/
















































































































Теги: фармакопея фармакология
Год: 1981
Похожие




Текст
THE INTERNATIONAL PHARMACOPOEIA
THIRD EDITION
PHARMACOPOEA INTERNATIONALIS
EDITIO TERTIA
Volume 1
General Methods of Analysis
WORLD HEALTH ORGANIZATION
GENEVA
1979
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
ИЗДАНИЕ ТРЕТЬЕ
PHARMACOPOEA INTERNATIONALIS
EDITIO TERTIA
Том 1
Общие методы анализа
51W
Выпущено издательством «Медицина» по поручению Министерства здравоохранения Союза Советских Социалистических Республик, которому ВОЗ вверила выпуск данного издания на русском языке
J/л-НИР'1
ВСЕМИРНАЯ ОРГАНИЗАЦИЯ ЗДРАВООХРАНЕНИЯ
ЖЕНЕВА
1981
Международная фармакопея. Третье издание. Т. 1. Общие методы анализа. Всемирная организация здравоохранения, Женева, 1981, ил., с. 242.
В отличие от предыдущих изданий третье издание Международной фармакопеи предполагается выпустить не в одном, а в нескольких томах. Первый том включает описание 42 общих методов анализа, последующие тома будут содержать частные статьи — спецификации для фармацевтических препаратов. В т. 1 изложены общие принципы физических, химических, физико-химических и биологических методов, перечень которых значительно расширен и дополнен за счет современных достижений в этой области; кроме того, в описание каждого метода включены рекомендуемые методики его применения в фармацевтическом анализе. Последний раздел книги содержит перечень реактивов и растворов, упоминаемых в Между-народнсй фармакопее. Все единицы измерения приведены в системе СИ.
Применение унифицированных требований к фармацевтическим препаратам и методов их контроля способствует повышению качества лекарственных средств и обеспечению их безопасности в международном масштабе.
Книга рассчитана на специалистов, занимающихся разработкой и сценкой качества новых лекарственных средств, а также контролем уже применяемых препаратов, на фармацевтов, лабораторных и научных работников.
4 рис., 4 табл.
© Всемирная организация здравоохранения, 1981
На публикации Всемирной организации здравоохранения распространяются положения протокола № 2 Всемирной конвенции об охране авторских прав. Заявления о разрешении на перепечатку или перевод публикаций ВОЗ частично или in toto следует направлять в Отдел публикаций и переводов Всемирной организации здравоохранения, Женева, Швейцария. Всемирная организация здравоохранения охотно удовлетворяет такие просьбы.
Наименования, используемые в настоящем издании, и приводимые в нем материалы не выражают мнения Секретариата Всемирной организации здравоохранения о юридическом статусе какой-либо страны, территории, города или района, их правительстве или другом органе власти, или о их государственных границах.
Упоминание некоторых компаний или продукции отдельных изготовителей не означает, что Всемирная организация здравоохранения отдает им предпочтение по сравнению с другими, не упомянутыми в тексте. Патентованные наименования выделяются начальными прописными буквами.
„ 50700—407
^039(01)—81 КБ—20—10—81. 4102000000
СОДЕРЖАНИЕ
Ведение ....................................................
Общие замечания . В
Единицы измерения 14
ФИЗИЧЕСКИЕ МЕТОДЫ
Измерение массы.............................................. 19
Определение температуры плавления, температурного интервала плавления, точки затвердевания, точки кипения и температурного интервала кипения ...................................... 22
Определение плотности и относительной плотности . . 31
Определение оптического вращения и удельного вращения . 32
Определение показателя преломления . . . . 36
Спектрофотометрия в видимой и ультрафиолетовой областях спектра 37
Спектрофотометрия в инфракрасной области спектра . 45
Атомная абсорбционная спектрофотометрия . 50
Флуоресцентная спектрофотометрия . 52
Турбидиметрия и нефелометрия . . .56
Окраска жидкостей............................................. 57
Радиофармацевтические препараты 59
Степень измельчения порошков и сита . 87
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
Хроматография 91
Определение pH 110
Электрофорез .... 114
Метод фазовой растворимости . .118
ХИМИЧЕСКИЕ МЕТОДЫ
Общие испытания на подлинность . 127
Испытание на хлориды . 132
Испытание на сульфаты 133
Испытание на тяжелые металлы 134
Испытание на железо 138
Испытание на мышьяк 139
Сульфатная зола ... . . 142
Метод сжигания в колбе с кислородом 142
Комплексонометрическое титрование . 145
Неводное титрование . , 149
Нитритометрия........................... . 153
Определение воды методом Карла Фишера........................ 154
Определение метоксильных групп 155
Определение азота ... 157
Определение йодного числа ... 158
Определение перекисей в жирных маслах 159
Определение числа омыления . . 159
Определение неомыляемых веществ . 160
Определение кислотного числа ................................ 161
БИОЛОГИЧЕСКИЕ МЕТОДЫ
Количественное определение микробиологической активности антибиотиков . ... .............165
Испытание антибиотиков на стерильность . 172
Неспецифическая токсичность . ... 176
Испытание на пирогенность............. . 176
Испытание на гистаминоподобные вещества (вазодепресспвные вещества) 178
ФАРМАКОГНОСТИЧЕСКИЕ МЕТОДЫ
Определение золы и золы, не растворимой в кислоте........183
МЕЖДУНАРОДНЫЕ ХИМИЧЕСКИЕ СТАНДАРТНЫЕ ОБРАЗЦЫ. РЕАКТИВЫ, РАСТВОРЫ
Международные химические стандартные образцы............ 187
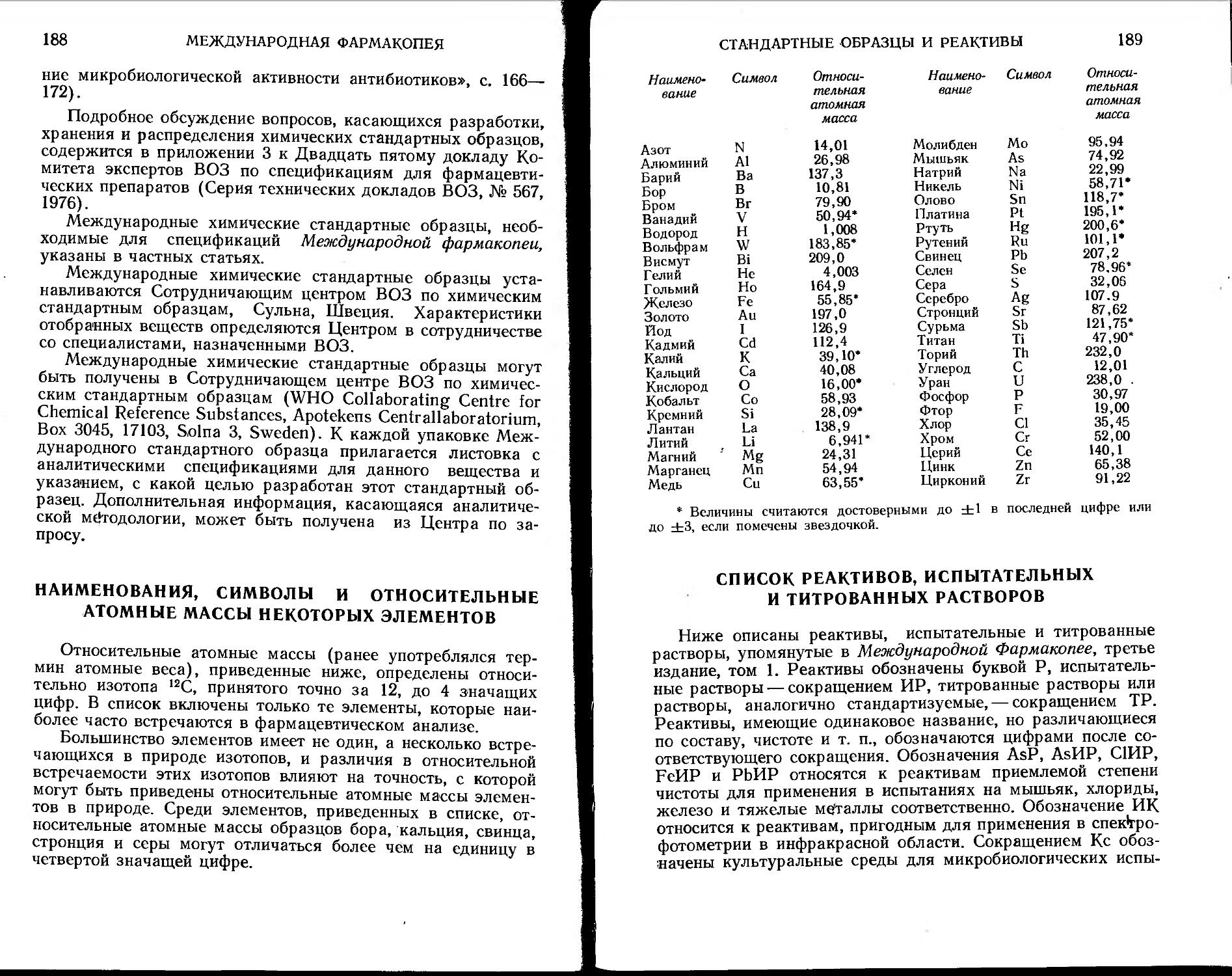
Наименования, символы и относительные атомные массы некоторых элементов........................................... .188
Список реактивов, испытательных и титрованных растворов . . . 189
ВВЕДЕНИЕ
Международная фармакопея издается Всемирной организацией здравоохранения в соответствии с резолюцией WHA3.101 Всемирной ассамблеи здравоохранения. Первое издание было опубликовано в двух томах, первый том — в 1951 г., второй — в 1955 г., затем было выпущено Дополнение 1959 г. Эти три тома были выпущены на английском, французском и испанском языках. Были опубликованы также переводы на немецкий и японский языки. Второе издание было выпущено в свет в 1967 г., а Дополнение к нему—-в 1971 г. Эти два тома были выпущены на английском, французском, русском и испанском языках.
Комитет экспертов ВОЗ по спецификациям для фармацевтических препаратов в своих 25-м и 26-м докладах рассмотрел вопросы организации работы по пересмотру Международной фармакопеи и по созданию и пересмотру спецификаций качества лекарственных средств, публикуемых Всемирной организацией здравоохранения. Комитет рекомендовал подходящие процедуры и установил порядок очередности выполнения положений, содержащихся в резолюции Всемирной ассамблеи здравоохранения WHA20.34* 2, в которой Генеральному директору предлагалось «...продолжать работу над спецификациями для аналитического контроля», и резолюции WHA28.663, в которой Генеральному директору предлагалось «...продолжать разрабатывать мероприятия, относящиеся к созданию и пересмотру международных стандартов, требований и инструкций в отношении профилактических и терапевтических средств».
Следуя этим рекомендациям, ВОЗ продолжала работу по подготовке третьего издания Международной фармакопеи, которая будет опубликована в нескольких томах. Том 1 содержит описание общих методов анализа; затем последуют тома, содержащие частные статьи, т. е. спецификации качества для отдельных лекарственных средств, преимущественно для тех из них, которые наиболее широко используются в медицинской практике.
’ Сборник резолюций и решений Всемирной ассамблеи здравоохранения и Исполнительного комитета, т. I, 1974, с. 143.
2 Там же, с. 149.
3 Сборник резолюций и решений Всемирной ассамблеи здравоохранения и Исполнительного комитета, т. П, третье издание, 1980, с. 65.
8
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
При выборе методов и процедур, включенных в том 1 третьего издания, учитывалась их полезность для целей обеспечения качества фармацевтических препаратов. В описания методов, содержавшиеся в предыдущих изданиях, были внесены многочисленные изменения для приведения их в соответствие с достижениями в разработке новых аналитических средств. При этом, однако, принимались в расчет различные технические и экономические ограничения и выбор рекомендованных методик опирался на оптимальное решение; надо надеяться, что это позволит использовать эти методики в лабораториях контроля качества лекарственных средств, находящихся в развивающихся странах.
Пересмотр общих методов анализа, включенных в том 1 третьего издания, был осуществлен с помощью членов Экспертно-консультативного совета ВОЗ по международной фармакопее и фармацевтическим препаратам, а также других специалистов. Сам процесс пересмотра проходил в форме серии совещаний в течение 1974—1977 гг. и путем переписки. В июле 1978 г. проект текста тома 1 третьего издания Международной фармакопеи был разослан для получения окончательных замечаний всем государствам — членам ВОЗ, членам Экспертно-консультативного совета ВОЗ по международной фармакопее и фармацевтическим препаратам и другим специалистам.
Следующие специалисты принимали участие лично или путем переписки в упомянутых выше дискуссиях и представили замечания к окончательному проекту: проф. Э. А. Бабаян, Министерство здравоохранения, Москва, СССР; д-р D. Banes, Фармакопея США, Роквилл, штат Мэриленд, США; д-р I. Bayer, Национальный институт фармации, Будапешт, Венгрия; д-р Т. Bican, Институт контроля лекарственных средств, Загреб, Югославия; г-н J. Y. Binka, Государственная химическая лаборатория, Аккра, Гана; проф. W. Н. Briner, Медицинский центр, Университета Дьюка, Дарем, штат Северная Каролина, США; г-н J. R. Burianek, Государственный институт контроля лекарственных средств, Прага, Чехословакия; д-р Т. Canback, Шведская фармакопейная комиссия, Стокгольм, Швеция; д-р J. С. Charlton, Радиохимический центр, Амершем, Англия; проф. Y. Cohen, Комиссия по атомной энергии, Сакле, Жив-сюр-Иветт, Франция; д-р D. Cook, Лаборатория по исследованию лекарственных средств, Оттава, провинция Онтарио, Канада; д-р N. Diding, Сотрудничающий центр ВОЗ по химическим стандартным образцам, Сульна, Швеция; д-р L. F. Dodson, Национальная лаборатория биологических стандартов, Министерство здравоохранения, Канберра, Австралия; д-р К. Florey, Институт медицинских на
ВВЕДЕНИЕ
9
учных исследований фирмы Скуибб, Нью-Брансуик, штат Нью-Джерси, США; г-жа М. A. Garth, Национальный центр по анализу антибиотиков, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, Вашингтон, округ Колумбия, США; д-р A. R. Gennaro, Филадельфийский колледж фармации, Филадельфия, штат Пенсильвания, США; д-р Т. George. Исследовательский центр акц. об-ва ЦИБА — ГЕЙГИ, Горегаон, Бомбей, Индия; г-н W. Hewitt, Челтнем, Англия; г-н Kang Hu, Пекинский институт контроля фармацевтических и биологических продуктов, Пекин, КНР; д-р Т. Inoue, Национальный институт гигиены, Токио, Япония; г-жа S. Johansson, Сотрудничающий Центр ВОЗ по химическим стандартным образцам, Суль-на, Швеция; г-н С. A. Johnson, Британская фармакопейная комиссия, Лондон, Англия; г-н Н. G. Kristensen, Датская фармакопейная комиссия, Бр^нсх^й, Дания; д-р К. Kristensen, Фармацевтический отдел изотопов, Бр^нсх^й, Дания; д-р С. S. Kumkumian, Бюро лекарственных средств, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, Роквилл, штат Мэриленд, США; д-р Е. Lang, акц. об-во ЦИБА— ГЕЙГИ, Базель, Швейцария; проф. J. Laszlovszky, Национальный институт фармации, Будапешт, Венгрия; д-р Т. Layloff, Национальный центр анализа лекарственных средств, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, Сент-Луис, штат Миннесота, США; д-р J. W. Ligtbown, Национальный институт биологических стандартов и контроля, Лондон, Англия; д-р A. J. Liston, Директорат лекарственных средств, Оттава, провинция Онтарио, Канада; г-н W. J. Mader, Корпорация Альза, Пало-Альто, штат Калифорния, США; проф. М. Д. Машковский, Фармакопейный комитет СССР, Москва, СССР; д-р Е. Nieminen, Лаборатория контроля лекарственных средств, Хельсинки, Финляндия; д-р А. Н. Обой-макова, Фармакопейный комитет СССР, Москва, СССР; г-н В. Ohrner, Сотрудничающий Центр ВОЗ по химическим стандартным образцам, Сульна, Швеция; д-р Т. Olawuyi Оке, Федеральная лаборатория контроля качества лекарственных средств, Аба, штат Лагос, Нигерия; проф. X. Perlia, Фармацевтический институт, Цюрих, Швейцария; д-р М. Pesez, акц. об-во «Руссель Уклаф», Роменвиль, Франция; проф. J. Richter, Институт лекарствоведения ГДР, Берлин, Вайсензе, ГДР; д-р G. Schwartzman, Бюро лекарственных средств, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, Вашингтон, округ Колумбия, США; проф. С. Д. Соколов, Всесоюзный научно-исследовательский химико-фармацевтический институт, Москва, СССР; д-р I. Suzuki,
10
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Национальный институт гигиены. Токио, Япония; г-н Tien Sung-chiu, Пекинский институт контроля фармацевтических и биологических продуктов, Пекин, КНР; г-н Tu Kuo-shih, Пекинский институт контроля фармацевтических и биологических продуктов, Пекин, КНР; д-р М. М. Tuckerman, Фармацевтический факультет, Университет Темпл, Филадельфия, штат Пенсильвания, США; проф. Н. Vanderhaeghe, Фармацевтический институт Рега, Левен, Бельгия; д-р R. Vasiliev, Государственный институт контроля лекарственных средств и фармацевтических исследований, Бухарест, Румыния; д-р A. Vegh, Венгерская фармакопейная комиссия, Будапешт, Венгрия; д-р М. J. Welsh, Отдел радиационной медицины, Оттава, провинция Онтарио, Канада; д-р В. A. Wills, Аллен и Ханберис лтд., Уэйр, Англия; д-р W. W. Wright, Бюро лекарственных средств, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, Вашингтон, округ Колумбия, США.
Кроме того, замечания были получены от МАГАТЭ, Австрийской фармакопейной комиссии, Датского фармакопейного совета, Французской национальной фармакопейной комиссии, Фармакопейной комиссии ФРГ, Венгерской фармакопейной комиссии, Фармакопеи США, от Министерств здравоохранения Болгарии, Румынии и Швеции, а также от Национальной лаборатории биологических стандартов, Канберра, Австралия, Лаборатории по исследованию лекарственных средств, Оттава, провинция Онтарио, Канада, и Департамента научных и промышленных исследований, Веллингтон, Новая Зеландия. Дополнительно замечания и предложения были представлены некоторыми профессиональными ассоциациями.
Всемирная организация здравоохранения пользуется возможностью выразить благодарность всем лицам и организациям, принимавшим участие в подготовке этого тома.
В качестве председателя на упомянутых выше совещаниях Комитета экспертов ВОЗ по спецификациям для фармацевтических препаратов выступал г-н С. A. Johnson. Функции секретаря Комитета выполняли г-н О. Wallen, руководитель фармацевтической секции ВОЗ, и д-р W. Wieniawski, главный специалист этой секции, которым помогала г-жа М. Schmid, технический помощник.
Том 1 третьего издания Международной фармакопеи содержит описание 42 общих методов анализа. Для большинства физических и физико-химических методов вначале приводится вводная часть, а затем излагаются рекомендованные методики. Такое общее изложение имеет целью облегчить использование этих методов для обеспечения качества лекарственных средств, даже если они используются для кон
ОБЩИЕ ЗАМЕЧАНИЯ
11
троля лекарственных средств, спецификации для которых не содержатся в Международной фармакопее.
В соответствии с резолюцией WHA30.39 Тридцатой Всемирной ассамблеи здравоохранения1 единицы4 измерения, используемые в третьем издании Международной фармакопеи, основаны на Международной системе единиц (СИ) (см. с. 14).
Общие замечания, которые предшествуют тому 1 третьего издания, в первую очередь касаются терминов и положений, применяемых в связи с общими методами анализа; этот раздел будет расширен в последующих томах Фармакопеи.
В соответствии с упомянутой выше резолюцией Всемирной ассамблеи здравоохранения WHA3.10 Международная фармакопея представляет собой сборник рекомендованных методов и спецификаций, которые ни в одной стране не должны носить законодательного характера, если они специально не введены в действие для этой цели соответствующим законодательным актом. Эти методы и спецификации предназначены для того, чтобы служить справочным материалом, на основании которого в любой стране могут быть разработаны национальные требования. Любое государство — член Всемирной организации здравоохранения может полностью или частично включать эти положения в свои национальные требования.
Все замечания и предложения, касающиеся содержания Международной фармакопеи, будут изучены; предложенные поправки будут рассмотрены с целью включения в последующие тома Международной фармакопеи.
ОБЩИЕ ЗАМЕЧАНИЯ
Количества и точность их измерения
Количества веществ и реактивов, используемых в испытаниях, количественных определениях и методиках, должны быть измерены с достаточной точностью. Требуемая степень точности обозначается числом десятичных знаков, приведенных в тексте. Например, 20 означает величину не менее 19,5 и не более 20,5; 2,0 — величину не менее 1,95 и не более 2,05; 0,20 — величину не менее 0,195 и не более 0,205.
Температуры и точность их измерения
Требуемая точность измерения температуры указывается так же, как точность измерения количества вещества. 1 *
1 Сборник резолюций и решений Всемирной ассамблеи здравоохране-
ния и Исполнительного комитета, т. II, третье издание, 1980, с. 118.
12
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Величины pH и точность их измерения
Требуемая точность величин pH указывается так же, как точность измерения количества вещества.
Вычисление результатов
Результаты количественных определений должны быть вычислены с точностью на один десятичный знак больше, чем указано в требовании, и затем округлены следующим образом: если последняя подсчитанная цифра 5 и более (до 9), то предшествующая цифра увеличивается на 1; если последняя цифра 4 или менее, предшествующая цифра остается без изменения. Другие вычисления, например, при стандартизации объемных растворов, проводятся так же.
Растворы
Если нет специальных указаний, все растворы, указанные в испытаниях и количественных определениях, готовят на дистиллированной или деминерализованной воде.
Растворимость
Данные о растворимости вещества означают приблизительную растворимость при температуре 20°C, если нет других указаний. Выражение «растворим в стольких-то частях» следует понимать как указание на число миллилитров растворителя (представленное указанным числом частей), в которых растворим 1 г твердого вещества.
Иногда для обозначения растворимости вещества используются описательные термины. Следующая таблица показывает значения этих терминов.
Описательные термины
Очень легко растворим
Легко растворим Растворим
Умеренно растворим
Мало растворим
Очень мало растворим
Практически нерастворим
Число миллилитров растворителя, необходимое для растворения 1 г твердого вещества
Менее От 1 до 1 10
От 10 до 30
От 30 до 100
От 100 до 1000
От 1000 до 10 000
Более 10 000
ОБЩИЕ ЗАМЕЧАНИЯ 13
Потеря при высушивании
При определении потери при высушивании, если не указано другое количество вещества, высушивают 1,0 г вещества в предписанных условиях.
Постоянная масса
Выражение «высушить до постоянной массы» означает, что процесс высушивания должен продолжаться до того момента, когда результаты двух последовательных взвешиваний будут отличаться не более чем на 0,5 мг на 1 г вещества, взятого для определения; второе взвешивание производят после дополнительного высушивания в течение 1 ч при предписанных условиях. Выражение «прокалить до постоянной массы» имеет аналогичное значение; второе взвешивание производят после повторного прокаливания.
Тара
Материал, из которого изготовлены тара и пробка, не должен взаимодействовать физически или химически с содержимым этой тары таким образом, чтобы это могло привести к изменению чистоты или активности вещества. Дополнительные требования в отношении проницаемости тары обозначаются следующими терминами.
Хорошо укупоренная тара. Хорошо укупоренная тара должна защищать содержимое от попадания посторонних веществ или от потери вещества при обычных условиях обращения, перевозки и хранения.
Плотно укупоренная тара. Плотно укупоренная тара должна защищать содержимое от попадания посторонних веществ, от потерн вещества, выветривания, расплывания вследствие поглощения влаги и испарения при обычных условиях обращения, перевозки и хранения. Эта тара допускает повторную плотную укупорку.
Защита от действия света
Вещество, которое согласно требованию должно храниться защищенным от действия света, должно содержаться в устойчивой к свету таре, защищающей содержимое от влияния света либо за счет свойств материала тары, либо благодаря специальному покрытию тары. Кроме того, тару можно поместить в подходящую устойчивую к свету (непрозрачную) оболочку.
14
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Патенты и товарные знаки
Включение в Международную фармакопею любого лекарственного средства, на которое выдан или может быть выдан, патент, или лекарственного средства, защищенного другим правом, или включение в нее какого-либо наименования, являющегося товарным знаком в какой-либо стране, не предполагает и не должно предполагать передачу разрешения, права или лицензии на пользование любым правом или привилегией, защищенной таким патентом или товарным знаком, в том числе лицензий на производство, без специально оговоренного согласия лица или лиц, которым принадлежат эти права и привилегии.
Использование торговых наименований
Упоминание конкретного торгового наименования в описании некоторых материалов, используемых в количественных: определениях и испытаниях, не означает, что другие, эквивалентные, материалы также не могут быть использованы.
Реактивы, испытательные растворы и титрованные растворы
Буквы Р, ИК, ИР и ТР после наименования реактивов, испытательных растворов и титрованных растворов указывают, что их описание содержится в списке на с. 189.
ЕДИНИЦЫ ИЗМЕРЕНИЯ
В Международной фармакопее использованы наименования и символы единиц измерения Международной системы единиц (СИ) (Systeme international d'Unites) \ эта практическая система единиц была разработана благодаря усилиям Генеральной конференции по мерам и весам (CGPM) и других международных организаций. На 11-й сессии Генеральной конференции (1960) было принято международное сокращение СИ для этой системы единиц1.
Единицы СИ, использованные в третьем издании Международной фармакопеи, так же, как их кратные и дольные единицы, во многих случаях идентичны единицам, использованным для соответствующих единиц измерения во втором 1
1 Полная информация о единицах системы СИ содержится в издании A guide to international recommendations on names and symbols for quantities and on units of measurement, D. A. Lowe, Geneva, WHO, 1975; более краткие сведения приводятся в публикации Единицы СИ в медицине, Женева, Всемирная организация здравоохранения, 1979.
ЕДИНИЦЫ ИЗМЕРЕНИЯ
15
издании. В других случаях, однако, система СИ ввела единицы, имеющие другое определение; последнее особенно справедливо для производных единиц. В таких случаях для лучшего понимания методик и пределов, относящихся к требованиям к качеству, дополнительно к единицам системы СИ в третьем издании Международной фармакопеи приведены единицы, ранее использованные во втором издании, вместе с соответствующим пересчетом числовых значений.
В Международной фармакопее используются следующие множительные приставки, которые указывают на десятичные кратные и дольные единицы СИ:
гига (Г) 100
мега (М) 106
кило (к) 103
санти (с) ю-2
милли (м) IO"3
микро (мк) IO"6
нано (Н) 10-8
ПИКО (п) Ю-12
Использование этих приставок показано на примере следующих единиц, кратных и дольных, которые применяются в третьем издании Международной фармакопеи:
Единицы длины Единицы массы
Метр (м) Килограмм (кг)
Сантиметр (см) Грамм ( г)
Миллиметр (мм) Миллиграмм (мг)
Микрометр (мкм) Микрограмм (мкг)
Нанометр (нм) Нанограмм (нг)
Единицы объема (емкости) Единицы времени
Литр (л) = 1 000 см3 Год ( [а)
Миллилитр (мл) =1 см3 Сутки | [сут)
Микролитр (мкл) =0,001 см3 Час I :ч)
Минута I [мин)
Секунда ( :<о
Миллисекунда 1 [мс)
Микросекунда ( [мкс)
Единицы температуры
Кельвин (К)
Градус Цельсия (°C)
Единицы давления
Килопаскаль (кПа)
Паскаль (Па)
В некоторых специальных случаях также используются следующие единицы давления, не входящие в систему СИ:
Фунт-сила на квадратный дюйм (lbf/in2 или psi) ==6,895 кПа
миллиметр ртутного столба (мм рт. ст.) «133 Па
16
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Единицы радиоактивности'
Гигабеккерель Мегабеккерель Беккерель Кюри Милликюри Микрокюри
(ГБк) (МБк) (Бк) (Ки) (мКи) (мкКи)
= 27,03 = 27,03 = 27,03
= 37 ГБк = 37 МБк
= 38 кБк
мКи мкКи
пКи
Единицы силы электрического тока
Ампер (А)
Миллиампер (мА)
Наноампер (нА)
Единицы электрического потенциала
Вольт (В)
Милливольт (мВ)
Единица электрического
сопротивления
Ом (Ом)
1 Определение единиц радиоактивности приведено в разделе «Радиофармацевтические преператы» (см. с. 59—63).
ФИЗИЧЕСКИЕ МЕТОДЫ
№
Лениетрад, . Есгтужегскак у , Зэнхимфа^игбъед. • -и „ -. лу/'Т :;оз -:г- -
ИЗМЕРЕНИЕ МАССЫ
Для испытаний, связанных с измерением массы, следует использовать весы соответствующей предельной нагрузки и чувствительности в зависимости от необходимой степени точности.
Чтобы получить «точную навеску» вещества в количестве 50 мг или более, требуются аналитические весы с предельной нагрузкой 100—200 г и чувствительностью 0,1 мг. Если взвешивают вещество в количестве менее 50 мг, для получения «точной навески» применяют аналитические весы с предельной нагрузкой 20 г и чувствительностью 0,001 мг, обычно называемые микроаналитическими весами.
Для других испытаний, связанных с измерением массы, используют весы меньшей чувствительности.
Приборы
Аналитические весы должны иметь достаточную предельную нагрузку и чувствительность. По конструкции они могут быть равноплечими с набором калиброванных гирь (разновес) или любого другого подходящего типа (например, микроаналитические весы, основанные на принципе магнитного измерения). Работу весов следует периодически проверять при помощи стандартного набора калиброванных гирь.
Аналитические весы должны быть сконструированы таким образом, чтобы выдерживать полную нагрузку без чрезмерного напряжения, а их чувствительность не должна изменяться при повторных взвешиваниях при полной нагрузке. Желательно, чтобы весы были снабжены демпфирующим устройством (например, магнитным или воздушным успокоителем колебаний), с помощью которого стрелка быстро возвращается в исходное положение (апериодические весы).
Аналитические весы могут быть сконструированы для ручного накладывания всех гирь, но предпочтительно, чтобы они были оборудованы специальным приспособлением для накладывания всех или части гирь в пределах допустимой нагрузки. В последнем случае весы должны быть оборудованы регистраторами нагрузки, четко показывающими приложенную нагрузку. Кроме того, аналитические весы могут ыть оборудованы оптическим устройством с подсветкой, которое проецирует рабочую часть шкалы на экран (например, когда смещение проецируемой части шкалы относительно ну-2*
20
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
левой линии дает прямое значение величины массы), или иметь устройство для отсчета показаний любого другого типа.
Одночашечные весы с постоянным противовесом относятся к типу весов, имеющих постоянную чувствительность во всем интервале нагрузок. Над чашкой весов на ее подвеске помещен набор навесок, соответствующий наибольшей нагрузке; при взвешивании их снимают с помощью управляемого вручную механического устройства до достижения равновесия.
Аналитические весы должны быть установлены в специальном корпусе с удобными дверцами, позволяющими накладывать взвешиваемый материал. Дверцы должны быть сконструированы таким образом, чтобы исключить влияние воздушных потоков. Для снижения уровня влажности в атмосфере корпуса в него можно помещать осушители (например, силикагель, безводный хлорид кальция).
Набор калиброванных гирь для весов, на которые гири помещают вручную, и набор гирь, используемых для проверки чувствительности весов другого типа, должны храниться в коробке, выполненной из подходящего материала и соответствующим образом обитой изнутри.
Установка весов
Аналитические весы следует установить на прочное основание, которое в минимальной степени подвержено механической вибрации; предпочтительно использовать для этого антивибрационный стол соответствующей конструкции. Кроме того, весы могут быть установлены на бетонной плите, покоящейся на столбах, которые либо укреплены в грунте, либо соединены с конструктивными элементами здания; весы могут быть также помещены на прочный стол или полку, защищенный амортизаторами, такими, как пробковые маты или листы резины.
Весы следует также защищать от влажности и паров кислот, для чего желательно установить их в отдельном помещении лаборатории. Весы не следует помещать около окна, радиатора или на прямом солнечном свету; нельзя их ставить на сквозняке.
Весы должны быть снабжены нивелиром и указателем правильного положения. Необходимо часто проверять регулировку весов по уровню.
Проверка чувствительности
Чувствительность весов должна периодически проверяться квалифицированным специалистом.
ФИЗИЧЕСКИЕ МЕТОДЫ
21
РЕКОМЕНДУЕМАЯ МЕТОДИКА '
Проверка стабильности положения равновесия
Прежде чем использовать весы, необходимо несколько раз проверить положение равновесия без нагрузки. После каждого испытания весы должны быть остановлены.
Время от времени следует также проверять положение равновесия весов под нагрузкой, например, с одной десятой полной нагрузки и при полной нагрузке. Разность между положениями равновесия, найденная в двух последовательных определениях при одинаковой нагрузке, не должна превышать 0,1 мг для аналитических весов и 0,001 мг для микроаналитических весов.
Работа с весами
Если весы не используются, коромысло весов и держатели чашек должны быть подняты. Дверцы корпуса весов должны быть всегда закрытыми.
Для освобождения весов следует очень осторожно опустить коромысло и чашки.
Прежде чем начать взвешивание, необходимо выдержать взвешиваемые предметы при температуре весов. Взвешиваемый предмет, а также гири следует всегда помещать, насколько это возможно, в центр чашки. Во время взвешивания и в любом другом случае, когда предметы добавляются на чашку или снимаются с нее, арретир коромысла и держатели чашек должны быть подняты. Взвешиваемые вещества следует помещать в подходящую тару, например в стаканы, склянки для взвешивания или тигли. Жидкости, летучие или гигроскопические твердые вещества следует взвешивать в плотно укупоренной таре, например в склянках для взвешивания с притертой пробкой (бюксы). Нельзя помещать непосредственно на чашки весов химические вещества или предметы, которые могут их повредить.
Если должно быть взвешено небольшое количество вещества (например, при определении сульфатной золы) в большом сосуде, а между двумя взвешиваниями проходит достаточно длительный промежуток времени, атмосферное давление и температура могут измениться настолько, что это может повлиять на устойчивость показаний весов и вызвать существенную ошибку. При взвешивании на двухчашечных весах этой ошибки можно избежать, если использовать для тарирования другой сосуд того же размера и массы.
Чашки весов следует периодически легко очищать кисточ
22
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
кой из верблюжьего волоса или аналогичной мягкой щеткой для удаления пыли, которая могла накопиться.
Гири следует брать только пинцетом, кончики которого имеют специальное покрытие.
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ, ТЕМПЕРАТУРНОГО ИНТЕРВАЛА ПЛАВЛЕНИЯ,
ТОЧКИ ЗАТВЕРДЕВАНИЯ, ТОЧКИ КИПЕНИЯ И ТЕМПЕРАТУРНОГО ИНТЕРВАЛА КИПЕНИЯ
Термодинамически истинная точка плавления вещества (тройная точка) представляет собой физическую константу, которая свидетельствует о подлинности и чистоте материала. Она определяется как температура, при которой твердая, жидкая и газовая фазы вещества находятся в равновесии в закрытой системе без воздуха. При обычном атмосферном давлении твердая и жидкая фазы вещества находятся в равновесии при температуре, которая несколько отличается от тройной точки, но, так как влияние давления на температуру перехода «твердое вещество — жидкость» минимально, эта разница в общем не превышает нескольких сотых градуса по шкале Цельсия.
Методы определения равновесных точек плавления трудоемки и требуют сложного оборудования. Поэтому в обычной практике точки плавления определяют чаще динамическими, а не равновесными методами. Точки плавления, определенные динамическим методом, обычно существенно отличаются от соответствующих тройных точек. Степень отклонения варьирует в зависимости от применяемого метода, критерия, принятого для определения «точка плавления», и, возможно, от испытуемого вещества. Точки плавления, определенные капиллярным методом, принятым в Международной фармакопее, как правило, на один градус выше, чем истинные термодинамические точки плавления.
Определение точек плавления (называемых в последующем температурами плавления) используется в фармакопейных спецификациях главным образом для установления подлинности данного вещества. Ценность такой идентификации в значительной степени возрастает, если используется методика определения так называемых смешанных точек плавления. Эта методика связана с дополнительным определением, призванным показать, что испытуемое вещество и смесь, приготовленная из равных частей этого вещества и аутентичного образца (стандартного образца) того же вещества, плавятся при одной и той же температуре. Если эти два вещества не
ФИЗИЧЕСКИЕ МЕТОДЫ
23
идентичны, смесь обычно плавится при значительно более низкой температуре, чем испытуемое вещество, и температурный интервал плавления относительно более широк.
Наличие примесей в веществе приводит к более или менее заметному снижению его точки плавления. Еще более существенным является тот факт, что примеси, присутствующие в веществе, могут вызывать расширение температурного интервала плавления. В большинстве случаев, когда поведение вещества в процессе плавления используется как критерий чистоты, Международная фармакопея предписывает определение температурного интервала плавления, а не точки плавления.
Точно так же для жидкостей определение точки кипения и температурного интервала кипения дает информацию, на основании которой можно судить о подлинности и чистоте жидких веществ. Практические соображения вновь диктуют необходимость применения методов, дающих кажущиеся константы, которые могут отличаться от истинных термодинамических величин. Однако, если предписанные экспериментальные условия строго соблюдаются, то полученные результаты отличаются значительной воспроизводимостью.
А. Определение температуры плавления и температурного интервала плавления измельчаемых веществ
Температурный интервал плавления вещества — интервал между скорректированной температурой, при которой вещество начинает спадаться или образовывать капли на стенках капиллярной трубки, и скорректированной температурой, при которой вещество полностью переходит в расплавленное состояние, о чем свидетельствует исчезновение твердой фазы.
Указание в статье «температурный интервал плавления °—Ь°С» означает, что температурный интервал плавления, определенный методом, описанным ниже, должен находиться в этих пределах.
Температура плавления вещества — скорректированная температура, при которой вещество полностью расплавлено, о чем свидетельствует исчезновение твердой фазы.
Прибор
Подходящий прибор для определения состоит из стеклянного сосуда с соответствующей жидкостью, контролируемого источника тепла, термометра, капиллярной трубки и увеличительного стекла.
Стеклянный сосуд должен иметь подходящую конструк
24
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
цию, содержать соответствующую жидкость и быть снабжен устройством для быстрого перемешивания жидкости (для этого подходят некоторые жидкие силиконы). Контролируемый источник тепла должен обеспечивать подъем температуры жидкой нагреваемой среды с требуемой скоростью.
Стандартизованные термометры должны иметь интервал определения от —10 до +360 °C при длине одного деления на шкале не менее 0,8 мм. Желательно использовать стеклянные ртутные термометры с твердым столбиком и резервуаром цилиндрической формы, выполненные из специального стекла, подходящего для данного интервала температур; каждый термометр должен быть снабжен защитной трубкой.
Термометры, используемые для определения температуры плавления, могут быть откалиброваны для полного или частичного погружения. Термометр для полного погружения — это термометр, показания которого будут правильными, если термометр погружен по крайней мере до конца столбика ртути в среду, температуру которой необходимо измерить. Термометр для частичного погружения — термометр, показания которого будут правильными, если термометр погружен на предписанную глубину и выступающий столбик ртути находится в предписанных условиях. Если термометры для полного погружения используются для частичного погружения, необходим вспомогательный термометр для определения поправки на выступающий столбик ртути. Выступающие над поверхностью нагреваемого материала части этих термометров должны быть заключены в стеклянную трубку.
Капиллярная трубка из боросиликатного стекла, закрытая с одного конца, должна иметь следующие размеры: толщина стенки около 0,10—0,15 мм; длина, подходящая для применяемого прибора; внутренний диаметр 0,9—1,1 мм.
Для наблюдения за капиллярной трубкой требуется подходящее увеличительное стекло. Может быть использован любой другой прибор и метод, обеспечивающие такую же точность определения и откалиброванные по методу Международной фармакопеи при помощи стандартных образцов ВОЗ для определения точки плавления1.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Распределяют небольшое количество мелкоизмельченного вещества тонким слоем и сушат его в вакуум-эксикаторе над
1 Набор веществ с точками плавления, которые согласно требованиям Международной фармакопеи находятся иа интервале от +69 до +263 °C. Эти вещества могут быть получены в сотрудничающем центре ВОЗ по химическим стандартным образцам.
ФИЗИЧЕСКИЕ МЕТОДЫ
25-
силикагелем-осушителем Р, пятиокисью фосфора Р или другим подходящим осушителем в течение 24 ч или при температуре, указанной в соответствующей статье.
Переносят высушенный порошок в сухую капиллярную трубку и уплотняют его постукиванием капилляра по твердой поверхности таким образом, чтобы получить плотный столбик вещества высотой около 3 мм. Вносят капилляр в баню, предварительно нагретую до температуры на 5 °C ниже ожидаемой температуры плавления; подъем температуры регулируют таким образом, чтобы он составлял 1 °C в минуту, если в статье нет других указаний относительно температуры внесения капилляра в баню или скорости подъема температуры. Капилляр должен быть укреплен в бане таким образом, чтобы его закрытый конец находился на уровне середины шарика стандартного термометра.
Если используют термометр, калиброванный на частичное погружение, надо проследить за тем, чтобы он был погружен точно до отметки в момент снятия показаний.
Если нет специальных указаний, отмечают температуру, при которой наблюдается спадение вещества или образование капель на стенках капилляра, и температуру, при которой вещество полностью расплавилось, на что указывает исчезновение твердой фазы.
К полученным температурным данным прибавляют поправку на отклонение стандартного термометра. Если термометры, калиброванные на полное погружение, используют частично погруженными, к данным стандартного термометра прибавляют также поправку на выступающий столбик ртути, которую получают описанным ниже способом.
Перед определением температуры плавления к стандартному термометру присоединяют вспомогательный термометр таким образом, чтобы его шарик ртути находился посредине между отметкой ожидаемой температуры плавления на стандартном термометре и поверхностью нагреваемого материала. Когда вещество расплавится, отмечают показания вспомогательного термометра. Поправку, которую следует, прибавить к показанию стандартного термометра, вычисляют по следующей формуле:
0,00015ЛЦТ — О,
гДе Т— показания стандартного термометра;
t — показания вспомогательного термометра;
N — число делений на шкале стандартного термометра между поверхностью нагреваемого материала и уровнем ртути.
26
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Если необходимо, поправку на выступающий столбик ртути для термометров, калиброванных на частичное погружение, рассчитывают по той же формуле, но заменяют Т на Ts — среднюю температуру на выступающем столбике ртути во время калибровки.
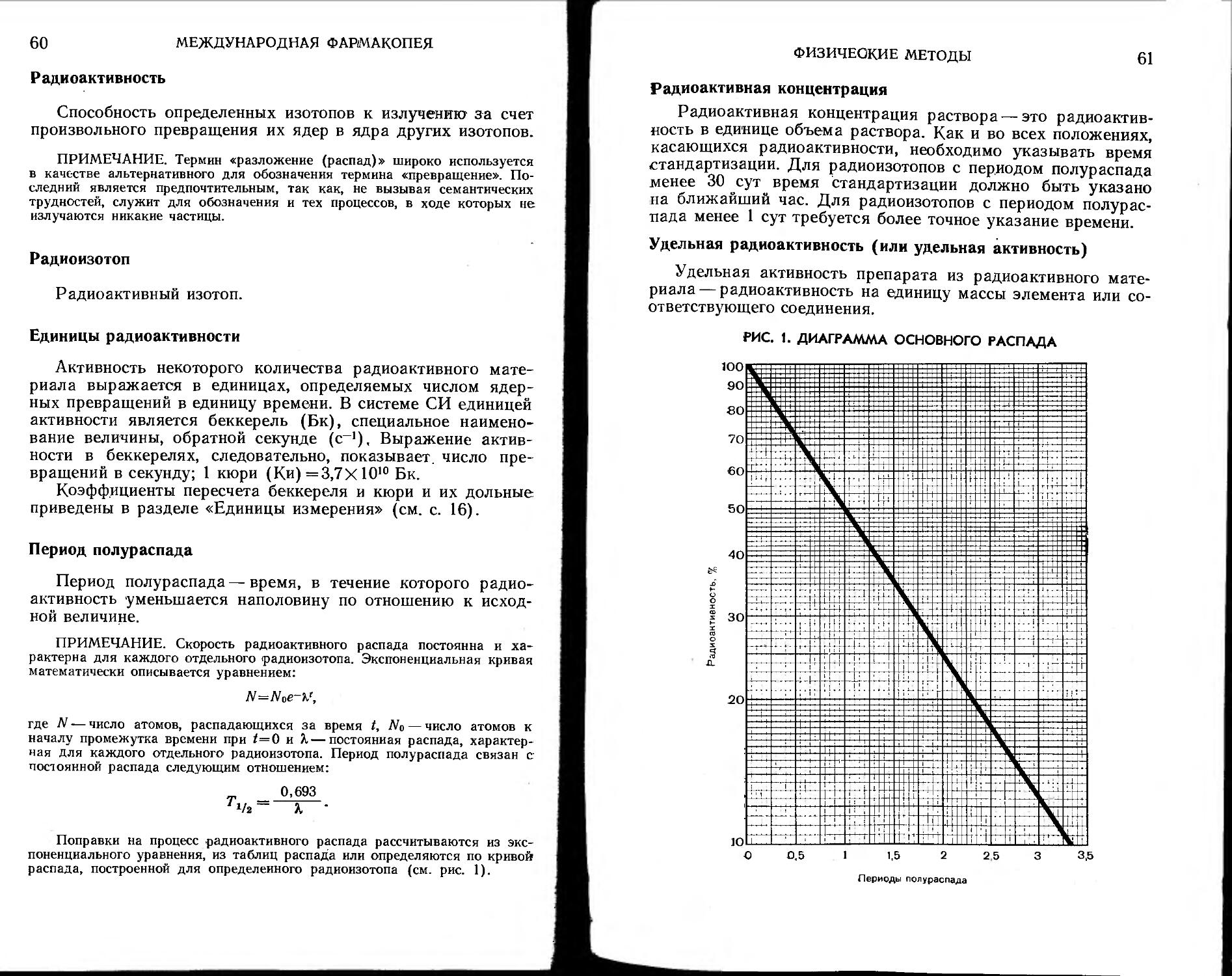
Для удобства обе упомянутые выше поправки на выступающий столбик ртути и любое отклонение от стандартного термометра могут быть заменены калибровкой прибора при помощи стандартных образцов ВОЗ для определения точки плавления.
Б. Определение точки плавления жиров, восков и т. п.
Точка плавления жиров, восков и т. п. — это скорректированная температура, при которой столбик вещества в капилляре становится прозрачным или внезапно поднимается, если определение проводят описанным ниже методом.
Прибор
Используют прибор, аналогичный описанному в разделе А для определения точки плавления и температуры плавления измельчаемых веществ, внося следующие изменения:
— в сосуде для нагревания используют воду;
— точно стандартизованный термометр должен охватывать интервал от —10 до +100 °C;
— стеклянная капиллярная трубка должна иметь те же размеры, что и трубка, описанная в разделе А, но оба ее конца должны быть открыты; можно использовать капиллярные трубки из мягкого стекла.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Если в статье нет других указаний, расплавляют вещество при самой низкой, насколько возможно, температуре и затем засасывают жидкость в капиллярную трубку до высоты около 10 мм. Охлаждают заполненную трубку при 10оС или при более низкой температуре в течение 24 ч. Указание в статье «определяют без предварительного плавления» означает, что капиллярную трубку следует наполнить погружением в нерасплавленное вещество так, чтобы в нижней части капилляра образовался столбик высотой около 10 мм. Затем сразу же проводят определение. Прикрепляют капилляр к термометру в водяной бане посредством резинового кольца или
ФИЗИЧЕСКИЕ МЕТОДЫ
27
другим способом так, чтобы нижний конец капиллярной трубки был на уровне середины ртутного шарика термометра, а расстояние между нижним концом капиллярной трубки и уровнем воды составляло около 20 мм. Затем нагревают баню при постоянном перемешивании, регулируя температуру таким образом, чтобы за 5 °C до ожидаемой температуры скорость подъема составляла около 1 °C в минуту7.
В. Определение точки затвердевания
Точка затвердевания жидкости или расплавленного твердого вещества — это наивысшая температура, при которой оно затвердевает. Точка затвердевания жидкости та же, что и точка плавления твердого вещества, но, так как жидкость может быть охлаждена до температуры ниже точки затвердевания без образования твердой фазы, для определения точки затвердевания жидкости или расплавленного твердого вещества используют описанный ниже метод.
Прибор
Подходящий прибор состоит из пробирки с внутренним диаметром около 2 см и длиной около 10 см, вставленной при помощи корковой пробки в большую по размерам пробирку диаметром около 3 см и длиной 12 см, сосуда с водой или другой охлаждающей смесью и точно стандартизованного термометра.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Если нет специальных указаний в статье, помещают около 10 мл испытуемой жидкости или 10 г расплавленного твердого вещества во внутреннюю пробирку и охлаждают вместе внутреннюю и наружную пробирки в воде или в подходящей охлаждающей смеси до температуры примерно на 5 °C ниже ожидаемой точки затвердевания жидкости; слегка перемешивают жидкость термометром до начала затвердевания жидкости. Вначале наблюдается значительное падение температуры. Затем, когда образуется твердая фаза, температура в течение некоторого времени остается постоянной или возрастает, прежде чем стать постоянной. Наивысшую отмеченную температуру принимают за точку затвердевания. Если жидкость не начинает затвердевать в пределах 2 °C от ожидаемой температуры, затвердевание может быть вызвано прибавлением небольшого кристалла вещества к жидкости или потиранием внутренней стенки пробирки термометром.
28
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Г. Определение точки кипения
Точка кипения жидкости—это скорректированная температура, при которой жидкость кипит при нормальном атмосферном давлении, если определение проводят описанным ниже методом.
Прибор
Подходящий прибор для определения состоит из сосуда с соответствующей жидкостью, источника тепла и термометра, описанного в разделе А для определения точки плавления и температуры плавления измельчаемых веществ; необходима также тонкостенная стеклянная пробирка с наружным диаметром около 4 мм и длиной, подходящей для применяемого прибора, а также тонкостенная стеклянная капиллярная трубка с внутренним диаметром не более 1 мм, которая заплав-лена с одного конца на 2 мм.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
В пробирку помещают 3—4 капли испытуемой жидкости (или эквивалентное количество твердого вещества). Капиллярную трубку вносят в пробирку (заплавленным концом вниз) и помещают пробирку в нагревательную баню таким образом, чтобы ее нижний конец находился на уровне середины шарика термометра. Быстро и при постоянном перемешивании нагревают баню до температуры около 10 °C ниже ожидаемой точки кипения, затем регулируют подъем температуры таким образом, чтобы он составлял 1—2 °C в минуту. Во время нагревания от нижнего конца капиллярной трубки начинают отделяться пузырьки воздуха, вначале медленно, а затем, по мере приближения к точке кипения, — быстрее. Отмечают температуру, при которой пузырьки отделяются в виде ровной быстрой струйки, и затем уменьшают нагревание,, чтобы температура бани понижалась со скоростью 1—2°C в минуту. Отмечают температуру, при которой отделение пузырьков прекращается. За точку кипения принимают среднюю величину этих двух температур, внося поправку на выступающий столбик термометра и отклонение от нормального атмосферного давления. Поправку на выступающий столбик термометра получают, как описано в разделе А для определения температуры плавления и температурного интервала плавления измельчаемых веществ. Если определение выполнено при барометрическом давлении, отличающемся от
ФИЗИЧЕСКИЕ МЕТОДЫ
29
101,3 кПа (760 мм рт. ст.), к значению температуры прибавляют следующую поправку:
kip — Pi), где р — стандартное барометрическое давление;
рг — барометрическое давление, отмеченное на ртутном барометре без учета температуры воздуха;
k — инкремент температуры кипения, указанный ниже.
Если величина давления отмечается по барометру, откалиброванному в килопаскалях, используют следующие данные:
р= 101,3;
£ = 0,3 (инкремент температуры кипения при повышении давления на 1 кПа), если в статье нет специальных указаний.
Если величина давления отмечается по барометру, откалиброванному в миллиметрах ртутного столба, используют -следующие данные:
р = 760;
£ = 0,04 (инкремент температуры кипения при повышении давления на 1 мм рт. ст.), если в статье нет специальных указаний.
Д. Определение температурного интервала кипения (интервала перегонки)
Температурный интервал кипения (интервал перегонки)— это скорректированный интервал температур, в котором перегоняется все вещество или его определенная часть при нормальном атмосферном давлении, если определение проводят описанным ниже методом.
Прибор
Подходящий прибор для определения состоит из перегонной колбы, холодильника, приемника, источника тепла с защитным экраном и термометра.
Перегонная колба емкостью 50—60 мл должна быть выполнена из термоустойчивого стекла. Удобны колбы следующих размеров: горло 10—12 см длиной и внутренним диаметром 14—16 мм; боковой отвод длиной 10—12 см и внутренним диаметром около 5 мм находится посредине горловины колбы и образует угол 70—75° с нижней частью горла.
Холодильник представляет собой прямой стеклянный холодильник из термоустойчивого стекла, длиной 55—60 см, с водяной рубашкой длиной около 40 см или холодильник
30
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
другой системы с такой же охлаждающей способностью. Нижний конец холодильника может быть изогнут, чтобы служить выходной трубкой, или к холодильнику для этой цели может быть присоединен изогнутый наконечник.
Приемник представляет собой мерный цилиндр емкостью 25—50 мл с делением шкалы 0,5 мл.
Источник тепла состоит из небольшой газовой горелки, предпочтительно горелки Бунзена, или электрического нагревателя, обеспечивающих такое регулирование нагрева, которое достигается с помощью газовой горелки. Если используют газовую горелку, основание колбы закрывают асбестовым экраном. Экран делают из листа асбеста толщиной 5—7 мм в форме квадрата со стороной 14—16 см и отверстием в центре. Диаметр последнего должен быть таким, чтобы часть вставленной в него колбы ниже верхней поверхности асбестового листа имела емкость 3—4 мл.
Желательно, чтобы термометр был откалиброван на частичное погружение на 100 мм, как описано в разделе А для определения температуры плавления и температурного интервала плавления измельчаемых веществ; в противном случае можно использовать термометр, откалиброванный на полное погружение, с соответствующей поправкой на выступающий столбик. После установления термометра столбик должен располагаться в центре горла колбы, а вершина шарика должна находиться непосредственно под основанием выхода бокового отвода.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Помещают в колбу 25 мл испытуемой жидкости, соблюдая меры предосторожности, чтобы жидкость не попала в боковой отвод, и прибавляют 0,3—0,5 г стеклянных шариков или другое подходящее вещество. Защищают горелку и колбу кожухом от движения воздушных потоков и нагревают таким образом, чтобы пары медленно поднимались в горло колбы и чтобы перед падением первой капли дистиллята из холодильника прошло 5—10 мин. Продолжают перегонку со скоростью 2—3 мл в минуту, собирая дистиллят в приемник. Отмечают температуру, при которой из холодильника падает первая капля, и затем температуру, при которой выпарится последнее количество жидкости в колбе или отгонится определенный процент жидкости.
Температурные интервалы кипения (интервалы перегонки), указанные в статьях, отнесены к барометрическому давлению 101,3 кПа (760 мм рт. ст.). Если определение проводят при другом барометрическом давлении, вносят поправку
ФИЗИЧЕСКИЕ МЕТОДЫ
31
в показания термометра на любую разницу в барометрическом давлении из расчета 0,1 °C на кждые 0,36 кПа (2,7 мм рт. ст.) разности, прибавляя соответствующую величину, если давление ниже, и вычитая ее, если давление выше.
ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ И ОТНОСИТЕЛЬНОЙ ПЛОТНОСТИ
Плотность (р) вещества есть масса одной единицы объема вещества. В соответствии с основными единицами системы СИ плотность выражают в килограммах на кубический метр. Однако в Международной фармакопее плотность выражается в килограммах на литр (что соответствует граммам на миллилитр) при температуре 20 °C (рго) с поправкой на выталкивающую силу воздуха (т. е. в условиях вакуума). Для фармакопейных целей плотность жидкостей не измеряется непосредственно, а вычисляется из их относительной плотности.
Относительная плотность d2O2o— это отношение массы вещества в воздухе при температуре 20к массе равного объема воды при той же температуре. Термин «относительная плотность с?2о2о» соответствует применявшемуся ранее термину «удельный вес, определенный при 20 °C».
Относительная плотность d „ обозначает отношение массы вещества в воздухе при 20°C к массе равного объема воды при 4 °C. Так как относительная плотность воды при 20 °C равна 0,998234, эти величины связаны между собой следующим уравнением:
d420 = 0,998234Д,,,2».
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Относительную плотность (d’o) определяют с помощью гидростатических весов (если точность, указанная в статье, составляет три десятичных знака) или с помощью пикнометра.
Если в статье указана величина плотности р20 (в кг/л или г/мл), проводят измерение относительной плотности и из полученной величины вычисляют плотность массы в соответствии с уравнением:
р20 == 0,99703d2020 -J- 0,0012.
Использование гидростатических весов
Используют прибор подходящей конструкции, установлен-и на горизонтальной стойке. Поплавок должен быть под-шен на тонкой проволоке, изготовленной предпочтительно
32
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
из платины. Для калибровки прибора устанавливают поплавок в положение равновесия на воздухе, затем погружают его в цилиндр, заполненный водой, и вновь устанавливают положение равновесия путем помещения рейтеров (гирь) в соответствующие углубления на коромысле. Поплавок должен свободно плавать в жидкости. Заполняют цилиндр испытуемой жидкостью и проводят измерение аналогичным путем. Следует проследить, чтобы погруженная часть проволоки, на которой подвешен поплавок, была одинаковой при всех измерениях. Величина массы, которая должна быть прибавлена для достижения равновесия в испытуемой жидкости (или вычтена, если плотность испытуемой жидкости ниже плотности воды), дает прямое значение относительной плотности этой жидкости.
Использование пикнометра
Используют пикнометр подходящей формы емкостью не менее 5 мл. Точно взвешивают пустой сухой цикнометр и заполняют его испытуемой жидкостью, температура которой предварительно доведена примерно до 20°C. Выдерживают заполненный пикнометр в течение примерно 30 мин при температуре 20 ± 1 °C, доводят объем жидкости до отметки, используя, если необходимо, небольшую полоску фильтровальной бумаги для удаления избытка жидкости и для протирания входного отверстия изнутри, и точно взвешивают. Вычисляют массу жидкости в пикнометре. Удаляют жидкость, очищают и высушивают пикнометр, повторяют измерение с водой, свободной от углекислоты, Р также при 20±1 °C и вычисляют массу воды в пикнометре. Отношение массы испытуемой жидкости к массе воды дает относительную плотность (с?|о)-
ОПРЕДЕЛЕНИЕ ОПТИЧЕСКОГО ВРАЩЕНИЯ И УДЕЛЬНОГО ВРАЩЕНИЯ
Многие вещества обладают свойством отклонять плоскость поляризациии при прохождении через них прямолинейно поляризованного света; это свойство называют оптической активностью. Измерение оптической активности используется в фармакопейных целях главным образом для установления подлинности вещества. Оно может также применяться как испытание на чистоту (отсутствие оптически неактивных посторонних веществ) и как метод количественного определения.
ФИЗИЧЕСКИЕ МЕТОДЫ
33
Оптическое вращение
Оптическое вращение — это угол, на который отклоняется плоскость поляризации при прохождении поляризованного света через слой жидкости. Вещества считаются правовращающими или левовращающими в зависимости от того, вращается ли плоскость поляризации по часовой стрелке или против нее, что устанавливается наблюдением в направлении источника света. Вращение вправо обозначается ( + ), а вращение влево (—).
В Международной фармакопее оптическое вращение (а) выражается в угловых градусах. В системе единиц СИ угол оптического вращения выражается в радианах (рад).
Оптическое вращение измеряют в слое жидкости подходящей толщины при длине волны, указанной в статье. Если указана D-линия спектра натрия, следует использовать линию спектра натрия с длиной волны 589,3 нм (средняя величина для дублета при 589,0 нм и 589,6 нм). Часто также используют зеленую линию спектра ртути с длиной волны 546,1 нм. Если указана длина волны, лежащая в ультрафиолетовой области, необходимо применять фотоэлектрический поляриметр.
Измерение оптического вращения следует проводить при температуре, указанной в статье, обычно при 20—25 °C. Некоторые вещества имеют большой температурный коэффициент. поэтому необходимо особо проследить за тем, чтобы были соблюдены указанные температурные условия.
Удельное оптическое вращение (удельное вращение)
Удельное оптическое вращение жидкого вещества — это угол вращения, измеренный, как указано в статье, вычисленный в пересчете на слой толщиной 100 мм и разделенный на относительную плотность (удельную массу), измеренную при температуре, при которой определено вращение.
Удельное оптическое вращение твердого вещества — это угол вращения, измеренный, как указано в статье, и вычисленный в пересчете на слой толщиной 100 мм раствора, содержащего 1 г вещества в 1 мл.
.. 10000а 10 000а
Удельное вращение = —,
где а наблюдаемое вращение, I—-длина наблюдаемого слоя в мм, с — число граммов вещества, содержащееся в 100 мл раствора, d — относительная плотность и р — число граммов вещества, содержащееся в 100 г раствора.
3—2025
34
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
В Международной фармакопее удельное оптическое вращение выражается как [а]' »где t — температура, а X — длина волны. Для твердых веществ указываются растворитель, если это не вода, и концентрация. Общие указания, касающиеся длин волн и температуры и приведенные выше для оптического вращения, также относятся к измерению удельного оптического вращения. В системе СИ удельное оптическое вращение (сила оптического вращения) приводится в м2-рад/кг, а сила молярного оптического вращения (ап) в м2-рад/моль.
Прибор
Оптическое вращение измеряют при помощи поляриметра. Нулевая точка поляриметра определяется при пустой, но закрытой трубке для жидких веществ и при наполненной определенным растворителем для растворов твердых веществ.
Обычно фармакопейным целям удовлетворяет поляриметр с точностью измерения вращения угла 0,05° и обеспечивающий отсчет с той же точностью; в некоторых случаях может потребоваться поляриметр с точностью измерения до 0,01° вращения угла и обеспечивающий ту же точность отсчета.
Поляриметры для визуального измерения: имеющиеся в продаже приборы обычно сконструированы для использования спектра натрия или ртутной лампы. Следует соблюдать указания изготовителя в отношении соответствующего источника света.
Фотоэлектрические поляриметры: если статья предписывает определение оптического вращения фотоэлектрически, используют фотоэлектрический поляриметр, обеспечивающий точность не менее 0,01°.
Измерение оптического вращения
Для повышения правильности и точности измерения оптического вращения следует учесть приведенные ниже общие указания.
Оптические элементы прибора должны быть безукоризненно чистыми и точно подогнанными. Точка соответствия должна находиться вплотную к обычной нулевой отметке. Источник света должен быть прочно установлен и точно выровнен по отношению к оптической скамье. Он должен быть дополнен системой фильтров, обеспечивающей пропускание света достаточно монохроматической природы. В конструкции прецизионных поляриметров обычно предусмотрены взаимозаменяемые диски для отделения D-линии спектра натрия от линии спектра ртути с. длиной волны 546,1 нм. Для поляри-
ФИЗИЧЕСКИЕ МЕТОДЫ
35
метоов имеющих другую конструкцию, в качестве фильтров можно’ использовать кюветы, содержащие соответственна окрашенные жидкости.
Степень точности и воспроизводимости наблюдении долж--на быть такой, чтобы разность между повторными измерен ниями или между наблюдаемой и истинной величиной вра--щения (последняя устанавливается калибровкой шкалы поляриметра с подходящими стандартами) не превышала */4 интервала, приведенного в статье для вращения испытуемого вещества.
Трубки поляриметра следует заполнять таким образом, чтобы в них не образовывались и не оставались пузырьки воздуха, которые мешают прохождению луча света. Влияние пузырьков уменьшается, если использовать трубку, у которой с одного конца расширено отверстие. Однако при наполнении трубок с одинаковым отверстием, таких, как полумикро- и микротрубки, следует соблюдать соответствующие меры предосторожности.
Закрывая трубки, имеющие съемные оконца с прокладками и колпачками, последние следует затягивать лишь настолько, насколько это нужно, чтобы жидкость не просачивалась между оконцем и самой трубкой. Избыточное давление на оконце может вызвать деформацию, что приводит к помехам при измерении. При определении оптического вращения веществ с низким оптическим вращением желательно освобождать колпачки и затягивать их снова между последовательными отсчетами при снятии показаний оптического вращения, а также при нулевой точке. Таким образом обычно устанавливают различия в показаниях, обусловленных деформацией от оконца, после чего производят соответствующую подстройку для устранения помех.
Во всех статьях, в которых приведены нормы в отношении потери при высушивании, воды или содержания растворителя, требования, касающиеся оптического вращения и удельного вращения, относятся к высушенному, безводному или не содержащему растворителя материалу. При вычислении результатов необходимо принимать во внимание содержание воды или растворителя и потери при высушивании, опреде-ленные методом, указанным в статье.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
ЕсЛИ иссладуется твердое вещество, взвешивают подходящую часть этого вещества и переносят в мерную колбу при оет°Т В°ДЫ ИЛИ ДРУГОГО растворителя, указанного в статье,.
ляя часть растворителя для контрольного определения..
3*
36
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Прибавляют растворитель в количестве, достаточном, чтобы мениск жидкости оказался вблизи, но все же ниже отметки, и доводят температуру содержимого колбы до постоянной, помещая колбу в баню с постоянной температурой. Добавляют растворитель до метки и перемешивают. Переносят раствор в трубку поляриметра, предпочтительно в течение 30 мин с момента растворения вещества, принимая меры для стандартизации времени, прошедшего с момента приготовления раствора, если известно, что вещества подвергаются рацемизации или мутаротации. Во время опыта поддерживают требуемую температуру раствора.
Если вещество представляет собой жидкость, доводят его температуру, если необходимо, до требуемой и переносят жидкость непосредственно в трубку поляриметра.
Если используется поляриметр для визуального измерения, снимают не менее 6 показаний наблюдаемого вращения при требуемой температуре. Берут половину показаний в направлении по часовой стрелке, а вторую половину — в направлении против часовой стрелки. Заменяют раствор оставшимся растворителем и проводят равное число измерений. Если исследуются жидкие вещества, проводят контрольное измерение с пустой, сухой трубкой. Нулевая поправка представляет собой среднюю величину контрольных измерений; ее вычитают из средней величины наблюдаемого вращения, если две цифры имеют один и тот же знак, или прибавляют, если они имеют противоположный знак; таким образом получают скорректированную величину наблюдаемого оптического вращения.
Если используется фотоэлектрический поляриметр, в зависимости от типа прибора снимают меньшее число показаний.
ОПРЕДЕЛЕНИЕ ПОКАЗАТЕЛЯ ПРЕЛОМЛЕНИЯ
Показатель преломления вещества (л) —отношение скорости распространения света в вакууме к скорости его распространения в испытуемом веществе. Этот показатель изменяется в зависимости от длины волны света, используемого при измерении, и температуры, поэтому необходимо указывать эти условия (пк). На практике обычно удобно измерять преломление по отношению к атмосферному воздуху и веществу, а не к вакууму и веществу, так как для фармакопейных целей это не оказывает существенного влияния на наблюдаемые величины.
Показатель преломления может быть также определен как отношение синуса угла падения к синусу угла преломления.
ФИЗИЧЕСКИЕ МЕТОДЫ 37
Измерение показателя преломления используется в фармакопейных целях в основном для установления подлинности жидких веществ. Он может также использоваться для испытания чистоты таких веществ.
Показатель преломления обычно выражают в безразмерных единицах при длине волны 589,3 нм (D-линия спектра натрия) при температуре 20±0,5°С (пЬ0).
Точность измерения должна соответствовать требованиям статьи. Для фармакопейных целей обычно достаточно выражать показатель преломления до трех десятичных знаков.
Прибор
Имеющиеся в продаже приборы обычно сконструированы для использования дневного света, но они калибруются для выражения показателя преломления в безразмерных единицах при длине волны 589,3 нм (D-линия спектра натрия).
Оптические части прибора должны сохраняться абсолютно чистыми. Рабочие поверхности призм не должны иметь царапин.
Помимо соблюдения приведенных выше указаний, следует выполнять инструкции изготовителя в отношении соответствующего источника света.
Прибор калибруют по отношению к стандарту, поставляемому изготовителем; постоянный температурный контроль испытуемой жидкости и проверка чистоты призм осуществляется путем определения показателя преломления дистиллированной воды, который равен 1,3330 при 20 °C и 1,3325 при 25 С.
СПЕКТРОФОТОМЕТРИЯ В ВИДИМОЙ И УЛЬТРАФИОЛЕТОВОЙ ОБЛАСТЯХ СПЕКТРА
Абсорбционная спектрофотометрия — измерение количества поглощенной веществами электромагнитной радиации определенной и узкой волновой области приближенного монохроматического излучения.
Спектральная область, используемая в описанных ниже измерениях, распространяется от коротковолновой ультрафиолетовой до видимой области спектра. Для удобства сравнения эту область можно рассматривать как состоящую из двух ^^« — ультрафиолетовой (190~380 нм) « видимой (380—
ПектР°Ф°тометрия в видимой области (ранее обычно ис-пг>гп3°ВаЛСЯ теРмин колориметрия) — измерение количества ощенного излучения в видимой области спектра, обычно
38
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
немонохроматического, но ограниченного применением окрашенных или интерференционных фильтров.
Ультрафиолетовые и видимые спектры вещества обычно не отличаются высокой степенью избирательности. Тем не менее они чрезвычайно удобны для количественных определений, а для многих веществ служат дополнительным средством установления подлинности.
Пока еще не достигнуто общее соглашение относительно определения терминов, применяемых в спектрофотометрии. Ниже приведены определения и символы, используемые в описании спектрофотометрических испытаний, применяемых в Международной фармакопее.
Поглощение (Д) —десятичный логарифм обратной величины пропускаемости (7). Как синоним поглощения может использоваться термин «плотность внутреннего пропускания»; ранее применявшиеся описательные термины — «оптическая плотность», «абсорбция» и «экстинкция».
Пропускаемость (Т)—частное от деления интенсивности света, прошедшего через вещество, на интенсивность света, падающего на вещество. Ранее применявшиеся термины — «пропускание» и «коэффициент пропускания».
Поглощаемость (а) — частное от деления поглощения (Д) на концентрацию вещества (с), выраженную в граммах на литр, и длину слоя поглощения в сантиметрах (Ь) (а—А/Ьс). С термином «поглощаемость» тесно связаны два других термина: «удельная экстинкция» и «удельный коэффициент поглощения». Термин «удельная экстинкция» (E'fa), обычно используемый в фармакопеях, представляет собой частное от деления поглощения (Д) на концентрацию вещества (с), выраженную в граммах на 100 мл, и длину слоя поглощения в сантиметрах (6); следовательно Е\%ы = 10а. Термин «удельный коэффициент поглощения», предварительно предложенный Комиссией по физико-химическим символам, терминологии и единицам Международного союза теоретической .и прикладной химии (ИЮПАК), представляет собой частное от деления поглощения (Д) на концентрацию (с) и длину слоя поглощения (/); когда для удельного коэффициента поглощения используется символ aSi, который в единицах системы СИ должен выражаться в квадратных метрах на килограмм, aSi= 100 а. Термин «поглощаемость» не следует смешивать с показателем поглощения или коэффициентом экстинкции.
Молярная поглощаемость (е) — частное от деления поглощения (Д) на концентрацию вещества (с), выраженную в
ФИЗИЧЕСКИЕ МЕТОДЫ 39
молях на литр, и длину слоя поглощения в сантиметрах (Ь). Она также является произведением поглощаемости (а) на молекулярную массу вещества. Термин «молярный коэффициент поглощения» (линейный), рекомендованный Комиссией по физико-химическим символам, терминологии и единицам ИЮПАК, представляет собой частное от деления плотности внутреннего пропускания (поглощения) вещества на концентрацию вещества и длину слоя поглощения и соответственно системе СИ должен выражаться в квадратных метрах на моль. Ранее применявшиеся термины — «показатель молярного поглощения» и «коэффициент молярной экстинкции».
Спектр поглощения — часто выражаемое графически отношение поглощения или любой функции поглощения к длине волны или любой функции длины волны.
Применение абсорбционной спектрофотометрии в видимой и ультрафиолетовой областях спектра для методик количественного определения основано на том факте, что поглощаемость вещества обычно является константой, независимой от интенсивности падающего излучения, длины кюветы и концентрации, вследствие чего концентрация может быть определена фотометрически.
Отклонения от приведенных выше величин могут быть обусловлены физическими, химическими или инструментальными переменными. Отклонения вследствие инструментальной ошибки могут быть вызваны влиянием ширины щели, рассеянием света или полихроматическим излучением. Очевидные ошибки могут также появиться в результате изменения концентрации растворенных молекул вследствие ассоциации между молекулами растворенного вещества, между молекулами растворенного вещества и растворителя, а также вследствие диссоциации или ионизации.
Прибор
По существу все типы спектрофотометров обеспечивают прохождение в основном монохроматического света через испытуемое вещество в подходящей форме и измерение интенсивности прошедшей части света. Спектрофотометры состоят из источника энергии, диспергирующего устройства со щелями для выбора волнового диапазона, кюветы или держателя для образца, детектора излучения и присоединенных усилителен, а также измеряющего и регистрирующего устройств, екоторые приборы имеют ручное управление, другие обору-Д ваны автоматическим устройством. Имеются приборы для пользования в видимой области спектра — обычно от 380
40
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
до примерно 700 нм и в видимой и ультрафиолетовой областях спектра — обычно от 190 нм до 700 нм.
Промышленность производит двухлучевые и однолучевые приборы, причем оба типа пригодны для применения. В зависимости от схемы применяемого прибора полученные результаты фиксируются на шкале, цифровом счетчике, регистрирующем или печатном устройстве.
При эксплуатации прибора должны поддерживаться установленные рабочие условия. Оптическая система должна быть размещена таким образом, чтобы любая возможность ошибок, обусловленных рассеянным светом, была сведена к минимуму; это особенно важно для области коротких волн спектра.
Кюветы, обычно используемые в указанной спектральной области, имеют толщину слоя 1 см и снабжены стеклянными или кварцевыми окнами. Могут использоваться кюветы другой толщины. Кюветы, применяемые для испытуемого и контрольного растворов, должны быть одинаковыми и иметь одну и ту же спектральную пропускаемость, если содержат только один растворитель. В ином случае необходимо внести соответствующую поправку.
Калибровка спектрофотометров
Спектрофотометры следует регулярно проверять на точность калибровок. Если используется непрерывный источник излучения, надо калибровать и шкалу длин волн, и фотометрическую шкалу; если используется источник спектральных линий, следует проверять только фотометрическую шкалу.
Ряд источников излучения имеет спектральные линии подходящей интенсивности, распределенные соответствующим образом в избранной спектральной области. Точные значения положения характерных линий кварцево-ртутной дуги — 253,7; 302,25; 313,16; 334,15; 365,48; 404,66 и 435,83 нм. Шкалу длин волн можно также калибровать при помощи соответствующих стеклянных фильтров, которые имеют приемлемые полосы поглощения в видимой и ультрафиолетовой областях. Широко используются стандартные стекла, содержащие дидимий (смесь празеодима и неодима). Лучшим считается стекло, содержащее гольмий. Точные значения положения характерных максимумов фильтров из гольмиевого стекла — 241,5±1; 281,5±1; 360,9±1 и 536,2±3 нм. Фильтры из гольмиевого стекла можно получить из некоторых национальных учреждений и коммерческих источников. Эксплуатационные качества непроверенного фильтра должны быть установлены по отношению к фильтру, подвергнутому правильной проверке.
ФИЗИЧЕСКИЕ МЕТОДЫ
41
Шкалу длин волн можно также калибровать при помощи перхлората гольмия ИР. Точные значения положения характерных максимумов этого раствора — 241,15; 278,2; 361,5 и 536,3 нм. Следует отметить, что положения характерных максимумов растворов перхлората гольмия и фильтров из гольмиевого стекла могут несколько отличаться.
Для калибровки фотометрической шкалы обычно допускается отклонение ± 1 % поглощаемости. Для проверки этой шкалы можно использовать бихромат калия ИР. Точные величины поглощения и удельной экстинкции раствора бихромата калия, содержащего точно 60,06 мг в 1000 мл серной кислоты (0,005 моль/л) ТР, при толщине слоя 1,000 см и допустимые отклонения для А приведены ниже:
Длина волны
А
Допустимое отклонение £1еМ’%
350 нм (максимум)
0,640
0,634—0,646-ЮЗ,56
313 нм (минимум) 0,292
0,289-0,295 48,62
257 им (максимум) 0,865
0,856-0,874
144,02
235 нм (минимум)
0,748
0,740-0,756
124,54
Для проверки фотометрической шкалы имеется ряд стандартных фильтров из неорганического стекла с известной про-пускаемостью; эти фильтры можно также получить из некоторых национальных учреждений и коммерческих источников,, но они могут нуждаться в периодической калибровке.
Работа со спектрофотометром
Изготовители снабжают спектрофотометры подробными инструкциями по их эксплуатации. Для получения значимых и обоснованных результатов оператор спектрофотометра должен быть осведомлен об ограничениях прибора и возможных, источниках ошибок и отклонений. Необходимо точно соблюдать указания инструкций по уходу за прибором, его калибровке и эксплуатации. Если используются регистрирующие-двухлучевые приборы, кювету, содержащую только растворитель, помещают в луч сравнения.
Особое внимание следует обращать на чистоту кювет. Обычно после обработки соответствующим очищающим средством кюветы споласкивают дистиллированной водой, а затем для ускорения высушивания — летучим органическим растворителем. Испытуемые растворы не следует оставлять в кюветах дольше, чем это требуется для выполнения измерения, аботая с кюветами, оператор никогда не должен касаться пальцами наружных поверхностей, через которые проходит луч света. Переносить в кюветы растворитель и испытуемый р створ надо осторожно, чтобы жидкость не попала на наружные поверхности кювет.
42
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Растворители, применяемые в ультрафиолетовой области
Для испытаний и количественных определений с использованием спектрофотометрии в ультрафиолетовой области спектра пригодны многие растворители: вода, спирты, хлороформ, низшие углеводороды, эфиры, разведенные растворы аммиака, гидроокиси натрия, серной и соляной кислот. Растворители различаются по той наименьшей длине волны, при которой снижение пропускаемости препятствует их применению. Следует соблюдать осторожность и использовать растворители, не содержащие примесей, поглощающих в данной спектральной области. В продаже имеются растворители специального спектрофотометрического качества, однако их следует применять только в тех случаях, когда спектральные характеристики растворителя обычного аналитического качества не соответствуют конкретной цели.
Поглощение кюветы для растворителя и его содержимого не должно превышать 0,4 на сантиметр оптического пути при измерении по отношению к воздуху при той же длине волны. Растворитель в кювете должен быть взят той же серии, что и растворитель, использованный для приготовления раствора, и не должен флуоресцировать при той длине волны, при которой проводится измерение. Поглощение этанола ( — 750 г/л), безводного этанола, метанола и циклогексана, используемых в качестве растворителей, измеренное в кювете с толщиной слоя 1 см при 240 нм, не должно превышать 0,10.
Испытания на подлинность в ультрафиолетовой
области спектра
В статьях, описывающих качественные испытания с использованием спектрофотометрии в ультрафиолетовой области спектра, содержатся указания относительно концентрации раствора и толщины слоя. При таких испытаниях удобнее пользоваться регистрирующим прибором. Если указанные условия не подходят для конкретного прибора, рекомендуется изменить толщину слоя раствора, но не его концентрацию.
Некоторые испытания на подлинность с использованием -спектрофотометрии требуют применения стандартных образцов (обычно это Международный химический стандартный образец). В этом случае проба стандартного образца для любых практических целей должна быть изготовлена и одновременно определена в тех же условиях, что и испытуемое вещество. Если в статье нет других указаний, раствор химического стандартного образца должен иметь приблизительно требуемую концентрацию (допускается отклонение в преде
ФИЗИЧЕСКИЕ МЕТОДЫ
43
лах 10%). К идентичным условиям измерения относятся установка длин волн, подстройка ширины щели, установка кювет и поправка на кювету и уровни пропускаемости.
Удобный прием для проведения испытаний на подлинность в ультрафиолетовой области спектра — определение отношения величин поглощения при двух максимумах. Такая методика уменьшает влияние переменных характеристик прибора на испытание и исключает необходимость использования стандартного образца.
Количественные определения в ультрафиолетовой
области спектра
Обычно проведение спектрофотометрических количественных определений требует сравнения величины поглощения, полученной для раствора испытуемого вещества, приготовленного, как указано в статье, с величиной поглощения стандартного образца. В таких случаях спектрофотометрические измерения вначале проводят с раствором стандартного образца, а затем с раствором испытуемого вещества. Второе измерение проводят как можно быстрее после первого и в тех же экспериментальных условиях.
Спектрофотометрические количественные определения обычно проводят в пике спектрального поглощения данного вещества. В статьях приводится общепринятая длина волны для пика спектрального поглощения исследуемого вещества. Известно, что различные спектрофотометры могут давать небольшие отклонения от длины волны этого пика. Практика показывает, что следует использовать длину волны пика, найденную на данном приборе, а не конкретную длину волны, приведенную в статье, при условии, что эти величины отличаются одна от другой не более чем на ±0,5 нм в области 240—280 нм, ±1 нм в области 280—320 нм и ±2 нм в области выше 320 нм. Если разница больше, прибор следует повторно калибровать.
Раствор стандартного образца, обычно Международного химического стандартного образца, должен быть приготовлен и определен так же, как описано в разделе «Испытания на подлинность в ультрафиолетовой области спектра». Вычисления следует производить на основе точно взвешенного количества, а если стандартный образец не был предварительно высушен — в пересчете на высушенное или безводное вещество. в отдельных статьях имеются специальные указания от-сительно того, как должен быть высушен стандартный об-Р зец или как с ним следует поступать перед использовани
44
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
ем. Эти указания необходимо соблюдать, если для отдельного испытания, количественного определения или на этикетке стандартного образца не приведены другие инструкции.
Для того чтобы можно было убедиться, что условия проведения испытания соответствуют установленным, в статье также может быть указана величина поглощения слоя стандартного образца толщиной 1 см. В этом случае определение, выполненное для стандартного образца, считается верным, если наблюдаемая величина поглощения находится в пределах величин, приведенных в статье.
Для количественных определений часто используют приборы с ручной разверткой длин волн. Если для этой цели применяется регистрирующий прибор, следует обратить внимание на правильность калибровки шкалы поглощения при используемой длине волны.
Количественные определения обычно проводят при длинах волн выше 235 нм. Если измерения должны выполняться в области длин волн 190—210 нм, необходимо соблюдать специальные меры предосторожности, такие, как продувание кюветного отделения азотом, использование растворителей специального спектрофотометрического качества и применение прозрачных в этой области кювет.
При измерении поглощения при его максимуме ширина спектральной щели должна быть меньше, чем половина полосы поглощения, иначе результаты будут ошибочно указывать на низкое поглощение. Некоторые вещества в этом отношении требуют особого внимания; во всех случаях ширина щели прибора должна быть такой, чтобы дальнейшее уменьшение щели не приводило к возрастанию величины поглощения. При ширине щели менее 0,01 мм из-за дифракции светового потока измерение может быть затруднено.
Если количественные измерения проводятся достаточно регулярно и часто, можно вместо стандартного образца использовать подходящий калибровочный график, полученный для соответствующего Международного химического стандартного образца. Таким графиком можно воспользоваться, когда для испытуемого вещества поглощение пропорционально концентрации в пределах примерно 75—125% от окончательной концентрации, используемой в количественном определении. При этих условиях величина поглощения, найденная при количественном определении, может быть интерполирована с помощью калибровочного графика, а отсюда рассчитывается результат количественного определения. Такие калибровочные графики надо часто проверять и каждый раз готовить заново для нового прибора и новой серии реактивов. Если результаты оказываются неопределенными или вызывают сомнения,
ФИЗИЧЕСКИЕ МЕТОДЫ
45
следует их непосредственно сравнить с результатами, полученными с Международным химическим стандартным образцом.
Количественные определения в видимой области спектра
Проведение спектрофотометрических количественных определений в видимой области спектра, как и в ультрафиолетовой области, обычно требует одновременного сравнения величины поглощения, полученной при количественном определении испытуемого препарата, с величиной поглощения, полученной со стандартным препаратом, содержащим приблизительно равное количество стандартного образца.
Для спектрофотометрических количественных определений в видимой области спектра должны соблюдаться рекомендации, приведенные в разделе «Количественные определения в ультрафиолетовой области спектра», в том числе применение калибровочных графиков; если необходимо, в эти рекомендации можно внести соответствующие изменения. В видимой области спектра наблюдаемые длины волн не должны отличаться более чем на 5 нм от длин волн, указанных в статье.
СПЕКТРОФОТОМЕТРИЯ В ИНФРАКРАСНОЙ ОБЛАСТИ СПЕКТРА
Инфракрасная область электромагнитного спектра, используемая в фармацевтическом анализе, охватывает интервал 4000—250 см-1 (2,5—40 мкм)1.
Спектрофотометрические измерения в инфракрасной области спектра используются в основном как испытания на подлинность. Инфракрасный спектр уникален для каждого данного химического соединения, за исключением оптических изомеров, имеющих идентичные спектры в растворе. Однако иногда разница в характере инфракрасного спектра данного вещества в твердом состоянии может быть обусловлена полиморфизмом и рядом других факторов, таких, как различия в размере кристаллов и их ориентации, методика растирания и возможное образование гидратов. Присутствие в небольших количествах примесей (до нескольких процентов) в испытуемом веществе обычно незначительно влияет на характер спектра. Для определения подлинности спектр можно срав-котопы еко®ках указаны длины воли; им предшествуют волновые числа, Р е являются обратными величинами длин волн.
46
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
кивать со спектром одновременно приготовленного стандартного образца или со стандартным образцом сравнения.
Термины спектр поглощения, поглощение, пропускаемое™, поглощаемость и удельная экстинкция описаны в разделе-«Спектрофотометрия в видимой и ультрафиолетовой областях спектра» (см. с. 33).
Прибор
Спектрофотометры для инфракрасной области спектра ж основном аналогичны приборам для видимой и ультрафиолетовой областей; они могут отличаться источником энергии,, оптическими материалами, детекторными устройствами. Кроме того, в некоторых приборах монохроматор может располагаться между испытуемым веществом и детектором.
Спектрофотометры, подходящие для испытаний на подлинность, должны работать в области 4000—670 см1 (2,5— 15 мкм). Приборы необходимо часто проверять, чтобы они отвечали условиям эксплуатации, указанным изготовителем; устойчивость шкалы длины волн проверяется с помощью» пленки полистирола.
Для применения методики нарушенного полного внутреннего отражения (НПВО) прибор должен иметь дополнительную приставку однократного или многократного отражения. Приставка состоит из отражающего элемента и подходящего» подъемного устройства, позволяющего установить элемент в: спектрофотометре в положении, обеспечивающем максимум: пропускания.
Применение растворителей
Растворитель, используемый в инфракрасной спектрофотометрии, не должен влиять на материал, обычно хлорид натрия, из которого изготовлены кюветы.
Ни один растворитель при достаточной толщине слоя полностью не прозрачен во всей области инфракрасного спектра. Четыреххлористый углерод Р практически прозрачен (толщиной до 1 мм) от 4000 до 1700 см-1 (от 2,5 до 6 мкм). Хлороформ Р, дихлорметан Р и дибромметан Р являются также подходящими растворителями. Сероуглерод ИК (толщиной до 1 мм) пригоден как растворитель до 250 см-1 (40 мкм), за исключением областей 2400—2000 см-1 (4,2—5,0 мкм) и 1800—1300 см-1 (5,5—7,5 мкм), в которых обнаруживает сильное поглощение. Следует также отметить слабое поглощение сероуглерода при 875—845 см-1 (11,4—11,8 мкм). Другие растворители имеют относительно узкие области пропускаемое™.
ФИЗИЧЕСКИЕ МЕТОДЫ
47
Приготовление испытуемого вещества
Для определения инфракрасного спектра поглощения вещества последнее должно быть соответствующим образом подготовлено. Жидкие вещества можно испытывать непосредственно или в подходящем растворе. Для подготовки твердых веществ обычно используют один из следующих методов: диспергирование мелко измельченного твердого образца в минеральном масле; включение его в прозрачный диск или шарик, который получают путем тщательного смешивания вещества с предварительно высушенным галогенидом калия и прессования смеси в матрице; приготовление раствора в подходящем растворителе. Приготовление вещества для методики нарушенного полного внутреннего отражения описано отдельно.
Для подготовки вещества можно использовать один из следующих методов.
Метод 1. Используют капиллярную пленку жидкости, помещенную между двумя пластинками хлорида натрия, или заполненную кювету подходящей толщины.
Метод 2. Растирают небольшое количество вещества с минимальным количеством подходящего минерального масла или другой подходящей жидкости до получения однородной кремообразной пасты; 2—5 мг испытуемого вещества обычно достаточно для приготовления требуемой пасты, которая должна быть полупрозрачной на свет. Сжимают часть пасты между двумя пластинками хлорида натрия или между другими подходящими пластинками.
Метод 3. Растирают твердое вещество с сухим мелкоиз-мельченным галогенидом калия (бромид калия ИК, хлорид калия ПК); соотношение вещества и галоида должно быть приблизительно 1:200, например 1,5 мг в 300 мг галоида для призменных приборов, и около 1:300, например 0,1 мг в 300 мг галоида для приборов с дифракционной решеткой. Взятое количество вещества должно быть таким, чтобы масса вещества, приходящаяся на единицу площади диска, составляла примерно 5—15 мкг на 1 мм2, изменяясь в зависимости от молекулярной массы и в известной мере от типа используемого прибора. Часть смеси помещают в специальную матрицу и в условиях вакуума прессуют. В продаже имеются матрицы, при использовании которых необходимо выполнять указания изготовителя. Укрепляют полученный диск в подходящем держателе. Неудовлетворительные диски °гУа получаться из-за неправильного или слишком интен-вного растирания, наличия влаги или других примесей в
48
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
галоиде. Если изготовление диска не связано с особыми трудностями, диск лучше отбраковать, когда при визуальном осмотре обнаруживается отсутствие гомогенности или пропус-каёмость примерно при 2000 см-1 (5 мкм) в отсутствие специфической полосы поглощения составляет менее 75% без компенсации.
Метод 4. Готовят раствор жидкости или твердого вещества в подходящем растворителе, устанавливая такую концентрацию и толщину слоя в кювете, чтобы получить удовлетворительный спектр в достаточно широком волновом диапазоне.
Установление подлинности по стандартному образцу
Готовят исследуемое вещество и стандартный образец одинаковым методом и регистрируют спектр каждого из них в интервале примерно от 4000 до 670 см-1 (от 2,5 до 15 мкм). Концентрация вещества должна быть такой, чтобы наиболее сильный пик, присущий веществу, имел пропускаемость между 5 и 25%.
Если положения и относительные интенсивности максимумов поглощения на спектре испытуемого вещества, подготовленного по методу 2 или 3, не согласуются с таковыми на спектре стандартного образца, это может быть вызвано различиями в -кристаллической форме. Если есть опасения, что могут возникнуть подобные трудности, вещество, когда это i возможно, исследуют в растворе. Если определение в растворе практически неосуществимо, следует попытаться получить путем перекристаллизации одинаковую кристаллическую форму стандартного образца и испытуемого вещества.
Если спектр минерального масла, использованного по методу 2, мешает определению в данной области, можно допол- i нительно приготовить пасту из испытуемого вещества, диспергированного в такой среде, как подходящее фторированное углеводородное масло или гексахлорбутадиен. Затем регистрируют спектр в тех областях, в которых минеральное масло обнаруживает сильное поглощение.
Установление подлинности по спектру сравнения
Готовят испытуемое вещество точно так, как описано в листовке, приложенной к Международному спектру сравнения, и регистрируют спектр в области примерно от 4000 до 670 см-1 (от 2,5 до 15 мкм), используя прибор, часто проверяемый на соответствие нормам эксплуатации, установленным изготовителем. Для того чтобы сделать допуск на возможную
ФИЗИЧЕСКИЕ МЕТОДЫ
49
разницу в калибровке шкалы длин волн между прибором, на котором был получен Международный спектр сравнения, и прибором, на котором должен быть зарегистрирован спектр данного вещества, на Международный спектр сравнения накладывают стандартный спектр максимума поглощения полистирола примерно при 2851 см-1 (3,51 мкм), 1601 см-1 (6,25 мкм) и 1028 см-1 (9,73 мкм). Аналогичный спектр максимума поглощения полистирола должен быть наложен на спектр исследуемого вещества. С учетом этих максимумов поглощения полистирола испытание на подлинность считают положительным, если основные максимумы поглощения на спектре испытуемого вещества согласуются с соответствующими максимумами Международного спектра сравнения. При сравнении двух спектров следует сделать допуск на возможные различия в разрешающей силе прибора, на котором был получен Международный спектр сравнения, и прибора, на котором получен спектр исследуемого вещества. Для оценки этих различий следует использовать Международный спектр сравнения полистирола, который снят на том же приборе, что и коллекция Международных спектров сравнения. Следует отметить, что наибольшее отклонение, возникающее из-за различий в разрешающей силе, может отмечаться в области между 4000 и 2000 см-1 (2,5—5 мкм).
Методика нарушенного полного внутреннего
отражения
Для определения инфракрасного спектра поглощения вещества с помощью методики нарушенного полного внутреннего отражения твердое вещество обычно следует тонко измельчить. Порошок можно поместить либо непосредственно против призмы приставки, либо для улучшения контакта может быть использована клейкая лента. Измельченное вещество распределяют на клеющей стороне ленты так, чтобы образовался почти прозрачный слой; ленту прижимают к отражающему элементу стороной, на которой находится порошок. Затем прикрепляют пластинку-подложку или на 1—2 мин слегка прижимают с помощью зажима. Наконец, отражающий элемент помещают в держатель. Для этой методики предпочтительно использовать ленту с клеем на основе натурального каучука. При исследовании некоторых пластических материалов их можно помещать непосредственно на отражающий элемент.
Необходимо тщательно контролировать правильную подгонку приставки в приборе.
4—2025
50
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
АТОМНАЯ АБСОРБЦИОННАЯ СПЕКТРОФОТОМЕТРИЯ
Пламенная атомная абсорбционная спектрофотометрия, или пламенная абсорбционная спектрофотометрия, — процесс, во время которого атомы, ионы или ионные комплексы элемента, распыленного до основного состояния в пламени, поглощают свет при длине волны, характерной для данного элемента.
Если процесс поглощения протекает в пламени при воспроизводимых условиях, величина поглощения (обратный логарифм пропускаемое™) пропорциональна числу поглощающих атомов. На этом основании можно построить калибровочные кривые, позволяющие оценивать неизвестные величины поглощения в единицах концентрации элемента в растворе.
Следует отметить, что, помимо пламенной атомной абсорбционной спектрофотометрии, разрабатывается методика беспламенной атомной абсорбционной спектрофотометрии.
Прибор
Пламенный атомный абсорбционный спектрофотометр состоит из источника излучения, который обеспечивает характерную спектральную линию определяемого элемента, системы распыления и сжигания для введения образца раствора в пламя и детекторной системы.
Источником излучения обычно служит газоразрядная лампа с полым катодом; катод при возбуждении испускает предусмотренное условиями испытания излучение. Так как излучение, поглощаемое элементом в испытуемом растворе, обычно имеет ту же длину волны, что и его линия излучения, элемент в лампе с полым катодом должен быть тем же, что и определяемый элемент. Как правило, для каждого элемента используется своя лампа, однако в настоящее время -в продаже имеются лампы, в которых совмещены комбинации некоторых элементов.
Система распыления и сжигания вводит испытуемый раствор в пламя, которое обычно образуется при сгорании смесей воздух — ацетилен и воздух — водород. Фактически пламя представляет собой как бы нагретую камеру для образца.
Детекторная система, служащая для регистрации сигнала, поступающего из камеры, состоит из оптического диспергирующего устройства, например монохроматора или фильтра, выделяющего резонансную линию элемента, и детектора излучения, например фотоумножителя, а также системы индикации для снятия величины поглощения.
ФИЗИЧЕСКИЕ МЕТОДЫ
51
Интерференционное излучение от пламени во время сгорания смеси может быть устранено фильтрацией сигнала определенной частоты от источника. Затем детектор настраивается на переменную составляющую сигнала, так что постоянная составляющая игнорируется. Поэтому такая детекторная система отмечает только изменения сигнала от источника с полым катодом; этот сигнал прямо пропорционален числу атомов, подлежащих определению в испытуемом веществе.
Применение растворителей
Идеальным растворителем является такой растворитель, который в минимальной степени мешает процессам поглощения и излучения и который дает в пламени нейтральные атомы. Если существует значительная разница между поверхностным натяжением или вязкостью испытуемого вещества и теми же свойствами стандартного раствора, растворы будут распыляться и распадаться с разной скоростью, что приведет к существенным различиям в генерируемых сигналах. Концентрация кислоты в растворах также влияет на процесс поглощения. Поэтому для приготовления испытуемого раствора и для приготовления стандартного раствора следует использовать либо один и тот же растворитель, либо эти растворители должны быть максимально сходными по указанным свойствам. При этом должны получаться растворы, легко распыляемые через трубку устройства для распыления и сжигания. Если в растворах имеется твердое вещество, оно может мешать определению, поэтому, когда возможно, содержание твердого вещества в растворах не должно превышать 2%.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Готовят не менее 3 стандартных растворов определяемого элемента в том интервале концентраций, который рекомендован изготовителем для элемента и используемого прибора. Любой реактив, используемый для приготовления раствора исследуемого вещества, следует прибавлять к стандартным образцам в той же концентрации.
Для измерений на приборах, откалиброванных по пропускаемое™, впрыскивают в пламя воду и подстраивают отсчет из всю шкалу; для такого типа приборов никаких других подстроек не требуется. Для приборов, откалиброванных по глощению, следует руководствоваться указаниями изгото-теля прибора. Впрыскивают каждый стандартный раствор пламя 3 раза, отмечая устойчивое значение полученного азания и промывая прибор водой после каждого впрыс-4*
52
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
кивания. Готовят калибровочную кривую зависимости среднего значения для каждой группы из 3 отсчетов от концентрации.
Готовят раствор испытуемого вещества, как указано в статье, но, если необходимо, подбирают концентрацию определяемого элемента таким образом, чтобы она находилась в интервале концентраций, рекомендованном для используемого прибора. Впрыскивают раствор в пламя 3 раза, отмечают показания и промывают прибор водой после каждого впрыскивания. Используют среднее значение показаний для определения содержания элемента по калибровочной кривой.
ФЛУОРЕСЦЕНТНАЯ СПЕКТРОФОТОМЕТРИЯ
Флуоресцентная спектрофотометрия — это измерение флуоресценции, т. е. фотолюминесценции, испускаемой веществом, подвергнутым воздействию света, ультрафиолетового или другого электромагнитного излучения. Как правило, максимум интенсивности света, испускаемого флуоресцирующим раствором, обнаруживается при длине волны, большей чем максимум возбуждающего света обычно на 20—30 нм.
Интенсивность света, испускаемого флуоресцирующим раствором, при определенных условиях находится в простой зависимости от концентрации растворенного вещества и, следовательно, может быть использована для анализа. Трудно, однако, измерить абсолютную интенсивность флуоресценции, и обычно измерения проводят по отношению к разведениям правильно выбранного стандартного образца. Общая схема флуоресцентной спектроскопии состоит в возбуждении флуоресценции излучением при длине волны максимума поглощения и в измерении или сравнении интенсивности флуоресци- . рующего света с флуоресценцией определенного стандартного раствора. Флуоресцирующий свет должен быть тщательно отделен от рассеянного света, падающего на вещество.
Термины
Интенсивность флуоресценции — эмпирическое выражение флуоресцентной активности, обычно приводимое в условных единицах, пропорциональных ответу детекторов.
Спектр флуоресцентного излучения — обычно представляемое в графической форме отношение интенсивности испускаемого излучения к длине волны.
ФИЗИЧЕСКИЕ МЕТОДЫ
53
Спектр флуоресцентного возбуждения — обычно представляемое в графической форме отношение максимума интенсивности излучения, испускаемого активированным веществом, к длине волны падающего излучения.
Прибор
Измерение интенсивности флуоресценции можно провести с помощью простого флуорометра с фильтрами (иногда прибор называют флуориметром). Такой прибор состоит из источника излучения, первичного фильтра, камеры для вещества вторичного фильтра и системы обнаружения флуоресценции. В большинстве таких флуорометров детектор располагается под углом 909 к падающему лучу, что позволяет падающему излучению проходить через испытуемый раствор без загрязнения выходного сигнала, получаемого детектором флуоресценции. Однако на детектор неизбежно попадает некоторое количество падающего излучения в результате внутреннего рассеивания — свойства, присущего самим растворам; таким же образом влияет присутствие пыли или других твердых веществ. Для удаления этого остаточного рассеивания используют фильтры. Первичный фильтр отбирает коротковолновое излучение, способное вызывать возбуждение испытуемого вещества, в то время как вторичный фильтр, обычно строго отсекающего типа, пропускает флуоресценцию при большей длине волны, но блокирует рассеянное возбуждающее излучение.
В большинстве флуорометров в качестве детекторов используются фотоумножители; существует много типов фотоумножителей со специальными характеристиками в отношении спектральной области максимальной чувствительности, электрического шума и усиления. После усиления фотоэлектронного тока его значение либо отсчитывается визуально на измерительном приспособлении, либо регистрируется.
Флуоресцентный спектрофотометр отличается от флуорометра с фильтрами тем, что фильтры в нем заменены монохроматорами призменного или решетчатого типа. Для аналитических целей флуоресцентный спектрофотометр имеет преимущества перед фильтрующим флуорометром в отношении избирательности длины волны, возможности быстрого приспособления к условиям опыта и удобства пользования.
Во флуорометрах и флуоресцентных спектрофотометрах используются различные источники излучения. Ртутные лампы относительно стабильны и излучают энергию в основном при дискретных длинах волн. Вольфрамовые лампы обеспе-вают достаточно равномерное излучение по всей видимой
54
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
области спектра. В флуоресцентных спектрофотометрах часто используется ксеноновая дуговая лампа высокого давления, поскольку она достаточно равномерно излучает энергию от ультрафиолетовой до инфракрасной области спектра.
Во флуоресцентных спектрофотометрах монохроматоры имеют щель. Узкая щель обеспечивает высокое разрешение и спектральную чистоту, большая щель — высокую интенсивность. Выбор ширины щели определяется разницей в длине волны возбуждающего и испускаемого излучения, а также требуемой степенью чувствительности.
Кюветы, используемые при измерении флуоресценции, могут быть прямоугольными, аналогичными кюветам, применяемым в абсорбционных спектрофотометрах; их отличие состоит в том, что все 4 вертикальные стенки и основание должны быть отполированы; можно применять также кюветы в виде пробирок с плоским отполированным основанием. Удобный размер кювет 2—3 мл, но некоторые приборы могут быть снабжены небольшими кюветами емкостью 0,1—0,3 мл или капиллярной кюветой, требующей еще меньше раствора.
Стандартизация j
Флуорометры и флуоресцентные спектрофотометры надо j ежедневно стандартизовать по стабильному флуорофору для обеспечения правильной воспроизводимости ответа. Изменения обусловлены факторами, связанными с самим прибором, такими, как различия в интенсивности -лампы и чувствительности фотоумножителя. Флуорофором может служить чистый образец флуоресцирующего вещества, подвергаемого анализу, или другое легко очищаемое флуоресцирующее вещество с полосами поглощения флуоресценции такими же, что и у ис- ] пытуемого вещества. Подходящим флуорофором для голубой флуоресценции является хинин в разведенной серной кислоте, для зеленой флуоресценции — флуоресцеин натрий и для красной — родамин.
Калибровка шкалы длин волн
Шкалу длин волн флуоресцентного спектрофотометра следует периодически калибровать.
Приготовление раствора
Необходимо правильно выбирать используемый для измерения растворитель. На интенсивность и спектральное распределение флуоресценции могут существенно влиять сам растворитель, его чистота и величина pH.
ФИЗИЧЕСКИЕ МЕТОДЫ
55
Концентрация испытуемых растворов для флуоресцентной спектрофотометрии обычно в 10—100 раз слабее концентрации растворов, применяемых в абсорбционной спектрофотометрии. Для аналитического применения необходимо, чтобы интенсивность флуоресценции была линейно связана с концентрацией вещества в области, используемой для измерений- однако если раствор слишком концентрированный, то значительная часть падающего света поглощается веществом у поверхности кюветы, что приводит к снижению интенсивности потока света, достигающего центра кюветы. В результате получается, что само вещество действует как «внутренний фильтр». Но поскольку флуоресцентная спектрофотометрия — высокочувствительный метод, часто можно использовать растворы в концентрации порядка 10-5—10~7 моль/л.
Область, в которой флуоресценция пропорциональна концентрации флуоресцирующего вещества, обычно очень узка; поэтому соотношение (с—d)[(a—b), где а — интенсивность флуоресценции для стандартного вещества, b — то же для соответствующего контрольного вещества, с — интенсивность флуоресценции для испытуемого вещества и d — то же для соответствующего контрольного раствора, должно быть не менее 0,40 и не более 2,50. Поэтому необходимо построить рабочую кривую зависимости интенсивности флуоресценции с поправкой на контрольный раствор от концентрации.
Методика измерения
Измерения флуоресценции чувствительны к присутствию в испытуемом растворе твердых частиц. Такие частицы могут снижать интенсивность возбуждающего луча или давать ошибочно высокие показания вследствие множественного отражения в кювете. Поэтому целесообразно удалять твердые частицы центрифугированием; можно применять и фильтрование, но некоторые виды фильтровальной бумаги могут содержать флуоресцирующие примеси.
Кислород, растворенный в растворителе, обладает сильным гасящим эффектом. Поэтому интенсивность флуоресценции возрастает при применении дегазирующей методики, такой, как барботирование через испытуемый раствор азота или Другого инертного газа.
Для флуоресцентной спектрофотометрии часто важны температурные условия. Для некоторых веществ интенсивность флуоресценции может снизиться до 1—2% на градус повышения температуры. В таких случаях, если необходимо получить максимальную точность, следует использовать термостатированные кюветы. Для рутинного анализа может быть
56
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
достаточным быстрое проведение измерений, чтобы испытуемый раствор не успевал нагреться под действием интенсивного источника света.
Многие флуоресцирующие вещества светочувствительны. Будучи помещены во флуоресцентный спектрофотометр, они могут подвергаться фотохимическому разложению на более или менее флуоресцирующие продукты. Такой эффект можно обнаружить путем наблюдения показаний детектора во времени и снизить его, уменьшив интенсивность источника света с помощью фильтров или экранов.
ТУРБИДИМЕТРИЯ И НЕФЕЛОМЕТРИЯ
Турбидиметрия — измерение степени ослабления луча света, падающего на частицы, суспендированные в среде; измерение проводят в прямо проходящем луче света. Измерения проводят при помощи стандартного фотоэлектрофотометра с фильтром или спектрофотометра с освещением при подходящей длине волны.
Нефелометрия — измерение интенсивности света, рассеиваемого суспендированными частицами; измерение обычно производят перпендикулярно к падающему свету.
Турбидиметрия и нефелометрия могут быть использованы для измерения осадков, образующихся при взаимодействии очень сильно разведенных растворов реактивов, или других частиц, таких, как суспендированные бактериальные клетки. Для получения постоянных воспроизводимых результатов следует тщательно проверять все варьирующие показатели. При измерении суспендированных бактериальных клеток могут возникнуть трудности, обусловленные явлением двойного лучепреломления. Если возможен правильный контроль, то можно измерять чрезвычайно сильно разведенные суспензии.
Термины
Пропускаемость (Т)—частное от деления интенсивности лучевого потока, прошедшего через вещество, на интенсивность лучевого потока, падающего на вещество.
Мутность (S) — мера эффекта рассеивания света суспендированными частицами.
Степень мутности (т) — при измерениях рассеивания света степень мутности определяется как уменьшение интенсивности падающего света на единицу толщины слоя данной суспензии.
ФИЗИЧЕСКИЕ МЕТОДЫ
57
Прибор
Степень мутности может быть измерена на стандартном Фотоэлектрическом фотометре с фильтрами или на спектрофотометре, предпочтительно с освещением в красно-оранжевой области спектра (например, за счет использования синего фильтра).
Для нефелометрических измерений требуется прибор с фотоэлементом, расположенным таким образом, чтобы получать рассеянный, а не проходящий через образец свет; это требование также относится к флуорометрам, так что, как правило, флуорометры при соответствующем выборе фильтров могут применяться в качестве нефелометров.
Инструментальное измерение
При проведении инструментального измерения следует удостовериться в том, что осаждение измеряемых частиц столь незначительно, что им можно пренебречь. Обычно для этого в жидкую суспендирующую среду добавляют защитный коллоид. Важно, чтобы результаты оценивались путем сравнения с показаниями, характеризующими известные концентрации суспендированного вещества и полученными при тех же условиях.
Визуальное сравнение
Проводят сравнение степени мутности в пробирках, имеющих одинаковый внутренний диаметр и максимально совпадающих по всем другим характеристикам. Для этого подходят плоскодонные сосуды из прозрачного стекла емкостью около 70 мл с внутренним диаметром около 23 мм. Для сравнения степени мутности пробирки следует рассматривать в горизонтальном положении, на черном фоне, с помощью источника света, расположенного сбоку от пробирок.
ОКРАСКА ЖИДКОСТЕЙ
Испытание для оценки окраски жидкостей проводится путем сравнения испытуемого раствора, приготовленного, как указано в статье, со стандартным окрашенным раствором, указанным в статье. Состав стандартного окрашенного раствора выбирается в зависимости от оттенка и интенсивности окраски испытуемого раствора, соответствующих пределам, Допускаемым спецификациями.
58
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Если в статье нет других указаний, сравнение проводят в плоскодонных пробирках из прозрачного стекла, имеющих одинаковый внутренний диаметр и максимально совпадающих по всем другим характеристикам (подходят пробирки с внутренним диаметром около 16 мм). Используют 10 мл испытуемого раствора и 10 мл стандартного окрашенного раствора; глубина слоя жидкости должна быть около 50 мм. Интенсивность окраски испытуемого раствора не должна превышать интенсивности окраски стандартного раствора при рассмотрении сверху вниз по вертикальной оси на белом фоне в рассеянном свете.
Исходные окрашенные стандартные растворы
Желтый исходный окрашенный стандарт ИР
К 9,5 мл окрашенного раствора кобальта ИР прибавляют 1,9 мл окрашенного раствора меди ИР, 10,7 мл окрашенного раствора бихромата ИР, 4,0 мл окрашенного раствора железа ИР, доводят до 100,0 мл серной кислотой (~10 г/л) ИР и перемешивают.
Красный исходный окрашенный стандарт ИР
К 40,5 мл окрашенного раствора кобальта ИР прибавляют 6,1 мл окрашенного раствора меди ИР, 6,3 мл окрашенного раствора бихромата ИР, 12,0 мл окрашенного раствора железа ИР, разводят до 100,0 мл серной кислотой (~10 г/л) ИР и перемешивают.
Зеленый исходный окрашенный стандарт ИР
К 3,5 мл окрашенного раствора кобальта ИР прибавляют 20,1 мл окрашенного раствора меди ИР, 10,4 мл окрашенного раствора бихромата ИР, 4,0 мл окрашенного раствора железа ИР, доводят до 100,0 мл серной кислотой (~10 г/л) ИР и перемешивают.
Коричневый исходный окрашенный стандарт ИР
К 35,0 мл окрашенного кобальта ИР прибавляют 17,0 окрашенного раствора меди ИР, 8,0 мл окрашенного раствора бихромата ИР, доводят до 100,0 мл раствором железа ИР и перемешивают.
ФИЗИЧЕСКИЕ МЕТОДЫ
59
Стандартные окрашенные растворы
Стандартный окрашенный раствор готовят соответствующим разведением исходных стандартных растворов (желтого, красного, зеленого и коричневого ИР) серной кислотой (~ 1.0 г/л) ИР. Обозначение стандартного окрашенного раствора состоит из двух букв, указывающих исходный стандартный раствор (Жл — желтый, Кр — красный, Зл — зеленый и Кч — коричневый), и цифры, указывающей разведение, как приведено ниже:
Номер разведения стандартного окрашенного раствора
0
1
2
3
4
5
6
7
Исходный стандартный раствор (мл)
0,78 1,56 3,12 6,25 12,50 25,00 50,00 100,00
Серная кислота (~10 г!л) ИР (мл)
99,22
98,44
96,88
93,75
87.50
75,00
50,00
-Стандартные окрашенные растворы номеров 4—7 можно хранить в стеклянных запаянных ампулах, защищенных от солнечного света; более разбавленные стандартные окрашенные растворы следует готовить по мере необходимости.
РАДИОФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ
Обращение с радиоактивными фармацевтическими препаратами и их испытания требуют применения специальных методов, обеспечивающих получение правильных результатов и сведение до минимума риска облучения для обслуживающего персонала. Все операции должны проводиться людьми, специально обученными обращению с радиоактивными веществами, или под их наблюдением.
Определения
Изотоп
Разновидность атома, характеризующаяся его массовым числом, атомным числом и состоянием энергии ядра, при условии, что среднее время существования в этом состоянии Достаточно длительно, чтобы быть отмеченным.
60
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Радиоактивность
Способность определенных изотопов к излучению за счет произвольного превращения их ядер в ядра других изотопов.
ПРИМЕЧАНИЕ. Термин «разложение (распад)» широко используется в качестве альтернативного для обозначения термина «превращение». Последний является предпочтительным, так как, не вызывая семантических трудностей, служит для обозначения и тех процессов, в ходе которых не излучаются никакие частицы.
Радиоизотоп
Радиоактивный изотоп.
Единицы радиоактивности
Активность некоторого количества радиоактивного материала выражается в единицах, определяемых числом ядер-ных превращений в единицу времени. В системе СИ единицей активности является беккерель (Бк), специальное наименование величины, обратной секунде (с-1). Выражение активности в беккерелях, следовательно, показывает, число превращений в секунду; 1 кюри (Ки) =3,7Х 1010 Бк.
Коэффициенты пересчета беккереля и кюри и их дольные приведены в разделе «Единицы измерения» (см. с. 16).
Период полураспада
Период полураспада — время, в течение которого радиоактивность уменьшается наполовину по отношению к исходной величине.
ПРИМЕЧАНИЕ. Скорость радиоактивного распада постоянна и характерна для каждого отдельного радиоизотопа. Экспоненциальная кривая математически описывается уравнением:
N=Noe~kt,
где N — число атомов, распадающихся за время t, No — число атомов к началу промежутка времени при /=0 и X—постоянная распада, характерная для каждого отдельного радиоизотопа. Период полураспада связан с постоянной распада следующим отношением:
„ 0,693
гч2 - X ‘
Поправки на процесс радиоактивного распада рассчитываются из экспоненциального уравнения, из таблиц распада или определяются по кривой распада, построенной для определенного радиоизотопа (см. рис. 1).
ФИЗИЧЕСКИЕ МЕТОДЫ
61
Радиоактивная концентрация
Радиоактивная концентрация раствора — это радиоактивность в единице объема раствора. Как и во всех положениях, касающихся радиоактивности, необходимо указывать время стандартизации. Для радиоизотопов с периодом полураспада менее 30 сут^ время стандартизации должно быть указано на ближайший час. Для радиоизотопов с периодом полураспада менее 1 сут требуется более точное указание времени.
Удельная радиоактивность (или удельная активность)
Удельная активность препарата из радиоактивного материала радиоактивность на единицу массы элемента или соответствующего соединения.
Периоды полураспада
62
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
ПРИМЕЧАНИЕ. Обычно принято указывать соответствующий радиоизотоп; кроме того, необходимо обозначать время: «1 мкюри йода-131 на 1 мг о-йодгиппуровой кислоты на 12 ч 00 мии по гринвичскому времени на 1' января 1979 г.», или «40 МБк селена-76 иа 1 мг селеиометионина на 1 января 1979 г.».
Удельная радиоактивность часто определяется не непосредственно, а вычисляется на основании известной радиоактивной концентрации раствора и химической концентрации радиоактивного вещества. Так, если раствор содержит х мкюри 13Ч в 1 мл и если ,3Ч целиком представлен в виде натриевой соли о-йодгиппуровой кислоты, концентрация которой у мг в 1 мл, то удельная активность на это время составляет:
х/у мкюри йода-131 на 1 мг о-йодгиппуровой кислоты.
Когда необходимо, следует учитывать радиохимическую чистоту препарата (см. ниже).
«Удельная активность» — термин, используемый в радиохимии. Так как слово «активность» применяется в фармакопее для обозначения других понятий, этот термин во избежание двойного толкования следует, когда необходимо, изменять иа «удельную радиоактивность».
Радиоизотопная чистота
Радиоизотопная чистота препарата — выраженная в процентах часть общей радиоактивности препарата, которая присутствует в форме указанного радиоизотопа.
ПРИМЕЧАНИЕ. Некоторые радиоизотопы распадаются на изотопы, которые сами являются радиоактивными: соответственно их называют материнскими или родительскими) и дочерними радиоизотопами. Такие дсчериие радиоизотопы часто исключают при расчетах радиоизотопной чистоты: например, йод-131 всегда будет содержать дочерний ксенои-131, но последний ие будет рассматриваться как примесь, потому что его присутствие неизбежно.
При определении радиохимической чистоты следует измерять радиоактивность в соответствующих единицах, т. е. в числе ядерных превращений, происходящих за единицу времени и выражаемых в кюри или беккерелях. Если, например, препарат йод-125 содержит 99 мкюри йода-125 и 1 мкюри йода-126 и не содержит других радиоизотопов, то такой препарат характеризуется 99% радиоизотопной чистотой. Следует отметить, что относительные количества йода-125 и йода-126, а следовательно, и радиоизотопная чистота будут меняться со временем. Поэтому выражение радиоизотопной чистоты должно содержать указание на время, например: «Не более 1% от общей радиоактивности приходится иа йод-126 на день, указанный на этикетке». Если период полураспада радиоизотопов менее 30 сут, следует также указывать час стандартизации.
Вполне ясно, что для установления радиоизотопной чистоты препарата надо иметь данные об активности (и, следовательно, об идентичности) каждого присутствующего радиоизотопа. Простых и надежных методов идентификации и измерения всех радиоизотопных примесей, которые могут присутствовать в препарате, не существует. Выражение радиоизотопной чистоты либо будет зависеть от заключения лица, устанавливающего эту величину, либо должно оцениваться с учетом примененного метода, например: «С помощью гамма-сцинтилляциоииой спектрометрии с использованием детектора йодида натрия не было обнаружено никаких радиоизотопных примесей».
ФИЗИЧЕСКИЕ МЕТОДЫ
63
Радиохимическая чистота
Радиохимическая чистота — процентное содержание указанного радиоизотопа, который находится в указанной химической форме. Так как радиохимическая чистота может меняться во времени, в основном за счет радиоактивного распада, должно быть указано время, к которому относится предел радиохимической чистоты.
ПРИМЕЧАНИЕ. Если, например, указано, что препарат цианокобаламин (57Со) имеет радиохимическую чистоту 99%, то это означает, что 99% кобальта-67 присутствует в форме цианокобаламина. Радиохимические примеси могут включать такие вещества, как ион кобальта (57Со) или ок-сикобаламии (57Со).
Возможное присутствие радиоизотопных примесей не т1ринимается во внимание при определении радиохимической чистоты. Если радиоизотопная примесь не изотопна по отношению к указанному радиоизотопу, тогда она не может иметь идентичную химическую форму. Если же радиоизотопная примесь изотопна по отношению к указанному радиоизотопу, то возможно, и даже весьма вероятно, что она будет иметь ту же химическую форму.
Радиохимические примеси могут появляться во время приготовления материала или во время хранения вследствие обычного химического разложения, или, что часто более важно, вследствие радиационного разложения (т. е. вследствие физического и химического действия радиации).
Производство радиоактивных фармацевтических препаратов и обращение с ними В * * * * * * * * * * * * * * * * * *
В следующих разделах изложены специальные вопросы,
которые касаются статей, содержащих спецификации для
радиоактивных лекарственных средств. Обычно материально-
техническая база производства, средства применения и хра-
нения радиоактивных фармацевтических препаратов подле-
жат лицензированию национальными органами. Часто для
выдачи лицензии эти средства должны удовлетворять требо-
ваниям двух сводов правил — правил, касающихся фармацев-
тических препаратов, и правил, касающихся радиоактивных
материалов. Каждый изготовитель и потребитель должен
быть детально осведомлен о национальных требованиях, со-
держащихся в соответствующих статьях этих правил.
Носители
Как правило, количество радиоактивного вещества в фар-
мацевтических радиоактивных препаратах слишком мало,
чтобы его можно было измерить обычными химическими или
физическими методами. Так как столь малые количества ве-
щества нельзя разделить или очистить обычными методами,
для удобства обращения с активным веществом к нему в
64 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
процессе производства может быть добавлен носитель в виде неактивного материала — изотопа данного элемента или неизотопа, но химически близкого к изотопу. Так, в препарате Nat г Н Phosphatis (32Р) Inject io [Натрия фосфата (32Р) раствор для инъекций] присутствует носитель фосфат натрия; в некоторых коллоидных препаратах технеция-99т в качестве носителя используется рений. Количество добавляемого носителя должно быть достаточно малым, чтобы избежать нежелательных физиологических эффектов. Масса элемента, образующегося при ядерной реакции, может быть увеличена за счет массы неактивного изотопа, присутствующего в облучаемом материале или в реактивах, используемых в процессах разделения.
Термин без носителя относится к радиоактивным препаратам, в которые в процессе производства не добавляется намеренно никаких носителей.
Обнаружение и измерение радиоактивности
Радиоактивные превращения могут быть связаны с излучением заряженных частиц, процессом электронного захвата или процессом изомерного перехода.* Заряженные частицы, излучаемые из ядер, могут быть альфа-частицами (ядра гелия с массовым числом 4) или бета-частицами (электроны с положительным или отрицательным зарядом, р— или §+ со-•ответственно; последние известны как позитроны). Излучение заряженных частиц из ядра может сопровождаться гамма-излучением, имеющим ту же физическую природу, что и рентгеновское излучение. Гамма-лучи испускаются также в процессе изомерного перехода (ИП). Рентгеновские лучи, которые могут сопровождаться гамма-лучами, испускаются в процессе электронного захвата (ЭЗ). Позитроны уничтожаются при взаимодействии с веществом, причем этот процесс сопровождается испусканием двух гамма-лучей, каждый из которых имеет энергию 0,511 мэВ.
Физические характеристики радиоизотопов приведены в табл. 1.
Методы, используемые для обнаружения и измерения радиоактивности, зависят от природы и энергии радиации. Радиоактивность может быть обнаружена и/или измерена различными приборами, принцип действия которых основан на улавливании и регистрации количества возникших ионов газов, на измерении флуоресценции отдельных твердых веществ и жидкостей или измерении эффекта воздействия излучения на фотоэмульсию.
ФИЗИЧЕСКИЕ МЕТОДЫ
65
Как правило, счетное устройство состоит из чувствительного элемента и электронного счетчика импульсов. В качестве чувствительного элемента может быть использована трубка Гейгера — Мюллера, пропорциональный счетчик или сцинтилляционный детектор, в котором в сочетании со сцинтиллятором используется фотоумножитель или твердый полупроводник.
Счетчики Гейгера — Мюллера и пропорциональные счетчики обычно применяются для измерения бета-излучателей. Сцинтилляционные счетчики, в которых используются жидкие или твердые соединения фосфора, могут быть применены для измерения альфа-, бета- и гамма-излучателей. Для альфа-, бета- и гамма-излучателей могут быть также использованы твердые полупроводниковые устройства. Электронная цепь, связанная с детекторной системой, обычно состоит из источника высокого напряжения, усилителя, амплитудного селектора импульсов и пересчетной схемы, интенсиметра или другого считывающего устройства. В результате замены электронного счетчика импульсов или пересчетной схемы электронным интегрирующим устройством получают интенсиметр, который используется для контроля и прослеживания радиоактивности; точность измерения с помощью этих устройств несколько ниже, чем с помощью упомянутых выше счетчиков.
Для измерения гамма-активности часто используются ионизационные камеры, пригодные также, если они имеют тонкие стенки, для измерения рентгеновских лучей.
Излучение радиоактивного источника распространяется во всех направлениях. Метод стандартизации и измерения таких источников, при которых излучение подсчитывается во всех направлениях, известен как 4л-расчет; метод, основанный на подсчете излучения в постоянном угловом интервале 180°, известен как 2л-расчет; методы, основанные на выделении доли излучений в определенном угловом интервале, определяемом взаимным расположением источника и противолежащего ему приемника излучений, известны как методы расчета с помощью фиксированной геометрии. Обычно количественное определение радиоактивности препарата проводится путем сравнения со стандартизованным образцом в идентичных геометрических условиях. Валидность такого количественного определения в значительной степени зависит от воспроизводимости пространственных отношений источника и детектора, а также от точности стандартизованного образца. Если схема распада изотопа позволяет, то для первичной стандартизации такого изотопа используют методику совпадений, а не простой 4л-расчет. Чаще всего применяется методика совпадений,
5—2025
ТАБЛИЦА 1. ФИЗИЧЕСКИЕ ХАРАКТЕРИСТИКИ РАДИОИЗОТОПОВ
Энергия частиц и вероятность переходов Электромагнитные переходы
Радиоизотоп Период полураспада Тип а распада энергия, МэВ вероятность переходов, проценты энергия фотона, МэВ излучаемые фотоны, проценты коэффициент внутренней конверсии, проценты
Цезнй-137 30,1 года р- 0,512 1,174 94,6 5,4 3 и * Та
Через 137тВа с периодом полураспада 2,6 мнн 0,662 0,032-0,038 85,1 8 (К-рентгеновская линия Ва) 9,5 =< I > S >
Хром-51 27,7 сут эз 100 0,320 0,005-0,006 9,83 ~22 (К-рентгеновская линия V) о >
Кобальт-57 270 сут эз 100 0,014 0,122 0,136 0,570 0,692 Другие 0,006—0,007 9,4 85,2 П,1 0,02 0,16 Низкая интенсивность ~55 (К-рентгеновская линия Fe) 78,0 2,0 1,5 о а m ►а
сл
Кобальт-58 70,8 сут Р+ ЭЗ 0,475 15,0 85,0 0,511 0,811 0,864 1,675 0,006—0,007 Из р + 99,4 0,7 0,5 ~26 (К-рентгеновская линия Fe)
Кобальт-60 5,27 года ₽- 0,318 1,491 99,9 0,1 1,173 1,133 Другие 99,86 99,98 <0,01 0,02 0,01
Галлий-67 78,3 ч ЭЗ 100 0,091 0,185 0,209 0,300 0,394 0,494 0,704 0,795 0,888 0,008-0,010 3,6 23,5 2,6 16,7 4,4 0,1 0,02 0,06 0,17 43 (К-рентгенов- 0,3 0,4 0,02 0,06 0,01
ская линия Zn)
G S w S £
О X S гп
m
О fa Е
Через 67mZn с пе-
риодом полураспада 9,2 мкс 0,093 0,008-0,010 37,6 13 (К-рентгеновская линия Zn) 32,4
Золото-198 2,70 сут Р- 0,285 1,32 0,412 95,45 4,3
0,961 98,66 0,676 1,06 0,03
1,373 0,02 1,088 0,23 05
ТАБЛИЦА 1 (продолжение)
Энергия частиц и вероятность переходов Электромагнитные переходы
Радиоизотоп Период полураспада Тип распада0 энергия, МэВ вероятность переходов, проценты энергия фотона, МэВ излучаемые фотоны, проценты коэффициент внутренней конверсии, проценты
Золото-199 3,13 сут ₽- 0,25 0,29 0,45 21 72 7 0,050 0,158 0,208 0,069-0,083 0,3 39,6 8,8 ~18 (К-рентгенов-ская линия Hg) 3,5 36,4 8,3 £ га S д
Индий-111 2,81 сут эз 100 0,172 0,247 89,6 94,0 10,4 6,0 > о
Индий-113m 99,5 мин ИИ 100 0,392 0,024—0,028 64,9 24 (К-рентгенов-ска ялнния In) 35,1 ж £ е >
Иод-123 13,2 ч эз 100 0,159 0,347 0,440 0,506 0,529 0,539 0,027-0,032 83,0 0,10 0,35 0,26 1,05 0,27 ~86 (К-рентгенов-ская линия Те) 16,3 га
Йод-125 60,0 сут эз 100 0,035 0,027—0,032 7 138 (К-рентгенов-ская линия Те) 93
йод-126 13 сут в- ₽+ ЭЗ 0,38 0,88 1,27 0,46 1,1 3 30 15 ~0,1 —0,4 51,5 0,389 0,491 0,511 0,666 0,754 0,880 1,420 Другие 0,027—0,032 32 2 из В + 30 4 0,8 0,3 <0,1 каждый ~38 (К-рентгенопекая линия Те) 0,5 0,1 &
Йод-131 (Ксенон-131m) 8,06 сут в- ип 0,247 1,8 0,080 2,4 0,304 0,6 0,284 5,9 0,334 7,2 0,364 81,8 0,606 89,7 0,637 7,2 0,806 0,7 0,723 1,8 1,3% 13Ч распадается через^131тХе с периодом полураспада 12 сут (Процент относится к распаду 131тХе) 3,8 0,3 1,7 98 S Q0 § S S га £ га н О ь tr
йод-132 2,29 ч в- 0,84 1,01 1,07 1,09 1,10 1,26 1,29 1,57 1,72 2,24 Другие 16,0 3,5 6,5 3,0 2,6 2,9 18,4 10,8 12,7 20,2 3,4 0,506 0,523 0,621 0,630 0,651 0,668 0,670 0,672 0,727 0,773 0,810 0,812 5,0 16,1 2,0 13,7 2,7 98,7 4,9 5,2 6,5 76,2 2,9 5,6 0,2 0,1 0,4 0,3 05 CD
ТАБЛИЦА 1 (продолжение) О
Радиоизотоп Период полураспада Тип а распада Энергия частиц и вероятность переходов Электромагнитные переходы
энергия, МэВ вероятность переходов, проценты энергия фотона, МэВ излучаемые фотоны, проценты коэффициент внутренней конверсии, проценты
0,955 1,136 1,295 1,372 1,399 1,433 1,921 2,002 Другие • 18,1 3,0 2,0 2,5 7,1 1,4 1,2 1,1 ’ <1‘,5 В: и Я Ж о $
Железо-55 2,69 года эз 0,006 ~28 (К-рентгенов-ская линия Мп) > е
Железо-59 44,6 сут р- 0,084 0,132 0,274 0,467 1,566 0,1 1,1 45,8 52,7 0,3 0,143 0,192 0,335 0,383 1,099 1,292 1,482 0,8 2,8 0,3 0,02 55,8 43,8 0,06 > § § 3 и ю
Ртуть-197 64,4 ч 33 100 0,077 0,192 0,268 0,067—0,080 19,2 ~1,1 ~0,1 ~73 (К-рентгенов-ская линия Au) 80,7 0,9
Ртуть-197m 24 ч ЭЗ ИП 6,5 93,5 0,134 0,165 0,067—0,083 31,8 0,3 36 (К-рентгенов-ская линия Au/Hg) 61,7 93,2
Дочерний I97Hg Через 197mAu с периодом полураспада 7,8 с О', 130 0,279 0,409 0,067—0,080 0,5 5,0 <0,005 ~2 (К-рентгенов-ская лнння Au) 6 1,5 е S ш S £ ж S m
Ртуть-203 46,6 сут Р- 0,212 100 0,279 0,071—0,085 81,5 12,8 (К-рентгенов-ская линия Т1) 18,5 га 8 §
Молнбден-99 66,2 ч Р- 0,454 0,866 1,232 Другие 18,3 1,4 80 0,3 0,041 0,141 0,181 0,366 0,412 0,529 0,621 0,740 0,778 0,823 0,961 1,2 5,4 6,6 1,4 0,02 0,05 0,02 13,6 4,7 0,13 о,1 4,8 0,7 1,0
ТАБЛИЦА 1 (продолжение)
Энергия частиц н ве- Электромагнитные переходы
роятность переходов
Радиоизотоп Период Полураспада Тип а распада энергия, вероятность энергия фотона, излучаемые фотоны, коэффициент внутренней
МэВ переходов, п роценты МэВ проценты конверсии, проценты
Через "“.Тс с периодом полураспада 6,02 ч в равновесии 0,002 0,141 0,143 ~0 83,9 0,03 93,9 10,0 0,8
Фосфор-32 14,3 сут Р- 1,709 100
Селен-75 118,5 сут ЭЗ 100 0,066 0,097 0,121 0,136 0,199 0,265 0,280 0,401 Другие 0,010—0,012 1,1 2,9 15,7 54,0 1,5 56,9 18,5 11,7 <0,05 каждый ~50 (К-рентгеновская лнння As) 0,3 3,0 0,7 1,6 0,4 0,2
Через 7SAs с перио-
дом полураспада 16,4 мс 0,024 0,280 0,304 0,010—0,012 0,03 5,4 1,2 ~2,6 (К-рентге-новская линия As) 5,5 0,1
Технеций-99т 6,02 ч ИП 100 0,002 ~0 99,1
0,141 88,5 10,6
0,143 0,03 0,87
Дочерннй "Тс
Таллий-201 73,5 ч ЭЗ 100 0,031 0,032 0,135 0,166 0,167 0,29 0,25 2,9 0,13 8,81 10,1 9,6 [8,9 1 0,2 16,0
Олово-113 115 сут ЭЗ 100 0,255 0,024—0,028 2,1 73 (К-рентгенов- 0,1
екая линия In)
е s w S ►с m о я S и 2 гп н о
Дочерний 131mIn
Тритий (3Н) 12,35 года Р- 0,0186 100
Ксенон-131т 11,9 сут ИП 100 0,164 2 98 0,029—0,035 ~52 (К-рентгенов- ская лнння Хе)
74
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
' ЭЗ — электронный захват; ИП — изомерный переход.
ФИЗИЧЕСКИЕ МЕТОДЫ
75
представляющая собой 4л-бета/гамма-расчет совпадений, используемый для изотопов, у которых некоторые или все распады сопровождаются быстрым излучением фотонов. Для измерения эффективности в 4л-счетчике тех распадов, с которыми совпадают фотоны, используют дополнительный смежный детектор, чувствительный только к фотонам. Для стандартизации чистых гамма-излучателей часто используют 4л-гамма/гамма-расчет совпадений.
Конструкция и принцип действия приборов и вспомогательных приспособлений различны. При использовании определенного вида аппаратуры методика подготовки образцов должна быть модифицирована для получения удовлетворительных результатов. Чтобы обеспечить работу приборов в оптимальном режиме, оператор должен неукоснительно следовать инструкциям фирмы-изготовителя и для повышения надежности результатов тщательно сравнивать исследуемые образцы с известными. Контроль за работой прибора и надежностью его показаний должен быть повседневным; для этого используются вторичные стандартные образцы.
Радиоактивность, обусловленная излучением материалов конструкции, действием космических лучей и спонтанным выбросом в атмосферу, создает так называемый радиоактивный фон. Во все измерения радиоактивности образцов должна вноситься поправка на радиоактивный фон, величина которого вычитается из полученных результатов.
При работе с образцами высокой радиоактивности должна вводиться поправка на просчеты совпадений вследствие того, что прибор не может разрешать импульсы, поступающие в узком интервале времени. Такая поправка на просчеты совпадений должна быть сделана до того, как вводят поправку на фон.
Исправленная скорость счета R определяется формулой:
1 —ГТ1
где г— наблюдаемая скорость счета импульсов, т — время разрешения.
Счет радиоактивных импульсов является статистической величиной, т. е. импульс есть мера вероятности радиоактивного распада и не является точной постоянной величиной вне заданного интервала времени. Величина стандартного отклонения приблизительно равна квадратному корню из числа счетов импульсов. Как правило, для того чтобы получить стандартное отклонение порядка 1%, счет импульсов должен составлять по крайней мере 10 000.
76
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Поглощение
Ионизирующее излучение поглощается материалом, окружающим радиоактивный источник. Это поглощение происходит в воздухе, в самом веществе (самопоглощение), в стенках устройства, экранирующего образец, в окошке обнаруживающего излучение прибора, а также во всех видах специальных поглотителей, монтируемых между образцом и детектором. Определение типа излучения и его энергии производится с помощью поглотителей различной толщины, так как известно, что альфа-частицы имеют очень небольшую глубину проникания, бета-частицы проникают в материал несколько глубже, а гамма-лучи могут проникать очень глубоко. На практике этот метод используется очень редко, и только в связи с бета-излучателями. Однако различия в счете импульсов, обусловленные различиями в толщине и плотности контейнеров образцов, могут создавать серьезные трудности, когда речь идет о бета-излучателях и источниках рентгеновского излучения, таких, как йод-125. Поэтому в этих случаях часто используют пластмассовые пробирки, у которых различия в толщине и плотности минимальны.
Для характеристики бета-излучения, испускаемого радиоизотопом, обычно определяется коэффициент поглощения (р)—величина, обратная «толщине», выражаемой в мг/см2, или толщина слоя половинного поглощения (толщина защитного поглотителя, необходимая для того, чтобы уменьшить интенсивность радиации вдвое по сравнению с ее первоначальной величиной).
Метод
В испытании на подлинность, описанном в статье Natrii Phosphatis (32Р) Injectio [Натрия фосфата (32Р) раствор для инъекций], для измерения бета-активности и расчета коэффициента поглощения или толщины слоя половинного поглощения применяется следующая методика.
Помещают образец радиоактивного вещества, соответствующим образом укрепленный для удобства счета, под выбранным для данного случая счетчиком. Проводят определение активности образца отдельно и последовательно, применяя по крайней мере 6 листков алюминиевой фольги различной толщины в пределах от 10 до 200 мг/см2 и отдельный поглотитель толщиной не менее 800 мг/см2. Для уменьшения эффекта рассеяния света образец и поглотители должны как можно ближе располагаться к детектору. Получают истинные значения бета-излучения при различных поглотите
ФИЗИЧЕСКИЕ МЕТОДЫ
77
лях, вычитая значение активности, найденное при поглотителе 800 мг/см2 или более. Наносят на график логарифм истинной активности бета-излучения как функцию общей толщины поглотителя. Общая толщина поглотителя складывается из толщины алюминиевой фольги, толщины окошка счетчика, указанной заводом-изготовителем, и поправки на величину воздушного зазора (расстояние в сантиметрах между образцом и окошком счетчика, умноженное на 1,205); все величины выражаются в мг/см2. На графике получается почти прямая линия.
Выбирают две толщины поглотителей (ti и t2), отличающиеся одна от другой на 20 мг/см2 или более и нанесенные на график, и рассчитывают коэффициент поглощения (р) по уравнению:
1 . Ак
Р = ' t —t 1п ~аГ’
где ti — толщина более тонкого поглотителя, t2— толщина более толстого поглотителя; и Д2—значения истинной активности бета-излучения при поглотителях с толщиной соответственно ti и t2. Толщина слоя половинного поглощения может быть найдена непосредственно из графика.
Выбор толщины поглотителя зависит от радиоизотопа. Для других радиоизотопов, кроме фосфора-32, которые имеют более высокую или более низкую энергию бета-излучения, необходимы поглотители большей или меньшей толщины.
Для характеристики радиоизотопа коэффициент поглощения радиоактивного образца не должен отличаться от коэффициента поглощения образца того же радиоизотопа известной чистоты более чем на ±5% при параллельном определении.
Радиоактивность при нулевом значении общей толщины поглотителя может быть определена построением кривой, которая строится, как это было описано выше, для определения коэффициента поглощения, и экстраполяцией прямой на нулевое значение толщины поглотителя; следует учитывать толщину покрытия образца, воздушный зазор и толщину окошка счетчика, выраженные в мг/см2.
Радиационная спектрометрия
Кристаллическая сцинтилляционная спектрометрия
Когда энергия бета- или гамма-излучения рассеивается в веществах, известных под названием сцинтилляторов, возни
78
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
кает свечение, интенсивность которого пропорциональна количеству рассеянной энергии. Интенсивность этого свечения может быть измерена соответствующими методами и она пропорциональна количеству энергии, поглощенной сцинтиллятором. Свет, испускаемый под воздействием гамма-фотонов или бета-частиц, превращается при помощи фотоумножителя в электрический выходной импульс. Развертка выходных импульсов при помощи соответствующих амплитудных анализаторов импульсов дает возможность регистрировать энергетический спектр источника излучений.
Сцинтилляторы, которые наиболее часто применяются для гамма-спектрометрии, представляют собой одиночные кристаллы йодида натрия, активированного таллием. Сцинтилляционные спектры гамма-излучения состоят из одного или более острых характерных фотоэлектрических пиков, соответствующих энергиям источника гамма-радиации. Поэтому эти спектры полезны для идентификации, а также для обнаружения гамма-излучающих примесей в препарате. Кроме характерных пиков, в спектре обычно имеются и другие пики, обусловленные вторичным воздействием радиации на сцинтиллятор и его окружение, таким, как обратное отражение, аннигиляция позитронов, суммирование совпадений и флуоресцентные рентгеновские лучи. Кроме того, в результате рассеяния гамма-фотонов в сцинтилляторе и окружающих материалах возникают широкие полосы, известные как спектры Комптона (эффект Комптона). Калибровка прибора производится с помощью известных образцов радиоактивных изотопов, энергетические спектры которых определены. Форма спектров будет различной в зависимости от используемых приборов; это определяется различной формой и размерами кристаллов, применяемыми защитными материалами, расстоянием между источником излучения и детектором, а также типами дискриминаторов, используемых в амплитудных анализаторах импульсов. При использовании спектра для установления подлинности радиоизотопов необходимо сравнивать спектр исследуемого образца со спектром известного вещества, радиоактивность которого измерена тем же прибором и при тех же условиях.
Некоторые радиоизотопы, например йод-125, испускают характерные рентгеновские лучи с четко выраженными энергиями, которые будут давать фотоэлектрические пики в соответствующем гамма-спектрометре. Бета-радиация также взаимодействует со сцинтилляторами, но эти спектры непрерывны и диффузны и обычно не могут быть использованы для идентификации радиоизотопа или для обнаружения бета-излучающих примесей в препарате.
ФИЗИЧЕСКИЕ МЕТОДЫ
79
Спектрометрия с помощью полупроводниковых детекторов
Спектры гамма- и бета-излучения могут быть получены с помощью твердых полупроводниковых детекторов. Получаемые пики не подвержены расширению полос в той же мере, в какой это наблюдается при кристаллической сцинтилляционной спектрометрии, и разрешение гамма-фотонов с аналогичными энергиями значительно лучше. Однако производительность таких детекторов существенно ниже.
Энергия, необходимая для создания электронной пары или для перемещения электрона от валентной полосы к проводниковой полосе в полупроводнике, значительно меньше, чем энергия, требуемая для образования фотона в сцинтилляционном кристалле. В спектрометрии гамма-излучения детектор из германия с добавкой лития может обеспечить для фотона кобальта-60 с энергией 1,33 МэВ энергетическое разрешение порядка 0,33% по сравнению с 5,9% —результатом, получаемым при использовании активированного таллием кристалла йодида натрия размером 7,6X7,6 см.
Жидкостный сцинтилляционный метод счета
Для слабых бета-излучателей, таких как 35S, 14С и 3Н, когда самопоглощение бета-частиц с низкой энергией значительно, предпочтительным методом счета является метод жидкостной сцинтилляции, который может быть иногда использован также для излучателей рентгеновских лучей, альфа-лучей и гамма-лучей. Если образец, подлежащий обсчету, растворяется в подходящем сцинтилляционном материале или смешивается с ним, энергия распада образца превращается в фотоны света. Последние воспринимаются фотоумножителем, превращающим их в электрический импульс, интенсивность которого пропорциональна энергии начальной радиации. Таким образом, одновременный обсчет нескольких радиоизотопов, отличающихся энергиями радиации, может быть осуществлен с помощью подходящих дискриминаторов (амплитудных анализаторов импульсов) при условии, что энергия разделения достаточна. Так как самопоглощение при этом минимально, можно добиться эффективности обнаружения, близкой к 95% для 14С и 60% для 3Н.
Сцинтилляционный растворитель обычно состоит из полициклического ароматического соединения, такого, как и-тер-фенил или 2,5-дифенилоксазол (первичный растворитель), к которому добавлен вторичный растворитель, например 1,4-ди [2-(4-метил-5-фенилоксазол] бензол (диметил-РОРОР); последний смещает длину волны излучаемого света так, что
80
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
она соответствует наивысшей чувствительности фотоумножителя. Могут быть использованы как несмешивающиеся с водой жидкости, например, толуол, так и смешивающиеся с водой растворители, например диоксан. Разработаны специальные растворители, облегчающие обсчет водных растворов. Кроме того, измерению можно подвергать водные образцы в виде суспензий в сцинтилляционном геле. Для достижения совместимости и смешения с анализируемыми водными образцами в сцинтиллятор можно вводить также многие дополнительные вещества, такие, как поверхностно-активные и солюбилизирующие агенты. Для того чтобы измерение радиоактивности образца было точным, необходимо обращать особое внимание на приготовление истинно гомогенного образца. Присутствие примесей в растворе и его окрашивание снижают выход фотонов в сцинтилляторе; такое уменьшение известно как явление гашения. Точное измерение радиоактивности требует внесения поправки на снижение счета импульсов вследствие гашения. Растворы, содержащие органические сцинтилляторы, склонны к возбуждению под действием света, поэтому может оказаться необходимым готовить образцы в рассеянном свете и перед обсчетом хранить их в темноте.
Защитные экраны
Для защиты лабораторного персонала от всех видов излучения и для защиты регистрирующей аппаратуры от радиоактивного фона следует применять соответствующие защитные экраны.
Защита от альфа- и бета-излучений легко осуществима благодаря их малой проникающей способности, хотя следует принимать во внимание тормозную радиацию (Bremsstrahlung), продуцируемую при поглощении бета-излучения (см. ниже). Глубина проникновения альфа- и бета-частиц изменяется в зависимости от их кинетической энергии. Альфа-излучение представляет собой поток моноэнергетических частиц и полностью поглощается воздушным слоем толщиной в несколько сантиметров. Поглощение бета-излучения в связи с его непрерывным энергетическим спектром и рассеянием подчиняется приблизительной экспоненциальной зависимости. Пробег бета-частиц в воздухе составляет расстояние от нескольких сантиметров до нескольких метров.
При поглощении бета-излучения экранирующим материалом возникает вторичная радиация — так называемое тормозное излучение (Bremmstrahlung), по проникающей способности подобное мягкому рентгеновскому излучению. Чем вы
ФИЗИЧЕСКИЕ МЕТОДЫ
81
ше атомный номер или плотность поглощающего материала, тем выше энергия тормозного излучения. Элементы с низким атомным номером продуцируют тормозное излучение низкой энергии, которое легко поглощается. Следовательно, при изготовлении защитных экранов для источников бета-излучения должны применяться материалы с небольшим атомным номером или невысокой плотностью, такие, как алюминий, стекло, прозрачные пластмассы.
Гамма-лучи проникают в материал на большую глубину. Ослабление гамма-излучения в материале подчиняется экспоненциальной зависимости и определяется количественно по слоям половинного поглощения. Слой половинного поглощения— это толщина защитного материала, необходимая для того, чтобы уменьшить интенсивность радиации вдвое по сравнению с ее первоначальной величиной. Экран, состоящий из 7 слоев половинного поглощения, имеет толщину, снижающую интенсивность радиации до величины, составляющей менее 1% интенсивности неэкранированного первоначального излучения. Защитным материалом для поглощения гамма-излучения обычно служит свинец.
Интенсивность гамма-излучения уменьшается обратно пропорционально квадрату расстояния между источником и точкой замера. Радиоактивные материалы, излучающие энергию в несколько десятков милликюри, могут быть использованы в лабораториях без опасения, если при этом применяются соответствующие экраны или с помощью приспособлений дистанционного управления устанавливается максимальное расстояние между источником и оператором.
Определение радиоизотопной чистоты
Наиболее широко применяемым методом оценки радиоизотопной чистоты для гамма-излучателей является гамма-спектрометрия. Однако этот метод не является абсолютным, так как:
а) бета-излучающие примеси, как правило, не обнаруживаются;
б) если применяются детекторы с йодидом натрия, фотоэлектрические пики, обусловленные примесями, могут маскироваться пиками основного радиоизотопа, т. е. другими словами, степень разрешения прибора будет недостаточна; эта проблема может быть решена за счет применения твердых полупроводниковых детекторов с высоким разрешением, таких, как литий-германиевый детектор (Ge: Li);
6—2025
82
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
в) если прибор не откалиброван по известному стандартному источнику с известной радиоизотопной чистотой в идентичных геометрических условиях, то трудно определить, обусловлены ли дополнительные пики примесями или являются результатом таких вторичных эффектов, как обратное рассеяние, суммирование совпадений или флуоресцентные рентгеновские лучи.
Диапазон гамма-спектрометрии может быть расширен двумя путями: во-первых, путем наблюдения изменений в спектре препарата, происходящих во времени (это особенно полезно для обнаружения присутствия долгоживущих примесей в препаратах короткоживущих радиоизотопов); во-вторых, применением химического разделения, при котором основной радиоизотоп может быть удален химическим путем, а остаток затем проверяется на содержание примесей, или специфические примеси могут удаляться химически и затем подвергаться количественной оценке. Однако совершенно очевидно, что с помощью химических средств нельзя отделить примесь, которая является изотопом основного радиоизотопа.
Требования в отношении радиоизотопной чистоты
Для выражения требований в отношении радиоизотопной чистоты существуют два способа.
1. Указание минимального уровня радиоизотопной чистоты. Если в частной статье нет других указаний, радиоизотопная чистота, определенная методом простой гамма-спектрометрии с применением детектора йодида натрия, не должна значительно отличаться от чистоты стандартизованного раствора радиоизотопа до окончания его срока годности. Как говорилось выше, трудно установить более точные требования для минимального уровня радиоизотопной чистоты.
2. Указание в частных статьях максимальных уровней специфических радиоизотопных примесей. Как правило, это такие примеси, которые, как известно, могут образовываться в процессе производства материала, например ртуть-203 в препаратах ртути-197.
Очевидно, что, хотя упомянутые выше требования необходимы, они сами по себе еще недостаточны, чтобы радиоизотопная чистота препарата считалась соответствующей требованиям применения в медицине. Обязанность изготовителя— более детально исследовать препараты короткоживущих радиоизотопов на присутствие в них долгоживущих примесей после соответствующего периода распада. Таким образом из
ФИЗИЧЕСКИЕ МЕТОДЫ
83
готовитель может убедиться, что производственные процессы обеспечивают получение материала приемлемой степени чистоты. Б частности, радиоизотопный состав некоторых препаратов определяется химическим и изотопным составом исходного материала мишени, подвергнутой бомбардировке нейтронами, поэтому когда используются новые партии материала мишени, рекомендуется получать новые опытные образцы препаратов.
Определение радиохимической чистоты
Радиохимическая чистота может быть исследована различными методами, но наиболее важными из них являются бумажная хроматография и тонкослойная хроматография (см. с. 92—97). После завершения разделения на хроматограмме определяют распределение радиоактивности. Количество вещества, наносимого на хроматограмму, часто крайне мало (вследствие высокой чувствительности обнаружения радиоактивности), и поэтому надо быть особенно осторожным в интерпретации результатов в связи с возможностью возникновения артефактов. Кроме хроматографии, для разделения может быть использован электрофорез (см. с. 114—118). Как упоминалось выше, иногда может оказаться полезным добавление к самому радиофармацевтическому соединению или к ожидаемым примесям носителей, т. е. соответствующих нерадиоактивных соединений. Существует, однако, опасность, что прибавленный неактивный носитель радиоактивного фармацевтического вещества может взаимодействовать с радиохимической примесью, что в свою очередь может привести к заниженной оценке этих примесей. Другой подходящий метод— наблюдение за биологическим распределением инъецированного радиофармацевтического вещества в испытании на животных.
Определение химической чистоты
Понятие химической чистоты относится к той части препарата, которая представлена конкретной химической формой, независимо от наличия радиоактивности; химическая чистота может быть определена обычными методами анализа.
Химическая чистота препарата не всегда может служить свидетельством его радиохимической чистоты. Препараты, особенно те, которые получаются в ходе обменных реакций (например, когда некоторые атомы йода в о-йодгиппуровой
6*
84
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
ФИЗИЧЕСКИЕ МЕТОДЫ
85
кислоте замещены атомами йода-131), могут обладать высокой степенью химической чистоты, но при этом содержать примеси с высокой удельной активностью (т. е. минимальная по весу химическая примесь может быть связана с большим количеством радиоизотопа).
Как правило, присутствие химических примесей в радиоактивных фармацевтических препаратах считается неприемлемым в тех случаях, когда они токсичны или изменяют исследуемые физиологические процессы.
Испытания на стерильность и пирогенность
Ряд статей для радиофармацевтических препаратов содержит требования, согласно которым продукт должен быть стерильным и свободным от пирогенных веществ. Период .полураспада некоторых радиоактивных фармацевтических препаратов так короток, что только испытания на пирогенность могут быть выполнены изготовителем до их выпуска. Испытания на стерильность, как правило, выполняются после выпуска препарата.
Испытания на стерильность
Изготовитель должен начать испытание на стерильность как можно скорее, а зарегистрировать результаты — после выпуска препарата.
С особой ответственностью изготовитель радиофармацевтических препаратов должен обеспечить надежность процесса .стерилизации всеми доступными средствами, в том числе с помощью тщательной и частой калибровки стерилизаторов и применения биологических и химических индикаторов, подтверждающих эффективность процесса стерилизации.
Испытания на пирогенность
Изготовитель также несет особую ответственность за использование в приготовлении радиоактивных продуктов только таких веществ, которые обрабатываются способами, гарантирующими чистоту конечных продуктов в отношении пирогенности. Испытания на пирогенность приводятся в некоторых частных статьях в тех случаях, когда пирогенность представляет собой особую опасность.
Прибавление бактериологических агентов
Растворы радиоактивных фармацевтических препаратов для инъекций обычно поставляются в таре (контейнерах), закрытой таким образом, чтобы можно было брать одну за другой отдельные дозы по мере необходимости. В соответствии с требованиями Международной фармакопеи такие растворы для инъекций обычно должны содержать подходящий бактериостатический агент в подобранной должным образом концентрации.
Многие известные бактериостатические агенты, например бензиловый спирт, постепенно разрушаются в водных растворах под действием радиации. Скорость разрушения зависит от ряда факторов, в том числе от природы радиоизотопа и радиоактивной концентрации раствора. Поэтому не всегда возможно определить эффективный бактериостатический агент для раствора радиофармацевтического препарата для инъекций, и для ряда препаратов добавление такого агента нежелательно; по этой причине включение бактериостатических агентов не является обязательным. Природа бактериостатического агента, если таковой присутствует, должна быть указана на этикетке; если бактериостатический агент не введен в препарат, это также должно быть указано на этикетке. Желательно, чтобы радиофармацевтические препараты со сроком годности более одних суток и не содержащие бактериостатический агент поставлялись в таре для одноразового применения.
Другие требования
Радиоактивные фармацевтические препараты, применяемые парентерально, должны отвечать соответствующим требованиям для инъецируемых лекарственных форм, указанным в Международной фармакопее, за исключением тех случаев, когда они должны удовлетворять требованиям, касающимся объема раствора для инъекций в таре для одноразового применения.
Срок годности
Ввиду особой природы радиоактивных фармацевтических препаратов для них должен быть установлен срок годности, после которого их дальнейшее применение не рекомендуется. Установленный срок годности начинается с того дня, который
86 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
указан на этикетке как день определения радиоактивности, и может выражаться в сутках, неделях или месяцах. Для долгоживущих радиоизотопов срок годности не превышает 6 мес. Срок годности зависит от радиохимической стабильности и от содержания долгоживущей радиоизотопной примеси в конкретном препарате. К концу срока годности радиоактивность уменьшится настолько, что станет недостаточной для применения в назначенных целях или дозу активного ингредиента надо будет увеличить до такой степени, что это вызовет нежелательную физиологическую реакцию. Кроме того, химическое или радиационное разложение может снизить радиохимическую чистоту до неприемлемого уровня, а содержание радиоизотопной примеси может оказаться настолько высоким, что больной получит нежелательную дозу радиации. Поэтому применять продукты после окончания срока годности не рекомендуется.
Этикетирование
Как травило, следующая информация должна быть приведена на таре (контейнере), непосредственно содержащей препарат (например, на флаконе):
1) наименование препарата;
2) указание, что продукт является радиоактивным;
3) наименование и адрес предприятия-изготовителя;
4) общая радиоактивность на указанный день и час (если период полураспада составляет более 30 сут, то указывают только день);
5) дата окончания срока годности пли период годности;
6) номер или другое указание, по которому можно восстановить по стадиям процесс производства продукта, например, номер серии или партии;
7) для растворов должен быть указан общий объем раствора;
8) специальные условия хранения >в отношении температуры и света.
ПРИМЕЧАНИЕ. Для растворов вместо указания общей радиоактивности может быть указана радиоактивная концентрация (например, в мкюри или МБк на 1 мл раствора).
При перевозке радиоактивных веществ следует руководствоваться специальными национальными и международными правилами, регламентирующими упаковку и наружное этикетирование.
ФИЗИЧЕСКИЕ МЕТОДЫ 87
Хранение
Радиоактивные фармацевтические препараты следует хранить в хорошо укупоренной таре (контейнерах), в местах, предназначенных для этой цели. Условия хранения должны быть такими, чтобы лица, которые могут подвергнуться действию радиации, не получили дозу, превышающую приемлемый уровень. Следует принимать меры для соблюдения национальных требований, касающихся защиты от ионизирующей радиации. Стеклянная тара может темнеть под воздействием радиации.
СТЕПЕНЬ ИЗМЕЛЬЧЕНИЯ ПОРОШКОВ И СИТА
А. Порошки
Порошки различают по степени измельчения, выражаемой размеров отверстия (меша) сита в микрометрах, через которое порошок может проходить.
При описании порошков используют следующие определения:
Крупный порошок (2000/355). Порошок, все частицы которого проходят через сито № 2000 и не более 40% проходит через сито № 355.
Среднекрупный порошок (710/250). Порошок, все частицы которого проходят через сито № 710 и не более 40% проходит через сито № 250.
Среднемелкий порошок (355/180). Порошок, все частицы которого проходят через сито № 355 и не более 40% через сито № 180.
Мелкий порошок (180). Порошок, все частицы которого проходят через сито № 180.
Очень мелкий порошок (125). Порошок, все частицы которого проходят через сито № 125.
Если степень измельчения порошка обозначается номером, это значит, что все частицы порошка должны проходить через сито указанного номера.
Б. Сита
Проволочные сита, применяемые для просеивания измельченных лекарственных средств, различают по номерам, которые соответствуют номинальному размеру отверстия, выраженному в микрометрах.
88
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Сита делают из проволки равномерного поперечного сечения в соответствии со спецификациями, приведенными в табл 2.
ТАБЛИЦА 2. ПРОВОЛОЧНЫЕ СИТА
Номер сита, мкм Номинальный размер отверстий, мм Номинальный диаметр проволоки, мм (Приблизительная доля площади сита, занятого отверстиями, %
2000 2,00 0,90 48
710 0,710 0,450 37
500 0,500 0,315 38
355 0,355 0,224 38
250 0,250 0,160 37
212 0,212 0,140 36
180 0,180 0,125 35
150 0,150 0,100 36
125 0,125 0,090 34
90 0,090 0,063 35
75 0,075 0,050 36
45 0,045 0,032 34
Номинальные размеры отверстий проволочных сит в основном были выбраны из размеров, рекомендованных стандартом Международной организации по стандартизации (ИСО) 565—1972.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
ХРОМАТОГРАФИЯ
Принцип хроматографических процессов, как правило, состоит в распределении растворенного вещества между двумя фазами, одна из которых является подвижной, а другая — неподвижной. Вещества могут распределяться на неподвижной фазе за счет адсорбции, распределения (когда, например, жидкость, не смешивающаяся с подвижной фазой, может быть нанесена на поверхность твердого носителя), ионного обмена или проникания в гель. Практически хроматографические процессы во многих случаях, будучи использованы в целях фармацевтического анализа, могут представлять собой сложное сочетание нескольких физических явлений; несомненно, на многие хроматографические методики, которые считаются распределительными, в значительной степени влияют и адсорбционные эффекты.
Таким образом, хроматография—‘просто метод разделения и сама по себе не может служить ни методом испытаний на подлинность, ни методом количественных измерений. Только сочетание хроматографии с подходящими методами определения и измерения представляет собой хотя и неточно, но удобно названный «хроматографический метод анализа».
Для удобства типы хроматографии, которые используются в фармацевтическом анализе, можно разделить на три большие группы. К плоскостным методам относится хроматография, которая осуществляется путем прохождения подвижной фазы через слой адсорбента (бумажная и тонкослойная хроматография). Вторая группа методов — хроматография на колонках. При использовании хроматографии на колонках колонку заполняют адсорбентом; колонка может быть либо обычного открытого типа либо закрытого; колонка закрытого типа должна выдерживать значительное давление, чтобы подвижную фазу можно было подавать насосом через колонку с большой скоростью (жидкостная хроматография высокого давления, иногда называемая высокоэффективной или высокоскоростной жидкостной хроматографией). Газовая хроматография— частный случай хроматографии на колонках; здесь подвижной фазой является газ, а не жидкость, а растворенное вещество должно быть либо летучим либо переведено в это состояние путем повышения температуры и/или превращения в летучие производные.
Ни один из хроматографических методов не может удовлетворять всем целям, так как каждый из них имеет свои
92 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
преимущества и недостатки. Большинство плоскостных методов просты и эффективны и требуют недорогого оборудования (хотя имеются и некоторые сложные приспособления); эти методы удобны для испытания на подлинность и скрининга, но менее пригодны для точных количественных измерений. Более старые колоночные методы не требуют больших затрат, но они часто трудоемки и отнимают много времени. Жидкостная хроматография высокого давления и газовая хроматография требуют специального оборудования, но позволяют достигать быстрого и эффективного разделения, что необходимо для точного количественного измерения компонентов. Эти методы особенно ценны для определения небольших количеств примесей. В настоящее время ценность метода жидкостной хроматографии высокого давления снижается из-за отсутствия универсально применимых методов определения;, применение метода газо-жидкостной хроматографии ограничивается из-за отсутствия летучести или термостабильности многих соединений.
Тонкослойная хроматография
В тонкослойной хроматографии адсорбентом служит тонкий, равномерный слой (обычно толщиной около 0,24 мм) сухого мелкоизмельченного материала, нанесенного на подходящую подложку, например на стеклянную пластинку, алюминиевую фольгу или пластмассовую пленку. Подвижная фаза движется по поверхности пластинки (обычно под действием капиллярных сил); хроматографический процесс может зависеть от адсорбции, распределения или комбинации обоих явлений, что в свою очередь зависит от адсорбента, его обработки и природы используемых растворителей. Во время хроматографирования пластинка находится в хроматографической камере (чаще всего изготовленной из стекла, чтобы можно было наблюдать движение подвижной фазы по пластинке), которая обычно насыщена парами растворителя. В качестве твердого носителя часто используются силикагель, кизельгур, окись алюминия и целлюлоза; для лучшего сцепления с носителем к нему можно прибавлять соответствующие вещества, например сульфат кальция (гипс). Для изменения свойств приготовленного слоя его можно пропитать буферными материалами, чтобы получить кислый, нейтральный или основной слой; можно использовать и другие вещества, такие, как нитрат серебра. В некоторых случаях слой может состоять из ионообменной смолы. Такой широкий диапазон различных слоев, используемых в сочетании с разными
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
93
системами растворителей, дает почти неограниченную возможность изменять силу разделения, что делает тонкослойную хроматографию столь полезным методом для фармацевтического анализа.
В испытаниях на подлинность тонкослойная хроматография служит для сравнения поведения идентифицируемого материала и стандартного образца, обычно аутентичного исследуемому веществу. Если оба вещества продвигаются во время хроматографического процесса на одинаковое расстояние и если оба вещества, смешанные и подвергнутые хроматографированию, движутся как единое вещество, можно предположить, что эти вещества идентичны. Это предположение может быть подтверждено повторением той же процедуры с использованием другой хроматографической системы; как правило, если два вещества ведут себя идентично в трех совершенно различных системах, предположение об их идентичности вполне обосновано.
Для установления подлинности удобно определять отношение расстояния, пройденного неизвестным веществом, к расстоянию, пройденному либо фронтом растворителя, либо стандартным образцом. На полученной хроматограмме отношение расстояния, пройденного на адсорбенте данным веществом, к расстоянию, пройденному передним краем растворителя или подвижной фазы (оба расстояния измеряют от точки нанесения испытуемого вещества), есть величина Rf, характерная для данного вещества в данной хроматографической системе. Отношение расстояний, пройденных испытуемым веществом и стандартным образцом, принимают за величину Rr. На практике величины Rf могут значительно варьировать в зависимости от конкретных экспериментальных условий, поэтому величина Rr, определенная по отношению к стандартному образцу, подвергнутому хроматографированию на той же пластинке, имеет более достоверное числовое значение. Еще более надежные результаты дает сравнение с аутентичным образцом, как описано выше, и именно эта методика используется для фармакопейных целей.
Для определения положения неокрашенного вещества на полученной хроматограмме обычно необходимо обрабатывать хроматограмму реактивом, который либо обугливает разделенные вещества, либо переводит их в окрашенные или флуоресцирующие производные. Часто применяют и другой удобный метод: проводят хроматографию на пластинке, пропитанной веществом, сильно флуоресцирующим под воздействием коротковолнового ультрафиолетового света. Площади на пластинке, занятые веществами, поглощающими при той же длине волны, выглядят как темные пятна на флуоресцирую
94
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
щем фоне. В особых случаях можно использовать и другие средства обнаружения, например, определять радиоактивность, если разделяются меченые соединения, или получить микробиологический ответ, когда речь идет об антибиотиках.
Наиболее ценные результаты дает применение тонкослойной хроматографии в качестве метода оценки низких уровней примесей в медицинских веществах. Для этой цели вещество наносят на хроматографическую пластинку и после хроматографирования любые вторичные пятна, которые могут быть видны на хроматограмме после соответствующего проявления, сравнивают по размеру и интенсивности с пятнами, которые дают небольшие количества ожидаемых примесей при одновременном хроматографировании на той же пластинке. Для этой методики нужно иметь в наличии ожидаемые примеси, поэтому в некоторых статьях предписывается использование аутентичных образцов примесей. Часто бывает, что в лабораториях этих примесей нет; в таких случаях можно сравнивать вторичные пятна, образующиеся от следовых количеств примесей, с пятном, полученным при хроматографировании на той же пластинке соответствующего небольшого количества испытуемого вещества. Этот прием не всегда возможно применить, так как примеси и испытуемое вещество могут по-разному реагировать на метод обнаружения, однако с его помощью можно получить приемлемый критерий, по которому можно судить об уровне примеси в веществе. Третья, иногда рекомендуемая методика состоит в нанесении такого количества испытуемого вещества, при котором после хроматографирования не появляется никаких вторичных пятен, если образец приемлемо чист. Это наименее удовлетворительный из всех трех методов, так как возможность увидеть вторичное пятно зависит от субъективных особенностей наблюдающего, а интенсивность пятен на хроматограмме может значительно варьировать в зависимости от конкретных условий хроматографирования.
Для количественных измерений пятно можно удалить с пластинки, вещество элюировать подходящим растворителем, а затем определить его достаточно чувствительным методом, таким, как спектрофотометрическое измерение, либо непосредственно, либо после какой-либо химической реакции. В некоторых случаях количественную оценку также можно провести путем измерения интенсивности пятна с помощью сканирующего денситометра и последующего сравнения этой интенсивности с интенсивностями пятен, полученных со стандартными количествами того же аналогично обработанного вещества.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
95
Методы разделения с применением тонкослойной хроматографии иногда могут быть усовершенствованы путем многократного хроматографирования (хроматограмме дают высохнуть и вновь хроматографируют в той же системе), непрерывного хроматографирования (подвижная фаза непрерывно испаряется с верхнего края поверхности адсорбента) или двухмерного хроматографирования (хроматограмме дают высохнуть, повопачивают под прямым углом и затем вновь хроматографицуют, часто в иной системе растворителей, чем та, что была использована первоначально). Однако интерпретировать результаты хроматографии, если используются такие процессы промежуточного высушивания, надо с осторожностью, так как во время хроматографирования на пластинке может происходить разрушение вещества, например вследствие окисления. Методика двухмерной хроматографии имеет особую ценность для заключения о химических изменениях, происходящих в процессе хроматографирования. Если смесь вначале хроматографируют в одном направлении, а затем под прямым углом в той же системе растворителя, пятна, соответствующие разделенным веществам, будут лежать на пластинке по диагонали при условии, что не возникнет никаких артефактов.
При тонкослойной хроматографии сорбент обычно распределяют тонким равномерным слоем на подложке. Это можно выполнить в аналитической лаборатории, но удобно пользоваться и имеющимися в продаже готовыми хроматографическими сорбентами, закрепленными на стеклянной пластинке, пластмассовой пленке или на металлической фольге. К сожалению, хроматографический процесс может быть настолько чувствителен к небольшим изменениям в условиях опыта, что эти имеющиеся в продаже различные материалы и аналогичные приготовленные в лаборатории пластинки не всегда взаимозаменяемы. Характеристики разделения, полученные с коммерческими пластинками силикагеля, и характеристики, полученные с пластинками, приготовленными в лаборатории с тем же сорбентом, который был использован фирмой-изготовителем, могут быть совершенно различными. Бывают случаи, когда вполне удовлетворительный метод разделения, разработанный с использованием приготовленных в лаборатории пластинок, не дает положительных результатов при использовании готовых пластинок, и наоборот. Поэтому, переходя с одного типа пластинок на другой, следует соблюдать особую осторожность и всегда предварительно проверять степень пригодности новой пластинки.
Хроматографическая камера, в которой проводят процесс хроматографирования, должна быть защищена от действия
<J6 международная фармакопея
света, если предполагается, что исследуемые материалы могут быть светочувствительны. В любом случае хроматографическая камера должна быть защищена от прямых солнечных лучей, так как лучи могут подвергаться различной степени преломления из-за дефектов стеклянных стенок камеры. В результате на пластинке образуются области повышенной температуры и нарушается правильное перемещение подвижной фазы.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Методика, приведенная ниже, предполагает применение хроматографической пластинки, приготовленной в лаборатории, однако можно использовать предварительно покрытые адсорбентом готовые и, если необходимо, активированные пластинки при условии, что показана их пригодность для данного конкретного случая.
Оборудование состоит из:
— устройства для распределения на пластинке равномерного слоя покрывающего вещества желаемой толщины;
— пластинок длиной 200 мм и шириной, достаточной для нанесения требуемого количества проб испытуемого раствора и раствора стандартного образца;
— хроматографической камеры из прозрачного материала, обычно из стекла, с притертой крышкой; размеры камеры должны соответствовать размеру используемых пластинок.
Готовят суспензию покрывающего вещества и, используя приспособление для распределения сорбента, покрывают тщательно очищенные пластинки слоем около 0,25 мм толщины, если в статье нет других указаний. Дают покрытым пластинкам высохнуть на воздухе и нагревают для активации, если нет других указаний, при 110° С в течение 30 мин, затем дают охладиться. Если пластинки сразу не используют, их хранят в эксикаторе, содержащем осушитель силикагель Р. С вертикальных сторон пластинки удаляют слой сорбента шириной 2—5 мм.
Если в статье нет других указаний, работу проводят в условиях насыщенной камеры. Для достижения таких условий в камеру помещают фильтровальную бумагу и вливают такое количество подвижной фазы, которое достаточно для насыщения фильтровальной бумаги и образования слоя глубиной около 5 мм. Закрывают камеру и оставляют стоять не менее чем на 1 ч при комнатной температуре.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
97
Метод
Все операции, во время которых пластинка подвергается воздействию воздушной среды, предпочтительно проводить при относительной влажности 50—60%. Наносят объем раствора, указанный в статье, в виде компактного пятна диаметром не более 4 мм. Для нанесения используют микропипетку, шприц или другой подходящий инструмент. Пятно должно находиться на расстоянии около 1,5 см от нижнего края и отстоять не менее чем на 2 см от вертикальной стороны пластинки. Если на одной пластинке получают несколько хроматограмм, пятна следует располагать на расстоянии не менее 1,5 см друг от друга на линии, параллельной нижнему краю пластинки. Когда растворитель выпарится, помещают пластинку в хроматографическую камеру, стараясь установить ее как можно точнее в вертикальном положении; стартовые точки должны находиться выше уровня подвижной фазы. Закрывают камеру и выдерживают при постоянной температуре. Дают подвижной фазе подняться на предписанное расстояние, обычно на 10—15 см, вынимают пластинку, отмечают положение фронта растворителя и высушивают, как указано в соответствующей статье.
Хроматография на бумаге
При хроматографии на бумаге неподвижной фазой служит лист бумаги подходящего строения и толщины, который иногда может быть пропитан жидкой фазой, не смешивающейся с подвижной фазой.
Хроматографическое разделение на бумаге обычно протекает значительно медленнее, чем на пластинке при тонкослойной хроматографии, а сам метод, как правило, не столь универсален, как тонкослойная хроматография, поскольку возможные вариации неподвижной фазы гораздо более ограничены. Нельзя также использовать для определения многие коррозирующие реактивы, которые обычно применяют, когда сорбентом служит нанесенный на стеклянную пластинку неорганический материал. Тем не менее хроматография на бумаге остается полезным методом, и некоторые весьма эффективные разделения, которые первоначально были осуществлены с использованием бумаги, не удавалось успешно перенести «а тонкослойную пластинку. Для пол у количественной и количественной оценки значительно легче и эффективнее вырезать нужную площадь бумаги и элюировать разделенный компонент, чем полностью снять слой порошка для
7—2G25
98
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
элюирования, как это необходимо сделать при тонкослойной хроматографии.
Определение значений и Rr, рассмотренное при обсуждении тонкослойной хроматографии, в равной мере относится к хроматографии на бумаге. Благодаря природе сорбента хроматографию на бумаге можно проводить и восходящим и нисходящим способом.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Нисходящая хроматография на бумаге
Прибор состоит из стеклянной камеры, размеры которой должны соответствовать размерам используемой бумаги; сверху камера плотно закрывается притертой стеклянной крышкой. Крышка имеет центральное отверстие диаметром около 1,5 см, закрытое массивной стеклянной палочкой или пробкой. В верхней части камеры находится лодочка с растворителем и приспособлением, обычно в виде стеклянной палочки, для удерживания хроматографической бумаги. С обеих сторон лодочки, параллельно и несколько выше ее краев имеются две стеклянные направляющие палочки для поддержания бумаги в таком положении, при котором она совершенно не будет касаться стенок камеры. Хроматографическая бумага представляет собой подходящую фильтровальную бумагу, нарезанную полосками достаточной длины и любой подходящей ширины — от 2,5 см до длины лодочки. Бумагу разрезают таким образом, чтобы подвижная фаза двигалась в направлении волокна бумаги.
Метод
На дно хроматографической камеры помещают слой подвижной фазы, указанной в статье, глубиной 2—3 см. Закрывают камеру и оставляют стоять на 24 ч при постоянной температуре. Все операции, во время которых бумага подвергается воздействию воздушной среды, должны проводиться при относительной влажности около 50%. Эти условия поддерживаются в камере в течение всего последующего опыта. На одном конце бумаги тонко отточенным карандашом проводят горизонтальную линию на таком расстоянии от края, чтобы она была на несколько сантиметров ниже направляющей палочки и параллельна ей, когда этот конец бумаги погружен в лодочку с растворителем, а оставшаяся часть свободно висит на направляющей палочке. С помощью микропипетки, шприца или другого подходящего инструмента наносят раствор, указанный в статье, на какую-либо точку на линии,
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
99
прочерченной карандашом. Если общий объем раствора, который надо нанести, дает пятно диаметром более 10 мм, раствор наносят частями, высушивая каждый раз перед нанесением следующей порции. Если на одной полоске бумаги необходимо получить несколько хроматограмм, то расстояния между точками нанесения растворов на карандашной линии должны быть не менее 3 см. Вносят бумагу в камеру, закрывают крышку и оставляют стоять на 90 мин. Затем в лодочку для растворителя через отверстие в крышке вносят в достаточном количестве подвижную фазу, указанную в статье, закрывают камеру и хроматографируют до указанного расстоя-нили или в течение определенного времени. Вынимают бумагу из хроматографической камеры и высушивают на воздухе. Во время хроматографирования и высушивания бумагу следует защищать от яркого света.
Восходящая хроматография на бумаге
Прибор состоит из стеклянной камеры, размеры которой должны соответствовать размерам используемой бумаги; сверху камера закрывается плотно притертой стеклянной крышкой. В верхней части камеры имеется приспособление, которое поддерживает хроматографическую бумагу и которое можно опустить, не открывая камеры. У основания камеры имеется чашка для подвижной фазы, в которую можно опустить бумагу. Хроматографическая бумага представляет собой подходящую фильтровальную бумагу, нарезанную (полосками достаточной длины и шириной не менее 2,5 см; бумагу разрезают таким образом, чтобы подвижная фаза двигалась в направлении волокон бумаги.
Метод
Помещают в чашку подвижную фазу, указанную в статье, слоем глубиной 2—3 см. Если в статье указано, между чапь кой и стенками камеры наливают неподвижную фазу. Закрыт вают камеру и оставляют на 24 чпри постоянной температуре. Поддерживают камеру в тех же температурных условиях в течение всего последующего опыта. Все операции, во время которых бумага подвергается действию воздушной среды, должны проводиться при относительной влажности около 50%. Проводят тонко отточенным карандашом горизонтальную линию на расстоянии 3 см от одного из концов бумаги. С помощью микропипетки, шприца или другого подходящего инструмента наносят раствор, указанный в статье, на какую-либо точку на линии, прочерченной карандашом. Если общий
7*
100
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
объем раствора, который надо нанести, дает пятно диаметром более 10 мм, его наносят частями, высушивая каждый раз перед нанесением следующей порции. Если на одной полоске бумаги необходимо получить несколько хроматограмм, то расстояния между точками нанесения растворов на карандашной линии должны быть не менее 3 см. Вносят бумагу в камеру, закрывают крышку и оставляют на 90 мин. Погружают бумагу в подвижную фазу, указанную в статье, и хроматографируют до указанного расстояния или в течение определенного времени. Вынимают бумагу из хроматографической камеры и высушивают на воздухе. Во время хроматографирования и высушивания бумагу следует защищать от яркого света.
Хроматография на колонках
При адсорбционной хроматографии на колонках адсорбент (например, активированная окись алюминия, порошок целлюлозы, кремневая кислота или кизельгур) в виде сухого твердого вещества или пасты укладывают в трубку (стеклянную, пластмассовую или из другого подходящего материала), имеющую ограниченное выходное отверстие (обычно защищенное стеклянной пористой пластинкой) для вытекания подвижной фазы. Раствор хроматографируемого вещества наносят на поверхность сорбента в колонке и дают ему протечь в сорбент; затем на вершину колонки наносят растворитель, представляющий собой подвижную фазу, помещают и дают ему протечь вниз либо под действием силы тяжести, либо под небольшим давлением. При выполнении этой методики надо следить за тем, чтобы вершина колонки не обсыхала. Анализируют протекающий раствор — элюент — либо непрерывно (например, с помощью проточной кюветы, в которой измеряется поглощение в ультрафиолетовой области), либо поэтапно (например, собирая фракции либо через определенные промежутки времени, либо определенного объема или массы элюата с последующим определением разделяемых компонентов в каждой фракции). Необходимость индивидуально анализировать много фракций для получения полной количественной оценки вещества привела к тому, что применение в последние годы классических методик хроматографии на колонках сократилось; там, где их продолжают использовать, существует естественная тенденция выбирать те методы обнаружения и определения, которые легко переводятся в автоматические процессы.
При распределительной хроматографии на колонках жидкая неподвижная фаза, которая не должна существенно смешиваться с подвижной фазой, адсорбируется на поверхности
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
101
твердого сорбента. Хроматографирование проводят так же, как описано для адсорбционной хроматографии на колонках. Прежде чем использовать подвижную фазу для элюирования, ее насыщают неподвижной фазой. Обычно твердый сорбент в распределительной хроматографии полярен, и адсорбированная неподвижная фаза также полярна по отношению к подвижной фазе, поэтому наиболее часто применяемым сорбентом является кремнезем, имеющий такие размеры частиц, которые позволяют легко протекать подвижной фазе. В некоторых случаях удобным методом является обращенно-фазовая распределительная хроматография; в этом случае полярный сорбент превращают в неполярный путем силаниза-ции или другим способом, например обработкой парафинами, а адсорбированная неподвижная фаза менее полярна, чем подвижная фаза.
В этих распределительных системах степень распределения вещества определяется коэффициентом его распределения между двумя жидкими фазами, а если вещества диссоциируют—величиной pH более полярной из двух фаз. Часто можно добиться избирательного элюирования компонентов смеси путем последовательных изменений в подвижной фазе или изменения pH неподвижной фазы за счет применения подвижной фазы, представляющей собой раствор соответствующей кислоты или основания в органическом растворителе.
Ионообменную хроматографию можно рассматривать как частный случай хроматографии, при которой твердая фаза содержит ионообменный материал, обычно называемый ионообменной смолой.
Ионный обмен определяется как обратимое взаимодействие иона, находящегося в растворе, с ионом противоположного знака смолы-полимера, модифицированной целлюлозы или связанного силикагеля-носителя; в качестве примеров можно привести обмен H+/Na+ на сильнокислотной катионообменной смоле:
RSO3H + Na+ «==> RSO3Na + Нх
и обмен С1_/ОН~ на сильноосновной анионообменной смоле: R'N(CH3)3OH + СИ > R'N(CH3)3C1+ОН-.
Выбор сильных или слабых смол любого типа в значительной мере зависит от величины pH, при которой должен проводиться обмен, и от характера катионов и анионов, которые будут обмениваться. Однако в большинстве случаев для аналитических целей используют сильнокислотные и сильноосновные обменные смолы. Их обменная емкость может варьировать от 2 до 5 миллимолей на 1 г (сухое вещество).
102
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
На практике применяется 'большой избыток смолы, на 200— 300% превышающий расчетное стехиометрическое требование.
Законы, по которым происходит реакция обмена, сложны: частично они могут 'быть объяснены законом действия масс и соотношением зарядов и активности ионов. На преимущественное свойство ионообменной смолы принимать из раствора два или больше ионов указывает коэффициент избирательности. Вообще говоря, смола поглощает преимущественно двухвалентные и 'более ионы, а не моновалентные, и в случае выбора между ионами одинаковой валентности смола поглощает более тяжелый ион.
Обработка ионообменной смолы и подготовка колонки. Обычно ионообменную смолу оставляют для набухания в воде на 24 ч; затем ее укладывают в подходящую колонку. Если это анионообменная смола, ее переводят в основную форму, пропуская гидроокись натрия (~80 г/л) ИР через колонку со скоростью около 3 мл в минуту до отрицательной реакции элюата на хлориды; затем промывают водой, свободной от углекислоты, Р для удаления щелочности. Если смола катионообмениая, переведение в кислотную форму достигается пропусканием через колонку соляной кислоты (~70 г/л) ИР с последующим промыванием водой, свободной от углекислоты, Р до нейтральной реакции.
Приготовленную колонку используют так же, как это описано для адсорбционной хроматографии на колонках, за исключением того, что обычно не надо наблюдать за элюатом. В зависимости от типа выбранной смолы и определяемого материала собирают определенный для конкретного случая объем элюата и титруют кислотой или основанием, применяя подходящий индикатор.
После окончания определения ионообменную колонку можно регенерировать промыванием гидроокисью натрия (~80 г/л) ИР для анионообменной колонки или соляной кислотой (~70 г/л) ИР для катионообменной колонки с последующим промыванием водой до получения нейтральной реакции.
Жидкостная хроматография высокого давления
Этот совсем недавно введенный метод хроматографии вновь выдвинул на передний край исследований хроматографию на колонках — самую старую форму аналитического искусства. Основное достижение, благодаря которому стало возможным применение нового метода, — это техника получения частиц.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
103
устойчивых к высокому давлению и .имеющих одинаковый диаметр менее 50 мкм. Эти частицы обычно имеют твердый центр, например из стекла, и тонкий тористый наружный слой, например из кремния; благодаря небольшому размеру и большой тлощади поверхности эти частицы, будучи применены в адсорбционной хроматографии, обеспечивают очень высокую эффективность метода. Если частицы покрыты (подходящей неподвижной фазой, жидкостную хроматографию высокого давления можно использовать как метод распределения.
Для обеспечения стабильности подготовленной колонки неподвижные фазы часто связывают химически с носителем (обычно с томощью сложного или простого эфира). Простая эфирная связь обеспечивает 'более устойчивый продукт, чем сложноэфирная связь, которая может гидролизоваться полярными растворителями; например, в покрытых октадецилсила-ном бусинках углеводородная цепь связана простой эфирной связью со стеклянными бусинками, покрытыми тонким слоем кремния, и это обеспечивает весьма эффективную систему с обращенной фазой, которая исключительно стабильна в пользовании. Для хроматографирования эти частицы укладывают в колонки с узким отверстием (внутренним диаметром обычно 2—4 мм); вполне очевидно, что такой мелкий материал, упакованный в колонки длиной до 1 м, будет создавать значительное сопротивление потоку подвижной фазы, в силу чего и должно применяться высокое давление. Типичная длина колонок составляет 20—30 см, а условия, обычные для количественного анализа, — скорость потока около 1—3 мл в минуту и давление до 28 000 кПа (4000 фунтов/дюйм2).
Совсем недавно появились кремниевые шарики с одинаковым диаметром около 5 мкм; они полностью пористые и могут иметь площадь поверхности до 300 м2/г. Соответственно они дают более эффективные разделения, чем частицы размером 30—50 мкм. Для приготовления колонок большие частицы можно укладывать сухими, но для частиц диаметром 5 мкм в основном используется суспензионный способ заполнения колонки.
Кроме адсорбционного и распределительного способов, рассмотренных выше, принцип метода высокого давления применим и к ионообменной хроматографии при условии, что имеются подходящие смолы в виде достаточно мелких, устойчивых к давлению частиц.
Естественно, что такое высокое давление требует и специального оборудования. К основным частям прибора относятся подходящий насос для подачи подвижной фазы из закрытого резервуара растворителя на колонку, приспособления для
104
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
введения испытуемого раствора в колонку (обычно это вид инжекторного клапана, предназначенного для работы при высоком давлении), сама колонка (часто определение происходит при комнатной температуре, но иногда колонка выдерживается при температуре до 100° С), соответствующая детекторная система и усилитель, связанный с подходящим регистрирующим устройством, таким, как ленточный самописец, позволяющий выстроить график зависимости сигналов от времени, или электронный интегратор.
В качестве детекторов на настоящей стадии разработки метода наиболее часто применяются устройства, основанные на ультрафиолетовой спектрофотометрии, на измерении показателя преломления или на измерениях флуоресценции. Для фармацевтических целей наиболее подходящим является ультрафиолетовый спектрофотометр, обладающий высокой чувствительностью (низший уровень обнаружения составляет 1—2 нг для материала, имеющего хорошие светопоглощающие свойства) и стабильностью (в частности он отличается низкой чувствительностью к контролируемым изменениям в составе растворителя и неравномерности потока); естественно, что такой детектор не может быть использован, если элюируется материал, не имеющий заметного поглощения в ультрафиолетовой области. Рефрактометр реагирует на разницу в показателе преломления чистой подвижной фазы и подвижной фазы, содержащей элюируемый материал; этот метод имеет более широкое применение, чем адсорбционная спектрофотометрия в ультрафиолетовой области, но он малочувствителен и в значительной степени зависит от небольших изменений в составе растворителя, от скорости потока и температуры.
В некоторых случаях, в частности, в опытах, имеющих целью определение оптимального состава растворителя для метода, который затем будет использоваться повседневно, удобна методика градиентного элюирования. Состав смеси растворителей, входящих в подвижную фазу, во время хроматографирования непрерывно меняют с предварительно установленной скоростью, что дает возможность решать при помощи одной хроматограммы проблему разделения сложной смеси веществ, имеющих совершенно различные коэффициенты распределения. Коэффициент распределения К, как указано в разделе «Газовая хроматография» (см. ниже), является мерой количества растворенного вещества в неподвижной фазе по отношению к концентрации вещества в подвижной фазе.
При использовании методов расчета, аналогичных описанным в разделе «Газовая хроматография», методика высокого давления дает возможность получать результаты высокой точности и поэтому она чрезвычайно удобна для количествен
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
105
ных определений. Эта методика отнимает мало времени и используется для осуществления многих высокоэффективных .разделений, однако для ее применения нужны специальные приборы и во многих случаях дорогостоящие материалы для колонок. Потенциальное преимущество этой методики перед газовой хроматографией состоит в том, что летучесть и термостабильность, факторы столь важные для последней, не имеют никакого значения при жидкостной хроматографии. К ее недостаткам в настоящее время относится отсутствие универсально применимых детекторных систем.
Газовая хроматография
Газовую хроматографию можно рассматривать как форму хроматографии на .колонках, при которой подвижной фазой является газ (газ-носитель), а не жидкий растворитель. Неподвижной фазой может служить либо активный сорбент, такой, как окись алюминия, силикагель или уголь (газоад-сорбционная хроматография), либо жидкость, которая в виде тонкой пленки покрывает тонко измельченный инертный твердый носитель, такой, как диатомовая земля, кирпич,, стеклянные бусинки или другой подходящий .материал (газожидкостная хроматография); если хроматографическая колонка имеет очень небольшой диаметр, неподвижной фазой может быть покрыта внутренняя стенка колонки; это так называемые открытые трубчатые, или капиллярные, колонки. Имеются некоторые материалы, которые не требуют покрытия жидкой фазой, например полиаромэтические пористые бусинки, что весьма ценно в случаях специального применения.
Вещество в выпаренном состоянии вводят в поток газа-носителя на вершину колонки и оно подвергается распределению между газом и жидкой или твердой неподвижной фазой аналогично тому, как это происходит при других видах хроматографии. Коэффициент распределения (К) определяется следующим отношением:
количество растворенного вещества в неподвижной фазе А ~ количество растворенного вещества в подвижной фазе
Коэффициент К зависит от природы растворенного вещества, природы и количества неподвижной фазы, температуры и скорости потока газа-носителя. Очевидно, что величина К будет также зависеть от конкретной колонки и точных условий выполнения анализа, а поскольку эти условия невозможно
106
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
точно воспроизвести в разных лабораториях и на разных .производствах, изготовляющих приборы, необходимо условия опыта, разработанные для фармакопейных целей, воспринимать с известной гибкостью, что не допускается в отношении других, не хроматографических, методик испытания.
Основными составными частями прибора для выполнения газожидкостной хроматографии (гораздо шире применяемой в фармацевтическом анализе, чем газоадсорбционная хроматография) являются источник газа-носителя (обычно содержится в сжатом виде в цилиндре, снабженном редуктором), расходомер, через который проходит газ, и выходное отверстие для ввода пробы, которое можно нагревать до подходящей температуры для испарения, но не разрушения вещества и через которое в поток газа-носителя вводится испытуемый раствор, предпочтительно непосредственно в колонку. Во время хроматографирования (поддерживается постоянная скорость потока газа-носителя, а компоненты испытуемого раствора разделяются в соответствии с величиной К для каждого компонента при данных условиях.
По мере выхода компонентов из колонки они попадают в детектор дифференциального типа, который обычно зависит от изменений ионизации в пламени или изменений термопроводимости. Существует много других типов детекторов; некоторые из них пригодны для специфических видов фармацевтического анализа, например электронзахватный детектор особенно ценен для чувствительного обнаружения галогенированных соединений. Электрические сигналы от детектора поступают в усилитель, связанный с подходящим регистрирующим устройством, таким, как ленточный самописец, который регистрирует сигналы в зависимости от времени. 'Весьма эффективным, но очень дорогим средством обнаружения является применение масс-спектрометра, присоединенного к газовому хроматографу. Это очень чувствительный метод, обеспечивающий точную идентификацию веществ, выходящих из колонки.
Колонка, изготовленная из стекла или металла (в последнем случае необходимо соблюдать осторожность, так как некоторые органические вещества при повышенной температуре подвергаются катализируемому металлом разрушению), заключена в термостатированную печь, температура в которой может поддерживаться от комнатной до примерно 300° С в соответствии с конкретным применением. Нагревание может контролироваться таким образом, что равномерное повышение температуры обеспечивается в течение определенного промежутка времени; такое «температурное программирование» очень удобно в тех случаях, когда исследуется сложная
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
107
смесь соединений, имеющих совершенно различные характеристики иопарения. Обычно применяют колонки различной длины; длина заполненных колонок может составлять от 0,5 почти до 3 м, капиллярных колонок — от 10 до 100 м; для многих фармацевтических анализов обычно применяют колонку длиной около- 1,5 м с внутренним диаметром 2— 5 мм.
Материал, заполняющий колонку, оказывает существенное влияние на .качество и эффективность разделений. Твердый носитель, размер частиц которого может варьировать примерно от 75 до 250 мкм (60—200 мешей; хотя в любой данной колонке размер частиц должен 'быть строго определен в пределах узкой области), должен быть, насколько это возможно, более инертным, особенно если полярные лекарственные вещества хроматографируются на носителях, покрытых малыми количествами жидкости низкой полярности. Наличие активных центров на твердом носителе может привести к удлинению пика растворенного вещества или даже к его разрушению или перегруппировке. Реактивность носителя может быть уменьшена путем обработки силанизирующим реактивом до того, как носитель покрывают неподвижной фазой. Остатки вводимых проб могут привести к тому, что во входной части колонки процесс сорбции будет нарушен.
В качестве жидких фаз обычно используют макроголы (полиэтиленгликоли) и сложные эфиры, высокомолекулярные амиды, силиконовые каучуки и жидкости, а также углеводороды. Силиконовые каучуки представляют собой замещенные полисилоксаны и относятся к наиболее .ценной группе неподвижных фаз. Следует обратить особое внимание на наивысшую температуру, при которой предназначенная неподвижная фаза должна использоваться, и тщательно следить за тем, чтобы она не была превышена, так как при избыточной температуре может произойти «утечка колонки», что приведет к искажению результатов. Перед применением любую новую колонку следует подогнать к условиям испытания, выдерживая ее в течение нескольких часов в токе газа-носителя, пропускаемого при температуре несколько более высокой, чем температура, при которой впоследствии колонка будет использоваться, но, естественно, не выше, чем самая высокая рекомендуемая температура.
В качестве газа-носителя следует выбирать инертный газ; наиболеее подходящим для пламенно-ионизационного метода определения, чаще других применяемого в фармацевтическом анализе, является азот. Для этих целей пригоден также гелий и его следовало бы предпочесть при работе с детектором по теплопроводности, поскольку гелий обладает высокой тепло
108
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
проводностью, однако он дорог .и не всегда имеется во всех странах мира, в то время как азот доступен повсюду.
Для количественных определений методом газожидкостной хроматографии в фармакопее обычно используют внутренний стандарт, так как результаты сравнения одной хроматограммы с другой, полученной после второго введения в колонку, могут быть ошибочными. Прибавление подходящего внутреннего стандарта к испытуемому раствору и к стандартному раствору исключает эту ошибку, так как на хроматограммах сравнивается отношение площади или высоты пика (см. ниже) определяемого вещества к аналогичным величинам, полученным с внутренним стандартом. При других определениях, в частности когда оценивается содержание примеси, удобнее использовать процесс нормализации. В этом случае площадь пика, относимая за счет предполагаемой примеси, выражается как процент от общей площади всех пиков, полученных с испытуемым веществом и его ожидаемыми примесями. Поскольку при этом величина пика основного компонента обычно на два порядка выше, чем величина пика наименьшей примеси, для таких определений необходимо использовать надежный автоматический интегратор и усилитель широкого диапазона, который обеспечивает линейное усиление сигнала и от большего и от меньшего компонентов. Площади пиков можно также измерять планиметром, графически или по массе 'бумаги, вырезанной по размерам пиков из хроматограммы. При определенных обстоятельствах более целесообразно измерять высоту пика, а не его площадь, хотя последняя величина более точна для количественных определений. Ширина пика определяется как отрезок нулевой линии, заключенный между точками пересечения линий, касательных к образующим пика.
Если для определения используют внутренний стандарт, можно применить методику, описанную ниже. Следует отметить, что применение этой методики требует приготовления 3 растворов. Первый из них (раствор 1) содержит внутренний стандарт и соответствующее количество определяемого вещества (в случае определения примеси это может быть сама примесь, если она имеется, или достаточно низкая нагрузка вещества, в котором определяются примеси). Полученная таким образом хроматограмма А позволяет установить отношение ответа внутреннего стандарта к ответу определяемого вещества. Второй раствор (раствор 2) состоит только из исследуемого вещества; хроматограмма Б дает возможность аналитику убедиться в том, что примесь, которая должна была бы элюироваться с тем же временем удерживания, что и внутренний стандарт, отсутствует или, если наблюдается
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
109
совпадающий пик, допустить, что имеется определенное количество примеси. Третий раствор (раствор 3) состоит из испытуемого вещества и внутреннего стандарта, причем последний присутствует в той же концентрации, что и в растворе 1. Данные, полученные на хроматограммах А и В, скорректированные, если необходимо, по результатам хроматограммы Б, дают возможность определять компоненты, содержащиеся в исследуемом веществе .в минимальном количестве.
Если используется метод нормализации, достаточно одного раствора, так как общая площадь всех минимальных пиков выражается как часть общей площади пиков. Если нужно оценить конкретный минимальный пик, необходимо иметь второй раствор, содержащий определяемый материал, чтобы можно было идентифицировать соответствующий пик на хроматограмме исследуемого вещества.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Длина хроматографической колонки, неподвижная фаза, твердый носитель, температура, газ-носитель, детектор и другие особенности определения указываются в соответствующей частной статье. Если в колонку должно вводиться нелетучее вещество, можно применить подходящую взаимозаменяемую колонку (предколонку).
В некоторых статьях может быть предусмотрена минимальная эффективность колонки. Она определяется выражением:
\Gt\iLy\
где tR — расстояние (мм) на нулевой линии между точкой введения определяемого вещества и перпендикуляром, опущенным из высшей точки пика, полученного с внутренним стандартом, указанным в соответствующей частной статье;
L — длина колонки (м);
у — ширина пика, полученного с внутренним стандартом (мм).
При исследовании водных растворов с использованием пламенного ионизационного детектора результаты недействительны, если одновременно с любым из определяемых компонентов элюируется вода.
Метод
Применяя раствор 1, экспериментально определяют чувствительность прибора и объем растворов, необходимый для получения приемлемого ответа. Если необходимо, подбирают
110
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
концентрацию внутреннего стандарта таким образом, чтобы ответ самописца, полученный с внутренним стандартом, был. приблизительно таким же, что и ответ, полученный с определяемым веществом. Получают дифференциальную хроматограмму, вводя выбранный объем раствора 1 через отверстие для ввода пробы в хроматографическую колонку, поддерживаемую при соответствующей температуре, и элюируют при помощи газа-носителя. Повторяют определение еще два раза. Таким же образом получают кривые, используя такие же объемы растворов 2 и 3. Измеряют площадь пика, или, если фактор симметрии находится в пределах 0,95—1,05, измеряют высоту пиков определяемого вещества или веществ и внутреннего стандарта. Если пик примеси имеет то же время удерживания, что и пик внутреннего стандарта, то при оценке результатов делается допуск на взаимное влияние етих компонентов друг на друга. На основании полученных данных: рассчитывают содержание определяемых веществ.
Фактор симметрии пика определяют по выражению ух/24, где ух — ширина пика на 1/20 его высоты;
А — расстояние между перпендикуляром, опущенным и» максимума пика, и точкой на образующей пика, соответствующей 1/20 его высоты.
Результаты определения считаются недействительными, если разрешение между измеренными пиками на хроматограмме превышает 1,0. Разрешение (степень полноты разделения) рассчитывают по формуле:
2(tKb~tKa)/(Ya + Yb),
где tRa и tRb— расстояния на нулевой линии между точкой введения и перпендикуляром, опущенным из максимумов двух близлежащих пиков;
и Уь — ширина соответствующих пиков.
ОПРЕДЕЛЕНИЕ pH
Величина pH является характеристикой водного раствора; эта константа условно обозначает его кислотность или щелочность.
pH раствора — отрицательный десятичный логарифм активности водородных ионов, которая может быть измерена потенциометрически. Ранее величина pH рассматривалась как отрицательный десятичный логарифм концентрации водородных ионов. Поскольку известно, что не все водородные ионы обязательно обладают равной Активностью, концентрация
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
111
водородных ионов может отличаться от их активности. Однако, если коэффициент активности близок к 1, что справедливо для разведенных растворов, величины активности водородных ионов и их концентрации становятся почти идентичными.
Величина pH определяется путем измерения разности потенциалов между электродами, погруженными в стандартный и .испытуемый растворы. Величина pH стандартных растворов условно принята постоянной.
При измерениях pH широко применяется стеклянный электрод, так как он дает немедленный ответ на быстрое изменение концентрации водородных ионов даже в растворах с небольшой буферной емкостью. Так как этот механизм не связан с электронным обменом, это единственный чувствительный к водородным ионам электрод, на показания которого не влияют окислительные и восстановительные агенты.
Для растворов или суспензий, которые являются только частично водными, величины pH могут рассматриваться только как «кажущиеся»; их также можно измерить путем применения подходящего электрода и соответствующим образом стандартизованного рН-метра.
Величины pH зависят от температуры, поэтому их измерение проводят при выбранных постоянных температурах.
Растворы, используемые при определениях pH, готовят на воде, не содержащей углекислоты, Р.
Шкала pH
Различие между pH двух растворов — X и S — при одинаковой температуре может быть определено следующим образом.
Электродвижущая сила Ех элемента
Pt | Н2 | раствор X | 3,5 моль/л КС11 электрод сравнения
и электродвижущая сила Es элемента
Pt | Н2 | раствор S | 3,5 моль/л КО | электрод сравнения
измеряется при одинаковой температуре обоих элементов в течение всего времени измерения; электроды сравнения и растворы в соединительных мостиках должны быть идентичными в обоих элементах.
Отношение pH раствора X, обозначенного pH (X), к pH раствора S, обозначенному pH (S), определяется по уравнению:
fv -— Es
pH (X) = pH (S) + 2,30267?T/F ’
112
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
где R— газовая постоянная, Т — термодинамическая (абсолютная) температура ( в кельвинах, К), F—число Фарадея. Таким образом, количественная величина pH не имеет размерности.
Числовые значения фактора 2,3026 RT/F три разных температурах указаны ниже:
Температура 2,3026 RT/F
(°C) (мВ)
10 56,18
15 57,17
20 58,17
25 59,16
30 60,15
Потенциометрическое определение pH
Для практического определения величины pH обычно используют потенциометрический метод. Если применяются стеклянные электроды, их необходимо хранить в подходящей жидкости, обычно в воде.
Как правило, удобнее производить измерения pH со стеклянным электродом, а не с водородным. В некоторых растворах, особенно в растворах, содержащих окислители, когда нельзя использовать водородный электрод, применяют стеклянный электрод. Однако точность и воспроизводимость определения, равная ±0,005 и обычно получаемая с водородным электродом, редко может 'быть получена со стеклянным электродом и никогда не может быть получена вне значений pH от 2 до 10.
Если используют стеклянный электрод, то наивысшая точность достигается при условии, что в узкой области pH имеет-
ТАБЛИЦА 3. СТАНДАРТНЫЕ БУФЕРНЫЕ РАСТВОРЫ И ИХ
ВЕЛИЧИНЫ pH
Значения pH при различных температурах
Стандартные буферные растворы 20 °C 25 °C 30 °C 35 °C 40 °C
Калия тетраоксалата ИР 1,675 1,679 1,683 1,688 1,694
Калия гидротартрата ИР — 3,557 3,552 3,549 3,547
Калия гидрофталата ИР 4,002 4,008 4,015 4,024 4,035
Фосфатный буферный раствор,
pH 6,8 ИР 6,881 6,865 6,853 6,844 6,838
Фосфатный буферный раствор,
pH 7,4 ИР 7,429 7,413 7,400 7,389 7,380
Натрия тетрабората ИР 9,225 9,180 9,139 9,102 9,068
Натрия карбоната ИР 10,062 10,012 9,966 9,925 9,889
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ Hfe
ся линейная зависимость между pH и измеряемой электродвижущей силой, но что фактор пропорциональности между ними не обязательно точно равен 2,3026 RF/Т. Этот метод с применением стеклянного электрода требует калибровки посредством двух растворов с известным значением pH, близким и предпочтительно .включающим значение pH испытуемого раствора. В настояще время имеются различные подходящие растворы, величина pH которых достоверно известна с точностью ±0,005 (см. табл. 3).
Калибровка прибора
Прибор калибруют по стандартным буферным растворам для проверки линейности показаний электрода при различных значениях pH и для обнаружения неисправного стеклянного электрода. Стандартизация прибора только по одному раствору может быть полностью ошибочной, поэтому для калибровки следует использовать не менее двух стандартных буферных растворов. Наличие неисправного электрода обнаруживается по невозможности получить приемлемую точную величину (±0,04 единицы) для pH второго стандартного раствора, если прибор был стандартизован в единицах первого стандарта. Разбитый электрод дает одинаковые значения pH для обоих растворов. Если различие между известным и наблюдаемым .pH для второго стандартного раствора превышает ±0,04, следует заменить стеклянный электрод. Если это различие сохраняется, следует приготовить свежие станг дартные растворы.
1 РЕКОМЕНДУЕМАЯ МЕТОДИКА
После калибровки лгрибора электроды и стаканчик тщательно промывают. Наполняют стаканчик частью испытуемого раствора и получают предварительное значение pH. Как правило, эта величина ‘будет смещаться, и ее следует рассматривать как приблизительную. Последующие показания, полу-ценные с остальными -порциями того же раствора, будут давать более постоянные значения pH. Если растворы хорошо забуферены, 3 порций может быть достаточно для получения значения pH, воспроизводимого с точностью до ±0,04 единицы и смещающегося менее чем на ±0,04 единицы за 1—2 мин. Если растворы сильно разведены или обладают слабыми буферными свойствами, может понадобиться до 6 порций испытуемого раствора, а значение pH будет продолжать смещаться и будет воспроизводимо с точностью только до ±0,05 единицы.
8—2025
114
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Если требуется получить значение pH с точностью более 0,1 единицы, температура стандартных растворов, стеклянного и каломельного электродов и испытуемых растворов может разниться не более чем на 2° С, а электроды, стандартные растворы, испытуемые растворы и промывная вода должны храниться при температуре измерения не менее 2 ч до проведения измерения, чтобы свести влияние термического или электрического гистерезиса электродов до величины, которой можно пренебречь.
Стандартные буферные растворы
Стандартные буферные растворы используют для определения величин pH. Их готовят на воде, свободной от углекислоты, Р. Растворы должны храниться в склянках из химически устойчивого стекла .или в сосудах из полиэтилена.
Если нет других указаний, стандартные буферные растворы не следует использовать позже 3 мес с момента их приготовления. Если начинается рост микроорганизмов, растворы тотчас выбрасывают; склянки тщательно очищают и стерилизуют перед новым наполнением.
ЭЛЕКТРОФОРЕЗ
Электрофорез — физический метод анализа, позволяющий проводить разделение веществ, способных приобретать электрический заряд в проводящем электролите. В этой среде ионизированные частицы более или менее быстро перемещаются под воздействием электрического поля.
Электрофоретическая подвижность — это скорость перемещения вещества, измеряемая в сантиметрах в секунду под влиянием градиента потенциала 1 В/см и выражаемая в см2-В-1-с-1.
Измерение величины электрофоретической подвижности дает значимый результат только в том случае, когда экспериментальные условия точно определены. Эта подвижность зависит от характеристик вещества, его природы, размера, формы и электрического заряда. Она также зависит от проводящей жидкости, ее природы, концентрации, pH, присутствия дополнительных растворителей и вязкости. Направление перемещения зависит от знака электрического заряда частицы, так как она движется к электроду с противоположным знаком.
В соответствии с применяемыми методами электрофоретическая подвижность либо измеряется непосредственно, либо сравнивается с подвижностью стандартного образца.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
115
Электрофорез с движущимся пограничным слоем (без носителя)
Эта методика, используемая исключительно для определения подвижности, особенно подходит для веществ с высокой молекулярной массой и слабыми диффузионными свойствами.
Границы обычно измеряются до и после наложения электрического поля каким-нибудь физическим методом, таким, как рефрактометрия или кондуктометрия. Концентрация вещества в проводящей жидкости, характеристики последней и подробности методики, в том числе количественная оценка фракций, описаны в соответствующих частных статьях.
Зонный электрофорез (электрофорез с использованием среды носителя)
При этом методе используют образцы только небольших размеров. Природа среды носителя (например, бумага, ацетат целлюлозы, гель крахмала, агаровый гель, полиметакриламид, смешанный гель) обусловливает дополнительные факторы, влияющие на подвижность. Скорость перемещения зависит от подвижности частиц, а также от электро-эндосмо-тического тока (в случае носителей с полярными свойствами), от токов, обусловленных испарением (вызванным теплом, генерируемым за счет эффекта Джоуля), и от градиента электрического поля.
На практике подвижностью электрофоретических зон и их знаками пренебрегают; зоны обнаруживают эмпирически или путем сравнения с поведением стандартного образца в тех же условиях.
После разделения составных частей положение бесцветных веществ может быть определено путем обработки электрофореграмм реактивом, который переводит их в окрашенные или флуоресцирующие производные. Для количественных целей пятно (зона) может быть тщательно выделено, вещество элюировано подходящим растворителем и затем определено непосредственно или после химической реакции достаточно чувствительным методом, таким, как спектрофотометрическое измерение. По другой 'количественной методике после превращения вещества в окрашенное производное интенсивность зоны можно измерить при помощи сканирующего денситометра.
Ниже перечислены составные части прибора для электрофореза на среде-носителе.
8*
1
J16 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Источник постоянного тока, предпочтительно со стабилизированным напряжением.
Камера для электрофореза, обычно в форме параллелепипеда, выполненная из стекла или прочного пластического материала, с плотной воздухонепроницаемой крышкой, обеспечивающей поддержание атмосферы насыщенной влажности. В противоположные стенки камеры запаяны два изолированных проводника, на конце каждого из них имеется внутренний соединитель, к которому присоединены электроды из платиновой проволоки. В целях безопасности камера должна быть снабжена приспособлением, обеспечивающим прерывание электропитания при снятии крышки. В камере у двух противоположных сторон вставлены две двойные кюветы, разделенные вдоль на две равные половины. В другом варианте эти кюветы могут быть частью самой камеры. Вдоль основания каждого из внешних отделений кювет проложен один платиновый электрод. Электроды соединены через наружный изолированный кабель с источником постоянного тока напряжением не менее 450 В при 150 мА. Источник энергии должен быть оборудован прибором, контролирующим и указывающим напряжение, а также потребление тока. Для стабилизации напряжения может быть встроена дополнительная цепь.
Держатель. Если используют бумагу или ацетат целлюлозы, полоски носителя, пропитанные проводящей жидкостью, укрепляют с помощью соответствующего приспособления в определенном положении, а их концы погружают в кюветы с электродами. При электрофорезе на геле ровный слой геля помещают клеющей стороной на стекло и к каждому концу слоя присоединяют электрические клеммы.
' ' Приспособление для обнаружения й измерения пятен.
, . РЕКОМЕНДУЕМАЯ МЕТОДИКА
•• ♦
Электрофорез на бумаге
Для определения .пригодна камера длиной около 50 см, шириной. 38 см и глубиной 4,5 см с кюветами, .внутренняя длина которых составляет около 37 см, ширина 5 см и внутренняя глубина 2 см.? з..._
г Бумага для электрофореза представляет собой., подходящую фильтровальную бумагу (пригодна бумагазВатманЗ ММ плиз аналогичная ей), промытая хроматографически по дходя-тцимрастворителем, если в статье есть на это указание. Бумагу разрезают на полоски подходящего размера и.ша. рас-стрннии около 13 см от одного конца карандашом прочерчивают базовую линию.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ 1 it
Заполняют кюветы прибора проводящей жидкостью, указанной в статье. Полоски бумага для электрофореза (около 30X5 см) помещают в кюветы так, чтобы между наружным и внутренним отделениями образовался мостик; электроды должны быть полностью погружены в проводящую жидкость во внутренних отделениях.
Наносят отдельно в точки вдоль базовой линии на бумаге на расстоянии не менее 1 см от края бумаги и не менее 2,5 см друг от друга объемы растворов, приготовленных, как указано в статье.
Дают пятнам высохнуть и затем помещают конец бумаги,, ближайший к базовой линии, во внутреннее отделение кюветы, связанное с анодом, а другой конец бумаги — во внутреннее отделение кюветы, связанное с катодом. С помощью кисти смачивают бумагу проводящей жидкостью, начиная с концов бумаги в направлении базовой линии. Не смачивают полоску, на которой находится нанесенное вещество. Закрывают крышку и дают жидкости диффундировать через базовую линию; если необходимо, закрывают прибор, чтобы защитить его от действия света, соединяют кабель с источником энергии и включают ток. Доводят напряжение примерно до 20 В на 1 см бумаги между кюветами и дают процессу протекать в течение указанного времени или до тех пор, пока маркерное вещество не пройдет определенное расстояние. Выключают ток, вынимают бумагу, высушивают в токе воздуха, защищая, если необходимо, от действия света, и оценивают полученную электрофореграмму в условиях, описанных в статье. Если статьей предписано применение маркерного вещества, результат испытания считается достоверным только в том случае, если это вещество продвинется от базовой линии на указанное расстояние. Если интенсивность любого дополнительного пятна, полученного с испытуемым веществом, меньше, чем интенсивность пятна, полученного с раствором стандартного образца, вещество соответствует требованиям. Если указано в статье, опрыскивают бумагу равномерно с обеих сторон реактивом, проводят дальнейшую предписанную обработку для завершения реакции и применяют те же критерии для оценки полученных пятен.
Электрофорез на полосках ацетата целлюлозы
Предпочтительно использовать камеру меньших размеров, чем при электрофорезе на бумаге; удобна камера размером около 25x24 см с кюветами 10x23 см.
Полоски полиацетата целлюлозы соответствующего качества размером 2,5х 17 см погружают в проводящую жидкость приблизительно за 1 ч до использования.
118 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
На полоски на расстоянии 8 см от их конца наносят растворы, приготовленные, как указано в статье, и в указанных объемах, затем проводят электрофорез, как описано в методике электрофореза на бумаге. Полосы окрашивают, промывают и, чтобы они стали прозрачными, обрабатывают методом, указанным в статье, где также описан метод оценки электрофореграмм.
Электрофорез на геле
Внутренний носитель представляет собой слой геля агара или крахмала подходящей консистенции толщиной 1—2 мм и имеет форму удлиненного прямоугольника.
Проводящую жидкость либо вводят в слой геля, либо после того, как гель сформировался, опрыскивают ею слой до полного смачивания. Раствор вещества помещают на поверхность слоя геля или внутрь отверстий, вырезанных в слое для этой цели. Слой геля соединен своими узкими концами с двумя кюветами, содержащими проводящую жидкость; соединение осуществляется при помощи фитилей из двойного слоя марли, смоченной проводящей жидкостью. Затем слой геля на его держателе и соединения помещают в камеру.
Электрофоретический процесс осуществляют с помощью постоянного электрического тока. Для устранения тепла, генерируемого за счет эффекта Джоуля, через пластинку держателя во время испытания должна циркулировать вода или другая подходящая охлаждающая жидкость.
После завершения процесса полученные пятна или площади миграции обнаруживают подходящим методом, указанным в частной статье; например, может быть определена зона угнетения после соответствующей инкубации, если испытуемое вещество является антибиотиком и соответствующий тест-организм был включен в слой геля, или применен какой-либо химический метод.
МЕТОД ФАЗОВОЙ РАСТВОРИМОСТИ
Метод фазовой растворимости — это количественное определение чистоты вещества путем точных измерений величины растворимости. Приданной температуре® определенном количестве растворителя растворяется определенное количество чистого вещества. Полученный раствор насыщен определенным веществом, но этот же раствор остается ненасыщенным в отношении других веществ, даже если эти вещества могут быть близки по химическому строению и физическим свойствам к
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ 119
данному исследуемому веществу. Постоянство величины растворимости указывает на то, что материал чист или свободен от посторонних веществ, за исключением единственного случая, когда процентный состав исследуемого вещества представляет собой прямое отношение величин растворимости соответствующих компонентов. И наоборот, различие в значениях растворимости указывает на наличие примеси или примесей.
Стандартный метод растворимости состоит из нескольких этапов: а) приготовление ряда отдельных систем, состоящих из возрастающих количеств материала и измеренных, постоянных количеств растворителя; б) установление равновесия для каждой системы при одинаковых постоянных температуре и давлении; в) отделение твердой фазы от растворов; г) определение концентрации материала, растворенного в различных растворах; д) построение графика, отражающего зависимость отношения концентрации растворенного материала к единице массы растворителя от отношения общей массы материала к единице массы растворителя, экстраполирование и вычисление. Кроме того, для оценки чистоты испытуемого вещества на основании полученных данных можно использовать статистическую методику расчета.
Растворители
При выборе подходящего растворителя для метода фазовой растворимости руководствуются следующими критериями.
1) Растворитель должен иметь такую летучесть, чтобы его можно было выпарить в условиях вакуума, но не должен быть настолько летучим, чтобы перенос и взвешивание самого растворителя и его растворов было сопряжено с трудностями. Обычно подходят растворители с температурами кипения от 60 до 150° С.
2) Растворитель не должен неблагоприятно влиять на образец. Нельзя использовать растворители, которые вызывают разрушение вещества или реагируют с ним. По возможности следует избегать растворителей, образующих сольваты или соли.
3) Степень чистоты и состав растворителя должны быть известны. Допускаются смешанные растворители. Следовые количества примесей могут существенно влиять на растворимость.
4) Для метода, описанного ниже, растворимость испытуемого вещества в избранном растворителе должна быть не
120
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
менее 4 мг/г и не более 50 мг/г. Оптимальной является растворимость 10—20 мг/г.
Приборы
Термостат. Для этих испытаний используют термостат, способный поддерживать заданную температуру в пределах ±0,1 °C. Обычно выбирают температуру от 25 до 30°С. Тер-
РИС. 2. АМПУЛА (СЛЕВА) И КОЛБЫ ДЛЯ РАСТВОРЕНИЯ (СПРАВА), ИСПОЛЬЗУЕМЫЕ ПРИ АНАЛИЗЕ МЕТОДОМ ФАЗОВОЙ РАСТВОРИМОСТИ
мостат оборудуют горизонтальным стержнем, способным вращаться со скоростью приблизительно 25 об/мин и имеющим зажимы для ампул. В другом варианте термостат может быть снабжен подходящим вибратором, обеспечивающим 100— 120 вибраций в секунду и имеющим стержень с зажимами для ампул или другое подходящее устройство для достижения равновесия в ампулах.
ФИЗИКО ХИМИЧЕСКИЕ МЕТОДЫ
121
Ампулы. Используют ампулы емкостью 15 мл (рис 2). Можно использовать и другие емкости при условии, что они герметичны и подходят во всех других отношениях.
Колбочки для растворения. Используют колбочки, .пригодные для лиофилизации. Подходящая колбочка с пробкой показана на рис. 2.
Весы. Используют весы и метод взвешивания, обеспечивающие точность взвешивания в пределах ±10 мкг.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Описанная ниже методика общепринята. Однако в некоторых случаях можно предпочесть и другие условия (объем растворителя и т. д.).
Состав системы
Точно взвешивают не менее 7 помеченных, тщательно вымытых ампул и в каждой из них точно взвешивают возрастающие количества исследуемого вещества. Массу вещества подбирают таким образом, чтобы первая ампула содержала немного меньше вещества, чем растворяется в 5 мл выбранного растворителя, а вторая и последующие ампулы — несколько больше, чем указанная величина растворимости. В каждую из ампул пипеткой вносят 5,0 мл растворителя, охлаждают в смеси сухого льда с ацетоном и запаивают с помощью двухструйной газовой горелки, следя за тем, чтобы сохранились все кусочки стекла. Дают ампулам вместе с содержимым остыть до комнатной .температуры и взвешивают отдельно каждую запаянную ампулу вместе с относящимися к ней кусочками стекла. Рассчитывают состав системы ,в миллиграммах вещества на грамм растворителя для каждой ампулы по формуле: 1000 (U72—Wi)/(W3—W2),rjie Wi —масса пустой ампулы, W2— масса ампулы вместе с исследуемым веществом и Ws— масса ампулы вместе с исследуемым веществом, растворителем и кусочками стекла.
Равновесие
Время, необходимое для достижения равновесия, зависит от исследуемого вещества, метода перемешивания (вибрация или вращение) и температуры. Обычно равновесие устанавливается быстрее с помощью вибрационного метода (1—7 сут), чем ротационного метода (7—14 сут).
Убедиться в том, что состояние равновесия достигнуто, можно следующим образом. В одной из ампул — предпослед
122
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
ней в этой серии—получают пересыщенный раствор нагреванием при температуре на 10° С выше, чем температура термостата, следя за тем, чтобы твердое вещество в ампуле не растворилось полностью. Затем с этой ампулой поступают так же, как с другими. Если величина растворимости, полученная для этой ампулы, будет находиться на одной прямой с другими величинами на графике, это указывает на то, что достигнуто равновесие. Однако, если величина растворимости, полученная для «пересыщенной» ампулы, окажется вне прямой, на которой лежат другие значения растворимости, это не обязательно означает, что в других ампулах не достигнуто равновесие, так как в ряде случаев это .может быть Обусловлено тенденцией некоторых веществ образовывать пересыщенные растворы. Для достижения состояния равновесия в таких случаях проводят ряд определений методом фазовой растворимости, подбирая различные отрезки времени, чтобы убедиться в том, что получены постоянные величины наклона кривой растворимости.
Состав раствора
После достижения состояния равновесия ампулы помещают вертикально в стойку в термостат, горлышками над уровнем воды, и дают содержимому осесть. «Соблюдая все меры предосторожности, чтобы снизить до минимума испарение растворителя, открывают ампулы и берут 2,0 мл из каждой ампулы пипеткой, на кончике которой укреплен комочек ваты или другого подходящего материала, служащего фильтром. Удаляют вату, переносят прозрачный раствор из каждой ампулы в помеченную, предварительно взвешенную колбочку и взвешивают каждую колбочку вместе с раствором; таким образом получают массу раствора. Охлаждают колбочки в смеси сухого льда с ацетоном и затем выпаривают растворитель в вакууме. Постепенно увеличивают температуру с 70 до 100° С и высушивают остаток до постоянной массы. Рассчитывают состав раствора в миллиграммах вещества на грамм растворителя по формуле: 1000 (Е3 — Fi)/(F2— F3), где Fl — масса колбочки, F2— масса колбочки вместе с раствором и F3—масса колбочки вместе с остатком.
Расчет
Результаты, полученные для каждой порции исследуемого вещества, представляют графически. Для этого на оси ординат откладывают отношение массы растворенного материала к единице массы растворителя (ось У, или состав раствора),
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
123
а на оси абсцисс откладывают отношение общей массы материала к единице массы растворителя (ось X, или состав системы). Как показано на рис. 3, точки для тех ампул, в которых получены истинные растворы, должны приближаться к прямой линии (АВ) с наклоном 1, проходящей через начало координат; точки, соответствующие насыщенным растворам, должны приближаться к другой прямой линии (ВС), наклон которой 5 представляет содержание суммы примесей в исследуемом веществе. Если точки не приближаются к прямой линии, это означает, что состояние равновесия достигнуто не было, хотя это может быть также обусловлено образованием твердого раствора или нарушением процесса растворения. Процентное содержание примеси в исследуемом веществе рассчитывают по формуле: 100—100 S. Наклон может быть рассчитан по уравнению: 5=(У2—У1)/(Х2—Xi), где У2 и У1 — составы растворов, Х2 и Х\ — составы систем, соответствующие точкам, взятым на второй прямой линии (ВС).
Точка В на диаграмме представляет систему, в которой основной компонент или, в редких случаях, наименее растворимые компоненты испытуемого вещества достигли предела своей растворимости. Значение 'растворимости этого компонента получают продолжением линии растворимости (ВС) до пересечения с осью У. Точка пересечения на оси У дает вели-
РИС. 3. ТИПИЧНАЯ ДИАГРАММА ФАЗОВОЙ
РАСТВОРИМОСТИ
Состав системы, мг/г (X)
124 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
чину растворимости в миллиграммах на грамм, которая должна быть постоянной для данного соединения.
В точке С на диаграмме следующий компонент испытуемого вещества достиг предела своей растворимости. Точка, полученная при продолжении линии растворимости (CD) до пересечения с осью У, дает величину общей растворимости’ компонентов, которые первыми достигли пределов своей растворимости. Таким образом, растворимость второго компонента может быть получена вычитанием.
Между точками D и Е на диаграмме раствор насыщен всеми компонентами испытуемого вещества и его состав остается постоянным.
В идеальных условиях число примесей в исследуемом веществе соответствует числу изломов кривой растворимости выше точки насыщения В, и значения растворимости соответствующих компонентов могут быть получены описанным выше способом.
ХИМИЧЕСКИЕ МЕТОДЫ
ОБЩИЕ ИСПЫТАНИЯ НА ПОДЛИННОСТЬ
Ацетилированные вещества
Помещают вещество в количестве, указанном в частной статье, в пробирку (максимальный диаметр 18 мм) и обрабатывают 3 каплями фосфорной кислоты (~1440 г/л) ИР. Закрывают пробирку пробкой, через которую проходит пробирка меньшего размера, заполненная водой; на конце пробирки висит капля нитрата лантана (30 г/л) ИР. Помещают прибор на 5 мин в кипящую водяную баню. Переносят каплю раствора нитрата лантана на белую фарфоровую пластинку и смешивают с каплей раствора йода (0,02 моль/л) ТР. На краю смеси помещают каплю аммиака (~ 100 г/л) ИР; на месте соприкосновения двух жидкостей медленно появляется синее окрашивание, исчезающее через короткое время.
Амины ароматические первичные
Растворяют вещество в количестве, указанном в частной статье, в 2 мл соляной кислоты (~70 г/л) ИР, если необходимо, при помощи нагревания. Охлаждают во льду, обрабатывают 4 мл нитрита натрия (10 г/л) ИР и выливают смесь в 2 мл раствора 2-нафтола ИР1, содержащего 1 г ацетата натрия Р; образуется тяжелый осадок, имеющий окраску, указанную в соответствующей частной статье.
Аммиак >и летучие алифатические амины
Растворяют вещество в количестве, указанном в частной статье, помещают полученный раствор в пробирку и прибавляют 1 г окиси магния Р; нагревают, если указано в статье. Постепенно выделяющиеся щелочные пары вызывают почернение реактивной марганцово-серебряной бумаги Р; реактивную бумагу вводят в верхнюю часть пробирки.
Аммоний
Проводят испытание в приборе, состоящем из пробирок с притертыми пробками, соединенных изогнутой стеклянной трубкой, позволяющей последовательно проходить потоку воздуха через пробирки А и Б.
Помещают раствор вещества, полученный, как указано в частной статье, и 0,2 г окиси магния Р в пробирку А, а в пробирку Б вносят 1 мл соляной кислоты (0,1 моль/л) ТР, со
128
ХИМИЧЕСКИЕ МЕТОДЫ
держащий 1 каплю раствора метилового красного в этаноле ИР. Пропускают через прибор воздух. Выделйющийся аммиак изменяет окраску раствора в пробирке Б на желтую. После прибавления к этому раствору 1 мл раствора кобальтнитри-та натрия (100 г/л) ИР образуется желтовато-коричневый осадок.
Висмут
, А. Готовят раствор вещества в соляной кислоте (~ 250 г/л) ИР, как указано в частной статье, и разводят водой в 10 раз; образуется белый осадок, который становится темно-коричневым при прибавлении раствора сульфида натрия ИР.
Б. Обрабатывают раствор вещества .в азотной кислоте (~-ilOOO г/л) ИР, приготовленный, как указано в частной статье, раствором йодида калия (80 г/л) ИР; образуется черный, осадок, растворимый в избытке реактива с образовав нием желтовато-коричневого или оранжевого раствора. Разводят этот раствор несколькими объемами воды и нагревают; образуется осадок оранжевого или медного цвета. Черный осадок, 'образовавшийся вначале при добавлении раствора йодида калия (80 г/л) ИР, также становится оранжевым или1 приобретает медную окраску при нагревании с водой.
Бромиды
А. Готовят раствор вещества, пак указано в частной статье, подкисляют азотной кислотой (~ 130 г/л) ИР и прибавляют раствор нитрата серебра (40 ;г/л) ИР; образуется желтоватый творожистый осадок, частично -растворимый в аммиаке (~260 г/л) ИР, но почти нерастворимый в аммиаке (~ 100 г/л) ИР и азотной кислоте (~1000 г/л) ИР.
Б. (Для испытания бромидов и гидробромидов нерастворимых и ;малораствбримых оснований:) Готовят раствор вещества, как указано в частной статье, прибавляют амм'иак (~ 100 г/л) ИР, фильтруют, подкисляют фильтрат азотной кислотой (~ 130 г/л) ИР и далее поступают, как указано в испытании А.
В. Готовят раствор вещества, как указано в частной статье,.подкисляют серной кислотой (~100 г/л) ИР и смешивают с раствором хлора ИР; образуется коричневый раст-pop,.который после взбалтывания с хлороформом Р становится бесцветным; хлороформный слой при 'этом окрашивается в красноватый цвет. . с
ХИМИЧЕСКИЕ МЕТОДЫ
129
Кальций
А. Готовят раствор вещества, как указано в частной статье, и прибавляют к нему раствор оксалата аммония (25 г/л) ИР; образуется белый осадок, растворимый в соляной кислоте (~ 250 г/л) ИР, но практически нерастворимый в уксусной кислоте (~ 300 г/л) ИР.
Б. Обрабатывают 1 каплю раствора вещества, приготовленного, как указано .в частной статье, 4 каплями раствора глиоксаля бис(2-оксианила) ИР и 1 каплей раствора гидроокиси натрия (~80 г/л) ИР; образуется красновато-коричневый осадок, растворимый в хлороформе Р с образованием красного раствора.
Хлориды
А. Готовят раствор вещества, как указано в частной статье, подкисляют азотной кислотой (~ 130 г/л) ИР и прибавляют раствор нитрата серебра (40 г/л) ИР; образуется белый творожистый осадок, растворимый ,в аммиаке (~ 100 г/л) ИР, но практически нерастворимый в азотной кислоте (~ 1000 г/л) ИР.
Б. (Для испытания хлоридов и гидрохлоридов нерастворимых и малорастворимых оснований.) Готовят раствор вещества, как указано в частной статье, прибавляют аммиак (—100 г/л) ИР, фильтруют, подкисляют фильтрат азотной кислотой (~130 г/л) и далее поступают, как указано в испытании А.
В. 'Смешивают вещество >в количестве, указанном в частной статье, с равным количеством двуокиси марганца Р, смачивают серной кислотой (~1760 г/л) ИР и слегка нагревают. Выделившийся хлор обнаруживают по его зеленоватому 'цвету и по синему окрашиванию влажной крахмал-йодидной бумаги Р. Желательно эту реакцию проводить под тягой.
Цитраты
А. Смешивают при комнатной температуре нейтральный раствор вещества, полученный, как указано в частной статье, с раствором хлорида кальция (55 г/л) ИР; осадок не образуется, но при кипячении появляется белый осадок, растворимый в уксусной кислоте (~300 г/л) ИР.
Б. Кипятят раствор вещества с раствором сульфата ртути ИР, как указано в частной статье, и, если необходимо.
9—2025
130
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
фильтруют. После прибавления к фильтрату нескольких капель раствора перманганата (10 г/л) ИР окраска исчезает и образуется 'белый осадок.
Соли железа (II)
А. Готовят раствор вещества, как указано в частной статье, и прибавляют раствор феррицианида калия (10 г/л) ИР; образуется темно-синий осадок, практически нерастворимый в соляной кислоте ( — 70 г/л) ИР.
Б. Готовят раствор вещества, как указано в частной статье, подкисляют серной кислотой (— 100 г/л) ИР и прибавляют раствор о-фенантролина (1 г/л) ИР; появляется интенсивное красное окрашивание, которое исчезает при добавлении раствора сульфата церия (35 г/л) ИР.
Йодиды
А. Готовят раствор вещества, как указано в частной статье, подкисляют азотной кислотой ( — 130 г/л) ИР и прибавляют раствор нитрата серебра (40 г/л) ИР; образуется желтый творожистый осадок, практически нерастворимый в аммиаке ( — 100 г/л) ИР и азотной кислоте (-4000 г/л) ИР.
Б. (Для испытания йодидов нерастворимых и малораство-римых оснований.) Готовят раствор вещества, как указано в частной статье, прибавляют аммиак (— 100 г/л) ИР, 'фильтруют, подкисляют фильтрат азотной кислотой (—130 г/л) ИР и далее поступают, как указано в испытании А.
В. Готовят раствор вещества, как указано в частной статье, подкисляют серной кислотой (—100 г/л) ИР и прибавляют раствор нитрита калия (100 г/л) ИР; образуется коричневый раствор, который после взбалтывания с хлороформом Р становится бесцветным; хлороформный слой при этом окрашивается в фиолетовый цвет.
Нитраты
А. Готовят раствор вещества, как указано в частной статье, и прибавляют к нему раствор сульфата железа (II) (15 г/л) ИР; появления коричневого окрашивания не наблюдается до тех пор, пока к раствору осторожно не прибавляют серную кислоту (— 1760 г/л) ИР для образования нижнего слоя. Коричневое окрашивание появляется на месте соприкосновения двух жидкостей.
ХИМИЧЕСКИЕ МЕТОДЫ
i3i
Б. Прибавляют 2 мг мелко измельченного испытуемого вещества к смеси 0,1 мл нитробензола Р и 0,2 мл серной кислоты (~1760 г/л) ИР. Оставляют стоять на 5 мин при комнатной температуре, охлаждают во льду и, перемешивая, медленно прибавляют 5 мл воды и 3 мл раствора гидроокиси натрия (~400 г/л) ИР. Прибавляют 5 мл ацетона Р, встряхивают и дают слоям разделиться; в верхней фазе появляется интенсивное фиолетовое окрашивание.
Ортофосфаты
А. Прибавляют по каплям азотную кислоту (~130 г/л) ИР и 5 мл раствора молибдата аммония (95 г/л) ИР до полного растворения любого осадка, который может появиться. Делят этот раствор на две порции, прибавляют к одной порции испытуемый раствор, подкисленный азотной кислотой (~ 130 г/л) ИР, 'как указано в статье, и кипятят обе порции. С испытуемым раствором образуется желтый осадок, в то время как в другой порции наблюдается лишь легкая опалесценция.
Б. Готовят нейтральный раствор вещества, как указано в частной статье, и прибавляют раствор нитрата серебра (40 г/л) ИР; образуется желтый осадок, который не темнеет при нагревании раствора до кипения. Осадок растворим в аммиаке (~ 100 г/л) ИР и азотной кислоте (~130 г/л) ИР.
Калий
Готовят щелочной раствор вещества, как указано в частной статье, и прибавляют раствор тетрафен ил бор а та натрия (30 г/л) ИР; образуется белый осадок.
Салицилаты
К нейтральному раствору вещества, полученному, как указано в частной статье, прибавляют раствор хлорида железа (III) (25 г/л) ИР; появляется интенсивное красно-фиолетовое окрашивание, которое сохраняется при добавлении небольшого количества уксусной кислоты (~300 г/л) ИР, но исчезает при прибавлении соляной кислоты (~70 г/л) ИР с образованием белого кристаллического осадка.
Натрий
А. Смачивают вещество соляной кислотой (~ 250 г/л) ИР; при внесении в бесцветное пламя полученный раствор окраг шивает его в желтый цвет.
9
132 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
ПРИМЕЧАНИЕ. Проводят испытание Б, если по техническим причинам нельзя выполнить испытание А.
Б. Подкисляют раствор вещества, как указано в частной статье, уксусной кислотой ( — 60 г/л) ИР, фильтруют, если необходимо, и прибавляют раствор ацетата уранила-цинка ИР; образуется желтый кристаллический осадок.
Сульфаты
А. Готовят раствор вещества, как указано в частной статье, и прибавляют раствор хлорида бария (50 г/л) ИР; образуется белый осадок, практически нерастворимый в соляной кислоте ( — 250 г/л) ИР.
Б. К раствору вещества, приготовленному, как указано в частной статье, прибавляют раствор ацетата свинца (80 г/л) ИР; образуется белый осадок, растворимый в ацетате аммония (80 г/л) ИР и гидроокиси натрия ( — 80 г/л) ИР, но практически нерастворимый в горячей воде.
Тартраты
А. Подкисляют раствор вещества, как указано в частной статье, уксусной кислотой (— 300 г/л) ИР, прибавляют 1 каплю раствора сульфата железа (II) (15 г/л) ИР, несколько капель перекиси водорода ( — 60 г/л) ИР и подщелачивают раствором гидроокиси натрия ( — 80 г/л) ИР; появляется пурпурное или фиолетовое окрашивание.
Б. Смешивают несколько миллилитров серной кислоты ( — 1760 г/л) ИР с несколькими каплями раствора резорцина (20 г/л) ИР и несколькими каплями раствора бромида калия (100 г/л) ИР, а затем прибавляют 2 или 3 капли раствора вещества, приготовленного, как указано в частной статье. Нагревают жидкость в водяной бане в течение 5—10 мин; появляется интенсивное синее окрашивание. Жидкость охлаждают и выливают в воду; раствор становится красным.
ИСПЫТАНИЕ НА ХЛОРИДЫ
Испытание на хлориды предусмотрено для доказательства того, что содержание хлоридов не превышает предел, указанный в частной статье и выраженный в микрограммах хлорид-•иона на 1 г испытуемого вещества. Стандартный раствор, с которым проводится сравнение опалесценции, содержит 250 мкг С1_.
ХИМИЧЕСКИЕ МЕТОДЫ 133
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Испытание проводят в подобранных для сравнения сосудах с плоским дном из прозрачного стекла емкостью около 70 мл и внутренним диаметром около 23 мм, имеющих отметки 45 и 50 мл. Для испытания пригодны цилиндры Несслера указанных выше размеров. Выражение «подобранные для сравнения сосуды» означает, «что эти сосуды 'максимально совпадают по внутреннему диаметру и по 'всем остальным характеристикам.
Готовят раствор вещества, как указано в частной статье, переносят в сосуд для сравнения, разводят водой до 50 мл и прибавляют 1 мл раствора нитрата серебра (40 г/л) ИР. Тотчас перемешивают стеклянной палочкой и оставляют стоять на 5 мин, защищая от прямого солнечного света. (При рассмотрении 'сверху вниз по вертикальной оси в рассеянном свете на черном фоне опалесценция, появившаяся в испытуемом растворе, не должна быть интенсивнее опалесценции в стандартном растворе.
Стандартная опалесценция
Отмеряют 5,0 мл соляной кислоты С1ИР и 10 мл азотной кислоты (—130 г/л) ИР в сосуд для сравнения. Доводят водой до 50 мл и прибавляют 1 мл раствора нитрата серебра (40 г/л) ИР. Тотчас перемешивают стеклянной палочкой и оставляют стоять на 5 мин, защищая от прямого солнечного света.
ИСПЫТАНИЕ НА СУЛЬФАТЫ
Испытание на сульфаты предусмотрено для доказательства того, что содержание сульфатов не превышает предел, указанный в частной статье и выраженный в микрограммах сульфатов на 1 г испытуемого вещества.
Раствор, с которым проводится сравнение мутности, содержит на 480 мкг SO4_- больше, чем стандартная суспензия сульфата бария.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Испытание проводят в подобранных для сравнения сосудах с плоским дном из прозрачного стекла емкостью около 70 мл и внутренним диаметром около 23 мм, имеющих отметки 45 и 50 мл. Для испытания пригодны цилиндры Несслера
134
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
указанных выше размеров. Выражение «подобранные для сравнения сосуды» означает, что эти сосуды максимально совпадают по внутреннему диаметру и по всем остальным характеристикам.
Готовят раствор вещества, как указано в частной статье, переносят в сосуд для сравнения, разводят водой до 45 мл и прибавляют 5 мл суспензии сульфата бария ИР. Тотчас перемешивают стеклянной палочкой и оставляют стоять на 10 мин. При рассмотрении сверху вниз по вертикальной оси в рассеянном свете на черном фоне мутность, появившаяся в испытуемом растворе, не должна быть интенсивнее мутности в стандартном растворе.
Стандартная мутность
Отмеряют 1,00 мл серной кислоты (0,005 моль/л) ТР и 3 мл соляной кислоты (~70 г/л) ИР в сосуд для сравнения. Разводят водой до 45 мл .и прибавляют 5 мл суспензии сульфата бария ИР. Тотчас перемешивают стеклянной палочкой и оставляют стоять на 10 мин.
ИСПЫТАНИЕ НА ТЯЖЕЛЫЕ МЕТАЛЛЫ
Испытание на тяжелые металлы предусмотрено для доказательства того, что содержание примесей (металлов, окрашивающихся сероводородом, не превышает предел, указанный в частной статье и выраженный в микрограммах свинца на 1 грамм испытуемого вещества.
Испытание состоит из двух последовательных операций: приготовления испытуемого раствора и получения окрашивания с помощью реакции с сероводородом, а затем сравнения полученной окраски с окраской стандартного раствора свинца.
Приготовление испытуемого раствора проводят, как указано в частной статье, в соответствии с методиками 1—4, описанными ниже. Контрольный раствор готовят таким же образом.
Реакцию с сероводородом проводят путем смешивания испытуемого раствора со свежеприготовленным раствором сероводорода ИР. Сравнение интенсивности полученной таким образом окраски проводят либо непосредственно, сравнивая окраску жидкости в подходящих сосудах для сравнения (метод А), либо сравнивая интенсивность окраски пятен, полученных при фильтровании жидкости через подходящий прибор (методБ).
ХИМИЧЕСКИЕ МЕТОДЫ
135
Обычно метод А применяется в тех случаях, когда количество тяжелых металлов в навеске испытуемого вещества превышает 5 мкг; если количество тяжелых металлов составляет 2—5 мкг, следует применять метод Б.
Используемый в испытании стандартный раствор свинца готовят разведением раствора свинца РЬИР до содержания 10 мкг свинца в 1 мл. Если 0,1 мл этого раствора берут для приготовления стандартного раствора, предназначенного для сравнения с раствором 1 г испытуемого вещества, приготовленный таким образом стандартный раствор содержит 1 мкг РЬ, что эквивалентно содержанию 1 мкг свинца на 1 г испытуемого вещества.
Прибор
Испытание тяжелых металлов по методу А проводят в подобранных для сравнения сосудах с плоским дном, из прозрачного стекла емкостью около 70 мл и внутренним диаметром около 23 мм, имеющих отметки 40 и 50 мл. Для испытания пригодны цилиндры Несслера указанных выше размеров. Выражение «подобранные для сравнения сосуды» означает, что эти сосуды максимально совпадают по внутреннему диаметру и по всем остальным характеристикам. Для перемешивания раствора используют стеклянную палочку, имеющую на нижнем конце петлю.
Для определения тяжелых металлов методом Б используют шприц емкостью 50 мл из подходящего материала (обычно из пластмассы) с удаляемым поршнем и прямым коническим соединением Люера внутренним диаметром у нижнего конца 9 мм (пригоден шприц фирмы Миллипор XX 11 050 05); к нижнему концу конуса Люера присоединена насадка для фильтрации.
Насадка изготовляется из подходящего материала (пригодна фильтрационная насадка фирмы Миллипор SXOO 013 00 из полипропилена) и имеет обратный шлиф для соединения со шприцем. Насадка сконструирована таким образом, чтобы ее можно 'было разделить на две части для замены фильтров; в нижней части насадки имеется прокладка для крепления мембранных фильтров диаметром 13 мм. Для фильтрования используют подходящий фильтр предварительной фильтрации (пригоден фильтр фирмы Миллипор АР 2001 300) и мембранный фильтр из смешанных эфиров целлюлозы диаметром 13 мм и величиной пор 3 мкм (пригоден фильтр фирмы Мил-липор SSWP 013 00).
136 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Приготовление испытуемого раствора
Методика 1. Взвешивают навеску вещества, указанную в частной статье, растворяют ,в 25 мл .воды, доводят pH раствора до 3—4 при помощи уксусной кислоты (60 г/л) РЬ ИР или аммиака (~ 100 г/л) РЬИР, в зависимости от необходимости, затем разводят водой до 40 мл и перемешивают.
Методика 2. Взвешивают навеску вещества, указанную в частной статье, растворяют примерно в 30 мл указанного растворителя [можно использовать этанол (~750 г/л) ИР, метанол Р, ацетон Р или диоксан Р], прибавляют 0,5 мл уксусной кислоты (~300 г/л) ИР и разводят растворителем до 40 мл.
Методика 3. Помещают навеску вещества, указанную в частной статье, в подходящий тигель, предпочтительно кварцевый, и осторожно сжигают при низкой температуре до полного обугливания содержимого. Во время обугливания тигель может быть неплотно прикрыт крышкой. Прибавляют к содержимому тигля 2 мл азотной кислоты (~ 1000 г/л) ИР и 5 капель серной кислоты (~ 1760 г/л) ИР и осторожно нагревают до выделения белых паров, а затем сжигают, предпочтительно в муфельной печи, при температуре 500 °C до полного сгорания органического вещества. Охлаждают, прибавляют 2 мл соляной кислоты (~ 250 г/л) ИР и медленно выпаривают на водяной бане досуха. Смачивают остаток 1 каплей соляной кислоты (~ 250 г/л ИР, прибавляют 10 мл горячей воды и настаивают в течение 2 мин. Прибавляют по каплям аммиак (~ 100 г/л) РЬИР, пока pH раствора не достигнет 8—8,5, а затем по каплям прибавляют уксусную кислоту (<—60 г/л) РЬИР до pH, равного 3—4. Фильтруют, если необходимо, промывают тигель и фильтр примерно 10 мл воды, разводят водой до 40 мл и перемешивают.
Методика 4. Помещают навеску вещества, указанную в частной статье, в подходящий тигель, предпочтительно кварцевый, хорошо смешивают примерно с 0,5 г окиси магния Р и прокаливают до получения белой однородной массы. Если через 15 мин после прокаливания остаток не теряет окраски, дают тиглю остыть, перемешивают содержимое 'Стеклянной палочкой и снова нагревают. Затем растворяют остаток в соляной кислоте ( — 70 г/л) ИР, прибавляют по каплям раствор аммиака (— 100 г/л) РЬИР, пока pH раствора не достигнет 8—8,5, а затем прибавляют также по каплям уксусную кислоту ( — 60 г/л) РЬИР до pH, равного 3—4, фильтруют, разводят водой до 40 мл и перемешивают.
ХИМИЧЕСКИЕ МЕТОДЫ
137
Окрашивание и измерение его интенсивности
Метод А
К 40 мл жидкости в сосуде для сравнения прибавляют 10 мл свежеприготовленного раствора сероводорода ИР, перемешивают и оставляют стоять на 5 мин.
В другой сосуд для сравнения вносят объем разведенного раствора свинца РЬИР, содержащий количество свинца, эквивалентное предельному .содержанию тяжелых металлов, указанному в частной статье, разводят водой, доводят pH аммиаком (~100 г/л) РЬИР и уксусной кислотой (~60 г/л) РЬИР до 3—4; доводят водой или использованным растворителем до 40 мл, перемешивают, прибавляют 10 мл свежеприготовленного раствора сероводорода ИР, перемешивают и оставляют стоять на 5 мин.
Сравнивают окраски, рассматривая растворы сверху вниз по вертикальной оси в рассеянном свете на белом фоне или любым подходящим методом. Окраска испытуемого раствора не должна быть интенсивнее, чем окраска стандартного раствора свинца.
Метод Б
Берут шприц для фильтрования, устанавливают фильтр предварительной фильтрации и мембранный фильтр, 'как показано на рис. 4, удаляют поршень из шприца, помещают испытуемый раствор в шприц, вставляют вновь поршень и медленно фильтруют испытуемый раствор при постоянном давлении на поршень. Собирают фильтрат в стакан или пробирку. Открывают насадку и проверяют, свободен ли 'мембранный фильтр от загрязнений. Если загрязнения имеются, заменяют фильтр и повторяют операцию таким же образом. Затем переставляют фильтр предварительной фильтрации и мембранный фильтр, как показано на рис. 4. Доводят pH фильтрата аммиаком (100 г/л) РЬИР и уксусной кислотой (60 г/л) РЬИР до 3—4, прибавляют- 100 мл свежеприготовленного раствора сероводорода ИР (все реактивы предварительно фильтруют через мембранный фильтр), перемешивают, оставляют стоять на 5 мин, вынимают поршень, вносят раствор в шприц и медленно фильтруют через мембранный фильтр при равномерном и умеренном давлении на поршень. Открывают насадку и вынимают мембранный фильтр.
Берут объем разведенного раствора свинца РЬИР, содержащий количество свинца, эквивалентное предельному содержанию тяжелых металлов, указанному в частной статье,
138
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
разводят водой, доводят pH аммиаком (~ 100 г/л) РЬИР и уксусной кислотой (~60 г/л) РЬИР до 3—4, доводят водой до 40 мл, перемешивают и поступают, как описано выше.
РИС. 4. ИСПЫТАНИЕ НА ТЯЖЕЛЫЕ МЕТАЛЛЫ: МЕТОД Б
Предварительная фильтрация
Фильтрация
А—Конические соединения Люера
Б—Соединения
В—Фильтр предварительной фильтрации
Г—Мембранный фильтр
Д—Держатель фильтров
Сравнивают интенсивность окраски пятен, полученных на мембранных фильтрах. Окраска ‘пятна, 1полученного с испытуемым раствором, не должна быть интенсивнее, чем окраска пятна, полученного со стандартным раствором свинца.
ИСПЫТАНИЕ НА ЖЕЛЕЗО
Испытание на железо предусмотрено для доказательства того, что содержание железа не превышает предел, указанный в частной статье и выраженный в микрограммах железа на 1 грамм испытуемого вещества.
Стандартный раствор, с которым сравнивают окраску, содержит 40 мкг Fe.
ХИМИЧЕСКИЕ МЕТОДЫ
139
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Испытание проводят в подобранных для сравнения сосудах с плоским дном, из прозрачного стекла емкостью около 70 мл и внутренним диаметром около 23 мм, имеющих отметки 45 и 50 мл. Для испытания пригодны цилиндры Несслера указанных выше размеров. Выражение «подобранные для сравнения сосуды» означает, что эти сосуды максимально совпадают по внутреннему диаметру и по всем остальным характеристикам.
Подготавливают .вещество, как указано в частной статье, или непосредственно растворяют указанное количество в 40 мл воды и переносят в цилиндр для сравнения. Прибавляют 2 мл раствора лимонной кислоты (180 г/л) FeHP и 2 капли меркаптоуксусной кислоты Р; перемешивают, подщелачивают аммиаком (~100 г/л) FeHP, разводят водой до 50 мл и оставляют стоять на 5 мин. Полученное окрашивание не должно быть более интенсивным, чем полученное аналогичным образом окрашивание стандартного раствора при рассмотрении сверху вниз по вертикальной оси в рассеянном свете на белом фоне.
Стандартная окраска
Отмеривают 2 мл стандартного раствора железа FeHP и 40 мл воды в цилиндр для сравнения. Прибавляют 2 мл раствора лимонной кислоты (180 г/л) FeHP и 2 капли меркаптоуксусной кислоты Р; перемешивают, подщелачивают аммиаком (—100 г/л) FeHP, разводят водой до 50 мл и оставляют стоять на 5 мин.
ИСПЫТАНИЕ НА МЫШЬЯК
Испытание на мышьяк предусмотрено для доказательства того, что содержание мышьяка не превышает предел, указанный в частной статье и выраженный в микрограммах мышьяка на 1 грамм испытуемого вещества.
Для проведения испытания на мышьяк раствор испытуемого вещества готовят согласно методике, описанной в соответствующей частной статье. Приготовление раствора по этой методике гарантирует, что в любом случае раствор содержит весь мышьяк (если таковой имеется), присутствующий в веществе.
Стандартное пятно, с которым проводится сравнение, содержит 10 мкг As.
140
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Описанная ниже методика также может быть использована для определения ’количества мышьяка в .веществе путем подбора интенсивности окраски пятна с интенсивностью окраски ряда стандартных пятен. Пятно, эквивалентное пятну, полученному с 1 мл стандартного раствора, и образовавшееся при обработке 10 г вещества, указывает, что содержание мышьяка равно 1 мкг/г.
При указании предельного содержания мышьяка допустимое количество примеси выражается как содержание элемента мышьяка.
Прибор
Ниже приводится описание прибора подходящего типа, хотя имеются и другие приемлемые конструкции приборов.
Широкогорлая колба емкостью около 120 мл снабжена резиновой пробкой, через которую проходит стеклянная трубка. Последняя, сделанная из обычной стеклянной трубки, имеет общую длину 200 мм и внутренний диаметр точно 6,5 мм (наружный диаметр около 6 мм); трубка вытянута с одного конца до диаметра примерно 1 мм и имеет в стенке отверстие диаметром не менее 2 мм недалеко от суженной части. Трубка проходит через резиновую пробку, закрывающую колбу, таким образом, что, когда трубка вставлена в колбу, содержащую 70 мл жидкости, суженный конец трубки находится над поверхностью жидкости, а боковое отверстие — под дном резиновой пробки. Верхний конец трубки прямоугольно срезан и немного закруглен или сглажен.
Две резиновые втулки (около 25x25 мм), каждая с центральным отверстием, имеющим точно 6,5 мм в диаметре, снабжены резиновой лентой или пружинным зажимом для прочного удерживания вместе. Две втулки могут быть заменены любым приспособлением, удовлетворяющим условиям испытания, описанным ниже.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
В стеклянную трубку легко набивают предварительно пропитанный раствором ацетат свинца (80 г/л) ИР и высушенный тампон ваты таким образом, чтобы верхний край тампона находился не менее чем на 25 мм ниже верхней части трубки.
Затем верхний конец трубки вставляют в отверстие одной из пары резиновых втулок либо на глубину около 10 мм, если трубка имеет закругленный конец, либо так, чтобы притертый конец трубки находился вровень с отверстием втулки.
ХИМИЧЕСКИЕ МЕТОДЫ
141
На верхнюю часть втулки ровно кладут ртуть-бромидную бумагу AsP и сверху укрепляют вторую втулку. Втулки скрепляют вместе прм помощи резиновой ленты или пружинного зажима так, чтобы отверстия двух втулок (или отверстия верхней втулки и стеклянной трубки), соединяясь, образовывали правильную трубку диаметром 6,5 мм, разделенную диафрагмой из ртуть-бромидной бумаги AsP.
Вместо этого 'метода укрепления ртуть-бромидной бумаги AsP можно применить любой другой метод, если будут соблюдены следующие условия: 1) весь выделяющийся газ должен проходить через бумагу; 2) часть бумаги, находящаяся в контакте с газом, должна иметь форму круга диаметром 6,5 мм; 3) бумага во время определения должна быть защищена от солнечного света.
Испытуемый раствор, приготовленный, как указано в частной статье, помещают в широкогорлую колбу, прибавляют 1 г йодида калия AsP и 10 г гранулированного цинка AsP и тотчас закрывают колбу пробкой со вставленной в нее приготовленной трубкой. Реакция должна протекать в течение 40 мин. Желтое пятно, которое получается на ртуть-бромидной бумаге AsP, сравнивают со стандартным пятном, полученным таким же образом с разведенным раствором мышьяка AsHP, взятым в известном количестве. Сравнение производят при дневном свете немедленно после получения испытуемого и стандартного пятен; при хранении цвет пятен блекнет.
Обычно наиболее подходящая температура для проведения определения 40 °C, но скорость выделения газа может быть различной в зависимости от разных партий гранулированного цинка AsP, поэтому температуру можно регулировать с таким расчетом, чтобы выделение газа было равномерным, но не слишком энергичным. Реакцию можно ускорить, если поместить прибор на теплую поверхность; при этом надо следить за тем, чтобы во время определения ртуть-бромидная бумага AsP оставалась совершенно сухой.
После каждого испытания трубку следует промыть соляной кислотой AsHP, сполоснуть водой и высушить.
Стандартное пятно
Готовят раствор, прибавляя к 50 мл воды 10 мл соляной кислоты (~250 г/л), содержащей олово, AsHP и 1 мл разведенного раствора мышьяка AsHP. При испытании полученного раствора по методу, описанному выше, на ртуть-бромидной бумаге AsP образуется пятно, которое принимается за стандартное.
142
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
СУЛЬФАТНАЯ ЗОЛА
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Точно взвешивают около 1 г вещества или такое количество, которое указано в частной статье, в подходящем тигле (обычно платиновом) и смачивают серной кислотой (~1760 г/л) ИР. Слегка нагревают для удаления избытка кислоты и прокаливают при температуре около 800° С до удаления всех черных частиц; снова смачивают серной кислотой (~1760 г/л) ИР и вновь прокаливают. Прибавляют небольшое количество карбоната аммония Р и прокаливают до постоянной массы.
МЕТОД СЖИГАНИЯ В КОЛБЕ С КИСЛОРОДОМ
Метод сжигания в колбе с кислородом для определения галогенов и серы в органических соединениях состоит из методики сжигания с последующим соответствующим титри-метрическим определением. Сжигание органического материала в кислороде дает водорастворимые неорганические продукты, которые определяются, если предписано, для каждого отдельного элемента.
Прибор
Сжигание проводят в подходящей конической колбе, в пробку которой вплавлен конец платиновой проволоки. Если в статье нет других указаний, используют колбу емкостью 500 мл. К другому концу проволоки прикреплен кусочек платиновой сетки для укрепления образца, который во время сгорания не должен соприкасаться с поглощающей жидкостью.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
ПРЕДОСТЕРЕЖЕНИЕ. Аналитик должен надеть защитные очки и использовать подходящий защитный экран, отделяющий его от прибора. Колба должна быть тщательно очищена от следов органических растворителей.
Заворачивают исследуемое вещество в кусочек свободной от галоидов фильтровальной бумаги длиной около 5 см и шириной 3 см, укрепляют пакетик ib платиновой сетке и вставляют в него конец узкой фильтровальной бумаги. Смачивают горло колбы водой, вносят в колбу указанную поглощающую жидкость, наполняют колбу кислородом, зажигают свободный конец узкой полоски фильтровальной бумаги и тотчас закры
ХИМИЧЕСКИЕ МЕТОДЫ
143
вают колбу пробкой. Плотно удерживают пробку в горле колбы. Когда начнется энергичное горение, наклоняют колбу, чтобы предотвратить попадание несгоревшего вещества в жидкость. Как только процесс горения закончится, энергично встряхивают колбу в течение примерно 10 мин, наливают несколько миллилитров воды на поверхность пробки, закрывающей колбу, осторожно вынимают пробку и споласкивают ее, платиновую проволоку, платиновую сетку и стенки колбы водой. Далее проводят анализ этого раствора, как описано в частной статье.
Измельчаемые вещества должны быть мелко растерты и тщательно перемешаны перед отвешиванием указанного количества.
Для испытания жидкостей используют капсулы из подходящего материала (например, из метилцеллюлозы). Кладут указанное количество вещества на кусочек беззольной фильтровальной бумаги весом около 15 мг, помещенный в одну часть капсулы подходящего размера. Закрывают капсулу, вставляют между двумя ее частями один конец узкой фильтровальной бумаги и укрепляют капсулу в платиновой сетке.
Определение брома и хлора
Используя метод сжигания в колбе с кислородом, сжигают количество вещества, указанное в частной статье. Поглощающая жидкость состоит из 17 мл перекиси водорода (~60 г/л) ИР и 3 мл воды. После завершения процесса споласкивают пробку, платиновую проволоку, платиновую сетку и стенки колбы 40 мл воды.
Прибавляют 5 капель раствора бромфенолового синего в этаноле ИР и затем по каплям раствор гидроокиси натрия (0,1 моль/л) ТР до изменения окраски с желтой на синюю. Затем прибавляют 1 мл азотной кислоты (3 г/л) ИР, 5 капель раствора дифенилкарбазона в этаноле ИР в качестве индикатора и титруют раствором нитрата ртути (0,01 моль/л) ТР до появления светло-фиолетового окрашивания.
Каждый миллилитр раствора нитрата ртути (0,01 моль/л) ТР соответствует 1,598 мг Вг или 0,709 мг С1.
Определение фтора
Используя метод сжигания в колбе с кислородом, описанный на с. 142, сжигают количество вещества, указанное в частной статье. Поглощающая жидкость состоит из 15 мл воды. После завершения процесса споласкивают пробку, платиновую проволоку, платиновую сетку и стенки колбы 40 мл воды.
144
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Прибавляют 0,6 мл раствора ализаринсульфоната натрия (1 г/л) ИР и затем по кашлям раствор гидроокиси иатрия (0,1 моль/л) ТР до изменения окраски с розовой на желтую. Прибавляют 5 мл ацетатного буфера, pH 3,0, ИР и титруют раствором нитрата тория (0,005 моль/л) ТР до перехода желтой окраски в розовато-желтую.
Каждый миллилитр .раствора нитрата тория (0,005 моль/л) ТР соответствует 0,380 мг F.
Если за изменением окраски индикатора трудно наблюдать, проводят предварительное испытание с раствором, содержащим известное количество неорганического фторида.
Определение йода
Используя метод сжигания в колбе с кислородом, описанный на с. 142, сжигают количество вещества, указанное в частной статье. Поглощающая жидкость состоит из 10 мл раствора гидроокиси натрия (0,2 моль/л) ТР. После завершения процесса споласкивают пробку, платиновую проволоку, платиновую сетку и стенки колбы 25 мл раствора ацетата калия ИР, к которому прибавлены 15 капель раствора брома ИР1. Затем споласкивают вое перечисленные части 40 мл воды и добавляют по каплям до обесцвечивания муравьиную кислоту (~1080 т/л) ИР; после этого прибавляют 20 мл серной кислоты (0,05 моль/л) ТР, 0,5 г йодида калия Р и оставляют стоять на 5 мин. Титруют (выделившийся йод тиосульфатом натрия (0,05 моль/л) ТР, прибавляя к концу титрования раствор крахмала ИР в качестве индикатора.
Каждый миллилитр раствора тиосульфата натрия (0,05 моль/л) ТР соответствует 1,06 мг I.
Определение серы
Используя метод сжигания в колбе с кислородом, описанный на с. 142, сжигают количество вещества, указанное в частной статье. Поглощающая жидкость состоит из 12,5 мл раствора перекиси водорода (~6() г/л) ИР. После завершения процесса споласкивают пробку, платиновую проволоку, платиновую сетку и стенки колбы 40 мл воды. Кипятят раствор в течение 10 мин, охлаждают, прибавляют 2 мл уксусной кислоты (~300 г/л) ИР и 20 мл этанола (~750 г/л) ИР. Титруют раствором нитрата бария (0,01 моль/л) ТР, используя в качестве индикатора 2 капли .раствора торина (2 г/л) ИР и 2 капли раствора метиленового синего (0,2г/л) ИР до перехода желтой окраски в розовую.
Каждый миллилитр раствора нитрата бария (0,01 моль/л) ТР соответствует 0,321 мг S.
ХИМИЧЕСКИЕ МЕТОДЫ
145
КОМПЛЕКСОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Комплексонообразующими агентами, используемыми в качестве титрантов, являются аминополикарбоновые кислоты, имеющие характерную группу
--N
^СНгСООН
Такие соединения способны образовывать хелатные комплексы со многими катионами, в которых катион связан в кольцевой структуре. Кольцо образовано солевыми связями катиона с карбоксильными группами, а также координационной связью за счет свободной пары электронов атома азота. Если кольцо пятичленное, то образованный хелат должен иметь высокую стабильность, так что наиболее удобные хелатные титранты это те, которые способны образовывать такие кольца. Это справедливо для этилендиаминтетрауксусной кислоты (ЭДТА), обычно применяемой в виде динатриевой соли, известной как эдетат динатрия. С большинством металлов, имеющих более одного положительного заряда, ЭДТА образует высокорастворимые в воде комплексы при соотношении 1 : 1. При этом образуется структура, содержащая не менее 3 пятичленных хелатных колец, что обеспечивает высокую устойчивость комплекса. В некоторых случаях, помимо связей, которые образуются за счет свободной пары электронов азота, могут образовываться координационные связи за счет карбонильных кислородов других карбоксильных групп. Так, комплексы, образованные кальцием и трехвалентным алюминием, могут быть представлены формулами:
и
^СН^-СО-0 о-со-сн2
N---------а-«----------—N
|\ /\
СН^СО-О’ -О-СО-СН2
С Н 2------------------С Н г
СНгСО-О о-со-сн2
/ 2 \ / \
N----------------N
\ /\ /
СН^СО-О' О-СО-СН2
с Н5--------------------с н 2
10—2025
146
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Стабильность таких комплексов в значительной степени зависит от pH раствора. Большинство двухвалентных металлов образует комплексы, устойчивые в щелочной среде, но хелаты щелочноземельных металлов разрушаются при pH примерно ниже 8; в то же время многие комплексы двухвалентных металлов (например, цинка и свинца) также устойчивы в достаточно кислом растворе. Комплексы трехвалентных металлов благодаря дополнительной стабильности, которая обеспечивается увеличенным числом хелатных колец, часто устойчивы даже в сильно кислых растворах. В щелочных растворах, однако, некоторые из этих металлов в присутствии ЭДТА осаждаются в виде гидроокисей, но не вследствие нестабильности комплекса, а 'вследствие более мощного влияния низкой растворимости гидроокиси металла.
Ниже приведены константы стабильности хелатов ЭДТА с некоторыми металлами, определенные Шварценбахом для 0,1 моль/л растворов при 20° С:
Na 1,7
Li 2,8
Mg 8,7
Са 10,6
Fe2+ 14,3
Al 15,5a
Zn 16,1
Pb 17,6
Hg2+ 20,4
Fe3+ 25,1
° Хелат алюминия образуется медленно, так что этот металл обычно определяют методом обратного титрования.
Для определения эквивалентной точки при титровании ионов металлов с ЭДТА необходимо использовать подходящий индикатор, который будет реагировать на присутствие свободных ионов металлов в растворе. Таким индикатором, первоначально использованным Шварценбахом для титрования ионов кальция, был мурексид (пурпурат аммония), который в настоящее время очень редко применяется. Наиболее часто из индикаторов используется протравной черный 11 (известный также под другими торговыми названиями). Этот индикатор имеет синюю окраску в аммиачном растворе, но дает красные комплексы со многими ионами металлов в этом растворе. Образуемые при этом комплексы металлов, как правило, менее прочные, чем соответствующие комплексы с ЭДТА, так что титрование с эдетатами легко удаляет металл из его комплекса с индикатором и изменение цвета на чисто синий свидетельствует о полном титровании металла,
ХИМИЧЕСКИЕ МЕТОДЫ
147
имеющегося в растворе. Протравной черный 11 часто используют -в смеси с метиловым оранжевым; на 'фоне последнего легче обнаруживается конечная точка титрования.
В качестве индикаторов для комплексонометр'ических титрований были предложены и использовались многие другие вещества, однако в этой работе приводятся лишь те индикаторы, которые имеют потенциальную ценность для фармацевтического анализа. Калькой и кальконкарбоновая кислота дают очень четкий переход окраски от винно-красной к чисто синей при титровании кальция эдетатом динатрия при pH 12—14. Если присутствует магний, то при этом значении pH он осаждается в виде гидроокиси, и если перед прибавлением 'индикатора добавляют щелочь, то он не мешает определению. Однако все эти индикаторы недостаточно устойчивы в щелочном растворе и поэтому желательно прибавлять их к концу титрования.
Другой широко применяемый индикатор — ксиленоловый оранжевый; это обычный кислотно-основный индикатор, в который введены группы пминодиуксусной кислоты, благодаря чему это вещество действует в качестве металл-комп-лексного индикатора. Этот индикатор дает четкий переход окраски от розово-фиолетовой к желтой в конце титрования алюминия, висмута, свинца, ртути и 'цинка и может применяться при pH 2—6 в зависимости от титруемого металла.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Алюминий
Растворяют точную навеску вещества, как указано в частной статье, в 2 мл соляной кислоты (1 моль/л) ТР и 50 <мл воды, если статья не предписывает другие условия для приготовления раствора. Прибавляют 50 мл эдетата динатрия (0,05 моль/л)ТР и нейтрализуют по раствору метилового красного в этаноле ИР гидроокисью натрия (1 моль/л) ТР. Нагревают раствор до кипения и поддерживают кипение в бане не менее 10 мин. Охлаждают, прибавляют около 50 мг индикаторной смеси ксиленолового оранжевого Р и 5 г метенамина Р и титруют избыток эдетата раствором нитрата свинца (0,05 моль/л) ТР до перехода окрашивания от желтого к розово-фиолетовому. Каждый миллилитр раствора эдетата динатрия (0,05 моль/л) ТР соответствует 1,349 мг А1.
Висмут
Растворяют точную навеску вещества, как указано в частной статье, в минимальном количестве азотной кислоты
10*
148 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
(-~ 130 г/л) ИР, прибавляют 50 мл воды и доводят pH до 1—2, добавляя по каплям либо азотную кислоту (~ 130 г/л) ИР, либо аммиак (-—'100 г/л) ИР. Прибавляют около 50 мг индикаторной смеси ксиленолового оранжевого Р и медленно титруют эдетатом динатрия (0,05 моль/л) ТР до перехода окрашивания от розово-фиолетового к чисто желтому. Каждый миллилитр раствора эдетата динатрия (0,05 моль/л) ТР соответствует 10,45 мг Bi.
Кальций
„Растворяют точную навеску вещества, как указано в частной статье, в нескольких миллилитрах воды, подкисленной, если необходимо, минимальным количеством соляной кислоты ( 70 г/л) ИР, и затем разводят водой примерно до 100 мл. Титруют раствором эдетата динатрия (0,05 моль/л) ТР до тех пор, пока до ожидаемой эквивалентной точки не остается добавить приблизительно 2 мл, и прибавляют 4 мл раствора гидроокиси натрия (—300 г/л) ИР и 0,1 г индикаторной смеси калькона Р или индикаторной смеси кальконкарбоно-вой кислоты Р и продолжают титрование до перехода окрашивания от розового к чисто синему. Каждый миллилитр раствора эдетата динатрия (0,05 моль/л) ТР соответствует 2,004 мг Са.
Свинец
„Растворяют точную навеску вещества, как указано в частной статье, в 5—10 мл воды, подкисленной, если необходимо, минимальным количеством уксусной кислоты (~300 г/л) ИР^ и затем доводят водой примерно до 50 мл. Затем прибавляют около 50 мг индикаторной смеси ксиленолового оранжевого Р и достаточное количество метенамина Р (около 5 г) для получения красного окрашивания. Титруют раствором эдетата динатрия (0,05 моль/л) ТР до перехода окрашивания от темно-фиолетового к чисто желтому. Каждый миллилитр раствора эдетата динатрия (0,05 моль/л) ТР соответствует 10,35 мг РЬ.
Магний
„Растворяют точную навеску вещества, как указано в частной статье, в 5—10 мл '.воды, подкисленной, если необходимо, минимальным количеством соляной кислоты ( — 70 г/л) ИР, и затем доводят водой примерно до 50 мл. Прибавляют 10 мл буферного раствора хлорида аммония, pH 10,0, ИР и 100 мг
ХИМИЧЕСКИЕ МЕТОДЫ ' 149
индикаторной смеси протравного черного 11 Р и титруют раствором эдетата динатрия (0,05 моль/л) ТР до перехода окрашивания от фиолетового >к зеленому. Каждый миллилитр раствора эдетата динатрия (0,05 моль/л) ТР соответствует 1,215 мг Mg.
Цинк
Растворяют точную навеску вещества, как указано в частной статье, в 5—10 мл воды, подкисленной, если необходимо, минимальным количеством уксусной кислоты (~300 г/л) ИР, и затем доводят водой примерно до 50 мл. Прибавляют около 50 мг индикаторной смеси ксиленолового оранжевого Р и достаточное количество метенамина Р (около 5 г) для получения красного окрашивания. Титруют раствором эдетата динатрия (0,05 моль/л) ТР до перехода окрашивания от розово-фиолетового к чисто желтому. Каждый миллилитр раствора эдетата динатрия (0,05 моль/л) ТР соответствует 3,268 мг Zn.
НЕВОДНОЕ ТИТРОВАНИЕ
Кислоты и основания в течение длительного времени определялись как вещества, которые при растворении в воде образуют соответственно ион водорода и гидроксильный ион. Это определение, введенное Аррениусом, не учитывает того факта, что, свойства, характерные для кислот и оснований, могут проявляться также в других растворителях.„Более общее определение принадлежит Брёнстеду, который рассматривает кислоту как вещество, выделяющее протоны (донор протонов), а основание как вещество, присоединяющее протоны (акцептор протонов). Еще более широкое 'определение дано Льюисом, считающим кислотой любое вещество, которое принимает пару электронов, а основанием — любое вещество, которое отдает пару электронов; нейтрализацию же он определяет как образование координационной связи между кислотой и основанием.
Кажущаяся сила кислоты или основания определяется степенью их реакции с растворителем. .В водных растворах все сильные кислоты являются одинаково сильными, потому что они реагируют с растворителем, подвергаясь почти полному превращению в ион гидроксония (Н3О+) и кислотный анион. В слабо протофильном растворителе, например в уксусной кислоте, степень образования мона ацетония (СН3СООН2+) вследствие присоединения протона обеспечивает более чувствительную дифференциацию силы кислот
150 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
и показывает следующий порядок уменьшения их силы: хлорная, бромистоводородная, серная, соляная и азотная.
Уксусная кислота реагирует с водой не полностью, образуя ион гидроксония и, следовательно, является слабой кислотой. В основании, например в этилендиамине, она, напротив, реагирует с растворителем так полно, что ведет себя как сильная кислота.
Этот так называемый эффект выравнивания силы кислот наблюдается также для оснований. В серной кислоте все основания имеют также одну и ту же силу. По мере уменьшения кислотных свойств растворителя в ряду серная кислота — уксусная кислота — фенол — вода — пиридин — бутиламин растворенные в них основания становятся постепенно слабее и разница 'Между ними становится более четко выраженной. В порядке уменьшения силы сильными основаниями для неводного титрования являются метилат калия, метилат натрия, метилат лития и гидроокись тетрабутиламмония.
Многие нерастворимые в воде соединения проявляют кислотные или основные свойства при растворении в органических растворителях. Таким образом, выбор подходящего растворителя позволяет определять многие такие соединения с помощью неводного титрования. Далее, в зависимости от того, какая часть соединения является физиологически активной, можно титровать эту часть путем правильного выбора растворителя и титранта. Чистые вещества можно титровать непосредственно, но часто бывает необходимо отделить активный ингредиент лекарственных форм от мешающих наполнителей и носителей.
К соединениям, которые можно титровать как кислоты, относятся кислотные галогениды, ангидриды кислот, карбоновые кислоты, аминокислоты, энолы, такие, как барбитураты и ксантины, имиды, фенолы, пирролы, сульфаниламиды. К соединениям, которые можно титровать как основания, относятся амины, азотсодержащие гетероциклические соединения, четвертичные аммониевые соединения, щелочные соли органических кислот, щелочные соли неорганических кислот и некоторые соли аминов. Многие соли галоидоводородных кислот можно титровать в уксусной кислоте или уксусном ангидриде после прибавления ацетата ртути, который удаляет ион галоида переведением в неионизированный комплекс галогенида ртути. Гидрохлориды слабых оснований, не содержащие группировок, способных ацетилироваться, можно также титровать в уксусном ангидриде без добавления ацетата ртути, используя в качестве индикатора малахитовый зеленый или кристаллический фиолетовый. Титрования, проводимые при избытке уксусного ангидрида, следует приме
ХИМИЧЕСКИЕ МЕТОДЫ 151
нять с осторожностью, так как любая реакция ангидрида с титруемым веществом может привести к заниженным результатам.
При титровании основных соединений обычно используют объемный раствор хлорной кислоты в ледяной уксусной кислоте, хотя в особых случаях удобнее использовать раствор хлорной кислоты в диоксане. При титровании кислых соединений часто применяют объемный раствор метилата лития в растворе метанол — толуол. Для многих случаев удобно использовать раствор гидроокиси тетрабутиламмония в толуоле; метилат натрия, ранее широко применявшийся, часто может давать вызывающий затруднения желатинообразный осадок.
Чтобы исключить влияние углекислого газа, растворители для кислотных соединений в процессе титрования должны быть защищены от избыточного действия воздуха подходящей пробкой или инертным газом. Следует провести контрольный опыт; обычно объем 0,1 моль/л титранта не должен превышать 0,01 мл на 1 мл растворителя.
Конец титрования можно определять визуально по изменению окраски или потенциометрически. Если применяется каломельный электрод сравнения, то удобнее заменить водный раствор хлорида калия ib солевом мостике на раствор перхлората лития в уксусной кислоте ИР для титрования в кислых растворителях и на раствор хлорида калия в метаноле для титрования в основных растворителях. Следует помнить, что некоторые обычно используемые индикаторы (например,'кристаллический фиолетовый) подвергаются постепенному изменению окраски, поэтому при оценке пригодности метода неводного титрования для конкретного случая необходимо проследить за тем, чтобы при потенциометрическом титровании вещества изменение окраски в конечной точке титрова-ния соответствовало максимальной величине d-E/dV (где Е электродвижущая сила, а V — объем титранта).
Если используют титранты, приготовленные с растворителями, имеющими относительно высокий коэффициент расширения (например, с ледяной уксусной кислотой, толуолом и т. п.), следует проследить за тем, чтобы были компенсированы различия в температуре между временем применения и временем стандартизации титранта.
РЕКОМЕНДУЕМЫЕ МЕТОДИКИ
Метод А (для оснований и их солей)
Готовят раствор, как указано в частной статье, или растворяют испытуемое вещество в подходящем объеме ледяной уксусной кислоты Р1, предварительно нейтрализованной по
152
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
раствору кристаллического фиолетового в уксусной кислоте ИР, нагревая и охлаждая, если необходимо. Кроме того, контрольную поправку на расход титранта на растворитель и индикатор можно установить в отдельном определении. Если вещество является солью галоидоводородной кислоты, прибавляют 10 мл раствора ацетата ртути в уксусной кислоте ИР. Если конечную точку определяют визуально по изменению окраски, прибавляют 2—3 капли раствора кристаллического фиолетового в уксусной кислоте ИР и титруют хлорной кислотой указанной концентрации ('Моль/л) до соответствующего изменения окраски индикатора. Если в частной статье указан другой индикатор, этот же индикатор следует использовать для нейтрализации ледяной уксусной кислоты Р1 и раствора ацетата ртути в уксусной кислоте ИР, а также для стандартизации титранта.
Если эквивалентные точки определяются потенциометрически, индикатор не применяют, а нейтрализацию раствора и стандартизацию титранта также проводят потенциометрически. Используют стеклянный электрод и насыщенный каломельный элемент [содержащий раствор хлорида калия (350 г/л) ИР] в качестве электрода сравнения. Соединительный мостик между каломельным электродом и титруемой жидкостью должен иметь достаточно низкое электрическое сопротивление, а перенос жидкости должен быть минимальным. Соединение потенциометра с системой электродов должно выполняться согласно инструкции изготовителя, иначе будут получены нестабильные данные.
Если температура (/г), <п<ри которой проводится титрование, отличается от температуры (!]), при которой стандартизовался титрант, умножают объем требуемого титранта на [1 + 0,001(1]—4)] и вычисляют результат количественного определения, исходя из найденного исправленного объема.
Метод Б (для кислот)
Титрант, растворитель и индикатор (если конечная точка определяется визуально), которые должны применяться для каждого вещества, указываются в соответствующей частной статье.
Во время титрования раствор и титрант защищают от углекислоты воздуха. Это может быть достигнуто вытеснением воздуха над титруемой жидкостью азотом.
Растворяют испытуемое вещество в подходящем объеме растворителя, предварительно нейтрализованного по индикатору, нагревая и охлаждая, если необходимо, или готовят раствор, как указано в частной статье. Титруют до соответ
ХИМИЧЕСКИЕ МЕТОДЫ
153
ствующего изменения окраски индикатора. Проводят контрольное титрование и вносят необходимые поправки. Титрант стандартизуют с тем же растворителем и индикатором, которые указаны для данного вещества.
Если эквивалентная точка устанавливается потенциометрически, то индикатор не применяют, а нейтрализацию раствора и стандартизацию титранта также проводят потенциометрически.
Используют стеклянный электрод и насыщенный каломельный электрод сравнения, в котором водный раствор хлорида калия (350 г/л) ИР заменен насыщенным раствором хлорида калия Р в метаноле Р. Соединительный мостик между каломельным электродом и титруемой жидкостью должен иметь достаточно низкое электрическое сопротивление, а перенос жидкости должен быть минимальным. Соединение потенциометра с системой электродов должно выполняться согласно инструкции изготовителя, иначе будут получены нестабильные данные.
НИТРИТОМЕТРИЯ
Нитритометрия — титриметрический метод, применяемый главным образом для количественного определения первичных ароматических аминов.
Прибор, обычно используемый ® методике электрометрического титрования нитритом, состоит из открытого сосуда для титрования, содержащего два платиновых электрода, соединенных подходящей цепью. Электроды должны иметь разность потенциалов порядка 50—100 мВ. Электрическая цепь должна включать прибор для измерения силы тока с чувствительностью от 0,1 до 1 нА, обычно со стрелкой-индикатором. Сосуд для титрования должен быть снабжен соответствующим механическим или магнитным перемешивающим устройством; для перемешивания раствора также может быть использован поток азота, проходящий через раствор. Электроды должны быть изготовлены из платиновой проволоки' диаметром 0,5 мм и длиной около 20 мм. Перед каждым применением электроды следует очищать погружением на несколько секунд в кипящую азотную кислоту (~1000 г/л) ИР, к которой предварительно прибавлен хлорид железа (III) Р в количестве 1мг/мл. Затем электроды тщательно промывают водой.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Помещают 20 мл соляной кислоты (~ 250 г/л) ИР и 50 мл воды в сосуд для титрования, прибавляют определенное количество исследуемого вещества, если предписано, катали
154
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
затор, указанный в частной статье, и растирают для (растворения; охлаждают приблизительно до 15° С и медленно титруют раствором нитрата натрия (0,1 моль/л) ТР, помещая кончик бюретки ниже поверхности раствора. Во время (прибавления титранта раствор непрерывно слегка перемешивают, избегая завихрений воздуха в самом растворе и поддерживая температуру раствора около 15° С.
Когда до конца титрования оставшийся расход титранта составит 1 мл, титрант прибавляют порциями по 0,1 мл с выдержкой между прибавлениями не менее 1 мин. Вначале стрелка измерительного прибора отклоняется при каждом прибавлении реактива, а затем возвращается в исходное положение. При достижении конечной точки стрелка остается в отклоненном положении.
ОПРЕДЕЛЕНИЕ ВОДЫ МЕТОДОМ КАРЛА ФИШЕРА
Титриметрическое определение воды методом Карла Фишера основано на количественной реакции между водой и реактивом, состоящим из двуокиси серы и йода в безводном пиридине и обычно метанола. Реакцию проводят в подходящем растворителе, таком, как метанол или уксусная кислота.
Реактивы и растворы, используемые при определении воды этим методом, чувствительны к воде, поэтому постоянно следует принимать меры, чтобы они не подвергались действию атмосферной влаги.
Титрационный сосуд снабжен двумя платиновыми электродами, если необходимо, трубкой для впуска газа, пробкой, в которой укреплен кончик бюретки, и выходной трубкой, защищенной осушителем. Титруемый образец вводят через входную трубку или боковое плечо, которое закрывается притертой пробкой. Реактив К. Фишера ИР защищают от действия света и хранят в склянке, снабженной автоматической бюреткой. Бюретку заполняют реактивом при помощи ручного сильфона, доступ влаги предупреждается подходящей системой осушительных трубок. Перемешивание производят с помощью магнитной мешалки или с помощью тока высушенного азота, пропускаемого через раствор во время титрования.
Конец титрования определяют при помощи 'электрической цепи, состоящей из микроамперметра, платиновых электродов и батареи напряжением 1,5—2 В, соединенной через переменное сопротивление порядка 2000 Ом. Сопротивление устанавливают таким образом, чтобы начальный ток проходил последовательно через платиновые электроды и микроамперметр.
ХИМИЧЕСКИЕ МЕТОДЫ
155
После каждого прибавления реактива стрелка ‘микроамперметра отклоняется, но быстро возвращается в исходное положение. Когда титрование окончено, стрелка остается в отклоненном положении 10—15 с. Кроме того, конец титрования может быть определен вольтаметрическим методом. К платиновым электродам прилагают разность потенциалов порядка 30—50 мВ для обеспечения постоянного поляризационного тока и раствор титруют реактивом. Разность потенциалов определяют при помощи микровольтметра. Конечную точку считают достигнутой, когда вольтметр показывает устойчивое падение напряжения. При вольтаметрическом методе конечная точка может быть установлена графически путем построения зависимости напряжения ют объема реактива и определения начала падения напряжения.
РЕКОМЕНДУЕМЫЕ МЕТОДИКИ
Прямое титрование (метод А)
В сосуд для титрования вносят около 2С мл безводного метанола Р, если в частной статье нет специальных указаний, и титруют до конечной точки реактивом К. Фишера ИР. Быстро вносят указанное количество исследуемого образца (точная навеска) в сосуд для титрования. Перемешивают в течение 1 мин и снова титруют реактивом К. Фишера ИР.
Обратное титрование (метод Б)
В сосуд для титрования вносят около 10 мл безводного метанола Р, если в частной статье нет специальных указаний, и титруют до конечной точки реактивом К. Фишера ИР. Быстро вносят указанное количество исследуемого образца (точная навеска) и затем точно отмеренный объем реактива К. Фишера ИР, достаточный для обеспечения избытка около 1 мл. 'Оставляют стоять в защищенном от света месте на 1 мин или на время, указанное в частной статье; время от времени раствор перемешивают.
Титруют избыток реактива К- Фишера ИР до конечной точки безводным метанолом Р, к которому прибавлено точно известное количество воды, обычно эквивалентное примерно 2,5 мг/мл.
ОПРЕДЕЛЕНИЕ МЕТОКСИЛЬНЫХ ГРУПП
Содержание метоксильных групп в органическом веществе количественно определяется путем реакции вещества с концентрированной йодистоводородной кислотой; образующийся
156
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
при реакции метилйодид отгоняют, поглощают в подходящую жидкость и количественно определяют титриметрически.
Прибор
Прибор для определения состоит из круглодонной колбы для кипячения емкостью 25 мл, в которую впаяна капиллярная боковая трубка диаметром 1 мм для впуска углекислого газа. Колба снабжена воздушным холодильником высотой около 25 см и диаметром около 9 мм. Над холодильником помещают подходящее газопромывное устройство, содержащее около 2 мл воды. В промывное устройство помещают 5 мл раствора тартрата натрия-сурьмы ( 50 г/л) ИР. Выход газопромывателя заканчивается трубкой, которая погружена в поглощающую жидкость в первой из двух поглотительных склянок, соединенных между собой. Для большего удобства при использовании и очистке отдельные части прибора соединены посредством притертых стеклянных конических или шаровых соединений.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Помещают точную навеску исследуемого вещества, как указано в частной статье, ,в колбу для кипячения с палочкой для равномерного кипения. Прибавляют 2,5 мл расплавленного фенола Р и 5 мл йодистоводородной кислоты ( — 970 г/л) ИР и соединяют колбу с холодильником. Прибавляют в каждую из поглотительных склянок раствор ацетата калия ИР, около 6 мл в первую и около 4 мл во вторую; затем в каждую из склянок прибавляют по 6 капель брома Р. Через капиллярную боковую трубку в колбе для кипячения равномерно пропускают медленный ток углекислого газа Р и жидкость слегка нагревают при помощи микрогорелки или другого подходящего устройства так, чтобы пары кипящей жидкости поднимались до половины холодильника. Для большинства веществ для завершения реакции и прохождения через прибор достаточно 30 мин. Затем содержимое обеих поглотительных склянок смывают в коническую колбу емкостью 250 мл, содержащую 5 мл раствора ацетата натрия (150 г/л) ИР.
Доводят объем жидкости примерно до 125 мл и прибавляют 6 капель муравьиной кислоты ( — 1080 г/л) ИР. Вращают колбу до исчезновения окраски брома, затем прибавляют 12 капель муравьиной кислоты ( — 1080 г/л) ИР, закрывают колбу пробкой и, тщательно перемешав содержимое для удаления избытка брома из паров над жидкостью, оставляют
ХИМИЧЕСКИЕ МЕТОДЫ
157
раствор стоять 1—2 мин; прибавляют 1 г йодида калия Р и 5 мл серной кислоты (~ 100 г/л) ИР и титруют выделившийся йод раствором тиосульфата натрия (0,1 моль/л) ТР, используя раствор крахмала ИР в качестве индикатора. Проводят определение без испытуемого вещества и вычитают объем раствора тиосульфата натрия (0,1 моль/л) ТР, пошедшего на контрольное определение, из объема, пошедшего на определение метоксильной группы.
Каждый миллилитр раствора тиосульфата натрия (0,1 моль/л) ТР соответствует 0,5172 мг метоксильной группы (СН3О).
ОПРЕДЕЛЕНИЕ АЗОТА
РЕКОМЕНДУЕМЫЕ МЕТОДИКИ
Методика для макроопределения (метод А)
Помещают указанную в частной статье навеску вещества в длинногорлую колбу емкостью 200 мл, прибавляют 1 г смеси, состоящей из 10 частей сульфата калия Р или безводного сульфата натрия Р и I части меди (II) Р, затем прибавляют указанное в статье количество серной кислоты (~ 1760 г/л), не содержащей соединений азота, ИР. Нагревают смесь на небольшом пламени до получения прозрачного зеленого раствора и слегка кипятят еще 30 мин, если в статье нет других указаний; необходимо следить за тем, чтобы верхняя часть колбы не перегревалась. Охлаждают, разводят водой до 75—80 мл, соблюдая соответствующие меры предосторожности, <и прибавляют кусочек гранулированного цинка Р и раствор 15 г гидроокиси натрия Р и 2 г тиосульфата натрия Р в 25 мл воды. Если необходимо, количество гидроокиси натрия можно увеличить для того, чтобы обеспечить перед отгонкой сильно щелочную реакцию. Тотчас соединяют колбу с перегонным аппаратом, перемешивают содержимое, отгоняют выделившийся аммиак в 16 мл раствора борной кислоты ('50 г/л) ИР и титруют серной кислотой (0,05 моль/л) ТР, используя в качестве индикатора раствор метилового красного в этаноле ИР. Повторяют определение без исследуемого вещества; разность между титрованиями соответствует количеству аммиака, образовавшегося из исследуемого вещества. Каждый миллилитр серной кислоты (0,05 моль/л) ТР соответствует 1,401 мг азота N.
158 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Методика для микроопределения (метод Б)
Помещают указанную в частной статье навеску вещества в подходящую пробирку для настаивания, прибавляют 3 капли раствора сульфата меди (II) (190 г/л) ИР и 1 мл серной кислоты (~1760 г/л), не содержащей соединений азота, ИР, слегка кипятят в течение 10 мин и охлаждают; прибавляют 1 г безводного сульфата натрия Р и 10 мг селена Р, слетка кипятят в течение 1 ч и охлаждают. Переносят содержимое пробирки в прибор для микроперегонки аммиака или присоединяют к нему, прибавляют 6 мл раствора гидроокиси натрия (~400 г/л) ИР и пропускают через колбу пар; отгоняют в течение 7 мин, собирая отгон в смесь 5 мл раствора борной кислоты (50 г/л) ИР, 5 мл воды и 1 капли раствора метилового красного и метиленового синего ИР и титруют соляной кислотой (0,015 моль/л) ТР. Повторяют определение без исследуемого вещества; разность между титрованиями соответствует количеству аммиака, образовавшегося из исследуемого вещества. Каждый миллилитр серной кислоты (0,015 моль/л) ТР соответствует 0,210 мг азота N.
ОПРЕДЕЛЕНИЕ ЙОДНОГО ЧИСЛА
Иодное число вещества — это масса галоидов, выраженная как йод, поглощаемый 100 частями массы исследуемого вещества. В определении следует использовать такое количество вещества, чтобы не менее 70% прибавленного йода, как предусмотрено в рекомендуемой методике, не поглощалось. Если в частной статье нет других указаний, то в зависимости от ожидаемого йодного числа, для его определения следует использовать навески вещества, указанные в следующей таблице.
Йодное число Навеска вещества, г
Менее 20 1,0
20— 60 0,5—0,25
60—100 0,25—0,15
Более 100 0,15—0,10
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Помещают точную навеску вещества, как указано в частной статье, в сухую колбу с притертой пробкой емкостью 300— 500 мл, прибавляют 15 мл четыреххлористого углерода Р и растворяют. Прибавляют 25 мл раствора бромистого йода ИР, закрывают пробкой, предварительно смоченной раствором
ХИМИЧЕСКИЕ МЕТОДЫ
159
йодида калия (80 г/л) ИР, слегка встряхивают и оставляют стоять в темном месте на 30 мин, если в частной статье нет других указаний. Прибавляют 20 мл раствора йодида калия (80 г/л) ИР и 150 мл воды; встряхивая содержимое колбы, титруют раствором тиосульфата натрия (0,1 моль/л) ТР; к концу титрования прибавляют индикатор — раствор крахмала ИР. Отмечают число миллилитров тиосульфата натрия (0,1 моль/л) ТР, пошедшее на титрование, (а). Одновременно таким же образом проводят определение без исследуемого вещества и отмечают число миллилитров тиосульфата натрия (0,1 моль/л) ТР, пошедшее на титрование, (б). Йодное число рассчитывают по следующей формуле:
„ (б— а)х0,01269x100
Йодное число Навеска вещества, г '
ОПРЕДЕЛЕНИЕ ПЕРЕКИСЕЙ В ЖИРНЫХ МАСЛАХ
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Растворяют навеску исследуемого вещества, как указано в частной статье, обычно около 3 г (точная навеска) в 15 мл хлороформа Р и 30 мл ледяной уксусной кислоты Р в колбе емкостью 250 мл с притертой пробкой. Прибавляют 1 мл свежеприготовленного раствора йодида калия Р в 1 мл воды, закрывают колбу пробкой, перемешивают путем легкого вращения и оставляют в темном месте на 3 мин. Прибавляют 100 мл воды, встряхивают и титруют раствором тиосульфата натрия (0,01 моль/л) ТР, используя в качестве индикатора раствор крахмала ИР. Повторяют операцию без исследуемого вещества и вычисляют разность между титрованиями; допустимый предел содержания перекисей указывается в частной статье.
ОПРЕДЕЛЕНИЕ ЧИСЛА ОМЫЛЕНИЯ
Число омыления — это количество миллиграммов гидроокиси калия, необходимое для нейтрализации жирных кислот, полученных при полном гидролизе 1 г вещества.
В описанной методике предпочтительно использовать для титрования бюретку емкостью 50 мл, так как в контрольном титровании используемый объем соляной кислоты (0,5 моль/л) ТР составляет точно 35,5 мл, если концентрация этанольного раствора гидроокиси калия равна точно 40 г/л.
160
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Точную навеску вещества (около 2 г) или количество, указанное в частной статье, помещают в колбу емкостью около 200 мл, прибавляют 25 мл раствора гидроокиси калия в этаноле ИР 1, присоединяют обратный холодильник и нагревают на кипящей водяной ‘бане в течение 30 мин или в течение времени, указанного в частной статье, часто вращая содержимое колбы; тотчас прибавляют 1 мл раствора фенолфталеина в этаноле ИР и титруют избыток щелочи соляной кислотой (0,5 моль/л) ТР. Отмечают число миллилитров соляной кислоты (0,5 моль/л) ТР, необходимое на титрование образца, (а). Повторяют операцию без исследуемого вещества и отмечают число миллилитров соляной кислоты (0,5 моль/л) ТР, требуемое для нейтрализации, (б). Число омыления рассчитывают по следующей формуле:
Число омыления =
(б — а)ХО,02805X1000
Навеска вещества, г '
ОПРЕДЕЛЕНИЕ НЕОМЫЛЯЕМЫХ ВЕЩЕСТВ
Определение «неомыляемые вещства» относится к веществам, которые присутствуют в маслах или жирах, не омы-ляются едкими щелочами и извлекаются в эфир.
РЕКОМЕНДУЕМАЯ МЕТОДИКА J
Помещают точную навеску вещества, как указано в частной статье, в колбу, снабженную обратным холодильником, и кипятят на водяной бане в течение 1 ч с 25 мл раствора гидроокиси калия в этаноле (0,5 моль/л) ТР, часто вращая содержимое. Смывают содержимое колбы в делительную воронку при помощи 50 мл воды и, пока жидкость еще теплая, извлекают, энергично встряхивая, 3 порциями, каждая по 50 мл, эфира Р; промывают колбу первой порцией эфира Р (ПРИМЕЧАНИЕ. Эфир не должен содержать перекисей). Часто и осторожно снижают давление, которое может возникнуть в делительной воронке. Объединяют эфирные растворы в другой делительной воронке, содержащей 20 мл воды. (Если эфирные растворы содержат твердое взвешанное вещество, их фильтруют в делительную воронку через фильтровальную бумагу, не содержащую жира, и промывают фильтр эфиром Р.) Слегка вращают делительную
ХИМИЧЕСКИЕ МЕТОДЫ
161
воронку в течение нескольких минут, избегая сильного встряхивания, дают жидкостям разделиться и отбрасывают водный слой. Промывают эфирные извлечения, энергично встряхивая с 2 порциями, каждая по 20 мл, воды; затем обрабатывают 3 раза 20 мл раствора гидроокиси калия (0,5 моль/л) ТР (ПРИМЕЧАНИЕ. Водный реактив), энергично встряхивая каждый раз и промывая 20 мл воды после каждой обработки. Наконец, промывают последовательными порциями, каждая по 20 мл, воды до нейтральной реакции на раствор фенолфталеина в этаноле ИР. Переносят эфирные извлечения во взвешенную колбу, промывают делительную воронку эфиром Р; отгоняют эфир, соблюдая необходимые предосторожности, и прибавляют 3 мл ацетона Р.
Умеренным продуванием воздуха полностью удаляют растворитель из колбы, которую лучше держать наклонно и вращать, почти полностью погрузив в водяную баню с температурой примерно 60° С. Высушивают до постоянной .массы при температуре не выше 80° С и растворяют содержимое колбы в 10 мл этанола ( — 750 г/л) ИР, предварительно нейтрализованного по .раствору фенолфталеина в этаноле ИР. Титруют раствором гидроокиси натрия (0,1 моль/л), не содержащим карбонатов, ТР, используя в качестве индикатора раствор фенолфталеина в этаноле ИР. Если на титрование идет не более 0,2 мл, то считают, что взвешенный остаток представляет собой неомыляемые вещества. Рассчитывают процентное содержание неомыляемых веществ в масле или жире. Если на титрование идет более 0,2 мл раствора гидроокиси натрия (0,1 моль/л) ТР, взвешенный остаток нельзя считать неомыляемым веществом п испытание следует повто-»ить.
ОПРЕДЕЛЕНИЕ КИСЛОТНОГО ЧИСЛА
Кислотное число — это количество миллиграммов гидроокиси калия, необходимое для нейтрализации свободной кислоты в 1 г вещества.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Точно взвешивают около 10 г исследуемого вещества или количество, указанное в частной статье, в колбе емкостью 250 мл и прибавляют 50 мл смеси равных объемов этанола ( — 750 г/л) ИР и эфира Р, предварительно нейтрализованной раствором гидроокиси калия (0,1 моль/л) ТР после до-
11—2025
162
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
бавления 1 мл раствора фенолфталеина в этаноле ИР. Если необходимо, нагревают до полного растворения вещества и охлаждают; титруют раствором гидроокиси калия (0,1 моль/л) ТР при постоянном встряхивании до появления розового окрашивания, не исчезающего в течение 15 с. Отмечают число миллилитров, пошедшее на титрование, (а). Кислотное число рассчитывают по формуле:
аХ0,00561Х1000
Кислотное число = -rj—~----------------
Навеска вещества, г
БИОЛОГИЧЕСКИЕ МЕТОДЫ
и
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ МИКРОБИОЛОГИЧЕСКОЙ АКТИВНОСТИ АНТИБИОТИКОВ
Активность образца антибиотика выражают как отношение дозы, которая угнетает рост соответствующего чувствительного микроорганизма, к дозе Международного биологического стандарта, Международного биологического эталонного препарата или Международного химического стандартного образца этого антибиотика, которая дает такую же степень угнетения. В количественном определении можно также использовать вторичные стандартные образцы, активность которых удостоверена соответствующим методом. Количественное определение основано на сравнении угнетения роста микроорганизмов, вызванного известными концентрациями стандартного образца, с угнетением, вызванным определенными разведениями испытуемого вещества. Этот эффект может быть измерен при помощи метода диффузии, как описано ниже, или турбидиметрическим методом.
Международная единица — это специфическая активность определенного количества (массы) Международного биологического стандарта или Международного биологического эталонного препарата. Комитет экспертов ВОЗ по стандартизации биологических препаратов время от времени указывает количество антибиотика, точно эквивалентное единице, принятой для международного использования. В некоторых случаях из-за определенных свойств материала взвешивание с достаточной точностью небольших количеств соответствующего Международного биологического стандарта или Международного биологического эталонного препарата сопряжено с трудностями, поэтому международные единицы определяются для всего материала, содержащегося в ампуле или флаконе, т. е. активность всего содержимого ампулы или флакона оценивается тем или иным количеством международных единиц; такой материал следует осторожно удалить при помощи подходящего растворителя, а полученный раствор довести до точного объема.
Международные химические стандартные образцы не не имеют единиц биологической активности. При количественном определении биологической активности таких препаратов последнюю выражают в эквивалентных весовых единицах чистого вещества.
БИОЛОГИЧЕСКИЕ МЕТОДЫ
167
166 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Используют чашки Петри или прямоугольные лотки, заполненные на глубину 3—4 мм, если нет других указаний в частной статье, культуральной средой, предварительно засеянной соответствующей культурой чувствительного тест-орга-низма, приготовленного, как описано ниже. Питательный агар может состоять из двух отдельных слоев, из которых только верхний слой может быть засеян. Концентрация тест-организ-ма должна быть подобрана таким образом, чтобы получить четкие зоны угнетения в соответствии с ответами на дозу с различными концентрациями стандарта. Если посев готовят, как описано ниже, засеянная среда обычно содержит 1 мл посевного материала на 100 мл культуральной среды. Если культура состоит из суспензии вегетативных клеток, температура расплавленной для засева агаровой среды не должна превышать 48—50° С. 'Следует специально отобрать чашки и лотки с плоским дном; во время заполнения их устанавливают на ровной горизонтальной поверхности для обеспечения одинаковой толщины слоя среды. При использовании некоторых тест-организмов методику можно усовершенствовать, перед употреблением высушивая засеянные лотки при комнатной температуре в течение 30 мин или помещая их в холодильник при 4°C на несколько часов.
Для нанесения испытуемого раствора на поверхности засеянной среды расставляют небольшие стерильные цилиндры одинакового размера высотой приблизительно 10 мм и внутренним диаметром около 5 мм, из стекла, фарфора или нержавеющей стали. Вместо цилиндров иногда в слое среды стерильным бором делают отверстия диаметром 5— 8 мм. Можно использовать и другие методы нанесения испытуемого раствора. Испытуемый раствор надо располагать так, чтобы избежать перекрывания (совпадения) зон.
Растворы стандартного образца известной концентрации и соответствующие растворы испытуемого вещества, имеющие концентрации предположительно того же порядка, готовят в стерильном буферном растворе с подходящим значением pH. Для оценки правильности количественного определения необходимо использовать не менее 3 различных доз стандартного образца вместе с равным количеством доз испытуемого вещества, имеющих предположительно ту же активность, что и растворы стандартного образца. Уровни доз должны соотноситься в геометрической прогрессии, например, готовят серию разведений в отношении 2:1. Если известно, что отношение между логарифмом концентрации антибиотика и диаметром зон угнетения является приблизительно прямолинейным
для используемой системы, рутинные анализы можно проводить, используя только 2 концентрации стандартного образца и 2 концентрации испытуемого вещества. Если в частной статье указано приготовление исходного раствора испытуемого вещества, этот раствор затем разводят, как необходимо, подходящим стерильным буферным раствором.
Если применяют прямоугольные лотки, растворы стандартного образца и испытуемого вещества предпочтительно располагать по .методу латинских квадратов. Если используют чашки Петри, растворы располагают так, чтобы на каждой чашке растворы стандартного образца и испытуемого вещества чередовались по кругу и были размещены таким образом, чтобы наивысшие концентрации стандартного образца и испытуемого вещества не были рядом. С помощью пипетки отмеривают и вносят в цилиндры или отверстия одинаковое количество растворов. Если используют отверстия, отмериваемый объем должен быть достаточным, чтобы почти доверху заполнить отверстие.
Чашки и лотки инкубируют при подходящей температуре, контролируемой с точностью ±0,5° С, приблизительно 16 ч; при помощи подходящего устройства точно измеряют диаметры или площади зон угнетения, вызванного различными концентрациями стандарта и испытуемого вещества, желательно до 0,1 мм фактического размера зоны. По результатам рассчитывают активность испытуемого образца. Ниже (см. с. 170) перечислены публикации по статистическим ме-. годам оценки биологических количественных определений.
Условия проведения количественного определения активности отдельных антибиотиков и подходящие тест-организмы указаны в соответствующих частных статьях. Решающее значение для количественного определения может иметь выбор подходящего штамма тест-организма. Для облегчения справочного поиска примеры подходящих тест-ортанизмов для ряда антибиотиков приведены в табл. 4, в которой использованы следующие обозначения штаммов.
NCTC — Национальная коллекция типов культур, Центральная лаборатория общественного здравоохранения, Колиндейл, Лондон, Англия
NCYC — Национальная коллекция дрожжевых культур, Фонд научных исследований пивоваренной промышленности, Натфилд, Ред-Хилл, Суррей, Англия
АТСС — Американская коллекция типов культур, Роквилл, штат Мэриленд 20852, США.
Можно использовать другие подходящие штаммы тест-организмов. Дополнительная информация об источниках
168
ТАБЛИЦА 4. ТЕСТ-ОРГАНИЗМЫ И УСЛОВИЯ ОПРЕДЕЛЕНИЯ ОТДЕЛЬНЫХ АНТИБИОТИКОВ
Антибиотик Тест-организм Культуральная среда: конечное значение pH 1 Фосфатный буферный раствор стерильный, рНа, ИР Концентрация (масса или ME в 1 мл)6 Температура инкубации (‘С,}
Бацитрацин Цефалексин Цефалотин Хлортетрацик-лин Клоксациллин Диклоксацил-лин Эритромицин Неомицин Micrococcus luteus NCTC 7743; АТСС 10240 Micrococcus luteus NCTC 7743 ATCC 10240 Staphylococcus aureus NCTC 6571 ATCC 9144 Staphylococcus aureus ATCC 6538-P Staphylococcus aureus NCTC 6571 ATCC 9144 Staphylococcus aureus ATCC 6538-P Bacillus pumilus NCTC 8241 ATCC 14884 Bacillus cereus ATCC 11778 Bacillus subtilis NCTC 8236 ATCC 11774 Staphylococcus aureus ATCC 6538-P Staphylococcus aureus NCTC 6571 ATCC 9144 Staphylococcus aureus ATCC 6538-P Bacillus pumilus NCTC 8241 ATCC 14884 Micrococcus luteus ATCC 9341 Bacillus pumilus NCTC 8241 ATCC 14884 Kcl 7,0—7,1 Kcl 6,5—6,6 Kcl 6,5—6,6 Kcl 6,5-6,6 Kcl 6,5—6,6 Kcl 6,5—6,6 Kcl 6,0—6,6 Kcl 5,9—6,0 Kcl 6,5—6,6 Kcl 6,5—6,6 Kcl 6,6 Kcl 6,5—6,6 Kcl 8,0—8,1 Kcl 8,0—8,1 Kcl 8,0—8,1 7,0 6,0 6,0 6,0 6,0 6,0 4,5 4,5 7,0 6,0 6,0 6,0 8,0 8,0 8,0 1—4 ME 1—4 ME 10—40 мкг 10—40 мкг 0,5—2 ME 0,5—2 ME 2—20 ME 0,05—0,2 ME 5—20 мкг 2—8 мкг 2,5— 10 мкг 2—8 мкг 5—25 ME 0,5—1,5 ME 2—14 ME 35—37 30—31 32—35 32—35 32—35 32-35 37—39 30—33 37—39 32—35 37—39 32—35 37—39 35—37 37—39
169
Антибиотик Тест-организм Культуральная среда: конечное значение pH Фосфатный буферный раствор стерильный, рН°, ИР Коиценгоация (масса или ME в 1 мл)6 Температура инкубации (°C)
Staphylococcus aureus ATCC 6538-P Kcl 7,8—8,0 8,0 2—20 ME 35—37
Staphylococcus epi-dermidis ATCC 12228 Kcl 8,0—8,1 8,0 0,5—2 ME 35—37
Ноиобиоцин Bacillus subtilis NCTC 10315 Kcl 6,6—6,6 6,0 1—5 ME 30—33
Micrococcus luteus ATCC 9341 Kcl 6,5—6,6 6,0 10—50 ME 30—35
Нистатин Saccl’aromyces ce-revisiae NCYC 87 ATCC 9763 Kc3 6,0—6,2 в 25—300 ME 35—37
Оксациллин Bacillus subtilis NCTC 8236 ATCC 11774 Kcl 6,5—6,6 7,0 2,5—10 мкг 37—39
Staphylococcus aureus ATCC 6538-P Kcl 6,5—6,6 6,0 2—8 мкг 32—35
Окситетрациклин Bacillus pumilus NCTC 8241 ATCC 14884 Kcl 6,5—6,6 4,5 2—20 ME 37—39
Полимиксин В Bacillus cereus ATCC 11778 Kcl 5,9—6,0 4,5 0,5—2 ME 30—33
ЛИН Bordetella bronchi-septica NCTC 8344 ATCC 4617 Kc2 7,2—7,3 6,0; ИРЗ 20—100 ME 35—37
Bordetella bronchi-septica NCTC 8344 ATCC 4617 Kc2 7,2—7,3 7,2 50—200 ME 35—37
Escherichia coli ATCC 10536 Kcl 6,5—6,6 7,2 5—100 ME 35—37
Стрептомицин Bacillus subtilis NCTC 8236 ATCC 11774 Kcl 7,9—8,0 8,0 5—20 ME 37—39
Bacillus subtilis ATCC 6633 Kcl 8,0—8,1 8,0 3—15 ME 35—37
Тетрациклин Bacillus pumilus NCTC 8241 ATCC 14884 Kcl 6,5—6,6 4,5 2—20 ME 37—39
Bacillus cereus ATCC 11778 Kcl 5,9—6,0 4,5 0,5—2 ME 30—33
аФосфатные буферные растворы стерильные, с подходящим значением pH. Можно использовать буферные растворы, обозначенные ИР, ИР1 или ИР2.
^Область, в пределах которой могут быть найдены подходящие концентрации.
6Приготовление растворов стандартного материала и испытуемого вещества описано в частной статье и производится при помощи днметилформамида Р и стерильного фосфатного буфер а,pH 6,0» ИРЗ.
170
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
подходящих штаммов может быть получена в секции стандартизации биологических препаратов, Всемирная организация здравоохранения, Женева, Швейцария.
Точность количественного определения
Для того чтобы определить, соответствует ли данное вещество требованиям в отношении активности, указанным в частной статье, количественное определение следует, если необходимо, повторить для достижения требуемой точности. Эта точность должна быть такова, чтобы пределы достоверности (Р = 0,95) средней величины найденной активности, выраженной в процентах средней найденной активности, находились в пределах требуемого интервала, приведенного в индивидуальной статье.
Вычисление результатов
В перечисленных ниже публикациях описаны подходящие методы, которые можно использовать для статистичской оценки микробиологических количественных определений антибиотиков.
1. Международная фармакопея, издание второе, Женева, Всемирная организация здравоохранения, 1969, приложение 45: Количественное определение и испытание биологической активности.
2. С. I. Bliss: Statistics of bioassay, New York. Academic Press, 1952.
3. С. I. Bliss: Statistics in biology, vol. I., New York, McGraw Hill, 1967.
4. С. I. Bliss: Statistics in biology, vol. II, New York, McGraw Hill, 1970.
5. D. J. Finney: Statistical methods in biological assays, London, Griffin, 1964.
6. W. Hewitt: Microbiological assay, New York, Academic Press, 1977.
7. J. Philippe: Les methodes statisstiques en pharmacie et en chimie, Paris, Masson, 1967.
Методы статистической оценки микробиологических количественных определений антибиотиков также описаны во многих национальных и региональных фармакопеях.
БИОЛОГИЧЕСКИЕ МЕТОДЫ
171
Культуральные среды
Состав культуральных сред (Кс), упомянутых в табл. 4, приведен в «Списке реактивов, испытательных и титрованных растворов» (см. с. 189). В каждом случае окончательное значение pH доводится до величины, указанной в таблице.
Приготовление тест-культур
Bacillus cereus; Bacillus pumilus; Bacillus subtilis. Тест-культуру выращивают ь течение 7 сут при температуре 37—39 °C на поверхности культуральной среды Кс1 (pH 6,5— 6.6 после стерилизации), к которой добавлен 1 мкг сульфата марганца Р на 1 мл. Культуру, состоящую в основном из спор, смывают стерильной водой, нагревают в течение 30 мин при 70° С и разводят соответствующим образом, например, до содержания 107—108 спор в 1 мл. Суспензию спор можно хранить в течение длительного времени при температуре не выше 4° С.
Bordetella bronchiseptica. Тест-культуру выращивают в течение ночи на культуральной среде Кс2 (pH 6,5—6,6 после стерилизации) при температуре 35—37 °C. Суспензию готовят смыванием культуры и разведением изотоническим стерильным раствором ИР или водой до определенной степени мутности взвеси, такой, например, при которой слой толщиной 1 см при 650 нм пропускает 50% падающего света. Суспензию можно хранить в течение 2 нед при температуре не выше 4 °C.
Свежеприготовленную тест-культуру можно заменить суспензией этой культуры в подходящем растворителе, таком, как стерильный пептон (1 г/л)ИР2, которая хранилась замороженной при —70 °C; перед употреблением суспензию соответствующим образом оттаивают.
Micrococcus luteus. Тест-культуру выращивают в течение ночи на культуральной среде Кс1 (pH 6,5—6,6 после стерилизации) при температуре 35—37 °C. Суспензию готовят смыванием и разведением изотоническим стерильным раствором ИР до определенной степени мутности взвеси, такой, например, при которой слой разведения (1 часть на 50) толщиной 1 см при 650 нм пропускает 80% падающего света. Суспензию можно хранить в течение 2 нед при температуре не выше 4 °C.
Свежеприготовленную тест-культуру можно заменить суспензией этой культуры в подходящем растворителе, таком, как стерильный пептон (1 г/л) ИР2, которая хранилась замороженной при —70 °C; перед употреблением суспензию соответствующим образом оттаивают.
172
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Saccharomyces cerevisiae. Тест-культуру выращивают в течение ночи на культуральной среде КсЗ (pH 6,0—6,2 после стерилизации) при температуре 35—37 °C. Суспензию готовят смыванием и разведением изотоническим стерильным раствором ИР до определенной степени мутности взвеси, такой, например, при которой слой толщиной 1 см при 650 нм пропускает 50 % падающего света. Суспензию можно хранить в течение 2 нед при температуре не выше 4 °C.
Свежеприготовленную тест-культуру можно заменить суспензией этой культуры в подходящем растворителе, таком, как стерильный пептон (1 г/л) ИР2, которая хранилась замороженной при —70 °C; перед употреблением суспензию соответствующим образом оттаивают.
Staphylococcus aureus. Тест-культуру выращивают в течение ночи на культуральной среде Кс1 (pH 6,5—6,6 после стерилизации) при температуре 35—37 °C. Суспензию готовят смыванием и разведением изотоническим стерильным раствором ИР до определенной степени мутности взвеси, такой, например, при которой слой толщиной 1 см при 650 нм пропускает 50% падающего света.
Свежеприготовленную тест-культуру можно заменить суспензией этой культуры в подходящем растворителе, таком, как стерильный пептон (1 г/л) ИР2, которая хранилась замороженной при —70 °C; перед употреблением суспензию соответствующим образом оттаивают.
ИСПЫТАНИЕ АНТИБИОТИКОВ НА СТЕРИЛЬНОСТЬ
Испытание предназначено для обнаружения загрязнения антибиотиков, предназначенных для парентерального введения или для других стерильных форм применения, живыми микроорганизмами.
Условия испытания
Испытание должно проводиться в асептических условиях в помещении, свободном от загрязнения в той мере, 'в какой этого возможно достичь с помощью дезинфицирующих агентов, бактерицидных ламп и воздушных фильтров. В процессе самого испытания бактерицидные лампы и дезинфицирующие аэрозоли применять не следует. Все манипуляции, предусмотренные испытанием, следует проводить в профильтрованном воздухе или под вытяжкой с ламинарным потоком; операторы должны быть одеты в стерильную антистатическую одежду, включающую колпак и бахилы. Давление воздуха в помещении, где проводится испытание, должно быть выше,
БИОЛОГИЧЕСКИЕ МЕТОДЫ
173
чем во внешней среде. Работу вытяжки с ламинарным потоком следует контролировать при помощи счетчика частиц, фильтра или щелевого устройства для отбора проб воздуха; работу фильтров и бактерицидных ламп также следует постоянно проверять.
Прибор для мембранной фильтрации
Подходящий прибор состоит из закрытого резервуара и вместилища, разделенных тщательно укрепленной мембраной соответствующей степени пористости. Мембраны, обычно применяемые для испытания на стерильность, имеют номинальный размер пор 0,45 мкм, диаметр около 47 мм и обеспечивают скорость потока воды 55—75 мл в минуту при давлении 90 кПа (700 мм рт. ст). Желательно весь прибор до применения собрать и простерилизовать с укрепленной в нем мембраной. Если ожидается, 'что каждая мембрана будет загрязнена культурой, следует использовать установку по меньшей мере с 2 фильтрами.
Весь воздух, входящий в фильтрующую установку, должен проходить через воздушный фильтр, удаляющий микроорганизмы.
Взятие пробы
Берут образец пробы таким образом, чтобы он был репрезентативным для испытуемого материала. Взятое количество должно быть достаточным для проведения испытаний и, в случае необходимости, повторения испытаний. При отборе пробы следует следить за тем, чтобы не нарушить стерильность материала.
Культуральная среда
Используемая для испытаний на стерильность грибов и бактерий культуральная среда должна поддерживать рост самых разнообразных микроорганизмов, аэробных и анаэробных; в том числе тех, которые обнаруживаются в производственных условиях. Для того чтобы удовлетворить этим критериям, обычно необходимо применять более чем одну культуральную среду. К средам, которые обычно дают удовлетворительные результаты, относятся жидкая меркаптоуксусная (тиогликолевая) среда (культуральная среда Кс4) и среда гидролизата соевой муки и казеина (культуральная среда Кс5). Однако можно применить любую другую среду, если подтверждена ее способность поддерживать рост микроорганизмов на том же уровне, что и указанные выше среды.
174 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Для проверки способности каждой культуральной среды поддерживать рост микроорганизмов необходимо использовать штаммы микроорганизмов с точно установленными питательными и аэробно-анаэробными потребностями в тест-культуре, содержащей лишь небольшое число организмов (менее 100). Среду следует инкубировать при той же температуре, при которой она будет использоваться в испытании на стерильность; наличие роста должно быть очевидным через 24 ч.
Каждую партию обезвоженной среды, полученной от специализированной фирмы-изготовителя, или каждую партию среды, полностью приготовленной в лаборатории, следует проверять на способность поддерживать рост, так как не каждая партия оказывается способной поддерживать рост микроорганизмов в желаемой степени^ Различия могут быть вызваны случайным присутствием в конкретной партии компонентов неудовлетворительного качества или разрушением некоторых компонентов при перегревании или чрезмерной стерилизации среды.
РЕКОМЕНДУЕМЫЕ МЕТОДИКИ
Методика мембранной фильтрации
Асептически переносят подходящее количество твердого испытуемого материала (0,3—6 г в зависимости от размера упаковки) в стерильную колбу, содержащую около 200 мл пептона (1 г/л) ИР1, закрывают колбу и вращают для быстрого растворения. Если испытуемый материал растворяется медленно или полученный раствор быстро не фильтруется, объем растворителя можно увеличить, но он не должен превышать 400 мл. Тотчас после растворения материала асептически фильтруют раствор при пониженном давлении через мембранный фильтр, смоченный стерильной водой или пептоном (1 г/л) ИР1. Для ускорения процесса фильтрования раствор можно фильтровать, используя одновременно две фильтровальные установки. Для удаления остаточного количества антибиотика из мембраны ее промывают достаточным количеством пептона (1 г/л) ИР1, к которому, если имеется указание в частной статье (для антибиотиков ряда пенициллина и цефалоспорина), прибавлено достаточное количество пенициллиназы ИР.
После завершения фильтрования мембрану асептически делят на две приблизительно равные части. Переносят одну часть мембраны в сосуд (для этого пригодна пробирка), содержащий 50—100 мл культуральной среды Кс4 (меркапто-уксусная среда), другую часть мембраны помещают в другой
БИОЛОГИЧЕСКИЕ МЕТОДЫ 157
сосуд, содержащий 50—100 мл культуральной среды Кс5 (гидролизат соевой муки и казеина).
Через определенные промежутки времени следует проводить контрольные испытания, чтобы показать, что остаточная антибиотическая активность снижена с помощью описанной выше методики промывания ниже уровня, при котором происходит рост посевного материала, содержащего 50—100 микроорганизмов, чувствительных к испытуемому антибиотику.
Методика прямого испытания
В зависимости от размера упаковки берут от 1 до 10 порций, каждая по 0,3 г испытуемого материала, и асептически переносят их в отдельные стерильные сосуды (для этого пригодны пробирки), содержащие 50—100 мл культуральной среды Кеб (меркаптоуксусная среда с пенициллиназой). Помещают порции такого же размера в другой набор отдельных стерильных сосудов, содержащих 50—100 мл культуральной среды Кс7 (гидролизат соевой муки и казеина с пенициллиназой).
Проверяют способность пенициллиназы, содержащейся в среде, инактивировать весь пенициллин в испытуемом материале; для этого добавляют в один сосуд, содержащий культуральную среду Кеб, количество материала, взятое из одной испытуемой упаковки. Затем прибавляют 1,0 мл разведения, содержащего 50—100 микроорганизмов подходящего штамма (пригоден штамм АТСС 6538-Р Staphylococcus aureus) в культуральной среде Кс4. Через 24 ч после инкубации при 30—32 °C должен наблюдаться типичный рост микроорганизмов.
Инкубирование
Инкубируют в течение 7 сут сосуды, содержащие жидкую меркаптоуксусную среду (культуральные среды Кс4 и Кеб), при 30—32 °C и сосуды, содержащие гидролизат соевой муки и казеина (среды Кс5 и Кс7) при 22—25 °C. При прямом испытании слегка встряхивают сосуды не менее одного раза в день или до полного растворения.
Если используют другие культуральные среды, то температура и длительность инкубации должны быть соответственно изменены.
Осматривают сосуды с инокулированной средой через регулярные промежутки времени и в последний день инкубации, чтобы выявить микробный рост. Если такой рост наблюдается, его следует подтвердить исследованием среды под микро
176 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
скопом. Желательно, чтобы аппарат для инкубации был снабжен регистрирующим температуру устройством.
Интерпретация результатов испытания
Если ни в одном из сосудов не отмечен рост микроорганизмов (за исключением контрольного сосуда с положительным ростом), материал отвечает требованиям испытания. Если, однако, рост отмечен, испытание следует повторить. Если при этом ни в одном из сосудов не отмечен рост микроорганизмов (за исключением контрольного сосуда с положительным ростом), материал отвечает требованиям испытания. Если в повторном испытании отмечен рост, материал не выдерживает испытания на стерильность.
Для того чтобы установить, связана ли неудача испытания с самим материалом или с возможными недостатками методики испытания, требуется компетентное заключение эксперта.
НЕСПЕЦИФИЧЕСКАЯ токсичность
Это испытание предназначено для установления отсутствия неспецифической токсичности антибиотиков, предназначенных для парентерального применения.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Для испытания берут здоровых мышей одной линии, которых ранее не использовали ни для какого опыта. Отбирают 5 мышей массой 18—22 г. Готовят раствор испытуемого вещества, как указано в частной статье. Тест-дозу вводят в хвостовую вену с постоянной скоростью; введение должно занимать 5 с. После инъекции наблюдают за мышами в течение 48 ч. Образец отвечает требованию в отношении неспецифической токсичности, если в течение 48 ч не погибнет ни одна из мышей.
Если в период наблюдения погибнет 1 или 2 мыши, повторяют опыт один раз, используя соответственно 5 или 15 мышей, здоровых, ранее не использованных ни для каких испытаний, массой 19,5—20,5 г. Образец отвечает требованию в отношении неспецифической токсичности, если во втором опыте в течение 48 ч не погибнет ни одна из мышей.
ИСПЫТАНИЕ НА ПИРОГЕННОСТЬ
Испытание на пирогенность предназначено для ограничения риска возникновения лихорадочного состояния после парентерального введения лекарственных средств. Это испыта
БИОЛОГИЧЕСКИЕ МЕТОДЫ 177
ние применяется для жидких препаратов, которые могут быть переносимы кроликами в дозе 10 мл на 1 кг массы при введении в вену, как правило, в течение не более 4 мин. Для испытания препаратов, введение которых требует предварительной подготовки или специальных условий введения, следует руководствоваться дополнительными указаниями, приведенными в частных статьях.
Животные, используемые в испытании
Используют здоровых, взрослых кроликов, предпочтительно одного вида. Каждого животного содержат отдельно при постоянной температуре (±2°C), по возможности при постоянной влажности и оберегают от раздражений, которые могут вызвать возбуждение. Животным дают неограниченное количество воды и пищи, обычно потребляемой лабораторными животными. За 1—Зсут до испытания животных, которых раньше не использовали в тестах на пирогенность, приучают к испытанию, проводят описанную ниже подготовку, но не делая инъекций.
Используют животных для испытания на пирогенность не чаще, чем один раз за 48 ч. После испытания на пирогенность, в ходе которого было отмечено повышение температуры кролика на 0,5 °C или более, или после введения кролику испытуемого вещества, которое было признано пирогенным, животное используют повторно не раньше, чем через 2 нед.
Регистрация температуры
Применяют точный медицинский термометр с ценой деления 0,1 °C, для которого установлено время, необходимое для достижения максимальной температуры, или любой другой прибор, регистрирующий температуру с такой же чувствительностью. Вводят устройство, чувствительное к температуре, в прямую кишку подопытного животного на глубину около 6 см. Если это устройство остается введенным все то время, которое необходимо для достижения максимальной температуры, кролика удерживают при помощи легкой шейной подставки, позволяющей ему занять естественную позу отдыха. Если используют термометр, то прежде, чем снять показания, выдерживают время, достаточное для достижения максимальной температуры и установленное предварительно.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Испытание проводят в помещении, где содержатся животные, или в других аналогичных условиях. За два часа перед испытанием и во время испытания животные не получают 12—2025
178
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
корма, но могут получать воду. Животные должны быть помещены в условия опыта по крайне мере за 1 ч до инъекции.
Перед испытанием, за 40 мин до введения испытуемого материала, дважды с интервалом в 30 мин определяют температуру тела каждого животного. Средняя из двух температур служит «контрольной температурой» животного. Контрольная температура, отмеченная для каждого кролика, представляет собой температуру, от которой отсчитывается подъем температуры после инъекции.
Для любого испытания используют только тех животных, контрольная температура которых колеблется не более чем на 1 °C. Тех животных, у которых 2 температуры, измеренные для определения контрольной температуры, отклоняются более чем на ±0,2 °C от средней величины, не следует использовать в испытании; также непригодны для испытания животные с контрольной температурой ниже 38,0 °C или выше 39,8 °C.
Шприц, иглы и изделия из стекла освобождают от пирогенных агентов нагреванием при 250 °C в течение не менее 30 мин или любым другим подходящим способом. Испытуемый раствор нагревают примерно до 38 °C.
Вводят в ушную вену каждого из 3 кроликов 10 мл раствора на 1 кг массы животного или количество, указанное в частной статье. На введение должно уходить не более 4 мин, если в частной статье нет других указаний.
После введения отмечают температуру животного в течение 3 ч, проводя измерения непрерывно или через каждые 30 мин. Максимальная температура, отмеченная для каждого кролика, рассматривается как ответ животного на введение препарата; если все температурные показания после введения ниже контрольной температуры, ответ считается нулевым повышением температуры.
Испытуемый препарат отвечает требованиям на отсутствие пирогенности, если ни у одного из кроликов «е наблюдается повышения температуры на 0,6° С или более по сравнению с соответствующей контрольной температурой и если сумма повышения температур у трех кроликов не превышает 1,4 °C. Если у одного или двух кроликов наблюдается повышение температуры на 0,6 °C или более и если сумма повышения их температур превышает 1,4 °C, повторяют испытание, используя 5 других кроликов. Если повышение температуры на 0,6 °C или более наблюдается не более чем у трех из восьми кроликов и если сумма повышения температур у восьми животных не превышает 3,7 °C, препарат выдерживает испытание на отсутствие пирогенности.
БИОЛОГИЧЕСКИЕ МЕТОДЫ
179
РЕКОМЕНДУЕМАЯ МЕТОДИКА
ИСПЫТАНИЕ НА ГИСТАМИНОПОДОБНЫЕ ВЕЩЕСТВА (ВАЗОДЕПРЕССИВНЫЕ ВЕЩЕСТВА)
Испытание на вазодепрессивные вещества проводится на кошках путем сравнения снижения артериального давления, вызванного испытуемым раствором, со снижением, обусловленным введением раствора гистамина. Используют здоровых взрослых кошек — самцов или небеременных самок.
Определяют массу животного и дают ему общий наркоз, вводя хлоралгидрат Р или подходящий барбитурат, поддерживающий равномерное кровяное давление. Защищают животное от охлаждения и поддерживают в таком состоянии, при котором ректальная температура остается в физиологических пределах. Вводят трубку в трахею. Хирургическим путем открывают сонную артерию и атравматично отделяют ее полностью от всех подлежащих структур, в том числе от блуждающего нерва. Вводят в артерию канюлю, заполненную гепаринизированным физиологическим раствором ИР, и соединяют ее с ртутным манометром или другим устройством, позволяющим постоянно регистрировать кровяное давление. Хирургически выделяют яремную или бедренную вену и вставляют вторую канюлю, которая заполнена гепаринизированным физиологическим раствором ИР и через которую можно вводить растворы гистамина и испытуемого вещества.
Определяют чувствительность животного к гистамину следующим образом. Включают кимограф или подобное регистрирующее устройство и контролируют запись амплитуды колебаний и относительной устойчивости кровяного давления. Вводят в яремную или бедренную вену раствор гистамина ИР в дозах 0,05 мкг (доза А), 0,1 мкг (доза Б), повторенных не менее 3 раз, и 0,15 мкг (доза В) гистамина основания на 1 кг массы животного. Вводят вторую и последующие дозы не менее чем через 1 мин после возвращения кровяного давления к уровню, зарегистрированному сразу после предшествующего введения. Повторяют эту серию введений до тех пор, пока не будет получено для доз Б гистамина относительно равномерное снижение давления; при этом результаты первой серии введений не принимаются в расчет. Животное можно использовать для испытания только в том случае, если снижение давления, вызванное дозами Б, составляет не менее 2,7 кПа (20 мм рт. ст.) и, кроме того, если доза А вызывает меньший ответ, чем доза Б, а доза В дает больший ответ, чем доза Б.
12
180
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Готовят испытуемый раствор, как описано в частной статье. Во время испытания следят за тем, чтобы скорость введения испытуемого и стандартного растворов была равномерной и одинаковой. Если растворы вводят в яремную вену, необходимо обратить внимание на то, чтобы испытуемый и стандартный растворы гистамина вводились в разных объемах во избежание влияния объема на кровяное давление. Если для стандартного и испытуемого растворов используется общая канюля, необходимо тотчас после введения каждого из растворов вводить приблизительно 2,0 мл изотонического раствора ИР для промывания трубки.
Вводят дозу Б стандартного раствора, затем указанное количество испытуемого раствора, а затем другую дозу Б стандартного раствора. Вторую и третью дозы вводят не менее чем через 1 мин после того, как кровяное давление возвратится к уровню, отмеченному непосредственно перед предшествующим введением. Если ответ на испытуемый раствор больше, чем ответ, ранее полученный на дозу А, повторяют серию введений дважды и заканчивают испытание, вводя дозу В стандартного раствора. Если ответ на дозу В не превышает ответ на дозу Б, испытание считают недействительным.
Животное можно использовать для испытаний до тех пор, пока оно остается относительно устойчивым и реагирующим на гистамин при условии, что а) введение испытуемого вещества не вызывает большего снижения, чем снижение, вызванное дозой В, и б) ответ на дозу В стандартного раствора, введенного после испытуемого вещества, не будет ниже среднего ответа на ранее введенные дозы Б.
Вещество выдерживает испытание, если ответ или средняя величина ответов после введения количества, указанного в частной статье, меньше средней величины соответствующих ответов на дозу Б стандартного раствора (0,1 мкг гистамина основания на 1 кг массы животного) и ни одна из отдельных доз испытуемого раствора не вызывает снижения давления больше, чем доза В стандартного раствора (0,15 мкг гистамина основания на 1 кг массы животного).
ФАРМАКОГНОСТИЧЕСКИЕ МЕТОДЫ
ОПРЕДЕЛЕНИЕ ЗОЛЫ И ЗОЛЫ, НЕ РАСТВОРИМОЙ В КИСЛОТЕ
РЕКОМЕНДУЕМАЯ МЕТОДЙКА
Определение золы
Помещают около 3 г измельченного материала (точная навеска) или количество, указанное в частной статье, в подходящий тигель (например, из кварца или платины), предварительно прокаленный, охлажденный и взвешенный. Сжигают вещество, постепенно повышая температуру нагревания, но не превышая 450 °C, до полного сгорания; охлаждают и взвешивают. Если таким образом не удается достичь полного сгорания, обугленную массу обрабатывают горячей водой, собирают остаток на беззольный фильтр, сжигают остаток вместе с фильтром, прибавляют фильтрат, выпаривают досуха и сжигают при температуре не выше 450 °C. Рассчитывают содержание золы в миллиграммах на 1 г воздушно-сухого сырья.
Определение золы, не растворимой в кислоте
Кипятят золу в течение 5 мин с 25 мл соляной кислоты (70 г/л) ИР; собирают нерастворившееся вещество на стеклянный или беззольный фильтр, промывают горячей водой и сжигают до постоянной массы при температуре около 500 °C. Рассчитывают содержание золы, не растворимой в кислоте, в миллиграммах на 1 г воздушно-сухого сырья.
МЕЖДУНАРОДНЫЕ ХИМИЧЕСКИЕ СТАНДАРТНЫЕ ОБРАЗЦЫ
РЕАКТИВЫ. РАСТВОРЫ
МЕЖДУНАРОДНЫЕ ХИМИЧЕСКИЕ СТАНДАРТНЫЕ ОБРАЗЦЫ
Химический стандартный образец — аутентичный гомогенный материал, предназначенный для использования в специальных химических, физических и иногда биологических испытаниях, в ходе которых его свойства сравниваются со свойствами исследуемого продукта; образец обладает степенью чистоты, достаточной для его предполагаемого применения.
Ниже перечислены аналитические методы, которые используются в настоящее время в спецификациях для фармацевтических веществ и могут требовать применения химических стандартных образцов:
— инфракрасная спектрофотометрия для установления подлинности или для количественного анализа;
— количественные методы, основанные на ультрафиолетовой абсорбционной спектрофотометрии;
• — количественные методы, основанные на получении окрашивания и измерения его интенсивности при помощи инструментальных методов или визуальным сравнением;
— методы, основанные на хроматографическом разделении для установления подлинности или для количественного анализа;
— количественные методы (в том числе автоматизированные), основанные на разделении, зависящем от распределения испытуемого материала между фазами растворителя; точная эффективность экстракционной методики может зависеть от окружающих условий, которые могут быть различными в разных лабораториях;
— количественные методы, часто титриметрические, но иногда весовые, основанные на нестехиометрическом отношении продуктов реакции;
— методы количественного определения, основанные на измерении оптического вращения;
— методы, основанные на полярографии;
— методы, основанные на флуоресцентной спектрофотометрии;
• — микробиологические количественные методы, основанные на измерении степени угнетения роста микроорганизмов антибиотиками, когда стандартный образец может быть соответствующим образом охарактеризован химическими и физическими методами (см. раздел «Количественное определе
188
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
ние микробиологической активности антибиотиков», с. 166— 172).
Подробное обсуждение вопросов, касающихся разработки, хранения и распределения химических стандартных образцов, содержится в приложении 3 к Двадцать пятому докладу Комитета экспертов ВОЗ по спецификациям для фармацевтических препаратов (Серия технических докладов ВОЗ, № 567, 1976).
Международные химические стандартные образцы, необходимые для спецификаций Международной фармакопеи, указаны в частных статьях.
Международные химические стандартные образцы устанавливаются Сотрудничающим центром ВОЗ по химическим стандартным образцам, Сульна, Швеция. Характеристики отобранных веществ определяются Центром в сотрудничестве со специалистами, назначенными ВОЗ.
Международные химические стандартные образцы могут быть получены в Сотрудничающем центре ВОЗ по химичес-ским стандартным образцам (WHO Collaborating Centre for Chemical Reference Substances, Apotekens Centrallaboratorium, Box 3045, 17103, Soina 3, Sweden). К каждой упаковке Международного стандартного образца прилагается листовка с аналитическими спецификациями для данного вещества и указанием, с какой целью разработан этот стандартный образец. Дополнительная информация, касающаяся аналитической методологии, может быть получена из Центра по запросу.
НАИМЕНОВАНИЯ, СИМВОЛЫ И ОТНОСИТЕЛЬНЫЕ АТОМНЫЕ МАССЫ НЕКОТОРЫХ ЭЛЕМЕНТОВ
Относительные атомные массы (ранее употреблялся термин атомные веса), приведенные ниже, определены относительно изотопа 12С, принятого точно за 12, до 4 значащих цифр. В список включены только те элементы, которые наиболее часто встречаются в фармацевтическом анализе.
Большинство элементов имеет не один, а несколько встречающихся в природе изотопов, и различия в относительной встречаемости этих изотопов влияют на точность, с которой могут быть приведены относительные атомные массы элементов в природе. Среди элементов, приведенных в списке, относительные атомные массы образцов бора, кальция, свинца, стронция и серы могут отличаться более чем на единицу в четвертой значащей цифре.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
189
Наименование Символ Относительная атомная масса Наименование Символ Относительная атомная масса
Азот N 14,01 Молибден Мо 95,94
Алюминий А1 26,98 Мышьяк As 74,92
Барий Ва 137,3 Натрий Na 22,99
Бор В 10,81 Никель Ni 58,71*
Бром Вг 79,90 Олово Sn 118,7*
Ванадий V 50,94* Платина Pt 195,1*
Водород Н 1,008 Ртуть Hg 200,6*
Вольфрам W 183,85* Рутений Ru 101,1*
Висмут Bi 209,0 Свинец Pb 207,2
Гелий Не 4,003 Селен Se 78,96*
Гольмий Но 164,9 Сера S 32.06
Железо Fe 55,85* Серебро Ag 107.9
Золото Au 197,0 Стронций Sr 87,62
йод I 126,9 Сурьма Sb 121,75*
Кадмий Cd 112,4 Титан Ti 47,90*
Калий К 39,10* Торий Th 232,0
Кальций Ca 40,08 Углерод c 12,01
Кислород О 16,00* Уран и 238,0 .
Кобальт Co 58,93 Фосфор p 30,97
Кремний Si 28,09* Фтор F 19,00
Лантан La 138,9 Хлор Cl 35,45
Литий Li 6,941* Хром Cr 52,00
Магний Mg 24,31 Церий Ce 140,1
Марганец Mn 54,94 Цинк Zn 65,38
Медь Cu 63,55* Цирконий Zr 91,22
* Величины считаются достоверными до ±1 в последней цифре или до ±3, если помечены звездочкой.
СПИСОК РЕАКТИВОВ, ИСПЫТАТЕЛЬНЫХ И ТИТРОВАННЫХ РАСТВОРОВ
Ниже описаны реактивы, испытательные и титрованные растворы, упомянутые в Международной Фармакопее, третье издание, том 1. Реактивы обозначены буквой Р, испытательные растворы—-сокращением ИР, титрованные растворы или растворы, аналогично стандартизуемые, — сокращением ТР. Реактивы, имеющие одинаковое название, но различающиеся по составу, чистоте и т. п., обозначаются цифрами после соответствующего сокращения. Обозначения AsP, AsHP, С1ИР, FeHP и РЬИР относятся к реактивам приемлемой степени чистоты для применения в испытаниях на мышьяк, хлориды, железо и тяжелые металлы соответственно. Обозначение ИК относится к реактивам, пригодным для применения в спектрофотометрии в инфракрасной области. Сокращением Кс обозначены культуральные среды для микробиологических испы
190
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
таний. Концентрации растворов выражены в соответствии с Международной системой единиц (СИ) и относятся к безводному веществу. Ссылка SRIP указывает на сборник Specifications for reagents mentioned in the International Pharmacopoeia (World Health Organization, Geneva, 1963). Обозначения, ранее использованные в SRIP, приведены в квадратных скобках. Обозначение d означает относительную плотность Д2О2о, т. е. плотноссть, измеренную при 20 °C в воздухе по отношению к воде при 20 °C.
Агар Р. (SRIP, 1963, с. 27).
Азот Р. N2 (SRIP, 1963, с. 129).
Азотная кислота (~ 1000 г/л) ИР {азотная кислота (70%)Р] (SRIP, 1963, с. 125); 1,41.
Азотная кислота (~130 г/л) ИР.
Методика приготовления. Разводят 130 мг азотной кислоты (~ 1000 г/л) ИР водой до получения 1000 мл (приблизительно 2 моль/л); d~ 1,07.
Азотная кислота (15 г/л) ИР. Азотная кислота (~1000 мл/л) ИР, разведенная водой до содержания HNO3 15,0 г/л.
Азотная кислота (~3 г/л) ИР. Азотная кислота (~ 1000г/л) ИР, разведенная водой до содержания HNO3 3,0 г/л.
Алюминия гидроокись Р. Водный А1(ОН)3.
Описание. Белый поршок без запаха.
Растворимость. Практически нерастворим в воде и этаноле (~750 г/л) ИР.
Аммиак (~260 г/л) ИР {аммиак концентрированный Р]. (SRIP, 1963, с. 31); d~ 0,894.
Аммиак (~100 г/л) ИР. Аммиак (~260 г/л) ИР, разведенный до содержания NH3 около 100 г/л (приблизительно 5 моль/л); d~0,956.
Аммиак (~100 г л) для испытания на железо FeHP. Аммиак (~ 100 г/л) ИР, который выдерживает следующее испытание: выпаривают 5 мл почти досуха на водяной бане, прибавляют 40 мл воды, 2 мл раствора лимонной кислоты (200 г/л) FeHP и 2 капли меркаптоуксусной кислоты Р, смешивают, подщелачивают аммиаком (~100 г/л) FeHP и разводят водой до 50 мл; не должно появляться розовое окрашивание.
Аммиак (~100 г/л) для испытания на свинец РЬИР. Аммиак (~ 100 г/л) ИР, который выдерживает следующее испытание: выпаривают 5 мл аммиака (~100 г/л) ИР досуха на водяной бане, прибавляют к остатку 1 мл соляной кислоты
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
191
(~70 г/л) ИР и выпаривают досуха. Растворяют остаток в 2 мл уксусной кислоты (~60 г/л) РЬИР, разводят водой до 25 мл и проводят испытание на тяжелые металлы. Аналогично готовят контрольный раствор. Предел содержания тяжелых металлов 2 мкг/мл.
Аммиачный буферный раствор ИР.
Методика приготовления. Растворяют 67,5 г хлорида аммония Р в 570 мл аммиака (~260 г/л) ИР и разводят водой до 1000 мл.
Аммония ацетат Р. C2H7NO2 (SRIP, 1963, с. 32).
Аммония ацетата раствор (80 г/л) ИР. Раствор ацетата аммония Р, содержащий C2H7NO2 около 77 г/л (приблизительно 1 моль/л).
Аммония карбонат Р. (МН^гСОз (SRIP, 1963, с. 33).
Аммония молибдат Р. (NH4)6Mo7O24-4H2O (SRIP, 1963, с. 34).
Аммония молибдата раствор (95 г/л) ИР. Раствор молибдата аммония Р, содержащий (NH4)6Mo7O24 около 95 г/л.
Аммония оксалат Р. C2H8N2O4-H2O (SRIP, 1963, с. 36).
Аммония оксалата раствор (25 г/л) ИР. Раствор оксалата аммония Р, содержащий C2H8N2O4 около 27 г/л.
Аммония хлорид Р. NH4CI (SRIP, 1963, с. 33).
Аммония хлорида буферный раствор, pH 10,0, ИР. Буферная смесь с pH 10,0.
Методика приготовления. Растворяют 7,0 л хлорида аммония Р в 57 мл аммиака (~260 г/л) ИР и разводят водой до получения 100 мл.
Аммония хлорида раствор (10 мкг/мл NH4) ИР.
Методика приготовления. Растворяют 0,296 г (точная навеска) хлорида аммония Р в воде до получения 1000 мл. Разводят 10 мл этого раствора до 100 мл.
Срок годности. Используют раствор в течение 2 нед после приготовления.
Аммония тиоцианат Р. NH4SCN (SRIP, 1963, с. 40).
Аммония тиоцианата раствор (~75 г/л) ИР. Раствор тиоцианата аммония Р, содержащий NH4SCN около 75 г/л (приблизительно 1 моль/л).
Аммония тиоцианата раствор (0,1 моль/л) ТР. Тиоцианат аммония Р, растворенный в воде до содержания 7,612 г NH4SCN в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,1 моль/л раствора следующим образом: помещают
192
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
30,0 мл раствора нитрата серебра (0,1 моль/л) ТР в колбу с притертой пробкой. Разводят 50 мл воды, прибавляют 2 мл азотной кислоты (~ 1000 г/л) ИР и затем титруют раствором тиоцианата аммония до первого появления красно-коричневого окрашивания, используя в качестве индикатора 2 мл раствора сульфата аммония железа(III) (45 г/л) ИР.
Аммония тиоцианата раствор (0,01 моль/л) ТР. Тиоцианат аммония Р, растворенный в воде до содержания 0,7612 г NH4SCN в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора тиоцианата аммония (0,1 моль/л) ТР.
Ацетатный буферный раствор, pH 3,3, ИР. Буферная смесь с pH 3,0.
Методика приготовления. Растворяют 12 г ацетата натрия Р в воде, прибавляют 6 мл ледяной уксусной кислоты Р и разводят водой до получения 100 мл.
Ацетон Р. С3Н6О (SRIP, 1963, с. 27).
Ацетонитрил Р. Метилцианид C2H3N.
Описание. Прозрачная бесцветная жидкость. Смешиваемость. Легко смешивается с водой.
Ацетонитрила раствор (400 г/л) ИР.
Методика приготовления. Смешивают 1 объем ацетонитрила Р с одним объемом воды. Полученный раствор содержит C2H3N около 400 г/л.
Бария нитрат Р. Ba(NO3)2 (SRIP, 1963, с. 47).
Бария нитрата раствор (0,01 моль/л) ТР. Нитрат бария Р, растворенный в воде до содержания 2,614 г Ва(МОз)2 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,01 моль/л раствора следующим образом: помещают 10,0 мл серной кислоты (0,01 моль/л) ТР в колбу, разводят 40 мл воды, прибавляют 2 капли раствора торина (2 г/л) ИР и 2 капли раствора хлорида метилтиониния (0,2 г/л) ИР и медленно титруют раствором нитрата бария до изменения желтой окраски на розовую.
Бария окись Р. ВаО.
Описание. Кусочки или порошок от белого до желтовато-белого цвета. На воздухе поглощает влагу и углекислый газ.
Хранение. Хранят в плотно укупоренной таре.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
193
Бария сульфата суспензия ИР.
Методика приготовления. Смешивают 15 мл раствора хлорида бария (0,5 моль/л) ТР с 55 мл воды и 20 мл не содержащего сульфатов этанола (~750 г/л) ИР, прибавляют 5 мл раствора сульфата калия (174 мг/Л) ИР и разводят водой до получения 100 мл.
Примечание. Суспензия сульфата бария ИР должна быть свежеприготовленной.
Бария хлорид Р. ВаС12-2Н2О (SRIP, 1963, с. 45).
Бария хлорида раствор (50 г/л) ИР. Раствор хлорида бария Р, содержащий около 52 г ВаС12 (приблизительно 0,25 моль/л).
Бария хлорида раствор (0,5 моль/л) ТР. Хлорид бария Р, растворенный в воде до содержания 104,2 г ВаС12 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,5 моль/л раствора следующим образом; помещают 10,0 мл серной кислоты (0,5 моль/л) ТР в колбу, разводят 40 мл воды, прибавляют 2 капли раствора торина (2 г/л) ИР и медленно титруют раствором хлорида бария до появления красноватой окраски.
Бензилпенициллин натрий Р. Ci6Hi7N2NaO4S. Содержит не менее 96,0 и не более 102,0% Ci6Hi7N2NaO4S в пересчете на высушенное вещество.
Описание. Белый или почти белый кристаллический порошок; без запаха или с легким характерным запахом.
Растворимость. Растворим примерно в 0,5 части воды; практически нерастворим в хлороформе Р и эфире Р.
Бензилпенициллина натрия раствор ИР.
Методика приготовления. Растворяют 0,03 г бензилпенициллина натрия Р в достаточном количестве фосфатного буфера, pH 7,0, ИР до получения 10 мл. Этот раствор содержит бензилпенициллина натрия не менее 3 мг/мл.
Бихромата раствор концентрированный окрашенный ИР.
Методика приготовления. Растворяют 6,0 г бихромата калия Р в 120 мл серной кислоты (~10 г/л) ИР, фильтруют раствор, если необходимо, и определяют концентрацию КгСг2О7.
Количественное определение. Разводят 5,0 мл водой до получения 50 мл. Помещают 10,0 мл этого раствора в колбу с притертой пробкой, прибавляют 10 мл воды, 2 г бикарбоната калия Р и 20 мл серной кислоты (~100 г/л) ИР. Слегка прикрывают колбу пробкой. Когда выделение газа
13—2025
194 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
прекратится, прибавляют 1 г йодида калия Р, помещают колбу на 5 мин в темное место и титруют выделившийся йод раствором тиосульфата натрия (0,1 моль/л) ТР, используя в качестве индикатора раствор крахмала ИР. Каждый миллилитр раствора тиосульфата натрия (0,1 моль/л) ТР соответствует 4,904 мг К2СГ2О7.
Бихромата раствор окрашенный ИР. Раствор, содержащий К2СГ2О7 4,904 г/л.
Методика приготовления. Готовят раствор, содержащий 490,35 мг К2СГ2О7 в 100 мл путем разведения концентрированного окрашенного раствора бихромата ИР серной кислотой ('-'10 г/л) ИР, как требуется.
Борная кислота Р. Н3ВО3. Содержит не менее 99,0 % Н3ВО3. Описание. Белые кристаллы, блестящие чешуйки или белый кристаллический порошок.
Растворимость. Растворим в 20 частях воды, в 3 частях кипящей воды и в 16 частях этанола (~ 750 г/л) ИР.
Нерастворимые в воде вещества. 1,0 г растворяется в 30 частях воды; полученный раствор прозрачный и бесцветный.
Нерастворимые в этаноле вещества. 1,0 г растворяется в 10 частях кипящего этанола ('-'750 г/л) ИР; полученный раствор лишь слегка мутный.
Количественное определение. Растворяют около 1 г (точная навеска) в 30 мл воды; прибавляют 50 мл глицерина Р, предварительно нейтрализованного по раствору фенолфталеина в этаноле ИР, и титруют раствором гидроокиси натрия (1 моль/л), не содержащим карбонатов, ТР, используя в качестве индикатора раствор фенолфталеина в этаноле ИР. Каждый миллилитр раствора гидроокиси натрия (1 моль/л), не содержащего карбонатов, ТР соответствует 61,83 мг Н3ВО3.
Борной кислоты раствор (50 г/л) ИР. Раствор борной кислоты Р, содержащий Н3ВО3 около 50 г/л.
Бром Р. Вг2 (SRIP, 1963, с. 51).
Брома раствор ИР1. Насыщенный раствор брома Р.
Брома раствор для испытания на мышьяк As ИР.
„ Методика приготовления. Растворяют 30 г бромида калия Р в 40 мл воды, прибавляют 30 г брома Р и разводят водой до получения 100 мл. Раствор выдерживает следующее испытание: выпаривают 10 мл почти досуха на водяной бане, прибавляют 50 мл воды, 10 мл соляной кислоты ('—250 г/л), AsHP и хлорид олова AsHP в количестве, ; достаточном для восстановления оставшегося брома, а за
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ 195
тем проводят испытание на мышьяк. Окраска полученного пятна не должна быть более интенсивной, чем окраска пятна, полученного с 1 мл стандартного раствора мышьяка, что указывает на содержание мышьяка не более 1 мкг/мл.
Бромтимоловый синий Р. C27H28Br2O5S (SRIP, 1963, с. 53).
Бромтимолового синего раствор в этаноле ИР.
Методика приготовления. Нагревают 0,1 г бромтимолового синего Р с 3,2 мл раствора гидроокиси натрия (0,05 моль/л) ТР и 5 мл этанола (~710 г/л) ИР; после получения раствора прибавляют этанол (-~ 150 г/л) ИР в количестве, достаточном для получения 250 мл.
Бромфеноловый синий Р. Ci9Hi0Br4O5S (SRIP, 1963, с. 52).
Бромфенолового синего раствор в этаноле ИР.
Методика приготовления. Нагревают 0,1 г бромфенолового синего Р с 3,2 мл раствора гидроокиси натрия (0,05 моль/л) ТР и 5 мл этанола (~710 г/л) ИР; после получения раствора прибавляют этанол (~150 г/л) ИР в количестве, достаточном для получения 250 мл.
Вода, свободная от углекислоты, Р. Вода, подвергшаяся энергичному кипячению в течение нескольких минут и защищенная от воздуха во время охлаждения и хранения.
Гелий Р. Не. Содержит не менее 999,95 мл/л Не.
Гепаринизированный изотонический раствор ИР. Стерильный изотонический раствор хлорида натрия ИР, содержащий 50 Международных единиц гепарина в 1 мл.
Гистамина раствор концентрированный ИР. Раствор, содержащий 1,00 г/л гистамина основания.
Методика приготовления. Растворяют 138,1 мг (точная навеска) фосфата гистамина Р или 82,8 мг (точная навеска) дигидрохлорида гистамина Р в воде до получения 50,0 мл. Хранение. Концентрированный раствор гистамина ИР следует хранить при температуре не выше 4—10 °C, в склянках темного стекла с притертой пробкой, защищенных от действия света.
Срок годности. Не более 30 сут.
Гистамина раствор ИР. Раствор, содержащий 1,0 мг/л гистамина основания.
Методика приготовления. Готовят раствор гистамина ИР путем разведения концентрированного раствора гистамина ИР достаточным количеством изотонического раствора ИР. Примечание. Раствор гистамина ИР должен быть свежеприготовленным.
14—2025
196 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Гистамина дигидрохлорид Р. C5H9N3-2HC1. Содержит не менее 98,0 и не более 101,0% C8H9N3-2HC1 в пересчете на высушенное вещество.
Описание. Бесцветные кристаллы или белый кристаллический порошок; без запаха.
Растворимость. Легко растворим в воде и метаноле Р; растворим в этаноле ( — 750 г/л) ИР.
Температурный интервал плавления. 244—246 °C.
Потеря при высушивании. Не более 5,0 мг/г.
Количественное определение. Растворяют около 0,15 г (точная навеска) в 10 мл воды. Прибавляют 5 мл хлороформа Р и 25 мл этанола ( — 750 г/л) ИР. Титруют раствором гидроокиси натрия (0,2 моль/л), не содержащим карбонатов, ТР, используя в качестве индикатора 0,5 мл раствора тимолфталеина в этаноле ИР. Каждый миллилитр раствора гидроокиси натрия (0,2 моль/л), не содержащего карбонатов, ТР соответствует 9,21 мг CsH9N3-2HCl.
Гистамина фосфат Р. CsH9N3-2H3PO4. Содержит не менее 98,0 и не более 101,0% CsH9N3-2H3PO4 в пересчете на безводное вещество.
Описание. Бесцветные, длинные, призматические кристаллы; без запаха; устойчивые на воздухе.
Растворимость. Растворим примерно в 5 частях воды; слабо растворим в этаноле ( — 750 г/л) ИР.
Температура плавления. Около 132 °C.
Вода. Определяют методом К. Фишера, используя около 1,0 г; содержание воды 50—60 мг/г.
Количественное определение. Растворяют около 0,15 г (точная навеска) в 10 мл воды. Прибавляют 5 мл хлороформа Р и 25 мл этанола ( — 750 г/л) ИР. Титруют раствором гидроокиси натрия (0,2 моль/л), не содержащим карбонатов, ТР, используя 0,5 мл раствора тимолфталеина в этаноле ИР в качестве индикатора. Каждый миллилитр раствора гидроокиси натрия (0,2 моль/л), не содержащего карбонатов, ТР соответствует 15,36 мг C5H9N3-2H3PO4.
Глиоксаль бис(2-оксианил) Р. 2,2'-(Этандиилидендинит-рил) дифенол Ci4Hi2N2O2.
Описание. Белые кристаллы.
Растворимость. Растворим в горячем этаноле ( — 750 г/л) ИР.
Температурный интервал плавления. 203—205 °C.
Глиоксаля бис(2-оксианила) раствор ИР. Раствор гликосаля бис(2-оксианила) в этаноле ( — 750 г/л) ИР, содержащий Ci4H12N2O2 около 10 г/л.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ 197
Глицерин Р. Пропан-1,2,3-триол с небольшим количеством воды С3Н8О3. Содержит С3Н8О3 не менее 970 г/кг.
Описание. Прозрачная, почти бесцветная, сиропообразная, гигроскопичная жидкость; без запаха.
Смешиваемость. Смешивается с водой и этанолом ( — 750 г/л) ИР; практически не смешивается с эфиром Р и хлороформом Р.
Плотность (ргс). Не менее 1,256 кг/л.
Показатель преломления (м20п). Не менее 1,469.
Акролеин и другие восстанавливающие вещества. Смешивают 1 мл с 1 мл аммиака (— 100 г/л) ИР и нагревают на водяной бане при 60 °C в течение 5 мин; жидкость не должна окрашиваться в желтый цвет. Снимают с водяной бани и прибавляют 3 капли раствора нитрата серебра (40 г/л) ИР; жидкость не должна окрашиваться в течение 5 мин.
Сульфатная зола. Не более 0,5 мг/мл.
Глюкозы гидрат Р. Моногидрат a-D-глюкопиранозы С6Н12О6-Н2О. Содержит не менее 99,0 и не более 101,5% С8Н120б в пересчете на высушенное вещество.
Описание. Бесцветные кристаллы, белый кристаллический или гранулированный порошок; без запаха.
Растворимость. Растворим примерно в 1 части воды и примерно в 60 частях этанола ( — 750 г/л) ИР; лучше растворим в кипящей воде и кипящем этаноле ( — 750 г/л) ИР. Кислотность. Растворяют 5 г в 50 мл воды, свободной от углекислоты, Р. На нейтрализацию полученного раствора требуется не более 0,5 мл раствора гидроокиси натрия (0,02 моль/л), не содержащего карбонатов, ТР; индикатор — раствор фенолфталеина в этаноле ИР.
Удельное оптическое вращение. Растворяют 100 мг, предварительно высушенные до постоянного веса, в 1 мл воды и прибавляют несколько капель аммиака (—100 г/л) ИР; [а]о20 оС= от +52 до +53°.
Растворимый крахмал и сульфиты. Растворяют 1 г в 10 мл воды и прибавляют 1 каплю раствора йода ИР; жидкость окрашивается в желтый цвет
Потеря при высушивании. Высушивают до постоянной массы при 105 °C; потеря составляет не менее 80 и не более 100 мг/г.
Сульфатная зола. Не более 1,0 мг/г.
Количественное определение. Растворяют около 0,1 г (точная навеска) в 50 мл воды, прибавляют 30 мл раствора йода (0,1 моль/л) ТР, 10 мл раствора карбоната натрия (50 г/л) ИР и оставляют стоять на 20 мин. Прибавляют
14!
198
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
15 мл соляной кислоты (~70 г/л) ИР и титруют избыток йода раствором тиосульфата натрия (0,1 моль/л) ТР, используя в качестве индикатора раствор крахмала ИР. Проводят контрольный опыт и вносят необходимые поправки. Каждый миллилитр раствора йода (0,1 моль/л) ТР соответствует 9,008 мг С6Н12Об-
Гольмия окись Р. Но2О3. Содержит не менее 99,9% НогОз, примеси состоят из Ег2О3 и Dy2O3.
Описание. Рыжевато-коричневый порошок.
Растворимость. Нерастворим в воде.
Гольмия перхлората раствор ИР.
Методика приготовления. Растворяют 40 г окиси гольмия Р в достаточном количестве хлорной кислоты (~ 140 г/л) ИР до получения 1000 мл.
Дибромметан Р. Метиленбромид СН2Вг2.
Описание. Бесцветная или желтоватая жидкость.
Смешиваемость. Смешивается с этанолом (—750 г/л) ИР, эфиром Р и ацетоном Р.
Дикалия гидрофосфат Р. К2НРО4 (SRIP, 1963, с. 81).
1,4-Ди[2-(4-метил-5-фенилоксазол)] бензол Р. Диметил-РОРОР C26H2qN2o02. Применяется для сцинтилляционного счета.
Диметилформамид Р. C3H7NO.
Описание. Прозрачная, бесцветная жидкость с характерным запахом.
Смешиваемость. Смешивается с водой и этанолом (-750 г/л ИР.
Температурный интервал кипения. Не менее 95% перегоняется между 152 и 156 °C.
Плотность (рго) 0,945—0,947 кг/л.
Кислотность или щелочность. Растворяют 1 г в 10 мл воды, прибавляют 2 капли раствора фенолфталеина в этаноле ИР; для получения красного окрашивания требуется не более 0,2 мл раствора гидроокиси натрия (0,01 моль/л), не содержащего карбонатов, ТР. Прибавляют 0,3 мл соляной кислоты (0,01 моль/л) ТР и 5 капель раствора метилового красного в этаноле ИР; раствор окрашивается в оранжевый цвет.
Динатрия гидрофосфат безводный Р [натрия фосфат безводный Р]. Na2HPO4 (SRIP, 1963, с. 193).
Динатрия эдетат [динатриевая соль этилендиаминтетрауксус-ной кислоты (ЭДТА)] Р. CioHi4N2Na208-2H20 (SRIP, 1963, с. 82).
стандартные образцы и реактивы
199
Динатрия эдетата раствора (0,05 моль/л) ТР. Эдетат динатрия Р, растворенный в воде до содержания 16,81 г Ci0Hi4N2Na2O8 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию подходящим методом. Пригоден следующий метод: около 200 мг карбоната кальция Р2 (точная навеска) помещают в стакан емкостью 400 мл, прибавляют 10 мл воды и перемешивают до получения суспензии. Накрывают стакан часовым стеклом и вводят при помощи пипетки 2 мл соляной кислоты (~70 г/л) ИР; при этом кончик пипетки должен находиться между краем стакана и краем часового стекла. Вращают содержимое стакана для растворения карбоната кальция и смывают раствор со стенок стакана, наружной части пипетки и часового стекла водой; разводят водой примерно до 100 мл. Размешивая раствор (предпочтительно магнитной мешалкой), прибавляют около 30 мл раствора эдетата динатрия из бюретки емкостью 50 мл. Прибавляют 10 мл раствора гидроокиси натрия (~80 г/л) ИР и 0,3 г индикаторной смеси калько-на Р или индикаторной смеси кальконкарбоновой кислоты Р и продолжают титрование раствором эдетата динатрия до появления синего окрашивания. Каждые 5,005 мг карбоната кальция соответствуют 1 мл раствора динатрия эдетата (0,05 моль/л) ТР.
Диоксан Р. 1,4-Диоксан С4Н8О2.
Предупреждение. Определять температуру кипения или остаток после выпаривания до проведения испытания на перекиси опасно.
Описание. Прозрачная, бесцветная жидкость.
. Смешиваемость. Смешивается с водой, этанолом (~750 г/л) ИР и эфиром Р.
Температурный интервал кипения. Не менее 95% перегоняется между 101 и 105 °C.
Температура плавления. Затвердевает при охлаждении во льду и полностью не плавится при температуре ниже 10 °C, Остаток после выпаривания. Выпаривают на водяной ба^ не и высушивают до постоянной массы при 105 °C; остаток должен составлять не более 0,1 мг/мл.
Плотность. (ip2o) Около 1,031 кг/л.
Вода. При определении методом К. Фишера не ’более 5,0 мг/мл.
Перекиси. Прибавляют 5 мл к смеси 1 г йодида калия Р, растворенного в 10 мл воды, 5 мл соляной кислоты (~70 г/л) ИР, 2 мл раствора крахмала ИР и перемешивают; появляется совсем легкое синее или коричневое ок-, рашивание.
200
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Дифенилбензидин Р. C24H20N2.
Описание. Белый или слегка зеленоватый кристаллический порошок.
Растворимость. Нерастворим в воде, мало растворим в этаноле (~750 г/л) ИР и ацетоне Р.
Температурный интервал плавления. 246—250 °C.
Сульфатная зола. Не более 1,0 мг/г.
Нитраты. Растворяют 8 мг в охлажденной смеси серной кислоты (~1760 г/л), не содержащей соединений азота, ИР и 5 мл воды; раствор бесцветный или совсем бледно-голубой.
Дифенил карбазон Р. C13H12N4O (SRIP, 1963, с. 81).
Дифенилкарбазона раствор в этаноле ИР. Раствор дифенил-карбазона Р в этаноле (~750 г/л) ИР, содержащий C13H12N4O около 1 г/л.
Дифениловый эфир Р. Фениловый эфир С^НюО.
Описание. Бесцветная жидкость.
Смешиваемость. Не смешивается с водой, легко смешивается с ледяной уксусной кислотой Рис большинством органических растворителей.
Температура кипения. Около 259 °C.
Температурный интервал плавления. 26—28 °C.
2,5-Дифенилоксазол Р. РРО, С15НцМО. Применяется для сцинтилляционного счета.
Дихлорметан Р. Метиленхлорид CH2CI2.
Описание. Прозрачная, бесцветная, подвижная жидкость. Смешиваемость. Легко смешивается с этанолом (~750 г/л) ИР и эфиром Р.
Температурный интервал кипения. Не менее 95% перегоняется между 39 и 41 °C.
Остаток после выпаривания. Оставляет после выпаривания на водяной бане и высушивания при 105 °C не более 0,5 мг/мл.
Диэтиленгликоль Р. С4Н10О3.
Описание. Бесцветная или слегка желтоватая жидкость со слабым запахом.
Смешиваемость. Легко смешивается с водой, этанолом (~750 г/л) ИР, эфиром Р и ацетоном Р.
Температурный интервал кипения. Между 240 и 250 °C.
Плотность (р2о) • 1,117—1,120 кг/л.
Кислотность. Помещают 60 г в коническую колбу емкостью 250 мл, прибавляют раствор фенолфталеина в этаноле ИР и титруют раствором гидроокиси калия' в этаноле
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
201
(0,02 моль/л) ТР до появления розового окрашивания, устойчивого в течение не менее 15 с. Должно расходоваться не более 2,5 мл титранта.
Древесный уголь Р. (SRIP, 1963, с. 64).
Дрожжей экстракт водорастворимый Р. (SRIP, 1963, с. 215).
Желатин Р. Желатин подходящей степени чистоты.
Желатина раствор ИР. Раствор желатина Р в фосфатном буфере, pH 7,0, ИР, содержащий желатина около 10 г/л.
Железа(Н) аммония сульфат Р. Fe(NH4)2(SO4)2-6H2O. SRTP, 1963, с. 89).
Железа (II) аммония сульфата раствор (1 г/л) ИР. Раствор сульфата аммония железа (II) Р, содержащий Fe(NH4)2(SO4)2 около 1 г/л.
Железа (III) аммония сульфат Р. FeNH4(SO4)2- 12Н2О (SRIP, 1963, с. 88).
Железа (Ш) аммония сульфата раствор (45 г/л) ИР. Раствор сульфата аммония железа (III) Р, содержащий FeNH4(SO4)2 около 45 г/л.
Железа II сульфат Р. FeSO4-7H2O (SRIP, 1963, с. 90).
Железа II сульфата раствор (15 г/л) ИР. Раствор сульфата железа (П) Р. содержащий FeSO4 в свежепрокипяченной и охлажденной воде около 15 г/л (приблизительно 0,1 моль/л).
Примечание. Железа (II) сульфата раствор (15 г/л) ИР должен быть свежеприготовленным.
Железа (II) сульфата раствор (0,1 моль/л) ТР.
Методика приготовления. Растворяют 2,8 г сульфата железа (II) Р в 90 мл свежепрокипяченной и охлажденной воды и доводят серной кислотой (~ 1760 г/л) ИР до получения 100 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,1 моль/л раствора следующим образом: к 40,0 мл раствора сульфата железа (II) Р прибавляют 5 мл фосфорной кислоты (~1440 г/л) ИР и тотчас титруют раствором перманганата калия (0,02 моль/л) ТР.
Примечание. Раствор стандартизуют непосредственно перед применением.
Железа (III) хлорид Р. FeCl3-6H2O (SRIP, 1963, с. 88).
Железа (III) хлорида раствор (25 г/л) ИР. Раствор хлорида железа (III) Р, содержащий FeCl3 около 27 г/л.
202
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Железа раствор окрашенный концентрированный ИР.
Методика приготовления. Растворяют 6,6 г хлорида железа (III) Р в 120 мл серной кислоты (~10 г/л) ИР, фильтруют раствор, если необходимо, и определяют концентрацию FeCl3-6H2O.
Количественное определение. Разводят 5,0 мл водой до получения 25,0 мл. Помещают 10,0 мл этого раствора в колбу и прибавляют 60 мл воды. Доводят pH до 2—3 соляной кислотой (1 моль/л) ТР и аммиаком (~100 г/л) ИР, используя бумагу конго красного Р. Нагревают раствор приблизительно до 45 °C и титруют раствором динатрия эдетата (0,05 моль/л) ТР, используя в качестве индикатора 2 мл раствора сульфосалициловой кислоты (175 г/л) ИР, до изменения окраски раствора от фиолетовой до соломенно-желтой. Каждый миллилитр раствора динатрия эдетата (0,05 моль/л) ТР соответствует 13,52 г FeCl3-6H2O.
Железа раствор окрашенный ИР. Раствор, содержащий FeCl3-6H2O 45,0 мг/мл.
Методика приготовления. Готовят раствор, содержащий 4,500 г FeCl3-6H2O в 100 мл, путем разведения концентрированного окрашенного раствора железа ИР серной кислотой (~10 г/л) ИР, как требуется.
Железа раствор стандартный для испытания на железо FeHP. Методика приготовления. Растворяют 0,173 г сульфата аммония железа (III) Р в 100 мл воды, прибавяют 5 мл соляной кислоты (~70 г/л) ИР и воды до получения 1000 мл. Каждый миллилитр этого раствора содержит 20 мкг железа.
Желтый исходный стандартный раствор ИР.
Методика приготовления. К 9,5 мл окрашенного раствора кобальта ИР прибавляют 1,9 мл окрашенного раствора меди ИР, 10,7 мл окрашенного раствора бихромата ИР, 4,0 мл окрашенного раствора железа ИР; разводят до 100,0 мл серной кислотой (~10 г/л) ИР и перемешивают.
Зеленый исходный стандартный раствор ИР.
Методика приготовления. К 3,5 мл окрашенного раствора кобальта ИР прибавляют 20,1 мл окрашенного раствора меди ИР, 10,4 мл окрашенного раствора бихромата ИР и 4,0 мл окрашенного раствора железа ИР; разводят до 100,0 мл серной кислотой (~10 г/л) ИР и перемешивают.
Изотонический раствор ИР. Стерильный раствор хлорида натрия Р, содержащий NaCl около 9 г/л. Стерилизовать мож
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
203
но нагреванием в паровом автоклаве при 120 °C в течение 30 мин.
Йод Р. I2 (SRIP, 1963, с. 101).
Йода раствор ИР.
Методика приготовления. Растворяют 2,6 г йода Р и 3 г йодида калия Р в воде до получения 100 мл (приблизительно 0,2 моль/л).
Йода раствор (0,02 моль/л) ТР. Йод Р и йодид калия Р, растворенные в воде до содержания 12,69 г I и 18,0 г KI в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора 0,1 моль/л путем титрования 25,0 мл раствором тиосульфата натрия (0,1 моль/л) ТР, используя в качестве индикатора раствор крахмала ИР.
Йода раствор (0,02 моль/л) ТР. Иод Р и йодид калия Р, растворенные в воде до содержания 2,538 г I и 3,6 кг KI в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора йода (0,1 моль/л) ТР.
Йода раствор (0,01 моль/л) ТР. Йод Р и йодид калия Р, растворенные в воде до содержания 1,269 г I и 3,6 г KI в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора йода (0,1 моль/л) ТР.
Йод бромистый Р. 1Вг.
Описание. Сине-черные пли коричневато-черные кристаллы.
Растворимость. Легко растворим в воде, этаноле ( — 750 г/л) ИР, хлороформе Р, эфире Р и ледяной уксусной кислоте Р.
Температура плавления. Около 40 °C.
Хранение. Хранят в холодном месте, в плотной укупоренной таре, защищенной от действия света.
Йода бромистого раствор ИР.
Методика приготовления. Растворяют 20 г бромистого йода Р в ледяной уксусной кислоте Р до получения 1000 мл.
Хранение. Хранят в плотно укупоренной таре, защищенной от действия света.
Йодистоводородная кислота ( — 970 г/л) ИР [йодистоводородная кислота Р]. HI (SRIP, 1963, с. 95).
204 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Калия ацетат Р. С2Н3КО2 (SRIP, 1963, с. 144).
Калия ацетата раствор ИР.
Методика приготовления. Растворяют 100 г ацетата калия Р в ледяной уксусной кислоте Р до получения 1000 мл.
Калия бикарбонат Р. КНСО3 (SRIP, 1963, с. 145).
Калия бихромат Р. К2СГ2О7 (SRIP, 1963, с. 154).
Калия бихромат Р1. Бихромат калия Р, содержащий не менее 99,9% К2Сг2О7.
Калия бихромата раствор ИР.
Методика приготовления. Растворяют около 60 мг (точная навеска) предварительно высушенного при 130 °C бихромата калия Р1 в серной кислоте (0,005 моль/л) ТР до получения 1000,0 мл.
Калия бихромата раствор (0,0167 моль/л) ТР. Бихромат калия Р, растворенный в воде до содержания 4,904 г К2Сг2О7 в 1000 мл.
Калия бромид Р. КВг (SRIP, 1963, с. 148).
Калия бромид И К. Бромид калия Р, который выдерживает следующее испытание: ИК-спектр диска, приготовленного, как описано в методе 3 в разделе «Спектрофотометрия в инфракрасной области спектра» (см. с. 45), с бромидом калия Р, предварительно высушенным при 250 °C в течение 1 ч, имеет в основном плоскую базовую линию в области 4000—650 см-1; максимальное поглощение составляет не более 0,1 выше базовой линии, за исключением максимумов, обусловленных присутствием воды при 3440 и 1630 см-1.
Калия бромида раствор (100 г/л) ИР. Раствор бромида калия Р, содержащего около 100 г КВг в 1 л.
Калия гидроокись (едкое кали) Р. КОН (SRIP, 1963, с. 159).
Калия гидроокиси раствор ( — 110 г/л) ИР. Раствор гидроокиси калия Р, содержащий КОН около 112 г/л (приблизительно 2 моль/л).
Калия гидроокиси раствор в этаноле ИР1.
Методика приготовления. Растворяют 40 г гидроокиси калия Р в 20 мл воды и прибавляют этанол ( — 750 г/л) ИР до получения 1000 мл. Оставляют стоять на ночь и сливают прозрачную жидкость.
Калия гидроокиси раствор (1 моль/л) ТР. Гидроокись калия Р, растворенная в воде до содержания 56,10 г КОН в 1000 мл.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ 205
Метод стандартизации. Устанавливают точную концентрацию 1 моль/л раствора следующим образом: высушивают около 5 г гидрофталата калия Р при 105 °C в течение 3 ч и точно взвешивают. Если гидрофталат калия имеет форму больших кристаллов, эти кристаллы следует измельчить перед высушиванием. Растворяют в 75 мл воды, не содержащей углекислоты, Р и титруют раствором гидроокиси калия, используя в качестве индикатора раствор фенолфталеина в этаноле ИР. Каждые 0,2042 г гидрофталата калия соответствуют 1 мл раствора гидроокиси калия (1 моль/л) ТР. Стандартные растворы гидроокиси калия следует часто стандартизовать.
Хранение. Растворы едких щелочей поглощают углекислоту воздуха, поэтому их следует хранить в сосудах с хорошо закрывающейся нестеклянной пробкой, в которой имеется трубка, заполненная натронной известью Р.
Калия гидроокиси раствор (0,5 моль/л) ТР. Гидроокись калия Р, растворенная в воде до содержания 28,05 г КОН в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора гидроокиси калия (1 моль/л) ТР.
Калия гидроокиси раствор (0,1 моль/л) ТР. Гидроокись калия Р, растворенная в воде до содержания 5,610 г КОН в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора гидроокиси калия (1 моль/л) ТР.
Калия гидроокиси раствор в этаноле (0,5 моль/л) ТР. Гидроокись калия, растворенная в этаноле ( — 710 г/л) ИР до содержания 28,05 г КОН в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,5 моль/л раствора следующим образом: разводят 25,0 мл соляной кислоты (0,5 моль/л) ТР 50 мл воды и титруют раствором гидроокиси калия в этаноле — раствор фенолфталеина в этаноле ИР.
Калия гидроокиси раствор в этаноле (0,02 моль/л) ТР. Гидроокись калия Р, растворенная в этаноле ( — 710 г/л) ИР до содержания 1,122 г КОН в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора гидроокиси калия в этаноле (0,5 моль/л) ТР.
Калия гидротартрат Р. С4Н5КО6 (SRIP, 1963, с. 158).
Калия гидротартрата стандартный раствор ИР.
Методика приготовления. Прибавляют 2 г гидротартрата
206
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
калия Р к 100 мл воды, не содержащей углекислоты, Р в-, колбе с притертой пробкой и энергично встряхивают колбу. Оставляют раствор, чтобы его температура достигла комнатной, дают твердому веществу осесть и удаляют его-фильтрацией или декантированием.
Примечание. Калия гидротартрата стандартный раствор-ИР должен быть свежеприготовленным.
Калия гидрофталат Р. С8Н5КО4 (SRIP, 1963, с. 157).
Калия гидрофталата стандартный раствор ИР.
Методика приготовления. Растворяют 10,21 г гидрофталата калия Р, предварительно высушенного при 120 °C, в воде, не содержащей углекислоты, Р до получения 1000 мл. Величина pH этого раствора принята за 4,000 при 15 °C.
Калия дигидрофосфат Р. КН2РО4 (SRIP, 1963, с. 155).
Калия йодид Р. KI (SRIP, 1963, с. 161).
Калия йодид для испытания на мышьяк AsP. Йодид калия Рг который выдерживает следующее испытание: растворяют 10 г йодида калия Р в 25 мл соляной кислоты ( — 250 г/л) AsHP и 35 мл воды, прибавляют 2 капли раствора хлорида олова AsHP и далее проводят испытание на мышьяк; не должно образовыватьсся видимое пятно.
Калия йодида раствор (80 г/л) ИР. Раствор йодида калия Р, содержащий KI около 83 г/л (приблизительно 0,5 моль/л).
Калия нитрат Р. KNO3 (SRIP, 1963, с. 162).
Калия нитрит Р. KNO2.
Описание. Белые или слегка желтые, расплывающиеся на воздухе гранулы или палочки.
Растворимость. Растворим в 0,35 части воды; мало растворим в этаноле ( — 750 г/л) ИР.
Калия нитрита раствор (100 г/л) ИР. Раствор нитрита калия Р, содержащий KNO2 около 100 г/л.
Калия перманганат Р. KMnO4 (SRIP, 1963, с. 165).
Калия перманганата раствор (10 г/л) ИР. Раствор перманганата калия Р, содержащий КМпО4 около 10 г/л.
Калия перманганата раствор (0,02 моль/л) ТР. Перманганат калия Р, растворенный в воде до содержания 3,161 г КМпО4 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,02 моль/л раствора следующим образом: растворяют около 0,2 г (точная навеска) оксалата натрия Р, предварительно высушенного до постоянной массы при ПО °C,
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ 207
в 250 мл воды. Прибавляют 7 мл серной кислоты ( — 1760 г/л) ИР, нагревают примерно до 70°С и затем медленно прибавляют из бюретки раствор перманганата калия при постоянном перемешивании, пока не появится в растворе розовое окрашивание, которое сохраняется в течение 15 с. Температура при завершении титрования должна быть не ниже 60 °C. Каждые 6,7 мг оксалата натрия соответствуют 1 мл раствора перманганата калия (0,02 моль/л) ТР. Растворы перманганата калия следует часто повторно стандартизовать.
Хранение. Раствор хранят в плотно укупоренной таре, защищенной от действия света.
Калия сульфат Р. K2SO4 (SRIP, 1963, с. 165).
Калия сульфата раствор (174 г/л) ИР.
Методика приготовления. Растворяют 174 мг (точная навеска) сульфата калия Р в воде до получения 1000 мл.
Калия тетраоксалат Р. С4Н3КО8-2Н2О (SRIP, 1963, с. 166).
Калия тетраоксалата стандартный раствор ИР.
Методика приготовления. Растворяют 25,42 г тетраоксалата калия Р в воде, не содержащей углекислоты, Р до получения 1000 мл.
Калия феррицианид Р. КзРе(СМ)6 (SRIP, 1963, с. 156).
Калия феррицианида раствор (10 г/л) ИР.
Методика приготовления. Промывают около 1 г кристаллического феррицианида калия Р небольшим количеством воды и растворяют промытые кристаллы в воде до получения 100 мл.
Примечание. Калия феррицианида раствор (10 г/л) ИР должен быть свежеприготовленным.
Калия хлорид Р. КС1 (SRIP, 1963, с. 151).
Калия хлорид И К. Хлорид калия Р, который выдерживает следующее испытание: ПК-спектр диска, приготовленного, как описано в методе 3 в разделе «Спектрофотометрия в инфракрасной области спектра» (см. с. 45), с хлоридом калия Р, предварительно высушенным при 250 °C в течение 1 ч, имеет в основном плоскую базовую линию в области 4000—670 см-1; максимальное поглощение составляет не более 0,1 Выше базовой линии, за исключением максимумов, обусловленных присутствием воды при 3440 и 1630 см-1.
Калия хлорида раствор (350 г/л) ИР. Насыщенный раствор хлорида калия Р, содержащий КС1 около 350 г/л.
208
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Калькой Р. Мононатриевая соль 2-окси-1-[(2-окси-1-нафтил)-азо]нафталин-4-сульфоновой кислсоты; C.I. Протравной черный 17, C.I. № 15705, Эрнохром сине-черный Р, Соло-хром темно-синий; C20Hi3N2NaO5S.
Калькона индикаторная смесь Р.
Методика приготовления. Смешивают 0,1 г калькона Р с 10 г безводного сульфата натрия Р.
Калькой карбоновая кислота Р. 2-Окси-1-(2-окси-4-сульфо-1-нафтил)-азо-3-нафтоевая кислота; C2iHuN2O7S -ЗН2О.
Описание. Темно-коричневый порошок с фиолетовым оттенком.
Растворимость. Практически нерастворим в воде; мало растворим в метаноле Р и этаноле (~750 г/л) ИР; легко растворим в растворах едких щелочей.
Кальконкарбоновой кислоты индикаторная смесь Р.
Методика приготовления. Смешивают 0,1 г кальконкарбоновой кислоты Р с 10 г безводного сульфата натрия Р.
Кальция карбонат Pl. СаСО3 (SRIP, 1963, с. 156).
Кальция карбонат Р2. СаСО3. Карбонат кальция Р1 такого качества, при котором он может служить первичным стандартом для стандартизации раствора эдетата динатрия.
Кальция хлорид безводный Р [кальция хлорид Р]. СаС12 (SRIP, 1963, с. 58).
Кальция хлорид водный Р. СаС12-6Н2О (SRIP, 1963, с. 58).
Кальция хлорида раствор (55 г/л) ИР. Раствор водного хлорида кальция Р, содержащий СаС12 около 55 г/л (приблизительно 0,5 моль/л).
Кобальта раствор окрашенный концентрированный ИР.
Методика приготовления. Растворяют 8,0 г хлорида кобальта Р в 120 мл серной кислоты 10 г/л) ИР, фильтруют раствор, если необходимо, и определяют концентрацию СоС12-6Н2О.
Количественное определение. Разводят 5,0 мл раствора водой до получения 100 мл. Переносят 10,0 мл этого раствора в колбу с притертой пробкой, прибавляют 10 мл воды, 0,5 мл перекиси водорода (~60 г/л) ИР и 10 мл раствора гидроокиси натрия (~80 г/л) ИР. Прибавляют несколько бусинок для кипячения и кипятят содержимое колбы до тех пор, пока полностью не будет разрушен избыток перекиси водорода (приблизительно 10 мин). Охлаждают колбу, прибавляют 20 мл воды, 1 г йодида ка
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
209
лия Р и 25 мл соляной кислоты (2 моль/л) ТР. Закрывают колбу пробкой и оставляют стоять до растворения осадка. Титруют выделившийся йод раствором тиосульфата натрия (0,01 моль/л) ТР, используя в качестве индикатора раствор крахмала ИР. Каждый миллилитр раствора тиосульфата натрия (0,01 моль/л) ТР соответствует 2,380 мг СоС12-6Н2О.
Кобальта раствор окрашенный ИР. Раствор, содержащий СоС12-6Н2О 60,0 г/л.
Методика приготовления. Готовят раствор, содержащий 6,000 г СоС12-6Н2О в 100 мл путем разведения концентрированного окрашенного раствора кобальта ИР серной кислотой (~10 г/л) ИР, как требуется.
Кобальта хлорид Р. СоС12-6Н2О (SRIP, 1963, с. 70).
Конго красного бумага Р. (SRIP, 1963, с. 72).
Коричневый исходный стандартный раствор ИР.
Методика приготовления. К 35,0 мл окрашенного раствора кобальта ИР прибавляют 17,0 мл окрашенного раствора меди ИР, 8,0 мл окрашенного раствора бихромата ИР, разводят окрашенным раствором железа ИР до 100,0 мл и перемешивают.
Красный исходный стандартный раствор ИР.
Методика приготовления. К 40,4 мл окрашенного раствора кобальта ИР прибавляют 6,1 мл окрашенного раствора меди ИР, 6,3 мл окрашенного раствора бихромата ИР, 12,0 мл окрашенного раствора железа ИР, разводят до 100,0 мл серной кислотой (~10 г/л) ИР и перемешивают.
Крахмал Р [картофельный или кукурузный крахмал Р]. (SRIP, 1963, с. 199).
Крахмал растворимый Р. (SRIP, 1963, с. 199).
Крахмала раствор ИР.
Методика приготовления. Смешивают 0,5 г крахмала Р или растворимого крахмала Р с 5 мл воды и прибавляют этот раствор при постоянном перемешивании к воде до получения примерно 100 мл; кипятят в течение нескольких минут, охлаждают и фильтруют.
Примечание. Крахмала раствор ИР должен быть свежеприготовленным.
Крахмал-йодидная бумага Р [крахмал-йодидная бумага Р]. (SRIP, 1963, с. 200).
Кристаллический фиолетовый Р. C25H3oC1N3 (SRIP, 1963,. с. 73).
210
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Кристаллического фиолетового раствора в уксусной кислоте ИР. Раствор кристаллического фиолетового Р в ледяной уксусной кислоте Р1, содержащий C25H30CIN3 около 5 г/л.
Ксиленоловый оранжевый Р. [3/7-2,1-Бензоксатиол-З-илиден бис[6-окси-5-метил-ж-фенил) метиленнитрил]] тетрауксус-ная кислота, S,S-диоксид, C31H32N2O13S.
Описание. Оранжевый порошок.
Растворимость. Растворим в воде и этаноле (~750 г/л) ИР.
Ксиленолового оранжевого индикаторная смесь Р.
Методика приготовления. Смешивают 0,1 г ксиленолового оранжевого Р и 10 г нитрата калия Р.
Культуральная среда Kcl.
Методика приготовления. Растворяют 6,0 г высушенного пептона Р, 4,0 г панкреатического гидролизата казеина Р, 3,0 г водорастворимого экстракта дрожжей Р, 1,5 г мясного экстракта Р, 1,0 г гидрата глюкозы Р и 10—20 г агара Р в воде до получения 1000 мл.
Примечание. Агар Р следует взять в таком количестве, которое обеспечит достаточную прочность среды, чтобы она выдерживала установленные на ней цилиндры и чтобы можно было вырезать в ней отверстия, не разрывая слой геля.
Культуральная среда Кс2.
Методика приготовления. Растворяют 17,0 г панкреатического гидролизата казеина Р, 3,0 г папаинового гидролизата соевой муки Р, 5,0 г хлорида натрия Р, 2,5 г гидрофосфата дикалия Р, 2,5 г гидрата глюкозы Р и 10—20 г агара Р в воде до получения примерно 500 мл. Нагревают раствор, прибавляют 10,0 г полисорбата-80 Р и тотчас разводят водой до получения 1000 мл.
Примечание. Агар Р следует взять в таком количестве, которое обеспечит достаточную прочность среды, чтобы она выдерживала установленные на ней цилиндры и чтобы можно было вырезать в ней отверстия, не разрывая слой геля.
Культуральная среда КсЗ.
Методика приготовления. Растворяют 9,4 г высушенного пептона Р, 4,7 г водорастворимого экстракта дрожжей Р, 2,4 г мясного экстракта Р, 10,0 г хлорида натрия Р, 10,0 г гидрата глюкозы Р и 15—25 г агара Р в воде до получения 1000 мл.
Примечание. Агар Р следует взять в таком количестве,
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
211
которое обеспечит достаточную прочность среды, чтобы она выдерживала установленные на ней цилиндры и чтобы можно было вырезать отверстия, не разрывая слой геля.
Культуральная среда Кс4. Жидкая меркаптоуксусная (тио-гликолевая) среда.
Методика приготовления. Тщательно растирают в ступке в следующем порядке: 0,5 г L-цистина Р, 2,5 г хлорида натрия Р, 5,5 г гидрата глюкозы Р, 0,75 г агара Р, 5,0 г водорастворимого экстракта дрожжей Р и 15,0 г панкреатического гидролизата казеина Р. Прибавляют немного горячей воды, переносят в подходящую посуду и прибавляют воды до получения 1000 мл. Растворяют все содержимое путем нагревания на кипящей водяной бане, следя за тем, чтобы полностью растворился L-цистин Р. Прибавляют 0,3 мл меркаптоуксусной кислоты Р или 0,5 г меркаптоацетата натрия Р (предпочтительно последний, так как он более устойчив) и прибавляют раствор гидроокиси натрия (1 моль/л) ТР в количестве, достаточном для получения окончательного значения pH простерили-зованной среды 7,0—7,2. Вновь нагревают раствор, но не до кипения, фильтруют, если необходимо, через кусочек ваты и прибавляют 1,0 мл раствора резазурина натрия (1 г/л) ИР. Разливают раствор в подходящие флаконы, стерилизуют автоклавированием в течение 18—20 мин при 121 °C и быстро охлаждают до 25 °C.
Хранение. При 20—30 °C; следует предохранять от яркого света.
Примечание. Если верхний слой среды более чем на Vs ее . глубины стал розовым, среда непригодна для применения. Один раз ее можно восстановить путем нагревания паром.
Культуральная среда Кс5. Среда гидролизата соевой муки и казеина.
Методика получения. Растворяют в воде 17,0 г панкреатического гидролизата казеина Р, 3,0 г папаинового гидролизата соевой муки Р, 5,0 г хлорида натрия Р, 2,5 г гидрофосфата дикалия Р и 2,5 г гидрата глюкозы Р. Слегка нагревают раствор, затем охлаждают до комнатной температуры и прибавляют воды до получения 1000 мл. Доводят, если необходимо, pH раствора раствором гидроокиси натрия (1 моль/л) ТР так, чтобы pH готовой и просте-рилизованной среды был 7,1—7,6. Фильтруют, если необходимо осветлить, разливают раствор в подходящие флаконы и стерилизуют в автоклаве в течение 18—20 мин при - 121 °C.
212
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Культуральная среда Кеб. Жидкая меркаптоуксусная (тио-гликолевая) среда с пенициллиназой.
Методика приготовления. Используют культуральную среду Кс4 с достаточным количеством стерильного раствора пенициллиназы ИР, прибавленной для инактивации пенициллина в испытуемом материале. Раствор пенициллиназы ИР прибавляют асептически в отдельные флаконы, содержащие стерильную культуральную среду Кс4. Перед применением или во время испытания отбирают из партии репрезентативное число флаконов с культуральной средой Кеб, инкубируют их при 30—32 °C в течение 42— 48 ч и проверяют на стерильность.
Культуральная среда Кс7. Среда гидролизата соевой муки и казеина с пенициллиназой.
Методика приготовления. Используют культуральную среду Кс5, к которой перед стерилизацией прибавляют 5,0 мл полисорбата-80 Р и достаточное количество стерильного раствора пенициллиназы ИР для инактивации пенициллина в испытуемом материале. Раствор пенициллиназы ИР прибавляют асептически в отдельные флаконы, содержащие стерильную культуральную среду Кс5.
Лантана нитрат Р. La(NO3)3-6H2O. Содержит не менее 97,0% La(NO3)3-6H2O.
Описание. Бесцветные кристаллы; на воздухе расплываются.
Растворимость. Легко растворим в воде.
Количественное определение. Растворяют около 0,75 г (точная навеска) в 25 мл воды, прибавляют 3 мл азотной кислоты (~ 130 г/л) ИР, 3 г метенамина Р, около 20 мг индикаторной смеси ксиленолового оранжевого Р и титруют раствором эдетата динатрия (0,05 моль/л) ТР до тех пор, пока цвет раствора не станет чисто желтым. Если к концу титрования окраска исчезает, следует прибавить еще метенамина Р. Каждый миллилитр раствора эдетата динатрия (0,05 моль/л) ТР соответствует 21,65 г La(NO3)3-6H2O.
Лантана нитрата раствор (30 г/л) ИР.
Методика приготовления. Растворяют 4,3 г нитрата лантана Р в 1 мл азотной кислоты (~130 г/л) ИР и доводят водой до 100 мл.
Лимонная кислота Р. С6Н8О7-Н2О (SRIP, 1963, с. 69).
Лимонная кислота для испытания на железо FeP. Лимонная кислота Р, которая выдерживает следующее испытание: растворяют 0,5 г лимонной кислоты Р в 40 мл воды, при
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
213
бавляют 2 капли меркаптоуксусной кислоты Р, перемешивают, подщелачивают аммиаком (~< 100 г/л) FeHP и разводят водой до 50 мл; не должно появляться розовое окрашивание.
Лимонной кислоты раствор (180 г/л) для испытания на железо FeHP. Раствор лимонной кислоты FeP, содержащий C6HgO7 около 183 г/л.
Лития перхлорат Р. ЫС1О4.
Описание. Небольшие кристаллы.
Растворимость. Легко растворим в воде; мало растворим в этаноле (~750 г/л) ИР, ацетоне Р, эфире Р и этилацетате Р.
Лития перхлората раствор в уксусной кислоте ИР.
Методика приготовления. Растворяют 10,64 г перхлората лития Р в ледяной уксусной кислоте Р1 до получения 1000 мл.
Магния окись Р. MgO.
Описание. Белый, очень мелкий порошок.
Растворимость. Очень мало растворим в воде; нерастворим в этаноле (~750 г/л) ИР.
Магния сульфат Р. MgSO4-7H2O (SRIP, 1963, с. 111).
Макрогол-400 Р. Полиэтиленгликоль-400. Макрогол-400 Р — полимер окиси этилена и воды, представленный формулой Н(ОСН2СН2)ПОН, в которой среднее значение п лежит в пределах 8,2—9,1.
Описание. Прозрачная бесцветная (или практически бесцветная) вязкая жидкость с легким характерным запахом; слабо гигроскопична.
Средняя молекулярная масса. Помещают в устойчивую к давлению колбу 2,1 г макрогола-400 Р (точная навеска) и прибавляют раствор фталевого ангидрида в пиридине ИР. Закрывают колбу пробкой, тщательно оборачивают колбу тканью и помещают в водяную баню с температурой 96—100 °C на глубину слоя жидкости в колбе на 1 ч. Вынимают колбу, оставляя ее обернутой тканью, и дают охладиться на воздухе до комнатной температуры. К содержимому колбы прибавляют 50 мл раствора гидроокиси натрия (0,5 моль/л), не содержащего карбонатов, ТР и 5 капель раствора фенолфталеина в пиридине ИР. Титруют раствором гидроокиси натрия (0,5 моль/л), не содержащим карбонатов, ТР до появления розовой окраски, сохраняющейся в течение не менее 15 с. Аналогично проводят контрольное титрование. Вычисляют среднюю моле
15—2025
214 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
кулярную массу, умножая на 4000 массу (в граммах) испытуемого вещества и деля результат на разность между объемом (в миллилитрах) раствора гидроокиси натрия (0,5 моль/л), не содержащего карбонатов, ТР, пошедшего на титрование испытуемого вещества, и объемом этого раствора, пошедшим на контрольное определение.
Средняя молекулярная масса составляет 380—420. Плотность (р2о) • 1,110—1,140 кг/л.
Точка затвердевания. Между 4 и 8 °C; точку затвердевания определяют по 4 последовательным измерениям температуры; максимальная и минимальная температура не должна отличаться более чем на 0,4 °C.
pH. В растворе 50 г/л между 4,5 и 7,5.
Кислотность или щелочность. Растворяют 5,0 г в 50 мл воды. Прибавляют несколько капель раствора фенолового красного в этаноле ИР. Если раствор становится желтым, титруют раствором гидроокиси натрия (0,01 моль/л) ТР; если раствор становится красным, титруют соляной кислотой (0,01 моль/л) ТР. В каждом случае требуется не более 2,0 мл титранта.
Сульфатная зола. Не более 10 мг/г.
Тяжелые металлы. Смешивают 4 г (точная навеска) с 1 мл соляной кислоты (~70 г/л) ИР и разводят водой до 25 мл. Предел содержания 50 мкг/г.
Содержание моноэтилен- и диэтиленгликолей. Растворяют 50 г в 75 мл дифенилового эфира Р в колбе для перегонки емкостью 250 мл. Медленно отгоняют при давлении 100— 250 Па (1—2 мм рт. ст.) в приемник, градуированный на 100 мл с ценой делений 1 мл, до получения 25 мл отгона. К отгону прибавляют 25,0 мл воды, энергично встряхивают приемник и дают слоям разделиться. Охлаждают приемник в ледяной бане для затвердевания и облегчения удаления слоя дифенилового эфира Р. Фильтруют водный слой через фильтровальную бумагу в градуированный цилиндр емкостью 50 мл с притертой пробкой. К фильтрату прибавляют равный объем свежеперегнанного ацетонитрила Р и встряхивают цилиндр до получения раствора. Вносят 10 мл этого раствора пипеткой в 15 мл раствора нитрата церия-аммония ИР, смешивают и через 2—5 мин определяют поглощение полученного раствора при 525 нм. Используют контрольный раствор, состоящий из 15 мл раствора нитрата церия-аммония ИР и 10 мл ацетонитрила (~400 г/л) ИР. Готовят стандартный раствор путем смешивания 10 мл ацетонитрила (400 г/л) ИР, к которому прибавлены 30 мг диэтиленгликоля Р и 15 мл нитрата церия-аммония ИР и определяют поглощение получение-
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ 215
го раствора через 2—5 мин при 525 нм, используя тот же контрольный раствор, что и в предыдущем испытании. Поглощение испытуемого раствора не должно превышать поглощение стандартного раствора.
Марганца двуокись Р. MnO2 (SRIP, 1963, с. 112).
Марганцово-серебряная бумага реактивная Р.
Методика приготовления. К смеси равных объемов раствора нитрата серебра (0,1 моль/л) ТР и сульфата марганца (15 г/л) ИР прибавляют по каплям раствор гидроокиси натрия (0,1 моль/л) ТР до образования постоянного остатка и фильтруют. В полученный раствор погружают на 15 мин полоски фильтровальной бумаги (например, бумага Ватман № 1), затем бумагу высушивают при комнатной температуре, защищая от действия света и паров щелочи или кислоты. Марганцово-серебряная реактивная бумага Р должна быть бесцветной.
Испытание чувствительности. В цилиндр емкостью около 40 мл (высотой около 80 мм и внутренним диаметром около 30 мм) помещают 1,0 мл раствора хлорида аммония (10 мкг/мл NH4) ИР. Прибавляют 9 мл воды и 1 г окиси магния Р. Тотчас закрывают цилиндр полиэтиленовой пробкой, под которой помещают марганцово-серебряную реактивную бумагу Р. Осторожно вращают раствор, чтобы частички окиси магния не касались реактивной бумаги. Цилиндр выдерживают при 50—60 °C в течение 1 ч. На реактивной бумаге появляется серое окрашивание.
Марганца сульфат Р. MnSO4-H2O.
Описание. Розовые, слегка выветривающиеся кристаллы. Растворимость. Растворим примерно в I части воды и в 0,6 части кипящей воды; практически нерастворим в этаноле ( — 750 г/л) ИР.
Марганца сульфата раствор (15 г/л) ИР. Сульфат марганца Р, растворенный в воде до содержания MnSO4 15,0 г/л.
Меди раствор окрашенный концентрированный ИР.
Методика приготовления. Растворяют 8,0 г сульфата меди (II) Р в 120 мл серной кислоты ( — 10 г/л) ИР, фильтруют раствор, если необходимо, и определяют содержание CuSO4-5H2O.
Количественное определение. Разводят 5,0 мл раствора водой до получения 100 мл. Переносят 10,0 мл этого раствора в колбу с притертой пробкой, прибавляют 20 мл воды, 1 г йодида калия Р и 5 мл ледяной уксусной кислоты Р. Через 10 мин титруют выделившийся йод раствором тиосульфата натрия (0,01 моль/л) ТР, используя в каче
15
216
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
стве индикатора раствор крахмала ИР. Каждый миллилитр раствора тиосульфата натрия (0,01 моль/л) ТР соответствует 2,497 мг CuSO4-5H2O.
Меди раствор окрашенный ИР. Раствор, содержащий 60,0 г/л CuSO4-5H2O.
Методика приготовления. Готовят раствор, содержащий 6,000 г CuSO4-5H2O в 100 мл, путем разведения концентрированного окрашенного раствора меди ИР серной кислотой (~ 10 г/л) ИР, как требуется.
Меди (II) сульфат Р. CuSO4-5H2O (SRIP, 1963, с. 72).
Меди (II) сульфата раствор (160 г/л) ИР. Раствор сульфата меди (И) Р, содержащий CuSO4 около 160 г/л.
Меркаптоуксусная кислота Р (тиогликолевая кислота). C2H4O2S (SRIP, 1963, с. 209).
Метанол Р. СН3ОН (SRIP, 1963, с. 117).
Метанол безводный Р. Метанол Р, выдерживающий следующее требование: содержание воды не более 1,0 мг/г.
Метенамин Р. Гексаметилентетрамин C6Hj2N4. Содержит не менее 99,0% C6Hi2N4.
Описание. Бесцветные кристаллы или кристаллический порошок; без запаха.
Растворимость. Растворим в воде и этаноле (~750 г/л) ИР.
Кислотность и щелочность. Растворяют 2,5 г в 25 мл воды. К Ю мл прибавляют 3 капли раствора фенолфталеина в этаноле ИР; появляется розовое окрашивание, которое переходит в красное после прибавления 1 капли раствора гидроокиси натрия (0,1 моль/л), не содержащего карбонатов, ТР. К остальным 10 мл прибавляют 3 капли раствора бромтимолового синего в этаноле ИР; появляется синее окрашивание, которое изменяется на сине-зеленое после прибавления 3 капель соляной кислоты (0,1 моль/л) ТР.
Сульфатная зола. Не более 0,5 мг/г.
Количественное определение. Растворяют около 1,5 г (точная навеска) в 10 мл воды, прибавляют 50 мл серной кислоты (0,5 моль/л) ТР и кипятят до исчезновения запаха формальдегида. Титруют избыток кислоты раствором гидроокиси натрия (1 моль/л) ТР, используя в качестве индикатора раствор метилового красного в этаноле ИР. Каждый миллиметр серной кислоты (0,5 моль/л) ТР соответствует 35,05 мг CgH]2N4.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
217
Метиловый красный Р. 4'-Диметиламиноазобензол-2-карбоно* вая кислота C15H15N3O2 (SRIP, 1963, с. 118).
Метилового красного раствор в этаноле ИР.
Методика приготовления. Растворяют 25 мг метилового красного Р в смеси 0,95 мл раствора гидроокиси натрия (0,05 моль/л) ТР и 5 мл этанола ( — 750 г/л) ИР, слегка нагревают раствор и после охлаждения разводят этанолом ( — 375 г/л) ИР до получения 250 мл.
Метилового красного и метилтиониния хлорида раствор ИР.
Методика приготовления. Смешивают 20 мл 0,5 мг/мл раствора метилового красного Р в этаноле (—150 г/л) ИР с 0,4 мл 20 мг/мл раствора хлорида метилтиониния Р в воде.
Метиловый оранжевый Р. Натриевая соль 4'-диметиламино-азобензол-4-сульфоновой кислоты Ci4Hl4N3NaO3S (SRIP, 1963, с. 118).
Метилового оранжевого раствор в этаноле ИР.
Методика приготовления. Растворяют 0,04 г метилового оранжевого Р в этаноле (—150 г/л) ИР до получения 100 мл.
Метилтиониния хлорид (метиленовый синий) Р. CieHieClNaS* •ЗН2О (SRIP, 1963, с. 119).
Метилтиониния хлорида раствор (0,2 г/л) ИР.
Методика приготовления. Растворяют 23 г хлорида метилтиониния Р в воде до получения 100 мл.
Муравьиная кислота ( — 1080 г/л) ИР [муравьиная кислота Р]. СН2О2 (SRIP, 1963, с. 92); d ~ 1,2.
Мышьяка раствор концентрированный для испытания на мышьяк AsHP.
Методика приготовления. Растворяют 0,132 г трехокиси мышьяка Р в 6 мл раствора гидроокиси натрия ( — 80 г/л) ИР при легком нагревании. Разводят охлажденный раствор 20 мл воды, прибавляют 50 мл соляной кислоты ( — 250 г/л) ИР и разводят водой до получения 100 мл.
Мышьяка раствор разведенный для испытания на мышьяк AsHP. В 1 мл раствора содержится 10 мкг вещества.
Методика приготовления. Разводят 1 мл концентрированного раствора мышьяка для испытания на мышьяк AsHP водой до получения 100 мл.
Примечание. Мышьяка раствор разведенный AsHP должен быть свежеприготовленным.
Мышьяка трехокись Р. As2O3 (SRIP, 1963, с. 44).
218
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Мясной экстракт Р. Остаток мясного бульона, полученный варкой в воде свежего доброкачественного постного мяса и выпариванием полученного бульона при низкой температуре, обычно при сниженном давлении, до получения густого пастообразного остатка.
Натрия ализаринсульфонат Р. Ализариновый красный С, натриевая соль 3,4-диокси-9,10-антрахинон-2-сульфоновой кислоты; СмНуЫаОуБ-НгО.
Описание. Желто-коричневый или оранжево-желтый порошок.
Растворимость. Легко растворяется в воде, образуя желтый раствор; умеренно растворим в этаноле (''-750 г/л) ИР.
Натрия ализаринсульфоната раствор (1 г/л) ИР.
Методика приготовления. Растворяют 0,11 г ализаринсульфоната натрия Р в воде до получения 100 мл.
Натрия ацетат Р. C2H3NaO2-3H2O (SRIP, 1963, с. 176).
Натрия ацетата раствор (~150 г/л) ИР. Раствор ацетата натрия Р, содержащий C2H3NaO2 около 150 г/л.
Натрия бикарбонат Р. NaHCO3 (SRIP, 1963, с. 177).
Натрия гидроокись (натр едкий) Р. NaOH (SRIP, 1963, с. 185).
Натрия гидроокиси раствора (~400 г/л) ИР. Раствор гидроокиси натрия Р, содержащий NaOH около 400 г/л.
Натрия гидроокиси раствор ('—300 г/л) ИР. Раствор гидроокиси натрия Р, содержащий NaOH около 300 г/л.
Натрия гидроокиси раствор ('—200 г/л) ИР. Раствор гидроокиси натрия Р, содержащий NaOH около 200 г/л.
Натрия гидроокиси раствор (~ 80 г/л) ИР. Раствор гидроокиси натрия Р, содержащий NaOH около 80 г/л (приблизительно 2 моль/л).
Натрия гидроокиси раствор (1 моль/л) ТР. Гидроокись натрия, растворенная в воде до содержания 40,01 г NaOH в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 1 моль/л раствора следующим образом: высушивают около 5 г гидрофталата калия Р при 105°C в течение 3 ч и точно взвешивают. Если кристаллы гидрофталата калия большие, их следует перед высушиванием измельчить. Растворяют в 75 мл воды, не содержащей углекислоты, Р и титруют раствором гидроокиси натрия, используя в каче
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
219
стве индикатора раствор фенолфталеина в этаноле ИР. Каждые 0,2042 г гидрофталата калия соответствуют 1 мл раствора гидроокиси натрия (1 моль/л) ТР. Стандартные растворы гидроокиси натрия следует часто повторно стандартизовать.
Хранение. Растворы едких щелочей поглощают углекислоту воздуха, поэтому их следует хранить в сосудах с хорошо закрывающейся нестеклянной пробкой с трубкой, заполненной натронной известью Р.
Натрия гидроокиси раствор (0,2 моль/л) ТР. Гидроокись натрия Р, растворенная в воде до содержания 8,001 г NaOH в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора гидроокиси натрия (1 Моль/л) ТР.
Натрия гидроокиси раствор (0,1 моль/л) ТР. Гидроокись натрия, растворенная в воде до содержания 4,001 г NaOH в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора гидроокиси натрия (1 моль/л) ТР.
Натрия гидроокиси раствор (0,05 моль/л) ТР. Гидроокись натрия Р, растворенная в воде до содержания 2,000 г NaOH в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора гидроокиси натрия (1 моль/л) ТР.
Натрия гидроокиси раствор (0,01 моль/л) ТР. Гидроокись натрия Р, растворенная в воде до содержания 0,4001 г NaOH в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора гидроокиси натрия (1 моль/л) ТР.
Натрия гидроокиси раствор (1 моль/л), не содержащий карбонатов, ТР. Гидроокись натрия Р, растворенная в воде до содержания 40,01 г NaOH в 1000 мл.
Методика приготовления. Растворяют гидроокись натрия Р в воде до получения раствора с содержанием NaOH 400— 600 г/л и оставляют стоять. Следя за тем, чтобы раствор не поглощал углекислоту, сифонируют прозрачную надосадочную жидкость и разводят, как требуется, водой, не содержащей углекислоты, Р.
Испытание на карбонаты. Титруют 45 мл соляной кислоты (1 моль/л) ТР раствором гидроокиси натрия, не содержа
220
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
щим карбонатов, используя в качестве индикатора раствор фенолфталеина в этаноле ИР. К концу титрования прибавляют кислоту в количестве, достаточном для исчезновения розовой окраски, и кипятят до уменьшения объема до 20 мл. Во время кипячения вновь прибавляют кислоту, чтобы удалить розовую окраску и предупредить ее появление при дальнейшем кипячении; требуется не более 0,1 мл кислоты.
Метод стандартизации. Устанавливают точную концентрацию 1 моль/л раствора следующим образом: высушивают около 5 г гидрофталата калия Р при 105 °C в течение 3 ч и точно взвешивают. Если кристаллы гидрофталата калия большие, их следует перед высушиванием измельчить. Растворяют в 75 мл воды, не содержащей углекислоты, Р и титруют раствором гидроокиси натрия, не содержащим карбонатов, используя в качестве индикатора раствор фенолфталеина в этаноле ИР. Каждые 0,2042 г гидрофталата калия соответствуют 1 мл раствора гидроокиси натрия (1 моль/л), не содержащего карбонатов, ТР. Стандартные растворы гидроокиси натрия следует часто повторно стандартизовать.
Хранение. Растворы едких щелочей поглощают углекислоту воздуха, поэтому их следует хранить в сосудах с хорошо закрывающейся нестеклянной пробкой с трубкой, заполненной натронной известью Р.
Натрия гидроокиси раствор (0,5 моль/л), не содержащий карбонатов, ТР. Гидроокись натрия Р, растворенная в воде до содержания 20,00 г NaOH в 1000 мл.
Методика приготовления, испытание на карбонаты и метод стандартизации. Готовят раствор, проводят испытание и устанавливают точную концентрацию методами, описанными для раствора гидроокиси натрия (1 моль/л), не содержащего карбонатов, ТР.
Натрия гидроокиси раствор (0,2 моль/л), не содержащий карбонатов, ТР. Гидроокись натрия Р, растворенная в воде до содержания 8,001 г NaOH в 1000 мл.
Методика приготовления, испытание на карбонаты и метод стандартизации. Готовят раствор, проводят испытание и устанавливают точную концентрацию методами, описанными для раствора гидроокиси натрия (1 моль/л), не содержащего карбонатов, ТР.
Натрия гидроокиси раствор (0,1 моль/л), не содержащий карбонатов, ТР. Гидроокись натрия, растворенная в воде до содержания 4,001 г NaOH в 1000 мл.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
221
Методика приготовления, испытание на карбонаты и метод стандартизации. Готовят раствор, проводят испытание и устанавливают точную концентрацию методами, описанными для раствора гидроокиси натрия (1 моль/л), не содержащего карбонатов, ТР.
Натрия гидроокиси раствор (0,02 моль/л) не содержащий карбонатов, ТР. Гидроокись натрия Р, растворенная в воде до содержания 0,8001 г NaOH в 1000 мл.
Методика приготовления, испытание на карбонаты и метод стандартизации. Готовят раствор, проводят испытание и устанавливают точную концентрацию методами, описанными для раствора гидроокиси натрия (1 моль/л), не содержащего карбонатов, ТР.
Натрия гидроокиси раствор (0,01 моль/л), не содержащий карбонатов, ТР. Гидроокись натрия Р, растворенная в воде до содержания 0,4001 г NaOH в 1000 мл.
Методика приготовления, испытание на карбонаты и метод стандартизации. Готовят раствор, проводят испытание и устанавливают точную концентрацию методами, описанными для раствора гидроокиси натрия (1 моль/л), не содержащего карбонатов, ТР.
Натрия карбонат Р. Na2CO3-10H2O (SRIP, 1963, с. 179).
Натрия карбонат безводный Р. Na2CO3 (SRIP, 1963, с. 179).
Натрия карбоната раствор (50 г/л) ИР. Раствор карбоната натрия Р, содержащий Na2CO3 около 50 г/л (приблизительно 0,5 моль/л).
Натрия карбоната стандартный раствор ИР.
Методика приготовления. Растворяют 2,64 г карбоната натрия Р и 2,093 г бикарбоната натрия Р в воде, свободной от углекислоты, Р до получения 100 мл.
Натрия кобальтнитрит Р. Na3CO(NO2)6 (SRIP, 1963, с. 182).
Натрия кобальтнитрита раствор (100 г/л) ИР. Раствор ко-бальтнитрита натрия Р, содержащий Na3CO(NO2)6 около 100 г/л.
Натрия меркаптоацетат (натрия тиогликолят) Р. C2H3NaO2S. Описание. Гигроскопичные кристаллы.
Растворимость. Легко растворим в воде; мало растворим в этаноле (~ 750 r/л) ИР.
Натрия нитрит Р. NaNO2 (SRIP, 1963, с 189).
Натрия нитрита раствор (10 г/л) ИР. Раствор нитрита натрия Р, содержащий NaNO2 около 10 г/л.
222
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Натрия нитрита раствор (0,1 моль/л) ТР. Нитрит натрия Р, растворенный в воде до содержания 6,900 г NaNO2 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,1 моль/л раствора следующим образом: помещают 50,0 мл раствора перманганата калия (0,02 моль/л) ТР в колбу с притертой пробкой, разводят 300 мл воды, прибавляют 25 мл серной кислоты (— 100 г/л) ИР и 20,0 мл раствора нитрита натрия. Оставляют раствор на 10 мин. Затем прибавляют 2 г йодида калия Р и титруют раствором тиосульфата натрия (0,1 моль/л) ТР, используя в качестве индикатора раствор крахмала ИР. Проводят контрольное титрование и вносят необходимые поправки.
Натрия оксалат Р. C2Na2O4 (SRIP, 1963, с. 190).
Натрия сульфат безводный Р. iNa2SO4 (SRIP, 1963, с. 195).
Натрия сульфид Р. Na2S-9H2O (SRIP, 1963, с. 195).
Натрия сульфида раствор ИР.
Методика приготовления. Растворяют 12 г сульфида натрия Р в 25 мл воды и доводят глицерином Р до получения 100 мл.
Натрия-сурьмы тартрат Р. C4H4NaO7Sb.
Описание. Гигроскопичные, прозрачные или беловатые чешуйки или порошок.
Растворимость. Растворим в 1,5 части воды; практически нерастворим в этаноле ( — 710 г/л) ИР.
Натрия-сурьмы тартрата раствор (50 г/л) ИР. Раствор тартрата натрия-сурьмы Р, содержащий C4H4NaO7Sb около 50 г/л.
Натрия тетраборат Р. Бура ,Na2B4O7- ЮН2О.
Описание. Прозрачные, бесцветные кристаллы или белый кристаллический порошок; без запаха.
Растворимость. Растворим в 20 частях воды и в 0,6 части кипящей воды; очень легко растворим в этаноле (-750 г/л) ИР.
pH 0,01 моль! л раствора. Растворяют 0,3814 г в воде и разводят до 100 мл, используя воду с pH 6,5—7,4. Величина pH должна быть 9,15—9,20 при 25 °C.
Хлориды. Растворяют 1,0 г в 20 мл воды, фильтруют, если необходимо, через фильтр, не содержащий хлоридов, прибавляют 1 мл азотной кислоты ( — 1000 г/л) ИР и далее поступают, как описано в разделе «Испытание на хлориды» (см. с. 132). Тетраборат натрия Р содержит хлориды не более 250 мкг/г.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ 223
Сульфаты. Растворяют 0,5 г в 20 мл воды, прибавляют 2 мл соляной кислоты (~70 г/л) ИР и фильтруют. Далее поступают, как описано в разделе «Испытание на сульфаты» (см. с. 133). Тетраборат натрия Р содержит сульфаты не более 1,0 мг/г.
Натрия тетрабората раствор стандартный ИР.
Методика приготовления. Растворяют 3,81 г тетрабората натрия Р в воде, не содержащей углекислоты, Р до получения 1000 мл.
Хранение. Хранят раствор, защищенным от углекислоты воздуха, всегда, кроме времени использования, в закрытом виде.
Натрия тетрафенилборат Р. C24H2oBNa.
^Описание. Пушистый, белый или почти белый порошок. Растворимость. Легко растворим в воде и ацетоне Р; нерастворим в петролейном эфире Р.
pH. pH 20 г/л раствора не менее 7,5.
Натрия тетрафенилбората раствор (30 г/л) ИР. Раствор тет-рафенилбората натрия Р, содержащий C24H20BNa около 30 г/л.
Примечание. Если необходимо, растворяют в течение 5 мин с 1 г окиси алюминия Р или с древесным углем Р и фильтруют для осветления.
Натрия тиосульфат Р. Na2S2O3-5H2O (SRIP, 1963, с. 197).
Натрия тиосульфата раствор (0,1 моль/л) ТР. Тиосульфат натрия Р, растворенный в воде до содержания 15,82 г Na2S2O3 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,1 моль/л раствора следующим образом: помещают 30,0 мл раствора бихромата калия (0,0167 моль/л) ТР в склянку с притертой пробкой и разводят 50 мл воды. Прибавляют 2 г йодида калия Р и 5 мл соляной кислоты (~250 г/л) ИР, закрывают и оставляют стоять на 10 мин. Разводят 100 мл воды и титруют выделившийся йод раствором тиосульфата натрия, используя в качестве индикатора раствор крахмала ИР. Растворы тиосульфата натрия следует часто повторно стандартизовать.
Натрия тиосульфата раствор (0,05 моль/л) ТР. Тиосульфат натрия Р, растворенный в воде до содержания 7,910 г Na2S2O3 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора тиосульфата натрия (0,1 моль/л) ТР.
224
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Натрия тиосульфата раствор (0,01 моль/л) ТР. Тиосульфат натрия Р, растворенный в воде до содержания 1,582 г Na2S2O3 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для раствора тиосульфата натрия (0,1 моль/л) ТР.
Натрия фторид Р. NaF (SRIP, 1963, с. 183).
Натрия хлорид Р. NaCl (SRIP, 1963, с. 181).
Натрия цитрат Р. C6H5Na3O7-2H2O.
Содержит не менее 99,0% C6H5Na3O7 в пересчете на безводное вещество.
Описание. Белые гранулированные кристаллы или кристаллический порошок; без запаха. Слегка расплывается на влажном воздухе.
Растворимость. Растворим менее чем в 2 частях воды, практически нерастворим в этаноле (~750 г/л) ТР.
Внешний вид раствора. Раствор 100 г/л прозрачный и бесцветный.
Вода. При определении методом К. Фишера после контакта вещества с безводным метанолом Р в течение 15 мин не менее ПО и не более 130 мг/г.
Количественное определение. Растворяют около 0,15 г (точная навеска) в 20 мл ледяной уксусной кислоты Р и титруют хлорной кислотой (0,1 моль/л) ТР, как описано в разделе «Неводное титрование», метод А (см. с. 151). Каждый миллилитр хлорной кислоты (0,1 моль/л) ТР соответствует 8,603 мг C6H5Na3O7.
Натронная известь Р. (SRIP, 1963, с. 176).
2-Нафтол Р. [р-нафтол Р]. С10Н8О (SRIP, 1963, с. 122).
2-Нафтола раствор ИР1.
Методика приготовления. Растворяют 5 г свежеперекри-сталлизованного 2-нафтола Р в 40 мл раствора гидроокиси натрия (~80 г/л) ИР и прибавляют воды до 100 мл. Примечание. 2-Нафтола раствор ИР1 должен быть свежеприготовленным.
Нитробензол Р. C6H5NO2 (SRIP, 1963, с. 128).
Олова хлорид Р. SnCl2-2H2O (SRIP, 1963, с. 198).
Олова хлорида раствор ИР.
Методика приготовления. Растворяют 330 г хлорида олова Р в 100 мл соляной кислоты (-—250 г/л) ИР и доводят водой до получения 1000 мл.
Олова хлорида раствор для испытания на мышьяк AsHP.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
225
Методика приготовления. Готовят из раствора хлорида олова Р прибавлением равного объема соляной кислоты (~250 г/л) ИР, кипятят до начального объема и фильтруют через тонкий фильтр.
Испытание на мышьяк. К 10 мл прибавляют 6 мл воды и 10 мл соляной кислоты (~250 г/л) AsHP и отгоняют 16 мл. К отгону прибавляют 50 мл воды и 2 капли раствора хлорида олова AsHP; далее проводят испытание на мышьяк. Окраска полученного пятна не должна быть интенсивнее, чем окраска пятна, полученного с 1 мл стандартного раствора, что указывает на содержание мышьяка не более 1 мкг/мл.
Панкреатический гидролизат казеина Р (SRIP, 1963, с. 132). Папаиновый гидролизат соевой муки Р. (S'RIP, 1963, с. 134).
Парафин жидкий Р. (SRIP, 1963, с. 135).
Пенициллиназа Р. Фермент, обычно получаемый из культурального фильтрата штамма Bacillus cereus; этот фермент обладает специфическим свойством инактивировать пенициллин путем расщепления связи азота тиазолидинового кольца с ближайшим карбонильным углеродом с высвобождением карбонильной группы. Осаждается из водных растворов ацетоном Р, этанолом (~750 г/л) ИР и диоксаном Р, но через несколько часов контакта с этими растворителями инактивируется; быстро инактивируется этилацетатом Р. Вместо пенициллиназы Р можно непосредственно использовать стерильный фильтрат, полученный ферментацией продуцирующего пенициллиназу организма в подходящей среде, как описано ниже («Приготовление пенициллиназы»).
Описание. Небольшие, коричневые, легко измельчаемые кусочки или гранулы.
Растворимость. Легко растворима в воде с образованием слегка опалесцирующего раствора.
Приготовление пенициллиназы. Растворяют 10 г панкреатического гидролизата казеина Р, 2,7 г дигидрофосфата калия Р и 5,9 г цитрата натрия Р в 200 мл воды, доводят щелочность до pH 7,2 раствором гидроокиси натрия 200 г/л) ИР и разводят водой до 1000 мл. Растворяют <3,4 г сульфата магния Р в 5 мл воды, прибавляют 1 мл раствора сульфата аммония железа (II) (~1 г/л) ИР и воды до 10 мл. Стерилизуют оба раствора нагреванием в автоклаве, охлаждают, перемешивают, распределяют тонким слоем в конических колбах и засеивают подходящим штаммом (пригоден штамм Bacillus cereus NCTC
226
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
9946). Оставляют колбы стоять при 18—37 °C до появления-роста и затем выдерживают при 35—37 °C в течение 16 ч„ постоянно встряхивая для максимальной аэрации. Центрифугируют и стерилизуют надосадочную жидкость путем фильтрации через подходящий мембранный фильтр.
Пенициллиназы раствор ИР. Стерильный водный раствор пенициллиназы Р.
Для проверки активности раствора пенициллиназы ИР проводят «Количественное определение пенициллиназы». Для обесцвечивания йода требуется не более 36 с.
Количественное определение пенициллиназы. Определение проводят в пробирках из боросиликатного стекла длиной. 15 см и внутренним диаметром около 20 мм, погруженных, в водяную баню при 30± 1 °C. Все реактивы перед применением должны иметь температуру 30 °C.
В пробирки вносят реактивы в следующем порядке: 1,6 мл раствора желатина ИР, 0,4 мл подлежащего испытанию' раствора пенициллиназы ИР, 1 каплю раствора крахмала ИР и 1 мл раствора бензилпенициллина натрия ИР, выдувая последний реактив из пипетки емкостью 1 мл. Включают секундомер и через 15 с прибавляют 2,0 мл раствора йода (0,01 моль/л) ТР, отмечая время обесцвечивания йода с момента прибавления раствора бензилпенициллина натрия ИР. Из результатов определения вычисляют активность раствора пенициллиназы ИР. Время обесцвечивания точно за 36 с соответствует активности пенициллиназы, эквивалентной скорости разрушения (при 30°C и pH 7,0) 220 мг бензилпенициллина натрия Р в час на 1 мл раствора пенициллиназы ИР.
Хранение. Хранят при температуре 0—2 °C и применяют в течение 2—3 сут. В лиофилизированном виде пенициллиназа ИР может храниться в запаянных ампулах в течение нескольких месяцев.
Пептон сухой Р. (SRIP, 1963, с. 137)
Пептон Р1. Сухой пептон Р, который выдерживает следующее требование: автоклавированный раствор, содержащий пептона 0,02 г/мл, должен быть прозрачным и нейтральным или почти нейтральным.
Пептона раствор (5 г/л) ИР.
Методика приготовления. Растворяют в воде при нагревании 5,0 г сухого пептона Р и 7 г хлорида натрия Р и разводят водой до получения 1000 мл. Доводят pH до 8,0— 8,4 и кипятят в течение 20 мин. Фильтруют, доводят pH до 7,2—7,4 и стерилизуют, выдерживая при 115°С в течение 30 мин.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
227
Пептона раствор (1 г/л) ИР1.
Методика приготовления. Растворяют 1,0 г пептона Р1 (или аналогичного гидролизата тканей животного) в воде до получения 1000 мл, фильтруют или центрифугируют для осветления, доводят pH до 7,1 ±0,2, помещают отдельные порции по 100 мл в сосуды и стерилизуют, выдерживая при 121 °C в течение 18—20 мин.
Пептона раствор (1 Г/л) ИР2.
Методика приготовления. Растворяют в воде при нагревании 1,0 г сухого пептона Р и 9 г хлорида натрия Р и доводят водой до получения 1000 мл. Доводят pH до 8,0— 8,4 и кипятят в течение 20 мин. Фильтруют, доводят pH до 7,2—7,4 и стерилизуют, выдерживая при 115 °C в течение 30 мин.
Перекись водорода (60 г/л) ИР. Раствор в воде, содержащий около 60 г Н2О2 в литре.
Петролейный эфир Р [эфир петролейный Р]. (SRIP, 1963, с. 108).
Пиридин Р. C5H5N (SRIP, 1963, с. 169).
Пиридин безводный Р. Пиридин, высушенный путем выдерживания над гидроокисью натрия Р.
Полисорбат-80 Р. Моносложный эфир олеиновой кислоты и триполиэтиленгликоль 300-сорбитанового простого эфира. Описание. Маслянистая жидкость от лимонного до янтарного цвета.
Смешиваемость. Смешивается с водой с образованием почти бесцветного раствора без запаха. Смешивается с этанолом (~750 г/л) ИР, этилацетатом Р и растительными маслами; смешивается с минеральными маслами.
Протравной черный ПР [эриохром черный Р].
C.I. Протравной черный 11, C.I. № 14645, Эриохром черный Т, Солохром черный; натриевая соль 2-(2-окси-6-нит-ро-4-сульфо-1-нафтилазо) -1-нафтола C20Hi2N3NaO7S (SRIP, 1963, с. 84).
Протравного черного 11 индикаторная смесь Р.
Методика приготовления. Смешивают 1 г протравного черного 11 со 100 г хлорида натрия Р.
Резазурин натрий Р. Ci2H6NNaO4 (SRIP, 1963, с. 170).
Резазурина натрия раствор (1 г/л) ИР. Раствор резазурина натрия Р, содержащий Ci2H6NNaO4 около 1 г/л.
Примечание. Резазурина натрия раствор (1 г/л) ИР должен быть свежеприготовленным.
228
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Резорцин Р. 1,3-Диоксибензол C6H6C>2 (SRIP, 1963, с. 171).
Резорцина раствор (20 г/л) ИР. Раствор резорцина Р, содержащий С6Н6О2 20 г/л.
Ртути ацетат Р. C4H6HgO4 (SRIP, 1963, с. 112).
Ртути ацетата раствор в уксусной кислоте ИР.
Методика приготовления. Растворяют 50 г ацетата ртути Р в ледяной уксусной кислоте Р1, нейтрализованной, если необходимо, по раствору индикатора кристаллического фиолетового в уксусной кислоте ИР хлорной кислотой (0,1 моль/л) ТР, до получения 1000 мл.
Ртути бромид Р. HgBr2 (SRIP, 1963, с. 113).
Ртути бромида раствор для испытания на мышьяк AsHP.
Методика приготовления. Растворяют 5 г бромида ртути Р в этаноле (~750 г/л) ИР до получения 100 мл.
Ртуть-бромидная бумага для испытания на мышьяк AsP.
Методика приготовления. Используют мягкую белую фильтровальную бумагу, весящую 65—-120 г/м2. Толщина бумаги в миллиметрах должна быть приблизительно равна указанной выше массе, деленной на 400. Погружают куски фильтровальной бумаги шириной не менее 25 мм в раствор бромида ртути AsHP, декантируют оставшуюся жидкость, помещают бумагу на неметаллический поднос и дают высохнуть, защищая от действия света.
Хранение. Ртуть-бромидную бумагу AsP хранят в закрытых склянках в темном месте.
Примечание. Бумага, подвергшаяся действию света или паров аммиака, не должна применяться, так как на ней либо образуются только светлые пятна либо пятна не появляются совсем.
Ртути нитрат Р. Hg(NO3)2-H2O.
Предостережение. Ртути нитрат Р ядовит.
Описание. Белый или слегка желтоватый расплывающийся кристаллический порошок.
Растворимость. Растворим в воде в присутствии небольшого количества азотной кислоты (~1000 г/л) ИР.
Ртути нитрата раствор (0,01 моль/л) ТР.
Методика приготовления. Растворяют около 3,5 г (точная навеска) нитрата ртути Р в смеси 5 мл азотной кислоты ( — 1000 г/л) ИР и 500 мл воды и доводят водой до 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,01 моль/л раствора следующим образом: помещают 20,0 мл раствора в коническую колбу, прибавляют 2 мл
стандартные образцы и реактивы 229
азотной кислоты (~1000 г/л) ИР и 2 мл сульфата аммония железа (III) (45 г/л) ИР. Охлаждают до температуры ниже 20 °C и титруют тиоцианатом аммония (0,01 моль/л) ТР до первого появления устойчивого коричневого окрашивания.
Ртути окись желтая Р. HgO (SRIP, 1963, с. 114).
Ртути сульфата раствор ИР.
Методика приготовления. Смешивают 5 г желтой окиси ртути Р с 40 мл воды и при перемешивании прибавляют 20 мл серной кислоты (~1760 г/л) ИР; затем прибавляют 40 мл воды и перемешивают до полного растворения.
Свинца ацетат Р. С4Н6О4РЬ• ЗН2О (SRIP, 1963, с. 105).
Свинца ацетата раствор (80 г/л) ИР. Раствор ацетата свинца Р в свежепрокипяченной воде, содержащий С4Н6О4РЬ около 80 г/л (приблизительно 0,25 моль/л).
Свинца нитрат Р. Pb(NO3)2 (SRIP, 1963, с. 107).
Свинца нитрата раствор (0,05 моль/л) ТР. Нитрат свинца Р, растворенный в воде до содержания 16,56 г Pb(NO3)2 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,05 моль/л раствора путем разведения 25,0 мл 200 мл воды, прибавляют 10 мл аммиачного буфера ИР и около 20 мг индикаторной смеси протравного черного ПР. Титруют раствором эдетата динатрия (0,05 моль/л) ТР.
Свинца раствор концентрированный для испытания на тяжелые металлы РЬИР. 1 мл раствора содержит 100 мкг свинца.
Методика приготовления. Растворяют 0,1598 г нитрата свинца Р в 5 мл азотной кислоты (~ 1000 г/л) ИР и доводят водой до 1000 мл.
Свинца раствор разведенный для испытания на тяжелые металлы РЬИР. 1 мл раствора содержит 10 мкг свинца.
Методика приготовления. Разводят 10 мл концентрированного раствора свинца РЬИР водой до получения 100 мл. Примечание. Свинца раствор разведенный РЬИР должен быть свежеприготовленным.
Селен Р. Se (SRIP, 1963, с. 172).
Предупреждение. Пары селена токсичны.
Серебра нитрат Р. AgNO3 (SRIP, 1963, с. 173).
Серебра нитрата раствор (40 г/л) ИР. Раствор нитрата серебра Р, содержащий AgNO3 около 42,5 г/л (приблизительно 0,25 моль/л).
16-2025
230
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Серебра нитрата раствор (0,1 моль/л) ТР. Нитрат серебра Р, растворенный в воде до содержания 16,99 г AgNO3 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,1 моль/л раствора следующим образом: разводят 40,0 мл раствора нитрата серебра 100 мл воды. Нагревают раствор и медленно прибавляют при постоянном перемешивании соляную кислоту ( — 70 г/л) ИР до полного осаждения серебра. Осторожно кипятят смесь около 5 мин, затем оставляют стоять в темноте до осаждения осадка и осветления надосадочной жидкости. Полностью переносят осадок во взвешенный стеклянный фильтр и промывают небольшими порциями воды, слегка подкисленной азотной кислотой ( — 1000 г/л) ИР. Высушивают осадок до постоянной массы при НО °C. Из массы хлорида серебра рассчитывают концентрацию раствора нитрата серебра в моль/л. Во время определения хлорид серебра, насколько это возможно, защищают от действия света.
Серная кислота (—1760 г/л) ИР [серная кислота Р]. (SRIP, 1963, с. 202); 1,84.
Серная кислота ( — 1760 г/л), не содержащая соединений азота, ИР. Серная кислота ( — 1760 г/л) ИР, содержащая H2SO4 не менее 1760 /л и выдерживающая испытание на нитраты.
Нитраты. Смешивают 45 мл с 5 мл воды, охлаждают и прибавляют 8 мг дифенилбензидина Р; раствор должен быть бесцветным или совсем бледно-голубым.
Серная кислота ( — 190 г/л) ИР.
Методика приготовления. Смешивают 1 объем серной кислоты ( — 1760 г/л) ИР с 9 объемами воды и охлаждают. Полученный раствор содержит H2SO4 около 190 г/л; d~ 1,12.
Серная кислота ( — 100 г/л) ИР.
Методика приготовления. Прибавляют 57 мл серной кислоты ( — 1760 г/л) ИР к воде до получения 1000 мл (приблизительно 1 моль/л); d—1,065.
Серная кислота ( — 10 г/л) ИР.
Методика приготовления. Смешивают 100 мл серной кислоты ( — 1760 г/л) ИР с водой до получения 1000 мл.
Серная кислота (0,5 моль/л) ТР. Серная кислота ( — 1760 г/л) ИР, разведенная водой до содержания 49,04 г H2SO4 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,5 моль/л раствора следующим образом: растворяют
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
231
около 1,5 г (точная навеска) безводного карбоната натрия Р, предварительно высушенного при 270 °C в течение 1 ч, в 50 мл воды и титруют раствором серной кислоты, используя в качестве индикатора раствор метилового оранжевого в этаноле ИР. Каждые 52,99 г безводного карбоната натрия соответствуют 1 мл серной кислоты
(0,5 моль/л) ТР.
Серная кислота (0,05 моль/л) ТР. Серная кислота
( — 1760 г/л) ИР, разведенная водой до содержания 4,904 г H2SO4 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для серной кислоты (0,5 моль/л) ТР.
Серная кислота (0,01 моль/л) ТР. Серная кислота (—1760 г/л) ИР, разведенная водой до содержания 0,9808 г. H2SO4 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для серной кислоты (0,5 моль/л) ТР.
Серная кислота (0,005 моль/л) ТР. Серная кислота (—1760 г/л) ИР, разведенная водой до содержания 0,4904 r-H2SO4 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для серной кислоты (0,5 моль/л) ТР.
Сероводород Р. H2S (SRIP, 1963, с. 98).
Сероводорода раствор ИР. Насыщенный раствор сероводорода Р в холодной воде.
Примечание. Сероводорода раствор ИР должен быть свежеприготовленным.
Сероуглерод Р. CS2 (SRIP, 1963, с. 62).
Сероуглерод И К. Сероуглерод Р, который выдерживает следующее испытание: инфракрасный спектр реактива в слое толщиной 1,0 мм, как описано в методе 4 в разделе «Спектрофотометрия в инфракрасной области спектра» (см. с. 45), в области 4000—670 см-1 имеет поглощение менее 0,1 в областях 4000—3030 см-1, 2635—2440 см-1, 2000—1755 см-1 и 1265—935 см-1 и поглощение менее 0,17 в области 800—715 см-1.
Серы двуокись Р. SO2 (SRIP, 1963, с. 202).
Силикагель-осушитель Р.
Описание. Аморфная, частично водная окись кремния SiO2 в виде стекловидных гранул различного размера. Часто
16
232
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
покрывается веществом, изменяющим окраску, когда способность поглощать влагу исчерпана. Такие окрашенные продукты могут быть регенерированы (т. е. могут восстановить способность поглощать воду) путем нагревания при 110 °C до восстановления начальной окраски.
Потери при прокаливании. Прокаливают 2 г (точная навеска) при 950+50 °C до постоянной массы; потеря составляет не более 60 мг/г.
Поглощение воды. Помещают около 10 г во взвешенный бюкс и взвешивают. Затем помещают бюкс со снятой крышкой на 24 ч закрытый сосуд, в котором поддерживается относительная влажность 80%, достигаемая с помощью серной кислоты, имеющей относительную плотность 1,19. Взвешивают снова; увеличение массы должно составлять не менее 310 мг/г.
Соляная кислота (~420г/л) ИР [соляная кислота насыщенная Р]. (SRIP, 1963, с. 96); d~ 1,18.
Соляная кислота (~250 г/л) ИР. Раствор соляной кислоты (~420 г/л) ИР в воде, содержащий НС1 приблизительно 250 г/л; d~ 1,12.
Соляная кислота (~70 г/л) ИР.
Методика приготовления. Разводят 260 мл соляной кислоты (~250 г/л) ИР водой до получения 1000 мл (приблизительно 2 моль/л); d~ 1,035.
Соляная кислота (2 моль/л) ТР. Соляная кислота (~250 г/л) ИР, разведенная водой до содержания 72,93 г НС1 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для соляной кислоты (1 моль/л) ТР.
Соляная кислота (1 моль/л) ТР. Соляная кислота (~250 г/л) ИР, разведенная водой до содержания 36,47 г НС1 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 1 моль/л раствора следующим образом: растворяют около 1,5 г (точная навеска) безводного карбоната натрия Р, предварительно высушенного при 270 °C в течение 1 ч, в 50 мл воды и титруют раствором соляной кислоты, используя в качестве индикатора раствор метилового оранжевого в этаноле ИР. Каждые 52,99 мг безводного карбоната натрия соответствуют 1 мл соляной кислоты (1 моль/л) ТР.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
233
Соляная кислота (0,5 моль/л) ТР. Соляная кислота (~250 г/л) ИР, разведенная водой до содержания 18,23 г НС1 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для соляной кислоты (1 моль/л) ТР.
Соляная кислота (0,1 моль/л) ТР. Соляная кислота (~250 г/л) ИР, разведенная водой до содержания 3,647 г НС1 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для соляной кислоты (1 моль/л) ТР.
Соляная кислота (0,015 моль/л) ТР. Соляная кислота (~250 г/л) ИР, разведенная водой до содержания 0,5470 г НС1 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для соляной кислоты (1 моль/л) ТР.
Соляная кислота (0,01 моль/л) ТР. Соляная кислота (~250 г/л) ИР, разведенная водой до содержания 0,3647 г НО в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию раствора методом, описанным для соляной кислоты (1 моль/л) ТР.
Соляная кислота (~250 г/л) для испытания на мышьяк AsHP. Соляная кислота (~250 г/л) ИР, которая выдерживает приведенные ниже испытания А и Б.
А. Разводят 10 мл водой до получения 50 мл, прибавляют 5 мл раствора тиоцианата аммония (75 г/л) ИР и немедленно перемешивают; раствор не окрашивается.
Б. К 50 мл прибавляют 0,2 мл раствора брома AsHP и выпаривают на водяной бане до 16 мл, добавляя раствор брома AsHP, если необходимо, для обеспечения избытка брома, о чем свидетельствует окраска раствора, сохраняющаяся все время выпаривания. Прибавляют 50 мл воды и 5 капель раствора хлорида олова AsHP и далее проводят общее испытание на мышьяк. Окраска полученного пятна не должна быть более интенсивной, чем окраска пятна, полученного с 0,2 мл стандартного раствора, что указывает на содержание мышьяка не более 0,05 мкг/мл.
Соляная кислота (~ 250 г/л) для испытания на мышьяк, содержащая олово, AsHP.
234
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Методика приготовления. Разводят 1 мл раствора хлорида олова AsHP соляной кислотой (~250 г/л) AsHP до получения 100 мл.
Соляная кислота для испытания на хлориды С1ИР. 1 мл содержит 50 мкг С1.
Методика приготовления. Разводят 14,3 мл соляной кислоты (0,1 моль/л) ТР водой до получения 1000 мл.
Сульфосалициловая кислота Р. C7H6O6S-2^0.
Описание. Белые или бледно-розовые игольчатые кристаллы.
Растворимость. Растворима в воде и этаноле (~750 г/л) ИР.
Нерастворимые вещества. Растворяют 5,0 г в 50 мл воды, нагревают до кипения и настаивают в закрытом стакане на водяной бане в течение 1 ч. Фильтруют через взвешенный стеклянный фильтр, тщательно промывают и высушивают при 105 °C. Масса остатка не должна превышать 1,0 мг.
Сульфатная зола. Слегка прокаливают 1,0 г во взвешенном тигле или чашке, не из платины, до обугливания. Охлаждают, смачивают остаток 1 мл серной кислоты , (~1760 г/л) ИР и прокаливают снова; остаток составляет
не более 1,0 мг/г.
Сульфосалициловой кислоты раствор (175 г/л) ИР. Раствор сульфосалициловой кислоты Р, содержащий C7H6O6S около 175 г/л.
п-Терпенйл Р. 1,4-Дифенилбензол Ci8Hi4. Применяется для сцинтилляционного счета.
Тимолфталеин Р. С28Н30О4 (SRIP, 1963, с. 207).
Тимолфталеина раствор в этаноле ИР.
Методика приготовления. Растворяют 0,1 г тимолфталеина Р в 100 мл этанола (~750 г/л) ИР и фильтруют, если необходимо.
Толуол Р.С7Н8 (SRIP, 1963, с. 209).
Торин Р. 2,7-Динатрий 4-[ (о-арсонофенил) азо]-3-окси-2,7-на-фталиндисульфонат С।AsN^Na2OioS2.
Торина раствор (2 г/л) ИР.
Методика приготовления. Растворяют 0,2 г торина Р в воде до получения 100 мл.
Хранение. Раствор хранят защищенным от действия света. Срок годности. Используют в течение 1 нед ссо дня изготовления.
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
235
Тория нитрат Р. ТЬ(МОз)4-4Н2О.
Описание. Белые, слегка расплывающиеся кристаллы.
Растворимость. Очень легко растворим в воде и этаноле ( — 750 г/л) ИР.
Тория нитрата раствор (0,005 моль/л) ТР. Тория нитрат Р, растворенный в воде до содержания 2,401 г Th(NO3)4 в 1000 мл.
Метод стандартизации. Устанавливают точную концентрацию 0,005 моль/л раствора следующим образом: помещают 0,050 г (точная навеска) фторида натрия Р, предварительно высушенного, в колбу и растворяют в воде до получения 250 мл. К 20,0 мл этого раствора прибавляют 0,6 мл раствора ализаринсульфоната натрия (1 г/л) ИР и затем по каплям раствор гидроокиси натрия (0,1 моль/л) ТР до изменения розовой окраски на желтую. Прибавляют 5 мл ацетатного буфера, pH 3,0, ИР и титруют раствором нитрата тория до изменения желтой окраски на розовато-желтую. Каждые 0,8398 г фторида натрия соответствуют 1 мл раствора нитрата тория (0,005 моль/л) ТР.
о-Фенантролин Р. 1,10-Фенантролин Ci2H8N2-H2O (SRIP, 1963, с. 138).
о-Фенантролина раствор (1 г/л) ИР.
Методика приготовления. Растворяют 0,11 г о-фенантро-лина Р в воде до получения 100 мл.
о-Фенантролина раствор ИР.
Методика приготовления. Растворяют 0,7 г сульфата железа (II) Р примерно в 70 мл воды, прибавляют около 1,5 г о-фенантролина Р и разводят водой до получения 100 мл.
Фенол Р. С6Н6О.
Описание. Бесцветные или бледно-розовые, сцепленные или отдельные игольчатые кристаллы или кристаллическая масса; запах характерный. Едкое вещество, вызывает ожоги кожи и слизистых оболочек.
Растворимость. Растворим примерно в 15 частях воды и примерно в 100 частях жидкого парафина Р; легко рас-створим в этаноле ( — 750 г/л) ИР, эфире Р и хлороформе Р.
Полнота растворения. 1,0 г растворяется полностью в 15 мл воды при 15 °C.
Температура затвердевания. Не ниже 40,5 °C.
Остаток после выпаривания. Выпаривают на водяной бане и высушивают до постоянной массы при 105 °C; остается не более 0,5 мг/г.
236 МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Феноловый красный Р. Фенолсульфонфталеин CigHuOgS (SRIP, 1963, с. 139).
Фенолового красного раствор в этаноле ИР.
Методика приготовления. Растворяют 0,05 г фенолового красного Р в смеси 2,85 мл раствора гидроокиси натрия (0,05 моль/л) ТР и 5 мл этанола (~710 г/л) ИР. Слегка нагревают раствор и после охлаждения разводят этанолом 150 г/л) ИР до получения 250 мл.
Фенолфталеин Р. С20Н14О4 (SRIP, 1963, с. 139).
Фенолфталеина раствор в этаноле ИР.
Методика приготовления. Растворяют 1,0 г фенолфталеина Р в этаноле (~750 г/л) ИР до получения 100 мл.
Фенолфталеина раствор в пиридине ИР.
Методика приготовления. Растворяют 1,0 г фенолфталеина Р в пиридине Р до получения 100 мл.
Фишера Карла (К. Фишера) реактив ИР. Свежеприготовленный раствор имеет титр до 5,0 мг/мл воды. Раствор не должен применяться, если титр по воде падает ниже 2,5 мг воды на 1 мл реактива.
Методика приготовления. Растворяют 63 г йода Р в 100 мл безводного пиридина Р, охлаждают во льду и пропускают через раствор двуокись серы Р до увеличения массы на 32 г, соблюдая меры предосторожности для предотвращения поглощения влаги воздуха. Добавляют безводный метанол Р до 500 мл и оставляют стоять на 24 ч. Реактив К. Фишера ИР можно также приготовить, смешав имеющиеся в продаже растворы двуокиси серы в пиридине и йода в метаноле, которые стабильны при правильном хранении, например при защите от действия света. Полученный раствор должен удовлетворять требованиям, приведенным ниже.
Метод стандартизации. Устанавливают точный титр по воде следующим образом: в сосуд для титрования прибавляют около 20 мл безводного метанола Р и титруют до конечной точки реактивом К. Фишера ИР, не отмечая объем, пошедший на титрование. Вносят в колбу в соответствующем виде точную навеску воды и снова титруют до конечной точки реактивом К. Фишера ИР, отмечая израсходованный объем. Воду можно внести, например, в виде раствора в сухом метаноле или в форме гидрата какого-либо соединения. Рассчитывают эквивалент воды в миллиграммах воды на 1 мл реактива. Реактив К. Фишера ИР постоянно разрушается, поэтому его следует стандартизовать непосредственно перед применением или ежедневно, как требуется.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
237
Примечание. Вместо безводного метанола Р при изготовлении реактива можно применять моноэтиловый эфир этиленгликоля Р.
Углекислый газ Р. СОг-
Описание. Бесцветный газ; без запаха.
Растворимость. Растворим примерно в 1,3 части воды.
Уксусная кислота ледяная Р. С2Н4О2 (SRIP, 1963, с. 25); rf-1,048.
Уксусная кислота ледяная Р1. Ледяная уксусная кислота Р, которая выдерживает следующее испытание: смешивают 10,0 г с 10 мл серной кислоты (—1760 г/л) ИР, охлаждают до 20 °C, прибавляют 1 мл раствора бихромата калия (0,0167 моль/л) ТР и оставляют стоять на 30 мин. Прибавляют 50 мл воды, охлаждают до 20°C, прибавляют 1,5 мл раствора йодида калия (80 г/л) ИР и титруют выделившийся йод раствором тиосульфата натрия (0,1 моль/л) ТР, используя в качестве индикатора раствор крахмала ИР. На титрование требуется не менее 0,6 мл раствора тиосульфата натрия (0,1 моль/л) ТР.
Уксусная кислота ( — 300 г/л) ИР. Раствор ледяной уксусной кислоты Р, содержащий С2Н4О2 около 300 г/л (приблизительно 5 моль/л); rf—1,037.
Уксусная кислота (—60 г/л) ИР. Уксусная кислота (300 г/л) ИР, разведенная до содержания С2Н4О2 около 60 г/л (приблизительно 1 моль/л); rf—1,008.
Уксусная кислота (—60 г/л) для испытания на свинец РЬИР. Уксусная кислота ( — 60 г/л) ИР, которая выдерживает следующее испытание: выпаривают 20 мл уксусной кислоты ( — 60 г/л) ИР почти досуха на водяной бане, прибавляют 25 мл воды и проводят испытание на тяжелые металлы. Предел содержания тяжелых металлов 3 мкг/мл.
Уксусный ангидрид Р. С4Н6О3 (SRIP, 1963, с. 26).
Уранила ацетат Р. C4H6O6U-2H2O (SRIP, 1963, с. 213).
Уранила-цинка ацетата раствор ИР.
Методика приготовления. Растворяют 10 г уранила ацетата Р при нагревании с 50 мл воды и 5 мл уксусной кислоты ( — 300 г/л) ИР; растворяют 30 г ацетата цинка Р при нагревании с 30 мл воды и 3 мл уксусной кислоты ( — 300 г/л) ИР. Смешивают два раствора, дают охладиться до комнатной температуры и удаляют путем фильтрации любое выделившееся твердое вещество.
238
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Фосфатный буферный раствор стерильный, pH 4,5, ИР.
Методика приготовления. Растворяют 13,6 г дигидрофосфата калия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120 °C и, если необходимо, доводят pH до 4,45—4,55 фосфорной кислотой ( — 1440 г/л) ИР или раствором гидроокиси калия (-110 г/л) ИР.
Фосфатный буферный раствор стерильный, pH 6,0, ИР1.
Методика приготовления. Растворяют 2,0 г гидрофосфата дикалия Р и 8,0 г дигидрофосфата калия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120°C и, если необходимо, доводят pH до 5,95—6,05 фосфорной кислотой ( — 1440 г/л) ИР или раствором гидроокиси калия ( — 110 г/л) ИР.
Фосфатный буферный раствор стерильный, pH 6,0, ИР2.
Методика приготовления. Растворяют 1,16 г безводного гидрофосфата динатрия Р и 7,96 г дигидрофосфата калия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120°C и, если необходимо, доводят pH до 5,95—6,05 фосфорной кислотой ( — 1440 г/л) ИР или раствором гидроокиси калия ( — 110 г/л) ИР.
Фосфатный буферный раствор стерильный, pH 6,0, ИРЗ.
Методика приготовления. Растворяют 20,0 г гидрофосфата дикалия Р и 80,0 г дигидрофосфата калия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120°C и, если необходимо, доводят pH до 5,95—6,05 фосфорной кислотой ( — 1440 г/л) ИР или раствором гидроокиси калия ( — 110 г/л) ИР.
Фосфатный буферный раствор стандартный, pH 6,8, ИР.
Методика приготовления. Растворяют 3,40 г дигидрофосфата калия Р и 3,53 г безводного гидрофосфата динатрия Р в воде, не содержащей углекислоты, Р до получения 1000 мл.
Фосфатный буферный раствор, pH 7,0, ИР.
Методика приготовления. Растворяют 5,76 г безводного гидрофосфата динатрия Р и 3,55 г дигидрофосфата калия Р в воде до получения 1000 мл.
Фосфатный буферный раствор стерильный, pH 7,0, ИР.
Методика приготовления. Растворяют 5,76 г безводного гидрофосфата динатрия Р и 3,55 г дигидрофосфата калия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120 °C и, если необходимо, доводят pH до 6,95—7,05 фосфорной кислотой
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
239
(~1440 г/л) ИР или раствором гидроокиси калия (~ 110 г/л) ИР.
Фосфатный буферный раствор стерильный, pH 7,2, ИР.
Методика приготовления. Растворяют 6,80 г дигидрофосфата калия Р и 1,4 г гидроокиси натрия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120 °C и, если необходимо, доводят pH до 7,1—7,3 фосфорной кислотой (~1440 г/л) ИР или раствором гидроокиси калия (~ 110 г/л) ИР.
Фосфатный буферный раствор стандартный, pH 7,4, ИР.
Методика приготовления. Растворяют 1,18 г дигидрофосфата калия Р и 4,30 г безводного гидрофосфата динатрия Р в воде, не содержащей углекислоты, Р до получения 1000 мл.
Фосфатный буферный раствор стерильный, pH 8,0, ИР1.
Методика приготовления. Растворяют 16,73 г гидрофосфата дикалия Р и 0,52 г гидрофосфата калия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120°C и, если необходимо, доводят pH до 7,9—8,1 фосфорной кислотой (~1440 г/л) ИР или раствором гидроокиси калия (~110 г/л) ИР.
Фосфатный буферный раствор стерильный, pH 8,0, ИР2.
Методика приготовления. Растворяют 8,95 г безводного гидрофосфата динатрия Р и 0,50 г дигидрофосфата калия Р в воде до получения 1000 мл. Стерилизуют раствор в течение 20 мин в автоклаве при 120°C и, если необходимо, доводят pH до 7,9—8,1 фосфорной кислотой (~1440 г/л) ИР или раствором гидроокиси калия (~ 110 г/л) ИР.
Фосфорная кислота (~1440 г/л) ИР [фосфорная кислота Р] (SRIP, 1963, с. 141); 1,7.
Фосфора пятиокись Р. Р2О5 (SRIP, 1963, с. 142).
Фталевый ангидрид Р. С8Н4О3.
Описание. Белые блестящие иглы.
Растворимость. Мало растворим в воде, лучше растворим в горячей воде; растворим в этаноле (~750 г/л) ИР и эфире Р.
Температура плавления. Около 130 °C.
Фталевого ангидрида раствор в пиридине ИР.
Методика приготовления. Прибавляют 42 г фталевого ангидрида Р (точная навеска) к 300 мл свежеперегнанного пиридина Р (нагретого с обратным холодильником с окисью бария Р), содержащего менее 1 мг/мл воды, в колбе емкостью 1000 мл с притертой пробкой. Используют
240
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
колбу из темного стекла или защищают раствор от действия света. Энергично встряхивают до полного растворения и оставляют стоять на ночь для завершения реакции.
Примечание. Фталевого ангидрида раствор в пиридине ИР должен быть свежеприготовленным.
Хлор Р. Cl2 (SRIP, 1963, с. 66).
Хлора раствор ИР. Насыщенный раствор хлора Р в воде. Примечание. Хлора раствор ИР должен быть свежеприготовленным.
Хлоралгидрат Р. С8НцС13О6.
Описание. Бесцветный кристаллический порошок. Температура плавления. Около 187 °C.
Удельное вращение. Используют 50 мг/мл раствор в этаноле ( — 750 г/л) ИР. [a]D20 оС= + 19°С.
Хлороформ Р. СНС13 (SRIP, 1963, с. 66).
Хлорная кислота ( — 1170 г/л) ИР [хлорная кислота
(70% м/м) Р]. (SRIP, 1963, с. 137); d-1,67.
Хлорная кислота (—140 г/л) ИР. Хлорная кислота
( — 1170 г/л) ИР, разведенная водой до содержания НС1О4 141 г/л; d—1,09.
Хлорная кислота (0,1 моль/л) ТР.
Методика приготовления. К 900 мл ледяной уксусной кислоты Р1 примерно при 25 °C прибавляют 8,2 мл хлорной кислоты ( — 1170 г/л) ИР, перемешивают, прибавляют 32 мл уксусного ангидрида Р и снова перемешивают. Охлаждают до комнатной температуры, прибавляют ледяную уксусную кислоту Р1 до получения 1000 мл и оставляют на 24 ч.
Вода. Определяют содержание воды методом К- Фишера. Если необходимо, прибавляют воду или уксусный ангидрид Р в количестве, достаточном для того, чтобы содержание воды составило 0,1—2,0 мг/мл, и оставляют стоять еще на 24 ч.
Метод стандартизации. Устанавливают точную концентрацию титрованием 0,5 г (точная навеска) гидрофталата калия Р, предварительно высушенного при 120 °C в течение 2 ч, используя прямой метод титрования по К- Фишеру (метод А, см. с. 155). Каждый миллилитр раствора хлорной кислоты (0,1 моль/л) ТР соответствует 20,42 мг С8Н5КО4. Отмечают температуру, при которой проводят стандартизацию.
Церия-аммония нитрат Р. Ce(NO3)4-2NH4NO3.
СТАНДАРТНЫЕ ОБРАЗЦЫ И РЕАКТИВЫ
241
Описание. Небольшие оранжево-красные моноклинические кристаллы.
Растворимость. Очень легко растворим в воде.
Нерастворимые вещества. К 5 г (точная навеска) прибавляют 10 мл серной кислоты ( — 1760 г/л) ИР, перемешивают и осторожно прибавляют 90 мл воды для растворения. Нагревают до кипения и настаивают на водяной бане в закрытом стакане в течение 1 ч. Фильтруют через предварительно взвешенный стеклянный фильтр, тщательно промывают и высушивают при 105 °C. Масса остатка не должна превышать 2,5 мг.
Количественное определение. Растворяют 2,5 г (точная навеска) нитрата церия-аммония, предварительно высушенного при 85 °C в течение 24 ч, в 10 мл серной кислоты ('—190 г/л) ИР и прибавляют 40 мл воды. Прибавляют несколько капель раствора о-фенантролина ИР и титруют раствором сульфата железа (II) (0,1 моль/л) ТР. Каждый миллилитр раствора сульфата железа (II) (0,1 моль/л) ТР соответствует 54,8 мг Се(ЫОз)4-2ЫН4ЫО3.
Церия-аммония нитрата раствор ИР.
Методика приготовления. Растворяют 6,25 г нитрата церия-аммония Р в 10 мл азотной кислоты (15 г/л) ИР.
Срок годности. Используют в течение 3 дней с момента приготовления.
Церия сульфат Р. Обычно Ce(SO4)2-4H2O (SRIP, 1963, с. 63).
Церия сульфата раствор (35 г/л) ИР. Раствор сульфата церия Р, содержащий Ce(SO4)2 около 33 г.
Циклогексан Р. C6Hi2 (SRIP, 1963, с. 74).
Цинк Р. Zn (SRIP, 1963, с. 216); гранулированный порошок или пыль.
Цинк гранулированный для испытаний на мышьяк AsHP. Гранулированный цинк Р, который выдерживает следующие испытания.
Содержание мышьяка. Прибавляют 10 мл раствора соляной кислоты ( — 250 г/л), содержащей олово, AsHP к 50 мл воды и далее проводят общее испытание на мышьяк; используют 10 г гранулированного цинка Р и дают реакции протекать в течение 1 ч; не должно появляться видимое пятно.
Испытание на чувствительность. Повторяют испытание на мышьяк с добавлением 0,1 мл разведенного раствора мышьяка AsHP; образуется бледное, но отчетливое желтое пятно.
242
МЕЖДУНАРОДНАЯ ФАРМАКОПЕЯ
Цинка ацетат Р. C4H6O4Zn-2H2O (SRIP, 1963, с. 216).
L-Цистин Р. C6Hi2N2O4S2 (SRIP, 1963, с. 75).
Этанол безводный Р. С2Н5ОН (SRIP, 1963, с. 85).
Этанол ( — 750 г/л) ИР [этанол (95%) Р] (SRIP, 1963, с. 84).
Этанол ( — 750 г/л), не содержащий сульфатов, ИР. Этанол (—750 г/л) ИР, выдерживающий следующее испытание: выпаривают 25 мл этанола ( — 750 г/л) ИР до объема около 2 мл, прибавляют смесь 3 мл соляной кислоты ( — 70 г/л) ИР и 42 мл воды и 5 мл суспензии сульфата бария ИР. Далее поступают, как описано в разделе «Испытание на сульфаты» (см. с. 133). Этанол ( — 750 г/л), не содержащий сульфатов, ИР содержит сульфаты не более 20 мкг/мл.
Этанол ( — 710 г/л) ИР. Раствор около 950 мл этанола ( — 750 г/л) ИР, разведенный водой до 1000 мл.
Этанол ( — 375 г/л) ИР. Раствор около 525 мл этанола ( — 750 г/л) ИР, разведенный водой до 1000 мл.
Этанол ( — 150 г/л) ИР. Раствор около 210 мл этанола ( — 750 г/л) ИР, разведенный водой до 1000 мл.
Этилацетат Р. С4Н8О2 (SRIP, 1963, с. 86).
Этиленгликоля моноэтиловый эфир Р. С4НюО2.
Описание. Прозрачная, бесцветная жидкость.
Смешиваемость. Смешивается с водой, этанолом (—750 г/л) ИР, эфиром Р и ацетоном Р.
Температурный интервал кипения. Не менее 95% перегоняется между 133 и 135 °C.
Плотность (р2о)- Около 0,93 кг/л.
Эфир Р. С4Н10О (SRIP, 1963, с. 85).
Перевод с английского А. П. АРЗАМАСЦЕВА
Ответственная за редактирование Н. А. КОЛЧИНСЦАЯ
Заказ. 2025.
Московская типография № 11 Союзполиграфпрома при Государственном комитете CGCP по делам издательств, полиграфии и книжной торговли. Москва, 1113105, Нагатинская ул., д. 1.