/





























































































































































Текст
ЛЬЛДКМИЯ НАУК СССР
CIHJIH'CKOE ОТДЕЛЕНИЕ
ИНСТИГУТ ХИМИИ ТИНГ Ь’ГО ТЕЛА
II iii.i’Epahotkh МИНЕРАЛЬНОГО СЫРЬЯ
В. Г. АВВАКУМОВ
МЕХАНИЧЕСКИЕ
МЕТОДЫ
АКТИВАЦИИ
ХИМИЧЕСКИХ
ПРОЦЕССОВ
2-е ИЗДАНИЕ, ПЕРИРАБОТЛИИОЕ
И ДОПОЛНЕННОЕ
Ответственный редактор
панд. хим. паук -1. С. Колосм
НОВОСПВП РСК
I! :: Д \ТЕ Л Ь С Т В О «И А У К А»
СШИ1РСКОЕ ОТДЕЛЕНИЕ
19 8 6
УДК 541.053
Аввакумов Е. Г. Механические методы
активации химических процессов.— 2-е над., ие-
рераб. и доп.— Новосибирск: Наука, 4986.
В монографии рассмотрены закономерности дисперги-
рования твердых тел н изменения их физико-химических
свойств нод влиянием механической обработки в зперго-
ианряженпых иамельчительпых аппаратах. Приведены ре-
зультаты исследовании изменения структуры и электрон-
ных свойств ряда практически важных неорганических
веществ (оксиды, сульфиды, соли и др.) с применением
современных физических методов. Изложены новые экс-
периментальные данные но кинетике и механизму механо-
химических реакций в неорганических системах. Обсуж-
даются возможности механических методов активации для
неорганического синтеза, катализа, ускорения гетероген-
ных реакции, регулирования свойств твердых тел и т. д.,
намечаются перспективные направления их использования.
Книга рассчитала па научных и пижеиерпо-техниче-
скпх работников, химиков, технологов.
Рецензенты А. С. Лсргер, 11. 10. Луптгин
А
1802000000—804.-г ,,
-1 - — -- — — <1 OV—“11
042(02)-86
© Издательство «Паука», 1986 г.
ПРЕДИСЛОВИЕ КО ВТОРОМУ ИЗДАНИЮ_____-
Первое издание настоящей книги вышло в 1979 г. Ввиду
интенсивного развития в последние годы исследований по
механической активации химических процессов интерес к
обобщающим работам в этой области чрезвычайно возрос.
Это сделало необходимым повторный выпуск монографии.
Для второго издания книга переработана, хотя в целом
ее научное направление и общая структура сохранены. Со-
держание отдельных глав не только дополнено новым мате-
риалом, отражающим развитие мехапохимии за последние
годы, по и углублено и расширено за счет новых теорети-
ческих и экспериментальных работ.
Как и в мерном издании, книга не содерлшт исчерпываю-
щего обзора литературы и изложения всех существующих в
мехапохимии неорганических веществ направлений. Опа
отражает в основном результаты работ, выполненных груп-
пой исследователей Института химии твердого тела и пере-
работки минералi.hoi'o сырья СО АН СССР под руководст-
вом члена-корреснондента АН СССР В. В. Болдырева.
Появление в 1983 г. монографии В. В. Болдырева
«Экспериментальные методы в мехапохимии твердых неорга-
нических веществ», в которой изложены наиболее важные
результаты, описана экспериментальная техника и дапы
методологические основы и области мехапохимических про-
цессов, существенно упростило задачи, стоявшие перед авто-
ром при подготовке настоящего издания.
В книгу внесены изменения, касающиеся как существа
затронутых вопросов, так и формы изложения. Более де-
тально рассмотрены положения, которые в нервом издании
намечены лишь схематично. К таковым относятся вопросы о
роли пластической деформации, электронных возбуждений
3
ii мехамохпмпчоскнх реакциях, об образовании пестехно-
метрпческнх дефектов, о сравнении термически п механически
акишпрхемых реакций и т. д. В,месте с тем исключены не-
которые разделы, например изложение .> темен гарны х пред-
ставлений о кристаллической структуре твердых тел, общие
сведения о реакциях с участием твердых тел и т. д. Появи-
лись новые разделы по вопросам, не нашедшим отражения
в первом издании, в частности ио мохапохпмическим реак-
циям твердых тол с жидкими средами, мехаиохнмическому
синтезу в многокомпонентных системах п т. д.
Автор надеется, что книга будет полезна химикам, инте-
ресующимся влиянием механических процессов па протека-
ние химических реакций, инженерам, конструкторам, тех-
нологам, а также молодым специалистам, приступающим к
исследованиям в дайной области.
Хочется выразить глубокую признательность члену-кор-
респонденту АН СССР В. В. Болдыреву за постановку ис-
следовании, результаты которых легли в основу монографии,
за постоянный интерес и поддержку в работе, а также благо-
дарность всем научным сотрудникам, в той или иной мере
участвовавшим в экспериментах и обсуждении затронутых
в книге проблем: В. В. Александрову, В. Ф. Ануфриенко,
В. А. Варпеку, А. И. Воробейчик, II. В. Косовой, О. И. Са-
марину, Т. И. Татаринцевой, Ф. X. Уракаеву, М. В. Чай-
киной и др. Автор благодарит сотрудников лаборатории
мехапохпмни неорганических соединений В. М. Тремасова,
II. 11. Николаева за большую помощь мри подготовке моно-
графии к печати, а также А. С. Колосова, Л. С. Бергер и
11. 10. Бутягипа, взявших па себя труд ио редактированию
и рецензированию монографии.
Все замечания и предложения по поводу содержания кни-
ги будут восприняты автором с признательностью.
ВВЕДЕНИЕ
Интерес к механическим методам ускорения химических
реакций, особенно между твердыми телами, начал прояв-
ляться и конце прошлого пека. Менделеев [11 указывал, что
для того чтобы между твердыми телами протекали реакции,
«необходимо сколь возможно мелко измельчить и переме-
шать их между собой. Через это взаимодействие значитель-
но ускоряется». Оствальд 12, 3], занимаясь систематикой
химических наук па основе энергетического подхода, отнес
к мехаиохимии широкий круг проблем, в частности разделы
хинин, изучающие взаимосвязи химической энергии с раз-
личными видами механической энергии, такими как «энергия
силы тяжести», «энергия движения», «энергия изменения
объема», «поверхностная энергия», «энергия формы» и т. д.
Что определение мехапохпмни значительно шире, чем совре-
менное, так как п него включаются п хемомехапика, н раз-
делы химии, изучающие поверхностные процессы, хими-
ческие превращения, идущие с изменением объема системы,
физико-химические процессы, протекающие иод влиянием
гравитации и т. и. Он считал, что механохимию следует рас-
сматривать как пауку, являющуюся одним из разделов фи-
зической химии, стоящую в одном ряду с такими науками,
как термохимия, электрохимия н фотохимия.
Впервые снедения о разложении неорганических веществ
и результате механических воздействий были систематизиро-
ваны в работе Кэри Ли [41. Нм было показано, что многио
соединения серебра, ртути, золота и платины при растира-
нии в ступке разлагаются с выделением металлов. Причем
такое растирание оказывается более аффективным, чем при-
менение стационарного, даже очень высокого давления.
Позднее были обнаружены другие особенности химических
5
реакций, имеющих место при истирании, paapyiilciiiiil и
измельчении. Большие исследования но твердофазным реак-
циям, протекающим при растирания порошков, выполнены
в конце прошлого века Флавицким [51, а несколько ранее
Паркером [б, 7]. Метод проведения реакций путем растира-
ния порошков впоследствии нашел применение для качест-
венного анализа природных смесей в геологической практи-
ке. Многое для этого сделали Воскресенский {81 и Иса-
ков [91.
Однако, несмотря па то, что приоритет в историческом
плане принадлежит мехапохпмип минеральных соединений,
научная оценка значимости мехапохимических явлений,
обоснование и установление их специфики сделаны для вы-
сокомолекулярных органических соединений. После того
как Ванетигом [101 было показано, что при помоле целлю-
лозы происходит механодеструкция молекул целлюлозы,
были выполнены большие исследования по механодеструк-
ции других органических соединений (полистирола, высших
полимеров и др. [111). Результаты исследований по механо-
химии полимеров и высокомолекулярных соединений обоб-
щены в обзорных работах Берлина 1121, Барамбойма [13 J,
Симиопеску и Опреа [14].
В 20-е годы Бриджменом были начаты систематические
исследования влияния высоких давлений па скорость твер-
дофазных превращений. Важным результатом его исследова-
ния явилось установление факта ускорения твердофазных
процессов (фазовых переходов, химических реакций и др.)
при наложении на твердые тела высоких давлений с дефор-
мацией сдвига [15, 161, что и положило начало широким
исследованиям этого эффекта в химии [17, 18].
В те же годы Тамман [19 J наблюдал увеличение скорости
растворения металлов, вызванное механической обработкой.
Он нашел, что не вся механическая энергия превращается
в тепло, а от 5 до 15% се остается в металле (потенциальная
энергия) и увеличивает термодинамический потенциал твер-
дого тела. В начале 40-х годов Кларк и Рован [201 показа-
ли, что механическая обработка материалов в мельницах
приводит к таким же фазовым переходам, как и при воз-
действии высокого давления и сдвиговых деформаций. Ре-
зультатом такого рода исследований стало формирование ме-
хапохпмии неорганических веществ как пауки. К ней отно-
сят процессы, происходящие в момент механической обработ-
ки и являющиеся ее следствием: разложение и синтез ве-
ществ, ускорение химических реакций, увеличение скорости
растворения, рост каталитических свойств, изменение
с।p,vhгуры ii физике химических снойстн исщести, пониже-
ние температуры, необходимой для реагирования п спека-
нии, и т. д. Объектом мехапохпмип являются исследования,
направленные па выяснение вопроса: каким образом механи-
ческая энергия переходит в химическую. Переходит ли сна-
чала механическая энергия в другие формы (тепло, свет
и т. д.) п только затем в химическую энергию или имеет мес-
то непосредственный переход из механической энергии в
химическую?
Согласно определению, данному академиком Ребиндером,
«цель мехапохпмип состоит в использовании или предот-
вращении тех химических реакций, которые вызываются
или ускоряются механической активацией» [21].
Особый интерес представляет механическая активация
твердых тел и реакций с их участием, так как установлено,
что часть механической энергии, подведенной к твердому
телу во время активации, усваивается им в виде новой по-
верхности, линейных и точечных дефектов. Кроме того,
известно, что химические свойства кристаллов определяются
наличием в них дефектов, их природой и концентрацией.
Таким образом, в исследованиях по механохимии органично
сливаются два потока знаний — по физико-химической ме-
ханике твердых тел и химии дефектных кристаллов. С по-
мощью механической активации удается поставить па служ-
бу химии ряд физических явлений, происходящих в твердых
телах при больших скоростях деформации. К ним относят-
ся: изменение структуры твердых тел, ускорение процессов
диффузии при пластической деформации, образование актив-
ных центров па свежеобразованпой поверхности, возникпо-
неиие импульсов высоких локальных температур и давлений
и т. д. Впервые к использованию этих эффектов в химии по-
дошли исследователи, изучавшие влияние высоких давлений
со сдвиговыми деформациями [22—24] н ударных воли па
свойства твердых тел [23, 261. Однако указанные эффекты
можно получить н с использованием измельчительного обо-
рудования, что с практической точки зрения более целесо-
образно и осуществимо, особенно для непрерывных процес-
сов. В результате совершенствования этого оборудования
появились аппараты с высокой интенсивностью подвода
энергии, и роль этих эффектов при измельчении сильно воз-
росла.
Одной из первых работ, обобщающих результаты иссле-
дований по мехапохпмип неорганических веществ, является
монография Тиссена с соавторами «Основы трибохнмин»
[27]. Авторы привели большой экспериментальный мате-
7
риал по физике и химии мехапохимнческпх процессии н рас-
смотрели его с точки зрения разниваемой ими «магма-плаз-
ма модели».
Ученые ГДР успению развивали исследования в этом
направлении. Коллективом авторов из Института физи-
ческой .химии ЛИ ГДР (Берлин)под руководством крупного
специалиста в области мехапохимии профессора Герхар-
да Хойнике, безвременно скончавшегося в 1982 г., подго-
товлена капитальная монография «Трнбохимия», которая
является в настоящее время самым обширным в мировой
литературе обобщающим трудом в области мехапохимии
[28]. В нем собраны сведения практически обо всех рабо-
тах по механохимии, опубликованных до 1980 г.
Свидетельством стремительного развития исследований
по мехапохимии является проведение различных совещаний,
появление большого числа статей, обзоров и монографий во
всех странах мира. Среди них следует отметить работы уче-
ных, представляющих различные научные школы и направ-
ления, сформировавшиеся и развивающиеся в странах со-
циалистического содружества: в СССР — Болдырева [29—
33], Сутягина 134, 35], Дерягина [36—38], Гутмана 139],
Власовой, Каказей 140], Зеликмапа 141, 42], Лаптевой,
Юсупова, Бергер 1431, Молоцкого 144, 45], Молчанова,
Юсупова 146], Павлюхина 147—49], Ходакова 1501; в
ГДР — П. Тиссена 128] и К. Тиссена 128], Хеннига 122],
Линке 128], Штайпеке 128], Поста ]28], Бернхарда, Хеег-
па 151, 52]; в НРБ — Чакырова, Господинова 153—55];
в ВНР — Юхаса 156], Опоцки 1571; в ЧССР —Ткачовой
[58], Гаска ]59], Ружека (601; в ПНР — Павельчнка (611;
в СФРЮ — Оцепека 162] и др. Обзоры, содержащие новые
результаты в различных направлениях мехапохимии неорга-
нических систем, опубликованы Гилманом, Бенджамином
[63], Лином, Надивом [64], Кубо [65], Питерсом [66 I, ‘Рок-
сом [67] и др.).
С помощью метода механической активации могут быть
решены самые разнообразные задачи: повышение реакцион-
ной способности твердых тел, изменение структуры, уско-
рение твердофазных реакций и г. д. Однако теоретических
знаний относительно механизма дефектообразования, а так-
же механизма и кинетики указанных процессов накоплено
еще недостаточно для того, чтобы прогнозировать возмож-
ности метода.
Суть научного направления, которое рассматривается в
настоящей монографии, составляют исследования механо-
химических превращений неорганических веществ при обра-
нотке ii\ и и iMe.ii.4iiT(','ii.iii.i\ аппаратах с noituiiifKKoii inrreil-
< niiiiocri.io подвода механический энергии. В таких динара
i;>\ iiMin п.< ui.ie нагрузки н скорость нагружения значитель-
но ni.iiiie. чем и опытных. I! них но1енцна.ты1н пилмо.киы ка-
чесгиенно новые эффекты, так как увеличении указанных
параметров может приводить к увеличению скорости разру-
шения п пластической деформации твердых тел.
В задачу наших исследований входило установление за-
кономерностей дефектообразования в некоторых неоргани-
ческих веществах, а также особенностей мехапохимнческпх
реакций под влиянием механической активации.
Многие исследователи связывают изменение реакцион-
ной способности в основном со структурными дефектами.
В то же время известно, что ряд вопросов реакционной спо-
собности твердых тел может быть решен с позиции теории
иестехнометрпи неорганических соединений. В связи с обна-
ружением в неорганических веществах протяженных дефек-
тов, образующихся ио механизму кристаллографического
сдвига, теория нестехиометрни учитывает структурные осо-
бенности исследуемых веществ. Поэтому в настоящей работе
для объяснения механизмов активации твердых тел привле-
чены обе эти точки зрения и сделана попытка показать их
взаимосвязь.
Механические методы активации перспективны для уско-
рения гетерогенных химических реакций. Нами изложены
результаты исследования различных типов мехаио.хпмн-
ческих реакций в зависимости от механических нагрузок
и других факторов и закономерности протекании таких ре-
акций.
В отношении обоих направлений исследований осуществ-
лен единый методологический подход, предложенный чле-
ном-корреспондентом АН СССР В. В. Болдыревым: струк-
турно-химические изменения в твердых телах н механо-
химические реакции иод влиянием механической активации
сравнивались с изменениями и реакциями, протекающими
при нагревании. Последовательная реализация такого под-
хода позволила выявить специфику процессов механической
активации и механохимических реакций, установить их
особенности и сделать определенные выводы об их механиз-
ме.
---------------Г ЛАВА 1
ОБЩИЕ ЗАКОНОМЕРНОСТИ ДИСПЕРГИРОВАНИЯ
И АКТИВАЦИИ ТВЕРДЫХ ТЕЛ
1.1. МЕХАНИЧЕСКИЕ СВОЙСТВА КРИСТАЛЛОВ
Механические свойства кристаллов как совокупности
многих частиц (атомов, ионов или молекул), образующих
правильную решетку, определяются его составом, силами
взаимодействия частиц, структурой кристалла и наличием
в нем разного рода дефектов. В зависимости от того, как
твердые тела реагируют на приложенную нагрузку, они
имеют три основные характеристики: первая—упругость,
отражающая способность твердого тела вернуть свою форму,
если к нему приложить на какое-то время силу, а потом
снять ее; вторая — пластичность, показывающая, как быст-
ро под действием длительной нагрузки тело изменяет свою -
форму или какой должна быть сила, чтобы это изменение
шло с определенной скоростью; третья — прочность, или
сопротивление разрушению. Упругость зависит в основном
от энергии взаимодействия составляющих кристалл частиц,
пластичность — от образования в твердых телах линейных
дефектов (дислокаций), а прочность — от совокупности фак-
торов разного масштаба: характера химической связи,
структуры кристалла, существования в реальном кристалле
структурных дефектов (точечных, линейных, поверхностных
и объемных) [68, 691. Эти дефекты возникают в процессе
роста кристалла или в резуль-
р тате различных воздействий на
D него.
д / Если к кристаллу прило-
________________-У жить механическое напряже-
ние, то прежде чем разрушить-
/_______________ся, он пройдет стадии упругой,
Pitc. 1. Диаграмма растяжения твер-
А с дого тела.
10
а затем пластической деформации. Снизь между деформа-
цией п приложенным напряжением изображена на рис. 1.
11|»п малых напряжениях кристалл деформируется вполне
упруго, а после разгрузки ею начальная форма полностью
восстанавливается. В области упругости связь между де-
формацией н напряжением подчиняется закону Гука:
/’ - Л'е, (1.1)
где /’ — напряжение, е — относительное удлинение, Е —
коэффициент пропорциональности, называемый модулем
Юнга (модуль нормальной упругости). Для кристаллических
твердых тел Е зависит от направления. Если от некоторого
центра отложить по разным направлениям векторы, харак-
теризующие модуль упругости, и через их окончания про-
вести поверхность, то получится фигура, отражающая сим-
метрию кристалла.
Предельное напряжение, до которого действует закон
Гука, называется пределом упругости {Ру). Область ВС
соответствует пластической деформации (область теку-
чести). И наконец, напряжение, при котором кристалл раз-
рушается (точка D), соответствует пределу прочности крис-
талла (Пир).
Вид кривой Р — в характеризует прочность, хрупкость
и пластичность кристалла. Эти физические свойства кристал-
лического вещества, в свою очередь, зависят от температу-
ры, скорости и типа деформирования, наличия примесей
в кристаллах, предварительной механической и термической
обработки, ориентировки кристалла и т. д.
11.масти чес кая деформация кристалла может происходить
пу гем скольжения или путем двойникования. При сколь-
жении топкие слон кристалла смещаются друг относитель-
но друга подобно соскальзывающей стопке книг. При меха-
ническом двойниковании происходит деформация кристалла
таким образом, чго две части его оказываются зеркально-
симметричными или повернутыми относительно оси второго
порядка.
Отличительной особенностью пластической деформации
кристалла является ее анизотропия. Кристалл деформирует-
ся не ио направлению действующей силы, а только в опре-
деленных кристаллографических плоскостях по определен-
ным кристаллографическим направлениям, зависящим от
его структуры.
Как правило, плоскостями и направлениями скольжения
служат плоскости и направления плотнейшей упаковки.
Для ионных кристаллов со структурой типа NaCl, для ко-
11
валентных кристаллов со структурой алмаза, для ОЦК-
металлов скольа.епие обычно идет ио плоскости {111}. Сис-
тема двойникования тоже зависит от тина структуры ]В9].
Тейлором 170] показано, что способность твердых тел к
пластическому деформированию определяется наличием в
их структуре дислокаций, которые могут перемещаться в
кристалле иод действием малых напряжений н генерировать
другие дислокации в процессе деформации. Законы, по ко-
торым движутся дислокации, исследовали Френкель и Кон-
торова 171], показавшие, что перемещения дислокаций в
кристалле являются элементарными актами пластической
деформации. Скорость деформации описывается соотно-
шением
е = бри, (1.2)
где b — вектор Бюргерса, риг — соответственно плотность
н средняя скорость движения дислокаций.
Дислокаций, которые возникают при росте в кристалли-
ческих твердых телах, сравнительно немного, поэтому они
не способны обеспечить макронластичиость кристалла, даже
будучи подвижными. Пластическая деформации определяет-
ся процессами возникновения и размножения подвижных
дислокаций непосредственно в момент механического воз-
действия. Существующие в твердом теле дефекты создают
препятствия для движения дислокаций. При малых его ско-
ростях торможение объясняется остановкой на барьерах,
которые преодолеваются термоактивируемым путем. Обыч-
но такие барьеры в кристалле подразделяют на два типа —
линейные и локальные. Характерным примером первого ти-
па является барьер Пайерлса — Иабарро, обусловленный
периодическим полем кристаллической решетки. Ко второ-
му типу барьеров относят разнообразные точечные дефекты
(примесные атомы, вакансии п др.), ступеньки на дислока-
циях и дислокации «леса». При больших скоростях тормо-
жение дислокаций является следствием взаимодействия их
с элементарными возбуждениями кристалла (фононами,
экситонами и т. д.).
Пока внешняя нагрузка создает силу, меньшую, чем
действие всех внутренних препятствий, дислокации остают-
ся в покос (участок АВ кривой растяжения). Когда силы
сравниваются, происходит освобождение дислокаций (пре-
дел текучести), и даже при незначительном повышении
нагрузки они, ускоряясь, начинают двигаться по кристаллу
(площадка текучести ВС). 13 разных точках кристалла дис-
локации встречают различные барьеры, и, срываясь с более
12
низких, они останавливаются у более высоких, еще сильнее
повышая их нолем своего заряда. Накапливаясь друг за
другом, дислокации искривляют кристалл настолько, что
дальнейшее распространение сдвига приостанавливается —
кристалл упрочняется. Новое повышение внешней нагрузки
оторвет часть дислокаций и еще больше испортит кристалл.
Так постепенно происходит подъем по кривой упрочнения
CD. Однако и скопление дислокаций у препятствий имеет
свой предел. Когда нагрузка достигает предела прочности,
первые дислокации, остановившиеся у границы зерна, под-
вергаются действию настолько мощного поля, что происхо-
дит своеобразная «ядсрпая реакция» — несколько дислока-
ций сливаются в одну. Заряды их складывается, н образует-
ся мнкротрещипа. Достаточно нагрузке немного превзойти
предел прочности, как такая трещина раскалывает кристалл.
Металлы и неметаллы отличаются условиями движения
дислокаций. В металлах, не подвергавшихся специальной
обработке, как правило, предел текучести невысок, по они
способны к значительному упрочнению при пластической
деформации. Неметаллы, наоборот, очень плохо деформи-
руются и обычно растрескиваются еще до предела текучести.
Таким образом, в металлах препятствия невысоки и дисло-
кации легко выходят из кристалла на поверхность, медлен-
но накапливаясь при упрочнении. В неметаллах дислокации
перемещаются с большим трудом при высоких напряжениях
и, застревая внутри кристалла, создают зародышевые тре-
щины.
Экспериментально дислокации выявляются путем трав-
ления кристаллов растворителями. Наблюдая в микроскоп
за ямками травления, возникающими в местах пересечения
плоскостей скольжения с поверхностью кристалла, можно
проследить за пластической деформацией кристалла. В ка-
честве примера па рис. 2 приведены результаты наблюде-
ний, выполненных Мейером 127], при единичном механи-
ческом воздействии на кристалл NaCl. Видно, что дислока-
ции распространяются вдоль плоскостей скольжения па зна-
чительные расстояния от моста единичного механического
воздействия. Объем зоны пластической деформации зави-
сит от кинетической энергии воздействующего тела.
Сами дислокации, возникшие при деформации, влияют
со своей стороны на внутреннее строение кристаллов,
в частности притягивают примесные атомы и образуют обла-
ко примесей вокруг дислокации («облака» Котрелла 172]).
Как показывает опыт, существенное влияние па_ироч-
ность кристаллов имеют их размеры. С уменьшением их
13
Рис. 2. Ямки травления, образовавшиеся в местах выхода дислока-
ций на поверхность кристалла NaCl при воздействии на него иглой
из карбида кремния [27]. X 250.
уменьшается н вероятность возникновения дефектных участ-
ков в кристалле, в результате чего прочность воз-
растает [73].
Рассмотрение твердых тел как своеобразных «конструк-
ций» из частиц, связанных между собой силами сцепления,
привело первоначально к созданию чисто механических тео-
рий разрушения твердого тела, согласно которым разруше-
ние наступает при достижении действующим напряжением
предельного «критического» значения. Задача теорий разру-
шения сводилась к нахождению энергетического критерия
для предельного значения исходя из расчетов на основе тео-
рий упругости локальных напряжений вблизи полостей и
трещин. Гриффитс [74] впервые показал, что разрыв идеаль-
но упругих тел возможен, если количество упругой энергии,
освобождающейся при росте трещин разрушения, достаточ-
но для компенсации затраты энергии на образование новой
поверхности.
14
Рассматривая разрушение материала как одновременный
разрыв связей между всеми атомами, расположенными по
обе стороны площадки в 1 см2, выбранной перпендикулярно
растягивающей силе, Орован [75] получил для расчета на-
пряжения отрыва одной части от другой уравнение
1
где а — постоянная решетки, о — поверхностное натяжение.
При выводе этого уравнения Орован считал, что накоплен-
ная упругая энергия твердого тела в момент разрушения
равна l/2/’е, поверхностная энергия каждой из двух вновь
образованных поверхностей на единицу площади равна о,
откуда 1/2/^е = 2о. Далее, согласно закону Гука, для упру-
гого материала Р — ч/аЕ, где а — межатомное расстоя-
ние. Исключив в пз этих уравнений, Орован получил урав-
нение (1.3).
При расчете напряжений разрушения твердых тел по
уравнению (1.3) необходимо знать их удельную поверхност-
ную энергию. Для ионных кристаллов Маделунгом, Бор-
ном, Френкелем, Ямада разработаны методы теоретического
расчета поверхностной энергии (см., например, [76]).
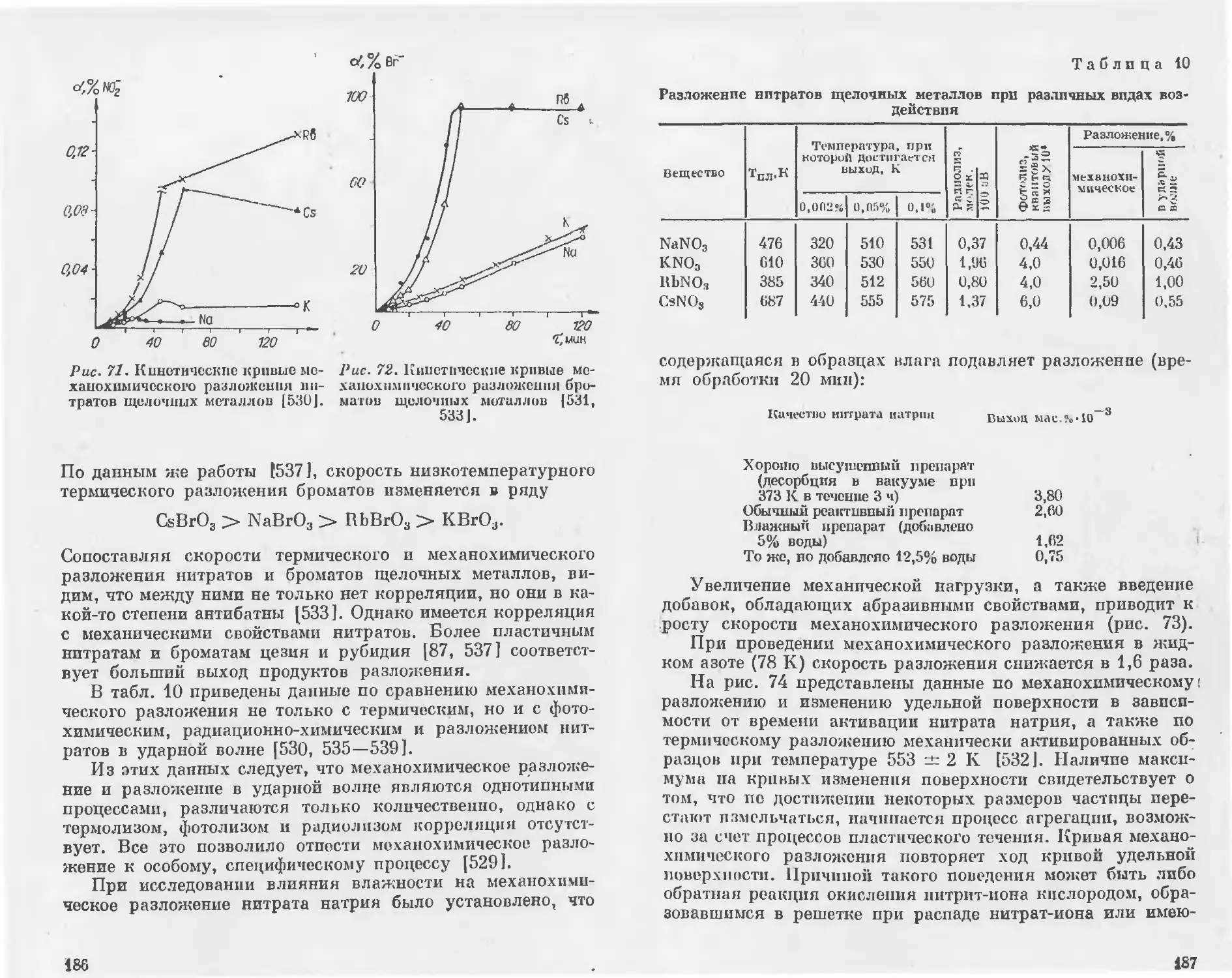
В частности, для соединений со структурой типа ХаС1
Френкелем [77] получены следующие уравнения:
2
а100 = 1,28.10"8^(Джсм=); (1.4)
и
о'ыо = 2,5о100; (1.5)
°ш ~ 5,8а100, (1.6)
где е — заряд электрона.
Согласно Ормонту [78], удельная поверхностная энергия
для широкого круга неорганических соединений, в том числе
и с ковалентной связью, связана с энергией ато.мпзацйи
кристалла уравнением
(Дж/см2), (1.7)
а
где Q — энергия атомизации, равная энергии образования
кристалла из атомов:
. DB
йдв — Qab + ЙА + ~2----Для ионных соединений, (1.8)
QAB = QAB + QA + —для ковалентных соединений, (1.9)
15
М — структурная постоянная, зависящая от структурного
типа и символа грани, равная
М = 0,624^—^. (1.10)
В уравнениях (1.8)—(1.10) — энергии атомизации
вещества А, В; QAB — тепловой эффект образования 1 моля
иэ простых веществ; Dt — энергия диссоциации вещества
на атомы; и — количество атомов в формуле; с — координа-
ционное число; б — кратпость элементарного параллело-
грамма на плоскости; т — множитель (равен 1 для грани
(100), в других случаях отличен от 1); а: — число разрывае-
мых связей. Пользуясь указанными уравнениями, можно
рассчитывать ряды твердости, как это сделано, например,
для карбидов и нитридов в работе [78].
В последнее время Русановым [79] и Ощериным [80]
разработаны новые методы расчета свободной поверхностной
энергии твердых тел, что позволяет более широко использо-
вать их для оценки прочностных свойств твердых тел.
Подстановкой обычных значений В, а, а в выражение
(1.3) получают теоретическое значение Ртеор « 1010 Н/м2,
которое выше экспериментальных значений на 2—3 по-
рядка.
Гриффитс [74] объяснил это несоответствие, предполо-
жив, что в хрупком теле имеются микротрещины, которые
вызывают концентрацию напряжений, достаточную для ло-
кального превышения теоретической прочности. Трещина в
твердом теле развивается, если упругая энергия в трещине,
равная РЧ2/2Е (I — длина трещины), превышает энергию,
необходимую для образования поверхности 2oZ, т. е. при
заданном значении I напряжение разрыва
j
Р=И2. (1.11)
Из сравнения (1.3) и (1.11) видно, что
р г I
* теор
(1.12)
Поскольку а 10"10 м, достаточно иметь в твердом теле
трещину длиной I = 10"® м, чтобы прочность тела понизи-
лась в 100 раз по сравнению с теоретическим значением.
Таким образом, отношение реальной и идеальной проч-
ностей твердого тела определяется соотношением между меж-
атомным расстоянием а и средним размером трещины I.
16
KA-&6-W9
Ребиндер [81 ] применил эти представления и для разру-
шения твердых тел, которое сопровождается заметным плас-
тическим деформированием. В таком случае связь прочности
тела с размером зародышевой трещины I может быть оппсапа
выражением, сходным с уравнением Гриффитса:
1
р = (о*£у (1 13)
Однако здесь величина о* — «эффективная поверхностная
энергия», представляющая собой удельную (на единицу
вновь образующейся поверхности) работу разрушения. ОнА
включает помимо истинной поверхностной энергии а и ра-
боту пластических деформаций на единицу поверхности тре-
щины, т. е. энергию искажений решетки, возникающих при
разаитип трещины. Величина о* может на несколько поряд-
ков превосходить истинное значение о, но, как отмечал Ре-
бнпдор, работа пластических деформаций спмбатна величи-
не о. При снижении о, например путем введения поверх-
ностно-активных веществ, понижается и работа пластической
деформации.
Систематический и детальный анализ характера времен-
ных зависимостей прочности, а также прямое изучение эле-
ментарных процессов, происходящих в напряженных твердых
телах, показали, что «критические» значения прочности,
рассчитанные на основе энергетической теории, не являются
стабильными, и привели к необходимости учета теплового
движения атомов в твердом теле. Этот подход составляет
основу кинетического подхода к проблеме прочности твер-
дых тел, разработанного Журковым с сотрудниками
[82, 83].
Согласно кинетической теории прочности, разъединение
атомов осуществляется при нагрузках, меньших прочности
межатомных связей, причем «дорывание» напряженных меж-
атомных связей осуществляют тепловые флуктуации. Кон-
станта скорости разрыва связей увеличивается по экспонен-
циальному закону:
к = к0 ехр
где U — энергия активации разрушения, Р — энергия де-
формации, у — коэффициент пропорциональности.
Результаты исследования долговечности различных ве-
ществ под нагрузкой показали, что энергия активации раз-
17
рушения совпадает с теплотой испарения веществ (кДж/моль) [831:
Вещество и Ецси B< шесто и Eucu
Si 473 465 Ад 260 277
Ge 382 382 Zn 126 130
LiF 310 268 Al 220 230
NaCl 276 243 Fe 419 407
KOI 232 222 Pt 503 532
Cu 340 336
Близость этих величин свидетельствует об общности пер-
вичных стадий процессов. В процессе испарения атомов с
поверхности веществ в каждом акте отрыва атома от кристал-
лической решетки, как это показано Френкелем [84], разры-
ваются связи, число которых соответствует примерно поло-
вине координационного числа. Для образования элементар-
ного разрыва в решетке также должны быть разорваны
единичные связи в количестве, близком к половине коорди-
национного числа.
Энергия активации разрушения, так же как теплота
испарения, не зависит от ориентации образца, предвари-
тельной обработки, примесей и т. д., т. е. является структур-
но-нечувствительной величиной. Коэффициент у в уравне-
нии (1.14) является коэффициентом перенапряжения, пока-
зывает неравномерность нагружения межатомных связей и
различных участках твердого тела и является в отличие от
U структурно-чувствительной величиной, т. е. зависит от
ориентации, примесей и т. д.
Таким образом, прочность твердых тел, согласно энерге-
тической теории прочности, можно оцепить, если известна
удельная поверхностная энергия твердого тела, а согласно
кинетической теории — энергия испарения вещества.
Хотя прочность не является термодинамической величи-
ной, тем не менее симбатность ее таковым имеется. Так, на-
пример, эффективные пределы прочности многих веществ
изменяются линейно в зависимости от температуры плав-
ления [85].
Однако такой корреляции может не наблюдаться между
механической прочностью и термической устойчивостью.
Причина такой неадекватности может быть связана с тем, что
процессы механического и термического разрушений опреде-
ляются прочностью различных связей: в первом случае —
прочностью связи катион — анион, во втором — прочностью
связей в сложном анионе. Это видно из табл. 1, в которой
приведены значения твердости (величина, пропорциопаль-
18
Таблица t
Механическая твердость и термическая устойчивость неко-
торых твердых веществ [85—89]
Соедине- ние Тисриосп. [87, 88J 'Герм пч ос к а и у сто П чивость [86]
ПО Моосу Микротвер- чость по Виккерсу (среди»»), кг/ым2 Температура разложения (давление газа достига- ет атмосфер- ного)^ Энергия активации разложения, кДж/моль
МЯСО, 4.5 — 923 150
СаС03 4,0 — 1073 205
SrCO3 4,0 — l'.8i 222
ВаСО3 3.5 — 1520 283
l’cS2 — 2181 1015 285
CoSj — 1073 1223 242
NiS2 — 811 1295 23!
ная прочности) и термической устойчивости для некоторых
карбонатов и сульфидов.
В работе 188] определены длины связей М—S и S—S для
указанных в таблице сульфидов методом ПК-спектроско-
пии. В ряду FeS2, CoS2, NiS2 длина связи М—S увеличи-
вается, a S—S уменьшается, соответственно прочность свя-
зи М—S уменьшается, a S—S увеличивается. Близость
энтальпий образования этих соединений есть проявление
двух противоположных тенденций: ослабления связи М—S
и упрочнения связи S—S в этом ряду сульфидов.
Из вышесказанного следует, что механические свойства
реальных твердых тел существенно отличаются от свойств,
определяемых из теории идеального кристалла. Так, значе-
ния прочности, близкие к рассчитанным по энергетической
теории, получены только для нитевидных бездислокацион-
ных кристаллов. Развитие современных теорий прочности
идет по пути учета дислокационных структур, различного
рода дефектов, примесей и других параметров, влияющих
па механические свойства.
19
1.2. ДИСПЕРГИРОВАНИЕ
II АКТИВАЦИЯ ТВЕРДЫХ ТЕЛ
1.2.1. Энергетика процессов
диспергирования
Измельчение веществ осуществляется путем последова-
тельного дробления частиц материала мелющими телами.
По размерам образующихся частиц различают грубое (0,1 —
1 мм), среднее (0,01—0,1 мм) ц топкое (< 0,01 мм) измель-
чение. Процесс получения тончайших порошков и высоко-
дисперсных суспензий определяют более узким понятием —
диспергирование.
Попытки создания теорий измельчения делались уже бо-
лее ста лет назад. Первая научная теория измельчения была
разработана Риттипгером (1867 г.) 190]. Он исходил из пред-
положения, что работа измельчения затрачивается на обра-
зование новой поверхности, т. е. в эквивалентных коли-
чествах переходит в свободную поверхностную энергию.
Закон выражается следующим образом:
А = /fps, (1.15)
где А — удельная работа измельчения, Ар — коэффициент,
определяющий работу, затрачиваемую на создание единицы
поверхности.
Согласно другой теории, созданной Киком (1883 г.), ра-
бота измельчения затрачивается на упругопластическую
деформацию, которую нужно произвести, чтобы разрушить
твердое тело:
А = kKV, (1.16)
где V — объем измельчаемого тела, А’|( — коэффициент про-
порциональности.
Имеются также другие теории процессов разрушения,
например теория Бонда, которая может рассматриваться как
промежуточная между законами Риттингера и Кика. В ней
предполагается, что энергия, передаваемая телу при сжа-
тии, распределяется сначала по его массе и, следовательно,
пропорциональна d9 (d — диаметр твердой частицы), но с
момента начала образования па поверхности трещины эта
энергия концентрируется на поверхности у краев трещины и
становится пропорциональной <Р. Па этом основании прини-
мается, что работа разрушения тела пропорциональна
Многочисленны более современные попытки развития тео-
рии измельчения с более полным учетом физических свойств
20
твердых тел, их реальной структуры, влияния среды, в ко-
торой идет измельчение [58, 91].
Сложность процесса измельчения, зависимость его от
многих факторов, часто неоднозначных для различных ма-
териалов, делают невозможным чисто теоретический расчет
минимума работы, необходимого для измельчения. Если по-
лезной считать только ту работу, которая затрачена на обра-
зование поверхности, то в известных нзмельчптельных аппа-
ратах большая часть подводимой механической энергии пре-
вращается в тепловую, рассеиваясь бесполезно. КПД аппа-
ратов по такому расчету составляет от долей процента до,
в лучшем случае, нескольких процентов [92]. Исследования
па калориметрических мельницах показали, что часть ме-
ханической энергии запасается измельчаемыми твердыми те-
лами. В первых исследованиях Шеллингера доля затрачен-^
пой энергии, которая не была обнаружена в виде тепловой,
составила 10—19% [197]. Последние исследования, выпол-
ненные в Институте горного дела Словацкой АИ [93] н в
Институте обогащения АП ГДР [94], подтверждают данные
Шеллингера. Общая аккумулированная энергия зависит от
условий измельчения: интенсивности подвода энергии,
свойств вещества, длительности и т. д., однако н в самых
оптимальных условиях она не превышает 25—30% от под-
веденной энергии [95].
Для определения минимума работы, необходимой для
измельчения, существуют специальные методы. Одним из
них является метод «свободного дробления», который разви-
вался Кэри п Стайрмаидом с сотрудниками [96]. Идея его
проста. Постулируется, что разрушение отдельной частицы
может быть произведено с реально возможным минимумом
затраты энергии, когда частица дробится без контакта с дру-
гими частицами (без затрат на трение) и использования ме-
лющих тел (без затрат на их трение и деформацию). «Свобод-
ное дробление» осуществлялось раздавливанием слоя частиц
определенного размера толщиной в одну частицу под прес-
сом между двумя плоскими поверхностями. Зная силу воз-
действия и путь продвижения сдавливающей плоскости,
легко определить удельный расход энергии на измельчение
данного вещества от исходного размера частиц до конечного.
Стайрмапд, используя этот метод, провел сравнение
энергетической эффективности ряда промышленных изме.ть-
чительпых аппаратов с различными типами воздействия на
измельчаемый материал [961. Анализ результатов привел
Стайрмапда к выводу, что известные мельницы в основном
далеки от реально достижимого предела энергетической эф-
21
фективностп. Возможно увеличение этого показателя в 5—
10 раз. Для этого необходимо выполнение как минимум двух
условии: 1) мельница должна измельчать в условиях, мак-
симально приближенных к «свободному дроблению», 2) по-
лученный продукт (мелочь) должен немедленно удаляться
из зоны дробления. Только при этом будут сведены к мини-
муму потерн энергии, связанные с взаимодействием частиц
между собой. Наиболее эффективны, с его точки зрения,
аппараты, приближающиеся по типу воздействия к «свобод-
ному дроблению», например валковые дробилки.
Развитием данного метода определения минимума энер-
гии, необходимой для измельчения, является метод «измель-
чения индивидуального зерна (частицы)», позволяющий так-
же моделировать элементарный акт измельчения. Метод
развивался в работах немецких ученых школы Румнфа 197).
Техника измельчения базируется в основном на двух ме-
ханизмах воздействия па отдельную частицу. В первом —
частица пли агломераты частиц подвергаются дроблению
между двумя поверхностями, чаще всего при наличии сдвига
(дробление раздавливанием). Во втором — частица разру-
шается при соударении с твердой поверхностью (свободное
ударное воздействие). Поэтому работы немецких ученых по-
священы в основном исследованию этих двух механизмов воз-
действия .
В случае дробления раздавливанием использовались
прецизионные прессы, дающие возможность вести экспери-
менты в широких пределах параметров (давление от 1 до
1000 кПа, нижний предел испытуемой частицы до 1 мкм,
точность замера пути деформации от 1/50 до 1/100 мкм, ско-
рость воздействия от 1 до 10-2 см/с). Для случая свободного
ударного воздействия применяли аппараты с воздействием ,
падающего тела (скорость падения до 10 см/с), а также аппа-
раты с дозвуковым потоком, увлекающим частицы, или с
пневматической пушкой (скорость частиц от 20 до 200 м/с).
Показано, что данные по энергетической эффективности
измельчения различных материалов методом «индивидуаль-
ного зерна» близки к тем, которые получены методом «сво-
бодного дробления». При сопоставлении первого и второго
методов воздействия установлено, что для хрупких тел энер-
гетически наиболее выгоден процесс медленного раздавли-
вания (прессования) [98, 99]. Так, для цементного клинкера
(для «индивидуальных» зерен) расход энергии на разруше-
ние частиц до поверхности 0,3 м2/г составляет 2,5—
5,8 кВт-ч/т, при свободном же ударе энергозатраты возрас-
тают до 23—58 кВт-ч/т, в то время как при измельчении до
22
этой же поверхности клинкера в лабораторной шаровой мель-
нице затраты составляют 32 кВт-ч/т.
Опыты с шарами из различных материалов на установ-
ках, позволяющих использовать не только давление, но и
разные варианты давления со сдвигом, свидетельствуют о
том, что использование энергии при этом значительно зави-
сит от свойств материала: для типично упругих (кварц, стек-
ло) степень использования при наличии сдвига остается по-
стоянной или повышается, а для таких материалов, как
известняк, цементный камень, снижается.
Один из важных аспектов процесса измельчения позволи-
ли выявить опыты Шонерта с сотрудниками [99, 101], кото-
рые моделировали измельчение частиц с различными разме-
рами (случай наиболее близкий к обычному процессу измель-
чения) на свинцовых шарах и смесях кварца разных фрак-
ций при прессовании в аппаратах, обычных для метода
«индивидуального зерна» [100, 102]. Были установлены при-
чины снижения эффективности использования энергии по
мере накопления мелкой фракции вещества в ходе измель-
чения: растет количество контактов между частицами и
при общей прежней нагрузке уменьшаются контактные си-
лы, причем до пределов ниже разрушающих нагрузок, при
этом увеличивается диссипация энергии.
С целью значительного повышения энергетической эф-
фективности измельчения Шонертом [103] предложен двух-
ступенчатый процесс: дробление прессованием хрупких
материалов при высоких давлениях (30—50 МПа), затем
доизмельченпе в дезагрегирование в шаровой мельнице.
Суммарный процесс потребляет 30—50% энергии от необ-
ходимого на одноступенчатое измельчение в шаровой мель-
нице.
В исследованиях немецких ученых показано, что проч-
ность частиц н работа, необходимая для их разрушения,
возрастают с уменьшением размера частиц [73, 102]. Этот
факт объясняется тем, что с уменьшением размера частиц
снижается их дефектность. Прп некотором предельно мини-
мальном размере частицы, зависящем от физико-механи-
ческих свойств материала, наблюдается переход от хрупко-
го разрушения к вязкому. На опытных кривых деформации
это прослеживается в переходе от кривых с пиками, харак-
терными для хрупкого разрушения, к кривым с медленным
плавным подъемом.
Госсом [102] приведены следующие данные для нижнего
предела размера частицы (мкм), когда прекращается хруп-
кое разрушение и начинается пластическое течение материа-
23
ла, полученные л одинаковых условиях эксперимента:
1. Ка.чпшшя соль (КСЬ 150—250
2. Сахар (песок: 50—100
3. Бурый уголь 23—35
4. Каменный уголь 20—30
5. Аморфный бор 10—20
(>. Мрамор 10—15
7. Известняк 5—9
8. Кварц 4—6
!). Кристаллический бор 2 — 3
10, Карбид бора 2—3
Эти размеры определены Гессом по относительно формаль-
ным признакам (наличие пиков на мпкродеформацпонной
кривой или плавный ее подъем). Одновременно микроскопи-
ческие наблюдения свидетельствуют о том, что в переходной
области еще имеются следы хрупкого разрушения, но обра-
зующиеся частицы уже включаются в процесс агрегирова-
ния (наблюдается своеобразное равновесие: разрушение —
агрегирование с преобладанием пластического течения).
Порядок размера частиц, определенный для начала' плас-
тического течения, близок к теоретическому пределу дроби-
мости частиц, рассчитанному Кендаллом [1041 с привлече-
нием условия развития трещины по Гриффитсу.
С пластической деформацией материала связаны процес-
сы дефектообразования в твердых телах. Поэтому детальное
изучение области критических размеров Частиц важно для
понимания механохимических процессов.
Рассматривая совокупность процессов при диспергирова-
нии, Ребипдер [105, 106] показал, что в принципе работа дис-
пергирования складывается из работы упругой деформации,
работы пластической деформации, работы разделения на
более мелкие части и работы на придание разделяющимся
частям кинетической энергии:
А = ^4упр + Ипл + ИПов 4~ -^к- (1.17)
Полезной в данном уравнении с точки зрения теории про-
цессов диспергирования является работа образования но-
вой поверхности (ЛПов)- Работа, затраченная на пласти-
ческую деформацию, может быть выше работы образования
поверхности в 10—103 раз и, согласно современным пред-
ставлениям [50], расходоваться на образование в твердом
теле различного рода дефектов. Исходя из (1.17), Ребиндер
предложил коэффициент полезного действия процесса дис-
пергирования выразить уравнением
л
Как указывалось выше, к. п. д. процесса диспергирования
различных неорганических материалов составляет 0,01 —
1% [92].
24
Рис. 3. К. п. д. процессов об-
разевания дефектов (/) н по- ?
иерхпости (2) [108]. с,‘с
С точки зрения химии де- <V-" Z X.
фектпых кристаллов твердое те- / тч.
ло, содержащее повышенное /
по сравнению с равновесным /
количество дефектов, имеет ? /
более высокую реакционную —1— Л
способность. Таким образом, ° ^0 120 200
если в последующих процес-
сах запасенную твердым телом энергию удается реализо-
вать, то к. п. д. следует относить не к процессу диспергиро-
вания, а к процессу активации в целом. К. п. д. процесса
активации уже следует выразить уравнением [107 ]
_______AlOB ~Ь At л___
А1ОВ + (А'пр "Ь Aia)
(1.19)
По оценкам ряда авторов, эта величина может составлять
5—1096 [52].
В качестве примера можно привести данные, полученные
Шрадером [108], по оценке поверхностного и объемного
вкладов в изменение свободной энергии карбоната кальция
после механической обработки его в вибромельнице. Изме-
нение свободной энергии в зависимости от размера частиц
рассчитано Шрадером по уравнению Томсона:
AG = (1.20)
/(ля расчета взято значение поверхностного натяжения а для
СаСО3, равное 0,23 Дж/м2. Изменение свободной энергии оп-
ределено по равновесному давлению термической диссоциа-
ции активированных и неактивированных образцов при тем-
пературе 873 К по уравнению
ДС = -ЛТ1н/^, (1.21)
Результаты расчетов приведены на рис. 3. бпдно, что энер
гни, расходуемая на образование дефектов, в 40—50 раз вы"
ше, чем энергия, расходуемая на образование поверхности-
Эти результаты свидетельствуют о структурных нару-
шениях, происходящих в кристаллической решетке при ак-
тивации и существенно облегчающих последующее термиче-
ское разложение веществ.
Кривая изменения реакционной способности карбоната
кальция в зависимости от длительности активации имеет яр-
ко выраженный максимум, который закономерно проявляет-
ся и при активации других веществ [501. Связан он, по-ви-
димому, с природой дислокационных взаимодействий, воз-
никающих при пластической деформации твердых тел. Так,
Смирнов с соавторами [631 ] наблюдал максимум скорости
растворения в зависимости от величины и скорости пластиче-
ской деформации монокристаллов хлористого натрия. Но
данным Гольдберга н др. [632—636], максимум обусловлен
скоплениями дислокаций, которые образуются, а затем ис-
чезают в ходе пластической деформации.
Таким образом, обработка в измельчительных аппаратах
является не только способом получения веществ в мелкодис-
персном состоянии, но и способом генерации различного
рода структурных дефектов в объеме и активных состояний
на поверхности кристалла, повышающих реакционную спо-
собность твердых тел.
1.2.2. Дефектообразование
при пластической деформации
При упругопластической деформации твердых тел проис-
ходит образование неравновесных структурных дефектов
различного типа [109, 1101. Это могут быть дефекты, лока-
лизующиеся в пределах микроструктуры (смещенные из
положения равновесия атомы, напряженные и деформиро-
ванные связи, точечные дефекты и т. д.), или дислокации и
макроскопические дефекты типа микротрещин и границ
раздела между элементами структуры (одномерные и дву-
мерные дефекты) и др. На образование дефектов первого ти-
па требуются значительные затраты энергии, при повыше-
нии температуры они сравнительно быстро исчезают. Напро-
тив, менее энергоемкие одномерные и двумерные дефекты
более устойчивы и играют большую роль в процессах пласти-
ческого течения (ползучести) твердых тел.
Типично двумерными дефектами являются области несо-
гласованности в местах соприкосновения соседних зерен.
Экспериментальные измерения энергии межзереиных границ
дают значения в пределах от 0,1 до 1 Дж/м3, в зависимости
от состава и ориентировки соседних зерен, которые несколь-
ко ниже, чем значения свободной поверхностной энергии для
неорганических материалов (0,1—3 Дж/м2) [111, 1121.
Предельно возможное количество энергии, запасенной за
счет поверхностной энергии и энергии межзеренных границ,
20
находится йа уровне теплоты плавления неорганических ве-
ществ (10—150 кДж/моль) 1112].
Согласно континуальной теории дислокаций, энергия кра-
евой дислокации равна [ИЗ]
ВИНТОВОЙ — = (1.23)
где Е — модуль сдвига, v — коэффициент Пуассона (отноше-
ние поперечной и продольной деформаций при упругом одно-
осном растяжении), Ъ — вектор Бюргерса дислокации, г —
расстояние от рассматриваемой точки до линии дислокации,
г„ — радиус ядра дислокации. Для большинства твердых
тел Е лежит в пределах от 1010до 1011 Н/м2, v — от 0,1 до 1,
Ъ — от 1010 до 10* 9 м. При г0 = 10”9 м, а г = 10*3 энергия
дислокации составляет величину порядка 10“2В — 10“28 Джм,
или 10*17— 10*19 Дж на одно межатомное расстояние.
В продельном случае, когда предполагается, что все атомы
находятся в зоне действия дислокаций, расчеты для запасен-
ной энергии дают значения порядка 100—1000 кДж/моль,
что существенно выше теплоты плавления твердых тел.
Однако, так как число дислокаций в твердых телах ограни-
ченно и, как правило, не превышает 1013 см*2 (что соответ-
ствует среднему расстоянию между дислокациями порядка
десяти постоянных решетки), значения запасенной энергии
значительно ниже. При высоких концентрациях наблюдает-
ся тенденция либо к взаимному уничтожению дислокаций с
образованием точечных дефектов, либо к группировке их в
сетки или регулярные ряды (что эквивалентно взаимодей-
ствию границ зерен). С макроскопической точки зрения такой
образец полигонизуется или превращается в микрокристал-
лический [632].
По вектору Бюргерса дислокации подразделяются на раз-
личные типы. Дислокация, имеющая вектор Бюргерса, рав-
ный вектору решетки, называется единичной и совершен-
ной — если ее вектор является кратным вектору решетки.
Несовершенные дислокации, у которых вектор Бюргерса
меньше вектора решетки, называются частичными.
Ilpii взаимодействии частичных дислокаций образуются
дефекты упаковки п двойники, являющиеся двумерными по-
верхностными дефектами. Возникают дефекты упаковки в
27
плотноупаковаппых гексагональных и кубических решетках
при изменении присущей данной структуре последователь-
ности слоев [ИЗ]. Бесконечная последовательность
слоев...АВАВ... соответствует гексагональной решетке, а по-
следовательность ...АВСАВС... — гранецентрированной ку-
бической. Нарушение этой последовательности и есть дефект
упаковки. Наличие дефектов упаковки в плотноупакованных
структурах не меняет ни числа ближайших соседей, ни рас-
стояний между ними, однако вносит изменения в последую-
щие за первой координационные сферы. Энергия дефектов
упаковки, например, для металлов лежпт в интервале 0,01 —
0,3 Дж/м2 [1171.
Из этих данных видно, что энергия образования поверх-
ностей, связанных с дефектами упаковки и двойниками, на
1—3 порядка ниже энергий образования поверхности и па
один порядок ниже энергии образования поверхности, раз-
деляющей отдельные зерна кристаллов. Поэтому в условиях
пластической деформации идет насыщение кристаллов де-
фектами упаковки и двойниками.
При пластической деформации может происходить и обра-
зование дефектов с более высокими энергиями, в частности
точечных (вакансий и междоузельных ионов), на образова-
ние которых необходимо затратить энергию от 10"18 до
10'18 Дж. Предложено несколько механизмов образования
точечных дефектов при пластической деформации 1109, 1131.
Наиболее часто встречающийся механизм — аннигиляция
дислокаций. Две краевые дислокации с противоположно на-
правленными векторами Бюргерса Ь± — —Ь2, находящиеся
в соседних плоскостях скольжения, под действием напряже-
ния двигаются навстречу друг другу и, соединяясь, образу-
ют сплошной ряд либо вакансий, либо междоузельных ато-
мов в зависимости от их векторов Бюргерса. Другим менее
эффективным механизмом считается образование точечных
дефектов при пересечении дислокации с «лесом» дислокаций.
В соответствии с диффузионным механизмом ползучести
возникающий при наложении внешней силы градиент упру-
гих напряжений вызывает дрейфовую подвижность вакансий
и междоузельных ионов. При этом вакансии стремятся в об-
ласть сжатия, междоузельные ионы — в область растяжения
и возникают их направленные потоки в поле иапряже-
ний [114].
Таким образом, из современных представлений о механи-
ческих свойствах кристаллов следует, что образование и пе-
ремещение дефектов в твердых телах предопределено при-
родой пластического течения материалов.
28
1.2.3. Уравнение кинетики
диспергирования
Степень диспергирования характеризуется грануломет-
рическим составом, который задается обычно в виде функ-
ций распределения частиц по фракциям, и удельной по-
верхностью порошка.
Функции распределения частиц являются сложными.
Экспериментально установлено, что в большинстве случаев
распределение порошков по фракциям пе укладывается в за-
кон нормального распределения случайных величин (распре-
деление Гаусса). Попытки нахождения закономерностей рас-
пределения порошков по фракциям предпринимались мно-
гими авторами, были получены формулы, пригодные, одна-
ко, только в частных случаях [90].
Наиболее удобна для характеристики удельная поверх-
ность, так как опа сравнительно легко поддается измерению
различными методами адсорбции газов. По этой причине ею
обычно и пользуются для оценки дисперсности порошков.
При диспергировании наряду с разрушением происходит
необратимое пластическое деформирование поверхностных
с.чоев. упрочнение частиц по мере уменьшения размеров,
а также их агрегирование, снижается энергия мелющего те-
ла ввиду появления у топко измельченных веществ вязкости.
В результате изменения условий диспергирования процесс
останавливается в некотором состоянии, в котором скорость
разрушения становится равной скорости агрегирования.
Ходаков [50, 115], развивая представления Ребнпдера о
необходимости учета при диспергировании энергии, затра-
чиваемой на необратимую пластическую деформацию^ теоре-
тически рассмотрел процесс диспергирования с учетом де-
формации поверхностных слоев. Им получено следующее
уравнение:
W = In А + + (PL + о) Sm] In ^2, (1.24)
где Де — энергия, затраченная па диспергирование, s0 и
sm — исходная и максимально достигаемая в заданных ус-
ловиях поверхность, с — коэффициент формы частицы, а —
постоянная, е — плотпость предельных упругих деформа-
ций, р — средняя по толщине плотность энергии предель-
ных пластических деформаций, L — толщина деформируе-
мого слоя, 1] — коэффициент полезного действия.
Ввиду того что первый член в уравнении (1.24) значи-
тельно меньше, чем второй (для кварца, например, е =
29
= 2-105 Дж/м3, a Р — 4,4'10® Дж/мя), им можно пренебречь.
Тогда из (1.24) дли случая su<Csm следует
А.Де = 1п—, (1.25)
sm s
ГДС К = —-------5-------•
— + (₽Л + о) sm
При условпп постоянства режима диспергирования Ас =
N
=—т, где N — мощность аппарата, v — объем обрабатыва-
емого материала, т — время диспергирования, из (1.25) сле-
дует уравнение кинетики диспергирования:
А’2т = 1п е • плп s = (1 — е-й“х), (1.26)
йт
к N
где к2 — —имеет смысл константы скорости диспергиро-
вания.
Для обработки экспериментальных данных в координатах
уравнения (1.26) необходимо знать sm. Ввиду того что при
больших временах измельчения возможна агрегация частиц
и значения могут быть заниженными, для нахождения sm
можно использовать начальную часть кривой, когда вклад
агрегации еще незначителен. При условии 2т, = т2 из урав-
нения (1.26) находим
= U-27)
ZS1 2
где s, и s2 — поверхности, соответствующие временам т,
п т2.
Затем можно определить к2, обработав эксперименталь-
ные данные в координатах
1g (
— Ayr 1g е.
Экспериментальные данные, полученные разными авто-
рами, достаточно хорошо соответствуют уравнению (1.26)
[50]. Августиником, Вигдергаузом и др. [116) было показа-
но наличие связи константы диспергирования к2 с такими
механическими свойствами веществ, как ударная вязкость,
модуль сдвига, амплитуда тепловых колебаний, микротвер-
дость и др.
30
Рис. 4. Кинетика диспергиро-
вания оксидов на воздухе в
лабораторной мнкромельпнцс.
1 — а-А1Д),; 2 — Tio2; з —
SnO2; 4 — а - Fe3Oa.
С целью определить, нас-
колько установленные законо-
мерности являются общими и
для аппаратов повышенной
мощности, нами исследована
кинетика диспергирования не-
которых оксидов и сульфидов с
использованием планетарных
мельниц, описание которых при-
ведено ниже.
Оксиды диспергировали на
лабораторной планетарной
мельнице с использованном од-
ного мелющего тела (шести-
гранник пз сплава ВК8) [214)
на воздухе, но во избежание
агрегации проводили дополнительную операцию — диспер-
гирование в этиловом спирте в течение 30 с.
Сульфиды диспергировали в планетарной мельнице
311-2 хХ 150 (частота вращения 12 и 5 с-1, мощность соответ-
ственно 200 и 10 Вт) в воздушной и водной среде [205]. Ис-
пользовались стальные барабаны и шары.
Изменение удельной поверхности в зависимости от дли-
тельности диспергирования приведено на рис. 4 и 5, а в
табл. 2 даны значения микротвердостп и теоретической проч-
ности оксидов и сульфидов, которые рассчитаны по урав-
нению (1.11).
Сопоставляя скорость диспергирования оксидов с проч-
ностью и микротвердостью, видим, что для наиболее проч-
ного а-А120^ получена наименьшая удельная поверхность,
для наименее прочного a-F2O3 — наибольшая.
В случае диспергирования сульфидов на воздухе такой
закономерности в явном виде не наблюдается, зато очень
сильно проявляется эффект агрегирования первичных час-
тиц. Максимальные удельные поверхности при диспергиро-
вании па воздухе пе превышают 10—15 м2/г и при времени
дцснергиродаппя 40—G0 миц становятся близкими к нсход-
31
Таблица 2
Микретвсрдоеть и теоретическая прочность некоторых оксидов и суль-
фидов
Соедине- ние Структура Микро-твер- дость, кг/мм1 [117] Постоянная решетки, нм [118] Модуль уп- ругости, дин/смМО [89] удельная поверхност- ная энергия, эр г/см2 (80] L 1 ~ О V л С ОКиХ ФЛОЙ Ьх ХИ
а-А12О3 Ромбоэдриче- ская 1750-2525 а = 0,512 3,9 905 39,8
TiO., (ру- тил) Тетра гояа ль- ва я 784—1278 а — 0,458 с = 0,295 2,8 3S0 21,6
SnO3 » 988- 1360 а = 0,472 с = 0,317 — — —
a-Fe2O3 Ромбоэдриче- ская 680-1098 а = 0,542 2,3 200 13,0
FeS2 Кубическая 963-2056 а = 0,540 1,65 1117 26,10
ZnS » 140 -276 а - 0,542 0,70 1000 16,0
FeS Гексагональ- ная 230 300 а о,:м*> с = 0,586 0,89 453 15,3
PbS Кубическая 56-116 а — 0,591 0,83 491 11,7
ним. Вода выступает как поверхностно-активное вещество,
и удельная поверхность при диспергировании в воде значи-
тельно превосходит таковые, полученные на воздухе (см.
рис. 5). Однако как в том, так п в другом случае скорость
диспергирования уменьшается в ряду FeS, ZnS, FeS, PbS,
Рис. 5. Кинетика диспергирования сульфидов в планетар-
ной мельнице (ш = 12,4 с~*).
и — сухое диспергироиаиие; б — диспергирование- в поде;
см, рис, 4.
32
Рис. 6. Кппотика дегидрировании
сульфидин в планетарной мельнице
(ы = 5 с-1).
1 — PbS; 2 — FeS,.
что ио согласуется с микро-
твердостью и прочностью, при-
веденными в табл. 2.
Диспергирование крайних
в указанном ряду сульфидов
на воздухе при малых энер-
гиях (<о — 5 с-1) показало,
что в этом случае (рис. G)
на начальном участке кинети-
ческой кривой PbS имеет
большую удельную поверх-
ность, чем FeS2, что уже соответствует их микротвердости
и прочности.
Таким образом, корреляция кинетических параметров
диспергирования с мпкротвердостыо в теоретически рассчи-
танной прочностью существует, по при использовании боль-
ших энергий она может быть скрыта вторичными явлениями,
имеющими место при диспергировании твердых тел. При
больших энергиях очень быстро достигается такой размер
частиц, при котором отсутствуют условия для образования
трещин, начинается пластическое течение материала, и про-
цесс диспергирования прекращается. Конечный результат
обработки твердых тел в измельчите.! ьном аппарате повышен-
ной мощности можно представить как результат действия
двух противоположных эффектов; уменьшение размеров
частиц при разрушении и образование вторичных агрегиро-
ванных частиц в результате перехода их в состояние пласти-
ческого течения. В зависимости от того, какой эффект пре-
обладает, удельная поверхность либо увеличивается с дли-
тельностью обработки, либо уменьшается. При их равенстве
возникает стационарное состояние, когда поверхность не из-
меняется.
1.2.4. Взаимосвязь процессов диспергирования
и активации
При диспергировании имеет место изменение как макро-л
так и микросвойств твердого тела. Поэтому в рамках одного
метода, в частности измерением удельной поверхности дне-
** 15. 1’. Авпакуыив
пергированных порошков, трудно получить полную карти-
ну происходящих изменений.
Для этих целей нами применены метод электронной мик-
роскопнн и метод ядерной гамма-резонансной спектроскопии
(ЯГРС); последний позволяет определить размер областей
дальнего порядка обменных взаимодействий в магнитоупо-
рядоченных веществах. В таких веществах материальным
носителем обменного взаимодействия является химическая
связь катион — катион через ион кислорода. Удобен для
таких исследований феррит цинка. Диамагнитный цинк в
ZnFe2O4 подавляет обменные взаимодействия между тетра- и
окта-катионами железа, а для структуры шпинели обменные
взаимодействия между окта-катионами железа слабы, так
как-угол связи Fe—О—Fe составляет 90°. Если происходят
какие-либо искажения структуры, то возникают магнитные
взаимодействия, которые проявляются в спектрах ЯГР.
С использованием указанного метода исследовали феррит
цинка [119, 120]. Диспергирование проводили в центробеж-
ной планетарной мельнице ЭИ-2 X 150, материал барабана
и шаров — керамика на основе А12О3. Диаметр шаров 5 мм,
масса шаров 90 г, масса загрузки 1 г. Определение поверх-
ности проводили газоадсорбциопным методом [218], элект-
ронно-микроскопические снимки получены на микроскопе
«Tesla».
При комнатной температуре ZnFe2O4 является парамаг-
нитным веществом и его мессбауэровский спектр имеет вид
дублета (рис. 7). Под влиянием диспергирования происходит
увеличение квадрупольного расщепления и ширины линии.
Наблюдаемое изменение квадрупольного расщепления свя-
зано с изменениями химического состояния железа, вызван-
ными искажением его ближайшего окружения, и в зависи-
мости от времени диспергирования имеет четко выраженный
поэтапный характер.
На рис. 8 показано изменение удельной поверхности в за-
висимости от длительности диспергирования. Кривая про-
ходит через максимум при времени диспергирования 12 мин.
Данные электронной микроскопии (рис. 9) согласуются с
данными измерения поверхности. Минимальные размеры
частиц соответствуют времени диспергирования 12—15 мин.
При большой длительности происходит образование агрега-
тов, которые хорошо видны на снимках.
На первом этапе (до 12—16 мин) квадрупольное расщеп-
ление изменяется незначительно. Как следует из предыдущих
рисунков, на этом этапе идет прогрессивное уменьшение
размера частиц. Пока частицы достаточно крупные, количе-
34
Относительная скорости счета
Рис. 7. Спектры ЯГР при 300 К
пеактпвированпого (1) и активи-
рованного в течение 30 мин (2)
феррита цапка.
Рис. 8. Измспеппе удельной по-
верхности (») п величины квадру-
польпого расщепления (е) ферри-
та цпнка в зависимости от време-
ни диспергирования.
£, мм/с
ство материала, оказавшегося в зоне пластического течения,
незначительно и вклад процессов дефектообразования не-
велик.
На втором этапе (16 мин) диспергирование характеризует-
ся быстрым ростом (в 2—3 раза) квадрупольного расщепле-
ния и ширины липни, снижением удельной поверхности п
началом образования агрегированных частиц. Причиной
агрегирования является переход вещества в состояние пла-
стического течения, и именно это состояние ответственно за
интенсивно протекающий здесь процесс дефектообразования.
На третьем этапе образование агрегированных частиц про-
должается, а квадрупольное расщепление достигает некото-
рого постоянного значения и дальше не изменяется.
Существование предельных значений параметров актива-
ции, таких как величина искажений решетки Аа/а и запасен-
ная энергия (определенная калориметрическими измерени-
ями), которые выходят на некоторое постоянное значение
и ие увеличиваются с ростом механической энергии, расхо-
дуемой на единицу массы активируемого вещества, для
кварца, известняка и магнезита наблюдали Бернхард и
Хеегп при активации их в вибрационной мельнице [52].
Эти предельные значения зависят от природы вещества и
составляют 0,013; 0,032 и 0,050 кВт-ч/кг соответственно для
2*
35
Рис. 0. ЭлектронЯо-мпкроскопвчесКпе сВпмкл Иорошков феррита цИН-
ка в зависимости от времени диспергирования. X15 500.
I цсходныП, 3 — 5 mud, 3 — 10; 4 Id; з — 20. 6—23 мин
известняка, магнезита и кварца. С таким же предельным зна-
чением, возникающим на третьем этапе актпвацпп, по-впди-
мому, имеем дело и в случае феррита цинка.
Представления, изложенные выше, согласуются с данны-
ми и других авторов. Так, Неверов и др. 11211 показали, что
механоактивирующая обработка оксида магния при воздей-
ствии давления и деформации сдвпга определяется поочеред-
но развитием следующих процессов: дробления, агрегации и
пластического течения, при котором создается наибольшая
активность.
Из приведенных данных следует, что для ускорения про-
цесса активации необходимо реализовать такие условия,
при которых максимально быстро наступало бы пластическое
течение материала. Этого можно достигнуть либо увеличени-
ем механических параметров (скорости нагружения, увеличе-
ния мощности и т. д.), либо использованием в качестве ис-
ходных более мелких частиц, которые сразу могут переходить
при активации в состояние пластического течения.
С точки зрения диспергирования наличие большого ко-
личества контактов между частицами и их пластическое
течение являклся вредными, так как повышают энергетиче-
ские затраты. С точки же зрения активации такие процессы
приводят к дефсктообразовапию, повышению реакционной
способности и мехаиохпмическим реакциям. Как показано
ниже, на соотношение между этими двумя процессами можно
влиять, либо изменяя температуру процесса, либо вводя по-
верхностно-активные добавки и т. д. Например, низкие тем-
пературы позволяют усилить процесс диспергирования, а вы-
сокие — соответственно пластического течения. Аналогич-
ным образом поверхностно-активные вещества (ПАВ) могут
ускорить процесс диспергирования и достижение частицами
минимальных размеров.
1.3. ФИЗИКО-ХИМИЧЕСКИЕ ЯВЛЕНИЯ,
СОПРОВОЖДАЮЩИЕ ДИСПЕРГИРОВАНИЕ ТВЕРДЫХ ТЕЛ
1.3.1. Выделение тепла
Процесс диспергирования — с южный физико-химиче-
ский процесс, при котором реализуется целый комплекс фи-
зических и химических явлений, которые могут служить
37
Таблица 3 Плавление некоторых веществ п месте контакта при скольжении стального шарика [124 ]
Вещество Темпера- тура плав- ления, К ^пл> кДж/моль Скорость скольжения шарика, при которой начинается плавле- ние, м/с
Сплав Еуца .338 80
AgNO» 483 I* — 190
Bi 544 11,30 150
Sn 505 7,13 220
Pb 601 5,10 320
Си 1353 13,00 800
причиной механохимпческих эффектов при активации твер-
дых тел.
При движении твердых тел, находящихся в контакте
друг с другом, большая часть работы, совершаемой против
сил трения, освобождается в виде тепла. В локально ограни-
ченных точках поверхности температура может достигать
весьма больших значений. В работах Боудена с сотрудни-
ками [122—124] показано, что при трении скольжения тем-
пература в местах контактов повышается до точки плав-
ления одного из веществ, по никогда не поднимается выше
нее. Для тугоплавких веществ в поверхностном слое может
быть достигнуто мгновенное возрастание температуры до
1300 К. Основная масса вещества в это время остается холод-
ной. Время существования таких температурных вспышек на
площади 10-7 — 10"8 м2 порядка 10-4 с.
Для оценки локальных температур, возникающих па кон-
такте трущихся тел, Боуденом предложена формула
Д7, = тт--г4т-> (128)
4а/ к1 + /с2 ’
где р. — коэффициент трения, Р — сила давления, действу-
ющая на поверхность контакта, v — скорость скольжения,
а — радиус контактной области, I — механический экви-
валент тепла, kv и к3 — коэффициенты теплопроводности
трущихся тел.
В качестве примера в табл. 3 приведены результаты Боу-
дена и Персона [124] по определению скоростей скольжения
стального шарика, при которых происходит плавление кон-
тактирующих с ним веществ.
Из работы Тейлора [70 ] известно, что нагревание твердо-
го тела обусловлено пластической деформацией. В частно-
сти, считается, что энергия пластической деформации в ме-
таллах превращается в тепло приблизительно иа 95%, ос-
тальная энергия связывается в решетке дефектами.
Эшелбп и Пратт [125 ] показали, что существенного повы-
шении температуры деформируемого тела можно достичь
только в том случае, когда скольжение имеет место одно-
временно по многим параллельно лежащим плоскостям.
Такая ситуация возникает при больших скоростях деформа-
ции, например при высокоскоростном кручении ковких ме-
таллов, таких как алюминий, когда достигаются температу-
ры, близкие к температурам плавления металла.
Один из механизмов выделения тепла при пластической
деформации рассмотрен Дубновым, Сухих, Томашевичем
[126 J. Согласно их гипотезе, в областях интенсивного сдви-
га отдельные пары дислокаций с антипараллельными век-
торами Бюргерса способны аннигилировать с восстановле-
нием совершенной структуры и выделением энергии порядка
энергии дислокации. Исходя из оценок Коттрелла [72 J
энергия дислокации на одно межатомное расстояние а равна
U^Eb'a. (1.29)
Для большинства твердых тел Е — 0,1 кг/м3, Ъ = 1,0 им,
а = 0,15 нм. Из соотношения (1.29) следует, что U = 1,5 X
ХЮ'19 Дж « 1 эВ.
Вследствие близкодепствня атомных сил зона выделения
энергии ограничивается радиусом порядка одного межатом-
ного расстояния. При плотноупакованной структуре это со-
ставляет 8—12 атомов, т. е. в среднем на каждый атом в зоне
аннигиляции приходится энергия 0,1 эВ, что соответствует
интенсивности локального разогрева 103К. ,
Современные представления о механизмах разогрева рас-
смотрены в работах по исследованию инициирования твер-
дых взрывчатых веществ ударом и связываются с пластиче-
ским течением твердых тел [127]. Согласно этим представле-
ниям, разогрев происходит тогда, когда тепло, выделяюще-
еся при пластической деформации, не успевает отводиться
из зоны деформации вследствие неоднородности деформации
но объему деформируемого тела, в то время как при одно-
родной деформации тело нагревается равномерно и незна-
чительно.
В соответствии с диаграммой Р — е деформация идеально-
пластического тела протекает при постоянном напряжении,
равном пределу текучести Рт, а упрочняющегося тела — прп
возрастающем напряжении. Величину dpldz = D называют
модулем пластичности твердого тела. Для идеального пласти-
39
р
Рис. 11. Самолокалпзацпя деформа-
ции в плоскости ЕР при увеличении
деформации сдвига выше предела те-
кучести (127].
Рис. 10. Деформация идеаль-
но-пластического и упрочняю- ческого тела D = 0, для уп-
щсгося твердого тела [127]. рочняющегося — Е > D > 0.
Для случая D < 0 твердое
тело должно разупрочняться самопроизвольно (рис. 10).
Известно, что пластические свойства твердых тел суще-
ственно зависят от температуры: предел текучести уменьша-
ется с ростом температуры. Это приводит к разупрочнению
идеально-пластического поля в адиабатических условиях
после участка упругой деформации (кривая II). Для упроч-
няющегося твердого тела разупрочнение наступает после
некоторого участка деформации с упрочнением (кривая Ill).
С переходом к разупрочнению деформация становится не-
однородной, что ведет к локальному выделению тепла, па-
пример, в плоскости EF, как показано на рис. 11. Выделение
тепла, в свою очередь, снижает предел текучести, и процесс
становится самопроизвольным. Энергетическим источником
разогрева служит упругая энергия, запасенная в образце.
Температура разогрева определяется характером зависимо-
сти предела текучести от температуры, величиной запасен-
ной упругой энергии и теплоотводом. Необходимый мини-
мум запасенной энергии для получения локального разогре-
ва, способного инициировать взрыв, определяется степенью
деформации частиц твердого тела и размерами образца.
Поэтому такие разогревы возникают только при достижении
определенного давления (Z-*K|1), которое значительно выше
предела текучести, а также при толщине образца выше не-
которой критической.
Экспериментальные данные, полученные разными иссле-
дователями, согласуются с приведенным выше механизмом
разогрева. Так, на возможность образования локального
разогрева за счет пластической деформации отдельных зерен,
разрушения пх и трения указывал Харитон [123]. Позднее
Холево [123], Сухих и Харитон [1301 пришли к выводу, что
определяющей при возбуждении взрыва является пеупругая
деформация заряда в целом. Болховитиновым [131, 132]
показано, что для веществ, температура вспышки которых
выше температуры плавления, необходимо повышенно тем-
пературы плавления под влиянием давления до температуры
вспышки.
Раскол кристаллов сопровождается «сбросом» упругой
энергии поверхностей разрушения, которая, не совершая
работы против внешних сил, превращается в тепло. Согласно
Вейхерту и Шонерту [133], во время разрушения кристаллов
в носке трещины температура может превосходить 2000 К.
Оценка температур, возникающих на поверхностях раз-
рушения, проведена Фоксом [134] путем определения коли-
чества продуктов разложения при расколе термически не-
устойчивых веществ в источнике масс-спектрометра. При этом
предполагалось, что разложение происходит по тому же
самому механизму, который имеет место при термическом
разложении. Время, в течение которого существует возбуж-
денно поверхностных атомов, сопоставимо со скоростью раз-
рыва связи (К)-13 с), поэтому тепловое равновесие установить-
ся не может, и речь идет о некой «эффективной» пли «экви-
валентной» температуре.
Метод вычисления следующий. Если N — число разло-
жившихся молекул на единице поверхности, ат — время
пребывания носка трещины на единичной элементарной ячей-
ке, то скорость процесса может быть записана так:
v -=4-
Скорость термического разложения выражается уравнением
Аррениуса:
н = Аехр(—(1.31)
где А — предэкспоненциальный (частотный) фактор, Ua —
энергия активации. Определив частотный фактор и энергию
активации из данных по термическому разложению, Фокс
выражает эффективную температуру с учетом (1.30) и (1.31):
т . _ ____1___________1 - (I 3-м
яф R 1в(.1/Л) И 1в (тЛ/Л )’ k 7
Конечные результаты для некоторых веществ, получен-
ные Фоксом при максимальной скорости движения трещины
через вещество (3—5-103 м'с), приведены в табл. 4.
41
Таблица 4
Результаты по определению эффективных темпера-
тур на поверхностях раскола [134J
Тип реакции Вещество Число разложив- шихся монослоев §2 5 «а - а» -5. eq rt rt Ь- Эффективная температура, К
Эндотермиче- ская СаСО3 0,09 160 1523
» РЬСО3 2,0 845 1123
Экза термиче- ская NaN3 0,02 10 1273
PbN 10,0 130 Отрицатель- ное значе- ние
Как указывалось выше, и поле механических напряже-
ний энергия активации разрыва связей снижается. Если бы
этот фактор можно было учесть, то значения эффективных
температур были бы получены более низкими.
Теоретическая оценка температурного пика на контакте
трущихся твердых частиц для скоростей движения мелющих
тел 5—10 м/с, имеющих место в планетарных мельницах,
сделана в работе Уракасва 11.35]. Рассмотрен случай, когда
в аппарате устанавливается квазиравновеспос распределе-
ние по дисперсности частиц, а главную роль в генерировании
тепла играют деформации и трепне частиц друг о друга.
При этом использованы труды Беляева [136], Динпика
[137], Ландау и Лифшица 1138], в которых разработана
теория взаимодействия двух упругих твердых тел.
В результате для максимальной температуры, возникаю-
щей при обработке смесей или одного вещества в планетар-
ной мельнице, Уракаевым получено [135]
_ р°’5Д (р^ + р2ь?) (8t + ej1-4
тах ~ К + ^)5 М2 VAM0’25 + ®)2’15’ ( ’ ?
где р, pi, р2 — плотности мелющего тела и реагирующих ве-
ществ соответственно; и Ьг — средняя кривизна частиц
1 и 2 Pi — радиус частицы); 0, 8П 02 и 0 — подат-
ливости шара, обрабатываемых частиц и смеси веществ (0 =
= —j - 0 = -1——Е — модуль Юнга; v — коэффи-
циент Пуассона); с и X — удельная теплоемкость и теплопро-
водность частиц; £ — коэффициент трепня частиц 1 н 2;
IP — скорость соударения шара со стенкой барабана.
В качестве конкретного примера теоретические расче-
ты Уракаевым проведены для хлористого натрия, измельча-
емого в планетарной мельнице при частоте вращения 10 с-1.
Скорость движения шаров в момент соударения со стенкой
составляет 4,2 м/с. Равновесный размер частиц NaCl (когда
скорость разрушения равна скорости рекристаллизации),
в соответствии с работой Шнейдера 1139], принят равным
1{ — 1,63-10-6 м. Значения pj = 2,136-1()3 кг/м3, Vj = 0,252,
— 0,369-1011 Н/м2 для NaCl, а также р = 7,86-103 кг'м3,
v = 0,285, Е = 2,232 -10й II м2 для стального шарика взя-
ты из работ Андерсона 1140], Маклпнтока и Аргона 1141).
В результате расчетов получено: Д7’тах — 1035 К, вре-
мя взаимодействия частиц т = б-10~® с, площадь контакта
частиц s= 7-10-4 м2, сила сдавливания частиц / = 11,8 X
Х10-6П. Как видно из результатов расчета, температура на
поверхностях трепня близка к температуре плавления NaCl.
Однако время существования температуры по расчетам
составляет лишь 6-10~9 с.
1.3.2. Появление высоких давлений
При исследовании фазовых переходов, происходящих
под влиянием измельчения и вызываемых высоким давлени-
ем, Дахнлем и Роу (142], Кларком и Рованом [20] установ-
лено, что между ними имеется много общего. Па основании
этих данных высказано предположение, что на контакте тру-
щихся частиц развиваются достаточно высокие давления и
происходят сдвиговые деформации. Возникающие при из-
мельчении давления оценивались косвенно в работе 1142].
Был взят ряд веществ с известными давлениями фазовых
превращений в (Па X 108):
Р1>О MnF, Sb20s SiOj BcFa В,О, ВаАвО.
5,5 9,5 10,8 13,5 17,5 18,5 30
После диспергирования веществ в мельнице в продуктах,
за исключением BaAsO4, обнаружены модификации, сущест-
вующие при высоких давлениях. На основании этих данных
сделан вывод, что давления, развивающиеся па контактиру-
ющих участках частиц, могут составлять (15—18)-10® Па.
Расчет сил, действующих па контакте трущихся частиц
[135], показал, что силы (10—12)-10’5 Н приходятся на
43
участки с площадью 5-10-14 м2 и дают давление 20-10s Па,
что согласуется с данными (1421.
Возникновение давлений, коюрые значительно больше
указанных, обнаружено Толочко, Шсромовым и др. [1431
ири исследовании деформированных срезом металлических
проволочек с использованием синхротронного излучения.
Так, в случае олова наблюдается появление фазы, которой
на диаграмме р — Т соответствует давление >-92-108 Па.
Результатом появления высоких давлений при механиче-
ском активации могут быть увеличение областей перекрыва-
ния между электронными орбиталями или относительная
замена их на орбитали, относящиеся к другому типу сим-
метрии, что приводит к делокализации электронов, ушире-
нию полос (зон) разрешенных энергий и уменьшению зап-
рещенной зоны [144].
Как и температуры на поверхностях раскола, возникаю-
щие давления в ряде случаев можно рассматривать как «эф-
фективные». Суммарный эффект может быть результатом
сдвиговых деформаций, приводящих в конечном итоге к об-
разованию фаз высокого давления.
1.3.3. Ускорение процессов переноса
В результате пластической деформации твердых тел про-
исходит ускорение процессов массопсреноса в них. Причем
реализоваться оно может двумя путями. Первый — переме-
щение частиц в объеме или на поверхности твердого тела по
механизмам, характерным для развития деформации. Твер-
дое вещество при пластическом течении приобретает свой-
ства «квази-жидкости», создаются благоприятные условия
для всех видов подвижности, необходимых для интенсифика-
ции диффузно контролируемых процессов и осуществления
элементарного химического акта. В режиме пластического
течения скорость процессов определяется в основном ско-
ростью развития деформации.
Второй путь — диффузионный. В результате пластической
деформации твердых тел, элементарными актами которой
являются генерация и размножение дислокаций, происхо-
дит разупорядочснис кристаллов, увеличивается свободный
объем, возникают вакансии анионов или катионов, растет ин-
тенсивность беспорядочного теплового движения частиц.
Резкое увеличение числа вакантных мест и движение их в
градиентах упругих напряжений приводит к возрастанию
электронной и ионной проводимости, а также к ускорению
процессов диффузии.
44
Экспериментально увеличение ионной проводимости в
процессе пластической деформации обнаружено Дыолап и
Хартли в 192b г. [145], а затем Степановым [146]. Они под-
вергали кристалл каменной соли сжатию между двумя элект-
родами. Проводимость возрастала в 10—100 раз пропорцио-
нально увеличению нагрузки. Степанов наблюдал еще боль-
шее возрастание проводимости при сжатии хлористого нат-
рия — в 1010 раз, причем он обнаружил, что прирост прово-
димости начинался лишь после приложения определенной
нагрузки, которая выше предела текучести. Степанов отме-
чал, что проводимость растет не во всем объеме образца,
а только в области полос скольжения. В дальнейшем этот ре-
зультат был подтвержден другими авторами. Обнаружено
также линейное возрастание эффекта с ростом деформации.
Во всех экспериментальных работах отмечается, что при-
рост ионной проводимости наблюдается только при импульс-
ном нагружении, под постоянной же нагрузкой проводи-
мость не возрастает, а даже значительно падает. Нагревание
снижает эффект или приводит к полному его исчезновению,
анионные примеси усиливают его, катионные ослабляют.
Степанов, основываясь на сходстве увеличения проводимо-
сти деформированной и расплавленной каменной соли, а так-
же на локальном характере возрастания проводимости,
считал, что ее причиной является локальное расплавление
кристалла вдоль полос скольжения.
Начиная с 1952 г. появилось значительное число публи-
каций, посвященных экспериментальному исследованию диф-
фузии в металлах при пластической деформации. Однако ре-
зультаты первых работ противоречивы, что объясняется ря-
дом методических ошибок. Только в последнее время доста-
точно убедительно показано ускорение диффузии при пласти-
ческой деформации металлов [147—150].
В работе [151] приведен коэффициент самодиффузин изо-
топа 65Fe в деформированном при сжатии железе. Общая ве-
личина деформации в = 0,2. При скорости деформации е =
= 2-10-3 с-1 и температуре 1015 К происходит увеличение
коэффициента самодиффузин в 2,5-103 раз. При повышении
температуры увеличение коэффициента диффузии наблюда-
ется в меньшей степени. Так, при 1158 К оно при той же ско-
рости деформации составило 2-103 раз. Было обнаружено
также, что увеличение коэффициента самодиффузин не зави-
сит от полной деформации, накопленной образцом. Величина
Dt, линейно возрастала с ростом скорости деформации.
Авторы [152] наблюдали эффект ускорения самодиффузин
изотопа e3Ni при сжатии и растяжении. Было установлено
4?
увеличение прироста коэффициента диффузии с понижением
температуры и отсутствие зависимости от знака деформа-
ции, а также, что ускорение диффузии определяется в основ-
ном скоростью деформирования металлов, а нс абсолютной
величиной относительной деформации [153—155].
В работах [156, 157] исследуется диффузия в условиях
соединения однородных и разнородных металлов при им-
пульсном нагружении со скоростью деформирования кон-
тактной зоны порядка 10—10~а с-1 в течение 10-2—10-3 с.
Сообщается о том, что при таких параметрах деформирова-
ния развивается диффузионная зона, протяженность ко-
торой описывается при значениях коэффициента диффузии
порядка Ю-2—10_3смг/с,т. е. на несколько порядков больше,
чем в жидком состоянии. Авторы считают, что имеет место
«аномальное» ускорение диффузии. Однако в работе [158] по-
казано, что для такого вывода ист оснований, так как авто-
ры допустили методическую ошибку. По условиям теплоот-
вода образцы за время импульса нагревались нс на 400—
500 К, а на 1100 К и длительность охлаждения от этой тем-
пературы до 700 К составляет не 10-3—10~3 с, а 103с. С уче-
том этих поправок получается значение коэффициента диффу-
зии 10—®—10~8 смг/с вместо 10~2—10-3 см2/с, что согласуется
с вышеприведенными данными по диффузии в пластически
деформируемых металлах.
В соответствии с работой [158] пределом скорости диффу-
зии в пластически деформируемых твердых телах является
скорость диффузии в жидкости, т. е. 10~4—10-6 см2/с.
Относительно механизма ускорения диффузии при пласти-
ческой деформации металлов существует две точки зрения.
Согласно первой, ускорение происходит в результате быст-
рой диффузии атомов вдоль дислокаций, образующихся при
деформировании [159—165]. При описании диффузии вдоль
дислокаций обычно используют следующую модель [165]:
дислокация в кристаллической решетке — это узкая область
(дислокационная трубка), в которой диффузионная подвиж-
ность на несколько порядков выше, чем в матрице, что теоре-
тически и экспериментально показано различными мето-
дами.
Было показано, что наблюдаемый в эксперименте коэффи-
циент диффузии Dp связан следующим соотношением [165]:
+ (1.34)
где D и Dg — коэффициенты диффузии соответственно в не-
деформированном металле и вдоль дислокаций; g — доля
атомов, находящихся в дислокациях.
АЛ
Когда температура опытов выше 0,5 Тпл, а плотность дис-
локации 107—10° см-3, то второй член в уравнении 1.
Отсюда следует, что ускорение диффузии при иовышенных
температурах нельзя объяснить быстрой диффузией атомов
вдоль дислокаций. Однако с понижением температуры
< 0,5 Тпл эффект, связанный с диффузней по отдельным
дислокациям, может превзойти решеточную диффузию и да-
же граничную.
Согласно второй точке зрения, ускорение диффузии при
пластическом деформировании обусловлено образованпем
при деформации подвижных вакансий [154, 164 J. В соответ-
ствии с теорией пластического течения [164] число «деформа-
ционных» вакансий равно
Дм — i|Tve, (1.35)
где 1] — коэффициент пропорциональности, tv — время
жизни вакансии, е — скорость деформации. Коэффициент
диффузии при деформировании металла в этом случае будет
равен [153]
Dp = D -| \n-Dv, (1.36)
ил к
где Dv — коэффициент диффузии вакансий.
Из уравнения (1.37) следует, что ускорение диффузии при
пластическом деформировании в значительной степени за-
висит от времени жизни вакансий tv, которые определяются
природой их стоков. Если принять, как это сделано в рабо-
те [163], что основными стоками вакансий являются либо
границы субзерен, либо поверхности образцов, то т0 =
= 20 4- 300 с [164]. В этом случае ускорение диффузии,
вычисленное по уравнению (1.37), соответствует наблюдае-
мому в экспериментах [153, 154, 164].
1.3.4. Электрические явления
Возникающие при расколе твердых тел свежеобразован-
ные поверхности несут на себе электрический заряд, который
не является однородным, а имеет микро- и макрорельеф, от-
ражающий существование различного типа дефектов в твер-
дых телах. Исследование пространственного распределения
47
заряда, его плотности методами зондирования [27] и деко-
рирования [1(1(5] позволяет выяснить природу активных эле-
ментов поверхности, таких как точечные дефекты, скопление
точечных дефектов, участки поверхности, в том числе и эле-
менты геометрического микрорельефа с различными электри-
ческими свойствами.
Под влиянием пластической деформации имеет место элек-
тризация поверхностей диэлектриков. Считается, что этот
эффект обусловлен образованием разности потенциалов из-
за движения заряженных дислокаций. Возникающий па по-
верхности ионных кристаллов под действием удара или де-
формации потенциал, как это показано Урусовской,
Добржанским и др. [167], коррелирует с пластичностью
кристалла: чем кристалл пластичнее, тем потенциал выше,
п наоборот.
При механической обработке смесей может происходить
электризация за счет контактной разности потенциалов.
Бредовым и Кшемяпской [168] показано, что заряды, возни-
кающие при контакте полупроводников и металлов, нахо-
дятся в линейной зависимости от разности работ выхода
электрона из контактирующих тел.
В присутствии растворов электролитов такие контакти-
рующие вещества образуют электрические пары, которые
работают как электрохимический элемент, вызывая ускоре-
ние или замедление процессов растворения но сравнению с
растворением индивидуальных компонентов [1691.
1.3.5. Экзоэмиссия
и механоэмисспя электронов
В твердых телах в момент и после механической обработ-
ки наблюдаются явления экзо- и механоэмиссии электронов.
Электроны экзоэмиссии, затухающей весьма медленно после
механической обработки, имеют энергию не выше электрон-
вольта, электроны же механоэмиссии (механоэлектроны)
возникают в основном при разрушении диэлектриков и об-
ладают энергией в десятки килоэлектронвольт. Впервые они
были обнаружены Дерягиным, Кротовой, Карасевым [381.
Механозлектроны получают такие большие энергии за счет
разгона в поле «мпкроконденсатора», пластинами которого
являются разъединяемые поверхности раскола. Механоэлек-
троны обнаруживают ряд эффектов, совершенно не свойствен-
ных экзоэлектропам. В частности, как это показано в работе
Анисимовой, Дерягина и др. [170], имеет место тормозное
излучение рентгеновских квантов, а также радиационное
48
действие, проявляющееся в химических реакциях радиаци-
онной прививки, исследованное в работе Капптанчука [171].
Однако энергетический выход таких химических реакций
мал п существен, по-видпмому, только для цепных процессов
(полимеризация и др.).
Относительно механизма экзоэмиссии существует ряд
гипотез [172]. Условно их можно разбить на две группы.
В первой считается, что экзоэмпсспя зависит от нарушений
структуры металла. Полагается, что интенсивность эмиссии
непосредственно связана с величиной работы выхода элект-
рона, т. е. проводится параллель между экзоэмиссией и клас-
сическими видами стационарной эмиссии. Однако представ-
ления об экзозмиссип как о «внутреннем» свойстве металла
оказываются недостаточными для объяснения всей совокуп-
ности экспериментальных данных. В связи с этим наиболь-
шее распространение получило объяснение экзоэмиссии с
металлов как явления, обусловленного внешними факто-
рами. Согласно мнению ряда исследователей, экзоэмпсспя
электронов с механически обработанных поверхностей объяс-
няется процессами сорбции или химическими реакциями,
которые протекают на свежих поверхностях непосредствен-
но после окончания обработки. В качестве источника энер-
гии выступает освобождаемая теплота адсорбции. Резуль-
таты исследования экзоэмпсспя в различных газовых средах,
а также в высоком и ультравысоком вакууме показали, что
между интенсивностью эмиссии и химической активностью
металлов существует прямая связь [1731.
1.3.6. Триболюминесценция
При деформировании, разрушении п тренпп твердых тел
возникает весьма интенсивная люминесценция. Свечение
может быть вызвано, как считают Линке и Гофман [174J,
прямым механическим воздействием, согласно Мейеру, Обри-
кату и Розбергу [175 J — тепловым возбуждением поверх-
ности, а также рядом других причин (рекомбинация заря-
женных центров при развитии микропластпческой деформа-
ции [176], возникновение электрических полей [177], газо-
разрядные явления [178, 179]). Стрелецким, Бутягпным и
Руфовым [181 ] показано, что при разрушении гомеополяр-
ных кристаллов в вакууме основную роль играет рекомбина-
ционное свечение, возникающее в результате гибели корот-
коживущих центров на поверхностях разрушения. При раз-
рушении в газе при высоких давлениях еще долго после рас-
кола проявляется адсорболюмииесценция, вызванная взаи-
49
модейстпием газов со стабилизированной поверхностью.
При невысоких давлениях имеет место газоразрядное свече-
ние. Так, Беляев, Мартынов, Набатов [1801 при разрушении
ионных кристаллов на воздухе наблюдали тот >ке спектр
азота, что и при электрическом разряде. Авторы приписы-
вают этот эффект газоразрядным явлениям.
1.4. ВЛИЯНИЕ ТЕМПЕРАТУРЫ, СРЕДЫ
И ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ
ПА ПРОЦЕССЫ ДИСПЕРГИРОВАНИЯ И АКТИВАЦИИ •
На эффективность диспергирования и активации можно
влиять, изменяя условия, в которых они проводятся, в част-
ности варьируя температуру, среду или вводя поверхностно-
активные вещества.
Для хрупких тел, к которым относится большинство не-
органических соединений (оксиды, сульфиды и т. д.), разру-
шение связывают с образованием и развитием зародыше-
вых трещин, которые, в свою очередь, зависят от подвижно-
сти дислокаций в твердом теле. С увеличением температуры
барьеры для дислокаций «размываются» и становятся легче
преодолимыми, соответственно предел текучести твердых тел
снижается. Требуется более значительная деформация кри-
сталла, чтобы возникли условия, необходимые для форми-
рования трещин разрушения, поэтому с повышением темпе-
ратуры эффективность диспергирования уменьшается.
11а рис. 12, а приведено изменение предела текучести для
металлического железа в зависимости от температуры, из ко-
торого следует, что эта зависимость в интервале температур
73—173 К очень резкая. При охлаждении железа до низких
температур происходит переход его из пластического состо-
яния в хрупкое. И действительно, при низких температурах
скорость диспергирования железа резко увеличивается
(рис. 12, б). На основании такого свойства материалов раз-
работаны эффективные способы диспергирования, при кото-
рых материалы охлаждаются до очень низкой температуры
жидким азотом (77 К) и подвергаются измельчению [182].
Большое влияние на процесс диспергирования оказывает
среда, в которой он проводится, а также добавки поверхност-
но-активных веществ. Исследования, начатые Ребипдсром в
1928 г., показали, что прочность как работа образования
единицы новой поверхности при раскалывании кристалла
может быть понижена при переходе от воздуха в качестве
* Раздел написан совместно с Г. Г. Кочегаровым.
50
Рис. 12. Снижение предела текучести железа в зависимости от темпе-
ратуры (а) и кинетика диспергирования железа (б) црп 8(J (7) и ЗОИ К(2).
среды к жидкости, возможно более близкой по полярности
к данному кристаллу, или к раствору поверхностно-актив-
ных веществ (органические кислоты, спирты, амины и др.),
адсорбирующихся на твердых телах. Было показано, что
понижение прочности в зависимости от концентраций ПАВ
идет параллельно их адсорбируемости и характеризуется
уравнением, аналогичным уравнению Лангмюра 1183]:
С -г
(1.38)
где Д/У — понижение прочности, с — концентрация поверх-
ностно-активного вещества, — наибольшее понижение
прочности при полном насыщении адсорбционного слоя,
а — константа.
Исследование этого явления (адсорбционного пониже-
ния прочности твердых тел) показало, что эффект не связан с
коррозией, так как он вызывается химически неактивными
средами или примесями, присутствующими в малых количе-
ствах, и не связан с растворением твердых тел в данных сре-
дах. Согласно Ребиндеру [183], изменение свойств материала
под влиянием поверхностно-активных веществ обусловлено
снижением свободной поверхностной энергии и, как след-
ствие, уменьшением работы, необходимой для увеличения
поверхности.
Дальнейшее развитие представления о механизме дейст-
вия ПАВ получили в свете теории дислокаций 1811 и с точки
зрения квантово-химических особенностей строения поверх-
ности твердых тел и электронной структуры адсорбирую-
щихся молекул [192].
Разрушение твердых тел в присутствии ПАВ, как н раз-
рушение в обычных условиях, связано с движением, размно-
жением и накоплением дислокаций перед барьерами. По-
верхность твердого тела является своего рода барьером,
51
перед которым скапливаются дислокации. Кроме того, дис-
локации вблизи поверхности в силу особенностей структуры
кристаллического твердого тела начинают работать как ис-
точники новых дислокаций. Если каким-либо способом
удалить поверхностный слой, например путем растворения,
то прочность твердых тел возрастает (эффект Иоффе 1184 I).
При адсорбции ПАВ происходит ослабление связей поверх-
ностных атомов с другими атомами, возникают индуцирован-
ные адсорбцией поверхностные заряды и поверхностные
структуры, происходит деформация энергетических полос
н т. д. Результатом этого является более легкое образование
зародышевых микротрещпн. ПАВ, попадая в область микро-
трещины, вследствие снижения им поверхностной энергии,
облегчает развитие микротрещипы в трещину разрушения.
По этой причине адсорбционное понижение прочности прояв-
ляется тем сильнее, чем выше дефектность структуры [192].
Кинетика процессов понижения прочности твердых тел в
присутствии ПАВ обусловлена скоростью их адсорбции па
поверхностях разрушения в момент их образования 1185].
Поэтому наиболее благоприятны такие механические воз-
действия, при которых деформация протекает медленно по
сравнению со скоростью миграции жидкости по поверхности,
а также периодические силовые воздействия, обеспечиваю-
щие проникновение ПАВ в глубь микротрещин [81, 191].
Систематические исследования действия ПАВ на неорга-
нические вещества проведены Ходаковым [50]. В результате
было установлено, что диспергирование твердых тел в адсорб-
ционно-активной среде (40—80% от массы материала) при-
водит к многократному увеличению удельной поверхности
по сравнению с диспергированием на воздухе. Причем удель-
ная поверхность порошков при диспергировании на воздухе
обычно достигает максимума, после которого она либо не
возрастает, либо даже уменьшается, что объяснено образо-
ванием молекулярно-плотных агрегатов [115, 186, 187].
При исследовании интенсивности диспергирования квар-
ца в зависимости от содержания воды Ходаковым [50] обна-
ружен очень важный с практической точки зрения эффект
влияния малых добавок воды. При малых содержаниях во-
ды, (от 0,04 до 1 %) интенсивность диспергирования резко воз-
растает (рис. 13), а затем при увеличении содержания воды
от 2 до 30% снижается, при 30—40% снова резко возрастает
и достигает оптимальных условий при переходе к диспер-
гированию с 50—80% воды. Расчеты, выполненные Ходако-
вым, показали, что первый максимум на кривой соответству-
ет образованию на поверхности порошков одного насыщен-
ного мономолекулярного слоя воды.
яа
Рис. 13. Удельная поверхность
кварца при диспергировании в
присутствии различных количеств
воды (50].
Преми лпсигргпропанпл, мин: 1 — 2;
2 — 4; а — В; J — 16.
Поскольку поверхность
является границей раздела
двух фаз, она находится во
взаимодействии с обеими фа-
зами н при изучении необходим комплексный подход, учи-
тывающий эти взаимодействия.
С использованием такого подхода в 1188, 189] рассмотре-
но взаимодействие адсорбент — адсорбат как единая кванто-
во-химическая система с привлечением современных пред-
ставлений о структуре и химическом строении поверхности
твердого тела. Исследовалось влияние ПАВ на диспергиро-
вание кварца. В качестве ПАВ были взяты гексил сульфат
натрия (ГСП) и цетплтрпметплам.моиинбромид (ЦТАВ).
С использованием метода ПК-сиектроскошш показано, что
адсорбция ГСП протекает по координационному механизму
па координационно-ненасыщенных атомах кремния с воз-
можным изменением локальной симметрии сульфогруппы
—SO^ с С3и на С3 [189], а хемосорбция ЦТАВ обусловлена
его взаимодействием с атомами кислорода на поверхности
SiO, [188].
На основе рассмотрения литературных данных о природе
силоксановой связи и проведенных экспериментальных ис-
следований но адсорбционному взаимодействию ГСП и
ЦТАВ с поверхностью SiO2 предложена гипотеза о механизме
адсорбционного понижения прочности кварца [189], который
заключается в том, что смещение электронной плотности
сульфогруппы на внешнюю вакантную Зй-орбиталь атома
кремния при адсорбционном взаимодействии ГСН с кварцем
снижает эффективный положительный заряд на кремнии, что
изменяет характер распределения электронной плотности
силоксановой связи — центр тяжести электронного облака
связи смещается по индуктивному механизму (Н-1-эффект)
к атомам кислорода по схеме
V
I:'
0
!)
-9281= Q
(I
58
вследствие смещения л-элсктроппой плотности. Смещение л-
электроппой плотности силоксановой связи к атому кисло-
рода в результате уменьшения элсктропоакцепторных
свойств кремния понижает степень ковалентности связи
вследствие уменьшения (р — <2)л-взаимодействпя между ато-
мами Si н О. Понижение прочности Si—О уменьшает величи-
ну ее постоянной, равновесное межатомное расстояние свя-
зи увеличивается, а энергия се разрыва понижается 11931.
Хемосорбция ЦТАБ на кварце вызывает смещение элект-
ронной плотности атома О к элсктропоакцепторпой группе
Вследствие этого нуклеофильные свойства атома О
понижаются, электронная плотность кремнскислородпой
связи смещается к кислороду по индуктивному механизму
(-1-эффект) по схеме
>n4<
' I х
I
I
Оо Sih
Смещение л-электропной плотности к атому кислорода при-
водит к понижению частичной двоссвязиости силоксановой
связи, чему способствуют и координационно-связанные с
атомами кремния молекулы воды, понижая акцепторные
свойства кремния. Повышение ненасыщенности связи Si—О
уменьшает энергию ее разрыва.
Таким образом, предполагается, что механизм адсорбци-
онного понижения прочности кварца заключается в измене-
нии прочности кремнекислородной связи при адсорбционном
взаимодействии со средой, электронное возбуждение переда-
ется по системе силоксановых связей (194), а уменьшение
энергии связей в твердом теле при адсорбции понижает уро-
вень локальной концентрации разрушающих напряжений.
Ходаковым в работе [50] отмечено, что достаточно дли-
тельное диспергирование кварца в присутствии ПАВ не толь-
ко способствует увеличению степени диспергирования, но и
может приводить к некоторому увеличению степени его
аморфизацпп.
Экспериментальные результаты по изучению параметров
тонкой кристаллической структуры — размеров областей
когерентного рассеяния (23) и мнкроискажспня (е) — методом
гармонического анализа профиля рентгеновских дифракци-
онных линий (па примере диспергированного кварца) под-
тверждают эти данные (195):
Среда
Do 1Д> X
Дистиллированная вода
Водные растворы ЦТАБ,
ГСП, ОП-Ю *
D, им
, нм
е* 10*
30 0,012 14,0
63 0,020 3,2
61-40 0,051—0,016 6,5-11.5
* ОП-10— полиэтиленовые эфиры моно- и диалкилфенолов; по-
пользовались растворы концентрации: ЦТАБ (0,34—2,5) lt)~« М
ГСП (1,23—9,8)10—‘ М и ОП-10 (1,25—2,5)-II)-э%.
Итак, видно, что у кварца, диспергированного па возду-
хе, наблюдается усложнение субструктуры, проявляющее-
ся в уменьшении блоков когерентного рассеяния и увеличе-
нии микроискажений по сравнению с кварцем, дисперги-
рованным в адсорбционно-активных средах; при этом уве-
личение числа элементов топкой кристаллической структу-
ры и возрастание элементарных искажений атомной кри-
сталлической решетки, которое связано со смещением цент-
ров колебаний атомов из узлов пространственной решетки,
должно сопровождаться возрастанием угла мозаичности
[195].
Использование теоретических и экспериментальных дан-
ных, полученных при изучении адсорбционного понижения
прочности твердых тел, открывает перспективу [195] воз-
действия на инициирование мехаиохимических реакций, для
чего па первой стадии диспергирования необходимо исполь-
зовать адсорбцпопио-активную среду, позволяющую полу-
чить материал в предельно дисперсном состоянии, при этом
достаточно такого количества активной среды, которое обес-
печит образование насыщенного молекулярного слоя на по-
верхности частиц. При достижении материалом предельно
развитой поверхности в системе возникает дефицит адсорб-
ционно-активной среды, что сводит к минимуму ее влияние
на вторую стадию процесса — собственно механохимиче-
скую реакцию или механическую активацию; не исключено,
однако, и положительное влияние адсорбционно-активной
среды на кинетику механохимнческой реакции, но здесь
необходимо учитывать влияние среды [195].
1.5. ВЫСОКОЭПЕРГОПАПРЯЖЕНПЫЕ АППАРАТЫ
ДЛЯ ДИСПЕРГИРОВАНИЯ
И АКТИВАЦИИ ТВЕРДЫХ ТЕЛ
Для диспергирования твердых тел находят применение
аппараты различных конструкций, различающиеся видом
механического воздействия па вещество: в одном случае это
55
может быть раздавливание, в Других — удар, раскалыва-
ние, истирапис и т. д. В современных измельчителях обычно
сочетаются два или более видов таких воздействий. Для
хрупких материалов, как правило, используют машины, в
которых преобладают раздавливающие и ударные воздей-
ствия, для мягких — истирание, для волокнистых материа-
лов эффективны разрывающие усилия и т. д.
К настоящему времени разработано большое количество
конструкций аппаратов для диспергирования. Классифика-
ция и детальное описание аппаратов приведены в моногра-
фиях Козулина и Горловского [196], Сиденко 1197], а так-
же в справочниках [198, 199]. Отдельные типы аппаратов —
вибрационные, струйные, с вихревым слоем, дезинтеграто-
ры — описаны в монографиях Роуза [200], Акунова [203],
Логвиненко и Шелякова [201], Хипта [202] и др.
В работе [204] по виду преимущественного нагружения
выделены три типа мельниц:
1. Мельницы с низкой скоростью нагружения, преиму-
щественно сжатием (шаровые, шарокольцевыс, стержневые,
бегуны, центробежные и т. д.).
2. Мельницы со средней скоростью нагружения, разру-
шающие в основном стесненным ударом (вибрационные, цент-
робежно-планетарные, магнитно-вихревые и т. д.).
3. Мельницы с высокой скоростью нагружения, измель-
чающие материалы преимущественно свободным ударом
(ударно-отражательного действия, ударные мельницы, де-
зинтеграторы и дисмембраторы, молотковые, струйные, ро-
торные и т. д.).
Как следует из вышесказанного, эффективная механиче-
ская активация твердого тела требует больших плотностей
энергии в рабочем пространстве и механохимические эффек-
ты реализуются в основном в аппаратах с высокой энерго-
напряженностью, которая характеризуется мощностью, при-
ходящейся на единицу рабочего объема мельницы. Необхо-
димость таких больших плотностей является самым жестким
требованием к технической реализации процессов актива-
ции в измельчительных аппаратах. Эти условия могут быть
реализованы в аппаратах со средней и высокой скоростью
нагружения: вибрационных, центробежно-планетарных,
ударно-отражательных, роторно-центробежных, дезиптегра-
торпых и других аппаратах.
Сравнительно новыми и перспективными высокоэнерго-
напряженными измельчитсльными аппаратами являются ап-
параты планетарного типа: планетарные и дифференциаль-
ные центробежные мельницы, разработанные Голосовым
[205, 206], Бушуевым [207, 208] и другими [209—214].
Рис.
ца е
13. Планетарная мсльпи-
гндростатпчсскпми обой-
мами 1212].
вид сбоку, б — ичд сиерху.
па измельчаемые частицы и
скоростью движения загрузки
в рабочем барабане. Опп ра-
ботают по принципу гравита-
ционного измельчения, который
реализуется за счет взаимодействия двух центробежных по-
лей. 11а рис. 14 представлена схема аппарата, а на рис. 15
в качестве примера — одна из конструкций с барабанами
в обоймах, в которые под давлением подается вода, при-
жимающая барабаны к стенке и выполняющая роль смазки
и охлаждающей среды [212].
Как видно из схемы планетарной центробежной мельни-
цы (ПЦМ), частица в барабане участвует в двух движениях:
относительном движении вокруг общей оси 0'0 и вращении
вокруг собственной оси М'М.
Для характеристики режима работы ПЦМ введены па-
раметры к и т. Величина к = (w, — число оборотов
вокруг собственной оси, — число оборотов вокруг об-
щей оси) называется кинематической характеристикой мель-
ницы и может быть как положительной, так и отрицатель-
ной. Положительное значение к принимает при одинаковом
направлении векторов скоростей относительного н собствен-
ного вращения барабанов. При противоположном направ-
57
лении векторов скоростей к отрицательно. Эксперименталь-
но установлено, что при отрицательном значении к дис-
пергирование наиболее эффективно. Величина т. = (/?,,
— радиусы общего и собственного вращений) является
геометрической характеристикой ПЦМ. Для выполнения
планетарпости движения необходимо, чтобы к 0 и т =# 0.
Варьируя эти величины, можно изменять динамические ус-
ловия диспергирования в широком диапазоне режимов.
Теоретическая скорость мелющего тела в момент отрыва
его от стенки барабана под углом 180° или близким к нему
(наиболее часто реализующаяся ситуация в планетарных
мельницах) раина
v = vY -f- v2 = 2л[ю1(7?1 -|- 11.,) -|- ю2/?2].
Выражая ее через характеристики мельницы, получим
и = -|- (ш |- 1)].
Планетарные центробежные мельницы отличаются от
дифференциальных центробежных мельниц тем, что в пла-
нетарной передача вращения барабана осуществляется с по-
мощью вращающегося водила и неподвижного элемента.
В дифференциальной мельнице барабан и водило вращаются
вокруг собственных осей независимо друг от друга. Приме-
нительно к центробежным мельницам (планетарным и диф-
ференциальным) Жирновым [209] выделены три режима
движения шаровой загрузки (рис. 16): а) скольжение (исти-
рание), при котором материал движется в направлении,
противоположном направлению вращения барабана вокруг
собственной оси; б) перекатывание (ударный), которое опи-
сывается криволинейной траекторией материала, располо-
женного в виде сегмента; в) вихревой, при котором мате-
риал, двигаясь по пологой криволинейной траектории, за-
полняет весь объем в барабане.
Режим скольжения характеризуется движением мате-
риала относительно стенки барабана в направлении, проти-
воположном движению стенки барабана. При этом скорость
шаров возрастает по мере удаления от стенки барабана. На-
личие градиента скоростей в движущейся загрузке создаст
условия для интенсивного истирания частиц, а противопо-
ложные направления движения стенки и загрузки приводят
к истиранию пристеночным слоем загрузки внутренней по-
верхности барабана.
В режиме перекатывания загрузка расположена в виде
сегмента, смещенного в направлении вращения барабана.
Рис. 1f>. Гржпмы двпжеппп загрузки в
барабане планетарной центробежной
мельницы.
а — скольжение (истирание); б — перекаты-
вание (ударный); в — вихревой.
Этот режим можно условно срав-
нить с одноименным режимом
движения шаровой загрузки, опи-
санным в теории гравитационных
шаровых мельниц. Согласии ос-
новным положениям этой теории,
принимается, что частица, поки-
нув круговую траекторию, про-
должает двигаться по параболе.
При этом мерой эффекта измель-
чения считают скорость шара по
отношению к скорости измельчае-
мого материала в точке, в кото-
рой шар встречается с измельчае-
мой частицей. При движении ша-
ровой загрузки в режиме перека-
тывания материал разрушается
ударом, истиранием и раздавли-
ванием.
Вихревой режим характери-
зуется распределением загрузки
по всем объему барабана. Движу-
щиеся шары описывают кривые,
близкие к эллиптической траекто-
рии. Свободный пробег шаров в
этом режиме значителен, и шар,
двигаясь с большой скоростью
до встречи со стенкой или другой
частицей, разрушает материал преимущественно ударом.
Поскольку имеющиеся конструкции планетарных и диф-
ференциальных мельниц пе удовлетворяли требованиям хи-
мического эксперимента (отсутствие термостатированпя, гер-
метизации и др.), специально была разработана конструк-
ция, учитывающая эти требования (211]. Пзмельчительный
аппарат рассматривали как химический реактор и принима-
ли соответствующие меры, чтобы свести к минимуму мешаю-
щие факторы: термостатпровалп рабочие емкости, изолиро-
вали обрабатываемое вещество от внешней атмосферы, ис-
пользовали химически инертные материалы и т. д.
59
Рис. 17. Горизонтальный вариант планетарной мельницы с термоста-
тированным барабаном [211].
Аппарат представлен на рис. 17. В отличие от существую-
щих аппаратов с целью обеспечения герметизации и термо-
статирования рабочих емкостей вращающаяся емкость с за-
грузочными устройствами устанавливается под углом к об-
щей осп вращения, а загрузочное устройство снабжено гиб-
кими шлангами и двумя пальцами, движущимися в пазах
втулки, жестко установленной на станине. При вращении
ротора рабочая емкость в результате кинематической связи
со станиной, осуществляемой пальцами, движущимися в па-
зах втулки, совершает планетарное движение. Последнее
складывается из кругового движения наклонной собствен-
ной оси емкости по конусной поверхности вокруг оси рото-
ра и собственного вращения емкости относительно ротора
вокруг наклонной собственной оси. При такой конструкции
аппарата все параметры планетарпой мельницы в нем со-
храняются.
Энергетические параметры активации в данном аппарате
можно варьировать в широких интервалах путем изменения
частоты вращения рабочей емкости вокруг общей оси. По-
скольку частота вращения вокруг .планетарной оси равна
частоте вращения вокруг общей оси, режим движения за-
грузки в барабане мельницы при этом является неизменным
(что подтверждается стробоскопическими исследованиями).
Расчет мощности мельницы может быть сделан по форму-
ле, выведенной Голосовым [206]:
117 __ IV 4л'ЛЛ' ".
1КПЛ - И Ш —ggj -
(1.39)
здесь IV — мощность мельницы в режиме работы ее как ша-
ровой:
Wm = (1.40)
т — масса шаровой загрузки, кг; D — диаметр барабана,
м; N— число оборотов ротора, с~*; — критическое чис-
ло оборотов центробежной мельницы:
/7Г
= 0,8iV у (1.41)
п.2 — критическое число оборотов центробежной мельницы
в условиях работы ее как шаровой:
11.2
32
УБ‘
(1.42)
Получены следующие результаты расчетов мощности ак-
тиватора в ааниспмости от числа оборотов барабана по урав-
нению:
Число оборотов, с*”1 7,4 9,0
Пил. Нт но уравнению (1.39) 18 35
il’n.i.ni.-c.n, Bl 10 31
10.3 11,2 12.0 14.1 15.5
54 71 100 13S 19о
98 143 235 330 427
Проведено также экспериментальное определение мощ-
ности аппарата, представленного на рис. 17, в зависимости
Рис. 18. Влияние количества («) и диаметра (б) шаров па степень ди-
спергирования ZrB2 в планетарной мелышце (время диспергирования
ООО с) (2151.
61
-~i---
80 ^мин
х2
Рис. 1П. Кинетика диспергиро-
вании в различных мельницах.
1 — планетарная мельница [205 ];
2 — впиромельница [217]; 2 — аппарат
вихревого слоя 1201].
от числа оборотов рабочей
емкости при заданных ус-
ловиях активации (масса ша-
ров 200 г, масса обраба-
тываемого вещества 8 г, диа-
метр шаров 3—4 мм, плот-
ность шаров 15,6 г/см3). Зат-
раты мощности регистриро-
вались электроизмерительны-
ми приборами. Затраченная
на диспергирование и акти-
вацию мощность определя-
лась как разность мощностей
при работе аппарата, загру-
женного шарами и веще-
ством Р7р, и при работе аппарата, когда вместо шаров и ве-
щества внутрь барабана поместили неподвижно пластину,
равную массе шаров и вещества (РГП), т- е-
^пл = IVp - Wn.
(1.43)
Видно, что теоретический расчет по уравнению (1.39)
при частотах вращения выше 9 с-1 дает заниженные'более
чем в 2 раза результаты, что может быть связано с пеуче-
том взаимодействия шаров друг с другом, возрастанием ко-
эффициента трения и т. д.
К параметрам, оказывающим влияние на эффективность
диспергирования и активации, относятся также плотность
и размер шаров, объем, заполняемый шарами, соотношение
массы шаров и массы обрабатываемого вещества и т. д. Эти
параметры неодинаковы для всех режимов и изменяются в
зависимости от характеристик мельницы (к, т, энергона-
пряженность и т. д.). Оптимальные значения некоторых из
них для центробежной планетарной мельницы с параметра-
ми = 15,2 с-1, «2 = М с-1, к = —0,453 даны в рабо-
те Мацеры и др. [215] и приведены на рис. 18. Видно, что
они проходят через максимум. Максимальное количество
шаров составляет 50 об.%, диаметр шаров — 10 мм.
По сравнению с другими аппараты планетарного типа об-
ладают более высокой эффективностью и создают наиболь-
62
1 а о л и ц а а
Размеры мнкронлокон когерентного рассеяния (первичных
кристаллитов) и значения относительных среднеквадра-
тичных мнкродеформацнп решетки кварца после актива-
ции п в аппаратах разных конструкции [216)
Наименование аппарата Время обработки, с </ им О 1 ||»
Планетарная мельница [205] 180 63 2.8
600 34 2.7
900 20 5.0
Вибрационная мельница [217] 600 44 1,8
2400 50 2.1
5400 50 2,3
Динара г с вихревым слоем [201] 2700 100 Не обна- руже- но
шпе структурные изменения в диспергируемых веществах
12161- Это можно видеть из рпс. 19, на котором приведена
кинетика измельчения кварца в оптимальных режимах ра-
боты аппаратов, и из табл. 5, где даны размеры первичных'
частиц и микро деформации решетки, определенные по ме-
тоду, описанному в работе [219].
Дифференциальная центробежная мельница (рис. 20)
предоставляет большую возможность для оптимизации про-
цесса диспергирования и активации, чем планетарная, так
как в отличие от нее позволяет изменять параметр к в широ-
ком интервале значений.
Однако для того, чтобы частота вращения барабана во-
круг планетарной оси отличалась от частоты вращения во-
круг общей оси, необходимы относительно сложные механи-
ческие системы. С целью упрощения системы нами предло-
жена конструкция дифференциальной центробежной мель-
ницы, в которой барабаны приводятся во вращение вокруг
своей осн гидродвпгателем с ротором, закрепленным на ба-
рабане [213]. Общее центральное вращение осуществляется
обычным способом пли также с помощью гидродвигателя.
Жидкость, которая обеспечивает работу гидродвигателей,
служит, кромо того, охлаждающей и смазывающей средой
(см. рис. 20). В результате снижаются энергопотерп на тре-
пне и повышается зффектпццость диспергирования и акти-
вации твердых тел.
63
Описанные аппараты включают в себя признаки, прису-
щие химическим реакторам смещения: реакционную ем-
кость, устройства для перемешивания, ввода и вывода ре-
агентов. Дополнительный признак — наличие эффективно-
го многократного механического воздействия мелющих тел
на смесь реагентов, находящихся в реакторе. Это обеспечи-
вает не только диспергирование, но и протекание гетероген-
ных химических реакций во время работы аппарата.
Отметим, что для диспергирования и активации могут
найти применение и аппараты других конструкций. В част-
ности, в отечественной и зарубежной практике разработаны
и используются различные аппараты центробежного типа
(аттриторы, бисерные мельницы и др.) [198].
Нами для исследований, результаты которых приведены
ниже, использовались в основном два аппарата: центробеж-
ная планетарная мельница ЭИ-2Х150 конструкции Голосо-
ва [205] (соответствует приведенной на рис. 15 схеме) и ап-
парат непрерывного действия [211], описанный выше (см.
рис. 17). Эксперименты проводились па нем в периодпче-
Рис. 20. Дифференциальная мельница с гидравлическим приводом п
гпдростатнческпмп обоймами [213].
а — вид сбоку, б — вид сверху.
ском режиме при термостатироваппп рабочей емкости и кон-
тролируемой газовой атмосфере (вакуум, инертпый газ
и др.). Параметры (объем барабана, мощность и др.) аппа-
ратов близки между собой. Барабаны и шары мельницы ЭИ-
2x150 выполнены из упорной керамики на основе а-А1203.
Эксперименты проводились по одинаковой методике, и в
дальнейшем при описании экспериментов будут указаны
только минимальные сведения (частоты вращения, мощность,
массы загрузки и мелющих шаров).
3 Е. Г, Лпвт.умов
---------------Г Л А В A 2------------
МЕХАНИЗМЫ ИНИЦИИРОВАНИЯ
МЕХАНОХИМНЧЕСКИХ РЕАКЦИЙ
2.1. ТЕПЛОВЫЕ ТЕОРИИ ИНИЦИИРОВАНИЯ
МЕХАНОХИМНЧЕСКИХ РЕАКЦИЙ
Несмотря на сложность происходящих при деформиро-
вании и разрушении процессов, в объяснении специфики
механохимическпх процессов наметилось несколько подхо-
дов, в каждом из которых отдается предпочтение одному пл
факторов.
Как указывалось выше, выделение тепла при механиче-
ской активации связано с превращением кинетической энер-
гии в* тепловую в момент столкновения мелющего тела со
стенкой барабана пли другим телом, с трением частиц, а так-
же с пластическим течением материала. Все эти три источ-
ника могут быть ответственны за тепловое инициирование
реакций.
Исследования локальных разогревов при трении начаты
Смекалом [220J. Им было развито представление об «атер-
мическом плавлении», согласно которому микропластич-
ность твердого тела, появляющаяся при царапании его дру-
гим телом, обусловлена возникновением состояния, близко-
го к расплаву. По Смекалу, теплота плавления есть верхняя
граница избыточной энергии, которая может возникать при
таких видах механических воздействий.
В дальнейшем представления о локальных разогревах
при тренип развивались школой Боудена и приведены вы-
ше [122—124]. В результате исследований установлено, что
площадь очагов разогрева составляет в среднем 10-2—
10-4 см2, продолжительность их существования 10~6—10~3 с,
а температурный скачок может достигать температуры плав-
ления низкоплавкого компонента, но не выше.
За счет трения твердых тел может реализоваться меха-
низм контактного плавления для веществ, образующих эн
тектическую пару. Жидкая пленка в мостах их контакта
появляется при температурах, значительно меньших, чем
температура плавления чистых кристаллов. Эксперимен-
тальные исследования, выполненные Савинцевым с сотр.
[221, 222], показали, что скорости контактного плавления
для ионных кристаллов составляют 10-3 см/с. Расчеты, вы-
полпсппыо Савицкой [223] для смеси 8п—РЬ, показывают,
что время, необходимое для проплавления металлов на глу-
бину 2,5—5,0 им (5—10 атомных слоев) после приведения
их в контакт при температуре 468 К (температура эвтектики
456 К), составляет 3-10”3 с.
Кинетические аспекты локального плавления для сме-
сей, обрабатываемых в планетарной мельнице, рассмотрены
и работе 1224]. Для кинетических расчетов необходимо зна-
ние скорости плавления твердых тел под действием тепловых
потоков разной интенсивности, по, поскольку в литературе
такие данные отсутствуют, авторы в соответствии с работой
Ландау [225] полагают, что стационарная скорость плавле-
ния целиком контролируется переносом тепла п определя-
ется выражением
Чч
ScT = р(л + сДГт) * (2Л)
где qe — тепловой поток к поверхности, р — плотность
твердого тела, А — теплота плавления, АГт — разность
между температурой плавления и начальной температурой,
с — теплоемкость твердого тела. Время, необходимое для
достижения температуры плавления на заданной поверхно-
сти, определяется в соответствии с работой [225] выра-
жением
где L — теплопроводность, к — температуропроводность.
Выше (см. гл. 1) были приведены результаты расчета
Для импульса температуры на контакте трущихся частиц
NaCl без учета плавления при обработке соли в планетарной
мельнице и показано, что температура на поверхностях тре-
ния близка к температуре плавления, а время ее существо-
вания составляет 6-10-8 с.
Температура плавления NaCl Тт = 1073 К. С учетом
фоновой температуры 7'ф, равной 373 К, Д71 = Тт —
— Тф = 700 К. Время достижения температуры Тт на по-
верхности скольжения, определяемое но формуле (2.2) при
тех же значениях параметров, для которых выше сделан
расчет импульса температуры, оказалось равным 2,4-10“8 с.
Время действия теплового потока qc составляет Д£ = т —
— тт — 3,6-10“8 с.
Определение величины qe для случая скольжения частиц
NaCl при существовании расплавленной зоны проведено ме-
тодом, аналогичным описанному в работе [226]. В пей рас-
3'
66
67
сматривается ламинарный сдвиг вязкой пленки жидкости
между двумя плоскими параллельными стенками бесконеч-
ной ширины, одна из которых движется в своей плоскости
с постоянной скоростью <о1. Величина (Оц равная 58,6 м/с,
как это видно из расчета (см. гл. 1), близка к скорости сколь-
жения, при которой Боуденом и Персоном [124] наблюда-
лось локальное плавление.
Если вязкость жидкости 1], расстояние между стенками
2d, то рассеяние тепла на единицу поверхности составит
<7е=^- (2.3)
Когда температура обеих стенок поддерживается посто-
янной (Тт), температура в жидком слое изменяется парабо-
лически с расстоянием х от стенки при стационарном режиме:
(2.4)
9 ,
2 ~Тт = (1 — 2d
Вязкость расплава NaCl при 1098 К 1,4-10-4 Па-с. Опре-
деление величины d может быть сделано с учетом уравнений
(2.1), (2.4), а также уравнений
/ ~
(2.5)
(2.6)
®стац, V
d — s(t Ти).
Уравнение (2.6) имеет графическое решение, приведен-
ное в работе [225]. Решение системы из четырех уравнений
позволяет определить d — 2-Ю"7 см. Таким образом, тол-
щина расплавленной зоны может достигать 5.10“7 см при
размерах частиц NaCl 10-4 см. Максимальная разность тем-
ператур по формуле (2.4) при значении х = d равна 0,5 К.
Это указывает на то, что локальные разогревы при учете
возможности плавления обрабатываемых частиц ограничи-
ваются сверху температурой их плавления, что согласуется
с экспериментальными данными Боудена с сотр. [124]. Вре-
мя кристаллизации расплава для индивидуальных веществ
определяется уравнением [227]
d
т,<~ /М?
и составляет величину порядка 1U'9 с. Вследствие существо-
вания достаточно больших переохлаждений во время кри-
сталлизации расплавов вероятность образования зародышей
(2-7)
очень велика и время кристаллизации также близко к это-
му значению и не будет лимитировать процесс в целом.
Тепловые модели, учитывающие пластическое течение
твердых тел, являются общепризнанными в теориях ини-
циирования взрывчатых веществ механическими воздей-
ствиями [127]. Попытки объяснить взрыв локальными ра-
зогревамп при трепни частиц друг о друга предпринимались
начиная с Боудена [12.">, 124]. При этом плавление одного
из твердых веществ считалось верхней границей разогрева.
Однако ряд фактов не укладывался в рамки этой модели.
Так, например, эксперимент показывал, что температуры
вспышки для некоторых ВВ лежат много выше их темпера-
тур плавлеппя, отсутствовал параллелизм между располо-
жением ВВ по чувствительности к механическому воздей-
ствию со стандартными температурами вспышки и т. д. По-
этому такая теория была признана неудовлетворительной.
Исследователи пришли к выводу, что необходимо учиты-
вать возрастание давления при механическом воздействии,
а также образование эффективного локального разогрева
за счет пластического течения твердых тел [131]. С ростом
давления температура плавления повышается и могут быть
достигнуты необходимые для вспышки температуры.
Согласно теоретическим представлениям о тепловом вос-
пламенении, основанным на нахождении условий, при ко-
торых нарушается равновесие между теплоотводом из зоны
химической реакции и теплопрпходом п последний начинает
превышать теплоотвод, критические температуры, необхо-
димые для инициирования взрыва, для большинства рас-
пространенных ВВ лежат в интервале 672—873 К, а крити-
ческий размер очага составляет 10~4 см.
Было показано [1271, что задача возбуждения взрыва при
механическом воздействии в общем случае сводится к проч-
ностной (непзотермической) задаче с условием, что давление
должно быть выше некоторого критического значения.
Взрыв происходит, если прилагаемое давление, изменяю-
щееся ио гиперболической зависимости от толщины образца
h (D — диаметр образца) и равное
+гй)'
превосходит критическое значение Pw (рис. 21). Необхо-
димым является условие, чтобы размер деформируемого за-
ряда был не меньше некоторого предельного (запас упругой
энергии должен быть достаточным для создания эффектив-
ного очага разогрева).
(2.8)
69
Prip L--1---------------
lWf h/n
Рис. 22. «Магма-цлазма»— модель
мехапохимнческпх процессов [27 J.
Puc. 21. Переход or при niocr-
ного разрушения заряда к
взрыву при повышении удар-
ной нагрузки па взрывчатое
вещество (127 j.
С целью объяснить в рамках единой модели большинство
явлений, сопровождающих механическую активацию, Тис-
сеп, Мейер, Хейнике создали модель «магма-плазма», кото-
рая позднее получила название модели «трибоплазма» 127,
281. Согласно этой модели, выделяющаяся при механиче-
ском воздействии энергия значительно превышает теплоту
плавления и вследствие слабой теплопроводности твердых
тел приводит не только к расплавлению вещества, но и к его
сублимации, к возникновению такого состояния, в котором
оно находится в виде ионов и электронов (плазма). Схема,
иллюстрирующая эту модель, приведена на рис. 22. Точные
характеристики плазменных состояний определить очень
трудно, хотя косвенные факты, подтверждающие их суще-
ствование, имеются. Например, при царапании пластинки
из какого-либо материала иглой на диске, расположенном,
на небольшом расстоянии от пластинки, осаждается веще-
ство, из которого изготовлена пластинка. Обнаруживаются
также эмиссия электронов, свечение и т. д. Авторы считают,
что все эти явления обусловлены тепловой энергией. Одна-
ко не удалось подтвердить, что этот процесс только терми
ческий. В частности, показано, что процесс возбуждения
связан и с электрическими эффектами (228].
Выше указывалось, что при разрушении кристаллов в
носке трещины имеет место тепловое возбуждение молекул,
температурный пик может превосходить 2000 К [133]. Од-
нако время жизни такого возбуждения мало' (~'10~и—
10~10 с), поэтому оно инициирует преимущественно хпмпче-
-кие превращения на поверхности твердых тел (разложение,
регистрируемое по выделению летучих продуктов).
Такое разложение исследовалось Фоксом [134], а также
Болдыревым, Регелем, Уракаевым и др. [229], которые про-
води hi масс-спектрометрические исследования газообразных
продуктов, образующихся при раскалывании некоторых
неорганических солен, размещенных вблизи источника в
вакуумной камере времянролетиого масс-спектрометра.
Анализируя особенности мехапохимнческпх реакции,
Болдырев [230] рассмотрел их с точки зрения лимитирую-
щей стадии процесса. Процесс распада твердого тела мо-
жет быть представлен как определенная последовательность
стадий активации, дезактивации и собственно химической
реакции. Б зависимости от того, какая из этих стадий явля-
ется лимитирующей, различают два крайних случая: распад
лимитируется процессами возбуждения н разрыва связи (на
пример, термическое разложение) или одной из вторичных
стадий (превращения промежуточных продуктов, образо-
вавшихся в результате первичного акта). Па основании со
поставления рядов устойчивоетп нитратов щелочных метал-
лов к воздействию тепла, у радиации н механической обра
боткн (в пзмельчительном аппарате пли в ударной волне)
Болдыревым сделан вывод о том. что мехапохпмпческое
разложение представляет собой'процесс, лимитируемый не
стадией возбуждения, а последующими химическими ста
днями, протекание которых так же, как и в случае радиоли-
за, определяется свободным объемом в решетке.
Специфика мехапохимнческпх процессов объяснена Бол-
дыревым па основании учета кинетических факторов. Оп
считает, что вследствие импульсного характера процесса
нагревания и охлаждения может происходить закалка обра-
зующихся продуктов. Если при обычном нагревании снача-
ла реализуются низкотемпературные стадии, а затем высо-
котемпературные, то при импульсном нагревании н быстром
охлаждении могут пройти только высокотемпературные про-
цессы. Если в системе протекают параллельные или после-
довательные реакции, то реализуются пз них только те,
которые по времени реакции «вписываются» в кратковремен-
ный импульс тепла, возникающий при воздействии па твер-
дое чело. Данные, подтверждающие предложенную Бол-'
дыревым кинетическую модель, приведены в работах Ура-
ваева, Позднякова, Болдырева [231, 232]. Ими изложены
результаты масс-споктрометрического изучения продуктов,
образующихся в носке движущейся трещины при разруше-
нии iiuipaioB щелочных металлов.
71
Для нитратов, в частности для нитрата натрия, имеется
несколько возможных путей распада:
^rNaNO2 + 1/2О2
NaNO3-----*1/2Nb2O + 1/2N2 + 5/4Oa
1/2.\аО + NO2 + 3/4Оа
АЯ288 = 109 кДж/моль I
11
А//28в — 206 кДж/моль
III
Первая из зтпх реакций обычно реализуется в интервале
температур 600—800 К, вторая — выше 800 К, третья воз-
можна лишь при температурах выше 1000 К. В газовых про-
дуктах, образовавшихся при раскалывании монокристаллов
нитратов, авторами наряду с кислородом обнаружены про-
дукты высокотемпературных реакций — азот и его оксиды
[231].
Кинетическая модель мехаиохпмпческого разложения
твердых' тел, учитывающая возникновение в момент удара
тепловых импульсов, предложена Ляховым 12331. Он счп
тает, что вследствие импульсного воздействия мелющих ша-
ров на обрабатываемое вещество кинетика реакции опреде-
ляется временем пребывания вещества под импульсом (rj.
Температура и скорость реакции в импульсе меняются по
разным законам. Падение температуры в импульсе про-
исходит в соответствии с уравнением
t
Т = Тте (2.9)
тт — максимальная температура в импульсе, тр — время
релаксации импульса. Скорость же реакции в соответствии
с уравнением
w = wm exp
Е
М'т
(2.10)
падает в несколько раз быстрее, чем температура. Из анали-
за уравнений следует, что средняя температура за время
тр может быть принята равной ТсР л; 0,62’м, а в скорость
реакции основной вклад вносит только вершина импульса,
т. е. может быть принято 7'сР « 0,97'м и соответственно
« 0,1тр.
Экспериментальная проверка модели сделана па реак-
ции разложения гидрита алюминия в устройстве с электро-
магнитным приводом, в котором ударное воздействие на ве-
щество осуществляется металлическим стержнем.
Время релаксация тр определялось во отклонению от
аддитивности химического выхода при увеличении частоты
импульсов, т. е. по началу перекрывания релаксационных
«хвостов» с последующими импульсами. По данным анализа
смена режима наблюдается при частоте 20 с-1, т. е. тр =
0,05 с.
Из этих данных с учетом того, что т, « 0,1тр, автор ио
лучил значение длительности импульса тх, равное 5-10-3 с.
Зная время импульса, химический выход, полагая, что ме-
ха нохимическое разложение идет через тепловой канал со
значением энергии активации, соответствующим термиче-
скому, автор определил эквивалентные температуры, воз-
никающие при ударных воздействиях, аналогично тому, как
это делалось Фоксом [134]. Полученные им значения соот-
ветствуют температуре 463—473 К.
2.2. МЕХАНИЗМЫ ФОРМИРОВАНИЯ НОВЫХ ФАЗ,
ОБУСЛОВЛЕННЫЕ ПЛАСТИЧЕСКИМ ТЕЧЕНИЕМ
ТВЕРДЫХ ТЕЛ
И ВЫХОДЯЩИМИ НА ПОВЕРХНОСТЬ ДИСЛОКАЦИЯМИ
Начиная с работ Бриджмена [15, 16], установлено, что
приложение сдвига во время действия высоких давлений
приводит к резкому ускорению твердофазных процессов.
1акое воздействие сопровождается различными необратимы-
ми явлениями: некоторые минералы переходят из термодина-
мически неустойчивых модификаций в устойчивые: напри-
мер, вюртцит превращается в сфалерит или красный фосфор
превращается в черный кристаллический (переход, который
в других условиях не наблюдается). Происходят п химиче-
ские превращения: синтез соединений из элементов, напри-
мер Cu.2S; разложение, например, BiaO8 до металлического
висмута или изменение валентности, например SnOj в SnO
[234]. Было обнаружено восстановление оксида свинца в
металлический свинец, оксида серебра до металла, трпокси-
да молибдена до диоксида, а также возможность прямого
синтеза сульфидов меди, никеля, кобальта, железа и фосфи-
дов магния и цинка из элементов [235].
Ларсен п Дрнкамер [236] обнаружили быстрое протека-
ние превращения K3Fe(CN)e в K4Fe(CiN)e при сочетании дав
ления от 0,3 до 5 МПа со сдвигом при комнатной температу-
ре. Гидростатическое давление без сдвига не приводит к за-
метным химическим изменениям в этих соединениях.
73
О е
®Щ«Ю®О©<' )®O®Ofe
©•oec®oeoeoeoeo®
•©•©•©•©•о о о о
Э®О®О®О@О®и©<'
ё)О®О®О®О®О®О®
0©0®0®0®0©0©С
©О©О®О®О@О@О@
@о©о©о®о®о@о@
J@O©O®O®O®O@C
s>0©0®0®0®0®0@
3@0®0®0®0®©®C
®о®о®о®о®о®о®
0®0®0@0®0®@®0
© атом А © атом А, поисоедикившийся к „ролику” Q атом В
Рис. 23. Механизм роста кристаллического зародыша в условиях* пла-
u ___ стическоги течении материала |25|.
При рассмотрении процессов синтеза или разложения
под воздействием высоких давлений со сдвигом маловероят-
ным представляется обычный механизм диффузия, при ко-
тором продвижение отдельных атомов может привести к об-
разованию частиц повой фазы.
Как указывалось выше, ускоренный массоиереиос под
влиянием пластической деформации может быть вызван дву-
мя причинами: перенос, связанный с существованием пла-
стических точений, п ускорением собственно диффузии и ре-
зультате образования линейных и точечных дефектов и пе-
ремещения их за счет градиента напряжений.
Качественная картина роста новой фазы при наличии
пластических течений рассмотрена Дремииым и Бреусовым
[25] па примере разложения соединения типа АВ. Предло-
женная ими модель процесса носит название «модель роли-
ка» (рис. 23).
Если два слоя вещества смещаются друг относительно
друга, то зародыш фазы А, находящийся между ними, мож-
но рассматривать как своеобразный ролик качения. Ввиду
большей прочности связи А—А, чем связи А—В, можно рас-
сматривать фазу А как твердое тело, а фазу АВ — как жид-
кость. При быстром смещении двух слоев фазы АВ относи-
тельно друг друга мимо зародыша проходит множество ато-
мов обоих сортов. Так как время перестройки электронных
оболочек (10-13—10-14 с) значительно меньше времени со-
прикосновения атомов друг с другом (расчеты показывают,
что при пластическом смещении па расстояние 1 мм, разме-
ре одного атома 0,2 нм и времени воздействия 10-ь с время
контакта двух атомов составит 2-10~12 с), то все атомы А,
проходящие в непосредственной близости от зародыша,
успевают присоединиться к нему, и именно таким путем
может происходить рост зародышей повой фазы.
Такны образом, в отлично от обычного диффузионного
роста центров кристаллизации, когда каждый из атомов
должен «пробираться» к повой фазе, нри росте зародышей
новых фаз за счет пластического течения материала нужные
атомы приносятся и присоединяются к зародышу сами.
Па рис. 23, б представлена развертка фазы АВ по плос-
кости скольжения при пластическом течении. Обозначены
атомы А, присоединяющиеся к исходному атому — заро-
дышу при движении фазы АВ около образующегося ролика.
Авторы дают оценку размера частиц новой фазы. Так, если
два атома смещаются друг относительно друга па расстоя-
ние //, а строение каждого из слоев соответствует рис. 23
и расстояние А — В равно L, то N — число атомов, захва-
ченных роликом при смещении слоев, будет определяться
выражением
¥=1 + 2 2 + 3+ ... +(т^-~ 1) +зг]~77
Например, для 77=1 мм и L — 0.1 нм, .V = 2,5-1013
атомов размер получающейся частицы равен -^О.СОЗ мм.
Такой же механизм роста частиц повой фазы может ра-
ботать и нри синтезе соединения из двух компонентов, а так-
же при полиморфных превращениях. При обработке .мелко-
кристаллических частиц в этих условиях они могут объеди-
няться, и под действием пластического течения из них могут
вырастать частицы более крупные, чем исходные.
Многочисленные эксперименты указывают па то, чю ме-
ста дислокаций па поверхность являются активными в хи-
мическом отношении центрами. В частности, выявление дис-
локаций основано именно на повышенной растворимости
твердого тела вблизи дислокации [237].
Шрадером, Стадтером и др. [238] исследовалась зависи-
мость каталитической активности никелевого порошка от
времени измельчения в аттрпторе в реакциях гидрирования
и конверсии орто-параводорода. Опп установили, что су-
ществует линейная связь между удельной скоростью реак-
ции п плотностью дислокаций в никеле. Показана четкая за-
висимость между энергией активации и плотностью дисло-
каций. С увеличением последней энергия активации реак-
ции резко падает. Повышение температуры приводит к сни-
жению плотности дислокаций н, как следствие, к снижению
скорости реакции.
75
Рис. 24. Взаимодействие сероводорода с никелем в местах выхода
дислокаций на поверхность в области, прилегающей к месту удара
шарика (черное пятно) [27]. X 135.
Мейером [27] показано, что па мостах выходов дислока
ций после удара шарика о поверхность электролитически
полированного никеля имеет место селективная реакция ни-
келя с сероводородом с образованием сульфида никеля. В ре-
зультате с ее помощью выявлена зона пластической дефор-
мации вокруг места удара и система плоскостей скольже-
ния, по которым двигаются дислокации в объемно центри-
рованной решетке никеля. Из рис. 24 видно, что в непосред-
ственной близости к зоне удара плотность дислокаций боль-
шая, п с увеличением расстояния она падает. Сульфидиро-
вание никеля вдоль линии скольжения наблюдалось Мейе-
ром и на пластинке никеля, которая не подвергалась удар-
ному воздействию, а деформировалась пластически растя-
гиванием.
Разницы между образцами, которые декорировались не-
посредственно в момент механического воздействия или че-
рез некоторое время выдержки в вакууме, не наблюдалось.
Это означает, что активность поверхности сохраняется до
тех пор, пока существуют упругие напряжения вокруг дис-
76
локации. После отжига деформированного никеля при тем-
пературе 673 К, когда дислокации исчезают, скорость вза-
имодействия никеля с сероводородом резко падает.
Микроскопические исследования поверхности никеля с
разной предварительной обработкой (отожженный при вы-
соких температурах в среде На, отожженный в этих же ус-
ловиях и выдержанный в среде СО, отожжеппып и подверг-
нутый механической обработке), проведенные Хепнике, Ха-
ренцом, Рихтер-Мендау [239], показали, что в не обрабо-
танных механически образцах образование карбоппла нике-
ля идет па границах зерен, а поверхность механически об-
работанных пластинок сильно нарушена. Морфология на-
рушенных областей характеризуется повышенной плотно-
стью дислокаций по сравнению с недеформированными участ-
ками, в центре же их проявляется квазпаморфная структура.
Механическая обработка пластпнок приводит не только к де-
формированию поверхностного слоя, но и к значительному
увеличению реакционной площади вследствие увеличения ее
шероховатости.
Удельная скорость образования карбоппла нпкеля на
исходной (выдержанной в водороде) пластинке составляет!
5- 10—° мо.чь/г-см2, а после механической обработки опа уве-
личивается более чем на два порядка. Причем активность
пластинок практически не падает после выдержки в вакууме
в течение нескольких часов, после выдержки в течение
8-101 с активность та же, что и после выдержки fi-102 с.
Поверхность механически активированной пластинки со-
— ставляет 400% поверхности исходной пластинки, а актив-
ность возрастает на 2-104%. Сравнивая эти величины, авто-
ры [239] приходят к выводу о том, что увеличение активно-
сти за счет увеличения поверхности составляет всего 2%.
Основной же вклад, таким образом, вносит энергетический
фактор, связанный с накоплением дислокаций.
Увеличение химической активности под влиянием нагру-
жения имеет место не только для металлов, но и для ионных
и ковалентных кристаллов. Изменение скорости растворе-
ния нагруженного образца кальцита в уксусной кислоте
наблюдали Гутман п Абдуллин [39]. Весьма интересным яв-
ляется тот факт, что химическая активность изменяется по-
разному в зависимости от характера механического воздей-
ствия (растяжение пли сжатие). Сжимающее напряжение
замедляет взаимодействие кварца с водой (гидролиз); рас-
тягивающее — ускоряет процесс, и, что особенно существен-
но, эффект возрастает с поппжеппем температуры гидроли-
за [240].
77
В работе Красулина [241], посвященной сварке алюми-
ния с кремнием под давлением, показано, что плотность ди-
слокаций на поверхности кремния равна примерно
10’ см~2. Сопоставив это значение с числом атомов на поверх-
ности кремния (-—-1014 см-2), автор приходит к выводу, что
сама дислокация еще не служит активным центром. Актин
ными центрами являются, согласно Красулину, атомы, на-
ходящиеся в области упругих искажений вокруг дислокации.
Каракозов [242] на основании уравнений теории упру-
гого континуума, в рамках которой обычно проводятся энер-
гетические расчеты дислокационных структур, построил
энергетическую модель состояний атомов вокруг дислокации
(рис. 25). Он предположил, что все атомы на поверхности в
поле упругих искажений вокруг дислокации, энергия кото-
рых достигла или превысила потенциальный барьер у по-
верхности U, разрывают старые и образуют новые межатом
ные связи. По уравнениям теории упругого континуума для
заданного значения U можно вычислить площадь активного
центра и число атомов на его поверхности.
Красулип [241] методом электронной микроскопии экс-
периментально установил, что диаметр активных участков
составляет в среднем около 3 мкм. При плотности дислока-
ций около 10’ см-2 площадь активных участков составляет
около 70% поверхности, т. е. почти вся поверхность крем-
ния становится активной.
В последпее время предпринимаются попытки количе-
ственно оцепить вклад дислокаций в реакционную способ-
ность твердых тел. Однако вычисление точных величин со-
пряжено с большими трудностями, и приходится пользо-
ваться различными приближениями. Об одном приближе-
нии как самом простом — теории упругого континуума —
говорилось выше. Молоцким сделаны полуэмпирические рас-
четы [243, 244] влияния дислокаций на химические реакции
в адсорбированных на поверхности твердых тел газах. Он
показал, что ступени и изломы существенно меняют спектр
локальных электронных состояний хемосорбированных ато-
мов, понижая в несколько раз положение локального уров-
ня основного состояния. В месте выхода краевой дислока-
ции на поверхность существует глубокая электронная ло-
вушка. Энергия связи электрона с этой ловушкой в ионных
кристаллах может превышать 0,5 эВ. Поверхностный заряд,
локализованный здесь, способен существенно снизить вы-
соту термодинамического барьера, который необходимо пре-
одолеть при образовании зародыша па поверхности. Расче-
ты, проведенные автором для образования зародыша па
78
Рис. 25. Модель активного центра, образованного при вы-
ходе дислокации на поверхность [241].
кристалле NaCl, показывают, что критический размер за-
родыша вблизи дислокации уменьшается на порядок, а тер-
модинамический барьер уменьшается на два порядка. Катали-
тическая активность мест выхода краевых дислокаций на
поверхность объясняется образованием прочной двухэлек-
тропиой связи хемосорбированного атома с электроном,
локализованным на ядре дислокации, уменьшением термо-
динамического барьера образования зародышей новой фа-
зы в поле локализованного электрона, повышением доли
реакционноспособной формы хемосорбции вблизи ядра дис-
локации, притяжением хемосорбированных частиц к ядру
дислокации и т. д.
В описанных выше работах рассматривались дислокации
с небольшими скоростями. Однако известно, что движущие-
ся дислокации имеют повышенную энергию [72]:
V 1 — и /с
где Q — энергия движущейся дислокации, Qo — энергия
неподвижной дислокации, и — скорость дислокации, с —
скорость звука. Поскольку скорость дислокаций достигает
околозвуковой (например, в LiF скорость составляет
~105 см/с), можно прийти к выводу, что движущаяся дисло-
кация способна возбуждать адсорбированные частпцы ана-
логично молекулярным столкновениям в ударных трубах.
Рассматривая модель адсорбции двухатомного газа, свя-
занного с двумя узлами решетки, Молоцкий [245] показал,
что при выходе дислокации па поверхность деформация ре-
шетки изменяет равновесное расстояние между атомами ад-
79
сорбироваппой молекулы. Это вызывает искажение углов
связи между атомами газа и кристалла, что, в свою очередь,
вызывает появление деформационной силы, действующей
вдоль осн молекулы. Колебания в молекуле адсорбата ло-
кализованы п в кристалл не проникают, поскольку силовая
константа молекулы примерно на порядок превышает де
формационную силовую константу связи молекулы с кри
сталлом. Движение дислокации со скоростью v приводит к
появлению переменной деформационной силы, возбуждаю-
щей внутримолекулярные колебания адсорбированной час-
тицы. При v с энергия возбуждения значительно растет.
Так, для молекулы азота, хемосорбированной на LiF, ве-
роятность одиокваптовых возбуждений движущейся дисло
нацией на три порядка выше вероятности одиокваптовых
молекулярных возбуждений в газовой фазе. В случае
двухквантовых возбуждений разница составляет уже пять
порядков.
Выше (см. гл. 1) указывалось, что аннигиляция дисло-
каций с аптипараллельпыми векторами Бюргерса может
приводить к локальному разогреву до 1000 К в объемах, не
превышающих объема одной координационной сферы. Ана-
логичная точка зрения развивается в работах Зубовом и
Апарнпкова [246], которые полагают, что такие дислокации
при встрече с другими дислокациями, дефектами, граница-
ми зереп генерируют высокочастотные фононы, способные
инициировать механохимпческое разложение твердых тел.
Они полагают, что в таком случае количество прореагиро-
вавшего вещества будет пропорционально деформации, так
как количество появившихся фононов с энергией, достаточ-
ной для разрыва связи, линейно зависит от деформации.
По подсчетам авторов, для разрыва связи необходима час
тота фононов v = 0,25-Ю15 Гц. Это на два порядка выше
частоты тепловых фононов, существующих при комнатной
температуре. Энергия дислокации составляет несколько
электронвольт на одно межатомное расстояние, и генерация
таких фононов возможна. Высокочастотные фононы не мо-
гут свободно перемещаться по решетке, так как средний
пробег их быстро уменьшается с частотой; поэтому фонон,
возникший при столкновении дислокации с другим дефек-
том, производит химическое воздействие в решетке рядом
с тем местом, где он появился.
Развивая концепцию фононного разрушения хрупких
тел, Бартенев и Разумовская [247] считают, что яри разру-
шении твердых тел необходимо рассматривать но только
тепловые колебания атомов, по и распространение, взаимо-
действие п генерирование фононов. Стадии разрушения
твердого тела (первая — образование дефектов, вторая —
рост из этих дефектов мнкротрещип) можно рассматривать
как независимые только в самом начальном периоде. 11о ме-
ре развития дефектов выделяющаяся при разрыве энергия
приводит к повышению плотности фононов определенных
частот (неравновесных фононов). Эта подкачка энергии вли-
яет как на развитие самого дефекта, генерирующего фоно-
ны, так и на развитие соседних дефектов. Переход к фонон-
ной концепции позволит, как утверждают авторы, ввести
для каждой структуры своеобразный спектр, который мож-
но получить экспериментально.
2.3. ТЕОРИЯ АКТИВНЫХ
ПОВЕРХНОСТНЫХ СОСТОЯНИИ
Свойства атомов на поверхности твердых тел отличны от
свойств атомов, находящихся в объеме кристалла. С точки
зрения зонной теории твердого тела возникновение поверх-
ностных состоянии электронов вызвано нарушением пери-
одичности потенциала взаимодействия при переходе от ре-
гулярного объема к его границе, т. е. к поверхности. Впер-
вые возможность появления особых поверхностных элек-
тронных состояний была показана Таммом и Шокли [248,
249], а в дальнейшем получила развитие в работах Коутец- .
кого н Томашека [250]. Состояния Тамма возникают из-за
искажения кулоновского потенциала на границе твердого
тела п представляют собой неглубокие электронные ловущ-
кп, концентрация которых может быть соизмерима с кон-
центрацией поверхностных атомов. Состояния Шокли воз-
можны на поверхности твердых тел, у которых связи между
атомами в решетке частично или полностью локализованы
(структуры с ковалентной связью). Интерпретация этих со-
стояний проста — они представляют собой локализованные
свободные валентности на поверхности кристалла. В лите-
ратуре их часто называют разорванными или висячими свя-
зями (dangling bonds) [251, 252]. Число их может быть рав-
но числу поверхностных атомов, и онп обнаруживаются
на поверхности пленок, приготовленных в сверхвысоком
вакууме, а также па атомарно-чистых поверхностях крис-
таллов.
В качестве метода получения атомарно-чистых поверхно-
стен твердого тела может служить раскол кристалла, при
котором происходит разрыв химических связей и возможно
81
появление валентно-ненасыщенных атомов. Сутягиным [34]
показано, что разрыв различных по ирироде химических
связей энергетически более выгоден до электронейтральных
продуктов распада, чем до ионов. В двух возможных путях
распада
А + В I (?а-в
А-в(
А+ -|- В II (Д_в -|- /д — Ав
затрата энергии в первом случае равна прочности связи
(>А-в, а во втором нужно дополнительно подвести энергию,
равную разности потенциала ионизации /А и сродства к
электрону Ав. Для органических соединений сродство к
электрону радикалов может достигать 1,5—2,0 эВ, но по-
тенциал ионизации их осколков редко оказывается ниже
6 эВ; разность (/А — Ав) практически пе может быть ниже
4—5 эВ. Поэтому распад органических соединений проис-
ходит в основном по радикальному или молекулярному,
а не ионному пути.
При распаде в вакууме типично ионного соединения
NaCl разность — Aci — 1,33 эВ — 126 кДж/моль, т. е.
даже в этом почти предельном случае ионной связи образо-
вание электроиейтралышх атомов Na и С1 остается более
выгодным, чем ионов Na+ и С1_.
Первые попытки обнаружить их при разрушении поли-
меров методом ЭПР относятся к 1959 г. [253].
Результаты опытов совпали с ожидаемыми только при
разрыве линейных цепочек химических связей: в полимерах
были обнаружены свободнорадикальные состояния «конце-
вых» граничных атомов углерода. Например, в полиэтилене
зафиксированы свободные радикалы ~СН2—СН2 и 'СН2—
—СН2—, образовавшиеся в месте разрыва полимерной цепи.
Парамагнитные центры возникают при расколе типичных
гомеополярных кристаллов (кварц, кремний и др.). Однако
число их во много раз (в 103—104) меньше, чем следовало бы
ожидать исходя из числа поверхностных атомов. Как уста-
новлено позднее, регистрируемые парамагнитные центры —
только доля существовавших в момент разрушения коротко-
живущих активных центров, являющихся валентно-ненасы-
щенными состояниями поверхностных атомов.
С точки зрения современной квантовой химии классиче-
ская ионная модель пе применима для описания характера
химической связи в окислах и сульфидах. На это прямо ука-
зывает тот факт, что эффективный заряд ионов кислорода
82
в окислах металлов близок к —I, а ионов халькогенов в
халькогенидах металлов — к 0,5—0,8 [254]. В кварце эф-
фективный заряд па ионе кислорода равен —0,62 [255].
Химическая связь в диоксиде кремния определяется в ос-
новном «р’-гибрнднымп орбиталями. Ко у кремния имеются
вакантные Зй-уронни, которые близки по энергии к х- и р-
орбиталям внешней электронной оболочки и также могут
принимать участие в образовании химических связей. Уча-
стием в образовании химических связей rf-орбпталей наряду
с s- и р-орбнталямн объясняется большая устойчивость тет-
раэдрического иона SiO4, который является основой кри-
сталлических структур силикатов.
При разрушении кварца в мельнице в инертной среде
сигнал ЭПР наблюдали впервые Бутягпн, Берлин и др.
[256]. Позднее было установлено, что при разрушении квар-
ца, плавленого кремнезема и силикагеля возникают ПМЦ
одного типа, спектр которых представляет узкую одиноч-
ную линию, а g-фактор близок к 2. Авторы работ [257—
260] пришли к выводу, что ПМЦ находятся в тонком при-
поверхностном слое. ПМЦ, образующиеся при разрушении
кварца, стабильны при комнатной температуре при храпе-
нии в вакууме и течение месяцев. Пх концентрация быстро
уменьшается со временем лишь при Т — 500 К; 80% ПМЦ
мгновенно гибнет при комнатной температуре после контак-
та с воздухом [252].
В [251, 252] ПМЦ были зарегистрированы после разру-
шения кварца и кварцевого стекла в сверхвысоком вакуу-
ме (~10-8 Па). Авторы этих работ отмечали близость пара-
метров спектров ЭПР «механических» и радиационных ПМЦ
в кварце. Согласно модели, предложенной Виксом [261,
262], образование радиационных (возникающих под дей-
ствием у-лучей) ПМЦ связано с образованием в SiO2 кисло-
родных вакансий. В работе Силсби [263] отмечается, что
образование радиационных ПМЦ обусловлено дефицитом
кислорода в кварцевых стеклах, и считается, что неспарен-
ный электрон находится на $р3-гпбрпдной орбптали атома
кремния.
Чтобы решить вопрос, является лн наличие дефицита
кислорода в кварцевых стеклах решающим условием обра-
зования «механических» ПМЦ, Ахмед-Заде, Баптпзмапскпй
и др. [264] изучили разрушение стекол и кристаллических
модификаций кремнезема различных типов. Ими установ-
лено, что при разрушении всех стекол п кристаллов регист-
рируются полностью подобные спектры ЭПР, не сильно от-
личающиеся но интенсивности. Концентрация ПМЦ во всех
ЬЗ
случаях близка к ~1017 г-1. Равенство ПМЦ для всех сте-
кол и кристаллических модификаций позволило авторам
сделать заключение, что сигнал нельзя связывать с дефек-
тами структуры (дефицитом кислорода). Образование ПМЦ
не связано также с присутствием примесей и гпдроокислов
и не зависит от количества легирующих добавок. Авторы
полагают, что «механические» ПМЦ возникают в результате
разрыва «нормальных» межатомных химических связей при
образовании новой поверхности, так как для обнажения
одного дефекта необходимо разорвать огромное число окру-
жающих его обычных связей.
Структура этих ПМЦ расшифрована в работе [252].
Авторы доказали, что спектр принадлежит радикалам типа
-° х
—О—Si', неспаренный электрон в этих ПМЦ занимает sp3-
-ОХ
гибридную орбиту (Л'5 — центры). Подробные исследования
свойств ПМЦ на поверхности кварца, выполненные в рабо-
тах [265—2681, показали, что помимо указанного радикала
при измельчении образуются и радикалы типа ==SiO'. Спектр
ЭПР этих радикалов зарегистрировать не удается из-за вы-
рождения основного орбитального состояния [269]. Нали-
чие этих радикалов доказано с помощью химических реак-
ций ПМЦ с газами.
Радикалы типа =Si" и =SiO' имеются на поверхности
примерно в равном количестве, и их общая концентрация
составляет 4-1016 м-2. Образование этих центров указывает
на гомолитический разрыв связей Si—О в процессе измель-
чения.
Распределение ПМЦ на поверхности кварца является рав-
номерным, на это указывают анализ крыльев спектра ЭПР
радикалов типа ==Si’ и регистрация запрещенного перехода
Am, = 2.
Парамагнитные центры на поверхности кварца отлича-
ются высокой реакционной способностью по отношению к
молекулам из газовой фазы, реагируют с ними с образова-
нием поверхностных соединений. Наряду с радикалами
==Si* и =SiO' образуются и другие активные состоя-
ния. К ним относятся атомы кремния с двумя разор-
ванными связями — силиленовые центры =Si: [270]. Су-
щественный вклад в химическую активность вносят дефор-
мированные связи, которые являются носителями остаточ-
ных внутренних напряжений [270]. Деформированные свя-
зи на поверхности кварца по своим химическим свойствам
84
похожи на радикальные пары =Si ... OSis=, Так, водород
реагирует с ними практически мгновенно при 80 К. анер-
гия активации реакции
£=SiO ... Si== 4- Н2 -> sSiOH + HSi=
не превышает 8—12 кДж моль 1270]. Такая низкая энергия
активации характерна для свободнорадикальных, а не для
молекулярных реакции. Помимо гидрирования, деформиро-
ванные связи легко гидролизуются и карбонизируются
1271-276].
Таким образом, в поверхностных слоях при разрушении
и трепни возникают разорванные п деформированные связи.
При комнатной температуре деформированные связи само-
произвольно сокращаются, а радикалы частично гибнут под
действием сил трения.
Согласно теории короткоживущих активных центров
1277—281], стабилизация свежей поверхности, образующей-
ся при разрушении твердых тел, не заканчивается релакса-
цией теплового возбуждения в течение 10-°—10-11 с, а про-
должается более длительное время, порядка 10-4—К)-7 с.
За это время происходят перегруппировка химических свя-
зей, формирование электрического рельефа поверхности и
другие процессы «химической» релаксации. Молекулы газа,
жидкости или твердого тела, соприкасающиеся в этот мо-
мент с поверхностью разрушаемого твердого тела, за данный
промежуток времени успеют прореагировать с активными
центрами, находящимися на поверхности, число которых
практически равно числу поверхностных атомов. Часть
из этих КЖЦ оказывается «замурованной» и недоступной
для внешней среды; они фиксируются методом ЭПР как
«механические» ПМЦ, свойства которых обсуждались выше.
Для количественного анализа закономерностей меха-
нохпмических реакций с точки зрения КЖЦ важно устано-
вить концентрации, время жизни и химические свойства ак-
тивных центров. Эти сведения можно получить, изучая хе-
мосорбцию газов в момент образования поверхности в за-
висимости от давления газа. Прп низком давлении, когдд
промежуток между ударами о единичный центр велик, по-
верхность успевает стабилизироваться и хемосорбция мала.
Ври высоком давлении, когда частота ударов велика, боль-
шинство поверхностных центров успевает прореагировать
с газом до перестройки. В этих условиях хемосорбция мак-
симальна и практически совпадает с концентрацией поверх-
ности атомов.
85
Для таких мехапохимнческпх реакций Бутягипым [34 |
рассмотрена кинетическая схема, основанная на следующих
допущениях:
1) активные центры локализованы па свежеобразованной
поверхности;
2) скорость образования центров пропорциональна ско-
рости образования поверхности, т. е. число центров на еди-
нице повой поверхности постоянно для данного твердою
тела;
3) в первом приближении все центры энергетически рав
неценны, и процессы с их участием можно описывать в рам-
ках модели однородном поверхности;
4) процесс самопроизвольной гибели центров, скорость
которого характеризуется временем их жизни ти. отожде
ствляется с релаксацией избыточной энергии свежей поверх
мости раскола.
Таким образом, коротко живущие активные центры в ва-
кууме гибнут самопроизвольно, а в химически активной
среде их гибель сопровождается взаимодействием с окру
жающпми молекулами. Для скоростей образования и рас-
ходования центров в процессе механического разрушения
твердых тел им записаны следующие выражения: скорость
образования центров
</.V (1) r/.s
Ш -- ----- — II.
<11 </т
скорость самопроизвольной гибели
_ ЛУ(П _ _l_ N
Нг т0
скорость взаимодействия с молекулами газа
(2-14)
В этих уравнениях: 7V(t) — концентрация центров в мо-
мент времени т (центров/г); s — удельная поверхность про-
дуктов разрушения (мг2/г); п — концентрация центров на
свежеобразовапной поверхности (центров/м2); р — давление
газа, кр—частота соударения молекул газа с активными
центрами (с-1); а — вероятность химической реакции при
соударении; константа (фактор соударений)
|/ ‘ZnmkT
(2.15)
где со — сечение соударения (~10"16см 2), m — масса моле-
кул газал Т — температура, к — константа Больцмана.
Изменение концентрации центров N (т) определяется
соотношением скоростей процессов (2.12)—(2.14):
==W°~\ N ~ akpN 16>
При измельчении твердого тела в мельнице скорость
роста его удельной поверхности со временем постепенно на-
дает, тем не менее при не слишком длительном измельчении
можно считать, что
ds
и'° = пШ = conpt-
(2.17)
После интегрирования уравнения (2.1G) при условии
it’u -- const получено
А (т) = Н’„тр Г1 — ехр /— -Ц1, (2.18)
L \ е /1
где
1
~ “j 1
— i-аАд»
о
пли при т > гр (стационарный режим)
М-ац = »’0Тр =
(2.19)
т. е. стационарная концентрация КЖЦ определяется кон-
куренцией процессов самопроизвольной дезактивации и хи-
мического взаимодействия с окружающей средой.
Для случая взаимодействия газа с поверхностью из (2.14)
и (2.19) следует
и’оато^ 1 1 , 1 [ 1 \
= 7-7 , ПЛИ ------------ =------------[----
1 + атокр wxeM wQ ат0А- ( р j
(2.20)
Измеряя скорость хемосорбции газа в момент механиче-
ской обработки в зависимости от давления газа по уравнению
(2.20), можно определить такие величины, как скорость воз-
никновения активных центров wa и время их жизни т0.
Экспериментальные данные но хемосорбции газов в мо-
мент обработки для кварца и дисульфида молибдена приве-
дены в работах Бутягпна с сотр. 1277, 282]. Ими измерялись
кинетика окисления оксида углерода до диоксида, рост
удельной поверхности, сорбция СО, концентрация ГШЦ.
Экспериментальные данные для скорости реакции обраба-
87
Рис. 26. Зависимость скорости реакции СО -J- О2 -*• СО2 па поверхно-
сти кварца в момент активации от давления окиси углерода (и); то же,
в координатах уравнения (2.20) (б).
мкг-с/мол.
6
тыпались в координатах уравнения (2.20) и представлены на
рис. 26, пз которого видно, что они хорошо укладываются
на прямую линию.
Сопоставляя скорость реакции окисления оксида угле-
рода кислородом, которая определялась по уравнению (2.20)
и оказалась равной (1,8 ± 0,1)-1015 ыол./(с-см2), со ско-
ростью образования поверхностных атомов, которая опреде-
лялась путем измерения удельной поверхности и равна (2 ±
± 0,2)-101ь ат./(с-см2), авторы пришли к выводу, что реак-
ция идет на КЖЦ, образующихся при измельчении кварца.
Полученная скорость возникновения ПМЦ и кварце в этих
же условиях в 2 раза меньше скорости реакции. Время жизни
активных центров равно 10“4 с.
Аналогичные данные получены для дисульфида молиб-
дена 12821. Известно, что при трении сульфидов в атмосфере
кислорода интенсивно идет окисление. Первой стадией окис-
ления сульфидов является хемосорбция кислорода на свеже
образованной поверхности сульфида. Проводя опыты по сори
ции кислорода в момент измельчении н вибрационной мель
нице, авторы установили, что сорбция идет по закону Лаш
мюре. Концентрация хемосорбированных атомов кислорода
равна (8 ± 1)-1014 см®, что близко к числу поверхностных
атомов сульфида. Предельное значение сорбции пе зависит
от температуры. При постоянном давлении скорость ее дли-
тельное время сохраняется постоянной. При 200 К поглоще-
ние необратимо, при 300 К заметна десорбция продуктов
окисления.
Экспериментальные данные по скорости сорбции и об-
работка их в координатах уравнения (2.20) приведены па
Рис. 27. Зависимость скорости поглощения 0а от давления при акти-
вации MoS2 (о); то мео, и координатах уравнения (2.20) (6).
'Гем)11т>,|тура, К: / —21Н>: 2— JOB.
риг. 27. Из этих данных найдено аги — (1-4-5)-10"7 с; если
а = 1 (предельный случай), то время жизни активного цент-
ра то же. Скорость образования активных центров <г(, —
1-I016” г"1 с"1.
Из полученных данных авторы делают выводы, что хе-
мосорбцию кислорода во время механической обработки мож-
но рассматривать как хемосорбцию кислорода на коротко-
живущих активных центрах, концентрация которых близка
к концентрации поверхностных атомов.
Прямое определение времени жизни К?КЦ было проведе-
но путем измерения характерного времени спада интенсив-
ности отдельных импульсов при разрушении кварца Бутя-
1ЛПЫМ и Стрелецким [280] и для кварца оказалось равным
10”2 с. Кроме кварца парамагнитные центры зарегистриро-
ваны методом ЭПР для большой группы веществ с ковалент-
ной связью: кремния, германия, графита и др. [283—289].
Для них теория КЖЦ достаточно хорошо объясняет наблю-
даемые экспериментальные данные.
Установлено, что поверхностные активные состояния воз-
никают пе только для ковалентпых кристаллов, но и для ион-
ных. Так, например, диспергирование оксида магния, имею-
щего температуру плавления 2900 К, сопровождается обра-
зованием химически активных центров, количество которых
составляет до 1 -10й см-2 в реакции с Н2 и 3—5-Ю11 см”3
в реакции с СО [35]. Исследования показывают, что в MgO
образуются F-центры, являющиеся анионными вакансиями,
захватившими по одному электрону [290]. Интенсивность
сигнала ЭПР от F-цеитров прямо пропорциональна эффек-
89
Рис. 28. Идеализированная ми-
дель дефектной структуры части-
цы, формирующейся в процессе ме -
химической обработки ворошков
[291J.
1 — поверхностный слой, содержащий
хемосорбированные газы; 2 — поверх-
ностный сильно разупорлдочеппый
слой; з — приповерхностный пласти-
чески деформированный слой, содер-
жащий мпнротреппш^; 4 — слабо ис-
каженное ядро частицы.
тинной поверхности 1291]. Эти результаты в совокупности
с тем фактом, что F-центры легко уничтожались травлением
порошков в слабых растворах кислот п даже промыванием
в воде, свидетельствовали об их локализации в поверхност-
ных слоях частиц. Последуя такие спектры ЭПР примесного
иона Мп2+ в кристаллах MgO, Власова и Каказей 1291] сде-
лали вывод, что дефектную структуру частиц порошка MgO
можно представить идеализированной моделью (рис. 28).
Она состоит из очень топкого чисто поверхностного слоя,
содержащего 80% F-цептрон, сильно разупоридочопиого по-
верхностного слоя, пластически деформированного припо-
верхностного слоя, в котором сосредоточены мелкие трещи-
ны, и слабо искаженного ядра частицы.
Берестецкая и Бутягпп [292] объясняют образование хи-
мически активной разупорядочениоп поверхности MgO тем,
что число соседей у каждого попа становится меньше, а меж-
атомные расстояния больше, чем в регулярной решетке.
В таких разупорядочеипых областях ослаблено электро-
статическое взаимодействие, обеспечивающее устойчивость
попов. Потеря устойчивости приводит к потере электрона,
т. е. к превращению нона О2- в химически активный ион —
радикал О".
В исходном кристалле MgO ширина запрещенной зоны
близка к 8 эВ. Для разупорядоченного кристалла край по-
лосы поглощения на спектре диффузионного отражения сме-
щается па 3—4 эВ, т. е. энергия ионизации снижается до
4 эВ; происходит электронный переход от аниона к катиону
п возникает активное возбужденное состояние Mg+O~, мета-
стабилытое при комнатной температуре. В том случае, когда
под влиянием механической активации происходит обра-
90
зовяппо кислородной вакансии, электрон переходит на ва-
кансию п образуется F-цептр.
Аналогичные эффекты обнаруживаются н нрн рааупоря-
доченпи других оксидов [293].
2.4. ЭЛЕКТРОННЫЕ ВОЗБУЖДЕНИЯ
НРН РАЗРУШЕНИИ КРИСТАЛЛОВ
Разрушение и пластическая деформация сопровождаются
рядом неравновесных процессов: эмиссией электронов
н ионов, триболюминесценцией, различного рода излуче-’
пнями и т. д. В основе всех механоэмнссионных явлений ле-
жат возникновение, взаимодействие и рассеивание электри-
ческих зарядов, и поэтому одним из основных направлений
исследований в этой области является расшифровка меха-
низмов ионизации вещества и создания электрических во-
лей. В литературе обсуждается несколько механизмов: тер-
мо- 11 автоионнзацпонпып, разделения двойного электрпче-i
ского слоя, поляризационный, дислокационный и др. [35].
Термо- и автоионизацпоиный механизм реализуется в ос-
новном для ионных и ковалентных кристаллов. Он заклю-
чается в захвате электронов п.чн дырок па донорные и акцеп-
торные электронные уровни, локализованные па структур-
ных дефектах.
Существование легко ионизуемых дефектов подтверждает-
ся частичной ионизацией их во время механической обработ-
ки твердых тел, а также аномально большим радиационным
выходом при действии ионизирующего излучения на меха-
нически разупорядоченные кристаллы.
В том случае, когда в состав ионных солей входят слож-
ные многоатомные анноны, разупорядоченпе структуры
приводит к увеличению склонности их к разложению. Дейст-
вительно, такие анионы, как, например, N07, С20з~, .\<Г
п др., после потери электронов переходят в нейтральную
пли однозарядную форму: как правило, такое состояние не-
устойчиво и потеря электрона инициирует дальнейший рас-
пад аннона на отдельные фрагменты. D результате процессы
разложения солен могут происходить по другому механизму,
отличному от обычного термического, что наблюдалось,
например, Хайретдпновым, Галицыным, Постом [294] для
оксалата серебра, который может разлагаться ио двум па-
раллельным стадиям:
f 2Ag I 2С.О.»
Ag2C2O4 ч
Ag2O + СО + СО.2
91
Под влиянием механической активации разложение окса-
лата серебра пошло ио второй реакции.
Таким образом, усиление донорных свойств анионов
под влиянием разупорядочения, возникающего при меха-
нической активации, следует рассматривать как один из
важнейших факторов в процессах мехапохимпчоского раз
ложепия неорганических соединений.
По Дерягину и др. [37 ], возникновение электрического
слоя обусловлено разделением двойного электрического слоя.
На границе соприкосновения двух разнородных по-
верхностей всегда существует двойной электрический слой,
и с этой точки зрения нарушение адгезионного контакта
идентично разделению обкладок конденсатора. Разряды,
возникающие при отрыве полимерной пленки от подложки,
а также при расщеплении пластинок слюды, являются при-
мерами электризации по этому механизму.
Известен также поляризационный механизм электриза-
ции [295]. Электрические ноля в этом случае возникают
благодаря ориентации полярных молекул при развитии де-
формаций. Аналогичный эффект достигается п при относи-
тельном смещении противоположно заряженных ионов. По-
ляризационный механизм играет основную роль в формиро-
вании электретов в полимерах.
В 1976 г. Молоцким [2961 был предложен дислокацион-
ный механизм электризации, в котором появление избыточ
ного заряда связывается с направленным перемещением за-
ряженных дислокаций. Дислокации в кристаллах электри-
чески заряжены, заряд их компенсируется облаком зарядов
противоположного знака, локализованным на точечных
дефектах. При деформировании и разрушении дислокации
выпосят свой заряд на поверхность. При несимметричном
расколе части кристалла деформируются по-разному, и в
устье растущей трещины возникает составляющая движения
дислокаций, направленная в сторону большей части крис-
талла: вместе с дислокациями привносится избыточный за-
ряд. В щелочно-галогенпдных кристаллах заряд достигает
10ь—107 ед. СГСЕ/м2.
Поверхностная плотность заряда, переносимая дислока-
циями в соответствии с теоретическими расчетами Молоцкого
[296], может быть выражена формулой
»-2')
где Ато — плотность подвижных дислокаций; Ь — величина
вектора Бюргерса; q — линейная плотность заряда дисло-
к.тцпп; В — вязкость кристалла, определяющая торможение
подвижных дислокаций; 1\ — предел текучести; сс — ско-
рость трещины; кг — коэффициент интенсивности напряже-
ний. Если плотность энергии разрушения равна с, то в ма-
i
черпале с модулем Юнга Е kj а функция
л
q (h., kA = (4-Y2 С -4---(2.22)
' 1 2' 2 ) Лз/2у> V —)
где h = ht 4- h2 зависит только от соотношения толщин от-
калываемых частей.
Наблюдается соответствие рассчитанных значений Q
экспериментальным. Так, при разрушении LiF, в котором
q ~ 10"4 ед. СГСЕ/см3, Ъ = 0,2 81 им, В = 0,25 мПз, Рт ~
~ 10® днн/см2, Е = 8,7 -10й дип/см2, h} — 0,Зй, v — 2,8X
Х104 см/с, Лт — 10® см“3, н наименьшем значении энергии
разрушения сел; 10® эрг/см2 теоретическое значение плот-
ности заряда равно 200 ед. СГСЕ/см2, а экспериментально
наблюдаемые значения находятся в интервале 500—100 ед.
СГСЕ/см2 [2971.
Заряд трещины, возникающей вблизи ее вершины, быстро
релаксирует в процессе скола. В то время как исходное зна-
чение плотности равно 103 ед. СГСЕ/см2, ее конечное значе-
ние не превышает 5—50 ед. СГСЕ/см2.
Как правило, возникновение электрических полей при
деформировании или при разрушении сопровождается его
рассеиванием. Основной путь рассеивания заряда на возду-
хе пли в атмосфере какого-либо газа — эго электрический
пробои, т. е. газовый разряд с сопровождающим его харак-
терным свечением, акустическими эффектами и т. д. В ва
кууме заряды могут рассеиваться несколькими путями.
Электроны могут стабилизироваться в структурных ловуш-
ках, выделяясь затем с возбуждением эмиссии пли рекомби-
национной люминесценции. В другом случае, когда кон-
центрация зарядов велика, а электроны не столь прочно
связаны с ловушками, основную роль в релаксации зарядов
играет эмиссия электронов, вызванная их отталкиванием
друг от друга. При локальной плотности зарядов 10® ед.
СГСЕ/см2 энергия эмитирующих ьтектропов может дости-
гать 101-юл кзВ [298].
Молоцкпм (299] предложен радиационный механизм рас-
сеивания зарядов для объяснения механоэмпссиопных эф-
фектов при раскалывании щелочно-галоидны.х кристаллов.
Заряженные стенки раст^ щей трещины представляют собой
' 93
быстро расходящийся конденсатор, в поле которого ускоря-
ются вылетающие с поверхности электроны и ионы. Уско-
ренные частицы бомбардируют противоположную стейку
трещины, возбуждая экситоны. Часть экситонов распадается
внутри кристалла по безызлучательному механизму с обра-
зованием дефекта в анионной подрешетке или путем пере-
дачи энергии к катиону, что приводит к выбиванию его в меж-
доузлие, другая часть экситонов распадается после выхода
на поверхность с образованием свободных катионов. Такие
катионы, ускоренные в поле трещины, бомбардируют отри-
цательно заряженную стенку и вызывают вторичную ионно-
электронную эмиссию, после чего цепочка процессов, вклю-
чающая электронную, экситонную и ионную стадии, повто-
ряется. Реализуется такой механизм в основном в условиях
вакуума. В присутствии газовой среды эти процессы проте-
кают с меньшей эффективностью.
Безызлучательный распад экситона может приводить
к созданию высоковозбуждепных колебательных состояний,
способных вызвать механохимическую диссоциацию ионных
солей. Экситон в ионных кристаллах представляет собой
Рц-центр, вблизи которого локализован электрон. При рас-
паде экситона образуется пара из F- и Н-центра (междоузель-
ные атомы из анионной подрешетки). Энергия возбужденного
состояния экситона по данным люминесценции составляет
5—8 эВ, а именно эта энергия передается колебательным со-
стояниям анионной подрешетки вблизи распадающегося
экситона. Такое колебательное возбуждение на четыре-пять
порядков выше термического возбуждения при разрушении
ионных кристаллов и может внести вклад в механохимиче-
скую диссоциацию.
В пользу экситонного механизма говорит ряд факторов,
из которых наиболее ярким является нестехиометричиость
разложения при разрушении большинства кристаллов,
аналогичная нестехиометрпчностп при их электронном
распылении [300, 3011. Так, например, наблюдается пре-
имущественный выход атома галоида из щелочных галогени-
дов, который можно объяснить локализацией в анионной под-
решетке колебательного возбуждения, вызванного электро-
ном, возникающим при расколе кристалла. Наблюдается
корреляция и в значениях нестехиометричпостн. Она велика
при электронном облучении LiF п значительно меньше в слу-
чае КС1. При расколе этих кристаллов возникает аналогич-
ная картина: выход атомов Li меньше, чем фтора, а выход
К и С1 примерно одинаков.
и4
На возможность мехаиохпмической диссоциации ГЦГК
по экситопному механизму первыми указали Гофман и Лин-
ке [302]. Однако до появления работ Молоцкого [296. 299]
неясен был механизм образования самих эксптопов. Отме-
тим, что роль электронного фактора в механолнзе впервые
обнаружена в 1969 г. 1303] при исследовании механохнмп-
ческого разложения нитратов щелочных металлов в присутст-
вии каталитических добавок.
В заключение отметим, что для установления каналов
усвоения механической энергии и соответственно механиз-
мов, по которым протекает процесс, крайне важна характе-
ристика выхода в зависимости от вида и интенсивности меха-
нических воздействий, скорости разрушения, характера на-
пряженного состояния, мощности аппарата и т. д. Таких
данных в литературе очень мало.
Сутягин [304] предложил характеризовать мехапохпмп-
ческпе процессы энергетическим выходом gM, равным числу
молей образовавшихся пли прореагировавших частпц при
затрате 1 МДж механической работы. Приводим значения
энергетических выходов, рассчитанные им для некоторых
механохпмических процессов [305 ]:
Процесс
моль/МДж
1 Образование свободных радикалов в
кварце и полимерах нри измельчении
2. Разложение неорганн"ескпх твердых
TejiAgsC,Oj, NaN03, СаСО-. GNH1kCr,O,
3. Генерация поверхностных атомов СаСОя.
Si О.,, BN
1. Хемосорбция СО на поверхности MgO,
хемосорбция Н.; на поверхности кварца
и графпта
5. Реакция между твердыми телами Ва( О,
+ W03 = BaWO, СО2
6. Лморфпзация кварца
7. Полиморфные переходы в оксидах свинка
и ниобия
1(1-
10--10-
10-
5.10-
10-
2-3
10г
Видно, что энергетические выходы механохпмических
процессов лежат в очень широком интервале — от 10"3 до
10s, что обусловлено различием в механизмах инициирова-
ния механохпмических реакций. Так, образование свобод-
ных радикалов относится к энергетическим процессам с ма-
лым выходом. Для реакций разложения неорганических ве-
ществ выход может достигать 10“’—IO"3 моль/МДж. Выход
порядка 10“1 моль/МДж характерен для различных реакций
95
газов с механически разупорядоченной поверхностью. Наи-
большие выходы характерны для твердофазных реакций,
аморфизации кварца и полиморфных переходов.
Поскольку различно в энергетических выходах свиде-
тельствует о различных механизмах активации, такие данные
могут служить начальной точкой для последующих, более,
детальных исследований механохпмпческих процессов.
--------------------Г Л А В А 3-------------------
ЗАКОНОМЕРНОСТИ СТРУКТУРНЫХ
II ХИМИЧЕСКИХ ИЗМЕНЕНИЙ В ТВЕРДЫХ ТЕЛАХ,
ПРОИСХОДЯЩИХ ПОД ВЛИЯНИЕМ
МЕХАНИЧЕСКОЙ АКТИВАЦИИ
3.1. ТЕРМОДИНАМИЧЕСКАЯ ХАРАКТЕРИСТИКА
АКТИВИРОВАННЫХ ТВЕРДЫХ ТЕЛ
Изменение реакционной способности твердых тел под
влиянием механической активации может быть следствием
изменения размера частиц, структуры, а также состава твер-
дого тела (например, в результате механохнмического раз-
ложения). Эти явления в какой-то степени взаимосвязаны:
переход в новую фазу может быть результатом уменьшения
частиц или изменения состава, фазовый прреход может при-
вести к диспергированию и т. д.
Общее изменение свободной энергии активированного ве-
щества может быть оценено путем измерения равновесия
химической реакции, в которую вступает активированное
вещество с другим веществом известной активности, напри-
мер газом, ио уравнению ,
ДС = - ПТ In (3.1)
Ai
где к} и к2 — константы скоростей прямой и обратной реак-
ции. Примером может служить реакция, исследованная виер-
вые Миташем [306 ]:
Ni + 4СО Ni(CO)4. (3.2)
Положение равновесия реакции ио мерс увеличения ак-
тивности никеля сдвигается вправо.
96
Изменение размера частиц приводит к изменению свобод-
ной энергии в соответствии с уравнением Томсона
ЛС_2пГ(Д_А). (3.3)
где ДС — изменение свободной энергии, и г2 — радиусы
мелкой и крупной частиц соответственно, Г — мольный
объем, и — поверхностное натяжение. Уравнение (3.3) мож-
но использовать, если известны размер частиц и поверхност-
ная энергия о. Существующие в настоящее время физиче-
ские методы, в частности электронная микроскопия, адсорб-
ция газов и др., позволяют определять размер частиц с боль-
шой точностью. Однако методы определения величины и
для твердых тел весьма приближенны. Экспериментальные
методы дают значения, различающиеся на Два-три порядка.
Теоретические расчеты, исходящие из энергии связи ато-
мов в кристаллах, также являются приближенными [307].
Свободная энергия связана с растворимостью, упру-
гостью пара, температурой и теплотой фазовых переходов
соотношениями
ДО =/77 In А ДС = /?7’1п—; ДС = -^, (3.4)
7'<» Гф.и
где сто и рос — равновесная растворимость вещества и упру-
гость пара; сир — эти же величины при заданном значении
г; X — скрытая теплота фазового перехода; Тф.п — темпера-
тура фазового перехода. В соответствии с уравнением (3.4)
изменяются кривые фазовых равновесий в системах
твердое — пар, твердое — жидкость, а также температура
плавления с уменьшением размера частиц. Эксперименталь-
ные определения возрастания теплового эффекта растворения
в зависимости от размера частиц приведены, например, в ра-
боте [307 ] для некоторых щелочных галогеппдов и оксидов.
По этим данным, возрастание энтальпии вещества при умень-
шении размера частиц до долей микрона составляет для ще-
лочных галогенидов 1—4 кДж/моль, а для оксидов 4 —
12 кДж/моль.
Очень мелкие частицы за счет действия поверхностных
сил подвергаются сжатию. В соответствии с формулой Лап-
ласа вблизи поверхности, имеющей некоторую кривизну,
создастся избыточное давление
p="(^ + i)- (35)
где //j и 7?2 — радиусы кривизны поверхности в дайной гоч-
4 Е. Г. Аииакумои
97
кс в двух взаимно перпендикулярных плоскостях. Для
ультрадисперспых частиц размером 1—10 им сжимающее
давление по уравнению равно 103—104 МПа, что может вызы-
вать изменение объема. Так, переходные металлы, имеющие
большую поверхностную энергию (ниобий, тантал, молиб-
ден, вольфрам), в массивных образцах образуют ОЦК-ре-
шетку, а в тонких пленках вследствие указанного эффекта
переходят в более плотно упакованные структуры (ГЦК
и ГПУ) с меньшей поверхностной энергией [308].
Относительное изменение параметра решетки Дя/я изо-
тропной сферической частицы диаметром D и сжимаемостью
Р связано соотношением [309]
Да 4 6а р.
- = ~tV М
Эта формула с определенным приближением справедлива
и для кубических частиц. Расчетные значения о в ряде слу-
чаев расходятся со значениями, полученными ио формуле
(3.6) с учетом kata, определенного методом дифракции рент-
геновских лучей. Не исключено, что влияют линейные и то-
чечные дефекты, содержащиеся внутри кристаллов. Однако
для малых частиц ионных солей (LiF, NaCl, NaBr, КС1),
полученных в вакууме, наблюдается обратно пропорцио-
нальная зависимость между Ьа!а и D [310].
Как следует из монографии Петрова [308], в которой дан
анализ состояния вопроса о влиянии размеров частиц па
физические свойства веществ, ощутимые изменепия пара-
метра Ьа!а, температур плавления и др. начинаются у частиц,
размер которых меньше 5 пм.
Одним из путей усвоения механической энергии, под-
водимой к твердому телу, является возникновение внешних
и внутренних поверхностей раздела, дислокаций, вакансий
и междоузельных атомов. Возникающие при упругом де-
формировании состояния кристалла с распределенными
в объеме дислокациями и дефектами, в которых сосредото-
чена избыточная энергия, термодинамически более выгод-
ны, чем состояния с равномерным распределением упругих
напряжений по всем межатомным связям. Стабилизирую-
щим фактором и в этом случае является вклад Конфигура-
ционной энтропии Sk в свободную энергию кристалла. При-
ращение свободной энергии при возникновении дефектов со-
ставит в этом случае
\G - Gn - Gn -= -кТ In [V„. (3.7)
Здесь Go — свободная энергия упруго деформированного
кристалла, Gn — то же для кристалла, и котором вся энергия
деформации сосредоточена и дефектах структуры, 1ИД —
термодинамическая вероятность, равная числу способов,
которыми эти дефекты можно разместить в решетке [351.
Таким образом, стремление сконцентрировать избыточ-
ную энергию в структурных дефектах характерно для крис-
таллического состояния вещества. Именно с этой способ-
ностью твердых тел и связана в значительной мере природа
механохимпческих превращений. Гутманом [39] для ха-
рактеристики твердых тел, находящихся под механическим
напряжением, введено понятие мехапохимпческой актив-
ности. Он исходил из общих положений термодинамики п
уравнения состояния твердых тел, согласно которым
а химический потенциал вещества
п
= щ = + (3.9)
где П[ — число молей, р.® — химический потенциал в стан-
дартном состоянии, at — активность. Из этих уравнений
следует
Лр;= ( V(p)dp. (3.10)
pi
Известно, что вследствие невысокой сжимаемости твер-
дых тел объем можно выразить из уравнения состояния кон-
денсированных фаз следующим образом:
V = Voe-w (3.11)
где х — коэффициент сжимаемости. Но поскольку для
твердых тел х « 10-0, величина хр <С 1, и вследствие этого
можно принять V = Уо. Тогда из (3.10) следует
Р< = ЮЛр=о + Д^’ <ЗЛ2>
где V — мольный объем вещества, Др — избыточное давле-
ние, которое в случае твердого тела представляет собой аб-
солютную величину шаровой части тензора напряжений и
не зависит от их знака.
Значение П<др-_0 соответствует значению химического
потенциала при отсутствии механического воздействия, от-
99
сюда
М1Др_0-+ &pV = Hi + КТ In«i + &pV = fi? F KT Ina. (3.13)
Величина
7= a exp (3.14)
является мехаиохпмической активностью вещества.
Обычно искажения структуры пластически деформирован-
ных материалов оценивают по увеличению ширины рентге-
новских интерференционных линий, определяя таким обра-
зом относительную величину мнкронскажений кристалли-
ческой решетки (Да/a) и размеры первичных кристаллов (d).
Однако из этих величин нельзя выделить вклад плоских
скоплений дислокаций, играющих роль в формировании ме-
хапохпмпческой активности материалов вследствие локаль-
ной концентрации напряжений. Вместе с тем, когда отсутст-
вуют компланарные скопления и имеет место хаотическое
распределение дислокаций, а статически усредненная ве-
личина Да/a пропорциональна числу элементарных лекаже-
А 771 До
пии решетки, можно принять, что ЕрязЕ--
С учетом этого на основании уравнения (3.14) и закона
действия масс может быть рассчитан коэффициент ускоре-
ния процессов (например, растворения) в зависимости от сте-
пени деформпрованпостп материала:
. “2 [(ДД2 — ДР1)И | Е ~ /о л г,
/г = = ехрр -ду' ] « exp)• (3.15)
Гутманом и Абдуллиным [39] для механически активи-
рованного карбоната кальция теоретически рассчитанное
значение к было сопоставлено со значением, полученным
из данных по кинетике растворения, и найдено, что вычис-
ленное значение к = 2,25 близко к измеренному, равно-
му 2,8.
3.2. МЕХАНИЧЕСКИ СТИМУЛИРОВАННЫЕ
ФАЗОВЫЕ ПЕРЕХОДЫ
Известно, что при механических воздействиях на вещест-
во имеют место фазовые переходы, в результате которых,
как правило, возникают фазы, имеющие более плотную упа-
ковку [312—343].
В соответствии с термодинамикой система, поддержива-
емая при постоянных давлении п температуре, стремится
к минимуму свободной энергии. Если дли двух фаз (7Л >
> GB, то фаза А должна превратиться в фазу В. Зависи-
мость между давлением, температурой п объемами фаз для
перехода одной фазы в другую определяется соотношением
Клайперона — Клаузиуса:
tip *''в *^д
>в-Кл’
(3.16)
где N — энтропия фазы, V — мольный объем. Из этого урав-
нения следует, что возможны различные варианты границ
фазового перехода в одиокомпоиентных системах. Если
УА > Ув, то dpIdT отрицательно (с увеличением темпера-
туры давление, при котором А переходит в В, падает). Если
УЛ < Ув, то dpIdT положительно (с увеличением темпера-
туры давление, при котором Л переходит в В, растет).
Скорость полиморфных превращений зависит от высоты
энергетического барьера, который разделяет две формы.
Для того чтобы попять происходящие при переходах изме-
нения, необходимо знать структуры исходных и конечных
форм и сделать разумное предположение о возможном меха-
низме превращения.
Вюргер |311) предложил классификацию структурных
изменений в твердых телах па основе крнсталлохпмнческих
признаков, учитывающих соотношения между начальной
и конечной структурами, и оценил относительные скорости
этих превращений. Оп выделил четыре основные группы,
которые могут быть охарактеризованы следующим образом.
1. Превращения в первой координационной сфере.
При этом изменяется число ближайших соседей, т. е. коор-
динационное число. Расположение ближайших соседей пол-
ностью нарушается и создается новый тип решетки с изме-
ненными условиями координации. Здесь различают дефор-
мационные и реконструктивные превращения. К первой
группе относится, например, превращение при нагреве до
718 К решетки CsCl (координационное число 8) в решетку
типа NaCl (координационное число 6) благодаря вытягиванию
решетки в направлении (111) (рис. 29). При реконструктив-
ном изменении, например при превращении арагонит—>
кальцит (координационные числа соответственно 9 и 6), об-
разуются различные промежуточные координации, которые
требуют определенных энергетических затрат. Поэтому де-
формационные превращения протекаю г, как правило, быст-
ро, а реконструктивные — медленно.
101
Рис. 29. Схема измене-
ния структуры при де-
формационном превра-
щении (переход структу-
ры CsCl (а) в структур}’
NaCl (б)) 1311].
2. Превращения во второй координационной сфере. Час-
то при полиморфных превращениях число ближайших со-
седей сохраняется, а изменяется только число дальних.
Прп этом также могут быть превращения со смещением
и реконструктивные. Схема превращений во второй коор-
динационной сфере как для того, так и для другого случая
представлена па рис. 30. Различные структуры имеют и ка-
честве общего конструктивного элемента октаэдр, в углах
которого находятся анионы, а в центре — катион. Коорди-
национное число 6 сохраняется во всех трех структурах,
в то время как расположение дальних соседей меняется.
В первом случае происходит только сдвиг, без разрыва свя-
зей, а во втором — новый координационный полиэдр может
возникнуть только при условии, если предварительно рвут-
ся связи между ближайшими соседними атомами. Так как
большая часть энергии решетки обусловлена силами связи
между ближайшими соседними частицами, то в первом слу-
чае различие в энергиях обеих модификаций незначительно,
переход совершается быстро. Во втором случае из-за на-
личия потенциального барьера переход происходит медлен-
нее, чем в первом.
3. Превращения с разупорядочением (переход порядок —
беспорядок). В этом случае превращения могут происходить
вследствие вращения определенных атомных группировок или
статического распределения атомов по эквивалентным местам
решетки. Превращения первого типа протекают очень быстро,
а последнего — медленно. Примером может служить пре-
вращение в твердом метане, происходящее вследствие вра-
щения молекулы СН4. Возникновение сверхструктур в спла-
вах, например в сплаве СпАи, при температуре выше 681 К
может служить примером второго типа.
4. Превращения с изменением типа связи. Примером та-
ких превращений является переход графита в алмаз, серого
олова в белое и т. п. Эти переходы совершаются с большими
затратами энергии и потому протекают медленно.
102
Рис. 30. Изменения структуры при превращениях во вто-
рой координационной сфере [311].
а — исходная структура; и — после превращения со смещением;
в — после реконструктивного превращения.
По типам процессов роста новой фазы кинетика полиморф-
ных превращений подразделяется на мартенситную, связан-
ную с координированным коллективным смещением атомов,
н нормальную, связанную с образованием зародышей и по-
следующим переходом одиночных атомов через межфазную
границу. Хотя в принципе полиморфные переходы не тре-
буют для своего развития переноса вещества на расстоянии
существенно больше межатомных, считается, что если такие
превращения совершаются путем переноса одиночных ато-
мов, то их следует относить к категории диффузионных.
Причины, по которым совершаются механически стиму-
лированные фазовые переходы, могут быть различными.
Прежде всего следует, по-видимому, отметить возникно-
вение высоких локальных давлений и температур на кон-
тактирующих участках механически активируемых частиц,
т. е. реализацию термодинамически необходимых для фазо-
вого перехода параметров. Подтверждением могут служить
данные табл. 6, из которых видно, что переходы, реализую-
щиеся при высоких статических давлениях и температурах,
реализуются и под действием обработки в измельчительных
аппаратах.
Другой причиной могут быть размерные эффекты. Как
указывалось выше, условия фазового равновесия с учетом
размеров частиц определяются значением свободной энергии:
G = GU + (3.17)
Шрадером и Гоффманом [108] показано, что под влиянием
механической активации кальцит переходит в арагонит, но
может совершаться п обратный переход. При большой дли-
тельности активации между ними устанавливается равновес-
ное состояние. Свободная энергия перехода кальцита в ара-<
гонит составляет —0,742 кДж/моль. Стрид [344] показал,
ЮЗ
Т а б л и ц a G
Механически стимулированные фазовые переходы в твердых
телах
Вещество Фаза A—»фаза D Плотность, г/с№ Направление пере- хода ио сравнению с термическим н барическим* Литера- тура
исход- ная конеч- ная
т, к j>, МПА
С. Гексагональная-* ромбическая Ч-, 1300 1312]
Р Со Трпклинпзя-* ‘ ромбическая Кубическая-^ гексагональная 2,4 8.79 2,69 88i о о £2 о 00 CD 1 1 1313] 1314]
РЬО Terpai опальная-*- орторомбпчс- 9,70 9,27 750—863 300 120,
1-е2О;1 скал Тетрагонал ып я-* ромбнческая 5.07 5,29 (0 1 МПа) (546 К) 315- 320] (321, 322]
Еи2Оа Sb..O3 КубН’КЧ'КПЯ-»- мопоклпиная Кубическая-* орторомбмче- 5,19 5,75 10 |323] 13271
TiO2 скал Тетрагонал ьняя-»- орюримбпче- 3,90 4,12 ->-, 1073 — ]324, 325]
ZK).. ска я Орторомбиче- ская -*- тетраго- нальная Млюклннная-*- тетрагэнальиая 4,12 5,74 4,25 5,86 1313 1473 — 1326]
РЬО,, Тетрагональная-* орторомбнче- 9,33 9,67 — -ь, 1100 |327, 329]
NlbO., скал Монок'нпныя— гексагональная 4,55 4,99 -г, 1273 1700 1330)
ZnS Гексагона л ьная-*- кубичсская 3,48 4,09 —, 1293 — (331, 334]
CdS Г ексагона л ьная-> кубпческая 4,83 4,83 800 — (335 ]
Agl Гексагонал ьная-> кубическая 5,68 5,68 408 — 1336]
CaCO3 Тригональная-* ромбическая 2,71 2,93 — — 1337, ЗЮ]
Гексагонал ьная-»- ро.мбическая — — — —
BaTiO3 Тетра гопал ышя-* кvonческая — — 393 — |336, 341]
Направление: -» совпадает» противоположное.
что при размерах частиц кальцита и арагонита, равных
10-’ м (100 им), и поверхностном натяжении кальцита, рав-
ном 1 Дж/м2,
ДС = (GoA - GoB) + = - 0,6^. (3.1S)
\ ГА 'В / МОЛЬ
Видно, что эти величины близки друг другу. Таким образом,
переход кальцита в арагонит н обратно термодинамически
возможен при температуре 298 К и очень малых размерах
частиц.
Наряду с размерными эффектами следует учитывать и за-
пасенную твердым телом упруго пластическую энергию, т. е;
G = G0 + y+GI(. (3.19)
С учетом этой энергии равновесие на диаграмме р—Т опре-
деляется уже не липнем, а широкой двухфазной областью.
Этим может быть объяснена наблюдаемая рядом авторов
обратимость механически стимулированных фазовых пере
ходов, например для РЬО [64] и других соединении.
Особенность механически стимулированных переходов
заключается в том, что в результате перестройки решетки
возникают более плотные фазы (см. табл. б). Повышенно же
температуры стимулирует обычно переход из более плотной
структуры в менее плотную, т. е. по направленности меха-
нически стимулированные переходы противоположны дейст-
вию высоких температур.
Иногда фазы, возникающие при диспергировании, ана-
логичны фазам высокого давления, в ряде же случаев ста-
тическим сжатием их получить не удается. Необходимо,
чтобы вмела место сдвиговая деформация. Статическая ком-
понента, при условии, что она достаточно большая, пере-
водит материал в область высокоплотной фазы, сдвиговая
же компонента вызывает прямое, сдвигом индуцированное
превращение через всевозможные когерентные и некогереьт-
пые переходы.
Дахплл и Рой [327] провели ряд исследований, целью
которых являлось отделение влияния .гидростатического
давления на превращения в твердой фазе от влияния напря-
жений сдвига. Исследовалось полиморфное превращение
Р-РЬО2 -+• сс-РЬО2. Результаты этой работы однозначно сви-
детельствуют о том, что наложение напряжений сдвига не
влияет заметным образом на условия термодинамическою
равновесия, во ускоряет протекание превращений. Приме-
няя напряжение сдвига, удается проводить процессы с за-
105
Рис. 31. Зависимость степени превращения ВЫ (грапоцеитрнровап-
ная -* объемно центрированная) от давления для различных скоро-
стей нагружения |345], МИа/с.
1 — И; г — 7,2; 3 — 6,8; 4 — 1,8; 5 — 0,57; 6 — 0,58; 7 — 0,76.
метнымп скоростями при температурах на 100—300 К более
низких, чем необходимо для осуществления этих процессов
в «статических условиях», т. е. в отсутствие сдвига. Так, на-
пример, ход превращения р-РЬО2 а-РЬО2 нрн 208 К
и 4 МПа со сдвигом аналогичен процессу при 473 К и том же
давлении, но без сдвига.
Следует отметить, что при равенстве термодинамических
параметров (Т и р) скорость фазового перехода зависит от
скорости ^нагружения. Это можно видеть из данных, при-
веденных в работе [345], для фазового перехода в Rbl, из
ГЦК в ОЦК структуру, в зависимости от скорости нагруже-
ния (рис. 31). Видно, что при больших скоростях нагруже-
ния кинетическая зависимость имеет другой характер. Авто-
ры объясняют указанные особенности различным механиз-
мом протекания процесса: при больших скоростях — мар-
тенситный, при малых — через образование и рост заро-
дышей.
Многочисленные деформации идеальной структуры, в ре-
зультате которых происходит направленное смещение атомов
решетки, вызывают фазовые переходы по различного рода
сдвиговым механизмам. В качестве примера можно привести
превращение а-графнта в 0-графпт, при котором плотпоупа-
кованная гексагональная упаковка ...z\13AB... превращает-
ся в кубическую плотноупакованную ...АВСАВС... путем
смещения промен;уточных слоев [346]. Аналогичным обра-
зом протекает фазовый переход в сульфидах цинка и кад-
мия (переход вюртцит — сфалерит) [333] и др.
Механически активируемые фазовые переходы не требуют
больших затрат механической энергии ввиду структурного
соответствия полиморфных модификаций. Однако образо-
вавшаяся новая фаза может существенно отличаться по ре-
акционной способности от исходной. В качестве примера
можно привести фазовый переход в РЬО, который исследова-
ли Ходаков и Редькина [316].
Моноксид свинца известен в двух модификациях — мета-
стабильной ромбической (массикот), термодинамически ус-
тойчивой при 7’>761,5 К, и стабильной тетрагональной
(глет), термодинамически устойчивой при Т <761,5 К.
Переход из глета в массикот (тетрагональная ромбическая
модификация) легко осуществляется при повышении темпе-
ратуры, однако обратная перестройка решетки обычно не
осуществляется из-за высокой устойчивости ромбической
фазы. Для перехода массикот —> глет необходимо примене-
ние высоких давлений. По этот же переход был реализован
практически нацело при обработке массикота в вибрацион-
ной мельнице. В зависимости от длительности измельчения
сначала происходит резкое уменьшение содержания масси-
кота и более медленное частичное превращение в тетраго-
нальную форму. Такое соотношение фаз обусловлено тем,
что ромбическая модификация переходит в тетрагональную
через промежуточное аморфное состояние, которое присутст-
вует на всех стадиях измельчения. При дальнейшем измель-
чении содержание ромбической фазы остается постоянным,
а тетрагональной — падает, что свидетельствует о процессе
аморфизации уже новой тетрагональной фазы. Максималь-
ная химическая активность (окисление РЬО до РЬ3О-.) реа-
лизуется при наиболее полном полиморфном превращении
ромбической формы в тетрагональную, нрн этом энергия ак-
тивации процесса окисления понижается примерно в 2 раза,
т. е. активной к окислению является полученная тетраго-
нальная фаза. Обычная же иеактпвпроваппая тетрагональ-
ная окись свинца подобно ромбической (той же дисперсности)
является малоактивной. Активный после измсльчеппя по-
рошок в результате нагревания иолностью теряет свою ак-
107
тпвность ij при повторном термическом процессе практиче-
ски в данных условиях опыта пе окисляется.
Оказалось, что па скорость фазовых переходов оказывают
сильное влияние поверхностно-активные вещества. Но дан-
ным работы [347], фазовый переход ромбическая — тетра-
гональная модификация РЬО может быть ускорен такими
ПАВ, как этиловый спирт (содержание тетрагональной фазы
62%), вода (75%), сульфанол (96%), триэтаноламин (96,2%),
и замедлен стеариновой кислотой (13%), стеаратом цинка
(5,5%), стеаратом кальция (5,4%). Поскольку ускоряют
переход вещества, обладающие донорными свойствами,
а замедляют — акцепторными свойствами, то авторы выска-
зывают мысль, что такое поведение связано с изменениями
в электронной подсистеме оксида. О том, что они влияют па
фазовые переходы, свидетельствуют данные по изменению
скорости перехода из тетрагональной фазы в кубическую
для BaTiO3 при возбуждении электронных переходов под
воздействием света [348].
Механическая активация оказывает влияние и на терми-
чески стимулированные фазовые переходы. Так, например,
изменяется последовательность полиморфных превращений
в кварце [349, 350]. Неактивироваииый кварц претерпевает
следующие превращения: а-кварц (гексагональная снпго-
,К
нпя Р3221, d = 2,65) —> [3-кварц (гексагональная —
Р6022, d = 2,53) [З-трндимнт (гексагональная —
P63/mmc, d = 2,22) l7J3K [3-кристобалит (кубическая —
Frf3m, d = 2,20).
Устойчивость полиморфных модификаций при нагрева-
нии определяется энергией: низкотемпературный кварц имеет
самую низкую симметрию и самую плотную структуру, с по-
вышением температуры симметрия решетки повышается,
а плотность уменьшается.
Переходы а-кварц —[3-кварц, а также [3-кварц -> (3-три-
димит относятся, по классификации Бюргерса, к превраще-
ниям с изменением во второй координационной сфере, про-
исходящим при незначительном смещении атомов без раз-
рыва связи Si—О, а переход [3-тридимит -> [3-кристобалит
относится к реконструктивному типу, т. е. с разрывом связи
Si—О. В механически активированном кварце последователь-
ность превращения при нагревании изменяется и а-кварц
сразу переходит в [3-крпстобалпт при температуре 1470 К, ко-
торая ниже, чем для инактивированного, почти на 300 К.
Экзоэффекты па кривых нагревания, соответствующие двум
108
первым переходам, исчезают, так как и результате разуно-
рядочення структуры претерпели необходимые для превра-
щения смещения атомов решетки.
Такого рода фазовые переходы очень сильно влияют на
кинетику высокотемпературных твердофазных процессов.
'Гак, например, температура получения такого керамиче-
ского материала, как CaTiSiO6, из CaTiO3 и механически
активированного SiO2 снижается до 1790 К, вместо 1900 К,
что существенно упрощает его технологию [350].
3.3. МЕХАНИЧЕСКАЯ АКТИВАЦИЯ
НЕОРГАНИЧЕСКИХ ТВЕРДЫХ ТЕЛ
И ЕЕ ПОСЛЕДСТВИЯ
3.3.1. Оксиды металлов
К настоящему времени исследовано влияние механиче-
ской активации на широкий круг окислов. В частности, ис-
следовались причины изменения каталитической активности
оксидов NiO [351], ZnO [352, 353], Fe2O3 [354, 355], CuO
[355], MgO [360], A12O3 [362] и других [355—359]. По дан-
ным Шрадера [3^>], активность a-Fea03 в точке максимума,
(в зависимости от длительности механической обработки она
проходит через максимум) в реакции окисления оксида угле-
рода до углекислого газа выше активности неактивирован-
ного катализатора в 5 раз. Имеет место и увеличение ката-
литической активности a-Fe2O3 в других реакциях: окисле-
ние SO2 до SO3 [363], сорбции 112S на a-Fe2O3 [354] и др.
Было показано, что изменение химической активности опре-
деляется в основном нарушениями решетки. Установлено
значительное увеличение . химической активности MgO по
отношению к воде после механической активации его в пла-
нетарных мельницах [360]. Механическая активация су-
щественно снижает температуру восстановления оксидов.
По данным [361], после 20 ч обработки в вибромельнице
температура начала восстановления снизилась: a-Fc2O3
на 80 К, Fe3O4 — 75, NiO — 50, CuO — 40 К. »
Особенно много работ выполнено по исследованию меха-
нической активации кварца (см. [28, 50]). Происходящая
при механической активации кварца аморфпзацпя сущест-
венным образом ускоряет процессы его твердофазного вза-
имодействия с-другими веществами [350], увеличивает его
100
растворимость в щелочных и нейтральных растворах ('50],
приводит к появлению у кварца вяжущих свойств [364, 365].
Ходаков [50], Сенна и Шонерт [366, 367] показали, что эф-
фекты механохимической активации являются следствием
пластической деформации кварца. Согласно Штайнике, Мюл-
леру и др. [367], пластическая деформация приводит к на-
рушению дальнего порядка в расположении тетраэдров, в то
время как сам тетраэдр S1O4 как структурная единица остает-
ся неизменным.
Влияние механической активации на свойства диоксида
олова исследовалось Лаудертом с сотр. [369, 370] и Юсу-
повым с сотр. [371, 372]. Было показано, что после длитель-
ной механической обработки в напряженных измельчитель-
ных аппаратах диоксид олова переходит в форму, раствори-
мую в кислотах и щелочах. Авторами [372] проведен анализ
структурных изменений касситерита после механической
активации его в водной и воздушной средах. Определялись
аморфизации касситерита как отношение интенсивности ос-
новных рефлексов исходного и измельченного касситерита,
полуширина рефлексов, а также размер кристаллитов в ве-
личина микроискажений. Установлено, что все структур-
ные характеристики меняются под влиянием измельчения.
Наибольшее изменение наблюдается после измельчения
в воздушной среде по сравнению с диспергированием в воде.
Минерал в рентгеноаморфном состоянии, полученном в ус-
ловиях водного и сухого измельчения, существенно отлича-
ется способностью к растворению. Амортизация, наступив-
шая при измельчении в воздушной среде, приводит к значи-
тельно более активному химическому состоянию касситери-
та. Размер кристаллитов при измельчении в воздухе значи-
тельно меньше, чем в условиях водного измельчения. Умень-
шение размера кристаллитов при сухом диспергировании
свидетельствует о непрерывности процесса измельчения,
хотя удельная поверхность, определяемая газоадсорбцион-
ным методом без дезагрегации частиц, при этом не увеличи-
вается, а в ряде случаев даже уменьшается. Авторы прихо-
дят к выводу, что величина микроискажений является одним
из главных факторов, определяющих превращение относи-
тельно инертного минерала касситерита в растворимую
форму.
Исследованы структурные изменения в механически акти-
вированных оксидных фазах типа корунда па примере а-
А12О3 и cc-Fe2O3 [373]. Показано, что преобладающей плос-
костью скольжения в структуре корунда является базис-
ная плоскость (0001). Скольжение дислокаций происходит
НО
при кооперативном смещении катионных и анионных сеток
с сохранением октаэдрической координации катионов. Рас-
щепление дислокации меняет последовательность чередова-
ния слоев, что соответствует образованию протяженных
(плоских) дефектов упаковки (ДУ). Прп прохождении поло-
винных дислокаций образуются катионные ДУ, четвертич-
ных — катионные и анионные ДУ. Последовательные по*
слойпые смещения анионных слоев за счет четвертичных
дислокаций с соответствующими синхросдвигами промежу-
точных катионов образуют двойниковые ДУ. Изменения
реакционной способности указанных оксидов есть следствие
образования остаточных микропскажений в решетке и ука-
занных дефектов упаковки. Это приводит к ускорению про-
цессов растворения [3741, спекания, твердофазного синтеза
и т. д.
При исследовании влияния механической активации на
свойства сложных оксидов, таких как тнтаномагнетит 1374,
375], хромит железа [355], феррит цинка [377], каолинит
[378—381], нефелин [382], боксит [383], гидраргиллит
[384—386] и др., было установлено увеличение их реакцион-
ной способности. Авторы указанных работ связывают это
увеличение в основном со структурными нарушениями, а не
с поверхностью, измеряемой газоадсорбцпонпым методом,
которое вследствие наличия сильной агрегации не дает ис-
тинного представления о размерах первичных частиц.
Лаптевой, Юсуповым, Бергер [43] показано, что меха-
ническая активация слоистых силикатов приводит к росту
сорбционной емкости по отношению к парам воды и кисло-
роду, повышению реакционной способности в процессах
гидрохимического взаимодействия с растворами неоргани-
ческих кислот и гидроксида натрия, а также к изменению
последовательности и природы фазовых превращений в про-
цессах термолиза активированных продуктов при их нагре-
вании до 1300—1500 К. Так, например, при нагревании као-
линита до 1300 К в продуктах разложения появляются:
из минерала, активированного в водной среде,— оксид алю-
миния -у-А12О3, после активации в воздушной среде — мул-
лит 3Al2O3-2SiO2, термолиз же неактивированного каолини-
та приводит к образованию смеси указанных фаз. Такое
различие обусловлено различными механизмами активации
в водной и воздушной средах. Активация слоистых силика-
тов в водной среде связана преимущественно с диспергиро-
ванием их частиц по направлениям, совпадающим с плоскос-
тями спайности, п разрывом связей, локализованных в ука-
занных плоскостях. Рентгенографически это проявляется
111
в уменьшении интенсивности всех рефлексов и возрастании
уровня фоиа. Однако даже после 30—60 мин на дифракто-
граммах активированных продуктов сохраняются наиболее
интенсивные максимумы, характерные для исходных струк-
тур. Это свидетельствует о сохранении основного структур-
ного мотива изученных слоистых силикатов даже при дости-
жении высоких значений удельной поверхности (~85—
220 м2/т ) у продуктов их активации.
Механическая активация слоистых силикатов в воздуш-
ной среде приводит к разупорядочепию исходных кристал-
лических решеток путем ориентированного или беспорядоч-
ного смещения слоев и сеток относительно друг друга, что
приводит к снижению кристалличности указанных си-
ликатов.
Характер структурных напряжений, которые обуслов-
ливают в конечном счете образование рептгеиоаморфпой
фазы, различен для разных слоистых силикатов. В струк-
туре каолинита (отношение тетра- и октаэдрических сеток
1 : 1) преимущественно нарушаются связи Si—О—A1V1
и A1VI—О в октаэдрическом слое при сохранении связей
Si—О тетраэдрического слоя и рудиментов первоначального
порядка в расположении тетраэдрических слоев.
Структурные нарушения в кристаллической решетке
в силикатах типа 2 : 1 (лепидолит) локализованы преи-
мущественно в межслоевом пространстве и в октаэдриче-
ском слое. Для слоистых структур силикатов типа 2 : 2
(хлорит) пе выявлено какого-либо преимущественного на-
правления структурных нарушений в процессе активации
минерала в воздушной среде: структура разрушается по всем
кристаллографическим направлениям за счет неориентиро-
ванного смещения слоев, и сеток.
Исследование структурных изменений в ферритах нике-
ля и цинка под влиянием механической активации проведено
Павлюхиным, Медиковым, Болдыревым и др. [47—49, 119,
120, 388—390]. Ими показано, что в основе изменения маг-
нитных и других свойств ферритов-шпинелей под влиянием
активации лежит процесс разупорядочения кислородной под-
решетив, в результате которого исчезает дальний порядок,
а катионы вытесняются в большие по размерам окта-пусто-
ты. Образование структур, содержащих плотиоупаконапную
кислородную подрешетку, в которой катионы находятся
только в окта-положениях, довольно необычно для ферри-
тов, так как для них наиболее характерным является отно-
шение тетра-катиоиов к окта-катионам, равное 1 : 2. Таким
образом, в результате активации образуется вещество с ио-
112
пой кристаллической структурой, оставаясь тем же самым
по химическому составу. Физико-химические свойства такого
вещества, в частности скорость взаимодействия с различными
реагентами, растворителями и др., отличаются от свойств
исходного. Рассматривая плотноупакованные структуры
в целом, авторы пришли к выводу, что к нарушению даль-
него порядка приводят сдвиги по плоскости (111), а сдвиги
по плоскостям (110) и (100) к такому нарушению пе приво-
дят. Это позволяет заранее предсказать, какие структуры
будут эффективно активироваться, а для каких активация
малоэффективна [48].
Результаты исследований механической активации пиро-
хлора — одного из важнейших минералов ниобия — изло-
жены в работе Бацуева, Карпухина и др. [391]. Теоретиче-
ская крнсталлохимпческая формула пирохлора (Na, Са) ,s
X(Nb, Ta)2OB(F, CI, Oil). По результатам химического ана-
лиза п минерале содержалось: 7,2% Na.,О, 16,7% СаО, 70%
Nb„O, 0,44% Та2О5, 2,47% TiO,.
По данным рентгеиофазового анализа, под влиянием меха-
нической активации происходит глубокая аморфпзацпя ми-
нерала, которая обусловливает повышение его реакционной
способности в растворах комилексообразователя. После 2-
часовой активации практически весь ниобий переходит в
сернокислотно-перекисный раствор.
Авторы предполагают, что в результате мехапохпмпче-
ской обработки происходит деструкция пирохлора на более
простые соединения и продуктом механохимической обработки
является смесь, состоящая из простых (бинарных) оксидов.
Основанием для такого предположения служат данные по
ИК-спектроскопии активированных и неактивированных
пирохлорных концентратов. На спектре поглощения акти-
вированного пирохлорного концентрата наблюдается появ-
ление полос в областях 850—950 и 1400 см-1, интенсивность
которых с увеличением активации растет. Этп полосы
(835—910 и 1400 см-1) отнесены авторами к аморфной пятп-
окпеи ниобия.
Механическая активация на стадиях подготовки сырья
к химическому переделу может быть эффективно использо-
вана для ускорения последующих процессов и снижения
их параметров.
Из краткого обзора работ по активации оксидов видно,
что в качестве причины появления новых свойств у оксидов
рассматриваются в основном структурные нарушения. Воп-
росы же изменения стехиометрии авторами не обсуждаются
и пе учитываются, хотя с позиций химии твердого тела от-
113
клонения от стехиометрии определяют изменение важней-
ших физико-химических свойств твердых тел.
Вопросы изменения стехиометрии под влиянием механи-
ческой активации рассмотрены ниже.
3.3.1.1. Современные представления
о дефектах в оксидах металлов
переменной валентности
В соответствии с типом структуры структурно-химиче-
ские последствия механической активации могут быть раз-
личными. Для установления закономерностей дефектообра-
зования мы выбрали оксиды металлов переменной валент-
ности, имеющие в основе кислородный октаэдр и усложняю-
щиеся по принципу соединения октаэдров (рис. 32).
В этом направлении осуществляется переход от менее
плотных каркасных структур, в которых октаэдры соедине-
ны вершинами, к более плотным, в которых большая часть
октаэдров соединена гранями.
Представления о строении дефектов в оксидах металлов
переменной валентности претерпели в последнее время су-
щественные изменения. Первоначально по аналогии с клас-
сическими диэлектриками считалось, что дефектами являют-
ся вакансии, междоузельпыс ионы, а также примеси заме-
щения и внедрения, которые взаимно компенсируются, обес-
печивая электронейтралыюсть (например, вакансия в узле —
междоузэлышй пои, две разноименного заряда вакансии в
узлах решетки — примесный ион-вакансия). Аналогия
с классическими полупроводниками привела к еще одной мо-
дели, когда примесный ион в междоузлии понижает валент-
ное состояние основного катиона (так называемая «контро-
лируемая» валентность). Однако эти модели не исчерпывают
всего разнообразия дефектов в оксидах металлов переменной
валентности.
При больших концентрациях точечных вакансий в резуль-
тате взаимодействия может произойти их упорядочение с об-
разованием сверхструктуры или структуры сдвига. Первая
возникает путем ассимиляции вакансий или внедренных ато-
мов (одинаковые по заряду дефекты стремятся занять более
удаленные друг от друга позиции, но по мере увеличения их
концентрации отталкивающие силы заставляют дефекты за-
нимать вполне определенные кристаллографические узлы,
т. е. образуется сверхструктура), вторая — при исключении
точечных дефектов путем локальной перестройки координа-
ционных многогранников. Так^ в оксидах переходных метал-
Рис. 32. Элементы структуры оксидов переменной валентно-
сти [396].
а —тип Rf'Oj, б — гик рутила, в —тип корунда.
лов упорядочение дефектов может происходить путем пере-
группировки октаэдров, в результате чего уменьшается от-
ношение кислород/металл внутри плоскостей кристалла
1392-4071.
Принцип образовании структур сдвига рассмотрим па
примере структур, состоящих из октаэдров, в центре кото-
рых находится металл, а в вершинах — кислород. Как ука-
зывалось выше, в зависимости от того как октаэдры соеди-
няются, образуются структуры различного типа. Когда окта-
эдры соединяются вершинами, образуются структуры типа
КеО3 (см. рис. 32). Если соседние октаэдры в разных направ-
лениях соединены между собой ребрами и вершинами так,
что образуются ленты октаэдров с общими ребрами, соеди-
ненными друг с другом вершинами, то возникают структуры
типа рутила. При соединении октаэдров гранями возникают
плотноупакованные структуры типа корунда.
При дефиците кислорода происходит сжатие структуры
таким образом, что вдоль некоторых направлений и плос-
костей октаэдры приходят в контакт уже не вершинами,
а ребрами или гранями. Эти плоскости называются «плос-
костями сдвига», или дефектами по Вадслн 1396]. Когда ко-
личество таких плоскостей возрастает, начинается взаимо-
действие между ними и упорядочение. Образуется новая
сверхструктура, соединяющая «родительские фазы», перио-
дически разделенные плоскостями сдвига. Так возникают
гомологические ряды соединений с общими формулами типа
Мп°2п-г- МпОзп-г. Мп0зп-2. где п = 1, 2, 3 и т. д. Впер-
вые понятие о гомологических рядах ввел Магпели [397].
С тех пор эти фазы получили название фазы Магнелп.
Структуры сдвига образуются не при всяком смещении
плоскостей друг относительно друга. Изменения в составе
US
Рис. 33. Изменение координации октаэдров в КеО3 нутом
кристаллографпческого сдви га.
а — исходная структура; б — после кристаллографического сдвига.
I
происходят в плоскости наклонного вектора сдвига (рис. 33,
направления а и с). Плоскости с параллельными векторами
сдвига (ио вертикали и горизонтали па рис. 33) не вызывают
изменений в составе.
Существует следующая классификация структур кристал-
лографического сдвига [39G, 398j: 1) пластинчатые структу-
ры, образуемые одномерным сдвигом; 2) столбчатые структу-
ры, образуемые двумерным сдвигом, 3) блочные структуры,
образуемые трехмерным сдвигом.
Следствием двумерного сдвига является разделение крис-
талла на прямоугольные колонки размером тХпХоо (где
тип — число элементарных октаэдров, образующих струк-
туры ReO3). В результате трехмерного сдвига кристаллы раз-
биваются на блоки размером тХпХр, соединенные друг
с другом плоскостями сдвига. Состав кристалла с трехмер-
ной структурой сдвига можно выразить общей формулой
Мптр0з„п1р-г(т+п)+4-
На рис. 34 в качестве примера приведена структура 11-
формы Nb2O5, образованной блоками из 12(3x4) или 15(3x5)
кислородных октаздров в результате их трехмерного сдвига
1408).
Перегруппировка координационных полиэдров может
происходить также путем вращения, скольжения и отраже-
ния. В этом случае в структурах возникают туннели вдоль
определенных кристаллографических осей. В частности,
такой механизм перестройки имеет место в соединениях со
структурой рутила. В результате образуются широкие тун-
нели, в которых могут размещаться крупные попы с низкой
валентностью.
Рис. 34. Фрагмент структуры II-формы Nb2O5 [408].
Штрихами обозначены онтаздры нпжпего слои, точками — атомы
ниобии, находящиеся в тетраэдрическом окружении.
Существует несколько моделей реальных диффузионных
процессов, приводящих к образованию структур сдвига.
В модели Гадо [399] предполагается, что анионные вакан-
сии, возникающие при потере кислорода, упорядочиваются
в стенки, вдоль которых осуществляется кристаллографи-
ческий сдвиг с аннигиляцией вакансий. Поскольку анниги-
ляция вакансий может происходить лишь при их высокой
концентрации (4%), сдвигу должно предшествовать образо-
вание сверхструктуры, что иногда наблюдают эксперимен-
тально.
В модели Андерсона и Хайде [400], как и в предыдущем
случае, первичным процессом является возникновение бес-
порядочно распределенных кислородных вакансий, затем
они исчезают путем взаимодействия с дислокациями. При не-
которой критической, возможно очень низкой, концентрации
вакансии собираются в плоские диски. Контуры этих дисков
представляют собой дислокации, которые благодаря дейст-
вующим вдоль них напряжениям распространяются, захва-
тывая новые вакансии, и собираются в определенной крис-
таллографической плоскости. Затем сдвиг решетки на не-
которое определенное расстояние уничтожает вакансии и
снова дает атомам металла окружение с координационным
числом б. В отличие от первой модели здесь для возникно-
вения структуры сдвига не требуется высокой концентрации
апнойных вакансий. Плоскости сдвига могут зарождаться
не только па поверхностях восстанавливаемого кристалла,
ио и в глубине его. '•
лли
ш
Согласно модели Андерсона и Вадсли 011, в процессе
восстановления оксида с поверхности кристалла «испаря-
ется») плоскость кислородных ионов, после чего соседние
ряды катионов и анионов коллективно мигрируют в окта-
эдрические пустоты структуры. При этом плоскость сдвига
зарождается на поверхностях кристалла, а последующая
кооперативная диффузия рядов металл — кислород при-
водит к проникновению плоскости сдвига в глубь кристалла.
Продолжающееся восстановление ведет к многократному
повторению этого процесса и, следовательно, к повышению
концентрации плоскостей сдвига, упорядочение которых
предопределено их происхождением.
Строгих доказательств в пользу той или иной модели
до сих пор нет, хотя Хайде и Барсил [402] на основании элек-
тронно-микроскопических исследований сделали вывод, что
при высоких температурах для состояний, близких к равно-
весному, в структурах типа БсОа и рутила доминирует меха-
низм Андерсона — Хайде. Нри высоких температурах н су-
щественных отклонениях от стехиометрии (т. е. сильном вза-
имодействии между плоскостями сдвига) предпочтительна
модель Андерсона—Вадсли, объясняющая эксперименталь-
но наблюдаемое упорядочение плоскостей сдвига.
В фазах сдвига наблюдается ускорение диффузионных
процессов. Об этом свидетельствует легкость изотопного
обмена кислорода в них, т. о. процесса, зависящего от само-
диффузии ионов кислорода. При повышении температуры
может происходить переход от сдвиговой структуры одного
типа к структуре другого типа. Эти процессы сопровож-
даются диффузионной перестройкой вместе с миграцией плос-
костей сдвига к новым правильным периодам по кристаллу.
Так, например, монокристалл H-Nb2O5 сохраняет свою це-
лостность при восстановлении, хотя и проходит при этом
следующие стадии [394]: Nb,8O70 -+• Nb35O87->- Nb25O62 ->
Nb32O7()-> Nb2,O61-> Nb12O29.
На диаграмме полной свободной энергии в зависимости
от состава каждая фаза имеет свою узкую кривую. Огибаю-
щая этих кривых, образованная касательными, в условиях
совместного существования аппроксимируется плавной кри-
вой, типичной для обычной фазы с переменным составом.
Между отдельными фазами сдвига существуют упорядочен-
ные фазы срастания, т. е. двухфазные смеси двух блочных
структур. Форма огибающей свободной энергии для после-
довательности фаз такова, что свободная энергия должна
лишь немного возрасти, чтобы соединение с составом j:1 пре-
терпело локальное разложение на две новые структуры с со-
118
ставами, соответствующими х2 и х3. Замечено, что такой рас-
пад в простых оксидах обнаруживается с меньшей вероят-
ностью, чем в сложных. Предполагается, что в простых ок-
сидах флуктуации могут перераспределяться не за счет диф-
фузии, а с помощью электронных переходов. Это указывает
на возможную роль очень малых концентраций атомов при-
меси в поведении пестехпометрпческнх оксидов [394].
Упорядоченные фазы не всегда возможно наблюдать. Как
правило, необходимо проводить предварительную термо-
обработку при не очень высокой температуре, чтобы избе-
жать полного разупорядоченпя. Известно, что в условиях
химического равновесия при заданной температуре крис-
талл содержит определенное количество точечных дефектов.
Однако, поскольку дислокация не является равновесным де-
фектом, кристалл, имеющий упорядочение с образованием
дислокационных петель, как в случае сдвиговой структуру,
нельзя считать находящимся в равновесии.
Сдвиговые структуры возникают при быстром охлаждении
и могут быть исследованы при комнатной температуре. Ре-
зультаты такого исследования для WO3, выполненного Бу-
том, Экстромом, Игучп, Тпллеем, приведены в работе [406].
Из нее следует, что па диаграмме Т — х могут быть выделены
области существования структур сдвига.
Механическая активация оксидов сопровождается интен-
сивной пластической деформацией твердых тел, элементар-
ными актами которой являются движение, генерация и вза-
имодействие дислокаций. Б результате концентрирования
большой энергии в ядрах дислокаций и на поверхностях
скола возможен отрыв кислорода от оксида, т. е. их частич-
ная механохимпческая диссоциация [407]. Поэтому была
выдвинута рабочая гипотеза о том, что в результате механи-
ческой активации оксидов металлов переменной валентности
с каркасной структурой наряду с другими каналами (дис-
локационный и др.) возможно запасание энергии путем об-
разования дефектов по механизму кристаллографического
сдвига [413].
С целью подтверждения данного предположения нами
проведено исследование влияния механической активации
на некоторые оксиды металлов переменной валентности IV,
V, VI, VIII групп Периодической системы элементов
Д. И. Менделеева. Решались вопросы о том, насколько глу-
боко идет при активации процесс механохимпческой диссо-
циации оксидов и как это отражается на изменении струк-
туры п свойств оксидов.
119
5.3.1.2. Структурно-химические изменения
в оксидах со структурой типа рутила
Структура типа рутила, в которой кристаллизуются ди-
оксиды некоторых металлов, в идеальном случае имеет тетра-
гональную элементарную ячейку, в которой атомы металлов
октаэдрически окружены атомами кислорода, а октаэдры со-
единены вершинами и ребрами (см. рис. 32). К этому струк-
турному типу принадлежат диоксиды титана, олова, свин-
ца и др.
Диоксид титана. Для диоксида титана известны четыре
полиморфные модификации: анатаз, брукит, рутил, фаза вы-
сокого давления [409]. Анатаз имеет тетрагональную решет-
ку с параметрами а — 0,378 нм, с — 0,949, пространственная
группа Ii/amd [409]. Плотность анатаза 3,84 г/см3. Эта мо-
дификация может иметь состав, отличный от стехиометриче-
ского, если ее отжечь в вакууме при Т — 773 К. Брукит
имеет ромбическую структуру с параметрами а = 0,544 нм,
b — 0,817, с = 0,513 им, пространственная группа Рсаъ-
Плотность брукита 4,06 г/см3. Рутил принадлежит к тетра-
гональной сингонии и при 25°С имеет элементарную ячейку
с параметрами а = 0,459, с — 0,295 нм, пространственная
группа РЩтпт. Параметр ячейки может несколько изме-
няться в зависимости от давления кислорода. Плотность
рутила 4,23 г/см3. Фаза высокого давления синтезируется
при давлениях 4—12-10я МПа и температуре 673—1773 К.
При нагревании до 873 К она переходит полностью в рутил.
Фаза принадлежит к ромбической сингонии с пространст-
венной группой D\h — Рьсп параметрами а = 0,453 нм,
b = 0,550, с — 0,490 нм. Рентгеновская плотность 4,35 г/см3.
Считается, что анатаз и брукит термодинамически неста-
бильны и существуют только при наличии примесей. Темпе-
ратуры перехода модификаций (в присутствии примесей):
к>7« к , ппак
анатаз--->брукит----► рутил.
В структуре рутила, которая является наиболее устойчивой
из трех модификаций, каждый октаэдр имеет по два ребра,
общих с соседними. В следующей по устойчивости модифика-
ции число общих ребер три, и, наконец, в наименее устойчи-
вом — анатазе таких ребер четыре. Общие ребра у полиэд-
ров короче, чем другие, а расстояния Ti—О увеличиваются,
что приводит к искажению идеальной структуры и, соот-
ветственно, к меньшей устойчивости структуры.
120
При действии высоких давлений превращение анатаза в
рутил ускоряется. При давлении 2,4-10s МПа равновесная
точка снижается до температуры около 673 К. Граница, раз-
деляющая область анатаза и рутила на графике в координа-
тах Т — р, представляется прямой линией, наклон которой
выражается формулой dT/dp = —0,02 град/102 МПа.
Область гомогенности TiO, лежит в интервале TiOE<Jg—
—TiO,. При меньших содержаниях кислорода происходит
образование фаз с более низкой симметрией по механизму
кристаллографического сдвига. Общая формула их имеет
вид TinO2„_1, где и — целые числа от 4 до 10 включитель-
но, а дифракционные параметры всех этих фаз весьма близ-
ки и сходны с дифракционными параметрами TiO, [410—
4121. При определении структуры, например, такого соеди-
нения, как Ti5Ofl, было установлено, что оно содержит до-
мены или блоки со структурой типа рутила. Блоки имеют
бесконечные размеры в двух направлениях, но толщина их
конечна и равна пяти октаэдрам. Каждая фаза в ряду имеет
свою собственную характеристическую толщину, равную
4, 5, 6, 7, 8, 9 или 10 октаэдрам (эго число соответствует чис-
лу п в формуле ряда), а при п — оо соответствует С4 и фор-
муле TiO,.
Влияние механической активации на реакционную спо-
собность ТЮ2 исследовано в работе [413].
Изменение поверхности, интенсивности основного рент-
геновского отражения и количества вещества, перешедшего
в состояние, растворимое в концентрированной серной кис-
лоте, в зависимости от времени активации для TiO,, взятого
в форме анатаза, представлено на рис. 35.
По ходу активации анатаза он претерпевает полиморф-
ные переходы, аналогичные переходам при термическом акти-
вации: анатаз—> брукит—► рутил. Па промежуточных ста-
диях возникают рентгеноаморфные формы анатаза, брукита,
а затем рутила.
Удельная поверхность в зависимости от времени МА
быстро растет на начальном этапе, а затем проходит через
максимум, значение удельной поверхности в котором равно
45 м4/г, что соответствует размеру зерна порядка 10—20 нм.
Снижение поверхности обусловлено, по-впдпмому, процес-
сом образования агрегированных частиц.
Степень взаимодействия диоксида титана с серной кисло-
той (температура 473 К, время взаимодействия 1 ч) в зави-
симости от времени активации, напротив, на начальном
этапе уменьшается, что свидетельствует об образовании на
поверхности зерен анатаза «корочки» из рутила, плохо раст-
121
Рис. 35. Изменение
удельной поверхности
(«) оксида титана (IV),
количества титана, пере-
шедшего в раствор (б),
интенсивности основно-
го рентгеновского отра-
жения (в) в зависимости
от времени активации.
Исходная форма — ана-
таз [414].
1 — анатаз (0,352 нм); 2 —
брукит (0,284 им); s — ру-
тил (0,325 им); 4 — анатаз
после актнкацип и иоде.
воримого в серной
кислоте. После дос-
тижения некоторого
минимума количество
растворенного диок-
сида титана начинает
возрастать, ио-види-
мому, как результат
начавшегося в рути-
ле процесса дефекто-
образовапия. Ход
кривых изменений
удельной поверхно-
сти и степени взаи-
модействия с кисло-
той может служить
примером того, что
изменение реакцион-
ной способности при
механической актива-
ции не может рассматриваться только как результат уве-
личения поверхности.
Отметим, что при механической активации аиатаза в во-
де перехода в рутил мы ие наблюдали. Это может свиде-
тельствовать о разном характере активации порошков в за-
висимости от среды. В частности, известно, что вода сущест-
венно снижает коэффициент трения твердых материалов.
На рис. 36 приведены данные для механически активиро-
ванного рутила. С увеличением времени активации рутила
наблюдается снижение интенсивности дифракционных ли-
ний, образец постепенно становится рентгецоаморфным, зпа-
122
Рис. 36. Степень взаимодействия с серной кислотой (7), удельная по-
верхность (,?), мнкродеформации (3) и размер блоков когерентного рас-
сеяния (-7) для рутила в зависимости от времени активации [414].
чонпя относительных среднеквадратичных мнкродеформа-
ций увеличиваются от 10-3 до 10“ “, а размер блоков коге- I
рентного рассеяния уменьшается до некоторой постоянной
величины (5—1U нм); удельная поверхность н степень взаи-
модействия с серной кислотой непрерывно увеличиваются.
Ход кривых на рпс. 36 показывает, что в начальный пе-
риод (до 4 мин обработки) при непрерывном увеличении сте-
пени взаимодействия образцов с серной кислотой (кривая 1)
микродеформации изменяются незначительно (кривая 3),
тогда как удельная поверхность возрастает примерно в 10
раз (кривая 2). К этому времени размер блоков когерентно-
го рассеяния достигает минимума (5—10 нм) для данных
условий эксперимента и далее практически не меняется
(кривая 4). После 4 мин обработки скорость увеличения
удельной поверхности значительно снижается, а скорость
роста микродеформацпн возрастает. Степень взаимодействия
образцов с серной кислотой увеличивается монотонно. Такой
ход кривых показывает, что в начальный период изменение
растворимости обусловлено увеличением поверхности;
в дальнейшем, при установившихся размерах блоков коге-
123
Рис. 37. Кривые ДТА для механически ак-
тивированного рутила [413].
Время активации, мин: 1 — исходный; 2 — 10;
Л' - 15; / - 20; ,5 — 40; 6 — 20 (и атмосфсро
аргона).
Рис. 38. Изменение параметра а в трап*
сформированной триклинной объемно
центрированной ячейке, по данным |411).
Отмечено значение а для активированною в тече-
ние 30 мин рутила.
рентного рассеяния, существенное значение начинает при-
обретать увеличение дефектности кристаллической структу-
ры, которое в данном случае выражается в изменении раз-
меров микродеформаций. Зто согласуется с приведенными
выше данными (см. гл. 1) о стадийности процессов дисперги-
рования и активации.
При исследовании механически активированных образ-
цов рутила методом термического анализа установлено, что
при нагревании в воздушной среде на кривых ДТА в области
температур 400—700 К появляются значительные экзотер-
мические эффекты. На исходных образцах такие экзоэффек-
ты отсутствуют, что позволяет утверждать, что они появ-
ляются в результате механической активации. И действи-
тельно, с увеличением времени активации они возрастают
(рнс. 37).
Для количественной оценки экзоэффектов нами опреде-
лены площади па кривых ДТА и сравнены с площадью из-
вестного теплового эффекта. После 20-минутной активации
экзоэффект для TiO2 составляет 23,2 кДж/моль, после 40-
минутной — 73 кДж/моль. Точность определения экзоэффек-
та по данным ДТА лежит в пределах 5—10%. Отметим, что
па используемом для сравнения образце а-А1.>03 (материал
барабана и шаров) за время активации 30 мни экзоэффект в
124
этом интервале температур практически отсутствует
(~ 8 кДж/моль). За это же время активации достигается
удельная поверхность 30 м2/г. Удельная поверхностная энер-
гия TiO3 380 эрг/см2, а общая поверхностная энергия состав-
ляет величину порядка 1 кДж/моль. Сопоставляя получен-
ное значение со значением экзоэффекта, видим, что оно со-
ставляет лишь незначительную его часть.
Известно, что аморфные оксиды, полученные, например,
из водных растворов солей металлов путем их гидролиза и
последующей дегидратации, при нагревании в области тем-
ператур 473—637 К выделяют теплоту 50—150 кДж/моль
|415]. Эти экзоэффекты связывают с перекристаллизацией
«реликтовой» аморфной структуры, которая остается после
удаления воды из гидроокисей. Образцы рутила после
активации также реитгеноаморфны. Однако из данных РФА
следует, что интенсивность рентгеновских отражений после
отжига диоксида титана при температуре до 773 К сущест-
венно пе увеличивается, поэтому экзоэффект нельзя объяс-
нить кристаллизацией рентгеноаморфного оксида.
Выло замечено, что экзоэффект па кривых ДТА при на-
гревании в среде аргона практически исчезает (остается
небольшой эффект в области 473 К). Это может свидетельст-
вовать о связи экзоэффекта с окислением на воздухе частично
восстановившегося при механической активации оксида.
И действительно, проводя активацию в вакуумной мельнице,
мы обнаружили значительное газовыделение {4131. Сопостав-
ляя давление с давлением кислорода при полном разложении
диоксида свинца в этих же условиях, провелп количествен-
ную оценку степени разложения TiO2. Ниже приведено
изменение состава рутила под влиянием активации в вакуум-
ной мельнице [413]:
Время активации,
мин 10 20 30 40 50 60
х в Ti()2-x 0,003 0,007 0,018 0,028 0,039 0,019
В работе [411] рассмотрено изменение параметра а в
трансформированной ячейке рутила как в триклинной объем-
но центрированной псевдоструктуре в зависимости от содер-
жания кислорода в TiO2_K. Полученному нами для механи-
чески активированного образца значению а, равному 0,553
(рис. 38), соответствует состав TiOb9l>.
Образование структур кристаллографического сдвига
при активации Т1О3 подтверждено памп в результате иссле-
дования активированных образцов методом ЭПР |416].
125
Таблица 7
Основные значении д-факторои парамагнитных ча-
стиц фаз Магнели TinO2n_j при 90 К ]424]
п вх eV fiz Ширина линии ДН, Гс Концентрация спинов на формульную единицу
5 1,9681 1,9791 1.9386 30 0,2
6 1,9721 1,9774 1,9436 23 0,3
7 1,9694 1,9737 1,9359 42 0,3
8 1,9694 1,9726 1.9422 53 0,4
Прежде всего отметим, что методом ЭПР в TiO2 регист-
рируется как продукт восстановления ион Ti3+ (сГ-ион),
параметры спектра которого зависят от координации. В ру-
тпльной модификации он может находиться, как это показа-
но в работе Гаджиевой и Ануфриенко [417], в узельных и
междоузельных позициях. Для узельных позиций характе-
рен немного вытянутый октаэдр, расстояния до аксиальных
лигандов несколько больше, чем до экваториальных (0,199 и
0,194 нм соответственно), а для междоузельных позиций ок-
ружение также октаэдрическое, но имеются две короткие
связи с аксиальными лигандами и длинные с экваториаль-
ными лигандами (0,160 и 0,220 им соответственно).
В первом случае спектры ЭПР ионов Ti3+ не наблюдают-
ся, во всяком случае, нет четких обоснований отнесения
наблюдаемых спектров к ионам в узельных позициях. Во
втором за счет ковалентных связей значительно уменьшает-
ся константа спин-орбптального взаимодействия и регист-
рируется спектр с параметрами g± = 1,99, gB = 1,96—1,97
[418—420]. При химическом восстановлении рутила на по-
верхности часто наблюдается ион Ti3+ в специфической коор-
динации с g0 — 1,93, который не исчезает при нагревании
до 300 К. Эта форма наблюдается в большинстве работ, по-
священных изучению восстановленного Т1О2 [420].
В соответствии с результатами Ферхурста, Инглиса и
др. [423, 424], которые изучили спектры ЭПР монокристал-
лов соединений, соответствующих фазам Магнели, для них
характерен спектр с близкими параметрами g-факторов
(табл. 7)
Авторы отнесли этот спектр к димерам Ti2+, в которых
ионы титана связаны через кислород. Они исходили из сле-
дующего наблюдения: g2-TCH3op почти аксиален и его миин-
126
мальное значение находится вдоль осн с рутила по направ-
лению самой короткой связи Ti—Ti. Искажение ^-тензо-
ра в пределах структуры рутила означает, что неспаренный
электрон связан более чем с одним ионом титана, и кристал-
лическая структура, например фазы Ti6Ou, подтверждает
это. Если бы образовалась пара Ti3+—Tis+, то для нее ха-
рактерно триплетное состояние с S = 1 и должен наблю-
даться сильно анизотропный триплетный спектр или широ-
кий сигнал ЭПР. Однако этого пе наблюдается.
Другой уникальной особенностью спектров ЭПР фаз
Магнели является наличие моттовского перехода диэлект-
рик — металл при температуре 120—150 К [395, 424]. Вы-
ше этой температуры спектр Э11Р не наблюдается.
Образцы Ti(J2 (в анатазной и рутпльной модификации)
активировали в среде аргона, и все дальнейшие операции
(измерение, отжиг н др.) проводили в запаянных стеклянных
ампулах. Спектры регистрировали на радиоспектрометре
JES-3BS-Q в х- и (^-диапазонах.
Результаты исследований представлены на рис. 39. Как
следует из приведенных данных, спектры ЭПР по ходу акти-
вации изменяются. Если на начальных этапах активации
анатаза (см. рнс. 39, а) наблюдается спектр с g0 = 1,98, то
затем появляется спектр с параметрами g± = 1,97 в g| =
= 1,94ч-1,93. Первый спектр, как показали более детальные
исследования, имеет параметры g. — 1,99, gj| = 1,96 и
строго совпадает с параметрами ЭПР изолированных нонов
Ti3- в междоузлиях рутила [417]. Второй спектр полностью
совпадает с параметрами для димера Ti2+, характерного для
фаз Магнели, которые приведены выше. Суммарная интен-
сивность сигнала ~ 10” спин/г. Таким образом, в ходе
активации анатаз переходит в рутил, а из рутила уже обра-
зуются фазы Магнели. Отметим, что впервые наблюдали
Ti5+ с g0 = 1,98 при механической активации анатаза Зы-
рянов, Ляхов, Болдырев [421].
При активации рутила (см. рис. 39, б) на начальной ста-
дии также регистрируется спектр с g0 = 1,98, соответствую-
щий Ti3+ в междоузлии, а затем появляется спектр с g± =
= 1,97 (g у — 1,94-4-1,93), принадлежащий димеру Ti2+, что
свидетельствует об образовании фаз Магнели.
Па рис. 4U показаны спектры фаз Магнели, образовав-
шихся в результате механической активации, в зависимости
от температуры. Выше 140 К спектр уширяется и становит-
ся изотропным. Это находится в полном соответствии с при-
веденными выше данными для перехода диэлектрик — ме-
127
Время активации и планетарной мелышце. мин: 1 — 6,3 — 10; з — 21); 4 — 0,5;
талл, наблюдаемого для фаз Магнели в области температур
120-140 К.
Отжиг активированных образцов в аргоне при темпера-
туре G73 К увеличивает многократно интенсивность спектра
ЭПР, принадлежащего фазам Магнели, а отжиг на воздухе
вызывает их полное исчезновение, что согласуется с приве-
денными выше данными ДТА.
Таким образом, при активации различных модификаций
TiO2 в аргоне происходит химическое восстановление окси-
да с образованием структурных дефектов, упорядоченных
по механизму кристаллографического сдвига (фаз Магнели).
При механической активации анатаз сначала переходит в ру-
тил с нарушенным катионным распределением (изолирован-
ные ионы Ti3+ в междоузлиях), а в последующем образуются
фазы Магнели. Для рутила последний процесс является
основным. Спектры ЭПР изолированных междоузельных
ионов Ti3+ в этом случае наблюдаются только при очень ма-
лой длительности активации.
Рис. 40. Изменение спектров ЭПР
и laiiiicibMoc.Tii от температуры (пе-
реход диэлектрик — металл) для
механически активированного ру-
тила [416 ].
Отметим, что образова-
ние протяженных дефектов
при деформировании моно-
кристаллов рутила недавно
обнаружено Блапчипом, Бар-
силлом, Смитом [638] мето-
дом высокоразрешающей
электронной микроскопии.
Па основании совокупности
данных следует считать до-
казанным механизм усвое-
ния механической энергии дефектами, образующимися по
механизму кристаллографического сдвига.
Блаичнп п Барсплл [425, 639] считают, что образование
протяженных дефектов происходит путем упорядочения
нопон в (Н.меры Ti’+ затем в изолированные плоскости
сдвига, одновременно с упорядочением этих плоскостей
сдвига в структуры сдвига.
Диоксид олова. Для изучения изменений в структуре и
составе при механической активации дпокспда олова нами
использован метод ядерной гамма-резонансной спектроско-
пии [426—429].
Как известно, SnO2 имеет тетрагональную структуру ти-
па рутила. Величина квадрупольного расщепления SnO2
определялась в ряде работ. Общепризнано значение 0,50 ±
± 0,07 мм/с [4301.
Мессбауэровский спектр SnO2 является, по существу,
суперпозицией двух линий — компонент квадрупольного
дублета (рис. 41). Вероятность резонансного поглощения ве-
лика при комнатной температуре и слабо зависит от темпе-
ратуры. Большая вероятность резонансного поглощения
означает, что средний квадрат амплитуды колебаний для
атомов олова мал, т. е. олово, находящееся в центре кисло-
родного октаэдра, связано с окружением достаточно прочно.
Механическая активация SnO2 для мессбауэровских
измерений проводилась с использованием керамических ба-
рабанов п шаров (а-А1.,0з). Мессбауэровские спектры снима-
лись па спектрометре MS = 10 1х при 293 К и источником
В aS иО 3.
128
5 К Г. Ami.ii
129
2 IT, ММ/'
На рис. 41
ческой активации, из «0Т°Р кающая рост нарушенное™
^“““pb,,₽“TpS..eM J™..»,,,,.. На ри-
Рис. 41. Спектры Я ГР (а) и за-
висимость ширины мессбауэров-
ской ливни, удельной поверхно-
сти и степени растворении оксида
олова (IV) от времени активации
(б) [426].
а) 1 исходный образец; г.ремн aiciii-
нацпн, мин: J — 5, э - 33. J — (io,
5 — 80; б) 1 — ширина .пикш (Гэ>;
2 — удельная поверхность (s); 3 — сте-
пень растворения (е).
супке приведены также ре-
зультаты измерений полуши-
рины мессбауэровских ли-
ний, удельной поверхности
порошков, определенной ме-
тодом тепловой десорбции
аргона, и количества олова,
перешедшего в раствор в
последова-
[431], в за-
определепного по стандартной методике путем
тельного выщелачивания в кислоте п щелочи
В11СПМОСТП от времени активации.
Значение Гэ увеличивается монотонно от 1,40 ± 0,03
до 1,73 ± 0,03 нм/с. Такое изменение спектра указывает на
то, что под влиянием механической активации заметно по-
нижается симметрия локального оружия вблизи мессбауэ-
ровских атомов, что связано непосредственно с изменением
расстояний Sn — О, т. е. с деформацией октаэдра SnO6.
Аналогичным образом изменяется и степень взаимодейст-
вия SnO2 с реагентами. Напротив, удельная поверхность
после достижения ею определенного значения (~ 8 м2/г)
существенно не увеличивается. По-видимому, имеет место
образование молекулярно-плотных агрегатов. Таким обра-
зом, увеличение количества перешедшего в раствор олова
связано с повышенной дефектностью решетки диоксида оло-
ва, которую характеризует величина Гэ. Отсутствие воз-
растания вероятности поглощения, которого следовало ожи-
дать для точечных дефектов в соответствии с работой [432],
может свидетельствовать о том, что дефектами являются
протяженные дислокации или протяженные лпнейные де-
фекты, возникшие в результате сдвига плоскостей.
., При активации в воде достигается высокая дисперсность
SnO3 (~ 30 м2/г), однако уширения мессбауэровской линии
пе наблюдалось. Пе увеличивается существенно при этом и
количество олова, перешедшего в раствор, что свидетельст-
5*
131
130
Pi.r. 42. Спектры ЯГР для Sn02
после .механической активации
1428].
1 — исходный образец; 2 — 45 мин,
300 К; 3 — 45 мни, 77 К; 4 — то же и
отжиг на воздухе 20 мин, 137 К; 3 —
отжиг в вакууме при 1175 К,
вует об отсутствии описан-
ных выше эффектов при дан-
ных условиях активации.
На рис. 42 представлены
результаты измерений для
активированных порошков, а
также порошков после отжи-
га на воздухе и в вакууме.
Па спектре активированного
образца выявляются линии
двухвалентного олова (6 =
= 3,06, е = 2,1 мм/q» два
минимума в правой части спектра). Появление ионов
Sn2+ связано с разрывом химических связей Sn—О. То,
что диссоциация оксида при механической активации
имеет место, подтверждают также полученные нами
данные по газовыделенпю при обработке оксида в вакуум-
ной мельнице [221]. Сопоставляя количество кислорода, по-
лученное при механической активации диоксида олова с
кислородом, выделившимся при разложении диоксида свин-
ца, который разлагается полностью, мы вычислили степень
разложения SnO2 [4131:
Время активации,
мня 10 20
аг в SnO4_x 0,010 0,014
30 40 50
0,018 0.024 0,032
GO
0,037
Так же как и для TiO
при исследовании SnO2 методом
термического анализа установлено, что при нагревании в
воздушной среде на кривых ДТА в области температур 400—
700 К для активированных образцов появляются значитель-
ные экзоэффекты. Экзоэффект возрастает со временем акти-
вации и составляет для 10-м и нут пой активации 72 кДж/моль,
для 30-минутной — 124 кДж/моль. Экзоэффекты для активи-
рованных образцов исчезают, если кривые ДТА снимаются
в среде аргона. При нагревании некоторых образцов SnO2
на кривых ТГ было зафиксировано небольшое увеличение
массы (1—2%). Отметим, что авторами [293] получено еще
более высокое значение теплового эффекта окисления оксида
после механической активации, а именно 176 кДж 'моль.
Сравнение его с тепловым эффектом окисления SnO показы-
вает, что он непропорционально велик (А/7 окисления SnO
до SnO2 составляет 295 кДж/моль). В таком случае можно
было бы ожидать, что восстановление ShO2 при механи-
ческой активации пройдет почти на 50%. Однако это не со-
гласуется с приведенными выше данными. Отсюда следует,
что изменение стехиометрии не является единственным ка-
налом усвоения механической энергии.
В результате отжига на воздухе линии, соответствующие
Sn2+, исчезают при температурах 600—800 К, а уширение
липни, обусловленное нарушением структуры, начинает
исчезать при более высоких температурах (800—1300 К).
Отжиг в вакууме (10~3 мм рт. ст.) при 1173 К приводит к
дальнейшему разложению SnO2 до SnO, которое началось
под влиянием механической активации (см. рис. 42, кри-
вая 5).
При исследовании изотермического отжига на воздухе
порошков SnO2 путем измерения ширины мессбауэровской
липин было установлено, что кривые отжига характеризуют-
ся спадом, а затем выходят на плато, не достигая минималь-
ной ширины липни, характерной для исходной активиро-
ванной двуокиси олова (рис. 43). Из полученных экспери-
ментальных данных мы попытались определить энергию
активации отжига дефектов. Поскольку в настоящее время
вид функциональной зависимости ширины линии от кон-
центрации неизвестен, энергию активации определили по
уравнению
RT Т
в котором значения температур и времени соответствуют
одинаковому значению АГ. Энергия активации составляет
450—500 кДж/моль.
Следует заметить, что кривые отжига имеют весьма за-
метное сходство с кривыми восстановления, приведенными в
литературе, например для запасенной энергии в щелочно-
галоидных соединениях после их радиационного облуче-
ния, а также с кривыми «ступепчато-образиой» кинетики
рекомбинации свободных радикалов в облученных ве-
ществах |433].
Согласно одной из гипотез, «ступенчато-образная» кинети-
ка гибели радикалов при отжиге веществ после радиационно-
л on
133
Рис. 43. Изменение ширины мессбау-
эровской линии при изотермическом
отжиге па воздухе [428].
Температура, К: 1 — 973; 2 — 1023; 3 —
1173; 4 — 1227; 5 — 1373.
Рис. 44. Изменение ширины
линии ИГР р зависимости от
температуры (время отжига
20 мин) [428].
го облучения объясняется наличием широкого спектра час-
тот молекулярной подвижности вблизи дефекта [434]. Так,
подвижность в тех частях объема вещества, которые нахо-
дятся в непосредственной близости от дефектов, может обна-
руживаться при более низких температурах по сравнению
с подвижностью в областях, находящихся в большем удале-
нии от дефектов.
Аналогично можно рассматривать изменение подвиж-
ности ионов при отжиге SnO2 вблизи наведенных при меха-
нической активации структурных дефектов. Таким образом,
поведение твердых веществ после механической активация
аналогично поведению их после радиационного облучения,
что проявляется в одинаковых кинетических закономерно-
стях отжига дефектов.
Отметим также, что при изучении кинетики спекания
активированного диоксида олова при разных температурах
путем измерения поверхности получены такие же, как и для
ширины мессбауэровской линии, кривые, т. е. с выходом
на плато при разных температурах. Так, при отжиге при
температурах 1073 и 1173 К поверхность с 22,4 м2/г сокра-
щается до 7,2 и 3,4 м2/г соответственно и не изменяется при
дальнейшем увеличении длительности отжига. Эти данные
свидетельствуют и о том, что отжиг дефектов связан с процес-
сом перекристаллизации диоксида олова, который начинает-
ся при температурах выше 1000 К (рис. 44).
В целом из полученных данных следует, что механи-
ческая активация приводит к изменению химической связи
в SnO2. Наряду с искажением элементарной ячейки и де-
134
формацией октаэдров происходит восстановитсяьный про-
цесс с образованием олова в форме Sn2+.
Бобышев и Радциг 1435] исследовали механическую
активацию диокепда олова с использованием целого комп-
лекса физических методов (ИК-, ЭПР-спектроскопии, элект-
ропроводности, мпкрокалорпметрпи и др.). Полученные ими
результаты подтверждают и дополняют вышеприведенные.
Так, при нагревании SnO2 после активации его в атмосфе-
ре гелия было обнаружено выделение с его поверхности
молекулярного кислорода в количествах, по абсолютному
значению почти в 200 раз больших, чем для неактивирован-
ного образца. При этом резко возрастает проводимость об-
разца, энергия активации которой снижается от 0,08 ± 0,01
до 0,03—0,04 эВ.
По данным ИК-спектроскоппи, исходный SnO2 имеет по-
лосу поглощения с краем около 350 нм, соответствующую
ширине запрещенной зоны оксида 3,8 эВ. Активация приво-
дит к появлению широкого крыла линии поглощения, тяну-
щегося от 350 до 850 нм. Это указывает на образование в ре-
зультате активации спектра электронных состояний, кото-
рые заполняют запрещенную зону вплоть до зоны проводи-
мости. Наряду с этим образуются и свободные носители,
о чем свидетельствует возрастание проводимости.
Методом ЭПР зарегистрировано появление сигнала, пред-
ставляющего анизотропную линию с = 2,003, g2 — 2,010
и g3 — 2,027. Форма сигнала не зависит от температуры,
а его амплитуда подчиняется закону Кюри. Сигнал исчезает
при прогреве в вакууме при 470 К. Сопоставление парамет-
ров с литературными данными позволило идентифицировать
наблюдаемые центры как 07, локализованные вблизи Sn4+.
Предполагается, что образование радикала 07 происходит
по реакции: О2- + О2-->-О7 + Зе, при этом электроны
переходят на Sn4+ с образованием Sns* и Sn2+. Прямыми ме-
тодами авторам [435] не удалось зарегистрировать появление
Sns+. Они считают, что присутствием Sns+ может быть обус-
ловлена длинноволновая часть ИК-спектра. Косвенно опре-
делено их содержание по образованию дополнительных 07
при хемосорбции кислорода на поверхности активированно-
го оксида. Количество Sn3+ близко к ожидаемому по указан-
ной выше реакции (т. е. примерно в 3 раза больше, чем 07-
центров). Данные же об обнаружении ионов Sn2+ методом
ЯГРС в механически активированных образцах SnOs при-
водились выше.
В целом совокупность структурных, электронных и хими-
ческих превращений и определяет изменение реакционной
135
способности диоксида олова в результате механической
активации.
Диоксид свинца. При обычных условиях диоксид синица
имеет две полиморфные модификации: [3-РЬО2 — тетраго-
нальная сингония (а — 0,494 нм, с = 0,377 нм) и а-РЬО., —
орторомбическая сингония (а = 0,499 нм, b = 0,590, с =
= 0,547 нм) [409]. Термическое разложение а-РЬО2 происхо-
дит при температурах примерно на 100 К ниже, чем |3-РЬО2,
который разлагается при 053 К [430]. Начальным продук-
том разложения как [3-, так и a-формы является РЬО1>57-
(3-РЬО2 переходит в а-РЬО2 при повышении давления
> 103 МПа. Этот переход слабо зависит от температуры (так,
при 273 К равновесная точка соответствует давлению
1,2-103 МПа, а при 873 К — давлению 1,4- 10э МПа). Ука-
занный переход реализуется и при обработке [3-РЬОа в
пзмельчнтельпых аппаратах. При продолжительном диспер-
гировании |3- и a-форм РЬО2 между ними устанавливается
стационарное состояние, в котором соотношение форм
остается неизменным, равным 90% a-формы и 10% [3-фор-
мы [328].
Сенна и Шоперт [329] исследовали изменение энтальпии
реакции
[3 - РЬО2 -> РЬО + -4- О2 ДЯ298 = + 14 кДж/моль
в зависимости от условий механической активации. Исследо-
вались процессы внброизмельченпя и прессования. Было по-
казано, что коэффициент полезного действия виброизмель-
чения составляет 4,3%, а прессования — 32%. Авторы счи-
тают ответственной за образование дефектов как в том, так
и в другом случае пластическую деформацию, но в случае
внброизмельченпя большое количество энергии расходуется
на трение и неэффективные соударения мелющих шаров.
Тем не менее максимальное значение запасенной энергии
значительно больше у оксида свинца после виброизмельче-
ния в течение 20 ч (Д7/ = 80 Дж/г), чем у оксида после 20
прессований (ДЯ — 24 Дж/г). Авторы также наблюдали
переход [3-РЬО2 в a-форму, но равновесие в данном случае
устанавливалось при содержании а-формы 60%. За увели-
чение энтальпии авторы считают ответственным разупорядо-
чение в решетке как в [3-, так и в а-формс. Они показали, что
имеется линейная зависимость между запасенной энергией
и параметром z:
Z == Хр/Ур -[“
где .г — мольная доля фазы, /3 — интенсивная ширина диф-
ракционной линии (Ш) для a-формы и (110) для [3-формы.
Однако возможность механохимнческого разложения окси-
да, особенно при большой длительности активации, ими пе
учитывалась.
В работе [437] приведены результаты исследования
влияния скорости нагревания на термическое разложение
диоксида свинца. Показано, что при низких скоростях на-
гревания (от 0 до 0,02 град/с) кислород выделяется в четыре
стадии:
о г>1 г, 663!!п1л 713К 7СЗК 873К _
[3- РЬО2 > РЬО4|97----> РЬО3144--> bO]t3y----* FЬО.
Указанным температурам соответствует максимальная ско-
рость выделения кислорода. При высоких скоростях нагре-
вания (2-102 — 2,5-103 град/с) число стадий уменьшается
до двух:
РЬ02^ГЬ01>44^ГЬ0.
Известно также, что соединение ГЬО133 при повышенных
температурах и давлениях распадается по реакции
РЬ3О4->РЬО | l’b2O3.
13 отличие от фазового перехода [3-РЬО2 —>- а-РЬО2 давле-
ние, при котором совершается реакция распада РЬ3О4, спль- !
по зависит от температуры. При температуре 473 К равно-
весная точка соответствует давлению 4-103 МПа, а при тем-
пературе 723 К — около 0,1 МПа [409].
Исследование механохимнческого разложения рРЬО2
проводилось в вакуумной термостатированной мельнпце
планетарного типа [438]. Использовались шары пз сплава
13К-8 диаметром 3—4 мм и стальные шары разных диаметров.
Мощность .мельницы в зависимости от числа оборотов приве-
дена выше (см. гл. 1).
За кинетикой разложения следили по увеличению дав-
ления в системе с помощью образцового вакуумметра. Точ-
ность определения давления 0,3—0,5 отн.%. Термический
анализ образцов выполняли на дерпватографе МОМ при ско-
рости нагревания 10 град/мин. Фазовый состав продуктов
разложения был определен путем сравнения значений рент-
геновских отражений с приведенными в справочнике данны-
ми [439]. Дифрактограммы снимались па приборе ДРОН-05
(излучение Cuxa, Ni — фильтр). 13 работе использовался
P-l’bOo с содержанием основного вещества 97% (производст-
во VEB «Apoldo», ГДР).
137
130
Рис. 45. Кинетика механохнмпческого разложения Р-РЬО2 в зависи-
мости от частоты вращения (а) и диаметра шаров (б) [438].
а — шары из сплава В К-8, диаметр 3—4 мм, стрелкой показано газовыделспие
после дополнительной откачки, частота вращения, с-1: 1 — 8,7, 2 — 10,1, 5—10,8,
4 — 12,4, 5 — 15,3; б — шары из стали, частота вращения 12,4 с”1, диаметр
шаров, мм: 1 — 2,2— 3, 4 — 5, 5 — 12.
Поскольку кинетическая энергия мелющего шара Е =
= ти*12, где т — масса, a v — скорость шара, изменять ее
можно путем изменения частоты вращения и радиуса шаров.
Кинетические кривые, полученные при разных частотах вра-
щения и разных диаметрах шаров, приведены на рис. 45.
Кинетические кривые имеют 5-образный характер, при
определенной длительности скорость разложения замедляет-
ся и кривые выходят на насыщение. Стрелкой на кривой 5
показано газовыделение после дополнительной откачки.
Видно, что ход кривой после откачки изменяется, т. е. на
стадиях насыщения имеется некоторое стационарное состоя-
ние, когда скорость прямой реакции сравнивается со ско-
ростью обратной. Откачка сдвигает «равновесие» в сторону
образования продукта диссоциации.
На рис. 45 могут быть выделены две области: область I
при частоте вращения пиже 10,8 с-1 и диаметре шаров мень-
ше 3 мм — скорость разложения здесь незначительна —
и область II (выше этих значений), где начинается эффектив-
ное разложение РЬО2.
Различным является поведение образцов, активирован-
ных с большими и малыми кинетическими энергиями шаров.
На рис. 46 приведены кривые ДТА для одинаковой степени
разложения РЬО2. После обработки р-РЬО2 в мельнице тем-
пература начала разложения как в том, как и в другом слу-
138
Рис. 46. Кривые ДТА исходного
f-PbO2 (1) п продуктов ого меха-
нической активации с частотой
вращения 12,4 с'1 (2) в течение
6 мин (а = 0,15) и 8,7 с-1 (3) в те-
чение 60 мин (а = 0,13).
чае снижается примерно на
100°, что объяснимо, так
как [3-форма перешла в
a-форму, которая менее тер-
мостабильна, чем [3-форма.
После обработки при Maj
Рис. 47. Дифрактограммы активи-
рованного (т = 60 мин, со =
= 8,7 с-1) Р-РЬОа (7) н после на-
гревания его в течение 30 мин
до 583 К (2).
х энергиях процесс раз-
ложения а-РЬО2 до РЬО1>67 сопровождается экзоэффек-
том. О том, что в результате разложения образуется пменно
PbOli57, свидетельствует дифрактограмма активированного
образца после нагревания его до 583 К (рис. 47). После обра-
ботки с большими энергиями экзоэффект на кривых ДТА
отсутствует.
Па рис. 48 представлены данные РФА по изменению
фазового состава образца в ходе активации при частоте вра-
щения 12,4 с-1. Установлена следующая последователь-
ность фаз:
₽-РЬО2 а-РЪО, -> PbOli57 -> PbOlil4 -> РЬО.
Из этих данных, а также данных ДТА следует, что фаза
РЬО1>за в кристаллическом виде не обнаруживается.
Как видно из рис. 45, степень разложения, при которой
кинетические кривые выходят на насыщение, различается в
..139
Рис. 48. Дпфрактограммы проме-
жуточных продуктов мехапохпми-
ческого разложения P-PbO2 (<и *=
= 12,4 с"1, шары из ВК-8, диа-
метр 3—4 мм).
1 —исходный образец; степень разло-
жении: 2 — 6,18, 3 — 0,40, 4 — 0,08,
5 — 0,84, в — 1,0.
зависимости от кинетической
энергии шаров, т. е. опреде-
ленному уровню подводимой
энергии соответствует свое
«равновесие». РФА образцов,
активированных при разных
энергиях, после выхода ки-
нетических кривых на насы-
щение показал, что они име-
ют одинаковый фазовый сос-
тав, различие только коли-
чественное. Основными фа-
зами являются РЬО, РЬО11Б,
и небольшие количества
PbOli44, Фаза РЬО1133 в этом
случае также не была обна-
ружена.
Отсюда следует, что прак-
тически полный переход из
P-формы в «-форму РЬО, наб-
людается при всех исследо-
ванных условиях активации
и сопровождается разложе-
нием на 10—15%. Таким об-
разом, полученная под влия-
нием активации «-форма PbOj
представляет собой дефицит-
ную по кислороду фазу, со-
держание кислорода в ко-
торой уменьшается до сос-
тава РЬО11ОО — РЬОЬ85. Сог-
ласно литературным данным,
а-РЬО2 имеет интервал гомо-
генности до РЬО1187 1409]. По-
теря кислорода связана с пе-
рестройкой структуры, кото-
рую претерпевает PbO2 в процессе механической обработки.
Координационный октаэдр РЬО6, лежащий в основе струк-
туры [5-РЬО.,, существенно искажается и приобретает непра-
вильную форму. Кроме того, меняется и структурный мо-
тив. Если в p-формс октаэдры имеют два общих ребра и укла-
дываются ровными рядами, то в а-РЬО2 оип образуют зиг-
загообразные цепи, по два октаэдра в каждом звене [440].
Наблюдаемое памп различие в кривых ДТА для образ-
цов |3-РЬО2, активированных с разной механической энер-
гией, может быть вызвано следующими причинами. При ма-
лых механических энергиях ы = 8,7 с~1 длительность обра-
ботки значительная и составляет 60 мни, при этом происхо-
дит аморфизация вновь образовавшейся фазы а-РЬО2, о чем
свидетельствует уширение линий на дпфрактограмме
(см. рпс. 47), а также наблюдаемое увеличение экзоэффекта
со временем активации. Пэ аморфной фазы а-РЬО2 кристал-
лизуется фаза РЬО1157 (см. рпс. 47); при этом освобождает-
ся энергия, которой достаточно, чтобы не только покрыть
затраты па отрыв кислорода (разложение РЬО2 идет в обыч-
ных условиях с поглощением тепла), по и привести к экзо-
термическому эффекту. Экзоэффект оценивается нами ~
~ 20 кДж/моль.
При больших механических энергиях для тех же степе-
ней разложения (15%) наблюдается также образование фа-
зы РЬО1157 (в какой-то степени аморфизованной, так как
линии являются уширенными), но экзоэффект на кривой
ДТА отсутствует.
Различие кривых ДТА может быть связано с различием
в образовании сдвиговых структур в зависимости от механи-
ческой энергии. Известно, что переход от а-РЬО2 к РЬО11Ь7
совершается по механизму кристаллографического сдвига
[441]. При нагревании после активации с малыми энергия-
ми, по-видимому, генерируются кислородные вакансии, при
объединении которых происходит выделение энергии. При
активации же с большими энергиями в структуре а-РЬОа
уже содержатся «заготовки» фазы РЬО1г67. Можно допустить,
что образуется своего рода «твердый раствор» РЬОЬ67 в
а-РЬО2, распад которого происходит эндотермически, и эк-
зоэффект при нагревании исчезает.
В исчезновении фазы РЬО1133 па дпфрактограммах про-
дуктов разложения р-РЬО2 проявляется сходство механо-
химического разложения с разложением при импульсном
нагреве, исследованным в работе [437]. Причиной того, что
не обнаружена фаза РЬО133, может быть возникновение та-
ких импульсов давления п температуры., при которых проис-
141
ходит распад РЬаО4, причем скорость его значительно выше,
чем скорость образования, вследствие чего концентрация
снижается ниже предела чувствительности РФА и ДТА.
Фазовый переход из р- в a-форму РЬОа от температуры
не зависит, для его реализации необходимо только давление
(1,2-1,4).103 МПа.
Для реализации же реакции распада РЬ3О4 необходимы
локальные температуры 473, 573, 650 К и давления соот-
ветственно 4-Ю3, 2-Ю3, 1-103 МПа. Факт протекания этой
реакции и фазового перехода _р —> а может свидетельство-
вать о том, что в импульсах давления а температуры при
ударе шаров о стенку эти значения (650 К, 1 103 МПа) до-
стигаются при частотах вращения барабанов > 10,1 с-1
и диаметре шаров > 3 мм.
З.З.1.З. Структурно-химические изменения
в оксидах со структурой типа НеО3
Образование структур кристаллографического сдвига ха-
рактерно для оксидов V и VI групп. Рассмотрим некоторые
экспериментальные данные, полученные для механически
активированных оксидов вольфрама, ниобия и тантала.
Оксиды ниобия и тантала. Для Nb2O5 известно большое
количество различных модификаций 1442]. Есть мнение, что
Nb2O5 существует при обычных давлениях в двух кристал-
лических модификациях — низкотемпературной и высоко-
температурной [408]. Все остальные формы представляют
разные степени упорядоченности этих модификаций.
Андерсоном и Вадсли [408] показано, что структуры
кристаллографического сдвига образуются в результате по-
явления протяженных дефектов па границах зерен и блоков
(дефекты Вадсли). Состав образующихся при изменении
стехиометрии соединений вписывается в гомологичный ряд
^1еап+1О8п _2.
Высокотемпературная форма //-Nb2O5 представляет со-
бой структуру, состоящую из прямоугольных блоков, со-
держащих 12(3x4) или 15(3x5) кислородных октаэдров,
соединенных вершинами. Внутри октаэдров размещаются
ионы ниобия. Блоки соединены вместе вдоль линий, преры-
вающихся ионами ниобия, находящимися в тетраэдрическом
положении (см. рис. 34).
Превращения высокотемпературной //-формы в другие
не наблюдалось даже при продолжительной температурной
обработке различных форм Nb2O6 и их смесей [445]. Однако
Ададуров, Бреусов и др. [446] отмечали переход из //-формы
142
в Т’-форму под действием высоких давлении и при ударпом
сжатии в интервале давлений (1,5 ± 2,5) -108 Па и Т —
— 1100 К. По данным Тимура [447], для перехода Н Т
АН = —12,6 кДж/моль.
Механическую обработку оксидов ниобия проводили в
планетарной мельнице с использованием барабанов и шаров
из стали, а в некоторых случаях из керамики на основе окси-
да алюминия. Скорость вращения и масса измельчаемого
вещества варьировали [448].
Днфрактограммы фаз, возникающих при механической
обработке пентаоксида ниобия в зависимости от времени,
приведены па рис. 49. В результате механической обработки
исходной 77-формы Nb2O6 в планетарной мельнице наблюда-
лось появление повой фазы, соответствующей Т’Т’-форме
Nb2O6 (спектр 3) с псевдогексагональпой решеткой, имеющей
параметры а — 0,3608, с = 0,3936, что согласуется с данны-
ми, приведенными в работе Д449]. Авторы ее считают, что
7'7'-форма представляет собоп^ плохо окристаллизованную
Т’-форму, которая изоструктурна подрешетке типа a-U03,
исследованной Захариазеном [450]. В образующейся новой
фазе октаэдры соединяются посредством ребер, в то время
как в исходной 77-форме они соприкасаются друг с другом
через вершины [451].
При объяснении протекания фазовых переходов под
влиянием механической обработки, как указывалось выше,
определенная роль отводится размерным факторам. Измель-
чением 77-Nb2O5 в присутствии воды получены частицы раз-
мером в 2—3 меньше, чем при сухом помоле, однако полного
перехода в 7’7'-форму не наблюдалось. Очевидно, в данном
случае размерные факторы не играют определяющей роли.
Мы полагаем, что в результате механического воздейст-
вия решетка уплотняется за счет вынужденной перестройки
октаэдров /7-формы. Уменьшение расстояния между атома-
ми в блочной структуре, а также замена тетраэдрической
координации ионов ниобия на октаэдрическую указывают
на то, что переход Н —> ТТ относится к превращению со
смещением во второй координационной сфере. Плотность
оксида при переходе из //-формы в ТТ’-форму изменяется
от 4,55 до 4,99 г/см3.
Перестройка октаэдров происходит в результате измене-
ния стехиометрии оксида, вызываемого рядом причин. Одна
из них — взаимодействие оксида с материалом барабана и
шаров (металлическое железо). Стехиометрия может изме-
няться и но другим причинам, в частности, при диспергиро-
вании в отсутствие восстановителя (керамические шары и
143
Рае. 49. Дифрактограммы П-формы Nb2O6, подвергаемой активации
в течение разного времени [448].
Условия активации: стальные шары и барабаны, ускорение 40g, масса шаров
0,2 кг. Продолжительность активации, мин: 1 — исходный образец, 2 — 5, 3 —
10, 4—25, 5 — 150.
барабаны) может происходить частичная механохимическая
диссоциация оксида. И действительно, как показано Андер-
соном и др. [407, 452], которые использовали высокоразре-
шающую электронную микроскопию и наблюдали пепос-
Рис. 50. Время полного превраще-
ния 7/-Nb2O5 -> 7’7’-Nb,O5 -> NbO2 в
зависимости от величины подводи-
мой механической энергии.
Области сущестношшия: 1 —фаза W-NbsOs;
JI — фаза TT-Nb2OB; III — NbO?.
редственно блоки Я-формы
Nb2O6, вдоль линии дислока-
ции, остановившейся у меж-
зеренной границы, возникает
новая структура со значитель-
ным изменением стехиометрии
(средний состав около NbO2i48).
При механической актива-
ции Та2О5 наблюдается переход
ромбической формы [3-Та2О5 в
псевдогексагоиальную б-Та2О5
Мощность на единицу
массы вещества, Вт/г
с параметрами а = 0,362, с = 0,387 им. В этом случае, так
же как и для Nb2O5, механически стимулированный пере-
ход, сопровождается изменением стехиометрии Та2О5 [448].
При длительной активации наряду с фазовым переходом
наблюдается механохимическое разложение Nb2O5. Конеч-
ным продуктом восстановления является NbO2 (рис. 49,
спектр 5). Получающийся диоксид ниобия имеет тетраго-
нальную решетку с параметрами а = 0,484, с = 0,298 нм,
что согласуется с данными Шафера и Роу [442].
Па рис. 50 представлена кривая зависимости времени
полного перехода Я-формы (область I) в 7'7'-форму (область
II) и далее в NbO2 (область III) от подводимой мощности
при обработке в стальных барабанах со стальными шарами.
Время полного перехода определялось по полному исчезно-
вению линий исходной фазы на рентгенограммах, получен-
ных для образцов после механической активации. При тех же
условиях обработки (мощность, масса шаров и др.) в кера-
мических барабанах с керамическими шарами переход в
П’-форму происходит в 2,5 раза медленнее. Это свидетельст-
вует о том, что материал шаров и барабана (Fe) выступает
как восстановитель, ускоряющий процесс восстановления
оксида.
Отметим, что ранее была получена фаза переменного со-
става NbxO2 (0,83 х 1,0) при воздействии сильных
ударных волн па оксид ппобия (V) [453]. Авторы полагают,
что это — метастабпльпые твердые растворы на базе Nb2O5.
Опп отмечают, что метастабпльпые фазы однородны по со-
145
144
Таблица 8
Области существования различных модификаций WOj.x [399]
Сингония Точечная группа Решеточ- ные пара- метры, нм Температур- ный интервал существования । фазы, К Состав фа- зы по кис- лороду Валент- ность вольфра- ма Плотность, г/см*
Триклинная Р\!тп —D1^ ggg 0" 0 0" II II II и 233—290 3-2,99 6-5,99 7,29
Моноклин- РтаЪ — D11 а=0,383 2,99- 5,99-
ная 6=0,752 290—600 7,1/
с=0,728 2,98 5,97
Орторомби- ческая P2a-C|h С- R 11 11 О О '^4 00 •ТОО—юос 2,98— 5,97- 5,94 7,17
с -0,384 2,97 7,14
Теграго- /', - С1 а 0,525 Выше 2,97 — 5,9'1—
нальная с-0,391 1050 2,89 5,78
ставу п хорошо окристаллизованы. По-видимому, авторы
также наблюдали образование структур кристаллографи-
ческого сдвига.
Оксид вольфрама (VI). WO3— соединение переменного
состава, имеющее несколько полиморфных модификаций.
Строительным элементом различных модификаций является
октаэдр, в центре которого находится атом металла, окру-
женный шестью атомами кислорода. Все модификации
более или менее упорядоченные вариации кубической решет-
ки типа Re03 (Pm3m) [399].
Полиморфизм WO3 был исследован разными авторами
[399], которые нашли области существования полиморфных
модификаций в зависимости от температуры и состава и
пришли к выводу, что состав изменяется в сторону умень-
шения кислорода в направлении триклинная —► тетраго-
нальная модификация (табл. 8).
Образование кубической решетки WO3, полного аналога
решетки Ве03, затруднено, так как валентность вольфрама
должна быть 5,78, а х выше 0,11. Ввиду такого дефицита
кислорода решетка трансформируется раньше, чем полу-
чается кубическая структура. И в самом деле, уже для со-
става ниже WO2>B образуются сдвиговые структуры как
наиболее устойчивые образования. Концентрация, при ко-
торой совершается переход беспорядочно распределенных
вакансий к сдвиговой структуре, по данным раооты [399 J,
составляет 2-1021 вакансий/см3.
Плоскости сдвига могут формироваться в разных на-
правлениях. Для структур типа ReO3 характерно образова-
ние гомологического ряда М„О3„_2 с /1 = 8, 9, 10, 11, 12 и
14, отдельные гомологи которого отличаются лишь частотой
повторения плоскостей сдвига. Так, сдвиговые структуры
WO3 имеют состав W20O58, W24O70 п т. д.
Механическая активация WO3, имеющего орторомби-
ческую структуру, проводилась в вакуумной планетарной
мельнице с термостатированным барабаном (284 К). Усло-
вия активации: загрузка вещества 6 г, шары стальные 200 г
частота вращения 740 об/мин. В ходе активации регистриро-
валось газовыделение образцовым вакуумметром. Сравни-
вая количество газа, выделившегося при активации WO3,
с количеством выделившегося при активации диоксида свин-
ца, который в данных условиях разлагается практически
полностью, мы рассчитали изменение стехиометрии WO3
[413]. Приводим результаты этого расчета:
т, мии 10 20 30 40 50 60
х 0,017 0,028 0,039 0,053 0,071 0,091
Из сопоставления с данными [454] видно, что диссоциа-
ция оксида WO3 идет в планетарной мельнице до более глу-
боких степеней восстановления, чем в вибрационной мель-
нице, в которой состав изменяется до WO., 98.
Поело механической активации образцы исследовали ме-
тодами рентгенофазового анализа, ПК-спектроскопии, опре-
деляли удельную поверхность газоадсорбцпонпым методом
и плотность пикнометрическим методом. И НК-спектре пос-
ти 6ОГ.М11П паблюдается сдвиг полосы валентного колебания
W — О в длинноволновую область, что характерно для W03,
имеющего изменения в составе я > 0,10 [455].
При исследовании ооразцов W03 методом термического
анализа оыло установлено, что при нагревании в воздушной
среде на кривых ДТА в области температур 400—700 К по-
является значительный экзотермический эффект. Для исход-
ного образца такой экзоэффект отсутствует. По сопоставле-
нию площади пика с площадью известного теплового эффекта
9апУитН0/ ЧТ° °Н составляет 77 кДж/моль для 10 мин и
2У0 кДж/моль до 30 мин активации. Замечено, что экзоэф-
фект исчезает, если кривая ДТА снимается в среде аргона.
По нашему мнению, исчезновение экзоэффекта связано с
окислением частично восстановившегося при механической
активации оксида.
Сравнение значения теплового эффекта для оксида после
активации с тепловым эффектом окисления оксида низшей
146
147
Рис. 51. Изменение удельной поверхности п плотности WO3 под влия-
нием механической активации.
А — триклинная, В — моноклинная, С — орторомбическая, D — тетрагональная.
валентности показывает, что он непропорционально велик,
Тепловой эффект окисления WO2 до W03 составляет
270 кДж/моль [456]. Сопоставляя эти данные с приведенны-
ми для механически активированного оксида, можно прийти
к выводу, что содержание низшего оксида должно находить-
ся на уровне 70%, что не согласуется с данными по газовы-
делепию. Нельзя получить такие значения и за счет кристал-
лизации аморфных частиц.
В рассматриваемом случае экзоэффект обусловлен, не-
видимому, другими причинами, в частности образованием
дефектов нестехиометрии сдвигового типа. На образование
таких сдвиговых дефектов затрачивается весьма значитель-
ная энергия. Из работы [457 ] следует, что для реакции
WO3 = WO2>81 (структ. сдвиг) + -^О2
148
требуется энергия порядка 240 кДж/моль, что близко к
значению, полученному памп по данным ДТА для W03
после активации.
Поскольку прн образовании сдвиговых структур проис-
ходят переориентация октаэдров и их уплотнение, плотность
сдвиговых структур должна быть выше плотности исходно-
го оксида [406, 458]. Эксперимент показывает (рис. 51), что
плотность в зависимости от времени активации проходит
через минимум. Снижение плотности WO3 может свидетельст-
вовать о том, что образованию сдвиговых структур предшест-
вует образование кислородных вакансий. Отметим, что, по
данным Вержбицкого и др. [630], наблюдается минимум
па кривой диэлектрических потерь в зависимости от времени
активации и для других неорганических веществ, что может
свидетельствовать о развитии вторичных процессов, в ре-
зультате которых образовавшиеся точечные дефекты исче-
зают или превращаются в дефекты другого типа.
3.3.1.4. Структурно-химические изменения
в оксидах со структурой типа корунда
Структуру типа корунда можно рассматривать как со-
стоящую из гексагонально плотноупакованпых ионов кисло-
рода и трехвалентпых атомов алюминия, занимающих 2/3
октаэдрических узлов. Поскольку атомы металла занимают
октаэдрические узлы, каждый из них окружен шестью ато-
мами кислорода, в то время как каждый атом кислорода
находится в окружении четырех атомов металла. Катионы
попарно занимают соседние октаэдрические узлы решетки,
и два соответствующих октаэдра А1О6 имеют общую грань
(см. рис. 32). Кроме а-А1303 структурой типа корунда обла-
дают оксиды <z-Fe3O3, Сг3О3, Ti,03 и др. Нами для исследова-
ния взят оксид a-Fe2O3v
Влияние активации на каталитическую активность п на
восстановление оксидов железа уже исследовалось [354, 355,
361, 374]. В основе изменения свойств лежат такие наруше-
ния структуры оксидов, которые рентгеноструктурпыми
методами определить трудно, так как вещества становятся
рентгеиоаморфными. Поэтому для исследования изменений,
происходящих в оксидах железа, нами применен метод
ИГР [461].
Дефекты кристаллической структуры, влияя на характер
распределения электронной и спиновой плотности вблизи
мессбауэровского ядра, могут приводить к изменению элект-
149
Рис. 52. Спектры гематита после механической активации в течение
15 (/), 30 (2), 60 (3) и 90 мип (4).
Температура измерения. К: а — 300, б — 78,
рического и магнитного сверхтонких взаимодействий ядра с
окружением [459]. В работе [460] показано, что остаточные
дефекты, возникающие при измельчении оксидов железа,
приводили к уширению компонентов мультиплетной струк-
туры мессбауэровских спектров, по положение линий оста-
валось прежним. Такое поведение связывалось с изменением
константы квадрупольпого взаимодействия и появлением
вариации угла между направлением градиента электри-
ческого поля и направлением эффективного магнитного' по-
ля на ядрах 57Fe.
Для механической активации a-Fe2O3 нами использова-
лась планетарная центробежная мельпица ЭИ-2 X150 с кера-
мическими барабанами и шарами [119, 461].
Мессбауэровские спектры a-Fe2O3 после механической
активации, снятые с источником из нержавеющей стали при
300 и 78 К, представлены на рис. 52.
Для активированного порошка <z-Fe2O3 линии в спектре
уширепы па 50—60% по сравнению со спектром исходного
порошка. Особенно заметен эффект уширения по изменению
интенсивности счета на крыльях линий. Кроме того, линии,
особенно крайние, проявляют асимметрию — внутренние
склоны более пологие. Уже после 15 мип активации гемати-
та средняя величина магнитного поля На уменьшается
до 506 кЭ (при 300 К), а температура Морина Ты лежит
ниже 78 К (в исходном образце Ты = 260 К). Такое пове-
дение Ты типично как для мелкодисперсного, так и для де-
фектного гематита [462—467]. Появление после 30 мин
активации в спектрах дублета и зависимость формы спектра
150
от температуры позволяют оцсппть размеры частиц порядка
20 нм [1101. Активация в течение 90 мин ведет к образованию
спектра в виде дублета с квадрупольным расщеплением
0,93 мм/с. Максимально достигнутое значение составляет
1,05 мм/с, и ему соответствует размер частиц ~ 5 нм. Если
учесть влияние дефектообразованпя па квадрупольное рас-
щепление, эти оценки следует считать несколько занижен-
ными.
Относительно природы несимметричности уширения ли-
ний в спектрах ЯГР a-Fe2O3 авторы [119] предположили, что
наблюдаемые спектры отвечают суперпозиции компонент,
связанных с исходным веществом и веществом, образовав-
шимся в результате активации и находящимся в магнито-
упорядоченном состоянии. При активации происходит изме-
нение относительного содержания таких компонентов. Прп
большой длительности активации в спектрах появляются
широкие линии се — 0,10 мм/с, в которых затруднительно
выделить сколько-нибудь интенсивную компоненту от исход-
ной окиси железа. Па этом этапе все вещество становит-
ся дефектным.
Для a-Fe2O3, так же как и для других оксидов, проводи-
ли активацию в вакууме, одпако газовыделепис в этом слу-
чае отсутствовало. Тем пе менее в спектре образца а-Ре^Оэ,
который после 30-мпнутной активации отжигали в среде
аргона при температуре 1000 К 30 мип, обнаруживается
двухвалентное железо (рис. 53). После отжига исходных
образцов этого не наблюдалось, что может свидетельствовать
об ослаблении связи Fe—О в результате разупорядочения
решетки прп активации.
С целью выяснения влияния условий механической акти-
вации на характер превращений cz-Fe2O3 активацию прово-
дили в стальных барабанах в водной и воздушной средах.
Здесь картина совершенно иная, чем при активации в кера-
мических барабанах. В обоих случаях имеет место хими-
ческое превращение, переход оксида железа в магнетит, оче-
видно, за счет восстановления материалом шаров и барабана
(сталь). Наряду с этим спектры полученного таким образом
магнетита свидетельствуют о существенных нарушениях
кристаллической структуры, особенно при пзмельчении в
воздушной среде, что выражается в уширении линий, умень-
шении Н3 и появлении в последнем случае парамагнитной
компоненты. В спектре образца, активированного в воде,
линия Fe2+ уширена сильнее, чем линия Fe3+. Интересно от-
метить, что химический сдвиг парамагнитной компоненты
в спектре после воздушной активации совпадает со сдвпгом
151
1
Рис 53 Спектп гематита после ЗО-мппутпой активации и отжига в сре-
£Гарина npu%W К В течение 30 мин (2) спектры исходного гемати-
г 1 та (1\ и магнетита (3).
липин l('e'2t самого магнетита (~ 0,8 мм с). Эго, ио-впдпмому,
подтверждает предположение о том, что часть атомов не на-
ходится в магнитном состоянии, а спектр представляет собой
суперпозицию исходного и механически активированного
образца.
Исследования механически активированного железа ме-
тодом ЯГРС позволили оценить размеры конечных частиц
(порядка 5—7 нм), установить изменение магнитных свойств
(снижение температуры Морина с 260 до 78 К), показать,
что, как и в других случаях, при механической активации
реализуются восстановительные условия, в результате ко-
торых имеют место частично механохпмпческое разложение
и переход Fe3+ в Fe3+. С этим может быть связано увеличение
каталитической активности механически активированного
a-Fe2O3 при использовании его как катализатора окисления
SO2 до SO3.
Таким образом, из совокупности данных, полученных
для оксидов металлов IV, V, VI и VIII групп, следует, что
иод влиянием активации в них в той или иной степени идет
процесс мехапохимпческоп диссоциации с образованием
нестехиометрических соединений с дефицитом кислорода.
Этот процесс выражен сильнее для оксидов с малой энергией
связи кислород—металл, имеющих неплотную упаковку
кислородных октаэдров, содержащих внутри ион металла.
Возникающие в результате мехапохимпческоп диссоциации
дефекты упорядочиваются в плоскости кристаллографи-
ческого сдвига, которые, в свою очередь, упорядочиваются
в структуры кристаллографического сдвига. Этот процесс
зависит от электронного строения катионов и в большей сте-
пени характерен для оксидов переходных металлов с запол-
няющейся d-оболочкой (Ti, W, Nb) и возможным образованием
связей металл—металл, а в меньшей мере — для оксидов
металлов, у которых на внешней оболочке находятся s- и
р-электроны (Sn, РЬ).
Это позволяет рассматривать механическую активацию
как простой метод получения нестехпометрнческпх соеди-
нений с дефектами по механизму кристаллографического
сдвига, обладающими новыми свойствами по сравнению с
исходными. В этих соединениях наблюдается ускорение диф-
фузионных процессов [468], изотопного обмена кислорода,
перестройка их приводит к эмиссии синглетного кислорода,
ответственного за специфичность действия катализаторов
]640], пт. д.
Реализации при механической активации высоких дав_
лопни приводит в итоге к отличным от термического процес.
153
са результатам. Это хорошо видно па примере Р-РЬО2, при
механохимнческом разложении которого образуются фазы
высокого давления, отсутствующие при термическом разло-
жении. Из этого следует, что развитие механохимического
процесса может быть в какой-то степени предсказано из
р — Т диаграмм состояния твердых тел. Однако наряду со
статической компонентой в этих процессах большую роль
играет сдвиговая компонента, вызывающая пластическое те-
чение материала и ускорение кинетики фазовых превра-
щений.
3.3.2. Сульфиды металлов
Вопросы механической активации сульфидов представ-
ляют интерес с точки зрения совершенствования и разработ-
ки новых методов переработки руд цветных металлов, в част-
ности гидрометаллургических.
Влияние топкого измельчения на свойства сульфидов ис-
следовано Малиновским [469] еще в начале 30-х годов. Им
доказано образование растворимых солей в ходе дисперги-
рования сульфидов в водной среде, что вызывает потери цен-
ных продуктов и в той или иной мере влияет на ход техноло-
гического процесса. Например, образующийся при окисле-
нии халькопирита сульфат меди преждевременно повышает
гидрофобность цинковой обманки при флотации.
Бочаровым, Голиковым и Митрофановым [4701 исследо-
вана кинетика окисления сульфидов в процессе измельчения
в условиях доступа кислорода. Опыты проводились в шаро-
вой фарфоровой мельнице при соотношении масс твердого
вещества, воды и шаров 1:2: 10. Скорость окисления конт-
ролировали по убыли кислорода. Было показано, что при
pH 1,5—8,4 в течение длительного времени измельчения по-
глощения кислорода не наблюдается. При pH выше 8,4
скорость окисления возрастает, одновременно происходит
значительное понижение pH раствора. Установлено", что в
процессе окисления пирита происходит образование сернис-
того железа (за 4,8-103 с до 3%), а в растворе появляются
ионы S2-, SOg“, S20g-, политиопаты (соответственно 4,4-10'J,
3-10'4, 7,3-10'3, 4,14-10'3 моль/дм8).
Высказано предположение, что окисление пирита при
измельчении происходит, по-видимому, путем первичной
диссоциации пирита с выделением одного атома серы и обра-
зованием сернистого железа с последующим окислением
серы до сульфатов, а затем до других окисленных состояний.
Считается, что реакция окисления сульфидной серы до суль-
154
фат-ионов протекает ступенчато, слагаясь из более про-
стых стадий, с образованием промежуточных кислородных
соединений серы низшей валентности. В нейтральных и ще-
лочных растворах окисление идет по схеме
S'2" -> s2or -* sor -> s2_eor -> SO;~,
а в кислых растворах
s2-->s0->sor.
Скорость окисления сульфидных минералов под влиянием
механической обработки в щелочной среде уменьшается
по ряду
CnS, ChFcS2, FeS2, ZnS, Cu2S.
Как результат окисления механически активированных
сульфидов водой в щелочной среде, Молчановым наблюда-
лось выделение водорода [471, 472]. Образование различных
сульфатов двух- и трехвалептного железа зафиксировано
Кулебакиным [473] при механической активации в водной
среде сульфидов железа (пирит, пирротин).
Тюленевым, Марксом и др. [474] методом ЯГРС иссле-
дованы продукты диспергирования пирита и пирротина в
различных мельницах, а также в разных условиях. Анализ
мессбауэровских спектров диспергированного пирита по-
казал, что активация его в агатовом барабане (при диспер-
гировании в воздухе и воде) не приводит к изменению спектра
исходного образца. Некоторое увеличение квадрупольпого
расщепления до е = 0,70 ± 0,03 мм/с в спектре образца,
диспергированного в стальном барабане с водой, по сравне-
нию с исходным свидетельствует о возможном образовании
y-FeOOH. Асимметрия дублета в этом случае указывает на
появление магнитной компоненты, соответствующей
Fe^S. Образование Fej_TS происходит за счет взаимодейст-
вия FeS_, с железом из стенок барабанов и шаров.
По данным [474], диспергирование пирротина в агато-
вом барабане практически не сказывается па структуре
части спектра, отвечающей исходному веществу. При дис-
пергировании в стальных барабанах (в воде) наблюдается
некоторое ухудшение разрешения спектров, а при сухом
измельчении, наряду с уширением линий, имеет место и
сдвиг крайних линий к центру, так что среднее расстояние
между крайними пиками становится равным — 21,6-10“ А/м.
Кроме таких изменений во всех случаях в спектрах по-
является новая дублетная асимметричная линия (в центре),
155
соответствующая Fe3* в парамагнитном состоянии. Парамет-
ры дублета — химический сдвиг б = 0/4 ± 0,05 мм/с и
квадруполыюе расщепление е == 0,72 ± 0,05 мм/с — не соот-
ветствуют параметрам для обычных кристаллических образ-
цов оксидов и гидроксидов железа (a-, y-Fe2O3, а-, [1-,
y-FeOOH, Fe(OH)3 [475]), а также Fe2(SO4)3 [475]. В то же вре-
мя наблюдается соответствие параметров дублетной линии
и спектров высокодпсперсного (~ 6 нм) слабокристаллизо-
ванного феррогеля y-FeOOH (б = 0,45 ± 0,02 мм/с, е =
= 0,77 мм/с [4761). Косвенным подтверждением того, что
дублетная линия в спектрах диспергированного пирротина
соответствует y-FeOOH в слабокристаллизованном состоя-
нии, является тот факт, что интенсивность линии максималь-
на при измельчении вещества в воде.
Наблюдаемые изменения в спектре пирротина, дисперги-
рованного в мельнице со стальными барабанами на воздухе
(т = 30 мни), авторы объясняют уменьшением х до значений
0,07 х 0,10 вместо 0,11 а. 0,13 для исходного
образца за счет взаимодействия сульфида с металлическим
железом.
Под влиянием механической активации возрастает сте-
пень взаимодействия сульфидов со слабыми кислотами.
В работе [477] приведены данные по кинетике растворения
гексагонального пирротина в солянокислом растворе
(pH 2), активированного в водной среде в различные проме-
жутки времени, и показано, что степень его взаимодействия
коррелирует с удельной поверхностью. Активированный
сульфид железа может быть использован для выделения пла-
тиновых металлов, меди, цинка и никеля из водных раство-
ров, так как произведение растворимости сульфидов этих
металлов значительно ниже, чем сульфида Железа: CuS —
3,2-10~38, а-ZnS - 6,9-Ю'26, р-ZnS - 1,1-IO'24, a-NiS-
3-10-21, 0-NiS - 1 -10“26, y-Ni'S - 2-10"28, FeS - 3,3-10~19.
Более растворимый в этих условиях сульфид железа будет
осаждать другие сульфиды.
Свежепзмельченный тонкодисперсный порошок пирроти-
на в качестве copt5eirra для платиновых металлов предложен
в работах [477, 478]. Достигнута максимальная емкость
пирротина по платиновым металлам, мг/г: осмий — 3000,
палладий — 2300, платина — 23,2, родий — 14,0, руте-
ний — 5,0, иридий — 2,2. Авторами высказано предполо-
жение, что выделение платиновых металлов из растворов в
осадок происходит за счет образования их сульфидов, вос-
становления катионов платиновых металлов двухвалент-
ным железом до металлического состояния, а также в ре-
156
зультате поверхностной адсорбции па нерастворпвшемся
пирротине и на свежеосажденных сульфидах.
Растворение смесей сульфидов отлично от растворения
индивидуальных сульфидов. В смесях имеют место электро-
химические процессы, возникающие вследствие соприкосно-
вения частиц сульфидов, обладающих различными электро-
химическими потенциалами. При совместном растворении
двух сульфидов растворение одного из них может ускорить-
ся, а другого — замедлиться. По данным работы Свешнико-
ва и Кедрипского (169], сульфиды в порядке уменьшения
электродного потенциала располагаются в ряд: FeS2, CuFeS,,
FeS, PbS, ZnS.
Наибольшее взаимное влияние оказывают друг па друга
крайние члены ряда. Болес электроположительный ускоряет
растворение более электроотрицательного. Добавка пирита
к чистому сфалериту ускоряет выщелачивание последнего в
кислых растворах с выделением элементарной серы, но в
то же время выщелачивание самого пирита замедляется.
Повышение дисперсности сульфидов будет увеличивать пло-
щадь соприкосновения сульфидов и растворителя, следова-
тельно, ускорять их переход в растворимые формы.
Механическая активация в инертной пли воздушной сре-
де приводит пе только к увеличению поверхности, но и к по-
вышению их химической активности, проявляющейся в уве-
личении степени термической диссоциации, окпслеппя, де-
сульфуризации, восстановления. При этом снижается тем-
пература начала этих процессов. Так, температура терми-
ческой диссоциации пирита существенно снижается, и дис-
социация становится уже весьма значительной при 473 К.
Данные по равновесной диссоциации, удельной поверхности
и растворимости в 3 н. H2SO4 механически активированного
в воздушной среде пирита приведены в работе [479]. Эти ве-
личины проходят через максимум, соответствующий одному
времени активации. Наличие максимума связано, по-види-
мому, с образованием молекулярно-плотных агрегатов, ко-
торые образуются при сухом диспергировании сульфидов.
При окислительном обжиге, как индивидуальных суль-
фидов железа и меди, так и медного концентрата, степень
десульфуризации повышается на 12—30?о, а температура
начала воспламенения сульфидов снижается па 70—100 К
после обработки в планетарной мелышце в течение 2—15 мип
[480, 481J.
По данным Смагупова, Рейнгольда, Молчанова [482],
предварительная механическая активация влияет па меха-
низм окисления пирога. Сопоставляя окисление механически
157
активированных и неактивированных образцов одинаковой
дисперсности, они показали, что для неактивированного об-
разца окисление начинается при температуре около 693 К
и идет до магнетита, а при нагревании выше 733 К — до ге-
матита, для активированного же окисление начинается при
температуре около 573 К и идет с образованием сульфатов
трех- и двухвалентного железа.
Весьма интенсивное окисление сульфидов идет в момент
механической обработки. Быстриков и Бутягип [282] пока-
зали, что первой стадией окисления дисульфида молибдена
является хемосорбция кислорода па короткоживущих актив-
ных центрах, образующихся при его разрушении и имеющих
концентрацию, близкую к таковой поверхностных атомов.
Ускорение стадии хемосорбции под влиянием механической
активации ускоряет процесс окисления в целом.
Предварительная активация халькопиритпых концентра-
тов в воздушной среде обеспечивает высокую степень извле-
чения меди при последующем автоклавном окислительном
выщелачивании концентрата [483—485].
Бьерлингом и Лесидренским [486] для получения более
растворимых простых сульфидов из сложных предложен ме-
тод механотермической активации, заключающийся в том,
что между сложными сульфидами и восстановителями про-
водят твердофазное взаимодействие во вращающейся печи с
одновременным ее нагреванием до 700 К. Реакции идут при
этом с большими скоростями и практически до конца. Таким
способом могут быть активированы сульфиды меди и железа
по реакциям: CuFeS2 + Си = Cu2S -f- FeS; FeS2 -f- Fe =
= 2FeS. Наиболее вероятным для этих реакций авторы
[486] считают стадийный механизм. Они полагают, что пер-
вой стадией являются реакции термического разложения ми-
нералов: 4CuFeS2 = 2Cu2S + 4FeS + S2; 2FeS2 = 2FeS -f-
+ S2. Обе эти реакции эндотермические. Второй стадией
являются экзотермические реакции: 4Cu + S2 = 2Cu2S;
2Fe + S2 = 2FeS.
Энтальпия суммарных реакций составляет уже отрица-
тельную величину, и реакция в условиях, когда в результате
трения при вращении печи сульфидизированный слой исти-
рается и обнажает металлическую поверхность для даль-
нейшей сульфпдизации, становится весьма вероятной.
Аналогичной точки зрения придерживаются Рейнгольд
и Смагупов [487], которые исследовали мехапохимическую
реакцию
FeS2 + 2Ag = Ag2S + FeS.
158
По их мнению, эта реакция также идет в две стадии, пер-
вая — диссоциация пирита, вторая — взаимодействие серы
с серебром.
Использование напряженных нзмельчитсльных аппара-
тов позволпло проводить эти реакции без внешнего источни-
ка тепла [479, 488]. Исследовалось взаимодействие пирита
с металлическим железом. Было показано, что стадия терми-
ческой диссоциации пирита не лимитирует реакцию, поэтому
реакция протекает без участия этой стадии путем прямого
взаимодействия указанных компонентов [488].
Активация заметно влияет на скорость и полноту возгон-
ки и восстановления сульфидов. Благодаря механической
активации восстановление сульфидов свинца или цинка по
реакции
MeS 4- СаО -|- С = Me -| CaS СО |- СО,
возрастает почти в два раза, а температура начала восста-
новления снижается па 100—150 К [481].
Из приведенного, весьма краткого, обзора работ по ме-
ханической активации сульфидов следует, что с ее помощью
могут быть существенно ускорены процессы окисления и
растворения сульфидов п может даже измениться их меха-
низм. Поэтому необходимы широкие теоретические исследо-
вания с целью более обоснованного применения методов ме-
ханической активации в технологии.
3.3.2.1. Химическая связь
и дефектообразование в сульфидах металлов
Химическая связь и образование дефектов в сульфидах
металлов имеют некоторые особенности по сравнению с ок-
сидами.
Как правило, химическая связь в сульфидах возникает
за счет образования различных типов sp-копфигурации ва-
лентных электронов атомов компонентов и различных ва-
риантов их перекрывания. Определяющим при образовании
сульфидов sp-элементов является высокая акцепторная спо-
собность атомов серы, которые имеют конфигурацию валент-
ных электронов $2р4, стремящуюся к достройке до стабиль-
ной конфигурации инертного газа s2p6. Пз-за многовариант-
ности возможностей образования устойчивых электронных
состояний sp-элементов в сульфидах возможно возникнове-
ние полиморфных фаз.
Согласно Белову [489], в отличие от оксидов и хлоридов, в
которых «благородногазовая» форма атомов достигается за счет
159
кулоновских взаимодействий, в сульфидах при образовании
связей реализуется другой принцип, а именно: после отда-
чи определенного количества s- и /^-электронов катионы фор-
мируют вокруг себя 18-электронную оболочку криптона
(4s24p63d10) с помощью донорно-акцепторной связи, кото-
рую «рыхлый» анион серы o6pa3veT легко.
Например, в ZnS (сфалерит) потерявший в пользу аниона
два валентных s-электрона Zu2*- сохраняет десять d-электро-
нов и для криптоподобной конфигурации нуждается в вось-
ми электронах: соответствующие четыре пары он получает по
донорпо-акцепториому механизму от четырех соседних, уже
сформировавшихся анионов р S :j2 По этой причине цинк
в ZnS имеет координационное число 4 в обеих кристалли-
ческих модификациях (сфалерит — кубическая решетка с
а = 0,542 нм; вюртцит — гексагональная решетка с а =
= 0,384 нм, с — 0,628 им [490]).
По-иному образуется химическая связь в сульфидах же-
леза (FeS и FeS2) [489]. У иона серы S2- наружная восьмер-
ка электронов 3s23p6 слабо связана с ядром, и при образова-
нии связи S2- анион выделяет не более четырех донорских
пар. Когда же этого числа пар для стабилизирующихся по
правилу «18» катионов металлов не хватает, тогда приходит
на помощь укрупнение анионов с большим числом донорских
пар, например с образованием S2~ с шестью парами донор-
ных электронов, либо обращение к металлической связи —
непосредственному обмену недостающим числом электронов
между сближающимися катионами.
Примером первой ситуации является FeS2, существующий
в двух кристаллических модификациях (марказит — ромби-
ческая решетка с а — 0,355 нм, Ъ = 0,440, с = 0,535 им,
пирит — кубическая решетка с а = 0,540 нм). Атомы серы
в FeS2 спариваются в анионную гантель р S • S , и уже
эти анионы S®- перекладываются одиночными катионами Fe2+
по типу NaCl (к. ч. 6). Расстояние S — Sb пирите состав-
ляет 0,212 нм, а в марказите — 0,221 нм. Переход от марка-
зита к пириту происходит при температуре 723 К.
Примером второй ситуации является структура пирро-
тина FeS. Уступивший атому серы два валентных s-электро-
иа катион Fe2+ с шестью электронами нуждается в шести па-
рах добавочных (акцепторных) и соответственно размещает-
ся в центре октаэдра, но каждый анион в состоянии предоста-
вить только четыре пары, а при невозможности организовать
Рис. 54. Структурный мотив в FeS. Черные
шары внутри октаэдров — атомы железа [489].
более сложный анион S2- в пирроти-
не октаэдры выстраиваются в колон-
ки, ложась один на другой общей
гранью (рис. 54). Этот прием, пе реко-
мендуемый правилами Полинга для ион-
ных соединений,в пирротине позволяет
атомам железа непосредственно обме-
ниваться собственными электронами,
что отражается на физических свойст-
вах пирротина. FeS является соедине-
нием переменного состава и меняет
физико-химические свойства в зависи-
мости от содержания серы. Формулу
соединения записывают обычно в виде
Foj-^S, где х — избыток серы по отношению к стехиометри-
ческому составу. При увеличении содержания серы в FeS
возникают вакансии железа, которые взаимодействуют по
типу отталкивания.
Для сульфидов характерно образование структур с плот-
нейшей упаковкой анионов серы, а катионы, которые по раз-
меру значительно меньше анионов, занимают пустоты меж-
ду анионами. Общее число пустот в 3 раза больше, чем анио-
нов, из них 2/3 составляют тетраэдрические пустоты, а 1/3 —
октаэдрические. Например, структура Р-ZnS (вюртцит) соот-
ветствует плотнейшей гексагональной упаковке анионов
АВАВАВ..., а структура a-ZnS (сфалерит) — плотнейшей
кубической ABCABGABC... [491, 492].
Однако периодичность формирования слоев по тппу
АВАВАВ... или АВСАВС... может быть нарушена, и в этом
случае возникают структуры, в которых гексагональная
упаковка чередуется с кубической. В таком случае говорят
об образовании политипных модификаций, которые отли-
чаются тем, что ближний порядок у них одинаков, а распо-
ложение атомов в далеких (третьей, четвертой п т. д.) сферах
различно. Размеры элементарной ячейки, параллельные
слоям, у них одинаковы, а различаются только длины осей
элементарных ячеек, перпендикулярных к упакованным
слоям, в кратное число раз. Это обусловливает близость
внутренних энергий политипов, тождественность пх физи-
ческих свойств н высокую вероятность их взапмопероходов.
160
Ь 13. Г Auuuhjmou
161
Таблица 9
Структура и параметры некоторых политипов
ZnS [4911
Политип Простран- ственная группа Последова- тельность слоев две (по Жданову)* Параметр гексаго- нальной ячейки, нм
а 1 с
2Н Р63тс И 0,381 0,24
ЗС F4mc оо 0,381 9,36
411 P63mc 22 0,382 12,48
GII Р63шс 33 0,381 18,72
811 Рб^тс 44 0,382 24,46
ЮН Р63тс . 55 0,382 31,20
9Н НЗт <21)я 0,382 28,08
12Н НЗт (13), 0,382 37,44
15R НЗт (З2)3 0,382 46,80
21Н НЗлп (31/2)3 0,382 65,52
* Последовательность слоев ЛВС, меняющихся в но-
рядке в+,с1 обозначается знаком «плюс», а в обратном
порядке — знаком «минус». Ciimbo.ii Жданова состоит из двух
чисел, первое из которых выражает количество следующих
непосредственно один за другим знаков «плюс», а второе —
знаков «.минус», следующих за знаком «плюс». Так, гекса-
гональная структура А_В+А_Вр будет иметь символ (11),
кубическая А_В_С_А_В-—«>, структура А_.В_С+Б+ —
(22), девятислойная ромбическая В_А+В+С_В+С+А_ — (21),
11 т. д.
Сульфид цинка имеет многочисленные политипы: 4П, 611,
8Н, ЮН, 12Н, 21R, ЗС, 6С и т. д. (цифра обозначает слои в
повторяющейся ячейке, а буква — тип решетки: II — гек-
сагональная, R — ромбическая, С — кубическая). Нор-
мальный вюртцит с ГПУ обозначается как политип тина
211, а сфалерит — типа 2С. Структура и параметры некото-
рых политипов приведены в табл. 9.
Считается, что образование столь большого числа ноли-
тпппых форм обусловлено дислокациями, возникающими
при росте кристаллов. Винтовые дислокации с различными
векторами Бюргерса создают различные политипы. Когда
вектор Бюргерса винтовой дислокации кратен целому чис-
лу высот исходной элементарной ячейки, получившаяся
структура будет такой же, как основная. Винтовая же дис-
локация с вектором Бюргерса, не кратным целому числу вы-
сот исходной ячейки (частичная дислокация), будет приво-
дить к новому политипу, период которого вдоль оси с опре-
деляется высотой выступа винтовой дислокации. Венд (4931,
предложивший такое объяснение явления политипизма, от-
мечал, что случайные ошибки упаковки в уступе винтовой
дислокации приведут к переменному политипу, поскольку
механизм спирального роста будет периодически повторять
их в структуре. К настоящему времени дислокационная тео-
рия политипизма получила экспериментальное подтвержде-
ние при наблюдении спиралей роста на поверхности иоли-
типпых структур и измерений высот ступенек спиралей.
Верма [491] показал, что для гексагональных политипов вы-
сота ступеньки спиралей роста (и, следовательно, вектор
Бюргерса дислокации) равна высоте рентгеновской элемен-
тарной ячейки, в то время как для ромбоэдрических поли-
типов она составляет одну треть от высоты, соответствующей
гексагональной ячейки.
Прп нагревании сульфида цинка до температуры 1293 К
происходит переход из кубической модификации в гексаго-
нальную (из более плотной в менее плотную модификацию,
плотность сфалерита 4,102 г/см3, плотность вюртцита
4,087 г/см3).
Установлено, что в превращении принимает участие по-
литип 3R, образующийся прп неодновременном термическом
расширении кубической модификации вдоль одного из четы-
рех направлений {111}. Кроме того, в качестве промежуточ-
ных форм выступают политипы со смешанной кубической и
гексагональной упаковкой типа 211 |491|.
Гексагональную структуру типа NiAs с плотнейшей
упаковкой ионов серы и заполненными октаэдрическими
пустотами имеет сульфид железа (элементы симметрии
D*h — СС[ттсу Размер тетраэдрических пустот мал и не-
достаточен для размещения в них двух- и трехвалептного
железа [494—502].
Наиболее интересная особенность этой структуры заклю-
чается в том, что в пей могут проявляться значительные ко-
лебания состава, образуя область составов с общей формулой
Предельными членами ряда составов являются,
с одной стороны, стехиометрический FeS (троилит), с дру-
гой — Fe7S8. Соединения, содержащие вакансии железа, ле-
жащие внутри этого интервала составов, называются пир-
роти нам и.
При высоких температурах вакапспп в ппрротппах разу-
порядочепы, а при низких они стремятся к максимальному
отталкиванию друг от друга, поэтому они упорядочивают-
ся. Например, в структуре состава Fc7S8 вакансии упоря-
дочены внутри слоев так, как показано па рис. 55, а для мак- ,
симального разделения слои укладываются таким образом,
а
• О • о •
• • • •
о »со • о
• • «Ъ« • •
• оа» о •
е
о
Рис. 55. Упорядоченное распределение вакансий в’структуре пир-
ротина [494 J.
а — размещение вакансий (белые кружки) и катнопиом слое структуры Fe-S
(указаны только атомы железа); б — размещение слоев в структуре 4с состава
Fe,s,; в — структура 5с состава Fe,S10; г — структура Сс состава F<',,S,..
чтобы вакансии не располагались одна над другой. В резуль-
тате возникает восьмпслойный блок, который вчетверо боль-
ше блока упорядоченной структуры NiAs. Так как этот
блок вытянут вдоль осп с элементарной ячейки, он назы-
вается сверхструктурой 4с.
Промежуточные пирротины в принципе могут представ-
лять либо смесь компонентов FeS и Fe7Sg, либо упорядочен-
ные структуры, которые можно построить путем прибавле-
ния дополнительных катионных слоев к структуре Fe7Sg
[494]. Если сверхструктуру 4с обозначить последователь-
ностью катионных слоев: FDaFDbFDcFDdF, где F — пол-
ный слой, a Di — слой, содержащий вакансию, то, вклады-
вая дополнительные полные слои, можно получить различ-
ные сверхструктуры и составы. Например, последователь-
ность слоев FDaFFDbFDcFFDdFDa приводит к сверхструк-
туре 5с и составу FeBS10. Последовательност!, слоев
FFDaFFDhFFDcFFDdFFDc даст сверхструктуру 6с и со-
став FeuS12 и т. д. Фактически любой состав может достичь
какой-либо степени упорядочения, хотя получающиеся
сверхструктуры очень громоздки и могут иметь периодич-
ность по оси с, не выражающуюся целым числом. Причем
степень упорядочения будет зависеть от скорости охлажде-
ния, так как наивысшая степень организации достигается
при самом медленном охлаждении. В хгриродпых ппрротн-
нах описано много различных сверхструктур или их тесное
когерентное срастание.
В каком виде будут существовать эти промежуточные
пирротины, определяется кинетикой конкурирующих про-
цессов: распад па более стабильные упорядоченные фазы
FeS и Fe7Ss пли упорядочение вакансий с образованпем ука-
занных выше сверхструктур. Упорядочение, требующее
только диффузии ближнего порядка, кинетически более *
благоприятно и может быть альтернативой по отношению
к распаду, требующему процессов более дальней диффузии.
Вследствие близости значений свободной энергии вероят-
ности этих процессов близки, поэтому природные пирротины
представляют собой метастабильпыс образования.
Формирование сверхструктуры типа Fe7S8 приводит к
изменению физических и химических свойств сульфида же-
леза. Они характеризуются более низкой скоростью само-
диффузпи железа [494], пониженной степенью взаимодейст-
вия с серной кислотой (2,32 М) — от 93% для стехиометри-
ческого троилита до 62% для пирротина Fe;Sg [503]. По
данным Ария п Морозовой [504], энтальпия их образова-
ния возрастает от 91 до 101 кДж7(моль • формулу) при изме-
нении ,т от 0 до 0,10.
3.3.2.2. Структурно-химические изменения
в сульфидах цинка, железа
и меди под влиянием механической активации
Сульфид цинка. Авторы работ [331—334] исследовали
влияние измельчения на структуру сульфида цинка и пока-
зали, что наблюдается механически стимулированный пере-
ход вюртцит -> сфалерит. Аналогичные данные получены и
нами при использовании планетарных мельниц ЭП-2х150.
Активацию проводили в следующих условиях: барабаны и
шары стальные, диаметр шаров 3 мм, загрузка сульфида 6 г,
масса шаров 200 г, частота вращения барабанов 12,4 с-1.
Использованы сфалеритная и вюртцитная модификации
сульфида цинка. После активации образцы исследовали ме-
тодом РФА.
В сфалерите под влиянием активации наблюдается толь-
ко явление аморфпзации; переход в вюртцнтпую фазу, как
это имеет место при нагревании, в данном случае отсутст-
вует.
165
Рис. 56. Фазовым переход в ZnS из втортцитной модификации в сфа-
леритпуго под влиянием механической активации.
1 — исходный вюртцит; 2 — после активации 30 мпн.
Напротив, при активации вюртцита происходит посте-
пенный переход к более плотной сфалсритной модификации
(рис. 56).
Отметим, что такой переход наблюдали и при пласти-
ческой деформации вюртцита (333, 334]. Согласно [333],
такой переход совершается через промежуточные политип-
ные модификации 6Н и ЮН, которые возникают при транс-
ляционных сдвиговых деформациях в решетке вюртцита.
Но направленности механически стимулированный фазо-
вый переход противоположен термическому. Это позволяет
считать, что движущей силой такого перехода является дви-
жение дислокаций. В результате движения и взаимодейст-
вия частичных дислокаций в плотнейшей упаковке анионов
происходит трансляционное перемещение слоев и изменяет-
ся их последовательность от типа АВ к типу АВС.
Сульфид железа. Для исследования последствий механи-
ческой активации в качестве исходных были взяты пирротин
состава Fe7Ss и сульфид железа, близкий по составу к трои-
литу (505]. Как указывалось выше, анионы S2- в структуре
Fe,S« плотно упакованы в близкую к гексагональной под-
решетку. Катионы железа в октаэдрических пустотах обра-
зуют простую гексагональную подрешетку железа с упоря-
доченными вакансиями. Причем слои, содержащие катион-
ные вакансии и не содержащие их, чередуются. Методом
ЯГР в пирротине Fe7S8 удается выделить ряд состояний ка-
тионов с различной степенью заполнения ближайших катион-
ных координационных сфер [506, 507]. Так, спектр исходно-
го пирротина представляет собой суперпозицию трех магнит-
ных сверхтонких структур с параметрами квадрупольпого
расщепления е (мм/с) и химического сдвига б (мм/с) соот-
ветственно: для позиции А (I) — 0,06 ± 0,05 и 0,72 ± 0,5;
для позиции В( II) — 0,08 ± 0,05 и 0,73 ± 0,05; для пози-
ции С (III) — 0,16 ± 0,05 и 0,74 ± 0,05.
Для позиции А в качестве ближайших соседей содержат-
ся 2 вакансии и 6 катионов, для В — 4 вакансии и 8 катио-
нов, для С — 1 ваканспя и 7 катионов (рис. 57).
Механическую активацию образцов пирротина проводи-
ли в керамических барабанах при частоте вращения 12,3 с-1.
Принимали меры, чтобы избежать окисления образцов. Мак-
симально наблюдаемое нами окисление образцов ио данным
химического анализа не более 6%.
На спектрах ЯГР пирротина (рис. 58) после активацпп
наблюдается уширение линий трех исходных структур. При
малых временах активации удается выделить две компонен-
ты: первая близка к А, вторая отвечает суперпозиции В п С.
При большой длительности активации присутствуют только
линии структуры, близкие к В-позпцпям. Наряду с этим
появляется квадрупольный дублет с расщеплением е]=
= 0,58 ± 0,05 мм/с и изомерным сдвигом 6 = 0,31 ±
± 0,05 мм/с, интенсивность которого увеличивается со вре-
менем активации.
Рис. 57. Структура пирротина.
Состояние ионов железа с раз-
личной степенью заполнения
ближайших катионных коор-
динационных сфер [506 J.
I — в ближайшем окружении на-
ходятся 2 вакансии и 6 катионов;
II — 4 вакансии, 8 катионов; III
I вакансия, 7 катионов.
По нашему мнению,
изменение спектров под
влиянием механической ак-
тивации происходит вслед-
ствие разупорядочения ка-
тионной подрешетки желе-
за [505]. Вакансии вмес-
то упорядоченного состоя-
ния распределяются слу-
чайным образом по всем
плоскостям катионной под-
решетки. Такой процесс
Рис. 58. Спектры ИГР пирротина
после механической активации [505].
идет сначала в части ве-
щества, а со временем доля
активированного вещества
возрастает. В результате
случайного распределения
вакансий в каком-то выделенном объеме возможно об-
1 — исходный образец; продолжитель-
ность ш.-гпиашш, мпп: 2 — 3, 4 — 30,
S — 45, 6 — 00.
разование области с большей концентрацией вакансий по
сравнению со средней концентрацией в исходной струк-
туре. В таких областях с большим дефицитом железа
по отношению к сере происходит изменение химического
состояния ионов железа. Этому соответствует появление
на спектрах новой компоненты дублета с б — 0,31 мм/с.
Рис. Г>9. Кинетика растворения пир-
ротина разного состава п 0,1 п. со-
лнпоп кислоте.
J — feSj ов; 2 — FeS112; а — исходный;
"б — активированный 30 мин.
Одновременно в оставшейся час-
ти вещества образуются облас-
ти с уменьшенной концент-
рацией вакансий. Такому сос-
тоянию вещества отвечают уши-
ренные линии на спектрах с
параметрами, близкими к па-
раметрам для катионов в В-по-
зицпях.
Возникновение разупорядоченпой структуры приводит к
изменению реакционной способности пирротина. Как ука-
зывалось выше, упорядоченные структуры пирротинового
ряда более устойчивы (за счет энергии упорядочения), при
взаимодействии с 0,1 н. соляной кислотой они действитель-
но растворяются хуже, чем стехиомотрпческий трон.тит
(рис. 59). Однако относительные эффекты активации на них
проявляются сильнее, чем на соединении, близком по со-
стоянию к троилиту. В последнем случае степень взаимо-
действия возрастает в 20 раз, а для пирротина — почти
в 40 раз.
Таким образом, можно считать, что усвоение механи-
ческой энергии при этом осуществляется за счет разупоря-
доченпя содержащей упорядоченные катионные вакансии
структуры пирротина.
На рис. 60 приведены данные по растворению пирротина
Fe,S8, активированного в разных средах. При активации на
воздухе количество пирротина, перешедшего в раствор, в
150 раз больше по сравнению с неактивировапным пирроти-
ном. Активация в воде менее эффективна, хотя достигается
более существенное увеличение удельной поверхности, чем
на воздухе (30 м2/г в воде и 7 м2/г на воздухе). Структурные
нарушения на воздухе значительно выше, чем в присутствии
воды (рис. 61).
Наряду с указанной выше причиной изменения реакцион-
ной способности пирротина (разупорядоченпе вакансий) мо-
жет иметь место изменение его состава в сторону троилита
под влиянием механической активации. В этом случае также
будет наблюдаться увеличение степени взаимодействия пир-
169
Рис. 60. Кинетика растворении активированного в разных условиях
пирротина Fe7Sg в 0,1 н. соляной кислоте [488).
а — активация на воздухе; б — в воде; продолжительность активации, мин; 1 —
5, г — 2, з — i, 4 — исходный образец.
ротина с кислотой. Однако экспериментально очень трудно
установить глубину процесса ввиду протекания на поверх-
ности пирротина окислительных процессов.
Халькопирит. Несмотря на то, что метод механической ак-
тивации успешно применяется для ускорения процессов выще-
'-----------J
лачивания меди из халь-
копирита |483—485], при-
чины изменения свойств
халькопирита недоста-
точно ясны. Ряд авто-
ров считают, что при ак-
тивации реализуются вы-
сокие локальные давления
и температуры, а меха-
нохимичсские эффекты яв-
ляются их следствием. В
частности, в работе [484]
указывается на возмож-
ность протекания в халько-
Рис. 61. Дифрактограмма пир-
ротина Fe7S8 (1 — исходный
образец), активированного в
воде (2) и на воадухе (3) [488].
170
пирите при больших энергиях фазовых переходов, аналогич-
ных таковым прп нагревании. Имеются также данные, полу-
ченные методом ЯГРС, по действию па халькопирит высоких
статических давлений [508]. Поэтому определенного по-
нимания механизма активации халькопирита можно до-
стичь, сопоставляя результаты, полученные при его механи-
ческой активации, с результатами, полученными при дейст-
вии на него высоких статических давлений и температур.
Согласно результатам мессбауэровских исследований
формула халькопирита может быть записана в виде
Cu1+Fo3+S2 [495]. Существует три полиморфные модифика-
ции халькопирита. Низкотемпературная a-форма имеет
тетрагональную решетку, близкую к кубической сингонии
типа сфалерита, с параметрами ячейки а — 0,525 и с =
= 1,03 нм [495]. При нагревании до 823 К a-форма перехо-
дит в тетрагональную у-форму, имеющую параметры ячейки
а — 1,057 и с — 0,537 нм, при этом наблюдается потеря се-
ры. Наиболее бедная по сере фаза наблюдается прп 993 К.
После охлаждения неупорядоченной у-формы до темпера-
тур ниже 503 К образуется 0-форма, имеющая кубическую
решетку (а = 1,06 нм), или происходит распад на соедине-
ния FeS, Cu2S и CuS в зависимости от дефицита серы [509].
а-Форма является магнитоупорядоченным минералом (анти-
ферромагнетиком), и мессбауэровский спектр его состоит
из шести линий сверхтонкой структуры. Наблюдаемые па-
раметры согласуются с представлениями о магнптосвязанных
атомах Fes+, находящихся в высокосппновом состоянии в
тетраэдрических позициях.
Под влиянием высоких давлений CuFeS2 переходят из
магнитоупорядоченной фазы в разупорядоченное состояние
при давлениях выше 0,5-105 МПа. Спектр из шести линий
превращается в дублет с теми же значениями изомерного
сдвига (0,23 мм/с) и квадрупольного расщепления (0,72 мм/с).
Переход обратим, и в определенной области давлений су-
ществуют упорядоченная и разупорядоченная фазы. Такой
магнитный переход авторы [508] связывают с переходом
тетраэдрически координированных ионов Fe8+ из высоко-
спинового состояния в низкоспиновое при уменьшении рас-
стояния Fe — S.
Как показано в работе [484], поведение халькопирита при
механической активации в большой степени зависит от ее
интенсивности. Если удельная энергия ниже 0,2 кВт • ч/кг,
то реализуется хрупкое разрушение халькопирита.
При повышении удельной энергии выше 0,2 кВт-ч/кг раз-
виваются процессы пластического деформирования, которые
хорошо просматриваются на электронно-микроскопических
снимках. При дальнейшем увеличении удельной энергии
наблюдается спекание мелких частиц, как при действии вы-
соких температур, при этом происходит потеря серы. При
1,5 кВт-ч/кг потеря серы доходит до 15%, а в продуктах
механической активации одновременно обнаруживаются он.
и P-фазы, а также и у-фаза, которая при хранении перехо-
дит в р-фазу.
Нами проводилась механическая активация «-халько-
пирита в вибрационной мельнице при удельной энергии
0,5 кВт-ч/кг [510, 511]. Условия активации следующие:
масса шаров 1110 г, диаметр 15 мм, загрузка халькопирита
25 г, амплитуда 3,4 нм, частота 18,5 с-1. Исходные образцы
имели размер частиц ~ 1 мм. После активации поверхность
образцов составляла 3,81; 4,47; 5,32; 4,72 м2/г при длитель-
ности активации 7,5; 15; 30; 60; 120 мин соответственно.
По данным рентгенофазового анализа, перехода структуры
a-CuFeS2 в другую модификацию не наблюдалось даже при
времени активации 120 мин. Образцы халькопирита после
активации растворяли в 0,1 н. НС1 при температуре 298 К
и интенсивном перемешивании раствора (навеска 0,5 г,
объем раствора 200 мл). В процессе растворения отбирали
пробы, в которых определялось содержание ионов меди и
железа атомно-адсорбционным методом. Точность определе-
ния составляла 3—5%. Отжиг образцов халькопирита про-
водили в вакууме в кварцевом сосуде. Мессбауэровские
спектры снимали на спектрометре ЯГРС-4, работающем в
режиме постоянных ускорений. Химические сдвиги приведе-
ны относительно a-Fe.
На рис. 62 представлены спектры ЯГР для исходного и
механически активированного халькопирита. Спектр исход-
ного образца полностью совпадает с известным в литературе
[512]. В зависимости от времени активации происходит
изменение спектра. Так же как и при действии высоких
давлений, при времени активации 30 мин и больше наблю-
дается переход от шести линий к дублету с квадрупольным
расщеплением 0,60 мм/с, т. е. имеет место переход из анти-
ферромагнитного состояния в парамагнитное. Спектры при
малых временах активации являются промежуточными,
т. е. упорядоченная и разупорядоченная фазы присутствуют
одновременно. Сопоставляя полученные данные с данными
[508], можно видеть, что в результате происходит изменение
магнитного состояния вещества, аналогичное наблюдаемому
при воздействии внешнего давления. При нагревании до
172
Рис. 62. ЯГР'Снектры халькопп
рита при 300 К.
7 — исходный; продолжительность ак-
тина! ши, мин: 2 — |г>; ;> — 30; 4 —
60; 5 - 120; (> — 60, отжиг при 873 К
в течение 120 мпп.
температуры выше 700 К
спектр изменяется и становят-
ся похожим на спектр исход-
ного халькопирита (рис. 62,
спектр 6‘).
Па рис. 63 приведены
данные по выщелачиванию
меди из халькопирита после
механической, термической и
совместной (механическая+
термическая) активации.
Для образцов после меха-
нической активации на на-
чальном этапе выщелачива-
ние идет с большой ско-
ростью, а затем замедляется.
Большая скорость вначале,
вероятно, связана с раство-
рением поверхностного слоя,
после удаления которого
начинается растворение пластически деформированного
объема частицы, степень дефектности которого, опре-
деляющая увеличение скорости выщелачивания, воз-
растает со временем активации. Нагревание в инертной
атмосфере прн температурах выше 800 К также приводит к
увеличению скорости выщелачивания меди из халькопири-
та. Особенно сильно она возрастает для халькоппрпта, на-
гретого до 1023 К, что, по-видимому, связано с образова-
нием новых фаз в результате термического разложения.
И действительно, на спектрах ЯГР и рентгенограммах образ-
цов халькопирита, нагреваемого в интервале температур
850—1000 К, наблюдаются линии новой, отличной от халь-
копирита фазы (рис. 64), что согласуется с данными, при-
веденными в [509].
Последовательная обработка халькопирита — сначала
механическая, а затем термическая при температуре 873 К —
приводит к дополнительному увеличению степени выщела-
173
Рис. 63. Кпнетпка выщелачивания меди из халькопирита в 0,1 в.
соляной кислоте после механической (а), термической (б) и совмест-
ной (в) активации [511}.
а) 1 — исходный образец; продолжительность активации, мин: а — 7,5, 3 — 15,
4 — 6н, 3 — 120, в — 150; б) 1 — исходный обра н-ц; а — 873 К, 120 мин, 3 —
1023 К, 120 мин; «) I — исходный образец; а — 873 К, 120 мин, 3 — 120 мин ак-
тивации, 1 — 120 мин активации и нагрев при 873 К. и течение 120 мин.
чивания по сравнению с халькопиритом после механической
активации (см. рис. 63).
Соотношение между количествами меди п железа, пере-
ходящими в раствор, как видно из рис. 65, изменяется в за-
висимости от времени активации и растворения. Для механи-
чески активированных образцов это отношение стремится
к единице, т. е. при длительной активации скорости выщела-
чивания меди и железа сравниваются. Для термически акти-
вированных образцов, а также если эти две обработки про-
изводить последовательно (механическая 4- термическая
активация), в раствор в основном переходит медь.
Из сопоставления полученных данных с известными [508]
следует, что химический сдвиг после механической актива-
ции не равен химическому сдвигу в халькопирите, который
подвергается действию высоких статических давлений
(0,33 ± 0,03 против 0,20 ± 0,03 мм/с). Это не позволяет го-
ворить об идентичности двух процессов.
Термообработка образцов после активации приводит к
разным эффектам в зависимости от того, при какой темпера-
туре она проводится. При невысоких температурах (700 К)
она приводит к отжигу образовавшихся дефектов, а при тем-
пературах выше 700 К — к образованию новых фаз. Именно
этим обусловлено увеличение скорости выщелачивания и
мвменение соотношения в сторону меди после термообработ-
ки механически активированных и неактивированных образ-
цов при температурах выше 800 К.
174
a
4
”i—
10
О, град
Рис- 64. ЯГР-спектры (а) и рентгенограммы (б) халькопирита после
термической активации.
1 — исходный образец; 2 — 873 К, 120 мин; 3 — 1023 К, 120 мин, 4 — СО мин ак-
тинидии п нагрей при 873 К, 120 мин.
Поскольку соотношение железо/медь при механической
вктивацип не изменяется в сторону меди* как под влиянием
термообработки при температурах выше 700 К, нельзя рас-
сматривать изменения в кристаллах халькопирита как
175
Рис. 65. Изменение соотношения
между количествами железа и ме-
ди в растворе прп растворении
халькопирита в 0,1 н. соляной
кислоте.
1— „сходный образец; продолжитель-
ность актипацип, мин: и — 15, з —
30; 4 — 120; 5 — 150; 6 — 120, отжиг
при 873 К в течение 120 мин; 7 —
образец нагрет при 873 К в течение
120 мин (без механической активации).
следствие высоких локаль-
ных температур, возникаю-
щих в местах трения обраба-
тываемых частиц. По-видимо-1
му, активация халькопирита
вызывается процессами со
специфическими механизма-
ми.
При исследовании последствий активации в некоторых
ферритах-шпинолях [47—49, 388], для которых основным
мотивом структуры является кубическая плотпоупаковапная
кислородная подрешетка, было обнаружено изменение в
магнитном состоянии шпинелей. Сделано предположение
[47], что в результате сдвиговых деформаций происходит
смещение кислородных плоскостей друг относительно друга,
плотноупаковапная кубическая кислородная подрешетка,
оставаясь плотпоупаковаиной, разупорядочивается, а ка-
тионы, находящиеся между ними, могут трапсляционпо сме-
щаться, и, в частности, тетра-катионы вытесняются в боль-
шие по размерам окта-положения. Следствием такого ка-
тионного перераспределения и является изменение магнит-
ных свойств вещества.
Такой же механизм активации, связанный с переходом
ионов железа и меди из тетраэдрического окружения в окта-
эдрическое, возможен и для халькопирита. Как и в шпине-
лях, атомы серы в халькопирите образуют плотнейшую упа-
ковку, но уже гексагональную. Под влиянием активации
здесь также возможно разупорядочепие плотноупакованной
подрешетки серы. Увеличение химического сдвига до 0,33 ±
± 0,003 мм/с характерно при изменении координационного
числа иона железа с четырех до шести [513]. На рис. 66
приведен спектр активированного халькопирита, получен-
ный в узком интервале скоростей измерения па спектрометре
176
N-IO3
-ItW
тетра-Ре5*’
Рис. 66. ЯГР-спектр халькопирита (120 мин активации в интервале
скоростей от —2 до +2 мм-'с).
Л ГР. Здесь же указаны пары линий, отвечающие дублету
окта-железа, а также третьей п четвертой линий шестерки
тетра-железа, в которой наблюдаемый параметр квадру-
полыюго расщепления близок к пулю.
Таким образом, механическая активация халькопирита в
первую очередь связана с трансляционными смещениями
слоев в илотноунаковаппой анионной подрешетке в резуль-
тате сдвиговых деформаций и переходом тетра-катнопов в
окта-положения [510]. Наблюдаемые иа этом соединении
последствия активации, как и в случае сульфида цинка,
не могут быть сведены к воздействию внешнего однородного
давления и температуры в локальных областях в момент ме-
ханического воздействия. В сульфиде железа механическая
активация связана с изменением распределения катпонпых
вакансий (от упорядоченного к случайному), что приводит
к изменению химического состояния ионов железа.
3.3.3. Соли
Механическая активация служит эффективным средством
воздействия на реакционную способность солей.
Вилчинский, Луни, Торпквист [514] показали, что пнтен- '
сивное диспергирование а-Т1С13 приводит к сильному (бо-
лее чем в 10 раз) увеличению его каталитической актив-
ности, причем наибольшая эффективность наблюдается прп
сухом диспергировании.
Авторы [514] пришли к выводу, что увеличение катали-
тической активности зависит от роста поверхности, умень-
шения размеров первичных кристаллитов и образования па ,
поверхности a-TiCl3 пепта-коордпннрованпых атомов ти-
гана,
Рис. 67. Структура TiCl3 [514].
а — а-TiCl, (слоистая структура); б — p-TiCI, (волокнистая структура).
С уменьшением размера первичных частиц активность
растет, но до определенного значения. При размерах частиц
ниже 5,0 нм активность падает. Авторы связывают такое
явление со структурными переходами a-TiCl3. Это соедине-
ние имеет слоистую структуру (рис. 67). Каждый атом в
этом слое связан с тремя атомами хлора. Последовательность
слоев соответствует гексагопальной плотной упаковке ато-
мов’ хлора. Атом титана координирован шестью атомами хло-
ра (октаэдрическая координация).
В процессе механической активации имеет место сколь-
жение слоев Cl— Ti—Cl друг относительно друга и а-фаза
переходит в б-форму, имеющую тоже слоистую структуру,
но с беспорядочной упаковкой слоев. Атом титана, находя-
щийся на поверхности, является не полностью координиро-
ванным. При уменьшении размера частиц до 5 нм число не
полностью координированных атомов становится весьма зна-
чительным (до 27%). При таком высоком содержании пента-
178
координированных атомов создаются условия для перехода
из 6- в |3-фазу, имеющую волокнистую структуру, которую
можно рассматривать как линейный полимер (см. рис. 67).
Поскольку в этой структуре не полностью координирован-
ные атомы отсутствуют, каталитическая активность ее не-
значительна.
Фазовый переход сопровождается изменением цвета: пур-
пурно-фиолетовый a-TiCl3 переходит в коричневый P-TiC)3,
что наблюдалось при измельчении неоднократно [515].
Получение катализатора высокой активности достигнуто
совместным диспергированием TiCl3 с MgCl. В результате
такой обработки содержание MgCl2 в катализаторе повыси-
лось от 0,4 до 4% [516].
Повышение реакционной способности CaF2 при взаимо-
действии его с H2SO4 для получения HF под влиянием ме-
ханической активации установлено Шрадером и Оезе [517]. '
Рост выхода связан с общим ростом неупорядоченности ре-
шетки. Как считают авторы, увеличение внутренней энергии
CaF2 при механической активации за счет уменьшения раз-
мера первичных частиц (т. с. роста внутренней поверхности)
составляет 16%, а вклад микродеформацнп и отклонений
атомов в решетке от равновесных положений 42%. Все
структурные эффекты сохраняются длительное время. Ме-
ханическая обработка позволяет снизить температуру раз-
ложения до 353 К. (вместо 453—573 К) и увеличить скорость _
разложения. Аналогичным образом увеличивается и раст-
воримость фторапатита после механической активации [518].
По данным работы [519], за 20 ч истирания в вибромель-
пице разлагаются карбонаты: магния на 28,9%, кальция —
13,9, стронция — 10,8, барпя — 8,4, натрия — 0,22, ка-
лия — 0,16%. Прямые масс-спектрометрические измерения,
сделанные Фоксом [154], показали, что среднее число раз-
лагающихся монослоев при раскалывании кристаллов кар-
бонатов свинца, магния и кальция равно соответственно
2; 0,37; 0,09. Из этих данных следует, что скорость механо-
химического разложения уменьшается с повышением тем-
пературы термической диссоциации карбоната (температура,
при которой давление углекислого газа достигает атмосфер-
ного, составляет: для MgCO3 — 923 К, СаСО3 — 1073,
SrCO3 — 1484, ВаСО3 — 1520 К). Эта закономерность под-
тверждена Молчановым и др. [520] при исследовании про-
стых и сложных карбонатов, подвергаемых механической
активации. Кроме того, ими показано, что сложные карбо-
наты при диспергировании в планетарной мельнице сначала
разлагаются на простые карбонаты: соответственно доло-
170
Рис. 68. Скорость реакции
в системе CdO + СО2
г* CdCOs в зависимости от
давления СО2 и состава
твердой фазы (х — мольная
доля CdCO3) [521].
мит — на кальцит и ма-
гнезит, анкерит — на
кальцит, магнезит и си-
дерит, которые потом
частично диссоциируют
с образованием окиси металла и углекислого газа.
Питерсом [66] показано, что в зависимости от условий,
в которых проводится механическая обработка карбонатов,
могут быть получены разные продукты. При разложении
карбоната железа (сидерита) на воздухе получается оксид
Fe2O3, а в присутствии влаги — гидроксид Fe(OII)3. При
измельчении карбонатов в вакууме количество выделяюще-
гося углекислого газа на порядок больше, чем при разложе-
нии на воздухе. Реакции,’протекающие при механической
обработке карбонатов, являются обратимыми. Например,
обработка оксида цинка в присутствии углекислого газа
приводит к образованию карбоната цинка, при обработке же
карбоната цинка в вакууме идет его диссоциация.
Если скорости прямой и обратной реакций при механи-
ческой обработке равны, то общее макроскопическое измене-
ние в системе отсутствует. В отличие от химического равно-
весия возникающее при механической обработке стационар-
ное состояние обозначается как «механохимическое равно-
весие» .
Количественные данные «но механохимическому равно-
весию» в системе CdO + СО2 CdCO3 получены Хайнике и
Сигристом [521]. На рис. 68 показана зависимость скорости
реакции от давления углекислого газа и состава твердой
фазы. Положительное значение ординаты означает прямую
реакцию, отрицательное — обратную. Для различных со-
ставов твердой фазы скорость реакции становится равной
нулю при разных давлениях СО2. Такого стационарного со-
стояния можно достигнуть с двух сторон. В отличие от хи-
мического равновесия, в котором независимо от состава твер-
дой фазы каждой температуре соответствует определенное
давление, «механохимическое равновесие» сильно зависит от
180
состава измельчаемого материала. При изменении содержа-
ния CdCO3 в смеси CdO—CdCO3 от 50 до 90 мол. % равно-
весное давление увеличивается примерно на два порядка.
Особенность «механохимического равновесия» заклю-
чается в том, что для его поддержания к системе должна
необратимо подводиться механическая энергия. В этом ме-
ханохимические процессы сходны с фото- и радиационно-
химическим процессами.
Для изучения реакций механохимического разложения
Уракаев, Савинцев и Болдырев [522] применили методику
времяпролетной масс-спектрометрип. Для NH4Br возможны
следующие пути распада:
НВт + NH3 I
NII4Br—*Br + NH4 (NH3 + Н) II
^>*‘Вг + NII^(b решетке) + е III
Механизм первой реакции объясняется возбуждением коле-
бательно-вращательных уровней основного терма катиона
NH4b, что в конечном итоге приводит к переносу протона
1523]. Реакции же II и III реализуются только за счет
электронных процессов [524] и требуют гораздо больших
энергетических затрат, чем реакция I.
Опыты по раскалыванию кристаллов NII4Br проводились
при температуре 200 К, что позволило добиться хрупкого
разрушения монокристаллов NH4Br, вымораживать влагу,
мешавшую корректному проведению опыта, п исключить
сублимацию NII4Br. Давление перед измерением было
G-10-5 Па, а чувствительность методики к потоку газа со-
ставляла 1011 мол./(с-мм шкалы).
Сопоставление полученного при раскалывании моно-
кристалла масс-спектра с масс-спектрами чистых NH3 и
ПВг, приведенных в атласе масс-спектров [525], показало,
что летучими продуктами механохимического разложения
NH4Br являются NH3 и НВг, которые выделяются примерно
в равных количествах (1014—1015 мол./с).
Масс-спектры не содержат в пределах чувствительности
методики никаких сведений о возможности реализации при
раскалывании монокристаллов NH.Br реакций типа II
и III.
Это дает основание предполагать, что так же, как п при
термическом разложении, механизм механохимического
разложения аммонийных солей заключается в переносе про-
тона от катиона к аниону вследствие возбуждения оптических
<81
колебаний решетки. Электронные же процессы не реализуют-
ся, поскольку имеется эффективный механизм поглощения
механохимической энергии на более низкой ступени энер-
гетической шкалы.
Процессы, происходящие в результате обработки веществ
в измельчительных аппаратах, имеют много общего с про-
цессами, происходящими при одновременном действии вы-
соких давлений со сдвиговой деформацией. Как для тех,
так и для других характерно пластическое течение вещест-
ва. При диспергировании реализуются ударные воздейст-
вия мелющих тел о вещество и имеют место локальные тем-
пературы и давления. При воздействии высоких давлений
со сдвигом локальные температуры можно свести до мини-
мума. Эти процессы, как правило, рассматривают вместе,
так как они приводят к сходным результатам. Из указанных
видов воздействия предпочтение как наиболее «чистому»
механохимическому воздействию отдают обработке твердых
тел высокими давлениями со сдвигом, ввиду отсутствия в
этом способе значительных локальных разогревов.
Ларсен и Дрикамер [236] исследовали разложение фер-
роцианида калия при действии высоких давлений со сдви-
гом на вещество и показали, что разложение идет но реак-
ции
4K3Fe(CN)e->3K4Fo (CN)„ + Fe + A (CN)„
причем степень разложения зависит только от угла поворо-
та наковальни, но не зависит от скорости, с которой этот
угол поворота достигнут. Они рассматривают различные ме-
ханизмы химического воздействия сдвиговых деформаций
на вещество. Отвергается роль общего разогревания мате-
риала вследствие пластической деформации, так как велик
теплоотвод через наковальни и скорость процесса нс зави-
сит от скорости вращения. Авторы работы [527] вводили
термопары в неглубокие канавки на наковальнях Бридж-
мена и исследовали возрастание температуры при сдвиге
как функцию давления, скорости деформации и температуры
окружающей среды. Они получили, что для оксида алюми-
ния и кварца такой разогрев не превышает 10 К. В резуль-
тате Ларсен и Дрикамер [236] считают, что за механизм
разложения в K3Fe(CN)e ответственны свободные электро-
ны, появляющиеся в процессе деформации, и передача их
катионам. Косвенным подтверждением своих взглядов Лар-
сен и Дрикамер считают данные Тилера [528], объясняюще-
го возрастание электропроводности кристаллов в процессе
пластической деформации образовавшимися прп »том сво-
бодными электронами.
Разложение бихромата аммония при одновременном воз-
действии высокого давления п сдвиговой деформации иссле-
довано Зубовой и Апарниковым [246]. Термическое разло-
жение бихромата аммония является топохимической реак-
цией и определяется двумя факторами: скоростью зарожде-
ния ядер продукта реакции п скоростью роста этих ядер.
Эксперименты проводили по такой методике: небольшое ко-
личество порошка (NH4)2Cr2O7 насыпали на наковальню и
несколько раз обдавливали до 4-108 Па для достижения по-
стоянной толщины таблетки. После этого образец подвер-
гался деформации кручения под давлением на определен-
ный угол при скорости вращения 6,1 -10-3 или 3,1 •
• 10~3 рад/с. Продукты разложения анализировали потен-
циометрическим титрованием по расходу исходного вещест-
ва. Предполагалось что разложение идет по реакции
(NH4)2Cr2O7 = Cr2O3 + N, + 4Н2О.
Было показано, что степень разложения бнхромата аммония
не зависит от скорости деформации, а только от ее величины
(угла поворота) (рис. 69). Исследована зависимость скорости
разложения от температуры и давления. Показано, что ка-
жущаяся энергия активации снижается от 1384-205 до 17 4-
4-30 кДж/мольи имеет тенденцию к росту с увеличением дав-
ления.
При измельчении бпхромата аммония в планетарной
мельнице также происходит его разложение. Данные по
механохимическому разложению в зависимости от размера
шаров при одинаковой их общей массе приведены на рис. 70.
С увеличением диаметра шаров возрастает механическая
нагрузка на вещество, а следовательно, и его деформация.
Это свидетельствует об общности процессов, протекающих
при высоком давлении с деформацией сдвига и в измельчи-
тельных аппаратах. На начальном участке кривых имеет
место всплеск, вызванный, по нашему мнению, появлением
переходных состояний хрома в начальный момент обра-
ботки.
Зубова и Апарннков объясняют причины разложения
(NH4)2Cr2O7 появлением высокочастотных фононов прп
взаимодействии дислокаций друг с другом или с границами
зерен. Появление фононов, с их точки зрения, вызывает
дополнительное возбуждение колебательных уровней в анио-
не Ci^O^j что приводит к снижению энергии^ необходимой
18%
183
V,pad
Рис. 70. Механохимическое раз-
ложение (NII4)2Cr2O7 в планетар-
ной мельнице.
Диаметр шаров, мм: 1 — 5, г — 15.
Рис. 69. Степень разложения
(NH4)aCr2O7 в зависимости от уг-
ла поворота наковальни [240].
для разрыва связей, поэтому скорость реакции слабо зави-
сит от внешней температуры.
Рассматривая процесс возбуждения колебательных уров-
ней химических связей в процессе рассеивания упругой энер-
гии в тепло, Болдырев [230] указывает, что при механо-
химическом разложении может происходить возбуждение
более высоких уровней, чем при термолизе, по более низ-
ких, чем при радиолизе и фотолизе, когда сразу происхо-
дит возбуждение электронных уровней. Наличие таких со-
стояний может приводить к химическим превращениям, ко-
торые обычно не реализуются при термическом разложении
и в то же время отличны от радиолиза и фотолиза [529].
Механохимическое разложение нитратов и броматов ще-
лочных металлов исследовалось с целью сравнения с терми-
ческим разложением и установления возможности направ-
ленного регулирования скорости этого процесса [529—534].
Известно, что разложение нитратов щелочных металлов
возможно по трем путям (см. гл. 1). Реакция с образованием
нитрита щелочного металла и кислорода реализуется прп
температурах ниже 800 К, остальные реакции — выше
800 К.
Аналогичным образом разлагаются броматы щелочных
металлов:
МеВг + -7- О2 I
МеВЮ< 1 5
Ме2О + -у Вг2 + — О2 II
4 ал
Реакция I реализуется при низких температурах, реакция
11 — при высоких.
Кинетика термического разложения нитратов щелочных
металлов до нитритов исследована Бордюшковой с соавт.
[5351 начиная от температур плавления до 1000 К. При ско-
рости термического разложения при температурах <900 К
нитраты располагаются в ряд
NaNO3 > KNO3 > RbN03 > CsNO3.
Уменьшение скорости низкотемпературного термического
разложения в ряде нитратов связывают с уменьшением по-
ляризующего действия катиона, которое ведет к ослабле-
нию связи внутри анионной группировки и ее раатожению.
Аналогичные выводы были сделаны в работе [536] прп ис-
следовании влияния междоузельных катионов па скорость
термического разложения нитрата натрия. Взаимодействие
с междоузелыпями катионами приводит к тому, что одна из
связен N—О в анноне становится неэквивалентной другим
и разрывается в первую очередь.
Механохимическое разложение нитратов щелочных ме-
таллов проводилось в планетарной мельнице. Использова-
лись кварцевые барабаны и кварцевая дробь. После актива-
ции в нитрате щелочного металла определялось содержание
нитрата калориметрическим методом с использованием реак-
тива Грисса [431]. Кинетические кривые механохимнческого
разложения (рис. 71) отличаются от кривых термического раз-
ложения тем, что через определенное время активации дости-
гается некоторое стационарное состояние, при котором ско-
рость разложения равна скорости обратной реакции. Самую
высокую начальную скорость разложения имеет нитрат ру-
бидия и самую малую — нитрат калия, и ряд скоростей раз-
ложения получается такой [530]:
RbNO3 > CsNO3 > KNO3 > NaNO3.
Сопоставляя этот ряд с вышеприведенным, видим, что
сходства между ними не наблюдается. Такое же сравнение
было сделано для броматов щелочных металлов. В продук-
тах активации определялся ион брома методом Фольгарта
14311. Кинетические кривые приведены на рис. 72. Из него
видно, что по скорости механохимнческого разложения бро-
маты располагаются в ряд [531, 533]
RbBrOg > CsBrO3 > KBrOg > NaBrOa.
185
Рис. 71. Кинетические кривые мс-
ханохимичсского разложения ни-
тратов щелочных металлов (530j.
el, % Вт"
Рис. 72. Квиетические кривые ме-
хаиохп.мического разложения бро-
матов щелочных металлов [531,
533 J.
По данным же работы (537 ], скорость низкотемпературного
термического разложения броматов изменяется в ряду
CsBrO3 > NaBrO3 > RbBrO3 > КВгО3.
Сопоставляя скорости термического и механохимического
разложения нитратов и броматов щелочных металлов, ви-
дим, что между ними не только нет корреляции, но они в ка-
кой-то степени антпбатны (533]. Однако имеется корреляция
с механическими свойствами нитратов. Более пластичным
нитратам и броматам цезия и рубидия [87, 537] соответст-
вует больший выход продуктов разложения.
В табл. 10 приведены данные по сравнению механохпми-
ческого разложения не только с термическим, но и с фото-
химическим, радиационно-химическим и разложением нит-
ратов в ударной волне [530, 535—539].
Из этих данных следует, что механохимическое разложе-
ние и разложение в ударной волне являются однотипными
процессами, различаются только количественно, однако с
термолизом, фотолизом и радиолизом корреляция отсутст-
вует. Все это позволило отпести механохимическое разло-
жение к особому, специфическому процессу [529].
При исследовании влияния влажности на механохими-
ческое разложение нитрата натрия было установлено, что
186
Таблица 10
Разложение нитратов щелочных металлов прп различных видах воз-
действия
Вещество ТПЛ.К Температура, при которой достигается выход, К I Радиолиз, молен. ЯС ()р( Фотолиз, квантовый ВЫХОДУ 10* Разложение, %
механохи- мическое в ударной волне
0,002?; 0,05% о.1“;,
NaNO3 476 320 510 531 0,37 0,44 0,006 0,43
KNO3 610 360 530 550 1,96 4,0 0,016 0,46
RbNOa 385 340 512 560 0,80 4,0 2,50 1,00
CsNO3 687 440 555 575 1,37 6,0 0,09 0,55
содержащаяся в образцах влага подавляет разложение (вре-
мя обработки 20 мин):
Качество нитрата натрин
Выход мае.%-10
Хорошо высушенный препарат
(десорбция в вакууме при
373 К в течение 3 ч) 3,80
Обычиый реактивный препарат 2,60
Влажный ирепарат (добавлено
5% воды) 1,62
То же, но добавлено 12,5% воды 0,75
Увеличение механической нагрузки, а также введение
добавок, обладающих абразивными свойствами, приводит к
росту скорости механохимического разложения (рис. 73).
При проведении механохимического разложения в жид-
ком азоте (78 К) скорость разложения снижается в 1,6 раза.
На рис. 74 представлены данные по механохимическому!
разложению и изменению удельной поверхности в зависи-
мости от времени активации нитрата натрия, а также по
термическому разложению механически активированных об-
разцов при температуре 553 ±2 К [532]. Наличие макси-
мума па кривых изменения поверхности свидетельствует о
том, что по достижении некоторых размеров частицы пере-
стают измельчаться, начинается процесс агрегации, возмож-
но за счет процессов пластического течения. Кривая механо-
химического разложения повторяет ход кривой удельной
поверхности. Причиной такого поведения может быть либо
обратная реакция окисления нитрит-иона кислородом, обра-
зовавшимся в решетке при распаде нитрат-иона или имею-
187
Рис. 73. Механохимическое раз-
ложение нитрата натрия в при-
сутствии абразивных добавок
(время активации 30 мин).
Рис. 74. Выход продуктов терми-
ческого (2) и механохимического
(2) разложения и изменения удель-
ной поверхности (3) нитрата нат-
рия в зависимости от времени ак-
тивации [532].
щпмея в воздухе, либо реали-
зация в этих условиях (плас-
тическое течение материала)
более высокотемпературной
реакции II. Анализ па содер-
жание щелочи в водном раст-
воре показал, что концентра-
ция ОН” в продуктах актива-
ции в 2—3 раза меньше, чем
нитрит-иона, причем идет
плавное накопление его со временем активации, без резко-
го увеличения концентрации после 20 мин (время максиму-
ма). Эти данные свидетельствуют в пользу первого предпо-
ложения.
На основе кривых рис. 74 была оценена энергия, запасен-
ная нитратом натрия под влиянием механической активации
по уравнению Томсона (см. гл. 3). При значении поверх-
ностной энергии и = 0,1 Дж/м2, взятом из работы [540]
для расплавленного нитрата натрия (вблизи температуры
плавления), изменение свободной энергии составляет
0,18 Дж/моль. Из данных по термическому разложению при
температуре 553 К в соответствии с уравнением
(3.20)
&G = — RT\nP\
получено AG = 7,04 Дж/моль. Сравнивая эти значения, мож-
но видеть, что они различаются почти в 40 раз. Полученные
Рис. 75. Зависимость степени раз-
ложения нитрата натрия от рабо-
ты выхода электрона из оксида
при механической активации сме-
си нитрата с оксидом.
результаты свидетельствуют о том, что структурные нару-
шения в решетке нитрата натрия после активации весьма
значительны и существенно облегчают его последующее
термическое разложение.
Разложение нитратов представляет собой типичную окис-
лительно-восстановительную реакцию, поэтому интересно
было выяснить, как па него действуют добавки, влияющие
на электронные процессы, а именно гетерофазные добавки,
обладающие каталитическим п ингибирующим действием
(оксиды никеля, марганца, железа, цинка и др.). Установ-
лено, что среди гетерофазных добавок па механохимическое
разложение больше влияют те, которые имеют более высо-
кую работу выхода электрона из оксида (рис. 75).
Чтобы убедиться в правильности сделанного предполо-
жения, были проведены опыты, в которых добавку закиси
никеля допировали ионами хрома и лития. Как известно,
введение в оксид нпкеля добавки хрома приводит к умень-
шению концентрации ионов, повышая уровень Фермп,
а добавки ионов лития, наоборот, увеличивая их концентра-
цию, понижают уровень Ферми. Выяснено, что оксид нике-
ля с добавкой хрома замедляет механохимическое разложе-
ние, а с добавкой лития ускоряет его [5291.
Таким образом, можно рассматривать мсхапохпмпческие
реакции как обратимый окислительно-восстановительный
процесс, на который можно воздействовать электронно-
активными добавками, например оксидами металлов пере-
менной валентности.
Отсутствие корреляции термической устойчивости с ме-
ханохимической для нитратов и броматов щелочных метал-
лов не единичный факт. Оказывается, что для щелочпо-гало-
пдпых солей, как это показано в работе Геллоиа с соавт. [541|,
также отсутствует такая корреляция. Молоцким [451 для
л агх
такого рода процессов предложен эксптоппый механизм
возбуждения (см. гл. 2). Для реализации его необходи-
мо накопление на поверхности электрических зарядов, кото-
рое связано с механическими свойствами кристаллов: чем
кристалл пластичнее, тем выше в нем подвижность дислока-
ций, которые прп своем движении захватывают заряженные
вакансии и выносят заряд па поверхность кристалла.
Теория релаксации заряда трещины в щелочпо-галопд-
ных кристаллах была построена для случая разрушения в
вакууме. Однако ее можно применять и для разрушения в
среде, поскольку внутренняя часть трещины вакуумирова-
на (см., например, 15421).
Исходя из общих положений, развитый Молоцким под-
ход, по-видимому, можно применить и к нитратам. Так',
возрастание выхода продукта в ряду питратов и броматов
можно объяснить усилением пластических свойств при паро-
ходе от соединений натрия к соединениям цезия.
Наличие влаги способствует стеканию заряда с поверх-
ности и соответственно вызывает уменьшение мехапохими-
ческого выхода.
Введение добавок, имеющих дырочную проводимость,
приводит к поглощению электронов и снижает вероятность
образования возбужденных экситонов, способных вызвать
колебательное возбуждение в нитрат-ионе. Введение доно-
ров электронов приводит к обратному эффекту. Эти факты
находятся в качественном согласии с экситонной теорией.
Однако имеющихся данных недостаточно, чтобы сделать
выводы о применимостп этой теории к нитратам.
-------------------Г Л А В А 4
МЕХАНОХИМПЧЕСКИЕ РЕАКЦИИ ТВЕРДЫХ ТЕЛ
С ГАЗАМИ И ЖИДКОСТЯМИ
4.1. МЕХАНОХПМПЧЕСКИЕ РЕАКЦИИ ТВЕРДЫХ ТЕЛ
С ГАЗАМИ
4.1.1. Реакции металлов с газами
В обычном состоянии поверхности твердых тол мало-
активны вследствие того, что они покрыты слоями адсорби-
рованных веществ или продуктами их взаимодействия с
190
a
Рис. 76. Скорость MOxauoxiiMiiuuCKvro окне линия металлов [543].
л — обработка н bi io роме; п>ц ине, барабан 1,1 дмя, 1,05 кг норуидоных шаров,
диаметр шарон 21 мм, 0,2 моли металла; б — обработка и нпбромельницс, барабан
0,05 дм3, 0.G5 км фарфоровых шарон, диаметр шарив И мм, 0,2 моля металла.
окружающей средой (воздухом, парами воды и т. д.). Нали-
чие такой пленки затрудняет химическое взаимодействие с
газами. Для устранения пассивации и удаления продуктов
реакции с поверхности применяется механическая активация
последних. При этом пленки разрушаются, обнажается све-
жая поверхность с содержащимися на ней активными цент-
рами, что облегчает протекание химических реакций. Таким
образом, можно проводить широкий круг химических реак-
ций тел с газами: окисление, хлорирование, получение кар-
бонилов и т. д.
Скорость механохимнческого окисления металлов ис-
следовалась Хеннике с соавт. [543]. Кривые зависимости
скорости окисления от продолжительности измельчения
имеют максимум (рис. 76), причем положение и значение
максимума для мельниц различной энергонапряженности
существенно различны. Для большинства порошков кривые
скорости окисления в более напряженной мельнице отли-
чаются не только большим значением максимума, но и боль-
шой крутизной. Экстремальный ход кривых зависимости
скорости окисления от времени диспергирования объясняет-
ся тем, что скорость образования свежей.поверхности, как и
степень нарушенное™ поверхностных слоев, в начальный
период диспергирования быстро растет, а затем диспергпро-
191
вание замедляется и даже прекращается. Возможно даже
уменьшение дисперсности вследствие агрегации первичных
частиц, особенно интенсивной для пластичных материалов.
Сопоставляя скорости реакций механохимического окис-
ления со свободной энергией образования оксидов для раз-
ных металлов, можно видеть, что за некоторым исключением
(Al, Si, Ст) термодинамические параметры соответствуют
кинетическим: чем меньше свободная энергия, тем больше
скорость реакции.
Однако не всегда, основываясь на термодинамических
расчетах, можно предсказать протекание некоторых механо-
химических реакций. К числу таких реакций относится окис-
ление золота, меди и никеля в атмосфере углекислого газа.
Как показано Хейнике с соавт. [544], в условиях механи-
ческой обработки идет реакция
Ан + 4- СО., = 4- Ач,О3 + 4 С,
изменение свободной энергии которой составляет
378 кДж/моль при температуре 571 К. Реакция Доказана
радиохимическими методами по меченому углероду 1ЯС,
а также с помощью ПК-спектроскопии. Протекание реакции
обусловлено, по-видимому, иоявлением атомов в высоко-
возбужденном состоянии, которое при термодинамических
расчетах не учитывается.
Многие вещества не реагируют с хлором при комнатной
температуре. Однако хлорирование можно провести, если
подвергать их механической активации в атмосфере хлора.
Костером [545] в этих условиях были подвергнуты хлори-
рованию кремний, ферросилиций, титан и кристаллический
бор. Так, при диспергировании на вибромельнице 5-10 м кг
кремния в барабане емкостью 0,8 дм3 в течение 2 ч полу-
чено 0,284 кг SiCl4 (96%) и 5.8-10"3 кг Si2Cl0. Скорости хло-
рирования при измельчении значительно больше, чем на
предварительно измельченных в аргоне порошках.
Некоторые металлы, к числу которых относятся никель,
железо, молибден, вольфрам, хром, взаимодействуют с окси-
дом углерода, образуя карбонилы металлов. Эти реакции
обратимы, что и обусловливает существование максимума
скорости карбонилирования в зависимости от температуры:
с повышением температуры скорость разложения карбонила
растет быстрее, чем скорость синтеза, т. е. увеличение тем-
пературы приводит к смещению равновесия в сторону обра-
зования . исходных продуктов. Эти реакции идут с умень-
шением числа молей газообразных продуктов, поэтому уве-
192
/'«с. 77. Скорость реакции образо-
вания карбоппла никеля в зависи-
мости от давления оксида углерода
[550—553J.
1 — Пей механической обработки при
2!>К К; 2 — с механической обработкой при
21>3 К; J — го же, при температуре 100 К;
I — то же, при температуре 323 К н прн-
сутстинн U.S.
Рис. 78. Температурная зави-
симость реакции образования ;
карбонила никеля [5501.
1 — пред;>Я|ште.1ьно механически
актнппроваиный никель; ? — и мо-
мент механической обработки.
лпчепие давления смеща-
ст равновесие реакции в
сторону образования кар-
бонилов.
Как показали исследо-
вания Хепнпке с сотр.
[546—553], механическая
обработка никеля в про-
цессе взаимодействия с
оксидом углерода коренным образом меняет кинетику
реакций. Изучалось влияние давления, температуры и до-
бавления газа H2S на кинетику механохпмической реакции.
Эксперименты с механической активацией никеля проводи-
ли в внбромельнпце с частотой 11 с-1, амплитудой 5,5 мм.
В качестве мелющих тел использовали шары из карбида
кремния (SiC марки К10) или корунда, диаметр шаров
1,5 мм. Обрабатывали никелевые пластинки размером
5x5x1 мм. Применяли оксид углерода высокой частоты с
содержанием следов кислорода (<10-4%).
Ранее Траутом 1549] было показано, что при термическом
синтезе карбонила никеля порядок реакции меняется от 1
до 2 в зависимости от давления оксида углерода: ниже
5,3-Ю1 Па порядок равен 1, выше 9,3-104 Па — 2, а в интер-
вале давлений (5,3—9,3)-10* Па порядок реакции носит про-
межуточный характер.
7 Е. Г. Лоиаиумоп
193
В реакции, протекающей при механической активации
никеля в том же интервале давлений, порядок реакций уже
незначительно меняется от давления и равен 0,2 (рис. 77,
составленный по данным работ [550—553]). Авторы объяс-
няют такое значение порядка реакции в процессе механи-
ческой активации наличием мехапохимической и терми-
ческой составляющих, действующих одновременно, что при-
водит к значению от 0 до 2. С уменьшением температуры
термическая составляющая уменьшается и реакция проте-
кает чисто механохимически, порядок реакции при 100 К
в этом случае равен 0.
В работе [550] приведены экспериментальные данные,
показывающие, что температурная зависимость этих двух
составляющих различна. Скорость реакции, обусловленная
термической составляющей, довольно быстро снижается, а
при температуре 238 К ничтожно мала. Механохимическая
составляющая, напротив, с понижением температуры воз-
растает, и скорость реакции при 100 К мало отличается от
таковой при комнатной температуре (рис. 78).
Отрицательный температурный коэффициент в области
низких температур авторы работы [550] объясняют двумя
эффектами: увеличением степени покрытия никеля при этих
температурах оксидом углерода и повышением микротвер-"
дости металлического никеля. Следует отметить, что первая
причина более важная, чем вторая. И и самом деле, степень
покрытия изменяется примерно в 26 раз при снижении тем-
пературы от 298 до 80 К, а микротвердость в этом же интер-
вале температур изменяется только в 1,5 раза. Таким обра-
зом, чем ниже температура проведения реакции, тем выше
степень покрытия поверхности твердого тела газом и выше
скорость мехапохимической реакции.
Сильное влияние на кинетику карбонилообразовапия
оказывает сероводород. На рис. 77 представлены кривые,
характеризующие скорости образования карбонила никеля
при механической активации в чистом оксиде углерода и с
добавлением сероводорода в зависимости от давления оксида
углерода [550]. Из этих данных следует, что в присутствии
H2S порядок реакции при 298 К равен 2, как и в случае
термически активируемой реакции. На основании этих ре-
зультатов авторы делают предположение, что H,S влияет
только на термическую часть совокупной реакции и при
понижении температуры до 195 К не оказывает сенсибили-
зирующего влияния па образование карбонила. Сенсибили-
зирующее влияние сероводорода па образование карбонила
никеля Хейпике с соавт. [532] объясняет образованием
1Q4
Т а о лица 11
Энергия активации реакция Ni+ 4С0 -* Ni(CO)4 (5521
Темпера- тура, К Характер реакции Кажущаяся энергия активации, кДж/моль
100—250 Механохимичгский 0
293-305 Термичсекий и мохаиохимиче-
скпй 14,6
291-305 Сенсибилизированный и терми-
ческин 22,4
291—305 Термический 55,0
промежуточного соединения — сульфида никеля по реакции
— и +5СО
Ni + II2S —4-NiS — Ni (CO),.
В табл. 11 представлены данные, характеризующие ка-
жущуюся энергию активации синтеза карбонила в зависи-
мости от температуры и характера реакции.
При исследовании сорбции оксида углерода на поверх-
ности никелевых пластин во время механической обработки
были выявлены две области (554]: а) область, в которой ме-
хаподесорбцпя не зависит от давления (при давлениях
> 0,66 Па); б) область, в которой механосорбция опреде-
ляется числом ударов молекул газовой фазы (при давле-
ниях < 0,66 Па).
На основании полученных результатов авторы делают
вывод о постоянном формировании сорбционной поверхности,
которая моментально покрывается молекулами окружаю-
щего ее газа уже при давлениях > 0,13 Па.
Хейппке и Бок [554] установили, что следы газов, обра-
зующих с никелем окисный слой, например кислорода, мо-
гут очень сильно затормозить реакцию окиси углерода с
никелем. Они наблюдали снижение удельной поверхности
металлического никеля в зависимости от времени измельче-
ния и в то же время уменьшение размера первичных частиц,
определяемого рентгенографически. По-видимому, на опре-
деление удельной поверхности сильно влияет образование
агрегатов из первичных частиц. При обработке никеля ими
наблюдалось увеличение искажении решетки примерно в
10 раз, в то время как удельная скорость увеличилась при-
мерно в 25 раз. Авторы связывают увеличение скорости не
только с искажением решетки, по и с эффектом свежей по-
верхности.
7*
195
Разработаппый Хейпике с соавторами па основе прове-
денных исследований механохнмический метод синтеза кар-
бонила оказался более экономичным и удобным, чем суще-
ствующие методы, он нашел промышленное применение для
изготовления изделий сложной конфигурации из металли-
ческого никеля. Синтез протекает прп нормальном давлении
и обычной температуре с выходами па два порядка выше,
чем у существующих способов. Поскольку массоперенос в
газовой фазе происходит быстрее, чем в жидкой, скорость
получения изделия из никеля в 5—7 рал выше, чем электро-
химическим способом (554].
Механохимическим методом можно проводить синтезы
карбонилов и других металлов, например вольфрама, мо-
либдена, хрома и др. [555, 556]. Однако эффективность их
значительно ниже, чем в случае синтеза карбонила никеля.
Для реакции образования карбонила вольфрама было ус-
тановлено, что скорость реакции на начальных стадиях вы-
ше при использовании шаров диаметром 2,5 мм, чем для ша-
ров 5 мм, при постоянстве других параметров (масса шаров,
загрузки, частота вращения и др.) [556].
4.1.2. Поверхностные реакции газов
с кварцем
Свойства атомов па поверхности твердых тел отличны от
таковых в объеме кристалла. Возникают особые поверхност-
ные электронные состояния Тамма и Шокли [248, 249]. На-
ряду с этим поверхность имеет геометрические неоднород-
ности, за которые ответственны микроскопические дефек-
ты — ступеньки роста, щели, поры, выходы дислокаций я
т. д., а также химические неоднородности по составу в на-
сыщенности связей. Существование таких состояний на по-
верхности твердых тел определяет повышенную адсорбцион-
ную и химическую активность твердых тел. Кроме того, по-
лученные при разрушении твердых тел свежеобразовапные
поверхности содержат разорванные и деформированные свя-
зи, которые практически безактивационно реагируют с ок-
ружающими твердое тело газами и жидкостями, с образова-
нием на поверхности различного рода соединений.
Работы в области исследования поверхностных реакций
неорганических соединений начались с исследования по-
верхностей раскола кремния и германия [283—289]. Накоп-
лены сведения о реакционной способности поверхности для
ряда неорганических соединений, различающихся по типу
196
химической связи. Детально исследованы поверхностные
химические соединения, образующиеся при n.iaiiMoдействии
поверхности кварца с различными газами.
Прп разрушении кварца на его поверхности образуются
парамагнитные центры = Si" и ss SiO примерно в равном
количестве. Эти центры отличаются высокой реакционной
способностью по отношению к молекулам из газовой фазы.
При этом радикалы =s SiO выступают в качестве окисляю-
щих центров, а радикалы ss Si — в качестве восстанавлива-
ющих (см. гл. 2).
Приведем результаты изучения свободнорадикальных
реакций, протекающих на поверхности измельченного квар-
ца, полученные Бутягпным и др. [265—2811:
Реакция Температур^
проведения
реакции, К
s=Si- + 02 -> sSiOO- 77,300
esSiO' + О2 -> ==SiOOO’ 77 '
ExSi- 4- СО (=SiCO)- 77
. EsSiO' + CO -» (=SiOCO)’ 77
(=SiOCO)' -> =sSi’ + CO2 >350
sSi' + CO2 (=SiOCO)' 300
=Si’ + NO2 -> (=SiONN)’ 300 <
(=SiONN)’ -> EiSiO- + N2 >400
sSiO' -I- H,(D2) -> =SiOII(Z>) + 11(7/) 77
-(k —0, .
>Si: 4-П (£>)-► >Si - П (D) 77
—(K —oz
=^Si О Si== + H(Z>) -> ==SiOII(/J) -j- Si s 77
=Si’ + II2 =SiIl + IT 400
(==Si()CO) -1- O . -> (=»SiCO4)- 77
(sESiCO,)- -> ==SiOO‘ + CO, >150
(EsSiOOO)- + CO (=SiCO\)- 77
(sSiONN)’ + O2 sSiOO’ 4- N,O 300
=SiOO' 4- CO -* SiO’ 4- CO2 . >320
3=Si’ 4- СП, = CH2 -* sa Si-СП,—CH, 300
=SiO 4- CII2 = CH2 -> ssSiO—СИ,—СП, 300
sSi-CH,CII -* =Si -CH-CH3 42C
=Si- + CII2 = C(CH) -> sSi—CH2-C(CH,)2 300
=3Si ’ 4- CH,0 sSi—О -CH2 300
sSi 4- Celle -> (^Si-CeHb)- 300
=SiO 4- C.,11,, -> (EsSi-O-Celle)’ 300
С помощью приведенных реакций путем соответствую-
щей обработки поверхности мо;кпо многократно переводить
центры из одного типа в другой, например:
со °,
= SiO’^sSi’ =SiOO’.
со
Методом микрокалориметрин были измерены тепловые
эффекты радикальных реакций и вычислена прочность свя-
197
зей в некоторых радикалах (2701. 13 частности, прочность
связи Si —О в радикале =SiO’ составляет 4GG кДж/моль,
а связи О—О в перекисном радикале szSiOO —
281 кДж/моль. Высокая прочность кремнекислородной свя-
зи приводит к стабилизации на поверхности кварца таких
радикалов, как =Si0C0‘, (==SiCO4)’, (==SiO3)‘ и др.
При обработке водородом поверхности SiO2, содержащей
радикалы =SiO', образуются атомы водорода, являющиеся
активными свободными радикалами, которые принимают
участие во вторичных поверхностных реакциях:
=SiO‘ + Н2-> ==SiOH + Н;
Н + (=Si‘, ==SiO", =SiOO’) -► гибель;
=Si: + II-> = Si - II;
H + (=Si - О—81=)деф-> =SiOH + =Si\
Атомы водорода в ходе таких превращений выявляют
другие непарамагнитные центры. К таковым относятся «си-
лиленовые» =Si:, которые образуются при одновременном
разрыве двух связей у одного атома кремния, и «напряжен-
ные связи» =Si—О—Si==, активные в хемосорбции газов.
Они представляют собой деформированные фрагменты
=Si—О—Si=, прочность связи Si—О в которых составля-
ет 340—380 кДж/моль против обычного значения
505 кДж/моль [270]. Для «напряженных» связей характер-
но их исчезновение при отжиге в результате релаксацион-
ных процессов. Количество напряженных связей составляе!
1,5-1017 м~г, т. е. почти в 3 раза больше числа ПМЦ. «Напря-
женные» связи являются центрами одноразового действия,
т. е. после обработки поверхности СО2 (или СО и N2O) мож-
но «закрыть» эти места хемосорбции и дальнейшая актив-
ность поверхности будет обусловлена только парамагнит-
ными состояниями.
Таким образом, на поверхности измельченного кварца
стабилизируются дефекты разных типов: разорванные свя-
зи (парамагнитные центры =Si и ==SiO'), «напряженные»
связи, «силиленовые» центры =Si:, которые активно всту-
пают в реакции с разными молекулами.
При диспергировании кварца в поверхностно-активных
средах (в воде при 293 К, в ацетоне при 200 К) парамаг-
нитные центры методом ЭПР не обнаружены [557 |.
198
4.1.3. Кинетические особенности реакций
твердых тел с газами
Из приведенных выше данных следует, что реакции га-
зов идут на активных центрах поверхности твердых тел.
Скорость таких реакций определяется скоростью образова-
ния н расходования активных центров. Считается, что ско-
рость образования активных центров пропорциональна ско-
рости роста поверхности
а расходование центров идет по двум каналам: самопроиз-
вольная гибель активных центров и собственно взаимодей-
ствие центров с молекулами газа.
Полученные на основе этих допущений уравнения по-
зволяют, измеряя скорость химической реакции в зависимо-
сти от давления газа, определить такие величины, как ско-
рость возникновения активных центров w0 и время их жиз-
ни (см. гл. 2).
Однако из экспериментальных данных, в качестве кото-
рых могут служить кривые изменения поверхности кварца
и накопления ПМЦ в зависимости от времени активации,
приведенные в работе Колбанева, Бутягнна [557], следует,
что по мере уменьшения размера частиц скорость роста
удельной поверхности падает, а накопление активных цент-
ров продолжается практически с постоянной скоростью.
На основании этих данных Стрелецким, Бутягпным (272 I
выделены два режима протекания реакции газа с поверх-
ностью: режим раскалывания и режим трения (поверхност-
ного разрушения). В первом механическая энергия расхо-
дуется на образование поверхности, активные центры обра-
зуются на свежей поверхности и скорость их возникновения
пропорциональна скорости роста новой поверхности в соот-
ветствии с уравнением (4.1), где п — коэффициент пропор-
циональности, равный концентрации центров на свежеоб-
разованной поверхности. Во втором режиме основная часть
механической энергии расходуется на пластические дефор-
мации в приповерхностных слоях материала на контактах
отдельных частиц друг с другом и с материалом измельчи-
тся ьной аппаратуры.
Скорость инициирования реакции трением является уже
функцией удельной поверхности, и суммарная скорость про-
цесса соответственно равна
«’ = «£ + /(*). М
1У*Э
Рис. 70. Два режима протекании реакции в системе твердое — газ
[272].
а) 1 — кинетика дпенергпроианин кварца; 2 — кинетика пакой.тения парамагнит-
ных центров; и) 1 — скорость реакции взаимодействия водорода с активными
центрами поверхности; 2 — вклад. обус.ювиеииый ростом поверхности; J —
вклад трения.
Режим раскалывания играет основную роль в иницииро-
вании процесса до тех нор, пока удельная поверхность квар-
ца не достигнет нескольких квадратных метров, затем оба
пути инициирования примерно равноценны, и при высо-
ких значениях удельной поверхности определяющая роль
принадлежит трению.
13 качестве примера на рис. 79 показана зависимость ско-
рости хемосорбции водорода от поверхности кварца, приве-
дена суммарная скорость реакции и вычислены скорости
реакции за счет раскалывания и трепня. Считается, что ско-
рость реакции, инициированной трением, равна разности
между суммарной скоростью, определенной эксперименталь-
но, и скоростью реакции, рассчитанной по уравнению (4.1).
Значения dsldx рассчитывали методом графического интег-
рирования кривой s(t),
2(J0
Таким образом, из этих данных следует, что кинетиче-
ские особенности мехапохимнческпх реакций газон с твер-
дыми телами обусловлены не только реакциими на све.не-
образованной поверхности, но н реакциями на сформиро-
ванной («старой») поверхности, генерация активных цент-
ров на которой происходит вследствие трения частиц друг
о друга.
4.2. МЕХАНОХПМПЧЕСКПЕ РЕАКЦИИ ТВЕРДЫХ ТЕЛ
С ЖИДКОСТЯМИ
4.2.1. Реакции металлов с жидкостями
Различного рода химические реакции протекают при ак-
тивации металлических порошков в жидких средах. С по-
мощью активации может быть получен широкий спектр ме-
таллоорганических соединении. В частности, были прове-
дены исследования с такими металлами, как никель, хром,
олово, алюминий, кремний и др.
Ариас [558] исследовал реакции между металлическими
никелем и хромом и различными органическими и неорга-
ническими жидкостями. j
Активацию порошков (150 г хрома и 200 г никеля) про-
водили в барабане шаровой мельницы, футерованном не-
ржавеющей сталью п заполненном жидкостью (500—750 мл)
и гелием под давлением 6,9 МПа. Длительность активации
384 ч при скорости вращения 100 об/мин. По ходу актива-
ции производили отбор проб газовой фазы, а по окончании
отделяли порошки от жидкости центрифугированием, су-
шили п производили анализ химического состава порошков
и жидкостей. Использовали органические жидкости: н-геп-
тан, метиловый спирт, этиловый спирт, диэтилэфир, ацетон,
я-бутплальдегид, уксусную кислоту, четыреххлористый уг-
лерод, бензол, толуол. При активации хрома, кроме того,
использовали жидкие неорганические хлориды: SiCl4,
SnCI4, TiCI4.
Ви всех случаях после активации порошки содержали
на поверхности новые соединения в пределах от 0,01 до
56%. Количество содержащихся на поверхности продуктов
связано с изменением удельной поверхности порошков: чем
больше удельная поверхность, тем выше содержание про-
дуктов. Минимальную удельную поверхность н минимум
продуктов имеет порошок хрома при активации в TiCl4 и
никеля при активации в я-гептане. Максимальное колпче-
201
ство продуктов взаимодействия с неорганическими веще-
ствами обнаружено при активации хрома в SnCl4. Содержа-
ние хрома достигает 33%.
Во всех жидкостях обнаружены продукты взаимодействия
жидкостей с металлическими хромом и никелем. Природа
их весьма разнообразна и зависит от типа органической жид-
кости. При активации в среде я-гептана образуются высоко-
кипящпе алканы, при активации в метиловом и этиловом
спиртах — металлоорганические соединения, в ацетоне —
дпацетоналкоголь. При активации в бутплальдегиде по-
лучаются вода и ряд ненасыщенных а- и [J-альдегидов, при-
сутствующих как в мономерной, так и в полимерной форме.
При активации хрома и никеля в бензоле и толуоле образу-
ется высококипящее соединение, ИК-спектр которого бли-
зок к ПК-спектру полистирола, а в четыреххлористом уг-
лероде — гексахлорэтап.
В газовой фазе обнаружено присутствие газообразных
продуктов взаимодействия хрома и никеля с жидкостями —
водорода, метана, углекислого газа.
В синтезах алгоминийорганических соединений очень
сильное тормозящее действие оказывает имеющаяся на по-
верхности алюминия окисная пленка. Механическое уда-
ление пленки оказывает ускоряющее действие на реакции
[559]. Авторами работы [560] успешно проведены реакции
3C4II8I + 2А1 -> (С4П8)3А1212;
ЗС4Н8Вг + А1 -> А1Вг3 + яС4П10 + полимерный материал.
Протекание реакции опи связывают с возникновением эк-
зоэлектронов при активации алюминия.
Ряд оловоорганических соединений получен Гроном, Па-
удертом [561 ] при обработке металлического олова с орга-
ническими жидкостями. Они же показали, что при диспер-
гировании кремния в четыреххлористом углероде происхо-
дит хлорирование кремния с образованием SiCl4 и ССГра-
дикалов, которые димеризущтся в С2С1в. Авторы полагают,
что реакция идет по радикальному механизму [562].
>.
4.2.2. Кинетические особенности реакций t
твердых тел в жидкой среде
Взаимодействие твердых тел с жидкостями имеет общее
для гетерогенных процессов свойство: оно включает в себя
несколько стадий. Первой из них является стадия переноса
реагента к поверхности, на которой происходит реакция,
202
на второй стадии идет собственно химическая реакция,
третья стадия заключается в отводе продуктов реакции ют
реакционной поверхности. Суммарная скорость процесса
определяется скоростями отдельных стадии, а лимитировать
процесс будет самая медленная из них. Таковой может быть
диффузионная стадия, когда скорость подвода реагентов н
отвода продуктов реакции определяется скоростью диффу-
зии, пли кинетическая, когда медленным является процесс
химического взаимодействия.
В свою очередь, эти стадии гетерогенного процесса подраз-
деляются на внешние и внутренние диффузионные и кинети-
ческие области. Кроме диффузии в жидкой фазе, процесс
может лимитировать диффузия реагента через слой продук-
та, образующегося на поверхности твердого вещества в ре-
зультате взаимодействия с реагентом (внутридиффузионное
сопротивление).
Кинетические особенности механохпмических реакций
твердых тел с жидкостями будут различными в зависимости
от того, образуется или не образуется на поверхности твер-
дых тел слой продукта.
Очевидно, что когда внутридиффузионное сопротивле-
ние отсутствует (слой продукта на поверхности твердого те-
ла не образуется) п химический реагент достаточно активен,
скорость реакции должна быть пропорциональна общей
поверхности твердого тела. Такой случай, как показали Кол-
банев и Бутягнн [567], имеет место прп механохимическом
растворении кремния в воде в соответствии с уравнением
Si + 4НаО -> H4SiO4 + 2Н2.
Продукт взаимодействия — мономерная кремниевая кисло-
та — хорошо растворима в водных растворах.
Однако скорость реакции в момент механической актива-
ции в 2—3 раза выше, чем на предварительно активирован-
ных образцах. Если перемешивание прп растворении пред-
варительно активированных образцов было достаточно эф-
фективным, то указанный эффект следует отнести за счет
образования более активной в химическом отношении по-
верхности в момент разрушения по сравнению с предвари-
тельно активированной п выдержанной некоторое время
поверхностью.
Па практике чаще встречаются реакции, осложненные
образованием на поверхности продуктов взаимодействия.
Хорошо известей пример замедления кислотного разложе-
ния шеелита:
CaWO4 + 2НС1 = H2WO4 + СаС12
203
н.з-за образования вольфрамовой кислоты, обволакивающей
частицы исходного вещества [565]. Применение механиче-
ской активации, при которой возникает свожая поверхность
и пленка продукта удаляется с поверхности, позволяет от
внутридиффузнонного режима перейти к кинетическому.
Исерсоном и Хавским [565] показано, что разложение шее-
лита соляной кислотой, а также монацита и вольфрамита
растворами щелочи интенсифицируется, а расход реагентов
сокращается при осуществлении операции в подогреваемой
шаровой мельнице.
Аналогичная картина наблюдается при выщелачивании"
вольфрамита содой по реакции
Fc(Mn)WO4 + Na2CO3 -> Na2WO4 + zFe2O3 + t/Mn3O4 + CO2,
продуктами которой являются растворимый вольфрамат
натрия и плохорастворимые, затрудняющие дальнейшее
растворение оксиды железа и марганца. Альбрехт и др. [566]
показали, что проведение данной реакции в шаровой мель-
нице существенно ускоряет процесс.
Таким образом, если торможение реакций твердых тел
с жидкостями вызвано образованием слоя продукта на по-
верхности, то скорость реакции будет пропорциональна уже
не всей поверхности, а только скорости образования актив-
ной в химическом отношении поверхности.
В качестве примера реакций такого типа нами [568] ис-
следована имеющая важное практическое значение обменная
механохимическая реакция между сульфидом железа и иона-
ми металла в растворе
FeSTB + Me^, -> MeSTB + Fe^,,
где Me — Си, Zn, Ni, Со, платиновые и благородные метал-
лы. По этой реакции сульфид железа взаимодействует с но-
нами других металлов с образованием менее растворимых
сульфидов, осаждающихся, по-видимому, на его поверхно-
сти. Необходимо отметить, что термодинамически реакция
возможна, но практически не протекает по кинетическим
причинам. Обычно эти реакции реализуются при наличии
свободной кислоты в растворе при нагревании и под давле-
нием, однако в момент интенсивного механического воздей-
ствия они протекают и в нейтральной среде [568].
Опыты проводились в планетарной мельнице ЭИ-2 X150.
Поскольку металлическое железо в соответствии с рядом
напряжений является осадителем для некоторых цветных
металлов, все дальнейшие эксперименты проводились в
204
Рис. 81. Кинетика диспергирова-
ния сульфида железа при исполь-
зовании шаров различного диа-
метра [508[.
Диаметр шаров, мм: 1 — 1,3; 2 — 20
3 — зи; 4 — 50; 3 — 70; 6 — 120.
Рис. 80. Изменение концентрации
никеля в растворе при протека-
нии обменной реакции FeS +
-h NiSO4 -> NiS + FeSO4 [568] в
условиях механической актива-
ции.
1 — остановка после активации в те-
чение 1 мни; 2 — то же, в течение
2 мин; 3 — длительная активации.
медных барабанах с медными шарами (или шарами из спла-
ва ВК-8).
Так же как и для реакций с газами, обменное взаимодей-
ствие происходит только в момент обработки (рис. 80). Пос-
ле остановки аппарата скорость реакции резко падает. Отсю-
да следует, что для механохимической реакции важна не
общая поверхность, а только свежеобразованная. (
Из практики диспергирования известно, что скорость его
в зависимости от диаметра мелющих шаров проходит через
максимум. В таких высоконапряженных аппаратах, как
планетарные мельницы, этот максимум сдвинут в область
очень малых шаров, так что практически с увеличением ди-
аметра шара скорость измельчения падает (рис. 81).
Па рис. 82 приведены данные по кинетике осаждения
цинка пирротином в зависимости от диаметра мелющих ша-
ров. Из рисунка видно, что скорость извлечения цинка из
раствора также падает с увеличением диаметра шаров.
Отношение скорости реакции к скорости измельчения г
зависимости от диаметра шаров растет незначительно и яв
ляется практически постоянной величиной (рис. 83).
301
Рис. 82. Кинетика обменной реак-
ции FeS + ZnSO4 -> ZnS (-FcSO4
в зависимости от диаметра мелю-
щих шарой [5С8].
Диаметр шаров, njij: 1 — |,5; 2 — 2;
3 — 3; 4 — 5; 5 — 7.
. _ Ус
I/S
о—V-
аг-
. 0,1 ----1-----!------!-------
О 2 4 6 #мм
Рис. 83. Отношение скоростей хи-
мической реакции и роста поверх-
ности сульфида железа прп ис-
пользовании шаров различного
диаметра [568].'
Таким образом, скорость реакций твердых тел с раство-
рами, протекающих с образованием продукта на поверхно-
сти, определяется скоростью образования самой поверх-
ности, т. е.
= (4.3)
На основании результатов исследования можно ре-
комендовать к использованию мехапохимические реакции
обменного разложения для извлечения никеля и платино-
вых металлов из промышленных растворов, цинка пз рас-
творов кучного выщелачивания и т. д. путем обработки
суспензии в измельчнтельных аппаратах, обеспечивающих
эффективное диспергирование.
------------------ГЛАВА 5--------------------
МЕХАНОХИМИЧЕСКИЕ РЕАКЦИИ
В СМЕСЯХ ТВЕРДЫХ ВЕЩЕСТВ
5.1. ОБЩАЯ ХАРАКТЕРИСТИКА ТВЕРДОФАЗНЫХ РЕАКЦИИ
Возможность проведения реакций непосредственно ме-
жду твердыми телами, минуя стадию их растворения, всег-
да была привлекательна для химиков. Разрабатывались и
разрабатываются методы их проведения с применением в ос-
новном термической активации. Метод механической акти-
вов
наций, позволяющий создавать активные состояния в твер-
дом теле, открывает определенную перспективу для ускоре-
ния таких реакций.
В принципе возможны несколько способов применения
механической активации для этих целей: 1) предваритель-
ная активация исходных (или одного из исходных) компо-
нентов перед термической обработкой; 2) предварительная
совместная активация смеси твердых веществ перед терми-
ческой обработкой; 3) проведение реакции непосредственно
в момент подвода механической энергии (механохнмический
способ); 4) проведение реакции с одновременной механиче-
ской активацией и термической обработкой (механотерми-
ческий способ).
Нельзя сказать заранее, применение какого из указан-
ных способов более целесообразно для той или иной реак-
ции. Тем не менее с точки зрения использования коротко-
живущих активных состояний непосредственно в момент
проведения реакции представляется, что последние два спо-
соба имеют определенные преимущества. Поэтому основное
внимание в настоящей работе уделено механохимическому
способу проведения реакций в смесях твердых веществ,
а также предварительной совместной активации. Стадия
предварительной активации исходных компонентов, приво-
дящая к ускорению последующих твердофазных реакций, де-
тально рассматривалась выше на примере конкретных не-
органических соединений (оксиды, сульфиды, соли и др.).
К исследованию более сложного механотермпческого спосо-
ба, по нашему мнению, следует переходить после установ-
ления основных закономерностей механохимического спо-
соба.
В соответствии с термодинамикой направление реакции
и степень превращения определяются изобарно-изотермиче-
ским потенциалом реакции AG. Реакция между твердыми ве-
ществами возможна, если
Sag
продуктов
-Sag
реагентов
AGpeaKmm
<0.
(5-1)
Для реакции
^тв GTB GTB ОтВ (или ?1Z?TB)
n flzid Г)
АСреакции = AGpeanni«il + GT1 In .
« ,«п
(5-2)
где ас, aD, аА, ав — активности конечных н начальных
продуктов. Поскольку твердые вещества находятся прп нор-
207
мальном давлении и обладают активностями, равными пли
близкими к единице, то
AGI)cai ции —
Л^реакцпи
(5.3)
Таким образом, для определения направления реакции до-
статочно знать значение AG реакции в стандартном состоя-
нии (Т — 298 К, р = 0,1 МПа). Если реакция термодина-
мически возможна (AGp<0), то теоретически в смеси твер-
дых веществ она должна идти до конца.
Если реакция сопровождается появлением жидкостей, га-
зов или образованием твердых растворов, то вклад T&S
в изменение изобарно-изотермического потенциала
AG = АЯ — TAS (5.4)
становится весьма существенным, и тогда трудно говорить
о связи между направлением реакции и значением величи-
ны AG, так как изменение энтропии при этих процессах мо-
жет быть как положительным, так и отрицательным.
Под влиянием механической активации в твердых телах
происходит накопление энергии за счет образования дефек-
тов, смещений атомов со своих равновесных положений и
т. д., что приводит к возрастанию энтальпии и энтропии твер-
дых тел. В ряде случаев изменения этих величин являются
значительными, и тогда общее изменение свободной энергии
будет таково, что реакция также может изменить свое на-
правление. Недоучет этих факторов может привести к ка-
жущемуся «противоречию» с термодинамическими расче-
тами.
Характерной особенностью химических реакций в сме-
сях твердых веществ является их стадийность и локализа-
ция процесса только в местах соприкосновения между ис-
ходными реагентами.
Основную роль в кинетике твердофазных реакций игра-
ют стадии собственно химического взаимодействия, зароды-
шеобразования и диффузионного переноса через слой обра-
зовавшегося продукта. Химическое взаимодействие начина-
ется на наиболее активных участках поверхности (выходы
дислокаций, точечные дефекты и т. д.), здесь образуются
ядра (зародыши) продукта реакции, рост и слияние кото-
рых приводят к появлению сплошной реакционной зоны.
После образования продукта устанавливается стационарный
диффузионный процесс переноса одного из компонентов к
другому через слой продукта. Диффузионный массопере-
нос может происходить по нескольким механизмам: по объ-
208
емному (по системе точечных дефектов), зерногранпчному
и поверхностному.
Большинство теоретических п экспериментальных ис-
следований, выполненных к настоящему времени, относят-
ся к рассмотрению в качестве лимитирующей стадии диффу-
зионного процесса. Это модели и соответствующие им урав-
нения Лидера [569], Дюнвальда-Вагнера [570] или Серина-
Эллпксопа [571], Гинстлинга-Броунштейна [572], Картера-
Валепси [573, 574], Коматцу-Уемура [575], Конопюка
[576], Вагнера [577]. Во всех моделях, за исключением мо-
дели Дюнвальда-Вагнера, предполагается, что профили кон-
центраций диффундирующих частиц квазистационарны. Ес-
ли же решается нестационарное уравнение диффузии, то
не учитывается изменение удельного объема в ходе реакции.
В наиболее совершенных моделях учтена встречная диффу-
зия реакционных частиц, а также различие удельных объе-
мов продукта реакции и одного из реагентов.
Одна из наиболее ранних и широко известных моделей
реакций в смесях порошков предложена Лидером [569].
Согласно этой модели, скорость реагирования лимитиру-
ется скоростью односторонней диффузии частиц «покрыва-
ющего» компонента через слой продукта. Применяя к сфе-
рическим зернам параболический закон Фика для стацио-
нарной диффузии из постоянного источника через плоские
поверхности, Лидер получил известное соотношение
—I2
1 — (1 — а) 3 = Z-jT. (5.5)
Имеются модели для реакций, которые лимитируются
собственно химической реакцией [86]. Одна из них носит
название модель «сжимающейся сферы». В соответствпп с
ней реакция протекает одновременно по всей поверхности
частицы, причем продукты реакции не обладают блокирую-
щим действием. Скорость реакции пропорциональна доступ-
ной поверхности, и в этом случае используется уравнение
= *2т, (5.6)
1
1 - (1 - а) 3
где а — степень превращения, т — время реакцпп, к —•
константа.
Воздействуя на лимитирующую стадию процесса, можно
изменять скорость твердофазных реакций. В частности, одпн
из способов ускорения предложен Хедваллом [578]. Он за-
ключается в том, что реакции проводят при температуре, со-
200
ответствующей температуре фазового перехода в продукте
реакции, при этом возникает разупорядоченность в кри-
сталле п процессы диффузии ускоряются. Механическая
активация смесей твердых веществ позволяет все время об-
новлять поверхности реагирующих веществ, т. е. самим
устранять диффузионные затруднения. Однако для приме-
нения модели «сжимающейся сферы» нет оснований, так как
реакция идет одновременно пе по всей поверхности зерна,
а только в некоторых определенных точках (в местах кон-
такта частиц в момент соударения с мелющим телом). Таким
образом, подобные реакции совершенно отличны от указан-
ных выше, для них характерны свои закономерности и дол-
жны быть созданы свои модели.
Для создания таких моделей необходимо знание факто-
ров, обусловливающих специфику механохимических ре-
акций. Выше указывалось, что к таким факторам относятся
локальные давления и температуры, возникающие в местах
соударения мелющих тел, и специфические явления, возни-
кающие при пластическом течении материала. Импульсный
характер локальных температур может приводить к отли-
чию механохимических реакций от реакции при медленном
нагревании. Так, согласно Болдыреву [230], при возможно-
сти протекания в системе параллельных или последователь-
ных реакций реализуются те из них, которые «вписываются»
в кратковременный импульс тепла в момент соударения.
Кроме того, если учесть, что большая часть механиче-
ской энергии превращается в тепло, то ясно, что тепловой
вклад весьма существен при активации смесей в пзмельчп-
тельных аппаратах, особенно в том случае, когда охлажде-
ние аппаратов отсутствует.
Имеется достаточно много теоретических и эксперимен-
тальных исследований, в которых рассмотрен вопрос о влия-
нии высоких статических давлений на скорость химических
реакций в твердой фазе. Так, многочисленными исследования-
ми подтверждено, что скорость диффузии всегда уменьша-
ется с увеличением давления. Это может быть обусловлено
как уменьшением равновесной концентрации вакансий, так
и уменьшением нх подвижности.
Эванс и Поляни [579] па основании теории переходного
состояния вывели уравнение, связывающее константу ско-
рости процесса, протекающего в твердой фазе, с давлением:
0 In к ДУ*
др ~ ПТ’
где ДV* — разность между объемами активированного комп-
(5.7)
210
лекса н веществ, вступающих в реакцию. Так как объем ак-
тивированного комплекса всегда больше объема исходных
компонентов, то из уравнения следует, что с увеличением
давления скорость реакции падает.
Эксперимент показывает, что скорость твердофазной ре-
акции, например CuSO4 РЬО = PbSO4 + СнО, при уве-
личении давления до 2,4-103 МПа замедляется в 2 раза [580].
Отсюда можно сделать вывод, что основную роль при ме-
ханической активации играет не статическая, а сдвиговая
компонента возникающих локальных давлении.
Из приведенных выше экспериментальных работ следу-
ет, что пластическое течение материала снимает многие ки-
нетические затруднения в химических реакциях. При пере-
ходе твердых тел к пластическому течению размораживают-
ся и становятся подвижными многие типы дефектов.
Таким образом, влияние механической активации на ско-
рость твердофазных химических реакций может быть связа-
но не только с возникновением локальных температур и
давлений, но и с образованием активной поверхности и уско-
рением массоперспоса вещества в условиях пластического те-
чения за счет генерации точечных дефектов подвижными
дислокациями.
5.2. ВОЗМОЖНОСТИ МЕТОДА
МЕХАНИЧЕСКОЙ АКТИВАЦИИ
ТВЕРДОФАЗНЫХ РЕАКЦИЙ
5.2.1. Окислительно-восстановительные реакции
В соответствии с имеющейся классификацией химические
реакции подразделяют на две большие группы: 1) с измене-
нием степени окисления реагирующих компонентов (окис-
лительно-восстановительные реакции) и 2) без изменения
степени окисления (реакции соединения, обмена, замещения
и т. д.). Следует ожидать, что влияние механической акти-
вации будет различным для разных типов реакций.
Грон и соавт. [581] исследовали восстановление некото-
рых оксидов активированным углем при обработке смесей
в вибромельнице. Условия активации: смесь из оксида
(20 г) и угля (8 г) загружалась в барабан вибромельницы,
содержащей 12 кг фарфоровых шаров диаметром 30 мм, час-
тота п амплитуда составляли 230 с-1 и 1,75 мм соответствен-
но. За кинетикой восстановления следили по выделению
211
СО и CO2. Анализ показал, что содержание СО было незна-
чительным.
За 150 ч активации прошло восстановление следующих
оксидов: СнО — 80%, ZnO — 53, Fe2O3 — 35, NiO — 30,
Fe2O3 — 27, CoO — 18, MgO — 2,5%. Указанная последо-
вательность совпадает co значениями изобарно-изотермиче-
скнх потенциалов реакции восстановления оксидов углеро-
дом, которые равны для восстановления СнО —268 кДж/моль,
а для MgO +177 кДж/моль.
Нами [581—586 J исследовано восстановление диоксида
олова различными восстановителями:
ддо
298
1п
SnO2 = SbO + 2°2
SnOo + С = Sn -f- СО,
2 2
SnO2 +-7TW Sn +^wo3
о о
+295
+126
+25
SnO2 + Si — Sn + SiU.. 4 2 ' SllO, +~.Л1 = Sn +~5 Al..O:| ~ «J <J “ -276 - 535
где AG"88 — изменение изобарно-изотермического потенциа-
ла при 298 К.
Использованы диоксид олова квалификации «ЧДА», гра-
фит с содержанием основного вещества 99,9%, кремний —
99,9, вольфрам — 99,9, алюминий — 98,8%. Средние раз-
меры исходных компонентов составляли SnO2 — 5 мкм,
С — 30, Si — 30, Al — 200, W — 40 мкм. Активацию сме-
сей проводили на планетарной мельнице со стальными ба-
рабанами и шарами (диаметр 8 мм). Частота вращения
13,3 с-1, что соответствует мощности ~100 Вт.
После механической активации определяли количества
Sn, SnO и SnO2 по известной методике [431J путем последо-
вательного выщелачивания пробы в щелочном и кислом раст-
воре и сплавления нерастворившегося остатка с перекисью
натрия. Степень превращения а определялась по количе-
ству как SnO2 в остатке после выщелачивания, так и олова,
перешедшего в растворимые формы. Наблюдалось совпаде-
ние указанных величин с точностью до 1—2%. Поверхность
порошков определяли газосорбцпоипым хроматографиче-
ским методом [218]. Рентгенофазовый анализ порошков вы-
полняли на аппарате ДРОН-2 (излучение медное).
212
к с
<0 V
oj *5
Рис. 84. Дифрактограммы сме-
сей оксида олива (IV) с крем-
нием после механической ак-
тивации [582].
/ — исходна л смесь; продолжитель-
ность активации, мни: 2 — 12,5;
3 — 30; I — 91); рефлексы 0,291,
0,279, 0,201 им принадлежат олову.
Рис. 85. Кинетика механохпмпческо-
го восстановлении оксида олова (IV)
различными восстановителями [586].
I — исходный образец; BoccranoiniT<?.iii:
2 — углерод: в — вольфрам; J — крем-
нии; а — алюминий.
Данные рентгенофазового анализа показывают, что из
продуктов реакции, как правило, хорошо регистрируется
0-Sn. Липпи |3-Sn начинают регистрироваться на рентгено-
граммах нрн содержании его в пробе 2—3% (рнс. 84).
На рис. 85 приведены кинетические данные для указан-
ных выше реакций по результатам химического анализа.
Видно, что скорость восстановления также возрастает по
мере уменьшения изобарно-изотермического потенциала ре-
акции.
Для выяснения роли активных поверхностных центров
в твердофазных механохимпческнх реакциях памп детально
исследована реакция между диоксидом олова и кремнием,
которые имеют очень высокие температуры плавления (Твл
диоксида олова выше 2300, кремния — 1700 К [5851).
Известно, что при разрушении кремния образуются па-
рамагнитные центры, которые регистрируются методом ЭПР.
Впервые их обнаружил Фехер [5871 после пескоструйной
полировки кремния. Сигнал ЭПР представляет собой оди-
ночную симметричную линию шириной 0,5 ЛО3 А/.м с g-фак-
213
тором, равным 2,0061. Позднее парамагнитные центры на
кремнии исследовались многими авторами. В частности,
Чанг и Ханеман [284] зарегистрировали интенсивный сиг-
нал ЭПР с такими же параметрами при дроблении кремния
в сверхвысоком вакууме. Поверхностная концентрация со-
ставляла 2-10й см--; сверхтонкой структуры и анизотро-
пии g-фактора не наблюдалось. При взаимодействии про-
дуктов разрушения с кислородом происходит изменение сиг-
нала.
Согласно Ханеману [283], неспарениые электроны воз-
никают в результате разрыва Si—Si-связей при образова-
нии новой поверхности. Строение вновь образованной по-
верхности отличается от строения идеальной поверхности,
вследствие чего неспаренные электроны не занимают sp3-
орбиты поверхностных атомов. В поверхностном слое часть
атомов приподнята, а другая опущепа по сравнению с их
«нормальным» расположением. Такая перестройка поверх-
ности, как считает автор, энергетически выгодна. При этом
парамагнитные свойства поверхности определяются верх-
ним слоем атомов, имеющих неспарениые электроны на час-
тично перекрывающихся s-орбиталях. Степень перекрыва-
ния в соответствии с поверхностной концентрацией спинов
равна 80%. Нижний слой атомов не содержит песпарениых
спинов вследствие сильного перекрывания р-орбиталей.
Как рассматривалось выше, при механической актива-
ции SnO2 на его поверхности возникают парамагнитные
центры ОГ в количестве 1012—1013 см-2, обладающие высо-
кой реакционной способностью [435]. Таким образом, про-
текание реакции между Si и SnO2 может быть связано с вза-
имодействием радикальных центров ^Si' и О^-
Из экспериментальных данных по взаимодействию ди-
оксида олова с кремнием, приведенных на рис. 85, видно,
что после 1,5 ч обработки смеси диоксида олова с кремнием
степень восстановления достигает 70%, причем до 50% пре-
вращения скорость восстановления практически постоянна
и равна 5-1017 мол./(г-с). Скорость измельчения в планетар-
ной мельнице составляет 120 м2/(г-ч) (рис. 86). Для смеси
состава SnO2: Si — 3 : 1 (по массе) и поверхностной кон-
центрации атомов кремния, равной 1015 см-3, следует, что
скорость их появления на поверхности ^2 • 1017 ат./(г-с).
Скорость восстановления в этих же условиях, как указано
выше, равна 5-1017 мол./(г-с). Таким образом, учитывая
неточность оценки начальной скорости роста поверхности
кремния в смеси с SnO2, можно считать, что эти величины
214
Рис. 86. Кинетика 'диспергиро-
вания оксида олова (7), крем-
ния (2) и их смеси (3),
^МИН
Рис. 87. Сорбция кислорода в про-
цессе диспергирования кремния
прп различных начальных давле-
ниях кислорода [585].
Давление кислорода, Па: 1 — 120: S >—
532; 3 —2.I-103; J — 1,4-10*.
совпадают, т. е. каждый поверхностный атом кремния всту-
пает в химическую реакцию.
Ранее было показано, что механическая активация твер-
дых тел сопровождается сорбцией газа, которая прекраща-
ется с окончанием механической обработки. Предполага-
ется, что за сорбцию газов ответственны короткоживущие
активные центры. Регистрируя количество поглощенного
в момент механической обработки газа, можно определить
число активных центров. Проведены опыты по сорбции кис-
лорода на кремнии под влиянием механической обработки.
Опыты проводили в стеклянной вакуумной мельнице. В про-
цессе диспергирования наблюдалось быстрое необратимое
поглощение кислорода, практически не зависящее от началь-
ного давления (рис. 87). Скорость сорбции равна 1,5-
• 101в мол./(г-с), а скорость появления поверхностных ато-
мов — 1,2-1015 ат./(г-с). Таким образом, валентно-ненасы-
щенные атомы поверхности кремния являются центрами не-
обратимого поглощения кислорода.
После механической обработки в Si методом ЭПР зафик-
сированы парамагнитные центры, причем скорость их обра-
зования примерно 3-1013 центров/(г-с), что па два порядка
меньше скорости разрыва связей прп возникновении по-
верхности. Следовательно, методом ЭПР регистрируются не
все валентно-ненасыщенные атомы, а только часть их, со-
хранившаяся после стабилизации поверхности. Содержание
215
Рис. 88. Изменение Концентрации Па-
рамагнитных центров в процессе ди-
спергирования Si (]) и SiiOa |-'Si (2).
ПМЦ после активации смеси
SnO2 -[- Si значительно ниже,
чем в чистом кремнии (рпс. 88).
По-видимому, валентно-ненасы-
щенные атомы кремния смеси
SnO2 + Si расходуются в реак-
ции восстановления SnO2, и кон-
центрация стабилизирующихся
центров резко падает.
Данные по тормозящему действию газов па реакцию, при-
веденные ниже, свидетельствуют о том, что газы подавляют
активные центры. Снижение скорости наблюдается даже в
случае оксида углерода, который, как известно, является
восстановителем по отношению к SnO.,. В данном случае
он подавляет активные центры на кремнии:
Вых щ растворимых
иролуктои, %
Аргон (0,1 МПа) 32,0
Воздух 26,2
Оксид углерода (0,1 МПа) 17,2
Вода (5 см*) 10,2
Исследования, проведенные нами па реакциях твердо-
фазного восстановления диоксида олова различными восста-
новителями с целью проверки и уточнения представлений
об общности химических процессов, протекающих в измель-
чительных аппаратах и при воздействии ударных волн, по-
казали, что сходство между ними проявляется только при
небольших интенсивностях ударного сжатия [586]. При
больших интенсивностях ударного сжатия высокие локаль-
ные температуры существуют длительное время и реакции
не отличаются от термически активируемых реакций.
Выше нами указывалось, что представления о коротко-
живущих активных центрах применимы в основном для сое-
динений с ковалентными химическими связями. К числу
таковых относятся и сульфиды. Возможность протекания
механохимических реакций сульфидов с газами и участием
короткоживущих активных центров показана в работе Быст-
рикова и Бутягина [282]. По-видимому, механохимическне
реакции сульфидов с твердыми веществами, которые проте-
216
Рис. 89. Спектры ЯГР смеси оксида олова (IV) с пиритом после ме-
ханической активации, снятые па ядрах 1I9Sn (я) и 5TFe (б) [4881.
кают с весьма значительными скоростями, также могут быть
объяснены участием короткоживущих активных центров.
В частности, взаимодействие сульфидов с оксидами проте-
кает по уравнениям
MeS -| МеО -> Me ф- SO.,;
Mcjbj ф- МсцО., —> Me4S ф- Ме^О -ф SO.,,
где Me = Pb, Си, Fe, Sn п др. [488]. Так, при механической
активации смеси PbS -|- РЬО наблюдалось увеличение дав-
ления в системе за счет образования SO2. При активации ин-
дивидуальных соединении в этих же условиях давление ос-
тавалось неизменным. Более сильные окислители в этой
реакции (РЬО2, КМпО4 и др.) вызывали взрывной процесс.
В качестве примера результатов для реакций второго
типа могут служить данные, полученные .методом ЯГРС
(рис. 89). В спектрах продуктов, снятых как на ядрах 11<JSn,
так и па ядрах 37Fe, в продуктах активации обнаружива-
ются соединения SnS и FeS. Вследствие образования этих
соединений после обработки активированной смеси по ука-
занной выше методике в растворимое состояние переходит
почти в 3 раза больше олова, чем при активации в аналогич-
ных условиях диоксида олова с кварцем, абразивные свой-
ства которого почти одинаковы со свойствами пирита
(рпс. 90).
Таким образом, разрыв под плпппнем механической ак-
тивации связей, возбуждение электронной подсистемы кри-
сталлов способствуют осуществлению различного рода окпе-
лительпо-восстаповительпых реакций. Причем большое влп-
217
Рис. 90. Переход олова в раство-
римую форму после активации
[488].
1 — в смеси с FeS; г — в смеси с Sil).
Рис. 91. Кинетика диспергирова-
ния хрома (1), железа (2) и их
эквиато.мнон смеси (3) [591 ].
янпе па эти реакции оказы-
вает среда, в которой они
проводятся, так как взаимо-
действие короткоживущих
активных состояний со средой является конкурирующим
процессом по отношению к твердофазному взаимодействию.
5.2.2. Реакции соединения
С применением механической активации были реализо-
ваны самые разнообразные синтезы: соединений металлов с
неметаллами (серой, фосфором, графитом, бором, иодом и
др.), интерметаллидов, сплавов, смешанных кристаллов,
сложных соединений и т. д. [17, 64 [.
Механохимические методы синтеза могут быть примене-
ны в том случае, если необходимы мелкодисперсные порош-
ки продуктов. Для боридов, например, получение порош-
ков с размером частиц порядка микрона затруднено, так
как при высокой температуре они спекаются. Для других ве-
ществ сплавление компонентов прп высокой температуре
иногда сопровождается частичным их испарением, что
может привести к нарушению стехиометричности соедине-
ния и снизить качество продукта. Это важно, например, для
полупроводниковых соединений типа Л11/?'1. В табл. 12
приведены результаты рентгепофазопого анализа, а в рабо-
218
Таблица 12
Результаты рентгеиофазового анализа механохимически
синтезированных соединении
Соедине- ние до0 288» кДж/моль т ! ]п.т соединения, _ к т„., низкоплавкой пптгктики, I* . Параметры ячейки, нм
Литературные данные Синтезирован- ные образцы
TiB2 -276,0 3143 — а -= 0.3025 с = 0,3213 0,3023 0,3220
NbB2 —176 3273 2123 а = 0,3095 с = 0,3306 0,3119 0,3288
МоВ -68,2 3093 — а = 0,3110 с = 1,695 0.312 1,696
SnTe —60,6 1063 505 а -= 0,6290 0,6287
1’bTe -69,0 1206 600 л — 0,6500 0,6502
—79,0 858 539 « - 0.4384 с = 3,045 0,4379 3,044
Sb2Te3 —56.5 889 697 а = 0,425 с ~ 3,000 0,424 2,998
те [588] — данные, полученные методом ЯГРС, для неко-
торых соединений, синтезированных в планетарной мель-
нице.
Способы получения механической активацией сложноле-
гнровапных металлических порошковых .материалов, днс-
иерснопио-упрочнепных сплавов, а также сплавов с сильно
ограниченной взаимной растворимостью компонентов твер-
дых смесей предложены Бенджамином [65, 589]. В качестве
примеров приводится получение сложных смесей на основе
никеля и железа (нержавеющие стали, инструментальная
сталь, содержащая20% W, 12% Со, 4% Сг, 2% Ni, 0,8% С),
сплавов Си—W, Си—Pb, Fe—Al, а также суперкорродирую-
щих сплавов на основе магния, содержащих до 5% Fe, Си,
Сг, С, Ti и др., активно реагирующих с водой и электролита-
ми с выделением тепла и водорода [590]. Для получения
указанных сплавов и порошков используется аттритор объе-
мом 380 л со скоростью вращения крестообразной меша.1-
ки 4,2 с-1.
Образование твердых растворов в системе Fe—Сг иссле-
довалось с использованием метода ЯГРС [591]. Механиче-
скую активацию порошков проводили в мельнице типа ЭП-
2x150. Загрузка смеси составляла 4 г, масса шаров 200 г.
Для исследования были взяты железо «карбонильное» мар-
ки «х. ч.» и хром марки «ч.». Температура плавления исход-
ных компонентов Fe — 1812 К, Сг — 2073 К, а температура
219
плавления твердого раствора состава 1 : 1—1648 К [4561.
Свободная энергия образования твердого раствора данного
состава равна —7,5 кДж/(г-атом) 1456].
Выбор системы Fe—Сг для исследования основан на хо-
рошо известном факте, что введение в металлическое желе-
зо хрома приводит к стабилизации гранецентрированной
модификации железа (y-Fe), которая является немагнитной.
Это приводит к появлению в мессбауэровском спектре оди-
ночной лпппп и позволяет надежно следить за степенью
перехода из объемно-центрированной исходной фазы ct-Fe в
y-Fe.
Результаты измерений поверхности продуктов диспер-
гирования газоадсорбционным методом при частоте враще-
ния 12 с-1 приведены на рис. 91. Видно, что удельная по-
верхность исходных компонентов растет практически линей-
но со временем активации, а удельная поверхность экви-
атомной смеси проходит через максимум. До максимума мож-
но рассматривать поверхность смеси как аддитивную, после
максимума она становится меньше поверхности железа.
Данные электронно-микроскопических исследований по-
казывают, что после 10 мин активации наблюдаются ска-
танные зерна размеро.м 2—3 мкм. После 20 мип образуются
агрегаты из разнородных частиц, которые из трехмерных на-
чинают превращаться в двумерные. В агрегатах происхо-
дит вторичная кристаллизация и далее — укрупнение их
с образованием совершенных кристаллических структур с
сохранением аморфного поверхностного слоя. Размер таких
агрегатов после 40 мип активации составляет 10—20 мкм.
При дальнейшем диспергировании структура вторичных кри-
сталлов становится более совершенной.
Данные мессбауэровских исследований показывают, что
агрегаты представляют собой твердый раствор Fe—Сг
(рис. 92).
На основании полученных данных для смеси Fe—Сг
проведена оцепка коэффициента диффузии хрома в железо
в условиях активации. В обычных условиях коэффициент
диффузии хрома в железо равен 10“12—10"14 см2/с, при на-
гревании до 1400—1600 К он возрастает до (2-4-8)-
•10"® см2/с. Если предположить, что диффузия прп актива-
ции протекает между сферическими частицами одинакового
радиуса Н, то для степени образования твердого раствора,
равной 0,5, справедливо соотношение где D — ко-
эффициент диффузии, 1„ — время нахождения частицы под
нагрузкой со стороны мелющего шара. Величина t„ про-
порциональна длительности удара мелющего тела о плоский
220
1
§ 0,8
й)
“ кт
о
§
I
й 0.97
5?
5 0.94
i
100
0.95
0,90
О 5 V, мм/с
Рис. 92. Спектры ЯГР смеси железа с хромом [591].
- исходили гмгсь; придо/ркнте. imhicti» антннащш, мин: 2 — 20; а — 40.
слой /уД, числу шаров Nm, числу ударов за время механи-
ческой активации Л\-д и отношению контактной площади
(площади деформации шара при ударе о плоский слой)
к площади плоского слон я:
S .
^11 — ^УД’Л'ш’Л’уд-— . (5.8)
В соответствии с работой [135] для нашего случая /уд ~
~ 10-в с, sK ~ 10“3 см2. Параметры Л'ш, Л'уд, s заданы ус-
ловиями эксперимента и соответственно равны 103, 104,
Ю2 см2.
Подставляя указанные значения в (5.8), получаем D »
л; 10“5 *—10“7 см2/с. Полученные результаты существенно
отличаются от приведенных выше и позволяют говорить об
ускорении диффузии па стадии пластического течения мате-
риалов прп обработке их в планетарных мельницах.
Прп механической активации смесей ионных солен про-
исходит образование смешанных кристаллов или слоимых
двойных солей. Уйбо, Паа 15921 и др. [5931 исследовали
получение твердых растворов поттпых солей при обработке
их смесей в дезинтеграторе. Им удалось получить смешан-
ные кристаллы для всех пар, приведенных ниже:
Исходные соли
Различие в
постоянных
решеток, %
ле .
sea
смеси 1:1,
КДж/моль
ВЫ + nh4i 1,11 —
KI + nh4i 2,69 —
KI+ вы 3,60 —1,29
CsCl + CsBr 4 22 —
KBr+ RbBr 4,31 —’1,19
RbCl + RbBr 4,42 -0,93
KC1 + RbCl 4,53 —0,945
NH4C1 + NH4Br 4,57 —
KC1 + KBr 4,64 —0,936
NaCl + NaBr 5,33
Пластическая деформация в ряде случаев оказывается
более эффективной, чем другие методы активации. Так, на-
пример, показано, что фаза Сп2Те при 523 К. образуется толь-
ко в результате приложения напряжений [155].
Проявляется избирательность метода, т. е. образуются
только некоторые из возможных в системе соединений. На-
ми показано, что при обработке смеси Ее и Sn, взятых в раз-
ных соотношениях, в мельнице образуется соединение FeSib,
хотя возможно л FeSii 1594, 595]. Кинетику образования
соединения FeSn., можно наблюдать на рпс. 93, па котором
приведены мессбауэровские спектры смесей после активации
через разные промежутки времени. Выяснилось, что синте-
зированные частицы имеют размер ~100 нм и проявляют
суперпарамагнитные свойства. Мы полагаем, что это явля-
ется не только следствием малого размера частиц, но и ре-
зультатом повышенной дефектности частиц, которая харак-
терна для механически активированных веществ.
Систематическое исследование образования промежуточ-
ных фаз — соединений и твердых растворов — при пласти-
ческом деформировании смесей порошков двух элементов
выполнено Неверовым, Буровым и др. [596, 597]. Опыты
проводились в условиях высокого давления со сдвигом. Фа-
зовый состав образцов определяли методом РФА.
Было показано, что механическое воздействие ускоряет
процессы фазообразоваиия и в деформированных образцах
появляются соединения и твердые растворы, соответствую-
щие равновесным диаграммам, а также пересыщенные кри-
сталлические и аморфные твердые растворы. Одпако обра-
зуются не все возможные фазы и не во всех системах.
222
Рис. 93. Спектры ЯГР для смеси
железа с оловом [594].
1 — исходная смесь; продолжитель-
ность aicniuuuun, мин. 2 — 30; 3 —
00; 4 — 90; 5—180.
“I I I I I I
25 20 15 28, град
Рис. 94. Рентгенограммы смеси
CaF3+ Са3(РО4)2 в зависимости от
времени механической активации
[598].
1 — исходная смесь; продолжитель-
ность аьтпвацип. мин: 2 — 10; 3 —
45; 1 — 50; 5 — 12о; 6 — 180.
По изменению фо рмы и положению пнков на дифракто-
граммах установлено преимущественное проникновение од-
ного из элементов в другой. Последовательность образования
фаз в смесях элементов, на равновесных диаграммах кото-
рых имеется несколько фаз, отличается от тон, которая на-
блюдается при термической активации. Это наблюдение,
а также ряд других данных показывают, что повышение тем-
пературы деформируемого образца невелико и существен-
ного влияния па фазообразование не оказывает [596].
Проведено сопоставление возможности образования со-
единений с рядом параметров, характеризующих смешивае-
мые элементы пли ожидаемые соединения. Наиболее полная
сепарация точек, соответствующих полученным и неполучен-
243
ным соединениям, наблюдается в координатах 0СМ — z,
У«е ©см= Люмн/Усм. T’cM=5^iTi, mi — атомная доля,
Ti — температура плавления элемента, z — число атомов в
элементарной ячейке соединения. Оба параметра характери-
зуют процесс зарождения: 0СМ— энергию активации па
один атом, z — число атомов в активируемом комплексе,
из которого формируется зародыш; поэтому авторы считают,
что стадия образования зародыша в этом процессе является
лимитирующей. В условиях рассматриваемых опытов сое-
динения образуются, если 0СМ > 0,25 и z < 20.
В целом полученные в работах [596, 597] результаты
объясняются предположением, что в деформируемых систе-
мах развиваются процессы двух типов. Процессы первого
типа вызываются механическим воздействием, и в резуль-
тате любая’ смесь приближается к состоянию аморфного
твердого раствора. Преимущественное растворение одного
из элементов и последовательность образования соединений
определяются механическими свойствами фаз. Чем мягче
фаза, тем больше степень ее деформации, тем быстрее идут
в пей диффузия и растворение другой фазы. Процессы вто-
рого типа являются релаксационными. Они термически ак-
тивируемы, и их развитие определяется необходимой энер-
гией активации. Многообразие наблюдаемых фазовых со-
стояний определяется степенью развития процессов релак-
сации. Поэтому если 0СМ < 0,25 и (или) z> 20, то в аморф-
ных образцах возможно сохранение пересыщенных твердых
растворов.
В работе Кубо [65] приводятся данные о возможности
механохимического синтеза фторапатита из CaFa и
Са3(РО4)2. Эта же реакция синтеза исследована в работе
[598]. Известно, что Са3(РО4)2, используемый для синтеза
фторапатита, является представителем ряда соединений пе-
ременного состава с общей формулой: Са10_х(НРО4)х-
•(РО4)6_Х(ОН)2_Х-Н2О. Эти соединения имеют апатитовую
структуру с частичным замещением ионов кальция водоро-
дом НРО4~-группы. Электронейтральность молекулы, как
показывает общая формула, компенсируется за счет ОН_-
групп, которых значительно меньше, чем в идеальной струк-
туре гидроксил апатита Calu(PO4)e(OH)2.
Данные рентгенофазового анализа смеси Са3(РО4)2 и
CaF2, подвергнутой механической обработке, приведены на
рис. 94. Они показывают следующее. В первые 7 мин меха-
нической активации па дифрактограммах можно видеть ос-
новные рентгеновские линии Са3(1’О4)2 — 0,279; 0,271;
Рис. 9~>. Количество Р2О5,
перешедшее в раствор ли-
монной кислоты, в зависи-
мости от времени актива-
ции [598].
I — емс ь CaF.-|- СаДРОРг,
J — фторапатит.
0,263 нм, а также линии
CaF2 — 0,317; 0,193 им,
которые уже через
10 мин почти исчезают,
и образец становится
С увеличением длительно-
практически реитгсноаморфным. С увеличением длительно-
сти механической активации смеси дифрактограмма стано-
вится более четкой, и через 60 мин четко проявляются ли-!
нии, характерные для фторапатита. Рентгеновские данные
подтверждаются данными по изменению растворимости сме-
си и механически активированного фторапатита в 2?6-ной
лимонной кислоте (рис. 95). Снижение растворимости синте-
тической смеси, подвергаемой механической обработке, го-
ворит об образовании фтораиатита. Наименьшее значение
растворимости наблюдается после 25 мин обработки, затем
оно изменяется мало. Кроме того, фторапатит, подвергае-
мый активации, становится рентгеноаморфным и увеличи-
вает свою растворимость. Кривые с двух сторон выходят на
одинаковое значение рентгеноаморфности п относительной
растворимости, которое сохраняется постоянным длитель-
ное время.
Таким образом, в результате механохимического синтеза
не происходит образования совершенной структуры, а воз-
никает некое стационарное состояние, при котором скорости
кристаллизации и разупорядочения сравниваются.
5.2.3. Обменные реакции
Широкий круг обменных реакций между ионными соля-
ми под влиянием механической обработки смесей в дезин-
теграторе исследован Уибо, Паэ и др. [392]. Ими было по-
казано, что все реакции кроме одной протекают в термоди-
намически разрешенном направлении, а для термодинами-
чески разрешенных реакций реагирование тем полнее (и
быстрее), чем меньше значение свободной энергии:
8 Е. Г. Лвиакумов
224
Реакция
дс298,
кДж/моль
LiCl + NH4F -> I.iF + NH4C1 —58,0
LiCl + NaF -> LiF Ц- NaCl —44,0
NaCl + NII4F -> NaF + NII4C1 —13,8
NaBr + NH4F -* N.iF + NH4Br —21,0
NaBr 4- BbCl -> NaCl + BbBr —13,8
NaBr -}- NH4Cl -► NaCl 4- NII4Br —6,7
NaBr 4- KC1 NaCl + KBr —6,7
NaBr 4- CsCl -> NaCl 4- CsBr —5,9
Nal 4- NII4F -> NaF 4- NH4I —23,0
Na I 4- BbCl -> NaCl 4- Bbl —17,6
Na I 4- KC1 NaCl 4- КI —1.3,4
Nal-j-KBr — NaBr 4-KI —9,5
Nal 4- NHjCl — NaCl 4- NH4I —9,2
Nal 4- BbBr -> NaBr 4- Bbl —7,5
Nal 4- NH4Br — NaBr 4- NH4I —2,4
KC14-N1IJ — KI4-NH4C1 —7,1
KC1 4- NH4Br — KBr 4- NH4C1 0
KBr 4- Bbl -> KI 4- BbBr —15,9
KBi 4- NH.I-> KI 4- NJl4Br —7,1
KBr 4- CsCl -> KC1 4- CsBr '—3,3
KI 4- CsCl -> KC1 4- CsI —9,2
KI4-CsBr->KBr4-CsI —5,9
KI + BbCl -> KCI 4-’ Bbl —1,3
BbBr 4- CsCl -> KCI 4- CsBr —0,42
BbBr 4- NI14F -> BbF 4- NII4Br 4-29,0
Bbl 4-CsBr-> BbBr 4-CsI —7,5
Bbl 4- CsCl — BbCl 4- Csl —7,5
CsCl 4- NH4I ->CsI4- NH4C1 —16,3
CsCl 4- NII4Br — CsBr 4- NH4C1 0
Авторы [592] с помощью скоростного рентгеновского ана-
лиза показали, что механохимические реакции осуществля-
ются не непосредственно в дезинтеграторе, а через некото-
рое время после диспергирования. В присутствии следов во-
ды скорость обменных реакций резко возрастает в отличие
от окислительно-восстановительных реакций, в которых она
замедляется. По нашему мнению, в результате обработки
смеси солей в дезинтеграторе могли возникнуть рентгено-
аморфные продукты, из которых в последующем идет их кри-
сталлизация, в присутствии же воды она ускоряется. Факт
существования ионных солей в рентгеноаморфном состоянии
доказан недавно Гусевым, Гагариной и др. в работе [599].
Выше уже приводились данные по образованию рентгено-
аморфных состояний в смесях металлов и солей.
При объяснении причин механохимических реакций не-
которые авторы отводили главную роль адсорбированной
па поверхности частиц влаге. Предполагалось, что пет прин-
ципиальной разницы между реакциями, происходящими в
226
концентрированных водных растворах, и реакциями при
растирании. Однако Болдыревым и Еремеевой 1600j было
установлено, что связи между скоростью реакции при рас-
тирании и растворимостью соли не существует:
Рс.ишмя
PacTuopuMOCTb, Скорость,
г/л C-1
PbCl + 2KI = РЫ2 + 2КС1
РЬВг2 + 2KI = РЫ, + 2КВг
Pb(NO3)2 + 2KI -= Pbl2 + 2KNO3
Pb(CH,COO)2+ 2KI = Pbb-h 2K(CH3COO)2
11,40
9,70
565
500
0.100
0.080
0.055
0,065
Как видно, труднорастворимые хлорид и бромид свинца
реагируют с KI быстрее, чем хорошо растворимые нитрат и
ацетат.
Одной из возможных причин протекания механохимиче-
ских обменных реакций между солями может быть контакт-
ное плавление веществ и последующая кристаллизация из
расплава конечных продуктов. В работах Савинцева с сотр.
[221—223] показано, что для контактного плавления необ-
ходимо при температуре, равной или выше эвтектической,
обеспечить контакт твердых тел, способных образовать эв-
тектику. Линейная скорость контактного плавления при
этих температурах совпадает со скоростью растворения ком-
понентов в расплаве при заданной температуре и составляет
величину порядка 10~3 см/с. Причем независимо от темпера-
туры перегрева в результате контактного плавления полу-
чаются расплавы одинакового эвтектического состава.
С точки зрения физико-химического анализа системы,
в которых идут реакции обмена, являются трехкомпонент-
ными и изображаются с помощью диаграммы Енека, пред-
ставляющей собой квадрат, каждой из вершин которого от-
вечает 10%-ное содержание одной из четырех солей. Про-
центные доли катионов, входящих в систему, откладывают-
ся по вертикальным сторонам квадрата, а процентные доли
анионов — по горизонтальным.
Составы, отвечающие точкам г и q (составы тройных эв-
тектик), являются самыми низкоплавкмми в системе. Чтобы
расплавить вещества на контакте, необходимо достигнуть
как минимум температуры плавления тропной эвтектики.
С целью установления возможности реализации меха-
низма контактного плавления в механохимических реакци-
ях памп был взят ряд обменных реакций типа АХ -f- BY —
— AY | ВХ, в которых температуры плавления эвтектик
изменялись в широком интервале — от 500 до 1100 К [601].
8*
227
Таблица 13
Обменные реакции в смесях ионных солен
Реакция АХ+ВУ=ВХ+АУ кДж/миль Температура тройной звтектнки, К Степень взанмо- дспствнн*
г Q
NaNO3 + КС1 = NaCl + К NO3 —1,3 517 568 100
NaNO3 -г2-ВаС12 = NaCl -Ну Ba(NO3)2 -9,8 681 589 100
1 1 2-К2СО3 + NaCl Na,CO3 + KC1 -32,3 823 873 100
1 1 NaF «j'SrClg = NaCl -F^SrFo -6,6 1018 983 100
PbSO4 -1- K»\VO4 = PbWO4 + K2SO4 -41,2 1059 1133 70
* С точностью рентгенофазового анализа ± 5—7%.
Предполагалось, что если будет наблюдаться замедление
реакции для смесей е высокой температурой эвтектики, то,
значит, температуры контактного плавления не достигнуты.
Результаты опытов для смесей после активации их в плане-
тарной мельнице (со = 12,6 с-1) приведены в табл. 13.
Из таблицы видно, что реакции имеют место даже для
смесп, температура плавления эвтектики которой превыша-
ет 1133 К. Но так как реакция для этой системы уже не идет
нацело, как для первых четырех, то, видимо, локальные
температуры лежат вблизи указанной температуры. Этот
вывод в какой-то мере согласуется с расчетом, приведенным
в гл. I для NaCl, где показано, что локальные температуры,
рассчитанные для этих же условий работы мельницы, со-
ставляют 1000—1300 К за время 10-8—10~9 с. Поскольку
условия динамические, зона плавления не развивается, по-
видимому, на большую глубину, а затрагивает только по-
верхностные области (5—10 ионных слоев), т. е. скорость
реакции будет определяться площадью контактирующих
участков разнородных частиц. Очевидно, за счет пластиче-
ской деформации материала эти точечные контакты могут
перейти в контакты по всей поверхности.
Возможно, что в этом случае, как и для металлов, вза-
имодействие протекает через стадию образования твердого
раствора с последующим распадом ею на конечные продукты.
Механохимический способ осуществления обменных ре-
акций имеет ряд преимуществ перед термическими метода-
228
ми: например, позволяет проводить синтезы между легко
диссоциирующими при нагревании солями, существенно
улучшать кинетические условия процесса. Благодаря это-
му метод можно считать перспективным в неорганическом
синтезе.
Весьма эффективными оказались мехапохимпческие ме-
тоды для обменных синтезов алюмпнпигидридов, боргпдри-
дов и др. Клазеном [602], а затем Захаркиным и Гавриленко
[603] с целью интенсификации обменной реакции
NaAlH4 + LiCl LiAlII4 + NaCl
были применены шаровые мельницы, выход LiAlH4 в кото-
рых за 4 ч работы составляет 30—90%.
Синтез цнрконийборпдгпдрида твердофазной обменной
реакцией между хлоридом циркония и литийборгпдридом
при интенсивном растирании смеси стальными шарами в
вакуумной шаровой мельнице при комнатной температуре
осуществлен Волковым и Мякишевым [604]. Максимальный
выход около 95% достигнут за 1,5 ч. Теми же авторами раз-
работаны методы синтеза уранборгпдрида обменной реакци-
ей между кристаллическими UC14 и LiBH4 с выходом 50%
[605] и тптанборгидрпда из тетрахлорпда титана и литий-
боргидрида и др. [606, 607]. Обменная механохпмическая
реакция между гексагидратом триполифосфата натрия ц
пентагидратом сульфата меди исследована недавно Прода-
ном и др. [608].
Замечено, что некоторые обменные реакции протекают
не при любой механической активации, а только прп ис-
пользовании мощных измельчптельных аппаратов. В каче-
стве примера можно привести данные работы Мпрсандова и
Курбонбекова [609], в которой показано, что обменная ре-
акция
LnCl3 + ЗМВН4 = Ln(BH4)3 + ЗМС1
(Ln — лантаноид, М — щелочной металл) реализуется толь-
ко в планетарной мельнице и практически не пдет в впбро-
мелышце.
Практически важная реакция получения боразпна пз
хлористого аммония и боргидрпда натрия реализована Вол-
ковым, Багрянцевым, Мякишевым и др. [610] при исполь-
зовании вибрационной мельницы с магнитным приводом
[217]. Реакция записывается следующим уравнением:
3NaBlI4 4- 3NII4C1 = B3N3II6(nap) -]- 3NaCI + 911,.
Реактор вибрационной мельницы и шары диаметром fi мм
изготовлены из нержавеющей стали, загрузка шаров 60%
от общего объема (1,5 л), амплитуда колебаний 20 мм, час-
тота 25—30 Гц. При этих параметрах процесса и загрузке
250 г ХаВП4, 500 т NII4C1 выход боразина составлял 40—.
50 г. По данным хроматографического анализа, примеси в
продукте отсутствуют.
Создание способа синтеза боразина, аппарата для его осу-
ществления, а также разработка системы хроматографиче-
ского контроля чистоты и методики очистки позволили ис-
пользовать боразин для получения высококачественных изо-
лирующих слоев из нитрида бора [217].
5.2.4. Кислотно-основные реакции
Известны попытки выделить среди твердофазных реак-
ций оксидов группу реакций, которые протекают по меха-
низму кислотно-основного взаимодействия [611]. К таким
взаимодействиям отно-
сятся реакции основ-
ных оксидов, таких как
BaO, CaO, SrO, MgO,
ZnO, РЬО, CdO, NiO,
FeO, CoO, СнО и др., с
каким-либо из кислот-
ных оксидов, таких как
WO3,,MoO3, SiO2, TiO2,
V2O6, Fe2O3 и др., по
реакциям типа
Мв]О -|- МецОп =
= MeiMenOn+1,
где п = 2, 3 и т. д.1
Эффективным средст-
вом ускорения реакций
в смесях, состоящих из
Рис. 96. И К-спсктры сме-
сей оксида кальция с квар-
цем.
/ — активированный кварц;
о — CaSiO,; 3 — акгииацин сме-
си 10 мин в присутствии вод-
ного раствора NaCl (5 нес. %);
4 — активация смеси 30 мин на
воздухе.
230
кислотного н основного оксидов, является механическая ак-
тивация в присутствии воды или растворов ионных солей.
1J качестве примера па рис. 96 приведены данные но вза-
имодействию оксидов кальция н кремния, взятых в соот-
ношении 1:1. Активацию проводили в планетарной мель-
нице ЭИ-2X150. Из результатов ПК-спектроскопии следует,
что активация сухих смесей в течение 30 мин к химическому
взаимодействию не приводит. В присутствии же раствора,
содержащего хлористый натрий, наблюдается образование
соединения CaSiO3. Аналогичный синтез реализован Лог-
виненко, Савпнкиной [612, 613] прп активации смесей окси-
да кальция и кварца в присутствии воды.
Рассматривая полученный результат в совокупности с
приведенными выше, можно сделать вывод, что среди раз-
личных факторов такой фактор, как влияние воды, проявляет
себя совершенно различным образом в разных типах реак-
ций — в одних случаях он тормозит процесс, в других ус-
коряет.
5.3. ЗАКОНОМЕРНОСТИ
МЕХАНОХИМНЧЕСКИХ РЕАКЦИЙ
В СМЕСЯХ ТВЕРДЫХ ВЕЩЕСТВ
5.3.1. Сравнение механически
и термически активируемых реакций
в смесях твердых веществ
Как правило, в твердофазных реакциях лимитирующая
стадия процесса изменяется по ходу реакции. Если на на-
чальных этапах твердофазная реакция лимитируется соб-
ственно химическим взаимодействием, происходящим в ме-
стах тесного контакта, то после образования сплошного слоя
продукта она переходит в диффузионный режим и лимити-
руется движением ионов или атомов через слой продукта.
В ходе диффузии может возникать микронеоднородность об-
разца вследствие различия типов дефектов в кристалличе-
ской решетке. Это, например, наблюдается прп синтезе
ВаТЮ3, когда к концу процесса частица продукта состоит
из двух слоев — один содержит кислородные, а другой —
катионные вакансии [614].
Кроме того, на завершающих стадиях наблюдается за-
медление скорости реакции, которое значительно больше,
чем определяемое диффузионной моделью. Многие авторы
231
Рис. 97. Кинетика термического
синтеза вольфрамата бария из
карбоната бария и оксида воль-
фрама (VI).
объясняют этот факт со-
вершенствованием продукта
реакции, которое приводит
к уменьшению потока ве-
щества по межкристаллит-
ному пространству, и переходом процесса во впутри-
диффузионную область. Однако данное явление наблю-
дается для самых различных систем, даже при доста-
точно низких температурах, когда процессы спекания про-
дуктов протекают еще недостаточно интенсивно. По-видимо-
му, основная причина замедления процессов заключается
в нарушении контактов между частицами. После взаимо-
действия близко расположенных частиц реакция практиче-
ски останавливается, так как оставшиеся непрореагпровав-
шие частицы должны преодолевать макрорасстояния, чтобы
прийти к непосредственному контакту. Подобные представ-
ления подтверждены микроскопическими наблюдениями. Бы-
ло показано, что в системах после практической остановки
реакций имеются отдельные иепрореагировавшие агрегаты
из частичек даже тех компонентов, которые были взяты в
недостатке по отношению к стехиометрическому составу.
Вероятно, такие агрегаты образуются в процессе подготов-
ки смесей благодаря способности мелкодисперсных частиц
к образованию агрегатов или в ходе спекания, когда идет
процесс укрупнения частиц за счет более мелких.
В качестве примера, иллюстрирующего изменения ско-
рости процесса по ходу реакции, на рис. 97 приведены дан-
ные по кинетике взаимодействия карбоната бария с триокси-
дом вольфрама при нагревании смесей при температурах
923, 1023 и 1083 К. На начальных этапах реакция прохо-
дит с максимальной скоростью, а затем наступает замедле-
ние, и для достижения 100%-ного превращения 'требуется
весьма длительное спекание. Для ускорения процесса при-
меняют размельчение не полностью прореагировавших сме-
сей, таблетирование и последующий отжиг.
При проведении реакций путем активации твердых сме-
сей в измельчптельных аппаратах не только измельчаются
частицы и возникают точки контакта между ними, но и бла-
годаря пластическому течению твердых тел точечные коп-
232
такты превращаются в контакты по некоторой поверхности.
Предельная контактная площадь определяется импульсом
давления и твердостью частиц, причем е уменьшением раз-
мера частиц она возрастает. Таким образом, обеспечиваются
хорошее контактирование частиц и активация поверхност-
ных атомов за счет локальных температур н давлении, а так-
же пластического течения материала. Можно говорить
о том, что в рамках представлений о лимитирующей стадии
процесса реализуется кинетический пли близкий к нему
режим твердофазной реакции. Вероятность перехода в дан-
ный режим возрастает с увеличением мощности измельчи-
тельного аппарата, поскольку пропорционально мощности
возрастают зона пластического течения твердых тел и зна-
чения локальных температур н давлений.
Кроме того, как указывалось выше, в этих условиях мо-
гут иметь место механически стимулированные фазовые пе-
реходы в реагирующих веществах и продуктах реакции. Это
принципиально новый момент для твердофазных реакций,
поскольку при нагревании реализуются только термически
стимулированные фазовые переходы. Появление новых фаз
в соответствии с правилом Хедвалла существенно сказывается
на кинетике твердофазных реакций.
Выяснение механизма механохпмических реакций воз-
можно путем сравнения их с термическими реакциями, ли-
митирующие стадии которых известны. Повтому нами было
проведено сравнение некоторых типов твердофазных реак-
ций, в частности карбонатов щелочноземельных металлов,
с оксидами элементов IV, V и VI групп Периодической сис-
темы [615]. Отметим, что Альбрехт и соавт. [616—619] ре-
ализовали механохимнческие синтезы большой группы
вольфраматов щелочных и щелочноземельных металлов. При
нагревании до высоких температур карбонаты щелочноземель-
ных металлов разлагаются без плавления до оксидов и угле-
кислого газа (—1200—1800 К),- а оксиды металлов имеют вы-
сокие температуры плавления (—1600 К) пли тоже диссо-
циируют с образованием низших оксидов без плавления,
т. е. реакции протекают по твердофазному механизму без
участия жидкой фазы.
Карбонаты щелочноземельных металлов реагируют с ок-
сидами с выделением СО2 и образованием металлатных со-
единений по общей брутто-схеме:
М^Оз -|- МПшО„ -> МДМнД + CO3f .
Считается, что на начальных участках (а ~ 0,1) термиче-
233-
ская кинетика взаимодействия таких реакций лимитируется
стадией химического взаимодействия и подчиняется урав-
нению «сжимающейся сферы». В дальнейшем лимитирую-
щей стадией процесса становится диффузионный перенос
ионов металла через слой продукта и кинетика подчиняется
диффузионным уравнениям (например, уравнению Лидера).
Механохимические синтезы проводили в аппарате пла-
нетарного типа, описанном выше (см. гл. 1). Скорость вра-
щения барабанов 12,4 с-1, мощность ~230 Вт, масса шаро-
вой загрузки 200 г, шары диаметром 2—3 мм изготовлены
из сплава ВК-8 (плотность 15,6 г/см3). Для синтезов исполь-
зовали реактивы «х. ч.», предварительно прокаленные на
воздухе при 700 К в течение 2 ч. Исходные вещества брали
в мольном отношении 1 : 1 так, что объем смеси был равен
1 см3.
Систему предварительно вакуумировали до 10~4 МПа
и кинетику реакции фиксировали по росту давления диок-
сида углерода образцовым вакуумметром с ценой деле-
ния 5-10~3.
Поскольку разложения карбонатов в данных условиях
практически не происходило (например, карбонат бария за
1,5 ч механической активации разлагается менее чем на 1%),
оио не учитывалось. Точность определения степени превра-
щения данным способом 0,3—0,5 отн.%. Продукты реакций
идентифицировали методом рентгенофазового анализа на
приборе ДРОН-05 (Снка-излучение, Ni-фильтр). Термиче-
ские синтезы проводили на дериватографе «МОМ» при ско-
рости прогрева 10 град/мин — фиксировалась скорость убы-
ли массы.
Кривые термического и механохимического взаимодей-
ствия карбоната бария с оксидами приведены на рис. 98.
Видно, что механохимические реакции для оксидов элемен-
тов VI группы (МоО3, WO3) протекают с большей скоростью,
чем для оксидов элементов V группы (Nb2O5, Та2О6). Окси-
ды же элементов IV группы Периодической Системы элемен-
тов (TiO2, ZnO2, SnO2) в выбранных условиях с карбоната-
ми бария практически не реагируют. Скорость реакций кар-
боната бария с оксидами, как в случае термической акти-
вации, так и в случае механической активации, изменяется
одинаковым образом, т. е. уменьшается в ряду МоО3, WO3?
Nb2O6, Та2О5. Однако следует отметить, что в случае реак-
ций с МоО3 и WO3 различие в кинетике при механической
активации значительно меньше, чем при термической. Бо-
лее высокая скорость реакции ВаСО3 с МоО, чем с VVO3B
в случае термической активации объясняется более высо-
234
a
Рис. 98. Термографические кривые (а) и кинетика механохимического
взаимодействия (б) оксидов элементов IV, и VI групп с карбонатом
бария [615].
ким значением коэффициента диффузии Мо6* в ВаМоО4
по сравнению с W®+ в BaWO4. При механической актива-
ции скорости этих реакции близки, так как в этом случае
на первый план выступает, по-видимому, не диффузионный
перенос, а перенос, обусловленный пластическими дефор-
мациями.
Выше указывалось, что имеется корреляция скорости
механохимического разложения карбонатов щелочноземель-
ных элементов со скоростью их термического разложения.
Так, при большой длительности активации (в вибромель-
нице 20 ч) степень разложения равна 28,9; 13,9; 10,8; 8,4%
соответственно для карбонатов Mg, Са, Sr, Ва. Температу-
ра, при которой давление углекислого газа достигает атмо-
сферного, составляет для MgCO3 923 К, СаСО3 1073, SrCO3
1484, ВаСО3 1520 К, а энергия активации термического раз-
ложения равна соответственно 150, 205, 222, 283 кДж/моль.
Термодинамические расчеты показывают, что взаимо-
действие триоксида вольфрама с карбонатом магния несколь-
ко выгоднее, чем с карбонатом бария (AG^e равно соответ-
ственно —41,4 и —35,8 кДж/моль).
Однако эксперимент показывает, что при механической
активации (со >10 с-1) скорость механохимического вза-
имодействия с карбонатом бария больше, чем с карбонатом
магния (рис. 99). В данных условиях активации на первый
план выступают механические свойства карбонатов, опре-
деляющие способность к образованию агрегированных час-
тиц, в частности твердость карбонатов. В ряду карбонатов
твердость по Моосу закономерно падает: MgCO3 — 4,5;
СаСО3 — 4,0; SrCO3 — 4,0; ВаСО3 — 3,5 [89J. Как видно
235
(VI) [622].
из рис. 99, в этом же направлении увеличивается и ско-
рость механохимического синтеза вольфраматов.
Отсюда следует, что полной аналогии между термиче-
ски и механически активируемыми реакциями не наблюда-
ется. Поскольку скорость реакции обусловлена механиче-
скими свойствами реагирующих веществ, это также подтвер-
ждает, что взаимодействие является твердофазным.
В случае чисто твердофазного механизма скорость ре-
акции пропорциональна числу точек контакта между реаги-
рующими частицами. Для идеальных систем, состоящих из
сферических частиц, число контактных точек между одной
центральной частицей В и соседними частицами А дается из
простых математических соотношений уравнением [620]
= <5-9)
где пв — общее число частиц А и В, окружающих частицу
В, х — отношение массы компонента А к массе компонента
р
В, а = -^-5, где гА и гв, рА и рв — радиусы и плотности
гвРл
компонентов А и В соответственно. По уравнению (5.9) ско-
рость реакции по твердофазному механизму должна возра-
стать с увеличением содержания одного из компонентов.
236
Рис. 100. Скорость мехапохими-
ческою взаимодействия карбона-
та бария с оксидом вольфрама
(VI) в зависимости от соотноше-
ния компонентов.
Частота upameiina, c—1: 1 — 7,0; г —
12.4.
Экспериментальные данные,
полученные при нагревании
смеси порошков, подтверж-
дают указанное соотношение
1620].
Нами была исследована скорость мехапохимической ре-
акции для смеси карбоната бария с оксидом вольфрама (VI)
при изменении соотношения компонентов в широком интер-
вале составов (от 25 до 95 мае.%). Причем исследования про-
водились прп разной мощности мельницы (10 и 230 Вт),
которая зависит от частоты вращения барабанов. При ма-
лых интенсивностях воздействия (ш =7 с-1) скорость воз-
растает так же, как и для термического процесса. Однако
при больших частотах вращения (я = 12,4 с-1), начиная
с некоторого состава (>75 мол.%), скорость реакции от со-
отношения реагентов не зависит (рис. 100). Данный эффект
обусловлен, ио нашему мнению, переходом карбоната ба-
рия в состояние пластического течения, скорость лимити-
руется не числом точечных контактов, а общей поверх-
ностью карбоната бария, по отношению к которой достигнуто
насыщение оксидом вольфрама (VI).
Таким образом, результаты анализа литературных дан-
ных и проведенных нами исследований по сравнению меха-
низмов реакций в условиях термической и механической
активации применительно к твердофазным механохпмпче-
ским реакциям позволяют сделать следующие выводы.
Окислительно-восстановительные реакции в условиях ме-
ханической активации протекают с участием короткоживу-
щих центров, образующихся на поверхностях разрушения
й при трении частиц и реагирующих практически безакти-
вационно с атомами другого компонента. Термическая ак-
тивация к образованию таких центров не приводит или да-
же способствует пх гибели. В таком случае эти два метода
активации принципиально различны.
В реакциях, протекающих без изменения степени окис-
ления, а принципе возможны механизмы, которые не нротн-
237
воречат термическому. Это может быть механизм контакт-
ного плавления на поверхностях трения, термически акти-
вированные диффузионные процессы и т. д. Однако и в этом
случае есть факты, не подтверждающие протекания реак-
ции по чисто термическому механизму. Для взаимодействия
металлов — это отсутствие последовательности в образова-
нии соединений при изменении состава смеси, определяемой
термическим механизмом [596]. Для реакций обмена и об-
менного разложения — зто отсутствие параллелизма в ско-
ростях термически и механически активируемых реакций.
Так, скорость взаимодействия карбонатов щелочноземель-
ных металлов при взаимодействии с оксидом вольфрама
(VI) по термическому механизму убывает от магния к барию,)
в случае механохимнческого взаимодействия наблюдается
обратная картина и т. д.
Все это подтверждает отличие твердофазных механохп-
мических реакций от чисто термических и делает необходи-
мым установление других факторов, обеспечивающих спе-
цифику механохпмических реакций.
В частности, выявлено, что одним из таких факторов яв-
ляются механические свойства реагирующих компонентов.
Вещества, обладающие ярко выраженными пластическими
свойствами, дают более значительные механохимические
эффекты (выше скорость реакций с их участием, скорость
разложения и т. д.). Причины такого поведения заложены
в физике процессов пластической деформации, которые в
настоящее время интенсивно исследуются.
Общая картина и последовательность процессов, разви-
вающихся при механической активации смесей твердых тел,
может быть сведена к следующим этапам: диспергирование
реагентов, механическое смешение, химическое взаимодей-
ствие в точках контакта частиц, увеличение точечных кон-
тактов по всей площади, разрушение слоя продукта и обра-
зование новых свежих контактов, химическое взаимодей-
ствие и т. д., до окончания взаимодействия. Может происхо-
дить смешение компонентов, близких по свойствам (метал-
лов и ионных солей), на атомном уровне по дислокационно-
диффузионным механизмам, обеспечиваемое подводом ме-
ханической энергии, и последующий распад твердого раство-
ра с образованием либо исходных соединений, либо заро-
дышей и ростом новой фазы. Процессы распада могут
протекать самопроизвольно или при нагревании.
С этой точки зрения выгоден процесс предварительной
механической активации с последующей термической акти-
вацией. В этом случае происходит образование конечного
продукта с более совершенной кристаллической структурой.
238
5.3.2. Кинетические особенности
твердофазных механохпмических реакции
5.З.2.1. О двух режимах твердофазных
механохимических реакций
В обычной твердофазной кинетике реакции описывают
уравнением
= (5.10)
и проводят исследование зависимости удельной скорости к
от температуры, давления и других интенсивных перемен-
ных [86]. Кинетическое же описание механохпмических
твердофазных реакций представляет собой трудную зада-
чу. Это связано с рядом методологических трудностей, обу-
словленных недостаточным пониманием механизмов иниции-
ровании механохпмических реакций и лимитирующих ста-
дий процесса. Одним из таких вопросов является вопрос
о соотношении времени импульсного воздействия и времени
релаксационных процессов в твердом теле [33, 621]. Важен
также вопрос о лимитирующей стадии химической реакции.
Так, например, показано, что в некоторых случаях хими-
ческая реакция лимитируется скоростью образования по-
верхности [34, 35]. Это положение доказано для реакций
с газами [277, 282].
Для выявления кинетических особенностей твердофаз-
ных механохимических реакций необходимо исследовать
влияние различных параметров процесса на скорость реак-
ции. Дли этого мы выбрали реакцию
ВаСО3 + WO3 BaWO4 + СО2,
которая удобна тем, что за ее кинетикой сравнительно лег-
ко можно следить по выделению диоксида углерода [622,
623]. Как показал рентгенофазовый анализ, единственным
продуктом в этой реакции является BaWO4 в мелкодисперс-
ном и существенно дефектном состоянии (наблюдается уши-
рение линий и снижение их интенсивности). Отметим, что
после отжига дефекты устраняются и получается хорошо
окристаллизованный продукт [615].
Как мы видели выше, тестом, позволяющим выявить
роль поверхности в механохимических реакциях твердых
тел с газами и жидкостями, являются результаты кинети-
ческих исследований в зависимости от диаметра- мелющих
шаров, которые мы провели и па этой реакции.
239-
Рис. 101. Изменение удельной по-
верхности смеси карбоиата бария
с оксидом вольфрама (VI) в ходе
реакции взаимодействия.
Диаметр шаров, мм: 1 — 2; 2 — 8,
При исследований этой
реакции в зависимости от диа-
метра шаров было показано,,
что, как и в случае реакций
твердое — газ, твердое—жид-
кость, удельная поверхность
смеси ВаС03 + W03 при обра-
ботке шарами малого диамет-
ра (2 мм) выше, чем при измельчении шарами диаметром
В мм (10,6 м2/г против 3,9 м2/г при времени обработки
6 мин), т. е. реализуется зависимость, согласно которой
с уменьшением диаметра шаров скорость диспергирования
возрастает (рис. 101). Однако скорость реакции с увеличе-
нием размеров шаров увеличивается (рис. 102), т. е. по срав-
нению с реакцией твердое — жидкость эта зависимость яв-
ляется антибатиой. Таким образом, реакции, протекающие
в смесях твердых веществ, принципиально отличаются от
реакций типа «твердое — газ», «твердое — жидкость» тем,,
что они в большей мере связаны с пластическим течением
материала, вызванным импульсным приложением давления
и температуры.
При исследовании зависимости скорости твердофазной
реакции синтеза вольфрамата бария от частоты вращения и
диаметра шаров (параметры, определяющие энергию соуда-
рения) нами установлено, что при определенных условиях
имеет место изменение режима протекания реакций. На
рис. 102 и 103 представлены кривые а — т (время работы
аппарата) для данной реакции в зависимости от ы и d. Вид-
но, что с уменьшением этих величин скорость реакции за-
медляется и, кроме того, при значениях со = 9 с-1 и d =
= 3 мм изменяется вид кинетических кривых.
Электронно-микроскопические исследования смесей, сде-
ланные нами по ходу механической активации, показали,
что в случае применения шаров с d — 8 мм происходит сна-
чала быстрое диспергирование частиц, которые затем соби-
раются в крупные агрегаты, причем частицы «замуровыва-
ются» внутрь более крупных частиц ВаС03, которые легче
поддаются пластическому течению, чем частицы WO3. Ре-
акция протекает внутри этих агрегатов, подвергающихся
240
Рис. 102. Кинетические кривые мехаиохпмпческого синтеза вольфрама-
та бария при использовании стальных шаров разного диаметра (ы =
= 12,6 с’1) [623].
Диаметр шаров, мм: I — 1,5: 2 — 3,2: J — 3: I — 5; 3 — 8.
Рис. 103. Кинетические кривые механохимического синтеза вольфрама-
та бария в зависимости от частоты вращения [623].
Частота вращения, с — |: 1 — I5.G; 2 — 12,6; 3 — 11,0; 4 — 10,1', 5 — 9,2; 6 —
9,0; 7 — 8.9; « — 8,5
9 Е Г. Аввакумов
в
Рис. 101. Элсктронио-мпкроскопические снимки смесей карбоната ба-
рия с оксидом вольфрама (VI). X 2(> 400.
а — исходили смесь; б — актпнацил 15 мин шарами 2 мм; « — акгикация 4 мин
шарами 8 мм
интенсивному механическому воздействию. В случае мелких
шаров (<7 = 1,5 мм) процесс агрегатообразования выражен
слабее и реакция идет в основном на контактирующих участ-
ках частиц. Это хорошо видно из рис. 104, на котором пред-
ставлены некоторые из полученных электронно-микроско-
пических снимков.
При больших энергиях шаров быстро происходит пере-
ход от хрупкого разрушения к пластическому течению и
усилению процессов рекристаллизации (увеличение «сред-
него» размера частиц).
При небольших значениях со и d энергии шаров недоста-
точно для активации процесса рекристаллизации и реак-
ция идет па контактирующих участках частиц. Кинетиче-
ские кривые имеют другой вид и сходны с кривыми при тер-
мической активации смесей, описываемыми параболическим
диффузионным законом или уравнением Лидера.
В ряде случаев кинетические кривые имеют перегибы, обу-
словленные переходом от одного режима реакции к другому
в ходе реакции. Связан такой переход, ио-внднмому, с тем.
9*
243
что в частицах твердого тела при достижении ими опреде-
ленных размеров происходит переход от хрупкого разру-
шения к их пластическому течению. Такой переход сопро-
вождается увеличением контактной поверхности, ускорени-
ем дефектообразованпя и соответственно ускорением реак-
ции, которая лимитируется концентрацией и подвижностью
дефектов в решетке твердых тел.
Это явление характерно и для других твердофазных ре-
акций. Так, например, оно наблюдалось прп взаимодей-
ствии карбоната калия с триоксидом вольфрама и прп син-
тезе сульфида цинка пз цинка и серы [53] и др.
5.3.2.2. Формально-кинетическая модель
для твердофазных-механохимических реакции
и ее соответствие эксперименту
Необходимым условием мехапохимпческой реакции в
смеси твердых веществ является наряду с термодинамиче-
скими условиями (температура, давление и т. д.) наличие
контакта между частицами компонентов, т. е. вероятность
образования таких контактов можно считать лимитирую-
щей стадией процесса. Так, в первом случае, когда реакция
протекает на точечных контактах между частицами, скорость
реакции определяется числбм и площадью этих контактов.
Во-втором точечные контакты переходят в контакты но всей
поверхности одного трудпоизмельчаемого вещества или же,
когда оба компонента перейдут в состояние пластического
течения, скорость реакции также будет определяться вели-
чиной поверхности контактирования их друг с другом.
Используя это обстоятельство, мы разработали формаль-
но-кинетическую модель, основанную на допущении, что
реакция между твердыми порошками осуществляется в мо-
мент удара между теми частицами, которые, находясь в зо-
не механического воздействия, контактируют между собой.
В соответствии с указанным допущением полагаем, что
общая скорость реакции в работающем пзмельчптелыюм
аппарате
w — kMxsK, (5.11)
где /гм — копстанта, характеризующая вероятность проте-
кания реакции при данном механическом воздействии па
единицу контактной поверхности (энергетическая кон-
станта, определяющая термодинамические условия реак-
ции); х — вероятность попадания контактов под удар;
244
Рис. 105. Воздействие меТютЦеГо Тела па
смесь веществ Л и В.
як — площадь контактирующих участ-
ков веществ А и В в смеси, содер-
жащей исходные вещества и про-
дукты их взаимодействия в момент
механического воздействия (рис. 105).
Рассмотрим, как изменяется хк в этом
процессе. Под влиянием диспергирова-
ния общая величина sK возрастает, по-
скольку возрастают поверхности реагирующих компонен-
тов. Известно, что зависимость удельной поверхности при
диспергировании какого-либо вещества от времени подчи-
няется закону 150]
* = ( 1 - е-'- ’1), (5.12)
где s,nax — максимально достигаемая прп заданных усло-
виях работы аппарата поверхность вещества, кг — кон-
станта диспергирования, зависящая от природы измельчае-
мого вещества. Предполагается, что исходная поверхность
sn мала п ею можно пренебречь.
Поскольку, как это следует из простых геометрических
соотношений, прп уменьшении размера частиц sK пропор-
циональна общей поверхности частиц, можно записать ана-
логичное уравнение
«« = .?к.,пах(1-<АгТ). (5.13)
На практике можно ограничиться начальным участком
этой зависимости, т. е. принято, что як зависит от времени
диспергирования линейным образом:
я„ - кг. (5.14)
По мере расходования исходных компонентов образу-
ется продукт, который оказывает тормозящее действие (эк-
ранирование) на реагенты. Для случая взаимодействия
двух компонентов будем считать, что скорость реакции про-
порциональна произведению объемов (количеств) реагиру-
ющих веществ (аналогично закону действующих масс).
Если степень взаимодействия веществ выразить через а,
то при стехиометрическом соотношении компонентов коли-
245
чество пепрорсагпровавгпого вещества
т, = mOi(l — а), (5.15)
где mOi — начальное количество;
при пестех иометр нческом
тл — /п()А(1 — а) n W[t /Пов(1 _ еа), (5.16)
где е — коэффициент, учитывающий отклонение от сте-
хиометрии и ранный к - сП (с — стехиометрический коэф-
фициент, I — мольное соотношение между Л и В в исходной
смеси). В результате можем записать
= ArMzsK (1 — а) (1 — еа). (5.17)
Интегрирование (5.17) с учетом (5.13) при условии (I -f-
+ е)2 >4 приводит к уравнению
где
/С = A’Mxs1(rnax. (5.19)
При стехиометрическом соотношении компонентов (т. е.
когда в = ти е = 1) интегрирование уравнения (5.17) при-
водит к уравнению
Когда удельная поверхность измельчаемых веществ име-
ет линейную зависимость от времени измельчения, из (5.14)
следует уравнение
—— = Кх2 (5.21)
1 — а ' '
илп при малых а
а = /<т2. (5.22)
Исходя из сделанных допущений, следует отметить, что’
уравнения будут применимы не только когда измельчаются
два сравнительно хрупких вещества, но и когда одно веще-
ство является трудноизмельчаемым, а другое переходит в
состояние пластическою течения. В этом случае поверхность
контакта равна поверхности трудпоизмельчаемого вещества.
Линейный закон роста поверхности трудноизмельчае-
мого вещества (поверхности контакта) может быть енравед-
246
ливым в большом интервале изменения а(т) как результат
совместного действия механического разрушения и хими-
ческого взаимодействия компонентов.
Выбор образования контактов в качестве лимитирующей
стадии приводит к тому, что слабо пли совсем не учитыва-
ются другие стороны процесса. Так, величина /г„ не может
отразить все термодинамические особенности процесса. Ви-
димо, существует какое-то критическое значение энергии,
ниже которого не обеспечивается протекание реакции.
Твердофазные реакции являются экзотермическими и
протекают с выделением тепла. В силу указанных выше
причин могут измениться условия теплоотвода. Если на ста-
диях мелкодисперсных частиц теплоотвод в окружающую
среду пе затруднен, то при возникновении укрупненных
частиц возникает тепловая изоляция их от внешней среды,
что может приводить к ускорению реакции и протеканию
ее в самоподдерживающемся режиме.
Пз этого следует, что предложенная модель имеет огра-
ниченную область применения для определенного интерва-
ла условии. I.
Полученные в соответствии с изложенной моделью урав-
нения сравнивались с экспериментальными кинетическими
данными, полученными нами при исследовании ряда твер-
дофазных механохимических реакции. В реакции твердофаз-
ного восстановления оксида олова ЦУ) кремнием 1584] оба
реагирующих компонента являются сравнительно малопла-
стичными, хорошо диспергирующими твердыми веществами.
Кинетические кривые и обработка их в координатах урав-
нения (5.18) приведены на рпс. 106, из которого видно, что
наблюдается удовлетворительное соответствие уравнений
экспериментальным данным.
Применимость модели к обменным реакциям проверя-
лась нами па реакции взаимодействия хлористого аммония
с патрпйборгпдридом 1624]. Механическая активация смеси
проводилась в вибрационной мельнице из нержавеющей
стали. Шары, изготовленные из этого же материала, имели
диаметр 8 мм. Объем рабочего пространства 60 см3, ампли-
туда 5 мм, частота 50 Гц. Вибромельница связана трубо-
проводом с вакуумной измерительной системой, что позво-
лило вести непрерывную регистрацию количества выделив-
шегося водорода. Образующийся в результате механохими-
ческоп реакции
NaBH4 + NH4C1 -* NaCl + NH4BH4
аммоипйборгидрпд оказался неустойчивым при комнатной
247
Рис. 106. Соответствие экспериментальных данных для реакции
SnO2+ Si Sn SiO2 кинетическому уравнению (5.18).
а — кинетика п.>аимодсйствия и зависимости от частоты вращения барабанов,
с-1: 1—10, 2 — 13,3, з — 16,6; б — обработка и координатах уравнения (5.18).
температуре продуктом. Методами ИКС и химического ана-
лиза было показано, что продуктами разложении NII4BH4
являются тетрагндрпдборатбисамипобороиий и водород:
Nil.ВИ.4 ВП., (NIL), ВП. + Н2,
а затем из него при разложении образуется боразпн B3N3II0.
Кинетика механохимпческой реакции изучалась следую-
щим образом: определялось количество водорода, выделив-
шегося в момент обработки, а затем при температуре 333 К
после остановки аппарата, проработавшего заданное время,
проводилось доразложение NH4BII4 до конца и определя-
лось суммарное количество водорода.
Поверхность измельчаемых по отдельности веществ из-
меряли газоабсорбционным методом [218]. Результаты при-
ведены на рис. 107. Исходная поверхность NaBH4 равна
11,2 м2/мкг и практически не зависит от времени измельче-
ния. Поверхность NH (C1 увеличивается со временем измель-
чения в соответствии с уравнением (5.13) со значениями
/,2 — 5-10-4 с-1 и А'щах = 7,14 м2/мкг. Здесь реализуется
случай, когда хлористый аммоний выступает как трудпо-
измельчаемое вещество и поверхность контактирующих
участков практически равна поверхности хлористого ам-
мония.
Экспериментальные данные по кинетике взаимодействия
и их обработка в координатах уравнения (5.20) приведены
па рис. 107. Как видно, экспериментальные данные также
248
Рис. 107. Соответствие экспериментальных данных для механохими-
ческоп реакция NaBH4+ NH4C1 -> NII4B1I4+ NaCl кинетическому
уравнению (5.20) [624].
и — удельная nom*u\ иоеть: 1 — NaBH4, 2 — XII.C.I; о — юпи-пничтыч i piiLi>:i n
<|Г>1>аГ><нт:а ее и коирдинатах yp.iaueiiii.'i (а.20).
b
t-~ (1 e *гг;
кг се
достаточно хорошо укладываются на прямую, идущую че-
рез начало координат. Константа К равна 1,3-10-3 г1 для
температуры опыта 333 К.
На рпс. 108 приведены кинетические данные для реак-
ции [615]
ВаС03 + WO3 = BaWO4 + СО2,
протекающей в режиме II, и пх обработка в координатах
Рис. 108. Соответствие экспериментальных данных для механохими-
ческой реакции ВаСО3-|- W03-* BaWO4+ СО, кинетическому yp.vue-
iiiiio (5.2В |615|.
п—i;n»R>TH*ii,<,Kiir кривые, с-1: 1 — 14,3, 2 — 11,7, — 14,2, -I — 4,0; 6 — об-
работка в координатах уравнения (5.21).
249
уравнения (5.21). Видно, что до степеней в&аимодействия
0,6—0,7 наблюдается достаточно хорошее соответствие урав-
нений с экспериментом. Выше этих степеней наблюдаются
отклонения в сторону увеличения скорости реакции по от-
ношению к скорости, определяемой принятой выше кинети-
ческой моделью. Одной из причин ускорения реакции мо-
жет быть повышение температуры внутри агрегированных 1
частиц за счет ухудшения условий отвода выделяющегося
прп реакцпп тепла.
5.4. МЕХАПОХИМИЧЕСКИП СИНТЕЗ
В МНОГОКОМПОНЕНТНЫХ СИСТЕМАХ
Принципиально новые возможности предоставляет метод
механической активации для проведения твердофазного син-
теза в многокомпонентных системах. Дело в том, что, по-
скольку тройные (и более) контакты между частицами мало-
вероятны, синтезы в этих системах протекают через ряд про-
межуточных реакций, в которых участвуют пары контакти-
рующих частиц (рис. 109, 1).
При механической активации происходит пластическое
течение твердых частиц и точечные контакты превращаются
в контакты по поверхности, возможно образование тройных
контактов, как это показано на рис. 113. Важной особенно-
стью является также то, что каждая частичка порошкообраз-
ной многокомпонентной смеси находится в контакте не с од-
ной и той же частичкой, как в случае твердофазного взаимо-
действия в порошках или таблетках, а периодически с раз-
ными частицами.
Все это в конечном итоге приводит к уменьшению числа
промежуточных стадий, ускорению синтеза и повышению
однородности получаемого продукта.
Имеются публикации об успешном использовании мето-
да для синтеза в многокомпонентных системах. Таким спо-
собом Чайкиной, Шапкиным и др. [625] проведен синтез
фтор- и хлорапатитов из стехиометрической смеси СаНРО4,
СаО и CaF2(CaCl2) [625], а Ткаченко, Летюк и Башкировым
[626] — синтез литий-марганцевого феррита из смеси порош-
ков Li2CO;i, МпСО;1, MgO, Fe2O;i п др.
Нами исследован мехапохимпческпй синтез в трехком-
попептпой системе:
ВаСО3 -]- 2La2(CO3)3 + 7WO3 = BaLa4(WO4)7 + 7СОа
25(1
Рис, 109. Схема, поясняющая образование контактов в трехкомно
neirnioii смеси.
1 — двойные контакты в трех компонентной смеси при термической активации;
2—4 — ovpfijonaiiiic тройных контактов при пластическом течении одного, двух,
трех шчцсстн в смеси; 5 — контактирование частицы вещества е твердым раство-
ром из двух других веществ, образующихся нри пластическом деформировании.
(в действительности система четырехкомпопептпая —
BaO/La.,O3/\VO3/CO2, однако мы рассматриваем только чис-
ло компонентов в твердой фазе [629]).
Термический синтез барнй-лаптапового вольфрамата из
карбоната бария и оксидов лантана и вольфрама проводят
в течение 100 ч при 1200—1250 К с последующим понп/ке-
ппем температуры до 1100 К и отжигом прп этой температу-
ре в течение 100 ч прп 4-кратном перетирании снека [628].
Исходные вещества (ВаСО3 марки «х. ч.», La.,(CO3)3 и
WO3 марки «ч.») брали в эквимолярных количествах соглас-
но уравнению реакции и предварительно перемешивали пе-
ретиранием в ступке. В ходе обработки смеси в вакуумной
термостатированной мельнице регистрировали давление вы-
деляющегося диоксида углерода. Рентгенофазовый анализ
(РФА) образцов проводили на приборе ДРОН-05 (Снка-
излучение, Ni-фильтр). Термический анализ (ДТА) образ-
цов выполнен на дериватографе «МОМ». Удельную поверх-
ность образцов определяли методом тепловой десорбции
аргоиа.
На рис. ПО представлены валовые кинетические кривые
газовыделения СО2 в процессе обработки реакционной сме-
си. Поскольку изменение давления СО2 в системе может
отражать как степень протекания синтеза BaLa4(WO4)7^
так и протекание реакций в двухкомпопентных системах
ВаСОз + \V03 и La.2(CO3)3 I W03 с образованием соответ-
ствующих вольфраматов (степень протекания механохими-
ческого разложения отдельных карбонатов мала п не пре-
вышает для ВаСО3 1%, а для La«(CO3)3 14%), полученные
после активации образцы (нумерация образцов дана в по-
рядке увеличения степени взаимодействия, определенной
но значению давления СО2 в момент остановки мельницы)
были исследованы методами РФА и ДТА. Термический ана-
251
Рис. 111. Дернватограммы ис-
ходного (7) п активированных
(2— 6) образцов. Нумерация об-
разцов соответствует рпс. 110.
лпз (рпс. 111) подтвердил
почти количественно по
ТГ согласие степени про-
текания реакции синтеза
с данными но газовыделе-
ппю (протекание реакции
нацело для образца 1 и
неполнота превращения
для образцов 2—6) и по-
казал радикальное разли-
чие кривых ДТА и ДТГ
исходной смеси (образец 7)
п активированных образ-
цов.
На рис. 111 видно, что
дернватограмма деактиви-
рованной смеси соответст-
вует эндотермическим ре-
акциям разложения кар-
бонатов [86, 627]:
670К »70К
Ьа2 (С03)., —► La„02 • СОЙ —> La,O3;
X of о и L л & о '
ВаС03 ВаО + СО2.
Экзоэффект, обнаруживающийся на кривой ДТА-1 выше
1170 К, отражает протекание реакции образования
BaLa4(WO4)7 и отвечает условиям его термического синтеза.
Для механически активированных образцов экзозффект сме-
щен в область более низких температур, а эндозффекты не
наблюдаются. Это дает основание предположить, что синтез
Рис. ПО. Кривые роста давления СО3 в процессе механической обра-
ботки образца 1 (механическая смесь порошков ВаСО3+ 2La2(CO3)34-
+ 7WO3) я условия получения активированных образцов.
2— обра.нц 1 актшшрошш 5 мин при частоте вращения 12,3 с-*1; 3— 9 мин
(12,3г 'О, 4 — 2110 (7,2); .5 — 16 (12,3); « — 90 (12,3); 7 — 30 (15,6); 9— 1 (12,3);
Jt) — <30 (8,9); S — изменение удедьпоО поверхности в процессе обработки исходной
емггн при 12,3 с”1; а — расчетное значение давлении СО2 в результате протека-
ния реакции синтеза BaLa4(VVO4)T в исходной смеси нацело; Ь, с, d, t — уровень
падения давления Р после остановки мелышин.
85Э
CM
Puc. 112. Дпфрактограммы продуктов реакции в смеси BaCOs +
-}- 2Еа2(СОз)3-{- 7WO3-»- BaWO4-2La2(WO4)s-|- СО2 в ходе механохи-
мической реакции.
а — частота вращения 7 с-1: 1 — 20 мин, 2 — 40, 3— 108, 4 — 180; б— частота
вращения 12,4 с~1. 1 — 2 мни, 2 — 4, 3 — 0, 1 — 12.
BaLa4(WO4)7 в случае активированных образцов протекает
сопряженно с разложением карбонатов в одну стадию в ин-
тервале температур 770—820 К, что более чем па 400 К ни-
же по сравнению с исходной смесью.
Данные РФЛ, полученные по ходу механической актива-
ции смесей при разных частотах вращения, приведены на
рис. 112. Видно, что чем выше подводимая механически
254
nncpritn, тем более окрнсталлпЗоваПпым получается конеч-
ный продукт BaLa(WO4)7.
Для получения кристаллических образцов и выяснения
влияния предварительной механической активации на темпе-
ратуру синтеза BaLa4(\VO4)7 был проведен отжиг активи-
рованных образцов при температурах 670 и 770 К в тече-
ние 4 ч.
Как видно из рис. 113, для исходной смеси реакция син-
теза при температурах отжига 673 и 773 К в течение 4 ч не
идет (незначительное отличие рентгенограммы объясняется
С70К
разложением Ьа2(СО3)3 —* Ьа.2О2«СО2). Если кристаллиза-
ция Ba2La4(WO4)7 в процессе отжига при 670 К образцов 7
и 6 представляется естественной, то резкое уменьшение ин-
тенсивности пиков WO3 для образца 2 можно объяснить толь-
ко начавшейся реакцией термического синтеза BaLa4(\VO4)7
при (570 К. Отжиг при 770 К в течение 4 ч для всех активи-
рованных образцов приводит к образованию практически
чистого BaLa4(WO4)7.
Отметим некоторые особенности протекания реакции син-
теза BaLa4(WO4)7. Во-первых, в процессе механической ак-
тивации после непродолжительного диспергирования про-
исходит «укрупнение» частиц, что видно по уменьшению
удельной поверхности образцов. Так, для исходной смеси
(образец 1) удельная поверхность s = 3,2 м3/г, для образца
9 - 10,9; 2 - 6,0; 3 - 2,3; 4 — 3.4; 5 — 2,0; 6 - 1.7; 7 —
3,4 м2/г (см. рис. НО). Причем скорость «укрупнения» тем
выше, чем интенсивнее подвод механической энергии. Ме-
ханизм «укрупнения» частиц заключается в образовании
агрегированных частиц, включающих в себя в виде вкрап-
лений или слоев все исходные и конечные фазы. Возникно-
вение молекулярно-плотных агрегатов в процессе интенсив-
ного пластического течения было показано еще Ходаковым
[50]. Выше были приведены данные по образованию их в
реакции синтеза BaWO4 из ВаСО3 и WO3 [623]. Однако роль
их в твердофазном синтезе неясна. По-впднмому, образова-
ние молекулярно-плотных агрегатов частиц является одним
из определяющих факторов в твердофазном механохпмиче*
ском синтезе. Поэтому механизм мехапохимического син-
теза в многокомпонентных системах может быть принци-
пиально тождествен таковому в двухкомпонентной системе,
а различие будет определяться только содержанием раз-
личных компонентов в агрегированной частице.
Естественно полагать, что агрегированные частицы, воз-
никающие при различных интенсивностях подвода мехапп-
255
673
Рис. 113. Рентгенограммы отжига исходной смеси (1) и активирован-
ных образцов при 073 и 773 К в течение 4 ч.
Нумерация соответствует рпс. ИО; Т — рентгенограмма термически синтезирован-
него BaLa4(WO«), [628]; цифры — значения межпло, костных рассгояиий.
ческой энергии, будут различаться по плотности, внутрен-
нему строению и т. д. Это, в свою очередь, может принести
к разному поведению активированных образцов в процессе
их термического отжига. Действительно (см. рис. ИЗ), бо-
лее крупные агрегаты частиц, отвечающие большей степе-
ни превращения (образцы 7 и б), дают в процессе отжига
рефлексы 1’ФЛ без дублетов, характерных для наиболее
25С
интенсивных рефлексов на рентгенограмме термически син-
тезированного BaLaJVVOj)?. Неразрешепность дублетов свя-
зана, ио-видимому, со значительным искажением решетки
механохнмпчески синтезированного BaLa4(WO4)7 по осн с
в моноклинной ячейке [G28], которое нс снимается прн ис-
пользованных температурах обжига. Напротив, в процес-
се термического синтеза из активированных образцов на-*
блюдается полное соответствие рефлексов табличным.
Таким образом, на примере трехкомпопентной системы
ВаСО3—La2(CO3)3—W03 показано, что синтез в ней можно
провести либо мехапохимпчески, либо путем термической об-
работки предварительно механически активированной сме-
си. В последнем случае температура синтеза BaLa4(WO4)7
снижается более чем на 400 К и продукт лучше окрнсталли-
зован, чем после механохимического синтеза. Ускорение '•
реакции и снижение температуры синтеза объясняются об-
разованием молекулярно-плотного контакта между части-
цами различных компонентов как результата их пластиче-
ского течения под влиянием активации.
Таким образом, открывается широкая перспектива ис-
пользования механической активации для реализации син-
тезов в многокомпонентных системах.
•------------ЗАКЛЮЧЕНИЕ--------------
Рассматривая результаты исследований, изложенные в
настоящей монографии, а также проблемы механохимии
неорганических соединений в целом, можно видеть, что ме-
ханохимия имеет свои закономерности и развивается как
самостоятельное направление физической химии.
В основе механохимических процессов, реализующихся
в специально разработанной для этих целей измельчитель-
ной аппаратуре повышенной мощности, лежат те же физи-
ческие явления, которые возникают при воздействии
на твердые тела высоких давлений со сдвигом или ударных 1-
нагрузок. Результатом их является образование структур-
ных и химических дефектов в объеме и на поверхности твер-
дых тел, что принципиально отличает эти процессы от про-
цессов диспергирования, в которых основную роль играет
образование поверхности.
Таким образом, механическая активация твердых тел
в измельчптельных аппаратах — это удобный в технологи-
ческом отношении прием, позволяющий реализовать им-
пульсный подвод механической энергии к обрабатываемым
веществам. Такой подвод энергии обеспечивает реализа-
цию условий, которые положительно отличают этот метод
от метода воздействия на твердые тела высоких статиче-
ских давлений или температур. Процесс дефектообразова-
ния, происходящий при этом, усиливается при переходе
от хрупкого разрушения к стадии пластического течения
твердых частиц материала.
Под влиянием механической обработки образуются де-
фекты в ряду пуль-мериые (точечные) -► одномерные (т-
нейные) —f двумерные (поверхностные). Энергетически бо-
лее выгодно образование последних, а среди поверхиост-
258
пых — дефектов упаковки и двойников. Поэтому прп ме-
ханической обработке сначала идет насыщение твердых тел
дефектами упаковки и двойниками, затем образование меж-
зерениых границ и поверхности. При увеличении длитель-
ности активации образуются точечные дефекты, а также де-
формированные и разорванные химические связи н др.
Прежде всего остановимся на выводах, которые были
сделаны после сравнения последствий механической и тер-
мической активации для некоторых неорганических сое-
динений п механохимических твердофазных реакций. От-
сутствие корреляции термических и механохимических про-
цессов в рядах однотипных соединений (а в некоторых слу-
чаях их полная антибатность) свидетельствует о несводи-
мости механохимических процессов к тепловым. Этот вы-
вод следует также из данных по механически стимулиро-
ванным фазовым переходам, когда они совершаются в на-
правлении, противоположном действию высоких темпера-
тур, и из данных по сравнению механически и термически
активируемых твердофазных реакций.
При механической активации включаются специфиче-
ские механизмы. Так, возникновение локальных разогре-
вов может быть не только результатом трепня частиц, по и,
установлено при исследовании инициирования взрывчатых
веществ ударом, результатом неоднородной пластической
деформации но объему твердых тел. В этом случае на пер-
вый план выступают механические свойства твердых тел.
И действительно, в ряде случаев имеется корреляция ме-
ханохимнческих эффектов с механическими свойствами.
Реализация некоторых полиморфных превращений также
может быть объяснена вызванными механической энергией
перемещениями: трансляционными смещениями решетки,
образованием дефектов упаковки, двойников и т. д.
В силу действия механических факторов происходят из-
менение расстояний между нонами кристаллической решет-
ки, смещение или переход их в другие положения, что при-
водит к возбуждению электронной подсистемы, изменению
прочности химических связей от возникновения «напря-
женных» или деформированных связей до полного их раз-
рыва. Содержащие такие дефекты вещества пли их смеси
находятся в метастабильном состоянии, из которого онп
самопроизвольно или под каким-либо воздействием (нагре-
вание, облучение и т. д.) могут перейти в стабильное сос-
тояние.
'Гак, па примере оксидов переходных металлов показа-
но, что механическая активация приводит к их диссоциа-
25V
ции с образованием пестех неметрических соединений, име-
ющих недостаток кислорода. Эти соединения в дальнейшем
упорядочиваются по механизму кристаллографического
сдвига. О реализации такого механизма свидетельствуют
данные, полученные методами ЭП1’, ДТА, РФА п др. для
оксидов титана, ниобия, вольфрама.
Напротив, в соединениях, уже содержащих в исходных
структурах упорядоченные дефекты (например, вакансии
ионов железа в EeS), под влиянием механических актива-
ций происходит их разунорядоченне с образованием ста-
тистически распределенных дефектов и их ассоциаций.
Существование электронных возбуждений при разру-
шении кристаллов дает возможность воздействовать на них
путем введения добавок, обладающих донорными или ак-
цепторными свойствами по отношению к электрону, и тем
самым повлиять на последующие механохимические эффек-
ты. В настоящей работе это продемонстрировано на приме-
ре механохимпческого разложения нитратов щелочных ме-
таллов в присутствии электронно-активных добавок окси-
дов переходных металлов.
Возникновение под влиянием механической активации
различных активных состояний, ускорение процессов мас-
сопереноса обеспечивают эффективное протекание гетеро-
генных механохимическнх реакций в момент механической
обработки. Однако закономерности, которым подчиняются
эти реакции, не одинаковы для всех типов реакций. Так,
из экспериментальных данных следует, что скорости реак-
ций в системах твердое — газ, твердое — жидкость опре-
деляются скоростью образования активных поверхностных
центров, которая пропорциональна скорости образования
поверхности в момент диспергирования.
В твердофазных реакциях наряду с образованием ак-
тивных центров большое значение имеют пластические
свойства веществ и скорость реакции определяется в основ-
ном пластической деформацией твердых частиц, которая
находится в прямой зависимости от энергии единичного
механического воздействия.
Для этих реакций характерно увеличение скорости с
увеличением диаметра мелющего чела в противоположность
реакциям твердое — газ, твердое — жидкость, в которых
фактор поверхности является превалирующим и скорость
с увеличением диаметра мелющего шара надает.
Поэтому результаты кинетических исследований в за-
висимости от диаметра мелющих тел (при постоянной об-
щей массе шаров) могут служить своего рода тестом, нозво-
260
ляющпм отличить нонерхностный механизм взаимодействия
от других.
13 результате проведенных нами исследований установ-
лены дна режима протекания реакций в смесях твердых ве-
ществ в зависимости от условий активации. При малых ин-
тенсивностях подвода механической энергии скорость ре-
акции зависит от числа и площади точечных контактов меж-
ду частицами: при больших интенсивностях реализуется
режим пластического течения для одного или обоих реаги-
рующих компонентов, контакты из точечных превращают-
ся в контакты по всей поверхности труднонзмельчаемого
вещества. Происходит формирование молекулярно-плот-
ных агрегатов, реакция в которых может развиваться как
самоподдерживающаяся за счет экзотермического эффекта.
Разработанная нами с учетом экспериментальных ре-
зультатов формально-кинетическая модель механохимиче-
ских реакций в смесях твердых веществ применена для йе-
которых твердофазных реакций и привела к удовлетвори-
тельному соответствию экспериментальных данных с рас-
четами.
Для анализа эффективности мехапохпмнческпх процес-
сов необходимы оценки выходов в зависимости от количест-
ва затраченной механической энергии. Однако такие рас-
четы не нашли пока широкого применения. Из предвари-
тельных выводов, сделанных на основе имеющихся немно-
гочисленных данных, следует, что наиболее выгодны реак-
ции, в которых механическое напряжение является лишь
инициирующим фактором, а далее они развиваются само-
произвольно, затем по эффективности следуют реакции в
смесях твердых веществ, полиморфные превращения, амор-
фпзация н т. д.
Поэтому следует считать наиболее обоснованными сле-
дующие возможные направления применения механиче-
ских методов активации в химических процессах:
1. Проведение различного рода гетерогенных реакций.
Осуществление реакции в момент механической активации
приводит к качественно новым эффектам, облегчающим про-
текание химического взаимодействия. Особенно это важно
для реакций в многокомпонентных системах, в кото-
рых тронные (и более) контакты между частицами в усло-
виях нагревания маловероятны.
2. Превращения твердых кристаллических тел в новые
полиморфные пли полнтпнпые модификации, обладающие
другими, чем исходные структуры, свойствами. Особенно
перспективно получение таких модификаций в тех случа-
261
ях, когда новая модификация обладает повышенной реак-
ционной способностью и получить ее другими способами
(например, термической активацией) трудно.
3. Получение разупорядочепных аморфных твердых тел,
содержащих структурные и стехиометрические дефекты,
с целью повышения их реакционной способности и приобре-
тения ими новых свойств. Практически это может позво-
лить решить многие важные задачи переработки минераль-
ного сырья методами гидрометаллургии, так как аморфные
вещества легче перевести в растворимые формы, чем крис-
таллические. Такие структуры перспективны в катализе,
неорганическом синтезе, материаловедении и т. д.
Безусловно, не следует считать, что возможности меха-
нических методов ограничиваются только этими направ-
лениями. Мы указали только на те из них, которые в той
или иной мере отражены в настоящей работе, не,претенду-
ющей на исчерпывающий охват всех вопросов механохи-
мии неорганических веществ. Многое еще предстоит сде-
лать, чтобы понять сущность происходящих при механи-
ческой активации явлений и целенаправленно использо-
вать их для стимулирования химических реакций.
---------------ЛИТЕРАТУРА-----------------
1. Менделеев Д. И. Основы химии. Т. 1.— М.: Госхпмиздат, 1947.—
662 с.
2. Ostwald W. Lehrbuch der allgemeinc Clieinie. Bd 2.— Leipzig,
1887, I Aullage.— 616 S.
3. Ostwald W. Die cheinische Literatiir und die Organisation der Wis-
senschaft.— Leipzig: Akadeniische Verlagsgesellschal't, 1919.—
85 S.
4. Carey Lea M. Transformations of mechanical into chemical energy.—
The London, Edinburgh and Dublin Philosophical Magazine and
Journal of Science, 1894, v. 37, N 228, p. 470—475.
5. Флавпцкпй Ф. M. О новом методе аналитических испытаний меж-
ду твердыми веществами.— Жури. Русского физ.-хнм. о-ва,
1902, т. 34, с. 8—11.
6. Parker L. II. Reactions by trituration.— J. Chem. Soc., 1914, v. 105,
p. 1504—1516.
7. Parker L. II. Reactions between solid substances.— J. Chem. Soc.,
1918, v. 113, p. 396—409.
8. Воскресенский П. И. Аналитические реакции между твердыми ве-
ществами н полевой химический анализ.— М.: Госгеолтехпздат,
1963,— 191 с.
9. Исаков М. П. Качественный химический анализ руд и минералов
методом растирания порошков.— М.: Госгеолтехпздат, 1955,—
183 с.
10. Wanetig Р. Zur Frage der Zahflussigkeitsanderung von Viskoselosun-
gen.— Kolloid Z., 1927, Bd 41, S. 152—158.
11. Staudinger H., Dreher E. Mitteilung uber hochpolymere Verbindun-
gen.— Ber. Dtsch. Chem. Ges., 1936, Bd A69, S. 1091—1099.
12. Берлин А. А. Механохимические превращения и синтез полиме-
ров.— Успехи химии, 1958, т. 27, с. 94—112.
J13. Барамбопм И. К. Мехапохимии высокомолекулярных соедине-
ний.— М.: Химия, 1971.— 363 с.
./14. Спмпопеску Л. К., Оиреа К. Мехапохимии высокомолекулярных
* соединений.— М.: Мир, 1970.— 357 с.
\1 15. Бриджмен И. В. Новейшие работы в области высоких давлений.—
. М.: 11.4, 1948,— 300 с.
J 16. Бриджмен П. В. Исследование больших пластических деформаций
п разрыва.— М.: ПЛ, 1955,— 444 с.
263
17. Гопикберг М. Г. Химическое равновесие и скорость реакции при
высоких даплепиях. - М.: Изд-no ЛИ СССР, 1960,— 340 с.
18. Капустин В. М., Жаров Л. Л., Еппколопли II. С. Полимеризация
мономеров в твердой фазе в условиях высоких давлений и напря-
жений сдвига.— Докл. АП СССР, 1968, т. 179, № 3, с. 627—1*32.
19. Taininaiin G. Der Einfluss der Kallbearbeitung auf die cliemischcn
Eigenschallen, insbesondere von Melallen.— Z. Electrocheui.,
1929, Bd 35, S. 21—29.
20. Clark J., Bowan It. J. Studies on lead oxides. Polymorphic trans-
formations by grinding, distortion and catalytic activity in PbO.—
J. Amer. Cliein. Soc., 1941, v. 63, p. 1302—1305.
21. Ребиндер П. Л. Влияние активных смазочных сред па деформиро-
вание сопряженных поверхностей трепня.— В кп.: О природе тре-
ния твердых тел. Минск: Наука и техника, 1971, с. 8—20.
22. Соловьева А. Б., Жорин В. Л., Епнколопян Н. С. Химические пре-
вращения малеиновой и фумаровой кислот под воздействием высо-
ких давлений, сочетаемых с деформацией сдвига.— Докл. АН
СССР, 1978, т. 240, с. 125—127.
23. Совместная полимеризация стирола и метилметакрилата при вы-
соких давлениях в сочетании с деформацией сдвпга/Жорин В. А.,
Жаров А. А., Кисспп 10. В. и др.— Докл. АН СССР, 1974, т. 219,
№ 3, с. 647—652.
24. Образование комплексов железа с графитом при высоких давлени-
ях и сдвиговых деформацпях/Жорин В. А., Нефедьев А. Е., Лин-
скип В. А. и др,— Докл. АН СССР, 1981, т. 256, №3, с. 598—600.
25. Дремли А. Н., Бреусоп О. II. Процессы, протекающие в твердых те-
лах под действием сильных ударных поли.—'Успехи химии, 1968,
т. 37, выи. 5, с. 898—916.
26. Бацанов С. С. Синтезы под действием ударного сжатия.— В кп.:
Препаративные методы в химии твердого тела. М.: Мир, 1976,
с. 155—170.
27. Thiessen Р. A., Meyer К., Ileinicke G. Griindlugeii der Tribochemie.—
Berlin: Akad.-Veri., 1966, N 1.— 194 S.
28. Ileinicke G. Tribochemistry.— Berlin: Aknd.-Verl., 19841—
495 S.
29. Болдырев В. В. Экспериментальные методы в мехаиохимпи твер-
дых неорганических веществ.— Новосибирск: Наука, 1983.—
65 с.
30. Болдырев В. В., Аввакумов Е. Г. Мехапохпмпя твердых неоргани-
ческих веществ.— Успехи химии, 1971, т. 40, с. 1835-1-1856.
31. Болдырев В. В. Механохпмия неорганических веществ.— Man.
СО АН СССР, 1978, № 14. Сер. хим. паук, выи. 6, с. 3—11.
32. Болдырев В. В. О некоторых проблемах мехаиохимпи неорганиче-
ских веществ.— Пзв. СО АП СССР, 1982, № 7. Сер. хим. наук,
выи. 3, с. 3—8.
33. Ляхов Н. 3., Болдырев В. В. Механохпмия неорганических ве-
ществ. Анализ факторов, пптенепфицпрующих химический про-
цесс.— Пзв. СО АН СССР, 1983, № 12. Сер. хим. паук, выи. 5,
с. 3—8.
34. Бутягпи П. IO. Кинетика и природа механохимических реакции.—
Успехи химии, 1971, т. 40, с. 1935—1959.
35. Бутлгип П. IO. Разупоридочепиые структуры и мехапохпмиче-
ские реакции в твердых телах.— Успехи химии, 1984, т. 53, пын.
И, с. 1769 — 1789.
36. Дерягин Б. В., Топоров IO. II. Современное <«стояние исследова-
ний механизм песни.— В кн.: Доклады VII Всесоюзного енмно-
264
зптма no механизм псе пн и мехаиохимпи твердых тел. Ташкент,
1981, с. 3—7.
37. Дерягин 1». В., Кротона II. Л., Смнлга В. II. Адгезия твердых
тел.— М.: Паука, 1973.— 279 с.
38. Кротова Н. А., Карасев В. В., Дерягин Б. В. Исследование элект-
ронной эмиссии при отрыве пленки высокополпмера от стекла в
вакууме.— Докл. АН СССР, 1953, т. 83, с. 777—780.
39. Гутман Э. М. Мехапохпмпя металлов и защита от коррозии. 2-е
над., исир. п доп.— М.: Металлургия, 1981.— 299 с.
40. Власова М. В., Каказеп II. Г. Электронный парамагнитный резо-
нанс в механически разрушенных твердых телах.— Киев: Пау-
кова думка, 1979.— 210 е.
41. Зелпкмап A. II., Ракова И. II., Арампсова Ф. А. Интенсификация
автоклавно-содового способа разложения шеелитовых концентра-
тов механическим активированном.— Цветные металлы, 1978,
№ 7, с. 57—59.
42. Вольдман Г. М., Зелпкмап А. Н., Ермилов А. Г. Оценка степени
воздействия при механическом активировании материалов.—
Изи. вузов. Цветная металлургия, 1979, Л" 4, с. 24—28.
43. Лаптева Е. С., Юсупов Т. С., Бергер А. С. Физико-химические из-
менения слоистых силикатов в процессе механической актива-
ции.— Новосибирск: Наука, 1981.— 87 с.
44. Молоцкий М. И. Электронные .возбуждения прп разрушении кри-
сталлов.— Пзв. СО АН СССР, 1983, А» 12. Сер. хим. наук, выи. 5,
с. 30—40.
45. Молодкин М. И. Экснтопиые п дислокационные процессы в меха-
похпмической диссоциации ионных кристаллов,— Кинетика и
катализ, 1981, т. 22, № 5, с. 1153 — 1101.
46. Молчанов В. И., Юсупов Т. С. Физические и химические свойства
тоикодпепергяроваппых минералов.— М.: Недра. 1981.— 157 с.
47. Навлю.хни Ю. Т., Медиков Я. Я., Болдырев В. В. Изменение ка-
тионного распределения в феррнтах-шпинелях в результате их
механической активации.— Докл. АП СССР, 1982, т. 206,
с. 1420—1423.
48. Павлюхпн IO. Т., Медиков Я. Я., Болдырев В. В. Исследование
магнитных свойств аморфных магнетиков ферритного состава с по-
мощью эффекта Мессбауэра.— Фцп. твердого тела, 1983, т. 5,
выи. 3, с. 630—638.
49. Павлюхип Ю. Т., Медиков Я. Я., Болдырев В. В. Механизм н ста-
дийность механической активации некоторых ферритов шпине-
лей.— Изв. СО АН СССР, 1983, ЛЬ 12. Сер. хим. наук, выи. 5,
с. 46—53.
50. Ходаков Г. С. Физика измельчения.— М.: Наука, 1972.— 307 с.
51. Bernhardt С., Heegn Н. Zur nieclianibchen Aktivicrung in Feinzer-
kleincrungsmaschinen.— In: Zerkleinein (4 Europaischen Sympo-
sium), Declienia Monogr. Weinheim: Chemie, 1976, S. 213—225.
52. Бернхард !{.. Xeern X. Связь между акп1вписгыо и расходом энер-
гии при механическом активировании твердых материалов.—
В кп.: Доклады VII Всесоюзного симпозиума но мехаиоэмпсспи
п мехаиохимпи твердых тел. Ч. 3. Ташкент, 1981, с. 145—153.
53. Tschakarov Chr. G., Gospodinov G. G., Bontschev Z. i'ber den Mecha-
uisiiius der mechaiiochemischeu Synlhcse auorganiseher Verbin-
diingen.— J. Solid State Chein., 1982, v. 41, p. 244—252.
54. Chakurov Kh., Gospodinov G. Mechanism of niechaiiocheinical syn-
thesis of some tin (IV) chalcogenides and halogenides studied by Mos-
sbauer spectroscopy.— Bulg. J. Phys., 1982, v. 9, N 3, p. 259—263.
55. Chakurov Kh., Gospodinov G. Studies он the iiiechanism of mechaiio-
cheniical synthesis of tin (II) chalcogenides in air medium by the
Mossbauer effect.— Bulg. J. Phys., 1982, v. 9, N 5, p. 483—487.
56. Juhasz Z. A mechanical Kemia hekyzele es ered inenyci a szakiro-
dalorn tukreben.— Tudomanyos kozlemenyek, Budapest!, 198(1,
N 33, p. 180—210.
57. Juhasz Z., Opoczky L. Mechanical activation of silicates by fine
grinding.— Budapest: Akademiai Kiado, 1982.— 213 p.
58. Tkaeova K. Zdrobnovanic a aktivacia v tiprave a spracovani neras-
tov.— Bratislava: VEDA, 1984.— 103 s.
59. Hajek B., Brozek V. Tribochemike reakce. IV Prispevek к probleinii
oxydace praskovych kovu pri trcni.— Pokroky praskove metalurgie,
I960, N 4, s. 3-19.
60. Buzek J. Increase in internal energy and mechanism of quartz grin-
ding.— Silikaty'c, 1974, N 4, p. 295—312.
61. Pawelezyk Л. Wplyw operacji mechaniczhych na rcaklywnosc i
przemiany chemicznc substancji nieorganicznych.— Wiadomosci
Chemiczne, 1981, t. 35, s. 161—185.
62. Ocepek D. Einige Entwickhingen der mechanischen Aktivierung.—
Banicke listy (Mimoriadnecislo), Bratislava: VEDA, 1980, s. 154—
159.
63. Gihnan P. S., Benjamin J. S. Mechanical alloying.— Ann. Bev.
Materials Sei., 1983, v. 13, p. 279—300.
64. Lin J. J., Nadiv S. Review of the phase transformation and synthe-
sis of inorganic solids obtained by mechanical treatment (mechano-
cheinical reactions).— Mater. Sci. and Eng., 1979, v. 39, N 2,
p. 193—203.
65. Kubo T. Mechanochemistry of inorganic substances.— .1. Clieni.
Soc. of Jap., 1968, v. 71, p. 1301 — 1309.
66. Питерс К. Механохимпческве реакции.— В кп.: Труды Европей-
ского совещания по измельчению. М.: Строппздат, 1966, с. 80—
97.
67. Fox Р. G. Review and mechanically initiated chemical reactions
in solids.— J. Materials Sci., 1975, v. 10, p. 340—360.
68. Юшкин II. П. Механические свойства минералов.— JI.: Наука,
1971,— 283 с.
69. Шаскольская М. II. Кристаллография.— М.: Высшая школа,
1976,— 308 с
70. Taylor G. I. 'lhe mechanism of plastic deformation of crystals.—
Proc. Hoy. Soc., 1934, v. Л145, p. 362—404.
71. Френкель Я. И., Конторова Т. А. К теории пластической деформа-
ции п двойникования.— Журн. эксиер. п техн, физики, 1938,
т. 8, с. 89—95.
72. Коттрелл Л. X. Дислокации п пластическое течение,— М.: ИЛ,
1958.— 606 с
73. Екобори Т. d>ii3iiKa и механика разрушении и прочности твердых
тел.— М.: Металлургия, 1971.— 263.
74. Griffith А. Л. The phenomena of rupture and flow in solids.— Phil.
Trans. Boy. Soc., 1920, v. A221, p. 163—197.
75. Orowan E. Fracture and strength of solids.— Hept. Progr; Phys.,
1949, v. 12, p. 185—230.
76. Ормонт Б. Ф. Введение в физическую химию и кристаллохимию
полупроводников.— М.: Высшая школа, 1968.— 485 с.
77. Френкель Я. И. Электрическая теория твердых тол,— М.: Наука,
1924.— 299 с.
266
78. Ормопт Б. Ф. О связи между химической и механической проч-
ностью очень твердых тел.— Докл. ЛИ СССР, 19(>6, т. 106, .V» 4,
с. 687—690.
79. Русанов А. И. О связи между теплотой испарения и поверхностной
энергией.— Докл. АН СССР, 1981, т. 261, № 3, с. 700—703.
80. Ощерив I». 11. О поверхностном натяжении кристаллов неметал-
лов.— Жури. физ. химии, 1970, т. 44, с. 547.
81. Щукин Е. Д., Перцов А. В., Амелина Е.Л. Коллоидная химия.—
М.: 11зд-но Моск, ун-та, 1982.— 352 с.
82. Журков С. II. Проблема прочности твердых тел.— Вести. АН
СССР, 1957, .№ И, с. 78—82.
83. Рсгель В. В., Слуцкер А. II., Томашевский Э. Е. Кинетическая
природа прочности твердых тел.— М.: Наука, 1974.— 559 с. ,
84. Френкель Я. II. Введение в теорию металлов.— М.: Физматгнз,”
1958.— 424 с.
85. Федоров В. В. Термодинамические аспекты прочности и разруше-
ния твердых тел.— Ташкент: Фаи, 1979.— 156 с.
86. Браун М., Доллимор Д., Галвей А. Реакции твердых тел.'Пер. С
англ.- - М.: Мир, 1983.— 360 с.
87. Минералогические таблицы. Справочник'Под ред. Е. II. Семено-
ва.— М.: Недра, 1981,— 399 с.
88. Nickel Е. П., Webster А. II., Ripley L. С. Bond strengths in disul-
phides of iron, cobalt and nickel.— Can. Mineralogist, 1971, v. 10;
p. 5, p. 773—777.
89. Справочник физических констант горных пород,'Под ред. С. Клар-
ка.— М.: Мир, 1969.— 544 с.
90. Гпйо Р. Проблема измельчения материалов п ее развитие.— М.:
Строппздат, 1964.— 111 с.
91. Verdes S., Nemeth J., Kiraly L. Effect of grinding parameters on
I lie kinetics of grinding.— Banieke listy (Mimoriadne cislo), Brati-
slava: VEDA, 1984, s. 88-95.
92. Itumpf II. Wirscliaftliehkeit und. Okonomisclie Bedeutung des
Zerkleinern.— In: Zerkleinern (4 Enropaischen Symposium), Deche-
ma Monogr. Weinheim: Chemie, 1976, Bd 79, S. 19—41.
93. Krupa V., Sekyla F., Merva M. Klassifikacia melitelnosti pomocou
encrgelikotraiisforinacnycli merani.— Banieke listy (Mimoriadne
cislo), Bratislava: VEDA, 1980, s. 208—213.
94. Bernhardt C., Ileegn II., Ilgen S. Zur Mahlung und Aktivierung in
einer Miihle mit Kalorimctcr.— Banieke listy (Mimoriadne cislo),
Bratislava: VEDA, 1980, s. 214—220.
95. Колосов А. С. Некоторые вопросы моделирования и оценки энер-
гетической эффективности процессов измельчения твердых тел.—
Изв. СО АН СССР, 1985, № 2. Сер. хим. паук, вып. 1, с. 26—39.
96. Stairmand С. The energy efficiency of milling processes. A review
of some fundamental investigations and their application to mill
design.— In: Zerkleinern (4 Enropaischen Symposium), Dechema
Monogr. Weinheim: Chemie, 1976, Bd 79, S. 1 —17.
97. Il uni pl II. Die Einzelkornzeikleinernng als Grundlage einer Zer-
kleinerung.— Wissenschaft. Chemie-Ing. Techn., 1965, Jhrg. 37,
N 3, S. 187—202.
98. Hess W., Schonert K. Die Zerkleinerung von Kalksteinkugel bei
koinbinierler Druck und Schubbeanspruchung.— In: Zerkleinern
(4 Enropaischen Symposium), Dechema Monogr. Weinheim: Chemie,
1976. Bd 79, S. 151 — 165.
99. Schonert K. Energetischo Aspekle des Zerkleinerns sproder Stoffe.—
Zeinenl-Kalk-Gips., 1972, Jhrg. 32, N 1, S. 1—9.
267
1/)0 . Hoffman N., Pingel P., Sehiinerf K. Dio Drnclistiick-CroPon vertei-
hing bei der Zerkleiiicriing von bhiaren und Irriiiimi Miscliinigmi.
Cheniie-liig. Techn., 197G, J Ing. 48, N 4, S. 329—331.
101. Scbonert K. Zwci Aspekte der Partikelzerstorung.— Banicke listy
(Mimoriadnc qislo), Bratislava: VEDA, 1980, s. 48—53.
102. Hess \V. EinfluB der Schubbeansprucliuiig und des Verforiinings-
verhalttens bei der Druckzerkleinernng von Kugeln und kleine Par-
tikeln. Dissertation.— Karlsruhe, 1980.
103. Sehonert J., Sleier K. Greuze der Zerkleineriing bei kleinen Korn-
grofien.—Cliemie-Ing. Teclin., 1979, .Ihrg. 43, N 13, S, 773—777.
104. Kendall K. The impossibility of coinuiiHiiting small particles by
compression.— Nature, 1978, v. 272, p. 710—711,
105. Ребиндер П. А. Физико-химические исследования процессов де-
формации твердых тел.— В кп.: Юбилейный сборник АН СССР к
ХХХ-лспио Великой Октябрьской социалистической револю-
ции. Т. 1. М.: Изд-во АП СССР, 1947, с. 333.
106. Ребппдер П. А., Шрейнер Л. А., Жигач К. Ф. Показатели твердо-
сти в бурении.— М.: Изд-во АН СССР, 1944.— 276 с.
107. Блпппчев В. 11., Бобков С. II., Клочков II. В. Распределение анер-
гии, подводимой к телу в процессе разрушения.— В кн.: Докла-
ды VII Всесоюзного симпозиума по мехапозмисспи и мехапохимии
твердых тел. Ч. II. Ташкент, 1981, с. 152—154.
108. Schrader R., Hoffman В. liber die mechanische Aklivierung von
Calciumcarbonat.— Z. anorg. allg. Chem., 1969, Bd 369, S. 41—42.
109. Ван Бюрен. Дефекты в кристаллах.— М.: ИЛ, 1962.— 584 с.
110. Основы фнзпко-хпмви вещества в метастабильном ультрадпеперс-
ном состоянии и перспектива их исиользоваппя/З'апанаев И. В.,
Федоров В. Б., Морохов И. Д. и др.— Изв. АН СССР. Неоргап.
материалы, 1984, т. 20, № 6, с. 1026—1033.
111. Вишняков Я. Д., Файпштеии Г. С. Превращения в металлах с раз-
личной анергией дефектов упаковки.-— М.: Металлургия, 1981.—
135 с.
112. Ileegn II. Die Veriindernng der Festkorpereigenschaflen bei der
Feinzerkleinerung und mechaiiischen Aklivierung.— Banicke listy
(Mimoriadne cislo), Bratislava: VEDA, 1976, s. 58—68.
ИЗ. Фридель Ж. Дислокации.— M.: Мир, 1967.— 644 с.
114. Косевич А. М. Как течет кристалл.— Усп. физ. паук, 1974, т. 117,
вып. 3, с. 509—525.
115. Ходаков Г. С., Ребпндер П. А. О механизме измельчечия кварца
в поверхностно-активных средах.— Коллоидный журз., 1961,
т. 23, № 4, с. 482-488.
116. О кинетике измельчения некоторых тугоплавких металлив/Ая-
густпннпк А. И., Випдергауз В. 111., Гропяпов В. М. и др.— По-
рошковая металлургия, 1963, А’г 2, с. 3—7.
117. Иванько А. А. Твердость. Справочник.— Киев: Изд-во АН УССР,
1971,— 150 с.
118. Ормопт Б. Ф. Структуры неорганических веществ,.— М.: Гос.
изд-во техп.-теор. лит., 1950.— 968 с.
119. Исследование методом ИГР ферритов никеля, цинка и окиси желе-
за после механической активацпп/Иаилюхип Ю. Т., Меди-
ков Я. Я., А ина кумов Е. Г. и др.— Изи. СО АП СССР, 1979, .№ 9.
Сер. хим. паук, вып. 4, с. 14--2O.
120. Исследование дефектообразования при механической активации и
окисных системах методом ЯГР/Паилюхип 10. Т., Медиков Я. Я.,
Аввакумов Е. Г. и др.— Изв. СО АН СССР, 1981, № 9. Сер. хим.
наук, вып. 4, с. 11 —16.
268
121. Пееледовапие топких слоен Периклаза при мехаПоактпвнругощей
обработке/Певсроп В. В., »Кпгникон II. II., Суппес В. Г. и др.
Изи. ЛИ СССР. Пеоргап. материалы. 1983, т. 19, Л» 11, е. 1917 —
1920.
122. Боуден Ф. П., Тейбор Л. Трение п смазка твердых тел.— М.:
Maiiirna, 19(10.— 202 с.
123. Bowden F. Р., Thomas Г. R. S. and Р. II. The surface temperature
о! sliding solids.— Proc. Boy. Soc., 1951. v. A223, p. 29—40.
124. Bowden !•’. I’., Persson P. A. Defornialion heating and melting of
solids in high-speed friction. - Proc Boy. Soc., 1961, v. /4260,
p. 433—451.
125. Eshelby J. D., Pratt P. L. Note on the healing effect of moving dislo-
cations.— Acta inetallurg., 1956, v. 4, p. 560—562.
126. Дубнов А. В., Сухих В. Л., Томашевпч II. И. К вопросу о при-
роде локальных микроочагов разложения в конденсированных
ВВ при механических воздействиях.— Физика горения п взрыва,
1972, т. 7, № 1, с. 147—149.
127. Афанасьев Г. Т., Боболев В. К. Инициирование твердых нарыв-
чатых веществ ударом.— М.: Наука. 1968.— 172 с.
128. Харитон IO. Б. К вопросу о детонации от удара.— В кн.: Сборник
по теории взрывчатых веществ.— М.: Оборонгнз, 1940, с. 177—
200.
129, Холево II. А. Чувствительность взрывчатых веществ к удару.—
М.: Машиностроение, 1974.— 136 с.
130. Сухих В. А., Харитон 10. Б. Возникновение вспышек во взрывча-
том веществе при кратковременных деформациях.— В кн.: Воп-
росы теории взрывчатых веществ. Вын. 1. М.—Л.: Пзд-во АН
СССР, 1947, с. 149—156.
131. Болховитинов Л. Г. К теории возбуждения взрыва ударом.—
Докл. АП СССР, 1959, т. 125, № 3, с. 570—573.
, 132. Болховитинов Л. Г. О связи между чувствительностью к удару и
температурой испышкп взрывчатых веществ.— 71{урн. фнз. химии,
1960, т. 34, с. 476—477.
133. Weichert R., Schonert К. On the temperature rise at the tip of a
fast running crack.— .1. Meeh. Phys. Solids, 1974, v. 22, p. 127—133.
134. Fox P. G., Soria-Ruiz J. Fracture-itidnced thermal decomposition
in brittle crystalline solids.— Proc. Boy. Soc., 1970, v. A317,
p. 79—91.
135. Уракаев Ф. X. Теоретическая оценка импульсов давления и тем-
пературы па контакте трущихся частиц в диспергирующих ап-
паратах,— Изв. СО АП СССР, 1978, Л» 7. Сер. хим. паук, вып. 3,
с. 5—10.
136. Беляев II. М. ’Груды по теории упругости и пластичности.— М.:
Гостехиздат, 1957.— 632 с.
137. Диипик А. И. Избранные труды. Т. 7.— Киев: Пзд-во АН УССР,
1952,— 152 с.
138. Ландау Л. Д., Лифшиц Е. М. Механика сплошных сред. Гидроди-
намика и теория упругости.— М.—Л.: Гостехиздат, 1944.— 624 с.
139. Schneider U. Makroskopische und mikroskopischo Eigenschaftsan-
deriingen von Feslstoffpulvern infolge starker rnechanischer Beans-
pruchung in Muhlen.— Aufbereit Techn., 1968, Bd 9, S. 567—573.
140. Андерсон О. Физическая акустика.— М.: Мир, 1968.— 391 с.
111. Маклипток <!•., Ариш А. Деформация и разрушение материалов. —
М.: Мир, 1970.— 433 с.
142. Dachille F., Roy R. High-pressure phase transformations in labo-
ratory mechanical mixers and mortars.— Nature, 1960, v. 186,
p. 39, 71.
269
143. Об использовании сппхротронпого п.зчучоппя для исследования
реакции в твердой фазе/То.точьо II. II., Шеронов М. Л., Ля-
хов II. 3., Болдырев В. В.— Док I. АН СССР, 1981, т. 260, № 6,
с. 1415—1417.
144. Твердые тела под высоким давлением/Под ред. В. Пола, Д. Вар-
шауэра,— М.: Мир, 1966,— 523 с.
145. Dyulai Z., Hartly D. Elcktrische Lcitl’ahigkeit verfonnler Steinsalz-
kristaile.— Z. Phys., 1928, Bd 51, S. 378—380.
146. Stepanov A. \V. liber den Mechanisinus der plastischen Deforma-
tion.— 7.. Phys., 1933, Bd 81, S. 56(1—564.
147. Герцрикеп С. Д., Дехтяр 11. Я. Диффузия в металлах и сплавах.—
М.: Фпзматгпз, I960,— 564 с.
148. Ромашкин 10. П. К теории диффузии в пластически деформируе-
мых металлах. 1. Обзор работ.— Физ. твердого тела, 1960, т. 2,
№ 12, с. 3050—3063.
149. Ромашкин 10. II. К теории диффузии в пластически деформируе-
мых металлах. II. Общие функциональные связи.— Физ. твердого
тела, 1960, т. 2, № 12, с. 3059—3064.
150. Ромашкин IO, П. К теории диффузии при пластической деформа-
ции металлов. III. Возможная природа явления.— Физ. твердого
тела, 1960, т. 2, № 12, с. 3(153—3076.
151. Self-diffusion in alpha-iron during compressive plastic flow/IIi-
rano K., Cohen M., Averbuch B. L. e. a.— AJME Traus. Met. Soc.,
1963, v. 227, N 4, p. 950—957.
152. Wassan A. R., Dorn J. E. Analysis of Enhanced Diffusivity in Nic-
kel.— .1. Appl. Phys., 1965, v. 36, N 1, p. 222—226.
153. Шестопалов Л. M., Ромашкин IO. П. Диффузия серебра в пласти-
чески деформируемую медь.— Физика твердого тела, 1960, т. 11,
№ 12, с. 2998—3006.
154. Земский С. В., Грузии II. Л., Тихонов А. С. Диффузия никеля в
сверхиластичны.х двухфазных сплавах систем никель — хром и
никель — молибден.— Физ. и хпм. обраб. материалов, 1971,
№ 5, с. 83—88.
155. Brown L. С., Sanderson С. С., John St. Stress-enhanced-diffnsion in
copper tellurium couples.— A.IME Trans. Met. Soc., 1966, v. 236,
N 7, p. 1589—1593.
156. Аномальное ускорение диффузии при импульсном нагружении ме-
таллов/Ларпков Л. Н., Фальченко В. М., Мазанко В. Ф. и др.—
Докл. АН СССР, 1975, т. 221, № 5, с. 1073-^1075.
157. Исследование аномального массопереноса в условиях ударного
нагружения разнородных металлов/Ларпков А. Н., Мазан-
ко В. Ф., Немошкалснко В. В. и др.— Физ. п хпм. обраб. матери-
алов, 1981, № 4, с. 128—132.
158. Красулпп 10. Л. Об «аномальной» диффузии в материалах при им-
пульсном нагружении.— Физ. п хпм. обраб. материалов, 1981,
№ 4, с. 133—135. 1
159. Balluffi В. W., Ruoff A. L. Strain-enhanced diffusion im metals.
I. Point defect models.— J. Appl. Phys., 1963, v. 34, N 6, p. 1634—
1642.
160. Ruoff A. I., Balluffi R. W. St ni ill-enhanced diffusion in metals.
II. Dislocation and grain-boundary short-circulating models.—
J. Appl. Phys., 1963, v. 34, N 7, p. 1848—1853.
161. Balluffi R. W. On Measurement of self-diffusion rates along dislo-
cations in F. С. C. Metals.— Phys. Slat. Sol., 1970, v. 42, N 1,
p. 11 — 19.
270
162. Клоцман С. М., Тимофеев А. II., Трахтенберг II. Ш. К вопросу о
механизме диффузии по структурным дефектам (тина дислокации
и межкристаллитных сочленении) в металлах.— Физ. металлов и
металловедение, 1967, т. 23, № 2, с. 257—267.
163. Павлов П. В., Майоров Л. В.. Пантелеев В. В. Диффузия сурьмы
и кремния вдоль дислокации.— Физ. твердого тела, 1964, т. 6,
.V- 2, с. 382 - 385.
161. Вее С. II., Maddin К. Effect of torsional deformation on self-diffu-
sion in silver. J. Appl. Phys., 1961, v. 32, N 10(1), p. 1848—
1851.
165. Hart E. W. On the role of dislocations in bulk diffusion.— Acta
melallurg.. 1957, v. 5, N t(l, p. 597—602.
166. Декорирование поверхности твердых тел/Дпстлер Г. И., Вла-
сов В. II., Герасимов |О. М. и др.— М.: Наука, 1976.— 112 с.
167. Урусовекап А. А. Электрические эффекты, связанные с пластиче-
ской деформацией ионных кристаллов.— Уси. физ. наук, 1968,
т. 96, с. 39—60.
168. Бредов М. М., Кшемлпская II. 3. Электризация, обнаруживаемая
после соприкосновения двух твердых тел.— Журн. техн, физики,
1957. т. 27. с. 921—925.
169. Свешников Г. Б., Кедрпнскип II. А. Об электрохимическом раст-
ворении.— Вести. ЛГУ. Сер. геол, и геогр., 1963, № 2, с. 62—71.
170. Исследование рентгеновского излучения при разрушении адгези-
онного коптакта/Анпснмоиа В. И., Дерягин II. В., Клюев В. А.
и др.— В кн.: Материалы V Всесоюзного симпозиума по механо-
эмпссин и мехаиохимпи твердых тел. Ч. 1. Таллин, 1977, с. 98—
103.
171. Капптапчук В. А. О радиационном действии мехапоэлектронов.—
В кн.: Механизм песня и мехапохпмпя твердых тел. Фрунзе, 1974,
с. 2S—33.
172. Евдокимов В. Д., Семов IO. II. Эьзоэ.тсктропиая эмиссия при тре-
нии,— М.: Паука, 1973.— 182 с.
173. Lohff I. Die Electroiienemission bei der Oxidation mechanise!)
bearbeileter Metalloberflachen.— Z. Phys., 1956, Bd 146, S. 346—
446.
174. Линке Э., Гоффман К. Влияние механически возбужденных цент-
ров па эмиссию фотонов п электронов при фрезеровании.— В кн.:
Материалы V Всесоюзного симпозиума по механоэмиссин и меха-
нохпмпп твердых тел. Ч. 1. Таллии, 1977, с. 167—171.
175. Meyer К., Obrikat D., Rossberg М. Progress in triboluminescence
of alkali halides and doped zinc sulphides.— Krist. und Techn.,
1970, Bd 5, S. 5—49, 181—205.
176. Семчуков Ф. Д., Шмурак С. 3. Исследование механизма деформиро-
ванном люминесценции.— Физ. твердого тела, 1970, т. 12, с. 9—12.
177. Sodomka L. Zur Theorie der Triboluminoszenz.— Krist. und Techn.,
1972. Bd 7, S. 975—981.
178. Мамбетов Д. M., Мусуралпев T. Исследование газоразрядных яв-
лении при нарушении адгезионной связи.— Докл. АН СССР,
1969, т. I85, с. 122—124.
179. Стрелецкий А. И., Бутягпп II. 10. Природа люминесценции, со-
провождающей деформирование, разрушение и трепне полиме-
ров. Высокомол. соед., 1973, т. А15, А» 3, с. 654—660.
180. Беляев Л. М., Мартынов IO. П., Набатов В. В. Исследование све-
чения при разрушении кристаллов. Времена высвечиваипя.—
В кп.: Физика щелочио-галопдиых кристаллов. Рига, 1965,
у. 179—183,
271
181. Стрелецкий Л. II., Бутлгин П. Ю., Руфов Ю. II. Возможные пути
возбуждения люминесценции при расколе гомополяриых крис-
таллов.— В ки.: Материалы V Всесоюзного симпозиума но меха-
ноэмисспи и механохимии твердых тел. Ч. 1\ Таллин, 1977,
с. 172—178.
182. Freese grinding.— Chem. Proc,. Eng., 1957, v. 38, N 6, p. 259.
183. Ребппдер II. А., Калиновская II. А. Понижения прочности поверх-
ностного слоя твердых тел при адсорбции поверхностно-актив-
ных веществ.— Журн. техн, физики, 1932, т. 2, с. 726—755.
18-1. Ioffe A. F., Kirpitschewa М. W., Lewitsky М. A. Deformation und
Festigkeit der Kristalle.— Z. Physiks, 1924, Bd 22, S. 286—302?'
185. Кочегаров Г. Г. Некоторые вопросы оптимизации процесса дис-
пергирования.— Пав. СО АП СССР, 1977, № 9. Сер. хим., вып. 4,
с. 23—26.
186. Ходаков Г. С., Ребнндер П. А. Исследование тонкого дисперги-
рования кварца и влияния добавок жидкостей на этот процесс.—
Докл. АН СССР, 1959, т. 127, № 5, с. 1070.
187. Эдельман Л. И., Ходаков Г. С. Роль среды а активизации диспер-
гируемого кварца.— Коллоидный жури., 1972, т. 34, JN» 3, с. 438.
188. Кочегаров Г. Г. Молекулярное взаимодействие на поверхности
раздела фаз при днспергпроваинп кварца.— Изв. СО АН СССР,
1985, № 5. Сер. хим. наук, вып. 2, с. 69.
189. Кочегаров Г. Г. Исследование адсорбции гекенлеульфата натрия
па природном кварце методом ИК-спектроекоини.— Изв. Сиб.
отд. АН СССР, 1977, № 2. Сер. хим. паук, вып. 1, с. 62.
190. Cruickshank D. W. J. The role of 3d-orbilals in-bonds between (a)
silicon, phosphorus, sulphur, or chlorine and (b) oxygon or nitro-
gen.— J. Chem. Soc., 1961, p. 5486.
191. Кочегаров Г. Г. Влияние среды на деформацию поверхностного
слоя кристаллографических плоскостей кварца.— Изв. Сиб. отд.
АН СССР, 1981. № 7. Сер. хим. паук, вып. 3, с. 39.
192. Лихтмап В. И., Щукин Е. Д. Физико-химические явления при
деформации металлов.— Успехи физ. наук, 1958, т. 63, вып. 2,
с. 213—245.
193. Жданов С. П., Киселев А. В. О химическом строении поверхности
кварца п силикагеля и ее гидратации.— Журн. физ. химии, 1957,
т. 31, № 10, с. 2213.
194. Hockey J. A., Pethica В. A. Surface hydration of silicas.— Trans.
Farad. Soc., 1961, v. 57, N 12, p, 2247.
195. Кочегаров Г. Г. Технологические аспекты механической актива-
ции твердых тел.— В кн.: Тезисы докладов Всесоюзного сове-
щания «Мехаиохпмия неорганических веществ».— Новосибирск,
1982, с. 98—99.
196. Козулин Н. А., Горловский И. А. Оборудование заводов лакокра-
сочной промышленности.— Л.: Химия, 1968.— 630 с.
197. Сиденко П. М. Измельчение в химической промышленности.—
М.: Химия, 1977.— 382 с.
198. Химико-технологическая аппаратура с использованием ^физиче-
ских методов.— 2-е изд., испр. п дои.— М.: изд. ЦИПТИХИМ-
НЕФТЕМАШ, 1983,— 95 е.
199. Справочник по обогащению руд,- Подготовительные процессы/Под
ред. О. С. Богданова, В. А. Олевского.— 2-е изд., испр. и дои. -
М.: Недра, 1982.— 366 с.
200. Rose II. Е., Sullivan К. М. S. Vibralion mills and vibration nul-
ling.— L., 1961.— 302 p.
272
201. Логвиненко Д. Д., Шеллков О. П. Ппгепспфпкацпя технологиче-
ских процессов в аппаратах с вихревым слоем.— Киев: Техника,
1976.— 144 с.
202. Хпнт II. А. Основы производства сплпкатных пзделпй.— М.—Л.:
Госстройиздат, 1962.— 601 с.
203. Акунов В. II. Струйные мельницы. Элементы теории и расчета.—
М.: Машиностроение, 1967.— 263 с.
204. Блпнпчев В. Н., Бобков С. П., Гуюмджан П. II. Влияние кон-
структивного оформления мельниц на удельные энергозатраты
и мехапохимическпе превращения измельчаемых материалов.—
В кп.: Доклады VII Всесоюзного симпозиума по механоэмиссии ц
мехапохпмип твердых тел. Ч. 1. Ташкент, 1981, с. 73—78.
205. А. с. 101874 (СССР). Центробежная барабанная мельница/Голо-
сов С. И.— Бюл. нзобр., 1955, № 11.
206. Голосов С. И., Молчанов В. II. Центробежная планетарная мель-
ница, ее технические возможности п применение в практике геоло-
гических исследований.— В кп.: <1>11:|ико-хпмическио изменения
минералов в процессе сверхтонкого измельчения. Новосибирск,
1966, с. 5—15.
207. Бушуев Л. П. О конструировании н применении планетарных
центробежных мельниц.— Изв. вузов. Горн, журн., 1960, № 2,
с. 17—20.
208. Бушуев Л. П. Мпогорежпмная планетарная мельница.— Изв.
вузов. Машиностроение, 1964, № 10, с. 17—20.
209. Жирнов Е. И. Современные измельчающие аппараты, основанные
па принципе планетарного движения, их классификация.—
В кп.: Фпзико-хпмпческие исследования механически активиро-
ванных веществ. Новосибирск: Паука, 1975, с. 3—12.
210. Козлов В. II., Козырев С. А., Редькин В. Ф. Модернизация плане-
тарных мельниц с целью их применения в механохпмпческой
технологии н создание специальных мельниц-активаторов.—
Изв. СО АН СССР, 1983, № 12. Сер. хим. наук, вып. 5, с. 25—30.
211. А. е. 433714 (СССР). Аппарат непрерывного депствпя/Болды-
рев В. В., Голосов С. И., Аввакумов Е. Г. п др.— О. И., 1975,
№ 22.
212. А. с. 975068 (СССР). Планетарная мельнпца/Аввакумов Е. Г.,
Поткип А. Р., Самарин О. И.— О. И., 1982, № 43.
213. А. с. 1024102 (СССР). Планетарная мельница/ Аввакумов Е. Г.,
Боткин А. Р., Самарин О. И.— О. И., 1983, № 23.
214. Клетенпк Ю. Б., Марьева II. Н., Полякин Л. 10. Лабораторный
микронзмельчитель.— Изв. СО АП СССР, 1982, № 14. Сер. хим.
наук, с. 125—127.
215. Измельчение порошков в планетарной центробежной мельнице.
I. Определение оптимальных условий измельчения/Мацера В. Е.,
Пугпн В. С., Добровольский А. Г. и др.— Порошковая металлур-
гия, 1973, № 6, с. 11—19.
216. Эффективность пзмельчптельных аппаратов для механического
активирования твердых тел/Болдырев В. В., Аивакумов Е. Г.,
Логвиненко А. Т.— В кн.: Обогащение нолезных ископаемых.—
Новосибирск: Наука, 1977, с. 3—10.
217. Герметичный вибрационный аппарат для механохимических син-
тезов и его применение для получения боразппа/Волков В. В.,
Ларионов И. Г., Пухов А. А. п др.— Изв. СО АН СССР, 1981,
Л" 12. Сер. хим. паук, вып. 5, с. 151—156.
218. Буянова П. Е., Карнаухов А. П. Определение удельной поверх-
ности твердых тел хроматографическим методом тепловой десорб-
ции аргона.— Новосибирск, 1965.— 60 с.
IO v
219. Дымчснко И. II., Шишляпипкова Л. М., Ярославцева Н. Я. К рас-
чету блочности п мпкродеформи1(пп поликристаллов методом гар-
монического анализа с использованием ЭВМ.— В кн.: Методы и
аппаратура рентгеновского анализа.— Вып. 15. Л.: Машиностро-
ение, 1974, с. 37—45.
220. Smekal A. Bitzvorgaiig unci uiolekulare Eestigkeit.— Nalurwis-
senschaften, 1942, Bd 30, S. 224 — 225.
221. Саратовкнн Д. Д., Савинцев П. А. Эффект контактного плавления
как причина нпзкоплавкостп эвтектик.— Докл. АИ СССР, 1947,
т. 58, № 9, с. 1943—1944.
222. Савинцев П. А., Аверпчева В. Е., Костюкевнч М. В. О скорости
контактного плавления щелочио-галопдных кристаллов.— Изв.
вузов. Физика, I960, № 4, с. 107—109.
223. Савицкая Л. К. Расчет скорости контактного плавления эвтекти-
ческих систем.— Изв. вузов. Физика, 1962, А« 6, с. 132—138.
224. Уракаев Ф. X., Аввакумов Е. Г. О механизме механохиПпческих
реакции в диспергирующих аппаратах.— Изв. СО АП СССР,
1978, А: 7. Сер. хим. паук, вып. 3, с. 18—23.
225. Landau II. G. Heat conduction in a inciting solid..— Quartz. Appl.
Math., 1950, v. 8. p. 81—94.
226. Schlicliting 11. Einige exakte Liisungen fur die Temperaturvertei-
lung in finer laniinaren Stronning.— Z. Angew. Math. Meeh.,-1951,
v. 31, p. 78—83.
227. Убеллоде А. Плавление н кристаллическая структура.— M.:
Мир, 1969.— 419 с.
228. Мейер К. Физико-химическая кристаллография.— М.: Металлур-
гия, 1972.— 479 с.
229. Исследование химических реакций при разрушении кристаллов
неорганических солей/Волды реи В. В., Регель В. Р., Поздня-
ков О. Ф. и др.—- Докл. АН СССР, 1975, т. 221, № 3, с. 634—637.
230. Волдырей В. В. О кинетических факторах, определяющих специ-
фику механохимических процессов в неорганических системах.—
Кинетика и катализ, 1972, т. 13, вып. (>, с. 1414—1421.
231. Изучение механизма механохимического разложения твердых не-
органических соедппеиип/Уракаев Ф. X., Болдырев В. В., Позд-
няков О. Ф. и др.— Кинетика н катализ, 1977, т. 18, выи. 2, с.
350—358.
232. Кинетика выделения летучих продуктов при раскалывании моно-
кристаллов неорганических сосдннеивн/Уракаеп Ф. X., Поздня-
ков О. Ф., Болдырев В. В. и др.— Кинетика н катализ, 1978,
т. 19, вып. 6, с. 1442—1448.
233. Ляхов II. 3. Кинетика мехапохимнческпх реакций.— Banicke
listy (Miinoriadne cislo), Bratislava: VEDA, 1984, s. 40—48.
234. Поведение окпелов при действии высокого давления с одновремен-
ным приложением напряжения сдиига/Верещагип Л. Ф., Зубо-
ва Е. В., Бурдина К. П. п др.— Докл. АН СССР, 1971, т. 196,
№ 5, с. 1057—1059.
235. Зубова Е. В., Коротаева Л. А. Явления химических превращении
нетвердой фазе под давлением 50 000 кг/см2’прп одновременном
действии сдвига.— /Кури. физ. химии, 1958, т. 32, с. 1576—1585.
236. Larsen II. A., Drickainer II.,G. Chemical effects of plastic deforma-
tion al high pressure.— J. Phys. Chem., 1957, v. 61, p. 1249—1254.
237. Пшеничной 10. И. Выявление топкой структуры металлов.— М.:
Металлургия, 1974.— 527 с.
238. Schrader IL, Stadter W., Oettel II. Unlersuchungen an niechanisch
aktivierten Konlakleii. XIII. Eeslkorperslruklur und katalytisehes
Verballen von Nickelpulver.— Z. Phys. Chem., 1972, Bd 249,
S. 87—100.
239. Ileinicke G., Harcnz II., Richter-Menday I. Tribomechanische Akti-
vierung dor Nickelcarbonylbildung dureh Erzeuguiig energetisell
angeregler Festkorperzuslande.— Krisl. und Techn., 1969, Bel 4,
S. 105—115.
240. Берштейн В. Л., Мовчан IO. II.. Никитин В. В. Влияние механи-
ческих напряжении на гидролиз cim.ieii поперхносгн стекла.
Физ. твердого тела, 1972, т. 14, вып. 9, с. 2792—2794.
241. Красулпн Ю. А. Дислокации как активные центры в топохимиче-
ских реакциях.— Теорет. и экспер. химия, 1907, т. 3, Л» 1, с. 58—!
62.
242. Каракозов Э. С. Соединение металлов в твердой фазе.— М.: Метал-
лургия, 1976.— 262 с.
243. Молоцкпп М. II. Каталитическая активность дислокаций.— Ки-
нетика п катализ, 1072, т. 13, с. 898—9(17.
244. Молоцкни М. II. Влияние краевых дислокаций па образование по-
верхностных зародышей.— Кристаллография, 1072, т. 17, .V 5.
с. 1015—1017.
245. Молоцкнп М. II. Возбуждение колебательных термов адсорбиро-
ванных молекул движущимися дислокациями.—К ристал.loi ра-
фия, 1975, т. 20, № 2, с. 371—374.
246. Зубова Е. В., Апарпнков Г. Л. Разложение бпхромата аммония
при высоком давлении и пластической деформации.— Докл. АП
СССР, 1974, т. 215, № 5, с. 1150—1153.
247. Бертенов Г. М., Разумовская II. В. Фононная концепция хрупкого
разрушения твердых тел.— Физ.-хим. .механика материалов,
1969, т. 5, с. 60—64.
248. Tamm Ig. Ober vine inogliclie Ari der Elect ronciibilthing an Ixri-
slalioherflachen. Z. 1’hys.. 1932. Bd 76. S. 819—850.
249. Seliokli \V. On the surface stales associated with a periodic poten-
tial.— Phys. Rev., 1939, v. 56, p. 317—323.
250. Koutecky J., Toinasek M. Study of the surface state of diamond and
graphite by a simple MO-LCAO method.— Phvs. Rev.. 1960. v. 120,
p. 1212-1223.
251. Hoclistrasser G.. Courvoisier J. C. Detection par k. p. e. de liaisons
compiles («dangling bonds») a la surface de la silice el du quartz.—
llelv. Phys. Acta, 1966, v. 39. p. 189—191.
252. Hoclistrasser G., Antonin! I. E. Surface stales ol pristine silica sur-
faces. I. ESR studies of Es dangling bonds and of CO7 adsorbed ra-
dicals.— Surface Sri., 1972, v. 32, p. 644—664.
253. Бутягип II. IO., Дубинская A. M., Радцпг В. А. Спектры ЭПР,
конформация п химические свойства свободных радикалов в твер-
дых полимерах.— Успехи химии, 1969, т. 38, с. 593—623.
254. Левин А. А., Сыркин Я. К., Дяткина М. Е. Проблема одноатомных
мпогозарядпых попов п характер химической cini.ni п неорганиче-
ских кристаллах.— Успехи химии, I960, т. 38, с. 193—219.
255. Бартенев Г. М. Сверхпрочные и высокопрочные неорганические
стекла.— М.: Строппздат, 1974.— 240 с.
25G. Об образовании при механической деструкция зас геклопаииых
иолпмероп/Бутигип И. 10., Берлин А.. Колмапсон А. Э. ндр.—
Высокомолск. соед., 1959. т. 1, с. S65 —869.
257. Arends I., Dekker A. I.. Perdock \V. I). Colour Centers in quartz
produced by crushing. Phvs. Stat. Solidi, 1963, v. 3, p. 2275 —
2279.
10*
258. Ruzek J., Elbert K. Formation of free radicals in the course of
grinding quartz glass and quartz.— Silikaty c, 1980, v. 24, p. 119—
131.
259. Колбанев II. В., Бутягин П. Ю. Изучение процессов диспергиро-
вания кварца методом ЭПР.— В кн.: Механоэмпссил и механохи-
мня твердых тел. Фрунзе: Илим, 1971, с. 215—218.
260. Walters G. К., Estle Т. L. Paramagnetic resonance of defects intro-
duced near the surface of solids by mechanical damage.— J. Appl.
Phys., 1961, v. 32, p. 1854—1858.
261. Wce.ks R. A. Paramagnetic spectra of E2 centres in crystalline
quartz.— Phys. Rev., 1963, v. 130, p. 570—976.
262. Weeks R. A., Nelson C. N. Trapped electrons in irradiated quartz
and silica. II. Electron spin resonance.— J. Arper. Ceram. Soc.,
1960, v. 43, p. 399—404.
263. Silsbee R. H. Electron spin resonance in neutron-irradiated quartz.—
J. Appl. Phys., 1961, v. 32, p. 1459—1462.
264. Ахмед-Заде К. А., Баптизмапский В. В., Зпкревскпп В. А. Пара-
магнитные центры, образующиеся при разрушении двуокиси крем-
ния.— Физ. твердого тела, 1972, т. 14, с. 422—430.
265. Радцпг В. А., Быстрнков А. В. Исследование химически актив-
ных центров на поверхности кварца методом ЭПР.— Кинетика и1
катализ, 1978, т. 19, № 3, с. 713—718.
266. Радцпг В. А. Парамагнитные центры на поверхности раскола
кварца. Взаимодействие с молекулами СО н N2O.— Кинетика и
катализ, 1979, т. 20, № 2, с. 448—455.
267. Радцпг В. А. Парамагнитные центры па поверхности раскола
кварца. Взаимодействие с молекулами П2 и В2.— Кинетика и
катализ, 1979, т. 20, № 2, с. 456—464.
268. Радцпг В. А., Халиф В. А. Изучение процессов хемосорбции га-
зов на поверхности измельченного кварца методами ЭПР-епектро-
скоппп п микрокалориметрпп.— Кинетика и катализ, 1979, т. 20,
№ 3, с. 705—772.
269. Jaffe Н. II. Studies in molecular orbital theory of valence. III.
Multiple bonds involving d-orbitals.— J. Phys. Chem., 1954, v. 58,
p. 185—190.
270. Радцпг В. А. Химически активные центры па поверхности измель-
ченного кварца.— В кн.: Доклады VII Всесоюзного симпозиума
но механоэмиссии п механохимпп твердых тел. Ч. I. Ташкент,
1981, с. 24—28.
271. Быстрнков А. В., Береетецкая И. В., Стрелецкий А. II., Бутя-
гнн П. Ю. Механохимпп поверхности кварца. I. Продукты реак-
ции с водородом.— Кинетика и катализ, 1980, т. 21, № 3, с. 765—
769.
272. Стрелецкий А. II., Бутягин П. 10. Мехапохпмпя поверхности
кварца. II. Роль трепня.— Кинетика и катализ, 1980, т. 21, № 3,
с. 770-775.
273. Быстрнков А. В., Стрелецкий А. Н., Бутягин П. 10. Механохимпп
поверхности кварца. III. Активные центры в реакции с водоро-
дом.— Кинетика и катализ, 1980, т. 21, № 4, с. 1013—1018.
274. Береетецкая И. В., Быстрнков А. В., Стрелецкий А. Н., Бутя-
гнн П. 10. Механохимия поверхности кварца. IV. Взаимодействие
с кислородом.— Кинетика п катализ, 1980, т. 21, № 4, с. 1019
1022.
275. Быстрнков А. В., Стрелецкий А. II., Бутягин П. IO. Механохимпп
поверхности кварца. V. Окисление окиси углерода.— Кинетика и
катализ, 1980, т. 21, № 5, с. 1148—1153.
276
270. Колбапев ГТ. В., Береетецкая II. В., Бутлгпп П. ТО. Механохимпп
поверхности кварца. VI. Свойства перекиси = Si—О—Si se.—
Кинетика и катализ, 1980, т. 21, № 5, с. 1154—1158.
277. Ярым-Агаев Ю. Н., Бутягин П. Ю. О короткоживущих активных
центрах в гетерогенных механохпмпческих реакциях.— Докл.
АН СССР, 1972, т. 207, с. 892—896.
278. Бутягин П. Ю. Первичные активные центры в механохнмпческих
реакциях,— Журн. ВХО им. Д. II. Менделеева, 1973, т. 18,
с. 90—95.
279. Колбапев И. В., Бутягин П. Ю. Исследование механохпмпческих
реакций с участием кварца методом ЭПР.— Жури. физ. хим.,
1974, т. 48, с. 1158—1161.
280. Стрелецкий А. И., Бутягин П. 10. Люминесценция и адсорбция
кислорода на кварце.— Докл. АН СССР, 1975, т. 225, с. 1118.
281. Бутягин II. 10., Быстрнков А. В. Об инициировании химических
реакций при разрушении твердых тел.— В кн.: Материалы V Все-
союзного симпозиума по механоэмиссии и механохимпп твердых
тел. Ч. 1. 'Галлин, 1977, с. 63—78.
282. Быстрнков А. В., Бутягин П. 10. Взаимодействие MoS2 с кислоро-
дом и процессе механической обработки.— Изв. АП СССР. Сер.
хим., 1977, № 2, с. 416—419.
283. Напешап D. Electron paramagnetic resonance from clean single
crystal cleavage surfaces of silicon.— Phys. Rev., 1968, v. 170,
p. 705—718.
284. Chung M. F., Ilaneinan D. Properties of clean silicon surfaces by
paramagnetic resonance.— J. Appl. Phys., 1966, v. 37, p. 1879—
1883.
285. Higginbotham J., Haneman D. Paramagnetic surface states of germa-
nium.— Surface Sci., 1973, v. 34, N 2, p. 450—456.
28G. Киселев В. Ф. Поверхностные пиления в полупроводниках и ди-
электриках.— М.: Паука, 1970.— 256 с.
287. Демидович Г. Б., Киселев В. Ф., Никитина О. В. О локализован-
ных электронных состояниях на атомарно чистой поверхности
графита, кремния и германия.— Докл. АН СССР, 1972, т. 205,
№ 2, с. 383—386.
288. Об образовании макрорадикалов при механической деструкции
застекловапных полимеров/Бутя гни П. 10., Берлин А. А., Кол-
мансон А. Э. п др.— Высокомолек. соед., 1959, т. 1, с. 865—869.
289. Радцпг В. А. Структура и реакционная способность дефектов в ме-
ханически активированных твердых телах. Автореф. докт. дне.—
М., 1985,— 36 с.
290. Теплое В. Г., Полежаев IO. М., Новиков Б. С. Эмиссия электро-
нов с уровней захвата, образующихся на сколах кристаллов окси-
да магния.— В кп.: Тезисы докладов VIII Всесоюзного симпози-
ума по механоэмиссии и механохимпп твердых тел. Таллин, 1981,
с. 197.
291. Власова М. В., Каказсп II. Т. Изучение процесса механического
активирования твердых тел методом ЭПР.— Изв. СО АН СССР, >•
1983, № 12. Сер. хим,- наук, вып. 5, с. 40—45.
292. Береетецкая И. В., Бутягин П. Ю., Колбанев И. В. Реакционная
способность поверхности трения MgO.— Кинетика и катализ,
1983, т. 24, № 2, с. 441—448.
293. Бобьинев А. А. Структура и реакционная способность поверхности
механически активированных оксидов германия, олова и магния.
Автореф. капд. дне.— М., 1983.— 23 с.
294. Хайретдппов 3. Ф., Галицин IO. Г., Пост Г. Влияние механиче-
ской обработки на последующее термическое разложенпе >
277
I
Ag2C2O4.— Пзв. СО ЛП СССР, 1979, № 14. Сер. хпм. паук, шли. 6,
с. 50—55.
295. Лущепкпп I’. Л. Полимерные электреты. М.: Химия, 1984.—
24 с.
290. Молоцкпп М. II. Дислокационный механизм электризации ионных
кристаллов прп расщеплении.— Физ. твердого тела, 1970, т. 18,
Л» 0, с. 1703—1708.
297. Электризация щелочно-галоидных кристаллов в процессе скола/
Финкель D. М., Тямии 10. 11., Муратова Л. II. и др.— Физ.твер-
I дого тела, 1979, т. 21, К: 7, с. 1943.
298. Эмиссия быстрых электронов при разрушении ионных крпстал-
лов/Кротова II. Л., Линке Э., Хрусталев 10. А. н др.— Докл.
АН СССР, 1973, т. 208, № 1, с. 138—141.
299. Молоцкпп М. И. Ионно-электронный механизм мехапоэмнеенн.—
Физ. твердого тела, 1977, т. 19, № 2, с. 042—047.
300. Energy dishibutions of atoms sputtered from alkali halides by
540 eV electrons/Ovcrlynder II., Szymnoski M., liarring A. e. a.—
I Hadiat. Elf., 1978, v. 30, N 1, p. 63 -05.
301. The (100) surfaces of alkali halides II Electron stimulated dissocia-
tion/Tokutoka II., Prution M., lliginbolhain J. e. a.— Surface Sci.,
1970, v. 21, N 2A, p. 233.
302. Hoffman K., Linke E. Mechanismus der Photonenemission bei
mechanischcr Anregung von Alkalilmlogenidkristallen.— Krist.
und Techn., 1977, Bd 12, N 5, S. 495—499.
303. Болдырев В. В., Аввакумов Е. Г., Гусев Г. М. Изменение скорости
механохимнческого разложения нитрата натрия с помощью ката-
литических добавок.— Докл. АН СССР, 1909, т. 184, № 1, с. 119 -
121.
30 4. Бутлпш П. 10. Энергетический выход механохимических процес-
сов.-— В кп.: УДА -технология. Таллии, 1983, с. 5 8.
305. Butjagin Р. Yu., Pavlychev .1. К. I lie eslnmilioii of the mecliaiioclie-
mical energy yields.— In: Extended Abstracts of lOlh International
Symposium on the reactivity of Solids. Dijon, 1984, p. 322—323.
300. Miltash A. Uber die chemische Dynaniik des Nickelkohleuoxyds.—
Z. Phys. Chem., 1902, Bd 4(1, S.’ 1— 84.
307. Бенсон Г., Юн К. Поверхностная энергия п поверхностное натяже-
ние кристаллических твердых тел.— В кп.: Межфазовая граница
газ — твердое тело. М., 1970, с. 172—229.
308. Петров 10. Н. Физика малых частиц.— М.: Паука, 1982.— 359 с.
309. Nicolson М. М. Surface tension in ionic crystals.— Proc. Boy. Soc.
L., 1955, v. A228, p. 490—495.
310. Boswell E. W. C. Precise determination of lattice constants by elect-
ron diffraction and variations in lattice constants of very small
crystallites.— Proc. Phys. Soc. L., 1951, v. A64, p. 465.
311. Burger M. I. Phase (raiisfornialions in solids.— N. Y.: John Wiley
and Sons, 1951.— 183 p.
312. Boehm 11. P., Hoffman U. Die rhomboedrische Modification des
Graphits.— Z. anorg. allg. Chem., 1955, Bd 278, S. 58—62.
313. Paiz K. Beitrag zur Unlersuchung des Charakterz der Uiiiwandlung
des xvciBen Phosphorus in seine schwarze Modifikation.— Z. anorg.
allg. Chem., 1959, Bd 299, N 5-6, S. 297—301.
314. Малютина T. В., Горбачева T. Б. Активирующее влияние размо-
ла па фазовые переходы при спекании порошков кобальта.— Изи.
СО АН СССР, 1983, А" 12. Сер. хим. паук, выи. 5, с. 72—74.
315. Senna М., Соппу П. Polymorphic transformation of PbO by isother-
mal wet ball-milling.— J. Amer. Ceram. Soc., 1971, v. 54, p.
I 259—264.
278
316. Редькппа IT. II., Ходаков Г. С. Механохимическое модифицирова-
ние структуры и актипиропаипе окислен синица. Коллоидный
жури., 1976, т. 38, № 3, с. 596 —598.
317. Jmamura К., Senna М. Difference between mechanocheniical and
thermal processes oj polymorphic transformation of ZnS and PbO.—
Mai. Kes. Bui., 198-4, v. 19. p. 59—65.
318. While W. B., Dachille I-'., Roy R. High-Pressure — high-tempera-
ture polymorphism of the Oxides of lead.— J. Amer. Ceram. Sue.,
1961, v. 44, p. 170—174.
319. Lewis D., Northwood D. O., Reeve R. C. Strain-induced phase trails^
formations in lead monoxide.— J. Appl. Crystallogr., 1962, v. 2j-
p. 156—164.
320. Lin J. J., Niedzwiedz S. Kinetics of the iiiassicotlithanie transfor-
mation during comminution.— J. Amer. Ceram. Soc., 1973, v, 56,
N 2, p. 62-64.
321. Senna M., Tojo S., Кино II. Polymorphic Irausforinalinn of y-Fi-.O,
by isothermal ball-milling and vacuum hot-pressing. — Nippon
Kagahn Zasslli, 1971, v. 92, N 9. p. 78(1 -784.
322. Imai II., Senna M, Energy storage and liberation of vibro-ntilled
V-l?e2O:i.— J. Appl. Phys. , 1978, v. 49, N 8, p. 4433—4437. >
323. Ross W., Gibby R. L. Polymorphic transformation of Еиг03 by ab-
rasion.— J. Amer. Ceram. Soc.. 1974, v. 57, p. 46—47.
324. Wankova J., Kochanovska A. Dutch mechaiiische Deformation
hervorherufenc Struktiirveranderung am Kristallgilter von Titano-
xid.— Krist. und Techn., 1966. Bd 1. 8. 319 .'.31.
325. О механической активации pyсильной n ап.ггазн.'й модификаций
диоксида титана и изменении их реакционной способности Воро-
бейчик Л. В.. Пряхина ’Г. Л., Колдыреп В. В. и др. Пан. СО АН
СССР, 1983, № 12. Сер. хим. паук. вы и. 5. с. 121 127.
326. Bailey .1. Е. Phase transformation in milled zirconia.— J.Brit. Ce-
ram. Soc., 1972, v. 71, N 1, p. 25—30.
327. Daehille F., Roy It. Phase transformation PbO,I — PbO2II.—
Proc. IV Intern. Symp. on Reactivity of Solids (Amsterdam), I960,
p. 501.
328. Schrader R., Weigelt D. Das niechanocheniische Gleichgewicht der
Phasen a- und [3-PbO2.— Z. anorg. allg. Chem., 1971), Bd 372,
S. 228-235.
329. Senna M., Schonert K. Change in the enthalpy and structure of
PbO., byr grinding and pressing.— Powder Technology, 1982, N 31,
p. 269—275.
330. Аввакумов E. Г., Разворотнева Л. 11. Механически стимулирован-
ные фазовые переходы в окислах ниобия н тантала,— Пзв. СО АП
СССР, 1977, № 9. Сер. хпм. наук, выи. 4. с. 19—22.
331. Schort М. A., Steward Е. G. The effect of grinding on the structure
and luminescence of zinc and zinc-cadmium sulphides.— Z. Phys.
Chem.. N. F., 1957, Bd 13, S. 298—315.
332. Imamura K., Senna M. Change in phase stability of zinc blende and
wurtzite on grinding.— J. Chem. Soc. Faraday Trans., 1982, v. 78,
N 1, p. 1131-1140,
333. Фазовый переход при пластической деформации крас галлов сер-
нистого цинка/Абдикамалов II. Л., Бредихин С. I!.. Кулаков Л. 11.
и др.— <1>ил. твердого тела, 1976, т. 18, с. 2463 2170.
334. Farkas-Jahnke М., Gates Р. Changes in structure of ZnS crystals due
to mechanical stresses.— Krist. und Techn., 1979, Bd 11, N 12,
S. 1475—1482.
335. Hashimoto K., Toda Y. On the grinding of CdS and CdSe. J. Chem.
Soc. Jap., 1968, v. 71, p. 1102—1418.
336. Sigrist К., IleinickcG., Steinicke U. Zu einigen energctischen Aspek-
tcn tribomechanisch bewirkter Modifications uinwaudlungcn.—
Krist. und Techn., 1973, Bd 8, S. 393—397.
337. Burns I. H., Bredig M. A. Transformation of calcite to aragonite by
grinding.— J. Chem. Phys., 1956, v. 25, p. 1281—1286.
338. Shrader R., Hoffmann B. Enantiotropic Transformation of Calcite-
Aragonite by Mechanic Forces.— Z. Chem., 1966, Bd 6, N 10,
S. 388—389.
339. Northwood D. O., Lewis D. Strain induced calcite-aragonite trans-
formation in calcium carbonate.— Can. Mineralogist, 1970, v. 10,
p. 216—224.
340. Criado J. M., Trillo J. M. Effect of mechanical grinding on the tex-
ture and structure of calcium carbonate.— J. Chem. Soc. Faraday
Trans., 1975, v. 7, p. 961—966.
341. Синяков E. В., Флерова С. А., Аршинов В. II. Влияние-одномер-
ных механических напряжений на доменную структуру монокри-
сталлов BaTiO3 в ромбической фале.— Физ. твердого тола, 1973,
т. 15, с. 1239—1246.
342. Зозуля П. В., Яковлева Л. А. Влияние ультразвука и впбропомола
на полиморфизм двухкальцпевого силиката.— Изв. АН СССР.
Неорганические материалы, 1973, т. 9, № 1, с. 159—160.
343. Uehara Y. Polymorphic transformation in copper ferrite and manga-
nite by grinding.— Bui. Chem. Soc. Jap., 1972; v. 45, p. 3209—
3211.
344. Strid K. G. On the concept of «inechanochemical equilibrium».—
Acta Chein. Scand., 1971, v. '25, p. 1157—1158.
345. Лифшиц Л. Д., Ларионов Л. В., Рябинин 10. И. Кинетические
особенности полиморфных переходов структуры тина NaCl к струк-
туре типа CsCl при высоком давлении (па примере полиморфных
превращений в Bbl и КС1).— Изв. АП СССР. Физика земли,
1972, № И, с. 28-37.
346. Эддисон У. Аллотропия химических элементов.— М.: Мир,
1966,- 165 с.
347. Редькина Н. И., Ходаков Г. С. Влияние ПАВ па модифицирование
структуры н активирование окислов свинца.— В кп.: Доклады
Всесоюзного симпозиума по механоэмиссии н механохимпп твер-
дых тел. Ч. III. Ташкент, 1981, с. 120—128.
348. Вайнштейн Б. К., Фридкин В. И., Инденбом В. Л. Современная
кристаллография. Т. 2,— М.: Наука, 1979.— 359 с.
349. Reaktivitat von inechanisch aktivierton Quarz/Steinike IL,
Ebert J., Geissler II. e. a.— Krist. und Techn., 1978, Bd 13, N 5,
S. 597—603.
350. Штайникс У. Механически ппдуцпроваиная реакционная способ-
ность кварца п ее связь с реальной структурой.— Изв. СО АН
СССР, 1985, № 8. Сер. хим. наук, вып. 3, с. 40—47.
351. Sadahiro Y., Shimidzy К. Effects of dry ball-milling on the physico-
chemical properties of NiO powder.— J. Chem. Soc. Jap., 1968,
v. 71, p. 1374—1378.
352. Takahashi H., Tsutsumi K. Correlation between the structural disor-
der and the catalytic activity of zinc oxide.— J. Chem. Soc. Jap.,
1968, v. 71, p. 1345—1349.
353. Takahashi If. Mechanochcmical effects on zinc oxide powdcrcrys-
tals.— Dechema Monogr., 1967, v. 57, p. 475—494.
354. Paudert R. SO2-Oxidation an mechanisch aktivierlen Eisenoxiden.—
Monatsberichte der Deutsch. Akad. Wissenschaflen zu Berlin, 1967,
Bd 9, S. 719-724.
355. Шрадер Р. Новые представления в области механохпмпп.— В кн.:
Мсханоэмпсспя и механохпмпя твердых тел. Фрунзе: Илим, 1974,
с. 57—65.
356. Schrader R., Rump Н. Die mechanische Aktivierung von Korund.—
Z. anorg. allg. Chem., 1967, Bd 350, S. 137—142.
357. Schrader R., Rump II., Sanner G. Die mechanische Aktivierung von
11 yd rargill it.— Z. anorg. allg. Chem., 1967, Bd 350, S. 130—136.
358. Mechanische Aktivierung von Ni — SiO2 Tragerkatalysatoren/
Schrader 11., Nobst P., Tetzner G. e. a.— Z. anorg. allg. Chem.,
1969, Bd 365, S. 255—262.
359. HeinickeG., Lise like I. Wirksanikeit von Katalysatoren unter Bedin-
gungen der inechanischen Bearbeitung.— Z. Chem., 1963, Bd 3,
S. 355—356.
360. Логвиненко A. T., Савннкнна M. А., Логвиненко В. А. Активность
топкоизмольченпого периклаза.— Изв. СО АН СССР, 1972, № 7.
Сер. хим. паук, вып. 3, с. 140—147.
361. NaeserG., Scholz W. Der Eiufluff einer mechauischen Bearbeitung
auf das Reaklionsvcrmiigen von feslen Sloffen.— Kolloid Zlselir.,
1958, Bil 156, S. 1-8.
362. Shirasaki T., Ochiai Y., Murnnaka K. Mechanocheniical phenomena
of fused a-alumina by grinding.— J. Chem. Soc. Jap., 1968, v. 71,
p. 1363—1367.
363. Schrader R. Uber die Darstcllung aktivierten Zustande an festen
Sloffen durch mechanische Energie.— Freiberger Forschungshefte,
1966, Bd A392, S. 82-86.
364. Пнвннскнй IO. E., Бевз В. А., Мнтякнн П. Л. Основные принципы
получения высокодиспергированных суспензий кварцевого пес-
ка.— Огнеупоры, 1979, № 3, с. 46—51.
365. Мнтякнн II. Л., Ливийский 10. Е. Свойства кварцевого керамзпт-
бетопа.— Огнеупоры, 1980, № 9, с. 55—62.
366. Senna М., Schonert К. Direct observation of inelastic deformation
and mechanocheniical activation of indented quartz single cry-
stals.— Powder Technol., 1982, v. 32, p. 217—221.
367. Ilofman F., Schonert K. Effect of strain energy and particle size on
mechanical activation of guartz and lead dioxide.— Powder Tech-
nol., 1984, v. 39, N 1, p. 77—81.
368. Zur Strnktur mechanisch erzeugter amorpher Quarzschichten/Stei-
nike U., Muller B., Henning E. e. a.— Krist. und Techn., 1979,
Bd 14, S. 696.
369. Pandert R., Liebscher S., Trinks W. Liislicher Zinnstein durch me-
clianischc Bearbeitung.— Chem. Techn., 1967, Bd 19, № 8, S. 355—
356.
370. Paudert R., Liebscher S., Trinks W. Die mechanische Aktivierung
von Zinnstein.— Monatsbericlite der Deutsch. Akad. Wissenschaf-
tun zu Berlin, 1967, Bd 9, S. 66—69.
371. Исследование растворимости и деструкция касситерита после ме-
ханической активации в центробежных мельнпцах/Юсупов Т. G.,
Молчанов В. И., Архипенко Д. К. и др.— В кн.: Механохпмиче-
скпе явления при сверхтонком измельчении. Новосибирск, 1971,
с. 135—147.
372. Юсупов Т. С., Крпвопуцкая Л. И., Кириллова Е. А. О взаимосвя-
зи структурных изменении п растворимости механически активи-
рованных минералов на примере касситерита.— В кн.: Физико-
химические исследования механически активированных минераль-
ных веществ. Новосибирск, 1975, с. 37—47.
I.
281
373. Панченко Л. Л. Исследования реальной структуры окспдпых фаз
тина корунда. Лвтореф. каид. дне.— М., 1982.— 19 с.
374. Широков 10. Г., Ильин Л. II., Кириллов II. II. Влияние дисперги-
рования на кинетику растворения трудпорастворнмых окпслов.—
Изв. СО All СССР, 1979, Л? 7. Сер. хим. наук, пып. 3, с. 45—49.
375. Szantho Е., Gerlach I., Gock Е. .Mechanische Aklivierung von Ti-
tanomagnetit (lurch Schwinginahlimg.— Chem. Ing. Techn., 1909,
Bd 41, S. 843—850.
376. Механическая активация титансодержащпх продуктов/Воробей-
чнк А. II., Пряхина Т. А., Болдырев В. В., Аввакумов Е. Г.
и др.— Изв. СО АН СССР, 1979, № 7. Сер. хпм. наук, вып. 3,
с. 37—45.
377. О возможностях пптепспфнкацпп переработки полезных ископа-
емых па основе сверхтонкого измельчения и механической актива-
цпи/Юсупов Т. С., Голосон С. II., Гусев Г. М. и др.— Физ.-техн,
проблемы переработки полезных ископаемых, 1972, А1» 4, с. 104—
110.
378. Takahashi 1Г. Effects of dry grinding on kaolin minerals. I. Kaoli-
nite.— Bui. Chem. Soc. Jap., 1959, v. 32, N 3, p. 235—245.
379. Schrader R., Kutzer II.-I., Hoffmann B. Uber die mechanische Ak-
tivierung von Kaolinit.— Toninduslrie Ztg., 1970, N 10, S. 410—
416.
380. Лапухова E. С., Столповская В. II., Юсупов T. С. Химические и
структурные особенности механически активированного каоли-
нита.— Изв. СО АН СССР, 1977, № 9. Сер. хим. паук, вып. 4,
с. 110—115.
381. Gregg S. I., Parker Т. W., Stephens М. I. The grinding of kaolinite.
II. A more detailed study. - .1. Appl. Chem., 1954, v. 4, p. 666—
673.
382. Об изменении нефелина в процессе сверхтонкого измельчен ня/Гу-
сев Г. М., Кляровскпй В. М., Ковалева Л. Т. п др.— В кн.: Меха-
нохп.мичсские явления при сверхтонком измельчении. Новоси-
бирск, 1971, с. 62—78.
383. Schrader R., Rump II., Kressncr It. Uber das Verhalten von Banxil
und seinen Einzelphasen nach mechanischer Aklivierung.— Chem.
Ing. Techn., 1967, Bd 39, S. 843—848.
384. Мальцева H. В., Белоцерковский Г. M. Получение формованного
активного оксида алюминия из тоикодпенергироваппого техниче-
ского гидроксида без его переосажденпя.— Журн. прпкл. химии,
1984, т. 57, № 12, с. 2641—2645.
385. Мальцева Н. В., Белоцерковский Г. М. Получение формованного
активного оксида алюминия из тоикодпенергироваппого техниче-
ского гидроксида без его переосажденпя.— Жури, прпкл. хим.,
1983, т. 56, № 5, с. 1009-1012.
386. Изучение продуктов мехапохимпческоп активации гпдраргнлли-
та/Парамзпп С. К., Криворучко О. П., Золотовскпп Б. П. ндр.—
Изв. СО АН СССР, 1984, № 17. Сер. хпм. паук, вып. 6, с. 36—38.
387. Механохпмнческая активация карбопатсодержащего гпдрокспл-
алюмппата/Исупов В. П., Пушпякова В. А., Коцупало Н. П.—
Изв. СО АП СССР, 1983, № 12. Сер. хим. паук, вып. 5, с. 88—91.
388. Mechanical activation of solids: Basic principles and a new method
of preparation of small particles/Pavlukhin Y. T., Boldyrev V. V.,
Avvakumov E. G. e. a.— Proc. Intern. Meet, on highly dispersed
iron oxides and corrosion, Leningrad, Febr. 24—27, 1981, Stock-
holm, 1981, p. 115—117.
282
389. Pavlukliin Y. T., Medikov Ya. Ya.. Boldyrev V. A’. Magnetic and
cheuiicai properties ol' nicchanically activated zine and nickel fer-
rites.— Mat. Res. Bull., v. IS. p. 1317—1327 (I9S3). IS\.
390. Pavlukliin Y. T., Medikov Ya. Ya.. Boldyrev V. V. On the Conse-
quences of mechanical activation of Zine and .Nickel Ferrites.—
.1. of Solid Slate Chemistry. 1984. v. 53, p. 155— ItiO.
391. Исследование мехапохимпческоп активации itnpoxnopa I’.a.ty-
ch А. А., Карпухин A. 1!.. Черник А. С. и др.— В кн.: Maiepua.iij
V Всесоюзного совещания по мехапозмпсспп и мехапохимии твер-
дых тел. Ч. III. Таллин, 1977, с. 91—96.
393. Третьяков 10. Д. Твердофазные реакции.— М.: Химия, 1978.—
359 с.
393. Колопг Р. Нестехпометрпя. Неорганические .матерпалы перемен-
ного состава.— М.: Мир. 1974.— 245 с.
394. Андерсон Дж. С. Термодинамика п теория песгехиометрпческпх
соединений.— В кн.: Проблемы пестехпомсгрпп. М.: Металлур-
гия, 1975, с. 11—93.
395. Бугаев А. А., Захарчспя Б. II., Чудповскпп Ф. А. (Разовый пере-
ход металл — полупроводник и его применение.— Л.: Паука,
1979.
396. The Chemistry of Extended Defects in Nou-metallic SolidsEd.
L. R. Eiring, M. O'Keeffe.— Amsterdam — London: Norlh-ilol-
land РнЫ. Co., 1970.— 374 p.
397. Magneli A. Structures of the ReO.rtype with recurrent dislocations
of atoms: «Homologous Series» of molibdenum and tungsten oxi-
des.— Acta Cryst., 1953, v. 6, p. 495—500.
398. Lundberg M., Sundberg M.„ Magneli A. The «pentagonol column» as
a building unit in crystal and defect structures of some groups of
transition metal compounds.— .1. Solid State Chem., 1982. v. 44.
p. 32—40.
399. Gado P. X-ray powder diffraction study of the \V20 0-,s shear,
transformation.— Acta Phvs. .Acad. Sci. Пипс.. 1965. v. IS. N 2,
p. 111 — 117.
400. Anderson J. S., Hyde B. G. On the possible role of dislocations in
generating ordered and disordered shear structures.— J. Phvs.
Chem. Solids, 1967, v. 28, N 8, p. 1393—1408.
401. Andersson S.. Wadsley A. D. Crystallographic shear and diffusion
paths in certain higher oxides of niobium, tungsten, molybdenum
and titanium.— Nature, 1966, v. 221, N 5049, p. 581 — 583.
402. Hyde В. C., Bursill L. A. Point, line and planar defects in some
non-sloichiomelrie compounds.— In: Chemistry of extended defects
in non-inelallic solids. Amsterdam — London, 1970, p. 347—374.
403. Landuyt J.. Van Amelinx S. On generation mechanism for shear planes
in shear structures.— J. Solid Stale Chem., 1973, N 6, p. 222—229.
404. Anderson J. S. Defects in oxides.— J. Solid State Chem., NBS
Spec. PubL, 1972, v. 364, p. 295—317.
405. Kroger F. A. Non-stoichiometry in shear structures.— J. Phys.
Chem. Solids, 1983, v. 44, N 4, p. 345—347.
406. Notes on phases occuring in the binary tungsten-oxygen system/
Booth J., Ekstrom T., Iguchi E.. Tilley R. J. D.— J. Solid Stale
Chem., 1982, v. 41, p. 293—307.
407. Anderson J. S., Hutchison I. L., Lincoln F. I. Dislocations and rela-
ted defects in niobium oxide structures.— Proc. Roy. Soc., L., 1977,
v. A352, p. 303—323.
408. Galehouse В. M., Wadsley A. D. The crystal structure of the high
temperature form of niobium pentoxide.— Aeta Crvslallogr., 1964,
v. 17, p. 1545-1554.
409. Диаграммы состояния силикатных систем. Справочник. Вып. 2/
Торопов Н. А., Барзаковскпй В. П., Бопдарь И. Л. и-'др. — Л.:
Наука, 1970, с. 168 181.
410. Phase analysis studies on titanium-oxygen system/Andersson S.,
Collen B., Knylenstierna U. c. a.— Acta Chem. Scand., 1957, v. 11
p. 1641 — 1652.
411. Le Page Y., Strobel P. Structural chemistry of Magueli phases
TinO2n—j(4 n 9). I. Cell and structural comparisons.— J. So-
lid State Chem., 1982, v. 43, p. 314—319.
412. Wood G. J., Bursill J. A. The formation energy of crystallographic
shear planes in TinO»n.— Proc. Boy. Soc. L., 1981, v. A375, p. 105 —
125.
413. Аввакумов E. Г., Косова H. В., Александров В. В. Дефектообра-
зование при механической активации оксидов титана, олова п
вольфрама.— Изв. АН СССР. Ыеоргап. материалы, 1983, т. 19,
№ 7, с. 1118—1121.
414. Механическая активация — перспективный способ повышения
реакционной способности трудновскрываемых тптансодержащпх
материалов/flряхипа Т. А., Воробейчик А. И., Аввакумов Е. Г.,
Болдырев В. В.— Изв. СО АН СССР, 1985, № 11. Сер. хим. паук,
вып. 4, с. 34—41.
415. Natarajan М., Chandrasherdar G. V., Вао С, N. R. Heals of crystal-
lization of some amorphous oxides.— J. Chem. Eng. Data, 1968,
v. 13, p. 235.
416. Исследование структурных изменений в механически активиро-
ванных оксидах титана методом ЭПР/Анвакумов Е. Г., Ануфриен-
ко В. Ф., Восель С. В. и др.— Изв. СО АН СССР, 1986, № 6. Сер.
хим. наук, вып. 17, с. 16—21.
417. Гаджиева Ф. С., Ануфриенко Е. Ф. Особенности состояния ионов
Ti3+ в узельиых п междоузельных позициях структуры рутила
по данным ЭПР.— Журн. структур, химии, 1982, т. 23, № 5,
с. 43—49.
418. Chester Р. F. Electron spin resonance in semiconducting rutil.—
J. Appl. Phys., 1961, Suppl. to v. 32, N 10, p. 2233—2236.
419. Kingsbury Ir. P. J., Ohlcn W. D., Johnston O. \V. Defects in rutile.
Electron paramagnetic resonance of interstitially doped n-type ruti-
le.— Phys. Rev., 1968, v. 175, p. 1091—1093.
420. Akse J. R., Whitehurst И. B. Diffusion of titanium in slightly redu-
ced rutile.— J. Phys. Chem. Solids, 1978, v. 39, p. 457—465.
421. Зырянов В. В., Ляхов Н. 3., Болдырев В. В. Исследование меха-
нолиза двуокиси титана методом ЭПР.— Докл. АН СССР, 1981,
т. 258, № 2, с. 394—396.
422. Chester Р. F. Cross-doping agents for rutile masers.— J. Appl. Phys.,
1961, v. 32, N 5, p. 866—868.
423. EPR Spectrum of Ti2+ in single crystals of TiflOn/Fairhurst S. S.,
Inglis A. D., Le Page Y. e. a.— Chem. Phys. Lett., 1983, v. 95,
N 4/5, p. 444-448.
424. Intrinsic paramagnetic dimers in Magneli phases of TinO,2n_J[/Fair-
hurst S. S., Inglis A D., Le Pago Y. o. a.— J. Magn. Res., 1983,
v. 54, p. 300—304.
425. Blanchin M.G., Bursill L. A., Smith D. J. Precipitation phenomena
in non-stoichiometric oxides.— Proc. Roy. Soc. L., 1984, v. 391,
p. 351—391.
426. Варнек В. А., Аввакумов E. Г., Ли Бон Твои. Квадруполыюе рас-
щепление ,wSn в тонкоизмельченных порошках SnO2.— Изв. СО
АН СССР, 1977, № 7. Сер. хим. наук, вып. 3, с. 88—94.
284
427. Аввакумов E. Г., Варнек В. А., Мазалов Л. Н. Исследование о,-
жига дефектов в механически активированных порошках двуокиси
олова методом ЯГРС.— Изв. СО АН СССР, 1980, № 2. Сер. хим.
наук, вин. 1, с. 119—123.
128 . Аввакумов Е. Г., Вариек В. А. Исследование отжига дефектов в
двуокиси олова после механической активации методом ЯГРС.—
В кн.: Доклады VII Всесоюзного симпозиума по механоэмиссии и
механохимпп твердых тел. Ч. III. Ташкент, 1981, с. 128—131.
429. Аввакумов Е. Г. Влияние механической активации на последую-
щие химические реакции твердой фазы.— Banieke listy (Momoriad-
пе cislo), Bratislava: VEDA, 1980, s. 228—234.
430. Митрофанов К. II., Плотников M. В., Шпинель В. С. Форма спект-
ров резонансного поглощения у-лучеп 23,8 кэВ изомера U9Sn в
окиси олова и металлическом белом олове.— Журн. экснер. н тео-
рет. физики, 1965, т. 48, с. 791—795.
431. Анализ минерального сырья/Под ред. Книпович 10. Н., Морачев-
ского 10. В.— М.: Изд-во хим. лит., 1959.— 1055 с.
432. Сабитов Р. М. Интенсивность >ффекта Мессбауэра в кристаллах
с вакансиями и в твердых растворах в случае алокальиых колеба-
ний.— Изв. вузов. Физика, 1976, вып. 6, с. 81—86.
433. Михайлов А. И., Лебедев Я. С., Бубен М. Я. Стуненчатообразная
рекомбинация свободных радикалов в облученных органических
веществах.— Кинетика и катализ, 1964, т. 1, выц. 6, с. 1020—
1027.
434. Толкачев В. А.. Соколов И. Б., Ковалевский В. И. Исследование
гибели свободных радикалов, в кристаллических феноле, анилине,
этпланплпно и о-нитро-Р-метиланилине.— Химия высоких энер-
гий, 1970, т. 4, с. 455—459.
435. Бобышев А. А., Радцпг В. А. О структуре дефектов, образующих-
ся при механической активации оксида олова.— Химическая фи-
зика, 1984, т. 3, № И, с. 1525—1531.
436. Малинин Г. В., Толмачев’Ю. М., Ядрпнцев'В. Б. Дернватографи-
ческое исследование разложения окпелов свинца.— Журн. неор-
ган. химии, 1966, т.. 13, № 7, с. 1746—1750.
437. Effect of heating rate on the thermal decomposition of lead dioxi-
de/Aleksandrov V, V., Boldyrev V. V., Marusin V. V. e. a.—
J. Therm. Anal., 1978, v. 13, p. 205—212.
438. Аввакумов E. Г., Косова II. В., Александров В. В. Влияние меха-
нической активации на разложение диоксида свинца.— Изв. СО
АН СССР, 1983, № 7. Сер. хим. наук, вып. 3, с.-25—30.
439. The powder diffraction file. American Society for testing and mate-
rials (ASTM).— Philadelphia, 1963—1984.
440. Заславский А. И., Толкачев С. С. Структура а-моднфнкацпи дву-
окиси свинца.— Журн. физ. химии, 1952, т. 26, вып. 5, с. 743—
752.
441. Eyring L. В., Icnng Так Tai. The structural chemistry of some
complex oxides: ordered and disordered extended defects.— In:
Treatise on solid state chemistry. Vol. 3. Ciystallme and noncrystal
line solids.—N. Y. —L„ 1979, p. 167—252.
442. Shafer 11., Bov R. The polviuorpiiisin of Nb2Oft.— Z. Kristallogr.
1958, Bd 110,’S. 241—248.’
443. Anderson I. S., Hutchison .1. L., Browne J. M. Ordering of the point
defects in non-stoichioiiielry crystals of Nbj.,0.,,.— Acta crystallogr.
1976, v. A32, N 4, p. 670—671.
444. Anderson J. S.. Hutchison .1. L., Lincoln F. J. Dislocations and re
lated defects in niobium oxide structures.— Proc. Roy. Soc. L.
1977, v. A352, p. 303—323.
285
445. Ланицкпп А. В., Симаков JO. В., Ярембат Е. И. О некоторых
свойствах пятиокпсп ниобия. /Кури. физ. химии, 11)53, т. 20,
с. 56 59.
446. О фазовых превращениях Т —i\b2O5 и II -Nb.2O5 при ударном
сжатпн/Ададуров Г. Л., Бреусов О. II., Дремин А. II. и др.1--
Фнзнка горения и взрыва. 1971, т. 7, с. 589—594.
447. Tamura S. IIigh-рге.ч.чнге phase research on Nb2O,,.— ,1. Mater. Sci.,
1972, v. 7, p. 298—302.
448. Дефектообразованпе в простых оксидах под влиянием механи-
ческой активации./ Аввакумов Е. Г., Ануфриенко В. Ф., Полу-
бояров Л. Л., Восель С. В. —Тезисы докладов IV' Всесоюзного
совещания по химии твердого тела. —Свердловск, 1985, с. 3.
449. Frevel J. К., Kinn С. N. Powder diffraction standards Гог niobium
pentoxide and tantalum pentoxide.— Anal. Chem., 1955, v. 27,
p. 1329—1330.
450. Zaeharisen W. M. Crystal chemical studies of the 5lh-series of ele-
ments. 1. New structure types.— Acta crystallogr., 1948, v. 1,
p. 265—268.
451. Wadsley A. 1). Crystallographic shear in the niobium oxides and
oxides fluorides in the composition region MX , 2,4 < x < 2,1.—
Perspective struct. Chem., 1970, v. 3, p. 1—58.
452. Anderson J. S., Browne J. M., Hutchison J. L. Electron microscopy
of niobium oxides. 1. Twinning and delects in 11—Nb2O5.— J. Solid
Stale Chem., 1972, v. 5, p. 419—431.
453. Бреусов О. II., Бробышев В. II. К вопросу образования метаста-
бильиы.х фаз переменного состава при ударном сжатии.— /Нури,
неорг. хил........, 1979, т. 24, е. 3128—3131.
454. Albrecht К., Hausler И., Mobius К. Beitrag znr Tribochemic des
Wolfram (V'I)-Oxides.— Z. anorg. allg. Chem., 1970, Bd 377,
S. 310—315.
455. Kiss A. B. Vibration fregnencies of WO3_ (1 > x > 0) reduced
tungsten oxides.— Acta Techn. Acad. Sci. Hung., 1974, v. 78,
N 3/4, p. 293—308.
456. Термодинамические свойства неорганических веществ. Снравоч-
ипк/Верятнн У. Д., Машпров В. В., Рябцев Н. Г. п др.— М.:
Атомпздат, 1965.
457. Ihuchi Е., Tilley J. D. The elastic strain energy-of crystallographic
shear planes in KeO3-relaled oxides. I. The formation energy of
isolated CS-planes. If. CS-plano arrays.— J. Solid Stale Chem.,
1978, v. 24, p. 121 — 141.
458. Sahle W. Electron microscopy studies of Wl8O49 .— J. Solid Stale
Chem., 1982, v. 45, p. 324—342.
459. Квашнина Л. Б., Кривоглаз M. А. Мессбауэровские спектры в
кристаллах, содержащих дефекты.— Физ. металлов и металлове-
дение, 1967, т. 23, с. 3—14.
460. Stockler Н. S., Sano If., Herber В. II. Mossbauer-effect studies of
weak nuclear quadrupole interactions in lieSn.— J. Chem. Phys.,
1966, v. 45, p. 1182-1186.
461. Эффект Мессбауэра в тонконзмельчеппых порошках окислов же-
леза/Аввакумов Е. Г., Кречман А. <9., Маркс Т. Л. и др.— Нэп.
СО АН СССР, 1977, № 4. Сер. хим. наук, вын. 2; с. 3—8.
462. Some properties of supported small a-l,’e2O3 particles determined
with the Mossbaner idfccl/Kiindig W., Bommel H., Constabaris C.,
Lindquist It. II,— Phys, liov., 1966, v. 142, p. 327—333.
463. Van der Kraan A. M. Mossbauer effect studies of surface ions of
ultrafine a-Fe2O3 particles.— Phys. Stat. Solidi (a), 1973, v. 18,
N 1, p. 215—226.
286
464. Суздалев II. II. О суперпарамагпетпзме ультрамалых частиц антп-
ферромагпетиков.— Физ. твердого тела, 1970, т. 12, вып. -i,
с. 988—990.
465. Крупянскцй 1О. Ф., Суздалев И. II. Магнитные свопегва ультра-
малых частиц окиси железа.— /Кури, экспер. п теорет. физики,
1973, т. 65, выи. 4, с. 1715—1725.
466. Крупяпекии 10. Ф., Суздалев 11. II. Некоторые особенное 1 и маг-
нитных свойств малых частиц a-FeaOs. Физ. твердого села,
1975, т. 17, выи. 2, с. 588—590.
4G7. Возпюк П. О., Дубинин В. II. Магнитная структура ультрамалых
антиферромагнитных частиц a-Fe«O3.— Физ. твердого тела, 1973,
т. 15, вын. 6, с. 1897—1899.
468. Изучение методом ЭПР процесса внедрения попов .меди (II; в ре-
шетку TiOs прп механической активации'Восель С. Л., Помошип-
ков 3. Е., Полубояроп В. А. н др,- Кинетика и катализ. 1984,
т. 25, выи. 6, е. 1501—1504.
469. Малиновский В. А. К вопросу об окислении сульфидных руд в
процессе топкого измельчения.— Цветные .металлы. 1933. Д4 4,
с. 33—35.
470. Бочаров В. Л., Голиков Л. А.. Митрофанов С. II. Кинетика окисле-
ния сульфидных минералов при измельчении.— В кн.: Физико-
химические основы комплексной переработки руд Средней Азин.
Душанбе: Дошип, 1970, с. 235—239.
471. Молчанов В. II. Осадконакопление и свободный водород. - Докл.
ЛИ СССР, 1968, т. 182. с. 445 —448.
472. Л. с. 264360 (СССР). Способ получения водорода Молча-
нов В. И.— О. 11.. 1970, № 9.
473. Кулебакнн В. Г. Превращения сульфидов прп активировании.—
Новосибирск: Наука, 1983.— 208 с.
474. Исследование методом ИГР превращений сульфидов железа при
топком измельчении,Тюленев Г. В., Маркс Г. .1.. Кречман А. Ф.
и др.— Пзв. СО АН СССР, 1976, № 14. Сер. хпм. наук, вын. 6,
с. 21—24. J (
475. Гольданский В. II. Химические применения мессбауэровской
спектроскопии.— М.: Мир, 1970.— 502 с.
476. Исследование перехода феррогеля a-FeOOH в y-Fe2O3 в гидро-
термальных условиях методом гамма-резонансной спектроско-
пии/ Плачпнда А. С., Чертов В. М., Суздалев II. П. и др.— Тео-
рет. и экспер. химия, 1974, т. 10, № 4, с. 545—549.
477. Драгавцева Н. А., Аввакумов Е. Г., Матвеева II. II. Извлечение
платиновых металлов из растворов механически активированным
пирротином.— Цветные металлы, 1977, № 6, с. 21—23.
478. Извлечение платиновых металлов из растворов мехаппчееюг акти-
вированными сульфидами железа и никеля'Матвеева II. 11., Дра-
гавцева И. А., Антипов Н. II. п др.— Цветные металлы, 1977,
№ 12, с. 19—21.
479. Аввакумов Е. Г., Болдырев В. В., Кособудскнн II. Д. Механиче-
ская активация твердофазных реакций. Сообщ. I. О взаимодей-
ствии пирита с железом.— Пзв. СО АН СССР, 1972, № 9. Сер.
хпм. паук, вып. 4, с. 45—50.
480. Исследование влияния механической активации минерального
сырья па скорость его обжига Колобердпп В. 1!.. Ражев В. М.,
Путников Н. А. и др.— Пзв. СО АН СССР. 1983, № 14. Сер. хпм.
наук, вып. 6, с. 42—45.
481. Влияние механической активации па процессы возгонки, окисле-
ния и восстановления сульфидных материалов/Векелер С. Ф.,
287
Скоков Г. Ф., Цптовпч Н. В. п др.— В кн.: Тезисы докладов
Всесоюзного совещания «Мехапохимии неорганических ве-
ществ», 26—29 окт. 1982 г. Новосибирск, 1982, с. 116—118.
482. Смагунов В. Н., Рейнгольд Б. Б., Молчанов В. И. Об особенностях
окисления пирита, активированного сверхтонким измельчением.—
}Курн. прпкл. химии, 1976, т. 49, с. 2339—2341.
483. Неустроев В. И., Иаионченко С. С., Худяков И. Ф. Автоклавное
выщелачивание механически активированного халькопирнтного
концентрата.— Изв. вузов. Цветная металлургия, 1982, № 3,
с. 11—15.
484. Gock Е. Beeinflussung ties Loseverhaltens von Kupfcrkies durch
Festkorperreactionen bei der Schwiiigmahlung.— Erzmetall./1978,
Bd 31, S. 262—268.
485. Ткачова К., Балаж П. Процесс выщелачивании механически ак-
тивированного халькопирита.— Banicke lisly (Minioriadne cislo),
Bratislava: VEDA, 1980, s. 235—240.
486. Бьерлинг Г., Лесидреиский П. Активация сульфидных минералов
для гидрометаллургической обработки.— В кн.: Труды VIII Меж-
дународного конгресса ио обогащению полезных ископаемых.
Е-2. Л., 1968, с. 1-8.
487. Рейнгольд Б. М., Смагунов В. II. Изучение механохимического
взаимодействия серебра с сульфидами в процессе планетарного
измельчения.— Изв. СО АН СССР, 1983, № 12. Сер. хим. паук,
вып. 5, с. 111—119.
488. Аввакумов Е. Г., Самарин О. И., Кулебакпп В. Г. Роль мехапохи-
мпческп.х реакций в переработке сульфидного сырья.— Изв. СО
АН СССР, 1981, № 7. Сер. хим. паук, нып. 3, с. 29—35.
489. Белов Н. В. Минералогия и периодический закон.— Вести. АП
СССР, 1976, № 2, с. 105—114.
490. Кребс Г. Основы кристаллохимии неорганических соединений.—
М.: Мир, 1971.— 304 с.
491. Верма А., Кришна П. Полиморфизм и политипизм в кристал-
лах.— М.: Мир, 1969.— 274 с.
492. Белов II. В. Структура ионных кристаллов п металлических
фаз,— М.: Изд-во АН СССР, 1947,— 237 с.
493. Vand V. Polytypism arising from screw dislocations.— Nature,
1953, v. 168, p. 1951.
494. Патнне А., Мак-Коннелн Дж. Основные черты поведения минера-
лов,— М.: Мир, 1983.— 304 с.
495. Воган Д., Крейг Дж. Химия сульфидных минералов.— М.: Мир,
1981,— 436 с.
496. Ward J. С. The structure and properties of some iron sulphides.—
Rev. Pure Appl. Chem., 1970, v. 20, p. 175—206.
497. Bertaut E. F. Contribution a 1'etude des structures lacunaires: la
pyrrhotine.— Acta cryslallogr., 1953, v. 6, p. 557—561.
498. Carpender В. 11., Desborongh G. A. Range in solid solution and
structure of naturally occuring troilite and pyrrhotite.— Amer.
Mineralogist, 1964, v. 49, p. 1350—1365.
499. Superstructure and honstoichiometry of the intermediate pyrrhoti-
te/Morimoto N., Tsukuma K. c. a.— Amer. Mineralogist, 1975,
v. 60, p. 240— 248.
500. Pyrrhotites: stoichiometric compounds with composition Fen_lSn ,
(n 8)/Morimoto N., Nakazawa II., Nisliiguchi K.— Science,
1970, v. 168, p. 964—966.
501. Nakazawa II., Morimoto N. Phase relations and superstructures ol
pyrrhotite Fe1_xS.— Mater. Res. Bull., 1971, v. 6, p. 345—358.
i
288
502. Tokonami M., Nishiguehi К., Morimoto N. Crystal structure of a
monoclinic pyrrhotite Fe-S».— Amer. Mineralogist, 1972, v. 57,
p. 1066—1081.
503. Влияние нестехиометрпп синтезированного пирротина на ere раст-
ворение в сернокислых растворах.— Цветные металлы, 1973,!
№ 12, с. 23—24.
504. Эитальиня образования сульфидов железа FeS1+x/Apun С. М.,
Морозова М. П., Столярова Т. А. и др.— 7Курн. фпз. химии, 1966,
т. 40, № 7, с. 1604—1607.
505. Исследование структурных изменении в механически активиро-
ванном пирротине методом ЯГРС/Павлюхпн 1О. Т., Авваку-
мов Е. Г., Садыков Р. Ш. и др.— В кн.: Тезисы докладов Всесо-
юзного совещания «Мсханохимия неорганических веществ», 26—
29 окт. 1982 г. Новосибирск, 1982, с. 103—105.
506. Levinson L. М., Treves D. Mossbauer study of the magnetic structure
of Fe7Sg.— J. Phys. Chem. Solids, 1968, v. 29, p. 2227—2231.
507. Mfissbauer effect in FeS1+x system/Gouciiarov G. N., Ostane-
vich Y. M., Tomilov S. B. e. n.— Phys. Stat. Solidi, 1970, v. 37,
p. 141 — 150.
508. Vaughan D. J., Tossel J. A. Magnetic transitions observed in sulfi-
de minerals at elevated pressures and their geological significance.—
Science, 1973, v. 179, p. 375—377.
509. Hiller Joh.-E., Probstain K. Thermische und rontgenographische
Untersuchungen an Kupfcrkies.— Z. Kristallogr., 1956, Bd 108,
S. 108-129.
510. Исследование структурных изменений в механически активиро-
ванном халькопирите методом ЯГРС/Болдырев В. В., Ткачова К.,
Паплюхин 10. Т. и др.— Докл. АН СССР, 1983, т. 273, .V 3,
с. 643—646.
511. Исследование структурных изменении в механически и термиче-
ски активированном халькопирите методом ЯГРС/Ткачова К.,
Болдырев В. В., ПавлюхннЮ. Т. и др.— Изв. СО АИ СССР, 1983,
№ 4. Сер. хим. наук, вып. 2, с. 9—13.
512. Piekoszewski I., Suwalski J., Lugenza S. Mossbauer effect study in
chalcopyrite.— Phys. Stat. Solidi, 1969, v. 29, p. K99 — K101.
513. Goodenongh J. B., Fatseas G. A. Mossbauer ?eFe isomer shift as a
measure valence in mixed-valence iron sulfides.— Solid State
Chem., 1982, v. 41, p. 1—2.
514. Wilchinsky Z. W., Looney R. W., Torngvist E. G. Dependence of
polymerisation activity on particle and crystallite dimensions in
ball milled TiCl3 and TiCl3-0,33AlCl3 catalyst components.— J. Ca-
talists, 1973, v. 128, p. 351—367.
515. Natta G., Corradini P., Allegra G. The different crystalline modifi-
cations of TiCl3, a catalyst component for the polymerization of
a-olefins.— J. Polymer Sci., 1961, v. 51, p. 399—410.
516. Kashiwa N. Super active catalyst for (olefin polymerization.—
J. Polym. Sci.,» 1980, v. 12, N 9, p. 603—608.
517. Schrader R., Oese W. Uber die Gewiuming von Fluorwasserstoff
aus mechanisch aktiviertein Calciunifluorid.— Chem. Teclin., 1971,
N 12, S. 741-745.
518. О механической активации апатита и апатптсодержащих пород/
Болдырев В. В., Колосов А. С., Чайкина М. В., Аввакумов Е. Г.—
Докл. АН СССР, 1977, т. 233, № 5, с. 892 —895.
519. Grohn И., Paudert R., Bisinger И. I. Uber die mechanische Anreguug
einiger cheinischer Reaclioneii anorgauisclier Feslsloffe.— Z. Che-
mie, 1962, Bd 2, S. 88—90.
520. Диссоциация карбонатов в процессе тонкого нзмельченпя/Мол-
чанов В. И., Гордеева В. 11., Корнева Т. А. и др.— В кн.: Меха-
нохимнческие явления при сверхтонком измельчении. Новоси-
бирск, 1971, с. 155- 161.
521. lleiiiieke G., Sigrisl К. 1)а.ч Iribocliemische Gloichwichl.— Monals-
ber. Otsch. Akail. Wiss. Berlin, 1969, Bd 11, S. 44—48.
522. Уракаев Ф. X., Сании цен 10. IL, Болдырев В. В. Изучение реак-
ций распада при раскалывании монокристаллов бромистого аммо-
ния.— В кн.: Совещание ио кинетике и механизму химических
реакций в твердом теле. Ч. 1. Новосибирск,, 1977, с. 125—127.
523. Khairetdinov Е. F., Boldyrev V. V., Burstein A. I. Mechanism of
charge transport in ammonium salts.— J. Solid State Chem., 1974,
v. 10, p. 288-301.
524. Беляев Л. M., Мартышев 10. 11., lOniiin IO. Я. Об электронных
процессах, сопровождающих механическое воздействие па поверх-
ность щелочио-галоидных кристаллов.— Acta Phys. Acad. Sci.
Hung., 1973, v. 33, p. 307—322.
525. Cornu A., Masset 11. Complication of mass spectral data.— L.: Ilej'-
den and Son Lind., 19G(>.
526. Larsen II. A., Driekainer II. G. Mechanical degradation and linking
ol polymers by plastic deformation.— .1. Phys, Chem., 1957, v. til,
N 9, p. 1249-1251.
527. Calvert P., Brown T. J., L’nlman D. R. Thermal effects of shear in
opposed-anvile high-pressure devices.— Amer. Mineralogist, 1969,
v. 54, p. 1732.
528. Tyler \V. \V. Plastic flow in alkali halide crystals.— Phys. Rev.,
1952, v. 86, p. 801—803.
529. Болдырев В. В., Аввакумов Е. Г., Гусев Г. М. Изменение скорости
мехапохп.мнческого разложения с помощью каталитических доба-
вок,— Докл. АН СССР, 1969, т. 184, № 1, с. 119—121.
530. Исследование мехаиохпмпческого разложении нитратов щелоч-
ных металлов/Болдырев В. В., Аввакумов Е. Г., Гусев Г. М. и
др,— В кн.: Мехаиохпмпческие явления при сверхтонком из-
мельчении. Новосибирск: Наука, 1972, с. 42—54.
531. О механизме механохпмпческого разложения галогенатов и пптра-
тов I и 11 групп Периодической спстемы/Болдыров В. В., Авваку-
мов Е. Г., Стругова Л. 11. и др.— В кп.: Механоэмпссня и меха-
нохпмпя твердых тел. Фрунзе, 1974, с. 43—49.
532. Механохпмпческое разложение нитрата патрня/Аввакумов Е. Г.,
Болдырев В. В., Стругова Л. И. и др.— Изв. СО АП СССР, 1971,
№ 9. Сер. хим. наук, вып. 4, с. 122—124.
533. Zur tribochcniischen Zersetzung von Alkali Brotnalen und Nitra-
ten/Boldyrew W. W., Awwakumow E. G., llarcnz H. c. a.—
Z. anorg. allg. Chem., 1972, Bd 393, S. 152—158.
534. Chodakov G. S., Avvakumov E. G. Mcchanochemische Zersetzung
anorganischer Festkorper.— In: Fcstkdrpcrchemic. Leipzig: Verlag
fur Grundstoffindustrie, 1973, S. 544—559.
535. Бордюшкова E. А., Проценко П. И., Венеровская Л. И. О кине-
тике термической диссоциации и устойчивости расплавов нитра-
тов щелочных металлов.— Журн. прикл. химии, 1967, т. 40,
с. 1438—1441.
536. Шмидт И. В., Аввакумов Е. Г., Болдырев В. В. О низкотемпера-
турном термическом разложении нитрата натрия.— Пзв. СО АН
СССР, 1974, № 7. Сер. хим. паук, вып. 3, с. 18—22.
537. Свиридов В. В. Фотохимия и радиационная химия твердых неор-
ганических веществ. Ч. 1.— Минск; Высшая школа, 1970,—
463 с.
290
538. Doigan P., Davis T. W. TIip photolysis of crystalline nitriles.—
J. Phys. Cliciii., 1952, v. 56. p. 764—766.
539. Болдырев В. В., Зырко '). A.. Дерибас А. А, I» вопросу о раз.тоже-
iiini броматов и нитрагин щелочных металлов под дебсгнием удар-
ной волны.— Хим. высоких энергии, 1967, т. 1, с. 177—180.
540. Осипов О. Л. О зависимости между поверхностным натяжением,
энергией связи и ионными радиусами.— Докл. АП СССР, 1955,
т. 102, с. 1171 — 1175.
541. The (100) surfaces of alkali halides. 1. The air and vacuum cleaved
surfaces/Callon T. E., Higginbotham J. C., Prutton M. e. a.— Sur-
face Sci., 1970. v. 21, N 2A. p. 224—233.
542. Финкель E. M. Физические основы торможении разрушения.—
М.: Металлургия, 1977.— 309 с.
543. Ileinieke G.. Riedel R., Ilarenz П. Oxydalionsreaktionen durch
Jnipaklbearheiliiiig.— Z. Phys. Chem., 1964, Bd 227. S. 62—80.
544. Thiessen P., Ileinieke G., Sehober E. Zur tribocheinischen Vniset-
zung von Gold und Cl).j mil Hille radioakliver Markierling.—
Z. anorg. allg. Chem., 1970, Bd 377, S. 20—28.
515. Koster A. liber cine nene praparalive Methode zur Darslelluug
lliiclitiger Chloride.— Angew. Chem., 1957, Ed 69, S. 563.
546. Ileinieke G., Sigrist K. Chemische Umselzungen im System Xi—CO
bei tribomechanischer Beanspruchiing der feslcii Phase.— Z. anorg.
allg. Chem.. 1967, Bd 350. S. 148—159.
547. Ileinieke G., Sigrist K. Tribochemische Umselzuiigeii von MetaHen
mil Kohlenstoff und Oxiden des KohieiislolTs.— Monatsber.
Disch. Akad. Wiss., I960, Bd 8. S. 654—656.
518. Ileinieke G., Boek N. Melalformen ails der Gaspiiase.— Wissen-
schaft und h'orschrill. 1975, Bd 25. N 1, S. 10—15.
549. Trautz M. Der Exist enzbereich der Slol'fe, Hire kinetische Analyse
und die Besliiuiiuiiig von Danipldruckeu.— Z. anorg. allg. Chem..
1918, Bd 104, S. 169-212.
550. Heinicke G., Ilarenz II., Sigrist K. Zur Kinelik der Beaktion Ni -r
+ 4CO = Ni(CO)4 bei tribomechanischer Bearbeitung des Nickels.—
Z. anorg. allg. Chem., 1967, Bd 352, S. 168—189.
551. Ileinieke G., Harcnz II. Bildnng von Nickeltetracarbonyl an tribo-
mechanisch und chemisch regenerierten Nickeloberflachen.—
Z. phys. Chem., 1969, Bd 240, S. 325—338.
552. Ileinieke G., Bock N., Ilarenz II. Zum Mcchanisnius der tribomecha-
nisch aktivierten Metallcarbouylbildting unter Einllup schwefel-
halliger Substanz.— Z. anorg. allg. Chem., 1970, Bd 372, S. 162—
170.
553. Ileinieke G., Hoffman II. Zur Wechsehvirkuug von Gasen au tribo-
mechanisch bearbeilcten Nickeloberflachen.— Surface Sci., 197.1,
v. 24, p. 484-494.
554. Хойнике Г., Бок H. О трпбохпмпческом приготовлении карбонила
никеля и его техническом использовании при изготовлении фор-
мовочных инструментов.— В кп.: Материалы V Всесоюзного сим-
позиума по механоэмиссии и механохимпп твердых тел, окт. 1975.
Ч. II. Таллин, 1977, с. 39—77.
' 555. Л. с. 509539 (СССР). Способ получения карбонила вольфрама'Ав-
вакумов Е. Г., Гимаутдппов 10. В., Болдырев В. В.— О. И., 1976,
№ 13.
556. Аввакумов Е. Г., Гпмаутдинов IO. В., Болдырев В. В. Механб-
химпческие реакции окиси углерода с тугоплавкими металлами.—
В кп.: Материалы V Всесоюзного симпозиума по механохимпп и
мехапоэмпсепп твердых тел, окт. 1975. Ч. 11. Таллин, 1977, с. 52—
55.
291
557. Колбанев И. В., Бутлгпи П. ТО. Изучение процесса диспергиро-
вания кварца методом ЭПР.— В кп.: Мехапозмпссия и мехапохи-
мия твердых тел. Фрунзе: Илим, 1971, с. 215—218.
558. Arias A. Metal powder reactions in ball milling.— Ind. Eng. Chem.
Prod. Res. Develop., 1976, v. 15, N 2, p. 142—152.
559. Корнеев II. H. Химия и технология алюмянийорганических со-
единений.— М.: Химия, 1979.— 256 с.
560. Mechanically initialed reaction of aluminium with alkyl halides/
Mori S., Kuriyana O., Maki Y., Tainai Y.— Z. anorg. allg. Chem.,
1983, Bd 492, S. 201-207.
561. Grohn II., Paudert R. Mechanochemische Reaktionen von Eleinen-
ten der IV. Hauptgruppe mit einigen organischen Verbindungen.—
Z. Chem., 1963, N 3, S. 89.
562. Grohn II., Paudert R. Uber die Unisetzurig von Silicium und Tetra-
clorhohlenstoff unter Schwinginahlung.— Chem. Techn., 1958,
Bd 10, S. 307.
563. Вигдорчнк E. M., Шейнин А. Б. Математическое моделирование
непрерывных процессов растворения.— М.: Химия, 1971.— 235 с.
564. Зеликмаи А. Н., Больцман Г. М., Белявская Л. В. Теория гидро-
металлургических процессов.— М.: Металлургия, 1975.— 504 с.
565. Меерсон Г. А., Хавский II. II. Изучение путей усовершенствования
кислотного метода переработки шеелита.— Цветные ' металлы,
1957, № 3, с. 41.
566. Albrecht R., Blechschinidt U., Mobius II. Einflufi der inechanischen
Aktivierung von Wolframerzen beim Aufschlufi mit Soda.— Chem.
Techn., 1971, Bd 23, N 8, S. 473—475.
567. Колбанев И. В., Бутягнн II. Ю.'Механохпмическпе реакции крем-
ния с водой.— Кинетика и катализ, 1982, т. 23, вып. 2, с. 327—
333.
568. Самарин О. И., Аввакумов Е. Г. Исследование кинетики механо-
хи.мпческой реакции сульфидов железа с растворами солей цин-
ка.— В кн.: Тезисы Всесоюзного совещания «Мехапохпмпя не-
органических веществ», 26—29 окт. 1982 г. Новосибирск, 1982,
с. 106—108.
569. Jander W. Reactionen im festen Zustande bei hiiheren Temperatu-
ren. 1. Mitteilung. Reaktiongeschwindigkeiten endotherm verlau-
fender Umsetzungen.— Z. anorg. allg. Chem., 1927, Bd 163, S. 1—
30.
570. Diinwald II., Wagner C. Methodik dor Messung von Diffusionsge-
schwindigkeiten bei Losungsvorgangen von Gasen in festen Phasen.—
Z. Phys. Chem., 1934, Bd 24, S. 53—59.
571. Serin B., Elickson R. T. Determination of diffusion coefficients.—
J. Chem. Phys., 1941, v. 9, N 10, p. 742—747.
572. Гпнстлпнг A. M., Броунштейн В. И. О диффузионной кинетике
реакций в сферических частицах.— Журн. прикл. химии, 1950,
т. 23, с. 1249—1256.
573. Carter R. Е. Kinetic model for solid-state reactions.— J. Chem.
Phys., 1961, v. 34, N 6, p. 2010—2015.
574. Valenci G. Cin'etiquo de e'oxydation do sphe'rules et de poudres
me'talliques.— С. r. Acad. Sci., 1936, v. 202, p. 309—312.
575. Komatsy W., Uemura T. Kinetic equations of solid state reactions
for counterdiffusion systems.— Z. Phys. Chem. (N. F.), 1970, Bd 72,
S. 59—75.
576. Kohoiiiok И. Ф. Об одной модели твердофазных реакций п смесях
порошков.— Жури. физ. химии, 1973, т. 47( с. 526—531.
577. Wagner C. Uber den Mechauisnius der Bildung von lonenverbindun-
gen hoherer Ordnung (Dopelsolze, Spinelie, Silikatc).— Z. Phys.
Chem., 1936, Bd 34, S. 309—316.
578. Iledvall I. A., Loffler L. Uber den EinfluP von Ubergangszustanden
aus die Bildungsgeschwindigkeit des Kobaltspihells aus festen
Oxyden.— Z. anorg. allg. Chem., 1937, Bd 234, S. 233—238.
579. Evans M. G., Polanyi M. Some applications of the transition state
method to the calculation of reaction velocities, especially in solu-
tion.— Trans. Faraday Soc., 1935, v. 31, p. 875—894.
580. Познн M. E., Гинстлннг A. M., Печковскнй В. В. О роли газовой
среды в реакциях между твердыми веществами.— Журн. нрпКл.
химии, 1954, т. 27, с. 376—380.
581. Grohn II., Paudert R., Friedrich II. Uber die mechanochemische
Redukliou einiger Melalloxide durch Kohlenstoff.— Z. anorg. allg.
Chem., 1966, Bd 348, S. 21—27.
582. Механическая активация твердофазных реакций. Сообщ. 4. Твер-
дофазное восстановление кассптерпта/Аввакумов Е. Г., Дьяко-
ва В. Е., Стругова Л. И. и др.— Пзв. СО АН СССР, 1974, As 2.
Сер. хим. наук, выи. 1, с. 26—29.
583. Механическая активация твердофазных реакций. Сообщ. 5. Изу-
чение твердофазного восстановления двуокиси олова методом
ЯГР/Варпек В. А., Аввакумов Е. Г., Стругова Л. II. и др.— Там
же, с. 29—33.
584. Аввакумов Е. Г., Стругова Л. II. Механическая активация твердо-
фазных реакций. Сообщ. 6. О применении уравнении бездиффу-
зиониой кинетики к механохимическпм реакциям в смесях твер-
дых веществ.— Там же, с. 34—38.
585. О механизме реакции механохимнческого восстановления дву-
окиси олова кремнием/Бутягин П. 10., Аввакумов Е. Г., Струго-
ва Л. И. и др.— Журн. физ. химии, 1974, № 12, с. 3009—3012.
586. Аввакумов Е. Г., Матыцнн Л. И., Ставер А. М. Твердофазное
восстановление SnO2 при ударном сжатии п механической актива-
ции.— Физ. горения и взрыва, 1975, -№ 6, с. 922—927.
587. Feher G. Electron spin resonance experiments on donor in silicon.
1. Electronic structure of donors by nuclear double resonance techni-
que.— Phys. Rev., 1959, v. 114, p. 1219—1244.
588. Варнек В. А., Аввакумов E. Г., Разворотнева Л. И. Исследование
методом Я1Р механохимнческого взаимодействия олова с теллу-
ром, селеном, серой.— Изв. СО АП СССР, 1979, № 9. Сер. хим.
наук, вып. 4, с. 20—24.
589. Patent 3723092 (USA). Composite metal powder and production
thereof/Benjamin J. S.— March 27, 1973, Official Gazette, 1973,
v. 908, N 5.
590. Black S. A. Development of supercorroding alloys for use as timed
releases for Ocean engineering applications.— Civil Eng. Lab.
(navy), Port Hneneine, 1979, p. 40.
591. Образование твердых растворов в системе Fe—Сг под влиянием
механической активацпп/Павлюхнн 10. Т., Манзанов Ю. Г.,
Аввакумов Е. Г. и др.— Пзв. СО АН СССР, 1981, № 14. Сер. хим.
наук, выи. 6, с. 84—89.
592. Химические реакции прп диспергировании твердых тел/Уп-
бо Л. Я., Паз А. Я., Мюйрсепп Т. К. и др.— В кп.: Материалы
V Всесоюзного симпозиума по мехапоэмпсспн и мехаиохимпи
твердых тел, окт. 1975. Ч. 11. Таллии, 1977, с. 15-25.
593. Pai Verneher V. R., Seetharaiuatharyulu D., Mallya R. M. Mecha-
noformation of ammonium perchlorale-potassium perchlorate solid
solutions.— J. Solid State Chem., 1981, v, 39, p. 154—160.
594. Варнек В. Л., Стругова JT. II., Аввакумов Е. Г. Магнитная струк-
тура частиц EeSn2, полученных при твердофазном взаимодействии
олова н железа.— Физ. твердого тела, 1974, т. 16, с. 1819—1818.
595. Изменение магнитных свойств PeSn2 при сверхтонком пзмельче-
инн/Варнск В. Л., Занижений В. Д., Аввакумов Е. Г. и др.— Изи.
СО ЛИ СССР, 1976, № 14. Сер. .хпм. паук, вып. (>, с. 17—20.
596. Неверов В. В., Буров В. II., Коротков А. И. Особенности диффу-
зионных процессов в пластически деформируемой смеси цинка и
меди.— Физ. металлов и металловедение, 1978, т. 46, вып. 5,
с. 978—983.
597. Неверов В. В., Буров В. II. Условии образования соединений при
механической активации.— Изв. СО All СССР, 1979, № 9. Сер.
хпм. паук, вып. 4, с. 3—8.
598. Мехапохпмпчсская активация апатита и его растворпмость/Чап-
кдна М. В., Колосов А. С., Аввакумов Е. Г. и др.— Изв. СО ЛИ
СССР, 1978, № 4. Сер. хпм. паук, вып. 2, с. 52—59.
599. Рентгенографическое исследование распределения солеи тина
«ионных кристаллов» и порах дисперсных носителей/Гусев В. Л.,
Гагарина В. А., Мороз Э. М. и др.— Кпнетпца и катализ, 1976,
т. 17, вып. 2, с. 500—507.
600. Болдырев В. В., Еремеева Ю. Е. Изучение скорости реакции подп-
дцв щелочных металлов с солями свинца при растирании.— Учен,
зап. Томск, ун-та, 1959, № 29, с. 27—30.
601. Уракаев Ф. X., Аввакумов Е. Г. О механизме механохимических
реакций в диспергирующих аппаратах.— Изв. СО АП СССР,
1978, № 7. Сер. хпм. паук, пып. 3, с. 18—23. t
602. Clasen II. Alanl-Synthese ans den Elemeiilen nnd ihre Bedeutung.—'
Angew. Chem., 1961, Bd 73, S. 322—331.
603. Захаркин Л. И., Гавриленко В. В. Взаимные переходы в ряду
алю.могидрпдов лития, натрии и калии.— Изв. ЛИ СССР. Сер.
хим., 1962, Л» 7, с. 1146—1150.
604. Волков В. В., Мякпшев К. Г., Югов С. И. Изучение синтеза тетра-
гпдробората циркония реакцией хлорида циркония с тетрагпдро-
боратом лития,— Журн. прпкл. химии, 1975, т. 48, с. 2109.
605. Волков В. В., Мякпшев К. Г. Синтез тетрагпдробората урана
(IV) обменной реакцией хлорида урана (IV) с тетрагидроборатамп
щелочных металлов.— Радиохимия, 1976, № 4, с. 512—513.
606. Волков В. В., Мякпшев К. Г. Синтез и исследование свойств тет-
рагидробората Т (III).— Изв. СО АН СССР, 1977, № 2. Сер. хпм.
наук, вып 1, с. 77—81
607. Волков В. В., Мякпшев К. Г., Горбачева II. II. О мехаиохпмиче-
скоп реакции тетрагпдроборатов щелочных металлов с двухва-
лентным оловом.— Изв. АН СССР. Сер. хим., 1983, № 6, с. 1442—
1447.
608. Механохимические превращения Na2P3O10, инициированные со-
единениями меди/Продап Е. А., Павлюченко М М., Песляк Г. В.
и др._ Докл. АН БССР, 1975, т. 19, № 9, с. 796-799.
609. Мнрсапдов У., Курбонбекон А., Хпкматов М. Получение п неко-
торые свойства боргидрпдов лантана и церия.— Журн. неорган.
химии, 1982, т. 27, № 9, с. 2436—2439.
610. Волков В. В., Пухов А. Л., Мякпшев К. Г. Получение боразина
реакцией тетрагпдробората натрия с хлоридом аммония после
предварительной механической активации реакционной смеси,—
Изв. СО АН СССР, 1983, № 7. Сер. хпм. наук, вып. 3, с. 116—123.
611. Будников П. П., Гппстлипг А. М. Реакции в смесях твердых
веществ.— М.: Стройпздат, 1965.— 473 с.
294
012. Логвиненко А. Т., Савиикипа М. Л.. Татаринцева М. II. Исследо-
вание свош-тп высокодпсиерспых GaO и SiO . - Изи. 10 ЛИ
СССР, 1973, 2. Сер. хим. наук, вып. 1. с. 121 —134.
613. Логвиненко А. Т., Савинкииа М. А. Свойства буроугольной золы
сверхтонкого диспергирования.— В кн.: Механохимические явле-
ния прн сверхтонком измельчения.— Новосибирск: Наука. 1971,
с. 79—85.
614. Particular crystalline deled, nou-sloichoimetry and activation of
barium metalitanate BaTiO3 of obtained by solid way synthesis/
/Balllo F., Cleval B., Mabie S. e. a.— Extended Abstracts of 10th
Intern. Symp. Reactiv. of Solids, Dijon, 1984, p. 348—349.
615. Копылов А. В., Аввакумов E. Г., Уракаев Ф. X. Механохимпче-
сное взаимодействие карбоната бария с окислами элементов IV,
V и VI групп Периодической системы элементов,— Изв. СО All
СССР, 1979, № 9. Сер. хим. паук, вып. 4, с. 45—50.
616. Albrecht R., Hausler И., Mobius 11. Darstellung von Bariumwolfra-
inat dnrch tribochemische Reaktion.— Z. anorg. allg. Chem., 1971,
Bd 384, S. 211-220.
617. Hausler II., Mobius It., Nau P. -E. Darstellung von Magnesium- und
Berylliumwolfrainat (lurch tribochemische Reaktion.— Z. anorg.
allg. Chem., 1971, Bd 386, S. 270—276.
618. Albrecht R., Mobius R. Bildung von Alkalimetallwolframaten dnrch
tribochemische Reaktion.— Z. Chem., 1972, N 11, S. 421—422.
619. Albrecht R., Hausler II., Mobius R. Darslellnng von Calcium- und
Slroiiliiimwolframat dutch tribochemische Reaktion.— Z. anorg.
allg. Chem., 1971, Bd 382, S. 177—187.
620. Konialsu W. Kinetics of solid-state reactions.— Proc. IV. Intern.
Symp. on Reactivity of Solids (Amsterdam), 1960, p. 182—186.
621. Ляхов II. 3., Болдырев В. В. Кинетика мехапохимнческпх реак-
ций. - Нан. Ct) ЛИ СССР, 1982, .V 12. Сер. хпм. паук. пып. 5,
с. 3 9.
622. Аввакумов Е. Г., Уракаев Ф. X. Кинетика твердофазных механо-
химических реакций в зависимости от условий механической об-
работки.— В кн.: Кинетика п механизм химических реакций в
твердой фазе,— Кемерово, 1982, с. 3—12.
623. Аввакумов Е. Г., Уракаев Ф. X., Татаринцева М. II. О двух режи-
мах протекания твердофазных .механохимических реакций в за-
висимости от условий диспергирования.— Кинетика и катализ,
1983, т. 24, вып. 1, с. 227 — 229.
624. Аввакумов Е. Г., Багрянцев Г. II., Волков В. В. Кинетика меха-
нохпмпческого взаимодействия тетрагпдробората натрия с хлори-
стым аммонием.— В кн.: Материалы V Всесоюзного симпозиума
по мохаиоэмисспп и мехапохимии твердых тел. Ч. II. Таллин,
1977, с. 26-32.
625. Механохпмнчоский синтез фтор- п х.торапатпгои/Чайкпиа М. В.,
Шапкин В. J1., Колосов Л. С. и др.— Изв. СО АН СССР, 1978,
№ 7. Сер. хпм. паук, вып. 3, с. 96—101.
626. Ткаченко В. А., Летюк Л. М., Башкиров Л. А. Об особенностях
механизма образования феррита в условиях термовибропомола.—
Пап. СО АН СССР, 1983, № 14. Сер. хпм. наук, вып. 6, с. 39—42.
627. Wakita II., Kinoshita S. Thermal decoinposition of lanthanum car-
bonate.— Bui. Chem. Soc. Jap., 1979, v. 52, N 2, p. 428.
628. Евдокимов А. А., Трунов В. К. К исследованию некоторых воль-
фраматных систем.— Жури, пеорг. химии, 1973, т. 18, вин. 10,
е. 2834—2840.
629. Механо.хнмнческпй синтез в многокомпонентных системах. Синтез
барий-лантанового вольфрамата ВаЕа4(\УО4)7/Уракаев Ф. X.,
295
Аввакумов Е. Г., Чумаченко Ю. В., Болдырев В. В,— Пзв. СО
АН СССР, 1985, № 17. Сер. хим. паук, вып. 6, с. 21- -28.
630. Влияние механической и термической обработки солей на их
структуру и закономерность протекания твердофазных реакций/
/Вержбицкий Ф. Р., Голдобина В. И., Зеленина И. В. и др.—
В кн.: Термический анализ и фазовые равновесия. Пермь, 1984,
с. 63—70.
631. Смирнов А. Е., Урусовская А. А., Регель В. Р. Мехапохимпческип
эффект в кристаллах NaCl.— Докл. All СССР, 1985, т. 280, № 5,
с. 1122—1124.
632. Займан Д;к. Модели беспорядка. Теоретическая физика однород-
но неупорядоченных систем.— М.: Мир, 1982.— 592 с.
633. Еремин А. Ф., Гольдберг Е. Л. Механическая активация фторпда
натрия. I. Заполнение каналов аккумулирования энергии.— Пзв.
СО АН СССР, 1985, № 17. Сер. хим. наук, вып. 6.
634. Гольдберг Е. Л., Рыков А. И., Еремин А. Ф. Механическая акти-
вация фторпда натрия. П. Дислокационная структура активиро-
ванного NaE.— Там же.
635. Еремин А. Ф., Гольдберг Е. Л., Павлов С. В. Механическая ак-
тивация фторида натрия. Ш. Особенности растворения активи-
рованного NaF.— Там же.
636. Гольдберг Е. Л., Еремин А. Ф. Механическая активация фторида
натрия. IV. Баланс аккумулированной энергии.— Там же.
637. Уракаев Ф. X., Гольдберг Е. Л., Еремин А. Ф., Павлов С. В. Ме-
ханическая активация фторида патрия. V. Критерии для описа-
ния скорости растворения активированного NaF в этаноле.—
Там же.
638. Blanchin М. G., Bursill L. A., Smith D. J. Extended defects in
deformed rutile.— Phys. Stat. Solidi (a), 1985, v. 89, N 2, p. 559—570.
639. Bursill L. A., Blanchin M. G. Structure of small oxygen vacancy
defects in nonsloichiometric rutile.— J. Solid Slate Chem., 1984,
v. 51, p. 321—355.
640. Завьялов С. А., Мясников И. А., Завьялова Л. M. Роль структур-
но-химических превращений па поверх пости твердых тел в обра-
зовании синглетного кислорода и его участие в каталитическом
окислении нафталина.— Докл. АП СССР, 1985, т. 284, № 2,
с. 378—381.
----------- ИМЕННОЙ УКАЗАТЕЛЬ -----------
Абдпкамалои Б. А. [333]
Аввакумов Е. Г. [30, 120, 211 —
213, 216, 224, 303, 325, 330,
376, 413, 414, 416, 426—429,
438, 448, 461, 477, 479, 488,
505, 510, 511, 518, 529—532,
536, 555, 556, 568, 582—586,
588, 591, 594, 595, 598, 601,
615, 622—624, 629]
Августниник А. И. [116]
Аверичсва В. Е. [222]
Ададуров Г. А. [446]
Акунов В. И. [203]
Александров В. В. [413, 438]
Амелина Е. А. [81]
Андерсон Дж. С. [394]
Андерсон О. [140]
Анисимова В. И. [170]
Антипов II. И. [478]
Ануфриенко Е. Ф. [417, 448, 46S]
Апарнпков Г. Л. [246]
Арамисов Ф. А. [41]
Аргон А. [141]
Ария С, М. [504]
Архипенко Д. К. [371]
Аршинов В. II. [341]
Афанасьев Г. Т. [127]
Ахмед-Заде К. А. [264]
Багрянцев Г. И. [624]
Балаж II. [485]
Бантизмаискпн В. В. [264]
Барамбопм II. К. [13]
Барзаковский В. П. [409]
Бацапов С. С. [26]
Бацуев А. А. [391]
Башкиров Л. А. [626]
Бевз В. А. [364]
Белов II. В. [489, 492]
Белоцерковский Г. М. [384, 385]
Белинская Л. В. 1564]
Беляев Л. М. 1180, 524]
Беляев И. М. [136]
Бенсон Г. [307]
Берестецкая II. В. [271, 274,
276, 292]
Берлин А. Л. [12, 256, 288]
Бернхард К. [52]
Бертенов Г. М. [247, 255]
Берштейн В. А. [240]
Блипичев В. И. [107, 204]
Бобков С. П. [107, 204]
Боболев В. К. [127]
Бобышев А. А. [293, 435) •
Бок Н.— [554]
Болдырев В. В. [29—33, 47—
49, 143, 211, 216, 229—23’1,
303, 325, 376, 414, 421, 479,
510, 511, 518, 522, 529-532,
536, 539, 555, 556, 600, 621, 629]
Болховитинов Л. Г. [131, 132]
Бондарь II. А. [4091
Бордюшкова Е. А. [535]
Боудон Ф. П. [122]
Бочаров В. А. [470]
Браун М. [86]
Бредихин С. И. [333]
Бредов М. М. [168]
Бреусов О. II. [25, 446, 453]
Бриджмен П. В. [15, 16]
Бробышев В. II. [453]
Броунштейн В. II. [572]
Бубен М. Я. [433]
Бугаев А. А. [395]
Будников П. П. [611]
Бурдипа К. П. [234]
Буров В. Н. [596 , 597]
Бутягин П. Ю. [34, 35, 179, 181,
253, 255, 259. 271—282, 288,
292, 304, 557, 567, 585]
Бушуев Л. П. [207, 208]
Буянова Н. Е. [218]
Быстрпков А. В. [265, 271, 273—
275, 281, 282]
Бьерлинг Г. [486]
Вайнштейн Б. К. [348]
Ван Бюрен [109]
Варпек В. А. [426—428, 583, 588,
594, 595]
Векслер С. Ф. [481]
Венеровская Л. И. [535]
Верещагин Л. Ф. [234]
Вержбицкий Ф. Р. [630]
297
Веря.з Л. [4911
Веригин У. Д. [456 ]
Внгдорчпк Е. М. [563[
Виидергауз В. Ш. |116]
Вишняков Я. Д. [Ill J
Власов В. II. [166]
Власова М. В. [40, 291J
Воган Д. [495]
Волиюк П. О. [467]
Волков В. В. ]217, 604—607,
610, 624]
Вольдмап Г. М. [42, 564]
Воробейчик А. И. [325, 376, 414]
Восель С. А. [448, 468]
Воскресенский А. II. [8]
Гавриленко В. В. [603]
Гагарина В. А. [599]
Гаджиева Ф. С. [416, 417]
Галвей А. [86]
Галицин Ю. Г. [294]
Герасимов 10. М. [166]
Герцрикен С. Д. [147]
Гпйо Р. [90]
Гимаутдипов 10. В. [555, 556]
Гиистлинг А. М. [572, 580, 611]
Голдобина В. II. [6301
Голиков А. А. [470]
Голосов С. И. [205, 20G, 211,
377]
Гольданскнй В. II. [475] .
Гольдберг Е. Л. [633—637]
Гоникберг М. Г. [17]
Горбачева II. И. [607]
Горбачева Т. В. [314]
Гордеева В. И. [520]
Горловский И. А. [196]
Гоффман К. [174]
Гропянов В. М. [116]
Грузин П. Л. [154]
Гусев В. А. [599]
Гусев Г. М. [303, 377, 382, 529,
530]
Гутман Э. М. [39]
Гуюмджап П. П. [204]
Демидович Г. В. [287]
Дерибас А. А. [539]
Дерягин Б. В. [36—38, 170|
Динпик А. II. [137]
Дистлер Г. II. [166]
Драгавцева И. А. [477, 478]
Дремип A. II. [23, 446]
Дубинин В. II. |467[
Дубинская А. М. [253]
Дубнов А. В. [126]
Д billion В. Е. 15821
Дымчош.о II. II. |219|
Диткппа М. Е. [254|
Евдокимов А. А. [628]
Евдокимов В. Д. [172]
Екоборн Т. [73]
Еппколопян II. С. [18, 22]
Еремеева IO. Е. [600]
Еремяп А. Ф. [633—637]
Ермилов А. Г. [42]
Жаров А. А. [18, 22]
Жданов С. П. [193]
Жнгач К. <9. |106]
Жирнов Е. II. [209]
Житников П. П. [121]
Жорин В. А. [22, 24]
Журков С. II. [82]
Завьялова Л. М. [640]
Займам Дж. [82]
Займам Дж. [632]
Закревскпй В. А. [264]
Заможский В. Д. [595]
Зарко 3. А. [539]
Заславский А. 11. [440]
Захаркин Л. И. [603|
Захарчепя Б. П. [395]
Зеленина И. В. [630]
Зеликмап А. Ц. [41, 42, 564]
Земский С. В. [154]
Зозуля И. В. [342]
Золотовскнй Б. П. [386]
Зубова Е. В. [234, 235, 246]
Зырянов В. В. [421]
Иванько А. А. [117]
Ильин А. П. [374]
Инденбом В. Л. [3481
Исаков М. И. [9]
Исупов В. П. [387]
Пост Г. [294]
Киселев В. Ф. [286, 287]
Кпсснп JO. В. [23]
Клетепнк Ю. В. [2141
Клоцмап С. М. |162|
Клочков II. В. [107]
Клюев В. А. [170]
Кляровскпй В. М. |382]
Ковалева Л. Т. [3821
Ковалевский В. II. [434]
Козлов В. И. [210]
Козулин II. А. [196]
Козырев С. А. [210]
298
I io лбапев II. II. |259, 276, 279.
292, l>;>7, 567 J
Колмапс.оп A. 3. (256 , 288]
Колоберднн В. II. [480]
I {слои г P [393]
Колосов А. С. (95, 518, 598]
Копоток 11. >1>. ]57(>]
Конгорова Т. А. [71]
Копылов А. В. (615]
Корпела Т. А. (520]
Корнеев II II. [559]
Коротаева Л. А. [235]
Коротков А. И. 1596]
Косевпч А. М. [114]
Кособудскпй И. Д. [479]
Косова II. В. (413, 438]
Каказеп II Г. (40 291]
Калиновская И. А [183]
Капптапчук В. А. ]171 ]
Капустин В. М. [18]
Каракозов Э. С. [242]
Карнаухов А. П. [218]
Карпухин А. И. [391]
Квашнина Л. В. [459]
Кедрнпс.кпн II. А. [169]
Кириллова Е. А. [372]
Кириллов II. [1. [374]
Костюкевич М. В. (222]
Коттрел А. X. [72]
Коцуиало II. II. [387]
Кочегаров Г. Г. [185, 188, 189,
191, 195]
Красулпп 10. А. [158, 241]
Кребс Г. [490]
Крейг Дж. [495]
Кречман А Ф. [461 474]
Кривоглаз М. А. [459]
Крпвоиуцкая Л. II. [372]
Криворучко О. П. [386]
Кришна П. [491 ]
Кротова II А. [37, 3S, 298]
Крупяпскпй В. Г [465, 466]
Кулаков А П. [333]
Ку.тебакип В. Г. [473, 488]
Курбопбеков А. [609]
Кшемяпскаи II. 3. [168]
Ландау Л. Д. [138]
Лаппцкип А. В. 1445]
Лаптева Е. С. [43]
Лопухина Е. С. [380]
Лариков Л. II. |156. 1571
Ларионов II. Г. |217|
Небеден Я С. [433]
Лепин А. А. |254]
Лесндрепский II. [4S6]
Легюк Л. М. [626]
.'hi Вон Гнои |42l’>|
Линке J. [174, 296]
Лпнскпи В. А. [24]
Лифшиц Е. М. [138]
Лифшиц Л. Д. [345]
Лихтмап В И. [192]
Логвиненко А. 'Г. [216, 360, 612,
613]
Логвиненко В. А. [360]
Логвиненко Д. Д. (201 ]
Лущейкпп Г А. [295]
Ляхов II. 3. [33, 143, 233, 421,
621]
Мазалов Л. II. [427]
Мазапко В Ф. |156, 157]
Маноров Л В. [163]
Мак-Коппе иг Дж. [494]
Маклпиток <1>. |141|
Малинин Г. В. [436]
Малиновский В. А. [469]
Мальцева Н. В. [384, 385]
Малютина Т. В. [314]
Мамбетов Д. М. [178]
Маннанов 10. Г. 1591 ]
Маркс Т. Л. [461. 474]
Мартынов IO. II. [180]
Маргышев 10. II. [524]
Марьева Н. Н. [214]
Матвеева II. II. [477, 478]
Матыции Л. II. [586]
Мацера В. Е. (215]
Мапшров В. В. [456]
Медиков Я. Я. (47—49, 119, 120]
Мсерсоп Г. А. [565]
Мейер К [110, 228]
Менделеев Д. II. [1]
Мирсапдов У. [609]
Митрофанов К.* Н. [430]
Митякип П. Л. [364. 365]
Михайлов А И. [433]
Мовчан IO. II. |24о]
Молоцкип М. II. (41, 45, 243—
245, 296, 299]
Молчанов В. И. [46, 206, 371,
471. 472, 4S2. 520]
Мороз 9. М. [599]
Морозова М. II. [504]
.Порохов II. Д. [110]
Муратова Л. И. ]297]
Мусуралпев 'Г. |178]
Mioiipceiin Т. К. [592]
Мвкпшеп К. Г. |6О4—607, 610]
Мисников II. A. |6iii]
Набатов В. В. (180|
Пабойченко С. С. [483]
299
Неверов В. В. [121, 596, 597]
Немошкаленко В. В. [157]
Неустроев В, И. [483]
Нефедьев А. Е. [24]
Никитин В. В. [240]
Никитина О. В. [287]
Новиков Б, С. [290]
Ормонт Б. Ф. [76, 78, 118]
Осипов О. А. [540]
Ощерил Б. Н. [80]
Павлов П. В. [163]
Павлов С. В. [637]
Павлюхпи 10. Т. [47—49, 119,
120, 505, 510, 511, 591]
Павлюченко М. М. [608]
Пантелеев В. В. [163]
Панченко Л. Л. [373]
Парамзин С. К. [386]
Патнис А. [494]
Паэ А. Я. [592]
Перцов А. В. [81
Песляк Г. В. [608
Петров Ю. И. [308
Пивинский Ю. Б. [364, 365]
Питерс К. [66]
Плачинда А. С. [476]
Плотников М. В. [430]
Поздняков О. Ф. [229, 231, 232]
Позин М. Е. [580]
Полежаев 10. М. [290]
Полубояров В. А. [416, 468]
Полякин Л. 10. [214]
Помощников Э. Е. [468]
Поткин А. Р. [212, 213]
Почковский В. В. [580]
Продан Е. А. [608]
Проценко П. И. [535]
Пряхина Т. А. 325, 376, 414]
Пугин В. С. [215
Путников Н. А. [480]
Пухов А. А. [217, 610]
Пушнякова В. А. [387]
Пшенпчнов IO. П. [237]
Радцпг В. А. [253, 265—268, 270,
435]
Ражев В. М. [480]
Разворотпива Л. II. [330, 448,
588]
Разумовская И. В. [247]
Ракова Н. II. [41]
Ребиндер П. А. [21, 105, 106,
115, 183, 186]
Регель В. Р. [83, 229, 631]
Редькин В. Ф. [210]
Родькипа II. И. [316, 347]
Рейнгольд Б. М. [482, 487]
Ромашкин 10. II. [148—150, 153]
Русанов А. И. [79]
Руфов 10. Н. [181]
Рыков А. И. [634]
Рябинин Ю. II. [345]
Рябцев 11. Г. [456]
Сабитов Р. М. [432]
Савипкпна М. А. [360, 612, 613]
Савинцев П. А. [221, 222]
Савинцев 10. II. [522]
Савицкая Л. К. [223]
Садыков Р. III. [505]
Самарии О. 11. [212, 213, 488,
568]
Саратовкнн Д. Д. [221]
Свешников Г. В. |169]
Свиридов В. В. [537]
Семов 10. И. [172]
Сенчуков Ф. Д. [176]
Сиденко II. М. [197]
Симаков Ю. В. [445]
Снмонеску А. К. [14]
Синяков Е. В. [341]
Скоков Г. Ф. [481]
Слуцкер А. И. [83]
Смагупов В. И. [482, 487]
Смилга В. II. [37]
Смирнов А. Е. [631]
Соколов И. В. [434]
Соловьева А. В. [22]
Ставер А. М. [586]
Столповская В. Н. [380]
Столярова Т. А. [504]
Стрелецкий А. Н. [179, 181, 271 —
275, 280]
Стругова Л. И. [531, 532, 582—
585]
Суздалев II. П. [464—466, 476]
Суппес В. Г. [121]
Сухих В. А. [126, 130]
Сыркин Я. К. [254]
Тананаев II. В. [110]
Татаринцева М. И. [612, 623J
Тейбор Л. [ 122J
Теилов В. Г. [290]
Тимофеев А. II. [162]
Тихонов А. С. [154]
Ткаченко В. А. [626]
Ткачова К. [485, 510, 511]
Толкачев В. А. [434]
Толкачев С. С. [440]
Толмачеа IO. М. [436]
300
Толочко Г>. П. [143]
Томашевпч И. II. [12G]
Томашевский Э. Е. [83]
Топоров Ю. 11. [36]
Торопов II. А. [409]
Трахтенберг II. III. [162]
Третьяков 10. Д. [392]
Трупов В. К. [628]
Тюмевеп Г. В. [474]
Тямпн IO. Н. [297]
Убеллоде А. [227]
У ибо Л. Я. [592]
Уракаев Ф. X. [135, 224, 231,
232, 522, 601, 615, 622, 623,
629, 6371
Урусопская А. А. [167, 631]
Фальчепко В. М. [156]
Федоров В. В. [110]
Федоров В. В. [85]
Финкель В. М. [297, 542]
Флавицний Ф. М. [5]
Флерова С. А. [341 ]
Френкель Я. II. [71, 77, 84]
Фридель Ж. [ИЗ]
Фридкин В. М. l[348]
Хавский II. II. [565]
Хайретдинов Э. Ф. [294]
Халиф В. A. [268]
Харитон Ю. Б. [128, 130]
Хееги X. [52]
Хейпике Г. [554]
Хикматов М. [G09]
Хинт И. А. [202]
Ходаков Г. С. [50, 115, 186, 187,
316, 347]
Холево Н. А. [129]
Хрусталев Ю. А. [298]
Худяков И. Ф. [483]
Цптовпч II. В. [481]
Чайкина М. В. [518, 598, 625]
Черняк А. С. [391]
Чертов В. М. [476]
Чудпопскпй Ф. А. [395] >
Чумачепко 10. В. [629]
Шапкин В. Л. [625]
Шаскольская М. II. [69]
Шейнин А. Б. [563]
Шеляков О. II. [201]
Шеромов М. А. [143]
Шестопалов А. М. [153]
Широков 10. Г. [374]
Шпшляпппкопа Л. М. [219]
Шмпдг II. В. [536
Шмурак С. 3. [176
Шпинель В. С. [430
Шрадер Р. [355]
Шрейнер Л. А. [106]
Щукин Е. Д. [81, 192]
Эддисон У. [34G]
Эдельман Л. II. [187]
Югов С. И. [604]
Юн К. [307]
Юсупов Т. С. [46, 371, 372, 377,
380]
Юшпп Ю. Я. [524]
Юшкин II. П. [68]
Ядрпнцев В. Б. [436]
Яковлева Л. А. [342]
Ярембаш Е. И. [445]
Ярославцева Н. Я. [219]
Ярым-Агаев IO. Н. [277]
Akse J. К. [420]
Albrecht 11. [454, 566, 616, 618,
619]
Aleksandrov V. V. [437]
Anderson J. S. [400, 404, 407,
443, 452]
Andersson S. [401, 410]
Antonini I. E. [252]
Arends J. [257 ]
Averbuch B. L. [151]
Avvakumov E. G. [388, 533 , 534]
Bailey J. E. [326]
Balluffi R. W. [159, 160, 161]
Battlo F. [614]
Benjamin j. S. [589]
Bernhardt C. [51, 94]
Bertant E. F. [497]
Bisinger H. I. [519]
Black S. A. [590]
Blanchin M. G. [425 . 638 , 639]
Blcchsclnnidt U. [566]
Bochin II. P. [312]
Bock N. [548, 552]
Boldyrev V. V. [338, 380, 390,
437, 523, 533]
Boinmel H. [462]
Bontschev Z. [53]
Booth J. [406]
Boswell E. W. C. [310]
Bowden F. P. [123, 124]
301
В redig м. Л. [337]
Brown L. С. [155]
Brown Т. J. [527]
Browne J. M. [452]
Brozek V. [59]
Burger M. 1. |311 |
Burns 1. 11. [337]
Bursill L. A.[4(12, 412, 425,638,639]
Burstein Л. I. [523]
Butjagin P. Yu. [305]
Calverb P. [527]
Carey Lea M. [4]
Carpentier R. II. [498]
Carter R. E. [573]
Chakurov Kh. [54, 55]
Chandrasherdar G. V. [415]
Chester P. F. [418, 422]
Chodakov G. S. [534]
Chung M. E. [284]
Clark J. [20]
Clasen II. [6021
Cleval B. [614]
Cohen M. [151]
Collen B. [410]
Conny H. [315]
Constabaris G. [462]
Cornu A. [525]
Corradini P. [515]
Courvoisier J. C. [251]
Criado J. M. [340]
Cruickshank D. VV. J. [190]
Dachille F. [142, 318, 327]
Davis T. W. [538]
Dekker A. I. [257]
Desborongh G. A. [498]
Doigan P. [538]
Dorn J. F. [152]
Drickamer II. G. [236, 526]
Dunvald H. [570|
Dyulaj L. [145]
Ebert J. [349]
Elickson R. T. [571]
Eshelby J. D. [125]
Estle T. L. [260. 289]
Evans M. G. [579]
Eyring I. It. [441]
Fairhurst S. S. [423, 424]
Farkas-Janke Л1. |334]
Fatseas G. A. ]513]
Feher G. [587]
Fox P. G. [67, 1341
Frevel J. K. [449]
Friedrich H. [581]
Gacs P. [334]
Gado P. [399]
Gallon T. E. [541]
Gatehouse В. M. [408]
Geissler II. [349|
Gerlach 1. [3751
Gibby It. J. [323]
Gilman P. S. [63]
Gock E. |375, 484]
Goncharov G. N. |5t)7|
Goodenongh J. B. [513]
Gospodinov G. G. [53, 54, 55]
Griffith A. A. [74]
Gregg S. I. [381]
Grohn JI. [519, 561, 581]
llajek B. [59]
Haneinan D. [283, 284, 285]
llarenz II. [239, 533, 543, 550,
551, 552]
llurring A. [300]
Hurl E. VV. 1165]
Hartly D. [145]
Hashimoto K. [335]
Hausler II. [454, 616, 617, 619]
Hedvall 1. A. [578]
lleegn 11. [51, 94, 112]
Ileinicke G. [27, 28, 239, 336,
359, 521, 543, 544, 546, 548,
550, 551, 552, 553]
Henning E. [368]
llerber В. II. [460]
Hess W. [98, 102]
Higginbotham J. [285, 301, 541]
Hiller Joh. [509]
Hirano K. [151]
Hochstrasser G. [251, 2521
Hockey J. A. [194]
lloffinan В. [108, 338, 377]
Hollman H. 1553]
Hollman F. [367 [
Hoffman K. ]302]
ПоГГтап N. [10O]
Hollman U. [312]
Hutchison J. L. [407, 444, 452]
Hyde B. G. [400, 402]
Ignchi E. [406, 457]
Ilgeu S. 194 |
Imai 11. [322]
Imaniura K. [317, 332]
Inglis Л. I). [423, 42'ij
Ioffe A. F. |1S4]
J а ГГе И. II. |269]
Jander VV. [569]
John St. [155]
302
Johnston О. VV. [415]
Juhasz Z. [56, 57]
Kashiwa N. [516]
Kendall K. |104]
Khaireldinov E. F. [523]
Kingsbury lr. P. J. [419]
Kinoshila S. [627]
Kiraly L. [91 [
Kirpilschewa M. VV. [184]
Kiss Л. B. [455]
К nyleiistierna U. [410]
Kochanovska A. [324]
Konialsy VV. [575, 620]
Koster A. [545]
Koutecky J. [250]
Kressuer It. [383]
Kroger F. A. [405]
Krupa V. [93]
Kubo T. [65]
Kiindig VV. | 462 ]
К uno It. |32l]
Kurijana O. [560]
Kutzer II.-I. [379]
Landau II. G. [225]
Landayl J. 1403|
Larsen IL A. [236, 526]
Lee С. 11. 1164]
LcIIler L. [578]
Leung Так Tai [441]
Le Page 1. [411, 425, 424]
Levinson L. M. [506]
Lewis D. [319, 339]
Lewitsky M. A. [184]
Liebscher S. [369, 370]
Lin J. J. [64, 320]
Lincoln E. I. [407, 444]
Linke E. [302]
Lisehke 1. |359]
LohlT 1. [173]
Looney It. VV. [514]
Lugeuza S. J512]
Lundberg M. [398]
Mabie S. [614]
Macklin R. [164]
Magneli A. [397, 398]
Maki J. [566]
Mallya It. M. [593|
Marusia V. V. [437]
Massol It. [525]
Medikov Ya. Y'a. [389, 3901
Merva M. [93]
Meyer K. [27, 175]
Mil lash A. [306]
Mobius It. [454, 566, 616, 617, 618,
619]
Mori S. [560]
Morimoto N. [499, 500, 501 ]
Midler II. 136S]
Muranaka V. [362]
N.uliv S. [641
Naeser G. [361]
Nakazawa II. [500, 501]
Nalarajan M. [415]
Natla G. [515|
Nau P.-E. [617]
Nelson C. N. [2ii2]
Nemeth J. [91]
Nickel E. II. [88]
Nicolson M. M. [309]
Niedzwiedz S. [320]
Nishidnchi K. [56(1, 502]
Nobst P. [358]
Northwood D. 0. [319, 339]
Obrikal I). [175]
Ocepek D. [62]
Ochiai I. [362]
Oesc N. [517]
Oettel II. [238]
Ohlen VV. P. [419]
Opoczky L. [57]
Orowan E. |75]
Ostancvich J. M. [507]
Ostwald VV. [2]
Overlynder 11. [300]
Pai Verneher V. It. [593]
Parker L. II. [6, 7]
Parker T. VV. [381]
Patz K. [313]
Paudert R. [354, 369, 370,
561, 562, 581]
Pavlukliin Ju. T. [388, 389.
Pavlychev J. K. [305]
Pawelezyk II. [61]
Perdock VV’. D. [2571
Persson P. A. [124]
Pethica 13. A. [194]
Petzold D. [358]
Piekoszewski I. [512]
Polanyi M. [579]
Pratt P. L. [125]
Probstain K. |5U9]
Prution M. [301, 541]
519,
39u ]
Rao C. N. 11. [415]
Reeve It. C. [319]
Richler-Meudav 1. [239]
Riedel R. [543]
Riun U. N. [449]
Ripley L. C. [88]
3o3
Rose H. E. Г2ОЛ]
Ross W. [323]
Rossberg M. [175]
Rowan R. I. [20]
Roy R. [142, 318, 327, 442]
Rumpf II. [92, 97, 356, 357, 383]
Ruof A. L. [159, 160]
Ruzek J. [60, 258]
Sadahiro I. [351]
Sable W. [458]
Sanderson С. C. [155]
Sanner G. |357]
Sano II. [460]
Schlichting H. [226]
Schneider II. [139]
Schober E. [544, 547]
Schoenert K. [367]
Schokli W. [249]
Scholz W. [361]
Scbonert J. [103]
Schonert K. [98, 99, 101, 133,
329, 366]
Schort M. A. [331]
Schrader R. [108, 238, 328, 338,
356, 357, 358, 363, 379, 383, 571 ]
Seetliaramatharyulu D. [593]
Sekyla F. [93]
Senna M. [315, 317, 321, 322, 329,
332, 366]
Shaler R. [442]
Serin B. [571]
Shimidzy K. [351]
Shirasaki T. [362]
Sigrist K. [521, 546, 550]
Sigrist K. [336]
Silsbee В. H. [263]
Smekal A. [220]
Smith D. J. [425, 638]
Sodomka L. [177]
Soria-Ruiz J. [134]
Stadter U. [238]
Staudinger H. [11]
Steier K. [103]
Steinicke U. [349, 350, 368]
Steirmand C. [96]
Stepanov A. W. [146]
Stephens M. I. [381]
Steward E. G. [331]
Stockler H. S. [460]
Strid K. G. [344]
Strobel P. [411]
Sullivan R. M. E. [200]
Sundberg M. [398]
Snwalsi J. [512]
Szantho E. [375]
Szymnoski M. [300]
Takahashi H. [352, 353, 378]
Tamai J. [560]
Tamm Ig. [248]
Tammann G. [19]
Tamura S. [447]
Taylor G, I. [70]
Tetzner G. [358]
Thiessen P. A. [27, 544]
Thomas F. P. S. [123]
Tilley R. J. D. [406, 457]
Tkacova K. [58]
Toda J. [335]
Tojo S. [321]
Tokonami M. [502]
Tokutoka U. [301]
Tomilov S. B. [504]
Torngwist E. G. [514]
Tossel J. A. [508]
Trautz M. [549]
Treves D. [506]
Trillo J. M. [340]
Trinks W. [369, 370]
Tschakarov Chr. G. [53]
Tsukuma K. [499]
Tsutsumi K. [3521
Tyler W. W. [528[
Uehara J. [343]
Uemura T. [575]
Ulbert K. [258]
UnIman D. R. [527]
Valenci G. [574]
Van Amelinx S. [403]
Vand V. [493]
Van der Kraan A. M. [463]
Vanghan D. J. [508]
Verdes S. [91]
Wadsley A. D. [401, 408, 451]
Wagner C. [570, 577]
Wakita H. [627]
Walters G. K. [260, 289]
Wanetig P. [10]
Wankova J. [324]
Ward J. C. [496]
Wassan A. R. [152]
Webster A. H. [88]
Weeks R. A. [261, 262]
Weichert R. [133]
Weigelt D. [328]
White W. B. [318]
Whitehurst H. B. [420]
Wilchinsky Z. W. [514]
Wood G. J. [412]
Zacharisen W. M. [450]
ОГЛАВЛЕНИЕ
Предисловие ко второму изданию ............................ 3
Введение .................................................. 5
Глава 1. Общие закономерности диспергирования и актива-
ции твердых тел....................................... 10
1.1. Механические свойства кристаллов................. —
1.2. Диспергирование и активация твердых тел .... 20
1.3. Физико-химические явления, сопровождающие дис-
пергирование твердых тел............................ 37
1.4. Влияние температуры, среды и поверхностно-актив-
ных веществ на процессы диспергирования и актива-
ции ................................................ 50
1.5. Высокоэнергонапряженные аппараты для дисперги-
рования и активации твердых тел..................... 55
Глава 2. Механизмы инициирования механохимических ре-
акций ................................................ 66
2.1. Тепловые теории инициирования механохимических
реакций ............................................. —
2.2. Механизмы формирования новых фаз, обусловленные
пластическим течением твердых тел и выходящими на
поверхность дислокациями............................ 73
2.3. Теория активных поверхностных состояний .... 31
2.4. Электронные возбуждения при разрушении кристал-
лов ................................................ 91
Глава 3. Закономерности структурных н химических измене-
ний в твердых телах, происходящих иод влиянием механи-
ческой активации...................................... 96
3.1. Термодинамическая характеристика актнвпровангых
твердых тел........................................... —
3.2. Механически стимулированные фазовые переходы Ifni
3.3. Механическая активация неорганических твердых
тел ее последствия................................ 169
Глава 4. Механохимические реакции твердых тел с газами
н жидкостями......................................... 190
4.1. Механохимические реакции твердых тел с газамп . . —
4.2. Механохимические реакции твердых тел с жидкостя-
ми ........................................... 201
Глава 5. Механохимические реакции в смесях твердых ве-
ществ ............................................... 206
5.1. Общан характеристика твердофазных реакций .... —
5.2. Возможности метода механической активации твердо-
фазных реакции..................................... 211
5.3. Закономерности .механохимических реакций в сме-
сях твердых веществ................................ 231
5.4. Механохпмический синтез в многокомпонентных си-
стемах........................................... 250
Заключение............................................... 25S
Литература...................... . ... 2<>3
Именной указатель ....................................... 297
Проект- ОТКРЫТЫЙ ДОСТУП
Над оцифровкой данной книги работали:
Ружинский С.И. rygmskifaiaport.ги
Ружинский Ю.И.
Раенко А.С.
август 2005, г. Харьков, Украина
г.Харьков, ул. Чкалова 1
МП «Городок»
Популяризация применения химических добавок и оригинальных технологий в строительной индустрии.
ryginski@aport.ru
+38(057)315-32-63
Здесь может быть Ваша реклама!
Закажи книгу по бетоноведению или строительству на оцифровку и размести в ней свою рекламу.
Дополнительная информация: ryginski@aport.ru