают им бумагу. После сушки бумага имеет желтый цвет. В присут-
ствии хлора она окрашивается в красный цвет вследствие образования эозина;
228
чувствительность пробы 0,03 мг/л. Пары брома вызывают такое же окраши-
вание.
3) о-Толидиновая бумага. Пропитывают бумагу раствором 0,1 г о-толидина
в 100 мл 10%-ного раствора НС1 и затем сушат. Хлор, окисляя о-толидин, вы-
зывает появление желтовато-коричневого окрашивания; чувствительность пробы
0,03 мг/л.
9.2.3.3. Индикаторные бумаги иа фосген. Приводим несколько
типов таких бумаг.
1) Растворяют 5 г дифениламина и 5 г n-диметиламинобензальдегида в
100 мл этилового спирта или четыреххлористого углерода и этим раствором
пропитывают бумагу и сушат в темном помещении. Пары фосгена, в зависи-
мости от их концентрации, окрашивают бумагу от желтого до оранжевато-
коричневого цвета, чувствительность пробы 0,003 ле/л. Пары соляной кислоты,
так же как и вещества, отщепляющие хлористый водород, вызывают такое же
окрашивание, а при действии хлора бумага приобретает желто-зеленую окраску.
Бумага может сравнительно долго сохраняться в закрытом, защищенном от про-
никновения света сосуде 13-13.
2) Аналогичную индикаторную бумагу получают при пропитывании бума-
ги 37 смесью равных частей насыщенного раствора п-диметнламинобензальдегида
и 25%-ного раствора диметиланилина в 95%-ном этиловом спирте. При концен-
трации фосгена не менее 0,01 мг/л свежеприготовленная бумага окрашивается в
синий цвет.
3) Растворяют 1,68 г М-этил-М-2-оксиэтиланилииа, 0,75 г п-диметиламино-
бензальдегида и 2,5 мл диэтилфталата в 25 мл этилового спирта, пропитывают
этим раствором бумагу и сушат. В присутствии паров фосгена белая бумага
приобретает окраску от светло-голубой до синей 14. Бумага мало чувствительна
к парам минеральных кислот и при ее помощи можно обнаружить фосген уже
при концентрации 0,001 мг/л.
4) Готовят раствор 0,1 г 1-нитрозо-3,6-диметиламинофенола в 50 мл горя-
чёго ксилола и раствор 0,25 г л-диэтнламинофенола в 50 мл ксилола. Перед
употреблением смешйвают 5 мл первого раствора и 2 мл второго раствора и
после выдерживания смеси в течение 2—3 суток пропитывают ею бумагу, кото-
рую затем сушат. Непосредственно перед применением бумагу смачивают
50%-иым этиловым спиртом. В присутствии паров фосгена при концентрации
0,0008 мг/л и экспозиции 3 мин бумага окрашивается в зеленый цвет 13-1в. Об
изменении окраски в присутствии других газов и паров ничего не известно.
5) Готовят раствор 2 г 4-(n-иитробеизил)-пиридина и 4 г N-беизилаиилина
в 100 мл бензола, пропитывают этим раствором бумагу и сушат. В закупорен-
ной склинке бумага может сохраниться в течение нескольких месяцев.
Фосген может быть обнаружен уже при концентрации паров 0,004 мг/л по
появлению красной окраски ”•13. Подобную окраску дают хлор, а также пары
хлористого ацетила и хлористого бензоила. Применение держателя (см. рис. 17,
стр. 228) позволяет предохранить индикаторную бумагу от попадания хлора.
Для этого перед держателем с иидикаториой бумагой помещают ловушку с ку-
сочками бумаги, пропитанной 10%-ным раствором NajSjOs и 4%-ным раствором
Nal.
9.2.3.4. Индикаторные бумаги на синильную кислоту. Для обна-
ружения синильной кислоты можно пользоваться следующими ин-
дикаторными бумагами.
1) Бензидин — медьацетатная бумага ”.20. Готовят раствор 2,86 г ацетата
меди в 1 л воды и раствор 475 мл насыщенного раствора ацетата бензидина в
525 мл воды и пропитывают бумагу смесью равных частей обоих растворов.
Для индикации применяют влажную бумагу. В присутствии 0,001 мг/л синиль-
ной кислоты через 1 мин бумага окрашивается в сиинй цвет. Можно считать,
229
что опасности отравления не существует, если в течение 7 мин синяя окраска
не появляется. Индикации мешает присутствие хлора, брома, сероводорода,
двуокиси серы и паров соляной кислоты.
Такую же индикаторную бумагу можно приготовить пропитыванием бумаги
смесью равных частей насыщенного водного раствора ацетата бензидина и
0,3%-ного водного раствора ацетата меди. Пропитанная бумага сохраняется
очень непродолжительное время, так как вскоре окрашивается в коричневый
цвет. Смесью обоих растворов (хранящихся в темноте) можно пользоваться
около двух недель.
При использовании вместо бензидина ацетата о-толидина повышается чув-
ствительность и стабильность бумаги, а также устойчивость возникающей при
индикации окраски.
2) Состав для индикаторных трубок, пригодный также для приготовления
индикаторной бумаги24-21. Готовят раствор 0,3 г ацетата меди в 100 мл воды
и раствор 0,5 г тетраметилдиаминодифенилметаиа в 100 л л ацетона. Бумагу по-
гружают в первый раствор, сушат иа воздухе и пропитывают вторым раствором.
Сухая бумага может сохраняться в темном месте в плотно закрытом сосуде в
течение 8 недель. При большем сроке хранения бумага постепенно синеет. В при-
сутствии 0,001 мг/л HCN появляется достаточно устойчивое синее окрашивание.
3) Индикаторная бумага на основе гваяковой смолы22. Готовят раствор
0,05 г CuSO4 в 100 мл воды и раствор 4 г гваяковой смолы в 100 мл этилового
спирта. После погружения в первый раствор бумагу сушат и затем пропиты-
вают вторым раствором. Синильная кислота вызывает появление на бумаге
синей окраски.
4) Индикаторная бумага на основе HgCl2 и метилового оранжевого23. Го-
товят раствор 1,25 г HgCl2 в 250 мл воды и раствор 4 г метилового оранжевого
в 250 мл воды. Непосредственно перед использованием смешивают 100 мл пер-
вого раствора с 50 мл второго раствора и 10 мл глицерина, пропитывают бу-
магу этой смесью и сушат в атмосфере, свободной от паров кислот. Присутствие
в воздухе уже 0,01 мг/л синильной кислоты обнаруживают по окрашиванию
бумаги в красный цвет. Пары кислот вызывают такую же цветную реакцию.
Бумагу сохраняют в течение 2 суток в затемненном месте.
5) Индикаторная бумага на основе AgNO3 и конго красного24. Готовят рас-
твор 0,05 г конго красного в 100 мл воды и раствор 5 г AgNO3 в 100 мл воды.
Бумагу опускают иа 1 мин в раствор конго красного и сушат на воздухе; Затем
ее пропитывают раствором AgNO3 и сушат в темном помещении. Используют
свежеприготовленную бумагу. В присутствии в воздухе уже 0,01 мг/л синильной
кислоты бумага из красной становится синей.
6) Инбикаторная бумага, на которой образуется берлинская лазурь 25- 23.
В профильтрованный раствор 5 г FeSO4 в 50 мл воды погружают на 5 мин
бумагу. Затем ее вынимают, сушат и на 15 сек погружают в 20%-ный раствор
NaOH, не содержащий карбонатов, и снова тщательно высушивают. В холодном
и темном помещении в вакуум-эксикаторе бумага может сохраняться несколько
недель. После воздействия на бумагу воздуха, содержащего пары синильной
кислоты, бумагу погружают в разбавленный раствор НС1 (1:4) или 30%-ный
раствор H2SO4. При растворении гидроокиси железа (II) через 30—60 мин по-
является сине-зеленое окрашивание. Минимально обнаруживаемое количество
HCN меньше 0,01 мг/л. Бумага отличается высокой специфичностью.
7) Индикаторную бумагу, предназначенную для немедленного использова-
ния, можно приготовить пропитыванием бумаги смесью 10 мл 10%-ного раствора
FeSO4, 20 мл раствора 30 г сегнетовой соли и 10 г КОН в 100 мл воды.
После помещения бумаги в атмосферу, содержащую ОВ, бумагу подвергают
действию паров НС1; при наличии HCN она окрашивается в сине-зеленый цвет.
8) Натрийпикратная бумага23-22. Готовят раствор 0,1 г Na2CO3 в 100 мл
воды и раствор 0,1 г пикриновой кислоты в 100 мл воды. Бумагу погружают
в раствор соды и после сушки на воздухе пропитывают раствором пикриновой
кислоты. При наличии HCN появляется красно-коричневая до красной окраска;
чувствительность пробы 1 мг/л. Альдегиды, ацетон, двуокись серы, сероводород
дают такую же реакцию.
230
9.2.3.5. Индикаторные бумаги на хлорпикрин. Приведены три
типа таких индикаторных бумаг.
1) Бумагу, пропитанную раствором 10 г диметиланилина в 90 мл бензо-
ла 30, сушат и помещают в атмосферу, содержащую ОВ. При содержании хлор-
пикрина в количестве, обнаруживаемом по запаху, бумага принимает окраску
от желтой до коричневой. Если вынести бумагу из атмосферы, зараженной хлор-
пикрином, окраска, обусловленная малыми концентрациями хлорпикрина, вскоре
исчезает.
2) Капли хлорпикрина на бумаге, пропитанной раствором 5 г п-диметил-
аминобензальдегида в 95 мл этилового спирта, вызывают образование желтых
пятен 1в.
3) Очень чувствительная реакция обнаружения хлорпикрина индикаторной
бумагой 37 основана на превращении хлорпикрина с помощью цианистого натрия
в хлорциан и индикации последнего по реакции Цинке — Кёнига (ср. раз-
дел 5.5.2.1),
Для приготовления индикаторной бумаги готовят два раствора — раствор
2,8 г NaCN в смеси 40 мл пиридина с 36 мл метилового спирта при охлаждении
льдом и 2%-ный раствор флороглюцина в метиловом спирте. Затем смешивают
19 мл раствора NaCN с 1 мл раствора флороглюцина и этой смесью пропиты-
вают бумагу. В зависимости от концентрации паров хлорпикрина бумага окра-
шивается от розового до красно-фиолетового цвета; чувствительность пробы
0,05 мг/л хлорпикрина.
9.2.3.6. Индикаторные бумаги на серный иприт. Приводим не*
сколько типов бумаги для обнаружения серного иприта.
1) Бумагу готовят непосредственно перед применением, пропитывая ее рас-
твором 2 г иодоплатината натрия Na2PtI6 в 100 мл воды, и используют во влаж-
ном состоянии. При наличии в воздухе паров серного иприта или капель его
раствора красноватая окраска бумаги изменяется на фиолетовую, пурпурную
или синюю, в зависимости от концентрации иприта 3i-59.
2) Для идентификации капель серного иприта пригодна бумага, пропитан-
ная 5%-ным раствором иатрийпентациаио-амииоферрата(П) Na3Fe(CN)s-NH3 и
осторожно просушенная. В присутствии иприта появляются сиие-зеленые пятиа 32.
3) На бумаге, пропитанной растворами кетоиа Михлера и хлориой ртути,
при нанесении капли испытуемого вещества иприт обнаруживается по появлению
красно-фиолетового окрашивания.
9.2.3.7. Индикаторные бумаги на галогеналкиламины (азотистые
иприты). Для обнаружения галогеналкиламинов пригодны следую-
щие индикаторные бумаги35.
1) В 9 мл концентрированной соляной кислоты растворяют 2,5 г основного
нитрата висмута и добавляют к раствору 10 мл глицерина, 3,5 г KI и 2,5 г
кристаллического СаС12. Этим коричневым раствором пропитывают бумагу и
сушат при 60 °C в течение 1 ч. Перед использованием ее ненадолго оставляют
на воздухе для увлажнения. Бумага сохраняется почти два года; устойчивость
по отношению к влаге можно повысить добавкой 8 г винной кислоты или
КН2РО4.
Капли 2-галогеналкиламинов оставляют на желто-оранжевой бумаге крас-
ные пятна, а люизит — синее пятно. Если зараженный воздух просасывать через
слой бумаги, то можно индицировать и меньшие количества 2-галогеналкилами-
нов; чувствительность пробы 25 мкг/л.
2) Бумагу пропитывают смесью следующих растворов, взятых каждый в
количестве 10 мл'. раствор 10 г Co(NO3)2 в 95 воды, раствор 5 г NaSCN в
95 мл воды, раствор 37 г ZnCL в 63 мл воды, раствор 60 г NH4NO3 в 50 мл
воды, раствор TiCh-
231
Раствор TiCI3 готовят смешением 50 мл 15%-ного раствора TiCl3, разбав-
ленного 25 мл воды и окисленного осторожным прибавлением по каплям 1 мл
концентрированной HNO3, с раствором 1,5 г окисных железоаммонийных квас-
цов в 25 мл воды, подкисленных 1 мл HNO3. К полученной смеси медленно до-
бавляют еще 4 мл концентрированной HNO3 и затем 10 мл глицерина. Пропи-
танную бумагу 2 ч сушат при 60 °C. При нанесении капель 2-галогеналкиламинов
бумага окрашивается в синий цвет; бумага очень стабильна при хранении.
9.2.3.8. Индикаторная бумага для обнаружения фосфорсодер-
жащих ОВ в воде. Реагентом служит диизонитрозоацетон н.
Бумагу пропитывают раствором 10 г тринатриевой соли диизонитрозоацетона
в 100 мл метилового спирта. При соприкосновении с водой, содержащей значи-
тельное количество нервно-паралитических ОВ типа зарин, бумага окраши-
вается в красно-фиолетовый цвет.
9.2.3.9. Индикаторная бумага для обнаружения фосфорсодер-
жащих ОВ в воздухе. Для обнаружения ОВ типа зарин пригодна
следующая индикаторная бумага.
Проба основана на реакции Шёнеманиа. Для ее осуществления пропиты-
вают бумагу 2%-ным раствором Na3PO4, высушивают и повторно пропитывают
непосредственно перед употреблением смесью из 2 ч. 1%-ного раствора Н2О2,
1 ч. 0,5%-кого водного раствора гидрохлорида о-дианизидина и 1 ч. изопропило-
вого спирта. При просасывании через индикаторную бумагу воздуха, заражен-
ного нервно-паралитическими ОВ типа зарин, бумага приобретает красно-корич-
невую окраску.
9.2.3.10. Индикаторные бумаги на этилдихлорарсин 36. Для про-
ведения испытания рекомендуются следующие индикаторные бу-
маги.
1) Бумагу пропитывают насыщенным раствором 2,4-динитрофеиола в
95%-ном этиловом спирте. После нанесения капли ОВ (или его раствора) опу-
скают на это же место 1 каплю 2,5 н. раствора NaOH. В присутствии этилди-
хлорарсина (при минимальной концентрации 1 мг/мл) первоначально желтая
окраска бумаги изменяется в оранжево-красную.
2) Несколько, более чувствительна бумага, пропитанная 2%-ным раствором
м-динитробензола в ацетоне и обработанная после воздействия ОВ Юн. раство-
ром NaOH. В присутствии 0,1 мг/мл этилдихлорарсина бумага окрашивается в
красно-коричневый цвет. Люизит вызывает серовато-зеленую окраску.
9.2.3.11. Индикаторная бумага на окись углерода. Такая бума-
га необходима для индикации в воздухе паров карбонилов метал-
лов. Приготовить ее можно следующим образом 39.
Бумагу пропитывают вначале 1%-ным раствором PdCl2, а затем 5%-ным
раствором ацетата натрия. Бумага чувствительна к воздействию света и восста-
новителей. Через 1 мин при концентрации 0,9 мг/л СО и через 20 мин при кон-
центрации 0,09 мг/л СО бумага окрашивается в черный цвет.
9.2.4. Индикаторные трубки
Индикаторные трубки наиболее широко применяются для индика-
ции ОВ в полевых условиях. Они находят также широкое примене-
ние при изучении гигиены труда, в промышленной токсикологии,
в промышленном контроле, в токсикологических исследованиях и,
наконец, в качестве детекторов в газовой хроматографии. Совре-
232
менные типы трубок представляют собой дальнейшее усовершен-
ствование трубок с силикагелем, предложенных еще в 1930 г. для
индикации ОВ. Общим для всех трубок является наличие наполни-
теля— сорбента (в основном белого силикагеля), на котором раз-
вивается цветная реакция ОВ с реагентом.
Стеклянные трубки обычно запаяны; выполненные в любом
ином варианте трубки закрыты так, чтобы исключалось случайное
Рис. 18. Индикаторные трубки:
а —индикаторная трубка на зарин; б—ин-
дикаторная трубка на нхгрнт; а—индика-
торная трубка на зоман; а—индикаторная
трубка на фосген, дифосген, хлорцнан и
синильную кислоту; 1— стеклянная трубка;
2 — перепускной канал; 3—ампула; 4—мар-
кировочные кольца.
Рис. 19. Поршневой насос прибора
химической разведки:
а—общий вид; б — разрез; / — коллектор;
2 — корпус насоса; 3 — рукоятка с вскрыва-
телем ампул; 4—шток поршня; 5 — манжета:
6— втулка; 7—направляющее кольцо;
8—защитный патрон; 9—вентиль; 10— седло
клапана; 11 — пружина; 12— пята пружины.
проникновение внутрь воздуха. Непосредственно перед использова-
нием их вскрывают путем отламывания кончиков или удалением
затворов и просасывают через них исследуемый воздух соответ-
ствующим насосом. В качестве примеров на рис. 18—20 изобра-
жено устройство некоторых индикаторных трубок и существующих
насосов. Применяются индикаторные трубки нескольких типов.
В одном типе трубок42 после просасывания отравленного воз-
духа, т. е. после поглощения ОВ, силикагель извлекают из трубки,
перемешивают и после деления на отдельные порции испытывают
каждую порцию на определенное ОВ. Можно также экстрагиро-
вать ОВ из силикагеля соответствующим растворителем. Части
полученного экстракта используют для определения различных
отравляющих веществ. В таких трубках, используемых главным
образом для отбора проб, можно в качестве сорбента применять
И активированный уголь 43-45.
233
В другом типе трубок после просасывания зараженного возду-
ха в трубку подают раствор реагента, который при взаимодействии
с группой ОВ 46’56 или специфично с одним ОВ 47-50 образует окра-
шенный продукт. Значительная часть способов обнаружения, за-
ключающихся в образовании окрашенных продуктов, может быть
приспособлена для осуществления в индикаторных трубках.
Имеются трубки, внутри которых помещены ампулы с раство-
рами реагентов, которые либо до, либо после просасывания зара-
женного воздуха
Рис. 20. Сильфон-
ный насос прибора
химической раз-
ведки фирмы Дре-
гер, модель 19/31.
разбивают при помощи соответствующих приспо-
соблений на трубках или на насосе, в результате
чего реагент вступает во взаимодействие с ОВ с
образованием окрашенного соединения 54’55. Од-
ним или несколькими компонентами реагента мо-
жет быть пропитан и силикагель.
В других трубках силикагель пропитан ре-
агентом; при просасывании зараженного воздуха
и адсорбции минимально необходимого или до-
статочного для цветной реакции количества ОВ
происходит соответствующее изменение окраски
силикагеля. С точки зрения простоты обращения
такие трубки являются идеальными, однако часто
из-за малой устойчивости реагентов такое техни-
ческое решение не всегда возможно. В этом типе
трубок применяют наполнители, не обладающие
большой адсорбционной способностью, например
стеклянный порошок или мелкозерненые поли-
мерные материалы, так как практически нежела-
тельно, чтобы адсорбция ОВ проходила на «вну-
тренней» поверхности носителя. Важно, чтобы
цветная реакция, по которой судят о присутствии ОВ, проходила на
поверхности адсорбента. Такие трубки пригодны для количествен-
ной оценки содержания ОВ51’52.
Количественное определение ОВ в индикаторных трубках
можно осуществить путем сопоставления полученного эффекта со
скоростью потока просасываемого воздуха40, например сопостав-
лением длины окрашенной зоны с объемом прошедшего воздуха
или сравнением полученной окраски с каким-то окрашенным этало-
ном. При калибровке трубок по воздуху, содержащему известную
концентрацию ОВ, важно установить зависимость длины окрашен-
ной зоны (или интенсивности окраски) от скорости воздушного по-
тока. Или, иначе говоря, полученный эффект зависит от времени
контакта испытуемого газа с реагентом, которое в свою очередь
является функцией скорости потока и скорости взаимодействия ОВ
с реагентом. Кроме того, на показания трубок влияют процессы
абсорбции и диффузии. Скорость потока измерить очень трудно.
Средняя его величина может быть рассчитана 40 по данным измере-
ний пористости слоя носителя (отношение пустот между зернами к
общему объему слоя) и интенсивности потока, пропорциональной
234
его скорости. Так, для трубки с внутренним диаметром 5,2 мм и по-
ристостью носителя 0,12 (размер частиц 0,3 мм) при интенсивности
потока 1 л/мин была получена скорость потока, равная 7 м/сек.
Это соответствует времени контакта от 10~4 до 10-5 сек в слое на-
полнителя, равного по высоте среднему диаметру частиц. На
рис. 21 изображена зависимость по-
казаний от интенсивности потока
для трубок, в которых проводятся
реакции разного типа.
Ниже дано краткое подразделе-
ние трубок для основных возможных
реакций.
Группа I
К этой группе относятся трубки,
предназначенные для ионных реак-
ций, протекающих быстро и стехио-
метрически. Примером реализации
такой реакции может служить труб-
Рис. 21. Зависимость показаний
индикаторных трубок, групп I—V
от 1 объемной скорости при рав-
ных объемах испытуемого газа и
одинаковой концентрации ОВ40.
ка для определения окиси углеро-
да * по реакции с пятиокисью иода
и серной кислотой (олеумом). Такие
трубки легко откалибровать, так как
в широком диапазоне их показания
не зависят от скоростей потока. Еди-
ница длины окрашенной зоны соответствует определенному количе-
ству ОВ.
Группа II
Этот тип трубок соответствует типу I, но вместо окрашенных
зон образуются окрашенные кольца. Примером таких трубок
является трубка для определения систокса 41 по реакции с хлорным
золотом и натриевой солью N-хлорамида п-толуолсульфокислоты
(хлорамин Т).
Группа III
Это трубки, в которых проводят нестехиометрические реакции
ОВ с органическими реагентами. Показания трубок в этом случае
зависят от скорости потока и только при очень больших скоростях
зависимость исчезает.
Группа IV
Зависимость показаний от скорости потока существует на всем
протяжении кривой, но так как она различна, то для каждого от-
дельного случая следует устанавливать соответствующую область
калибровки. Примером может служить трубка для определения
фосгена с n-диметиламинобензальдегидом и диметиланилином.
* Взаимодействие окиси углерода с пятиокисью иода правильнее было бы
рассматривать как радикальную реакцию. — Прим. ред.
235
Группа V
Такие трубки калибруются путем сравнения появившейся окра-
ски. На каком-то небольшом отрезке кривой наблюдается зависи-
мость длины окрашенной зоны от концентрации ОВ. При увеличе-
нии скорости потока реакция перестает быть количественной —
количество ОВ, сорбирующееся из единицы объема, начинает
уменьшаться.
Имеются трубки, принцип действия которых основан на одно-
временном или последовательном течении двух реакций. Например,
определение четыреххлористого углерода после его превращения в
фосген.
Представление о характерных особенностях этих типов трубок
позволяет сделать определенные выводы, которые могут быть по-
лезны как при конструировании новых трубок, так и при примене-
нии уже существующих, поскольку можно определить, к какому
типу принадлежит данная трубка.
Калибровочные отметки на трубках могут быть использованы
только при работе с определенным насосом. Так, для трубок при-
бора химической разведки (см. рис. 19, стр. 233) насос с мехом из
прибора «Дрегер» непригоден. Если для трубок групп I и II можно
пренебречь ошибками, возникающими при применении любого на-
соса, то для трубок групп III, IV и V они становятся уже значитель-
ными. Кроме требований, предъявляемых к герметичности насосов,
следует обращать внимание и на правильное обращение с ними.
Так, при работе с насосом прибора химической разведки нельзя
производить слишком быстро качания, т. е. нельзя допускать сле-
дующий ход поршня, пока полностью не выравняется разрежение
в насосе. При изготовлении трубок важно точно выдерживать до-
пуски по сопротивлению.
При контроле качества изготовления и испытании имеющихся
в запасе трубок необходимо проверять все приведенные показатели.
Здесь будут описаны только индикаторные трубки и наборы с
трубками, специально предназначенными для обнаружения ОВ.
Первой такой трубкой была трубка из прибора Дрегера — Шрё-
тера, имевшегося на вооружении немецкой армии во время второй
мировой войны. Адсорбентом служил силикагель. После просасы-
вания зараженного воздуха в трубку засасывали несколько капель
разбавленного раствора КМпО4. Образование коричневого кольца
свидетельствовало о присутствии фосгена и других ОВ, обладающих
восстановительными свойствами, например иприта. Таким образом,
трубка эта была мало специфичной; чувствительность ее составляла
0,015 мг/л иприта при просасывании 2л отравленного воздуха46.
Позднее в трубку стали вводить несколько капель раствора хлор-
ного золота — специфичного реагента на серный иприт 47’53-60. Для
достижения лучшего цветового контраста избыток хлорного золота
восстанавливали перекисью водорода до металлического золота или
с помощью тиосульфата натрия превращали его в бесцветное ком-
плексное соединение61.
236
Эти опыты положили начало развитию средств индикации и
привели к созданию набора индикаторных трубок, принятого на
снабжение фашистских вооруженных сил во время второй мировой
войны. Это были трубки из полистирола, у которых реагенты были
частично нанесены на слой носителя, а частично находились в
ампулах. Для улавливания газов, мешающих обнаружению, в труб-
ках имелись слои предварительной очистки, которые часто приме-
няются и в современных индикаторных трубках.
В этих трубках использовались следующие реагенты:
на серный иприт (одно желтое кольцо) —хлорное золото на
силикагеле, хлорамин в ампуле;
на азотистый иприт (два желтых кольца) — калийвисмутиодид
(реагент Драгендорфа) в ампуле;
на синильную кислоту (одно черное кольцо) — ампула с раство-
ром ацетата меди, ампула с раствором ацетата бензидина;
на фосген (одно зеленое кольцо) — ампула с раствором п-диме-
тиламипобензальдегида в этиловом спирте, ампула с диметилани-
лином;
Таблица 23. Результаты индикации некоторых ОВ трубками
газоопределителя М18 (США)
Отравляющее вещество Маркировка трубки Число качаний насоса Операции после прокачи- вания воздуха Характерная окраска
Нервно-пара- литические Белая точка 20 Добавляют 1 каплю реагента с маркиров- кой «зеленое кольцо» Через 5 мин зеленовато- синяя
Хлорциаи Синяя точка 2 Отсутствуют' Через 1 мин желтая до оранжевой
Синильная кислота Коричневая точка 2 Отсутствуют Синяя
Серный иприт Синяя точка 5 На обогревательный кожух трубки нано- сят несколько капель специального рас- твора, охлаждают, удаляют кожух и добавляют 1 каплю реагента «синее кольцо» Синее кольцо
Люизит и этил- дихлорарсин Желтая точка 5 Наносят 1 каплю реа- гента «синее кольцо» Синее кольцо
Азотистый иприт Белая точка 5 Наносят 1 каплю реа- гента «красное коль- цо» Через 1 мин оранжево- красное
Фосген Зеленая точка 2 Отсутствуют КиЛЬци Зеленая
Отбор пробы Белая точка 30 Отправляют в лабора- торию с бланком до- несения
237
на хлорпикрин (два зеленых кольца) — ампула с цианистым
калием, ампула с пиридином.
Наиболее известными в настоящее время приборами химиче-
ской разведки с индикаторными трубками являются: прибор Дре-
гера модель 19/31 бундесвера, газоопределители М15, М18 и М9А2
(США), прибор химической разведки ПХР-54.
В индикаторных трубках Дрегера используются следующие
комбинации реагентов 40’57’ 58'62’63:
на серный иприт — хлорное золото на носителе и в качестве
«проявителя» онти-фенилдиазотат калия или хлорамин в ампуле; в
присутствии серного иприта появляется оранжево-красное окраши-
вание;
на азотистый иприт — калийвисмутиодид на носителе; пары
азотистого иприта вызывают образование оранжево-красного
кольца;
на синильную кислоту — хлорная ртуть и кислотно-основной ин-
дикатор на носителе; в присутствии HCN носитель окрашивается в
красный цвет; чувствительность реакции 0,002 мг/л\
на хлорциан— сернистый натрий и соль железа (III) или 4-бен-
зплпиридин и барбитуровая кислота;
на люизит — порошкообразный цинк, кислота и AgNO3 (проба
по Гутцепту) или тпокетон Мнхлера;
на фосген — 4-(п-нитробензил)-пиридин, N-фенилбепзиламин и
Хта2СОз пли п-дпметпламинобензальдегид и дпметпланилпн. Уже
концентрация фосгена 0,001 мг/л вызывает появление сине-зеленой
окраски.
Таблица 24. Индикаторные трубки к прибору химической разведки ПХР-54
Маркировка трубки ОВ, для индикации которого предназначена трубка Чувстви- тельность, мг'л Окраска слоя адсорбента
исходная после проведения реакции
Красное кольцо и красная точка Зоман 0,00005 Белая Красная, превра- щающаяся в желтую
Красное кольцо Зарин, зоман, табун 0,0002 Белая Желтая до жел- то-оранжевой
Желтое кольцо Серный иприт 0,002 Лимонно- желтая Оранжевая до красно-коричне- вой
Два желтых коль- ца Азотистый иприт 0,001 Желтая Оранжевая
Три зеленых кольца Фосген, дифосген 0,005 Белая Зеленая до сине- зеленой
— Синильная ки- слота 0,005 Белая Красная до фио- летовой
Хлорциан 0,008 Белая Красная до фио- летовой
238
Некоторые данные о работе с трубками М18 (США) приведены
в табл. 23, чтобы можно было провести сравнение с прибором
ПХР-54.
Набор трубок этого прибора химической разведки приведен
в табл. 24. Новый тип прибора химической разведки ВПХР снаб-
жен химическим нагревательным патроном, позволяющим кратко-
временный нагрев трубок при температуре воздуха ниже 15 °C. Это
устранило значительный недостаток старого прибора.
9.3. АВТОМАТИЧЕСКИЕ ПРИБОРЫ
Применение противником боевых химических веществ может при-
вести к тяжелым потерям в тех случаях, когда оно происходит вне-
запно. Своевременное предупреждение войск о появлении в воз-
духе минимальных, возможно еще и не опасных, концентраций ОВ
является поэтому важной задачей, решением которой занимаются
лица, работающие в области анализа и индикации боевых химиче-
ских веществ. Необходимость соответствия используемых средств
индикации все возрастающей токсичности ОВ затрудняет постав-
ленную задачу и в то же время заставляет постоянно заниматься
этой проблемой.
В современных условиях ведения химической войны недопустимо
пренебрегать субъективными методами оценки боевых действий
противника, хотя эффективность этих методов предупреждения
опасности крайне сомнительна, тем более, что обнаружение по за-
паху минимальных концентраций опаснейших фосфорорганических
соединений просто невозможно. Известные до сих пор индикатор-
ные бумаги на фосфорорганические ОВ слишком мало чувстви-
тельны, а индикаторные трубки конструктивно несовершенны. Зна-
чительная часть их не приспособлена для непрерывного контроля
заражения. После просасывания определенного объема воздуха
для обнаружения ОВ нужно вскрыть ампулу с реагентом. При на-
личии опасных концентраций ОВ затрачиваемое на эти операции
время настолько задержит предупреждение, что это может при-
вести к большим потерям.
Другим недостатком трубок является необходимость постоян-
ного наблюдения за их показаниями. По этой причине даже те
трубки ограниченно пригодны для своевременного предупреждения
об опасности, в которых на слое носителя уже имеются стабильные
реагенты и которые присоединены к автоматическим приспособле-
ниям для просасывания воздуха.
Поэтому во всех армиях существует стремление к снабжению
войск автоматическими приборами, которые обеспечивают непре-
рывную индикацию, работают продолжительное время без спе-
циального контроля и при появлении в атмосфере ОВ подают
звуковой или световой сигнал. Естественно, особенно большое зна-
чение придается приборам, которые легко транспортируются, имеют
небольшую массу, просты в обращении и надежны по конструкции.
Между разработкой автоматических приборов для индикации ОВ
239
и разработкой приборов автоматического контроля воздуха на
предприятиях химической промышленности существует, конечно,
тесная связь; в обоих случаях измерения основаны на тех же
принципах.
Действие приборов может быть основано на химическом, фи-
зико-химическом, физическом, биохимическом или биологическом
принципах. Действие большей части до сих пор разработанных при-
боров для определения ОВ основано на химическом принципе. Речь
идет исключительно о приборах для индикации самых токсических
боевых химических веществ — фосфорорганических соединений
типа зарин. В этих приборах при появлении определенной мини-
мальной концентрации ОВ, соответствующей диапазону срабаты-
вания аппарата, подается сигнал оповещения. Приборы, действую-
щие на физической, физико-химической, биохимической или биоло-
гической68 основе, пока еще описаны мало.
Наиболее важными критериями пригодности того или иного
автоматического газоанализатора, наряду с названными требова-
ниями к конструкции и надежности действия, являются специфич-
ность и чувствительность индикации. Помехи в работе прибора в
условиях боевой обстановки могут вызвать такие примеси в воз-
духе, как выхлопные газы двигателей внутреннего сгорания и ра-
кетных установок, пороховые газы и дымовые средства, концентра-
ции которых могут на порядки превосходить концентрации ОВ.
Отделять эти примеси до того, как'они попадут в зону реакции, с
помощью соответствующих фильтров практически очень трудно, так
как большинство фильтрующих материалов в той или иной степени
адсорбирует и ОВ, что приводит к их потере и снижению чувстви-
тельности. Поэтому имеется тенденция использовать специфические
или очень селективные средства индикации. Наиболее подходя-
щими для этих целей являются биохимические и химические реак-
ции ОВ. Возможно также применение адсорбционной спектроско-
пии в УФ- и ИК-областях. Специфичность других физических и
физико-химических способов, основанных, например, На измерении
показателя преломления, магнитной восприимчивости, электропро-
водности растворов или ионизации газов, на определении теплового
эффекта при сгорании или теплопроводности, — надо считать недо-
статочной.
Конструкция приборов должна быть такой, чтобы они были в
состоянии мгновенно оповещать о внезапном появлении в воздухе
ОВ в больших концентрациях, которые могут очень быстро при-
вести к смертельному исходу, а также своевременно (раньше, чем
истечет нужное время экспозиции) предупреждать о наличии малых
концентраций ОВ, которые могут вызывать отравление только
после продолжительной экспозиции. Это может исключить любую
степень поражения. Основываясь на данных чувствительности газо-
анализатора для нервно-паралитических ОВ типа Е21 (США),
можно определить зависимость между токсичностью, выражаемой
произведением ct (концентрация на время экспозиции), и необхо-
240
димой чувствительностью прибора. Чувствительность этого прибора
такова, что он может сигнализировать о наличии 0,1 мкг!л зарина
через 2—3 мин, а о наличии 10 мкг/л зарина через 5 сек.
Так как дозы зарина около 0,5 мкг-мин!л вызывают легкие по-
ражения, а дозы от 150 мкг-мин!л и выше приводят к тяжелым
поражениям, вплоть до летального исхода, не трудно видеть, что
чувствительность прибора обеспечивает в случае заражения воз-
духа парами зарина своевременное предупреждение только для за-
щиты от тяжелого поражения. Более токсичный зоман вызывает
легкие поражения уже при ct, равном 0,2 мкг • мин!л, а тяжелые по-
ражения при дозе 75 мкг-мин!л. Ввиду того, что чувствительность
прибора считается одинаковой как для зарина, так и для зомана,
то в случае зомана не может быть и речи о своевременном преду-
преждении. Это очень важное обстоятельство надо иметь в виду, так
как отсутствие предупреждающего сигнала о появлении малых
концентраций ОВ, вызывающих легкие поражения, может привести
к продолжительной утрате боеспособности личного состава. Кон-
центрации ОВ, образующиеся очень быстро во время внезапного
химического нападения, таковы, что они могут быть практически
мгновенно обнаружены как прибором Е21, так и другими аналогич-
ными приборами. Размеры потерь в районе цели и в направлении
распространения облака отравленной атмосферы зависят от вре-
мени, необходимого для надевания противогазов и защитной
одежды. В этой связи интересно привести выполненные Ге-Жако66
расчеты значений ct, которые в зависимости от условий защиты и
тренированности личного состава надо умножить на соответствую-
щие коэффициенты, чтобы получить размеры потерь, возможные
для незащищенных, непредупрежденных и нетренированных вой-
сковых частей.
Коэффициент
Незащищенные....................... 1
Защищенные, плохо тренированные, не-
предупрежденные .................. 4
Защищенные, тренированные, непредупре-
жденные ............................ 10
Защищенные, тренированные, предупре-
жденные ......................... 20
Из приведенных данных можно заключить о значении мер за-
щиты, своевременного оповещения, а также обученности и дисцип-
линированности войск при химических нападениях.
9.3.1. Приборы, действие которых основано
на химических реакциях
Основой действия известных приборов индикации фосфороргани-
ческих ОВ является колориметрическая или флуориметрическая
оценка эффекта реакции Шёнеманиа (ср. раздел 5.1,1). В амери-
241
канской армии разработан64 газоанализатор нервно-паралитиче-
ских ОВ Е21, изображенный на рис. 22 и 23. В этом приборе дви-
жущаяся бумажная лента, периодически (через каждые 5 мин)
смачиваемая реагентом, проходит в индикаторную головку. Реаген-
том служит раствор гидрохлорида о-дианизидина, содержащего
пероксопирофосфат натрия (pH 9), устойчивый в течение 24 ч. Он
подается на бумажную ленту с помощью дозирующего устройства,
соединенного с приводом бумажной ленты, через которую затем
Рис. 22. Автоматический газо-
анализатор Е21 для нервно-пара-
литических ОВ со снятой передней
стенкой.
Рис. 23. Автоматический газоанали-
затор Е21 для нервно-паралитиче-
ских отравляющих веществ со сня-
той задней стенкой.
просасывается зараженный воздух. В присутствии фосфорсодержа-
щих ОВ бумага окрашивается в красный цвет. В индикаторной
головке находятся два фотоэлемента, из которых один измеряет
интенсивность света, отраженного от смоченной реактивом бумаж-
ной ленты, освещенной лампой накаливания 6 в, а другой — интен-
сивность света, отраженного от бумажной ленты после просасыва-
ния отравленного воздуха. Появляющаяся на ленте красная окра-
ска нарушает равновесие световых потоков обоих фотоэлементов.
Возникающий в результате этого ток, проходя через усилитель, при-
водит в действие звуковой и световой сигналы. Прибор размещен в
водонепроницаемом алюминиевом корпусе размером 430 X 400 X
X 175 мм и весит 10,9 кг. Потребляемая энергия, равная 19 вт, по-
дается от батареи емкостью 24 в или от сети переменного тока
через выпрямитель. Прибор может работать непрерывно в течение
12 ч, после чего нужно менять бумажную ленту, раствор реагента
и батарею. Он работает безотказно при температурах от 0 до 38 °C.
При добавлении в раствор реагента в качестве антифриза изопро-
пилового спирта область температур, при которых прибор может
работать, увеличивается до —18 °C. Более совершенной моделью
этого же типа является прибор Е41, который вместе с батареей, по-
242
мещенной в корпусе прибора, весит до 14 кг. Корпус этой модели
теплоизолирован, вследствие чего область рабочих температур до-
стигает от —30 до +38 °C. Вследствие уменьшения габаритов де-
талей и введения в усилитель транзисторов значительно снижено
потребление энергии, прибор работает непрерывно 24 ч без пере-
зарядки в интервале температур от —30 до +40 °C, его масса 10 кг.
Продолжительность непрерывной работы 8 ч.
Рис. 24. Схема автоматического флуоресцентного индикатора ОВ
в воздухе:
/—резервуары для реагентов; 2—дозатор; 3 —смеситель; 4—абсорбционная
колонна; 5 —фотометр; 6 — сборник прореагировавших растворов; 7—всасываю-
щий насос; 3—размыкатель; 9—электропитание.»
В американской армии разработано несколько типов аппаратов-
индикаторов ОВ нервно-паралитического действия, предназначен-
ных для контроля за воздухом на заводах, производящих ОВ, и на
складах военного имущества. В них использован наиболее чувстви-
тельный флуоресцентный вариант реакции Шёнеманиа65. Принцип
работы такой установки по контролю за чистотой воздуха показан
на рис. 24. Так как раствор реагента не может храниться продол-
жительное время, то для его приготовления имеются два раздельно
хранящихся раствора, которые смешиваются в приборе автомати-
чески, непосредственно перед употреблением. Раствор 1 представ-
ляет собой раствор Юг Na2B4O7 и 12г Na2CO3 в 1000мл воды, к
которому добавлен индол, 150 мл изопропилового спирта и 100 мл
ацетона; раствор 2 — раствор 3,5 мл 13,3%-ной Н2О2 в 1000 мл
воды, к которому добавлено 250 мл изопропилового спирта. Равные
количества обоих растворов под постоянным давлением поступают
из сосудов 1 и протекают по капиллярам, проходящим внутри до-
затора 2, в смеситель 3. Смесь реагентов (pH 10,7) при прохожде-
243
ОВ, падает под прямым углом
Раствор
Рис. 25. Измерительная ячейка
флуоресцентного фотометра:
1 — кювета (сравнительная); 2—фильтр;
5—конденсатор; 4—УФ-лампа; 5—измери-
тельная кювета.
нии по абсорбционной колонне 4 поглощает из воздуха, поступаю-
щего противотоком, ОВ нервно-паралитического действия и посту-
пает в измерительную ячейку флуоресцентного фотометра 5.
Схема устройства измерительной ячейки фотометра показана
на рис. 25.
__Свет флуоресценции индоксила, образующегося в присутствии
к возбуждающему лучу света на
фотоэлектронный умножитель и
включает сигнальное устрой-
ство.
Более мощный прибор подоб-
ной конструкции (модель В25)
предназначен для стационарного
применения в полевых лаборато-
риях. Он состоит из двух частей.
В одной из них размещены на-
сосы, растворы реагентов, фото-
метр и фотоэлектронный умножи-
тель, в другой — источник энер-
гии, электроуправление и сигналь-
ное устройство. Более усовер-
шенствованной моделью аппара-
тов этого типа является газоана-
лизатор В38, предназначенный
для работы в лабораторных условиях. Он оборудован модифици-
рованным узлом отбора проб воздуха при разном давлении; в ка-
честве стандарта для измерения флуоресценции используется
кювета с раствором хинина. При изменении интенсивности флуо-
ресценции измерительного элемента в цепи анодного тока фото-
электронного умножителя возникает переменное напряжение, кото-
рое посредством усилителя и вспомогательного мотора приводит в
действие диафрагму до тех пор, пока снова не установится равно-
весие между измерительным элементом и выравнивателем. Измене-
ние диафрагмы является мерой концентрации ОВ нервно-паралити-
ческого действия.
Чувствительность прибора В38 около 0,01 мкг) л.
9.3.2. Приборы, основанные на физических
принципах индикации
Из автоматических приборов индикации токсических веществ, дей-
ствие которых основано на физических измерениях, для анализа
боевых химических веществ в полевых условиях пригодны только
такие селективные приборы, которые измеряют поглощение отрав-
ляющими веществами излучения в УФ- и ИК-областях спектра
Так, Клотц и Дол67 описали УФ-фотометр, предназначенный для
определения ОВ в лабораторных и полевых условиях, чувствитель-
244
ность которого оценивается в 3 р. р. гл. по иприту и 1 р. р. гл. по
фосгену и хлорпикрину.
Сигнальное устройство американской армии под названием
«лопаир» (сокращение от «long path infra-red») состоит из индика-
торной головки, укрепленной на штативе, в которой находятся
источник ИК-излучения, зеркало для направления излучения, де-
тектор излучения и зеркала, смонтированного также на штативе на
расстоянии 400 м от головки, которое возвращает излучение на
индикаторную головку. Все пространство между головкой и зерка-
лом служит как бы огромной «кюветой» для определяемого газа.
Индикация осуществляется селективно при помощи монохроматора,
установленного на определенные, характерные для определяемого
ОВ линии поглощения. Вес индикаторной головки равен 17,6 кг, а
отражающего зеркала — 7,4 кг. О чувствительности этого прибора
сказано, что с его помощью можно обнаружить зарин, капля кото-
рого величиной с булавочную головку испарилась в комнате сред-
них размеров; при грубом подсчете это соответствует концентрации
около 0,01 мкг!л.
По-видимому, принципиально возможно также применение ав-
томатических индикаторных приборов с использованием пламенных
ионизационных детекторов, разработанных в последнее время для
газовой хроматографии и специфично определяющих галоген- и
фосфорсодержащие соединения. Чувствительность этих детекторов
порядка нанограммов, но пока никаких сведений об этом не
имеется.
9.3.3. Приборы, действие которых основано
на биохимическом принципе
Создание автоматических газоанализаторов,' в которых принцип
индикации явлаетбя биохимическим, дает право надеяться на до-
стижение высокой чувствительности и селективности. Вместе с тем
необходимость своевременного предупреждения требует опреде-
ленной быстроты' проведения индикации, что очень трудно дости-
жимо вследствие времени, требующегося на угнетение фермента
отравляющим веществом. В одном голландском патенте68 описан
газоанализатор, действие которого основано на таком принципе.
В нем индикация зарина при концентрации 5 мкг/л происходит
через 5 сек, а при концентрации 1 мкг/л — через 60 сек.
Существуют две возможности работы газоанализатора на био-
химической основе. В соответствии с одной из них воздушный по-
ток нагнетается насосом в абсорбер, где он тщательно смешивается
с раствором фермента. Количество субстрата, разложенного рас-
твором фермента за определенный промежуток времени, изме-
ряют фотометрически, по изменению флуоресценции или электро-
химически. При прохождении через прибор чистого незараженного
воздуха количество субстрата, разлагаемого данным ферментом,
постоянно; если же в воздухе находится ингибитор фермента,
разложение субстрата уменьшается и получаемое в результате
245
этого отклонение от исходной величины приводит в действие си-
гнальное устройство.
Вторая возможность состоит в разделении воздушного потока
на две части, причем одна часть перед поступлением в абсорбер
с измерительным элементом проходит через специальный фильтр,
поглощающий ингибитор фермента, вторая же попадает сразу во
второй абсорбер с измерительным элементом. Разница в показа-
ниях обоих измерительных элементов указывает на присутствие
ингибитора фермента в воздухе, а возникшее отклонение приводит
в действие сигнальное устройство.
В качестве источника фермента используют сыворотку крови,
а для выбора подходящего субстрата имеются различные возмож-
ности. Фотометрическое измерение расщепления карбоксихолинов
с помощью pH-индикатора чрезвычайно чувствительно, но при
этом необходимо улавливать соответствующими фильтрами из воз-
душного потока все кислые газы и пары. Вполне пригодны такие
флуорогенные субстраты, как индоксилацетат, который дает воз-
можность с большой чувствительностью определять активность фер-
мента. Недостаток, связанный с их применением, заключается в не-
обходимости более сложного аппаратурного оформления для изме-
рения флуоресценции.
9.4. ИНДИКАТОРНЫЕ НАБОРЫ
И НОСИМЫЕ ПОЛЕВЫЕ ЛАБОРАТОРИИ
Наряду с приборами химической разведки, имеющими набор ин-
дикаторных трубок, автоматическими газоанализаторами, пере-
движными и стационарными полевыми лабораториями на снабже-
нии большинства армий имеются еще и простейшие индикаторные
наборы и носимые полевые лаборатории. Ввиду множества таких
устройств, а также многообразия их выполнения описать их до-
вольно трудно. В простейшем случае речь идет о наборе элемен-
тарных индикаторных средств — индикаторных бумаг и карандашей
в какой-то удобной упаковке, который разведчик-химик носит или
возит с собой.
Например, таким индикаторным набором является набор, со-
стоящий из двух индикаторных бумаг на капельно-жидкие ОВ
(серный иприт и фосфорсодержащие ОВ), и двух индикаторных
бумаг для паров синильной кислоты и фосгена. Этот набор заклю-
чен в пластмассовую коробку размером 9 X 12 X 1 см,. Основными
преимуществами этого индикационного набора, как и других ана-
логичных, являются простота обращения, скорость и однозначность
индикации. Такой набор разрешает разведчику-химику идентифи-
цировать капли стойких ОВ на местности или на поверхности
предметов техники или вооружения, облегчает принятие быст-
рого решения по организации необходимых мероприятий, связан-
ных с преодолением зараженных участков местности или с дегаза-
цией техники.
246
Доставка проб в передвижные или стационарные полевые ла-
боратории и получение результатов анализа занимают очень много
времени. Стремление исключить эту стадию работы приводит к
ограничению числа проб, анализируемых в лабораториях, несколь-
кими наиболее важными. Поэтому стремятся создать такой носи-
мый набор реагентов и приборов, который позволил бы в кратчай-
ший срок провести анализ некоторых проб непосредственно на
месте их отбора. В наборах этого типа обычно имеются специально
подобранные реагенты и приборы для осуществления качественного,
а в ряде случаев и количественного определения важнейших ОВ и
диверсионных ядов.
Так, в одном из комплектов для проверки пищевых продуктов
имеется простейший экстракционный прибор и набор реагентов
для индикации фосфорсодержащих ОВ, иприта, мышьяка, а также
для групповой индикации алкалоидов и токсичных солей тяжелых
металлов. Комплект для исследования воды должен быть составлен
с учетом выполнения этой задачи. Для решения вопроса, будет ли
вода какого-нибудь источника пригодна для питья после ее хлори-
рования, вполне достаточно качественного исследования, чувстви-
тельность которого подобрана так, чтобы определить концентрации
ОВ выше допустимых. Если ОВ содержится в количествах, превы-
шающих количества, которые могут быть устранены хлорированием,
то это указывает на необходимость проведения специальной очист-
ки воды. Для более подробного испытания воды, например для
контроля водоочистительйых установок, следует пользоваться ин-
дикаторным набором, содержащим приборы и реагенты для
полуколичественного определения основных ОВ и для определе-
ния pH.
Фосфорорганические ОВ можно количественно определять био-
химическим методом при помощи индикаторной бумаги, содержа-
щей субстрат, причем время реакции можно определять по подго-
товленной контрольной пробе сравнения. Иприт и цианиды м5жно
определять путем визуально-колориметрической оценки цветных
реакций, с использованием соответствующих цветных стандартов;
мышьяк обнаруживают пробой Гутцейта, сравнивая полученные на
бумаге окраски с таблицей цветовых эталонов.
Кроме индикаторных наборов можно еще использовать ком-
плект для проверки полноты дегазации, в котором наряду со сред-
ствами, необходимыми для индикации ОВ, должны быть тампоны и
растворители для выполнения проб на мазки.
В основном известные индикаторные наборы рассчитаны на про-
ведение в среднем 20—50 испытаний, после чего аппараты и реа-
генты следует заменить свежими, из запасов службы снабжения.
Очень удобными являются расфасованные реагенты в ампулах или
таблетках, содержащие уже отвешенные количества веществ, не-
обходимых для проведения того или иного анализа или для при-
готовления определенных объемов нужных растворов.
247
Реактивы и аппараты в запасных наборах следует разделять
на менее и более стабильные. Некоторые виды определений тре-
буют обязательного нагревания. Для этого в наборах должны быть
предусмотрены бензиновые или спиртовые горелки с подставками.
Нагревание может быть также осуществлено таблетками твердого
спирта или электрическими нагревательными приборами, которые
можно питать от аккумуляторных батарей автомобилей.
9.5. ПЕРЕДВИЖНЫЕ ПОЛЕВЫЕ ЛАБОРАТОРИИ
Передвижные полевые лаборатории создают практически неогра-
ниченные возможности для анализа боевых химических веществ в
непосредственной близости к фронту. Эти лаборатории, смонтиро-
ванные в кузове крытого грузового автомобиля, чрезвычайно под-
вижны, что зависит от проходимости автомобиля), и могут быть
приведены в рабочее состояние в кратчайший срок.
Приводим возможный вариант оснащения лаборатории: пита-
ние током осуществляется транспортируемыми агрегатами, обеспе-
чивающими отопление, внутреннее -освещение, вентиляцию вытяж-
ных шкафов, работу настенных вентиляторов, вакуум-насоса, водя-
ного насоса для наполнения водяных баков, сушильного шкафа,
УФ-лампы, подогрев резервуара для теплой воды. Источниками
тепла, необходимымидля аналитических работ, являются установка
с пропаном для обслуживания газовых горелок и спиртовые горел-
ки. Дистиллированную воду можно получать на ионообменных ко-
лонках.
Лаборатории обычно имеют достаточно большой запас дегази-
рующих средств для дегазации использованной посуды и рабочего
места. Каждый полевой лаборант должен иметь отдельное рабо-
чее место на столе и в вытяжном шкафу. Место для работы дол-
жно быть покрыто такими легко дегазируемыми материалами, как
поливинилхлорид или листы жести. Стеклянные приборы, рабочие
и запасные реактивы во время переездов находятся в специальных
шкафах и ящиках, закрепленные в держателях, обеспечивающих
их сохранность. На каждом рабочем месте имеются штативы из
дерева или пластмассы с рабочим набором реактивов в простых
капельницах или капельницах с пипетками. Они должны обычно
храниться в шкафах, а перед началом работы — выставляться на
столы на специально для них предназначенные места.
Задачи, разрешаемые в передвижных полевых химических ла-
бораториях, зависящие от величины и оборудования этих, лабора-
торий, очень велики. В основном, по-видимому, этими задачами
являются:
1. индикация всех известных ОВ в воздухе, воде и почве (про-
бы земли), а также на обмундировании, снаряжении и технике;
2. количественное определение фосфорорганических ОВ, ипри-
248
job, цианидов и мышьяксодержащих соединений в воде и пищевых
продуктах;
3. индикация диверсионных ядов (алкалоидов и токсичных ани-
онов и катионов) в воде и продуктах питания;
4. качественный элементный анализ и индикация определенных
функциональных групп для идентификации неизвестных ОВ;
5. контроль за полнотой дегазации обмундирования и снаря-
жения;
6. химический анализ питьевой воды и контроль за водоподго-
товкой;
7, качественный и количественный анализ дегазирующих ве-
ществ;
8. определение важнейших физических констант ОВ.
Кроме того, оборудование большинства передвижных полевых
автолабораторий дает возможность отбирать любые пробы для
анализа. При помощи средств, предназначенных для биохимиче-
ского определения фосфорорганических ОВ, в этих лабораториях
можно определять активность холинэстеразы в крови.
В основном все анализы проводятся полумикрометодом. Для
количественного определения ОВ, установления pH и содержания
хлора в воде в автолабораториях многих армий имеются специ-
альные компараторы.
9.6. ОТБОР ПРОБ ДЛЯ АНАЛИЗА
Анализ ОВ начинается с отбора проб. От навыка и умения лица,
отбирающего пробы, и от соблюдения указаний и соответствующих
предписаний зависит скорость и точность анализа. Общим прави-
лом является стремление отобрать пробу так, чтобы ОВ попадало
по возможности в чистом виде. Так, если при разведке заражен-
ного участка местности- найдены явно различимые остатки ОВ на
осколках боеприпасов или капли на растительности или предметах
вооружения, то вместо образцов почвы и растительности берут про-
бы мазком. Это дает возможность избежать последующей экс-
тракции ОВ из образцов почвы или растений и обесцвечивания
этих экстрактов. Все эти операции, как известно, отнимают много
времени и, таким образом, увеличивают продолжительность ана-
лиза и задерживают выдачу результатов.
Другим общим правилом является отбор наименьшего числа
проб для отправки в лабораторию. Если химик-разведчик имею-
щимися у него средствами (индикаторные трубки, индикаторные
бумаги) установит, что различные участки местности заражены
одинаковым ОВ, то вполне достаточно отобрать одну пробу, по
которой можно убедиться в применении химического оружия.
В случае, когда возникают сомнения, приходится отбирать не-
сколько проб в различных местах.
Лицо, отбирающее пробы, должно следить за тем, чтобы в пробу
попало как можно больше ОВ. Это необходимо для того, чтобы
249
в химической лаборатории при обследовании неизвестного ОВ мо-
жно было провести его качественный элементный анализ или опре-
делить его физические константы, для чего требуется значительное
количество вещества. При малой плотности заражения взятые про-
бы необходимо до проведения аналитических работ подвергнуть
соответствующей обработке, способствующей повышению концен-
трации ОВ, на что, естественно, затрачивается известное время.
В следующих разделах приведены некоторые рекомендации по по-
вышению концентрации веществ в пробах.
9.6.1. Отбор проб почвы
Пробы следует отбирать в местах, где предполагается повышен-
ная плотность заражения. К таким местам в первую очередь от-
носятся края и непосредственно примыкающая к воронкам от
взрыва химических боеприпасов местность. Кроме того, следует по-
стараться отыскать осколки боеприпасов, на которых может остать-
ся ОВ.
Проба почвы для количественного определения плотности за-
ражения отбирается при помощи шаблона площадью 50 или
100 см2. В зависимости от времени, прошедшего с момента зара-
жения, снимают слой почвы глубиной 1—5 см. По возможности
следует избегать брать на анализ влажную почву, так как пробы
анализируют не сразу и до проведения анализа содержание ОВ
Таблица 25. Стойкость ОВ на местности
Отравляющее вещество СТОЙКОСТЬ
летом ЗИМОЙ
Серный иприт 3—4 суток на открытом месте, 1 не- Несколько недель
Люизит деля в лесу 1 сутки на открытом месте, Около 1 недели
Зарин 2—3 суток в лесу От 10 мин до 12 ч То же
Табун От 10 мин до 24 ч »
Фосген 5 мин на открытом месте 10 мин на открытом
Синильная кисло- От 1 до 10 мин месте Несколько часов
та Хлорцнан От 5 до 10 мин —
может сильно понизиться вследствие гидролиза. При отборе проб
почвы большое значение имеет стойкость ОВ, которая может за-
висеть от давления паров, устойчивости к гидролизу и многих дру-
гих факторов (см. табл. 25).
250
Брызги и капли стойких ОВ дольше удерживаются на листьях
кустов и на траве, и их легко снимать фильтровальной бумагой,
тампонами ваты или целлюлозы. Если же с момента заражения
прошло длительное время и на местности следы ОВ не заметны,
то в качестве проб следует собрать те части растений, на которых
ясно заметно заражение (иная окраска, увядшие части).
9.6.2. Отбор пробы воды
При отборе проб воды необходимо учитывать различное поведе-
ние ОВ в воде. Серный иприт мало растворим в воде и имеет
большую плотность, чем вода, поэтому относительно крупные кап-
ли иприта опускаются на дно водоема и могут сохраняться там в
течение долгого срока. Некоторое его количество распределяется
тонким слоем по поверхности воды, обра-
зуя маслянистые пятна. В стоячих водое- j
мах следует такие маслянистые пятна 6»
обязательно собрать и отправить на ана- ,а
лиз. Растворенный иприт сравнительно ( \ те
быстро гидролизуется. Длительные зара- \ //\\\
жения водоемов ипритом объясняются \ \ Rob
тем, что по мере его гидролиза свежие \ \ '
порции поступают из находящихся на
дне капель иприта.
Фосфорорганические ОВ по своему
поведению резко отличаются от иприта. Cr'YiS'
Зарин очень хорошо растворим в воде и
поэтому его концентрации быстро дости- РиС 26 Б для 0
гают высоких значении. Табун и зоман Проб воды,
растворяются хуже и капли их также мо-
гут оседать на дно водоема и постепенно растворяться в течение
длительного времени. Синильная кислота и ее соли очень хорошо
растворимы в воде, вследствие чего вода остается зараженной дли-
тельное время. Аналогично ведут себя гидрохлорид азотистого
иприта, фторацетат и другие диверсионные яды (алкалоиды и то-
ксичные неорганические соли).
Отбирать пробы воды следует как с поверхности, так и на раз-
личной глубине в заранее подготовленные бутылки, изображенные
иа рис. 26. После погружения бутылки на требуемую глубину
рывком веревки открывают пробку, и вода заполняет бутылку.
В специальных приборах для отбора проб воды имеются пружин-
ные пробки, закрывающиеся после ослабления веревки. Для про-
ведения полного анализа требуется около 2 л воды. Пробу из ко-
лодца отбирают после тщательного перемешивания в нем воды при
помощи ведра на канате или подобного приспособления. Особое
внимание следует уделять осмотру окрестности места отбора проб
с целью обнаружения каких-либо других признаков воздействия
отравляющего вещества.
251
9.6.3. Отбор проб ОВ с военной техники,
обмундирования и снаряжения
С металлических поверхностей, не имеющих лакокрасочного по-
крытия, заметные капли ОВ снимают тампонами из ваты или
целлюлозы. Если мельчайшие капельки ОВ или его аэрозольные
частицы распределены по всей поверхности, то пробу отбирают
тампонами, смоченными этиловым ^спиртом или петролейным эфи-
ром. Таким же образом поступают и при отборе проб с окрашен-
ных поверхностей. При длительном воздействии ОВ на лакокра-
сочную поверхность после удаления капель соскребывают также
и покров находившейся под ними краски. При отборе проб с дере-
вянных поверхностей снимают ножом верхний слой древесины тол-
щиной 0,5—1 см.
Заметные капли ОВ на обмундировании также снимают тампо-
нами, смоченными'спиртом или петролейным эфиром. Если же ви-
зуально капли не обнаружены, то отдельные части обмундирования
погружают в растворитель, а раствор затем упаривают. В некото-
рых случаях необходимо даже вырезать отдельные куски обмунди-
рования для экстракции ОВ; так поступают при отборе выборочных
проб и при определении зараженности резинового защитного ко-
стюма.
9.6.4. Отбор проб продовольствия и фуража
Прежде чем отбирать пробы продовольствия, тщательно осматри-
вают местность вокруг склада и упаковку продуктов питания. В за-
крытых помещениях складов при помощи индикаторных трубок
устанавливают наличие ОВ в воздухе. Продукты питания можно
считать полностью защищенными от воздействия жидких или га-
зообразных ОВ, если они были упакованы в металлическую или
стеклянную тару. Для того чтобы убедиться, не заражена ли упа-
ковка, нужно смоченными тампонами взять пробу с ее поверхно-
сти, как это было указано раньше при описании отбора проб с
поверхности военной техники. Упаковка из искусственных материа-
лов пропускает ОВ, и через некоторое время находящиеся в ней
продукты будут заражены. 'Жидкие ОВ очень быстро проникают
через бумагу и текстильные материалы (мешки). То же относится
и к парообразным веществам, легко заражающим продукты в
такой упаковке, если только упаковка ничем не была защищена
снаружи.
Для установления зараженности продуктов, находящихся в про-
ницаемой для ОВ упаковке, кроме пробы с поверхности тары бе-
рется проба слоя продукта, находящегося под зараженным местом
тары. Такая проба в количестве 50—100 г используется для каче-
ственного обнаружения ОВ. Если упаковка сильно нарушена, то
требуется взять среднюю пробу от всего содержимого тары. Для
этого сначала удаляют сильно зараженные слои, а остальные тща-
252
тельно перемешивают и отбирают пробу. Даже для сыпучих про-
дуктов (зерно, мука, крупа и сахар), верхние слои которых наибо-
лее сильно адсорбируют пары ОВ, берут пробу с поверхности не
глубже 1—3 см. При видимом проникновении капель ОВ внутрь
продукта пробу следует отбирать и с большей глубины. Особенно
опасно заражение жиров или жирных колбас и мяса. Хорошая
растворимость большей части ОВ в жирах при их длительном воз-
действии приводит к полному заражению продуктов этого типа.
Чтобы исключить возможные ошибки, рекомендуется наряду
с пробами зараженных продуктов отправлять в лабораторию и
пробы чистых продуктов, чтобы там, при необходимости, могли
для сравнения обследовать их поведение при взаимодействии с
реагентами.
9.6.5. Отбор проб воздуха
Качественное и количественное определение газов и паров в воз-
духе обычно производится предназначенными для этой цели сред-
ствами— автоматическим газоанализатором, индикаторными труб-
ками и бумагами. В сомнительных же случаях (при несовпадении
показаний) необходимо отбирать пробы воздуха для отправки на
анализ в лабораторию. Способ отбора проб, широко применяемый
в промышленности, непригоден в боевых условиях при очень малом
содержании ОВ в воздухе. Необходимо создавать в пробах такое
содержание ОВ, которое бы отвечало чувствительности аналитиче-
ских реакций. Для этого существуют различные методы, как-то:
абсорбционное и адсорбционное улавливанйе ОВ на сорбентах и
растворителями, а при отборе проб ядовитых дымов и аэрозолей
используют различные фильтры. В общем случае обычно бывает
достаточйо качественно установить наличие того или иного ОВ, гак
как концентрация последнего в воздухе так быстро изменяется, что
количественное определение не имеет большого значения. Количе-
ственные определения требуются для лабораторных исследований,
например при калибровке индикаторных приборов, когда концен-
трация ОВ в испытуемом воздухе камеры или динамического' дози-
рующего прибора должна быть точно известна. В этих случаях на-
ряду с соблюдением постоянства таких факторов, как температура
и скорость воздушного потока, объем воздушной пробы, нужно так-
же иметь сведения об адсорбции или абсорбции данного ОВ, а так-
же о десорбции его с сорбента.
9.6.5.1. Адсорбция. Накопление ОВ в пробе достигается про-
сасыванием с помощью резинового баллона, ручного насоса (от
прибора химической разведки) или насоса с электроприводом за-
раженного воздуха через твердый сорбент, помещенный в стеклян-
ную трубку, подобную индикаторной (рис. 27). Объем воздуха,
проходящего через трубку, зависит от мощности насоса и времени,
имеющегося в распоряжении лица, отбирающего пробу. Для систе-
матического анализа, который приходится проводить при полном
253
Вата Адсорбент
Вата
Рис. 27. Трубка для адсорбции газо-
образных ОВ.
отсутствии данных о типе ОВ, требуется пропустить не менее 50 —
100 л воздуха со скоростью около 2 л/мин.
В качестве адсорбентов применяют обычно активированный
уголь и силикагель. Мелкозерненый активированный уголь обла-
дает высокой адсорбционной способностью. Однако это свойство
является и недостатком, так как сорбированное вещество настолько
прочно удерживается углем, что извлечение его (особенно это от-
носится к фосфорорганическим ОВ) связано с большими труд-
ностями. Другим недостатком активированного угля является его
черная окраска, исключающая
возможность проведения цветной
реакции непосредственно на copJ
бенте. Извлечение ОВ проводят
пропусканием через трубку соот-
ветствующего растворителя. Чаще
всего для этой цели применяют
этиловый спирт. Более полное из-
влечение достигается при кипяче-
нии сорбента с этиловым спиртом. После фильтрования горячего рас-
твора и охлаждения фильтрата проводят реакции обнаружения ОВ.
Для увеличения активности силикагеля, предназначенного для
снаряжения трубок, его подвергают специальной обработке.
В колбе с обратным холодильником в течение 2 ч кипятят 100 г мелкозер-
неного (0,25—0,5 мм) силикагеля в 0,5 л разбавленной НС1 (1 : 1). Кислоту
сливают, а силикагель сначала промывают водой до нейтральной реакции, а за-
тем 0,5 л этилового спирта и сушат на воздухе. После этого сухой силикагель
прокаливают в печи при 300—400 °C. Охлажденный силикагель хранят в герме-
тичной стеклянной посуде*й перед употреблением смачивают небольшим коли-
чеством воды.
Силикагелевые трубки с адсорбированным ОВ следует подвер-
гать обработке не позднее чем через 24 ч после отбора пробы, так
как устойчивость ОВ, особенно эфиров фосфорной кислоты, очень
невелика вследствие гидролитического разложения под влиянием
влаги, всегда присутствующей на силикагеле. Устойчивость метил-
фторфосфонатов можно повысить, если предварительно пропитать
силикагель раствором гидрофосфата калия, доведенным до pH 4
добавлением фосфорной кислоты69. Адсорбированное ОВ лучше
всего экстрагировать ацетоном, что благоприятствует дальнейшему
проведению реакции Шёнеманна для обнаружения фосфороргани-
ческих ОВ.
9.6.5.2. Абсорбция. Для эффективной абсорбции ОВ требуется
соответствующая аппаратура. Обычно для этого применяют раз-
личного типа поглотительные склянки. Наиболее удобны такие кон-
струкции склянок, которые обеспечивают быстрое обновление слоя
жидкости на поверхности раздела фаз газ — жидкость. Склянки
различных конструкций показаны на рис. 28. Первая склянка пред-
ставляет собой склянку Дрекселя, в которой газ перед попаданием
в жидкость проходит через пористую стеклянную пластинку, В этой
254
склянке происходит хорошее перемешивание, однако в нее прихо-
дится наливать большой объем поглощающей жидкости. Для ра-
боты в полевых условиях больше подходят две другие склянки. Их
преимущество заключается в том, что в них достаточно налить
только несколько миллилитров растворителя. После завершения
поглощения вынимают барботирующее устройство и, не переливая
раствор в другой срсуд, добавляют реагент для обнаружения ОВ.
Во время поглощения воздух пропускают через склянки со скоро-
стью примерно 0,5—1 л)мин.
Вторым не менее важным фактором эффективного поглощения
является выбор подходящего растворителя, при подборе которого
Рис. 28. Поглотительные склянки различных конструкций.
следует учитывать такие его свойства: физические константы, спо-
собствующие поглощению (летучесть, вязкость); устойчивость ОВ
в данном растворителе; смешиваемость с реагентами, применяе-
мыми для обнаружения ОВ; чувствительность реакции в данном
растворителе; действие солнечного света на растворитель, напри-
мер образование перекисей, мешающих проведению реакции Шёне-
манна; чистота растворителя.
Часто применяемые этиловый и изопропиловый спирты обла-
дают слишком большой летучестью, что ограничивает их примене-
ние только для случаев, когда для анализа достаточно пропустить
небольшие объемы воздуха. Для поглощения фосфорорганических
ОВ и последующего определения их по реакции Шёнеманна при-
годны (2-метилпентадиол-2,4) 69 и циклогексанол70. Если же инди-
кацию фосфорорганических ОВ осуществляют биохимическим ме-
тодом, то следует пользоваться водным спиртом или водным рас-
твором таких веществ, которые совместимы с сывороткой крови
лошади. При обнаружении фосфорорганических ОВ по продуктам
их разложения — фосфат- и фтор-ионам (зарин, зоман) или фос-
фат- и цианид-ионам (табун) — в качестве растворителя используют
2%-ную щелочь.
255
При идентификации заранее известного ОВ в поглотительную
склянку можно наливать устойчивый, мгновенно реагирующий с
ОВ реагент в соответствующем растворителе. Такие реакции и
реагенты были описаны в гл. 5, посвященной качественным и коли-
чественным методам определения отдельных ОВ.
9.6.5.3. Фильтрация. Некоторые малолетучие кристаллические
вещества с высокой температурой плавления, такие, как раздра-
жающие глаза хлорацетофенон и бромбензилцианид или раздра-
жающие носоглотку и органы дыхания дифенилхлорарсин и адам-
сит, могут быть применены в виде аэрозолей. Коэффициент осажде-
ния частиц аэрозоля размером меньше 8 мкм в поглотительных
склянках очень непостоянен и в большинстве случаев недостаточен,
Рис. 29. Устройство мембранного фильтра:
/—мембранный фильтр; 2—металлическое сито; 5 —резиновое уплот-
нение; 4—муфта.
вследствие чего частицы размером 0,4 мкм, обладающие наиболь-
шим физиологическим действием, практически поглощаются очень
слабо. По этой причине аэрозоли ОВ улавливают из воздуха при
помощи соответствующих фильтрующих материалов. Насосу при-
бора химической разведки придана специальная насадка, в кото-
рой имеется противодымный фильтр или кусочек фильтр-картона
соответствующей формы (картон производится Народным пред-
приятием Нидершлаг, ГДР). Частицы ОВ извлекают обработкой
фильтра кипящим спиртом. Для труднорастворимых ОВ, таких, как
адамсит, экстракцию приходится повторять многократно.
Для улавливания частиц аэрозоля из воздуха могут быть ис-
пользованы также трубки для отбора проб воздуха, в которых
слой силикагеля заменен ватным тампоном или мембранным филь-
тром 73. Последний представляет собой листочки фильтра из спе-
циально подготовленной целлюлозы, легко пропускающей воздух
и газы, но задерживающей частицы аэрозоля. Диаметр пор такого
фильтра рассчитан так, чтобы на нем задерживались частицы, раз-
мер которых характерен для частиц ядовитого дыма. Удобный
фильтр изображен на рис. 29. Преимуществом мембранных фильт-
ров является их растворимость в ряде органических растворителей,
что в значительной степени облегчает количественное определение
осевшего аэрозоля.
256
9.6.6 Маркировка проб
Большое значение для дальнейшего испытания проб имеет точное
их обозначение перед отправкой в лабораторию. Часть необходи-
мых данных можно написать иа этикетке, наклеиваемой на упа-
ковку пробы. Более подробные сведения заносятся в формуляр до-
несения.
Основными пунктами описания пробы являются: место и объект
отбора пробы (для проб, взятых на местности, указывается геогра-
фическое положение на карте или приложенной кроке); время от-
бора пробы; наблюдения, которые могут помочь определить способ
применения ОВ противником; средство применения; метеорологи-
ческие условия в период времени между химическим нападением
и отбором пробы; результаты индикации ОВ простейшими сред-
ствами (индикаторные бумаги, порошки, трубки) или исследова-
ния, проведенного индикаторным набором.
В зависимости от вида пробы приводятся некоторые-специаль-
ные данные: тац, например, для проб пищевых продуктов — опи-
сание упаковки, для проб воды — скорость течения, глубина и
ширина водного источника.
9.7. ПОДГОТОВКА ПРОБ К АНАЛИЗУ
Из различных проб, доставляемых в полевую химическую лабора-
торию, непосредственно анализу можно подвергать только пробы
воды, пробы воздуха, полученные поглощением специальным рас-
творителем, и пробы чистых отравляющих веществ. Пробы почвы,
пищевых продуктов и других подобных материалов должны быть
предварительно соответствующим образом подготовлены. Задачей
подготовки пробы является перевод ОВ из материала пробы в та-
кую среду, которая была бы удобна для индикации или количе-
ственного анализа.
Основными способами решения этой задачи являются экстрак-
ция ОВ растворителями или вытеснение их воздухом. Оба способа
приводят в конечном счете к увеличению концентрации ОВ, что в
большинстве случаев дает возможность установить присутствие
даже следов ОВ в исходной пробе. По этой же причине всегда
нужно стремиться экстрагировать ОВ минимальным количеством
растворителя.
Обработку растворителями производят в химических стаканах,
заливая измельченную пробу растворителем так, чтобы уровень
растворителя был на несколько миллиметров выше пробы. Вопрос
о том, при какой температуре производить экстракцию, зависит от
материала пробы и растворимости ОВ. Экстракцию можно прово-
дить- и в специальных приборах, например в аппарате Сокслета.
Преимущество таких приборов заключается в малом расходе
растворителя, а также в том, что одновременно с извлечением
идет и фильтрация раствора. Однако для этих приборов требуется
довольно много места и, кроме того, обогрев. Существенным
9 Зак. 677
257
затруднением в работе с подобными приборами является трудность
дегазации их после работы с ОВ. Дегазировать химические стака-
ны, конечно, гораздо проще. Все это является причиной того, что
эти приборы используются только при обширных исследованиях.
Растворителями для экстракции ОВ служат обычные органиче-
ские растворители — этиловый спирт, петролейный эфир, диэтило-
вый эфир, дихлорэтан, хлороформ и др. Часто используют также
и воду. Выбор растворителя прежде всего зависит от вида ОВ и его
растворимости, материала пробы и предполагаемого метода ана-
лиза. Хлорсодержащие растворители — дихлорэтан и хлороформ—
реагируют, например, с тимолфталеином — реагентом для обнару-
жения иприта, поэтому в данном случае эти растворители не могут
быть применены, но для определения фосфорорганических ОВ или
для экстракции алкалоидов они вполне пригодны. Во всех случаях,
где только это возможно, следует применять растворители, хорошо
смешивающиеся с водой, что обеспечивает получение гомогенных
смесей экстракта с водными растворами реагентов.
При выборе того или иного растворителя нужно руководство-
ваться следующим правилом: растворитель должен как можно
меньше (если совсем не должен) извлекать из материала пробы
такие вещества, которые могут помешать определению. Конечно,
это не всегда достижимо. В том случае, если не удается подыскать
подходящий растворитель и нет возможности удалить такие при-
меси путем соответствующей обработки экстракта, приходится
прибегать к продуванию воздухом. Спиртовые экстракты можно
обесцвечивать обработкой их активированным углем с последую-
щим фильтрованием горячего раствора, а в более крупных иссле-
довательских лабораториях можно отделять примеси пропуска-
нием экстракта через хроматографическую колонку. В том случае,
когда приходится использовать достаточно большой объем раство-
рителя, следует предпочесть растворитель с низкой температурой
кипения. Такой растворитель можно удалить осторожным нагрева-
нием на воздухе или при разрежении, не теряя при этом значитель-
ного количества ОВ. Если для извлечения выбран не смешиваю-
щийся с водой растворитель (эфир, бензол, толуол, дихлорэтан или
петролейный эфир), то при попадании в него воды раствор мутнеет.
Экстракт сушат при энергичном встряхивании над небольшим ко-
личеством безводного NajSOi. Сохранение помутнения свидетель-
ствует о наличии взвеси, т. е. частиц, прошедших через фильтр, не
соответствовавший материалу пробы, поэтому его следует профиль-
тровать еще раз через более плотный фильтр.
Для извлечения ОВ воздухом взрыхленную пробу помещают в
Ъ’-образиую трубку, которая погружена в водяную баню с темпе-
ратурой ~50°С. Воздушный поток, пройдя эту трубку, поступает
в поглотительную склянку с растворителем, поглощающим ОВ,
Воздух продувают через систему при помощи резиновой груши или
насоса. Во избежание потерь ОВ все сосуды установки должны
быть герметично присоединены друг к другу.
258
Извлечение ОВ воздухом применимо только для ОВ, обладаю-
щих большой летучестью. Обычно на этот процесс расходуется
много времени — от 30 до 120 мин. Однако он совершенно непри-
меним для малолетучих ОВ, таких, как дифенилхлорарсин, дифе-
нилцианарсин и адамсит, а также алкалоиды и токсичные анионы
и катионы. Воздушный поток можно направить не в поглотитель-
ные склянки, а в индикаторные трубки или в склянки, в которых
вместо растворителя налит специфически устойчивый реагент на
данное ОВ. В этом случае сразу получают нужный ответ.
При необходимости провести только качественное обследование
выбирают для экстракции наиболее зараженную часть пробы. Для
количественного определения тщательно перемешивают всю пробу
и используют определенные навески для экстракции разными рас-
творителями.
Конечно, нет возможности предусмотреть и описать условия
работы для каждого отдельного практического случая. Поэтому
ниже приводятся некоторые общие указания по подготовке различ-
ных проб, которые могут служить своеобразным руководством при
проведении подобных работ.
Пробы почвы. В качестве растворителей для экстракции приме-
няются этиловый спирт, петролейный эфир и вода. При окрашива-
нии спиртовой вытяжки к ней прибавляют измельченный активиро-
ванный уголь (на кончике шпателя), тщательно перемешивают, на-
гревают и горячим фильтруют. Фосфорорганические ОВ могут быть
извлечены из водных растворов дихлорэтаном.
Пробы растений. Для извлечения ОВ из свежих растении ре-
комендуется кратковременная экстракция петролейным эфиром без
нагревания. Экстракция этиловым спиртом проводится на холоду
в течение 1 мин. Так как при этом невозможно избежать растворе-
ния зеленого пигмента листьев — хлорофилла, то после этого тре-
буется обесцвечивание активированным углем.
Пробы воды. При достаточно высокой концентрации ОВ в воде
пробы ее можно анализировать без всякой обработки. Однако в
большинстве случаев в воде находятся только следы ОВ, поэтому
приходится проводить обогащение раствора. Для этой цели при-
годно экстрагирование отдельных частей пробы различными рас-
творителями, не смешивающимися с водой. Экстракцию проводят в
делительной воронке, используя 50—100 мл пробы и 2—5 мл рас-
творителя. После разделения слоев органический слой можно ис-
пользовать для обработки другой части пробы воды. Для экстрак-
ции из воды серного иприта пригоден петролейный эфир, а для
экстракции фосфорорганических ОВ—дихлорэтан. Алкалоиды и
азотистый иприт извлекаются из проб воды хлороформом. Более
обогащенный образец для анализа получают путем пропускания
определенного объема исследуемой воды через трубку с мелкозер-
неным активированным углем, после чего ОВ извлекают из угля
этиловым спиртом.
9*
259
Способ концентрирования проб на активированном угле приго-
ден для извлечения серного иприта, однако при этом возможны
значительные потери иприта, объясняемые ускорением его гидро-
лиза при адсорбции на активированном угле.
При количественных определениях следует учитывать, что в
процессе концентрирования ОВ переходит в растворитель не ко-
личественно, а в соответствии с его коэффициентом распределения
в водной и органической фазах. Так, например, при экстракции
серного иприта из 100 мл воды и 5 мл петролейного эфира в ор-
ганическую фазу переходит 75% иприта. Из этой величины и нуж-
но исходить при расчете результатов анализа, т. е. при обнару-
жении 0,06 мг серного иприта в экстракте петролейного эфира, по-
лученного из пробы воды объемом 100 мл, истинное содержание
иприта в воде будет равно
0,06-100 1П по .
---— • 10 = 0,8 мг/л
Пробы пищевых продуктов. Выделение ОВ из проб пищевых
продуктов является наиболее трудной задачей. Это связано со
сравнительно прочным поглощением (при абсорбции и адсорбции)
большинства ОВ продуктами питания, а в случае жиров или жир-
ных продуктов — даже с равномерным распределением ОВ по
всему объему продукта, а отсюда и пробы. Кроме того, при эк-
стракции ОВ в экстракт переходит значительное количество ве-
ществ, мешающих анализу или искажающих полученные резуль-
таты. В связи с этим при подготовке проб пищевых продуктов
большое значение приобретает способ извлечения ОВ воздухом.
Алкалоиды и токсичные анионы и катионы экстрагируют горя-
чей водой. При извлечении алкалоидов рекомендуется добавлять
в воду небольшое количество НС1, а из полученного экстракта из-
влекать их хлороформом после подщелачивания раствора NaOH.
Мышьяксодержащие ОВ и продукты их разложения определяют
в части пробы после ее полной минерализации (ср. раздел
5.4.2.1).
9.8. СИСТЕМАТИЧЕСКИЙ КАЧЕСТВЕННЫЙ АНАЛИЗ ПРОБ ОВ
В ПОЛЕВЫХ ЛАБОРАТОРИЯХ
В большинстве случаев пробы ОВ, поступающие для анализа в
полевую лабораторию, имеют такую маркировку или сопровож-
даются таким донесением, что по этим данным уже можно сделать
предварительные выводы о типе ОВ. Поэтому после соответству-
щей подготовки пробы делается попытка идентифицировать пред-
полагаемое вещество с помощью специфичной или селективной ре-
акции. Однако если такая попытка не приводит к ожидаемым ре-
зультатам или если в описании пробы не содержится указаний
на присутствие какого-то определенного ОВ, то такую пробу под-
вергают систематическому анализу. При этом рекомендуется при-
260
держиваться определенного порядка, так как при бессистемных
поисках получение достоверного результата будет зависеть от сча-
стливой случайности. Конечно, очень трудно систематизировать
обнаружение разных типов ОВ, принадлежащих к самым разным
классам химических соединений. Кроме того, возможности
полевых лабораторий (о чем уже упоминалось) определенным
образом ограничивают осуществление такой работы. Поэтому
предлагаемые указания могут носить только общий характер и
представлять собой лишь исходные пункты для последующей систе-
матизации- и рационализации работ, проводимых в полевых лабо-
раториях.
Схема 1. Испытание ОВ в полевых лабораториях.
Сначала нрн помощи индикаторных трубок необходимо уста-
новить присутствие летучих ОВ, поскольку если это испытание и
производилось, то не самим лаборантом, и полезно лично про-
вести наблюдение за поведением индикаторных трубок под дей-
ствием имеющегося в пробе ОВ. При отрицательном результате
такого испытания вещество извлекают из пробы просасыванием
воздуха и повторно испытывают в индикаторных трубках эту более
Таблица 26. Групповое разделение О В
№ группы Биохимический способ * Реакция с тимолфталеином Реакция Гутцейта
Группа 1: зарин, зоман, +
табун, V-газы Группа 2: серный и азо- — +
тистый иприты Группа 3: мышьяксодер- — — +
жащие ОВ Группа 4: прочие ОВ — — —
261
Зарин Зоман Табун Тетрам и
аналогичные
соединения
Группа 2
Азотистый иприт
Серный иприт
'i'
Дифенилхлорарсин
(кларк I)
нитрил i
Бром-
ацетон
Схема 2. Идентификация ОВ после разделения их на группы.
концентрированную пробу. Кроме того, при нагревании пробы во
время продувки воздухом из нее могли выделиться и менее лету-
чие ОВ.
Капли ОВ, если они имеются на материале пробы, подвергают
испытанию при помощи индикаторных бумаг. При положительном
результате часть пробы экстрагируют растворителем и в получен-
ном растворе для подтверждения предварительных данных прово-
дят обнаружение вещества специальным методом. Если же пред-
варительное определение дает отрицательный результат, то готовят
экстракты с различными растворителями и используют их для
дальнейших исследований.
В том случае, если с материала пробы были сняты капли
или кристаллы ОВ или путем экстракции этиловым спиртом был
получен раствор достаточно высокой концентрации, рекомендуется
провести качественный элементный анализ. Предположение о на-
263
личии того или иного ОВ, сделанное на основании результатов
элементного анализа, можно затем подтвердить проведением спе-
цифических реакций. Если же концентрация ОВ в пробе слишком
мала для проведения качественного элементного анализа, следует
попытаться идентифицировать ОВ при помощи отдельных харак-
терных реакций.
На схеме 1 приведена последовательность различных этапов
анализа ОВ в полевых лабораториях.
Последовательность индикации отдельных ОВ диктуется сте-
пенью их токсичности. Обычно начинают с индикации фосфорорга-
нических ОВ, затем обнаруживают иприты, мышьяксодержащие и
раздражающие вещества.
Следующие три важнейших метода индикации позволяют раз-
делить все отравляющие вещества иа четыре группы: биохимиче-
ское обнаружение ингибиторов холинэстеразы, реакция с тимол-
фталеином, реакция Гутцейта.
Из табл. 26 видно, какие ОВ попадают в каждую из четырех
групп.
После такого разделения в каждой группе идентифицируют от-
дельные ОВ, пользуясь схемой 2.
Разумеется, что, используя реакции обнаружения, описанные
в разделе 5, можно составить и другие подобные схемы. В табл. 27
сопоставлены реагенты и реакции для индикации основных ОВ.
Приводим список использованных в ней под номерами реакций и
реагентов.
1 Биохимическое обнаружение ингибиторов холинэстеразы, см. стр. 178.
2. Реакция' Шёнеманиа (амин/перекись), см. стр. 60, реакция I и стр. 69.
3. 4-(п-Нитробензил)-пиридин, см. стр. 62, 87, 114, 117.
4. Тимолфталеин, см. стр. 78.
5. Тиомочевина, аммиачный раствор соли никеля, см. стр. 76.
6. Ацетат меди (II), о-дианизидин, см. стр. 84.
7. Реагент Драгендорфа, см. стр. 85.
8. Этилат натрия, нитропруссид натрия, см. стр. 28.
9. Сероводород, см. стр. 146.
10. Реагент Илосвая, см. стр. 93.
11. Азотная кислота, КОН, см. стр 98.
12. Пиридин — барбитуровая кислота (для синильной кислоты предва-
рительная обработка хлорамином; для хлорпикрина—KCN), см. стр. 109.
13. Образование берлинской лазури (FeSO4+FeCls), см. стр. 101.
14 Анилиновая вода, см. стр. 111.
15. .и-Динитробеизол, КОН, см. стр. 116.
16. Ванилин, H2SO4, см. стр. 116.
17. Т-иосалициловая кислота, см. стр. 130.
9.9. СПЕЦИАЛЬНЫЕ ЗАДАЧИ ПОЛЕВОГО АНАЛИЗА ОВ
9.9.1. Анализ смесей О В
В табл. 28, составленной по литературным данным, приведен ряд
ставших известными тактических ОВ и их загущенных смесей. Но
в лабораторию, кроме того, могут попаеть на испытание и необыч-
ные смеси ОВ, образовавшиеся в результате последовательного
264
Таблица 27. Сводка реагентов для индикации ОВ
Знак (+) обозначает, что реакция яд^г .медленно или при нагревании; знак [+]—что реакция идет только
лосле'гидролитического отщеплении HCN
Отравляющее вещество Реагенты
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
Зоман + 4- 4-
Зарии Табун 4- + + 4* 4- 4- — — — —. — — — — .[+] [+] — — —
Тетрам и его аналоги + (+) + — — . — 4- 4-'
Серный иприт — — + 4- -1- — — 4- — — — — — — — — —
Сесквииприт — 4- 4- 4- +
Азотистый иприт — — 4- 4- +• —- 4-
Люизит 4- —• —- — 4- + — — —
Метилдихлора рсин — — 4- — —— — — 4- — — — — —• — —
Зтилдихлорарсин — — 4- —• —- —- — + — —• —• —
Дифенилцианарсин — — 4- —* —— — — — (+) — [+1 1+] —
Дифеиилхлорарсин — — + — — — (+) + — —
Адамсит — + — — —• (+) — —• —• ““ —
Галогенциаи — —“ 4- —— — —• — —• + —•
Синильная кислота + 4- — —
Фосген —— — 4- — —— —— — — — —• — — 4- —-
Дифосген — 4- 4- — — — — — — — — — 4- — —•
Фосгеноксим — —“ -1- —• —• —• — — —• + — —• —
Хлорацетофенон — — + — — — — — — — — — — 4- —
Бромбензилцианид + — — — — — — —• — + —— 4-
Хлорпикрин — —- — —• —“ — — — — —— + —— 1
Бромацетон — — 4- —— —- — — — — — — + —“ —— -Г —
Некоторые алкалоиды — *— —— — — + — — —• —
Некоторые соли тяже- +
лых металлов Фторацетат 4-
Таблица 28. Тактические ОВ, стабилизованные н загущенные смеси ОВ
№№ Состав Обозначение смеси Государство
1 90% табуна, 10% хлорбензола — Германия (до 1945 г.)
2 97% зарина, 3% ацетобутирата целлюлозы — США
3 50% ДФФ, 50% серного иприта — Англия
4 75—90% серного иприта, 10—25% СС14, хлор- или нитробензола — Германия (до 1945 г.)
5 66% серного иприта, 34% люизита —
6 — 70% серного иприта, ~25% полисульфидов,'5% серы Иприт по Левин- штейну
7 95% серного иприта, 5% дитиана Перегнанный иприт по Ле- винштейну
8 99% иприта Левинштейна, 1% гек- саметилентетрамина или броми- стого тетраметиламмония — США
9 92—93% серного иприта, 2—3% полиметилметакрилата, 5% пи- ридина, хинолина или пиколина — США
10 80% этилдихлорарсина, 20% ди- хлордиметилового эфира — Германия (до 1945 г.)
11 50% дифеннлцианарсина, 50% этил- карбазола — Германия (до 1945 г.)
12 Дифенилцианарсин, фенилдихлор- арсин — Германия (до 1945 г.)
13 50% синильной кислоты, 35% AsCl3, 15% SnCl4, 5% хлороформа 50% HCN, 50% AsCl3 Винсенит Франция
14 Англия
15 46% HCN, 54% AsCl3 Манганит Франция
16 50% HCN, 25% AsCl3, 25% хлоро- форма Англия
17 Хлорциан, AsCl3 57,5% фосгена, 42,5% хлорпикрина Виврит Франция
18 — Германия (до 1945 г.)
19 60% фосгена, 5% хлорпикрина, 35% SnCl4 — Англия
20 25% фосгена, 75% хлорпикрина 60% фосгена, 40% SnCl4 Смесь PG Англия
21 Коллоргит Франция
22 71% фосгена, 29% SnCl4 __ Франция
23 50% фосгена, 50% AsCl3 — Англия
24 62% фосгена, 34% дифосгена, 2% хлорметилового эфира хлоруголь- ной кислоты — Германия (до 1945 г.)
25' 87% фосгена, 13% хлора Франция
26 91% фосгена, 5% CS2, 4% хлори- рованных углеводородов — Италия
27 50% фосгена, 50% хлора Уайт стар Англия
28 80% бромацетона, 20% хлораце- тона Мартонит Франция
29 80% бромметилэтилкетона, 20% хлорметнлэтилкетона Гомомартонит Франция
26§
Продолжение
№№ Состав Обозначение смеси Государство
30 20% бромацетона, 20% хлорацето- фенона, 30% СС14, 30% тетра- лина — Франция
31 80% хлорпикрина, 20% SnCl4 Аквинит Смесь NC Франция Англия
32 65% хлорпикрина, 35% H2S Рвотный газ, грин стар Англия
33 30% хлорпикрина, 70% хлора Клоп Еллоу стар Германия (до 1945 г.) Англия
34 Хлорпикрин, дифосген Грюнкрейц 1 Германия (до 1945 г.)
35 75% диметилсульфата, 25% хлор- метилсульфата Рационит Франция
36 Хлорметилхлоркарбонат (смесь мо- иохлор- и дихлорметилового эфи- ров) Ци палит Франция
37 75% эфира иодуксусной кислоты, 25% этилового спирта — Англия
38 Ксилилбромид, ксилиленбромид Вещество Т Германия (до 1945 г.)
39 Ксилилбромид, ксилиленбромид, полухлорированный метиловый эфир муравьиной кислоты Вещество Т-зе- леное Германия (до 1945 г.)
40 Ксилилбромид, ксилиленбромид, бромацетон — Германия (до 1945 г.)
41 H2S, CS2 2-Ред стар Англия
применения ряда ОВ. Методика испытания в этом случае зави-
сит от имеющегося количества вещества пробы, типа смеси и,
конечно, от поставленной задачи. При значительном количестве
смеси, например если проба взята из трофейных боеприпасов,
можно поставить задачу определения состава смеси. Для этого
смесь подвергают фракционной вакуумной разгонке, что воз-
можно лишь в условиях передвижных и стационарных полевых ла-
бораторий. Разведчик может определить только качественный со-
став смеси с помощью селективных методов индикации, используя
для этого различные индикаторные наборы или носимые полевые
лаборатории. При каких-либо затруднениях проба передается в
следующую по инстанции полевую аналитическую группу. Там
после разделения смеси разгонкой или при малом количестве про-
бы— хроматографическом разделении, устанавливают состав смеси.
Основные сведения о типе смеси дает качествнный элементный ана-
лиз. При наличии серы, мышьяка и хлора можно предположить, что
смесь состоит из серного иприта и люизита, а при наличии серы,
фосфора, хлора и фтора — из серного иприта и фосфорфтороргани-
ческих ОВ.
8
267
9.9.2. Анализ загущенных Об
Идентификация загущенных ОВ обычными способами весьма за-
труднительна, так как индикаторные бумаги очень медленно их
впитывают, а показания индикаторных трубок из-за пониженного
давления пара замедлены. Для анализа ОВ этого типа следует
отделить загуститель, так как, во-первых, его присутствие мешает
индикации, а во-вторых, представляет интерес также тип приме-
ненного полимера. Последнее, например, необходимо хотя бы для
того, чтобы знать, какое средство будет эффективно для дегазации
даного ОВ. Отделить загуститель можно относительно просто —
подбором растворителя или смеси растворителей, в которых ОВ
растворяется, а загуститель не растворяется.
Если нет никаких данных о загустителе, то растворитель под-
бирают экспериментально, проведением ряда опытов. Пробу делят
на несколько частей, помещают в химические стаканы и приливают
различные растворители, такие, например, как этиловый спирт,
ацетон, петролейный эфир, бензол, дихлорэтан, и устанавливают,
в каком из них загуститель растворяется, а в каком нет. Далее,
чтобы отделить загуститель, пробу обрабатывают смесью из 2—3 ч.
растворителя, не растворяющего загуститель, и 1 ч. растворяю-
щего. Загуститель, оставшийся в стакане, после декантации рас-
твора ОВ многократно промывают не растворяющим его раствори-
телем. В полученном растворе обнаруживают ОВ обычными спо-
собами. Для идентификации загустителя используют наблюдения,
полученные при изучении его растворимости, поведения при пиро-
литическом разложении и при пробе на пламя.
9.9.3. Исследование проб пищевых продуктов
Разрешение на использование пищевых продуктов дается только
после того, как наряду с химическим анализом на присутствие
ОВ и диверсионных^ядов будут проведены также радиохимические
и бактериологические испытания. При химическом исследовании
особое внимание надо уделять обнаружению алкалоидов и токсич-
ных катионов и анионов, отравление которыми очень трудно уста-
новить по малозаметным внешним признакам.
9.9.4. Исследование проб воды
В идеальном случае анализ проб воды следует проводить сразу
после их отбора, так как некоторые ОВ, такие, как серный иприт
и фосфорфторорганические соединения, в значительной степени ги-
дролизуются и после продолжительного контакта с водой не могут
быть определены количественно. Более достоверные результаты мо-
гут быть получены при анализе даже после продолжительного
хранения проб воды, содержащих трудногидролизуемые ОВ, такие,
как азотистый иприт, V-газы, мышьяксодержащие вещества и
большая часть диверсионных ядов.
268
Количественный анализ воды, требующийся при контроле ра-
боты установок для очистки воды, в случае быстро гидролизую-
щихся ОВ имеет смысл проводить только непосредственно после
отбора пробы.
Наряду с индикацией ОВ и диверсионных ядов при испытании
проб питьевой воды не следует пренебрегать общими химическими
наблюдениями. Так, если pH воды меньше 6, расход КМпО< пре-
вышает 12 мг/л и содержание хлоридов более 100 мг/л, то это
позволяет предполагать возможное заражение воды и присутствие
в ней продуктов гидролиза ОВ.
Максимально допустимые концентрации ОВ и диверсионных
ядов в питьевой воде, которые могут в ней содержаться до ее хло-
рирования, были опубликованы в США. Эти цифры подвергаются
обсуждению в армейских кругах и организациях гражданской обо-
роны различных западных стран. Сводка этих величин- приведена
в табл. 29.
Таблица 29. Максимально допустимое содержание ОВ в воде
71 72
до ее очистки *’
Отравляющее вещество или яд Допусти- мая концен-- трация, мг/л При потреб- лении около 3 Л/суТКЦ спустя Отравляющее вещество или яд Допусти- мая концен- трация, мг/л При потреб- лении около 3 л/сутки спустя
Серный иприт 1 1 неделю Зоман 0,5 3 суток
Азотистый иприт 5 1 неделю Мышьяк 5 —
Люизит 2 1 неделю Свинец 0,1 —
Синильная кислота 25 1 неделю Ртуть 5
Табун 2-3 3 суток Сурьма 5 —
Зарин 0,5 3 суток Фтор 1,5 —
9.9.5. Контроль полноты дегазации
Методы контроля полноты дегазации имеют очень большое значе-
ние. Контроль после дегазации техники, обмундирования, снаря-
жения и др. дает возможность командиру в зависимости от полу-
ченных результатов отдать распоряжение о необходимости про-
должения пребывания в защитной одежде при работе с техникой
или о возможности снятия противогаза. Кроме того, методы кон-
троля дегазации необходимы для определения эффективности раз-
рабатывающихся способов дегазации.
При введении новых способов, дегазации их так’отрабатывают
в лабораторных и войсковых испытаниях, чтобы в дальнейшем при
точном соблюдении режима дегазации можно было достичь допу-
стимой (и даже более низкой) концентрации ОВ, в результате
чего Контроль дегазации сможет быть ограничен взятием отдель-
ных выборочных проб.
Допустимые нормы остаточного заражения выражают в мил-
лиграммах ОВ на 1 м2 поверхности или, для концентрации ОВ в
атмосфере над зараженной поверхностью, — в миллиграммах ОВ
269
на 1 м3 воздуха; они представляют такую зараженность, которая
при соблюдении определенных мер защиты не представляет ника-
кой опасности для человеческого организма.
Установление таких норм обосновано тем, что в большинстве
случаев абсолютная дегазация в полевых условиях связана с не-
оправданной тратой дегазирующих средств и времени и, в конце
концов, даже и не требуется.
По аналогии с методами работы исследователей в области про-
мышленной токсикологии и фармакологии при установлении допу-
стимых норм остаточного 'заражения должны быть проведены
контрольные опыты на животных.
В случае кожно-нарывных ОВ и веществ общеядовитого дей-
ствия, резорбирующихся через кожу, подопытных животных при-
ходилось подвергать контакту с испытуемым материалом, напри-
мер материалом обмундирования; в случае ОВ, обладающих
ингаляционным действием, подопытные животные и испытуемые
материалы помещались в закрытую камеру, в которой контакт
между ними был исключен и животные только вдыхали ОВ, испа-
ряющееся из материала пробы. Само собой разумеется, что для
контроля полноты дегазации в войсковых подразделениях опыты
на животных мало пригодны; для этой цели применяются химиче-
ские и биохимические методы.
Из самого определения понятия «допустимые нормы остаточ-
ного заражения» следует, что применение качественного анализа
имеет смысл лишь тогда, когда по чувствительности реакции обна-
ружения подобраны таким образом, что с их помощью можно обна-
руживать плотности заражения, превышающие допустимые нор-
мы. В случае отрицательного результата можно считать, что
принятые меры дегазации дали удовлетворительный результат. Од-
нако практически не всегда удается подобрать такие реакции, и
приходится прибегать к количественным определениям. К прибо-
рам, применяемым для этих целей, предъявляются опять те же
повышенные требования в смысле чувствительности реакции. Если
нельзя количественно определить содержание вещества на поверх-
ности и внутри материала, то при исследовании эффективности
дегазации прибегают к опытам на животных и, кроме того, парал-
лельно проводят химический анализ проб после их обогащения.
Другой способ оценки эффективности дегазации по результа-
там ее контроля заключается в установлении пересчетных коэф-
фициентов, дающих возможность по результатам химического ана-
лиза оценить фактическую плотность остаточного заражения.
Приводимые ниже описания важнейших методов полевого и
лабораторного контроля полноты дегазации должны дополнить
все высказанные выше предварительные соображения. Наиболь-
шие трудности представляет отбор проб. Что же касается методов
определения, то в каждом отдельном случае следует выбирать наи-
более чувствительный метод, например для фосфорорганических
ОВ — биохимический. Отметим, что остатки дегазирующих веществ
270
мешают индикации, и перед проведением анализа их необходимо
разрушить.
Контроль полноты дегазации применяется в двух случаях —
при дегазации военной техники и вооружения и при дегазации об-
мундирования.
При контроле дегазации поверхностей отбор проб преимуще-
ственно осуществляется мазком. Измеренный шаблоном участок
поверхности -протирают одним или несколькими тампонами из
ваты или целлюлозы, пропитанными растворителями. В лабора-
тории тампоны обрабатывают растворителем и в полученном экс-
тракте определяют ОВ. При таком методе отбора проб с неокра-
шенных металлических поверхностей ОВ полностью переходит в
пробу. При отборе проб с металлических окрашенных поверхно-
стей эффективность взятия пробы мазком зависит от вида лако-
красочного покрытия, типа ОВ и продолжительности воздействия,
типа и количества растворителя и продолжительности экстракции,
а также от растворимости ОВ в соответствующем растворителе,
температуры и других факторов. От этих же факторов зависит
эффективность отбора пробы с окрашенных и неокрашенных дере-
вянных и всяких других поверхностей, впитывающих ОВ.
При применении в качестве растворителя воды зарин и V-газы
извлекаются только с поверхности лакокрасочного покрытия. Впи-
тавшееся в покрытие ОВ можно извлечь только после нескольких
минут обработки органическим растворителем. Таким образом, сте-
пень извлечения ОВ из лакокрасочного покрытия нужно устанав-
ливать опытным путем для каждого типа покрытия, соблюдая при
этом одинаковые условия. В этих опытах определяют соотношение
количества нанесенного на поверхность ОВ и затем обнаруженного
после извлечения. При этом, применительно к количеству и вре-
мени воздействия растворителя, выбирают такую методику работы,
которая обеспечивает степень извлечения ОВ, не зависящую от
продолжительности его воздействия. Выполнение этого требова-
ния имеет определенные границы, зависящие от скорости проника-
ния ОВ и растворителя в глубину лакокрасочного слоя и от тол-
щины слоя покрытия.
Другая возможность отбора проб для контроля дегазации за-
ключается в сдирании лакокрасочного покрытия с определенного
участка поверхности или в снятии стружки с деревянной неокра-
шенной поверхности на необходимую глубину и в обработке полу-
ченного материала растворителем. При таком методе отбора пробы
из материала извлекается все имеющееся в нем ОВ.
Важным методом контроля дегазации в случае применения ОВ
ингаляционного действия является определение их содержания в
слое воздуха над дегазированной поверхностью. Для этого нужно
поместить на соответствующую поверхность колоколообразный со-
суд и через некоторое время отсасывать из-под него воздух, про-
пуская его затем через трубки, заполненные силикагелем, индика-
торные трубки или поглотительную склянку с поглотителем или
271
соответствующим реагентом. Для получения сопоставимых резуль-
татов нужно, чтобы продолжительность испарения вещества, объем
пропускаемого через поглотители воздуха и температура испыта-
ния были постоянными. По полученным данным делают выводы
о том, требуется ли ношение противогаза при обращении с дегази-
рованной техникой.
При контроле дегазации обмундирования в полевых лаборато-
риях отбор проб производится экстракцией ОВ растворителями или
вытеснением его воздухом. Обозначив места наиболее заметного
заражения, их дегазируют, после чего эти участки ткани вырезают
и обрабатывают растворителем. Суждение об эффективности дега-
зации может быть высказано по результатам анализа выборочных
проб.
При контроле дегазации прорёзииенных тканей очень трудно
подобрать такой растворитель, который, не растворяя резины, из-
влекал бы все ОВ из материала. В случае текстильных тканей в
раствор вместе с ОВ могут перейти импрегнирующие средства, что
может помешать последующему определению ОВ.
Эти помехи исключаются в способе, использующем воздух для
извлечения ОВ. Степень извлечения вещества при воздушной экс-
тракции значительно меньше, что часто обусловлено весьма устой-
чивой сорбцией ОВ материалом обмундирования. Поэтому для извле-
чения малолетучих ОВ, таких, как серный иприт, зом.ан или V-газы,
требуется просасывать воздух в течение продолжительного времени.
При воздушной экстракции подлежащие испытанию предметы
обмундирования обрабатывают в закрытой камере теплым возду-
хом, который по прошествии определенного времени отсасывают,
пропуская его затем через силикагелевые или индикаторные труб-
ки или через сосуд с поглотителем. В качестве такой камеры мож-
но использовать мешок из синтетического материала, большой
ящик или просто камеру автодегазационной станции. Для полу-
чения воспроизводимых результатов нужно всегда проводить испы-
тания при определенной температуре, одинаковом объеме просасьв
ваемого воздуха, продолжительности испарения, а также одинако-
вом числе подвергаемых испытанию предметов обмундирования.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. По каким внешним признакам могут быть обнаружены отравляющие
вещества на местности?
2. Какими простыми средствами располагает разведчик для индикации бое-
вых отравляющих веществ?
3. Какие существуют типы индикаторных порошков?
4. Каковы преимущества и недостатки индикаторных порошков для раз-
ведки на местности?
5. Каковы преимущества и недостатки индикаторных бумаг для разведки
на местности?
6. Какие известны типы индикаторных бумаг для обнаружения отравляю-
щих веществ?
7. Как различаются индикаторные трубки по виду их применения?
8. Каким путем можно количественио оценить показания индикаторных
трубок?
272
9. Как подразделяют индикаторные трубки на основе зависимости их пока-
заний отскорости просасываемого через них воздуха и какое это имеет значение?
10. Какие требования предъявляются к автоматическим приборам индика-
ции боевых отравляющих веществ?
11. Какие принципы лежат в основе действия автоматических индикаторных
приборов?
12. Какая химическая реакция использована в автоматическом газоанализа-
торе для индикации фосфорорганических ОВ типа зарин?
13. Из чего должен состоять комплект для контроля очистки питьевой воды?
14. Какие испытания могут быть осуществлены в полевой автолаборатории?
15. На что следует обращать внимание при отборе проб?
16. Как отбираются пробы почвы?
17. Как отбираются пробы воды? Какое значение имеет растворимость ОВ
при отборе проб воды?
18. Как отбираются пробы с поверхностей военной техники и предметов об-
мундирования?
19. Какие существуют способы повысить содержание ОВ в пробах воздуха?
20. Какими свойствами должен обладать растворитель, используемый для
поглощения ОВ?
21. Какие растворители пригодны для поглощения фосфорорганических ОВ?
22. Что представляют собой мембранные фильтры и для чего они приме-
няются?
23. Какие данные должны быть приведены в донесениях, сопровождающих
пробы почвы н пробы воды?
24. Каково назначение подготовки проб к анализу?
25. Какие соображения -должны учитываться при выборе применяемых для
экстракции растворителей?
26. Когда применяется способ воздушной экстракции?
27. На что следует обращать внимание при экстракции ОВ из проб растений?
28. Как осуществляется концентрирование ОВ в пробах воды при малой их
концентрации?
29. Поясните схему этапов исследования ОВ в полевой лаборатории.
30. Какими способами можно установить присутствие определенных групп
ОВ?
31. Какие методы индикации пригодны для идентификации мышьяксодержа-
щих ОВ?
32. Каким способом отделяют загуститель при исследовании загущенных ОВ?
33. Что такое допустимые нормы остаточной зараженности и как их опре-
деляют?
34. Как определить остаточную плотность заражения на окрашенных по-
верхностях и на что при этом следует обращать внимание?
ЛИТЕРАТУРА
1. Muntsch, Gasschutz und Luftschutz, 1932, 273.
2. N i e 1 s e n A., Gasschutz und Luftschutz, 1935, 48, 214.
3. T h e m m e F., Gasschutz und Luftschutz, 1934, 321.
4. H 1 e b e г E„ Gasschutz und Luftschutz, 1938, 96, 135, 169.
5. Нем. пат. 722293 (1942).
6. L i g t e n b e г g H. C„ Chem. Weekbl., 34, 321 (1937).
7. Нем. пат. 710532 (1941).
8. Пат. ФРГ 1198590 (1963).
9. Witten В., Prost ak A., Anal. Chem., 29, 885 (1957).
10. Пат. США 2926072 (1960).
11. Пат. США 2867509 (1959).
12. S и сh I е г A., Z. anal. Chem., 79, 183 (f929).
13. Дубинин М. М„ ЖПХ, 4, 1109 (1931).
14. L i d d е 1 Н. F., Analyst, 82, 375 (1957);
15. Кретов А. Е„ ЖПХ, 2, 483 (1929).
16. TnommannJ., Schweiz. Apoth. Ztg., 75, 41 (1937).
17- D i х о п В. Е., Н a n d s G. C., Analyst, 84, 463 (1959).
273
18. Methods for the detection of toxic substances in air. Phosgene, № 8. London,
Ministry of Labour, 1961.
19. P er t u s i C„ G a s t a 1 d I E., Chem. Ztg., 37, 609 (1913).
20. Sievert A., Hermsdorf A., Z. angew. Chem., 34, 3 (1921).
21. Пат. США 2534229 (1950).
22. Katz S. H., Longfellow E. S., J. Ind. Hyg., 5, 97 (1923/1924).
23. Sherrard G. C., U. S. Publ. Health Service, 1928, p. 1224.
24. Pieters H. A. J., Safety in the Chemical Laboratorys. London, 1951, 165.
25. G e 111 e r A. O., G о 1 d b a u m L., Anal. Chem., 19, 270 (1947).
26. Dixon В. E., Hands G. C., Bartlett A. F., Analyst, 83, 199 (1958).
27. Пат. США 2855289 (1958).
28. G u i g n a r d, Compt. rend., 142, 552 (1906).
29. S tey n D„ J. S.-Afric. Vet. Med. Assoc., 10, 65 (1939).
30. Decker t W., Hyg. Infect.-Krankh., 109, 485 (1929).
31. Spica, Gazz. chim. ital., 49, 299 (1919).
32. В i e d e b a c h, Siiddeutsch. Apoth. Ztg., 1949, 17.
33. Пат. ФРГ 710533 (1949).
34. Нем. пат. 710531 (1941).
35. С г u i к s h а п к A. J. et al., Anal. Chem., 19, 849 (1947).
36. Y о e J. H., С о g b i 11 E. C., Microchem. verein. Microchim. Acta, 38, 492 (1951).
37. Moureu H., Chovin P. et al., Arch. Malad. Prof., 11, 445 (1950).
38. Cut a F., Chem. Listy, 34, 216 (1940).
39. Winkler L. W„ Z. anal. Chem., 97, 18 (1934).
40. G г о s s к о p f K-, Chem.-Ztg., 87, 270 (1963).
41. Grosskopf K, Chem.-Ztg., 83, 115 (1959).
42. Fenton P. F„ J. Chem. Educ., 21, 488 (1944).
43. D i j ks t r a D. W., Chem. Weekbl., 34, 351 (1937).
44. H о о g e v e n A. P. J., Chem. Ind., 1940, 550.
45. H a g 1 u n d H., Sillen L. G., Svensk. Kem. Tidskr., 69, 440 (1957)
46. Stampe G., S ch rot er G. A., Gasschutz und Luftschutz,- 1934, 16.
47. S c h г о t e r G. A„ Angew. Chem., 49, 164 (1936).
48. Drager-Hefte, 186, 3297 (1936).
49. F e n t о n P. F., J. Chem. Educ., 20, 564 (1943).
50. Z a i s A. M., J. Chem. Educ., 21, 489 (1944).
51. Kitagawa T., C A., 1950, 1851c; 7718c; 1951, 4874g, 4974c, 5183e, 7832e;
1952 1920c
52. Grosskopf K-, Angew. Chem., 63, 306 (1954).
53. Нем. пат. 691442 (1936).
54. Нем. пат. 712827 (1941); 713378 (1941); 713658 (1941).
55. Нем. пат. 749526 (1941).
56. Пат. ФРГ 903041 (1954).
57. Франц, пат. 1273205 (1962).
58. Пат. ФРГ 1183279 (1964).
59. В га d ley Т. F., Chem. Eng. News, 20, 893 (1942)
60. Нем. пат. 708669 (1941).
61. Нем. пат. 692375 (1940).
62. Пат. ФРГ 865382 (1953).
63. Пат. ФРГ 1077457 (1958).
64. Y о u п g J. С. et al„ Anal. Chem., 30, 1236 (1958).
65. Cherry Е. Н. et al., Anal. Chem., 30, 1239 (1958).
66. G у e - J a q u о t, L’Armee, 49, 38 (1965).
67. Klotz J. M., Dole M., Ind. Eng. Chem., Anal. Ed., 18, 741 (1946).
68. Франц, пат. 1192780 (1959).
69. Koblin A., Epstein J., Armed Forces Chem. J., 3 (Sept./Okt.), 24 (1957).
70. N e a 1 e E., P e г г у D. J., Analyst, 84, 226 (1959).
71. Reber H„ Vol kart W., Vierteljaresschrift fiir Schweizer Sanitatsoffiziere,
1961, S. 16.
72. Mutschin A„ Zivilschutz, 27, 164 (1963).
73. Lohs K-, Nachweisgerate fiir giftige Gase, Dampfe u. Staube, Berlin, 1962.
S. 124.
ДЕГАЗАЦИЯ
И СРЕДСТВА
ДЕГАЗАЦИИ
10. РОЛЬ ДЕГАЗАЦИИ В СИСТЕМЕ МЕРОПРИЯТИЙ
ЗАЩИТЫ ОТ ХИМИЧЕСКОГО ОРУЖИЯ
10.1. ПРИЧИНЫ НЕОБХОДИМОСТИ ДЕГАЗАЦИИ
На основании изложенного в т. 1 настоящей книги можно с уве-
ренностью утверждать, что некоторые империалистические дер-
жавы стремятся создавать все более эффективные боевые химиче-
ские вещества и увеличивать их производство. В связи с этим для
обеспечения защиты населения и армии важнейшей задачей всех
миролюбивых государств является дальнейшее развитие средств
и способов дегазации боевых химических веществ. Это особенно
важно потому, что стойкость ОВ постоянно возрастает (см. табл. 30).
В результате химического нападения сможет произойти долговре-
менное заражение огромных территорий и самых различных
объектов.
Таблица 30. Стойкость некоторых ОВ
Отравляющее вещество' Дата получения применения ИЛИ Стойкость на местности при нормальных условиях
Фосген Впервые применен в 1915 г. Нестоек
Синильная кислота Впервые применена в 1916 г. Нестойкая
Сериый иприт Впервые применен ь 1917 г. Около 20 «
Азотистый иприт Впервые получен в 1935 г. Около 30 ч
Зарин Впервые получен в 1939 г. Около 4 ч
Зоман Впервые получен в 1944 г. Около 20 ч
V-Газы Впервые получены в 1955 г. Около 5 суток
Высокая точсичность ОВ и их способность проникать через
кожу и различные материалы вынуждают людей, находящихся вне
коллективных убежищ, надевать средства защиты органов дыха-
ния и кожи, снимать которые можно только после выхода из зоны
заражения и полной дегазации техники.
Таким образом, дегазация играет существенную роль в вос-
становлении боеспособности войск, подвергшихся химическому на-
падению. Наряду с дегазацией боевой техники важное значение
имеет и дегазация обмундирования, местности, воды и находяще-
гося в упаковке продовольствия.
Многообразие упомянутых задач вынуждает иметь специальные
дегазационные средства и приборы, отработанные способы дега-
зации и хорошо обученный персонал.
277
10.2. ОПРЕДЕЛЕНИЕ ПОНЯТИЯ «ДЕГАЗАЦИЯ»
Удаление в минимальные сроки максимально возможного количе-
ства боевых отравляющих веществ или ядов с поверхностей раз-
личных объектов, из воды и воздуха, с обмундирования и других
предметов и сред (покрытые пленкой лака поверхности, дерево,
почва и др.) называется дегазацией. Для этого применяются раз-
личные способы, в которых используются многочисленные сред-
ства, приборы и приемы, предназначенные для дегазаций того или
иного объекта (см. табл. 31, стр. 281).
В основе того или иного способа дегазации лежит определен-
ный химический или физический процесс. В основе химической
дегазации лежит процесс, в результате которого ОВ превращается
в нетоксичные или малотоксичные соединения. Физическая дега-
зация заключается в удалении ОВ (без изменения его состава)
с какой-либо поверхности или из какой-либо среды, что и устра-
няет опасность поражения.
Большая часть способов дегазации представляет собой опреде-
ленное сочетание химических и физических процессов, что в общем
случае позволяет рассматривать дегазацию как комбинированный
процесс.
Следует также учитывать и естественную дегазацию, которая
всегда протекает наряду с указанными способами дегазации. Есте-
ственная дегазация протекает самопроизвольно, независимо от
вмешательства человека, и представляет собой также сочетание
химических и физических процессов. К физическим процессам есте-
ственной дегазации относятся: испарение, разбавление или меха-
ническое удаление боевых ОВ, в результате которых происходит
их местное обезвреживание. При этом возможны такие случаи,
когда устранение опасности поражения в одном месте создает или
усиливает ее в другом. Естественная дегазация называется также
самодегазацией.
Кроме классификации приемов и способов дегазации по тому
или иному процессу, существует и тактическая классификация.
В соответствии с определенными сроками возможных мероприя-
тий, связанными с действиями войск для ликвидации последствий
химического нападения, производится частичная или полная дега-
зация. Частичная дегазация предназначена для возможно более
быстрого удаления боевых ОВ со всех поверхностей, с которыми
в последующих боевых действиях личный состав подразделений
должен обязательно соприкасаться. В результате частичной дега-
зации в основном предотвращается проникновение ОВ через ма-
териал средств защиты, снимать которые после частичной дегаза-
ции нельзя, так как в воздухе еще могут содержаться пары ОВ;
кроме того, не исключен контакт с еще зараженными поверхно-
стями снаряжения и вооружения. В зависимости от условий, сро-
ков и применяемых средств частичная дегазация постепенно за-
вершается полной. Противогаз можно снимать только после про-
ведения полной дегазации. Остальные средства защиты можно
278
снимать по мере того, как дегазация будет приближаться к пол-
ной. Эффективность дегазации можно оценить по тому, насколько
устранена опасность поражения как активной, так и естественной
дегазацией. Необходимые меры предосторожности и защиты об-
условливаются достигнутой степенью дегазации (полной или ча-
стичной).
Из изложенного выше вытекает, что дегазацией можно назвать
все процессы, которые служат цели удаления боевых ОВ или ядов
с поверхности различных объектов или из любой среды.
Исходя из этого определения, становится ясным, что к дегаза-
ции можно относить не только специальные методы обработки
после химического нападения (частичная или полная дегазация);
понимать это следует более широко. Так, например, в фильтре
противогаза очистка воздуха происходит за счет адсорбции — это
тоже является дегазацией.
Для проведения дегазации нужны такие средства, которые, по-
глощая или вступая во взаимодействие с ОВ, одновременно с уда-
лением их с дегазируемого объекта могут превращать их в нето-
ксичные соединения.
Дегазирующими веществами являются . все соединения, способ-
ные удалять ОВ или яды с поверхности различных объектов или
из той или иной среды.
К средствам дегазации относятся: дегазирующие вещества
(твердые, жидкие или газообразные), растворители, растворы де-
газирующих средств в растворителях, моющие средства, моющие
растворы (моющие вещества и вода), адсорбционные средства.
По-видимому, нецелесообразно относить к дегазирующим сред-
ствам только те вещества, которые вызывают химическое превра-
щение ОВ. К дегазирующим следует отнести также и те вещества,
с помощью которых дегазация ОВ осуществляется в результате
физических процессов, например растворители, вызывающие рас-
творение дегазирующих веществ. Поэтому принятое выражение
«дегазирующие средства и растворители» следовало бы изменить
на более правильное «дегазирующие вещества и растворители».
Вместе с тем дегазирующие вещества необходимо отличать от
антидотов. Антидотами называют вещества, которые применяются
для противодействия ядам в организме человека.
Дегазирующие вещества, применяемые для обработки открытых
кожных покровов человека, занимают промежуточное положение.
Целью применения этих веществ является не только дегазация ОВ
на поверхности кожных покровов, но и активное воздействие на
токсичное вещество, успевшее проникнуть в верхние слои кожных
покровов (т. е. внутрь организма).
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Дайте определение понятий «способ дегазации» и «принцип дегазации».
2. Как происходит естественная дегазация?
3. Что такое «дегазация»?
4. Какие вещества относят к средствам дегазации?
11. КОМБИНИРОВАННАЯ ДЕГАЗАЦИЯ
При подробном рассмотрении многообразия способов дегазации
становится очевидным, что в большинстве случаев невозможно
провести границу между способами, основанными на чисто хими-
ческих или чисто физических процессах. Только в том случае, когда
речь идет о специфичности действия того или иного дегазирующего
средства, можно допустить известное разграничение между хими-
ческими и физическими процессами дегазации.
В последующем при обсуждении и описании дегазирующих
средств такое разграничение будет использовано, поскольку одни
средства вызывают химическое преобразование, а другие дегази-
руют на основе физических процессов. Специфическое действие
того или иного дегазирующего средства можно рассматривать по-
этому только в тесной связи с их характерными свойствами и
свойствами ОВ. При этом нельзя упускать из виду, что большин-
ство способов дегазации основано на комбинированных процессах.
В табл. 31 на ряде'примеров показаны взаимосвязи между от-
дельными способами, средствами и процессами дегазации.
Из таблицы видно, что химическая дегазация никогда не пред-
ставляет собой индивидуальный процесс. Причина этого заклю-
чается в том, что дегазацию веществами, обусловливающими хими-
ческое превращение ОВ в нетоксичные продукты, почти всегда про-
водят в среде, которая обеспечила бы их контакт с ОВ. Эта среда
наряду со способностью смешиваться с дегазирующими веществами
должна также растворять ОВ, что практически уже удовлетворяет
одному из условий физической дегазации. Таким образом, раство-
рители выполняют двойную роль. Исходя из соображений систе-
матизации, мы помещаем их рассмотрение в раздел 13.2, где трак-
туются вопросы физической дегазации.
При различных способах дегазацци особое значение имеют ве-
щества, образующие с ОВ соли. Так, азотистый иприт образует
с бисульфатом натрия легко растворимую в воде соль. Аналогич-
ные реакции идут и с минеральными кислотами. Вместе с тем они
возможны только для ОВ, обладающих основными свойствами,
т. е кроме азотистого иприта подобные реакции могут идти еще
только с веществами типа V-газов.
При дегазации веществами, приводящими к образованию со-
лей соответствующих органических оснований, токсичность этих
ОВ не утрачивается. Но так как соли не способны проникать че-
28Q
Таблица 31. Примеры дегазации различных объектов
Дегазируемый объект . Способ дегазации Средство дегазации Вид дегазации
Поверхности техники Обработка дега- зирующей жид- костью Раствор дегазирую- щего вещества в органическом раст ворителе или воде Комбинированная
Поверхности техники Обработка раство- рителем или моющим сред- ством Растворитель или раствор моющего средства в воде Физическая
Поверхности техники Обработка горя- чим воздухом Горячий воздух Физическая
Поверхности техники Обработка водя- ным паром ' Водяиой пар Комбинированная
Местность Обработка дега- зирующей жид- костью Раствор дегазирую- щего вещества в . органическом раст- ворителе или воде Комбинированная
Местность Рассылание дега- зирующего ве- щества Твердое дегазирую- щее вещество Преимущественно химическая
Местность Покрытие ней- тральным мате- риалом — Физическая
Обмундирование Дегазация кипя- чением Дегазирующая жид- кость Комбинированная
Обмундирование Обработка паром Водяиой пар с газо- образным дегази- рующим веществом Комбинированная
Обмундирование Дегазация эк- стракцией Растворитель с опре деленным количест- вом дегазирующего вещества Комбинированная
Вода Обработка дегази- рующими ве- ществами Твердые, жидкие (или парообраз- ные) дегазирую- щие средства Химическая
Вода Обработка адсор- бентами (филь- трация) Адсорбенты Физическая
Воздух Распыление дега- зирующей жид- кости Дегазирующая жид- кость Комбинированная
Воздух Распыление ' ад- сорбентов Адсорбенты Физическая
Воздух Просасывание че- рез фильтр,- со- держащий адсор бейт и химичес кий поглотитель Адсорбент и дегази- рующее вещество Комбинированная
рез материал защитной одежды или через кожные покровы чело-
века и легко смываются с поверхности объектов просто водой, то
при известных условиях этот способ дегазации может быть до-
статочно эффективным. Образование соли является химическим
процессом, а смывание соли — физическим, поэтому такой способ
дегазации следует считать комбинированным. Особенность состоит
в том, что опасность поражения, обусловленная действием ОВ, в
результате химической реакции устраняется не полностью, только
физический процесс полностью освобождает зараженные поверх-
ности объектов от присутствия ОВ.
Некоторые другие взаимосвязи между физическими процессами
и химической дегазацией приведены ниже, в разделах 12.1.3 и
12.1.4.
В связи с тем, что естественная дегазация протекает незави-
симо от способов дегазации и дегазирующих средств, мы не бу-
дем в дальнейшем останавливаться на этом вопросе.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Дайте определение термину «комбинированная дегазация»,
2. Какую роль в дегазации играют солеобразователи?
3. Какое значение для дегазации имеют растворители?
12. ХИМИЧЕСКАЯ ДЕГАЗАЦИЯ
12.1. ОБЩИЕ ПОЛОЖЕНИЯ
В химических процессах дегазации ОВ не только удаляется с за-
раженной поверхности или из зараженной среды, но за счет хими-
ческой реакции утрачивает и свою токсичность. В этом основное
преимущество химической дегазации. Кроме того, некоторые дега-
зирующие вещества производятся химической промышленностью
в большом масштабе и сравнительно недороги.
Вместе с тем можно привести и ряд недостатков, присущих
дегазирующим веществам. К ним относятся:
плохая устойчивость при хранении (пример — хлорная известь);
сильное корродирующее действие при хранении (пример — хло-
ристый сульфурил);
сильное корродирующее действие на дегазационные приборы
и дегазируемые объекты (пример — гипохлориты);
большой расход растворителей для водонерастворимых дега-
зирующих веществ (пример — дихлорамин);
отсутствие универсальности действия по отношению ко всем
ОВ (пример — хлорамины);
непригодность водных дегазирующих жидкостей для дегазации
ОВ, проникших в слой лакокрасочного покрытия поверхностей
(пример — растворы гипохлорита кальция);
более слабое действие при низких температурах (пример —
растворы монохлорамина);
повреждение лаковых покрытий и некоторых деталей боевой
техники (пример—дегазирующие растворы, содержащие дихлор-
этан);
сравнительно высокая токсичность (дихлорэтан), раздражаю-
щее действие на органы дыхания (хлористый сульфурил) или кожу
(едкие щелочи).
Указанные недостатки характерны в большей или меньшей сте-
пени для всех дегазирующих веществ. Идеальное дегазирующее
вещество, которое не имело бы ни одного из перечисленных недо-
статков, а обладало бы всеми перечисленными выше преимуще-
ствами, пока не найдено. Стремление найти такое вещество будет,
очевидно, продолжаться до тех пор, пока будут существовать ОВ
и будет необходимость в их обезвреживании.
В последующем наложении будут разбираться основные проб-
лемы, которые стоят на пути изыскания такого идеального дегази-
рующего вещества и решение которых связано, помимо всего про-
чего, с учетом следующих общих положений.
283
Современные ОВ представляю^ собой исключительйо органи-
ческие соединения*; при низких температурах они реагируют, как
правило, очень медленно.
Некоторые ОВ (иприт и зоман) плохо растворимы в воде. По-
этому скорость реакций таких веществ с водными растворами де-
газаторов зависит от величины граничащих поверхностей реаги-
рующих фаз и от того, с какой скоростью они растворяются в рас-
творителе дегазирующего раствора. По этим причинам ОВ лучше
всего дегазируются твердым или жидким дегазирующими веще-
ствами в среде органического растворителя.
Действие дегазирующего вещества в глубине материала может
проявляться только при применении растворов дегазирующих ве-
ществ в органических растворителях. Однако широкое использо-
вание таких средств связано с необходимостью перевозок боль-
ших объемов органических растворителей. Твердые неорганические
дегазирующие вещества лишены этого недостатка; для их приме-
нения требуется обычно только вода.
Применение водных растворов дегазирующих веществ особенно
проблематично зимой. Во-первых, химические процессы протекают
очень медленно, а во-вторых, в растворы необходимо добавлять
до 30% антифриза.
Чем большей реакционной способностью обладает дегазирую-
щее вещество, тем более вероятно, что его применение будет свя-
зано с вредным физиологическим действием на человека и что оно
агрессивно по отношению к обрабатываемым поверхностям или
предметам.
Некоторые ОВ сравнительно легко гидролизуются, другие —
более легко окисляются. Дегазирующие вещества, которые обла-
дали бы универсальным действием по отношению к разным типам
ОВ, встречаются весьма редко. Учитывая сложность проблемы,
можно рекомендовать следующие варианты возможного обеспечен
ния мероприятий по дегазации.
Вариант 1. Снабжение армии Двумя типами дегазирующих ве-
ществ, один из которых используется для дегазации сравнительно
легко окисляющихся ОВ (например, иприта), а другой — для дега-
зации легко гидролизующихся ОВ. Оба эти типа веществ должны
быть пригодными для применения как в зимних, так и в летних
условиях.
Вариант 2. Снабжение одним типом дегазирующих веществ.
Если речь идет о водорастворимом дегазирующем веществе, то
для использования его водных растворов в условиях низких темпе-
ратур необходимо дополнительно предусмотреть либо их подогрев,
либо специальные добавки для понижения температуры замерза-
ния растворов.
* Утверждение не совсем точное. К ОВ относятся также синильная кислота,
фосген, мышьяковистый водород и другие неорганические вещества. Дегазиро-
вать же на каких-либо поверхностях, действительно, нужно только ОВ органиче-
ского происхождения. — Прим, ред.
284
Если в качестве универсального дегазирующего вещества вы-
брано органическое соединение, то вследствие необходимости в
растворителе повысится стоимость дегазации; кроме того, потре-
буется транспорт для доставки больших количеств растворителя.
Все современные армии в прошлом в той или иной степени при-
держивались какого-нибудь одного из этих вариантов. Все же
предпочтение отдавалось универсальному дегазирующему сред-
ству. Однако это стремление не должно привести к недооценке
многих других дегазирующих средств, не обладающих свойствами
абсолютной универсальности. Следует подчеркнуть, что такие
средства при определенных обстоятельствах могут сыграть решаю-
щую роль. Чем основательнее будут изучены свойства и границы
применения этих неуниверсальных веществ, тем большего эффекта
можнб достигнуть при их применении и тем меньше они будут
уступать высокоактивным веществам.
Предпосылкой успешного решения проблем дегазации является
понимание того, что при этом нельзя ограничиваться «собиранием
рецептрв», а необходимы научно обоснованные решения. Для этого
требуется понимание механизма реакций органических соединений.
В пределах настоящей книги может быть обсужден лишь минимум
необходимых теоретических положений. Для основательного изу-
чения этих вопросов рекомендуется обратиться к специальной ли-
тературе *>4.
12.1.1. Некоторые вопросы теории процессов дегазации
Аналогично тому, как пионеры в области применения взрывчатых
веществ должны были сопоставлять количество взрывчатого веще-
ства и его бризантное действие со статической прочностью разру-
шаемого препятствия, так и военные химики должны сопоставлять
реакционную способность конкретного дегазирующего вещества с
прочностью разрываемой химической связи. Для этого нужно уметь
оценивать взаимоотношения атомов в молекулах ОВ и дегазирую-
щих веществ. Эти отношения характеризуются типом химических
связей и распределением электронов в молекулах.
Для неорганических'соединений в основном характерна ионная
(электровалентная) связь между атомами, возникающая в резуль-
тате передачи электронов от одного атома к другому. Природа
связи в органических соединениях иная, осуществляется она не
путем передачи, а путем обобщения электронов и носит название
ковалентной. Полярность связи указывает на характер распреде-
ления электронов между связанными атомами и на имеющийся на
атомах заряд. Это распределение никогда не бывает совершенно
симметричным. Всегда наблюдается небольшое смещение в сторойу
более электроотрицательного атома. Например:
R. 6-
n\e+ в- в+ -
R'—р=о! R—Н2С—С1:
р?/
285
Частичные заряды б+ и 6" выражают дефицит или избыток элек-
тронной плотности. Это различие в электронной плотности является
следствием большей электрофильности атомов кислорода и хлора
по сравнению с атомами фосфора или углерода. Какой из атомов
несет частичный отрицательный заряд б-, а который — положи-
тельный б+, можно оценить исходя из электроотрицательности того
или иного элемента.
Значения электроотрицательности, приведенные в табл. 32, рас-
считаны на основании энергии простых связей между элементами2.
Таблица 32. Шкала электроотрицательности элементов
н 2,1
Li Be В с N 0 F
1,0 1,5 2,0 2,5 3,o 3,5 4,0
Na Mg Ai Si p s Ci
Р,9 1,2 1,5 1,8 2,1 2,5 3,0
К Са Sc . Ge As Se Br
0,8 1,0 1,3 1,8 2,0 2,4 2,8
С помощью этой шкалы можно получить любые разности электро-
отрицательности. Чем выше разность электроотрицательности двух
взаимно связанных элементов, тем бблее полярна, и, в ряде слу-
чаев, более устойчива между ними связь. Например:
С-F С-С1 С-Вг
1,5 0,5 0,3
Это утверждение находится в соответствии с экспериментом,
например фторуксусная кислота значительно устойчивее, чем хлор-
уксусная. Тот факт, что различные, почти неполярные соединения
(парафины, силоксаны) относительно стабильны, находится в про-
тиворечии с только что высказанным положением. Однако это про-
тиворечие объясняется стремлением атомов к симметричному рас-
пределению зарядов. Согласно Полингу2 наиболее стабильное со-
стояние достигается тогда, когда в каждом атоме все устойчивые
орбитали родственны по связям или каждая занята одной парой
электронов. Это положение объясняет также сравнительную не-
устойчивость ковалентных связей с атомами, обладающими более,
чем одной свободной парой электронов:
:с) — с): [ :о—ci: ] ’.ci—о—ci.
Примеры ясно показывают, что симметричные связи только
тогда стабильны, когда в их образовании участвует возможно боль-
шее число пар внешних электронов. Свободные пары электронов,
не участвующие в связи, вызывают известное отталкивание атомов.
286
Недостаточная стабильность различных молекул во многих слу-
чаях может быть также объяснена энергетическим выигрышем при
образовании связей в продуктах реакции. Так, например, кислород
стремится к стабильной молекулярной форме
•• ••
0=0
•• ••
По этой же причине молекула окиси хлора очень лабильна и
даже реакционную способность иона гипохлорита можно объяснить
и связать с этим положением.
Следующий пример должен показать, что оценка связи с по-
мощью понятия электроотрицательности не может быть формально
перенесена на оценку реакционной способности молекул. Так, азо-»
тистый иприт I гидролизуется значительно медленнее, чем серный
иприт II:
✓СН2СН2С1
С1СН2СН2—С1СН2СН2—S—СН2СН2С1
^СН2СН2С1
I п
Несмотря на то, что в обоих соединениях имеются связи С—С1,
реакционная способность веществ различна. Отсюда можно сде-
лать вывод, что нельзя рассматривать связи отвлеченно от всей
молекулы.
Для того, чтобы составить ясное представление о молекуле ор-
ганического соединения, необходимо использовать понятия индук-
тивного эффекта и эффекта мезомерии. Индуктивный эффект
(/-эффект) связан с электроотрицательностью. Сообразно этому
атомы или функциональные группы обладают положительным
^-/-эффектом, если они менее электроотрицательны (отталкивают
электроны) и отрицательным —/-эффектом, если они более элек-
троотрицательны (т. е. сильнее притягивают электроны, более элек-
трофильны), чем водород, который служит стандартом. Все атомы
и группы, располагающиеся справа от атома водорода, вызывают
4-/-эффект, а расположенные слева —/-эффект *.
F > Cl > Вг > I > OR > NR2 > Н > О > СН3 > С2Н5 > СН(СН3)2
— /-эффект + /-эффект
Особый интерес для реакций дегазации представляет то, что
•-{-/-эффект способствует отщеплению отрицательных ионов от мо-
лекулы (например, ионов галогена), так как группа Y подает элек-
троны.
в- в+
R3C ч— Y
+ /-эффект
Эффект мезомерии также является результатом перераспреде-
ления электронов, особенно в соединениях с двойной связью. Мезо-
мерия— это взаимодействие свободной пары электронов с сосед-
ней кратной связью. В этой трактовке реальная молекула может
287
быть изображена так называемыми предельными структурами
(двойная стрелка обозначает, что истинная структура лежит где-то*
посередине):
С1\ _ + Ск
/As—СН—СН=С1 /As—СН=СН—С!
СГ СК
или, пользуясь другой символикой, выравнивание зарядов и связей
можно схематично показать так:
С1
С1
^As-CH=CH-Q1
Для химии Дегазации особый интерес представляет то, что ме-
зомерия сопровождается стабилизацией той или иной системы. Это
справедливо, например, для продуктов гидролиза эфиров фторфос-
фоновых кислот. В этом случае эффект мезомерии ОН-группы
(Af-эффект) сильнее, чем фтора, что приводит к легкому замеще-
нию фтора на гидроксил:
он"
--
R СЬ
X
RO Ср—Н
Рассмотренные до сих пор сведения о поляризации связей в
молекулах служили в первую очередь для того, чтобы понять и
оценить направление течения реакции между двумя реагентами.
В дальнейшем должны быть рассмотрены основные типы реакций,
имеющие значение для химии дегазации. К ним относятся нуклео-
фильное и электрофильное замещение и окисление.
В реакциях дегазации как окислители выступают преимуще-
ственно электрофильные реагенты (например, С1+ и О), а в реак-
циях гидролиза и алкоголиза нуклеофильные реагенты (например,
ОН- и RO"). Приведем в качестве примеров реакции дегазации
иприта и фосфонатов (реакции 1 и 2):
,СН2СН2С1
С1+ + s;
кислота \СН2СН2С1
осаовааае
+ /СН2СН2С1
ci—s;
\СН2СН2С1
(1)
+ /СН2СН2С1 /СН2СН2С1
Cl—SZ 4- Н2о —> O=S^ + НС1 + Н+
\сН2СН2С1 \СН2СН2С1
R\«/Л R\ А
+ОН' —/Р' 1 +Х‘ (2)
R'tA 'X основание R'O' ОН
кислота
288
Эти реакции представляют собой кислотно-основные системы
Льюиса. В зависимости от того, окисляется ли молекула ОВ или
подвергается нуклеофильному замещению, реагент можно рассмат-
ривать как кислоту Льюиса (электрофил) или как основание
Льюиса (нуклеофил).
Полная оценка той или иной возможности использования реак-
ции дегазации возможна на основании кинетических исследова-
ний.3. Очень важной характеристикой реакций является константа
равновесия, которая может быть выведена на основании закона
действия масс:
аА + 6В cC + dD
„ [С]с [D]d
[А]» [В]6
где [А] и [BJ — мольные концентрации веществ А н В.
Определение исходит из того, что в зависимости от темпера-
туры в каждой химической реакции устанавливается равновесие
между исходными и конечными продуктами. В связи с этим до-
стижение полной дегазации может показаться противоречивым.
Однако это противоречие кажущееся, так как практически во мно-
гих реакциях исходные продукты по окончании процесса остаются
в реакционной смеси в неизмеримо малых количествах. Такие ус-
ловия наступают тогда, когда обратная реакция протекает неиз-
меримо медленно. Вследствие того, что важным условием дегаза-
ции яв’ляется практически полное превращение ОВ, закон действия
масс применим лишь к сравнительно немногим процессам дегаза-
ции. Например, протекающий с измеримой скоростью равновесный
процесс лежит в основе первой фазы гидролиза иприта:
,СН2СН2С1 zCH2CH2OH
+ОН‘ «=> +сг
\ch2ch2ci \ch2ch2ci
[С1СН2СН2—S—СН2СН2ОН] [СГ]
К= [S(CH2CH2C1)2] [ОН‘]
Равновесие устанавливается независимо от того, растворен ли
в воде иприт или в содержащей хлор-ионы воде растворен 2-окси-
2-хлордиэтилсульфид. Повышение концентрации ионов ОН- способ-
ствует дальнейшему гидролизу иприта, так как иначе произведе-
ние [S(CH2CH2C1)2][OH“] будет возрастать и значение константы
не останется постоянным. Наоборот, избыточная концентрация
хлор-ионов сдвигает равновесную реакцию влево, т. е. хлор-ионы
затрудняют гидролиз.
Равновесные процессы имеют особенное значение для понима-
ния поведения дегазирующих жидкостей. Равновесные реакции,
представленные следующими примерами, могут протекать в среде
ю Зак. 677
289
дегазирующих жидкостей или под воздействием дегазирующих ве-
ществ в присутствии воды:
NaOCl «=h Na+ + ОСГ
ОСГ + Н2О «=> НОС1 + ОН"
(1)
(2)
RONa «=> Na* + RO"
R— NCI24-2H2O ч=* R—NH2 + 2HOC1
(3)
(4)
Приведенные примеры интересны тем, что они показывают, что
равновесие той или иной реакции дегазации должно быть сдви-
нуто максимально вправо, так как активные компоненты находятся
в правой части уравнений (С1О~, НОС1, RO"). Некоторые из них
действуют как окислители, а некоторые выступают в роли нуклео-
фильных агентов. Так как равновесные реакции характеризуются
константами равновесия, то из этого следует, что при определенной
температуре и заданных концентрациях исходных веществ или не-
диссоциированных соединений (реакция 2) эти ионы присутствуют
в совершенно определенных концентрациях. Изменить концентра-
цию ионов возможно только введением посторонних ионов. Напри-
мер, при введении в равновесную систему (реакция 2) ионов во-
дорода возникает недостаток ионов ОН" вследствие образования
недиссоциированных молекул воды. В соответствии с законом дей-
ствия масс в реакцию с водой должны вступать новьРе ионы СЮ",
что приведет к увеличению концентрации недиссоциированных мо-
лекул хлорноватистой кислоты:
[НОС1][ОН~]
Л [С1О'][Н2О]
{ОН"] [Н*]
Нг0 [Н2О]
(НОСИ кНг0
[ОСГ1 [Н+1
[0H-J—ЧоВД
1н+]
Из этого уравнения следует, что рост концентрации водородных
ионов приводит к изменению соотношения концентраций иона гипо-
хлорита и хлорноватистой кислоты в пользу увеличения кислоты.
Рост образования свободной хлорноватистой кислоты интересен
тем, что это вещество оказывает более сильное окисляющее дей-
ствие, чем гипохлорит-ион. С другой стороны, эта кислота очень
неустойчива. Поэтому всегда надо стремиться к поддержанию оп-
тимального значения pH в дегазирующем растворе4’7.
Примером нежелательного образования хлорноватистой кис-
лоты является реакция 4. В этом случае вода вызывает разложе-
ние хлорамина, что должно учитываться при его хранении:
„ [HOCl]2 [RNH2]
[RNC12] [Н2ор
290
Из уравнения следует, что с уменьшением концентрации воды
уменьшается и концентрация продуктов реакции. Константа гид-
ролиза хлорамина Т равна 4,9-10-8, а дихлорамина Т — 8,0-Ю-7.
Отсюда следует, что дихлорамин разлагается водой с образова-
нием хлорноватистой кислоты в большей степени, чем хлорамин Т.
Приведенные выше реакции 2 и 4 представляют собой равновес-
ные реакции гидролиза, тогда как реакции 1 и 3 являются равно-
весными реакциями диссоциации. В качестве среды для реакции 1
служит вода, а для реакции 3 — спирт или подобные органические
растворители. Соли типа NaOCl диссоциируют практически пол-
ностью, поэтому знак обратимости в реакции 1 излишен. Вместе
с тем, в реальных растворах (в противоположность идеальным —
разбавленным) в результате взаимного влияния ионов, в зависи-
мости от чдсла их зарядов, устанавливается кажущаяся степень
диссоциации. Так, например, Са(ОС1)2 в водных растворах менее
диссоциирован, чем NaOCl.
Существует общее правило, гласящее, что чем более полярно
соединение, тем больше оно склонно к диссоциации. Из данных,
приведенных в табл. 32, видно, что по разности электроотрицатель-
ности атомов можно оценить ионный характер той или иной связи:
Разность Ионный характер
электроотрицатель- связи, %
ности
0,2 1
1,0 22
2,0 63
3,0 89
С этой точки зрения легко можно понять различие в степени
диссоциации гипохлорита натрия и свободный хлорноватистой кис-
лоты.
Стремление к практически количественному завершению про-
цесса дегазации в кратчайший срок требует оценивать дегазирую-
щие вещества и по скорости их реакции с ОВ. Если подходить к
проблеме с чисто практической точки зрения, то интерес представ-
ляет лишь способность изучаемого дегазирующего вещества в те-
чение определенного времени вызвать такое превращение ОВ, ко-
торое полностью исключало бы опасность любой степени пораже-
ния. Для достижения такого эффекта требуется провести ряд
опытов по дегазации.
Для научного исследования реакций между дегазирующими и
отравляющими веществами нужно изучать кинетику процессов.
Результаты такого исследования представляют не только теорети-
ческий интерес, они позволяют сделать вывод об оптимальных ус-
ловиях проведения реакций.
Наиболее важными кинетическими критериями реакции яв-
ляются: порядок реакции, константа скорости реакции К, время
полупревращения 6/,.
Ю* 291
Порядок реакции получают установлением характера зависи-
мости скорости реакции от концентрации реагирующих веществ.
Скорость реакции первого порядка определяется изменением кон-
центрации одного из компонентов во времени. В реакциях второго
порядка определяющими скорость реакции будут, напротив, изме-
нения концентраций двух реагирующих веществ. Для химии дега-
зации характерны реакции второго порядка. Между скоростью
реакции и концентрацией для процесса
А + в C + D
существует следующая зависимость: — dcjdt пропорциональна
сАсв, т. е. концентрации обоих исходных веществ оказывают влия-
ние на скорость процесса. В качестве коэффициента пропорцио-
нальности вводится константа скорости k, тогда выражение при-
мет следующий вид:
Интегрированием этого уравнения получают:
2,303 Лс.
b sss_____1 а - D А
/(4-4)‘Ч<в
где с°— исходная концентрация; с — концентрация ко времени t.
Ниже приводятся константы скорости гидролиза ряда ОВ (см.
сс.5 стр. 197 и210) в щелочной среде при 25°C (в л-моль~1 -м.ин~1):
Зарин...................................... 1550
ДФФ.......................................... 50
0-Этнл-5-(р-диметиламиноэтил)-метилтиофос-
фонат..................................... 0,32
При использовании подобных данных следует иметь в виду, что
они правильны лишь для тех температур, при которых они опре-
делялись. При кинетических исследованиях очень легко поддер-
живать постоянную температуру, а добиться воспроизводимых кон-
центраций реагирующих веществ весьма трудно, особенно вслед-
ствие колебания чистоты исходных веществ. Доказательством этого
является разноречивость кинетических данных, опубликованных
для одних и тех же веществ в разных источниках.
Для исследования скорости гидролиза в щелочной среде суще-
ствуют две принципиальные возможности.
1. Смешивают определенные количества компонентов, а ионы
гидроксила вводят в несколько более высокой концентрации. Из-
меряют убыль концентрации одного из компонентов или рост кон-
центрации одного из продуктов реакции в единицу времени, про-
водя измерение непрерывно или через определенные промежутки
времени. Скорость процесса отвечает реакции второго порядка.
2. Исследование проводится при постоянном значении pH,
т. е. изменяется концентрация лишь одного компонента. Благодаря
292
этому процесс идет как реакция первого порядка, так как его ско-
рость определяется изменением концентрации только одного ком-
понента. Концентрация гидроксил-ионов, определяемая значением
pH, входит в величину константы.
Скорость реакции выражается следующим уравнением:
dcA
— = ^сасв ~ k'c\
где Сд — концентрация ОВ; св — концентрация ионов ОН".
Интегрированием этого уравнения получают:
2,303 Л
В зависимости от того, протекает ли реакция по первому или
второму порядку, для расчета времени полупревращения * исполь-
зуют следующие уравнения:
, 0,692 , _ 1
tl!*—Г
реакция первого порядки реакция второго порядка
Например, при гидролизе зарина и ДФФ при pH 7 А/2 равно
300 и 3000 мин соответственно. Сравнение скоростей гидролиза
свидетельствует о том, что низкая константа скорости отвечает
большому времени полупревращения, т. е. процесс идет с малой
скоростью.
В заключение следует обратить внимание на зависимость ско-
рости реакции от температуры; для реакций дегазации это имеет
большое значение. Широко распространенное правило гласит: ско-
рость простой гомогенной реакции при увеличении температуры
на 10° возрастает в 2—2,5 раза (см. т. 1, стр. 275).
Другим чрезвычайно важным фактором увеличения скорости
реакций дегазации является влияние катализаторов и растворите-
лей.
Эти возможные воздействия можно представить в виде мате-
матических зависимостей между кинетическими величинами, из ко-
торых следует, что описанные выше закономерности нельзя приме-
нять чисто формально.
12.1.2. Каталитическое ускорение реакций дегазации
До настоящего времени только немногие реакции дегазации могли
быть ускорены каталитически. Как это уже отмечалось (т. 1, раз-
дел 7.5.5.3), такая возможность существует преимущественно для
реакций гидролиза эфиров фосфоновой и фосфорной кислот.
* Время полупревращения — это время, за которое начальная концентрация
уменьшается наполовину.
293
Принцип описанных катализируемых реакций основан на том,
что поляризацией связи Р—О достигается увеличение дефицита
электронной плотности на атоме фосфора и тем самым облегчается
нуклеофильная атака гидроксил-ионом:
а- в*
W°\в+ • \’+/°
Р ^Катализатор] или Р' + [Катализатор] —►
/ / \F
в-
\6V°
—+ F
' ^Катализатор]
Для реакции решающим является не только первоначально су-
ществовавшее распределение электронов в молекуле, но и в зна-
чительно большей степени способность сдвига электронов.
Способность молекулы подвергаться внешнему воздействию,
результатом которого является оттягивание или нагнетание элек-
тронов относительно того или иного реакционного центра, назы-
вается поляризуемостью. Реакционная способность той или иной
молекулы, кроме того, повышается за счет поляризации.
Молекула поляризуется тем сильнее, чем меньше она поляри-
зована вначале. Катализаторы ускоряют преимущественно те реак-
ции, в которых участвуют легко или относительно легко поляри-
зуемые молекулы.
Механизмы действия катализаторов чрезвычайно многообраз-
ны. Решающим является влияние какого-либо вещества на энергию
активации реакции, которую следует понимать как энергетический
барьер между энергиями исходного и конечного состояний. Пони-
жение энергии возможно, например, вследствие разделения про-
цесса на две последовательно протекающие реакции, энергия ак-
тивации которых меньше, чем энергия активации общего процесса.
Примером может быть катализ гидролиза зарина гипохлоритом
(ср. т. 1, стр. 307).
Однако часто мы имеем дело с такими реакциями, снижение
энергии активации которых происходит за счет воздействия ката-
лизатора на исходное или промежуточное соединение без участия
катализатора в самой реакции замещения.
Такое действие может быть вызвано, например, достаточно по-
лярным растворителем. Однако эти случаи ограничены щелочным
или кислотным катализом.
12.1.3. Влияние растворителей на реакции дегазации
Полярные растворители могут воздействовать на скорость гомо-
генных реакций дегазации в двух направлениях.
Они способны повлиять на течение процесса за счет уже упо-
мянутой поляризации. Так, например, гидролиз иприта в смеси воды
294
и спирта (1:1) протекает примерно в. 10 раз медленнее, чем в
воде (вода более полярна, чем спирт). При помощи веществ более
полярных, чем вода, и поэтому оказывающих большее поляризую-
щее воздействие на молекулу иприта, можно было бы весьма эф-
фективно осуществить дегазацию иприта за счет реакции нуклео-
фильного замещения.
Полярные растворители оказывают решающее влияние на де-
газационные реакции еще и потому, что они способны сольватиро-
вать ионы и молекулы с образованием вокруг них сольватной обо-
лочки.
Сольватная оболочка возникает вследствие расположения полярных моле-
кул растворителя на нонах или полярных молекулах. Например, вода гидрати-
рует катионы за счет взаимодействия с кислородом, несущим частичный отри-
цательный заряд.
Образование сольватной оболочки может влиять на скорость
реакции как положительно, так и отрицательно.
Положительное влияние сказывается тогда, когда сольватация
высвобождающегося в реакции иона затрудняет его вступление в
обратную реакцию (например, при дегазации иприта американским
дегазирующим средством DS-2). Вследствие того, что сольвата-
ции подвержены и полярные молекулы, сольватная оболочка может
образоваться как вокруг молекул ОВ, так и вокруг возникаю-
щих в процессе реакций активных комплексов (переходных со-
стояний). При сольватации молекула совершает работу притяже-
ния молекул растворителя. Связанное с этим снижение энергии
приводит к стабилизации системы. В соответствии с этим влияние
растворителя на скорость реакции зависит от того, становится ли
молекула в переходном состоянии более или менее полярной, чем
в исходном.
Если переходное состояние более полярно, то в результате соль-
ватации оно становится более стабильным, чем исходное состояние.
Так как реакция предпочтительно проходит в направлении
большей стабильности, то стабилизация переходного состояния вы-
зывает увеличение скорости реакции. При стабилизации исходного
вещества происходит снижение скорости реакции.
12.1.4. Влияние физических факторов
на химические способы дегазации
На химическую дегазацию большое влияние оказывают темпера-
тура и физические свойства ОВ, дегазирующей жидкости и др.
Особенно большое значение это имеет при проведении дегазации
различных поверхностей, так как, например, внешние условия мо-
гут быть чрезвычайно разнообразны и изменить их практически
невозможно.
К таким свойствам относятся: растворимость ОВ в дегазирую-
щих жидкостях; смачиваемость дегазируемой поверхности и по-
верхностное натяжение на границе раздела ОВ и дегазирующей
295
жидкости; вязкость дегазирующей жидкости и связанный с этим
диффузионный эффект.
Эти вопросы частично обсуждаются р разделах 12.2, 12.3 и 13,
поэтому здесь мы рассмотрим только наиболее важные положе-
ния, которые имеют отношение к химической дегазации.
Основная проблема этого комплекса вопросов состоит в том,
что высокая реакционная способность дегазирующего средства ста-
новится бесполезной, если не созданы условия для наиболее пол-
ного его взаимодействия с ОВ. В случае применения водных дега-
зирующих растворов возникают затруднения, которые удается
преодолеть, используя простые физические закономерности.
Известно, что растворимость вещества в воде при заданной тем-
пературе является величиной постоянной. При применении высоко-
эффективных дегазирующих растворов, таких, как растворы гипо-
хлорита натрия или кальция, дегазация часто протекает быстрее,
чем растворение ОВ. Так, в 1 л воды растворяется лишь 0,5 г
иприта и менее Юг зомана24. Поэтому при плотности заражения
ипритом какой-либо поверхности, равной 10г/лг2, и применении
дегазирующей жидкости в количестве 1 л/л»2 теоретически в гомо-
генных условиях могут прореагировать только 5% от исходного ко-
личества иприта. В этом случае, чтобы процесс дегазации не пре-
кратился, необходимо постоянно механически поддерживать про-
цесс растворения.
Взаимодействие с дегазирующей жидкостью молекул ОВ, диф-
фундирующих из образованного ими поверхностного слоя, приво-
дит к снижению концентрации дегазирующего вещества вблизи
границы раздела фаз. Вследствие этого возникает постепенно рас-
ширяющаяся зона перепада концентрации дегазирующего веще-
ства, что, в свою очередь, вызывает непрестанное удлинение пути,
проходимого диффундирующими молекулами ОВ и дегазирующего
вещества. Влияние этих факторов особенно проявляется при низ-
ких температурах, когда резко снижается скорость растворения,
тормозится процесс диффузии и уменьшается скорость химических
реакций.
. В связи с описанными явлениями нужно стремиться создавать
максимально высокие концентрации дегазирующего вещества в
растворе с тем, чтобы и без постоянного механического вмешатель-
ства в зоне реакции не происходило уменьшение концентрации
дегазирующего вещества. Идеальным считается случай прохожде-
ния реакции на поверхности раздела фаз. Независимо от того, до-
стигнуты ли такие идеальные условия или нет, целесообразно, как
и во всех гетерогенных реакциях, создавать максимально большие
поверхности раздела, так как количество ОВ, переходящее в рас-
твор за единицу времени, зависит от площади поверхности.
Наряду с механическим воздействием на процесс химической
дегазации путем обработки поверхности щетками или другими
средствами особое значение для увеличения поверхности раздела
между водным дегазирующим раствором и слоем нерастворенного
296
ОВ приобретает применение поверхностно-активных веществ. Дей-
ствие этих веществ заключается в сильном уменьшении поверх-
ностного натяжения на границе раздела водной и органической
фаз, и приводит к переводу ОВ в эмульсию. Следствием этого яв-
ляется значительное увеличение поверхности раздела фаз, на кото-
рой протекает дегазация, и облегчение взаимной диффузии между
фазами. Кроме того, поверхностно-активные вещества способны в
большей или меньшей степени препятствовать оседанию капель ОВ,
удаленных с дегазируемой поверхности.
Структура дегазируемых поверхностей, как правило, не являет-
ся однородной. Поэтому необходимо как-то создавать тесный кон-
такт между дегазирующим веществом и ОВ при проникновении
последнего в Щели, трещины и поры. Увеличивая смачивающую
способность водной фазы, поверхностно-активные вещества и в
этом случае оказываются весьма полезными и способствуют успеш-
ному течению процесса дегазации.
Выбор того или иного моющего средства диктуется совмести-
мостью отдельных компонентов раствора с дегазирующим веще-
ством. Для смешения с водными растворами дегазирующих
средств, содержащих активный хлор, наиболее пригодны анионо-
активные моющие вещества, например вторичные алкилсульфо-
наты (мерзоляты).
Некоторые моющие средства могут даже затруднить процесс
дегазации. Например, изготовленные на основе жирных кислот
моющие средства — мыла, мыльная стружка и др., образуют при
дегазации хлопьевидные осадки, которые оседают на каплях ОВ
и тем самым мешают дегазации (сс.5, стр. 130). Естественно, такие
моющие средства для дегазации не подходят.
При попадании ОВ на поверхности, имеющие лакокрасочные
или другие покрытия, ОВ весьма быстро проникают внутрь орга-
нических материалов (резина, пластические массы и др.) и про-
ведение дегазации в этих случаях становится весьма затруднитель-
ным. Водные дегазирующие растворы практически вообще не при-
годны для этой цели. Проникшее в такие покрытия ОВ может быть
обезврежено только в случае принятия мер для его извлечения из
органической пленки покрытия.
Огромное значение в этой связи имеет устойчивость красок и
других покрытий к прониканию ОВ. Полностью исключить диффу-
зию ОВ в органические материалы невозможно. Поэтому, чтобы
свести диффузию к минимуму, очень важно сразу же после напа-
дения приступать к дегазации.
Вместе с тем, целесообразно использовать разные возможности
для обезвреживания ОВ, проникших в лаковые, резиновые и дру-
гие подобные покрытия. Например, хорошие результаты можно
ожидать от применения дегазирующих растворов на основе орга-
нических растворителей, которые способны проникать в органи-
ческие материалы. Однако в таком высоковязком материале, как
резина или слой лакокрасочного покрытия, дегазирующее вещество
297
диффундирует довольно медленно, и поэтому очень трудно себе
представить, как будет проходить дегазация внутри слоя такого
покрытия при постоянном уменьшении концентрации дегазирую-
щего вещества. Кроме того, следует также учитывать разрушающее
действие органических растворителей на органические материалы.
В неблагоприятном случае, например, лак может полностью от-
слоиться от дегазированной поверхности. Правда, успех дегазации
при этом гарантирован, однако по возможности таких радикальных
методов следует избегать.
Менее агрессивный метод состоит в применении горячих вод-
ных дегазирующих жидкостей, подогретых до 60—80 °C. При этом
не только значительно ускоряется реакция, лежащая в основе де-
газации, но и увеличивается растворимость и скорость растворе-
ния ОВ, а также диффузия его из покрытия.
Таким образом, можно при повышенных температурах про-
дегазировать за сравнительно короткое время ОВ, не слишком
глубоко проникшее в какое-либо покрытие органического проис-
хождения.
Вследствие зависимости скорости диффузии, от температуры
способность ОВ проникать в лаковые и прочие покрытия в зимних
условиях очень низка. Например, при —10 °C практически ОВ не
проникают в лакокрасочные покрытия, В этом случае можно огра-
ничиться поверхностной дегазацией.
12.2. ДЕГАЗАЦИЯ НУКЛЕОФИЛЬНЫМИ РЕАГЕНТАМИ
12.2.1. Общие сведения
На реакционную способность нуклеофильных<реагентов, применяе-
мых для дегазации зараженных материалов, действуют многие
факторы. Этими факторами являются, основность и нуклеофиль-
ность самого реагента и замещаемых анионов; влияние раствори-
теля на сольватацию реагирующих веществ, в особенности продук-
тов реакции; влияние катализаторов; влияние температуры; обра-
зование стабильных продуктов реакции; растворимость ОВ или
тактических смесей ОВ в реакционной среде; поверхностное натя-
жение на границе фаз между плохо смешивающимися компонен-
тами реакции.
На скорость дегазации, кроме того, в значительной степени
влияет соотношение концентраций реагирующих веществ.
Для теории и практики дегазации нуклеофильными реагентами
особое значение имеют основность и нуклеофильность раствори-
теля и катализатора. Поэтому в данном разделе им уделено значи-
тельное внимание.
Понятия основности и нуклеофильности не тождественны. С точ-
ки зрения «статической» электроотрицательности различия в по-
нятиях действительно нет, если же привлечь и поляризуемость, то
основность является мерой сродства вещества к протону. Нуклер-
298
фильная активность характеризует стремление частицы ’ отдавать
электроны положительно поляризованному атому углерода *.
Нуклеофильные реагенты располагаются по активности в сле-
дующий ряд
F" < Н2О < СГ < С6Н6О' < ВГ < ОН' « OR' <
< NH3 (амины) < I” < S2~
Применительно к использованию реакции нуклеофильного за-
мещения для дегазации иприта этот ряд показывает, что способ-
ность вытеснять хлор из молекулы иприта увеличивается от иона
фенолята к сульфид-иону.
Различие между нуклеофильностью и основностью следует еще
раз наглядно представить на примере фенолят- и бромид-ионов.
Как видно из приведенного ряда, эти два иона по нуклеофильности
незначительно отличаются друг от друга, однако основность фено-
лят-иона значительно выше, чем бромид-иона.
Рассмотрим дегазацию эфира фторфосфоновой кислоты на при-
мере нуклеофильного замещения фенолят- и бром-ионами. Оказы-
вается, что фенолят-ионы легко реагируют с ОВ, тогда как бром-
ионы не действуют на молекулу этого ОВ. Таким образом, в этом
случае решающей является основность, а не нуклеофильная актив-
ность реагента. Несомненно, это явление находится в связи с тем,
что замещение идет не по углероду, а по пятивалентному атому
фосфора.
Общая математическая зависимость между экспериментальной
и расчетной константой скорости реакции выведена Эдвардсом5:
feb
Kw
где kb — константа скорости реакции между реагентом и субстратом (в данном
случае OB);.few—константа скорости реакции между водой и субстратом;
Еп — константа нуклеофильности реагента; Н — Константа основности реагента;
а и р — константы субстрата (здесь константы ОВ).
Для иприта коэффициент а = 2,45, а коэффициент 0 = 0,074,
для' зарина а = 0; ₽ (при ОН") = 0,53. Из этого следует, что
нуклеофильное замещение в молекуле иприта зависит от нуклео-
фильности, а в молекуле зарина — от основности применяемых
реагентов.
Таким образом, при выборе вещества для дегазации иприта
предпочтение должно быть отдано реагенту с высокой нуклеофиль-
ной активностью, а для дегазации зарина — с высокой основностью.
Сделать такой выбор помогают данные, приведенные в табл. 33.
"* Автор рассматривает только частный случай нуклеофильности, под кото-
рой обычно понимают сродство к ядру атома, у которого происходит замеще-
ние. Приведенный ниже ряд нуклеофильных агентов относится главным обра-
зом к реакциям замещения (у насыщенного атома углерода) в водных раство-
рах. — Прим. ред.
299
Таблица 33. Нуклеофильная активность и основность важнейших
нуклеофильных реагентов
(сс. s, стр: 117)
Основание Константа нуклео- фильности *я Константа основности Н Основание Константа нуклео- фильности Еп Константа основности Н
сг 1,24 -3,00 СбН5О 1,46 11,74
НО" 1,65 17,48 s2o2- 2,57 9,00
NH3 1,84 11,22 s2- 3,08 14,88
F" 0,25 3,45 НОО" — 13,36
СгО2" 4 — 8,23 СЮ" — 9,27
При анализе данных этой таблицы обращает на себя внимание,
что сульфидная сера имеет благоприятные значения констант ну-
клеофильности и основности. Однако в действительности реаген-
ты, содержащие сульфид-, тиосульфат- или тиофенолят-ионы, не
вступают во взаимодействие с зарином, несмотря на высокую их
основность. Причину этого следует искать в пространственных за-
труднениях, возникающих из-за сравнительно большого размера
атома серы.
Щелочные сульфиды пригодны для дегазации ОВ типа зарин
только потому, что они повышают концентрацию гидроксил-ионов
в водных растворах.
Следует подчеркнуть, что величины, приведенные в табл. 33,
выражают в каждом случае только основность и нуклеофильность
по отношению к воде. Значения основности могут быть рассчитаны
по следующему уравнению:
Я = рКа+ 1,74
Константы нуклеофильности получают 1 из уравнения:
5п = аР4- ЬН
где Р — поляризуемость, а и b —коэффициенты.
Чаще оперируют рАа, нежели величиной поляризуемости; р/<а
представляет собой отрицательный логарифм константы диссоциа-
ции кислоты (—1gЛа)- Значение рЛа тем меньше, чем более силь-
ными кислотными свойствами обладает соединение.
12.2.2. Дегазация едким натром
Чистый едкий натр NaOH представляет собой белое гигроскопиче-
ское вещество, выпускаемое в продажу в виде палочек или табле-
ток; технический продукт — белые или серые чешуйчатые пластин-
ки. Едкий натр производится промышленностью в больших коли-
чествах.
300
Чистый твердый NaOH имеет плотность 2,13г/с.н3, т. пл. 318 °C.
Растворимость едкого натра в воде при разных температурах сле-
дующая:
Температура, Растворимость,
°C г/ЮО г
О 42
20 109
100 342
При растворении едкого натра выделяется значительное коли-
чество тепла, около 10 ккал/моль. Это значит, что при приготовле-
нии 10%-ного раствора NaOH выделяется 25 ккал тепла на 1 л
жидкости. Принимая, что теплоемкость раствора близка к тепло-
емкости воды, можно в среднем считать, что при растворении
NaOH температура раствора повышается на 25°C. Поэтому во
избежание возможных перегревов требуется хорошее перемешива-
ние; последнее, кроме того, препятствует спеканию твердого NaOH
и способствует равномерному растворению.
В водном растворе NaOH сильно диссоциирован:
NaOH <=± Na+ + OH~
Едкий натр растворяется также в метиловом и этиловом спир-
тах. Наряду с образованием спиртовых растворов идет еще и хи-
мическая реакция — образование алкоголятов:
ROH + ОН" «=> RO' + H2O
RO" + Na+ ч=> RONa
Присутствие воды сильно сдвигает равновесие этой реакции
влево. Чем больше воды содержит спирт, тем меньше в нем содер-
жится алкоголята. Основность ионов гидроксила и ионов аклого-
лята мало отличается друг от друга.
Как твердый NaOH, так и его растворы весьма агрессивно дей-
ствуют на кожу человека. Особенно опасно попадание едкого натра
в глаза, что может привести к потере зрения. Кроме того, едкий
натр и. его растворы разрушают многие ткани. Поэтому при ра-
боте с едким натром необходимо надевать защитные очки и сред-
ства защиты кожи, а при приготовлении дегазирующих жидкостей
с использованием едкого натра — полную защитную одежду.
Едкий натр и. его растворы по-разному реагируют с многими
материалами. Так, при взаимодействии с алюминием и сплавами
легких металлов происходит очень бурная реакция с выделением
водорода. В стальных сосудах растворы едкого натра можно хра-
нить длительное время. Действие едкого натра на лакокрасочные
покрытия в течение более 20 мин приводит к их набуханию и раз-
рушению.
Технический едкий натр хранят в жестяных барабанах (50 кг),
жестяных банках или в пластикатовых мешках, уложенных в кар-
тонные'парафинированные емкости (0,5 кг). При хранении едкого
301
натра его необходимо предохранять от попадания влаги, углекис-
лоты воздуха, под влиянием которой образуется карбонат. Поэто-
му упаковка должна быть воздухонепроницаемой. Образование
карбоната, так же как и поглощение малых количеств воды, при-
водит к слеживанию едкого натра. Едкое кали, обладающее ана-
логичными свойствами, редко применяется для дегазации. Про-
дукт выпускается в меньшем количестве промышленностью и более
дорог.
Едкий натр применяется в первую очередь для дегазации-ОВ
типа зарин. Для этого применяют 10%-ные водные растворы, а
также растворы, содержащие 5% NaOH и 5% моноэтаноламина
HOCH2CH2NH2 (дегазирующее действие моноэтаноламина см.
стр. 317). Дегазировать растворами едкого натра можно до тем-
ператур —5° или —8 °C. При более низких температурах необхо-
димо добавлять антифриз или увеличенное количество моноэтанол-
амина (см. раздел 13.2.4).
Об эффективности растворов NaOH при дегазации ДФФ см.
т. 1, раздел 7.3.3.3. При этом следует учитывать, что ДФФ является
одним из наиболее стабильных веществ типа зарин. При концен-
трациях дегазирующих растворов, применяемых в полевых усло-
виях, достигаются более высокие скорости гидролиза. На 1 м2 за-
раженной поверхности следует расходовать около 1 л дегазирую-
щей жидкости.
Если 1 л’• жидкости содержит 100 г NaOH, а на поверхности
находится 10 г ДФФ, то мольное соотношение реагентов равно
примерно 45:1. При таком мольном соотношении реагентов про-
должительность дегазации значительно ниже 30 мин, требующихся
для дегазации ДФФ при соотношении реагентов 4:1 и темпера-
туре 17 °C.
Если дегазацию едким натром производят в лаборатории, то
рекомендуется применять спиртовые или водноспиртовые растворы
щелочи, которые обладают тем преимуществом, что в них ОВ более
легко растворимы. При использовании в лабораторных условиях
водных растворов щелочи создаются затруднения, связанные с раз-
личием в плотностях дегазирующего раствора и ОВ и невозмож-
ностью использовать такие вспомогательные средства, как щетки
и др. Так, например, плотность 10%-ного водного раствора NaOH
при 20°C равна 1,11 г!см3. При смешении этого раствора сзоманом,
плотность которого равна 1,01 г!см3, последний будет находиться
над щелочью и может заражать атмосферу.
Растворы едкого натра можно применять не только для дегаза-
ции ОВ типа зарин, но в ограниченных размерах и для дегазации
иприта. При этом, однако, следует пользоваться более концентри-
рованными водноспиртовыми растворами. Так как в результате до-
бавления спиртов, обладающих незначительной полярностью, ско-
рость гидролиза уменьшается, то добавлять более 50% спирта не
следует. Для дегазации азотистого иприта и V-газов едкий натр не
пригоден8.
302
12.2.3. Дегазация содой
Сода или карбонат натрия Na2CO3 представляет собой белые кри-
сталлы с различным содержанием кристаллизационной воды (одна,
семь и десять молекул воды на одну молекулу карбоната натрия).
Практическое значение имеет только кальцинированная сода (без-
водная) и кристаллическая Na2CO3-ЮН2О, т. пл. 32°С. При нагре-
вании до 107 °C она теряет воду и переходит в кальцинированную.
В воде сода очень хорошо растворима. Как соль очень сильного
основания и слабой кислоты она легко гидролизуется в водных
растворах7:
Na2CO3 + Н2О NaOH + NaHCO3
NaHCO3 + H2O NaOH + H2CO3
Концентрация ионов ОН" в 0,1 н. растворе Na2CO3 (что отве-
чает 1,2%-ному раствору) при 18°С равна примерно 3,5-10-3моль/л
(pH 11—12); концентрация ионов ОН" в таком же растворе
NaHCO3 равна 1,5-10"6 моль/л.
Водными растворами соды пользуются главным образом для
дегазации обмундирования. Так как дегазацию производят при по-
вышенных температурах, то по сути процесс дегазации представ-
ляет собой гидролиз ОВ водой. Исходя из правила, упоминавшегося
в разделе 12.1.1, при температуре 100 °C скорость гидролиза в 28
раз больше, чем при нормальной температуре. Содержание щелочи
в растворе при таких высоких скоростях гидролиза имеет для са-
мой реакции дегазации подчиненное значение, она необходима для
нейтрализации освобождающихся при гидролизе ОВ кислот, кото-
рые могут способствовать разрушению дегазируемого материала.
Сравнительно невысокая щелочность растворов соды не разрушает
текстильные изделия.
Гидролиз большинства ОВ при 100°C заканчивается уже через
несколько минут. Тем не менее, вследствие того, что ОВ до начала
гидролиза должно перейти Из ткани или из защитной одежды в
раствор, продолжительность дегазации составляет не менее 1 ч.
Способ дегазации кипячением с содой эффективен для обезврежи-
вания почти всех основных типов ОВ.
Дегазация кипячением. Этот способ обладает как преимущест-
вами, таК и недостатками. Основной проблемой этого вида дегаза-
ции является малая устойчивость к кипячению текстильных изде-
лий и других материалов. Так, например, некоторые синтетические
волокна нельзя нагревать выше 60 °C; в полевых установках для
дегазации кипячением практически невозможно постоянно поддер-
живать высокую температуру; скорости процессов дегазации (рас-
творение, диффузия, химические реакции) при 60 °C значительно
ниже, чем при 100 °C.
Применение моющих средств, содержащих незначительные
количества активного хлора (2,5% активного хлора в пересчете на
твердый гипохлорит натрия), почти не улучшает качество дегазации.
303
Добавки гипохлорита натрия ускоряют только гидролиз ОВ типа
зарин (см. раздел 12.2.4). Учитывая высокое pH моющих растворов,
трудно предположить, что незначительные концентрации активного
хлора, содержащегося в гипохлорите, могут повлиять на дегазацию
иприта и V-газов (см. раздел 12.3.2).
Улучшение дегазации текстильных материалов возможно путем
повышения моющего эффекта, для чего используют обычные сти-
ральные установки (см. раздел 13.3). Если же зараженные мате-
риалы после пребывания в моющей ванне в течение допустимого
срока (не более 30 мин) не будут дегазированы, то, естественно,
такой метод дегазации не пригоден и им следует пользоваться
лишь в крайних случаях; при этом приходится несколько раз сти-
рать и полоскать материалы.
Из рассмотрения данного вопроса вытекает, что при некоторых
условиях выгоднее применять для дегазации текстильных материа-
лов экстракцию органическими растворителями (см. раздел 13.2).
С другой стороны, способ кипячения имеет то преимущество, что
при этом достигается высокая степень дегазации с помощью
дешевых средств и подсобной аппаратуры.
12.2.4. Дегазация другими кислородсодержащими
нуклеофильными реагентами
В качестве нуклеофильных заместителей наряду с ионом гидро-
ксила имеют значение и другие кислородсодержащие ионы, кото-
рые по существу можно рассматривать как производные гидрок-
сила "О—Н
"О—С1 'О — алкил "О — арил “О—ОН
и которые являются анионами слабых кислот. Часть этих ионов
можно считать катализаторами нуклеофильной реакции гидроли-
тической дегазации ОВ типа зарин. Для нуклеофильного замеще-
ния в молекулах иприта или V-газов они, за исключением алкок-
сидных ионов и в известной мере ионов ОН-, представляют лишь
незначительный интерес.
Ионы гипохлорита СЮ- и гидроперекиси НОО- как носители
активного хлора или активного кислорода имеют очень большое
значение для дегазации окислением и хлорированием. Генерирую-
щие их соединения будут более подробно рассмотрены в разделе
12.3. Роль иона гидроксила уже была описана в разделе 12.2.1, по-
этому здесь следует более детально остановиться на алкоголягах
и фенолятах; при рассмотрении гипохлорит- и гидропероксил-ионов
речь будет идти об их действии как катализаторов (см. также т. 1,
разделы 7.3.3.3 и 7.5.5.3).
Алкоголяты. Не содержащие воды или спирта алкоголяты пред-
ставляют собой порошкообразные вещества. Они хорошо раство-
римы в спиртах и других безводных полярных органических рас-
творителях.
304
Как было уже отмечено в разделе 12.2.2, в воде алкоголяты
гидролизуются:
RONa + Н2О «=* ROH + NaOH
Однако при добавлении малых количеств воды часть алкоголята
остается неизменной. На этом же основании можно сказать, что в
растворах едких щелочей в спиртах всегда содержится некоторое
количество алкоголята. В этом заключается и дегазирующее дей-
ствие жидкости DS-2 (США), представляющей собой смесь спирто-
вого раствора щелочи с амином (ср. стр. 317).
В разделе 12.2.1 отмечалось также, что нет существенной раз-
ницы между нуклеофильностью гидроксила и алкоксид-иона.
Вместе с тем основность алкоголятов в значительной мере зависит
от свойств алкильного радикала. В общем виде можно сказать, что
благодаря влиянию +/-эффекта с увеличением длины углеводо-
родной цепи радикала основность алкоксид-аниона возрастает, а
при наличии групп, обладающих — /-эффектом, основность падает.
Так как спирты являются очень слабыми кислотами, то в качестве
меры основности можно использовать относительную кислотность
спиртов, которая тем выше, чем ниже основность.
Ниже приведена относительная кислотность ряда спиртов, из-
меренная по отношению к изопропиловому спирту, основность ко-
торого принята равной нулю10.
Спирт Относительная
кислотность
(СНз)зСОН.............................. <0,2
СН3-СН2-СНОН-СН3....................... <0,2
СН?-СН2-СН2ОН.......................... >0,5
(СН3)2СН-СН2ОН......................... >0,5
СН3-СН2-СН2-СН2ОН...................... >0,6
СН3-СН2ОН............................. 0,95
Н20............................... 1,20
СН3О-СН2-СНОН-СН3................. 1,80
СН2=СН-СН2ОН ........................... 2,7
СН3ОН................................... 4,0
СН3О-СН2-СН2ОН.......................... 8,0
СН3-СН2-О-СН2-СН2ОН.................... 12,0
Рассматривая этот ряд с точки зрения пригодности соответст-
вующих алкоголятов для дегазации, следует помнить, что дегаза-
цию нужно проводить в почти безводной среде, так как алкоголяты
в воде тотчас гидролизуются с образованием спиртов и щелочи.
Исходя из соображений, что для дегазации наиболеее пригодны
соединения, обладающие сильной основностью, следовало бы ожи-
дать наилучшего дегазирующего действия от алкоголята третич-
ного бутилового спирта.
При нуклеофильном замещении, кроме основности какого-либо
спирта, имеет значение и его способность сольватировать освобож-
дающийся в этом процессе анион (см. раздел 12.1.3).
Эффективность спирта следует оценивать наряду с эффектив-
ностью соответствующего алкоголята, так как практически при
305
употреблении, например, этилата натрия в метаноле тотчас проис-
ходит образование метилата:
C2H5ONa + СН3ОН CH3ONa + С2Н6ОН
Установлено, что способность спиртов сольватировать анионы
увеличивается с ростом их кислотности. Если рассматривать моле-
кулу спирта как диполь, то ассоциация его молекул между собой
возникает в результате электростатических дипольных взаимодей-
ствий атома водорода гидроксильной группы (положительный
конец диполя) и атома кислорода второй молекулы спирта (отри-
цательный конец диполя):
б- б+ б- б+ б- б+ б- 6j-
о—н-. о—н- • -о—н- - о—н
I I I I
СН3СН2 сн,сн2 СН3СН2 СН3СН2
Ассоциация молекул спирта и аниона обусловлена также обра-
зованием водородных связей между атомами водорода гидроксила
с положительным частичным зарядом и отрицательными ионами
или молекулами с отрицательными частичными зарядами:
6- 6+ в+ &-
О—Н---[аниои]""Н—О
СН3—СН2 СН2—СН3
Величина частичного положительного заряда водорода группы
ОН характеризуется кислотностью спирта. В растворе алкоголята
третичного бутилового спирта, в третичном бутиловом спирте вы-
сокой основности алкоголята противостоит низкая сольватирующая
способность спирта, йлияние которой на скорость реакции обратно
влиянию основности.
Следует ожидать, что высокой сольватирующей способностью
обладает 2-этоксиэтанол (относительная кислотность 12,0). Исходя
из этих соображений для создания смеси с оптимальным дегази-
рующим действием необходимо подобрать и алкоголят с соответ-
ствующей основностью. Однако влияние сольватации заключается
не только в этом. Речь идет о влиянии сольватации на стабилиза-
цию исходных полярных продуктов или промежуточных состояний
в связи с возможным торможением или ускорением реакции. В на-
стоящее время роль этих факторов в развитии процессов дегазации
может быть оценена пока только на основании общих тееоретиче-
ских положений. И это особенно трудно потому, что структуры про-
межуточных соединений могут быть чрезвычайно разнообразными.
Конкретные данные о влиянии сольватации на дегазацию до сих
пор еще не публиковались.
Что касается дегазирующего действия смесей спирт — алкого-
ля?, то можно считать установленной пригодность этих смесей для
дегазации почти всех ОВ за счет реакций нуклеофильного замеще-
ния (см. т. 1, стр. 156).
306
В противоположность реакциям различных других кислород-
содержащих нуклеофильных реагентов с ОВ типа зарин, катализ в
данном случае не наблюдается. Большие скорости реакции обу-
словлены в первую очередь сильной основностью алкоголятов,
сольватацией спиртом выделяющегося аниона и действием возмож-
но присутствующей при этом щелочи. Как видно из раздела 12.2.6.5,
эти реакции способны ускорять амины.
Феноляты. Феноляты являются ароматическими аналогами алко-
голятов. Этот класс соединений назван так по их простейшему
представителю — фенолу.
Фенолы представляют собой по большей части низкоплавкие,
хорошо растворимые в воде вещества, обладающие характерным
неприятным запахом., Их водные растворы имеют кислую реакцию.
Уже в этом проявляется меньшая основность фенолят-ионов по
сравнению с -алкоголят-ионами. Они гораздо более устойчивы в
водной среде, чем алкоголяты. Присутствие фенолят-ионов даже в
слабо кислых растворах фенолов объясняется чрезвычайно высокой
способностью последних к диссоциации. Вследствие гидролиза рас-
творы фенолятов имеют сильно щелочную реакцию:
CeH6ONa + Н2О CeH6OH + NaOH
Как видно из уравнения реакции, между фенолятом и фенолом
устанавливается равновесие. Концентрация фенолят-ионов, от кото-
рой собственно зависит дегазирующее действие, остается постоянно
достаточно высокой.
Растворы фенолятов в противоположность растворам фенола
не действуют раздражающе на кожные покровы. Для дегазации
иприта и V-газов феноляты не пригодны. Вместе с тем их высокая
дегазирующая способность по отношению к ОВ типа зарин опре-
деляет их значение в качестве дегазирующего средства для кожных
покровов.
Как следует из механизма реакции (см. т. 1, раздел 7.3.3.3), наи-
более пригодными для дегазации являются феноляты многооснов-
ных фенолов с группами ОН* в положении 1,2.
Таковыми являются — 1,2-диоксибензол (пирокатехин), 1,2,3-три-
оксибензол (пирогаллол). Приведенные ниже периоды полураспада
ДФФ в растворах фенолов (см. сс. 11, стр. 319) являются доказа-
тельством этого положения:
tij2, мин fy, мин
Без фенола .... ~3000 С пирокатехином . . 27
С фенолом........ ~320 С пирогаллолом . . 6
Реакции проводились с растворами ДФФ, содержащими
4,5-10"3 моль/л, и растворами фенолов, содержащими 22,5-10-3
моль!л, при pH 7,6 и -температуре 38 °C.
В соответствии с механизмом реакции способность нуклеофиль-
ного реагента поляризовать связь Р=О или Р—F является решаю-
щей для проявления каталитического действия реагента.
307
Другие нуклеофильные реагенты, обладающие каталитическим
действием. Как удалось установить, катализ гидролитического рас-
щепления ОВ типа зарин происходит, главным образом, с помощью
анионов, образующихся в некоторых системах. Ниже приведены си-
стемы, в которых образуются такие анионы, и константы диссоциа-
ции соответствующих им кислот4> 7j
К = [анион ] [н+] = j [н+р
[кислота] bi j i j
Система К при 20 °C
НО~ + Н+ «=> Н2О..................0,86-10-и (/св)
НОСГ + Н+ Н2О2..................1,5-10“12
СвН5О~ + Н+ 4=2= . С6Н5ОН............1,28- 10-1°
С1О~ + Н+ нею..................2,95 • 10-8 (при 18 СС)
Очевидно, для того, чтобы иметь право называть эти анионы
«катализаторами дегазации», едва ли могут быть выдвинуты какие-
либо другие соображения, кроме их поляризующего воздействия на
молекулу ОВ.
Из сравнения констант диссоциации видно, что в этом ряду
хлорноватистая кислота обладает наибольшей, а вода наименьшей
кислотностью. Основность же падает от ОН- к С1" (см. табл. 33,
стр. 300).
Если же этот факт сопоставить с реальным временем дегазации
(см. т. 1, раздел 7.3.3.3 и 7.5.5.3), то обнаруживается некоторое про-
тиворечие. Дегазация зарина перекисью водорода происходит в
50 раз быстрее, чем водой. Фенол ускоряет гидролиз ДФФ при-
мерно в 10 раз. Причину этого следует искать, как это уже указы-
валось вт. 1, в различном поляризующем действии анионов. При
желании обобщить механизм действия катализирующих ионов
удается лишь установить, что во всех реакциях вначале с большой
скоростью возникают промежуточные соединения, реагирующие за-
тем с ионами гидроксила или с водой. Для реакционного процесса
такого типа характерен катализ промежуточными продуктами. При
этом на скорость реакции влияет концентрация каталитически дей-
ствующих анионов и ионов гидроксила. Дегазация ОВ типа зарин
гипохлоритами должна протекать, например, при значении pH 10
и концентрации активного хлора не менее 0,5%.
В практике дегазации указанные катализаторы применяются
главным образом для обработки кожных покровов и военной тех-
ники.
При дегазации кожных покровов эти вещества дают возмож-
ность работать в области pH 7—8, что исключает агрессивное дей-
ствие их на кожу человека. Различные производные фенола, внутри-
комплексные соли меди (см. раздел 12.2.6.6) или перекись водорода
получили широкое распространение в качестве компонентов инди-
видуальных химических пакетов.
308
Для дегазации военной техники особенно важно каталитическое
действие гипохлоритов, в результате чего достигается достаточная
скорость дегазации ОВ типа зарин даже при температурах ниже
нуля.
12.2.5. Сульфид натрия и другие
сернистые щелочные соединения
Технический сульфид натрия содержит 60—62% Na2S, а относи-
тельно чистый кристаллический продукт состава Na2S-9H2O— от
30 до 32%. В зависимости от степени чистоты вещество может быть
бесцветным или окрашенным в желтый цвет. Желтая окраска обу-
словлена образованием под действием кислорода воздуха поли-
сульфидов, а также примесями железа. Продукт гигроскопичен и
пахнет сероводородом, который образуется в результате вытесне-
ния его из соли углекислотой воздуха (H2S сильный яд!):
Na2S + СО2 —> Na2CO3 + H2S
Сульфид натрия находит разнообразное применение в ряде
производств, например как депилятор в кожевенном производстве.
Транспортируют и хранят сульфид натрия в гофрированных жестя-
ных барабанах. Он очень хорошо растворим в воде. Например, при
20°C в 1 л воды растворяется 186 г Na2S. Его водные растворы
гидролизуются с образованием едкого натра и гидросульфида на-
трия:
Na2S + Н2О +=> NaHS + NaOH
Состав 1 М раствора Na2S следующий (в .моль)9:
s2- HS— н2з н+ он-
0,09 0,91 1,3 10-7 1,3-10-н 0,91
Плотность 5%-ного раствора Na2S равна 1,056 г!см\ Концент-
рированные растворы Na2S в герметичной упаковке сохраняются
до двух месяцев. В этиловом спирте сульфид натрия растворим не-
сколько хуже, чем в воде, и подвергается алкоголизу:
Na2S + С2Н8ОН NaOC2H6 + NaHS
Сульфид натрия агрессивно действует на кожные покровы чело-
века, он вызывает образование темных пятен. Текстильные мате-
риалы при действии растворов сернистого натрия -также разру-
шаются. Поэтому при обращении с- сульфидом натрия следует при-
менять те же меры предосторожности, что и в случае едкого
натра.
Высокая основность сульфид-иона не может быть использована
для дегазации ОВ типа зарин из-за большого объема этого иона.
То же относится, очевидно, и к реакциям с V-газами. Вместе с тем
значительная щелочность его растворов дает возможность приме-
нять сульфид натрия анало'гично едкому натру для дегазации
309
чувствительных к гидролизу эфиров фосфорной и фосфоновой КИС-
ЛОТ. В реакциях с ипритом пространственные затруднения не на-
блюдаются. Высокая нуклеофильность иона сульфида (см. табл. 33)
приводит в этом случае к более полной дегазации, чем при приме-
нении NaOH (течение реакции см. т. 1, стр. 159). Для эффективной
дегазации азотистого иприта растворами Na2S необходимо рабо-
тать при температуре 80—90°C.
Высокая нуклеофильность сульфид-иоиа делает его пригодным
для дегазации ОВ раздражающего типа, таких, как хлорацетофе-
нон или мышьяксодержащие вещества. В этом случае в качестве
растворителя для приготовления растворов используют спирты.
Для дегазации чувствительных поверхностей кожи сульфид натрия
не пригоден. Вместо сульфида иатрия можно применять сульфид
калия.
Кроме сульфидов для дегазации можно использовать и другие
соединения серы, например:
О
_ II .
S—S—О S—алкил S—арнл
О
тиосульфат тиолят тиофеиолят
В этом ряду практическое значение имеет тиосульфат натрия
ЫагЗгОз. Он-используется, главным образом, для дегазации азоти-
стого иприта.
Высокая нуклеофильность названных соединений видна при
сравнении с аналогичными кислородсодержащими (см. сс1.,
стр. 162). Так, для реакций н-бутилбромида в этиловом спирте с
бутилат-, тиофенолят- и бутилмеркапто-аиионами при 25 °C полу-
чены следующие величциы относительных скоростей — 1:1260:1830.
Так как основность серусодержащих реагентов ниже, чем алко-
голятов, эти данные еще раз доказывают особое значение поляри-
зуемости. Высокая нуклеофильность сернистых реагентов связана
с легкой поляризуемостью серы. Больший, по сравнению с кисло-
родом, атомный объем серы является причиной более легкого сме-
щения электронов в этих соединениях, что дает выигрыш энергии.
На основании приведенного примера следует ожидать, что тиоляты,
тиофенолят и тиосульфат так же пригодны для'дегазации, как и
сульфиды. Они не вступают в реакции нуклеофильного замещения
с ОВ типа зарин, но очень быстро разрушают связь С—С1, напри-
мер, в иприте.
Ни тиоляты, ни тиофеноляты не нашли до настоящего времени
применения в практике дегазации. Это происходит возможно потому,
что они не являются универсальными дегазирующими средствами и,
кроме того, их производят-значительно в меньшем количестве, чем
кислородсодержащие соединения. Еще одним недостатком можно
считать то, что продукты гидролиза этих соединений (тиол и тио-
фенол) обладают чрезвычайно неприятным запахом.
310
12.2.6. Аммиак и амины
12.2.6.1. Общие сведения. В азотистых основаниях азот имеет
степень окисления 3 и, кроме того, свободную пару электронов. Из
всего класса этих соединений для дегазации представляют интерес
преимущественно аммиак, первичные и вторичные амины. Эти ве-
щества характеризуются наличием в молекуле одной или несколь-
ких связей N—Н.
В соответствии с положением азота в периодической системе
элементов эта связь менее поляризована, чем, например, связи
И—С1 или Н—S. Поэтому протоны могут быть оторваны от азота
только при особых условиях. Вместе с тем полярность связи N—Н
такова, что атомы водорода аммиака, а также первичных и вторич-
ных аминов способны образовывать водородные связи. Это прояв-
ляется в ассоциации молекул
в- в+ в- в+
N—H..-N—Н
что подтверждается тем, что амины имеют более высокие темпера-
туры кипения, чем сопоставимые углеводородные соединения.
Существенное влияние оказывают амины на реакции, в резуль-
тате которых получаются анионы. Эти анионы сольватируются
молекулами аминов в результате образования водородных связей:
в- б+ в+ 6-
N—Н- • -[анион-]- • -Н—N
Вызванная сольватацией стабилизация аниона затрудняет об-
ратную реакцию и способствует замещению.
В том же направлении действует способность аммиака и аминов
связывать катионы или протоны
H+4-NH3 [Н—NH3]+
и образовывать с кислотами соли. Ион аммония NHJ является
простейшим из большого числа комплексных ионов, которые могут
образоваться вследствие основных свойств аммиака.
Водные растворы аммиака и аминов имеют щелочную реакцию,
что объясняется их способностью связывать протоны:
RNH24-H2O RNHJ + OH"
NH3 + H2O NHt + OH"
Основность азотсодержащих оснований уменьшается в следующей
последовательности:
R2NH > RNH2 > R3N > NH3
Приведенные в табл. 34 величины рКь азотсодержащих основа-
ний для равновесной реакции
В + Н2О ВН+ + ОН"
311
рассчитывают по следующей формуле:
_ [ВН+] [ОН"]
Л и — 11 --1 . 1 "
Чем ниже значение р/(ь, тем выше основность амина
(рКь = - 1g Кь).
Таблица 34. Значение рКь различных азотсодержащих оснований
(сс.12, т. XI/1)
Основание РКь РКа Основание PKb P*a
NH3 4,73 9,3 N(CH2CH2OH)3 6,23 7,8
h2n-ch3 3,36 10,4 2,9 H,1
h2n—сн2—СН2ОН 4,56 9,4 O~NH2 9,24 4,8
Значения р/(ь дают возможность оценить способность аминов
повышать концентрацию ионов гидроксила в водных растворах, а
также притягивать протоны.
Амины обладают способностью связывать катионы тяжелых
металлов с образованием хелатных или внутрикомплексных соеди-
нений, у которых место центрального атома занимают катионы, а
аддендами служат молекулы органических соединений, имеющие
по крайней мере две комплексообразующие группы. Например:
СН2—СН2
H2N^ ^nh2
\u2+
Различные внутрикомплексные соединения меди являются пре-
восходными катализаторами гидролиза ОВ типа зарин.
Исходя из описанных свойств, возможность использования ам-
миака, и особенно аминов, определяется следующим:
а) способностью этих соединений в водных растворах повышать
pH, что благоприятствует гидролитическому разложению ОВ;
б) сольватацией аминами выделяющихся при дегазации анио-
нов, что оказывает влияние на скорость дегазации;
в) способностью внутрикомплексных солей аминов с катионами
вызывать катализ гидролиза ОВ типа зарин (см. также раздел
12.2.6.6).
12.2.6.2. Аммиак. Аммиак NH3 представляет собой бесцвет-
ный газ, обладающий резким запахом; его температура кипения
—33,4 °C. Его производят в больших количествах в промышленном
масштабе.
312
Основное применение аммиак находит в производстве удобре-
ний, а также для получения азотной кислоты и различных пластиче-
ских масс. В продажу аммиак поступает в сжиженном виде (в бал-
лонах под давлением 6 или 12 ат) или в виде водных растворов
(в стеклянных сосудах).
Водные растворы аммиака (аммиачная вода, нашатырный
спирт) обычно содержат 25% NH3 (при 20 °C в воде растворяется
34,2%, а при, 40 °C — 23,7% NH3). Часть растворенного газа реаги-
рует с водой, образуя гидроокись аммония:
NH3 4- Н2О NH4OH
Однако равновесие этой реакции сдвинуто влево (pH 1 н. рас-
твора аммиака равно 11,77).
Концентрированные растворы аммиака раздражают кожные по-
кровы человека, а при содержании в воздухе 0,5 г/м3 паров амми-
ака воздух делается опасным для вдыхания. Запах аммиака ощу-
щается уже при концентрации 0,04 г/м3. Смеси аммиака с воздухом
взрывоопасны в пределах 19—25% NH3.
Аммиак применяется в различных способах дегазации. Для
обезвреживания поверхностей военной техники, зараженных ОВ
типа зарин, применимы 15—25%-ные водные растворы аммиака,
содержащие, кроме того, 5% моноэтаноламина. Моноэтаноламин
повышает скорость дегазации (влияние сольватации и основности),
а также понижает температуру замерзания раствора. Такую дега-
зирующую жидкость можно применять во всех климатических усло-
виях. Ее преимущество по сравнению с растворами едких щело-
чей состоит в том, что она меньше, чем последние, разрушает раз-
личные покрытия.
В то время как едкие щелочи можно удалить с обработанных
ими поверхностей только тщательным • промыванием, аммиак и
амины настолько летучи, что при сравнительно высокой темпера-
туре самопроизвольное испарение приводит к полному удалению
остатков дегазирующих средств. Так как пары аммиака почти не
разрушают текстильные материалы и легко удаляются простым
проветриванием, то кроме дегазации поверхностей аммиак приме-
ним также и для дегазации обмундирования. В этом случае дега-
зация производится в дегазационных камерах смесью аммиака и
водяного пара. Камеры могут находиться на специальных машинах;
однако для проведения дегазации могут быть использованы и дру-
гие подручные средства.
Принцип дегазации в камерах состоит в том, что при пропуска-
нии через концентрированный раствор аммиака или бикарбоната
аммония перегретого водяного пара последний насыщается аммиа-
ком, и образовавшаяся паро-аммиачная смесь дегазирует заражен-
ное обмундирование в камере. Процесс дегазации аналогичен про-
цессу, происходящему при кипячении, и основан на гидролитиче-
ском разложении ОВ при высокой температуре (см. раздел 12.2.3).
313
Аммиак лишь нейтрализует освобождающуюся кислоту (например,
НС1 или HF) и тем самым предохраняет дегазируемые материалы
от разрушения.
12.2.6.3. Бикарбонат аммония. Бикарбонат аммония NH4HCO3
представляет собой белую кристаллическую соль (в нем содержит-
ся и некоторое количество карбамата аммония). Его растворимость
в воде при 20 °C составляет 17,8%, а при 60 °C — 37,2%. Разло-
жение соли на NH3, СО2 и НгО начинается уже при 33 °C. В выде-
ляющейся воде расплываются кристаллы соли. Такое разложение
можно предотвратить герметичной упаковкой и поддержанием тем-
пературы хранилища ниже 20 °C.
Как уже было указано, для получения паров аммиака вместо
его растворов можно, использовать бикарбонат.аммония. Это видно
из реакции гидролиза этой соли в воде и является следствием вы-
сокой летучести освобождающегося аммиака:
NH4HCO3 + H2O nh4oh + h2co3
NH4OH NH3 4- H2O
H2CO3 CO2 + H2O
При термическом разложении бикарбоната аммония водяным
паром выделяется также и двуокись углерода, которая, однако, не
мешает нейтрализации таких кислот, как HF или НС1, образую-
щихся при дегазации зараженного обмундирования.
12.2.6.4. Продукты конденсации аммиака и гидроксиламина.
Аммиак и другие азотсодержащие основания, имеющие не менее
двух атомов водорода при азоте, дают в результате реакции с кар-
бонильными соединениями продукты, ряд которых имеет значение
для специальных областей дегазации или используется как анти-
доты. Таким продуктом, в частности, является продукт конденсации
аммиака и формальдегида, гексаметилентетрамин (уротропин)
2NH, + енс^
3 хн
Он представляет собой белые, хорошо растворимые в воде кри-
сталлы и находит применение в дегазации, особенно для обработки
кожных покровов человека сразу после заражения ипритом (см.
т. 1, стр. 164).
Ряд веществ, представляющих интерес как антидоты против ин-
гибиторов холинэстеразы, получается при взаимодействии гидр-
314
оксиламина с карбонильными соединениями. При .взаимодействии
с альдегидами или кетонами получаются оксимы:
R\ R\
V=O + h2noh —► ^C=N—он + H2O
R'/ Rz/
Гидроксамовые кислоты могут быть получены из гидроксил-
амина и эфиров карбоновых кислот:
R— +H2NOH —► R—с' + нх
\nhoh
/он
R-с; R-C'
XNHOH "ЧюН
Действие этих соединений, подобно гидроксиламину, основано
на реакционной способности функциональной группы =N—О—Н
(см. т. 1. стр. 281).
12.2.6.5. Общие свойства ал кил аминов. Свойства аминов зави-
сят как от числа атомов углерода в радикале, так и от структуры
молекулы. С увеличением молекулярного веса амина ослабевает
типичный, напоминающий аммиак запах и уменьшается его рас-
творимость в воде. Низкомолекулярные соединения (различные
метиламины и этиламин) при нормальных условиях представляют
собой газы, а амины со средним молекулярным весом (пропиламин
или диэтиламин) являются жидкостями. Температуры вспышки
низших ш средних аминов сравнительно низки. Пары аминов обра-
зуют с воздухом взрывчатые смеси. Амины сравнительно чувстви-
тельны к действию окислителей.
Все амины более или менее раздражающе действуют на кожу
человека. У людей с повышенной чувствительностью может воз-
никнуть экзема.
Многие амины широко используются в качестве растворителей
и исходных продуктов для получения поверхностно-активных
веществ, красителей и др.
Об эффективности амина как дегазирующего вещества судят
по его основности (см. табл. 34), а также по его способности соль-
ватировать анионы или полярные молекулы.
Водные растворы аминов пригодны для дегазации только ОВ
типа зарин. В этих- реакциях принимают участие непосредственно
амины в виде свободных оснований, а также освобождающиеся
гидроксильные ионы, что зависит от растворимости амина в воде и
величины р/(а. которая у аминов может быть выше, чем у аммиака.
В качестве примера можно назвать моноэтаноламин (см. табл. 34).
Благодаря значению, которое это вещество приобрело в практике
дегазации, оно будет в последующем рассмотрено более подробно.
315
Влияние, которое амин в виде свободного основания оказывает
на течение той или иной реакции, может быть оценено также с по-
мощью рКа. Как уже упоминалось в т. 1 стр. 160, 282, в безводной
среде амины реагируют непосредственно с такими ОВ, как иприт
и ДФФ. Так как рКа, приведенные в табл. 34, относятся к водным
средам, то в данном случае они не могут непосредственно характе-
ризовать эффективность аминов. Однако, исходя из того, что рКа
амина характеризует электронную плотность на атоме азота, можно
все же с некоторым ограничением судить по ним и об эффектив-
ности аминов в неводных средах. Очевидно, что высокая основ-
ность (высокие рКа) в водной среде означает и высокую эффектив-
ность в других средах.
Другим свойством аминов, очень важным для дегазации,
является их способность сольватировать анионы и полярные моле-
кулы. Это свойство для некоторых соединений можно связать так-
же с рКа на основании следующих .соображений.
Сольватация возникает в результате образования водородных
связей. Последние обладают тем большей прочностью, чем более
полярна связь N—Н. Полярность этой связи зависит от электронной
плотности на атоме азота, а высокая электронная плотность озна-
чает слабую тенденцию отдавать протоны и, соответственно, малую
склонность к образованию водородных связей. Так как высокая
основность идентична слабой тенденции к отдаче протонов, то спо-
собность аминов к образованию водородных связей снижается при
увеличении рКа. Здесь имеется противоречие, заключающееся в
том, что высокая основность, необходимая для нуклеофильного за-
мещения, связана с уменьшением сольватационной способности
амина. Это затруднение можно обойти, применяя для дегазации
смесь аминов с сильными нуклеофильными реагентами. Вообще
амины слишком дороги для того, чтобы использовать их для дега-
зации как таковые. В частности, особенно пригодны их смеси с
едкими щелочами и спиртами (ср. стр. 317, жидкость DS-2).
Как мы уже знаем, из спиртов и щелочей образуются алкого-
ляты, основность которых высока. Такие смеси, обладающие одно-
временно высокой основностью и хорошей сольватирующей способ-
ностью, пригодны для дегазации даже таких ОВ, которые в водных
средах очень устойчивы к нуклеофильным атакам. При рассмотре-
нии процессов, происходящих при дегазации различных ОВ этими
дегазирующими жидкостями, возникает вопрос о том, как влияет
полярность растворителей на скорость нуклеофильного замещения.
С возрастанием полярности растворителей возрастает их склон-
ность к образованию водородных связей; если ОВ обладает высо-
кой полярностью, то скорость реакции уменьшается. С такими от-
ношениями столкнулись при дегазации V-газов 8.
При составлении дегазирующих растворов нужно учитывать не
только полярность аминов, но и спиртов. Исходя из указанных
выше теоретических соображений, можно в общих чертах оценить
возможную эффективность дегазирующих смесей, составленных из
316
аминов и алкоголятов. Очевидно, что скорость дегазации будет
снижаться в ряду иприт, V-газы и отравляющие вещества типа
зарин.
Моноэтаноламин (2-аминоэтанол, H2NCH2—СН2ОН) представ-
ляет собой бесцветную или желтоватую жидкость с характерным
запахом, т. кип. 171 °C, т. пл. 10°С, плотность 1,02 г!см3.
Это вещество применяется главным образом в качестве исход-
ного продукта для получения поверхностно-активных веществ. Не-
которые сведения о применении моноэтаноламина в водных дега-
зирующих жидкбстях были приведены в разделе 12.2.6.2, а теоре-
тические соображения о пригодности аминов для дегазации — в
этом разделе (см. стр. 315).
Пригодность вещества для использования в водных дегазирую-
щих жидкостях определяется его основностью, хорошей раствори-
мостью в воде и свойством понижать температуру замерзания
раствора.
Моноэтаноламин применим и в неводных системах. Так, напри-
мер, его можно добавлять к хлорированным углеводородам, таким,
как трихлорэтилен или дихлорэтан, которыми пользуются для
дегазации обмундирования экстракцией. При этом одновременно
с экстракцией ОВ типа зарин идет и их химическая дегазация.
Моноэтаноламин мало растворим в алифатических углеводородах.
Дегазирующая жидкость DS-2. Описание состава этой дегази-
рующей жидкости (дегазирующий раствор 2) приведено в амери-
канском патенте 13. Она состоит из 2 вес. % NaOH, 28 вес. % моно-
метилового эфира этиленгликоля (СН3ОСН2СН2ОН, метилцелло-
зольв), 70 вес.% диэтилентриамина (H2NCH2CH2NHCH2CH2NH2)
или этилендиамина (H2NCH2CH2NH2).
Раствор перевозят в виде готовой дегазирующей жидкости, он
должен применяться в неразбавленном виде. Преимущество рас-
твора заключается в универсальности его действия даже при низ-
ких температурах. Обработка им требует незначительного механиче
ского воздействия, так как все ОВ в нем хорошо растворимы. Он
не разрушает или весьма незначительно разрушает резину, лако-
красочные покрытия и металлы (за исключением алюминия).
Эта дегазирующая жидкость имеет и некоторые недостатки.
Они заключаются в значительном увеличении вязкости жидкости
при низких температурах, токсичности аминов, высокой стоимо-
сти их по сравнению с другими дегазирующими средствами и,
наконец, перегрузке транспортных средств.
12.2.6.6. Комплексные соединения аминов. В разделах т. 1 —
7.3.3.3 и 7.5.5.3 подробно изложено значение и эффективность при-
менения хелатных комплексов, образуемых медью с различными
аминами, которые ускоряют гидролиз ОВ типа зарин. Эти комплек-
сные соединения находят практическое применение в качестве де-
газирующих средств для кожных покровов. Так, например, при
помощи дегазирующей мази, содержащей комплексное соединение
меди и бис-1,2-(N-диметил амино)-этана, достигается прекрасный
317
эффект дегазации кожных покровов. Состав этой мази должен
иметь pH не выше 8. Получают комплексные соединения меди
простым смешением реагентов. Мази, приготовленные на этой
основе, сохраняют свое действие практически неограниченно долго.
Однако так как V-газы не дегазируются медными внутрикомплекс-
ными солями, то в последние годы эти соединения утратили свое
значение и являются лишь вспомогательными средствами. В со-
временных условиях требуются средства, обладающие универсаль-
ным действием.
12.3. ДЕГАЗАЦИЯ ОКИСЛЕНИЕМ И ХЛОРИРОВАНИЕМ
12.3.1. Общие сведения
Окисление нельзя рассматривать только как реакцию с кислородом.
В более широком смысле можно сказать, что вещество окисляется,
если оно отдает электроны; одновременно с процессом окисления
всегда идет и процесс восстановления — прием электронов окисли-
телем, т. е. весь процесс в целом, называется окислительно-восста-
новительным процессом. Таким образом сущность окислительно-
восстановительного процесса заключается в переносе электронов
от восстановителя (или окисляемого вещества) на окислитель (или
восстанавливаемое вещество). Стремление атома в молекуле при-
тянуть к себе электроны характеризуется положением атома в
шкале электроотрицательностей. При рассмотрении этой шкалы
становится ясной превалирующая роль галоидов и кислорода как
окислителей или составной части окислителей (см. стр. 286, табл. 32).
Однако по значениям электроотрицательности формально
нельзя судить об окислительном действии элементов или каких-либо
их ионов. Уже тот факт, что кислород в обычных условиях суще-
ствует в относительно неактивной молекулярной форме, является
подтверждением этому. Точное суждение об окислительной способ-
ности элементов или соединений в водной среде возможно по зна-
чению окислительно-восстановительного потенциала. Эти величины
получают, измеряя напряжения между двумя электродами, из ко-
торых один соединен с восстановителем, а другой с окислителем
или изготовлен из него.
При применении водородного электрода (платиновый электрод,
омываемый газообразным водородом) и соблюдении стандартных
условий (при 18—25 °C, давлении 1 ат и концентрации или актив-
ности растворенных компонентов, равной 1) получают нормальные
потенциалы Ео.
Нормальные потенциалы, представленные в табл. 35, распола-
гаются в последовательный ряд, аналогичный шкале электроотри-
цательности. Для дегазационной практики больший интерес пред-
ставляют не элементы, а соединения с высоким окислительным
потенциалом. На основании закона действия масс и из данных
табл. 36 следует, что повышение концентрации водородных ионов
318
Таблица 35. Нормальные потенциалы некоторых элементов4
Восстановленная Окисленная форма форма £*о> & (при 18 °C)
СЧ я, + 1 04 м « 4- * + 4- 4-0 ^7 g йТ 1 « Г* Ur —•* CN оо CQ О U. ТИИШИ! 1 1 1 1 1 1 е* >—< и—мГт. оэ О CQ О й сч сч —0,51 +0,40 +0,54 + 1,07 + 1,36 +2,85
приводит к увеличению окислительной способности кислородсодер-
жащих окислителей. Этот факт можно объяснить тем, что при пере-
ходе атома, связанного с кислородом, в более низкую валентность
Таблица Зв. Нормальные потенциалы важнейших
окислительно-восстановительных реакций 4
м Восстановленная форма Окисленная форма Ео, в (при 18 °C)
NO + 2H2O 5= * NOJ + 4Н+ + Зе +0,96
г + нго ± НЮ + Н++2е +0,99
Вг' + Н2О 5= ♦ НВгО + Н++ 2е + 1,33
Вг"т-ЗН2О ♦ ВгО; + 6Н+ + 6е + 1,42
СГ + ЗН2О 4= ± С1О; + 6Н+ + 6е + 1,47
сг + н2о ;= ± НСЮ + Н++2е + 1,49
Mn2++ 4Н2О ♦ MnOJ4-8H++5e + 1,52
2Н2О 5= t Н2О2 + 2Н+ + 2е + 1,77
2SO*" 5= ± S2O|” + 2е +2,05
о2 + н2о ± О3 + 2Н*4-2е +2,07
кислород освобождается, образуя воду. Количественно это явление
можно описать при помощи уравнения Нернста.
Например, для перекиси водорода получаем:
Е = Ео +
0,059 [Н2О2] [Н+]2
п g [Н2О]
где Ео — нормальный потенциал; Е — потенциал при заданной концентрации;
п — число электронов, участвующих в окислительно-восстановительном процессе;
0,059 — константа.
В нейтральной среде для 1 М раствора Н2О2 получаем:
£= 1,77 + 1g 1 (10~Т -= 1,77 + 0,029 (-14) = 1,36
319
Устойчивость кислородных кислот галоидов наглядно иллюст-
рируется значениями их окислительно-восстановительных потен-
циалов при 25 °C в равновесии со свободным хлором 6:
*/2С124-Н2О 5=^ нею 4-Н+ 4-е
>/2С124-2Н2О 5=^: НСЮ24-ЗН+ 4-Зе
*/2 С12 4- ЗН2О «=> НС1О3 4-5Н+4-5е
*/2 С12 4- 4Н2О ==> НСЮ44-Н+ 4- 1е
(-1,63 в)
(-1,63 в)
(-1,47 в)
(-1,34 в)
Как видно из величин окислительно-восстановительных потен-
циалов, хлораты и перхлораты непригодны в качестве дегазирую-
щих веществ, хлориты могут быть использованы условно, а гипо-
хлориты — пригодны безусловно. Исходя из изложенного, можно
подобрать ряд веществ окислительного действия, принципиально
годных для дегазации. Например: хлор, хлорноватистая кислота и
ее соли, хлористая кислота и ее соли, перманганаты, перекись во-
дорода, персульфаты, озон, фтор.
Весьма высокая окислительная способность свободного фтора
представляет чисто теоретический интерес, так как с этим газом
настолько трудно обращаться и он настолько агрессивен, что не мо-
жет иметь значение в качестве дегазирующего вещества.
Совершенно по-иному ведут себя хлор и озон. Оба газа широко
применяются для очистки воды, а потому имеют определенное зна-
чение и для ее дегазациии (из хлора при этом образуется хлорнова-
тистая кислота). В результате сопоставления свойств различных
окислителей можно заключить, что наиболее пригодными для дега-
зации военной техники и местности являются соединения хлора.
Химическая промышленность располагает достаточной сырьевой
базой для их широкого производства. Они сравнительно легко по-
лучаются, а часть из них вполне устойчива при хранении.
Перманганаты и перекиси имеют значение только для специаль-
ных областей дегазации, особенно для дегазации кожных покровов,
причем недостаточная универсальность действия перманганатов
еще более ограничивает возможность их применения.
12.3.2. Гипохлориты
12.3.2.1. Общие сведения. Гипохлориты — соли хлорноватистой
кислоты. Эта кислота устойчива только в разбавленных водных
растворах (не выше 1%-ных). Ее получают пропусканием хлора
или окиси хлора (С12О) через воду или вытеснением из ее солей бо-
лее сильной кислотой. Из ее производных в качестве дегазирующих
средств находят применение главным образом хлорная известь,
гипохлориты кальция и натрия.
Гипохлориты являются дегазирующими веществами универсаль-
ного действия. Как уже было указано в разделе 12.2, они оказы-
вают каталитическое действие на гидролиз ОВ типа зарин, а высо-
кая окислительная способность хлорноватистой кислоты (см. 12.3.1)
имеет значение для дегазации иприта и V-газов.
320
Окислительная Способность Гипохлоритов й меньшей степени
зависит от наличия свободного аниона С1О~, а в большей степени
связана с образованием хлорноватистой кислоты вследствие гидро-
лиза гипохлоритов
NaClO + H2O NaOH + HOCl
или разложения их другими кислотами:
NaClO + Н+ Na+ + HOCl
Этот факт находится в соответствии с описанной ранее способ-
ностью водородных ионов повышать окислительный потенциал кис-
лородсодержащих кислот. Сравнение хлорноватистой и хлорнова-
той кислот указывает на то, что водородные ионы повышают потен-
циал независимо от того, получаются ли они в результате диссоциа-
ции сильной кислоты или связаны с анионами слабых кислот. Кон-
станта диссоциации хлорноватистой кислоты равна 4-10~8. Эта кис-
лота значительно слабее хлорноватой кислоты, которая в водных
растворах диссоциирована практически полностью. Так как способ-
ность -хлорноватистой кислоты к диссоциации очень низка, то ионы
водорода, необходимые для реакций окисления, как правило, свя-
зываются в растворе гипохлорит-ионами:
С1О“ + Н+ НОС1
Однако это никак не ограничивает окислительного действия
хлорноватистой кислоты. Важно, чтобы водородные ионы, необхо-
димые для образования воды, всегда присутствовали в реакцион-
ной среде, а связаны ли они с гипохлорит-ионами или находятся
в свободном состоянии, — значения не имеет.
Хлорноватистая кислота не только очень реакционноспособна, но
и (что аналогично) очень неустойчива. Она способна самоокислять-
ся, т. е. диспропорционировать:
(1+) (1+) (3+) (1—)
нею 4-нею —> НСЮ2 + НС1
(3+) (1+) (5+) (1-)
НСЮ2 + НСЮ —► НСЮз + НС1
В то время как хлор со степенью окисления 3 и 5 известен толь-
ко в соединениях (хлориты, хлористая кислота, хлораты, хлорная
кислота), а. со степенью окисления —1 представляет собой устой-
чивый хлор-анион (хлориды, соляная кислота), хлор в степени
окисления +1 занимает совершенно особое положение. Он встре-
чается в соединениях (гипохлориты, хлорноватистая кислота, хлор-
амины), но, кроме того, может иногда существовать и в свободном
виде. Возможность диссоциации по схеме
НОС1 Ч=* НО“ + С1+
следует полностью отвергнуть, так как энергия гидратации катио-
она хлора в отличие от водорода Н+очень низка (см. сс.7). Другими
И Зак. 677
321
словами, за счет освобождающейся энергии Гидратации не проис-
ходит стабилизации катиона хлора. В соответствии с вышеприве-
денной константой диссоциация хлорноватистой кислоты протекает
исключительно по уравнению:
НОС1 Н+Ч-С1О'
Однако если хлорноватистая кислота или раствор гипохлорита
приходят в соприкосновение со способными окисляться веществами,
то в этом случае можно ожидать образования катиона хлора. При
этом, видимо, происходит следующая реакция1:
НОС1 + Н+ Н—б—Cl (быстро)
Н
Н—О—С1 С1+ + Н2О (медленно)
Н
Этот механизм снова указывает на роль ионов водорода в реак-
ции окисления.
Количество катионов хлора, которое может быть отдано гипо-
хлоритом или хлорамином, соответствует содержанию активного
хлора. Катион хлора (активный хлор) характеризуется тем, что
он в состоянии принять два электрона:
С1+ + 2е «=> СГ
Молекула элементарного хлора может тоже связать два элек-
трона (по одному каждым атомом):
С1—С1+2е 2СГ
В поведении элементарного хлора и соединений, содержащих
активный хлор, существует известное сходство, которое заключает-
ся в том, что многие окислительные процессы и реакции электро-
фильного замещения с участием элементарного хлора могут быть
объяснены только в том случае, если допустить образование ка-
тиона хлора. В простейшем случае этот процесс следует представ-
лять себе таким образом:
С12 ^=> СГ+ С1+
Отсюда следует, что процессы, обычно называемые хлорирова-
нием, в принципе являются реакциями с хлор-катпоном, исключая
хлорирование атомарным хлором (см. раздел 12.3.5). Поэтому не
так уж неверно и окислительные реакции гипохлоритов частично
рассматривать как реакции хлорирования. Примером таких реак-
ций являются — замещение водорода при атоме углерода или
дегазация иприта хлорирующими агентами (см. т. 1, стр. 162). При
этом, особенно в водных средах, просходят также и процессы, ко-
торые не совсем точно могут быть отнесены к процессу хлорирова*
322
ния. Так, например, образование 2,2-дихлордиэтилсульфоксида мо-
жет происходить прямым окислением (действием активного кисло-
рода) :
zCHjCH2Cl XHjCHjCl
О + —> O=S\
\ch2ch2ci ^СН2СН2С1
Вместе с тем оно в равной степени может произойти и под воз-
действием хлор-катиона (см. сс.5, стр. 135):
Cl+ +S(CH2CH2C1)2 —> Cl—S(CH2CH2C1)2 (1)
Cl—S(CH2CH2C1)2 + OH’ —> O=S(CH2CH2C1)2 + HC1 (2)
Принимая во внимание, что хлорноватистая кислота легко от-
дает элементарный кислород
НС1О —> НС1 + О
следует считать, что и в данном случае происходит прямое окисле-
ние иприта (реакция 1, см. сс.6, т. 5, стр. 504).
Действие тяжелых металлов (особенно кобальта, никеля, меди,
марганца, железа и их солей), а также воздействие света может
ускорить отдачу кислорода хлорноватистой кислотой и ее солями.
Как известно, кислород in statu nascendi обладает очень сильными
окислительными свойствами. Он может принять два электрона, до-
полнив свою электронную оболочку до оболочки инертного газа.
Однако непосредственное окисление может быть вызвано не толь-
ко атомарным кислородом. Известную роль в этом процессе
может играть и ангидрид хлорноватистой кислоты, окись хлора.
Реакция между окисью хлора и водой, приводящая к образова-
нию хлорноватистой кислоты
С12О+Н2О 2НОС1
в значительной мере сдвинута вправо, тем не менее окись хлора,
образующаяся при обратной реакции, вследствие своей высокой
реакционной способности все же принимает участие в процессе де-
газации. Убыль окиси хлора постоянно пополняется за счет сдвига
реакции влево.
Окись хлора считается более сильным окислителем, чем хлорно-
ватистая кислота5; это подтверждается тем, что жидкая окись хло-
ра при переливании разлагается со взрывом.
Таким образом, подытоживая все сказанное, можно считать, что
гипохлориты окисляют ОВ как с помощью хлор-катиона, так и пу-
тем непосредственной передачи кислорода (прямое окисление).
Важнейшим исходным веществом для получения гипохлоритов
является хлор. В связи с тем, что хлор используется и в полевых
способах получения растворов гипохлорита, сначала будут кратко
описаны его свойства.
12.3.2.2. Хлор. Хлор при нормальных условиях — газ зелено-
ватого цвета с резким запахом. Он в 2,5 раза тяжелее воздуха и
И* 323
довольно хорошо растворим в холодной воде (в одном объеме во-
ды при 20°C растворяется 2,3 объема хлора). Жидкий хлор кипит
при —34,05 °C. В баллонах со сжиженным хлором в зависимости
от температуры создается соответствующее давление:
Температура, °C Давление» ат Температура, °C Давление, ат
-20 1,8 10 5,1
-10 2,7 20 6,8
0 3,7 30 9,0
Сухой жидкий хлор не корродирует сталь. Влажный хлор, на-
против, очень агрессивен (образование хлорноватистой кислоты’).
Поэтому попадание воды в баллоны с хлором должно быть абсо-
лютно исключено. Вследствие коррозии вентили баллонов «зае-
дают». Строго запрещается открывать вентили с применением силы
или подогреванием на огне. Если такой вентиль невозможно от-
крыть после обработки горячей водой, то баллон следует возвра-
тить на станцию снаряжения.
Хлор поражает легкие. Минимально ощутимая концентрация
хлора равна 0,002 мг/л, раздражающее действие хлора наблюдает-
ся при концентрации около 0,01 мг/л-, концентрации выше 0,1 мг/л
опасны для жизни.
12.3.2.3. Хлорная известь. Хлорной известью называют продукт,
образующийся при действии хлора на гашеную известь (гидро-
окись кальция). Действующее начало хлорной извести — гипохло-
рит кальция. Наряду с ним хлорная известь содержит большее или
меньшее количество непрореагировавшей гидроокиси кальция. Эти
два основные вещества только в незначительном количестве нахо-
дятся в свободном состоянии в виде кристаллов; обычно они обра-
зуют друг с другом, а также с хлористым кальцием и водой двой-
ные, тройные и более сложные соли, обладающие характерной
кристаллической структурой.
Существовавшее ранее представление о хлорной извести как о
соединении типа С1—Са—ОС1 ныне полностью отвергнуто. Хлорная
известь не содержит и свободного хлористого кальция. Если бы он
присутствовал в продукте, то гигроскопичность хлорной извести
была бы более сильной, чем это есть на самом деле. Гигроскопич-
ность хлорной извести связана в первую очередь с присутствием
двухосновного гипохлорита кальция IV (см. табл. 37).
Состав хлорной извести зависит от способа ее получения. Сле-
дует считать, что гипохлорит кальция находится преимущественно
в форме продуктов IV и III, содержащих в среднем 36—38% актив-
ного хлора. Содержание воды (в основном кристаллической) в
продукте, не подвергавшемся дополнительной сушке, составляет
около 10%. В некоторых непрерывных методах получения хлорная
известь содержит меньшее количество воды. После вакуум-сушки
получается устойчивая и сухая хлорная известь, содержащая около
0,5% влаги. Чем ниже содержание воды в продукте, тем выше со-
держание гидроокиси кальция (16—26%).
324
Хлорная известь обычно содержит также 2—3% связанного.
СаС12 (преимущественно в виде продукта VI), 3—4% СаСОз и 1%
различных окислов (Fe2O3, А12О3, SiO2 и др.).
Таблица 37. Состав хлорной извести
Компонент Формула Кристаллическая форма
I. Гипохлорит каль- ция нейтральный Са(ОС1)2 Очень тонкие пла- стинки непра- вильной формы
II. Гипохлорит каль- ция дигидрат Са(ОС1)2 • 2Н2О Тонкие театраго- иальные пла- стинки
III. Гипохлорит каль- ция дветрети- основиой ЗСа(ОС1)2 • 2Са(ОН)2 • 2Н2О Острые палочки
IV. Гипохлорит каль- ция двухоснов- ной Са(ОС1)2 • 2Са(ОН)2 Г ексагональные . пластинки
V. Гипохлорит каль- ция . четырехос- новной Са(ОС1)2 • СаС12 • 4Са(ОН)2 • 24Н2О Длинные нглы
VI. Хлористый каль- ций однооснов- ной СаС12 • Са(ОН)2 • Н2О Мелкокристалли- ческая масса
VII. Гидроокись каль- ция Са(ОН)2 Гексагональные пластинки
Промышленный способ получения хлорной извести заключает-
ся в хлорировании гашеной извести смесью воздуха с хлором; про-
цесс осуществляется в многополочных камерах методом противо-
тока с перемешиванием продукта на полках.'
Сложный состав хлорной извести (см. табл. 37) позволяет пред-
положить, что при прохождении хлора через гашеную известь про-
ходит множество процессов. Вначале, до образования в реакцион-
ной смеси продукта с содержанием 28% активного хлора, преиму-
щественно протекает следующая реакция 6:
5Са(ОН)2 + 2С12 —► СаС12 • Са(ОН)2 • Н2О + Са(ОС1)2 • 2Са(ОН)2 + Н2О
VI IV
При последующем хлорировании протекают конкурирующие ре-
акции поглощения хлора продуктами IV и VI:
7Са(ОС1)2 • 2Са(ОН)2 + 6С12 —► ЮСа(ОС1)2- >/2Са(ОН)2 +
IV
+ ЗСаС12 • Са(ОН)2 • Н2О + 2Н2О
5СаС12 • Са(ОН)2 • Н2О + 4С12 —► 7СаС12-Н2О+
VI
+ 2Са(ОС1)2 • >/2Са(ОН)2 + 2Н2О
325
Главным образом реагирует продукт IV, продукт VI поглощает
хлор очень медленно. Именно этим обусловлено то, что технический
продукт содержит не более 38% активного хлора. Так как в гото-
вом продукте отсутствует свободный хлористый кальций, то сле-
дует предположить, что СаС12, взаимодействуя с еще непрореагиро-
вавшей Са(ОН)2, образует продукт VI; возможно, что при поглоще-
нии реакционной воды образуются более гидратированные продукты.
Хлорная известь представляет собой белый порошок, обладаю-
щий запахом хлора, насыпной вес 0,6—0,8 кг/л. В воде она плохо
растворима. При растворении остается осадок, состоящий в основ-
ном из нехлорированных продуктов и некоторого количества про-
дуктов, содержащих активный хлор, которые переходят в раствор
только после продолжительного перемешивания с водой. Таким
путем можно получить растворы, содержащие до 15% активного
хлора. Растворы хлорной извести имеют щелочную реакцию.
Хлорная известь относительно неустойчива: при 45 °C в стан-
дартной упаковке она за 50 суток практически полностью теряет
свою активность. При 22,5 °C за то же время содержание активного
хлора падает с 38 до 36—35%. После двух лет хранения хлорная
известь уже совсем непригодна для дегазации. Снижение содержа-
ния активного хлора в хлорной извести происходит еще быстрее под
воздействием влаги воздуха, углекислого газа или'солнечного света.
В присутствии влаги образуются комки, углекислый газ вытесняет
хлорноватистую кислоту и таким образом ускоряет снижение со-
держания активного хлора. После того как было установлено, что
высокое содержание влаги отрицательно влияет на стабильность
хлорной извести, проводилось немало опытов по изготовлению су-
хого устойчивого продукта. Такие продукты, как так называемая
«сухая хлорная известь», способны храниться много лет. Однако
они обладают меньшей реакционной способностью, чем обычная
хлорная известь. Из-за малого содержания влаги стабилизованная
хлорная известь почти не разрушает металлы. Обычно получаемая
хлорная известь, напротив, вызывает сильную коррозию.
Хлорную известь помещают в покрытые изнутри парафиниро-
ванной бумагой деревянные бочки или в покрытые лаком металли-
ческие барабаны и хранят в закрытых, темных, хорошо проветри-
ваемых помещениях при температуре не выше 25 °C и влажности не
выше 20 г/м3. Не следует хранить в одном помещении с хлорной
известью взрывчатые и огнеопасные вещества, аппараты с метал-
лическими частями, баллоны со сжатыми газами и продукты пи-
тания.
При обращении с хлорной известью необходимо защищать гла-
за, чувствительные части кожных покровов и особенно слизистые
оболочки. В связи с тем, что текстильные ткани также в значитель-
ной степени разрушаются, при работе с хлорной известью необхо-
димо надевать защитную одежду.
12.3.2.4. Гипохлорит кальция. Недостатки хлорной извести уже
давно привели к попыткам создать более активный и более устой-
326
йивый при хранении препарат, не вызывающий коррозии металлов.
Чистый гипохлорит кальция Са(ОС1)2 был получен в лаборатории
еще в 1875 г. путем выщелачивания хлорной извести и последую-
щей переработки раствора. Впоследствии во всех странах возника-
ли производства гипохлорита кальция. Продукт выпускался под
разными торговыми названиями и с разным содержанием активного
хлора. Например, концерном «ИГ-Фарбениндустри» был выпущен
нейтральный продукт перхлорон, содержавший 70—80% активного
хлора; в США — препарат High Test Hypochlorit (НТН), в
Англии — МахсЫог, а в Японии — пархлорин, антипул и ниполит.
Содержание активного хлора в техническом продукте состав-
ляет 60—80%, чаще всего около 70%. Так как гипохлорит-ион по
своему окислительному действию эквивалентен двум атомам хлора
(С1+ может принять два электрона, а С1° — только один), то каче-
ство Са(ОС1)2 определяют по содержанию активного хлора. Моле-
кулярный вес гипохлорита кальция 142,99, вес 4 г-экв хлора со-
ставляет 141,83; отсюда следует, что приблизительное содержание
гипохлорита кальция можно считать равным содержанию актив-
ного хлора. Кроме Са(ОС1)2 в продукте содержится 10—20% не-
прореагировавшего Са(ОН)2, 5—10% СаС12 и до 1% воды.
Как видно из ранее сказанного, продукт состоит не только из
нейтрального гипохлориту кальция, он содержит в более или менее
значительных количествах основные соли (IV и III), в которые
входит непрореагировавшая гидроокись кальция.
Продукты, содержащие в основном соединения IV и III, также
можно называть гипохлоритом кальция. Вещество IV, известное
под названием двухосновной гипохлорит, содержит до 40% актив-
ного хлора, а вещество III, известное под названием дветретиоснов-
ной гипохлорит, содержит не менее 52% активного хлора.
Содержание активного хлора в различных продуктах является
решающим фактором при оценке их рентабельности, так как боль-
шое значение имеют средства, затрачиваемые на их перевозку.
В табл. 38 сравниваются масса и активность хлорной извести, ги-
похлорита кальция и жидкого хлора.
Гипохлорит кальция получают путем пропускания жидкого хло-
ра через кашицу гидроокиси кальция. При этом протекает сле-
дующая реакция:
2Са(ОН)2 + 2С12 —► Са(ОС1)2 + СаС12 + 2Н2О
Как видно из табл. 37, получаемые соединения могут давать
с непрореагировавшей гидроокисью кальция основные соли типа
III, IV и VI (соль V имеет второстепенное значение). Так как соль
IV наиболее трудно растворима, она первой выпадает в осадок
(при температуре ниже 35 °C выпадает и соль VI). Если на этом
остановить хлорирование и отфильтровать осадок, то в качестве
главного продукта получается двухосновной гипохлорит кальция IV
(базогрелит).
327
Таблица 38. Содержание активного хлора в различных
хлорсодержащих продуктах
Продукт Масса (брутто), г Эквивалентное, количество активного хлора, г
Хлориая известь 3,5 1
Высокопроцентный гипохлорит кальция 1,9 1
Жидкий хлор в стальных баллонах 2,0 1
При желании получить продукт с большим содержанием хлора
хлорирование следует продолжить, осадок при этом переходит в
раствор (при температуре 50°C осадок вообще не образуется). По-
сле поглощения хлора в количестве, отвечающем продукту III, вы-
падают его кристаллы, которые при продолжении хлорирования
постепенно переходят в раствор. В щелоке должно содержаться
примерно 0,5% Са(ОН)2, чтобы не образовывалась свободная хлор-
новатистая кислота, которая будет окислять продукт до хлората.
При охлаждении массы из нее выпадает нейтральный гипохло-
рит кальция. Его отделяют от маточного раствора на специальных
фильтр-прессах высокого давления. Отфильтрованный кек измель-
чают и после предварительной сушки окончательно сушат при раз-
режении.
Основная трудность этого процесса, разработанного в Биттер-
фельде, заключается в отделении продукта от маточного раствора.
Фильтрация продуктов IV и III значительно проще. В других спо-
собах получения это затруднение обходят различными путями,
однако при этом теряются значительные количества хлора.
Вообще говоря, все способы получения гипохлорита кальция
сравнительно сложны. ~
Гипохлориты кальция представляют собой белые порошкообраз-
ные или зерненые продукты, обладающие запахом хлора. Насып-
ной вес 70%-ного (по содержанию активного хлора) гипохлорита
кальция равен примерно 0,9 кг)л\ дветретиосновная соль (продукт
III), как и нейтральные продукты, слабо растворяется в воде. При
изготовлении 10%-ного раствора из продукта III нерастворимый
осадок составляет 10—15%, тогда как при использовании 70%-ного
продукта количество нерастворимых или труднорастворимых про-
дуктов составляет около 6% (в расчете на исходный продукт).
В 10%-ном растворе, приготовленном из 70%-ного гипохлорита
кальция, содержание активного хлора составляет ~6,5%.
Двухосновной*'гипохлорит кальция (продукт IV) растворяется
в воде еще хуже, чем хлорная известь. Нерастворимый осадок со-
ставляет приблизительно 20—25% от массы растворяемого веще-
ства. Устойчивость отдельных продуктов в значительной степени
зависит от их состава. Двухосновная соль гипохлорита кальция го-
раздо более устойчива, чем хлорная известь. Из дветретиосновного
328
гипохлорита при соблюдении правильного режима хранения за
три года утрачивается не бол'ее 8% активного хлора. Для 70%-ных
продуктов установлен срок хранения до 10 лет; однако они очень
чувствительны к влаге.
Поведение высокопроцентного гипохлорита кальция при повы-
шенных температурах в значительной степени зависит от содержа-
ния влаги и размеров расфасовки. При содержании влаги более
2% гипохлорит, содержащий 70% активного хлора, разлагается.
Чувствительность его к влаге можно значительно снизить добав-
лением около 2% окиси кальция. Продукт, содержавший 1,5%
влаги и упакованный в барабан емкостью 50 кг, при 85 °C так
бурно разлагался, что это привело к разрыву барабана. При содер-
жании 2% влаги это явление наступило раньше. В банках емкостью
1 кг продукт выдерживает нагревание до ПО—120 °C.
По-видимому, решающее значение в этих процессах имеет обра-
зование окиси хлора, легко разлагающейся с выделением тепла.
В малых емкостях теплообмен осуществляется легче, в больших же
емкостях за счет теплоты реакции происходит местный перегрев до
температуры разложения, что в конце концов приводит к разложе-
нию всего продукта.
При нормальных температурах 70%-ный гипохлорит кальция
реагирует с органическими веществами (древесные опилки, масло,
дизельное или карбюраторное топливо) не столь энергично, чтобы
это могло стать опасным. При повышенных температурах происхо-
дит бурная реакция вплбть до возгорания.
Для предотвращения замерзания растворов гипохлорита каль-
ция его растворяют в смеси воды и метилового спирта. Состав
смеси заранее подбирают так, чтобы он соответствовал требованию
раствора при нужной температуре. Если же поступают наоборот —
добавляют метанол к готовому раствору гипохлорита, то происхо-
дит повышение температуры. Так, добавка 30% метилового спирта
вызывает повышение температуры раствора на 10 °C. При недоста-
точно низкой начальной температуре раствора такое разогревание
может привести к разложению раствора с выделением газообраз-
ных продуктов.
Вблизи температуры замерзания раствора, содержащего 30%
метанола, концентрация активного хлора за 24 ч изменяется очень
незначительно. Однако постепенно процесс разложения становится
более активным и за 48 « общее содержание активного хлора сни-
жается с 6,5 до 0,2%.
Свойства гипохлорита кальция определяют и требования, предъ-
являемые к упаковке. Самым главным является герметизация гипо-
хлорита от доступа влаги воздуха. Поэтому продукт загружают в
жестяные барабаны с привинчиваемой или другим путем гермети-
зированной крышкой. Барабаны либо оцинкованы, либо покрыты
битумом. Их емкость обычно не превышает 50 кг.
Герметизация тары представляет собой специальную проблему.
Так как нельзя полностью исключить выделение кислорода из
329
продукта, то барабаны должны выдерживать возникающее внутри
избыточное давление. Металлические барабаны и банки выдержи-
вают давление, а сварные пластмассовые емкости раздуваются и
их герметичность нарушается. Кроме того, гипохлорит кальция ре-
агирует с полиэтиленом, в результате чего разрушается материал
тары, особенно если он представляет собой тонкую пленку. Для ма-
териалов большей толщины агрессивное действие гипохлорита ме-
нее опасно.
При хранении гипохлорита кальция необходимо строго соблю-
дать те же условия, которые установлены для хлорной извести.
В присутствии влаги температура разложения гипохлорита каль-
ция понижается до 50 °C, а в присутствии органических примесей
она еще ниже.
12.3.2.5. Гипохлорит натрия. Гипохлорит натрия NaOCl значи-
тельно менее устойчив, чем гипохлорит кальция. Из водных рас-
творов он кристаллизуется в виде тонких игл, представляющих
кристаллогидраты состава NaOCl-бНгО. Вследствие небольшого
содержания воды кристаллы плавятся уже при 20 °C. Соль можно
сравнительно легко обезводить в токе сухого воздуха, после чего
она не отличается по устойчивости от хлорной извести, но более
чувствительна к температурным воздействиям. Так, моногидрат
NaOCl-Н2О разлагается со взрывом уже при 70 °C. Сухие продук-
ты, содержащие 40—60% активного хлора, плавятся при 45 °C.
Существует много патентов на получение твердого гипохлорита
натрия. Некоторые способы заключаются в обработке хлором твер-
дого едкого натра или его смеси с безводной содой *4’ 15. Продукт
стабилизуют добавлением тринатрийфосфата, жидкого стекла или
мыла 15’16. В других способах рекомендуется вначале получать рас-
твор хлорноватистой кислоты в четыреххлористом углероде 17 или
же исходить из третичного алкилгипохлорита 18. Однако вследствие
крайне низкой устойчивости твердые гипохлориты щелочных метал-
лов так и не получили применения в технике.
В основном твердый гипохлорит натрия широко применяется
только как составная часть моющих средств, используемых для
отбеливания грубых тканей, и в промышленных дезинфекционных
установках. Содержание активного хлора в таких продуктах со-
ставляет около 3% и снижается при хранении в течение года
до 1,5%.
Гораздо большее значение для промышленного использования
имеют растворы гипохлорита натрия. Они широко применяются в
качестве отбеливающих щелоков и обладают тем преимуществом
перед растворами гипохлорита кальция, что их проще получать,
легче с ними обращаться и, кроме того, они представляют собой
раствор индивидуального вещества, а не смеси продуктов.
Растворы гипохлорита натрия главным образом получают, дей-
ствуя хлором на раствор едкого натра:
2NaOH 4- С12 —» NaOCl + NaCl + HSO
330
Щелока, годные к перевозкам, содержат до 13% активного хло-
ра и 0,25—О,35°/о свободной щелочи (2,5—3,5 г/л). Если вместо
раствора едкого натра применяют растворы соды, то получается
хлорноватистая кислота:
Na2CO3 + С12 + Н2О —> NaCl + NaHCO3 + НОС1
В смесях растворов едкого натра и соды вначале с хлором
реагирует NaOH, а затем NajCOs.
Устойчивость растворов и их окисляющее действие определяют
по содержанию в них свободной щелочи (или по pH) и по темпе-
ратуре разложения.
Из табл. 39 видно, что стойкость гипохлорита в присутствии не-
связанной щелочи зависит в первую очередь от температуры хра-
нения (даже в отсутствие свободной щелочи раствор гипохлорита
натрия имеет щелочную реакцию).
Таблица 39. Снижение содержания активного хлора в растворах
гипохлорита натрия в зависимости от содержания щелочи
и температуры
(см. сс.9, Syst. № 6, стр. 276)
NaOH, а Продолжительность хранения Темпера- тура. °C Активный хлор, г Хлор-ион, г Потери активного хлора иа образование
хлората, % кислорода, %
— Свежеприготовленный 20 5,0 0,0
— 4 недели 18 4,15 0,71 14,2 2,8
— Свежеприготовленный 20 4,90 0,03 — —
—- 1 час 50 4,67 0,19 3,3 1.4
— Свежеприготовленный 20 4,60 0,26 — —
—- 1 час , 90 2,61 2,09 39,8 3,7
4,6 Свежеприготовленный 20 4,85 0,03 — —
4,6 2 недели 20 4,25 0,31 6,1 1,5
4,6 Свежеприготовленный 20 4,85 0,03 — —
4,6 1 час 90 1,93 2,30 50 7,6
10 Свежеприготовленный 20 3,71 0,13 — —
10 2 часа 90 — — 57,2 19,7
Выделение кислорода из гипохлорита протекает по следующей
реакции:
2NaC10 —► 2NaCl + О2
Причины неустойчивости хлорноватистой кислоты уже обсуж-
дались в разделе 12.3.2.1.
Соотношение гипохлорит-ионов и свободной кислоты зависит
от pH и может быть определено из следующего уравнения21 (Ка—
константа диссоциации НОС1):
[С1О'] =
*а[С12]
[Н+] + К А
831
В табл. 40 приведено содержание СЮ' в 1,41 М раСТвбрак ги-
похлорита натрия при разном pH. На основании этих данных мож-
но считать, что растворы гипохлорита натрия при pH ниже И дис-
пропорционируют. Образовавшийся хлор, реагируя с водой, дает
хлорноватистую и соляную кислоты, но если при этом избыток
щелочи в значительной степени израсходован на образование ги-
похлорита, то pH растворов гипохлорита натрия будут всегда ни-
же, чем это соответствует содержанию свободной NaOH в рас-
творе.
Таблица 40. Содержание CIO' и НС1О в растворах гипохлорита натрия
при разных pH раствора
pH [осг], моль!л ОСГ, % носи % pH [осг], моль!л ОСГ, К носи %
6 0,054 4,2 95,8 9 1,375 97,6 2,4
7 0,403 28,7 71,3 10 1,407 99,7 0,3
8 1,13 80,2 19,8 11 ' 1,41 100 0
Таким образом, при приготовлении растворов гипохлорита нат-
рия для повышения содержания гипохлорита в растворе и улучше-
ния стойкости необходимо соблюдать следующие условия:
температура раствора NaOH, через который пропускают хлор,
должна быть около 25 °C, но не выше 40 °C;
раствор нужно перемешивать во избежание местного обогаще-
ния раствора хлором или гипохлоритом;
соотношение хлора и раствора едкого натра должно быть та-
ким, чтобы избыток щелочи не превышал 0,25%.
Отбеливающие щелока, приготовленные по этому методу, могут
сохраняться в течение многих месяцев.
При наличии соответствующих приспособлений в дегазационных
машинах растворы гипохлорита натрия могут быть приготовлены
и в полевых условиях. Такая возможность представляет большой
интерес вследствие значительного сокращения перевозок. Так, вме-
сто доставки цистерны 1%-ного раствора гипохлорита емкостью
2500 л можно было бы привезти два стальных баллона емкостью
по 12,5 кг жидкого хлора каждый, 50 кг едкого натра и пригото-
вить дегазирующий раствор, содержащий около 9 г/л свободной
щелочи. Корродирующее действие такой жидкости соответствует
допустимым пределам.
Для приготовления дегазирующих растворов в полевых усло-
виях необходимо особенно тщательно придерживаться приведенных
выше требований. Этого можно.достичь, применяя непрерывное пе-
ремешивание раствора едкого натра и точно дозируя хлор. Наибо-
лее трудно точно поддерживать нужную температуру. Принимая во
внимание теплоту реакции взаимодействия хлора со щелочью
332
(25,31 ккал]моль), температура раствора может подниматься во
время реакции примерно на 4 °C.
Вследствие большого избытка щелочи растворы, полученные
таким путем, -обладают, как и белильные щелока, сравнительно
низкой окисляющей способностью. Белильные щелока, применяе-
мые в промышленности, приходится хранить продолжительное
время, но и дегазирующие жидкости часто приходится готовить за
несколько часов до употребления. Избыток щелочи в этом случае
обеспечивает устойчивость при хранении и снижает корродирую-
щее действие. Поэтому рекомендуется снижать избыток щелочи до
необходимого уровня лишь непосредственно перед употреблением
раствора или даже в процессе дегазации. Такая практика принята
в промышленности и в несколько измененном виде в полевых ус-
ловиях (ср. раздел 12.3.2.6).
Если предполагают приготовить дегазирующую жидкость с по-
ниженной температурой замерзания, то метиловый спирт следует
добавлять только после пропускания хлора, иначе значительная
часть хлора будет расходоваться на взаимодействие со спиртом
(образование формальдегида). Предварительно нужно соответ-
ственно уменьшить количество добавляемой воды.
Стабильность растворов гипохлорита натрия, содержащих ме-
тиловый спирт, при достаточном избытке щелочи соответствует
стабильности подобных растворов гипохлорита кальция..
12.3.2.6. Дегазация гипохлоритами. В предыдущих разделах
уже были рассмотрены катализ гидролитического разложения ОВ
(раздел 12.2.4) и зависимость окислительного потенциала от pH
(разделы 12.3.1 и 12.3.2.1). Поэтому здесь не требуется объяснять,
почему при использовании гипохлоритов в качестве универсального
дегазирующего средства необходимо присутствие в их растворах
известного количества щелочи, которое, однако, ие должно быть
слишком большим, так как иначе окислительная способность рас-
твора будет недостаточной. Количество щелочи в значительной сте-
пени будет зависеть от применяемых продуктов.
Установлено, что окислительная способность растворов и сус-
пензий хлорной извести и гипохлорита кальция тем меньше, чем
меньше содержание активного хлора в исходных твердых продук-
тах. Малое содержание активного хлора в этих продуктах всегда
связано с присутствием в них значительных количеств непрореаги-
ровавшей гидроокиси кальция, повышающей pH растворов.
Таким образом, для дегазации нужно готовить суспензии, со-
держащие большие количества продуктов. Обычно берут 1 ч. сухой
хлорной извести на 1 или 2 ч. воды. Для того, чтобы предотвра-
тить осаждение твердого вещества, можно применять различные
добавки. Например, в одной из американских инструкций22 ука-
зывается на возможность сохранения стабильной кашицы из 1 ч.
хлорной извести и 1,5 ч. воды путем добавления 0,25% лимонной
кислоты. Можно также предотвратить осаждение твердых частиц
введением жидкого стекла.
333
В целом следует считать, что использование хлорной извести
можно рекомендовать только как вынужденное мероприятие. Ее
применение с помощью дегазационных приборов весьма затрудни-
тельно, а расход 1 кг сухого вещества для приготовления 2 кг
дегазирующей кашицы не оправдан с точки зрения службы снаб-
жения. То же следует сказать и о применении сухой хлорной из-
вести для дегазации местности, что предусматривалось в инструк-
циях, действовавших еще после второй мировой войны. В современ-
ных условиях это мероприятие утратило всякое значение. Легко
представить, каков будет расход хлорной извести при норме около
0,5 кг)м2, например, для дегазации 5 км дороги, зараженной аэро-
золем ОВ. Кроме того, дегазация сухой хлорной известью ОВ типа
зарин не эффективна. Иприт же реагирует с сухой хлорной изве-
стью настолько интенсивно, что это приводит к воспламенению.
Эту реакцию можно проводить лишь при определенном содержа-
нии влаги в дегазирующем веществе.
Во время второй мировой войны на предприятиях концерна
«ИГ-Фарбениндустри» был выпущен продукт на основе гипохло-
рита кальция под названием лозантин 12, содержавший 42—45%
активного хлора, 28—30% СаО и 6—9% воды. Продукт давал воз-
можность дегазировать иприт, не вызывая воспламенения. Тот же
продукт в таблетках предназначался в качестве средства дегаза-
ции кожи и личного оружия. Лозантин 12 более устойчив, чем
хлорная известь, — срок хранения около 5 лет.
Другие препараты, содержащие гипохлорит кальция, еще ме-
нее пригодны для использования их в сухом виде, во-первых, из-за
того, что действуют они только в присутствии воды, и, во-вторых,
потому, что высокопроцентные сорта с большим содержанием ак-
тивного вещества реагируют слишком бурно. С продуктами .типа
двухосновного гипохлорита кальция Са(ОС1)2 • 2Са(ОН)2 иприт
даже и после увлажнения реагирует сравнительно медленно.
Таким образом, для дегазации современных ОВ могут иметь
значение лишь высокопроцентные растворы нейтрального гипо-
хлорита кальция и растворы гипохлорита натрия. Эти растворы
пригодны для универсальной дегазации8, что особенно важно, по-
скольку ОВ типа зарин и V-газы трудно различимы с помощью
обычных биохимических методов индикации.
Растворы для дегазации иприта и V-газов отличаются только
по содержанию щелочи. Так, pH растворов гипохлорита кальция
обычно равно 12,5, вследствие чего они обладают недостаточным
дегазирующим действием; их окислительные свойства при темпе-
ратурах ниже 5°C не обеспечивают достаточной скорости дегаза-
ции. При более низких температурах воздуха раствор необходимо
подогревать (например, используя теплоту реакции с амальгами-
рованной алюминиевой фольгой) или снижать pH. При распыле-
нии дегазирующей жидкости выхлопными газами • достигаются
сразу оба эффекта — идет нагревание выхлопными газами и зна-
чительное снижение pH раствора за счет двуокиси углерода, со-
334
держащейся в этих газах. Количества двуокиси углерода, обра-
зующейся при сгорании 10—20 г топлива, достаточно, например,
для полного вытеснения всей-хлорноватистой кислоты из 1 л 10%-
ного раствора гипохлорита кальция:
Са(ОС1)2 + СО2 + Н2О —>• СаСО3 + 2НОС1
При pH 8,5 (ср. табл. 40) раствор настолько реакционноспосо-
бен, что даже при температуре ниже 0°С возможна быстрая дега-
зация и иприта, и V-газов. Такое pH гарантирует и достаточную
скорость дегазации ОВ типа зарин путем гидролиза.
Так как в выхлопных газах содержится также значительное
количество окиси углерода, описанный способ дегазации связан
и с некоторым снижением концентрации активного хлора. Так,
например, если исходная концентрация активного хлора равна
2%, то в результате контакта с выхлопными газами она снизится
до 1%.
При приготовлении универсальной дегазирующей жидкости на
основе гипохлорита натрия полученный раствор необходимо также
доводить до такого оптимального pH, для чего в растворе гипо-
хлорита, содержащем 1% активного хлора, концентрацию свобод-
ной щелочи нужно доводить до 1 г/л. Жидкость такого состава яв-
ляется очень эффективным дегазирующим средством для всех
важнейших ОВ.
Эффективность дегазации растворами гипохлоритов может быть
повышена путем прибавления синтетических поверхностно-актив-
ных веществ, которые, однако, не должны подвергаться окислению.
Такому требованию полностью удовлетворяют вторичные алкил-
сульфонаты (мерзолят), а также алкиларилсульфонаты и алкил-
сульфаты. Введение этих веществ в количестве 0,3% дополняет
химическую дегазацию еще и моющим процессом.
Норма расхода растворов гипохлорита при дегазации равна
примерно 1 л/м2. Для дегазации азотистого иприта нужно расхо-
довать 1,5—2лДи2; при дегазации серного иприта 1%-ным раство-
ром гипохлорита натрия норма расхода также 2 л/м2. Растворы
гипохлоритов пригодны не только для дегазации, но и для дезин-
фекции. Норма расхода в этих случаях такая же (1—2 л/м2).
Кроме того, гипохлориты используют для дегазации и дезин-
фекции воды. При добавлении гипохлорита кальция из расчета
50 мг/л достигается и тот и другой эффект. Избыток гипохлорита
удаляют добавлением тиосульфата и последующим фильтрованием.
Вследствие корродирующего действия гипохлоритов после дега-
зации следует тщательно промыть водой все предметы, особенно
металлические поверхности, и после сушки обработать обычными
для таких материалов средствами.
В заключение напомним, что гипохлориты оказывают более
или менее раздражающее действие на кожные покровы (особенно
на слизистые оболочки) и на одежду. При пересыпании их, при-
готовлении на их основе дегазирующих растворов и при любом
335
другом обращении с ними необходимо надевать полную защитную
одежду. Особую осторожность нужно соблюдать при изготовлении
растворов гипохлорита натрия из хлора и едкого натра. В частно-
сти, при выборе.места для работы, учитывая возможность утечки
хлора, нужно по возможности исключить поражение людей и жи-
вотных.
12.3.3. Хлориты
За последние 20 лет наибольшее значение приобрел хлорит натрия
NaC102. Это вещество, так же как и гипохлориты, находит приме-
нение в качестве отбеливающего агента в текстильной и целлю-
лозной промышленности. Широкие возможности применения этого
продукта компенсируют его-высокую стоимость. В США выпущен
в продажу препарат текстов,. Он представляет собой безводную
соль или тригидрат. Хлорит натрия получают из двуокиси хлора,
которая в свою очередь может быть получена, например, из хло-
ратов.
Хлорит натрия не является универсальным дегазирующим ве-
ществом— он не способен ускорять гидролиз ОВ типа зарин. Иприт
реагирует с растворами хлорита только в кислой среде. Так как
раствор 5 г NaC102 -,ЗН2О в 100 мл воды имеет pH 9,8, то, оче-
видно, и в этом случае возникают те же вопросы, что и при работе
с гипохлоритами, с той лишь разницей, что для получения положи-
тельного эффекта pH должно быть сдвинуто в кислотную область.
Это наглядно иллюстрируют следующие значения окислительных
потенциалов:
Д.в
NaC102 (pH 4-9)........0,79-0,66
NaOCl (pH 7-10)......... 1.2-0,9
Дегазирующее действие хлоритов в кислой среде следует от-
нести главным образом за счет выделения двуокиси хлора:
5НС1О2 4С1О2 + НС1 + 2Н2О
В последние годы двуокись хлора находит все большее приме-
нение для очистки воды и поэтому особый интерес приобретает
ее использование для дегазации воды.
Двуокись хлора представляет собой желто-зеленый газ со спе-
цифическим раздражающим запахом. Она легко сжижается в крас-
ную жидкость с т. кип. —11 °C. При +4 °C в воде растворяются
20 объемн. ч. С1О2. Ввиду того, что двуокись хлора как в виде
сжатого газа, так и жидкая легко взрывается, ее нельзя траспор-
тировать в баллонах. Поэтому ее приходится получать на месте
применения, используя, например, следующие процессы:
2КС1ОЭ + H2SO4 + SO2 —► 2С1О2 4-2KHSO4
K2S2O84- 2NaC102 —► K2SO4 4- Na2SO4 4- 2C1O2
336
Для полевых условий более удобен способ получения из смеси
персульфата калия и хлорита натрия. Для этого, смешивают кон-
центрированные растворы этих солей в аппарате из поливинил-
хлорида; образующуюся двуокись хлора удаляют и дозируют по-
током воздуха.
Вода, обработанная двуокисью хлора, имеет лучшие вкусовые
качества, чем прщобычном. хлорировании.
Растворы, содержащие С1О2 в количестве 0,3 г/л, имеют pH ~4.
Легко окисляемые ОВ в этих условиях очень легко дегазируются.
Несмотря на то, что ОВ типа зарин реагируют с двуокисью хлора,
полная дегазация воды таким путем невозможна. На дегазацию
воды требуется больше времени, чем для ее дезинфекции, поэтому
для обеспечения дегазации необходимы дополнительные мероприя-
тия. После дезинфекции или дегазации следует удалить избыток
двуокиси хлора из воды. Для этого достаточно пропустить воду че-
рез активированный уголь или обработать ее восстановителем типа
тиосульфата.
12.3.4. Хлорамины
12.3.4.1. Общие сведения. Хлораминами называют соединения, в
которых хлор связан непосредственно с азотом амино- или амидо-
группы. Такие N-хлорпроизводные известны для аммиака, аминов,
амидов, мочевины, гидантоинов, уретанов и других соединений.
Хлорамины можно представить как производные амида хлорнова-
тистой кислоты:
. иоравияеи- шиит—.
кислота
В этой связи следует кратко рассмотреть, какие типы соедине-
ний способны отдавать С1+. Сравнивая электроотрицательности
элементов, мы видим, что хлор и азот имеют одинаковую электро-
отрицательность (3,0), а кислород большую (3,5), что связи С1—С1
и F—С1 способны генерировать катионы хлора. Однако соединения,
содержащие группу F—С1, непригодны в качестве дегазирующего
вещества.
Известен ряд соединений, содержащих группу S—С1. В соот-
ветствии со значениями электроотрицательности соединения типа
сульфенхлорида I не могут давать положительно' поляризованный
хлор. Аналогично этому для тионилхлорида II также неизвестны
реакции, в которых хлор мог бы быть положительно поляризован.
Этот вопрос более сложно решается для сульфурилхлорида III.
/С1 Ох /С1
R—s—ci o=s^
\ci (X \с1
I II ш
337
Легко себе представить, что остаток O2SC1 притягивает элек-
троны подобно остатку C12N. Однако здесь не может уже идти
речь об активном хлоре, т. е. хлоре, имеющем положительную сте-
пень окисления. Как это будет более подробно освещено в раз-
деле 12.3.5, реакции с сульфурилхлоридом должны сопровождаться
отщеплением радикалов или-элементарного хлора.
Само собой разумеется, что свойства соединений со связью
N—С1 будут зависеть от атомов или групп атомов, связанных с
азотом. Как было установлено, особенно хорошая активность на-
блюдается у тех хлораминов, которые содержат при азоте группы,
относительно сильно притягивающие электроны, такие, как SO2 или
СО. Во всяком случае, до сих пор для дегазационной практики
имеют значение только хлорамины с такими группами. В после-
дующем изложении другие хлорамины обсуждаться не будут.
12.3.4.2. Хлорамин Б. Это соединение представляет собой натрие-
вую соль N-хлорамида бензолсульфокислоты
/С1
C6HBSO2fZ • ЗН2О
'Na
Получается в виде бесцветных кристаллов, которые после обез-
воживания разлагаются при т. пл. 180—185 °C со взрывом.
Хлорамин Б получают хлорированием бензолсульфамида и по-
следующим высаливанием продукта реакции хлористым натрием.
Содержание активного хлора в препарате составляет примерно
30%. Свойства продукта во многих отношениях сходны со свой-
ствами хлорамина Т.
12.3.4.3. Хлорамин Т. Хлорамин Т является натриевой солью
хлорамида п-толуолсульфокислоты.
,С1
CH3C6H4SO2N^ • ЗН2О
'Na
Он образует бесцветные кристаллы, которые после обезвожива-
ния разлагаются при т. пл. 175—180°C со взрывом.
Содержание активного хлора в продукте составляет около 26%.
Хлорамин Т особенно широко применяется в качестве дезинфици-
рующего средства. Синтез его сравнительно прост. Его получают
из га-толуолсульфохлорида — побочного продукта производства са-
харина.
Вследствие своего мягкого действия и высокой стабильности
хлорамин Т как дезинфицирующее средство всегда серьезно кон-
курировал с гипохлоритом кальция. Устойчивость при хранении
зависит от степени чистоты продукта. Кондиционные продукты со-
храняются не менее трех лет, однако и после этого срока содержа-
ние активного хлора заметно не меняется. Выпускают продукт в
деревянных барабанах весом 50 кг, выложенных изнутри поливи-
нилхлоридной пленкой, или в бумажных пакетах весом 500 г, ко-
торые укладывают в картонные коробки,
338
Как и все хлорамины, хлорамин Т следует хранить в сухих, за-
крытых, темных и хорошо проветриваемых помещениях, при тем-
пературе не выше 20°C. В одном помещении с хлораминами нельзя
хранить огнеопасные, материалы, чувствительные к коррозии ме-
таллические предметы, а также реагенты, выделяющие кислотные
или щелочные пары.
Монохлорамин Т очень хорошо растворим в воде и спирте;
в 100 мл воды при 25 °C растворяется около 14 г. Для дегазации
применяют 10%-ный раствор. Так как хлорамин Т представляет
собой соль сильного основания и весьма слабой кислоты, его рас-
творы в результате гидролиза имеют щелочную реакцию:
XI
CH3C6H4SO2N^ +Н2О CH3C6H4SO2NH2 + NaOCl
'Na
Чрезвычайно низкая константа гидролиза — 4,9-10“8 (см. сс. ®,
т. 5, стр. 382)— указывает, что это равновесие сильно сдвинуто
влево. В соответствии с этим концентрация гипохлорит-иона в рас-
творе недостаточна для каталитического ускорения гидролиза ОВ
типа зарин. Поэтому растворы хлорамина Т непригодны для дега-
зации этих веществ.
Водные, водно-спиртовые или спиртовые растворы хлораминов
Б и Т пригодны только для дегазации кожно-нарывных ОВ, таких,
как иприт и люизит.
В связи с тем, что хлорамин Т не поражает кожу человека и
не разрушает текстильные ткани, его растворы особенно успешно
могут быть применены для обработки одежды и кожных покровов.
Распространены также мази на основе хлорамина Т. Слабая окис-
лительная способность хлораминов, не обеспечивающая достаточно
быстрого воздействия на V-газы, заставляет считать эти дегази-
рующие вещества не удовлетворяющими современным требова-
ниям.
Окислительную способность растворов монохлораминов можно
повысить добавлением кислот. Особенно эффективно добавление
соляной кислоты или солей, гидролизующихся с образованием со-
ляной кислоты (например, А1С1з или ZnCh). Применение растворов
с pH 4 дает возможность достигать высоких степеней дегазации.
Вместе с тем не следует сильно подкислять раствор монохлорамина,
так как при этом выделяется нерастворимый монохлорамид толуол-
сульфокислоты
XI XI
CH3C6H4SO2N^ +н+ —> CH3CeH4SO2Nf + Na*
'Na 'Н
который в результате диспропорционирования образует толуол-
сульфамид и М,М-дихлор-п-толуолсульфамид (дихлорамин Т):
XI XI
2CH3CeH4SO2N^ —> CH3C6H4SO2N^ + CH3CeH4SO2NH2
'H 'Cl
339
Так как при этом процессе в конце концов образуются только
нерастворимые продукты, то ясно, что повышенное (в кислой сре-
де) окислительное действие не может быть длительным. Исходя
из этих же соображений, использование хлорамина для дегазации
военной техники, даже в случае подкисления его растворов, сле-
дует рассматривать как вспомогательное мероприятие. Принимая
во взимание, что окислительная способность хлорамина Т зависят
от pH, нецелесообразно применять для дегазации смеси растворов
NaOH и хлорамина Т. Хотя большое содержание щелочи способ-
ствует дегазации ОВ типа зарин, но практически при этом полно-
стью исключаются какие-либо превращения иприта и V-газов.
12.3.4.4. Дихлорамин Б и дихлорамин Т. Дихлорамин Б, или
М,М-дихлорбензолсульфамид, и дихлорамии Т, или М,М-дихлор-п-
толуолсульфамид; представляют- собой кристаллы белого или кре-
мового цвета, обладающие запахом хлора. Характеристики этих
соединений приведены в табл. 41.
Таблица 41. Свойства дихлораминов Б и Т
Название Т. пл.. °C Т. разл.. Активный хлор, %
Дихлорамин Б (ДТ-2) 69-72 200 До 60
Дихлорамин Т (ДТ-2Т) 80—83 150-160 57-59
Получение дихлорамина Б протекает по следующей схеме:
С6Н6 + 2HOSO2C1 —> CBHBSO2C1 + H2SO4 + НС1
50 °C
CBHBSO2C1 + 2NH3 ---► CBHBSO2NH2+NH4C1
C3HbSO2NH2 + 2NaOH + 2C12 —>
/С1
—► C6HbSO2N^ + 2NaCl + 2H2O
xi
Для получения дихлорайина T, как и в случае монохлорамина
Т, из n-толуолсульфохлорида получают п-толуолсульфамид
CH3C6H4SO2C1 + 2NH3 —> ch3cbh4so2nh2 + NH4C1
который при растворении в едком натре дает соль:
/И
CH3CBH4SO2NH2 + NaOH —> CH3CBH4SO2N^ + Н2О
'Na
Далее эту соль хлорируют, пропуская хлор:
,Н XI
CH3CbH4SO2N^ +2Cl2 + NaOH —> CH3CeH4SO2N^ + 2NaCl + H2O
'Na 'Cl
340
Можно хлорировать белильными щелоками, из которых при
действии HCI выделяется хлорноватистая кислота.
Дихлорамин Т выпадает в осадок, его центрифугируют, промы-
вают и сушат.
Если хлорирование проводят при 90 °C, то выделяется жидкий
дихлорамин Т и центрифугирование отпадает.
Готовый продукт упаковывают в деревянные ящики (50 кг)
или в деревянные бочки (40 кг), выложенные парафинированной
бумагой. Срок хранения не менее трех лет.
Начальное содержание активного хлора й'дихлорамине Т вы-
сокого качества, равное 58%, после 10 лёт хранения снижается до
50%. Условия хранения те же, что и для монохлорамина.
При загорании хлорамина Т он бурно разлагается (примерно
так же, как черный порох) с выделением пламени и дыма.
Дихлорамины не растворяются в воде, поэтому применяют ис-
ключительно их растворы в органических растворителях. Лучше
всего для этих целей пригоден 1,2-дихлорэтан, в котором даже при
низких температурах растворяется значительное количество ди-
хлорамина. Ниже приведена растворимость дихлорамина Т в ди-
хлорэтане при разных температурах:
Температура, °C Растворимость Температура, Растворимость дихлорамина Т, °C дихлорамина Т,
мол. % мол. %
40 60 0 25
30 50 —10 18
20 41 —20 14
10 32 —30 10
Четыреххлористый углерод несколько хуже растворяет дихлор-
амин, но полученные растворы более устойчивы, чем растворы в
дихлорэтане. В бензине и других нефтепродуктах дихлорамин
почти не растворим. Также недостаточна его растворимость и в
различных спиртах. Для дегазации применяется 10%-ный раствор
дихлорамина в дихлорэтане. За 4 ч раствор утрачивает около 10%
от начального содержания активного хлора. Процесс разложения
инициируется светом, влагой и воздействием железа. С помощью
растворов дихлорамина в дихлорэтане можно дегазировать иприты
и V-газы при любых температурах. Преимущество этой дегазирую-
щей жидкости состоит главным образом в том, что в ней очень хо-
рошо растворяется ОВ и, кроме того, она легко проникает во все
щели и поры материалов.
Вместе с тем растворитель может довольно сильно повредить
лакокрасочные покрытия. Разрушение лакокрасочных покрытий в
значительной степени зависит от температуры. При низких темпе-
ратурах разрушения незначительны; так, при —10 °C различные
типы лакокрасочных покрытий практически не разрушаются дега-
зирующими Жидкостями. При таких температурах и ОВ почти
не проникают внутрь покрытий, поэтому в этих условиях дости-
гается полная дегазация. Норма расхода дегазирующей жидкости
341
составляет 0,5 л/м2, однако и при расходе 0,3 л/м2 вполне дости-
жимы хорошие результаты.
Растворы дихлорамина в дихлорэтане не дегазируют ОВ типа
зарин.
12.3.4.5. Гексахлормеламин (ДТ-6). Гексахлормеламин — жел-
товатое мелкокристаллическое вещество, т. пл. 149 °C.
N
C12N—\С—NC12
I II
N4 /N
С
I
NC12
Как видно из формулы, он представляет собой полностью хло-
рированный меламин. Последний производят в больших количествах
и применяют для производства термореактивных пластмасс. Тех-
нический гексахлормеламин плавится при 125—135 °C. Вследствие
высокого содержания активного хлора (124%) с этим продуктом
еще труднее обращаться, чем с дихлорамином. При пробивании
пулей стального сосуда с гексахлормеламином продукт воспламе-
няется. Бризантные свойства чистого гексахлормеламина при-
мерно соответствуют свойствам пикриновой кислоты. При контакте
различных органических веществ с гексахлормеламином уже при
незначительном разогревании происходит бурная реакция с вос-
пламенением.
Для повышения безопасности продукт смешивают с твердыми
высокохлорированными углероводородами. Флегматизированный
таким образом гексахлормеламин содержит не менее 91% актив-
ного хлора. Он не загорается от прикосновения накаленного до-
красна металлического стержня.
При хранении гексахлормеламина необходимо придерживаться
требований, описанных в разделе 12.3.4.3.
Гексахлормеламин применяется для дегазации аналогично ди-
хлорамину. Растворимость его также близка к растворимости ди-
хлорамина. Обычно применяются его 5%-ные растворы.
Дегазирующее действие гексахлормеламина вследствие более
высокого содержания активного хлора более эффективно, чем ди-
хлорамина.
12.3.4.6. Изоцианурхлорид. Изоцианурхлорид является произ-
водным тримера изоциануровой кислоты, из которой его получают
хлорированием. О
ь
Cl—US' \N—Cl
O=(L /С=О
N
изоцианурхлорид
342
Он представляет собой порошок, обладающий раздражающим
запахом хлора./В армии бывшей фашистской Германии он состоял
на снабжении в виде 50%-ной сухой смеси с кварцевым песком
(дегазатор 40) и предназначался для дегазации азотистого иприта
и других ОВ. Вещество хранилось в стальных бочках емкостью
30 и 60 л.
В настоящее время изоцианурхлорид находит все большее при-
менение в качестве исходного продукта для получения средств за-
щиты растений; он может снова приобрести известное значение и
как дегазирующее вещество. Его устойчивость при хранении хуже,
чем устойчивость описанных ранее хлораминов.
12.3.4.7. Дихлорамид метансульфокислоты. Дихлорамид метан-
сульфокислоты CH3SO2NCI2 — белое кристаллическое вещество, об-
ладающее запахом хлора. Технический продукт плавится при 75—
77 °C и содержит около 86% активного хлора. Он хорошо раство-
ряется в бензине, плохо — в дихлорэтане и совсем не растворяется
в воде.
Производство этого продукта началось в 1935 г. на заводе кра-
сок в Вольфене, после того как фашистское высшее командование
дало задание разработать средство для дегазации боевой техники.
Получали этот хлорамид по следующей схеме.
CH3SH + ЗС12 + 2Н2О —> CH3SO2C1 + 5НС1
CH3SO2C1 + 2NH3 —> CH3SO2NH2 + NH4C1
CH3SO2NH2 + 2HOC1 —> CH3SO2NC12 + 2H2O
Сухой дихлорамид метансульфокислоты является взрывоопас-
ным веществом. Поэтому конечный продукт сразу же после филь-
трования растворяли в трихлрртриэтилфосфате. Растворы сушили
в вакууме и поставляли с содержанием 10—12% дихлорамида
метансульфокислоты в качестве «средства для дегазации боевой
техники». С 1939 г. этот дегазатор находился на снабжении фа-
шистской армии; он (по патенту концерна «ИГ-Фарбениндустри»)
пригоден для дегазации азотистого иприта 20.
12.3.4.8. 1,3-Дихлор-5,5-диметилгидантоии. На вооружении ар-
мии США продолжительное время находилось дегазирующее
средство DANC. Оно представляет собой 1,3-дихлор-5,5-диметил-
гидантоин
(СН3)2С---N-C1
О=С\ /С=о
N
I
С1
Вещество плавится при 132°С, содержит около 33% активного
хлора и хорошо растворяется в дихлорэтане и тетрахлорэтане.
Для дегазации кожно-нарывных ОВ используют 6—7%-ные рас-
творы в тетрахлорэтане. По имеющимся данным, длительность де-
газации должна составлять 30 миц.
343
12.3.4.9. N-Хлоргликольурилы. Общая формула этих соединений
следующая:
. Cl R С1
Ji—С—ik
о=с/ I V=o
\n—с—nz
Cl R Cl
По своей устойчивости они превосходят все до сих пор изве-
стные хлорамины23. Так, нагревание в течение 100 ч при 100°C
не приводит к какому-либо снижению активности. При более про-
должительном кипячении в -воде содержание активного хлора
незначительно снижается. Различные N-хлоргликольурилы обла-
дают чрезвычайно эффективным дегазирующим действием по от-
ношению к кожно-нарывным ОВ. Вследствие относительно слож-
ного получения эти вещества применяются главным образом только
для дегазации кожных покровов,
12.3.5. Сульфурилхлорид
Сульфурилхлорид представляет собой жидкость, которая в зави-
симости от степени чистоты имеет окраску от бледно-желтой до
коричневой. Вследствие взаимодействия с влагой вещество на воз-
духе дымит:
SO2C12 + 2H2O —> H2SO4 + 2НС1
Пары обладают резким раздражающим действием на носоглот-
ку, Сульфурилхлорид кипит при 69,3 °C и плавится при —54 °C.
Плотность вещества при 20 °C равна 1,65—1,66 г/см?.
Продукт получают, действуя хлором на двуокись серы:
SO2 4“ С12 < * SO2C12
Нагревание до 160 °C приводит к разложению на первоначаль-
ные продукты..
Сухое вещество не разрушает сталь. Присутствие даже самых
незначительных количеств влаги вызывает сильную коррозию.
Кожные покровы и обмундирование также сильно разъедаются
сульфурилхлоридом. Из-за того, что он весьма агрессивен, а его
пары очень сильно раздражают дыхательные пути, при работе с
этим веществом необходимо надевать полную защитную одежду.
Сульфурилхлорид можно хранить и транспортировать в толсто-
стенных стальных сосудах или в вагонах-цистернах.
Сульфурилхлорид служит «зимним» дегазирующим веще-
ством. Вследствие низкой температуры кипения его применение
при 4-5 °C и выше нецелесообразно.
Сильное агрессивное действие этого вещества еще более огра-
ничивает возможности его применения. Оно не должно соприка-
344
саться с боевой техникой и поэтому пригодно только для дегазации
местности (дороги, мосты и т. п.). Перед применением сульфурил-
хлорида рекомендуется счищать с поверхностей снег, так как он в
значительной степени снижает эффективность этого вещества.
Сульфурилхлорид для дегазации применяют либо в смеси с ди-
хлорэтаном (1:1 или 1:2), либо неразбавленный.
Дегазация ОВ сульфурилхлоридом основана на реакциях хлори-
рования, поэтому ОВ типа зарин этим веществом не дегазируются.
Хлорирование протекает в основном по ионному и радикальному
механизмам. Ионный механизм основан на действии хлор-катиона
(см. раздел 12.3.2), причем совершенно не важно, образовался ли
CI+ из молекулы сульфурилхлорида или из молекулы хлора. Так
как сульфурилхлорид сравнительно легко распадается на SO2 и С12,
то можно предположить, что катионы хлора в обоих случаях обра-
зуются из молекулы хлора.
Чтобы подчеркнуть принципиальную разницу между вещества-
ми, содержащими активный хлор, и сульфурилхлоридом, мы рас-
смотрим здесь и радикальный механизм хлорирования.
В результате воздействия энергии (свет или тепло) сульфурил-
хлорид способен распадаться на радикалы (см. сс1, стр. 461). При
этом ковалентная связь разрывается таким образом, что каждый
осколок становится обладателем свободного электрона. Образую-
щиеся радикалы не несут на себе заряда и обладают большой ре-
акционной способностью:
SO2C12 —> SO2C1« + C1«
Радикал хлора может реагировать с углеводородами следую-
щим образом:
С1 • + R—Н —► R. + HC1
Радикал Р«, взаимодействует с SO2C12 с образованием новых
радикалов:'
R- + SO2C12 —> RC1 + SO2C1.
SO2C1 • —> SO2 + Cl • и т. д.
В конечном счете в результате этой цепной реакции между
сульфурилхлоридом и углеводородом образуются RC1, НС1 и SO2.
Само собой разумеется, что в этом случае невозможно решить,
какой из механизмов преобладает; в общем случае по энергети-
ческим соображениям все же следует считать, что при более низ-
ких температурах преобладает ионный механизм.
12.3.6. Дегазирующие вещества, разлагающиеся
с выделением кислорода
12.3.6.1. Общие сведения. В разделах 12.3.2.—12.3.5. обсужда-
лись дегазирующие вещества,-действие которых определяется на-
личием активного хлора или способностью разлагаться с выделением
элементарного хлора. Дегазирующее действие генерирующих
345
кислород средств основано на окислительной способности актив-
ного кислорода. Понятие активный кислород идентично понятию
атомарный.
Для приобретения конфигурации инертного газа кислороду не-
достает двух электронов:
O-f-2e —> О2-
Способность вещества отщеплять кислород определяется не
только наличием последнего, но главным образом прочностью той
Или иной разрываемой связи. Из шкалы электроотрицательности
(ср. табл. 32) следует, что элементы, обладающие низкой электро-
отрицательностью (например, Na, С, Р), образуют с кислородом
сравнительно прочную связь. Относительно легко отщепляется кис-
лород из его соединений с элементами, имеющими равную, более
высокую или несколько меньшую электроотрицательность. Это под-
тверждается тем, что сильными окислителями являются соедине-
ния кислорода с азотом, хлором, а также вещества, имеющие
связь О—О.
Пригодность того или иного окислителя как дегазирующего
средства определяется не только его способностью отщеплять кис-
лород; решающей является его окислительная способность при
возможно более низких температурах. Этим свойством обладают
лишь очень немногие окислители. С таких позиций хорошими де-
газирующими веществами следует считать озон, перекисные сое-
динения, окислы азота и хлора. Свойства двуокиси хлора уже об-
суждались в разделе 12.3.3. Другие окислы хлора неустойчивы и
поэтому непригодны для дегазации.
Многие окислы азота представляют собой высокотоксичные
газы. Из этого типа соединений некоторое значение для дегазации
имеет тетраокись азота N2O4, а также азотная кислота, получаю-
щаяся при растворении окислов азота в воде. Однако вследствие
агрессивности эти соединения вряд ли пригодны для полевой де-
газации. Поэтому далее более подробно будут рассмотрены только
озон и перекисные соединения. Такие мягкие окислители, как пер-
манганат и бихромат калия, обсуждаться не будут, так как эти
вещества только условно могут применяться даже для дегазации
иприта.
12.3.6.2. Озон. Озон Оз представляет собой газ, обладающий
специфическим приятным запахом. Получается он в озонаторах
из кислорода или воздуха действием электрического разряда. Об-
разующаяся смесь озона с кислородом содержит 10% и более
озона.
Как видно из табл. 36, озон принадлежит к наиболее сильным
окислителям. Он разрушает многие органические соединения, об-
ладает сильной отбеливающей способностью, дезинфицирующим
действием и поэтому находит широкое применение для уничтоже-
ния бактерий в воде. В этой связи ему отводится известная роль
и как средства для дегазации воды.
346
Озон очень незначительно растворим в воде. Поэтому при де-
газации воды озоном требуется тщательное перемешивание. Эф-
фективность озона в основном зависит от pH воды; его окислитель-
ная способность уменьшается при переходе от кислой среды к ще-
лочной. Устойчивость озона меняется в обратном порядке.
В щелочных растворах вследствие разложения озона образуется
главным образом ион Ог:
О3 + 2ОН“ —> 2О2 + Н2О
При низкой щелочности эти ионы с ионами кислорода образуют
перекись водорода и молекулярный кислород:
О2 + Н+ —> НО2.
2НО2. —> Н2О2 + О2
В связи с тем, что в щелочной среде перекись водорода ката-
лизирует гидролиз ОВ типа зарин, а сам озон является сильным
окислителем, то, используя последний, можно было бы создать
универсальный дегазатор, обладающий как дегазирующей, так и
дезинфекционной способностью. Поскольку озон растворим, и то
незначительно, только в органических растворителях, таких, на-
пример, как четыреххлористый углерод, и устойчив лишь в этих
растворителях, то для применения его в полевых условиях потре-
бовалось бы создать портативные озонаторы. Исходя из всего из-
ложенного применение озона возможно только для дегазации воды.
12.3.6.3. Перекись водорода. Чистая безводная перекись пред-
ставляет собой бесцветную сиропообразную жидкость, с которой
очень трудно работать. Поэтому она поступает в продажу в виде
3 и 30%-ных водных растворов; 30%-ный раствор перекиси водо-
рода известен под названием пергидрол. Его получают электроли-
тическим окислением сульфатов с последующей отгонкой перекиси
водорода из смеси с серной кислотой:
2SO^’-2e —> S2O*~
S2O|_ + 2H2O —> 2SOj“ + 2Н++ Н2О2
Стойкость растворов перекиси водорода зависит от чистоты
продукта. Следы металлов, щелочей и даже просто пыли катали-
зируют разложение перекиси:
2Н2О2 —> 2Н2О4-О2
Разложение протекает также под воздействием света и тепла.
Растворы перекиси водорода можно стабилизовать незначитель-
ными добавками силикатов, мочевины, а также фосфорной или
барбитуровой кислот.
Хранить растворы перекиси водорода лучше всего в полиэтиле-
новых сосудах. Сосуды из темного стекла пригодны для хранения
347
только стабилизованных растворов; это обусловлено тем, что ма-
лейшего количества щелочи (из стекла) уже достаточно для раз-
ложения перекиси водорода. Окислительная способность перекиси
водорода, как и всех рассмотренных окислителей, в значительной
мере зависит от pH. В кислой среде концентрированные растворы
перекиси водорода дегазируют иприт и V-газы. Для дегазации
азотистого иприта окислительная способность таких растворов не-
достаточна.
Каталитическое действие перекиси водорода на гидролиз ОВ
типа зарин было уже разобрано в разделе 12.2.4. Оно наблюдается
только в щелочной среде. Так, период полураспада зарина при
25°С и концентрации перекиси водорода 0,1% при pH 11 равен
1,8 сек-, при pH ниже 7 увеличение концентрации перекиси водо-
рода не приводит к ускорению реакции.
Так как для дегазации перекисью водорода очень большое
значение имеет pH среды,'то перекись водорода также нельзя счи-
тать универсальным дегазирующим веществом.
При работе с 30%-ным раствором перекиси водорода нужно
защищать глаза, кожные покровы и одежду. При попадании НгО2
на кожу образуются белые пятна и маленькие пузыри. Пергидрол
разрушает даже резиновую одежду.
12.3.6.4. Другие перекисные соединения. Значительная часть пе-
рекиси водорода, получаемой промышленностью, идет на изготов-
ление комплексных соединений. Наиболее важными представите-
лями этого типа веществ являются:
NaBO2 • Н2О2 • Н2О 2Na4CO3 • ЗН2О2
Na2B4O7 • Н2О2 • 9Н2О ' Na2PO4e2H2O2
CO(NH2)2-H2O2
Вещества на основе неорганических перекисей находят очень
широкое применение в производстве моющих средств. Количество
перборатов, добавляемых в моющие средства, может составлять
10% и более. Эти же вещества являются хорошими дегазирующи-
ми средствами.
Соединение перекиси водорода с мочевиной широко применяет-
ся в косметике (гидропирит).
Преимуществом этих комплексных соединений является их вы-
сокая стойкость — они могут храниться годами, а их кристалличе-
ская структура и хорошая растворимость в воде определяют удоб-
ство обращения с ними.
Эти продукты используются в тех случаях, что и перекись во-
дорода; их применение лимитируется сравнительно высокой ценой.
В дегазационной практике перекисные соединения используются
главным образом как компоненты дегазирующих средств для кож-
ных покровов.
12.3.6.5. Персульфаты. Как видно из табл. 36, персульфаты
принадлежат к наиболее сильным окислителям..
348
Их получают электролитическим окислением серной кислоты
или сульфатов на платиновых электродах при высокой плотности
тока.
Пероксодисерную кислоту
°Ч /°1
О—S—О—О—S—о
Хх
и ее соли следует рассматривать как производные перекиси во-
дорода.
Практическое значение имеют персульфаты калия и аммония.
В то время как свободная кислота сравнительно неустойчива, соли
сохраняются хорошо. Значение их в производстве двуокиси хлора
уже было отмечено в разделе 12.3.3.
Пока в литературе нет упоминаний о практическом применении
их для дегазации различных поверхностей. Однако в соответствии
с принципами, изложенными в разделе 12.3.6.3, по-видимому, это
возможно. Следует иметь в виду, что каталитическое действие при
дегазации ОВ типа зарин наступает только после образования пе-
рекиси водорода. Для этого необходимо, чтобы сначала прошел
гидролиз пероксодисерной кислоты в кислой среде, и только после
добавления щелочи возможен каталитический гидролиз ОВ.
Из этого следует, что персульфаты нельзя рассматривать как
универсальные дегазирующие вещества.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. -Назовите четыре важные критерия, определяющие понятие «дегазирую-
щее вещество».
2. Какие преимущества и недостатки имеют химические способы дегазации?
3. На примере связей О—Cl, N—С1 и С—С1 поясните, как зависит их устой-
чивость от положения элементов в Периодической системе элементов и от их
электроотрицательности.
4. Чем можно объяснить различную устойчивость кислот НС1О, НС1О2,
НС1О3 и НС1О4?
5. Объясните, какое влияние оказывает /-эффект иа гидролиз хлористого
метила, хлористого пропила и хлористого бутила.
6. Приведите примеры кислот и оснований Льюиса.
7. Почему реакция зарина с водой рассматривается как реакция первого
порядка?
8. На примере первой ступени гидролиза иприта поясните применимость
закона действия масс.
9. Что такое сольватация?
10. Как влияет сольватация образующихся в реакции анионов иа скорость
реакции дегазации?
11. Назовите различные возможности ускорения реакции между зоманом и
едким натром.
12. Каким путем можно преодолеть затруднения, возникающие при дегаза-
ции резины?
13. Что такое поверхность раздела фаз и какое влияние оказывает ее вели-
чина на дегазацию иприта водными дегазирующими растворами?
14. Как увеличить поверхности раздела фаз?
349
15. Как сказывается на дегазации иприта добавление этанола в раствор
сульфида натрия?
16. На примере реакций дегазации иприта и зарина поясните различие ме-
жду нуклеофильностью и основностью дегазирующих веществ.
17. Назовите основания, обладающие высокой нуклеофильностью.
18. Почему иприт легче дегазировать растворами сульфида натрия, чем ед-
ким натром?
19. Почему сода и аммиак наиболее пригодны для дегазации кипячением?
20. Какие преимущества дегазации ОВ смесями спиртов с алкоголятами?
21. Какое влияние на скорость реакции оказывает образование водородных
связей?
22. На примере спиртов и аминов объясните, какая имеется связь между
основностью и сольватирующим действием дегазирующих растворов.
23. Назовите и обоснуйте главные области применения катализаторов де-
газации.
24. Почему серусодержащие нуклеофильные реагенты реагируют с ОВ типа
зарин хуже, чем кислородсодержащие?.
25. Опишите возможности' применения для дегазации моноэтаноламина.
26. Назовите преимущества и недостатки дегазирующих жидкостей на осно-
ве смесей алкоголятов с аминами.
27. Какие меры предосторожности следует принимать при работе с дегази-
рующими средствами основного характера?
28. Что такое окисление?
29. Сопоставьте значение процессов окисления и хлорирования для дегаза-
ции ОВ.
30. Что такое нормальный потенциал?
31. Можно ли иа основании уравнений окислительно-восстановительных
реакций сделать выводы б влиянии pH?
32. Перечислите основные окислители, имеющие военно-химическое значение.
33. Почему хлораты не пригодны как дегазирующие средства?
34. Что такое «активный хлор»?
35. Что такое «активный кислород»?
36. На чем основано универсальное дегазирующее действие гипохлоритов?
37. Какова связь между дегазирующим действием и pH растворов гипохло-
ритов?
38. На что следует обращать внимание при приготовлении растворов гипо-
хлорита натрия?
39. Для чего нужно добавлять метиловый спирт в растворы гипохлоритов?
40. Что Вы знаете об устойчивости гипохлоритов?
41. Что такое хлорамины?
42. Какова растворимость различных хлораминов?
43. В чем заключается дегазирующее действие важнейших хлораминов?
44. Каковы условия хранения хлораминов?
45. Назовите дегазирующие вещества, пригодные для дегазации воды.
46. Почему перекись водорода не является универсальным дегазирующим
средством?
47. Назовите дегазирующие вещества, применяемые для дегазации кожных
покровов. Какими свойствами должны обладать вещества, применимые для де-
газации кожных покровов?
ЛИТЕРАТУРА
1. Becker Н. Einftihrung in die Elektronentheorie organischchemischer Reak-
tionen. Berlin, 1961.
2. Pauling L. Die Natur der chemischen Bindung. Weinheim, 1962.
3. Frost A. A., Pearson R. G. Kinetik und Mechanismen homogener chemi-
scher Reaktionen. Weinheim, 1964.
4. N a s e г К- H., Physikalische Chemie fur Techniker und Ingenieure. Leipzig,
1966.
360
5. Matousek J., TomeCek I. Analyse synthetischer Gifte. Berlin, 1965.
6. F о e r s t W. In: «Ullmanns Enzyklopadie der technischen Chemie». Miinchen-
Berlin, 1954.
7. Реми Г. Курс неорганической химнн. Т. 1. Пер. с нем., под ред. академика
АН СССР А. В. Новоселовой, М., «Мир», 1972 г. 824 с.
8. Traebert Н., J. Militarwesen, 11, 657 (1967).
9. Gmelin-Krauts Handbuch der anorganischen Chemie, Weinheim.
10. H i n e J., H i n e H„ J. Am. Chem. Soc., 74, 5266 (1952).
11. Lohs K- Synthetische Gifte. Berlin, 1967.
12. M u 11 e r E. Methoden der organischen Chemie. Stuttgart, 1963.
13. Пат. США 3079346.
14. Нем. пат. 338962 (1919).
15. Нем. пат. 330192 (1919).
16. Нем. пат. 566762 (1931).
17. Нем. пат. 615963 (1934).
18. Winnacker u. Weingaertner. Chemische Technologie, Sammelwerk in
5 Banden, B2. Anorganische Technologie. Mfinchen, 1950. S. 495.
19. Пат. США 1632483 (1925); 1632485 (1925).
20. IG-Farbenindustrie G-Patent 273/36 (1936).
21. Epstein J., Bauer V. E. et al., J. Am. Chem. Soc., 78, 4068 (1956).
22. Vorschrift der US-Marine, ABC-Verteidigung auf dem Land, 1960.
23. Пат. США 2649389 (1953).
24. Stohr R. Die chemischen Kampfstoffe. Berlin, 1961.
13. ФИЗИЧЕСКИЕ СПОСОБЫ ДЕГАЗАЦИИ
13.1. ОБЩИЕ СВЕДЕНИЯ
При физических способах устранения ОВ после химического напа-
дения находящееся на поверхности или в окружающей среде ОВ
удаляется без каких-либо химических изменений его структуры.
Средства и методы» применяемые для физической дегазации, пред-
ставлены в табл. 31. Так как в этих способах дегазации ОВ пере-
ходит в неизмененном виде в среду дегазирующего средства, то это
означает, что последнее (растворитель, моющий раствор, адсорбент)
после проведения дегазации становится опасным для человека и
животных. Вследствие этого в большинстве случаев возникает не-
обходимость завершать физическую дегазацию химической или
комбинировать оба эти способа. Точно так же химическая дегаза-
ция немыслима без привлечения физических процессов. Недостатки
различных дегазирующих веществ, перечисленные в разделе 12.1,
особенно четко подчеркивают эту необходимость. Но несмотря на
все это дегазация с использованием исключительно физических ме-
тодов для ряда конкретных случаев целесообразна, например де-
газация воздуха адсорбентами, дегазация поверхностей с помощью
горячего воздуха или отработанными газами двигателей.
Ниже будут обсуждены средства и методы физической дегаза-
ции, независимо от того, применяются ли они самостоятельно или
главным образом в комбинации с химическими средствами и мето-
дами.
13.2. ДЕГАЗАЦИЯ РАСТВОРИТЕЛЯМИ
13.2.1. Общие сведения
Растворы представляют собой твердые или жидкие гомогенные
смеси веществ.
Для дегазации в первую очередь представляет интерес процесс
растворения твердых и жидких органических или неорганических
веществ в жидкостях. Жидкость, в которой растворяется вещество,
называется растворителем.
Считается, что раствор образуется тем легче, чем ближе поляр-
ность участвующих в этом процессе веществ (ср. раздел 12.2.1). По
полярности важнейшие растворители и группы растворителей мож-
но расположить следующим образом:
362
Неполярные растворители
Алифатические углеводороды
Ароматические углеводороды
Галогенированные углеводороды
Полярные растворители (в порядке увеличения полярности)
Эфиры Ацетонитрил
Спирты Нитрометан
Амииы Диметилформамид
Диметилсульфокснд Вода
Полярность наиболее важных ОВ увеличивается в ряду: азоти-
стый иприт, зоман, зарин, V-газы.
Растворимость основных видов ОВ в перечисленных раствори-
телях следующая.
В воде — хорошо растворяются V-газы и зарин, очень плохо —
зоман, иприт и азотистый иприт (свободное основание).
В спиртах — хорошо растворимы V-газы, зарин и зоман; хуже —
иприты.
В галогенированных углеводородах все ОВ растворяются хоро-
шо или даже очень хорошо, в алифатических углеводородах — хуже.
Способность органических растворителей растворять ОВ исполь-
зуется как для приготовления дегазирующих жидкостей, так и для
дегазации растворением (растворителями).
Как видно из раздела 12, для приготовления дегазирующих жид-
костей, действующих как нуклеофильные агенты, в качестве раство-
рителей особенно пригодны вода, спирты и амины. К этой группе
растворителей, действие которых основано на их нуклеофильных
свойствах, а также на способности связывать анионы (образование
водородных связей), относятся и карбоновые кислоты.
Для растворения органических дегазирующих веществ, содер-
жащих активный хлор, требуются растворители, достаточно устой-
чивые к окислению. К таким относятся главным образом галогени-
рованные углеводороды, а также ацетонитрил (CH3CN) и нитро-
метан (CH3NO2).
В качестве растворителей для физической дегазации можно при-
менять углеводороды, эфиры и диметилформамид. Для приготовле-
ния дегазирующих растворов они мало пригодны.
Некоторые растворители способны повышать растворимость ОВ
в водных дегазирующих растворах. Примерами являются спирты,
прибавляемые к едким щелочам и растворам сульфидов щелочных
металлов (ср. разделы 12.2.2 и 12.2.5). Ацетонитрил (нитрил уксус-
ной кислоты), диметилсульфоксид и диоксан также обладают этим
свойством.
Полярность этих растворителей ниже полярности воды, но выше
полярности ОВ, плохо растворяющихся в воде. Хорошо смешиваясь
с обоими компонентами, эти растворители способствуют смешению
неполярного вещества с сильнополярным растворителем и являют-
ся как бы «посредниками» при растворении.
у212 Зак. 677
353
Такие растворители, как диоксан, ацетонитрил, диметилсульф-
оксид и диметилформамид, сравнительно дороги, что ограничивает
их широкое применение для дегазации. Хлорированные углеводо-
роды и низшие спирты более дешевы, поэтому для их применения
нет экономических препятствий. Однако некоторые их свойства,
как, например, токсичность хлорированных углеводородов и мета-
нола, или легкую воспламеняемость низших спиртов, следует учи-
тывать при их применении.
По опасности, обусловленной их воспламеняемостью, раствори-
тели могут быть подразделены на следующие группы:
Группа А —жидкости, не смешивающиеся или частично смешивающиеся
с водой
Группа AI — вещества с температурой вспышки до 21 °C
Группа АП—вещества с температурой вспышки 21—55“С
Группа АШ — вещества с температурой вспышки 55—100 “С
Группа Б —жидкости, смешивающиеся с водой во всех отношениях
В группы Б1—БШ входят вещества, воспламеняющиеся в тех
же температурных интервалах, что и вещества групп AI—АШ, но
смешивающиеся с водой во всех отношениях. Хорошая раствори-
мость спиртов в воде, наряду с их сравнительно низкой стоимостью,
позволила использовать их в качестве антифризных добавок к вод-
ным дегазирующим растворам. Кроме того, эти растворители при-
годны и для дегазации рас/гворением.
13.2.2. Свойства важнейших растворителей
13.2.2.1. Дихлорэтан. Это бесцветная жидкость, обладающая
сладковатым, напоминающим хлороформ запахом; т. кип. 83—84 °C;
т. застыв, около —35 °C; т. самовоспл. (на воздухе) около 449 °C;
т. вспышки ~18°С; плотность (d20) 1,25 г/сж3; давление пара 62 ат;
пределы взрываемости в смеси с воздухом составляют от 6,1 до
16,9 объемн. %.
Дихлорэтан получают хлорированием этилена:
СН2=СН2+С12 —► СН2С1—СН,С1
В качестве побочного продукта образуется и трихлорэтан. В во-
де при 20 °C растворяется 0,1 % дихлорэтана, а в дихлорэтане рас-
творяется 0,5% воды. Дихлорэтан медленно гидролизуется водой
с образованием соляной кислоты:
СН,С1—СНаС1 + НаО —> СН,С1—сн,он + НС1
В связи с тем, что дихлорэтан хранится в стальных бочках,
чрезвычайно чувствительных к действию соляной кислоты, в рас-
творитель не должна попадать влага. Тем не менее при длитель-
ном хранении в отсутствие стабилизаторов бочки с дихлорэтаном
все же подвергаются коррозии. Поэтому, во избежание затруднений
при вскрытии бочек из-за ржавчины на запорных устройствах,
рекомендуется время от времени открывать бочки. Стабилизовать
354
дихлорэтан и другие хлорированные углеводороды можно добав-
лением алкил аминов, спиртов или фенолов в количестве 0,01—
0,1%. Хорошим стабилизатором является смесь триэтиламина и
тимолфталеина. Пригодны также дифениламин1 и диизопропил-
амин 2.
При индикациях иприта тимолфталеиновым реагентом («синий
реагент») дихлорэтан в смесях с водой -дает аналогичное иприту
окрашивание. :
Дихлорэтан является сильным ядом. В любых условиях следует
избегать вдыхания его паров. Вещество это вызывает поражение
нервной системы, а также действует на деятельность печени и
сердца.
В случае отравления пострадавшим необходимо немедленно ока-
зать врачебную помощь. Все работы с дихлорэтаном должны про-
изводиться в полной защитной одежде.
13.2.2.2. Четыреххлористый углерод (тетрахлорметан). Пред-
ставляет собой бесцветную жидкость, обладающую сладковатым
запахом; т. кип. 77°C; т. пл. —24 °C; плотность (d20) 1,59 г!см3.
Его получают хлорированием метана или сероуглерода в присут-
ствии серы или двуххлористой серы:
2С8а + бС1, —► 2СС1< + 2saci,
CSj + 2SjC1j —► CCI4 + 6S
Растворы хлораминов в четыреххлористом углероде более ста-
бильны, чем в дихлорэтане, что объясняется невозможностью даль-
нейшего хлорирования четыреххлористого углерода соединениями,
содержащими активный хлор. Однако растворимость дихлорамина
и гексахлормеламииа в четыреххлористом углероде при низких
температурах значительно уменьшается, вследствие чего эти рас-
творы непригодны для работы в зимних условиях.
При действии света и тепла пары четыреххлористого углерода
могут разлагаться с образованием фосгена.
13.2.2.3. Метиловый и этиловый спирты. Оба спирта представ-
ляют еобой бесцветные жидкости, обладающие характерным запа-
хом. Метанол кипит при 65—66 °C, этиловый спирт — при 78 °C.
Это полярные жидкости, смешивающиеся с водой в любых отноше-
ниях. По степени опасности воспламенения они принадлежат к
группе Б. Метиловый спирт восплчменяется от искры при 6,5 °C, а
этиловый — при 12 °C.
Метиловый спирт получают в больших количествах из водяного
газа в присутствии катализатора
катализатор
СО + 2Н, --------»- СНзОН
Этиловый спирт получают методом брожения и гидратацией эти-
лена.
По физиологическому действию эти два спирта довольно сильно
отличаются друг от друга. Этиловый спирт относительно нетоксичен,
355
а метиловый — сильный яд, вызывающий слепоту; отравление мо-
жет иметь летальный исход (ЛД для человека составляет примерно
Юг). Даже пары метанола могут представлять значительную опас-
ность, они резорбируются через кожу. Поэтому при обращении с
метиловым спиртом нужно использовать средства защиты.
Метиловый и этиловый спирты, вследствие того, что они хорошо
смешиваются с водой и относительно дешевы, удобны в качестве
антифризов для водных дегазирующих и дезактивирующих раство-
ров. В табл. 42 показано, как влияют добавки метилового спирта на
температуру замерзания дезактивирующего раствора, содержащего
0,5% мерзолята, и на 10%-ный водный раствор NaOH.
Таблица 42. Влияние метилового спирта иа температуру застывания
растворов мерзолята и едкого натра
Состав раствора Температура замерзания растворов при добавлении СН3ОН. °C
5% 10% 15% 20% 25% 30% 35% 40%
Мерзолят —4 -7 -11 -14 -18 -20 —24 -28
NaOH, 10%-иый -6 -10 -15 -18 —21 -26 -30 -35
13.2.2.4. Прочие растворители. Наряду с дихлорэтаном и четы-
реххлористым углеродом можно применять в качестве раствори-
телей: хлороформ СНС13 (т. кип. 61—62°C), трихлорэтилен
СНС1=СС12 (т. кип. 87 °C), тетрахлорэтан СНС12—СНС12
(т. кип. 146 °C). Эти растворители, также как и дихлорэтан, ядовиты.
Хлорированные углеводороды, содержащие по одному атому во-
дорода и атом углерода, считаются негорючими. Однако правиль-
нее было бы считать их веществами с пониженной горючестью, так
как при достаточно высокой температуре эти жидкости также го-
рючи. При горении хлорированных углеводородов образуется много
дыма; при неполном сгорании — возможно образование фосгена.
Здесь не рассматривались многие другие растворители, исполь-
зуемые в промышленности или в повседневной жизни. Зная основ-
ные требования, предъявляемые к растворителям ОВ, можно в каж-
дом отдельном случае решать, насколько пригоден имеющийся в
распоряжении растворитель. Необходимо особо подчеркнуть, что
применение для дегазации моторного топлива представляет большую
опасность, не говоря уже о том, что моторное топливо дефицитно.
Вообще дегазация растворителями как самостоятельный тип де-
газации имеет второстепенное значение.
13.2.3. Дегазация зараженных поверхностей растворителями
В этом способе поверхность, подлежащая дегазации, должна подвер-
гнуться по меньшей мере двукратной обработке обильным количе-
ством растворителя. Более мелкие приборы и предметы можно погру-
жать в растворитель. Расход растворителя при таком способе обра-
356
ботки больший, чем при использовании дегазирующих растворов.
При дегазации поверхностей растворителями довольно трудно
достигнуть допустимых плотностей остаточного заражения. Так как
для высокотоксичных ОВ эти значения достаточно низки, то необхо-
дима особенно тщательная обработка поверхностей. При известных
условиях, в соответствий с результатами химического контроля,
может потребоваться повторная обработка.
Ниже перечислены растворители, пригодность которых для де-
газации падает в приведенной последовательности:
I Галогенированные углеводороды
I Этанол
ф Метанол
Карбюраторное топливо
Дизельное топливо
. - Реактивное топливо
Особые трудности связаны с полным удалением ОВ с поверх-
ностей, покрытых лакокрасочными покрытиями или воском. В то
время как большая часть растворителей способна удалить слой
воска, а вместе с ним и проникшие в него ОВ, лакокрасочные по-
крытия при кратковременной обработке растворяются только в
хлорированных углеводородах (при низких температурах их рас-
творимость резко падает). При повторной обработке окрашенной
поверхности другим растворителем можно более полно извлечь ОВ
из слоя краски.
Особое внимание следует обратить на то, что после дегазации
ОВ переходит в неизменном виде в растворитель. Для обезврежи-
вания отработанного растворителя его следует сжечь. Исключение
составляют негорючие хлорированные углеводороды. Исходя из
этого, следует считать, что дегазация зараженных поверхностей
растворителями целесообразна только тогда, когда нет в распоря-
жении подходящего дегазирующего раствора, или когда свойства
подлежащих дегазации объектов исключают возможность их обра-
ботки сравнительно сильно корродирующими средствами.
Нужно строго следить за тем, чтобы не проливать на землю не
смешивающиеся с водой отработанные растворители, так как за-
раженные таким образом участки представляют опасность в тече-
ние длительного времени.
Кроме того, следует-обращать внимание на то, что под влия-
нием различных растворителей сильно снижается непроницаемость
защитной одежды для ОВ. Особенно это относится к хлорирован-
ным углеводородам.
13.2.4. Экстракционные способы дегазации обмундирования
Вопрос дегазации обмундирования уже рассматривался в разделе
12.2.3. Агрессивность большинства дегазирующих веществ настоль-
ко велика, что они не могут быть использованы для обработки
текстильных материалов.
С другой стороны, такие вещества, как сода и аммиак, в мень-
шей степени снижающие прочность текстильных тканей, приме-
няются при таких температурных режимах дегазации, которце син-
]2 Зек. 677
357
тетические волокна не выдерживают. Одним из возможных решений
этого вопроса является использование экстракционных процессов.
Принцип экстракционного способа дегазации обмундирования
состоит в удалении ОВ из материала обмундирования путем рас-
творения их растворителем. Повышение температуры способствует
этому процессу; уже при 50—60 °C достигается хороший эффект.
В качестве растворителей могут быть использованы те же, что и
при химической чистке одежды, т. е. различные фракции бензина и
хлорированные углеводороды. Установлено, что разные сорта бен-
зина хорошо подходят для обработки таких недостаточно устойчивых
текстильных материалов, как дедерон, в то время как хлорирован-
ные углеводороды их разрушают. Однако поскольку хлорированные
углеводороды обладают лучшей растворяющей способностью, их
рекомендуется применять в первую очередь (трихлорэтилен или
дихлорэтан).
Иприт и V-газы, обладающие высокими температурами кипе-
ния, можно отделять от растворителей перегонкой. Отравляющие
вещества типа зарин следует переводить в нетоксичные соединения,
так как их давление пара достаточно высокое. Это достигается
добавлением к растворителю 0,5% моноэтаноламина (см. раз-
дел 12.2.6). В качестве добавок к хлорированным углеводородам
пригодны спирты, улучшающие растворимость таких полярных со-
единений, как зарин.
Дегазацию экстракцией растворителями можно проводить на
предприятиях химической чистки или в специальных передвижных
установках. В этих способах в среднем на 1 кг ткани расходуется
примерно от 0,2 до 0,5 кг растворителя. Часть растворителя после
экстракции удерживается дегазированным материалом, а часть те-
ряется при регенерации.
Само собой разумеется, что прорезиненную одежду и, особенно,
защитную одежду этими методами дегазировать нельзя.
13.3. ДЕГАЗАЦИЯ СТИРКОИ
13.3.1. Общие сведения
О значении моющего действия при химической дегазации и о воз-
можности совместного использования различных дегазирующих и
моющих средств отмечалось уже в разделах 12.1 и 12.3.2.
Однако помимо этого моющие растворы также и сами по себе
могут быть использованы для де!азации.
Действие моющего раствора объясняется способностью поверх-
ностно-активного вещества снижать поверхностное натяжение на
границе раздела фаз. Поверхностное натяжение на границе раздела
фаз равно работе, которую нужно затратить, чтобы увеличить гра-
ницу раздела двух соседних фаз на единицу поверхности. Поверх-
ностное натяжение жидкости следует рассматривать как частный
случай поверхностного натяжения на границе раздела фаз. В пер-
вую очередь оно имеет значение для системы жидкость — газ (пар).
358
Под поверхностным натяжением жидкости понимают работу, кото-
рую нужно затратить, чтобы увеличить поверхность на единицу
длины.
В соответствии с известным физическим принципом поверхност-
ное натяжение можно также рассматривать как силу, с которой
участок поверхности шириной в 1 см сжимается в продольном на-
правлении. Это можно наглядно проиллюстрировать опытным пу-
тем. Так, если в находящейся в проволочной рамке пленке жидко-
сти уменьшается поверхностное натяжение, то подвижная перего-
родка стремится подняться вверх (рис. 30).
Поверхностное натяжение о представляет собой работу, необхо-
димую для образования единицы поверхности, и измеряется в
эрг/см? или дин/см. Возникновение
поверхностного натяжения объяс-
няется силами межмолекулярного
взаимодействия. В жидкости эти
Рис, 30. Опыт, иллюстрирую-
щий явление поверхностного
натяжения.
Рис. 31. Ориентирование мо-
лекул поверхностно-активного
вещества в поверхностном слое
воды.
силы действуют во всех направлениях, а в поверхностном слое их
действие направлено параллельно поверхности или внутрь жидко-
сти. Это объясняет тот факт, что жидкости на несмачиваемых по-
верхностях стремятся принять форму шара или капли.
Поверхностное натяжение тем выше, чем более полярны молеку-
лы жидкости. В соответствии с этим вода имеет высокое поверх-
ностное натяжение. В результате этого силы, действующие в по-
верхностном слое, уменьшают возможность проникновения в него
неполярных молекул.
Поэтому нельзя очищать водой поверхности, на которых нахо-
дятся жиры или подобные им неполярные вещества. Так как вода
все же наиболее доступный растворитель, чтобы использовать ее
для целей очистки, необходимо понизить .поверхностное натяжение
воды. Это достигается повышением температуры, а также введе-
нием в воду поверхностно-активных веществ.
Молекулы таких веществ содержат полярные и неполярные
группы. По отношению к воде полярные группы проявляют гидро-
фильные свойства, а неполярные — гидрофобные; например, обыч-
ные мыла, представляющие собой соли щелочных металлов и выс-
ших жирных кислот, состоят из гидрофильной карбоксильной груп-
пы и гидрофобного алкильного радикала CH3(CH2)i3—СН2—.
При растворении такого поверхностно-активного вещества в
воде (т. е. оно должно быть растворимо в воде) гидрофильные
359
участки ориентированы вовнутрь объема воды, гидрофобные участки
стремятся ориентироваться на поверхности воды. При этом поверх-
ностный слой воды обогащается поверхностно-активным веществом
(рис. 31), за счет чего прочность этого слоя снижается. Сосредото-
ченные в поверхностном слое воды неполярные радикалы облег-
чают смачивание гидрофобных веществ (жиров, масел, а также
различных ОВ). При помощи микрофотографий-удалось обнару-
жить, что благодаря воздействию раствора поверхностно-активного
вещества капли неполярной жидкости иа твердой поверхности по-
степенно принимают форму щара3>4, а краевой угол смачивания
О при этом сильно уменьшается (рис. 32).
Рис. 32. Капли неполярной жидкости на твердой поверхности.
Как только капли жидкости приобретут шарообразную форму,
они практически перестают быть прочно связанными с твердой
поверхностью и могут быть с нее смыты. Для достижения этого
эффекта на стальных поверхностях, зараженных ипритом, был при-
менен4 моющий раствор, содержащий менее 0,2% поверхностно-
активного вещества.
13.3.2. Важнейшие поверхностно-активные вещества
Для приготовления водных моющих растворов нужно располагать
растворимыми в воде поверхностно-активными веществами. Так как
такие вещества должны содержать гидрофобный радикал, то требо-
вание их растворимости в воде может быть выполнено только при
условий1, что они находятся в виде соли или наряду с гидрофобным
радикалом имеют большое число спиртовых или других групп, спо-
собствующих растворению в воде.
Различают анионоактивные, катионоактивные и неионогенные
поверхностно-активные вещества. В табл. 43 приведены основные
группы поверхностно-активных веществ. Они представляют почти
все типы выпускаемых в мире моющих веществ.
Между натуральными и синтетическими моющими веществами
часто проводится различие. Натуральными моющими веществами
являются обычные жировые мыла, представляющие собой соли
щелочных металлов и карбоновых кислот и получаемые омылением
натуральных жиров. Представленные в табл. 43 другие соединения
являются синтетическими моющими веществами. В то время как
в жировых мылах гидрофильную часть молекулы образует карб-
оксильная группа СОО", в синтетических веществах такими группа-
ми являются сульфатная, сульфонатная, гидроксильная и четвер-
тичная аммониевая группы, .
360
Таблица 43. Важнейшие поверхностно-активные вещества
Тип соединения Формула Примерное моющее вещество
А н н он о а к ти в н ы е вещества
Жировые мыла
Первичные алкилсульфаты
Вторичные алкилсульфаты
Сульфаты замещенных полиглико-
левых эфиров
Первичные алкнлсульфонаты
Вторичные алкнлсульфонаты
Алкилбензолсульфонаты
Алкилиафтилсульфонаты
RCOQMe (где R от С,4 до С20) Me — К илн Na ROSO3Me Гардинол
R\
\ZHOSOsMe Теепол
R'Z
RCOO(CH2O)nSO3Me
RNH(CH2O)„SO3Me
R— SOaMe
Ri\ TH— SOaMe Мерзолят
RZ
К а ти он о а к т и в и ы e вещества
Соли четвертичных аммониевых ос-
нований
Соли алкилпиридиния
Неионогенные вещества
Эфиры полигликолей
Алкнларилполиглнколевые эфиры
Замещенные полиамнны
RO(CH2CH5O)„H
4-О(СН2СН2О)пН
R—СО—NH—(СН2СН2ЫН)цН
Жировые мыла получают омылением жиров в варочных коТлаХ
или нейтрализацией едким натром некоторых жирных кислот:
сн3(сн2)п—о—сн2
СН3(СН2)п—О—CH -|-3NaOH —► 3CH3(CH2)„COONa+
СН3(СН2)П—О—СН2 + СН2ОН—СНОН—СН2ОН
CH3(CH2)nCOOH + NaOH —► CH3(CH2)nCOONa + Н2О
Для получения алкилсульфатов действуют на жирные спирты
серной кислотой и затем нейтрализуют смесь едким натром:
СНз(СН2)„ОН 4- H2SO4 —CH3(CH2)nOSO3H + Н2О
СНз(СН2)пО5ОзН + NaOH -Л СН3(СН2)пОЗОзНа + Н2О
В основном используют жирные спирты, содержащие в углерод-
ной цепи от 6 до 22 атомов углерода. Такие спирты получают ката-
литическим гидрированием под высоким давлением сложных эфи-
ров жирных кислот:
CH3(CH2)nCOOR + 2Н2 —> СН3(СН2)„СН2ОН + ROH
Одним из наиболее важных методов получения поверхностно-
активных веществ является сульфохлорирование. Оно осущест-
вляется действием на углеводороды хлором и двуокисью серы при
облучении УФ-светом:
R\
Vh2 + so2 + ci2
R'z
R\
Vhso2ci + HC1
R'Z
При нейтрализации продуктов реакции образуются первичные
или вторичные алкилсульфонаты. Ароматические сульфонаты полу-
чают сульфированием алкилбензола или алкилнафталина концен-
трированной серной кислотой.
Катионоактивные вещества получают главным образом из ами-
нов путем их ацилирования или превращения в четвертичные ам-
мониевые основания:
/СН2СН2ОН /С1 ХСН2СН2ОН
HN^ +R——> N—СН2СН2ОН + НС1
\сн2сн2он ЧО \с—R
А
/СНз +/СНз
R—N^ + Н2С—СН2 + Н2О —> R—N—СНз + ОН”
^сНз \ / \сн2сн2он
362
Важнейшим сырьем для Производства нейоногенных Поверх-
ностно-активных веществ является окись этилена. В результате ее
конденсации, например, со спиртами, образуются различные поверх-
ностно-активные полигликолевые эфиры:
R—ОН + пН2С—СН2 —> RO(CH2CH2O)nH
Из твердых жировых мыл готовят кусковое мыло. Их недостат-
ком является то, что их действие зависит от жесткости воды (обра-
зуются нерастворимые кальциевые мыла). Поэтому для очистки
текстильных материалов они используются только при наличии
умягченной воды (например, с помощью соды).
Устойчивость к образованию кальциевых мыл, высокая сма-
чивающая способность, пенообразование, способность к удержанию
загрязнений — все это важнейшие свойства, которыми должно об-
ладать хорошее моющее средство. Для специальных задач тек-
стильной промышленности требуется, кроме того, чтобы моющее
средство было кислотоустойчивым.
Основные моющие вещества в той или иной степени обладают
всеми этими свойствами. Комбинацией отдельных веществ, а так-
же добавками других соединений можно составлять композиции
специальных моющих средств для самых различных областей при-
менения. В табл. 44 приведены важнейшие области применения этих
Таблица 44. Основные типы моющих средств, выпускаемых в ГДР
Наименование препарата Вид применения Характеристика
Fewa Тонкая стирка (т-ра раствора до 50 °C, pH 7-8) До 35% моющего вещества, осталь- ное — фосфаты, Na2SO4, неиоио- генные вещества
Gentfna special Грубая стирка (т-ра раствора выше 50° С, pH 9-10) До 35% моющего вещества, осталь- ное — Na2CO3, силикаты, фосфаты, Na2SO4, неионогеиные вещества Добавка NaHCO3 обеспечивает раз- ную величину pH при разной тем- пературе стирки (образование соды!)
Универсальный мою- щий препарат (при т-ре раствора до 50 °C, pH 7-7,5, при более высокой т-ре PH 9)
Wok Препарат для быстрой стирки (pH такой же, как для тонкой и грубой стирки) Высокое содержание окислителя (перборатов)
Fit Смывающее средство Высокое содержание моющего веще- ства (например, 10% мерзолита), остальное — вода, полифосфаты, эфиры гликолей
Gr.-Bakto Промышленная очист- ка Щелочные соли, моющее средство, возможны дезинфицирующие до- бавки гипохлорита натрии
363
Средств. Распространение того или иного моющего средства в дай-
ной стране определяется возможностями производства поверх-
ностно-активных веществ в этой стране, а область применения
зависит от специфических свойств определенного моющего сред-
ства. Так, алдилсульфонаты с сульфогруппой, расположенной по-
середине молекулы, обладают преимущественно смачивающим
действием; расположение же сульфогруппы на конце молекулы
обеспечивает хорошую способность удерживать загрязнения и хоро-
шее смачивающее действие.
По сравнению с сульфонатами алкилсульфаты обладают лучши-
ми моющими свойствами, при этом вторичные алкилсульфаты луч-
ше первичных. Для того чтобы получить моющий препарат с хоро-
шими смачивающими и моющими свойствами, часто комбинируют
алкнлсульфонаты с алкилсульфатами или алкиларилсульфонатами,
так как последние обладают особой способностью удерживать за-
грязнения.
Перечисленные синтетические моющие вещества в основном
представляют собой гигроскопичные пастообразные вещества. При
смешении с солями они становятся твердыми.
13.3.3. Дегазация моющими растворами
Применение для дегазации только моющих растворов связано с
теми же вопросами, которые возникали при дегазации растворите-
лями. В большинстве случаев ОВ при этом не разлагается, а если
и разлагается, то медленно. Разложению способствуют имеющиеся
в моющих средствах щелочь или перборат. Таким образом, моющий
раствор, после того как он образовал эмульсию с ОВ или частично
его растворил (например, зарин), должен быть тщательно смыт с
поверхности, а затем подвергнут химической или естественной де-
газации.
Если, например, подлежит дегазации самолет, зараженный зо-
маном, и в распоряжении имеются только относительно агрессив-
ные дегазирующие вещества, то дегазация проводится моющим
раствором с последующей химической дегазацией стекшего рас-
твора. Можно также заранее рассыпать дегазирующие вещества
под дегазируемой техникой.
Моющие растворы обладают тем же недостатком, что и водные
дегазирующие жидкости, — они не могут обеспечить глубинной де-
газации. Поэтому при использовании моющих растворов трудно до-
стигнуть даже максимальной допустимой остаточной плотности
заражения, особенно когда ОВ уже успело проникнуть в глубь
материала. В этом случае выходом из положения может явиться
применение горячих моющих растворов. При известных обстоятель-
ствах следует считаться с необходимостью многократной обработ-
ки. При этом неизбежна основательная обработка щетками. В сом-
нительных случаях по окончании работы необходим контроль эф-
фективности дегазации.
364
Помимо дегазации поверхностей военной техники моющие сред-
ства могут быть использованы также и для дегазации обмундиро-
вания. дегазация текстильных материалов стиркой производится
обычно при 50—60 °C. При этом нужно часто менять моющий рас-
твор. Так, в способе дегазации кипячением более целесообразно
вместо соды применять моющее средство для грубой стирки. Осно-
вой дегазирующего действия в этом случае является гидролиз ОВ,
который проходит примерно при 90 °C, чему, само собой разумеется,
способствует поверхностно-активное вещество (см. раздел 12.2.3).
13.4. ДЕГАЗАЦИЯ АДСОРБЦИЕЙ
13.4.1. Общие сведения
Под адсорбцией понимают оседание молекул газа или жидкости на
поверхности твердого или жидкого вещества.
Следует различать физическую и химическую адсорбцию. В то
время как физическая адсорбция осуществляется под влиянием Ван-
дервальсовых сил, химическая ад-
сорбция есть результат действия
сил химических связей.
В отличие от адсорбции, за-
ключающейся в удержании веще-
ства поверхностью, при абсорбции
происходит его гомогенное рас-
пределение по всему объему ад-
сорбента.
Так как адсорбция осуществи-
ма только на поверхности адсор-
бента, то, очевидно, все пористые
Рис. 33. Изотермы адсорбции в ста-
тических условиях:
/—•ипрнт на активированном угле; 2—ипрнт
иа силикагеле; 3—зарин на активированном
угле; 4—зарнн на силикагеле.
вещества с развитой поверхностью
обладают способностью к сорб-
ции. Количество вещества, сорби-
руемое при данной температуре
адсорбентом, зависит от парциального давления вещества (у газов)
или от его концентрации (у растворов). Эта зависимость математи-
чески выражается изотермой сорбции Лангмюра:
где а»д — количество сорбированного вещества; а — максимальное сорбируемое
количество при условии образования мономолекуляриого слоя (при бесконечном
давлении); р—парциальное давление (может быть заменено концентрацией);
b — константа.
Из уравнения следует, что при повышении концентрации рас-
твора или парциального давления газа возрастает количество сор-
бированного вещества. При этом устанавливается зависящее от
температуры равновесие между числом сорбирующихся и десорби-
рующихся молекул в единицу времени.
365
Эти отношения наблюдаются сравнительно просто в статических
условиях, т. е. тогда, когда адсорбент и адсорбат находятся в по-
кое. На рис. 33 представлены изотермы сорбции иприта на угле и
силикагеле, снятые в статических условиях.
Более сложна динамическая сорбция. Она зависит не только от
давления, температуры и сорбционной емкости, но также и от объ-
емной скорости адсорбата, длины и поперечного сечения слоя ад-
сорбента.
13.4.2. Основные адсорбенты
Адсорбенты должны при небольшом объеме иметь большую разви-
тую поверхность, т. е. более пористый материал обладает большей
сорбционной емкостью. Нужный диаметр пор должен определяться
величиной молекул сорбируемого вещества. Селективное погло-
щение при помощи адсорбентов с очень малым диаметром пор
(молекулярные сита) может быть использовано для разделения
веществ, для дегазации ОВ оно значения не имеет.
Активированный уголь незаменим для различных способов де-
газации. Он является составной частью фильтров противогазов, но,
кроме того, используется для дегазации воды.
Активированный уголь получают термической и химической
обработкой естественных органических материалов растительного
и животного происхождения, а также бурого и каменного угля.
Свойства и области применения получаемых сорбентов определяют-
ся как исходным веществом, из которого они получены, так и спе-
циальными способами последующей обработки.
Так, для физической сорбции решающим являются пористость и
зернение полученных углей. Химическая же сорбция зависит, кро-
ме того, от числа активных центров, имеющихся в единице объема.
Последние представляют собой места, на которых возможно обра-
зование химической связи между адсорбентом и сорбируемым ве-
ществом. Способность активированного угля к химической сорбции
можно изменять специальной обработкой растворами солей тяже-
лых металлов.
Вообще известно, что температура по-разному влияет на хими-
ческую и физическую сорбцию. В то время как при повышении тем-
пературы физическая сорбция уменьшается, хемосорбция, напро-
тив, при этом возрастает. Наглядным доказательством этого яв-
ляется применение активированного угля в качестве катализатора
различных синтезов, осуществляемых при высоких температурах.
Так же как и в процессах растворения, способность активиро-
ванного угля поглощать вещества из воздуха или воды обусловлена
его полярностью и полярностью извлекаемого вещества.
Активированный уголь, являясь гидрофобным сорбентом, очень
легко сорбирует из воздуха или водных растворов неполярные со-
единения. Сродство к воде у него незначительное. Вместе с тем по
отношению к органическим соединениям средней полярности, а сле-
довательно, также и к ОВ, он обладает достаточным сродством.
366
Совершенно иными свойствами обладает силикагель. Проявляя
сильное сродство к полярным или легко поляризуемым соедине-
ниям, он не пригоден для дегазации воды, так как вся его сорбцион-
ная емкость будет исчерпана сорбцией только воды, а из воздуха
он способен извлекать даже малополярные вещества. Это свойство
силикагеля широко используется в индикаторных трубках и труб-
ках для отбора проб из воздуха.
Силикагель получают сплавлением минерального сырья — пес-
ка, соды и др. Желаемая степень зернения достигается в результате
помола и просеивания.
13.4.3. Дегазация воздуха
Извлечение ОВ из воздуха осуществимо различными способами.
Основным способом очистки воздуха, необходимого для дыхания,
является применение фильтров, которые используют как для инди-
видуальной, так и для коллективной защиты.
Фильтры состоят из нескольких слоев. Первым слоем обычно
служит волокнистый материал, который предназначен для задер-
жания более грубых частиц, способных проходить через каналы
пор активированного угля, не удерживаясь на поверхности стенок.
За ним располагается слой пропитанного активированного угля,
способного извлекать из газовоздушной смеси только частицы, об-
ладающие достаточной скоростью движения. Такими являются
частицы газообразных веществ.
Емкость фильтра зависит от количества адсорбента, от того, как
долго он был в употреблении, а также от температуры. Пары ве-
ществ, подвергающихся на поверхности активированного угля ка-
талитическому разложению уже при низких температурах, могут,
в результате выделения тепла реакции, сильно снижать емкость
фильтра противогаза. Такой эффект проявляется только после про-
хождения большого количества таких веществ.
К средствам индивидуальной защиты относится также импре-
гнированное обмундирование, применяемое как для защиты от па-
ров, так и от капель ОВ. В защитном действии пропиток адсорбция
имеет второстепенное значение. Пары, адсорбированные обмунди-
рованием, во избежание их проникновения через ткань должны не-
обратимо дегазироваться активными химическими компонентами
пропитки. Такие активные вещества должны обладать высокой
реакционной способностью по отношению ко всем ОВ, оставаясь
одновременно устойчивыми на воздухе и на волокне материала.
Все эти требования довольно трудно выполнимы.
В качестве веществ, способствующих осуществлению адсорб-
ции и в то же время выполняющих роль реакционной среды по
отношению к ОВ, применимы труднолетучие органические раство-
рители (см. т. 1, стр. 212). В ткани, пропитанной такими раствори-
телями, наряду с адсорбцией происходит также и имеющая боль-
шое значение абсорбция. Благодаря абсорбции ОВ прочно удер-
367
Живаются пропиткой и действие химических реагентов протекает в
этой среде. Активными химическими реагентами обычно являются
уже рассмотренные ранее хлорамины и феноляты.
Подобно пропитке действуют также и распыляемые адсорбен-
ты, которые после прохождения облака ОВ должны снижать опас-
ность попадания в воздух ОВ, десорбирующихся из пористых по-
верхностей или из обмундирования. Их следует в случае необходи-
мости наносить на пористые поверхности.
Другим способом очистки воздуха, основанным на адсорбции,
абсорбции и химических превращениях ОВ, является дегазация при
помощи аэрозолей. Этот способ в первую очередь имеет значение
для дегазации воздуха в закрытых помещениях (например, кабин
автомобилей). В качестве сорбентов ОВ здесь могут применяться
как труднолетучие растворители, так и высокодисперсные твердые
адсорбенты.
13.4.4. Дегазация воды
Воду можно дегазировать при помощи сорбентов. Исходя из опи-
санного, наиболее подходящим для этого является активированный
уголь, наименее пригодным — силикагель.
В зависимости от имеющегося оборудования применим один из
двух основных способов. Один способ состоит в том, что подлежа-
щую дегазации воду перемешивают определенное время с порошко-
образным активированным углем и затем фильтруют. По второму
способу очистку воды ведут непрерывно.
Мучин5 описывает опыты по удалению из воды гидрохлорида
трихлортриэтиламина, люизита, зарина, табуна, мышьяковистого
водорода и хлорпикрина пропусканием ее через колонки с активи-
рованным углем. Данные из этой статьи, приводимые в табл. 45,
определяют границы применения такого метода. Достигнутые
после очистки концентрации ОВ удовлетворяют принятым в ар-
миях НАТО максимально допустимым нормам, вместе с тем они
Таблица 45. Дегазация воды в колонках с активированным углем
(количество активированного угля 100 мл, высота слоя 33 см,
температура 20 °C)
Отравляющее вещество Концен- трация. мг/л Фильтрат, Л Средняя скорость пропускания воды через колонку, м/ч Адсорб- ция» вес. % Конечная концентрация, р. р. т.
Азотистый иприт 680 6,6 4 5,5 20
(солевая форма) 150 Н.о 4 3,7 20
68 18,7 4 2,7 20
Зарин 10 2 0,01
100 4 0,04-0,07
368
остаются еще рискованно высокими. Это обстоятельство может
быть надежно преодолено применением больших количеств активи-
рованного угля, что, однакб, приведет к увеличению и без того зна-
чительных материальных затрат.
Если предположить, что в среднем на обработку 10 л воды тре-
буется 100 мл активированного угля (около 50 г), то при очистке
1000 л воды это уже составит 5 кг сорбента. Для того, чтобы со-
держание зарина достигло предельно допустимой концентрации,
обнаруживаемой биохимическим способом, требуется еще больше
активированного угля независимо от того, проводится ли очистка
непрерывно или периодически. Поэтому использование для дега-
зации воды одного только активированного угля нецелесообразно
и часто рекомендуется комбинировать адсорбционный и химический
способы очистки (см. разделы 12.3.2 и 12.3.3).
Наряду с активированным углем для сорбционного удаления
ОВ пригодны также коагулянты. Хотя бы из того факта, что на
водоочистных станциях коагуляцию используют только для предва-
рительной очистки воды, можно уже заключить о ее малой эффек-
тивности для дегазации воды. Очевидно и при дегазации воды ко-
агуляция может быть применена для предварительной обработки
воды. Важно, что на это расходуется мало химикалиев — от 10
до 30 мг А12(5О4)з или FeCl3 на 1 л в зависимости от pH воды6. Эго
составляет только двухсотую часть расхода активированного угля.
Исходя из этих данных, целесообразно при дегазации воды комби-
нировать коагуляцию, химические методы и адсорбцию активиро-
ванным углем. При этом более дорогой активированный уголь сле-
дует использовать для окончательной тонкой очистки воды, тогда
угольные фильтры будут удалять из воды и остатки химических
реагентов.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Что представляют собой растворители?
2. Поясните на конкретных примерах связь между структурой молекулы
вещества и его растворимостью.
3. Расположите известные вам ОВ в ряд по их растворимости в воде.
4. Используя некоторые примеры, опишите преимущества и недостатки спо-
собов дегазации растворителями.
5. Классифицируйте важнейшие растворители по нх воспламеняемости.
6. Какие меры предосторожности должны соблюдаться при проведении
дегазации растворителями?
7. Что такое поверхностное натяжение?
3. Назовите основные поверхиостно-активные вещества.
9. Как могут быть использованы поверхностно-активные вещества для де-
газации?
10. Какие моющие вещества не могут быть использованы совместно с соеди-
нениями, содержащими активный хлор?
11. Какова смачивающаяся способность и способность удерживать загряз-
нения поверхностно-активных веществ?
12. Какой тип моющего средства особенно удобен для использования в спо-
собе дегазации обмундирования кипячением?
13. Назовите области вренио-химического применения адсорбентов.
369
14. Что выражает изотерма адсорбции?
15. Каково различие между адсорбцией и абсорбцией?
16. Назовите преимущества и недостатки применения активированного угля
для дегазации воды.
17. Какую роль играют коагулянты при очистке и дегазации воды?
ЛИТЕРАТУРА
1. Mitt. chem. Forsch.-Inst. Wirtsch. Osterreiche, 9, 149—152 (1955).
2. Пат. ФРГ 1046003 (1957).
3. Ga wa lek G., Wasch- und Netzmittel, Berlin, 1962.
4. Szczucki E„ Durka K., Biuletyn WAT (Польша), Heft IV u. VIII (1966),
5. M u t s c h i n A., Zivilschutz, Heft 12, 405—407 (1966).
6. Dietrich K- R-, Chemiker-Zeitung, 16, 563 (1966).
14. ИСПЫТАНИЕ СРЕДСТВ ДЕГАЗАЦИИ
14.1. ОБЩИЕ СВЕДЕНИЯ
К числу средств дегазации относятся собственно дегазирующие ве-
щества, растворители, моющие средства, адсорбенты. Обычно спо-
соб испытания каждого вещества определяется в первую очередь
целью этого испытания. ___
Так, если необходимо установить только качество находящегося
на снабжении дегазирующего средства (например, после его хране-
ния в течение нескольких лет), можно ограничиться проведением
обычного химического анализа (например, определением активного
хлора в гипохлоритах). При определенных обстоятельствах доста-
точно будет даже визуальной оценки продукта (например, оценка
качества едкого натра). Иногда нужно установить, годится ли дан-
ное вещество в качестве дегазирующего средства. В .этом случае
проводят его идентификацию и определяют содержание в нем дей-
ствующего начала. При этом прежде всего следует пользоваться
стандартными методами исследования. Само собой разумеется, все
эти методы пригодны и для исследования неизвестного соединения.
14.2. ОТБОР ПРОБ
Отбирать пробу нужно всегда так, чтобы данные анализа пробы
характеризовали качество всего продукта в целом. Если речь идет
об анализе поступающих на склад продуктов, то необходимо при
отборе проб руководствоваться Правилами, имеющимися в стан-
дарте, разработанном для этого вещества. Этими же стандартами
пользуются при контроле продуктов, находившихся на складе в те-
чение определенного времени.
Если же контролю подвергается состояние целой партии по-
ставляемого дегазирующего средства (например, для того, чтобы
вынести суждение о дальнейшей его применимости), то в каждом
случае работники лаборатории устанавливают по своему усмотре-
нию число отбираемых по выбору проб. Способ отбора проб за-
висит при этом от общего объема партии и вида ее упаковки. Если
дегазирующее средство хорошо упаковано (например, гипохлорит
кальция в герметичных барабанах), то пробы отбирают из неболь-
шого числа барабанов, например из 10 барабанов, расположенных
в разных местах всей партии общим числом 1000 барабанов. Кроме
того, подвергают визуальному контролю не менее 10% партии; при
этом особое рнимание уделяют герметичности упаковки.
371
Если же дегазирующее средство упаковано плохо, то качество
его в каждой отдельной упаковке может очень сильно отличаться.
В этом случае число выборочных проб должно быть увеличено. Уста-
новленное в каждом конкретном случае число проб зависит также
и от периодичности контроля. В случае веществ долгосрочного
хранения (например, 10 лет) достаточно ежегодно отбирать пробы
максимально из 1 % упаковок. Хлорная известь или другие быстро
изменяющиеся вещества испытывают по меньшей мере каждые
полгода.
Из разных мест выбранной для отбора проб упаковки отбирают
несколько проб и составляют из них среднюю пробу. Так, например,
технический стандарт предусматривает для дихлорамина Т отбор
при помощи щупа трех проб из мест, отстоящих на 150 мм от края
упаковки и по возможности удаленных друг от друга, после чего
все пробы перемешивают в ступке. Щуп при отборе проб должен
доставать до дна упаковки; в качестве щупа может быть использо-
вана обычная стеклянная трубка.
Некоторые дегазирующие вещества и растворители так мало
изменяются при хранении, что нет необходимости каждый раз от-
бирать и исследовать пробы. Вместе с тем для принятия своевре-
менных мер против коррозии или других повреждений тары тре-
буется периодически контролировать упаковку.
14.3. ИСПЫТАНИЕ ДЕГАЗИРУЮЩИХ СРЕДСТВ ПЕРЕГОНКОЙ
Качество растворителей, жидких дегазирующих веществ или дега-
зирующих растворов, составленных из жидких компонентов (на-
пример, смеси аминов и алкоголятов), может быть определено
фракционной перегонкой.
Так как большинство растворителей и жидких дегазирующих
средств кипят ниже 100 °C, то перегонные колбы рекомендуется
нагревать на водяной бане. Неперегоняемый остаток после пере-
гонки определяют по разности между весом колбы после перегонки
и весом пустой колбы. При перегонке термически неустойчивых
или неизвестных веществ следует принять все меры предосторож-
ности.
Для установления веса фракции, отогнанной в определенном
температурном интервале, следует предварительно подготовить та-
рированные приемники с пробками; если дистиллят гигроскопичен
или чувствителен к влаге воздуха, приемник должен быть защи-
щен хлоркальциевой трубкой, предупреждающей доступ влаги из
воздуха.
Некоторые дегазирующие вещества (моноэтаноламин и другие
амины) лучше всего перегонять в вакууме.
14.4. ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ И НАСЫПНОЙ МАССЫ
Плотность жидкостей можно определять ареометром. Для этого
жидкость наливают в мензурку и погружают в нее ареометр так,
372
чтобы он не прикасался к стенкам. Показания ареометра на уров-
не зеркала жидкости и являются плотностью, выражаемой в грам-
мах на кубический сантиметр. Относительная плотность жидкости
может быть точно определена также пикнометром по разности
взвешиваний испытуемой жидкости и воды.
Определение насыпной массы твердого вещества наиболее про-
сто осуществляется по разности масс мерного цилиндра до и после
наполнения его сыпучим веществом.
14.5. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ
Для определения температуры плавления вещество, подлежащее
испытанию, тонко измельчают, а само определение проводят в при-
борах, описанных в разделе 8.2. Очень многие дегазирующие ве-
щества разлагаются при температуре плавления или даже при бо-
лее низкой температуре.
Неорганические дегазирующие вещества имеют в большинстве
случаев высокие температуры плавления, которые описанными ме-
тодами не могут быть установлены.
14.6. ОПРЕДЕЛЕНИЕ НЕРАСТВОРИМОГО ОСТАТКА
ТВЕРДЫХ ДЕГАЗИРУЮЩИХ ВЕЩЕСТВ
Для этого определения готовят при 20 °C небольшое количество де-
газирующего раствора определенной концентрации и пропускают
его через предварительно взвешенный стеклянный фильтр, после
чего фильтр сушат и повторно взвешивают.
14.7. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВЛАГИ
Выбор метода, который может быть использован для определения
влаги, зависит от свойств исследуемого вещества.
В случае дихлорамина и стабилизованного гексахлормелами-
на содержание влаги определяют сушкой до постоянного веса точно
отвешенных количеств вещества (1 — 1,5 а). Дихлорамин лучше всего
сушить при комнатной температуре в эксикаторе, а стабилизо-
ванный гексахлормеламин — в сушильном шкафу при температуре
70 °C, но не выше 75 °C.
Не следует определять содержание влаги в хлорной извести, ги-
похлорите кальция и монохлорамине сушкой, так как при этом
могут возникать различные нежелательные реакции. В данном слу-
чае наиболее удобным является карбидный способ определения
влажности.
Способ заключается в том, что точную навеску вещества (1—2 г) смеши-
вают в колбе емкостью 150 мл с 10 г битого стекла или фарфоровых бус, за-
тем туда добавляют примерно 4—5 г тонко растертого карбида кальция. В ре-
зультате реакции выделяется ацетилен, который собирается в газовой бюретке,
где измеряют его объем. Перед добавлением карбида проверяют, герметично ли
соединена колба е газовой бюреткой, что можно установить по неизменному
373
уровню жидкости в бюретке. До этого момента карбид находится в колбе в
маленькой пробирке. Затем колбу встряхивают, в результате чего карбид сме-
шивается с веществом. Объем выделившегося газа определяют примерно через
5—7 мин после смешения вещества с карбидом. Содержаиие влаги рассчитывают
по формуле:
V I 760 (1 + 0.00367Т) ] 0,1633
— ----------------------------— /о
А
где V — объем ацетилена, мл; р — атмосферное давление, мм рт. ст.-, А — иавеска
вещества, г; Т — температура воздуха, °C.
Другой метод определения влажности основан на перегонке.
Для этого 10 г вещества взвешивают в колбе и смешивают с 50—100 мл рас-
творителя. Гипохлориты смешивают с ксилолом, дихлорамииы — с дихлорэтаном.
При определении влажности дихлорамииа берут иавеску 50 г. Колбу, снабжен-
ную иасадкой для определения влаги и обратным холодильником, осторожно
нагревают (в присутствии пористых шариков!). При выборе насадки для сбора
дистиллята следует иметь в виду, что дихлорэтан тяжелее, а ксилол легче воды.
Кипячение продолжают примерно 2 ч. Содержание влаги в веществе устанавли-
вают по объему отогнанной воды.
14.8. АНАЛИЗ ЩЕЛОЧНЫХ ДЕГАЗИРУЮЩИХ ВЕЩЕСТВ
Содержание щелочи в дегазирующем веществе определяют титро-
ванием 0,1 н. раствором соляной кислоты.
Для контроля качества едкого натра берут иавеску, содержащую примерно
ОД г NaOH. После прибавления 2—3 капель метилового оранжевого пробу ти-
труют до слабо-красного окрашивания. Расход 0,1 и. раствора НС1 составляет
примерно 50 мл.
Содержаиие NaOH в продукте рассчитывается по следующей формуле:
0,0040 ^00 _%Na0H
где о — количество 0,1 и. раствора НС1, пошедшего иа титрование, мл; А — на-
веска вещества, е.
Если необходимо установить содержание NaOH или Са(ОН)2 в
гипохлоритах, то перед титрованием гипохлориты разлагают.
Точную навеску гипохлорита кальция (около 1 г) тщательно растирают с не-
большим количеством воды и количественно переносят в мерную колбу ем-
костью 250 мл. Затем добавляют 3%-ный раствор Н2О1 до тех пор, пока не
прекратится синее окрашивание иод-крахмальной бумаги.
Для определения содержания NaOH проводят ацидиметриче-
ское титрование 25 мл приготовленного раствора так же, как и при
испытании едкого натра. Для определения содержания Са(ОН)2
раствор титруют в присутствии фенолфталеина до обесцвечивания
красной окраски.
Содержание аммиака в дегазирующих растворах определяют от-
гонкой.
Для этого раствор, подлежащий испытанию, подкисляют и затем добавляют
коицеитрироваииый раствор NaOH. Газообразный аммиак отгоняют водяным
паром с применением насадки Кьельдаля и вертикального холодильника и улав-
374
ливают определенным количеством 4%-иого раствора борной кислоты. Отгонку
ведут примерно 15 мин, после чего дистиллят титруют 0,1 и. раствором НС1 в
присутствии индикатора метилового оранжевого. Содержание аммиака рассчи-
тывают по формуле:
Л
Навеска испытуемого вещества должна составлять примерно ОД г.
Бикарбонат аммония анализируют непосредственным титрова-
нием навески вещества 1 и. раствором HjSO4 или НС1. При этом
навеска должна составлять примерно 2 а. Расчет ведут по формуле:
----—------- % бикарбоната аммония
Карбонат натрия можно анализировать аналогично (коэффи-
циент пересчета 0,053) . Таким же путем можно определять аммиак
в его растворах, не прибегая к перегонке. Удобно для взятия на-
вески аммиака пользоваться ампулами.
Для этого взвешивают пустую ампулу, затем шарик ампулы нагревают и
капилляр ее погружают в раствор аммиака. После охлаждения шарика капил-
ляр вынимают из раствора, запаивают и ампулу снова взвешивают. Для анализа
берут примерно 1,5 мл аммиака. Содержание аммиака устанавливают обратным
титрованием. Для этого ампулу опускают в стакан, содержащий 40 мл 1 и. рас-
твора HiSO< или НС1, и оттитровывают избыток кислоты 1 и. раствором NaOH
(индикатор метиловый красный). Расчет проводят по формуле:
(40-о)*0,017* 100 _
--------д 70 Г*
Этаиоламии можно также анализировать объемным способом.
Путем разгонки в вакууме получают ряд фракций: первая фракция
содержит в основном моноэтаноламин, другие фракции содержат
ди- и триэтаноламины. Каждую фракцию титруют 0,1 н. раствором
НС1 илн H2SO4 в присутствии метилового красного.
Расчет содержания моноэтанол амина в 1-й фракции ведут по
формуле
v • 0,0061 * 100
А
а во 2-й фракции по формуле:
д. inn
Лом— м’100
где Л41 и Мг — молекулярные веса моно- и диэтаноламина, равные 61,0 и 105,1
соответствеиио; А — навеска этаиоламииа (около 0,2 г).
14.9. ИСПЫТАНИЕ ДЕГАЗИРУЮЩИХ ВЕЩЕСТВ,
СОДЕРЖАЩИХ АКТИВНЫЙ ХЛОР
Выбор способа испытания соединений, содержащих активный хлор,
зависит от их растворимости. В остальном способы основаны иа
одном и том же принципе. Иод, образующийся в реакции между
С1* + 2Г —> 1, + СГ
375
определяют*титрованием. Из реакции следует, что один атом ак-
тивного хлора окисляет два иона иода с образованием молекулы
иода.
Для испытания водорастворимых продуктов достаточно 1 г вещества. На-
веску помещают в мерную колбу емкостью 250 мл и доливают до метки водой
(гипохлориты предварительно растирают в ступке с -небольшим количеством
воды). После энергичного встряхивания из полученного раствора пипеткой от-
бирают 50 мл, смешивают с 7—8 мл 10%-ного раствора KI и 3—5 мл концентри-
рованной уксусной кислоты и титруют 0,1 и. раствором Na2S2O3. Незадолго до
конца титрования прибавляют 1 мл 0,5%-ного раствора крахмала и титруют до
исчезновения синей окраски. Для расчета содержания активного хлора поль-
зуются следующей формулой:
о И7 0,003546- 100
Aw
где v — количество 0,1 н. раствора Na2S2O3, пошедшее на титрование, мл; W—
объем мерной колбы, мл; w — объем пробы, взятой для определения, мл; А —
навеска, г.
В случае высокоактивных соединений берут несколько меньшую
навеску, а в случае хлорамина вдвое увеличенную. При определе-
нии хлорамина для подкисления раствора вместо уксусной кисло-
ты добавляют 1 мл 10%-ной НС1 и перед титрованием выдержи-
вают реакционную смесь 10 мин в темном месте.
Определение активного хлора в нерастворимых в воде хлор-
аминах проводят в ледяной уксусной кислоте.
Взвешивают 0,2 г вещества в колбе Эрлеимейера, снабженной пришлифо-
ванной пробкой, смешивают с 10 мл ледяной уксусной кислоты и 15 мл свеже-
приготовленного 10%-ного раствора KI и титруют 0,1 н. раствором Na2S2O3 сна-
чала до желтого окрашивания и затем, после прибавления 1 мл 0,5%-ного рас-
твора крахмала, до обесцвечивания. Расчет ведут по формуле:
v 0,003546 • 100
А
14.10. ИСПЫТАНИЕ МОЮЩИХ СРЕДСТВ
Свойства моющих средств, особенно состоящих из синтетических
поверхностно-активных веществ, в процессе хранения изменяются
незначительно.
Увеличение содержания влаги можно определять, в зависимости
от состава средства, при помощи одного из методов, описанных в
разделе 14.7.
Иногда возникает необходимость установить примерный состав
неизвестного моющего средства. Прежде всего следует оценить его
пенообразующую способность. Для этого приготавливают прибли-
зительно 0,1%-ный раствор средства в воде и энергично взбалты-
вают.
Если хотят отделить поверхностно-активное вещество от не-
органических компонентов, то для этого пользуются экстракцией.
Отвешивают примерно 5 г вещества, переносят навеску в колбу, содержа-
щую 100 мл этилового спирта и снабженную обратным холодильником, и нагре-
вают примерно 30 мин на водяной бане. После фильтрования остаток еще два-
376
жды обрабатывают двумя порциями спирта по 75 мл каждая. Нерастворимый
остаток после сушки и взвешивания может быть подвергнут другим испыта-
ниям.
Для выделения спирторастворимой части спирт отгоняют и остаток сушат
при 105 °C.
Для исследования твердых веществ (илй" паст) прежде всего
используется элементный анализ. Жировые мыла можно отличить
от большинства анионоактивных веществ по отсутствию серы. При-
сутствие азота указывает на катионоактивные вещества-или на
присутствие карбамидной функциональной группы.
Для того, чтобы установить или исключить присутствие неионо-
генных веществ, можно провести испытания на присутствие катио-
не- или анионоактивных веществ. Если прикапывать 1%-ный рас-
твор поверхностно-активного вещества к разбавленному раствору
ацетата алюминия, то образование белого или желтоватого осадка
указывает на наличие анионоактивного вещества. Присутствие ка-
тионоактивных веществ подтверждается тем, что при прикапыва-
нии к испытуемому раствору 1%-ного раствора мерзолята выпа-
дает плотный белый осадок.
Присутствие неорганических компонентов устанавливают ка-
чественными реакциями на карбонат (бурное выделение СОг при
действии разбавленной НС1), сульфаты (осаждение BaSO4 при до-
бавлении ВаСЬ к солянокислой среде) и фосфаты.
14.11. ИССЛЕДОВАНИЕ НЕИЗВЕСТНОГО
ДЕГАЗИРУЮЩЕГО ВЕЩЕСТВА
Подобно тому, как это было уже описано при испытании моющих
средств, прежде всего следует установить присутствие неорганиче-
ских и органических веществ.
Если, например, речь идет о веществе, пахнущем хлором, то
уже осторожным 'нагреванием малого количества вещества в труб-
ке для прокаливания можно вызвать его разложение. При этом та-
кие органические вещества, как хлорамин, дихлорамин или гекса-
хлормеламин, разлагаются с выделением черного дыма, а неорга-
нические вещества — гипохлорит кальция или хлорит натрия более
устойчивы к воздействию температуры. Кроме того, при их разло-
жении черный дым не выделяется.
Отношение исследуемого вещества к вод^ также дает определен-
ные сведения о его природе. При растворении вещества в воде мож-
но ориентировочно установить pH раствора при помощи универ-
сальной индикаторной бумаги. Кальциевые и натриевые соли легко
различить при помощи ручного спектроскопа. Грубо отличить их
можно по более слабой растворимости кальциевых солей. Специаль-
ные способы исследования см. сс.1.
Кроме запаха, предварительные данные о наличии содержа-
щих активный хлор продуктов дают реакции с KI или проба с
иод-крахмальной бумагой (коричневое или синее окрашивание).
13 Зак. 677
377
Если должна быть установлена идентичность двух твердых ор-
ганических веществ, то это можно сделать, определив температуру
плавления смешанной пробы (смешение двух идентичных веществ
не приводит к изменению температуры плавления).
Жидкости лучше всего исследовать методом фракционной раз-
гонки. Однако предварительно следует провести качественный эле-
ментный анализ. Если обнаружен азот, но отсутствует запах амина
и исключено наличие продуктов, содержащих активный хлор,
то это может означать присутствие опасных нитросоединений (взры-
воопасно!). Если невозможно провести идентификацию по извест-
ным температурам кипения, то в зависимости от обстоятельств на-
до попробовать получить твердые производные. Температуры плав-
ления этих производных можно сравнить с табличными данными
для температур плавления различных органических соединений.
Так, например, амины могут быть идентифицированы превраще-
нием их в бензолсульфамиды.
Для этого 2 г вещества смешивают с 40 мл 10%-ного NaOH и 4 г бензол-
сульфохлорида н недолго нагревают на водяной бане. После подкисления раз-
бавленной НС1 образовавшийся осадок отфильтровывают, промывают холодной
водой и после перекристаллизации из разбавленного спирта определяют его
температуру плавления.
Другие методы исследования неорганических и органических ве-
ществ в случае необходимости можно найти в специальной лите-
ратуре '•2.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Как определяют плотность жидкостей?
2. Как определить насыпную массу твердого вещества?
3. Как определить нерастворимый остаток твердого дегазирующего вещества?
4. Какое значение имеет определение содержания влаги в дихлорамине и гекса-
хлормел амине?
5. Как определить содержание аммиака в дегазирующем растворе?
6. Как устанавливается состав неизвестного моющего средства?
7. Как можно исследовать неизвестное дегазирующее вещество?
ЛИТЕРАТУРА
1. Jander G., Wend Н., Lehrhuch der analytischen praparativen Chemie,
Leipzig.
2. Organikum, Organisch-chemisches Grundpraktikum (AutorenkoIIektiv), Berlin,
1962.
ПРИЛОЖЕНИЕ
МЕРЫ ПЕРВОЙ ПОМОЩИ ПРИ ПОРАЖЕНИЯХ
И ОТРАВЛЕНИЯХ ДЕГАЗИРУЮЩИМИ ВЕЩЕСТВАМИ
И РАСТВОРИТЕЛЯМИ
При всех несчастных случаях, особенно при поражении глаз и отравлениях, не-
обходимо тотчас вызвать врача. Люди, ие имеющие медицинского образования,
могут оказывать только первую помощь.
Степень поражения или отравления далеко ие во всех случаях можно оце-
нить сразу. К несчастным случаям надо всегда относиться серьезно.
Желательно сразу предотвратить развитие поражения или отравления.
Пораженного следует немедленно отправить в больницу. В качестве сопро-
вождающего должен быть назначен сведущий человек.
Человеку, находящемуся в бессознательном состоянии, нельзя давать ни-
каких жидкостей.
Так как судьба отравленного зависит от быстроты оказания помощи, то
действовать надо быстро и в соответствии с правилами.
Принятые внутрь яды, медленно разрушающие оболочки, должны быть уда-
лены из желудка промыванием или рвотой. Рвоту вызывают щекотаиием глотки
птичьим пером или пальцами.
Промывать желудок вливанием теплой воды через рот шлангом длиной
150 см. При повторном промывании к воде добавляют около 2% медицинского
активированного угля. Таким же путем можио ввести в качестве слабительного
Na^SOf. Для этого применяют раствор 30 г NajSCU в 500 мл воды, к которому
следует добавить 30 г медицинского активированного угля.
Щелочи
Кожные покровы. Места попадания брызг щелочи промыть большим количе-
ством воды или 1%-ной уксусной кислотой.
Глаза. Пострадавшего положить иа спину и для удаления остатков щелочи
промывать глаза 10—15 мин струей воды. После промывания глаз покрыть их
чистым белым платком. Для промывания глаз нельзя применять никакие кис-
лоты.
Рот, желудок. Немедленно принять внутрь 1%-иый раствор лимонной или
уксусной кислоты. В случае крайней необходимости следует дать молоко с сы-
рым яйцом. Не давать средств, вызывающих рвоту, желудок ие промывать.
Кислоты, галогеиаигидриды кислот
Кожные покровы. Брызги кислоты смыть обильным количеством воды или
нейтрализовать мелом, содой или бикарбонатом натрия.
Глаза. Поступать так, как при попадании щелочей. Для промывания глаз
нельзя употреблять щелочные растворы.
Рот, желудок. Давать охлажденную льдом воду, в которой суспендирована
окись магния (Magnesia usta). В крайнем случае дать раствор бикарбоната
натрия или суспензию мела (карбоиата кальция). При этом следует помнить, что
обильное образование СО» может стать опасным.
13* 379
Аммиак, гидроокись аммония
При вдыхании. Немедленно вынести на свежий воздух. Никакого искусствен-
ного дыхания, ингаляция кислорода, покой, транспортировать в лежачем поло-
жении, пораженного держать в тепле, дать вдыхать пары уксусной кислоты,
спирта или эфира. Немедленно эвакуировать в больницу.
При попадании аммиака на кожные покровы, в глаза, рот и желудок посту-
пать аналогично случаям ожогов щелочью.
Хлор
При вдыхании. Немедленно вынести на свежий воздух. Запретить самостоя-
тельно передвигаться, транспортировать в лежачем положении, глубокое дыха-
ние в состоянии покоя, никакого искусственного дыхания. Пораженного держать
в тепле, никаких жидких средств внутрь. Дать дышать парами спирта илн воды,
для смягчения раздражения вдыхание аэрозоля 0,5%-ного раствора бикарбоната
натрия. Немедленно эвакуировать в больницу.
Гипохлориты кальция и щелочных металлов
Кожные покровы и глаза обрабатывать так же, как при поражении щелочами.
Рот, желудок. Немедленно дать внутрь молоко, ввести обильное количество
медицинского активированного угля, в остальном действовать так же, как и в
случаях отравления щелочами или кислотами.
Дихлорэтан, четыреххлЬристый углерод и подобные соединения
При вдыхании. Немедленно вынести на свежий воздух, держать в тепле и
покое. Вдыхание кислорода, искусственное дыхание. Дать внутрь крепкий горя-
чий кофе.
Кожные покровы. Хороший уход за кожей, жировые мази.
Глаза. Промывать глаза не менее 10—15 мин струей воды. Вливать по кап-
лям парафиновое масло.
Рот, желудок. Немедленно промыть желудок водой, дать внутрь обильное
количество медицинского активированного угля.
Метанол
Пострадавшему оказать первую помощь даже при подозрении на отравление.
Обильно поить. В течение часа и более через каждые 15 мин вводить внутрь
4 г бикарбоната натрия с обильным количеством воды. Промыть желудок, до-
бавляя в раствор медицинский активированный уголь. Вводить внутрь горячее
питье, лучше всего крепкий кофе. Пострадавшего предохранять от охлаждения.
Углеводороды (бензин и подобные ему вещества)
При вдыхании. Немедленно вынести на свежий воздух. Вводить внутрь
крепкий кофе. Никаких спиртных напитков и молока.
Рот, желудок. Немедленно промыть желудок с добавлением медицинского
активированного угля.
ПРЕДМЕТНЫЙ
УКАЗАТЕЛЬ
Адамсит (Дифеииламинохлорарсии,
10-Хлор-9,10-дигидрофенар-
сазинхлорид)
индикация
в воздухе 256
по запаху 222
обнаружение 91 сл., 94 сл., 119,
262, 265
определение
колориметрическое 100
методом УФ-спектроскопии 199
объемное 98
улавливание аэрозоля 256
Адренохром 142
2-Азобеизол-1 -нафтилацетат
обнаружение фосфорорганических
ОВ 208
определение
зарина 73
фосфорорганических ОВ 73, 177
Азот
обнаружение 27, 28
окислы, пригодность как дегаза-
торов 346
определение 36, 40
Азотистые иприты см. также Азоти-
стый иприт
обнаружение 78, 85 сл.
определение
колориметрическое 86 сл.
объемное 88
Азотистый иприт [Трис-(2-хлорэтил) -
амин, 2,2',2"-Трихлорэтил-
амин, Р,^'Р"-Трихлорэтил-
амин]
дегазация
активированным углем 368
бисульфатом натрия 280
гипохлоритами 335
дихлорамидом метансульфокис-
лоты 343
изоциаиурхлоридом 343
растворителями 353
Азотистый иприт
дегазация
тиосульфатом натрия 310
допустимая концентрация в воде
269
индикация
бумагами 231, 232
в полевых лабораториях 261,
262, 265
по запаху 222
порошками 225
трубками 237, 238
обнаружение 85 сл., 237, 238
определение
колориметрическое 86, 87
объемное 88, 89
стойкость
в воде 251
на местности 277
экстракция растворителями 259
Азотная кислота
как дегазирующее вещество 346
обнаружение
адамсита 96
алкалоидов 134, 136, 137
мескалина 145
пикротоксина 139
свйица 125
определение адамсита 100
Аквииит 267
Аконитин, обнаружение 133, 135, 136,
138, 139, 210
цис-Аконитовая кислота 90
Активированный уголь как дегази-
рующее вещество 366, 368,
369
Алкалоиды 132 сл.
групповое разделение 132, 133,
135 сл.
обнаружение 50
индикаторным набором 247,
249, 265
по температуре плавления 132,
133
цветными реакциями 132, 134
сл., 265
383
Алкалоиды
обнаружение
цветными реакциями 132, 134
с л., 265
спорыньи 143
стойкость в воде 251
экстракция из проб 258 сл.
Алкиламины как дегазирующие ве-
щества 315, 316
Алкоголяты как дегазирующие ве-
щества 304 сл.
Алюминий
как, восстановитель 150
обнаружение ртути 149
Алюминия сульфат, дегазация воды
369
Алюмогидрид литня, обнаружение
фосфора 32
Амальгама натрия, обнаружение
хлорпикрина 119, 120
6-Амино-1-нафтол-3-сульфокислота
(И-кислота), определение
хлорфеноксиуксусных кис-
лот 142
Амины как дегазирующие вещества
311 сл., 375
Амитон см. Тетрам
Аммиак
как дегазирующее вещество 311
сл., 357
обнаружение
бромбензилцианида 118, 119
параоксона 65
паратиона 65
помощь при отравлениях 379, 380
Аммоний сульфаминовокислый, обна-
ружение тетрама 68
Аммония бикарбонат как дегазирую-
щее вещество 314
Аммония гидроокись, помощь при от-
равлениях 379, 380
Аммония молибдат, обнаружение
окиси углерода 127
фосфорорганических ОВ 206
Аммония нитрат, Индикация галоген-
алкиламинов 231
Аммония персульфат, обнаружение
тетраэтилпирофосфата 66
Аммония сульфид, обнаружение
окиси углерода 127
холинэстеразы 169
Аммония тиокарбаминат, обнаруже-
ние токсичных катионов 146
Аммония хлорпалладит, определение
фосфорилтиохолинов 76
Анабазин, обнаружение галогенциа-
иов 106
Анализ ОВ
азотистых ипритов см. Азотистые
иприты
бактериального 140 происхождения
биологиечский 184 сл.
биохимический см. Биохимический
анализ ОВ
животного происхождения 140
загущенных 266 сл.
запись результатов 20, 21, 33
извлечение из проб воздухом 259
ИК-спектроскопический 127, 200,
244, 245
маркировка проб 257
минерализация (сожжение) .образ-
цов 28, 30, 35 сл., 40, 41
мышьяксодержащих см. Мышьяк-
содержащие ОВ
нейтронный активационный 191
неорганических 145 сл.
цервно-паралитических см. Нерв-
но-паралитические ОВ
оборудование рабочего места
21 сл.
организация работы 19 сл.
по давлению пара 194 сл.
по функциональным группам 46 сл.,
100, 191
подготовка проб 257 сл.
полевой 260 сл., 264 сл.
психохимических 142 сл.
раздражающих см. Раздражаю
щие О В
свинецорганических 124 сл.
смесей 264 сл.
стабилизованных 266, 267
тактических 266, 267
техника безопасности 23 сл.
УФ-спектроскопический 199 сл.
физико-химический 189 сл.
физический см. Физические мето-
ды анализа ОВ
фитотоксических 141, 142
флуоресцентный 70, 104, НО, 191,
198
фосфорорганических см. Фосфор-
органические ОВ
фторорганических 129 сл.
хемолюминесцентный 103
хроматографический см. Хромато-
графический анализ ОВ
чувствительность определения 25,
26
экспресс-методом 72
экстракция растворителями 257 сл.
элементный 27 сл., 30 сл., 249
Анилин в анализе
галогенцианов 107
фосгена 111
хлорпикрина 121
384
Анилиновая вода
индикация ОВ 264, 265
обнаружение фосгена 111
определение
ди- и трифосгена 115
фосгена 113
Антидоты 279, 314, 315
Антипул 327
Арсенат-ион, обнаружение 146, 150
Арсенит-ион, обнаружение 146, 150
Атропин, обнаружение 49, 50, 133 сл.,
210
Ахолест 168
Ацетгидроксамовая кислота, опреде-
ление ацетилхолина 168
Ацетилхолин
гидролиз 167
, определение 168, 169
Ацетилхолинэстераза в анализе ОВ
158 сл., 164 сл.
Ацетобутират целлюлозы в смесях
ОВ 266
Ацетонитрил как дегазирующее ве-
щество 353
Ацетоуксусный эфир, обнаружение
галогенцианов 107
Ацилхолины, обнаружение 59
Базогрелит (Двухосновной гипохло-
рит кальция) 327, 328, 334
Барбитуровая кислота
индикация
галогенцианов 226
хлорциана 238
обнаружение
галогенцианов 107, 108, 226
никотина 139
хлорпикрина 122
Барий, обнаружение 146, 147, 211
Бария перхлорат, определение серы
39
Бария сульфат, индикация ОВ 226,
227
Бензальдегид, обнаружение цианид-
ионов 104
Бензгидроксамовая кислота, обнару-
жение фосфорорганических
соединений 63
Бензидин
индикация синильной кислоты 102,
229, 230, 287
обнаружение
Галогенцианов 107
цианид-иоцов 102
Бензидин-медьацетатная индикатор-
ная бумага 229, 230
N-Бензиланилии, индикация фосгена
229
4-Беизилпиридин
индикация
галогеициаиов 226
хлорциана 238
обнаружение галогенцианов 106,
226
Бензин как дегазирующее вещество
357, 358 ?
помощь при отравлениях 380
Бензоин, обнаружение цианид-ионов
104
Бензоиновая конденсация 104, 105
Беизолсульфохлорид, обнаружение
перекисью водорода 59
третичных аминов 49
п-Беизохиион, определение цианид-
ионов НО
Бензохинонмонооксимсульфокислоты
эфир, определение цианид-
ионов 110
Берберин, обнаружение 135 сл.
Бериллий, обнаружение 147, 211
Берлинская лазурь
индикация
ОВ в полевых лабораториях
264, 265
синильной кислоты 230
обнаружение
азота 28
бромбензилциаиида 118
цианид-ионов 101
Беттендорфа реакция 93
Биологический анализ ОВ 184 сл.
Биофан С 168
Биохимический анализ ОВ 158 сл.
в полевых лабораториях 261, 262,
264, 265
ДФФ 159
зарина 159
инсектицидов 159
приборы 245, 246
фосфорорганических 159, 207, 208,
247, 255
«число оборотов» 158
Бис- (п-диметнламино-о-амииофенил) -
кетой, индикация фосгена
226
Бис-1,2-(N-диметиламино)-этан, дега-
зация кожных покровов 317
Бис-2-дихлорэтиловый тиоэфир, опре-
деление 83
385
4,4'-Бис- (диэтнламиио) -бензофенон-
оксим, индикация нервно-па-
ралитических ОВ 227
Бис- (n-нитрофенил) -дисульфид, обна-
ружение ТИОЛОВ ЬУ
Бис- (фенилцианметил) -сульфид, опре-
деление бромбензилциаиида
119
Бис-2-хлорэтиловый тиоэфир см. Ип-
рит
1,2-Бис-(2-хлорэтилтио)-этан см. Се-
сквииприт
Ботулизма токсин 140
Бром
обнаружение 211
фосфорорганических ОВ 206
определение 43
Бромацетон
в смесях ОВ 266, 267
индикация ОВ 263, 265
обнаружение 77, 116
определение 199
Бромбензилцианид
индикация
в воздухе 256
в полевых лабораториях 263,
265
по запаху 222
порошками 224
обнаружение 50, 116, 118,'119, 123
определение 119, 199
улавливание аэрозоля 256
Бромметилэтилкетон
в смесях ОВ 266
обнаружение 116
определение 199
6-Бром-2-нафтилацетат, определение
фосфорорганических ОВ 177
Бромная вода, обнаружение синиль-
ной кислоты 106
N-Бромсукциннмид, обнаружение
фосфорорганических ОВ 206
Бромтион, определение 203
5-Бром-4-хлориндоксилацетат, опре-
деление ферментов 170
Бромциан
определение 111, 121
отличие от хлорциана 108
Бруции, обнаружение 49, 50, 133 сл.,
210
Буфотенин 142, 143
Валди метод обнаружения алкалои-
дов 208
Ванилин, обнаружение галогениро-
ванных кетонов 116, 263, 265
Ван Урка — Смита реакция 143, 144
Вариаминовый синий 108
Вератрин, обнаружеине 50, 135 сл.
Винсенит, индикация 266
Вилларда и Винтера метод определе-
ния фторидов 41
Винсеиит, нинднкация 266
Висмут, обнаружение 211
Висмута нитрат, индикация галоген-
алкиламинов 231
Вода
анализ 259, 260, 268, 269
дегазация 281, 335 сл., 346, 347,
366 сл.
отбор проб 251
Водорода перекись 347, 348
; дегазация
V-газов 348
зарина 308, 348
иприта 348
индикация ОВ 60
дихлофоса 60
зарина 232
серного иприта 236
фосфорорганических ОВ 60, 243
обнаружение
ацилхолииов 59
бензолсульфохлорида 59
дифосгена 59
зарина 59
пентакарбонила железа 128
табуна 59
фосфорорганических ОВ 52 сл.
хлорацетофенона 117
хлорпикрина 121
циаиид-ионов 103
окислительно-восстановительный
потенциал 319
определение фосфорорганических
ОВ 69 сл., 72
Воздух
дегазация 281, 367, 368
извлечение ОВ 253—256
Вуртцшмитта универсальная бомба
36
Газоанализаторы 240 сл.
Газоопределители 238
V-Газы
дегазация
гипохлоритами 334, 335
дихлораминами 341
минеральными кислотами 280
перекисью водорода 348
растворителями 353, 358
хлорноватистой кислотой 320
индикация
р полевых лабораториях 261
386
V-Газы.
индикация по запаху 222
стойкость на местности 277
Галогенангидриды кислот, помощь
при отравлениях 379
Галогенированные кетоны 116 сл.
Галогенированные тиоэфиры см. Сер-
ные иприты
Галогенпроизводиые угольной кисло-
ты см. Фосген, Дифосген
Галоген цианы
индикация
в полевых лабораториях 265
карандашами 226
обнаружение 101 сл., 106 сл.
определение 111, 112
хлорпикрина 122
Галогены
обнаружение 27, 29,-30, 211
определение 100
Гваяковая смола
индикация синильной кислоты 230
обнаружение циаиид-ионов 103
Гексаметилентетрамин (Уротропин)
в смесях ОВ 266
как дегазирующее вещество 314
определение фосгена 114
Гексанол, поглощение фторорганиче-
ских ОВ 130
Гексахлормеламин (ДТ-6) как дега-
зирующее вещество 342, 373,
377
Гидрастин, обнаружение 138
Герони, обнаружение 135 сл.
Гидроксамовые кислоты
как антидоты 315
обнаружение фосфорорганических
ОВ 63, 64, 73
2-Гидроксиамиио-3,5-дициаи-4,6-ди-
иитрофенол (Изопурпуровая'
кислота) 103
Гидроксиламин солянокислый, опре-
деление ацетилхолина 168
Гидропирит 348
Гиосциамин, обнаружение 138
Гипобромит, определение азота 40
Гипохлориты
как дегазирующие вещества 283,
291, 296, 320 сл., 333 сл., 376
помощь при отравлениях 380
Глиоксалевая кислота, обнаружение
алкалоидов спорыньи 143
Глиоксалевокислый натрий, определе-
ние производных индола
144
Глутакоиовый диальдегид, обнару-
жение галогенцианов 107,
108
Гоматропин, обнаружение 210
Гомомартонит, индикация 266
Гопкалит 128
Грии стар 267
Гриньяра реакция 93
Грюнкрейц 1 267
Гутцейта реакция 32, 150, 238, 261,
262, 338
Далапон см. а,а-Дихлорпропионовая
кислота
Дегазатор 40 343
Дегазация
агенты см. Дегазирующие веще-
ства
адсорбентами 281, 365 сл., 368
активированным углем 366, 368, 369
алкиламинами 315, 316
алкоголятами 304 сл.
аминами 311 сл.
аммиаком 311 сл., 357
аммоний бикарбонатом 314
ацетонитрилом 353
бензином 357, 358
в камерах 313
в лаборатории 22 сл.
в полевых условиях 332
в фильтрах 367
влияние растворителей 294, 295
— физических факторов 295 сл.
водой 353
воды 281, 335 сл., 346, 347. 366 сл.
водяным паром 281
военной техники 281, 309, 364
воздуха 281, 367, 368
воздухом 281
галогенированными углеводорода-
ми 353
гексаметилентетрамином (уротро-
пином) 314
гексахлормеламином (ДТ-6) 342,
373, 377
гипохлоритами 283, 291, 296, 320
сл., 333 сл., 376
двуокисью хлора 336, 337
диметилсульфоксидом 353
диметилформамидом 353
дихлорамидом метансульфокисло-
ты 343
дихлораминами 283, 291, 340 сл.,
372, 373
дихлорэтаном 283, 354, 355, 358,
едкими щелочами 283, 300 сл.
естественная (Самодегазация) 278
387
Дегазация
зараженных поверхностей 356, 357
изоцианурхлоридом 342, 343
катализаторы 293, 294
кипячением 303, 304
коагулянтами 369
кожно-нарывных ОВ 343
кожных покровов 308, 317, 318,
320, 339, 344, 348
комбинированная 278, 280 сл.
комплексными соединениями ами-
нов 317, 318
методы контроля 269 сл.
местности 281
минеральными кислотами 280
моющими средствами 281, 297,
335, 358 сл., 364, 376, 377
нитрометаном 353
нуклеофильными реагентами 288,
289, 298 сл., 304 сл.
обмундирования 281, 303, 304 , 313,
339, 357, 358, 365
озоном 320, 346, 347
окислением 288, 318 сл.
отбор проб 270, 271, 371, 372
перборатами 348
перекисью водорода 308, 319, 320,
348
перманганатами 320
пероксидисерной кислотой 349
персульфатами 320, 348, 349
полная 278
растворителями 281, 352 сл., 356,
357
силикагелем 367, 368
содой 303, 304, 357, 375
соляной кислотой 339
спиртами 353
стиркой 358 сл.
сульфидом натрия 300, 309, 310
сульфурилхлоридом 345
тетраокисью азота 346
тиолятами 310
тиосульфатом натрия 310
тиофенолятами 310
углеводородами 353
фенолятами 307, 368
физическая 278, 280, 281, 352 сл.
фосфатов 293, 294, 310
фосфонатов 288, 293, 294, 310
фтором 320
химическая 278, 280, 281, 283 сл.
хлораминами 283, 290, 291, 337 сл.,
376
N-хлоргликольурнлами 344
хлорированием 288, 289, 318 сл.
хлористым сульфурилом 283
хлоритами 320, 336, 337
хлорноватистой кислотой 320 сл.
Дегазация
хлорной известью 283, 324 сл., 334,
372, 373
хлором 320
частичная 278
экстракцией 281, 357, 358
электрофильными реагентами 288,
289
этаноламииами 375
этиловым спиртом 355, 356
Дегазирующие вещества 279, 283 сл.,
296, 371 сл.
анализ 372 сл.
отбор проб 371, 372
хлорсодержащие 375, 376
щелочные 374, 375
р-Диалкиламиноэтилтиолфосфонаты,
обнаружение 64
2,7-Диаминодифениленоксид, обнару-
жение цианид-ионов 102
о-Дианизидин
в анализе фосфорорганических ОВ
69, 232, 242
определение
дифосгена 115
сесквииприта 84
2,6-Дибром_-М-хлор-п-хинонимин, об-
наружение ОВ 206, 208
Дигидрофенарсазинхлорид см. Адам-
сит
Дигиталин, обнаружение 137
Дигитонин, обнаружение 137
Диизонитрозоацетон
индикация
нервно-паралитических ОВ 227
фосфорорганических ОВ 62, 232
Диизопропилфторфосфат см. ДФФ
Диизопропоксифосфорилтиохолин 163
Димедон см. 5,5-Диметилдигидроре-
зорцин
М,М-Диметиламидо-О-этилцианфос-
фат см. Т^бун
л-Диметиламинобензальдегид
индикация
фосгена 229, 235, 237, 238
хлорпикрина 231
обнаружение
алкалоидов спорыньи 143, 144
фосгена 111, 112
LSD-25
определение производных индола
144
п-Диметиламинобеизилнденроданин,
определение цианид-ионов
108
о-Диметиламиноэтилдиэтилтионфос-
фат
обнаружение 205
определение 209
388
Диметиланилии
индикация
фосгена 229, 235, 237, 238
хлорпикрина 231
обнаружение
фосгена 111, 112
хлорпикрина 121
Диметилглиоксим
обнаружение цианид-ионов 104
определение карбонила никеля 129
N.N'-Диметилдиакридиния динитрат
(Люцигении) 58
5,5-Диметилдигидрорезорцин (Диме-
дон)
обнаружение галогенциаиов 107
определение хлорпикрина 122
Диметилсульфат в смесях ОВ. 267
Диметилсульфоксид как дегазирую-
щее вещество 353
Диметилтриптамин 142, 143
Диметилформамид
как дегазирующее вещество 353
определение хлорацетофенона 117
Диметоат см. Фосфамид
Дииитрил малоновой кислоты, обна-
ружение 123, 124
Динитробензойная кислота, обнару-
жение хлорацетофенона 116,
117
л-Дииитробеизол
индикация ОВ
в полевых лабораториях 263 сл.
этилдихлорарсина 232
обнаружение
бромбензилциаиида 116, 119
Си Эс 123
хлорацетофенона 116, 123
о-Динитробензол, обнаружение циа-
нид-ионов 104, 105
Динитро-о-крезол, определение 142
2,4-Динитрофенол, индикация этилди-
хлорарсина 232
1,8-Диоксинафталин-3,6-дисульфокис-
лота, см. Хромотроповая
кислота
4,4'-Диокси-3,5,3',5'-тетраметоксиди-
фенил, обнаружение цианид-
ионов 103
Дипикриламин, обнаружение фосфор-
органических ОВ 206
Диптерекс см. Хлорофос
Дитиан (Диэтилен дисульфид)
в смесях ОВ 266
обнаружение иприта 81
Дитизон 108
обнаружение
свинца 125, 148, 211
токсичных катионов 148, 211
определение цианид-ионов 109
5,5'-Дитио-бис- (2-нитробензойная)
кислота см. ДТНБ
Дитиогликолят калня, обнаружение
хлорпикрина 121
Дифениламин
индикация фосгена 229
обнаружение фосфорорганических
ОВ 65
Дифениламинохлорарсин см. Адамсит
Дифеиилдисульфид, обнаружение
хлорпикрина 121
Дифенилкарбазид 108
обнаружение фосгена 112
Дифенилмочевина
обнаружение фосгена 111
определение
ди- и трифосгена 115
фосгена 113
Дифенилхлорарсин (Кларк I)
индикация
в воздухе 256
в полевых лабораториях 262,
265
по запаху 222
обнаружение 50, 91 сл., 95, 96
определение
йодометрическое 98 сл.
спектрооскопическое 199
улавливание аэрозоля 256
Дифенилцианарсин (Кларк II)
в смесях ОВ 266
индикация
в полевых лабораториях 262,
265
по запаху 222
обнаружение 50, 91 сл., 119
определение
йодометрическое 98
спектроскопическое 199
Дифосген (Трихлорметиловый эфир
хлоругольной кислоты)
в смесях ОВ 266
индикация
в полевых лабораториях 263
трубками 233, 238
обнаружение 59
определение 114, 115
Дихлорамид метансульфокислоты 343
Дихлорамин Б (ДТ-2, N.N-Дихлор-
бензолсульфамид) как дега-
зирующее вещество 340 сл.
389
Дихлорамин Т (ДТ-2Т, N.N-Дихлор-
n-толуолсульфамид) как де-
газирующее вещество 283,
340, 372, 373
определение бис-2-хлорэтилового
тиоэфира 83
Ди-(2-хлорвинил)-хлорарсин (f-Люи-
зит) 91 сл.
1,3-Дихлор-5,5-диметилгидантоин
(DANC), дегазация кожио-
нарывных ОВ 343
Дихлордиметиловый эфир в смесях
ОВ 266
а,а-ДихлорпропиоИовая кислота (Да-
лапон), обнаружение 209,
212
М,М-Дихлор-л-толуолсульфамид см.
Дихлорамин Т
2,4-Дихлорфеноксимасляная кислота
•(2,4-DB) 209, 212
2,4-Дихлорфеноксиуксусная кислота
(2,4-D) 141, 209, 212
Дихлорфлуоресцеии, определение серы
и хлора 39
Дихлорэтан
как дегазирующее вещество 283,
354, 355, 358
помощь при отравлениях 380
экстракция ОВ 258, 259
Дихлофос, индикация 60
Диэтиламид (+)-лизергиновой кисло-
ты см. LSD
.и-Диэтиламинофенол, индикация фос-
гена 229
Диэтилбарбитурат натрия, обнаруже-
ние фосфорорганических ОВ
178
Диэтилдитиокарбамат серебра, опре-
деление мышьяковистого во-
дорода 97, 98
Диэтилдитиокарбамииат меди, опре-
деление цианидов 109
Диэтилеидисульфид см. Дитиан
Диэтил-4-иитрофенилфосфат см. Па-
раоксон
Диэтилфталат, индикация фосгена
229
Диэтоксифосфорилтиохолин
действие иа ферменты 163, 165
обнаружение 50. 205
Допустимые нормы остаточного зара-
жения 269, 270
Драгера трубки 236, 238
ДТ-6 см. Гексахлормеламин
890
ДТНБ [5,5'-Дитио-бис- (2-иитробен-
зойиая) кислота] 169, 173
обнаружение тиолов 68
определение
активности холинэстеразы 173
тиохолина 169
ДФФ (Диизопропилфторфосфат)
биохимический анализ 159
в смесях ОВ 266
гидролиз 292, 293
дегазация 302, 308
действие иа ферменты 165
индикация 227
обнаружение 50, 64
определение 199
стойкость в присутствии перекиси
водорода 54
2,4-D (2,4-Дихлорфеиоксиуксусиая
кислота) 141, 209, 212
DANC 343
2,4-DB (2,4-Дихлорфеноксимасляиая
кислота) 141, 209
DS-2 295, 305, 316
Едкий иатр
как дегазирующее вещество 283,
300 сл.
помощь при отравлениях 379
Едкое кали
как дегазирующее вещество 283,
300 сл.
помощь при отравлениях 379
Еллоу стар 267
Железа пеитакарбоиил, обнаружение
128
Железа сульфат, индикация синиль-
ной кислоты 230
Железо, обнаружение 211
Железо хлорное
дегазация воды 369
обнаружение
алкалоидов спорыньи 143
колхицина 139
фосфорорганических ОВ 206
определение производных индола
144
Загущенные смеси ОВ 264, 268
Зарин (О-Изопропилметилфторфос-
фонат, Трилон 46)
биологическая активность 162
биохимический анализ 159
в смесях ОВ 266
гидролиз 292, 293
дегазация
активированным углем 368, 369
алкиламинами 315
аммиаком 313
Зарин
дегазация
гипохлоритами 308, 320
едким натром 302
комплексами аминов с медью
317
моиоэтаиоламииом 313, 316
моющими средствами 364
нуклеофильными реагентами
299, 300, 304
перекисью водорода 308, 348
персульфатами 349
растворителями 353, 358
фенолятами 307
хлоритом натрия 336
действие на ферменты 162, 163, 165
допустимая концентрация в воде
269
индикация 179
автоматическими приборами
240, 241, 245
биохимическая 245
бумагами 232
в воздухе 255
в полевых лабораториях 261,
262, 265
карандашами 227
иа животных 186, 187
по запаху 222
порошками 224, 225
трубками 233, 238
химическими реакциями 232
обнаружение 50, 58, 59, 62, 64, 200,
205
фтора 30
определение
биологическое 186, 187
летальной дозы 187
ферментами 176 сл.
химическими методами 71, 73,
88
хроматографическое 203
стойкость
в присутствии перекиси водо-
рода 54
иа местности 250, 277
Золото хлориое
индикация серного иприта 79, 80,
236 сл.
обнаружение
мышьяковистого водорода 91
окиси углерода 128
Золотохлористоводородная кислота,
обнаружение алкалоидов
138
Зоман (втор-О-Неогексилметилфтор-
фосфонат)
биологическая активность 162
дегазация
моющими средствами 364
Зоман
дегазация
растворителями 353
действие иа ферменты 162, 163
допустимая концентрация в воде
269
индикация 179
автоматическими приборами
241
в воздухе 254
в полевых лабораториях 261,
262, 265
по запаху 222, 223
трубками 233, 238
обнаружение 50, 62, 64
фтора 30
определение
ферментами 179
химическими методами 71, 88
стойкость
в воде 251, 284
иа местности 277
Изоиитрозобензоилацетои, обнаруже-
ние цианид-ионов 105
Изонитрозокетоны, обнаружение фос-
форорганических ОВ 61, 62
О-Изопропилметилфторфосфоиат см.
Зарин
Изопропоксиметилфосфорилтиохолин,
обнаружение 50
Изопурпуровая кислота (2-Гидрокси-
амиио-3,5-дициаи-4,6-дииит-
рофеиол) 103
Изопурпуровой кислоты соль, обна-
ружение бромбеизилциа-
иида 119
Изосистокс
действие на ферменты 165
обнаружение 50, 66, 205
определение 75
Изоцианурхлорид как дегазирующее
вещество 342, 343
И-кислота см. 6-Амино-1-иафтол-3-
сульфокислота
ИК-спектроскопия ОВ 127, 200 сл.,
244, 245
Ингаляционные ОВ, определение ле-
тальной дозы 185
Ингибирующее действие ОВ, опреде-
ление 164 сл.
Индикаторные бумаги 227 сл.
беизидии-медьацетатная 229, 230
в наборе 246
иодкрахмальиая 228
иа зарин 232
иа люизит 231
иа мышьяксодержащие ОВ 91, 97
иа неорганические ОВ 146
на окись углерода 232
391
Индикаторные бумаги
на серный иприт 228, 231, 232
на синильную кислоту 229, 230
на сурьму 149
на ферменты 168
на фосген 229
на фосфор 32
иа фосфорорганические ОВ 176,
178, 179, 232, 247
на хлор 228, 229
иа хлорпикрин 231
на цианид-ионы 102
на этилдихлорарсин 232
натрийпикратная 230
о-толидиновая 229
флуоресцеиновая 228
цирконазоарсоновая 30
Индикаторные карандаши 226, 227
Индикаторные наборы 246 сл.
Индикаторные порошки 223 сл.
Индикаторные трубки 232 сл.
Дрегера 238
маркировка 237, 238
на карбонил никеля 128
на нервцо-паралнтические ОВ 232
на окись углерода 127
на синильную кислоту 230, 233,
237, 238
на тетраэтилсвинец 125
на цианид-ионы 102
Индикаторный гель NBS, обнаруже-
ние окиси углерода 127
Индикация ОВ 17
автоматическими приборами
239 сл.
биохимическими методами 245,
246
бумагами см. Индикаторные бу-
маги
в носимых лабораториях 246, 247
в полевых лабораториях 248, 249
в полевых условиях 221 сл., 260 сл,
на животных 185 сл.
неорганических 146
нервно-паралитических 60
отбор проб 249 сл.
по запаху 221 сл.
подготовка проб 257 сл.
порошками 232 сл.
трубками 93 сл., 232 сл.
физическими методами 244, 245
фосфорорганических 60
цветными реакциями 60, 226, 227,
241 сл., 263 сл.
Индоксилацетат
биохимическая индикация ОВ 246
обнаружение фосфорорганических
ОВ 207, 208
определение ферментов 170
Индол
индикация фосфорорганических
ОВ 243
обнаружение хлорацетофенола 117
производные 142 сл.
Р-Иидолилуксусная кислота, обнару-
жение 143
Индофенилацетат, определение фер-
ментов 171
Инсектициды фосфорорганические
индикация
на животных 186
перекисью водорода 60
обнаружение 50, 65
биохимическое 159
хроматографическое 209, 212
определение
биологическое 186, 187
биохимическое 159
на животных 187
спектроскопическое 200
ферментами 174 сл.
Иод
обнаружение 211
мышьякорганических ОВ 93
тетраэтилсвинца 125
циаиид-ионов 106
определение 43
карбонила никеля 129
цианид-ионов 109
Иода пятиокись
обнаружение окиси углерода 127,
235
определение окиси углерода 128
Иод-азидная реакция 28, 206, 207
Иодкрахмальная индикаторная бу-
мага 228
Иодуксчсной кислоты эфир в смесях
ОВ 267
Иодциан
обнаружение 108
определение цианидов 109
Иприт (Бис-2-хлорэтиловый тиоэфир.
Серный иприт)
биохимический анализ 159
В смесях ОВ 266
гидролиз 294, 295
дегазация 284, 287 сл., 295
гексаметилентетрамином 314
гипохлоритами 322, 335
дихлораминами 341
1,3-дихлор-5,5-диметилгиданто-
„ином 343
нуклеофильными реагентами
299
перекисью водорода 348
растворителями 353, 358
392
Иприт
дегазация
хлораминами 339
хлоритом натрия 336
хлорноватистой кислотой 320
хлорной известью 334
допустимая концентрация в воде
269
индикация
автоматическими приборами 245
бумагой 228, 231, 232
в полевых условиях 261, 262,
265
на животных 185, 187
набором 246, 247
по запаху 222
порошками 224, 225
трубками 233, 236 сл.
химическими методами 228,
231, 236-—238
обнаружение 29, 50, 66, 80, 81, 92,
258
определение 82, 83, 85
биологическое 185, 187
в полевых лабораториях 248
в полевых условиях 247
УФ-спектроскопическое 199
химическими методами 81 сл.
по Левинштейну 266
стойкость
в воде 251, 284
на местности 250, 277
экстракция растворителями 259 сл.
Кадмий, обнаружение 147, 149, 211
Кадмия ацетат, обнаружение серы 28
Калий, обнаружение 211
Калий иодистый
индикация
галогеналкиламинов 231
хлора 228
обнаружение
галогенцианов 108
таллия 148
хлорпикрина 120
определение
азота 40
серных ипритов 82
Калий цианистый
индикация ОВ
в полевых лабораториях
263 сл.
хлорпикрина 238
обнаружение
динитрила малоновой кислоты
123
кадмия 149
никотина 139
свинца 148
хлорпикрина 121, 122
Калийвисмутиодид
индикация азотистого иприта 237,
238
обнаружение азотистых ипритов
85, 86
определение азотистого иприта 88,
89
Калия бихромат, обнаружение
свинца 148
стрихнина 139
фтора 149
Калия бромид
индикация хлора 228
обнаружение хлорпикрина 121
Калия гексаиодоплатинат, обнаруже-
ние
алкалоидов 208—210
фосфорорганических ОВ 206, 207
Калия меркаптиды, обнаружение
хлорпикрина 121
Калия метапериодат, обнаружение
фторкарбоновых кислот 130
Калия перманганат
индикация
фосгена 236
иприта 236
обнаружение
бария 147
фосфорорганических ОВ 208
Калия роданид, обнаружение пента-
карбонила железа 128
Калия феррицианид
обнаружение фторацетатов 130
определение
фторуксусной кислоты 131
цианиД-ионов 109
Калия хлорат, обнаружение свинца 125
Кальций 211
обнаружение хлорпикрина 119, 120
Кальция гипохлорит 326 сл., 371 сл.
двухосновной (Базогрелит) 327,
328, 334
Кальция карбонат, обнаружение ди-
нитрила малоновой кислоты
123, 124
Кальция окись, обнаружение дини-
трила малоновой кислоты
123, 124
Кантаридин, обнаружение 140
Карбоксилаза, определение фосфор-
органических ОВ 159, 180
Карбоксихолины
определение фосфорорганических
ОВ 175 сл.
393
Карбоксихолины
разложение 167 сл.
Карбокситиохолии, определение
169 сл.
фосфорорганических ОВ 176
Карбонилы металлов 128, 129
Каталаза, определение фосфорорга-
нических ОВ 159, 180
Келлера реакция 143
Кетон Михлера, индикация
серного иприта 231
фосгена 226, 227
Кларк I см. Дифеиилхлорарсии
Кларк II см. Дифенилцианарсин
Клоп 267
Кобальт, обнаружение 211 ,
Кодеин, обнаружение 138
Кожно-нарывные ОВ
дегазация 343
определение летальной дозы 185
Кокаин, обнаружение 138, 210
Коллоргит, индикация 266
Колхицин, обнаружение 133, 135 сл.,
210
Конго красный, индикация сниильиой
кислоты 230
Кониин, обнаружение 135 сл.
Кофеин, обнаружение 138
Крезолфталии, обнаружение цианид-
ионов 103
Кремиевольфрамовая кислота, обна-
ружение
азотистого иприта 86
алкалоидов 139
Ксилилбромид
в смесях ОВ 267
обнаружение 119
определение 199
Ксилилеибромид в смесях ОВ 267
Кураре яд, обнаружение 136
Кьельдаля метод минерализации ОВ
37
Лабатша проба 120
Лантана соли, обнаружение фтораце-
татов 130
Лассеня проба 27
Лейперта метод, определения йоди-
дов 43
Либиха способ определения цианнд-
иоиов 108
Лизергиновая кислота, производные
142, 144
Литий, обнаружение 211
394
Лозантин 12, 334
Лост 388
а-Люизит (2-Хлорвинилдихлорарсии)
биохимический анализ 159
в смесях ОВ 266
дегазация
активированным углем 368
1,3-дихлор-5,5-диметилгидан-
тоииом 343
хлораминами 339
допустимая концентрация в воде
269
индикация
бумагами 231
в полевых лабораториях 265
карандашами 226, 227
по запаху 222
порошками 225
трубками 237, 238
цветными реакциями 226, 227,
238
обнаружение 90 сл.
определение 88, 100, 199
стойкость иа местности 250
Р-Люизит Щи-(2-хлорвииил)-хлор-
арсин] 91 сл.
у-Люизит [Трис-(2-хлорвинил)-арсин]
91 сл.
Люмииол, обнаружение цианид-ионов
103
Люцигении (Дииитрат N.N'-диметил-
диакридиния) 58
LSD (Диэтиламид лизергиновой кис-
лоты)
обнаружение' 49, 143, 144, 211
определение 144, 145
Магний, обнаружение 211
Мазора метод анализа ОВ 34
Манганит, индикация 266
Марганец
двуокись, обнаружение дииитрила
малоновой кислоты 124
обнаружение 211
Мартонит, индикация 266
Maxchlor 327
Меди ацетат
индикация ОВ
в полевых лабораториях 264,
265
синильной кислоты 229, 230,
237
обнаружение
азота 28
дииитрила малоновой кислоты
124
цианнд-ионов 102
Меди ацетат
определение сесквииприта 84
Меди сульфат, обнаружение
фосгена 113
цианид-ионов 103, 230
Меркаптофос см. Систокс
Мескалин (3,4,5-Триметоксифеиил-
2-аминоэтаи), обнаружение
145
Метилалкоксифосфорилхолины, дей-
ствие иа ферменты 163
Метилалкоксифторфосфонаты, дей-
ствие иа ферменты 163
Метилалкоксициаифосфонаты, дей-
ствие иа ферменты 163
М-Метил-Ы.Ы-бис- (2-хлорэтил) -амии
50, 89
Метилдихлорарсии
индикация
в полевых лабораториях 262,
265
по запаху 222
обнаружение 77, 90 сл.
определение 88, 97
Метиленовый синий, определение се-
ры 39
Метиловый спирт (Метаиол) 355,
356, 380
З-Метил-1 -фенил-5-пиразолон, обна-
ружение галогеициаиов 107
Метилфторацетат, определение 88
Метилфторфосфорилгомохолии, дей-
ствие на ферменты 165
Метилфторфосфорилтиохолин, дей-
ствие иа ферменты 164
Метилфторфосфорилхолин
действие иа ферменты 164, 165
обнаружение 50
определение 174, 176
Метилэтоксифосфорилтиохолин
действие иа ферменты 163, 165
обнаружение 205
Метилэтоксифосфорилхолии, действие
иа ферменты 164
Молекулярная активность при биохи-
мическом анализе 158
Молибденовая кислота, обнаружение
мышьякорганических ОВ 93
Моиоизонитрозоацетои, обнаружение
фосфорорганических ОВ 61
Моиохлорамиц
как дегазирующее вещество 373
обнаружение иикотииа 139
Моиоэтаиоламии как дегазирующее
вещество 302, 313, 316
Морфии, обнаружение 49, 133 сл„ 210
Морфолид пеларгоновой кислоты,
обнаружение 124
Морфолин, обнаружение
морфолида пеларгоновой кислоты
124
третичных аминов 49
Мурексид (Аммониевая соль пурпу-
ровой кислоты) 109
Мышьяк
допустимая концентрация в воде
269
обнаружение 30, 32, 211
определение 36, 43
Мышьяк треххлористый в смесях ОВ
266
Мышьяковистая кислота, обнаруже-
ние окиси углерода 128
Мышьяковистый водород
дегазация активированным углем
368
обнаружение 91
определение 97, 98
Мышьякоргаиические ОВ
восстановление до AsHs 96, 150
индикация 91, 97, 261, 262
минерализация проб 96, 150
обнаружение 88, 90 сл.
определение 88, 96 сл.
в полевых лабораториях 249
галогенов 100
циаиид-иоиов 100
Наркеин, обнаружение 138
Наркотин, обнаружение 135 сл., 210
Натрий, обнаружение 211
Натрий иодистый
обнаружение галогеициаиов 108
определение
ди- и трифосгена 115
фосгена 113, 114
Натрий сернистый, индикация хлор-
циана 238
Натрий цианистый, индикация хлор-
пикрина 231
Натрийпеитацианоаминоферрат, инди-
кация серного иприта 231
Натрийпикратная индикаторная бу-
мага 230
Натрия бикарбонат, определение
азотистых ипритов 86, 87
дииитро-о-крезола 142
Натрия гипохлорит как дегазирую-
щее вещество 330 сл., 337
395
Натрия иодоплатинат в анализе сер-
ных ипритов 81, 231
Натрия карбонат (Сода)
индикация
окиси углерода 232
синильной кислоты 2 О
фосгена 226, 238
фосфорорганических ОВ 243
как дегазирующее вещество 303,
304, 357, 375
Натрия молибдат, определение фос-
фора 38
Натрия нитрит
обнаружение
тетрама 68
хлорпикрина 119, 120
определение
паратиона 74
параоксона 74
Натрия нитропруссид
индикация ОВ 264, 265
обнаружение
бромацетона 77
метилдихлорарсина 77
морфолида пеларгоновой кис-
лоты 124
серных ипритов 77
серы 28, 29
систокса 66
тиолов 69
третичных аминов 49
хлорацетофенона 77
хлорпикрина 77
этилдихлорарсина 77
определение тиохолина 169
Натрия перборат
индикация фосфорорганических
ОВ 243
обнаружение фосфора 31
определение
дифосгена 115
фосфорорганических ОВ 70
Натрия перекись, определение хлор-
пикрина 122
Натрия пероксопирофосфат, индика-
ция фосфорорганических
ОВ 242
Натрия пикрат, обнаружение
бромбеизилцианида 119
дифенилцианарсина 119
табуна 119
циаиид-ионов 103
Натрия роданид, индикация галоген-
алкиламинов 231
Натрия роднзонат, обнаружение ток-
сичных катионов 146, 147
Натрия сульфид
как дегазирующее вещество 309,
310
обнаружение
кадмия 149
селенит-ионов 150
определение
бромбензилциаиида 119
окиси углерода 129
хлорацетофенона 118
Натрия тиосульфат
индикация серного иприта 236
как дегазирующее вещество 310
обнаружение
окиси углерода 128
таллия 148
определение
азота 40
азотистых ипритов 89
бромциана 111
двуокиси углерода 128
серных ипритов 82
фосфорорганических ОВ 72
Натрия фенолят, определение хлор-
ацетофенона 118
Натрия этилат
индикация ОВ 264, 265
обнаружение хлорпикрина 120
а-Нафтиламин, обнаружение хлор-
пикрина 120
(З-Нафтилацетат, определение фос-
форорганических ОВ 171,
176
а-Нафтилдиазо-1,5-нафталиндисуль-
фокислота, определение
ферментов 171
N- (1 -Нафтил) -этилендиамина гидро-
хлорид, определение
параоксона 74
паратиона 65, 74
хлорпикрина 123
втор-О-Неогексилметилфторфосфонат
см. Зоман
Неорганические ОВ
индикация 146
определение 190
Нервно-паралитические ОВ
индикация
автоматическими приборами
241, 242
бумагой 232
карандашами 227
трубками 237
цветными реакциями 60, 227
определение летальной дозы 185
Никель, обнаружение 211
Никеля карбонил
обнаружение 128
3%
Никеля карбонил
определение 129
Никеля соли, индикация ОВ 264, 265
Никеля сульфат
обнаружение серных ипритов 77
определение цианид-ионов 109
Никотин, обнаружение 50, 135 сл.
Никотинамид, определение цианид-
ионов НО
Ниполит 327
n-Нитробензальдегид, обнаружение
цианид-ионов 104, 105
4-л- (Нитробензил) -пиридин
индикация ОВ 1
в полевых лабораториях 264,
265
фосгена 226, 229, 238, 265
обнаружение
серного иприта 80
фосфорорганических ОВ 62
хлорацетофенона 117
определение
азотистых ипритов 87, 88
люизита 88
метилдихлорарсина 88
метилфторацетата 88
мышьяксодержащих ОВ 88
серных ипритов 85, 88
сесквииприта 83, 84
фенилдихлорарсина 88
фосгена 114
фосфорорганических ОВ 72, 88
хлорацетофенона 88
этилдихлорарсина 88
Нитробензол
в смесях ОВ 266
определение цианид-ионов 109
п-Нитрозодиметиланилин 76
1-Нитрозо-3,6-диметиламинофенол,
индикация фосгена 229
Нитрометан как дегазирующее веще-
ство 353
4- (n-Нитрофенил) -пиридин, обнару-
жение табуна 62
о-Нитрофенола эфиры, определение
ферментов 171
Озон как дегазирующее вещество
320, 346, 347
8-Окси-7-иод-6-хинолинсульфокис-
лота, определение цианид-
ионов НО
Оксимы как антидоты 315
8-Оксихинолин (Оксин)
обнаружение
серного иприта 80
токсичных катионов 211
8-Оксихинолин (Оксин)
обнаружение
цианид-ионов 104
определение
азотистых ипритов 86, 87
иприта 85
Оксихинолинат меди, обнаружение
цианид-ионов 104
8-Оксихинолинсульфокислота, опре-
деление цианид-ионов НО
Олово, обнаружение 211
Олово хлористое, обнаружение
мышьякорганических ОВ 93
Олово хлорное в смесях ОВ 266, 267
Осмия четырехокись, обнаружение
мышьякорганических ОВ 92
Отбеливающие щелока 330, 331
Отбор проб
воды 251, 268, 269
воздуха 253 сл.
пищевых продуктов, фуража 252,
253, 260, 268
почвы 250, 259
при дегазации 270, 271, 371, 372
с военной техники, обмундирова-
ния 252
с растений 259
Открываемый минимум ОВ 24
Палладия комплексы, определение
цианид-ионов НО
Палладия сульфат, обнаружение оки-
си углерода 127
Палладия хлорид, обнаружение
окиси углерода 126, 127, 232
фосфорорганических ОВ 207, 208
Папаверин, обнаружение 135 сл., 210
Параоксон (Диэтил-4-иитрофенил-
фосфат)
биохимический анализ 159
индикация карандашами 227
обнаружение 65, 205
определение 65, 73, 74
спектроскопическое 199
хроматографическое 203
стойкость в присутствии перекиси
водорода 54
Паратион (Тиофос)
действие иа ферменты 165, 207
индикация
карандашами 227
иа животных 186, 187
обнаружение 50, 65, 205, 207, 209
определение
биохимическое 186
колориметрическое 73, 74
спектроскопическое 199, 200
хроматографическое 203
397
Пахлорин 327
Пербораты как дегазирующие веще-
ства 348
Пергидрол 347, 348
Пероксодисериая кислота как дега-
зирующее вещество 349
Перхлорон 327
Петролейиый эфир, экстракция ОВ
258 сл.
Пиколин
в смесях ОВ 266
обнаружение галогенциаиов 107
Пикриновая кислота
индикация синильной кислоты 230
обнаружение алкалоидов 138
Пикролоновая кислота, обнаружение
алкалоидов 138, 139
Пикротоксин, обнаружение 133 сл.
Пирамидон, обнаружение цианид-
иоиов 103
Пиридин
в смесях ОВ 266
индикация хлорпикрина 238
обнаружение
галогенциаиов 106
хлорпикрина 121
определение
хлорацетофенона 117
хлорпикрина 122
Пиридин — барбитуровая кислота,
индикация ОВ 263 сл.
Пирофосфоиаты, определение 70 сл.
Платииохлористоводородная кислота,
обнаружение
алкалоидов 138
мескалина 145
Полиалкиленгликолевый эфир, опре-
деление мышьяксодержа-
щих ОВ 100
Полисульфиды в смесях ОВ 266
Полярографический анализ фермен-
тов 170
Прегля метод сожжения 39
Предельная концентрация ОВ 25
Приборы при анализе ОВ 21, 22,
35 сл.
Аббе рефрактометр 197
автоматические 238 сл.
Бетге 36
Варбурга 166, 171
Викболта 37
ВПХР 239
Приборы при анализе ОВ
Джелли микрорефрактометр 197
для индикации ОВ 233 сл., 238 сл.,
245, 246
для отбора проб 251, 253, 254
для химической разведки 233, 234,
238, 239
Дрегера — Шрётера трубка 236
Дрюса для определения давления
195
Е21 241
Кофлера баня для определения
температуры плавления 192
ПХР-54 238
Тиле для определения темпера-
туры плавления 192
фильтры для удержания аэрозо-
лей 256
УФ-фотометр Клотца и Дола 244
Хенниона пикнометр 192
Шпреигеля — Оствальда пикно-
метр 189, 192
Эмиха трубки 193, 194
Пробы при анализе ОВ
Бельштейна 29
Гутцейта 32, 150, 238, 261, 262,
238
Лабатша 120
Лассеня 27, 28
подготовка для анализа 257 сл.
Хинсберга 49
Псилобиции 142, 143
Психохимические ОВ, обнаружение
142 сл.
Пурпуровой кислоты аммониевая
соль (Мурексид) 109
Раздражающие ОВ 123, 124
биохимический анализ 159
определение летальной дозы 185
Рациоиит 267
Рвотный газ 267
Реагенты при анализе ОВ
Вагиера 135, 138
Ваи Урка 212
Васицкого 134, 136, 137
Витали 134, 136, 137, 139, 145
Годффройя 139
Гриньяра 79
Грисса 46, 120, 123, 149
Гроте 206
о-диаиизидиимолибдатный 31
Драгендорфа 69, 135, 138, 145,206,
237, 262, 264, 265
Драгендорфа — Краута 86
«желтое кольцо» 237, 238
«зеленое кольцо» 237, 238
Зёренсеиа 178
398
Реагенты
Зонненштейна 135, 136
Илосвая 262, 264, 265
иод-азядиый 28
Келлера 144
Кларка и Лабса 179
<красное кольцо» 237, 238
Майера 66, 135, 138, 145
Манделина 134, 136, 137
Марки 145
Марме 135, 138
Мекке 134, 136, 137
Миллона 65
Михеля 172, 178
Несслера 86
пиридин-барбитуровый 109
Рингера 171
<синее кольцо» 237
Фолина — Циокалтё 66
$>рмальдегид-сернокислотный 48
реде 134, 136, 137
Цинцадзе 31
<чериое кольцо» 237
Шарграфа 206
Шейблера 135, 138, 145
Эрдмана 134, 136, 137
2-Ред стар 267
Резорцин, обнаружение
галогенцианов 106
хлорпикрина 120
Рейнеке соль 86
Робертсона метод определения
мышьякорганических ОВ 99
Роджерса метод определения мышь-
якорганических ОВ 99
Ртути бромид, обнаружение мышья-
ковистого водорода 91
Ртути нитрат, обнаружение мышьяк-
органических ОВ 92
Ртути окись, определение окисй уг-
лерода 129
Ртуть
допустимей концентрация в воде
269
обнаружение 148, 149, 211
Ртуть хлорная
индикация
серного иприта 231
синильной кислоты 230, 238
обнаружение цианид-ионов 103
определение цианид-ионов 109, 110
Руппа метод определения мышьяк-
содержащих ОВ 98, 99
Самодегазация (Дегазация естествен-
ная) 278
Свинец
допустимая концентрация в воде
269
обнаружение 125, 146 сл., 211
Свииецоргаиические ОВ 124 сл.
минерализация проб 124, 126
Свинца ацетат
обнаружение серы 28
определение
изосистокса 75
мышьяксодержащих ОВ 97
Сегнетова соль
индикация синильной кислоты
230
обнаружение свинца 148
Селенистая кислота, обнаружение
кантаридина 140
Селениты, обнаружение 150, 211
Селеновая кислота, обнаружение 150
Сера
в смесях ОВ 266
обнаружение 27 сл., 211
дииитрила малоновой кислоты
124
определение 38, 39
Серебра.иитрат
индикация
люизита 238
синильной кислоты 230
обнаружение
арсенат-иоиов см. Гутцейта ре-
акция
мышьяковистого водорода 91
окиси углерода 127
определение
синильной кислоты 108
фосгена 114
цианидов 108
Серебра перхлорат
обнаружение фторкарбоновых кис-
лот 130
определение серы и хлора 39
Серная кислота
индикация ОВ
в полевых лабораториях 264,
1 265
окиси углерода
обнаружение
адамсита 119
аконитина 139
алкалоидов 134, 136, 137, 143
бромбензилцианида 119
кантаридина 140
карбонила железа 128
ксилилбромида 119
мескалина 145
стрихнина 139
фторуксусной кислоты 131
399
Серная кислота
обнаружение
хлорфеноксиуксусных кислот
141, 209, 212
определение
мышьяксодержащих ОВ 97
производных индола 144
хлорфеноксиуксусных кислот
142
помощь при отравлениях 379
Серные иприты (Галогенированные
тиоэфиры)
обнаружение 76—80, 116
определение 81 сл., 88
Серный иприт см. Иприт
Сероводород
в смесях ОВ 267
индикация ОВ 262, 264, 265
обнаружение
мышьякорганических ОВ 91
токсичных катионов 146
Сероуглерод в смесях ОВ 266, 267
Сесквииприт [1,2-Бис- (2-хлорэтил-
тио)-этан]
индикация 83, 84, 265
определение 83, 84
Си Эс см. о-Хлорбензилиденмалоно-
динитрил
Силикагель
как дегазирующее вещество 367,
368
обнаружение токсичных • катионов
211
Синильная кислота
в смесях ОВ 266
допустимое содержание в воде
269
индикация
бумагой 229, 230
в полевых лабораториях 263,
265
карандашами 226
на животных 185
набором 246
по запаху 222
трубками 230, 233, 237, 238
химическими реакциями 226,
229, 230, 237, 238
обнаружение 101 сл.
определение
биологическое 185
летальной дозы 185
химическими методами 108 сл.
стойкость
в воде 251
на местности 250, 277
Систокс (Меркаптофос)
индикация 60, 235
обнаружение 50, 66, 205
Систокс
определение 74, 75, 176, 180, 200
Скополамин, обнаружение 137
Смеси ОВ
загущенные 264, 268
NG 267
PG 266
Сода см. Карбонат натрия
Соланин, обнаружение 135 сл.
Соляная кислота
как дегазирующее вещество 339
обнаружение
колхицина 139
селенит-ионов 150
помощь при отравлениях 379
Спорыньи алкалоиды 142 сл.
Средняя смертельная доза ОВ 184
Средняя эффективная доза ОВ 184
Стаса — Отто способ анализа алка-
лоидов 132, 133
Стеапсинлипаза, определение фос-
форорганических ОВ 180
Стрептоцид красный, определение
хлорпикрина 123
Стрихнин, обнаружение 49, 50,
133 сл., 210
Стронций, обнаружение 147, 211
Судан красный, индикация серного
иприта 228
Сульфамин, обнаружение иприта 81
Сульфаминовая кислота, определение
ОВ 74
Сульфаниламид, определение хлор-
пикрина 123
Сульфаниловая кислота, обнаружение
хлорпикрина 120
Сульфоксид 81
Сульфосалициловая кислота, обнару-
жение фосфорорганических
О В 206
Сульфурилхлорид 344, 345
Сурьма
допустимая концентрация в воде
269
обнаружение 149, 211
Т-вещество 267
Т-зеленое вещество 267
2,4,5-Т(2,4,5-Трихлорфеноксиуксусная
кислота) 131, 141, 209, 212
Табун (М,М-Днметиламидо-о-этил-
цианфосфат, Трилон, 83)
биологическая активность 162
биохимический анализ 159
40Q
Табун
в смесях ОВ 266
дегазация 368
действие на ферменты 162, 165
допустимая концентрация в воде
269
индикация
в воздухе 255
карандашами 227
по запаху 222
обнаружение 50, 59, 62, 64, 119,
179
в полевых условиях 261, 262,
265
определение 176, 179, 200
стойкость
в воде 251
на местности 250
Тактические ОВ 264, 266
Таллий, обнаружение 147, 148, 211
Таннин, обнаружение алкалоидов
138, 139
«Твердый сероводород» 146
Текстон 336
Тетраалкил (арил) аммония соли, оп-
ределение 90
Тетразолий синий, обнаружение фос-
форорганических ОВ 206
Тетралин в смесях ОВ 267
Тетрам (Амитон), обнаружение 50,
67 с л., 262
Тетраметиламмоний бромистый в сме-
сях ОВ 266
Тетраметилдиаминодифенилметан,
обнаружение цианид-ионов
102, 230
1,2,5,8-Тетраоксиантрахинон см. Хина-
лизарин
Тетрахлординитроэтан, обнаружение
хлорпикрина 121
Тетрахлорэтан 356
Тетраэтилпирофосфат (ТЭПФ)
действие на ферменты 165
индикация карандашами 227
обнаружение 66
стойкость в присутствии перекиси
водорода 54
Тетраэтилсвинец 125
Тимол, обнаружение хлорпикрина 120
Тимолфталеин
обнаружение иприта 78, 79, 258,
261 сл.
определение иприта 85
Тиоацетамид, обнаружение токсич-
ных катионов 146
Тиодихлорарсин, определение 98
Тиоиндиго, обнаружение фторацета-
тов 130, 131
Тиокетон Михлера, индикация, люи-
зита 226, 227, 238
Тиолы, обнаружение 68, 69
Тиоляты как дегазирующие вещества
310
"Тиомочевина
обнаружение
ипритов 76 сл., 264, 265
токсичных катионов 146
определение
иприта 85
хлорацетофенона 118
Тиосалициловая кислота
обнаружение фторацетатов 130,
131, 264, 265
определение фторуксусной кисло-
ты 131
Тиофенол, обнаружение хлорпикрина
121
Тиофеноляты • как дегазирующие ве-
щества 310
Тиоформанилид, обнаружение ток-
сичных катионов 146
Тнофос см. Паратион
Тиохолин, определение 169, 170
активности холинэстеразы 173
Тиоцианаты, обнаружение цианид-
ионов 102
Титан хлористый
индикация
галогеналкиламинов 231
параоксона 65
определение параоксона и пара-
тиона 74
о-Толидин
индикация
синильной кислоты 230
хлора 229
обнаружение
азота 28
галогенциаиов 106
динитрила малоновой кислоты
124
цианид-ионов 102
определение
иприта 83
фосфорорганических ОВ 70
о-Толидиновая индикаторная бумага
229
Толуидин, обнаружение галогенциа-
нов 107
Торин, определение серы 39
Тория нитрат, определение фтора 42
401
Триалкилсвинецсульфамиды 126
Трилон 46 см. Зарин
6,7,8-Триметоксиизохииолин, обнару-
жение мескалина 145
3,4,б-Триметоксифенил-2-амииоэтаи см.
Мескалин
Три-н-пропилсвинец, соли 126
Трис-(2-хлорвниил)-арсин см. у-Люи-
зит
Трис-(2-хлорэтил)-амин см. Азоти-
стый иприт
Т рифенилтетразолийхлорид, обнару-
жение
фосфорорганических ОВ 206
цианид-ионов 106
Трифенилформазан, обнаружение
цианид-ионов 106
Трифосген, определение 115
Трихлорметиловый эфир хлоруголь-
ной кислоты см. Дифосген
Трнхлорнитрометаи см. Хлорпикрин
2,4,5-Трнхлорфеноксиуксусная кислота
(2,4,5-Т) 141, 209, 212
2,2',2"-Трихлорэтиламии см. Азоти-
стый иприт
Трихлорэтилен 356, 358
Трнэтнлсвннец, соли 126
ТЭПФ см. Тетраэтнлпирофосфат
Уайт стар 266
Углерода окись
индикация
бумагой 232
на животных 185
трубками 127, 235
химическими методами 232,235
обнаружение 126 сл., 235
определение 128, 129, 185
Уксусная кислота
обнаружение алкалоидов спо-
рыньи 143
определение
адамсита 100
серных ипритов 81
Уксусный альдегид, обнаружение
морфолида пеларгоновой
кислоты 124
Уреаза, определение фосфороргани-
ческих ОВ 180
Уротропин см. Гексаметилентетрамин
УФ-спектроскопия ОВ 199 сл.
Феиацил-4- (л-иитробеизил) -пириди-
ния хлорид, обнаружение
хлорацетофенона 117
ФенапилПирндиний, определений
хлорацетофенона 117
Фениламиноалканы 145
4-Фенил-2-амниотиазола гидрохлорид,
определение хлорацетофено-
на 118
N-Феиилбеизиламин, индикация фос-
гена 226, 238
Фенилбензоат, определение фермен-
тов 171
Фенилгидразин, обнаружение фосге-
на 112, 113
анти-Фенилдиазотат калия, индика-
ция серного иприта 238
Фенилдихлорарсин
в смесях ОВ 266
обнаружение 50, 91 сл.
определение 88, 98, 99
л-Фенилеиднамин, обнаружение га-
логенцианов 107
л-Феиилеидиамина гидрохлорид, оп-
ределение хлорпикрина 123
Ы-Феинл-1-нафтиламии, индикация
фосгена 226
N-Феннлфенол, определение азоти-
стых ипритов 89
Феноксиуксусиые кислоты хлориро-
ванные, обнаружение 131,
141, 209, 212
Фенолфталеин, обнаружение цианид-
нонов 103
Феноляты как дегазирующие веще-
ства 307, 368
Ферменты при анализе ОВ 158 сл.
биохимическая индикация ОВ 245.
246
обнаружение фосфорорганических
ОВ 207
определение 167, 169 сл.
активности 165 сл.
фосфорорганических ОВ 175 сл.,
180
Физико-химический анализ ОВ
189 сл.
Физический анализ ОВ 189 сл.
индикаторными приборами 244,
245
по плотности 189, 192
по показателю преломления 196,
197
по температуре кипеиия 193,
194
-------плавления 192, 193
Физостигмин, обнаружение 135 сл.,
210
402
Фитотоксические ОВ 141, 142
Флороглюцин, анализ хлорпикрина
122, 231
Флуоресцеин, индикация хлора 228
Флуоресцеиновая индикаторная бу-
мага 228
Флуоресцентный анализ ОВ 70, 104,
ПО, 144, 145, 191, 198
Флуориметрический анализ фосфор-
органических ОВ 70, 241 сл.
Фосген
в смесях ОВ 266
индикация
автоматическими приборами
245
бумагой 229
в полевых лабораториях 263,
265
карандашами 226
иа животных 185
набором 246
по запаху 222
трубками 233, 235 сл.
химическими методами 226,
227, 229, 235 сл.
обнаружение 111 сл.
определение 113 сл.
летальной дозы 185
стойкость иа местности 250, 277
Фосгеноксим, индикация 265
Фосфамид (Диметоат), индикация 60
Фосфаты, дегазация 293, 294, 310
Фосфонаты, дегазация 288, 293, 294,
310
Фосфор
обнаружение 28, 30 сл.
определение 36 сл.
Фосфор-18-вольфрамовая кислота,
обнаружение тиолов 69
Фосфорилтиохолины
действие иа ферменты 162, 163
обнаружение 50
определение 75, 76, 174
Фосфорновольфрамовая кислота, об-
наружение
азотистых ипритов 86
мескалина 145
Фосфорномолнбденованадиевая кис-
лота, обнаружение карбони-
ла никеля 128
Фосфорномолибдеиовая кислота, об-
наружение
мескалина 145
окиси углерода 126, 127
сурьмы 149
фосфорорганических ОВ 206.
Фосфорорганические ОВ
биологическая активность 159 сл.,
180, 207
индикация
автоматическими приборами
241 сл.
биохимическая 255
бумагой 176, 178, 179, 232,
247
в воздухе 254, 255
на животных 186, 187
набором 246, 247
трубками 239
химическими методами 60,
170, 171, 224, 225, 232, 242,
243
обнаружение
биохимическое 207, 208
фосфора 31, 32
фтора 30
химическими методами 30 сл.,
52 сл., 61 сл., 176 сл., 187,
206 сл.
определение
биологическое 186
биохимическое 159, 175 сл.,
207, 208, 247, 255
в полевых лабораториях 248
спектроскопическое 199 сл.
флуоресцентное 241 сл.
химическими методами 69 сл.,
88
хроматографическое 203 сл.,
207 сл., 217
стойкость в воде 251
экстракция растворителями 258,
259
Фритца и Ямамура метод определе-
ния серы 39
Фтор
допустимая концентрация в воде
269
обнаружение 29, 30, 149, 211
определение 40 сл.
Фтор ангидриды метилфосфоиатов,
определение 71, 72
Фторангидриды фосфоновой кислоты,
определение 70
Фторацетаты
индикация 265
обнаружение 130, 131
определение 131
разложение 129
стойкость в воде 251
Фторкарбоновые кислоты
минерализация проб 129, 130
обнаружение 129 сл.
Фтороргаиические ОВ 129 сл.
403
Фторуксусная кислота 129 сл.
Фторфосфоновой кислоты эфиры, де-
газация 288, 299
Фторфосфорилхолин, действие на
ферменты 162
Хинализарин (1,2,5,8-Тетраоксиантра-
хинон), обнаружение берил-
лия 147
Хинидина сульфат, определение ак-
тивности холинэстеразы 173
Хинии, обнаружение 138
Хинолин в смесях ОВ 266
Хинолина гидрохлорид, определение
фосфора 38
Кинсберга проба 49
Хлор 323, 324
в смесях ОВ 266, 267
индикация
бумагой 228, 229
на животных 186
химическими методами 228
как дегазирующее вещество 320
обнаружение 211
определение 39, 43
биологическое 186
мышьяксодержащих ОВ 100
помощь при отравлениях 380
Хлора двуокись, дегазация воды 336,
337
Хлорамин Б (Натриевая соль N-хлор-
амида бензолсульфокисло-
ты) 109, 338
Хлорамин Т (N-Хлорамид п-толуол-
сульфокислоты, натриевая
соль)
индикация ОВ
в полевых лабораториях 264,
265
синильной кислоты 226
систокса 235
как дегазирующее вещество
338 сл.
обнаружение серных ипритов 80
определение
иприта 82
карбонила никеля 129
цианид-ионов НО
Хлорамины
индикация серного иприта 237, 238
как дегазирующие вещества 283,
290, 291, 337 сл., 368, 376
обнаружение синильной кислоты
106
определение цианид-ионов 109
Хлоранил (Тетрахлор-п-бензохинои),
анализ аминов 49, 90
Хлораниловая кислота, определение
цианид-ионов 111
Хлорацетон
в смесях ОВ 266
индикация по запаху 222
обнаружение 123
индикация
в воздухе 256
в полевых лабораториях 263,
265
по запаху 222
обнаружение 50, 116, 117, 123
бромбензилциаиида 119
определение 77, 117, 118
спектроскопическое 199
улавливание аэрозоля 257
о-Хлорбензальдегид, обнаружение Си-
Эс 123
о-Хлорбензилиденмалонодинитрил
(Си Эс, о-Хлорбензальмало-
нодинитрил)
индикация в полевых лаборато-
риях 263
обнаружение 123, 124
Хлорбензол в смесях ОВ 266
л-Хлорбензолсульфофторид, обнару-
жение 59
2-Хлорвинилдихлорарсин см. а-Люи-
знт
N-Хлоргликольурилы, дегазация
кожно-нарывных ОВ 344
10-Хлор-9,10-дигидрофенарсазинхло-
рид см. Адамсит
Хлористый сульфурил как дегазирую-
щее вещество 283
Хлориты как дегазирующие вещества
336, 337, 377
Хлорметиловый эфир хлоругольной
кислоты (Хлорметилхлор-.
карбонат) 266, 267
Хлорметилсульфат в смесях ОВ 267
у-(4-Хлор-2-метилфенокси)-масляная
кислота, обнаружение 209,
212
Хлорметилэтилкетон в смесях ОВ
266
Хлорная известь как дегазирующее
вещество 283, 324 сл., 334,
372, 373
Хлорная кислота, обнаружение фто-
ра 130
Хлорноватистая кислота как дегази-
рующее вещество 320 сл.
Хлороформ 356
в смесях ОВ 266
404
Хлороформ
обнаружение колхицина 139
экстракция ОВ 258, 259
Хлорофос (Диптерекс), индикацця 60
Хлорпикрин (Трихлорнитрометан)
в смесях ОВ 266, 267
восстановление 119, 120, 123
дегазация 368
индикация
автоматическими приборами
245
бумагой 231
в полевых лабораториях 263,
265
по запаху 222
трубками 239
химическими методами 231,
238
обнаружение 77, 119 сл.
определение 120, 122, 123, 199* 231
разложение 121
Хлортион
обнаружение 65
определение 203
Хлорциан
в смесях ОВ 266
индикация 238
в полевых лабораториях 263
трубками 233, 237, 238
обнаружение 108
определение 111
стойкость на местности 250
Холинэстераза
в анализе ОВ 159, 160 сл.,
164 сл., 171 сл.
как антидот ингибиторов 314, 315
обнаружение 169
фосфорорганических ОВ 207
определение
активности 165 сл., 171 сл.
ингибиторов 173 сл.
фосфорорганических ОВ 174сл.
Хром, обнаружение 211
Хроматографический анализ 191,
203 сл.
алкалоидов 208, 209
в колонках 203, 204
газовый 212
w-диметиламиноэтилтионфосфа-
та 205
днэтоксифосфорилтиохолина 205
зарина 203, 205
инсектицидов фосфорорганиче-
ских 209, 212
метилэтоксифосфорилтиохолина
на бумаге 204 сл.
Хроматографический анализ
параоксона 203, 205
паратиона 203, 205
систокса 205
2,4,5-Т 209, 212
токсичных ионов 209, 211
тонкослойный 207 сл.
фосфорорганических ОВ 187,
203 сл., 207 сл., 217
у-4-хлор-2-метилиденоксимасля-
ной кислоты 209, 212
хлорфеноксиуксусных кислот 142,
209, 212
2,4-DB 209, 212
LSD 212
Хромотроповая кислота (1,8-Диокси-
нафталин-3,6-дисульфокис-
лота)
обнаружение 46
фторуксусной кислоты 131
хлорфеноксиуксусиой кислот
141
Хуламит 127
Цейзеля метод определения алкокси-
групп 50
Цианиды
обнаружение 101 сл., 211
определение 108 сл.
в полевых условиях 247
Циммермана метод минерализации
ОВ 36
Цинк
обнаружение 211
определение мышьяксодержащих
ОВ 97
Цинке —Кенига реакция 106 сл„ 231
Цинцадзе реакция 81
Ципалит 267
Цирконализариновый лак, обнаруже-
ние фтора 30
Четыреххлористый углерод (Тетра-
хлорметан) 355
в смесях ОВ 266, 267
помощь при отравлении 380
«Число оборотов» при биохимиче-
ском анализе 158
Чувствительность определения ОВ 25
Шёнбейна реакция 103
Шенеманна реакция 52 сл., 69 сл.,
169, 232, 242, 243, 254, 255,
264, 265
Шёнигера метод минерализация ОВ 34
405
Эвинса метод определения мышьяк-
оргаиическнх ОВ 99
Элементный анализ ОВ 27 сл.,
30 сл., 249
Эметин, обнаружение 138, 210
Этаноламииы как дегазирующие ве-
щества 375
Этилацетондикарбоиат, обнаружение
галогенциаиов 107
М-Этил-1Ч,М-бис-(хлорэтил)-амины,
обнаружение 50
О-Этил-S- (р-диметиламиноэтил) -
метнлтиофосфоиат, гидролиз
292, 293
Этилдитиокарбимниат, обнаружение
токсичных катионов 146
Этилдихлорарсии
в смесях ОВ 266
индикация
бумагой 232
в полевых лабораториях 262,
265
по запаху 222
трубками 237
обнаружение 77, 90 сл.
определение 88, 99, 199
Этилкарбазол в смесях ОВ 266
Этиловый спирт (Этанол)
как дегазирующее вещество 355,
356
экстракция ОВ 258, 259
N-Этил-N -2-оксиэти лани лин, индика-
ция фосгена 229
ФРАНКЕ ЗИГФРИД, ФРАНЦ ПЕТЕР,
ВАРНКЕ ВЕРНЕР
ХИМИЯ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ
Том 2
Редактор Ф. В. Рабинович
Технический редактор В. М. Скитина
Художник Е. В- Бекентов
Корректор М. С. Хрипунова
Сдано в наб. 6/VI 1973 г. Подп. в печ. 28/IX 1973 г. Формат
бумаги 60Х90’/|«. Бумага тип. № 2. Усл. веч. л. 25,5.
Уч.-нзд. л. 30,19. Тираж 11 000 экз. Зак. 677. Изд. № 192.
Ц. 2 р. 31 к.
Издательство „Химия". 107076, Москва, Стромынка, 23.
Ордена Трудового Красного Знамени
Ленинградская типографии № 2 имени Евгении Соколовой
Союзполнграфпрома прн Государственном комитете Совета
Министров СССР по делам издательств, полиграфии и книжной
торговли
198052, Ленинград, Л-52, Измайловский проспект, 29